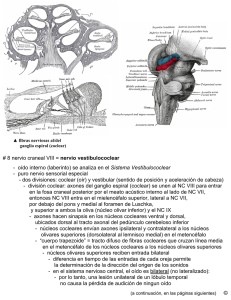
Distribución y maduración de las estructuras nerviosas del oído
Anuncio

UNIVERSIDAD DE GRANADA FACULTAD DE MEDICINA DEPARTAMENTO DE CIRUGÍA Y SUS ESPECIALIDADES Distribución y maduración de las estructuras nerviosas del oído interno en pacientes con malformación coclear Tesis Doctoral presentada por Elena Fernández Pascual Granada, 2009 Editor: Editorial de la Universidad de Granada Autor: Elena Fernández Pascual D.L.: GR. 3519-2009 ISBN: 978-84-692-6399-0 DISTRIBUCION Y MADURACION DE LAS ESTRUCTURAS NERVIOSAS DEL OIDO INTERNO EN PACIENTES CON MALFORMACION COCLEAR TESIS DOCTORAL ELENA FERNANDEZ PASCUAL GRANADA, SEPTIEMBRE 2009 DON MANUEL SAINZ QUEVEDO. PROFESOR TITULAR DE OTORRINOLARINGOLOGIA DE LA FACULTAD DE MEDICINA DE LA UNIVERSIDAD DE GRANADA Y JEFE DE SERVICIO DE OTORRINOLARINGOLOGIA DEL HOSPITAL UNIVERSITARIO SAN CECILIO CERTIFICA: Que la Tesis Doctoral que presenta a la consideración del Tribunal Dª. Elena Fernández Pascual bajo el título “Distribución y maduración de las estructuras nerviosas del oído interno en pacientes con malformación coclear“ ha sido realizada con mi dirección, siendo expresión de la capacidad técnica y científica de su autora, en condiciones tan aventajadas que le hacen acreedora del título de Doctora, siempre que así lo considere el referido Tribunal. Granada, a 1 de Septiembre de 2009 Fdo. D. Manuel Sainz Quevedo DON JUAN GARCIA-VALDECASAS BERNAL, DOCTOR POR LA UNIVERSIDAD DE GRANADA Y FACULTATIVO ESPECIALISTA DE AREA EN OTORRINOLARINGOLOGIA CERTIFICA: Que la Tesis Doctoral que presenta a la consideración del Tribunal Dª. Elena Fernández Pascual bajo título “Distribución y maduración de las estructuras nerviosas del oído interno en pacientes con malformación coclear“ ha sido realizada con mi dirección, siendo expresión de la capacidad técnica y científica de su autora, en condiciones tan aventajadas que le hacen acreedora del título de Doctora, siempre que así lo considere el referido Tribunal. Granada, a 1 de Septiembre de 2009 Fdo. D. Juan García-Valdecasas Bernal. Agradecimientos: Quisiera agradecer a los Doctores Don Manuel Sainz y Don Juan GarcíaValdecasas, directores de esta Tesis Doctoral, la confianza y dedicación depositadas en este estudio. A la Doctora Doña María Teresa Pascual, por compartir conmigo su conocimiento y sus horas de trabajo. Al Departamento de Anatomía y Embriología Humana de la Facultad de Medicina de la Universidad de Granada, por poner a nuestra disposición el material de su embrioteca. A todo el equipo de implantes cocleares del Hospital Universitario San Cecilio de Granada, en especial a su programador y logopedas, por su inestimable colaboración en la recopilación de datos. A todos mis compañeros del Servicio de Otorrinolaringología del Hospital Universitario San Cecilio y a todos aquellos que en mayor o menor medida han participado en la consecución de este trabajo. A mis padres INDICE 1.- INTRODUCCION………………………………………………………………………1 1.1.-EMBRIOLOGIA OIDO INTERNO……………..………..…………..........……...3 1.2.- MALFORMACIONES DE OIDO INTERNO 1.2.1.- Aspectos generales……………………………………………...…..19 1.2.2.- Clasificación malformaciones cocleares……………………...…...20 1.2.2.1.- Jackler………………………………………………..……..20 1.2.2.2.- Otras clasificaciones……………………………..………..24 1.2.3.- Otras malformaciones de oído interno……………………….........30 1.2.4.-Etiología de las malformaciones cocleares…………………..........37 1.2.4.1.- Causas no genéticas………………………………..……..37 1.2.4.2.- Causas genéticas………………………………….………39 1.2.5.- El nervio coclear………………………………………………..........53 1.3.-IMPLANTES COCLEARES 1.3.1.- Reseña histórica…………………………………………………......55 1.3.2.- Criterios de implantación…………………………………….……...56 1.3.3.- Composición y estructura de los implantes cocleares……..........58 1.3.4.- Tipos de implantes………………………………………….….........58 1.3.5.- Funcionamiento básico del implante coclear…………….….........61 1.3.6.- Evaluación preoperatoria…………………………………….……...62 1.3.7.- Actuación postoperatoria……………………………..……...……..64 1.3.8- Programación implante coclear………………………….....……….66 1.4.- IMPLANTES COCLEARES EN MALFORMACIONES 1.4.1.- Contraindicaciones absolutas…………………………….………...73 1.4.2.- Malformaciones mayores……………………………….……..........74 1.4.3.- Malformaciones menores……………………………………………74 1.4.4.- Cirugía según el tipo de malformación…………………..………...75 1.4.5.- Tipos especiales de implantes cocleares………...……................83 1.4.6.- Postoperatorio………………………………………………………..84 2.- ANTECEDENTES Y ESTADO ACTUAL DEL TEMA…………………...87 3.- OBJETIVOS…………………………………………………………….........93 4.- PARTE EXPERIMENTAL………………………………………………..….97 4.1.- ESTUDIO EMBRIOLOGICO DEL OIDO INTERNO 4.1.1.- Objetivos y metodología……………………………............99 4.1.2.- Resultados…………………………………………….........103 4.2.- ESTUDIO ESTADISTICO DE PACIENTES IMPLANTADOS 4.2.1.- Tipo de diseño………………………………………………137 4.2.2.- Muestra………………………………………………...........137 4.2.3.- Método estadístico……………………………………........138 4.2.4.- Variables clínicas…………………………………………...138 4.2.5.- Resultados…………………………………………………..146 5.- DISCUSION………………………………………………………………....179 6.- CONCLUSIONES…………………………………………………….........203 7.- BIBLIOGRAFIA………………………………………………………….....207 1.-INTRODUCCION 1.1 EMBRIOLOGIA DEL OIDO INTERNO La formación del oído interno es uno de los hechos más significativos en la organogénesis de los animales vertebrados. A pesar de que nuestro conocimiento acerca del desarrollo celular y morfológico es aún limitado, durante los últimos 15 años, ha progresado de forma considerable tanto en el plano morfológico, debido a las aportaciones de estudios con microscopía electrónica como en el plano de los mecanismos de diferenciación, gracias al conocimiento de los mecanismos que inducen las citocinas1. Con el fin de presentar los conocimientos adquiridos durante los últimos 15 años, de forma clara y esquemática, se describen a continuación los hitos más importantes del desarrollo embrionario de los distintos componentes del sistema auditivo. A) Placoda auditiva. Su aparición constituye el primer signo visible de desarrollo del oído interno. A los 22 días surge un engrosamiento del ectoblasto superficial de cada lado del rombencéfalo por encima del primer surco branquial que recibe el nombre de placoda auditiva y dará lugar al epitelio del oído interno y las neuronas sensoriales. Diferentes factores o mecanismos de inducción determinarán el desarrollo de la placoda. La inducción primitiva depende de la notocorda (cordamesodermo), la secundaria del mesodermo paraaxial, siendo el rombencéfalo el responsable de la tercera inducción, estimulando el engrosamiento del ectodermo superficial adyacente que da lugar a la placoda auditiva2. La literatura relaciona fuertemente a las rombómeras 4, 5 y 6 con la formación del oído interno, estando los mecanismos moleculares subyacentes a este efecto bajo estudio en la actualidad3. Son los errores durante la inducción los responsables de las malformaciones de oído interno. Mientras el ectodermo situado hacia fuera del borde del tubo neural comienza a afinarse, las placodas auditivas se engruesan y luego se invaginan, formando la fosita y posteriormente la vesícula auditiva (otocisto), que queda aislada del ectodermo superficial y se rodea por completo del mesénquima. Para la aparición de la vesícula ótica a finales de la cuarta semana parece ser de gran importancia el factor de crecimiento fibroblástico 3 (FGF-3) secretado por el rombencéfalo adyacente a la placoda2, siendo por tanto éste último una estructura imprescindible en el desarrollo de la vesícula y del conducto endolinfático4. El gen homeobox Pax-2 está muy implicado en los primeros momentos de desarrollo de la vesícula ótica, hasta el punto que cuando existe una alteración en dicho gen o en su trascripción, no se formarán ni la cóclea ni el ganglio espiral2. Figura 15.- Cortes transversales a nivel de la región del rombencéfalo. A: 24 días, B: 27 días, C: 32 días. B) Ganglio estatoacústico. El ganglio estatoacústico tiene un doble origen puesto que las células de Schwann y las células satélites derivan de las crestas neurales mientras que las neuronas derivan de la pared del canal auditivo. La migración de estas células desde la porción ventral de la vesícula auditiva resulta aparente a la cuarta semana de gestación. C) Laberinto membranoso. Hacia el día 25, en el embrión de 4-6mm, cada placoda auditiva ya está transformada en una vesícula, y cada vesícula se dividirá en una parte ventral, a partir de la cual se originará el sáculo y el conducto coclear con el órgano de Corti, y una parte dorsal que formará el utrículo, los conductos semicirculares y el canal endolinfático Al finalizar la cuarta semana, la vesícula auditiva aumenta de tamaño formando dos bolsas, una ventral y otra dorsal, siendo el punto de contacto entre ambas la zona que dará lugar al sáculo y al utrículo, y que recibe el nombre de atrio. De la bolsa dorsal se originarán los tres canales semicirculares y la bolsa ventral se prolonga e incurva hacia delante formando el caracol. El ganglio estatoacústico se forma en el mesénquima, justo hacia adentro y por delante de la vesícula auditiva. Durante la quinta semana las fibras nerviosas procedentes del ganglio estatoacústico comienzan a penetrar la cara anteromedial de la vesícula ótica, a la vez que se van formando los conductos semicirculares, con claras diferencias, ya que el canal lateral se desarrolla más tarde y a menor velocidad que el superior y el posterior6. Las ampollas se forman al mismo tiempo que ellos. A la octava semana deben haber alcanzado su forma definitiva. Tres genes son importantes en el desarrollo del laberinto posterior. El gen del factor de transcripción Nkx 5-1 que actúa en la formación de todo el vestíbulo, el gen Otx-1 que es responsable de la formación del conducto semicircular lateral, mientras que para el posterior y el anterior es imprescindible la expresión del factor de transcripción con homeobox Dlx-5 2. Existe cierta controversia en la literatura en lo referente al crecimiento de los canales semicirculares. Mientras que Hyrtl (1845) y Tremble (1929) coinciden en que los canales semicirculares experimentan un crecimiento constante en longitud en el periodo postnatal, Siebenmann (1890) y Schönemann (1906) no hallaron ningún cambio tras el nacimiento en la morfología de dichas estructuras6. Durante la sexta semana se desarrollan el sáculo y el utrículo tras dividirse el atrio en una porción superior y otra inferior, lo cual provoca el principio del canal endolinfático que dará lugar a la formación del canal utriculosacular. También en esta semana comienza a formarse el acueducto coclear a partir de la superficie posteroventral del sáculo. Figura 22.- Desarrollo del oído interno. A: 28 días. B: 33 días. C: 38 días. D: 41 días. Hacia el día 36, en la porción ventral o inferior de la llamada cámara utriculosacular de la vesícula ótica, empieza a aparecer una evaginación tubular que se va alargando progresivamente. Primero es rectilínea y recibe el nombre de conducto coclear, pero hacia el día 49, dicho conducto comienza a enrollarse sobre sí mismo, tomando forma de espiral a causa de la desigualdad de crecimiento de las caras externa e interna. Sobre el día 56, ya están formadas una vuelta y media de espira coclear y al 70 día de gestación, la cóclea ya presenta su morfología definitiva con sus dos vueltas y media de espiral y una longitud coclear de 20mm. A las 16 semanas se alcanza la longitud completa del conducto coclear (33 a 37mm). Figura 32.- E: embrión de 50 días (7semanas) con una vuelta y media de espira coclear. F: embrión de 56 días (8 semanas) con dos vueltas y media de espira. El laberinto membranoso definitivo se forma paralelamente al crecimiento del ganglio estatoacústico, lo que determina que en aquellos casos que resultan en una malformación coclear por detención del desarrollo embrionario, exista un menor número de neuronas. Al principio, el ganglio forma una porción superior y una porción inferior cuando la vesícula auditiva se separa en una bolsa vestibular y una bolsa coclear. Después la porción inferior origina la porción vestibular y el ganglio espiral, y la porción superior del ganglio da lugar a las conexiones nerviosas de las ampollas superiores y externas y del utrículo, mientras que la porción inferior inerva el sáculo y la ampolla posterior. El ganglio espiral envía sus fibras al epitelio sensorial adyacente1. D) Epitelio sensorial. Durante el desarrollo del laberinto membranoso, las paredes de la vesícula auditiva están compuestas de un epitelio pseudoestratificado con dos o tres capas de núcleos. Estas células presentan sus superficies basales hacia la superficie externa de la vesícula ótica y su vertiente luminar se sitúa hacia la luz de la vesícula auditiva. Mientras el laberinto membranoso adquiere su estructura definitiva, las diversas regiones sensoriales se diferencian a partir del epitelio de la pared interna, formando las tres ampollas de los canales semicirculares, las máculas de utrículo y sáculo y el órgano de Corti. Sobre las máculas se depositan finas partículas calcáreas u otolitos, que se forman al depositarse calcio sobre una matriz glicoproteica. En dicho proceso parece jugar un papel importante la anhidrasa carbónica4. 