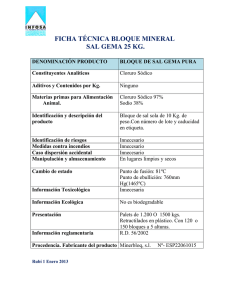
procedimiento para la fabricacion de complejos medicamentosos.
Anuncio

19 OFICINA ESPAÑOLA DE PATENTES Y MARCAS 11 Número de publicación: 2 229 355 51 Int. Cl. : A61K 47/48 7 ESPAÑA 12 TRADUCCIÓN DE PATENTE EUROPEA T3 86 Número de solicitud europea: 97924326 .8 86 Fecha de presentación: 05.06.1997 87 Número de publicación de la solicitud: 0955064 87 Fecha de publicación de la solicitud: 10.11.1999 54 Título: Procedimiento para la fabricación de complejos medicamentosos. 30 Prioridad: 06.06.1996 JP 14452296 73 Titular/es: DAIICHI PHARMACEUTICAL Co., Ltd. 14-10, Nihonbashi 3-chome Chuo-ku, Tokyo 103, JP 45 Fecha de publicación de la mención BOPI: 16.04.2005 72 Inventor/es: Inoue, Kazuhiro; Susaki, Hiroshi y Ikeda, Masahiro 45 Fecha de la publicación del folleto de la patente: 74 Agente: Ibañez González, José Francisco ES 2 229 355 T3 16.04.2005 Aviso: En el plazo de nueve meses a contar desde la fecha de publicación en el Boletín europeo de patentes, de la mención de concesión de la patente europea, cualquier persona podrá oponerse ante la Oficina Europea de Patentes a la patente concedida. La oposición deberá formularse por escrito y estar motivada; sólo se considerará como formulada una vez que se haya realizado el pago de la tasa de oposición (art. 99.1 del Convenio sobre concesión de Patentes Europeas). Venta de fascículos: Oficina Española de Patentes y Marcas. C/Panamá, 1 – 28036 Madrid ES 2 229 355 T3 DESCRIPCIÓN Procedimiento para la fabricación de complejos medicamentosos. 5 Campo técnico de la invención La presente invención se refiere a un procedimiento para la fabricación de un complejo medicamentoso en el cual están ligados entre sí un derivado de polisacárido y un compuesto medicamentoso, tal como un agente neoplásico. 10 15 20 Antecedentes de la técnica Los agentes antineoplásicos, utilizados para el tratamiento de cánceres sólidos tales como cáncer de pulmón o carcinomas de órganos digestivos, y cánceres de sangre tal como leucemia, se administran sistémicamente a través de rutas como la administración intravenosa u oral y luego se distribuyen hasta localizaciones tumorales específicas, e inhiben o suprimen la proliferación de células cancerosas para mostrar su eficacia terapéutica. Sin embargo, los agentes neoplásicos administrados sistémicamente son rápidamente asimilados por el hígado y órganos retículoendoteliales desde la sangre, o rápidamente excretados en la orina y, por tanto, su concentración en sangre puede reducirse en ocasiones hasta ser insuficiente para su distribución en áreas tumorales. Además, los agentes neoplásicos comunes presentan escasa capacidad de distribución hacia áreas tumorales (selectividad tumoral) y, por tanto, los agentes neoplásicos se distribuyen uniformemente en diversos tejidos y células de todo el cuerpo y actúan como citotoxinas también contra células y tejidos normales, lo cual se traduce en problemas de aparición de efectos adversos, por ejemplo emesis, pirexia o alopecia, en una proporción muy alta. Por tanto, era deseable desarrollar un medio de distribución eficiente y selectiva de agentes neoplásicos hacia áreas tumorales. 25 El documento EP 757049 A1 describe, según el artículo 54.3 del CPE, un derivado de camptotecina y un procedimiento para su preparación. Los derivados de camptotecina presentan una actividad antitumoral incrementada y se preparan mediante combinación del hidrocloruro del compuesto medicamentoso con la sal sódica de un polisacárido en un sistema equivalente. 30 El documento EP 059221 A1 describe composiciones medicinales para el tracto gastrointestinal. La composición comprende una sal hidrosoluble del ácido algínico como su ingrediente activo. Como sal hidrosoluble se utiliza sal amónica y sal amina. 35 40 45 50 55 60 65 El documento JP 57046920 A describe en forma similar un medicamento para los órganos digestivos que contiene como ingrediente activo una sal hidrosoluble del ácido algínico. Las sales preferibles son las sódicas, potásicas, amónicas y aminas. El medicamento presenta actividad protectora de la mucosa del órgano digestivo y actividad hemostática del órgano digestivo. Como uno de tales medios, se ha propuesto un procedimiento en el cual se utiliza como transportador-suministrador del medicamento un derivado de polisacárido que presenta grupos carboxilo, y un agente antineoplásico está ligado al derivado polisacárido para retrasar la desaparición del agente antineoplásico de la sangre y mejorar la selectividad hacia tejidos tumorales. Por ejemplo, la publicación internacional W094/19376 describe un complejo medicamentoso en el cual una cadena peptídica (número de residuos aminoácidos: 1 a 8) está ligada a un grupo carboxilo de un polisacárido que presenta grupos carboxilo, y adicionalmente está ligada doxorrubicina, daunorrubicina, mitomicina C, bleomicina, o similares, por medio de la cadena peptídica. Además, la publicación de la patente japonesa (KOKOKU) nº (Hei) 7-84481/1995 describe un complejo medicamentoso en el cual el agente antineoplásico antes mencionado se introduce en un derivado de manoglucano carboximetilado por medio de una base Schiff o un enlace ácido-amida. Estos complejos medicamentosos se caracterizan porque poseen una actividad antitumoral mejorada en comparación con el agente antitumoral aislado, que no está ligado a un transportador-suministrador del medicamento, y toxicidad y efectos adversos reducidos. En cuanto a las tecnologías relativas a complejos medicamentosos utilizando derivados polisacáridos polialcoholizados como transportadores-suministradores del medicamento, están disponibles algunos informes, por ejemplo, “Researches on polysaccharide-peptide-doxorubicin complexes - Correlations between stabilities of polysaccharide carriers in blood and their anti-neoplastic activities” (Abstracts of 10th Meeting of the Japan Society of Drug Delivery System, 279, 1994); “Researches on polysaccharide-peptide-doxorubicin complexes - Pharmacokinetics and anti-neoplastic activity” (Abstracts of 9th Annual Meeting of Japanese Society for the study of xenobiotics, 292, 1994); Abstracts of 19th Seminar of Trends in Research and Development (organizada por The Organization for Drug A D R Relief. R&D Promotion and Product Review), D-9, 1995; y “Researches on drug delivery to a tumor tissue by polysaccharide carriers” (Abstracts of 12th Colloid and Interface Technology Symposium, The Chemical Society of Japan, 51, 1995). Estos complejos medicamentosos se han fabricado convencionalmente preparando la sal sódica de un derivado de polisacárido que presenta grupos carboxilo, por ejemplo carboximetilpululano o carboximetilmanoglucano, y luego enlazando un grupo carboxilo del derivado polisacárido a un grupo amino de un agente antitumoral (o un grupo amino N-terminal de una cadena peptídica enlazado al agente antitumoral) por medio de un enlace ácido-amida. La sal sódica de derivados polisacáridos es totalmente insoluble al menos en disolventes orgánicos y, por tanto, cuando se realiza la reacción anterior, se ha adoptado el procedimiento que comprende preparar la sal sódica de un derivado de polisacárido 2 ES 2 229 355 T3 como una solución en agua o un disolvente orgánico acuoso que contenga agua, y luego disolver en la solución un agente condensador y un agente antitumoral (o un agente antitumoral que posea una cadena peptídica) en forma de hidrocloruro o similar. 5 10 15 Sin embargo, en la reacción, la condensación por deshidratación se realiza en un disolvente que contiene agua y, por tanto, era imposible obtener el producto deseado con altos rendimientos. Los agentes antitumorales utilizados como materiales de reacción son caros generalmente, y resultaba deseable desarrollar un procedimiento para producir eficientemente los complejos medicamentosos antes mencionados desde el punto de vista del coste de fabricación. Además, los compuestos descritos en la publicación de la patente japonesa sin examinar (KOKAI) nº (Hei) 6-87746/1994, que son útiles como agentes antitumorales, presentan un anillo lactona en sus moléculas y entonces aparecen problemas, ya que cuando estos compuestos, cada uno ligado a una cadena peptídica, se someten a la reacción antes mencionada, el anillo lactona se abre en presencia de una base y agua, y el grupo carboxilo resultante puede reaccionar con el grupo amino N-terminal de la cadena peptídica ligada al agente antitumoral para reducir apreciablemente el rendimiento del producto deseado y fracasar en la obtención del deseable complejo medicamentoso uniforme. Por tanto, se ha buscado desarrollar un método de reacción que pueda evitar la generación de compuestos cuyo anillo lactona se abra. Descripción de la invención 20 25 30 35 40 45 50 55 Un objetivo de la presente invención es proporcionar un procedimiento para producir eficientemente complejos medicamentosos que son capaces de hacer llegar con selectividad de localización un ingrediente activo, tal como agentes antitumorales o agentes anti-inflamatorios, hacia áreas tumorales o similares. Más específicamente, el objetivo de la presente invención es proporcionar un procedimiento capaz de producir los complejos medicamentosos en gran escala y a bajo coste haciendo reaccionar eficientemente un derivado de polisacárido que presenta grupos carboxilo con un compuesto medicamentoso, tal como un agente antitumoral, o con un espaciador que comprende un oligopéptido o similar que está ligado a un compuesto medicamentoso. Para lograr tales objetivos, los inventores de la presente realizaron intensivas investigaciones, y como resultado hallaron que cuando se hace reaccionar un derivado de polisacárido que presenta grupos carboxilo con un compuesto medicamentoso o con un espaciador ligado a un compuesto medicamentoso, utilizando una sal de amina orgánica del derivado polisacárido en lugar de la sal sódica utilizada convencionalmente, la sal del derivado polisacárido puede disolverse con una alta concentración en un disolvente orgánico sustancialmente libre de agua, de forma que la reacción de los derivados polisacáridos con el compuesto medicamentoso o con el espaciador ligado al compuesto medicamentoso puede realizarse con rendimientos extraordinariamente altos, y que los subproductos o similares pueden ser reducidos. La presente invención se logró con base en estos hallazgos. La presente invención proporciona así un procedimiento para la fabricación de un complejo medicamentoso en el cual un derivado de polisacárido que presenta grupos carboxilo y un residuo de un compuesto medicamentoso están ligados entre sí por medio de un espaciador que comprende un aminoácido o un espaciador que comprende 2 a 8 aminoácidos enlazados peptídicamente, o un complejo medicamentoso en el cual un derivado de polisacárido que presenta grupos carboxilo y un residuo de un compuesto medicamentoso están ligados entre sí sin el espaciador, caracterizado porque una sal de amina orgánica del derivado polisacárido que presenta grupos carboxilo se hace reaccionar con el compuesto medicamentoso o el compuesto medicamentoso ligado al espaciador en un sistema no acuoso; y un procedimiento para la fabricación del antes mencionado complejo medicamentoso, que comprende las etapas de (1) convertir una sal de metal alcalino del derivado polisacárido que presenta grupos carboxilo en una sal de amina orgánica de dicho derivado, y (2) hacer reaccionar la sal de amina orgánica con el compuesto medicamentoso o el compuesto medicamentoso ligado al espaciador en un sistema no acuoso. De acuerdo con realizaciones preferibles de la presente invención, se proporciona el procedimiento anterior en el cual el derivado de polisacárido que presenta grupos carboxilo es un polialcohol carboxi(C1−4 )alquildextrano; el procedimiento anterior en el cual el polialcohol dextrano que constituye el polialcohol carboxi(C1−4 )alquildextrano es un polialcohol dextrano obtenido mediante tratamiento de un dextrano bajo condiciones que permiten una polialcoholización sustancialmente completa; el procedimiento anterior en el cual el polialcohol carboxi(C1−4 )alquildextrano es un polialcohol carboximetildextrano; el procedimiento anterior en el cual el polialcohol carboxi(C1−4 )alquildextrano es un polialcohol carboximetildextrano que presenta un peso molecular en el intervalo de 5.000 a 500.000, preferiblemente en el intervalo de 50.000 a 450.000, y más preferiblemente en el intervalo de 200.000 a 400.000, y el grado de carboximetilación por cada residuo sacárido constitutivo está en el intervalo de 0,01 a 2,0, preferiblemente en el intervalo de 0,1 a 1,0, y más preferiblemente en el intervalo de 0,3 a 0,5; y el procedimiento anterior en el cual el compuesto medicamentoso es un agente antineoplásico o un agente anti-inflamatorio. 60 65 De acuerdo con realizaciones preferibles adicionales de la presente invención, se proporciona el procedimiento anterior en el cual el compuesto medicamentoso es un compuesto que puede formar un anillo lactona; el procedimiento anterior en el que un compuesto medicamentoso en el cual está formado un anillo lactona o un espaciador ligado a un compuesto medicamentoso en el cual está formado un anillo lactona es utilizado para la reacción de la sal de amina orgánica con el compuesto medicamentoso o con el espaciador ligado al compuesto medicamentoso; y el procedimiento anterior en el cual el compuesto medicamentoso que puede formar un anillo lactona es (1S,9S)1-amino-9-etil-5-fluoro-2,3-dihidro-9-hidroxi-4-metil-1H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolina10,13(9H,15H)-diona. 3 ES 2 229 355 T3 Breve descripción de los dibujos La Figura 1 representa el diagrama GPC (cromatografía de permeación de gel) del complejo medicamentoso del Ejemplo 8 preparado mediante el procedimiento de la presente invención. 5 La Figura 2 representa el espectro de absorción ultravioleta del complejo medicamentoso del Ejemplo 8 preparado mediante el procedimiento de la presente invención. 10 La Figura 3 representa el diagrama GPC del complejo medicamentoso del Ejemplo 9 preparado mediante el procedimiento de la presente invención. La Figura 4 representa el espectro de absorción ultravioleta del complejo medicamentoso del Ejemplo 9 preparado mediante el procedimiento de la presente invención. 15 La Figura 5 representa el espectro de absorción ultravioleta del complejo medicamentoso del Ejemplo 10 preparado mediante el procedimiento de la presente invención. La Figura 6 representa el diagrama GPC del complejo medicamentoso del Ejemplo 15 preparado mediante el procedimiento de la presente invención. 20 La Figura 7 representa el espectro de absorción ultravioleta del complejo medicamentoso del Ejemplo 15 preparado mediante el procedimiento de la presente invención. 25 La Figura 8 representa el diagrama GPC del complejo medicamentoso del Ejemplo 28 preparado mediante el procedimiento de la presente invención. La Figura 9 representa el espectro de absorción ultravioleta del complejo medicamentoso del Ejemplo 28 preparado mediante el procedimiento de la presente invención. 30 La Figura 10 representa el diagrama GPC del complejo medicamentoso del Ejemplo 29 preparado mediante el procedimiento de la presente invención. La Figura 11 representa el espectro de absorción ultravioleta del complejo medicamentoso del Ejemplo 29 preparado mediante el procedimiento de la presente invención. 35 La Figura 12 representa el diagrama GPC del complejo medicamentoso del Ejemplo 34 preparado mediante el procedimiento de la presente invención. 40 La Figura 13 representa el espectro de absorción ultravioleta del complejo medicamentoso del Ejemplo 34 preparado mediante el procedimiento de la presente invención. La Figura 14 representa el diagrama GPC del complejo medicamentoso del Ejemplo 39 preparado mediante el procedimiento de la presente invención. 45 La Figura 15 representa el espectro de absorción ultravioleta del complejo medicamentoso del Ejemplo 39 preparado mediante el procedimiento de la presente invención. La Figura 16 representa el diagrama GPC del complejo medicamentoso del Ejemplo 41 preparado mediante el procedimiento de la presente invención. 50 La Figura 17 representa el espectro de absorción ultravioleta del complejo medicamentoso del Ejemplo 41 preparado mediante el procedimiento de la presente invención. 55 La Figura 18 representa el diagrama GPC del complejo medicamentoso del Ejemplo 44 preparado mediante el procedimiento de la presente invención. La Figura 19 representa el espectro de absorción ultravioleta del complejo medicamentoso del Ejemplo 44 preparado mediante el procedimiento de la presente invención. 