Hemostasia P r
Anuncio

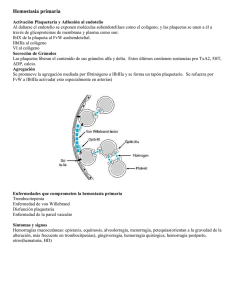
Resumen del Dr. Aparicio (pasado por Irela Soto y Ariel Francis) + documento de la charla. por Graciela Libertad Dixon Hemostasia Primaria Generalidades de la Hemostasia 1ª La hemostasia consta de tres partes: 1. Hemostasia primaria 2. Fase plasmática de la coagulación 3. Fase fibrinolítica La hemostasia primaria se divide en: 1. Fase vascular: integridad de los vasos sanguíneos. 2. Factores que afectan el endotelio vascular: traumáticos, infecciones, medicamentos, etc. 3. Número y función plaquetaria. En una lesión del endotelio, primero entran en juego los mecanismos locales de vasoconstricción y atracción de las plaquetas al sitio de la lesión (mediado por el endotelio). Las plaquetas también participan ya que en su superficie tienen glicoproteínas que tienen receptores correspondientes en el endotelio. Se produce entonces lo que se conoce como ADHESIVIDAD PLAQUETARIA. Las plaquetas también tienen sustancias como el ADP, que al liberarse atrae más plaquetas al sitio de la lesión y les permite interaccionar con otras. Esto contribuye a la AGREGABILIDAD PLAQUETARIA. 1. 2. 3. 4. Entonces se forma el TAPON PLAQUETARIO PRIMARIO, lo cual depende de los siguientes factores: Número adecuado de plaquetas. Plaquetas íntegras con las GP en su superficie, necesarias para la adhesividad y agregabiliidad. Plaquetas con suficientes sustancias: ADP. Exposición al colágeno. Pruebas para valorar la hemostasia 1ª 1. Tiempo de sangría: se coloca un esfigmomanómetro al paciente con una presión constante. Se realiza una incisión en el antebrazo y se mide el tiempo. El valor normal oscila entre 5 y 10 minutos. Aumenta cuando hay menos de 100,000 plaquetas. 2. Adhesividad plaquetaria 3. Agregabilidad plaquetaria: si se tiene un plasma rico en plaquetas y se coloca frente a una fuente de luz, ésta refracta. Al colocar sustancias proagregantes plaquetatias (ADP, colágeno, adrenalina y un AB llamado ristocetina), las plaquetas se unen unas a otras; la liberación de sustancias de sus gránulos trae más plaquetas. Entonces cambia la refracción de la luz, lo cual se puede graficar resultando en curvas características para cada sustancia. A mayor agregación mayor pendiente; todas terminan en un plano. Agregabilidad y sus trastornos La GP IIb-IIIa es el receptor del fibrinógeno que le permite a la plaqueta interaccionar con otras. Su deficiencia produce Trombastenia de Glanzman. Es congénita. Lo que se produce es una disminución de la agregabilidad plaquetaria que conlleva a equimosis, hematomas o traumas pequeños. En la menarquia 1 Resumen del Dr. Aparicio (pasado por Irela Soto y Ariel Francis) + documento de la charla. por Graciela Libertad Dixon lleva a hipermenorrea o aumento de la menstruación. En las pruebas de laboratorio hay disminución del número de plaquetas (ligera; mayor de 100000) y tiempo de sangría aumentado. La GP 1b es el receptor del factor de von Willebrand. Su deficiencia produce el Síndrome de Bernard Soulier, donde hay una disminución de plaquetas pero con macroplaquetas en sangre periférica. El tiempo de sangría está aumentado. El factor plasmástico de la coagulación es el factor de vW, que es importante para la interacción y agregación plaquetaria. Sus funciones son agregar plaquetas y transportar el factor VIII. Su deficiencia produce Enfermedad de vW. Se caracteriza por deficiencia de la función plaquetaria, trastornos de la fase plasmática y una disminución del factor VIII. Puede producir en ambos sexos manifestaciones clínicas de hemorragias (altera la fase plasmática y la primaria) como equimosis, sangrado incontrolable en extracción dentaria, etc. EL tiempo de sangría está aumentado. En la historia clínica pueden aparecer datos como: cordón umbilical, fácilmente se hacen moretones por traumas, caída de los primeros dientes con mucha sangre, etc. La fase plasmática es activada por las plaquetas con el factor de vW; así las plaquetas también participan en la fase plasmática de la coagulación. En conjunto la agregación depende de; GP IIIb-IIa, GP 1b y F vW. La prueba de agregabilidad nos permite hacer el diagnostico. Los resultados pueden ser los siguientes: Síndrome de Bernard-Soulier: la curva de ristocetina es plana (anormal), por que a pesar de ser AB se requiere la presencia del factor de Von Willebrand para agregar las plaquetas, y estos pacientes no tienen los receptores de este factor. El ADP, colágeno y adrenalina van a ser normales. Tromboastenia de Glazman: si sale la curva de ristocetina normal y ADP, colágeno, adrenalina planos. Enfermedad de Von Willebrand: La curva de ristocetina sale plana (anormal) pero se corrige al agregar al agregar plasma normal. La de ADP, colágeno y adrenalina van a ser normales. También tiene TP aumentado por deficiencia de factor VIII. Nota: La curva de agregabilidad es igual a la de Síndrome de BernardSoulier, pero el diagnóstico se hace además por la disminución del factor VIII y la presencia de macroplaquetas en sangre periférica. Otras consideraciones generales Metabolismo de las plaquetas con relación al ácido araquidónico (AAQ): La principal enzima es la ciclooxigenasa (COX), que determina la formación de tromboxano (que produce agregabilidad plaquetaria y vasoconstricción). La inhibición de la COX lleva a la producción de prostaciclinas (que causan vasodilatación y desagregación plaquetaria) Importancia: manipulación de este metabolito con fármacos. Disfunción plaquetaria por medicamentos: Aspirina (ASA): inhibe COX durante toda la vida media de la plaqueta. Se puede dar en profilaxis de trombosis venosa profunda, tromboembolismo. No usar en pacientes con riesgo de IAM o ECV. Versantin: Dipridanol, inhibe el AMPc. Ticlopidina, ticlin: actúa en el peróxido de las plaquetas, es inhibidor de la función plaquetaria. 2 Resumen del Dr. Aparicio (pasado por Irela Soto y Ariel Francis) + documento de la charla. por Graciela Libertad Dixon Medicamentos que inhiben las GP: Clopidogrel. AINES: Naproxeno, voltaren, motrin. Disfunción (inhibición de la función plaquetaria) por alteraciones no hematológicas: IRC: debido a toxinas circulantes. Mieloma múltiple: las proteínas se pegan a la superficie de las plaquetas. En síndromes mieloproliferativos: Trombocitosis primaria LMC Policitemia Vera Metaplasma mieloide. Trastornos cualitativos de las plaquetas Síndrome de Bernard-Soulier Autonómico recesivo Conocido tbn como síndrome de plaquetas gigantes; se ven macroplaquetas (2.5-8 m)1 y aumento de los gránulos densos. Tendencia a la hemorragia de inicio quizá en la lactancia. Síntomas principalmente en piel y mucosas. Síntomas comunes: Epistaxis Hemorragias menstruales excesivas Diátesis hemorrágica (equimosis o petequias). Hemorragia excesiva después de traumatismos. Causas: disminución en la cantidad o fx normal de las fracciones de glucoproteína, incluso gp Ib/IX. Esto impide la interacción de las PLT con el factor de von Willebrand. Diagnóstico: Recuento de PLT normal o ligeramente disminuido. Tiempo de sangría aumentado. Retracción del coágulo normal. Agregación con ristocetina anormal; no se corrige al agregar el factor de von Willebrand. Tratamiento: medidas de soporte; concentrado de PLT y GR cuando sea necesario. Enfermedad de von Willebrand Trastorno hemorrágico hereditario + común. Autonómica dominante. En muchos pt con la enfermedad, tbn se ve afectado el factor VIII. Funciones fisiológicas de la proteína (factor): Mediar la adhesión de las PLT a los sitios de lesión vascular, mediante la