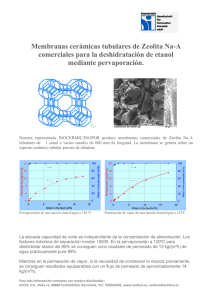
papel del sistema endocannabinoide en el alcoholismo
Anuncio

MENÚ SALIR DEPARTAMENTO DE BIOLOGÍA CELULAR, GENÉTICA Y FISIOLOGÍA UNIDAD DE INVESTIGACIÓN HOSPITAL CIVIL PAPEL DEL SISTEMA ENDOCANNABINOIDE EN EL ALCOHOLISMO: ESTUDIO EN MODELOS ANIMALES Irene Sánchez Vera MÁLAGA, 2007 MENÚ SALIR MENÚ SALIR A la memoria de mi abuela Encarna, A mi madre, A mi familia “La vida sólo se puede comprender mirando hacia atrás pero sólo puede vivirse mirando hacia delante” Kierkegaard MENÚ SALIR MENÚ SALIR ÍNDICE MENÚ SALIR MENÚ SALIR INTRODUCCIÓN 1. LA ADICCIÓN A ALCOHOL.................................................................................1 1.1. PROBLEMAS CLÍNICOS Y SOCIALES..................................................1 1.2. FARMACOLOGÍA DEL ALCOHOL ........................................................2 1.2.1. Metabolismo del Etanol en el Cerebro ......................................2 1.2.2. Interacciones del Etanol con los Sistemas de Neurotransmisión ......................................................................3 1.2.2.1. Efectos del Etanol sobre el Sistema de Neurotransmisión GABAérgico ..................................5 1.2.2.2. Efectos del Etanol sobre el Sistema de Neurotransmisión Glutamatérgico...............................7 1.2.2.3. Efectos del Etanol sobre el Sistema de Neurotransmisión Dopaminérgico.............................10 1.2.2.4. Efectos del Etanol sobre el Sistema de Neurotransmisión Opioide.........................................11 1.2.2.5. Efectos del Etanol sobre el Sistema de Neurotransmisión Serotoninérgico ............................12 1.2.2.6. Efectos del Etanol sobre el Neuromodulador Adenosina......................................14 1.3. SISTEMAS NEUROBIOLÓGICOS IMPLICADOS EN LA ADICCIÓN A ALCOHOL........................................................................15 MENÚ SALIR 1.4. MODELOS ANIMALES DE ALCOHOLISMO: líneas msP y AA/ANA....................................................................................................20 1.5. PERSPECTIVAS DE TRATAMIENTO DE LA ADICCIÓN A ALCOHOL ............................................................................................22 2. EL SISTEMA ENDOCANNABINOIDE ..............................................................25 2.1. HISTORIA ................................................................................................25 2.2. LIGANDOS EXÓGENOS ........................................................................26 2.3. LIGANDOS ENDÓGENOS .....................................................................27 2.4. 2.3.1. Biosíntesis de Anandamida .....................................................29 2.3.2. Biosíntesis de 2 araquidonilglicerol ........................................31 RECEPTORES CANNABINOIDES ........................................................33 2.4.1. 2.5. Mecanismos de Transducción de Seńales ...............................35 NEUROFARMACOLOGÍA DEL SISTEMA ENDOCANNABINOIDE .........................................................................37 3. ALCOHOL Y SISTEMA ENDOCANNABINOIDE ...........................................43 3.1. Efectos Agudos del Alcohol sobre el Sistema Endocannabinoide .....................................................................................43 3.2. Efectos Crónicos del Alcohol sobre el Sistema Endocannabinoide .....................................................................................43 MENÚ SALIR 3.3. Interacción de Cannabinoides y Alcohol: una Nueva Diana para el Tratamiento....................................................................................45 OBJETIVOS MATERIALES Y MÉTODOS 1. ANIMALES DE EXPERIMENTACIÓN..............................................................55 1.1. RATAS WISTAR......................................................................................55 1.2. MODELOS ANIMALES DE ALCOHOLISMO......................................55 1.3. 1.2.1. Ratas AA .................................................................................55 1.2.2. Ratas msP ................................................................................56 ADMINISTRACIÓN DE ALCOHOL......................................................57 1.3.1. Tratamientos Intraperitoneales de Etanol ................................57 1.3.2. Entrenamiento Operante y Autoadministración de Alcohol ....................................................................................58 1.4. CIRUGÍA ..................................................................................................60 1.5. ADMINISTRACIÓN DE DROGAS ........................................................60 1.6. MICRODIÁLISIS IN VIVO .....................................................................61 1.7. MEDIDA DE LA ACTIVIDAD LOCOMOTORA Y LA TEMPERATURA CORPORAL ...............................................................62 1.8. DISECCIONES DE CEREBROS DE RATA ...........................................63 MENÚ SALIR 2. ANÁLISIS DE EXPRESIÓN GÉNICA DEL SISTEMA ENDOCANNABINOIDE........................................................................................65 2.1. PCR CUANTITATIVA A TIEMPO REAL .............................................65 2.1.1. Puesta a Punto de la PCR cuantitativa.....................................66 2.1.1.1. Diseño de los Cebadores .....................................66 2.1.1.2. Determinación de la Temperatura de Alineamiento de los Cebadores ...........................66 2.1.1.3. Secuencia de los Cebadores y Temperatura de Alineamiento .............................67 2.1.1.4. Producción y Purificación del Estándar ..............................................................68 2.2. 2.1.2. Extracción de ARN total..........................................................69 2.1.3. Limpieza del ARN total...........................................................69 2.1.4. Cuantificación del ARN ..........................................................70 2.1.5. Electroforesis de ARN.............................................................70 2.1.6. Transcripción Reversa ...........................................................171 2.1.7. PCR Cuantitativa a Tiempo Real...........................................172 HIBRIDACIÓN in situ DEL ARNm DEL RECEPTOR CANNABINOIDE CB1 EN MODELOS ANIMALES DE ALCOHOLISMO (líneas AA y msP) .......................................................75 2.2.1. Procesamiento de los Cerebros................................................75 2.2.2. Producción de la ribosonda de CB1 ........................................76 2.2.3. Hibridación in situ ...................................................................76 MENÚ SALIR 2.2.4. Obtención de datos ..................................................................77 3. CUANTIFICACIÓN DE LA EXPRESIÓN PROTEICA MEDIANTE INMUNOHISTOQUÍMICA............................................................78 3.1. Procesamiento del Tejido ..........................................................................78 3.2. Inmunohistoquímica ..................................................................................79 3.3. Cuantificaciones ........................................................................................80 4. ANÁLISIS DE LA FUNCIONALIDAD PROTEICA..........................................83 4.1. Extracción de Membranas .........................................................................83 4.2. Cuantificación de Proteínas .......................................................................83 4.3. Ensayo de la Actividad Enzimática de la Amidohidrolasa de Ácidos Grasos (FAAH) .............................................................................85 4.4. Ensayo de la Actividad Enzimática de la Transferasa de grupo N-acilo (NAT) ...........................................................................................86 4.5. Ensayo de Unión del receptor CB1 ...........................................................86 4.6. Ensayo de Acoplamiento del receptor CB1...............................................87 5. MEDIDAS BIOQUÍMICAS ...................................................................................87 5.1. MEDIDA DE LOS NIVELES DE ETANOL EN PLASMA....................87 5.2. ANÁLISIS DEL CONTENIDO ENDOCANNABINOIDE .....................88 5.2.1. Ratas Wistar.............................................................................88 MENÚ SALIR 5.2.2. 5.3. Ratas AA y ANA.....................................................................89 ANÁLISIS DEL CONTENIDO DE GLUTAMATO ...............................90 6. ANÁLISIS ESTADÍSTICO ....................................................................................92 7. PROTOCOLOS EXPERIMENTALES ................................................................93 7.1. Extracción de ARN total ...........................................................................93 7.2. Purificación del ARN total ........................................................................94 7.3. Transcipción Reversa ................................................................................95 7.4. PCR Cuantitativa a Tiempo Real...............................................................96 7.5. PCR para la determinación de la Temperatura de Alineamiento de los Cebadores........................................................................................97 7.6. Producción del Estándar mediante PCR....................................................97 7.7. Purificación del Estándar...........................................................................98 7.8. Inmunohistoquímica de CB1 .....................................................................99 7.9. Inmunohistoquímica de FAAH ...............................................................100 7.10. Inmunohistoquímica de MAGL ..............................................................101 7.11. Inmunohistoquímica de NAPE-PLD .......................................................101 7.12. Ensayo de la Actividad Enzimática FAAH .............................................102 7.13. Ensayo de la Actividad Enzimática NAT................................................103 7.14. Ensayo de Unión del receptor CB1 .........................................................104 MENÚ SALIR 7.15. Ensayo de Acoplamiento del receptor CB1.............................................105 7.16. Medida de Etanol en ...............................................................................105 7.17. Soluciones de Uso Común.......................................................................106 RESULTADOS 1. TRATAMIENTO AGUDO DE ETANOL ..........................................................111 1.1. Medida de los Niveles de Etanol y Anandamida en Plasma y Tejidos Periféricos...................................................................................111 1.2. Medida de los Niveles de Endocannabinoides en Cerebro......................112 1.3. Medida de la Actividad Enzimática FAAH.............................................115 1.4. Medida de la Actividad Enzimática NAT ...............................................117 1.5. Medida de los niveles de ARNm del Sistema Endocannabinoide ...................................................................................117 1.6. Inmunohistoquímica de CB1 y FAAH ....................................................119 1.7. Medida de los niveles de Anandamida y Glutamato en Estriado Ventral.......................................................................................129 1.8. Medida de la Temperatura Corporal y Actividad Locomotora ...............131 2. TRATAMIENTO CRÓNICO DE ETANOL......................................................132 2.1. Medida de los niveles de ARNm del Sistema Endocannabinoide ...................................................................................132 MENÚ SALIR 2.2. Inmunohistoquímica de CB1 y FAAH ....................................................134 3. DISCUSIÓN PARCIAL........................................................................................144 4. MODELOS ANIMALES DE ALCOHOLISMO ...............................................145 4.1. Ratas AA y ANA.....................................................................................145 4.1.1. Medida de los niveles de ARNm del Sistema Endocannabinoide .................................................................145 4.1.2. Medida de la Actividad Enzimática FAAH...........................146 4.1.3. Medida de los Niveles de Endocannabinoides ......................147 4.1.4. Estado Funcional del Receptor Cannabinoide CB1 ..............147 4.1.5. Medida de la Expresión Génica del Receptor CB1 mediante Hibridación in situ .................................................148 4.1.6. Efecto del antagonista SR141716A sobre la Autoadministración de Alcohol.............................................150 4.2. Ratas msP y Wistar..................................................................................152 4.2.1. Medida de los niveles de ARNm de CB1 mediante Hibridación in situ .................................................................152 4.2.2. Medida de los niveles de ARNm del Sistema Endocannabinoide .................................................................155 4.2.3. Medida de la Expresión Proteica del Sistema Endocannabinoide .................................................................157 MENÚ SALIR DISCUSIÓN 1. Papel General del Sistema Endocannabinoide en el Alcoholismo ....................161 2. Acciones Agudas del Etanol y Adaptación Crónica ...........................................163 2.1. Acciones Agudas del Etanol....................................................................163 2.2. Acciones Crónicas del Etanol..................................................................165 3. Selección de Preferencia Alcohólica y Sistema Endocannabinoide ..................168 4. Implicaciones Terapéuticas ..................................................................................173 4.1. Dependencia Severa a Alcohol y Sistema Endocannabinoide ................173 4.2. Recaídas y Sistema Endocannabinoide ...................................................173 4.3. Futuro: Tratamiento del Alcoholismo con Fármacos del Sistema Endocannabinoide......................................................................177 CONCLUSIONES BIBLIOGRAFÍA MENÚ SALIR MENÚ SALIR ABREVIATURAS MENÚ SALIR MENÚ SALIR A: adenina DEPC: dietil pirocarbonato ADH: alcohol deshidrogenasa dGTP: desoxiguanosina trifosfato ADN: ácido desoxirribonucleico DMSO: dimetil sulfóxido ADNc: ADN complementario dNTP: dinucleótidos trifosfato AEA: anandamida o araquidoniletanolamida D.O.: densidad óptica AM404: N-araquidonoilfenolamina; inhibidor del transportador de anandamida Dpm: desintegraciones por minuto AMPA: ácido α-amino 3-hidroxi 4-metil 5ixazol propiónico DSE: supresión de la excitación inducida por despolarización 1-AG: 1-araquidonilglicerol DSI: supresión de la inhibición inducida por despolarización 2-AG: 2-araquidonilglicerol DTT: ditiotreitol ALDH: aldehído deshidrogenasa dTTP: desoxitimidina trifosfato AMPc: 3’,5’-adenosínmonofosfato cíclico DV: dorso-ventral AT: transportador de anandamida 2E1: subtipo 2E1 del citocromo P450 ARNm: ácido ribonucleico mensajero FAAH: amidohidrolasa de ácidos grasos ARNt: ácido ribonucleico transferente G: guanina ATP: trifosfato de adenosina GABA: ácido γ-aminobutírico ATV: área tegmental ventral GABAA: subtipo A del receptor del ácido γaminobutírico AP: antero-posterior BAC: concentración de alcohol en sangre BZD: benzodiacepinas GABAB: subtipo B del receptor del ácido γaminobutírico C: citosina GAPDH: deshidrogenasa de gliceraldehído 3fosfato Ca2+: ión calcio Gi/o: proteína G inhibidora/estimuladora CB: receptor cannabinoide Glu: glutamato CB1: receptor cannabinoide tipo 1 GTPγS: guanosina 5’(γ-tio)trifosfato CB2: receptor cannabinoide tipo 2 GUS: β-glucuronidasa Cl- : iones cloro HB-β-CD: hidroxipropil-β-ciclodextrina Cpm: cuentas por minuto HCl: ácido clorhídrico CP-55,940: 3-(2-hidroxi-4-(1,1-dimetil heptil)fenil)-4-(3-hidroxipropil)ciclohexanol; agonista de los receptores cannabinoides HEPES: ácido N-2-hidroxietil-piperazin-N’-2etanosulfónico CPF: corteza prefrontal CPu: núcleos caudado y putamen CT: ciclo umbral CYP450: citocromo P450 DA: dopamina DAB: tetracloruro de diaminobenzidina DAG: diacilglicerol DAGL: lipasa de diacilglicerol dATP: desoxiadenosina trifosfato dCTP: desoxicitosina trifosfato HRP: peroxidasa de rábano 5-HT1A: subtipo 1A del receptor de 5hidroxitriptamina o serotonina 5-HT1B: subtipo 1B del receptor de 5hidroxitriptamina o serotonina 5-HT2: subtipo 2 del receptor hidroxitriptamina o serotonina de 5- 5-HT3: subtipo 3 del receptor hidroxitriptamina o serotonina de 5- HU-210: (6aR,10aR)-3-(1,1’-dimetilheptil) 6a,7,10,10a-tetrahidro-1-hidroxi-6,6-dimetil6H-dibenzo[b,d]piran-9-metanol; agonista de receptores cannabinoides MENÚ SALIR SEC: sistema endocannabinoide i.p.: intraperitoneal + K : ión potasio SEM: error estándar de la media LTD: depresión a largo plazo SNC: sistema nervioso central LTP: potenciación a largo plazo SR141716A ó Rimonabant: N-(piperidin-1-il) 5-(4-clorofenil)-1-(2,4-diclorofenil)-4-metil1H-pirazol-3-carboxamida hidroclorida; agonista inverso del receptor cannabinoide CB1 Lyso-PLD: lyso fosfolipasa D MAGL: lipasa de monoacilgliceroles MAPK: proteína quinasa activada por mitógeno MeOH: metanol MEOS: sistema microsomal de oxidación de etanol Mg2+: ión magnesio SR147778: [5-(4-bromofenil)-1-(2,4diclorofenil)-4-etil-N-(1-piperidinil)-1H-pirazol -3-carboxamida]; antagonista del receptor cannabinoide CB1 T: timina TBE: tampón Tris-EDTA-Borato ML: medio-lateral TE: tampón Tris-EDTA + Na : ión sodio Δ8THC: delta-8 tetrahidrocannabinol NADH: dinucleótido de nicotinamida y adenina reducido THC ó Δ9THC: delta-9 tetrahidrocannabinol Tm: temperatura de fusión NAPE: N-araquidonilfosfatidiletanolamida URB597: [3-(3-carbamoilfenil)fenil] Nciclohexilcarbamato; inhibidor de la enzima FAAH NAPE-PLD: fosfolipasa D de Naraquidonilfosfatidiletanolamida NAT: transferasa de grupo N-acilo UTP: uridintrifosfato NMDA: N-metil D-aspartato WIN 55,212-2: (R)-(+)-[2,3-dihidro-5-metil-3(4-morfolinilmetil)pirrolo-[1,2,3-de]-1,4benzoxazin-6-il]-1-naftalenilmetanonemesilato; agonista de receptores cannabinoides pb: pares de bases PB: tampón fosfato PBS: tampón fosfato salino PC: fosfatidilcolina PCR: reacción en cadena de la polimerasa PE: fosfatidiletanolamina PEA: palmitoiletanolamida PES: polietersulfona PKA: proteína quinasa A Ratas AA: ‘Alko-Alcohol’, preferencia al alcohol ratas con Ratas ANA: ‘Alko, Non-Alcohol’, ratas con aversión al alcohol Ratas sP: ‘Sardinian alcohol-Preferring’; ratas con preferencia al alcohol Ratas msP: ‘Marchigian Sardinian alcoholPreferring’; ratas con preferencia al alcohol Rpm: revoluciones por minuto RT: transcripción reversa RT-PCR: reacción en cadena de la polimerasa mediante transcripción reversa SCC: tampón citrato sódico salino SDS: dodecilsulfato sódico MENÚ SALIR INTRODUCCIÓN MENÚ SALIR MENÚ SALIR Introducción 1. LA ADICCIÓN A ALCOHOL 1.1 - PROBLEMAS CLÍNICOS Y SOCIALES La adicción podría definirse como la pérdida de control sobre la droga de abuso o como la búsqueda y consumo compulsivo, a pesar de sus consecuencias adversas (Nestler, 2001). El consumo de alcohol es una de las principales causas que generan problemas de salud en los países desarrollados (Mokdad et al., 2005). La mayoría de los efectos adversos a la salud son resultado de un consumo crónico, consecuencia de la dependencia al alcohol (alcoholismo). El alcoholismo es un desorden clínicamente definido donde el individuo que lo padece pierde el control de su comportamiento, de modo que tiende a perpetuar el consumo, desarrollando a su vez tolerancia a medida que consume más y abstinencia cuando deja de consumir. El consumo agudo se refiere a los primeros contactos con la droga, en los cuales se dan tanto los efectos a corto plazo del etanol, que llevan al estado de embriaguez, como los que activan al sistema de recompensa, que van a promover un consumo posterior más frecuente. No obstante, el consumo crónico de alcohol expone también al individuo diariamente a las consecuencias agudas de su consumo, esto es, sedación, hipnosis, euforia, alteración de la conducta motora y de la memoria, etc. Estos efectos son de una enorme importancia pues afectan a la actividad diaria del sujeto (su capacidad para conducir, la relación con su entorno familiar y laboral,…). La exposición prolongada y/o excesiva al alcohol lleva además a padecer problemas de salud secundarios (alteraciones neurológicas y cardiovasculares, hepatitis, cáncer, síndrome de alcoholismo fetal, etc.). Las condiciones que llevan al consumo excesivo de alcohol en unos individuos y no en otros son complejas ya que implican interacciones entre factores genéticos y ambientales (Enoch y Goldman, 2001). Existe el convencimiento de que determinados factores genéticos pueden contribuir a la predisposición al alcoholismo, y que diferentes subtipos de alcohólicos estarían afectados, a diferentes niveles, por los factores genéticos y ambientales (Cloninger, 1987). -1- MENÚ SALIR Introducción Al contrario que la mayoría de drogas de abuso, el alcohol actúa sobre una gran variedad de sistemas receptores y neuroquímicos (Glutamato, GABA, Glicina, Serina, etc.), tanto para provocar la sintomatología típica del consumo agudo como para actuar de reforzante positivo e inducir dependencia. 1.2 - FARMACOLOGÍA DEL ALCOHOL El alcohol se metaboliza en el organismo en una reacción en dos pasos: una inicial en la que se genera acetaldehído, proceso catalizado por la enzima alcohol deshidrogenasa (ADH) y otra secundaria que transforma el acetaldehído en ácido acético, proceso catalizado por la enzima acetaldehído deshidrogenasa (ALDH). En la presente tesis se analizarán las acciones farmacológicas del alcohol en cerebro. NADH NADH Ácido Acético Acetaldehído Etanol 1 2 Mitocondria Metabolismo del etanol. El etanol es oxidado a acetaldehído por acción de la ADH (alcohol deshidrogenasa) (1) en el citosol, reacción en la que se genera NADH. El acetaldehído difunde al interior de la mitocondria donde es sustrato de la enzima ALDH (acetaldehído deshidrogenasa) (2) que lo convierte en ácido acético generándose en dicha reacción NADH. 1.2.1 – Metabolismo del Etanol en el Cerebro En el cerebro la enzima ADH clase I, que en el hígado es la principal oxidante del etanol a concentraciones bajas y moderadas, posee una actuación muy limitada (Raskin y Sokoloff, 1972). En el cerebro de humanos y ratones, la isoforma más abundante es la clase III (Rout, 1992), aunque tiene baja afinidad por el etanol y -2- MENÚ SALIR Introducción difícilmente es activada por éste ya que, incluso en intoxicaciones etílicas severas, no se alcanzan las concentraciones necesarias para que su contribución sea relevante. Se ha descrito también la presencia de citocromos pertenecientes al complejo enzimático Meos y se ha demostrado que el CYP450 cerebral es inducido por el etanol, como ocurre en el hígado (Upadhya et al., 2000). La presencia e inducción de esta enzima en cerebro reviste mucha importancia ya que, al existir evidencias de que el subtipo 2E1 hepático es normalmente inducido por los sustratos que metaboliza, su inducción cerebral es una prueba indirecta para la hipótesis de la oxidación cerebral del etanol a acetaldehído. De esta forma, aunque sólo sean oxidadas cantidades muy pequeñas de alcohol en el cerebro, la generación local de acetaldehído podría tener consecuencias funcionales importantes: esta inducción ha sido asociada con los efectos tóxicos el etanol y la alteración de las membranas neuronales (Montoliu et al., 1994). Finalmente, existe un gran número de pruebas que constatan que el sistema catalasa-peróxido de hidrógeno se halla presente y activo en el Sistema Nervioso Central (SNC; Zimatkin y Deitrich, 1995). Algunas investigaciones han presentado pruebas indirectas de la oxidación de etanol a acetaldehído en el cerebro de rata, vía este sistema enzimático (Aragón et al., 2002). Estos datos nos sugieren que, aunque la cantidad total de acetaldehído que pueda producirse en el encéfalo a través de la catalasa sea pequeña, existe la posibilidad de que se produzcan acumulaciones de acetaldehído suficientes para provocar cambios en la fisiología y la actividad de determinados grupos neuronales (Aragón et al., 2002). 1.2.2 – Interacciones del Etanol con los Sistemas de Neurotransmisión Antes de los años ’70, se aceptaba que el etanol ejercía sus efectos centrales a través de una fluidificación inespecífica de las membranas neuronales (Seeman, 1972) ya que se consideraba dudoso, dada la simplicidad de su estructura química, que pudiera existir un receptor. Esta hipótesis se basaba en el hecho de que el etanol es soluble tanto en agua como en lípidos y proponía que los efectos agudos del etanol serían debidos a un aumento de la fluidez de la membrana neuronal mientras que el consumo crónico, de -3- MENÚ SALIR Introducción manera compensatoria, aumentaría la rigidez de la membrana con la consiguiente alteración de las funciones (Tabakoff y Hoffman, 1996). A medida que empezaron a comprenderse el papel de los neurotransmisores y receptores en el SNC, comenzó a admitirse que el etanol podría llegar a alterar la neurotransmisión central. Ahora está claro que el etanol no sólo afecta a un sistema de neurotransmisión sino que tiene, de hecho, múltiples dianas en el SNC (tabla 1). A partir de los años ’80 muchos estudios se centraron en definir, para los neurotransmisores aminoacídicos, el papel inhibidor y excitador de las acciones centrales del etanol. En la actualidad se sabe que el etanol interactúa con determinadas proteínas situadas en la membrana neuronal y que son responsables de la transmisión de señales. Entre las dianas sobre las que el etanol actúa se encuentran canales iónicos, transportadores, receptores, proteínas G y proteínas-quinasas. La administración forzada, en ratas, de dosis intoxicantes de etanol está asociada con cambios neuroadaptativos en muchos sistemas de neurotransmisores y mecanismos de transducción (Diamond y Gordon, 1997). Los sistemas de GABA y glutamato sufren estas alteraciones funcionales, siendo éstos los dos mayores neurotransmisores inhibitorios y excitadores respectivamente, y las neuronas que los utilizan constituyen más del 80% de las neuronas del cerebro. Sistema Efecto Farmacodinámico Efecto Clínico NMDA - Sedación, amnesia GABAA + Sedación, activación, euforia, ansiólisis 5- HT3 + Ansiólisis, náuseas DA + Activación, euforia OPIOIDE + Euforia MUSCARÍNICO + Amnesia ADENOSINA - Incoordinación, sedación Tabla 1. Efectos agudos del etanol sobre diferentes Sistemas de Neurotransmisión a nivel farmacodinámico y clínico. (-) = inhibidor; (+) = activador. -4- MENÚ SALIR Introducción 1.2.2.1 - Efectos del Etanol sobre el Sistema de Neurotransmisión GABAérgico El ácido γ-aminobutírico (GABA) es el principal neurotransmisor inhibidor en el cerebro y actúa a través de dos receptores (GABAA y GABAB) los cuales están ampliamente distribuidos en cerebro, cerebelo y tronco cerebral. Las neuronas sensibles a GABA generalmente también tienen receptores de glutamato en su membrana, de forma que GABA regula la liberación de glutamato permitiendo así un equilibrio que mantiene la excitabilidad neuronal en general (Fadda y Rosseti, 1998). El receptor GABA es un canal de iones cloro que contiene múltiples sitios de unión destacando: el sitio al que se une GABA, el sitio donde interaccionan benzodiacepinas (BZD) y un tercero donde actúan, entre otros, los barbitúricos. Aún no se ha descubierto la existencia de un sitio específico de unión para el etanol. Este receptor está formado por diferentes subunidades proteicas, cada una de la cuales tiene entre 1 y 6 variantes, cuya combinación genera las diferentes isoformas existentes. Esquema del receptor GABAA donde se muestran los sitios de unión a GABA y a benzodiacepinas. En la intoxicación aguda por etanol se potencia la acción del GABAA, lo cual favorece el flujo masivo de aniones Cl- en el interior de las neuronas. Ello conlleva una hiperpolarización neuronal y disminución de la excitabilidad, que explicaría en parte los efectos depresores del alcohol sobre el SNC. El etanol actúa sobre el receptor GABAA potenciando el efecto de cualquiera de los compuestos que interaccionan con este último (BZD, barbitúricos), aunque por -5- MENÚ SALIR Introducción mecanismos diferentes. Esto explicaría la tolerancia cruzada que se observa entre ellos, su capacidad de paliar la sintomatología de abstinencia así como, en el caso del alcohol, los efectos ansiolíticos, la ataxia, disminución de reflejos y, con dosis mayores, la amnesia y el coma (Nutt, 1999). Buen ejemplo de ello es que las BZD pierden su capacidad de incrementar los efectos producidos por GABA en ratones continuamente expuestos a alcohol (Buck y Harris, 1990). El incremento de GABA, por inhibición de su degradación, también incrementa los efectos que tiene sobre el comportamiento del etanol y, de acuerdo con esto, la reducción de la actividad de GABAA reduciría los signos de intoxicación del alcohol así como sus efectos ansiolíticos (Grobin et al., 1998). Sin embargo, debido a la heterogeneidad de las subunidades del receptor GABAA, el efecto estimulante del etanol varía según la región del cerebro estudiada. Así, en cerebelo, el etanol interacciona con las células de Purkinje, encargadas de la posición corporal en el espacio y la coordinación motora, y con las células granulares concretamente con un subtipo de receptor GABAA que posee la subunidad a6. La inhibición, debida a la potenciación que el etanol ejerce sobre GABA, induce los síntomas cerebelosos de intoxicación alcohólica como aumento del balanceo corporal e incoordinación motora. El consumo crónico de alcohol tiene el efecto opuesto al del consumo a corto plazo (Devaud, 2001) ya que lleva a una disminución de la hiperpolarización por GABA, reduciendo la inhibición neuronal, lo que podría contribuir a la aparición de tolerancia a etanol. Esta regulación a la baja se da tras exposiciones prolongadas a los ligandos de este receptor y el grado de dicha regulación depende de la dosis y duración de la exposición. Diversos estudios han demostrado que el consumo crónico altera diferencialmente la expresión del receptor GABAA en la corteza cerebral y el cerebelo (Devaud et al., 1995). Estos hallazgos apoyan la hipótesis de que la tolerancia a etanol implica la reducción en el número o función del receptor GABAA, lo que conlleva a una disminución en la sensibilidad hacia los neurotransmisores. Dichas alteraciones podrían contribuir a los síntomas de la abstinencia como hiperexcitabilidad y temblores (Dahchour y De Witte, 2000). -6- MENÚ SALIR Introducción No Alcohol Exposición Aguda a Etanol Exposición Crónica a Etanol Extracelular Receptor GABAA Intracelular Los receptores GABAA, en presencia de GABA, se abrirán para permitir la entrada de iones Cl-, hiperpolarizando la membrana (figura de la izquierda). La administración aguda de etanol aumenta lo efectos del GABA, permitiendo una mayor entrada de Cl- hacia la célula (figura del centro). Tras exposición prolongada a etanol, disminuyen los efectos sobre el receptor, disminuyendo la inhibición y el flujo de Cl- (figura derecha). De esta forma, el tono inhibitorio de la neurona aumenta agudamente con etanol pero se reduce tras la exposición prolongada a etanol. 1.2.2.2 - Efectos del Etanol sobre el Sistema de Neurotransmisión Glutamatérgico El glutamato es el principal neurotransmisor excitador en el SNC y ejerce su acción a través de tres subtipos de receptores: el receptor metabotrópico kainato y los receptores ionotrópicos N-metil D-aspartato (NMDA) y ácido α-amino 3-hidroxi 4metil 5-ixazol propiónico (AMPA). Los receptores NMDA están compuestos por la subunidad NR1 y al menos una subunidad NR2, siendo las subunidades identificadas en dicho receptor: NR1, NR2 (AD) y NR3 (A y B). Estos receptores están acoplados a canales iónicos sensibles a voltaje permeables a iones Ca2+ y cationes monovalentes como Na+ y K+. La unión del glutamato al receptor NMDA promueve la entrada de calcio en la célula, cuya activación da lugar a un aumento en la permeabilidad de Na+, K+ y Ca2+, lo cual se traduce en una despolarización de la membrana neuronal. Al igual que en el caso del receptor GABAA, existe una gran variabilidad local y regional en las acciones del etanol -7- MENÚ SALIR Introducción sobre el receptor NMDA. Parte de esta diferente sensibilidad depende de las subunidades que componen el receptor. Esquema del receptor ionotrópico NMDA donde se muestran los sitios de unión de glutamato, glicina y Zn2+ así como el lugar donde se une el ión Mg2+ gracias al cual se bloquea el flujo de iones a través del canal. El receptor NMDA es el más afectado por el alcohol, inhibiendo la entrada de iones calcio, lo cual puede contribuir a la liberación de otros neurotransmisores tales como la dopamina o noradrenalina. Esta acción antagónica del etanol es responsable, en parte, de sus efectos sedantes. La actividad de los receptores NMDA está implicada en el aprendizaje asociativo y juega un importante papel en el daño cerebral producido por un golpe o una excesiva actividad glutamatérgica tras continuados ataques. Dichos receptores también participan en otros efectos del alcohol como la pérdida de memoria asociada con la intoxicación, el daño cerebral tras el consumo crónico y el efecto rebote responsable del comportamiento hiperactivo durante la retirada. El etanol reduce la liberación de glutamato en el hipocampo alterando la memoria espacial. La pérdida de efectividad en los receptores y la disminución de liberación de neurotransmisores podrían generar pérdida de memoria transitoria durante la intoxicación (Diamond y Gordon, 1997). -8- MENÚ SALIR Introducción No se sabe exactamente cómo se produce el efecto del etanol sobre el receptor NMDA, ya que la acción bloqueante no parece ejercerse ni en el sitio de unión del glutamato ni en ninguno de los sitios moduladores hasta ahora conocidos, como son el de la glicina o el de la poliamina. El etanol tampoco interacciona con el lugar donde el Mg2+ bloquea el canal y por sí mismo no es capaz de cerrar los canales abiertos. No obstante, algunos autores describen el dominio M3 de la subunidad NR1 y el dominio M4 de la subunidad NR2A como posibles lugares de acción del alcohol (Ren et al., 2003, 2007; Honse et al., 2004). Durante el tratamiento crónico, los cambios en la neurotransmisión excitadora son importantes en el desarrollo de dependencia, tolerancia y síndrome de abstinencia. En adultos el consumo crónico conduce a un incremento en el número de receptores NMDA en respuesta a la bajada en la actividad del glutamato, observándose en áreas cerebrales como hipocampo y cerebelo. Estudios con ratas muestran que la liberación de glutamato, normalmente inhibida por etanol, incrementa drásticamente tras 10 horas del cese a la exposición a etanol (Fadda y Rossetti, 1998). Estos hechos explicarían los ataques y la hiperexicitabilidad que tienen los alcohólicos durante la retirada. Un exceso de la actividad de glutamato, durante la retirada, contribuye a la muerte celular lo cual, frecuentemente, conlleva daños cerebrales irreversibles. Por otro lado, existen evidencias que implican a los receptores metabótropicos en el consumo y comportamiento de búsqueda de etanol ya sea mediante la activación de determinados subtipos de receptores (mGlu2/3 y mGlu8) o por su inhibición (mGlu5) (Cowen et al, 2005; Bäckström e Hyytiä, 2005; Adams et al., 2007). Está descrito también que la potenciación de los receptores AMPA revierte los efectos centrales de la intoxicación aguda del etanol (Jones et al., 2007). Todo ello lleva a pensar que los cambios neuroadaptativos en el sistema glutamatérgico podrían tener relevancia en las alteraciones neuropatológicas, inducidas por la exposición crónica a etanol, en el cerebro de animales de experimentación y en humanos. Por estas razones el alcoholismo se ha considerado como un desorden neuropsiquiátrico causado por el glutamato (Fadda y Rossetti, 1998). -9- MENÚ SALIR Introducción 1.2.2.3 – Efectos del Etanol sobre el Sistema de Neurotransmisión Dopaminérgico Existen varias evidencias que convergen en demostrar que el sistema mesolímbico dopaminérgico interviene de manera significativa en los mecanismos motivacionales y de refuerzo relacionados con el comportamiento (Dulawa et al., 1999; Giros et al., 1996). De hecho, el efecto reforzador positivo de las sustancias psicotropas está relacionado con su capacidad de activar a este sistema. Parte de él pertenece a la red cerebral que integra las emociones, mediante respuestas hormonales, y a la actividad del sistema nervioso simpático. El core del núcleo accumbens es una de dichas estructuras (Heimer et al., 1997) encargada de modular el movimiento y de combinar procesos implicados en los estados de motivación, contenido emocional y respuestas motoras (DiChiara, 1997). El papel de esta área en producir el refuerzo, que conduce a la repetida ingestión de drogas, ayuda a explicar la naturaleza adictiva del alcohol. El etanol aumenta la liberación de dopamina en áreas como el núcleo accumbens (DiChiara, 1997) y, administrado de manera aguda, actúa sobre el ATV incrementando la frecuencia de descarga de las neuronas dopaminérgicas mediante la atenuación de las interneuronas GABAérgicas. En este sentido, se ha demostrado que la inyección de inhibidores de dopamina disminuye la autoadministración de alcohol en ratas. Un posible mecanismo responsable del significado anormal de los incentivos asociados al alcohol sería la naturaleza no adaptativa de la estimulación dopaminérgica, accionada por el alcohol, en el núcleo accumbens. Los reforzadores naturales (como la comida) inducen rápidamente habituación y la presencia repetida de estímulos relacionados no provoca un aumento de la dopamina. Por el contrario, tras el consumo repetido de etanol, no ocurre habituación y como resultado de la persistente liberación de dopamina, en el núcleo accumbens, los estímulos asociados adquirirían un significado emocional y motivacional anormal que resultaría en un control excesivo sobre la conducta del bebedor (DiChiara, 1997). Por el contrario, durante la retirada, en ratas que consumen crónicamente alcohol, disminuye la tasa de disparo y la liberación de dopamina en el core del núcleo accumbens (Diana et al., 1993) lo que conduce al típico bajo estado anímico en personas alcohólicas. De hecho, la retirada de alcohol se relaciona con emociones negativas como el malestar y depresión, demostrable sólo mediante la elevación de los umbrales de autoestimulación intracraneal en ratas (Schulteis et al., 1995). - 10 - MENÚ SALIR Introducción Diversos estudios describen que la abstinencia a etanol está asociada a una denervación temporal dopaminérgica, tal y como lo demuestran los signos de catatonia y temblores que acompañan al síndrome de abstinencia en ratas (Lal et al., 1998). De hecho, en alcohólicos, existen evidencias de que puede estar asociada a síntomas parkinsonianos que desaparecen tras varios meses de abstinencia (Fadda y Rossetti, 1998). La destrucción de los terminales mesolímbicos dopaminérgicos no anula completamente la autoadministración de alcohol en ratas, lo cual sugiere la existencia de otros mecanismos independientes de la dopamina que contribuirían a las propiedades reforzadoras del alcohol. Uno de esos mecanismos podría ser el sistema opioide. 1.2.2.4 – Efectos del Etanol sobre el Sistema de Neurotransmisión Opioide El sistema opioide de neurotransmisores peptídicos lo constituyen endorfinas y encefalinas las cuales son sustancias endógenas que modulan el dolor, la alimentación, el humor, el control de la voluntad y las respuestas ante el estrés. El etanol y los opiáceos presentan efectos farmacológicos y adictivos similares por lo que podrían tener un sustrato neurobiológico común. De hecho, las endorfinas y encefalinas están también relacionadas con el refuerzo al alcohol (Froelich, 1997). La administración de alcohol incrementa la actividad opioide endógena tanto en ratas como en humanos, mostrando una mayor liberación en sangre y en secciones cerebrales así como un incremento de la expresión génica de encefalinas y endorfinas en cerebro (Guardia, 2001). En bebedores ocasionales la liberación de dopamina, inducida por el alcohol, podría producir refuerzo debido a que tal liberación está regulada por opioides (Herz, 1997). A su vez, se piensa que la activación del sistema opioide podría estar relacionada con la recompensa. Por el contrario, el consumo crónico suele conducir a una reducción de los niveles de endorfinas en cerebro lo que se percibe como abstinencia opioide. Ello lleva a desarrollar estados emocionales negativos, que se dan en la retirada de alcohol, y promueve su consumo. - 11 - MENÚ SALIR Introducción La disminución de la respuesta opioide en el núcleo accumbens refleja la existencia de una respuesta neuroadaptativa al aumento de liberación de dopamina mientras que el aumento de respuesta de los receptores opioides, en la amígdala, está asociado a las acciones ansiolíticas inducidas por el alcohol (Bodnar y Hadjimarkou, 2002). Se ha demostrado que el bloqueo de los receptores opioides disminuye significativamente la autoadministración en animales. En humanos, que abusan del alcohol, las sustancias que interfieren con la actividad opioide disminuyen la ingesta, la recaída, el fuerte deseo de consumo y la euforia subjetiva producida por el alcohol (Meyer y Quenzer, 2005). Por último, las cepas genéticas de animales que difieren en su preferencia por el alcohol también lo hacen en sus sistemas opioides. Las cepas con preferencia por el alcohol tienen sistemas opioides que responden más ante el alcohol. A bajas dosis de morfina incrementan el consumo de alcohol, teóricamente a través de un efecto principal, mientras que con dosis de moderadas a altas de morfina producen una disminución del consumo de alcohol debido a que se anularía la función de los opioides endógenos (Herz, 1997). 1.2.2.5 – Efectos del Etanol sobre el Sistema de Neurotransmisión Serotoninérgico La serotonina juega un importante papel en la regulación del humor, la alimentación, excitación, sueño, dolor, entre muchos otros comportamientos (Carlson, 1998). Muchas neuronas serotonérgicas están localizadas en el núcleo del raphe el cual está implicado en funciones como la atención, emoción y motivación. Los axones de las neuronas de dicho núcleo se proyectan a numerosas regiones del cerebro como la amígdala y el núcleo accumbens (Guerri, 2000). El consumo agudo de alcohol incrementa la liberación de serotonina en el sistema nervioso lo que tiene efectos a nivel comportamental (emociones, humor y pensamientos) además de tener un efecto reforzador relacionado con el alivio de estados - 12 - MENÚ SALIR Introducción emocionales desagradables, disforia, trastornos afectivos y control de impulsos (Guerri, 2000). Existen varios subtipos de receptores de serotonina cada uno de los cuales tiene una influencia específica sobre el comportamiento relacionado con el consumo de alcohol (Lovinger, 1999). Uno de estos receptores (5-HT1A) podría controlar la conducta consumidora, incluyéndose la de alcohol, mientras que otro subtipo (5-HT1B) podría no sólo contribuir a los efectos intoxicantes del alcohol sino también influir en el desarrollo de tolerancia. Un tercer receptor (5-HT2) juega un papel en los efectos placenteros del alcohol y, por consiguiente, contribuiría al desarrollo del síndrome de abstinencia. Por último, el subtipo 5-HT3 estaría implicado en regular el consumo de alcohol (tabla 2). La exposición aguda de etanol aumenta las señales eléctricas generadas por el receptor 5-HT3 lo cual, probablemente, causa una excesiva estimulación en neuronas que reciben información de las neuronas serotoninérgicas. Ello llevaría a un aumento de la liberación de los neurotransmisores que participan en la intoxicación etílica. La serotonina también afecta a otros neurotransmisores como, por ejemplo, al sistema GABA, implicado en la toma de decisiones (Samson y Harris, 1992). De manera similar, la serotonina también estimula la producción de dopamina y, así, aumenta el comportamiento emocional (Campbell et al., 1996). El consumo crónico de etanol produce una disminución de los niveles de serotonina en el cerebro lo cual está asociado con trastornos de control de impulsos, trastornos afectivos y agresividad (Guerri, 2000). Se ha descrito que ratas con preferencia por el alcohol tienen menores niveles de serotonina en sus cerebros en comparación con las que no tienen dicha preferencia (Zhou et al., 1994). Esta observación apoya la noción de que, en los grandes consumidores, se podrían intentar normalizar los niveles de serotonina en regiones cerebrales claves ya que, durante los consumos agudos de alcohol, se elevan dichos niveles. - 13 - MENÚ SALIR Introducción Receptor Posible Papel 5-HT 1A Conducta consumidora, incluyendo el consumo de alcohol. 