60 La Figura 20 representa la farmacocinética del complejo medicamentoso del Ejemplo 15 preparado mediante el procedimiento de la presente invención. Cada punto en la figura representa un valor promedio de tres experimentos. Mejor modo de realizar la invención 65 El complejo medicamentoso preparado mediante el procedimiento según la presente invención se caracteriza porque un derivado de polisacárido que presenta grupos carboxilo y un residuo de un compuesto medicamentoso están ligados entre sí por medio de un espaciador que comprende un aminoácido o un espaciador que comprende 2 a 8 aminoácidos enlazados peptídicamente, o un derivado de polisacárido que presenta grupos carboxilo y un residuo de un 4 ES 2 229 355 T3 5 10 15 20 25 compuesto medicamentoso están ligados entre sí sin el espaciador. El residuo de un compuesto medicamentoso contenido en el complejo se deriva de un compuesto medicamentoso utilizado como un medicamento para el tratamiento terapéutico y/o preventivo de enfermedades de mamíferos incluyendo el humano, por ejemplo, un agente antineoplásico, un agente anti-inflamatorio, un agente antibacteriano, o similares, y el residuo está compuesto de una estructura parcial del compuesto medicamentoso. Sin embargo, el compuesto medicamentoso del cual se deriva el residuo no se limita a los mencionados anteriormente. Además, como compuesto medicamentoso, pueden ser utilizados cualesquiera compuestos siempre que posean uno o más grupos funcionales reactivos capaces de participar en la formación de enlace con un derivado de polisacárido o un espaciador (por ejemplo, un grupo amino, grupo carboxilo, grupo hidroxilo, grupo tiol, grupo éster o similares). En la presente descripción el término “compuesto medicamentoso” también incluye un profármaco que contiene, como parte de él, una estructura principal de un compuesto medicamentoso que posee actividad farmacológica propia y puede reproducir el compuesto in vivo. Más específicamente, en la presente descripción el término “residuo del compuesto medicamentoso” significa una estructura parcial derivada del compuesto medicamentoso existente en el compuesto después de la formación del enlace, presumiendo que se ha formado un enlace entre el derivado de polisacárido o el espaciador y el residuo de un compuesto medicamentoso, a través de una reacción de un grupo funcional reactivo del compuesto medicamentoso y un grupo funcional reactivo del derivado polisacárido o el espaciador (p.ej. condensación por deshidratación etc.). Cuando el compuesto medicamentoso se representa, por ejemplo, mediante D-NH2 , D-COOH, D-COOR, D-OH, DSH, D-CONH2 , o D-NH-COOR (R es un grupo alquilo inferior o similar), el residuo del compuesto medicamentoso se representa por D-NH-(D-NH-CO-Q, etc.), D-CO-(D-CO-NH-Q, D-CO-O-Q, D-CO-S-Q, etc.), D-CO-(D-CO-NHQ, D-CO-O-Q, D-CO-S-Q, etc.), D-O-(D-O-CO-Q, D-O-Q, etc.), D-S-(D-S-CO-Q, D-S-Q, etc.), D-CONH-(D-CONH-CO-Q, etc.) y D-NH-CO-(D-NH-CO-O-Q, D-NH-CO-NH-Q, etc.), respectivamente (el paréntesis representa un enlace entre el espaciador o el derivado de polisacárido y el residuo del compuesto medicamentoso, en el cual Q representa una estructura parcial remanente del espaciador y el derivado polisacárido excluyendo un grupo funcional reactivo y un grupo carboxilo, respectivamente). Sin embargo, el tipo de enlace entre el espaciador o el derivado de polisacárido y el residuo del compuesto medicamentoso no se limita a los mencionados anteriormente. El residuo del compuesto medicamentoso puede estar ligado a un grupo carboxilo del derivado polisacárido, al grupo amino Nterminal o el grupo carboxilo C-terminal del espaciador, o a un grupo funcional reactivo existente en un aminoácido que constituye el espaciador. 30 35 Por ejemplo, como residuo del compuesto medicamentoso, pueden utilizarse preferiblemente residuos de agentes neoplásicos tales como doxorrubicina, daunorrubicina, mitomicina C, bleomicina, ciclocitidina, vincristina, vinblastina, metotrexato, agentes neoplásicos platinados (cisplatino o sus derivados), taxol o sus derivados, camptotecina o sus derivados (agentes neoplásicos descritos en la publicación de la patente japonesa sin examinar (KOKAI) nº (Hei) 687746/1994, preferiblemente (1S,9S)-1-amino-9-etil-5-fluoro-2,3-dihidro-9-hidroxi-4-metil-1H,12H-benzo[de]pirano [3’,4’: 6,7]indolizino[1,2-b]quinolina-10,13-(9H,15H)-diona descrito en la reivindicación 2, o similares). Además, son también preferibles residuos de agentes anti-inflamatorios esteroides, tales como succinato de hidrocortisona y succinato de prednisolona y agentes anti-inflamatorios no esteroides, tales como ácido mefenámico, ácido flufenámico, diclofenac, ibuprofeno y tinoridina. 40 45 El residuo del compuesto medicamentoso puede ligarse directamente al polisacárido, o puede ligarse por medio de un espaciador. Como espaciador, pueden utilizarse aquellos que comprenden un aminoácido ó 2 a 8 aminoácidos enlazados peptídicamente. Más específicamente, cuando el residuo del compuesto medicamentoso y el derivado polisacárido están ligados por medio de un espaciador, el espaciador adopta una forma de residuo de un aminoácido, lo cual significa un residuo obtenido mediante eliminación de un átomo de hidrógeno y un grupo hidroxilo de un grupo amino y un grupo carboxilo del aminoácido, respectivamente, o un residuo de un oligopéptido que comprende 2 a 8 aminoácidos enlazados peptídicamente, lo que significa un residuo obtenido mediante eliminación de un átomo de hidrógeno y un grupo hidroxilo del grupo amino N-terminal y el grupo carboxilo C-terminal del oligopéptido, respectivamente. 50 55 60 65 Espaciadores preferibles son los residuos de oligopéptidos que comprenden 2 a 6 aminoácidos. El tipo del aminoácido constitutivo del espaciador no está particularmente limitado y, por ejemplo, pueden ser utilizados L- o Daminoácidos, preferiblemente L-aminoácidos, y también pueden ser utilizados β-alanina, ácido ε-aminocapróico, ácido γ-aminobutírico, o similares, así como α-aminoácidos. Estos aminoácidos, distintos de los α-aminoácidos, están situados preferiblemente próximos al derivado de polisacárido en el espaciador. Por ejemplo, cuando se utiliza un espaciador oligopeptídico, la dirección de enlace no está particularmente limitada y, generalmente, la N-terminación del espaciador puede estar ligada a un grupo carboxilo del polialcohol carboxi(C1−4 ) alquildextrano por medio de un enlace ácido-amida, y la C-terminación del espaciador puede estar ligada a un grupo amino del compuesto medicamentoso. Alternativamente, por ejemplo, cuando un residuo lisina se incorpora como una unidad constitutiva del espaciador peptídico, se permite al grupo α-amino y al grupo ε-amino del residuo lisina formar respectivos enlaces ácido-amida con grupos carboxilo de otros aminoácidos para formar N-terminaciones en ambos extremos del espaciador peptídico, lo cual posibilita la formación de enlaces con grupos carboxilo de los compuestos medicamentosos. Además, incorporando en un espaciador uno o más residuos de compuestos diamina o compuestos de ácido dicarboxílico (residuos de compuestos diamina tales como etilenodiamina o compuestos de ácido dicarboxílico tal como ácido succínico) como unidades constitutivas, puede ser utilizado un espaciador que posee tanto N-terminaciones como C-terminaciones en ambos extremos. 5 ES 2 229 355 T3 5 10 15 20 25 30 35 40 45 Cuando se utiliza un espaciador que comprende un oligopéptido, la secuencia aminoácida no está particularmente limitada. Los espaciadores preferiblemente utilizados incluyen, por ejemplo, un espaciador que es un residuo de un dipéptido representado por -X-Z-, en el cual X representa un residuo de un aminoácido hidrofóbico y Z representa un residuo de un aminoácido hidrofílico; y X-Z- significa un residuo que consta de un dipéptido que está formado por un enlace peptídico entre un aminoácido hidrofóbico (X) y un aminoácido hidrofílico (Z) en el extremo N-terminal y el extremo C-terminal, respectivamente, y cuyo átomo de hidrógeno y grupo hidroxilo son eliminados del grupo amino en la N-terminación y el grupo carboxilo en la C-terminación, respectivamente, y un espaciador que contiene un residuo del dipéptido como una secuencia peptídica parcial. Por ejemplo, como aminoácido hidrofóbico puede utilizarse fenilalanina, tirosina, leucina o similares, y como aminoácido hidrofílico pueden ser utilizados, por ejemplo, glicina, alanina o similares. El espaciador puede presentar una secuencia repetida del residuo dipeptídico (por ejemplo, X-Z-X-Z-, -X-Z-X-Z-X-Z-, etc.). Utilizando el espaciador que contiene tal estructura dipeptídica, el espaciador puede ser hidrolizado en áreas tumorales o zonas inflamatorias, que se consideran abundantes en peptidasa, para liberar el compuesto medicamentoso con alta concentración en dichos lugares y, por tanto, la estructura formada mediante enlace entre el espaciador que contiene el anterior dipéptido y el compuesto medicamentoso es una estructura parcial preferible del complejo medicamentoso, de acuerdo con el procedimiento de la presente invención. Cuando se utiliza un residuo de un agente antineoplásico que presenta actividad antineoplásica dependiente de su concentración (agentes neoplásicos que poseen una más potente actividad antineoplásica a mayor concentración: agentes neoplásicos dependientes de la concentración, p.ej. doxorrubicina, etc.) como el residuo del compuesto medicamentoso, puede utilizarse preferiblemente un espaciador compuesto del anterior residuo dipeptídico representado por -X-Z- o un espaciador que contiene el anterior residuo dipeptídico como una secuencia peptídica parcial. Además, cuando se utiliza como residuo del compuesto medicamentoso un tipo de agente antineoplásico dependiente del espacio de tiempo, el cual requiere un tiempo de actividad sostenida bajo determinada concentración, puede obtenerse algunas veces una actividad antineoplásica mejorada utilizando el espaciador anterior. Los ejemplos incluyen los agentes neoplásicos descritos en la publicación de la patente japonesa sin examinar (KOKAI) nº (Hei) 687746/1994, preferiblemente el agente antineoplásico descrito en la reivindicación 2. Generalmente, el espaciador no se limita a los mencionados anteriormente y es necesario elegir un espaciador apropiado desde los puntos de vista del modo de actuación del agente antineoplásico, características farmacocinéticas o aparición de toxicidad, liberación in vivo del agente antineoplásico y otros. Para carcinomas que presentan rápida proliferación, es preferible generalmente elegir el espaciador anterior capaz de liberar el compuesto medicamentoso con alta concentración en un corto intervalo. En la tabla siguiente se representan ejemplos específicos del espaciador. Sin embargo, el espaciador utilizado en el procedimiento para la fabricación de los complejos medicamentosos de la presente invención no se limita a los mencionados más adelante, y se comprende que un experto medio en la materia puede seleccionar apropiadamente un espaciador para lograr un índice óptimo de liberación de un compuesto medicamentoso. En la tabla, los extremos izquierdos de las secuencias peptídicas son N-terminaciones y los residuos de compuestos medicamentosos están ligados a C-terminaciones. D-Phe representa un residuo D-fenilalanina y los otros aminoácidos representan L-aminoácidos. Los grados del índice de liberación fueron estimados a partir del grado de aparición de eficacia de los complejos medicamentosos ligados con doxorrubicina experimentados en ratas portadoras de tumores Walker 256, o a partir de la concentración de doxorrubicina libre en áreas tumorales de ratas portadoras de tumores Walker 256. Entre estos espaciadores, para doxorrubicina se utiliza preferiblemente un espaciador que puede liberar el compuesto medicamentoso con alta concentración en un corto intervalo, por ejemplo (N-terminación)-Gly-Gly-Phe-Gly-. TABLA 1 (a) Espaciadores que presentan alto índice de liberación 50 -Leu-Gly-Tyr-Gly55 -Phe-Gly-Gly-Phe-Gly-Gly-Gly-Phe-Gly- 60 -Gly-Phe-Gly-Gly-Phe-Gly-Gly-Gly65 -Phe-Phe-Gly-Gly-Gly-Gly-Gly-Phe-Gly6 ES 2 229 355 T3 (b) Espaciadores que presentan índices de liberación relativamente altos -Gly-Gly-Phe-Phe5 -Gly-Gly-Gly-Gly-Gly-Gly(c) Espaciadores que presentan índices de liberación relativamente bajos -Phe-Phe- 10 -Ala-Gly-Pro-Gly15 -Gly-Gly-Gly-Phe(d) Espaciadores que presentan bajo índice de liberación -Gly- 20 -D-Phe-Gly-Gly-Phe25 -Ser-Gly-Gly-Gly-Gly-Gly-Gly- 30 -Gly-Gly-Gly-Gly- 35 40 45 50 55 60 65 Como derivado polisacárido que presenta grupos carboxilo y que constituye la fracción derivado de polisacárido del complejo medicamentoso pueden ser utilizados, por ejemplo, cualquier polisacárido y sus derivados química o biológicamente modificados siempre que posean grupos carboxilo en sus moléculas. Por ejemplo, pueden ser utilizados polisacáridos tales como ácido hialurónico, ácido péctico, ácido algínico, condroitina y heparina; y polisacáridos tales como pululano, dextrano, manano, quitina, inulina, levan, xylan, araban, manoglucano y quitosano, en los cuales todos o una parte de los grupos hidroxilo se introducen con grupos funcionales que presentan un grupo carboxilo. Por ejemplo, pueden utilizarse preferiblemente aquellos que presentan grupos hidroxilo carboxi(C1−4 )alquilados o aquellos que poseen grupos hidroxilo esterificados con uno de los grupos carboxilo de un ácido polibásico. Además, también pueden utilizarse los obtenidos mediante polialcoholización de los anteriores polisacáridos y posterior introducción de grupos funcionales que poseen un grupo carboxilo. Entre tales derivados polisacáridos, se prefiere un polialcohol carboxi(C1−4 )alquildextrano. Aunque no está particularmente limitado el grado de polialcoholización del polialcohol carboxi(C1−4 )alquildextrano, es preferible que el polialcohol dextrano que constituye el polialcohol carboxi(C1−4 )alquildextrano sea un polialcohol dextrano obtenido mediante tratamiento de un dextrano bajo condiciones que posibiliten una polialcoholización sustancialmente completa. El tipo de dextrano utilizado para la preparación del polialcohol carboxi(C1−4 )alquildextrano no está particularmente limitado, y el dextrano puede contener α-D-1,6-enlaces en cualquier cantidad. Por ejemplo, pueden ser utilizados los dextranos que contienen α-D-1,6-enlaces en una tasa del 85% o más, 90% o más, y 95% o más. El peso molecular del dextrano no está particularmente limitado y, por ejemplo, pueden ser utilizados los dextranos que presentan un peso molecular desde aproximadamente 10.000 hasta aproximadamente 2.000.000, preferiblemente desde aproximadamente 50.000 hasta aproximadamente 800.000. Como grupo C1−4 alquilo que constituye el grupo carboxi(C1−4 )alquilo del polialcohol carboxi(C1−4 )alquildextrano, pueden ser utilizados un grupo C1−4 alquilo linear o ramificado, específicamente, grupo metilo, grupo etilo, grupo n-propilo, grupo isopropilo, grupo n-butilo, grupo sec-butilo o similares, y puede utilizarse preferiblemente un grupo metilo. El procedimiento de la presente invención se caracteriza porque para preparar el complejo medicamentoso antes mencionado, se hace reaccionar una sal de amina orgánica del derivado polisacárido que presenta grupos carboxilo con el compuesto medicamentoso aislado, o una sal de amina orgánica del derivado polisacárido que presenta grupos carboxilo se hace reaccionar con un espaciador ligado al compuesto medicamentoso en un disolvente orgánico sustancialmente libre de agua, por ejemplo un sistema no acuoso. Como una realización preferible del procedimiento de la presente invención, un procedimiento que utiliza un polialcohol carboxi(C1−4 )alquildextrano como el derivado de polisacárido será explicado específicamente más adelante. Sin embargo, el objeto de la presente invención no está limitado a dicha realización. 7 ES 2 229 355 T3 5 Cuando se utiliza un dextrano como un material de partida, el dextrano puede ser tratado sucesivamente con un gran exceso de periodato sódico y borohidruro sódico para obtener un polialcohol dextrano sometido a una polialcoholización sustancialmente completa. Sin embargo, el procedimiento para la polialcoholización de dextranos no está limitado al procedimiento mencionado anteriormente, y puede ser adoptado cualquier procedimiento utilizable por los expertos en la materia. La carboxi(C1−4 )alquilación puede ser realizada, por ejemplo, haciendo reaccionar un ácido (C1−4 )alquilcarboxílico halogenado tal como ácido cloroacético, ácido bromoacético, ácido α-cloropropiónico, ácido α-metil-α-cloropropiónico, ácido β-cloropropiónico, ácido α-metil-β-cloropropiónico, ácido α-clorobutírico, ácido βclorobutírico o ácido γ-clorobutírico, preferiblemente ácido cloroacético, con grupos hidroxilo del polialcohol dextrano para alcanzar una carboxi(C1−4 )alquilación parcial o completa de los grupos hidroxilo. 