Alteraciones epigenéticas en la leucemia linfoblástica aguda del
Anuncio

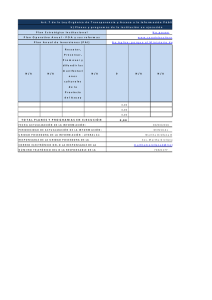
Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. Alteraciones epigenéticas en la leucemia linfoblástica aguda del adulto José Román-Gómeza, Antonio Jiménez-Velascob, Antonio Torresa, Felipe Prosperc, Anabel Heinigerb y Xabier Agirrec a Servicio de Hematología. Hospital Reina Sofía. Córdoba. Servicio de Hematología. Hospital Carlos Haya. Málaga. c Servicio de Hematología. Área de Terapia Celular. Clínica Universitaria de Navarra. Pamplona. Navarra. España. b El sello característico de la leucemia linfoblástica aguda (LLA) es la aparición progresiva de una conducta celular maligna generada por una función alterada de los genes. Aunque la LLA tradicionalmente se ha considerado como una enfermedad de base genética, es cada vez más evidente que las alteraciones epigenéticas también desempeñan un papel central en la patogenia y la progresión de esta enfermedad. La metilación de las regiones promotoras de los genes se asocia con la pérdida de función de éstos y es el episodio epigenético mejor caracterizado de la LLA. La metilación de múltiples genes que controlan la proliferación, la adhesión y la apoptosis es un fenómeno común en las células de la LLA y constituye el mecanismo más importante para inactivar genes relacionados con el cáncer en esta enfermedad. Este perfil de metilación sirve como marcador de riesgo y constituye una base racional para el empleo de agentes desmetilantes en el tratamiento de la LLA. Palabras clave: Leucemia linfoblástica aguda. Metilación. Promotores. Hipometilación. Pronóstico. Epigenetic abnormalities in adult acute lymphoblastic leukemia The hallmark of acute lymphoblastic leukemia (ALL) is the progressive appearance of malignant cell behavior triggered by altered gene function. ALL has traditionally been viewed as a genetic disease but epigenetic defects also play an important role in its pathogenesis and progression. Methylation of the promotor regions of genes is associated with functional loss in these genes and constitutes the best-characterized epigenetic event in ALL. Hypermethylation-associated inactivation affects virtually all of the pathways in the ALL cellular network, such as the cell cycle, apoptosis and adhesion. The identification of these methylation abnormalities and elucidation of the events surrounding them are important as the methylation status of ALL cells can be used as a prognostic biomarker and can also be manipulated in vivo with demethylating agents. Key words: Acute lymphoblastic leukemia. Methylation. Promotors. Hypomethylation. Prognosis. La leucemia linfoblástica aguda (LLA) es el tipo de cáncer más prevalente, así como la forma de leucemia más común en los niños, aunque no es infrecuente en la población adulta1. Las células leucémicas de la mayoría de los pacientes con LLA expresan en su superficie antígenos proteicos que también se encuentran en los diferentes estadios de maduración de las células precursoras linfoides T y B nor- Este trabajo ha sido financiado en parte por ayudas de: FIS 06/0003, Junta de Andalucía 06/0356, Beca Ortiz Landázuri 2006 (Dpto. Salud-Gobierno de Navarra), Fundación IMABIS, Fundación de Investigación Médica Mutua Madrileña Automovilistica, CIMA y Asociación Medicina e Investigación (AMI). Correspondencia: Dr. J. Román-Gómez. Servicio de Hematología. Hospital Reina Sofía. Avda. Menéndez Pidal, s/n. 14004 Córdoba. España. Correo electrónico: [email protected] males. De este modo, los clones leucémicos de la LLA parecen estar originados en células linfoides bloqueadas en un estadio temprano de su maduración2. La característica central de la LLA y de otros tipos de neoplasias es la aparición progresiva de una conducta celular maligna generada por una función genética alterada3. Las translocaciones cromosómicas constituyen un indudable escalón oncogénico en la LLA y los genes estructuralmente alterados por ellas desempeñan papeles cruciales en la proliferación, la diferenciación y la apoptosis4. Sin embargo, estas alteraciones sólo aparecen en un porcentaje bajo de casos y, además, en subtipos muy específicos de LLA5. Por otra parte, las mutaciones en oncogenes y genes supresores de tumores (p53, RAS), tan frecuentes en tumores sólidos, rara vez aparecen en las neoplasias linfoides agudas6. Junto a estas anomalías genéticas, es cada vez más evidente que las alteraciones epigenéticas también desempeñan un papel central en la patogenia y la progresión de este tipo de leucemias7-9. Metilación del ADN La epigenética hace referencia a los cambios hereditarios en los patrones de expresión génica que no dependen de alteraciones en la secuencia normal de los nucleótidos que componen el genoma. Entre todos los episodios epigenéticos existentes, el que está mejor caracterizado es la denominada metilación del ADN. La metilación