5-HT 1B Efectos intoxicantes del alcohol. Desarrollo de tolerancia. 5-HT2 Desarrollo de síntomas de abstinencia a alcohol. Efectos reforzadores del alcohol. 5-HT3 Regulación del consumo de alcohol. Contribución a sus efectos reforzadores. Tabla 2. Subtipos de receptores serotoninérgicos y sus potenciales papeles en el desarrollo del abuso de alcohol. Tomado de Lovinger, 1997. 1.2.2.6 - Efectos del Etanol sobre el Neuromodulador Adenosina La adenosina altera la actividad neuronal afectando a la conducta aunque no reúne todos los requisitos como neurotransmisor ya que, por ejemplo, no excita o inhibe por sí misma a células postsinápticas sino que regula o modula la actividad de neuronas en respuesta a otros neurotransmisores. Diversas evidencias indican que la adenosina media muchos de los efectos del alcohol. El alcohol puede aumentar sus niveles extracelulares, por un lado, inhibiendo su recaptación y aumentando, por otro, su producción en el organismo como resultado del metabolismo del alcohol en el hígado. Se cree que muchos de los efectos agudos y crónicos del alcohol en el SNC están mediados por este neuromodulador. Por ejemplo, estudios realizados con ratones han demostrado que la adenosina modula la incapacidad de coordinar movimientos musculares voluntarios inducidos por el alcohol (ataxia) (Dar, 1990). Es más, el agonista del receptor de adenosina aumenta y el antagonista disminuye la descoordinación inducida por etanol. - 14 - MENÚ SALIR Introducción 1.3 - SISTEMAS NEUROBIOLÓGICOS IMPLICADOS EN LA ADICCIÓN A ALCOHOL Las drogas adictivas presentan una gran diversidad molecular y actúan sobre diversos receptores y estructuras existiendo un factor común a las mismas: la activación de la vía mesocorticolímbica dopaminérgica (también denominado sistema de recompensa), crítica en el proceso de dependencia y adicción (Leshner, 1997; Shippenberg y Elmer, 1998; Melichar et al., 2001). Existen numerosos trabajos neurofarmacológicos (Goeders y Smith, 1983; Gardner et al., 1988) que implican al sistema dopaminérgico en los efectos reforzadores positivos de las drogas de abuso. El circuito cerebral de recompensa está estrechamente relacionado con la vía ascendente dopaminérgica mesocorticolímbica, originada en el área tegmental ventral (ATV), y que proyecta a estructuras corticales anteriores y subcorticales del sistema límbico (Corbett y Wise, 1980). Este sistema está compuesto por dos proyecciones principales: - la nigroestriatal, que desde la pars compacta de la substancia nigra proyecta al núcleo caudado de los ganglios basales y - la mesocorticolímbica, que desde el ATV proyecta al núcleo accumbens, tubérculo olfatorio, córtex frontal y amígdala. El sistema dopaminérgico mesolímbico (que incluye las áreas de proyección del núcleo accumbens, amígdala e hipocampo) ha estado tradicionalmente asociado a las propiedades reforzadoras de la administración aguda de las drogas de abuso así como con los procesos de memoria necesarios para el desarrollo de respuestas condicionadas (o aprendidas) que, a su vez, determinan el deseo por la droga. Los cambios, inducidos por las drogas, sobre la homeostasis de este sistema determinan los cambios emocionales y motivacionales que se ponen de manifiesto en la abstinencia. El circuito dopaminérgico mesocortical (corteza prefrontal, corteza orbitofrontal y cíngulo anterior) parece estar involucrado en la experiencia consciente de la intoxicación aguda pero también en primar la droga como incentivo y, por consiguiente, en el deseo por la sustancia y en su administración compulsiva. - 15 - MENÚ SALIR Introducción Mediante la interacción de ambos circuitos nerviosos se configura un sustrato neurobiológico que explicaría las propiedades reforzadoras de las drogas (área tegmental ventral, núcleo accumbens, tálamo, corteza prefrontal), tras su ingesta aguda, su integración como experiencia consciente (corteza prefrontal), la implicación de la memoria y las respuestas condicionadas (amígdala, hipocampo), la expectación o el deseo por la droga (circunvolución del cíngulo, corteza prefrontal), el desarrollo y percepción de los síntomas de la abstinencia (disminución de actividad en los circuitos límbicos y alteración del control superior de la corteza prefrontal), conduciendo así a perpetuar el consumo de la droga. La activación del sistema de recompensa, durante el consumo agudo, induce un incremento en la tasa de liberación de dopamina, en el ATV y áreas de proyección, y una regulación al alza en los niveles de AMPc en el núcleo accumbens y amígdala extendida, áreas que se relacionan decisivamente con la recompensa y con el aprendizaje para el consumo (Nestler y Aghajanian, 1997; Wise, 2000). Se cree que la vía mesocorticolímbica dopaminérgica participa, a nivel fisiológico, en la creación de hábitos de conducta tras estímulos reforzadores naturales (comida, bebida, sexo) modulando el circuito córtico-estriato-palidal donde se generan los patrones motores. Este circuito incluye a la corteza prefrontal, al núcleo accumbens o estriado ventral, al estriado dorsal y al globo pálido. La adicción sería, por tanto, una perturbación crónica de la vía mesocorticolímbica inducida por la droga, creándose un hábito patológico cuyo fin es el consumo de la droga. En presencia de un reforzador natural, por ejemplo, se sabe que el ATV cambia su disparo desde una frecuencia irregular a “ráfagas” regulares. Se postula que la estimulación de esta vía ha de ejercer un efecto placentero en los animales (Olds, 1956) y que las drogas potencian los efectos reforzadores de la estimulación eléctrica, en esta vía, como si acrecentaran el placer producido por un nivel determinado de estimulación cerebral (Wise et al., 1992). La asociación de este estímulo con una sensación es un proceso que Berridge denomina relevancia incentiva o exaltación del incentivo (Gardner, 1997; Wise, 1981; Robbins y Everitt, 1999). El alcohol actúa como un reforzador positivo activando los circuitos locales opioides de encefalinas del ATV, estimulando a las neuronas dopaminérgicas del área tegmental ventral y actuando sobre receptores tipo GABAA en el núcleo accumbens, en el núcleo central de la amígdala y en la corteza prefrontal. A su vez, los sistemas - 16 - MENÚ SALIR Introducción glutamatérgicos y serotoninérgicos parecen participar también en el refuerzo positivo que produce el etanol. Centros y circuitos nerviosos claves en la adicción a drogas. Las líneas de puntos indican las aferencias límbicas hacia el núcleo accumbens (NAc). Las líneas azules representan las eferencias desde el NAc. Las líneas rojas indican las proyecciones del sistema mesolímbico dopaminérgico, sustrato crítico para la recompensa. Las neuronas dopaminérgicas se originan en el área tegmental ventral (VTA) y proyectan hacia el NAc y otras estructuras límbicas, que incluyen al tubérculo olfatorio (OT), los dominios ventrales del caudado-putamen (C-P), la amígdala (AMG) y la corteza prefrontal (PFC). Las líneas verdes indican las neuronas que contienen péptidos opioides, implicadas en la recompensa de los opiáceos y etanol. Estos sistemas de péptidos opioides incluyen a los circuitos locales de encefalinas (segmentos cortos) y al circuito hipotalámico de las β-endorfinas (segmento largo). El sombreado azul indica la distribución aproximada del receptor GABAA que contribuiría a la recompensa del etanol. ARC: núcleo arqueado; Cer: cerebelo; DMT: tálamo dorsomedial; IC: colículo inferior; LC: locus coeruleus; LH: hipotálamo lateral; PAG: sustancia gris periacueductal; SC: colículo superior; SNr: substancia nigra pars reticulata; VP: pálido ventral. Modificado de Nestler, 2001. Se ha observado que, tras un consumo crónico de drogas, hay cambios críticos a nivel celular, conocidos como neuroadaptaciones, en los que se desarrolla una sensibilización en la liberación de dopamina mesocorticolímbica y se dan cambios permanentes en el circuito cortico-estriato-palidal, hechos que son de gran importancia en el establecimiento de la dependencia crónica y de la abstinencia tras el cese del consumo. Ello se debe a que generan un refuerzo negativo al cese de la administración de la droga. El refuerzo positivo parece ser el responsable del inicio al consumo y el refuerzo negativo, que se desarrolla como consecuencia de las neuroadaptaciones, del - 17 - MENÚ SALIR Introducción mantenimiento en el consumo. Esto produce comportamientos motivados que conducen a la ingesta de la droga, cursando con impulsividad, compulsividad y ansiedad. A nivel de integración superior, parece ser que la adicción produce cambios en el punto de ajuste del procesamiento hedónico de modo que la experiencia diaria sin presencia de la droga se percibe, en ese estado, como negativa. El estriado es una región importante para el control motor y la generación de planes motores, presentando una zona ventral o núcleo accumbens y otra dorsal o caudado-putamen. Ambas poseen neuronas GABAérgicas espinosas de mediano tamaño como neuronas de proyección que se interconectan con diferentes áreas. El núcleo accumbens se considera una interfase límbico-motora pues recibe información de áreas límbicas, como la amígdala y la corteza prefrontal además del hipocampo, y la envía al pálido ventral y tálamo integrándose en los circuitos motores. A su vez, su actividad está modulada por neuronas dopaminérgicas procedentes del ATV. Las drogas afectan a la actividad del núcleo accumbens a través de los cambios en la vía mesocorticolímbica ya comentados y también a través de acciones más directas sobre las aferencias de la corteza prefrontal y sobre las propias neuronas GABA estriatales. Inicialmente los cambios agudos por la acción de las drogas se localizan , entre otras regiones, en el núcleo accumbens. El desarrollo de LTP (potenciación a largo plazo) o LTD (depresión a largo plazo) en estriado parece estar relacionado con la consolidación de la dependencia y, en estos efectos, estarían involucrados cambios en la actividad dopaminérgica de origen nígrico y la acción glutamatérgica de origen cortical. Ambos fenómenos modifican, de modo permanente, la actividad sináptica ya que también subyacen a los cambios neuronales relacionados con el aprendizaje. La formación hipocampal es una estructura cerebral compuesta de diversas áreas como el giro dentado, el hipocampo y el subiculum, existiendo en todas ellas tres capas neuronales. El flujo de información es, en gran parte, unidireccional y se sabe que participa en los fenómenos de consolidación de la memoria. No parece ser el lugar de almacenamiento de la memoria pero sí donde se genera la memoria a largo plazo o memoria declarativa (hechos, imágenes y nombres que se aprenden). Al menos parte - 18 - MENÚ SALIR Introducción del mecanismo de consolidación de la memoria recae en los procesos de LTP, que se producen en la formación hipocampal. Dicha estructura cerebral forma parte del sistema límbico junto con la corteza de asociación límbica, la amígdala, el estriado ventral y algunos núcleos diencefálicos y mesencefálicos. Dicha macroestructura está implicada en el procesamiento de la información emocional y de los comportamientos motivados. Además participa activamente en el procesamiento de señales reforzadoras, positivas y negativas, asociadas al consumo de drogas o al cese de éste tras un consumo crónico. El alcohol interacciona con sistemas receptores como los de glutamato y GABA, entre otros. Las neuronas de proyección del hipocampo (las neuronas piramidales) y del giro dentado (las células granulares) utilizan al glutamato como neurotransmisor y las interneuronas (las células polimórficas) de estas regiones, GABA. Por tanto, estas neuronas de la formación hipocampal son dianas del alcohol participando, al menos parcialmente, en los efectos de la inhibición de la consolidación de la memoria que provoca la ingesta aguda de etanol. Por otro lado, el refuerzo positivo, que provoca posteriores ingestas de alcohol, hasta hacerlas crónicas, y el alivio al refuerzo negativo, que provoca la retirada, necesita de un soporte de memoria que permita la asociación del estímulo con su efecto de recompensa. De esta manera se deduce que la formación hipocampal juega también un papel en la ingesta crónica de etanol. El cerebelo es una estructura cerebral cuya corteza se encuentra plegada y organizada en capas neuronales. En la porción medular se encuentran los denominados núcleos profundos que reciben aferencias de la corteza. Gran parte de las neuronas cerebelares son GABAérgicas y están implicadas en la coordinación de movimientos y en el aprendizaje motor. La interferencia del alcohol en las neuronas GABAérgicas provoca los síntomas típicos de incoordinación psicomotora y de marcha, imprecisión de gestos y vértigo que aparecen tras la ingesta aguda de alcohol. Pese a que el cerebelo no participa en el proceso de adicción, el consumo crónico de etanol puede tener efectos farmacológicos en esta estructura aunque no está claro el papel que pueden jugar dichos cambios. - 19 - MENÚ SALIR Introducción 1.4 - MODELOS ANIMALES DE ALCOHOLISMO: líneas msP y AA/ ANA En la presente tesis doctoral se ha planteado usar modelos animales de alcoholismo que permitiesen realizar estudios bioquímicos y neurofarmacológicos. Los modelos analizados han sido los correspondientes a líneas de roedores seleccionados en base a su preferencia alcohólica. Los requisitos mínimos establecidos (McMillen, 1997) para la aceptación de un modelo animal ideal de alcoholismo humano son: Alcanzar niveles farmacológicos de concentración de alcohol en sangre (BAC: blood alcohol concentration). Mantener dichos niveles de BAC. Desarrollar dependencia física. Mantener la dependencia mediante autoadministración oral de etanol. El problema básico es que los animales no beben voluntariamente suficiente etanol como para intoxicarse y, en consecuencia, no se vuelven dependientes. Algunas especies animales, como los hamsters “Syrian Golden” y las ratas “Fawn-Hooded”, beben espontáneamente grandes cantidades de etanol aunque, al tener un elevado metabolismo de etanol, no se convierten físicamente dependientes a etanol a pesar de estar sometidos a largas exposiciones (McMillen y Shore, 1997). Por tanto, el desarrollo de preferencia por etanol no se puede considerar un criterio para un modelo animal de dependencia alcohólica. Los modelos animales seleccionados y criados durante generaciones, por su excesiva ingesta de etanol, han demostrado ser de gran utilidad para la identificación y validación de nuevos tratamientos (McBride y Li, 1998). Las crías de ratas que eligen voluntariamente consumir grandes cantidades de alcohol llegan a ser consumidores consolidados respecto a aquellas crías que tienden a evitarlo. Esta cría selectiva de animales, durante muchas generaciones, se ha usado para desarrollar, desde 1963, la línea de ratas AA (Alko Alcohol), las cuales prefieren una solución de alcohol al 10% frente al agua, y la línea de ratas ANA (Alko Non-Alcohol), que prefieren agua (Eriksson, 1968). Además de su preferencia por el alcohol, las líneas AA y ANA presentan desviaciones bidireccionales respecto a las ratas Wistar, genéticamente heterogénas, en relación a rasgos del comportamiento que son anormales - 20 - MENÚ SALIR Introducción en humanos alcohólicos con una elevada carga genética como, por ejemplo, una elevada impulsividad (Möller et al., 1997). Las líneas de ratas AA y ANA son los modelos mejor establecidos y seleccionados. Estas líneas se han usado para demostrar el posible papel de los factores genéticos en el consumo de alcohol y establecer las correlaciones neuroquímicas, metabólicas y de comportamiento que pudieran explicar las diferencias en la ingesta de alcohol. Algunas de las diferencias encontradas están implicadas en los efectos reforzadores del etanol, factores nutricionales y cambios producidos en el consumo por privación del alcohol, las reacciones de las monoaminas adrenales y cambios de comportamiento frente a un estrés severo, el desarrollo de tolerancia, la acumulación de acetaldehído durante el metabolismo del etanol y, a nivel de sistema nervioso, en los niveles cerebrales de serotonina (Sinclair et al, 1989), del sistema opioide y elevaciones en los niveles de dopamina en ratas AA respecto a las ANA (Gianoulakis et al., 1992; Nylander et al., 1994; Soini et al., 1997). Las ratas ANA poseen un metabolismo del etanol, en hígado y ciertas áreas cerebrales, disminuido respecto a las ratas AA lo que les lleva a tener mayores niveles de acetaldehído responsable del rechazo de la ingesta de alcohol. Otro modelo animal seleccionado durante generaciones son la línea de ratas msP las cuales tienen una preferencia natural por el etanol. Han sido criadas a partir de la 13ª generación de las ratas sP (sardinian alcohol-preferring) procedentes del Departamento de Neurociencias de la Universidad de Cagliari (Agabio et al., 1996; Colombo, 1998; Lobina et al., 1997). Se caracterizan por beber espontáneamente y de manera exagerada lo cual les lleva a presentar niveles de alcohol en sangre farmacológicamente significativos. Esta línea es muy vulnerable a la recaída y al estrés mostrando un fenotipo de ansiedad con síntomas, semejantes a los de la depresión, que desaparecen tras el consumo de etanol (Ciccocioppo et al., 2006). - 21 - MENÚ SALIR Introducción 1.5 - PERSPECTIVAS DE TRATAMIENTO DE LA ADICCIÓN A ALCOHOL Los principales objetivos del tratamiento de la adicción son la facilitación de la abstinencia y la prevención de la recaída. Tales objetivos han dificultado el tratamiento de muchas formas de adicción. El tratamiento farmacológico que se utiliza habitualmente es útil para reducir los síntomas de abstinencia aunque no es efectivo en la prevención de la recaída. La potencialidad de recaída dura más que los síntomas de abstinencia e implica a mecanismos complejos como el aprendizaje asociativo. De esta manera, los medicamentos que bloquean los efectos reforzantes de las drogas o la plasticidad inducida por la droga, podrían reducir el deseo por la droga y la recaída. Dichos medicamentos serían efectivos si pudiesen actuar sin producir, por sí mismos, efectos reforzadores o de anhedonia, ejerciendo sus efectos beneficiosos sin interferir en las respuestas del organismo hacia los reforzadores naturales. Durante la desintoxicación del alcohol típicamente se administran benzodiacepinas, como clordiacepoxide, ya que son menos reforzantes que el propio etanol en los alcohólicos y muestran dependencia cruzada con el etanol en los receptores GABAA. Algunos fármacos se han utilizado, junto con terapias psicosociales, para prevenir la recaída. Dos fármacos en particular - naltrexona y acamprosato - han mostrado resultados prometedores en ensayos clínicos y en la práctica. Desafortunadamente, a excepción de la naltrexona que es parcialmente eficaz en el tratamiento del alcoholismo, no se han desarrollado aún tratamientos que reduzcan el refuerzo producido por la droga. En estudios animales donde se han asociado el refuerzo del etanol y el sistema opiode endógeno, se ha utilizado naltrexona nalmefene) para prevenir (y al menos un antagonista opioide: la adicción a etanol. En muchos ensayos clínicos, la naltrexona disminuye tanto el riesgo de volver a consumir alcohol como la frecuencia del consumo aunque no existe un aumento del grado de abstinencia total. Reduce sentimientos positivos como la euforia, inducida por el alcohol, mediante el bloqueo de la liberación de endorfinas. Diversos estudios europeos sugieren que el acamprosato reduce la frecuencia del consumo y la recaída siendo su mecanismo de acción desconocido. En roedores se sabe - 22 - MENÚ SALIR Introducción que modula la función del receptor NMDA de forma que se da facilitación del receptor en ciertas regiones y efectos antagonistas en otras (Dachour y Dewitte, 2000). Otras farmacoterapias han mostrado menor eficacia clínica. El disulfiram es un inhibidor de la enzima aldehído deshidrogenasa, con lo que su administración conlleva toxicidad debido al acetaldehído generado tras el consumo de etanol. La razón por la que se usa disulfiram es que devalúa el consumo de alcohol al estar asociado a una experiencia fuertemente repulsiva (náuseas, vómitos, dolores en el pecho, disminución de la presión sanguínea, etc.; Garbutt et al., 1999) aunque, a pesar de ello, no previene el retorno al consumo de alcohol. La administración de citalopram (Naranjo et al., 1992), un antidepresivo, disminuye los efectos reforzadores del alcohol reduciendo así el deseo por dicha droga. Este fármaco actúa facilitando la disponibilidad de serotonina. Sin embargo, uno de cada seis pacientes que lo toman padecen náuseas, insomnio, sudoración excesiva, dolor de cabeza o convulsiones lo que reduce su atractivo como tratamiento para el alcoholismo por lo que finalmente se emplean otras drogas para tratar la depresión y la ansiedad que ayudan a reducir el consumo de alcohol (Johnson et al., 1993; Pettinati, 1996). Se ha descrito, en varios estudios en los que se ha utilizado el antagonista cannabinoide SR141716A, que el sistema endocannabinoide podría jugar un papel fundamental en las propiedades reforzadoras no sólo del cannabis sino también de otras sustancias de abuso como la cocaína, los opiáceos y el alcohol. En otros estudios, en los que se han utilizado diferentes protocolos experimentales, se ha demostrado la habilidad del SR141716A para reducir el consumo y el deseo por el alcohol (Arnone et al., 1997; Colombo et al., 1998; Rodríguez de Fonseca et al., 1999; Gallate y McGregor, 1999; Gallate et al., 1999). Es interesante destacar que existen datos que sugieren que la activación del sistema cannabinoide endógeno, por un agonista, promueve el deseo por etanol (Gallate et al., 1999). Recientemente se ha descrito que el antagonista SR147778 disminuye tanto la ingesta como la preferencia a etanol en ratas tras ser sometidas a tratamientos crónicos de etanol. Estos resultados refuerzan la hipótesis que implicaría al receptor CB1 como mediador de la ingesta de etanol y en las propiedades motivacionales del alcohol. - 23 - MENÚ SALIR Introducción Además este antagonista suprime aún más que SR141716A (Rimonabant) la ingesta de etanol en ratas sometidas a tratamiento crónico de etanol (Lallemand y De Witte, 2006). Hay que destacar que los receptores μ opioides y los receptores CB1 se coexpresan en varias zonas cerebrales, incluyendo el núcleo accumbens (Navarro et al., 1998). Estos datos en conjunto nos indican que el receptor CB1 podría ser importante no sólo para los efectos reforzadores de la marihuana, sino también en la modulación de los efectos reforzadores del alcohol y la morfina. Estas características neurobiológicas sugieren un papel del sistema cannabinoide endógeno en el procesamiento del refuerzo y abren un nuevo campo para el desarrollo de estrategias terapéuticas para el tratamiento del alcoholismo y la adicción en general. - 24 - MENÚ SALIR Introducción 2. EL SISTEMA ENDOCANNABINOIDE 2.1 - HISTORIA La planta Cannabis sativa es originaria, probablemente, de las estepas de las regiones que se extienden desde Asia Central al Norte del Himalaya, África y Europa Meridional desde donde fue difundida por los griegos y especialmente los romanos. Se ha cultivado durante siglos tanto por la fibra del cáñamo como por sus propiedades psicoactivas y su supuesta utilidad clínica. El término marihuana, de origen mejicano, se refiere a una forma particular del cannabis que deriva principalmente de las hojas de la planta. Los datos más antiguos sobre el uso del cannabis datan de hace 10.000 años. Entre los años 2700 y 2000 a.C. el cannabis se usaba en China para tratar los dolores reumáticos y otras patologías (Adams y Martins, 1996) y en la India el cannabis jugaba un papel muy importante en la religión. En Europa, esta planta llamó la atención de los científicos cuando las tropas de Napoleón volvieron de la campaña de Egipto introduciendo su uso como psicotrópico. Actualmente se sabe que los efectos del cannabis en humanos incluyen la alteración de la memoria a corto plazo, daños cognitivos, descoordinación, insomnio, taquicardia refleja, hipotermia y alteraciones del carácter con euforia o disforia según la experiencia previa del consumidor, etc. (Pertwee, 1988). En 1964, Gaoni y Mechoulam aislaron el principal componente psicoactivo del cannabis y determinaron su estructura química. El compuesto era un derivado de un dibenzopirano, el Δ9- tetrahydrocannabinol (Δ9 - THC), presente en la resina amarilla que cubre las hojas y flores de la planta femenina madura. A partir de ese momento, se realizaron grandes esfuerzos por averiguar el modo de acción molecular de las preparaciones de Cannabis sativa. En los últimos 50 años, se han dado grandes avances en el conocimiento de la fisiología y farmacología del sistema cannabinoide así como la posible utilidad de sus efectos terapéuticos en ciertas patologías. - 25 - MENÚ SALIR Introducción 2.2 - LIGANDOS EXÓGENOS La planta Cannabis sativa posee más de 60 cannabinoides, siendo el Δ9-THC el principal responsable de las propiedades psicoactivas aunque existen también otros cannabinoides como el Δ8-THC, el cannabinol y el cannabidiol. Estos dos últimos cannabinoides son además responsables de algunas propiedades antiinflamatorias del cannabis. A partir de la estructura química de estos cannabinoides naturales se han desarrollado una serie de cannabinoides sintéticos que han resultado muy importantes para avanzar en el conocimiento de la farmacología de estos compuestos y en la fisiología del Sistema Endocannabinoide. Existe una amplia variedad de moléculas que se comportan como agonistas de los receptores cannabinoides, es decir, activadores de las respuestas mediadas por éstos. Entre los agonistas de receptores de cannabinoides destacan análogos bicíclicos (CP55,940), derivados de aminoalquilindoles (WIN 55,212-2) y análogos sintéticos del THC (HU-210: 11-hidroxi-Δ8-THC-dimetilheptil). En cuanto a la capacidad de interacción de los compuestos cannabimiméticos, el THC se une con una afinidad similar a los receptores CB1 y CB2, funcionando como agonista parcial en ambos casos. Sin embargo, su eficacia es mayor en la interacción con CB1 llegando incluso a comportarse como un antagonista en determinados sistemas CB2. Los compuestos CP-55,940 y WIN 55,212-2 muestran afinidades por los dos receptores y su eficacia es relativamente alta. Por último, el compuesto HU-210 es un potente agonista cannabinoide clásico, cuya eficacia es similar a la de CP-55,940 y WIN 55,212-2 y su afinidad, por ambos receptores, superior. HU-210 WIN 55,212-2 - 26 - CP – 55,940 MENÚ SALIR Introducción 2.3 - LIGANDOS ENDÓGENOS El sistema cannabinoide endógeno o endocannabinoide está constituido por un conjunto de moléculas, con función moduladora sobre otros sistemas de señalización, entre las que se incluyen: receptores endógenos, transportadores, endocannabinoides y enzimas implicadas en la síntesis y degradación de éstos. Los endocannabinoides cumplen las condiciones necesarias de todo neurotransmisor ya que son sintetizados y liberados a partir de las neuronas, son capaces de unirse y activar receptores de membrana siendo, finalmente, inactivados por recaptación y degradación enzimática en el interior de la célula. A diferencia de los demás neurotransmisores, tienen naturaleza lipofílica por lo que no se almacenan en el interior de vesículas sinápticas sino que se sintetizan y liberan a demanda desde la membrana celular. El hecho de que CB1 se identificase como receptor del THC condujo a la búsqueda de cannabinoides endógenos. En 1992, se identificó al primer ligando endógeno de CB1 y se definió su estructura como una araquidonil-etanolamida, denominándose anandamida (AEA) (Devane et al., 1992). Se encontró que la AEA se unía a los receptores cannabinoides y mimetizaba muchos de los efectos farmacológicos y de comportamiento del Δ9-tetrahydrocannabinol (THC), el principal componente psicoactivo de la mariguana. La AEA es una amida, entre el ácido araquidónico y la etanolamina, que se comporta como un agonista parcial de CB1 y un agonista muy débil de CB2. Pertenece a la familia de las N-acil-etanolaminas (NAEs) y, al igual que otros componentes de esta familia como la homo-γ-linoleniletanolamida (HEA) y la docosatetraeniletanolamina (DEA), son producidas por neuronas y se unen a CB1. Las concentraciones de anandamida en cerebro son muy bajas ya que no se almacena en las células, en su forma biológicamente activa, sino que es sintetizado en respuesta a un determinado estímulo o a demanda a partir de un precursor presente en la membrana celular. En general, los niveles más altos de AEA en cerebro se corresponden con áreas que también presentan una elevada densidad de receptores para cannabinoides tales como el hipocampo, la corteza o el estriado (Felder et al., 1996). Esta correlación no es completa ya que en otras áreas como el tálamo o el tallo cerebral - 27 - MENÚ SALIR Introducción existen pocos receptores y un alto nivel de AEA (Felder et al., 1996; Bisogno et al., 1999). Posteriormente, se identificó a un segundo cannabinoide endógeno, el 2araquidonil-glicerol (2-AG), tanto en cerebro como intestino, siendo su concentración en cerebro de 2 a 3 órdenes de magnitud mayor que el de AEA (Mechoulam et al., 1994; Sugiura et al., 1995). En células y tejidos no estimulados, los niveles de 2-AG son mucho mayores que los de AEA aunque probablemente estén sobreestimados debido, por ejemplo, al rápido incremento de la formación de 2-AG que se da tras la decapitación de la rata (Sugiura et al., 2001). Se han descrito a otros tres candidatos como agonistas de receptores cannabinoides, denominándose: 2-araquidonil-gliceril-eter (Noladina, 2-AGE), que se comporta como agonista selectivo de CB1, O-araquidonil-etanolamina (Virodamina) antagonista de CB1 y agonista parcial de CB2 y N -araquidonil-Dopamina (NADA) agonista selectivo de CB1 y potente agonista de los receptores vaniloides (Hanus et al., 2001; Bisogno et al., 2000; Porter et al., 2002). Δ9- tetrahydrocannabinol (THC) Anandamida (AEA) 2- Araquidonil-glicerol (2-AG) N - araquidonil-Dopamina (NADA) Virodamina Noladin-eter Estructura química del principal componente psicoactivo del cannabis Δ9-tetrahydrocannabinol (THC) y de los principales endocannabinoides descritos. - 28 - MENÚ SALIR Introducción Otro miembro de esta familia de lípidos, la palmitoiletanolamida (PEA), comparte con los cannabinoides endógenos diversos efectos fisiológicos aunque no es capaz de unirse a ninguno de los dos subtipos de receptores de cannabinoides (CB1 y CB2), conocidos hasta el momento. 2.3.1 - Biosíntesis de Anandamida La AEA se produce a demanda mediante la hidrósilis de una N-araquidonil fosfatidiletanolamina (NAPE), presente en la membrana plasmática celular, proceso que es catalizado por una fosfolipasa D (PLD). Dicho precursor se genera a partir de fosfolípidos de membrana como la fosfatidilcolina y fosfatidiletanolamina gracias a la actividad de una transferasa de grupo N-acilo (NAT), dependiente de Ca2+, que transfiere el grupo acilo desde la posición sn-1 de los fosfolípidos al grupo amino de la fosfatidiletanolamina (PE). Esta vía no parece ser capaz de generar grandes cantidades de AEA ya que los niveles de ácido araquidónico, esterificado en la posición sn-1, de los fosfolípidos son normalmente muy bajos. Esto concuerda con el hecho de que los niveles de AEA son menores que los de aquellas otras N-aciletanolaminas en la mayoría de los tejidos analizados. De hecho, tanto NAPE-PLD como NAT no parecen ser selectivas para un determinado motivo de ácido graso. Se ha demostrado que la actividad enzimática de NAPE-PLD es dependiente de la concentración de Ca2+ y puede ser estimulada por poliaminas. Es diferente al resto de las fosfolipasas D ya que no cataliza la típica reacción de transfofatidilación de las PLDs, reacción por la que la cadena alifática de un alcohol primario se transfiere al motivo fosfatidil de un ácido fosfatídico (Ueda et al., 2001; Liu et al., 2002; Petersen y Hansen., 1999). - 29 - MENÚ SALIR Introducción Formación e inactivación de anandamida. La anandamida se genera por hidrólisis de la Naraquidonil fosfatidiletanolamina (N-araquidonil PE), la cual es catalizada por una fosfolipasa D (PLD) (1). La síntesis de N-araquidonil PE está mediada por una actividad N-acil transferasa (NAT) (2), la cual separa el motivo araquidonato (rojo) de la posición sn-1 de los fosfolípidos, tales como la fosfatidilcolina (PC), y lo transfiere al grupo amino de la PE. La anandamida recién formada se libera al espacio extracelular, donde se une y activa a receptores cannabinoides, acoplados a proteínas G, localizados en las células vecinas (3) o en las mismas células que producen anandamida. La anandamida puede ser interiorizada gracias a un transportador de anandamida (4). Dentro de las células es hidrolizada por una amidohidrolasa de ácidos grasos (FAAH) unida a membrana (5), liberándose ácido araquidónico y etanolamina. El ácido araquidónico producido puede ser reincorporado a los fosfolípidos y es improbable que sufran otro metabolismo. In vitro, FAAH puede catalizar la reacción contraria, la formación de anandamida a partir de ácido araquidónico y etanolamina, aunque el sentido fisiológico de esta reacción no está aún claro. Abreviatura: R, grupo ácido graso. Modificado de Piomelli et al., 2000. - 30 - MENÚ SALIR Introducción Otros estudios, en los que se ha inactivado la enzima NAPE-PLD, revelan la existencia de vías de síntesis independientes por las que otras enzimas participarían en la generación de N-aciletanolamidas poliinsaturadas como la AEA (Leung et al., 2006). De hecho, está descrito que existe una vía alternativa, independiente de NAPE-PLD, donde la N-acil-PE es primero hidrolizada liberándose N-acil-lyso-PE, junto con un ácido graso libre, gracias a una enzima miembro del grupo IB de las fosfolipasas A2. Posteriormente se libera la N-aciletanolamida (NAE) desde la N-acil-lyso-PE gracias a la actividad enzimática de una lyso-PLD (Sun et al., 2004). Rutas alternativas de síntesis de AEA. La AEA se puede formar a partir de la hidrólisis directa, catalizada por la enzima PLD, de la N-araquidonil-PE mayoritariamente o bien mediante la formación de un N-araquidonil-lysoPE gracias a la actividad enzimática de la PLA1/A2 para posteriormente generar AEA gracias a la lysoPLD, liberándose ácido lisofosfatídico (LPA). 2.3.2 – Biosíntesis de 2-araquidonilglicerol (2-AG) Este endocannabinoide es un importante precursor y/o producto de degradación de las vías de fosfo-, di- y triglicéridos. Se ha demostrado que varios estímulos pueden provocar la formación de 2-AG en células en reposo tales como lipopolisacáridos, - 31 - MENÚ SALIR Introducción endotelina, factor activador plaquetario, ionomicina, carbacol, trombina, etc. (Bisogno et al., 1997; Basavarajappa et al., 2000; Stella et al., 1997; Sugiura et al., 1998; 2002; Di Marzo et al., 1999; Berdyshev et al., 2001; Mechoulam et al., 1998; Stella y Piomelli, 2001; Walter y Stella, 2003). Sin embargo, poco se sabe acerca de la ruta de biosíntesis del 2-AG, la cual implicaría probablemente a la misma cascada enzimática que cataliza la formación de segundos mensajeros tales como el inositol (1,4,5)-trifosfato y 1,2-diacilglicerol (DAG). El DAG es el sustrato de la lipasa de diacilglicerol (DAGL), enzima que cataliza la producción de 2-AG. Tras su liberación, el 2-AG interaccionaría con los receptores cannabinoides y podría entrar en la célula, gracias al mismo sistema de transporte que la AEA, y ser hidrolizada por una lipasa de monoacilgliceroles (MAGL). Debido a su producción no sináptica y a su rápida inactivación, se piensa que los endocannabinoides podrían actuar como moduladores a corto plazo de la actividad sináptica y celular (Piomelli et al. 2000; Di Marzo et al. 1998; Fride, 2002). Formación e inactivación del 2-araquidonilglicerol (2-AG). La hidrólisis del fosfatidilinositol (4,5)-bifosfato [PtdIns (4,5) P2] por una fosfolipasa C (PLC) genera la liberación de los segundos mensajeros 1,2-diacilglicerol (DAG) e inositol (1,4,5)-trifosfato [Ins(1,4,5)P3] (1). El DAG sirve como sustrato para la DAG lipasa (DAGL), la cual cataliza la producción del 2-AG (2). El 2-AG puede liberarse al medio externo, tras lo cual interaccionaría con los receptores cannabinoides y sería transportado al interior celular. Sin embargo, la liberación extracelular del 2-AG no se ha descrito aún in vivo. Una vez dentro de la célula, el 2-AG puede ser hidrolizado a ácido araquidónico y glicerol por una esterasa como la lipasa de monoacilglicerol (MAGL). Tomado de Piomelli et al., 2000. - 32 - MENÚ SALIR Introducción 2.4 - RECEPTORES CANNABINOIDES Los receptores endocannabinoides, CB1 y CB2, pertenecen a una superfamilia de receptores, acoplados a proteínas G, con siete dominios transmembrana y con una secuencia altamente conservada entre especies. El receptor CB1 tiene un 44% de homología en su secuencia aminoacídica con el receptor CB2, aunque dicha cifra incrementa hasta un 68% si se comparan sólo las regiones transmembranales. El receptor CB1 se localiza predominantemente en el sistema nervioso central (SNC), donde se le atribuye un importante papel en la modulación de la liberación de neurotransmisores (Herkenham et al., 1991; Glass et al., 1997), pero también en la mayoría de los tejidos periféricos como células inmunes, el sistema reproductor, el tracto gastrointestinal, el hígado, páncreas endocrino y pulmón. La mayor densidad de CB1 en el SNC se da en áreas relacionadas con el control de la actividad motora tales como la corteza, ganglios basales y cerebelo. Son también abundantes en áreas importantes para la memoria y el aprendizaje (hipocampo) y la regulación neuroendocrina (núcleos hipotalámicos). Su localización es predominantemente presináptica, lo que concuerda en buena medida con el efecto inhibitorio que los cannabinoides ejercen sobre la liberación de varios neurotransmisores (Pertwee, 1997), aunque, en algunos casos, también se localiza en menor proporción en somas y dendritas neuronales así como en oligodendrocitos, - 33 - MENÚ SALIR Introducción astrocitos y células madre neurales (Rodríguez et al., 2001; Molina-Holgado et al., 2002; Aguado et al., 2005). El receptor CB2 se localiza principalmente en determinados componentes del sistema inmune (amígdala, bazo, macrófagos, linfocitos B y células NK) (Devane et al., 1988; Matsuda et al., 1990; Munro et al., 1993) aunque también en células de la microglía, astrocitos e incluso en ciertas subpoblaciones neuronales, aunque siempre en menor proporción que CB1 (Gong et al., 2006). Experimentos llevados a cabo en ratones carentes del receptor CB2 (CB2-/-) muestran cambios en la respuestas inmune, especialmente a nivel de la activación, inducida por macrófagos, de la célula T- helper (Munro et al., 1993; Galiègue et al., 1995; Herring y Kaminski, 1999). En algunas de las poblaciones celulares del sistema inmune, los niveles de expresión de los receptores CB2 son equiparables, cuantitativamente, a los de receptores CB1 en el SNC, por lo que se considera que CB2 podría desempeñar importantes funciones en los procesos de inflamación. La proporción de CB2, en dichas células inmunes, también podría explicar buena parte de los efectos moduladores que ejercen los cannabinoides naturales y sintéticos sobre la respuesta inmune. A este respecto, entre los cannabinoides naturales, parece ser más eficaz el cannabinol que el THC (Felder et al., 1995) con lo que sería el principal responsable de las propiedades antiinflamatorias de C. sativa. Se ha descrito que en la microglía presente en tejido del SNC sano, se expresa muy poco o nada CB2 mientras que las células de microglía activadas, tales como las encontradas en modelos de ratón de esclerosis múltiple y Alzheimer, se expresan niveles significativos de CB2 (Maresz et al., 2005; Benito et al., 2003). Esto concuerda con el hecho de que, en condiciones patológicas, se induce la expresión de CB2 en leucocitos (Carlisle et al., 2002). Estudios llevados a cabo en ratones CB1-/- y/o CB2-/-, sugieren la existencia de receptores cannabinoides cerebrales distintos a CB1 y CB2. Estudios farmacológicos han revelado la presencia de otras dianas de los endocannabinoides que incluyen al receptor vaniloide (Zygmunt et al., 1999) y a dos receptores “CB-like” distintos del CB1 y CB2, uno en el lecho vascular y otro en los terminales axónicos de glutamato (Hajos et al., 2001; Howlett et al., 2002; Kunos et al., 2002). En varios tejidos se ha - 34 - MENÚ SALIR Introducción descrito a un receptor acoplado a proteína G, denominado GPR55, como un posible nuevo receptor cannabinoide (Baker et al., 2006). La existencia de estos y otros posibles receptores para cannabinoides, y su función en la fisiología endocannabinoide sólo se verá clara tras su caracterización molecular. En relación con estos datos, se ha sugerido la presencia de sitios de fijación con perfil cannabimimético, distintos a CB1 y CB2, en cerebro de ratones en los que se ha generado genéticamente la ausencia de receptores CB1 (‘Knock-out’) (Berrendero et al., 1998). Sin embargo, la existencia de receptores cannabinoides diferentes a CB1 /CB2 en cerebro humano no ha sido confirmada. 