unión a la gp Ib/IX y al colágeno en el subendotelio. Facilitar la agregación mediante la unión a la go IIb/IIIa. Unirse al factor VIII y protegerlo de su degradación proteolítica. La ausencia del FvW funcional tbn puede llevar a problemas menores de la coagulación (por deficientia del factor VIIIc). Clasificación: 1 Lo normal es de 2-3m. 3 Resumen del Dr. Aparicio (pasado por Irela Soto y Ariel Francis) + documento de la charla. por Graciela Libertad Dixon Tipo I: déficit cuantitativo parcial. Deficiencia de F vW pero el poco que hay funciona Se caracteriza por aumento del tiempo de sangría y TP, más una curva plana de agregación plaquetaria con ristocetina. 70% casos. Tipo II: deficiencia de F vW pero el que existe es disfuncional, por lo que hay hipoagregabilidad plaquetaria aumento del tiempo de sangría y TP, agregabilidad con ristocetina normal o un poco aumentada. Tipo IIA: cualitativo; más común de los tipo II. Multímeros grandes e intermedios del plasma y de las PLT disminuidos. Hemorragia leve a moderada. Tipo IIB: cualitativo; disminución de los multímeros grandes SOLO del plasma. Aumento en la agregación de PLT con ristocetina xq’ el defecto hace a los multímeros más reactivos, de modo q’ se sintetizan e inmediatamente son retirados de la circulación. Hay trombocitopenia. Tipo IIC: cualitativo; estructura molecular normal. Autonómico recesivo. Tipo III: deficiencia completa del FvW; homocigoto o doblemente heterocigoto. Pacientes con síntomas graves. El FvW está disminuido, el factor VIII casi indetectable y la actividad de ristocetina reducida. Se comporta como una hemofilia, hay aumento del tiempo de sangría y el TP. La curva de agregabilidad plaquetaria con ristocetina es plana, hay menos de 1% de factor VIII lo que produce muchas hemorragias. Síntomas: Variantes leves: 2ª década; se parecen a los de los tr de las PLT. Variantes intensas: inicia temprano y disminuye con la edad; hemartrosis y hemorragia profunda espontánea. Stx + comunes: epistaxis, gingivorragia, hipermenorrea, equimosis fácil. A veces: sangrado GI, sangrado prolongado luego de procedimientos dentales, etc. Los cambios hormonales, el estrés, el embarazo, inflamación, infecciones pueden estimular la producción del FvW y mejorar temporalmente el cuadro. Pt del grupo sanguíneo 0 tienen valores 20-25% menores del FvW. Diagnóstico: Hx personal de hemorragias mucocutáneas excesivas. Hx familiar de hemorragias excesivas. Hallazgos de laboratorio: defecto cuantitativo y/o cualitativo del FvW. 1. Tiempo de sangría elevado. 6. Número de plaquetas normal 2. FC VIII 7. TPR anormal 3. FvW disminuido 8. TT anormal 4. Agregabilidad disminuido 9. TP normal 5. Adhesividad disminuida 10. Ristocetina anormal. Pruebas para cuantificar el FvW: análisis del antígeno de von Willebrand (Ac heterófilos), análisis del plasma. Pruebas cualitativas: inmunoelectroforesis cruzada y electroforesis del plasma. Geles dodecil sulfato sódico (DSS). El FvW normal se separa en tripletos. Tratamiento: reemplazo vs. Inducción de la liberación del complejo de las reservas titulares Tx con desmopresina (DDAVP): es un análogo sintético de la vasopresina que aumenta el factor VII y el FvW sin importantes efectos colaterales. Tx transfusional: concentrados de factor VIII/FvW. Antifibrinolíticos: ácido tranexámico IV en dosis de 30-50 mg/kg/día dividido c/8 horas; tbn tópico. Estrógenos: aumentan los niveles de Fvw; para reducir la severidad de la menorrgia. Concentrados de FvW recombinante: no se ha probado en humanos. Tipo I + hemorragia que no es severa (como en extracción de dientes o en menstruación) dar desmopresina 4 Resumen del Dr. Aparicio (pasado por Irela Soto y Ariel Francis) + documento de la charla. por Graciela Libertad Dixon Tipo II NO DAR