Estudio por imagen del desarrollo de tumores linforreticulares en la
Anuncio

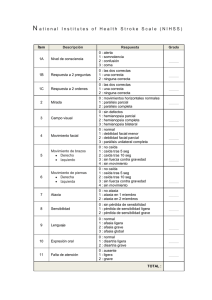
Documento descargado de http://www.elsevier.es el 01/12/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. COMUNICACIONES BREVES Estudio por imagen del desarrollo de tumores linforreticulares en la ataxia telangiectasia y el síndrome de Nijmegen María I. Martínez-León* • Luisa Ceres-Ruiz* • M.a Angeles Cuesta** • Francisco Jesús García-Martín*** * Servicio de Radiodiagnóstico, Radiología Pediátrica. **Servicio de Hematología, Hematología Pediátrica. ***Departamento de Pediatría. Unidad de Inmunodeficiencias. Hospital Materno-Infantil del C.H.U. Carlos Haya. Málaga. España. Imaging study of lymphoreticular tumor development in ataxiatelangiectasia and Nijmegen breakage syndrome La ataxia telangiectasia de Louis Barr (AT) es una enfermedad autosómica recesiva caracterizada por ataxia cerebelosa progresiva, telangiectasias oculocutáneas, inmunodeficiencia combinada con susceptibilidad a infecciones sinopulmonares y alta incidencia de desarrollo neoplásico. Ataxia-telangiectasia (AT), or Louis-Bar syndrome, is an autosomal recessive illness characterized by progressive cerebellar ataxia, oculocutaneous telangiectasias, immunodeficiency combined with susceptibility to sinopulmonary infections and high incidence of neoplastic development. El síndrome de Nijmegen (SNJ) es una variante de la AT, también autosómico recesivo, que presenta ataxia cerebelosa, inmunodeficiencia combinada y tendencia al desarrollo tumoral. A diferencia de la AT no muestra telangiectasias y exhibe un fenotipo característico (talla corta, cara «de pájaro» y microcefalia). Nijmegen breakage syndrome (NBS) is a variant of AT, is also an autosomal recessive illness that presents cerebellar ataxia, as well as combined immunodeficiency and a tendency toward tumor development. Contrary to Louis-Bar syndrome, it doesn’t present telangiectasia and exhibits a characteristic phenotype (short stature, bird-like face and microcephaly). Ambas entidades se incluyen dentro de la clasificación de los síndromes con fragilidad o inestabilidad cromosómica que agrupan además al síndrome de Bloom y la anemia de Fanconi. Todos ellos muestran un aumento en la frecuencia de presentación neoplásica, principalmente tumores del sistema linfoide. Both entities are classified as syndromes of chromosomal instability or chromosomal fragility, a group which also includes Bloom syndrome and Fanconi anemia. All of these show an increase in the frequency of neoplastic pathologies, mainly lymphoid tumors. Presentamos tres pacientes, dos con AT y uno con SNJ, que han desarrollado diferentes tipos de linfoma en el curso de la enfermedad, indicando los aspectos más relevantes desde el punto de vista clinicorradiológico. We present three patients, two with AT and one with NBS, who developed different lymphoma types in the course of the illness. We highlight the most outstanding aspects from a clinical-radiological point of view. Palabras clave: Ataxia telangiectasia. Síndrome de Nijmegen. Inestabilidad cromosómica. Fragilidad cromosómica. Sensibilidad a la radiación. Key words: Ataxia-telangiectasia. Nijmegen breakage syndrome. Chromosomal uncertainty. Chromosomal fragility. Radiation sensitivity. L — Telangiectasias oculocutáneas tardías. a ataxia telangiectasia (AT) es una enfermedad genética rara, multisistémica y autosómica recesiva. El gen se localiza en la región cromosómica 11q22-23. Sus características se resumen en: — Degeneración neuronal que desarrolla ataxia cerebelosa progresiva. — Inestabilidad genómica por defectos de reparación del ADN motivando sensibilidad aumentada a las radiaciones. — Inmunodeficiencia combinada con desarrollo de infecciones sinusales y pulmonares, recurrentes y graves. Martínez-León MI, Ceres-Ruiz L, Cuesta MA, et al. Estudio por imagen del desarrollo de tumores linforreticulares en la ataxia telangiectasia y el síndrome de Nijmegen. Radiologia 2003; 45(3):151-5. Correspondencia: MARÍA I. MARTÍNEZ-LEÓN. Avda. Pintor Sorolla, 16, 1o. 29016 Málaga. España. E-mail: [email protected] Recibido: 26-VIII-2002. Aceptado: 28-I-2003. 