2.4.1 - Mecanismos de Transducción de Señales Una vez liberada, la AEA puede actuar sobre los receptores cannabinoides y/o ser transportada al interior celular gracias a un sistema de transporte independiente de Na+ y energía. Dentro de las células, puede ser hidrolizada por una amidohidrolasa de ácidos grasos (FAAH) liberándose ácido araquidónico y etanolamina. Los principales mecanismos intracelulares, en los que están implicados los receptores CB1, incluyen la inhibición de la adenilato ciclasa, la regulación de diferentes canales iónicos y la activación de la vía de las MAP quinasas (Berrendero et al., 1998). Ambos receptores, CB1 y CB2, son sensibles a la inhibición de la formación de AMPc por la toxina pertusis, lo cual implica a proteínas Gi/o como transductores, y a la estimulación de la actividad kinasa de proteínas p42/p44 activadas por mitógenos (Vogel et al., 1993; Bouaboula et al., 1995). De hecho, el acoplamiento de estos receptores a proteínas Gi/o constituye la base de todos los efectos. La inhibición de la vía de la adenilato ciclasa da lugar a un descenso en los niveles de AMPc intracelular, así como a una inhibición de los canales de Ca2+ tipo N y P/Q y un aumento de la conductancia del K+ (Mackie y Hille, 1992; Mackie et al., 1995; McAllister et al., 1999). El efecto combinado sobre ambos tipos de canales parece ser la base de la inhibición que los cannabinoides ejercen en la liberación de neurotransmisores. Finalmente, también se activa la ruta de las MAP quinasas, vía involucrada en la regulación de fenómenos de proliferación y diferenciación (Breivogel et al., 2001). - 35 - MENÚ SALIR Introducción Esquema de las vías bioquímicas de síntesis/degradación y acciones celulares de la anandamida. La anandamida es liberada desde un precursor de membrana lipídico (N-araquidonil-fosfatidil etanolamida, NAPE) por la acción de una fosfolipasa D específica (PLD) activada por despolarización o estimulación de receptores acoplados a proteínas G (GPCR). La biosíntesis de NAPE es catalizada por una enzima, N-aciltransferasa (NAT) activada por calcio (Ca2+) y AMP cíclico. La anandamida actúa como un mensajero retrógrado en los receptores presinápticos cannabinoides (CB1) donde regula la liberación de neurotransmisores (NT) a través de sistemas de transducción secundarios (principalmente el Ca2+ incorporado a través de canales de calcio unidos a voltaje (VGCC) o receptores de glutamato NMDA). La anandamida también actúa como un neuromodulador de los principales sistemas de neurotransmisores, incluyendo dopamina, en las células postsinápticas, donde regula la excitabilidad y la plasticidad sináptica a través de la modulación de canales de potasio (K+) y la regulación de un amplio espectro de proteínas kinasas (PK), incluyendo la proteína kinasa A y las proteínas kinasas activadas por mitógeno (MAPK). La acción de la anandamida termina a través de un proceso de dos etapas que incluye, primero, su recaptación celular a través de un transportador específico de anandamida (AT) y segundo, la degradación enzimática a ácido araquidónico (AA) y etanolamida por la enzima de membrana amidohidrolasa de ácidos grasos (FAAH). Tomado de Rodríguez de Fonseca et al., 2005. - 36 - MENÚ SALIR Introducción En cuanto al receptor CB2, su activación también conduce a una inhibición de la adenilato ciclasa y una activación de la vía de las MAP quinasas. Sin embargo, a diferencia del CB1, el receptor CB2 no es capaz de modificar las corrientes de los canales de Ca2+ y K+ (Herkenham, 1995). Se ha propuesto que los endocannabinoides podrían funcionar como neuromoduladores reduciendo la liberación de ciertos neurotransmisores durante periodos de intensa estimulación. La actuación de los endocannabinoides sobre receptores presinápticos CB1 reduciría la actividad neuronal a través de la inhibición de canales de Ca2+ sensibles a voltaje, cuya apertura es necesaria para producir la liberación del neurotransmisor. A su vez, la activación de los receptores CB1 podría incrementar la entrada de K+, lo que reduciría la intensidad de eventuales despolarizaciones, la generación de los potenciales de acción, y, en definitiva, la propagación del impulso nervioso. 2.5 - NEUROFARMACOLOGÍA DEL SISTEMA ENDOCANNABINOIDE El Sistema Endocannabinoide (SEC) está ampliamente distribuido en todo el cuerpo, lo cual se correlaciona con su papel modulador de múltiples procesos fisiológicos. En los tejidos periféricos, la localización de los componentes del sistema endocannabinoide refleja la distribución de los tipos celulares donde se ubican (por ejemplo, linfocitos B en nódulos linfáticos y bazo). Sin embargo, en el sistema nervioso su distribución es mucho más compleja y estructurada, y claramente refleja la importancia de dicho sistema en la transmisión sináptica. En algunas regiones, tales como el hipocampo, hay una distribución complementaria de receptores cannabinoides, transportadores y enzimas de degradación. Sin embargo, en otras áreas del cerebro, como el tálamo, hay discrepancias (por ejemplo, donde existen transportadores y expresión de MAGL están ausentes los receptores CB1) en su distribución, lo cual refleja el desconocimiento existente acerca de este sistema. En cuanto a la bioquímica del SEC, los endocannabiniodes se liberan a demanda tras la despolarización celular o estimulación del receptor siendo, en ambos casos, de forma dependiente de calcio. Una vez producidos, actúan sobre los receptores - 37 - MENÚ SALIR Introducción cannabinoides como mediadores locales en las células cercanas al lugar de producción, lo que indica que actúan de manera similar a los autacoides. En el SNC, la distribución altamente organizada de los elementos de señalización endocannabinoide en las sinapsis glutamatérgicas y GABAérgicas y su conservación a lo largo de la evolución, demuestra su papel fundamental en la transmisión sináptica. Debido a la inhibición de la adenil ciclasa, la activación de las corrientes de K+ y la inhibición de la entrada de Ca2+ en la célula, el efecto neto de la estimulación del receptor CB1 es una hiperpolarización local que conduce a los efectos inhibitorios generales descritos. Si los endocannabinoides actúan postsinápticamente, contrarrestarán la activación existente en las células postsinápticas. Este mecanismo de interacciones postsinápticas se ha propuesto para el caso de la transmisión dopaminérgica (Felder et al., 1998; Rodríguez de Fonseca et al., 1998; Giuffrida et al., 1999). A pesar de su importancia, la modulación postsináptica en cuantitativamente menor a las importantes acciones presinápticas, lo cual se basa en dos hechos: la concentración de receptores CB1 en los terminales presinápticos y los efectos inhibidores descritos, con agonistas de CB1, sobre la liberación de GABA, glutamato, acetilcolina y noradrenalina (Schlicker y Kathmann, 2001; Piomelli, 2003). Este efecto inhibidor se ha demostrado también con neuropéptidos como el factor liberador de corticotropina y la colecistoquinina (Rodríguez de Fonseca et al., 1997; Beinfeld y Connolly, 2001). El efecto de inhibición de la liberación de neurotransmisores presinápticos se asocia con la acción inhibidora de los endocannabinoides sobre los canales de Ca2+ presinápticos, a través de la activación de CB1. Dicha inhibición podría dar lugar a dos formas diferentes de plasticidad a corto plazo, dependiendo de la implicación, ya sea de la transmisión de GABA o glutamato: supresión de la inhibición inducida por despolarización (DSI) y supresión de la excitación inducida por despolarización (DSE), respectivamente (Wilson y Nicoll, 2002; Diana y Marty, 2004). Ambas formas de plasticidad implican la activación inicial de una neurona de proyección postsináptica (célula piramidal o de Purkinje) que envía un mensajero retrógrado al terminal presináptico GABAérgico (DSI) o glutamatérgico (DSE), induciendo una supresión transitoria ya sea de la inhibición o excitación presináptica. La contribución de los endocannabinoides a estas formas de plasticidad a corto plazo se ha descrito en - 38 - MENÚ SALIR Introducción hipocampo (Wilson y Nicoll, 2001; Wilson et al., 2001) y en cerebelo (Diana et al., 2002). El papel del DSI o DSE, inducido por endocannabinoides, parece ser resultado de la coordinación de elementos neurales, dentro del hipocampo y el cerebelo, que están implicados en procesos fisiológicos relevantes, tales como la memoria o la coordinación motora. Otras formas adicionales de modulación endocannabinoide de la transmisión sináptica implican la inducción de plasticidad sináptica a largo plazo, denominada como potenciación a largo plazo (LTP) y depresión a largo plazo (LTD). Ambas formas de plasticidad implican cambios a largo plazo en la eficacia de la transmisión sináptica en las neuronas glutamatérgicas, las cuales tienen un mayor impacto sobre la consolidación y remodelación de la sinapsis. La activación de los receptores cannabinoides previene la inducción de LTP en las sinapsis hipocampales (Stella et al., 1997) y la facilitación de LTD en el estriado (Gerdeman et al., 2002) y núcleo accumbens (Robbe et al., 2002). En el hipocampo, los mensajeros endocannabinodes regulan una forma de LTD que afecta a las neuronas GABAérgicas (Chevaleyre y Castillo, 2003). En general, los endocannabinoides actúan como mensajeros locales que ajustan el valor sináptico y contribuyen significativamente a la eliminación del flujo de información a través de sinapsis específicas en un amplio rango temporal. El hecho de que la estimulación del receptor cannabinoide tenga impacto sobre los segundos mensajeros implicados no sólo en la remodelación sináptica (Derkinderen et al., 1996; Piomelli, 2003) sino también en la diferenciación neuronal (Rueda et al., 2002) y la supervivencia neuronal (Panikashvili et al., 2001; Marsicano et al., 2003), indica que el sistema de señalización es un importante mecanismo homeostático que garantiza un fino ajuste del procesamiento de la información en el cerebro y provee mecanismos contrarreguladores dirigidos a preservar la estructura y función de los principales circuitos cerebrales. Ambos procesos son relevantes para el comportamiento homeostático como es el comportamiento motivado (alimentación, reproducción, relajación, sueño) y emociones, así como para la cognición, ya que el aprendizaje y memoria requieren cambios morfológicos y en las dinámicas funcionales de los circuitos cerebrales. Una confirmación experimental de este hipotético papel del sistema - 39 - MENÚ SALIR Introducción endocannabinoide fue la demostración de su implicación en el control de la extinción de la memorias aversivas (Marsicano et al., 2002; Terranova et al., 1996). A pesar de la modulación periférica del sistema inmune, lechos vasculares, órganos reproductores, el metabolismo y motilidad gastrointestinal, el SEC también regula los procesos de percepción incluyendo la nocicepción (los cannabinoides son potentes analgésicos, Martin y Litchman, 1998) y el proceso visual en la retina (Straiker et al., 1999). Otras funciones ejercidas por el SEC implican la regulación de los circuitos cerebelares y de los ganglios basales, donde actúa en la modulación del aprendizaje implícito de las rutinas motoras (Rodríguez de Fonseca et al., 1998). Entre las variadas funciones en las que el SEC está implicado, el control homeostático de las emociones y la regulación del comportamiento motivado merece una atención especial debido a su impacto sobre enfermedades humanas, incluyendo la adicción. Los efectos positivos de los endocannabinoides sobre la motivación parecen estar mediados no sólo por los sistemas sensoriales periféricos en los que los receptores cannabinoides están presentes (por ejemplo, el fomento del consumo de alimentos mediante agonistas de receptores CB1, Gómez et al., 2002), sino también por la acción de los endocannabinoides sobre el sistema de recompensa. El SEC está ampliamente distribuido en la amígdala extendida (conjunto de núcleos telencefálicos localizados en las neuronas del septo medio, el shell del núcleo accumbens y el complejo amigdalar) y está implicado en el control del comportamiento motivado, en las respuestas condicionadas y en las respuestas emocionales asociadas. Esta hipótesis se apoya en dos hechos: la inhibición del comportamiento motivado tras la administración de antagonistas cannabinoides (Colombo et al., 1998a; Navarro et al., 2001) y los déficits de recompensa observados en el ratón ‘knockout’ del receptor CB1 (Ledent et al., 1999; Maldonado y Rodríguez de Fonseca, 2002; Sanchis-Segura et al., 2004). La investigación de las bases neurobiológicas de los efectos de los endocannabinoides sobre el comportamiento motivado se ha focalizado sobre la interacción endocannabionide-dopamina así como en el papel del SEC en el aprendizaje y condicionamiento. La amígdala extendida es una diana de las proyecciones mesocorticolímbicas ascendentes de las neuronas dopaminérgicas del área tegmental ventral (ATV), un subconjunto de neuronas mesencefálicas que dan lugar a una - 40 - MENÚ SALIR Introducción respuesta contundente a las drogas que causan dependencia (Maldonado y Rodríguez de Fonseca, 2002). La mayoría de las drogas activan a las neuronas dopaminérgicas del ATV, en términos de liberación de dopamina en las áreas terminales, especialmente en el núcleo accumbens y la corteza prefrontal, o mediante las tasas de disparo de dichas neuronas del ATV. El THC y otros agonistas de CB1, incrementan el eflujo de dopamina en el núcleo accumbens y en la corteza prefrontal e incrementan la tasa de disparo de dopamina en el ATV (Gardner y Vorel, 1998). Este efecto no está causado por la activación directa de las neuronas dopaminérgicas ya que no expresan CB1 (Julián et al., 2003). Aunque los efectos de los agonistas sobre la liberación de dopamina, en las áreas de proyección, pueden bloquearse con antagonistas opioides, como la naloxona, no se puede bloquear el incremento de la tasa de disparo en la célula. Esta discrepancia podría sugerir la existencia de un papel diferente para los sistemas opioides endógenos, como moduladores de las acciones cannabinoides, en los cuerpos celulares dopaminérgicos con respecto a sus terminaciones axonales. Los efectos cannabinoides podrían también implicar a las aferencias glutamatérgicas y GABAérgicas del núcleo accumbens y del ATV, ya que los receptores CB1 presinápticos regulan la liberación de GABA y glutamato en estas áreas, incluyendo el LTD (Schlicker y Kathman, 2001; Robbe et al., 2002). De acuerdo con estas acciones en los circuitos de recompensa, la exposición continuada a cannabinoides puede inducir sensibilización similar a la producida por otras drogas que causan dependencia. La administración crónica de cannabinoides también produce una sensibilización cruzada con los efectos locomotores de los psicoestimulantes (Maldonado y Rodríguez de Fonseca, 2002). Debido a que los endocannabinoides inducen LTD en el núcleo accumbens (que afecta a las aferencias procedentes de la corteza prefrontal), probablemente regulen la adquisición del hábito aprendido y las respuestas condicionadas relevantes para la pérdida progresiva del control, que caracterizan a la adicción a alcohol (Maldonado y Rodríguez de Fonseca, 2002). La administración de un antagonista de CB1 bloquea la recaída en el consumo de heroína y cocaína (De Vries et al., 2001, 2003). La importancia del SEC en el control - 41 - MENÚ SALIR Introducción del comportamiento motivado va más allá del control del procesamiento de las señales de recompensa constantes (Sanchis-Segura et al., 2004). Además, cuando los mecanismos homeostáticos cannabinoides no son adecuados para restaurar la pérdida de equilibrio en el control de la recompensa derivada de la exposición contínua y no controlada al reforzador (por ejemplo, opiáceos o alcohol), los cambios alostáticos que implican a CB1 contrarrestan la espiral de daños sobre el circuito de recompensa. Esto se ha demostrado en roedores expuestos a ciclos de dependencia-abstinencia a alcohol y morfina (Navarro et al., 2001; Rimondini et al., 2002). En este modelo, se asocia un historial de dependencia con una permanente sobrexpresión de CB1, en áreas relacionadas con la recompensa, y con una incrementada sensibilidad a la interrupción de la recompensa, inducida por antagonistas de los receptores cannabinoides (Rodríguez de Fonseca et al., 1999; Rimondini et al., 2002). Los receptores cannabinoides no sólo se asocian con alteraciones motivacionales, sino también se relacionan con el procesamiento de las emociones. Un punto clave para la regulación endocannabinoide de las emociones es el complejo amigdalar. Los endocannabinoides son capaces de disminuir la liberación de glutamato y del factor liberador de corticotropina, reduciendo la señalización vía eferencias amigdalares y la actividad de las proyecciones inhibidoras basolaterales de GABA hacia el núcleo central de la amígdala, con lo cual se activa la vía amigdalofugal (Rodríguez de Fonseca et al., 1996, 1997; Navarro et al., 1997; Marsicano et al., 2002; Piomelli, 2003). El balance final conducirá a la ansiedad o ansiolisis, dependiendo de la tasa de activación de las proyecciones descendentes del núcleo central de la amígdala hacia el hipotálamo (respuestas endocrinas) y el tronco cerebral (respuestas autónomas y de comportamiento). Sin embargo, estudios recientes indican que la ansiolisis es una respuesta normal a un incremento de la transmisión cannabinoide en el sistema límbico, como se refleja en el fenotipo de ratones ‘knockout’ de FAAH y con los efectos de los inhibidores de FAAH (Cravatt y Lichtman, 2003; Kathuria et al., 2003). La inducción de ansiedad por antagonistas cannabionides (Navarro et al., 1997) apoya también esta hipótesis. - 42 - MENÚ SALIR Introducción 3. ALCOHOL Y SISTEMA ENDOCANNABINOIDE 3.1 - Efectos Agudos del Alcohol sobre el Sistema Endocannabionoide Actualmente no existen estudios que describan los efectos centrales de la administración aguda de etanol. En la presente tesis, se expondrán los efectos observados tras dicho tratamiento sobre la síntesis, degradación y señalización mediada por anandamida y sus congéneres sobre sistemas de transmisión lipídicos como los endocannabinoides. 3.2 - Efectos Crónicos del Alcohol sobre el Sistema Endocannabionoide Gran número de artículos científicos relacionan la señalización a través del receptor CB1 con el comportamiento de búsqueda de alcohol. La administración de agonistas de CB1 promueve el consumo de etanol (Colombo et al., 2002) mientras que los antagonistas reducen la autoadministración de etanol (Arnone et al., 1997; Freedland et al., 2001), especialmente en animales con un historial de dependencia a etanol (Rodríguez de Fonseca et al., 1999) o en líneas de ratas con preferencia al alcohol (Colombo et al., 1998; Gessa et al., 2005). Mediante hibridación in situ se ha demostrado que, durante el consumo crónico de etanol, la expresión génica del receptor CB1 disminuye en áreas cerebrales de rata como el núcleo caudado-putamen, el núcleo ventromedial del hipotálamo y ciertas regiones del hipocampo (CA1, CA2) (Ortiz et al., 2004). El desarrollo de ratones deficientes en el gen del receptor CB1 ha permitido demostrar que la señalización endocannabinoide, actuando sobre CB1, está implicada en la preferencia a etanol. Se ha demostrado que los ratones CB1-/- muestran un consumo de etanol voluntario drásticamente disminuido (Hungund et al., 2003; Poncelet et al., 2003; Wang et al., 2003; Naassila et al., 2004). Por otro lado, un rastreo génico ha identificado al gen del receptor CB1 como uno de los que presentan alteraciones en su expresión durante los ciclos de exposición a etanol (Rimondini et al., 2002) y estudios genéticos en humanos han encontrado relación entre el alcoholismo y - 43 - MENÚ SALIR Introducción polimorfismos y/o mutaciones de dicho gen (Comings et al., 1997; Schmidt et al., 2002). Otros estudios han puesto de manifiesto que la administración crónica de etanol está asociada con un incremento en la formación de AEA y de su precursor la Naraquidonilfofatidiletanolamina (NAPE) en el linaje de células SK-N-SH de neuroblastoma humano (Basavarajappa y Hungund, 1999; Ortiz et al., 2004). A su vez, la exposición crónica a etanol también estimula la formación del endocannabinoide 2araquidonilglicerol (Basavarajappa et al., 2000). Se ha demostrado que el incremento encontrado en las concentraciones de anandamida extracelular en cultivos de neuronas granulares de cerebelo, tras someterse a tratamiento crónico de etanol, podrían deberse a una inhibición indirecta de FAAH y a una disminución en la recaptación de AEA (Basavarajappa et al., 2003). El transportador de anandamida es, además, independiente de CB1 ya que su actividad no se altera ante la presencia de antagonistas de CB1 (SR141716A) así como tampoco existen tales diferencias en ratones ‘knockout’ de CB1 disminuyendo, tanto en éstos como en ratones silvestres, el transporte de anandamida tras ser sometidos a tratamientos crónicos de etanol (Basavarajappa et al., 2003). Estos resultados apoyarían la hipótesis de que la ingesta prolongada de etanol incrementa los niveles de ligandos endocannabinoides cerebrales (Basavarajappa y Hungund, 1999), lo cual llevaría a un decremento de la expresión génica y función de CB1 (Basavarajappa et al., 1998; Basavarajappa y Hungund, 1999; Ortiz et al., 2004). Se sabe también que animales genéticamente predispuestos a un mayor consumo de etanol, poseen niveles elevados de endocannabinoides en la corteza prefrontal (área relacionada con la recompensa) y un menor número y activación de receptores CB1 (Hansson et al., 2007) Varias evidencias sugieren que la enzima FAAH actúa como un regulador catabólico primario de anandamida y que está relacionado con la degradación de otras amidas de ácidos grasos bioactivas. Concretamente, ratones FAAH -/- están seriamente afectados en su capacidad de degradar anandamida y muestran un comportamiento exagerado a esta amida de ácido graso, tales como hipomovilidad, hipotermia, analgesia y catalepsia. Además, los niveles endógenos de anandamida en cerebro y de amidas de ácidos grasos relacionadas, están incrementados unas 10 veces en ratones FAAH-/correlacionándose con el aumento de la analgesia, dependiente de CB1, en estos animales (Cravatt et al., 1996). - 44 - MENÚ SALIR Introducción Todos estos hechos, sugieren que FAAH podría controlar la magnitud y duración de la señalización in vivo de las amidas de ácidos grasos mediante el establecimiento de un tono endocannabinoide en el cerebro así como, posiblemente, en otras partes del sistema nervioso. De hecho, el patrón de distribución de FAAH es muy parecido al que presenta el receptor CB1, con altas concentraciones en hipocampo, cerebelo y corteza cerebral. En humanos se ha descrito que un polimorfismo en un solo nucleótido del gen de la enzima FAAH, 385A, está fuertemente asociado con el consumo de drogas, entre ellas el alcohol (Sipe et al., 2002). El polimorfismo encontrado en FAAH produce una mutación sin sentido (385C→A) que resulta en la conversión de un residuo de Pro por Thr (P179T). Curiosamente, este residuo está altamente conservado en todos los mamíferos caracterizados hasta la fecha. Aunque la mutación P179T no se ha encontrado que altere las propiedades catalíticas o la estabilidad estructural de FAAH, sí produciría una variante de FAAH con una incrementada sensibilidad a la degradación proteolítica (Sipe et al., 2002). Por tanto, los polimorfismos en el gen FAAH humano que generasen enzimas alteradas funcionalmente también se esperaría pudiesen cambiar los niveles tónicos de señalizadores lipídicos del SEC, lo cual podría repercutir sobre las vías cerebrales de adicción/recompensa a drogas. En ratones FAAH -/- está descrito un incremento en la preferencia y el consumo de alcohol (Walker et al., 2005; Basavarajappa et al., 2006; Blednov et al., 2007) y recientemente se han encontrado diferencias tanto en la expresión como en la actividad de FAAH en la corteza prefrontal de ratas genéticamente alcohólicas. Todo ello parece sugerir un importante papel para esta enzima en los procesos adictivos. 3.3 - Interacción de Cannabinoides y Alcohol: una Nueva Diana para el Tratamiento De todos los cannabinoides, sólo el THC posee una interacción farmacodinámica con el etanol significativa (Hollister y Gillespie, 1970), siendo sus efectos comportamentales y farmacológicos como hipotermia, euforia, analgesia, y ataxia además del desarrollo de tolerancia y dependencia similares entre sí (Kalant y LeBlanc, 1974). - 45 - MENÚ SALIR Introducción Un gran número de evidencias sugieren la existencia de interacciones funcionales entre los efectos del cannabis y el etanol. Primero, la capacidad de esta pequeña molécula para interaccionar con numerosos sistemas fisiológicos. Los efectos iniciales del etanol darían lugar a la facilitación de receptores GABAA y a la inhibición de la transmisión de receptores de glutamato NMDA. Segundo, el cannabis y el etanol muestran algunos perfiles de comportamiento similares: a bajas dosis ambos producen estimulación de la actividad locomotora y a altas dosis producen sedación aunque, obviamente, los niveles de dosis a los que estos efectos se dan son muy diferentes. Por último, ambos activan las mismas vías de recompensa y el receptor CB1 juega un importante papel en la regulación de sus propiedades de refuerzo positivo. Está bien establecido que el sistema mesolímbico dopaminérgico juega un papel central en los circuitos de recompensa, ruta que es común en el refuerzo producido por la mayoría de las drogas de abuso. Se ha demostrado que el tratamiento crónico con anandamida produce tolerancia a una determinada dosis y tolerancia cruzada al THC en ratones, de manera similar a la observada tras el tratamiento crónico de THC (Fride y Mechoulam, 1993). Ya que muchos de estos efectos comportamentales son similares a los observados en el etanol, se sugiere que estos compuestos (THC, etanol y anandamida) podrían compartir una diana común para inducir tolerancia y podrían tener un mecanismo común para sus acciones. Está descrito que la administración del antagonista SR141716A inhibe la ingesta de etanol en animales consumidores de etanol como los ratones C57Black/6 (Arnone et al., 1997) y las ratas sP (Serra et al., 2001), incrementando la ingesta voluntaria de dicha solución en estas últimas cuando se administran agonistas del receptor cannabinoide (Colombo et al., 2002). Por otro lado, usando cerveza (la cual las ratas beben fácilmente) como solución alcohólica, Gallate et al. (1999) han descrito que el agonista CP-55,940 incrementa, de manera dependiente de la dosis, la respuesta por la cerveza, efecto que revertía cuando se administraba SR141716A. Un estudio de autoadministración ha demostrado que el antagonista SR141716A reduce las respuestas operantes en ratas Wistar, que se hicieron dependientes de etanol mediante una exposición a vapores de etanol durante 14 días, aunque no ocurría lo mismo en ratas que no eran dependientes de etanol (Rodríguez de Fonseca et al., 1999). - 46 - MENÚ SALIR Introducción El hecho de que los receptores CB1 estén implicados en mediar los aspectos consumidores y de apetencia por el etanol (Freedland et al., 2001), sugiere que el antagonista SR141716A podría tener una gran utilidad para el tratamiento del abuso de alcohol y podría ser también efectivo en la prevención de la recaída ya que se ha descrito que dicho compuesto es eficaz en inhibir la restauración del comportamiento de búsqueda de droga provocado por la cocaína (De Vries et al., 2001). - 47 - MENÚ SALIR MENÚ SALIR OBJETIVOS MENÚ SALIR MENÚ SALIR Objetivos 1.- Caracterizar las acciones agudas del etanol, en ratas Wistar, sobre la dinámica de señalización cannabinoide mediante estudios de: a) Expresión de ARNm y de proteínas del Sistema Endocannabinoide. b) Actividades enzimáticas de síntesis y degradación de endocannabinoides. c) Producción de endocannabinoides. d) Relación con la actividad de neurotransmisores implicados en las acciones del alcohol. 2.- Caracterizar las acciones crónicas del etanol, en ratas Wistar, sobre la dinámica de señalización cannabinoide mediante estudios de: a) Expresión de ARNm y de proteínas del Sistema Endocannabinoide. b) Actividad enzimática de síntesis y degradación de endocannabinoides. 3.- Caracterizar el Sistema Endocannabinoide en líneas animales con preferencia alcohólica, mediante estudios de: a) La expresión de ARNm y de proteínas. b) La actividad enzimática de síntesis y degradación. c) La producción de endocannabinoides. d) Relación de la señalización endocannabinoide con la actividad de los neurotransmisores implicados en las acciones del alcohol. e) Respuesta de estas líneas a la administración de fármacos que interfieren con la señalización endocannabinoide. 4.- Formular propuestas de terapia del alcoholismo en base a los hallazgos en las acciones del alcohol sobre el Sistema Endocannabinoide. - 51 - MENÚ SALIR MENÚ SALIR MATERIALES Y MÉTODOS MENÚ SALIR MENÚ SALIR Materiales y Métodos 1. ANIMALES DE EXPERIMENTACIÓN 1.1 – RATAS WISTAR En la realización de esta tesis doctoral se han utilizado ratas adultas macho Wistar (Charles River, Wilmington, MA y Barcelona, España) con un peso entre 250400 gr, las cuales fueron sometidas a diferentes tratamientos de etanol tal y como se detalla en el apartado 1.3.1. Los animales se mantuvieron a una temperatura de 20 + 2º U U C, una humedad relativa de 40 + 5 % y a ciclos de luz-oscuridad de 12 horas de U U duración (encendido de luz a las 8:00 a.m.), teniendo libre acceso a comida (dieta comercial para roedores SAFE A04; Panlab, Barcelona, España) y agua en el animalario de la Facultad de Medicina de la Universidad de Málaga. Para el manejo y sacrificio de los animales se siguió la normativa europea vigente 86/609/ES que regula la investigación animal. 1.2 - MODELOS ANIMALES DE ALCOHOLISMO 1.2.1. Ratas AA En esta tesis se han estudiado modelos animales con preferencia al consumo de etanol como son las ratas AA (Alko Alcohol) las cuales fueron comparadas con ratas sin preferencia, denominadas ANA (Alko Non-Alcohol). Las ratas AA y ANA tenían un peso comprendido entre 180- 200 y 220-250 gr, respectivamente, y procedían del Instituto Nacional de Salud Público de Helsinki (Finlandia). En los experimentos de autoadministración de etanol se agruparon dos o cuatro animales en cada caja bajo condiciones controladas de temperatura (20 + 2º C) y U U humedad (55 + 5 %) y con ciclos de luz-oscuridad invertidos (fase de luz iniciada al U U mediodía). El agua y la comida estaban disponibles ad libitum en la caja, a excepción del inicio del entrenamiento (apartado 1.3.2). Todas las sesiones de entrenamiento se realizaron durante la fase oscura. - 55 - MENÚ SALIR Materiales y Métodos Para los estudios in vitro e in situ los animales fueron sacrificados por decapitación, sus cerebros fueron rápidamente extraídos y congelados a – 40º C en isopentano y almacenados a – 70º C. Todos los procedimientos experimentales llevados a cabo con estos animales se hicieron bajo la aprobación de los comités éticos locales (Nacional Public Health Institute’s Animal Care and Use Committee; Stockholm South Animal Ethics Committee) y cumpliendo la normativa europea vigente 86/609/ES que regula la investigación animal. 1.2.2. Ratas msP Las ratas msP (Marchigian Sardinian alcohol-preferring) son otro modelo animal de alcoholismo, por su preferencia al consumo de etanol, las cuales han sido comparadas con ratas Wistar no tratadas. Tenían un peso entre 175-225 gr y procedían del Departamento de Ciencias Farmacológicas y Medicina Experimental de la Universidad de Camerino (Italia). Fueron criadas durante más de 42 generaciones a partir de la 13ª generación de ratas sP (Sardinian alcohol-Preferring) procedentes del Departamento de Neurociencias de la Universidad de Cagliari (Agabio et al., 1996; Colombo, 1998; Lobina et al., 1997). Se agruparon dos animales en cada caja, en condiciones de temperatura y humedad controladas y bajo ciclos de luz-oscuridad invertidos (luz a partir de 6:00 pm; oscuridad a partir de 6:00 am), disponiendo de agua y comida ad libitum. Las sesiones de entrenamiento se realizaron durante la fase oscura del ciclo. Para el manejo y sacrificio de los animales se siguió la normativa europea vigente 86/609/ES que regula la investigación animal. - 56 - MENÚ SALIR Materiales y Métodos 1.3 – ADMINISTRACIÓN DE ALCOHOL 1.3.1. Tratamientos intraperitoneales de Etanol Se realizaron dos tipos de tratamientos a las ratas Wistar: - un tratamiento agudo y - un tratamiento crónico de etanol. Se establecieron, en ambos casos, dos grupos experimentales: 1. Grupo Etanol, a los que se les inyectó intraperitonealmente (i.p.) etanol al 20%, disuelto en solución fisiológica salina, a razón de 4 y 2 gr/kg. 2. Grupo Control, a los que se les inyectó i.p., proporcionalmente a su peso, el mismo volumen de solución fisiológica salina que el grupo etanol. En el tratamiento agudo, los animales fueron sacrificados a diferentes tiempos (0, 45, 90, 120, 240 y 720 minutos) tras haber recibido una inyección i.p. con la solución correspondiente. En el tratamiento crónico, los animales fueron inyectados i.p. cada 12 horas, durante 15 días, tras lo cual fueron sacrificados. En ambos tratamientos las inyecciones se realizaron entre las 9:00 y 13:00 a.m. de la fase de luz con el fin de evitar variaciones debidas al ritmo circadiano. En el tratamiento agudo de etanol se formaron varios grupos de animales los cuales fueron anestesiados con metoxiflurano y sacrificados mediante decapitación. Con el fin de evaluar el efecto agudo del etanol sobre el contenido endocannabinoide se recogieron muestras de sangre y vísceras (hígado, intestino delgado y grasa perirrenal) de un primer grupo de animales y, de un segundo grupo de animales, se extrajeron los cerebros como a continuación se detalla. La sangre fue extraída con una jeringa que contenía 1 ml de tampón Krebs-Tris (NaCl 136 mM; KCl 5 mM; MgCl2 1.2 mM; CaCl2 2.5 mM; glucosa 10 m; Trizma base B B B B 20 mM; pH 7.4) y posteriormente se centrifugó en tubos ‘accuspin’ (Sigma), con el fin - 57 - MENÚ SALIR Materiales y Métodos de obtener el plasma donde se analizó su contenido de AEA. Las muestras de hígado, grasa perirrenal e íleon se congelaron inmediatamente en hielo seco. Siguiendo el mismo protocolo descrito anteriormente, se extrajeron y congelaron rápidamente, en hielo seco, los cerebros de un segundo grupo de animales con el fin de evaluar la expresión de los ARNm del sistema endocannabinoide (tiempos: 0, 2 y 12 horas) y el contenido endocannabinoide de los mismos. Todas las muestras se mantuvieron a – 80º C hasta su posterior procesamiento. Tras el tratamiento crónico de etanol los animales fueron sacrificados y se tomaron muestras de sangre así como los cerebros, que fueron rápidamente congelados en hielo seco y almacenados a -80º C hasta su posterior procesamiento. Por otro lado, se estudió el posible efecto de la inyección i.p. sobre los niveles de endocannabinoides ya que el estrés podría afectar a los niveles de anandamida en determinadas áreas cerebrales (Patel et al., 2005). Se analizó el efecto de una única inyección de solución salina sobre los niveles de anandamida en el estriado ventral, cerebelo e hipocampo dorsal. Se anestesiaron un grupo de animales, habituados a las inyecciones, con metoxiflurano (Scherin-Plough, Union, NJ) a los 0, 30 y 60 minutos tras la inyección de solución salina al 0.9% (1 ml/kg). Se sacrificaron por decapitación y se extrajeron los cerebros de los animales y rápidamente se congelaron en hielo seco y se almacenaron a -80º C. 1.3.2. Entrenamiento Operante y Autoadministración de Etanol El entrenamiento y las pruebas realizadas se llevaron a cabo en cámaras operantes estandarizadas, localizadas en cubículos ventilados y con atenuación del sonido. Los estímulos visuales y auditivos se presentaban a través de un altavoz y un foco de luz colocados en el panel frontal de la cámara. Ésta estaba equipada con un reservorio de bebida (capacidad de volumen de 0.1 ml), posicionado a 4 cm del suelo en el centro del panel frontal, y con dos palancas colocadas a 3 cm, a la derecha e izquierda, del receptáculo de bebida. La palanca activaba una bomba que liberaba una gota con 0.1 ml de líquido en el recipiente donde bebían. Para controlar las cámaras y la - 58 - MENÚ SALIR Materiales y Métodos obtención de los datos se utilizó el programa de comportamiento MED-PC (MED Associates Inc., Georgia, VT). Las ratas AA se entrenaron para la autoadministración oral de etanol usando un protocolo de entrenamiento con sacarina consistente en la privación de agua durante 12 horas, 3 días consecutivos, y en el entrenamiento para responder a una gota de 0.1 ml de sacarina al 0.2% (p/v) en ambas palancas, con un programa de refuerzo de ratio fijo 1. Tras la solución de sacarina al 0.2% (p/v), la concentración se disminuyó al 0.1% y posteriormente al 0.05%. Se les permitió responder a esta solución durante 2 semanas antes de hacerlo a otros procedimientos. Las sesiones de entrenamiento tenían una duración de 30 minutos diarios. De manera similar, las ratas msP fueron entrenadas para autoadministrarse soluciones de etanol al 10% (v/v), sacarina al 0.2% (p/v), sacarosa al 10% (p/v) o agua en sesiones de 30 minutos diarios con un esquema de refuerzo de ratio fijo 1 (Weiss et al., 1993). En este caso, el entrenamiento consistía en la restricción de agua durante 2 horas diarias de los 3 primeros días. Durante este tiempo, se estableció el refuerzo mediante la presión de la palanca con solución de sacarosa al 10% (p/v) o sacarina al 0.2% (p/v). El entrenamiento con sacarosa/sacarina continuó hasta que los animales alcanzaron la estabilidad en sus respuestas. Tras el entrenamiento inicial los animales tuvieron libre acceso a comida y agua en sus cajas. Las ratas AA y un subconjunto de ratas msP, separadas de las entrenadas con sacarina, fueron posteriormente entrenadas para autoadministrarse etanol usando una modificación del procedimiento de sacarosa (Samson, 1986) ya que usaba sacarina en vez de sacarosa (Weiss et al., 1993). Durante los 6 primeros días de esta fase de iniciación al etanol, las respuestas sobre la palanca derecha daban lugar a la liberación de una solución de etanol al 5% (p/v) y sacarina al 0.2% (p/v) mientras que las respuestas sobre la otra palanca servían de recordatorio ya que no tenían consecuencias programas. A partir del día 7, la concentración de etanol se incrementó gradualmente desde el 5% al 8% y finalmente hasta el 10% (p/v), así como la concentración de sacarina fue disminuyendo hasta ser completamente eliminada de la solución. Las posiciones de las palancas activas e inactivas se mantuvieron constantes durante el entrenamiento y las pruebas. Las respuestas a la palanca inactiva fueron - 59 - MENÚ SALIR Materiales y Métodos grabadas como medida de la activación no específica del comportamiento. Todas las sesiones de entrenamiento y pruebas se realizaron en sesiones de 30 minutos diarios durante 5 días a la semana. A los animales se les permitió responder a soluciones de etanol al 10% durante 4-5 semanas antes de la cirugía. 1.4 – CIRUGÍA Las ratas AA fueron anestesiadas con una mezcla de halotano-aire y se posicionaron en un aparato estereotáxico con la barra incisiva ajustada a 3.3 mm debajo de la línea interaural. Se implantaron 23 cánulas guía bilaterales a 2 mm sobre las áreas diana en la corteza prefrontal (CPF) (AP 2.7 desde Bregma; ML + 0.6; DV 3.2 desde la U U superficie del cráneo) y estriado (AP 1.0 desde Bregma; ML + 3.0; DV 5.0) de acuerdo U U con Paxinos y Watson (1986). Las cánulas se fijaron al cráneo con tres tornillos de anclaje y cemento dental y se sellaron con hilos metálicos para evitar su oclusión. Con el fin de aliviar el dolor postoperatorio, se les administró una dosis de buprenorfina (0.03 mg/kg subcutáneamente) inmediatamente después de la cirugía. Los animales se recuperaron durante una semana, antes de las sesiones de autoadministración de etanol y sacarina previamente descritas. 1.5 – ADMINISTRACIÓN DE DROGAS El antagonista SR141716A del receptor cannabinoide CB1, administrado mediante inyecciones intraperitoneales a las ratas AA, estaba disuelto en una mezcla de propilen glicol, Tween-80 y solución salina en una proporción 1:1:18. Se administró 30 minutos antes de testarlo mediante un diseño experimental de cuadrados latinos. Para las microinyecciones intracerebrales (0.5 μl), se disolvió el antagonista en DMSO y se diluyó al 75% con solución salina. Las soluciones de la droga se inyectaron a través de 30 cánulas conectadas a microjeringas de 10 μl que se extendían hasta 2 mm bajo las guías. Las microinyecciones se realizaron con una bomba de inyección, durante - 60 - MENÚ SALIR Materiales y Métodos un periodo de 45 segundos, 20 minutos antes de las sesiones experimentales. Antes de retirar los inyectores, se dejó que difundiera durante 60 segundos. En la primera inyección se les suministró vehículo (DMSO al 75%) con el fin de habituarlos a las inyecciones intracraneales, seguido de dosis de 0, 3 y 6 μg de SR141716A usando un diseño experimental de cuadrados latinos. Se realizaron al menos dos sesiones con vehículo (sesiones control) entre las inyecciones. Por otro lado, se inyectó el vehículo (DMSO al 75%) a un grupo de ratas con cánulas en la CPF (n = 7) y estriado (n = 7) con el fin de poder excluir los posibles efectos del vehículo sobre la autoadministración de etanol. Para las microinyecciones intra-CPF (0.5 μl) del inhibidor de FAAH: URB597 (Kathuria et al., 2003; Cayman Chemicals, MI, USA), se disolvió la droga en DMSO al 75%. Se inyectó a través de 30 cánulas conectadas a microjeringas de 10 μl que se extendían hasta 2 mm bajo las guías. Las microinyecciones se realizaron, mediante una bomba, 30 minutos antes de las sesiones experimentales y durante un periodo de unos 60 segundos, dejándose difundir durante 60 segundos antes de retirar los inyectores. Se utilizó un diseño entre sujetos y cada animal recibió sólo una inyección de la droga. 1.6 - MICRODIÁLISIS IN VIVO A las ratas Wistar (n = 11; 300- 350 g), tras ser anestesiadas con vapor de isofluorano al 1-2%, se les implantó una cánula guía de microdiálisis en el shell del núcleo accumbens (+1.6 mm AP, +/- 0.8 mm ML y – 5.7 mm DV, según coordenadas Bregma; Paxinos y Watson, 1997; Giuffrida et al., 1999), dejándose siete días de recuperación antes de iniciar la microdiálisis. Un día antes de la misma, se anestesiaron a los animales (con vapor de isofluorano al 1-2%) y se insertó una sonda de microdiálisis (membrana PES de 2 mm, peso molecular de 15 kDa) que se fijó a la cánula guía previamente implantada. Las sondas fueron perfundidas, durante toda la noche, con líquido cerebroespinal artificial (145 mM NaCl, 2.8 mM KCl, 1.2 mM MgCl2, 1.2 mM CaCl2, 5.4 mM DB B B B glucosa, 0.25 mM ácido ascórbico, pH = 7.2 – 7.4) a una tasa de 0.2 μl/ min. A la mañana siguiente, se perfundió con líquido cerebroespinal artificial que contenía - 61 - MENÚ SALIR Materiales y Métodos hidroxipropil-β-ciclodextrina (HB-β-CD) al 30% (p/v) y se incrementó la tasa a 0.6 μl/min. La inclusión de HB-β-CD en el medio de perfusión permitió un sustancial incremento en la recuperación, mediante diálisis, de endocannabinoides (similar a los descritos por Walker et al., 1999). Tras un periodo de 90 min de equilibrado, se recolectaron las muestras dializadas en intervalos de 10 min, durante un periodo inicial de 40 minutos, una vez transcurridos 30 minutos después de la inyección del vehículo (etanol:emulfor:salino, 1:1:18; 1 ml/kg) y finalmente durante los 120 minutos posteriores a la administración i.p. de etanol al 20% (p/v) en dosis de 0.75 ó 2 g/kg. Las muestras se congelaron inmediatamente después de su recolección y se almacenaron a -80º C hasta el análisis del contenido de endocannabinoides mediante cromatografía líquida acoplada a espectrometría de masas (LC-MS) tal y como se describe en el apartado 5.2.1. A un segundo grupo de ratas macho Wistar (n = 5) se les implantó quirúrgicamente unas cánulas guía de microdiálisis en el núcleo accumbens para el análisis de las posibles alteraciones, inducidas por el etanol, sobre los niveles de glutamato (Glu). Se siguió el mismo protocolo descrito anteriormente a excepción de que el medio de perfusión no contenía HB-β-CD, ya que este aditivo no es necesario para la recuperación de monoaminas y aminoácidos. En esta ocasión, se usó una tasa de flujo mayor (1 μl/min) necesaria para la separación muestral y el análisis de monoaminas y aminoácidos (apartado 5.3). 1.7 - MEDIDA DE LA ACTIVIDAD LOCOMOTORA Y LA TEMPERATURA CORPORAL En este experimento se realizaron varios grupos de ratas Wistar (n = 8-10) a cada uno de los cuales se les inyectó una de las siguientes soluciones: - Vehículo: Tween-80, dimetilsulfóxido y agua 10:10:80 (v/v). - AM404 disuelto en la solución vehículo a una dosis de 2 mg/kg - Etanol (20% v/v en solución salina) a una dosis de 2 g/kg - 62 - MENÚ SALIR Materiales y Métodos - Combinación de AM404 (2 mg/kg) y Etanol (2 g/kg). En este caso, el AM404 se inyectó 30 minutos antes de administrar i.p. la solución de etanol. La temperatura corporal se midió utilizando una sonda rectal acoplada a un sistema telemático de registro, realizándose medidas a los 5, 30, 60 y 120 segundos de la inyección i.p. con la solución correspondiente. La actividad locomotora se midió en campo abierto (100 x 100 cm, divido en 25 cuadrados de 20 x 20 cm) grabándose automáticamente en un ordenador (SMART, Panlab) al que la cámara estaba conectada. Los animales se situaron en arena durante 10 minutos antes de dicha medida para que se habituasen y a continuación se ubicaron en el centro del campo abierto midiéndose la actividad horizontal (número de veces que los animales cruzaban los cuadrados en un tiempo dado). Esta medida se llevó a cabo en una habitación aislada de ruidos e iluminada con una luz halógena indirecta. 