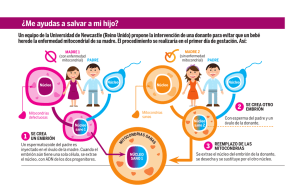
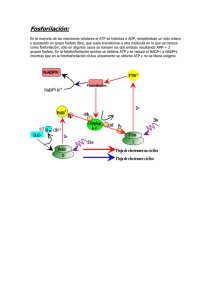
Tesis - Universidad de Colima
Anuncio

UNIVERSIDAD DE COLIMA FACULTAD DE MEDICINA CENTRO UNIVERSITARIO DE INVESTIGACIONES BIOMÉDICAS TESIS PARA OBTENER EL GRADO DE MAESTRO EN CIENCIAS FISIOLÓGICAS CON ESPECIALIDAD DE FISIOLOGÍA “PARTICIPACIÓN DE LOS CANALES MITOCONDRIALES DE K+ SENSIBLES A ATP EN LA FATIGA EN MÚSCULO ESQUELÉTICO DE POLLO” Que presenta la BIOL. ROCÍO DEL CARMEN MONTOYA PÉREZ ASESORA: DRA. XÓCHITL A. R. TRUJILLO TRUJILLO COASESOR: DR. ALFREDO SAAVEDRA MOLINA IIQB-UMSNH COLIMA, COL. JULIO DE 2006 Estar preparado es importante, saber esperar lo es aún más, pero aprovechar el momento adecuado es la clave de la vida. Arthur Schnitzler Dramaturgo austriaco. -2- DEDICATORIA A Rosita, Karina (Juan), Millo y Sinhué (Enano). AGRADECIMIENTOS Al Consejo Nacional de Ciencia y Tecnología, por otorgarme una beca para realización de mi maestría. A la Dra. Xochitl A. R. Trijillo Trujillo por fungir como mi asesora y brindarme su apoyo para el desarrollo de la maestría y la conclusión de la presente tesis. De igual forma al apoyo que recibe el proyecto que dirige: FRABA 378/05-XT. Al Dr. Alfredo Saavedra Molina, por recibirme en su laboratorio y apoyarme en la realización de la parte experimental del esta tesis y fungir como mi coasesor y a los apoyos de su proyecto: CONACyT (43705) y CIC-UMSNH (216). A SantanderUniversia en su convocatoria 2005-2, por otorgarme una beca de estancia de investigación en el Laboratorio de Bioquímica del Instituto de Investigaciones Químico-Biológicas de la Universidad Michoacana de San Nicolás de Hidalgo. A la Universidad de Colima por brindarme apoyo económico para presentar los resultados de esta tesis en la II Conferencia Mujer Ciencia 2006, 3-5 de Mayo de 2006, México, D.F. A mis compañeros de la maestría: Bety, Christina, Isaac y el Ing. Arturo; y del laboratorio: Enrique y Mónica, por su compañía y apoyo, pero sobre todo por esas pláticas amenas interminables. A Jersahí por estar conmigo en la buenas y en las malas. -3- PRESENTACIÓN DE RESULTADOS Los resultados de la presente tesis también se presentaran en el XLIX Congreso Nacional de Ciencias Fisiológicas que se realizará en la cuidad de Querétaro del 4-8 de Septiembre de este año y en The Annual Meeting Society for Neuroscience 2006 que se llevará a cabo en la cuidad de Atlanta, Georgia, USA del 14-18 de Octubre. -4- ÍNDICE RESUMEN………………………………………………………………………………..11 ABSTRACT……………………………………………………………………………….12 INTRODUCCIÓN.………………………………………………………………………..13 EL MÚSCULO……………...…………………………………………………….13 Clasificación de las fibras musculares esqueléticas………………….16 Fibras musculares de sacudida lenta o tipo I………………….17 Fibras musculares de sacudida rápida o tipo IIa……………...17 Fibras musculares de sacudida rápida o tipo IIb……………...18 LA FATIGA MUSCULAR………………..………………………………………20 MITOCONDRIA…………………………………………………………………..23 La cadena respiratoria colecta y oxida equivalentes reducidos…….25 Los componentes de la cadena respiratoria se colocan en orden creciente de su potencial redox…………………………………26 La cadena respiratoria aporta la mayor parte de la energía capturada en el metabolismo…………………………………………...30 El control respiratorio asegura el suministro constante de ATP…….32 Inhibición de la cadena respiratoria…………………………………….34 La teoría quimiosmótica explica el mecanismo de la fosforilación oxidativa y la formación de ATP…………………………36 Una ATP sintasa localizada en la membrana forma el ATP………...38 La teoría quimiosmótica puede explicar el control respiratorio……..40 La teoría quimiosmótica explica la acción de los desacopladores…40 El transporte iónico de la mitocondria requiere de energía………….42 Génesis mitocondrial…………………………………………………….42 Control de la expresión de DNA mitocondrial…………………46 Coordinación del DNA mitocondrial y nuclear………………...47 EL PAPEL DEL ATP EN LA ENERGÉTICA MUSCULAR…………………..49 LOS CANALES DE K+ SENSIBLES A ATP…………………………………..49 Regulación de los canales KATP………………………………………...50 Canales KATP mitocondriales……………………………………………52 -5- Composición de los canales KATP mitocondriales…………………….53 El antiportador de K+/H+…………………………………………………55 Fatiga muscular y canales KATP………………………………………...55 Precondicionamiento isquémico………………………………………..57 Relación entre el canal KATP mitocondrial y la PKC…………………..58 Relación entre el canal KATP mitocondrial y el óxido nítrico (NO)…..59 OBJETIVO GENERAL…………………………………………………………………..61 OBJETIVOS PARTICULARES…………………………………………………………61 MÉTODOS………………………………………………………………………………..62 Material biológico………………………………………………………………...62 Disección………………………………………………………………………….62 Extracción…………………………………………………………………………62 Soluciones………………………………………………………………………...63 Oximetría………………………………………………………………………….64 Análisis de datos…………………………………………………………………65 RESULTADOS…………………………………………………………………………..66 Efecto de la glibenclamida sobre el consumo de oxigeno en el estado 3 de la respiración………………………………………………..66 Efecto del pinacidil sobre el consumo de oxigeno en el estado 3 de la respiración………………………………………………..69 DISCUSIÓN………………………………………………………………………………73 CONCLUSIONES………………………………………………………………………..79 PERSPECTIVAS…………………………………………………………………………80 REFERENCIAS…………………………………………………………………………..81 -6- ÍNDICE DE CUADROS Y FIGURAS Cuadro 1. Clasificación de las fibras musculares esqueléticas…………………….17 Cuadro 2. Potenciales redox estándar de reacción en un solo sentido de los componentes principales de la cadena respiratoria……..………27 Cuadro 3. Estados de control respiratorio de la mitocondria………………………..32 Figura 1. Organización estructural y de origen de las células musculares………..15 Figura 2. Contracción rápida o lenta de diferentes músculos………………………19 Figura 3. Tipos de respuesta a la fatiga de diferentes músculos esqueléticos…...20 Figura 4 Comparación de las tres vías de producción de ATP……………………..22 Figura 5. Estructura de las membranas mitocondriales……………………………..24 Figura 6. La función de la cadena respiratoria es la transformación de la energía de los alimentos en ATP…………………………………….25 Figura 7. Transporte de equivalentes reducidos a través de la cadena respiratoria…………………………………………..26 Figura 8. Componentes de la cadena respiratoria…………………………………...28 Figura 9. Esquema de la disposición de los componentes de una cadena transportadora de electrones de la membrana interna mitocondrial……………………………………….30 Figura 10. Sitios de inhibición de la cadena respiratoria…………………………...32 Figura 11. Participación del ADP en el control de la respiración…………………...33 Figura 12. Control de la respiración mitocondrial…………………………………….36 Figura 13. Ilustra los principios de la teoría quimiosmótica…………………………37 Figura 14. Estructura de la ATP sintasa………………………………………………38 Figura 15. Hipótesis de "cambio de enlace"…………………………………………..40 Figura 16. Biogénesis mitocondrial en la célula muscular…………………………..48 Figura 17. Interacción de lo abridores y bloqueadores del canal de potasio sensible a ATP………………………………………54 -7- Figura 18. Efecto de la glibenclamida sobre el consumo de O2 en estado 3 de la respiración de mitocondrias aisladas de músculo esquelético de pollo………………………………………………67 Figura 19. Curva dosis-respuesta del efecto de la glibenclamida………………….68 Figura 20. Registro representativo del efecto de la glibenclamida sobre el consumo de oxígeno……………………………………………...68 Figura 21. Efecto del pinacidil sobre el consumo de O2 en estado 3 de la respiración de mitocondrias aisladas de músculo esquelético de pollo………………………………………………70 Figura 22. Curva dosis-respuesta del efecto del pinacidil…………………………..71 Figura 23. Registro representativo del efecto del pinacidil sobre el consumo de oxígeno……………………………………………..71 -8- ABREVIATURAS 5-hidroxidecanoato (5-HD) Ácido desoxirribonucleíco; deoxyribonucleic acid (DNA) Ácido ribonucleíco de transferencia; transference ribonucleic acid (tRNA) Ácido ribonucleico mensajero; messenger ribonucleic acid (mRNA) Ácido sulfhídrico (H2S) Ácido ribonucleíco ribosomal; ribosomal ribonucleic acid (rRNA) Adenosín difosfato (ADP) Adenosín monofosfato (AMP) Adenosín trifosfato (ATP) Agua (H2O) Anterior latissimus dorsi (ALD) Canales de potasio dependientes de ATP (KATP) Canales KATP mitocondriales (mito- KATP) Canales rectificadores entrantes (Kir6.x) Citocromo (Cit) Concentración media (EC50) Constante de disociación (Km) DNA mitocondrial (mtDNA) Electrón (e-) Energía libre (∆G) Energía libre estándar (∆G°) Equivalentes reducidos (2H) Flavín adenín dinucleótido (FAD) Flavín mononucleótido (FMN) Flavoproteína (Fp) Flavoproteína transferradora de electrones (ETF) Fosfatidilinositol 3-cinasa (PI3 cinasa) Fosfatidilinositol 4,5 bifosfato (PIP2) Fosfato inorgánico (Pi) Guanidín difosfato (GDP) -9- Guanidín trifosfato (GTP) Hidrógeno (H+) Hidroxilos (OH-) Ion dihidrógeno fosfato (H2PO4) Mitocondrias intermiofribrilares (IMFmt) Mitocondrias subsarcolemales (SSmt) Nanoátomos (nat) Nicotín adenín dinucleótido oxidado (NAD) Nicotín adenín dinucleótido reducido (NADH) Oxido nítrico (NO) Oxigeno (O2) Posterior latissimus dorsi (PLD) Potencial electroquímico a través de la membrana (∆µh+) Precondicionamiento isquémico (IPC) Presión parcial de oxígeno (PO2) Proteína cinasa C; protein kinase C (PKC) Proteínas cinasas activadas por mitógenos; mitogen activated protein kinases MAP kinase (MAP cinasa) Receptores a sulfonilureas (SUR) Retículo sarcoplásmico (RS) Succinato deshidrogenasa (SDH) Sulfoproteína férrica (FeS) Superfamilia cassette de unión a ATP; ATP-binding cassette (ABC) Transcripcion inversa de la reacción en cadena de la polimerasa; Reverse transcription polymerase chain reaction (RT-PCR) Ubiquinona (Q) - 10 - RESUMEN Los canales KATP fueron descritos por Noma (1983) en la membrana plasmática de células cardiacas y se han reportado en la membrana de células β-pancreáticas, neuronas, músculo liso y esquelético. También, se ha determinado su presencia en la membrana interna mitocondrial (Inoue et al, 1991; Hegazy et al., 1991) y ambos tipos de canales tienen la habilidad de acoplar el potencial de membrana plasmático (y/o el potencial redox mitocondrial y/o la liberación de radicales libres) al estado metabólico de la célula (Nichols et al., 1991; Pain et al., 2000; O’Rourke, 2000). Los canales KATP son heteromultímeros de dos tipos de subunidades: canales rectificadores entrantes, Kir6.x, y receptores a sulfonilureas, SUR; ordenados en tetrámeros (SUR/Kir). Garlid y cols. (1997) demostraron que el mitoKATP juega un papel importante en la cardioprotección puesto que está relacionado con la regulación del volumen mitocondrial, teniendo consecuencias en el metabolismo energético (Dos Santos et al., 2002; Belisle et al., 2002). Experimentos previos sugieren que el canal KATP sarcolemal en músculo lento de pollo contribuye a proteger al músculo de la fatiga, puesto que la glibenclamida, un bloqueador de estos canales incrementa la tensión de sacudida y tetánica de músculo fatigado hasta un 30%. También estudiamos el efecto de estos fármacos sobre el consumo de oxígeno de mitocondrias aisladas (con sustratos diferentes) y nuestros resultados muestran una disminución (concentración-dependiente) de la respiración al aplicar la glibenclamida, mientras que no se encontró diferencia estadísticamente significativa cuando se usó el pinacidil y el 5-hidroxidecanoato (5-HD) en presencia de succinato como sustrato. Por otro lado, se encontraron diferencias significativas en el consumo de oxígeno al aplicar pinacidil en presencia de glutamato-malato como sustrato, más no así cuando se utilizó glibenclamida y 5-HD. El no tener efecto con el 5-HD nos sugiere que el canal mito-KATP de pollo podría estar formado por subunidades diferentes, por lo que su comportamiento farmacológico difiere de lo descrito hasta ahora en preparaciones de músculo cardiaco. Los efectos de la glibenclamida y el pinacidil sobre la respiración mitocondrial al diferir con resultados previamente descritos, nos permiten observar nuevas perspectivas que requieren de más estudios. - 11 - ABSTRACT KATP channels were described by Noma (1083) in the plasmatic membrane of cardiac, β-pancreatic, neurons, smooth and skeletal muscle cells. In addition, these channels has been identified in the internal mitochondrial membrane (Inoue et al., 1991; Hegazy et al., 1991), where they have the ability to couple transmembrane potential (and/or the mitocondrial transmembrane potential redox and/or the production of reactive oxygen species) to the metabolic state of the cell (Nichols et al., 1991; Pain et al., 2000; O’Rourke, 2000). Structural conformation of KATP channels consists of a heteromultimer of an inward rectifying potassium channel (Kir 6.x) and a sulfonylurea receptor SUR, organized in a tetrameric form (SUR/Kir). Garlid et al., (1997) demonstrated that mito-KATP plays an important role in cadioprotection, because they are related with mitochondrial volume regulation, having effects over energetic metabolism (Dos Santos et al., 2002; Belisle et al., 2002). Previous experiments suggest that sarcolemmal KATP channels in chicken slow skeletal muscle contributes to protect the muscle of fatigue, since glibenclamide, a selective blocker of these channels, increases twitch and tetanic tension in post-fatigued muscle up to 30%. We also studied the effects of these drugs on oxygen consumption of isolated mitichondria (with differt substrates). The results showed a diminution (concentration-dependent) of the mitocondrial respiration when glibenclamide was present, while any significant effect with pinacidil and 5-hidroxidecanoate with succinate as a substrate. On the other hand, we found in mitochondrial respiration significative differences in the effects of pinacidil with glutamate-malate as substrate and no effects with glibenclamide and 5-hydroxydecanote. The negative effects with 5-hydroxydecanoate suggests that the mito-KATP channels could be formed with different subunits, while its pharmacologic behavior is different for what it has been described so far for cardiac muscle. Our results with glibenclamide and pinacidil are different also from which has been described previously. It allows us new perspectives for which more studies are required. - 12 - INTRODUCCIÓN EL MÚSCULO Los músculos están especializados para contraerse y proporcionan fuerza y movimiento a los animales. Los músculos por su aspecto al microscopio se clasifican tomando como base la presencia o ausencia de estriaciones o bandas. Eckert (1989) clasifica a los músculo en dos grandes grupos: A) Músculo liso: se denomina liso porque carece de estriaciones o bandas. En este músculo los filamentos se hallan distribuidos aleatoriamente en el mioplasma. Forman parte de este tipo el músculo retractor del bazo y los músculos aductores de los moluscos bivalvos, asimismo, el músculo que forma la pared de los órganos viscerales en los vertebrados (tubo digestivo, vejiga urinaria, uréteres, arterias y arteriolas). Las células musculares lisas están conectadas en grupos unas con otras, por uniones comunicantes que permiten la propagación eléctrica de la corriente iónica de una célula a otra. Por consiguiente, estas células forman unidades funcionales acopladas eléctricamente en haces de alrededor de 100 µm de espesor y de varios milímetros de largo (sincitio). La inervación del músculo liso es discreta y ocurre en plexos o agrupamientos de neuronas sensoriales, en interneuronas y en neuronas motoras. B) Músculo estriado: dentro de esta clasificación encontramos al cardiaco (en corazón) y al músculo esquelético (asociado al esqueleto). El músculo cardiaco está inervado difusamente por fibras nerviosas simpáticas (excitadoras, con adrenalina) y parasimpáticas (inhibidoras, con acetilcolina), ambas del sistema nervioso autónomo. La inervación cardiaca es solo moduladora de la contracción del corazón y sus acciones están dirigidas a incrementar o reducir la fuerza de la contracción. En contraste, la fibra muscular esquelética se halla inervada individualmente por un axón motor excitador y el neurotransmisor es la acetilcolina. - 13 - La unidad básica de todo músculo es la miofibrilla, estructura filiforme muy pequeña formada por proteínas complejas. La célula muscular contiene varias miofibrillas, compuestas de miofilamentos de dos tipos, gruesos y delgados, que adoptan una disposición regular (Véase figura 1). Cada miofilamento grueso contiene varios cientos de moléculas de la proteína miosina. Los filamentos delgados contienen dos cadenas de la proteína actina. Las miofribrillas están formadas de hileras que alternan miofilamentos gruesos y delgados con sus extremos traslapados. Durante las contracciones musculares, estos tipos de filamentos interdigitados se deslizan uno sobre otro por medio de puentes cruzados que actúan como ruedas. La energía que requiere este movimiento procede de mitocondrias que rodean las miofibrillas (http://es.encarta.msn.com). - 14 - Figura 1. Organización estructural y origen de las células musculares. (Tomado de Eckert, 1998). - 15 - Clasificación de las fibras musculares esqueléticas Un criterio de clasificación del tipo de las fibras musculares esqueléticas radica en su duración de la respuesta mecánica al ser estimuladas eléctricamente. En este sentido, las fibras musculares que responden con una sacudida única que relaja rápidamente se denominan fibras musculares rápidas. Aquellas fibras musculares que responden con una sacudida de seis a ocho veces más lenta que la de las fibras rápidas, se denominan fibras musculares lentas (Page, 1969). Finalmente, aquellas fibras musculares que no responden con una sacudida se denominan fibras musculares tónicas. Generalmente estos dos tipos de fibras musculares se hallan entremezcladas en los músculos esqueléticos y existen pocos músculos con fibras exclusivamente lentas, como es el músculo Anterior Latissiumus dorsi (ALD) de las aves. Otra manera de clasificar a las fibras musculares se basa en sus propiedades metabólicas, el número de mitocondrias que presentan, el tipo de la isoenzima lactato deshidrogenasa, la actividad de la miosina ATPasa y el contenido de glucógeno (Eckert, 1989). Como se dijo anteriomente, ante un estímulo eléctrico, las fibras musculares lentas generan una sacudida mas prolongada que aquella de las fibras musculares rápidas (Page, 1969). En el caso de las fibras musculares tónicas, éstas desarrollan una contracción graduada (Kuffler y Vaughan-Williams, 1953 a, b). A la respuesta mecánica (sacudida única) que las fibras musculares