63 — Aumento del desarrollo de cáncer con especial predisposición a tumores linfáticos. En esta enfermedad es imprescindible evitar al máximo las radiaciones, dada la elevada radiosensibilidad que presenta, por lo que la ecografía tiene un papel primordial en el diagnóstico y seguimiento. Presentamos la evolución clínica y radiológica de dos pacientes con AT y uno con síndrome de Nijmegen (SNJ). Radiología 2003;45(3):151-5 151 Documento descargado de http://www.elsevier.es el 01/12/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. Martínez-León MI, et al. Estudio por imagen del desarrollo de tumores linforreticulares en la ataxia telangiectasia y el síndrome de Nijmegen Fig. 2.—Ecografía abdominal del paciente 1, en la que se aprecian adenopatías retroperitoneales, principalmente, y mesentéricas (no mostradas) observadas de novo en el control anual que sugirieron posible desarrollo neoplásico. Los padres rehusan tratamiento agresivo, por lo que se mantiene control evolutivo cercano sin quimioterapia. Caso 2 Fig. 1.—Radiografía de tórax anteroposterior del paciente 1, con presencia de numerosas bronquiectasias secundarias a infecciones pulmonares de repetición, típica manifestación pulmonar debida a la inmunodepresión de estos pacientes. Caso 1 Niña de 12 años y 3 meses de edad diagnosticada de AT. Presenta síndrome dismórfico compatible con síndrome de Goldenhar incompleto (displasia óculo-aurículo vertebral) del cual la paciente manifiesta anotia; inmunodeficiencia combinada con historia secundaria de frecuentes infecciones pulmonares que dan como resultado insuficiencia respiratoria crónica y numerosas bronquiectasias (fig. 1), telangiectasias oculares, habla disártrica, temblor intencional y de reposo e hipotrofia muscular que impide la deambulación. Además requiere gastrostomía percutánea por malnutrición grave. En la analítica destaca inmunodeficiencia humoral y celular, con aumento marcado de la alfa-fetoproteína. En el control ecográfico anual se encuentran por primera vez adenopatías de tamaño considerable en el retroperitoneo y los hilios hepático y esplénico (fig. 2). También presenta crecimiento adenopático en las cadenas linfáticas cervicales bilaterales. La biopsia de la adenopatía cervical de mayor tamaño permite el diagnóstico de proceso linfoproliferativo difuso de células pequeñas con alto índice proliferativo e inmunofenotipo polimorfo compatible con linfoma B inclasificado de células pequeñas. 152 Varón de ocho años de edad diagnosticado con un año y ocho meses de SNJ. Presenta antecedente de hermano fallecido tras trasplante de médula ósea realizado en el curso del tratamiento de una leucemia mieloide aguda M3 de FAB, asociada a síndrome de Bloom. Para el diagnóstico de nuestro paciente se confirma el fenotipo (microcefalia con «cara de pájaro», talla baja y retraso psicomotor leve), y el estudio cromosómico, que revela rotura de los cromosomas 7 p.13, 7 q.35, 14 q.11.3 en el 16% de las células estudiadas. Además, presenta trastornos de la inmunidad humoral y celular. Desarrolla proceso condensativo pulmonar que tras tres meses no remite a pesar del tratamiento. La radiografía de tórax mantiene cambios compatibles con aumento de densidad basal izquierda (figs. 3A y B). La ecografía muestra hepatización del LII y la língula con escaso broncograma y alveolograma. Es ecogénico, heterogéneo, sin necrosis ni cavitación en el interior. En el estudio Doppler se encuentra una vascularización prominente en el interior y la periferia de la masa con vasos que provienen del parénquima y de la