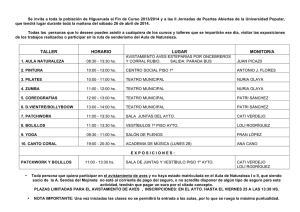
Evaluación comparativa de herramientas de análisis de número de
Anuncio

Evaluación comparativa de herramientas de análisis de número de copia a partir de datos NGS Estudiante: Felipe Were Eduardo MÁSTER EN BIOINFORMÁTICA Y BIOLOGÍA COMPUTACIONAL ESCUELA NACIONAL DE SALUD- INSTITUTO DE SALUD CARLOS III 2013-2015 Centro Nacional de Investigaciones Oncológicas (CNIO) Unidad de Bioinformática Directores: David G. Pisano y Gonzalo Gómez Tutores: David G. Pisano y Gonzalo Gómez Fecha: 15/09/2014 AGRADECIMIENTOS: Me gustaría agradecer a las siguientes personas, sin las cuales no habría sido posible la realización de estas prácticas: En primer lugar a Gonzalo Gómez y a David Pisano por ofrecerme la posibilidad de hacer las prácticas del máster en su laboratorio y por su asesoramiento durante todo el proceso. Agradecerle a Ángel Carro por toda su ayuda con el clúster de Ahsoka, incluida la instalación de muchos de los programas que he utilizado. A Miriam Rubio su ayuda en la primera etapa del Máster, principalmente con el pipeline de RuBioSeq. A Fátima Al-Sharour y a Elena Piñeiro por haberme proporcionado los “datasets reales”, fundamentales para la realización del proyecto. A Federico Abascal y a Enrique Carrillo por todos los consejos útiles que me han proporcionado a lo largo de estos meses de prácticas. A Juan y a José Manuel por su ayuda en la parte informática. A Jon y a Fernando, a quienes he acribillado a preguntas. Y en general a todo el equipo que trabaja al final del ala oeste de la planta 0 del CNIO. OBJETIVOS: – Revisión bibliográfica de los métodos de análisis de variaciones de número de copia sobre datos NGS disponibles hasta la fecha. – Selección de un subconjunto manejable de entre los métodos encontrados e implementación en el laboratorio. – Evaluación de la sensibilidad, especificidad y precisión en la asignación de los números de copia de los métodos seleccionados utilizando datos sintéticos y reales. – Visualización mediante circos.plot de los resultados de la ejecución de los métodos seleccionados sobre datos reales. INTRODUCCION : Variaciones de número de copia, definición y clasificación: Las variaciones de número de copia (en adelante, CNVs, “Copy Number Variations”) se definen como deleciones o amplificaciones de segmentos del genoma con un tamaño mínimo de entre 50bp y 1kb (Alkan et al., 2011 , Banerjee et al., 2011; Stranger et al., 2007; Feuk et al., 2006). Forman parte del grupo de mutaciones denominado variaciones estructurales (SV), que incluye también trasposiciones e inversiones. El tamaño mínimo de segmento que define una CNVs no está consensuado del todo, pues disminuye a medida que se va incrementando la resolución de los métodos de detección de CNVs (Liu et al., 2013). Las CNVs se pueden clasificar de distintas formas según el criterio utilizado: -Según origen: Germinales (CNVs propiamente dichas). Se originan en la línea germinal y por tanto se transmiten a la descendencia. Somáticas (SCNA, Somatic Copy Number Alterations). Se originan en células somáticas. Son particularmente frecuentes en tumores. -Según tamaño (Brosens et al., 2010; Koboldt et al., 2012): Grandes, (”Broad”): Afectan a más del 25% del tamaño de uno de los brazos de un cromosoma. Focales: Más pequeñas, típicamente de tamaño menor a 5 Mb -Según el tipo de modificación: Las variaciones de número de copia se definen por comparación con el número de copia normal de 2 de los autosomas de un genoma diploide y se clasifican en: a) Pérdidas Deleciones Heterocigotas (se pierde una de las dos copias) Deleciones Homocigotas (se pierden las dos copias) b)Ganancias Amplificaciones (ganancias de 1 o 2 copias) Grandes Amplificaciones (ganancias de más de 2 copias) Importancia de los CNVs: Tanto si afectan directamente a regiones codificantes como si afectan a regiones reguladoras, los CNVs pueden cambiar los niveles de expresión génica. Gran número de CNVs se han relacionado o implicado directamente en enfermedades, con una gran incidencia en trastornos neuropsiquiátricos y en cáncer. -CNVs en cáncer: Una de las características más comunes de los procesos tumorales es la presencia de reordenaciones estructurales en el genoma de las células tumorales. Las alteraciones somáticas de número de copia (SCNAs) son ubicuas en células tumorales (Futreal et al., 2004; Negrini et al., 2010; Kim et al., 2013, Albertson et al., 2003; Diskin et al., 2009; Shlien et al, 2009; Beroukhim et al., 2010; Frank et al., 2007) y pueden afectar de forma crítica los patrones de expresión de determinados genes (Stratton et al., 2009; Zack et al., 2013). Una prueba de la importancia de los SCNAs en cáncer es la existencia de patrones de SCNA que permiten diferenciar entre tipos de cáncer (Stratton et al., 2009;, Beroukhim et al) y analizar la progresión del cáncer y su complejidad (Liu et al., 2013). Los análisis de SCNA pueden llevar a la identificación de genes “directores” del cáncer (Louhimo et al ) y pueden incluso ayudar a establecer criterios sobre el tratamiento (Curtis et al, Dancey et al). Actualmente, la detección de CNVs se ha convertido en una parte esencial del análisis de los genomas de pacientes con cáncer. -CNVs en trastornos del sistema nervioso Se han relacionado diversas CNVs con la susceptibilidad de desarrollar diversos trastornos del sistema nervioso (Merikangas et al), como la enfermedad de ALzheimer (Rovelet-Lecruz et al,), la enfermedad de Parkinson (Ibanez et al, Singleton et al), la epilepsia (Helbig et al) y diversos trastornos psiquiátricos, incluyendo la esquizofrenia (Xu et al, Stefansson et al), el retraso mental (McMullan et al, Edelmann et al, Bijlsma et al), el autismo (Weiss et al, Szatmari et al, Paterson et al, Zwaigenbaum et al, Marshall et al, Kumar et al) y el trastorno depresivo (major depressive disorder, Glessner et al). Mediante la generación de mapas de morbilidad de CNVs, un estudio reciente ha logrado identificar 10 genes presuntamente implicados en el desarrollo de trastornos neurocognitivos y del desarrollo (Coe et al). Además del cáncer y los trastornos del sistema nervioso, se han implicado CNVs en enfermedades autoinmunes (McKinney et al, Yang et al, Fellermann et al, Hollox et al) y en la susceptibilidad a la infección por HIV-1 (Gonzalez et al). Tecnologías aplicadas al estudio de CNVs: La necesidad de establecer una identificación precisa de los eventos de modificación de número de copia ha llevado al desarrollo de numerosas técnicas para el análisis de CNVs. Tradicionalmente se utilizaban técnicas citogenéticas para la identificación de CNVs, como por ejemplo la hibridación de fluorescencia “in situ” (FISH)(Speicher et al, Schaaf et al). Posteriormente, y hasta la llegada de las tecnologías de NGS, las plataformas más utilizadas para el análisis de CNVs eran los arrays de hibridación genómica comparada (aCGH, Pinkel et al ) y los SNParrays (Bignell et al). En la actualidad, se está produciendo una gran proliferación de herramientas de análisis de número de copia basadas en datos NGS. El análisis de las variaciones de número de copia mediante NGS presenta numerosas ventajas potenciales frente al análisis mediante arrays (Klambauer et al): 1) La estimación del número de copia a partir de datos NGS es más precisa para números de copia grandes, ya que la profundidad de lectura escala de forma aproximadamente lineal con el número de copia (Alkan et al., 2011). 2) Los breakpoints de los segmentos de número de copia determinado pueden ser estimados con mayor precisión, ya que no dependen de sondas predefinidas. 3) Se pueden estimar números de copia correspondientes a alelos específicos, mientras que las técnicas basadas en arrays están restringidas a alelos predefinidos. Esto puede ser interesante para la identificación de mutaciones implicadas en el desarrollo de cáncer (Stratton et al., 2009;) Estrategias de análisis de CNVs a partir de datos NGS: Se han descrito 4 métodos para la detección de CNVs a partir de datos NGS, resumidas en la fig1: a) Pair-End Mapping o “mapeo de lecturas pareadas” (PEM). Este método requiere del uso de lecturas pareadas (”paired-end”) y se basa en que los fragmentos de DNA secuenciados mediante NGS tienen un tamaño que sigue una determinada distribución (Chen et al., 2010; Hormozdiari et al., 2009; Xi et al. (b), 2011). De manera que si las lecturas secuenciadas, que corresponden a los extremos de estos fragmentos, mapean a la referencia a una distancia mayor de la esperada, significa que hubo una deleción en el genoma estudiado. Por el contrario, si la distancia entre las lecturas es más pequeña que la esperada, se entiende que se ha producido una inserción en el genoma estudiado, por ejemplo una duplicación. b) Split Read o “lecturas interrumpidas” (SR). Este método también requiere del uso de lecturas pareadas (”paired-end”) y se basa la idea de que una lectura no mapeada en el genoma de referencia puede estar localizando un extremo o “breakpoint” de un CNVs (Ye et al., 2009). c) Assembly-Based o “métodos basados en ensamblaje” (AS). Este método, que se basa en el ensamblaje “de novo” del genoma, se utiliza de forma casi exclusiva en genomas bacterianos, ya que da problemas con genomas grandes como los humanos (Ye et al., 2009), asi que no nos vamos a extender más sobre él. d) Depth Of Coverage o “Profundidad de lectura” (DOC). Este método asume una correlación lineal entre la profundidad de lectura y el número de copia (Abyzov et al., 2011, Yoon et al., 2009). Una profundidad menor a la esperada representará una pérdida o deleción y una profundidad mayor que la esperada representará una ganancia o amplificación. En realidad esta relación no es tan directa porque el proceso de secuenciación no es uniforme, sino que existen distintos biases como los asociados a contenido GC y la mapeabilidad del genoma. Sin embargo, se han desarrollado mecanismos para corregir algunos de estos biases de forma eficiente (ver apartado “Importancia del pre-procesamiento de los datos”). Además, los métodos basados en DOC permiten la asignación precisa de número de copia, mientras que los anteriores solo permiten la asignación de breakpoints. Por todo lo anterior, los métodos basados en DOC son los más utilizados en la actualidad en los estudios de CNVs (Zhao et al., 2013). La profundidad de lectura suele ser procesada para obtener la LRR (Log of Read depth/count Ratio), calculada como el logaritmo en base dos de las lecturas encontradas, relativas a algún valor utilizado como referencia (Liu et al., 2013). Las herramientas de análisis de CNV que usan datos provenientes de un par de muestras normal/tumor, buscan identificar aquellos intervalos para los que el (log) del número de lecturas (corregidas) entre la muestra tumoral y la muestra normal se desvía significativamente de 1. -Análisis de exomas vs análisis de genoma completo: Una de las primeras características distintivas de los algoritmos de detección de alteraciones de número de copia basada en datos de NGS es el tipo de secuenciación utilizado: secuenciación masiva de exomas (WES) o de genomas completos (WGS). El análisis de CNVs a partir de datos procedentes de WGS presenta numerosas ventajas frente al de datos procedentes de WES: a) Los procesos de alteración de número copia se pueden dar en cualquier parte del genoma y no solo en las regiones exónicas. Además, las alteraciones en regiones no exónicas pueden tener efectos fundamentales en la expresión génica, caso ocurran en regiones promotoras o reguladoras de los genes, o en regiones implicadas en la regulación de la estructura de la cromatina. b) Se ha hecho una evaluación comparada de algoritmos de análisis de número de copia con datos de WGS y WES, encontrándose que las herramientas que usan datos de WGS tienen un rendimiento muy superior al de las herramientas que usan datos WES (Alkodsi et al, 2014). Esto se debe en gran parte a que los algoritmos WES presentan lo que se ha denominado como el “bias exónico”, derivado de la distribución no uniforme de los exones a lo largo del genoma. Esta es una limitación tecnológica, que no puede ser solucionada mediante algoritmos. Además, la identificación de los breakpoints está sujeta a un error derivado de las distancias inter-exónicas, error que puede ser muy grande (Alkodsi et al., 2014). c) El abaratamiento de los costes de la secuenciación masiva hará con que cada vez predomine más la secuenciación de genomas completos, frente a la secuenciación limitada a exomas. Esquema del funcionamiento de una herramienta de análisis CNVs: En la fig 2 se resume el mecanismo general de funcionamiento de las herramientas de análisis de CNVs , en la que se pueden identificar los siguientes pasos: a) Recogida de los datos. La profundidad de lecturas a lo largo del genoma (RC o RD), se suele obtener mediante la división del genoma completo en ventanas no solapantes de un tamaño determinado, en las que se cuenta el número de lecturas. Los datos de profundidad de lectura se pueden complementar con datos de frecuencia de alelo B (ver descripción más adelante en el texto) y, en ocasiones, alguna otra información adicional, como la de lecturas discordantes (lecturas paired-end que alinean al genoma de referencia de forma anómala (ver sección métodos de análisis de CNVs, apartado de métodos basados en PEM). b) Preprocesamiento de los datos crudos y, cuando posible, normalización (mediante la utilización de una muestra normal). El preprocesamiento permite corregir problemas técnicos o biológicos, por ejemplo mediante el filtrado de lecturas de baja calidad y la corrección del bias por contenido GC (ver sección “importancia del pre-procesamiento de los datos”) c) Segmentación: Se particiona la señal generada en segmentos. Estos segmentos se generan mediante la agrupación de ventanas consecutivas que presentan números de copia suficientemente similares