rápidas y lentas desarrollan se le puede medir la amplitud al pico, la velocidad de contracción, la resistencia a la fatiga, etc. Con estas características y las mencionadas en el párrafo anterior, Burke et al., (1971) clasificaron a las fibras musculares en tres tipos que se describen en el Cuadro 1. - 16 - Cuadro 1. Clasificación de las fibras musculares esqueléticas de acuerdo a Burke et al., (1971). Tipo I Tipo IIa Tipo IIb Fibras de Fibras de sacudida Fibras de sacudida lenta rápida (a) sacudida rápida (b) Metabolismo Metabolismo Metabolismo oxidativo lento oxidativo y glucolítico glucolítico Fibras rojas Fibras pálidas Fibras blancas Fibras musculares de sacudida lenta o tipo I Son fibras musculares cuya velocidad de contracción es lenta; desarrollan poca fuerza y presentan resistencia a la fatiga. Desde el punto de vista Bioquímico, son fibras ricas en enzimas oxidativas, pero bajas en enzimas glucolíticas y presentan baja actividad de ATPasa. Son llamadas también fibras rojas por su alto contenido de mioglobina y poseen alto número de mitocondrias; por lo que son más dependientes del metabolismo aeróbico. Su diámetro es menor en comparación con las fibras rápidas. Presentan escaso desarrollo del retículo sarcoplásmico (RS), por lo que son más dependientes del calcio extracelular que los otros tipos de fibras musculares (Page, 1969; Huerta y Stefani, 1981). Fibras musculares de sacudida rápida o tipo IIa Son fibras de velocidad de contracción más rápida que la de las fibras lentas, pero a diferencia de las fibras tipo IIb, éstas presentan una resistencia a la fatiga intermedia, puesto que pueden mantener la fuerza aún después de un gran número de contracciones. Estas fibras tienden a tener una gran actividad - 17 - glucolítica y oxidativa y además, presentan una gran actividad de ATPasa. Son llamadas también fibras pálidas; presentan menor contenido de mioglobina, alto número de mitocondrias y moderada capacidad oxidativa. Son fibras de diámetro intermedio y poseen un buen desarrollo del RS, teniendo hasta tres veces más disponibilidad de Ca2+ que las fibras tipo I. Fibras musculares de sacudida rápida o tipo IIb Este grupo de fibras musculares tiene como característica una velocidad de contracción muy rápida (6 a 8 veces más rápida que las fibras musculares lentas) y desarrollan mayor cantidad de fuerza que las fibras tipo I y tipo IIa. Sin embargo, son incapaces de mantenerla; es decir, se fatigan fácilmente. Tienen alta velocidad de consumo de ATP y presentan baja capacidad oxidativa. Son llamadas también fibras blancas por su bajo contenido de mioglobina. Poseen pocas mitocondrias y son muy dependientes del metabolismo glucolítico. Son fibras de diámetro grueso y al igual que las fibras tipo IIa, poseen un buen desarrollo del retículo sarcoplásmico (RS) y por lo tanto poseen una gran capacidad de almacenamiento de Ca2+. Como ya se mencionó, en los músculos esqueléticos de los vertebrados generalmente coexisten los tres tipos de fibras y la proporción de un tipo u otro está genéticamente regulada. Una característica importante del músculo esquelético es la capacidad de adaptarse para cambiar las propiedades funcionales de las fibras musculares (Punkt, 2002). En los músculos rápidos de los mamíferos, esa mezcla de fibras tipo I y II confiere la velocidad de contracción del músculo completo y la contracción es rápida o lenta dependiendo de la mayor proporción del tipo de fibras que el músculo tenga (véase figura 2). Además, la actividad de la fibra muscular depende de su inervación; es decir, éstas adquieren las características que les confiere el patrón de disparo de la neurona que las inerva. En el ser humano, la proporción de fibras IIa y IIb varía con la edad y el - 18 - entrenamiento las hace más resistentes a la fatiga, una actividad que ha sido aprovechada por los atletas (Wicks y Hood, 1991). Figura 2. Contracción rápida o lenta de diferentes músculos. A, frontalis/orbicularis oculi (15% de fibras lentas); B, Interóseo del primer dedo (57% de fibras lentas); C, sóleo (80% de fibras lentas); D, extensor digitorum brevis (60% de fibras lentas). Nótese que cuanto mayor es la proporción de fibras lentas, la duración de la sacudida se incrementa (Modificado de McArdle et al., 1996). En las aves, la parte anterior del músculo latissimus dorsi se caracteriza por poseer exclusivamente fibras lentas y la parte posterior de dicho músculo latissimus dorsi (PLD) está constituido exclusivamente de fibras rápidas (Ginsborg, 1960; Ginsborg y Mackay, 1961). Esta disposición y composición del músculo latissimus dorsi posibilita estudiar en detalle características propias de los tipos de fibras musculares de nuestro interés. Además, las contracturas inducidas por K+ en ambos tipos de fibras están bien caracterizadas, ya que las fibras rápidas del PLD en soluciones con K+ o cafeína, desarrollan una contractura transitoria; es decir, relajan espontáneamente. En contraste, las fibras musculares lentas del ALD desarrollan una contractura sostenida que depende de la entrada de Ca2+ (Page, 1969; Huerta y Stefani, 1981; Trujillo et al., 2002). Cuando estas fibras musculares son estimuladas repetitivamente presentan una gran resistencia a la fatiga, una - 19 - característica propia de las fibras musculares lentas tipo I de acuerdo a la clasificación de Burke et al., 1971 (véase Figura 3). Figura 3. Tipos de respuesta a la fatiga de diferentes músculos esqueléticos. En A, se ilustra la respuesta mecánica (sacudida única) de las fibras musculares que se fatigan facilmente. En B, se muestra la respuesta de las fibras musculares resistentes a la fatiga. En C, la respuesta de las fibras musculares muy resistentes a la fatiga (Modificado de Burke et al., 1971). LA FATIGA MUSCULAR La fatiga se describe como la reducción en la potencia física o como una falla en el mantenimiento de la producción de energía (Edwards, 1981); cualquier factor que reduzca la velocidad del desarrollo de la fuerza puede contribuir a la fatiga en el periodo inicial (pico) seguido de la activación del músculo. En el músculo esquelético coexisten las fibras musculares fatigables rápidas y aquellas resistentes a la fatiga; estando en menor proporción las segundas. - 20 - Considerando la clasificación de Burke et al., (1971), el hecho de que las fibras musculares sean o no resistentes a la fatiga es una característica que es atribuida a las propiedades metabólicas propias del tipo de fibra muscular (Véase Figura 3). Así, se ha mencionado que la fatiga metabólica ocurre por acumulación de metabolitos (principalmente, adenosina difosfato (ADP) y fosfato inorgánico (Pi)), por depleción de sustratos (adenosina trifosfato (ATP), fosfato de creatina y otros) o a la disminución del pH intracelular (Enoka y Stuart, 1992). Estudios de Westerblad y Allen (2002), sugieren que el Pi como metabolito acumulado, tiene efectos muy importantes como causa de fatiga. En las células musculares son dos las vías por las cuales se produce el ATP necesario para la contracción muscular. Una de ellas es la glucólisis y la otra la fosforilación oxidativa, la cual es más eficiente puesto que mientras en la primera solo se producen 2 ATP por cada glucosa que se consume, durante la fosforilación oxidativa que se realiza en la mitocondria, se producen 28 ATP por cada glucosa (Mitchell, 1961 citado por Solis, 2003(b)) (ver figura 4). De ahí, la eficiencia de las fibras musculares metabólicamente activas de tener un alto contenido de mitocondrias que les permiten generar la energía necesaria para realizar la contracción muscular. Es importante señalar, que la creatina es capaz de fosforilarse y formar el complejo fosfato de creatina y esto se denomina fosforilación directa a nivel de sustrato, en la cual ocurre el consumo de energía o bien desfosforilarse (cuando cede un fosfato inorgánico al ADP para formar ATP). A este fenómeno bioenergético se le denomina fosforilación directa (Véase figura 4). - 21 - Figura 4. Comparación de las tres vías de producción de ATP (Modificado de Berne y Levy, 1992). Durante el desarrollo de alguna actividad se deben mantener ciertos niveles en la concentración de ATP en el citosol, porque este es el sustrato que suple inmediatamente la energía para la generación de fuerza en el puente cruzado. También es necesario el ATP para el funcionamiento de la bomba Na+ - K+ que es muy importante para el mantenimiento del potencial de reposo de la membrana. El ATP es el sustrato que se requiere para la recaptación del Ca2+ por el RS, y como ya se había mencionado, la intervención de este proceso puede ser una causa potencial de la fatiga. En un trabajo extenuante, la depleción del glucógeno muscular se da después de un periodo largo de duración del trabajo cuando este se desdobla a glucosa. Al agotarse la glucosa circulante en el torrente sanguíneo, la fatiga se produce por la falta de carbohidratos que posteriormente entran al ciclo del ácido cítrico. - 22 - LA MITOCONDRIA La mitocondria es la responsable de la producción de ATP en las células, el cual es el compuesto que provee de energía al músculo durante la contracción. En general, en la mitocondria se produce la mayor parte de energía del músculo bajo condiciones aeróbicas. Las diferencias en la capacidad oxidativa entre los diferentes tejidos se debe principalmente al contenido mitocondrial, medido bioquímicamente (actividad enzimática), ultraestructuralmente (densidad) o genéticamente (la cantidad de DNA (siglas en inglés deoxyribonucleic acid) mitocondrial)) (Moyes et al., 1998). Las mitocondrias son organelos membranosos que como ya se dijo son importantes en la regulación de la energética celular. Estas poseen una membrana exterior permeable a la mayor parte de los metabolitos, ya que a través de porinas, permiten el paso de moléculas de hasta 10000 daltones (Karp, 1998), y una membrana interna con permeabilidad selectiva que se dobla en pliegues con crestas. En esta, es necesaria la presencia de transportadores para el acceso de iones y moléculas (Bernardi, 1999) (figura 5). La membrana externa se puede remover mediante el tratamiento con digitonina, y se caracteriza por la presencia de las enzimas monoaminooxidasas, acil-CoA sintetasa, glicerolfosfato aciltransferasa, monoacilglicerol fosfato aciltransferasa y fosfolipasa A2. Mientras que el fosfolipido cardiolipina se concentra en la membrana interna (Murray et al., 2001). - 23 - Figura 5. Estructura de las membranas mitocondriales. Los complejos proteícos de la membrana se encuentran dirigidos hacia la matriz de la membrana mitocondrial interna. La aplicación de ultrasonido permite la formación de mitosomas, favoreciendo el estudio de los componentes de la membrana interna. Véase texto para detalle. (Tomada de Murray et al., 2001). Las enzimas solubles responsables de llevar a cabo el ciclo del ácido cítrico y las enzimas de la β-oxidación de los ácidos grasos están en la matriz, por lo cual se requieren mecanismos para el transporte de los metabolitos y los nucleótidos necesarios a través de la membrana interna. La succinato deshidrogenasa se ubica en la superficie interna de la membrana mitocondrial, sitio del cual transporta los equivalentes reductores hacia las enzimas de la cadena respiratoria. En la superficie exterior de la membrana interna se localiza la glicerol 3-fosfato deshidrogenasa, en posición adecuada para su participación en la lanzadera de glicerolfosfato. En el lado matricial de la membrana interna mitocondrial también se enlaza la 3-hidroxibutirato deshidrogenasa. (Murray et al., 2001). - 24 - La cadena respiratoria colecta y oxida equivalentes reducidos La energía liberada durante la oxidación de los ácidos grasos y los aminoácidos, así como la liberada a partir de la oxidación de los carbohidratos, queda disponible en el interior de la mitocondria en forma de equivalentes reducidos (2H). Las mitocondrias contienen una serie de catalizadores conocida como cadena respiratoria, la cual colecta y transporta a los equivalentes reductores y los conduce hacia su reacción final con el oxígeno para formar agua. También, presentan la maquinaria para acoplar la unión de fosfato al ADP con la energía liberada de la cadena respiratoria. Estos organelos también contienen los sistemas enzimáticos responsables de la producción de la mayor parte de los equivalentes reducidos, como las enzimas de la β-oxidación y del ciclo del ácido cítrico. Este último constituye la vía metabólica común para la oxidación de los principales alimentos (Murray et al., 2001) (figura 6). Figura 6. La función de la cadena respiratoria es la transformación de la energía de los alimentos en ATP. La oxidación de los alimentos genera 2H, que se colectan por la cadena respiratoria para su oxidación y la generación acoplada de ATP y agua. (Tomada de Murray et al., 2001). - 25 - Los componentes de la cadena respiratoria se colocan en orden creciente de su potencial redox El hidrógeno y los electrones que se generaron en el ciclo de Krebs fluyen a lo largo de la cadena respiratoria en etapas, es decir, a partir de los componentes de mayor potencial redox negativo hacia los componentes de mayor potencial redox positivo, a través de un intervalo de 1.1 V que abarca desde el nicotín adenín dinucleótido oxidado/reducido (NAD+/NADH) hasta el oxígeno/agua (O2/2H2O). En la figura 7, se muestran los componentes principales de la cadena respiratoria. (Véase también Cuadro 2). Figura 7. Transporte de equivalentes reducidos a través de la cadena respiratoria. (Tomada de Murray et al., 2001). La cadena respiratoria en la mitocondria consta de varios acarreadores redox, que proceden de la enzima NADH+ deshidrogenasa enlazada al NAD+ hasta el oxígeno molecular, con las flavoproteínas y los citocromos en las etapas intermedias. No todos los sustratos se vinculan a la cadena respiratoria mediante las deshidrogenasas específicas para NAD+; algunos, debido a que poseen potenciales redox más positivos (por ejemplo, el fumarato/succinato), se vinculan directamente con las deshidrogenasas de flavoproteínas, las cuales, a su vez, se vinculan con los citocromos de la cadena respiratoria (Murray et al., 2001). - 26 - Cuadro 2. Potenciales redox estándar de reacción en un solo sentido de los componentes principales de la cadena respiratoria. (Tomado de Karp, 1998). Ecuación de electrodo Acetato + 2H+ + 2e- ⇔ acetaldehído 2H+ + 2e- ⇔ H2 α-cetoglutarato + CO2 + 2H+ + 2e- ⇔ isocitrato Acetoacetato + 2H+ + 2e- ⇔ β- hidroxibutirato NAD+ + 2H+ + 2e- ⇔ NADH + H+ NADP+ + 2H+ + 2e- ⇔ NADPH + H+ Acetaldehído + 2H+ + 2e- ⇔ etanol Piruvato + 2H+ + 2e- ⇔ lactato Oxalacetato + 2H+ + 2e- ⇔ malato FAD + 2H+ + 2e- ⇔ FADH2 (flavoproteínas) Fumarato + 2H+ + 2e- ⇔ succionato 2 citocromo bK(ox) + 2e- ⇔ 2 citocromo bK(red) Ubiquinona + 2H+ + 2e- ⇔ Ubiquinol 2 citocromo cox + 2e- ⇔ 2 citocromo cred 2 citocromo a3(ox) + 2e- ⇔ 2 citocromo a3(red) ½ O2 + 2H+ + 2e- ⇔ H2O E′0(V) -0.58 -0.421 -0.38 -0.346 -0.32 -0.324 -0.197 -0.185 -0.166 +0.031 +0.031 +0.030 +0.01 +0.254 +0.385 +0.816 En la actualidad, queda clara la presencia de un acarreador adicional en la cadena respiratoria, que vincula a las flavoproteínas con el citocromo b. Este integrante de la cadena de citocromos con menor potencial redox, que bajo condiciones aerobias, se le ha denominado ubiquinona o coenzima Q. Existe en las mitocondrias en forma de quinona oxidada, y bajo condiciones anaerobias existe la forma reducida, quinol. La coenzima Q forma parte de los lípidos mitocondriales; el resto, corresponde de manera predominante, a los fosfolípidos de la membrana mitocondrial. Un componente adicional presente en las preparaciones mitocondriales que contiene la cadena respiratoria corresponde a la proteína ferrosulfurada (ferrosulfo-proteínas) (FeS; hierro no hem). Estas proteínas se vinculan con las flavoproteínas y con el citocromo b. Se estima que el azufre y el hierro participan en el mecanismo de oxido-reducción entre la flavina y la coenzima Q, que involucra el cambio de un solo electrón (e-), el del átomo de hierro, objeto de la oxido reducción entre el Fe2+ y el Fe3+. - 27 - En la figura 8 se muestra un punto de vista actual de la secuencia de los componentes principales de la cadena respiratoria. Las enzimas deshidrogenasas catalizan la transferencia de electrones hacia el NAD de la cadena a partir de los sustratos. Existen algunas diferencias en la forma en que esto se realiza. Los αcetoácidos piruvato y cetoglutarato, no disponen de sistemas complejos y de deshidrogenasas, los cuales involucran el lipoato y el flavín adenín dinucleótido (FAD), previos al paso de electrones hacia el NAD de la cadena respiratoria. La transferencia de electrones a partir de estas deshidrogenasas se acopla de manera directa con el NAD de la cadena respiratoria, como los correspondientes a la L(+)-3-hidroxiacil-CoA, D(-)-3-hidroxibutirato, prolina, glutamato, malato e isocitrato (Murray et al., 2001). Figura 8. Componentes de la cadena respiratoria. Se muestran los puntos de colección de equivalentes reductores a partir de sustratos importantes. La FeS se genera en las secuencias correspondientes al lado del O2 de la Fp o del Cit b. (Tomada de Murray et al., 2001). A su vez, el NADH reducido de la cadena respiratoria se oxida mediante una enzima metaloflovoprotéica, la NADH deshidrogenasa. Esta enzima contiene FeS y flavin mononucleótido (FMN), está fuertemente enlazada a la cadena respiratoria y pasa los equivalentes reductores a la coenzima Q. Esta reacción - 28 - constituye el punto de recolección en la cadena respiratoria, de los equivalentes reductores provenientes de otros sustratos que están directamente vinculados a dicha cadena a través de la flavoproteína deshidrogenasa. Entre estos sustratos se incluye el succinato, la colina, el glicerol 3-fosfato, la sarcosina, la dimetilglicina y la acil CoA. La porción flavina de todas estas deshidrogenasas corresponde al FAD. Los electrones fluyen desde la coenzima Q hasta el oxígeno molecular a través de la serie de citocromos mostrados en la figura 8. Estos citocromos se colocan en orden creciente de potencial redox. El citocromo aa3 terminal (citocromo oxidasa) es el responsable de la combinación final de los equivalentes reductores con el oxígeno molecular. Se ha observado que este sistema enzimático contiene cobre, un componente de varias enzimas oxidasas. La citocromo oxidasa posee alta afinidad por el oxígeno, y esto permite que la cadena respiratoria funcione a la máxima velocidad hasta que el tejido queda prácticamente libre de oxígeno. Debido a que esta reacción resulta irreversible (la única en la cadena), esto proporciona direccionalidad al movimiento de los equivalentes reductores en la cadena respiratoria y en la producción de ATP, a la cual está acoplada (Murray et al., 2001). La organización estructural de la cadena respiratoria ha sido objeto de considerables estudios. Las proporciones molares de sus componentes son casi constantes; es decir, funcional y estructuralmente, dichos componentes se representan en la membrana interna mitocondrial en forma de cuatro complejos proteínico-lipídicos que atraviesan la membrana denominados Complejo I, II, III y IV. El citocromo c, es el único citocromo soluble y junto con la coenzima Q, parece ser un componente de la cadena respiratoria, con mayor movilidad, que conecta los complejos fijos. Todo lo anterior se ilustra en la figura 9. - 29 - Figura 9. Esquema de la disposición de los componentes de una cadena transportadora de electrones de la membrana interna mitocondrial. (Tomada de Karp, 1998). La cadena respiratoria aporta la mayor parte de la energía capturada en el metabolismo El ADP es una molécula que captura, en forma de fosfato de alta energía, una proporción importante de la energía libre resultante de los procesos catabólicos. El ATP resultante de dicha captura transfiere esta energía libre para impulsar los procesos que la requieren. Por lo tanto, el ATP se ha denominado "moneda energética" de la célula. En las reacciones glucolíticas tiene lugar la captura directa neta de dos grupos fosfato de alta energía, equivalente a cerca de 103. 