10 15 20 25 30 35 40 45 50 55 60 65 Por ejemplo, el polialcohol dextrano se disuelve en un disolvente inerte que no participa en las reacciones (p.ej. agua, N,N-dimetilformamida, o dimetil sulfóxido) y la solución se añade con un ácido (C1−4 )alquilcarboxílico halogenado o una sal de éste en presencia de una base (p.ej. hidróxido sódico o hidróxido potásico), y entonces se deja reaccionar la mezcla desde varios minutos hasta varios días a una temperatura desde enfriamiento con hielo hasta aproximadamente 100ºC. El grado de introducción del grupo carboxi(C1−4 )alquilo puede ser fácilmente controlado, por ejemplo mediante adecuada elección de la temperatura de reacción de la carboxi(C1−4 )alquilación o la cantidad de ácido (C1−4 )alquilcarboxílico halogenado o las bases utilizadas como reactivos, y estos medios son bien conocidos para los expertos en la materia. El grado de carboxi(C1−4 )alquilación para los grupos hidroxilo del polialcohol dextrano no está particularmente limitado y, por ejemplo, el grado puede estar en el intervalo de 0,01 a 2,0, preferiblemente de 0,1 a 1,0, y más preferiblemente de 0,3 a 0,5 por cada residuo del sacárido constitutivo. El peso molecular del polialcohol carboxi(C1−4 )alquildextrano es desde aproximadamente 5.000 a 500.000, preferiblemente desde aproximadamente 50.000 a 450.000, y más preferiblemente desde aproximadamente 200.000 a 400.000, cuando se determina por el método de filtración de gel. El polialcohol carboxi(C1−4 )alquildextrano preparado según lo anterior está previsto como una solución acuosa de una sal de metal alcalino, tal como la sal sódica o potásica. El procedimiento de la presente invención se caracteriza porque se utiliza una sal de amina orgánica del derivado polisacárido, en lugar del anterior derivado polisacárido en forma de una sal de metal alcalino, para la formación del enlace entre el compuesto medicamentoso o el espaciador ligado al compuesto medicamentoso y los grupos carboxilo del derivado polisacárido. La sal de amina orgánica de los derivados polisacáridos puede disolverse con una alta concentración en un disolvente orgánico sustancialmente libre de agua y posibilita que la reacción se realice en un sistema no acuoso y, por tanto, puede ser notablemente mejorada la eficiencia de la reacción. Por ejemplo, como sal de amina orgánica pueden ser utilizadas sales de aminas alifáticas, tales como trietilamina, trimetilamina, o trietanolamina; sales de aminas alicíclicas y aromáticas tales como N-metilpirrolidina, N-metilpiperidina, N-metilmorfolina, o dimetil-aminopiridina; o sales de amonio cuaternario tales como cloruro tetrametilamónico o cloruro tetraetilamónico. La conversión desde la sal sódica del derivado de polisacárido en la sal de amina orgánica puede ser realizada utilizando una resina de intercambio fónico, o similar. Por ejemplo, la sal sódica de polialcohol carboximetildextrano puede disolverse en agua, aplicarse a una columna cargada con resina Bio-Rad AG 50W-X2 (trama 200-400, tipo H+ ), eluirse con agua, y luego el efluente resultante puede ser adicionado con una amina orgánica, tal como trietilamina, y ser liofilizado. Alternativamente, también es posible realizar la conversión en una etapa, por ejemplo disolviendo la sal sódica de un polialcohol carboximetildextrano en agua y pasando la solución a través de resina tipo trietilamónica. El enlace entre el compuesto medicamentoso aislado, y el grupo carboxilo del polialcohol carboximetildextrano, o el enlace entre el espaciador ligado al compuesto medicamentoso y el grupo carboxilo del polialcohol carboximetildextrano, puede estar formado generalmente enlazando un grupo amino reactivo del compuesto medicamentoso, o el grupo amino N-terminal del espaciador, a un grupo carboxilo del polialcohol carboximetildextrano por medio de un enlace ácido-amida. Sin embargo, el enlace entre el compuesto medicamentoso o el espaciador y el grupo carboxilo del polialcohol carboximetildextrano no está limitado al enlace antes descrito, y pueden ser utilizados otros enlaces químicos y enlaces utilizando uno o más espaciadores. Por ejemplo, puede estar formado un anhídrido ácido entre el grupo carboxilo C-terminal del espaciador o un grupo carboxilo del compuesto medicamentoso y un grupo carboxilo del polialcohol carboximetildextrano, o utilizando un compuesto diamina, tal como etilenodiamina, como un espaciador. Cada uno de los grupos carboxilo puede estar ligado por medio de un enlace ácido-amida a cada uno de los grupos amino del compuesto diamina. Cuando el grupo amino reactivo sustituido en el compuesto medicamentoso, o el grupo amino N-terminal del espaciador, está ligado a un grupo carboxilo del polialcohol carboximetildextrano por medio de un enlace ácido-amida, pueden ser utilizados agentes de condensación por deshidratación ordinariamente utilizados para síntesis de cadenas peptídicas, por ejemplo, N,N’-dicicloalquilcarbodiimidas, tal como N,N’-diciclohexilcarbodiimida (DCC), derivados de carbodiimida, tal como 1-etil-3-(3-dimetilaminopropil)carbodiimida (EDAPC), derivados de benzotriazol, tal como 1-hidroxibenzotriazol (HOBT), 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (EEDQ), y similares. Además, la reacción también puede ser realizada mediante el procedimiento de éster activado o el procedimiento de ácido haluro. La reacción puede ser realizada con cualquier disolvente orgánico siempre que esté sustancialmente libre de agua y pueda disolver los reactantes (la sal de amina orgánica de polialcohol carboximetildextrano y el compuesto medicamentoso o el espaciador ligado al compuesto medicamentoso). Por ejemplo, pueden utilizarse preferiblemente N,N-dimetilformamida, dimetil sulfóxido, acetoamida, N-metilpirrolidona, sulfolano y similares. Aunque no está 8 ES 2 229 355 T3 5 10 15 particularmente limitada la cantidad del residuo del compuesto medicamentoso, que se introduce en el polialcohol carboximetildextrano mediante la reacción con el compuesto medicamentoso o el espaciador ligado al compuesto medicamentoso, dicha cantidad debe ser adecuadamente seleccionada desde los puntos de vista de las propiedades físicoquímicas del residuo del compuesto medicamentoso, farmacocinética, eficacia y toxicidad del complejo medicamentoso. Generalmente, puede ser elegido el intervalo desde 0,1 a 30% en peso aproximadamente, y preferiblemente desde 1 a 15% en peso aproximadamente. La proporción del residuo del compuesto medicamentoso introducido en el polialcohol carboximetildextrano puede ser fácilmente determinada, por ejemplo, mediante análisis espectrométrico de absorción. Como una realización preferible del procedimiento de fabricación de la presente invención, el siguiente esquema representa el procedimiento para introducir el oligopéptido ligado al compuesto medicamentoso, que es el agente antineoplásico descrito en la reivindicación 2 de la publicación de la patente japonesa sin examinar (KOKAI) nº (Hei) 6-87746/1994, en un polialcohol carboximetildextrano. Sin embargo, los procedimientos de la presente invención no se limitan a los representados. En el esquema siguiente, la cantidad introducida del residuo del compuesto medicamentoso es, por ejemplo, desde aproximadamente 1 a 15% en peso, preferiblemente desde 4 a 8% en peso aproximadamente. Además, entre las unidades constitutivas de los polialcoholes, solamente una unidad constitutiva, que se introduce con uno o dos grupos carboximetilo, se representa como ejemplo en el esquema siguiente. Sin embargo, debe entenderse que la fracción derivado de polisacárido del complejo medicamentoso no está formada por la repetición de la antes mencionada unidad constitutiva. 20 (Esquema pasa a página siguiente) 25 30 35 40 45 50 55 60 65 9 ES 2 229 355 T3 5 10 15 20 25 30 35 40 45 50 55 60 65 10 ES 2 229 355 T3 5 10 15 20 25 30 35 40 45 50 55 60 Es conocido que el equilibrio del compuesto medicamentoso en el esquema anterior se sitúa en el compuesto cuyo anillo lactona está cerrado (el compuesto de anillo cerrado) en un medio acuoso ácido (por ejemplo, a pH 3 aproximadamente), mientras que el equilibrio se sitúa en el compuesto cuyo anillo lactona está abierto (el compuesto de anillo abierto) en un medio acuoso básico (por ejemplo, a pH 10 aproximadamente), y el complejo medicamentoso introducido con el residuo correspondiente al compuesto de anillo cerrado o de anillo abierto posee similar actividad antineoplásica. Sin embargo, cuando un reactante, cuyo anillo lactona está abierto, está presente en el sistema para la reacción entre el polialcohol carboximetildextrano y el espaciador ligado al antes mencionado compuesto medicamentoso (p.ej. un espaciador oligopeptídico), la reacción de condensación progresará entre un grupo carboxilo derivado del anillo lactona y un grupo amino derivado del espaciador, lo cual resulta en un apreciable descenso del rendimiento de la reacción y, además, algunas veces no puede obtenerse un deseable complejo medicamentoso uniforme. Dicha reacción colateral puede ser evitada utilizando el compuesto de anillo cerrado como reactante en un sistema no acuoso que no permite el equilibrio. Por tanto, el procedimiento de la presente invención es particularmente adecuado para preparar el complejo medicamentoso que contiene el compuesto medicamentoso antes mencionado. El complejo medicamentoso antes mencionado preparado según el procedimiento de la presente invención se caracteriza porque puede presentar una actividad farmacológica específicamente deseada en una localización definida, tal como áreas tumorales o zonas inflamatorias, dependiendo del tipo de residuo del compuesto medicamentoso (p.ej. el residuo del compuesto medicamentoso como agente antineoplásico o anti-inflamatorio), y puede reducir la toxicidad inherente al compuesto medicamentoso aislado. Por ejemplo, el polialcohol carboximetildextrano al ser la fracción derivada de polisacárido posee una excelente retención en sangre y alcanza elevada acumulación en zonas tumorales o inflamatorias como un transportador-suministrador del medicamento y permite al complejo medicamentoso presentar selectividad de localización neoplásica y selectividad de localización inflamatoria. Además, se considera que la proteasa (peptidasa) se expresa en áreas tumorales o zonas inflamatorias y, por tanto, el complejo medicamentoso que presenta un espaciador que comprende un oligopéptido es fácilmente hidrolizado en la fracción espaciador para permitir mostrar su eficacia al compuesto medicamentoso liberado. Un medicamento que contiene el complejo medicamentoso preparado de acuerdo con el procedimiento de la presente invención puede generalmente envasarse en viales o similares en forma de un producto liofilizado u otro, y estar previsto para uso clínico como preparaciones para administración parenteral, tales como inyecciones o infusiones por goteo que se disuelven al tiempo de utilizarse. Sin embargo, la presentación de las preparaciones farmacéuticas del medicamento no está limitada a las formas antes mencionadas. Para la fabricación de las anteriores preparaciones farmacéuticas, pueden ser utilizadas composiciones farmacéuticas preparadas utilizando aditivos farmacéuticos disponibles en el estado de la técnica, por ejemplo, solubilizantes, modificadores de pH, estabilizadores y similares. Aunque la dosis del medicamento anterior no está particularmente limitada, deberá decidirse normalmente en función de la dosis del compuesto medicamentoso que constituye el residuo del compuesto medicamentoso, la cantidad del residuo del compuesto medicamentoso introducida en el complejo medicamentoso, la condición del paciente, el tipo de enfermedad, y similares. Por ejemplo, cuando se administra parenteralmente un complejo medicamentoso que se introduce con aproximadamente 6% en peso del residuo del agente antineoplásico descrito en la reivindicación 2 de la publicación de la patente japonesa sin examinar (KOKAI) nº (Hei) 6-87746/1994, puede administrarse generalmente una vez al día aproximadamente 1 a 500 mg, preferiblemente 10 a 100 mg aproximadamente por cada m2 de superficie corporal por día, y la administración puede repetirse preferiblemente cada 3 a 4 semanas. Ejemplos 65 La presente invención se describirá más específicamente mediante ejemplos. En los ejemplos, “A-NH-” representa un residuo de un compuesto medicamentoso en el cual el compuesto medicamentoso presenta un anillo lactona, tal como el compuesto medicamentoso descrito en la reivindicación 2 de la publicación de la patente japonesa sin examinar (KOKAI) nº (Hei) 6-87746/1994 (en ocasiones designado como “DX-8951” en los ejemplos), y el compuesto medi11 ES 2 229 355 T3 camentoso que posee un anillo lactona cerrado se representa por A-NH2 . Un ejemplo incluye el grupo representado por A-NH- en el esquema antes descrito, en el cual está formado un anillo lactona. Además, A’-NH- representa que el anillo lactona del residuo de un compuesto medicamentoso representado por A-NH- está en forma de anillo cerrado o de anillo abierto o, alternativamente, una mezcla de ambos. 5 10 15 En los ejemplos, a no ser que se mencione específicamente, el grado de carboximetilación en polialcohol carboximetildextrano (el grado de sustitución con grupo carboximetilo por cada residuo sacárido constitutivo) se determinó mediante conversión en forma de ácido libre de la sal sódica del polialcohol carboximetildextrano, disolviendo el ácido resultante en una solución acuosa de hidróxido sódico 0,1 N, y luego valorando (neutralizando) con ácido clorhídrico 0,1 N. Una solución acuosa de la sal sódica del polialcohol carboximetildextrano se aplicó a una columna Bio-Rad AG 50W-X2 (tipo H+ ) y el efluente fue liofilizado y luego utilizado como muestra. La muestra se disolvió en un exceso determinado de solución acuosa de hidróxido sódico 0,1 N y fue neutralizada con ácido clorhídrico 0,1 N utilizando fenolftaleína como un indicador. El grado de carboximetilación se calculó utilizando la siguiente ecuación: Grado de carboximetilación = 13,4(a-b)/[s-5,8(a-b)] en la cual “s” es el peso de la muestra aplicada (mg), “a” es el exceso determinado de solución acuosa de hidróxido sódico 0,1 N (ml), y “b” es el volumen de ácido clorhídrico 0,1 N consumido para la valoración (ml). La cantidad de medicamento introducida (porcentaje en peso) se determinó mediante análisis espectroscópico de absorción utilizando absorciones características del compuesto medicamentoso (aproximadamente 362 nm). La filtración de gel se realizó bajo las siguientes condiciones: columna: TSK Gel G4000 PWXL ; eluyente: NaCl 0,1 M; índice de flujo: 0,8 ml/min; y temperatura de columna: 40ºC. 20 Ejemplo 1 3’-N-(Boc-Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) 25 30 35 40 45 50 55 Se disolvieron Boc-Gly-Gly-Phe-Gly (600 mg) y N-hidroxisuccinimida (160 mg) en N,N-dimetilformamida (20 ml), se enfrió a 4ºC, y luego se adicionó con N,N’-diciclohexilcarbodiimida (280 mg). A esta solución, se añadió una solución de metanosulfonato del compuesto medicamentoso descrito en la reivindicación 2 de la publicación de la patente japonesa sin examinar (KOKAI) nº (Hei) 6-87746/1994 (600 mg del compuesto descrito en el Ejemplo 50 de la publicación de patente antes mencionada) y trietilamina (0,16 ml) disueltos en N,N-dimetilformamida (30 ml), y se dejó reaccionar la mezcla a temperatura ambiente durante 16 horas con agitación bajo condiciones protegidas de la luz. Esta mezcla de reacción fue evaporada hasta sequedad bajo presión reducida, y el residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: diclorometano:metanol = 10:1 solución conteniendo ácido acético 0,5%) para obtener el compuesto del enunciado (1,0 g). H-NMR (DMSO-d6 ) δ: 8,40 (d,1H,J=8,3 Hz), 8,10-8,17 (m,2H), 7,91-8,01 (m,1H), 7,78 (d,1H,J=10,75 Hz), 7,32 (s,1H), 6,94-6,96 (m,1H), 6,50 (s,1H), 5,57 (t,1H,J=4,5 Hz), 5,43 (s,2H), 5,23 (s,2H), 3,77 (dd,2H,J=5,85 Hz,J=8,80 Hz), 3,70 (d,2H,J=4,40 Hz), 3,65 (d,2H,J=5,35 Hz), 3,56 (d,2H,J=5,85), 3,15-3,25 (m,2H), 2,40 (s,3H), 2,05-2,25 (m,1H), 1,86 (m,2H), 1,35 (s,9H), 0,88 (t,3H,J=7,35). 1 Masa (FAB); m/e 854 (M+1). Ejemplo 2 Síntesis de 3’-N-(Boc-Gly-Gly-Gly-Phe)-NH-A (A-NH2 =DX-8951) 60 65 Se disolvieron Boc-Gly-Gly-Gly-Phe (600 mg) y N-hidroxisuccinimida (160 mg) en N,N-dimetilformamida (20 ml) y se enfrió a 4ºC, luego se adicionó con N,N’-diciclohexilcarbodiimida (280 mg). A esta solución, se añadió una solución de metanosulfonato de DX-8951 (600 mg) y trietilamina (0,16 ml) disueltos en N,N-dimetilformamida (30 ml) y se dejó reaccionar la mezcla a temperatura ambiente durante 16 horas con agitación bajo condiciones protegidas de la luz. Esta mezcla de reacción fue evaporada hasta sequedad bajo presión reducida, y el residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: diclorometano:metanol = 10:1 solución conteniendo ácido acético 0,5%) para obtener el compuesto del enunciado (700 mg). 12 ES 2 229 355 T3 1 H-NMR (DMSO-d6 ) δ: 8,57 (d,1H,J=7,8 Hz), 8,19 (d,1H), 8,05-8,07 (m,2H), 7,79 (d,1H,J=11,2 Hz), 7,32 (s,1H), 7,10 (d,2H,J=7,8 Hz), 6,93-7,03 (m,4H), 6,51 (s,1H), 5,52-5,55 (m,1H), 5,44 (s,2H), 5,18 (d,1H,J=18,5 Hz), 4,84 (d,1H,J=18,5 Hz), 4,57-4,59 (m,1H), 3,57-3,71 (m,6H), 3,15-3,25 (m,2H), 3,00-3,02 (m,1H), 2,80-2,90 (m,1H), 2,40 (s,3H), 2,05-2,25 (m,1H), 1,86 (m,2H), 1,35 (s,9H), 0,88 (t,3H,J=7,35 Hz). 5 Masa (FAB); m/e 854 (M+1). Ejemplo 3 10 Síntesis de 3’-N-(Boc-Gly-Gly-Gly-Gly)-NH-A (A-NH2 =DX-8951) 15 Se disolvieron Boc-Gly-Gly-Gly-Gly (120 mg) y N-hidroxisuccinimida (39 mg) en N,N-dimetilformamida (20 ml), se enfrió a 4ºC, y luego se adicionó con N,N’-diciclohexilcarbodiimida (70 mg). A esta solución, se añadió una solución de metanosulfonato de DX-8951 (150 mg) y trietilamina (0,039 ml) disueltos en N,N-dimetilformamida (10 ml), y se dejó reaccionar la mezcla a temperatura ambiente durante 16 horas con agitación bajo condiciones protegidas de la luz. Esta mezcla de reacción fue evaporada hasta sequedad bajo presión reducida, y el residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: solución diclorometano:metanol = 10:1) para obtener el compuesto del enunciado (100 mg). 20 25 H-NMR (DMSO-d6 ) δ: 8,40 (d,1H,J=8,3 Hz), 8,10-8,17 (m,2H), 7,91-8,01 (m,1H), 7,78 (d,1H,J=10,75 Hz), 7,32 (s,1H), 6,94-6,96 (m,1H), 6,50 (s,1H), 5,57 (t,1 H,J=4,5 Hz), 5,43 (s,2H), 5,23 (s,2H), 3,77 (dd,2H,J=5,85 Hz,J=8,80 Hz), 3,70 (d,2H,J=4,40 Hz), 3,65 (d,2H,J=5,35 Hz), 3,56 (d,2H,J=5,85 Hz), 3,15-3,25 (m,2H), 2,40 (s,3H), 2,05-2,25 (m,1H), 1,86 (m,2H),1,35(s,9H), 0,88 (t,3H,J=7,35 Hz). 