Aterosclerosis y trombosis - Sociedad Mexicana de Hipertensión
Anuncio

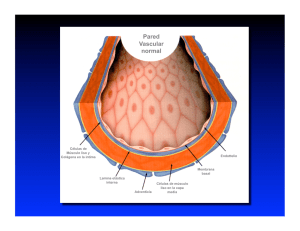
Aterosclerosis y Trombosis Editor responsable Angel F. González Caamaño Sociedad Mexicana para el Estudio de la Hipertensión Arterial Consejo Mexicano de Aterosclerosis Dr. Angel F. González Caamaño Miembro de la Sociedad Mexicana para el Estudio de la Hipertensión Miembro del Consejo Mexicano de Hipertensión Miembro de la Sociedad Mexicana de Hipertensión Miembro de la Sociedad Interamericana de Hipertensión Miembro de la Sociedad Internacional de Hipertensión Miembro de la Sociedad Mexicana de Cardiología Miembro del American College of Cardiology Miembro del American Heart Association Miembro del Consejo Mexicano de Aterosclerosis Profesor Titular Facultad de Medicina UNAM Editor en Jefe de la revista "Lancet" México Aterosclerosis y Trombosis Revisión general del proceso evolutivo a aterosclerosis es una enfermedad inflamatoria de las arterias de mediano y gran calibre, cuya consecuencia final es la disminución u obstrucción del flujo sanguíneo; de hecho, las interacciones celulares identificadas en la aterogénesis no difieren mucho de aquellas observadas en enfermedades inflamatorias fibroproliferativas crónicas de diversos órganos, como hígado (cirrosis), articulaciones (artritis reumatoide), riñones (glomerulonefritis), pulmones (fibrosis pulmonar), páncreas (pancreatitis) y por supuesto, arterias (aterosclerosis). Las respuestas inflamatorias crónicas a menudo se relacionan con tipos específicos de agentes nocivos o inductores de granulomas. L La hipertensión no representa tan solo un factor de riesgo cardiovascular sino también un aumento para el riesgo de la aterosclerosis, las pruebas clínicas han demostrado que, el punto más alto de la presión diastólica, tomando en cuenta los riesgos adicionales del colesterol alto y del cigarro, la hipertensión contribuye de manera significativa de presentar aterosclerosis. La fisiopatología de la aterosclerosis se caracteriza por la presencia de disfunción endotelial, inflamación vascular y acumulación de lípidos, células de músculo liso y de tejido conectivo. Las manifestaciones clínicas de la aterosclerosis son: enfermedad de arterias coronarias, enfermedad cerebrovascular (accidentes hemorrágicos o embólicos o mixtos), enfermedad vascular periférica y aneurisma de aorta abdominal. Un recurso clínico de gran utilidad y que se utiliza cada vez con más frecuencia en el consultorio como parte de la evaluación del paciente con factores de riesgo cardiovascular es el índice tobillo-branquial*. Otros estudios útiles y accesibles en caso de sospecha de enfermedad aterosclerótica periférica son: arteriografía, para identificar una obstrucción ateromatosa; electrocardiograma en reposo, placa de rayos X de tórax, prueba de esfuerzo, ecocardiografía y angiografía coronaria, para diagnosticar y vigilar la progresión de enfermedad de arterias coronarias; ultrasono- grafía Doppler o dúplex de arterias carótidas, para identificar y vigilar una enfermedad cerebrovascular. El médico puede optar por cualquiera de estas modalidades diagnósticas con base en su criterio y las características clínicas de cada paciente; sin embargo, el monitoreo periódico del control metabólico a través de indicadores específicos, particularmente glucemia y perfil lipídico (por ejemplo con AccuChek, Accutrend GC / GCT y Reflotron) no es un rasgo prescindible dentro del esquema profiláctico-terapéutico de la enfermedad cardiovascular. La primera alteración fisiopatológica en la aterosclerosis es la disfunción endotelial ocasionada por la presencia excesiva de moléculas de LDL modificadas, radicales libres (generados por el hábito de fumar, hipertensión y diabetes, entre otras causas), alteraciones genéticas, concentraciones plasmáticas elevadas de homocisteína, microorganismos infecciosos (p.ej. virus del herpes y Chlamydia pneumoniae), y la combinación de éstos y otros factores.1 Sin importar la causa de la disfunción endotelial, la aterosclerosis representa una manera particular de responder (mecanismos compensatorios) de ciertas arterias. Los primeros cambios patológicos observados en el endotelio son el incremento de la adhesividad (mayor circulación local de leucocitos y plaquetas) y la permeabilidad, adquiriendo así propiedades procoagulantes en lugar de anticoagulantes; asimismo, el endotelio comienza a producir moléculas vasoactivas, citocinas y factores de crecimiento. Si la respuesta inflamatoria no neutraliza o repele de manera efectiva a los agentes agresores, entonces el proceso continuará indefinidamente, cuya consecuencia a mediano o largo plazo será el engrosamiento de la pared arterial