1) Órgano de Kölliker: es el precursor embrionario del Órgano de Corti. El epitelio sensorial del canal coclear comienza a desarrollarse durante la séptima semana, momento en el que el canal comienza a enrollarse para formar la espiral. El órgano de Kölliker está compuesto de dos crestas que se enrollan a lo largo de la cóclea (columna externa y columna interna). Las células ciliadas internas (CCI) se desarrollarán a partir de las células más externas de la columna interna, las células de los pilares lo harán a partir de la unión entre la columna externa e interna, y las células ciliadas externas (CCE) y las células de Deiters se formarán a partir de la porción más interna de la columna externa. Células ciliadas externas diferenciadas pueden ser identificadas en los extremos basales y apicales de la cóclea a la 16ª semana, existiendo en este periodo tres filas de células ciliadas externas, y a veces en la zona apical, hasta cuatro. En la semana 17ª, la cóclea ha alcanzado el número definitivo de células ciliadas (3.400 células ciliadas internas y 13.400 externas). Las células ciliadas comienzan a diferenciarse a la semana 11, momento en el que la superficie celular está cubierta únicamente de pequeños estereocilios que presentan un tamaño casi idéntico al de las microvellosidades de las células de sostén adyacentes. Un único kinocilio se sitúa en la periferia de cada una de las células. Cerca de la semana 13, los estereocilios que están más cerca del kinocilio aumentan de tamaño y comienzan a formarse las filas individuales cada vez más alejadas del kinocilio. A los 5 meses de gestación los estereocilios están formados, por lo que en la región basal las células ciliadas internas poseen una estructura próxima a la definitiva hacia la semana 20, mientras que las externas no lo hacen hasta la semana 22 ó 251. 2) Células de sostén: comienzan a desarrollarse en paralelo con las células ciliadas. Este paralelismo resulta de especial importancia en los casos de hipoacusia por mutaciones en los genes de las conexinas, pequeñas proteínas transmembrana implicadas en la formación de uniones comunicantes entre las células adyacentes. El túnel de Corti aparece primero en la región basal a las 20 semanas. 3) Membrana tectoria: cubre al Órgano de Corti. Su desarrollo comienza a la novena semana, cuando las células de la columna interna secretan una sustancia que será su componente principal. Células de la columna externa formarán el resto de la membrana tectoria hacia la semana 10 ó 121. Figura 42.- Cortes transversales del Órgano de Corti en desarrollo. La cóclea no alcanza su estructura definitiva hasta el octavo mes de gestación1, lo que explica que se incluya a la prematuridad entre los principales factores de riesgo de hipoacusia. E) Inervación del epitelio sensorial. El enrollamiento del acueducto coclear hace que queden en contacto el epitelio sensorial y las células del ganglio espiral en el modiolo. Poco después de la semana 9 pueden verse fibras nerviosas penetrando el epitelio sensorial. Alrededor de la semana 11 las células ciliadas internas presentan terminaciones sinápticas de fibras aferentes y eferentes, mientras que las células ciliadas externas sólo reciben conexiones de fibras aferentes, hasta que alrededor de la semana 20, momento en el que comienza la audición, progresivamente van siendo alcanzadas por las eferentes que van desplazando a las anteriores. En este momento del desarrollo las células ciliadas internas sólo se conectan a fibras aferentes. La función auditiva aparece cuando la maduración sináptica se ha producido sobre las células ciliadas internas, mucho antes de que la inervación de las células ciliadas externas haya alcanzado su estructura definitiva1. F) Membrana basilar. Es la estructura que separa la rampa timpánica del conducto coclear, delimitado por el limbo espiral en su parte interna y por el ligamento espiral en su parte externa. En la época embrionaria, la membrana basilar es gruesa y tiene varias capas de células pero conforme va madurando la cóclea va perdiendo grosor, aunque sigue manteniendo las células timpánicas. G) Membrana de Reissner. Se sitúa entre el limbo espiral y el ligamento espiral por encima de la estría vascular, separando el conducto coclear de la rampa vestibular. H) Estría vascular. Localizada en la pared externa del canal coclear. Es la estructura donde se produce la endolinfa. Está formada por tres tipos de células: las marginales, que derivan del epitelio del acueducto coclear; las intermedias desarrolladas probablemente a partir de las crestas neurales y las basales, que derivan del mesénquima subyacente1. La existencia de tres tipos de células puede explicar la pérdida de función que ocurre en determinadas patologías como la presbiacusia, en la que la pérdida de audición puede diferir de forma importante de un paciente a otro, tanto en grado de hipoacusia como en frecuencias afectadas. I) Cápsula auditiva. Los datos de los que disponemos hasta ahora son los publicados por Anson y Donalson1, que establecen que la cápsula auditiva tiene su origen en el mesoblasto que rodea al laberinto membranoso. Alrededor del día 36, el mesénquima se condensa rodeando la vesícula y dando lugar a dos capas: - Precartílago, más superficial, que dará lugar a la cápsula ótica. Crece al mismo tiempo que el laberinto membranoso amoldándose a su forma hasta la mitad de la gestación, momento en el que comienza su osificación en catorce puntos que irán confluyendo progresivamente. - Otra capa más profunda que tras reabsorberse parcialmente da lugar a los espacios perilinfáticos y a su contenido. A la semana 22, la cápsula auditiva tiene ya su conformación adulta, es decir, hueso encondral recubierto en su totalidad por hueso pericondral. Recientemente, gracias a los resultados de estudios histoquímicos e in vitro, se sugirió que el factor beta de transformación del crecimiento (FTC-B1) puede tener cierto papel en la modulación de la interacción entre el epitelio y el mesénquima en el oído interno y en la dirección del desarrollo de la cápsula ótica7. Hacia la 8ª semana la capa superficial del molde cartilaginoso es tapizada superficial y profundamente por una hoja de tejido conjuntivo pericondral. En cuanto al resto del hueso temporal, hacia el 5º ó 6º mes, en la masa cartilaginosa aparecen cuatro centros, a partir de los cuales se osifican las regiones petrosa y mastoidea. Hacia la semana 23 tiene lugar la osificación del ápex de la cóclea y del canal semicircular lateral, pero algunas zonas como el contorno de la ventana oval y la región de la fístula ante-fenestram continuarán siendo cartilaginosas. El anillo timpánico se une con la región escamosa poco antes del nacimiento, fusión que prosigue durante el primer año de vida. La parte distal de la apófisis estiloides no se une con el resto del hueso hasta después de la pubertad y en algunos cráneos no lo hace nunca. Tras el nacimiento, en el hueso temporal, además de un aumento de tamaño, tiene lugar la neumatización y el crecimiento de la apófisis mastoides. A nivel de la cóclea, el órgano de Corti se enrolla alrededor de un eje vasculonervioso. Las células mesenquimatosas que rodean a este eje sufren una osificación hacia la semana 20, y el espacio perilinfático que se forma alrededor del canal coclear membranoso queda dividido en dos partes por una lámina fina mesenquimatosa, que una vez se diferencie, dará lugar a la hoja ósea espiral, a la membrana basilar y al ligamento espiral, quedando así conformadas las rampas vestibular y timpánica. El laberinto humano experimenta importantes cambios durante la época fetal, incluyendo la reorientación espacial de la cóclea. A pesar de la diferente velocidad a la que se desarrollan sus distintas estructuras, el laberinto alcanza su tamaño adulto alrededor de las semanas 17 a 19, y se osifica por completo algunas semanas más tarde6. En cuanto a su forma, no cambia más a partir de la osificación, pero el hecho de encontrar diferencias entre la forma fetal y la adulta, hace pensar que puedan deberse a diferencias de población o a que quizás, esos cambios se produzcan en épocas de la vida posteriores a la osificación laberíntica6. J) Embriología de los espacios perlinfáticos. Los espacios perilinfáticos se encuentran entre el laberinto óseo y el membranoso. Derivan del mesénquima produciéndose a partir de la cuarta semana un proceso de multiplicación celular en el mesénquima que rodea al laberinto membranoso, adquiriendo un aspecto distinto al resto del tejido circundante. Sobre la 8ª semana, este mesénquima comienza a sufrir un fenómeno de involución, transformándose en un retículo con pequeñas lagunas, cuya confluencia dará lugar al espacio perilinfático definitivo ocupado por un líquido, fluido periótico. K) Formación de las estructuras nerviosas. Los pares craneales séptimo y octavo se forman a partir del primordio estatoacústico inmediatamente rostral al otocisto a partir del que se desarrolla el conducto coclear. El primordium estatoacústico comienza a separarse en los nervios séptimo y octavo cuando la longitud mayor del feto es aproximadamente de 8 a 10mm. Cuando éste alcanza los 14mm, los dos nervios ya están totalmente separados y a este mismo tiempo el conducto coclear comienza a desarrollarse, primero como una pequeña bolsa en la parte ventral del otocisto y después enrollándose rostralmente hacia el primordio facioacústico8. Esquema 12.- Principales sucesos inductivos y transformaciones tisulares en el desarrollo del oído. Las flechas discontinuas indican fenómenos inductivos. Streeter estudió el desarrollo embrionario dividiendo las diferentes etapas en Horizontes4; así, tabla 1: Tabla 14.- Horizontes de Streeter. HORIZONTE TAMAÑO EDAD ESTRUCTURAS XIII 5mm 28-30 días Vesícula ótica XV 7-8mm 31-32 días Esbozo conducto endolinfático Esbozo utrículo y canales XVI 9-10mm 32-34 días semicirculares Comienza la XVII 11-14mm 34-36 días XVIII 14-16mm 36-38 días individualización de los canales Aislamiento canales Más separación entre XIX 17-20mm 38-40 días sáculo y utrículo La cóclea crece hacia XX 20-22mm 42-45 días arriba y horizontalmente Mayor longitud y apunta XXI 23-25mm 46-49 días abajo Crece más y apunta XXII 25-27mm 50-54 días arriba Adquiere las 2,5 vueltas XXIII 28-30mm 54-58 días definitivas. Figuras 5 y 64.- Representación gráfica de los Horizontes descritos por Streeter. Tabla 24.- Descripción de las estructuras representadas en las figuras 5 y 6. 1, 11 y 13 Conducto endolinfático 6 Conducto semicircular posterior 7 Conducto semicircular lateral Ampolla Zona de adosamiento y 2 reabsorción 3 y 10 Utrículo 8 4 Sáculo 12 y 14 Segmento ventral (saculococlear) del otocisto Segmento dorsal (utriculocanalicular) del 5y9 Cóclea 15 otocisto 1.2.- MALFORMACIONES DEL OIDO INTERNO 1.2.1.- Aspectos generales. Entre todos los defectos sensoriales presentes en el momento del nacimiento, el más frecuente es la hipoacusia profunda, con una incidencia estimada entre 0,8 y 3 casos por cada 1.000 nacimientos en EE.UU. y entre 1,5 y 2 por cada 1.000 nacimientos en Europa9. Actualmente se estima que la mitad de los casos de hipoacusia congénita se debe a causa genética, atribuyendo el otro 50% a las causas ambientales como las infecciones, la prematuridad, la eritroblastosis fetal, la exposición del feto a drogas ototóxicas y los traumatismos. Aunque topográficamente, la hipoacusia puede tener su origen en cualquier punto de la vía auditiva, la mayoría de los casos de hipoacusia neurosensorial en niños resulta de una malfunción coclear, ya sea congénita o adquirida, y más específicamente interesando a las células ciliadas. Además, estudios recientes sugieren que la disfunción del nervio coclear y sus conexiones con las células ciliadas (neuropatía auditiva) está presente en el 10% de los déficit de audición10. Si bien, ésta puede no tener relación con la ausencia de nervio coclear, sino más bien con la funcionalidad de éste, pues la ausencia del nervio coclear como causa de hipoacusia congénita es considerada como poco probable por parte de algunos autores10. Las malformaciones de oído interno están presentes en aproximadamente el 20% de los pacientes con pérdida auditiva neurosensorial congénita11, pudiendo o no estar asociadas a otras malformaciones no relacionadas con el oído. El déficit funcional de estas malformaciones congénitas de oído es muy variable, siendo mayor cuanto mayor es el grado de malformación12. 