2 kJ/mol de glucosa. Se ha calculado que la energía libre (∆G) de la síntesis in vivo del ATP, a partir del ADP, es alrededor de 51.6 kJ/mol, que es congruente con la concentración de los reactantes existentes en la célula. Esta ∆G es mayor que la energía libre estándar (∆G°) para la inhibición del ATP obtenido en concentraciones estándar de 1.0 mol/L. Toda vez que la combustión completa de 1 mol de glucosa genera aproximadamente 2870 kJ, sin embargo, la energía capturada por la fosforilación - 30 - durante la glucólisis es menor. Las reacciones del ciclo del ácido cítrico, la vía final para la oxidación completa de la glucosa incluyen una etapa de fosforilación en la cual la conversión de la succinil-CoA en succinato, permite la captura de solo 2 fosfatos de alta energía adicionales por cada mol de glucosa. Todas las fosforilaciones descritas a la fecha acontecen a nivel de sustrato. El estudio de las mitocondrias aisladas intactas y que funcionalmente respiran, revela que cuando los sustratos se oxidan vía una deshidrogenasa unida al NAD+ y la cadena respiratoria, aproximadamente 3 moles de Pi se incorporan a 3 moles de ADP para formar 3 moles de ATP por medio mol de oxígeno consumido (½ O2), es decir, el índice P:O=3. Por otra parte, con la oxidación del sustrato vía una deshidrogenasa vinculada a una flavoproteína solo se forman 2 moles de ATP; es decir, el índice P:O=2. A estas reacciones en conjunto se les conoce como fosforilación oxidativa que ocurre a nivel de la cadena respiratoria. Las deshidrogenaciones (oxidaciones) en la vía del catabolismo de la glucosa que acontecen en la glucólisis y en el ciclo del ácido cítrico, más las fosforilaciones a nivel de sustrato, representan el 68% de la energía libre resultante de la combustión de la glucosa, capturada en forma de fosfato de alta energía. Es evidente que la cadena respiratoria es responsable de una gran proporción del ATP total formado. En la figura 10 se ilustran los sitios de inhibición de la cadena respiratoria y la inhibición de la formación de sustratos específicos. - 31 - Figura 10. Sitios de inhibición de la cadena respiratoria (Ө). Los desacopladores inhiben principalmente los complejos I, III y IV. (Tomada de Murray et al., 2001). El control respiratorio asegura el suministro constante de ATP La velocidad de la respiración de las mitocondrias puede controlarse mediante la concentración del ADP. Esto se debe al estrecho acoplamiento entre la oxidación y la fosforilación; es decir la oxidación no puede proseguir a través de la cadena respiratoria sin la fosforilación concomitante del ADP. Chance y Williams (1955), identificaron cinco condiciones capaces de controlar la velocidad de respiración de las mitocondrias, las cuales se ilustran en el cuadro 3. Cuadro 3. Estados de control respiratorio de la mitocondria, con base en la disponibilidad de sustrato y/o ADP. - 32 - Por lo general, la mayor parte de las células en estado de reposo se encuentran en el estado cuatro, y la respiración se controla por la disponibilidad de ADP. Cuando se realiza un trabajo, el ATP se convierte en ADP y facilita la respiración, lo que a su vez repone las reservas de ATP (figura 11). Figura 11. Participación del ADP en el control de la respiración. Los procesos que demandan energía promueven el consumo de ATP, la fosforilación de ADP durante la cadena respiratoria y la producción de calor y consumo de O2 para la formación de ATP durante la respiración. (Tomada de Murray et al., 2001). Parecería que bajo ciertas condiciones, la concentración de fosfato inorgánico también podría afectar la velocidad de funcionamiento de la cadena respiratoria. Es decir, conforme se incrementa la respiración (como ocurre en el ejercicio), la célula se aproxima al estado 3 o al estado 5, ya sea porque se sature la capacidad de la cadena respiratoria o que la presión parcial de oxígeno (PO2) disminuya por debajo de la constante de disociación (Km) para el citocromo a3. También existe la posibilidad de que el transportador ADP/ATP (figura 5), que facilita el ingreso de ADP citosólico al interior de la mitocondria y la salida de ésta del ATP, se convierta en la limitante de la velocidad de la cadena respiratoria. Por lo tanto, la forma en la que los procesos oxidativos biológicos permiten la disponibilidad y captura de la energía libre resultante de la oxidación de los - 33 - alimentos, constituye un proceso eficiente por etapas (alrededor de 68%) y controlado, en lugar de un explosivo, ineficiente y descontrolado, como es el caso de muchos procesos no biológicos. El resto de la energía libre, no capturada como fosfato de alta energía, se libera en forma de calor. A éste no debe considerársele un "desperdicio", ya que se asegura que el sistema respiratorio en su conjunto sea suficientemente un proceso exergónico para apartarse del equilibrio, lo que permite el flujo unidireccional continuo y la provisión constante de ATP. En los animales de sangre caliente (aves y mamíferos) este calor contribuye a la conservación de la temperatura corporal. Inhibición de la cadena respiratoria El uso de inhibidores de la cadena respiratoria ha proporcionado información sobre el mecanismo de acción de algunos venenos, y sobre la misma cadena respiratoria. Con propósitos descriptivos, los venenos se pueden clasificar en: 1) inhibidores de la propia cadena respiratoria, 2) inhibidores de la fosforilación oxidativa y 3) desacopladores de la fosforilación oxidativa. Los inhibidores que interrumpen la respiración mediante el bloqueo de la cadena respiratoria actúan en tres sitios. El primero de estos sitios se inhibe con los barbitúricos, como el amobarbital, con el antibiótico piericidina A y con el insecticida rotenona que también es tóxico para los peces. Estos inhibidores evitan la oxidación de los sustratos que se comunican directamente con la cadena respiratoria, mediante una deshidrogenasa enlazada al NAD al impedir la transferencia de la FeS a la coenzima Q. A dosis altas, estos inhibidores resultan mortales para los animales in vivo. El dimercaprol y la antimicina A inhiben la cadena respiratoria en el sitio entre el citocromo b y el citocromo c. Los venenos típicos como el ácido sulfhídrico, el monóxido de carbono y el cianuro inhiben a la citocromooxidasa y, por tanto, pueden detener por completo la respiración. La carboxina y la tenoyltrifluoroacetona inhiben específicamente la transferencia de equivalentes reducidos, de la deshidrogenasa succínica a la coenzima Q, en tanto - 34 - que el malonato es un inhibidor competitivo de la deshidrogenasa succínica. El antibiótico oligomicina impide por completo la fosforilación oxidativa y la fosforilación en las mitocondrias intactas. Sin embargo, en presencia del desacoplador dinitrofenol la oxidación se produce pero sin fosforilación, lo cual indica que la oligomicina no actúa de modo directo sobre la cadena respiratoria, sino de manera subsecuente sobre un paso de la fosforilación. El atractilósido inhibe la fosforilación oxidativa que depende del transporte de nucleótidos de adenina a través de la membrana mitocondrial interna. Se considera que el compuesto inhibe al cotransportador que introduce ADP a la mitocondria, con la salida simultánea de ATP (Slater, 1967). (Véase figura 12). La acción de los desacoplantes en la cadena respiratoria consiste en disociar la oxidación en la cadena respiratoria, de la fosforilación. Esta acción puede explicar el efecto tóxico de estos compuestos in vivo. El desacople se traduce en una respiración descontrolada, debido a que las concentraciones de ADP o/y del Pi ya no limitan la velocidad de la respiración. El desacoplante de uso más frecuente es el 2,4-dinitrofenol, pero otros compuestos entre los cuales incluyen el dinitrocresol, el pentacloro fenol y el CCCP (m-clorocarbonilcianuro fenilhidrazona), actúan de manera similar; este último es 100 veces más activo que el dinitrofenol. - 35 - Figura 12. Control de la respiración mitocondrial. En A se muestra el estado basal de la respiración (estado 4) al adicionar ADP se incrementa la velocidad de consumo de oxígeno (estado 3) y una vez que al ADP se fosforila a ATP regresa al estado 4. Al adicionar un desacoplador en el estado 4 en este caso, por ejemplo el dinitrofenol, desacopla la respiración de la fosforilación. En B, se muestra como al adicionar oligomicina se impide la fosforilación del ADP y por lo tanto también la respiración. (Tomada de Murray et al., 2001). La teoría quimiosmótica explica el mecanismo de la fosforilación oxidativa y la formación de ATP Para explicar el acoplamiento entre la oxidación y la fosforilación se han propuesto dos hipótesis principales, la química y la quimiosmótica. La hipótesis química postula el acoplamiento químico directo en todas las etapas del proceso, de manera similar a las reacciones generadas del ATP en la glucólisis. Sin embargo, nunca se han aislado los intermediarios ricos en energía presuntamente vinculando las reacciones redox con la fosforilación oxidativa, por lo que esta hipótesis se ha desacreditado. La teoría quimiosmótica propuesta por Mitchell (1961; 1979) postula que la energía proveniente de la oxidación de los componentes en la cadena respiratoria se acopla al traslado al interior de la membrana interna mitocondrial de iones hidrógeno (H+), procedentes del interior de la mitocondria. La diferencia resulta en - 36 - un potencial electroquímico debido a la distribución asimétrica de los iones de hidrógeno, el cual se utiliza para impulsar el mecanismo responsable de la formación de ATP durante la cadena respiratoria (figura 13). Figura 13. Ilustra los principios de la teoría quimiosmótica. El principal circuito de protones se crea mediante el acoplamiento de la oxidación en la cadena respiratoria con el traslado de protones del interior al exterior de la membrana, impulsado por los complejos I, III y IV, donde cada uno de estos complejos actúa como una bomba de protones. La ubiquinona, el citocromo c y la ATPsintasa utilizan este gradiente para promover la fosforilación. Los desacopladores permiten la fuga de H+ de la membrana y asi colapsan el gradiente electroquímico protónico. La oligomicina, específicamente, impide la conducción de H+ a través de la subunidad F0 de la ATPsintasa. Véase texto. (Tomada de Murray et al, 2001). Cada uno de los complejos I, III y IV de la cadena respiratoria actúan como una bomba de protones. La membrana interna es impermeable a los iones en general, pero en particular a los protones, los cuales se acumulan en el exterior de la membrana y crean una diferencia de potencial electroquímico a través de la - 37 - membrana (∆µH+). Esta diferencia consta de un potencial químico (diferencia en el pH por los protones) y un potencial eléctrico debido a las cargas. Una ATP sintasa localizada en la membrana forma el ATP La diferencia de potencial electroquímico se utiliza para impulsar una ATP sintasa localizada en la membrana, la cual forma ATP en presencia de Pi + ADP. Por lo tanto, no existe ningún intermediario de alta energía común a la oxidación y a la fosforilación, como se indica en la hipótesis química (Murray et al., 2001). Figura 14. Estructura de la ATP sintasa. La enzima consta de 2 porciones principales: Cabeza o F1, que consta de 5 subunidades diferentes en proporción 3α: 3β: 1δ: 1γ y 1ε y la pieza basal o F0, la cual se integra a la membrana y consta de 3 subunidades diferentes en la relación 1a:2b:12c. Las subunidades b de F0 se extienden hacia adentro de la pieza cefálica para formar un tallo. Según Soper et al., (1979) Tomado de Karp, 1998. Los complejos fosforilantes responsables de la producción de ATP se encuentran diseminados en la superficie de la membrana interna. Estos complejos constan de varias subunidades proteícas comúnmente conocidas como F1, que se proyectan al interior de la matriz y contienen la ATP sintasa. Estas entidades se fijan, mediante un tallo, al complejo proteíco de la membrana conocido como F0, que atraviesa la membrana y que consta de varios subunidades (figura 14). El paso de protones a través de los complejos lleva a la formación de ATP a partir de - 38 - ADP y Pi. Es interesante notar que en el interior de la membrana plasmática de las bacterias se presentan unidades fosforilantes similares, pero que estas se localizan en el exterior de la membrana de los tilacoides en los cloroplastos. En la perspectiva de la hipótesis que postula el origen de las mitocondrias por evolución a partir de las bacterias, es importante que el gradiente protónico tenga una dirección desde el exterior hacia el interior de las mitocondrias y las bacterias, y que tenga un sentido inverso en los cloroplastos (Karp, 1998). El mecanismo del acoplamiento del transporte de los protones con el sistema de la ATP sintasa es una conjetura. La evidencia sugiere que la síntesis del ATP que podría darse mientras permanece unido a la enzima, no representa el paso principal que requiere energía, y que el paso que consume energía corresponde a la liberación del ATP del sitio activo. Esto puede implicar cambios en la conformación de las subunidades F1 generados por el gradiente protónico. Se desconoce la cantidad precisa de protones bombeados por cada complejo por mol de NADH oxidado, pero las estimaciones actuales sugieren que el complejo I traslada 4 y los complejos III y IV trasladan en conjunto 6. También tres o cuatro protones se incorporan al interior de la mitocondria por cada ATP exportado. Por lo tanto, el índice P:O no necesariamente resultada en un número entero, es decir 3, sino tal vez 2.5. Sin embargo, por razones de simplificación, se utiliza el valor de 3 para la oxidación del NADH + H+ y de dos para la oxidación del flavín mononucleótido reducido (FADH2) (Mitchell, 1961; 1979) Por su parte, Boyer, (1989) sugiró una hipótesis en relación a la producción de ATP llamada de “cambio de enlace”, donde cada subunidad F1, contiene 3 sitios catalíticos (uno por cada subunidad β) (figura 15) que varían progresivamente a través de 3 diferentes conformaciones, según su afinidad por los nucleótidos. A estas conformaciones se les conoce como, conformación “apretada” o T, en la cual los nucleótidos (ADP + Pi o ATP) están firmemente unidos; la conformación “laxa” o L, en donde el ADP y Pi están laxamente unidos; y la conformación “abierta” u O, en la cual debido a su muy escasa afinidad por los nucleótidos se considera que esta vacía. - 39 - Figura 15. Hipótesis de "cambio de enlace" para explicar el mecanismo por el cual el gradiente de protones afecta en la síntesis de ATP. El enlace de protones a la enzima en el lado citoplásmico de la membrana provoca un cambio de conformación (desde el paso 1 hasta el 2), que da como resultado el enlace firme de ATP y de Pi en el sitio catalítico de la enzima. La formación de ATP ocurre de manera espontánea. La liberación subsiguiente de protones al interior de la matriz causa un segundo cambio de conformación (pasos 3 a 4) que libera el ATP recién formado. (Tomada de Karp, 1998). La teoría quimiosmótica puede explicar el control respiratorio Como se mencionó anteriormente, el resultado del traslado de los protones establece la diferencia del potencial electroquímico a través de la membrana, el cual, inhibe el transporte subsecuente de equivalentes reductores a lo largo de la cadena respiratoria a menos que dicho potencial se descargue mediante el traslado retrógrado de los protones a través de la membrana a cargo de la ATP sintasa vectorial. Esto, a su vez, depende de la disponibilidad de ADP y Pi (Murray et al., 2001). La teoría quimiosmótica explica la acción de los desacoplantes Los desacoplantes son compuestos anfipáticos que incrementan la permeabilidad de los protones por la membrana interna mitocondrial lipídica y, por tanto, disminuyen el potencial electroquímico impidiendo la activación de la ATP sintasa. De esta manera, la oxidación puede proceder sin que se produzca la fosforilación. Dada la impermeabilidad relativa de la membrana mitocondrial interna se requiere de un transportador de este intercambio. En la membrana existen sistemas de - 40 - intercambio por difusión para el intercambio de aniones e hidroxilos (OH-) y de cationes por y H+. Estos sistemas se requieren para la captura y salida de los metabolitos ionizados al tiempo que se conserva el equilibrio eléctrico y osmótico (Murray et al., 2001). La membrana mitocondrial interna, es libremente permeable a las moléculas pequeñas sin carga, como el oxígeno, el agua, el dióxido de carbono y el amoniaco al igual que a los ácidos monocarboxílicos, como el 3- hidroxibutírico, el acetoacético y el acético. Los ácidos grasos de cadena larga se transportan al interior de las mitocondrias mediante el sistema de carnitina; también existe