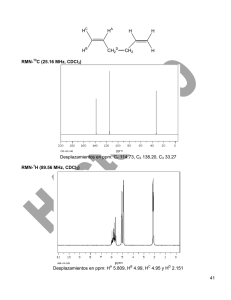
Santiago Barroso Ivars
Anuncio

DEPARTAMENT DE QUIMICA ORGÀNICA CATÁLISIS ASIMÉTRICA EN REACCIONES DE CICLOADICIÓN MEDIANTE COMPLEJOS QUIRALES DE Cu (II). SANTIAGO BARROSO IVARS UNIVERSITAT DE VALÈNCIA Servei de Publicacions 2009 Aquesta Tesi Doctoral va ser presentada a València el dia 29 de juliol de 2009 davant un tribunal format per: - Dr. Santos Fustero Lardiés Dr. Pedro Miguel Carda Usó Dr. Juan-Enrique Oltra Ferrero Dr. Adriaan Jacobus Minnaard Dra. Isabel Fernández Picot Va ser dirigida per: Dr. José Ramón Pedro Llinares Dr. Gonzalo Blay Llinares ©Copyright: Servei de Publicacions Santiago Barroso Ivars Dipòsit legal: V-4146-2010 I.S.B.N.: 978-84-370-7633-1 Edita: Universitat de València Servei de Publicacions C/ Arts Gràfiques, 13 baix 46010 València Spain Telèfon:(0034)963864115 I II Facultat de Química Departament de Química Orgànica CATÁLISIS ASIMÉTRICA EN REACCIONES DE CICLOADICIÓN MEDIANTE COMPLEJOS QUIRALES DE CU(II) Santiago Barroso Ivars Tesis Doctoral València, 2009 III IV Memoria presentada para optar al grado de Doctor en Ciencias Químicas Santiago Barroso Ivars DIRECTORES Dr. D. José Ramón Pedro Llinares Dr. D. Gonzalo Blay Llinares V VI An die Musik VII VIII Agradecimientos / Agraïments / Acknowledgements Agradecer en primer lugar a la Universitat de València la concesión de la beca predoctoral V Segles, sin cuya financiación hubiera sido muy duro realizar esta tesis doctoral. Després d’estar 7 anys amb ells, incloent l’últim any de carrera, agrair decididament als meus directors, Gonzalo i José Ramón, la seua ajuda i l’intens treball realitzat per a que aquesta tesi arribara a bon port. Estendre l’agraïment a la resta de professores del grup d’investigació, Isabel i Mari Llum, pel seu suport, les hores de conversa i les sessions de RMN. He d’agrair també l’ajuda que m’han prestat tots els companys que han passat pel laboratori durant tots aquests anys, en especial Carlos (també gran company de pis), Alícia Marco, Alicia Monleón, Víctor H., Estela, Eva, Belén i Lina. Vull incloure en aquesta llista als companys del “laboratori 3” Nacho, Rosa, Xavi, Antonio, David i Ismael. Cal dir que m’emporte una gran estima i amistat de molts d’ells, incloent en aquest grup a Anna Ibàñez i a Vio. Sense eixir del treball, recordar per la seua atenció al personal de Secretaria, Vicenta en particular, als tècnics del departament i a la professora Ana Cuñat. També al servei de masses, Sales i Isabel, pel seu esforç i la seua simpatia. I would like to express my gratitude to Professor Adri Minnaard for giving me the opportunity to stay 3 months in his group and for his kindly attention. I did not only learn quite a lot about research and English, but I also had a good time and found very good friends. Thanks to Tatiana, Núria, Johan, Robert, Mahthild, Lorena, Marta and all the nice people who I met there. Take care… I go back! També vull recordar a Joves Investigadors, la Federación de Jóvenes Investigadores i a tota la gent que he conegut en els anys d’activitat dintre d’aquestes organitzacions. La seua labor ha permès dignificar la figura del jove investigador i avançar poc a poc en els seus drets. Investigar es Trabajar. Ningún Investigador Sin Contrato. Por una Carrera Investigadora Digna. I, on ja el campus és una difusa referència, queden les (altres) amistats. La tesi és una cosa dura i va fortament associada a la vida d’estos anys. Tindre els amics al costat facilita les coses i cal agrair-ho. A Anna, que des de primer de carrera continua estant molt a prop meu. A Mariola, sempre disposta als bons consells i la conversa. A la “porta nº 19”: Almu, Sara, Marta, la Mari i Ivan, que sempre tenen sa casa oberta per a mi. A Ignasi, que des de lluny encara se’n recorda. A Sergio, que sempre troba un lloc en la seua agenda per veure’ns i xerrar. I a tots els amics, inestimables, de “I Hui On Anem”: Raül, Pepo, Tonyo, Víctor i un llarg etc. Deixe molta gent i molts detalls per anomenar, però m’he marcat el límit (injust) d’una pàgina... I no puc oblidar de cap manera a la meua família, que han estat, estan i estaran sempre al meu costat. Anava a dir que sense la beca no haguera sigut possible fer la tesi, però ells sí que ho hagueren fet possible, com tot. Gràcies a ma mare, mon pare, la meua germana, la iaia i les meues ties, als quals estime molt, encara que siga un poc “desapegat”. ... i a la música. IX X Índice Í NDICE ÍNDICE ........................................................................................................................ 1 ABREVIATURAS / ABBREVIATIONS ............................................................................. 5 a) En español ............................................................................................................................................. 5 b) In English ............................................................................................................................................... 6 1. INTRODUCCIÓN ...................................................................................................... 9 2. ANTECEDENTES BIBLIOGRÁFICOS ......................................................................... 11 2.1. LA REACCIÓN DE DIELS-ALDER ................................................................................................................ 11 2.2. REACCIONES DE CICLOADICIÓN CATALIZADAS POR COMPLEJOS CU(II)-BOX ..................................................... 15 2.2.1. Ligandos tipo bis(oxazolina) .................................................................................................... 15 2.2.2. Reacciones de Diels-Alder enantioselectivas .......................................................................... 18 2.2.2.1. Reacciones de Diels-Alder enantioselectivas con 3-alquenoil-2-oxazolidonas como dienófilos ..... 18 2.2.2.2. Reacciones de Diels-Alder enantioselectivas con otros dienófilos .................................................. 22 2.2.3. Reacciones de oxo-hetero-Diels-Alder con demanda electrónica inversa.............................. 25 2.2.4. Cicloadiciones [3+2] ................................................................................................................ 31 2.3. EL ESQUELETO DE CANFANO EN CATÁLISIS ASIMÉTRICA................................................................................. 35 3. OBJETIVOS ............................................................................................................ 47 4. RESULTADOS Y DISCUSIÓN ................................................................................... 49 4.1. DISEÑO DE LIGANDOS CON ESTRUCTURA DE HIDROXIOXAZOLINAS A PARTIR DEL ÁCIDO (1S)-CETOPÍNICO. APLICACIÓN EN REACCIONES DE DIELS-ALDER ENANTIOSELECTIVAS ........................................................................................ 49 4.1.1. Introducción ............................................................................................................................ 49 4.1.2. Síntesis de las hidroxioxazolinas 2-6 y 21 ............................................................................... 50 4.1.3. Síntesis de 3-alquenoil-2-oxazolidonas ................................................................................... 57 4.1.4. Síntesis de los dienos .............................................................................................................. 58 4.1.5. Reacciones de Diels-Alder catalizadas por complejos de hidroxioxazolinas y Cu(II) .............. 59 4.1.5.1. Optimización de las condiciones de reacción .................................................................................. 59 4.1.5.2. Alcance y limitaciones de la reacción .............................................................................................. 61 4.2. NUEVAS REACCIONES DE CICLOADICIÓN CATALIZADAS POR COMPLEJOS DE CU(II)-BOX...................................... 65 4.2.1. N-Óxidos de 2-alquenoilpiridina en reacciones de cicloadición ............................................. 65 4.2.1.1. Antecedentes .................................................................................................................................. 65 4.2.1.2. Síntesis de N-óxidos de 2-alquenoilpiridina .................................................................................... 70 4.2.1.3. N-Óxidos de 2-alquenoilpiridina como dienófilos en reacciones de Diels-Alder ............................. 73 a) Optimización ....................................................................................................................................... 73 b) Alcance y limitaciones de la reacción.................................................................................................. 74 1 Índice c) Determinación de la estereoquímica absoluta y modelo estereoquímico .......................................... 78 d) Transformaciones sintéticas ............................................................................................................... 80 4.2.1.4. N-Óxidos de 2-alquenoilpiridina como heterodienos en reacciones de hetero-Diels-Alder ........... 83 a) Optimización ....................................................................................................................................... 83 b) Alcance y limitaciones de la reacción.................................................................................................. 85 c) Determinación de la estereoquímica absoluta y modelo estereoquímico .......................................... 91 d) Transformaciones sintéticas ............................................................................................................... 93 4.2.1.5. N-Óxidos de 2-alquenoilpiridina como dipolarófilos en cicloadiciones [2+3] con nitronas............. 94 a) Síntesis de las nitronas ........................................................................................................................ 94 b) Optimización ....................................................................................................................................... 95 c) Alcance y limitaciones de la reacción .................................................................................................. 98 d) Determinación de la estereoquímica absoluta y modelo estereoquímico ....................................... 102 4.2.2. Reacciones de Diels-Alder asimétricas con α’-arilsulfonil enonas catalizadas por Cu(II)-BOX ........................................................................................................................................................ 104 4.2.2.1. Antecedentes ................................................................................................................................ 104 4.2.2.2. Síntesis de α’-arilsulfonil enonas ................................................................................................... 106 4.2.2.3. α’-Arilsulfonil enonas como dienófilos en reacciones de Diels-Alder ........................................... 108 a) Optimización ..................................................................................................................................... 108 b) Alcance y limitaciones de la reacción................................................................................................ 110 c) Determinación de la estereoquímica y modelo estereoquímico ...................................................... 113 d) Transformaciones sintéticas ............................................................................................................. 115 5. EXPERIMENTAL SECTION .................................................................................... 117 5.1. GENERAL EXPERIMENTAL METHODS ...................................................................................................... 117 5.1.1. General Procedures .............................................................................................................. 117 5.1.2. Solvents and reagents ........................................................................................................... 117 5.1.2.1. Solvents ......................................................................................................................................... 117 5.1.2.2. Reagents ........................................................................................................................................ 117 5.1.3. Instrumentation .................................................................................................................... 117 5.1.3.1. Melting points ............................................................................................................................... 117 5.1.3.2. Nuclear magnetic resonance (NMR) ............................................................................................. 118 5.1.3.3. Polarimetry .................................................................................................................................... 118 5.1.3.4. Mass spectrometry ........................................................................................................................ 118 5.1.3.5. HPLC analyses ................................................................................................................................ 118 5.1.3.6. Gas Chromatography analyses ...................................................................................................... 118 5.2. SYNTHESIS OF LIGANDS WITH HYDROXYOXAZOLINE MOIETY BASED ON (1S)-(+)-KETOPINIC ACID. APPLICATION TO ENANTIOSELECTIVE DIELS-ALDER REACTION. .................................................................................................. 119 5.2.1. Synthesis and characterization of (1S)-(+)-ketopinic acid (1) ............................................... 119 5.2.2. Synthesis and characterization of amides 13-16 .................................................................. 120 5.2.3. Synthesis and characterization of ketooxazolines 17-20 ...................................................... 122 5.2.4. Synthesis and characterization of hydroxyoxazolines 2-6 and 21 ........................................ 124 2 Índice 5.2.5. Synthesis and characterization of 3-alkenoyl-2-oxazolidones .............................................. 126 5.2.6. Synthesis and characterization of dienes.............................................................................. 128 5.2.7. Enantioselective Diels-Alder reaction with 3-alkenoyl-2-oxazolidones catalyzed by Cu(II)hydroxyoxazoline complexes .......................................................................................................... 130 5.2.7.1. General procedure for the catalytic enantioselective Diels-Alder reaction .................................. 130 5.2.7.2. Characterization of the products................................................................................................... 130 5.3. ENANTIOSELECTIVE CU(II)-BOX CATALYZED CYCLOADDITION REACTIONS OF 2-ALKENOYLPYRIDINE N-OXIDES....... 134 5.3.1. Synthesis and characterization of 2-alkenoylpyridine N-oxides ........................................... 134 5.3.2. Enantioselective catalyzed Diels-Alder reaction ................................................................... 138 5.3.2.1. General procedure for the catalytic enantioselective Diels-Alder reaction .................................. 138 5.3.2.2. Characterization of the products................................................................................................... 139 5.3.2.3. Synthetic transformations ............................................................................................................. 147 5.3.3. Enantioselective catalyzed oxo-hetero-Diels-Alder reaction ................................................ 151 5.3.3.1. General procedure for the catalytic enantioselective oxo-hetero-Diels-Alder reaction ............... 151 5.3.3.2. Characterization of the products................................................................................................... 152 5.3.3.3. Synthetic transformations ............................................................................................................. 164 5.3.4. Enantioselective catalyzed 1,3-dipolar cycloadditions whit nitrones ................................... 164 5.3.4.1. General procedure for the synthesis of nitrones .......................................................................... 164 5.3.4.2. General procedure for the catalytic enantioselective 1,3-dipolar cycloadditions ......................... 165 5.3.4.3. Characterization of the products................................................................................................... 165 5.4. ENANTIOSELECTIVE CU(II)-BOX CATALYZED DIELS-ALDER REACTIONS OF Α’-ARYLSULFONYL ENONES ................. 173 5.4.1. Synthesis and characterization of α’-arylsulfonyl enones .................................................... 173 5.4.2. Enantioselective catalyzed Diels-Alder reaction ................................................................... 182 5.4.2.1. General procedure for the catalytic enantioselective Diels-Alder reaction .................................. 182 5.4.2.2. Characterization of the products................................................................................................... 182 5.4.2.3. Synthetic transformations ............................................................................................................. 193 6. SUMMARY AND CONCLUSIONS .......................................................................... 197 7. REFERENCES ....................................................................................................... 201 ANEXO: FIGURAS .................................................................................................... 209 3 Índice 4 Abreviaturas / Abbreviations A BREVIATURAS / A BBREVIATIONS a) En español Å AcOEt Ar Bn BOX BSA Bu CCF COD d dba DCC DIPEA DMAP DMF DMSO DMSO-d6 ee eq. EI Et g h HPLC Hz i-Pr L M m mCPBA Me mg MHz min mL mm ångström acetato de etilo arilo bencilo bis(oxazolina) N,O-bis(trimetilsilil)acetoamida butilo cromatografía de capa fina 1,5-ciclooctadieno doblete dibencilidenacetona N,N’-diciclohexilcarbodiimida N,N-diisopropiletilamina 4-(dimetilamino)piridina N,N-dimetilformamida dimetilsulfóxido dimetilsulfóxido deuterado exceso enantiomérico equivalente impacto electrónico etilo gramo hora cromatografía líquida de alta eficacia hercio isopropilo litro molar multiplete ácido m-cloroperbenzóico metilo miligramo megahercio minuto mililitro milímetro 5 Abreviaturas / Abbreviations mmol MNBA n-Bu n.d. NOE NOESY Ph ppm q RMN s t T t. a. TADDOL t-Bu Tf THF TMS Tol Ts UHP [α] δ μL milimol alcohol m-nitrobencilíco n-butilo no determinado efecto nuclear Overhauser espectroscopía de efecto nuclear Overhauser fenilo partes por millón cuadruplete resonancia magnética nuclear singlete triplete temperatura temperatura ambiente α, α, α’,α’-tetraaril-1,3-dioxolan-4,5-dimetanol terc-butilo trifato (trifluorometanosulfonilo) tetrahidrofurano trimetilsililo tolil (p-metilfenil) tosilo (p-toluenosulfonilo) urea-peróxido de hidrógeno rotación específica desplazamiento químico microlitro b) In English Å Ar Bn BOX br s BSA Bu COD d dba DCC DIPEA DMAP 6 ångström aryl benzyl bis(oxazoline) broad singlet N,O-bis(trimethylsilyl)acetoamide butyl 1,5-cyclooctadiene doublet dibenzylideneacetone N,N’-dicyclohexylcarbodiimide N,N-diisopropylethylamine 4-(dimethylamino)pyridine Abreviaturas / Abbreviations DMF DMSO DMSO-d6 ee EI eq. Et EtOAc eV FAB g h HPLC HRMS Hz i-Pr L MNBA NMR M m M+ mCPBA Me mg MHz min. mL mm mmol mp MS n-Bu nd NOE NOESY Ph ppm q rt s N,N-dimethylformamide dimethylsulfoxide deuterated dimethylsulfoxide enantiomeric excess electron impact equivalent ethyl ethyl acetate electron volts fast atom bombardment gram hour high performace liquid chromatography high resolution mass spectrum hertz isopropyl liters m-nitrobenzyl alcohol nuclear magnetic resonance molar multiplet molecular ion m-chloroperbenzoic acid methyl milligram megahertz minutes milliliter millimeter millimol melting point mass spectroscopy n-butyl not determined nuclear Overhauser effect nuclear Overhauser effect spectroscopy phenyl parts per million quartet room temperature singlet 7 Abreviaturas / Abbreviations t T t-Bu Tf THF TLC TMS Tol Ts UHP [α] δ μL 8 triplet temperature tert-butyl triflate (trifluoromethansulfonate) tetrahydrofuran thin layer chromatography trimethylsilyl tolyl (p-methylfenil) tosyl (p-toluenesulfonyl) urea-hydrogen peroxide specific rotation chemical shift microliter Introducción 1. I NTRODUCCIÓN Uno de los principales objetivos de la Química Orgánica ha sido, desde sus inicios, la síntesis de compuestos orgánicos de la manera más eficiente posible, optimizando el tiempo de reacción y las cantidades de reactivos necesarios. La reacción de Diels-Alder representa un buen ejemplo de eficiencia, pues permite construir anillos de ciclohexeno en un solo paso con una gran economía atómica. a Desde su descubrimiento en 1928 por Otto Diels y Kurt Alder,1 la utilidad de la reacción ha sido ampliamente demostrada, habiendo sido utilizada en la síntesis de numerosos compuestos, especialmente de origen natural.2,3 En 1960 Yates y Eaton iniciaron una revolución en el campo de las reacciones de cicloadición al descubrir que en presencia de un ácido de Lewis éstas se podían desarrollar en condiciones mucho más suaves y también se podía cambiar la diastereoselectividad.4 Pese a la antigüedad de esta reacción de cicloadición [4+2], todavía hoy se siguen desarrollando nuevas metodologías sintéticas relacionadas con la misma, así como con otros tipos de cicloadiciones tales como las reacciones de tipo hetero-Diels-Alder o la cicloadiciones [3+2].5,6 Podemos decir pues, que la reacción de Diels-Alder ha tenido, y tiene todavía, un alto impacto en la Química y en la sociedad. En las últimas décadas la síntesis de compuestos quirales de manera enantioselectiva ha atraído la atención de los químicos orgánicos, debido principalmente a leyes más restrictivas con la generación de residuos y con las patentes farmacéuticas, el aumento del interés en fármacos enantioméricamente puros desde la tragedia de la talidomida y la importancia de nuevos materiales quirales. Así, se han desarrollado diversos métodos para la obtención compuestos enantioméricamente enriquecidos que implican bien la resolución de mezclas racémicas o procedimientos de síntesis enantioselectiva mediante el uso de auxiliares, reactivos o catalizadores quirales. De todos ellos, el uso de catalizadores quirales (catálisis asimétrica) es el más eficaz y conveniente ya que permite la reducción del número de pasos sintéticos y de la cantidad de inductor quiral, así como la disminución de los residuos producidos.7 No es de extrañar pues el gran interés que ha tenido el desarrollo de nuevas metodologías de catálisis asimétrica aplicadas a las reacciones de cicloadición, dado el elevado número de centros estereogénicos que pueden formarse y su importancia a La economía atómica (expresada como eficiencia) describe la relación entre el número de átomos que intervienen en una reacción y el número de átomos del compuesto resultante de interés. En el caso de las reacciones de cicloadición la eficiencia atómica es, por tanto, del 100%.222 9 Introducción sintética. Ya en 1976 Guseinov publicó un primer intento de catálisis asimétrica de una reacción de Diels-Alder,8 si bien hubo que esperar hasta 1979 y 1987 para encontrar ejemplos con una enantioselectividad aceptable en sendos artículos publicados por Koga.9,10 Desde entonces han sido muchos los sistemas catalíticos asimétricos descritos, basados tanto en el uso de ácidos de Lewis como en organocatalizadores.1113 Entre los ácidos de Lewis, el cobre ha jugado papel prominente en el campo de la catálisis asimétrica, habiendo sido ampliamente utilizado.14 Sin embargo, todavía hoy existen grandes limitaciones en cuanto a ligandos utilizados y respecto a la variedad de sustratos empleados En esta tesis abordaremos estas dos limitaciones mediante el diseño de nuevos ligandos quirales a partir de fuentes naturales asequibles, por un lado, y mediante la búsqueda de sustratos que presenten plantillas coordinantes adecuadas compatibles con sistemas catalíticos disponibles. 10 La reacción de Diels-Alder 2. A NTECEDENTES B IBLIOGRÁFICOS 2.1. La reacción de Diels-Alder Las reacciones de cicloadición son aquellas donde dos moléculas reaccionan para dar lugar a la formación de una estructura cíclica. La más importante de todas ellas, la reacción de Diels-Alder, es una cicloadición de tipo [4+2] donde intervienen un dieno conjugado y un alqueno, llamado comúnmente dienófilo. Como resultado de la reacción se forma un anillo de ciclohexeno (Esquema 1).1 Esquema 1 Se acepta de manera general que la reacción transcurre de manera concertada, sin intermedios de reacción. Sin embargo, en el caso de dienos y alquenos sustituidos la formación de los enlaces puede estar más avanzada en un extremo que en el otro en un proceso asíncrono que puede implicar un mecanismo polar. La interacción entre los orbitales del dieno y del dienófilo se muestra en la Figura 1. El dieno adopta una conformación s-cis, aproximándose al dienófilo en planos aproximadamente paralelos. Las propiedades de simetría de los orbitales π permiten estabilizar las interacciones entre el dienófilo y las posiciones C1 y C4 del dieno. LUMO del dienófilo HOMO del dieno Figura 1 Usualmente las interacciones más fuertes se dan entre el HOMO del dieno y el LUMO del dienófilo. En este caso hablamos de reacciones con demanda electrónica normal. De esta manera, cuando el dienófilo tiene grupos electrón-atrayentes, tal como ocurre en compuestos carbonílicos α,β-insaturados o en nitroalquenos, la reacción experimenta un aumento de la velocidad, debido a la disminución de energía del orbital LUMO del dienófilo y, por tanto, la aproximación energética entre éste y el HOMO del dieno. También se puede observar un aumento de la velocidad en reacciones entre dienos con baja densidad electrónica y dienófilos con grupos electrón-dadores como pueden ser los éteres vinílicos. En este caso hay una mayor aproximación energética 11 Antecedentes Bibliográficos entre el LUMO del dieno y el HOMO del dienófilo, hablándose entonces de reacciones con demanda electrónica inversa (Figura 2). Demanda electrónica normal Demanda electrónica normal activada Demanda electrónica inversa EA ED + dieno dienófilo dieno dienófilo dieno dienófilo LUMO LUMO HOMO LUMO HOMO HOMO EA = electrón-atrayente; ED = electrón-donante Figura 2 La mayoría de las reacciones de Diels-Alder son estereoespecíficas supra respecto a ambos componentes, formándose un único aducto a partir de una aproximación determinada entre el dieno y el dienófilo. Sin embargo, dado que pueden existir diversos modos de aproximación, se plantea una cuestión de estereoselectividad y de regioselectividad. En primer lugar, cuando el dieno y el dienófilo no están sustituidos simétricamente encontramos la posibilidad de formación de dos regioisómeros. La preferencia en la formación de un regioisómero determinado viene marcada por la naturaleza de los sustituyentes así como por la posición de éstos en el dieno. Esta preferencia se encuentra reflejada en el Esquema 2, predominado la formación de aductos 1,2-disustituidos y 1,4-disustituidos. Este comportamiento puede ser explicado a partir de los coeficientes de los orbitales frontera implicados, según la reacción sea con demanda electrónica normal o con demanda electrónica inversa, predominando los productos con solapamiento de los lóbulos con mayor coeficiente de los orbitales frontera implicados. 12 La reacción de Diels-Alder Esquema 2: Regioselectividad de la reacción de Diels-Alder. Reacciones con demanda electrónica normal Reacciones con demanda electrónica inversa ED ED EA EA EA EA + + ED ED EA = electrón-atrayente; ED = electrón-donante En segundo lugar, se encuentra la posibilidad de formación de dos diastereoisómeros endo y exo según la orientación entre los sustituyentes del dienófilo o el sistema de dieno (Figura 3). En el caso de dienófilos con sustituyentes insaturados se suele observar una preferencia hacia la formación de aductos endo, hecho conocido como regla de Alder. Este fenómeno se debe a una interacción secundaria de los orbitales π de los sustituyentes con los orbitales π del dieno, la cual estabiliza el estado de transición pese a la mayor congestión estérica. Sin embargo, es usual la formación mayoritaria de aductos exo en el caso de aldehídos α,β-insaturados tales como la metacroleína. Figura 3 En el ejemplo representado en el Esquema 3 podemos ver como la combinación de regioselectividad y estereoselectividad da lugar a un único producto 1,2disustituido, con disposición cis de los sustituyentes debido a la aproximación endo. Esquema 3 NEt2 CO2Et + NEt2 20 ºC CO2Et 13 Antecedentes Bibliográficos En 1960 Yates y Eaton observaron un factor de aceleración de 105 en la reacción entre el antraceno y el anhídrido maleico en presencia de tricloruro de aluminio.4 Este descubrimiento inició uno de los mayores avances en el área de la reacción de DielsAlder: el uso de ácidos de Lewis como catalizadores. Los ácidos de Lewis, al coordinarse con grupos carbonilos conjugados enfatizan la disminución de energía del LUMO del dienófilo, provocando un descenso de la energía de activación y, por tanto, acelerando la reacción (Figura 4). Todo esto permite llevar a cabo la reacción en condiciones mucho más suaves. Figura 4: Efecto de la protonación sobre la energía y los coeficientes orbitálicos en la acroleína. Uno de los aspectos a destacar en la reacción de Diels-Alder es la posibilidad de formación de hasta cuatro centros estereogénicos contiguos en la molécula (Figura 5). Es por esto que uno de los principales puntos de atención es el control estereoquímico de la reacción: por un lado el dominio de la diastereoselectividad endo/exo y por otro el de la enantioselectividad de la reacción, de gran utilidad en la síntesis de productos bioactivos. Figura 5 La sola utilización de un ácido de Lewis como catalizador puede implicar ya de por sí un cambio en la diastereoselectividad respecto a la reacción no catalizada. Pero hace falta un modificador quiral para llevar a cabo la reacción de forma enantioselectiva. Si bien puede incorporarse un auxiliar quiral al dieno o al dienófilo, resulta más interesante cuando estos se incorporan al catalizador. Esta vía permite reducir el número de etapas de la secuencia sintética y disminuir la cantidad necesaria de material quiral a la hora de sintetizar aductos enantioméricamente puros o enriquecidos. 14 Reacciones de cicloadición catalizadas por complejos Cu(II)-BOX 2.2. Reacciones de cicloadición complejos Cu(II)-BOX catalizadas por 2.2.1. Ligandos tipo bis(oxazolina) Los ligandos de tipo bis(oxazolina) (BOX) con simetría C2 son una de las clases más populares de ligandos quirales, habiendo recibido una gran atención tanto en química de coordinación como en catálisis asimétrica. Estos ligandos presentan dos anillos de oxazolina unidos directamente o a través de un espaciador. Las bis(oxazolinas) en las que el espaciador está formado por un único átomo de carbono con dos sustituyentes idénticos sobre el mismo son las más habituales y las que utilizaremos en esta tesis (Figura 6). El primer ejemplo de catálisis asimétrica con este tipo de ligandos fue publicado por Masamune en 1990 en reacciones de ciclopropanación asimétrica de alquenos.15 Sin embargo, podemos encontrar precedentes con ligandos similares, pues en 1986 Pfaltz ya había utilizado derivados de semicorrina en este mismo tipo de reacción y en 1989 Nishiyama había utilizado ligandos de tipo PyBOX en la hidrosililación enantioselectiva de cetonas.16,17 Figura 6: Ligandos BOX y otros ligandos C2 similares. Desde entonces se han ampliado enormemente la gama y el número de reacciones enantioselectivas que utilizan los ligandos de tipo BOX (Figura 7) en combinación con diferentes metales con muy buenos resultados.18,19 Por todo ello, los BOX han sido incluidos dentro del grupo de los llamados “ligandos privilegiados”.20 Una de las características más destacables de las bis(oxazolinas) es la presencia de un eje de simetría binario C2. Es conocido que los ligandos con este tipo de simetría permiten reducir el número de geometrías complejo-sustrato posibles y también, por tanto, los posibles caminos de reacción y estados de transición, conduciendo a estereoselectividades elevadas. Este hecho también simplifica y facilita los estudios estructurales y mecanísticos. 15 Antecedentes Bibliográficos 70 60 50 40 30 20 10 2008 2007 2006 2005 2004 2003 2002 2001 2000 1999 1998 1997 1996 1995 1994 1993 1992 1991 1990 0 Figura 7: Evolución en el número de publicaciones donde se emplean ligandos BOX. Otra de las ventajas de los ligandos de tipo BOX reside en la facilidad de su síntesis y en su modularidad. Básicamente los ligandos BOX se componen de una unidad derivada del ácido malónico y de dos unidades de un mismo β-aminoalcohol. Dado que en general es posible tener acceso a ambos enantiómeros del βaminoalcohol resulta posible obtener las 2 formas enantioméricas del ligando, lo que permite controlar el sentido de la enantioselectividad en la reacción.19 En el Esquema 4 se muestra el procedimiento más habitual para la síntesis de estos ligandos. En una primera etapa se forma la correspondiente bis(β-hidroxiamida), generalmente por reacción del aminoalcohol con el cloruro de ácido dialquilmalónico requerido o bien del malononitrilo equivalente. La segunda etapa es la ciclación de la agrupación βhidroxiamida a un anillo de oxazolina para dar lugar al ligando BOX. Los métodos y condiciones aplicados a esta etapa son muy variados. En algunos casos consiste en un solo paso de deshidratación directa, mientras que en otros la reacción requiere la transformación del hidroxilo en un grupo saliente como cloruro o sulfonato, el cual se elimina en una etapa posterior. No obstante, los ligandos de este tipo utilizados con mayor frecuencia, los llamados Ph-BOX (R=Ph, R’=Me) y t-Bu-BOX (R=t-Bu, R’=Me), se encuentran disponibles comercialmente. Esquema 4: Procedimiento general de síntesis de bis(oxazolinas). R' R' X NH2 HO R' R' R X NH HN HO R X= O CCl , NH COR'' ó R' R' O O NuOH NH HN Nu R R - HNu -H2O R' R' C N O O N R 16 O O N R Nu R Reacciones de cicloadición catalizadas por complejos Cu(II)-BOX Los ligandos BOX han sido usados en reacciones catalizadas por una gran variedad de metales, aunque el cobre, en particular la especie Cu(II), es el más utilizado con diferencia. Como podemos ver en la Figura 8, para el caso de Ph-BOX, el uso de Cu(II) supone cerca del 50% de reacciones desarrolladas con este ligando que consiguen al menos un 50% de exceso enantiomérico.19 En el caso del ligando t-BuBOX el porcentaje alcanza casi un 80% respecto al conjunto de otros metales. Figura 8: Porcentaje de reacciones desarrolladas con un ee de al menos un 50%. Así pues, el cobre es un ácido de Lewis que funciona de forma efectiva. El Cu(II) en particular forma los complejos ligando-metal más estables de todos los cationes divalentes de la primera serie de transición, según la serie de Irving-Williams (Mn < Fe < Co < Ni < Cu > Zn).21,22 Así mismo, aunque parezca contradictorio, la disociación de ligandos quelantes no es difícil en los complejos de Cu(II). En efecto, el complejo [Cu(H2O)6]+2 tiene la ratio de intercambio más alta de toda la serie anterior de cationes divalentes. Esto se debe al efecto Jahn-Teller de distorsión, que permite la formación de enlaces lábiles en las posiciones axiales. El Cu(II) tiende a formar complejos de geometría plano-cuadrada o tetraédrica alargada. En el caso de los complejos [Cu(II)-(BOX)]X2 donde X son aniones débilmente o no coordinantes se favorece la coordinación ecuatorial de los sustratos bidentados, dejando las posiciones axiales para el anión X en caso de que coordine. Todo esto lleva a que los complejos Cu(II)-BOX tengan una geometría bien definida y pueda haber un adecuado intercambio sustrato-producto, características indispensables para que el complejo actúe como catalizador. A continuación llevaremos a cabo una revisión sobre la aplicación de complejos Cu(II)-BOX en reacciones de cicloadición, donde han sido utilizados ampliamente desde 1991. 17 Antecedentes Bibliográficos 2.2.2. Reacciones de Diels-Alder enantioselectivas 2.2.2.1. Reacciones de Diels-Alder enantioselectivas con 3-alquenoil-2oxazolidonas como dienófilos Desde su introducción por Narasaka en la reacción de Diels-Alder,23 las 3alquenoil-2-oxazolidonas se han erigido como los dienófilos estándar para comprobar la eficiencia de nuevos sistemas catalíticos enantioselectivos aplicados a esta reacción. Estas imidas resultan por una parte sintéticamente útiles, al ser equivalentes sintéticos de ácidos carboxílicos y, por otra parte, su coordinación bidentada permite la formación de complejos sustrato-catalizador rígidos (facilitando la discriminación facial del doble enlace), mayores velocidades de reacción y mejores relaciones endo/exo. En 1991 Corey et al. describieron por primera vez el uso de ligandos tipo BOX en la catálisis asimétrica de la reacción de Diels-Alder, utilizando ciclopentadieno y 3acriloil-2-oxazolidona (Esquema 5). Emplearon complejos de Fe(III)-Ph-BOX, alcanzando una enantioselectividad del 86% de exceso enantiomérico con un selectividad endo/exo 99:1.24 Destacar como anécdota que en la página anterior a este artículo Evans detallaba el uso de ligandos tipo BOX en reacciones de ciclopropanación con Cu(I) en un trabajo simultaneo25 al ya citado de Masamune.15 En 1992 Corey amplió su estudio a complejos Mg(II)-BOX con resultados similares, pero sin ampliar la gama de dienos ni de dienófilos.26 Esquema 5 Fueron Evans y colaboradores quienes describieron en 1993 la utilización por primera vez de complejos quirales de Cu(II)-BOX en la reacción de Diels-Alder.27 También en este caso los dienófilos utilizados fueron de tipo 3-alquenoil-2oxazolidona, así como los análogos de 2-tioazolidintiona, con diversas sustituciones, llegando a obtener una enantioselectividad mayor del 98% ee y ratios endo/exo superiores a 90:10 (Esquema 6). Estudios posteriores del mismo autor han permitido una completa caracterización del sistema,28-33 detallando los diferentes efectos que el sustituyente del ligando BOX, el tipo de contraión, aditivos, disolventes y temperatura ejercen sobre la reacción. 18 Reacciones de cicloadición catalizadas por complejos Cu(II)-BOX Esquema 6 2+ O O N Y O + N t-Bu Cu 2 X- N R t-Bu Y Y O R Y = O, S X = OTf, SbF6 N Y >98% ee, >90:10 endo/exo La reacción es válida para un gran número de dienófilos sustituidos en β y de dienos (no limitándose al ciclopentadieno). Además, la utilidad del sistema se demostró con la síntesis de diversos productos naturales como el THC o el ácido shikímico, tal como ilustra el Esquema 7. Esquema 7: Ejemplos de productos naturales sintetizados usando el sistema Cu(II)-BOX. O O O + N [Cu(II)-t-Bu-BOX] (SbF6)2 O HO O O O N O CO2H HO 97% ee OH ácido shikimico El contraión de la sal de Cu(II) ejerce una gran influencia sobre la reacción, siendo los aniones triflato y hexafluoroantimoniato los más convenientes, sobretodo éste último. Esta influencia se ejerce en varios ámbitos: velocidad de reacción, diastereo- y enantioselectividad, causando el SbF6- un incremento de la velocidad de reacción de hasta 20 veces. Sin embargo, el anión hexafluoroantimoniato es incompatible con las 2-alquenoil-3-tioazolidintionas por el envenenamiento que se produce del catalizador. Por lo que respecta al ligando, el t-Bu-BOX es el más eficiente, con gran diferencia sobre otros ligandos tipo BOX con sustituyente alquílico o el PhBOX, que conducía a buenos resultados en el caso de los complejos descritos por Corey con Fe(III) y Mg(II). Por lo que respecta al disolvente, el diclorometano resulta por lo general el más adecuado. En el caso de disolventes con capacidad coordinante, el THF y el nitrometano no modifican sustancialmente el comportamiento, mientras que el acetonitrilo y la adición de isopropanol rebajan la efectividad del catalizador de forma importante. En un estudio paralelo de Jørgensen utilizando ciclohexadieno como dieno 19 Antecedentes Bibliográficos este autor aprecia una ligera mejora en la enantioselectividad al emplear nitrometano como disolvente, especialmente con el complejo con triflato como contraión, produciendo además una gran aceleración de la reacción.34 Las estructuras de los complejos Cu(II)-(S)-t-Bu-BOX obtenidas por difracción de rayos X y por cálculos computacionales revelan que la reacción debería transcurrir, en este caso, a través de complejos plano-cuadrados ligeramente distorsionados, con el dienófilo adoptando una conformación s-cis. La aproximación del dieno debería ser por tanto a través de la cara α-Re, lo cual concuerda con la estereoquímica de los productos obtenida experimentalmente (Figura 9a). (a) (b1, matched) (b2, mismatched) Figura 9: Geometría del complejo y discriminación facial. Esta propuesta mecanística se refuerza a partir de ensayos de doble estereodiferenciación, utilizando dienófilos quirales. Cuando el carbono estereogénico en el anillo de oxazolidona tiene configuración R (Figura 9b1, matched) la reacción transcurre con una conversión y selectividad próxima al 100%, mientras que si la estereoquímica de dicho carbono es S (Figura 9b2, mismatched), la selectividad y la conversión disminuyen de manera drástica. Esto concuerda con un complejo planocuadrado, donde en el primer caso tanto el grupo t-Bu como el bencilo cubren la misma cara del doble enlace, mientras que en el segundo caso el grupo bencilo pasa a cubrir la cara α-Re, siendo impedida la entrada del dieno por ambas caras, afectando negativamente al transcurso de la reacción. En 1996 Davies y Ghosh, de forma independiente, investigaron el papel del espaciador que une ambos anillos de oxazolina en el caso de los ligandos BOX derivados de indanol (Figura 10, Esquema 8).35-37 A diferencia del ligando Ph-BOX, estos ligandos permiten obtener enantioselectividades elevadas en la reacción entre ciclopentadieno y 3-acriloil-2-oxazolidona. En los resultados se observa que, frente al ligando dimetilado (X=CMe2), la introducción de una estructura espiránica en el puente favorece la enantioselectividad de la reacción, en especial cuando el número de eslabones es más bajo. Por el contrario, con ligandos con un puente más largo (X=CH2CH2) se reduce la enantioselectividad de forma importante. Por lo que respecta al ligando con un puente no sustituido (X=CH2) presenta una alta enantioselectividad, 20 Reacciones de cicloadición catalizadas por complejos Cu(II)-BOX superando en algunos casos el sistema Cu(II)-t-Bu-BOX de Evans. Otra ventaja de este último catalizador es su compatibilidad con la presencia de moléculas de agua, lo cual permite utilizar Cu(ClO4)2·6H2O como fuente de Cu(II), conservando una alta eficacia, al contrario de lo que se observa con los ligandos t-Bu-BOX y Ph-BOX, en los que se produce una disminución drástica de la enantioselectividad.38 X O N n O O N N L1 X = CH2, CMe2, CH2CH2 O N L2 n = 1, 2, 3, 4 Figura 10: Ligandos BOX derivados del indanol utilizados por Davies y Ghosh. Esquema 8 L1, X = CH2, ee = 98%, endo/exo >99:1 (-78 °C) L1, X = CMe2, ee = 92%, endo/exo >99:1 (-65 °C) L1, X = CMe2, ee = 82%, endo/exo = 98:2 (-50°C) L1, X = CH2CH2, ee = 13%, endo/exo = 89:11 (-50 °C) L2, n = 1, ee = 96%, endo/exo = 98:2 (-50 °C) L2, n = 2, ee = 92%, endo/exo >97:3 (-50 °C) L2, n = 3, ee = 90%, endo/exo = 97:3 (-50 °C) L2, n = 4, ee = 83%, endo/exo = 98:2 (-50 °C) En la bibliografía se pueden encontrar otros ligandos de tipo BOX de estructura más compleja.19 En algunos casos se han utilizado con diferente éxito ligandos BOX enlazados a soportes sólidos39-42 o ligandos con un grupo catiónico para ser utilizado en disolventes iónicos, aprovechando las ventajas de estos sistemas.43-45 Uno de los ligandos tipo BOX con estructura más compleja ha sido preparado por Schulz et al. En este caso el propio ligando forma un complejo de transferencia de carga con un derivado de fluorenona (Figura 11).46,47 Los complejos formados a partir de este ligando y triflato de cobre(II) permiten obtener enantioselectividades entre el 77 y el 94% de exceso enantiomérico en la reacción de Diels-Alder entre ciclopentadieno y 3-alquenoil-2oxazolidonas. Una de las características más reseñables de este catalizador es que puede precipitarse con pentano una vez terminada la reacción, separarse de los productos y, después de sucesivos lavados, puede volver a utilizarse, manteniendo su actividad inalterada hasta después de 11 usos. 21 Antecedentes Bibliográficos O2N O O NO2 O2N O O N N Figura 11 2.2.2.2. Reacciones de Diels-Alder enantioselectivas con otros dienófilos Aparte de las 3-alquenoil-2-oxazolidonas existen otros compuestos utilizados con éxito como dienófilos en reacciones de Diels-Alder catalizadas por complejos de Cu(II)-BOX. La mayoría de ellos posee, al igual que las citadas 3-alquenoil-2oxazolidonas, una agrupación de tipo imida. En 2002, Renau estudió diversas 1,3benzoxazol-2-(3H)-onas sustituidas en la posición 4 utilizando el ligando (S)-Ph-BOX, observando un aumento de la enantioselectividad al aumentar el volumen del grupo R unido al anillo aromático, si bien los excesos enantioméricos obtenidos son moderados (Esquema 9).48 Esquema 9 O + O O O [Cu(II)-Ph-BOX] (OTf)2 N R CH2Cl2 O N O R R = H, ee = 44%, endo/exo >100:1 R = Me, ee = 55%, endo/exo >100:1 R = Bn, ee = 64%, endo/exo >100:1 Sibi y colaboradores introdujeron en 2001 el uso de N-alquenoilimidas derivadas de 5,5-dimetil-3-pirazolidonas 1-substituidas (Esquema 10)49,50 En este caso el sustituyente sobre el nitrógeno no amídico también ejerce una gran influencia sobre la reacción, permitiendo obtener altos excesos enantioméricos cuando éste es muy voluminoso, como es el caso de grupos bencilo o 1-naftilmetilo. Los dos metilos en posición 5 del anillo obligan al sustituyente sobre el nitrógeno, por repulsión estérica, a adoptar la conformación que lo sitúe próximo al doble enlace. Como ventaja, este sistema permite utilizar el ligando i-Pr-BOX, más asequible que t-Bu-BOX o Ph-BOX. 22 Reacciones de cicloadición catalizadas por complejos Cu(II)-BOX Esquema 10 R = H, ee = 8%, endo/exo = 95:5 R = Et, ee = 56%, endo/exo = 96:4 R = Bn, ee = 71%, endo/exo = 93:7 R = 2-CH2Naftil, ee = 65%, endo/exo = 91:9 R = 1-CH2Naftil, ee = 92%, endo/exo = 90:10 Sibi ha estudiado el comportamiento como dienófilos de diversas imidas α,βinsaturadas análogas de N-alquenoilpirrolidina en reacciones de Diels-Alder catalizadas por complejos de Cu(II) y una bis(oxazolina) ciclopropánica (Esquema 11).51 En este caso, las imidas en las que el anillo de cinco miembros está completamente saturado (Figura 12) conducen a elevados excesos enantioméricos, mientras que la presencia de insaturaciones en dicho anillo (Figura 13) disminuye drásticamente la enantioselectividad. Esquema 11 Figura 12 R = Me, ee = 95% R = H, ee = 88% R = Me, R’ = Bn, ee = 67% R = H, R’ = Ph, ee = 78% R = Me, R’ = 1-CH2Naftil, ee = 88% R = Ph, ee = 86% R = Bn, ee = 65% Figura 13 O O S ee = 0% ee = 5% ee = 32% O N ee = 0% 23 Antecedentes Bibliográficos El mismo autor ha demostrado que en el caso de alquenos con un grupo aciloxi en el carbono β del doble enlace, con una reactividad más moderada por su mayor densidad electrónica, la plantilla N-alquenoilpirrolidona es más eficiente que la 3alquenoil-2-oxazolidona.52 También es destacable en el caso de estos dienófilos la obtención de resultados significativamente mejores con el ligando Ph-BOX que con tBu-BOX, tanto en lo que se refiere a los tiempos de reacción como a la estereoselectividad. Murai et al. describieron en 2002 el uso de N-carboxibencil metilenocaprolactamas (Esquema 12) como dienófilos en reacciones de Diels-Alder utilizando complejos de Cu(II)-t-Bu-BOX. El sistema conduce a los aductos exo de forma mayoritaria, con una diastereoselectividad prácticamente total cuando se usan dienos diferentes al ciclopentadieno y una elevada enantioselectividad.53 Esquema 12 Brimble y McEwan han descrito el uso de 2-acetilnaftoquinonas (Esquema 13) como dienófilos en reacciones de Diels-Alder catalizadas por complejos de Cu(II)-BOX, obteniendo en todos los casos muy bajas enantioselectividades.54 Esquema 13 Por último, cabe señalar dos tipos de dienófilos utilizados con los sistemas Cu(II)-BOX que se caracterizan por no presentar una agrupación β-dicarbonílica. Por un lado Aggarwal ha descrito el uso de α-tioacrilatos (Esquema 14a) como equivalentes sintéticos de cetenas. Los complejos Cu(II)-BOX catalizan la reacción de estos dienófilos con ciclopentadieno con alta enantioselectividad, aunque la diastereoselectividad es baja.55,56 Por otro lado Palomo y colaboradores han descrito el uso de α’-hidroxienonas (Esquema 14b), demostrando su eficiencia como dienófilos en reacciones enantioselectivas de Diels-Alder.57 Este tipo de dienófilos son equivalentes sintéticos 24 Reacciones de cicloadición catalizadas por complejos Cu(II)-BOX de acrilatos, ya que la agrupación α’-hidroxicetona puede ser transformada en un ácido carboxílico por ruptura oxidativa del enlace carbono-carbono con NaIO4. Esquema 14 (a) (b) 2.2.3. Reacciones de oxo-hetero-Diels-Alder con demanda electrónica inversa Las reacciones de hetero-Diels-Alder son excelentes herramientas sintéticas para la síntesis de dihidropiranos y dihidropiranonas a partir de compuestos carbonílicos. Estos sistemas heterocíclicos se encuentran presentes en muchos productos bioactivos y naturales.58,59 Consecuentemente se han desarrollado diversos procedimientos de catálisis asimétrica para este tipo de cicloadiciones [4+2],60 muchos de los cuales utilizan complejos de Cu(II)-BOX.14,19 Podemos distinguir dos tipos de reacciones de oxo-Diels-Alder. Por un lado las llamadas reacciones con demanda electrónica normal en las que aldehídos y cetonas actúan como heterodienófilos (Esquema 15a).5,61-63 Por otro lado tenemos las reacciones con demanda electrónica inversa, en las que aldehídos y cetonas α,βinsaturados actúan como heterodienos frente a alquenos, generalmente con elevada densidad electrónica (Esquema 15b). En ambos casos, los grupos carbonilo son los puntos de coordinación al ácido de Lewis. Esquema 15 (a) O + O (b) En los antecedentes bibliográficos sólo encontramos dos ejemplos de reacciones de hetero-Diels-Alder con demanda electrónica inversa catalizadas por complejos de Cu(II)-BOX, que utilizan acilfosfonatos (Figura 14a) y α-cetoésteres 25 Antecedentes Bibliográficos (Figura 14b) como heterodienos, respectivamente. En ambos casos destaca la presencia de grupos electrón-atrayentes unidos al grupo carbonilo de cetona. O (a) O P OMe OMe (b) R Figura 14 En 1998 Evans y colaboradores publicaron sus primeros estudios sobre reacciones de hetero-Diels-Alder de acilfosfonatos α,β-insaturados (Esquema 16).64,65 Los complejos de Cu(II)-BOX dan lugar a excelentes enantioselectividades y diastereoselectividades que se acercan al 100% de producto endo. Estos sustratos son muy reactivos, pudiendo utilizarse temperaturas muy bajas, tanto con enoles cíclicos como acíclicos, aunque un aumento de la temperatura no conlleva grandes variaciones en la enantioselectividad. Esquema 16 (a) (b) El sustituyente R puede ser variado sin afectar sensiblemente la estereoselectividad de la reacción. La reactividad del sistema es tan alta que permite disminuir la carga catalítica hasta sólo un 0,2 % molar. Sin embargo, las reacciones con éteres de enol derivados de cetonas, es decir, alquenos disustituidos en posición geminal, transcurren con resultados uniformemente mediocres, en palabras de los propios autores. El fuerte carácter electrón-atrayente del grupo fosfonato parece facilitar la reacción de hetero-Diels-Alder. Así, la reacción con ciclopentadieno del fosfonato sustituido con un metilo en posición β del doble enlace proporciona sólo un 35% de 26 Reacciones de cicloadición catalizadas por complejos Cu(II)-BOX producto de Diels-Alder, frente al 65% de producto de hetero-Diels-Alder, reaccionando el ciclopentadieno preferentemente como dienófilo (Esquema 17). Esquema 17 2+ O O N O + O P OMe OMe t-Bu 2 Cu N SBF6- t-Bu O O P OMe OMe + Me O Me Me 65:35 OMe OMe P O La reacción se ve poco afectada por un cambio en el contraión de la sal de cobre. En este caso es el triflato el que conduce a los mejores resultados, si bien el SbF6- aún ofrece una ligera ventaja en cuanto a tiempos de reacción. A diferencia de las reacciones de Diels-Alder con 3-alquenoil-2-oxazolidona, la reacción de hetero-Diels-Alder proporciona buenos resultados tanto con el ligando tBu-BOX como con Ph-BOX, si bien con este último se consiguen enantioselectividades ligeramente más altas y mayor reactividad. Un hecho muy interesante observado en esta reacción es la inversión en el sentido de la inducción asimétrica con estos dos ligandos. Así, (S,S)-t-Bu-BOX y (S,S)Ph-BOX conducen a enantiómeros opuestos, a pesar de que ambos ligandos tienen la misma configuración en el carbono estereogénico de la oxazolina, tal como se pone de manifiesto en el Esquema 16. En el caso de los ligandos i-Pr-BOX y Bn-BOX la inducción tiene el mismo sentido que con Ph-BOX, aunque la enantioselectividad es menor. Este fenómeno ya había sido encontrado por Jørgensen y Johannsen en reacciones de hetero-Diels-Alder con demanda electrónica normal con glioxilatos de alquilo y se produce en otras reacciones catalizadas por Cu(II)-BOX como, por ejemplo, reacciones énicas entre alquenos y glioxilatos de alquilo.66 La explicación de esta inversión ha sido objeto de cierta controversia entre los grupos de Evans y Jørgensen.32,67 Evans propone la participación de un complejo plano-cuadrado distorsionado como especie catalítica activa. De hecho, las estructuras de rayos X de diversos complejos [Cu(BOX)(H2O)2](SbF6)2 son todas ellas plano-cuadradas ligeramente distorsionadas (Figura 15), siendo mayor esta distorsión en el caso del ligando t-BuBOX, con las moléculas de agua repeliendo el efecto estérico del grupo t-Bu. Sin embargo, en el caso del ligando Ph-BOX la distorsión, aunque ligera, es de sentido contrario y, además, las moléculas de agua coordinadas poseen un ángulo que las acerca a los grupos fenilos del ligando, en lugar de alejarlos. 