circulación sistémica, preferentemente ramas diafragmáticas, aunque también se observan algunas ramas pequeñas procedentes de la aorta descendente. Todo ello indica neovascularización sospechosa de neoplasia (figs. 4A y B). En la tomografía computarizada (TC) de tórax sin contraste se observan lesiones similares. El lavado broncoalveolar es positivo al virus de Epstein Barr y en la fibrobroncoscopia se observa edema, friabilidad de la mucosa y obstrucción subsegmentaria en las ramas bronquiales inferiores izquierdas. Ante los hallazgos de imagen, la persistencia de la lesión y la base patológica del paciente, se decide biopsia mediante videotoracoscopia, terminando intraoperatoriamente en toracotomía. El resultado es linfoma no hodgkiniano de células B grandes. Se inicia tratamiento con quimioterapia y el control a los tres meses muestra excelentes resultados con marcada disminución del tamaño tumoral. Radiología 2003;45(3):151-5 64 Documento descargado de http://www.elsevier.es el 01/12/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. Martínez-León MI, et al. Estudio por imagen del desarrollo de tumores linforreticulares en la ataxia telangiectasia y el síndrome de Nijmegen A B Fig. 3A y B.–Radiografía de tórax anteroposterior del paciente 2 pasados tres meses de tratamiento antibiótico, con aumento de densidad basal izquierda y persistencia de la enfermedad en base, lo que sugirió una etiología pulmonar no infecciosa en el contexto de un paciente con SNJ. La biopsia indicó desarrollo tumoral (linfoma no hodgkiniano pulmonar). Caso 3 Paciente varón de siete años de edad, diagnosticado de AT desde los cuatro años y cinco meses. Desde el inicio de la deambulación presentó marcha inestable con caídas frecuentes, babeo continuo y dificultad para el lenguaje. Posteriormente desarrolla coreoatetosis y varios años después se hace evidente la presencia de telangiectasias conjuntivales. Tiene antecedente de otitis de repetición y procesos infecciosos respiratorios frecuentes. En la analítica destaca déficit de IgA y elevación de alfa-fetoproteína. En la resonancia magnética (RM) de cráneo existe atrofia cerebelosa moderada (fig. 5). En el control ecográfico anual se observa crecimiento del tamaño del bazo y una adenopatía de 1,5 cm en el hilio hepático. En el seguimiento a los tres meses continúa el crecimiento esplénico y se observan, mediante ecografía, adenopatías en el retroperitoneo además de las ya visualizadas previamente. La TC de abdomen confirma la esplenomegalia y las numerosas adenopatías de tamaño considerable, principalmente retroperitoneales altas y en el saco menor. La TC de tórax encuentra adenopatías de 1 cm y 1,5 cm en los espacios prevascular, supracarinal, paratraqueal derecho y en receso pleuroacigoesofágico. Además existen numerosos nódulos pequeños dispersos por ambos parénquimas pulmonares (figs. 6A y B). Se descarta enfermedad infecciosa pulmonar. Se desestima el acceso a las adenopatías mediastínicas por mediastinoscopia o fibrobroncoscopia, y también se descarta el abordaje de las adenopatías retroperitoneales y del saco menor mediante cirugía laparoscópica. Ante la necesidad de laparotomía abierta para el diagnóstico de desarrollo linfoproliferativo, los padres rehusan la intervención. En el control a los tres meses persisten las adenopatías mediastínicas, mesentéricas y retroperitoneales y ha aumentado el número de nódulos en el parénquima pulmonar así como la esplenomegalia. El aspirado de médula ósea es negativo. DISCUSIÓN La AT es una afectación neurodegenerativa que se define por la asociación de ataxia progresiva junto a otras alteraciones neu- A B Fig. 4A y B.