un acarreador especial para el piruvato por el simporte que utiliza el gradiente de H+ desde el exterior hasta el interior de la mitocondria. Sin embargo, los aniones monocarboxilato, dicarboxilato y tricarboxilato, y los aminoácidos requieren de un transportador de estos sistemas específicos para facilitar su paso a través de la membrana. Al parecer, los ácidos monocarboxílicos penetran más fácilmente debido a su menor grado de disociación. Se estima que los ácidos sin disociar y mas liposolubles constituyen la especie molecular que penetran la membrana lipídica (Palmeri et al., 1992; Kuan y Saier, 1993; Palmeri, 1994; Sluse, 1996). El transporte de los aniones dicarboxilato y tricarboxilato está estrechamente vinculado al de fosfato inorgánico, el cual penetra con rapidez como ion dihidrógeno fosfato (H2PO4) en el intercambio por OH-. La captura neta de malato, a cargo del transportador dicarboxilato, requiere de fosfato inorgánico para su intercambio en dirección opuesta. La captura neta de citrato, isocitrato o cis-aconitato, a cargo del transportador tricarboxilato, requiere intercambiarse con el malato. El transporte del α-cetoglutarato también requiere de intercambio con malato. Por tanto, el uso de los mecanismos de intercambio contribuye a conservar el equilibrio osmótico. Sin embargo, el transporte del citrato a través de la membrana mitocondrial no depende sólo del transporte del malato, sino que requiere también del transporte del fosfato inorgánico. El transportador de nucleótidos de adenina permite intercambio de ATP y ADP, pero no de adenosina monofosfato (AMP). Esto es fundamental para la salida del ATP de las - 41 - mitocondrias hacia los sitios de su utilización extramitocondrial, y para permitir el regreso del ADP al interior de la mitocondria. Impulsado por el gradiente protónico. El sodio puede intercambiarse por hidrógeno. Se estima que la captura de calcio por las mitocondrias tiene lugar con una carga neta de transferencia de 1 (uniportador de Ca2+), posiblemente a través del antiportador Ca2+/H+. La liberación de calcio a partir de las mitocondrias se facilita mediante intercambio con sodio. El transporte iónico de la mitocondria requiere de energía Las mitocondrias con respiración activa (acopladas), en las cuales tiene lugar la fosforilación oxidativa, no acumulan algunos cationes como el potasio, sodio, calcio y magnesio, así como el Pi. El desacoplamiento por dinitrofenol causa pérdida de iones en las mitocondrias, pero la captura de iones no se inhibe por la oligomicina, lo cual sugiere que la energía necesaria no procede de la fosforilación del ADP. Se estima que una bomba primaria de protones impulsa el intercambio de cationes (Murray et al., 2001). Génesis mitocondrial Se ha demostrado que el tamaño de la mitocondria, su número y/o volumen puede aumentar en determinado tejido como es en el músculo esquelético, en respuesta a un estímulo fisiológico, por ejemplo con el entrenamiento (Holloszy, 1967; Gollnick y King, 1969; Kiessling et al., 1971; Hoppeler et al., 1973). El término biogénesis mitocondrial, implica un proceso celular que involucra la síntesis y la degradación del organelo (Larsson y Luft, 1999; Wallace, 2000). Dentro de las condiciones fisiológicas que pueden producir la biogénesis mitocondrial existen diferentes ejemplos, desde aquellos que se pueden ocasionar de una manera "artificial", como los modelos de ejercicio, en aquellos donde se induce una contracción crónica producida por estimulación eléctrica. Este modelo produce - 42 - grandes cambios en la biogénesis mitocondrial, ya que el cambio se da en un periodo relativamente breve de tiempo, es decir, de 1 a 3 semanas (Pette et al., 1973; Eisenberg y Salmons, 1981; Takahashi y Hood, 1993). Por otra parte, la biogénesis también se da a consecuencia de condiciones más intrínsecas, como ocurre por la acción hormonal (Winder, 1980). Aunque el cambio en la biogénesis, como se mencionó anteriormente, se da en un periodo relativamente corto de tiempo; se requiere de aproximadamente de seis semanas para alcanzar un cambio en el estado general mitocondrial como respuesta al entrenamiento. Esto no se ve reflejado en cambios moleculares que nos pudieran permitir obtener cambios morfológicos medibles. De hecho, la vida media de las proteínas se reduce a la mitad una vez que el músculo ha encontrado una nueva actividad de su patrón contráctil (Henriksson y Reitman, 1977; Booth, 1977; Terjung, 1979; Hickson y Rosenkoetter, 1981). Esta actividad debe mantenerse así, ya que de otra manera, esto se ve reflejado en la pérdida de la capacidad oxidativa. Es interesante también hacer notar que el contenido de fosfolípidos cambia en un periodo aún más corto, en un tiempo menor a cuatro días (Wicks y Hood, 1991; Takahashi y Hood, 1993), sugiriendo que el ensamble y/o la degradación del organelo puede ser iniciada por cambios en la composición de los fosfolípidos. El oxígeno respirado es consumido en el paso final de la cadena de fosforilación oxidativa en las mitocondrial por la citocromo oxidasa. La máxima velocidad de consumo de oxígeno del músculo esquelético en la mayoría de los mamíferos está relacionada con el volumen mitocondrial (Suárez, 1996). La densidad mitocondrial depende de un proceso de adaptación; se sabe que diferentes vertebrados como los colibríes (Suárez et al., 1991), los antílopes (Lindstedt et al., 1991), y los atunes (Moyes et al., 1992) poseen de dos a tres veces mayor número de mitocondrias por cm3 que cualquier otro mamífero cuadrúpedo previamente estudiado. Aunque se sabe que esta cifra se puede conservar entre individuos y especies, también existen muchos ejemplos de diferencias cuantitativas y cualitativas de las mitocondrias de los músculos entre las diferentes especies incluso aún en el mismo tejido (Moyes et al., 1998). - 43 - En el músculo esquelético se encuentran diferentes tipos de fibras que difieren en sus propiedades bioquímicas incluyendo el número de mitocondrias que contienen por unidad de volumen. Las fibras rápidas glucolíticas tienen menos mitocondrias que las fibras lentas oxidativas. Existe evidencia de que las mitocondrias que poseen son diferentes. Es decir, las mitocondrias de las fibras rápidas glucolíticas tienen bajas velocidades de oxidación de los sustratos lipídicos, lo que se correlaciona con la baja actividad enzimática de oxidación lipídica (Baldwin et al., 1972; Moyes et al., 1992). Los estudios estructurales muestran la presencia de dos poblaciones de mitocondrias en el músculo (tanto esquelético como cardiaco). Las mitocondrias subsarcolemales (SSmt), las cuales están localizadas justo por debajo de la membrana, mientras que el segundo tipo o población se encuentra dentro de las miofribillas (mitocondrias intermiofribrilares o IMFmt). Las SSmt ocupan del 5 al 40% del total del volumen muscular dependiendo de la especie y el tipo de fibra (Suárez et al., 1996). La ventaja de tener dos tipos diferentes de mitocondrias puede estar relacionada con su localización con respecto a la cercanía de los metabolitos que utilizan, lo que beneficia el incremento de la concentración de oxígeno aún cuando los dos tipos de mitocondrias sean idénticas cuando son observadas in vitro (Suárez et al., 1991). Sin embargo, existen algunas diferencias bioquímicas, en general las IMFmt tienen mayor PO2 y se ha demostrado que tienen gran capacidad para oxidar ácidos grasos (e.g. Palmer et al., 1985; Cogswell et al., 1993). Se ha demostrado que la actividad especifica del complejo de la succinato deshidrogenasa y de la citrato sintasa es mayor en las mitocondrias intermiofribrilares, mientras que la actividad de la carnitina palmitoiltransferasa y de la α-glicerolfosfato deshidrogenasa es prácticamente la misma, por lo que se puede considerar que estos dos tipos de poblaciones de mitocondrias pudieran jugar papeles metabólicos diferentes que contribuyen a economizar las reservas energéticas de la célula (Palmer et al., 1977). Existe evidencia de que se puede incrementar el número de mitocondrias de una de las poblaciones bajo determinadas condiciones patológicas (Gustafsson et al., 1965; McCallister y Brown, 1969; Kubista et al., 1971; Jones et al., 1975; Muller, 1976; - 44 - Tandler y Hoppel, 1972). Sin embargo, las diferencias entre los diferentes tipos de mitocondrias musculares no siempre son evidentes (e.g. McKean, 1990; Sillau et al., 1990; Manneschi y Federico, 1995), por lo que no se ha demostrado que tengan propiedades diferentes in situ. Los orígenes de los dos tipos de poblaciones mitocondriales, en caso de que existan, no es clara ya que la biogénesis de ambos debe estar bajo el control del mismo núcleo. La distribución intracelular del núcleo, los ribosomas libres en el citosol, y las mitocondrias pueden crear la heterogeneidad de las poblaciones de mitocondrias a través de una difusión diferencial de factores regulatorios y pre-proteínas mitocondriales. Las diferentes poblaciones en una sola fibra pueden originar un proceso diferencial de los organelos existentes, más que diferencias intracelulares durante la biogénesis (Moyes et al., 1998). Las propiedades de las mitocondrias del músculo cambian con la edad del organismo. Bajo una adecuada influencia hormonal, los mioblastos entran en un programa miogénico, el cual va acompañado de la proliferación mitocondrial, incrementándose con ello el número de mitocondrias. Una vez que el programa miogénico está completo, el músculo adulto muestra una impresionante capacidad para adaptarse a los cambios, en donde preserva la mayor parte de las relaciones de los elementos funcionales y estructurales (Puntschart et al., 1995). El ejercicio y la estimulación eléctrica crónica de los nervios que inervan al músculo producen paralelamente un incremento en la actividad de las enzimas mitocondriales, lo cual, se puede ver con la cuantificación del mRNA (siglas en inglés messenger ribonucleic acid) y el número de copias del DNA mitocondrial (mtDNA) (Hood et al., 1994). Aunque las propiedades oxidativas de mitocondrias aisladas de individuos entrenados y sedentarios no difiere (Davies et al., 1981). La edad, conlleva a un cambio en las enzimas mitocondriales que origina el decline de las propiedades respiratorias (Ames et al., 1995). Se cree que el decline de la capacidad de las mitocondrias se debe principalmente a la acumulación de radicales libres, lo que produce mutaciones en el mtDNA (Linnane et al., 1989). - 45 - Así pues, la biogénesis mitocondrial se da en respuesta a un estímulo fisiológico que requiere de la apropiada expresión de todos los genes nucleares que codifican para los productos mitocondriales y de la coordinación regulada de la expresión de los genes. Se sabe que un gran número de proteínas son las que coordinan la expresión de genes nucleares y de respiración mitocondrial (Moyes et al., 1998). Control de la expresión de DNA mitocondrial El DNA mitocondrial es de estructura circular y se encuentra densamente empacado. El mtDNA de vertebrados codifica para 13 proteínas, 22 tRNAs (siglas en inglés transference ribonucleic acid) y 2 rRNAs (siglas en inglés ribosomal ribonucleic acid) (Attardi y Schatz, 1988). El mtDNA codifica para proteínas que constituyen 4 de los 5 pasos de la fosforilación oxidativa, incluyendo al dinucleótido de nicotinamida (ND) ND-1 a ND-6 (complejo 1), citocromo b (complejo III), citocromo oxidasa 1, 2 y 3 (complejo IV o citocromo oxidasa c) y ATP6 y ATP8 (subunidades de la ATPsintasa o complejo V). Solo el complejo II es completamente codificado por el núcleo (Moyes et al., 1997). En general, la transcripción del mtDNA es asimétrica con la velocidad de transcripción del rRNA mitocondrial, casi en un orden de magnitud (Gelfand y Attardi, 1981). Se creía que la primera respuesta a un estímulo fisiológico era la replicación del mtDNA, es decir, el incremento en el número de copias de este, más que ocurrir cambios en la transcripción. La estimulación eléctrica crónica en un modelo de entrenamiento extremo resulta en el incremento del número de copias de cuatro a cinco veces más (Williams, 1986). Sin embargo, el número de copias puede no ser el único factor que controla la transcripción del mtDNA en un tejido; también lo pudiera ser la cantidad relativa de los tripletes de mtDNA lo que se correlaciona con el incremento del mRNA (citocromo b). Los tejidos con alta capacidad aeróbica tienen una gran proporción de mtDNA en la forma de triplete (Annex y Williams, 1990). - 46 - Coordinación del DNA mitocondrial y nuclear Aunque le coordinación de los distintos tipos de DNA aún no es muy clara se sabe que los factores de transcripción nucleares tienen el potencial "colectivo" de controlar la expresión de todos los genes nucleares relevantes. Estos factores pueden tener acceso a la matriz mitocondrial e interaccionar con el DNA de este. Diversos estudios utilizando enfoques diferentes han concluido que la expresión de los genes mitocondriales durante la biogénesis se encuentra altamente coordinada (Hood et al., 1989). El descubrimiento de los factores de regulación ha reforzado la percepción de que la expresión de genes de la respiración está estrechamente coordinada pero de manera temporal. Sin embargo, cuando los intervalos de estimulación son suficientemente frecuentes, asíncronos cambios en los niveles del mRNA de determinados genes respiratorios son observados durante dicha estimulación (Annex et al., 1991), como ocurre en el hipertiroidismo (Luciakova y Nelson, 1992) y en la diferenciación celular, los cuales inducen la biogénesis mitocondrial (Moyes et al., 1997). La eficacia de los factores de transcripción en cualquier estado fisiológico es influenciado por su concentración y actividad, así como por la presencia de represores que pueden competir por el mismo sitio de unión o en otro diferente, del mismo modo, la función de la proteínas mitocondriales puede alterarse por periodos relativamente cortos mediante un proceso de regulación alostérica o covalente, lo que permite la diferenciación y desarrollo de isoformas lo que le provee a la célula de plasticidad para responder adecuadamente al estimulo externo y por lo tanto constituye una ventaja evolutiva (Véase figura 16) (Moyes et al., 1997). - 47 - Figura 16. Biogénesis mitocondrial en la célula muscular. Las señales que desencadenan el proceso son provenientes de la paca neuromuscular incluyendo la propagación del potencial de acción y la liberación de sustancias tróficas que interactúan con la membrana postsináptica. La actividad eléctrica en el sarcolema esta acoplada con la liberación de calcio del retículo sarcoplásmico. El calcio actúa como un segundo mensajero que activa las fosfatasas y/o cinasas, las cuales son llevadas al núcleo para afectar la activación de los factores de transcripción cuya influencia determina la expresión de genes nucleares que codifican para proteínas mitocondriales. El mRNA producido por la transcripción es acompañado de una proteína en el citosol, que puede llevarlo tanto de nuevo al núcleo (como factor de transcripción) o llevarlo a la maquinaria de importación de proteínas e introducirlo dentro del organelo. Una vez dentro de la mitocondria puede actuar como una simple subunidad o combinarse con otra para formar una holoenzima (e.g., citocromo c oxidasa). Algunas veces las subunidades pueden derivar del genoma mitocondrial (mtDNA), el mismo que controla la transcripción, síntesis y transporte del número limitado de proteínas que son componentes esenciales de la cadena respiratoria. Modificada de Hood, (2001). - 48 - EL PAPEL DEL ATP EN LA ENERGÉTICA MUSCULAR La falta de ATP o la reducción de sus niveles intracelulares se ha relacionado con la fatiga muscular. La concentración intracelular de ATP durante una actividad muscular intensa no cae más allá del 70% de los niveles preexistentes; sin embargo, se ha especulado que el ATP intracelular está restringido a la mitocondria, es decir, está “almacenado” y que aunque haya suficiente en el medio intracelular éste no se encuentra en el sitio donde es requerido, como sería en el caso de los miofilamentos (Fitts et al., 1981) Además, también se sabe que durante el ejercicio exhaustivo, durante una estimulación constante y durante la fatiga, lo que se reduce es la velocidad de hidrólisis del ATP. En las células musculares al igual que en otras células (del páncreas y corazón), la actividad de los canales de potasio dependientes de ATP (KATP) ha sido atribuida a censar los niveles intracelulares de ATP y ADP y a controlar la excitabilidad de la membrana (véase más adelante). LOS CANALES DE K+ SENSIBLES A ATP El descubrimiento de los canales KATP fue hecho en 1983 por Noma en células cardíacas. Poco después, los canales KATP se encontraron y describieron en la membrana plasmática de las células β-pancreáticas, en las neuronas, en la aurícula, en las células de marcapaso, en el músculo esquelético tanto de anfibio como de mamífero y en el músculo liso. Posteriormente, se encontraron también a nivel de la membrana interna mitocondrial (Inoue et al., 1991). Ambos tipos de canales tienen la habilidad única de acoplar el potencial de membrana plasmático (y/o el potencial redox mitocondrial, y/o la liberación de radicales libres) al estado metabólico de la célula (Nichols et al., 1991; Liss et al., 1999a; Pain et al., 2000; O’Rourke, 2000). Existen evidencias de que este canal es regulado por cambios en los niveles de nucleótidos de adenina, aumentos en la relación ATP/ADP, y que - 49 - es un blanco para un tipo de fármacos denominados sulfonilureas. Estos hipoglucemiantes orales son utilizados ampliamente en el tratamiento de la diabetes mellitus no dependiente de la insulina (diabetes tipo II). Regulación de los canales KATP Como se mencionó anteriormente, los canales KATP son regulados por nucleótidos intracelulares ATP/ADP, sin embargo, recientemente se ha puesto de manifiesto que la regulación de estos canales es mas complicada. Los canales KATP también son regulados por el pH (Wu et al., 2002), la proteína cinasa A (Lin et al., 2000; Beguin et al., 1999) y la proteína cinasa C (Light et al., 2000; Thorneloe et al., 2002). También se sabe que el fosfatidilinositol 4,5 bifosfato (PIP2), el cual está relacionado con las cascadas de señalización por segundos mensajeros mediadas por proteínas G (Shyng y Nichols, 1998; Baukrowitz y Fakler, 2000) regula también la actividad del canal KATP. Ya que la concentración de