1.8 – DISECCIONES DE CEREBROS DE RATA Los cerebros fueron cortados coronalmente en secciones de 1 mm de grosor (6.70 mm a -9.30 mm coordenadas Bregma), a excepción del cerebelo. Las secciones se manipularon sobre un soporte de nieve carbónica que los mantenía congelados y permitía el aislamiento de las regiones cerebrales de interés. Las regiones aisladas fueron: - Corteza Prefrontal (5.20 a 4.20 mm, coordenadas Bregma). - Cuerpo Estriado y Núcleo accumbens (1.70 a -2.12 mm, coordenadas Bregma) - Formación hipocampal (entre -2.12 y -5.80 mm, coordenadas Bregma), - Cerebelo (desde -9.30 a -14.60 mm, coordenadas Bregma), - 63 - MENÚ SALIR Materiales y Métodos CORTEZA PREFRONTAL (Bregma 4.20 mm) ESTRIADO (Bregma 0.70 mm) CPu, núcleos caudado y putamen; AcbC, core del núcleo accumbens; AcbSh, shell del núcleo accumbens. FORMACIÓN HIPOCAMPAL (Bregma -3.80 mm) DG, Giro Dentado; áreas CA1, CA2, CA3 y CA4 del hipocampo. - 64 - MENÚ SALIR Materiales y Métodos CEREBELO (Bregma -10.04 mm) Una vez aisladas las regiones cerebrales de interés, se procedió a su procesamiento ya fuese para la obtención de extractos proteicos o de ARN total. 2. ANÁLISIS DE LA EXPRESIÓN GÉNICA DEL SISTEMA ENDOCANNABIONIDE 2.1 – PCR CUANTITATIVA A TIEMPO REAL La cuantificación de los niveles de expresión de los genes del sistema endocannabinoide mediante PCR cuantitativa a tiempo real. Para ello, realizaron una secuencia ordenada de técnicas experimentales que a continuación se detallan: 1. Extracción de ARN total 2. Cuantificación del ARN total 3. Purificación del ARN total 4. Cuantificación y electroforesis del ARN purificado 5. Transcripción reversa (RT) 6. PCR cuantitativa a tiempo real - 65 - MENÚ SALIR Materiales y Métodos 2.1.1 – Puesta a punto de la PCR cuantitativa 2.1.1.1 - Diseño de los Cebadores El programa informático empleado para el diseño de los cebadores fue Primer3 versión 0.3.0 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). Las variables T T establecidas fueron, entre otras, que el rango de tamaño de nuestro producto de PCR estuviese entre 150 y 200 pares de bases (pb), el rango de temperatura de fusión de los cebadores estuviese comprendida entre 57 y 63º C, el porcentaje de nucleótidos G-C oscilara entre el 40 y 60% y el tamaño de los cebadores entre 18 y 23 nucleótidos. En el diseño de los cebadores se tuvo además en cuenta que la secuencia de los cebadores no tuviesen un nucleótido T ni más de tres nucleótidos G ó C en el extremo 3’. También se evitaron la posible complementariedad de dos o más bases en los extremos 3’ de la pareja de cebadores para así reducir la formación de dímeros así como la complementariedad dentro de la secuencia. Una vez diseñada y elegida la pareja de cebadores se realizó un análisis de comparación de secuencias con la base de datos del genoma de rata, mediante el programa informático Nucleotide BLAST (http://www.ncbi.nlm.nih.gov/BLAST/). Ello T T permitió discriminar aquellas parejas de cebadores que se pudieran unir a los ADNc de otros genes evitándose, de esta forma, su amplificación durante la PCR. 2.1.1.2 – Determinación de la Temperatura de Alineamiento de los Cebadores Tras el diseño de la secuencia de los cebadores específicos de cada gen se hace necesario determinar la temperatura de alineamiento más adecuada. Para ello se realizó una PCR con diferentes temperaturas de alineamiento y, a continuación, se corrieron las muestras en un gel de agarosa al 1%, seleccionándose aquella temperatura a la que la muestra no presentaba productos de PCR de tamaño diferente al esperado. El protocolo seguido para realizar esta PCR se muestra en el apartado 7.5. Las secuencias de los genes analizados así como las temperaturas de alineamiento empleadas en la PCR a tiempo real se encuentran en el siguiente apartado (2.1.1.2). - 66 - MENÚ SALIR Materiales y Métodos Electroforesis en gel de agarosa de los productos de PCR de una muestra sometida a diferentes temperaturas de alineamiento (calle 1 = 48º C; calle 2 = 50º C; calle 3 = 52.8º C; calle 4 = 55º C; calle 5 = 57.2º C). De esta forma se determina que la más adecuada para generar el estándar de ciclofilina-A de rata es de 55º C. PM = pesos moleculares de ADN desde 100 a 1000 pb, en intervalos de 100 pb respecto al fragmento anterior. 2.1.1.3 - Secuencias de los Cebadores y Temperatura de Alineamiento Gen Secuencia Cebador Sentido Secuencia Cebador Antisentido Temperatura Alineamiento β-Actina 5’-TACAGCTTCACCACCACAGC-3’ 5’-AAGGAAGGCTGGAAGAGAGC-3’ 43.5º C CB1 5’-AGACCTCCTCTACGTGGGCTCG-3’ 5’-GTACAGCGATGGCCAGCTGCTG-3’ 58º C Ciclofilina-A 5’-AGAAGGCATGAGCATTGTGG-3’ 5’-TTACAGGGTATTGCGAGCAG-3’ 55º C DAGL-α 5’- GGGTACCTAATGGCTGCTCA- 3’ 5’- AGGACTGACCATCCAACCTG- 3’ 60.5º C FAAH 5’-GTTACAGAGTGGAGAGCTGTC-3’ 5’-GAGGGTTACTGCAGTCAAAGC-3’ 41º C GAPDH 5’-GGTGATGCTGGTGCTGAGTA-3’ 5’-ACTGTGGTCATGAGCCCTTC-3’ 66º C GUS 5’- TCCTGTACACCACCCCTACC- 3’ 5’- GCCATCCTCATCCAGAAGAC- 3’ 61º C MAGL 5’-CATGGAGCTGGGGAACACTG-3’ 5’-GGAGATGGCACCGCCCATGGAG-3’ 58º C NAPE-PLD 5’-GGAGCTTATGAGCCAAGGTG-3’ 5’- ACTCTCCGTGCTTCAGGATG-3’ 57º C - 67 - MENÚ SALIR Materiales y Métodos 2.1.1.4 - Producción y Purificación del Estándar El estándar de un determinado gen permite la cuantificación de los niveles de expresión de dicho gen en una muestra, gracias a que previamente se conoce la concentración del primero. Para generar un estándar se debe hacer un adecuado diseño de los cebadores (sentido y antisentido) así como establecer su temperatura de alineamiento (apartado 2.1.1.3). Una vez determinada, se procede a la producción del estándar mediante la amplificación de ADNc procedente de un tejido de rata rico en el ARNm que se desea cuantificar (protocolo en apartado 7.6). Una vez generado el estándar, se purifica con el fin de eliminar los productos de menos de 100 pb (protocolo en apartado 7.7). Para la purificación de los estándares se utilizó un kit de purificación de alta pureza de productos de PCR (Roche), el cual se basa en la selectividad de unión de éstos, en presencia de sales de tiocianato de guanidina, a las fibras de vidrios especiales pre-empaquetadas que contiene el tubo con membrana suministrado con el kit. El ADN es purificado en una serie de pasos rápidos de lavados y centrifugaciones que permiten eliminar los cebadores, nucleótidos y sales contaminantes. Posteriormente se eluye el ADN en una solución baja en sales. Tras la purificación del estándar, se cuantifica su concentración mediante espectrometría (a una longitud de onda de 260 nm) y se visualiza mediante electroforesis en gel de agarosa al 1% (p/v) junto con el estándar no purificado para comprobar la eficiencia de la purificación. Electroforesis en gel de agarosa al 1% (p/v) del estándar sin purificar (1) y purificado (2) de ciclofilina-A de rata. Se puede apreciar cómo tras la purificación se han eliminado los productos con un tamaño inferior a 100 pb. PM = pesos moleculares de ADN de 100 a 1000 pb. - 68 - MENÚ SALIR Materiales y Métodos 2.1.2 - Extracción de ARN total La extracción de ARN total se realizó homogeneizando las muestras en reactivo Trizol (Invitrogen). Dicho reactivo, constituido por fenol e isotiocianato de guanidina, permite mantener la integridad del ARN cuando se rompen las células y se disuelven sus componentes celulares. La adición de cloroformo, seguida de la centrifugación, permite la separación de una fase acuosa, donde se encuentra el ARN exclusivamente, de otra fase orgánica. Tras separar la fase acuosa, se precipitó el ARN con isopropanol y posteriormente se realizaron lavados con etanol al 75% (v/v). Por último se dejaron secar los precipitados y posteriormente se disolvieron en agua, tratada con DEPC, libre de RNasas y DNasas (apartados 7.1 y 7.17). 2.1.3 - Limpieza del ARN total La purificación permite la eliminación de posibles contaminantes (ADN, ARN de pequeño tamaño, etc.) procedentes de la extracción de ARN total y, una vez realizada, que el ARN se encuentre en un ambiente libre de sales. La limpieza del ARN se realizó siguiendo las instrucciones del manual del RNeasy Mini Kit de Qiagen (apartado 7.2). La adición de etanol a la muestra genera las condiciones necesarias para la unión selectiva del ARN a la membrana de sílica-gel, suministrada con el kit, eliminándose los contaminantes y diluyéndose posteriormente el ARN en agua libre de RNasas. El tratamiento con la enzima DNasa permite la digestión de posibles restos de ADN contaminante en la muestra, eliminándose dicha enzima en lavados posteriores. El ARN total purificado se conservó a -80º C hasta su posterior uso. - 69 - MENÚ SALIR Materiales y Métodos 2.1.4 - Cuantificación del ARN La concentración del ARN se obtuvo tras medir la absorbancia, a una longitud de onda de 260 nm, del ARN diluido en agua DEPC. Los ratios ARN/proteína se obtuvieron midiendo las absorbancias a 260 nm y 280 nm de la muestra (diluida en solución de Tris-HCl 10mM, EDTA 1mM) y dividiendo los valores obtenidos para ambas absorbancias. Se consideraron valores óptimos a aquellos comprendidos entre 1.8 - 2.1. RATIO = Abs (260 nm)/ Abs (280 nm) La cuantificación del ARN se realizó antes y después de su limpieza, lo cual permitía conocer cuál había sido la eficiencia de la purificación. 2.1.5 - Electroforesis de ARN Con el fin de conocer la integridad de los ARNs purificados se realizó la visualización de los ARNs ribosómicos 18S y 28S. Se cargaron, en cada pocillo, 1 μg por muestra junto con 2 μl de tampón de carga de ARN (apartado 7.17) en un gel de agarosa al 1% (p/v), preparado con tampón Tris-EDTA 1x diluido en agua DEPC. Se corrieron a un voltaje constante de 68 V durante los primeros 10 minutos y a 80 V posteriormente. Todo el material empleado fue previamente limpiado con una solución de SDS al 10% en agua DEPC y con RNAse AWAY (Invitrogen). Una vez finalizada la electroforesis, las bandas de ARN ribosomal 18S y 28S fueron visualizadas y capturadas con el programa informático GelSuperII (TDI, Madrid, España). - 70 - MENÚ SALIR Materiales y Métodos Gel de agarosa al 1% (p/v) con muestras de ARN total purificado (1 μg/ pocillo). Se pueden observar dos bandas correspondientes a los ARNr de 28S y 18S. 2.1.6 – Transcripción Reversa (RT) Los ARNs purificados fueron sometidos a una transcripción reversa, usando cebadores de secuencia azarosa, lo que permitió obtener ADNc de todos los ARN mensajeros de la muestra. Se partió de un volumen correspondiente a 4 μg de ARN de cada muestra, completándose hasta un volumen final de 54 μl con agua libre de RNasas. Posteriormente se añadieron 4 μl de los cebadores de secuencia azarosa (2 mM) y se sometieron a una temperatura de 65º C con la finalidad de facilitar la alineación de los cebadores al ARN. Una vez finalizada, a cada tubo se le añadió 22 μl de una solución que contenía la enzima que cataliza la transcripción reversa, los desoxinucleótidos trifosfato (dATP, dGTP, dTTP y dCTP), el tampón de la enzima y el protector de ARN (RNAsín, Roche) y se corrió la RT (protocolo en apartado 7.3) en el termociclador Opticon 2 (MJ Research). Paralelamente, se incluyeron controles negativos de la PCR de cada una de las muestras, procesadas en iguales condiciones, a excepción de la no inclusión de la enzima que cataliza la transcripción reversa. Una vez concluida la RT, las muestras de ADNc se conservaron a -20º C hasta su posterior cuantificación mediante PCR a tiempo real. - 71 - MENÚ SALIR Materiales y Métodos 2.1.7 - PCR Cuantitativa a Tiempo Real Los ADNc, obtenidos a partir de la transcripción reversa, fueron de nuevo amplificados mediante PCR con cebadores de secuencia específica al del ARNm del gen a cuantificar (protocolo en apartado 7.4). En este caso, al final de cada ciclo de amplificación se cuantificaban los niveles de ADN, de doble cadena, gracias al reactivo SYBR-Green. El reactivo SYBR-Green tiene la capacidad de unirse a las dobles cadenas de ADN, emitiendo, al unirse, una señal de fluorescencia. La máxima excitación y emisión de SYBR-Green se da a longitudes de onda de 494 y 521 nm respectivamente, las cuales son compatibles con el uso de cualquier termociclador a tiempo real. En nuestro caso el termociclador a tiempo real empleado fue el Opticon 2 (MJ Research) junto con el programa informático Opticon Monitor 2, versión 2.0.0.1 (MJ Research). Los datos de fluorescencia obtenidos generan, cuando la escala es lineal, una curva de puntos con forma sigmoidal en la que la fluorescencia está representada frente al número de ciclos. El ciclo umbral (CT) sirve como herramienta para el cálculo de las B B cantidades iniciales de ADNc en cada muestra. Este es el ciclo en el que hay un primer incremento detectable en la fluorescencia y en el que, por tanto, el valor alcanza el umbral. Fase de Saturación Fluorescencia Muestra A Muestra B Fase Log-Lineal Umbral Fase Lineal CTA B 10 B 20 CTB B B 30 40 Número de Ciclos Esta curva presenta diferentes fases: - Fase Lineal: en ella se visualiza la fluorescencia debida al ruido de fondo. Se da en los primeros ciclos de amplificación (ciclos 3 a 15), donde no es aún - 72 - MENÚ SALIR Materiales y Métodos detectable el incremento en fluorescencia debida a los productos de PCR. Los valores de esta fase son sustraídos de los debidos a la fluorescencia de los productos de PCR. - Fase Log-Lineal. Se da un incremento exponencial en el número de copias de ADN. - Fase de Saturación, en ella se alcanzan los máximos valores de fluorescencia y se estabilizan a lo largo de los ciclos ya sea debido a un cese en la síntesis de ADN, ya que alguno de los reactivos (nucleótidos, polimerasa de ADN, etc.) se haga limitante, o a causa del agotamiento de moléculas disponibles del reactivo SYBR-Green. El umbral se ajusta a un valor sobre la fase lineal, debiéndose localizar en la fase log-lineal de la PCR y nunca en la fase de saturación. Existen dos formas distintas de calcular el CT ya sea mediante el método de B B ajuste de dicho punto o por el método del máximo de la segunda derivada. En el método de ajuste el umbral se establece manualmente en la fase log-lineal de la PCR y de esta manera se define el ciclo umbral (CT). En el segundo método, el B B punto en el que se da el máximo incremento de la fluorescencia, dentro de la fase loglineal, se calcula determinando el máximo de la segunda derivada de la curva de amplificación. El programa informático calcula cuál es el ciclo en el que se alcanza dicho valor, de manera que no es necesario establecer dónde se encuentra el umbral. En esta tesis se ha empleado el primer método para establecer el CT. B B Para poder determinar cuál es la concentración de ADN correspondiente a un determinado valor de fluorescencia es necesario el uso de muestras estándares de ADN del gen de interés de concentración conocida. Los resultados obtenidos, tras la PCR de dichos estándares, permiten generar una curva estándar donde se representan los CT B B frente al logaritmo de la concentración de ADN, lo cual genera una recta. Los valores CT de las muestras y de los estándares son posteriormente usados para calcular la B B cantidad inicial del ADNc, específico del gen a determinar, en las muestras experimentales. - 73 - MENÚ SALIR Materiales y Métodos Es posible, además, saber cuál ha sido la eficiencia (E) de la PCR mediante la aplicación de la siguiente fórmula: E = 10 (-1/m) - 1 P P donde m es la pendiente de la recta que se obtiene cuando se comparan las CT frente al B B logaritmo de la concentración. y = mx + a Con el fin de corroborar la especificidad de los productos generados por PCR, se hace necesario realizar una curva de fusión (“Melting Curve”) consistente en incrementar gradualmente la temperatura (0.5ºC/ciclo) desde una temperatura de 56º C hasta 95º C, realizándose una lectura de fluorescencia cada 0.5º C. A bajas temperaturas, todos los productos de PCR se encuentran como doble cadena, con lo que el SYBR Green se une a ellos y la fluorescencia es elevada. Sin embargo, a medida que incrementa la temperatura, se van alcanzando valores de temperatura correspondientes a las temperaturas de fusión de los productos de PCR, lo que provoca descensos en la fluorescencia. Este fenómeno se puede visualizar en una gráfica donde se represente la fluorescencia frente a la temperatura. Los sistemas de detección calculan la primera derivada de la curva generándose un máximo correspondiente a la temperatura de fusión (Tm) del producto de PCR. Las B - 74 - B MENÚ SALIR Materiales y Métodos curvas que presenten máximos relativos a una Tm más baja que la del producto B B específico de PCR podrían indicar bien la formación de dímeros de cebadores o de productos inespecíficos de pequeño tamaño y/o bajo contenido G-C. Aquellos máximos relativos con Tm diferentes indicarían la generación de productos inespecíficos de mayor B B tamaño y/o contenido G-C. Fluorescencia – dF/dT Productos Específicos Dímeros de cebadores Muestra A Muestra B Temperatura (º C) Curva de fusión. La muestra A tiene un único máximo resultante de la amplificación de un producto específico por PCR. Sin embargo, la muestra B muestra un máximo relativo a una temperatura que podría corresponderse con la temperatura de fusión de los dímeros de cebadores. 2.2 – HIBRIDACIÓN in situ DEL ARNm DEL RECEPTOR CANNABINOIDE CB1 EN MODELOS ANIMALES DE ALCOHOLISMO (Líneas AA y msP) 2.2.1 - Procesamiento de los Cerebros En este experimento se utilizaron ratas msP y Wistar no tratadas así como ratas msP que, durante 18 días, tuvieron libre acceso a agua y a una solución de etanol al 10% (n = 8 por grupo). Posteriormente fueron sacrificadas, mediante decapitación, se extrajeron rápidamente los cerebros, se congelaron a – 40º C en isopentano y se almacenaron a – 70º C hasta su procesamiento. - 75 - MENÚ SALIR Materiales y Métodos Se realizaron secciones cerebrales de 10 μm de grosor a los siguientes niveles Bregma: (1) + 2.5 a + 1.7 mm; (2) – 0.3 a – 0.4 mm y (3) – 2.3 a – 3.3 mm, de acuerdo con el atlas de Paxinos y Watson (1986). Las ratas AA y ANA, no sometidas a ningún tratamiento, fueron sacrificadas por decapitación y sus cerebros fueron tratados en las mismas condiciones descritas anteriormente para la línea msP. Las secciones cerebrales (10 μm de grosor) se tomaron (1) desde + 2.2 a + 1.7 mm; (2) + 1.0 a + 0.2 mm; (3) - 0.4 a – 1.8 mm y (4) – 2.3 a – 3.3 mm, coordenadas Bregma (Paxinos y Watson, 1986). 2.2.2 – Producción de la Ribosonda de CB1 La ribosonda del receptor CB1 fue generada mediante PCR y se corresponde con los nucleótidos 1232-1272 de la secuencia de ADNc patentada NM_012784 (Matsuda et al., 1990). La síntesis de la sonda de ARN y la hibridación in situ se realizó tal y como está descrito por Caberlotto et al., 2004. Antes de iniciar la transcripción, los plásmidos fueron cortados con enzimas de restricción con el fin de obtener una secuencia lineal y, así, poder sintetizar las ribosondas sentido y antisentido. Las sondas, complementarias a las secuencias codificantes, se transcribieron con 150 μCi α-[35S]UTP (Dupont NEN, Boston, USA) P P gracias a las polimerasas de ARN T3, T7 y SP6. La transcripción se realizó en presencia de 10 mM de ditiotreitol (DTT), 0.5 mM de cada nucleótido (ATP, GPT, CTP) y 1 μg del plásmido lineal en tampón de transcripción 1x, durante 60 minutos a 37º C. Las sondas marcadas se separaron de los nucléotidos no incorporados usando columnas spin (Pharmacia Biotech). 2.2.3 – Hibridación in situ La hibridación in situ comenzó con la fijación de las secciones de cerebro en paraformaldehído al 4% / tampón fosfato salino 1x durante 10 minutos, seguido de dos - 76 - MENÚ SALIR Materiales y Métodos lavados en PBS 1x y el tratamiento, durante 5 minutos, con anhídrido acético al 0.25% / trietanolamina 0.1 M/ cloruro sódico al 0.9%. Posteriormente, se lavaron los cortes dos veces en tampón SSC 2x, se deshidrataron en una serie graduada de etanol (70, 80, 95, 100%) y finalmente pasaron por cloroformo. Se dejaron secar antes de ser usados o congelados a -70º C hasta su uso. Todas las soluciones acuosas fueron pretratadas con 0.1% de DEPC antes de ser utilizadas. El tampón de hibridación contenía 0.5 mg/ml de ADN monocadena cortado, 250 μg/ml de ARNt de levadura, solución de Denhardt 1x, sulfato de dextrano al 10%, SSC 4x y formamida al 50%. Antes de la hibridación, se añadió la sonda marcada al cóctel de hibridación a una concentración de 20x103 cpm/μl. Se pusieron 150 μl de esta mezcla a cada una de P P las secciones de cerebro y se cubrieron con el fin de evitar su evaporación. La hibridación se realizó en una cámara húmeda durante toda la noche a 55º C. Una vez finalizada la incubación, se lavaron las secciones en una serie graduada de SSC (2x, 1x, 0.5x, 0.5x/formamida al 50%, 0.1x), que contenían DTT 1mM, a temperatura ambiente a excepción del lavado con la solución de SSC 0.1x que se realizó a 53º C. La deshidratación se llevó a cabo con soluciones de etanol graduadas que contenían acetato amónico 300 mM. Se dejaron secar las secciones y se expusieron ante películas Biomax Kodak (Amersham) junto con estándares de 14C durante un periodo de P P 3-7 días. 2.2.4 – Obtención de datos Se escanearon las autorradiografías, usando el aparato ScanMaker III (Microtek) a una resolución de 400 dpi, y se midieron los valores de transmitancia de la luz, a partir de las imágenes digitalizadas, con el programa informático IMAGE. Posteriormente, dichas imágenes se analizaron usando el programa AIS (Imaging Research Inc.). Las regiones de interés se marcaron mediante trazado individual de las estructuras con un cursor y de acuerdo con el atlas de Paxinos (Paxinos y Watson, 1986). En base a la radiactividad conocida de los estándares de 14C, se convirtieron los P - 77 - P MENÚ SALIR Materiales y Métodos valores de transmitancia de las imágenes en nCi/g para lo cual se usó una curva de calibración Rodbard. Las secciones hibridadas se expusieron ante una película BiomaxTMMR (Kodak, P P NY, USA), definiéndose para ello las regiones de interés con determinadas marcas anatómicas. Se midieron los valores medios de grises del autorradiograma usando el sistema analizador de imágenes Biovision SAS (Avanti, Milan, Italia). Los valores de densidad óptica (medias + SEM) se obtuvieron como describen Hansson et al (2003). U U Los artefactos de radiactividad cercanos al tejido fueron eliminados de dichas medidas. Se realizaron tres medidas por cada región cerebral: (1) el valor total, es decir, las medidas en las secciones hibridadas de la región con ARN antisentido marcada con α-[35S]UTP; (2) el valor inespecífico, es decir, las medidas en las secciones control P P hibridadas con el ARN sentido marcado con α-[35S]UTP de la región correspondiente y P P (3) el valor del fondo, es decir, las medidas del fondo de la película fuera de las secciones. Los valores de porcentaje de transmitancia (%T) del marcaje específico e inespecífico se obtuvieron tal y como describen Benfati et al. (1986) y Zoli et al. (1991). A partir de los valores de %T se obtuvieron los valores de densidad óptica (D.O.) mediante la fórmula D.O. = - log (%T). 3. CUANTIFICACIÓN DE LA EXPRESIÓN PROTEICA MEDIANTE INMUNOHISTOQUÍMICA 3.1 – Procesamiento del tejido Los animales fueron anestesiados con una inyección i.p. de pentobarbitol a una dosis de 60 mg/kg, tras lo cual fueron perfundidos intracardialmente con 100 ml de tampón fosfato salino (PBS) 0.1M pH = 7.3, seguido de 400 ml de paraformaldehído al 4% en tampón fosfato (PB) 0.1M pH = 7.3. - 78 - MENÚ SALIR Materiales y Métodos Los cerebros y cerebelos fueron extraídos y permanecieron en la misma solución fijadora durante 12 h a temperatura ambiente y en agitación. Fueron lavados en PBS tres veces, durante 10 minutos, en agitación a temperatura ambiente, tras lo cual se incubaron en solución crioprotectora de sacarosa al 30% (p/v), azida sódica 0.02% (p/v) en PBS, a 4º C hasta que decantaron. Los cerebros fueron cortados, rostro-caudalmente, en secciones de 30 μm de grosor en un microtomo de congelación y depositados seriadamente sobre placas multipocillos con azida sódica 0.02% en PBS. Se conservaron a 4º C hasta ser inmunoteñidos. 3.2 - Inmunohistoquímica Se seleccionaron aquellos cortes que contenían al núcleo accumbens (1.70 a 0.7 mm, coordenadas Bregma), pálido ventral (-0.26 a -0.4 mm, coordenadas Bregma) hipocampo dorsal (-2.30 a -3.80 mm, coordenadas Bregma) y distintas regiones del cerebelo (-9.68 a -12.80 mm, coordenadas Bregma) según Paxinos y Watson (1997). Todos los cortes fueron lavados tres veces en PBS, durante 10 minutos, en agitación y posteriormente se siguió el protocolo específico para cada marcador (apartados 7.8, 7.9, 7.10, 7.11). La inmunotinción se realizó mediante el método ABC (complejo avidinabiotina) aunque, en nuestro caso, se utilizó estreptavidina en vez de avidina con el que se consiguió un marcaje inmunohistoquímico indirecto para CB1, FAAH, MAGL y NAPE-PLD. Este método se fundamenta en la aplicación, sobre el tejido, de un anticuerpo primario que es reconocido por un segundo anticuerpo, secundario, que se une a la región Fc del primario. El anticuerpo secundario está unido a una molécula de biotina, por la que tiene gran afinidad la estreptavidina que, a su vez, va unida al marcador peroxidasa (HRP). - 79 - MENÚ SALIR Materiales y Métodos El cromógeno empleado fue el tetracloruro de la diaminobenzidina (DAB), el cual forma un precipitado de color marrón e insoluble cuando se oxida, en presencia de agua oxigenada, por acción de la enzima peroxidasa (HRP). Para incrementar la señal, se añadió sal de níquel a la solución de DAB, formándose un precipitado de color más intenso. Este método es mucho más sensible que los directos ya que al anticuerpo primario se pueden unir más de un anticuerpo secundario y a éste más de un complejo estreptavidina-HRP, incrementándose así la señal. Las inmunohistoquímicas de todos los animales de cada tratamiento, para un determinado anticuerpo, se realizaron simultáneamente con el fin de evitar diferencias no debidas al tratamiento recibido. 3.3 - Cuantificaciones La inmunorreactividad de cada marcador fue medida mediante análisis de imágenes bidimensionales por densitometría digital. Para ello se tomaron fotografías de las regiones cerebrales de interés con una cámara fotográfica digital (Olympus BX41) y empleando el programa informático Olympus Micro DP70 DP Manager. Todas las imágenes fueron obtenidas en idénticas condiciones de luz, tiempo de exposición, apertura del diafragma (ajuste manual) y aumentos. - 80 - MENÚ SALIR Materiales y Métodos Posteriormente las fotografías se pasaron a escala de grises con el programa Adobe Photoshop 7.0 y fueron cuantificadas con el programa informático Visilog 6 (Noesis Vision Inc. Quebec PQ, Canada). Con este programa se puede delimitar, de cada fotografía, el área de interés a analizar y obtener los valores de intensidad de grises en cada punto, oscilando éstos entre 0 (negro) y 255 (blanco). De esta manera se obtuvieron los valores medios, desviación estándar, máximo y mínimos de intensidad de las áreas seleccionadas. Las distintas áreas cerebrales fueron seleccionadas según su participación, primero, en los procesos de recompensa y dependencia a drogas de abuso y, segundo, según su grado de expresión. Las áreas a cuantificar, en ratas Wistar, para el marcador CB1 fueron: - la capa molecular y de Purkinje del cerebelo, - la capa granular del giro dentado hipocampal, - el accumbens shell del estriado ventral y - el pálido ventral Para el marcador FAAH: - la capa molecular y de Purkinje del cerebelo, - la capa granular del giro dentado hipocampal, - la capa piramidal de CA4 (hipocampo), - el accumbens shell del estriado ventral y - el pálido ventral En la corteza prefrontal de las ratas msP se realizaron inmunohistoquímicas para los marcadores CB1, FAAH, MAGL y NAPE-PLD. - 81 - MENÚ SALIR Materiales y Métodos Capas Molecular, Purkinje y Granular de la Corteza Cerebelar Capas Molecular, Granular y Polimórfica del Giro Dentado (DG); Capas Molecular, Granular y Polimórfica de CA1 y CA4 en Hipocampo. - 82 - MENÚ SALIR Materiales y Métodos 4. ANÁLISIS DE LA FUNCIONALIDAD PROTEICA Previo al análisis de la funcionalidad proteica, se realizó una extracción de membranas cuantificándose su contenido proteico. Posteriormente, se realizaron los análisis de actividad enzimática así como los ensayos de unión y funcionalidad del receptor CB1. 4.1 - Extracción de Membranas Las muestras se homogeneizaron en solución HEPES 50 mM pH = 8 sacarosa 0.32 M en un volumen, en ml, 10 veces el peso (gr) de la muestra, manteniéndose durante todo el proceso a 4º C. Se centrifugaron a 800 xg durante 10 minutos a 4º C y se descartó el precipitado. Se centrifugó el sobrenadante a 40.000 xg durante 30 minutos a 4º C y, tras ello, se desechó el sobrenadante. El precipitado se resuspendió y homogeneizó en solución HEPES 50 mM pH = 8 en un volumen que era un cuarto del añadido inicialmente de HEPES 50 mM pH = 8 sacarosa 0.32 M. Posteriormente se realizó la cuantificación de la cantidad total de proteínas mediante el método de Bradford tal y como se detalla en el siguiente apartado. 4.2 - Cuantificación de Proteínas La cuantificación de proteínas se realizó mediante el método de Bradford empleándose para ello el reactivo Coomasie Blue G-250. Este reactivo interacciona principalmente con residuos de arginina y levemente con otros residuos de aminoácidos básicos (histidina y lisina) y residuos aromáticos (tirosina, triptófano y fenilalanina). En ausencia de proteínas, tiene color rojo palo y cuando interacciona con ellas toma color azul, teniendo su máximo de absorbancia a una longitud de onda (λ) de 595 nm. - 83 - MENÚ SALIR Materiales y Métodos Reactivo de Coomasie Blue G-250 De cada muestra (por duplicado) se mezclaron 5 μl de la solución proteica con 250 μl de reactivo de Coomasie, dejándose incubar 2-3 minutos a temperatura ambiente. Posteriormente se midió la absorbancia a λ = 595 nm. Se construyó una curva patrón con diferentes concentraciones de albúmina (0, 75, 100, 250, 400, 500, 1000 y 1500 μg/ml). Para las muestras de concentración desconocida se realizaron diferentes diluciones (1, 1/10 y 1/100) de forma que sólo se tomaron aquellos valores que estuviesen comprendidos entre los valores extremos Densidad óptica (595 nm) generados con los estándares de albúmina en la curva patrón. 0,7 0,6 0,5 0,4 0,3 0,2 0,1 0 0 200 400 600 800 1000 1200 1400 Concentración (μg/ ml) Curva patrón de albúmina (0, 75, 100, 250, 400, 500, 1000 y 1500 μg/ml). y = - 6x10-3 + 10 -3 x – 6.42 x10-7 x2; r2 = 0.999. P P P P P - 84 - P P P P P MENÚ SALIR Materiales y Métodos 4.3 -Ensayo de Actividad Enzimática de la Amidohidrolasa de Ácidos Grasos (FAAH) La actividad de la enzima FAAH se midió empleando como sustrato araquidonil-[1-3H]-etanolamina la cual, tras ser metabolizada, libera [3H]-AEA (en P P P P forma de [3H]- etanolamina) en la fase acuosa, tras la extracción con cloroformo, tal y P P como está descrito (Desarnaud et al, 1995; Rodríguez et al, 2001). La mezcla se dejó incubar a 37º C en tampón TRIS 50 mM, pH = 7.5 que contenía además de la fracción de membranas, una concentración saturante de [3H]P P AEA (10.000 dpm/ml) (protocolo en apartado 7.12). Cada muestra se realizó por triplicado. Los blancos fueron incubados en idénticas condiciones, a excepción de que no contenían membranas, siendo su valor sustraído al de las muestras con membranas. Bajo estas condiciones está descrito que existe una linealidad en la hidrólisis de AEA con respecto al tiempo y la concentración de proteínas (Desarnaud et al, 1995). Medida de la cantidad de [3H]-etanolamina liberada, a lo largo del tiempo, y de la actividad amidohidrolasa (medido como cantidad de [3H]-etanolamina formada) respecto a la cantidad de proteína existente en la mezcla de incubación en condiciones estándares. Tomado de Desarnaud et al (1995). P P P P - 85 - MENÚ SALIR Materiales y Métodos 4.4 - Ensayo de Actividad Enzimática de la Transferasa de grupo N-Acilo (NAT) La actividad de la tranferasa de grupo N-acilo (NAT) se midió empleando como sustrato L-3 Fosfatidilcolina 1,2-di(1 C14) palmitoil (Cadas H. et al., 1997) la cual, junto P con fosfatidiletanolamina, puede P dar lugar al precursor lipídico N-acil- fosfatidiletanolamina (NAPE) marcado con C14 gracias a la actividad NAT, reacción en P P la que necesita como cosustrato al ión Ca2+. P P A cada tubo se añadieron, por duplicado, la mezcla de incubación que contenía: la muestra de fracción de membrana, tampón HEPES, el sustrato marcado (L-3 fosfatidilcolina 1,2-di(1 C14) palmitoil), cloruro cálcico o un quelante de calcio (EGTA) P P (protocolo en apartado 7.13). Los datos de actividad obtenidos en presencia del quelante de calcio fueron sustraídos a los obtenidos, en las correspondientes muestras, cuando se añadía cloruro cálcico. La mezcla se dejó incubar a 37º C y, tras parar la reacción, se realizó una extracción de ácidos grasos con cloroformo. Posteriormente se separaron los ácidos grasos de las NAPEs, mediante cromatografía líquida de alta resolución (HPLC), y se cuantificaron por centelleo. 4.5 - Ensayo de Unión del receptor CB1 El ensayo de unión de CB1 (Hirst et al, 1996) se llevó a cabo en membranas de tejido cerebral usando como ligando [3H]SR141716A (Amersham, 59 Ci/mmol). P P Está demostrado que la unión de [3H]SR141716A, en membranas cerebelares, es P P dependiente de la dosis y saturable (Hirst et al., 1996). A una concentración de 0.4 nM se une a un 71.1 + 1.5 % (n = 11) respecto de la unión total a receptores y dicha unión U U puede desplazarse con SR141716A de forma dependiente de la dosis, con una pKi = B B 8.37 + 0.07. U U Cada muestra se realizó por triplicado y la mezcla contenía la fracción de membrana junto con una concentración saturante de SR141716A, la cual se incubó a 30º C (protocolo en apartado 7.14). Se lograron uniones inespecíficas con la inclusión de WIN-55,212-2 a 10 nM. Los resultados se expresan como fmol/mg de proteína. - 86 - MENÚ SALIR Materiales y Métodos 4.6 - Ensayo de Acoplamiento del receptor CB1 Esta técnica se basa en la unión de [35S]GTPγS, estimulada por un agonista de P P CB1, a las membranas (Hilf et al., 1989; Traynor y Nahorski, 1995). Cada muestra se realizó por triplicado (con y sin el agonista WIN 55,212-2) y se añadieron a la mezcla de incubación: proteínas de las fracciones membranales, el agonista WIN 55,212-2 (a los tubos estimulados), GDP y [35S]GTPγS en tampón P P (protocolo en apartado 7.15). Se midió la unión basal en ausencia del agonista, y la unión inespecífica en presencia de guanidil imidodifosfato, el cual desplaza al GTP marcado. La radiactividad de unión se determinó mediante espectrofotometría de centelleo, con un 95% de eficiencia para [35S]. Los datos se presentan como media + P P U U SEM del porcentaje de estimulación respecto a los niveles basales. 5. MEDIDAS BIOQUÍMICAS 5.1 - MEDIDA DE LOS NIVELES DE ETANOL EN PLASMA En ratas Wistar sometidas a tratamientos agudo y crónico de etanol, se realizó la medida de los niveles de etanol en el plasma mediante un método basado en la oxidación del alcohol a acetaldehído. En esta reacción, que cataliza la enzima alcohol deshidrogenasa (ADH), se genera NADH cuyos niveles son medidos posteriormente por espectrofotometría (protocolo en apartado 7.16). ADH B + C2H5OH + NAD B B B B P P CH3CHO + NADH + H+ B B P En ratas AA, con el fin de verificar la correlación entre las concentraciones de etanol en sangre y la autoadministración de dicha solución, se tomaron muestras de sangre (10 μl) de la vena lateral de la cola del animal justo tras la sesión de - 87 - MENÚ SALIR Materiales y Métodos autoadministración (30 minutos, n = 20). Las concentraciones de etanol se determinaron mediante cromatografía gaseosa en espacio abierto. 5.2 - ANÁLISIS DEL CONTENIDO ENDOCANNABINOIDE 5.2.1 – Ratas Wistar La determinación de los niveles de AEA en la solución resultante de la microdiálisis (apartado 1.6) se realizó mediante cromatografía líquida acoplada a espectrometría de masas (LC-MS). Se tomaron alícuotas de 5 μl de la solución microdializada junto con 5 μl de (S)(+)-araquidonil-2’-hidroxi-1’-propilamida (S-2 metanandamida) 100 nM y se cargó sobre una precolumna (0.5 x 2.5 mm, Haisil HL C18 5 μm; Higgins Analytical Inc., Mountain View, CA) usando una fase móvil de metanol (MeOH) al 30% (v/v) a una tasa de 70 μl/min. Tras un lavado de 2 minutos, se invirtió el flujo de la fase móvil a través de la precolumna gracias a una válvula y de esta manera se liberaron los endocannabinoides en una columna analítica microperforada (0.3 x 50 mm; Haisil HL C18 3μm; Higgins Analytical Inc., Mountain View, CA) usando una fase móvil isocrática formada por MeOH al 70% (v/v) que descargaba 5 μl/min. La columna eluyente se descargaba en el interior del espectrómetro de masas (Agilent 1100MSD), que se corría en modo iónico positivo con el fin de maximizar la sensibilidad. Al igual que se ha descrito anteriormente (Giuffrida et al., 2000) se encontró que los aductos de sodio de estas moléculas permitían tener una mayor sensibilidad respecto a la de sus formas protonadas. Los ratios masa/carga (m/z) usados fueron: AEA, 370.3 (M + 1Na); S-2 metanandamida, 384.3 (M + 1Na). Las curvas de calibración se construyeron a partir de un mínimo de 3 concentraciones estándar (por duplicado) y se realizaron diariamente. Bajo estas condiciones, los límites de cuantificación eran aproximadamente 0.1 nM. Por otro lado, se determinaron los niveles de endocannabinoides (AEA y PEA) en muestras de plasma, en ciertas áreas cerebrales (cerebelo, hipocampo dorsal y estriado) y tejidos periféricos (grasa perirrenal, hígado e íleon). Los endocannabinoides, - 88 - MENÚ SALIR Materiales y Métodos se fraccionaron mediante cromatografía en columna y se cuantificaron por HPLC/MS mediante el método de dilución del isótopo (Giuffrida et al., 2000). Se realizaron separaciones en fase reversa usando soluciones con concentraciones crecientes de metanol en agua (25% agua, 75% metanol durante 2 minutos; 15% agua, 85% metanol durante 3 minutos; 5% agua, 95% metanol durante 20 minutos; 100% metanol durante 5 minutos) con un flujo de 0.5 ml/min manteniéndose la temperatura de la columna a 20º C. Bajo estas condiciones, se eluyeron los estándares de la columna con los siguientes tiempos de retención: 15.4 minutos para AEA y 17.3 minutos en el caso de PEA. El análisis mediante espectrometría de masas (MS) se llevó a cabo con una fuente iónica de electrospray situado en modo iónico negativo o positivo. El voltaje capilar se ajustó a 3.5 kV y el voltaje de fragmentación varió desde 80 a 100 V. Para secar se usó nitrógeno a una tasa de 12 litros/minuto ajustándose su temperatura a 350º C y la presión del nebulizador a 50 psi. Para los análisis cuantitativos, los iones de diagnóstico (ión molecular protonado [M+H]+ y aductos de sodio de los iones P P moleculares [M+Na]+) se detectaron en modo de monitorización iónica seleccionada P P (SIM). El control del sistema completo y la evaluación de los datos se realizaron con el programa informático HP Chemstation. 5.2.2 - Ratas AA y ANA La medida del contenido de AEA y 1-AG en el cerebro de las ratas AA y ANA se realizó según el método descrito por Wang et al., 2003. Las áreas cerebrales aisladas (corteza prefrontal, núcleos caudado-putamen e hipocampo), de peso entre 30-60 mg, fueron homogeneizadas en 0.5 ml de solución fría de metanol:tampón Tris (50 mM, pH = 8), 1:1, con 7 ng de d4-anandamida. A cada muestra se le añadió 2 ml de una mezcla enfriada de cloroformo:metanol (1:1) y 0.5 ml de tampón Tris 50 mM, pH = 8. Dicho homogeneizado se centrifugó a 4º C (500 xg durante 2 minutos), se recuperó la fase de cloroformo y se transfirió a un tubo de borosilicato. La fase acuosa se extrajo, dos veces más, con cloroformo refrigerado. - 89 - MENÚ SALIR Materiales y Métodos Se evaporó la muestra a 32º C con nitrógeno y el residuo seco resultante se reconstituyó con 110 μl cloroformo añadiéndose posteriormente 2 ml de acetona fría. Se desecharon las proteínas precipitadas mediante centrifugación (1800 xg, 10 minutos), y el sobrenadante fue de nuevo evaporado hasta su desecación. Los residuos secos resultantes se reconstituyeron en 50 μl de cloroformo frío, de los cuales se usaron 35 μl para realizar análisis mediante cromatografía líquida/espectrometría de masas con el aparato LC-MSD Agilent serie 1100. La separación mediante cromatografía líquida de los endocannabinoides se llevó a cabo mediante una columna guardia (Discovery HS C18, 2 cm x 4 mm, 3 μm, 120A) y una columna analítica (Discovery HS C18, 7.5 cm x 4.6 mm, 3 μm) a 32º C con una fase móvil de metanol: agua: ácido acético (85: 15: 0.1; vol: vol: vol) a un flujo de 1 ml/min durante 12 minutos seguido de 8 minutos de metanol: ácido acético (100: 0.1; vol: vol). El MSD (modelo LS) fue ajustado para ionización química bajo presión atmosférica, polaridad positiva, y monitorización iónica seleccionada para los iones de ratio m/z de 348 para AEA, 352 para d4-AEA y 379 para 2-araquidonilglicerol (2-AG). Los ajustes de la cámara de spray fueron los siguientes: vaporizador, 400º C; temperatura del gas, 350º C; secado del gas, 5 litros/min; el nitrógeno fue empleado como gas nebulizante a una presión de 60 psig. Las curvas de calibración se hicieron usando anandamida sintética y 2-AG (Cayman Chemical, Ann Arbor, MI). Las cantidades de AEA y de 2-AG en las muestras se hallaron mediante regresión lineal invertida de las curvas estándar. Los valores se expresan como fmol o pmol por mg de tejido húmedo. 