1.2.2.- Clasificación de las malformaciones cocleares Son muchas los autores que han clasificado las malformaciones cocleares, pero es sin duda la clasificación de Jackler13, en 1987, tabla 3, basada en el desarrollo embriológico y el diagnóstico radiológico por politomografía11, la más usada y conceptualmente más clarificante. Ésta, clasifica las malformaciones en función del momento en el que se detiene el desarrollo embriológico13,14, provocado por cualquier noxa. Según el momento de detención del desarrollo embriológico, se obtendrá una malformación diferente, siendo menos diferenciada a menor edad gestacional. 1.2.2.1.- Jackler Diferencia entre malformaciones con la cóclea ausente o malformada, y otras con cóclea normal: Con cóclea ausente o malformada: - Aplasia completa o de Michel: descrita por este autor en 1863 al realizar la autopsia a un niño sordo y mudo de 11 años de edad, es una ausencia completa de oído interno y nervio auditivo15, mientras que el nervio facial suele ser normal en morfología y funcionalidad. Estos pacientes padecen con frecuencia malformaciones traqueoesofágicas, de las extremidades y curso aberrante de los grandes vasos cervicales15. Surge como resultado de una detención en el desarrollo embrionario en la tercera semana de gestación. - Cavidad común: la pérdida total de todo el septo interescalar hace que no exista separación entre la cóclea y el vestíbulo, que forman conjuntamente una única cavidad. Los canales semicirculares pueden ser normales o no. Puede existir comunicación con el CAI, con el consiguiente peligro de fístula de líquido cefalorraquídeo (LCR) durante la intervención14. Su frecuencia está alrededor del 25% del total de las malformaciones cocleares según algunas publicaciones16 y resulta de la detención del desarrollo embrionario del oído interno alrededor de la cuarta semana de gestación. - Aplasia coclear: cóclea no desarrollada. Los canales semicirculares y el vestíbulo pueden ser normales o no. El desarrollo embrionario se detiene sobre la quinta semana. .- Hipoplasia coclear: la cóclea carece de espira apical y media, y sólo existe la basal. En este caso el desarrollo embrionario no evoluciona desde la sexta semana de vida intrauterina. - Partición incompleta: también conocida como displasia de Mondini, se caracteriza por presentar una cóclea pequeña con menos de 2,5 vueltas de espira. Resulta de la pérdida del septum interescalar entre las espiras apical y media de la cóclea, con la consiguiente comunicación entre ambas12. En esta clasificación, el término partición incompleta incluye casos en los que además de la falta del septum interescalar, existe alguna anomalía de la espira basal de la cóclea12. Como en las malformaciones descritas anteriormente, el vestíbulo y los canales semicirculares pueden ser normales o malformados. El desarrollo embrionario se detiene hacia la séptima semana de vida intrauterina. La audición es variable ya que el Órgano de Corti puede ser más o menos normal14. Según los hallazgos de Jackler, la clásica displasia de Mondini supone el 55% de todos los casos de malformaciones cocleares16. Con cóclea normal: -Displasia del canal semicircular lateral y vestíbulo: asocia un canal semicircular lateral dilatado con un vestíbulo agrandado. -Acueducto vestibular agrandado, con canales semicirculares normales. Diferentes malformaciones resultarían si en lugar de producirse una detención del desarrollo embrionario, éste se continuara de forma aberrante16. La pérdida auditiva va irremediablemente asociada al grado de malformación, siendo la regla general, que cuanto más severa es la malformación, peor será la función auditiva de ésta. Así, Jackler obtuvo de su estudio unos umbrales medios para cada malformación, siendo de 94 decibelios (dB) en el grupo de cavidades comunes, de 75 dB en las particiones incompletas y de 53 dB en las hipoplasias cocleares16, sin tener en cuenta eso sí, la discriminación verbal. La clasificación de Jackler sigue vigente y ha sido la matriz de las nuevas clasificaciones, basadas en las nuevas técnicas de imagen, la tomografía axial computadorizada (TAC) y la resonancia magnética nuclear (RMN), que han permitido un mayor conocimiento de las malformaciones. Tabla 3.- Clasificación de Jackler. Jackler y cols. (1987) Aplasia de oído interno (3ª semana de gestación) Cavidad común (4ª semana de gestación) Agenesia coclear (5ª semana de gestación) Hipoplasia coclear (6ª semana de gestación) Partición incompleta (7ª semana de gestación) Desarrollo coclear normal (8ª semana de gestación) Figura 713,17.- Malformaciones cocleares según Jackler. Composición secuencial de los cortes derivados de un examen anteroposterior con politomografía. 1.2.2.2.-Otras clasificaciones De entre las múltiples clasificaciones que han surgido desde la elaborada por Jackler, es sin duda la propuesta por Sennaroglu (2002)17 tabla 4 la más exhaustiva. Basándose en la Tomografía Computarizada y en la Resonancia Magnética como pruebas de imagen más actuales que la politomografía usada por Jackler, este autor clasifica las malformaciones cocleares en: -Deformidad de Michel: caracterizada por la inexistencia total de cualquier estructura del oído interno. -Aplasia coclear: malformación en la que no existe cóclea. Estas dos malformaciones suponen el 1-3% de todas las malformaciones del oído y es frecuente encontrarlas asociadas a otras malformaciones del peñasco14. -Cavidad común: no existe una separación entre la cóclea y el vestíbulo sino que forman una única cavidad. -Hipoplasia coclear: Supone el 15 % de las alteraciones. La cóclea tiene sólo la espira basal y algún resto de las estructuras medias14. -Partición incompleta tipo I: a la cóclea le falta el área cribiforme y el modiolo, acompañándose de un vestíbulo quístico grande. -Partición incompleta tipo II (clásica malformación de Mondini): la cóclea presenta una vuelta y media, con las espiras medial y apical unidas en una formación quística, acompañada de un vestíbulo mínimamente dilatado y un acueducto vestibular agrandado. Esta malformación corresponde a la descrita por Carlo Mondini en 179118. Además clasifica las malformaciones vestibulares, incluyendo en este grupo la deformidad de Michel, la cavidad común y el vestíbulo ausente, hipoplásico o dilatado. Los canales semicirculares pueden ser ausentes, hipoplásicos o agrandados. El conducto auditivo interno puede estar ausente, estrechado o agrandado, así como el acueducto coclear y vestibular que pueden ser agrandados o normales. Tabla 4.- Clasificación de Sennaroglu y cols. Sennaroglu y cols. (2002) Deformidad de Michel Aplasia coclear Cavidad común Cavidad única (partición incompleta tipo I) Hipoplasia coclear Partición incompleta tipo II Desarrollo normal La principal diferencia de esta clasificación respecto a la de Jackler, es que considera la cavidad única como una forma grave de partición incompleta (tipo 1). Pensó que como la diferenciación en cóclea y vestíbulo no ha tenido lugar en la cavidad única, debe por tanto preceder a la hipoplasia coclear, en la que la separación entre cóclea y vestíbulo ya se ha producido12. Una cavidad única enormemente dilatada es difícil de emplazar en una línea continua de desarrollo cocleovestibular. Como apuntaron Romo y cols.18, el acueducto del vestíbulo es una de las últimas estructuras del oído interno en desarrollarse, por lo que si en la mayoría de los casos de partición incompleta tipo I no se encuentra, será signo de que es una malformación más temprana que la tipo II. Otros autores describieron también las dos formas diferentes de partición incompleta basándose en estudios histológicos. A la partición incompleta tipo I, Reissner la definió como “cóclea vacía”, por ser aquella con aspecto quístico y ausencia de estructuras internas; y Phelps utilizó primero el nombre de “pseudo-Mondini” y más tarde el de “displasia” para definirla. Recientemente, Davidson y cols. acuñaron el término “dismorfia coclear grave” para esta malformación18. De las consideraciones previas resalta el mal uso realizado por numerosos especialistas del concepto “malformación de Mondini”. Desde que Carlo Mondini describiera el primer caso de malformación coclear congénita en 1791, el término Mondini ha sido usado de forma genérica para designar casi cualquier malformación congénita de la cóclea. Jensen reconoció la malformación de Mondini en politomografía en 1969 y posteriormente describió un tipo de malformación más severa que consistía en una cóclea amorfa con forma de saco unida a un vestíbulo dilatado, y para la que propuso el nombre de “displasia”19. Desde entonces, cualquier anomalía congénita del laberinto óseo que pueda verse en pruebas radiográficas, puede designarse con el nombre de “Displasia de Mondini”, lo que lleva a confusiones. Las características histopatológicas de la malformación de Mondini incluyen una espira basal totalmente desarrollada, con una base ósea en el modiolo y una cóclea que está aplanada y/o acortada, con 1,5 vueltas de espira en lugar de las 2,5 habituales. La vuelta superior forma una cavidad común conocida como “escala común”. Esta es la clásica descripción de Mondini, y se corresponde con la partición incompleta de la clasificación de Jackler, debida a la detención del desarrollo embrionario a la séptima semana19. Al estar conservada la espira basal, alguna función auditiva es posible y el espacio subaracnoideo se encuentra separado de la cóclea19. Estos pacientes presentan diferentes grados de hipoacusia, desde audición normal o conservación en frecuencias agudas a cofosis, y puede tener su explicación en las diferentes lesiones laberínticas membranosas que pueden aparecer asociadas a las consabidas malformaciones óseas16. Así lo constataron Magabeira-Albernaz y cols.19 al estudiar once pacientes con displasia de Mondini, de los cuales siete presentaban hipoacusia progresiva y cuatro hipoacusia fluctuante. La anomalía de Mondini puede manifestarse de dos formas: una grave, de herencia autosómica recesiva, que cursa con hipoplasia coclear y dilatación del vestíbulo y los canales semicirculares; y otra forma más leve, de herencia autosómica dominante, que se caracteriza por un aplanamiento de la cápsula ótica17. Además de la clasificación de Jackler y la de Sennaroglu, que sin duda son las de mayor difusión, existen otras muchas. Phelps (1992)12 tabla 5, basa su clasificación en el aspecto de la espira basal de la cóclea que parece tener más relación con los resultados tras la implantación y con la probabilidad de fuga de perilinfa durante la cirugía del implante coclear. Así, la cóclea malformada con una espira basal anómala fue designada “displasia laberíntica severa”, mientras que los casos en los que se conservaba una espira basal normal, eran llamados “displasia de Mondini”. Phelps incluso describe un subgrupo del tipo Mondini en el que la espira basal es normal pero la longitud de la cóclea se acorta a 1,5-2 vueltas12. Tabla 5.- Clasificación de Phelps. Phelps (1992) Displasia laberíntica severa Deformidad de Mondini En el mismo sentido, Slattery y Luxford20, teorizan que las células ganglionares en los humanos están presentes en la vuelta y media más basal de la cóclea, por lo que si ésta está presente, la probabilidad de que haya terminaciones nerviosas es mayor que en los casos de partición incompleta tipo I, y por tanto la audición y la ganancia auditiva tras la implantación coclear serán mucho mejores. Una espira basal presente aunque rudimentaria, implica la posibilidad de cierta función auditiva12. Por otro lado, Schuknecht17 utiliza el término Mondini para describir dos situaciones diferentes: 1) Una cóclea sin modiolo con aspecto quístico al igual que el vestíbulo y 2) una cóclea con modiolo corto, espira basal normal, y las espiras media y apical formando una cavidad quística. Además Schuknecht describió cócleas de 23mm de longitud, en lugar de los 32mm normales, aunque con una densidad de células ciliadas y de neuronas dentro de la normalidad12. Por otro lado, Zheng y cols. (2002) tabla 6 llegan más allá en su clasificación de las deformidades tipo Mondini y las subclasifican en base a la presencia o ausencia de una completa base ósea del modiolo, denominando tipo A a las que la tienen y tipo B a las que no. Esta subclasificación trata de correlacionar las anomalías radiológicas con la probabilidad de que aparezcan fístulas perilinfáticas o de líquido cefalorraquídeo o meningitis recurrentes tras la realización de la cocleostomía12. Tabla 6.