PIP2 esta altamente regulada por la fosfolipasa C (PLC) y la fosfatidilinositol 3-cinasa (PI3 cinasa), la modulación del PIP2 juega un papel importante en la regulación del canal KATP (Xie et al., 1999; Haruina et al., 2002). La PLC y la PI3 cinasa son reguladas vía proteínas Gq. Los canales KATP también son regulados por el acido fosfatídico, producida en la cascada de la fosfolipasa D (Fan et al., 2003). Desde el descubrimiento de estos canales, diversos trabajos se han enfocado a definir sus propiedades electrofisiológicas y su farmacología, incluyendo sus propiedades biofísicas a nivel de canal unitario, la regulación por los nucleótidos y su respuesta a abridores y bloqueadores del canal. Actualmente, se empiezan a definir estos canales en términos de su composición molecular. Los canales KATP son heteromultímeros de dos tipos de subunidades, los canales rectificadores entrantes, Kir6.x, y los receptores a las sulfonilureas, SUR, que son miembros de la superfamilia cassette de unión a ATP (siglas en inglés ATPbinding cassette: ABC) (Bryan et al., 2004). La evidencia disponible indica que estos canales son tetrámeros (SUR/Kir)4. La clasificación que hay de estos - 50 - canales muy probablemente es provisional ya que es posible que otras isoformas no hayan sido aún identificadas. Su identificación se basa fundamentalmente en criterios farmacológicos y se requiere de más trabajo en cuanto a su localización celular y tisular para confirmarla. Es importante mencionar que el comportamiento del canal es dependiente del órgano en donde se localiza, por lo que su activación y mecanismo de acción varía en cada tipo celular. En el corazón, por ejemplo, los canales KATP de los cardiomiocitos están usualmente cerrados, se abren bajo condiciones de hipoxia o isquemia, lo que provoca un acortamiento del potencial de acción y a consecuencia de esto hay una disminución de la fuerza de contracción debido a la falta de disponibilidad de energía. Se cree que este es el mecanismo de protección mediante el cual la célula se adapta a la falta de energía y protege sus reservas durante los periodos de flujo sanguíneo interrumpido (Nerbonne et al., 2001; Flagg et al., 2001; Suzuki et al., 2001; Rajashree et al., 2002; Seino, 2003). En el páncreas, en contraste, la apertura de los KATP esta regulada por la concentración de glucosa en la sangre (lo que modula el metabolismo energético de estas células). Durante la hipoglucemia, la concentración de ATP intracelular decrece aumentando la concentración de ADP lo que ocasiona el cierre de los canales, esto a su vez despolariza la membrana y se incrementa la secreción de insulina, por lo que los KATP juegan un papel importante en la homeostasis de la glucosa (Gopel et al., 2000; Rorsman et al., 2000; Grimberg et al., 2001; AguilarBryan et a., 2001; Kanno et al., 2002a,b; Renstrom et al., 2002). En el cerebro, la hipoglucemia o hipoxia local induce la apertura del canal, lo cual reduce la velocidad de disparo de una serie de neuronas en conexión las cuales también expresan el canal KATP; por ejemplo en el hipotálamo (Miki et al., 2001), en el núcleo del nervio vago (Muller et al., 2002) y en la neuronas dopaminérgicas (Liss y Roeper, 2001a,b). - 51 - En los vasos sanguíneos, los KATP modulan el tono vascular del músculo liso regulando el potencial de membrana (Daut et al., 1990; Daut et al., 1994a,b; Suzuki et al., 2001; Miki et al., 2002). La hiperpolarización normalmente lleva a la vasoconstricción debido a la entada de Ca2+ a través de los canales de Ca2+ tipo L en estos tipos celulares. Canales KATP mitocondriales Los canales KATP mitocondriales (mito- KATP) fueron descubiertos en 1991 (Inoue et al., 1991). Inoue y cols. (1991) evidenciaron electrofisiológicamente estos canales registrando corrientes unitarias en mitoplastos fusionados. El grupo de Garlid (Paucek et al., 1992) por su parte, mediante reconstitución de estos canales altamente purificados en liposomas, describieron algunas propiedades cinéticas y algunos mecanismos de regulación de estos canales. Por ejemplo, la Km para el K+ es de 32 mM, es altamente selectivo para K+, es inhibido con alta afinidad por ATP y ADP y la inhibición tiene un requerimiento absoluto de cationes divalentes. El ATP inhibe el flujo de K+ con una concentración media (EC50) entre 20 y 40 µM. De estos datos surge la pregunta de cómo puede abrirse el canal en condiciones fisiológicas con las concentraciones normales de ATP, que en el caso del músculo esquelético en reposo son de alrededor de 5 mM. Paucek et al., (1996), usando también la técnica de fijación de voltaje en liposomas, encontraron que el flujo de K+ inhibido por ATP podía reactivarse completamente en presencia de guanidina trifosfato (GTP), (EC50= 7 µM) y guanidina difosfato (GDP) (EC50 = 140 µM) pero estos no tenían efecto en ausencia de ATP. Ellos también encontraron que tanto el palmitoil-CoA como el oleil-CoA inhibían el flujo de K+ con EC50 de 260 nM y 80 nM, respectivamente. Esta inhibición fue revertida por GTP (EC50=232 µM). Es interesante hacer notar que el efecto de estos últimos es opuesto al que tienen sobre el canal KATP de la membrana plasmática, lo que sugiere que el KATP mitocondrial es diferente al de la superficie celular. - 52 - Composición de los canales KATP mitocondriales La composición molecular de los canales KATP mitocondriales ha sido el objetivo de varios trabajos y los resultados han sido controversiales, fundamentalmente en cuanto al tipo de canal rectificador. Suzuki et al., (1997), encontraron por técnicas de Western blot que las fracciones que contenían mitocondrias provenientes tanto de músculo esquelético de rata, como de hígado, mostraban una banda individual que correspondía a Kir6.1. En ese trabajo estos investigadores encontraron además, tanto por tinción inmunofluorescente como por microscopía electrónica, una distribución dispersa de Kir6.1 que se localizaba en la membrana interna mitocondrial. Esto les hizo sugerir que Kir6.1 era una de las subunidades del canal KATP mitocondrial. Seharaseyon et al., (2000), utilizando técnicas de transferencia de genes a través de virus, encontraron que ni Kir6.1, ni Kir6.2 tendrían un papel funcional en el canal KATP mitocondrial. Mientras que, la disminución de Kir6.2, suprimía la corriente del canal KATP del sarcolema. Además, por técnicas de inmunohistoquímica encontraron que Kir6.1 se expresa en corazón, pero aparentemente no en colocalización con las mitocondrias. La misma existencia del canal KATP mitocondrial fue cuestionada (Lim et al., 2002; Das et al., 2003), con base en que los inhibidores de la enzima succinato deshidrogenasa (SDH), son capaces de imitar el proceso del precondicionamiento isquémico (IPC) (Véase mas adelante). Por otra parte, es sabido que los canales KATP del sarcolema y los mitocondriales se activan por pinacidil y son inhibidos por la glibenclamida (Paucek et al., 1992; Garlid et al., 1997). Sin embargo, el 5- hidroxidecanoato (5HD) y el diazoxido son más específicos para el canal KATP mitocondrial (Garlid et al., 1997; Sato et al., 1998). Estas diferencias en cuanto a su respuesta a los fármacos les hicieron concluir que estos dos canales son entidades diferentes. Es decir, algunos fármacos que activan a los canales KATP mitocondriales también inhiben a la SDH (Hanley et al., 2002), por ejemplo, el diazoxido inhibe a la SDH - 53 - con una concentración de 100 µM, la cual no es mucho mas alta que aquella utilizada para abrir el canal (30 µM) (Kowaltowski et al., 2001). Todo esto llevó a que algunos autores concluyeran que una inhibición respiratoria, más que una actividad del canal es lo que estaría relacionado con el precondicionamiento isquémico. Sin embargo, en el 2005 Ardehali y O’Rourke encontraron, a partir de una fracción purificada de la membrana interna de mitocondrias con actividad del canal KATP, que éste forma parte de un complejo macromolecular formado, entre otros componentes, por la misma SDH. Con base en sus resultados proponen además que la SDH modula la actividad del canal por un proceso de interacción física y no por su papel en la fosforilación oxidativa. Estos datos permiten sugerir que el KATP mitocondrial tiene una estructura molecular diferente al de la superficie celular y que la actividad de la SDH es fundamental para la función del canal (véase figura 17). Figura 17. Muestra la interacción de lo abridores y bloqueadores del canal de potasio sensible a ATP mitocondrial así como su relación con algunos otros componentes de la cadena respiratoria y del citosol. Modificada de Szewczyk y Wojtczak, (2002). - 54 - Ardehali et al., (2005) continuando con la exploración de este complejo, han estudiado la mABC1, única proteína de este complejo con función desconocida, y encontraron que tiene un papel en la resistencia celular al stress oxidativo. Recientemente, Zhou et al., (2005) en contraste con Ardehali y O’Rourke (2005) sugieren que el canal Kir6.1 es la subunidad del canal KATP mitocondrial. Ellos propusieron que Kir6.1 sería un buen candidato para explicar los mecanismos de cardioprotección observados en el precondicionamiento isquémico, por lo que trataron de localizarlo con precisión con anticuerpos específicos. Así, encontraron que las mitocondrias contienen ambos tipos de canales, tanto Kir6.1 como Kir6.2, aunque consideran que Kir6.1 está principalmente localizado en la mitocondria. Sin embargo, todavía no se sabe con precisión si estos canales se coexpresan con las subunidades SUR2A o SUR2B en la mitocondria. Con base en lo anteriormente descrito, a la fecha, la identidad molecular del canal KATP mitocondrial está todavía en discusión y es un tema importante de investigación. El antiportador de K+/H+ Los antiportadores mitocondriales son necesarios para mantener la integridad del organelo, ya que previenen crecimiento excesivo de la matriz mitocondrial debido a la entrada de K+ acompañado de agua. Los iones de K+ entran a la matriz mitocondrial en relación al potencial de membrana y la forma de regular la salida del mismo es mediante el antiportador K+/H+, el cual usa el potencial de H+ este mismo gradiente para expulsar el K+ (Garlid y Paucek, 2003). La fatiga muscular y los canales KATP Durante el ejercicio, aumenta la concentración extracelular de K+, sobre todo en el caso de fatiga. Se ha propuesto que este hecho es un factor importante en la etiología de la fatiga muscular extrema ya que suprimiría la excitabilidad de la membrana y, eventualmente, el desarrollo de fuerza (Sen et al., 1995). En ese - 55 - sentido, se ha sugerido que los canales KATP del sarcolema se activarían al disminuir los niveles de ATP o cuando los niveles de varios metabolitos aumentaran, de tal forma que permitirían un incremento del K+ extracelular para suprimir el desarrollo de fuerza. Sin embargo, algunos estudios recientes demuestran que se logra el aumento en el K+ extracelular, siempre y cuando se mantenga el gradiente de Na+ a través de la bomba Na+/ K+, esto, optimizaría el desarrollo de fuerza durante el ejercicio. Una forma de estudiar el posible papel de estos canales en la fatiga es la utilización de ratones que carecen del gen para la expresión de Kir6.2. Con este modelo encontraron que no se presentan cambios en el desarrollo de fatiga (Gong et al., 2000). Los autores concluyeron que el canal KATP con esta subunidad no interviene en la disminución de la tensión durante el desarrollo de la fatiga y que probablemente tiene alguna importancia en la protección ante los cambios que se producen en el desarrollo de tensión durante el envejecimiento. Como se describió previamente, en los canales KATP mitocondriales, la subunidad Kir6.2 no parece encontrarse por lo que los animales que experimentalmente carecen de la subunidad Kir6.2 en los canales KATP del sarcolema no parecen mostrar el papel fundamental en la fatiga. Por su parte Tricarico et al., (2006) mediante experimentos de RT-PCR (siglas en inglés Reverse transcription-polymerase chain reaction) han encontrado que la subunidad más abundante de las fibras musculares esqueléticas es Kir6.2, de igual forma lo corroboraron con la medición de corrientes unitarias en las mismas preparaciones y vieron que la corriente aumentaba conforme aumentaba la expresión de la subunidad. También, se han utilizado fármacos que afectan a los canales KATP del sarcolema encontrándose que no tienen efecto, o sus efectos son pequeños en el desarrollo de la fatiga en músculo esquelético de rata (Burton et al., 1997). Sin embargo, en algunos trabajos se han probado algunos de ellos, específicamente la glibenclamida, fármaco no específico para los diferentes tipos de KATP, en condiciones de ‘hipoxia’ (~alrededor 3.8% de O2), y en ese caso, sí parece haber una atenuación de la fatiga inducida experimentalmente, así como una disminución en la velocidad de relajación de la tensión de sacudida (Aggugini et - 56 - al., 1996). El efecto sobre la relajación también se encontró en condiciones de saturación de O2 (95% de O2). Esto último estaría de acuerdo en que hay efectos que sólo se observan en ciertas condiciones de concentración de O2 (Van Lunteren et al., 1998). Precondicionamiento isquémico El fenómeno protector del miocardio ante un periodo breve de isquemia/hipoxia conocido como precondicionamiento isquémico se ha estudiado extensamente por casi ya 20 años. Sin embargo, las vías precisas de transducción de señales involucradas en el disparo y mantenimiento de la protección son todavía tema de intensa investigación. Se ha demostrado en estudios de IPC que el canal KATP juega un papel central en dicho proceso. Se ha sugerido que en el corazón se activarían mecanismos de cardioprotección contra varios tipos de estrés durante el IPC, como la isquemia y la hipoxia. Por ejemplo, durante un período de isquemia prolongada en el tejido cardíaco se produce una caída de los niveles de ATP, y el corazón no sobrevive a la reperfusión. Sin embargo, se ha encontrado que con un precondicionamiento, consistente en breves períodos de isquemia, o con un tratamiento previo con abridores de canales de KATP, se protege al corazón del daño irreversible. Así, durante la isquemia subsecuente la pérdida de ATP se reduce, y el corazón se recupera a una función más cercana a la normal con la reperfusión. Esta cardioprotección, tanto la producida con el tratamiento farmacológico como por el precondicionamiento, se bloquea con fármacos que bloquean el canal KATP mitocondrial como el gliburide (glibenclamida) y el 5hidroxidecanoato (5-HD), lo cual apunta al canal de K+ mitocondrial modulado por ATP como un elemento central en la cardioprotección. Esto ha producido un gran entusiasmo por estudiar el potencial terapéutico de estos canales en el tratamiento de la isquemia aguda del miocardio. O´Rourke (2004) propone que la apertura de los canales KATP mitocondriales tiene un doble efecto, por un lado, disminuye la sobrecarga de calcio en la mitocondria, preservando la integridad de la misma, y por otro lado, estos canales también regulan el volumen de la mitocondria. Estos - 57 - cambios son importantes porque modifican el flujo de energía a través del sistema de electrones, influyendo de esta manera en la transferencia eficiente de energía entre la mitocondria y las ATPasas celulares. Por su parte Czyż et al., (1995) atribuyen al canal KATP mitocondrial la capacidad para compensar la transferencia de carga eléctrica producida por las bombas de protones y de esta manera permitir la formación del gradiente de pH que genera el potencial de membrana. Y finalmente Pain et al., (2000) sugirieron que la apertura del canal mitocondrial no es el efector final de la cascada del precondicionamiento, si no que actúa como el disparador inicial induciendo la liberación de especies reactivas de oxígeno (siglas en inglés reactive oxigen species: ROS). Relación entre el canal KATP mitocondrial y la PKC La activación de la proteína cinasa C (PKC), la apertura de los canales KATP mitocondriales, la liberación de adenosina, son algunos de los elementos comunes en la mayoría de los modelos de IPC. Se ha descrito (Sato et al., 1998) que la actividad de los canales KATP mitocondriales en miocitos cardíacos puede ser regulada por la PKC. Se sabe asimismo que los abridores y bloqueadores del canal KATP mitocondrial afectan la actividad de la PKC (Wang y Ashraf, 1999a,b). La activación de esta enzima incrementa la inactivación de los canales de Ca2+ tipo L del músculo esquelético, lo cual disminuye importantemente el influjo de calcio a la célula (Zaldivar et al., 2005). Por lo tanto, una posibilidad es que los abridores y bloqueadores del canal KATP mitocondrial modulen la actividad de los canales de Ca2+ tipo L del músculo esquelético, lo cual contribuye a regular la concentración intracelular de Ca2+. La administración de 5-HD, un efectivo bloqueador del canal mitoKATP, o la aplicación de queleritrina o calfostina C, o de inhibidores de la PKC, ambos fármacos inhiben completamente el efecto benéfico que tiene el diazoxido sobre la isquemia/IPC de mitocondrias pretratadas (Wang et al., 1999b). - 58 - Iwai et al., 2000 observaron que el diazoxido preserva la función mitocondrial durante isquemia sostenida. Se ha visto que la hipoxia reduce el consumo de oxígeno en fibras desnudas del miocardio en un 40% del valor prehipóxico. En contraste, el tratamiento con diazoxido preserva el consumo normal de oxígeno durante la hipoxia. Este efecto fue completamente inhibido al aplicar glibenclamida o 5-HD. Relación entre el canal KATP mitocondrial y el óxido nítrico Se ha encontrado en estudios electrofisiológicos de canal unitario con el canal KATP mitocondrial un bloqueo importante de su actividad en respuesta a cantidades fisiológicas de oxido nítrico (NO) (Dahlem et al., 2004). Sin embargo, estos resultados están en contradicción con la respuesta obtenida en estudios previos, en donde el canal es abierto por NO y la presencia de éste potencia el efecto del diazóxido (Ockaili et al., 1999). Dahlem et al., (2004) sugirieron que las discrepancias pueden deberse a que el NO puede estar en varios estados redox cuando se utilizan donadores, así como a la formación de peroxinitrito y otras especies reactivas de nitrógeno cuando la cadena respiratoria está activa. Otra posibilidad es que se presenten efectos duales con base en la concentración de NO, sin embargo, es evidente que el NO tuvo efectos directos sobre el canal KATP mitocondrial, al igual que el ATP, el Ca2+ y el 5-HD. Por otro lado, entre los aspectos más estudiados, hasta el momento, son los mecanismos subyacentes al precondicionamiento del corazón. Se ha encontrado que períodos cortos de isquemia y reperfusión llevan a la liberación de substancias tales como el NO, bajas dosis de intermediarios de oxígeno reactivo, adenosina, bradicinina, y prostaciclina. Estas sustancias ‘disparadoras’ pueden producir la activación de cascadas de cinasas, fundamentalmente la PKC, y probablemente río abajo la tirosina cinasa y miembros de la cascada de la MAP cinasa (proteínas cinasas activadas por mitogenos) que son importantes para la señalización celular puesto que, las proteínas cinasas pueden activar factores de - 59 - transcripción. También se ha encontrado un incremento del mRNA para la óxido nítrico sintasa inducible (Li et al., 2004). Con el fin de establecer un modelo experimental de estudio de la función mitocondrial de músculo esquelético, llevamos a cabo procesos de estandarización de aislamiento de mitocondrias de músculo esquelético de pollo y exploramos el efecto de abridores (pinacidil) y bloqueadores (glibenclamida y 5HD) de los canales KATP mitondriales sobre el consumo de oxígeno de mitocondrias aisladas. - 60 - OBJETIVO GENERAL Estudiar el efecto de abridores (pinacidil) y bloqueadores (glibenclamida y 5-HD) de los canales KATP sobre la respiración de mitocondrias aisladas de músculo esquelético de pollo. OBJETIVOS PARTICULARES 1. Estandarizar la técnica de asilamiento de mitocondrias de músculo esquelético de pollo. 