27 Antecedentes Bibliográficos Figura 15: Estructuras de rayos X de cristales [Cu(BOX)(H2O)2](SbF6)2 y su distorsión.65 Es conocido que para obtener aductos endo, la conformación del heterodieno debe ser preferentemente s-trans.68 Sin embargo, si suponemos una aproximación endo del éter de enol al heterodieno s-trans, un complejo tetraédrico ejercería, mediante los sustituyentes de la bis(oxazolina) en posición ecuatorial, un impedimento estérico que dificultaría la entrada del éter de enol (Figura 16a). Según Evans, este hecho es contradictorio con la uniformidad de altas diastereoselectividades observadas, pues éteres de enol muy voluminosos comprometerían la diastereoselectividad de la reacción. En el caso del complejo plano-cuadrado propuesto por Evans, este impedimento no tendría lugar, tal como vemos en la Figura 16b. Para justificar la inversión de configuración observada en el caso del ligando PhBOX, Evans sugiere finalmente que un grupo fenilo del ligando estabilizaría mediante una interacción tipo π la carga positiva del dienófilo, surgida de un estado de transición asíncrono.65 O H - N O N R H O R P OMe R' O OMe O Cu + Me Me (a) (b) O N OR O Cu N O O P MeO OMe (c) Figura 16: Interacciones estéricas del dienófilo con un complejo tetraédrico, plano-cuadrado y plano-cuadrado con estabilización π. 28 Reacciones de cicloadición catalizadas por complejos Cu(II)-BOX Sin embargo a partir de resultados experimentales y un estudio teórico con glioxilatos como sustratos coordinantes, Jørgensen mantiene que la inversión de configuración entre los ligandos se debe a un cambio en la geometría de los complejos de cobre de plano-cuadrada a tetraédrica.67 Jørgensen fundamenta su propuesta en lo siguiente: el complejo con i-Pr-BOX expresa el mismo sentido de inducción que el ligando Ph-BOX, aunque la interacción π con el sustrato no es posible. Además, sus cálculos teóricos muestran que la barrera energética entre complejo plano-cuadrado y complejo tetraédrico es bastante más baja para el ligando Ph-BOX que para que el ligando t-Bu, con lo cual no sería difícil que se produjera ese cambio estructural en las condiciones de reacción y, por tanto, la inversión en el sentido de la enantioselectividad. Jørgensen además observa que la reacción de hetero-Diels-Alder de glioxilatos con ciclohexeno no está influenciada por el disolvente al usar el complejo Cu(II)-t-Bu-BOX, mientras que sí se observa una fuerte dependencia lineal de la enantioselectividad obtenida con el complejo Cu(II)-Ph-BOX con respecto a la constante dieléctrica del disolvente, de manera que con disolventes de alta constante dieléctrica (EtNO2, MeCN) se llega a invertir la enantioselectividad, obteniéndose el mismo enantiómero mayoritario que con t-Bu-BOX. Jørgensen concluye que el factor determinante no sería la geometría estática de los complejos, por otra parte tendentes a grandes distorsiones que los alejan de la perfección, sino su (poca o mucha) flexibilidad y la dinámica estructural de los ligandos y de los complejos. En 1998 Jørgensen et al. publicaron por primera vez el uso de α-cetoésteres β,γ-insaturados en reacciones de oxo-hetero-Diels-Alder con demanda electrónica inversa (Esquema 18).69 Desde entonces han sido publicados diversos estudios por parte del mismo grupo así como por el de Evans de forma paralela.65,70-72 Esquema 18 Las reacciones de hetero-Diels-Alder con α-cetoésteres β,γ-insaturados son de gran utilidad sintética pues permiten acceder a un gran número de carbohidratos y aminoazúcares ópticamente activos (Esquema 19).71,72 Existen reseñables diferencias entre la reactividad de los acilfosfonatos y los αcetoésteres β,γ-insaturados. Los segundos resultan más reactivos, como se pone de manifiesto al hacer reaccionar una mezcla a partes iguales de ambos sustratos con etil vinil éter, obteniendo de forma mayoritaria (3,5:1) los aductos derivados del αcetoéster. 29 Antecedentes Bibliográficos Esquema 19: Ejemplo de síntesis de un carbohidrato a partir de un α-cetoéster β,γ-insaturado.72 Su comportamiento frente a complejos de [Cu(II)-BOX]X2 también es diferente. En el caso de los α-cetoésteres también se produce una inversión de la enantioselectividad con los ligandos t-Bu-BOX y Ph-BOX, si bien con el primero se obtienen excelentes resultados mientras que con el segundo la enantioselectividad es moderada. Otra diferencia es que los ligandos i-Pr-BOX y Bn-BOX conducen al mismo enantiómero mayoritario que el ligando t-Bu-BOX, si bien dan resultados pobres. Por lo que respecta a los contraiones, el más reactivo SBF6- da lugar a una diastereoselectividad muy elevada, contrariamente a lo que ocurre con el empleo del anión triflato. Resulta también interesante que el diclorometano no da lugar a los mejores resultados, siendo superiores los resultados con THF, éter etílico o tolueno, por ejemplo. La alta reactividad de los α-cetoésteres permite llevar a cabo las reacciones con sólo un 2 % molar de carga catalítica a 0 °C en menos de 1 hora, conservando una excelente enantioselectividad. Resulta también interesante la posibilidad de utilizar aqua-complejos como los caracterizados por rayos X (Figura 15) junto a tamiz de 3 Å, al ser estos complejos sólidos estables, evitando así la etapa de formación a partir de ligando y sal metálica. Evans ha diseñado un protocolo por el cual el catalizador puede ser reciclado. La reacción se lleva a cabo en hexano en presencia de Florisil®, recuperándose el catalizador por decantación. En estas condiciones el catalizador puede ser reutilizado varias veces sin pérdidas en la enantioselectividad y el rendimiento, aunque sí que se produce una merma de la reactividad.70 Wada ha publicado una interesante modificación de la reacción, empleando alcoholes olefínicos como dienófilos y γ-metoxi-α-cetoésteres β,γ-insaturados. En la misma reacción, catalizada por un complejo Cu(II)-t-Bu-BOX se produce una transeterificación seguida de reacción intramolecular de hetero-Diels-Alder. Este 30 Reacciones de cicloadición catalizadas por complejos Cu(II)-BOX proceso transcurre con una elevada eficacia por lo que respecta al rendimiento y a la enantioselectividad (Esquema 20).73 Esquema 20 Kurosu ha obtenido muy buenos resultados con α-cetoésteres utilizando ligandos tipo BOX soportados sobre polímeros.74 Kim et al. han documentado la utilización de líquidos iónicos como disolventes, permitiendo la reutilización del complejo Cu(II)-BOX hasta 8 veces sin apenas pérdidas de reactividad o eficacia.75 Para terminar este apartado, cabe indicar que las 3-alquenoil-2-oxazolidonas, empleadas en las reacciones de Diels-Alder, no resultan sustratos adecuados, al tratarse de compuestos con un carbonilo de imida, con mayor densidad electrónica que los compuestos anteriores. Las reacciones de 3-alquenoil-2-oxazolidonas con alquenos de elevada densidad electrónica dan lugar a productos de MukaiayamaMichael. El aducto de hetero-Diels-Alder sólo ha sido observado de forma espectroscópica como intermedio de reacción y únicamente ha sido aislado con bajo rendimiento en el caso descrito por Evans que se muestra en el Esquema 21.76,77 Esquema 21 2.2.4. Cicloadiciones [3+2] Las cicloadiciones 1,3-dipolares son, después de las reacciones de Diels-Alder en todas sus variantes, las reacciones pericíclicas más importantes. Estas reacciones se producen entre un compuesto que presenta dos cargas opuestas en disposición 1,3- en alguna de sus formas resonantes (dipolo) y un compuesto insaturado (dipolarófilo). Se clasifican como reacciones de cicloadición [3+2]. Estas reacciones son una herramienta sintética muy importante en la síntesis directa de heterociclos de cinco miembros que se encuentran presentes en un gran número de productos bioactivos.78-82 Existe una gran variedad de compuestos 1,3-dipolares que han sido utilizados en reacciones de cicloadición [3+2] enantioselectivas. En la Figura 17 se muestran los 31 Antecedentes Bibliográficos más estudiados: nitronas, iminas de azometino, iluros de azometino, iluros de carbonilo, iminas de nitrilo, óxidos de nitrilo y diazoalcanos.83-87 Figura 17: Compuestos 1,3-dipolares utilizados en métodos asimétricos. Nos centraremos aquí en las reacciones con nitronas. Las nitronas se preparan fácilmente a partir de aldehídos, aminas, iminas y oximas. Otra ventaja reside en su estabilidad, que permite guardarlas por un tiempo prolongado, en lugar de tener que generarse in situ como es el caso de otros compuestos 1,3-dipolares. Las nitronas pueden reaccionar tanto con alquenos con baja densidad electrónica (demanda electrónica normal) como con alquenos con elevada densidad electrónica (demanda electrónica inversa). Al contrario que las cicloadiciones [4+2], en las cicloadiciones 1,3-dipolares el cobre(II) no es el ión metálico de elección preferente con ligandos BOX, particularmente en el caso de Ph-BOX, siendo más utilizados complejos de Zn(II) y de Mg(II).19 El primer ejemplo de cicloadición 1,3-dipolar con demanda electrónica normal catalizada por un complejo Cu(II)-BOX fue publicado por Saito et al. en 2004 utilizando 3-alquenoil-2-oxazolidonas como dipolarófilos y el ligando BOX derivado de indanol con un puente no sustituido (Esquema 22).88 Con este sistema se obtienen elevados rendimientos y enantioselectividades, con diastereoselectividades entre buenas y moderadas. Curiosamente la reacción de la 3-acriloil-2-oxazolidona, al contrario que sus equivalentes sustituidos, da lugar a una mezcla en la que predomina mayoritariamente el diastereoisómero exo. La misma reacción catalizada por complejos Cu(II)-Bn-BOX tiene lugar con bajos rendimientos y enantioselectividades.89 Esquema 22 Sibi y colaboradores han documentado el uso de complejos Cu(II)-BOX con otros sustratos de tipo imida.90 Las imidas derivadas de 5,5-dimetil-3-pirazolidonas 1substituidas favorecen la formación de aductos exo de forma preferente y con elevada enantioselectividad, especialmente con el ligando inda-BOX con un puente ciclopropánico (Esquema 23). La adición de tamiz molecular provoca una reducción en 32 Reacciones de cicloadición catalizadas por complejos Cu(II)-BOX la diastereoselectividad. Con el ligando Me-BOX se produce una completa inversión de la diastereoselectividad pero manteniendo la enantioselectividad. Esquema 23 Además, Sibi et al. han utilizado N-acilacrilimidas acíclicas como dipolarófilos (Esquema 24).91 En este caso, utilizando el ligando inda-BOX con puente ciclopropílico, se obtienen rendimientos moderados con sustratos altamente impedidos como son los α,β-disustituidos, dando lugar a altas enantioselectividades. Por lo que respecta a la diastereoselectividad también es alta, proporcionando el aducto exo mayoritariamente, incluso con tamiz molecular. Esquema 24 Me O N Me Me Ph + Me O HN O R Cu(II)-BOX Me + N O Ph Me O Me O N HN Ph Me O Me BOX = O O N N HN O R R O R = alquil, aril El grupo de Palomo también ha utilizado las α’-hidroxienonas como dipolarófilos en cicloadiciones 1,3-dipolares (Esquema 25).92 La reacción transcurre con altos rendimientos y excelentes enantioselectividades y diastereoselectividades en presencia del complejo Cu(II)-t-Bu-BOX. La reacción tiene lugar con elevada regioselectividad (90:10), incluso con el derivado de la acroleína, la cual suele conducir a mezclas de regioisómeros. Esquema 25 Por lo que respecta a reacciones enantioselectivas con demanda electrónica inversa catalizadas por Cu(II), sólo se ha descrito un ejemplo, por Jørgensen, utilizando nitronas derivadas del glioxilato de etilo, que transcurre con resultados variables (Esquema 26). No obstante, se han descrito otros ejemplos con sistemas catalíticos que no utilizan Cu(II) que proporcionan mejores resultados.93-95 33 Antecedentes Bibliográficos Esquema 26 EtO O + O N OR Bn Bn [Cu(II)-t-Bu-BOX](OTf)2 RO O N O Bn OEt + RO N O OEt O La presencia de tamiz molecular juega un decisivo papel en muchas reacciones de cicloadición 1,3-dipolares, siendo necesaria en muchos procedimientos descritos en la literatura. En muchas reacciones catalizadas por ácidos de Lewis la presencia de tamiz es requerida para eliminar trazas de agua. Sin embargo, Jørgensen y colaboradores han demostrado que en el caso de cicloadiciones 1,3-dipolares catalizadas por complejos Mg(II)-BOX el papel del tamiz va más allá, jugando un papel activo en la reacción.96 Así, la presencia de tamiz en esta reacción da lugar a una inversión en la enantioselectividad respecto a la reacción llevada a cabo en ausencia de este aditivo. Esta inversión en la enantioselectividad se observa incluso cuando el tamiz está saturado de agua. Por lo expuesto anteriormente resulta difícil establecer un modelo estereoquímico de reacción en el caso de las cicloadiciones [3+2] asimétricas catalizadas con ácidos de Lewis quirales, dado que puede haber una interacción entre el complejo metálico y el tamiz. Hay que tener en cuenta además que las nitronas son especies que también pueden coordinar al centro metálico, complicando el mecanismo por el cual transcurre la reacción. 34 El esqueleto de canfano en catálisis asimétrica 2.3. El esqueleto de canfano en catálisis asimétrica El alcanfor es un compuesto natural abundante, quiral y estructuralmente rígido. Tanto este compuesto como otros derivados están disponibles comercialmente a un precio asequible. Estas características han hecho que los compuestos con esqueleto de canfano hayan sido ampliamente utilizados como auxiliares quirales en síntesis asimétrica.97 Con el auge de la catálisis enantioselectiva también se han sintetizado muchos compuestos derivados del alcanfor capaces de actuar como ligandos para la formación de complejos metálicos utilizados como catalizadores en reacciones enantioselectivas, muchas de ellas en reacciones de adición de reactivos de dialquilcinc a aldehídos. En 1986 Noyori y colaboradores introdujeron el (-)-3-exo(dimetilamino)isoborneol (DAIB, Figura 18a) como ligando para la adición de reactivos de dialquilcinc a aldehídos, consiguiendo hasta el 99% de exceso enantiomérico con aldehídos aromáticos y el 60% con alifáticos (Esquema 27.98,99 Por otra parte, Nugent ha utilizado el ligando (2R)-(+)-3-exo-morfolinoisoborneol (MIB), una modificación del DAIB que incorpora un anillo de morfolina, en esta misma reacción (Figura 18b).100 Este ligando mejora notablemente las adiciones a aldehídos alifáticos, llegando a un 99% de exceso enantiomérico en estos casos. (a) (b) OH N O Figura 18 Esquema 27 Oppolzer y Radinov documentaron en 1988 tres ligandos de tipo aminoalcohol e hidroxiamida derivados del ácido cetopínico (Figura 19a).101 El ligando tridentado proporciona buenos rendimientos en la adición de dietilcinc y divinilcinc a benzaldehído y hexanal, con excesos enantioméricos del 90%. Resulta destacable que para la adición de divinilcinc sólo sea necesario el uso de una carga molar del ligando del 2%. Por lo que respecta a los otros dos ligandos, la enantioselectividad es semejante, pero los rendimientos obtenidos son menores, en especial con el aminoalcohol, aunque la inducción asimétrica es inversa respecto a la obtenida con el ligando tridentado. 35 Antecedentes Bibliográficos OH OH N MeN OH O N NMe2 (a) (b) Figura 19 Aoyama y Hari han presentado recientemente otro aminoalcohol (Figura 19b) funcionalizado en la posición 10 con el que se consiguen enantioselectividades de hasta el 95% en la adición de dietilcinc a aldehídos aromáticos.102 En 1997 Dimitrov et al. describieron el uso de aminoalcoholes procedentes de la alquilación o arilación del alcanfor en la adición de dietilcinc a benzaldehído (Figura 20a), obteniendo excesos enantioméricos no mayores del 64%.103 El mismo grupo ha preparado una serie de aminodioles que proporcionan resultados similares en cuanto a enantioselectividad (Figura 20b).104 (a) (b) Figura 20 También se han utilizado ligandos derivados del ácido canforsulfónico para reacciones de adición de reactivos de organocinc. En 1997 Yus y Ramón presentaron ligandos de tipo canforsulfonamida (Figura 21a), consiguiendo excesos enantioméricos de hasta el 80% mediante la activación con alcóxidos de titanio, tanto en la adición a aldehídos como a cetonas.105-107 En 1998 el grupo de Uang describió otro ejemplo de adición de dietilcinc a benzaldehídos mediada por Ti(IV) utilizando la bis(sulfonamida) con simetría C2 representada en la Figura 21b.108 En estas condiciones llegaron a obtener el producto de reacción con un 91% de exceso enantiomérico. El uso del ligando diastereoisomérico preparado a partir de la (S,S)-ciclohexanodiamina proporciona el enantiómero de configuración opuesta aunque con una merma importante en la enantioselectividad. Walsh y Balsells profundizaron en el estudio de estos ligandos, determinando que la diamina controlaba el sentido de la inducción quiral, mientras 36 El esqueleto de canfano en catálisis asimétrica que el fragmento de canfano controlaba la velocidad de reacción (TOF b ).109 Este hecho permite la obtención de hasta un 84% de exceso enantiomérico utilizando una mezcla diastereoisomérica del ligando sintetizado a partir de la diamina racémica, pese la inducción de quiralidad opuesta de ambos ligandos diastereoisoméricos. OH (b) (a) O O O2S NH HN SO2 OH (c) O2S NH HN SO2 R = Bn, 1-naftilmetil Figura 21 Tanto Yus como Walsh, de forma independiente, han utilizado también la bis(hidroxisulfonamida) resultante de la reducción de los grupos carbonilos de este mismo ligando C2 (Figura 21c) en adiciones a grupo carbonilo. Con este ligando se han conseguido excelentes excesos enantioméricos en la adición de una amplia variedad de reactivos de dialquilcinc y diarilcinc tanto a aldehídos como a cetonas.110-117 Así mismo, también se obtiene una alta enantioselectividad en la adición de ácidos borónicos a cetonas α,β-insaturadas por transmetalación con dietilcinc.118 Walsh también ha ensayado un ligando similar pero sustituyendo el anillo de ciclohexano por otro de ciclopentano, obteniendo resultados ligeramente inferiores.119 Posteriormente, Yus ha sintetizado otro ligando C2 derivado del ácido canforsulfónico (Figura 22a) con el que se mejoran los resultados anteriores en diversas reacciones de adición de reactivos organometálicos de cinc a cetonas, llegando en algún caso a obtener un 90% de exceso enantiomérico.118 Más recientemente Wang y colaboradores han utilizado este mismo ligando para adiciones de fenilacetileno mediadas por dietilcinc y triflato de cobre con resultados notables.120 (a) (b) Figura 22 b TOF = Turnover frecuency. Expresa el número de moléculas transformada por el catalizador por unidad de tiempo. 37 Antecedentes Bibliográficos Éste último grupo había publicado con anterioridad la adición de fenilacetileno a aldehídos con el uso de otro tipo de hidroxisulfonamida de estructura similar al DAIB (Figura 22b) con resultados de hasta un 97% de exceso enantiomérico, empleando alcóxidos de titanio y dietilcinc.121 De la Moya et al. han descrito recientemente un grupo de bis(hidroxiamidas) con simetría C2 derivadas del ácido cetopínico y diaminas (Figura 23).122 Estos ligandos dan lugar a mejores rendimientos que el ligando C1 de Oppolzer (Figura 19a) en la adición de dietilcinc a aldehídos, con una enantioselectividad semejante (90% de exceso enantiomérico) en el caso del derivado de piperazina. El mismo autor ha comparado estos ligandos con otros ligandos de tipo hidroxiamida similares con simetrías C1, C2 y C3, derivados de aminas cíclicas y ácido cetopínico, así como de sus aminoalcoholes derivados.123 Los rendimientos obtenidos son similares en todos los casos, pero con enantioselectividades variables que llegan hasta el 72% de exceso enantiomérico. OH O O N N OH Figura 23 Una de las últimas incorporaciones a la lista de ligandos con esqueleto de canfano aplicados en adiciones a grupo carbonilo de reactivos organometálicos de cinc es el aminotiol de la Figura 24 descrito por Uang. Este ligando da lugar a altas enantioselectividades en la adición de ácidos arilborónicos y reactivos de divinilcinc a aldehídos. En el caso de adiciones de dimetilcinc a cetoésteres los resultados son moderados.124-126 Figura 24 Los ligandos derivados de canfano se han utilizado también en otras reacciones. Los ligandos C2 descritos por Uang derivados del ácido cetopínico ( Figura 25), han sido utilizados en la reacción de adición de cianotrimetilsilano a aldehídos catalizada por Ti(IV) (Esquema 28), llegando a obtener una elevada enantioselectividad (99% de exceso enantiomérico).127,128 38 El esqueleto de canfano en catálisis asimétrica Figura 25 Esquema 28 Uang también ha descrito el uso de N-hidroxiamidas derivadas del ácido cetopínico (Figura 26) como ligandos en epoxidaciones de alcoholes alílicos catalizadas por complejos de vanadio(V) con una enantioselectividad moderada.129 O R NOH O O R NOH R' Figura 26 Los derivados del alcanfor también han sido utilizados como ligandos en la reducción asimétrica de cetonas con borano (Esquema 29). Así, Kagan et al. publicaron en 1998 el uso de una serie de hidroxitioles (Figura 27a,b).130 En este caso la enantioselectividad conseguida sólo llega hasta al 64% de exceso enantiomérico. Poco después Yang et al. consiguieron mejorar el resultado del ligando de la Figura 27a, optimizando las condiciones de reacción y mejorando ligeramente la enantioselectividad.131 También Chan et al. presentaron casi al mismo tiempo otro ligando, derivado del ácido cetopínico, pero sin mejora sustancial de la enantioselectividad (Figura 27c).132 Esquema 29 (a) (b) (c) Figura 27 39 Antecedentes Bibliográficos En el año 2000, Santhi et al. obtuvieron una enantioselectividad ligeramente mayor en esta reacción haciendo uso de aminoalcoholes con una orientación endo (Figura 28a).133 Ambos ligandos conducen a resultados similares, con enantioselectividades entre el 60 y el 90% de exceso enantiomérico. Recientemente Xie et al. han modificado uno de estos ligandos formando una amida del ácido escuárico (Figura 28b), que ha permitido en algunos casos incrementar la enantioselectividad hasta el 99% de exceso enantiomérico aunque los resultados son generalmente inferiores con la mayoría de sustratos.134 (a) (b) Figura 28 En el mismo año 2000 Chan y colaboradores describieron un ligando de tipo aminofosfina-fosfinito derivado del ácido cetopínico (Figura 29a).135 Los complejos de rodio catalizan la hidrogenación de aminoácidos α,β-insaturados (Esquema 30a) con excesos enantioméricos entre el 62 y el 90%. En esta misma reacción, así como en la hidrogenación de iminas y β-iminoácidos (Esquema 30b,c), Kadyrov y colaboradores han alcanzado una enantioselectividad que supera el 99% de exceso enantiomérico con otra serie de ligandos de tipo fosfina (Figura 29b). La hidrogenación también es muy enantioselectiva en el caso de N-acetil enaminas.136 Knochel describió en 2004 otros ligandos de tipo P,N (Figura 29c) para la formación de complejos de iridio, obteniendo altas enantioselectividades en reacciones de hidrogenación de alquenos, substituciones de acetatos alílicos (Esquema 32) y, con resultados más modestos, en hidroboraciones de alquenos.137-139 PAr2 (a) OPPh2 MeN PPh2 (b) PPh2 (c) Ar2P N S Figura 29 Esquema 30 (a) Continúa en la página siguiente… 40 El esqueleto de canfano en catálisis asimétrica Esquema 30 (continuación) (b) (c) Andersson et al. describieron en 2004 el uso de un ligando de tipo aminotiol para la formación de complejos de iridio como catalizadores de hidrogenación por transferencia de hidruro con resultados que llegan al 70% de exceso enantiomérico (Esquema 31).140 Ligando = Esquema 31 El esqueleto de canfano se encuentra presente también en numerosos ligandos cuyos complejos de paladio han sido utilizados como catalizadores en diversos tipos de reacciones. Nakano y colaboradores han presentado un fosfinooxatiano (Figura 30a) que da lugar a enantioselectividades de hasta el 94% en reacciones de substitución de acetatos alílicos con malonato de dimetilo en medio básico (Esquema 32) así como con bencilamina o ftalimida potásica.141,142 También han aplicado este tipo de ligandos en reacciones de Diels-Alder utilizando 3-acriloil-2-oxazolidona como dienófilo, llegando a un exceso enantiomérico máximo del 93%.143 Otro ligando similar utilizado para formar complejos de paladio ha sido descrito por Mino et al. (Figura 30b). En este caso los excesos enantioméricos obtenidos en substituciones alílicas con alquilmalonatos se sitúan entre el 64 y el 95%.144 S (a) O N (b) Ph Ar2P O PAr2 Figura 30 Esquema 32 41 Antecedentes Bibliográficos Gilbertson y Fu han sintetizado un ligando de tipo fosfinooxazolina (Figura 31a) que han utilizado en la reacción de Heck asimétrica entre triflatos olefínicos o arílicos y dihidrofurano (Esquema 33).145 La enantioselectividad inducida por el ligando se sitúa entre el 75 y el 96% de exceso enantiomérico. Hiroi et al. han utilizado otros ligandos con esqueleto de canfano (Figura 31b) para la catálisis con paladio de reacciones de Diels-Alder entre ciclopentadieno y 3-alquenoil-2-oxazolidonas, obteniéndose los cicloaductos correspondientes con un exceso enantiomérico entre el 80 y el 93%%.146 (a) O N PPh2 (b) t-Bu Figura 31 Esquema 33 C.-T. Chen y Barhate han usado un complejo de oxovanadilo con una imina derivada del ácido cetopínico en el acoplamiento oxidativo de naftoles para dar binaftoles quirales (BINOLes) (Esquema 34).147 La enantioselectividad conseguida con este ligando varía entre el 60 y el 87% de exceso enantiomérico. Esquema 34 complejo = complejo, O2 OH CCl4, t.a. OH OH O O O N t O O Bu V Otra serie de ligandos de tipo imina derivados del ácido cetopínico ha sido desarrollada por K. Chen y colaboradores (Figura 32). Una primera aplicación de estos ligandos ha sido la formación de N-ftalimidoaziridinas a partir de 3-alquenoil-2oxazolidonas mediante tetraacetato de plomo, obteniendo excesos enantioméricos entre el 67 y el 90% (Esquema 35a.148 También se han aplicado estos ligandos junto a triflato de lantano en la reacción de Baylis-Hillman (Esquema 35b), con una inducción asimétrica entre el 10 y el 95% de exceso enantiomérico, dependiendo del acrilato utilizado.149 42 El esqueleto de canfano en catálisis asimétrica HO N O O N OH Figura 32 Esquema 35 (a) (b) Kataoka et al. también han utilizado el ligando de la Figura 27a, así como sus derivados S-bencil, S-metil y O,S-dimetil, en reacciones de Baylis-Hillman.150 Los resultados en general son muy pobres, aunque en el caso de ligando S-metilado se llega a obtener el producto esperado con un 71% de exceso enantiomérico aunque con bajo rendimiento. Nuestro grupo de investigación ha desarrollado una serie de ligandos modulares con estructura de iminopiridina por condensación de diversas cetonas monoterpénicas con piridinalquilaminas. Los complejos de Cu(II) con iminopiridinas derivadas de alcanfor y del ácido cetopínico catalizan la reacción de Henry entre aldehídos y nitrometano (Esquema 36) con excesos enantioméricos moderados (hasta el 86%).151-154 Cabe destacar que los ligandos derivados de alcanfor conducen a enantiómeros mayoritarios opuestos a los obtenidos con los ligandos derivados de ácido cetopínico. Por otra parte la aminopiridina obtenida por reducción del doble enlace C=N en el primer ligando, más flexible que la imina, permite utilizar otros nitroalcanos superiores con excelentes resultados en cuanto rendimiento y exceso enantiomérico (hasta el 98%). Los ligandos anteriores de tipo iminopiridina también catalizan la adición de nitrometano a cetoésteres, aunque en esta reacción es una imina derivada de la fenchona la que conduce a los mejores resultados.155 43 Antecedentes Bibliográficos Esquema 36 L = H N N R N N R R = Me, COOH Por último, cabe mencionar la utilización de compuestos con esqueleto de canfano como organocatalizadores. Kokotos ha preparado una serie de ésteres de 4-hidroxiprolina, dos de los cuales son los ésteres de los ácidos (R)-canforsulfónico y (S)-canforsulfónico (Figura 33) que han sido utilizados como catalizadores en reacciones aldólicas, proporcionando los productos correspondientes con enantioselectividades notables que en algún caso alcanzan el 90% de exceso enantiomérico.156 OH HO O S O O O S O O N N O H O H HO HO Figura 33 Ogilvie y Lemay han descrito un derivado del ácido cetopínico que cataliza la reacción de Diels-Alder (Figura 34a) entre ciclopentadieno y diversos aldehídos α,βinsaturados en medio acuso con excesos enantioméricos entre el 69 y el 94%.157 Sigman y Rajaram consiguieron enantioselectividades entre el 70 y el 90% en reacciones de hetero-Diels-Alder organocatalíticas entre diversos aldehídos y 3-(tbutildimetilsililoxi)-1-(dimetilamino)-1,3-butadieno empleando el ligando de la Figura 34b que contiene un resto canforsulfonamida.158 Cuando este resto sulfonamida se cambia por otros tipos de amidas se obtienen resultados peores. 44 El esqueleto de canfano en catálisis asimétrica Ph O N O NH · TfOH Ph Ph N O O S N H O Ph Ph OH (a) (b) Figura 34 Sin embargo, en un intento de utilizar el ácido canforsulfónico como catalizador en reacciones de Friedel-Craft entre enonas y indoles, Xu et al. obtuvieron resultados muy poco satisfactorios.159 Lee ha descrito en 2008 un organocatalizador de tipo canforsulfonilhidracinas (Figura 35) para reacciones de Diels-Alder en agua, obteniendo enantioselectividades similares a las obtenidas por Ogilvie.160 Este mismo año Langlois ha publicado un trabajo similar, utilizando otra serie de canforsulfonilhidracinas y nitrometano como disolvente pero con resultados inferiores.161 NH SN O O Et Figura 35 Por último, Chen et al. han descrito recientemente diferentes tioureas unidas a una unidad de ácido cetopínico y a una unidad de prolina (Figura 36).162 Estos compuestos catalizan reacciones aldólicas entre ciclohexanona y diversos benzaldehídos en medio acuoso con una elevada enantioselectividad. Figura 36 45 Antecedentes Bibliográficos 46 Objetivos 3. O BJETIVOS Las reacciones de cicloadición tienen gran importancia en síntesis orgánica ya que permiten preparar sistemas cíclicos y heterocíclicos con una gran eficacia y economía atómica. El desarrollo de versiones enantioselectivas de estas reacciones, especialmente las catalíticas, ha atraído un interés considerable en años recientes. La búsqueda de nuevos ligandos quirales capaces de formar complejos metálicos con carácter ácido de Lewis que puedan actuar como catalizadores en estas reacciones presenta un gran interés. Como se ha mostrado en la introducción, el esqueleto de canfano ha sido ampliamente utilizado como plataforma quiral en el desarrollo de auxiliares y ligandos quirales. El ácido (1S)-(+)-cetopínico presenta doble funcionalización sobre esta plataforma, lo que lo convierte en un material de partida muy atractivo para la preparación de ligandos quirales. Por otra parte, la enantioselectividad de las reacciones depende en gran manera de la unión del sustrato orgánico al centro metálico del catalizador. La formación de un quelato entre ambos suele imponer serias restricciones conformacionales en la especie catalítica activa, por lo que los sustratos quelantes con dos puntos de unión al catalizador suelen conducir a enantioselectividades muy elevadas. De acuerdo con estas consideraciones, en la presente tesis se plantearon los siguientes objetivos: 1. Desarrollo de nuevos ligandos de tipo hidroxioxazolina derivados del ácido cetopínico y su aplicación en reacciones de Diels-Alder enantioselectivas catalizadas por Cu(II). 2. Desarrollo de nuevas reacciones de cicloadición enantioselectivas utilizadas por complejos de Cu(II)-BOX con sustratos quelantes. 2.1. N-óxidos de 2-alquenoilpiridina en reacciones de cicloadición. 2.1.1. Reacciones de Diels-Alder. 2.1.2. Reacciones de hetero-Diels-Alder con demanda electrónica inversa. 2.1.3. Reacciones de cicloadición 1,3-dipolares. 2.2. α’-Arilsulfonil enonas en reacciones de cicloadición. 2.2.1. Reacciones de Diels-Alder. 47 Objetivos 48 Diseño de ligandos con estructura de hidroxioxazolinas a partir del ácido (1S)-cetopínico. Aplicación en Reacciones de Diels-Alder enantioselectivas 4. R ESULTADOS Y D ISCUSIÓN 4.1. Diseño de ligandos con estructura de hidroxioxazolinas a partir del ácido (1S)-cetopínico. Aplicación en reacciones de Diels-Alder enantioselectivas 4.1.1. Introducción La catálisis asimétrica mediante complejos metálicos es uno de los campos de investigación más importantes y de desarrollo más rápido en química orgánica. En este contexto la búsqueda de nuevos compuestos que puedan ser utilizados como ligandos quirales resulta particularmente interesante. Los ligandos basados en el anillo de oxazolidona han jugado un papel fundamental en reacciones asimétricas catalizadas por metales, especialmente con Cu(II) y otros metales de transición. Sin lugar a dudas, las bis(oxazolinas) con simetría C2 son el grupo más importante de estos ligandos.19 Sin embargo, también se han descrito ligandos con simetría C2 que presentan una agrupación oxazolina y que se han utilizado con éxito en diversas reacciones enantioselectivas. Por ejemplo, Bolm et al. han preparado una serie de ligandos con estructura de hidroxioxazolina a partir de aminoalcoholes e hidroxiácidos que han sido utilizados en reacciones de transferencia de grupo fenilo a aldehídos y cetoésteres (Figura 37).163-165 Figura 37 Tal como se ha visto en los antecedentes, muchos compuestos con esqueleto de canfano han sido utilizados como auxiliares quirales y ligandos en diversas reacciones enantioselectivas debido a su rigidez y a la elevada estereodiferenciación que este esqueleto impone. En el diseño de nuevos ligandos con estructura de hidroxioxazolina se planteó la posibilidad de utilizar ácido (1S)-(+)-cetopínico (1), un βcetoácido derivado del alcanfor que, dada su doble funcionalización, debe permitir preparar, por modificación del ácido carboxílico y/o del grupo cetona, una gran variedad de ligandos útiles en química de coordinación, entre ellos β-hidroxioxazolinas. En este trabajo se han preparado las siguientes hidroxioxazolinas 2-6 a partir del ácido 49 Resultados y Discusión (1S)-(+)-cetopínico con la finalidad de utilizarlas como ligandos en la reacción de DielsAlder enantioselectiva catalizada por ácidos de Lewis (Figura 38). OH N O 1 2 OH OH N O 3 N O 4 OH N O 5 6 Figura 38 Estas hidroxioxazolinas son ligandos bidentados con simetría C1. Se diferencian en la sustitución y estereoquímica del anillo de oxazolina. Todos ellos presentan un grupo hidroxilo derivado de la reducción de la cetona, siendo su orientación exo, a excepción del ligando 6, el cual presenta una orientación endo. 4.1.2. Síntesis de las hidroxioxazolinas 2-6 y 21 Aunque el ácido (1S)-(+)-cetopínico (1) es comercial, para el uso a escala de gramos resulta más ventajoso sintetizarlo a partir del ácido (1S)-(+)-canforsulfónico (7), disponible comercialmente en forma enantioméricamente pura a un precio mucho más asequible. c La síntesis del ácido (1S)-(+)-cetopínico (1) se llevó a cabo siguiendo un proceso que consta de dos pasos (Esquema 37). El primero de ellos es la preparación del cloruro de (1S)-(+)-canforsulfonilo por tratamiento del ácido 7 con cloruro de tionilo a reflujo de cloroformo según el procedimiento experimental descrito por Towson et al.166 En la segunda etapa se oxidó el cloruro de (1S)-(+)-canforsulfonilo con KMnO4 en medio básico acuoso, siguiendo el procedimiento aportado por Bartlett y Knox.167 Después de una purificación por recristalización de agua se obtuvo el ácido 1 con un rendimiento global del 35%. c Según el catálogo de Sigma-Aldrich 1 g de ácido (1S)-(+)-cetopínico cuesta 72,7 €, mientras que 500 g del ácido (1S)-(+)-canforsulfónico cuestan 127,0 €. 50 Diseño de ligandos con estructura de hidroxioxazolinas a partir del ácido (1S)-cetopínico. Aplicación en Reacciones de Diels-Alder enantioselectivas Esquema 37 7 8 1 Una vez sintetizado el ácido (1S)-(+)-cetopínico (1), la síntesis de las hidroxioxazolinas 2-6 se realizó en tres etapas de acuerdo con el Esquema 38. Así, el primer paso es la formación de una hidroxiamida entre el ácido (1S)-(+)-cetopínico (1) y los correspondientes β-aminoalcoholes 9-12. La segunda etapa es la ciclación de la hidroxiamida con deshidratación para formar el anillo de oxazolina. Finalmente la reducción de la cetona en la posición 2 del anillo de canfano conduce a las hidroxioxazolinas. Esquema 38 En el Esquema 39 se muestra la preparación de las β-hidroxiamidas 13-16. En primer lugar se llevó a cabo la formación del cloruro del ácido (1S)-(+)-cetopínico por reacción con cloruro de tionilo a reflujo durante 2 horas. Después de eliminar a presión reducida el exceso de SOCl2, el cloruro de ácido resultante se disolvió en diclorometano y se inyectó gota a gota sobre otra disolución del correspondiente βaminoalcohol y trietilamina en diclorometano a 0 °C. Los rendimientos obtenidos están recogidos en la Tabla 1. Esquema 39 51 Resultados y Discusión Tabla 1: Formación de las β-Hidroxiamidas. β-Aminoalcohol 1 9 βhidroxiamida Rendimiento (%) 13 90 14 84 15 90 16 80 (S)-(+)-2-amino-3-metil-1-butanol 2 10 (S)-(+)-fenilglicinol NH2 3 HO 11 (R)-(-)-fenilglicinol 4 12 (1R,2S)-(+)-cis-1-amino-2-indanol El paso siguiente en la síntesis de los ligandos fue la ciclación de la agrupación β-hidroxiamida para formar el anillo de oxazolina. En el caso de las amidas 13, 14 y 15 la ciclación se llevó a cabo siguiendo el procedimiento descrito por Gilbertson y Zu (Esquema 40).145 Este procedimiento implica la disolución de la amida y trietilamina en diclorometano con la posterior adición de cloruro de mesilo, dejando la mezcla agitando durante una noche a temperatura ambiente. Esquema 40: Formación del anillo de oxazolina. O O HO NH R 13 R = (S) i-Pr 14 R = (S) Ph 15 R = (R) Ph 52 O MsCl, Et3N CH2Cl2 N O R 17 R = (S) i-Pr 18 R = (S) Ph 19 R = (R) Ph Diseño de ligandos con estructura de hidroxioxazolinas a partir del ácido (1S)-cetopínico. Aplicación en Reacciones de Diels-Alder enantioselectivas Tabla 2: Formación del anillo de oxazolina. Cetoamida 1 13a 2 14a 3 4 a Rendimiento (%) Cetooxazolina N 17 90 18 81 15a 19 96 16b 20 64 O N O MsCl, Et3N, CH2Cl2. b (NH4)6Mo7O24·4H2O, tolueno, reflujo. El mecanismo de reacción de este método probablemente implica la mesilación del grupo hidroxilo seguido de desplazamiento nucleofílico del mesilato por parte de la amida con inversión de configuración (Esquema 41a).168-170 En las amidas 13 a 15 esta inversión no tiene ninguna consecuencia estereoquímica, pero sí en el caso de la amida 16, donde existe un centro estereogénico sobre el carbono que soporta el grupo hidroxilo. Por esta razón, para la ciclación de este sustrato se prefirió aplicar directamente un método sintético que transcurriera con retención de configuración (Esquema 41b). El procedimiento que se utilizó fue el descrito por Ishihara et al. (Esquema 42).171,172 Una mezcla del sustrato y del catalizador (NH4)6Mo7O24·4H2O se calentó a reflujo de tolueno hasta que se consumió el compuesto 16. Los rendimientos obtenidos se encuentran expresados al final de la Tabla 2. 53 Resultados y Discusión Esquema 41 (a) Ciclación con inversión de configuración: (b) Ciclación con retención de configuración: Esquema 42: Formación del anillo de oxazolina catalizada por molibdeno. Tal como se explica más adelante, experimentos de RMN con el ligando 5 confirmaron que la reacción transcurre con retención de configuración. Reducción de la cetona en C2 del esqueleto de canfano Una vez formado el anillo de oxazolina se procedió a la reducción del grupo carbonilo de las diferentes cetooxazolinas. Se ensayaron diversos métodos de reducción con el compuesto 17. Los intentos de reducción con NaBH4 en metanol, así como con LiBH4, más reactivo, no tuvieron éxito, recuperándose en ambos casos el sustrato de partida sin reaccionar. El uso de LiAlH4 en THF dio lugar a la descomposición del material de partida. No obstante, se consiguió llevar a cabo la reducción de la cetona haciendo uso del complejo [LiAlH4·2THF] en tolueno a 0 °C, obteniéndose la hidroxioxazolina deseada 2 como único producto con un 57% de rendimiento. Finalmente se pudo mejorar el rendimiento disminuyendo la temperatura de reacción hasta -40 °C (Esquema 43). El resto de cetooxazolinas 18-20 se redujeron bajo idénticas condiciones. En el caso de las cetooxazolinas 19 y 20, además de los alcoholes 4 y 5, también se aislaron sus epímeros 21 y 6 con el grupo hidroxilo con orientación endo. Hay que destacar la baja diastereoselectividad observada en la 54 Diseño de ligandos con estructura de hidroxioxazolinas a partir del ácido (1S)-cetopínico. Aplicación en Reacciones de Diels-Alder enantioselectivas reducción de 20 en estas condiciones. Esta diastereoselectividad se pudo mejorar reduciendo la cetooxazolina 20 con NaBH4 en metanol en presencia de CeCl3·7H2O. De esta forma se aumentó la proporción endo/exo de los alcoholes, aunque el rendimiento total de la reacción disminuyó apreciablemente. Los resultados obtenidos en la reducción de las diferentes cetooxazolinas 17-20 se muestran en la Tabla 3. Esquema 43 Tabla 3: Reducción del carbonilo de cetona. Cetooxazolina Hidroxioxazolina Rendimiento (%) 1 17a 2 63 2 18 a 3 70 3 19a 4 62 21 (endo) 15 4 20a 5 45 6 (endo) 47 5 20b 5 50 a 6 (endo) [LiAlH4·2THF] 1 M, tolueno, -40 °C. MeOH, -10 °C. 10 b NaBH4, CeCl3·7H2O, De los resultados se puede desprender que los sustituyentes en el anillo de oxazolina con configuración R dan lugar a un mayor impedimento estérico por la cara Si de la cetona, dificultando la formación preferencial del alcohol con orientación exo. La estereoquímica del carbono que soporta el grupo hidroxilo en los compuestos 4 y 5 se verificó mediante experimentos de tipo NOE empleando dimetilsulfóxido como disolvente (Figura 39). Este disolvente permite obtener señales definidas del protón hidroxílico. En el caso del compuesto 4 se observó NOE entre el protón hidroxílico (δ 5,27 ppm) y el metilo situado a δ 1,26 ppm. Por lo que respecta al compuesto 5 también se observó NOE entre el protón hidroxílico (δ 5,33 ppm) y el metilo a δ 1,15 ppm, mientras que al irradiar la señal correspondiente al protón geminal al oxígeno (δ 3,78 ppm) no se observó respuesta en ninguno de los metilos del esqueleto de canfano. Estos resultados confirmaron la orientación exo esperada para el grupo hidroxilo en ambos compuestos. 55 Resultados y Discusión 1,15 O H 5,33 N O 4 5 Figura 39 La comparación de las constantes de acoplamiento teóricas de un sistema de norbornano con las medidas experimentalmente para el protón geminal a oxígeno en el ligando 5 [δ 3,9 ppm (dd, J = 3,5 Hz, J = 7,8 Hz)] y en el ligando 6 [δ 4,7 ppm (ddd, J = 1,3 Hz, J = 3,7 Hz, J = 10,2 Hz)] apoya la asignación exo y endo respectiva para el grupo hidroxilo en ambos compuestos (Figura 40). Para el caso de las hidroxioxazolinas 2 y 3 se asignó la orientación exo para el grupo hidroxilo de acuerdo con los valores de las constantes de acoplamiento siguiendo este mismo criterio. Figura 40 Además, los experimentos de RMN realizados con la hidroxioxazolina 5 permitieron comprobar que la reacción de ciclación de la β-hidroxiamida 16 para obtener la cetooxazolina 20 catalizada por molibdato amónico tiene lugar con retención de la configuración del carbono que soporta el grupo hidroxilo. Así, irradiando la señal correspondiente al protón del anillo de oxazolina (HO) que aparece a δ 5,29 ppm se observó NOE intenso con la señal que aparece a δ 3,47 ppm (dd, J = 7,0 Hz, J = 18,0 Hz), que se asignó al protón Hcis (Figura 41). Además, se observó otro efecto NOE más débil con la señal a δ 3,23 ppm (dd, J = 1,6 Hz, J = 18,0 Hz) que se asignó al protón Htrans. Las constantes de acoplamiento respectivas para dichos protones coinciden con las calculadas a partir de la ecuación de Karplus con los ángulos de torsión obtenidos computacionalmente (PM3, disolvente: CHCl3) para la hidroxioxazolina 5 derivada del cis-aminoindanol 12 (Tabla 4). 56 Diseño de ligandos con estructura de hidroxioxazolinas a partir del ácido (1S)-cetopínico. Aplicación en Reacciones de Diels-Alder enantioselectivas R R O Inversión de configuración N HN HO Htrans Hcis O NOE Retención de configuración N HN HO Hcis Htrans Figura 41 Tabla 4. Comparación de los acoplamientos con HO teóricos y experimentales. HX Inversión (HO-HN en trans) Ángulo (°) JHo-Hx (Hz) Retención (HO-HN en cis) Ángulo (°) JHo-Hx (Hz) Valores Experimentales J (Hz) HN 173 ∼9 2 ∼ 8,5 8,1 Hcis 46 ∼3 9 ∼8 7,0 Htrans 171 ∼8 110 ∼1 1,8 4.1.3. Síntesis de 3-alquenoil-2-oxazolidonas Para ensayar la actividad de los ligandos preparados anteriormente como precatalizadores en reacciones de Diels-Alder se utilizaron dienófilos bidentados de tipo 3alquenoil-2-oxazolidona. Los dienófilos 22a-d (Figura 42) fueron sintetizados siguiendo o adaptando procesos descritos en la literatura. 22a, R1 = H , R2 = H 22b, R1 = Me, R2 = H 22c, R1 = H, R2 = Me 22d, R1 = Ph, R2 = H Figura 42 La 3-acriloil-2-oxazolidona 22a se sintetizó haciendo reaccionar cloruro de acriloilo con 2-oxazolidona previamente desprotonada con hidruro sódico, obteniéndose el producto con un rendimiento del 46% (Esquema 44). Este mismo procedimiento se utilizó en la preparación de la 3-crotonoil-2-oxazolidona 22b y la 3metacriloil-2-oxazolidona 22c, obteniéndose un rendimiento del 80 y del 56 % respectivamente. Esquema 44 22a, R1 = H , R2 = H 46% 22b, R1 = Me, R2 = H 80% 1 2 22c, R = H, R = Me 56% 57 Resultados y Discusión La 3-cinamoil-2-oxazolidona 22d se sintetizó según el procedimiento descrito por Andrade et al. (Esquema 45),173 que implica la reacción entre ácido cinámico y 2oxazolidona en presencia de diciclohexilcarbodiimida (DCC) como agente activante y de una cantidad subestequiométrica de dimetilaminopiridina (DMAP) como catalizador, obteniéndose un rendimiento del 65%. Esquema 45 22d 4.1.4. Síntesis de los dienos Si bien la mayoría de los dienos utilizados (Figura 43) en las reacciones de DielsAlder fueron adquiridos de fuentes comerciales (23a-e), algunos tuvieron que ser sintetizados (23f-h). 