—Ecografía del paciente 2 que muestra una condensación en LII y la língula con parénquima de ecogenicidad heterogénea, alveolograma, mínimo broncograma y pequeño derrame. El estudio Doppler del parénquima hepatizado manifiesta vascularización importante e irregular en e l 65 Radiología 2003;45(3):151-5 153 Documento descargado de http://www.elsevier.es el 01/12/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. Martínez-León MI, et al. Estudio por imagen del desarrollo de tumores linforreticulares en la ataxia telangiectasia y el síndrome de Nijmegen formar parte del grupo de síndromes caracterizados por fragilidad o inestabilidad cromosómica que incluyen la AT de Louis Barr, el síndrome de Nijmegen, la anemia de Fanconi y el síndrome de Bloom. Fig. 5.—Resonancia magnética de cráneo del paciente 3, plano sagital, secuencia T1, localización en la línea media, aumento de la profundidad de las folias cerebelosas con disminución del espesor del parénquima por atrofia, en un paciente con clínica de ataxia establecida. rológicas, inmunodeficiencia combinada y telangiectasias cutáneo-mucosas. Descrita en 1926 por Syllab y Henner1, difundida posteriormente en 1941 por Louis Barr2, es denominada también con el sinónimo de síndrome de Boder-Sedgwich, autores que la estudiaron en 19583. Es una genopatía rara, con una incidencia aproximada de 1/30.000-100.000 nacimientos4,5, multisistémica, con herencia autosómica recesiva. El gen de la AT es una mutación del gen que produce la cinasa ATM (ataxia telangiectasia mutada)6 y corresponde a una proteincinasa «señalante» que interviene en el control del ciclo celular, la recombinación del ADN, la apoptosis y otras respuestas celulares al daño del ADN4,5, provoca una mayor incidencia de rupturas y reordenamientos relacionados con los receptores de las células T, la inmunoglobulina de cadena pesada y el desarrollo de malignidad hematológica7. El locus del gen de la AT fue mapeado en 1998 en la región cromosómica 11q22-238. Entra a El cuadro clínico inicial corresponde a alteraciones del sistema nervioso con hipotonía muscular y retraso de las funciones motoras a partir de los dos años de edad, hasta que se establece en más del 85% de los pacientes la marcha atáxica aproximadamente a los cuatro años. Es una ataxia cerebelosa progresiva, estática pero acentuada con el movimiento, a la que se van sumando con la edad temblor intencional, disinergia, coreoatetosis, mioclonías y disminución del cociente intelectual pasados los diez años. Entre los cuatro y ocho años desarrollan las telangiectasisas en escleróticas y piel, y se les denomina «tardías» por aparecer después de la ataxia. Por el cuadro de inmunodepresión combinada, humoral y celular, las infecciones sinopulmonares son muy habituales, repetidas y graves. Entre las manifestaciones clínicas adicionales se incluyen retraso del crecimiento, hipogonadismo, retraso puberal y diabetes no cetósica resistente a insulina5. Además de las infecciones y el deterioro neuropsíquico progresivo hay que destacar la especial predisposición al desarrollo de tumores malignos, que aparecen en más de la tercera parte de los pacientes. El 80% son leucemias y linfomas no hodgkinianos4,6,9-12, les siguen otros tumores sólidos del tracto gastrointestinal, especialmente adenocarcinomas, y por último, linfoma Hodgkin6. Actualmente los procesos malignos son la causa más frecuente de mortalidad en estos pacientes, sobre todo desde el uso extendido de antibióticos e inmunoterapia para el control de las infecciones7. Debido a la reparación defectuosa del ADN las células de estos pacientes son altamente sensibles a las radiaciones ionizantes, observándose también inestabilidad cromosómica inducida por fármacos como la bleomicina7. La variabilidad del cuadro clínico y la tardanza en la presentación de algunas de las manifestaciones características, hacen difícil el diagnóstico antes de los cuatro años13. Clínicamente se puede confundir con otras enfermedades como parálisis cerebral atáxica o discinética, ataxia-apraxia oculomotora, enfermedad de A B Fig. 6A y B.