2. Cuantificar consumo de oxígeno de la función respiratoria de las mitocondrias aisladas. 3. Realizar una curva dosis-respuesta para los fármacos explorados sobre el consumo de oxigeno. - 61 - MÉTODOS Material biológico: Se utilizaron los músculos Pectoralis (rápido) y Anterior Latissimus Dorsi (lento) de pollos de la variedad Arbor acres, de entre 2 y 4 semanas de edad. La razón de usar pollos de estas edades es por su facilidad de manejo, además de que el músculo es suficientemente grande para proporcionar una cantidad suficiente de mitocondrias aisladas, con las cuales posteriormente se realizaron las mediciones de consumo de oxígeno (véase mas adelante). Disección: Los pollos se sacrificaron por decapitación y enseguida se disecaron los músculos Pectoralis y el ALD, éstos fueron colocados en vasos de precipitados que contenían solución Ginsborg normal (Véase soluciones). Posteriormente se pesaron, y según el peso del músculo se aplicó proteasa (nagarasa 1µg/1mg) necesaria para la extracción de la mitocondrias intermiofibrilares (Talbot et al., 2003, 2004). Extracción: Una vez pesado el músculo, este se colocó en un nuevo vaso el cual contenía la solución 1 de extracción (véase mas adelante), previamente enfriada a 4 °C. El músculo se fraccionó en secciones de aproximadamente 1mm y se pasó a un mortero de un homogenizador que contenía la solución 1, previamente enfriado a 4 °C, con el fin de mantener en buen estado las mitocondrias. El pistón de teflón - 62 - del homogenizador estaba sujeto a un “taladro”, a fin de deshacer la estructura fibrosa del músculo con mayor rapidez y eficacia y favorecer que la nagarasa penetrase hasta las miofibrillas mas internas para permitir la extracción de las mitocondrias. Después de haber homogenizado el tejido este fue centrifugado en una centrifuga refrigerada a 4 °C (Beckman J6 MI) en varias ocasiones (centrifugación diferencial) a 2500, 3500, 7500 y 9000 rpm por 10 min respectivamente, con el fin de ir eliminando tejido y partículas de mayor tamaño hasta obtener solo las mitocondrias. Una vez obtenida la pastilla final, en la que solo se tuvieron mitocondrias, estas se resuspendieron en la solución 2 (véase en soluciones). Soluciones: La solución Ginsborg normal tenía la siguiente composición (en mM): NaCl 167; KCl 5; MgCl2 2; CaCl2 5; Manitol 2 g/L. Esta solución fue ajustada a un pH de 7.4 con Imidazol-Cl 2 mM (Modificada de Huerta y Stefani, 1981) La solución 1 contenía: Sacarosa 100; Tris base 50; KCl 50; EDTA 5. La solución 2 contenia: Sacarosa 250: EGTA 1; Tris base 20. A ambas soluciones se les ajustó el pH a 7.4 con HCl (Barre et al., 1989) Para cuantificar el consumo de oxígeno (oximetría) la solución contenía: KCl 120; KH2PO4 5; Hepes 3; EGTA 1; MgCl2 1. Se le ajustó el pH a 7.4 con Tris base (Barre et al., 1989). Se utilizaron soluciones de succinato 10 mM, glutamato-malato 10 mM y ADP 10 mM, a partir de las cuales se tomaron los volúmenes necesarios para ser - 63 - disueltos en la solución de oximetría teniendo una concentración de 0.26 nM, 1.5 nM y 0.078 nM respectivamente (la concentración de ADP que fue adicionada a la cámara de registro del oxímetro en cada análisis fue dependiente del consumo de oxígeno presentado por las mitocondrias en respuesta al ADP, de 0.039 a 0.156 nM). Las soluciones de los fármacos explorados de glibenclamida (0.5, 1, 5 10 y 20 µM), pinacidil (0.3, 1, 5, 10 y 50 µM) y 5-HD (100 – 500 µM) fueron obtenidas a partir de soluciones iniciales (2mM para la glibenclamida y 1 mM para el pinacidil y el 5-HD) de los fármacos, diluidos en dimetil sulfoxido (glibenclamida y pinacidil) y agua (5-HD), a partir de las cuales se tomaron los volúmenes necesarios que fueron disueltos en las soluciones de registro. Oximetría: Para la medición de consumo de oxígeno se utilizó un oxímetro marca YSI-5300, conectado a una computadora Pentium III por medio de un voltímetro con interfase a PC. Se utilizó 1mg/mL de proteínas (mitocondrias) el cual se determino mediante el método de Biuret (Gornall el at. 1949) con un espectofotómetro marca Perkin Elmer y el sofware Lambda 18. Se le proveyó de succinato y rotenota así como de malato y glutamato como sustratos (véase resultados). Estos experimentos se realizaron el laboratorio de Bioquímica del Instituto de Investigaciones Químico-Biológicas de la Universidad Michoacana de San Nicolás de Hidalgo bajo la supervisión del Dr. Alfredo Saavedra Molina. - 64 - Análisis de datos: Se midió el consumo de oxígeno con una escala normalizada (100%) y se vió la disminución de consumo relativo bajo las diferentes condiciones experimentales. Posteriormente se obtuvo la cantidad de oxígeno consumido acorde para la ciudad de Morelia, Mich. a partir de los registros, ciudad en donde la cantidad de oxígeno disuelta es de 800 nanoátomos (nat) de O2/mL. Se realizó una curva dosis respuesta (ajustada a la ecuación de Hill) para los fármacos utilizados, en donde se graficó el porcentaje de inhibición del consumo de oxígeno contra el logaritmo de la concentración del fármaco (véase resultados). El análisis se realizó con una computadora con un microprocesador Celeron a 1.5 MHz, equipada con el “software” Microsoft Excel (Microsoft Office 2000), Microcal Origin ver. 5.0, Sigmaplot ver. 8.0 e Illustrator 9.0, con los cuales se prepararon las gráficas y figuras finales. Los valores son presentados como medias ± error estándar de la media de al menos 4 experimentos. La significancia de los promedios se determinó empleando la prueba estadística “t” de Student, considerando como significativos aquellos valores de p<0.05. El número de observaciones se presenta entre paréntesis. - 65 - RESULTADOS Efecto de la glibenclamida sobre el consumo de oxigeno en el estado 3 de la respiración. Una vez obtenidas la fracción de mitocondrias por el método de centrifugación diferencial y determinado la concentración de éstas por el método de Biuret (Gornall el at. 1949), se colocó 1 mg/mL de mitocondrias en la cámara de registro del oxímetro, el cual tiene una capacidad de 3 mL, también se le adicionó rotenona, la cual es un insecticida que evita el transporte de electrones del NADH a la coenzima Q, inhibiéndose así a el complejo I, evitando la formación de anión superóxido, se les proporcionó succinato como sustrato (complejo II) (Hanley et al., 2002) y se les proveyó de ADP para inducir su respiración (estado 3), una vez que se determinaba el buen estado de las mitocondrias se observó su comportamiento en este estado, al adicionar glibenclamida 0.5, 1, 5, 10 y 20 µM, el cual, es un bloqueador no selectivo para el canal KATP (Beavis et al., 1993) (Véase la figura 18). - 66 - Figura 18. Muestra el efecto de la glibenclamida sobre el consumo de O2 en estado 3 de la respiración de mitocondrias aisladas de músculo esquelético de pollo. En A se muestra el consumo de O2 en nat de O2/min·mg, en donde, para la ciudad de Morelia fue de 74.27 ± 5.4, lugar en donde se realizaron los experimentos. En B se muestra el consumo de O2 en una escala normalizada al 100%, en ambas gráficas de observa una disminución del consumo de O2 en relación al aumento de la concentración de glibenclamida (efecto dependiente de la concentración) Se muestran con un asterisco lo valores que fueron estadísticamente significativos (p< 0.05). - 67 - 100 Inhibición (%) 80 60 40 20 0 0.1 1 10 Glibenclamida [µM] Figura 19. Muestra la curva dosis-respuesta ajustada a la ecuación de Hill, donde se encontró una EC50 de 0.989 ± 0.06 µM la cual es la concentración media en la cual el fármaco tiene efecto (n=4). Figura 20. Muestra un registro representativo del efecto de la glibenclamida sobre el consumo de oxígeno. Al aplicar concentraciones más altas del fármaco, la modificación de la pendiente es más abrupta, lo que refleja una inhibición de la respiración y una disminución del consumo de O2. - 68 - Ajustamos la curva con la ecuación de Hill, ya que esto nos permite conocer la EC50 (mencionada en el pie de figura), todo esto con el Software Origin 5.0. Además de la glibenclamida, también se probaron los fármacos 5-HD y el pinacidil proporcionando a las mitocondrias succinato como sustrato. En el caso del 5-HD (100 – 500 µM), no se encontró efecto, a pesar de que se le considera a éste un bloqueador selectivo del KATP mitocondrial (Jaburek et al., 1998) (Véase discusión). Por otra parte, tampoco se encontró efecto al aplicar el pinacidil el cual es un abridor no selectivo (Hanley et al., 2002), el cual abre tanto el KATP sarcolemal como el mitocondrial (Liu et al., 1998). Se sabe que algunos abridores de los canales KATP, además de tener efecto sobre el canal mismo pueden afectar algún componente de la cadena respiratoria, como es el caso del diazoxido (bloqueador específico del KATP mitocondrial) sobre el complejo II (Garlid et al., 1997), y la subunidad F0 de la ATP sintasa (Contessi et al., 2005), por lo que también probamos otros sustratos en conjunto con los fármacos mencionados (véase mas adelante). Efecto del pinacidil sobre el consumo de oxigeno en el estado 3 de la respiración. En este caso, se les proporcionó glutamato-malato como sustrato (complejo I) (Debska et al., 2002), y vimos que el pinacidil (0.3, 1, 5, 10 y 50 µM) inhibó el consumo de oxígeno de manera dependiente de la concentración (véase figura 21). - 69 - Figura 21. Ilustra el efecto del pinacidil sobre el consumo de O2 en estado 3 de la respiración de mitocondrias aisladas de músculo esquelético de pollo. En A se muestra el consumo de O2 en nat de O2/min·mg, y fue de 107.2 ± 10.53. En B se muestra el consumo de O2 en una escala normalizada al 100%, en ambas gráficas se observa una disminución del consumo de O2 en relación al aumento de la concentración de pinacidil (efecto similar a lo descrito para la glibenclamida). Se muestran con un asterisco lo valores que fueron diferentes y estadísticamente significativos (p< 0.05). - 70 - 90 80 70 Inhibición (%) 60 50 40 30 20 10 0 -10 0.1 1 10 Pinacidil [µM] Figura 22. Muestra la curva dosis-respuesta ajustada a la ecuación de Hill, donde encontramos un EC50 de 3.5 ± 1.14 µM (n=4). Figura 23. Muestra un registro representativo del efecto del pinacidil sobre el consumo de oxígeno. Aunque el pinacidil como ya se mostró, inhibe la respiración, no lo hace tan eficazmente como la glibenclamida, incluso se probó con este fármaco una concentración más alta, y no se observó el efecto de inhibición total, como en el caso de la glibenclamida (véase discusión). - 71 - Además, también se probaron la glibenclamida y el 5-HD con este sustrato, y no encontramos efecto alguno (resultados no mostrados). Los resultados obtenidos difieren de lo descrito a hasta ahora para el canal KATP mitocondrial en distintas preparaciones, por lo que, discutiremos a fondo nuestras observaciones en la siguiente sección. - 72 - DISCUSIÓN En el presente trabajo se realizaron pruebas de consumo de oxigeno de mitocondrias aisladas tanto del músculo Pectoralis, como del ALD en presencia de glibenclamida y 5-HD. En el caso del músculo ALD, se utilizaron el bloqueante específico para canales KATP mitocondriales 5-HD (Jaburek et al., 1998) y el abridor de canales KATP pinacidil (Hanley et al., 2002) teniendo como sustrato succinato. Para ambos fármacos, no encontramos efecto alguno sobre el consumo de oxígeno por las mitocondrias comparando las respuestas antes y después de la adición de los fármacos. Se realizó la observación empleando el 5-HD teniendo como sustrato glutamato y el consumo de oxígeno por las mitocondrias tampoco mostró algún cambio significativo en las respuestas antes y posterior a la adición del bloqueante de los canales KATP mitocondriales (datos no mostrados). Sin embargo, los resultados presentados en el presente trabajo, representan los efectos observados en el consumo de oxigeno de las mitocondrias aisladas de músculo Pectoralis, el cual fue seleccionado por aportar una mayor densidad de mitocondrias por volumen de músculo y posibilitó obtener un mayor número de registros de mitocondrias aisladas. Con respecto al efecto de la glibenclamida sobre el consumo de oxígeno, Jaburek y cols. (1998) reportaron que la inhibición de la respiración mitocondrial por la glibenclamida al usar succinato como sustrato es debido a un efecto inespecífico, puesto que la glibenclamida no es un bloqueante selectivo para el canal KATP mitocondrial (Beavis et al., 1993) cuando fue explorado en mitocondrias de corazón desacopladas. En nuestro caso, empleando mitocondrias aisladas de músculo esquelético, la glibenclamida indujo la disminución de la respiración mitocondrial cuando éstas no fueron expuestas a desacoplantes, lo cual pudiera establecer los resultados contrastantes con aquellos reportados para mitocondrias aisladas de corazón. Por otra parte, al igual que lo descrito por Jaburek y cols. (1998), no se observó ningún efecto del 5-HD sobre la respiración mitocondrial cuando se - 73 - usaron sustratos tanto para complejo I y II de la respiración. Sin embargo, estos autores argumentan que esto puede ser debido a la necesidad de algún ligando que se una al canal y modifique su conformación para que el fármaco ejerza su efecto, lo que se cree que suceda en situaciones in vivo y no en las condiciones in vitro y usando succinato como sustrato. Estos resultados son acordes con lo descrito para el mito-KATP de corazón (Garlid et al., 1997; Grover, 1997). Sin embargo, Debska et al. (2002), reportaron el efecto inhibidor del 5-HD (500 µM) en mitocondrias de músculo esquelético de rata solo de un 20% en los experimentos analizados, por lo que sugirieron que el fármaco solo es efectivo para mitocondrias de corazón. Por otro lado, es importante mencionar que en los estudios realizados con sistemas de expresión heterólogos, donde no se tienen las condiciones del canal in vivo, es posible tener una aproximación de las condiciones para conocer el funcionamiento del canal y su susceptibilidad a los fármacos que hemos utilizado como son los estudios realizados por Liu y cols. (1999), quienes transfectaron en ovocitos de Xenopus todas las combinaciones posibles de las subunidades del canal KATP y utilizaron varios fármacos abridores y bloqueadores del canal, tanto específicos como no específicos; encontrando que el diazóxido (100 µM) abre el canal en la combinación Kir6.1/SUR1 (situación no descrita en ningún tejido hasta esa fecha); así como con la combinación Kir6.1/SUR2B descrito para tejido vascular y Kir6.2/SUR1 descrito en células β-pancreáticas, así como es conocido que el diazóxido también abre el canal mitocondrial. Por otra parte, encontraron que la glibenclamida (10 µM) bloquea al canal en todas sus combinaciones. Y, finalmente, encontraron que el 5-HD (200 µM) cierra el canal con la composición estructural Kir6.1/SUR1, Kir6.1/SUR2B y el canal mitocondrial, sugiriendo que la composición del canal mito-KATP pudiera estar formada por Kir6.1/SUR1, estructura que fue confirmada por Suzuki y cols. (1997), quienes con técnicas de tinción con anticuerpos específicos localizaron la subunidad Kir6.1 en la membrana interna de mitocondrias aisladas del músculo esquelético. Sin embargo, esta estructura no concuerda para el canal de mitocondrias de músculo cardiaco (Seharaseyon et al., 2000). - 74 - Aunque se sabe de la inespecificidad de la glibenclamida sobre los canales KATP dado que bloquea también otros tipos de canales de potasio, así como las corrientes acarreadas por TEA+ (Jaburek et al., 1998), su inespecificidad es debida a las altas concentraciones empleadas y a la baja afinidad que el fármaco tiene sobre las enzimas transductoras de energía (Debeer at al., 1974; McGuines y Cherrington, 1990; Somogyi et al., 1995a,b), así que su efecto refleja en una disminución del flujo de K+ que pudiera no estar directamente relacionada con el KATP. Sin embargo, no se descarta que el efecto pudiera estar relacionado con el bloqueo del canal KATP mitocondrial y se requieren de mayores estudios y la utilización de diferentes técnicas como la medición de la corriente de potasio en canales unitarios aislados de membrana interna mitocondrial que ofrezcan información adicional para concluir con mayor precisión sobre la acción de estos fármacos sobre el funcionamiento del canal KATP en mitocondrias aisladas de músculo esquelético. Para el caso de los experimentos en los cuales se utilizó el pinacidil, se decidió usar un sustrato diferente al usado con glibenclamida puesto que en trabajos previos fue mostrado que el pinacidil no tuvo efecto sobre el consumo de oxígeno en presencia de succinato como sustrato (Hanley et al., 2002). En este caso, Hanley y cols. (2002) usaron NADH (800 µM) como sustrato para partículas submitocondriales aisladas de mitocondrias de corazón de cobayo, en las cuales característicamente queda invertida la membrana interna mitocondrial que es el lugar donde se encuentra la NADH