Presentación de PowerPoint
Anuncio

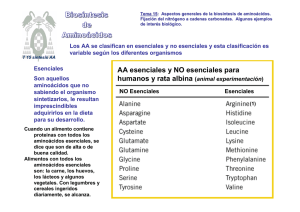
Metabolismo de aminoácidos El nitrógeno, presente en la biosfera como nitrato (NO3-) o dinitrógeno (N2), debe ser reducido a amonio (NH4+) para su incorporación a proteínas. El hombre adquiere el nitrógeno mayoritariamente de las proteínas de la dieta. El ciclo del nitrógeno Proteínas de la dieta Aminoácidos en la sangre Aminoácidos Proteínas Esquema general del metabolismo de aminoácidos Compuestos nitrogenados Carbono Nitrógeno Glucógeno Triaglicéridos CO2 + H2O Energía Urea y otros Orina Coenzimas vedette: Piridoxal fosfato Folato Tetrahidrobiopterina Cobalamina Digestión de proteínas La digestión de las proteínas comienza en el estómago, donde el pepsinógeno se convierte en pepsina. La llegada del bolo alimenticio al intestino dispara la liberación de las hormonas colecistoquinina y secretina, las cuales promueven la secreción de las proenzimas pancreáticas. La enteropeptidasa activa el tripsinógeno y la tripsina activa el resto de las proteasas. Los péptidos y los aminoácidos son transportados a través de los enterecitos a la circulación portal, por transporte activo o difusión facilitada. Aminoácidos esenciales (o indispensables) Esenciales Histidina Isoleucina Leucina Lisina Metionina Fenilalanina Treonina Triptofano Valina Arginina (en crecimiento) No Esenciales Alanina Asparagina Aspartato Cisteína Glutamato Glutamina Glicina Prolina Serina Tirosina BALANCE DE NITRÓGENO El nitrógeno no tiene fuentes de almacenamiento especiales en el organismo. En los adultos sanos la degradación y la síntesis de proteínas ocurren a la misma velocidad y se mantiene el balance nitrogenado, donde el nitrógeno que ingresa y el que se excreta son similares. Niños en crecimiento, adultos en recuperación de enfermedades o embarazadas tienen balance de nitrógeno positivo porque hay síntesis neta de proteína. Cuando se excreta más nitrógeno del que se incorpora, estamos en balance de nitrógeno negativo. Esto ocurre cuando falta algún aminoácido esencial en la dieta, o en el ayuno. Las proteínas del organismo están en continuo recambio Todo el tiempo se están degradando y sintetizando proteínas. En la especie humana, 1-2% de las proteínas, fundamentalmente musculares, están recambiándose. De los aminoácidos que se liberan, 75% se reciclan. El resto forma urea. Las enzimas glutamato deshidrogenasa, glutamina sintetasa y las aminotransferasas (transaminasas) tienen un rol muy importante en el metabolismo de aminoácidos. TRANSAMINACIÓN Los grupos α-amino se transfieren de un aminoácido a un alfa-cetoácido con las enzimas transaminasas o aminotransferasas. Suele participar el par glutamato/α-cetoglutarato. Reacciones reversibles, participan en la síntesis y degradación. Existen transaminasas para casi todos los aminoácidos. Tienen piridoxal 5'fosfato como grupo prostético. El piridoxal fosfato deriva de la vitamina B6. El PLP se une covalentemente a la enzima, a través de una base de Schiff o imina con una lisina del sitio activo Aspartato aminotransferasa Aspartato aminotransferasa Sitio activo con el piridoxal fosfato formando una base de Schiff con una lisina de la enzima Sitio activo con el piridoxal fosfato unido a un análogo de sustrato. El aumento en el suero de determinadas aminotransferasas es utilizado como marcador clínico de daño tisular. SGOT: glutamato-oxaloacetato aminotransferasa o aspartato transaminasa SGPT: glutamato-piruvato aminotransferasa o alanina transaminasa Por ejemplo, luego de un ataque cardíaco aumenta en primer lugar la creatina quinasa, luego aumenta SGOT, y más tarde SGPT. Como el par α-cetoglutarato/glutamato participa en muchas transaminaciones, el glutamato es un intermediario prominente en la eliminación de amonio así como en vías anabólicas. El glutamato está en un “nudo” entre el amonio libre y los grupos amino de los aminoácidos. ¿Qué opina acerca de la siguiente afirmación? ¿Es verdadera o falsa? "Dado que el nitrógeno del glutamato puede ser redistribuído por transaminación, el glutamato es un buen suplemento para proteínas pobres desde el punto de vista nutricional." Glutamato deshidrogenasa utiliza NADPH, incorpora amonio al glutamato para sintetizar este aminoácido y otros por transaminación, activada por ATP y GTP utiliza NAD+, reacción anaplerótica, provee de un intermediario oxidable y NADH, activada por ADP y GDP El glutamato recluta grupos amino. En el hígado se libera amonio. Éste va al ciclo de la urea. Glutamina La glutamina transporta amonio en la sangre. El amonio es tóxico para el sistema nervioso central. Los tejidos lo transforman en glutamina gracias a la