como para asumir que son idénticos. Existen diversos métodos de segmentación, como por ejemplo el método de segmentación circular binario (CBS, Olshen et al., 2004; Popova et al., 2009; Olshen et al (b), 2011), métodos basados en modelos ocultos de Markov (HMM, Yau et al., 2010; Sun et al., 2009) y métodos basados en criterios de información Bayesiana (BIC, Xi et al (a), 2011), entre otros. d) Interpretación: Se interpretan los datos procedentes del proceso de segmentación, a partir de los cuales se asignan números de copia o estados a los segmentos identificados. Este paso no es necesario en los programas basados en HMM, ya que éstos procesan de forma simultánea la clasificación de ventanas en estados y la agrupación de ventanas consecutivas en segmentos. Además de la asignación de números de copia o de estados, algunos algoritmos diseñados para el análisis de muestras tumorales pueden llegar a identificar el nivel de pureza, el grado de poliploidia y la heterogeneidad tumoral de dichas muestras. e) Output: Presentación de los resultados obtenidos. Importancia del pre-procesamiento de los datos: Corrección por contenido GC y por “mapeabilidad”: Como ya se ha mencionado, el proceso de secuenciación no es uniforme a lo largo del genoma, sino que presenta “biases”. Los dos “biases” más estudiados son la mappbilidad y el contenido GC (Liu et al., 2013). -mapeabilidad: La mapeabilidad de una región de un genoma de referencia se define como la probabilidad de que una lectura procedente de esa región se vuelva a mapear sobre ella sin ambigüedades (Teo et al., 2012). Algunas herramientas de análisis de CNVs disponen de programas que calculan los valores de mapeabilidad a lo largo de un genoma, y los utilizan para implementar una corrección asociada a la mapeabilidad (Lai et al., 2012). -Corrección por contenido GC La profundidad de lectura suele presentar una distribución unimodal respecto de su contenido en nucleótidos GC, que es independiente del tamaño de la región o ventana considerada y de la profundidad media de lecturas (Abyzov et al., 2011; Benjamini et al., 2012; Yoon et al., 2009). Regiones con un contenido GC medio (40 a 55% GC) tienen una profundidad de lectura media mayor que las de contenido GC más alto o más bajo. Al igual que las correcciones de mapeabilidad, algunas herramientas de análisis de CNVs han inlcuido en su software algoritmos de corrección del bias asociado al contenido GC (Teo et al., 2012; Yoon et al., 2009) Importancia de la inclusión de un control normal (“matched normal”) cuando se analizan muestras procedentes de tumores: El uso de un control normal diploide permite asegurar que cualquier artefacto que aparezca simultáneamente en tumor y normal, como por ejemplo biases específicos de la plataforma de secuenciación utilizada, regiones no secuenciables, etc, sean corregidas o eliminadas de forma eficiente ( Xi R (a), 2011). Se facilita también la corrección de los efectos de bias que provienen del contenido GC o de mapeabilidad del genoma, ya que se pueden comparar directamente las profundidades de lectura de tumor y normal. La presencia de matched normal también permite la identificación de locus de SNPs heterocigotos para calcular el valor BAF o de imbalance alélico (ver apartado “información sobre frecuencia del alelo B” (BAF)) y filtrar los CNVs benignos del paciente (Liu et al., 2013) Problemas asociados al análisis de muestras tumorales: El análisis de CNVs en tumores presenta sus propias complicaciones, derivadas de la presencia de contaminación en las muestras y de anomalias genómicas difíciles de identificar. Es frecuente encontrar que las muestras de tejidos tumorales presentan infiltraciones de estroma normal (notumoral), lo que resulta en una inevitable contaminación con DNA normal y la dilución de las señales correspondientes a aberraciones somáticas (Boeva et al., 2011, 2012; Gusnanto et al., 2012; Ha et al., 2012; Mayrhofer et al., 2013). La presencia de impurezas en muestras tumorales puede alterar significativamente los datos WGS, particularmente cuando las células normales dominan sobe las tumorales. Sin embargo, muy pocas herramientas de análisis de CNVs tienen la capacidad de abordar este problema. Entre los que sí lo hacen se incluyen FREEC (Boeva et al., 2011, 2012), APOLLOH (Ha et al., 2012), CLImAT (Yu et al., 2014) y Patchwork (Mayrhofer et al., 2013). Un problema similar surge cuando se utilizan modelos de xenotransplantes en ratón, es decir, transplantes de muestras de tumores humanos en ratones. En estos casos el problema deriva de la contaminación de las muestras tumorales con tejido de ratón (Huynh et al., 2011). WaveCNVs es una herramienta de análisis de número de copia de genomas secuenciados mediante NGS que se ha desarrollado para el análisis de muestras de tumores incluyendo modelos de xenotransplantes (Holt et al). Además de la presencia de impurezas en los tumores, éstos presentan con frecuencia fenómenos de aneuploidia, derivados de las anomalias estructurales y numéricas que con frecuencia se presentan en los cromosomas de genomas tumorales (Carter et al., 2012). La interpretación de los datos procedentes de NGS se hace especialmente complicada en muestras tumorales con presencia de impurezas y de aneuploidia (Oesper et al., 2013). La herramientas Patchwork y CLImAT, de análisis de número de copia en tumores, toman en consideración tanto la presencia de impurezas como el fenómeno de aneuploidia (Mayrhofer et al., 2013, Yu et al., 2014). A todos estos problemas técnicos hay que añadir el hecho de que los propios tumores pueden ser internamente heterogéneos, subdividiéndose en subclones con propiedades diferentes (Liu et al., 2013). Información sobre frecuencia del alelo B (BAF) o “imbalance” alélico: El análisis de las alteraciones de número de copia basado exclusivamente en la profundidad de lectura puede presentar problemas, debido a los diversos biases que pueden presentar los datos, a las características intrínsecas de las muestras analizadas y a variaciones experimentales (Liu et al., 2013). Muchos algoritmos añaden la información de frecuencia del alelo B “BAF” o “imbalance” alélico, basada en la fracción alélica presente en el locus de cada SNP. Si llamamos 'alelo A' al alelo que se corresponde con el mismo nucleótido que se encuentra en el genoma de referencia, y 'alelo B' al que presenta un nucleótido diferente, el valor de “imbalance” alélico se calcula como b/(a+b). En un genoma normal diploide, los valores de “imbalance” alélico correspondientes a los genotipos AA, AB, BA y BB son 0, 0.5, 0.5 y 1 respectivamente. Como se puede ver, solo se consideran las proporciones alélicas, de forma que los genotipos AB y BA son indistinguibles. Si un evento CNV altera el número de copias, el valor de “imbalance” alélico puede variar, dependiendo del número de copias. Por ejemplo, si hay m copias del homólogo 1 y n copias del homólogo 2 en una región de un genoma tumoral, el valor de BAF puede ser alguno de los siguientes, 0, m/(m+n), n/(m+n) y 1. Diversos factores, como la contaminación de muestras tumorales por células normales, puede alterar estos valores teóricos. El análisis de estas variaciones sobre el valor teórico puede aportar información sobre el grado de contaminación de una muestra tumoral con células normales (Liu et al., 2013, Boeva et al 2012). La ploidia también se puede revelar mediante información procedente de BAF. Por ejemplo, la diploidia admite las posibilidades 0, 0.5 y 1, y la tetraploidia añade las posibilidades 0.25 y 0.75. Principales características de los algoritmos Patchwork y HMMcopy: En el presente trabajo se ha hecho una recopilación de las herramientas de análisis de CNVs descritas en la literatura hasta la fecha. Se seleccionó un subconjunto para su implementación en el laboratorio. Dos de ellas, “BIC-seq” y “seqCNA”, dieron diversos errores durante el proceso de implementación y ejecución, por lo que tuvieron que ser descartadas. Las otras dos herramientas seleccionadas, “Patchwork” y “HMMcopy”, fueron implementadas con éxito, y a continuación se procedió a la evaluación de su capacidad de detección de CNVs, con datos simulados y reales. HMMcopy HMMcopy es una colección de herramientas para la detección de CNVs o SCNAs a partir de datos de secuenciación de genoma completo (WGS)(Ha et al., 2012; Dempster et al., 1977; Lai et al., 2012). El algoritmo empieza computando el número de lecturas en muestras tumoral y normal, utilizando ventanas con un tamaño fijo, que puede ser especificado por el usuario. Sobre esas mismas ventanas de tumor y control, HMMcopy obtiene un perfil de contenido GC y mapeabilidad. Tras filtrar las ventanas con contenido GC extremo y las de baja mapeabilidad, el algoritmo normaliza el contenido GC y la mapeabilidad de las muestras tumor y normal por separado. Finalmente, se normalizan las lecturas de tumor frente a las normales y empieza el proceso de segmentación. El proceso de segmentación usa un modelo oculto de Markov (HMM) de 6 estados donde en el primer paso se estiman los parámetros óptimos de segmentación mediante un algoritmo EM (”Expectation-Maximization”) y en el segundo paso ejecuta la segmentación propiamente dicha mediante el algoritmo Viterbi (Forney et al., 1973), el cual asigna uno de seis posibles estados de número de copia a cada segmento (0, 1, 2, 3, 4 y 5 o más copias, para los estados 1 a 6, respectivamente). En resumen: -Input de HMMcopy: Ficheros BAM de lecturas Tumor/Normal y fichero fasta del genoma utilizado para el alineamiento. -Parámetros de HMMcopy: a) Tamaño de ventana con la que dividir el genoma (default: 1 kb). b) Parámetros de segmentación: Los Parámetros de segmentación se dividen en 2 categorias: • Parámetros iniciales: e, mu, lambda, un, kappa • Parámetros de flexibilidad: strength, m, eta, gamma, S Los parámetros iniciales fijan los parámetros de partida para el algoritmo de optimización y los parámetros de flexibilidad definen el grado de variación que se admite sobre los parámetros iniciales durante el proceso de optimización. La modulación de estos parámetros por el usuario permite controlar tanto el proceso de segmentación como el de asignación de número de copia a los segmentos generados. Todos los parámetros de HMMcopy tienen un valor asignado por defecto. -Output de HMMcopy: HMMcopy genera un fichero excel que entre su información incluye las coordenadas de los segmentos no solapantes identificados, el cromosoma al que pertenecen y el estado asignado a cada segmento. Patchwork: La herramienta Patchwork ha sido diseñada para el análisis de variaciones de número de copia en tejido tumoral. Su principal característica es la incorporación de información sobre el “imbalance” alélico o BAF, que complementa la información basada en profundidad de lectura. En la fig 3 se representa el diagrama de flujo correspondiente al funcionamiento de Patchwork, que consta de los siguientes pasos: 1) Se alinean las lecturas al genoma de referencia. 2) Se extraen las variantes de copia única (o, de forma opcional, los Indels) que no coincidan con el genoma de referencia. 3) Se normaliza por contenido GC y otros efectos de posición de naturaleza desconocida. Para llevar a cabo este proceso, se dividen los datos de lecturas alineadas en ventanas con un tamaño fijo de 200pb. La normalización por contenido GC se efectúa mediante la generación de grupos de ventanas con contenido GC similar y posterior normalización de la profundidad de lectura en cada ventana según contenido GC del grupo al que pertenece. Para la normalización de otros efectos de posición se utiliza la información de profundidad de lectura de muestras que hayan sido secuenciadas con el mismo método que el utilizado para la muestra tumoral. 4) Se segmenta el genoma en base a la profundidad de lectura normalizada y resumida en ventanas de 10kb. La segmentación se lleva a cabo por el método de segmentación circular binario (CBS). 5) Se identifican las variantes heterozigotas informativas. 6) Se calcula el ratio de imbalance alélico para cada segmento, de acuerdo con la siguiente fórmula: (∑ mayor - ∑ menor)/(∑ mayor), donde ∑ mayor y ∑ menor representan el número de lecturas correspondientes a los alelos mayoritarios y minoritarios, respectivamente, sumados para todos los SNPs heterocigotos que cubren ese segmento. 7) Se visualiza en un plot el ratio de imbalance alélico vs profundidad de lectura normalizada en los segmentos genómicos. 8) El usuario interpreta el plot anterior y determina los parámetros/argumentos a utilizar en el siguiente paso (más adelante se explica el procedimiento). 