del nucleótido citosina es la única modificación conocida que ocurre de modo endógeno en el ADN de los mamíferos y se debe a la adición enzimática, mediada por las ADN metiltransferasas (DNMT), de un grupo metilo al carbono en posición 5 de la citosina10 (fig. 1A). La mayor parte de las 5-metilcitosinas (5mC) en el ADN humano están presentes en los dinucleótidos 5’-CpG-3’ (citosina-fosfatoguanina [CpG]), los cuales no se encuentran distribuidos de modo uniforme en el genoma, ya que se han deplecionado progresivamente de éste en el transcurso de la evolución10-11. En el 98% del genoma, los dinucleótidos CpG aparecen una vez cada 100 dinucleótidos y se encuentran fuertemente metilados con objeto de estructurar la cromatina nuclear en un estado represivo que impida la transcripción de regiones poco útiles y potencialmente peligrosas del ADN, como las secuencias repetitivas Alu o transposones. En contraste, regiones pequeñas del ADN, que van desde 200 pb hasta varias kilobases, denominadas «islas CpG», contienen la frecuencia esperada de dinucleótidos CpG (1 por cada 10 dinucleótidos)10-13. Estas áreas están protegidas de la metilación y se encuentran localizadas en las regiones promotoras proximales de cerca del 50-60% de todos los genes10-13. Esta ausencia de metilación es un requisito básico para la transcripción activa y el funcionamiento normal de éstos. La consecuencia principal de la metilación de los promotores es el silenciamiento en la expresión del gen correspondiente10-13. Gracias a este mecanismo, se impide la transMed Clin (Barc). 2007;129(Supl 1):15-22 15 Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. ROMÁN-GÓMEZ J ET AL. ALTERACIONES EPIGENÉTICAS EN LA LEUCEMIA LINFOBLÁSTICA AGUDA DEL ADULTO A B N N O Citosina CH3 NH2 N N O 5-metil-citosina Célula sana NH2 Inactivación del cromosoma X Organización de la cromatina Represión secuencias parásitas Metilación específica de tejido Impresión génica Célula tumoral Metilación de ADN Inactivación de genes supresores de tumores Persistencia de residuos 5-metilcitosina Alteración de las siguientes vías moleculares: apoptosis, angiogénesis, ciclo celular, diferenciación, reparación de ADN, metástasis, resistencia a fármacos, señales de transducción Generación espontánea de: Mutaciones C → T Mutaciones CC → TT Mutaciones G → T Fig. 1. (A) La metilación del ADN consiste en la adición de un grupo metilo al carbono en posición 5 del anillo del nucleótido citosina. (B) Papel de la metilación del ADN en la biología de la célula normal y neoplásica. cripción de secuencias parásitas del ADN, potencialmente dañinas para la integridad celular, como retrovirus endógenos o transposones. Además, la metilación es muy útil para la célula en la inactivación de uno de los cromosomas X en la mujer, la impresión génica o la expresión de genes específicos de tejidos (fig. 1B)10-13. Aunque la importancia funcional de la metilación en la pérdida de función génica está claramente establecida, los mecanismos moleculares implicados en este proceso no están totalmente dilucidados. Recientemente, se ha demostrado la implicación funcional de la asociación entre metilación y una cromatina nuclear inactiva desde el punto de vista transcripcional14-21. Los nucleosomas, constituidos por las proteínas denominadas histonas, se configuran de forma diferente dependiendo del estado de metilación; en los genes metilados, estos nucleosomas se encuentran fuertemente compactados e impiden el acceso de los activadores de la transcripción del gen. Además, las histonas se presentan acetiladas en los genes activos, pero presentan deacetilación en los genes metilados. Esta reacción es mediada por las deacetilasas de las histonas (HDAC). En esta última situación, los genes metilados son accesibles a proteínas que se fijan de forma preferente a las 5mC capaces de reprimir la transcripción del gen. La interacción entre metilación e histonas es incluso más estrecha, ya que los cambios en el estado de metilación de las propias histonas influyen en el estado de transcripción génica: es el llamado código de las histonas. Así, la metilación de lisina 9 en la histona H3 indica silencio del gen, mientras que la metilación de lisina 4 en la histona H3 se asocia a actividad génica. 16 Med Clin (Barc). 