23a 23b 23e 23f 23d 23c 23h 23g Figura 43 El 1-fenil-1,3-butadieno 23f se sintetizó de acuerdo al procedimiento descrito por Okamoto et al. (Esquema 46).174 La síntesis consiste en una reacción de Wittig entre el cinamaldehído y el iluro derivado de bromuro de metiltrifenilfosfonio en THF, que en nuestro caso transcurrió con un rendimiento del 62%. Esquema 46 23f El 1,2-dimetilenociclohexano (23g) se sintetizó a partir del anhídrido del ácido cis-1,2-ciclohexanodicarboxílico (24) (Esquema 47). Después de reducir 24 con LiAlH4 (93% de rendimiento), el diol resultante 25 se trató con cloruro de mesilo en diclorometano y trietilamina como base para dar lugar a 26 de forma cuantitativa. Este se trató finalmente con carbonato de litio y bromuro de litio a 140 °C en 58 Diseño de ligandos con estructura de hidroxioxazolinas a partir del ácido (1S)-cetopínico. Aplicación en Reacciones de Diels-Alder enantioselectivas dimetilformamida, aislando después de 6 horas de reacción el producto 23g y el posible intermedio 27 con un 15% y un 10% de rendimiento respectivamente. Esquema 47 Para la síntesis del silil enol éter 23h se siguió el procedimiento de Jung y McCombs (Esquema 48).175 La metilvinilcetona se hizo reaccionar con cloruro de trimetilsililo utilizando trietilamina como base en dimetilformamida a 85 °C. El producto 23h se aisló por destilación con un 43% de rendimiento. Esquema 48 4.1.5. Reacciones de Diels-Alder catalizadas complejos de hidroxioxazolinas y Cu(II) por 4.1.5.1. Optimización de las condiciones de reacción Una vez preparados los ligandos 2-6, se estudió su capacidad para inducir enantioselectividad en reacciones de Diels-Alder entre dienos y 3-alquenoil-2oxazolidonas como dienófilos catalizadas por complejos de metales de transición. Como reacción estándar para la optimización de las condiciones se eligió la reacción de Diels-Alder entre 3-acriloil-2-oxazolidona (22a) y ciclopentadieno (23a) para dar lugar al aducto 28aa. En primer lugar se evaluaron los diferentes ligandos, empleando una carga molar de los mismos del 10% respecto 22a, 10% molar de triflato cobre(II) como sal metálica y diclorometano como disolvente. Los resultados se recogen en la Tabla 5. Tal como se puede observar, la estereoquímica absoluta del producto de DielsAlder está determinada primariamente por la estereoquímica del centro estereogénico en el anillo de oxazolina. Así, los ligandos procedentes de aminoalcoholes con configuración S en el carbono unido al nitrógeno dan lugar al aducto 28aa con configuración R (entradas 1-4), mientras que los ligandos con configuración R en dicho 59 Resultados y Discusión carbono (entradas 5-11) conducen a productos de Diels-Alder con configuración S. Igualmente se observa que la presencia de un sustituyente fenilo en el anillo de oxazolina mejora la enantioselectividad así como la velocidad de reacción respecto al ligando con un sustituyente isopropilo. Tabla 5: Evaluación de los ligandos.a Ligando Disolvente T (°C) tiempo (h) endo:exo ee endo (%)b Configuraciónc 1 2 CH2Cl2 -30 45 85:15 19 R 2 3 CH2Cl2 -30 0,6 80:20 46 R 3 3 Acetona -20 20 90:10 24 R 4 3 Tolueno -30 1 85:15 47 R 5 4 CH2Cl2 -30 1 89:11 70 S 6 4 CH2Cl2 -65 1 89:11 75 S 7 5 CH2Cl2 -65 0,8 97:3 90 S 8 5 CH2Cl2 0 0,2 87:13 73 S 9 5 CH2Cl2 -78 1 98:2 90 S 10 5 CHCl3 -65 2 96:4 90 S 11d 5 CH2Cl2 -20 24 90:10 24 S 12 6 CH2Cl2 -78 1 94:6 6 S a Todos los experimentos se llevaron a cabo bajo nitrógeno, Cu(OTf)2 (0,025 mmol), ligando (0,025 mmol), disolvente (1,5 mL), 22a (0,25 mmol) y 23a (1,8 mmol). b Determinado por HPLC con una columna chiralpak AD-H. c Asignada por comparación con los datos descritos en la literatura. d Se utilizó Zn(OTf)2 en lugar de Cu(OTf)2. La reacción no funciona a temperaturas más bajas La enantioselectividad, tanto en su sentido como en su valor, varía según la orientación del grupo fenilo, siendo mayor cuando el carbono del anillo de oxazolina tiene configuración R. La máxima enantioselectividad, un 90%, se obtuvo con el ligando 5, donde el grupo fenilo forma parte de una estructura rígida tipo indeno y no tiene libertad de rotación (entradas 7-9). Como se observa, no hay diferencia significativa cuando se disminuye la temperatura de reacción de -65 a -78 °C. También es destacable la importancia de la estereoquímica del grupo hidroxilo presente en anillo de canfano. Así, el ligando 6 (entrada 12), con el grupo hidroxilo con orientación endo, condujo a un aducto 28aa prácticamente racémico, aunque la diastereoselectividad endo/exo inducida fue alta. 60 Diseño de ligandos con estructura de hidroxioxazolinas a partir del ácido (1S)-cetopínico. Aplicación en Reacciones de Diels-Alder enantioselectivas También se realizó una evaluación de diferentes disolventes. El uso de acetona comporta tiempos de reacción mucho más largos que con disolventes no coordinantes como tolueno o diclorometano. Este último da lugar a enantioselectividades mayores que el tolueno en el caso del ligando 5, si bien son comparables con el ligando 3. Así, el diclorometano resulta ser el más conveniente de todos los disolventes ensayados, con una enantioselectividad comparable al uso de cloroformo, pero con tiempos de reacción más cortos. El uso de triflato de Zn(II) no resulta satisfactorio, dando lugar a tiempos de reacción más elevados y una caída drástica de la enantioselectividad (entrada 11). 4.1.5.2. Alcance y limitaciones de la reacción Una vez constatado que los mejores resultados se obtenían con los complejos formados por el ligando 5 y triflato de Cu(II), utilizando diclorometano como disolvente, se procedió a evaluar la actividad de este sistema catalítico con otros sustratos. Tabla 6: Reacción del ciclopentadieno (23a) con diversos dienófilos.a Dienófilo (22) 1 T (°C) tiempo (h) Rendimientob endo:exo ee endo (%)c,d ee exo (%)c 0 0,2 28aa (99%) 87:13 (S) 73 54 -78 1 28aa (98%) 98:2 (S) 90 52 a 2 b 0 20 28ba (78%) 85:15 (S) 74 53 3 c 25 96 28ca (58%) 81:19 n.d. 58 4 d 25 44 28da (92%) 78:22 (S) 76 58 a Todos los experimentos se llevaron a cabo bajo nitrógeno, Cu(OTf)2 (0,025 mmol), ligando 5 (0,025 mmol), CH2Cl2 (1,5 mL), dienófilo 22 (0,25 mmol) y 23a (1,8 mmol). b Purificado por cromatografía. c Determinado por HPLC con columnas de fase estacionaria quiral. d Configuración asignada por comparación con los datos descritos en la literatura. 61 Resultados y Discusión Primeramente se estudió la reacción del ciclopentadieno con los diferentes dienófilos 22a-d (Tabla 6). La presencia de un sustituyente adicional sobre el doble enlace del dienófilo provocó una esperada disminución de la reactividad, por lo cual fue necesario llevar a cabo la reacción a temperaturas más altas y se necesitaron tiempos de reacción más largos. Este hecho es especialmente significativo cuando el sustituyente está en el carbono α del doble enlace. Con dienófilos sustituidos también se observó una disminución tanto de la enantioselectividad como de la diastereoselectividad. Sin embargo, esta disminución es achacable al aumento de temperatura, pues los resultados son similares a los obtenidos con 22a a 0 °C. En el caso de aducto 28ca no se ha podido determinar el exceso enantiomérico del aducto endo con los medios disponibles en el laboratorio. Tabla 7: Reacción del dienófilo 22a con diversos dienos.a Dieno T (°C) tiempo (h) Rendim. (%)b Relación de isómeros ee mayor. (%)c,d ee minor. (%)c 1 23a -78 1 28aa (98%) 98:2 (S) 90e 52e 2 23b 25 18 28ab (87%) 98:2 (S) 77e 78e 3 23c 0 90 28ac (60%) - (S) 55 - 25 24 28ad (94%) 96,5:3,5 (S) 72f 47f 0 24 28ad (72%) 98,5:1,5 (S) 79f 56f -25 120 28ad (25%) 98,5:1,5 (S) 84f 38f 4 23d 5 23e 25 48 28ae (83%) 67:19:11:2g 44g 32g 6 23f -20 21 28af (52%) 92:8g 0g 0g 7 23g 0 7,5 28ag (93%) - (-) 46 - 8 23h -20 16 28ah (57%) 100:0 (+) 50f - a OTMS Todos los experimentos se llevaron a cabo bajo nitrógeno, Cu(OTf)2 (0,025 mmol), ligando 5 (0,025 mmol), CH2Cl2 (1,5 mL), dienófilo 22a (0,25 mmol) y dieno 23 (1,8 mmol). b Purificado por cromatografía. c Determinado por HPLC con columnas de fase estacionaria quiral. d Configuración asignada por comparación con los datos descritos en la literatura. e mayoritario = endo, minoritario = exo. f Mayoritario = 1,4-disustituido, minoritario = 1,3-disustituido. g mayoritario = 1,2-cis ( = endo), minoritario = 1,2-trans ( = exo), otros: 1,3-cis( = endo) y 1,3-trans ( = exo). 62 Diseño de ligandos con estructura de hidroxioxazolinas a partir del ácido (1S)-cetopínico. Aplicación en Reacciones de Diels-Alder enantioselectivas N endo-28aa O O endo-28ab N cis-(1,2)-28ae O Ph O O cis-(1,2)-28af 28ac (1,4)-28ad 28ag 28ah O Finalmente, se estudió la reacción entre el dienófilo 22a y diferentes dienos (Tabla 7). Estos dienos se mostraron menos reactivos que el ciclopentadieno y las reacciones requirieron tiempos de reacción más prolongados y temperaturas más altas, lo cual condujo probablemente a los menores excesos enantioméricos obtenidos respecto al ciclopentadieno. No obstante, con ciclohexadieno (entrada 2) e isopreno (entrada 4) la reacción proporciona los correspondientes productos de Diels-Alder con excesos enantioméricos más elevados que los obtenidos con el ciclopentadieno a 0 °C. Cabe destacar que en el caso del isopreno (entrada 4), la enantioselectividad y la diastereoselectividad fueron mejor que las obtenidos por Evans para esta misma reacción con t-Bu-BOX (60% de exceso enantiomérico, relación endo/exo 97:3), uno de los ligandos considerados privilegiados.31 En el caso de los dienos 1-sustituidos 23e y 23f la diastereoselectividad obtenida fue moderada (entrada 5 y 6). La reacción con piperileno (23e) condujo a los dos regioisómeros 1,2- y 1,3-disustituidos. El regioisómero mayoritario se obtuvo como una mezcla 67:19 de los diastereoisómeros 1,2-cis y 1,2-trans con 44% y 32% de exceso enantiomérico, respectivamente (entrada 5). Por otra parte, la reacción con 1-fenilbutadieno (23f) proporcionó el aducto esperado con una diastereoselectividad 1,2-cis / 1,2-trans elevada (92:8), pero en forma racémica para los dos diastereoisómeros (entrada 6). La reacción con 1,2-dimetilenociclohexano (23g) condujo al correspondiente producto de Diels-Alder con elevado rendimiento y exceso enantiomérico del 46% (entrada 7). Cabe señalar que su esta es la primera vez que se describe el uso de este dieno en reacciones de catálisis asimétrica. En cuanto al silil enol éter 23h, tampoco se ha encontrado un símil en la bibliografía, aunque sí con otros derivados más sustituidos. No se llevó a cabo la hidrólisis del producto de Diels-Alder 28h, ya que la cetona resultante no posee centros estereogénicos, aislándose cromatográficamente el aducto con la agrupación silil enol éter con un 57% de rendimiento. La disminución del exceso enantiomérico en este caso podría estar relacionada con un mecanismo no estrictamente de Diels-Alder 63 Resultados y Discusión sino en uno de tipo Michael-Mukaiyama con posterior ciclación por adición de Michael intramolecular. 64 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina en reacciones de cicloadición. Antecedentes 4.2. Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX 4.2.1. N-Óxidos de 2-alquenoilpiridina en reacciones de cicloadición A pesar del gran número de estudios realizados en reacciones de cicloadición enantioselectivas catalizadas por complejos metálicos, existe un número muy restringido de tipos de sustratos adecuados que puedan utilizarse de manera eficaz en este tipo de reacciones, incluido aquellos que utilizan ligandos privilegiados como los BOX. Como segundo objetivo de la tesis se planteó el desarrollo de nuevas reacciones de cicloadición catalizadas por complejos metálicos con ligandos BOX utilizando diferentes sustratos bidentados que no han sido usados previamente en estas reacciones. En este apartado se estudia el uso de N-óxidos de 2-alquenoilpiridina en tres tipos de cicloadiciones: reacción de Diels-Alder, reacción de oxo-hetero-Diels-Alder con demanda electrónica inversa y, por último, cicloadiciones 1,3-dipolares. 4.2.1.1. Antecedentes El anillo heterocíclico de piridina resulta de gran importancia dada su extensa presencia en la naturaleza, en compuestos de uso agrícola, en fármacos y en ligandos para química de coordinación, así como por la versatilidad de sus derivados en síntesis orgánica.176 Las 2-alquenoilpiridinas son sustratos bidentados que podrían formar quelatos con el catalizador por coordinación del centro metálico con los pares de electrones libres del grupo carbonilo y del nitrógeno de la piridina. La utilización de este tipo de sustratos en reacciones enantioselectivas es de gran interés ya que permiten obtener derivados de piridina quirales que pueden tener un potencial aplicación tanto a nivel biológico como en la química de coordinación o química de materiales donde los derivados de este heterociclo son muy utilizados. Como se ha visto en la introducción general, los compuestos bidentados, con dos puntos de coordinación, resultan sustratos adecuados para reacciones catalizadas por Cu(II). La formación de un quelato entre el sustrato y el centro metálico restringe los grados de libertad del complejo formado, facilitando así la discriminación facial y, por tanto, la enantioselectividad. Engberts et al. publicaron por primera vez en 1996 el uso de estos sustratos en reacciones de Diels-Alder no enantioselectivas catalizadas por ácidos de Lewis en medio acuoso (Esquema 49).177 En este caso los resultados siguen la serie de Irving65 Resultados y Discusión Williams (Co+2 < Ni+2 < Cu+2 >> Zn+2), obteniendo los mejores resultados con Cu(II).21,22 Posteriormente el mismo grupo observó una aceleración mayor en esta reacción al combinar el ácido de Lewis con un medio micelar.178-180 Schreiner y Wittkopp han documentado otro ejemplo de catálisis en esta reacción mediante la formación de puentes de hidrógeno, tanto en disolventes orgánicos como en agua.181 Esquema 49: Reacción de Diels-Alder entre ciclopentadieno y 2-cinamoilpiridina. Domingo et al. han estudiado computacionalmente esta reacción catalizada por cationes Zn+2. De las conclusiones de este estudio se puede establecer que la reacción no catalizada transcurre por un mecanismo concertado con formación asíncrona de los nuevos enlaces. Sin embargo, en presencia de Zn+2 el mecanismo implica dos etapas, actuando el ciclopentadieno como nucleófilo que ataca a la posición β de la 2alquenoilpiridina coordinada al catión Zn+2.182,183 La primera versión asimétrica de esta reacción fue documentada por Engberts en 1998, utilizando aminoácidos naturales junto a sales de Cu(II).184,185 Los mejores resultados se obtiene con el aminoácido abrina (N-metil triptófano), consiguiendo hasta un 74% de exceso enantiomérico. Entre los diversos disolventes empleados, el agua es el que da lugar a los mejores resultados. Feringa y Roelfes, han utilizando complejos aquirales de Cu(II) anclados a cadenas de ADN en medio acuoso (Figura 44) en la reacción entre 2-alquenoilpiridinas y ciclopentadieno, obteniendo excelentes resultados.186-188 Sin embargo, tanto estos autores como los anteriores no describen el uso de otros dienos diferentes al ciclopentadieno. Figura 44: Catalizadores quirales basados en ADN. Jitsukawa et al. describieron en 2005 el uso de complejos Cu(II)-(BOX)2 en la reacción entre 2-cinamoilpiridina y ciclohexadieno, utilizando para ello tres ligandos de tipo BOX. En todos los casos el exceso enantiomérico conseguido supera el 99%, aunque los rendimientos obtenidos son extremadamente bajos excepto en el caso en el que se utiliza un ligando que presenta grupos amida capaces de establecer puentes 66 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina en reacciones de cicloadición. Antecedentes de hidrogeno adicionales con el dienófilo, con el que se consigue un 51% de rendimiento (Figura 45).189 Figura 45 Reetz y Jiao han desarrollado otro sistema con complejos aquirales Cu(II)ftalocianina conjugados con varios tipos de proteína albúmina sérica como catalizadores de esta reacción de Diels-Alder en medio acuoso, consiguiendo inducciones asimétricas entre altas y moderadas, pero que se reducen al incrementar la escala de la reacción.190 Bergman et al. publicaron en 2002 un artículo describiendo el uso de 2alquenoilpiridinas en una reacción de hetero-Diels-Alder con etil vinil éter para la síntesis racémica de los correspondientes dihidropiranos, que fueron utilizados como intermedios para la síntesis de bipiridinas aquirales (Esquema 50).191 La reacción está catalizada por hexafluoroacetilacetonato de itrio(III) y transcurre con rendimientos entre el 50 y el 86%. Esquema 50 Por último, Sibi y Yang ha utilizado 2-alquenoilpiridinas en adiciones conjugadas radicalarias catalizadas por complejos Zn(II)-Ph-BOX, obteniendo excesos enantioméricos moderados (Esquema 51).192 Esquema 51 En resumen, podemos ver como las aplicaciones sintéticas de 2alquenoilpiridinas han sido hasta el momento bastante limitadas. Si bien se obtienen buenos resultados con algunos sistemas catalíticos, la mayoría están restringidos a 67 Resultados y Discusión condiciones no enantioselectivas o catalizadores muy complejos y en todos los casos el número de sustratos utilizados es muy limitado. Por otra parte, el uso de compuestos bidentados con la plantilla quelante Nóxido de 2-acilpiridina en catálisis asimétrica ha sido poco estudiado. Jørgensen et al. fueron los primeros en constatar que los N-óxidos de 2-piridincarboxaldehído eran sustratos más eficientes que los correspondientes 2-piridincarboxaldehídos no oxidados en reacciones aldólicas de tipo Mukaiyama catalizadas por complejos de Cu(II)-t-Bu-BOX, permitiendo obtener los correspondientes productos de condensación aldólica con buenos rendimientos y enantioselectividades excelentes, superando en algún caso el 99% de exceso enantiomérico (Esquema 52a).193 Esquema 52 (a) (b) Este mismo tipo de sustratos, así como el N-óxido de 2-acetilpiridina fueron utilizados por el mismo grupo en reacciones de hetero-Diels-Alder con demanda electrónica normal con buenos excesos enantioméricos, si bien los rendimientos son modestos (Esquema 52b).194 Las condiciones optimas de reacción implican el uso de una mezcla tolueno/diclorometano como disolvente, así como el uso del ligando cis-diPh-BOX, con dos sustituyentes fenilo. La reacción parece transcurrir en dos etapas vía adición de Mukaiyama seguida de adición de Michael intramolecular. El dieno de Brassard no da lugar al aducto de hetero-Diels-Alder sino al producto de reacción de Mukaiyama. Tanto en las reacciones de hetero-Diels-Alder como en la reacción de Mukaiyama se observa una inversión en el sentido de la enantioselectividad entre el ligando t-Bu-BOX y los ligandos Ph-BOX o cis-di-Ph-BOX. Cabe señalar que con posterioridad a la publicación de nuestro trabajo con Nóxidos de 2-alquenoilpiridinas en reacciones de Diels-Alder que se describe a continuación,195 otros autores han reconocido la gran eficacia de los mismos sustratos en reacciones catalizadas por complejos de Cu(II)-BOX. Así, Singh y Singh han descrito 68 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina en reacciones de cicloadición. Antecedentes la reacción de Friedel-Crafts de indoles con N-óxidos de 2-alquenoilpiridinas.196 Esta reacción está catalizada por un complejo de Cu(OTf)2 con un ligando PyBOX altamente sustituido, dando lugar a excelentes rendimientos y enantioselectividades (Esquema 53). Esquema 53 69 Resultados y Discusión 4.2.1.2. Síntesis de N-óxidos de 2-alquenoilpiridina Los N-óxidos de 2-alquenoilpiridinas fueron sintetizados por condensación aldólica entre el N-óxido de 2-acetilpiridina (30) y diversos aldehídos siguiendo el Esquema 54. El N-óxido de 2-acetilpiridina (30) se preparó previamente por oxidación de N-acetilpiridina (29) con el complejo urea-peróxido de hidrogeno (UHP) en presencia de anhídrido ftálico.197 La reacción transcurrió con un rendimiento del 50%, recuperándose un 36% de 2-acetilpiridina sin reaccionar. Esquema 54: Esquema sintético para la formación de N-óxidos de 2-alquenoilpiridina. O N UHP anhídrido ftálico O O O N R-CHO O N base CH2Cl2 R 29 30 31 Los compuestos 31 se prepararon por condensación aldólica de 30 con los aldehídos adecuados. Como método general (procedimiento A) la reacción se llevó a cabo en metanol a 0 °C en presencia de 2 equivalentes del aldehído correspondiente y 0,2 equivalentes de KOH respecto a 30. La condensación de 30 con trimetilacetaldehído se llevó a cabo a reflujo de THF-etanol 1:1 saturado de KOH para dar lugar a 31k (procedimiento B). Ninguno de estos dos métodos dio rendimientos satisfactorios en la síntesis de 31l a partir de glioxilato de etilo. Tras ensayar diversas condiciones fue posible obtener 31l con un rendimiento del 18% llevando a cabo la condensación de 30 en tolueno a 90 °C en presencia de tamiz molecular y ácido p-toluenosulfónico como catalizador (procedimiento C). Los resultados para la síntesis de los compuestos 31 se resumen en la Tabla 8. Tabla 8: Condensación de 30 con diversos aldehídos. Aldehído Rendimiento (%)a Enona 1 31a O 2 O N 31b 64 50 MeO Continúa en la página siguiente… 70 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina en reacciones de cicloadición. Síntesis de N-óxidos de 2-alquenoilpiridina Aldehído Rendimiento (%)a Enona 3 31c 52 31d 91 31e 30 31f 58 31g 69 31h 53 9 31i 47 10 31j 58 11 31k 64b 12 31l 18c O 4 O N O2N 5 O 6 O N O O 7 O N O O 8 O N S a Excepto donde se indiquen otras condiciones, la síntesis se llevó a cabo mediante el procedimiento A. N-Óxido 30 (7,9 mmol), aldehído (15,8 mmol), KOH 1 M (1,6 mL, 1,6 mmol), MeOH (40 mL) a 0 °C. b Procedimiento B: N-Óxido 30 (400 mg, 2,9 mmol), trimetilacetaldehído (0,63 mL, 5,8 mmol), mezcla THF/EtOH 1:1 saturada de KOH (0,1 mL), THF (5 mL) a reflujo. c Procedimiento C: N-Óxido 30 (805 mg, 5,9 mmol), glioxilato de etilo (50% en tolueno, 2,3 mL, 11,76 mmol), tolueno (5 mL), tamiz molecular de 4 Å (100 mg), TsOH·H2O (25 mg). La enonas obtenidas mostraron señales olefínicas en el espectro de RMN por lo general a campo más bajo de δ 7,50 ppm, con una constante de acoplamiento característica (J ≈ 16 Hz) de protones olefínicos en trans. En ningún caso se observó la 71 Resultados y Discusión formación de las enonas cis, si bien en algunos casos se pudo aislar de la mezcla de reacción el producto de adición 1,4 de metanol a la enona. 72 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina como dienófilos en reacciones de Diels-Alder 4.2.1.3. N-Óxidos de 2-alquenoilpiridina como dienófilos en reacciones de Diels-Alder a) Optimización Como se ha visto en los antecedentes bibliográficos, la reacción de Diels-Alder entre 2-alqueonilpiridinas y dienos se ha llevado a cabo de manera enantioselectiva en algunos casos con baja enantioselectividad, bajo rendimiento o, en otros, utilizando sistemas catalíticos poco convencionales. Dada la elevada eficacia mostrada por los complejos Cu(II)-BOX en múltiples reacciones enantioselectivas decidimos estudiar el uso de estos complejos en dicha reacción. Para el proceso de optimización se eligió la reacción entre 2-cinamoilpiridina (32) y ciclopentadieno (23a). Sin embargo, en presencia del complejo formado por triflato de Cu(II) y el ligando comercial (S)-Ph-BOX (33a), la reacción transcurrió lentamente para dar el producto de Diels-Alder correspondiente (35) con diastereoselectividad moderada (endo/exo = 86:14) y baja enantioselectividad (19% y 11% de exceso enantiomérico para los aductos endo y exo respectivamente). Resultados similares se obtuvieron a partir del complejo formado a partir de triflato de cinc y el ligando 33a (Tabla 9, entrada 1). Por esta razón, y teniendo en mente los resultados obtenidos por Jørgensen con N-óxidos de 2-piridincarboxaldehído en reacciones de Mukaiayama,193 se decidió sustituir el dienófilo 32 por el correspondientes N-óxido 31a. Un primer ensayo utilizando el sustrato 31a en idéntica condiciones a las utilizadas previamente con 32 condujo al aducto de Diels-Alder 34aa con una excelente diastereoselectividad (endo/exo = 98:2) y un exceso enantiomérico del 96 % en el aducto endo en tan solo 20 minutos (Tabla 9, entrada 5). El proceso de optimización continuó con el compuesto 31a, con el cual se ensayaron diferentes temperaturas y ligandos de tipo BOX. Como se puede ver en la Tabla 9, los N-óxidos de 2-alquenoilpiridina son mucho más reactivos que las 2alquenoilpiridinas no oxidadas, necesitando tiempos de reacción muchísimo más cortos. Por lo que respecta al ligando, se obtuvieron resultados similares con los dos ligandos BOX utilizados (entradas 5 y 7), si bien Ph-BOX proporciona mayor diastereoselectividad a expensas de una disminución del exceso enantiomérico del diastereoisómero minoritario exo. Al igual que en otras reacciones catalizadas por complejos Cu(II)-BOX, se observa una inversión en el sentido de la inducción quiral, proporcionando ambos ligandos enantiómeros mayoritarios opuestos. 73 Resultados y Discusión Tabla 9: Optimización de la reacción de Diels-Alder.a O O N N R R 33a R = Ph 33b R = t-Bu Dienófilo M(OTf)2 Ligando T (°C) tiempo (h) Conv. (%) endo/exo ee endo (%)b,c ee exo (%)b 1 32 Cu(OTf)2 33a 0 22 87 86:14 (S) 19 11 2 32 Zn(OTf)2 33a 0 22 98 82:18 (S) 23 41 3 31a Zn(OTf)2 33a 0 2,5 100 96:4 (S) 91 54 4 31a Zn(OTf)2 33a -40 28 47 96:4 (S) 90 14 5 31a Cu(OTf)2 33a 0 0,3 100 98:2 (S) 96 81 6 31a Cu(OTf)2 33a -40 3 100 99:1 (S) 95 76 7 31a Cu(OTf)2 33b 0 1 100 92:8 (R) 92 (-) 90c a Todos los experimentos se llevaron a cabo bajo nitrógeno, M(OTf)2 (0,025 mmol), ligando 33 (0,025 mmol), CH2Cl2 (1,5 mL), dienófilo (0,25 mmol) y ciclopentadieno (23a) (1,8 mmol). b Determinado por HPLC con una columna chiralpak AD-H. c Entre paréntesis la configuración absoluta del carbono α a carbonilo (ver apartado c). En cuanto al ión metálico, el Cu(II) proporcionó mayor aceleración de la reacción que el Zn(II), dando lugar al producto de Diels-Alder con diastereo- y enantioselectividad ligeramente superiores (entradas 3 y 5). Una disminución de la temperatura desde 0 °C hasta -40 °C no mejoró la enantioselectividad de la reacción (entrada 5 y 6) y en el caso de la reacción catalizada por el complejo Zn(II)-33a se produjo una ralentización apreciable, observándose una conversión de sólo el 47% después de 28 horas (entradas 3 y 4). b) Alcance y limitaciones de la reacción Una vez optimizadas las condiciones de reacción, se estudió la aplicación a otras 2-alquenoilpiridinas y dienos conjugados, utilizando como catalizador el complejo Cu(II)-33a preparado in situ a partir del ligando 33a y Cu(OTf)2 en diclorometano a 0 °C. En primer lugar se estudió la reacción de ciclopentadieno con N-óxidos de 2alquenoilpiridina con diferente sustitución en el carbono β del doble enlace. Como puede observarse en la Tabla 8, todos los sustratos dieron lugar a los productos de Diels-Alder con excelentes enantioselectividades, con excesos enantioméricos cercanos o superiores al 95%. En el caso de los dienófilos 31a-d, con un anillo de fenilo 74 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina como dienófilos en reacciones de Diels-Alder para-sustituido (entradas 1, 3-5), no se observan prácticamente diferencias en los resultados, independientemente de la naturaleza electrón-dadora o electrónatrayente del sustituyente en para, si bien el compuesto con el grupo nitro 31d mostró una mayor reactividad. En el caso del dienófilo 31e (entrada 6), el cual posee un anillo de piridina unido al doble enlace, se observó un transcurso más lento de la reacción, una pequeña disminución de la enantioselectividad (90% ee) y del rendimiento, posiblemente debido a la capacidad del heterociclo para coordinarse al centro metálico, interfiriendo en el modo de coordinación normal del dienófilo a la especie catalítica activa. Sin embargo, la diastereoselectividad obtenida es prácticamente total. La reacción también permite utilizar N-óxidos de 2-alquenoilpiridina que presentan un anillo heterocíclico π-excedentario de cinco miembros en el carbono β del alqueno (entradas 7-10). La reacción transcurre en todos los casos con buen rendimiento, diastereoselectividad prácticamente total y excelente enantioselectividad. Resulta reseñable la compatibilidad de un grupo furilo, que podría llegar a actuar como dieno. El dienófilo también admite sustituyentes alifáticos en el doble enlace (entradas 11 y 12). El compuesto 31k, con un grupo t-butilo, mantuvo la reactividad y la enantioselectividad de los sustratos anteriores, aunque se produjo una notable disminución de la diastereoselectividad, que fue mayor con el ligando 33b. Por último, se ensayó el dienófilo dicarbonílico 31l, fuertemente electrófilo (entrada 13). Éste reaccionó muy rápidamente, obteniéndose el aducto correspondiente con la mayor enantioselectividad observada (98% de exceso enantiomérico). Tabla 10: Reacción de Diels-Alder entre diferentes dienófilos (31) y ciclopentadieno (23a).a tiempo (h) Dienófilo 31 1 0,3 a 3 O N endo:exo ee endo (%)c ee exo (%)c 98:2 96 81 0,4 34aa (94) 97:3 95 76 b 0,3 34ba (95) 98:2 95 65 c 0,25 34ca (100) 97:3 94 95 2d O Rendim. (%)b 34aa (98) MeO 4 Continúa en la página siguiente… 75 Resultados y Discusión tiempo (h) Rendim. (%)b endo:exo ee endo (%)c ee exo (%)c d 0,1 34da (93) 96:4 96 n.d. 6 e 5 34ea (52) >99:1 90 n.d. 7 f 0,2 34fa (89) >99:1 95 n.d. 8 g 0,5 34ga (96) >99:1 97 n.d. 9 h 0,2 34ha (94) >99:1 97 n.d. 10 i 0,5 34ia (96) >99:1 94 n.d. 0,3 34ka (92) 34ka (91) 78:22 93 70 60:40 (-) 91f (-) 84f 34la (97) 97:3 98 89 dienófilo 31 O 5 O N O2N 11 k 12e 13 0,5 l 0,05 a Excepto donde se indique lo contrario, todos los experimentos se llevaron a cabo bajo nitrógeno, Cu(OTf)2 (0,025 mmol), 33a (0,025 mmol), CH2Cl2 (1,5 mL), dienófilo 31 (0,25 mmol) y ciclopentadieno (23a) (1,8 mmol) a 0 °C. b Purificado por cromatografía. c Determinado por HPLC con columnas de fase estacionaria quiral. d Llevada a cabo con Cu(OTf)2 (0,0067 mmol), 33a (0,0067 mmol), 31 (0,67 mmol), ciclopentadieno (23a) (2,33 mmol) y CH2Cl2 (1,5 mL). e Llevada a cabo con el ligando 33b. f Se obtuvo el enantiómero opuesto. Una vez establecida la generalidad de la reacción con diversos dienófilos se estudió la aplicación con diferentes dienos, utilizando el compuesto 31a como dienófilo. Los resultados se recogen en la Tabla 11. Aunque los experimentos anteriores se llevaron a cabo con una carga catalítica del 10% molar, un experimento adicional con el compuesto 31a mostró que la carga de catalizador puede reducirse hasta el 1% molar, sin pérdidas apreciables en rendimiento y estereoselectividad (entrada 2). 76 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina como dienófilos en reacciones de Diels-Alder Tabla 11: Resultados de la reacción entre el dienófilo 31a y diversos dienos.a Dieno tiempo (h) Rendimiento (%)b Ratio Diastereo. ee mayor. (%)c ee minor. (%)c 1 23a 0,3 34aa (98) 98:2d 96 81 2 23b 30 34ab (100) 85:15d 96 47 3 23c 21 34ac (95) - 93 - 4 23d 20 34ad (93) 96:4e 92 46 5 23e 7,5 34ae (97) 90:10d 94 n.d. 6 23f 0,5 34af (100) 99:1d 94 n.d. 7 23h 0,15 34ahf (83) 100:0 95 - 8 23i 2,5 34ai (95) 99:1d 96 72 OTMS AcO a Todos los experimentos se llevaron a cabo bajo nitrógeno, Cu(OTf)2 (0,025 mmol), 33a (0,025 mmol), CH2Cl2 (1,5 mL), dienófilo 31a (0,25 mmol) y dieno (1,8 mmol) a 0 °C. b Purificado por cromatografía. c Determinado por HPLC con columnas de fase estacionaria quiral. d ratio cis (= endo)/trans(= exo) para (1,2)-34ae. e ratio 1,4-34ad/1,3-34ad. f 34ah es la cetona resultante después de la hidrólisis con ácido cítrico 0,01 M en H2O:MeOH 1:1. O O O N Ph endo-34aa Me Me endo-34ab Ph cis-(1,2)-34ae O Ph 34ac O N O Me O N Ph (1,4)-34ad O N Ph cis-(1,2)-34af 34ah cis-(1,2)-34ai Como se puede observar, la reacción transcurrió con elevados rendimientos con todos los dienos utilizados, proporcionando los productos con excesos enantioméricos superiores al 92% y diastereoselectividad variable. Por lo que respecta a la reacción con ciclohexadieno (23b), el tiempo de reacción requerido fue mucho mayor debido a la menor reactividad de este dieno (entrada 2). En este caso también 77 Resultados y Discusión se observó una diastereoselectividad endo/exo moderada (85:15), aunque el aducto mayoritario endo se obtuvo con elevado exceso enantiomérico (96%). El 2,3-dimetilbutadieno (23c) también reaccionó lentamente (21 horas) proporcionando el producto de Diels-Alder con excelente enantioselectividad (entrada 3). Más significativos fueron los resultados obtenidos con dienos más problemáticos. Con isopreno (entrada 4), la reacción condujo a una mezcla de regioisómeros de la que el isómero mayoritario 1,4-substituido se obtuvo con el 92% de exceso enantiomérico. Similares niveles de enantioselectividad (94% de exceso enantiomérico) se obtuvieron con piperileno y 1-fenilbutadieno. Con el piperileno (entrada 5) se obtuvo el aducto cis-1,2-34ae (equivalente a una aproximación endo) con una relación 90:10 respecto al isómero trans. En el caso del 1-fenilbutadieno (entrada 6) la diastereoselectividad fue mayor obteniéndose el isómero cis casi de manera exclusiva. Los dienos 23h y 23i (entradas 7 y 8), sustituidos con grupos electrón-donantes, mostraron una elevada reactividad, especialmente 23h. El dieno 23i proporcionó mayoritariamente el producto 1,2-cis con elevada diastereo- y enantioselectividad. Hay que reseñar que en el caso del acetato 23i se parte de una mezcla cis/trans del dieno, siendo este último el que da lugar a la reacción. Otros dienos poco reactivos como el furano no dieron reacción de Diels-Alder con el dienófilo 31a. En su lugar, se produjo una reacción de adición conjugada de Michael del furano al doble enlace, extremadamente lenta a temperatura ambiente. c) Determinación de la estereoquímica absoluta y modelo estereoquímico La estereoquímica relativa del diastereoisómero mayoritario del aducto de Diels-Alder 34aa se determinó mediante experimentos de RMN de tipo NOESY. Así, al irradiar la señal del protón en α al grupo carbonilo (δ 4,50 ppm) se obtuvo una clara señal de NOE con uno de los dos protones (δ 1,88 ppm) del metileno puente del esqueleto de norbornano y al mismo tiempo con las señales correspondientes al grupo fenilo, que demostraron la orientación endo de la agrupación N-óxido de acilpiridina respecto al sistema bicíclico de norbornano (Figura 46). Figura 46: Experimento NOE del compuesto 34aa. Así mismo, a partir de la mezcla de diastereoisómeros del compuesto 34ca obtenida por la reacción del dienófilo bromado 31c con ciclopentadieno, se pudieron 78 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina como dienófilos en reacciones de Diels-Alder obtener cristales del aducto endo-34ca adecuados para su análisis mediante difracción de rayos X. El refinamiento de los cálculos permitió obtener la estructura mostrada en la Figura 47 con un parámetro de Flack de 0,036(13), que muestra que la configuración absoluta del estereocentro que soporta el grupo carbonilo es S. (S) Br (S) O N O endo-34ca Figura 47: Estructura del compuesto endo-34ca elucidada por difracción de rayos X. De acuerdo con esta estereoquímica se propone un modelo de trabajo según el cual la reacción transcurre a través de un complejo de geometría tetraédrica, con el dienófilo adoptando la conformación s-cis de acuerdo con la propuesta de Jørgensen (vide supra). Esta estructura en la que un grupo fenilo del ligando 33a bloquearía el acceso del dieno a la cara Re del alqueno estaría estabilizada por una interacción π entre el doble enlace del dienófilo y el grupo fenilo del ligando (Figura 48a). La obtención del enantiómero opuesto con el ligando t-Bu-BOX (33b) se explicaría suponiendo la formación de un complejo plano-cuadrado en la que el ataque a la cara Si del doble enlace estaría bloqueada por un grupo t-butilo. (a) O N (b) O O Cu N (S) O N cara Re Figura 48: Modelos estereoquímicos propuestos para la reacción catalizada por Cu(II)-33a y Cu(II)-33b. 79 Resultados y Discusión d) Transformaciones sintéticas Un aspecto importante relacionado con los productos enriquecidos enantioméricamente obtenidos en las reacciones de Diels-Alder anteriores es la posibilidad de llevar a cabo modificaciones sintéticas sobre los mismos manteniendo su integridad estereoquímica. En particular, la desoxigenación del anillo heterocíclico una vez aprovechado el efecto beneficioso del N-óxido sobre la reactividad y enantioselectividad, permitiría obtener los correspondientes aductos de Diels-Alder con el anillo de piridina. Esta transformación se llevó a cabo con el aducto endo-34aa (91% de exceso enantiomérico) mediante un tratamiento con indio metálico en polvo en una mezcla de etanol y disolución acuosa saturada de NH4Cl a reflujo (Esquema 55). Se obtuvo el compuesto 35 con un 75% de rendimiento y con idéntica pureza óptica a la del sustrato de partida. Esquema 55 O Ph 34aa O N O In/NH4Cl (aq) EtOH, reflujo 75% N Ph 35 El compuesto 35 mostró las mismas características espectrales que las descritas por Engberts.184,185 La comparación de los tiempos de retención en HPLC del producto 35 obtenido a partir de 34aa con los descritos por Engberts para el producto obtenido a partir de ciclopentadieno y 2-cinamoilpiridina en presencia del complejo Cu(II)-Nabrina indica que ambos poseen la misma estereoquímica absoluta. Cabe señalar que esta es la primera vez que se ha podido asignar la estereoquímica absoluta de uno de los enantiómeros del producto 35, ya que aunque este producto había sido preparado en forma enantioméricamente pura por varios autores, ninguno de ellos había sido capaz de determinar su estereoquímica absoluta. Por otra parte la presencia del N-óxido en los aductos de Diels-Alder puede ser ventajosa ya que es posible aprovechar la reactividad química de dicha agrupación para llevar a cabo modificaciones del anillo heterocíclico que no serían posibles de otra forma.198 Por ejemplo, es posible llevar a cabo la cianación regioselectiva en la posición 6 del anillo de piridina a través de una modificación de la reacción de ReissertHenze,199 con tratamiento del aducto 34aa con cianuro de trimetilsililo y cloruro de dimetilcarbamoilo, obteniendo 36 con un 83% de rendimiento (Esquema 56). 80 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina como dienófilos en reacciones de Diels-Alder Esquema 56 También se estudió la reacción de oxidación de Baeyer-Villiger sobre la agrupación acilpiridina. Para evitar la posible epoxidación del doble enlace primero se procedió a la hidrogenación del aducto 34aa catalizada con Pd/C. En estas condiciones no sólo se hidrogenó el doble enlace, sino que además se produjo la desoxigenación del anillo de piridina (Esquema 57a), resultando el compuesto 37 con un rendimiento cuantitativo. La oxidación se llevó a cabo mediante ácido m-cloroperbenzoico, seguido de un tratamiento posterior de la mezcla de reacción con metanol para dar el alcohol 38 con un 55% de rendimiento (Esquema 57b). La reacción transcurrió de manera regioselectiva y con retención de configuración, como se demuestra por la comparación de los datos espectroscópicos del producto 38 con los descritos en la literatura.200 Esquema 57 (a) (b) Esta transformación resulta de gran interés, dado que en la secuencia global los N-óxidos de 2-alquenoilpiridinas estarían actuando como dienófilos equivalentes de enoles en reacciones de Diels-Alder enantioselectivas. Este hecho debe ser convenientemente subrayado, pues permite obtener aductos de Diels-Alder que retrosintéticamente derivan de dienófilos específicos para reacciones con demanda electrónica inversa y dienos propios de reacciones con demanda electrónica normal. Por otra parte se llevó a cabo la oxidación del compuesto 37 con NaIO4 y tricloruro de rutenio, en un intento de transformar la cetona en un ácido carboxílico con eliminación del anillo de piridina, invirtiendo la regioselectividad de la reacción de Baeyer-Villiger. Después de tratar la mezcla de oxidación, previamente filtrada y 81 Resultados y Discusión extraída, con trimetilsilildiazometano, se obtuvo un único compuesto identificado como el éster 39, resultante de la oxidación del grupo fenilo, electrónicamente más rico que el anillo de piridina. La reacción transcurrió con un rendimiento del 30%, recuperándose parte del sustrato de partida (39% de conversión). Esquema 58 Por último, se aplicó la metodología de Diels-Alder enantioselectiva con Nóxidos de 2-alquenoilpiridina a la síntesis del compuesto bioactivo 40, que se ha descrito como inhibidor de la cisteína proteasa para el tratamiento de infartos cerebrales, enfermedad de Alzeihmer y otras dolencias.201 La secuencia sintética se resume en el Esquema 59. El primer paso consistió en la reacción de Diels-Alder entre 1,3-butadieno (23j) y el dienófilo 31l catalizada por el complejo Cu(II)-33a, obteniéndose 34lj con un rendimiento del 71% y un 98 % de exceso enantiomérico. La hidrogenación catalítica del aducto 34lj con paladio sobre carbono condujo al compuesto 41 con un rendimiento del 85%. Finalmente, el éster 41 se hidrolizó con hidróxido potásico en etanol para dar 40 con un 80% de rendimiento. Esquema 59 O + 82 O Cu(OTf)2 + 33a O N CH2Cl2, 0 ºC EtO2C 23j O N 31l CO2Et 34lj 71% 98% ee Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina como heterodienos en reacciones de hetero-Diels-Alder 4.2.1.4. N-Óxidos de 2-alquenoilpiridina como heterodienos en reacciones de hetero-Diels-Alder Después de haber probado la eficacia de los N-óxidos de 2-alquenoilpiridina como dienófilos en reacciones de Diels-Alder enantioselectivas catalizadas por complejos de metal-BOX, se decidió estudiar su uso como heterodienos en reacciones de oxo-hetero-Diels-Alder con demanda electrónica inversa con alquenos de elevada densidad electrónica, empleando el mismo tipo de sistema catalítico. Las reacciones de oxo-hetero-Diels-Alder constituyen un instrumento muy eficaz para la síntesis de sistemas heterocíclicos oxigenados de 6 miembros tales como dihidropiranos y dihidropiranonas. La reacción de 2-alquenoilpiridinas (o de sus Nóxidos) con alquenos permitiría la preparación de dihidropiranos con un sustituyente 2-piridilo en la posición 2 del heterociclo oxigenado. Este tipo de compuestos son precursores sintéticos de bipiridinas191 y también son isósteros flexibles de compuestos biológicamente activos.202-204 Hasta el momento sólo existe un ejemplo descrito de esta reacción, como se recoge en los antecedentes bibliográficos de esta memoria,191 no habiéndose desarrollado ninguna versión enantioselectiva de la misma. a) Optimización La investigación se inició comprobando la reacción entre la 2-alquenoilpiridina 32 y etil vinil éter (42a) en presencia del complejo Cu(II)-33a en diclorometano. Como se anticipaba, la reacción transcurrió de forma lenta, proporcionando el compuesto 44 como una mezcla 85:15 de los diastereoisómeros endo/exo con excesos enantioméricos del 16 y del 15% respectivamente. Una vez llevada a cabo esta comprobación procedimos a estudiar la reacción utilizando los correspondientes N-óxidos de 2-alquenoilpiridina como heterodienos. Los resultados se recogen en la Tabla 12. Utilizando la reacción entre el compuesto 31a y etil vinil éter (42a) se ensayaron los complejos de Cu(OTf)2 con los ligandos 33a, 33b y 33c en las condiciones optimizadas anteriormente para la reacción de Diels-Alder, en diclorometano como disolvente (Tabla 12, entradas 2-4). En todos los casos la reacción transcurrió rápidamente completándose en menos de una hora. El ligando 33a proporcionó los mejores resultados en términos de rendimiento, diastereo- y enantioselectividad 83 Resultados y Discusión (entrada 2). Al igual que con la reacción de Diels-Alder, también se produce una inversión de la inducción asimétrica por parte del ligando 33a respecto a 33b y 33c. d Tabla 12: Optimización de la reacción de hetero-Diels-Alder.a X O EtO X + O disolvente Ph 42a EtO metal + BOX Ph 43aa X = N-O 44 X = N 31a X = N-O 32 X = N MX2 Ligando Disolvente T (°C) tiempo (h) Conver. (%) endo/exo (% ed)b ee endo (%)c,d ee exo (%)c 1e Cu(OTf)2 33a CH2Cl2 25 90 100f 71 16 15 2 Cu(OTf)2 33a CH2Cl2 0 0,5 100 98 (S) 91 86 3 Cu(OTf)2 33b CH2Cl2 0 0,5 100 88 (R) 75 7 4 Cu(OTf)2 33c CH2Cl2 0 1 100 96 (S) 79 0 5 Cu(OTf)2 33a Tolueno 0 3 100 >99,9 (S) 88 - 6 Cu(OTf)2 33a MeNO2 0 5 100 92 (S) 46 43 7 Cu(OTf)2 33a THF 0 2,5 100 99,1 (S) 86 - 8 Cu(SbF6)2 33a CH2Cl2 0 48 100 97 (S) 59 37 9 Zn(OTf)2 33a CH2Cl2 0 120 100 99 (S) 51 25 10 Mg(OTf)2 33a CH2Cl2 0 77 100 99,3 0 - 11 Cu(OTf)2 33a CH2Cl2 -20 4 100 99,5 (S) 94 73 12 Cu(OTf)2 33a CH2Cl2 -40 20 100 99,8 (S) 96 - a Todos los experimentos se llevaron a cabo bajo nitrógeno, MX2 (0,025 mmol), ligando (0,025 mmol), CH2Cl2 (1,5 mL), heterodieno 31a (0,25 mmol) y 42a (0,75 mmol). b Endo = 2,4-cis, exo = 2,3trans. c Determinado por HPLC con una columna chiralpak AD-H. d Entre paréntesis la configuración absoluta del carbono acetálico de 43aa (ver apartado c). e Se utilizó 32 como heterodieno. f El producto resultante es 44. d El ligando 33c tiene configuración R en lugar de la configuración S de los ligandos 33a y 33b. Es por ello que, a pesar de que el enantiómero mayoritario obtenido con 33c es el mismo que con 33a, se considera la existencia de una inducción quiral inversa entre 33c y 33a. 84 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina como heterodienos en reacciones de hetero-Diels-Alder También se evaluó el efecto de diversos disolventes (entradas 5-7), sin obtener ninguna mejora respecto al diclorometano. Por lo que respecta a otras sales metálicas (entradas 8-10), el uso de Cu(SbF6)2, triflato de cinc o triflato de magnesio dio lugar a una brusca disminución de la reactividad, necesitándose tiempos de reacción que superaron los 2 días para una conversión total y produciéndose un descenso acusado de la enantioselectividad en todos los casos. Ya por último, disminuyendo la temperatura de reacción a -20 °C o a -40 °C fue posible mejorar la estereoselectividad de la reacción, alcanzándose un 96% de exceso enantiomérico y con excelente diastereoselectividad (99,8% ed). b) Alcance y limitaciones de la reacción Una vez establecidas las condiciones optimas de reacción (diclorometano como disolvente, Cu(OTf)2 y el ligando 33a), se procedió a estudiar el comportamiento como heterodienos de los diversos N-óxidos de 2-alquenoilpiridina (31) utilizando etil vinil éter (42a) como dienófilo (Tabla 13). Igualmente, este estudio se extendió a la reacción de diversos alquenos (42) frente al heterodieno 31a (Tabla 14). En la mayoría de los casos la reacción se llevó a cabo a -40 °C aunque en otros la temperatura debió elevarse para mantener tiempos de reacción aceptables. En general, tal como puede observarse en la Tabla 13, la reacción transcurrió con excelente estereoselectividad proporcionando los dihidropiranos 43 con excesos enantioméricos superiores al 96% en la mayoría de los casos y con una alta diastereoselectividad, que en muchos casos se eleva prácticamente al 100%. Tabla 13: Resultados de la reacción entre etil vinil éter (42a) y diferentes heterodienos 31.a O EtO O O N + Cu(OTf)2 + 33a EtO 42a Heterodieno 31 2 3 Ph R 43 31 O O CH2Cl2 R 1 N O N a T (°C) tiempo (h) Rendim. (%)b endo/exo (% ed)c ee endo (%)d ee exo (%)d 0 0,5 43aa (100) 98 91 86 -20 4 (100) 99,5 94 73 -40 20 (100) 99,8 96 - Continúa en la página siguiente… 85 Resultados y Discusión heterodieno 31 4 T (°C) tiempo (h) Rendim. (%)b endo/exo (% ed)c ee endo (%)d ee exo (%)d -20 20 43ba (100) >99,5 92 - -40 48 (80)d >99,9 94 - -40 14 43ca (100) >99,5 96 - -20 3,3 43da (96) >99,9 96 - -40 16 (85)e >99,9 90 - b 5 c 6 O 7 8 O N d O2N 9 f -40 70 43fa (98) 97 96 50 10 g -40 70 43ga (99) 98 96 n.d. 11 h -20 70 43ha (92) 98 95 n.d. 12 i -20 70 43ia (92) 98 95 n.d. 13 j 0 10 43ja (100) >99,9 88 - k -40 18 43ka (100) 90 96 37 O 14 O N a Todos los experimentos se llevaron a cabo bajo nitrógeno, Cu(OTf)2 (0,025 mmol), 33a (0,025 mmol), CH2Cl2 (1,5 mL), heterodieno 31 (0,25 mmol) y 42a (0,75 mmol). b Purificado por cromatografía. c Endo = 2,4-cis, exo = 2,4-trans. d Determinado por HPLC con columnas de fase estacionaria quiral. e En este caso se trata de conversión (%). En el caso de los sustratos sustituidos en el carbono β del doble enlace con un anillo aromático (entradas 1-8), se observa una disminución de la reactividad con la presencia de un grupo metoxilo electrón-donante en para que apenas afecta a la enantioselectividad. En el caso del heterodieno 31d que presenta un grupo p-nitrofenil se observa una disminución significativa de la enantioselectividad y del rendimiento a 40 °C que podría ser atribuido a la baja solubilidad del compuesto a esta temperatura. 