9) Se calcula el número de copia específico de alelo para cada segmento genómico. Los pasos 3 a 7 y el paso 9 se llevan a cabo con los módulos Patchwork.plot() y Patchwork.copynumbers() respectivamente. En la fig 4 se muestra un ejemplo del procedimiento a emplear para la asignación de parámetros al módulo Patchwork.copynumbers(). El plot de la izquierda muestra como a cada nº de copia, posicionada sobre el eje horizontal según su profundidad de lectura, le corresponde una distribución concreta de estados de imbalance alélico, siendo mayor el número de estados posibles a mayor número de copia. A partir de estos datos, se pueden establecer los argumentos a utilizar en el módulo Patchwork.copynumbers y que son los siguientes: El argumento cn2 es la posición del número de copia 2 en el eje profundidad de lectura. En este ejemplo, cn2 es ~0.8. El argumento delta es la diferencia entre dos números de copia consecutivos en el eje de profundidad de lectura. En este ejemplo se toman los números de copia 2 y 3. En este ejemplo es ~0.28. El argumento het es la posición del número de copia 2 heterozigoto en el eje de “imbalance” de alelos. En este ejemplo het es ~0.21. El argumento hom es la posición del número de copia 2 homocigoto en el eje de “imbalance” de alelos. En este ejemplo hom es ~0.79. Aunque el plot de la fig 4 muestra una situación ideal, la presencia conjunta de impurezas, aneuploidia y heterogeneidad tumoral, entre otros, pueden dificultar la interpretación de este tipo de plots, como se discutirá en la sección de resultados. Además del análisis de números de copia, Patchwork también permite calcular la ploidia media (definida como el número de copia medio de todos los segmentos genómicos, ponderado por el tamaño de segmento) y el grado de pureza de las muestras tumorales. La ploidia y la pureza se calculan mediante fórmulas basadas en variaciones entre las profundidades de lectura normalizadas encontradas y las que se esperan para muestras tumorales puras de células diploides. En resumen: -Input de Patchwork: Fichero BAM de lecturas y ficheros mpileup y VCF con información de SNPs e Indels, de Tumor y Normal -Output de Patchwork: se genera un fichero excel que entre su información incluye las coordenadas de los segmentos no solapantes identificados, el cromosoma al que pertenecen, el número de copia asignado a cada segmento y porcentaje de células tumorales presente en la muestra -Parámetros de Patchwork: Los dos módulos de Patchwork, Patchwork.plot y Patchwork.copynumbers, admiten parámetros que se pueden utilizar para modular el proceso de segmentación y la asignación de número de copias. Figura 1: Aproximaciones metodológicas para la detección de CNVs a partir de lecturas procedentes de NGS (figura tomada de Min Zao et al). Figura 2: Diagrama de flujo que muestra el mecanismo de funcionamiento de los métodos de análisis de variaciones de número de copia a partir de datos NGS (figura tomada de Liu et al., 2013. ) Figura 3: Diagrama de flujo del mecanismo de funcionamiento de Patchwork (Figura tomada de Mayrhofer et al., 2013). Figura 4: Representación esquemática del típico plot de “imbalance” de alelos vs profundidad de lectura generado por el módulo Patchwork.plot() de Patchwork. A) Se representan sobre las manchas del plot las posibles combinaciones de dos alelos (verde y morado) que les pueden corresponder. CN = número de copia. B) Se representan sobre el plot los valores de los argumentos a utilizar durante la ejecutación del módulo Patchwork.copynumbers. Figura procedente del tutorial online de Patchwork. MATERIALES Y METODOS En el presente estudio, se evaluaron los algroritmos de análisis de número de copia “Patchwork” y “HMMcopy” utilizando datasets artificiales y reales. Generación y análisis de datasets artificales: En la fig5 se resume el proceso utilizado para crear los genomas artificiales y la introducción de CNVs, mientras que en la fig 6 se presenta esquemáticamente el pipeline completo de generación de los datasets artificiales. El proceso completo consta de los siguientes pasos: 1) Generación de “minigenomas artificiales” En un principio, se había planeado partir exclusivamente del cromosoma 22 para la generación de genomas artificiales, ya que es el autosoma más pequeño del genoma humano y por tanto, el más manejable. Sin embargo, Patchwork no permite el análisis de cromosomas individuales, y utiliza como input ficheros que contienen información procedente del genoma completo. Con el objetivo de construir un genoma mínimo capaz de ser procesado por Patchwork, se generó un genoma artificial que consta del cromosoma 22 completo, flanqueado por una concatenación ordenada de pequeños segmentos procedentes de la región 3' de cada uno de los cromosomas que componen el genoma humano (Ver fig5). El genoma artificial resultante tiene un tamaño aproximado de 80Mb, frente a los 3000Mb del genoma completo. 2) Introducción de SNPs Para reproducir de forma fidedigna las propiedades de los genomas reales se han introducido SNPs e indels en el genoma. Para ello se ha utilizado la herramienta Genome-simulator (CovalSimulate), que incorpora SNPs e Indels de 1 a 6 pb de tamaño de forma aleatoria en genomas de referencia, siguiendo una distribución uniforme. Este programa introduce las mutaciones respetando las frecuencias naturales que ocurren en el genoma humano (en el caso de SNPs, 4 veces más transiciones que transversiones (Zhao et al., 2013) y en el caso de los indels, frecuencias de 66%, 17%, 7%, 7%, 2%, 1% para los indels de 1pb, 2pb, 3pb, 4pb, 5pb y 6pb respectivamente (Fujimoto A)). El programa permite que el usuario controle la cantidad total de SNPs y de Indels introducidos. En nuestro caso, se introdujeron los valores default del programa, 0.1% de SNPs y 0.01% de Indels, valores que coinciden con los predichos por algunos autores para el genoma humano (Pang et al., 2010). 3) Generación de las variaciones de número de copia: El siguiente paso consistió en la generación de las variaciones de número de copia en el genoma tumoral. Para simplificar el modelo, se decidió no introducir CNVs en el control, no alterando así el sistema de coordenadas del genoma de tumor relativo al del genoma de referencia. Los CNVs se introdujeron mediante la función “simulateSV” del paquete RSVSim de R/Bioconductor (version 1.6.1, Bartenhagen C, 2014). Esta función permite la introducción de variaciones estructurales en genomas de referencia, con tamaño de segmento fijado por el usuario. Aplicando esta función sobre los ficheros fasta generados en el paso anterior, se introdujeron deleciones y amplificaciones de segmentos de 3 tamaños (20kb, 200kb y 1Mb), en coordenadas aleatorias del genoma. Las deleciones heterocigotas y las ganancias de una sola copia se produjeron mediante la introducción de las modificaciones correspondientes en uno solo de los dos genomas artificiales utilizados como referencia para la generación de lecturas. Las deleciones homocigotas se obtuvieron mediante la introducción de pérdidas de segmentos en la misma posición (es decir, utilizando la misma semilla en la función simulateSV) en los dos genomas artificiales. Se ha visto que muchas veces las variaciones estrucurales co-ocurren con mutaciones mucho más pequeñas (Bartenhagen C, 2014). Para simular esta situación en nuestros datasets, se han introducido SNPs e Indels de hasta 10pb de tamaño en la regiones flanqueantes proximales (hasta una distancia de 50pb) de los breakpoints de los CNVs introducidos. Para dar valor estadístico al estudio, se realizaron 10 copias de cada genoma artificial, cada uno con una colección de CNVs situadas en coordenadas distintas. En total, se generaron 20 genomas artificiales simulando genomas tumorales, divididos en 2 grupos. El primero incluye deleciones y pequeñas amplificaciones (3 y 4 copias) y el segundo incluye grandes amplificaciones (6, 8 y 10 copias). En cada grupo las coordenadas de los segmentos con número de copia alterado se variaron mediante la asignación de un número de semilla diferente en la función simulateSV. 4) Generación de las lecturas simuladas: Para las primeras pruebas, se utilizó el software Sherman Artificial Dataset Generator, un simulador de lecturas cortas. Sin embargo, para los análisis definitivos se seleccionó el software ArtificialFastqGenerator (Frampton et al). Este software permite la generación de lecturas pairedend con una profundidad de lectura que simula el bias por contenido GC del genoma (ver apartado “Corrección por contenido GC” en introducción). Esto es particularmente importante en nuestro caso porque permite evaluar la capacidad de corrección por contenido GC de las herramientas de análisis de NGS que hemos evaluado. Utilizando los ficheros fasta procedentes del paso anterior, se generaron lecturas de 90 pb, paired-end, con un tamaño medio de fragmento de 210 pb y una desviación típica sobre la media de 60pb (los dos últimos valores son valores default del programa). El número de lecturas se ajustó para generar una profundidad media de lecturas de aproximadamente 6x. La elección de un valor de profundidad relativamente bajo se justifica porque las lecturas artificiales generadas presentan características muy optimizadas respecto de las lecturas que se generan por las plataformas NGS, como por ejemplo la ausencia en ellas de errores de secuenciación. 5) Procesamiento de los ficheros FASTQ de lecturas. Las lecturas se alinearon al genoma de referencia humano correspondiente al ensamblaje “hg19” de UCSC. Para el alineamiento, la ordenación e indexación de las lecturas se utilizaron los softwares BWA y Samtools. La generación de ficheros mpileup y VCF a partir de los ficheros BAM, requeridos por el algoritmo Patchwork, se ha llevado a cabo con el software bcftools y el programa de Perl “vcfutils.pl”. El funcionamiento de HMMcopy empieza mediante la subdivisión del genoma en ventanas que contendrán información de número de lecturas, asi como el perfil GC y la mapeabilidad del genoma (ver introducción). En este proceso, se utilizó un tamaño de ventana de 1kb, que es el valor default del programa. 7) Evaluación de los Algoritmos: Los segmentos estimados por HMMcopy y Patchwork se compararon con los segmentos reales, cuyo tamaño y número de copia se había definido durante la generación de los datos simulados. La sensibilidad y especificidad de los algoritmos se calculó en base al grado de solapamiento de las lecturas, a una resolución de 1pb. Aunque en algunos estudios previos no se habían impuesto restricciones en cuanto al grado mínimo de solapamiento entre los segmentos estimados y reales para el cálculo de los valores de sensibilidad y especificidad (Alkodsi et al., 2014; Duan et al., 2013), nosotros hemos pensado que sería más correcto preseleccionar como positivos aquellos segmentos estimados para los que el solapamiento con los reales fuese mayor del 70%. Para ser considerados positivos, los segmentos estimados también tenían que compartir el “estado de número de copia”, deleción o amplificación, con los correspondientes segmentos reales. Las intersecciones se generaron utilizando la función “bedtools intersect” del software bedtools. Una vez obtenidas las intersecciones se calcularon los siguientes parámetros: TP: número total de pb solapantes entre los segmentos estimados y los reales (70% de solapamiento mínimo entre segmentos) . FP: número total de pb en los segmentos estimados que no solapan con los segmentos reales. FN: número total de pb en los segmentos reales que no solapan con los segmentos estimados. La sensibilidad y especificidad de los algritmos HMMcopy y Patchwork se calculó utilizando las siguientes fórmulas: Sensibilidad: TP/TP+FN Especificidad: TP/TP+FP Procesamiento y evaluación de datasets reales: Para evaluar los algoritmos de análisis de número de copia sobre datasets reales, se utilizaron datos de secuenciación WGS de dos muestras de tumores primarios procedentes de dos pacientes varones con Carcinoma Adenoide Cístico (Adenoid cystic carcinoma (ACC)). Se utilizó una de las dos muestras tumorales para simular la correspondiente muestra normal en los algoritmos de análisis de CNVs, ya que no disponíamos de muestras de tejido normal de estos pacientes. Las muestras fueron secuenciadas en una plataforma Illumina, generańdose lecturas paired-end de 100 bases, con un tamaño medio de fragmento de 324 bases y una desviación estándar de 65 bases. Las lecturas fueron alineadas al genoma Humano de referencia NCBI37 de UCSC. Los ficheros BAM de lecturas procesadas nos fueron generosamente cedidos por Elena Piñeiro y Fátima Al-Sharour. Al igual que en el caso de los datasests simulados, se generaron ficheros mpileup y VCF a partir de los ficheros BAM, utilizados como input por el algoritmo Patchwork. Para la comparación entre los resultados obtenidos por Patchwork y HMMcopy se estudió el número de pb solapantes entre los segmentos identificados por ambos algoritmos. Para ello se utilizó la función “bedtools.intersect” del software de Bedtools. Parámetros elegidos durante la ejecución de los algoritmos Patchwork y HMMcopy: El óptimo funcionamiento de los algoritmos de CNVs depende en gran medida del valor de los parámetros utilizados. Cuando posible, se han utilizado los parámetros “default” o recomendados. Sin embargo, como se ha descrito en la introducción, el comando responsable de la asignación de número de copia a los segmentos generados por Patchwork depende de argumentos que tienen que ser asignados manualmente por el usuario. En la sección “resultados” se detalla el proceso de selección de los argumentos elegidos en cada caso. Recursos computacionales: Se ha utilizado un cluster de computadores, con 24 núcleos gestionados por el sistema operativo Darwin, Version 11.4.0. Todos los algoritmos utilizados en este proyecto se han lanzado en este cluster, para asegurar resultados comparables entre las diversas ejecuciones. Figura 5. Representación esquemática del proceso de generación partir del ensamblaje hg19 del genoma humano. Las cajas de representadas sobre el cromosoma 22 indican segmentos sujetos heterocigota y amplificación, respectivamente. Los símbolos (|) y en los genomas normal y tumor respectivamente) de los genomas tumor y normal a color rojo, naranja y azul a deleción homocigota, deleción (0) representan los SNPs introducidos Introducción de las alteraciones (Coval, RVSim) FASTA Generación de las lecturas simuladas (FastqArtificial Generator) FASTQ Alineamiento, ordenación e indexación (Bwa, Samtools) Evaluación de mappabilidad y contenido GC por ventanas del genoma BAM Identificación de SNPs e Indels (Bcftools, vcfutils) (Map.Counter) (GC.Counter) map.WIG gc.WIG Generar fichero de lecturas por ventanas del genoma mpileup VCF (Read.Counter) Reads.WIG HMMcopy CNVs detectados Patchwork CNVs detectados Evaluación comparativa de los resultados Figura 6: Diagrama de flujo del pipeline de análisis por HMMcopy y Patchwork de los datasets simulados RESULTADOS y DISCUSIÓN Herramientas de análisis de CNVs en la literatura : Se ha llevado a cabo una revisión exhaustiva en la literatura sobre algoritmos de análisis de CNVs a partir de datos