- Clasificación de Zheng y cols. Zheng y cols. (2002) Deformidad de Mondini Espira basal completa (tipo A) Espira basal incompleta (tipo B) 1.2.2.3.- Malformaciones membranosas Existen otras malformaciones laberínticas que comprometen a los tejidos blandos, sin afectar al hueso, y por tanto no siendo detectables en la TAC; estas son: - Malformación de Bing-Seibenman: la malformación afecta al laberinto membranoso anterior y posterior (cóclea y canales semicirculares). Puede asociarse a retinitis pigmentosa y retraso mental17. Su origen puede ser factores genéticos o teratógenos. - Malformación de Scheibe: afecta a la cóclea y al sáculo y el Órgano de Corti muestra ausencia de células ciliadas externas. Fue descrita por este autor en 189220 y es la malformación más frecuente de oído interno, representando el 70% de los casos con sordera hereditaria. Suele ser unilateral y se asocia a enfermedades como las infecciones por Citomegalovirus (CMV) o Rubéola, y también a síndromes de causa genética como Usher y Waardenburg17. La membrana sacular se encuentra colapsada con degeneración del neuroepitelio. También existe atrofia del nervio sacular y ampular del canal semicircular posterior, de la estría vascular y de la espira basal de la cóclea21. Al estar las lesiones mayores en la espira basal de la cóclea, pueden quedar restos auditivos únicamente en las frecuencias graves. - Displasia de Alexander: únicamente afecta al conducto coclear, a la espira basal membranosa. En la audiometría la hipoacusia se limita a las frecuencias agudas17. 1.2.3.- Otras malformaciones de oído interno 1.2.3.1.- Dilatación del acueducto del vestíbulo: El acueducto vestibular es un canal óseo que se extiende desde la pared medial del vestíbulo hasta la superficie posterior de la pirámide petrosa. Su longitud media en la persona adulta es de 10mm, adquiriendo una forma de “j” invertida determinada por la posición definitiva del saco endolinfático. Su forma también ha sido comparada con un árbol de Navidad22. En el interior de este canal óseo transcurre el conducto endolinfático23. El diámetro del conducto vestibular oscila en un rango entre 0,4 y 1mm según los estudios de Wibrand y cols. en huesos temporales23, pudiendo considerarse dilatado cuando su anchura, medida entre el punto medio de la cruz comunis y la apertura externa, supera los 1,5mm12,24. Subjetivamente, la comparación puede hacerse con los canales semicirculares superior y posterior, que suelen ser del mismo tamaño o más grandes que el acueducto del vestíbulo25. La dilatación del acueducto del vestíbulo fue descrita por primera vez en 1978 por Valvassory y Clemis26, siendo la malformación de oído interno que con mayor frecuencia puede encontrarse en la TAC o en la RMN de alta definición con cortes de 1mm en el plano sagital27. Asimismo es la malformación más frecuentemente asociada a hipoacusia neurosensorial con una herencia autosómica recesiva23. Esta malformación puede ser uni o bilateral y la hipoacusia asociada suele ser pre o perilocutiva, neurosensorial o mixta y normalmente progresiva28. Este síndrome suele manifestarse alrededor de los 3 años de edad en niños con hipoacusia neurosensorial progresiva con o sin síndrome vertiginoso o acúfenos que pueden estar presentes en más del 50% de los pacientes25. La dilatación del acueducto del vestíbulo es una malformación que se descubre en un 7% de las hipoacusias neurosensoriales. Además, la incidencia de dilatación del acueducto del vestíbulo se encuentra entre el 1% y el 1,3%12 y del 11,8% al 60,9% de los casos presentan una pérdida auditiva progresiva. Varios autores han sugerido que el ensanchamiento del acueducto del vestíbulo no es realmente el responsable del déficit auditivo, sino un marcador de otras anomalías laberínticas25, como puede ser la hipoplasia coclear, la partición incompleta o la cavidad común, a las que invariablemente acompaña y en función de las cuales serán clasificadas y no por el tamaño del acueducto del vestíbulo12. Existen numerosas teorías acerca del origen del síndrome de dilatación del acueducto del vestíbulo. Una de ellas defiende que tanto el conducto como el saco endolinfático continúan ensanchándose durante la infancia, lo que justifica la progresión de la hipoacusia. Según Pyle18, el origen del síndrome estaría en un mal desarrollo postnatal y en la primera infancia, no en la época fetal29. Otras teorías abogan por una detención del desarrollo alrededor de la quinta semana de gestación18, antes del alargamiento y estrechamiento del acueducto del vestíbulo12. A veces, aparece acompañado de una malformación de Mondini y otras veces existe una anatomía cocleovestibular normal, sugiriendo que la detención del desarrollo tiene lugar hacia la semana 25 en la que termina la formación del oído interno. Por tanto, la presunción de que el fallo puede producirse en el periodo postnatal parece improbable18. Además de la presencia de factores embriológicos, es factible que existan factores de tipo hidrodinámico, en la producción o reabsorción de la endolinfa, que condicionen en cierto modo la dilatación del acueducto del vestíbulo, así como factores genéticos como la mutación SLC26A4 (PDS) o factores ambientales28. Si la evolución lo requiere el tratamiento final de la hipoacusia neurosensorial es el implante coclear. En el año 1999 Bent y cols.23 publicaron 8 casos de implantados con dilataciones del acueducto vestibular encontrando como única complicación la aparición de salida de LCR (gusher) en 5 de los 8 pacientes. 1.2.3.2.- Malformaciones del conducto auditivo interno (CAI) 1.2.3.2.1.-Conducto auditivo interno estrecho: Se considera que el CAI está estenosado cuando presenta un diámetro de 1-2mm en la TAC de alta resolución30. El tamaño y la forma del CAI han sido estudiados anatómica, histológica y radiológicamente30. En los diferentes estudios se han medido una o todas las dimensiones del CAI (longitud, altura, diámetro y volumen), considerándose la longitud desde la pared anterior a la posterior en el plano axial, aunque en ocasiones se hace desde el techo hasta el suelo en el plano coronal30. Su presencia duplicada es extremadamente rara, con sólo cuatro casos recogidos en la bibliografía31. El primer caso fue recogido en 1997 por Casselman y cols. que, basándose en imágenes de resonancia magnética describieron un CAI estrecho y duplicado unilateral, con una porción superior que contenía un nervio facial normal y una porción inferior sin ningún contenido31 , sin afectación laberíntica. El segundo caso lo describieron Vilain y cols. en 199931, como un CAI estenosado y vacío, unilateral, y un segundo canal en posición anterior e inferior con respecto al primero en el cual discurren los nervios facial y vestibulococlear. El tercer caso lo publicaron Cho y cols. en 200031 y era de características similares al de Casselman y cols. Shelton y cols., Gray y cols. y Casselman y cols. basándose en imágenes radiológicas de RMN, mostraron que un CAI con una anchura de 1 ó 2mm podría asociarse con un nervio cocleovestibular ausente o subdesarrollado30. La presencia de un canal auditivo interno duplicado y estrechado suele ser unilateral y puede aparecer de forma aislada o formando parte de un síndrome, junto a malformaciones cocleares, de oído externo o medio, o incluso cardíacas, renales, óseas o digestivas31. Está presente en el 20% de los pacientes con hipoacusia neurosensorial congénita unilateral, aunque también hay casos en los que el paciente presenta una audición normal bilateral. Una hipótesis sugiere que el origen del problema está en la estenosis ósea y que esto causa la aplasia o hipoplasia del nervio, pero el hecho es que el nervio suele tener una apariencia normal, lo que pone en duda esta teoría. La otra hipótesis más aceptada defiende que la cóclea y el vestíbulo en el embrión inducen el crecimiento del octavo par craneal, y el canal óseo se desarrolla a su alrededor junto con el nervio facial por la osificación del mesodermo en la octava semana de gestación. Así cuando el nervio cocleovestibular es aplásico o hipoplásico, el CAI se desarrolla de forma anómala31. La TAC tiene una alta sensibilidad a la hora de evaluar el CAI, mientras que para conocer su contenido, la RMN en cortes coronales nos da una información mucho más detallada31, ya que como puntualizaron Casselman y cols.30, un CAI de diámetro normal visto en TAC puede presentar ausencia de octavo par craneal, siendo la RMN el único medio para conocer este evento. Por tanto, por su alta resolución y avanzadas técnicas de reconstrucción, la RMN podría sustituir a la TAC como prueba de imagen de elección en la valoración de niños con hipoacusia neurosensorial. 1.2.3.2.2.- CAI dilatado: Existe consenso en considerar esta patología a partir de los 8mm de diámetro30. Birman y Gibson30 publicaron que un CAI ensanchado estaría asociado con una dehiscencia parcial del fondo lateral del CAI, aumentando la comunicación entre el CAI y el oído interno y por tanto propiciando la aparición de una hipoacusia neurosensorial progresiva o fluctuante. Sin embargo, la presencia de un CAI ensanchado no es causa por si sola de hipoacusia, como demostraron Weinberg y cols.30 y Tomura y cols.30 al publicar casos de pacientes normooyentes con CAI ensanchado. Raramente encontramos un CAI dilatado de forma aislada, sin asociar otras malformaciones. Por ello, ante su presencia, se debe hacer un estudio minucioso de todo el oído interno. Otro parámetro tenido en cuenta es la forma del CAI, que se presenta recta en el 35-60% de los casos y oval en el 13-19% del total en los estudios publicados30. La sordera ligada al sexo con gusher se ha relacionado con un CAI con forma de bulbo30. Otras formas anómalas de presentación del CAI son: muy estrecho de forma que sólo contiene el nervio facial; doble, con una cresta falciforme prominente; adelgazándose cerca de la lamina cribosa en su extremo lateral (frecuentemente asociado con cavidades comunes y fístulas espontáneas de liquido cefalorraquídeo); y ensanchado o con forma ovalada32. En otras ocasiones, un CAI dilatado en su extremo lateral puede estar asociado a patologías como un neurinoma del acústico, neurofibromatosis o hidrocefalia crónica; siendo en estos casos la dilatación consecuencia del aumento de presión local y no de un defecto congénito. Los síndromes de Goldenhar, Apert y Patau, así como la asociación CHARGE y otras malformaciones congénitas no clasificables pueden cursar junto a dilataciones del CAI, dando lugar a una alta incidencia de fístulas de LCR en las intervenciones quirúrgicas que requieren manipular el estribo32. Aunque parece conocerse el origen de un CAI estrecho, no hay una explicación embriológica convincente para el CAI dilatado. 1.2.3.3.- Ausencia congénita de la ventana oval Es otra malformación menor descrita y que se encuentra a caballo entre el oído medio y el interno. Se produce por falta de desarrollo de estructuras derivadas del segundo arco branquial, a menudo incluyendo el canal del facial. Esta malformación consiste en una obliteración ósea completa de la ventana oval, ya sea por un estrechamiento concéntrico que produce una depresión con forma de hoyuelo a lo largo de la pared medial timpánica o bien por la presencia de una platina gruesa33. El estribo puede estar o no presente, pero no existe la platina ni el ligamento anular, a diferencia de la otosclerosis congénita, donde estas dos últimas estructuras sí se desarrollan33. Como consecuencia de estas lesiones, esta malformación cursa con una hipoacusia de transmisión. A diferencia de las malformaciones descritas anteriormente y que afectan exclusivamente al oído interno, la ausencia congénita de ventana oval tiene tratamiento quirúrgico, consistente en realizar una fenestración con fresa de diamante en la zona donde presumimos que se situaría la ventana oval, cubriéndola posteriormente con un injerto de fascia temporal sobre el que se apoya una prótesis de pistón anclada por el otro extremo a la rama larga del yunque. Los resultados de esta intervención parecen optimistas a corto plazo, no siendo tales a largo plazo. 