deshidrogenada y consecuentemente usa NADH como sustrato. La aplicación de pinacidil (23-230 µM) mostró una disminución concentración-dependiente del consumo de oxigeno con una EC50 de 90 µM, lo que sugiere que el pinacidil inhibe al complejo I (NADH: ubiquinona oxidoreductasa). En nuestro estudio, se utilizó glutamato-malato como sustrato (complejo I) encontrando que efectivamente, el pinacidil inhibe de manera concentración-dependiente el consumo de oxígeno (EC50= 3.5 ± 1.14 µM). Sin embargo, aunque nuestros resultados están de acuerdo con lo descrito por Hanley y cols. (2002), es importante resaltar que las concentraciones que utilizamos son menores a las reportadas por estos investigadores y a aquellas que - 75 - Holmuhamedov y cols (1998) reportaron como tóxicas. Es decir, se ha descrito que el efecto de algunos fármacos puede ser de tipo farmacológico o tóxico, dependiendo de la concentración que se utilice, este es el caso del pinacidil, que inhibe la respiración e incrementa la permeabilidad de la membrana, lo que causa una disminución del potencial de membrana y un decremento en la entrada de Ca2+. Se considera una dosis con efecto farmacológico aquella menor a 50 µM (Holmuhamedov et al., 1998; 1999), y como se mostró en nuestros resultados, esta fue la concentración mayor que se utilizó. También, como ya se mostró, se obtuvo inhibición del consumo de oxígeno (p= 0.012) (figura 21) a partir de 1 µM. Por otra parte, los experimentos realizados por estos investigadores son consistentes con el hecho de que observaron el efecto del fármaco después de haber expuesto a las mitocondrias a desacoplantes como el FCCP (carbonilcianida-p-trifluorometoxifenilhidrazona), y atribuyen el efecto observado a la naturaleza hidrofóbica de los fármacos usados. Por otra parte, Holmuhamedov y cols. (1998) también estudiaron el efecto de los abridores de los canales KATP (pinacidil, cromakalim y levocromakalim) sobre el potencial de membrana, la respiración y la generación de ATP en mitocondrias aisladas de corazón de rata y observaron que estos fármacos ocasionaban una despolarización de 10 ± 7, 25 ± 9 y 24 ± 10 mV, respectivamente, lo cual está relacionado con un incremento en la velocidad de respiración (en el estado 2 de la respiración) y una disminución en la síntesis de ATP. En experimentos en donde midieron el transporte de calcio, observaron que en mitocondrias pre-cargadas con este ión el efecto del abridor era dependiente de la concentración de potasio extra mitocondrial y, que se presentaba una inhibición la cual fue atribuida al canal mito-KATP, ya que en la pruebas de hinchamiento que realizaron observaron que tanto el cromakalim como el pinacidil incrementaron el volumen mitocondrial. Debska y cols. (2002) reportaron resultados similares en mitocondrias de músculo esquelético y de líneas celulares de rata con diazóxido y cromakalim. - 76 - Con respecto a los mecanismos relacionados con la fatiga del músculo esquelético, se han propuesto diversos que ocurren a nivel periférico, relacionados con la membrana de las células musculares y con el metabolismo celular (Fitts, 1994), por lo que no es sencillo dar una explicación para el fenómeno que describimos en la presente tesis, donde fue analizada la respiración mitocondrial de manera aislada del músculo in vivo, donde se analizó de manera simultánea la tensión, por eso es necesario correlacionar nuestras observaciones con los mecanismos ya descritos que se les considera responsables de fenómeno de fatiga. En estudios previos Andrade (2004) utilizando fibras musculares lentas registró la tensión de la sacudida e indujo la inhibición metabólica con cianuro (10 mM), mostrando que la glibenclamida incrementaba la tensión post-inhibición. Andrade (2004) concluyó que el cianuro al impedir la producción de ATP y la disminución intracelular de este, censada por los canales KATP presentes en el sarcolema, se verían activados en estas condiciones, ligando así la excitabilidad de la membrana con el metabolismo y la adición de la glibenclamida en esas condiciones de inhibición metabólica inhibió el efecto producido por el cianuro. Este modelo de inhibición metabólica inducida por esta droga fue utilizado por Gramolini y Renaud (1997) con el fin de que no se produjera ATP afectado el transporte de electrones en la cadena respiratoria, de tal forma que decrece la concentración intracelular de este metabolito. Por otra parte, se ha mostrado en el músculo esquelético que después de un periodo de contracciones fatigantes, el músculo entra en un periodo de disminución de su capacidad contráctil (Fitts y Metzger, 1993), lo cual también ha sido asociado a una disminución en la concentración intracelular de calcio (Allen et al., 2001), lo cual a su vez podría también afectar el acople entre la excitación y la contracción. Westerblad y Allen (2001) proponen que el cianuro previene la recuperación del almacenamiento del calcio en el retículo sarcoplásmico ya que la recaptura de calcio por el retículo depende del ATP, lo cual pudiera explicar el - 77 - incremento de la tensión basal en registros de tensión de estudios previos realizados en músculo esquelético lento (Andrade, 2004). De esta manera, la disminución del ATP juega un papel muy importante en el desarrollo de la fatiga muscular, por lo que cualquier suceso que altere la producción del mismo puede determinar la reacción del músculo a la fatiga. En nuestro caso, a nivel de la mitocondria, si el canal mitocondrial de potasio sensible a ATP censa los niveles de este metabolito permitiendo la entrada del ión potasio acompañado de agua, provoca el hinchamiento del organelo y de esta forma altera el proceso de producción de ATP, lo que a su vez provocaría una disminución del metabolito en el citosol activando los canales de potasio sensibles a ATP sarcolemales. Así, queda claro que existe una correlación muy estrecha entre el funcionamiento de la mitocondria y la respuesta del músculo a la fatiga, razón por la cual es de nuestro interés el tratar de relacionar estos fenómenos y como ya se menciono anteriormente, dado que las condiciones fisiológicas in vitro difieren de lo que sucede in vivo, sugerimos que se requiere de análisis más completo para aportar una conclusión mas contundente de la relación fatiga muscular-respiración mitocondrial. - 78 - CONCLUSIONES 1. Se estandarizó el método para el aislamiento de mitocondrias de músculo esquelético de pollo. 2. La glibenclamida (bloqueador de los canales KATP) inhibió el consumo de oxígeno de forma dependiente de la concentración en el estado 3 de la respiración de mitocondrias (no expuestas a desacoplantes) de músculo esquelético de pollo teniendo como sustrato succinato. 3. El 5-HD (bloqueador de los canales KATP mitocondriales) no tuvo efecto a las concentraciones exploradas (100 – 500 µM) sobre las mitocondrias aisladas de músculo esquelético de pollo, sugiriendo una composición diferente al KATP sarcolemal del mismo músculo esquelético y de mitocondrias de músculo cardiaco. 4. El pinacidil (abridor de los canales KATP) inhibió el consumo de oxígeno de forma dependiente de la concentración en el estado 3 de la respiración de mitocondrias (no expuestas a desacoplantes) de músculo esquelético de pollo teniendo como sustrato glutamato-malato. - 79 - PERSPECTIVAS Como se mencionó en la discusión, es necesario hacer más pruebas, con diferentes técnicas para tener resultados concluyentes, por lo que se propone analizar las siguientes metodologías: 1. Con el fin de determinar el efecto de los abridores y bloqueadores del canal mito-KATP, hacer pruebas de hinchamiento de las mitocondrias con espectrofotometría. 2. Para determinar la toxicidad de los fármacos medir transporte de Ca2+ a la mitocondria, mediante espectrofotometría. 3. Medir cambios en el potencial de membrana al aplicar los fármacos, esto en relación con el volumen mitocondrial y la medición de algún ion como el tetrafenilfosfonio (TPP+). 4. Realizar experimentos en presencia de ATP y Mg2+ con el fin de garantizar el estado cerrado de los canales mito-KATP. 5. Utilizar abridores más selectivos para el canal mito-KATP como el diazóxido, tanto en pruebas de consumo de oxígeno como las que ya realizamos. 6. Hacer pruebas de los fármacos usados en diferentes estados de la respiración. 7. Reconstruir el canal mito-KATP en liposomas y en bicapas lipídicas con el fin de medir corrientes unitarias. 8. Reconstruir el canal mito-KATP en liposomas y en bicapas lipídicas con el fin de observar su estructura con microscopia de fuerza atómica. 9. Realizar experimentos de medición de tensión utilizando el abridor pinacidil y el bloquedor 5-HD. - 80 - REFERENCIAS Aguggini G., Dimori M., Vanelli G., Albertini M. (1996) The effects of glibenclamide, a blocker of K+ ATP-sensitive potassium channels, on diaphragmatic fatigue during endotoxaemia in pigs. Vet Res Commun. 20(2):183-90. Aguilar-Bryan L., Bryan J. and Nakazaki, M. (2001). Of mice and men: K(ATP) channels and insulin secretion. Recent Prog Horm Res 56, 47-68. Allen D. G., Lee J. A. y Westerblad H. (2001). Intracellular calcium and tensión during fatigue in isolated single muscle fibres from Xenopus laevis. J. Physiol. 415:433-358. Ames B.N., Shigenaga M. K. and Hagen T.M. (1995). Mitochondrial decay in aging. Biochem Biophys Acta 1271, 165–170. Andrade F. (2004) Efecto de la glibenclamida sobre la fatiga en músculo esquelético lento de pollo. Tesis doctoral. Facultad de Medicina. Universidad de Colima. México. Annex B.H. And Williams R.S. (1990). Mitochondrial DNA structure and expression in specialized subtypes of mammalian striated muscle. Molec Cell Biol 10, 5671–5678. Annex B.H., Kraus W.E., Dohm G.L. and Williams R.S. (1991). Mitochondrial biogenesis in striated muscles: rapid induction of citrate synthase mRNA by nerve stimulation. Am J Physiol 260, c266– c270. Ardehali H., O’Rourke B. (2005) Mitochondrial K(ATP) channels in cell survival and death. J. Mol. Cell Cardiol. 39, 7-16. Ardehali H., O’Rourke B., Marbán E. (2005) Cardioprotective role of the mitochondrial ATP-binding cassette protein 1. Circ. Res. 97, 740-742. Attardi G. and Schatz G. (1988). Biogenesis of mitochondria. A. Rev. Cell biol. 4, 289–333. Baldwin K.M., Klinkerfuss G.H., Terjung R.L., Mole P.A. and Holloszy J.O. (1972). Respiratory capacity of white, red and intermediate muscle: adaptive response to exercise. Am J Physiol 222, 373–378. Barre H., Berne G., Brebion P., Cohen-Adad F., Rouanet J.L. (1989) Loose-coupled mitochondria in chronic glucagon-treated hyperthermic ducklings. Am J Physiol. 256(6 Pt 2):R1192-9. - 81 - Baukrowitz T. and Fakler, B. (2000). KATP channels gated by intracellular nucleotides and phospholipids. Eur J Biochem 267, 5842-5848. Beavis A.D., Lu Y., Garlid K.D. (1993) On the regulation of K+ uniport in intact mitochondria by adenine nucleotides and nucleotide analogs. J Biol Chem. 15;268(2):997-1004. Beguin P., Nagashima K., Nishimura M., Gonoi, T. and Seino S. (1999). PKA-mediated phosphorylation of the human K(ATP) channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. Embo J 18, 4722-4732. Bernardi, P. (1999) Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 79, 1127-1155. Berne R.M., Levy M.N., Koeppen B.M. y Stanton B.A. 1992. Physiology. 5ª ed. Mosby, St. Louis, Missouri. Booth F.W. (1977) Effects of endurance exercise on cytochrome cturnover in skeletal muscle. Ann NY Acad Sci 301: 431–439. Boyer P.D. (1989) A perspective of the binding change mechanism for ATP synthesis. FASEB J. 1989 Aug;3(10):2164-78. Review. Bryan J., Vila-Carriles W.H., Zhao G., Babenko A.P, Aguilar-Bryan L. (2004) Toward linking structure with function in ATP-sensitive K+ channels. Diabetes. 53 Suppl 3:S104-12. Burke R.E., Levine D.N. y Zajac F.E. (1971). Mammalian motor units: physiological-histochemical correlation in three types in cat gastrocnemius. Science. 174:709-12. Burton F.L. and Smith G.L. (1997) The effect of cromakalim on intracellular [Ca2+] in isolated rat skeletal muscle during fatigue and metabolic blockade. Exp. Physiol. 82; 469-483. Chance B., Williams G.R., (1955) A simple and rapid assay of oxidative phosphorylation. Nature. 175 (4469):1120-1. Cogswell A.M., Stevens R.J. and Hood D.A. (1993). Properties of skeletal muscle mitochondria isolated from sub-sarcolemmal and interfibrillar regions. Am. J Physiol 264, c383–c389. Contessi S, Metelli G, Mavelli I, Lippe G.(2004) Diazoxide affects the IF1 inhibitor protein binding to F1 sector of beef heart F0F1ATPsynthase. - 82 - Biochem Pharmacol. 15; 67(10):1843-51. Czyż, A., Szewczyk, A., Nałęcz, M.J., & Wojtczak, L. (1995) The role of mitochondrial potassium fluxes in controlling the protonmotive force in energized mitochondria. Biochem. Biophys. Res. Commun. 210, 98-104. Dahlem Y.A., Horn T.F.W, Buntinas L., Gonoi T., Wolf G., Siemen, D. (2004) The human mitochondrial KATP channel is modulated by calcium and nitric oxide: a patch-clamp approach. Biochim. Biophys. Acta. 1656, 46-56. Das M., Parker J.E., Halestrap A.P. (2003) Matrix volume measurements challenge the existence of diazoxide/glibencamide-sensitive KATP channels in rat mitochondria. J Physiol 547, 893-902. Daut J., Maier-Rudolph W., von Beckerath N., Mehrke G., Gunther K. and Goedel-Meinen, L. (1990). Hypoxic dilation of coronary arteries is mediated by ATP-sensitive potassium channels. Science 247, 1341-1344. Daut J., Klieber H. G., Cyrys S. and Noack, T. (1994a). KATP channels and basal coronary vascular tone. Cardiovasc Res 28, 811-817. Daut J., Standen N. B. and Nelson, M. T. (1994b).The role of the membrane potential of endothelial and smooth muscle cells in the regulation of coronary blood flow. J Cardiovasc Electrophysiol 5, 154-181. Davies K.J.A., Packer l. and Brooks G.A. (1981). Biochemical adaptation of mitochondria, muscle and whole animal respiration to endurance training. Archs biochem. Biophys. 209, 539–554. Debeer L., Mannaerts G., De Schepper .PJ. (1974) Metabolic effects of hypoglycemic sulfonylureas. In vitro effects of sulfonylureas on mitochondrial adenosine triphosphatase activity. Biochem Pharmacol 23(2):251-8. Debska G., Kicinska A., Skalska J., Szewczyka A., May R., Elger C., Kunz W., (2002) Opening of potassium channels modulates mitochondrial function in rat skeletal muscle. Biochim Biophys Acta 1556; 97–105. Edwards R.H.T (1981) Human muscle function and fatigue. En: R. Porter y J. Whelan (Eds.), Human muscle fatigue: Physiological mechanism 1–18. London: Pitman medical. Eisenberg B.R. and Salmons S. (1981) The reorganization of subcellular structure in muscle undergoing fast-to-slow type transformation. Cell Tissue Res 220: 449–471. - 83 - Enoka R.M. y Stuart D.G. (1992). Neurobiology of muscle fatigue. J Appl Physiol 72:1631-1648. Fan Z., Gao L., and Wang W. (2003). Phosphatidic acid stimulates cardiac KATP channels like phosphatidylinositols, but with novel gating kinetics. Am J Physiol Cell Physiol 284, C94-102. Fitts, R.H., Kim D.H., Witzmann F.A. (1981) The development of fatigue during high intensity and endurance exercise. Exercise in Health and Disease. 118–135. Fitts, R. H. y Metzger, J. M. (1993). Mechanisms of muscular fatigue. In: Principles of exercise biochemistry (2nd Ed. Vol. 38). J. R. Poortmans (Ed.) Basel, Switzerland: Karger.1993. Flagg T.P. and Nichols C.G. (2001). Sarcolemmal K(ATP) channels in the heart: molecular mechanisms brought to light, but physiologic consequences still in the dark. J Cardiovasc Electrophysiol 12, 1195-1198. Garlid K.D., Paucek P. Mitochondrial potassium transport: the K+ cycle, Biochim. Biophys. Acta 1606 (2003) 23–41. Garlid K.D., Paucek P., Yarov-Yarovoy H.N., Murray R.B., Darbenzio A.J., D´Alonzo N.J., Lodge N.J., Smith M.A., Grober G.J. (1997) Cardiprotective effect of diazoxide and its interaction with mitonchondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ. Res. 81, 1072 – 1082. Gelfand R. and Attardi G. (1981). Synthesis and turnover of mitochondrial ribonucleic acid in hela cells: the mature ribosomal and messenger ribonucleic acid species are metabolically unstable. Molec. Cell. Biol. 1, 497–511. Ginsborg B.L. (1960). Some properties of avian skeletal muscle fibres with multiple neuromuscular junctions. J Physiol 154: 581–598. Ginsborg B.L. y Mackay B. (1961). A histochemical demonstration of two types of motor innervation in avian skeletal muscle. Bibl. Anat. 2:174-180. Gollnick P.D. and King D.W. (1969) Effect of exercise and training on mitochondria of rat skeletal muscle. Am J Physiol 216: 1502–1509. Gong B., Miki T., Seino S., Renaud J.M. (2000) A K(ATP) channel deficiency affects resting tension, not contractile force, during fatigue in skeletal muscle. Am J Physiol Cell Physiol. 279(5):C1351-8. - 84 - Gopel S. O., Kanno T., Barg S., Weng X. G., Gromada J. and Rorsman P. (2000). Regulation of glucagon release in mouse -cells by KATP channels and inactivation of TTX-sensitive Na+ channels. J Physiol 528, 509-520. Gornall A.G., Bardawill C.J., David M.M. (1949) Determination of serum proteins by means of the biuret reaction. J Biol.Chem 177, 751-766. Gramolini, A., Renaud, J. M. (1997). Blocking ATP-sensitive K+ channel during metabolic inhibition impairs muscle contractility. Am. J. Physiol. 272:1936-1946. Grimberg A., Ferry R. J., Jr., Kelly A., Koo-McCoy S., Polonsky K., Glaser B., Permutt M. A., Aguilar-Bryan L., Stafford D., Thornton P. S., Baker L. and Stanley C. A. (2001). Dysregulation of insulin secretion in children with congenital hyperinsulinism due to sulfonylurea receptor mutations. Diabetes 50, 322-328. Grover G.J. (1997) Review. Pharmacology of ATP-sensitive potassium channel (KATP) openers in models of myocardial ischemia and reperfusion. Can J Physiol Pharmacol. 