2007;129(Supl 1):15-22 Metilación del ADN en el cáncer humano La metilación de las regiones promotoras de los genes es, en la actualidad, el episodio epigenético mejor caracterizado de las células tumorales; se encuentra virtualmente en todos los tipos de neoplasias humanas y se asocia con el inapropiado silenciamiento transcripcional y pérdida de función de estos genes10. Sorprendentemente, esta metilación es, al menos, tan frecuente como las mutaciones o las deleciones que se presentan en los genes supresores de tumores clásicos. La metilación de genes que desempeñan papeles críticos en la oncogenia y que se encuentran no metilados en los tejidos sanos, se observa de forma temprana en el proceso de tumorigenia. Estos episodios epigenéticos tempranos ocasionan la pérdida de control del ciclo celular, de la regulación de los factores de transcripción, de los mecanismos de reparación del ADN, de la apoptosis y de la angiogenia, y crean un fenotipo de inestabilidad genómica típico de las células tumorales (fig. 1B). Adicionalmente, el patrón de metilación génico aporta información importante sobre la célula neoplásica: a) cada tipo de tumor presenta un grupo de genes con especial predilección para presentar metilación. Este patrón de metilación es diferente para cada tipo de tumor, y determina un perfil específico de metilación para la neoplasia22, y b) cada tumor individual en un paciente determinado presenta un único mapa epigenético reflejo de su evolución y que difiere del patrón encontrado en otro paciente con el mismo tipo de tumor23. La detección de estos perfiles específicos de neoplasia y de paciente permite conocer mejor la metilación Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. ROMÁN-GÓMEZ J ET AL. ALTERACIONES EPIGENÉTICAS EN LA LEUCEMIA LINFOBLÁSTICA AGUDA DEL ADULTO coordinada de diversos genes en los sucesivos estadios de la enfermedad y pueden usarse como marcadores biológicos para mejorar el diagnóstico y determinar el pronóstico del proceso maligno. Metilación del ADN en la patogenia de la LLA Esta metilación específica de tumor también es evidente en las neoplasias hematológicas, y es en la LLA donde se encuentra mejor caracterizada (tabla 1). En los últimos años, varios grupos de trabajo, incluido el nuestro, han mostrado que la metilación de múltiples genes es un fenómeno común en las células de la LLA humana y es el mecanismo más importante para inactivar genes relacionados con el cáncer en esta enfermedad24-39; entre el 70-90% de casos presentan, al menos, un gen metilado, mientras que el 2540% presentan más de 2 genes metilados, ya sea al diagnóstico24-39 o en la recaída40-41. La metilación en la LLA participa en la inactivación de 5 vías moleculares esenciales: a) episodios que desregulan la proliferación celular, alteran el punto de control tardío en la transición G1-S del ciclo celular, bien directamente (inactivando p21, p15, p16 y p57) o indirectamente (inactivando p73, PTEN, NES1 y LATS2) y también alteran la transición G2-M (inactivando LATS1 y REPRIMO)24-41; b) el programa apoptótico mediante la inactivación de DAPK, p14, TMS1, APAF1, DIABLO, DBC1 y ASPP124-41; c) inhiben los antagonistas de la vía de señales WNT/beta-cate- nina permitiendo la activación constitutiva de ésta (metilación de DKK3, sFRP1, sFRP2, sFRP4, WIF1 y HDPR1)42; d) la adhesión celular, con la inactivación de la familia de las cadherinas (H-cadherina y E-cadherina) y la familia de las metaloproteasas (ADAMTS1 y ADAMTS5)24-41, y e) inactivación de varias proteínas con actividad cinasa o fosfatasa, las cuales se modulan mediante diversos receptores de citocinas (SHP1 y SYK1)43,44. Todas estas anomalías no deben sorprendernos ya que, subyacente a la complejidad de cada tumor, todos ellos presentan un cierto número de «misiones críticas» que conducen a la célula tumoral y a su progenie hacia la expansión incontrolada. Una de éstas es la desregulación de la proliferación celular, que junto a la supresión obligada de la apoptosis crean una plataforma mínima necesaria para el desarrollo neoplásico. Nuestros datos demuestran que en la LLA esta plataforma común se obtiene mediante un mecanismo epigenético de metilación. Más aún, aunque las anomalías del gen supresor p53 son las observadas con más frecuencia en los tumores sólidos3, estas lesiones son raras en la LLA6. Una hipótesis que nace a la luz de nuestros resultados es que la metilación de los promotores en la LLA inactiva las respuestas apoptóticas y de control de la proliferación, bien por encima (p14, DAPK, ASPP1) o por debajo (p21, APAF1) de p53. Esto implica que en la LLA también hay una fuerte selección celular para perder la funcionalidad de p53, pero en este caso mediante una vía epigenética alternativa45. TABLA 1 Principales genes metilados en la leucemia linfoblástica aguda Locus Función Pacientes con metilación % ADAMTS1 ADAMTS5 APAF1 ARTS ASPP1 CDH1 CDH13 DAPK DBC1 DIABLO DKK3 FHIT HDPR1 hRFC LATS1 LATS2 LHX2 NES1 P14 P15 P16 P57 P73 PACRG PARK2 PTEN 21q21.2 21q21.3 12q23 17q22-23 14q32-33 16q22 16q24 9q34 9q32-33 12q24.31 11p15 3p14.2 14q23.1 21q22.3 6q23-25 13q11-12 9q33-34.1 19q13 9p21 9p21 9p21 11p15 1p36 6q26 6q26 10q23 45 36 23 22 22 37 35 12 13 22 33 19 26 18 34 28 12 56 8 18 15 9 19 27 27 16 PGR REPRIMO RIZ sFRP1 sFRP2 sFRP4 sFRP5 SHP1 SMC1L1 SMC1L2 SYK TMS1 WIF1 11q22-23 2q23.3 1p36 8p12-11.1 4q31.3 7p14.1 10q24.1 12p13 Xp11.22 22q13.31 9q22 16p11-12 12q14.3 Metaloproteasa Metaloproteasa Regulación de la apoptosis Regulación de la apoptosis Regulación de la apoptosis