1.2.4.- Etiología de las malformaciones congénitas de la cóclea 1.2.4.1.- Causas no genéticas 1.2.4.1.1.- Infecciosas: la infección congénita por Toxoplasma gondii, Citomegalovirus (CMV) y por el virus de la Rubéola son causantes de una u otra malformación coclear según el momento del desarrollo embrionario en el que actúen, siempre que sea antes de la octava semana de gestación17. Para poder afirmar el origen infeccioso de una hipoacusia, es necesario que exista asociación clínica de la hipoacusia con el microorganismo o que haya sido demostrado un efecto similar sobre la audición en animales9. La infección congénita por CMV es la infección intrauterina más frecuente, con una incidencia estimada de 0,4-2,3% de los nacidos vivos en los Estados Unidos9. Aproximadamente el 10-15% de los recién nacidos infectados presentan clínica evidente en el momento del nacimiento, y tienen más probabilidades de presentar secuelas tales como síntomas neurológicos e hipoacusia neurosensorial. Esta última secuela es la más común y afecta al 3065% de los infectados sintomáticos y al 8-15% de los asintomáticos28. Se ha calculado que la infección congénita por CMV puede ser la causa del 20 al 30% de todos los casos de hipoacusia9, lo que justificaría un estudio de infección por CMV en todos aquellos niños en los que se sospecha una causa no genética de su hipoacusia. Cerca de la mitad de los recién nacidos sintomáticos desarrollan hipoacusia neurosensorial, que en la mayoría de los casos será progresiva. La terapia con ganciclovir en el periodo neonatal a niños con síntomas de afectación del sistema nervioso central, ha demostrado ser capaz de prevenir el deterioro de la audición9. La infección de la madre por el virus de la rubéola al comienzo del embarazo puede transmitirse al feto y provocar en él la rubéola congénita que se caracteriza por cataratas, cardiopatía y sordera34. La embriopatía rubeólica, así como la infección por CMV se relacionan, entre otras, con la malformación de Scheibe17. 1.2.4.1.2.-Metabólicas: Existen patologías metabólicas maternas que afectan al normal desarrollo embrionario, como el hipotiroidismo17. El déficit de yodo es más peligroso durante el periodo crítico de diferenciación neural, pudiendo producir graves y permanentes daños cerebrales. En el cretinismo endémico, por carencia de yodo, más de la mitad de los casos presentaran una hipoacusia mixta y progresiva por afectación de las células ciliadas, estría vascular y conductos semicirculares, así como una osificación incompleta del estribo, y una malformación del yunque y de las ventanas, redonda y oval. 1.2.4.1.3-Tóxicas: La ingesta de drogas ototóxicas como la difenilhidantoína, los aminoglicósidos, la talidomida, el alcohol o la isotretinoina entre otras, por parte de mujeres embarazadas y sobre todo en el primer trimestre, puede derivar en una hipoacusia congénita. Las malformaciones de oído interno atribuibles a este tipo de drogas serían la aplasia del oído interno, malformaciones de la cadena osicular y la ausencia del séptimo o el octavo par craneal9. El estudio anatomopatológico de los huesos temporales de dos fetos humanos expuestos a isotretinoina en las primeras fases de la gestación, mostró anomalías en la cadena osicular, desarrollo incompleto de la cavidad timpánica y un nervio facial dehiscente. Asimismo, la cóclea presentaba un número menor de vueltas de espira y un sáculo dilatado35. 1.2.4.1.4.- Físicas: la radiación ionizante17. 1.2.4.2.- Causas genéticas En los últimos años se han publicado numerosos estudios genéticos sobre las malformaciones de oído interno. MacKay y cols.20 publicaron que el factor de crecimiento fibroblástico (FGF-3) era necesario para un correcto patrón de diferenciación de la vesícula ótica. Torres y cols.20 demostraron que en el oído interno, mutaciones de Pax-2 producen agenesia de la cóclea y del ganglio espiral. Por tanto, el normal desarrollo de la cóclea depende de la correcta expresión de Pax-2, mientras que Nkx5 es necesario para la formación de los canales semicirculares. Las hipoacusias de origen genético pueden ser sindrómicas (30%) o no sindrómicas (70%), siendo en este último caso el oído interno el único órgano afectado9. En los últimos años ha habido un importante desarrollo en la genética de las hipoacusias no sindrómicas y sabemos que existen hasta 70 genes responsables de alguna forma de hipoacusia y han sido identificados 33 de estos genes y cuatro mutaciones mitocondriales36. En más del 33% de los casos de hipoacusia infantil, el síntoma es aislado y no existe ningún antecedente que nos ponga en la pista del problema. En la mayoría de los casos se trata de enfermedades monogénicas. Puede haber una gran variación en las malformaciones que un único gen puede producir, incluso en la misma familia o en diferentes oídos de la misma persona, indicando que un gen no produce un efecto concreto sobre una estructura concreta. No obstante, determinadas características fenotípicas son observadas con más frecuencia en determinados síndromes o en familias37. Futuros estudios moleculares determinarán los mecanismos precisos por los que los genes intervienen en el desarrollo del oído interno junto con los factores no genéticos37. 1.2.4.2.1.- Hipoacusias no sindrómicas Suelen ser neurosensoriales y responden a todos los patrones de herencia, siendo más frecuentes las autosómicas recesivas (DFNB) representando hasta el 77% del total, seguidas de las autosómicas dominantes (DFNA) con un 22% y por último un 1% de ligadas al cromosoma X (DFN). Además existe mitocondrial. un pequeño porcentaje de hipoacusias de herencia Más de la mitad de estas hipoacusias no sindrómicas se deben a una mutación en el gen GJB2, responsable de codificar la conexina 26, una proteína encargada de la difusión celular y del reciclaje de pequeñas moléculas9. En estos pacientes se pueden encontrar anomalías de la cóclea y dilatación del acueducto del vestíbulo. 1.2.4.2.2.- Hipoacusias sindrómicas Del total de las hipoacusias de causa genética, sólo un 10-15% son sindrómicas, habiéndose descrito ya varios centenares de síndromes que asocian hipoacusia. Debido a esto, ante un niño con cualquier tipo de malformación debemos hacer un estudio sistemático de la audición, para descartar que la hipoacusia forme parte de su síndrome. A continuación enumeramos las hipoacusias sindrómicas de obligado conocimiento, bien sea por su frecuencia o por su potencial gravedad, y siempre guardando relación con las distintas malformaciones de oído interno. Cualquier tipo de hipoacusia puede aparecer formando parte de un síndrome, ya sea unilateral o bilateral, perceptiva, de conducción o mixta, simétrica o asimétrica. Existen cerca de 400 síndromes que asocian hipoacusia, y que siguen todos los patrones de herencia conocidos. a) Autosómico recesivo: Como otras enfermedades genéticas de carácter autosómico recesivo, los progenitores portadores del alelo mutado son normooyentes mientras que los descendientes tienen un 25% de probabilidades de padecer hipoacusia. De entre ellos destacan: -Síndrome de Pendred: descrito por Vaughan Pendred en 18969, asocia hipoacusia coclear y un defecto en la organificación del yodo que produce bocio multinodular, que suele aparecer en la infancia o la adolescencia pero que rara vez es congénito30. Los pacientes suelen ser eutiroideos, aunque algunos pueden presentar diferentes grados de hipotiroidismo. Algunos pacientes pueden presentar vértigo con anomalías en los test vestibulares38. Tiene una prevalencia de 7-10 casos por cada 100.000 nacimientos. Es la forma más frecuente de hipoacusia sindrómica y supone más del 10% de los casos de sordera hereditaria38. El desorden se produce por una mutación en el gen PDS (SLC26A4) que produce un defecto en una proteína llamada pendrina, la cual se cree que tiene un papel fundamental en la producción de hormonas tiroideas y en el mantenimiento de la homeostasis del líquido endolinfático9. Hasta hoy se han podido identificar más de 50 mutaciones del gen PDS. La hipoacusia suele ser profunda y prelingual38, y tiene de particular el hecho de evolucionar con episodios de agravación intensa, con períodos de recuperación parcial posteriores. Un suceso traumático, una infección o un edema endolinfático pueden propiciar la hipoacusia38. Este síndrome asocia siempre malformaciones del oído interno que pueden ser vistas con tomografía computarizada, tales como dilatación del acueducto del vestíbulo y malformación coclear tipo Mondini, siendo ambas consideradas casi de obligada existencia para poder establecer el diagnóstico38. La hipoacusia progresiva con episodios de fístula perilinfática hace pensar que estos pacientes puedan tener también una dilatación de la espira basal de la cóclea37. Estudios radiológicos más recientes realizados con TAC y RMN, hallaron ausencia de la espira apical (debido a la falta de septum interescalar) en sólo el 20% de los pacientes con síndrome de Pendred estudiados38. Estudios histológicos en un paciente mostraron evidencia de hydrops, cambios posicionales en la membrana de Reissner, degeneración de la estría vascular y células ciliadas y cambios hidrópicos en el sáculo y el utrículo37. Estas anomalías pueden presentarse de forma uni o bilateral. Esta mutación puede producir hipoacusia aislada del síndrome de Pendred, de hecho se estima que las mutaciones en el gen SLC26A4 están presentes en el 78% de los pacientes con hipoacusia neurosensorial y dilatación del acueducto del vestíbulo, sin que presenten bocio9. -Síndrome de Fountain: es un síndrome muy raro que asocia retraso mental, anomalías óseas, adelgazamiento progresivo de algunos rasgos como los labios e hipoacusia profunda debida a malformaciones como partición incompleta de cóclea. La patogenia de este síndrome se desconoce por completo37. b) Autosómico dominante Los estudios realizados sugieren que este tipo de herencia produce un amplio rango de malformaciones, lo cual se confirma por los variados fenotipos encontrados; y que se cree que pueda deberse a la interacción con posibles variaciones de los alelos homólogos o de otros genes, o bien a efectos aleatorios del desarrollo37. En este tipo de transmisión la expresividad es variable. -Síndrome de Waardenburg: Presenta una incidencia de aproximadamente 1-2% de entre los pacientes con hipoacusia profunda9 y asocia hipoacusia y anomalías de la pigmentación (heterocromía del iris y mechones blancos entre otros). La hipoacusia varía entre leve y profunda incluso dentro de la misma familia y la disfunción vestibular puede estar presente incluso en sujetos normooyentes37. En él se han descrito malformaciones tanto óseas como membranosas del laberinto anterior y del posterior aunque no siempre asocia malformación del oído interno. Mientras que en otros cuadros las malformaciones más frecuentemente halladas son las que interesan al canal semicircular lateral, en el síndrome de Waardenburg es característica la afectación del posterior siendo también común la degeneración cocleosacular que puede existir con o sin hallazgos morfológicos37. Los genes involucrados en este síndrome intervienen en el desarrollo de la cresta neural y afectan a la migración y diferenciación de los melanocitos, por lo que estos pacientes presentarán defectos de las estructuras que derivan de la cresta neural y un déficit de melanocitos9. Existen cuatro tipos de este síndrome en función de los signos asociados: -Tipo I: asocia distopia cantal y de un 20 a un 58% de los pacientes presentan hipoacusia neurosensorial37. El gen causal es el PAX3. -Tipo II: carece de distopia cantal. Es el más frecuente. Los genes causantes son MITF y SLUG9. En la TAC de estos pacientes se puede ver aplasia del canal semicircular posterior con subdesarrollo del vestíbulo9. Otras anomalías que podemos ver son ensanchamiento del acueducto del vestíbulo, conducto auditivo interno estenosado e hipoplasia del modiolo9. Cerca del 70% de estos pacientes presentan hipoacusia neurosensorial37. -Tipo III: es como el tipo I pero añadiendo malformaciones de las extremidades. El gen responsable es el PAX39. -Tipo IV: es como el tipo II con enfermedad de Hirschsprung. Es de herencia autosómica recesiva y sus genes causantes son EDNRB, EDN3 y