—Tomografía computarizada de tórax sin contraste del paciente 3, ventana de mediastino, adenopatías cercanas a 1 cm de diámetro en localización prevascular, supracarinal y en el receso pleuroacigoesofágico (las mostradas). En la ventana de parénquima se observan numerosos nódulos de pequeño tamaño dispersos por ambos pulmones. Estos dos hallazgos, junto a adenopatías abdominales y esplenomegalia, sugieren desarrollo de enfermedad linforreticular en el marco de un paciente con AT. 154 Radiología 2003;45(3):151-5 66 Documento descargado de http://www.elsevier.es el 01/12/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. Martínez-León MI, et al. Estudio por imagen del desarrollo de tumores linforreticulares en la ataxia telangiectasia y el síndrome de Nijmegen Friedreich, algunas enfermedades metabólicas, degeneración espinocerebelosa y otras4. Algunos signos radiológicos diagnósticos en la AT son: atrofia cerebelosa difusa de predominio en vermis en el SNC; ausencia de sombra tímica en la radiografía de tórax; escasez de tejido linfoide, por ejemplo en nasofaringe, tan habitual en los niños normales; desarrollo de bronquiectasias y cambios parenquimatosos crónicos debidos a enfermedad infecciosa pulmonar recurrente; cambios sinusales crónicos por las infecciones de repetición, y alerta ante el desarrollo de adenopatías, esplenomegalia e infecciones con manifestaciones inhabituales, pues pueden ser signos indicativos de desarrollo neoplásico14,15. El diagnóstico de la enfermedad se confirma con estudios inmunológicos y citogenéticos7. Con frecuencia se observan concentraciones séricas de alfa-fetoproteína (AFP) y de antígeno carcinoembrionario altas4,5,7. Es importante considerar el diagnóstico de AT en cualquier niño con enfermedad linforreticular maligna a una edad menor a la esperada, con afectación tumoral atípica, o con una ataxia preexistente, para anticipar la grave toxicidad de algunos antineoplásicos usados en el tratamiento (especialmente bleomicina), para evitar la confusión entre el desarrollo de ataxia por el cuadro en sí mismo o por la toxicidad secundaria a la terapia, para modificar los protocolos de radiación y con el fin de realizar un consejo genético6,7,13. Se han descrito casos aislados en los que la neoplasia linfoide puede ser el signo inicial de presentación de la AT16. Por otro lado, en un paciente conocido con AT, el radiólogo y el pediatra deben estar alertas en el control del desarrollo tumoral, pues a veces el diagnóstico de la presencia de neoplasias se retrasa por la confusión entre las complicaciones infecciosas y los tumores6. En conclusión, la importancia del diagnóstico temprano se basa en poder realizar un consejo genético y el control y tratamiento adecuados de los procesos malignos y las infecciones7. El tratamiento no es curativo y se limita a los cuidados paliativos. El trasplante de médula ósea, terapia utilizada en gran parte de las inmunodeficiencias primarias pediátricas, no es efectivo en estos pacientes14. No se debe abusar de las radiaciones en el diagnóstico y seguimiento del paciente ya que pueden contribuir a las lesiones de radiodermitis y a la aparición de neoplasias. Por ello, la ecografía representa un papel tan importante para el diagnóstico y seguimiento, al ser una técnica carente de radiación con la que pueden controlarse periódicamente las manifestaciones ganglionares cervicales y abdominales de estos pacientes predispuestos al desarrollo de neoplasias sanguíneas. El síndrome de rotura de Nijmegen (SNJ) al igual que la AT, pertenece al grupo de cuadros con inestabilidad cromosómica, y es también una enfermedad autosómica recesiva rara. El defecto genético del SNJ se localiza en el cromosoma 8q21 y ha sido clonado recientemente; el producto de este gen, la nibrina, es una proteína similar a ATM interviniendo en la reparación del ADN y el control del ciclo celular5,17. Muestra un fenotipo propio que lo caracteriza y diferencia de la AT, con presencia de microcefalia, «cara de pájaro» y talla corta; no hay telangiectasias pero sí ataxia progresiva y mayores concentraciones de alfa-fetoproteína que en la AT5. Como la AT, se asocia a alteraciones inmunitarias celulares y humorales (alteración del número y función de los linfocitos T y déficit de inmunoglobulinas IgG e IgA) y existe aumento de la sensibilidad a las radiaciones y predisposición a padecer procesos linfoproliferativos malignos10. 