5.3 - ANÁLISIS DEL CONTENIDO DE GLUTAMATO Las muestras microdializadas fueron subdivididas en dos alícuotas de 5 μl para la cuantificación del contenido de glutamato mediante electroforesis capilar acoplada a un sistema de detección de fluorescencia inducida por láser (CE-LIF). - 90 - MENÚ SALIR Materiales y Métodos La electroforesis capilar (CE) permite la separación en un capilar de las moléculas de una muestra, sometidas a un campo eléctrico, en función de su movilidad electroforética. La muestra se introduce en el capilar (ya sea por capilaridad, aplicando presión, etc.), situando uno de sus extremos en el interior de una cubeta que contiene la muestra. El capilar se rellena con tampón y cada extremo se sitúa en una cubeta (de origen y destino) que contiene el mismo tampón electrolito. Se aplica un campo eléctrico entre la cubeta origen y de destino, que provoca la migración de las moléculas de la muestra según la relación carga/coeficiente de fricción (movilidad electroforética) de cada una de ellas. Las diferencias en la movilidad electroforética permiten la separación de las moléculas de la muestra. En el otro extremo del capilar cada uno de los analitos separados se detecta y cuantifica mediante fluorescencia inducida por láser. Este modo de detección ofrece una elevada sensibilidad y requiere que el láser enfoque hacia el capilar directamente. Los límites de detección de esta técnica son del orden de 10-18 a 10-21 mol. P P P P Los detalles específicos de esta técnica han sido previamente publicados (Hansen et al., 1997). Capilar Detector Ánodo + Tampón Cubeta Origen Tampón Muestra - Cátodo Cubeta Destino Fuente de Voltaje Electroforesis capilar acoplada a sistema de detección de fluorescencia inducida por láser (CE-LIF). Los diferentes componentes de una muestra, sometidos a un campo eléctrico, pueden ser separados, en función de su movidad electroforética, y detectados mediante fluorescencia inducida por láser. Los datos de las cuantificaciones son recogidos y almacenados en un sistema informático. - 91 - MENÚ SALIR Materiales y Métodos 6. ANÁLISIS ESTADÍSTICO El análisis estadístico de las PCR cuantitativas se realizó mediante ANOVA de una vía seguido del cálculo del valor F, que en caso de ser significativo, llevaba al análisis post hoc (Student-Newman- Keuls). El análisis de los datos obtenidos de las hibridaciones in situ en las ratas AA y ANA se realizó mediante ANOVA de una cola separando los datos de cada región ya que las varianzas eran homogéneas dentro de las regiones pero no entre ellas. Debido a la gran cantidad de regiones examinadas, las probabilidades computadas por ANOVA, para un efecto del tratamiento en cada región, fueron corregidas mediante el método de Bonferroni (Holm, 1979). En las ratas msP el análisis estadístico se realizó mediante el test t seguido de la corrección de Bonferroni. Los datos de la cuantificación de las inmunohistoquímicas se analizaron estadísticamente mediante la prueba t de Student para dos muestras. Se compararon los valores medios de las ratas control frente al de las ratas tratadas con etanol, tanto para el tratamiento agudo como para el crónico. Previo a ello, se realizó la prueba F de Snedecor. El análisis de los datos obtenidos de la medición de los niveles de endocannabinoides en las ratas AA y ANA se realizó mediante el test de MannWhitney. Los efectos del antagonista SR141716A sobre la autoadministración de sacarina y etanol se expresan como la media del número total de respuestas sobre la palanca activa e inactiva durante la sesión de 30 minutos. Dichos datos se analizaron mediante ANOVA de una vía con varias repeticiones de las medidas por dosis. Tras la significación del efecto de la dosis, cada dosis de la droga fue comparada con la condición vehículo usando una comparación post hoc de medias con corrección de Bonferroni. Los efectos de URB597 se analizaron usando ANOVA de una vía seguido del test post hoc de múltiples comparaciones de Tukey. - 92 - MENÚ SALIR Materiales y Métodos 7. PROTOCOLOS EXPERIMENTALES 7.1 - Extracción de ARN total Las áreas cerebrales, que pesaban entre 50 - 100 mg, fueron homogeneizadas en 1 ml de Trizol (Invitrogen) en tubos DNA LoBind 2,0 ml (Eppendorf). En cada extracción se manipularon un máximo de 24 muestras, de forma que hubiese el mismo número de muestras de cada grupo experimental. La superficie de trabajo era limpiada, previa a la extracción de ARN, con RNaseAWAY (Invitrogen) y, entre muestra y muestra, el homogeneizador se limpió en agua DEPC y solución de NaOH 0.5 N alternativamente finalizando siempre en agua DEPC. Los pasos que siguieron fueron: ¾ Centrifugar a 12000 xg durante 10 minutos a 4º C. ¾ Transferir el sobrenadante a otro tubo y añadir 200 μl de cloroformo. Agitar manualmente, por inversión, durante 15 segundos e incubar a temperatura ambiente durante 2 minutos. ¾ Centrifugar a 12000 xg durante 15 minutos a 4º C. ¾ Transferir la fase orgánica (incolora) a otro tubo (DNA LoBind 1,5 ml) y añadir 500 μl de isopropanol mezclando de nuevo por inversión. Incubar a temperatura ambiente durante 10 minutos. ¾ Centrifugar a 12000 xg durante 10 minutos a 4º C. ¾ Descartar el sobrenadante y añadir al precipitado 500 μl de etanol al 75% (v/v). ¾ Centrifugar a 7500 xg durante 5 minutos a 4º C. Repetir el lavado con etanol y la centrifugación. ¾ Tras descartar el sobrenadante, dejar secar los precipitados durante 10 minutos a temperatura ambiente. ¾ Añadir 50 μl de agua libre de RNasas a cada tubo e incubar 10 minutos a 55º C para disolver el precipitado. Resuspender con la micropipeta para obtener una solución homogénea de ARN. - 93 - MENÚ SALIR Materiales y Métodos 7.2 - Purificación del ARN total La purificación del ARN total se realizó según las instrucciones contenidas en el kit de purificación ‘RNeasy Mini Kit’ (Qiagen). El protocolo se detalla a continuación: ¾ Tomar el volumen correspondiente a 100 μg de ARN total, o todo si hay menos de 100 μg, y llevar a un volumen final de 100 μl con agua libre de RNasas (DEPC). ¾ Añadir 350 μl de tampón RLT (al que previamente se le ha incorporado βMercaptoetanol en proporción 1:100) a los 100 μl de muestra de ARN, mezclando bien. ¾ Añadir 250 μl de etanol 100%. Mezclar bien con la pipeta y transferir la muestra (700 μl) a una columna de sílica-gel del kit. ¾ Centrifugar durante 15 segundos a 8000 xg, descartando el filtrado y el tubo de recolección. ¾ Para el tratamiento con DNasa I, lavar con 350 μl del tampón RW1 en la columna y centrifugar 15 segundos a 8000 xg. Descartar el filtrado y reutilizar el tubo de recolección. ¾ Añadir por cada 10 μl de la solución ‘stock’ de DNasa I, 70 μl de tampón RDD (por muestra). Mezclar suavemente por inversión ya que la DNasa es sensible a la degradación física. ¾ Pipetear la mezcla de incubación de DNasa I (80 μl) directamente en la membrana de sílica-gel de la columna e incubar durante 15 minutos a 20-30º C. ¾ Añadir 350 μl de tampón RW1 a las columnas y centrifugar durante 15 segundos a 8000 xg. ¾ Descartar el filtrado y el tubo de recolección. Añadir 500 μl de tampón RPE. ¾ Centrifugar durante 15 segundos a 8000 xg. Descartar el filtrado. ¾ Añadir 500 μl de tampón RPE. ¾ Centrifugar durante 2 minutos a máxima velocidad. ¾ Descartar el filtrado y el tubo. Tomar otro tubo de recolección nuevo y centrifugar de nuevo a máxima velocidad, durante 1 minuto. - 94 - MENÚ SALIR Materiales y Métodos 7.3 – Transcripción Reversa (RT) ¾ Añadir el volumen correspondiente a 4 μg de ARN de cada una de las muestras y 1 μg para los blancos (controles negativos) en tubos de PCR. ¾ Completar con agua DEPC hasta un volumen final de 54 μl para las muestras y 13.5 μl para los blancos. ¾ Añadir a los tubos con las muestras 4 μl de cebadores de secuencia azarosa (Roche) y 1 μl a los blancos. ¾ Correr el programa 1 de la RT. ¾ Añadir, en función del número de muestras, el volumen necesario de los siguientes reactivos de forma que por cada muestra haya: - 16 μl de Tampón de reacción 5x (Roche) - 4 μl de dNTPs - 1 μl de RNasín - 20 unidades de la enzima Transcriptasa Reversa (Roche) Repartir 22 μl de esta solución a cada muestra. ¾ Preparar la solución para los blancos que contiene por cada muestra: - 4 μl de Tampón de reacción 5x - 1 μl dNTPs - 0.25 μl RNasín - 0.25 μl agua DEPC Repartir 5.5 μl de esta solución a cada tubo de blanco y correr junto con las muestras el programa 2 de la RT. Programa 1: - 65º C durante 5 minutos - 4º C durante 5 minutos - Mantener a 4º C - 25º C durante 10 minutos - 42º C durante 1 hora - 70º C durante 30 minutos - Mantener a 4º C Programa 2: - 95 - MENÚ SALIR Materiales y Métodos 7.4 - PCR Cuantitativa a Tiempo Real ¾ Añadir en un tubo DNA-Lobind (Eppendorf), el volumen de cada reactivo proporcional al número de muestras. Para una muestra sin réplica se añade: ¾ 3.38 μl de agua libre de DNasas y RNasas 0.26 μl de cebador sentido 20 μM 0.26 μl de cebador antisentido 20 μM 5.6 μl de Master Mix (Qiagen) Repartir en tubos de PCR la mezcla anterior a razón de 19 μl por tubo. Añadir a cada uno de ellos 3.36 μl del ADNc de cada muestra. Se incluye un control negativo (que llevará agua libre de DNasas y RNasas en vez de ADNc) y las diluciones del estándar del gen cuya expresión se quiere cuantificar. ¾ Pasar el contenido de cada tubo de PCR a dos pocillos (10 μl por pocillo) de la placa (de 96 pocillos) donde se realizará la PCR a tiempo real. Tapar los pocillos y centrifugar la placa hasta llegar a 3500 rpm. ¾ Meter la placa en el Opticon 2 y correr el programa de PCR específico de cada gen, con la temperatura de alineamiento adecuada. Los programas de PCR difieren en el valor de la temperatura de alineamiento y el número de ciclos, pero comparten pasos en común como son: 1. Incubar a 95º C durante 15 minutos. 2. Incubar a 94º C durante 15 segundos. 3. Incubar a la temperatura de alineamiento de cada gen durante 30 segundos. 4. Incubar a 72º C durante 30 segundos. 5. Lectura de la fluorescencia. 6. Volver al paso 2 el número de veces más adecuado según las diluciones del estándar utilizadas. 7. Hacer curva de fusión (‘Melting Curve’) incubando desde 56º C a 96º C, con incrementos de 0.5º C, manteniendo la temperatura durante 10 segundos, y realizando una lectura de la fluorescencia cada 0.5º C. 8. Disminución progresiva de la temperatura hasta 4º C. - 96 - MENÚ SALIR Materiales y Métodos 7.5 - PCR para la determinación de la Temperatura de Alineamiento de los Cebadores Los pasos a seguir para realizar la PCR son: ¾ Poner en un tubo de PCR la siguiente mezcla: - 27.5 μl de Master mix (Qiagen) - 1.1 μl de cebador sentido 20 μM - 1.1 μl de cebador antisentido 20 μM - 16.8 μl de agua grado de PCR - 8.5 μl de ADNc de rata ¾ Repartir la mezcla anterior a razón de 10 μl por tubo en un total de 5 tubos. ¾ Hacer una centrifugación rápida a todos los tubos para que la mezcla esté en el fondo del tubo. ¾ Poner a cada tubo en el hueco del Opticon 2 (MJ Research) que le corresponda en función de la temperatura del gradiente deseada. ¾ Correr el programa con los siguientes pasos: 1. Incubación a 95º C durante 15 minutos 2. Incubación a 94º C durante 15 segundos 3. Incubación a la temperatura del gradiente elegida durante 30 seg 4. Incuabación a 72º C durante 30 seg 5. Lectura de la fluorescencia 6. Volver al paso 2 durante 44 ciclos 7. Curva de Fusión (Melting Curve) con lectura de fluorescencia cada 0.5ºC, manteniéndose la temperatura durante 10 seg , incubando desde 56º C a 95º C. 8. Incubar a 4º C 7.6 - Producción del Estándar mediante PCR - Añadir a cada tubo de PCR, que posteriormente se dividirá en 2: ¾ 25 μl de Master Mix (Qiagen) ¾ 1.5 μl de cebador sentido 20 μM - 97 - MENÚ SALIR Materiales y Métodos ¾ 1.5 μl de cebador antisentido 20 μM ¾ 14.5 μl de agua grado de PCR ¾ 7.5 μl de ADNc procedente de un tejido de rata que exprese el ARNm que se desea cuantificar o de agua grado de PCR en el control negativo de la PCR. - Dar una centrifugación rápida al tubo de PCR para que todo el volumen se vaya al fondo del mismo. - Poner la PCR en el Opticon 2 (MJ Research) siguiendo el siguiente protocolo: 1. Incubación a 95º C durante 15 minutos 2. Incubación a 94º C durante 30 segundos 3. Incubación a la temperatura de alineamiento de los cebadores durante 1 minuto 4. Incubación a 72º C durante 1 minuto 5. Lectura de la fluorescencia 6. Volver al paso 2 durante 44 ciclos 7. Curva de Fusión (Melting Curve) con lectura de fluorescencia cada 0.5ºC, manteniéndose la temperatura durante 10 seg , incubando desde 56º C a 95º C. 8. Incubar a 4º C De los 50 μl de la mezcla de PCR, 10 μl se reservaban para realizar la visualización de las bandas de ADN, del estándar sin purificar, en gel de agarosa al 1% (p/v). Los 40 μl restantes se purificaron tal y como se detalla en el apartado 7.7. 7.7 - Purificación del Estándar La purificación de los estándares se realiza con los reactivos del kit ‘High Pure PCR Product Purification Kit’ (Roche) según el protocolo que se detalla a continuación: ¾ Llevar el estándar hasta un volumen final de 100 μl, completado con agua DEPC, y añadir 500 μl de tampón de unión. Pasar los 600 μl a un tubo con membrana (suministrado con el kit) e introducir éste en un tubo de recolección. - 98 - MENÚ SALIR Materiales y Métodos ¾ Centrifugar 1 minuto a máxima velocidad a 25º C. ¾ Descartar el filtrado y añadir 500 μl de tampón de lavado. ¾ Centrifugar 1 minuto a máxima velocidad a 25º C. ¾ Descartar el filtrado y el tubo de recolección. Meter el tubo con membrana en un tubo eppendorf libre de DNasas y RNasas y añadir 50 μl de tampón de elución. ¾ Centrifugar 1 minuto a máxima velocidad a 25º C. ¾ Descartar el tubo con membrana y centrifugar el tubo eppendorf durante 2 minutos a máxima velocidad a 25º C. 7.8 - Inmunohistoquímica de CB1 ٭ Realizar tres lavados a los cortes en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en citrato sódico 50 mM pH = 9, durante 30 minutos a 80º C. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en H2O2 al 3% en PBS, 10 minutos a temperatura ambiente en B B B B agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en anti-CB1 (cabra) a 1/100 en PBS 0.1 M pH=7.3, 0.2% Tritón TX100, 0.1% NaN3 (azida sódica), toda la noche a temperatura ambiente y en B B agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en anticuerpo secundario (anti-IgG biotinilado de cabra) a 1/500 en PBS 0.1M, 0.2% Tritón TX-100, 0.1% Azida Sódica, durante 1 hora a temperatura ambiente. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en estreptavidina-HRP (peroxidasa de rábano) diluida en PBS 0.1M, 0.2% Tritón TX-100 (sin azida ya que ésta inhibe a la peroxidasa) a 1/2000 durante 1h a temperatura ambiente y en agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. - 99 - MENÚ SALIR Materiales y Métodos ٭ Pasar los cortes a pocillos con solución de DAB (0.2 mg/ml), Níquel (0.08%) en PBS. Añadir 5-10 μl de H2O2 al 30% y agitar manualmente, B B B B parando la reacción pasando de nuevo los cortes a PBS. ٭ Montar los cortes sobre portaobjetos gelatinados. ٭ Deshidratar y montar en DPX. 7.9 - Inmunohistoquímica de FAAH ٭ Realizar a los cortes tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en citrato sódico 50 mM pH = 9, durante 30 minutos a 80º C. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en H2O2 al 3% en PBS, 10 minutos a temperatura ambiente en B B B B agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en anti- FAAH a 1/75 en PBS 0.1 M pH=7.3, 0.2% Tritón TX-100, 0.1% NaN3 (azida sódica), toda la noche a temperatura ambiente y en B B agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en anticuerpo secundario (anti-IgG biotinilado de conejo) a 1/500 en PBS 0.1M, 0.2% Tritón TX-100, 0.1% Azida Sódica, durante 1 hora a temperatura ambiente. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en estreptavidina-HRP diluida en PBS 0.1M, 0.2% Tritón TX-100 (sin azida ya que ésta inhibe a la peroxidasa) a 1/2000 durante 1h a temperatura ambiente y en agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Pasar los cortes a pocillos con solución de DAB (0.2 mg/ml), Níquel (0.08%) en PBS. Añadir 5-10 μl de H2O2 al 30% y agitar manualmente, B B B B parando la reacción pasando de nuevo los cortes a PBS. ٭ Montar los cortes sobre portaobjetos gelatinados. ٭ Deshidratar y montar en DPX. - 100 - MENÚ SALIR Materiales y Métodos 7.10 - Inmunohistoquímica de MAGL ٭ Realizar a los cortes tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en citrato sódico 50 mM pH = 9, durante 30 minutos a 80º C. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en H2O2 al 3% en PBS, 10 minutos a temperatura ambiente en B B B B agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en anti- MAGL a 1/200 en PBS 0.1 M pH=7.3, 0.2% Tritón TX-100, 0.1% NaN3 (azida sódica), toda la noche a temperatura ambiente y en B B agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en anticuerpo secundario (anti-IgG biotinilado de conejo) a 1/500 en PBS 0.1M, 0.2% Tritón TX-100, 0.1% Azida Sódica, durante 1 hora a temperatura ambiente. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en estreptavidina-HRP diluida en PBS 0.1M, 0.2% Tritón TX-100 (sin azida ya que ésta inhibe a la peroxidasa) a 1/2000 durante 1h a temperatura ambiente y en agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Pasar los cortes a pocillos con solución de DAB (0.2 mg/ml), Níquel (0.08%) en PBS. Añadir 5-10 μl de H2O2 al 30% y agitar manualmente, B B B B parando la reacción pasando de nuevo los cortes a PBS. ٭ Montar los cortes sobre portaobjetos gelatinados. ٭ Deshidratar y montar en DPX. 7.11 - Inmunohistoquímica de NAPE-PLD ٭ Realizar a los cortes tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en citrato sódico 50 mM pH = 9, durante 30 minutos a 80º C. - 101 - MENÚ SALIR Materiales y Métodos ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en H2O2 al 3% en PBS, 10 minutos a temperatura ambiente en B B B B agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en anti-NAPE-PLD a 1/400 en PBS 0.1 M pH=7.3, 0.2% Tritón TX100, 0.1% NaN3 (azida sódica), toda la noche a temperatura ambiente y en B B agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en anticuerpo secundario (anti-IgG biotinilado de conejo) a 1/500 en PBS 0.1M, 0.2% Tritón TX-100, 0.1% Azida Sódica, durante 1 hora a temperatura ambiente. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Incubar en estreptavidina-HRP diluida en PBS 0.1M, 0.2% Tritón TX-100 (sin azida ya que ésta inhibe a la peroxidasa) a 1/2000 durante 1h a temperatura ambiente y en agitación. ٭ Realizar tres lavados en PBS 0.1 M pH = 7.3, cada uno durante 10 minutos. ٭ Pasar los cortes a pocillos con solución de DAB (0.2 mg/ml), Níquel (0.08%) en PBS. Añadir 5-10 μl de H2O2 al 30% y agitar manualmente, B B B B parando la reacción pasando de nuevo los cortes a PBS. ٭ Montar los cortes sobre portaobjetos gelatinados. ٭ Deshidratar y montar en DPX. 7.12 - Ensayo de la Actividad Enzimática FAAH Las muestras se realizaron por triplicado incluyéndose un control negativo (que no llevaría membranas). La mezcla de incubación, con un volumen final de 0.5 ml por tubo, contenía: 50 μg de la fracción de membranas 10 μM de [3H]-AEA (10000 dpm/ml) Tris-HCl 50 mM, pH = 7.5 hasta completar un volumen de 0.5 P P ml. - 102 - MENÚ SALIR Materiales y Métodos ¾ Incubar durante 10 minutos a 37º C con agitación. ¾ Parar la reacción añadiendo 1 ml de cloroformo:metanol (1:1) a temperatura ambiente, mezclándolo vigorosamente y dejando reposar durante 2-3 minutos. ¾ Centrifugar a 500 xg durante 5 minutos a temperatura ambiente. ¾ Tomar 600 μl de la fase acuosa y llevar a viales de centelleo. ¾ Añadir líquido de centelleo (3 ml), tapar y agitar. Dejar incubar toda la noche en oscuridad a temperatura ambiente. ¾ Agitar vigorosamente. ¾ Añadir otros 3 viales de plástico con 50 μl de [3H]-AEA y 3 ml del líquido P P de centello con el fin de ver las cuentas totales. ¾ Realizar el contaje de la radiactividad en un contador de emisiones beta. 7.13 - Ensayo de la Actividad Enzimática NAT Las muestras se realizaron por duplicado, conteniendo uno de los tubos CaCl2 y B B otro EGTA, en un volumen final de 500 μl. Cada tubo contenía: 300 μg de fracción de membranas. 50 μl de solución de CaCl2 100 mM disuelta en HEPES 50 mM B B pH = 8 ó 50 μl de EGTA 100 mM disuelto en HEPES 50 mM pH = 8. 10 μl de L-3 fosfatidilcolina, 1,2-di(1 14C) palmitoil. Tampón HEPES 50 mM pH = 8 hasta completar el volumen P P final de 0.5 ml. ¾ Incubar a 37º C durante 1 hora con agitación. ¾ Parar la reacción con 1 ml de etanol. ¾ Añadir 2 ml de cloroformo para realizar la extracción. ¾ Centrifugar a máxima velocidad para separar las fases. ¾ Tomar la fase de cloroformo (en el fondo del tubo) y pasarlo a tubos limpios. ¾ Evaporar el cloroformo, en una centrífuga de vacío, a 55º C aproximadamente en 1 hora. - 103 - MENÚ SALIR Materiales y Métodos ¾ Añadir 400 μl de cloroformo a las muestras secas y resuspender el precipitado. ¾ Realizar la cromatografía añadiendo entre 180-200 μl de la muestra a cada columna. Cada muestra se divide en dos columnas. ¾ Correr 2 ml de metanol:cloroformo 9:1 y recoger, en viales de centelleo, las amidas de ácidos grasos. ¾ Correr 2 ml de metanol:cloroformo 5:5 y recoger, en viales de centelleo, las N-acil-fosfatidiletanolaminas (NAPEs). ¾ Añadir 4 ml de líquido de centelleo a cada vial, agitarlos y dejar incubar toda la noche en oscuridad. ¾ Realizar el contaje en un contador de emisiones beta. 7.14 - Ensayo de Unión del receptor CB1 Previo a la preparación de la mezcla de incubación, se realizan las diluciones de los compuestos desplazantes, los cuales se encuentran almacenados a 4º C, a una concentración de 10 mM en DMSO. Cada muestra se realiza por triplicado en un volumen final de 500 μl que contiene: - 350 μl de tampón (Tris-HCl 50 mM, pH = 7.4 – 7.7; BSA al 0.1% (p/v)) - 50 μl de la dilución del compuesto en sus correspondientes tubos - 50 μl de solución de membranas - 50 μl de [3H]SR141716A, 3.4 μM, diluido en tampón Tris-HCl 50 P P mM pH = 7.4 - 7.7; BSA al 0.1% (p/v). ¾ Incubar durante 1 hora a 30º C. ¾ Parar la reacción con la adición de tampón frío. ¾ Centrifugar durante 8 minutos a 1500 xg. ¾ Descartar el sobrenadante y añadir 2 ml de líquido de centelleo. ¾ Realizar el contaje de radiactividad en un contador de emisiones beta. - 104 - MENÚ SALIR Materiales y Métodos 7.15 - Ensayo de Acoplamiento del receptor CB1 El ensayo se realizó en un volumen total de 1 ml, haciéndose cada muestra por triplicado y en dos series: una serie con el agonista WIN 55,212-2 y otra con el agente desplazante guanidil imidodifosfato. La mezcla de incubación contenía: 20 μg de proteínas de la fracción de membranas 20 μM de GDP 0.05 nM de [35S]GTPγS 10 μM de WIN 55,212-2 (sólo a los tubos estimulados) ó 10 P P μM de guanidil imidodifosfato (sólo a los tubos basales). Tampón Tris-HCl 50 mM, MgCl2 3 mM, EGTA 0.2 mM, NaCl 100 mM, BSA 0.1 mg/ml; pH = 7.4, completando hasta un volumen final de 1 ml. ¾ Incubar a 30º C durante 1 hora. ¾ Parar la reacción centrifugando a 20000 xg a 4º C. ¾ Lavar dos veces el precipitado con tampón Tris frío. ¾ Añadir 5 ml de líquido de centelleo Ecolite y dejar toda la noche incubando en oscuridad. ¾ Realizar el contaje de radiactividad en un contador de emisiones beta. 7.16 - Medida de Etanol en plasma La medida de etanol en plasma se realizó en placas de 96 pocillos según el protocolo detallado en el kit de detección de etanol (Randox). Los pasos a seguir fueron: ¾ Añadir 150 μl del tampón (Pirofosfato 75 mM pH = 8.7, semicarbazida 75 mM, glicina 21 mM) y 50 μl de la solución de NAD+ a cada pocillo. P P ¾ Añadir 5 μl de la muestra correspondiente. ¾ Mezclar bien y añadir 10 μl de la solución de ADH. ¾ Mezclar bien e incubar la placa a 37º C durante 25 minutos. ¾ Leer la absorbancia a una longitud de onda de 340 nm. - 105 - MENÚ SALIR Materiales y Métodos 7.17 - Soluciones de uso común - Tampón de Carga de ARN Esta solución contiene: glicerol al 50%, EDTA 1mM, azul de bromofenol al 0.4% y bromuro de etidio al 4%. - Tampón de Carga de ADN Para un volumen final de 10 ml añadir: 5 ml de glicerol 4 mg de EDTA 40 mg de azul de bromofenol - Agua DEPC ¾ Añadir a un litro de agua mili-Q, 1 ml de Dietil-pirocarbonato (Sigma, D5758) - ¾ Agitar vigorosamente e incubar a 37º C toda la noche. ¾ Autoclavar. Solución Tris-EDTA (TE) La solución de TE 1x (pH= 7.5) está compuesta por: - Tris-HCl 10 mM pH=7.5 - EDTA 1 mM pH=8 ¾ La solución stock de Tris-HCl 100 mM pH=7.5 se preparó de la siguiente manera: - Disolver 1.211 gr de Tris Base en 80 ml de agua DEPC. - Ajustar pH hasta 7.5. - Enrasar hasta 100 ml con agua DEPC. - Guardar a 4ºC - 106 - MENÚ SALIR Materiales y Métodos ¾ La solución stock de EDTA 100 mM (pH=8) se preparó de la siguiente forma: ¾ - Disolver 1.861 g de EDTA en unos 40 ml de agua DEPC. - Ajustar el pH a 8. - Enrasar hasta 50 ml con agua DEPC. - Guardar a 4º C. Preparar solución de TE 1x pH = 7.5 Si se desea preparar, por ejemplo, un volumen final de 10 ml de TE 1x, se deben mezclar: - 8.9 ml de agua DEPC - 1 ml de Tris- HCl 100 mM pH = 7.5 y - 0.1 ml de EDTA 100 mM pH = 8 El pH de la solución debería ser de 7.5; en caso contrario se debe ajustar a dicho valor. - Tampón fosfato (PB) 0.4 M, pH = 7.3 Para 1000 ml: - NaH2PO4- H2O, 13.8 gr - Na2HPO4- 2H2O, 53.4 gr B B B B B B B B B B B B Disolver en agua destilada y ajustar el pH a 7.3 con HCl 1N ó NaOH 1N. - PBS (tampón fosfato salino), pH = 7.3 Para 1000 ml: - 250 ml de PB 0.4 M pH = 7.3 - 750 ml de agua destilada - 9 gr de NaCl Ajustar, si es necesario, el pH a 7.3 con HCl 1N ó NaOH 1N. - 107 - MENÚ SALIR MENÚ SALIR RESULTADOS MENÚ SALIR MENÚ SALIR Resultados TRATAMIENTO AGUDO DE ETANOL 1.1 - Medida de los Niveles de Etanol y Anandamida en Plasma y Tejidos Periféricos La administración aguda de etanol (4 gr/kg), mediante inyección intraperitoneal (i.p.), provoca un fuerte incremento de los niveles de etanol en plasma, permaneciendo estables durante las primeras cuatro horas y disminuyendo posteriormente, a partir de las 8 horas, hasta no ser detectables a las 24 horas. Los niveles de anandamida (AEA) se alteran, tras la administración de etanol, de la siguiente manera: no son detectables a los 45 minutos de la inyección de etanol, aumentan a partir de los 90 minutos alcanzando valores normales, siendo máximos a las 4 horas y volviendo a niveles control a las 8 horas (1.3 + 0.6 pmol/ml, n = 8) y 24 horas (0.7 + 0.5 pmol/ml, n = 8). A B 8 AEA (pmol/ml) ETANOL (mM) 50 40 30 20 10 0 N.D. 0 6 4 * 2 N.D. 45 90 0 240 480 1440 TIEMPO (min) N.D. 0 45 90 TIEMPO (min) 240 Figura 1. Efectos a lo largo del tiempo de la administración aguda de etanol (4 gr/kg) sobre los niveles de etanol (A) y anandamida (B) en plasma. Los datos son las medias + SEM de al menos 5 muestras por grupo. (*) P < 0.01 versus grupo del tiempo 0. ANOVA de una vía con análisis post-hoc de Student-Newman-Keuls. En tejidos periféricos, la administración de etanol provoca una disminución de los niveles de AEA en grasa perirrenal, aunque aumentan en intestino delgado e hígado tras 90 minutos de la inyección (ver tabla 1). - 111 - MENÚ SALIR Resultados Tiempo tras la inyección (min): 0’ 45’ 90’ 240’ Grasa 16.2 ± 1.7 11.9 ± 0.9 19.1 ± 1.2 6.4 ± 0.6 * Hígado 3.7 ± 1.8 3.7 ± 1.8 21.9 ± 3.7 * 4.8 ± 2.9 Intestino Delgado 33.9 ± 13.2 63.4 ± 23.5 285.1 ± 13.2 * 62.8 ± 12.8 Tabla 1. Efectos de la administración aguda de etanol (4 gr/kg i.p.) sobre los niveles de anandamida (pmol/gr tejido) en la grasa perirrenal, hígado e intestino delgado a los 0, 45, 90 y 240 minutos tras la inyección. Los datos son las medias + SEM de 5-8 muestras por grupo. (*) P < 0.01 versus grupo del tiempo 0. Newman-Keuls. En base a la estabilidad de los niveles de etanol y los cambios de los niveles de AEA en plasma, se han seleccionado los tiempos de 0, 45, 90 y 240 minutos para el análisis bioquímico de AEA y PEA (palmitoiletanolamida) y la medida de las actividades enzimáticas de NAT y FAAH en el cerebro. 1.2 - Medida de los Niveles de Endocannabinoides en Cerebro Previamente a la medida de los efectos del etanol, se comprobó si la administración aguda de solución salina, mediante inyección i.p., afectaba a los niveles de anandamida en estriado ventral, cerebelo o hipocampo, resultando negativo (tabla 2). Debido a que la inyección de solución salina en animales habituados provoca un estrés moderado, la ausencia de efectos por este procedimiento sobre el sistema endocannabinoide en cerebro concuerda con lo descrito previamente en la bibliografía (Patel et al., 2005): existe un decremento de AEA sólo tras estrés severo, inducido por inmovilización activa, localizándose exclusivamente en la amígdala (no existiendo ni en cerebelo ni prosencéfalo). - 112 - MENÚ SALIR Resultados Tiempo tras la inyección (min): 0’ 30’ 60’ Estriado Ventral 119 ± 20 98 ± 40 82 ± 29 Cerebelo 16.5 ± 1.4 14.7 ± 1.8 26.7 ± 1.9 Hipocampo Dorsal 137 ± 51 134 ± 63 119 ± 41 Tabla 2. Efecto de la administración aguda del vehículo (solución salina al 0,9%, en dosis de 1 ml/kg i.p.) sobre los niveles de anandamida (pmol/g tejido) en el estriado ventral (núcleo accumbens), cerebelo e hipocampo dorsal. Medidas realizadas a los 0, 30 y 60 minutos en animales habituados a inyección. Los datos son las medias + SEM de 5-8 muestras por grupo. (*) P < 0.01 versus grupo del tiempo 0. Newman-Keuls. Una vez comprobado que la manipulación experimental no afecta a los niveles de endocannabinoides, se procedió a su determinación tras la administración de alcohol. La inyección aguda de etanol produce una disminución en los niveles de AEA en estriado ventral, cerebelo e hipocampo, tal y como se muestra en la figura 2. Este efecto se observa en todas las áreas cerebrales estudiadas tras 90 minutos de la administración de etanol. En el hipocampo, la reducción de los niveles de AEA se observa en todos los tiempos seleccionados (figura 2C). 140 120 100 80 60 40 20 0 Estriado Ventral B. Cerebelo 2 AEA (pmol/g) AEA (pmol/g) A. * 0 45 90 1 1 5 0 240 TIEMPO (min) * 0 45 90 TIEMPO (min) - 113 - 240 MENÚ SALIR Resultados C. Hipocampo Dorsal AEA (pmol/g) 200 150 100 50 * * 45 90 * 0 0 240 TIEMPO (min) Figura 2. Efecto a lo largo del tiempo de la administración aguda i.p. de etanol (4 gr/kg) sobre los niveles titulares de AEA en: A. Estriado Ventral-núcleo accumbens; B. Cerebelo y C. Hipocampo Dorsal. Los datos son las medias + SEM de al menos 5 muestras por grupo. (*) P< 0.01, versus grupo del tiempo 0. De manera similar, se observa una disminución de los niveles de PEA en estriado ventral tras 45 min de la inyección, en cerebelo a los 45 y 90 min y en hipocampo en todos los tiempos estudiados. A. Estriado Ventral B. 800 PEA (pmol/g) PEA (pmol/g) 500 400 300 * 200 600 400 200 100 0 Cerebelo 0 45 90 0 240 TIEMPO (min) 0 * * 45 90 240 TIEMPO (min) - 114 - SALIR Resultados C. PEA (pmol/mg) MENÚ Hipocampo Dorsal 2500 2000 1500 1000 * 500 0 0 45 * * 90 240 TIEMPO (min) Figura 3. Efectos a lo largo del tiempo de la administración aguda de etanol (4 gr/kg) sobre los niveles titulares de palmitiletanolamida (PEA) en A. Estriado Ventral- Núcleo Accumbens, B. Cerebelo y C. Hipocampo dorsal. Los datos son las medias + SEM de al menos 5 muestras por grupo. (*) P< 0.01, versus grupo del tiempo 0. Newman-Keuls. Dado que los niveles de endocannabinoides están regulados a nivel de síntesis (actividades NAT y NAPE-PLD) y de degradación, estudiamos tanto las actividades de estas enzimas como su expresión. Comenzamos por FAAH dado que esta enzima ha sido vinculada al alcoholismo (Sipe et al., 2002). 1.3 - Medida de la Actividad Enzimática FAAH Se ha estudiado si la actividad enzimática FAAH podría estar alterada y con ello justificarse la disminución, en cerebro, de los niveles de AEA y PEA observados debido a la actividad de degradación de dicha enzima sobre estas aciletanolamidas. Se ha encontrado que no existen variaciones en la actividad FAAH ni en estriado ventral ni en cerebelo. Sin embargo, en hipocampo dicha actividad sí varía, disminuyendo a los 45 minutos de la administración de alcohol y normalizándose posteriormente. Otros estudios in vitro, usando membranas cerebelares, indican que la actividad FAAH no se ve afectada por la presencia de etanol en el tampón de ensayo a - 115 - MENÚ SALIR Resultados concentraciones en el rango de aquellas encontradas en el plasma tras la administración aguda de etanol (10-9 a 10-2 M). Estriado Ventral B. FAAH (pmol/min/mg) 300 200 100 0 0 45 90 Cerebelo 50 40 30 20 10 0 240 TIEMPO (min) 0 45 90 240 TIEMPO (min) C. FAAH (pmol/min/mg) FAAH (pmol/min/mg) A. Hipocampo Dorsal 50 40 * 30 20 10 0 0 45 90 240 TIEMPO (min) Figura 4. Efecto a lo largo del tiempo de la administración aguda i.p. de etanol (4 gr/kg) sobre la actividad de la amidohidrolasa de ácidos grasos (FAAH) en membranas de: A. Estriado VentralNúcleo Accumbens; B. Cerebelo y C. Hipocampo Dorsal. Los datos son las medias + SEM de al menos 7 muestras por grupo. (*) P< 0.01, versus grupo del tiempo 0. Newman-Keuls. - 116 - MENÚ SALIR Resultados 1.4 - Medida de la Actividad Enzimática NAT La actividad de la enzima implicada en la síntesis del precursor de la anandamida, N-araquidonil fosfatidiletanolamina, no se encuentra afectada en las membranas cerebelares tras la administración aguda de etanol. No se pudo medir la actividad NAT sobre hipocampo ya que la técnica requiere la recolección de varias muestras para obtener suficiente cantidad de proteína, por lo que no se dispuso de material suficiente. NAT (pmol/mg/min) 0’ 45’ 90’ 120’ 452 ± 108 449 ± 104 687 ± 234 385 ± 122 Tabla 3. La administración aguda de etanol (4 gr/kg i.p.) no produce ningún efecto sobre la actividad de la transferasa de grupo N-acilo (NAT) en las fracciones de membrana de cerebelo a los 0, 45, 90 y 240 minutos tras la inyección. Los datos son las medias + SEM de 5-8 muestras por grupo. 1.5 - Medida de los niveles de ARNm del Sistema Endocannabinoide Con el fin de averiguar si la disminución observada en los niveles de endocannabinoides podría ser debida a la alteración de la expresión de los genes que codifican para las enzimas de síntesis y degradación y/o el receptor CB1, se analizaron los niveles de ARNm de cada uno de dichos componentes. La expresión de ninguno de los genes relacionados con los endocannabinoides está alterada tras la administración aguda de etanol ni a las 2 ni a las 12 horas de su inyección vía intraperitoneal (figura 5A-C). Sin embargo, la expresión de GAPDH se encuentra claramente incrementada (figura 5D) tanto en hipocampo como en cerebelo. Estos resultados apuntan en contra de una activación de la degradación como la principal causa del descenso de los niveles de endocannabinoides en cerebro, inducido por etanol. - 117 - MENÚ SALIR Resultados A B 3000 RATIO FAAH/GUS RATIO CB1/GUS 200 100 0 0 2 12 0 2 2000 1000 0 12 0 HIPPOCAMPUS CEREBELLUM Tiempo tras inyección i.p. (h) 0 2 12 D 2500 RATIO GAPDH/GUS 8 RATIO NAPE-PLD/GUS 12 HIPPOCAMPUS CEREBELLUM Tiempo tras inyección i.p. (h) C 6 4 2 0 2 * 2000 1500 * * * 1000 500 0 2 12 0 2 0 12 HIPPOCAMPUS CEREBELLUM Tiempo tras inyección i.p. (h) 0 2 12 0 2 12 HIPPOCAMPUS CEREBELLUM Tiempo tras inyección i.p. (h) Figura 5. Niveles de expresión de ARNm, medidos por PCR a tiempo real, de: A. receptor cannabinoide CB1, B. amidohidrolasa de ácidos grasos (FAAH), C. fosfolipasa D específica de Nacilfosfatidil-etanolamina (NAPE-PLD) y D. deshidrogenasa de gliceraldehído 3-fosfato (GAPDH) en hipocampo y cerebelo de animales control (tiempo = 0) y animales tratados con alcohol (tiempos = 2 y 12 horas tras inyección i.p.). Los datos son las medias + SEM de al menos 7 muestras por grupo. (*) P < 0.01, versus grupo del tiempo 0. Newman-Keuls. - 118 - MENÚ SALIR Resultados 1.6 - Inmunohistoquímica de CB1 y FAAH Para comprobar si la ausencia de cambios a nivel de expresión génica tenía su correlato en cuanto a la presencia de la proteína, se procedió a la medición de la expresión de CB1 y FAAH en cortes histológicos. Las cuantificaciones realizadas para el receptor endocannabinoide CB1 revelan la no existencia de diferencias estadísticamente significativas, entre los grupos etanol y control, en ninguna de las áreas estudiadas, esto es, ni en la capas molecular y de Purkinje del cerebelo (Figura 6), ni en la capa granular del giro dentado hipocampal (Figura 7), ni en el accumbens shell del estriado ventral (Figura 8), ni en el pálido ventral (Figura 9). La inmunohistoquímica de FAAH tampoco reveló diferencias estadísticamente significativas entre los grupos etanol y control en ninguna de las áreas estudiadas, esto es, ni en las capa molecular y de Purkinje del cerebelo (Figura 10), ni en la capa granular del giro dentado hipocampal (Figura 11), ni en la capa piramidal de CA4 en hipocampo (Figura 12), ni en el accumbens shell del estriado ventral (Figura 13), ni en el pálido ventral (Figura 14). - 119 - MENÚ SALIR CB1 en Cerebelo Etanol CP CM Control Densidad Óptica (ua) 175 150 Control Etanol 125 100 75 50 25 0 Capa Molecular Capa Purkinje Figura 6. Cuantificación mediante densitometría digital del receptor CB1 en el cerebelo (capas molecular, CM, y de Purkinje,CP) de ratas con tratamiento agudo de etanol versus ratas control. U.a.= unidades arbitrarias. - 120 - SALIR CB1 en Giro Dentado Etanol CG Control Capa Granular Densidad Óptica (ua) MENÚ 60 50 40 30 20 10 0 CONTROL ETANOL Figura 7. Cuantificación mediante densitometría digital del receptor CB1 en la capa granular (CG) del giro dentado hipocampal de ratas con tratamiento agudo de etanol versus ratas control. U.a.= unidades arbitrarias. - 121 - SALIR CB1 en Estriado Ventral Etanol AcbSh Control Accumbens Shell Densidad Óptica (ua) MENÚ 125 100 75 50 25 0 CONTROL ETANOL Figura 8. Cuantificación mediante densitometría digital del receptor CB1 en el accumbens shell (AcbSh) del estriado ventral de ratas con tratamiento agudo de etanol versus ratas control. U.a.= unidades arbitrarias. - 122 - SALIR CB1 en Pálido Ventral Etanol PV Control 75 Densidad öptica (ua) MENÚ 60 45 30 15 0 CONTROL ETANOL Figura 9. Cuantificación mediante densitometría digital del receptor CB1 en el pálido ventral (PV) de ratas con tratamiento agudo de etanol versus ratas control. U.a.= unidades arbitrarias. - 123 - MENÚ SALIR FAAH en Cerebelo Etanol CP CM Control Densidad Óptica (ua) 200 175 Control Etanol 150 125 100 75 50 25 0 Capa Molecular Capa Purkinje Figura 10. Cuantificación mediante densitometría digital de FAAH en el cerebelo (capas molecular, CM, y de Purkinje, CP) de ratas con tratamiento agudo de etanol versus ratas control. U.a.= unidades arbitrarias. - 124 - SALIR FAAH en Giro Dentado Etanol CG Control Capa Granular Densidad Óptica (ua) MENÚ 75 60 45 30 15 0 CONTROL ETANOL Figura 11. Cuantificación mediante densitometría digital de FAAH en capa granular (CG) del giro dentado de ratas con tratamiento agudo de etanol versus ratas control. Diferencia estadísticamente significativa para p< 0.05 (*). U.a.= unidades arbitrarias. - 125 - SALIR FAAH en CA4 Etanol CP Control Capa Piramidal Densidad Óptica (ua) MENÚ 75 60 45 30 15 0 CONTROL ETANOL Figura 12. Cuantificación mediante densitometría digital de FAAH en la capa piramidal (CP) de CA4 de ratas con tratamiento agudo de etanol versus ratas control. U.a.= unidades arbitrarias. - 126 - SALIR FAAH en Estriado Ventral Etanol AcbSh Control Accumbens Shell Densidad Óptica (ua) MENÚ 100 80 60 40 20 0 CONTROL ETANOL Figura 13. Cuantificación mediante densitometría digital de FAAH en el accumbens shell (AcbSh) del estriado ventral de ratas con tratamiento agudo de etanol versus ratas control. U.a.= unidades arbitrarias. - 127 - SALIR FAAH en Pálido Ventral Etanol PV Control Densidad Óptica (ua) MENÚ 100 80 60 40 20 0 CONTROL ETANOL Figura 14. Cuantificación mediante densitometría digital de FAAH en pálido ventral (PV) de ratas con tratamiento agudo de etanol versus ratas control. U.a.= unidades arbitrarias. - 128 - MENÚ SALIR Resultados 1.7 - Medida de los niveles de Anandamida y Glutamato en Estriado Ventral Si la disminución de endocannabinoides no se debe ni a la enzima FAAH ni a NAT, cabe pensar que el candidato que explicaría tales cambios sería la enzima NAPEPLD. Esta enzima es activada por neurotransmisores excitadores como el glutamato, de forma que una disminución en la liberación de dicho neurotransmisor podría generar una bajada en la actividad de NAPE-PLD. Con el fin de dilucidar si la alteración en los niveles de AEA podría deberse a cambios en la actividad NAPE-PLD se han analizado, mediante diálisis, los niveles de AEA y glutamato en estriado ventral tras la administración aguda de etanol. Las concentraciones basales de AEA en estriado ventral son de 2.03 + 0.5 y 2.12 + 1.1 nM para los grupos etanol inyectados a una dosis de 0.75 g/kg (n = 6) y 2 g/kg (n = 5), respectivamente (figura 15A). La inyección del vehículo no altera los niveles de AEA pero cuando se administra etanol disminuye de forma dosis-dependiente (F(1.141) = 25.094; p < 0.001). La administración de una dosis de 0.75 g/kg de etanol genera un leve descenso, aunque no significativo, en los niveles de AEA. Sin embargo, la inyección de dosis de 2 g/kg de etanol produce una disminución significativa en los niveles de AEA (F12, 52 = 2.621; p < 0.001) transcurridos 40 minutos de la administración de etanol y persiste aproximadamente un 60% por debajo del nivel basal de AEA, medido antes de la administración, hasta el final del experimento (tiempo = 120 min). La concentración basal de glutamato, en las mismas muestras, es 602 + 21 nM (figura 15B). A los 30 min, de la administración de dosis de 2 g/kg de etanol, disminuye significativamente hasta aproximadamente un 70% respecto al nivel basal, manteniéndose hasta el final del experimento (t = 120 min). - 129 - SALIR Resultados AEA (% respecto a nivel basal) A 120 100 80 60 40 VEH EtOH ** 0.75 g/kg 2 g/kg -60 -40 -20 0 *** ** * 20 40 60 80 100 120 Tiempo (min) B GLU (% respecto nivel basal) MENÚ 160 140 120 100 80 60 40 * ** *** * * * * VEH EtOH -60 - 40 -20 0 20 40 60 80 100 120 Tiempo (min) Figura 15. La administración aguda de etanol disminuye la liberación de anandamida y glutamato en el estriado ventral. A. Efectos producidos a lo largo del tiempo sobre los niveles de AEA. La administración de dosis de 0.75 g/kg de etanol (n = 6) producen un leve pero no significativo descenso en el contenido de AEA, mientras que dosis de 2 g/kg (n = 5) sí generan una disminución significativa que persiste al menos 120 minutos tras la administración de etanol. Los datos son las medias + SEM del porcentaje de la concentración de AEA respecto a los niveles basales. B. Efectos sobre los niveles de glutamato tras la administración de etanol. Dosis de 2 g/kg (n = 5) generan un descenso significativo en los niveles de glutamato. Los datos son las medias + SEM del porcentaje de las concentraciones de glutamato respecto a niveles basales. (*) p < 0.05 determinado mediante análisis post hoc tras ANOVA. - 130 - MENÚ SALIR Resultados 1.8 - Medida de la Temperatura Corporal y la Actividad Locomotora Tanto los cannabiniodes como el alcohol producen una disminución de la temperatura corporal y la actividad locomotora. Con el fin de comprobar la existencia de posibles efectos aditivos tras la administración de etanol y del inhibidor del transportador de anandamida, AM404, sobre la hipotermia e hipolocomoción, se ha inyectado i.p. el inhibidor AM404 (2 mg/kg) a ratas 30 minutos antes de inyectarles i.p. la solución de etanol (2 g/kg). Los resultados obtenidos muestran que, al administrarse ambas soluciones, se obtienen resultados similares a cuando se inyecta individualmente el compuesto AM404, generándose hipolocomoción (figura 16A) e hipotermia (figura 16B). El pretratamiento con AM404 no potencia la hipolocomoción inducida por etanol aunque sí incrementa levemente la hipotermia. B Temperatura Rectal (ºC) Distancia Recorrida (cm) A 10000 8000 6000 4000 2000 0 5 30 60 120 Tiempo tras inyección (seg) 38 37 36 35 34 33 5 30 60 120 Tiempo tras inyección (seg) Vehículo AM404 (2 mg/kg) Etanol (2 g/kg) Etanol (2 g/kg) + AM404 (2 mg/kg) Figura 16. Efectos a lo largo del tiempo de la administración aguda de AM404 (2 mg/kg), 30 minutos antes de la de etanol (2 gr/kg), sobre la actividad locomotora (A) y la temperatura corporal (B) de ratas Wistar. El inhibidor AM404 no potencia ninguno de los efectos del etanol (hipotermia e hipomovilidad). Los datos son las medias + SEM de 8-10 muestras por grupo. - 131 - SALIR Resultados TRATAMIENTO CRÓNICO DE ETANOL 2.1 - Medida de los niveles de ARNm del Sistema Endocannabinoide Varios trabajos describen que, tras el tratamiento crónico de etanol, se produce una elevación de los niveles de endocannabinoides (Basavarajappa y Hungund, 1999) lo cual podría deberse bien a alteraciones en su síntesis o degradación. Es por ello que se han analizado los niveles de expresión de ARNm de enzimas implicadas en ambos procesos: NAPE-PLD, en el caso de la síntesis, y FAAH y MAGL, en el caso de la degradación. Por otro lado, también se han estudiado los niveles de expresión del receptor cannabinoide CB1 ya que existen trabajos que describen una regulación a la baja de sus niveles así como una activación en cerebro de ratón (Basavarajappa et al., 1998; Basavarajappaa y Hungund, 1999) tras tratamiento crónico de etanol. La cuantificación de los niveles de ARNm, tras la administración crónica de etanol, reveló la existencia de una disminución significativa de la expresión de CB1 en cerebelo. Sin embargo, en hipocampo no varía y en estriado se observa un aumento de su expresión (figura 17A). En cuanto a la expresión de las enzimas de degradación de endocannabinoides, no se observan cambios en la expresión de FAAH (figura 17B) ni de MAGL a excepción del estriado ventral donde están incrementados los niveles de ARNm de MAGL (figura 17C). Por último, el análisis de los niveles de NAPE-PLD (figura 17D) revela un incremento en su expresión tanto en hipocampo como en estriado siendo más pronunciada en esta última área cerebral. A 300 Ratio CB1/ GUS MENÚ 240 ** Salino Etanol 180 120 60 * 0 CEREBELO ESTRIADO - 132 - HIPOCAMPO SALIR Resultados B Ratio FAAH/ GUS 1500 1200 Salino Etanol 900 600 300 0 CEREBELO ESTRIADO HIPOCAMPO C Ratio MAGL/ GUS 500 Salino ** Etanol 250 0 CEREBELO ESTRIADO HIPOCAMPO D 7 Ratio NAPE- PLD/ GUS MENÚ Salino Etanol ** 5.25 * 3.5 1.75 0 CEREBELO ESTRIADO HIPOCAMPO Figura 17. Medida de los niveles de expresión del ARNm de CB1 (A), FAAH (B), MAGL (C) y NAPE-PLD (D) en cerebelo, hipocampo y estriado de ratas sometidas a tratamiento crónico de etanol (2 g/kg) mediante inyección intraperitoneal cada 12h durante 15 días. Los datos están expresados como medias + SEM y relativizados según los niveles de expresión de la β-glucoronidasa (GUS). (*) p < 0.05; (**) p < 0.01; análisis realizado mediante test t-Student; n = 6 / grupo. - 133 - MENÚ SALIR Resultados 2.2 - Inmunohistoquímica de CB1 y FAAH Con la finalidad de dilucidar si existe correlación entre los datos obtenidos de expresión génica y la expresión proteica, tras la administración crónica de etanol, se realizaron inmunohistoquímicas sobre secciones histológicas. Las cuantificaciones realizadas para el receptor CB1 revelan la existencia de un aumento, del grupo etanol respecto al control, en cerebelo (capas molecular y de Purkinje; figura 18) y en el giro dentado hipocampal (capa granular; figura 19). No se encontraron diferencias estadísticamente significativas ni en el accumbens shell del estriado ventral (figura 20) ni en el pálido ventral (figura 21). Las cuantificaciones realizadas para la enzima FAAH revelaron la existencia de una disminución, del grupo etanol respecto al control, en la capa molecular de la corteza cerebelar (figura 22) aunque no se aprecian diferencias en la capa de Purkinje. Se observa también una disminución en el accumbens shell del estriado ventral (figura 25) y en el pálido ventral (Figura 26) y no se observan diferencias en la formación hipocampal (Figuras 23 y 24) aunque sí haya una fuerte tendencia a disminuir tras el tratamiento crónico en la capa granular del giro dentado (p = 0.0506) y una ligera tendencia a disminuir en la capa piramidal de CA4 (p = 0.1396). - 134 - MENÚ SALIR CB1 en Cerebelo Etanol CP CM Control Control Densidad Óptica (ua) 150 125 *** * Etanol 100 75 50 25 0 Capa Molecular Capa Purkinje Figura 18. Cuantificación mediante densitometría digital del receptor CB1 en el cerebelo (capas molecular, CM, y de Purkinje, CP) de ratas con tratamiento crónico de etanol versus ratas control. Diferencias estadísticamente significativas para p< 0.05(*) y p< 0.001 (***). U.a.