procedentes de WGS (secuenciación masiva de genomas completos). La tabla 1 presenta una lista, ordenada por fechas, de los algoritmos de análisis de número de copia encontrados, junto con un resumen de algunas de sus principales características. Se han recogido un total de 51 herramientas distintas de análisis de variaciones de número de copia, tanto somáticas como de línea germinal. Estas herramientas presentan numerosas diferencias entre sí, como por ejemplo en cuanto al modelo estadístico utilizado, sus parámetros, el lenguaje de programación con el que han sido implementados, el sistema operativo, o los requisitos de input y el formato de output, entre otros. La gran proliferación de algoritmos de análisis de CNVs en los últimos años pone de manifiesto la complejidad del problema del análisis de CNVs a partir de datos NGS, una tecnología que a día de hoy todavía no está estandarizada. Criterios de selección de métodos: El primer objetivo de este trabajo ha consistido en la selección de un conjunto de herramientas de análisis de CNVs, para su implementación en el laboratorio y posterior evaluación. Se han utilizado los siguientes criterios de selección: a) Se seleccionaron métodos que permitían el análisis de datos procedentes de muestras tumorales. Estos métodos, que identifican SCNAs, utilizan algunas estrategias y algoritmos que no están presentes en los métodos de análisis de CNVs de línea germinal (Biao Liu et al., 2013.) . b) El software con el que se han implementado los algoritmos tenía que ser de libre acceso (por ejemplo, algunos algoritmos, como WaveCNV, incluyen Matlab entre los lenguajes utilizados para su implementación, por lo que tuvieron que ser descartados). c) Por diversas razones, ya comentadas en la introducción, se descartaron los métodos que solo utilizan estrategias de análisis basadas en mapeo de lecturas pareadas, en lecturas interrumpidas, o en ensamblaje “de novo”, d) Se dio preferencia a los algoritmos más recientes y a los métodos que habían recibido las evaluaciones más favorables en estudios previos (Duan et al., 2013; Pabinger et al., 2014; Alkosi et al., 2014). f) Se seleccionaron métodos que fueran sencillos de implementar en el laboratorio, bien documentados y citados en la literatura, y que admitieran ficheros de tipo BAM o SAM como input. Selección y primeras pruebas: Basados en estos criterios, se eligieron los siguientes 4 métodos para su implementación en el laboratorio, HMMcopy, Patchwork, BIC-seq y seqCNA, todos ellos desarrollados para la evaluación de variaciones de número de copia en tumores, utilizando como input datasets procedentes de parejas de muestras tumor/normal. En un estudio previo en el que se han comparado diversas herramientas de análisis de variaciones de número de copia sobre tumores, HMMcopy y BIC-seq han sido las herramientas mejor evaluadas sobre datasets reales, mostrando una gran concordancia entre los SCNAs encontrados por estos algoritmos y los datos proporcionados por SNP arrays (Alkosi et al., 2014). Patchwork es una herramienta desarrollada recientemente para el análisis específico de muestras tumorales, con mecanismos que permiten averiguar contaminación de las muestras tumorales por células normales, el número de copia específico de alelo o la presencia de aneuploidias (Mayrhofer et al., 2013), y seqCNA es una de las herramientas de análisis en tumores más reciente descrito hasta la fecha e incluye un método de filtrado de ventanas propio que, según los desarrolladores, reduce el número de falsos positivos (Mosen-Ansorena et al., 2014). Tras la instalación del software correspondiente a estos 4 algoritmos, se comprobó el funcionamiento de los 3 primeros mediante la ejecución sobre pares de pequeños datasets de prueba tumor/normal, cuyas lecturas proceden de cromosomas individuales, e incluidos en los propios paquetes de instalación de algunos de los algoritmos utilizados. En un primer intento, se obtuvieron errores de ejecución en Patchwork y BIC-seq, mientras que HMMcopy funcionó correctamente. En el caso de Patchwork, los desarrolladores nos informaron de que este algoritmo sólo funciona con datasets procedentes de genomas completos. Por otro lado BIC-seq dio un error de segmentación (“segmentation fault”) que no fué posible resolver, presumiblemente por alguna incompatibilidad de BIC-seq con el sistema operativo Darwin del cluster de computadores sobre el que se ejecutaron los algoritmos, lo que nos obligó a descartar esta herramienta en estudios posteriores. Seq-CNA se ejecutó con datasets de prueba artificiales cuyas lecturas paired-end fueron generadas por el algoritmo “FastqArtificialGenerator”, que se ha descrito con detalle en la sección de Materiales y Métodos. Un primer error durante la ejecución de Seq-CNA se corrigió mediante la utilización de una versión actualizada del algoritmo “FastqArtificialGenerator”, que permitía la generación de lecturas paired-end con orientación invertida en vez de orientación directa. Sin embargo, un segundo problema durante la ejecución resultó derivar de un error de software (“bug”). Aunque los desarrolladores proporcionaron después una versión actualizada del programa, el error no se solucionó y finalmente se descartó la herramienta Seq-CNA. En el siguiente paso, y tras descartar los algoritmos BIC-seq y Seq-CNA , se procedió a la evaluación de los algoritmos Patchwork y HMMcopy. Evaluación los algoritmos seleccionados: a) Datastes artificiales La evaluación de las herramientas Patchwork y HMMcopy empezó mediante el análisis de su funcionamiento sobre datasets simulados. El proceso de generación de los datasets se describe con detalle en la sección de Materiales y Métodos y se ha esquematizado en las figs. 5 y 6. Se obtuvieron un total 21 genomas artificiales, 20 de ellos simulando genomas tumorales y uno simulando un genoma normal. 10 de los genomas tumorales contenían deleciones homocigotas, deleciones heterocigotas, y amplificaciones de 1 y 2 copias y los otros 10 contenían amplificaciones de más de 2 copias. Las variaciones de número de copia se introdujeron en segmentos de 20kb, 200kb y 1Mb de tamaño. Tras la generación de lecturas a partir de los genomas artificiales y su alineamiento al genoma humano (hg19) se obtuvieron los ficheros BAM correspondientes. Para verificar que los datasets artificiales habían sido correctamente generados, se tomó al azar el fichero BAM correspondiente a uno de los 10 datasets que contenían tanto deleciones como amplificaciones y se visualizó mediante IGV. En la fig. 7 se puede ver como las modificaciones de número de copia de los segmentos de 20kb se habían introducido en las coordenadas previstas. La comprobación se extendió al conjunto de todos los segmentos modificados, verificándose en todos los casos que las coordenadas eran correctas (datos no mostrados). -Ejecución de HMMcopy y Patchwork: Los datastets artificiales generados se analizaron con los algoritmos HMMcopy y Patchwork, utilizando los parámetros default. HMMcopy genera un plot en el que se visualiza el proceso de corrección por contenido GC y mapeabilidad llevado a cabo durante el pre-procesamiento de las muestras (fig. 8A). Se puede apreciar la notable corrección llevada a cabo por HMMcopy sobre el bias por contenido GC introducido por el algoritmo FastqArtificialGenerator sobre la profundidad de lectura a lo largo del genoma. HMMcopy genera otro plot que permite visualizar la influencia que tienen dichos procesos de corrección sobre las estimaciones de número de copia (fig. 8B). Además, se genera un plot que permite visualizar en cada cromosoma los segmentos estimados y los estados de número de copia asociados a cada segmento, mediante un código cromático (fig. 8C). El primer módulo de Patchwork, patchwork.plot (), genera un plot por cromosoma, que representa el valor de imbalance alélico frente a la profundidad de lectura. Como se ha descrito en la introducción, la interpretación de dicho plot permite al usuario estimar los argumentos necesarios para la ejecución del segundo módulo de Patchwork, Patchwork.copynumbers (). La fig. 9 muestra la estimación de parámetros realizada a partir del plot correspondiente al cromosoma 22, obtenido tras la ejecución de patchwork.plot () sobre uno de los datasets artificales. Comparando con el plot modelo discutido en la introducción (fig. 4), se puede ver que la calidad de los plots obtenidos con nuestros datasets fué muy baja, hecho que ha dificultado enormemente la adecuada estimación de los argumentos de Patchwork.copynumbers (). La baja calidad de los plots podría deberse a un error de planteamiento en la generación de los datasets artificiales, ya que solo se introdujeron SNPs homocigotos (ver sección Materiales y Métodos). Un segundo problema podría derivar del tamaño relativamente pequeño de las alteraciones de número de copia introducidas. El escaso número de SNPs incluidos en los segmentos alterados podría explicar el hecho de que no se visualicen en el plot puntos de imbalance alélico correspondientes a segmentos con número de copia distinto a 2. El valor de delta, la distancia en el eje X entre dos números de copia sucesivos, fué el argumento de Patchwork.copynumbers más difícil de estimar, ya que casi todos los puntos del plot se correspondían con un valor de profundidad de lecturas correspondiente a un número de copia 2. Por ello se decidieron probar dos valores distintos de delta, 0.15 y 0.3, elegidos tras una serie de pruebas preliminares con Patchwork.copynumbers. En la fig. 10 se puede ver el conjunto de plots generado por Patchwork.copynumbers con argumento delta 0.3, para el cromosoma 22. Se incluye el número de copia específico de alelo y los valores de imbalance alélico a los largo del cromosoma, nótese en este último la escasa densidad de puntos, derivada de la escasez de SNPs informativos en los datasets. -Evaluación de sensibilidad y especificidad: En la sección de Materiales y Métodos se describe cómo se calcularon los valores de sensibilidad y especificidad con los que se ha evaluado el funcionamiento de HMMcopy y Patchwork sobre datasets artificiales. La fig. 11 muestra los valores medios de sensibilidad y especificidad calculados para las 10 muestras estudiadas de cada tipo. Tanto Patchwork como HMMcopy presentan valores superiores al 80% de sensibilidad, bastante altos si se tiene en cuenta la baja profundidad media de lecturas (aproximadamente 6x) de los datasets utilizados. En cambio, la especificidad en la detección de CNVs por parte de HMMcopy y Patchwork ha sido relativamente baja, rondando el 50%. En términos generales, HMMcopy presenta unos niveles de sensibilidad global algo mayores que los de Patchwork, mientras que Patchwork presenta una especificidad global ligeramente superior. Se ha calculado la sensibilidad de forma separada para los 3 tamaños de segmento utliizados en este estudio, 20 kb, 200 kb y 1 Mb. La fig. 12 muestra los valores medios de sensibilidad en 10 muestras para los distintos tamaños de segmento. Patchwork fué incapaz de detectar los segmentos de 20 kb, mientras que HMMcopy los ha detectado con una sensibilidad similar a la de detección de los segmentos más grandes, cercana al 90%. La sensibilidad no se ha medido en función del número de segmentos detectados correctamente sino a nivel del número total de nucleótidos que solapaban entre los segmentos predichos y los reales (ver materiales y métodos). Esto explica el hecho de que Patchwork no mostrase una caída más notable respecto de HMMcopy en sus valores de sensibilidad global (fig. 11), ya que los segmentos de 20 kb son los más pequeños que se han estudiado, y por lo tanto los que menos aportan al valor de sensibilidad global. Se ha calculado también la sensibilidad de forma separada para los CNVs de las distintas clases de estado de número de copia estudiadas (deleciones homocigotas, deleciones heterocigotas, amplificaciones (ganancias de 1 y 2 copias) y grandes amplificaciones (ganancias de 3 copias o más). La fig. 13 muestra las medias de los valores de sensibilidad de detección de CNVs de distintas clases de estado de número de copia. Es notable el hecho de que ni HMMcopy ni Patchwork han detectado las deleciones homocigotas. Aunque inesperado, este resultado ya se había descrito en el caso de HMMcopy (Alkosi et al., 2014) y podría deberse a que, a diferencia de lo que ocurre en el caso de datasets reales, absolutamente ninguna lectura de los datasets artificiales realinea al genoma de referencia en aquellos segmentos que se corresponden con deleciones homocigotas. Para comprobar el efecto de esta situación en Patchwork y HMMcopy se rastrearon los objetos que contienen la información de profundidad de lecturas por ventana del genoma y la interpretación correspondiente de número de copias. En el caso de HMMcopy el objeto “tumor_corrected_copy” recoge el número corregido de lecturas por ventana del genoma y les asocia la columna “copy”, con el valor de LRR normalizado. HMMcopy asocia un valor “NA” a la columna “copy” cuando el número de lecturas de la ventana asociada es 0 y este “valor” es traducido a “estado 3, número de copia 2” durante el proceso de segmentación, situación que se refleja en el output grafico de HMMcopy (fig 14, nótese la asignación de estado 3 a la región entre las coordendas 4.35 exp7 y 5.35 exp7, correspondiente a una deleción homocigota). Para comprobar si se podía corregir esta situación se decidió modificar el fichero wig de lecturas procedente de uno de los datastets artificiales para los que HMMcopy no había identificado ninguna de las deleciones homocigotas introducidas. Se modificó de 0 a 1 el número de lecturas correspondiente a las ventanas que cubrían los segmentos que habían sufrido una deleción homocigota y se volvió a ejecutar HMMcopy con el nuevo input, dejando todas las demás condiciones iguales. Cuando se analizaron los segmentos identificados