SOX109. -Síndrome braquio-oto-renal (BOR): El síndrome BOR fue descrito por primera vez en 1978 por Melnick-Fraser como un desorden autósomico dominante de alta penetrancia y expresividad variable39, con una prevalencia estimada en uno de cada 40.000 nacidos vivos39. Asocia hipoacusia neurosensorial o mixta, malformaciones renales y fístulas branquiales. La hipoacusia se acompaña de malformaciones de las tres partes de oído (aplasia de la oreja, estenosis del conducto auditivo externo, malformaciones cocleovestibulares…). Entre las malformaciones que puede producir en el oído interno destacamos la presencia de una espira basal afilada con una hipoplasia de las espiras media y apical, lo cual requiere hacer diagnóstico diferencial con la malformación de Mondini. Asimismo, malformaciones de la cadena osicular, ensanchamiento del acueducto del vestíbulo, hipoplasia bilateral del nervio coclear o ausencia o hipoplasia de los canales semicirculares37 también pueden presentarse en estos pacientes9. El 80% de los afectados presentan hipoacusia que puede ser neurosensorial, transmisiva o mixta, y en ocasiones progresiva9,37. Se debe a la mutación en heterocigosis del gen EYA1 en el brazo largo del cromosoma 8q13.337. Se ha identificado el gen SIX1 como otro involucrado en la génesis de este síndrome. Es conveniente el estudio mediante ecografía renal ante un paciente que asocie hipoacusia y fístula branquial o malformación de oído externo. -Síndrome de Down: se caracteriza por ser la trisomía del cromosoma 21, con una incidencia de aproximadamente 1 caso por cada 600-800 nacimientos, es la anomalía de causa genética más vista en la práctica otorrinolaringológica40. La hipoacusia aparece en el 80% o más de los niños afectados con este síndrome40, siendo en la mayoría de los casos de tipo conductivo, pero dejando un amplio margen de 4 a 55% de casos de hipoacusia neurosensorial o mixta, siendo en estos casos consecuencia de la laberintización de una hipoacusia de transmisión previa. Las malformaciones de oído interno son menos frecuentes que las de oído externo o medio, y menos frecuentes y graves que las asociadas a otras trisomías40, y aunque en la literatura se encuentran numerosos estudios anatomopatológicos, no ocurre lo mismo con los referentes a la radiología. En estudios postmortem de huesos temporales de pacientes con Síndrome de Down se han observado hallazgos compatibles con enfermedad de Mondini además de otras alteraciones histopatológicas cocleares como menor longitud de la cóclea, del modiolo, alteraciones del estribo, dehiscencias del canal del nervio facial, hipoplasia o duplicación del CAI, crítica estenosis del canal del nervio coclear, dilatación del saco endolinfático, ensanchamiento del acueducto del vestíbulo, así como también pueden existir rutas de comunicación a través de la membrana que cubre la ventana redonda y del ligamento anular en la ventana oval que explicarían probables vías de infección y fístulas. Las estructuras del oído interno aparecen en general de un tamaño menor a la normalidad, siendo las malformaciones vestibulares y la presencia de una pequeña isleta ósea del canal semicircular lateral muy típicas40. -Síndrome de Edwards: trisomía del cromosoma 18. Se relaciona con hipoplasia o agenesia del nervio coclear. -Síndrome de Turner: la trisomía del cromosoma 13 se asocia a anomalía de Mondini. c) Ligada al cromosoma X Las alteraciones genéticas ligadas al cromosoma X se caracterizan por ser transmitida por la madre y únicamente padecerla los hijos varones. -Hipoacusia neurosensorial ligada al cromosoma X: también se conoce como DFN39. Es una anomalía cromosómica que produce hydrops perilinfático debido a una fístula entre el CAI y la cóclea25. Se manifiesta en varones jóvenes con hipoacusia profunda neurosensorial o mixta que es a menudo progresiva y puede estar asociada a disfunción vestibular y fijación congénita del estribo, responsable de la otorrea perilinfática que se produce al intentar una estapedectomía9. Las mujeres portadoras pueden presentar un cuadro mucho menos severo25. Los hallazgos radiológicos que acompañan a este síndrome con más frecuencia son el ensanchamiento del CAI que es habitualmente bilateral y simétrico25 así como la hipoplasia de la espira basal de la cóclea, un modiolo ausente o deficiente y un déficit del septum interescalar coclear9. Se supone que esta es la causa de que exista una comunicación entre el espacio subaracnoideo en el CAI y la perilinfa de la cóclea, produciéndose un hydrops perilinfático si el estribo es manipulado9. También es frecuente encontrar un ensanchamiento del canal proximal del nervio facial y el ensanchamiento del acueducto del vestíbulo. La mutación que se asocia a esta anomalía se ha localizado en el cromosoma X, y en los varones afectados se ha localizado una mutación en un gen regulador ADN vinculante llamado POU3F49. d) Otros síndromes: -Síndrome de Goldenhar: o displasia oculoariculovertebral presenta un amplio abanico de malformaciones de oído externo y medio bien conocidas, mientras que no lo son tanto las anomalías que comprometen al oído interno41. .Entre otras podemos encontrar una cóclea hipoplásica, ausencia de canales semicirculares, conducto endolinfático anómalo, alargamiento del acueducto del vestíbulo, ausencia del canal del nervio facial, CAI duplicado... No se conoce bien la etiología de este síndrome, ni su patrón de herencia, pero Poswillo en 1975, propuso la formación de un hematoma en el embrión debido a hipoxia, hipertensión, anticoagulantes u otros factores. Poswillo no encontró afectación del oído interno, por lo que presumió que la cápsula ótica era una eficiente barrera que lo protegía del hematoma41. Esta teoría no consiguió muchos adeptos, por lo que finalmente se pensó en una detención del desarrollo a edades más tempranas (cuarta semana gestacional) en la etapa de los signos neuroectodérmicos y mesodérmicos, que toman parte en la formación de la placoda ótica así como de los arcos faríngeos primero y segundo. Realizar exámenes audiológicos y de imagen es obligatorio en pacientes afectos de síndrome de Goldenhar para descartar anomalías de oído medio o interno. -Síndrome de DiGeorge: incluye inmunodeficiencia e hipocalcemia debida a una malformación del timo y las paratiroides, defectos cardíacos, unas características faciales concretas y malformaciones de oído externo, medio o interno, destacando la malformación de Mondini entre estas últimas37. -Síndrome de Wildervanck: asocia anomalía de Klippel-Feil (fusión de vértebras cervicales), retracción de Duane (los ojos se van hacia atrás cuando miran lateralmente) e hipoacusia neurosensorial, aunque en algunos casos se ha presentado de forma mixta. Se han observado varias malformaciones de cóclea, vestíbulo, canales semicirculares o conducto auditivo interno; mientras que en otros casos la anatomía resultó completamente normal. La gran variedad de malformaciones de oído interno se debe probablemente a la heterogeneidad genética. Es característico de este síndrome la gran diferencia que hay en cuanto a la presentación por género, siendo 10/1 la relación mujeres / hombres37. -Tríada de Robin: se caracteriza por hendidura palatina, micrognatia, glosoptosis y problemas de alimentación, y puede aparecer tanto de forma esporádica como sindrómica, sobre todo en el síndrome de Stickler y en el síndrome velocardiofacial. La hipoacusia es variable y las malformaciones de oído más frecuentemente encontradas son: pabellones de implantación baja, anomalías del estribo, nervio facial dehiscente, malformaciones cocleares y del laberinto posterior y CAI de pequeño tamaño9. -Síndrome CHARGE: asocia coloboma, malformaciones cardíacas, atresia de coanas, retraso mental, anomalías genitourinarias y auditivas. Otras anomalías adicionales han sido descritas, tales como parálisis o asimetría facial, anomalías laríngeas o esofágicas, malformaciones renales o grietas faciales42. Tiene una incidencia de 1 caso por cada 8.500-12.000 nacimientos9. La asociación de hipoacusia, malformaciones de oído, atresia de coanas, colobomas y otras malformaciones fue reconocida por primera vez por Hittner y cols.9 y Hall y cols.9 en los años 70. Los criterios diagnósticos clásicos de Pagon y cols. establecidos en 198142, incluyen: una característica mayor (coloboma, microftalmia o atresia de coanas) y cuatro de las seis características que conforman el acrónimo CHARGE. Más recientemente, para confirmar el diagnóstico de síndrome CHARGE se han establecido criterios mayores: 1) coloboma, 2) atresia de coanas y 3) canales semicirculares hipoplásicos; y menores: 1) disfunción rombencefálica, 2) disfunción hipotálamo-hipofisaria, 3) malformaciones de oído medio externo, 4) malformaciones cardíacas y/o esofágicas y 5) retraso mental. Deberán estar presentes tres criterios mayores, o dos mayores y dos menores para hacer el diagnóstico9. Otra clasificación publicada por Blake diferencia cuatro criterios mayores y siete menores42: Tabla 742.- Criterios de Blake para el sdme. CHARGE MAYORES MENORES + Coloboma + Defectos cardíacos + Atresia de coanas - Grietas orofaciales + Anomalías auditivas + Hipoplasia de genitales + Alteraciones de pares craneales + Déficit de crecimiento + Retraso del desarrollo - Fístula traqueoesofágica + Anomalías faciales +: Hallazgos positivos necesarios; -: hallazgos negativos necesarios. Según esta clasificación deberían estar presentes cuatro criterios mayores o tres mayores y tres menores para establecer el diagnóstico de CHARGE. Recientes estudios sugieren que estos pacientes están en riesgo de padecer anomalías del octavo par craneal, tales como ausencia o hipoplasia43, así como hipoplasia de la espira apical de la cóclea, trayecto anómalo del nervio facial, malformaciones de la cadena osicular, hipoplasia de los canales semicirculares o ausencia de estos, siendo esta última la anomalía más específica9. A pesar de que muchos autores han descrito las anomalías auditivas asociadas al síndrome CHARGE, Lemmerling y cols.42 proporcionaron una de las más explicativas descripciones de estas anomalías basándose en imágenes de TAC. Wright y cols.42 también describieron la ausencia de músculo estapedial en todos sus casos. A.K. Morimoto y cols.42 realizaron un estudio a 13 pacientes con asociación CHARGE evaluando en TAC y RMN las malformaciones de oído interno asociadas, encontrando un 11,5% de CAIs de pequeño tamaño, un 11,5% de espiras basales anómalas, ensanchadas o con falta de segmentación, coincidiendo con sus respectivas espiras apicales con la misma anomalía, y encontrando un 69,2 % de estas ultimas hipoplásicas. En cuanto a los canales semicirculares, se constató su ausencia en el 96,1% de los casos, 23% de ventanas redondas y 80,7% de ventanas ovales aplásicas y nervios faciales con una disposición posterior a lo normal en el 88,4 % de los casos. Tellier42 también describió anomalías de la cóclea, incluyendo malformación de Mondini, displasias del vestíbulo, acueductos del vestíbulo dilatados, atresia de la ventana oval, yunque hipoplásico, oído medio de pequeño tamaño y anomalías del tendón del músculo estapedial. El conocimiento de estas malformaciones asociadas al síndrome CHARGE es vital para el éxito de la cirugía del implante coclear o del tronco cerebral, que son actualmente de las pocas opciones de tratamiento a las que optan estos pacientes. Niños con malformaciones cocleares, frecuentemente presentan asociadas anomalías como la asociación CHARGE y retraso psicomotor, lo que hace que la causa de sus malos resultados tras la implantación no quede clara43. 1.2.5.