1 Masa (FAB); m/e 764 (M+1). Ejemplo 4 Síntesis de trifluoroacetato de 3’-N-(Gly-Gly-Gly-Gly)-NH-A(A-NH2 =DX-8951) 30 35 40 45 50 55 Se disolvió 3’-N-(Boc-Gly-Gly-Gly-Gly)-NH-A (A-NH2 =DX-8951) (79 mg) en ácido trifluoroacético (3 ml) y se dejó reposar durante una hora. Se evaporó el disolvente, el residuo fue sometido a destilaciones azeotrópicas dos veces con metanol (30 ml) y dos veces con etanol (30 ml), y entonces el residuo fue lavado con éter para obtener el compuesto del enunciado (80 mg). 1 H-NMR (DMSO-d6 ) δ: 8,59-8,61 (m,1H), 8,50 (d,1H,J=8,3 Hz), 8,21-8,27 (m,2H), 7,91-8,01 (br,3H), 7,81 (d,1H,J=11,2 Hz), 7,32 (s,1H), 6,50-6,52 (br,1H), 5,57-5,59 (m,1H), 5,43 (s,2H), 5,23 (s,2H), 3,80-3,82 (m,3H), 3,703,75 (m,3H), 3,15-3,25 (m,2H), 2,41 (s,3H), 2,05-2,25 (m,1H), 1,86-1,88 (m,2H), 0,88 (t,3H,J=7,35 Hz). Ejemplo 5 Síntesis de sal trietilamónica de polialcohol carboximetildextrano 60 65 Se disolvió Dextran T2000 (10 g, Pharmacia, peso molecular medio: 2.000.000) en tampón acetato 0,1 M (pH 5,5, 1.000 ml) y se añadió con una solución acuosa (1000 ml) de periodato sódico (33,0 g). Después de agitar a 4ºC durante 10 días con protección de luz, la mezcla se añadió con glicol etileno (7,0 ml) y fue agitada durante la noche. La mezcla de reacción se ajustó a pH 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. Se añadió y disolvió borohidruro sódico (14 g), y la mezcla fue entonces agitada a temperatura ambiente durante la noche. La mezcla de reacción fue enfriada con hielo, ajustada a pH 5,5 con ácido acético, y agitada a 4ºC durante una hora, y entonces ajustada a pH 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. La solución acuosa resultante se sometió a ultrafiltración utilizando una membrana Biomax-30 (Millipore) para eliminar la fracción de bajo peso molecular. La fracción polimérica fue liofilizada para obtener polialcohol dextrano. Después de tratar este polialcohol dextrano a pH 3,0 durante una hora, la fracción de bajo peso molecular fue eliminada con una membrana 13 ES 2 229 355 T3 Biomax-50, y seguidamente, la fracción polimérica fue eliminada con una membrana Biomax-100, y el resultado fue liofilizado para obtener polialcohol dextrano purificado (2,0 g). El peso molecular de esta sustancia fue 220K (filtración de gel, estándar dextrano). 5 10 15 20 Este polialcohol dextrano purificado (1,8 g) se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (10,5 g) en agua (45 ml) y se disolvió a temperatura ambiente. A esta solución, se añadió ácido monocloroacético (15 g), se disolvió bajo enfriamiento con hielo, y luego se dejó reaccionar la mezcla a temperatura ambiente durante 20 horas. Después de ajustar esta mezcla de reacción a pH 8 con ácido acético, la fracción de bajo peso molecular fue eliminada mediante ultrafiltración utilizando una membrana Biomax-10. La fracción polimérica fue liofilizada para obtener la sal sódica de polialcohol carboximetildextrano (1,8 g). El peso molecular de esta sustancia fue 330K (filtración de gel, estándar dextrano) y el grado de carboximetilación fue 0,8. Se disolvió en agua la anterior la sal sódica de polialcohol carboximetildextrano (300 mg), se aplicó a una columna Bio-Rad AG 50W-X2 (trama 200-400, tipo H+ ) (1,5x8,6 cm) y fue eluida con agua. Este efluente se añadió con trietilamina (0,5 ml) y fue liofilizado para obtener sal trietilamónica de polialcohol carboximetildextrano (380 mg). Fueron tratadas porciones de la sal sódica de polialcohol carboximetildextrano (300 mg cada una) con la columna, según se describió anteriormente, para obtener sal trietilamónica de polialcohol carboximetildextrano (380 mg, 400 mg). Ejemplo 6 Síntesis de sal sódica de polialcohol carboximetildextrano 25 30 35 La sal sódica de polialcohol carboximetildextrano (0,15 g) obtenida en el Ejemplo 5 anterior se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (1,05 g) en agua (4,5 ml) y luego se disolvió a temperatura ambiente. A esta solución, se añadió y disolvió ácido monocloroacético (1,5 g) bajo enfriamiento con hielo, y se dejó reaccionar la mezcla a temperatura ambiente durante 18 horas. Esta mezcla de reacción se ajustó a pH 8 con ácido acético, se añadió por goteo en 90 ml de metanol, y se añadió con cloruro sódico acuoso 3 M (0,15 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados fueron lavados con metanol y luego disueltos en agua (5 ml), y se añadieron con cloruro sódico acuoso 3 M (0,15 ml). Esta solución acuosa se filtró a través de un filtro Millipore (0,45 µm), y el filtrado se añadió por goteo a 35 ml de etanol y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados fueron lavados con etanol, disueltos en agua y dializados con agua purificada utilizando una membrana de diálisis (Spectrapore 1, peso molecular de corte; 6.000-8.000). La solución dializada interna se filtró a través de un filtro Millipore (0,22 µm) y fue liofilizada para obtener sal sódica de polialcohol carboximetildextrano (0,18 g). El grado de carboximetilación de esta sustancia fue 1,2 por cada residuo sacárido (alcalimetría). Ejemplo 7 40 45 50 Síntesis de sal sódica de polialcohol carboximetildextrano El polialcohol dextrano purificado (0,2 g) obtenido en el Ejemplo 5 se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (0,84 g) en agua (6 ml) y se disolvió a temperatura ambiente. A esta solución, se añadió y disolvió ácido monocloroacético (1,2 g) bajo enfriamiento con hielo, y se dejó reaccionar la mezcla a temperatura ambiente durante 18 horas. La mezcla de reacción se ajustó a pH 8 con ácido acético, se añadió por goteo a 120 ml de metanol, y luego se adicionó con cloruro sódico acuoso 3 M (0,2 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados fueron lavados con metanol y luego disueltos en agua (5 ml), y se añadieron con cloruro sódico acuoso 3 M (0,2 ml). Esta solución acuosa se filtró a través de un filtro Millipore (0,45 µm), el filtrado se añadió por goteo a 35 ml de etanol y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados fueron lavados con etanol, disueltos en agua y dializados con agua purificada utilizando una membrana de diálisis (Spectrapore 1, peso molecular de corte; 6.000-8.000). La solución dializada interna se filtró a través de un filtro Millipore (0,22 µm) y fue liofilizada para obtener sal sódica de polialcohol carboximetildextrano (0,20 g). El grado de carboximetilación de esta sustancia fue 0,4 por cada residuo sacárido (alcalimetría). 55 Ejemplo 8 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Gly-Phe-NH-A’ (A-NH2 =DX-8951) 60 65 La sal trietilamónica de polialcohol carboximetildextrano obtenida en el Ejemplo 5 (380 mg, grado de carboximetilación: 0,8) se disolvió en N,N-dimetilformamida (30 ml). A esta solución, se añadieron sucesivamente una solución de sal de ácido trifluoroacético 3’-N-(Gly-Gly-Gly-Phe)-NH-A (A-NH2 =DX-8951) (49 mg) en N,N-dimetilformamida (5 ml), trietilamina (0,017 ml) y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (380 mg), y entonces se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Esta mezcla de reacción se ajustó a pH 10 con hidróxido sódico acuoso 1 M y cada una de las porciones de 5 ml de la mezcla se añadió por goteo a 25 ml de etanol. Esta mezcla se añadió con cloruro sódico acuoso 3 M (1 ml) y éter dietilo (5 ml) y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). 14 ES 2 229 355 T3 5 10 Los precipitados fueron disueltos en agua y dializados con agua purificada utilizando una membrana de diálisis (Spectrapore 1, peso molecular de corte; 6.000-8.000), y la solución dializada interna se filtró a través de un filtro Millipore (0,22 µm) y fue liofilizada. El producto crudo resultante se disolvió en agua (30 ml), se ajustó a pH 9 con hidróxido sódico acuoso 0,1 M, y tratado a 37ºC durante una hora. Esta solución tratada se dializó como se ha descrito anteriormente, y entonces la solución dializada interna se filtró a través de un filtro Millipore (0,22 µm) y fue liofilizada para obtener el compuesto del enunciado (289 mg). El resultado obtenido mediante análisis GPC (cromatografía de permeación de gel) después de disolver este compuesto en cloruro sódico acuoso 0,1 M (columna: TSK Gel PW4000XL, Tosoh, disolvente: NaCl 0,1 M, índice de flujo: 0,8 ml/min), y el espectro de absorción ultravioleta del compuesto (solución tampón Tris 0,1 M, pH 9,0, 0,25 mg/ml) se representan en las Figuras 1 y 2, respectivamente. El contenido del residuo del compuesto medicamentoso en el compuesto fue 5,3% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 9 15 20 25 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Phe-Gly-NH-A’ (A-NH2 =DX-8951) El compuesto del enunciado (300 mg) fue sintetizado de acuerdo con una forma similar a la del Ejemplo 8 mediante introducción de sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Phe-Gly)-NH-A, la cual se había obtenido mediante eliminación del grupo Boc del 3’-N-(Boc-Gly-Gly-Phe-Gly)-NH-A (A-NH2 =-DX-8951) (50 mg) de una manera similar a la del Ejemplo 4, en la sal trietilamónica de polialcohol carboximetildextrano (380 mg) obtenida en el Ejemplo 5. El resultado obtenido mediante análisis GPC después de disolver este compuesto en cloruro sódico acuoso 0,1 M (columna: TSK Gel PW-4000XL, Tosoh, disolvente: NaCl 0,1 M, índice de flujo: 0,8 ml/min), y el espectro de absorción ultravioleta del compuesto (solución tampón Tris 0,1 M, pH 9,0, 0,19 mg/ml) se representan en las Figuras 3 y 4, respectivamente. El contenido del residuo del compuesto medicamentoso en el compuesto fue 5,3% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 10 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Gly-Gly-NH-A’ (A-NH2 =DX-8951) 30 35 El compuesto del enunciado (190 mg) fue sintetizado según una forma similar a la del Ejemplo 8 mediante introducción de sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Gly-Gly)-NH-A, la cual se había obtenido a través de eliminación del grupo Boc del 3’-N-(Boc-Gly-Gly-Gly-Gly)-NH-A (A-NH2 =DX-8951) (41 mg) de una manera similar a la del Ejemplo 4, en la sal trietilamónica de polialcohol carboximetildextrano (380 mg) obtenida en el Ejemplo 5. El espectro de absorción ultravioleta de este compuesto (solución tampón Tris 0,1 M, pH 9,0, 0,34 mg/ml) se representa en la Figura 5. El contenido del residuo del compuesto medicamentoso en el compuesto fue 5,3% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 11 40 Actividad antitumoral del complejo medicamentoso de la presente invención 45 50 55 Se prepararon ratones portadores de tumor Meth A (7 ratones por grupo) mediante trasplante subcutáneo de 1x106 células de fibrosarcoma Meth A de ratón en las regiones inguinales derechas de ratones macho BALB/c (de 7 semanas). El 7º día, el complejo medicamentoso del Ejemplo 9 disuelto en agua destilada para inyecciones, se inyectó en la vena de cola de los ratones portadores de tumor Meth A 4 veces cada 4 días. El 21º día después del trasplante, se extirparon y pesaron masas tumorales para calcular la tasa de inhibición de crecimiento tumoral, de acuerdo con la siguiente ecuación: tasa de inhibición de crecimiento tumoral (%) = [1-(peso tumoral medio del grupo administrado con la muestra de prueba/peso tumoral medio del grupo de control)] x 100. Como resultado, se halló que el complejo medicamentoso de la presente invención obtenido en el Ejemplo 9 presentaba una actividad antitumoral considerablemente mejorada en comparación con el compuesto medicamentoso aislado, sin el espaciador y el derivado polisacárido, mientras que no mostraba toxicidad (pérdida de peso). El derivado polisacárido (Ejemplo 5) aislado, y el compuesto medicamentoso introducido con el espaciador solamente (sal de ácido trifluoroacético de H2 N-Gly-Gly-Phe-Gly-NH-A (A-NH2 =DX8951) obtenida mediante eliminación del grupo Boc del compuesto del Ejemplo 1 según el procedimiento del Ejemplo 4) no mostraron efectividad. TABLA 2 60 65 15 ES 2 229 355 T3 Ejemplo 12 Actividad antitumoral del complejo medicamentoso de la presente invención 5 10 Se prepararon ratones portadores de tumor Meth A (6 ratones por grupo) de acuerdo con una forma similar a la del Ejemplo 11, y la actividad antitumoral fue comparada con la obtenida por administración simple, una vez el 7º día, de los complejos medicamentosos de los Ejemplos 8 y 9. Como resultado, el grado de actividad antitumoral fue el siguiente: (Derivado polisacárido)-Gly-Gly-Phe-Gly-NH- A’ > (Derivado polisacárido)-Gly-Gly-Gly-Phe-NH-A’ > el compuesto medicamentoso aislado. No mostró efectividad el compuesto que comprende el residuo del compuesto medicamentoso directamente enlazado a un grupo carboxilo del polialcohol carboximetildextrano del Ejemplo 5 sin espaciador (cantidad de residuo del compuesto medicamentoso introducida: 6,2% en peso). TABLA 3 15 20 25 Ejemplo 13 Síntesis de sal trietilamónica de polialcohol carboximetildextrano 30 35 40 45 Se disolvió en tampón acetato 0,1 M (pH 5,5, 1000 ml) Dextran T500 (10 g, Pharmacia, peso molecular: 500K) y se añadió con una solución acuosa (1000 ml) de periodato sódico (33 g). Después de agitar a 4ºC durante diez días con protección de luz, la mezcla se añadió con glicol etileno (7,0 ml) y fue agitada durante la noche. La mezcla de reacción se ajustó a pH 7,5 con hidróxido sódico acuoso 8 M. Se añadió y disolvió borohidruro sódico (14 g), y luego la mezcla fue agitada durante la noche. La mezcla de reacción fue enfriada con hielo, ajustada a pH 5,5 con ácido acético y agitada a 4ºC durante una hora, y luego ajustada a pH 7,5 con hidróxido sódico acuoso 8 M para obtener la Solución 1. Separadamente, se realizaron una serie de procesos descritos anteriormente utilizando Dextran T500 (10 g, Pharmacia, peso molecular 500K) para obtener la Solución 2. Adicionalmente, se repitió una serie de procesos descritos anteriormente utilizando Dextran T250 (10 g cada vez, Pharmacia, peso molecular 250K) para obtener las Soluciones 3 y 4. Estas Soluciones 1 a 4 fueron combinadas y sometidas a ultrafiltración utilizando una membrana Biomax-50 para eliminar la fracción de bajo peso molecular. La fracción polimérica fue liofilizada para obtener polialcohol dextrano (25 g). El peso molecular de esta sustancia fue 163K (filtración de gel, estándar pululano). Este polialcohol dextrano (11 g) se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (46,2 g) en agua (330 ml) y se disolvió a temperatura ambiente. A esta solución, se añadió bajo enfriamiento con hielo ácido monocloroacético (66 g), se disolvió y se dejó reaccionar la mezcla a temperatura ambiente durante la noche. Esta mezcla de reacción se ajustó a pH 9 con ácido acético y fue desalada mediante ultrafiltración utilizando una membrana Biomax-30. La solución restante que no había pasado a través de la membrana fue liofilizada para obtener la sal sódica de polialcohol carboximetildextrano (13 g). El peso molecular de esta sustancia fue 228K (filtración de gel, estándar pululano) y el grado de carboximetilación fue 0,4. 50 Esta sal sódica de polialcohol carboximetildextrano (600 mg) se disolvió en agua, se aplicó a una columna BioRad AG 50W-X2 (trama 200-400, tipo H+ ) (diámetro: 44 mm, longitud: 210 mm) y fue eluida con agua. Este efluente se añadió con trietilamina (0,93 ml) y luego liofilizado para obtener el compuesto del enunciado (690 mg). 55 Ejemplo 14 Síntesis de sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) 60 65 El 3’-N-(Boc-Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) (79 mg) obtenido en el Ejemplo 1 se disolvió en ácido trifluoroacético (3 ml) y se dejó reposar durante una hora. Se evaporó el disolvente, y el residuo fue sometido a destilación azeotrópica dos veces con metanol (30 ml) y dos veces con etanol (30 ml), y luego lavado con éter para obtener el compuesto del enunciado (80 mg). 1 H-NMR (DMSO-d6 ) δ: 8,53 (d, 1H, J=8,3 Hz), 8,40-8,48 (m, 2H), 8,28 (d, 1H, J=8,3 Hz), 7,95-8,07 (br, 3H), 7,81 (d, 1H, J=10,2 Hz), 7,30-7,37 (m, 2H), 7,15-7,30 (m, 5H), 6,50-6,55 (br, 1H), 5,50-5,57 (m, 1H), 5,41 (d, 2H, J=7,82 Hz), 5,25 (s, 2H), 4,55-4,62 (m, 1H), 3,55-3,92 (m, 6H), 3,15-3,25 (br, 2H), 2,98-3,03 (m, 1H), 2,73-2,82 (m, 1H), 2,40 (s, 3H), 2,05-2,25 (m, 1H), 1,84-1,92 (m, 2H), 0,88 (t, 3H, J=7,35 Hz). 16 ES 2 229 355 T3 Ejemplo 15 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Phe-Gly-NH-A’ (A-NH2 =DX-8951) 5 10 15 20 La sal sódica de polialcohol carboximetildextrano obtenida en el Ejemplo 13 (400 mg) se convirtió en la sal trietilamónica (470 mg) y se disolvió en N,N-dimetilformamida (30 ml). A esta solución, se añadieron sucesivamente una solución de sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) obtenida en el Ejemplo 14 (62 mg) en N,N-dimetilformamida (5 ml), trietilamina (0,02 ml), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (470 mg), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación y protección de luz. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. Se añadieron a la mezcla cloruro sódico acuoso 3 M (2,5 ml) y éter dietilo (20 ml), y se recogieron los precipitados depositados mediante centrifugación. Los precipitados se disolvieron en cloruro sódico acuoso 0,5 M, se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M bajo enfriamiento con hielo y luego fueron dializados con agua purificada utilizando una membrana de diálisis (Spectrapore 1, peso molecular de corte; 6.000-8.000). La solución dializada interna se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (600 mg). El resultado obtenido mediante análisis GPC después de disolver este compuesto en cloruro sódico acuoso 0,1 M (columna: TSK Gel PW-4000XL, Tosoh; disolvente: NaCl 0,1 M; índice de flujo: 0,8 ml/min) y el espectro de absorción ultravioleta del compuesto (solución tampón Tris 0,1 M, pH 9,0, 0,1 mg/ml) se representan en las Figuras 6 y 7, respectivamente. El contenido del residuo del compuesto medicamentoso en el compuesto fue 5,8% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 16 Síntesis de sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Gly-Phe)-NH-A (A-NH2 =DX-8951) 25 El 3’-N-(Boc-Gly-Gly-Gly-Phe)-NH-A (A-NH2 =DX-8951) (79 mg) obtenido en el Ejemplo 2 se disolvió en ácido trifluoroacético (3 ml) y se dejó reposar durante una hora. Se evaporó el disolvente, y el residuo fue sometido a destilación azeotrópica dos veces con metanol (30 ml) y dos veces con etanol (30 ml), y entonces el residuo fue lavado con éter para obtener el compuesto del enunciado (80 mg). 30 H-NMR (DMSO-d6 ) δ: 8,62-8,66 (m, 2H), 8,23 (d, 1H, J=8,3 Hz), 8,18-8,20 (m, 1H), 7,98-8,10 (br, 2H), 7,79 (d, 1H, J=10,7 Hz), 7,32 (s, 1H), 7,09 (d, 2H, J=7,3 Hz), 6,93-7,03 (m, 4H), 6,50-6,60 (br,1H), 5,52-5,55 (m, 1H), 5,44 (s, 2H), 5,18 (d, 1H, J=18,5 Hz), 4,80 (d, 1H, J=18,5 Hz), 4,57-4,59 (m, 1H), 3,57-3,71 (m, 6H), 3,15-3,25 (m, 2H), 3,00-3,02 (m, 1H), 2,80-2,90 (m, 1H), 2,50 (s, 3H), 2,05-2,25 (m, 1H), 1,86-2,00 (m, 2H), 0,88 (t, 3H), J=7,35 Hz). 1 35 Ejemplo 17 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Gly-Phe-NH-A’ (A-NH2 =DX-8951) 40 45 50 55 La sal sódica de polialcohol carboximetildextrano obtenida en el Ejemplo 13 (1,0 g) se convirtió en la sal trietilamónica (1,2 g) y se disolvió en N,N-dimetilformamida (90 ml). A esta solución, se añadieron sucesivamente una solución de sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Gly-Phe)-NH-A (A-NH2 =DX-8951) obtenida en el Ejemplo 16 (158 mg) en N,N-dimetilformamida (15 ml), trietilamina (0,05 ml), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (1,2 g), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación y protección de luz. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. La mezcla se añadió con cloruro sódico acuoso 3 M (2,5 ml) y éter dietilo (20 ml), y luego los precipitados depositados se recogieron mediante centrifugación. Los precipitados se disolvieron en cloruro sódico acuoso 0,5 M, se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M bajo enfriamiento con hielo y fueron dializados con agua purificada utilizando una membrana de diálisis (Spectrapore 1, peso molecular de corte; 6.000-8.000). La solución dializada interna se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (1,4 g). El contenido del residuo del compuesto medicamentoso en este compuesto fue 5,2% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 18 Síntesis de Boc-Gly-Phe-Leu-OH 60 Se añadió H-Gly-Phe-Leu-OH (3,0 g) a dioxano acuoso 50% (48 ml) y se enfrió con hielo. A esta solución, se añadieron hidróxido sódico acuoso 1 N (9,45 ml) y una solución de dioxano (24 ml) conteniendo (Boc)2 O (2,27 g), y la mezcla fue agitada durante la noche. Se añadió ácido clorhídrico 1 N (9,45 ml) a la mezcla de reacción y se evaporó el disolvente. El residuo resultante se purificó mediante cromatografía en columna de gel de sílice (eluyente: solución diclorometano:metanol = 5:1) para obtener el compuesto del enunciado (2,5 g). 65 17 ES 2 229 355 T3 Ejemplo 19 Síntesis de Boc-Gly-Phe-Leu-Gly-OBzl 5 10 El Boc-Gly-Phe-Leu-OH obtenido en el Ejemplo 18 (2,4 g) y N-hidroxisuccinimida (656 mg) se disolvió en N,Ndimetilformamida (50 ml), se enfrió a 4ºC, y luego se adicionó con N,N’-diciclohexilcarbodiimida (1,17 g), y se agitó durante 2 horas. A esta solución, se añadió una solución de N,N-dimetilformamida (40 ml) en la cual se habían disuelto tosilato de H-Gly-OBzl (1,9 g) y trietilamina (0,79 ml), y se dejó reaccionar la mezcla con agitación a temperatura ambiente durante 16 horas. Esta mezcla de reacción fue evaporada hasta sequedad bajo presión reducida, y el residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: solución diclorometano:metanol = 50:1) para obtener el compuesto del enunciado (2,0 g). H-NMR (DMSO-d6 ) δ: 8,20-8,30 (m, 1H), 8,12 (d, 1H, J=8,3 Hz), 7,83 (d, 1H, J=8,3 Hz), 7,32-7,37 (m, 5H), 6,89-6,95 (m, 1H), 5,12 (s, 1H), 4,52-4,59 (br, 1H), 4,34 (dd, H, J=7,3 Hz, J=15,1 Hz), 3,93 (dd, 1H, J=5,5 Hz, J=17,2 Hz), 3,84 (dd, 1H, J=5,5 Hz, J=17,2 Hz), 3,54 (dd, 1H, J=5,9 Hz, J=16,7 Hz), 3,42 (dd, J=5,9 Hz, J=16,7 Hz), 3,00 (dd, 1H, J=4,4 Hz, 13,7 Hz), 2,78 (dd, 1H, J=8,8 Hz, J=13,2 Hz), 1,50-1,65 (m, 1 H), 1,45 (t, 2H, J=7,3 Hz), 1,36 (s, 9H), 0,86 (d, 3H, J=6,4 Hz), 0,82 (d, 3H, J=6,4 Hz). 1 15 Ejemplo 20 20 Síntesis de Boc-Gly-Phe-Leu-Gly-OH 25 El Boc-Gly-Phe-Leu-OBzl (1,7 g) obtenido en el Ejemplo 19 se disolvió en una solución mixta de acetato de etilo (30 ml) y metano) (30 ml), y se añadió con 5% Pd-C (1,5 g) para realizar reducción catalítica. La mezcla de reacción se filtró y el filtrado fue evaporado hasta sequedad bajo presión reducida para obtener el compuesto del enunciado (1,15 g). Ejemplo 21 30 35 40 45 Síntesis de 3’-N-(Boc-Gly-Phe-Leu-Gly)-NH-A (A-NH2 =DX-8951) El Boc-Gly-Phe-Leu-Gly-OH obtenido en el Ejemplo 20 (200 mg) y N-hidroxisuccinimida (58 mg) se disolvieron en N,N-dimetilformamida (5 ml). Después de enfriar a 4ºC, se añadió N,N’-diciclohexilcarbodiimida (104 mg) a la solución y se disolvió. A esta solución, se añadió una solución de N,N-dimetilformamida (5 ml) en la cual se habían disuelto metanosulfonato de DX-8951 (224 mg) y trietilamina (0,059 ml), y se dejó reaccionar la mezcla con agitación a temperatura ambiente durante 16 horas bajo condiciones protegidas de la luz. Esta mezcla de reacción fue evaporada hasta sequedad bajo presión reducida, y el residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: diclorometano:metanol = 10:1 solución conteniendo ácido acético 0,5%) para obtener el compuesto del enunciado (200 mg). 1 H-NMR (DMSO-d−6) δ: 8,35 (d, 1H, J=7,8 Hz), 8,08-8,18 (m, 2H), 7,75-7,85 (m, 2H), 7,32 (s, 1H), 7,10 (d, 2H, J=6,8 Hz), 7,08-7,13 (m, 3H), 6,85-6,95 (br, 1H), 6,40-6,65 (br, 1H), 5,52-5,55 (m, 1H), 5,46 (d, 1H, J=18,5 Hz), 5,37 (d, 1H, J=18,5 Hz), 5,24 (s,2H), 4,44-4,52 (m, 1H), 4,15-4,25 (m, 1H), 3,68-3,72 (m, 2H), 3,40-3,52 (m, 2H), 3,15-3,25 (br, 2H), 2,85-2,95 (m, 1H), 2,65-2,75 (m, 1H), 2,40 (s, 3H), 2,05-2,25 (m, 1H), 1,80-1,91 (m, 2H), 1,501,65 (m, 1H), 1,45 (t, 2H, J=7,3 Hz), 1,35 (s, 9H), 0,88 (t, 3H, J=7,4),0,86 (d, 3H, J=6,4 Hz), 0,82 (d, 3H, J=6,4 Hz). Ejemplo 22 50 Síntesis de sal de ácido trifluoroacético de 3’-N-(Gly-Phe-Leu-Gly)-NH-A (A-NH2 =DX-8951) 55 Se disolvió el 3’-N-(Boc-Gly-Phe-Leu-Gly)-NH-A (A-NH2 =DX-8951) (97 mg) obtenido en el Ejemplo 21 en ácido trifluoroacético (3 ml) y se dejó reposar durante una hora. Se evaporó el disolvente, y el residuo fue sometido a destilación azeotrópica dos veces con metano) (30 ml) y dos veces con etanol (30 ml), y luego lavado con éter para obtener el compuesto del enunciado (95 mg). H-NMR (DMSO-d6 ) δ: 8,57 (d, 1H, J=8,3 Hz), 8,47 (d, 1H, J=8,3 Hz), 8,32 (d, 1H, J=7,8 Hz), 8,17 (t, 1H, J=5,5 Hz), 7,81-7,91 (br, 3H), 7,79 (d, 1H, J=10,7 Hz), 7,32 (s, 1H), 7,21-7,23 (m, 5H), 7,12-7,17 (m, 1H), 6,45-6,55 (br, 1H), 5,57 (q, 1H, J=4,4 Hz), 5,43 (d, 1H, J=16,1 Hz), 5,34 (d, 1H, J=16,1 Hz), 5,23 (s, 2H), 4,67 (dt, 1H, J=4,0 Hz, J=9,0 Hz), 4,31 (dd, 1H, J=8,5 Hz, J=15,0 Hz), 4,0-4,4 (br, 1H), 3,74-3,76 (m, 2H), 3,56 (dd, 1H, J=6,0 Hz, J=16,0 Hz), 3,41 (dd, 1H, J=6,0 Hz, J=16,0 Hz), 3,17-3,19 (br, 2H), 3,02 (dd, 1H, J=4,0 Hz, J=14,0 Hz), 2,70 (dd, 1H, J=10,0 Hz, J=14,0 Hz), 2,40 (s, 3H), 2,05-2,15 (m, 1H), 1,85 (dt, 2H, J=7,0 Hz, J=14,0 Hz), 1,50-1,55 (m, 1H), 1,45 (t, 2H, J=6,0 Hz), 1,35 (s, 9H), 0,88 (t, 3H, J=7,4), 0,85 (d, 3H, J=6,4 Hz), 0,80 (d, 3H, J=6,4 Hz). 1 60 65 18 ES 2 229 355 T3 Ejemplo 23 Síntesis de polialcohol carboximetildextrano-Gly-Phe-Leu-Gly-NH-A’ (A-NH2 =DX-8951) 5 10 15 La sal trietilamónica de polialcohol carboximetildextrano obtenida en el Ejemplo 13 (690 mg) se disolvió en N,Ndimetilformamida (50 ml). A esta solución, se añadieron sucesivamente una solución de la sal de ácido trifluoroacético de 3’-N-(Gly-Phe-Leu-Gly)-NH-A (A-NH2 =DX-8951) (95 mg) obtenida en el Ejemplo 22 en N,N-dimetilformamida (10 ml), trietilamina (0,03 ml), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (690 mg), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. La mezcla se añadió con cloruro sódico acuoso 3 M (2,5 ml) y éter dietilo (20 ml), y los precipitados depositados se recogieron mediante centrifugación. Los precipitados se disolvieron en cloruro sódico acuoso 0,5 M, se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M, y fueron dializados con agua purificada utilizando una membrana de diálisis (Spectrapore 1, peso molecular de corte; 6.000-8.000). La solución dializada interna se filtró a través de un filtro Millipore (0,22 µm), y entonces el filtrado fue liofilizado para obtener el compuesto del enunciado (600 mg). El contenido del residuo del compuesto medicamentoso en este compuesto fue 4,8% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 24 20 Síntesis de sal trietilamónica de polialcohol carboximetildextrano 25 30 35 Se disolvió en tampón acetato 0,1 M (pH 5,5, 5000 ml) Dextran T500 (50 g, Pharmacia, peso molecular: 500K), y se añadió una solución acuosa (5000 ml) de periodato sódico (165,0 g). Después de agitar a 4ºC durante diez días con protección de luz, la mezcla se añadió con glicol etileno (35,0 ml) y fue agitada durante la noche. La mezcla de reacción se ajustó a pH 7,5 con hidróxido sódico acuoso 8 M. Se añadió y disolvió borohidruro sódico (70 g), y luego la mezcla fue agitada durante la noche. La mezcla de reacción fue enfriada con hielo, ajustada a pH 5,5 con ácido acético y agitada a 4ºC durante una hora, y entonces ajustada a pH 7,5 con hidróxido sódico acuoso 8 M. La solución resultante se sometió a ultrafiltración utilizando una membrana Biomax-50 para eliminar la fracción de bajo peso molecular. La fracción polimérica fue liofilizada para obtener polialcohol dextrano (27,1 g). El peso molecular de esta sustancia fue 140K (filtración de gel, estándar pululano). Este polialcohol dextrano (5 g) se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (21 g) en agua (150 ml), y se disolvió a temperatura ambiente. A esta solución, se añadió bajo enfriamiento con hielo y se disolvió ácido monocloroacético (30 g), y luego se dejó reaccionar la mezcla a temperatura ambiente durante la noche. Esta mezcla de reacción se ajustó a pH 8 con ácido acético y entonces fue desalada mediante ultrafiltración utilizando una membrana Biomax-50. La solución restante que no había pasado a través de la membrana fue liofilizada para obtener la sal sódica de polialcohol carboximetildextrano (5,6 g). El peso molecular de esta sustancia fue 263K (filtración de gel, estándar pululano) y el grado de carboximetilación fue 0,4. 40 Esta sal sódica de polialcohol carboximetildextrano (2,0 g) se disolvió en agua, se aplicó a una columna Bio-Rad AG 50W-X2 (trama 200-400, tipo H+ ) (diámetro: 44 mm, longitud: 210 mm) y fue eluida con agua. Este efluente se añadió con trietilamina (4 ml) y fue liofilizado para obtener el compuesto del enunciado (2,2 g). 45 Ejemplo 25 Síntesis de sal trimetilamónica de polialcohol carboximetildextrano 50 La sal sódica de polialcohol carboximetildextrano (1,0 g) obtenida en el Ejemplo 24 se disolvió en agua, se aplicó a una columna Bio-Rad AG 50W-X2 (trama 200-400, tipo Me3 N H+ ) y fue eluida con agua. Este efluente fue liofilizado para obtener el compuesto del enunciado (950 mg). Ejemplo 26 55 Síntesis de hidrocloruro de 3’-N-(Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) 60 De una manera similar a la del Ejemplo 14, sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Phe-Gly)-NH-A obtenida desde 3’-N-(Boc-Gly-Gly-Phe-Gly-(NH-A (A-NH2 =DX-8951) (400 mg) se disolvió en agua/MeOH (1:4), se aplicó a una columna Bio-Rad AG 1-X8 (trama 200-400, tipo Cl− ) (1,5 cm x 8,6 cm) y fue eluida con el anterior disolvente. Este efluente fue concentrado y luego liofilizado para obtener el compuesto del enunciado (310 mg). H-NMR (DMSO-d6 ) δ: 8,53 (d, 1H, J=8,5 Hz), 8,46-8,48 (m, 1H), 8,37-8,39 (m, 1H), 7,95 (d, 1H, J=8,0 Hz), 7,80 (s, 3H), 7,78 (d, 1H, J=11,1 Hz), 7,34 (s, 1H), 7,14-7,24 (m, 5H), 6,50(s, 1H), 5,56-5,60 (m, 1H), 5,35-5,40 (m, 2H), 5,24 (s, 2H), 4,51-4,56 (m, 1H), 3,86 (dd, J=4,8, 13,5 Hz, 1H), 3,68-3,79 (m, 3H), 3,54 (s, 2H), 3,15-3,22 (m, 2H), 3,01 (dd, J=5,6, 13,5 Hz, 1H), 2,78 (dd, J=9,6, 3,5 Hz, 1H), 2,41 (s, 3H), 2,12-2,23 (m, 2H), 1,81-1,89 (m, 2H), 0,88 (t, 3H, J=7,2 Hz). 1 65 Masa (FAB); m/e 753 (M+1). 19 ES 2 229 355 T3 Ejemplo 27 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Phe-Gly-NH-A’ (A-NH2 =DX-8951) 5 10 15 La sal trimetilamónica de polialcohol carboximetildextrano obtenida en el Ejemplo 25 (0,1 g) se disolvió en N,Ndimetilformamida (6 ml). A esta solución, se añadieron sucesivamente una solución del hidrocloruro de 3’-N-(GlyGly-Phe-Gly)-NH-A (A-NH2 =DX-8951) (24 mg) obtenido en el Ejemplo 26 en N,N-dimetilformamida (10 ml), trietilamina (5 µl), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (0,1 g), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. La mezcla se añadió con cloruro sódico acuoso 3 M (2,5 ml) y éter dietilo (20 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados se disolvieron en cloruro sódico acuoso 0,5 M y se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M bajo enfriamiento con hielo. La solución acuosa resultante fue desalada mediante ultrafiltración utilizando una membrana Biomax30. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (90 mg). El contenido del residuo del compuesto medicamentoso en este compuesto fue 11% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 28 20 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Phe-Gly-NH-A’ (A-NH2 =DX-8951) 25 30 35 40 La sal trimetilamónica de polialcohol carboximetildextrano obtenida en el Ejemplo 25 (0,1 g) se disolvió en N,Ndimetilformamida (6 ml). A esta solución, se añadieron sucesivamente una solución del hidrocloruro de 3’-N-(GlyGly-Phe-Gly)-NH-A (A-NH2 =DX-8951) (36 mg) obtenido en el Ejemplo 26 en N,N-dimetilformamida (10 ml), trietilamina (8 µl), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (0,1 g), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. La mezcla se añadió con cloruro sódico acuoso 3 M (2,5 ml) y éter dietilo (20 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados se disolvieron en cloruro sódico acuoso 0,5 M y se ajustaron a pH 12 con hidróxido sódico acuoso 0,1 M bajo enfriamiento con hielo. La solución acuosa resultante fue desalada mediante ultrafiltración utilizando una membrana Biomax30. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (80 mg). El resultado obtenido mediante análisis GPC después de disolver este compuesto en cloruro sódico acuoso 1,0 M (columna: TSK Gel PW-4000XL, Tosoh, disolvente: NaCl 0,1 M, índice de flujo: 0,8 ml/min), y el espectro de absorción ultravioleta del compuesto (solución tampón Tris 0,1 M, pH 9,0, 36 µg/ml) se representan en las Figuras 8 y 9, respectivamente. El contenido del residuo del compuesto medicamentoso en el compuesto fue 15% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 29 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Gly-Phe-NH-A’ (A-NH2 =DX-8951) 45 50 55 60 Se disolvió Dextran T250 (20 g, EXTRASYNTHESE, peso molecular medio: 250K) en tampón ácido acético 0,1 M (pH 5,5, 2000 ml) y se añadió con una solución acuosa (2000 ml) de periodato sódico (66,0 g). Después de agitar a 4ºC durante diez días con protección de luz, la mezcla se añadió con glicol etileno (14,0 ml) y fue agitada durante la noche. La mezcla de reacción se ajustó a pH 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. Se añadió y disolvió borohidruro sódico (28 g) y entonces la mezcla fue agitada durante la noche a temperatura ambiente. La mezcla de reacción fue enfriada con hielo, ajustada a pH 5,5 con ácido acético y agitada a 4ºC durante una hora, y luego ajustada a pH 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. La fracción de bajo peso molecular se eliminó en la solución acuosa resultante mediante ultrafiltración utilizando una membrana Biomax-30 para obtener la Solución Retenida 1 que no pasó a través de la membrana. Separadamente, se disolvió Dextran T250 (50 g, EXTRASYNTHESE, peso molecular medio: 250K) en tampón acetato 0,1 M (pH 5,5, 5000 ml) y se añadió con una solución acuosa (5000 ml) de periodato sódico (165 g). Después de agitar a 4ºC durante diez días con protección de luz, la mezcla se añadió con glicol etileno (35,0 ml) y fue agitada durante la noche. La mezcla de reacción se ajustó a pH 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. Se añadió y disolvió borohidruro sódico (70 g), y entonces la mezcla fue agitada durante la noche a temperatura ambiente. La mezcla de reacción fue enfriada con hielo, ajustada a pH 5,5 con ácido acético y agitada a 4ºC durante una hora, y luego ajustada a pH 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. La fracción de bajo peso molecular se eliminó en la solución acuosa resultante mediante ultrafiltración utilizando una membrana Biomax-30 para obtener la Solución Retenida 2 que no pasó a través de la membrana. Las Soluciones Retenidas 1 y 2 fueron combinadas, sometidas a ultrafiltración utilizando una membrana Biomax-30 para eliminar la fracción de bajo peso molecular de la fracción que había pasado a través de membrana Biomax-50, y fueron liofilizadas para obtener polialcohol dextrano (25,7 g). El peso molecular de esta sustancia fue 47K (filtración de gel, estándar pululano). 