glutamina sintetasa. En el hígado y en el riñón, la glutamina libera el amonio. A su vez, el amonio, en el hígado, forma urea. El riñón puede excretar directamente el amonio. Glutamato Glutamina sintetasa glutamato + NH4+ + ATP → glutamina + ADP + Pi + H+ Produce glutamina, uno de los 20 aminoácidos de las proteínas. La glutamina sirve como dador de nitrógeno en varias vías biosintéticas (purinas, citosina). La glutamina es muy abundante en la circulación, pues sirve como una forma de transporte inocua del amoníaco, que es tóxico, hacia el hígado y el riñón. Glutaminasa El hígado y el ríñón tienen glutaminasa, enzima mitocondrial que libera el amonio. glutamina + H2O → glutamato + NH4+ Destinos del amonio En el hígado, el amonio liberado de la glutamina por la glutaminasa se utiliza para sintetizar urea. En el riñón, la formación de amonio está relacionada con la eliminación de ácido (H+), puesto que el amoníaco (NH3) liberado por la glutaminasa se protona a amonio (NH4+) gracias a su pKa de 9.3. La glutamina sintetasa es una enzima regulada. En E. coli es regulada mediante los siguientes mecanismos: Alosterismo Modificación covalente Regulación de la expresión génica y la síntesis proteica (control de la cantidad de enzima) Regulación alostérica de la glutamina sintetasa • Nueve inhibidores “feedback”: Gly, Ala, Ser, His, Trp, CTP, AMP, carbamoil-P y glucosamina-6-P • Gly, Ala, Ser son indicadores del metabolismo de aminoácidos en las células • Los otros seis son producto final de vías metabólicas. Modificación covalente de la glutamina sintetasa • Cada subunidad es adenililada en la tirosina 397 •La adenililación inactiva la enzima • La adenilil transferasa cataliza la adenililación y la desadenililación •Una proteína reguladora (PII) controla ambas actividades •La actividad de PII se controla por uridililación. •La uridililación se inhibe por glutamina y se estimula por α-cetoglutarato y ATP. • En suma, la actividad se estimula cuando la glutamina desciende y cuando predominan α-cetoglutarato y ATP (sustratos). Regulación covalente de la glutamina sintetasa Adenililación de la tirosina Ciclo de la urea 80% del nitrógeno que se excreta, lo hace en forma de urea. Parte de las enzimas están en la matriz mitocondrial y parte en el citosol. La arginina, con la arginasa, genera urea y ornitina en el citosol. Luego, las siguientes enzimas regeneran la arginina. La enzima carbamoil fosfato sintetasa I mitocondrial cataliza el primer paso regulado de la síntesis de urea. Ciclo de la urea Destino de los esqueletos carbonados Todos los tejidos tienen cierta capacidad para síntesis y remodelación de aminoácidos. El hígado es el sitio principal de metabolismo de los aminoácidos. En tiempos de buena suplementación dietaria, el nitrógeno es eliminado vía transaminación, desaminación y síntesis de urea. Los esqueletos carbonados pueden conservarse como glucógeno o como ácidos grasos. Los aminoácidos pueden ser glucogénicos, cetogénicos o ambos. Los glucogénicos son los que generan piruvato o intermediarios del ciclo de Krebs como α-cetoglutarato o oxaloacetato. Los cetogénicos (Lys y Leu) generan sólo acetil-CoA o acetoacetil-CoA. En períodos de ayuno, los esqueletos carbonados se utilizan como fuente de energía, rindiendo CO2 y H2O. Biosíntesis de los aminoácidos no esenciales Aminoácidos esenciales y no esenciales Esenciales Histidina Isoleucina Leucina Lisina Metionina Fenilalanina Treonina Triptofano Valina Arginina (en crecimiento) No Esenciales Alanina Asparagina Aspartato Cisteína Glutamato Glutamina Glicina Prolina Serina Tirosina Los esqueletos carbonados de los aminoácidos no esenciales pueden sintetizarse a partir de metabolitos intermediarios derivados, por ejemplo, de glucosa. Dos aminoácidos no esenciales derivan directamente de aminoácidos esenciales (tirosina y cisteína). Coenzimas importantes: piridoxal fosfato, folato y tetrahidrobiopterina. Síntesis de glutamato El glutamato se sintetiza a partir de su precursor alfa-cetoácido con la enzima glutamato deshidrogenasa. Síntesis de aspartato El aspartato se sintetiza por transaminación del oxalaceto. glutamato + oxalacetato O O O O O O O O O O NH3+ alfa-cetoglutarato + aspartato O O O O O O O O NH3+ También se sintetiza aspartato por desaminación de la asparagina con la asparaginasa, análoga a la glutaminasa asparagina + H2O → aspartato + NH3 Síntesis de asparagina y glutamina La asparagina sintetasa y la glutamina sintetasa catalizan la producción de asparagina y glutamina glutamato + NH4+ + ATP → glutamina + ADP + Pi + H+ aspartato + NH4+ + ATP → asparagina + ADP + Pi + H+ Síntesis de alanina La alanina se sintetiza