desmopresina porque produce trombocitopenia con predisposición a hemorragias. Tipo III dar factor VIII (crioprecipitado). En todas las anteriores, si es hemorragia excesiva se tiene que dar factor VIII rico en f vW, que se obtiene del plasma fresco congelado que contiene IX, X, f VW, fibrinógeno (no es recomendable dar si no es tratado al calor y con solvente detergente, como ocurre en el interior, si no más bien usar el producido mediante fase industrial del plasma en donde se prepara el compoennte plasmático y se separa por el VIH+. Trombastenia de Glanzman Trastorno de la agregación plaquetaria Autonómico recesivo. Solo los homocigotos muestran síntomas. Deficiencia del complejo proteínico gp IIb/IIIa. Tampoco pueden unirse al fibrinógeno. Episodios repetidos de hemorragias. Sangrado en áreas superficiales, características de las anormalidades plaquetarias. Gravedad moderada. Laboratorios: Recuento plaquetario normal. Tiempo de sangría prolongado. Retracción del coágulo anorma No hay respuesta con ADP, adrenalina o colágena en la prueba de agregación plaquetaria. Agregación con ristocetina normal. Tratamiento: transfusión de PLT normales. En algunos casos transplante de médula ósea2. Síndrome de la Plaqueta Gris Trastorno de la secreción de las PLT: Deficiencia proteínica en el contenido de los gránulos alfa; FP4, -tromboglobulina, fibrinógeno, PDGF. Frotis: plaquetas agrandadas y grises. Aumento del tiempo de sangría, hemorragias mucosas, trombocitopenia con PLT grandes. Trastornos Cuantitativos de las Plaquetas Trombocitopenia Generalidades Se define como una cifra de PLT menor de 150mil/L. Causa más común de hemorragia excesiva o anormal. No se presentan stx clínicos a menos q’ la cifra caiga por debajo de 50mil/L. El tiempo de sangría está aumentado. Las pruebas de lab para los factores de la coagulación no se afectan. Si hay < 20 mil plaquetas hay riesgo de presentar sangrado espontáneo. 2 Según el documento del grupo, el resumen del Dr. Aparicio solo habla de transfusión. 5 Resumen del Dr. Aparicio (pasado por Irela Soto y Ariel Francis) + documento de la charla. por Graciela Libertad Dixon Si hay < 100 mil plaquetas, y el paciente tiene trauma o cirugía, no tiene el número suficiente para resolver la hemorragia pero sin embargo no sangra espontáneamente. Si hay > 100 mil plaquetas, la hemostasis es normal en cuanto al número de plaquetas y entonces depende de su funcionamiento. La trombocitopenia inmunitaria se presenta en varias situaciones clínicas: variantes idiomáticas, trombocitopienia transplacentaria, aloanticuerpos por transfusión o embarazo, vinculado a fármacos. Causas Disminución o falta en la producción en la médula ósea: Aplasia medular (Anemia aplásica) Mieloptisis: Infiltración medular neoplásica por hongos, VIH, etc. Reemplazo de la médula ósea. Mielofibrosis: fibrosis de la médula ósea. Trastornos de maduración Anemia megaloblástica Trombopoyesis ineficaz Síndrome mielodisplásico con hematopoyesis ineficaz. Aumento de la destrucción o utilización3 Por causas inmunológicas A. Mediada por anticuerpos: Púrpura trombocitopénica inmune (PPI) asociada a infección viral (VIH), neoplasia, fármacos, enfermedades autoinmunes (LES, etc), Sd. hemolítico-urémico. Por causas no inmunológicas A. Hiperesplenismo (secuestro esplénico, que es muy diferente a esplenomegalia): se caracteriza por un aumento en la función del bazo captando elementos de la sangre periférica. Puede ocurrir en patologías como: 1. Esferocitosis hereditaria 2. Metaplasia mieloide agnogénica aguda 3. Cirrosis hepática 4. Leucemia de células vellosas 5. Crisis de anemia falciforme: paciente falcémico doble heterocigoto en niños 6. Síndrome de Felty: Donde hay hiperesplenismo asociado a artritis reumatoide. 