se encontró que todas las deleciones homocigotas eran detectadas correctamente (datos no mostrados). En el caso de Patchwork, el problema parece ser muy similar. Patchwork también genera un objeto que recoge el número de lecturas por ventana, pero considera como outliers las ventanas en las que no se encuentra ninguna lectura mapeada, excluyéndolas de los análisis posteriores. Si se eliminan los datos correspondientes a las deleciones homocigotas del cómputo global de sensibilidad, su valor pasa a superar el 95% en el caso de Patchwork y el 98% en el caso de HMMcopy (datos no mostrados). Al estar en dos grupos de genomas distintos, se ha podido estudiar por separado la especificidad de detección de grandes amplificaciones frente a la de las restantes modificaciones (ver Materiales y Métodos). Sorprendentemente, se vio que la especificidad en la detección de las grandes amplificaciones era sensiblemente inferior a la de detección de las otras modificaciones, tanto en el caso de HMMcopy como en el de Patchwork (fig. 15). De hecho, casi todos los falsos positivos detectados por HMMcopy y Patchwork se correspondían con segmentos situados en el grupo de genomas que contenía las grandes amplificaciones, lo que sugiere que los parámetros de segmentación no estaban bien ajustados en este caso. En los tutoriales de estos algoritmos se subraya la importancia de que los usuarios evalúen los outputs gráficos obtenidos tras la ejecución y ajusten los parámetros de segmentación y asignación de número de copia en función de los resultados obtenidos (ver sección “Prinicipales características de los algoritmos Patchwork y HMMcopy” en introducción). -Evaluación de la precisión en la detección de breakpoints: Se ha comparado a máxima resolución (1 pb) la precisión en la asignación de las posiciones de los breakpoints predichos por Patchwork y HMMcopy. La precisión se ha determinado como la distancia en bases entre las coordenadas de los segmentos reales y la de los segmentos detectados, considerándose solo como positivos los segmentos con más del 70% de solapamiento. La fig. 16 muestra los boxplots de distancias para los 2 algoritmos evaluados. Se ha encontrado una importante diferencia entre la precisión de estimación de breakpoints por ambos métodos, que superaba el orden de magnitud (mediana de las distancias de 260 pb en el caso de HMMcopy y de 5049 pb en el caso de Patchwork). Una posible razón para explicar la baja precisión de Patchwork podría ser la baja profundidad de lectura de los datastets artificiales utilizados (6x de media). Sería interesante hacer una prueba comparativa de HMMcopy y Patchwork utilizando datasets con una profundidad de lectura más cercana a la que se obtiene en las plataformas actuales de secuenciación (30-60x), para la comprobación de este punto. -Evaluacion de la precisión en la determinación de número de copias: La fig 17 muestra los valores de número de copia (o de estados de número de copia, en el caso de HMMcopy) asignados a los segmentos encontrados, comparada con los valores reales (segmentos de colores). El código de colores de los segmentos permite establecer una equivalencia entre números de copia (output de Patchwork) y estados de número de copia (output de HMMcopy, ver Introducción). Un primer examen de esta figura sugiere que HMMcopy es más preciso que Patchwork en la asignación de números de copia. Sin embargo es importante recordar que, debido a problemas técnicos, no fué posible la asignación de parámetros óptimos al módulo Patchwork.copynumbers () de Patchwork, responsable de la asignación de números de copia a los segmentos identificados. Probablemente sea ésta la causa de que Patchwork haya identificado como homocigotas las deleciones heterocigotas. Más sorprendente es que el software HMMcopy también haya cometido el mismo error (fig. 17). Como se explica en el correspondiente tutorial, el ajuste de los parámetros de HMMcopy podría permitir la corrección de este tipo de asignaciones incorrectas. Los datos de Patchwork y HMMcopy correspondientes a las deleciones homocigotas carecen de valor estadístico, ya que, como se ha mencionado, la identificación de estos segmentos fue extremadamente ineficiente. Ambos algoritmos tendieron a asignar un número de copia superior al real en el caso de las amplificaciones débiles, de 3 o 4 copias. La asignación por Patchwork de 8 copias a los segmentos de 10 copias se debe probablemente a que no se asignó el valor de 10 al parámetro “maxCn” de Patchwork.copynumbers, en vez del valor 8 que tiene por defecto. Este parámetro le indica al algoritmo el máximo valor de número de copia que se quiere estimar. -Conclusiones: Nuestros resultados con datasets artificiales sugieren que HMMcopy es más eficiente que Patchwork, tanto en la localización de CNVs como en la asignación de números de copia. Sin embargo, es importante recordar que no hemos testado algunas de las propiedades más relevantes de Patchwork, como su capacidad de evaluar número de copias en muestras tumorales que presentan contaminación con tejido normal o ejemplos de aneuploidia. b) Datastes reales: Puesto que los datasets artificiales carecen de muchos de los niveles de complejidad que caracterizan los procesos de secuenciación masiva de muestras tumorales, se procedió a la evaluación del funcionamiento de HMMcopy y Patchwork con datasets reales. -Elección del dataset: El proceso de obtención de estos datasets no fué fácil. Nosotros estábamos interesados en obtener datasets con las siguientes características: -Que procediesen de estudios de secuenciación masiva de genoma completo de muestras tumorales, o de líneas celulares derivadas de tumores. -Que estuviese disponible el correspondiente control de muestras de tejido normal del mismo paciente. Como ya se ha mencionado en la introducción, la mayoría de las herramientas de análisis SCNAs a partir de muestras tumorales requieren de este control, o es muy recomendable. -Que se dispuesiese de datos de SCNAs procedentes de estudios de arrays sobre las mismas muestras, para utilizarlos como “gold Standard” de control sobre los SCNAs identificados por los algoritmos evaluados. El acceso a datos de secuenciación masiva procedentes de muestras de pacientes con tumores se encuentra bastante restringido hoy en día, por lo que nos hemos centrado en datasets procedentes de líneas celulares. En un principio se pensó utilizar datasets procedentes de la secuenciación WGS (de genoma completo) de la línea celular de cáncer de mama HCC1954 (profundidad de lectura aproximada de 4x) y de la correspondiente línea celular normal HCC1954BL (profundidad de lectura aproximada de 5x). Estos datasets habían sido utilizados en la evaluación del algoritmo Patchwork por sus desarrolladores (Mayrhofer et al., 2013), los cuales nos proporcionaron generosamente una lista completa de las variaciones de número de copia encontradas por la herramienta TAPS, desarrollada por ellos, a partir de datos de SNP arrays. Esta lista podía ser utilizada por nosotros como control “gold standard”. Se descargaron de SRA (“sequence read archive”, [SRA:SRA001246] ) los ficheros fastq correspondientes a un total de 4 parejas de muestras tumor/normal, con lecturas de 32pb y 36 pb de tamaño. Un análisis mediante Fastqc de la calidad de las lecturas contenidas en los ficheros fastq (fig suplementaria 1, incluida en anexos), mostró que la calidad de las lecturas, tanto de 32pb como de 36pb, era demasiado baja como para garantizar un alineamiento correcto a un genoma de referencia, por lo que fueron descartadas. En un segundo intento de obtener los datasets reales para la evaluación de Patchwork y HMMcopy, se solicitó a “Cancer Genome Project” acceso a los datos de secuenciación de genoma completo de las líneas celulares COLO-829 y COLO-829BL, depositados en “EGA” (European Genome-Phenome Archive, EGAS00000000052). Estas líneas celulares derivan de un melanoma maligno y de linfoblastos normales, respectivamente (Pleasance et al, 2010), y fueron secuenciadas en una plataforma Illumina GAII, obteniéndose lecturas pareadas de 75pb de tamaño, con una profundidad de lectura aproximada de 40x. Se disponían además de datos de número de copia obtenidos mediante arrays, para ser utilizados como “gold standard” en la evaluación de nuestros algoritmos. Una vez concedido el citado permiso, se comprobó que los datastes a los que habíamos tenido acceso no se correspondian con los que se habían solicitado, y no incluían datasets de NGS. Se recurrió pues a una tercera opción (la que finalmente fue válida), en la que se utilizaron datos de secuenciación de genoma completo de dos muestras tumorales procedentes de dos pacientes varones con Carcinoma Adenoide Cístico (Adenoid cystic carcinoma (ACC)). En materiales y métodos se ha descrito el proceso de secuenciación y posterior procesamiento de las lecturas para la generación de los ficheros input de HMMcopy y Patchwork. Una de las dos muestras tumorales se utilizó para simular la correspondiente muestra normal, ya que no disponíamos de muestras de tejido normal de estos pacientes. -Ejecución y evaluación de HMMcopy y Patchwork sobre datasets reales: La ejecución de HMMcopy y Patchwork sobre los datastets procedentes de los pacientes con Carcinoma Adenoide Cístico se llevó a cabo utilizando los parámetros default. La fig. 18 muestra el output gráfico correspondiente al cromosoma 1 generado por HMMcopy durante su ejecución, incluyendo el proceso de corrección por contenido GC y mapeabilidad (nótese la importante corrección por mapeabilidad llevada a cabo por el algoritmo), la visualización de los segmentos generados y el estado de número de copia asignado a cada uno. En la fig 19 se visualiza el output gráfico correspondiente al cromosoma 1 del módulo Patchwork.plot de Patchwork y en la fig 20 se muestra la asignación de los argumentos requeridos por el módulo Patchwork.copynumbers, que determina la interpretación por dicho módulo del “cariotipo del genoma” (número de copias y distribución de alelos, fig. 20B). El output gráfico del módulo Patchwork.copynumbers es básicamente idéntico al de Patchwork.plot, excepto por la inclusión adicional de un plot que representa la distribución en el cromosoma de los valores de número de copia total y del alelo minoritario (fig 21). Se incluyen en el anexo las tablas “segmentosHMMcopy.csv” y “segmentosPatchwork”, generadas por HMMcopy y Patchwork respectivamente, y que incluyen la lista de eventos de CNV encontrados. Se han identificado un total de 799 y 293 segmentos con el número de copia alterado en las muestras tumorales analizadas con HMMcopy y Patchwork respectivamente. Los tamaños de los segmentos identficados fué muy variable (de 7 kb a 55 Mb en el caso de HMMcopy y de 20kb a 38Mb en el caso de Patchwork). La fig 22 muestra la distribución de tamaños encontrada. En general, el tamaño medio de los segmentos identficados por Patchwork ha sido sensiblemente mayor que el de los identficados por HMMcopy. Además, HMMcopy ha identificado un gran número de segmentos de tamaño inferior a 20kb. La fig 23 muestra un circos.plot con la dsitribución en el genoma de los segmentos identificados por HMMcopy y Patchwork. Se ve una importante coincidencia entre los segmentos identificados por ambos algrotimos, sobretodo en el caso de los segmentos de mayor tamaño. La principal incongruencia se ha encontrado en los cromosomas sexuales, donde por ejemplo HMMcopy y Patchwork han predicho respectivamente una importante ganacia y pérdida de material genético del cromosoma Y. Patchwork admite el parámetro Male = True en su módulo Patchwork.copynumbers, pensado para el correcto procesamiento de los cromosomas sexuales de muestras procedentes de varones. La ausencia de un parámetro equivalente en HMMcopy sugiere que este algoritmo podría ser menos preciso a la hora de determinar variaciones de número de copia en los cromosomas sexuales. Muchas de las CNVs encontradas por los dos algoritmos se corresponden con deleciones (fig. 23). Curiosamente, casi todas estas deleciones han sido interpretadas como deleciones homocigotas por HMMcopy, mientras que Patchwork las ha interpretado como deleciones heterocigotas. Asimismo, el número de copia asignado por HMMcopy a las amplificaciones fue en general mayor que el asignado por Patchwork. HMMcopy había mostrado mayor precisión en la asignación de número de copias en el caso de los datatets artificiales, pero estos resultados no fueron definitivos debido a los problemas técnicos encontrados en el proceso de asignación de argumentos al módulo que estima los números de copia, de forma que no es posible extraer conclusiones definitivas a este respecto. El circos.plot de la fig. 23 también muestra el gran número de segmentos pequeños que han sido identificados exclusivamente por HMMcopy. Para conocer con más precisión la diferencia entre los resultados obtenidos por HMMcopy y Patchwork, se determinó el número exacto de bases identificados por ambos algoritmos, así como el número de pares de bases identificados exclusivamente por cada uno de ellos. Los resultados se muestran en el diagrama de Venn de la fig. 24. Más de un 90% de los pb correspondientes a segmentos con alteraciones de número de copia identificados por Patchwork han sido también identificados por HMMcopy. Sin embargo, un 18% de los pares de bases correspondientes a segmentos identificados por HMMcopy no fueron identificados por Patchwork. El análisis del tamaño de los segmentos solo identificados por HMMcopy muestra que en general se corresponden con los más pequeños, como se puede ver en los boxplots representados en la fig24. Al carecer de un “gold standard” para establecer cuáles de las variaciones de número de copia encontradas por cada algoritmo son reales, no se puede deducir si los pequeños segmentos identificados sólo por HMMcopy reflejan una mayor sensibilidad de este