Adhesión celular Adhesión celular Regulación de la apoptosis Regulación de la apoptosis Regulación de la apoptosis Inhibidor de la vía WNT Metabolismo de las purinas Inhibidor de la vía WNT Transportador del folato Control del ciclo celular en G2-M Control del ciclo celular en G1-S Control de la diferenciación celular Control de la diferenciación celular Regulación de la apoptosis Control del ciclo celular en G1-S Control del ciclo celular en G1-S Control del ciclo celular en G1-S Control del ciclo celular en G1-S Ubicuitinación Ubicuitinación Regulación de la apoptosis Control de la angiogénesis Control del ciclo celular Receptor de progestágenos Control del ciclo celular en G2-M Vía del retinoblastoma Inhibidor de la vía WNT Inhibidor de la vía WNT Inhibidor de la vía WNT Inhibidor de la vía WNT Inhibidor de la vía JAK/STAT Reparación del ADN Reparación del ADN Señales de transducción Regulación de la apoptosis Inhibidor de la vía WNT Gen 40 28 26 38 17 21 28 12 8 59 14 10 30 Med Clin (Barc). 2007;129(Supl 1):15-22 17 Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. ROMÁN-GÓMEZ J ET AL. ALTERACIONES EPIGENÉTICAS EN LA LEUCEMIA LINFOBLÁSTICA AGUDA DEL ADULTO B 1,1 1,0 0,9 0,8 0,7 0,6 0,5 0,4 0,3 0,2 0,1 0,0 CIMP– CIMP+ p < 0,0001 0 20 40 60 80 100 120 140 160 180 200 Meses Supervivencia global Supervivencia libre de enfermedad A 1,1 1,0 0,9 0,8 0,7 0,6 0,5 0,4 0,3 0,2 0,1 0,0 CIMP– CIMP+ p < 0,0002 0 20 40 60 80 100 120 140 160 180 200 Meses Fig. 2. Curvas de supervivencia libre de enfermedad (A) y supervivencia global (B) en pacientes afectados de leucemia linfoblástica aguda (LLA) (n = 307) de acuerdo al perfil de metilación génico. CIMP-: pacientes con 0-2 genes metilados; CIMP+: pacientes con más de 2 genes metilados. Metilación del ADN en las variantes clínicas de la LLA Influencia de la edad Puesto que el pronóstico de la LLA pediátrica es significativamente superior a la de los adultos, incluso si se emparejan por datos de mal pronóstico46, algunos autores han propuesto la existencia de diferencias en las bases biológicas de la enfermedad, entre las que destacarían las diferentes variaciones en el patrón de metilación. Nuestro grupo ha mostrado que la LLA del adulto presenta una frecuencia mayor de metilación de los genes LATS1, CDH1, p15, p14 y p57, así como, un número mayor de genes metilados simultáneamente que la LLA del niño36. Este mapa epigenético diferente podría explicar, en parte, la diferencia en el pronóstico entre ambos grupos de edad. Influencia del linaje inmunológico El análisis comparativo entre la metilación de la LLA de células precursoras B (LLA-B) y de células precursoras T (LLA-T) muestra un grupo de genes con mayor frecuencia de metilación en las LLA-T (SYK1, ASPP1, sFRP2, sFRP3 y WIF1), lo que indica que desempeñan un papel determinante en la leucemogenia de estirpe T38. Además, hay una metilación global mayor en LLA-T comparada con LLA-B. De hecho, el 76% de las LLA-T muestra más de 2 genes metilados comparado con el 60% entre las LLA-B (p = 0,03)38. Metilación de promotores como factor pronóstico en la LLA Está claramente demostrado que las características clínicas de la LLA (edad, número de leucocitos al diagnóstico) constituyen factores pronósticos relevantes en estos pacientes. Sin embargo, a pesar de una correcta clasificación en grupos de riesgo, hasta un 20% de los pacientes con LLA pediátrica de riesgo estándar y más del 50% de los adultos que reciben un tratamiento optimizado acabarán recayendo1. Otros marcadores biológicos, como el inmunofenotipo o las alteraciones genéticas, se han incorporado más recientemente como indicadores de un curso más radical y su uso se ha generalizado en la mayoría de los protocolos de tratamiento empleados en la actualidad1-2. 18 Med Clin (Barc). 2007;129(Supl 1):15-22 Junto a estos marcadores tradicionales, la metilación de los promotores de genes relacionados con el cáncer, y que son importantes desde el punto de vista funcional, también pueden afectar a la conducta tumoral e influenciar en el pronóstico final de los pacientes. El silencio epigenético de genes que controlan la invasión neoplásica, el crecimiento y la muerte celular pueden determinar el patrón de recurrencia tumoral tras la quimioterapia y, de este modo, determinar la supervivencia. Puesto que cada tumor presenta un grupo de genes susceptibles de experimentar metilación, cada tumor individual podría mostrar un patrón característico de metilación potencialmente predictivo del curso clínico de éste. Con objeto de confirmar esta hipótesis en la LLA, nuestro grupo ha estudiado el estado de metilación de 39 genes pertenecientes a las principales vías de transformación e inmortalización celular (tabla 1) y ha comparado el pronóstico de los pacientes (n = 201) con un fenotipo metilador positivo (CIMP+, más de 2 genes metilados) con los pacientes (n = 106) que presentan un fenotipo