-El nervio coclear Existe una gran controversia sobre las malformaciones congénitas cocleares y su asociación con agenesias del nervio coclear y del paquete neural del Conducto Auditivo Interno. Algunos autores sugieren que la cóclea no puede desarrollarse con normalidad si no tiene una adecuada inervación por parte del nervio coclear10, por lo que si las pruebas de diagnóstico por imagen mediante tomografía axial computadorizada muestran una cóclea de características normales, el nervio coclear debe estar presente. En contra de esta teoría, Nelson e Hinojosa10 demostraron la ausencia de nervio coclear a pesar de existir un desarrollo normal del oído interno y del CAI. Además, un estudio reciente realizado por Buchman, CA y cols.10 ha documentado las características electrofisiológicas de la neuropatía auditiva en niños con ausencia de nervio coclear. En ese estudio, el 70% de los oídos con déficit de nervio coclear demostraron la presencia de microfónicos cocleares con ausencia de ondas en los PEATC. Estos hallazgos implican que la función de las células ciliadas externas puede persistir a pesar de no tener nervio coclear. La inervación del oído interno depende de los factores neurotróficos secretados por las células ciliadas10, por lo que la existencia de microfónicos cocleares en ausencia de nervio coclear no es raro. Por estos motivos, Incesulu y cols.16 sugirieron que ante la existencia de CAIs con un diámetro menor de 2mm, la realización de RMN complementaria a la TAC es decisión obligada, con el fin de estudiar mejor el nervio auditivo. Por el contrario, puede darse el caso de un CAI ensanchado que contenga solamente el nervio coclear, lo que fue diagnosticado por RMN a un paciente con una malformación de Mondini y que fue publicado por Sennaroglu y cols.44. De todas formas no todas las agenesias de nervio coclear pueden ser sospechadas, pues Casselman y cols.16 estudiaron dos pacientes con CAIs normales pero nervio coclear ausente confirmado mediante RMN. Asimismo, describieron un paciente con una cavidad común cuya reconstrucción de imágenes del octavo par craneal descubrió un nervio coclear ausente, pero que curiosamente obtuvo umbrales dentro del rango normal tras la implantación. Esto les hizo lanzar la hipótesis de que la rama vestibular del octavo par craneal podría tener algunas fibras que proyectaran a la corteza auditiva, y sugirieron que la estimulación eléctrica del promontorio y la RMN de la corteza auditiva, no deberían ser ignoradas como herramientas diagnósticas para estos pacientes. Asimismo, debemos tener siempre en cuenta la posibilidad de la existencia de un trayecto del nervio coclear anómalo, que transcurra por fuera del CAI, y por tanto no sea detectable en los cortes radiológicos de éste. 1.3.- IMPLANTES COCLEARES Los implantes cocleares son dispositivos electrónicos implantables intracocleares, capaces de convertir las señales acústicas que reciben del exterior, en señales eléctricas y estimular así las terminaciones auditivas del nervio coclear45. 1.3.1.-Breve reseña histórica Los implantes cocleares pueden ser conceptuados como tecnología emergente, al ser su historia muy reciente y sus evoluciones trepidantes. El primer intento de estimulación eléctrica coclear fue desarrollado por Volta en el año 1800, al introducir en el conducto auditivo externo unas varillas metálicas conectadas a una fuente eléctrica de 50 voltios, apreciando sólo durante el tiempo de la estimulación eléctrica un ruido. Diversos científicos intentan la estimulación eléctrica del nervio auditivo, no obteniendo resultados satisfactorios a largo plazo. André Djourno46 realizó en 1957 el considerado primer implante coclear. Es al final de la década de los 60 cuando el Prof. William F. House (L.A) realiza la primera implantación coclear monocanal con electrodos conectados a una computadora externa. Él mismo consigue desarrollar en 1973 el primer implante portátil (3M/House). En la Universidad de Melbourne (Australia), el profesor Clark realiza el primer implante coclear multicanal, que permite discriminar la palabra, colaborando con la empresa Nucleus para comercializar este primer dispositivo (Nucleus 22), aprobado por la Food and Drugs Association (FDA) en 1985 para su uso en adultos y posteriormente en 1990, para niños45. En 1998 se aprueba la implantación coclear en menores de 2 años. En los últimos 5 años, los avances tecnológicos han permitido implementar los implantes cocleares con tecnología capaz de realizar pruebas telemétricas, como la obtención de impedancias eléctricas, potenciales de acción compuestos evocados eléctricamente y la realización de potenciales auditivos evocados eléctricamente, así como la posibilidad de la programación de los dispositivos a distancia a través de Internet. 1.3.2.-Criterios de Implantación coclear Los criterios de implantación de dispositivos electrónicos multicanales intracocleares (implantes cocleares), ha sido ampliamente estudiados, evaluados y consensuados en varias ocasiones desde que, en el año 1995, The National Institut of Health los estableciera en función de los beneficios y perspectivas futuras del implante coclear. En España, desde el año 2003, y siguiendo los criterios establecidos en el Pleno del Consejo Interterritorial de Salud del Ministerio de Sanidad y Consumo, la implantación coclear se realiza bajo los siguientes criterios, debiendo cumplir todos y cada uno de ellos: - Hipoacusias neurosensoriales bilaterales severas o profundas, entendiendo como tales las que presentan un umbral de audición promedio mayor de 70 dBHL para las frecuencias de 0,5 Khz., 1KHz., 2KHz. y 4KHz47. - Pacientes que no obtengan beneficio con la adaptación de prótesis auditivas. Se considera como no obtención de beneficio el presentar un umbral de audición tonal de 55 dBHL y alcanzar una inteligibilidad máxima de palabras bisílabas del 40%, a una intensidad de 65 dBHL47. Estos umbrales auditivos han de explorarse en campo libre, con prótesis auditivas y ajustes en posición de uso con y sin ruido de competencia. - Haber adquirido lenguaje verbal o estar aún, por edad, en condiciones de adquirirlo. Son, por lo tanto, buenos candidatos a la implantación coclear aquellos pacientes menores de 5 años con hipoacusia prelocutiva, y por tanto con posibilidad de adquirir lenguaje, y los mayores de esta edad con hipoacusia peri o postlocutiva con una correcta adquisición del lenguaje. Además, los pacientes prelocutivos que han adquirido el lenguaje a través de métodos tradicionales, también son buenos candidatos. - Convicción del paciente de que la mejoría auditiva aportada por el implante coclear le reportará beneficios personales y sociales. - No presentar las siguientes condiciones patológicas: a) Malformaciones cocleares: únicamente las agenesias cocleares bilaterales son consideradas como contraindicación. Otras malformaciones cocleares, tales como aplasias, hipoplasias y particiones incompletas, han de ser ampliamente estudiadas desde un punto de vista morfológico y funcional, ajustando los beneficios y complicaciones del proceso de implantación coclear. b) Enfermedades que causen hipoacusia de tipo central. Actualmente, las enfermedades que causan disfunción de la vía auditiva deben ser evaluadas individualmente, al mostrarse ciertos tipos de neuropatía auditiva como una entidad muy beneficiada de la implantación coclear. c) Enfermedades psiquiátricas graves. d) Enfermedades que contraindiquen la anestesia general. 1.3.3.- Composición y estructura de los implantes cocleares: Los implantes cocleares se componen de: 1.3.3.1.- Dispositivo externo o Procesador de sonido: es el encargado de recibir el estímulo acústico a través de micrófonos o de entradas auxiliares, separar los componentes frecuenciales y detectar las intensidades de dicha entrada para posteriormente codificarlos de tal manera que la estimulación eléctrica sea eficaz. La estrategia de codificación consiste en convertir las señales acústicas, analógicas, en una señal digital que posteriormente será transferida al dispositivo implantable a través de transmisores de radiofrecuencia. Las baterías, recargables o no, se situarán en este componente externo. 1.3.3.2.- Sistema implantable: estimulador receptor. Se implanta en el hueso temporal. Recibe la señal digital procedente del procesador de sonido y envía pulsos eléctricos controlados, a través de electrodos independientes intracocleares situados en la rampa timpánica, hasta la proximidad de los terminales nerviosos y células ganglionares situadas en el modiolo coclear. La disposición de estos electrodos permite mantener la tonotopía coclear. Estos estímulos pasan a través del nervio auditivo hasta los centros neurales superiores donde se reconocen. 1.3.4.- Tipos de implantes cocleares Las antiguas clasificaciones de implantes cocleares, basadas en la ubicación de los electrodos (intra o extracocleares) y el número de canales de estimulación (mono o multicanales), han quedado en desuso al ser todos los implantes cocleares, multicanalaes e intracocleares. De este modo, hoy día existen diferencias menores entre los distintos implantes cocleares, que pueden clasificarse según: -1.3.4.1.- Carcasa del dispositivo implantable: existen modelos de implantes con carcasas de cerámica, más resistentes a la formación de biofilms y menos resistentes a traumatismos mecánicos, y carcasas de titaniosilicona, más resistentes a traumatismos mecánicos aunque más sensibles a la formación de biofilms. -1.3.4.2.- Tipos de estimulación eléctrica: -Monopolar, en la que los electrodos estimulan compartiendo un electrodo de referencia extracoclear y distal al lugar de estimulación, disminuyendo la cantidad de corriente necesaria para estimular. -Bipolar, en la que los electrodos se estimulan en parejas, estando la pareja compuesta por electrodos muy próximos, siendo uno el electrodo estimulante y el otro el de referencia. Esta estrategia de estimulación resulta en una menor interacción entre electrodos. -1.3.4.3.- Estrategias de procesamiento del sonido: Los distintos fabricantes de implantes cocleares aportan diferentes estrategias de procesamiento del sonido, siendo éstas el resultado de los avances en la tecnología de los implantes y en el mejor conocimiento de la estimulación eléctrica del oído. Actualmente, existen diversas estrategias: + Analógica: ondas continuas que preservan el origen sinusoidal de la señal de entrada al procesador. Las estrategias analógicas, con el fin de obtener ondas de estimulación continuas, deben activar los electrodos de forma simultánea. + Pulsátil: ondas bifásicas discretas convertidas desde la señal acústica analógica de entrada. La estimulación se realiza de forma que en cada momento hay un solo canal activo para evitar las interferencias entre canales2. a) Estrategia Spectral-Peak (SPEAK): se basa en la extracción de los rasgos principales de la palabra persiguiendo conseguir una perfecta transmisión de los formantes seleccionados que representen más fidedignamente el mensaje hablado48. Los pulsos se liberan en una secuencia no simultánea sobre los electrodos seleccionados. b) Estrategia analógica-comprimida (CA): envía las señales de forma simultánea por lo que se pueden producir interacciones entre los canales generando la sumación de los campos eléctricos de cada electrodo. Así la respuesta neural quedará distorsionada48. c) Estrategia basada en el muestreo secuencial continuo (CIS): los pulsos llegan a los electrodos de forma no simultánea, estimulando un solo electrodo en cada momento. La velocidad en que se estimulan los electrodos ejerce un papel importante en el reconocimiento del habla. A mayor velocidad, mejores resultados48. d) Estrategia analógica simultánea (SAS): puede usarse con ondas pulsátiles o analógicas, ocurriendo en todos los canales al mismo tiempo. La tasa de estimulación ocurre a velocidades muy altas (91.000 muestras/s), por lo que estas estrategias son ricas en información temporal detallada48. e) Estrategia advanced combination encoders (ACE): combina elementos de las estrategias SPEAK y CIS. Esta estrategia permite elegir qué electrodos estimular, de qué manera estimular los canales y la tasa de estimulación, pudiendo adaptar la programación a cada paciente de una forma más individualizada y personal48. Tabla 8. Evolución de los diferentes tipos de estrategias de procesamiento del sonido. Mono canal multicanal Extracción características 1970 Información completa 1980 F0/F2 1990 F0/F1/F2 (CA) MPEAK SPEAK (CIS) 2000 ACE SAS Aún siendo multicanales todos los implantes cocleares disponibles, presentan diferencias en cuanto al número de canales de estimulación. No es quizás un hecho diferencial pues no se han encontrado diferencias importantes respecto a este hecho, siendo precisos como mínimo entre 4 y 6 canales para la discriminación del lenguaje. A más canales útiles, mayor colorido y mejor discriminación, siendo el factor limitante la interacción eléctrica entre canales, limitando así el número máximo de ellos48. 1.3.5.