7. Enfermedad de Gaucher4 B. Coagulación Intravascular diseminada C. Inducida por fármacos5 Púrpura Trombocitopénica Inmune o Idiopática (PTI) Variante + común de trombocitopenia. Tipos principales: aguda, crónica, intermitente. En lactantes nacidos de madres con PTI se conoce como púrpura trombocitopénica autoinmunitaria transplacentaria. En el examen de médula ósea: para evaluar los megacariocitos y descartar otras causas. Los megacariocitos aumentan; la MO puede aumentar la produx hasta 5 veces lo normal. Loa megacariocitos también aumentan su masa citoplasmática. El dx es por exclusión. La PTI aguda se presenta con frecuencia siguiendo a una infex viral (luego de 1-3 semanas); infex de vías respiratorias superiores inespecíficas. No hay esplenomegalia ni fiebre. Sin recidivas. La PTI recidiva en el 10-12% a 1-5 años. 3 El tiempo de supervivencia de la PLT está diminuido según lo determinan las pruebas Cr 51. de Minutos a 2-3 días. De acuerdo con el cuadro del documento del grupo. 5 Extra de los apuntes 4 6 por Graciela Libertad Dixon Resumen del Dr. Aparicio (pasado por Irela Soto y Ariel Francis) + documento de la charla. Cuadro comparativo Característica Edad de incidencia máxima Predilección por sexo Cifra de plaquetas Inicio de hemorragia Duración Bulas hemorrágicas en la boca PTI aguda Niños: 2 a 6 años Ninguna < 20 x 109/L Abrupta Comúnmente 1 a 3 semanas antes del comienzo. 2 a 6 sem, pocas veces más Presente en casos graves Remisiones En 80% de los casos Eosinofilia y linfocitosis Común Tratamiento Prevención de traumatismos, corticoesteroides. Antecedentes infecciosos PTI crónica Adultos, de 20 a 40 años Más común en mujeres (3:1) 30 a 80 x 109/L Insidiosa Inusual Meses o años (toda la vida) Ordinariamente ausentes No comunes, el curso de la enfermedad fluctúa. Rara Corticoesteroides, esplenectomía; alternative de dosis altas de gammaglobulina. Causas: Causas de PTI Aguda 2aria a infecciones virales (niños) 2aria a fármacos: Heparina, sales de oro Neonatal Hijo de madre con PTI Post-transfusional Crónica LES Síndrome de Evans: PTI con anemia hemolítica HIV LLC Hepatopatías crónicas Transplante de m.o. Hipertiroidismo Diagnóstico general: Trombocitopenia de moderada a severa. Sólo una línea celular afectada: plaquetas GB y Hb normal En la M.O. hay hiperplasia megacariocítica sin producción suficiente de plaquetas (no se liberan). Historia clínica: Pacientes pediátricos: Trombocitopenia inmunológica es autolimitada y no requiere tratamiento. Pródromo de infección faríngea viral: ↓ plaquetas y hay petequias en las extremidades. Su curso es mayor y se cree que es producida por cualquier virus como rubéola, varicela, VEB. Adolescentes y adulto joven: Mujer: enf. autoinmune LES (historia de eritema facial, caída del cabello, artralgias). Hombre o mujer sexualmente activo con factores de riesgo: VIH + Adulto mayor: preguntar por medicamentos. AINES Sulfas Sales de oro: pacientes con artritis reumatoide. Antiarrítmicos: quinidina, procainamida en pt de mayor edad. Tercera edad Trastornos linfoproliferativos: LLC, hipogamaglobulinemia, AHAI. La médula ósea es hipercelular con ↑ de megacariocitos de superficie lisa (poca producción de PLT). 7 Resumen del Dr. Aparicio (pasado por Irela Soto y Ariel Francis) + documento de la charla. por Graciela Libertad Dixon Linfoma de bajo grado de malignidad. Hepatitis viral. Tratamiento inicial: Esteroides: prednisona 1-2mg/Kg x 4-6 sem; 1/3 responde bien al tx único con esteroides (↑ PLT). Algunos se tratan con dosis elevadas de metilprednisolona. Si tiene recaída o necesita esteroides constantemente. Tx fase 2: esplenectomía si ha tenido recaída con los esteroides (hay aumento de plaquetas o se produce trombocitopenia) de los cuales 1/3 responden bien. Tx en embarazada: Gammaglobulina humana hiperinmune que disminuye sangrado, no hay efecto colateral de los esteroides y disminuye la púrpura neonatal. Al 1/3 que no responde a fase inicial y 2º, se le da Tx inmunosupresor: Vincristina, Vinblastina, Gammaglobulina humana (reduce púrpura trombocitopénica neonatal) Danazol (andrógeno que también se usa en Anemia aplásica) Azatriopina, ciclofosfamida Trombocitopenia de causa no inmunológica En embarazo-preeclampsia, síndrome de Hell Aumento de la destrucción de plaquetas (por HT + aumento de enzimas hepáticas). CID por consumo de plaquetas. Púrpura Trombocitopénica Trombótica: Aumento en el consumo de plaquetas. Laboratorios: La prueba de torniquete: no se usa porque depende de demasiados factores (fase plaquetaria, fase plasmática, fibrinolítica). Adhesividad plaquetaria: generalmente no se usa porque depende de demasiados factores. Las más útiles y específicas son: Recuento plaquetario: por el método de Coulter (en los labs automáticos) puede ser una pseudotrombocitopenia cuando se usa EDTA en el tubo (plaquetas agregadas) por lo que hay que ver el frotis de sangre periférica y correlacionar con el volumen medio de plaquetas. Tiempo de sangría Retracción del coágulo: depende de varios factores (# de PLT, fx plaquetaria, fase plasmática, fase fibrinolítica). Púrpura trombótica trombocitopénica (PTT) Se caracteriza por fiebre, insuficiencia renal y déficit neurológico* pasajero (cefaleas, paresias, afasia, síncope, disartria, etc.) Está dentro de los síndromes conocidos como microangiopatía trombótica, junto con el sd. hemolítico urémico del cual se distingue porque en el segundo no hay stx neurológicos. Síntoma ppal: aparición de trombos hialinos en la microcirculación. Conteos de PLT y GR bajos (hemólisis microangiopática; microesferocitos y esquizocitos). Los microtrombos están compuestos ppmte por PLT y fibrina en menor proporción, contrario al CID. Disfunción isquémica de los órganos; usualmente cerebro, riñón, vísceras abdominaes y corazón. Usualmente es idiomática, pero puede estar asociada o ser secundaria a infecciones, medicamentos, vacunas, enfermedades autoinmunes y oncológicas. Puede agravarse en el embarazo y puerperio. Laboratorio: anemia hemolítica grave con Coombs negativo, esquizocitos o eritrocitos fragmentados, complemento e IgG normales. Tratamiento: plasmaféresis intensiva con plasma fresco congelado. Coagulación intravascular diseminada (CID) Alteración en el equilibrio normal de la hemostasis. Formación inadecuada y no controlada de fibrina dentro de los vasos (microcirculación ppmte). 8 Resumen del Dr. Aparicio (pasado por Irela Soto y Ariel Francis) + documento de la charla. por Graciela Libertad Dixon Se consumen varias proteínas de la coagulación + rápido de lo q’ se sintetizan. Las PLT quedan atrapadas, se agregan y se retiran de la circulación. Cuadro clínico: coagulación y fibrinolisis. Etiología: infecciones, complicaciones del embarazo (retención placentaria, émbolo del líquido amniótico, muerte fetal IU, aborto séptico), neoplasias, lesión tisular masiva (quemaduras, etc.), lesión vascular, mordeduras de serpiente, shock térmico, etc. Más común en jóvenes y en edade avanzadas. Variante + común: inicio súbito de hemorragia intensa. CID aguda: abrupta; hematuria, hemorragia GI, equimosis, petequias, etc. CID crónica: insidiosa; stx leves o ausentes. Usualmente trombosis. Anoxia tisular y microinfartos (corazón, riñón, cerebro, hígado, páncreas) estados de shock. Laboratorios: PLT disminuidas; TP, TTP, TT aumentados (a veces disminuidos); fibrinógeno, plasminógeno, ATIII disminuidos; fibrinopéptido A aumentado. Pruebas de sulfato de protamina y degradación de fibrina positivas. Tratamiento: hemorragia (plasma fresco congelado), trombosis (heparina IV). Alteraciones de la fase vascular Causas del daño al endotelio Vasculitis Leucoplástica Púrpura de Henoch-Scholein: caracterizada por púrpura en las extremidades, nefritis, sangrado por mucosas en pacientes pediátricos después de un cuadro faríngeo con un número de plaquetas normal6, pero con lesiones a los vasos sanguíneos (vasculitis). LES Reacción alérgica a medicamentos: AINES Deficiencia de vitamina C (Escorbuto): causa disminución del soporte al endotelio. Trastornos en el endotelio: el síndrome de hiperelasticidad de Erhles Danlos produce hemorragia. Síndrome de Rendu-Osler-Weber: se asocia con trastornos como telangectasias, con fragilidad y se rompe en el pulmón, TGI, Piel y mucosas. Trastornos de causa infecciosa a. Bacterianas: meningococcemia (produce lesión hemorrágica EC) b. Virales: dengue hemorrágico. c. Endocarditis infecciosa productora de émbolos sépticos que dañan al endotelio vascular. Trastornos hereditarios del sistema vascular Telangiectasia Hereditaria o Enfermedad de Rendu-Osler-Weber Autosómica dominante. Se asocia a falta de fibras elásticas en una pared vascular delgada y sin soporte perivascular por la presencia de tejido conectivo anormal. Las uniones endoteliales tbn sond efectuosas. Lesiones vasculares del tamaño de una cabeza de alfiler (hasta 3 mm), color rojo púrpura, ligeramente elevadas, palidecen con la presión. En cualquier parte del cuerpo; ppmte en la piel de la cara, orejas, manos, pies, mucosa de nariz y boca. Manifestación + común: epistaxis. Sospechar diagnóstico: hemorragia recurrante, telangiectasia, antecedentes familiares de hemorragia. 6 A diferencia de la PTI aguda ( 20mil/L). 9 Resumen del Dr. Aparicio (pasado por Irela Soto y Ariel Francis) + documento de la charla. por Graciela Libertad Dixon Complicaciones: hemoptisis, epistaxis, riesgo aumentado dederrames e infecciones cerebrales, insuficiencia cardiaca, insuficiencia respiratoria, HT pulmonar. Diagnóstico: laboratorio de anemia, ferropenia en hemorragias crónicas, estudios de coagulación normales. Tratamiento sintomático y de soporte. Sd. de Ehlers-Danlos (SED) Enfermedades hereditarias del tejido conectivo por anormalidades de la estructura, procesamiento o síntesis de la colágena. Más frecuente en individuos de etnia negra. Características clínicas: hiperlaxitud articular, hiperextensibilidad de la piel, fragilidad de los tejidos. Manifestaciones hemorrágicas: magulladuras, hematomas SQ, púrpura, sangrado de la mucosa tras procedimientos dentales, hemoptisis, sangrado GI. Anormalidades en las fibras de colágeno perivasculares. Pueden presentar tiempo de sangría y pruebas de agregación plaquetaria anormales. El único que tiene características clínicas de carácter hematológico es el tipo IV. Gen COL3A1, 2q. se produce un colágeno tipo 3 anormal (abundante en las paredes arteriales). Los pt son propensos a aneurismas arteriales y disección de la aorta. Complicaciones hematológicas: prolapso de la válvula mitral, dificultad para las heridas quirúrgicas o para retirar puntos; ruptura de grandes vasos, aneurisma aórtico, u órgano hueco; ruptura prematura de membranas. Diagnóstico: criterios clínicos. Examenes de laboratori incluyen tipificación del colágeno, nivel sérico de Cu y ceruloplasmina, actividad de lisisl oxidasa o lisil hidroxilasa, examen de agregación plaquetaria, etc. No existe tx específico. Se deben hacer ecografías a intervalos regulares para detectar precozmente los aneurismas, aunque su tx quirúrgico es difícil. Sd. de Marfan Trastorno hereditario del tejido conectivo en el cual se fortalecen las estructuras corporales. Se afectan los sistemas esquelético y cardiovascular, la piel y los ojos. Autonómico dominante; en el 30% no hay hx familiar “esporádicos”. Mutaciones