86 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina como heterodienos en reacciones de hetero-Diels-Alder Los heterodienos 31f-j, sustituidos con anillos heterocíclicos de cinco miembros π-excedentarios requieren mayores tiempos de reacción (entradas 9-12) y en particular los derivados de tiofeno requieren una temperatura de reacción más alta. En todos los casos los productos de reacción se obtienen con elevada enantioselectividad. Nuevamente, la presencia de un grupo furilo, que podría actuar como dienófilo, no supone ninguna interferencia para la reacción. También cabe señalar que el compuesto 31e, sustituido con un anillo de piridina, fue totalmente inactivo en las condiciones de reacción, recuperándose inalterado. El sustrato 31j, que presenta dobles enlaces conjugados, se preparó para evaluar la tendencia de los N-óxidos de 2-alquenoilpiridina a actuar competitivamente como dienos en reacciones de Diels-Alder o heterodienos en reacciones de heteroDiels-Alder. En este caso la reacción requirió una temperatura de 0 °C y proporcionó el dihidropirano 43ja con total diastereoselectividad y con un 88% de exceso enantiomérico, no observándose la formación del producto de Diels-Alder (Esquema 60). Esquema 60 O Ph OEt O N O EtO Diels-Alder Ph O N OEt EtO O hetero-DA Ph 31j O N 43ja Por último, el heterodieno 31k, con un grupo alifático t-butilo sigue la tendencia general en cuanto a enantioselectividad, proporcionando el aducto esperado con un 96 % de exceso enantiomérico, aunque con un ligero descenso de la diastereoselectividad, situándose en una relación endo/exo 95:5 (90% de exceso diastereoisomérico). La reacción permite utilizar una gran variedad de alquenos sustituidos con grupos electrón-donantes. En el estudio de la reacción de 31a frente a diversos alquenos se puede observar una mayor variabilidad de resultados, si bien por lo general la enantioselectividad es excelente. Además del etil vinil éter utilizado anteriormente con los diferentes heterodienos, se ensayaron diferentes vinil éteres tanto cíclicos como acíclicos (entradas 1-6). El dihidrofurano 42b se mostró más reactivo incluso que el etil vinil éter, proporcionando el producto 43ab como un único diastereoisómero con un exceso enantiomérico del 99% (entrada 2). Por el contrario, el dihidropirano 42c se mostró poco reactivo, necesitando una temperatura más elevada y requiriendo 2 días para completar la reacción. Aún así el producto 43ac se obtuvo con una diastereoselectividad apreciable (93% ed) y con un exceso enantiomérico del 95% para el diastereoisómero mayoritario endo. 87 Resultados y Discusión Tabla 14: Resultados de la reacción entre el heterodieno 31a y diversos alquenos.a Alqueno T (°C) tiempo (h) Rendimiento (%)b endo/exo (% ed) ee endo (%)c ee exo (%)c 1 42a -40 20 43aa (100) 99,8 96 n.d. 2 42b -40 7 43ab (100) >99 99 n.d. 3 42c 0 48 43ac (90) 93 95 23 O O 4 42d -40 2 43ad (88) 64 90 58 5 42e -40 7 43ae (92) 32 94 95 6 42f -20 72 43af (53) 83 49 93 7 42g -40 5,5 43ag (100) 97 99 59 -40 25 43ah (93) 99 99,6 n.d. -40 7 43ai (98) 100 96 n.d. -40 5 43ci (100) 96 97 53 20 21 43aj (100) 95 77 52 20 120 43jj (72) 71 77 n.d. 0 168 43ak (52) 99 86 n.d. O 8 42h 9 10 d 42i N EtS 11 42j 12 e 13 a MeO 42k Todos los experimentos se llevaron a cabo bajo nitrógeno, Cu(OTf)2 (0,025 mmol), 33a (0,025 mmol), CH2Cl2 (1,5 mL), 31a (0,25 mmol) y alqueno 42 (0,75 mmol) b Rendimiento del producto purificado por cromatografía. c Determinado por HPLC con columnas de fase estacionaria quiral. d En este caso la reacción es con la enona 31c. e En este caso la reacción es con la dienona 31j. 88 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina como heterodienos en reacciones de hetero-Diels-Alder O O Me OMe Ph H O X Ph X 43af 43ae 43ad NH O Ph X OMe OMe O O Ph X 43aj 43ai 43ah 43ag 43ci X 43ac O OMe O Ph 43ab 43aa Ph X 43jj 43ak El más reactivo de todos los vinil éteres fue 42d (entrada 4). Pese a estar disustituido geminalmente, el hecho de ser un doble enlace exocíclico potencia la reactividad de este alqueno, finalizándose la reacción en tan solo 2 horas. Sin embargo, este tipo de substitución sí afecta a la enantioselectividad, que disminuye hasta el 90% de exceso enantiomérico, y, especialmente, a la diastereoselectividad (64% de exceso diastereoisomérico). Igualmente, el alqueno 42e, sustituido geminalmente reaccionó con baja diastereoselectividad (32% de exceso diastereoisomérico), si bien los excesos enantioméricos para ambos diastereoisómeros fueron elevados (entrada 5). El vinil éter 42f con un doble enlace trisustituido (entrada 6), mostró baja reactividad proporcionando el cicloaducto 43af con una diastereoselectividad considerable (83% de exceso diastereoisomérico) y con un exceso enantiomérico bajo (49%) para el aducto endo mayoritario. Algunas N-vinilamidas también reaccionaron con 31a con buenos resultados Con la N-vinilformamida (42g) la reacción fue completa en 5,5 horas, proporcionando el compuesto 43ag con elevada diastereoselectividad (97%) y enantioselectividad (99% de exceso enantiomérico). En el espectro de RMN, tanto en CDCl3 como en DMSO-d6, de 43ag se observaron las señales correspondientes a una mezcla de dos rotámeros scis y s-trans (Esquema 61). El aumento gradual de la temperatura al registrar el espectro de RMN 1H mostró una coalescencia de las señales, confirmando que los dos 89 Resultados y Discusión compuestos observados son rotámeros. Este hecho es debido a la baja velocidad de rotación del enlace amida. Esquema 61 s-cis-43ag s-trans-43ag Por otra parte, la N-vinilpirrolidona, que presenta mayor sustitución sobre el nitrógeno, requirió tiempos de reacción más largos (20 horas), si bien proporcionó el correspondiente producto 43ah con total diastereo- y enantioselectividad. El etil vinil sulfuro (42i) reaccionó de forma más rápida que el etil vinil éter, completándose la reacción en tan sólo 7 horas a -40 °C, proporcionando exclusivamente el diastereoisómero endo con un 96% de exceso enantiomérico (entrada 9). La eficacia del sistema catalítico se demostró además mediante la reacción con alquenos poco activados. Así, el 4-metoxiestireno reaccionó con 31a para dar el correspondiente producto con una alta diastereoselectividad (95% ed) y un meritorio 77% de exceso enantiomérico. Con el fin de comprobar si la dienona 31j podría actuar como dieno en la reacción de Diels-Alder frente a alquenos menos activos que el etil vinil éter se ensayó también la reacción entre esta dienona y p-metoxiestireno (42j) (entrada 12). Sin embargo, de nuevo el compuesto 31j prefirió actuar como heterodieno y se obtuvo el aducto de hetero-Diels-Alder, manteniendo la enantioselectividad, pero con peor rendimiento y diastereoselectividad que con el heterodieno 31a. En el caso del alqueno 42k hay que subrayar que, dado que se trata de un carbodieno, podría experimentar una reacción de Diels-Alder competitiva, actuando 31a como dienófilo (Esquema 62). Sin embargo, tras varios días de reacción, se obtuvo el producto de reacción de hetero-Diels-Alder con un 52% de rendimiento y 86% de exceso enantiomérico. Esquema 62 90 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina como heterodienos en reacciones de hetero-Diels-Alder Por último se ensayó la reacción con el silil éster 42l, con un doble enlace tetrasustituido fuertemente activado. Sin embargo, en este caso no se obtuvo el producto de cicloadición esperado. En su lugar se obtuvo el producto 45 resultante de una reacción de adición de Michael-Mukaiyama con un exceso enantiomérico relativamente bajo (74%) (Esquema 63). Esquema 63 Este hecho podría deberse a dos causas. Por un lado es posible que el impedimento estérico propio de un alqueno tetrasustituido dificulte una adecuada aproximación al heterodieno para dar lugar a la reacción de hetero-Diels-Alder a través de un mecanismo pericíclico. Por otro lado, también es posible que la reacción de hetero-Diels-Alder transcurra de manera general a través de un intermedio de adición de Michael-Mukaiyama seguido de una rápida ciclación, pero que en este caso la transferencia del grupo trimetilsililo desde el carbonilo al enolato en el intermedio de Michael-Mukaiyama sea más veloz que la propia ciclación (Esquema 64).65 Esquema 64 c) Determinación de la estereoquímica absoluta y modelo estereoquímico Experimentos NOESY de RMN con varios cicloaductos mostraron que el diastereoisómero mayoritario obtenido en la reacción es el que presenta una disposición cis entre los sustituyentes 2 y 4 del anillo heterocíclico y que proceden de una aproximación endo entre el alqueno y el heterodieno. Los ejemplos más significativos de NOE observados con algunos de los productos se exponen en la Figura 49. 91 Resultados y Discusión N O O OMe O O O O H H 5,22 H 5,04 3,86 O N O O 5,04 H H H Ph 1,71 H 1,71 43ka 43ba O N N O MeO Ph Me H 1,56 3,80 43ae 43ab 43af Figura 49 Además, fue posible conseguir cristales del compuesto endo-43ci adecuados para el análisis de difracción de rayos X que permitieron determinar su configuración absoluta. En la Figura 50 se muestra el grafico ORTEP correspondiente al compuesto endo-43ci, donde se aprecia la configuración (2R,4S) del aducto (parámetro de Flack = 0,005(9)). e O N EtS O (R) (S) Br 43ci Figura 50 La estereoquímica obtenida estaría de acuerdo con un estado de transición similar al descrito en la reacción de Diels-Alder, en la que el catalizador formaría un e Para los derivados de etil vinil éter la misma configuración absoluta es (2S,4S) por aplicación de las reglas de Cahn-Ingold-Prelog. 92 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX N-Óxidos de 2-alquenoilpiridina como heterodienos en reacciones de hetero-Diels-Alder complejo tetraédrico con el sustrato en el que la cara Si del heterodieno estaría más accesible al alqueno (Figura 51). Figura 51 d) Transformaciones sintéticas Como ejemplo de potenciales transformaciones de los aductos de hetero-DielsAlder obtenidos, se procedió a la hidrogenación catalítica del aducto 43aa en presencia de paladio sobre carbono (Esquema 65). La hidrogenación del doble enlace, que ocurre conjuntamente con la desoxigenación del anillo de piridina, tiene lugar con una elevada diastereoselectividad, proporcionando una mezcla de diastereoisómeros 92:8, en la que el tetrahidropirano 46 con los tres sustituyentes en cis resulta muy mayoritario. La reacción transcurrió además sin pérdida del exceso enantiomérico en el producto mayoritario. Esquema 65 93 Resultados y Discusión 4.2.1.5. N-Óxidos de 2-alquenoilpiridina como dipolarófilos en cicloadiciones [2+3] con nitronas Para finalizar el apartado dedicado al uso de N-óxidos de 2-alquenoilpiridina en reacciones de cicloadición se estudió su aplicación en reacciones de cicloadición 1,3dipolares con nitronas. Al contrario que las reacciones de Diels-Alder y hetero-DielsAlder, hasta la fecha no se encuentra en la literatura ningún ejemplo que utilice 2alquenoilpiridinas o sus correspondientes N-óxidos como dipolarófilos en reacciones [3+2]. a) Síntesis de las nitronas Como procedimiento general la preparación de las nitronas se realizó haciendo reaccionar cantidades equimolares de los correspondientes aldehídos e hidroxilaminas sustituidas en etanol, de acuerdo a los procedimientos que se encuentran descritos en la bibliografía.205-208 Tabla 15: Síntesis de las diversas nitronas.a Nitrona O 1 N Ph Rendimiento (%) 47a 73 47b 78 47c 75 47d 70 Ph 2 O 3 N Ph Br 4 Continúa en la página siguiente… 94 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX. N-Óxidos de 2-alquenoilpiridina como dipolarófilos en cicloadiciones [2+3] con nitronas nitrona Rendimiento (%) 5 47e 72 6 47f 80c 47g 85c 7 O N Ph Ph Todos los experimentos se llevaron a cabo con aldehído (20 mmol), hidroxilamina sustituida (20 mmol) y etanol (5 mL). C Se utilizó el clorhidrato de la hidroxilamina sustituida, añadiendo K2CO3 (20 mmol) a la mezcla de reacción. a b) Optimización El proceso de optimización se inició haciendo reaccionar el sustrato 31a (ahora como dipolarófilo) con la difenil nitrona 47a en las condiciones previamente optimizadas para las reacciones de Diels-Alder: 10% molar de Cu(OTf)2, 10% molar de ligando Ph-Box (33a), diclorometano como disolvente a 0 °C. Sin embargo, en estas condiciones, la reacción transcurrió con menos de un 10% de rendimiento después de 24 horas (entrada 1, Tabla 16), resultando aún inferior el rendimiento conseguido con el complejo Zn(II)-33a. Como se ha descrito en la introducción, la presencia de tamiz molecular puede jugar un papel fundamental en reacciones de cicloadición. Por esta razón se repitió la reacción en presencia de tamiz molecular de 4 Å (400 mg/mmol), observándose un aumento muy importante en la reactividad y el rendimiento (94%), aunque el producto se obtuvo con sólo un 10% de exceso enantiomérico. Este resultado animó a seguir el proceso de optimización. Se ensayaron los ligandos 33b, 33c, 33d con estructura de bis(oxazolina) y el ligando 49 con estructura 2,6-piridin bis(oxazolina) (PyBOX), obteniéndose el mejor resultado con el ligando 33b, que permitió una notable enantioselectividad (89% de exceso enantiomérico), así como también una mayor diastereoselectividad, con sólo un 8% de producto exo. Los ligandos 33d y 49 95 Resultados y Discusión condujeron a productos racémicos. Hay que señalar aquí que nuevamente se produce una inversión en la inducción quiral entre el ligando 33a y los ligandos 33b y 33c. f Tabla 16: Optimización del ligando en la reacción de cicloadición 1,3-dipolar.a MX2 Ligando MS (Å/mg) T (°C) tiempo (h) Rendim. (%)b endo/exo ee endo (%)c,d ee exo (%)c 1 Cu(OTf)2 33a - 0 24 <10 n.d. 0 0 2 Zn(OTf)2 33a - 0 24 <5 n.d. 0 0 3 Cu(OTf)2 33a 4/100 t.a. 5 94 62:38 (S) 10 8 4 Cu(OTf)2 33c 4/100 t.a. 5 90 80:20 (S) 59 n.d. 5 Cu(OTf)2 33b 4/100 t.a. 5 85 92:8 (R) 89 12 6 Cu(OTf)2 33d 4/100 t.a. 5 79 82:18 0 0 7 Cu(OTf)2 49 4/100 t.a. 5 86 85:15 0 0 a Todos los experimentos se llevaron a cabo bajo nitrógeno, MX2 (0,025 mmol), ligando (0,025 mmol), CH2Cl2 (1,5 mL), 31a (0,25 mmol) y 47a (0,3 mmol). b Purificado por cromatografía. c Determinado por HPLC con una columna chiralcel OD-H. d Entre paréntesis la configuración absoluta en el carbono α al carbonilo (ver apartado d). Una vez se determinó que 33b es el mejor ligando para la reacción, se procedió a optimizar otras variables de la reacción (Tabla 17). Se ensayaron varios disolventes clorados y no clorados. Nitrometano y acetato de etilo proporcionaron los mejores resultados en términos de enantioselectividad, si bien con acetato de etilo se obtuvo un mejor rendimiento (100%). En estas condiciones el producto de reacción se obtuvo con una elevada diastereoselectividad (relación endo/exo = 94:6) y enantioselectividad (92% de exceso enantiomérico para el aducto endo). f El ligando 33c tiene configuración R en lugar de la configuración S de los ligandos 33a y 33b. Es por ello que, a pesar de que el enantiómero mayoritario obtenido con 33c es el mismo que con 33a, se considera la existencia de una inducción quiral inversa entre 33c y 33a. 96 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX. N-Óxidos de 2-alquenoilpiridina como dipolarófilos en cicloadiciones [2+3] con nitronas Tabla 17: Optimización de la cicloadición 1,3-dipolar con el ligando 33b.a MX2 Disolvente MS (Å/mg) T (°C) tiempo (h) Rendim. (%)b endo/exo ee endo (%)c,d ee exo (%)c,d 1 Cu(OTf)2 CH2Cl2 4/100 t.a. 5 85 92:8 89 12 2 Cu(OTf)2 CHCl3 4/100 t.a. 5 80 92:8 86 8 3 Cu(OTf)2 (ClCH2)2 4/100 t.a. 5 93 90:10 88 18 4 Cu(OTf)2 Tolueno 4/100 t.a. 5,5 85 87:13 85 18 5 Cu(OTf)2 CH3NO2 4/100 t.a. 5 86 93:7 92 28 6 Cu(OTf)2 THF 4/100 t.a. 5,5 93 97:3 90 (+)2 7 Cu(OTf)2 AcOEt 4/100 t.a. 5 100 94:6 92 9 8 Cu(OTf)2 CH3CN 4/100 t.a. 29 20 79:21 78 32 9 Cu(OTf)2 EtOH 4/100 t.a. 24 <3 n.d. n.d. n.d. 10 Zn(OTf)2 AcOEt 4/100 t.a. 48 88 66:34 7 (+)17 11 Mg(OTf)2 AcOEt 4/100 t.a. 48 86 32:64 0 0 12 Cu(SbF6)2 AcOEt 4/100 t.a. 6 64 77:23 75 (+)4 13 Cu(ClO4)2 AcOEt 4/100 t.a. 6 83 81:19 81 22 14 Cu(OTf)2 AcOEt - t.a. 5 59 80:20 83 29 15 Cu(OTf)2 AcOEt 3/100 t.a. 5 81 89:12 96 33 16 Cu(OTf)2 AcOEt 5/100 t.a. 5 92 86:13 91 32 17 Cu(OTf)2 AcOEt 4/50 t.a. 21 97 90:10 95 36 18 Cu(OTf)2 AcOEt 4/75 t.a. 10 94 94:6 95 61 19 Cu(OTf)2 AcOEt 4/200 t.a. 21 86 91:9 93 17 20 Cu(OTf)2 AcOEt 4/75 0 24 96 91:9 98 41 21 Cu(OTf)2 AcOEt 4/75 -20 48 51 91:9 99 44 a Todos los experimentos se llevaron a cabo bajo nitrógeno, MX2 (0,025 mmol), ligando 33b (0,025 mmol), disolvente (1,5 mL), 31a (0,25 mmol) y 47a (0,3 mmol). b Purificado por cromatografía. c Determinado por HPLC con una columna chiralcel OD-H. d Excepto donde se señala lo contrario, el enantiómero mayoritario es el (-). Utilizando acetato de etilo como disolvente se ensayaron diversas sales metálicas. Los triflatos de cinc y magnesio dieron lugar a mezclas racémicas, después de largos tiempos de reacción. Sin embargo, resulta interesante que con triflato de magnesio se invierte la diastereoselectividad, dando lugar al diastereoisómero exo como producto mayoritario. También se ensayaron otras sales de Cu(II) variando el contraión (entradas 12 y 13). Sin embargo, tanto el hexafluoroantimoniato como el 97 Resultados y Discusión perclorato de Cu(II) dieron peores resultados en todos los aspectos, especialmente el primero. Por último se varió el tipo de tamiz molecular y la cantidad añadida (entradas 13-19). Como puede verse (entrada 14), en acetato de etilo y con el ligando 33b la presencia de tamiz molecular no es esencial para que se produzca la reacción, si bien los resultados son inferiores a cuando se utiliza el mismo. El uso de tamiz molecular de 3 Å así como el de 5 Å no supuso ninguna mejora respecto al tamiz de 4 Å ya que, aunque el uso de tamiz molecular de 3 Å permitió incrementar el exceso enantiomérico del producto, fue a expensas de la diastereoselectividad y del rendimiento de la reacción. Además, se varió la cantidad de tamiz encontrándose la proporción óptima en 300 mg/mmol (75 mg de tamiz en la escala a la que se desarrollaron los experimentos). Finalmente, fue posible incrementar la enantioselectividad hasta el 98% de exceso enantiomérico disminuyendo la temperatura de reacción a 0 °C (entrada 20). Una disminución mayor de la temperatura produjo una ralentización excesiva de la reacción que condujo a un bajo rendimiento (entrada 21). c) Alcance y limitaciones de la reacción Después del proceso de optimización se pasó a estudiar el comportamiento de los diferentes N-óxidos de 2-alquenoilpiridina frente a la nitrona 47a (Tabla 18). En general, todas las reacciones transcurrieron con elevada enantioselectividad en el diastereoisómero mayoritario aunque con diastereoselectividad y rendimientos variables. Al igual que en la reacción de Diels-Alder y hetero-Diels-Alder estudiada anteriormente, el sistema permite la presencia de un anillo aromático sustituido en para con grupos electrón-donantes o electrón-atrayentes en el carbono β del alqueno. En todos los casos el diastereoisómero mayoritario se obtiene con un exceso enantiomérico cercano al 95% aunque la diastereoselectividad disminuye ligeramente cuando el grupo fenilo presenta un grupo electrón-atrayente (bromo o nitro) en para. Los sustratos que poseen un anillo heteroaromático de cinco miembros πexcedentario, 31g y 31i, resultan menos reactivos y dan lugar a rendimientos moderados. En el caso del compuesto 31g la diastereoselectividad es casi total, mientras que en el caso del compuesto 31i es ligeramente inferior. En ambos casos la enantioselectividad es elevada. La dienona 31j, con un doble enlace conjugado adicional, reacciona con la nitrona 47a con buena diastereo- y enantioselectividad aunque con rendimiento moderado. 98 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX. N-Óxidos de 2-alquenoilpiridina como dipolarófilos en cicloadiciones [2+3] con nitronas Tabla 18: Resultados de la reacción entre la nitrona 47a y diferentes dipolarófilos 31.a Dipolarófilo 1 2d 31a 3 T (°C) tiempo (h) t.a. 10 t.a. 24 t.a. 24 t.a. 10 t.a. 1,5 t.a. 48 t.a. 48 t.a. 24 Rendim. (%)b 48aa (94) (100) 48ba (69) endo/exo ee endo (%)c ee exo (%)c 94:6 95 61 90:10 92 35 93:7 96 n.d. 90:10 96 22 87:13 96 21 >99:1 95 - 94:6 91 n.d. 93:7 93 33 31b 4 48ca (82) 31c 5 48da (56) 31d 6 48ga (43) 31g O 7 O N S 48ia (68) 31i 8 48ja (52) 31j Continúa en la página siguiente… 99 Resultados y Discusión dipolarófilo O 9 O N T (°C) tiempo (h) t.a. 1 Rendim. (%)b 48ka (98) endo/exo ee endo (%)c ee exo (%)c 92:8 86 45 31k a Todos los experimentos se llevaron a cabo bajo nitrógeno, Cu(OTf)2 (0,025 mmol), ligando 33b (0,025 mmol), tamiz molecular de 4 Å (75 mg), acetato de etilo (1,5 mL), dipolarófilo 31 (0,25 mmol) y 47a (0,3 mmol). b Purificado por cromatografía. C Determinado por HPLC con columnas de fase estacionaria quiral. d Cu(OTf)2 (0,125 mmol), ligando 33b (0,125 mmol), tamiz molecular de 4 Å (750 mg), acetato de etilo (7 mL), 31a (2,5 mmol) y 47a (3,0 mmol). Por último, el dipolarófilo 31k, con un grupo t-butilo alifático, mostró una elevada reactividad, completando la reacción en tan sólo 1 hora con un rendimiento excelente, aunque con una pequeña disminución en la enantioselectividad (86% de exceso enantiomérico) respecto al resto de sustratos aromáticos y heteroaromáticos. Además, la escala de la reacción puede multiplicarse por 10 o la carga catalítica disminuirse hasta el 5% molar sin que la estereoselectividad de la reacción se vea gravemente afectada, manteniendo además un rendimiento prácticamente cuantitativo (entrada 2). Seguidamente se pasó a estudiar la aplicación a otras nitronas utilizando el compuesto 31a como dipolarófilo (Tabla 19). Se ensayaron diversas nitronas 47a-d derivadas de N-fenilhidroxilamina y aldehídos aromáticos para-sustituidos con grupos electrón-donantes y electrónatrayentes (entradas 1-4). En la mayoría de los casos la reacción transcurrió con rendimientos entre moderados y altos, buena diastereoselectividad y alta enantioselectividad. Únicamente con la nitrona 47d, derivada del p-nitrobenzaldehído, se obtuvo un bajo rendimiento (entrada 4). La nitrona 47e, derivada de un aldehído alifático, reaccionó par a dar el correspondiente cicloaducto con bajo rendimiento y estereoselectividad inferior a la obtenida con las nitronas de aldehídos aromáticos (entrada 5). Por último se ensayaron las nitronas 47f y 47g derivadas de Nalquilhidroxilaminas que proporcionaron resultados inferiores a las derivadas de Nfenilhidroxilamina (entrada 6 y 7). 100 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX. N-Óxidos de 2-alquenoilpiridina como dipolarófilos en cicloadiciones [2+3] con nitronas Tabla 19: Resultados de la reacción entre 31a y diversas nitronas.a T (°C) Tiempo (h) 47a t.a. 10 47b t.a. 48 47c t.a. 48 4 47d t.a. 48 5 47e t.a. 24 6 47f t.a. 48 47g t.a. 48 Nitrona O 1 N Ph Rendim. (%)b 48aa (94) endo/exo ee endo (%)c ee exo (%)c 94:6 95 61 86:14 90 42 91:9 94 45 94:6 89 79 74:26 60 29 66:34 88 78 61:39 80 75 Ph 2 O N Ph 3 48ab (77) 48ac (81) Br 7 O N Ph 48ad (36) 48ae (16) 48ag (59) 48af (62) Ph Todos los experimentos se llevaron a cabo bajo nitrógeno, Cu(OTf)2 (0,025 mmol), ligando 33b (0,025 mmol), tamiz molecular de 4 Å (75 mg), acetato de etilo (1,5 mL), 31a (0,25 mmol) y nitrona 47 (0,3 mmol). b Purificado por cromatografía. C Determinado por HPLC con columnas de fase estacionaria quiral. a 101 Resultados y Discusión d) Determinación de la estereoquímica absoluta y modelo estereoquímico En todos los casos la identificación de los dos diastereoisómeros resultantes se realizó por espectroscopía de RMN 1H, a partir de las constantes de acoplamiento de las señales correspondientes a los protones en α al grupo carbonilo que aparecen como un doble doblete. Así, los aductos endo muestran unos valores de J ≈ 6 Hz y J ≈ 8 Hz, mientras que las constantes de acoplamiento para dicho protón en el diastereoisómero exo son J ≈ 9 Hz y J ≈ 10 Hz, tal y como se ha descrito en la bibliografía para compuestos similares (Figura 52).90,208 5,36 J=5,8 Hz 5,15 J=7,6 Hz, J=5,8 Hz Ph H H O Ph N O H X Ph 5,46 J=7,6 Hz endo-48aa 5,47 J=10,3 Hz 5,15 J=9,3 Hz, J=10,3 Hz Ph H H O Ph N O H X Ph O N X= 5,98 J=9,3 Hz exo-48aa Figura 52: constantes de acoplamiento en los productos endo y exo. Además, se han podido obtener cristales de los productos endo-48ba y endo48ja adecuados para su análisis mediante difracción de rayos X. Para el caso de endo48ba, al no poseer elementos pesados que favorezcan el fenómeno de dispersión anómala se obtuvo una estructura con un valor de 0,6(2) para el parámetro de Flack que no permitió establecer la estereoquímica absoluta de dicho producto aunque sí fue posible determinar que se trataba del diastereoisómero endo (Figura 53). Para el producto 48ja, que posee un átomo de azufre en la molécula, se obtuvo una estructura con un parámetro de Flack de 0,1(12), adecuado para establecer su configuración absoluta, como se muestra en la Figura 54. endo-48ba Figura 53 102 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX. N-Óxidos de 2-alquenoilpiridina como dipolarófilos en cicloadiciones [2+3] con nitronas O N O (R) Ph (S) N S (R) O Ph endo-48ja Figura 54 La configuración absoluta de endo-48ja es compatible con la formación de un complejo plano-cuadrado entre el catión metálico Cu(II), el ligando 33b y el dipolarófilo, tal como se representa en la Figura 55 y siguiendo el ejemplo de lo representado anteriormente par la reacción de Diels-Alder ( Figura 48b, página 79). Sin embargo, al contrario que en las reacciones Diels-Alder y de hetero-Diels-Alder, en este caso resulta más difícil establecer un mecanismo de reacción por dos motivos. El primero de ellos es el papel activo del tamiz molecular en la catálisis, que puede no limitarse a la simple absorción de agua, tal como se ha visto en la introducción y como parece deducirse también de los resultados en el proceso de optimización anterior. El segundo motivo es que la nitrona también podría coordinarse al ión Cu(II), por lo que la esfera de coordinación en el complejo metálico podría diferir de lo representado en la Figura 55. Figura 55 103 Resultados y Discusión 4.2.2. Reacciones de Diels-Alder asimétricas con α’arilsulfonil enonas catalizadas por Cu(II)-BOX En este apartado se estudia la aplicación de α’-arilsulfonil enonas como dienófilos en reacciones de Diels-Alder catalizadas por complejos de Cu(II)-BOX. Las α’arilsulfonil enonas son sustratos bidentados que pueden unirse al centro metálico del catalizador a través del oxígeno de la sulfona. Una de las ventajas de estos sustratos es que conducen a productos de reacción con una agrupación β-cetosulfona la cual permite llevar a cabo posteriores modificaciones de los mismos aprovechando la variada reactividad química de los grupos carbonilo y sulfona.209 En particular, es posible llevar a cabo la alquilación del metileno activo y la hidrogenólisis del enlace carbono-azufre dando lugar a cetonas. En este caso las α’arilsulfonil enonas actuarían como dienófilos equivalentes a enonas (Esquema 66). Esto es remarcable porque existen muy pocos ejemplos de reacciones de Diels-Alder que utilicen enonas sencillas no quelantes como dienófilos.210-216 Además, en la mayoría de los mismos el resultado varía notablemente con el radical unido al grupo carbonilo, permitiendo poca variación sobre el mismo. Esquema 66 O O O S Aril RX O O O S Aril R O - SO2Aril R 4.2.2.1. Antecedentes Las α’-arilsulfonil enonas han sido poco estudiadas como sustratos en reacciones enantioselectivas. Como antecedentes previos a esta tesis sólo se encuentran los trabajos de Wada et al., quienes utilizaron por primera vez α’arilsulfonil enonas, precisamente en reacciones de cicloadición. En su trabajo inicial estos autores describen la reacción de hetero-Diels-Alder con demanda electrónica inversa diastereoselectiva, empleando diversos ácidos de Lewis tales como TiCl2(i-PrO)2 y Eu(fod)3 como catalizadores, con los que obtuvieron los aductos endo (cis) en una proporción mayor del 98%.217,218 Posteriormente desarrollaron una versión enantioselectiva de esta reacción utilizando TADDOLatos de titanio (IV) como catalizadores ácidos de Lewis, obteniendo enantioselectividades entre el 60 y el 97% de exceso enantiomérico (Esquema 67).219 Por último, Wada aplicó también este 104 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX. Reacciones de Diels-Alder asimétricas con α’-arilsulfonil enonas catalizadas por Cu(II)-BOX sistema catalítico en reacciones de Diels-Alder, utilizando exclusivamente ciclopentadieno como dieno, llegando a obtener los aductos de Diels-Alder correspondientes enantioméricamente puros (Esquema 68).220 Esquema 67 Esquema 68 Por otra parte, cuando el trabajo descrito en este capítulo estaba a punto de concluir apareció publicado un trabajo de Kim y Yang describiendo el uso de estos sustratos en reacciones de Michael-Mukaiyama catalíticas asimétricas con 2(trimetilsililoxi)furano como nucleófilo y catalizadas por complejos Cu(II)-BOX (Esquema 69).221 La reacción transcurre generalmente con altos rendimientos, con excelentes enantioselectividades y notable diastereoselectividad. Los ligandos trans-diPh-BOX y t-Bu-BOX dan lugar a bajos excesos enantioméricos y compuestos racémicos, respectivamente. Por el contrario, Ph-BOX y cis-di-Ph-BOX dan lugar a una excelente inducción asimétrica, mientras que con el ligando inda-BOX se obtiene el producto con un 64% de exceso enantiomérico. Esquema 69 2+ O O Ph O O O S Ph TMSO O + R N Ph Cu CHCl3 Ph 2 -OTf N Ph O O R O S Ph O O 105 Resultados y Discusión 4.2.2.2. Síntesis de α’-arilsulfonil enonas Las α’-arilsulfonil enonas utilizadas en este trabajo se sintetizaron siguiendo la metodología descrita por Wada (Esquema 70).218 Se utilizaron dos procedimientos diferentes, ambos de dos etapas. El procedimiento A implica en primer lugar la adición de dienolato de una 1-(arilsulfonil) propanona a un aldehído. La β-hidroxicetona resultante se somete en un segundo paso a una eliminación mediante ácido ptoluenosulfónico en tolueno a reflujo para dar la correspondiente enona. En ocasiones se observa en este segundo paso una reversión parcial de la β-hidroxicetona a los compuestos de partida. El método B consta de una primera etapa de adición del enolato de una arilmetilsulfona a un aldehído α,β-insaturado. En una segunda etapa se lleva a cabo la oxidación del alcohol alílico resultante mediante el reactivo de Jones o dióxido de manganeso para obtener la α’-arilsulfonil enona deseada. En la Tabla 20 se resumen los resultados obtenidos en la síntesis de las diversas enonas 66a-h. Esquema 70 Procedimiento A: Procedimiento B: 106 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX. Reacciones de Diels-Alder asimétricas con α’-arilsulfonil enonas catalizadas por Cu(II)-BOX Tabla 20: Síntesis de α’-arilsulfonil enonas. Enona Procedimiento 1 66a A 2 67 Ba 68 A 4 66b A 5 66c A 6 66d A 7 66e A 8 66f Ba 9 66g A 10 66h A 11 66i Ba 12 66j Bb 3 Ph O O O S Rendimiento Rendimiento 1r paso (%) 2º paso (%) 54 (85) 55 (95) 56 (73) 50 65 62 Cl a 57 (60) 58 (55) 59 (55) 60 (64) 61 (88) 62 (96) 63 (72) 64 (61) 65 (88) 61 40 72 88 55 66 87 68 77 Se utilizó reactivo de Jones como oxidante. b Se utilizó dióxido de manganeso como oxidante. 107 Resultados y Discusión 4.2.2.3. α’-Arilsulfonil enonas como dienófilos en reacciones de Diels-Alder a) Optimización El proceso de optimización se llevó a cabo utilizando la reacción entre la α’arilsulfonil enona 66a y ciclopentadieno (23a) (Tabla 21). En primer lugar se ensayaron los diferentes ligandos 33a, 33b, 33c y 49, utilizando triflato de Cu(II) como sal metálica en diclorometano a 0 °C (entradas 1-4). Los mejores resultados se obtuvieron con el ligando 33a (entrada 1), que dio lugar a la formación del producto 69aa con alto rendimiento, notable diastereoselectividad endo y elevado exceso enantiomérico (95%). De nuevo se encontró una inversión en el sentido de la inducción asimétrica entre el ligando 33a y el resto de los ligandos. g El cambio en el contraión de la sal de cobre de triflato a hexafluoroantimoniato provocó una disminución en la estereoselectividad. El uso de triflato de magnesio como ácido de Lewis dio lugar al producto con una moderada diastereoselectividad y disminución de la enantioselectividad (73% de exceso enantiomérico), aunque la conversión no fue total después de 24 horas de reacción. En presencia de triflato de cinc se obtuvieron resultados similares a los obtenidos con triflato de cobre(II), aunque se requirió mayor tiempo de reacción. También se ensayaron diferentes disolventes para la reacción (entradas 8 -13) que no mejoraron los resultados. Con tolueno se produjo una pequeña mejora en la diastereoselectividad de la reacción a expensas de la enantioselectividad y la conversión. Para aprovechar el efecto positivo de ambos disolventes se ensayó la reacción utilizando una mezcla de diclorometano y tolueno 1:1, lo que permitió maximizar la eficacia de la reacción (entrada 13). Finalmente se observó que la temperatura afecta ligeramente la estereoselectividad de la reacción, obteniendo mejores resultados a temperaturas más bajas. g El ligando 33c tiene configuración R en lugar de la configuración S de los ligandos 33a y 33b. Es por ello que, a pesar de que el enantiómero mayoritario obtenido con 33c es el mismo que con 33a, se considera la existencia de una inducción quiral inversa entre 33c y 33a. 108 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX. Reacciones de Diels-Alder asimétricas con α’-arilsulfonil enonas catalizadas por Cu(II)-BOX Tabla 21: Optimización de la reacción de Diels-Alder entre 66a y ciclopentadieno catalizada por Cu(II)-33a.a MX2 Ligando Disolvente T (°C) tiempo (h) Conversión (%) endo/exo ee endo (%)b,c 1 Cu(OTf)2 33a CH2Cl2 0 5 100 91:9 (S) 95 2 Cu(OTf)2 33b CH2Cl2 0 70 29 76:24 (R) 7 3 Cu(OTf)2 33c CH2Cl2 0 70 94 89:11 (S) 42 4 Cu(OTf)2 49 CH2Cl2 0 24 9 82:18 (R) 34 5 Mg(OTf)2 33a CH2Cl2 t.a. 24 64 80:20 (S) 73 6 Zn(OTf)2 33a CH2Cl2 0 20 100 87:13 (S) 93 7 Cu(SbF6)2 33a CH2Cl2 0 22 100 86:14 (S) 56 8 Cu(OTf)2 33a Tolueno 0 20 84 95:5 (S) 91 9 Cu(OTf)2 33a MeNO2 0 4,5 100 80:20 (S) 61 10 Cu(OTf)2 33a MeCN 0 20 10 76:24 (R) 8 11 Cu(OTf)2 33a THF t.a. 24 20 79:21 (S) 69 Cu(OTf)2 33a CH2Cl2:tolueno 1:2 0 3 100 94:6 (S) 94 Cu(OTf)2 33a CH2Cl2:tolueno 1:1 0 4 100 93:7 (S) 95 Cu(OTf)2 33a CH2Cl2:tolueno 1:1 t.a. 1,2 100 91:9 (S) 93 Cu(OTf)2 33a CH2Cl2:tolueno 1:1 -20 24 100 95:5 (S) 96 12 13 14 15 a Todos los experimentos se llevaron a cabo bajo nitrógeno, MX2 (0,025 mmol), ligando (0,025 mmol), disolvente (1,5 mL), 66a (0,25 mmol) y 23a (1,8 mmol). b Determinado por HPLC con una columna chiralpak AD-H. c Entre paréntesis la estereoquímica absoluta del carbono α a carbonilo (ver apartado c). 109 Resultados y Discusión b) Alcance y limitaciones de la reacción Con las condiciones optimizadas (Cu(OTf)2 como sal metálica, ligando 33a y una mezcla 1:1 diclorometano-tolueno como disolvente) se procedió a estudiar el comportamiento de las diversas α’-arilsulfonil enonas frente a ciclopentadieno. Los resultados obtenidos se encuentran recogidos en la Tabla 22. En primer lugar se analizó la naturaleza del grupo arilsulfona (entradas 1-3), variando el sustituyente en la posición 4 de anillo de benceno. Las características electrónicas de dicho sustituyente mostraron tener poco efecto sobre la estereoselectividad de la reacción, si bien el derivado de 4-clorofenilsulfona 68 se mostró un poco más reactivo. A continuación se estudió el efecto de los sustituyentes sobre el doble enlace del dienófilo. Con los dienófilos con un anillo de benceno en el carbono β del doble enlace se obtuvieron altos excesos enantioméricos (por encima del 95% en todos los casos) y buenas diastereoselectividades con independencia de las características electrónicas del sustituyente en para del anillo de benceno (entradas 1-6). El dienófilo 66e reaccionó por el doble enlace α,β, tal como cabía esperar. Aunque este sustrato necesitó mayores tiempos de reacción, el producto se obtuvo con buen rendimiento y buena estereoselectividad (entrada 7). El dienófilo 66f con un doble enlace monosustituido reaccionó de manera extremadamente rápida con excelente diastereo- y enantioselectividad pero con bajo rendimiento. La alta reactividad de este sustrato probablemente da lugar a reacciones secundarias que son responsables de la disminución del rendimiento. El sistema también tolera la presencia de grupos alquilo en el carbono β del doble enlace tales como un grupo metilo o el más voluminoso t-butilo (entradas 9 y 10). En ambos casos la reacción mantiene una buena enantioselectividad, aunque la diastereoselectividad para el dienófilo con un grupo t-butilo es muy baja. Sin embargo, cuando el grupo metilo se encuentra en posición α del doble enlace el rendimiento de la reacción es moderado, sin discriminación exo/endo, si bien ambos diastereoisómeros se obtienen con buenos excesos enantioméricos (entrada 11). La reacción también funcionó con alquenos trisustituidos, como es el caso del dienófilo 66j con sendos metilos en las posiciones α y β. La reacción requirió un significativo aumento del tiempo de reacción y de nuevo se observó una nula diastereoselectividad. Sin embargo, el aducto endo se obtuvo con un excelente exceso enantiomérico del 96% (entrada 12). 110 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX. Reacciones de Diels-Alder asimétricas con α’-arilsulfonil enonas catalizadas por Cu(II)-BOX Tabla 22: Resultados de la reacción entre ciclopentadieno (23a) y diversos dienófilos catalizada por Cu(II)-33a.a Dienófilo tiempo (h) Rend. (%)b endo/exo ee endo (%)c ee exo (%)c 1 66a 4 69aa (100) 93:7 95 n.d. 2 67 2 70 (92) 93:7 95 n.d. 68 0,75 71 (96) 93:7 97 98 4 66b 5 69ba (95) 96:4 95 n.d. 5 66c 1 69ca (93) 92:8 95 94 6 66d 1 69da (90) 90:10 95 95 7 66e 24 69ea (86) 94:6 92 n.d. 8 66f 0,08 69fa (50) 92:8 96 n.d. 9 66g 0,25 69ga (93) 92:8 91 n.d. 10d 66h 5 69ha (99) 43:57 90 83 3 Ph O O O S Cl Continúa en la página siguiente… 111 Resultados y Discusión dienófilo tiempo (h) Rend. (%)b endo/exo ee endo (%)c ee exo (%)c 11 66i 0,33 69ia (65) 49:51 94 90 12d 66j 9 69ja (93) 50:50 96 88 a Todos los experimentos se llevaron a cabo bajo nitrógeno a 0 °C (excepto donde se indique lo contrario), Cu(OTF)2 (0,025 mmol), 33a (0,025 mmol), CH2Cl2 (0,75 mL), tolueno (0,75 mL), dienófilo (0,25 mmol) y 23a (1,8 mmol). b Purificado por cromatografía. c Determinado por HPLC con columnas de fase estacionaria quiral. d En este caso la reacción se desarrolló a temperatura ambiente. Una vez analizado el comportamiento de la reacción de Diels-Alder con las diversas α’-arilsulfonil enonas y ciclopentadieno, se pasó a estudiar la aplicación a otros dienos. La enona 66a mostró baja reactividad frente a dienos diferentes a ciclopentadieno. Las enonas 66f y 66g se mostraron más reactivas y fueron utilizadas en esta parte del estudio. Los resultados se resumen en la Tabla 23. Tabla 23: Reacción de Diels-Alder de los compuestos 66f y 66g con diversos dienos.a Dieno Dienófilo tiempo (h) Rend. (%)b Ratio diastereo. ee mayor (%)c ee minor (%)c 1 23a 66f 0,08 69fa (50) 92:8d 96 n.d. 2 23b 66f 8 69fb (45) >99:1d 96 n.d. 3 23c 66f 24 69fc (56)f - 76 - 4 23d 66f 21 69fd (35) 96:4e 79 69 5g 23f 66g 96 69gf (22)h 56:44i 71 15 6 23h 66g 0,5 69gh (67)j 100 92 - a Todos los experimentos se llevaron a cabo bajo nitrógeno a 0 °C (excepto donde se indique lo contrario), Cu(OTf)2 (0,025 mmol), 33a (0,025 mmol), CH2Cl2 (0,75 mL), tolueno (0,75 mL), dienófilo (0,25 mmol) y dieno (1,8 mmol) a 0 °C. b Purificado por cromatografía. c Determinado por HPLC con columnas de fase estacionaria quiral. d ratio endo/exo. e ratio 1,4-69fd/1,3-69fd. f Se obtuvo un 7% adicional de producto de reacción énica. g Se llevó a cabo a 25 °C. h La conversión fue del 33%. i Ratio cis(= endo)/trans (= exo) 1,2-69gf. j 69gh es la cetona resultante después de la hidrólisis de la mezcla de reacción con ácido cítrico 0,01 M en H2O:MeOH 1:1. 112 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX. Reacciones de Diels-Alder asimétricas con α’-arilsulfonil enonas catalizadas por Cu(II)-BOX O O O S Tol 69fb 69fa Ph 69fc 69fd O O O S Tol O O O S Tol Me 69gf O Me 69gh El dienófilo 66f y ciclohexadieno (23b) condujeron al producto esperado 69fb como un único diastereoisómero (endo/exo > 99:1) con elevada enantioselectividad (96% de exceso enantiomérico). La reacción de 66f con 2,3-dimetilbutadieno (entrada 3) transcurrió con alta regioselectividad (76% de exceso enantiomérico). Cabe señalar que con este dieno se observó también la formación de un pequeño porcentaje del producto 72, resultado de una reacción énica con el 2,3-dimetilbutadieno (Figura 56). Una enantioselectividad similar se obtuvo con isopreno (entrada 4), así como una alta regioselectividad, favoreciendo el regioisómero 1,4-disustituido con exceso enantiomérico moderado (79%). 72 Figura 56 El 1-fenilbutadieno (23f) reaccionó con 66g de manera muy lenta (38% de conversión después de 6 horas), dando el producto de Diels-Alder con baja diastereoselectividad y enantioselectividad moderada. Por último, el éter de enol 23h se mostró muy reactivo con la enona 66g, dando lugar al aducto 69gh con una completa diastereoselectividad y un gran exceso enantiomérico en tan sólo 30 minutos. c) Determinación de la estereoquímica y modelo estereoquímico Se realizaron experimentos de RMN de tipo NOE y NOESY para determinar la estereoquímica relativa endo y exo de los diferentes aductos. En la Figura 57 se muestran las interacciones entre protones más significativas para los productos 69aa, 69ia, 69ga, 69ja y 69ha. 113 Resultados y Discusión H 1,61 1,85 H H 3,44 H H H Ph 7,23 Me 2,80 1,14 O SO2Tol endo-69aa SO2Tol endo-69ga Me 1,69 H Me Me 1,02 exo-69ia H O exo-69ja SO2Tol 6,05 H 1,02 O H O 1,66 H H 3,03 t-Bu 0,84 O SO2Tol endo-69ja SO2Tol endo-69ha Figura 57 endo-66b Figura 58: Estructura del compuesto endo-66b elucidada por difracción de rayos X. Además, la estructura del aducto endo-66b fue elucidada mediante análisis cristalográfico de difracción de rayos X. Dicho análisis permitió obtener la estructura que se muestra en la Figura 58, con un valor para el parámetro de Flack de -0,09(7). Como se puede observar, el aducto 66b posee una configuración S para el carbono estereogénico que soporta el grupo carbonilo. Para explicar el curso estereoquímico de la reacción se proponen dos posibles modelos, similares a los propuestos anteriormente para otras reacciones catalizadas por complejos Cu(II). En ambos casos el dienófilo se coordinaría al átomo de Cu(II) de 114 Nuevas reacciones de cicloadición catalizadas por complejos de Cu(II)-BOX. Reacciones de Diels-Alder asimétricas con α’-arilsulfonil enonas catalizadas por Cu(II)-BOX forma bidentada a través de los átomos de oxígeno del grupo carbonilo y del grupo sulfona. En el primer modelo (Figura 59a) se formaría un complejo plano-cuadrado distorsionado en el cual la cara Si del doble enlace, en la conformación s-trans de la enona, estaría disponible para la aproximación endo del dieno. Alternativamente, un complejo de geometría tetraédrica (Figura 59b) y con la enona en conformación s-cis, normalmente más estable, conduciría al mismo resultado estereoquímico. Aunque la geometría plano-cuadrada por lo general es más favorable para los complejos Cu(II), en este caso el complejo tetraédrico podría estar estabilizado por una interacción de tipo π-π entre el doble enlace de la enona y el grupo fenilo de la oxazolina. Ar O N Ar O Cu O S N O O S O O N Cu N O O O (a) (b) Figura 59 d) Transformaciones sintéticas Para demostrar las posibilidades sintéticas de los aductos de Diels-Alder obtenidos con α’-arilsulfonil enonas, se llevaron a cabo algunas transformaciones, consistentes en la alquilación del metileno activo en posición α’ y la posterior eliminación del grupo arilsulfona por reducción (Esquema 71 y Esquema 72). Esta transformaciones prueban la utilidad de las α’-arilsulfonil enonas como equivalentes sintéticos de cetonas α,β-insaturadas en reacciones de Diels-Alder enantioselectivas. Esquema 71 115 Resultados y Discusión Esquema 72 Las alquilaciones se llevaron a cabo con el haluro de alquilo correspondiente en DMF y en presencia de carbonato potásico como base. El producto de reacción es una mezcla de diastereoisómeros no separables por cromatografía flash, sin embargo este hecho es poco relevante pues ambos diastereoisómeros dan lugar al mismo producto después de la eliminación reductiva del grupo sulfona. Para esta reducción del enlace carbono-azufre se utilizó amalgama de sodio en metanol. Las reacciones transcurrieron en todos los casos con buenos rendimientos y sin pérdida de pureza óptica. 116 Experimental section 5. E XPERIMENTAL SECTION 5.1. General Experimental Methods 5.1.1. General Procedures All catalytic reactions were carried out in Schlenck tubes oven-dried overnight, under nitrogen atmosphere. Reactions were monitored by TLC analysis using Merck Silica Gel 60 F-254 thin layer plates. Flash chromatography was performed on Merck silica gel 60, 0.040-0.063 mm. Racemic products for comparison were prepared using an equimolar amount of copper(II) triflate (in the absence of chiral ligand or in the presence of 2,2’-dipyridyl) in CH2Cl2 or magnesium perchlorate in CH2Cl2-THF 9:1. 5.1.2. Solvents and reagents 5.1.2.1. Solvents CH2Cl2 and toluene were distilled from CaH2 and stored on 4 Å molecular sieves. EtOAc was whased with saturated aqueous NaHCO3, dried over K2CO3, distilled from P2O5 and stored on 4 Å molecular sieves. THF was freshly distilled from Na/benzophenone. Chloroform was washed with water, then washed with brine, dried over Na2SO4 and distilled from P2O5 prior to use. 5.1.2.2. Reagents Most reagents were commercially available and used as purchased. Copper(II) triflate and ligands (S,S)-Ph-BOX (33a), (S,S)-t-Bu-BOX (33b) and (S,S)Ph-PyBOX (49) were commercially available and used without further purification. (1R,2S)-Inda-Box (33c) was synthesized according to a procedure described in the literature.74 Cyclopentadiene was freshly distilled from its dimer. All dienes were commercially available, except 23f, 23g, 23h and 23j which were synthesized according to procedures described in the literature, as is related in the following pages. 5.1.3. Instrumentation 5.1.3.1. Melting points Melting points were measured in a “Thermopan” microscope instrument and are uncorrected. 117 Experimental section 5.1.3.2. Nuclear magnetic resonance (NMR) NMR spectra were run at 300 MHz for 1H and at 75 MHz for 13C NMR in a Bruker Avance 300 spectrometer (299.95 MHz for 1H NMR and 75.43 MHz for 13C NMR). In some cases a Bruker Avance 400 spectrometer was used, especially for NOE and NOESY experiments. Samples were dissolved in deuterated solvents as stated, using TMS (δ 0 ppm for both 1H and 13C experiments) or the residual non-deuterated solvent as internal standard (δ 7.26 ppm for 1H experiments and δ 77.0 ppm for 13C experiments in the case of CDCl3). Chemical shifts are given in δ values (ppm). The carbon multiplicity was determined by DEPT experiments. 5.1.3.3. Polarimetry Specific optical rotations were measured in a Perkin-Elmer polarimeter using sodium light (D line 589 nm) and a 1 dm cell. Concentrations (c) are given in g/100 mL. 5.1.3.4. Mass spectrometry Mass spectra were recorded on a Fisons Instruments VG Autospec GC 8000 series. Mass spectra were run at 70 eV. FAB Mass spectra were carried out at 30 kV in a MNBA matrix. Data are given in mass units and values in parentheses express the relative intensity with respect to base peak. 5.1.3.5. HPLC analyses Chiral HPLC analyses were performed in an Agilent 1100 series instrument equipped with a refraction index detector or in a Hitachi Elite Lachrom instrument equipped with a Hitachi UV diode-array L-4500 detector using chiral stationary columns from Daicel. 5.1.3.6. Gas Chromatography analyses Chiral gas chromatography analyses were performed in a Termoquest Trace GC 2000 Series instrument equipped with a Supelco Beta-DEX 225 30 m × 0.25 mm × 0.25 μm column. N2 was used as carrier at 1 mL/minute. Injector and detector temperature was set at 220 °C. 118 Experimental section 5.2. Synthesis of ligands with hydroxyoxazoline moiety based on (1S)-(+)-ketopinic acid. Application to enantioselective Diels-Alder reaction. 5.2.1. Synthesis and characterization of (1S)-(+)-ketopinic acid (1) Ketopinic acid was synthesized according to previous procedures described in the literature.166,167 Into a two-necked, round-bottomed flask, equipped with a dropping funnel, a magnetic stirring bar, and a reflux condenser was placed of (1S)-(+)-camphorsulfonic acid (7, 100 g, 1 0.43 mol) and chloroform (640 mL). The suspension was heated to reflux and thionyl chloride (38 mL, 0.52 mol) was added dropwise over a 1 hour period. Heating was continued until gas evolution had ceased (approximately 7 hr). The solution was concentrated under reduced pressure and the resultant camphorsulfonyl chloride 8 was used without further purification in the following step. A 4 L beaker containing a solution of anhydrous sodium carbonate (100 g, 0.95 mol) in water (900 mL) was placed in a bath, provision being made for efficient mechanical stirring. The stirrer was started and, when the solution was hot (50-60 °C), one-third of a solution of potassium permanganate (100 g, 0.63 mol) in hot water (600 mL) was added all at once, followed by a portion of chloride 8 (34 g). After an interval of 5–10 minutes, half the remaining permanganate was poured in, followed by another portion of the chloride 8 (33 g). After a similar interval, the remaining permanganate solution and a final portion of the chloride (33 g) were added and heating was continued for an hour. The excess of permanganate was destroyed by adding a few milliliters of an acidified solution of sodium sulfite. The reaction mixture was cooled and made strongly acidic by cautious addition (foaming may occur) of 20% sulfuric acid. The mixture was heated (50-60 °C), and the precipitated manganese dioxide was dissolved by stirring in powdered sodium sulfite (usually 70–80 g. is required). The resulting solution was cooled and extracted with one 200 mL, two 150 mL, and one 100 mL portions of ether. The combined ether extracts were dried over anhydrous sodium sulfate and the bulk of the ether removed under reduced pressure. The residue was recrystallized from hot water to give pure 1 in 35% yield. mp 233-234 °C; [α]D25 +25.8 (c 0.65, MeOH); 1H NMR (300 MHz, CDCl3) δ 10.90 (1H, br s), 2.58 (1H, ddd, J = 3.1 Hz, J = 4.6 Hz, J = 18.7 Hz), 2.39 (1H, ddd, J = 4.6 Hz, J = 10.9 Hz, J = 14.5 Hz), 2.14 (1H, dd, J = 3.4 Hz, J = 5.6 Hz), 2.08 (1H, m), 2.00 (1H, d, J = 18.7 Hz), 1.79 (1H, ddd, J = 4.5 Hz, J = 9.2 Hz, J = 13.7 Hz), 1.44 (1H, ddd, J = 3.6 Hz, J = 119 Experimental section 9.2, J = 12.8Hz), 1.17 (3H, s), 1.12 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 213.0 (s), 174.6 (s), 66.9 (s), 49.7 (s), 44.0 (d), 43.6 (t), 26.9(t), 26.7(t), 20.8 (q), 19.8 (q). 5.2.2. Synthesis and characterization of amides 13-16 General procedure for the synthesis of amides 13-16 A solution of (S)-(+)-ketopinic acid (1, 1.3 g, 7.3 mmol) in SOCl2 (6 mL) was refluxed for 2 h and the excess SOCl2 was removed under reduced pressure. The residue dissolved in CH2Cl2 (5 mL) was added dropwise to a solution of aminoalcohol (7.3 mmol) and Et3N (1 mL, 7.3 mmol) in CH2Cl2 (4 mL) at 0 °C under nitrogen. After 1 h, the mixture was diluted with EtOAc (150 mL), washed with 2 M HCl (2 × 30 mL), saturated aqueous NaHCO3 (2 × 30 mL) and brine (30 mL), dried over MgSO4, concentrated under reduced pressure and purified. (1S,4R)-N-((S)-1-Hydroxy-3-methylbutan-2-yl)-7,7-dimethyl-2-oxobicyclo[2.2.1] heptane-1-carboxamide (13). Yield: 90 % (flash chromatography); mp 58-60 °C; [α]D25 +34.5 (c 0.79, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.64 (1H, d, J = 7.4 Hz), O 3.73 (1H, m), 3.57 (1H, dd, J = 4.1 Hz, J = 11.0 Hz), 3.48 (1H, dd, J = 6.6 Hz, J = 11.0 Hz), 3.43 (1H, br s), 2.43 (2H, m), 2.05 (1H, m), 2.01 O NH HO (1H, m), 1.89 (1H, d, J = 18.8 Hz), 1.87 (1H, m), 1.52 (1H, ddd, J = 4.3 Hz, J = 9.2 Hz, J = 13.6 Hz), 1.35 (1H, ddd, J = 3.8 Hz, J = 9.3 Hz, J = 13 12.7 Hz), 1.14 (3H, s), 0.90 (3H, s), 0.88 (3H, d, J = 6.5 Hz), 0.86 (3H, d, J = 6.5 Hz); 13C NMR (75.5 MHz, CDCl3) δ 217.2 (s), 170.0 (s), 64.5 (s), 64.2 (t), 56.8 (d), 49.9 (s), 43.5 (t), 42.9 (d), 28.6 (d), 28.0 (t), 27.5 (t), 20.7 (q), 20.1 (q), 19.5 (q), 17.9 (q); MS (EI) m/z (%): 267 (M+, 0.2), 206 (12), 236 ( 83), 165 (100), 123 (13), 95 (10); HRMS: 267.1822, C15H25NO3 required 267.1834. (1S,4R)-N-((S)-2-Hydroxy-1-phenylethyl)-7,7-dimethyl-2-oxobicyclo[2.2.1]heptane-1carboxamide (14). 14 120 Yield: 84% (flash chromatography); mp 108-109 °C; [α]D25 +85.4 (c 0.80, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.27 (1H, d, J = 6.5 Hz), 7.31 (5H, m), 5.13 (1H, dt, J = 4.8 Hz, J = 6.5 Hz), 3.83 (1H, dd, J = 3.4 Hz, J = 10.0 Hz), 3.78 (1H, dd, J = 5.0 Hz, J = 10.0 Hz), 2.67 (1H, br s), 2.57-2.44 (2H, m), 2.18-2.09 (2H, m), 1.98 (1H, d, J = 18.8 Hz), 1.60 (1H, ddd, J = 3.3 Hz, J = 6.8 Hz, J = 13.6 Hz), 1.42 (1H, ddd, J = 4.0 Hz, J = 9.4 Hz, J = 13.0 Hz), 1.24 (3H, s), 1.04 (3H, s); Experimental section 13 C NMR (75.5 MHz, CDCl3) δ 217.2 (s), 169.8 (s), 138.8 (s), 128.7 (d), 127.7 (d), 126.6 (d), 67.3 (t), 64.6 (s), 55.8 (d), 50.2 (s), 43.6 (t), 43.1 (d), 28.1 (t), 27.6 (t), 20.8 (q), 20.3 (q); MS (EI) m/z (%): 302 (M++1, 1.2), 283 (7), 270 (100), 165 (52), 106 (11); HRMS: 302.1754, C18H24NO3 required 302.1756. (1S,4R)-N-((R)-2-Hydroxy-1-phenylethyl)-7,7-dimethyl-2-oxobicyclo[2.2.1]heptane-1carboxamide (15). Yield: 90% (recrystallized from hexane-CH2Cl2); mp 140-142 °C; [α]D25 -6.2 (c 0.78, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.37 (1H, d, J = 6.4 Hz), 7.32 (5H, m), 5.15 (1H, td, J = 5.4 Hz, J = 7.0 Hz), 3.87 (2H, m), 2.81 (1H, s), 2.58 (1H, m), 2.51 (1H, dd, J = 4.7 Hz, J = 8.5 Hz), 2.17 (1H, m), 2.09 (1H, t, J = 4.4 Hz), 2.00 (1H, d, J = 18.8 Hz), 1.69 (1H, ddd, J = 4.6 Hz, J = 9.2 Hz, J = 13.8 Hz), 1.45 (1H, m), 1.22 (3H, s), 0.94 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 217.3 (s), 169.8 15 (s), 139.0 (s), 128.7 (d), 127.6 (d), 126.5 (d), 67.1 (t), 64.5 (s), 55.7 (d), 50.3 (s), 43.6 (t), 43.2 (d), 28.4 (t), 27.7 (t), 20.8 (q), 20.3 (q); MS (FAB) m/z (%): 302 (M++1, 100), 154 (71), 137 (58); HRMS: 302.1754, C18H24NO3 required 302.1756. (1S,4R)-N-((1R,2S)-2-Hydroxy-2,3-dihydro-1H-inden-1-yl)-7,7-dimethyl-2-oxobicyclo [2.2.1]heptane-1-carboxamide (16). Yield: 80% (recrystallized from hexane-CH2Cl2); mp 163-165 °C; [α]D25 +28.5 (c 0.69, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.85 (1H, d, J = 7.6 Hz), 7.25 (4H, m), 5.47 (1H, dd, J = 5.0 Hz, J = 8.1 Hz), 4.70 (1H, dt, J = 2.1 Hz, J = 5.1 Hz), 3.19 (1H, dd, J = 5.2 Hz, J = 16.6 Hz), 3.00 (1H, dd, J = 2.0 Hz, J = 16.6 Hz), 2.63 (1H, m), 2.53 (1H, m), 2.23-2.13 (3H, m), 2.00 (1H, d, J = 18.7 Hz), 1.67 (1H, m), 1.46 (1H, m), 1.30 (3H, s), 1.10 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 16 216.6 (s), 169.7 (s), 140.6 (s), 140.1 (s), 128.0 (d), 127.0 (d), 125.3 (d), 124.2 (d), 73.5 (d), 65.4 (s), 57.5 (d), 50.1 (s), 43.7 (t), 43.4 (d), 39.6 (t), 27.9 (t), 27.5 (t), 20.8 (q), 20.7 (q); MS (FAB) m/z (%): 314 (M++1, 100), 282 (45), 154 (40); HRMS: 314.1749, C19H24NO3 required 313.1751. 