por transaminación con la enzima alanina transaminasa α-cetoglutarato + alanina glutamato + piruvato O O O O O NH3+ O O O O O O O O O NH3+ La concentración de alanina en la circulación es alta, más baja solo que la de glutamina. CICLO DE LA GLUCOSA – ALANINA La alanina transaminasa participa en el transporte de esqueletos carbonados y nitrógeno del músculo al hígado. alanina + cetoglutarato COO− COO− CH2 CH2 CH2 CH3 HC piruvato + glutamato NH3+ COO− alanine + C CH3 O COO− C CH2 O COO− + HC NH3+ COO− α-ketoglutarate pyruvate glutamate Aminotransferase (Transaminase) Músculo esquelético: el piruvato de la glucólisis se transforma en alanina a expensas de glutamato. Hígado: se regenera el piruvato para la gluconeogénesis y el amonio del glutamato puede ir al ciclo de la urea. Síntesis de prolina La prolina se sintetiza a partir de glutamato con glutamato semialdehído como intermediario La prolina también se sintetiza a partir de arginina de la dieta. Primero, la arginina se transforma en ornitina vía arginasa. La enzima puede operar en dirección opuesta para sintetizar ornitina y arginina a partir de prolina. Síntesis de serina La serina se sintetiza a partir de 3-fosfoglicerato. Se oxida la función alcohólica para formar un alfacetoácido que se transamina y desfosforila. La glicina se sintetiza a partir de serina. La enzima utiliza tetrahidrofolato y piridoxal fosfato. En el hígado de vertebrados, la glicina también puede sintetizarse a partir de dióxido de carbono y amonio con la glicina sintasa O sea que la serina y la glicina son interconvertibles gracias a la serina hidroximetiltransferasa, que opera en los dos sentidos y utiliza folato (THF/MTHF) y PLP La conversión de serina en glicina es la entrada principal de unidades monocarbonadas al pool de folato. Ácido fólico Los derivados del folato sirven como donadores de unidades monocarbonadas en diferentes estados de oxidación intermedios (metil, metilen, metenil, formil o formimino) Síntesis de cisteína El azufre para la cisteína proviene de la metionina y los carbonos de la serina. En primer lugar, ocurre la síntesis de S-adenosil metionina, un importante agente metilante. ++ La homocisteína condensa con la serina para formar cistationina, la cual es clivada por la cistationasa para dar cisteína y alfa-cetobutirato Esto es conocido como la vía de transulfuración La cistationina beta-sintasa y la cistationina gama-liasa ambas utilizan PLP La cisteína se utiliza para la síntesis de proteínas y de glutatión Las deficiencias genéticas en la cistationina betasintasa causan enfermedades Los aumentos en la homocisteína del plasma se asocian a problemas vasculares. Síntesis de tirosina La tirosina se sintetiza a partir de fenilalanina Las deficiencias genéticas en la fenilalanina hidroxilasa llevan a la fenilcetonuria Retardo mental La acumulación de fenilalanina depleta de αcetoglutarato por transaminación La depleción de α-cetoglutarato compromete el ciclo de Krebs y el metabolismo aeróbico del cerebro Se encuentra fenilpiruvato, fenilacetato y fenillactato en la orina Se debe suplementar la dieta con tirosina y restringir la fenilalanina "contiene fenilalanina" Aspartamo: L-Aspartil-L-fenilalanina metil éster ¿Qué productos le parece que se forman de su catabolismo? Aspartamo: L-Aspartil-L-fenilalanina metil éster Se metaboliza dando aspartato, fenilalanina y metanol. El aspartato es inocuo, la fenilalanina también (excepto para los fenilcetonúricos!) y el metanol puede ser metabolizado por el hígado en pequeñas cantidades. Provee de 4 kcal/g, como otros aminoácidos. Pero es 200 veces más dulce que el azúcar, por lo tanto endulza en cantidades muy bajas. Bebida cola: 0.06 % de aspartamo contra 12 % de sacarosa. Fue descubierto por Jim Schlatter en 1965, un químico trabajando en nuevas drogas peptídicas para el tratamiento de úlceras. En sus experimentos, sintetizó el aspartato-fenilalanina metil éster. Casualmente, lo tocó. Más tarde, sin querer, llevó el dedo a la boca y notó el gusto dulce. ¡¿Cómo, si se había lavado las manos después del desayuno?! Sospechó del aspartato-fenilalanina metil éster y se animó a probarlo. Años más tarde, la curiosidad de Schlatter redundó en un negocio billonario. Antes de poder utilizar el esqueleto carbonado de los aminoácidos, el nitrógeno debe removerse en forma de amonio. Como el amonio derivado del grupo amino de los aminoácidos es tóxico, los tejidos convierten el amonio en glutamina. En el hígado, la glutamina se convierte en urea. En el hombre, el exceso de nitrógeno se excreta como urea. Peces: excretan amonio. Pájaros: ácido úrico. Humanos y otros animales terrestres: urea.