algoritmo o por el contrario, indican que su especificidad en la detección de CNVs es menor. Sin embargo, los estudios previos realizados sobre datastets artificiales mostraron que sólo HMMcopy era capaz de detectar los CNVs más pequeños, en el orden de 20kb de tamaño, hecho que apoya fuertemente la hipótesis de que las pequeñas variaciones de número de copia identificadas por HMMcopy son reales y reflejan la mayor sensibilidad de este algoritmo. Es importante resaltar que para establecer definitivamente la sensibilidad y especificidad de los dos algoritmos evaluados utilizando datastets reales, sería necesario ejecutarlos con una verdadera pareja de datastets tumor/normal y comparar los resultados con un “gold standard”, como el que se podría obtener mediante un análisis de CNVs sobre las mismas muestras utilizando arrays. -Problemas en la ejecución de Patchwork con muestras tumorales procedentes de pacientes de sexo femenino Además de las muestras procedentes de pacientes varones con Carcinoma Adenoide Cístico que se han utilizado en el análisis descrito arriba, también disponíamos de dos muestras procedentes de dos pacientes de sexo femenino. Sorprendentemente, la ejecución de Patchwork sobre estas muestras se vió interrumpida por el error “referencia del cromosoma Y inválida”. Se averiguó posteriormente que el error parece haberse debido a que las muestras se habían alineado a una referencia de la que se había retirado el cromosoma Y, para evitar la producción de alineamientos ambiguos. Este error refleja la falta de flexibilidad de Patchwork a la hora de analizar muestras en las que pueda faltar información de algún cromosoma, así como la imposibilidad de estudiar cromosomas individuales con este algoritmo. -Ejecución de Patchwork con un control normal Para los casos en los que no se dispone de un control normal, los desarrolladores de Patchwork han puesto a disposición del usuario un fichero de referencia estándar, que se puede utilizar como alternativa cuando el genoma de referencia al que se han alineado las lecturas de la muestra tumoral es UCSC hg19. Se ha ejecutado Patchwork utilizando dicha referencia control junto con la muestra que se había asignado como tumor en el experimento anterior, descrito arriba. La fig. 25 muestra el plot de los valores de imbalance alélico versus profundidad de lecturas generado por el módulo Patchwork.plot, y los valores asignados para los argumentos de Patchwork.copynumbers. La tabla “segmentosPatchwork_refNormal.csv”, incluida en Anexos, contiene los datos generados por Patchwork sobre la muestra tumoral analizada, y en la fig26 se puede ver, mediante circos.plot, la distribución a lo largo del genoma de los segmentos identificados por Patchwork así como los estados de número de copia asignados. Comparando la fig. 23 con el anillo interior de la fig 26 se puede ver que el patrón de variaciones de número de copia a lo largo del genoma fué muy similar al que se había encontrado cuando se utilizó como control una segunda muestra tumoral del mismo paciente. El análisis de Patchwork sobre la pareja tumor/normal también ha revelado la ausencia total de contaminación de la muestra tumoral por células normales (tabla “segmentosPatchwork_refNormal.csv”, en anexos). -Recursos computacionales: En general, el tiempo de ejecución de Patchwork es sensiblemente más elevado que el de HMMcopy. Patchwork requiere para su funcionamiento de la generación previa de ficheros mpileup y VCF con información sobre SNPs e Indels, necesarios para el proceso de segmentación y de asignación de números de copia por Patchwork. La generación de estos ficheros, a partir de ficheros BAM de alrededor de 300Gb - que es el tamaño aproximado de los datasets reales que se han utilizado en nuestro estudio - ha tardado entre 30 y 35 horas. A estos tiempos hay que sumar otras 40 horas para ejecutar los comandos “patchwork.plot” y “patchwork.copynumbers” de Patchwork. Por el contrario, la ejecución completa de HMMcopy sobre los mismos datasets ha tardado del orden de 4 a 5 horas, a las que hay que añadir, eso sí, otras 30 horas aproximadamente para la generación del fichero de mapeabilidad del genoma, requerido como input de HMMcopy. a b c Figura 7: Visualización por IGV de la densidad de lecturas en regiones del cromosoma 22 que incluyen segmentos de 20 kb con las siguientes modificaciones del número de copia: a) deleción homocigota b) deleción heterocigota c) amplificación. Las rayas horizontales indican las coordenadas de los punto de corte (breakpoints) de los 3 segmentos modificados. Herramienta Revista Metodo Input Lenguaje Evaluaciones Previas Comentarios BreakDancer Max Chen et al, 2009 PEM BAM/SAM Perl, C++ PEMer Korbel et al, 2009 PEM FASTA Perl, Python Pindel Ye et al, 2011 SR BAM /FASTQ C++ RDXplorer Yoon et al, 2009 RD BAM Duan et al(2013) Python, Shell Pabinger et al(2014) CNV-seq Xie et al, 2009 RD BAM/SAM Perl, R SegSeq mrCaNaVar GASV Chiang et al, 2009 Alkan et al, 2009 Sindi et al, 2009 RD RD PEM BAM/SAM SAM BAM Matlab C Java VariationHunter Hormozdiari et al, 2010 PEM DIVET (específico) C SLOPE RSW-seq a Abel et al, 2010 Kim et al, 2010 SR RD SAM/FASTQ/MAQ PLA** C++ C CNAseg CMDS b Ivakhno et al, 2010 Zhang et al, 2010 RD RD BAM PLA** R C, R SVDetect CNVer NovelSeq Zeitouni et al, 2010 Medvedev et al, 2010 Hajirasouliha et al, 2010 PEM+RD PEM+RD PEM+AS SAM/BAM/ELAND BAM/PLA** FASTA/SAM Perl Perl, C++ C HYDRA Quinlan et al, 2010 PEM+AS ALD* Python SOAPdenovo Li et al, 2010 AS N/A CopySeq CnD Waszak et al, 2010 Simpson et al, 2010 RD RD RCM*** SAM/BAM N/A Java R D SVmerge commonLAW Wong et al, 2010 Hormozdiari et al, 2011 RD PEM N/A BAM/SAM N/A C++ AGE SRiC Abyzov et al, 2011 Zhang et al, 2011 PEM SR FASTA BLAT output C++ N/A ReadDepth Miller et al, 2011 RD BED files R CNVnator Abyzov et al, 2011 RD BAM C++ Duan et al(2013) Duan et al(2013) Pabinger et al(2014) BIC-seq JointSLM Xi et al, 2012 Magi et al, 2011 RD + BAF RD BAM SAM/BAM Perl, R, C R, Fortran Alkodsi et al(2014) Duan et al(2013) Spanner Mills et al, 2011 PEM N/A N/A Genome STRiP inGAP-sv Handsaker et al, 2011 Qi et al, 2011 PEM+RD PEM+RD BAM SAM Java, R Java SVseq Zhang et al, 2011 PEM+SR FASTQ/BAM CNVnorm a CNVeM cn.MOPS Gusnanto et al, 2012 Wang et al, 2012 Klambauer et al, 2012 RD RD RD BAM N/A BAM/ C R Perl N/A R Cortex assembler Iqbal et al, 2012 AS FASTQ/FASTA C Magnolya GASVPro SeqCBS ControlFREEC Nijkamp et al, 2012 Sindi et al, 2012 Shen et al, 2012 AS PEM+RD RD Python C++ N/A Boeva et al, 2012 RD FASTA BAM N/A SAM,BAM, PileUp, Eland, BED y otros C++, R Alkodsi et al(2014) Duan et al(2013) HMMCopy Ha et al, 2012 RD BAM R, Perl Alkodsi et al(2014) COPS CONSERTING Golden Helix Krishnan et al, 2012 Chen et al, 2012 Golden Helix Inc. (2012) RD RD RD SAM/BAM N/A N/A Perl, Bash R N/A Alkodsi et al(2014) OncoSNP-SEQ Yau C (2013) RD + BAF N/A Patchwork Mayrhofer et al, 2013 RD + BAF N/A BAM (patchwork) CompleteGenomics (patchworkCG) CNV-TV Duan J et al, 2013 RD N/A WaveCNV m-HMM Holt et al, 2014 RD N/A Fichero pileup estándar generado a partir de ficheros SAM/BAM Wang et al, 2014 RD N/A N/A RD + PEM SAM/BAM R RD + BAF BAM Matlab C++ seqCNA CLImAT Mosen-Ansorena et al, 2014 Yu et al, 2014 N/A Matlab Pabinger et al(2014) Usado en el proyecto 1000 Genomas. Solo detecta CNVs de línea germinal. Usado en el proyecto 1000 Genomas. Solo detecta CNVs de línea germinal. Usado en el proyecto 1000 Genomas. Solo detecta CNVs de línea germinal Usado en el proyecto 1000 Genomas. Solo detecta CNVs de línea germinal. Duan et al(2013) Alkodsi et al(2014) Duan et al(2013) Solo detecta CNVs de línea germinal. Detecta SCNAs. Solo admite lecturas Single-end como input. Pabinger et al(2014) Solo detecta CNVs de línea germinal. Usado en el proyecto 1000 Genomas. Solo detecta CNVs de línea germinal Duan et al(2013) Alkodsi et al(2014) Duan et al(2013) Detecta SCNAs Duan et al(2013) Solo detecta CNVs de línea germinal. Detecta SCNAs. Usado en el proyecto 1000 Genomas. Solo detecta CNVs de línea germinal. Duan et al(2013) Duan et al(2013) Pabinger et al(2014) Alkodsi et al(2014) Duan et al(2013) Solo detecta CNVs de línea germinal. Solo detecta CNVs de línea germinal Pipeline con varias herramientas. Da problemas en la instalación (Pabinger et al(2013). Solo detecta CNVs de línea germinal Usado en el proyecto 1000 Genomas. Solo detecta CNVs de línea germinal. Detecta SCNAs. Junto con HMMcopy, El mejor evaluado sobre datasets reales y Muy preciso en la detección de breakpoints (Alkodsi et al, 2014). Solo detecta CNVs de línea germinal. Usado en el proyecto 1000 Genomas. Solo detecta CNVs de línea germinal. Usado en el proyecto 1000 Genomas. Solo detecta CNVs de línea germinal. Detecta SCNAs. Duan et al(2013) Usado en el proyecto 1000 Genomas. Solo detecta CNVs de línea germinal. Detecta SCNAs. Detecta SCNAs. Junto con HMMcopy, El mejor evaluado sobre datasets reales (Alkodsi et al et al). Detecta SCNAs. Detecta SCNAs. Pipeline con varias herramientas. Detecta SCNAs. Detecta SCNAs y estima el grado de ploidia, el porcentaje de células tumorales en Tumores primarios. No se ha encotntrado el software asociado A esta publicación. Detecta SCNAs y estima el grado de ploidia, el porcentaje de células tumorales en tumores primarios y la contaminación por Células de ratón en xenotransplantes. Utilizado fundamentalmente con Genomas de plantas Detecta SCNAs. Su novedoso método de filtrado de ventanas reduce el número de falsos positivos, respecto de métodos anteriores. Detecta SCNAs y estima el grado de ploidia, el porcentaje de células tumorales en tumores primarios. No requiere de pareja normal. Tabla1: Resumen de las herramientas de anaĺisis de número de copia a partir de datos WGS (secuenciación de genoma completo) disponibles en la literatura. RD: Método basado en profundidad de lecturas; PEM: Método basado en mapeo de lecturas pareadas SR: Método basado en lecturas interrumpidas ; AS: Método basado en ensamblaje “de novo”; BAF: Frecuencia del alelo B; * ALD: Alineamientos de lecturas paired-end discordantes; **PLA: Posiciones de lecturas alineadas; N/A: Información no disponible o no encontrada. Figura 8: Output gráfico de HMMcopy correspondiente al cromosoma 22. Resultados obtenidos del procesamiento de uno de los datasets artificiales utilizados en la evaluación de Patchwork y HMMcopy. A: Efecto de la corrección por mapeabilidad y contenido GC del genoma sobre la distribución de lecturas. B: Evolución en las estimaciones del número de copias en cada ventana del cromosoma tras la corrección por mapeabilidad y contenido GC. C:Visualización de los segmentos generados por HMMcopy y de los estados asignados a cada segmento. HOMD: Deleción homocigota; HETD: Deleción heterocigota; NEUT: 2 copias; GAIN: 3 copias; AMPL: 4 copias; HLAMP: más de 4 copias. Figura 9: Ejemplo de selección de argumentos para el módulo Patchwork.copynumbers de Patchwork. Se muestra en la figura el plot de imbalance alélico vs profundidad de lectura correspondiente al cromosoma 22, generado por el módulo Patchwork.plot tras el procesamiento de uno de los datasets artificiales utilizados en la evaluación de Patchwork y HMMcopy. Se han indicado sobre el plot los argumentos seleccionados para la ejecución de Patchwork.copynumbers. Figura 10: Output gráfico del comando Patchwork.copynumbers de Patchwork correspondiente al cromosoma 22. A. Representación del genoma de tumor completo con las correspondientes etiquetas de número de copia y contenido de alelos que han sido asignadas. 2m1: el nº de copias es 2, heterocigoto; 2m0: el nº de copias es 2, homocigoto, 4m1: el nº de copias es 4, heterocigoto; 4m0: el nº de copias es 4, homocigoto. B. Panel Superior: Valores estimados de número de copia total y número de copia asociado al alelo minoritario para el cromosoma 22.; Panel Intermedio: Plot de profundidad de lectura a lo largo del cromosoma; Panel Inferior: Valores de imbalance alélico a lo largo del cromosoma. 0,8 0,6 HMMcopy PATCHWORK 0,4 0,2 0 Sensibilidad Valor de sensibilidad Valor de sensibilidad/especificidad 1 Figura 11: Valores globales de sensibilidad y especificidad de HMMcopy y Patchwork. Especificidad Figura 12: Sensibilidad de detección por HMMcopy y Patchwork de CNVs con distinto HMMcopy tamaño de PATCHWORK segmento. 1 0,8 0,6 0,4 0,2 0 20000 200000 1000000 TamañoDeSegmento Figura 13: Sensibilidad de detección por HMMcopy y Patchwork de CNVs con distintos números de copia. Valor de sensibilidad 1,2 1 0,8 0,6 HMMcopy PATCHWORK 0,4 0,2 0 0 1 '3-4' '>4' Nº De Copia Figura 14: Ampliación de las coordenadas 4.1exp7 a 4.5 exp7 del cromosoma 22 de la figura 8C, que incluyen una deleción homocigota. 