121 Experimental section 5.2.3. Synthesis and characterization of ketooxazolines 17-20 General procedure for the synthesis of ketooxazolines 17-19 Mesyl chloride (6.75 mmol) was added dropwise to a solution of βhydroxyamide (3.4 mmol), Et3N (5.8 mL) and diisopropylethylamine (1.2 mL) in CH2Cl2 (17 mL) under nitrogen at 0 °C. The reaction was stirred overnight. After this time, reaction mixture was diluted with EtOAc (200 mL), washed with water (2 × 25 mL) and brine (25 mL), dried over MgSO4, concentrated under reduced pressure and the product purified by flash chromatography. (1R,4R)-1-((S)-4-Isopropyl-4,5-dihydrooxazol-2-yl)-7,7-dimethylbicyclo[2.2.1]heptan2-one (17). Yield: 90% (from hydroxyamide 13); mp 70-72 °C; [α]D25 -45.0 (c 1.0, CHCl3,); 1H NMR (300 MHz, CDCl3) δ 4.15 (1H, m), 3.94 (2H, m), 2.49 O (1H, td, J = 4.0 Hz, J = 18.3 Hz), 2.36 (1H, ddd, J = 4.1 Hz, J = 11.7 Hz, J = 14.8 Hz), 2.08 (1H, t, J = 4.4 Hz), 2.00 (1H, m), 1.90 (1H, d, J = 18.3 N O Hz), 1.75 (2H, m), 1.37 (1H, ddd, J = 4.0 Hz, J = 9.3 Hz, J = 13.1 Hz), 1.11 (3H, s), 1.03 (3H, s), 0.91 (3H, d, J = 6.8 Hz), 0.85 (3H, d, J = 6.8 17 Hz); 13C NMR (75.5 MHz, CDCl3) δ 211.7 (s), 163.2 (s), 71.7 (d), 69.1 (t), 62.7 (s), 49.1 (s), 44.1 (d), 43.7 (t), 32.2 (d), 27.0 (t), 26.4 (t), 21.2 (q), 19.6 (q), 18.6 (q), 17.5 (q); MS (EI) m/z (%): 249 (M+, 9.5), 234 (4.4), 221 (8), 206 (100), 178 (11), 162 (30); HRMS: 249.1722, C15H23NO2 required 249.1729. (1R,4R)-7,7-Dimethyl-1-((S)-4-phenyl-4,5-dihydrooxazol-2-yl)bicyclo[2.2.1]heptan-2one (18). Yield: 81% (from hydroxyamide 14); mp 100-105 °C; [α]D25 -68.6 (c 0.75, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.31 (1H, m), 5.22 (1H, dd, J = 7.8 Hz, J = 10.1 Hz), 4.63 (1H, dd, J = 8.4 Hz, J = 10.2 Hz), 4.13 (1H, t, J = 8.1 Hz), 2.59 (1H, ddd, J = 3.4 Hz, J = 4.4 Hz, J = 18.4 Hz), 2.49 (1H, ddd, J = 4.0 Hz, J = 11.6 Hz, J = 14.9 Hz), 2.18 (1H, t, J = 4.4 Hz), 2.10 (1H, m), 2.00 (1H, d, J = 18.4 Hz), 1.89 (1H, ddd, J = 4.8 Hz, J = 9.3 Hz, J = 14.1 Hz), 1.46 (1H, ddd, J = 4.1 Hz, J = 9.3 Hz, J = 13.0 18 Hz), 1.21 (3H, s), 1.15 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 211.7 (s), 165.3 (s), 142.3 (s), 128.6 (d), 127.4 (d), 126.7 (d), 74.7 (t), 69.6 (d), 63.0 (s), 49.3 (s), 44.4 (d), 43.8 (t), 27.2 (t), 26.5 (t), 21.4 (q), 19.8 (q); MS (EI) m/z (%): 283 (M+, 58), 122 Experimental section 268 (27), 255 (100), 240 (60), 214 (35), 210 (21), 120 (24), 104 (44), 91 (63), 77 (43), 67 (39); HRMS: 283.1571, C18H21NO2 required 283.1572. (1R,4R)-7,7-Dimethyl-1-((R)-4-phenyl-4,5-dihydrooxazol-2-yl)bicyclo[2.2.1]heptan-2one (19). Yield: 96% (from hydroxyamide 15); mp 122-125 °C; [α]D25 +88.5 (c 0.73, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.29 (5H, m), 5.23 (1H, dd, J = 8.0 Hz, J = 10.2 Hz), 4.67 (1H, dd, J = 8.5 Hz, J = 10.1 Hz), 4.07 (1H, t, J = 8.1 Hz), 2.64-2.49 (2H, m), 2.16 (1H, t, J = 4.4 Hz), 2.07 (1H, m), 1.98 (1H, d, J = 18.3 Hz), 1.84 (1H, ddd, J = 4.7 Hz, J = 9.2 Hz, J = 13.9 Hz), 1.43 (1H, ddd, J = 4.0 Hz, J = 9.2 Hz, J = 12.9 Hz), 1.19 (3H, s), 1.13 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 211.9 (s), 19 165.4 (s), 142.4 (s), 128.6 (d), 127.4 (d), 126.5 (d), 74.8 (t), 69.3 (d), 63.0 (s), 49.4 (s), 44.4 (d), 43.8 (t), 27.3 (t), 26.6 (t), 21.4 (q), 19.8 (q); MS (EI) m/z (%): 283 (M+, 73), 268 (26), 255 (99), 240 (60), 214 (28), 210 (19), 165 (24), 120 (27), 104 (60), 91 (100), 77 (67), 67 (81); HRMS: 283.1571, C18H21NO2 required 283.1572. (1R,4R)-1-((3aR,8aS)-8,8a-Dihydro-3aH-indeno[1,2-d]oxazol-2-yl)-7,7dimethylbicyclo[2.2.1]heptan-2-one (20). O N O A solution of 16 (2.49 g, 7.9 mmol) and (NH4)6Mo7O24·4H2O (988 mg, 0.8 mmol) in toluene (160 mL) was refluxed with a Dean–Stark trap for 4 h. The reaction mixture was filtered through a short pad of silica gel (2 cm), concentrated and the residue was chromatographed on silica gel using hexane–EtOAc mixtures to give 1.6 g of 20. Yield: 64%; mp 118-121 °C; [α]D25 +228.3 (c 0.69, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.47 (1H, m), 7.21 (3H, m), 5.53 (1H, d, J = 7.9 Hz), 5.39 (1H, ddd, J = 1.8 Hz, J = 6.8 Hz, J = 8.5 Hz), 3.41 (1H, dd, J = 6.8 Hz, J = 17.8 Hz), 3.18 (1H, dd, J = 1.5 Hz, J = 17.8 Hz), 2.54-2.39 (2H, m), 2.06 (1H, t, J = 4.4 Hz), 1.96 (1H, m), 1.91 (1H, d, J = 18.3 Hz), 1.74 (1H, ddd, J = 4.8 Hz, J = 9.3 Hz, J = 14.0 Hz), 1.36 (1H, ddd, J = 4.1 Hz, J = 9.2 Hz, J = 12.8 Hz), 1.02 (3H, s), 0.73 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 211.8 (s), 164.4 (s), 142.2 (s), 139.1 (s), 128.1 (d), 127.2 (d), 125.3 (d), 125.0 (d), 83.2 (d), 75.8 (d), 62.5 (s), 49.6 (s), 44.3 (d), 43.7 (t), 39.8 (t), 27.1 (t), 26.5 (t), 21.5 (q), 19.3 (q); MS (EI) m/z (%): 295 (M+, 50), 280 (10), 267 (29), 252 (39), 226 (15), 165 (16), 130 (62), 115 (100), 104 (52), 77 (39), 67 (31); HRMS: 295.1551, C19H21NO2 required 295.1572. 20 123 Experimental section 5.2.4. Synthesis and characterization of hydroxyoxazolines 2-6 and 21 General procedure for the synthesis of hydroxyoxazolines 2-4 and 21. A solution of [LiAlH4·2THF] complex in toluene (1 M, 1.5 mL, 1.5 mmol) was added dropwise to a solution of ketooxazoline (1.67 mmol) in toluene (16.7 mL) under nitrogen at -40 °C. The reaction was stirred until the starting material was consumed (TLC). The reaction mixture was then diluted with EtOAc (150 mL), washed with brine (3 x 10 mL), dried over MgSO4, concentrated under reduced pressure and the product purified by flash chromatography. (1S,2R,4R)-1-((S)-4-Isopropyl-4,5-dihydrooxazol-2-yl)-7,7-dimethylbicyclo[2.2.1] heptan-2-ol (2). Yield: 63% (from ketooxazoline 17); mp 51-53 °C; [α]D25 -75.8 (c 0.72, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.19 (1H, m), 3.99 (1H, dd, J = 3.4 OH Hz, J = 7.7 Hz), 3.93 (1H, dd, J = 5.7 Hz, J = 7.9 Hz), 3.88 (1H, d, J = 7.9 N Hz), 2.13 (1H, dd, J = 3.2 Hz, J = 11.0 Hz), 1.94-1.81 (2H, m), 1.80-1.69 O (3H, m), 1.58 (1H, br s), 1.18 (3H, s), 1.15 (2H, m), 1.02 (3H, s), 0.95 (3H, d, J = 6.8 Hz), 0.91 (3H, d, J = 6.8 Hz); 13C NMR (75.5 MHz, CDCl3) 2 δ 168.9 (s), 77.5 (d), 71.2 (d), 68.7 (t), 52.6 (s), 49.7 (s), 45.6 (d), 39.5 (t), 32.5 (d), 30.1 (t), 27.6 (t), 22.0 (q), 20.6 (q), 18.5 (q), 18.4 (q); MS (EI) m/z (%): 251 (M+, 0.8), 236 (100), 223 (53), 208 (96), 190 (45), 168 (49), 140 (15); HRMS: 251.1877, C15H25NO2 required 251.1885. (1S,2R,4R)-7,7-Dimethyl-1-((S)-4-phenyl-4,5-dihydrooxazol-2-yl)bicyclo[2.2.1]heptan2-ol (3). Yield: 70% (from ketooxazoline 18);[α]D25 -44.6 (c 0.72, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.30 (5H, m), 5.81 (1H, br s), 5.19 (1H, dd, OH J = 8.5 Hz, J = 10.2 Hz), 4.56 (1H, dd, J = 8.4 Hz, J = 10.3 Hz), 4.04 N (1H, dd, J = 3.4 Hz, J = 7.7 Hz), 3.98 (1H, t, J = 8.5 Hz), 2.21 (1H, m), O 1.98-1.91 (2H, m), 1.85-1.72 (2H, m), 1.30 (3H, s), 1.27-1.10 (2H, m), 1.09 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 170.4 (s), 142.2 (s), 128.6 (d), 127.4 (d), 126.3 (d), 77.5 (d), 73.4 (t), 68.9 (d), 52.9 (s), 3 50.0 (s), 45.7 (d), 39.5 (t), 30.2 (t), 27.6 (t), 22.1 (q), 20.8 (q); MS (EI) m/z (%): 285 (M+, 2.4), 270 (72), 257 (91), 242 (100), 216 (23), 202 (55), 174 (17), 151 (18), 120 (19), 106 (34), 91 (32), 77 (20); HRMS: 285.1727, C18H23NO2 required 285.1729. 124 Experimental section (1S,2R,4R)-7,7-Dimethyl-1-((R)-4-phenyl-4,5-dihydrooxazol-2-yl)bicyclo[2.2.1]heptan2-ol (4) and (1S,2S,4R)-7,7-dimethyl-1-((R)-4-phenyl-4,5-dihydrooxazol-2-yl) bicyclo[2.2.1]heptan-2-ol (21). Yield: 62% (from ketooxazoline 19); [α]D25 +14.3 (c 1.02, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.25 (5H, m), 5.53 (1H, br s), 5.20 (1H, dd, OH J = 7.7 Hz, J = 10.1 Hz), 4.53 (1H, dd, J = 8.4 Hz, J = 10.2 Hz), 4.06 (1H, dd, J = 3.5 Hz, J = 7.8 Hz), 4.00 (1H, t, J = 8.1 Hz), 2.20 (1H, m), N O 1.96-1.83 (2H, m), 1.80-1.70 (2H, m), 1.27 (1H, m), 1.22 (3H, s), 1.14 (1H, m), 1.06 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 170.2 (s), 142.0 (s), 128.5 (d), 127.4 (d), 126.2 (d), 77.5 (d), 73.4 (t), 68.4 (d), 4 53.0 (s), 49.7 (s), 45.6 (d), 39.3 (t), 30.3 (t), 27.6 (t), 21.9 (q), 20.5 (q); MS (EI) m/z (%): 285 (M+, 1.0), 270 (71), 257 (100), 242 (88), 216 (22), 202 (50), 174 (16), 151 (24), 120 (25), 104 (46), 91 (73), 77 (57); HRMS: 285.1718, C18H23NO2 required 285.1729. Yield: 15%; 1H NMR (300 MHz, CDCl3) δ 7.31 (5H, m), 5.17 (1H, ddd, J = 3.6 Hz, J = 7.3 Hz, J = 7.5 Hz), 4.93 (1H, br s), 4.04 (1H, dd, J = 3.6 Hz, J = 7.6 Hz), 3.86 (1H, dd, J = 3.9 Hz, J = 11.3 Hz), 3.73 (1H, dd, J = 7.3 Hz, J = 11.1 Hz), 2.21 (1H, m), 1.92-1.75 (4H, m), 1.22 (3H, s), 1.21 (1H, m), 1.05 (1H, m), 0.97 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 174.6 (s), 139.0 (s), 128.7 (d), 127.6 (d), 126.5 (d), 77.5 (d), 66.3 (t), 58.2 (s), 55.3 (d), 49.4 (s), 45.8 (d), 41.1 (t), 29.9 (t), 26.8 (t), 21 21.4 (q), 21.0 (q); MS (EI) m/z (%): 285 (M+, 2.1), 270 (65), 257 (100), 242 (75), 216 (29), 202 (60), 174 (13), 151 (32), 120 (25), 104 (40), 91 (77), 77 (61); HRMS: 285.1719, C18H23NO2 required 285.1729. (1S,2R,4R)-1-((3aR,8aS)-8,8a-Dihydro-3aH-indeno[1,2-d]oxazol-2-yl)-7,7dimethylbicyclo[2.2.1]heptan-2-ol (5) and (1S,2S,4R)-1-((3aR,8aS)- 8,8a-dihydro-3aHindeno[1,2-d]oxazol-2-yl)-7,7-dimethylbicyclo[2.2.1]heptan-2-ol (6). NaBH4 (22.6 mg, 0.6 mmol) was added in portions to a solution of ketooxazoline 20 (178 mg, 0.6 mmol) and CeCl3·7H2O (223 mg, 0.6 mmol) in MeOH (4 mL) at -10 °C. After 10 min, saturated aqueous NH4Cl (5 mL) was added, the mixture was diluted with EtOAc (100 mL) and washed with brine (2 × 10 mL), dried over MgSO4, concentrated under reduced pressure and the product purified by flash chromatography (hexane– EtOAc, 8:2) to give 88 mg of 5 (50% yield) and 18 mg of 6 (10% yield). 125 Experimental section Yield: 50%; mp 98-101 °C; [α]D25 +214.2 (c 0.80, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.47 (1H, m), 7.27 (3H, m), 5.79 (1H, br s), 5.58 OH (1H, d, J = 8.1 Hz), 5.29 (1H, ddd, J = 1.8 Hz, J = 7.2 Hz, J = 8.1 Hz), N 3.90 (1H, dd, J = 3.5 Hz, J = 7.8 Hz), 3.47 (1H, dd, J = 7.0 Hz, J = 18.0 O Hz), 3.23 (1H, dd, J = 1.6 Hz, J = 18.0 Hz), 2.07 (1H, m), 1.91 (1H, m), 1.81 (2H, m), 1.71 (1H, dd, J = 7.8 Hz, J = 12.9 Hz), 1.24 (3H, s), 1.10 (2H, m), 1.07 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 169.2 (s), 141.7 5 (s), 139.4 (s), 128.3 (d), 127.3 (d), 125.5 (d), 125.1 (d), 81.5 (d), 77.3 (d), 75.4 (d), 52.7 (s), 50.0 (s), 45.6 (d), 39.6 (t), 39.4 (t), 29.9 (t), 27.5 (t), 21.9 (q), 20.6 (q); MS (EI) m/z (%): 297 (M+, 1.3), 282 (50), 269 (69), 254 (61), 228 (17), 214 (42), 186 (13), 151 (14), 132 (20), 115 (100), 104 (23), 77 (24); HRMS: 297.1723, C19H23NO2 required 295.1729. Yield: 10%; mp 130-135 °C; [α]D25 +230.9 (c 1.03, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.48 (1H, m), 7.24 (3H, m), 5.49 (1H, d, J = 7.9 OH Hz), 5.30 (1H, ddd, J = 1.6 Hz, J = 6.9 Hz, J = 7.9 Hz), 4.70 (1H, ddd, J N = 1.3 Hz, J = 3.7 Hz, J = 10.2 Hz), 3.43 (1H, dd, J = 6.9 Hz, J = 17.9 O Hz), 3.18 (1H, dd, J = 1.6 Hz, J = 17.8 Hz), 2.67 (1H, br s), 2.27 (1H, m), 2.16 (1H, t, J = 8.8 Hz), 1.83 (2H, m), 1.66 (1H, t, J = 4.2 Hz), 1.32 (1H, t, J = 8.8 Hz), 1.04 (1H, dd, J = 3.9 Hz, J = 13.4 Hz), 1.00 (3H, s), 6 0.98 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 169.5 (s), 142.1 (s), 139.3 (s), 128.3 (d), 127.3 (d), 125.4 (d), 125.1 (d), 82.6 (d), 75.6 (d), 73.9 (d), 54.4 (s), 50.7 (s), 46.4 (d), 39.8 (t), 36.6 (t), 28.4 (t), 23.8 (t), 21.5 (q), 19.6 (q); MS (EI) m/z (%): 297 (M+, 2.5), 282 (80), 269 (100), 254 (79), 228 (24), 214 (54), 186 (16), 151 (14), 132 (20), 115 (75), 104 (18), 77 (11); HRMS: 297.1720, C19H23NO2 required 295.1729. 5.2.5. Synthesis and characterization of 3-alkenoyl-2oxazolidones General procedure for the synthesis of compounds 22a-c. 2-Oxazolidone (5.0 g, 57 mmol) was added to a suspension of NaH (60% dispersion in mineral oil, 4.6 g, 115 mmol) in 250 mL of CH2Cl2 under nitrogen at 0 °C. After gas evolution finished, the reaction was placed in a bath at -78 °C and alkenoyl chloride (62 mmol) was added. After 1 h, the reaction mixture was allowed to react at rt and stirred for 18 h. The excess of NaH was destroyed by addition of some drops of saturated aqueous NH4Cl and the reaction was filtered through a short pad of silica gel. The filtrate was concentrated under reduced pressure and the product recrystallized from hexane-CH2Cl2. 126 Experimental section 3-Acryloyloxazolidin-2-one (22a). Yield: 46%; 1H NMR (300 MHz, CDCl3) δ 7.48 (1H, dd, J = 10.5 Hz, J = 17.0 Hz), 6.54 (1H, dd, J = 1.8 Hz, J = 17.0 Hz), 5.89 (1H, dd, J = 1.8 Hz, J = 10.5 Hz), 4.44 (2H, t, J = 8.0 Hz), 4.08 (2H, t, J = 8.0 Hz); 13C NMR (75.5 MHz, CDCl3) δ 165.1 (s), 153.4 (s), 131.9 (d), 126.8 (t), 62.2 (t), 42.6 (t). 22a 3-Crotonoyloxazolidin-2-one (22b). O O O N Me 22b Yield: 80%; 1H NMR (300 MHz, CDCl3) δ 7.17 (1H, dd, J = 1.2 Hz, J = 15.3 Hz), 7.09 (1H, qd, J = 6.6 Hz, J = 15.3 Hz), 4.37 (2H, ddd, J = 1.2 Hz, J = 7.9 Hz, J = 8.1 Hz), 4.00 (2H, ddd, J = 1.5 Hz, J = 7.9 Hz, J = 8.1 Hz), 1.90 (3H, d, J = 6.6 Hz); 13C NMR (75.5 MHz, CDCl3) δ 164.9 (s), 153.3 (s), 146.4 (d), 121.2 (d), 61.9 (t), 42.4 (t), 18.2 (q). 3-Metacryloyloxazolidin-2-one (22c). O O Me N 22c O Yield: 80%; 1H NMR (300 MHz, CDCl3) δ 5.44 (1H, s), 5.41 (1H, s), 4.44 (2H, t, J = 7.9 Hz), 4.02 (2H, t, J = 7.9 Hz), 2.03 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 170.9 (s), 152.7 (s), 139.0 (s), 120.9 (t), 62.2 (t), 42.9 (t), 19.1 (q). 3-Cinnamoyloxazolidin-2-one (22d). According to the procedure described by Andrade et al.,173 dicyclohexylcarbodiimide (1.56, 7.56 mmol) was added to a suspension of 2-oxazolidone (0.5 g, 5.75 mmol), cinnamic acid (1.12 g, 7.54 mmol) and 4-(N,N-dimethylamino)pyridine (93 mg, 22d 0.76 mmol) in CH2Cl2 (8 mL) under nitrogen at 0 °C. After 10 minutes, the temperature was raised to rt and stirring was continued for 6 h. After this time, reaction was filtered and precipitate washed with CH2Cl2 (20 mL). The filtrate was diluted with diethyl ether (200 mL), washed with saturated aqueous NaHCO3 (2 x 20 mL) and brine (20 mL) and dried over anhydrous MgSO4. After evaporation of the solvent under reduced pressure the residue was chromatographed. The product was finally recrystallized from ethanol to give 810 mg of pure 22d (85% yield). 1 H NMR (300 MHz, CDCl3) δ 7.91 (1H, d, J = 15.8 Hz), 7.85 (1H, d, J = 15.8 Hz), 7.62 (2H, m), 7.39 (3H, m), 4.45 (2H, t, J = 8.1 Hz), 4.13 (2H, t, J = 8.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 165.3 (s), 153.5 (s), 146.2 (d), 134.4 (s), 130.6 (d), 128.8 (d), 128.5 (d), 116.5 (d), 62.0 (t), 42.7 (t). 127 Experimental section 5.2.6. Synthesis and characterization of dienes (E)-1-Phenyl-1,3-butadiene (23f). 23f Compound 23f was prepared according to the procedure described by Okamoto et al.174 n-Butyllithium (18.8 mL of a 1.6 M solution in hexane, 30 mmol) was added dropwise in 15 min to a suspension of methyltriphenylphosphonium bromide (10.7 g, 30 mmol) and THF (100 mL) in a flask fitted with a dropping funnel and a reflux condenser under nitrogen. The mixture was stirred for 1.5 h at rt and cinnamaldehyde (3.8 ml, 30 mmol) was then added and the resulting mixture was stirred for 1 h at rt. After this time, the solution was stirred for 5 h further at 65 °C and then it was allowed to cool to rt. The subsequent addition of diethyl ether (25 mL) and hexane (25 mL) gave a precipitate, which was removed by filtration. The filtrate was purified through a short flash chromatography, and diene 23f was isolated by distillation in the presence of a small amount of hydroquinone. Yield: 62%; 1H NMR (300 MHz, CDCl3) δ 7.33 (2H, d, J = 7.8 Hz), 7.24 (2H, dd, J = 7.2 Hz, J = 7.8 Hz), 7.14 (1H, t, J = 7.2 Hz), 6.72 (1H, dd, J = 10.3 Hz, J = 15.6 Hz), 6.49 (1H, d, J = 15.6 Hz), 6.44 (1H, td, J = 10.3 Hz, J = 16.8 Hz), 5.26 (1H, dd, J = 0.7 Hz, J = 16.8 Hz), 5.10 (1H, dd, J = 0.7 Hz, J = 10.3 Hz); 13C NMR (75.5 MHz, CDCl3) δ 137.1 (d), 137.0 (s), 132.8 (d), 129.5 (s), 128.5 (d), 127.5 (d), 126.3 (d), 117.5 (t). cis-Cyclohexane-1,2-diyldimethanol (25). Cis-1,2-cyclohexanedicarboxylic acid anhydride was slowly added to a suspension of LiAlH4 (9.0 g, 24 mmol) in 250 mL of dry THF under nitrogen at 0 °C. The mixture was stirred for 1 h and then heated to 25 reflux for 2 h. After this time, the reaction was cooled to rt and methanol was added dropwise until hydrogen evolution ceased. The reaction mixture was poured into a vessel containing saturated aqueous Na2SO4 (400 mL) and after stirring for 10 minutes it was filtered by suction. The aqueous phase was partitioned with EtOAc (200 mL) and phases separated. The organic phase was washed with water (2 x 30 mL) and the aqueous phase extracted with EtOAc (3 x 150 mL). Both organic phases were put together and dried over anhydrous MgSO4, filtered and concentrated to give 13.1 g of diol 25 (93% yield) which was used in the next step without further purification. 1 H NMR (300 MHz, CDCl3) δ 3.84 (2H, s), 3.71 (2H, dd, J = 8.0 Hz, J = 10.0 Hz), 3.53 (2H, dd, J = 3.9 Hz, J = 11.0 Hz), 1.88 (2H, m), 1.53-1.26 (8H, m); 13C NMR (75.5 MHz, CDCl3) δ 64.0 (t), 39.8 (d), 27.1 (t), 23.9 (t). 128 Experimental section cis-Ccyclohexane-1,2-diylbis(methylene) dimethanesulfonate (26). Diol 25 (3.5 g, 24 mmol) and Et3N (10 mL) were dissolved in CH2Cl2 (39 mL). The mixture was cooled to -78 °C and mesyl chloride (5.8 mL, 74 mmol) was added dropwise. After completion of the 26 reaction (TLC), water (25 mL) was added and the mixture was quickly extracted with EtOAc (200 mL). The organic phase was dried under anhydrous MgSO4, filtered and concentrated under reduced pressure, providing dimesylate 26. 1 H NMR (300 MHz, CDCl3) δ 4.26 (2H, dd, J = 7.2 Hz, J = 10.0 Hz), 4.18 (2H, dd, J = 6.8 Hz, J = 10.0 Hz), 3.03 (6H, s), 2.23 (2H, m), 1.48 (8H, m); 13C NMR (75.5 MHz, CDCl3) δ 69.7 (t), 37.3 (q), 36.6 (d), 25.8 (t), 23.0 (t). 1,2-Dimethylenecyclohexane (23g) and 1-(bromomethyl)-2-methylenecyclohexane (27). Crude dimesylate 26 (7.2, 24 mmol), Li2CO3 (4.9 g, 66 mmols) and LiBr (6.5 g, 76 mmols) in 110 mL of dimethylformamide were heated at 140 °C for 6 h. The reaction mixture was extracted with pentane (3 x 75 mL), dried over anhydrous MgSO4 and concentrated under reduced pressure. Purification by flash chromatography afforded 392 mg of diene 23g (15% yield) and bromide 27 (10% yield). 1 23g H NMR (300 MHz, CDCl3) δ 4.92 (1H, d, J = 2.0 Hz), 4.64 (1H, d, J = 2.0 Hz), 2.26 (2H, m), 1.63 (2H, m); 13C NMR (75.5 MHz, CDCl3) δ 149.6 (s), 107.8 (t), 35.2 (t), 26.7 (t). 1 H NMR (300 MHz, CDCl3) δ 4.77 (1H, s), 4.62 (1H, s), 3.72 (1H, dd, J = 6.1 Hz, J = 10.7 Hz), 3.54 (1H, dd, J = 7.9, J = 10.7 Hz), 2.37 (1H, m), 2.24 (1H, m), 2.05 (1H, m), 1.94 (1H, m), 1.72-1.63 (2H, m), 1.53-1.33 27 (3H, m); 13C NMR (75.5 MHz, CDCl3) δ 149.3 (s), 106.9 (t), 46.8 (t), 45.2 (d), 35.1 (t), 31.5 (t), 28.3 (t), 24.2 (t). 2-Trimethylsilyloxybuta-1,3-diene (23h). Silyl enol ether 23h was prepared according to the procedure described by Jung and McCombs.175 Methyl vinyl ketone (25.0 g, 0.357 mol) in DMF (25 mL) and chlorotrimethylsilane (43.4 g, 0.4 mol) in DMF (25 mL) were added over 30 minutes to a stirred solution of Et3N (40.5 g, 0.40 mol) in DMF (200 mL) in a threenecked round-bottomed flask under nitrogen at 80-90 °C. The reaction was stirred overnight and then cooled to rt, filtered and transferred to a separatory funnel containing pentane (300 mL). This mixture was washed with cold saturated NaHCO3 (1 23h 129 Experimental section L). The pentane layer was separated, and the aqueous layer extracted with pentane (2 x 300 mL). The pentane extracts were combined, washed with cold water (200 mL) and dried over Na2SO4 and filtered. The pentane and other volatiles were removed by fractional distillation heating at 70 °C and then the mixture was distillated over reduced pressure, affording 22.6 g of the diene 23h (43% yield). 1 H NMR (300 MHz, CDCl3) δ 6.20 (1H, dd, J = 10.4 Hz, J = 16.9 Hz), 5.47 (1H, dd, J = 1.4 Hz, J = 16.9 Hz), 5.08 (1H, d, J = 10.4 Hz), 4.36 (1H, s), 4.35 (1H, s), 0.23 (9H, s). 5.2.7. Enantioselective Diels-Alder alkenoyl-2-oxazolidones reaction catalyzed by with 3- Cu(II)- hydroxyoxazoline complexes 5.2.7.1. General procedure for the catalytic enantioselective Diels-Alder reaction Cu(OTf)2 (9.0 mg, 0.025 mmol) contained in a dry Schlenk tube was heated at 90 °C under vacuum for 1 h. After this time ligand 5 (7.5 mg, 0.025 mmol) and CH2Cl2 (1.5 mL) were added under nitrogen atmosphere and the mixture was stirred for 1 h at rt. Then, dienophile 22 (0.25 mmol) was added and the mixture stirred for 0.5 h. After this time, the solution was placed in a bath at the proper temperature and the diene 23 (1.8 mmol) was added. After completion of the reaction (TLC), the Diels-Alder product 28 was isolated directly by flash chromatography on silica gel eluting with hexane-EtOAc mixtures. 5.2.7.2. Characterization of the products See Tabla 6(page 61) and Tabla 7 (page 62) for yields, enantiomeric excesses and isomeric ratios. 3-((1S,2S,4S)-Bicyclo[2.2.1]hept-5-enecarbonyl)oxazolidin-2-one (28aa). N O 28aa O O [α]D25 -149.0 (c 1.0, CHCl3) (for a 90% ee and endo:exo 98:2 mixture). 1H NMR (300 MHz, CDCl3) δ 6.23 (1H, dd, J = 3.1 Hz, J = 5.6 Hz), 5.86 (1H, dd, J = 2.8 Hz, J = 5.6 Hz), 4.40 (2H, m), 3.95 (3H, m), 3.30 (1H, br s), 2.93 (1H, br s), 1.94 (1H, ddd, J = 3.7 Hz, J = 9.2 Hz, J = 12.1 Hz), 1.50-1.38 (3H, m). Exo-28aa (significant peaks, taken from an endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 6.16 (1H, br s), 2.98 (1H, br s). 130 Experimental section Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 7% isopropanol-93% hexane, 1.0 mL/min). Minor exo tR = 16.5 min, major endo-(-) tR = 17.6 min, major exo tR = 21.5 min, minor endo-(+) tR = 22.5 min. Endo/exo = 98:2. Ee(endo) = 90%. Ee(exo) = 52%. 3-((1S,2S,3R,4R)-3-Methylbicyclo[2.2.1]hept-5-enecarbonyl)oxazolidin-2-one (28ba). 1 H NMR (300 MHz, CDCl3) δ 6.37 (1H, dd, J = 3.1 Hz, J = 5.7 Hz), 5.78 (1H, dd, J = 2.8 Hz, J = 5.7 Hz), 4.40 (2H, t, J = 8.1 Hz), 4.043.91 (2H, m), 3.53 (1H, dd, J = 3.4 Hz, J = 4.4 Hz), 3.28 (1H, br s), 2.53 (1H, br s), 2.09 (1H, m), 1.71 (1H, d, J = 8.7 Hz), 1.46 (1H, qd, J = 1.7 Hz, J = 8.7 Hz), 1.13 (3H, d, J = 7.1 Hz). 28ba Exo-28ba (significant peaks, taken from an endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 6.32 (1H, dd, J = 3.1 Hz, J = 5.6 Hz), 6.16 (1H, dd, J = 2.9 Hz, J = 5.6 Hz), 2.89 (1H, br s), 2.73 (1H, br s), 0.85 (1H, d, J = 6.8 Hz). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 5% isopropanol-95% hexane, 1.0 mL/min). Major exo tR = 12.5 min, minor exo tR = 13.5 min, major endo tR = 13.5 min, minor endo tR = 15.6 min. Due to the overlap between minor exo and major endo peaks, enantiomeric excess was calculated by an equation system using HPLC data and NMR integrations. Endo/exo = 85:15. Ee(endo) = 74%. Ee(exo) = 53%. 3-((1S,2S,4S)-2-Methylbicyclo[2.2.1]hept-5-enecarbonyl)oxazolidin-2-one (28ca). Endo-28ca (significant peaks, taken from an endo/exo mixture) 1H NMR (300 MHz, CDCl3) δ 6.36 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.78 (1H, dd, J = 2.8 Hz, J = 5.7 Hz), 3.27 (1H, br s), 2.52 (1H, br s). Exo-28ca (significant peaks, taken from an endo/exo mixture) 1H NMR (300 MHz, CDCl3) δ 6.26 (1H, dd, J = 3.0 Hz, J = 5.7 Hz), 3.34 (1H, br s), 3.08 (1H, br s). 28ca Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 5% isopropanol-95% hexane, 1.0 mL/min). Major endo tR = 20.3 min, minor endo tR = 20.4 min, minor exo tR = 21.6 min, major exo tR = 26.6 min. Endo/exo = 81:19. Ee(endo) = nd. Ee(exo) = 58%. 3-((1S,2R,3R,4R)-3-Phenylbicyclo[2.2.1]hept-5-enecarbonyl)oxazolidin-2-one (28da). 1 Ph N O O H NMR (300 MHz, CDCl3) δ 7.28-7.15 (5H, m), 6.52 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.91 (1H, dd, J = 2.8 Hz, J = 5.6 Hz), 4.37 (2H, m), 4.19 (1H, dd, J = 3.4 Hz, J = 5.2 Hz), 3.98 (2H, m), 3.46 (1H, br O 28da 131 Experimental section s), 3.34 (1H, dd, J = 1.7 Hz, J = 5.2 Hz), 2.99 (1H, br s), 1.95 (1H, d, J = 8.8 Hz), 1.57 (1H, dd, J = 1.8 Hz, J = 8.8 Hz). Exo-28da(significant peaks, taken from an endo/exo mixture) 1H NMR (300 MHz, CDCl3) δ 6.46 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 6.03 (1H, dd, J = 2.8 Hz, J = 5.6 Hz), 3.71 (1H, dd, J = 1.0 Hz, J = 5.3 Hz), 3.11 (1H, br s), 3.09 (1H, br s), 1.83 (1H, d, J = 8.6 Hz), 1.47 (1H, dd, J = 1.5 Hz, J = 8.6 Hz). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 5% isopropanol-95% hexane, 1.0 mL/min). Minor exo tR = 18.9 min, minor endo tR = 22.0 min, major exo tR = 33.1 min, major endo tR = 49.4 min. Endo/exo = 78:22. Ee(endo) = 76%. Ee(exo) = 58%. 3-((1S,2S,4S)-Bicyclo[2.2.2]oct-5-enecarbonyl)oxazolidin-2-one (28ab). 1 H NMR (300 MHz, CDCl3) δ 6.34 (1H, t, J = 7.4 Hz), 6.15 (1H, t, J = 6.9 Hz), 4.38 (2H, m), 3.97 (2H, t, J = 8.3 Hz), 3.75 (1H, ddd, J = 1.9 N O Hz, J = 5.7 Hz, J = 9.9 Hz), 2.82 (1H, br s), 2.61 (1H, br s), 1.85 (1H, O O ddd, J = 2.7 Hz, J = 9.9 Hz, J = 12.6 Hz), 1.73-1.49 (3H, m), 1.26 28ab (2H, m); 13C NMR (75.5 MHz, CDCl3) δ 175.5 (s), 153.2 (s), 134.9 (d), 131.2 (d), 61.8 (t), 42.9 (d), 41.9 (t), 32.7 (t), 30.1 (d), 29.4 (d), 25.6 (t), 23.9 (t). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 5% isopropanol-95% hexane, 1.0 mL/min). Minor exo tR = 16.0 min, major exo tR = 16.9 min, minor endo tR = 19.3 min, major endo tR = 21.9 min. Endo/exo = 98:2. Ee(endo) = 77%. Ee(exo) = 78%. (S)-3-(3,4-Dimethylcyclohex-3-enecarbonyl)oxazolidin-2-one (28ac). 1 H NMR (300 MHz, CDCl3) δ 4.40 (2H, t, J = 8.1 Hz), 4.02 (2H, t, J = 8.0 Hz), 3.69 (1H, dddd, J = 2.8 Hz, J = 5.2 Hz, J = 10.5 Hz, J = 11.6 Hz), 2.28-1.88 (6H, m), 1.61 (6H, s). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 5% isopropanol-95% hexane, 1.0 mL/min). Major tR = 29.2 min, minor tR = 33.2 min. Ee = 55%. 28ac (S)-3-(4-Methylcyclohex-3-enecarbonyl)oxazolidin-2-one (28ad). 40 132 [α]D25 -26.5 (c 1.2, CHCl3) (for a 79% ee and (1,4-)/(1,3-) isomers 98.5:1.5 mixture); 1H NMR (300 MHz, CDCl3) δ 5.38 (1H, br s), 4.40 (2H, t, J = 8.1 Hz), 4.01 (2H, t, J = 8.1 Hz), 3.64 (1H, m), 2.50-2.05 (3H, m), 2.00-1.90 (2H, m), 1.70 (1H, m), 1.65 (s, 3H); 13C NMR (75.5 MHz, CDCl3) δ 176.6 (s), 153.1 Experimental section (s), 133.6 (s), 119.0 (d), 61.8 (t), 42.7 (t), 38.1 (d), 29.4 (t), 27.3 (t), 25.8 (t), 23.3 (q). Assay of enantiomeric excess: Chiral GC analysis (Supelco Beta-DEX 120, 30 m x 0.25 mm x 0.25 μm, Tcolumn = 100 °C (12 min) to 200 °C at 3 °C/min). Major 1,3- tR = 54.8 min, minor 1,3- tR = 55.1 min, major 1,4- tR = 55.5 min, minor 1,4- tR = 55.9 min. 1,4-/1,3- = 97:3. Ee(1,4-) = 72%. Ee (1,3-) = 47%. 3-((1S,2R)-2-Methylcyclohex-3-enecarbonyl)oxazolidin-2-one (28ae). cis-(1,2)-28ae 1H NMR (300 MHz, CDCl3) δ 5.64 (br s, 2H), 4.41 (2H, t, J = 8.3 Hz), 4.02 (2H, m), 3.84 (1H, ddd, J = 2.8 Hz, J = 5.5 Hz, J = 11.8 Hz), 2.73 (1H, m), 2.09-2.03 (2H, m), 1.91-180 (1H, m), 1.73-1.63 (1H, m), 0.87 (3H, d, J = 7.2 Hz). 28ae Significant peaks, taken from the isomers mixture: trans-(1,2)-28ae δ 0.95 ppm (3H, d, J = 7.0 Hz); cis-(1,3)-28ae δ 1.00 ppm (3H, d, J = 7.0 Hz); trans-(1,3)-28ae δ 1.06 ppm (3H, d, J = 7.1 Hz). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 5% isopropanol-95% hexane, 1.0 mL/min). Minor trans-1,2- tR = 11.8 min, major trans-1,2tR = 12.7 min, minor cis-1,2- tR = 14.2 min, major cis-1,2- tR = 21.9 min. Cis-1,2-/ trans1,2-/Cis-1,3-/ trans-1,3- = 69:19:11:2. Ee(cis-1,2-) = 44%. Ee (trans-1,2-) = 32%. 3-((1S,2R)-2-Phenylcyclohex-3-enecarbonyl)oxazolidin-2-one (28af). cis-(1,2)-28af 1H NMR (300 MHz, CDCl3) δ 7.24 (3H, m), 7.12 (2H, m), 5.97 (1H, ddd, J = 2.1 Hz, J = 4.6 Hz, J = 9.3 Hz), 5.72 (1H, m), N O 4.36 (2H, t, J = 8.1 Hz), 4.11 (1H, ddd, J = 3.0 Hz, J = 6.2 Hz, J = Ph O O 11.7 Hz), 4.02 (1H, m), 3.90 (1H, m), 3.60 (1H, td, J = 7.5 Hz, J = 28af 11.0 Hz), 2.32 (1H, m), 2.20 (1H, m), 1.99 (1H, m), 1.72 (1H, m); 13 C NMR (75.5 MHz, CDCl3) δ 174.3 (s), 141.1 (s), 129.4 (d), 128.8 (s), 128.5 (d), 128.2 (s), 128.1 (d), 127.5 (d), 62.5 (t), 44.1 (t), 43.02 (d), 42.3 (d), 24.5 (t), 20.0 (t). trans-(1,2)-29 (significant peaks, taken from an endo/exo mixture) 1H NMR (300 MHz, CDCl3) δ 5.85 (1H, ddd, J = 2.4 Hz, J = 4.9 Hz, J = 9.9 Hz), 5.64 (1H, ddd, J = 2.1 Hz, J = 4.2 Hz, J = 10.0 Hz). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 5% isopropanol-95% hexane, 1.0 mL/min). tR = 18.5 min, tR = 22.9 min. Ee = 0%. (S)-3-(1,2,3,4,5,6,7,8-Octahydronaphthalene-2-carbonyl)oxazolidin-2-one (28ag). mp 78-82 °C; [α]D25 -19.3 (c 0.91, CHCl3) (46% ee mixture); 1 H NMR (300 MHz, CDCl3) δ 4.40 (2H, dd, J = 8.1 Hz), 4.02 28ag 133 Experimental section (2H, t, J = 8.1 Hz), 3.71 (1H, dddd, J = 2.7 Hz, J = 5.1 Hz, J = 11.4 Hz), 2.23-1.85 (9H, m), 1.71-1.45 (5H, m); 13C NMR (75.5 MHz, CDCl3) δ 177.1 (s), 153.5 (s), 128.0 (s), 126.5 (s), 62.3 (t), 43.2 (t), 39.3 (d), 32.6 (t), 30.5 (t), 30.3 (t), 30.2 (t), 26.4 (t), 23.4 (t); MS (EI) m/z (%): 249 (M+, 27), 162 (100), 134 (67), 119 (14), 91 (34); HRMS: 249.1364, C14H19NO3 required 249.1365. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 10% isopropanol-90% hexane, 0.5 mL/min). Major-(+) tR = 29.5 min, minor-(+) tR = 33.5 min. Ee = 46 %. (S)-3-(4-(Trimethylsilyloxy)cyclohex-3-enecarbonyl)oxazolidin-2-one (28ah). [α]D25 +11.9 (c 1.67, CHCl3) (50% ee mixture); 1H NMR (300 MHz, CDCl3) δ 4.84 (1H, m), 4.41 (2H, t, J = 8.0 Hz), N O 4.02 (2H, t, J = 8.1 Hz), 3.65 (1H, m), 2.32-2.16 (3H, m), O O 2.01-1.93 (2H, m), 1.76 (1H, m), 0.18 (s, 9H); 13C NMR 28ah (75.5 MHz, CDCl3) δ 176.2 (s), 153.1 (s), 149.8 (s), 101.9 (d), 61.9 (t), 42.7 (t), 38.0 (d), 29.1 (t), 26.0 (t), 25.9 (t), 0.2 (q); MS (EI) m/z (%): 283 (M+, 13), 196 (45), 168 (100), 127 (14), 86 (11), 73 (71); HRMS: 283.1245, C13H21NO4Si required 283.1240. TMSO Assay of enantiomeric excess: Chiral HPLC analysis (Two bonded Chiralpak AD-H columns, 10% isopropanol-90% hexane, 0.33 mL/min). Major-(+) tR = 45.2 min, minor(-) tR = 46.9 min. Ee = 50%. 5.3. Enantioselective Cu(II)-BOX catalyzed cycloaddition reactions of 2-alkenoylpyridine N-oxides 5.3.1. Synthesis and characterization of 2- alkenoylpyridine N-oxides 2-Acetylpyridine N-oxide was prepared by oxidation of 2-acetylpyridine with the H2O2-urea complex and phthalic anhydride.197 General procedure for the synthesis of 2-alkenoyl pyridine N-oxides 31a-j. 1 M Potassium hydroxide (1.6 mL, 1.6 mmol) was added to a solution of 2acetylpyridine N-oxide (1.08 g, 7.9 mmol) and aldehyde (15.8 mmol) in methanol (40 mL) at 0 °C. The reaction mixture was stirred at this temperature until completion (TLC). The mixture was neutralized with 2 M HCl (0.78 mL, 1.6 mmol), diluted with 134 Experimental section water (80 mL) and extracted with CH2Cl2 (3 × 60 mL). The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. The product was isolated by flash chromatography. (E)-2-Cinnamoylpyridine N-oxide (31a). Yield: 64%; mp 104-107 °C; 1H NMR (300 MHz, CDCl3) δ 8.24 (1H, dd, J = 1.3 Hz, J = 6.2 Hz), 7.80 (1H, d, J = 15.9 Hz), 7.70 (1H, d, J = 15.9 Hz), 7.65 (3H, m), 7.38 (5H, m); 13C NMR (75.5 MHz, CDCl3) δ 186.2 (s), 147.0 (s), 144.1 (d), 140.3 (d), 134.4 (s), 130.7 31a (d), 128.7 (d), 128.7 (d), 127.6 (d), 127.1 (d), 125.7 (d), 124.1 (d); MS (EI) m/z (%): 225 (M+, 1.2), 209 (18), 196 (22), 209 (24), 180 (29), 168 (30), 106 (53), 78 (100); HRMS: 225.0793, C14H11NO2 required 225.0790. (E)-2-(3-(4-Methoxyphenyl)acryloyl)pyridine N-oxide (31b). Yield: 50%; mp 113-116 °C; 1H NMR (300 MHz, CDCl3) δ 8.24 (1H, m), 7.76 (1H, d, J = 15.8 Hz), 7.66 (1H, m), 7.60 (1H, d, J = 15.8 Hz), 7.60 (2H, d, J = 8.8 Hz), 7.36 (2H, m), 6.89 (2H, d, J = 8.8 Hz), 3.85 (3H, s); 13C NMR 31b (75.5 MHz, CDCl3) δ 186.1 (s), 161.9 (s), 147.4 (s), 144.5 (d), 140.4 (d), 130.7 (d), 127.46 (d), 127.3 (d), 127.2 (d), 125.8 (d), 122.0 (d), 114.3 (d), 55.4 (q); MS (EI) m/z (%): 255 (M+, 5.6), 239 (50), 210 (34), 161 (31), 149 (13), 132 (20), 121 (59), 106 (33), 78 (100); HRMS: 255.0889, C15H13NO3 required 255.0895. (E)-2-(3-(4-Bromophenyl)acryloyl)pyridine N-oxide (31c). Yield: 52%; mp 165-166 °C; 1H NMR (300 MHz, CDCl3) δ 8.22 (1H, dd, J = 1.4 Hz, J = 5.8 Hz), 7.7-7.66 (3H, m), 7.52-7.45 (4H, m), 7.41-7.34 (2H, m); 13C NMR (75.5 MHz, CDCl3) δ 186.0 (s), 147.0 (s), 142.5 (d), 140.4 (d), 31c 133.5 (s), 132.1 (d), 130.1 (d), 127.8 (d), 127.3 (d), 125.9 (d), 125.0 (s), 124.7 (d); MS (EI) m/z (%): 303 (M+, 0.9), 287 (9), 258 (12), 183 (14), 167 (14), 106 (79), 78 (100); HRMS: 302.9890, C14H10BrNO2 required 302.9895. (E)-2-(3-(4-Nitrophenyl)acryloyl)pyridine N-oxide (31d). O O2N O N Yield: 91%; mp 192-194 °C (decomposition); 1H NMR (300 MHz, CDCl3) δ 8.16 (1H, dd, J = 2.9 Hz, J = 4.6 Hz), 8.12 (2H, d, J = 8.8 Hz), 7.68 (1H, d, J = 8.8 Hz), 7.65 (2H, m), 7.61 (1H, m), 7.43 (2H, m); 13C NMR (75.5 31d 135 Experimental section MHz, CDCl3) δ 185.6 (s), 148.6 (s), 146.6 (s), 140.8 (d), 140.5 (d), 140.1 (d), 129.2 (d), 128.3 (d), 127.9 (d), 127.6 (d), 126.0 (d), 124.1 (d); MS (EI) m/z (%): 270 (M+, 0.1), 254 (7), 225 (8), 180 (3), 167 (6), 106 (19), 103 (13), 89 (8), 78 (100); HRMS: 270.0635, C14H10N2O4 required 270.0641. (E)-2-(3-(Pyridin-2-yl)acryloyl)pyridine N-oxide (31e). Yield: 30%; mp 125-127 °C (decomposition); 1H NMR (300 MHz, CDCl3) δ 8.64 (1H, d, J = 4.3 Hz), 8.23 (1H, dd, J = 1.4 Hz, J = 6.2 Hz), 8.00 (1H, d, J = 15.7 Hz), 7.75 (1H, d, J = 15.7 Hz), 7.71 (dt, 1H, J = 1.8 Hz, J = 7.7 Hz), 7.64 (1H, dd, J = 2.5 31e Hz, J = 7.5 Hz), 7.54 (1H, d, J = 7.8 Hz), 7.42-7.24 (2H, m), 7.26 (1H, ddd, J = 1.1 Hz, J = 4.8 Hz, J = 7.4 Hz); 13C NMR (75.5 MHz, CDCl3) δ 186.8 (s), 153.1 (s), 150.2 (d), 147.1 (s), 142.3 (d), 140.3 (d), 136.7 (d), 127.9 (d), 127.8 (d), 127.0 (d), 125.6 (d), 124.5 (d), 124.4 (d); MS (FAB) m/z (%): 227 (M++1, 100), 211 (13); HRMS: 227.0818, C13H11N2O2 required 227.0821. (E)-2-(3-(Furan-3-yl)acryloyl)pyridine N-oxide (31f). Yield: 58%; mp 109-111 °C; 1H NMR (300 MHz, CDCl3) δ 8.23 (1H, m), 7.70 (1H, d, J = 15.8 Hz), 7.65 (1H, m), 7.42 (td, 1H, J = 1.8 Hz, J = 4.7 Hz), 7.41 (1H, d, J = 15.7 Hz), 7.41-7.32 (1H, m), 6.67 (1H, d, J = 1.9 Hz); 13C NMR (75.5 MHz, CDCl3) δ 31f 186.0 (s), 147.1 (s), 146.0 (d), 144.5 (d), 140.4 (d), 134.6 (d), 127.6 (d), 127.2 (d), 126.0 (d), 124.2 (d), 123.2 (s), 107.7 (d); MS (FAB) m/z (%): 216 (M++1, 100), 200 (4); HRMS: 216.0666, C12H10NO3 required 216.0661. (E)-2-(3-(Furan-2-yl)acryloyl)pyridine N-oxide (31g). Yield: 69%; mp 107-110 °C; 1H NMR (300 MHz, CDCl3) δ 8.22 (1H, m), 7.63 (1H, m), 7.53 (2H, s), 7.49 (1H, d, J = 1.7 Hz), 7.39-7.29 (2H, m), 6.73 (1H, dd, J = 0.6 Hz, J = 3.5 Hz), 6.47 (1H, dd, J = 1.8 Hz, J = 3.5 Hz); 13C NMR (75.5 MHz, CDCl3) δ 31g 185.8 (s), 151.4 (s), 147.2 (s), 145.4 (d), 140.4 (d), 130.2 (d), 127.6 (d), 127.1 (d), 125.8 (d), 121.8 (d), 116.8 (d), 112.7 (d); MS (FAB) m/z (%): 216 (M++1, 100), 201 (5); HRMS: 216.0663, C12H10NO3 required 216.0661. 136 Experimental section (E)-2-(3-(Thiophen-3-yl)acryloyl)pyridine N-oxide (31h). Yield: 53%; mp 105-107 °C; 1H NMR (300 MHz, CDCl3) δ 8.24 (1H, m), 7.77 (1H, d, J = 15.8 Hz), 7.66 (1H, m), 7.61 (1H, dd, J = 1.2 Hz, J = 2.9 Hz), 7.50 (1H, d, J = 15.7 Hz), 7.40-7.35 (3H, m), 7.32 (1H, ddd, J = 0.5 Hz, J = 2.9 Hz, J = 5.1 Hz); 13C NMR 31h (75.5 MHz, CDCl3) δ 186.4 (s), 147.2 (s), 140.4 (d), 137.9 (s), 137.9 (d), 129.9 (d), 127.6 (d), 127.2 (d), 126.9 (d), 126.1 (d), 125.6 (d), 124.0 (d); MS (FAB) m/z (%): 232 (M++1, 100), 217 (5), 154 (13), 137 (12); HRMS: 232.0429, C12H10NO2S required 232.0432. (E)-2-(3-(Thiophen-2-yl)acryloyl)pyridine N-oxide (31i). Yield: 47%; mp 100-103 °C; 1H NMR (300 MHz, CDCl3) δ 8.24 (1H, m), 7.91 (1H, d, J = 15.5 Hz), 7.66 (1H, m), 7.50 (1H, d, J = 15.5 Hz), 7.42 (1H, d, J = 5.1 Hz), 7.40-7.32 (3H, m), 7.06 (1H, dd, J = 3.7 Hz, J = 5.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 185.6 31i (s), 147.1 (s), 140.4 (d), 140.1 (s), 136.8 (d), 132.5 (d), 129.7 (d), 128.3, 127.6 (d), 127.2 (d), 126.0 (d), 123.0 (d); MS (FAB) + m/z (%): 232 (M +1, 100), 217 (5), 204 (2), 149 (3); HRMS: 232.0432, C12H10NO2S required 232.0432. 2-((2E,4E)-5-Phenylpenta-2,4-dienoyl)pyridine N-oxide (31j). Yield: 58%; mp 108-111 °C; 1H NMR (300 MHz, CDCl3) δ 8.24 (1H, m), 7.65 (1H, m), 7.58 (1H, ddd, J = 3.9 Hz, J = Ph 6.4 Hz, J = 15.1 Hz), 7.47 (2H, dd, J = 1.6 Hz, J = 7.9 Hz), 7.40-7.31 (5H, m), 7.25 (1H, d, J = 15.2 Hz), 7.01 (1H, s), 31j 7.00 (1H, d, J = 2.6 Hz); 13C NMR (75.5 MHz, CDCl3) δ 186.1 (s), 147.2 (s), 144.6 (d), 142.7 (d), 140.4 (d), 135.9 (s), 129.3 (d), 128.8 (d), 127.6 (d), 127.4 (d), 127.2 (d), 126.9 (d), 125.9 (d); MS (FAB) m/z (%): 252 (M++1, 100), 236 (5), 222 (5) 154 (9); HRMS: 252.1023, C16H14NO3 required 252.1025. O O N (E)-2-(3-tert-Butylacryloyl)pyridine N-oxide (31k). A saturated solution of KOH in 1:1 THF-EtOH (0.1 mL) was added to a solution of 2-acetylpyridine N-oxide (400 mg, 2.9 mmol) and trimethylacetaldehyde (0.63 mL, 5.8 mmol) in THF (5 mL). The reaction mixture was refluxed overnight, and then 31k diluted with EtOAc (80 mL), washed with aqueous saturated NH4Cl solution (2 × 10 mL) and brine (10 mL), and dried over Na2SO4. After filtration 137 Experimental section and evaporation of the solvent under reduced pressure, flash chromatography (EtOAc) afforded 364 mg (62% yield) of 31k. mp 85-88 °C; 1H NMR (300 MHz, CDCl3) δ 8.34 (1H, dd, J = 2.7 Hz, J = 5.0 Hz), 7.61 (1H, dd, J = 3.7 Hz, J = 6.2 Hz), 7.44 (2H, dd, J = 2.7 Hz, J = 6.2 Hz), 7.07 (1H, d, J = 15.8 Hz), 6.90 (1H, d, J = 15.8 Hz), 1.13 (9H, s); 13C NMR (75.5 MHz, CDCl3) δ 187.3 (s), 159.3 (d), 147.1 (s), 140.1 (d), 127.3 (d), 126.6 (d), 125.5 (d), 123.2 (d), 34.0 (s), 28.4 (q); MS (FAB) m/z (%): 206 (M++1, 100), 190 (4), 174 (4), 148 (8); HRMS: 206.1179, C12H16NO2 required 206.1181. (E)-2-(4-Ethoxy-4-oxobut-2-enoyl)pyridine N-oxide (31l). 2-Acetylpyridine N-oxide (805 mg, 5.9 mmol) and 2.3 mL of ethyl glyoxalate (50% in toluene, 11.76 mmol) were dissolved in 5 mL of toluene. After this, 4 Å molecular sieves (100 mg) and 25 mg of p-toluenesulfonic acid monohydrate 31l were added and the mixture stirred at 90 °C until no more evolution of the reaction was observed (TLC). Then, the reaction mixture was filtered through a short pad of silica gel eluting with EtOAc. The solvent was removed under reduced pressure and the reaction mixture was purified by flash chromatography (hexane-EtOAc 40:60) to give 232 mg of enone 31l (18% yield). mp 79-81 °C; 1H NMR (300 MHz, CDCl3) δ 8.17 (1H, dd, J = 0.7 Hz, J = 6.4 Hz), 7.82 (1H, d, J = 15.6 Hz), 7.60 (1H, dd, J = 2.3 Hz, J = 7.5 Hz), 7.39 (1H, ddd, J = 2.3 Hz, J = 6.4 Hz, J = 7.6 Hz), 7.31 (1H, dt, J = 1.2 Hz, J = 7.7 Hz), 6.76 (1H, d, J = 15.6 Hz), 4.19 (2H, q, J = 7.1 Hz), 1.25 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 186.0 (s), 165.3 (s), 146.0 (s), 140.2 (d), 137.7 (d), 130.5 (d), 128.6 (d), 127.3 (d), 125.8 (d), 61.2 (t), 14.1 (q); MS (EI) m/z (%): 221 (M+, 2.4), 160 (3), 176 (15), 148 (33), 132 (11), 119 (3), 106 (19), 92 (8), 78 (100); HRMS: 221.0684, C11H11NO4 required 221.0688. 5.3.2. Enantioselective catalyzed Diels-Alder reaction 5.3.2.1. General procedure for the catalytic enantioselective Diels-Alder reaction Cu(OTf)2 (9.0 mg, 0.025 mmol) contained in a dry Schlenk tube was heated at 90 °C under vacuum for 1 h. After this time, ligand 33a (7.5 mg, 0.025 mmol) and CH2Cl2 (1.5 mL) were added under nitrogen atmosphere and the mixture was stirred for 1 h at rt. Then, dienophile 31 (0.25 mmol) was added and the mixture stirred for 0.5 h. After this time, the solution was placed in a bath at 0 °C and the diene 23 (1.8 138 Experimental section mmol) was added. After completion of the reaction (TLC), the Diels-Alder product 34 was isolated directly by flash chromatography on silica gel. 5.3.2.2. Characterization of the products See Tabla 10 (page 75) and Tabla 11 (page 77) for yields, enantiomeric excesses and isomeric ratios. 2-((1R,2S,3S,4S)-3-Phenylbicyclo[2.2.1]hept-5-enecarbonyl)pyridine N-oxide (34aa). mp 96-100 °C; [α]D25 +262.4 (c 0.94, CHCl3) (for a recrystallyzed mixture with 99.7% ee, 97% de); 1H NMR (300 MHz, CDCl3) δ 8.15 (1H, dd, J = 1.8 Hz, J = 5.7 Hz), 7.41 (1H, dd, J = 2.6 Hz, J = 7.3 Hz), 7.30 (6H, m), 7.17 (1H, m), 6.46 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 34aa 5.87 (1H, dd, J = 2.7 Hz, J = 5.6 Hz), 4.50 (1H, dd, J = 3.4 Hz, J = 5.0 Hz), 3.38 (1H, br s), 3.35 (1H, d, 5.4 Hz), 3.09 (1H, br s), 1.88 (1H, d, J = 8.6 Hz), 1.56 (1H, ddd, J = 1.7 Hz, J = 3.5 Hz, J = 8.6 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.5 (s), 147.3 (s), 143.8 (s), 140.2 (d), 139.8 (d), 133.0 (d), 128.3 (d), 127.5 (d), 127.3 (d), 126.2 (d), 125.9 (d), 125.4 (d), 58.1 (d), 49.0 (d), 47.5 (t), 46.3 (d), 46.3 (d); MS (EI) m/z (%): 291 (M+, 0.8), 274 (11), 226 (100), 209 (24), 185 (40), 180 (30), 168 (30), 106 (42), 78 (59); HRMS: 291.1263, C19H17NO2 required 291.1259. Exo-34aa (significant peaks, taken from an endo/exo mixture): 1H NMR (CDCl3, 300 MHz) δ 7.53 (1H, dd, J = 2.6, 7.4 Hz), 6.41 (1H, dd, J = 3.2, 5.6 Hz), 6.05 (1H, dd, J = 2.8, 5.6 Hz), 3.22 (2H, m). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Minor exo tR = 10.3 min, major exo tR = 11.6 min, major endo-(+) tR = 12.4 min, minor endo-(-) tR = 13.8 min. Endo/exo = 98:2. Ee(endo) = 96%. Ee(exo) = 81%. 2-((1R,2S,3S,4S)-3-(4-Methoxyphenyl)bicyclo[2.2.1]hept-5-enecarbonyl)pyridine N-oxide (34ba). [α]D25 + 177.5 (c 0.96, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.17 (1H, d, J = 5.8 Hz), 7.41 (1H, dd, J = 2.7 Hz, J = 7.1 Hz), 7.30 (m, 4H), 6.84 (2H, d, J = 8.8 Hz), 6.45 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.87 (1H, dd, J = 2.6 Hz, J = 5.6 Hz), 4.44 (1H, dd, J = 3.4 Hz, J = 4.9 Hz), 3.79 (3H, s), 3.36 (1H, br s), 3.28 (1H, d, J = 3.9 Hz), 3.04 OMe (1H, br s), 1.87 (1H, d, J = 8.5 Hz), 1.55 (1H, dd, J = 1.7 Hz, J = 8.6 34ba Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.7 (s), 157.7 (s), 147.4 (s), 140.2 (d), 139.8 (d), 135.8 (s), 132.8 (d), 128.4 (d), 127.3 (d), 126.1 (d), 125.6 (d), 113.7 (d), 58.2 (d), 55.2 (q), 49.3 (d), 47.4 (t), 46.2 (d), 45.6 (d); MS O O N 139 Experimental section (EI) m/z (%): 321 (M+, 0.9), 304 (9), 239 (67), 210 (30), 185 (25), 161 (22), 132 (23), 121 (58), 106 (47), 78 (100), 66 (24); HRMS: 321.1377, C20H19NO3 required 321.1365. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Minor exo tR = 24.8 min, major exo tR = 30.1 min, major endo-(+) tR = 35.4 min, minor endo-(-) tR = 36.2 min. Endo/exo = 98:2. Ee(endo) = 95%. Ee(exo) = 65%. 2-((1R,2S,3S,4S)-3-(4-Bromophenyl)bicyclo[2.2.1]hept-5-enecarbonyl)pyridine N-oxide (34ca). mp 98-99 °C; [α]D25 +202.9 (c 0.89, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.09 (1H, dd, J = 1.8 Hz, J = 5.7 Hz), 7.32 (3H, m), 7.24 (2H, m), 7.14 (2H, dd, J = 1.9 Hz, J = 8.7 Hz), 6.37 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.78 (1H, dd, J = 2.7 Hz, J = 5.6 Hz), 4.35 (1H, dd, J = 3.4 Hz, J = 5.0 Hz), 3.27 (1H, s), 3.22 (1H, d, J = 3.8 Hz), 2.98 (1H, s), 1.74 (1H, d, J = 8.6 Hz), 1.48 (1H, ddd, J = 1.6 Hz, J = 3.4 Hz, J = 34ca 8.6 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.2 (s), 147.1 (s), 142.9 (s), 140.2 (d), 139.7 (d), 133.0 (d), 131.3 (d), 129.3 (d), 127.5 (d), 126.1 (d), 125.7 (d), 119.6 (s), 58.2 (d), 48.8 (d), 47.5 (t), 46.3 (d), 45.6 (d); MS (EI) m/z (%): 369 (M+, 0.9), 304 (16), 288 (24), 287 (67), 258 (49), 185 (21), 106 (62), 102 (45), 78 (100); HRMS: 369.0367, C19H16BrNO2 required 369.0364. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Minor exo tR = 17.9 min, major exo tR = 23.2 min, major endo-(+) tR = 32.7 min, minor endo-(-) tR = 37.8 min. Endo/exo = 97:3. Ee(endo) = 94%. Ee(exo) = 95%. 2-((1R,2S,3S,4S)-3-(4-Nitrophenyl)bicyclo[2.2.1]hept-5-enecarbonyl)pyridine N-oxide (34da). [α]D25 +215.0 (c 0.99, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.15 (1H, t, J = 8.2 Hz), 8.12 (2H, d, J = 8.8 Hz), 7.48 (2H, d, J = 8.7 Hz), 7.37 (3H, m), 6.44 (1H, dd, J = 3.2 Hz, J = 5.5 Hz), 5.85 (1H, dd, J = 2.7 Hz, J = 5.6 Hz), 5.85 (1H, dd, J = 2.7 Hz, J = 5.6 Hz), 4.46 (1H, dd, J = 3.5 Hz, J = 5.0 Hz), 3.43 (1H, d, J = 4.7 Hz), 3.36 (1H, br s), NO2 3.12 (1H, br s), 1.79 (1H, d, J = 8.7 Hz), 1.59 (1H, dd, J = 1.7 Hz, J 34da = 8.8 Hz); 13C NMR (75.5 MHz, CDCl3) δ 197.7 (s), 151.9 (s), 146.96 (s), 146.1 (s), 140.3 (d), 139.5 (d), 133.2 (d), 128.3 (d), 127.6 (d), 126.2 (d), 125.8 (d), 123.5 (d), 58.4 (d), 48.4 (d), 47.6 (t), 46.3 (d), 45.9 (d); MS (EI) m/z (%): 336 (M+, 0.1), 319 (15), 271 (45), 254 (100), 225 (40), 179 (15); HRMS: 336.1092, C19H16N2O4 required 336.1110. O 140 O N Experimental section Exo-34da (significant peaks, taken from an endo/exo mixture): 1H NMR (CDCl3, 300 MHz) δ 8.05 (1H, d, J = 8.8 Hz), 6.38 (1H, dd, J = 3.1, 5.8 Hz), 5.99 (1H, dd, J = 2.8, 5.4 Hz). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Exo (both enantiomers) tR = 20.6 min, major endo-(+) tR = 24.9 min, minor endo-(-) tR = 35.4 min. Endo/exo = 96:4. Ee(endo) = 96%. Ee(exo) = nd. 2-((1R,2S,3S,4S)-3-(Pyridin-2-yl)bicyclo[2.2.1]hept-5-enecarbonyl)pyridine N-oxide (34ea). [α]D25 +220.2 (c 0.76, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.46 (1H, ddd, J = 0.8Hz, J = 1.8Hz, J = 4.9 Hz), 8.10 (1H, dd, J = 1.1Hz, J = 6.1 Hz), 7.52 (1H, dt, J = 1.9Hz, J = 7.7 Hz), 7.34 (1H, dd, J = 2.5Hz, J = 7.2 Hz), 7.25 (3H, m), 7.03 (1H, ddd, J = 1.1Hz, J = 4.9Hz, J = 7.4 Hz), 6.40 (1H, dd, J = 3.2Hz, J = 5.6 Hz), 5.87 (1H, dd, J = 2.7Hz, J = 5.6 Hz), 4.74 (1H, dd, J = 3.5Hz, J = 4.9 Hz), 3.36 (1H, dd, 34ea J = 1.6Hz, J = 4.9 Hz), 3.31 (1H, s), 3.11 (1H, s), 2.03 (1H, d, J = 8.4 Hz), 1.44 (1H, qd, J = 1.8Hz, J = 8.4 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.7 (s), 162.6 (s), 148.9 (d), 147.5 (s), 140.2 (d), 139.5 (d), 136.2 (d), 133.7 (d), 127.2 (d), 126.0 (d), 125.4 (d), 123.2 (d), 121.1 (d), 57.0 (d), 49.5 (d), 48.6 (d), 47.3 (t), 46.2 (d); MS (FAB) m/z (%): 293 (50, M++1), 281 (36), 266 (19), 227 (21), 221 (35), 207 (50), 147 (100); HRMS: 293.1287, C18H17N2O2 required 293.1290. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Minor endo-(-) tR = 14.5 min, major endo-(+) tR = 15.7 min. Endo/exo >99:1. Ee(endo) = 90%. Ee(exo) = nd. 2-((1R,2S,3S,4S)-3-(Furan-3-yl)bicyclo[2.2.1]hept-5-enecarbonyl)pyridine N-oxide (34fa). [α]D25 +253.2 (c 0.62, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.18 (1H, ddd, J = 0.8 Hz, J = 1.4 Hz, J = 6.0 Hz), 7.34 (5H, m), 6.39 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 6.36 (1H, t, J = 1.4 Hz), 5.83 (1H, dd, J = 2.8 Hz, J = 5.7 Hz), 4.34 (1H, dd, J = 3.4 Hz, J = 4.9 Hz), 3.30 (1H, br O s), 3.08 (1H, dd, J = 1.8 Hz, J = 4.8 Hz), 2.93 (1H, s), 1.74 (1H, d, J = 34fa 8.6 Hz), 1.51 (1H, qd, J = 1.8 Hz, J = 8.6 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.4 (s), 147.3 (s), 142.8 (d), 140.2 (d), 139.1 (d), 138.6 (d), 132.4 (d), 128.2 (s), 127.3 (d), 126.0 (d), 125.6 (d), 110.6 (d), 57.5 (d), 49.3 (d), 47.7 (t), 46.1 (d), 37.9 (d); MS (FAB) m/z (%): 282 (100, M++1), 217 (92), 112 (10); HRMS: 282.1138, C17H16NO3 required 282.1130. O O N 141 Experimental section Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Minor endo-(-) tR = 18.7 min, major endo-(+) tR = 20.1 min. Endo/exo >99:1. Ee(endo) = 95%. Ee(exo) = nd. 2-((1R,2S,3S,4S)-3-(Furan-2-yl)bicyclo[2.2.1]hept-5-enecarbonyl)pyridine N-oxide (34ga). [α]D25 +253.8 (c 0.68, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.18 (1H, dd, J = 1.6 Hz, J = 6.0 Hz), 7.42 (1H, dd, J = 2.4 Hz, J = 7.6 Hz), 7.32 (3H, m), 6.37 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 6.26 (1H, dd, J = 1.9 Hz, J = 3.2 Hz), 6.13 (1H, td, J = 0.9 Hz, J = 3.2 Hz), 5.85 (1H, dd, J = 2.8 Hz, J = 5.6 Hz), 4.56 (1H, dd, J = 3.4 Hz, J = 5.0 Hz), 3.37 (1H, 34ga s), 3.25 (1H, d, J = 4.9 Hz), 3.06 (1H, d, J = 1.6 Hz), 1.84 (1H, d, J = 8.6 Hz), 1.51 (1H, qd, J = 1.8 Hz, J = 8.6 Hz); 13C NMR (75.5 MHz, CDCl3) δ 197.7 (s), 157.4 (s), 147.2 (s), 141.1 (d), 140.3 (d), 138.8 (d), 133.0 (d), 127.5 (d), 126.4 (d), 125.5 (d), 110.1 (d), 105.1 (d), 56.3 (d), 48.8 (d), 47.9 (t), 45.9 (d), 40.7 (d); MS (FAB) m/z (%): 282 (75, M++1), 265(13), 217 (100), 109 (41); HRMS: 282.1125, C17H16NO3 required 282.1130. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 3% isopropanol-97% hexane, 0.5 mL/min). Minor endo-(-) tR = 96.3 min, major endo-(+) tR = 102.0 min. Endo/exo >99:1. Ee(endo) = 97%. Ee(exo) = nd. 2-((1R,2S,3S,4S)-3-(Thiophen-3-yl)bicyclo[2.2.1]hept-5-enecarbonyl)pyridine N-oxide (34ha). [α]D25 +238.0 (c 0.70, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.17 (1H, dd, J = 1.7 Hz, J = 5.7 Hz), 7.34 (3H, m), 7.24 (1H, dd, J = 3.0 Hz, J = 4.9 Hz), 7.09 (1H, td, J = 1.2 Hz, J = 2.8 Hz), 7.05 (1H, dd, J = 1.3 Hz, J = 4.9 Hz), 6.41 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.84 (1H, dd, J = 2.8 Hz, J = 5.7 Hz), 4.47 (1H, dd, J = 3.4 Hz, J = 4.9 Hz), 3.31 34ha (2H, m), 3.06 (1H, d, J = 1.6 Hz), 1.79 (1H, d, J = 8.6 Hz), 1.53 (1H, qd, J = 1.8 Hz, J = 8.6 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.4 (s), 147.3 (s), 145.0 (s), 140.2 (d), 139.3 (d), 132.6 (d), 128.0 (d), 127.3 (d), 126.1 (d), 125.6 (d), 125.4 (d), 119.6 (d), 57.9 (d), 49.4 (d), 47.7 (t), 46.3 (d), 42.4 (d); MS (FAB) m/z (%): 298 (95, M++1), 232 (100), 110 (40); HRMS: 298.0902, C17H16NO2S required 298.0902. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Major endo-(+) tR = 20.9 min, minor endo-(-) tR = 23.1 min. Endo/exo >99:1. Ee(endo) = 97%. Ee(exo) = nd. 142 Experimental section 2-((1R,2S,3S,4S)-3-(Thiophen-2-yl)bicyclo[2.2.1]hept-5-enecarbonyl)pyridine N-oxide (34ia). [α]D25 +214.5 (c 0.71, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.17 (1H, dd, J = 1.6 Hz, J = 6.0 Hz), 7.41 (1H, dd, J = 2.4 Hz, J = 7.5 Hz), 7.32 (2H, m), 7.11 (1H, dd, J = 1.4 Hz, J = 4.9 Hz), 6.93 (1H, td, J = 1.2 Hz, J = 3.1 Hz), 6.90 (1H, dd, J = 3.5 Hz, J = 4.9 Hz), 6.41 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.86 (1H, dd, J = 2.8 Hz, J = 5.6 Hz), 4.54 (1H, dd, J = 3.4 Hz, J = 4.9 Hz), 3.48 (1H, d, J = 4.9 Hz), 3.39 (1H, s), 34ia 3.05 (1H, br s), 1.92 (1H, d, J = 8.8 Hz), 1.58 (1H, qd, J = 1.8 Hz, J = 13 8.8 Hz); C NMR (75.5 MHz, CDCl3) δ 197.8 (s), 148.3 (s), 147.1 (s), 140.3 (d), 139.0 (d), 132.9 (d), 127.5 (d), 126.9 (d), 126.3 (d), 125.5 (d), 123.8 (d), 123.1 (d), 59.5 (d), 51.2 (d), 47.9 (t), 46.2 (d), 42.5 (d); MS (FAB) m/z (%): 298 (29, M++1), 232 (100), 133 (14), 110 (18); HRMS: 298.0902, C17H16NO2S required 298.0902. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Minor endo-(-) tR = 16.9 min, major endo-(+) tR = 20.7 min. Endo/exo >99:1. Ee(endo) = 94%. Ee(exo) = nd. 2-((1R,2S,3S,4S)-3-tert-Butylbicyclo[2.2.1]hept-5-enecarbonyl)pyridine N-oxide (endo-34ka) and 2-((1S,2S,3S,4R)-3-tert-butylbicyclo[2.2.1]hept-5-enecarbonyl) pyridine N-oxide (exo-34ka). [α]D25 -251.1 (c 0.78, CHCl3); 1H NMR (400 MHz, CDCl3), δ 8.19 (1H, dd, J = 0.9 Hz, J = 6.5 Hz), 7.37 (1H, dd, J = 2.4 Hz, J = 7.4 Hz), 7.33 (1H, d, J = 6.0 Hz), 7.31 (1H, dd, J = 1.3 Hz, J = 7.4 Hz), 6.43 (1H, dd, J = 3.3 Hz, J = 5.6 Hz), 5.75 (1H, dd, J = 2.7 Hz, J = 5.6 Hz), 4.14 (1H, dd, J = 3.2 Hz, J = 6.0 Hz), 3.17 (1H, br s), 2.77 (1H, dd, J exo-34ka = 1.4 Hz, J = 2.9 Hz), 1.87 (1H, dd, J = 1.7 Hz, J = 6.0 Hz), 1.66 (1H, d, J = 8.5 Hz), 1.35 (1H, ddd, J = 1.6 Hz, J = 3.3 Hz, J = 8.5 Hz), 0.94 13 (9H, s); C NMR (75.5 MHz, CDCl3) δ 199.5 (s), 147.6 (s), 141.3 (d), 140.3 (d), 131.4 (d), 127.2 (d), 126.2 (d), 125.7 (d), 53.3 (d), 51.6 (d), 47.8 (t), 46.7 (d), 44.5 (d), 32.5 (s), 28.9 (q); MS (FAB) m/z (%): 272 (M++1, 35), 206 (100); HRMS: 272.1646, C17H22NO2 required 272.1651. O O N mp 110-115 °C; [α]D25 +8.0 (c 0.55, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.23 (1H, d, J = 5.8 Hz), 7.53 (1H, dd, J = 2.7 Hz, J = 7.2 Hz), 7.36 (2H, m), 6.19 (1H, dd, J = 3.1 Hz, J = 5.5 Hz), 6.16 (1H, dd, J = 2.9 Hz, J = 5.4 Hz), 3.38 (1H, dd, J = 0.9 Hz, J = 6.3 Hz), 3.02 (1H, s), 2.98 (1H, s), 2.64 (1H, dd, J = 3.0 Hz, J = 6.3 Hz), 1.73 (1H, d, J = exo-34ka 8.3 Hz), 1.35 (1H, dd, J = 1.3 Hz, J = 8.3 Hz), 0.80 (9H, s); 13C NMR (75.5 MHz, CDCl3) δ 200.9 (s), 147.7 (s), 140.5 (d), 137.0 (d), 134.9 (d), 127.4 (d), 126.8 O O N 143 Experimental section (d), 126.1 (d), 55.3 (d), 52.0 (d), 49.1 (d), 48.1 (t), 45.3 (d), 32.6 (s), 29.3 (q); MS (FAB) m/z (%): 272 (M++1, 66), 206 (100); HRMS: 272.1649, C17H22NO2 required 272.1651. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 2% isopropanol-98% hexane, 1.0 mL/min). Minor exo-(-) tR = 20.2 min, major exo-(+) tR = 23.4 min, minor endo-(+) tR = 25.7 min, major endo-(-) tR = 29.1 min. Endo/exo = 78:22. Ee(endo) = 93%. Ee(exo) = 70%. 2-((1R,2S,3S,4S)-3-(Ethoxycarbonyl)bicyclo[2.2.1]hept-5-enecarbonyl)pyridine N-oxide (34la). [α]D25 +180.0 (c 1.59, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.17 (1H, dd, J = 1.3 Hz, J = 6.2 Hz), 7.41 (1H, dd, J = 2.2 Hz, J = 7.7 Hz), 7.31 (2H, m), 6.25 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.86 (1H, dd, J = 2.8 Hz, J = 5.6 Hz), 4.59 (1H, dd, J = 3.5 Hz, J = 4.8 Hz), 4.14 (2H, q, J = 7.1 Hz), 3.36 (1H, br s), 3.13 (1H, br s), 2.77 (1H, dd, J = 1.6 Hz, 34la J = 4.8 Hz), 1.79 (1H, d, J = 8.7 Hz), 1.45 (1H, qd, J = 1.7 Hz, J = 8.7 Hz), 1.23 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 197.3 (s), 174.3 (s), 146.9 (s), 140.2 (d), 137.9 (d), 134.3 (d), 127.6 (d), 126.5 (d), 125.4 (d), 60.7 (t), 55.4 (d), 47.6 (t), 47.5 (d), 46.1 (d), 45.5 (d), 14.1 (q); MS (FAB) m/z (%): 287 (M+, 84), 221 (81), 165 (13), 151 (23), 134 (27), 125 (84), 109 (100); HRMS: 288.1231, C16H18NO4 required 288.1236. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Minor exo tR = 12.8 min, minor endo-(-) tR = 15.2 min, major endo-(+) tR = 22.4 min, major exo tR = 44.0 min. Endo/exo = 97:3. Ee(endo) = 98%. Ee(exo) = 89%. 2-((1R,2S,3S,4S)-3-Phenylbicyclo[2.2.2]oct-5-enecarbonyl)pyridine N-oxide (34ab). mp 134-137 °C; [α]D25 +126.4 (c 0.72, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.07 (1H, dd, J = 2.5 Hz, J = 5.2 Hz), 7.40 (1H, dd, J = 3.1 Hz, J = 6.8 Hz), 7.27 (6H, m), 7.17 (1H, m), 6.55 (1H, t, J = 7.4 Hz), 6.10 (1H, t, J = 7.2 Hz), 4.43 (1H, d, J = 6.8 Hz), 3.35 (1H, d, J = 6.8 34ab Hz), 3.02 (1H, br s), 2.62 (1H, br s), 1.88 (1H, m), 1.69 (1H, m), 1.42 (1H, tt, J = 3.7 Hz, J = 12.1 Hz), 1.03 (1H, m); 13C NMR (75.5 MHz, CDCl3) δ 198.7 (s), 146.8 (s), 141.9 (s), 140.1 (d), 137.0 (d), 130.6 (d), 128.2 (d), 128.0 (d), 127.2 (d), 126.6 (d), 126.1 (d), 125.5 (d), 52.7 (d), 44.8 (d), 37.3 (d), 32.7 (d), 26.3 (t), 18.2 (t); MS (EI) m/z (%): 305 (M+, 0.2), 288 (10), 210 (52), 209 (50), 180 (19), 103 (12), 78 (77); HRMS: 305.1396, C20H19NO2 required 305.1416. Exo-34ab (significant peaks, taken from an endo/exo mixture): 1H NMR (CDCl3, 300 MHz) δ 8.23 (1H, d, J = 6.5 Hz), 7.83 (1H, dd, J = 2.0, 8.2 Hz), 6.03 (2H, m), 4.64 (1H, s), 4.24 (1H, dd, J = 2.5, 6.8 Hz). 144 Experimental section Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Minor endo-(-) tR = 10.9 min, major endo-(+) tR = 12.7 min, exo (both enantiomers) tR = 20.9 min. Chiral HPLC analysis (Chiralcel OD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Major exo tR = 10.5 min, endo (both enantiomers) tR = 15.9 min, minor exo tR = 19.6 min. Endo/exo = 85:15. Ee(endo) = 96%. Ee(exo) = 47%. 2-((1S,6S)-3,4-Dimethyl-6-phenylcyclohex-3-enecarbonyl)pyridine N-oxide (34ac). mp 116-119 °C; [α]D25 -221.4 (c 0.79, CHCl3); 1H NMR (300 Me MHz, CDCl3) δ 8.04 (1H, d, J = 6.4 Hz), 7.13 (7H, m), 6.89 (1H, dt, J = 1.0 Hz, J = 7.8 Hz), 6.22 (1H, dd, J = 1.8 Hz, J = 7.9 Hz), Me Ph 4.02 (1H, ddd, J = 6.8 Hz, J = 9.0 Hz, J = 11.5 Hz), 3.02 (1H, dt, 34ac J = 5.6 Hz, J = 11.3 Hz), 2.55 (2H, d, J = 6.6 Hz), 2.29 (1H, m), 2.16 (1H, dd, J = 4.7 Hz, J = 17.0 Hz), 1.71 (3H, s), 1.64 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 203.5 (s), 148.1 (s), 143.8 (s), 139.2 (d), 128.3 (d), 127.8 (d), 126.6 (d), 126.5 (d), 125.3 (d), 124.9 (d), 124.7 (s), 124.3 (s), 51.8 (d), 45.5 (d), 40.3 (t), 35.1 (t), 18.6 (q), 18.6 (q); MS (EI) m/z (%): 307 (M+, 0.6), 290 (100), 272 (28), 186 (26), 169 (20), 106 (43), 78 (99); HRMS: 307.1564, C20H21NO2 required 307.1572. O O N Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Minor (+) tR = 16.2 min, major (-) tR = 22.9 min. Ee = 93%. 2-((1S,6S)-4-Methyl-6-phenylcyclohex-3-enecarbonyl)pyridine N-oxide (34ad). mp 111-115 °C; [α]D25 -221. (c 0.79, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.04 (1H, d, J = 6.5 Hz), 7.14 (7H, m), 6.90 (1H, dt, J = 0.9 Hz, J = 7.8 Hz), 6.23 (1H, dd, J = 2.0 Hz, J = 7.9 Hz), Me Ph 5.54 (1H, br s), 3.98 (1H, dt, J = 5.3 Hz, J = 10.7 Hz), 3.06 (1H, 34ad dt, J = 5.9 Hz, J = 11.0 Hz), 2.58 (2H, m), 2.20 (2H, m), 1.69 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 203.6 (s), 148.0 (s), 143.8 (s), 139.2 (d), 132.6 (s), 128.3 (d), 127.8 (d), 126.6 (d), 126.5 (d), 125.3 (d), 125.0 (d), 119.9 (d), 50.9 (d), 45.1 (d), 38.5 (t), 29.2 (t), 23.1 (q); MS (EI) m/z (%): 293 (M+, 1.0), 276 (100), 258 (26), 172 (12), 155(14), 106 (41), 91 (30), 78 (81); HRMS: 293.1416, C19H19NO2 required 293.1416. O O N Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 5% isopropanol-95% hexane, 1.0 mL/min). Major 1,3- tR = 26.5 min, minor 1,3- tR = 27.8 min, minor 1,4-(+) tR = 37.8 min, major 1,4-(-) tR = 45.3 min. 1,4-/1,3- = 96:4. Ee(1,4-) = 92%. Ee (1,3-) = 46%. 145 Experimental section 2-((1S,2S,6S)-2-Methyl-6-phenylcyclohex-3-enecarbonyl)pyridine N-oxide (34ae). [α]D25 -32.6 (c 0.64, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.11 (1H, d, J = 6.5 Hz), 7.19 (7H, m), 7.07 (1H, d, J = 7.6 Hz), 6.76 (1H, dd, J = 2.1 Hz, J = 7.9 Hz), 5.78 (1H, m), 5.71 (1H, m), 4.51 (1H, dd, J = 5.2 Hz, J = 11.5 Hz), 3.28 (1H, dt, J = 5.8 Hz, J = 11.0 Hz), 3.01 (1H, 34ae dd, J = 5.6 Hz, J = 11.0 Hz), 2.37 (1H, td, J = 5.2 Hz, J = 18.0 Hz), 2.23 (1H, ddd, J = 2.1 Hz, J = 10.6 Hz, J = 18.1 Hz), 1.05 (3H, d, J = 7.2 Hz); 13C NMR (75.5 MHz, CDCl3) δ 200.2 (s), 147.7 (s), 144.8 (s), 139.9 (d), 132.0 (d), 128.2 (d), 128.1 (d), 127.2 (d), 126.3 (d), 126.1 (d), 125.4 (d), 124.5 (d), 53.9 (d), 38.3 (d), 34.4 (t), 31.7 (d), 17.3 (q); MS (EI) m/z (%): 293 (M+, 0.1), 276 (80), 258 (10), 172 (25), 106 (48), 78 (100); HRMS: 291.1415, C19H19NO2 required 293.1416. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Major cis-(1,2)-(-) tR = 12.9 min, minor cis-(1,2)(+) tR = 15.7 min. cis-(1,2)/trans-(1,2) > 99:1. Ee(cis-(1,2)) = 94%. 2-((1R,2S,6S)-2,6-Diphenylcyclohex-3-enecarbonyl)pyridine N-oxide (34af). mp 60-62 °C; [α]D25 +270.5 (c 0.90, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.11 (1H, d, J = 6.5 Hz), 7.17 (11H, m), 6.94 (1H, dt, J = 1.0 Hz, J = 7.9 Hz), 6.29 (1H, dd, J = 2.1 Hz, J = 7.9 Hz), 6.08 (1H, ddd, J = 2.3 Hz, J = 4.3 Hz, J = 10.0 Hz), 5.88 (1H, ddd, J = 2.1 Hz, J 34af = 4.2 Hz, J = 9.9 Hz), 4.59 (1H, dd, J = 6.0 Hz, J = 10.3 Hz), 4.31 (1H, s), 3.43 (1H, dt, J = 5.9 Hz, J = 9.8 Hz), 2.61 (1H, td, J = 5.1 Hz, J = 18.3 Hz), 2.38 (1H, m); 13C NMR (75.5 MHz, CDCl3) δ 199.3 (s), 147.7 (s), 144.4 (s), 141.0 (s), 139.5 (d), 130.0 (d), 128.2 (d), 128.2 (d), 128.0 (d), 127.7 (d), 127.1 (d), 126.7 (d), 126.6 (d), 126.2 (d), 125.4 (d), 55.5 (d), 42.9 (d), 38.8 (d), 32.7 (t); MS (EI) m/z (%): 355 (M+, 0.7), 338 (100), 320 (28), 234 (52), 152 814), 115 (42), 106 (69), 78 (47); HRMS: 355.1583, C24H21NO2 required 355.1572. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Minor cis-(-) tR = 18.8 min, major cis-(+) tR = 19.9 min. cis-(1,2)/trans-(1,2)>99:1. Ee(cis-(1,2)) = 94%. 2-((1S,2S)-4-Oxo-2-phenylcyclohexanecarbonyl)pyridine N-oxide (34ah). O O Ph 34ah 146 O N In this case, the Diels-Alder reaction mixture was filtered through a short pad of silica gel (2 cm), eluted with 50 mL of EtOAc and concentrated under reduced pressure. The DielsAlder adduct was then hydrolyzed by dissolving the mixture in a 0.01 M solution of citric acid in H2O/MeOH 1:1 (2 mL) to give 34ah. Experimental section [α]D25 -194.2 (c 0.61, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.10 (1H, d, J = 6.4Hz), 7.22 (1H, ddd, J = 2.1Hz, J = 6.6Hz, J = 7.6Hz), 7.13 (5H, m), 6.98 (1H, t, J = 7.7Hz), 6.46 (1H, dd, J = 2.0Hz, J = 7.9Hz), 4.30 (1H, dt, J = 2.6Hz, J = 11.3Hz), 3.28 (1H, dt, J = 5.3Hz, J = 11.6Hz), 2.63 (5H, m), 2.15 (1H, dt, J = 5.8Hz, J = 11.9Hz); 13C NMR (75.5 MHz, CDCl3) δ 209.0 (s), 201.1 (s), 147.3 (s), 141.4 (s), 139.5 (d), 128.7 (d), 127.3 (d), 127.2 (d), 127.2 (d), 125.9 (d), 125.4 (d), 53.0 (d), 48.3 (d), 48.2 (t), 40.0 (t), 28.3 (t); MS (EI) m/z (%): 295 (M+, 0.4), 278 (40), 260 (13), 232 (15), 221 (28), 146 (18), 106 (68), 95 (26), 78 (100); HRMS: 295.1200, C18H17NO3 required 295.1208. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Minor (+) tR = 28.1 min, major (-) tR = 37.8 min. Endo/exo >99:1. Ee(endo) = 95%. 2-((1R,2S,6S)-2-Acetoxy-6-phenylcyclohex-3-enecarbonyl)pyridine N-oxide (34ai). [α]D25 +107.977 (c 0.701, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.12 (1H, d, J = 6.5 Hz), 7.24 (5H, m), 7.10 (3H, m), 6.08 (1H, ddd, J = 2.2 Hz, J = 5.1 Hz, J = 9.0 Hz), 5.90 (2H, m), 4.88 (1H, dd, J = 3.3 Hz, J = 12.2 Hz), 3.51 (1H, dt, J = 5.5 Hz, J = 11.7 Hz), 2.50 (1H, 34ai dtd, J = 1.2 Hz, J = 5.2 Hz, J = 18.7 Hz), 2.23 (1H, dd, J = 11.3 Hz, J = 18.7 Hz), 2.00 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 195.8 (s), 170.2 (s), 146.2 (s), 144.0 (s), 140.3 (d), 132.5 (d), 128.3 (d), 127.7 (d), 127.6 (d), 127.0 (d), 126.2 (d), 125.3 (d), 123.5 (d), 66.1 (d), 53.1 (d), 37.5 (d), 34.8 (t), 21.0 (q); MS (FAB) m/z (%): 338 (M++1, 73), 279 (100), 148 (14); HRMS: 338.1392, C20H20NO4 required 338.1392. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Minor endo-(-) tR = 14.2 min, major endo-(+) tR = 16.8 min, major exo tR = 27.4 min, minor exo tR = 31.8 min. Endo/exo = 99:1. Ee(endo) = 96%. Ee(exo) = 72%. 5.3.2.3. Synthetic transformations ((1R,2S,3S,4S)-3-phenylbicyclo[2.2.1]hept-5-en-2-yl)(pyridin-2-yl)methanone (35). A mixture of endo-34aa (58 mg, 0.2 mmol, endo/exo 96:4, 91% ee), indium powder (30 mg, 0.30 mmol) and saturated aqueous NH4Cl (0.5 mL) in EtOH (0.8 mL) was refluxed for 1 h. The reaction mixture was filtered through Celite® eluting with diethyl ether (30 35 mL). The organic layer was washed with brine (2 x 5 mL) and dried over anhydrous Na2SO4. Purification by column chromatography (hexane/EtOAc 95:5) gave 41 mg of endo-35 (75% yield).177,185 147 Experimental section [α]D25 +176.6 (c 0.81, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 8.67 (1H, d, J = 4.8 Hz), 8.00 (1H, d, J = 7.8 Hz), 7.81 (1H, dt, J = 1.7, 7.7 Hz), 7.44 (1H, ddd, J = 1.2, 4.8, 7.5 Hz), 7.37-7.22 (m, 4H), 7.15 (tt, J = 1.7, 6.9Hz), 6.49 (1H, dd, J = 3.2, 5.6 Hz), 5.82 (1H, dd, J = 2.8, 5.6 Hz), 4.53 (1H, dd, J = 3.4, 5.2 Hz), 3.54 (1H, s), 3.45 (1H, dd, J = 1.3, 5.0 Hz), 3.08 (1H, s), 2.06 (1H, d, J = 8.4 Hz), 1.61 (1H, dd, J = 1.7, 8.5 Hz). Exo-35 (significant peaks, taken from the endo/exo mixture) 1H NMR (CDCl3, 300 MHz) δ 8.06 (1H, d, J = 7.8 Hz), 6.09 (1H, dd, J = 2.8, 5.5 Hz), 4.20 (dd, J = 1.3, 5.3 Hz), 4.02 (1H, dd, J = 3.4, 5.3 Hz), 3.21 (1H, d, J = 1.3 Hz), 1.86 (1H, d, J = 8.5 Hz), 1.47 (dd, J = 1.5, 8.5 Hz). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 2% isopropanol-98% hexane, 1.0 mL/min). Major exo tR = 6.5 min, minor exo tR = 7.2 min, minor endo-(-) tR = 7.9 min, major endo-(+) tR = 9.5 min. Endo/exo = 96:4, Ee(endo) = 90%. 6-((1R,2S,3S,4S)-3-Phenylbicyclo[2.2.1]hept-5-enecarbonyl)picolinonitrile (36). Dimethylcarbamoyl chloride (64 μL, 0.70 mmol) was added to a solution of endo-34aa (50 mg, 0.17 mmol, 95% ee, >95% N CN de) and cyanotrimethylsilane (100 μL, 0.75 mmol) in CH2Cl2 Ph (0.8 mL) at 0 °C under nitrogen. The reaction mixture was 36 stirred for 3 days. The volatiles were removed under reduced pressure and the residue chromatographed on silica gel (hexane/EtOAc 9:1) to give 43.3 mg of endo-13 (83% yield). O [α]D25 +111.9 (c 0.87, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.20 (1H, dd, J = 1.1Hz, J = 8.0Hz), 7.99 (1H, t, J = 7.8Hz), 7.85 (1H, dd, J = 1.1Hz, J = 7.7Hz), 7.30 (4H, m), 7.19 (1H, m), 6.50 (1H, dd, J = 3.2Hz, J = 5.6Hz), 5.82 (1H, dd, J = 2.8Hz, J = 5.6Hz), 4.46 (1H, dd, J = 3.4Hz, J = 5.2Hz), 3.56 (1H, br s), 3.41 (1H, dd, J = 1.7Hz, J = 5.2Hz), 3.09 (1H, d, J = 1.5Hz), 2.10 (1H, d, J = 8.6Hz), 1.65 (1H, ddd, J = 1.6Hz, J = 3.4Hz, J = 8.6Hz); 13C NMR (75.5 MHz, CDCl3) δ 199.3 (s), 154.3 (s), 144.0 (s), 139.7 (d), 138.2 (d), 133.0 (s), 132.6 (d), 131.1 (d), 128.4 (d), 127.5 (d), 126.0 (d), 125.0 (d), 116.7 (s), 54.3 (d), 49.4 (d), 48.7 (d), 48.2 (t), 45.8 (d); MS (EI) m/z (%): 300 (M+, 0.7), 234 (100), 205 (32), 152 (8), 131 (54), 115 (13), 103 (39), 66 (37); HRMS: 300.1263, C20H16N2O required 300.1263. ((1S,2S,3S,4R)-3-Phenylbicyclo[2.2.1]heptan-2-yl)(pyridin-2-yl)methanone (37). 37 148 A solution of compound 34aa (69.5 mg, 0.24 mmol) in EtOH (4.5 mL) and 5% Pd/C (15.5 mg) were stirred under hydrogen at atmospheric pressure for 3 h. After this time, the mixture was filtered through a short pad of silica gel to remove the catalyst. The solvent was removed under reduced pressure to give 66.4 Experimental section mg (100% yield) of compound 37, which was used without need of further purification. mp 54-57 °C; [α]D25 +62.9 (c 1.33, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.63 (1H, ddd, J = 0.7 Hz, J = 1.5 Hz, J = 4.7 Hz), 8.03 (1H, td, J = 0.9 Hz, J = 7.9 Hz), 7.78 (1H, dt, J = 1.7 Hz, J = 7.7 Hz), 7.39 (1H, ddd, J = 1.2 Hz, J = 4.8 Hz, J = 7.6 Hz), 7.24 (4H, m), 7.10 (1H, m), 4.34 (1H, ddd, J = 1.5 Hz, J = 4.0 Hz, J = 5.7 Hz), 3.53 (1H, dd, J = 1.0 Hz, J = 5.9 Hz), 2.92 (1H, br s), 2.52 (1H, d, J = 3.9 Hz), 2.04 (1H, d, J = 9.8 Hz), 1.58 (2H, m), 1.44 (1H, ddd, J = 1.5 Hz, J = 3.1 Hz, J = 9.8 Hz), 1.31 (1H, dtt, J = 1.6 Hz, J = 4.5 Hz, J = 11.9 Hz), 1.18 (1H, m); 13C NMR (75.5 MHz, CDCl3) δ 201.8 (s), 153.7 (s), 148.8 (d), 146.2 (s), 136.7 (d), 128.2 (d), 126.9 (d), 126.8 (d), 125.5 (d), 122.0 (d), 58.2 (d), 46.9 (d), 43.2 (d), 42.3 (d), 39.3 (t), 30.1 (t), 23.7 (t); MS (EI) m/z (%): 277 (M+, 100), 249 (23), 78 (19); HRMS: 277.1471, C19H19NO required 277.1467. (1S,2S,3R,4R)-3-Phenylbicyclo[2.2.1]heptan-2-ol (38). A solution of mCPBA (77.4 mg, 0.315 mmol) in CH2Cl2 (1.5 mL) was added to a solution of compound 37 (35 mg, 0.126 mmol) in CH2Cl2 (1.5 mL). Reaction was stirred at rt for 3 h and an additional amount of mCPBA (77.4 mg, 0.315 mmol) was added and stirring was 38 continued for 4 h. Then, an additional amount of mCPBA (77.4 mg, 0.315 mmol) was added. After stirring for additional 10 h, MeOH (1.5 mL) was added and the reaction was stirred for 24 h. After this time, the reaction mixture was diluted with EtOAc (40 mL), washed with 5% aqueous NaOH (2 x 10 mL) and brine (10 mL). The organic phase was dried over MgSO4, concentrated under reduced pressure and purified by flash chromatography (hexane-EtOAc 90:10) to give 13.0 mg of alcohol 38 (55% yield). mp 42-45 °C; [α]D25 +22.4 (c 0.65, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.30 (4H, m), 7.19 (1H, m), 4.18 (1H, dt, J = 1.4 Hz, J = 4.3 Hz), 2.41 (1H, d, J = 3.9 Hz), 2.35 (2H, m), 1.96 (1H, m), 1.72 (3H, m), 1.50 (2H, m), 1.37 (1H, ddd, J = 1.7 Hz, J = 4.2 Hz, J = 10.3 Hz); 13C NMR (75.5 MHz, CDCl3) δ 145.4 (s), 128.4 (d), 126.7 (d), 125.8 (d), 81.9 (d), 56.3 (d), 42.9 (d), 42.8 (d), 35.7 (t), 31.1 (t), 19.5 (t); MS (EI) m/z (%): 188 (M+, 61), 170 (33), 131 (13), 129 (21), 117 (57), 104 (21), 91 (100), 79 (22); HRMS: 188.1203, C13H16O required 188.1201. (1R,2S,3S,4S)-Methyl 3-picolinoylbicyclo[2.2.1]heptane-2-carboxylate (39). 39 RuCl3 (2.1 mg, 0.01 mmol) and NaIO4 (427 mg, 2 mmol) were added to a mixture of compound 37 (58.6 mg, 0.20 mmol), CCl4 (2 mL), acetonitrile (2 mL) and water (3 mL). Reaction was stirred at 40 °C until consumption of starting material was observed (TLC). Then, the reaction mixture was filtered through a short pad of Celite®, diluted with water (10 mL) and extracted with 149 Experimental section EtOAc (3 x 10 mL). The organic phase was washed with brine (10 mL), dried over MgSO4 and concentrated under reduced pressure. The concentrated was dissolved in 4 mL of CH2Cl2 and 4 mL of MeOH. 2 M trimethylsilyldiazomethane in diethyl ether (400 μL) was added to the solution and the reaction mixture was stirred for 1 h. After this time, the reaction mixture was diluted in EtOAc (40 mL), washed with brine (10 mL), dried over MgSO4 and the solvent removed under reduced pressure. Purification by flash chromatography (hexane-EtOAc 80:20) gave 15.0 mg of ester 39 (30% yield) and 22.9 mg of starting material 37 (61% conversion). 1 H NMR (300 MHz, CDCl3) δ 8.70 (1H, ddd, J = 0.9 Hz, J = 1.7 Hz, J = 4.7 Hz), 8.04 (1H, m), 7.83 (1H, dt, J = 1.7 Hz, J = 7.7 Hz), 7.46 (1H, ddd, J = 1.3 Hz, J = 4.7 Hz, J = 7.6 Hz), 4.51 (1H, ddd, J = 1.7 Hz, J = 4.2 Hz, J = 5.7 Hz), 3.64 (3H, s), 3.13 (1H, dd, J = 1.3 Hz, J = 5.5 Hz), 2.93 (1H, dt, J = 1.2 Hz, J = 4.2 Hz), 2.65 (1H, d, J = 4.3 Hz), 1.83 (1H, dd, J = 1.7 Hz, J = 9.9 Hz), 1.58 (1H, tt, J = 4.5 Hz, J = 12.3 Hz), 1.38 (2H, m), 1.26 (1H, dqd, J = 1.8 Hz, J = 4.2 Hz, J = 10.6 Hz), 1.08 (1H, dddd, J = 2.3 Hz, J = 4.5 Hz, J = 9.0 Hz, J = 13.1 Hz). 2-((1S,6S)-6-(Ethoxycarbonyl)cyclohex-3-enecarbonyl)pyridine N-oxide (34lj). O O N Adduct 34lj was synthesized according to general procedure for the Diels-Alder reaction, using 31l as dienophile and 1,3butadiene as diene. Yield: 71%. CO2Et mp 60-62 °C; [α]D25 -7.0 (c 1.03, CHCl3); 1H NMR (300 MHz, CDCl3) 34lj δ 8.14 (1H, dd, J = 1.5 Hz, J = 6.1 Hz), 7.57 (1H, dd, J = 2.6 Hz, J = 7.4 Hz), 7.31 (2H, m), 5.74 (1H, m), 5.65 (1H, m), 4.03 (2H, q, J = 7.1 Hz), 3.95 (dt, 1H, J = 5.2 Hz, J = 10.7 Hz), 3.06 (dt, 1H, J = 5.7 Hz, J = 11.1 Hz), 2.87 (1H, m), 2.49 (1H, m), 2.14 (2H, m), 1.16 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 201.9 (s), 174.8 (s), 147.3 (s), 140.1 (d), 127.5 (d), 127.4 (d), 125.9 (d), 125.3 (d), 124.2 (d), 60.6 (t), 46.0 (d), 42.8 (d), 28.4 (t), 28.3 (t), 14.0 (q); MS (FAB) m/z (%): 276 (M++1, 100), 207 (11), 154 (32), 136 (43); HRMS: 276.1235, C15H18NO4 required 276.1236. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Minor endo-(+) tR = 12.2 min, major endo-(-) tR = 13.3 min. Ee(endo) = 96%. Ee(exo) = 98%. (1S,2S)-Ethyl 2-picolinoylcyclohexanecarboxylate (41). A solution of compound 34lj (49.8 mg, 0.18 mmol) in acetone (3 mL) and 10% Pd/C (30 mg) were stirred under hydrogen at atmospheric pressure for 3 h. After this time, the mixture was filtered through a short pad of silica gel to remove the catalyst. The filtrate was concentrated under reduced pressure and 41 chromatographed on silica gel eluting with hexane-EtOAc (93:7 to 90:10) to give 39.9 mg (85%) of compound 41. 150 Experimental section [α]D25 +21.5 (c 0.63, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.72 (1H, d, J = 4.6 Hz), 8.04 (1H, d, J = 7.9 Hz), 7.85 (1H, dt, J = 1.6 Hz, J = 7.7 Hz), 7.47 (1H, ddd, J = 1.0 Hz, J = 4.8 Hz, J = 7.5 Hz), 4.22 (1H, m), 3.99 (2H, m), 2.89 (1H, ddd, J = 3.7 Hz, J = 11.4 Hz, J = 12.2 Hz), 2.17 (2H, ddd, J = 2.8 Hz, J = 12.9 Hz, J = 22.8 Hz), 1.83 (2H, m), 1.44 (3H, m), 1.22 (1H, m), 1.09 (1H, m); 13C NMR (75.5 MHz, CDCl3) δ 203.5 (s), 175.1 (s), 152.3 (s), 148.6 (d), 137.3 (d), 127.0 (d), 122.5 (d), 60.3 (t), 45.1 (d), 44.6 (d), 29.5 (t), 28.9 (t), 25.6 (t), 25.4 (t), 14.0 (q); MS (EI) m/z (%): 261 (M+, 30), 233 (4), 266 (19), 188 (74), 159 (20), 106 (33), 78 (100); HRMS: 261.1365, C15H19NO3 required 261.1365. (1S,2S)-2-Picolinoylcyclohexanecarboxylic acid (40). Ethyl ester 41 (12.6 mg, 0.048 mg) was dissolved in 1 mL of 1 M KOH in ethanol and 3 drops of water were added. The reaction was stirred for 1.5 h. After this time, the solution was diluted in water (20 mL), acidified with aqueous 2 M HCl until pH = 8, extracted with CH2Cl2 (10 mL), acidified until pH = 5 and 40 extracted again with CH2Cl2 (10 mL) and finally acidified until pH = 3 and extracted again with CH2Cl2 (10 mL). The combined organic phase was dried over MgSO4, concentrated under reduced pressure and purified by flash chromatography to give 9.0 mg of 40 (80 % yield). [α]D25 +16.5 (c 1.15, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.72 (1H, dd, J = 0.8 Hz, J = 4.8 Hz), 8.05 (1H, d, J = 7.9 Hz), 7.88 (1H, dt, J = 1.6 Hz, J = 7.7 Hz), 7.50 (1H, ddd, J = 1.1 Hz, J = 4.8 Hz, J = 7.5 Hz), 4.11 (1H, dt, J = 3.4 Hz, J = 11.8 Hz), 2.89 (1H, ddd, J = 3.7 Hz, J = 11.2 Hz, J = 12.4 Hz), 2.18 (2H, ddd, J = 3.2 Hz, J = 12.9 Hz, J = 19.3 Hz), 1.83 (2H, m), 1.33 (4H, m); 13C NMR (75.5 MHz, CDCl3) δ 203.1 (s), 180.8 (s), 152.0 (s), 148.6 (d), 137.3 (d), 127.1 (d), 122.7 (d), 45.1 (d), 44.1 (d), 29.4 (t), 28.9 (t), 25.6 (t), 25.4 (t); MS (EI) m/z (%): 233 (M+, 50), 215 (45), 189 (75), 170 (20), 159 (44), 137 (25), 106 (42), 79 (100); HRMS: 233.1058, C13H15NO3 required 233.1052. 5.3.3. Enantioselective catalyzed oxo-hetero-Diels-Alder reaction 5.3.3.1. General procedure for the catalytic enantioselective oxo-heteroDiels-Alder reaction Cu(OTf)2 (9.0 mg, 0.025 mmol) contained in a dry Schlenk tube was heated at 90 °C under vacuum for 1 h. After this time ligand 33a (7.5 mg, 0.025 mmol) and CH2Cl2 (1.5 mL) were added under nitrogen atmosphere and the mixture was stirred for 1 h at rt. Then, heterodiene 31 (0.25 mmol) was added and the mixture stirred for 0.5 h. After this time, the solution was placed in a bath at the reaction temperature and the 151 Experimental section alkene 42 (0.75 mmol) was added. After completion of the reaction (TLC), the dihydropyrane 43 was isolated directly by flash chromatography on silica gel. 5.3.3.2. Characterization of the products See Tabla 13 (page 85) and Tabla 14 (page 88) for yields, enantiomeric excesses and isomeric ratios. 2-((2S,4S)-2-Ethoxy-4-phenyl-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43aa). [α]D25 +98.8 (c 1.5, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.23 (1H, dd, J = 0.8 Hz, J = 6.5 Hz), 7.80 (1H, dd, J = 2.1 Hz, J = 8.2 Hz), 7.40 (1H, dd, J = 1.1 Hz, J = 2.9 Hz), 7.24 (6H, m), 7.12 (1H, ddd, J = 2.1 Hz, J = 6.6 Hz, J = 7.4 Hz), 5.24 (1H, dd, J = 1.9 Hz, J = 9.0 Hz), 4.07 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 3.93 (1H, ddd, J = 43aa 2.9 Hz, J = 6.9 Hz, J = 10.1 Hz), 3.71 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 2.40 (1H, dddd, J = 1.3 Hz, J = 1.9 Hz, J = 6.9 Hz, J = 13.2 Hz), 1.99 (1H, ddd, J = 9.0 Hz, J = 10.8 Hz, J = 13.2 Hz), 1.29 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 143.6 (s), 143.1 (s), 141.6 (s), 141.1 (d), 128.4 (d), 127.4 (d), 126.5 (d), 125.0 (d), 124.3 (d), 123.4 (d), 112.7 (d), 100.3 (d), 64.7 (t), 38.9 (d), 36.9 (t), 15.1 (q); MS (FAB) m/z (%): 298 (M++1, 100), 226 (47), 191 (23), 161 (58); HRMS: 298.1457, C18H20NO3 required 298.1443. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Exo tR = 10.3 min, Exo tR = 11.6 min, major endo-(+) tR = 16.5 min, minor endo-(-) tR = 24.7 min. Endo/exo >99.9:0.1. Ee(endo) = 96%. Ee(exo) = nd. 2-((2S,4S)-2-Ethoxy-4-(4-methoxyphenyl)-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ba). [α]D25 +87.3 (c 0.82, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.23 (1H, dd, J = 0.8 Hz, J = 6.5 Hz), 7.80 (1H, dd, J = 2.1 Hz, J = 8.2 Hz), 7.36 (1H, dd, J = 1.1 Hz, J = 2.9 Hz), 7.26 (1H, dt, J = 1.3 Hz, J = 8.4 Hz), 7.21 (2H, d, J = 8.6 Hz), 7.12 (1H, ddd, J = 2.1 Hz, J = 6.6 Hz, J = 7.4 Hz), 6.84 (2H, d, J = 8.8 Hz), 5.23 (1H, dd, J = 1.9 Hz, J = 43ba 9.0 Hz), 4.06 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 3.88 (1H, ddd, J = 2.9 Hz, J = 6.9 Hz, J = 10.5 Hz), 3.78 (3H, s), 3.70 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 2.37 (1H, dddd, J = 1.3 Hz, J = 1.8 Hz, J = 6.9 Hz, J = 13.2 Hz), 1.95 (1H, ddd, J = 9.0 Hz, J = 10.7 Hz, J = 13.2 Hz), 1.29 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 158.2 (s), 143.2 (s), 141.4 (s), 141.1 (d), 135.7 (s), 128.4 (d), 125.0 (d), 124.3 (d), 123.4 (d), 113.9 152 Experimental section (d), 113.2 (d), 100.4 (d), 64.7 (t), 55.2 (q), 38.1 (d), 37.2 (t), 15.2 (q); MS (FAB) m/z (%): 328 (M++1, 100), 313 (13), 256 (35), 191 (80); HRMS: 328.1542, C19H22NO4 required 328.1549. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Exo tR = 16.6 min, exo tR = 17.8 min, major endo-(+) tR = 24.7 min, minor endo-(-) tR = 40.4 min. Endo/exo >99.9:0.1. Ee(endo) = 94%. Ee(exo) = nd. 2-((2S,4S)-4-(4-Bromophenyl)-2-ethoxy-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ca). [α]D25 +77.9 (c 0.94, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.23 (1H, dd, J = 1.2 Hz, J = 6.6 Hz), 7.79 (1H, dd, J = 2.1 Hz, J = 8.2 Hz), 7.40 (3H, m), 7.26 (1H, dt, J = 1.3 Hz, J = 7.5 Hz), 7.17 (2H, d, J = 8.5 Hz), 7.13 (1H, m), 5.22 (1H, dd, J = 2.0 Hz, J = 8.5 Hz), 4.04 (1H, qd, J = 7.1 Hz, J 43ca = 9.5 Hz), 3.87 (1H, ddd, J = 3.0 Hz, J = 7.0 Hz, J = 10.1 Hz), 3.67 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 2.36 (1H, dddd, J = 1.2 Hz, J = 1.9 Hz, J = 7.0 Hz, J = 13.3 Hz), 1.93 (1H, ddd, J = 8.5 Hz, J = 10.2 Hz, J = 13.3 Hz), 1.26 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 143.0 (s), 142.8 (s), 141.8 (s), 141.2 (d), 131.5 (d), 129.3 (d), 125.1 (d), 124.3 (d), 123.6 (d), 120.2 (s), 112.0 (d), 100.0 (d), 64.7 (t), 38.1 (d), 36.7 (t), 15.2 (q); MS (FAB) m/z (%): 376 (M++1, 100), 304 (22), 239 (32), 176 (27), 108 (13); HRMS: 376.0546, C18H19BrNO3 required 376.0548. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Exo tR = 14.0 min, exo tR = 16.2 min, major endo-(+) tR 21.2 min, minor endo-(-) tR = 30.6 min. Endo/exo >99.5:0.5. Ee(endo) = 96%. Ee(exo) = nd. 2-((2S,4S)-2-Ethoxy-4-(4-nitrophenyl)-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43da). [α]D25 +94.2 (c 0.82, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.24 (1H, dd, J = 1.0 Hz, J = 6.4 Hz), 8.13 (2H, O O d, J = 8.8 Hz), 7.81 (1H, dd, J = 2.1 Hz, J = 8.2 Hz), 7.47 N (2H, d, J = 8.7 Hz), 7.46 (1H, m), 7.29 (1H, dt, J = 1.4 Hz, J = 7.6 Hz), 7.16 (1H, ddd, J = 2.2 Hz, J = 6.6 Hz, J = 7.5 O2N Hz), 5.25 (1H, dd, J = 2.1 Hz, J = 7.6 Hz), 3.99 (1H, qd, J 43da = 7.1 Hz, J = 9.4 Hz), 3.97 (1H, m), 3.64 (1H, qd, J = 7.1 Hz, J = 9.4 Hz), 2.41 (1H, dddd, J = 0.8 Hz, J = 2.0 Hz, J = 7.2 Hz, J = 13.2 Hz), 1.98 (1H, ddd, J = 7.7 Hz, J = 9.0 Hz, J = 13.4 Hz), 1.21 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, OEt 153 Experimental section CDCl3) δ 151.5 (s), 146.5 (s), 142.8 (s), 141.9 (s), 140.9 (d), 128.4 (d), 126.8 (d), 124.6 (d), 124.1 (d), 123.6 (d), 110.9 (d), 99.3 (d), 64.7 (t), 37.7 (d), 35.8 (t), 14.8 (q); MS (FAB) m/z (%): 343 (M++1, 100), 271 (31), 206 (13); HRMS: 343.1296, C18H19N2O5 required 343.1294. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Exo tR = 20.7 min, exo tR = 21.6 min, major endo-(+) tR = 31.2 min, minor endo-(-) tR = 56.2 min. Endo/exo >99.9:0.1. Ee(endo) = 96%. Ee(exo) = nd. 2-((2S,4S)-2-Ethoxy-4-(furan-3-yl)-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43fa). [α]D25 +71.2 (c 0.70, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.22 (1H, dd, J = 1.0 Hz, J = 6.5 Hz), 7.76 (1H, dd, J = 2.0 Hz, J O O = 8.2 Hz), 7.34 (3H, m), 7.24 (1H, dt, J = 1.2 Hz, J = 8.0 Hz), N 7.11 (1H, dt, J = 2.1 Hz, J = 7.1 Hz), 6.37 (1H, s), 5.20 (1H, dd, O J = 2.0 Hz, J = 8.7 Hz), 4.05 (1H, qd, J = 7.1 Hz, J = 9.4 Hz), 43fa 3.83 (1H, ddd, J = 2.9 Hz, J = 6.9 Hz, J = 10.0 Hz), 3.68 (1H, qd, J = 7.1 Hz, J = 9.4 Hz), 2.37 (1H, dd, J = 7.0 Hz, J = 12.9 Hz), 1.95 (1H, ddd, J = 8.7 Hz, J = 10.3 Hz, J = 13.2 Hz), 1.28 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 143.1 (s), 143.0 (d), 141.1 (d), 140.9 (s), 138.7 (d), 127.2 (s), 125.0 (d), 124.3 (d), 123.5 (d), 112.1 (d), 109.8 (d), 99.9 (d), 64.8 (t), 35.1 (t), 29.3 (d), 15.2 (q); MS (FAB) m/z (%): 288 (M++1, 100), 216 (19), 154 (26), 151 (24), 136 (20); HRMS: 288.1237, C16H18NO4 required 288.1236. OEt Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak OD-H, 10% isopropanol-90% hexane, 0.7 mL/min). Major exo tR = 23.6 min, minor exo tR = 25.9 min, major endo-(+) tR = 29.3 min, minor endo-(-) tR = 39.7 min. Endo/exo = 98.5:1.5. Ee(endo) = 96%. Ee(exo) = 50%. 2-((2S,4S)-2-Ethoxy-4-(furan-2-yl)-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ga). [α]D25 +73.1 (c 0.71, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.22 (1H, ddd, J = 0.5 Hz, J = 1.3 Hz, J = 6.5 Hz), 7.76 (1H, dd, O O J = 2.1 Hz, J = 8.2 Hz), 7.41 (1H, dd, J = 1.1 Hz, J = 3.1 Hz), N 7.31 (1H, dd, J = 0.8 Hz, J = 1.8 Hz), 7.24 (1H, ddd, J = 1.3 Hz, O J = 7.5 Hz, J = 8.2 Hz), 7.12 (1H, ddd, J = 2.2 Hz, J = 6.5 Hz, J = 43ga 7.5 Hz), 6.27 (1H, dd, J = 1.6 Hz, J = 3.2 Hz), 6.12 (1H, td, J = 0.9 Hz, J = 3.2 Hz), 5.21 (1H, dd, J = 2.1 Hz, J = 8.6 Hz), 4.03 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 4.01 (1H, m), 3.68 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 2.42 (1H, dddd, J = 1.1 Hz, J = 2.0 Hz, J = 6.9 Hz, J = 13.2 Hz), 2.10 (1H, ddd, J = 8.6 Hz, J = 10.2 Hz, J = 13.2 Hz), 1.27 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 155.9 (s), 143.1 (s), 141.5 (s), 141.3 (d), 141.1 (d), 125.0 (d), 124.4 (d), 123.6 (d), 110.1 (d), 109.46 OEt 154 Experimental section (d), 104.7 (d), 99.7 (d), 64.7 (t), 32.9 (t), 32.2 (d), 15.1 (q); MS (FAB) m/z (%): 288 (M++1, 100), 216 (23), 151 (32); HRMS: 288.1229, C16H18NO4 required 288.1236. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak OD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Exo (both) tR = 13.6 min, major endo-(+) tR = 14.4 min, minor endo-(-) tR = 18.1 min. Endo/exo = 99:1. Ee(endo) = 96%. Ee(exo) = nd. 2-((2S,4S)-2-Ethoxy-4-(thiophen-3-yl)-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ha). [α]D25 +78.6 (c 0.70, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.22 (1H, dd, J = 0.9 Hz, J = 6.5 Hz), 7.77 (1H, dd, J = 2.1 Hz, J O O = 8.2 Hz), 7.42 (1H, dd, J = 1.1 Hz, J = 3.0 Hz), 7.25 (2H, m), N 7.11 (2H, m), 7.04 (1H, dd, J = 1.3 Hz, J = 5.0 Hz), 5.21 (1H, S dd, J = 2.0 Hz, J = 8.8 Hz), 4.05 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 43ha 4.02 (1H, m), 3.69 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 2.42 (1H, dddd, J = 1.3 Hz, J = 1.9 Hz, J = 6.9 Hz, J = 13.1 Hz), 2.01 (1H, ddd, J = 8.8 Hz, J = 10.5 Hz, J = 13.2 Hz), 1.28 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 143.8 (s), 143.1 (s), 141.1 (d), 141.0 (s), 127.0 (d), 125.7 (d), 125.0 (d), 124.3 (d), 123.5 (d), 120.3 (d), 112.4 (d), 100.1 (d), 64.8 (t), 35.7 (t), 34.0 (d), 15.2 (q); MS (FAB) m/z (%): 304 (M++1, 100), 232 (23), 167 (21); HRMS: 304.1003, C16H18NO3S required 304.1007. OEt Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak OD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Exo tR = 15.0 min, major endo-(+) tR = 18.0 min, exo tR = 21.0 min, minor endo-(-) tR = 27.4 min. Endo/exo = 99:1. Ee(endo) = 95%. Ee(exo) = nd. 2-((2S,4S)-2-Ethoxy-4-(thiophen-2-yl)-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ia). [α]D25 +109.5 (c 0.70, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.23 (1H, dd, J = 0.8 Hz, J = 6.5 Hz), 7.77 (1H, dd, J = 2.1 Hz, J O O = 8.2 Hz), 7.41 (1H, dd, J = 1.1 Hz, J = 3.0 Hz), 7.25 (1H, dt, J = N 1.3 Hz, J = 7.8 Hz), 7.13 (2H, m), 6.93 (2H, m), 5.23 (1H, dd, J S = 2.0 Hz, J = 8.8 Hz), 4.23 (1H, ddd, J = 2.9 Hz, J = 6.9 Hz, J = 43ia 10.1 Hz), 4.05 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 3.69 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 2.49 (1H, dddd, J = 1.3 Hz, J = 1.9 Hz, J = 6.9 Hz, J = 13.2 Hz), 2.11 (1H, ddd, J = 8.8 Hz, J = 10.4 Hz, J = 13.2 Hz), 1.29 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 147.0 (s), 143.1 (s), 141.2 (s), 141.1 (d), 126.6 (d), 125.0 (d), 124.5 (d), 123.8 (d), 123.6 (d), 123.5 (d), 112.1 (d), 99.9 (d), 64.8 (t), 36.9 (t), OEt 155 Experimental section 33.9 (d), 15.2 (q); MS (FAB) m/z (%): 304 (M++1, 100), 260 (10), 232 (21); HRMS: 304.1000, C16H18NO3S required 304.1007. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Exo (both) tR = 12.1 min, major endo-(+) tR = 18.2 min, minor endo-(-) tR = 21.9 min. Endo/exo = 99:1. Ee(endo) = 95%. Ee(exo) = 50%. 2-((2S,4R)-2-Ethoxy-4-styryltetrahydro-2H-pyran-6-yl)pyridine N-oxide (43ja). [α]D25 +40.6 (c 0.81, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.24 (1H, dd, J = 1.3 Hz, J = 6.5 Hz), 7.77 (1H, dd, J = 2.1 Hz, J = 8.2 Hz), 7.30 (6H, m), 7.19 (1H, m), 7.11 (1H, ddd, J = 2.1 Hz, J = 6.7 Hz, J = 7.4 Hz), 6.49 (1H, d, J = 15.9 Hz), 6.27 (1H, dd, J = 7.8 Hz, J = 15.8 Hz), 5.21 (1H, dd, J = 2.1 Hz, J = 7.5 Hz), 4.03 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 3.68 (1H, qd, J = 7.1 Hz, J 43ja = 9.5 Hz), 3.45 (1H, dq, J = 3.4 Hz, J = 7.7 Hz), 2.27 (1H, ddd, J = 2.2 Hz, J = 7.0 Hz, J = 13.2 Hz), 1.89 (1H, ddd, J = 7.6 Hz, J = 8.5 Hz, J = 13.3 Hz), 1.29 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 143.2 (s), 141.1 (d), 140.7 (s), 137.2 (s), 132.0 (d), 129.7 (d), 128.4 (d), 127.1 (d), 126.1 (d), 125.1 (d), 124.2 (d), 123.4 (d), 111.9 (d), 99.5 (d), 64.6 (t), 35.8 (d), 33.9 (t), 15.2 (q); MS (FAB) m/z (%): 324 (M++1, 100), 253 (19), 188 (27), 176 (11), 154 (19), 136 (15); HRMS: 324.1599, C20H22NO3 required 324.1600. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak OD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Exo tR = 17.1 min, exo tR = 21.1 min, major endo-(+) tR = 23.6 min, minor endo-(-) tR = 28.5 min. Endo/exo >99.9:0.1. Ee(endo) = 88%. Ee(exo) = nd. 2-((2S,4S)-4-tert-Butyl-2-ethoxy-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ka). [α]D25 +100.1 (c 0.69, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.20 (1H, dd, J = 1.2 Hz, J = 6.5 Hz), 7.72 (1H, dd, J = 2.1 Hz, J = 8.2 Hz), 7.36 (1H, dd, J = 1.4 Hz, J = 2.6 Hz), 7.22 (1H, dt, J = 1.3 Hz, J = 7.5 Hz), 7.08 (1H, dt, J = 2.1 Hz, J = 6.5), 5.03 (1H, dd, J = 1.9 Hz, J = 9.7 Hz), 4.08 (1H, qd, J = 7.1 Hz, J = 9.5 Hz), 3.67 (1H, qd, 43ka J = 7.1 Hz, J = 9.5 Hz), 2.44 (1H, ddd, J = 2.7 Hz, J = 6.7 Hz, J = 11.8 Hz), 2.05 (1H, tdd, J = 1.7 Hz, J = 6.7 Hz, J = 12.8 Hz), 1.68 (1H, ddd, J = 9.8 Hz, J = 11.0, J = 12.8 Hz), 1.30 (3H, t, J = 7.1 Hz), 0.94 (9H, s); 13C NMR (75.5 MHz, CDCl3) δ 143.2 (s), 141.5 (s), 141.1 (d), 125.0 (d), 124.1 (d), 123.1 (d), 112.2 (d), 101.4 (d), 64.8 (t), 43.6 (d), 33.0 (s), 30.5 (t), 27.1 (q), 15.2 (q); MS (FAB) m/z (%): 278 (M++1, 100), 232(2), 206 (10), 171 (4), 141 (3); HRMS: 278.1749, C16H24NO3 required 278.1756. 156 Experimental section Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Minor exo tR = 11.3 min, major exo tR = 12.3 min, major endo-(+) tR = 13.2 min, minor endo-(-) tR = 15.5 min. Endo/exo = 95:5. Ee(endo) = 96%. Ee(exo) = 37%. 2-((3aR,4R,7aS)-4-Phenyl-3,3a,4,7a-tetrahydro-2H-furo[2,3-b]pyran-6-yl)pyridine Noxide (43ab). mp 140-143 °C; [α]D25 +130.5 (c 0. 76, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.25 (1H, dd, J = 1.2 Hz, J = 6.5 Hz), 7.88 (1H, dd, J = 2.1 Hz, J = 8.2 Hz), 7.58 (1H, dd, J = 1.0 Hz, J = 2.8 Hz), 7.31 (4H, m), 7.24 (2H, m), 7.13 (1H, dt, J = 2.1 Hz, J = 6.8 Hz), 5.75 (1H, d, J = 4.1 Hz), 4.27 (1H, dd, J = 2.9 Hz, J = 6.4 Hz), 4.10 (1H, 43ab dt, J = 2.4 Hz, J = 9.1 Hz), 3.85 (1H, dd, J = 8.8 Hz, J = 16.3 Hz), 2.77 (1H, m), 1.84 (1H, tt, J = 9.5 Hz, J = 12.1 Hz), 1.41 (1H, dtd, J = 2.5 Hz, J = 7.5 Hz, J = 12.3 Hz); 13C NMR (75.5 MHz, CDCl3) δ 142.9 (s), 141.7 (s), 141.6 (s), 141.1 (d), 128.5 (d), 127.7 (d), 126.6 (d), 125.2 (d), 124.7 (d), 123.5 (d), 109.9 (d), 101.3 (d), 68.4 (t), 44.5 (d), 38.6 (d), 24.8 (t); MS (FAB) m/z (%): 296 (M++1), 226 (35), 176 (17), 159 (28), 154 (45), 137 (39); HRMS: 296.1285, C18H18NO3 required 296.1287. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak OD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Exo tR = 15.9 min, major endo-(+) tR = 24.2 min, exo tR = 19.9 min, minor endo-(-) tR = 30.0 min. Endo/exo >99.5:0.5. Ee(endo) = 99%. Ee(exo) = nd. 2-((4R,4aR,8aS)-4-Phenyl-4,4a,5,6,7,8a-hexahydropyrano[2,3-b]pyran-2-yl)pyridine N-oxide (43ac). [α]D25 +14.0 (c 0.65, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.24 (1H, dd, J = 1.2Hz, J = 6.5Hz), 7.88 (1H, dd, J = 2.1Hz, J = 8.2Hz), O O 7.42 (1H, dd, J = 1.2Hz, J = 2.4Hz), 7.28 (3H, m), 7.21 (3H, ddd, J N Ph = 2.1Hz, J = 5.1Hz, J = 8.3Hz), 7.12 (1H, ddd, J = 2.1Hz, J = 6.5Hz, J = 7.4Hz), 5.58 (1H, d, J = 1.4Hz), 4.17 (1H, dd, J = 2.4Hz, J = 6.3Hz), 3.97 (1H, dt, J = 3.7Hz, J = 11.3Hz), 3.71 (1H, tdd, J = 43ac 1.7Hz, J = 3.1Hz, J = 9.7Hz), 2.18 (1H, m), 1.54 (1H, m), 1.41 (1H, ddd, J = 3.9Hz, J = 12.5Hz, J = 16.9Hz), 0.93 (1H, m); 13C NMR (75.5 MHz, CDCl3) δ 142.7 (s), 141.8 (s), 141.2 (d), 140.4 (s), 128.2 (d), 128.2 (d), 126.5 (d), 125.1 (d), 124.4 (d), 123.5 (d), 109.5 (d), 97.6 (d), 61.4 (t), 42.5 (d), 37.9 (d), 24.7 (t), 18.9 (t); MS (FAB) m/z (%): 310 (M++1, 100), 292 (25), 226 (33), 173 (49), 154 (79), 137 (59); HRMS: 310.1441, C19H20NO3 required 310.1443. O 157 Experimental section Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Minor endo-(-) tR = 17.4 min, major endo-(+) tR = 20.7 min, minor exo tR = 28.8 min, major exo tR = 35.8 min. Endo/exo = 96.5:3.5. Ee(endo) = 95%. Ee(exo) = 23%. 2-((4S,6S)-4-Phenyl-1,7-dioxaspiro[5.5]undec-2-en-2-yl)pyridine N-oxide (43ad). Endo and exo adducts could not be separated. [α]D25 +41.2 (c 0.71, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.25 (1H, dd, J = 1.2 O Hz, J = 6.5 Hz), 7.90 (1H, endo, dd, J = 2.1 Hz, J = 8.2 Hz), 7.88 O O N (1H, exo, dd, J = 2.1 Hz, J = 8.1 Hz), 7.58 (1H, exo, dd, J = 1.7 Hz, Ph J = 2.1 Hz), 7.52 (1H, endo, d, J = 3.1 Hz), 7.29 (5H, m), 7.20 (1H, m), 7.13 (1H, ddd, J = 2.1 Hz, J = 6.6 Hz, J = 7.4 Hz), 4.04 (1H, 43ad endo, dt, J = 3.2 Hz, J = 11.4 Hz), 3.95 (1H, exo, ddd, J = 2.5 Hz, J = 6.4 Hz, J = 12.3 Hz), 3.78 (2H, m), 2.18 (2H, m), 2.00 (1H, dd, J = 9.1 Hz, J = 13.4 Hz), 1.85 (1H, m), 1.67 (3H, m), 1.52 (1H, dt, J = 4.4 Hz, J = 13.1 Hz); 13C NMR (75.5 MHz, CDCl3) (endo) δ 144.1 (s), 143.4 (s), 141.3 (d), 140.9 (s), 128.4 (d), 127.8 (d), 126.4 (d), 125.1 (d), 124.1 (d), 123.3 (d), 112.2 (d), 98.8 (s), 62.3 (t), 41.2 (t), 37.4 (d), 31.2 (t), 24.8 (t), 18.7 (t); 13C NMR (75.5 MHz, CDCl3) (exo) δ 144.3 (s), 143.4 (s), 141.3 (d), 139.8 (s), 128.5 (d), 127.7 (d), 126.4 (d), 125.0 (d), 123.9 (d), 123.1 (d), 113.8 (d), 96.9 (s), 62.1 (t), 41.3 (t), 35.5 (d), 34.8 (t), 25.1 (t), 18.7 (t); MS (FAB) m/z (%): 324 (M++1, 100), 306 (28), 226 (80), 218 (55), 187 (43), 147 (27), 109 (24); HRMS: 324.1590, C20H22NO3 required 324.1600. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Major exo tR = 10.7 min, minor exo tR = 16.8 min, major endo tR = 19.7 min, minor endo tR = 21.7 min. Endo/exo = 82:18. Ee(endo) = 90%. Ee(exo) = 58%. 2-((2S,4S)-2-Methoxy-2-methyl-4-phenyl-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ae). Cis (major) and trans (minor) adducts could not be separated. [α]D25 +14.5 (c 0.68, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.24 (1H, ddd, J = 0.5 Hz, J = 1.3 Hz, J = 6.5 Hz), 7.84 (1H, dd, J = 2.3 Hz, J = 8.2 Hz), 7.47 (1H, d, J = 3.2 Hz), 7.27 (6H, m), 7.12 (1H, m), 3.94 (1H, minor diastereomer, ddd, J = 2.5 Hz, J = 6.4 Hz, J = 43ae 12.4 Hz), 3.80 (1H, major diastereomer, dt, J = 3.2 Hz, J = 8.0 Hz), 3.36 (3H, minor diastereomer, s), 3.35 (3H, major diastereomer, s), 2.28 (1H, major diastereomer, ddd, J = 1.6 Hz, J = 6.4 Hz, J = 13.3 Hz), 2.13 (2H, minor diastereomer, d, J = 8.1 Hz), 1.79 (1H, major diastereomer, dd, J = 12.6 Hz, J = 13.1 Hz), 1.56 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 144.2 (s), 144.0 (s), 143.3 (s), 158 Experimental section 141.6 (s), 141.3 (d), 141.2 (d), 140.1 (s), 128.5 (minor diastereomer, d), 128.4 (major diastereomer, d), 127.7 (major diastereomer, d), 127.7 (minor diastereomer, d), 126.4 (major diastereomer, d), 126.4 (minor diastereomer, d), 125.1 (major diastereomer, d), 125.0 (minor diastereomer, d), 124.3 (major diastereomer, d), 124.1 (minor diastereomer, d), 123.4 (major diastereomer, d), 123.3 (minor diastereomer, d), 113.7 (minor diastereomer, d), 111.4 (major diastereomer, d), 101.2 (major diastereomer, s), 99.1 (minor diastereomer, s), 49.3 (minor diastereomer, q), 49.2 (major diastereomer, q), 41.2 (minor diastereomer, t), 39.1 (major diastereomer, t), 38.0 (major diastereomer, d), 35.8 (minor diastereomer, d), 23.0 (minor diastereomer, q), 22.1 (major diastereomer, q); MS (FAB) m/z (%): 298 (M+-1, 100), 266 (53), 226 (8), 191 (15), 176 (18), 137 (11); HRMS: 298.1436, C18H20NO3 required 298.1443. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Major exo tR = 9.9 min, minor exo tR = 10.9 min, major endo tR = 14.1 min, minor endo tR = 15.6 min. Endo/exo = 66:34. Ee(endo) = 94%. Ee(exo) = 95%. 2-((1S,5S)-1-Methoxy-5-phenyl-2-oxaspiro[5.5]undec-3-en-3-yl)pyridine N-oxide (43af). [α]D25 -29.8 (c 0.92, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.23 (1H, d, J = 6.3 Hz), 7.87 (1H, dd, J = 2.1 Hz, J = 8.2 Hz), 7.45 (1H, O O d, J = 4.7 Hz), 7.40 (2H, dd, J = 1.6 Hz, J = 7.9 Hz), 7.22 (4H, m), N Ph 7.10 (1H, ddd, J = 2.1 Hz, J = 6.7 Hz, J = 7.4 Hz), 4.99 (1H, s), 3.45 (3H, s), 3.38 (1H, d, J = 4.7 Hz), 1.79 (1H, m), 1.38 (9H, m); 13 43af C NMR (75.5 MHz, CDCl3) δ 143.4 (s), 141.3 (d), 140.5 (s), 137.3 (s), 131.4 (d), 127.1 (d), 126.2 (d), 125.1 (d), 124.1 (d), 123.2 (d), 114.3 (d), 104.0 (d), 56.2 (q), 48.1 (d), 38.7 (s), 36.0 (t), 31.4 (t), 25.9 (t), 22.0 (t), 21.6 (t); MS (FAB) m/z (%): 352 (M++1, 80), 225 (100), 149 (19), 126 (12); HRMS: 352.1907, C22H26NO3 required 352.1913. OMe Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak OD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Major exo tR = 11.8 min, major endo-(-) tR = 13.4 min, minor exo tR = 15.9 min, minor endo-(+) tR = 26.9 min. Endo/exo = 91.5:8.5. Ee(endo) = 49%. Ee(exo) = 93%. 159 Experimental section 2-((2S,4S)-2-Formamido-4-phenyl-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ag). mp 78-82 °C; [α]D25 +75.0 (c 0.78, MeOH); 1H NMR (400 MHz, DMSO-d6) two rotamers in a 62:38 ratio, δ 9.11 (1H, H NH major rotamer, d, J = 9.1 Hz), 8.89 (1H, minor rotamer, t, J O O = 9.4 Hz), 8.36 (1H, minor rotamer, d, J = 11.0 Hz), 8.27 (9H, N Ph both rotamers, dd, J = 1.1 Hz, J = 6.4 Hz), 8.19 (1H, major rotamer, s), 7.76 (1H, minor rotamer, dd, J = 2.3 Hz, J = 8.0 Hz), 7.68 (1H, major rotamer, dd, J = 2.2 Hz, J = 8.1 Hz), 43ag 7.31 (8H, m), 5.77 (1H, major rotamer, t, J = 9.1 Hz), 5.52 (1H, minor rotamer, t, J = 9.4 Hz), 4.02 (1H, both rotamers, m), 2.31 (1H, minor rotamer, dd, J = 6.8 Hz, J = 13.2 Hz), 2.25 (1H, major rotamer, dd, J = 6.8 Hz, J = 13.0 Hz), 1.85 (1H, both rotamers, m); 13C NMR (75.5 MHz, CDCl3) δ 164.6 (minor rotamer, d), 161.0 (major rotamer, d), 143.1 (major rotamer, s), 142.9 (minor rotamer, s), 142.7 (major rotamer, s), 142.6 (minor rotamer, s), 142.6 (major rotamer, s), 142.5 (minor rotamer, s), 141.1 (minor rotamer, d), 141.0 (major rotamer, d), 128.8 (minor rotamer, d), 128.7 (major rotamer, d), 127.2 (major rotamer, d), 127.2 (minor rotamer, d), 127.0 (minor rotamer, d), 126.9 (major rotamer, d), 125.7 (major rotamer, d), 125.5 (minor rotamer, d), 125.0 (major rotamer, d), 124.6 (minor rotamer, d), 124.0 (minor rotamer, d), 123.9 (major rotamer, d), 112.2 (minor rotamer, d), 112.0 (major rotamer, d), 80.3 (minor rotamer, d), 75.4 (major rotamer, d), 39.3 (minor rotamer, d), 38.7 (major rotamer, d), 36.8 (minor rotamer, t), 36.4 (major rotamer, t); MS (FAB) m/z (%): 297 (M++1, 100), 281 (5), 226 (7), 109 (9); HRMS: 297.1243, C17H17N2O3 required 297.1239. O Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Exo tR = 23.7 min, exo tR = 24.4 min, major endo-(+) tR = 35.3 min, minor endo-(-) tR = 40.7 min. Endo/exo = 98.5:1.5. Ee(endo) = 99%. Ee(exo) = 59%. 2-((2S,4S)-2-(2-Oxopyrrolidin-1-yl)-4-phenyl-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ah). mp 167-170 °C; [α]D25 +31.1 (c 0.85, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.22 (1H, dd, J = 1.3 Hz, J = 6.5 Hz), 7.72 (1H, dd, J = 2.1 Hz, J = 8.2 Hz), 7.35 (1H, dd, J = 1.5 Hz, J = 2.3 Hz), 7.31 (4H, m), 7.23 (2H, m), 7.11 (1H, ddd, J = 2.2 Hz, J = 6.6 Hz, J = 7.5 Hz), 6.04 (1H, dd, J = 1.9 Hz, J = 11.2 Hz), 4.00 (1H, ddd, J = 2.5 Hz, J = 6.4 Hz, J = 11.5 Hz), 3.59 (1H, ddd, J = 6.2 Hz, J = 8.1 43ah Hz, J = 9.3 Hz), 3.42 (1H, ddd, J = 6.2 Hz, J = 8.1 Hz, J = 9.4 Hz), 2.48 (1H, t, J = 8.3 Hz), 2.22 (1H, tdd, J = 1.8 Hz, J = 6.4 Hz, J = 12.9 Hz), 2.07 (3H, m); 13C NMR (75.5 MHz, CDCl3) δ 176.2 (s), 143.8 (s), 143.6 (s), 143.2 (s), 141.6 (d), 129.2 (d), 127.7 (d), 127.3 (d), 125.5 (d), 125.2 (d), 124.1 (d), 112.6 160 Experimental section (d), 78.9 (d), 42.8 (t), 40.1 (d), 35.8 (t), 31.8 (t), 18.5 (t); MS (FAB) m/z (%): 337 (M++1, 100), 226 (37), 200 (14); HRMS: 337.1555, C20H21N2O3 required 337.1552. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 25% isopropanol-75% hexane, 1.0 mL/min). Minor endo-(-) tR = 14.4 min, exo tR = 19.1 min, exo tR = 24.2 min, major endo-(+) tR = 40.1 min. Endo/exo = 99.5:0.5. Ee(endo) = 99.6%. Ee(exo) = nd. 2-((2R,4S)-2-(Ethylthio)-4-phenyl-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ai). mp 128-130 °C; [α]D25 +119.6 (c 0.62, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.24 (1H, dd, J = 1.2 Hz, J = 6.5 Hz), 7.81 (1H, dd, O O J = 2.1 Hz, J = 8.2 Hz), 7.40 (1H, dd, J = 1.4 Hz, J = 2.7 Hz), 7.26 N Ph (m, 6H), 7.14 (1H, ddd, J = 2.1 Hz, J = 6.5 Hz, J = 7.5 Hz), 5.38 (1H, dd, J = 1.8 Hz, J = 11.2 Hz), 3.95 (1H, ddd, J = 2.7 Hz, J = 6.8 43ai Hz, J = 10.9 Hz), 2.88 (1H, qd, J = 7.4 Hz, J = 12.8 Hz), 2.80 (1H, qd, J = 7.4 Hz, J = 12.8 Hz), 2.49 (1H, tdd, J = 1.6 Hz, J = 6.8 Hz, J = 13.5 Hz), 2.07 (1H, td, J = 11.1 Hz, J = 13.5 Hz), 1.37 (3H, t, J = 7.4 Hz); 13C NMR (75.5 MHz, CDCl3) δ 143.7 (s), 143.4 (s), 143.0 (s), 141.1 (d), 128.6 (d), 127.3 (d), 126.7 (d), 125.0 (d), 124.5 (d), 123.6 (d), 112.6 (d), 81.2 (d), 39.9 (d), 37.6 (t), 24.7 (t), 15.1 (q); MS (FAB) m/z (%): 314 (M++1, 100), 253 (22), 226 (91), 207 (12); HRMS: 314.1216, C18H20NO2S required 314.1215. SEt Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Exo tR = 15.1 min, exo tR = 17.3 min, major endo-(+) tR = 18.0 min, minor endo-(-) tR = 25.1 min. Endo/exo >99.9:0.1. Ee(endo) = 96%. Ee(exo) = nd. 2-((2R,4S)-4-(4-Bromophenyl)-2-(ethylthio)-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ci). mp 125-129 °C; [α]D25 +99.3 (c 0.79, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.22 (1H, dd, J = 1.2 Hz, J = 6.5 Hz), O O 7.79 (1H, dd, J = 2.1 Hz, J = 8.2 Hz), 7.41 (2H, d, J = 8.3 N Hz), 7.37 (1H, dd, J = 1.2 Hz, J = 2.6 Hz), 7.26 (1H, dt, J = 1.2 Hz, J = 7.8 Hz), 7.17 (2H, d, J = 8.4 Hz), 7.12 (1H, dd, Br J = 2.1 Hz, J = 7.4 Hz), 5.35 (1H, dd, J = 1.9 Hz, J = 11.0 43ci Hz), 3.89 (ddd, 1H, J = 2.7 Hz, J = 6.8 Hz, J = 10.2 Hz), 2.85 (qd, 1H, J = 7.5 Hz, J = 13.0 Hz), 2.77 (ddd, 1H, J = 7.5 Hz, J = 12.9 Hz, J = 14.9 Hz), 2.45 (1H, tdd, J = 1.5 Hz, J = 6.8 Hz, J = 13.5 Hz), 1.99 (1H, td, J = 10.9 Hz, J = 13.5 Hz), 1.35 (3H, t, J = 7.4 Hz); 13C NMR (75.5 MHz, CDCl3) δ 143.9 (s), 142.8 (s), 142.5 (s), 141.1 (d), 131.6 (d), 129.1 (d), 125.1 (d), 124.4 (d), 123.7 (d), 120.4 (s), 111.8 (d), 81.1 SEt 161 Experimental section (d), 39.2 (d), 37.4 (t), 24.7 (t), 15.1 (q); MS (FAB) m/z (%): 392 (M++1, 60), 330 (16), 304 (42), 283 (12), 176 (25), 154 (98), 137 (100), 107 (42); HRMS: 392.0315, C18H19BrNO2S required 392.0320. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Minor exo tR = 17.8 min, major endo-(+) tR = 21.4 min, major exo tR = 25.5 min, minor endo-(-) tR = 35.8 min. Endo/exo = 98:2. Ee(endo) = 97%. Ee(exo) = 53%. 2-((2S,4S)-2-(4-Methoxyphenyl)-4-phenyl-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43aj). [α]D25 -14.3 (c 0.90, CHCl3); 1H NMR (300 MHz, CDCl3) N δ 8.21 (1H, d, J = 6.4 Hz), 7.79 (1H, dd, J = 2.1 Hz, J = O 8.2 Hz), 7.47 (1H, s), 7.32 (6H, m), 7.19 (2H, m), 7.07 (1H, dt, J = 2.1 Hz, J = 7.1 Hz), 6.90 (2H, d, J = 8.6 Hz), Ph 5.14 (1H, dd, J = 1.3 Hz, J = 11.4 Hz), 4.02 (1H, ddd, J = 43aj 2.4 Hz, J = 6.5 Hz, J = 11.3 Hz), 3.79 (3H, s), 2.38 (1H, tdd, J = 1.5 Hz, J = 6.6 Hz, J = 13.7 Hz), 2.04 (1H, td, J = 13 11.5 Hz, J = 13.5 Hz); C NMR (75.5 MHz, CDCl3) δ 159.3 (s), 144.2 (s), 144.1 (s), 143.3 (s), 141.1 (d), 133.1 (s), 128.5 (d), 127.5 (d), 127.3 (d), 126.5 (d), 125.1 (d), 124.6 (d), 123.4 (d), 113.8 (d), 112.4 (d), 78.2 (d), 55.2 (q), 40.0 (d), 39.7 (t); MS (FAB) m/z (%): 360 (M++1, 100), 307 (22), 189 (11), 253 (13), 226 (27), 154 (100), 137 (74); HRMS: 360.1596, C23H22NO3 required 360.1600. MeO O Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Minor exo tR = 26.9 min, minor endo-(+) tR = 30.8 min, major exo tR = 36.5 min, major endo-(-) tR = 47.6 min. Endo/exo = 92.5:2.5. Ee(endo) = 77%. Ee(exo) = 52%. 2-((2S,4R)-2-(4-Methoxyphenyl)-4-styryl-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43jj). [α]D25 -41.4 (c 0.64, CHCl3); 1H NMR (300 MHz, CDCl3) N δ 8.25 (1H, d, J = 6.3 Hz), 7.78 (1H, dd, J = 1.8 Hz, J = O 8.1 Hz), 7.26 (9H, m), 7.08 (1H, dt, J = 1.9 Hz, J = 7.4 Hz), 6.92 (2H, d, J = 8.7 Hz), 6.54 (1H, d, J = 15.8 Hz), 6.20 (1H, dd, J = 7.4 Hz, J = 15.8 Hz), 5.04 (1H, dd, J = 1.3 Hz, J = 11.4 Hz), 3.80 (s, 3H), 3.59 (1H, m), 2.27 Ph (1H, dd, J = 6.5 Hz, J = 13.5 Hz), 1.93 (1H, td, J = 11.3 43jj Hz, J = 13.4 Hz); 13C NMR (75.5 MHz, CDCl3) δ 159.3 (s), 143.6 (s), 143.3 (s), 141.1 (d), 137.2 (s), 133.2 (s), 131.8 (d), 129.8 (d), 128.4 (d), MeO 162 O Experimental section 127.4 (d), 127.1 (d), 126.1 (d), 125.5 (d), 124.6 (d), 123.4 (d), 113.9 (d), 111.7 (d), 77.9 (d), 55.3 (q), 37.3 (s), 36.9 (t); MS (FAB) m/z (%): 384 (M+-1, 100), 368 (11), 252 (94), 249 (40), 236 (13), 134 (19); HRMS: 386.1749, C25H24NO3 required 386.1756. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Exo tR = 18.6 min, exo tR = 22.4 min, minor endo -(+) tR = 25.9 min, major endo-(-) tR = 28.2 min. Endo/exo = 85.5:14.5. Ee(endo) = 77%. Ee(exo) = nd. 2-((2S,4S)-2-(2-Methylprop-1-enyl)-4-phenyl-3,4-dihydro-2H-pyran-6-yl)pyridine N-oxide (43ak). [α]D25 +35.0 (c 0.79, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.22 N (1H, dd, J = 0.8 Hz, J = 6.5 Hz), 7.79 (1H, dd, J = 2.1 Hz, J = 8.2 O Hz), 7.31 (4H, m), 7.23 (3H, m), 7.10 (1H, ddd, J = 2.2 Hz, J = 6.7 Hz, J = 7.4 Hz), 5.32 (1H, tdd, J = 1.4 Hz, J = 2.8 Hz, J = 8.4 Ph Hz), 4.89 (1H, ddd, J = 1.6 Hz, J = 8.4 Hz, J = 10.3 Hz), 3.91 43ak (1H, ddd, J = 2.5 Hz, J = 6.6 Hz, J = 11.4 Hz), 2.17 (1H, tdd, J = 1.7 Hz, J = 6.6 Hz, J = 13.6 Hz), 1.84 (1H, q, J = 11.4 Hz), 1.79 13 (3H, s), 1.78 (3H, s); C NMR (75.5 MHz, CDCl3) δ 144.4 (s), 144.1 (s), 143.7 (s), 141.1 (d), 137.5 (s), 128.5 (d), 127.3 (d), 126.4 (d), 125.0 (d), 124.7 (d), 124.3 (d), 123.4 (d), 112.0 (d), 73.8 (d), 39.5 (d), 38.1 (t), 25.8 (q), 18.5 (q); MS (EI) m/z (%): 307 (M+, 3.2), 226 (11), 210 (16), 188 (20), 91 (40), 78 (100), 69 (50); HRMS: 307.1572, C20H21NO2 required 307.1572. O Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak OD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Major endo-(+) tR = 14.7 min, minor endo-(-) tR = 24.2 min, exo tR = 27.3 min, exo tR = 30.5 min. Endo/exo = 99.5:0.5. Ee(endo) = 86%. Ee(exo) = nd. (S)-2-(5-Methoxycarbonyl-4,4-dimethyl-5-oxo-3-phenylpentanoyl)pyridine N-oxide (45). mp 102-105 °C; [α]D25 -33.4 (c 0.76, MeOH); 1H NMR (300 MHz, CDCl3) δ 8.12 (1H, d, J = 6.5Hz), 7.25 (1H, ddd, J = 2.2Hz, J = 6.3Hz, J = 7.4Hz), 7.15 (3H, m), 7.08 (3H, m), 3.88 (1H, dd, J = 11.4Hz, J = 16.5Hz), 3.66 (1H, dd, J = 45 3.6Hz, J = 11.5Hz), 3.62 (3H, s), 3.46 (1H, dd, J = 3.6Hz, J = 16.5Hz), 1.20 (3H, s), 1.10 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 197.1 (s), 177.3 (s), 146.9 (s), 140.0 (d), 139.6 (s), 129.3 (d), 127.8 (d), 127.4 (d), 126.9 (d), 126.4 (d), 125.3 (d), 51.8 (q), 48.2 (d), 46.2 (s), 43.6 (t), 23.6 (q), 21.9 (q); MS (FAB) m/z (%): 328 (M+-1, 100), 227 (15), 154 (14), 136 (13); HRMS: 328.1558, C19H22NO4 required 328.1549. 