67 Las técnicas de imagen para el diagnóstico y seguimento deben ajustarse al patrón de radiosensibilidad de estos cuadros, entre ellas la ecografía y la RM son las que representan un papel primordial. Signos como esplenomegalia, presencia de adenopatías que aumentan en número y tamaño en diferentes localizaciones, y patrón nodular pulmonar, son datos que en nuestros pacientes hicieron sospechar el desarrollo de enfermedad linforreticular en el contexto de enfermedades que presentan predisposición al desarrollo de tumores hematológicos, y que se manifestaron antes de que otras pruebas de laboratorio confirmaran la enfermedad neoplásica. De este modo se programa el seguimiento terapéutico con la necesidad intrínseca de modificar la quimioterapia y los protocolos de radiación, y se mantiene el control radiológico cercano con pruebas no radiantes. BIBLIOGRAFÍA 1. Syllaba L, Henner K. Contribution a l’independance de l’athetose double idiopathique et congenitale. Rev Neurol (Paris) 1926;1: 541-62. 2. Louis Bar D. Sur un syndrome progressif comprenant des telangiectasies capillaries cutanees et conjonctivales symetriques a disposition naevoide et de troubler cerebelleux. Confin Neurol (Basel) 1941;4: 32-42. 3. Boder E, Sedgwick RP. Ataxia telangiectasis. A familial síndrome of progressive cerebelar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics 1958;21:526-54. 4. Cruz M, Bosch J. Síndromes con malformación del SNC: ataxia-telangiectasia (Louis-Bar) En: Atlas de síndromes pediátricos. Barcelona: Espaxs S.A., 1998; p. 578-9. 5. Elder ME. Inmunodeficiencias de células T. Clin Ped North Am 2000;6:1295-1306. 6. Murphy KC, Berdon E, Ruzal-Shapiro, et al. Malignancies in pediatric patients with ataxia telangiectasia. Pediatr Radiol 1999;29: 225-30. 7. Valbuena O, Póo P, Campistol J, et al. Ataxia telangiectasia: revisión de 13 observaciones. Rev Neurol (Barc) 1996;24(125):77-80. 8. Gatti RA. Ataxia Telangiectasia. Dermatol Clin 1995;13(1):1-6. 9. Boultwood J. Ataxia telangiectasia gene mutations in leukaemia and lymphoma. J Clin Pathol 2001,54:512-6. 10. Seidemann K, Henze G, Beck JD, et al. Non-Hokgkin’s lymhoma in pediatric patients with chromosomal breakage syndromes (AT and NBS): Experience grom the BFM trials. Annals of Oncology 2000;11(suppl 1):S141-S5. 11. Taylor AMR, Metcalfe JA, Thick J, Mak YF. Leukemia and lymphoma in ataxia telangiectasia. Blood 1996;87(2):423-38. 12. Schulte U, Wegner RD, Baumgarten E, et al. Low-grade non-Hodgkin’s lymphoma after high-grade non-Hodgkin’s lymphoma in a child with ataxia telangiectasia. Cancer 1994;73:1522-5. 13. Cabana, MD, Crawford TO, Winkelstein JA, et al. Consequences of the delayed diagnosis of ataxia-telangiectasia. Pediatrics 1998;102 (1):98-100. 14. Yin EZ, Frush DP, Donnelly LF, Buckley RH. Primary immunodeficiency disorders in pediatric patients: clinical features and imaging findings. AJR 2001;176:1541-52. 15. Farina L, Uggetti C, Ottolini A, et al. Ataxia telangiectasia: MR and CT findings. J Comput Assist Tomogr 1994;18(5):724-7. 16. Loeb DM, Lederman HM, Winkelstein JA. Lymphoid malignancy as a presenting sign of ataxia-telangiectasia. J Pediatr Hematol Oncol 2000;22(5):464-7. 17. The International Nijmegen Breakaje Syndrome Study Group. Nijmegen Breakage Syndrome. Arch Dis Child 2000;82(5):400-6. Radiología 2003;45(3):151-5 155