metilador negativo (CIMP-, menos de 3 genes metilados)36,38-39. Las tasas de remisión completa (RC) de los pacientes CIMP- y CIMP+ fueron del 91 y el 87%, respectivamente, lo que indica que el patrón de metilación no influye en la respuesta al tratamiento de inducción a la remisión. Sin embargo, los pacientes CIMP- presentaron una tasa de recaídas (26 frente a 58%, p < 0,0001) y de mortalidad (34 frente a 58%, p < 0,001) significativamente más bajas que los pacientes del grupo CIMP+. La supervivencia libre de enfermedad (SLE) estimada a los 14 años fue del 68% para el grupo CIMP- y del 32% para el grupo CIMP+ (p < 0,0001, fig. 2A). La supervivencia global (SG) calculada a los 14 años fue del 63% para los pacientes CIMPy 32% para el grupo CIMP+ (p = 0,0002, fig. 2B). El análisis multivariable puso de manifiesto que el patrón de metilación constituía un factor pronóstico independiente tanto para la SLE (p < 0,0001) como para la SG (p = 0,003). Un aspecto muy interesante es que el patrón de metilación fue capaz de redefinir el pronóstico clínico de pacientes con LLA y que presentaban factores pronósticos claramente establecidos y aceptados por la comunidad internacional. De este modo, la existencia de un fenotipo CIMP- mejoró significativamente los resultados clínicos de pacientes considerados clásicamente como de mal pronóstico, especialmente Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. ROMÁN-GÓMEZ J ET AL. ALTERACIONES EPIGENÉTICAS EN LA LEUCEMIA LINFOBLÁSTICA AGUDA DEL ADULTO A A LLA de células T 1,0 1,2 0,8 CIMP– 0,6 0,5 0,2 CIMP+ p = 0,001 0,0 0 20 40 60 Meses 80 100 120 Supervivencia libre de enfermedad Supervivencia libre de enfermedad LLA con cromosoma Filadelfia 1,0 CIMP– 0,8 0,6 0,4 CIMP+ 0,2 p = 0,0006 0,0 B 0 1,0 50 75 100 125 150 175 200 B LLA de células T 0,8 1,2 CIMP– 0,6 1,0 0,5 0,2 CIMP+ p = 0,05 0,0 0 20 40 60 Meses 80 100 120 C CIMP– 0,8 0,6 0,4 0,2 LLA TEL-AML1 positiva Supervivencia libre de enfermedad 25 Meses Supervivencia global Supervivencia libre de enfermedad LLA leucocitósica 1,2 CIMP+ 1,2 p = 0,003 0,0 1,0 0 CIMP– 25 50 75 100 125 150 175 200 Meses 0,8 Fig. 4. Curvas de supervivencia libre de enfermedad (A) y supervivencia global (B) en pacientes afectados de leucemia linfoblástica aguda (LLA) de células precursoras T (n = 50) de acuerdo al perfil de metilación génico. CIMP-: pacientes con 0-2 genes metilados; CIMP+: pacientes con más de 2 genes metilados. 0,6 CIMP+ 0,5 0,2 p = 0,002 0,0 0 20 40 60 80 100 Meses Fig. 3. Curvas de supervivencia libre de enfermedad para pacientes con leucemia linfoblástica aguda (LLA) con cromosoma Filadelfia (n = 47, A), leucocitosis al diagnóstico (n = 73, B), y reordenamiento TEL-AML1 (n = 44, C) de acuerdo al perfil de metilación génico. CIMP-: pacientes con 0-2 genes metilados; CIMP+: pacientes con más de 2 genes metilados. los que presentaban el cromosoma Filadelfia (fig. 3A)36, un fenotipo celular de estirpe T (fig. 4)38 o un número elevado de leucocitos al diagnóstico (fig. 3B)36. En contraste, la presencia de un fenotipo CIMP+ empeoró el pronóstico del grupo de pacientes con reordenamiento TEL-AML1, considerados clásicamente como de buen pronóstico (fig. 3C)39. Todos estos resultados demuestran que el patrón de metilación tiene importantes implicaciones clínicas en la LLA y, junto con los estudios inmunológicos, citogenéticos y moleculares, podría emplearse para la elaboración de una estrategia terapéutica adaptada al riesgo en los pacientes con LLA. Hipometilación del ADN en la LLA La hipometilación del ADN asociada al cáncer humano es tan prevalente como la hipermetilación, pero estos 2 tipos de anormalidades epigenéticas afectan secuencias genómicas diferentes. La metilación se observa con mayor frecuencia en las islas CpG de los promotores de los genes, mientras que la hipometilación afecta a las regiones repetitivas del ADN, especialmente a retrotransposones y retrovirus endógenos (los cuales constituyen la mayor parte de todas Med Clin (Barc). 2007;129(Supl 1):15-22 19 Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. ROMÁN-GÓMEZ J ET AL. ALTERACIONES EPIGENÉTICAS EN LA LEUCEMIA LINFOBLÁSTICA AGUDA DEL ADULTO B 1,1 1,1 1,0 1,0 0,9 0,9 0,8 Supervivencia global Supervivencia libre de enfermedad A L1 hipometilado 0,7 0,6 0,5 L1 metilado 0,4 0,3 0,2 0,8 0,7 L1 hipometilado 0,6 0,5 L1 metilado 0,4 0,3 0,2 0,1 0,1 p = 0,02 0,0 p = 0,03 0,0 0 20 40 60 80 100 120 140 160 180 200 0 20 40 60 80 Meses 100 120 140 160 180 200 Meses Fig. 5. Curvas de supervivencia libre de enfermedad (A) y supervivencia global (B) en pacientes afectados de leucemia linfoblástica aguda (LLA) (n = 307) de acuerdo al estado de hipometilación del retrotransposon LINE1 (L1). A B LLA CIMP– LLA CIMP 1,1 1,0 1,0 L1 hipometilado 0,9 0,9 0,8 Supervivencia global Supervivencia libre de enfermedad 1,1 0,7 0,6 L1 metilado 0,5 0,4 0,3 0,2 L1 hipometilado 0,8 0,7 L1 metilado 0,6 