163 Experimental section Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak OD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Major (-) tR = 18.8 min, minor (+) tR = 24.0 min. 5.3.3.3. Synthetic transformations 2-((2R,4R,6S)-6-Ethoxy-4-phenyltetrahydro-2H-pyran-2-yl)pyridine (46). A solution of compound 43aa (119.0 mg, 0.40 mmol, 96% ee) in EtOH (5.5 mL) and 5% Pd/C (50 mg) were stirred under hydrogen at atmospheric pressure for 9h. After this time, the mixture was filtered through a short pad of silica gel to remove the catalyst. The filtrate was concentrated under reduced 46 pressure and chromatographed on silica gel eluting with hexane-EtOAc (93:7 to 90:10) to give 62 mg of compound 46 (61% yield). [α]D25 +94.5 (c 1.23, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.49 (1H, ddd, J = 0.9 Hz, J = 1.7 Hz, J = 4.9 Hz), 7.70 (1H, dt, J = 1.8 Hz, J = 7.7 Hz), 7.60 (1H, d, J = 7.9 Hz), 7.21 (6H, m), 4.79 (1H, dd, J = 2.1 Hz, J = 9.5 Hz), 4.73 (1H, dd, J = 2.1 Hz, J = 11.3 Hz), 4.06 (1H, qd, J = 7.1 Hz, J = 9.6 Hz), 3.63 (1H, qd, J = 7.1 Hz, J = 9.6 Hz), 3.06 (1H, tt, J = 3.7 Hz, J = 12.5 Hz), 2.34 (1H, tdd, J = 2.0 Hz, J = 3.9 Hz, J = 13.2 Hz), 2.12 (1H, tdd, J = 1.9 Hz, J = 3.8 Hz, J = 12.8 Hz), 1.78 (1H, dt, J = 9.5 Hz, J = 12.8 Hz), 1.55 (1H, ddd, J = 11.4 Hz, J = 12.4 Hz, J = 13.1 Hz), 1.28 (3H, t, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 160.9 (s), 148.3 (d), 144.2 (s), 136.8 (d), 128.5 (d), 126.7 (d), 126.4 (d), 122.3 (d), 120.0 (d), 102.1 (d), 77.9 (d), 64.4 (t), 40.5 (d), 39.8 (t), 37.8 (t), 15.3 (q); MS (EI) m/z (%): 283 (M+, 5), 238 (10), 209 (82), 194 (18), 178 (16), 104 (100), 78 (16); HRMS: 283.1579, C18H21NO2 required 283.1572. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Major (+) tR = 7.4 min, minor (-) tR = 14.5 min. 5.3.4. Enantioselective catalyzed cycloadditions whit nitrones 1,3-dipolar 5.3.4.1. General procedure for the synthesis of nitrones Nitrones were synthesized following procedures found in the literature.205-208 For the synthesis of nitrone 47a, to a stirred solution of N-phenylhydroxylamine (2.07 g, 19 mmol) in EtOH was added benzaldehyde (1.93 mL, 19 mmol). After overnight stirring at rt in the dark, the mixture were cooled in ice-water bath and filtered. The collected solid were recrystallized from EtOH to give diphenyl nitrone 47a 2.62 g (73% yield). 164 Experimental section Nitrones 47b-g were synthesized following the same procedure for the synthesis of 47a. Yields are summarized in Tabla 15 (page 94). 5.3.4.2. General procedure for the catalytic enantioselective 1,3-dipolar cycloadditions Cu(OTf)2 (9.0 mg, 0.025 mmol) contained in a dry Schlenk tube was heated at 90 °C under vacuum for 1 h. After this time, ligand 33b (7.5 mg, 0.025 mmol), 4 Å molecular sieves (75 mg) and CH2Cl2 (1.5 mL) were added under nitrogen atmosphere and the mixture was stirred for 1 h at rt. Then, dipolarophile 31 (0.25 mmol) was added and the mixture stirred for 0.5 h. After this time, the solution was placed in a bath at the reaction temperature and nitrone 47 (0.3 mmol) was added. After completion of the reaction (TLC), the isoxazolidine 48 was isolated directly by flash chromatography on silica gel. 5.3.4.3. Characterization of the products See Tabla 18 (page 99) and Tabla 19 (page 101) for yields, enantiomeric excesses and isomeric ratios. In case of additional purification by recrystallization or chiral HPLC, ee of the used sample is indicated for the measured [α]D25. 2-((3S,4R,5R)-2,3,5-Triphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48aa). mp 120-123 °C; [α]D25 -33.9 (c 1.32, CHCl3) (ee 98%); 1H NMR O (300 MHz, CDCl3) δ 7.86 (1H, d, J = 5.8 Hz), 7.59 (2H, d, J = 7.1 Ph N Hz), 7.35 (4H, m), 7.28 (m, 6H), 7.17 (1H, ddd, J = 2.7, J = 6.1, N Ph J = 7.2), 7.12 (1H, dd, J = 6.2 Hz, J = 7.4 Hz), 7.07 (3H, m), 6.99 O O endo-48aa (1H, tt, J = 1.1, J = 7.3 Hz), 5.46 (1H, d, J = 7.6 Hz), 5.36 (1H, d, J = 5.8 Hz), 5.15 (1H, dd, J = 5.7 Hz, J = 7.6 Hz); 13C NMR (75.5 MHz, CDCl3) δ 195.8 (s), 151.0 (s), 146.1 (s), 140.7 (s), 139.6 (d), 136.5 (s), 128.9 (d), 128.6 (d), 128.5 (d), 128.3 (d), 127.6 (d), 127.6 (d), 127.0 (d), 126.7 (d), 125.9 (d), 125.2 (d), 121.8 (d), 114.4 (d), 83.3 (d), 73.3 (d), 69.9 (d); MS (FAB) m/z (%): 423 (M++1, 87), 405 (100), 226 (57), 198 (44), 154 (42); HRMS: 423.1706, C27H23N2O3 required 423.1709. Ph mp 146-148 °C; [α]D25 -58.2 (c 0.53, CHCl3) (ee 35 %); 1H NMR (300 MHz, CDCl3) δ 8.01 (1H, d, J = 6.4 Hz), 7.59 (1H, dd, J = 1.5 Hz, J = 8.0 Hz), 7.48 (2H, m), 7.35 (3H, m), 7.16 (6H, m), 7.03 (3H, m), 6.95 (1H, m), 5.98 (1H, d, J = 9.3 Hz), 5.47 (1H, exo-48aa d, J = 10.3 Hz), 5.33 (1H, dd, J = 9.3 Hz, J = 10.3 Hz); 13C NMR (75.5 MHz, CDCl3) δ 192.5 (s), 149.7 (s), 145.0 (s), 140.1 (d), 138.9 (s), 137.5 (s), 128.5 (d), 128.5 (d), 128.4 (d), 128.4 (d), 128.0 (d), 128.0 (d), 128.0 (d), 127.4 (d), 127.3 (d), 125.1 (d), 122.2 (d), 116.1 (d), 80.4 (d), 71.7 (d), 67.8 (d); MS (FAB) m/z (%): 423 (M++1, 165 Experimental section 35), 314 (22), 226 (15), 198 (17), 154 (100), 136 (74); HRMS: 423.1693, C27H23N2O3 required 423.1709. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Major exo-(-) tR = 13.6 min, minor exo-(+) tR = 14.8 min, minor endo-(+) tR = 16.2 min, major endo-(-) tR = 23.8 min. Endo/exo = 94:6. Ee(endo) = 95%. Ee(exo) = 61%. 2-((3S,4R,5R)-5-(4-Methoxyphenyl)-2,3-diphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48ba). mp 150-152 °C; [α]D25 -20.1 (c 1.00, CHCl3) (93.7% ee); 1H NMR (300 MHz, CDCl3) δ 7.86 (1H, d, J = 6.0 Hz), 7.61 (2H, d, J O = 7.0 Hz), 7.37 (2H, t, J = 7.3 Hz), 7.27 (5H, m), 7.15 (2H, m), Ph N 7.05 (3H, m), 6.97 (1H, t, J = 7.3 Hz), 6.79 (2H, d, J = 8.8 Hz), N Ph O O 5.38 (1H, d, J = 5.8 Hz), 5.36 (1H, d, J = 8.0 Hz), 5.14 (1H, dd, J endo-48ba = 5.9 Hz, J = 8.1 Hz), 3.76 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 195.9 (s), 159.7 (s), 151.2 (s), 146.2 (s), 140.9 (s), 139.6 (d), 128.9 (d), 128.6 (d), 128.6 (d), 128.0 (s), 127.6 (d), 127.5 (d), 126.6 (d), 125.8 (d), 125.2 (d), 121.7 (d), 114.2 (d), 113.7 (d), 83.4 (d), 73.3 (d), 69.7 (d), 55.2 (q); MS (EI) m/z (%): 452 (M+, 1.1), 435 (16), 327 (6), 239 (19), 227 (11), 197 (32), 149 (15), 121 (48), 106 (33), 91 (100), 78 (71); HRMS: 452.1729, C28H24N2O4 required 452.1736. OMe [α]D25 -11.6 (c 0.34, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.06 (1H, d, J = 6.5 Hz), 7.51 (5H, m), 7.16 (6H, m), 7.01 (4H, m), 6.89 (2H, d, J = 8.8 Hz), 5.91 (1H, d, J = 9.5 Hz), 5.45 (1H, d, J = 10.3 Hz), 5.29 (1H, dd, J = 9.6 Hz, J = 10.2 Hz), 3.79 (3H, s); Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 15% isopropanol-80% hexane, 1.0 mL/min). Minor endo-(+) tR = 31.3 min, major endo-(-) tR = 46.6 min. Endo/exo = 93:7. Ee(endo) = 96%. Ee(exo) = nd. exo-48ba 2-((3S,4R,5R)-5-(4-Bromophenyl)-2,3-diphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48ca). endo-48ca 166 mp 170-172 °C; [α]D25 -67.7 (c 1.01, CHCl3) (100% ee); 1H NMR (300 MHz, CDCl3) δ 7.92 (1H, ddd, J = 0.6 Hz, J = 1.1 Hz, J = 6.4 Hz), 7.51 (2H, d, J = 6.8 Hz), 7.40 (2H, d, J = 8.5 Hz), 7.28 (8H, m), 7.16 (1H, dt, J = 1.2 Hz, J = 7.6 Hz), 7.11 (1H, ddd, J = 0.5 Hz, J = 2.4 Hz, J = 7.7 Hz), 7.00 (3H, m), 5.53 (1H, d, J = 6.7 Hz), 5.23 (1H, d, J = 5.4 Hz), 5.06 (1H, dd, J = 5.4 Hz, J = 6.7 Hz); 13C NMR (75.5 MHz, CDCl3) δ 195.7 (s), 150.6 (s), 146.0 Experimental section (s), 140.2 (s), 139.7 (d), 136.6 (s), 131.5 (d), 128.9 (d), 128.72 (d), 128.71 (d), 127.8 (d), 127.7 (d), 126.7 (d), 126.2 (d), 125.3 (d), 122.3 (d), 122.2 (d), 114.9 (d), 81.9 (d), 73.4 (d), 69.8 (d); MS (FAB) m/z (%): 500 (M+, 42), 484 (78), 377 (13), 317 (28), 304, (28), 222 (16), 198 (100), 180 (41), 154 (11), 136 (18); HRMS: 501.0807, C27H22BrN2O3 required 501.0814. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 30% isopropanol-70% hexane, 1.0 mL/min). Major endo-(-) tR = 15.3 min, major exo tR = 17.1 min, minor endo-(+) tR = 22.8 min, minor exo tR = 25.9 min. Endo/exo = 90:10. Ee(endo) = 96%. Ee(exo) = 22%. 2-((3S,4R,5R)-5-(4-Nitrophenyl)-2,3-diphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48da). mp 136-137 °C; [α]D25 -132.2 (c 0.86, CHCl3) (100% ee); 1H NMR (300 MHz, CDCl3) δ 8.15 (2H, d, J = 8.9 Hz), 7.98 (1H, O ddd, J = 0.6 Hz, J = 1.1 Hz, J = 6.4 Hz), 7.65 (2H, d, J = 8.4 Hz), Ph N 7.39 (2H, m), 7.25 (8H, m), 7.02 (3H, m), 5.82 (1H, d, J = 5.2 N Ph O O Hz), 5.11 (1H, d, J = 5.0 Hz), 5.02 (1H, t, J = 5.1 Hz); 13C NMR endo-48da (75.5 MHz, CDCl3) δ 195.6 (s), 149.9 (s), 147.4 (s), 146.6 (s), 145.8 (s), 139.7 (d), 139.4 (s), 128.9 (d), 128.7 (d), 128.1 (d), 127.9 (d), 127.5 (d), 126.7 (d), 126.7 (d), 125.5 (d), 123.5 (d), 122.8 (d), 115.7 (d), 80.3 (d), 73.5 (d), 69.9 (d); MS (FAB) m/z (%): 468 (M++1, 52), 450 (50), 317 (14), 307 (15), 289 (11), 271 (9), 210 (25), 198 (63), 180 (20), 154 (100), 136 (73); HRMS: 468.1560, C27H22N3O5 required 468.1560. NO2 Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 30% isopropanol-70% hexane, 1.0 mL/min). Major endo-(-) tR = 19.2 min, minor exo tR = 25.6 min, minor endo-(+) tR = 39.9 min, major exo tR = 46.5 min. Endo/exo = 87:13. Ee(endo) = 96%. Ee(exo) = 21%. 2-((3S,4R,5R)-5-(Furan-2-yl)-2,3-diphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48ga). mp 118-121 °C; [α]D25 -80.1 (c 0.99, CHCl3) (99.0% ee); 1H NMR (300 MHz, CDCl3) δ 7.99 (1H, d, J = 6.4 Hz), 7.62 (2H, d, J = 7.1 Hz), 7.30 (7H, m), 7.14 (1H, dt, J = 1.1 Hz, J = 7.7 Hz), 7.04 (3H, m), 6.97 (1H, tt, J = 1.1 Hz, J = 7.3 Hz), 6.32 (1H, d, J = 3.3 Hz), 6.28 (1H, dd, J = 1.8 Hz, J = 3.3 Hz), 5.61 (1H, d, J = endo-48ga 6.7 Hz), 5.30 (1H, d, J = 5.3 Hz), 5.26 (1H, dd, J = 5.3 Hz, J = 6.7 Hz); 13C NMR (75.5 MHz, CDCl3) δ 195.3 (s), 150.5 (s), 149.8 (s), 146.0 (s), 143.1 (d), 140.3 (s), 139.7 (d), 128.9 (d), 128.6 (d), 127.8 (d), 127.6 (d), 167 Experimental section 126.9 (d), 126.1 (d), 125.2 (d), 122.2 (d), 115.0 (d), 110.5 (d), 109.7 (d), 75.8 (d), 73.3 (d), 66.7 (d); MS (FAB) m/z (%): 413 (M++1, 37), 395 (38), 317 (21), 307 (23), 290 (21), 215 (33), 210 (21), 154 (100), 136 (76); HRMS: 413.1498, C25H21N2O4 required 413.1501. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 30% isopropanol-70% hexane, 1.0 mL/min). Major endo-(-) tR = 13.6 min, minor endo-(+) tR = 19.9 min. Endo/exo >99:1. Ee(endo) = 95%. Ee(exo) = nd. 2-((3S,4R,5R)-2,3-Diphenyl-5-(thiophen-2-yl)isoxazolidine-4-carbonyl)pyridine N-oxide (48ia). mp 140-142°C; [α]D25 -90.1 (c 1.01, CHCl3) (99.0% ee); 1H NMR (300 MHz, CDCl3) δ 7.98 (1H, d, J = 6.4 Hz), 7.60 (2H, d, J = 7.2 Hz), 7.30 (7H, m), 7.15 (1H, dt, J = 1.1 Hz, J = 7.6 Hz), 7.07 (4H, m), 6.99 (1H, tt, J = 1.1 Hz, J = 7.3 Hz), 6.91 (1H, dd, J = 3.6 Hz, J = 5.1 Hz), 5.82 (1H, d, J = 6.6 Hz), 5.30 (1H, d, J = endo-48ia 5.4 Hz), 5.13 (1H, dd, J = 5.4 Hz, J = 6.6 Hz); 13C NMR (75.5 MHz, CDCl3) δ 195.5 (s), 150.4 (s), 146.0 (s), 140.6 (s), 140.2 (s), 139.7 (d), 128.9 (d), 128.6 (d), 127.8 (d), 127.7 (d), 126.9 (d), 126.6 (d), 126.5 (d), 126.4 (d), 126.2 (d), 125.2 (d), 122.2 (d), 115.1 (d), 78.7 (d), 73.3 (d), 70.3 (d); MS (FAB) m/z (%): 429 (M++1, 4.1), 401 (11), 341 (15), 327 (25), 281 (40), 221 (31), 207 (48), 193 (20), 147 (100), 136 (30); HRMS: 429.1273, C25H21N2O3S required 429.1273. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, t0 = 10% isopropanol-90% hexane, t30 = 30% isopropanol-70% hexane, 1.0 mL/min). Exo (both) tR = 22.1 min, minor endo-(+) tR = 25.3 min, major endo-(-) tR = 41.5 min. Endo/exo = 94:6. Ee(endo) = 91%. Ee(exo) = nd. 2-((3S,4R,5S)-2,3-Diphenyl-5-styrylisoxazolidine-4-carbonyl)pyridine N-oxide (48ja). [α]D25 -74.6 (c 1.00, CHCl3) (100% ee); 1H NMR (300 MHz, O CDCl3) δ 7.94 (1H, m), 7.63 (2H, d, J = 6.9 Hz), 7.38 (2H, t, J = Ph N 7.3 Hz), 7.26 (8H, m), 7.15 (3H, m), 7.04 (2H, d, J = 7.7 Hz), N 6.97 (1H, t, J = 7.3 Hz), 6.57 (1H, dd, J = 0.6 Hz, J = 15.9 Hz), Ph O O 6.31 (1H, dd, J = 7.5 Hz, J = 15.9 Hz), 5.35 (1H, d, J = 5.3 Hz), endo-48ja 5.06 (1H, dt, J = 0.7 Hz, J = 7.4 Hz), 4.98 (1H, dd, J = 5.3 Hz, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 195.2 (s), 151.1 (s), 146.1 (s), 140.8 (s), 139.8 (d), 135.9 (s), 134.2 (d), 128.9 (d), 128.7 (d), 128.4 (d), 128.1 (d), 127.8 (d), 127.5 (d), 126.7 (d), 126.6 (d), 126.4 (d), 125.4 (d), 125.1 (d), 121.8 (d), 114.7 (d), 82.4 (d), 72.6 (d), 68.5 (d); MS (FAB) m/z (%): 449 (M++1, 47), 431 (63), 355 (10), 340 (20), 317 (16), 301 (10), 281 (38), 267 (15), 252 (68), 221 (41), 207 (41), 197 Ph 168 Experimental section (37), 180 (23), 154 (48), 147 (100), 136 (59); HRMS: 449.1862, C29H25N2O3 required 449.1865. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 30% isopropanol-70% hexane, 1.0 mL/min). Major endo-(-) tR = 12.7 min, minor exo tR = 14.3 min, minor endo-(+) tR = 22.4 min, major exo tR = 23.7 min. Endo/exo = 93:7. Ee(endo) = 93%. Ee(exo) = 33%. 2-((3S,4R,5R)-5-tert-Butyl-2,3-diphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48ka). [α]D25 -161.4 (c 1.00, CHCl3) (87.6% ee); 1H NMR (300 MHz, CDCl3) δ 8.03 (1H, ddd, J = 0.6 Hz, J = 1.2 Hz, J = 6.4 Hz), 7.53 (2H, d, J = 6.9 Hz), 7.26 (7H, m), 7.14 (1H, m), 6.96 (3H, m), 5.02 (1H, d, J = 4.6 Hz), 4.89 (1H, dd, J = 4.6 Hz, J = 7.7 Hz), 4.58 (1H, d, J = 7.7 Hz), 0.97 (9H, s); 13C NMR (75.5 MHz, endo-48ka CDCl3) δ 197.8 (s), 150.9 (s), 146.5 (s), 141.1 (s), 139.9 (d), 128.9 (d), 128.4 (d), 127.7 (d), 127.3 (d), 126.6 (d), 125.4 (d), 121.7 (d), 114.4 (d), 89.3 (d), 74.9 (d), 63.0 (d), 33.1 (s), 26.2 (q); MS (FAB) m/z (%): 403 (M++1, 11), 385 (100), 281 (15), 222 (18), 206 (29), 198 (31), 180 (34), 147 (47); HRMS: 403.2026, C25H27N2O3 required 403.2022. [α]D25 -42.3 (c 0.64, CHCl3) (44.8% ee); 1H NMR (300 O MHz, CDCl3) δ 8.04 (1H, d, J = 6.5 Hz), 7.32 (2H, m), 7.15 (6H, Ph N m), 6.92 (4H, m), 6.72 (1H, dd, J = 2.1 Hz, J = 8.0 Hz), 5.49 (1H, N dd, J = 8.6 Hz, J = 10.4 Hz), 4.95 (1H, d, J = 10.4 Hz), 4.85 (1H, Ph O O d, J = 8.6 Hz), 1.08 (9H, s); 13C NMR (75.5 MHz, CDCl3) δ 194.7 exo-48ka (s), 149.3 (s), 145.7 (s), 140.0 (d), 137.8 (s), 128.6 (d), 128.3 (d), 128.3 (d), 127.9 (d), 127.5 (d), 127.4 (d), 125.0 (d), 122.4 (d), 117.2 (d), 85.6 (d), 72.5 (d), 59.0 (d), 33.4 (s), 26.3 (q). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak IC, 20% isopropanol-80% hexane, 1.0 mL/min). Minor exo-(+) tR = 7.5 min, major exo-(-) tR = 8.8 min, minor endo-(+) tR = 15.6 min, major endo-(-) tR = 17.7 min. Endo/exo = 92:8. Ee(endo) = 86%. Ee(exo) = 45%. 169 Experimental section 2-((3S,4R,5R)-3-(4-Methoxyphenyl)-2,5-diphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48ab). [α]D25 -28.0 (c 0.99, CHCl3) (99.9% ee); 1H NMR (300 MHz, O CDCl3) δ 7.87 (1H, ddd, J = 0.6 Hz, J = 1.3 Hz, J = 6.4 Hz), 7.48 Ph N N (2H, d, J = 8.5 Hz), 7.38 (2H, m), 7.26 (5H, m), 7.11 (5H, m), O O 6.97 (1H, t, J = 7.3 Hz), 6.88 (2H, d, J = 8.8 Hz), 5.47 (1H, d, J = 7.5 Hz), 5.26 (1H, d, J = 5.8 Hz), 5.12 (1H, dd, J = 5.8 Hz, J = 7.5 Hz), 3.80 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 196.0 (s), MeO 159.0 (s), 151.0 (s), 146.2 (s), 139.6 (d), 136.9 (s), 132.6 (s), endo-48ab 128.9 (d), 128.4 (d), 128.4 (d), 127.9 (d), 127.6 (d), 127.0 (d), 125.9 (d), 125.1 (d), 121.9 (d), 114.6 (d), 114.0 (d), 83.1 (d), 73.1 (d), 69.9 (d), 55.2 (q); MS (FAB) m/z (%): 453 (M++1, 26), 435 (100), 347 (19), 328 (11), 281 (18), 240 (28), 228 (92), 210 (24), 154 (16), 147 (46), 136 (24); HRMS: 453.1821, C28H25N2O4 required 453.1814. Ph [α]D25 -48.8 (c 0.65, CHCl3) (32.6% ee); 1H NMR (300 MHz, CDCl3) δ 8.01 (1H, d, J = 6.4 Hz), 7.58 (2H, dd, J = 1.4 Hz, J = Ph N N 8.0 Hz), 7.35 (5H, m), 7.18 (3H, m), 7.11 (1H, dd, J = 2.1 Hz, J = O O 8.1 Hz), 7.01 (3H, m), 6.93 (1H, t, J = 7.3 Hz), 6.66 (2H, d, J = 8.8 Hz), 5.96 (1H, d, J = 9.3 Hz), 5.42 (1H, d, J = 10.3 Hz), 5.30 (1H, dd, J = 9.3 Hz, J = 10.2 Hz), 3.71 (3H, m); 13C NMR (75.5 MeO MHz, CDCl3) δ 192.6 (s), 159.2 (s), 149.8 (s), 145.1 (s), 140.1 exo-48ab (d), 137.7 (s), 130.8 (s), 129.2 (d), 128.48 (d), 128.48 (d), 128.3 (d), 127.9 (d), 127.3 (d), 124.8 (d), 122.1 (d), 116.1 (d), 113.7 (d), 80.2 (d), 71.2 (d), 67.7 (d), 55.1 (q). O Ph Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 30% isopropanol-70% hexane, 1.0 mL/min). Minor exo-(+) tR = 21.5 min, major exo-(-) tR = 24.1 min, major endo-(-) tR = 26.5 min, minor endo-(+) tR = 35.0 min. Endo/exo = 86:14. Ee(endo) = 95%. Ee(exo) = 61%. 2-((3S,4R,5R)-3-(4-Bromophenyl)-2,5-diphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48ac). endo-48ac 170 [α]D25 -77.85 (c 1.01, CHCl3) (94.1% ee); 1H NMR (300 MHz, CDCl3) δ 7.89 (1H, d, J = 6.2 Hz), 7.50 (4H, m), 7.29 (7H, m), 7.09 (6H, m), 5.43 (1H, d, J = 7.5 Hz), 5.36 (1H, d, J = 5.3 Hz), 5.07 (1H, dd, J = 5.3 Hz, J = 7.5 Hz); 13C NMR (75.5 MHz, CDCl3) δ 195.6 (s), 150.6 (s), 145.9 (s), 139.9 (s), 139.6 (d), 136.3 (s), 131.7 (d), 129.0 (d), 128.6 (d), 128.5 (d), 128.4 (d), 127.8 (d), 126.9 (d), 126.0 (d), 125.3 (d), 122.0 (d), 121.4 (d), Experimental section 114.3 (d), 83.4 (d), 72.3 (d), 69.8 (d); MS (FAB) m/z (%): 501 (M++1, 29), 485 (30), 391 (15), 307 (32), 226 (47), 154 (100), 136 (86); HRMS: 501.0806, C27H22BrN2O3 required 501.0814. [α]D25 -42.6 (c 0.69, CHCl3) (44.1% ee); 1H NMR (300 MHz, CDCl3) δ 8.08 (1H, d, J = 6.4 Hz), 7.54 (2H, dd, J = 1.6 Hz, J = 7.9 Hz), 7.41 (2H, d, J = 8.6 Hz), 7.30 (6H, m), 7.20 (3H, m), 7.12 (1H, ddd, J = 1.1 Hz, J = 7.5 Hz, J = 8.2 Hz), 6.97 (3H, m), 5.92 (1H, d, J = 9.5 Hz), 5.47 (1H, d, J = 10.2 Hz), 5.26 (1H, dd, J = 9.5 Hz, J = 10.2 Hz). exo-48ac Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Minor exo-(+) tR = 11.5 min, major exo-(-) tR = 13.0 min, minor endo-(+) tR = 15.2 min, major endo-(-) tR = 19.2 min. Endo/exo = 91:9. Ee(endo) = 94%. Ee(exo) = 45%. 2-((3S,4R,5R)-3-(4-Nitrophenyl)-2,5-diphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48ad). [α]D25 -15.5 (c 0.83, CHCl3) (88.9% ee); 1H NMR (300 MHz, CDCl3) δ 8.23 (2H, d, J = 8.9 Hz), 7.91 (1H, d, J = 6.2 Hz), 7.85 Ph N N (2H, d, J = 8.6 Hz), 7.27 (8H, m), 7.17 (1H, dt, J = 1.1 Hz, J = 7.7 O O Hz), 7.08 (1H, dd, J = 2.1 Hz, J = 7.8 Hz), 7.01 (3H, m), 5.54 (1H, d, J = 5.0 Hz), 5.43 (1H, d, J = 7.4 Hz), 5.06 (1H, dd, J = 5.0 Hz, J = 7.4 Hz); 13C NMR (75.5 MHz, CDCl3) δ 195.2 (s), 150.3 O2N (s), 148.4 (s), 147.3 (s), 145.8 (s), 139.6 (d), 135.8 (s), 129.2 endo-48ad (d), 128.8 (d), 128.5 (d), 128.0 (d), 127.8 (d), 126.8 (d), 126.2 (d), 125.6 (d), 123.9 (d), 122.3 (d), 114.2 (d), 83.7 (d), 72.0 (d), 69.9 (d); MS (FAB) m/z (%): 468 (M++1, 4), 307 (66), 289 (33), 226 (11), 154 (100), 137 (97); HRMS: 468.1547, C27H22N3O5 required 468.1559. O Ph O Ph Ph N O N O [α]D25 +4.7 (c 0.25, CHCl3) (79.4% ee); 1H NMR (300 MHz, CDCl3) δ 8.40 (1H, d, J = 8.8 Hz), 8.08 (2H, dd, J = 0.9 Hz, J = 8.9 Hz), 7.80 (2H, d, J = 8.8 Hz), 7.50 (4H, m), 7.32 (5H, m), 7.16 (3H, m), 6.98 (2H, m), 5.88 (1H, d, J = 9.9 Hz), 5.72 (1H, d, J = 9.9 Hz), 5.18 (1H, t, J = 9.9 Hz). O2N Assay of enantiomeric excess: Chiral HPLC analysis exo-48ad (Chiralcel OD-H, 50% isopropanol-50% hexane, 1.0 mL/min). Major endo-(-) tR = 14.8 min, minor endo-(+) tR = 19.5 min, minor exo-(-) tR = 23.0 min, major exo-(+) tR = 31.9 min. Endo/exo = 94:6. Ee(endo) = 89%. Ee(exo) = 79%. 171 Experimental section 2-((3R,4R,5R)-3-Cyclohexyl-2,5-diphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48ae). [α]D25 -6.7 (c 0.64, CHCl3) (60.3% ee); 1H NMR (300 MHz, CDCl3) δ 7.87 (1H, dd, J = 2.9 Hz, J = 4.6 Hz), 7.34 (2H, m), 7.29 (4H, m), 7.17 (2H, m), 7.11 (3H, m), 6.96 (1H, t, J = 7.3 Hz), 5.14 (1H, d, J = 9.0 Hz), 5.03 (1H, dd, J = 4.6 Hz, J = 9.0 Hz), 4.15 (1H, dd, J = 4.7 Hz, J = 7.5 Hz), 2.12 (1H, m), 1.82 (5H, m), 1.22 (5H, m); 13C NMR (75.5 MHz, CDCl3) δ 196.4 (s), 151.7 endo-48ae (s), 146.4 (s), 139.7 (d), 135.9 (s), 129.0 (d), 128.6 (d), 128.4 (d), 127.5 (d), 127.0 (d), 125.9 (d), 125.4 (d), 121.2 (d), 113.6 (d), 84.0 (d), 75.1 (d), 64.4 (d), 42.7 (d), 30.4 (t), 29.9 (t), 26.3 (t), 26.2 (t), 26.1 (t); MS (FAB) m/z (%): 429 (M++1, 74), 411 (32), 307 (33), 289 (18), 226 (28), 204 (26), 154 (100), 136 (82); HRMS: 429.2176, C27H29N2O3 required 429.2178. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Minor endo-(+) tR = 10.0 min, major endo-(-) tR = 11.4 min. Endo/exo = 74:26. Ee(endo) = 60%. Ee(exo) = nd. 2-((3S,4R,5R)-2-Methyl-3,5-diphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48af). [α]D25 -129.9 (c 0.99, CHCl3) (100% ee); 1H NMR (300 MHz, CDCl3) δ 7.82 (1H, m), 7.53 (2H, d, J = 6.9 Hz), 7.35 (2H, t, J = 7.3 Hz), 7.24 (7H, m), 7.14 (2H, m), 5.57 (1H, d, J = 6.0 Hz), 5.04 (1H, dd, J = 6.0 Hz, J = 8.7 Hz), 3.96 (1H, d, J = 8.6 Hz), endo-48af 2.70 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 196.8 (s), 146.5 (s), 141.6 (s), 139.6 (d), 136.9 (s), 128.49 (d), 128.426 (d), 128.1 (d), 127.8 (d), 127.5 (d), 127.4 (d), 126.1 (d), 125.5 (d), 125.0 (d), 80.6 (d), 77.8 (d), 69.2 (d), 43.1 (q); MS (FAB) m/z (%): 361 (M++1, 69), 342 (100), 326 (17), 226 (82), 209 (14), 154 (52), 136 (51); HRMS: 361.1543, C27H29N2O3 required 361.1552. [α]D25 -201.3 (c 0.99, CHCl3) (100% ee); 1H NMR (300 MHz, CDCl3) δ 7.87 (1H, d, J = 6.5 Hz), 7.55 (2H, d, J = 6.9 Hz), 7.33 (5H, m), 7.09 (4H, m), 6.89 (1H, dt, J = 1.1 Hz, J = 7.7 Hz), 6.76 (1H, dd, J = 2.2 Hz, J = 8.0 Hz), 5.83 (1H, d, J = 8.1 Hz), 5.30 exo-48af (1H, dd, J = 8.1 Hz, J = 10.3 Hz), 4.36 (1H, d, J = 10.3 Hz), 2.74 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 194.4 (s), 145.5 (s), 139.8 (d), 138.6 (s), 136.5 (s), 128.6 (d), 128.4 (d), 128.15 (d), 128.12 (d), 128.0 (d), 127.4 (d), 127.0 (d), 126.8 (d), 124.8 (d), 80.6 (d), 75.7 (d), 67.2 (d), 43.2 (q). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AS-H, 15% isopropanol-85% hexane, 1.0 mL/min). Minor exo-(+) tR = 11.2 min, major exo-(-) tR = 172 Experimental section 12.5 min, minor endo-(+) tR = 14.0 min, major endo-(-) tR = 20.0 min. Endo/exo = 66:34. Ee(endo) = 88%. Ee(exo) = 78%. 2-((3S,4R,5R)-2-Benzyl-3,5-diphenylisoxazolidine-4-carbonyl)pyridine N-oxide (48ag). [α]D25 -84.2 (c 1.00, CHCl3) (99.0% ee); 1H NMR (300 MHz, CDCl3) δ 7.84 (1H, m), 7.40 (4H, m), 7.26 (12H, m), 7.16 (2H, m), 5.57 (1H, d, J = 6.0 Hz), 5.02 (1H, dd, J = 6.0 Hz, J = 8.1 Hz), 4.29 (1H, d, J = 8.1 Hz), 4.09 (1H, d, J = 14.0 Hz), 3.96 (1H, d, J endo-48ag = 14.0 Hz); 13C NMR (75.5 MHz, CDCl3) δ 197.0 (s), 146.6 (s), 141.3 (s), 139.7 (d), 137.7 (s), 137.3 (s), 128.7 (d), 128.4 (d), 128.2 (d), 128.0 (d), 128.0 (d), 127.8 (d), 127.5 (d), 127.4 (d), 127.1 (d), 126.1 (d), 125.5 (d), 125.1 (d), 80.9 (d), 74.9 (d), 69.2 (d), 59.7 (t); MS (FAB) m/z (%): 437 (M++1, 9), 419 (16), 400 (11), 341 (12), 327 (18), 280 (23), 267 (14), 221 (32), 207 (31), 154 (154), 147 (100), 135 (48), 133 (17); HRMS: 437.1867, C28H25N2O3 required 437.1865. [α]D25 -160.7 (c 1.00, CHCl3) (99.0% ee); 1H NMR (300 MHz, CDCl3) δ 7.88 (1H, d, J = 6.1 Hz), 7.28 (10H, m), 7.08 (4H, m), 6.88 (1H, dt, J = 1.1 Hz, J = 7.7 Hz), 5.85 (1H, d, J = 8.3 Hz), 5.26 (1H, dd, J = 8.3 Hz, J = 10.4 Hz), 4.64 (1H, d, J = 10.4 Hz), exo-48ag 4.07 (1H, d, J = 14.3 Hz), 3.95 (1H, d, J = 14.3 Hz); 13C NMR (75.5 MHz, CDCl3) δ 194.6 (s), 145.9 (s), 140.2 (d), 139.4 (s), 137.4 (s), 137.3 (s), 129.4 (d), 129.2 (d), 128.7 (d), 128.5 (d), 128.4 (d), 128.4 (d), 128.3 (d), 127.8 (d), 127.5 (d), 127.4 (d), 127.3 (d), 125.2 (d), 80.8 (d), 73.0 (d), 67.5 (d), 59.9 (t). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 30% isopropanol-70% hexane, 1.0 mL/min). Minor exo-(+) tR = 10.4 min, major endo-(-) tR = 11.1 min, minor endo-(+) tR = 15.5 min, major exo-(-) tR = 17.0 min. Endo/exo = 61:39. Ee(endo) = 80%. Ee(exo) = 75%. 5.4. Enantioselective Cu(II)-BOX catalyzed Diels-Alder reactions of α’-arylsulfonyl enones 5.4.1. Synthesis and characterization of α’-arylsulfonyl enones General procedure for the synthesis of α’-arylsulfonyl enones 66a-h. α’-Arylsulfonyl enones were prepared according to procedures described in the literature.218 Compounds were prepared according by two different methods described in the following scheme: 173 Experimental section Method A: 1. LDA (2 eq.) O O O S 2. R2 O O O S O THF 1 R 50 R1 = Me 51 R1 = Cl 2 R OH 54, 56-60, 62, 63 O O O S TsOH·H2O 1 R Toluene reflux R2 R1 66a-e,g,h R1 = Me 68 R1 = Cl Method B: In the case of Method B, oxidation was carried out using Jones reagent in acetone or MnO2 in dichloromethane until completion. 4-Hydroxy-4-phenyl-1-tosylbutan-2-one (54). Method A, Yield: 85%; mp 65-70 °C; 1H NMR (300 MHz, CDCl3) δ 7.72 (2H, d, J = 8.4 Hz), 7.37-7.33 (7H, m), 5.15 (1H, dd, J = 3.5 Hz, J = 8.9 Hz), 4.22 (1H, d, J = 13.3 Hz), 4.17 (1H, d, J = 13.3 Hz), 3.17 (1H, dd, J = 8.9 Hz, J = 17.4 54 Hz), 3.04 (1H, dd, J = 3.5 Hz, J = 17.5 Hz), 2.48 (1H, br s), 13 2.46 (3H, s); C NMR (75.5 MHz, CDCl3) δ 198.1 (s), 145.5 (d), 142.2 (s), 135.4 (s), 130.0 (d), 128.6 (d), 128.2 (d), 127.8 (d), 125.6 (d), 69.7 (d), 67.5 (t), 52.7 (t), 21.7 (q); MS (FAB) m/z (%): 301 (M++1-H2O, 98), 155 (34), 136 (57), 115 (60), 104 (100); HRMS: 301.0912, C17H17O3S required 301.0898. (E)-4-Phenyl-1-tosylbut-3-en-2-one (66a). Method A, Yield: 50%; mp 122-125 °C; 1H NMR (300 MHz, CDCl3) δ 7.78 (2H, d, J = 8.4 Hz), 7.61 (1H, d, J = 16.0 Hz), 7.56 (2H, m), 7.42 (3H, m), 7.34 (2H, d, J = 7.9 Hz), 6.92 (1H, d, J = 16.0 Hz), 4.37 (2H, s), 2.43 (3H, s); 13C 66a NMR (75.5 MHz, CDCl3) δ 187.0 (s), 146.3 (d), 145.4 (s), 135.5 (s), 133.7 (s), 131.4 (d), 129.9 (d), 129.0 (d), 128.9 (d), 128.4 (d), 124.7 (d), 66.4 (t), 21.7 (q); MS (EI) m/z (%): 300 (M+, 1.6), 236 (6), 194 (3), 145 (47), 144 (46), 131 (100), 103 (32), 91 (30), 77 (19); HRMS: 300.0819, C17H16O3S required 300.0820. 174 Experimental section (E)-1-(4-Methoxyphenylsulfonyl)-4-phenylbut-3-en-2-ol (55). Method B, Yield: 95%; 1H NMR (300 MHz, CDCl3) δ 7.87 (2H, d, J = 9.0 Hz), 7.30 (4H, m), 7.23 (1H, m), 7.03 (2H, d, J = 9.0 Hz), 6.65 (1H, dd, J = 1.4 Hz, J = 15.9 Hz), 6.05 (1H, dd, J = 6.0 Hz, J = 15.8 Hz), 4.84 55 (1H, dddd, J = 1.4 Hz, J = 3.1 Hz, J = 6.0 Hz, J = 8.7 Hz), 3.88 (3H, s), 3.37 (1H, dd, J = 8.6 Hz, J = 14.3 Hz), 3.30 (1H, dd, J = 3.1 Hz, J = 14.3 Hz); 13C NMR (75.5 MHz, CDCl3) δ 164.0 (s), 135.9 (s), 131.7 (d), 130.6 (s), 130.3 (d), 128.6 (d), 128.1 (d), 127.7 (d), 126.6 (d), 114.6 (d), 67.1 (d), 62.2 (t), 55.7 (q); MS (EI) m/z (%): 318 (M+, 1.1), 300 (79), 236 (22), 210 (26), 194 (12), 155 (40), 146 (47), 128 (100), 77 (14); HRMS: 318.0936, C17H18O4S required 318.0926. (E)-1-(4-Methoxyphenylsulfonyl)-4-phenylbut-3-en-2-one (67). Method B (oxidation with Jones reagent), Yield: 65%; mp 112-116 °C; 1H NMR (300 MHz, CDCl3) δ 7.81 (2H, d, J = 9.0 Hz), 7.62 (1H, d, J = 16.0 Hz), 7.55 (2H, ddd, J = 2.4 Hz, J = 5.5 Hz, J = 9.2 Hz), 7.42 (3H, 67 m), 6.99 (2H, d, J = 9.0 Hz), 6.93 (1H, d, J = 16.0 Hz), 4.37 (2H, s), 3.85 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 187.2 (s), 164.1 (s), 146.3 (d), 133.7 (s), 131.4 (d), 130.7 (d), 130.0 (s), 129.0 (d), 128.9 (d), 124.7 (d), 114.4 (d), 66.6 (t), 55.7 (q); MS (EI) m/z (%): 316 (M+, 1.0), 252 (4), 210 (18), 171 (5), 144 (40), 131 (100), 124 (16), 103 (40), 77 (38); HRMS: 316.0766, C17H16O4S required 316.0769. 1-(4-Chlorophenylsulfonyl)-4-hydroxy-4-phenylbutan-2-one (56). Method A, Yield: 73%; mp 116-119°C; 1H NMR (300 MHz, CDCl3) δ 7.76 (2H, d, J = 8.8 Hz), 7.52 (2H, d, J = 8.8 Hz), 7.34 (5H, m), 5.15 (1H, dd, J = 3.6 Hz, J = 8.9 Hz), 4.21 (2H, s), 3.20 (1H, dd, J = 8.9 Hz, J = 17.4 Hz), 56 3.04 (1H, dd, J = 3.6 Hz, J = 17.4 Hz), 2.73 (1H, br); 13C NMR (75.5 MHz, CDCl3) δ 197.7 (s), 142.1 (s), 141.3 (s), 136.7 (s), 129.9 (d), 129.7 (d), 128.7 (d), 128.1 (d), 125.7 (d), 69.9 (d), 67.3 (t), 52.7 (t); MS (FAB) m/z (%): 321 (M++1-H2O, 6), 307 (45), 288 (17), 154 (100), 149 (24), 137 (68), 107 (17); HRMS: 321.0346, C16H14ClO3S required 321.0352. 175 Experimental section (E)-1-(4-Chlorophenylsulfonyl)-4-phenylbut-3-en-2-one (68). Method A, Yield: 62%; mp 141-144 °C; 1H NMR (300 MHz, CDCl3) δ 7.84 (2H, d, J = 8.8 Hz), 7.64 (1H, d, J = 16.0 Hz), 7.58 (2H, m), 7.53 (2H, d, J = 8.8 Hz), 7.42 (3H, m), 6.92 (1H, d, J = 16.0 Hz), 4.40 (2H, s); 13C NMR (75.5 68 MHz, CDCl3) δ 186.8 (s), 146.8 (d), 141.2 (s), 136.8 (s), 133.6 (s), 131.6 (d), 130.0 (d), 129.6 (d), 129.1 (d), 128.9 (d), 124.5 (d), 66.1 (t); MS (EI) m/z (%): 320 (M+, 3.7), 256 (5), 214 (2), 145 (97), 144 (37), 131 (100), 129 (16), 103 (36), 77 (13); HRMS: 320.0272, C17H16ClO4S required 320.0274. 4-Hydroxy-4-(4-methoxyphenyl)-1-tosylbutan-2-one (57). Method A, Yield: 60%; 1H NMR (300 MHz, CDCl3) δ 7.72 (2H, d, J = 8.4 Hz), 7.35 (2H, d, J = 8.0 Hz), 7.27 (2H, d, J = 8.2 Hz), 6.88 (2H, d, J = 8.7 Hz), 5.09 (1H, dd, J = 3.5 Hz, J = 8.9 57 Hz), 4.21 (1H, d, J = 13.3 Hz), 4.15 (1H, d, J = 13.3 Hz), 3.80 (3H, s), 3.17 (1H, dd, J = 9.0 Hz, J = 17.4 Hz), 3.03 (1H, dd, J = 3.6 Hz, J = 17.4 Hz), 2.45 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 198.2 (s), 159.3 (s), 145.6 (s), 135.5 (s), 134.4 (s), 130.0 (d), 128.3 (d), 127.0 (d), 114.0 (d), 69.5 (t), 67.7 (d), 55.5 (q), 52.7 (t), 21.7 (q); MS (EI) m/z (%): 330 (M+H2O, 88), 224 (24), 175 10), 161 (100), 133 (11), 91 (15); HRMS: 330.0931, C18H18O4S required 330.0926. (E)-4-(4-Methoxyphenyl)-1-tosylbut-3-en-2-one (66b). Method A, Yield: 61%; mp 120-124 °C; 1H NMR (300 MHz, CDCl3) δ 7.77 (2H, d, J = 8.3 Hz), 7.58 (1H, d, J = 15.9 Hz), 7.52 (2H, d, J = 8.6 Hz), 7.34 (2H, d, J = 7.9 Hz), 6.92 (2H, d, J 66b = 8.8 Hz), 6.80 (1H, d, J = 15.9 Hz), 4.35 (2H, s), 3.85 (3H, s), 2.43 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 186.8 (s), 162.3 (s), 145.2 (d), 145.3 (s), 135.6 (s), 130.8 (d), 129.8 (d), 128.4 (d), 126.4 (s), 122.4 (d), 114.5 (d), 66.3 (t), 55.4 (q), 21.6 (q); MS (EI) m/z (%): 330 (M+, 51), 224 (30), 175 (14), 161 (100), 133 (11), 91 (10); HRMS: 330.0921, C18H18O4S required 330.0926. 176 Experimental section 4-(4-Bromophenyl)-4-hydroxy-1-tosylbutan-2-one (58). Method A, Yield: 55%; mp 103-107 °C; 1H NMR (300 MHz, CDCl3) δ 7.69 (2H, d, J = 8.4 Hz), 7.47 (2H, d, J = 8.5 Hz), 7.35 (2H, d, J = 8.0 Hz), 7.23 (2H, d, J = 8.6 Hz), 5.11 (1H, dd, J = 3.9 Hz, 58 J = 8.4 Hz), 4.20 (1H, d, J = 13.2 Hz), 4.15 (1H, d, J = 13.2 Hz), 3.14 (1H, dd, J = 8.4 Hz, J = 17.8 Hz), 3.05 (1H, dd, J = 3.9 Hz, J = 17.8 Hz), 2.65 (1H, br), 2.46 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 198.0 (s), 145.7 (s), 141.2 (s), 135.4 (s), 131.7 (d), 130.0 (d), 128.2 (d), 127.4 (d), 121.7 (s), 69.1 (d), 67.6 (t), 52.5 (t), 21.7 (q); MS (EI) m/z (%): 378 (M+-H2O, 11), 224 (15), 212 (20), 209 (81), 183 (62), 170 (45), 148 (41), 139 (21), 105 (16), 102 (22), 91 (100), 77 (10), 65 (22). HRMS: 377.9935, C17H15BrO3S required 377.9925. (E)-4-(4-Bromophenyl)-1-tosylbut-3-en-2-one (66c). Method A, Yield: 40%; mp 123-125 °C; 1H NMR (300 MHz, CDCl3) δ 7.76 (2H, d, J = 8.2 Hz), 7.55 (1H, d, J = 16.0 Hz), 7.54 (2H, d, J = 8.6 Hz), 7.41 (2H, d, J = 8.4 Hz), 7.34 (2H, d, J = 66c 8.0 Hz), 6.92 (1H, d, J = 16.0 Hz), 4.36 (2H, s), 13 2.43 (3H, s); C NMR (75.5 MHz, CDCl3) δ 186.9 (s), 145.5 (s), 144.8 (d), 135.5 (s), 132.6 (s), 132.3 (d), 130.1 (d), 129.9 (d), 128.4 (d), 125.8 (d), 125.1 (d), 66.5 (t), 21.7 (q); MS (EI) m/z (%): 378 (M+, 16), 314, (11), 272 (11), 224 (22), 222 (21), 212 (10), 209 (100), 183 (11), 144 (24), 139 (16), 128 (17), 102 (54), 91 (52), 65 (22); HRMS: 377.9936, C17H15BrO3S required 377.9925. 4-Hydroxy-4-(4-nitrophenyl)-1-tosylbutan-2-one (59). Method A, Yield: 55%; mp 109-113 °C; 1H NMR (300 MHz, CDCl3) δ 8.20 (2H, d, J = 8.8 Hz), 7.72 (2H, d, J = 8.3 Hz), 7.54 (2H, d, J = O2N Me 8.8 Hz), 7.36 (2H, d, J = 8.3 Hz), 5.28 (1H, dd, J 59 = 5.8Hz, J = 6.3Hz), 4.20 (2H, s), 3.16 (1H, d, J = 1.4 Hz), 3.14 (1H, s), 2.78 (1H, br), 2.46 (3H, 13 s); C NMR (75.5 MHz, CDCl3) δ 197.7 (s), 149.3 (s), 147.4 (s), 145.9 (s), 135.3 (s), 130.1 (d), 128.2 (d), 126.5 (d), 123.8 (d), 68.7 (d), 67.6 (t), 52.4 (t), 21.7 (q); MS (EI) m/z (%): 345 (M+-H2O, 0.1), 281 (2), 239 (2), 212 (5), 176 (14), 170 (17), 151 (32), 91 (100), 77 (31), 65 (30); HRMS: 345.0669, C17H15NO5S required 345.0671. OH O O O S 177 Experimental section (E)-4-(4-Nitrophenyl)-1-tosylbut-3-en-2-one (66d). Method A, Yield: 72%; mp 153-156 °C; 1H NMR (300 MHz, CDCl3) δ 8.24 (2H, d, J = 8.7 Hz), 7.76 (2H, d, J = 8.2 Hz), 7.71 (2H, d, J = 8.8 Hz), 7.64 (1H, d, J = 16.1 Hz), 7.35 (2H, d, J 66d = 8.5 Hz), 7.07 (1H, d, J = 16.1 Hz), 4.41 (2H, s), 2.43 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 186.7 (s), 148.9 (s), 145.6 (s), 142.7 (d), 139.8 (s), 135.4 (s), 130.0 (d), 129.3 (d), 128.3 (d), 128.0 (d), 124.1 (d), 66.5 (t), 21.6 (q); MS (EI) m/z (%): 345 (M+, 2.2), 281 (34), 239 (18), 189 (48), 176 (100), 155 (8), 91 (33). HRMS: 345.0669, C17H15NO5S required 345.0671. (E)-4-Hydroxy-6-phenyl-1-tosylhex-5-en-2-one (60). Method A, Yield: 68%; mp 64-67 °C; 1H NMR (300 MHz, CDCl3) δ 7.74 (2H, d, J = 8.3 Hz), 7.38-7.25 (7H, m), 6.63 (1H, d, J = 16.0 Hz), 6.19 (1H, dd, J = 6.1 Hz, J = 16.0 Hz), 4.75 (1H, q, J = 5.8 Hz), 4.21 (2H, s), 3.06 60 (1H, dd, J = 7.0 Hz, J = 17.5 Hz), 3.00 (1H, dd, J = 4.9 Hz, J = 17.8 Hz), 2.51 (1H, s), 2.43 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 198.0 (s), 145.6 (s), 136.2 (s), 135.4 (s), 130.9 (d), 130.0 (d), 129.6 (d), 128.6 (d), 128.3 (d), 127.9 (d), 126.5 (d), 68.4 (d), 67.7 (t), 50.8 (t), 21.7 (q); MS (EI) m/z (%): 326 (M+-H2O, 5.2), 212 (7), 171 (30), 155 (36), 131 (52), 105 (18), 91 (100), 77 (26); HRMS: 326.0978, C19H18O3S required 326.0977. (3E,5E)-6-Phenyl-1-tosylhexa-3,5-dien-2-one (66e). Method A, Yield: 88%; mp 135-139 °C; 1H NMR (300 MHz, CDCl3) δ 7.77 (2H, d, J = 8.4 Hz), 7.50-7.33 (8H, m), 7.03 (1H, d, J = 15.5 Hz), 6.91 (1H, dd, J = 10.5 Hz, J = 15.5 Hz), 6.49 (1H, d, J = 15.2 Hz), 4.31 (2H, s), 66e 2.43 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 186.9 (s), 146.4 (d), 145.3 (s), 144.0 (d), 135.6 (s), 135.6 (s), 129.9 (d), 129.7 (d), 128.9 (d), 128.4 (d), 128.0 (d), 127.5 (d), 126.1 (d), 66.2 (t), 21.7 (q); MS (EI) m/z (%): 326 (M+, 35), 249 (2), 220 (4), 171 (100), 157 (38), 128 (54), 105 (39), 91 (27); HRMS: 326.0977, C19H18O3S required 326.0977. 178 Experimental section 1-Tosylbut-3-en-2-ol (61). Method B, Yield: 88%; 1H NMR (300 MHz, CDCl3) δ 7.80 (2H, d, J = 8.3 Hz), 7.37 (2H, d, J = 7.9 Hz), 5.75 (1H, ddd, J = 5.4 Hz, J = 10.4 Hz, J = 17.1 Hz), 5.35 (t, 1H, J = 1.3 Hz), 5.29 (t, 1H, J = 1.3 Hz), 5.18 (t, 1H, J = 1.3 Hz), 5.15 (t, 1H, J = 1.3 61 Hz), 4.66 (1H, ddd, J = 2.9 Hz, J = 5.4 Hz, J = 7.8 Hz), 3.27 (1H, dd, J = 7.8 Hz, J = 13.7 Hz), 3.20 (1H, dd, J = 2.9 Hz, J = 13.7 Hz), 3.24 (1H, br), 2.45 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 145.2 (s), 136.8 (d), 136.1 (s), 130.0 (d), 128.0 (d), 116.6 (t), 66.9 (d), 61.8 (t), 21.6 (q); MS (EI) m/z (%): 226 (M+, 0.7), 208 (20), 170 (50), 157 (65), 155 (42), 139 (74), 105 (45), 91 (100), 71 (40), 65 (59); HRMS: 226.0664, C11H14O3S required 226.0664. 1-Tosylbut-3-en-2-one (66f). Method B (oxidation with Jones reagent), Yield: 55%; mp 35-38 °C; 1H NMR (300 MHz, CDCl3) δ 7.73 (2H, d, J = 8.4 Hz), 7.32 (2H, d, J = 7.9 Hz), 6.50 (1H, dd, J = 10.4 Hz, J = 17.5 Hz), 6.31 (1H, dd, J = 0.7 Hz, J = 17.5 Hz), 5.97 (1H, dd, 66f J = 0.7 Hz, J = 10.4 Hz), 4.31 (2H, s), 2.42 (3H, s); NMR 13C (75.5 MHz, CDCl3) δ 187.8 (s), 145.3 (s), 135.5 (s), 135.2 (d), 132.1 (t), 129.8 (d), 128.2 (d), 64.7 (t), 21.5 (q); MS (EI) m/z (%): 224 (M+, 0.6), 170 (15), 170 (15), 155 (28), 139 (42), 108 (6), 91 (100), 77 (11), 65 (29); HRMS: 224.0502, C11H12O3S required 224.0507. 4-Hydroxy-1-tosylpentan-2-one (62) Method A, Yield: 96%; mp 62-65 °C; 1H NMR (300 MHz, CDCl3) δ 7.75 (2H, d, J = 8.2 Hz), 7.36 (2H, d, J = 8.2 Hz), 4.23 (1H, ddt, J = 3.2 Hz, J = 5.4 Hz, J = 6.4 Hz), 4.20 (1H, d, J = 15.1 Hz), 4.15 (1H, d, J = 13.3 Hz), 2.88 62 (1H, dd, J = 3.7 Hz, J = 17.8 Hz), 2.80 (1H, dd, J = 8.1 Hz, J = 17.8 Hz), 2.50 (1H, br), 2.45 (3H, s), 1.20 (3H, d, 13 J = 6.4 Hz); C NMR (75.5 MHz, CDCl3) δ 199.0 (s), 145.6 (s), 135.5 (s), 130.0 (d), 128.2 (d), 67.5 (t), 63.7 (d), 52.4 (t), 22.5 (q), 21.7 (q); MS (EI) m/z (%): 238 (M+-H2O, 3), 212 (14), 174 (18), 170 (29), 155 (60), 148 (30), 139 (32), 105 (20), 91 (100); HRMS: 238.0660, C12H14O3S required 238.0664. 179 Experimental section (E)-1-Tosylpent-3-en-2-one (66g). Method A, Yield: 66%; mp 60-63 °C; 1H NMR (300 MHz, CDCl3) δ 7.74 (2H, d, J = 8.4 Hz), 7.34 (2H, d, J = 8.0 Hz), 6.98 (1H, qd, J = 6.9 Hz, J = 15.7 Hz), 6.31 (1H, qd, J = 1.6 Hz, J = 15.7 Hz), 4.25 (2H, s), 2.44 (3H, s), 66g 1.95 (dd, 3H, J = 1.6 Hz, J = 6.9 Hz); 13C NMR (75.5 MHz, CDCl3) δ 187.0 (s), 147.8 (d), 145.3 (s), 135.6 (s), 130.9 (d), 129.8 (d), 128.3 (d), 65.3 (t), 21.6 (q), 18.6 (q); MS (EI) m/z (%): 238 (M+, 4), 174 (29), 155 (8), 108 (11), 91 (43), 69 (100); HRMS: 238.0660, C12H14O3S required 238.0664. 4-Hydroxy-5,5-dimethyl-1-tosylhexan-2-one (63). Method A, Yield: 72%; mp 44-47 °C; 1H NMR (300 MHz, CDCl3) δ 7.76 (2H, d, J = 8.4 Hz), 7.36 (2H, d, J = 7.9 Hz), 4.24 (1H, d, J = 13.3 Hz), 4.18 (1H, d, J = 13.3 Hz), 3.71 (1H, dd, J = 2.7 Hz, J = 9.7 Hz), 2.83 (1H, dd, J 63 = 2.7 Hz, J = 17.1 Hz), 2.73 (1H, dd, J = 9.7 Hz, J = 17.1 Hz), 2.44 (3H, s), 2.27 (1H, br), 0.89 (9H, s); 13C NMR (75.5 MHz, CDCl3) δ 199.6 (s), 145.5 (s), 135.6 (s), 130.0 (s), 128.3 (d), 75.0 (d), 67.7 (t), 46.2 (t), 34.4 (s), 25.4 (q), 21.7 (q); MS (EI) m/z (%): 280 (M+-H2O, 0.6), 265 (2), 212 (6), 170 (20), 155 (43), 125 (28), 91 (100); HRMS: 280.1126, C15H20O3S required 280.1133. (E)-5,5-Dimethyl-1-tosylhex-3-en-2-one (66h). Method A, Yield: 87%; mp 97-101 °C; 1H NMR (300 MHz, CDCl3) δ 7.75 (2H, d, J = 8.4 Hz), 7.34 (2H, d, J = 8.0 Hz), 6.89 (1H, d, J = 16.0 Hz), 6.12 (1H, d, J = 16.0 Hz), 4.28 (2H, s), 2.43 (3H, s), 1.06 (9H, s); 13C NMR 66h (75.5 MHz, CDCl3) δ 187.8 (s), 161.7 (d), 145.3 (s), 135.8 (s), 129.8 (d), 128.4 (d), 124.6 (d), 65.5 (t), 34.2 (s), 28.4 (q), 21.6 (q); MS (EI) m/z (%): 280 (M+, 1.1), 265 (7), 216 (11), 155 (16), 139 (12), 125 (100), 111 (51), 109 (65), 91 (64), 83 (37); HRMS: 280.1123, C15H20O3S required 280.1133. 3-Methyl-1-tosylbut-3-en-2-ol (64). Method B, Yield: 61%; 1H NMR (300 MHz, CDCl3) δ 7.82 (2H, d, J = 8.4 Hz), 7.38 (2H, d, J = 7.9 Hz), 5.03 (1H, q, J = 1.0 Hz), 4.89 (1H, dp, J = 0.6 Hz, J = 1.5 Hz), 4.57 (1H, dd, J = 2.4 Hz, J = 9.1 Hz), 3.29 (1H, dd, J = 9.1 Hz, J = 14.3 Hz), 64 180 Experimental section 3.21 (1H, dd, J = 2.5 Hz, J = 14.3 Hz), 2.95 (1H, br), 2.46 (3H, s), 1.67 (3H, t, J = 1.0 Hz); 13 C NMR (75.5 MHz, CDCl3) δ 145.2 (s), 143.6 (s), 136.1 (s), 130.0 (d), 128.0 (d), 112.9 (t), 69.6 (d), 61.1 (t), 21.7 (q), 17.8 (q); MS (EI) m/z (%): 240 (M+, 0.5), 222 (16), 170 (11), 157 (30), 155 (27), 139 (70), 105 (24), 91 (100), 84 (26), 65 (23); HRMS: 240.0831, C12H16O3S required 240.0820. 3-Methyl-1-tosylbut-3-en-2-one (66i). Method B (oxidation with Jones reagent), Yield: 68%; 1H NMR (300 MHz, CDCl3) δ 7.76 (2H, d, J = 8.4 Hz), 7.35 (2H, d, J = 7.9 Hz), 6.07 (1H, d, J = 0.8 Hz), 6.02 (q, 1H, J = 1.5 Hz), 4.47 (2H, s), 2.44 (3H, s), 1.85 (1H, dd, J = 0.9 Hz, J = 66i 1.4 Hz); 13C NMR (75.5 MHz, CDCl3) δ 189.5 (s), 145.3 (s), 144.1 (s), 135.8 (s), 129.8 (d), 129.5 (t), 128.5 (d), 62.6 (t), 21.7 (q), 17.3 (q); MS (EI) m/z (%): 238 (M+, 7), 226 (4), 212 (4), 197 (4), 174 (15), 155 (60), 139 (54), 132 (17), 123 (13), 105 (17), 91 (100), 69 (26); HRMS: 238.0669, C12H14O3S required 238.0664. (E)-3-Methyl-1-tosylpent-3-en-2-ol (65). Method B2, Yield: 88%; mp 67-70 °C; 1H NMR (300 MHz, CDCl3) δ 7.80 (2H, d, J = 8.4 Hz), 7.37 (2H, d, J = 7.9 Hz), 5.53 (1H, q, J = 6.7 Hz), 4.51 (1H, dd, J = 1.6 Hz, J = 9.5 Hz), 3.31 (1H, dd, J = 9.6 Hz, J = 14.2 Hz), 65 3.16 (1H, dd, J = 2.1 Hz, J = 14.3 Hz), 2.98 (1H, br s), 2.45 (3H, s), 1.54 (6H, m); 13C NMR (75.5 MHz, CDCl3) δ 145.0 (s), 136.2 (s), 134.3 (s), 129.9 (d), 127.9 (d), 122.6 (d), 71.3 (d), 61.1 (t), 21.6 (q), 12.9 (q), 11.3 (q); MS (FAB) m/z (%): 255 (M++1, 5), 237 (100), 157 (30), 153 (30), 137 (30). HRMS: 255.1057, C13H19O3S required 255.1055. (E)-3-Methyl-1-tosylpent-3-en-2-one (66j). Method B (oxidation with MnO2), Yield: 77%; mp 7679 °C; 1H NMR (300 MHz, CDCl3) δ 7.74 (2H, d, J = 8.4 Hz), 7.33 (2H, d, J = 7.9 Hz), 6.83 (1H, qq, J = 1.2 Hz, J = 6.9 Hz), 4.44 (2H, s), 2.43 (3H, s), 1.90 (3H, qd, J = 1.0 66j Hz, J = 6.9 Hz), 1.74 (3H, qd, J = 1.0 Hz, J = 1.2 Hz); 13 NMR C (75.5 MHz, CDCl3) δ 188.8 (s), 145.0 (s), 143.4 (d), 138.2 (s), 135.9 (s), 129.7 (d), 128.4 (d), 62.4 (t), 21.6 (q), 15.3 (q), 10.9 (q); MS (FAB) m/z (%): 253 (M++1, 100), 154 (30), 137 (32); HRMS: 253.0891, C13H17O3S required 253.0898. 181 Experimental section 5.4.2. Enantioselective catalyzed Diels-Alder reaction 5.4.2.1. General procedure for the catalytic enantioselective Diels-Alder reaction Cu(OTf)2 (9.0 mg, 0.025 mmol) contained in a dry Schlenk tube was heated at 90 °C under vacuum for 1 h. After this time ligand 33a (7.5 mg, 0.025 mmol), CH2Cl2 (0.75 mL) and toluene (0.75 mL) were added under nitrogen atmosphere and the mixture was stirred for 1 h at rt. Then, α’-arylsulfonyl enone 66 (0.25 mmol) was added and the mixture stirred for 0.5 h. After this time, the solution was placed in a bath at 0 °C and the diene 23 (1.8 mmol) was added. After completion of the reaction (TLC), the Diels-Alder product 69 was isolated directly by flash chromatography on silica gel. 5.4.2.2. Characterization of the products See Tabla 22 (page 111) and Tabla 23 (page 112) for yields, enantiomeric excesses and isomeric ratios. 1-((1R,2S,3S,4S)-3-Phenylbicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (69aa). mp 120-123 °C; [α]D25 +121.7 (c 0.92, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.68 (2H, d, J = 8.3 Hz), 7.31 (4H, m), 7.21 (3H, m), 6.36 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.92 (1H, dd, J = 2.7 Hz, J = 5.7 Hz), 4.31 (1H, d, J = 13.7 Hz), 69aa 4.07 (1H, d, J = 13.7 Hz), 3.44 (1H, dd, J = 3.4 Hz, J = 5.0 Hz), 3.39 (1H, s), 3.10 (1H, dd, J = 1.3 Hz, J = 4.9 Hz), 2.99 (1H, d, J = 1.5 Hz), 2.43 (3H, s), 1.85 (1H, d, J = 8.7 Hz), 1.60 (1H, ddd, J = 1.6 Hz, J = 3.3 Hz, J = 8.7 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.0 (s), 145.2 (s), 143.1 (s), 139.5 (d), 135.7 (s), 132.7 (d), 129.8 (d), 128.5 (d), 128.33 (d), 127.4 (d), 126.2 (d), 65.8 (t), 60.9 (d), 49.0 (d), 47.4 (t), 46.5 (d), 45.6 (d), 21.6 (q); MS (FAB) m/z (%): 367 (M++1, 4), 301 (100), 155 (24); HRMS: 367.1371, C22H23O3S required 367.1362. Exo-69aa (significant peaks, taken from the endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 6.01 (1H, dd, J = 2.9 Hz, J = 5.6 Hz), 4.22 (1H, d, J = 13.6 Hz), 3.54 (1H, dd, J = 3.4 Hz, J = 5.7 Hz), 3.15 (1H, d, J = 1.3 Hz), 1.49 (1H, ddd, J = 1.6 Hz, J = 3.3 Hz, J = 8.8 Hz); 13C NMR (75.5 MHz, CDCl3) δ 199.4 (s), 141.9 (s), 136.4 (d), 136.3 (d), 128.2 (d), 127.8 (d), 126.6 (d), 66.6 (t), 59.6 (d), 49.3 (d), 47.6 (t), 47.3 (d), 46.6 (d). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Exo (both) tR = 15.8 min, major endo-(+) tR = 17.6 min, minor endo-(-) tR = 22.5 min. Endo/exo = 93:7. Ee(endo) = 95%. Ee(exo) = nd. 182 Experimental section 2-(4-Methoxyphenylsulfonyl)-1-((1R,2S,3S,4S)-3-phenylbicyclo[2.2.1]hept-5-en-2yl)ethanone (70). mp 95-98 °C; [α]D25 +134.4 (c 1.007, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.71 (2H, d, J = 9.0 Hz), 7.31 (2H, m), 7.21 (3H, m), 6.94 (2H, d, J = 9.0 Hz), 6.37 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.93 (1H, dd, J = 2.7 Hz, J = 5.7 70 Hz), 4.30 (1H, d, J = 13.6 Hz), 4.06 (1H, d, J = 13.6 Hz), 3.86 (3H, s), 3.44 (1H, dd, J = 3.4 Hz, J = 5.0 Hz), 3.39 (1H, s), 3.10 (1H, dd, J = 1.4 Hz, J = 5.0 Hz), 2.99 (1H, d, J = 1.6 Hz), 1.86 (1H, d, J = 8.7 Hz), 1.61 (1H, ddd, J = 1.7 Hz, J = 3.4 Hz, J = 8.7 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.2 (s), 164.00 (s), 143.2 (s), 139.5 (d), 132.8 (d), 130.6 (d), 130.1 (s), 128.6 (d), 127.4 (d), 126.3 (d), 114.3 (d), 66.0 (t), 60.9 (d), 55.6 (q), 49.1 (d), 47.5 (t), 46.5 (d), 45.6 (d); MS (FAB) m/z (%): 383 (M++1, 1.2), 317 (76), 307 (45), 288 (17), 154 (100), 146 (19), 135 (57); HRMS: 383.1321, C22H23O4S required 383.1312. Exo-70 (significant peaks, taken from the endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 6.01 (1H, dd, J = 2.9 Hz, J = 5.6 Hz), 4.22 (1H, d, J = 13.5 Hz), 4.11 (1H, d, J = 8.1 Hz), 3.54 (1H, dd, J = 3.4 Hz, J = 5.7 Hz), 3.14 (1H, d, J = 1.4 Hz), 2.96 (1H, d, J = 1.5 Hz), 1.49 (1H, ddd, J = 1.7 Hz, J = 3.4 Hz, J = 8.8 Hz); 13C NMR (75.5 MHz, CDCl3) δ 199.5 (s), 167.0 (s), 136.4 (d), 136.3 (d), 130.1 (s), 128.2 (d), 127.8 (d), 126.6 (d), 66.8 (t), 59.5 (d), 49.3 (d), 47.7 (t), 47.3 (d), 46.3 (d). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Exo (both) tR = 35.0 min, major endo-(+) tR = 39.7 min, minor endo-(-) tR = 48.2 min. Endo/exo = 93:7. Ee(endo) = 95%. Ee(exo) = nd. 2-(4-Chlorophenylsulfonyl)-1-((1R,2S,3S,4S)-3-phenylbicyclo[2.2.1]hept-5-en-2yl)ethanone (71). mp 107-110 °C; [α]D25 +142.1 (c 0.93, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.73 (2H, d, J = 8.8 Hz), 7.47 (2H, d, J = 8.8 Hz), 7.33 (2H, m), 7.23 (3H, m), 6.38 (1H, dd, J = 3.2 Hz, J = 5.7 Hz), 5.94 (1H, dd, J = 2.5 Hz, J = 5.6 Hz), 4.31 71 (1H, d, J = 14.0 Hz), 4.10 (1H, d, J = 14.0 Hz), 3.41 (1H, dd, J = 3.4 Hz, J = 4.9 Hz), 3.39 (1H, s), 3.08 (1H, dd, J = 1.4 Hz, J = 4.8 Hz), 2.99 (1H, d, J = 1.5 Hz), 1.87 (1H, d, J = 8.8 Hz), 1.62 (1H, ddd, J = 1.6 Hz, J = 3.3 Hz, J = 8.6 Hz); 13C NMR (75.5 MHz, CDCl3) δ 197.9 (s), 143.0 (s), 141.0 (s), 139.7 (d), 137.0 (s), 132.7 (d), 129.9 (d), 129.5 (d), 128.7 (d), 127.4 (d), 126.4 (d), 65.6 (t), 61.1 (d), 49.1 (d), 47.5 (t), 46.4 (d), 45.8 (d); MS (EI) m/z (%): 386 (M+, 0.1), 321 (10), 256 (5.5), 214 (2.1), 145 (93), 131 (100), 103 (27), 66 (13); HRMS: 386.0735, C21H19ClO3S required 386.0743. 183 Experimental section Exo-71 (significant peaks, taken from the endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 6.03 (1H, dd, J = 2.9 Hz, J = 5.5 Hz), 4.23 (1H, d, J = 13.9 Hz), 4.13 (1H, d, J = 13.8 Hz), 3.53 (1H, dd, J = 3.4 Hz, J = 5.8 Hz), 3.15 (1H, d, J = 1.6 Hz), 2.93 (1H, dd, J = 1.4 Hz, J = 5.7 Hz), 1.51 (1H, ddd, J = 1.6 Hz, J = 3.2 Hz, J = 9.0 Hz); 13C NMR (75.5 MHz, CDCl3) δ 136.4 (d), 136.3 (d), 128.3 (d), 127.8 (d), 66.4 (t), 59.8 (d), 49.4 (d), 47.8 (t), 47.4 (d), 46.3 (d). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Major exo tR = 17.4 min, minor exo tR = 19.8 min, minor endo-(-) tR = 21.3 min, major endo-(+) tR = 31.1 min. Endo/exo = 93:7. Ee(endo) = 97%. Ee(exo) = 98%. 1-((1R,2S,3S,4S)-3-(4-Methoxyphenyl)bicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (69ba). mp 112-114 °C; [α]D25 +146.7 (c 0.94, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.67 (2H, d, J = 8.4 Hz), 7.29 (2H, d, J = 8.0 Hz), 7.14 (2H, d, J = 8.4 Hz), 6.85 (2H, d, J = 8.8 Hz), Me 6.35 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.92 (1H, dd, J = 2.7 OMe Hz, J = 5.6 Hz), 4.28 (1H, d, J = 13.6 Hz), 4.05 (1H, d, J = 69ba 13.6 Hz), 3.80 (3H, s), 3.41 (1H, dd, J = 3.4 Hz, J = 5.1 Hz), 3.36 (1H, d, J = 0.7 Hz), 3.01 (1H, dd, J = 1.3 Hz, J = 5.0 Hz), 2.92 (1H, d, J = 1.6 Hz), 2.43 (3H, s), 1.83 (1H, d, J = 8.7 Hz), 1.59 (1H, ddd, J = 1.7 Hz, J = 3.4 Hz, J = 8.7 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.1 (s), 158.0 (s), 145.2 (s), 139.4 (d), 135.7 (s), 135.1 (s), 132.8 (d), 129.8 (d), 128.4 (d), 128.4 (d), 113.9 (d), 65.8 (t), 61.1 (d), 55.3 (q), 49.4 (d), 47.4 (t), 46.4 (d), 45.1 (d), 21.7 (q); MS (FAB) m/z (%): 397 (M++1, 0.7), 331 (76), 306 (38), 288 (15), 154 (100), 149 (42), 136 (66); HRMS: 396.1384, C23H24O4S required 396.1395. O O O S Exo-69ba (significant peaks, taken from the endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 7.06 (2H, d, J = 8.5 Hz), 6.01 (1H, dd, J = 2.8 Hz, J = 5.5 Hz), 4.19 (1H, d, J = 13.5 Hz), 3.12 (1H, s), 1.48 (1H, ddd, J = 1.7 Hz, J = 3.4 Hz, J = 8.9 Hz). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 20% isopropanol-80% hexane, 1.0 mL/min). Exo (both) tR = 18.8 min, major endo-(+) tR = 21.6 min, minor endo-(-) tR = 26.3 min. Endo/exo = 96:4. Ee(endo) = 95%. Ee(exo) = nd. 184 Experimental section 1-((1R,2S,3S,4S)-3-(4-Bromophenyl)bicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (69ca). mp 127-130 °C; [α]D25 +129.9 (c 1.01, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.67 (2H, d, J = 8.4 Hz), 7.41 (2H, d, J = 8.5 Hz), 7.30 (2H, d, J = 8.0 Hz), 7.08 (2H, d, J = 8.4 Hz), Me 6.36 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.90 (1H, dd, J = 2.6 Br Hz, J = 5.6 Hz), 4.33 (1H, d, J = 13.5 Hz), 4.04 (1H, d, J = 69ca 13.5 Hz), 3.40 (1H, s), 3.38 (1H, dd, J = 3.4 Hz, J = 4.8 Hz), 3.06 (1H, dd, J = 1.1 Hz, J = 4.7 Hz), 2.97 (1H, d, J = 1.5 Hz), 2.43 (3H, s), 1.79 (1H, d, J = 8.8 Hz), 1.61 (1H, ddd, J = 1.7 Hz, J = 3.3 Hz, J = 8.8 Hz); 13 C NMR (75.5 MHz, CDCl3) δ 197.7 (s), 145.4 (s), 142.3 (s), 139.5 (d), 135.6 (s), 132.6 (d), 131.5 (d), 129.8 (d), 129.1 (d), 128.3 (d), 120.0 (s), 65.8 (t), 61.4 (d), 48.8 (d), 47.5 (t), 46.6 (d), 44.9 (d), 21.7 (q); MS (EI) m/z (%): 444 (M+, 0.1), 380 (32), 314 (17), 274 (14), 224 (22), 208 (100), 206 (5), 183 (9), 105 (15), 65 (5); HRMS: 444.0410, C22H21BrO3S required 444.0395. O O O S Exo-69ca (significant peaks, taken from the endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 7.63 (2H, d, J = 8.3 Hz), 7.01 (2H, d, J = 8.4 Hz), 5.99 (1H, dd, J = 2.8 Hz, J = 5.6 Hz), 4.26 (1H, d, J = 13.4 Hz), 3.55 (1H, dd, J = 3.3 Hz, J = 5.6 Hz), 3.14 (1H, d, J = 1.9 Hz), 2.92 (1H, dd, J = 1.4 Hz, J = 5.6 Hz), 1.48 (1H, ddd, J = 1.5 Hz, J = 3.1 Hz, J = 9.0 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.9 (s), 136.6 (s), 136.1 (s), 131.3 (d), 129.5 (d), 66.7 (t), 59.6 (d), 49.1 (d), 46.5 (d). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak IC, 25% isopropanol-75% hexane, 1.0 mL/min). Minor exo tR = 38.5 min, major exo tR = 41.0 min, minor endo-(-) tR = 42.8 min, major endo-(+) tR = 45.0 min. Endo/exo = 92:8. Ee(endo) = 95%. Ee(exo) = 94%. 1-((1R,2S,3S,4S)-3-(4-Nitrophenyl)bicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (69da). mp 141-143 °C; [α]D25 +141.3 (c 0.09, CHCl3); 1H NMR (300 MHz, CDCl3) δ 8.14 (2H, d, J = 8.8 Hz), 7.68 (2H, d, J = 8.4 Hz), 7.36 (2H, d, J = 8.5 Hz), 7.32 (2H, d, J = 8.0 Hz), 6.39 (1H, dd, J = 3.3 Hz, J = 5.6 Hz), 5.93 (1H, dd, J = 2.6 Hz, J = 5.7 Hz), 4.40 (1H, d, J = 13.4 Hz), 4.06 (1H, d, J = 13.4 Hz), 3.48 (1H, d, J = 1.4 Hz), 3.45 (1H, dd, J = 3.5 Hz, 69da J = 4.8 Hz), 3.26 (1H, dd, J = 1.0 Hz, J = 4.7 Hz), 3.09 (1H, d, J = 1.6 Hz), 2.44 (3H, s), 1.80 (1H, d, J = 8.8 Hz), 1.68 (1H, ddd, J = 1.6 Hz, J = 3.3 Hz, J = 8.8 Hz); 13C NMR (75.5 MHz, CDCl3) δ 197.3 (s), 151.3 (s), 146.3 (s), 145.5 (s), 139.5 (d), 135.6 (s), 132.7 (d), 129.9 (d), 128.2 (d), 123.7 (d), 65.8 (t), 61.3 (d), 48.3 (d), 47.8 185 Experimental section (t), 46.9 (d), 45.2 (d), 21.7 (q); MS (EI) m/z (%): 411 (M+, 0.1), 346 (8), 281 (15), 239 (10), 189 (31), 176 (100), 91 (40), 66 (16); HRMS: 411.1137, C22H21NO5S required 411.1140. Exo-69da (significant peaks, taken from the endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 7.64 (2H, d, J = 8.4 Hz), 5.99 (1H, dd, J = 2.8 Hz, J = 5.6 Hz), 3.19 (1H, d, J = 1.5 Hz), 3.16 (1H, s), 3.06 (1H, d, J = 1.2 Hz), 2.42 (3H, s), 1.62 (1H, d, J = 8.9 Hz), 1.52 (1H, ddd, J = 1.4 Hz, J = 3.0 Hz, J = 8.8 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.4 (s), 150.1 (s), 136.9 (d), 136.0 (d), 128.6 (d), 123.4 (d), 66.6 (t), 59.5 (d), 49.0 (d), 47.3 (t), 47.0 (d), 46.6 (d). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak IC, 55% isopropanol-45% hexane, 1.0 mL/min). Minor exo tR = 42.1 min, major exo tR = 46.4 min, major endo-(+) tR = 50.7 min, minor endo-(-) tR = 78.0 min. Endo/exo = 90:10. Ee(endo) = 95%. Ee(exo) = 95%. 1-((1R,2S,3S,4S)-3-Styrylbicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (69ea). mp 114-117 °C; [α]D25 +189.6 (c 0.85, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.61 (2H, d, J = 8.4 Hz), 7.29-7.12 (7H, m), 6.37 (1H, d, J = 15.7 Hz), 6.15 (1H, dd, J = 3.2 Hz, J = 5.7 Hz), 6.07 (1H, dd, J = 8.7 Hz, J = 15.7 Hz), 5.83 69ea (1H, dd, J = 2.8 Hz, J = 5.6 Hz), 4.15 (1H, d, J = 13.5 Hz), 4.01 (1H, d, J = 13.5 Hz), 3.19 (1H, s), 3.07 (1H, dd, J = 3.4 Hz, J = 4.7 Hz), 2.65 (1H, d, J = 1.5 Hz), 2.45 (1H, dd, J = 4.8 Hz, J = 8.6 Hz), 2.29 (3H, s), 1.61 (1H, d, J = 8.8 Hz), 1.45 (1H, qd, J = 1.6 Hz, J = 8.8 Hz); 13C NMR (75.5 MHz, CDCl3) δ 197.8 (s), 145.2 (s), 138.1 (d), 137.0 (s), 135.7 (s), 133.3 (d), 132.4 (d), 130.7 (d), 129.8 (d), 128.6 (d), 128.3 (d), 127.3 (d), 126.1 (d), 65.8 (t), 60.3 (d), 49.3 (d), 47.0 (t), 46.0 (d), 45.0 (d), 21.6 (q); MS (EI) m/z (%): 392 (M+, 0.1), 326 (34), 249 (2), 220 (4), 171 (100), 157 (36), 128 (45), 105 (34), 91 (25), 66 (24); HRMS: 392.1447, C24H24O3S required 392.1446. Characteristic signals for exo adduct: 1H NMR (300 MHz, CDCl3) δ 6.22 (1H, dd, J = 3.1 Hz, J = 5.6 Hz), 5.72 (1H, dd, J = 9.1 Hz, J = 15.7 Hz), 2.97 (1H, d, J = 1.3 Hz), 2.83 (1H, s); 13C NMR (75.5 MHz, CDCl3) δ 199.5 (s), 136.0 (d), 135.0 (s), 132.6 (d), 131.1 (d), 66.5 (t), 59.4 (d), 48.7 (d), 47.2 (t), 45.4 (d). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralcel OD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Exo (both) tR = 20.9 min, minor endo-(-) tR = 25.1 min, major endo-(+) tR = 31.4 min. Endo/exo = 94:6. Ee(endo) = 92%. Ee(exo) = nd. 186 Experimental section 1-((1R,2R,4R)-Bicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (69fa). mp 70-79 °C; [α]D25 +35.1 (c 0.71, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.76 (2H, d, J = 8.4 Hz), 7.35 (2H, d, J = 8.0 Hz), 6.13 (1H, dd, J = 3.1 Hz, J = 5.7 Hz), 5.73 (1H, dd, J = 2.8 Hz, J = 5.7 Hz), 4.30 (1H, d, J = 13.6 Hz), 4.05 (1H, d, J 69fa = 13.6 Hz), 3.37 (1H, td, J = 3.9 Hz, J = 8.8 Hz), 3.28 (1H, s), 2.92 (1H, s), 2.44 (3H, s), 1.80 (1H, ddd, J = 3.7 Hz, J = 8.8 Hz, J = 12.0 Hz), 1.45 (2H, m), 1.35 (1H, d, J = 8.1 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.6 (s), 145.3 (s), 138.2 (d), 135.9 (s), 130.9 (d), 129.8 (d), 128.4 (d), 66.0 (t), 53.1 (d), 49.9 (t), 46.0 (d), 42.8 (d), 27.8 (t), 21.7 (q); MS (EI) m/z (%): 290 (M+, 12), 225 (100), 160 (48), 155 (42), 139 (17), 91 (71), 66 (24); HRMS: 290.0981, C16H18O3S required 290.0977. Exo-69fa (significant peaks, taken from the endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 6.17 (1H, dd, J = 2.9 Hz, J = 5.6 Hz), 4.28 (1H, d, J = 13.5 Hz), 4.20 (1H, d, J = 13.5 Hz), 3.00 (1H, s), 2.67 (1H, dd, J = 3.8 Hz, J = 9.2 Hz); 13C NMR (75.5 MHz, CDCl3) δ 138.6 (d), 52.4 (d), 45.7 (d), 41.8 (d), 29.2 (t). Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak IC, 25% isopropanol-75% hexane, 1.0 mL/min). Exo (both) tR = 33.4 min, major endo-(+) tR = 43.1 min, minor endo-(-) tR = 47.8 min. Endo/exo = 92:8. Ee(endo) = 96%. Ee(exo) = nd. 1-((1R,2R,3S,4S)-3-Methylbicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (69ga). mp 95-97 °C; [α]D25 +73.1 (c 0.71, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.75 (2H, d, J = 8.3 Hz), 7.34 (2H, d, J = 8.0 Hz), 6.19 (1H, dd, J = 3.2 Hz, J = 5.6 Hz), 5.77 (1H, dd, J = 2.7 Hz, J = 5.7 Hz), 4.25 (1H, d, J = 13.6 Hz), 4.05 (1H, d, J 69ga = 13.6 Hz), 3.17 (1H, s), 2.80 (1H, dd, J = 3.4 Hz, J = 4.4 Hz), 2.47 (1H, d, J = 1.0 Hz), 2.43 (3H, s), 1.83 (1H, ddq, J = 1.6 Hz, J = 4.6 Hz, J = 7.0 Hz), 1.61 (1H, d, J = 8.7 Hz), 1.46 (1H, ddd, J = 1.7 Hz, J = 3.4 Hz, J = 8.7 Hz), 1.14 (3H, d, J = 7.0 Hz); 13C NMR (75.5 MHz, CDCl3) δ 198.6 (s), 145.2 (s), 139.0 (d), 135.8 (s), 131.8 (d), 129.8 (d), 128.3 (d), 65.8 (t), 61.9 (d), 49.0 (d), 46.4 (t), 46.3 (d), 35.8 (d), 21.6 (q), 20.6 (q); MS (EI) m/z (%): 304 (M+, 0.1), 239 (4), 174 (13), 155 (8), 139 (8), 108 (9), 91 (47), 69 (100), 66 (48); HRMS: 304.1136, C17H20O3S required 304.1133. Exo-69ga (significant peaks, taken from the endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 6.13 (1H, dd, J = 2.9 Hz, J = 5.7 Hz), 4.25 (1H, d, J = 13.4 Hz), 4.17 (1H, d, J = 13.5 Hz), 2.90 (1H, s), 2.70 (1H, s), 2.11 (1H, dd, J = 1.4 Hz, J = 5.1 Hz), 1.67 (1H, s), 1.38 (1H, m), 0.91 (3H, d, J = 6.9 Hz); 13C NMR (75.5 MHz, CDCl3) δ 136.2 (d), 136.0 (d), 66.8 (t), 60.4 (d), 47.5 (d), 47.3 (t), 37.5 (d), 19.1 (q). 187 Experimental section Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak IC, 12% isopropanol-88% hexane, 1.0 mL/min). Exo (both) tR = 55.4 min, major endo-(+) tR = 68.4 min, minor endo-(-) tR = 74.4 min. Endo/exo = 92:8. Ee(endo) = 91%. Ee(exo) = nd. 1-((1R,2S,3S,4S)-3-tert-Butylbicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (endo69ha) and 1-((1S,2S,3S,4R)-3-tert-Butylbicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (exo-69ha). [α]D25 +18.4 (c 0.90, CHCl3)(mixture of diastereomers endo/exo = 43:57). Endo-69ha (peaks taken from an endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 7.74 (2H, d, J = 8.0 Hz), 7.32 (2H, d, J = 8.4 Hz), 6.29 (1H, dd, J = 3.3 Hz, J = 5.6 Hz), 5.63 (1H, dd, J = 2.7 Hz, J = 5.6 Hz), 4.37 (1H, d, J = 13.8 Hz), 4.08 (1H, d, J = 13.8 Hz), 3.23 (1H, s), 3.03 (1H, dd, J Endo-69ha = 3.3 Hz, J = 5.7 Hz), 2.69 (1H, dd, J = 1.4 Hz, J = 3.0 Hz), 2.41 (3H, s), 1.66 (1H, dd, J = 1.7 Hz, J = 5.7 Hz), 1.60 (1H, d, J = 8.5 Hz), 1.38 (1H, t, J = 1.7 Hz), 0.84 (9H, s); 13C NMR (75.5 MHz, CDCl3) δ 198.4 (s), 145.0 (s), 141.2 (d), 135.9 (s), 130.9 (d), 129.7 (d), 128.3 (d), 65.5 (t), 55.2 (d), 51.5 (d), 47.8 (t), 46.6 (d), 44.3 (d), 32.3 (s), 28.7 (q), 21.6 (q). Exo-69ha (peaks taken from an endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 7.77 (2H, d, J = 8.2 Hz), 7.33 (2H, d, J = 7.9 Hz), 6.13 (1H, dd, J = 3.2 Hz, J = 5.4 Hz), 6.09 (1H, dd, J = 3.4 Hz, J = 5.3 Hz), 4.37 (1H, d, J = 13.8 Hz), 4.22 (1H, d, J = 13.8 Hz), 2.87 (2H, s), 2.47 (2H, s), Exo-69ha 2.42 (3H, s), 1.35 (1H, t, J = 1.6 Hz), 1.25 (1H, d, J = 8.4 13 Hz), 0.73 (9H, s); C NMR (75.5 MHz, CDCl3) δ 199.3 (s), 145.1 (s), 137.4 (d), 136.0 (s), 133.8 (d), 129.7 (d), 128.3 (d), 66.2 (t), 55.7 (d), 52.8 (d), 48.0 (t), 47.4 (d), 45.0 (d), 32.3 (s), 29.1 (q), 21.6 (q); MS (EI) m/z (%): 346 (M+, 0.2), 281 (19), 216 (13), 155 (23), 139 (22), 125 (100), 111 (48), 109 (55), 66 (89); HRMS: 346.1605, C20H26O3S required 346.1603. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 5% isopropanol-95% hexane, 1.0 mL/min). Minor exo tR = 14.8 min, minor endo tR = 20.8 min, major endo tR = 23.5 min, major exo tR = 26.1 min. Endo/exo = 43:57. Ee(endo) = 97%. Ee(exo) = 83%. 188 Experimental section 1-((1R,2R,4R)-2-Methylbicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (endo-69ia) and 1-((1S,2R,4S)-2-methylbicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (exo-69ia). [α]D25 +5.1 (c 0.80, CHCl3) (mixture of diastereomers) Endo-69ia (peaks taken from an endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 7.80 (2H, d, J = 8.2 Hz), 7.34 (2H, d, J = 8.4 Hz), 6.05 (1H, dd, J = 3.1 Hz, J = 5.6 Hz), 5.86 (1H, dd, J = 2.8 Hz, J = 5.7 Hz), 4.40 (1H, d, J = 15.1 Endo-69ia Hz), 4.09 (1H, d, J = 15.4 Hz), 2.82 (1H, s), 2.70 (1H, s), 2.44 (3H, s), 1.90 (1H, dd, J = 2.7 Hz, J = 12.0 Hz), 1.60 (1H, d, J = 8.8 Hz), 1.47 (1H, ddd, J = 1.9 Hz, J = 4.4 Hz, J = 9.0 Hz), 1.36 (1H, dd, J = 3.6 Hz, J = 12.0 Hz), 1.32 (3H, s); 13C NMR (75.5 MHz, CDCl3) δ 201.2 (s), 145.0 (s), 138.5 (d), 136.5 (s), 133.0 (d), 129.7 (d), 128.7 (d), 62.7 (t), 57.2 (s), 50.5 (d), 47.1 (t), 42.7 (d), 36.2 (t), 24.3 (q), 21.6 (q). Exo-69ia (peaks taken from an endo/exo mixture): 1H NMR (300 MHz, CDCl3) δ 7.82 (2H, d, J = 8.3 Hz), 7.36 (2H, d, J = 8.0 Hz), 6.25 (1H, dd, J = 3.0 Hz, J = 5.6 Hz), 6.05 (1H, dd, J = 3.1 Hz, J = 5.6 Hz), 4.37 (1H, d, J = 8.9 Hz), 4.32 (1H, d, J = 8.5 Hz), 2.94 (1H, d, J = 1.5 Hz), 2.79 Exo-69ia (1H, s), 2.44 (3H, s), 2.34 (1H, dd, J = 3.9 Hz, J = 11.9 Hz), 1.36 (1H, dd, J = 3.6 Hz, J = 12.0 Hz), 1.11 (1H, d, J = 8.8 Hz), 1.02 (3H, s), 0.77 (1H, dd, J = 2.7 Hz, J = 11.9 Hz); 13C NMR (75.5 MHz, CDCl3) δ 202.2 (s), 145.2 (s), 139.6 (d), 136.5 (s), 133.8 (d), 129.6 (d), 128.7 (d), 62.4 (t), 57.9 (s), 48.6 (t), 48.4 (d), 42.9 (d), 35.6 (t), 22.4 (q), 21.6 (q); MS (EI) m/z (%): 304 (M+, 3.5), 239 (70), 174 (23), 155 (65), 148, (44), 139 (54), 132 (28), 91 (100); HRMS: 304.1137, C17H20O3S required 304.1133. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AS-H, 25% isopropanol-75% hexane, 1.0 mL/min). Exo (both) tR = 25.7 min, minor endo tR = 28.9 min, major endo tR = 30.6 min. (Chiralpak AD-H, 25% isopropanol-75% hexane, 1.0 mL/min). Major exo tR = 10.3 min, minor exo tR = 11.9 min, endo (both) tR = 12.9 min. Endo/exo = 49:51. Ee(endo) = 94%. Ee(exo) = 90%. 1-((1R,2R,3S,4S)-2,3-Dimethylbicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (endo69ja) and 1-((1S,2R,3S,4R)-2,3-dimethylbicyclo[2.2.1]hept-5-en-2-yl)-2-tosylethanone (exo-69ja). Endo-69ja mp 120-122 °C; [α]D25 +53.9 (c 0.53, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.80 (2H, d, J = 8.4 Hz), 7.35 (2H, d, J = 8.0 Hz), 6.10 (1H, dd, J = 3.1 Hz, J = 5.7 Hz), 5.90 (1H, dd, J = 2.7 Hz, J = 5.8 Hz), 4.34 (1H, d, J = 15.5 Endo-69ja Hz), 4.08 (1H, d, J = 15.4 Hz), 2.65 (1H, s), 2.44 (3H, s), 2.39 (1H, s), 2.04 (1H, dq, J = 1.9 Hz, J = 7.3 Hz), 1.69 (1H, d, J = 9.1 Hz), 1.40 (1H, ddd, J 189 Experimental section = 1.7 Hz, J = 3.5 Hz, J = 9.0 Hz), 1.16 (3H, s), 1.02 (3H, d, J = 7.3 Hz); 13C NMR (75.5 MHz, CDCl3) δ 201.6 (s), 145.0 (s), 139.0 (d), 136.5 (s), 133.9 (d), 129.6 (d), 128.8 (d), 62.4 (t), 59.5 (s), 51.6 (d), 49.6 (d), 44.0 (t), 37.0 (d), 21.6 (q), 19.6 (q), 17.1 (q); MS (FAB) m/z (%): 319 (M++1, 11), 307 (28), 289 (18), 253 (68), 154 (100), 136 (69); HRMS: 319.1360, C18H23O3S required 319.1368. Exo-69ja mp 90-92 °C; [α]D25 +6.3 (c 0.47, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.83 (2H, d, J = 8.3 Hz), 7.36 (2H, d, J = 7.9 Hz), 6.24 (1H, dd, J = 2.9 Hz, J = 5.7 Hz), 6.14 (1H, dd, J = 3.2 Hz, J = 5.7 Hz), 4.34 (1H, d, J = 15.2 Hz), 4.28 Exo-69ja (1H, d, J = 15.2 Hz), 2.97 (1H, d, J = 1.2 Hz), 2.66 (1H, s), 2.45 (3H, s), 2.46 (1H, dq, J = 3.5 Hz, J = 7.2 Hz), 1.36 (1H, td, J = 1.8 Hz, J = 8.8 Hz), 1.13 (1H, d, J = 8.8 Hz), 0.85 (3H, s), 0.80 (3H, d, J = 7.2 Hz); 13C NMR (75.5 MHz, CDCl3) δ 202.4 (s), 145.0 (s), 137.6 (d), 136.5 (s), 135.3 (d), 129.6 (d), 128.7 (d), 62.7 (t), 59.0 (s), 49.6 (d), 48.8 (d), 47.6 (t), 37.6 (d), 21.6 (q), 17.1 (q), 15.1 (q); MS (FAB) m/z (%): 319 (M++1, 11), 307 (28), 289 (18), 253 (68), 154 (100), 136 (69); HRMS: 319.1360, C18H23O3S required 319.1368. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AS-H, 10% isopropanol-90% hexane, 0.5 mL/min). Minor exo-(-) tR = 63.9 min, major exo -(+) tR = 70.2 min, major endo-(+) tR = 75.2 min, minor endo-(-) tR = 79.9 min. Endo/exo = 50:50. Ee(endo) = 96%. Ee(exo) = 88%. 1-((1R,2R,4R)-Bicyclo[2.2.2]oct-5-en-2-yl)-2-tosylethanone (69fb). mp 78-81 °C; [α]D25 +13.1 (c 1.25, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.74 (2H, d, J = 8.4 Hz), 7.34 (2H, d, J = 8.1 Hz), 6.24 (1H, ddd, J = 0.8 Hz, J = 6.6 Hz, J = 7.8 Hz), 5.99 (1H, ddd, J = 0.9 Hz, J = 6.5 Hz, J = 7.9 Hz), 4.22 (1H, d, J 69fb = 13.7 Hz), 4.10 (1H, d, J = 13.7 Hz), 3.00 (1H, ddd, J = 1.9 Hz, J = 5.7 Hz, J = 9.7 Hz), 2.87 (1H, m), 2.60 (1H, m), 2.44 (3H, s), 1.72-1.45 (4H, m), 1.37-1.19 (2H, m); 13C NMR (75.5 MHz, CDCl3) δ 199.0 (s), 145.2 (s), 135.9 (s), 135.4 (d), 130.6 (d), 129.8 (d), 128.4 (d), 65.0 (t), 51.9 (d), 31.5 (d), 29.3 (d), 28.3 (t), 25.6 (t), 24.2 (t), 21.7 (q); MS (EI) m/z (%): 304 (M+, 1.5), 225 (22), 208 (12), 155 (20), 139 (16), 107 (18), 91 (66), 80 (100), 65 (19); HRMS: 304.1148, C17H20O3S required 304.1133. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Minor endo-(-) tR = 15.6 min, major endo-(+) tR = 18.9 min. Endo/exo = 92:8. Ee(endo) = 96%. Ee(exo) = nd. 190 Experimental section (R)-1-(3,4-Dimethylcyclohex-3-enyl)-2-tosylethanone (69fc). mp 104-107 °C; [α]D25 +47.7 (c 0.83, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.70 (2H, d, J = 8.4 Hz), 7.29 (2H, d, J = 8.1 Hz), 4.19 (1H, d, J = 13.6 Hz), 4.12 (1H, d, J = 13.6 Hz), 2.83 (1H, dddd, J = 2.9 Hz, 69fc J = 5.9 Hz, J = 9.3 Hz, J = 11.2 Hz), 2.38 (3H, s), 2.011.80 (5H, m), 1.54 (3H, s), 1.52 (3H, s), 1.40 (1H, dtd, J = 5.8 Hz, J = 10.8 Hz, J = 12.8 Hz); 13 C NMR (75.5 MHz, CDCl3) δ 201.2 (s), 145.3 (s), 135.9 (s), 129.9 (d), 128.4 (d), 125.4 (s), 123.4 (s), 65.0 (t), 48.4 (d), 32.5 (t), 30.9 (t), 24.8 (t), 21.7 (q), 19.0 (q), 18.8 (q); MS (EI) m/z (%): 306 (M+, 5.1), 238 (6), 178 (3), 150 (80), 135 (89), 123 (100), 109 (88), 107 (70), 91 (98); HRMS: 306.1286, C17H22O3S required 306.1290. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 15% isopropanol-85% hexane, 1.0 mL/min). Major (1R)-(+) tR = 13.5 min, minor (1S)-(-) tR = 14.6 min. Ee(endo) = 76%. 7-Methyl-6-methylene-1-tosyloct-7-en-2-one (72). 1 H NMR (300 MHz, CDCl3) δ 7.75 (2H, d, J = 8.3 Hz), 7.36 (2H, d, J = 8.0 Hz), 5.08 (1H, d, J = 13.0 Hz), 4.95 (1H, d, J = 14.5 Hz), 4.12 (2H, s), 2.72 (1H, t, J = 7.1 Hz), 2.45 (3H, s), 2.26 (1H, t, J = 7.3 72 Hz), 1.89 (3H, s), 1.75 (1H, tt, J = 7.1 Hz, J = 7.3 13 Hz); C NMR (100 MHz, CDCl3) δ 198.2 (s), 146.8 (s), 145.4 (s), 142.2 (s), 135.7 (s), 129.9 (d), 128.2 (d), 112.8 (t), 112.8 (t), 67.0 (t), 43.8 (t), 32.5 (t), 22.1 (q), 21.6 (t), 21.0 (q); MS (EI) m/z (%): 306 (M+, 0.2), 279 (15), 167 (20), 149 (55), 91 (100), 65 (29); HRMS: 306.1298, C17H22O3S required 306.1290. (R)-1-(4-Methylcyclohex-3-enyl)-2-tosylethanone (69fd). mp 68-71 °C; [α]D25 +33.8 (c 0.87, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.76 (2H, d, J = 8.3 Hz), 7.36 (2H, d, J = 8.0 Hz), 5.36 (1H, br), 4.25 (1H, d, J = 13.6 Hz), 4.19 (1H, d, J = 13.6 Hz), 2.86 (1H, m), 2.45 (3H, 69fd s), 2.23-1.90 (5H, m), 1.63 (3H, s), 1.55 (1H, m); 13C NMR (75.5 MHz, CDCl3) δ 201.3 (s), 145.3 (s), 135.9 (s), 133.9 (s), 129.9 (d), 128.4 (d), 118.6 (s), 65.0 (t), 47.4 (s), 29.2 (t), 26.6 (t), 24.4 (t), 23.3 (q), 21.7 (q); MS (EI) m/z (%): 292 (M+, 0.5), 224 (3), 155 (13), 136 (100), 121 (14), 118 (99), 109 (88), 95 (42), 91 (84); HRMS: 292.1129, C16H20O3S required 292.1333. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 10% isopropanol-90% hexane, 1.0 mL/min). Major 1,3- tR = 16.1 min, minor 1,3- tR = 18.5 191 Experimental section min, minor 1,4-(-) tR = 26.8 min, major 1,4-(+) tR = 28.2 min. 1,3-/1,4- = 96:4. Ee((1,4)-) = 79%. Ee((1,3)-) = 69%. 1-((1R,2S,6S)-6-Methyl-2-phenylcyclohex-3-enyl)-2-tosylethanone (69gf). (mixture of diastereomers cis/trans = 56:44) [α]D25 -25.7 (c 1.03, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.64 (2H, minor diastereomer, d, J = 8.3 Hz), 7.63 (2H, major diastereomer, d, J = 8.4 Hz), 7.32-7.18 (5H, m), 7.02 (2H, 69gf m), 5.90 (1H, major diastereomer, dddd, J = 1.9, J = 2.7, J = 4.5, J = 9.7), 5.81 (1H, minor diastereomer, tdd, J = 2.4 Hz, J = 5.1 Hz, J = 9.9 Hz), 5.71 (1H, major diastereomer, tdd, J = 2.3 Hz, J = 4.3 Hz, J = 10.0 Hz), 5.55 (1H, minor diastereomer, ddd, J = 2.0 Hz, J = 4.1 Hz, J = 9.8 Hz), 4.09 (1H, major diastereomer, d, J = 13.3 Hz), 3.81 (1H, major diastereomer, m), 3.70 (1H, major diastereomer, d, J = 13.3 Hz), 3.54 (1H, minor diastereomer, d, J = 15.3 Hz), 3.38 (1H, m), 3.33 (1H, minor diastereomer, d, J = 15.3 Hz), 2.69 (1H, minor diastereomer, t, J = 10.6 Hz), 2.45 (3H, minor diastereomer, s), 2.42 (3H, major diastereomer, s), 2.36 (1H, m), 2.22-2.04 (1H, m), 1.95-1.78 (1H, m), 0.97 (3H, major diastereomer, d, J = 6.5 Hz), 0.91 (3H, minor diastereomer, d, J = 6.4 Hz); 13C NMR (75.5 MHz, CDCl3) δ 203.2 (s), 198.8 (s), 145.1 (s), 144.9 (s), 142.7 (s), 140.0 (s), 136.1 (s), 135.5 (s), 129.7 (d), 129.6 (d), 129.5 (d), 129.0 (d), 128.8 (d), 128.4 (d), 128.3 (d), 128.3 (d), 127.7 (d), 127.25 (d), 127.23 (d), 127.1 (d), 126.9 (d), 126.8 (d), 68.1 (t), 66.6 (t), 62.6 (d), 58.8 (d), 47.8 (d), 42.3 (d), 33.5 (t), 32.1 (t), 31.5 (d), 25.0 (d), 21.67 (d), 21.65 (d), 19.8 (d), 19.4 (d); MS (FAB) m/z (%): 369 (M++1, 93), 305 (49), 283 (20), 239 (77), 213 (16), 193 (29), 167 (24), 153 (100), 150 (58), 137 (86); HRMS (EI): 368.1420, C22H24O3S required 368.1446. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 5% isopropanol-95% hexane, 1.0 mL/min). Major cis-1,2- tR = 16.0 min, minor cis-1,2- tR = 20.1 min, minor trans-1,2- tR = 26.9 min, major trans-1,2- tR = 28.6 min. Cis/trans = 56:44. Ee(endo) = 71%. Ee(exo) = 15%. (3S,4S)-3-Methyl-4-(2-tosylacetyl)cyclohexanone (69gh). [α]D25 +9.8 (c 1.03, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.76 (2H, d, J = 8.4 Hz), 7.36 (2H, d, J = 8.0 Hz), 4.29 (1H, d, J = 13.4 Hz), 4.19 (1H, d, J = 13.4 Hz), 2.97 (1H, ddd, J = 3.6 Hz, J = 9.3 Hz, J = 10.7 Hz), 2.44 (3H, 69gh s), 2.42-2.05 (6H, m), 1.68 (1H, dtd, J = 5.3 Hz, J = 11.5 Hz, J = 13.5 Hz), 0.97 (3H, d, J = 6.3 Hz); 13C NMR (75.5 MHz, CDCl3) δ 208.9 (s), 200.5 (s), 145.6 (s), 135.7 (s), 130.0 (d), 128.2 (d), 66.7 (t), 55.7 (d), 47.7 (t), 39.5 (t), 34.6 (d), 27.3 (t), 21.6 (q), 20.3 (q); MS (EI) m/z (%): 308 (M+, 0.4), 293 (1), 197 (2), 155 192 Experimental section (24), 153 (100), 139 (15), 125 (11), 111 (22), 91 (32); HRMS: 308.1068, C16H20O4S required 308.1082. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 25% isopropanol-75% hexane, 1.0 mL/min). Major (+) tR = 22.4 min, minor (-) tR = 38.5 min. Ee(endo) = 92%. 5.4.2.3. Synthetic transformations 1-((1R,2R,3S,4S)-3-Methylbicyclo[2.2.1]hept-5-en-2-yl)-2-tosylpropan-1-one (74). MeI (0.052 mL, 0.83 mmol) was added to a solution of compound 69ga (63.2 mg, 0.21 mmol) and K2CO3 (30 mg, 0.22 mmol) in DMF (1 mL) under nitrogen atmosphere. The mixture was stirred at rt for 3 h. After 74 this time, water (100 mL) was added, followed by 2 M HCl (20 mL) and the solution was extracted with dichloromethane (3 × 20 mL). The organic layer was washed with aqueous saturated NaHCO3 (20 mL) and brine (20 mL), and dried over Na2SO4. After evaporation of the solvent under reduced pressure, column chromatography eluting with hexane-EtOAc (9:1) gave 59.6 mg of compound 74 (90% yield) as a 67:33 diastereomeric mixture. Mixture of diastereomers 33:66. 1H NMR (300 MHz, CDCl3) δ 7.67 (1H, minor diastereomer, d, J = 8.4 Hz), 7.63 (2H, major diastereomer, d, J = 8.4 Hz), 7.34 (2H, minor diastereomer, d, J = 8.6 Hz), 7.33 (2H, major diastereomer, d, J = 8.0 Hz), 6.24 (1H, major diastereomer, dd, J = 3.2 Hz, J = 5.7 Hz), 6.14 (1H, minor diastereomer, dd, J = 3.2 Hz, J = 5.7 Hz), 5.96 (1H, minor diastereomer, dd, J = 2.8 Hz, J = 5.6 Hz), 5.63 (1H, major diastereomer, dd, J = 2.7 Hz, J = 5.7 Hz), 4.37 (1H, major diastereomer, q, J = 6.9 Hz), 4.20 (1H, minor diastereomer, q, J = 7.0 Hz), 3.20 (1H, major diastereomer, s), 3.14 (1H, minor diastereomer, s), 3.01 (1H, major diastereomer, dd, J = 3.5 Hz, J = 4.2 Hz), 2.91 (1H, minor diastereomer, dd, J = 3.1 Hz, J = 5.0 Hz), 2.49 (1H, major diastereomer, s), 2.44 (3H, minor diastereomer, s), 2.43 (3H, major diastereomer, s), 2.0-1.92 (1H, m), 1.66 (1H, both diastereomers, d, J = 8.6 Hz), 1.59 (1H, minor diastereomer, d, J = 8.7 Hz), 1.49 (1H, minor diastereomer, q, J = 1.8 Hz), 1.46 ( 1H, major diastereomer, q, J = 1.8 Hz), 1.34 (3H, minor diastereomer, d, J = 7.1 Hz), 1.28 (3H, major diastereomer, d, J = 7.0 Hz), 1.27 (3H, minor diastereomer, d, J = 6.9 Hz), 1.12 (3H, major diastereomer, d, J = 7.1 Hz); 13C NMR (75.5 MHz, CDCl3) Major diastereomer δ 202.4 (s), 145.3 (s), 139.7 (d), 132.8 (s), 130.5 (d), 129.6 (2d), 68.4 (d), 62.1 (d), 49.2 (d), 46.8 (d), 46.7 (t), 34.9 (d), 21.6 (q), 20.2 (q), 12.5 (q). Minor diastereomer δ 203.7 (s), 145.2 (s), 137.3 (d), 134.2 (d), 133.2 (s), 129.6 (d), 129.5 (d), 68.9 (d), 62.0 (d), 48.8 (d), 47.4 (d), 46.0 (t), 45.9 (d), 37.6 (q), 21.2 (q), 12.8 (q); MS (EI) m/z (%): 318 (M+, 1.2), 253 (98), 211 (6), 193 Experimental section 188 (29), 155 (100), 139 (28), 91 (33), 69 (100); HRMS: 318.1293, C18H22O3S required 318.1290. 1-((1R,2R,3S,4S)-3-Methylbicyclo[2.2.1]hept-5-en-2-yl)propan-1-one (73). To a solution of compound 74 (59.6 mg, 0.187 mmols) in methanol (8.3 mL), Na2HPO3 (107 mg, 0.77 mmol) was added and the mixture cooled to 0 °C. 20% Na(Hg) was added and the mixture was stirred for 1 h. After this time, the reaction was 73 quenched with water (100 mL), extracted with dichloromethane (3 × 20 mL), washed with brine (20 mL) and dried over MgSO4. Column chromatography eluting with hexane-Et2O (98:2) gave 27.7 mg of compound 73 (90% yield). [α]D25 +113.5 (c 0.92, CHCl3); 1H NMR (300 MHz, CDCl3) δ 6.21 (1H, dd, J = 3.1 Hz, J = 5.7 Hz), 5.89 (1H, dd, J = 2.8 Hz, J = 5.7 Hz), 3.12 (1H, d, J = 1.7 Hz), 2.44 (1H, m), 2.42 (1H, q, J = 7.3 Hz), 1.88 (1H, ddq, J = 1.7 Hz, J = 4.6 Hz, J = 7.0 Hz), 1.58 (1H, td, J = 1.5 Hz, J = 8.5 Hz), 1.43 (1H, qd, J = 1.7 Hz, J = 8.5 Hz), 1.15 (3H, d, J = 7.0 Hz), 1.01 (3H, t, J = 7.3 Hz); 13C NMR (75.5 MHz, CDCl3) δ 211.6 (s), 138.5 (d), 132.5 (d), 60.6 (d), 49.0 (d), 46.2 (t), 35.7 (d), 34.7 (t), 21.0 (q), 7.8 (q); MS (EI) m/z (%): 164 (M+, 22), 145 (6), 135 (9), 107 (16), 139 (16), 107 (18), 99 (67), 91 (21), 79 (19), 69 (70), 66 (100), 57 (50); HRMS: 164.1208, C11H16O required 164.1201. Assay of enantiomeric excess: Chiral GC analysis (Supelco Beta-DEX 120, 30 m x 0.25 mm x 0.25 μm, Tcolumn = 80 °C (10 min) to 200 °C at 8 °C/min ), minor (-)tR = 20.9 min, major (+) tR = 21.1 min. 1-((1R,2R,3S,4S)-3-Methylbicyclo[2.2.1]hept-5-en-2-yl)-3-phenyl-2-tosylpropan-1-one (76). Following the same procedure described for the synthesis of 74, compound 69ga (50 mg, 0.164 mmol) was treated with benzyl chloride to give 54.7 mg of compound 76 (84% yield) as a 74:26 diastereomeric mixture. Complex mixture of diastereomers. MS (EI) m/z (%): 394 (M+, 0.6), 329 (53), 287 (4), 238 (11), 173 (100), 139 (16), 91 (57), 69 (59); HRMS: 394.1617, C24H26O3S required 394.1603. 76 194 Experimental section 1-((1R,2R,3S,4S)-3-Methylbicyclo[2.2.1]hept-5-en-2-yl)-3-phenylpropan-1-one (75). Following the same procedure described for the synthesis of 73, compound 76 (47.1 mg, 0.120 mmol) was treated with Na(Hg) to give 28.3 mg of compound 75 (98% yield). [α]D25 +86.4 (c 1.03, CHCl3); 1H NMR (300 MHz, CDCl3) δ 7.29 (2H, m), 7.19 (3H, m), 6.21 (1H, dd, J = 3.2 Hz, J = 5.7 Hz), 5.78 (1H, dd, J = 2.8 Hz, J = 5.7 Hz), 3.09 (1H, s), 2.86 (2H, td, J = 2.6 Hz, J = 9.9 Hz), 2.73 (2H, ddd, J = 2.7 Hz, J = 6.4 Hz, J = 10.3 Hz), 75 2.45 (1H, d, J = 1.5 Hz), 2.41 (1H, dd, J = 3.4 Hz, J = 4.4 Hz), 1.89 (1H, m), 1.56 (1H, d, J = 8.6 Hz), 1.42 (1H, qd, J = 1.7 Hz, J = 8.6 Hz), 1.13 (3H, d, J = 7.0 Hz); 13C NMR (75.5 MHz, CDCl3) δ 210.1 (s), 141.4 (s), 138.5 (d), 132.4 (d), 128.4 (d), 128.4 (d), 126.0 (d), 61.1 (d), 49.0 (d), 46.2 (t), 46.2 (d), 43.3 (t), 35.5 (d), 29.8 (t), 21.0 (q); MS (EI) m/z (%): 240 (M+, 18), 175 (48), 174 (49), 159 (62), 105 (40), 91 (70), 79 (17), 69 (100), 66 (60); HRMS: 240.1513, C17H20O required 240.1514. Assay of enantiomeric excess: Chiral HPLC analysis (Chiralpak AD-H, 5% isopropanol-95% hexane, 1.0 mL/min). Major (+) tR = 4.8 min, minor (-) tR = 7.2 min. 195 Experimental section 196 Summary and conclusions 6. S UMMARY AND CONCLUSIONS 1. A new family of ligands which combine a hydroxyl group with an oxazoline ring as coordinating groups has been synthesized and characterized from (1S)ketopinic acid. 2. The Cu(II) complexes of these ligands generated in situ with Cu(OTf)2 catalyze the enantioselective Diels-Alder reaction between 3-alkenoyl-2-oxazolidones and different dienes. 3. The absolute stereochemistry of Diels-Alder adduct is primarily determined by the stereochemistry of the stereogenic center on the oxazoline moiety. Furthermore, only ligands having the C(2)-hydroxyl group oriented exo afford acceptable enantioselectivity. 4. Ligand 5, derived from (1S)-ketopinic acid and (1R,2S)-aminoindanol gave the best results in the Diels-Alder reaction between cyclopentadiene and 3-acryloyl-2oxazolidone (90% ee, endo/exo = 98:2). Dienes less reactive than cyclopentadiene or bulkier dienophiles could be also used with this catalytic system although the stereoselectivities attained were lower. 5. 2-Alkenoylpyridine N-oxides have been introduced by the first time as a new kind of bidentate substrates in catalytic enantioselective reactions. 6. 2-Alkenoylpyridine N-oxides are much more reactive than the corresponding non-oxidized 2-alkenoylpyridines in enantioselective Cu(II)-BOX catalyzed cycloadditions, affording higher diastereo- and enantioselectivities. 7. 2-Alkenoylpyridine N-oxides achieve high conversions, diastereomeric ratios and enantiomeric excesses (up to 98%) in the Cu(II)-33a catalyzed Diels-Alder reaction with cyclopentadiene, regardless the nature of the substituent on the double bond allowing either aryl or alkyl groups. High efficiency of the reaction is also maintained with dienes other than cyclopentadiene. In most of the cases, the resulting products were obtained with enantiomeric excess higher than 90%. 8. Diels-Alder adducts derived from 2-alkenoylpyridine N-oxides can be deoxygenated to the corresponding pyridine adducts without loss of optical purity. It is also possible to take advantage of the characteristic pyridine N-oxide chemistry to carry out transformations on the heteroaromatic ring which could be otherwise difficult to perform. For instance, the regioselective cyanation of 34aa in the position 6 of the pyridine ring has been carried out by a modified Reissert-Henze reaction. 197 Summary and conclusions 9. Besides the 2-pyridinylcarbonyl moiety of the Diels-Alder adducts can be replaced by a hydroxyl group through a Baeyer-Villiger oxidation and transesterification. As a result of this transformation, 2-alkenoylpyridine N-oxides can be regarded as synthetic equivalents of enols as dienophiles in Diels-Alder reactions. 10. 2-Alkenoylpyridine N-oxides can be also used as heterodienes in inverse electron-demand hetero-Diels-Alder reactions catalyzed by Cu(II)-33a. High conversions, diastereomeric ratios and enantiomeric excesses (up to 99.6%) are obtained regardless of the nature of the substituent on the double bond of the heterodiene, allowing either aryl, heteroaryl or alkyl groups. The high efficiency of the 2-alkenoylpyridine N-oxides in the Cu(II)-33a catalyzed hetero-Diels-Alder is also demonstrated by the reaction with less reactive alkenes, regardless the nature of the substituent on the double bond allowing either aryl or alkyl groups. 11. 2-Alkenoylpyridine N-oxides have been used as dipolarophiles in [3+2] cycloaddition reactions with nitrones. Conversions and stereoselectivities from good to excellent are achieved in the reaction catalyzed with Cu(II)-33b complex. The presence of molecular sieves is essential to obtain good results. Unlike in the Diels-Alder and the hetero-Diels-Alder reactions, ligand 33a gave poor results. 12. α’-Arylsulfonyl enones, previously used in Ti(IV)-catalyzed Diels-Alder reactions, are also efficient dienophiles in Cu(II)-33a catalyzed Diels-Alder reaction with dienes. Conversions from good to excellent, good diastereoselectivities and high enantiomeric excesses (up to 97%) are obtained in the presence of ligand 33a for a range of dienes. The resulting Diels-Alder adducts in these reactions can be alkylated and the sulfone group removed. Thus, α’-arylsulfonyl enones can be used as bidentate surrogates of simple monodentate enones, which are poor dienophiles, in enantioselective Cu(II)-BOX-catalyzed D-A reactions. 13. In all the studied reactions using 2-alkenoylpyridine N-oxides or α’arylsulfonyl enones as substrates, ligands 33a and 33b provided opposite enantiomers, in agreement with the results reported with other reactions catalyzed by these ligands. 14. The absolute stereochemistry of compounds 34ca, 43ci, 48ja and 66b have been established by X-ray diffraction crystallographic analysis. 15. The stereochemical pathway of these reactions has been explained in similar terms to other Cu(II)-BOX catalyzed reactions. For the reactions catalyzed with the Cu(II)-33a complex a distorted tetrahedral complex intermediate stabilized by a ππ interaction between the enone double bond and the phenyl group of the bis(oxazoline) is proposed. 198 Summary and conclusions 16. In these complexes the 2-alkenoylpyridine N-oxide will coordinate the Cu(II) atom in a bidentate fashion through the carbonyl group and the O atom of the Noxide, leaving the Si face of the double bond in the s-cis conformation of the enone available for the approach of the diene (Diels-Alder) or dienophile (hetero-Diels-Alder). A similar intermediate would be formed with α’-arylsulfonyl enones, with the substrate coordinating the Cu(II) atom through the carbonyl and sulfone oxygen atoms. 17. On the other hand, for the 1,3-dipolar cycloaddition with 2-alkenoylpyridine N-oxides catalyzed with Cu(II)-33b, a distorted square-planar complex with the dipolarophile similarly coordinated to the metal center would account for the absolute stereochemistry of the products 18. In conclusion: a. We have contributed to the development of new hydroxyoxazoline ligands to be used in Cu(II)-catalyzed enantioselective reactions, particularly Diels-Alder. b. We have enlarged the range of chelating substrates that can be used as highly efficient substrates in Metal-BOX complexes-catalyzed enantioselective reactions, particularly Cu(II)-catalyzed cycloadditions. 199 Summary and conclusions 200 References 7. R EFERENCES (1) (2) (3) (4) (5) (6) (7) (8) (9) (10) (11) (12) (13) (14) (15) (16) (17) (18) (19) (20) (21) (22) (23) (24) (25) (26) Diels, O.; Alder, K. Ann. 1928, 460, 98-122. Anand, N.; Bindra, J. S.; Ranganathan, S. Art in Organic Synthesis; Hoden-Day: San Francisco, 1970; Nicolaou, K. C.; Snyder, S. A.; Montagnon, T.; Vassilikogiannakis, G. Angew. Chem., Int. Ed. 2002, 41, 1668-1698. Yates, P.; Eaton, P. J. Am. Chem. Soc. 1960, 82, 4436-4437. Jørgensen, K. A. Catalytic Enantioselective Cycloaddition Reactions of Carbonyl Compounds In Cycloaddition Reactions in Organic Synthesis; Kobayashi, S., Jørgensen, K. A. Eds.; Wiley-VCH: Weinheim, 2002. Francesco, F.; Aldo, T. The Diels Alder reaction: selected practical methods; John Wiley and Sons: Chichester; New York, 2002. Green Chemistry: Frontiers in Benign Chemical Syntheses and Processes; Anastas, P. T.; Williamson, T. C. Eds.; Oxford University Press: Oxford, 1998. Guseinov, M. M.; Akhmedov, I. M.; Mamedov, E. G. Azerb. Khim. Zh. 1976, 46-48. Hashimoto, S.; Komeshima, N.; Koga, K. J. Chem. Soc., Chem. Commun. 1979, 437-438. Takemura, H.; Komeshima, N.; Takahashi, I.; Hashimoto, S.; Ikota, N.; Tomioka, T.; Koga, K. Tetrahedron Lett. 1987, 28, 5687-5690. Kagan, H. B.; Riant, O. Chem. Rev. 1992, 92, 1007-1019. Corey, E. J. Angew. Chem., Int. Ed. 2002, 41, 1650-1667. Evans, D. A.; Johnson, J. S. Diels-Alder Reactions In Comprehensive Asymmetric Catalysis; Jacobsen, E. N., Pfaltz, A., Yamamoto, H. Eds.; Springer: Berlin, 1999; Vol. 3. Reymond, S.; Cossy, J. Chem. Rev. 2008, 108, 5359-5406. Lowenthal, R. E.; Abiko, A.; Masamune, S. Tetrahedron Lett. 1990, 31, 6005-6008. Fritschi, H.; Leutenegger, U.; Pfaltz, A. Angew. Chem. 1986, 98, 1028-1029. Nishiyama, H.; Sakaguchi, H.; Nakamura, T.; Horihata, M.; Kondo, M.; Itoh, K. Organometallics 1989, 8, 846-848. Ghosh, A. K.; Mathivanan, P.; Cappiello, J. Tetrahedron: Asymmetry 1998, 9, 1-45. Desimoni, G.; Faita, G.; Jørgensen, K. A. Chem. Rev. 2006, 106, 3561-3651. Yoon, T. P.; Jacobsen, E. N. Science 2003, 299, 1691-1693. Irving, H. M.; Williams, R. J. P. J.Chem.Soc. 1953, 3192-3210. Johnson, J. S.; Evans, D. A. Acc. Chem. Res. 2000, 33, 325-335. Narasaka, K.; Inoue, M.; Okada, N. Chem. Lett. 1986, 1109-1112. Corey, E. J.; Imai, N.; Zhang, H. Y. J. Am. Chem. Soc. 1991, 113, 728-729. Evans, D. A.; Woerpel, K. A.; Hinman, M. M.; Faul, M. M. J. Am. Chem. Soc. 1991, 113, 726-728. Corey, E. J.; Ishihara, K. Tetrahedron Lett. 1992, 33, 6807-6810. 201 References (27) Evans, D. A.; Miller, S. J.; Lectka, T. J. Am. Chem. Soc. 1993, 115, 6460-6461. (28) Evans, D. A.; Murry, J. A.; von Matt, P.; Norcross, R. D.; Miller, S. J. Angew. Chem., Int. Ed. 1995, 34, 798-800. (29) Evans, D. A.; Kozlowski, M. C.; Tedrow, J. S. Tetrahedron Lett. 1996, 37, 74817484. (30) Evans, D. A.; Barnes, D. M. Tetrahedron Lett. 1997, 38, 57-58. (31) Evans, D. A.; Barnes, D. M.; Johnson, J. S.; Lectka, T.; von Matt, P.; Miller, S. J.; Murry, J. A.; Norcross, R. D.; Shaughnessy, E. A.; Campos, K. R. J. Am. Chem. Soc. 1999, 121, 7582-7594. (32) Evans, D. A.; Johnson, J. S.; Burgey, C. S.; Campos, K. R. Tetrahedron Lett. 1999, 40, 2879-2882. (33) Evans, D. A.; Miller, S. J.; Lectka, T.; von Matt, P. J. Am. Chem. Soc. 1999, 121, 7559-7573. (34) Johannsen, M.; Jørgensen, K. A. J. Chem. Soc., Perkin Trans. 2 1997, 1183-1185. (35) Davies, I. W.; Senanayake, C. H.; Larsen, R. D.; Verhoeven, T. R.; Reider, P. J. Tetrahedron Lett. 1996, 37, 1725-1726. (36) Davies, I. W.; Gerena, L.; Castonguay, L.; Senanayake, C. H.; Larsen, R. D.; Verhoeven, T. R.; Reider, P. J. J. Chem. Soc., Chem. Commun. 1996, 1753-1754. (37) Ghosh, A. K.; Mathivanan, P.; Cappiello, J. Tetrahedron Lett. 1996, 37, 3815-3818. (38) Ghosh, A. K.; Cho, H.; Cappiello, J. Tetrahedron: Asymmetry 1998, 9, 3687-3691. (39) Rechavi, D.; Lemaire, M. Chem. Rev. 2002, 102, 3467-3493. (40) Rechavi, D.; Lemaire, M. J. Mol. Catal. A: Chem. 2002, 182-183, 239-247. (41) Rechavi, D.; Albela, B.; Bonneviot, L.; Lemaire, M. Tetrahedron 2005, 61, 69766981. (42) Wang, H.; Liu, X.; Xia, H.; Liu, P.; Gao, J.; Ying, P.; Xiao, J.; Li, C. Tetrahedron 2006, 62, 1025-1032. (43) Meracz, I.; Oh, T. Tetrahedron Lett. 2003, 44, 6465-6468. (44) Doherty, S.; Goodrich, P.; Hardacre, C.; Knight, J. G.; Nguyen, M. T.; Parvulescu, V. I.; Paun, C. Adv. Synth. Catal. 2007, 349, 951-963. (45) Goodrich, P.; Hardacre, C.; Paun, C.; Parvulescu, V. I.; Podolean, I. Adv. Synth. Catal. 2008, 350, 2473-2476. (46) Chollet, G.; Rodriguez, F.; Schulz, E. Org. Lett. 2006, 8, 539-542. (47) Chollet, G.; Guillerez, M.; Schulz, E. Chem. Eur. J. 2007, 13, 992-1000. (48) Quaranta, L.; Corminboeuf, O.; Renaud, P. Org. Lett. 2002, 4, 39-42. (49) Sibi, M. P.; Venkatraman, L.; Liu, M.; Jasperse, C. P. J. Am. Chem. Soc. 2001, 123, 8444-8445. (50) Sibi, M. P.; Stanley, L. M.; Nie, X.; Venkatraman, L.; Liu, M.; Jasperse, C. P. J. Am. Chem. Soc. 2007, 129, 395-405. (51) Sibi, M. P.; Chen, J.; Stanley, L. Synlett 2007, 298-302. (52) Sibi, M. P.; Matsunaga, H. Tetrahedron Lett. 2004, 45, 5925-5929. (53) Ishihara, J.; Horie, M.; Shimada, Y.; Tojo, S.; Murai, A. Synlett 2002, 403-406. 202 References (54) Brimble, M. A.; McEwan, J. F. Tetrahedron: Asymmetry 1997, 8, 4069-4078. (55) Aggarwal, V. K.; Anderson, E. S.; Elfyn Jones, D.; Obierey, K. B.; Giles, R. Chem Commun. 1998, 1985-1986. (56) Aggarwal, V. K.; Jones, D. E.; Martin-Castro, A. M. Eur. J. Org. Chem. 2000, 29392945. (57) Palomo, C.; Oiarbide, M.; Garcia, J. M.; Gonzalez, A.; Arceo, E. J. Am. Chem. Soc. 2003, 125, 13942-13943. (58) Boger, D. L.; Weinreb, S. M. Hetero Diels-Alder Methodology in Organic Synthesis; Academic press: London, 1987. (59) Comprehensive Organic Synthesis: Selectivity, Strategy and Efficiency in Modern Organic Chemistry; Paquette, L. A.; Editor 1992. (60) Pellissier, H. Tetrahedron 2009, 65, 2839-2877. (61) Jørgensen, K. A. Angew. Chem. 2000, 39, 3558-3588. (62) Jørgensen, K. A. Eur. J. Org.Chem. 2004, 2093-2102. (63) Lin, L.; Liu, X.; Feng, X. Synlett 2007, 2147-2157. (64) Evans, D. A.; Johnson, J. S. J. Am. Chem. Soc. 1998, 120, 4895-4896. (65) Evans, D. A.; Johnson, J. S.; Olhava, E. J. J. Am. Chem. Soc. 2000, 122, 1635-1649. (66) Johannsen, M.; Jørgensen, K. A. J. Org. Chem. 1995, 60, 5757-5762. (67) Thorhauge, J.; Roberson, M.; Hazell, R. G.; Jørgensen, K. A. Chem. Eur. J. 2002, 8, 1888-1898. (68) Liu, J.; Niwayama, S.; You, Y.; Houk, K. N. J. Org. Chem. 1998, 63, 1064-1073. (69) Thorhauge, J.; Johannsen, M.; Jørgensen, K. A. Angew. Chem., Int. Ed. 1998, 37, 2404-2406. (70) Evans, D. A.; Olhava, E. J.; Johnson, J. S.; Janey, J. M. Angew. Chem., Int. Ed. 1998, 37, 3372-3375. (71) Zhuang, W.; Thorhauge, J.; Jørgensen, K. A. Chem. Commun. 2000, 459-460. (72) Audrain, H.; Thorhauge, J.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem. 2000, 65, 4487-4497. (73) Wada, E.; Koga, H.; Kumaran, G. Tetrahedron Lett. 2002, 43, 9397-9400. (74) Kurosu, M.; Porter, J. R.; Foley, M. A. Tetrahedron Lett. 2004, 45, 145-148. (75) Shin, Y. J.; Yeom, C.; Kim, M. J.; Kim, B. M. Synlett 2008, 89-93. (76) Evans, D. A.; Willis, M. C.; Johnston, J. N. Org. Lett. 1999, 1, 865-868. (77) Evans, D. A.; Scheidt, K. A.; Johnston, J. N.; Willis, M. C. J. Am. Chem. Soc. 2001, 123, 4480-4491. (78) 1,3-Dipolar Cycloaddition Chemistry; Padwa, A. Ed.; Wiley: New York, 1984; Vol. 1. (79) 1,3-Dipolar Cycloaddition Chemistry; Padwa, A. Eds; Wiley: New York, 1984; Vol. 2. (80) Torssell, K. B. G. Natural Product Chemistry; VCH: Weinheim, 1988. 203 References (81) Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Padwa, A.; Pearson, W. H. Eds.; John Wiley and Sons: Hoboken, 2002. (82) Patil, N. T.; Yamamoto, Y. Chem. Rev. 2008, 108, 3395-3442. (83) Gothelf, K. V.; Jørgensen, K. A. Chem. Rev. 1998, 98, 863-909. (84) Gothelf, K. V.; Jørgensen, K. A. Chem. Commun. 2000, 1449-1458. (85) Gothelf, K. V. Asymmetric metal-catalyzed 1,3-dipolar cycloaddition reactions In Cycloaddition Reactions in Organic Synthesis; Kobayashi, S., Jørgensen, K. A. Eds.; Wiley-VCH: Weinheim, 2002. (86) Stanley, L. M.; Sibi, M. P. Chem. Rev. 2008, 108, 2887-2902. (87) Pellissier, H. Tetrahedron 2007, 63, 3235-3285. (88) Saito, T.; Yamada, T.; Miyazaki, S.; Otani, T. Tetrahedron Lett. 2004, 45, 95819584. (89) Zhu, Y.; Liu, B.; You, J.; Wang, Y. J. Chem. Res. (S). 2007, 662-664. (90) Sibi, M. P.; Ma, Z.; Jasperse, C. P. J. Am. Chem. Soc. 2004, 126, 718-719. (91) Sibi, M. P.; Ma, Z.; Itoh, K.; Prabagaran, N.; Jasperse, C. P. Org. Lett. 2005, 7, 2349-2352. (92) Palomo, C.; Oiarbide, M.; Arceo, E.; Garcia, J. M.; Lopez, R.; Gonzalez, A.; Linden, A. Angew. Chem., Int. Ed. 2005, 44, 6187-6190. (93) Simonsen, K. B.; Bayon, P.; Hazell, R. G.; Gothelf, K. V.; Jørgensen, K. A. J. Am. Chem. Soc. 1999, 121, 3845-3853. (94) Jensen, K. B.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem. 1999, 64, 2353-2360. (95) Jensen, K. B.; Roberson, M.; Jørgensen, K. A. J. Org. Chem. 2000, 65, 9080-9084. (96) Gothelf, K. V.; Hazell, R. G.; Jørgensen, K. A. J. Org. Chem. 1998, 63, 5483-5488. (97) Oppolzer, W. Tetrahedron 1987, 43, 1969-2004. (98) Kitamura, M.; Suga, S.; Kawai, K.; Noyori, R. J. Am. Chem. Soc. 1986, 108, 60716072. (99) Kitamura, M.; Okada, S.; Suga, S.; Noyori, R. J. Am. Chem. Soc. 1989, 111, 40284036. (100) Nugent, W. A. Chem Commun. 1999, 1369-1370. (101) Oppolzer, W.; Radinov, R. N. Tetrahedron Lett. 1988, 29, 5645-5648. (102) Hari, Y.; Aoyama, T. Synthesis 2005, 583-587. (103) Genov, M.; Kostova, K.; Dimitrov, V. Tetrahedron: Asymmetry 1997, 8, 18691876. (104) Philipova, I.; Dimitrov, V.; Simova, S. Tetrahedron: Asymmetry 1999, 10, 13811391. (105) Ramon, D. J.; Yus, M. Tetrahedron: Asymmetry 1997, 8, 2479-2496. (106) Ramon, D. J.; Yus, M. Tetrahedron 1998, 54, 5651-5666. (107) Ramon, D. J.; Yus, M. Tetrahedron Lett. 1998, 39, 1239-1242. (108) Hwang, C.; Uang, B. Tetrahedron: Asymmetry 1998, 9, 3979-3984. (109) Balsells, J.; Walsh, P. J. J. Am. Chem. Soc. 2000, 122, 3250-3251. 204 References (110) Yus, M.; Ramon, D. J.; Prieto, O. Tetrahedron: Asymmetry 2002, 13, 2291-2293. (111) Prieto, O.; Ramon, D. J.; Yus, M. Tetrahedron: Asymmetry 2003, 14, 1955-1957. (112) Garcia, C.; LaRochelle, L. K.; Walsh, P. J. J. Am. Chem. Soc. 2002, 124, 1097010971. (113) Jeon, S.; Walsh, P. J. J. Am. Chem. Soc. 2003, 125, 9544-9545. (114) Li, H.; Garcia, C.; Walsh, P. J. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 5425-5427. (115) Jeon, S.; Li, H.; Garcia, C.; LaRochelle, L. K.; Walsh, P. J. J. Org. Chem. 2005, 70, 448-455. (116) Li, H.; Walsh, P. J. J. Am. Chem. Soc. 2005, 127, 8355-8361. (117) Forrat, V. J.; Prieto, O.; Ramon, D. J.; Yus, M. Chem. Eur. J. 2006, 12, 4431-4445. (118) Yus, M.; Ramon, D. J.; Prieto, O. Tetrahedron: Asymmetry 2003, 14, 1103-1114. (119) de Parrodi, C. A.; Walsh, P. J. Synlett 2004, 2417-2420. (120) Liu, L.; Wang, R.; Kang, Y.; Cai, H.; Chen, C. Synlett 2006, 1245-1249. (121) Xu, Z.; Chen, C.; Xu, J.; Miao, M.; Yan, W.; Wang, R. Org. Lett. 2004, 6, 1193-1195. (122) de las Casas Engel, T.; Lora Maroto, B.; Garcia Martinez, A.; de la Moya Cerero, S. Tetrahedron: Asymmetry 2008, 19, 646-650. (123) de las Casas Engel, T.; Maroto, B. L.; Martinez, A. G.; de la Moya Cerero, S. Tetrahedron: Asymmetry 2008, 19, 2003-2006. (124) Wu, P.; Wu, H.; Uang, B. J. Org. Chem. 2006, 71, 833-835. (125) Wu, H.; Wu, P.; Uang, B. J. Org. Chem. 2007, 72, 5935-5937. (126) Wu, H.; Wu, P.; Shen, Y.; Uang, B. J. Org. Chem. 2008, 73, 6445-6447. (127) Hwang, C.; Hwang, D.; Uang, B. J. Org. Chem. 1998, 63, 6762-6763. (128) Chang, C.; Yang, C.; Hwang, C.; Uang, B. Chem. Commun. 2002, 54-55. (129) Wu, H.; Uang, B. Tetrahedron: Asymmetry 2002, 13, 2625-2628. (130) Fiaud, J.; Maze, F.; Kagan, H. B. Tetrahedron: Asymmetry 1998, 9, 3647-3655. (131) Yang, T.; Lee, D. Tetrahedron: Asymmetry 1999, 10, 405-409. (132) Li, X.; Yeung, C.; Chan, A. S. C.; Yang, T. Tetrahedron: Asymmetry 1999, 10, 759763. (133) Santhi, V.; Rao, J. M. Tetrahedron: Asymmetry 2000, 11, 3553-3560. (134) Zou, H.; Hu, J.; Zhang, J.; You, J.; Ma, D.; Lue, D.; Xie, R. J. Mol. Catal. A: Chem. 2005, 242, 57-61. (135) Li, X.; Lou, R.; Yeung, C. -.; Chan, A. S. C.; Wong, W. K. Tetrahedron: Asymmetry 2000, 11, 2077-2082. (136) Kadyrov, R.; Ilaldinov, I. Z.; Almena, J.; Monsees, A.; Riermeier, T. H. Tetrahedron Lett. 2005, 46, 7397-7400. (137) Bunlaksananusorn, T.; Polborn, K.; Knochel, P. Angew. Chem., Int. Ed. 2003, 42, 3941-3943. (138) Bunlaksananusorn, T.; Luna, A. P.; Bonin, M.; Micouin, L.; Knochel, P. Synlett 2003, 2240-2242. (139) Bunlaksananusorn, T.; Knochel, P. J. Org. Chem. 2004, 69, 4595-4601. (140) Gayet, A.; Bolea, C.; Andersson, P. G. Org. Biomol. Chem. 2004, 2, 1887-1893. 205 References (141) Nakano, H.; Okuyama, Y.; Hongo, H. Tetrahedron Lett. 2000, 41, 4615-4618. (142) Nakano, H.; Okuyama, Y.; Yanagida, M.; Hongo, H. J. Org. Chem. 2001, 66, 620625. (143) Nakano, H.; Suzuki, Y.; Kabuto, C.; Fujita, R.; Hongo, H. J. Org. Chem. 2002, 67, 5011-5014. (144) Mino, T.; Hata, S.; Ohtaka, K.; Sakamoto, M.; Fujita, T. Tetrahedron Lett. 2001, 42, 4837-4839. (145) Gilbertson, S. R.; Fu, Z. Org. Lett. 2001, 3, 161-164. (146) Hiroi, K.; Watanabe, K. Tetrahedron: Asymmetry 2001, 12, 3067-3071. (147) Barhate, N. B.; Chen, C. Org. Lett. 2002, 4, 2529-2532. (148) Yang, K.; Chen, K. Org. Lett. 2002, 4, 1107-1109. (149) Yang, K.; Lee, W.; Pan, J.; Chen, K. J. Org. Chem. 2003, 68, 915-919. (150) Iwama, T.; Tsujiyama, S.; Kinoshita, H.; Kanematsu, K.; Tsurukami, Y.; Iwamura, T.; Watanabe, S.; Kataoka, T. Chem. Pharm. Bull. 1999, 47, 956-961. (151) Blay, G.; Climent, E.; Fernandez, I.; Hernandez-Olmos, V.; Pedro, J. R. Tetrahedron: Asymmetry 2007, 18, 1603-1612. (152) Blay, G.; Climent, E.; Fernandez, I.; Hernandez-Olmos, V.; Pedro, J. R. Tetrahedron: Asymmetry 2006, 17, 2046-2049. (153) Blay, G.; Domingo, L. R.; Hernandez-Olmos, V.; Pedro, J. R. Chem. Eur. J. 2008, 14, 4725-4730. (154) Blay, G.; Hernandez-Olmos, V.; Pedro, J. R. Chem. Commun. 2008, 4840-4842. (155) Blay, G.; Hernandez-Olmos, V.; Pedro, J. R. Org. Biomol. Chem. 2008, 6, 468-476. (156) Bellis, E.; Kokotos, G. Tetrahedron 2005, 61, 8669-8676. (157) Lemay, M.; Ogilvie, W. W. Org. Lett. 2005, 7, 4141-4144. (158) Rajaram, S.; Sigman, M. S. Org. Lett. 2005, 7, 5473-5475. (159) Zhou, W.; Xu, L.; Lyi, L.; Yang, L.; Xia, C. Eur. J. Org. Chem. 2006, 5225-5227. (160) He, H.; Pei, B.; Chou, H.; Tian, T.; Chan, W.; Lee, A. W. M. Org. Lett. 2008, 10, 2421-2424. (161) Langlois, Y.; Petit, A.; Remy, P.; Scherrmann, M.; Kouklovsky, C. Tetrahedron Lett. 2008, 49, 5576-5579. (162) Tzeng, Z.; Chen, H.; Huang, C.; Chen, K. Tetrahedron Lett. 2008, 49, 4134-4137. (163) Bolm, C.; Martin, M.; Simic, O.; Verrucci, M. Org. Lett. 2003, 5, 427-429. (164) Bolm, C.; Zani, L.; Rudolph, J.; Schiffers, I. Synthesis 2004, 2173-2180. (165) Bolm, C.; Schmidt, F.; Zani, L. Tetrahedron: Asymmetry 2005, 16, 1367-1376. (166) Towson, J. C.; Weismiller, M. C.; Lal, G. S.; Sheppard, A. C.; Davis, F. A. Org. Syntheses 1990, 69, 158-168. (167) Bartlett, P. D.; Knox, L. H. Org. Synth. 1965, 45, 55-56. (168) Johnson, W. S.; Schubert, E. N. J. Am. Chem. Soc. 1950, 72, 2187-2190. (169) Holerca, M. N.; Percec, V. Eur. J. Org. Chem. 2000, 2257-2263. (170) Somanathan, R.; Aguilar, H. R.; Rivero, I. A.; Aguirre, G.; Hellberg, L. H.; Yu, Z.; Thomas, J. A. J. Chem. Res. (S). 2001, 92, 348-353. 206 References (171) Sakakura, A.; Kondo, R.; Ishihara, K. Org. Lett. 2005, 7, 1971-1974. (172) Sakakura, A.; Kondo, R.; Umemura, S.; Ishihara, K. Tetrahedron 2009, 65, 21022109. (173) Andrade, C. K. Z.; Rocha, R. O.; Vercillo, O. E.; Silva, W. A.; Matos, R. A. F. Synlett 2003, 2351-2352. (174) Okamoto, T.; Kobayashi, K.; Oka, S.; Tanimoto, S. J. Org. Chem. 1988, 53, 48974901. (175) Jung, M. E.; McCombs, C. A. Org. Synth. 1978, 58, 163-168. (176) Pyridine and Its Derivatives, Suppl., Pt.1; Abramovitch, R. A. Ed.; Wiley & Sons: New york, 1974; (177) Otto, S.; Bertoncin, F.; Engberts, J. B. F. N. J. Am. Chem. Soc. 1996, 118, 77027707. (178) Otto, S.; Engberts, J. B. F. N.; Kwak, J. C. T. J. Am. Chem. Soc. 1998, 120, 95179525. (179) Rispens, T.; Engberts, J. B. F. N. Org. Lett. 2001, 3, 941-943. (180) Rispens, T.; Engberts, J. B. F. N. J. Org. Chem. 2002, 67, 7369-7377. (181) Wittkopp, A.; Schreiner, P. R. Chem. Eur. J. 2003, 9, 407-414. (182) Alves, C. N.; da Silva, A. B. F.; Marti, S.; Moliner, V.; Oliva, M.; Andres, J.; Domingo, L. R. Tetrahedron 2002, 58, 2695-2700. (183) Domingo, L. R.; Andres, J.; Alves, C. N. Eur. J. Org. Chem. 2002, 2557-2564. (184) Otto, S.; Boccaletti, G.; Engberts, J. B. F. N. J. Am. Chem. Soc. 1998, 120, 42384239. (185) Otto, S.; Engberts, J. B. F. N. J. Am. Chem. Soc. 1999, 121, 6798-6806. (186) Roelfes, G.; Feringa, B. L. Angew. Chem., Int. Ed. 2005, 44, 3230-3232. (187) Roelfes, G.; Boersma, A. J.; Feringa, B. L. Chem. Commun. 2006, 635-637. (188) Oltra, N. S.; Roelfes, G. Chem. Commun. 2008, 6039-6041. (189) Matsumoto, K.; Jitsukawa, K.; Masuda, H. Tetrahedron Lett. 2005, 46, 5687-5690. (190) Reetz, M. T.; Jiao, N. Angew. Chem., Int. Ed. 2006, 45, 2416-2419. (191) Cordaro, J. G.; McCusker, J. K.; Bergman, R. G. Chem. Commun. 2002, 1496-1497. (192) Sibi, M. P.; Yang, Y. Synlett 2008, 83-88. (193) Landa, A.; Minkkila, A.; Blay, G.; Jørgensen, K. A. Chem. Eur. J. 2006, 12, 34723483. (194) Landa, A.; Richter, B.; Johansen, R. L.; Minkkilae, A.; Jørgensen, K. A. J. Org. Chem. 2007, 72, 240-245. (195) Barroso, S.; Blay, G.; Pedro, J. R. Org. Lett. 2007, 9, 1983-1986. (196) Singh, P. K.; Singh, V. K. Org. Lett. 2008, 10, 4121-4124. (197) Kaczmarek, L.; Balicki, R.; Nantka-Namirski, P. Chem. Ber. 1992, 125, 1965-1966. (198) Katritzky, A. R.; Lagowski, J. M. Chemistry of the Heterocyclic N-Oxides; Academic Press: London, 1971. (199) Fife, W. K. J. Org. Chem. 1983, 48, 1375-1377. 207 References (200) Kleinfelter, D. C.; Dye, T. E.; Mallory, J. E.; Trent, E. S. J. Org. Chem. 1967, 32, 1734-1741. (201) Hiratate, A.; Tatsuzuki, M.; Busujima, T. PCT Int. Appl. 2005 CODEN: PIXXD2 WO 2005000793 A1, CAN 142:114079, AN 2005:14357 (202) Chandrasekhar, S.; Harvey, A. K.; Dell, C. P.; Ambler, S. J.; Smith, C. W. J. Pharmacol. Exp. Ther. 1995, 273, 1519-1528. (203) Dell, C. P.; Williams, A. C. Eur. Pat. Appl. 1994, CODEN: EPXXDW EP 599514 A2, CAN 121:108765, AN 1994:508765. (204) Williams, A. C. Eur. Pat. Appl. 1994, CODEN: EPXXDW EP 618206 A1, CAN 122:9869, AN 1995:231221. (205) Kamm, O. Org. Synth. 1925, IV, 57-58. (206) Bruening, I.; Grashey, R.; Hauck, H.; Huisgen, R.; Seidl, H. Org. Synth. 1966, 46, 127-130. (207) Bruening, I.; Grashey, R.; Hauck, H.; Huisgen, R.; Seidl, H. Org. Synth. 1966, 46, 96-98. (208) Evans, D. A.; Song, H.; Fandrick, K. R. Org. Lett. 2006, 8, 3351-3354. (209) Najera, C.; Yus, M. Tetrahedron 1999, 55, 10547-10658. (210) Singh, R. S.; Adachi, S.; Tanaka, F.; Yamauchi, T.; Inui, C.; Harada, T. J. Org. Chem. 2008, 73, 212-218. (211) Rickerby, J.; Vallet, M.; Bernardinelli, G.; Viton, F.; Kündig, E. P. Chem. Eur. J. 2007, 13, 3354-3368. (212) Liu, D.; Canales, E.; Corey, E. J. J. Am. Chem. Soc. 2007, 129, 1498-1499. (213) Singh, R. S.; Harada, T. Eur. J. Org. Chem. 2005, 3433-3435. (214) Futatsugi, K.; Yamamoto, H. Angew. Chem., Int. Ed. 2005, 44, 1484-1487, S1484/1-S1484/8. (215) Zhou, G.; Hu, Q.; Corey, E. J. Org. Lett. 2003, 5, 3979-3982. (216) Hawkins, J. M.; Nambu, M.; Loren, S. Org. Lett. 2003, 5, 4293-4295. (217) Wada, E.; Yasuoka, H.; Kanemasa, S. Chem. Lett. 1994, 145-148. (218) Wada, E.; Pei, W.; Yasuoka, H.; Chin, U.; Kanemasa, S. Tetrahedron 1996, 52, 1205-1220. (219) Wada, E.; Yasuoka, H.; Kanemasa, S. Chem. Lett. 1994, 1637-1640. (220) Wada, E.; Pei, W.; Kanemasa, S. Chem. Lett. 1994, 2345-2348. (221) Yang, H.; Kim, S. Synlett 2008, 555-560. (222) Trost, B. M. Angew. Chem., Int. Ed. 1995, 34, 259-281. 208 Anexo: Figuras A NEXO : F IGURAS OH OH OH N N N O O OH N O O 1 2 3 5 4 OH NH2 HO O N O SO3H 7 9 8 10 6 NH2 NH2 HO O HO O HO 12 11 NH 13 O O HO 14 O O NH N N O O 17 16 18 N N 22a 19 20 O O OH O 15 Me 22b O 22c 22d 21 209 Anexo: Figuras 23a 23d 23b 23e 23f 23h 23g 23c O O 23i 23j 26 25 O 27 24 N O O O 28aa N Me Ph N O N O O 28ca 28ba O O O 28da N O O O 28ab Me 28ac 28ad O O 28ae TMSO N Ph O N O O 28af O 28ag 30 O 29 28ah 31a 31c O 31b 31e 31d O S 31f 31i 210 31g 31j 31h 31k O N O Anexo: Figuras O O N N 31l N N Ph Ph 32 O O t-Bu t-Bu 33a 33b 33d 34aa 33c O O O N O N NO2 OMe 34ca 34ba O 34ea 34da O N O 34ga 34fa O O N O O O Ph 34ah O N Ph 34ab 34la 34ka 34ac 34ia 34ha 34ad 34ae 34ai 34lj 34af O N 35 211 Anexo: Figuras O O N N CN Ph 37 Ph 36 38 39 O O N 42a CO2H 40 O H N H 42e 42f OMe EtS OSiMe3 O 42g 42d 42b 42c 41 42k 42i 42h 42l 42j OEt O O N Ph 43aa 43ca 43ba OEt O OEt O N O O O 43fa 43da OEt O O N O O N S 43ha O Ph 43ac 43ka 43ja 43ia O 212 43ga OEt S 43ab O N O N O O Ph 43ad O N 43ae Anexo: Figuras O H SEt NH O Ph O O N O N Ph 43af 43ai 43ah 43ag OMe SEt O O N O N O Br 43ci Ph 43aj 43jj 45 44 43ak O N O Ph Ph N Ph 47c 47b O N 47d 47e Ph O Ph O N 47f Ph Ph N Ph Ph O2N Ph Br 47a 46 O N 47g O endo-48aa N O NO2 O Ph N exo-48aa Ph 48ba 48ca O N O 48da 213 Anexo: Figuras O S O O Ph N Ph Ph O Ph N Ph N O 48ga N O Ph N O O 48ia Ph O 48ja N O 48ka Ph O O Ph Ph N Ph N O N O O N O O2N MeO 48ac 48ab 48ad 48ag 48af 48ae O O O S O O S OMe Cl 50 49 52 51 O O S Me 53 54 55 56 57 OH O O O S O2N 58 214 Me 59 Anexo: Figuras 60 61 63 62 64 65 66b 66a O O O S O2N 66c 66d 66e 66f 66h 66g 66i 66j 68 67 O O O S 69aa O O O S Me Me OMe 69ba Me Br 69ca 69da 215 Anexo: Figuras 69ea 69fa 69ia 69ga 69ja 69ha 69fc 69fb 69gh 69fd 69gf 70 72 74 73 75 216 71 76 CCXXVII CCXXVIII