que, inicialmente, 1 se compensa mediante una dilatación gradual el lumen aún permanece inalterado, un fenómeno llamado “remodelación”-, hasta que finalmente deja de haber dilatación y el lumen del vaso se obstruye en grados variables. Contrario a lo que sucede en cualquier padecimiento inflamatorio, los granulocitos se presentan en cantidades mínimas durante cualquier fase de la aterogénesis; en cambio, son los macrófagos y subtipos específicos de linfocitos T quienes median la respuesta endotelial durante todo el proceso. Tales células son cada vez más abundantes debido a su multiplicación dentro de la lesión. La activación de estos tipos celulares lleva a la liberación de enzimas hidrolíticas, citocinas, quimiocinas y factores de crecimiento, los cuales inducen mayor daño y finalmente, causan necrosis focal. Entonces, los ciclos repetitivos de acumulación de células mononucleares, migración y proliferación de células de músculo liso y formación de tejido fibroso conducen al crecimiento y reestructuración de la lesión, la cual queda cubierta por una película fibrosa que envuelve un núcleo de lípidos y tejido necrótico llamada en conjunto placa complicada o avanzada. Es así que en algún momento impredecible, la arteria ya no puede compensar el daño mediante dilatación y la lesión protruye hacia el lumen, alterando el flujo de la sangre. 2 La hipercolesterolemia se relaciona con un incremento de la densidad espacial de los vasa vasorum de las arterias coronarias alrededor de las lesiones ateroscleróticas. Tal proceso de neovascularización se presenta en etapas muy tempranas del proceso aterosclerótico y antes de la disfunción endotelial epicárdica. Al parecer, la neovascularización coronaria se puede prevenir mediante el tratamiento con simvastatina o algún antagonista selectivo del receptor de endotelina A. Por otra parte, las moléculas LDL pueden sufrir modificación por medio de oxidación, glucosilación (en diabetes), agregación, unión a proteoglucanos o incorporación a complejos inmunes. Las LDL son la causa principal del daño endotelial y músculo liso subyacente. Cuando las partículas de LDL quedan atrapadas en una arteria, pueden sufrir una oxidación progresiva y ser ingeridas por macrófagos a través de receptores específicos localizados en la superficie celular y de esta manera ser introducidas en el endotelio; esto tiene como consecuencia la formación de peróxidos lipídicos y acumulación de ésteres de colesterol, resultando en la formación de células espumosas.2 El grado de modificación de las LDL puede variar significativamente. El secuestro de las moléculas LDL modificadas es un componente esencial de la función protectora inicial de los macrófagos en la respuesta inflamatoria, cuyo objetivo es minimizar los efectos de las LDL modificadas sobre el endotelio y células de músculo liso. En la mayoría de los pacientes, el infarto de miocardio se suscita como resultado de la erosión o adelgazamiento no uniforme y ruptura de la película fibrosa en la periferia de la lesión, justo donde los macrófagos ingresan, se acumulan y se activan y donde la apoptosis puede presentarse. La degradación de la película fibrosa puede ser secundaria a la elaboración de metaloproteinasas, como colagenasas, elastasas y estromelisinas. Los linfocitos T activados pueden estimular a los macrófagos para producir metaloproteinasas en las lesiones, lo cual promueve la inestabilidad de la placa ateromatosa, implicando una respuesta inmune. Tales cambios pueden también acompañarse de la producción del factor tisular procoagulante y otros factores hemostáticos, incrementándose así la posibilidad de trombosis. La interacción entre la placa aterosclerótica vulnerable y la formación de trombos, proceso conocido como aterotrombosis, es la piedra angular de los síndromes coronarios agudos. Una La aterosclerosis y la hipertensión se refieren a entidades de afecciones diferentes; cada una de las personas que tienen hipertensión no manifiestan una aterosclerosis extensa sino después de varios años. Ni tampoco puede decirse que en un principio la aterosclerosis vaya acompañada de la hipertensión. Placa vulnerable ■ Gran depósito excéntrico rico en lípidos. ■ Infiltración del centro lipídico por células espumosas secretoras de factor tisular. ■ Cubierta fibrosa delgada. ■ Medio de intensa inflamación local con presencia de neutrófilos, linfocitos T, macrófagos, células de músculo liso y citocinas que promueven la ruptura de la cubierta fibrosa mediante la secreción de metaloproteinasas. Los riesgos patológicos cardinales del desarrollo de la lesión aterosclerótica son: la presencia de monocitos, macrófagos y células T. Su localización en las arterias largas