0,5 0,4 0,3 0,2 0,1 0,1 p = 0,004 0,0 p = 0,08 0,0 0 20 40 60 80 100 120 140 160 0 20 40 60 Meses 80 100 120 140 160 Meses Fig. 6. Curvas de supervivencia libre de enfermedad (A) y supervivencia global (B) en pacientes afectados de leucemia linfoblástica aguda (LLA) y CIMP- (pacientes con 0-2 genes metilados, n = 106) de acuerdo al estado de hipometilación del retrotransposon LINE1 (L1). nuestras secuencias genómicas)47. El significado biológico de la hipometilación en el cáncer es menos conocido que el de la hipermetilación. Sin embargo, estudios realizados en ratones demuestran que la hipometilación inducida es capaz de producir tumores en éstos48. En humanos, la alta frecuencia de hipometilación ligada al cáncer, la naturaleza de las secuencias afectadas y la ausencia de una asociación clara con la hipermetilación hablan en favor de un papel importante e independiente de la hipometilación en la génesis y progresión tumoral49. Nuestro grupo ha analizado recientemente la frecuencia de hipometilación e hipermetilación en una serie amplia de pacientes con LLA (n = 307)50 con objeto de determinar si ambos episodios son independientes y, por tanto, reflejan subtipos de LLA con características clínicas y biológicas 20 Med Clin (Barc). 2007;129(Supl 1):15-22 diferentes. Nuestros resultados demuestran que la hipometilación del retrotransposon LINE1 está presente en el 24% de las LLA y que no hay ninguna asociación entre esta hipometilación y la hipermetilación de 39 islas CpG estudiadas. Esto indica que ambos cambios epigenéticos contribuyen a la leucemogenia linfoide de un modo separado e independiente. Además, al contrario de lo que sucede con el patrón de hipermetilación, la hipometilación del ADN es un factor pronóstico favorable (fig. 5), y establece un grupo de muy bajo riesgo entre los pacientes LLA con CIMP- (fig. 6). Perspectivas terapéuticas A diferencia de los cambios genéticos que ocurren en el cáncer, los cambios epigenéticos son potencialmente rever- Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. ROMÁN-GÓMEZ J ET AL. ALTERACIONES EPIGENÉTICAS EN LA LEUCEMIA LINFOBLÁSTICA AGUDA DEL ADULTO sibles y, por tanto, hay la posibilidad de reactivar los genes silenciados por metilación con objeto de obtener un beneficio terapéutico. Los agentes desmetilantes, como la azacitidina o la 5-aza-2-deoxicitidina (decitabina), son capaces de inducir la expresión de genes silenciados por metilación en cultivos de líneas celulares malignas. Estos compuestos deben incorporarse al ADN como un análogo de las citosinas y, por tanto, pueden inducir efectos tóxicos inespecíficos cuando se utilizan a altas dosis, probablemente por la inducción de lesión en el ADN. Sin embargo, su principal mecanismo de acción es por una inhibición irreversible de las DNMT51. Estos fármacos se han empleado con éxito en síndromes mielodisplásicos y leucemias agudas mieloblásticas52. En asociación con otros agentes terapéuticos, los fármacos desmetilantes pueden actuar sinérgicamente y favorecer la respuesta hematológica en leucemias previamente refractarias53. El hallazgo de que la desacetilación de las histonas está ligada a los procesos de metilación, como un factor coadyuvante, ha permitido el empleo combinado de inhibidores de las DNMT y HDAC en el tratamiento de las neoplasias, y muestra un importante efecto antileucémico tanto in vitro como in vivo54-56. Nuestro grupo ha demostrado recientemente que la regulación epigenética de los antagonistas WNT contribuye de una forma esencial a la activación de la vía de señales WNT en la LLA42. La demostración de que la inhibición de la beta-catenina intranuclear induce apoptosis en la LLA, establece esta vía como una diana muy atractiva para el tratamiento con agentes específicos, como la quercetina57, o antagonistas farmacológicos del complejo proteico beta-catenina/TCF58. Además, el hecho de que la azacitidina inactive la vía WNT, tal como demuestra nuestro estudio, constituye una base racional para el empleo de los agentes desmetilantes en los pacientes con LLA59. REFERENCIAS BIBLIOGRÁFICAS 1. Pui CH, Evans WE. Acute lymphoblastic leukemia. N Engl J Med. 1998;339:605-15. 2. Pui CH, Behm FG, Crist WM. Clinical and biologic relevance of immunologic marker studies in childhood acute lymphoblastic leukemia. Blood. 1993;82:343-62. 3. Ponder BAJ. Cancer genetics. Nature. 2001;411:336-41. 4. Pui CH, Crist WM, Look AT. Biology and clinical significance of cytogenetic abnormalities in childhood acute lymphoblastic leukemia. Blood. 1990;76:1449-63. 5. Faderl S, Kantarjian HM, Talpaz M, Estrov Z. Clinical significance of cytogenetic abnormalities in adult acute lymphoblastic leukemia. Blood. 1998;91:3995-4019. 6. Wada M, Bartram CR, Nakamura H, Hachiya M, Chen DL, Borenstein J, et al. Analysis of p53 mutations in a large series of lymphoid hematologic malignancies of childhood. Blood. 1993;82:3163-9. 