65 Este polialcohol dextrano (5 g) se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (35 g) en agua (150 ml) y se disolvió a temperatura ambiente. A esta solución, se añadió ácido monocloroacético (50 g) bajo enfriamiento con hielo y se disolvió, y entonces se dejó reaccionar la mezcla a temperatura ambiente durante 18 20 ES 2 229 355 T3 5 10 15 20 25 30 horas. Esta mezcla de reacción se ajustó a pH 8 con ácido acético y fue desalada mediante ultrafiltración utilizando una membrana Biomax-50. La solución restante que no había pasado a través de la membrana fue liofilizada para obtener la sal sódica de polialcohol carboximetildextrano (7,2 g). El peso molecular de esta sustancia fue 127K (filtración de gel, estándar pululano) y el grado de carboximetilación fue 0,8. Esta sal sódica de polialcohol carboximetildextrano (2,2 g) se disolvió en agua, se aplicó a una columna Bio-Rad AG 50W-X2 (trama 200-400, tipo H+ ) (diámetro: 44 mm, longitud: 210 mm) y fue eluida con agua. Este efluente se añadió con trietilamina (4 ml) y luego fue liofilizado para obtener sal trietilamónica de polialcohol carboximetildextrano (2,69 g). Esta sal trietilamónica de polialcohol carboximetildextrano (2,67 g) se disolvió en N,N-dimetilformamida (200 ml). A esta solución, se añadieron sucesivamente una solución obtenida mediante disolución de la sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Gly-Phe)-NH-A, la cual había sido obtenida mediante eliminación del grupo Boc, según el procedimiento similar al del Ejemplo 16 de 3’-N-(Boc-Gly-Gly-Gly-Phe)-NH-A (A-NH2 =DX-8951) (350 mg) preparada de una manera similar a la del Ejemplo 2, y trietilamina (0,116 ml) en N,N-dimetilformamida (10 ml), y una solución obtenida mediante disolución de 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (2,67 g) en N,N-dimetilformamida (10 ml), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Esta mezcla de reacción se añadió con cloruro sódico acuoso 3 M (100 ml) y cada una de las porciones de 8 ml de la mezcla se añadió por goteo a lotes de 30 ml de etanol. A cada mezcla, se añadieron cloruro sódico acuoso 3 M (1 ml) y éter dietilo (5 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados fueron lavados con acetona y luego disueltos en agua, y se añadieron con cloruro sódico acuoso 3 M (10 ml), y luego se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M, y seguidamente tratados a 37ºC durante 1 hora. La solución tratada fue desalada mediante ultrafiltración utilizando una membrana Biomax-10. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y fue luego liofilizada para obtener el compuesto del enunciado (2,30 g). El resultado obtenido mediante análisis GPC después de disolver este compuesto en cloruro sódico acuoso 0,1 M (columna: TSK Gel PW-4000XL, Tosoh, disolvente: NaCl 0,1 M, índice de flujo: 0,8 ml/min) y el espectro de absorción ultravioleta del compuesto (solución tampón Tris 0,1 M, pH 9,0, 0,20 mg/ml) se representan en las Figuras 10 y 11, respectivamente. El contenido del residuo del compuesto medicamentoso en el compuesto fue 5,8% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 30 Síntesis de sal trietilamónica de polialcohol carboximetildextrano 35 40 A una solución (2000 ml) de Dextran T10 (20 g, Pharmacia, peso molecular medio: 10K) en tampón ácido acético 0,1 M (pH 5,5) se añadió una solución acuosa (2000 ml) de periodato sódico (66,0 g). Después de agitar a 4ºC durante diez días con protección de luz, la mezcla se añadió con glicol etileno (14,0 ml) y fue agitada durante la noche. La mezcla de reacción se ajustó a pH 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. Se añadió y disolvió borohidruro sódico (28 g), y la mezcla fue agitada durante la noche a temperatura ambiente. La mezcla de reacción fue enfriada con hielo, ajustada a pH 5,5 con ácido acético y agitada a 4ºC durante una hora, y luego ajustada a pH 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. La solución acuosa resultante se sometió a ultrafiltración utilizando una membrana Biomax-5 (Millipore) para eliminar la fracción de bajo peso molecular, y la solución restante que no había atravesado la membrana fue pasada a través de una membrana Biomax-30. El filtrado resultante fue liofilizado para obtener polialcohol dextrano (8,0 g). El peso molecular de esta sustancia fue 13K (filtración de gel, estándar pululano). 45 50 55 Este polialcohol dextrano (3,7 g) se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (25,9 g) en agua (111 ml) y se disolvió a temperatura ambiente. A esta solución, se añadió ácido monocloroacético (37 g) bajo enfriamiento con hielo y se disolvió, y entonces se dejó reaccionar la mezcla a temperatura ambiente durante 20 horas. Esta mezcla de reacción se ajustó a pH 8 con ácido acético y fue desalada mediante ultrafiltración utilizando una membrana Biomax-5. La solución restante que no había pasado a través de la membrana fue liofilizada para obtener la sal sódica de polialcohol carboximetildextrano (6,2 g). El peso molecular de esta sustancia fue 37K (filtración de gel, estándar pululano) y el grado de carboximetilación fue 0,9. Esta sal sódica de polialcohol carboximetildextrano (6,0 g) se disolvió en agua, se aplicó a una columna Bio-Rad AG 50W-X2 (trama 200-400, tipo H+ ), y luego fue eluida con agua. Este afluente se añadió con trietilamina (9,3 ml) y entonces liofilizado para obtener el compuesto del enunciado (7,2 g). Ejemplo 31 60 Síntesis de sal trietilamónica de polialcohol carboximetildextrano 65 El polialcohol dextrano (3,9 g) obtenido en el Ejemplo 30 se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (16,3 g) en agua (117 ml) y se disolvió a temperatura ambiente. A esta solución, se añadió ácido monocloroacético (23,4 g) bajo enfriamiento con hielo y se disolvió, y luego se dejó reaccionar la mezcla a temperatura ambiente durante 18 horas. Esta mezcla de reacción se ajustó a pH 8 con ácido acético y fue desalada mediante ultrafiltración utilizando una membrana Biomax-5. La solución restante que no había pasado a través de la membrana fue liofilizada para obtener la sal sódica de polialcohol carboximetildextrano (5,0 g). El peso molecular de esta sustancia fue 28K (filtración de gel, estándar pululano) y el grado de carboximetilación fue 0,5. Esta sal sódica de 21 ES 2 229 355 T3 polialcohol carboximetildextrano (4,8 g) se convirtió en la sal trietilamónica de una manera similar a la del Ejemplo 30 para obtener el compuesto del enunciado (5,6 g). Ejemplo 32 5 Síntesis de sal trietilamónica de polialcohol carboximetildextrano 10 15 20 25 Una solución acuosa (2000 ml) de periodato sódico (66,0 g) se añadió a una solución (2000 ml) de Dextran 4 (20 g, Funakoshi, peso molecular medio: 4K-6K) en tampón ácido acético 0,1 M (pH 5,5). Después de agitar a 4ºC durante diez días con protección de luz, la mezcla se añadió con glicol etileno (14,0 ml) y fue agitada durante la noche. La mezcla de reacción se ajustó a pH 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. Se añadió y disolvió borohidruro sódico (28 g), y la mezcla fue agitada durante la noche a temperatura ambiente. La mezcla de reacción fue enfriada con hielo, ajustada a pH 5,5 con ácido acético y agitada a 4ºC durante una hora, y luego ajustada a pH 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. La solución acuosa resultante se sometió a ultrafiltración utilizando una membrana Biomax-3 (Millipore) para eliminar la fracción de bajo peso molecular. El filtrado obtenido fue liofilizado para obtener polialcohol dextrano (6,0 g). El peso molecular de esta sustancia fue 9K (filtración de gel, estándar pululano). Este polialcohol dextrano (2,7 g) se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (18,9 g) en agua (81 ml) y se disolvió a temperatura ambiente. A esta solución, se añadió ácido monocloroacético (27 g) bajo enfriamiento con hielo y se disolvió, y entonces se dejó reaccionar la mezcla a temperatura ambiente durante 20 horas. Esta mezcla de reacción se ajustó a pH 8 con ácido acético y fue desalada mediante ultrafiltración utilizando una membrana Biomax-5. La solución restante que no había pasado a través de la membrana fue liofilizada para obtener la sal sódica de polialcohol carboximetildextrano (4,2 g). El peso molecular de esta sustancia fue 20K (filtración de gel, estándar pululano), y el grado de carboximetilación fue 0,9. Esta sal sódica de polialcohol carboximetildextrano (4,0 g) se convirtió en la sal trietilamónica de una manera similar a la del Ejemplo 30 para obtener el compuesto del enunciado (4,8 g). Ejemplo 33 30 35 40 Síntesis de sal trietilamónica de polialcohol carboximetildextrano El polialcohol dextrano (2,7 g) obtenido en el Ejemplo 32 se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (11,3 g) en agua (81 ml) y se disolvió a temperatura ambiente. A esta solución, se añadió ácido monocloroacético (16,2 g) bajo enfriamiento con hielo y se disolvió, y luego se dejó reaccionar la mezcla a temperatura ambiente durante 18 horas. Esta mezcla de reacción se ajustó a pH 8 con ácido acético y fue desalada mediante ultrafiltración utilizando una membrana Biomax-5. La solución restante que no había pasado a través de la membrana fue liofilizada para obtener la sal sódica de polialcohol carboximetildextrano (2,7 g). El peso molecular de esta sustancia fue 16K (filtración de gel, estándar pululano) y el grado de carboximetilación fue 0,5. Esta sal sódica de polialcohol carboximetildextrano (2,7 g) se convirtió en la sal trietilamónica de una manera similar a la del Ejemplo 30 para obtener el compuesto del enunciado (3,1 g). Ejemplo 34 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Phe-Gly-NH-A’ (A-NH2 =DX-8951) 45 50 55 60 La sal trietilamónica de polialcohol carboximetildextrano obtenida en el Ejemplo 30 (1,5 g) se disolvió en N,Ndimetilformamida (90 ml). A esta solución, se añadieron sucesivamente una solución de trietilamina (0,07 ml) y sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) (210 mg) en N,N-dimetilformamida (40 ml), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (1,5 g), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. Cada lote se añadió con cloruro sódico acuoso 3 M (2,5 ml) y éter dietilo (20 ml), y entonces los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados se disolvieron en cloruro sódico acuoso 0,5 M y se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M bajo enfriamiento con hielo. La solución acuosa resultante fue desalada mediante ultrafiltración utilizando una membrana Biomax-3. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (1,3 g). El resultado obtenido mediante análisis GPC después de disolver este compuesto en cloruro sódico acuoso 0,1 M (columna: TSK Gel PW-4000XL, Tosoh, disolvente: NaCl 0,1 M, índice de flujo: 0,8 ml/min) y el espectro de absorción ultravioleta del compuesto (solución tampón Tris 0,1 M, pH 9,0, 65 µg/ml) se representan en las Figuras 12 y 13, respectivamente. El contenido del residuo del compuesto medicamentoso en el compuesto fue 6,4% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 35 65 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Phe-Gly-NH-A’ (A-NH2 =-DX-8951) La sal trietilamónica de polialcohol carboximetildextrano obtenida en el Ejemplo 31(1,2 g) se disolvió en N,Ndimetilformamida (90 ml). A esta solución, se añadieron sucesivamente una solución de trietilamina (0,056 ml) y sal de 22 ES 2 229 355 T3 5 10 ácido trifluoroacético de 3’-N-(Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) (168 mg) en N,N-dimetilformamida (30 ml), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (1,2 g), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. Cada lote se añadió con cloruro sódico acuoso 3 M (2,5 ml) y éter dietilo (20 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados se disolvieron en cloruro sódico acuoso 0,5 M y se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M bajo enfriamiento con hielo. La solución acuosa resultante fue desalada mediante ultrafiltración utilizando una membrana Biomax-3. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (1,0 g). El contenido del residuo del compuesto medicamentoso en este compuesto fue 4,8% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 36 15 20 25 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Phe-Gly-NH-A’ (A-NH2 =DX-8951) La sal trietilamónica de polialcohol carboximetildextrano obtenida en el Ejemplo 32 (1,2 g) se disolvió en N,Ndimetilformamida (90 ml). A esta solución, se añadieron sucesivamente una solución de trietilamina (0,056 ml) y sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) (168 mg) en N,N-dimetilformamida (30 ml), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (1,2 g), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. Cada lote se añadió con cloruro sódico acuoso 3 M (2,5 ml) y éter dietilo (20 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados se disolvieron en cloruro sódico acuoso 0,5 M y se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M bajo enfriamiento con hielo. La solución acuosa resultante fue desalada mediante ultrafiltración utilizando una membrana Biomax-3. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (1,0 g). El contenido del residuo del compuesto medicamentoso en este compuesto fue 5,9% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). 30 Ejemplo 37 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Phe-Gly-NH-A’ (A-NH2 =DX-8951) 35 40 45 La sal trietilamónica de polialcohol carboximetildextrano obtenida en el Ejemplo 33 (1,5 g) se disolvió en N,Ndimetilformamida (90 ml). A esta solución, se añadieron ssucesivamente una solución de trietilamina (0,07 ml) y sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) (210 mg) en N,N-dimetilformamida (40 ml), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (1,5 g), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. Cada lote se añadió con cloruro sódico acuoso 3 M (2,5 ml) y éter dietilo (20 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados se disolvieron en cloruro sódico acuoso 0,5 M y se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M bajo enfriamiento con hielo. La solución acuosa resultante fue desalada mediante ultrafiltración utilizando una membrana Biomax-3. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (1,3 g). El contenido del residuo del compuesto medicamentoso en este compuesto fue 4,6% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 38 50 Síntesis de Boc-Gly-Gly-Phe-Gly-NH-A (A-NH2 =DW-8286) 55 Se disolvieron Boc-Gly-Gly-Phe-Gly (42 mg) y N-hidroxisuccinimida (12 mg) en N,N-dimetilformamida (2 ml), se enfrió a 4ºC, y luego se adicionó con N,N’-diciclohexilcarbodiimida (22 mg). A esta solución, se añadió una solución de N,N-dimetilformamida (6 ml), en la cual se disolvieron el hidrocioruro del compuesto representado por la siguiente formula: 60 65 23 ES 2 229 355 T3 5 10 15 20 25 [(1 S,9S)-1-amino-5-cloro-9-etil-2,3-dihidro-9-hidroxi-1H,12H-benzo[de]pirano-[3’,4’:6,7]indolizino[1,2-b]quinolina-10,13(9H,15H]-diona: DW-8286] (50 mg) y trietilamina (0,01 ml), y se dejó reaccionar la mezcla con agitación y protección de luz a temperatura ambiente durante 16 horas. Esta mezcla de reacción fue evaporada hasta sequedad bajo presión reducida, y el residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: diclorometano:metanol = 10:1 solución conteniendo ácido acético 0,5%) para obtener el compuesto del enunciado (27 mg). H-NMR (CDCl3 ) δ: 8,10-8,20 (br, 1H), 7,95-8,05 (br, 1H), 7,70-7,80 (br, 2H), 7,50-7,60 (br, 1H), 7,40-7,50 (br, 1H), 7,10-7,25 (m, 5H), 7,05-7,15 (br, 1H), 5,85-5,95(br, 1H), 5,50-5,60 (br, 1H), 5,40-5,50 (m, 1H), 5,25-5,3 5 (m, 1H), 5,05-5,15 (m, 1H), 4,90-5,00 (m, 1H), 4,70-4,80 (br, 1H), 4,10-4,25 (br, 2H), 3,60-3,90 (m, 4H), 3,10-3,40 (m, 3H), 2,95-3,05 (br, 1H), 2,15-2,30 (br, 1H), 1,75-1,90 (br, 2H), 1,39 (s, 9H), 0,80-1,00 (m, 3H). 1 30 Ejemplo 39 35 40 45 50 55 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Phe-Gly-NH-A’ (A-NH2 =DW-8286) La sal trietilamónica de polialcohol carboximetildextrano (175 mg) obtenida en el Ejemplo 24 se disolvió en N,Ndimetilformamida (20 ml). A esta solución, se añadieron sucesivamente una solución de sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DW-8286) (29 mg), la cual había sido obtenida de 3’-N-(Boc-Gly-GlyPhe-Gly)-NH-A (27 mg) preparado en el Ejemplo 38 eliminando el grupo Boc de una manera similar a la del Ejemplo 4, y trietilamina (9 µl) en N,N-dimetilformamida (5 ml), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (175 mg), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. La mezcla se añadió con cloruro sódico acuoso 3 M (2,5 ml) y éter dietilo (20 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados se disolvieron en cloruro sódico acuoso 0,5 M y se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M bajo enfriamiento con hielo. La solución acuosa resultante fue desalada mediante ultrafiltración utilizando una membrana Biomax-30. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (135 mg). El resultado obtenido mediante análisis GPC después de disolver este compuesto en cloruro sódico acuoso 0,1 M (columna: TSK Gel PW-4000XL, Tosoh, disolvente: NaCl 0,1 M, índice de flujo: 0,8 ml/min) y el espectro de absorción ultravioleta del compuesto (solución tampón Tris 0,1 M, pH 9,0, 99 µg/ml) se representan en las Figuras 14 y 15, respectivamente. El contenido del residuo del compuesto medicamentoso en el compuesto fue 6,1% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 40 Síntesis de 3’-N-(Boc-(Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DW-8089) 60 Se disolvieron Boc-Gly-Gly-Phe-Gly (163 mg) y N-hidroxisuccinimida (45 mg) en N,N-dimetilformamida (10 ml), se enfrió a 4ºC, y luego se adicionó con N,N’-diciclohexilcarbodiimida (79 mg). A esta solución, se añadió una solución de N,N-dimetilformamida (30 ml), en la cual se disolvieron tosilato del compuesto representado por la siguiente formula: 65 24 ES 2 229 355 T3 5 10 15 20 25 30 35 [(1 S,9S)-1-amino-9-etil-2,3-dihidro-9-hidroxi-1 H,12H-benzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolina-10, 13(9H,15H)-diona: DW-8089] (170 mg) y trietilamina (0,054 ml), y se dejó reaccionar la mezcla con agitación a temperatura ambiente durante la noche bajo condiciones protegidas de la luz. Esta mezcla de reacción fue evaporada hasta sequedad bajo presión reducida, y el residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: diclorometano:metanol = 94:6 solución conteniendo ácido acético 0,5%) para obtener el compuesto del enunciado (100 mg). 1 H-NMR (DMSO-d6 ) δ: 8,51 (d, 1H, J=8,5 Hz), 8,41 (t, 1H, J=5,6 Hz), 8,29 (s, 1H), 8,17 (d, 1H, J=8,0 Hz), 8,03 (d, 1H, J=8,0 Hz), 7,90 (dd, 1H, J=4,8, 5,6 Hz), 7,79 (t, 1H, J=5,6 Hz), 7,53 (d, 1H, J=7,2 Hz), 7,36 (s, 1H), 7,13-7,25 (m, 5H), 6,94-6,95 (m, 1H), 5,60-5,63 (m, 1H), 5,36-5,47 (m, 2H), 5,21-5,30 (m, 2H), 4,42-4,47 (m, 1H), 3,63-3,96 (m, 3H), 3,51-3,59 (m, 3H), 3,31-3,40 (m, 1H), 3,09-3,21 (m, 1H), 3,02 (dd, 1H, J=4,8, 13,5 Hz), 2,76-2,81 (m, 1H), 2,13-2,17 (m, 2H), 1,85-1,90 (m, 2H), 1,37 (s, 9H), 0,89 (t, 3H, J=8,0 Hz). Masa (FAB); m/e 822 (M+1). Ejemplo 41 40 45 50 55 60 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Phe-Gly-NH-A’ (A-NH2 =DW-8089) Se disolvió la sal trietilamónica de polialcohol carboximetildextrano (1,6 g) obtenida en el Ejemplo 24 en N,Ndimetilformamida (60 ml). A esta solución, se añadieron sucesivamente una solución obtenida mediante disolución de sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Phe-Gly)-NH-A (A-NH2 =DW-8089), la cual había sido obtenida de 3’-N-(Boc-Gly-Gly-Phe-Gly)-NH-A (200 mg) preparado en el Ejemplo 40 eliminando el grupo Boc de una manera similar a la del Ejemplo 4, y trietilamina (0,07 ml) en N,N-dimetilformamida (20 ml), y 1-etoxicarbonil-2-etoxi-1,2dihidroxiquinolina (1,6 g), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. Cada lote se añadió con cloruro sódico acuoso 3 M (2,5 ml) y éter dietilo (25 ml), y los precipitados depositados se recogieron mediante centrifugación (2500 rpm, 8 minutos). Los precipitados fueron lavados con etanol, luego disueltos en agua, se añadieron con cloruro sódico acuoso 3 M (20 ml), y se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M. Esta solución fue desalada mediante ultrafiltración utilizando una membrana Biomax-10. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (1,20 g). El resultado obtenido mediante análisis GPC después de disolver el compuesto en cloruro sódico acuoso 0,1 M (columna: TSK Gel PW-4000XL, Tosoh, disolvente: NaCl 0,1 M, índice de flujo: 0,8 ml/min) y el espectro de absorción ultravioleta del compuesto (solución tampón Tris 0,1 M, pH 9,0, 0,26 mg/ml) se representan en las Figuras 16 y 17, respectivamente. El contenido del residuo del compuesto medicamentoso en el compuesto fue 5,0% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 42 Síntesis de sal trietilamónica de polialcohol carboximetildextrano 65 Se disolvió Dextran T150 (20 g, Pharmacia, peso molecular medio: 150K) en tampón acetato 0,1 M (pH 5,5, 2000 ml) y se añadió con una solución acuosa (2000 ml) de periodato sódico (66,0 g). Después de agitar a 4ºC durante diez días con protección de luz, la mezcla se añadió con glicol etileno (14,0 ml) y fue agitada durante la noche. La mezcla de reacción se ajustó a pH 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. Se añadió y disolvió 25 ES 2 229 355 T3 5 10 15 20 borohidruro sódico (28 g), y entonces la mezcla fue agitada a temperatura ambiente durante la noche. La mezcla de reacción fue enfriada con hielo, ajustada a pH 5,5 con ácido acético, y agitada a 4ºC durante 1 hora. El pH de la mezcla se ajustó a 7,5 con hidróxido sódico acuoso 8 M bajo enfriamiento con hielo. La solución acuosa resultante fue concentrada hasta 500 ml mediante ultrafiltración utilizando una membrana Biomax-5 (Millipore) para obtener la Solución 1. Separadamente, se realizaron una serie de procesos descritos anteriormente utilizando Dextran T110 (20 g) para obtener la Solución 2. Las Soluciones 1 y 2 fueron combinadas, y la solución combinada se ajustó a pH 3,0 y fue incubada a 40ºC durante 4 horas, y luego ajustada a pH 7 para obtener una solución conteniendo el polialcohol dextrano con peso molecular rebajado. La solución fue pasada a través de una membrana Biomax-30 y fue desalada mediante ultrafiltración utilizando una membrana Biomax-5, y luego fue liofilizada para obtener polialcohol dextrano (4,6 g). El peso molecular de esta sustancia fue 17K (filtración de gel, estándar pululano). Este polialcohol dextrano (2,5 g) se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (17,5 g) en agua (75 ml) y se disolvió a temperatura ambiente. A esta solución, se añadió ácido monocloroacético (25 g) bajo enfriamiento con hielo y se disolvió, y luego se dejó reaccionar la mezcla a temperatura ambiente durante 20 horas. Esta mezcla de reacción se ajustó a pH 9 con ácido acético y luego fue desalada mediante ultrafiltración utilizando una mmembrana Biomax-5. La solución restante que no había pasado a través de la membrana fue liofilizada para obtener la sal sódica de polialcohol carboximetildextrano (4,0 g). El peso molecular de esta sustancia fue 45K (filtración de gel, estándar pululano), y el grado de carboximetilación fue 0,9. Esta sal sódica de polialcohol carboximetildextrano (3,7 g) se disolvió en agua, se aplicó a una columna Bio-Rad AG 50W-X2 (trama 200-400, tipo H+ ) y fue eluida con agua. Este eluyente se añadió con trietilamina (5,8 ml) y luego fue liofilizado para obtener el compuesto del enunciado (4,4 g). Ejemplo 43 25 Síntesis de polialcohol carboximetildextrano-Gly-Gly-Gly-Phe-NH-A’ (A-NH2 =DX-8951) 30 35 Se disolvió la sal trietilamónica de polialcohol carboximetildextrano obtenida en el Ejemplo 42 (4,4 g) en N,Ndimetilformamida (300 ml). A esta solución, se añadieron sucesivamente una solución de trietilamina (0,19 ml) y sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Gly-Phe)-NH-A (A-NH2 =DX-8951) (580 mg) en N,N-dimetilformamida (45 ml) y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (4,4 g), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación y protección de luz. Esta mezcla de reacción se ajustó a pH 10 con hidróxido sódico acuoso 1 M, y entonces cada una de las porciones de 5 ml de la mezcla se añadió por goteo a lotes de 25 ml de etanol. La mezcla se añadió con cloruro sódico acuoso 3 M (1 ml) y éter dietilo (5 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados fueron disueltos en agua y dializados con agua purificada utilizando una membrana de diálisis (Spectrapore 1, peso molecular de corte; 6.000-8.000), y la solución dializada interna se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (3,4 g). El contenido del residuo del compuesto medicamentoso en este compuesto fue 4,6% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). 40 Ejemplo 44 Síntesis de polialcohol α-metilcarboximetildextrano-Gly-Gly-Gly-Phe-NH-A’ (A-NH2 =DX-8951) 45 50 55 60 65 El polialcohol dextrano (2 g) obtenido en el Ejemplo 42 se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (14 g) en agua (60 ml) y se disolvió a temperatura ambiente. A esta solución, se añadió y disolvió ácido α-bromopropiónico (19 ml) bajo enfriamiento con hielo, y luego se dejó reaccionar la mezcla a temperatura ambiente durante 18 horas. La mezcla de reacción se ajustó a pH 8 con ácido acético y fue desalada mediante ultrafiltración utilizando una membrana Biomax-50. La solución restante que no había pasado a través de la membrana fue liofilizada para obtener la sal sódica de polialcohol α-metilcarboximetildextrano (2,95 g). El peso molecular de esta sustancia fue 45K (filtración de gel, estándar pululano). El grado de α-metilcarboximetilación por cada residuo sacárido se obtuvo de acuerdo con los casos de polialcohol carboximetildextrano que siguen. Una solución acuosa de la sal sódica de polialcohol α-metilcarboximetildextrano se aplicó a una columna Bio-Rad AG 50W-X2 (tipo H+ ), y el efluente fue liofilizado y utilizado como muestra. Esta muestra se disolvió en un exceso determinado de solución acuosa de hidróxido sódico 0,1 N y fue neutralizada con ácido clorhídrico 0,1 N utilizando fenolftaleína como un indicador. El grado de α-metilcarboximetilación se obtuvo según la ecuación: grado de α-metilcarboximetilación = 13,4(a-b)/[s-7,2(a-b)] en la cual “s” es la cantidad de muestra utilizada (mg), “a” es el exceso determinado de solución acuosa de hidróxido sódico 0,1 N (ml), y “b” es la cantidad de ácido clorhídrico 0,1 N consumida para la valoración (ml). Como resultado, el grado de α-metilcarboximetilación se determinó en 0,8. Esta sal sódica de polialcohol α-metilcarboximetildextrano (2,2 g) se disolvió en agua, se aplicó a una columna Bio-Rad AG 50W-X2 (trama 200-400, tipo H+ ) (diámetro: 44 mm, longitud: 210 mm), y luego fue eluida con agua. Este efluente se añadió con trietilamina (4 ml) y luego fue liofilizado para obtener sal trietilamónica de polialcohol αmetilcarboximetildextrano (2,69 g). Esta sal trietilamónica de polialcohol α-metilcarboximetildextrano (2,68 g) se disolvió en N,N-dimetilformamida (60 ml). A esta solución, se añadieron sucesivamente una solución obtenida mediante disolución de sal de ácido trifluoroacético de 3’-N-(Gly-Gly-Gly-Phe)-NH-A (A-NH2 =DX-8951), la cual había sido obtenida de una manera 26 ES 2 229 355 T3 5 10 15 similar a la del Ejemplo 16 eliminando el grupo Boc de 3’-N-(Boc-Gly-Gly-Gly-Phe)-NH-A (350 mg) sintetizado similarmente al del Ejemplo 2, y trietilamina (0,116 ml) en N,N-dimetilformamida (10 ml), y una solución obtenida mediante disolución de 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (2,68 g) en N,N-dimetilformamida (10 ml), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Esta mezcla de reacción se añadió con cloruro sódico acuoso 3 M (40 ml), y cada una de las porciones de 6 ml de la mezcla se añadió por goteo a lotes de 30 ml de etanol. Cada lote se añadió con cloruro sódico acuoso 3 M (1 ml) y éter dietilo (5 ml), y el precipitado depositado fue recogido mediante centrifugación (3500 rpm, 8 minutos). Este precipitado fue lavado con acetona, luego disuelto en agua, añadido con cloruro sódico acuoso 3 M (10 ml), ajustado a pH 9 con hidróxido sódico acuoso 0,1 M, y tratado a 37ºC durante 1 hora. Esta solución tratada fue desalada mediante ultrafiltración utilizando una membrana Biomax-10. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (2,15 g). El resultado obtenido mediante análisis GPC después de disolver este compuesto en cloruro sódico acuoso 0,1 M (columna: TSK Gel PW4000XL, Tosoh, disolvente: NaCl 0,1 M, índice de flujo: 0,8 ml/min) y el espectro de absorción ultravioleta del compuesto (solución tampón Tris 0,1 M, pH 9,0, 0,21 mg/ml) se representan en las Figuras 18 y 19, respectivamente. El contenido del residuo del compuesto medicamentoso en el producto resultante fue 5,9% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 45 20 25 Síntesis de sal de ácido trifluoroacético de 3’-N-(Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) Una mezcla de sal de ácido p-toluenosulfónico de Phe-Gly-OBzl (3,06 g), Boc-Gly-OH (1,10 g), N-hidroxisuccinimida (941 mg), N-metilmorfolina (0,725 ml), y N,N-dimetilformamida (40 ml) se enfrió a 4ºC, y se añadió con N,N’-diciclohexil-carbodiimida (1,56 g). Se dejó reaccionar la mezcla durante la noche a temperatura ambiente con agitación, y entonces fue evaporada hasta sequedad bajo presión reducida. El residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: solución diclorometano:metanol = 98:2) para obtener Boc-Gly-Phe-GlyOBzl (1,93 g). H-NMR (DMSO-d6 ) δ: 8,52 (dd, 1H, J=5,6, 6,4 Hz), 7,97 (d, 1H, J=8,8 Hz), 7,30-7,39 (m, 5H), 7,15-7,26 (m, 5H), 6,83 (t, 1H, J=5,6 Hz), 5,14 (s, 1H), 4,52-4,57 (m, 1H), 3,87-3,96 (m, 2H), 3,57 (dd, 1H, J=5,6, 16,7 Hz), 3,43 (dd, 1H, J=5,6, 16,7 Hz), 3,01 (dd, 1H, J=4,8, 14,3 Hz), 2,77 (dd, 1H, J=5,6, 14,3 Hz), 1,37 (s, 9H). 1 30 35 El Boc-Gly-Phe-Gly-OBzl resultante (1,78 g) se disolvió en acetato de etilo (60 ml) y fue sometido a reducción catalítica durante 24 horas en presencia de 5%-Pd-C (1,8 g). El catalizador se eliminó mediante filtración y el filtrado fue concentrado bajo presión reducida para obtener Boc-Gly-Phe-Gly-OH (1,41 g). H-NMR (DMSO-d6 ) δ: 8,35 (t, 1H, J=5,6 Hz), 7,94 (d, 1H, J=8,8 Hz), 7,15-7,26 (m, 5H), 6,85 (dd, 1H, J=5,6, 6,4 Hz), 4,52-4,58 (m, 1H), 3,76 (d, 2H, J=5,6 Hz), 3,56 (dd, 1H, J=6,4, 16,7 Hz), 3,43 (dd, 1H, J=5,6, 16,7 Hz), 3,03 (dd, 1H, J=5,0, 13,5 Hz), 2,79 (dd, 1H, J=9,5, 13,5 Hz), 1,37 (s, 9H). 1 40 45 Se disolvieron el Boc-Gly-Phe-Gly-OH (500 mg) obtenido anteriormente y N-hidroxisuccinimida (161 mg) en N,N-dimetilformamida (10 ml). A esta solución, se añadió una solución de N,N-dimetilformamida (50 ml), en la cual se disolvieron metanosulfonato de DX-8951 (530 mg) y trietilamina (0,146 ml). La mezcla se enfrió a 4ºC, se añadió con N,N’-diciclohexilcarbodiimida (268 mg), y se dejó reaccionar durante la noche con agitación a temperatura ambiente bajo condiciones protegidas de la luz. Esta mezcla de reacción fue evaporada hasta sequedad bajo presión reducida y el residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: solución diclorometano:metanol = 96:4) para obtener 3’-N-(Boc-Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) (100 mg). H-NMR (DMSO-d6 ) δ: 8,39 (d, 1H, J=8,0 Hz), 8,34 (t, 1H, J=5,6 Hz), 7,98 (d, 1H, J=7,2 Hz), 7,78 (d, 1H, J=10,3 Hz), 7,33 (s, 1H), 7,13-7,24 (m, 5H), 6,80 (dd, 1H, J=5,6, 6,4 Hz), 5,55-5,61 (m, 1H), 5,44 (d, 1H, J=16,0 Hz), 5,41 (d, 1H, J=16,0 Hz), 5,25 (s, 2H), 4,43-4,46 (m, 1H), 3,69-3,79 (m, 2H), 3,50 (dd, 1H, J=5,6, 16,7 Hz), 3,41 (dd, 1H, J=5,6, 16,7 Hz), 3,16-3,19 (m, 2H), 2,98 (dd, 1H, J=4,8, 14,3 Hz), 2,79 (dd, 1H, J=9,5, 14,3 Hz) 2,41 (s, 3H), 2,192,25 (m, 1H), 2,10-2,15 (m, 1H), 1,82-1,90 (m, 2H), 1,35 (s, 9H), 0,88 (t, 3H, J=8,0 Hz). 1 50 55 Masa (FAB); m/e 797 (M+1). 60 El 3’-N-(Boc-Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) resultante (100 mg) se disolvió en ácido trifluoroacético (3 ml) y se dejó reposar durante una hora. Se evaporó el disolvente, y el residuo fue sometido a destilaciones azeotrópicas dos veces con metanol (30 ml) y dos veces con etanol (30 ml), y luego lavado con éter para obtener el compuesto del enunciado (80 mg). 65 H-NMR (DMSO-d6 ) δ: 8,52-8,62 (m, 1H), 7,94 (s, 3H), 7,79 (t, 1H, J=11,1 Hz), 7,34 (s, 1H), 7,15-7,27 (m, 5H), 6,52 (s, 1H), 5,57-5,61 (m, 1H), 5,36-5,46 (m, 2H), 4,66-4,70 (m, 1H), 3,69-3,81 (m, 2H), 3,61-3,68 (m, 1H), 3,403,47 (m, 1H), 3,15-3,23 (m, 1H), 3,01 (dd, 1H, J=4,0, 13,5 Hz), 2,77 (dd, 1H, J=9,5, 13,5 Hz), 2,12-2,23 (m, 2H), 1,81-1,91 (m, 2H), 0,89 (t, 3H, J=7,2 Hz). 1 Masa (FAB); m/e 697 (M+1). 27 ES 2 229 355 T3 Ejemplo 46 Síntesis de sal de ácido trifluoroacético de 3’-N-(Phe-Gly)-NH-A (A-NH2 =DX-8951) 5 10 Se disolvieron Boc-Phe-Gly (771 mg) y N-hidroxisuccinimida (300 mg) en N,N-dimetilformamida (10 ml). A esta solución, se añadió una solución de N,N-dimetilformamida (50 ml), en la cual se disolvieron metanosulfonato de DX8951 (1058 mg) y trietilamina (0,293 ml). La mezcla se enfrió a 4ºC, y luego se adicionó con N,N’-diciclohexilcarbodiimida (494 mg), y se dejó reaccionar con agitación a temperatura ambiente durante la noche bajo condiciones protegidas de la luz. Esta mezcla de reacción fue evaporada hasta sequedad bajo presión reducida, y el residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: solución diclorometano:metanoi = 98:2) para obtener 3’-N-(Boc-Phe-Gly)-NH-A (A-NH2 =DX-8951) (1,20 g). H-NMR (DMSO-d6 ) δ: 8,29 (d, 1H, J=8,0 Hz), 8,21 (t, 1H, J=4,8 Hz), 7,76 (d, 1H, J=10,3 Hz), 7,32 (s, 1H), 7,137,25 (m, 5H), 6,92 (d, 1H, J=7,2 Hz), 6,49 (s, 1H), 5,56-5,61 (m, 1H), 5,44 (d, 1H, J=15,9 Hz), 5,38 (d, 1H, J=15,9 Hz), 5,25 (s, 2H), 4,08-4,12 (m, 1H), 3,78 (d, 1H, J=4,8 Hz), 3,16-3,25 (m, 2H), 2,99 (dd, 1H, J=4,0, 13,5 Hz), 2,72 (dd, 1H, J=10,3, 13,5 Hz), 2,40 (s, 3H), 2,09-2,35 (m, 2H), 1,80-1,91 (m, 2H), 1,16 (s, 9H), 0,88 (t, 3H, J=8,0 Hz). 1 15 Masa (FAB); m/e 741 (M+1). 