1,2 Valor de especificidad 1 0,8 0,6 HMMcopy PATCHWORK 0,4 0,2 0 grupo”A” grupo”B” Figura 15: Valores de especificidad de HMMcopy y Patchwork para los CNVs incluidos en los 2 grupos de genomas artificiales utilizados. Grupo A: incluye segmentos con nº de copias: 0, 1, 3 y 4. Grupo B: incluye segmentos con nº de copias: >4 Figura16: Boxplots de la precisión en la asignación de las posiciones de los breakpoints de los CNVs estimados por HMMcopy y Patchwork . En el eje Y se representa la distancia entre las coordenadas de los breakpoints estimado y real. Figura 17: Histogramas de precisión en la evaluación del número de copias por HMMcopy (panel de arriba) y por Patchwork, ejecutado con dos argumentos distintos de delta (ver sección Resultados). La altura de las cajas representa la posición de los números de copia estimados. Los segmentos coloreados representan la posición de los números de copia reales y sus colores representan los estados de número de copia, según clasificación por HMMcopy (no se representa el estado 2, de 2 copias). Rojo oscuro: estado1 (deleción homocigota); Rojo claro: estado2 (deleción heterocigota); Azul claro: Estado 4 (3 copias); Azul intermedio: Estado 5 (4 copias); Azul oscuro: Estado 6 (más de 4 copias). Figura 18: Output gráfico correspondiente al cromosoma 1 del procesamiento por HMMcopy de datasets reales procedentes de pacientes con Carcinoma Adenoide Cístico. A: Corrección por mapeabilidad y contenido GC B: Efecto de la corrección por mapeabilidad y contenido GC sobre la estimación del número de copias. C: Segmentos generados y estado de número de copias asignado a cada segmento Figura 19: Output gráfico del comando Patchwork.plot de Patchwork correspondiente al cromosoma 1. Comando ejecutado sobre datasets reales procedentes de pacientes con Carcinoma Adenoide Cístico. Panel Superior:Plot de Imbalance Alélico frente a profundidad de lectura Panel Intermedio: Plot de profundidad de lectura a lo largo del cromosoma Panel Inferior: Valores de imbalance alélico a lo largo del cromosoma. Figura 20: Selección de argumentos para el módulo Patchwork.copynumbers de Patchwork. A: Se muestra el plot de imbalance alélico vs profundidad de lectura correspondiente al cromosoma 1, generado por el módulo Patchwork.plot tras el procesamiento de datasets reales procedentes de pacientes con Carcinoma Adenoide Cístico. Se han indicado sobre el plot los argumentos seleccionados para la ejecución de Patchwork.copynumbers. B:Plot generado por Patchwork.copynumbers que muestra el genoma de tumor completo con las correspondientes etiquetas de número de copia y contenido de alelos que han sido asignadas. 1m0: el nº de copias es 1, homocigoto; 2m1: el nº de copias es 2, heterocigoto; 2m0: el nº de copias es 2, homocigoto, y asi sucesivamente. Figura 21: Plot generado por el módulo Patchwork.Copynumbers que muestra los valores estimados de número de copia total y número de copia asociado al alelo minoritario para el cromosoma 1. Figura 22: Histograma de distribución de tamaños de los segmentos generados por Patchwork y HMMcopy en el procesamiento de datastets reales. En el panel inferior se muestra ampliada la región correspondiente a tamaños inferiores a 20 kb. Density: Proporción del total de segmentos presente en cada clase de tamaño de segmento. Figura 23: Resumen de los resultados obtenidos por HMMcopy y Patchwork sobre los datasets procedentes de tumores. En este “Circos plot” se resumen todos los CNVs detectados por HMMcopy (exterior) y Patchwork (interior). Los segmentos CNVs se distinguen por color como deleciones homocigotas (naranja intenso), deleciones heterocigotas (naranja claro), amplificaciones de una o dos copias (azul claro) y amplificaciones de más de dos copias (azul oscuro). Figura 24: Diagrama de Venn de la detección por HMMcopy y Patchwork de variaciones del número de copia tras el procesamiento de datasets reales. Los valores numéricos en cada sector indican pares de bases. Se representan también los boxplots de distribución de tamaños de los segmentos correspondientes a cada sector del diagrama de Venn. HMM_NI: Segmentos identificados exclusivamente por HMMcopy. HMM_I:Segmentos identificados por HMMcopy y Patchwork. PatchW: Segmentos identificados por Patchwork. Hom = 1 Delta = 5 Het = 0.3 Cn2 = 1.05 Figura 25: Selección de argumentos para el módulo Patchwork.copynumbers de Patchwork. A: Se muestra en la figura el plot de imbalance alélico vs profundidad de lectura correspondiente al cromosoma 1, generado por el módulo Patchwork.plot tras el procesamiento del dataset tumoral procedente de paciente con Carcinoma Adenoide Cístico y usando una referencia estándar de Patchwork como control normal. Se han indicado sobre el plot los argumentos seleccionados para la ejecución de Patchwork.copynumbers. B:Plot generado por Patchwork.copynumbers que muestra el genoma de tumor completo con las correspondientes etiquetas de número de copia y contenido de alelos que han sido asignadas.1m0: el nº de copias es 1, homocigoto; 2m1: el nº de copias es 2, heterocigoto; 2m0: el nº de copias es 2, homocigoto, y así sucesivamente. figura 26: Resumen de los resultados obtenidos por Patchwork sobre los datasets procedentes de tumor versus referencia normal de Patchwork. En este “Circos plot” se resumen todos los CNVs detectados por Patchwork. Los segmentos CNVs se distinguen por color como deleciones homocigotas (naranja intenso), deleciones heterocigotas (naranja claro), amplificaciones de una o dos copias (azul claro) y amplificaciones de más de dos copias (azul oscuro), CONCLUSIONES: - Se han recogido de la literatura más de 50 herramientas de análisis de variaciones de número de copia a partir de datos de secuenciación de genoma completo. Estas herramientas presentan numerosas diferencias entre sí, como por ejemplo en cuanto al modelo estadístico utilizado, sus parámetros, el lenguaje de programación con el que han sido implementados, los requisitos de input o el formato de output. - La ejecución de las herramientas HMMcopy y Patchwork con datasets artificiales ha mostrado que HMMcopy presenta una mayor sensibilidad de detección de CNVs que Patchwork, particularmente para aquellos segmentos de tamaño más pequeño. - La identificación de los CNVs introducidos en los genomas artificales fué algo más específica por parte de Patchwork que por HMMcopy. La especificidad de ambos algoritmos mostró ser muy dependiente del tipo de CNVs introducido en el genoma estudiado. - HMMcopy fué mucho más preciso que Patchwork en la identificación de los breakpoints de los segmentos con número de copia alterado en los genomas artificiales, con una diferencia de precisión que ha superado el orden de magnitud. - La precisión en la asignación de número de copias a los CNVs identificados por Patchwork depende en gran medida que el usuario pueda introducir correctamente los argumentos que requiere la función del programa que lleva a cabo dicha interpretación. - Mediante la utilización de datasets reales se confirma que HMMcopy presenta una mayor sensibilidad que Patchwork para la deteccción de CNVs de pequeño tamaño. -Patchwork utilliza más recursos computacionales y requiere más tiempo para su ejecución que HMMcopy, aunque por otro lado proporciona una información más completa sobre las muestras utilizadas, incluyendo la pureza de la muestra tumoral y la presencia de aneuploidias. BIBLIOGRAFIA Abel HJ, Duncavage EJ, Becker N, Armstrong JR, Magrini VJ, Pfeifer JD (2010) SLOPE: a quick and accurate method for locating non-SNP structural variation from targeted next-generation sequence data. Bioinformatics, 26:2684-2688. Abyzov A, Urban AE, Snyder M, Gerstein M. (2011) CNVsnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome research; 21(6):974–984. Abyzov A, Gerstein M (2011) AGE: defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision. Bioinformatics, 27:595-603. Albertson DG, Collins C, McCormick F and Gray JW. (2003) Chromosome aberrations in solid tumors. Nature genetics; 34(4):369-376. Alkan C, Coe BP, Eichler EE. (2011) Genome structural variation discovery and genotyping. Nature reviews Genetics;12(5):363–376. Alkan C, Kidd JM, Marques-Bonet T, Aksay G, Antonacci F, Hormozdiari F, Kitzman JO, Baker C, Malig M, Mutlu O, et al. (2009) Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet, 41:1061-1067. Alkodsi A, Louhimo R, Hautaniemi S. (2014) Comparative analysis of methods for identifying somatic copy number alterations from deep sequencing data. Briefings in Bioinformatics, 5. Bartenhagen C (2014). RSVSim: RSVSim: an R/Bioconductor package for the simulation of structural variations. Banerjee S, Oldridge D, Poptsova M, Hussain WM, Chakravarty D, Demichelis F (2011) A computational framework discovers new copy number variants with functional importance.PLoS ONE, 6:3. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, et al. (2010) The landscape of somatic copy-number alteration across human cancers. Nature, 463(7283):899-905. Benjamini Y, Speed TP. (2012) Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic acids research;40(10):e72. Bignell GR, Huang J, Greshock J, et al. (2004) High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res;14(2):287–95 . Bijlsma EK, Gijsbers ACJ, Schuurs-Hoeijmakers JHM, van Haeringen A, van de Putte DEF, et al. (2009) Extending the phenotype of recurrent rearrangements of 16p11.2: Deletions in mentally retarded patients without autism and in normal individuals. European Journal of Medical Genetics 52: 77–87. Boeva V, Zinovyev A, Bleakley K, Vert JP, Janoueix-Lerosey I, Delattre O, Barillot E. (2011) Control-free calling of copy number alterations in deepsequencing data using GC-content normalization. Bioinformatics; 27(2):268-9. Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O, Barillot E. (2012) Control-FREEC: a tool for assessing copy number and allelic content using next generation sequencing data. Bioinformatics. 28:423-5. Brosens RP, Haan JC, Carvalho B, Rustenburg F et al, (2010). Candidate driver genes in focal chromosomal aberrations of stage II colon cancer. The Journal of Pathology 221, 4, 411–424. Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LE, SchuursHoeijmakers JH, et al (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nature Genetics 46, 1063–1071 . Carter SL, et al. (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol.30:413–421. Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang QY, Locke DP, et al. (2009) BreakDancer: an algorithm for high resolution mapping of genomic structural variation. Nat. Methods 6:677-681. Chen X, Wang J, Roberts K, Pounds S, Dyer M, Mullighan C , Downing J, and Zhang J (2012) CONSERTING: an accurate method for detecting focal and gross somatic copy number alterations in cancer genome by next generation sequencing Cancer Res 72; 2487. Chiang DY, Getz G, Jaffe DB, O'Kelly MJ, Zhao X, Carter SL, Russ C, Nusbaum C, Meyerson M, Lander ES (2009) High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods, 6:99-103. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, et al.(2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 486(7403):346–352. Dancey JE, Bedard PL, Onetto N, Hudson TJ. (2012) The genetic basis for cancer treatment decisions. Cell;148(3):409–420 Dempster AP, Laird NM, Rubin DB. (1977) Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Series B Stat Methodol; pages 1–38. Diskin SJ, Hou C, Glessner JT, Attiyeh EF, Laudenslager M, Bosse K, Cole K, Mosse YP, Wood A, Lynch JE, Pecor K, Diamond M, Winter C, Wang K, Kim C, Geiger EA, et al. (2009) Copy number variation at 1q21.1 associated with neuroblastoma. Nature.; 459(7249):987-991. Duan J, Zhang J-G, Deng H-W, Wang Y-P (2013) Comparative Studies of Copy Number Variation Detection Methods for Next-Generation Sequencing Technologies. PLoS ONE 8(3): e59128. Duan J, Zhang J, Deng H and Wang Y (2013) CNV-TV: A robust method to discover copy number variation from short sequencing reads BMC Bioinformatics, 14:150. Eddie SR. (2004) What is a hidden Markov model? Nature biotechnology;22(10):1315–1316. Edelmann L, Hirschhorn K (2009) Clinical Utility of Array CGH for the Detection of Chromosomal Imbalances Associated with Mental Retardation and Multiple Congenital Anomalies. Year in Human and Medical Genetics 1151: 157–166. Fellermann K, Stange DE, Schaeffeler E, Schmalzl H, Wehkamp J, et al. (2006) A chromosome 8 gene-cluster polymorphism with low human betadefensin 2 gene copy number predisposes to Crohn disease of the colon. American Journal of Human Genetics 79: 439–448. Feuk L, Carson AR, Scherer SW. (2006) Structural variation in the human genome. Nature reviews Genetics;7(2):85–97. Forney Jr GD. (1973). The viterbi algorithm. Proceedings of the IEEE; 61(3):268–278. Frampton M, Houlston R (2012) Generation of Artificial FASTQ Files to Evaluate the Performance of Next-Generation Sequencing Pipelines. PLoS ONE 7(11): e49110. Frank B, Hemminki K, Meindl A, Wappenschmidt B, Sutter C et al.. (2007) BRIP1 (BACH1) variants and familial breast cancer risk: a case-control study. Bmc Cancer 7 Fujimoto A, Nakagawa H, Hosono N, Nakano K, Abe T, et al. (2010) Whole-genome sequencing and comprehensive variant analysis of a Japanese individual using massively parallel sequencing. Nat Genet 42: 931–936. Futreal PA, Coin L, Marshall M, et al. (2004) A census of human cancer genes. Nat. Rev. Cancer;4(3):177–83. Glessner JT, Wang K, Sleiman PMA, Zhang H, Kim CE et al.. (2010) Duplication of the SLIT3 Locus on 5q35.1 Predisposes to Major Depressive Disorder. PLOS ONE 5. Gonzalez E, Kulkarni H, Bolivar H, Mangano A, Sanchez R, et al. (2005) The influence of CCL3L1 gene-containing segmental duplications on HIV1/AIDS susceptibility. Science 307: 1434–1440. Gusnanto A, et al. (2012) Correcting for cancer genome size and tumour cell content enables better estimation of copy number alterations from nextgeneration sequence data. Bioinformatics;28:40–47. Ha G, Roth A, Lai D, et al. (2012). Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res;22(10):1995–2007. Hajirasouliha I, Hormozdiari F, Alkan C, Kidd JM, Birol I, Eichler EE, Sahinalp SC (2010). Detection and characterization of novel sequence insertions using paired-end next-generation sequencing. Bioinformatics , 26:1277-1283. Handsaker RE, Korn JM, Nemesh J, McCarroll SA (2011) Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet, 43:269-276. Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, et al. (2009) 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nature Genetics 41: 160–162. doi: 10.1038/ng.292 Hollox EJ, Huffmeier U, Zeeuwen PLJM, Palla R, Lascorz J, et al. (2008) Psoriasis is associated with increased beta-defensin genomic copy number. Nature Genetics 40: 23–25. Holt C, Losic B, Pai D, Zhao Z, Trinh Q, Syam S, Arshadi N, Jang GH, Ali J, Beck T, McPherson J, Muthuswamy LB. (2013). Wave CNVs: Allele specificCopy Number Alterations in primarytumors and xenograft modelsfromnext-generation sequencing. Bioinformatics. 32:1–7 Huynh, A.S., et al. (2011) Development of an orthotopic human pancreatic cancer xenograft model using ultrasound guided injection of cells, PLoS One, 6, e20330. Hormozdiari F, et al. (2009). Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res.19:1270-1278. Hormozdiari F, Hajirasouliha I, Dao P, Hach F, Yorukoglu D, Alkan C, Eichler EE, Sahinalp SC (2010) Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics, 26:i350-357. Hormozdiari F, Hajirasouliha I, McPherson A, Eichler EE, Sahinalp SC (2011) Simultaneous structural variation discovery among multiple pairedend sequenced genomes. Genome Res, 21:2203-2212. Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, et al. (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet 364: 1169–1171. Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G (2012) De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat Genet, 44:226-232. Ivakhno S, Royce T, Cox AJ, Evers DJ, Cheetham RK, Tavare S (2010) CNAseg--a novel framework for identification of copy number changes in cancer from second-generation sequencing data. Bioinformatics, 26:3051-3058. Kim TM, Luquette LJ, Xi R, Park PJ (2010) rSW-seq: algorithm for detection of copy number alterations in deep sequencing data. BMC Bioinformatics, 11:432. Kim TM, Xi R, Luquette LJ, et al (2013). Functional genomic analysis of chromosomal aberrations in a compendium of 8000 cancer genomes. Genome Res.;23(2):217–27. Klambauer G, Schwarzbauer K, Mayr A, Clevert DA, Mitterecker A, Bodenhofer U, Hochreiter S (2012). cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res, 40:e69 Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. (2012). VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22: 568-576. Korbel JO, Abyzov A, Mu XJ, Carriero N, Cayting P, Zhang ZD, Snyder M, Gerstein MB (2009) PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol, 10:R23. Kosugi S, Natsume S, Yoshida K, MacLean D, Cano L, et al. (2013). Coval: Improving Alignment Quality and Variant Calling Accuracy for NextGeneration Sequencing Data. PLoS ONE 8(10): e75402. Krishnan NM, Gaur P, Chaudhary R, Rao AA, Panda B. (2012) COPS: a sensitive and accurate tool for detecting somatic CopyNumber Alterations using short-read sequence data from paired samples. PLoS One. 7(10) Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, et al. (2008). Recurrent 16p11.2 microdeletions in autism. Human Molecular Genetics 17: 628–638. Lai D, Ha G, Shah S. (2012). HMMcopy, Copy number prediction with correction for GC and mappability bias for HTS data. R package version 1.2.0. Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, Li S, Yang H, Wang J, Wang J. (2010 ) De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. Feb;20(2):265-72. Liu B, Morrison CD, Johnson CS, Trump DL, Qin M, Conroy JC, Wang J, Liu S. (2013) Computational methods for detecting copy number variations in cancer genome using next generation sequencing: principles and challenges.Oncotarget; 4(11):1868-81. Liu P, Lacaria M, Zhang F, Withers M, Hastings P, Lupski JR. (2011). Frequency of nonallelic homologous recombination is correlated with length of homology: evidence that ectopic synapsis precedes ectopic crossing-over. Am J Hum Genet, 89(4):580-588. Louhimo R, Lepikhova T, Monni O, et al. (2012). Comparative analysis of algorithms for integration of copy number and expression data. Nat. Methods;9(4):351–5. Magi A, Benelli M, Yoon S, Roviello F, Torricelli F (2011) Detecting common copy number variants in high-throughput sequencing data by using JointSLM algorithm. Nucleic Acids Res 39:e65. Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, et al. (2008). Structural variation of chromosomes in autism spectrum disorder. American Journal of Human Genetics 82: 477–488. Mayrhofer M, et al. (2013). Patchwork: allele-specific copy number analysis of whole genome sequenced tumor tissue. Genome Biol. 2013;14:R24. McKinney C, Merriman ME, Chapman PT, Gow PJ, Harrison AA, et al. (2008). Evidence for an influence of chemokine ligand 3-like 1 (CCL3L1) gene copy number on susceptibility to rheumatoid arthritis. Annals of the Rheumatic Diseases 67: 409–413. McMullan DJ, Bonin M, Hehir-Kwa JY, de Vries BBA, Dufke A, et al. (2009). Molecular Karyotyping of Patients with Unexplained Mental Retardation by SNP Arrays: A Multicenter Study. Human Mutation 30: 1082–1092. Merikangas AK, Corvin AP, Gallagher L (2009). Copy-number variants in neurodevelopmental disorders: promises and challenges. Trends in Genetics 25: 536–544. Medvedev P, Fiume M, Dzamba M, Smith T, Brudno M (2010) Detecting copy number variation with mated short reads. Genome Res, 20:1613-1622. Miller CA, Hampton O, Coarfa C, Milosavljevic A (2011) ReadDepth: a parallel R package for detecting copy number alterations from short sequencing reads. PLoS One, 6:e16327. Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. (2011) Mapping copy number variation by population-scale genome sequencing. Nature, 470:59-65. Negrini S, Gorgoulis VG, Halazonetis TD (2010). Genomic in-stability–an evolving hallmark of cancer. Nat. Rev. Mol. Cell. Biol. 11(3):220–8. Nijkamp JF, van den Broek MA, Geertman JM, Reinders MJ, Daran JM, de Ridder D (2012) De novo detection of copy number variation by coassembly. Bioinformatics. Mosen-Ansorena D, Telleria N, Veganzones S, De la Orden V, Maestro M and Aransay A (2014). “seqCNA: an R package for DNA copy number analysis in cancer using high-throughput sequencing.”BMC Genomics, 15(1), pp. 178. Olshen AB, Venkatraman ES, Lucito R, Wigler M. (2004). Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 5(4):557–572. Olshen AB (b), Bengtsson H, Neuvial P, Spellman PT, Olshen RA, Seshan VE. (2011) Parent-specific copy number in paired tumor-normal studies using circular binary segmentation. Bioinformatics;27(15):2038–2046. Qi J, Zhao F (2011) inGAP-sv: a novel scheme to identify and visualize structural variation from paired end mapping data. Nucleic Acids Res, 39:W567-575 Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, Efremova M, Krabichler B, Speicher MR, Zschocke J, Trajanoski Z. (2014) A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 15(2):256-78. Pang, A., MacDonald, J., Pinto, D., Wei, J., Rafiq, M. et al. (2010) Towards a comprehensive structural variation map of an individual human genome. Genome Biology, 11:R52 Pinkel D, Segraves R, Sudar D, et al. (1998) High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet; 20(2):207–11. Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ , Humphray SJ, Greenman CD ,Varela I, et al (2010). A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463, 191-196. Popova T, Manie E, Stoppa-Lyonnet D, Rigaill G, Barillot E, Stern MH. (2009) Genome Alteration Print (GAP): a tool to visualize and mine complex cancer genomic profiles obtained by SNP arrays. Genome biology;10(11):R128. Quinlan AR, Clark RA, Sokolova S, Leibowitz ML, Zhang Y, Hurles ME, Mell JC, Hall IM (2010) Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome.Genome Res, 20:623-635. Rovelet-Lecruz A, Hannequin D, Raux G, Le Meur N, Laquerriere A, et al. (2006) APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nature Genetics 38: 24–26. Schaaf CP, Wiszniewska J, Beaudet AL. (2011) Copy number and SNP arrays in clinical diagnostics. Annual review of genomics and human genetics;12:25–51. Shen J, Zhang N. (2012) Change-point model on nonhomogeneous Poisson processes with application in copy number profiling by next-generation DNA sequencing. 6:476-496. Shlien A and Malkin D. (2009) Copy number variations and cancer. Genome medicine; 1(6):62. Simpson JT, McIntyre RE, Adams DJ, Durbin R. (2010) Copy number variant detection in inbred strains from short read sequence data. Bioinformatics. Feb 15;26(4):565-7. Sindi S, Helman E, Bashir A, Raphael BJ (2009) A geometric approach for classification and comparison of structural variants.Bioinformatics,25:i222-230. Sindi SS, Onal S, Peng LC, Wu HT, Raphael BJ (2012) An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol, 13:R22. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, et al. (2003) alpha-synuclein locus triplication causes Parkinson's disease. Science 302: 841. Speicher MR, Carter NP. (2005) The new cytogenetics: blurring the boundaries with molecular biology. Nature reviews Genetics;6(10):782–792. Stefansson H, Rujescu D, Cichon S, Pietilainen OPH, Ingason A, et al. (2008) Large recurrent microdeletions associated with schizophrenia. Nature 455: 232–U61. Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi Grassi, Lee C, Tyler-Smith C, Carter N, Scherer SW, Tavare S, Deloukas P, Hurles ME, et al. (2007) Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science;315(5813):848–853. Stratton MR, Campbell PJ, Futreal PA (2009): The cancer genome. Nature, 458(7239):719-724. Sun W, Wright FA, Tang Z, Nordgard SH, Van Loo Loo, Yu T, Kristensen VN, Perou CM. (2009) Integrated study of copy number states and genotype calls using high-density SNP arrays. Nucleic acids research;37(16):5365–5377. Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, et al. (2007) Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nature Genetics 39: 319–328. Teo SM, Pawitan Y, Ku CS, Chia KS, Salim A. (2012) Statistical challenges associated with detecting copy number variations with next-generation sequencing. Bioinformatics;28(21):2711–2718. Wang H, Nettleton D and Ying K (2014). Copy Number Variation Detection Using Next Generation Sequencing Read Counts, BMC Bioinformatics 2014, 15:109. Wang Z, Hormozdiari F, Yang W-Y, Halperin E, Eskin E (2012) CNVeM: Copy Number Variation Detection Using Uncertainty of Read Mapping. In Research in Computational Molecular Biology Edited by Chor B: Springer Berlin/Heidelberg., 7262:326-340. Lecture Notes in Computer Science. Waszak SM, Hasin Y, Zichner T, Olender T, Keydar I, Khen M, Stütz AM, Schlattl A, Lancet D, Korbel JO. (2010) Systematic inference of copynumber genotypes from personal genome sequencing data reveals extensive olfactory receptor gene content diversity. PLoS Comput Biol. Nov 11;6(11). Weiss LA, Shen YP, Korn JM, Arking DE, Miller DT, et al. (2008) Association between microdeletion and microduplication at 16p11.2 and autism. New England Journal of Medicine 358: 667–675. Wong K, Keane TM, Stalker J, Adams DJ. (2010) Enhanced structural variant and breakpoint detection using SVMerge by integration of multiple detection methods and local assembly. Genome Biol.;11(12):R128. Xi R (a), Hadjipanayis AG, Luquette LJ, Kim TM, Lee E, Zhang J, Johnson MD, Muzny DM, Wheeler DA, Gibbs RA, Kucherlapati R, Park PJ. (2011) Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proceedings of the National Academy of Sciences of the United States of America;108(46):E1128–1136 Xi R (b), et al. (2011) Detecting structural variations in the human genome using next generation sequencing. Brief. Funct. Genomics;9:405-415. Xie C, Tammi MT (2009) CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics, 10:80. Xu B, Roos JL, Levy S, Van Rensburg EJ, Gogos JA, et al. (2008) Strong association of de novo copy number mutations with sporadic schizophrenia. Nature Genetics 40: 880–885. Yang Y, Chung EK, Wu YL, Nagaraja HN, Zhou B, et al. (2007) Complement C4 gene copy number variation in human autoimmune disease systemic lupus erythematosus (SLE). Molecular Immunology 44: 261. Yau C, Mouradov D, Jorissen RN, Colella S, Mirza G, Steers G, Harris A, Ragoussis J, Sieber O, Holmes CC. (2010) A statistical approach for detecting genomic aberrations in heterogeneous tumor samples from single nucleotide polymorphism genotyping data. Genome biology;11(9):R92. Yau C (2013) OncoSNP-SEQ: a statistical approach for the identification of somatic copy number alterations from next-generation sequencing of cancer genomes. Bioinformatics 29(19):2482-4. Ye K, et al. (2009) Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics;25:2865-2871. Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. (2009) Sensitive and accurate detection of copy number variants using read depth of coverage. Genome research;19(9):1586–1592. Yu Z, Liu Y, Shen Y, Wang M, Li A. (2014) CLImAT: accurate detection of copy number alteration and loss of heterozygosity in impure and aneuploid tumor samples using whole-genome sequencing data. Bioinformatics. 30(18):2576-83. Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang C-Z, Wala J, Mermel CH, Sougnez C, Gabriel SB, Hernandez B, Shen H, Laird PW, Getz G, Meyerson M, Beroukhim R (2013) Pan-cancer patterns of somatic copy number alteration. Nat Genet, 45(10):1134-1140. Zhang J, Wu Y (2011). SVseq: an approach for detecting exact breakpoints of deletions with low-coverage sequence data.Bioinformatics, 27:32283234. Zhang Q, Ding L, Larson DE, Koboldt DC, McLellan MD, Chen K, Shi X, Kraja A, Mardis ER, Wilson RK, et al. (2010) CMDS: a population-based method for identifying recurrent DNA copy number aberrations in cancer from high-resolution data. Bioinformatics, 26:464-469. Zhang ZD, Du J, Lam H, Abyzov A, Urban AE, Snyder M, Gerstein M (2011) Identification of genomic indels and structural variations using split reads. BMC Genomics, 12:375. Zeitouni B, Boeva V, Janoueix-Lerosey I, Loeillet S, Legoix-ne P, Nicolas A, Delattre O, Barillot E (2010). SVDetect: a tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics, 26:1895-1896. Zhao M, Wang Q, Wang Q, Jia P, Zhao Z (2013) Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics, 14(Suppl 11):S1 Zhao Z, Boerwinkle E (2002) Neighboring-nucleotide effects on single nucleotide polymorphisms: a study of 2.6 million polymorphisms across the human genome. Genome Res 12: 1679–1686. Zhenhua Yu, Yuanning Liu, Yi Shen,1 Minghui Wang, and Ao Li (2014) CLImAT: accurate detection of copy number alteration and loss of heterozygosity in impure and aneuploid tumor samples using whole-genome sequencing data. Bioinformatics. Sep 15;30(18):2576-83.


![[MAD]vie25. La autoridad cuestionada](http://s2.studylib.es/store/data/003930021_1-ac9b47a903fd189ed19a937972401d6d-300x300.png)