7. Jones PA, Laird PW. Cancer epigenetics comes of age. Nature Genet. 1999;21:163-7. 8. Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286:481-6. 9. Tycko B. Epigenetic gene silencing in cancer. J Clin Invest. 2000;105:401-7. 10. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042-54. 11. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245-54. 12. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143-53. 13. Singal R, Ginder GD. DNA methylation. Blood. 1999;93:4059-70. 14. Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599-606. 15. Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386-9. 16. Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass KU, Landsberger N, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187-91. 17. Mannervik M, Nibu Y, Zhang H, Levine M. Transcriptional coregulators in development. Science. 1999;284:606-9. 18. Hendrich B, Bird A. Mammalian methyltransferases and methyl-CpG binding domains: proteins involved in DNA methylation. Curr Top Microbiol Immunol. 2000;249:55-74. 19. Rountree MR, Bachman KE, Herman JG, Baylin SB. DNA methylation, chromatin inheritance, and cancer. Oncogene. 2001;20:3156-65. 20. Im H, Grass JA, Christensen HM, Perkins A, Bresnick EH. Histone deacetylase-dependent establishment and maintenance of broad low-level histone acetylation within a tissue-specific chromatin domain. Biochemistry. 2002;41:15152-60. 21. El Osta A. DNMT cooperativity—the developing links between methylation, chromatin structure and cancer. Bioessays. 2003;25:1071-84. 22. Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225-9. 23. Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, et al. Aberrant CpG-island methylation has non-random and tumourtype-specific patterns. Nat Genet. 2000;24:132-8. 24. Roman-Gomez J, Castillejo JA, Jimenez A, Gonzalez MG, Reina ML, Rodriguez MC, et al. Hypermethylation of the calcitonin gene in acute lymphoblastic leukemia is associated with unfavourable clinical outcome. Br J Haematol. 2001;113:329-38. 25. Roman-Gomez J, Castillejo JA, Jimenez A, Gonzalez MG, Moreno F, Rodriguez MC, et al. 5’ CpG island hypermethylation is associated with transcriptional silencing of the p21CIP1/WAF1/SDI1gene and confers poor prognosis in acute lymphoblastic leukemia. Blood. 2002;99:2291-6. 26. Agirre X, Vizmanos JL, Calazans MJ, Garcia-Delgado M, Larrayoz MJ, Novo FJ. Methylation of CpG dinucleotides and/or CCWGG motifs at the promoter of TP53 correlates with decreased gene expression in a subset of acute lymphoblastic leukemia patients. Oncogene. 2003;22:1070-2. 27. Roman-Gomez J, Jimenez-Velasco A, Agirre X, Castillejo JA, Barrios M, Andreu EJ, et al. The normal epithelial cell-specific 1 (NES1) gene, a candidate tumor suppressor gene on chromosome 19q13.3-4, is downregulated by hypermethylation in acute lymphoblastic leukemia. Leukemia. 2004;18:362-5. 28. Roman-Gomez J, Jimenez-Velasco A, Agirre X, Castillejo JA, Navarro G, Barrios M, et al. Transcriptional silencing of the Dickkopfs-3 (Dkk-3) gene by CpG hypermethylation in acute lymphoblastic leukaemia. Br J Cancer. 2004;91:707-13. 29. Jimenez-Velasco A, Roman-Gomez J, Agirre X, Barrios M, Navarro G, Vazquez I, et al. Downregulation of the large tumor suppressor 2 (LATS2 / KPM) gene confers poor prognosis in acute lymphoblastic leukemia. Leukemia. 2005;19:2347-50. 30. Agirre X, Roman-Gomez J, Vazquez I, Jimenez-Velasco A, Garate L, Montiel-Duarte C, et al. Abnormal methylation of the common PARK2 and PACRG promoter is associated with downregulation of gene expression in Acute Lymphoblastic Leukemia (ALL) and Chronic Myeloid Leukemia (CML). Int J Cancer. 2006;118:1945-53. 31. Agirre X, Roman-Gomez J, Jimenez-Velasco A, Garate L, Montiel-Duarte C, Navarro G, et al. ASPP1, a common activator of TP53, is inactivated by aberrant methylation of its promoter in acute lymphoblastic leukemia. Oncogene. 2006;25:1862-70. 32. San Jose-Eneriz E, Agirre X, Roman-Gomez J, Cordeu L, Garate L, Jimenez-Velasco A, et al. Downregulation of DBC1 expression in acute lymphoblastic leukaemia is mediated by aberrant methylation of its promoter. Br J Haematol. 2006;134:137-44. 33. Garcia-Manero G, Daniel I, Smith TL, Kornblau SM, Kantarjian HM, Issa J-P. DNA methylation of multiple promoter-associated CpG islands in adult acute lymphocytic leukemia. Clin Cancer Res. 2002;8:2217-24. 34. Gutierrez MI, Siraj AK, Bhargava M, Ozbek U, Banavali S, Chaudhary MA, et al. Concurrent methylation of multiple genes in childhood ALL: Correlation with phenotype and molecular subgroup. Leukemia. 2003;17:1845-50. 35. Shen L, Toyota M, Kondo Y, Obata T, Daniel S, Pierce S, et al. Aberrant DNA methylation of p57KIP2 identifies a cell-cycle regulatory pathway with prognostic impact in adult acute lymphocytic leukemia. Blood. 2003;101:4131-6. 36. Roman-Gomez J, Jimenez-Velasco A, Castillejo JA, Agirre X, Barrios M, Navarro G, et al. Promoter hypermethylation of cancer-related genes: a strong independent prognostic factor in acute lymphoblastic leukemia. Blood. 