en el gen fibrilina-1, cromosoma 15. Cuadro clínico: extremidades largas y finas, disminución de la visión (por luxación del cristalino “ectopia lentic”) y aneurismas aórticos. Alteraciones cardiovasculares: dilatación de la raiz de la aorta con regurgitación aórtica y prolapso de la válvula mitral. Pueden desarrollar un aneurisma aórtico disecante. Diagnóstico: cuadro clínico; ecocardiograma, exploración ocular con lámpara de hendidura. Hay q’ descartar homocistinuria mediante un análisis de cianuronitroprusiato negativo para la presencia de disulfuros en orina. Tratamiento (cardiovascular): betabloqueadores para prevenir el estrés en la aorta, evitar actividades atléticas competitivas y deportes de contacto. Ecocardiogramas cada año. En algunos casos se requiere el reemplazo quirúrgico de la válvula y raiz aórticas. Antibióticos para prevenir endoccarditis, vigilancia cercana en el embarazo. Trastornos adquiridos del sistema vascular Púrpura Vascular Daño al endotelio vascular Infecciones de bacterias productoras de endotoxinas: émbolos sépticos Complejos inmunes circulantes: LES 10 Resumen del Dr. Aparicio (pasado por Irela Soto y Ariel Francis) + documento de la charla. por Graciela Libertad Dixon Accidentes y mordeduras de ofidios Daño a la estructura de soporte de la microcirculación Escorbuto Amiloidosis Trastornos del tejido conectivo hereditario Exceso de corticoides La más típica de todas las púrpuras es la púrpura senil: lesión por daño en endotelio vascular por la edad en miembros inferiores Vasculitis leucocitoclásica Enfermedad reumática Disproteinemia Enfermedad mediada por complejos inmunes: Púrpura de Henoch-Schonlein, caracterizada por ser en pacientes pediátricos después de un cuadro infeccioso faríngeo, produce nefritis por vasculitis y el Tx es la disminución de los complejos inmunes con esteroides Daño a la microcirculación por émbolos sépticos Púrpura autoeritrocítica El Tx es igual: corticoides, esplenectomía o inmunosupresión. Correlación de VMP y RDW RDW 20 + VCM 60: déficit de Fe RDW 14 + VCM 65: Talasemia VMP ↑ y trombocitopenia: lo más probable es que sea por aumento de la destrucción porque se envían plaquetas jóvenes o macroplaquetas a sangre periférica. VMP ↓ y trombocitopenia: lo más probable es que sea por la falta de producción: Anemia aplásica y trombocitopenia secundaria a quimioterapia VMP ↑ y pseudotrombocitopenia: anemia megaloblástica, Síndrome mieloproliferativo, Enfermedad de Bernard-Soulier. VMP ↑: Anemia megaloblástica Trombocitopenia Síndrome Mieloproliferativo Enfermedad de Bernard-Soulier Caso clínico Mujer de 32 años, dos abortos, dolor torácico, disnea, sensación de pesadez en el miembro inferior. Signo de Homans en miembro inferior derecho, ruidos respiratorios normales, equimosis y no es diabética. Signos vitales: Fc: 130/min Fr: 28cpm afebril Laboratorio: Hb 11g/dl ; Plt 90000 (trombocitopenia) ; WBC N ; TP 15/normal 14 ; TPT 50/normal 30. Posibles diagnósticos: Tromboembolismo Pulmonar, PTI, Déficit de algunos inhibidores fisiológicos, Déficit de un factor de la coagulación, Anticuerpos antifosfolípidos: 1º , 2º a LES Diagnóstico: Abortos recurrentes, Embolismo pulmonary, TPT pronlongados, Ac antifosfolípidos 1º(Trombocitopenia), Ac antifosfolípidos 2º a LES Tratamiento: Fraxiheparina no se monitoriza, Heparina IV y si se monitoriza con TPT: 5000-1000/h por 7-10 días; Cumarin, monitorizar con INR con 2.5-3(TP); Corticoides para Ac. Antifosfolípidos 2º a LES; Aspirina para: Ac antifosfolípidos 1º(trombocitopenia); Si se quiere embarazar debe pasar 3-6 meses con warfarina (teratogénica), se le quita la warfarina y se mantiene con aspirina. Dar heparina subcutánea durante todo el embarazo y si el paro es por cesárea quitar aspirina 7 días antes. 11