= unidades arbitrarias. - 135 - SALIR CB1 en Giro Dentado Etanol CG Control Capa Granular Densidad Óptica (ua) MENÚ 100 * 80 60 40 20 0 CONTROL ETANOL Figura 19. Cuantificación mediante densitometría digital del receptor CB1 en la capa granular (CG) del giro dentado hipocampal de ratas con tratamiento crónico de etanol versus ratas control. Diferencia estadísticamente significativa para p< 0.05 (*). U.a.= unidades arbitrarias. - 136 - SALIR CB1 en Estriado Ventral Etanol Control AcbSh Accumbens Shell Densidad Ó ptica (ua) MENÚ 150 125 100 75 50 25 0 CONTROL ETANOL Figura 20. Cuantificación mediante densitometría digital del receptor CB1 en el accumbens shell (AcbSh) del estriado ventral de ratas con tratamiento crónico de etanol versus ratas control. U.a.= unidades arbitrarias. - 137 - SALIR CB1 en Pálido Ventral Etanol PV Control 100 Densidad Óptica (ua) MENÚ 80 60 40 20 0 CONTROL ETANOL Figura 21. Cuantificación mediante densitometría digital del receptor CB1 en el pálido ventral (PV) de ratas con tratamiento crónico de etanol versus ratas control. U.a.= unidades arbitrarias. - 138 - SALIR FAAH en Cerebelo Etanol CP CM Control Control 120 Densidad Óptica (ua) MENÚ Etanol 100 80 * 60 40 20 0 Capa Molecular Capa Purkinje Figura 22. Cuantificación mediante densitometría digital de FAAH en el cerebelo (capas molecular, CM, y de Purkinje, CP) de ratas con tratamiento crónico de etanol versus ratas control. Diferencia estadísticamente significativa para p< 0.05 (*). U.a.= unidades arbitrarias. - 139 - SALIR FAAH en Giro Dentado Etanol CG Control Capa Granular Densidad Óptica (ua) MENÚ 100 80 60 40 20 0 CONTROL ETANOL Figura 23. Cuantificación mediante densitometría digital de FAAH en la capa granular (CG) del giro dentado de ratas con tratamiento crónico de etanol versus ratas control. U.a.= unidades arbitrarias. - 140 - SALIR FAAH en CA4 Etanol CP Control Capa Piramidal Densidad Óptica (ua) MENÚ 125 100 75 50 25 0 CONTROL ETANOL Figura 24. Cuantificación mediante densitometría digital de FAAH en la capa piramidal (CP) de CA4 de ratas con tratamiento crónico de etanol versus ratas control. U.a.= unidades arbitrarias. - 141 - SALIR FAAH en Estriado Ventral Etanol AcbSh Control Accumbens Shell Densidad Óptica (ua) MENÚ * 100 80 60 40 20 0 CONTROL ETANOL Figura 25. Cuantificación mediante densitometría digital de FAAH en el accumbens shell (AcbSh) del estriado ventral de ratas con tratamiento crónico de etanol versus ratas control. Diferencia signficativa para p< 0.05 (*). U.a.= unidades arbitrarias. - 142 - SALIR FAAH en Pálido Ventral Etanol PV Control Densidad Óptica (ua) MENÚ * 80 60 40 20 0 CONTROL ETANOL Figura 26. Cuantificación mediante densitometría digital de FAAH en pálido ventral (PV) de ratas con tratamiento crónico de etanol versus ratas control. Diferencia significativa para p< 0.05 (*). U.a.= unidades arbitrarias. - 143 - MENÚ SALIR Resultados 3. DISCUSIÓN PARCIAL El tratamiento agudo de etanol, según los resultados obtenidos, va acompañado de una bajada de los niveles de endocannabinoides, lo cual no podría deberse ni a la activación de FAAH ni a cambios en la actividad de NAT, por lo que cabe pensar que se deba a una disminución en la actividad de la enzima NAPE-PLD como principal fuente de liberación de aciletanolamidas (Giuffrida et al., 1999; Hansen et al., 1997; Stella y Piomelli, 2001). La acción del tratamiento agudo de etanol podría afectar a la liberación de neurotransmisores capaces de interaccionar con receptores acoplados a NAPE-PLD y, de esta manera, modificar su actividad sin que ello deba llevar asociado un cambio en la producción de dicha enzima. El etanol podría afectar a la actividad de NAPE-PLD por inhibición de la liberación presináptica de glutamato y acetilcolina los cuales son responsables de la activación de PLD (Maejima et al., 2001; Wilson y Nicoll, 2001). En este sentido los resultados obtenidos en los experimentos de microdiálisis confirman esta hipótesis ya que disminuyen paralelamente los niveles extracelulares de Glu y AEA en el estriado ventral. Por el contrario, está descrito que el tratamiento crónico de etanol está asociado con un aumento en los niveles de AEA y de su precursor NAPE en el linaje celular de neuroblastoma humano SK-N-SH (Basavarajappa y Hungund, 1999) y con un incremento en los niveles de otro endocannabinoide, el 2-araquidonilglicerol (2-AG), en neuronas granulares cerebelares (Basavarajappa et al., 2000). Esto concuerda con la disminución encontrada en los niveles proteicos de FAAH y el incremento en la expresión del ARNm en la enzima NAPE-PLD, tanto en estriado como hipocampo tras el tratamiento crónico de etanol realizado en la presente tesis y se podría explicar adicionalmente, según los resultados obtenidos, por una inhibición de la enzima MAGL. Otra posibilidad podría ser que el alcohol modificase la actividad de síntesis del 2-AG de las dos isoformas, α y β, de la lipasa de diacilglicerol (DAGL) aunque dicho estudio no se ha abordado en la presente tesis. - 144 - MENÚ SALIR Resultados MODELOS ANIMALES DE ALCOHOLISMO Una vez identificadas las acciones del alcohol sobre los elementos del sistema endocannabinoide, se procedió al análisis funcional de dicho sistema en dos modelos de ratas con preferencia alcohólica, las líneas AA/ANA y la línea msP. 4.1. Ratas AA y ANA Las ratas que tienen preferencia por el alcohol (AA) y las que lo rechazan (ANA) fueron proporcionadas por el Dr. Petri Hyytia (Instituto Nacional de la Salud, Helsinki). 4.1.1 - Medida de los niveles de ARNm del Sistema Endocannabinoide En la corteza prefrontal (CPF) de las ratas AA se encuentran disminuidos los niveles de las enzimas FAAH (62% respecto a la línea ANA; F[1,12] = 18.1; p < 0.001), y MAGL (19% respecto a la línea ANA; F[1,139] = 4.81; p < 0.05). A su vez, se observa la existencia de especificidad regional ya que ni en cerebelo ni en estriado existen diferencias en su expresión (figura 27). La expresión del ARNm de NAPE-PLD o del receptor CB1 no varía en ninguna de las áreas cerebrales estudiadas (esto es, ni en cerebelo, ni estriado, ni hipocampo ni corteza prefrontal) entre las líneas comparadas. A B 5000 AA ANA 600 400 200 0 Ratio FAAH/ GUS Ratio CB1/ GUS 800 4000 3000 2000 ** 1000 0 CPF Cerebelo CPF - 145 - Cerebelo MENÚ SALIR Resultados D Ratio MAGL/ GUS 120 AA ANA * 90 60 30 0 CPF Ratio NAPE-PLD/ GUS C Cerebelo 10 7.5 5 2.5 0 CPF Cerebelo Figura 27. Medida de los niveles de expresión de ARNm mediante PCR a tiempo real de CB1 (A), FAAH (B), MAGL (C) y NAPE-PLD (D) en la corteza prefrontal (CPF) y cerebelo de las ratas AA y de las ratas ANA. Los datos están relativizados respecto a la expresión del ARNm de la β-glucoronidasa (GUS) y son la media + SEM; (*) p < 0.05; (**) p < 0.01 versus ratas ANA; n = 7-8 por grupo. 4.1.2 - Medida de la Actividad Enzimática FAAH La actividad de la enzima FAAH, en condiciones saturantes, es aproximadamente un 30% (F[1,10] = 9.63; p < 0.01) menor en la corteza prefrontal (CPF) de las ratas AA respecto de las ANA, lo cual concuerda con los datos de expresión de ARNm obtenidos. Sin embargo, no varía su actividad ni en el estriado ni en el cerebelo de estos animales (figura 28). Figura 28. Análisis bioquímico de la actividad FAAH en membranas cerebrales de ratas AA (barras negras) y ANA (barras blancas). Se aprecia un descenso en la corteza prefrontal (CPF) de las ratas AA, usando como sustrato araquidonil-[1-3H]-etanolamida (media + SEM en pmol/mg proteína/min), (*) p < 0.05 versus ratas ANA; n = 6-8 por grupo. - 146 - MENÚ SALIR Resultados 4.1.3 - Medida de los Niveles de Endocannabinoides Los niveles de 1-AG y 2-AG están significativamente elevados en la corteza prefrontal de las ratas AA respecto a las ANA aunque no varían los de AEA (tabla 4). Tampoco se encuentran diferencias en los niveles de ninguno de los endocannabinoides ni en estriado ni en cerebelo (datos no mostrados). Niveles endocannabinoides (pmol/mg tejido) AA ANA 1 - AG 5.3 + 0.6 2 - AG ** 3.5 + 0.31 AEA 0.47 + 0.04 * 0.367 + 0.05 0.31 + 0.03 0.44 + 0.02 Tabla 4. Medida de los niveles de endocannabinoides en la corteza prefrontal de las ratas AA y ANA. Los niveles de 1-AG (araquidonilglicerol) y 2-AG están significativamente elevados en las ratas AA respecto a las ANA no existiendo diferencias en los de AEA (1-AG: F[1,14] = 14.3, (**) p < 0.002; 2-AG: F[1,15] = 11.8, (*) p < 0.005; AEA: F[1,17] = 1.8). En el resto de áreas cerebrales (cerebelo y estriado) no se aprecian diferencias en ninguno de los endocannabinoides analizados; n = 8-10. 4.1.4 – Estado Funcional del Receptor Cannabinoide CB1 Con el fin de valorar el estado funcional del receptor CB1 se midieron el número de sitios de unión y su capacidad de acoplamiento a proteínas G. Las preparaciones de membranas de las ratas AA, a concentraciones saturantes de SR141716A (10 mM), muestran un 20% menos de sitios de unión en la corteza prefrontal respecto a las ratas ANA (F[1,13] = 5.6, p < 0.05), y no varían ni en cerebelo ni en estriado dorsal. A su vez, la incorporación de [35S]GTPγS (mediante estimulación con WIN 55,212-2) se encuentra disminuida en la corteza prefrontal de las ratas AA no variando ni en el estriado ni en el cerebelo de las mismas. - 147 - MENÚ SALIR Resultados Densidad Acoplamiento Regiones Cerebrales ANA AA ANA AA Corteza Prefrontal 680.0 + 52.0 546.0 + 27.0* 203.1 + 12.9 170.5 + 9.6* Estriado Dorsal 2040.0 + 156.0 1742.0 + 132.0 195.8 + 18.4 161.2 + 8.9 Cerebelo 1462.0 + 190.0 1825.0 + 240.0 193.4 + 21.1 187.4 + 17.3 Tabla 5. Medida de los sitios de unión a CB1 con [3H]-SR141716A (10 nM). Los datos son las medias + SEM en fmol/mg proteína (columnas de la izquierda). Medida de la incorporación de [35S]GTPγS inducida por WIN 55,212-2 (10 mM) como porcentaje de unión estimulada (medias de % + SEM). (*) p < 0.05 versus ratas ANA; n = 6 - 8 por grupo. 4.1.5 - Medida de la Expresión Génica del Receptor CB1 mediante Hibridación in situ El patrón de expresión del ARNm de CB1 observado en las ratas AA y ANA concuerda con estudios previos realizados en otras cepas de ratas (Mailleux y Vanderhaeghen, 1992; Hohmann y Herkenham, 2000; Matsuda et al., 1990), no existiendo diferencias entre ambas líneas en la corteza prefrontal. En hipocampo, existen diferencias significativas aumentando sus niveles de expresión en la subregión CA4 de las ratas AA (tabla 6). Sin embargo, en los núcleos caudado-putamen se observa un gradiente de intensidad de ARNm de CB1 dorso-mediolateral o dorsoventrolateral, dependiendo del nivel Bregma de referencia. La región con mayor intensidad de señal en el caudado-putamen se encuentra rostralmente en la porción mediolateral (+ 1.0 a + 0.2 mm, coordenadas Bregma; figura 29) y caudalmente en la porción ventrolateral (- 0.4 a - 1.8 mm, coordenadas Bregma). Existe una expresión diferencial entre ambas líneas de ratas en el caudado-putamen a nivel + 1.0 a + 0.2 mm, coordenadas Bregma (media de densidad óptica x103 + SEM, n = 8; AA: 78 + 3 vs ANA: 106 + 6; F[1,14] = 17.39, ajuste de Bonferroni p < 0.002). No se encontraron diferencias en la expresión de CB1 ni en la corteza cingulada, ni frontal ni frontoparietal. - 148 - MENÚ SALIR Resultados Región Cerebral ANA AA Corteza Cingulada 120 ± 5 128 ± 6 Corteza Frontal 86 ± 2 91 ± 4 Corteza Frontoparietal 63 ± 1 64 ± 5 Caudado-putamen 106 ± 6 78 ± 3** Caudado-putamen Ventrolateral 182 ± 10 157 ± 6 Septum medial-Banda diagonal 76 ± 3 68 ± 2 Núcleo Amigdaloide Central 105 ± 3 113 ± 4 Núcleo Amigdaloide Basolateral 212 ± 5 193 ± 8 CA1 153 ± 6 138 ± 6 CA3 113 ± 5 124 ± 5 CA4 121 ± 3 145 ± 5** Giro Dentado 126 ± 5 129 ± 6 Subregiones de Hipocampo Dorsal: Tabla 6. Cuantificación de los niveles de expresión del ARNm de CB1 mediante hibridación in situ. Se observa una disminución en la expresión del ARNm de CB1 en los núcleos caudado-putamen (CPu) y un aumento en la subregión CA4 del hipocampo dorsal de ratas AA comparadas con ratas ANA. Los datos están expresados como valores de densidad óptica (x103, media + SEM). Análisis estadístico realizado mediante test t seguido de corrección de Bonferroni/Holm. (**) p < 0.01; n = 8 por grupo. Figura 29. Fotomicrografías en campo oscuro de películas autorradiográficas de hibridaciones in situ que muestran una disminución de los niveles de expresión del receptor CB1 en el caudado-putamen (CPu) de ratas AA (derecha) comparadas con las ratas ANA (izquierda). Corte a + 1.0 mm, coordenadas Bregma. - 149 - MENÚ SALIR Resultados 4.1.6 - Efecto del antagonista SR141716A sobre la Autoadministración de Alcohol Las ratas AA respondieron al condicionamiento operante tanto con etanol al 10% (v/v) como con sacarina al 0.05% (p/v). Durante una sesión inicial de 30 minutos, la autoadministración de alcohol se produjo en cantidades de 0.86 + 0.07 g/kg (media + SEM, n = 20), lo cual llevó a una concentración media de etanol en sangre de 5.85 + 0.85 mM tras finalizar la sesión. La concentración de etanol en sangre se correlacionaba con la ingesta de etanol durante la sesión (Pearson r = 0.84; p < 0.01), mostrando que el etanol liberado del recipiente era de hecho consumido. Las inyecciones intraperitoneales (i.p.) de SR141716A suprimen la respuesta a etanol de forma proporcional a la dosis administrada del antagonista (F[3,33] = 21.44, p < 0.0001; Figura 30). Las inyecciones en la corteza prefrontal (Figura 31a) reproducen el efecto sistémico y la respuesta se encuentra significativamente suprimida (F[2,16] = 16.13, p < 0.0001), mientras que las disminuciones por vía intraestriatal (Figura 31b) no llegan a ser significativas (F[2,18] = 3.28). Las zonas microinyectadas de la corteza prefrontal y estriado se muestran en las figuras 31c y d, respectivamente. Para las inyecciones de SR141716A en la corteza prefrontal medial, se verificaba que la cánula estuviese ubicada dentro de la región desde 2.7 a 3.7 mm anterior a bregma (según el atlas de Paxinos y Watson, 1997) y las posiciones para las inyecciones intraestriatales se verificaron durante el consumo de etanol. La inyección del inhibidor URB597 de FAAH en la corteza prefrontal, de ratas Wistar no seleccionadas, incrementa la respuesta a etanol (F[3,39] = 5.2, p < 0.01; Figura 32). Análisis posteriores muestran diferencias significativas entre la dosis más elevada (4 μg) y el grupo control (p < 0.05) o la dosis más baja (0.4 μg; p < 0.05). Figura 30. Efecto del tratamiento previo con SR141716A sobre la autoadministración de etanol. Los datos están expresados como media (+ SEM) de respuestas a la solución de etanol al 10% durante sesiones de 30 min. (***) p < 0.001 vs vehículo. - 150 - MENÚ SALIR Resultados Figura 31. Efectos de las microinyecciones de SR141716A en la corteza prefrontal (a) y estriado (b) sobre la respuesta a etanol. Los datos están expresados como medias (+ SEM) de las respuestas a solución de etanol al 10% durante sesiones de 30 minutos. (*) p < 0.05, (**) p < 0.001 vs vehículo. La zonas inyectadas con cánula de la corteza prefrontal y estriado se muestran en (c) y (d), respectivamente. Los números indican la posición (en mm) de las secciones coronales según las coordenadas Bregma. - 151 - MENÚ SALIR Resultados Figura 32. Efectos de la inyección local del inhibidor de FAAH URB597 (0, 0.4 y 4 μg) en la corteza prefrontal sobre la autoadministración de etanol. Los datos están expresados como media (+ SEM) de las respuestas a la solución de etanol al 10% durante sesiones de 30 minutos. (*) p < 0.05; test de Tukey; n = 7- 9 por grupo. 4.2. Ratas msP y Wistar Las ratas msP son una cepa de ratas con preferencia al alcohol derivadas de la línea de ratas sP (Sardinian-Preferring, Universidad de Cagliari) que han sido criadas durante más de 30 generaciones en la Universidad de Camerino (Italia) y puestas a disposición de nuestro grupo de colaboración. 4.2.1 - Medida de los niveles del ARNm de CB1 mediante Hibridación in situ Los niveles de expresión del ARNm de CB1, medidos mediante hibridación in situ, muestran fuertes diferencias entre las ratas msP y Wistar en áreas cerebrales como la porción rostral de los núcleos caudado-putamen (Cpu), la corteza frontoparietal (fpCx) y las subregiones CA1 y CA4 del hipocampo (13-26% menores en Wistar) tal y como se deduce del análisis estadístico mediante el test t con correción de Bonferroni/Holm (CPu rostral: F[1,14] = 24.0, p < 0.01 corregida; fpCx: F[1,13] = 19.6, p < 0.01 corregida; CA1: F[1,14] = 18.9, p < 0.01 corregida; CA4: F[1,14] = 7.8, p < 0.05 corregida; Figura 33A). En ratas msP, que consumieron voluntariamente etanol durante periodos de 18 días (ingesta de etanol que aumentaba de forma progresiva durante este periodo y entre - 152 - MENÚ SALIR Resultados rangos de 4.93 + 0.39 g/kg en el día 1 y de 8.36 + 0.36 g/kg en el día 18), se observa una regulación a la baja de la expresión del receptor CB1 en las regiones del prosencéfalo analizadas. Sin embargo, este efecto sólo era significativo en la porción rostral de los núcleos caudado-putamen (un 27% de regulación a la baja; test t con corrección de Bonferroni/Holm: Cpu rostral: F[1,14] = 18.0, p < 0.01 corregida; Figura 33B, Tabla 7). A ARNm de CB1 (% msP) 125 msP Wistar 100 75 50 25 0 ccg cf cfp CP CP CP ml (+ 2.2) (- 0.4) (- 0.4) CA1 CA3 CA4 GD ARNm de CB1 (% msP) B msP msP + EtOH 125 100 75 50 25 0 ccg cf cfp CP CP CP ml (+ 2.2) (- 0.4) (- 0.4) CA1 CA3 CA4 GD Figura 33. Niveles de expresión relativos del ARNm de CB1 en diferentes áreas cerebrales de las ratas mSP y Wistar (A), y de las ratas mSP control y tras 18 días de ingesta de etanol (B). Los datos están expresados como medias de porcentaje + SEM, n = 7-8. Los análisis estadísticos se han realizado mediante el test t con correción de Bonferroni/Holm (* p < 0.05; ** p < 0.01; *** p < 0.001 vs ratas mSP). Corteza frontal (cf); corteza cingulada (ccg); corteza frontoparietal (cfp); núcleos caudado-putamen (CP) a nivel de +2.2 coordenadas Bregma (CP, +2.2), a nivel -0.4 mm (CP, -0.4) y la porción mediolateral de CP a nivel -0.4 mm (CP ml, -0.4); subregiones del hipocampo dorsal (CA: áreas Cornus Ammon, de CA1 a CA4; giro dentado, GD). - 153 - MENÚ SALIR Resultados CP CP CP ml (+2.2) (– 0.4) (– 0.4) 2.7 ± 0.1 9.6 ± 0.3 20.1 ± 0.8 50.1 ± 2.6 7.9 ± 0.5 3.7 ± 0.2 12.8 ± 0.6 20.4 ± 0.7 54.7 ± 1.9 6.9 ± 0.6 3.1 ± 0.2 9.3 ± 0.6 17.4 ± 1.1 44.4 ± 2.7 Ratas Tratamiento ccg cf cfp Wistar Ninguno 11.8 ± 0.6 6.8 ± 0.4 msP Ninguno 11.9 ± 0.5 Etanol (10 días) 10.5 ± 0.4 msP Ratas Tratamiento CA1 CA3 CA4 GD Wistar Ninguno 11.2 ± 0.4 16.4 ± 1.6 7.4 ± 0.4 20.9 ± 1.1 msP Ninguno 15.0 ± 0.8 19.1 ± 1.8 8.9 ± 0.4 23.8 ± 0.9 Etanol (10 días) 11.8 ± 0.8 18.4 ± 1.1 7.6 ± 0.6 21.5 ± 0.5 msP Tabla 7. Niveles de ARNm de CB1 en diferentes áreas cerebrales de las ratas mSP y Wistar. Los datos se expresan como valores de densidad (nCi/g, medias + SEM; n = 7-8). Corteza frontal (cf); corteza cingulada (ccg); corteza frontoparietal (cfp); núcleos caudado-putamen (CP) a nivel de +2.2 coordenadas Bregma (CP, +2.2), a nivel -0.4 mm (CP, -0.4) y la porción mediolateral de CP a nivel -0.4 mm (CP ml, -0.4); subregiones del hipocampo dorsal (CA: áreas Cornus Ammon, de CA1 a CA4; giro dentado, GD). - 154 - MENÚ SALIR Resultados 4.2.2 - Medida de los niveles de ARNm del Sistema Endocannabinoide Existe una disminución en la expresión del ARNm de FAAH (figura 34C) en el estriado y de MAGL (figura 34D) en corteza prefrontal (CPF) de las ratas msP respecto a las Wistar. No se aprecian diferencias en los niveles de ARNm ni de CB1 (figura 34A), ni de NAPE-PLD (figura 34E) ni de DAGL-α (lipasa α de diacilglicerol, enzima que produce 2-AG; figura 34B) entre las ratas Wistar y msP en ninguna de las áreas cerebrales analizadas. A Ratio CB1 / Ciclofilina-A 14 Wistar 12 msP 10 8 6 4 2 0 Cerebelo Estriado Hipocampo CPF Ratio DAGL- α / Ciclofilina-A B 0.3 Wistar 0.25 msP 0.2 0.15 0.1 0.05 0 Cerebelo Estriado - 155 - Hipocampo CPF MENÚ SALIR Resultados C Wistar Ratio FAAH / Ciclofilina-A 30 msP 25 20 15 ** 10 5 0 Cerebelo Estriado Hipocampo CPF D Wistar Ratio MAGL / Ciclofilina-A 30 msP 25 ** 20 15 10 5 0 Cerebelo Estriado - 156 - Hipocampo CPF MENÚ SALIR Resultados Ratio NAPE-PLD / Ciclofilina-A E Wistar 0.12 msP 0.1 0.08 0.06 0.04 0.02 0 Cerebelo Estriado Hipocampo CPF Figura 34. Medida de los niveles de ARNm de CB1 (A), DAGL-α (B), FAAH (C), MAGL (D) y NAPE-PLD (E) en cerebelo, estriado, hipocampo y corteza prefrontal (CPF) de ratas mSP y Wistar. Los resultados se expresan como medias + SEM relativizadas a la expresión del ARNm de Ciclofilina-A. (*) p < 0.05; (**) p < 0.01 vs ratas Wistar; n = 8. 4.2.3 - Medida de la Expresión Proteica del Sistema Endocannabinoide Con el fin de dilucidar si existe una correlación entre los datos de expresión génica y proteica se realizaron inmunohistoquímicas para los marcadores CB1, FAAH, MAGL y NAPE-PLD a las secciones de cerebro de ratas msP y Wistar. A raíz de los resultados obtenidos en las ratas AA y ANA, se ha estudiado la corteza prefrontal de las ratas msP y Wistar, no encontrándose diferencias estadísticamente significativas para ninguno de los marcadores analizados (figura 35). - 157 - MENÚ SALIR Corteza Prefrontal Wistar msP CB1 FAAH MAGL NAPE-PLD Wistar 250 Densidad Óptica (ua) msP 200 150 100 50 0 CB1 FAAH MAGL NAPE-PLD Figura 35. Cuantificación, mediante densitometría digital, de la expresión proteica de CB1, FAAH, MAGL y NAPE-PLD en la corteza prefrontal de ratas msP y Wistar. - 158 - MENÚ SALIR DISCUSIÓN MENÚ SALIR MENÚ SALIR Discusión 1.- Papel General del Sistema Endocannabinoide en el Alcoholismo En la presente tesis se ha tratado establecer cuáles son las alteraciones que se dan en los componentes del Sistema Endocannabinoide (SEC) en varios modelos animales de alcoholismo, con el fin de intentar dilucidar el papel de dicho sistema en esta adicción. Varios trabajos científicos han demostrado que la administración crónica de alcohol promueve cambios en los niveles de endocannabinoides (Basavarajappa y Hungund, 1999; González et al., 2002) y en esta tesis se demuestra cuáles son los componentes del SEC que podrían ser responsables de tales variaciones. A su vez, se ha podido analizar cuáles son los efectos de la administración aguda de etanol, no descritos previamente, y completar las acciones crónicas de etanol sobre el sistema endocannabinoide (SEC). Por último, se ha caracterizado y analizado las alteraciones en el SEC de modelos animales seleccionados y criados por su preferencia al consumo de etanol (líneas AA y msP) en áreas cerebrales relacionadas con el proceso de adicción a drogas. No existe ningún trabajo científico que describa las alteraciones sufridas tras la exposición aguda de etanol sobre el SEC. En la presente tesis se ha abordado dicho análisis y se ha encontrado, a diferencia de los efectos crónicos del etanol, una disminución en los niveles de endocannabinoides (AEA y PEA) no asociada a alteraciones en la expresión de ninguna de las enzimas implicadas en la síntesis y degradación de endocannabinoides, así como la inexistencia de alteraciones en la expresión génica o proteica de CB1, aunque se desconoce la posible existencia de alteraciones a nivel funcional lo cual sería objeto de futuros estudios. Los receptores CB1 son abundantes en regiones cerebrales que juegan un papel importante en la regulación de los circuitos de recompensa, comúnmente activados por drogas de abuso como el etanol (Rodríguez de Fonseca y Navarro, 1998). El papel de dicho receptor durante la intoxicación crónica de etanol ha sido muy investigado, describiéndose una desensibilización de CB1 (Basavarajappa et al., 1998) asociada a un incremento en los niveles de endocannabinoides. Ello concuerda con el hecho de que la administración de agonistas de CB1 estimule el consumo, la autoadministración y las propiedades motivacionales del etanol, mientras que la administración de antagonistas las suprima (Colombo et al., 2005; Maldonado et al., 2006; Cippitelli et al., 2005). - 161 - MENÚ SALIR Discusión Los estudios en los que se ha bloqueado (Arnone et al., 1997; Rodríguez et al., 1999; Colombo et al., 1998) o inactivado genéticamente el receptor CB1 (Hungund et al., 2003; Wang et al., 2003; Naasila et al., 2004) implican a la señalización endocannabinoide en la regulación de la autoadministración de etanol. El perfil de expresión en cerebro, mas allá de indicar la posibilidad de una desregulación en la expresión de los genes endocannabinoides y las señales de transducción bajo su control, podría contribuir a una elevada preferencia al alcohol (Rimondini et al., 2002; Arlinde et al., 2004). En la presente tesis, se ha abordado un estudio más completo de las acciones de crónicas del etanol sobre el SEC en el que se ha podido comprobar que existen otras enzimas relacionadas con la degradación de endocannabinoides (MAGL, FAAH), que podrían ser, al menos en parte, responsables de dichos cambios. Los estudios con animales modelos han demostrado que las drogas, que activan a los receptores cannabinoides en cerebro, son capaces de regular la autoadministración de etanol, cannabinoides y opiáceos. El análisis del SEC de dos líneas de animales, seleccionados y criados por su preferencia al consumo de etanol, permitiría reafirmar la existencia de variaciones a nivel del SEC de la misma índole a las observadas tras la administración crónica de etanol, estableciéndose un vínculo entre dichas alteraciones y la preferencia y consumo de etanol así como facilitando la identificación de las áreas cerebrales relevantes en estos procesos. Los resultados obtenidos en estos animales tienen una gran relevancia en aquellos estudios en humanos sobre la retirada de alcohol ya que los receptores CB1 de rata y humano muestran una gran homología (93% y 97% de identidad nucleotídica y aminoacídica, respectivamente; Ameri, 1999). La identificación de las alteraciones sobre el SEC ayudará al diseño de estrategias farmacológicas específicas dirigidas al tratamiento del alcoholismo y al establecimiento, de nuevo, de la homeostasis del sistema endocannabinoide. - 162 - MENÚ SALIR Discusión 2.- Acciones Agudas del Etanol y Adaptación Crónica 2.1. Acciones Agudas del Etanol Los resultados obtenidos, tras el tratamiento agudo de etanol, sugieren que el etanol afecta a la formación de AEA en cerebro y tejidos periféricos sin que ello se deba a una inducción de la degradación por parte de la enzima FAAH. Los efectos sobre los niveles de AEA tienen su réplica en una segunda aciletanolamida, PEA, cuyas vías de síntesis y degradación se asemejan a las de AEA, lo que sugiere la existencia de un mecanismo común para la disminución de los niveles de ambas aciletanolamidas. El etanol induce una disminución en la formación de AEA en cerebro, plasma y tejido adiposo, limitado a los primeros 90 minutos del experimento. Sin embargo, en hígado y, especialmente, en intestino delgado se observa un remarcable incremento en su liberación tras 90 minutos de la administración de etanol. Estos efectos sugieren la existencia de una selección de dianas a nivel de tejido en el sistema endocannabinoide. La especificidad local de los mecanismos reguladores de la formación de AEA, podría basarse en el papel activador del etanol en el intestino delgado. Recientemente se ha descrito que el intestino delgado produce grandes cantidades de AEA en animales en ayunas (Gómez et al., 2002). Los efectos agudos del etanol sobre la producción de AEA se asemejan a los encontrados tras 18 horas de la privación de comida, probablemente debido a que el etanol podría actuar como una droga hipoglucemiante por su interferencia con el metabolismo de la glucosa (Williams, 1984). Esta hipótesis se vería apoyada por la existencia de un rápido incremento en la expresión génica de la enzima GAPDH, que es crucial para la regulación de los procesos de glucólisis y neoglucogénesis hepática. Los resultados obtenidos tras la administración aguda de etanol y/o el inhibidor del transportador de AEA, AM404, sugieren que el etanol no es responsable directo de la elevación de los niveles de endocannabinoides al no existir efecto aditivo cuando se inyectan ambos compuestos ni sobre la hipolocomoción ni hipotermia, efectos típicos generados por endocannabinoides (Dewey, 1986). Por tanto, cabría esperar que dichas alteraciones se deban a un efecto directo del etanol sobre los sistemas de neurotransmisión, los cuales podrían afectar al metabolismo endocannabinoide. Los experimentos realizados, mediante microdiálisis in vivo, apoyarían dicha hipótesis al - 163 - MENÚ SALIR Discusión disminuir paralelamente tanto la liberación de glutamato como los de AEA tras la administración aguda de etanol. El etanol provocaría una disminución en los niveles de neurotransmisores como glutamato y acetilcolina (Maejima et al., 2001; Wilson y Nicoll, 2001), lo que llevaría a un descenso en la rotura del precursor de anandamida (AEA) y palmitoiletanolamida (PEA), NAPE, mediada por la enzima NAPE-PLD. La exposición crónica a etanol resultaría en el desarrollo de tolerancia y, en consecuencia, en un aumento de la liberación de ambos neurotransmisores (AEA y glutamato) (Dahchour y De Witte, 2000; Imperato et al., 1998), lo cual contribuiría a un incremento en la formación de AEA (Hungund et al., 2002), tal y como está descrito en la bibliografía. El etanol podría inhibir directamente a los receptores metabotrópicos y NMDA de glutamato implicados en la liberación de AEA, dependiente de la enzima PLD, lo que conduciría a una disminución en la rotura del precursor NAPE (Lovinger et al., 1989; Maejima et al., 2001; Hansen et al., 1997). Sin embargo, ciertas evidencias directas apuntan a que el etanol podría alterar la señalización dependiente de PLD, actuando como sustrato metabólico lo que conduciría a la actividad de PLD hacia la producción de fosfatidiletanol (Kötter y Klein, 1999). Dicha hipótesis no podría explicarse según los efectos observados ya que está descrito que NAPE-PLD no usa al etanol como sustrato (Okamoto et al., 2004). Los efectos observados son específicos de tejido lo que indica la existencia de una compleja trama de señales regulando la producción endocannabionide. El hecho de que el hipocampo dorsal sea más sensible a los efectos inhibidores del etanol sobre la formación de AEA y PEA podría ser dependiente de la coexistencia de una importante inervación glutamatérgica y colinérgica, neurotransmisores que estimulan cooperativamente la liberación de AEA (Stella y Piomelli, 2001). Además, todos estos mecanismos podrían interaccionar juntos de manera selectiva según el tejido, de acuerdo con el papel propuesto para las aciletanolamidas como mediadores locales. Un factor adicional que podría contribuir a la disminución de la formación de anandamida sería la respuesta al estrés tras la exposición aguda a etanol (Rivier, 1993). Debido a que el etanol puede activar la actividad del eje hipotalámico-pituitario-adrenal, la elevación en los niveles de corticosterona afectaría a la producción endocannabinoide. Sin embargo, el hecho de que la inmovilización activa, una forma severa de estrés inducida en ratas, sólo disminuya la formación de AEA en la amígdala - 164 - MENÚ SALIR Discusión y no se altere en otras áreas, como el núcleo accumbens y cerebelo, sugiere que la contribución del estrés a la inhibición de la liberación de aciletanolamidas podría ser de menor importancia que la generada por la inhibición, inducida por etanol, sobre la liberación de glutamato y acetilcolina. Dicha hipótesis experimental necesitaría confirmarse en futuras experimentaciones y no se ha abordado como objetivo de la presente tesis. Todos los efectos sobre la formación de AEA y PEA descritas en cerebro se observaron cuando aún existían cantidades sustanciales de etanol sin metabolizar, tal y como se muestra en los niveles plasmáticos (Figura 1A, sección de Resultados). Su rápida desaparición sugiere la existencia de una fuerte regulación en la producción de aciletanolamidas, lo cual concuerda con el papel como mediador de la señalización intercelular. 2.2. Acciones Crónicas del Etanol La inhibición de la formación de anandamida estaría regulada por respuestas homeostáticas contrarreguladoras, que tras repetidas exposiciones a etanol, conduciría a un aumento en producción de precursores (NAPE) y a la liberación de AEA, tal y como está descrito que ocurre en cultivos de neuroblastoma humano (SK-N-SH) y en cerebro (Basavarajappa y Hungund, 1999; Basavarajappa et al., 2000; Hungund et al. 2002, González et al., 2002). La medida de los niveles de expresión génica de las diferentes enzimas de síntesis y degradación de endocannabinoides, tras tratamiento crónico de etanol, revela la existencia de un aumento en los niveles de NAPE-PLD, lo cual podría contribuir a dicho efecto en áreas como estriado e hipocampo. La disminución en los niveles proteicos de FAAH podría contribuir también a la elevación de los niveles de endocannabinoides en el shell del núcleo accumbens, corteza cerebelar (capa molecular) y pálido ventral. Sin embargo, en cultivos de neuronas granulares cerebelares, sometidas a tratamiento crónico de etanol, el incremento en los niveles de AEA y su precursor NAPE no está asociado a una inhibición directa de la actividad de FAAH aunque sí a una inhibición en el transporte de AEA (Basavarajappa - 165 - MENÚ SALIR Discusión et al., 2003) y a una inducción de las enzimas implicadas en su síntesis. Por otro lado, otros trabajos describen la no existencia de alteraciones en los niveles de endocannabinoides (AEA y 2-AG) ni en cerebelo ni estriado (González et al., 2002). En ambos casos ello no eximiría que dicha alteración se produjera en la capa molecular de la corteza cerebelar o en el shell del núcleo accumbens. De manera similar, si se comparan en cerebellum los niveles de proteínas con los de ARNm se observa que, en el caso de CB1, existe un aumento en sus niveles proteicos en dos capas de la corteza cerebelar (capa Molecular y de Purkinje) lo cual, aparentemente, no concordaría con la disminución en los niveles de ARNm de CB1. Esta aparente incongruencia podría tener su explicación en el hecho de que, para la medida de los niveles de ARNm, se toma el cerebelo completo y no se discrimina cuáles son los efectos del etanol en cada capa. Ello podría sugerir la existencia de un posible efecto de ‘dilución’ de los cambios en los niveles de ARNm, en la neuroanatomía cerebelar, ya que podrían darse efectos opuestos en cada estructura cerebelar. Estudios adicionales, mediante hibridaciones in situ, podrían ayudar a desvelar esta incógnita. Otros investigadores sí han acometido dicho estudio en áreas diferentes al cerebelo. Por ejemplo, las hibridaciones in situ de CB1 realizadas en secciones de cerebro de ratas sometidas a tratamiento crónico de etanol (Ortiz et al. 2004), revelan la existencia de diferencias en los niveles de ARNm en la formación hipocampal: existe un aumento en regiones como CA1 y CA2 y una disminución en giro dentado lo que, primero, concuerda con el incremento observado, en la presente tesis, en los niveles proteicos de CB1 en giro dentado (capa granular) y, segundo, vuelve a sugerir la existencia de dianas específicas del etanol, a nivel del SEC, en la neuroanatomía de la formación hipocampal. Está descrito que la exposición crónica a etanol induce un incremento en la liberación neurotransmisores como glutamato y acetilcolina (Dahchour y De Witte, 2000b; Imperato et al., 1998), responsables de la activación de los receptores NMDA y colinérgicos, así como un incremento en los niveles de 2-AG (Basavarajappa et al., 2000) probablemente asociado a la activación de dichos receptores (Dinh et al., 2002, Stella y Piomelli, 2001). La elevación en los niveles de 2-AG estimularía la expresión de MAGL, enzima responsable de la degradación de 2-AG y no de anandamida. Está descrito que la sobreexpresión de dicha enzima afectaría a la inactivación del - 166 - MENÚ SALIR Discusión endocannabinoide 2-AG aunque no a su síntesis mediada por receptor (Dinh et al., 2002). Por tanto, en striatum, sería necesario analizar los niveles de expresión de las enzimas implicadas en la síntesis de 2-AG, como la fosfolipasa-C (PLC) y la lipasa de diacilgliceroles (DAGL), así como realizar una medida de los niveles de 2-AG en dicha área. En la presente tesis se ha abordado principalmente el análisis de una de las dos vías de síntesis de endocannabinoides, la de aciletanolamidas, no realizándose el estudio de la vía de síntesis de 2-AG, la cual podría ser objeto de estudio en futuras investigaciones. El tratamiento crónico de etanol revela también la existencia, en striatum, de un aumento en la expresión génica de CB1, lo cual no concuerda con lo descrito hasta el momento. Teniendo en cuenta que los núcleos caudado y putamen son los que mayor área representan, serían, por tanto, en mayor parte los responsables de dicha alteración. Otros autores describen bien una disminución en la expresión génica de CB1 en caudado-putamen (Ortiz et al., 2004) o bien ningún cambio ni en la expresión génica ni en el número de sitios de unión de CB1 en ninguna de las áreas cerebrales analizadas, entre las que se incluían NAcb y Caudado-Putamen (González et al.; 2002a). No está claro cuáles son los motivos para estas incongruencias en estriado, a nivel del receptor CB1. Quizás se deba a la existencia de diferencias en el protocolo en cuanto a la duración del tratamiento (52 días según Ortiz et al. y 15 días según González et al.), a la concentración de la solución de etanol o al modo de administración de la misma. Ello podría sugerir la necesidad de ampliar la duración del tratamiento para poder observar estos cambios adaptativos en el SEC a nivel de estriado. En resumen, este primer conjunto de experimentos demuestra que la administración aguda de etanol suprime los niveles de endocannabinoides en cerebro y que las neuroadaptaciones, debidas a la exposición crónica, conducen a un incremento permanente de los mismos. Este hecho está de acuerdo con la teoría general homeostática y la teoría de los procesos opuestos (Solomon, 1980). Esta última se basa en que los organismos reaccionan, ante los cambios producidos a causa del medio, intentando mantener cierto equilibrio (principio de homeostasis). Solomon mantiene que cuando un evento externo produce una reacción en los sujetos (Proceso A), los organismos reaccionan produciendo una respuesta contraria (Proceso B) para amortiguar los efectos de la primera. Estos procesos B aparecen un poco más tarde, ya que son una reacción a los primeros, por lo que tardan un poco más en desaparecer - 167 - MENÚ SALIR Discusión dejando una sensación opuesta a la que generó inicialmente el evento externo. Todo ello ratificaría la veracidad de los efectos del etanol, descritos en la presente tesis, sobre el Sistema Endocannabinoide. 3.