20 25 El 3’-N-(Boc-Phe-Gly)-NH-A (170 mg) obtenido anteriormente se disolvió en ácido trifluoroacético (4 ml) y se dejó reposar durante una hora. Se evaporó el disolvente, y el residuo fue sometido a destilaciones azeotrópicas dos veces con metano) (10 ml) y dos veces con etanol (10 ml), y luego lavado con éter para obtener el compuesto del enunciado (100 mg). 1 H-NMR (DMSO-d6 ) δ: 8,88 (t, 1H, J=4,8 Hz), 8,68 (d, 1H, J=8,7 Hz), 8,05-8,15 (m, 3H), 7,79 (d, 1H, J=11,1 Hz), 7,26-7,36 (m, 5H), 6,52 (d, 1H, J=7,2 Hz), 5,57-5,62 (m, 1H), 5,43 ((d, 1H, J=15,9 Hz), 5,38 (d, 1H, J=15,9 Hz), 5,19-5,28 (m, 1H), 4,10-4,18 (m, 1H), 3,93 (dd, 1H, J=4,8, 16,7 Hz), 3,82 (dd, 1H, J=4,8, 16,7 Hz), 3,17-3,24 (m, 2H), 3,14 (dd, 1H, J=4,8, 13,5 Hz), 2,95 (dd, 1H, J=8,0, 13,5 Hz), 2,42 (s, 3H), 2,14-2,25 (m, 2H), 1,83-1,91 (m, 2H), 0,89 (t, 3H, J=8,0 Hz). 30 Masa (FAB); m/e 640 (M+1). Ejemplo 47 35 40 Síntesis de sal de ácido trifluoroacético de 3’-N-Gly-NH-A (A-NH2 =DX-8951) Se disolvieron metanosulfonato de DX-8951 (530 mg) y trietilamina (0,28 ml) en N,N-dimetilformamida (10 ml), se enfrió a 4ºC, y se añadió con éster N-hidroxisuccinimida de Boc-Gly (327 mg). Se dejó reaccionar la mezcla con agitación a temperatura ambiente durante la noche bajo condiciones protegidas de la luz. Esta mezcla de reacción fue evaporada hasta sequedad bajo presión reducida, y entonces el residuo se purificó mediante cromatografía en columna de gel de sílice (eluyente: solución diclorometano:metanol = 98:2) para obtener 3’-N-(Boc-Gly)-NH-A (A-NH2 =DX8951) (500 mg). H-NMR (DMSO-d6 ) δ: 8,38 (d, 1H, J=8,3 Hz), 7,77 (d, 1H, J=10,7 Hz), 7,31 (s, 1H), 6,89-6,91 (m, 1H), 6,49 (s, 1H), 5,55-5,59 (m, 1H), 5,45 (d, 1H, J=16,1 Hz), 5,38 (d, 1H, J=16,1 Hz), 5,27 (d, 1H, J=19,0 Hz), 5,18 (d, 1H, J=19,0 Hz), 3,50-3,62 (m, 2H), 3,15-3,19 (m, 2H), 2,41 (s, 3H), 2,18-2,24 (m, 1H), 2,08-2,12 (m, 1H), 1,81-1,91 (m, 2H), 1,31 (s, 9H), 0,87 (t, 3H, J=8,0 Hz). 1 45 Masa (FAB); m/e 593 (M+1). 50 El 3’-N-(Boc-Gly)-NH-A (100 mg) obtenido anteriormente se disolvió en ácido trifluoroacético (2 ml) y se dejó reposar durante una hora. Se evaporó el disolvente, y el residuo fue sometido a destilaciones azeotrópicas dos veces con metanol (10 ml) y dos veces con etanol (10 ml), y luego lavado con éter para obtener el compuesto del enunciado (70 mg). 55 1 H-NMR (DMSO-d6 ) δ: 8,88 (d, 1H, J=8,8 Hz), 8,08 (s, 3H), 7,81 (d, 1H, J=11,2 Hz), 7,34 (s, 1H), 6,52 (s, 1H), 5,63-5,67 (m, 1H), 5,45 (d, 1H, J=16,7 Hz), 5,40 (d, 1H, J=16,7 Hz), 5,36 (d, 1H, J=19,1 Hz), 5,25 (d, 1H, J=19,1 Hz), 3,56 (s, 2H), 3,11-3,19 (m, 2H), 2,43 (s, 3H), 2,23-2,28 (m, 1H), 2,11-2,19 (m, 1H), 1,81-1,91 (m, 2H), 0,88 (t, 3H, J=8,0 Hz). 60 Masa (FAB); m/e 493 (M+1). Ejemplo 48 65 Síntesis de sal trimetilamónica de polialcohol carboximetildextrano Se disolvió Dextran T500 (50 g, Pharmacia, peso molecular: 500K) en tampón acetato 0,1 M JpH 5,5, 5000 ml) y se añadió con una solución acuosa (5000 ml) de periodato sódico (165,0 g). Después de agitar a 4ºC durante diez 28 ES 2 229 355 T3 5 10 15 20 días con protección de luz, la mezcla se añadió con glicol etileno (35,0 ml) y fue agitada durante la noche. La mezcla de reacción se ajustó a pH 7 con hidróxido sódico acuoso 8 M. Se añadió y disolvió borohidruro sódico (70 g), y la mezcla fue agitada durante la noche. La mezcla de reacción fue enfriada con hielo, ajustada a pH 5,5 con ácido acético y agitada a 4ºC durante una hora, y luego ajustada a pH 7,5 con hidróxido sódico acuoso 8 M. La solución acuosa resultante fue desalada mediante ultrafiltración utilizando una membrana Biomax-50. La solución restante que no había pasado a través de la membrana fue liofilizada para obtener polialcohol dextrano (20,2 g). El peso molecular de esta sustancia fue 159K (filtración de gel, estándar pululano). Este polialcohol dextrano (7,5 g) se añadió a una solución acuosa obtenida mediante disolución de hidróxido sódico (31,5 g) en agua (225 ml) y se disolvió a temperatura ambiente. A esta solución, se añadió ácido monocloroacético (45 g) bajo enfriamiento con hielo, se disolvió, y se dejó reaccionar la mezcla a temperatura ambiente durante la noche. Esta mezcla de reacción se ajustó a pH 8 con ácido acético y luego fue desalada mediante ultrafiltración utilizando una membrana Biomax-50. La solución restante que no había pasado a través de la membrana fue liofilizada para obtener la sal sódica de polialcohol carboximetildextrano (8,5 g). El peso molecular de esta sustancia fue 274K (filtración de gel, estándar pululano), y el grado de carboximetilación fue 0,4. Esta sal sódica de polialcohol carboximetildextrano (2,0 g) se disolvió en agua, se aplicó a una columna Bio-Rad AG 50W-X2 (trama 200-400, tipo H+ ) (diámetro: 44 mm, longitud: 210 mm) y fue eluida con agua. Este efluente se añadió con trietilamina (4 ml) y luego fue liofilizado para obtener el compuesto del enunciado (2,2 g). Ejemplo 49 Síntesis de polialcohol carboximetildextrano-Gly-Phe-Gly-NH-A’ (A-NH2 =DX-8951) 25 30 35 La sal trietilamónica de polialcohol carboximetildextrano obtenida en el Ejemplo 48 (200 mg) se disolvió en N,Ndimetilformamida (7 ml). A esta solución, se añadieron sucesivamente una solución de la sal de ácido trifluoroacético de 3’-N-(Gly-Phe-Gly)-NH-A (A-NH2 =DX-8951) obtenida en el Ejemplo 45 (41 mg) en N,N-dimetilformamida (5 ml), trietilamina (0,014 ml), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (100 mg), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Cada una de las porciones de 5 mi de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. La mezcla se añadió con cloruro sódico acuoso 3 M (2,0 ml) y éter dietilo (25 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los precipitados se disolvieron en cloruro sódico acuoso 0,5 M y se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M bajo enfriamiento con hielo. La solución acuosa resultante fue desalada mediante ultrafiltración utilizando una membrana Biomax-50. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (190 mg). El contenido del residuo del compuesto medicamentoso en este compuesto fue 4,5% (w/w) cuando se determinó con base en la absorción a 362 nm en tampón Tris 0,1 M (pH 9,0). Ejemplo 50 40 Síntesis de polialcohol carboximetildextrano-Phe-Gly-NH-A’ (A-NH2 =DX-8951) La sal sódica de polialcohol carboximetildextrano obtenida en el Ejemplo 24 (2,5 g) se disolvió en agua, se aplicó a una columna Bio-Rad AG 50W-X2 (trama 200-400, tipo Et3 N H+ ) y fue eluida con agua. Este efluente fue liofilizado para obtener sal trietilamónica de polialcohol carboximetildextrano (2,5 g). 45 50 55 Esta sal trietilamónica de polialcohol carboximetildextrano (200 mg) se disolvió en N,N-dimetilformamida (12 ml). A esta solución, se añadieron sucesivamente una solución de la sal de ácido trifluoroacético de 3’-N-(Phe-Gly)NH-A (A-NH2 =DX-8951) (42 mg) obtenida en el Ejemplo 46 y trietilamina (0,016 ml) en N,N-dimetilformamida (5 ml), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (200 mg), y se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación y protección de luz. Esta mezcla de reacción se añadió con agua (300 ml) y fue sometida a ultrafiltración utilizando una membrana de ultrafiltración 10K (Filtron). La solución restante que no había pasado a través de la membrana se ajustó a pH 10 con hidróxido sódico acuoso 0,1 N, y se pasó a través de una membrana de filtración (0,16 µm, Filtron). El filtrado fue desalado mediante ultrafiltración utilizando una membrana Biomax-50, y entonces se filtró a través de un filtro Millipore (0,22 µm) y fue liofilizado para obtener el compuesto del enunciado (180 mg). El contenido del residuo del compuesto medicamentoso en este compuesto fue 6,1% (w/w) cuando se determinó con base en la absorción a 362 nm en solución tampón Tris 0,1 M (pH 9,0). Ejemplo 51 60 Síntesis de polialcohol carboximetildextrano-Gly-NH-A’ (A-NH2 =DX-8961) 65 La sal trietilamónica de polialcohol carboximetildextrano obtenida en el Ejemplo 48 (370 mg) se disolvió en N,Ndimetilformamida (10 ml). A esta solución, se añadieron sucesivamente una solución de la sal de ácido trifluoroacético de 3’-N-Gly-NH-A (A-NH2 =DX-8951) obtenida en el Ejemplo 47 (57 mg) en N,N-dimetilformamida (3 ml), trietilamina (0,027 ml), y 1-etoxicarbonil-2-etoxi-1,2-dihidroxiquinolina (185 mg), y luego se dejó reaccionar la mezcla a temperatura ambiente durante la noche con agitación. Cada una de las porciones de 5 ml de esta mezcla de reacción se añadió por goteo a lotes de 10 ml de etanol. La mezcla se añadió con cloruro sódico acuoso 3 M (2,0 ml) y éter dietilo (25 ml), y los precipitados depositados se recogieron mediante centrifugación (3500 rpm, 8 minutos). Los pre29 ES 2 229 355 T3 5 cipitados se disolvieron en cloruro sódico acuoso 0,5 M y se ajustaron a pH 9 con hidróxido sódico acuoso 0,1 M bajo enfriamiento con hielo. La solución acuosa resultante fue desalada mediante ultrafiltración utilizando una membrana Biomax-50. La solución restante que no había pasado a través de la membrana se filtró a través de un filtro Millipore (0,22 µm) y luego fue liofilizada para obtener el compuesto del enunciado (290 mg). El contenido del residuo del compuesto medicamentoso en este compuesto fue 0,5% (w/w) cuando se determinó con base en la absorción a 362 nm en tampón Tris 0,1 M (pH 9,0). Ejemplo 52 10 15 Actividad antitumoral del complejo medicamentoso de la presente invención Se prepararon ratones portadores de tumor Meth A (6 ratones por grupo) de acuerdo con una forma similar a la del Ejemplo 11 y se examinó la actividad antitumoral del complejo medicamentoso del Ejemplo 15 mediante administración simple, de una manera similar a la del Ejemplo 12. Como resultado, el complejo medicamentoso del Ejemplo 15 presentaba una actividad antitumoral considerablemente mejorada y campo de dosificación efectiva más amplio en comparación con el compuesto medicamentoso del Ejemplo 12 aislado. 20 25 Ejemplo 53 30 35 Actividad antitumoral del complejo medicamentoso de la presente invención Se prepararon ratones desnudos portadores de tumor SC-6 (5 ratones por grupo) mediante trasplante subcutáneo de una porción de bloque de tumor gástrico humano SC-6 en las regiones inguinales derechas de ratones desnudos (BALB/c-nu/nu, machos). El 27º día después del trasplante, el complejo medicamentoso del Ejemplo 15 disuelto en agua destilada para inyecciones, se inyectó como una administración intravenosa simple y su actividad antitumoral fue comparada con la del compuesto medicamentoso aislado. Como resultado, el complejo medicamentoso del Ejemplo 15 presentó mayor actividad antitumoral en comparación con el compuesto medicamentoso aislado, mientras que no hubo muertes debidas a toxicidad. 40 45 50 Ejemplo 54 55 60 Actividad antitumoral del complejo medicamentoso de la presente invención Se prepararon ratones desnudos portadores de cáncer de pulmón humano QG-90 (5 ratones por grupo) según una forma similar a la del Ejemplo 53. El 16º día después del trasplante, el complejo medicamentoso del Ejemplo 15 disuelto en agua destilada para inyecciones, se inyectó como una administración intravenosa simple y su actividad antitumoral fue comparada con la del compuesto medicamentoso por sí mismo. Como resultado, el complejo medicamentoso del Ejemplo 15 presentó una actividad antitumoral considerablemente mejorada y un campo de dosificación efectiva más amplio en comparación con el compuesto medicamentoso aislado. 65 30 ES 2 229 355 T3 5 10 Ejemplo 55 Actividad antitumoral del complejo medicamentoso de la presente invención 15 20 Se prepararon ratones portadores de tumor Meth A (6 ratones por grupo) de acuerdo con una forma similar a la del Ejemplo 11, y se examinó la actividad antitumoral del complejo medicamentoso del Ejemplo 41 en los casos de administración simple de una manera similar a la del Ejemplo 12, y su actividad antitumoral fue comparada con la del compuesto medicamentoso aislado. Como resultado, el complejo medicamentoso del Ejemplo 41 presentó una actividad antitumoral considerablemente mejorada y un campo de dosificación efectiva más amplio en comparación con el compuesto medicamentoso aislado. 25 30 35 Ejemplo 56 Actividad antitumoral del complejo medicamentoso de la presente invención 40 45 Se prepararon ratones portadores de tumor Meth A (6 ratones por grupo) de acuerdo con una forma similar a la del Ejemplo 11, y se examinó la actividad antitumoral de acuerdo con un procedimiento similar al del Ejemplo 12 mediante administración simple de los complejos medicamentosos de los Ejemplos 29, 43 y 44, respectivamente. Como resultado, todos los complejos medicamentosos presentaron mayor actividad antitumoral y campo de dosificación efectiva más amplio. 50 55 60 65 31 ES 2 229 355 T3 Ejemplo 57 Farmacocinética del complejo medicamentoso de la presente invención 5 10 Se prepararon ratones portadores de tumor Meth A según una forma similar a la del Ejemplo 11. Se inyectó el complejo medicamentoso del Ejemplo 15 como administración simple de una manera similar a la del Ejemplo 12 (10 mg/kg: calculado como el compuesto medicamentoso), y se determinó el cambio de concentración del complejo medicamentoso en diversos tejidos. Como resultado, el complejo medicamentoso del Ejemplo 15 se mostró poseedor de un nivel de retención en sangre considerablemente más largo, mayor distribución en tejidos tumorales, y más alta selectividad tumoral en hígado e intestino delgado. Los resultados se representan en la Figura 20. Aplicación industrial 15 La reacción entre el derivado de polisacárido que presenta grupos carboxilo y el compuesto medicamentoso ligado con el espaciador o similar puede ser realizada con alto rendimiento, y cuando un compuesto medicamentoso que presenta un anillo lactona se somete a la reacción, pueden reducirse las reacciones colaterales y, por tanto, el procedimiento de la presente invención es completamente útil como procedimiento para preparar el complejo medicamentoso. 20 25 30 35 40 45 50 55 60 65 32 ES 2 229 355 T3 REIVINDICACIONES 5 10 15 1. Un procedimiento para la fabricación de un complejo medicamentoso, en el cual un derivado de polisacárido que presenta grupos carboxilo y un residuo de un compuesto medicamentoso están ligados entre sí mediante un espaciador que comprende un aminoácido o un espaciador que comprende 2 a 8 aminoácidos enlazados peptídicamente, o en el cual un derivado de polisacárido que presenta grupos carboxilo y un residuo de un compuesto medicamentoso están ligados entre sí sin el espaciador, caracterizado porque una sal de amina orgánica del derivado polisacárido que presenta grupos carboxilo se hace reaccionar con el compuesto medicamentoso o con el espaciador ligado al compuesto medicamentoso en un sistema no acuoso. 2. Un procedimiento para la fabricación de un complejo medicamentoso, en el cual un derivado de polisacárido que presenta grupos carboxilo y un residuo de un compuesto medicamentoso están ligados entre sí mediante un espaciador que comprende un aminoácido o un espaciador que comprende 2 a 8 aminoácidos enlazados peptídicamente, o en el cual un derivado de polisacárido que presenta grupos carboxilo y un residuo de un compuesto medicamentoso están ligados entre sí sin el espaciador, cuyo procedimiento comprende las etapas de: (1) convertir una sal de metal alcalino del derivado polisacárido que presenta grupos carboxilo en una sal de amina orgánica de dicho derivado, y 20 25 (2) hacer reaccionar la sal de amina orgánica con el compuesto medicamentoso o con el espaciador ligado al compuesto medicamentoso en un sistema no acuoso. 3. El procedimiento según la reivindicación 1 ó 2, en el cual el derivado polisacárido que presenta grupos carboxilo es un polialcohol carboxi(C1−4 )alquildextrano. 4. El procedimiento según la reivindicación 3, en el cual el polialcohol dextrano que constituye el polialcohol carboxi(C1−4 )alquildextrano es un polialcohol dextrano obtenido mediante tratamiento de un dextrano bajo condiciones que permiten una polialcoholización sustancialmente completa. 30 35 5. El procedimiento según la reivindicación 3 ó 4, en el cual el polialcohol carboxi(C1−4 )alquildextrano es un polialcohol carboximetildextrano. 6. El procedimiento según una cualquiera de las reivindicaciones 1 a 5, en el cual el compuesto medicamentoso es un agente antineoplásico o un agente anti-inflamatorio. 7. El procedimiento según una cualquiera de las reivindicaciones 1 a 5, en el cual el compuesto medicamentoso es un compuesto medicamentoso que puede formar un anillo lactona. 40 45 8. El procedimiento según una cualquiera de las reivindicaciones 1 a 7, en el cual un compuesto medicamentoso que presenta un anillo lactona formado o un espaciador ligado a un compuesto medicamentoso que presenta un anillo lactona formado, se utiliza para la reacción de la sal de amina orgánica con el compuesto medicamentoso o con el espaciador ligado al compuesto medicamentoso. 9. El procedimiento según la reivindicación 8, en el cual el compuesto medicamentoso que puede formar un anillo lactona descrito en la reivindicación 7 es (1S,9S)-1-amino-9-etil-5-fluoro-2,3-dihidro-9-hidroxi-4-metil-1H,12Hbenzo[de]pirano[3’,4’:6,7]indolizino[1,2-b]quinolina-10,13(9H,15H)-diona. 50 55 60 65 33 ES 2 229 355 T3 34 ES 2 229 355 T3 35 ES 2 229 355 T3 36 ES 2 229 355 T3 37 ES 2 229 355 T3 38 ES 2 229 355 T3 39 ES 2 229 355 T3 40 ES 2 229 355 T3 41 ES 2 229 355 T3 42 ES 2 229 355 T3 43 ES 2 229 355 T3 44