2004;104:2492-8. 37. Takahashi T, Shivapurkar N, Reddy J, Shigematsu H, Miyajima K, Suzuki M, et al. DNA methylation profiles of lymphoid and hematopoietic malignancies. Clin Cancer Res. 2004;10:2928-35. 38. Roman-Gomez J, Jimenez-Velasco A, Agirre X, Prosper F, Heiniger A, Torres A. Lack of CpG island methylator phenotype defines a clinical subtype of T-cell acute lymphoblastic eukemia associated with good prognosis. J Clin Oncol. 2005;23:7043-9. 39. Roman-Gomez J, Jimenez-Velasco A, Agirre X, Castillejo JA, Navarro G, Calasanz MJ, et al. CpG island methylator phenotype redefines the prognostic effect of t(12;21) in childhood acute lymphoblastic leukemia. Clin Cancer Res. 2006;12:4845-50. 40. Garcia-Manero G, Bueso-Ramos C, Daniel J, Williamson J, Kantarjian HM, Issa JP. DNA methylation patterns at relapse in adult acute lymphocytic leukemia. Clin Cancer Res. 2002;8:1897-903. 41. Matsushita C, Yang Y, Takeuchi S, Matsushita M, Van Dongen JJ, Szczepanski T, et al. Aberrant methylation in promoter-associated CpG islands Med Clin (Barc). 2007;129(Supl 1):15-22 21 Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. ROMÁN-GÓMEZ J ET AL. ALTERACIONES EPIGENÉTICAS EN LA LEUCEMIA LINFOBLÁSTICA AGUDA DEL ADULTO of multiple genes in relapsed childhood acute lymphoblastic leukemia. Oncol Rep. 2004;12:97-9. 42. Roman-Gomez J, Cordeu L, Agirre X, Jimenez-Velasco A, San Jose-Eneriz E, Garate L, et al. Epigenetic regulation of WNT signaling pathway in acute lymphoblastic leukemia. Blood. 2007;109:3462-9. 43. Oka T, Ouchida M, Koyama M, Ogama Y, Takada S, Nakatani Y, et al. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 2002;62:6390-4. 44. Goodman PA, Burkhardt N, Juran B, Tibbles HE, Uckun FM. Hypermethylation of the spleen tyrosine kinase promoter in T-lineage acute lymphoblastic leukemia. Oncogene. 2003;22:2504-14. 45. Roman-Gomez J, Castillejo JA, Jimenez A, Barrios M, Heiniger A, Torres A. The role of DNA hypermethylation in the pathogenesis and prognosis of acute lymphoblastic leukemia. Leuk Lymphoma. 2003;44:1855-64. 46. Plasschaert SL, Kamps WA, Vellenga E, De Vries EG, De Bont ES. Prognosis in childhood and adult acute lymphoblastic leukaemia: a question of maturation? Cancer Treat Rev. 2004;30:37-51. 47. Roman-Gomez J, Jimenez-Velasco A, Agirre X, Cervantes F, Sanchez J, Garate L, et al. Promoter hypomethylation of the LINE-1 retrotransposable elements activates sense/antisense transcription and marks the progression of chronic myeloid leukemia. Oncogene. 2005;24:7213-23. 48. Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400-13. 49. Kaneda A, Tsukamoto T, Takamura-Enya T, Watanabe N, Kaminishi M, Sugimura T, et al. Frequent hypomethylation in multiple promoter CpG island is associated with global hypomethylation, but not with frequent promoter hypermethylation. Cancer Sci. 2004;95:58-64. 50. Roman-Gomez J, Jimenez-Velasco A, Agirre X, Castillejo JA, Navarro G, Garate L, et al. Promoter hypermethylation and global hypomethylation 22 Med Clin (Barc). 2007;129(Supl 1):15-22 are independent epigenetic events in lymphoid leukemogenesis with opposing effects on clinical outcome. Leukemia. 2006;20:1445-8. 51. Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85-93. 52. Ruter B, Wijermans PW, Lubbert M. DNA methylation as a therapeutic target in hematologic disorders: recent results in older patients with myelodysplasia and acute myeloid leukemia. Int J Hematol. 2004;80:12835. 53. Avramis VI, Mecum RA, Nyce J, Steele DA, Holcenberg JS. Pharmacodynamic and DNA methylation studies of high-dose 1-beta-D-arabinofuranosyl cytosine before and after in vivo 5-azacytidine treatment in pediatric patients with refractory acute lymphocytic leukemia. Cancer Chemother Pharmacol. 1989;24:203-10. 54. Shaker S, Bernstein M, Momparler LF, Momparler RL. Preclinical evaluation of antineoplastic activity of inhibitors of DNA methylation (5-aza2’-deoxycytidine) and histone deacetylation (trichostatin A, depsipeptide) in combination against myeloid leukemic cells. Leuk Res. 2003;27:437-44. 55. Lemaire M, Momparler LF, Farinha NJ, Bernstein M, Momparler RL. Enhancement of antineoplastic action of 5-aza-2’-deoxycytidine by phenylbutyrate on L1210 leukemic cells. Leuk Lymphoma. 2004;45:147-54. 56. Claus R, Lubbert M. Epigenetic targets in hematopoietic malignancies. Oncogene. 2003;22:6489-96. 57. Park CH, Chang JY, Hahm ER, Park S, Kim HK, Yang CH. Quercetin, a potent inhibitor against beta-catenin/Tcf signaling in SW480 colon cancer cells. Biochem Biophys Res Commun. 2005;328:227-34. 58. Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, et al. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell. 2004;5:91-102. 59. Fenaux P. Inhibitors of DNA methylation: beyond myelodysplastic syndromes. Nat Clin Pract Oncol. 2005;2 (Suppl 1):S36-44.