- Selección de Preferencia Alcohólica y Sistema Endocannabinoide En la presente tesis se ha caracterizado el Sistema Endocannabionide de dos modelos animales seleccionados y criados por su preferencia al alcohol en diferentes laboratorios. Ambas líneas, AA y msP, muestran una disminución en la expresión de las enzimas de degradación de endocannabinoides lo cual podría ser una de las causas que llevarían a la elevación de los niveles de endocannabinoides. Todo ello sugiere la existencia de una fuerte asociación de la preferencia al alcohol con una elevación en los niveles de endocannabinoides en áreas cerebrales implicadas en la recompensa y formación del hábito de consumo, concordando con los efectos descritos tras tratamiento crónico de etanol (Basavarajappa y Hungund, 1999). En la corteza prefrontal (CPF) de las ratas AA se observa una pronunciada disminución en la expresión génica de FAAH y en menor grado de MAGL, no variando la expresión de la enzima NAPE-PLD. Todo ello, junto con el incremento en los niveles de los endocannabinoides (1-AG y 2-AG), sugiere que la degradación de dichos compuestos está alterada en esta área cerebral. Ello conduciría a una elevación local del tono endocannabinoide lo cual sería un efecto esperable para compensar la desensibilización del receptor CB1. De hecho, existe una disminución en la densidad y acoplamiento de dicho receptor en la CPF de las ratas AA, aunque su expresión génica no muestra diferencias con las ratas ANA. Esta disociación entre la unión de CB1 y su expresión, en ratas AA, podría tener su explicación en que los receptores CB1 se localizan normalmente en los terminales axónicos de las neuronas de proyección (Tsou et al., 1998), por lo que la medida de la unión de CB1 sería debida, en gran parte, a los receptores de fibras aferentes procedentes de otras áreas cerebrales. En el estriado dorsal de las ratas AA se ha encontrado una disminución en los niveles de ARNm de CB1, no variando la expresión de enzimas de degradación como FAAH o MAGL entre ambas líneas. La menor expresión de CB1 no se acompaña con una disminución en su unión. Gran parte de la expresión del ARNm del receptor CB1 - 168 - MENÚ SALIR Discusión en el estriado dorsal depende de su expresión en las neuronas de proyección estriatales (Hohmann y Herkenham, 2000). Por tanto, las diferencias observadas en los niveles de ARNm de CB1 podrían afectar principalmente a la síntesis de receptores CB1 ubicados en los terminales eferentes que proyectan desde las regiones estriatales. Varios trabajos relacionan las diferencias en el Sistema Endocannabinoide con las encontradas en los fenotipos relacionados con el consumo de alcohol. Los ratones C57BL/6 tienen menor número de sitios de unión de CB1 aunque una mayor afinidad y acoplamiento que los ratones con baja preferencia al alcohol DBA/2 (Basavarajappa y Hungund, 2002; Hungund y Basavarajappa, 2000). En ratas Wistar, se han descrito gran variedad de cambios adaptativos en el Sistema Endocannabinoide tras ser sometidos a tratamiento crónico de etanol (González et al., 2004; Basavarajappa y Hungund, 2005). Existen trabajos que describen, en ratones nulos para el gen de FAAH (Walker et al., 2005; Basavarajappa et al., 2006; Blednov et al., 2007), un incremento en el consumo de etanol. Los resultados presentados en esta tesis, muestran una disminución en la expresión y actividad de FAAH en la CPF de ratas AA y un incremento de respuestas al etanol, en ratas Wistar, tras la administración intraprefrontal del inhibidor de FAAH, URB597. Todas estas evidencias apoyarían la hipótesis de que una disminución en la degradación endocannabinoide, en ciertas regiones cerebrales, serían claves para incrementar el consumo de etanol. Las diferencias encontradas en el Sistema Endocannabinoide, a nivel de la CPF, se añaden a la lista de aspectos neuroquímicos y genéticos que se han segregado mediante la cría selectiva de las líneas AA y ANA (Sinclair et al., 1989; Gianoulakis et al., 1992; Arlinde et al., 2004), siendo probablemente, la mayoría de ellos, resultado de una cosegregación al azar. El análisis de la expresión del receptor CB1 reveló la existencia de una disminución en los niveles de ARNm en los núcleos caudado-putamen, de las ratas AA respecto a las ratas ANA, no existiendo diferencias en su porción ventrolateral. De la misma manera, en hipocampo sólo se observa un aumento en su expresión en el área CA4 de ratas AA, no existiendo diferencias en las demás regiones del hipocampo analizadas. Todo ello sugeriría la existencia de una posible compartimentalización en ambas áreas cerebrales y una posible selectividad de dianas del etanol a nivel de Sistema Endocannabionide, lo cual debería ser estudiado en más profundidad en futuras investigaciones. - 169 - MENÚ SALIR Discusión Con el fin de demostrar que una sola diferencia entre las líneas podría contribuir casualmente a la preferencia por el alcohol, se ha realizado un tratamiento farmacológico que revierte dicho fenotipo. La administración sistémica del antagonista de CB1, SR141716A, produce una notable reducción en la autoadministración de etanol en las ratas AA. Las dosis necesarias para provocar dicho efecto son menores a las empleadas en ratas no seleccionadas (Rodríguez et al., 1999), lo que sugiere la existencia de un incremento de la sensibilidad a los efectos de este inhibidor en ratas AA. Este resultado concuerda con estudios previos en los que se describen una disminución en la ingesta voluntaria de alcohol o de autoadministración tras la administración de SR141716A (Arnone et al., 1997; Colombo et al., 1998b; Rodríguez et al., 1999). En esta línea, se ha demostrado que los ratones ‘knockout’ de CB1 tienen reducido tanto el consumo como la preferencia por el etanol (Hungund et al., 2003; Wang et al., 2003; Naasila et al., 2004). Estos estudios apoyan la noción de que el Sistema Endocannabinoide está implicado en la regulación del consumo de etanol. En la presente tesis, se demuestra por primera vez que las diferencias existentes en el metabolismo endocannabinoide contribuyen a la preferencia al alcohol y que dicho fenotipo puede revertir si se trata la diana que causa esta anormalidad preexistente. El inhibidor SR141716A, en las dosis utilizadas, antagoniza gran variedad de comportamientos relacionados con la ingesta. Por ejemplo, suprime la autoadministración de heroína (Navarro et al., 2001; Rodríguez et al., 1999), las respuestas ante la comida (Freedland et al., 2000) y disminuye también tanto la ingesta de etanol como de sacarina (Colombo et al., 1998 a,b). Sin embargo, los efectos de SR141716A sobre la autoadministración de etanol podrían separarse de los debidos a la ingesta de comida, ya que la administración central de dicho inhibidor suprime la autoadministración de etanol pero no la ingesta de comida. Ello sugeriría la posibilidad de que los antagonistas de receptores cannabinoides podrían mediar las acciones supresoras de la ingesta de comida periféricamente (Gómez et al., 2002). El análisis bioquímico de las líneas AA y ANA sugiere que la CPF y el estriado dorsal podrían ser las dianas implicadas en el efecto de SR141716A sobre la ingesta de etanol. Las inyecciones intracerebrales de SR141716A, en la CPF, revelaron la posibilidad de que dicha área, y no el estriado dorsal, podría estar implicada en mediar el efecto de supresión de la ingesta de etanol, por parte de SR141716A, en las ratas AA. - 170 - MENÚ SALIR Discusión Sin embargo, las microinyecciones suprimen la autoadministración de alcohol en menor grado que las inyecciones sistémicas, lo que sugeriría la implicación de otras regiones cerebrales en las que el receptor CB1 también se encuentra (Tsou et al., 1998), varias de las que se saben participan en la regulación de la ingesta de etanol, como por ejemplo el hipocampo (Tierney et al., 2004) y otras estructuras corticales y subcorticales (Carr y Sesack, 1998). La administración sistémica de SR141716A podría contribuir a que dichas áreas participasen en el efecto funcional observado. La CPF y el estriado dorsal se han asociado con la formación y expresión del hábito de consumo (Killcross y Coutureau, 2003). Al contrario que los reforzadores naturales, la autoadministración de etanol rápidamente se convierte en un hábito de estímulo-respuesta (Dickinson et al., 2002). Por tanto, la observación, en la presente tesis, de que el antagonismo del receptor CB1 suprime el comportamiento reforzador del etanol de manera más eficiente que la mantenida por sacarina, podría ser atribuible a los efectos sobre la motivación y/o al hábito de respuesta. Los análisis bioquímicos y farmacológicos demuestran la existencia de una elevación en el tono endocannabinoide en la CPF, debido a la inhibición de la degradación, lo cual conduciría a una regulación a la baja de la densidad y transducción del receptor para compensar dichos cambios. La compensación parece ser incompleta y el continuo y elevado tono endocannabinoide, en la CPF, contribuiría a un aumento en la preferencia y autoadministración de alcohol, reversible con la administración local de antagonistas. En ratas msP, comparadas con ratas Wistar, se observa una disminución en la expresión génica de enzimas implicadas en la degradación de endocannabinoides, esto es, FAAH en estriado y MAGL en CPF, no variando la expresión de las enzimas de síntesis NAPE-PLD o DAGL-α. Ello sugeriría la existencia de una alteración en la degradación de endocannabinoides lo cual podría llevar a la elevación del tono endocannabinoide en dichas áreas cerebrales. El análisis, por inmunohistoquímica, de la expresión proteica de CB1, FAAH, MAGL y NAPE-PLD no revela la existencia de cambios dramáticos en las áreas cerebrales analizadas. Sin embargo, la compartimentalización de estas proteínas hacia los terminales axónicos o dendríticos exigiría realizar un análisis en las áreas de - 171 - MENÚ SALIR Discusión proyección de las neuronas de la CPF y cuerpo estriado, en los que se han encontrado cambios en la expresión del ARNm. Los mecanismos neurales subyacentes implicados en la regulación endocannabinoide del consumo de etanol, en la CPF, son desconocidos pero probablemente esté relacionada con, o bien, las posibles dianas primarias del etanol, esto es, los sistemas glutamatérgicos y GABAérgicos, o con las vías dopaminérgicas u opioidérgicas de los circuitos de recompensa (Pistis et al., 2002). El consumo de etanol en ratas msP disminuye la expresión génica de CB1, siendo dicha alteración más patente en el núcleo caudado-putamen al ser la región con mayor diferencia respecto a las ratas Wistar cuando no se administra etanol. La restricción de este efecto a la región más rostral del caudado-putamen, apunta a una posible compartimentalización dentro de dicho núcleo. Sin embargo, la medida de expresión de ARNm, mediante PCR cuantitativa, en estriado no revela la existencia de ninguna diferencia. Ello puede ser debido a un efecto de dilución, al no discriminarse ninguno de los núcleos que la constituyen cuando se realiza dicho análisis. Por otro lado, en la línea msP se ha caracterizado una desregulación del sistema de señalización del péptido regulador de la respuesta a estrés del factor liberador de corticotropina (Hansson et al., 2006), por lo que el fenotipo de preferencia alcohólica no sólo parece centrarse en el Sistema Endocannabinoide. El análisis realizado, en ambos modelos de alcoholismo (ratas AA y msP), revela la existencia de alteraciones tanto en el receptor CB1 como en la degradación de endocannabinoides. La selección y cría de estos animales generaría una segregación de genes al azar a pesar de lo cual ambos comparten las mismas alteraciones en el SEC en las mismas áreas cerebrales, lo que reforzaría aún más la hipótesis que asociaría el alcoholismo con la alteraciones del SEC en regiones cerebrales claves. - 172 - MENÚ SALIR Discusión 4. Implicaciones Terapéuticas: 4.1 - Dependencia Severa a Alcohol y Sistema Endocannabinoide La dependencia es resultado de una serie de cambios adaptativos tras el consumo crónico de drogas, entre los que pueden incluirse la sensibilización y la tolerancia. La sensibilización causa una progresiva adicción a la droga, relacionada con alteraciones en el metabolismo de la misma, siendo el etanol una de las sustancias que puede provocar este fenómeno. La administración crónica de sustancias adictivas conduce a una progresiva y duradera tolerancia. El organismo intenta recuperar y preservar su homeostasis mediante una serie de cambios adaptativos de diversa índole. El desarrollo de tolerancia es un fenómeno complejo donde los factores ambientales juegan un importante papel. La dependencia al alcohol suele ir acompañada por una tolerancia a sus efectos intoxicantes y por los síntomas de retirada, como temblores y confusión, cuando cesa el consumo. El desarrollo de tolerancia tras un abuso continuado y el síndrome de abstinencia tras su cese, se consideran como el núcleo principal de la dependencia a alcohol (Maldonado, 2006). En el cerebro, la presencia del sistema de señalización endocannabinoide en áreas como hipocampo, corteza, estriado, sustancia negra o cerebelo apoya el papel de este sistema en las respuestas motoras y cognitivas. La distribución anatómica y las acciones de los endocannabinoides concuerdan con los efectos del alcohol sobre el comportamiento, incluyéndose alteraciones en la memoria, disminución en la actividad motora, catalepsia, antinocicepción e hipotermia (Ryan y Butters, 1980; Brandt et al., 1983; Gebhart et al., 1984; Herkehham et al., 1991; Compton et al., 1993; Fadda y Rossetti, 1998). La adaptación, en varios pasos, del Sistema Endocannabinoide en el cerebro podría jugar un importante papel en el desarrollo de tolerancia y dependencia al alcohol (Basavarajappa et al., 1999; Basavarajappa y Hungund, 1998,1999; Hungund y Basavarajappa 2000). En la presente tesis se ha demostrado que los diferentes modelos animales con preferencia al consumo a etanol, así como los tratados crónicamente con etanol, tienen alteradas las vías de degradación de endocannabinoides lo cual está fuertemente asociado con un incremento en el tono endocannabinoide. - 173 - MENÚ SALIR Discusión Varios estudios han relacionado el aumento en los niveles de endocannabinoides con la degeneración tisular y daño celular así como con el periodo postmortem (Schmid et al., 1995; Felder et al., 1996; Kempe et al., 1996). En ratas tolerantes a Δ9-THC (Di Marzo et al., 2000) se ha observado un incremento en la síntesis de AEA en el prosencéfalo límbico, efecto también descrito en cultivos de células de neuroblastoma de ratón tratadas con Δ9-THC (Hunter y Burstein, 1997). Ello sugiere la implicación del Sistema Endocannabinoide en los cambios neuroadaptativos en dichas células y en el cerebro. El cambio en la neurotransmisión, mediada por endocannabinoides, podría ser responsable, por tanto, de la activación del sistema de recompensa por parte del alcohol. Se ha encontrado que los ratones DBA/2, que evitan la ingesta de alcohol, tienen reducida la funcionalidad de los receptores CB1 en cerebro, lo que concuerda con otros estudios en los que el antagonista SR141716A bloquea la ingesta de alcohol en roedores. De manera similar, la activación de CB1 induce el fuerte deseo por el alcohol, sugiriendo la implicación del receptor CB1 en el comportamiento asociado con el consumo de alcohol y el desarrollo de alcoholismo (Basavarajappa et al., 2005). Varios trabajos científicos describen que ratones nulos para el gen de FAAH tienen incrementados tanto la preferencia como el consumo de alcohol (Blednov et al., 2007; Basavarajappa et al., 2006). En humanos con dependencia severa a diversas drogas de abuso, entre ellas el alcohol, existe una fuerte asociación de dicho estado adictivo con una mutación sin sentido (C385A) en el gen de la enzima FAAH, cuya consecuencia es una disminución tanto en la actividad como en los niveles proteicos de dicha enzima (Chiang et al., 2004). En la presente tesis, se ha demostrado que la reducción en la expresión y actividad de la enzima FAAH podría ser la principal responsable de la elevación en los niveles de endocannabinoides en la CPF de ratas AA y estriado de las ratas msP. En la CPF de ratas msP sería la disminución en la expresión de MAGL, y no de FAAH, la responsable de ese esperable incremento en los niveles de endocannabinoides. Ello sugiere la existencia de diferentes dianas del alcohol, implicadas en los mecanismos de degradación de endocannabinoides, en las diferentes áreas cerebrales analizadas y su relación con la dependencia severa a alcohol (ver tabla 1). - 174 - MENÚ SALIR Discusión Modelo Animal de Alcoholismo Área Cerebral CPF Línea AA Niveles Endocannabinoides (1-AG, 2-AG) CB1: Densidad y Acoplamiento FAAH: Expresión Génica y Actividad Enzimática MAGL: Expresión Génica CB1: Expresión Génica (CPu +1.0 a +0.2 mm Bregma) CPF MAGL: Expresión Génica Estriado CB1: FAAH: Expresión Génica (CPu +2.2 mm, Bregma) Expresión Génica CB1: Expresión Génica (CA1, CA4) Estriado Línea msP Alteración en Sistema Endocannabinoide Hipocampo Niveles Endocannabinoides (AEA) (Basavarajappa et al., 1999) Cerebelo CB1: FAAH: Ratas Wistar tras Tratamiento Crónico de Etanol Expresión Génica Afinidad, Sitios de Unión (Basavarajappa et al., 1998) Niveles Proteicos (Capa Molecular) Estriado CB1, MAGL, NAPE-PLD: Expresión Génica FAAH: Niveles Proteicos (Shell del núcleo accumbens) Hipocampo CB1: Niveles Proteicos (capa granular Giro Dentado) NAPE-PLD: Expresión Génica Tabla 1. Alteraciones en el Sistema Endocannabinoide en diversas áreas cerebrales de ratas Wistar tras tratamiento crónico de etanol y en los modelos animales con preferencia al alcohol (líneas AA y msP). CPF = corteza prefrontal. - 175 - MENÚ SALIR Discusión 4.2 - Recaídas y Sistema Endocannabinoide Uno de los principales problemas en el tratamiento del abuso de drogas es la reiniciación del fuerte deseo y búsqueda de la droga. Aún no se han identificado los mecanismos exactos que subyacen a la reinstauración del consumo de drogas tras un periodo de abstinencia, aunque cada vez existen más evidencias que apuntan a la estimulación del receptor CB1 en el fenómeno de la recaída. El Sistema Endocannabinoide se ha relacionado con los episodios de recaída, especialmente con el receptor CB1 el cual está asociado con el estrés inducido por el consumo y la retirada de alcohol en ratones ‘knockout’ de CB1 (Racz et al., 2003) así como en los mecanismos que median la recaída al alcohol. La exposición a agonistas cannabinoides WIN 55,212-2 ó a THC promueve la recaída en ratas abstinentes al alcohol (López-Moreno et al., 2004; McGregor et al., 2005) mientras que la administración del antagonista SR141716A reduce la reinstauración del comportamiento de búsqueda de etanol en ratas (Cippitelli et al., 2005). Ello concuerda con la elevación del tono endocannabinoide observado tras la administración crónica de etanol, tanto en cultivos de células granulares de cerebelo (Basavarajappa y Hungund, 1999) como en el modelo animal con preferencia alcohólica (línea AA) analizada en la presente tesis, lo que sugeriría la existencia de una fuerte probabilidad de recaída en dichos animales y que las alteraciones experimentadas, en el SEC, se mantendrían durante un largo periodo de tiempo tras el cese del consumo. El Sistema Endocannabinoide parece participar en las propiedades de recompensa del alcohol mediante la modulación de sus efectos sobre la activación de la transmisión mesolímbica dopaminérgica. Estudios de microdiálisis in vivo, revelan que el alcohol no incrementa los niveles extracelulares de dopamina en el núcleo accumbens (NAc) de ratones ‘knockout’ para CB1 (Hungund et al., 2003). Resultados similares se obtuvieron cuando se pretrataton a los ratones silvestres con el antagonista SR141716A antes de la administración de etanol (Hungund et al., 2003). En la actualidad no existe ningún dato clínico de la posible eficacia del bloqueo del receptor CB1 en humanos en el tratamiento de la adición a alcohol. El SEC puede modular la motivación que lleva a la búsqueda de la droga mediante un mecanismo independiente de la liberación de dopamina en el NAc. Los receptores CB1 están presentes en la CPF, lo cual constituye un nexo para la integración - 176 - MENÚ SALIR Discusión sensorial, el procesamiento de las emociones y la experiencia hedónica. Esta área cerebral es un importante componente en el fenómeno adictivo ya que procesa la recompensa hasta llegar a ser una ‘experiencia hedónica’ (Kringelbach, 2005). De ahí que los endocannabinoides pudieran estar implicados en la motivación para obtener la droga actuando como unión de la recompensa a la ‘experiencia hedónica’ en la CPF. Los mecanismos que subyacen al papel del SEC en la recaída del comportamiento de búsqueda de droga, producido por estímulos ambientales relacionados con la droga y la reexposición a la misma, parecen estar relacionados con la regulación del impacto sobre las memorias relacionadas con la recompensa. Además, los endocannabinoides, actuando como mensajeros retrógrados, median LTP y/o LTD de la transmisión sináptica en varias áreas cerebrales relacionadas con la memoria y la adicción, como el NAc, CPF, amígdala e hipocampo (De Vries y Schoffelmeer, 2005). Los efectos de los endocannabinoides sobre la plasticidad sináptica podrían consolidar el comportamiento que lleva a la recompensa necesaria para establecer los procesos adictivos. Existen otros circuitos neuroquímicos implicados en los efectos del SEC sobre la recompensa. Por ejemplo, los endocannabinoides podrían facilitar los efectos de las neuronas liberadoras de orexina en el hipotálamo, el cual proyecta hacia el NAc y el área tegmental ventral. De esta forma, las orexinas hipotalámicas junto con los endocannabinoides, estarían directamente implicadas en los efectos de recompensa de las drogas y en la recaídas del comportamiento de búsqueda de droga (Harris et al., 2005). Por tanto, el SEC representaría un elemento clave en el sustrato neurobiológico de la adicción a drogas, y las variaciones genómicas podrían determinar la vulnerabilidad en humanos a esta adicción (Zhang et al., 2004). 4.3 - Futuro: Tratamiento del Alcoholismo con Fármacos del Sistema Endocannabinoide De forma similar a otros desórdenes de recaída crónica, hay sólidas evidencias que demuestran que los tratamientos farmacológicos pueden mejorar clínicamente el alcoholismo. Los meta-análisis descritos demuestran la capacidad que tienen ciertos tratamientos actuales de prolongar la duración de la abstinencia, tras el cese del - 177 - MENÚ SALIR Discusión consumo, o de disminuir el número de días de fuerte consumo aunque ninguno de ellos evita la recaída (Bouza et al., 2004). Hay evidencias que apuntan a una fuerte asociación entre la dependencia de alcohol y los desórdenes psiquiátricos sobre todo los referentes a la ansiedad, cambios de humor y sueño. Estos efectos se añaden a los del síndrome de abstinencia y su tratamiento farmacológico es muy efectivo aunque no útiles para tratar el aspecto principal de la dependencia al alcohol. La detoxificación puede lograrse con agonistas opioides (metadona, buprenorfina) o agonistas α2-adrenérgicos (clonidina, lofexidina) y la prevención de la recaída puede apoyarse con el uso de naltrexona. Sin embargo, estas estrategias tienen una baja efectividad y éxito así como una serie de efectos colaterales, como la depresión y anhedonia (van der Brink y van Ree, 2003). Las nuevas estrategias de tratamiento buscan prevenir la recaída a otras drogas de abuso en individuos abstinentes, en vista de que lo anterior sólo es beneficioso en aquéllos fuertemente motivados. En consecuencia, la recurrencia de episodios de recaída en individuos abstinentes durante largos periodos es uno de los problemas más significativos durante la detoxificación. Numerosos trabajos científicos apuntan la posibilidad de tratar el alcoholismo con fármarcos dirigidos al Sistema Endocannabinoide. Se ha demostrado que el bloqueo total del receptor CB1 en ratones ‘knockout’ disminuye la preferencia e ingesta de alcohol cuando se comparan con ratones silvestres (Hungund et al., 2003; Poncelet et al., 2003; Wang et al., 2003; Naasila et al., 2004; Lallemand y De Witte, 2004; Racz et al., 2003). Por tanto, el bloqueo y la delección genética del receptor cannabinoide generan una marcada reducción en la ingesta, preferencia y propiedades motivacionales del alcohol. El consumo crónico de etanol provoca una disminución en la funcionalidad y acoplamiento del receptor CB1, los cuales no parecen ser cambios suficientemente compensatorios ante el aumento de los niveles de endocannabinoides. Por tanto, el tratamiento farmacológico más eficaz contra el alcoholismo podría ir dirigido hacia una disminución en la funcionalidad del receptor CB1 que produzca su bloqueo total aunque ello no excluiría otras posibilidades que generasen una modificación bien en la producción o en la degradación de endocannabinoides. - 178 - MENÚ SALIR Discusión En la literatura científica hay descritos varios compuestos que actúan como antagonistas del receptor cannabinoide CB1 como son: SR141716A (Rimonabant), SR147778, AM251, AM281 y LY320135. SR141716A LY320135 AM251 AM281 Estructura química de antagonistas y agonistas inversos del receptor CB1. En esta línea, se ha demostrado que la administración de antagonistas como SR141716A contribuye a la disminución de ingesta de etanol así como evita la recaída (Cippitelli et al., 2005; Economidou et al., 2006). Este compuesto inhibe la liberación de dopamina, inducida por etanol, en el NAc (Weiss et al., 1993; Campbell y McBride, 1995) lo cual contribuiría a la supresión de sus efectos reforzadores. En la presente tesis, se ha demostrado que la administración de SR141716A en ratas AA suprime la ingesta de etanol. Otros estudios describen que este antagonista de CB1 inhibe el consumo de etanol así como la reinstauración del comportamiento de búsqueda de alcohol en la línea de ratas msP (Cippitelli et al., 2005). En la línea de ratas sP, no expuestas anteriormente al alcohol, se suprime la ingesta de dicha solución cuando se someten a un régimen de libre elección entre dos botellas: una con alcohol y otra con agua (Serra et al., 2001). La administración de SR141716A reduce la ingesta voluntaria de alcohol en ratones C57BL/6J (Arnone et al., 1997), ratas sP (Colombo et al., 1998b) y ratas Wistar (Lallemand et al., 2001) que normalmente bebían alcohol (modelo de fase de - 179 - MENÚ SALIR Discusión mantenimiento o de bebedor activo del alcoholismo humano), suprime el incremento temporal en la ingesta de etanol en las ratas sP tras un periodo de privación de alcohol (fenómeno denominado ‘efecto de privación alcohólica’ el cual se propone como modelo de los episodios de recaída que se dan en los humanos alcohólicos; Serra et al., 2002), disminuye la autoadministración oral de alcohol en ratas no seleccionadas (Freedland et al., 2001) y suprime las respuestas al alcohol en ratas sP (Colombo et al., 2004). Existen estudios que sugieren el uso de una dosis inicial de SR141716A de 10 mg/kg/día, en el periodo inicial de retirada de alcohol, seguido de una dosis de mantenimiento de 3 mg/kg/día que evitaría el incremento en la preferencia de etanol asociado con la dosis inicial (Lallemand et al., 2004). Actualmente se están realizando estudios para testar la seguridad y fiabilidad del antagonista SR141716A en el Instituto Nacional de Abuso de Alcohol y Alcoholismo. Otros estudios apuntan al uso del antagonista SR147778 para el tratamiento del alcoholismo. Se ha demostrado que este compuesto disminuye el desarrollo de la preferencia por el alcohol en ratas sP que no han ingerido alcohol con anterioridad (Doggrell, 2005). En ratas sP que han ingerido alcohol, la administración de SR147778 (2.5, 5 y 10 mg/kg) reduce la ingesta de etanol. Cuando se les priva de alcohol, durante 15 días, se produce una gran ingesta de alcohol, comportamiento que es prácticamente suprimido cuando se administra SR147778. Por tanto, este compuesto, similar a su análogo estructural rimonabant, suprime la adquisición y mantenimiento del comportamiento de bebedor, la recaída y la motivación al consumo de alcohol en ratas sP con preferencia al alcohol (Gessa et al., 2005). Desde el punto de vista preclínico, actualmente no existen diferencias entre el uso de SR141716A (Rimonabant) y SR147778 para el tratamiento del alcoholismo. Otros posibles candidatos serían los agonistas inversos AM251, AM281 y LY320135 que producen efectos opuestos a los agonistas al interaccionar con los receptores CB1. El compuesto LY320135 es capaz de revertir la inhibición, mediada por AEA, de la adenilato ciclasa. Su estructura es más accesible para ser modificada que SR141716A por lo que podría ser menos tóxico que este último. - 180 - MENÚ SALIR Discusión Los compuestos AM251 y AM281 son estructuralmente muy parecidos a SR141716A. Tienen la capacidad de bloquear a los receptores CB1 y producir efectos inversos a los cannabimiméticos, aunque se han observado varias diferencias en cultivos in vitro con tejido cardiovascular (Pertwee, 2004) y en secciones de hipocampo de rata (Hájos y Freund, 2002; Hajos et al., 2001) respecto a los efectos de SR141716A. Algunos artículos describen que AM251 y AM281 tienen efectos opuestos en la señalización retrógrada estimulada eléctricamente en el Sistema Nerviosos Central. AM281 incrementa la liberación de [3H]D-aspartato en cultivos primarios de células granulares cerebelares e inhibe la unión basal de [35S]GTPγS, antagonizando las respuestas de los cannabinoides liberados endógenamente (Breivogel et al., 2004; Kreitzer y Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson y Nicoll, 2001). Tanto SR141716A como AM251 tienen la capacidad de actuar como antagonistas neutros de CB1 a bajas concentraciones y de comportarse como agonistas inversos con concentraciones mayores. Estos ligandos podrían, por tanto, tener dos sitios de acción en el receptor CB1: uno que desplace a los agonistas produciendo antagonismo y otro donde se induce un agonismo inverso, quizás debido a un mecanismo alostérico (Sim-Selley et al., 2001). Sin embargo, no existen trabajos que describan los efectos de estos compuestos (AM251, AM281 y LY320135) sobre la dependencia al alcohol. Existen trabajos que apuntan que el compuesto AM251 inhibe el desarrollo de tolerancia a los efectos analgésicos de la morfina, lo que sugiere la implicación de los receptores CB1 en el desarrollo y mantenimiento de la tolerancia a esta droga (Trang et al., 2007), dejando una vía abierta a futuras investigaciones en la dependencia a otras drogas. Otra posible terapia farmacológica sería aquella que fuera dirigida a incrementar la degradación de endocannabinoides con el fin de restablecer sus niveles normales. Está descrito que la administración de progesterona provoca un incremento en la expresión tanto proteica como génica de FAAH, en linfocitos T humanos, de hasta un 270% respecto a los controles, sin afectar ni a NAPE-PLD, ni a NAT, ni al tranportador de anandamida ni a la unión a los receptores cannabinoides. Ello provoca un descenso en los niveles de AEA de hasta un 60% (Maccarrone et al., 2003a). La leptina también es capaz de disminuir los niveles de AEA y 2-AG, en linfocitos T humanos, hasta un 300% respecto a los controles mediante la activación del - 181 - MENÚ SALIR Discusión factor de transcripción STAT3 el cual induciría la expresión génica de FAAH (Maccarrone et al., 2003b). Otra posible estrategia farmacológica podría consistir en disminuir la síntesis de endocannabinoides. En la literatura científica no está descrita la existencia de fármacos que actúen directamente sobre las enzimas de síntesis (NAT, NAPE-PLD y DAGL). Sin embargo, existen nuevos fármacos, como la safinamida, dirigidos a la inhibición de la liberación de glutamato, neurotransmisor responsable de la estimulación la actividad de la enzima NAPE-PLD. La safinamida es una sal de metanosulfonato muy estable y soluble en agua cuya administración está dirigida a tratar tanto la epilepsia como la fase III de la enfermedad del Parkinson ya que se cree que podría inhibir a la monoaminooxidasa-B (MAO-B), responsable de metabolizar y recaptar aminas como la dopamina. Está descrito que, en ratas sometidas a tratamiento crónico de etanol, disminuye la liberación de dopamina en el NAc lo que contribuye a la aparición de síntomas del síndrome de abstinencia (malestar, depresión, catatonia, temblores, etc.; Diana et al., 1993; Schulteis et al., 1995; Lal et al., 1988; Fadda y Rossetti, 1998). Dichos efectos podrían revertir con la administración de dicho fármaco al incrementar los niveles de dopamina en dicha área. La safinamida se concentra en mayor proporción en cerebro, donde tiene efectos neuroprotectores, que en plasma por lo que su efecto podría ser central. No posee efectos ni sobre el comportamiento, la función cardíaca, actividad locomotora, cognición, función renal ni tránsito intestinal ni siquiera a dosis muy superiores a las que se considerarían terapéuticas. Sería, por tanto, un buen candidato para el tratamiento del alcoholismo aunque dicha hipótesis necesitaría ser confirmada. Como corolario, los trabajos presentados en la presente tesis doctoral permiten confirmar el papel del Sistema Endocannabinoide en el alcoholismo, así como sugerir la necesidad de terapias experimentales basadas en la interferencia farmacológica con este sistema de señalización. - 182 - MENÚ SALIR Discusión Tratamiento Diana Efecto SR141716A CB1 Antagonista. Disminuye el consumo y las recaídas. SR147778 CB1 Antagonista. Disminuye la ingesta de etanol, la motivación al consumo y las recaídas. AM251, AM281, LY320135 CB1 Antagonistas o agonistas inversos. Posible papel supresor de AM251 en el desarrollo de tolerancia (Trang et al., 2007) Progesterona FAAH Incremento en la expresión génica y proteica de FAAH; disminución en los niveles de endocannabinoides. FAAH Incremento en la expresión génica y actividad de FAAH; disminución de los niveles de endocannabinoides. NAPE-PLD Acción central. Inhibición de la liberación de glutamato, bloqueo de los canales de Ca2+; posible papel inhibidor de la actividad NAPE-PLD. Leptina Safinamida Tabla 2. Posibles fármacos dirigidos al tratamiento del alcoholismo, dianas del Sistema Endocannabinoide sobre las que actuarían y efectos farmacológicos. - 183 - MENÚ SALIR MENÚ SALIR CONCLUSIONES MENÚ SALIR MENÚ SALIR Conclusiones 1.- La administración aguda de etanol afecta a los niveles de endocannabinoides, existiendo especificidad tisular en dicho efecto: incrementan en hígado e intestino delgado y disminuyen en cerebro, plasma y tejido adiposo. 2.- En cerebro, la exposición aguda a etanol disminuye los niveles de endocannabionides (AEA, PEA) sin actuar directamente sobre los mecanismos de síntesis y degradación de endocannabinoides aunque sí sobre otros sistemas de neurotransmisión, como el glutamatérgico, responsable directo de la estimulación de la actividad de la enzima de síntesis de anandamida, NAPE-PLD. Se descarta que la respuesta al estrés, tras la administración de etanol, sea responsable de la disminución en los niveles de endocannabinoides en las áreas cerebrales analizadas. 3.- La administración crónica de etanol genera un aumento en la expresión de enzimas de síntesis (NAPE-PLD) y una disminución en las enzimas de degradación (FAAH), lo que contribuiría al aumento en los niveles de endocannabinoides, previamente descritos por otros autores. La expresión génica y proteica del receptor cannabinoide CB1 está afectada, existiendo una especificidad en la neuroanatomía del Sistema Nervioso para dicha alteración. 4.- La administración aguda de etanol tiene efectos opuestos sobre el Sistema Endocannabinoide a los encontrados, y descritos previamente por otros autores, tras la administración crónica de etanol como consecuencia de la presión adaptativa a la que se ve sometida el organismo al estar continuamente expuesto al etanol. 5.- Los análisis bioquímicos de las líneas de roedores con preferencia alcohólica, AA y msP, ponen de manifiesto la presencia de alteraciones en la degradación de endocannabinoides asociadas con la preferencia y consumo del etanol. 6.- El análisis de la corteza prefrontal de la línea AA revela la existencia de una elevación en el tono endocannabinoide acompañado por una regulación a la baja en la densidad y transducción del receptor, cambio que no es suficientemente compensatorio para suprimir el consumo y la preferencia por el alcohol. - 187 - MENÚ SALIR Conclusiones 7.- El tratamiento local con el antagonista SR141716A (Rimonabant) extingue el consumo y la preferencia al alcohol, lo que abre nuevas expectativas futuras al tratamiento del alcoholismo con fármacos bloqueantes del Sistema Endocannabinoide en cerebro. Los tratamientos farmacológicos dirigidos a restablecer la homeostasis cannabinoide bien modificando al receptor cannabinoide CB1 y/o la producción o degradación de endocannabinoides, apuntan a la implicación de una prolongada elevación del tono endocannabinoide en las recaídas al consumo de alcohol. Los antagonistas de CB1 (SR141716A y SR147778) han demostrado ser eficaces en disminuir el consumo y evitar las recaídas al alcohol por lo que podrían ser candidatos ideales en el tratamiento del alcoholismo en humanos. - 188 - MENÚ SALIR BIBLIOGRAFÍA MENÚ SALIR MENÚ SALIR Bibliografía - Adams C.L., Cowen M.S., Short J.L., Lawrence A.J. (2007): Combined antagonism of glutamate mGlu5 and adenosine A2A receptors interact to regulate alcohol-seeking in rats. Int J Neuropsychopharmacol:1-13. - Adams I.B., Martin B.R. (1996): Cannabis: pharmacology and toxicology in animals and humans. Addiction 91, 1985-1614. - Agabio R., Cortis G., Fadda F., Gessa G.L., Lobina C., Reali R., Colombo G. (1996): Circadian drinking pattern of Sardinian alcohol-preferring rats. Alcohol Alcohol 31(4):385-8. - Aguado T., Monory K., Palazuelos J., Stella N., Cravatt B., Lutz B., Marsicano G., Kokaia Z., Guzman M., Galve-Roperh I. (2005): The endocannabinoid system drives neural progenitor proliferation. FASEB J; 19: 1704 - 1706. - Ameri A. (1999): The effects of cannabinoids on the brain. Prog Neurobiol. 58(4):315-48. - Aragón C., Miquel M., Correa M., Sanchís-Segura C. (2002): Alcohol y metabolismo humano. Adicciones 14(1); 23-42. - Arlinde C., Sommer W., Björk K., Reimers M., Hyytiä P., Kiianmaa K., Heilig M. (2004): A cluster of differentially expressed signal transduction genes identified by microarray analysis in a rat genetic model of alcoholism. Pharmacogenomics J. 4(3):208-18. - Arnone M., Maruani J., Chaperon F., Thiebot M.H., Poncelet M., Soubrie P., Le Fur G. (1997): Selective inhibition of sucrose and ethanol intake by SR141716A, an antagonist of central cannabinoid (CB1) receptors. Psychopharmacology 132, 104-6. - Bäckström P., Hyytiä P. (2005): Suppression of alcohol self-administration and cue-induced reinstatement of alcohol seeking by the mGlu2/3 receptor agonist LY379268 and the mGlu8 receptor agonist (S)-3,4-DCPG. Eur J Pharmacol 528(1-3):110-8. - Baker D., Pryce G., Davies W.L., Hiley C.R. (2006): In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol Sci 27(1):1-4. - Basavarajappa B.S., Cooper T.B., Hungund B.L. (1998): Chronic ethanol administration down-regulates cannabinoid receptors in mouse brain synaptic plasma membrane. Brain Res 793(1-2): 212- 8. - Basavarajappa B.S., Hungund B.L. (1999): Chronic ethanol increases the cannabinoid receptor agonist anandamide and its precursor Narachidonoylphosphatidyl-ethanolamine in SK-N-SH cells. J Neurochem 72, 522-8. - 191 - MENÚ SALIR Bibliografía - Basavarajappa B.S., Hungund B.L. (2002): Neuromodulatory role of the endocannabinoid signaling system in alcoholism: an overview. Prostag Leukot Essent Fatty Acids 66:287-299. - Basavarajappa B.S., Hungund B.L. (2005): Role of the endocannabinoid system in the development of tolerance to alcohol. Alcohol Alcohol 40(1):15-24. - Basavarajappa B.S., Saito M., Cooper T.B., Hungund B.L. (2000): Stimulation of cannabinoid receptor agonist 2-arachidonylglycerol by chronic ethanol and its modulation by specific neuromodulators in cerebellar granule neurons. Biochem Biophys Acta 1535, 78-86. - Basavarajappa B.S., Saito M., Cooper T.B., Hungund B.L. (2003): Chronic ethanol inhibits the anandamide transport and increases extracellular anandamide levels in cerebellar granule neurons. Eur J Pharmacol 466(1-2): 7383. - Basavarajappa B.S., Yalamanchili R., Cravatt B.F., Cooper T.B., Hungund B.L. (2006): Increased etanol consumption and preferente and decreased ethanol sensivity in female FAAH knockout mice. Neuropharmacology 50(7):834-44. - Beinfeld M.C., Connolly K. (2001): Activation of CB1 cannabinoid receptors in rat hippocampal slices inhibits potassium evoked cholecystokinin release, a possible mechanism contributing to the spatial memory defects produced by cannabinoids. Neuroscience Letters 301, 69–71. - Benfenati F., Cimino M., Agnati L., Fuxe K. (1986): Quantitative autoradiography of central neurotransmitter receptors: methodological and statistical aspects with special aspects with special references to computerassisted image analysis. Acta Physiol Scand 128:129–146. - Benito C., Núnez E., Tolón R.M., Carrier E.J., Rabano A., Hillard C.J., Romero J. (2003): Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaques-associated glia in Alzheimer’s disease brains. J Neurosci .2003 ; 23 : 11136 - 11141 . - Berdyshev E.V., Schmid P.C., Krebsbach R.J., Hillard C.J., Huang C., Chen N., Dong Z., Schmid H.H. (2001): Cannabinoid-receptor-independent cell signaling by N-acylethanolamines. Biochem J 360(Pt 1): 67-75. - Berrendero F., García-Gil L., Hernández M., Romero J., Cebeira M., De Miguel R., Ramos J., Fernández-Ruiz J. (1998): Localization of mRNA expression and activation of signal transduction mechanism for cannabinoid receptor in rat brain during fetal development. Development. 125: 3179-3188. - Bisogno T., Sepe N., Melck D., Maurelli S., De Petrocellis L., Di Marzo V. (1997): Biosíntesis, release and degradation of the novel endogenous cannabimimetic metabolite 2-arachidonoylglycerol in mouse neuroblastoma cells. Biochem J 322(2): 671-7. - 192 - MENÚ SALIR Bibliografía - Bisogno T., Melck D., Bobrov M.Y., Gretskaya N.M., Bezuglov V.V., De Petrocellis L., Di Marzo V. (2000): N-acyl-dopamines: novel synthetic CB(1) cannabinoid-receptor ligands and inhibitors of anandamide with cannabimimetic activity in vitro and in vivo. Biochem J 351:3:817-24. - Blednov Y.A., Cravatt B.F., Boehm S.L., Walker D., Harris R.A. (2006): Role of endocannabinoids in alcohol consumption and intoxication: studies of mice lacking fatty acid amide hydrolase. Neuropsychopharmacology 32(7):1570-82. - Bodnar R.J., Hadjimarkou M.M. (2002): Endogenous opiates and behavior: 2001. Peptides 23(12):2307-65. Bouaboula M., Poinot-Chazel C., Bourrie B., Canat X., Calandra B., RinaldiCarmona M., Le Fur G., Casellas P. (1995): Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J 312(2): 637- 41. - - Bouza C., Angeles M., Muñoz A., Amate J.M. (2004): Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction 99(7):811-28. - Brandt J., Butters N., Ryan C., Bayog R. (1983): Cognitive loss and recovery in long-term alcohol abusers. Archives of General Psychiatry 40:435- 442. - Breivogel C., Griffin G., Di Marzo V., Martín B. (2001): Evidence for a new G protein cannabinoid receptor in mouse brain. Mol Pharmacol 60:155-163. - Breivogel C.S., Walker J.M., Huang S.M., Roy M.B., Childers S.R. (2004): Cannabinoid signaling in rat cerebellar granule cells: G-protein activation, inhibition of glutamate release and endogenous cannabinoids. Neuropharmacology 47(1):81-91. - Buck, K.J., Harris R.A. (1990): Benzodiazepine agonist and inverse agonist actions on GABAA receptor-operated chloride channels. Acute effects of ethanol. Journal of Pharmacology and Experimental Therapeutics, 253, 706-712. - Caberlotto L., Rimondini R., Hansson A., Eriksson S., Heilig M. (2004): Corticotropin-Releasing Hormone (CRH) mRNA expression in rat central amygdala in cannabinoid tolerance and withdrawal: Evidence for an allostatic shift? Neuropsychopharmacology. 2004; 29(1): 15-22. - Cadas H., Tomaso E., Piomelli D. (1997): Occurrence and biosynthesis of endogenous cannabinoid precursor, N-arachidonoyl phosphatidylethanolamine, in rat brain. J Neurosci 17(4): 1226- 1242. - Campbell A.D., Kohl R.R., McBride W.J. (1996): Serotonin-3 receptor and ethanolstimulated somatodendritic dopamine release. Alcohol, 13, 569-574. - Campbell A.D., McBride W.J. (1995): Serotonin-3 receptor and ethanolstimulated dopamine release in the nucleus accumbens. Pharmacol Biochem Behav 51(4):835-42. - 193 - MENÚ SALIR Bibliografía - Carlisle S.J., Marciano-Cabral F., Staab A., Ludwick C., Cabral G.A. (2002): Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int Immunopharmacol 2: 69 – 82. - Carlson N. R. (1998): Physiology of behavior Boston, MA; Aliyn & Bacon. - Carr D.B., Sesack S.R. (1998): Callosal terminals in the rat prefrontal cortex: synaptic targets and association with GABA-inmunoreactive structures. Synapse 29:193-205. - Chevaleyre V., Castillo P.E. (2003): Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38, 461–472. - Chiang K.P., Gerber A.L., Sipe J.C., Cravatt B.F. (2004): Reduced cellular expression and activity of the P129T mutant of human fatty acid amide hydrolase: evidence for a link between defects in the endocannabinoid system and problem drug use. Human Molecular Genetics 13(18):2113-2119. - Ciccocioppo R., Economidou D., Cippitelli A., Cucculelli M., Ubaldi M., Soverchia L., Lourdusamy A., Massi M. (2006): Genetically selected Marchigian Sardinian alcohol-preferring (msP) rats: an animal model to study the neurobiology of alcoholism. Addict Biol 11(3-4):339-55. - Cippitelli A., Bilbao A., Hansson A.C., del Arco I., Sommer W., Heilig M., Massi M., Bermúdez-Silva F.J., Navarro M., Ciccocioppo R., de Fonseca F.R. (2005): Cannabinoid CB1 receptor antagonism reduces conditioned reinstatement of etanol-seeking behaviour in rats. Eur J Neurosci 21(8):2243-51. - Cloninger C.R. (1987): Neurogenetics adaptive mechanisms in alcoholism. Science 236 (4800):410-6. - Colombo G., Agabio R., Díaz G., Lobina C., Reali R., Gessa G.L. (1998a): Appetite suppression and weight loss after the cannabinoid antagonist SR141716. Life Sci 63:113-7. - Colombo G., Agabio R., Fa M., Guano L., Lobina C., Loche A., Reali R., Gessa G.L. (1998b): Reduction of voluntary ethanol intake in ethanol-preferring sP rats by the cannabinoid antagonist SR-141716. Alcohol Alcohol 33:126-30. - Colombo G., Serra S., Brunetti G., Gómez R., Melis S., Vacca G., Carai M. M., Gessa G. L., (2002): Stimulation of voluntary ethanol intake by cannabinoid receptor agonists in ethanol- preferring sP rats. Psychopharmacology 159, 18187. - Colombo G., Serra S., Vacca G., Carai M.A., Gessa G.L. (2005): Endocannabinoid system and alcohol addiction: pharmacological studies. Pharmacol Biochem Behav 81(2):369-80. - 194 - MENÚ SALIR Bibliografía - Colombo G., Vacca G., Serra S., Carai M.A., Gessa G.L. (2004): Suppressing effect of the cannabinoid CB1 receptor antagonist, SR141716, on alcohol’s motivational properties in alcohol-preferring rats. Eur J Pharmacol 498(13):119-23. - Comings D.E., Muhleman D., Gade R., Johnson P., Verde R., Saucier G., MacMurray J. (1997): Cannabinoid receptor gene (CNR1): association with i.v. drug use. Mol Psychiatry 2, 161-8. - Compton D.R., Rice K.C., De Costa B.R., Razdan R.K., Melvin L.S., Johnson M.R., Martin B.R. (1993): Cannabinoid structure-activity relationships: correlation of receptor binding and in vivo activities. Journal of Pharmacology and Experimental Therapeutics 265, 218-226. - Corbett D., Wise R.A. (1980): Intracraneal self-stimulation in relation to the ascending dopaminergic systems of the midbrain: a moveable electrode mapping study. Brain Research, 185:1. - Cowen S., Djouma E., Lawrence A.J. (2005): The metabotropic glutamate 5 receptor antagonist 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine reduces ethanol self-adminstration in multiple strains of alcohol-preferring rats and regulates olfactory glutamatergic systems. J Pharmacol Exp Ther 315(2):590600. - Cravatt B.F., Giang D.K., Mayfield S.P., Boger D.L., Lerner R.A., Gilula N.B. (1996): Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384, 83-87. - Cravatt B.F., Lichtman, A.H. (2003): Fatty acid amide hydrolase: an emerging therapeutic target in the endocannabinoid system. Current Opinion in Chemical Biology 7, 469–475. - Dahchour A., De Witte P. (2000a): Ethanol and amino acids in the central nervous system: assessment of the pharmacological actions of acamprosate. Prog Neurobiol; 60(4):343-62. - Dahchour A., De Witte P. (2000b) Taurine blocks the glutamate increase in the nucleus accumbens microdialysate of ethanol-dependent rats. Pharmacol Biochem Behav 65, 345-350. - Dar M.S. (1990): Central adenosinergic system involvement in ethanol-induced motor incoordination in mice. J Pharmacol Exp Ther 255(3):1202-9. - De Vries T.J., Homberg J.R., Binnekade R., Raaso H., Schoffelmeer A.N. (2003): Cannabinoid modulation of the reinforcing and motivational properties of heroin and heroin-associated cues in rats. Psychopharmacology 168, 164–169. - De Vries T.J., Schoffelmeer A.N. (2005): Cannabinoid CB1 receptors control conditioned drug seeking. Trends Pharmacol Sci 26, 420-426. - 195 - MENÚ SALIR Bibliografía - De Vries T.J., Shaham Y., Homberg J.R., Crombag H., Schuurman K., Dieben J., Vanderschuren L.J., Schoffelmeer A.N. (2001): A cannabinoid mechanism in relapse to cocaine seeking. Nature Medicine 7, 1151–1154. - Derkinderen P., Toutant M., Burgaya F., Le Bert, M., Siciliano J.C., De Franciscis V., Gelman M., Girault J.A. (1996): Regulation of a neuronal form of focal adhesion kinase by anandamide. Science 273, 1719–1722. - Desarnaud F., Cadas H., Piomelli D. (1995): Anandamide amidohydrolase activity in rat brain microsomes. Identification and partial characterization. J Biol Chem 270: 6030–6035. - Devane W.A., Dysarz F.A., Johnson M.R., Melvin L.S., Howlett A.C. (1988): Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol 34(5): 605- 13. - Devane W.A., Hanus L., Breuer A., Pertwee R.G., Stevenson L.A., Griffin G., Gibson D., Mandelbaum A., Etinger A., Mechoulam R. (1992): Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258: 1946-1949. - Devaud L.L. (2001): Ethanol dependence has limited effects on GABA or glutamate transporters in rat brain. Alcohol Clin Exp Res; 25(4):606-11. - Devaud L.L., Smith F.D., Grayson D.R., Morrow A.L. (1995): Chronic ethanol consumption differentially alters the expression of gamma-aminobutyric acid. A receptor subunit mRNAs in rat cerebral cortex: competitive, quantitative reverse transcriptase-polymerase chain reaction analysis. Mol Pharmacol. 