75(4):309-15. Gustafsson R., Tata J.R., Lindberg O., and Ernster L. (1965) The relationship between the structure and activity of rat skeletal muscle mitochondria after thyroidectomy and thyroid hormone treatment. J. Cell Biol. 26, 555-578. Hanley P.J., Mickel M., Loffler M., Brandt U., Daut J. (2002) K(ATP) channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J Physiol 542, 735-741. Haruna T., Yoshida H., Nakamura T. Y., Xie L. H., Otani H., Ninomiya T., Takano M., Coetzee, W. A. and Horie M. (2002). Alpha1-adrenoceptor-mediated breakdown of phosphatidylinositol 4,5bisphosphate inhibits pinacidil-activated ATP-sensitive K+ currents in rat ventricular myocytes. Circ Res 91, 232-239. Henriksson J. and Reitman J.S. (1977) Time course of changes in human skeletal muscle succinate dehydrogenase and cytochrome oxidase activities and maximal oxygen uptake with physical activity and inactivity. Acta Physiol Scand 99: 91–7. Wicks KL and Hood DA. (1991) Mitochondrial adaptations in denervated muscle: relationship to muscle performance. Am J Physiol Cell Physiol 260: C841–C850. Hickson R.C. and Rosenkoetter M.A. (1981) Separate turnover of cytochrome c and myoglobin in the red types of skeletal muscle. Am J Physiol Cell Physiol 241: C140–C144. - 85 - Holloszy J.O. (1967) Biochemical adaptations in muscle. J Biol Chem 242: 2278–2282. Holmuhamedov E.L., Wang L., and Terzic A. (1999) ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J Physiol (Lond) 519: 347–360. Holmuhamedov E.L., Jovanovic S., Dzeja P.P., Jovanovic A., Terzic A. (1998) Mitochondrial ATPsensitive K+ channels modulate cardiac mitochondrial function. Am J Physiol 275, H1567-H1576. Hood D. A., Zak R. and Pette D. (1989). Chronic stimulation of rat skeletal muscle induces coordinate increases in mitochondrial and nuclear mRNAs of cytochrome-c-oxidase subunits. Eur. J. Biochem. 179, 275–280. Hood D. A., Balaban A., Connor M. K., Craig E. E., Nishio M. L., Rezvani M. and Takahashi M. (1994). Mitochondrial biogenesis in striated muscle. Can J Appl Physiol 19, 12–48. Hood D.A. (2001) Plasticity in skeletal, Cardiac, an Smooth muscle. Invited Review: Contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol. 90_1137 – 1157. Hoppeler H., Luthi P., Claassen H., Weibel E.R. and Howald H. (1973) The ultrastructure of the normal human skeletal muscle. Pflügers Arch 344: 217–232. Huerta M., y Stefani E. (1981) Potassium and caffeine contractures in fast and slow muscles of the chicken. J Physiol. 318:181-9. Inoue I., Nagase H., Hishi K., Higuti, T. (1991) ATP-sensitive K+ channel in the mitochondrial inner membrana. Nature 352(6332), 244-247. Iwai T., Tanonaka K., Koshimizu M., Takeo S. (2000) Preservation of mitochondrial function by diazoxide during sustained ischaemia in the rat heart. Br. J. Pharmacol. 129, 1219-1227. Jaburek M., Yarov-Yarovoy V., Paucek P., Garlid K.D. (1998) State-dependent inhibition of the mitochondrial KATP channel by glyburide and 5-hydroxydecanoate. J Biol Chem. 1998 May 29;273(22):13578-82. Jones M., Ferrans V.J., Morrow A.G., and Roberts W. (1975) Ultrastructure of crista ventricularis muscle in patients with congenital heart disease associated with right ventricular outflow tract obstruction. Circulation 51, 39-67. - 86 - Kanno T., Gopel S. O., Rorsman P. and Wakui M. (2002). Cellular function in multicellular system for hormone-secretion: electrophysiological aspect of studies on alpha-, beta- and delta-cells of the pancreatic islet. Neurosci Res 42, 79-90. Kanno T., Rorsman P., and Gopel, S.O. (2002) Glucose-dependent regulation of rhythmic action potential firing in pancreatic beta-cells by K(ATP)-channel modulation. J Physiol 545, 501-507. Karp G. (1998) Biología Celular y Molecular. 1 Edición. Ed. McGraw-Hill Interamericana. México, D.F. Kiessling K.H., Piehl K. and Lundquist C.G. (1971) Effect of physical training on ultrastructural features in human skeletal muscle. In: Muscle Metabolism During Exercise, edited by Pernow B and Saltin B. New York: Plenum, p. 97–101. Kowaltowski, A.J. Seetharaman, S. Paucek, P. Garlid, K.D. (2001). Bioenergetic consequences of opening the ATP-sensitive K+ channel of heart mitochondria, Am J Physiol 280 H649–H657. Kuan J. and Saier M.H. Jr. (1993) The mitochondrial carrier family of transport proteins: structural, functional, and evolutionary relationships. Crit Rev Biochem Mol Biol 28:209–233. Kubista V, Kubistova J, Pette D. (1971) Thyroid hormone induced changes in the enzyme activity pattern of energy-supplying metabolism of fast (white), slow (red), and heart muscle of the rat. Eur J Biochem. 18(4):553-60. Kuffler S. W. y Vaughan-Williams E. M. (1953a). Small-nerve junction potentials. The distribution of small motor nerves to frog skeletal muscle and the membrane characteristics of the fibres they innervate. J Physiol 121:289-317. Kuffler S. W. y Vaughan-Williams E. M. (1953b). Properties of the slow skeletal muscle fibres of the frog. J Physiol 121:318-340. Li G., Labruto F., Sirsjo A., Chen F., Vaage J., Valen G. (2004). Myocardial protection by remote preconditioning: the role of nuclear factor kappa-B p105 and inducible nitric oxide synthase. Eur J Cardiothorac Surg. 26, 968-73. Light P. E., Bladen C., Winkfein R. J., Walsh M. P. and French, R. J. (2000). Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc Natl Acad Sci U S A 97, 9058-9063. - 87 - Lim K.H., Javadov S.A., Das M., Clarke S.J., Suleiman M.S., Halestrap A.P. (2002). The effects of ischaemic preconditioning, diazoxide and 5-hydroxydecanoate on rat heart mitochondrial volume and respiration. J Physiol 545, 961- 974. Lin Y. F., Jan Y. N. and Jan, L. Y. (2000). Regulation of ATP-sensitive potassium channel function by protein kinase A-mediated phosphorylation in transfected HEK293 cells. Embo J 19, 942-955. Linnane A. W., Marzuki S., Ozawa T. and Tanaka M. (1989). Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 8639, 642–644. Linstedt S. L., Hokanson J. F., Wells D. J., Swain S. D., Hoppeler H. and Navarro V. (1991) Running energetics in the pronghorn antelope. Nature 353, 748–750. Liss B. and Roeper, J. (2001a). Review. Molecular physiology of neuronal K-ATP channels. Mol Membr Biol 18, 117-127. Liss B. and Roeper, J. (2001b). ATP-sensitive potassium channels in dopaminergic neurons: transducers of mitochondrial dysfunction. News Physiol Sci 16, 214-217. Liss B., Bruns R., and Roeper J. (1999) Alternative sulfonylurea receptor expression defines metabolic sensitivity of K-ATP channels in dopaminergic midbrain neurons The EMBO Journal 18, 833–846, Liu Y., Ren G., O'Rourke B., Marban E., Seharaseyon J. (2001) Pharmacological comparison of native mitochondrial K(ATP) channels with molecularly defined surface K(ATP) channels. Mol Pharmacol. 59(2):225-30. Liu Y., Sato T., O’Rourke B. and Marban E. (1998) Mitochondrial ATP-dependent potassium channels: Novel effectors of cardioprotection? Circulation 97:2463–2469. Luciakova, K. and Nelson, B. D. (1992). Transcript levels for nuclear-encoded mammalian mitochondrial respiratory-chain components are regulated by thyroid hormone in an uncoordinated Fashion. Eur. J. Biochem. 207, 247–251. Manneschi, l. and Federico, A. (1995). Polarographic analyses of subsarcolemmal and interfibrillar mitochondria from rat skeletal and cardiac muscle. J. Neurol. Sci. 128, 151–156. McArdle W.D., Katch F.I. & Katch V.L. (1996). Exercise physiology: Energy, nutrition and human performance (4th ed.). Philadelphia: Lea & Febiger. USA. - 88 - McCallister, B. D., and Brown, A. L. (1969) A quantitative study of myocardial mitochondria in experimental cardiac hypertrophy. Ann. N. Y. Acad. Sci. 156, 469-479. McGuinness O.P.; discussion Cherrington A.D. (1990) Review Effect of glyburide on hepatic glucose metabolism. Am J Med. 89(2A):26S-37S. McKean, T. A. (1990). Comparison of respiration in rat, guinea pig and muskrat heart mitochondria. Comp. Biochem. Physiol. 97b, 109–112. Miki T., Suzuki M., Shibasaki T., Uemura H., Sato, T., Yamaguchi, K., Koseki, H., Iwanaga T., Nakaya H. and Seino S. (2002). Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat Med 8, 466-472. Mitchell P. (1979) Keilin's respiratory chain concept and its chemiosmotic consequences. Sience 206(4423):1148-59. Mitchell P. (1961) Coupling of phosphorylation to electrón and hydrogen transfer by a chemiosmotic type of mechanism. Nature (London) 191:144 – 148. Moyes C. D., Mathieu-Costello O. A., Brill R. W. and Hochachka P. W. (1992). Mitochondrial metabolism of cardiac and skeletal muscles from a fast (Katsuwonis pelamis) and a slow (Cyprinus carpio) fish. Can. J. Zool. 70, 1246–1253. Moyes C.D., Mathieu-Costello O.A., Tsuchiya N., Filburn C. and Hansford R.G. (1997) Mitochondrial biogenesis during Cellular differentiation. Am. J Physiol 272, c1345–c1351. Moyes C.D., Battersby B.J., Leary S.C. (1998) Review. Regulation of muscle mitochondrial desig. The Journal of experimental biology 201, 299 – 307. Muller W. (1976) Subsarcolemmal mitochondria and capillarization of soleus muscle fibers in young rats subjected to an endurance training. A morphometric study of semithin sections. Cell Tissue Res. 174, 367-389. Murray R.K., Mayes P.A., Granner D.K., Rodwell V.W. (2001) Bioquímica de Harper. 15 Edición. Ed. El manual moderno. México, D.F. - 89 - Nerbonne J. M., Nichols C. G., Schwarz T. L., and Escande D. (2001). Genetic manipulation of cardiac K(+) channel function in mice: what have we learned, and where do we go from here? Circ Res 89, 944-956. Nichols C. G., Lederer W. J. (1991) Adenosine triphosphate-sensitive potassium channels in the cardiovascular system. Am J Physiol 261,H1675-H1686. Noma A. (1983) ATP-regulated K+ channels in cardiac muscle. Nature 305:147-148. O’Rourke, B. (2000) Myocardial KATP channels in preconditioning. Circ. Res. 87,845-855. O´Rourke (2004) Evidence for Mitochondrial K+ Channels and Their Role in Cardioprotection . Circ Res. 94, 420-432. Page S. G. (1969). Structure and some contractile properties of fast and slow muscles of the chicken. J Physiol 205:131–145. Pain T., Yang X.M., Critz S.D., Yue Y., Nakano A., Liu G.S., Heusch G., Cohen M.V., Downey J.M. (2000) Opening of mitochondrial KATP channels triggers the preconditioned state by generating free radicals. Circ Res. 87(6):460-6. Palmer J.W., Tandler B. and Hoppel C.L. (1977) Biochemical properties of subsarcolemmal e intermiofrillar mitochondria isolated from rat cardiac muscle. J Biol Chem 252, 8731 – 8739. Palmer J.W., Tandler B. and Hoppel C.L. (1985). Biochemical differences between subsarcolemmal and interfibrillar mitochondria from rat cardiac muscle: effects of procedural manipulations. Archs Biochem Biophys 236, 692–702. Palmieri F. (1994) Mitochondrial carrier proteins. FEBS Lett 346:48–54. Palmieri F., Bisaccia F., Lacobazzi V., Indiveri C., and Zara V. (1992) Mitochondrial substrate carriers. Biochem Biophys Acta 1101:223–227. Paucek P., Mironova G., Mahdi F., Beavis A.D., Woldegiorgis G., Garlid, K.D. (1992) Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. J Biol Chem 267, 26062-9. - 90 - Paucek P., Yarov-Yarovoy V., Sun X., Garlid K.D. (1996) Inhibition of the mitochondrial KATP channel by long-chain acyl-CoA esters and activation by guanine nucleotides. J Biol Chem 271, 32084-32088. Pette D., Smith M.E., Staudte H.W. and Vrbova G. (1973) Effects of long-term electrical stimulation on some contractile and metabolic characteristics of fast rabbit muscles. Pflügers Arch 338: 257– 272. Punkt K. (2002). Changes of muscle fibre properties under physiological and pathological conditions. En: Fibre types in skeletal mucles. Ed. Springer-Verlag. Germany. Puntschart A., Claassen H., Jostarndt K., Hoppeler H. and Billeter R. (1995) mRNAs of enzymes involved in energy metabolism and mtDNA are increased in endurance-trained Athletes. Am J Physiol 269, c619–c625. Rajashree R., Koster J. C., Markova K. P., Nichols C. G., and Hofmann P. A. (2002). Contractility and ischemic response of hearts from transgenic mice with altered sarcolemmal K(ATP) channels. Am J Physiol Heart Circ Physiol 283, H584-590. Renstrom E., Barg S., Thevenod F. and Rorsman, P. (2002). Sulfonylurea-mediated stimulation of insulin exocytosis via an ATP-sensitive K+ channel-independent action. Diabetes 51 Suppl 1, S3336. Rorsman P., Eliasson L., Renstrom E., Gromada J., Barg S. and Gopel S. (2000). The Cell Physiology of Biphasic Insulin Secretion. News Physiol Sci 15, 72-77. Sato T., O'Rourke B., Marbán E. (1998) Modulation of mitochondrial ATP-dependent K+ channels by protein kinase. C Circ Res 83, 110-114. Seharaseyon J., Ohler A., Sasaki N., Fraser H., Sato T., Johns D.C., O’Rourke B., Marbán E. (2000) Molecular composition of mitonchondrial ATP-sensitive potessium channels proved by viral Kir gene tranfer. J Mol Cell Cardiol 32, 1923-1930. Seino S. (2003). Review. Physiology and pathophysiology of K(ATP) channels in the pancreas and cardiovascular system. J Diabetes Complications 17, 2-5. Sen C.K., Kolosova I., Hanninen O., Orlov S.N. (1995) Inward potassium transport systems in skeletal muscle derived cells are highly sensitive to oxidant exposure. Free Radic Biol Med 18(4):795-800. - 91 - Shyng S. L. and Nichols, C. G. (1998) Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282, 1138-1141. Sillau A. H., Ernst V. and Reyes N. (1990) Oxidative capacity and distribution in the cardiac myocytes of hypermetabolic rats. Respir Physiol 79, 279–292. Slater E. (1967) Application of inhibitors and uncouplers for a study of oxidative phosphorilation. Meth Enzymol 10: 48-57. Sluse F.E. (1996) Mitochondrial metabolite carrier family, topology, structure and functional properties: an overview. Acta Biochim Polon 43:349–360. Solis J.M. (2003) Acoplamiento quimiosmótico. Termodinámica de los gradientes de iones: potencial electroquímico. Energética del bombeo de protones. Energética de la formación de ATP. Departamento de Física. Universidad Carlos III de Madrid. (b) Somogyi J., Ver A., Troja G., Vegh E., Banyasz T., Csermely P., Popovic S., Kovacs T. (1995a) Interference of sulphonylurea antidiabetica with mitochondrial bioenergetics under in vivo conditions. Acta Physiol Hung. 83(4):313-21. Somogyi J., Ver A., Troja G., Vegh E., Buhler C., Hatfaludi F., Csermely P., Popovic S. (1995b) Interference of the sulphonylurea antidiabeticum gliquidone with mitochondrial bioenergetics in the rat under in vitro conditions. Acta Physiol Hung. 83(4):299-312. Soper J.W, Decker G.L, Pedersen P.L. (1979) Mitochondrial ATPase complex. A dispersed, cytochrome-deficient, oligomycin-sensitive preparation from rat liver containing molecules with a tripartite structural arrangement. J Biol Chem. 254(21):11170-6 Suárez R. K. (1996). Upper limits to mass-specific metabolic rates. A Rev Physiol 58, 583–605. Suárez R. K., Lighton J. R. B., Brown G. S. and Mathieucostello O. A. (1991). Mitochondrial respiration in hummingbird flight muscles. Proc Natn Acad Sci USA 88, 4870–4873. Suzuki M., Kotake K., Fujikura K., Inagaki N., Suzuki T., Gonoi T., Seino S., Takata K. (1997) Kir6.1: a possible subunit of ATP-sensitive K+ channels in mitochondria. Biochem Biophys Res Commun 241, 693-697. - 92 - Suzuki M., Li R. A., Miki T., Uemura H., Sakamoto N., Ohmoto-Sekine Y., Tamagawa M., Ogura T., Seino S., Marban E. and Nakaya, H. (2001). Functional roles of cardiac and vascular ATP-sensitive potassium channels clarified by Kir6.2-knockout mice. Circ Res 88, 570-577. Takahashi M., and Hood D.A. (1993) Chronic stimulation-induced changes in mitochondria and performance in rat skeletal muscle. J Appl Physiol 74: 934–941. Talbot D.A, Duchamp C., Rey B., Hanuise N., Rouanet J.L., Sibille B., Brand M.D. (2004) Uncoupling protein and ATP/ADP carrier increase mitochondrial proton conductance after cold adaptation of king penguins. J Physiol 558 (Pt 1):123-35. Talbot D.A, Hanuise N., Rey B., Rouanet J.L, Duchamp C., Brand M.D. (2003) Superoxide activates a GDP-sensitive proton conductance in skeletal muscle mitochondria from king penguin (Aptenodytes patagonicus). Biochem Biophys Res Commun. 26;312(4):983-8. Tandler B., and Hoppel C.L. (1972) Possible division of cardiac mitochondria. Anat. Rec. 173, 309323. Terjung R.L. (1979) The turnover of cytochrome c in different skeletal - muscle fibre types of the rat. Biochem J 178: 569–574. Thorneloe K. S., Maruyama Y., Malcolm A. T., Light P. E., Walsh M. P. and Cole W. C. (2002). Protein kinase C modulation of recombinant ATP-sensitive K+ channels composed of Kir6.1 and/or Kir6.2 expressed with SUR2B. J Physiol 541, 65-80. Tricarico D., Mele A., Lundquist A.L., Desai R.R., George A.L. Jr, Conte Camerino D. (2006) Hybrid assemblies of ATP-sensitive K+ channels determine their muscle-type-dependent biophysical and pharmacological properties. Proc Natl Acad Sci U S A. 103(4):1118-23. Trujillo X., Huerta M., Vásquez C. y Andrade F. (2002) Adrenaline diminishes K+ contractures and Ba2+-current in chicken slow skeletal muscle fibres. J. Muscle Res. Cell Motil. 23:157-65. Van Lunteren E., Moyer M., Torres A. (1998). ATP-sensitive K+ channel blocker glibenclamide and diaphragm fatigue during normoxia and hypoxia. J Appl Physiol. 85(2):601-8. Wang Y, and Ashraf M. (1999a) Activation of mitochondrial ATP-sensitive K+ channel for cardiac protection against ischemic injury is dependent on protein kinase C activity. Circ Res 84, 11561165. - 93 - Wang Y. and Ashraf M. (1999b) Role of protein kinase C in mitochondrial KATP channel-mediated protection against Ca2+ overload injury in rat myocardium. Circ Res 84, 1156-1165. Westerblad H, y Allen D. G. (2002). Recent advances in the understanding of skeletal muscle fatigue. Curr Opin Rheumatol 14:648-652. Wicks K.L. and Hood D.A. (1991) Mitochondrial adaptations in denervated muscle: relationship to muscle performance. Am J Physiol Cell Physiol 260: C841–C850. Williams R. S. (1986). Mitochondrial gene expression in mammalian striated muscle: evidence that variation in gene dosage Is the major regulatory event. J Biol Chem 261, 12390–12394. Winder W., Fitts R., Holloszy J.O., Kaiser K. and Brooke M. (1980) Effects of thyroid hormone on different types of skeletal muscle. In: Plasticity of Muscle, edited by Pette D. Berlin: Walter de Gruyter, p. 581–591. Wu J., Cui N., Piao H., Wang Y., Xu H., Mao J. and Jiang, C. (2002). Allosteric modulation of the mouse Kir6.2 channel by intracellular H+ and ATP. J Physiol 543, 495-504. Xie L. H., Horie M., and Takano M. (1999). Phospholipase C-linked receptors regulate the ATPsensitive potassium channel by means of phosphatidylinositol 4,5-bisphosphate metabolism. Proc Natl Acad Sci USA 96, 15292-15297. Zaldivar D., García M.C., Sánchez J.A. (2005). Ciliary neutrophic factor promotes inactivation of muscle Ca2+ channels via PKC. Biochem. Biophys. Res. Commun. 338, 1572-1577. Zhou M., Tanaka O., Sekiguchi M., He H-J, Yasuoka Y., Itoh H., Kawahara K., Abe H. (2005) ATPsensitive K+-channel Subunits on the Mitochondria and Endoplasmic Reticulum of Rat Cardiomyocytes. J Histochem Cytochem. 53(12):1491-500. (a) http://es.encarta.msn.com/encyclopedia_761553270/M%C3%BAsculo.html (b) http://bacterio.uc3m.es/docencia/doctorado/sistemas_complejos/biofisica/ - 94 -