elásticas de conducción. La proliferación y migración hacía la íntima de células musculares lisas de tipo medio. El depósito de una cantidad aumentada de tejido conectivo y por último la nevascularización. Ruptura y Disrupción de la placa Factores desencadenantes: esfuerzo físico, estrés mecánico debido a un incremento de contractilidad cardiaca, frecuencia cardiaca, presión sanguínea y posiblemente, vasoconstricción Formación de trombo ■ Trombogenicidad sistémica. ■ Activación, adhesión y agregasión plaquetaria. ■ Activación de la vía de la coagulación y formación de trombina. ■ Conversión de fibrinógeno a fibrina con entrecruzamiento de bandas de unión. Oclusión coronaria completa Infarto agudo de miocardio Lisis espontánea, reparación y remodelación de la pared vascular Resolución temporal de la inestabilidad. Lesión coronaria futura de alto riesgo Oclusión coronaria incompleta Angina inestable o infarto de miocardio sin onda Q Cuadro 1. Formación y ruptura de la placa 3 vez formada la placa aterosclerótica, ésta pasará por un largo trayecto que finalmente la llevará a la ruptura. La disrupción de la placa parece depender de fenómenos pasivos y activos relacionados con las fuerzas físicas. La disrupción pasiva frecuentemente inicia en el sitio donde la capa fibrosa es más delgada y densamente infiltrada con células espumosas. No obstante, el proceso de disrupción no es puramente mecánico, ya que la inflamación se relaciona directamente con todos los estadios del desarrollo de las placas ateroscleróticas, desde su formación inicial hasta la formación de trombos. Inmediatamente después de la disrupción o ruptura, el contenido altamente trombogénico de la placa queda expuesto a la circulación sanguínea, desencadenándose así la formación del trom3 bo.(Figura 1). Cambios de la geometría de una placa en disrupción, así como la organización del trombo parietal recién formado por parte del tejido conectivo, llevan a una rápida progresión de la placa que finalmente dará como resultado lesiones más oclusivas y fibróticas. 4 Por otra parte, también puede originarse trombosis en placas sin disrupción. En 33% de los síndromes coronarios agudos (SCA), particularmente en la muerte súbita coronaria, se identifican placas (vulnerables) ricas en lípidos íntegras (sin disrupción), pero notablemente estenóticas y fibróticas con una erosión superficial. La trombosis causada por disrupción suele observarse en placas con estenosis inicial de menor grado, la cual no puede identificarse mediante angiografía coronaria (Cuadro 1). En cambio, la trombosis causada por erosión endotelial suele localizarse en sitios de estenosis de alto grado preexistentes. Este segundo tipo de trombosis se reporta más comúnmente en mujeres jóvenes y en hombres con ciertos factores de riesgo protrombótico (tabaquismo, diabetes, hipercolesterolemia). Entonces, la formación de trombos en estos casos donde no existe disrupción de la placa, depende de un estado hipertrombogénico de- sencadenado por factores sistémicos, como niveles altos de lipoproteínas de baja densidad, tabaquismo, hiperglucemia, hemostasis y otros, los cuales también se relacionan con estados de mayor hipercoagulabilidad sanguínea. Por ejemplo, la diabetes mellitus se relaciona con hiperagregación plaquetaria, niveles altos del inhibidor del activador plasminógeno 1, mayor circulación de fibrinógeno y el factor von Willebrand, así como una menor actividad de la antitrombina III. Además, un mejor control glucémico se vincula con una menor trombogenicidad sanguínea. De igual manera, la hiperlipidemia se relaciona con un estado de hipercoagulabilidad y protrombótico, el cual suele disminuir significativamente o revertir por completo mediante el tratamiento de la hipercolesterolemia. Figura . Formación y ruptura de la placa Referencias Blibliográficas 1. 2. 3. González Caamaño AF. Hipertensión Arterial sexta edición. Ediciones Médicas Actualizadas. 2006;229241. González Caamaño AF. El Endotelio. McGrow Hill. En prensa. González Caamaño AF. Factores de Riesgo Cardiovascular. McGrow Hill. En prensa. La hipertensión ocasiona un aumento en el grosor de las arterias y con el tiempo suele haber un incremento de la masa de las células de tipo muscular lisas y/o del número de células y crecimiento en el depósito en el tejido conectivo. Tomando en cuenta la interacción de ambas afecciones, será muy útil considerar los mecanismos y las consecuencias que tengan ambas en común. XICANO D CA ME XI SOCIED AD HI P ER T RIAL RTE LE CO STUDIO DE LA EL E A ON SI NÚMERO DE REGISTRO PÚBLICO DEL DERECHO DE AUTOR: 04-2008-1115112380346794510-01 ISBN: 754-14782-38 AMECE EAN13: 3486501284593 NA RA PA EN ATEROSC NSEJO E ME ROSIS •