48(5):861-8. - Dewey W.L. (1986): Cannabinoid pharmacology. Pharmacol Rev 38(2):151-78. - Diamond I., Gordon A.S. (1997): Cellular and molecular neuroscience of alcoholism. Physiol Rev.; 77(1):1-20. - Diana M.A., Levenes C., Mackie K., Marty A. (2002): Short-term retrograde inhibition of GABAergic synaptic currents in rat Purkinje cells is mediated by endogenous cannabinoids. Journal of Neuroscience 22, 200–208. - Diana M.A., Marty A. (2004): Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). British Journal of Pharmacology 142, 9–19. - Diana M., Pistis M., Carboni S., Gessa G.L., Rossetti Z.L. (1993): Profound decrement of mesolimbic dopaminergic neuronal activity during ethanol withdrawal syndrome in rats: Electrophysiological and biochemical evidence. Proceedings of the National Academy of Sciences, 90, 7966-7969. - DiChiara G. (1997): Alcohol and dopamine. Alcohol Health & Research World, 21, 108-114. - 196 - MENÚ SALIR Bibliografía - Dickinson A., Wood N., Smith J.W. (2002): Alcohol seeking by rats: action or habit? Q J Exp Psychol B 55:331-348. - Dinh T.P., Carpenter D., Leslie F.M., Freund T.F., Katona I., Sensi S.L., Kathuria S., Piomelli D. (2002): Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci 99(16):10819-24. - Di Marzo V., Bisogno T., De Petrocellis L., Melck D., Orlando P., Wagner J.A., Kunos G. (1999): Biosíntesis and inactivation of the endocannabinoid 2arachidonoylglycerol in circulating and tumoral macrophages. Eur J Biochem 264(1): 258- 67. - Di Marzo V., Bisogno T., Sugiura T., Melck D., De Petrocellis L. (1998): The novel endogenous cannabinoid 2-arachidonoylglycerol is inactivated by neuronal- and basophil-like cells: connections with anandamide. Biochem J 331(1): 15- 9. - Di Marzo V., Hill M.P., Bisogno T., Crossman A.R., Brotchie J.M. (2000): Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson’s disease. The FASEB Journal 14:1432-1438. - Doggrell S.A. (2005): Will the new CB1 cannabinoid receptor antagonist SR147778 have advantages over rimonabant? Expert Opin Investig Drugs 14(3):339-42. - Dulawa S.C, Grandy D.K., Low M.J., Pauius M.P., Geyer M.A. (1999): Dopamine D4 receptor-knock-out mice exhibit reduced exploration of novel stimuli. Journal of Neuroscience, 19, 9550-9556. - Economidou D., Mattioli L., Cifani C., Perfumi M., Massi M., Cuomo V., Trabace L., Ciccocioppo R. (2006): Effect of the cannabinoid CB1 receptor antagonist SR-141716A on etanol self-administration and etanol-seeking behaviour in rats. Psychopharmacology 183(4):394-403. - Enoch M.A., Goldman D. (2001): The genetics of alcoholism and alcohol abuse. Curr Psychiatry Rep 3(2):144-51. - Eriksson K. (1968): Genetic selection for voluntary alcohol consumption in the albino rat. Science 159:739–741. - Fadda F., Rosseti Z. (1998): Chronic ethanol consumption: From neuroadaptation to neurodegeneration. Progress in Neurobiology, 56, 385-431. - Felder C.C., Joyce K.E., Briley E.M. Glass M., Mackie K.P., Fahey K.J., Cullinan G.J., Hunden D.C., Johnson D.W., Chaney M.O., Koppel G.A., Brownstein M. (1998): LY320135, a novel cannabinoid CB1 receptor antagonist, unmasks coupling of the CB1 receptor to stimulation of cAMP accumulation. Journal of Pharmacology and Experimental Therapeutics 284, 291–297. - 197 - MENÚ SALIR Bibliografía - Felder C.C., Joyce K.E., Briley E.M., Mansouri J., Mackie K., Blond O., Lai Y., Ma A.L., Mitchell R.L. (1995): Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol 48(3): 443-50. - Felder C.C., Nielsen A., Briley E.M., Palkovits M., Priller J., Axelrod J., Nguyen D.N., Richardson J.M., Riggin R.M., Koppe G.A., Paul S.M., Becker G.W. (1996): Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 393: 231-235. - Freeland C.S., Poston J.S., Porrino L.J. (2000): Effects of SR141716A, a central cannabinoid receptor antagonist, on food-maintained responding. Pharmacol Biochem Behav 67:265-270. - Freedland C.S., Sharpe A.L., Samson H.H., Porrino L.J. (2001): Effects of SR141716A on ethanol and sucrose self-administration. Alcohol Clin Exp Res 25(2):277-82. - Fride E. (2002): Endocannabinoids in the central nervous system-an overview. Prostaglandins Leukot Essent Fatty Acids 66(2-3): 221-33. - Fride E., Mechoulam R. (1993): Pharmacological activity of the cannabinoid receptor agonist, anandamide, a brain constituent. Eur J Pharmacol. 231, 313314. - Froelich J.C. (1997): Opioid peptides. Alcohol Health Research World, 21, 132143. - Galiègue S., Mary S., Marchand J., Dussossoy D., Carrière D., Carayon P., Bouaboula M., Shire D., Le Fur G., Casellas P. (1995): Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem 232 : 54 - 61 . - Gallate J.E., McGregor I.S. (1999): The motivation for beer in rats: effects of ritanserin , naloxone and SR141716A. Psychopharmacology 142, 302-8. - Gallate J.E., Saharov T., Mallet P.E., McGregor I.S. (1999): Increased motivation for beer in rats following the administration of a cannabinoid receptor agonist. Eur J Pharmacol 370, 233-240.´ - Gaoni Y., Mechoulam R. (1964): Isolation, structure and partial synthesis of an active constituent of hashish. J. Am. Chem. Soc. 86, 1646-164. - Garbutt J.C., West S.L., Carey T.S., Lohr K.N., Crews F.T. (1999): Pharmacological treatment of alcohol dependence: A review of the evidence. Journal of the American Medical Association, 281, 1318-1325. - Gardner E.L. (1997): Brain reward mechanisms. En: Lowinson JH, Ruiz P, Milman RB, Langrod JG, eds. Substance abuse: A comprehensive textbook. 3° ed. Baltimore: Williamns and Wilkins; pág: 51-85. - 198 - MENÚ SALIR Bibliografía - Gardner E.L., Paredes W., Smith D., Donner A. Milling C., Cohen D., Morrison D. (1988): Facilitation of brain stimulation reward by delta-9tetrahydrocannabinol. Psychopharmacology, 96: 142-144. - Gardner E.L., Vorel S.R. (1998): Cannabinoid transmission and reward-related events. Neurobiology of Disease 5, 502–533. - Gebhart C.A., Naeser M.A., Butters N. (1984): Computerized measures of CT scan of alcoholics: thalamic region related to memory. Alcohol 1, 133-140. - Gerdeman G.L., Ronesi J., Lovinger D.M. (2002): Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nature Neuroscience 5, 446–451. - Gessa G.L., Serra S., Vacca G., Carai M.A., Colombo G. (2005): Suppressing effect of the cannabinoid CB1 receptor antagonist, SR147778, on alcohol intake and motivational properties of alcohol in alcohol-preferring sP rats. Alcohol Alcohol 40(1):46-53. - Gianoulakis C., de Waele J.P., Kiianmaa K. (1992): Differences in the brain and pituitary beta-endorphin system between the alcohol-preferring AA and alcoholavoiding ANA rats. Alcoholism, Clinical and Experimental Research 16: 453– 459. - Giros B., Jabe M., Jones S.R., Wightman R.M., Carol M.G. (1996): Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature, 379, 606-612. - Giuffrida A., Parsons L.H., Kerr T.M., Rodriguez de Fonseca F., Navarro M. y Piomelli D. (1999): Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 2, 358-63. - Giuffrida A., Rodriguez de Fonseca F., Piomelli D. (2000): Quantification of bioactive acylethanolamides in rat plasma by electrospray mass spectrometry. Anal Biochem 280(1): 87-93. - Glass M., Brotchie J.M., MAneuf Y.P. (1997): Modulation of neurotransmission by cannabinoids in the basal ganglia. Eur J Neurosci; 9:199-203. - Goeders N.E., Smith J.E. (1983): Cortical involvement in cocaine reinforcement. Science, 221: 773-775. - Gómez R., Navarro M., Ferrer B., Trigo J.M., Bilbao A., Del Arco I., Cipetelli A., Nava F., Piomelli D., Rodriguez de Fonseca F. (2002): A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. Journal of Neuroscience 22, 9612–9617. - Gong J.P., Onaivi E.S., Ishiguro H., Liu Q.R., Tagliaferro P.A., Brusco A., Uhl G.R. (2006): Cannabinoid CB2 receptors: inmunohistochemical localization in brain. Brain Res 1071(1):10-23. - 199 - MENÚ SALIR Bibliografía - González S., Cascio M.G., Fernandez-Ruiz J., Fezza F., Di Marzo V., Ramos J.A. (2002): Changes in endocannabinoid contents in the brain of rats chronically exponed to nicotine, ethanol or cocaine. Brain Research 954(1): 7381. - González S., Valenti M., de Miguel R., Fezza F., Fernández-Ruiz J., Di Marzo V., Ramos J.A. (2004): Changes in endocannabinoid contents in reward-related brain regions of alcohol-exposed rats, and their possible relevance to alcohol relapse. Br J Pharmacol 143:455-464. - Grobin A.C., Matthews D.B., Devaud L.L., Morrow, A.L. (1998): The role of GABAA receptors in the acute and chronic effects of ethanol. Psychopharmacology, 139, 2-19. - Guardia J. (2001): Neurobiología del alcoholismo. Curso de especialización en alcoholismo. Madrid: Fundación de Ayuda contra la Drogadicción; Capítulo 3. - Guerri C. (2000): Cómo actúa el alcohol en nuestro cerebro. Trastornos adictivos; 2 (1): 14-25. - Hajos N., Freund T.F. (2002): Pharmacological separation of cannabinoid sensitive receptors on hippocampal excitatory and inhibitory fibers. Neuropharmacology 43(4):503-10. - Hajos N., Ledent C., Freund T.F. (2001): Novel cannabinoid-sensitive receptor mediates inhibition of glutamaergic synaptic transmisión in the hippocampus. Neuroscience. 106, 1- 4. - Hansen H.S., Lauritzen L., Strand A.M., Vinggaard A.M., Frandsen A., Schousboe A. (1997): Characterization of glutamate-induced formation of Nacylphosphatidyl-ethanolamine and N-acylethanolamine in cultured neocortical neurons. J Neurochem. 69, 753-761. - Hansson A.C., Bermudez-Silva F.J., Marinen H., Hyytia P., Sanchez-Vera I., Rimondini R., Rodríguez de Fonseca F., Kunos G., Sommer W.H., Heilig M. (2007): Genetic impairment of frontocortical endocannabinoid degradation and high alcohol preference. Neuropsychopharmacolgy 32(1): 117- 26. - Hannson A.C., Cippitelli A., Sommer W.H., Fedeli A., Björk K., Soverchia L., Terasmaa A., Massi M., Heilig M., Ciccocioppo R. (2006): Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc Natl Acad Sci USA 103(41):45236-41. - Hansson A.C., Sommer W., Rimondini R., Andbjer B., Strömberg I., Fuxe K. (2003): c-fos reduces corticosterone-mediated effects on neurotrophic factor expression in the rat hippocampal CA1 region. J Neurosci 23(14): 6013-6022. - Hanus L., Abu-Lafi S., Fride E., Breuer A., Vogel Z., Shalev D.E., Kustanovich I., Mechoulam R. (2001): 2-arachidonylglycerylether, an endogenous agonist of the cannabinoid CB1 receptor. Proc Natl Acad Sci USA 98(7): 3662-5. - 200 - MENÚ SALIR Bibliografía - Harris G.C., Wimmer M., Aston-Jones G. (2005): A role for lateral hypothalamic orexin neurons in reward seeking. Nature 437(7058):556-9. - Heimer L., Harlan R. E., Alheid G. R, Garcia M., De Olmos J. (1997): Substantia innominata: A notion which impedes clinical-anatomical correlations in neuropsychiatric disorders. Neuroscience, 76, 957-1006. - Herkenham M. (1995): Localization of cannabinoid receptors in brain and periphery. In: Pertwee RG ed. Cannabinoid Receptors. Academic Press Limited: 145-166 - Herkenham M., Lynn A., Johnson M., Melvin L., De Costa B. y Rice K. (1991): Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 11:563-583. - Herring A.C., Kaminski N.E. (1999): Cannabinol-mediated inhibition of nuclear factor- κB, cAMP response element-binding protein, and interleukin-2 secretion by activated thymocytes. J Pharmacol Exp Ther 291 : 1156 - 1163. - Herz A. (1997): Endogenous opioid systems and alcohol addiction. Psychopharmacology, 99, 99-1 l t. - Hilf G., Gierschik P., Jakobs G.H. (1986): Muscarinic acetylcholine receptorstimulated binding of guanosine 5'-O-(3-thiotriphosphate) to guaninenucleotide-binding proteins in cardiac membranes. Eur J Biochem. 22; 186(3):725-31. - Hirst R.A., Almond S.L., Lambert D.G. (1996): Characterisation of the rat cerebellar CB1 receptor using SR141716A, a central cannabinoid receptor antagonist. Neurosci Lett 220: 101–104. - Hohmann A.G., Herkenham M. (2000): Localization of cannabinoid CB(1) receptor mRNA in neuronal subpopulations of rat striatum: a double-label in situ hubridization study. Synapse 37(1): 71-80. - Hollister L.E., Gillespie H.K. (1970): Marijuana, ethanol, and dextroamphetamine. Mood and mental function alterations. Arch Gen Psychiatry. 23, 199-203. - Holm S., (1979): A simple sequentially rejective multiple test procedure. Scand J Stat 6:65-70. - Honse Y., Ren H., Lipsky R.H., Peoples R.W. (2004): Sites in the fourth membrane-associated domain regulate alcohol sensitivity of the NMDA receptor. Neuropharmacology 46(5):647-54. - Howlett A.C., Barth F., Bonner T.I., Cabral G., Casellas P., Devane W.A., Felder C.C., Herkenham M., Mackie K., Martin B.R., Mechoulam R., Pertwee R.G. (2002): International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacological Reviews. 54, 161-202. - 201 - MENÚ SALIR Bibliografía - Hungund B.L., Basavarajappa B.S. (2000): Distinct differences in the cannabinoid receptor binding in the brain of C57BL/6 and DBA/2 mice, selected for their differences in voluntary ethanol consumption. J Neurosci Res 60:122128. - Hungund B.L., Basavarajappa B.S., Vadasz C., Kunos G., Rodriguez de Fonseca F., Colombo G., Serra S., Parsons L., Koob G.F. (2002): Ethanol, endocannabinoids, and the cannabinoidergic signaling system. Alcohol Clin Exp Res. 26, 565-74. - Hungund B.L., Szakall I., Adam A., Basavarajappa B.S., Vadasz C. (2003): Cannabinoid CB1 receptor knockout mice exhibit markedly reduced voluntary alcohol consumption and lack alcohol-induced dopamine release in the nucleus accumbens. J Neurochem 84(4):698-704. - Hunter S.A., Burstein S.H. (1997): Receptor mediation in cannabinoid stimulated arachidonic acid mobilization and anandamide synthesis. Life Sciences 60:1563-1573. - Imperato A., Dazzi L., Carta G., Colombo G., Biggio G. (1998): Rapid increase in basal acetylcholine release in the hippocampus of freely moving rats induce by withdrawal from long-term ethanol intoxication. Brain Res 784, 347-350. - Johnson B.A., Campling G.M., Griffiths P., Cowen P.J. (1993): Attenuation of some alcohol-induced mood changes and the desire to drink by 5-HT3 receptor blockade: A preliminary study in healthy male volunteers. Psychopharmacology, 112, 142-144. - Jones N., Messenger M.J., O’neill M.J., Oldershaw A., Gilmour G., Simmons R.M., Iyengar S., Libri V., Tricklebank M., Williams S.C. (2007): AMPA receptor potentiation can prevent ethanol-induced intoxication. Neuropsychopharmacology - Julian M.D., Martin A.B., Cuellar B., Rodriguez de Fonseca F., Navarro M., Moratalla R., Garcia-Segura L.M. (2003): Neuroanatomical relationship between type 1 cannabinoid receptors and dopaminergic systems in the rat basal ganglia. Neuroscience 119, 309–318. - Kalant H., LeBlanc A.E. (1974): Effect of acute and chronic pretreatment with delta1- tetrahydrocannabinol on motor impairment by ethanol in the rat. Can J Physiol Pharmacol. 52, 291-297. - Kathuria S., Gaetani S., Fegley D., Valiño F., Duranti A., Tontini A., Mor M., Tarzia G., La Rana G., Calignano A., Giustino A., Tattoli M., Palmery M., Cuomo V., Piomelli D. (2003): Modulation of anxiety through blockade of anandamide hydrolysis. Nature Medicine 9, 76 – 81. - 202 - MENÚ SALIR Bibliografía - Kempe K., Hsu F., Bohrer A., Turk J. (1996): Isotope dilution mass spectrometric measurements indicate that arachidonylethanolamide, the proposed endogenous ligand of the cannabinoid receptor, accumulates in rat brain tissue post mortem but is contained at low levels in or is absent from fresh tissue. Journal of Biological Chemistry 271, 17287-17295. - Killcross S., Coutureau E. (2003): Coordination of actions and habits in the medial prefrontal cortex of rats. Cereb Cortex 13:400-408. - Kötter K., Klein J. (1999): Ethanol inhibits astroglial cell proliferation by disruption of phospholipase D-mediated signaling. J Neurochem 73, 2517-2523. - Kreitzer A.C., Regerhr W.G. (2001): Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29(3):717-727. - Kringelbach M.L. (2005): The human orbitofrontal cortex: linking reward to hedonic experience. Nat Rev Neurosci 6, 691-702. - Kunos G., Batkai S., Offertaler L., Mo F., Liu J., Karcher J., Harvey-White J. (2002): The quest for a vascular endothelial cannabinoid receptor. Chem Phys Lipids. 121, 45-56. - Lal H., Harris C.M., Benjamin D., Springfield A.C., Bhadra S., Emmett-Oglesby M.W. (1988): Characterization of a pentylenetetrazol-like interoceptive stimulus produced by ethanol withdrawal. J Pharmacol Exp Ther; 47(2):508-18. - Lallemand F., De Witte P. (2006): SR147778, a CB1 cannabinoid receptor antagonist, suppresses ethanol preference in chronically alcoholized Wistar rats. Alcohol 39:125-34. - Lallemand F., Soubrie P.H., De Witte P.H. (2001): Effects of CB1 cannabinoid receptor blockade on ethanol preference after chronic ethanol administration. Alcohol Clin Exp Res 25(9):1317-23. - Lallemand F., Soubrié P., De Witte P. (2004): Effects off CB1 cannabinoid receptor blockade on ethanol preference after chronic alcohol administration combined with repeated re-exposures and withdrawals. Alcohol Alcohol 39(6):486-92. - Ledent C., Valverde O., Cossu G., Petitet F., Aubert J.F., Beslot F., Bohme G.A., Imperato A., Pedrazzini T., Roques B.P., Vassart G., Fratta W., Parmentier M. (1999): Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 283, 401–404. - Leshner A.I. (1997): Drug abuse and addiction treatment research. The next generation. Arch Gen Psychiatry 54(8):691-4. - 203 - MENÚ SALIR Bibliografía - Leung D., Saghatelian A., Simon G.M., Cravatt B.F. (2006): Inactivation of Nacyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry 45(15):4720-4726. - Liu Q., Tonai T., Ueda N. (2002): Activation of N-acylethanolamine-releasing phospholipase D by polyamines. Chem Phys Lipids 115(1-2): 77-84. - Lobina C., Agabio R., Diaz G., Fa M., Fadda F., Gessa G.L., Reali R., Colombo G. (1997): Constant absolute ethanol intake by Sardinian alcohol-preferring rats independent of ethanol concentrations. Alcohol Alcohol 32(1):19-22. - López-Moreno J.A., González-Cuevas G., Rodríguez de Fonseca F., Navarro M. (2004): Long-lasting increase of alcohol relapse by the cannabinoid receptor agonist WIN 55,212 during alcohol deprivation. J Neurosci 24(38):8245-52. - Lovinger D.M. (1997): Alcohols and neurotransmitter gated ion channels: past, present and future. Naunyn Schmiedebergs Arch Pharmacol 356(3):267-82. - Lovinger D.M. (1999): The role of serotonin in alcohol's effects on the brain. Current Separations, 18, 23-28. - Maccarrone M., Bari M., Di Rienzo M., Finazzi-Agro A., Rossi A. (2003a): Progesterona activates fatty acid amide hydrolase (FAAH) promoter in human T lymphocytes through the transcription factor Ikaros. Evidence for a synergistic effect of leptin. J Biol Chem 278(35):32726-32. - Maccarrone M., Di Rienzo M., Finazzi-Agro A., Rossi A. (2003b): Leptin activates the anandamide hydrolase promoter in human T lymphocytes throug STAT3. J Biol Chem 278(15):13318-24. - Mackie K., Hille B. (1992): Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA 89(9): 3825- 9. - Mackie K., Lai Y., Westenbroek R., Mitchell R. (1995): Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci 15(10):6552- 61. - Maejima T., Hashimoto K., Yoshida T., Aiba A. y Kano M. (2001): Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31, 463-75. - Mailleux P., Vanderhaeghen J.J. (1992): Localization of cannabinoid receptor in human developing and adult basal ganglia. Higher levels in the striatonigral neurons. Neurosci Lett 148 (1-2): 173-6. - Maldonado R., Rodriguez de Fonseca F. (2002): Cannabinoid addiction: behavioral models and neural correlates. Journal of Neuroscience 22, 3326– 3321. - 204 - MENÚ SALIR Bibliografía - Maldonado R., Valverde O., Berrendero F. (2006): Involvement of the endocannabinoid system in drug addiction. Trends Neurosci 29(4): 225-32. - Maresz K., Carrier E.J., Ponomarev E.D., Hillard C.J., Dittel B.N. (2005): Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem 95: 437 - 445. - Marsicano G., Goodenough S., Monory K. Hermann H., Eder M., Cannich A., Azad S.C., Cascio M.G., Gutierrez S.O., van der Stelt M., Lopez-Rodriguez M.L., Casanova E., Schutz G., Zieglgansberger W., Di Marzo V., Behl C., Lutz B. (2003): CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302, 84–88. - Marsicano G., Wotjak C.T., Azad S.C., Bisogno T., Rammes G., Cascio M.G., Hermann H., Tang J., Hofmann C., Zieglgansberger W., Di Marzo V., Lutz B. (2002): The endogenous cannabinoid system controls extinction of aversive memories. Nature 418, 530–534. - Martin B.R., Lichtman A.H. (1998): Cannabinoid transmission and pain perception. Neurobiology of Disease 5, 447–461. - Matsuda L.A., Lolait S.J., Brownstein M.J., Young A.C., Bonner T.I. (1990): Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346: 561–564. - McAllister S.D., Griffin G., Satin L.S., Aboos M.E. (1999): Cannabinoid receptors can activate and inhibit G protein-coupled inwardly rectifying potassium channels in a xenopus oocite expression system. J Pharmacol Exp Ther 291(2): 618- 26. - McBride W.J., Li T.J. (1998): Animals models of alcoholism: neurobiology of high alcohol-drinking in rodents. Crit Rev Neurobiol 12 (4): 339-69. - McGregor I.S., Dam K.D., Mallet P.E., Gallate J.E. (2005): Delta9-THC reinstates beer- and sucrose-seeking behaviour in abstinent rats: comparison with midazolam, food deprivation and predator odour. Alcohol Alcohol 40(1): 35- 45. - McMillen B.A. (1997): Toward a definition of a valid model of alcoholism: multiple animal models for multiple diseases. Alcohol 14(4):409-19. - McMillen B.A., Shore P.A. (1977): The relative functional availability of brain noradrenaline and dopamine storage pools. J Pharm Pharmacol 29(12):780-1. - Mechoulam R., Fride E., Ben-Shabat S., Meiri U., Horowitz M. (1998): Carbachol, an acetylcholine receptor agonist, enhances production in rat aorta of 2-arachidonoyl glycerol, a hypotensive endocannabinoid. Eur J Pharmacol 362(1): 1- 3. - 205 - MENÚ SALIR Bibliografía - Mechoulam R., Hanus L., Martin B. R. (1994): Search for endogenous ligands of the cannabinoid receptor. Biochem Pharmacol 48: 1537-1544. - Melichar J.K., Daglish M.R., Nutt D.J. (2001): Addiction and withdrawal: current views. Curr Opin Pharmacol 1(1):84-90. - Meyer J.S. y Quenzer L.F. (2005): Psychopharmacology: Drugs, the brain, and behavior. Sunderland, MA: Sinauer Associates. - Mokdad A.H., Marks J.S., Stroup D.F. y Gerderding J.L. (2005): Actual causes of death in the United States, 2000. JAMA 19;293(3):298. - Molina-Holgado E., Vela J.M., Arévalo-Martin A., Almazan G., MolinaHolgado F., Borrell J. y Guaza C. (2002): Cannabinoids promote oligodendrocyte progenitor survival: involvement of cannabinoid receptor and phosphatidyllinositol-3 kinase/Akt signaling. J Neurosci; 22: 9742 - 9753. - Möller C., Wiklund L., Thorsell A., Hyytia P., Heilig M. (1997): Decreased measures of experimental anxiety in rats bred for high alcohol preference. Alcoholism, Clinical and Experimental Research 21: 656–660. - Montoliu C., Valles S., Renau-Piqueras J., Guerri C. (1994): Ethanol-induced oxygen radical formation and lipid peroxidation in rat brain: effect of chronic alcohol consumption. J Neurochem.; 63(5):1855-62. - Moranta D., Esteban S., García-Sevilla J.A. (2006): Ethanol desensitizes cannabinoid CB1 receptor modulating monoamine synthesis in the rat brain in vivo. Neurosci Lett 392: 58- 61. - Munro S., Thomas K.L., Abu-Shaar M. (1993): Molecular characterization of a peripheral receptor for cannabinoids. Nature 365(6441): 61-5. - Naassila M., Pierrefiche O., Ledent C., Daoust M. (2004): Decreased alcohol self-administration and increased alcohol sensitivity and withdrawal in CB1 receptor knockout mice. Neuropharmacology 46(2):243-53. - Naranjo C.A., Paulos C.X., Bremner K.E., Lanctot K.L. (1992): Citalopram decreases desirability, liking, and consumption of alcohol in alcohol-dependent drinkers. Clinical Pharmacology and Therapeutics, 51, 729-739. - Navarro M., Hernandez E., Munoz R.M., del Arco I., Villanua M.A., Carrera, M.R., Rodríguez de Fonseca F. (1997): Acute administration of the CB1 cannabinoid receptor antagonist SR 141716A induces anxiety-like responses in the rat. Neuroreport 8, 491–496. - Navarro M., Carrera M.R., Fratta W., Valverde O., Cossu G., Fattore L., Chowen J.A., Gomez R., del Arco I., Villanua M.A., Maldonado R., Koob G.F., Rodríguez de Fonseca F. (2001): Functional interaction between opioid and cannabinoid receptors in drug self-administration. Journal of Neuroscience 21, 5344–5350. - 206 - MENÚ SALIR Bibliografía - Navarro M., Chowen J., Rocio A., Carrera M., del Arco I., Villanua M.A., Martin Y., Roberts A.J., Koob G.F., de Fonseca F.R. (1998): CB1 cannabinoid receptor antagonist-induced opiate withdrawal in morphine-dependent rats. Neuroreport 9(15):3397- 402. - Nestler E.J. (2001): Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2(3):215. - Nestler E.J., Aghajanian G.K. (1997): Molecular and cellular basis of addiction. Science 3; 278(5335): 58-63 - Nutt D. (1999): Alcohol and the brain. Pharmacological insights for psychiatrists. Br J Psychiatry; 175:114-9. - Nylander I., Hyytia P., Forsander O., Terenius L. (1994): Differences between alcohol-preferring (AA) and alcoholavoiding (ANA) rats in the prodynorphin and proenkephalin systems. Alcoholism, Clinical and Experimental Research 18: 1272–1279. - Ohno-Shosaku T., Maejima T., Kano M. (2001): Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynapic terminals. Neuron 29(3):729-738. - Okamoto Y., Morishita J., Tsuboi K., Tonai T., Ueda N. (2004): Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem 279, 5298-5305. - Olds J. (1956): A preliminary mapping of electrical reinforcing effects in the rat brain. J Comp Physiol Psychol 49(3):281-5. - Ortiz S., Oliva J.M., Perez-Rial S., Palomo T., Manzanares J. (2004): Chronic ethanol consumption regulates cannabinoid CB1 receptor gene expression in selected regions of rat brain. Alcohol Alcohol 39(2):88-92. - Panikashvili D., Simeonidou C., Ben-Shabat S., Hanus L., Breuer A., Mechoulam R., Shohami E. (2001): An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 413, 527–531. - Patel S., Roelke C.T., Rademacher D.J., Hillard C.J. (2005): Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur J Neurosci 21, 1057-1069. - Paxinos G. y Watson C. (1986): The rat brain in stereotaxic coordinates. Second edition. New York, Academic Press. - Paxinos G. y Watson C. (1997): The rat brain in stereotaxic coordinates. Compact Third Edition. San Diego, Academic Press. - Pertwee R.G. (1988): The central pharmacology of psychotropic cannabinoids. Pharmacol. Therapeut. 36, 189-261. - 207 - MENÚ SALIR Bibliografía - Pertwee R.G. (1997): Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther 74; 129-180. - Pertwee R.G. (2004): Novel pharmacological targets for cannabinoids. Current Neuropharmacology 2, 9-29. - Petersen G., Hansen H.S. (1999): N-acylphosphatidylethanolamine-hydrolysing phospholipase D lacks the ability to transphosphatidylate. FEBS Lett 455(1-2): 41- 4. - Pettinati H.M. (1996): Use of serotonin selective pharmacotherapy in the treatment of alcohol dependence. Alcoholism: Clinical and Experimental Research, 20, 23A-29A. - Piomelli D. (2003): The molecular logic of endocannabinoid signalling. Nature Reviews Neuroscience 4, 873–884. - Piomelli D., Giuffrida D., Calignano A., Rodriguez de Fonseca F. (2000): The endocannabinoid system as a target for therapeutic drugs. Trends Pharmacol Sci 21: 218-224. - Pistis M., Ferraro L., Pira L., Flore G., Tanganelli S., Gessa G.L., Devoto P. (2002): Delta (9)-tetrahydrocannabinol decreases extracellular GABA and increases extracellular glutamate and dopamine levels in the rat prefrontal cortex: an in vivo microdialysis study. Brain Res 948:155-158. - Poncelet M., Maruani J., Calassi R., Soubrie P. (2003): Overeating, alcohol and sucrose consumption decrease in CB1 receptor deleted mice. Neurosci Lett 343(3):216-8. - Porter A.C., Sauer J.M., Knierman M.D., Becker G.W. Berna M.J., Bao J., Nomikos G.G., Carter P., Bymaster F.P., Leese A.B., Felder C.C. (2002): Characterization of a novel endocannabinoid, virodhamina, with antagonist activity at the CB1 receptor. J Pharmacol Exp Ther 301(3): 1020-4. - Racz I., Bilkei-Gorzo A., Toth Z.E., Michel K., Palkovits M., Zimmer A. (2003): A critical role for the cannabinoid CB1 receptors in alcohol dependence and stress-stimulated ethanol drinking. J Neurosci 23(6):2453-8. - Raskin N.H. y Sokoloff L. (1972): Ethanol-induced adaptation of alcohol dehydrogenase activity in rat brain. Nat New Biol.; 236(66):138- 40. - Ren H., Honse Y., Peoples R.W. (2003): A site of alcohol action in the fourth membrane-associated domain of the N-methyl-D-aspartate receptor. J Biol Chem 278(49):48815-20. - Ren H., Salous A.K., Paul J.M., Lipsky R.H., Peoples R.W. (2007): Mutations at F637 in the NMDA receptor NR2A subunit M3 domain influence agonist potency, ion channel gating and alcohol action. Br J Pharmacol 151(6):749-57. - 208 - MENÚ SALIR Bibliografía - Rimondini R., Arlinde C., Sommer W., Heilig M. (2002): Long-lasting increase in voluntary ethanol consumption and transcriptional regulation in the rat brain after intermittent exposure to alcohol. The FASEB Journal 16, 27–35. - Rivier C. (1993): Acute interactions between cytokines and alcohol on ACTH and corticosterone secretion in the rat. Alcohol Clin Exp Res. 17, 946-950. - Robbe D., Kopf M., Remaury A., Bockaert J., Manzoni O.J. (2002): Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proceedings of the National Academy of Sciences of the United States of America 99, 8384–8348. - Robbins T.W. y Everitt B.J. (1999): Drug addiction: bad habits add up. Nature, 398: 567-570. - Rodríguez de Fonseca F., del Arco I., Bermúdez-Silva F.J., Bilbao A., Cippitelli A., Navarro M. (2005): The endocannabinoid system: physiology and pharmacology. Alcohol Alcoholism 40(1):2-14. - Rodríguez de Fonseca F., Carrera M.R., Navarro M., Koob G.F., Weiss F. (1997): Activation of corticotropin-releasing factor in the limbic system during cannabinoid withdrawal. Science 276, 2050–2054. - Rodríguez de Fonseca F., Del Arco I., Martin-Calderon J.L., Gorriti M.A., Navarro M. (1998): Role of the endogenous cannabinoid system in the regulation of motor activity. Neurobiology of Disease 5, 483–501. - Rodríguez de Fonseca F., Navarro M. (1998): Role of the limbic system in dependence on drugs. Ann Med 30(4):397-405. - Rodríguez D.F., Navarro M., Gomez R., Escuredo L., Nava F., Fu J., MurilloRodríguez E., Giuffrida A., LoVerme J., Gaetani S., Kathuria S., Gall C., Piomelli D. (2001): An anorexic lipid mediator regulated by feeding. Nature 414: 209–212. - Rodríguez de Fonseca F., Roberts A.J., Bilbao A., Koob G.F., Navarro M. (1999): Cannabinoid receptor antagonist SR141716A decreases operant ethanol self administration in rats exposed to ethanol-vapor chambers. Zhongguo Yao Li Xue Bao. 20, 1109-14. - Rodríguez de Fonseca F., Rubio P., Menzaghi F., Merlo-Pich E., Rivier J., Koob G.F., Navarro M. (1996): Corticotropin-releasing factor (CRF) antagonist [DPhe12,Nle21,38,C alpha MeLeu37]CRF attenuates the acute actions of the highly potent cannabinoid receptor agonist HU-210 on defensive–withdrawal behavior in rats. Journal of Pharmacology and Experimental Therapeutics 276, 56 – 64. - Rodríguez J.J., Mackie K., Pickel V.M. (2001): Ultrastructural localization of the CB1 cannabinoid receptor in µ-opioid receptor patches of the rat caudate putamen nucleus. J Neurosci; 21: 823 - 833. - 209 - MENÚ SALIR Bibliografía - Rout U.K. (1992): Alcohol dehydrogenases in the brain of mice. Alcohol Clin Exp Res.;16(2):286-9. - Rueda D., Navarro B., Martinez-Serrano A., Guzman M., Galve-Roperh I. (2002): The endocannabinoid anandamide inhibits neuronal progenitor cell differentiation through attenuation of the Rap1/B-Raf/ERK pathway. Journal of Biological Chemistry 277, 46645–46650. - Ryan C., Butters N. (1980): Learning and memory impairments in young and old alcoholics: evidence for the premature-aging hypothesis. Alcoholism: Clinical and Experimental Research 4, 288-293. - Samson H.H. (1986): Initiation of ethanol reinforcement using a sucrosesubstitution procedure in food- and water-sated rats. Alcohol Clin Exp Res 10(4):436-42. - Samson H.H. y Harris R.A. (1992): Neurobiology of alcohol abuse. Trends in Pharmacological Science, 13, 206-211. - Sanchis-Segura C., Cline B.H., Marsicano G., Lutz B., Spanagel R. (2004): Reduced sensitivity to reward in CB1 knockout mice. Psychopharmacology 176(2):223-32. - Schlicker E., Kathmann M. (2001): Modulation of transmitter release via presynaptic cannabinoid receptor. Trends in Pharmacological Sciences 22, 565– 572. - Schmid P.C., Reddy P.V., Natarajan V., Schmid H.H. (1983): Metabolism of Nacylethanolamine phospholipids by a mammalian phosphodiesterase of the phospholipase D type. Journal of Biological Chemistry 258, 9302-9306. - Schmidt L.G., Samochowiec J., Finckh U., Fiszer-Piosik E., Horodnicki J., Wendel B., Rommelspacher H., Hoeche M.R. (2002): Association of a CB1 cannabinoid receptor gene (CNR1) polymorphism with severe alcohol dependence. Drug Alcohol Depend. 65: 221-4. - Schulteis G., Markou A., Cole M., Koob G.E. (1995). Decreased brain reward produced by ethanol withdrawal. Proceedings of the National Academy of Sciences, 92, 5880-5884. - Seeman P. (1972): The membrane actions of anesthetics and tranquilizers. Pharmac. Rev. 24, 583-655. - Serra S., Brunetti G., Pani M., Vacca G., Carai M.A., Gessa G.L., Colombo G. (2002): Blockade by the cannabinoid CB(1) receptor antagonist, SR141716, of alcohol deprivation effect in alcohol-preferring rats. Eur J Pharmacol 443(13):95-7. - 210 - MENÚ SALIR Bibliografía - Serra S., Carai M.A., Brunetti G., Gomez R., Melis S., Vacca G., Colombo G., Gessa G.L. (2001): The cannabinoid receptor antagonist SR 1417416A prevents acquisition of drinking behaviour in alcohol-preferring rats. Eur J Pharmacol 430(2-3): 369-71. - Shippenberg T.S., Elmer G.I. (1998): The neurobiology of opiate reinforcement. Crit Rev Neurobiol 12(4):267-303. - Sim-Selley L.J., Brunk L.K., Selley D.E. (2001): Inhibitory effects of SR141716A on G-protein activation in rat brain. European Journal of Pharmacology 414(2-3):135-143. - Sinclair J.D., Le A.D., Kiianmaa K. (1989): The AA and ANA rat lines, selected for differences in voluntary alcohol consumption. Experientia 45: 798 – 805. - Sipe J.C., Chiang K., Gerber A.L., Beutler E., Cravatt F. (2002): A missense mutation in human fatty acid amide hydrolase associated with problem drug abuse. PNAS 99 (12), 8394-8399. - Soini S.L., Honkanen A., Hyytia P., Korpi E.R. (1999): [3H] ethylketocyclazocine binding to brain opioid receptor subtypes in alcoholpreferring AA and alcohol-avoiding ANA rats. Alcoholism 18: 27–34. - Solomon R.L. (1980): Recent experiments testing an opponent-process theory of acquired motivation. Acta Neurobiol Exp (Wars); 40(1):271-89. - Stella N., Piomelli D. (2001): Receptor-dependent formation of endogenous cannabinoids in cortical neurons. Eur J Pharmacol 425, 189-196. - Stella N., Schweitzer P., Piomelli D. (1997): A second endogenous cannabinoid that modulates long-term potentiation. Nature 21; 388(6644): 773-8. - Straiker A., Stella N., Piomelli D., Mackie K., Karten H.J., Maguire G. (1999): Cannabinoid CB1 receptors and ligands in vertebrate retina: localization and function of an endogenous signaling system. Proceedings of the National Academy of Sciences of the United States of America 96, 14565–14570. - Sugiura T., Kodaka T., Nakane S., Kishimoto S., Kondo S., Waku K. (1998): Detection of an endogenous cannabimimetic molecule, 2-arachidonoylglycerol, and cannabinoid CB1 receptor mRNA in human vascular cells: is 2arachidonoylglycerol a possible vasomodulator? Biochem Biophys Res Commun 243(3): 838-43. - Sugiura T., Kondo S., Sukagawa A., Nakane S., Shinoda A., Itoh K., Yamashita A., Waku K. (1995): 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Comu 215: 89-97. - Sugiura T., Yoshinaga N., Waku K. (2001): Rapid generation of 2arachidonoylglycerol, an endogenous cannabinoid receptor ligand, in rat brain after decapitation. Neurosci Lett 19; 297(7): 175-8. - 211 - MENÚ SALIR Bibliografía - Sugiura T., Waku K. (2002): Cannabinoid receptor and their endogenous ligands. J Biochem (Tokyo) 132(7): 7- 12. - Sun Y.X., Tsuboi K., Okamoto Y., Tonai T., Murakami M., Kudo I., Ueda N. (2004): Biosynthesis of anandamide and N-palmitoylethanolamide by sequential actions of phospholipase A2 and lysophospholipase D. Biochem J 380:749- 56. - Tabakoff B. y Hoffman P.L. (1996): Alcohol addiction: an enigma among us. Neuron. May; 16(5):909-12. - Terranova J.P., Storme J.J., Lafon N., Perio A., Rinaldi-Carmona M., Le Fur G., Soubrié, P. (1996): Improvement of memory in rodents by the selective CB1 cannabinoid receptor antagonist, SR 141716. Psychopharmacology 126, 165– 172. - Tierney P.L., Degenetais E., Thierry A.M., Glowinski J., Gioanni Y. (2004): Influence of the hippocampus on interneurons of the rat prefrontal cortex. Eur J Neurosci 20:514-524. - Trang T., Sutak M., Jhamandas K. (2007): Involvement of cannabinoid (CB1)receptors in the development and maintenance of opioid tolerance. Neuroscience 146(3):1275-88. - Traynor J.R. y Nahorski S.R. (1995): Modulation by m-opioid agonists of guanosine-59-O-(3-[35S]thio)triphosphate binding to membranes from human neuroblastoma SH-SY5Y cells. Mol Pharmacol 47:848–854. - Tsou K., Brown S., Sanudo-Pena M.C., Mackie K., Walker J.M. (1998): Inmunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83:393- 411. - Ueda N., Liu Q., Yamanaka K. (2001): Marked activation of the Nacylphosphatidylethanolamine-hydrolyzing phosphodiesterase by divalent cations. Biochim Biophys Acta 31;1532(1-2): 121-7. - Upadhya S.C., Tirumalai P.S., Boyd M.R., Mori T., Ravindranath V. (2000): Cytochrome P4502E (CYP2E) in brain: constitutive expression, induction by ethanol and localization by fluorescence in situ hybridization. Arch Biochem Biophys.; 373(1):23-34. - Van der Brink W., van Ree J.M. (2003): Pharmacological treatments for heroin and cocaine addiction. Eur Neuropsychopharmacol 13(6):476-87. - Vogel Z., Barg J., Levy R., Saya D., Heldman E., Mechoulam R. (1993): Anandamide, a brain endogenous compound, interacts specifically with cannabinoid receptors and inhibits adenylate cyclase. J Neurochem 61(1):352- 5. - Walker D., Blednov B.F., Cravatt B.F., Harris R.A. (2005): Increased ethanol consumption in mice lacking fatty acid amide hydrolase: implications for endocannabinoids. Alcohol Clin Exp Res 29:99A. - 212 - MENÚ SALIR Bibliografía - Walker J.M., Huang S.M., Strangman N.M., Tsou K., Sanudo-Pena M.C. (1999): Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci U S A. 96, 12198-203. - Walter L., Stella N. (2003): Endothelin-1 increases 1-arachidonoyl glycerol (2AG) production in astrocytes. Glia 44(1): 85- 90. - Wang L., Liu J., Harvey-White J., Zimmer A., Kunos G. (2003): Endocannabinoid signaling via cannabinoid receptor 1 is involved in ethanol preference and its age-dependent decline in mice. Proc Natl Acad Sci USA 100: 1393–1398. - Weiss F., Lorang M.T., Bloom F.E., Koob G.F. (1993): Oral alcohol selfadministration stimulates dopamine release in the rat nucleus accumbens: genetic and motivational determinants. J Pharmacol Exp Ther 267(1):250-8. - Williams H.E. (1982): Alcoholic hypoglycemia and ketoacidosis. Med Clin North Am 68, 33-8. - Wilson R.I., Kunos G., Nicoll R.A. (2001): Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31, 453–462. - Wilson R.I., Nicoll R.A. (2001): Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410, 588-92. - Wilson R.I., Nicoll R.A. (2002): Endocannabinoid signaling in the brain. Science 296, 678–682. - Wise R.A. (1981): Brain dopamine and reward. En: Theory in psychopharmacology, Vol. 1, Cooper SJ. Ed. Academic Press, New York, pág.: 103 - Wise R.A. (2000): Interactions between medial prefrontal cortex and mesolimbic components of brain reward circuitry. Prog Brain Res 126:255-62. - Wise R. A., Bauco P., Carlezon W. A. Jr., Trojniar W. (1992): Self-stimulation and drug reward mechanisms. Ann N Y Acad Sci 28(654):192-8. - Zhang P.W., Ishiguro H., Ohtsuki T., Hess J., Carillo F., Walher D., Onaivi E.S., Arinami T., Uhl G.R. (2004): Human cannabinoid receptor 1: 5’ exons, candidate regulatory regions, polymorphisms, haplotypes and association with polysubstance abuse. Mol Psychiatry 9(10):916-31. - Zhou E.C., Pu C.K., Murphy J., Lumeng L., Li T.K. (1994): Serotonergic neurons in alcohol preferring rats. Alcohol, 11, 397-403. - Zimatkin S.M. y Deitrich R.A. (1995): Aldehyde dehydrogenase activities in the brains of rats and mice genetically selected for different sensitivity to alcohol. Alcohol Clin Exp Res.; 19(5):1300-6. - 213 - MENÚ SALIR Bibliografía - Zoli M., Bettuzzi S., Ferraguti F., Ingletti M., Zini I., Fuxe K., Agnati L., Corti A. (1991): Regional increases in ornithine decarboxylase mRNA levels in the rat brain after partial mesodiencephalic hemitransection as revealed by in situ hybridization histochemistry. Neurochem Int 18:347–352. - Zygmunt P.M., Petersson J., Andersson D.A., Chuang H., Sφrgard M., Di Marzo V., Julius D. y Högestätt E.D. (1999): Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 400, 452-457. - 214 -