-Funcionamiento básico del implante coclear Como ya se dijo anteriormente, el implante coclear es un dispositivo encargado de captar la energía sonora y transformarla en energía eléctrica para estimular así las fibras aferentes del nervio coclear y producir en el paciente una sensación sonora. La señal auditiva es captada por el micrófono y amplificada y analizada por el procesador. Parte de este análisis es el control automático de ganancia, de donde pasa la señal a un banco de filtros. Éstos sirven para eliminar las HIRes frecuencias que no son útiles para la comprensión de la voz y se divide la señal acústica en bandas de frecuencia para enviarlas a los diferentes electrodos; así, cada banda estimulará una región concreta de la cóclea. Los electrodos del implante coclear estimulan eléctricamente en forma de pulsos el nervio coclear, formado por neuronas que discurren desde el Órgano de Corti hasta los núcleos cocleares del tronco del encéfalo, pasando por el ganglio espiral, el modiolo coclear, el conducto auditivo interno y el ángulo pontocerebeloso48, pudiendo el implante estimular estas neuronas en cualquiera de estos puntos. 1.3.6.-Evaluación preoperatoria Durante la fase de diagnóstico, y con el fin de poder valorar los criterios de inclusión previamente descritos, se realizan las siguientes exploraciones: 1.3.6.1.- Adultos: a. Exploración física otológica. b. Audiometría tonal con y sin audífonos; Logoaudiometría con y sin audífonos. c. Exploraciones neurofisiológicas: Impedanciometría y reflejo estapedial, otoemisiones acústicas, potenciales evocados auditivos con o sin estimulación eléctrica y electrococleografía en situaciones especiales. e. Pruebas de imagen: En cuanto a la TAC, es importante destacar que no es necesaria la aplicación de contrastes para el estudio radiológico previo al implante coclear49, siendo de cara a esta intervención los cortes en el plano de la espira basal, paralelo al eje mayor del peñasco, los más útiles para mostrar la cóclea al completo50. Se realizan con programa de “ventana ósea” en cortes de 1mm y con desplazamiento de 1mm47. Aunque algunos autores han sugerido que la TAC del hueso temporal es la única prueba diagnóstica capaz de determinar la causa de la hipoacusia neurosensorial en niños, otros creen que la RMN también desempeña un papel importante en este campo51. Con la RMN se completa el estudio de los pacientes candidatos a implante coclear ya que suple la carencia de la TAC en lo referente a estudiar los tejidos blandos del oído (VII y VIII pares craneales, líquidos laberínticos etc.)49. El diagnóstico por imágenes utilizado habitualmente en la clínica se basa en la resonancia de protones. El principio es obtener una imagen después de colocar un órgano en un campo magnético intenso y enviar impulsos electromagnéticos según un ángulo variable52. 1.3.6.2.- Niños: a. Exploración física otológica. b. Impedanciometría timpanoosicular. c. Otoemisiones acústicas (OEA). d. Audiometría infantil. e. Potenciales Evocados Auditivos del Tronco Cerebral (PEATC) y Potenciales de Estado Estable (PEE). La electrococleografía (ECoG) debe ser realizada en situaciones especiales. f. Pruebas de imagen: TAC. En caso de malformaciones y osificaciones cocleares se acompaña de la realización de Resonancia Nuclear Magnética. Además de todas la exploraciones anteriores, el equipo de psicologíalogopedia realizará evaluaciones con el objetivo de ajustar individualmente las expectativas del paciente y familiares, valorar el grado de motivación hacia la implantación del paciente y de su entorno familiar, así como de informar al paciente y sus familiares del proceso de implantación y seguimientos postimplantación (Guía informativa del proceso de implantación). 1.3.7.- Actuación postoperatoria Tras el acto quirúrgico, la comprobación radiológica del correcto posicionamiento del dispositivo y de su buen funcionamiento mediante pruebas objetivas (Impedanciometría eléctrica y potenciales de acción compuesto evocados eléctricamente), se procede al alta hospitalaria y control de la herida quirúrgica durante al menos 1 mes, momento en que se inicia la programación del dispositivo y rehabilitación auditiva inmediata del paciente, preferentemente acompañado de un familiar, con los siguientes objetivos: 1. Obtener unos niveles de estimulación eléctrica iniciales adecuados al paciente. 2. Realizar las primeras estimulaciones auditivas, comprobando en todo momento el correcto funcionamiento de la programación del dispositivo. 3. Informar al programador de las respuestas auditivas observadas en el paciente al objeto de poder ir ajustando durante este periodo el dispositivo electrónico. 4. Valorar con test logopédicos de percepción auditiva las habilidades auditivas observadas postimplante, utilizando los mismos protocolos valorativos que en la fase preimplante. 5. Orientar a los pacientes y formar a los padres o familiares cercanos sobre pautas de actuación. 6. Emitir informes valorativos y orientativos para los logopedas de intervención. 7. Detectar posibles problemas o dificultades en el proceso. La duración de este periodo de adaptación varía entre una y dos semanas dependiendo de la evolución y las características de cada paciente. Posteriormente se procede a la fase de seguimiento, en la que el equipo de psicología-logopedia realiza los siguientes objetivos: 1. Comprobar la evolución perceptivo-auditiva y lingüística. 2. Obtener datos auditivos y lingüísticos indicadores del progreso. 3. Determinar variables influyentes en la evolución del paciente. 4. Dar información periódica de estos resultados a los logopedas de intervención y al entorno familiar, de cara a orientar posibles actuaciones. 5. Detectar posibles problemas o dificultades en el proceso. Este período de seguimiento consta de 7 episodios en niños (1, 3, 6, 12, 18, 24 y 36 meses) y de 6 en adultos (1, 3, 6, 12, 24, 36 meses). Todas las revisiones hasta los 12 meses, inclusive, tienen lugar en el Hospital Universitario San Cecilio de Granada, siendo realizadas las revisiones posteriores en los hospitales de todas las provincias de Andalucía, por el mismo equipo multidisciplinario. A partir del tercer año de uso de implante las revisiones serán bajo petición de los pacientes y en el Hospital Universitario San Cecilio de Granada. 1.3.8.- Programación implante coclear La implantación coclear no finaliza con la cirugía. Una vez concluida ésta, tiene lugar un procedimiento de gran importancia que es la programación o adaptación del procesador del implante. La programación consiste en establecer los parámetros que proporcionen al paciente un mejor aprovechamiento del implante y por tanto una mejor percepción de la señal sonora; para lo cual es primordial tener en cuenta las características del paciente, del implante empleado así como del procesador53. El implante coclear produce una estimulación eléctrica del nervio auditivo a través de unos electrodos que se insertan en el interior de la cóclea, y cuya actividad eléctrica es regulada por el procesador. Los implantes cocleares dividen el sonido en canales o bandas de frecuencia, correspondiendo cada banda a uno o varios electrodos intracocleares, de forma que los canales de frecuencias más graves estimulan los electrodos de las zonas más apicales, mientras que los de frecuencias más agudas hacen lo propio con los electrodos de las zonas más basales. Todo ello basándose en la teoría tonotópica según la cual la sensación de tono se percibe a través del lugar a lo largo de la cóclea en el que se produce la estimulación. Otro principio tenido en cuenta a la hora de la estimulación es el de codificación temporal, según el cual, las variaciones temporales de las características del sonido se perciben a través del patrón temporal de actividad en las fibras del nervio auditivo. Por ello, la estimulación generada por cada electrodo varía en el tiempo de acuerdo con la energía que hay en cada instante de tiempo en la correspondiente banda de frecuencia, y permite la percepción de la evolución temporal de las características del sonido. La calidad de sonido que va a percibir el paciente implantado va a depender de la resolución espectral, temporal y en intensidad que le proporciona el sistema, siendo parámetros variables durante la programación. -Resolución espectral: es la capacidad para distinguir la frecuencia de los sonidos, y en principio será mayor cuanto mayor sea el número de canales54, siempre teniendo en cuenta que la población de células ganglionares influye del mismo modo en dicha capacidad. La falta de resolución espectral no afecta de forma importante a la inteligibilidad de la voz54, pero sí afecta a la percepción de la voz en condiciones de ruido. -Resolución temporal: es la capacidad de percibir cambios temporales en las propiedades de la señal de audio54. Se modifica en función de la tasa de estimulación o número de pulsos por segundo. -Resolución en intensidad: determina la capacidad de percibir diferencias de intensidad de dos sonidos54 y depende fundamentalmente del estado de las terminaciones nerviosas; siendo mejor para un mayor porcentaje de terminaciones supervivientes. Con la programación se persigue obtener niveles adecuados de resolución espectral, temporal y en intensidad realizando ajustes en función de las características del implante y sobre todo del paciente54. Aproximadamente al mes de la intervención45 tiene lugar el encendido o primera programación del implante, debiendo decidir qué electrodos encender, qué niveles de estimulación requiere el paciente y qué estrategia de codificación va a seguirse entre otros parámetros. Normalmente la estrategia más apropiada a cada implante viene predeterminada por el fabricante. Sin embargo existen ciertos parámetros que pueden ser modificados por el programador, variando pues dicha estrategia. 1.3.9.1.- Parámetros de configuración de la estrategia a) Modo de estimulación: éste determina la configuración eléctrica de los electrodos, siendo necesario para insertar corriente la presencia de un electrodo activo y de otro de referencia. Diferenciamos entre estimulación monopolar cuando el electrodo de referencia es extracoclear, y de estimulación bipolar cuando tanto el electrodo activo como el de referencia son intracocleares. El modo monopolar es incompatible con una estimulación simultánea en varios canales, ya que produciría un fenómeno de suma de campos. Pero en cambio tiene la ventaja de requerir niveles de estimulación más bajos que el bipolar. El modo bipolar requiere niveles de estimulación muy altos, por lo que no se aconseja su uso en pacientes con lesiones que afecten a gran parte de la cóclea, ya que la guía de electrodos y las terminaciones supervivientes se encontrarán a una gran distancia. El modo bipolar se aconseja sólo si se va a realizar una estimulación simultánea en todos los canales. Los modelos con guías perimodiolares mejoran el rendimiento en el caso de la estimulación bipolar54. b) Estrategia de codificación: determina el conjunto de operaciones que se realizan con la señal de audio para generar los estímulos en los distintos electrodos del implante. Existen dos grupos de estrategias: analógicas y pulsátiles. Ambas comparten el hecho de separar la señal de audio en varias bandas de frecuencia mediante un banco de filtros. En la estrategia analógica se presenta en cada electrodo una corriente que varía de forma ininterrumpida, acorde con la señal a la salida del filtro correspondiente. Supone un procesamiento de la señal muy simple. En la estrategia pulsátil, presentando pulsos breves se pueden estimular los electrodos de forma secuencial, de modo que en cada momento sólo haya un electrodo activo. Al elegir una estrategia que nos permita establecer el número de canales activados en cada ciclo, como la ACE o la N-of-M, es conveniente activar el mayor número de ellos para presentar al paciente la mayor información posible54. c) Tasa de estimulación: es el número de pulsos por segundo que suministra cada electrodo, y se encuentra en relación directa con la resolución temporal que proporciona el implante, así como con el número de electrodos activos en cada ciclo de estimulación. Para aumentar la sensación de volumen podemos aumentar la intensidad o la duración, teniendo que recurrir a esto último cuando se alcanza el máximo de intensidad que permite el implante, pero recordando que a mayor duración de los pulsos, menor tasa de estimulación; no debiendo caer ésta nunca por debajo de los 800 a 1.000 pulsos por segundo. Para reducir la duración del ciclo de estimulación a veces conviene apagar electrodos, reduciendo así la resolución espectral, aumentando la tasa de estimulación y mejorando la resolución temporal54. d) Configuración del banco de filtros: ésta viene determinada por el rango espectral procesado por el procesador y por la distribución de los filtros en dicho rango. Cuando se dispone de un número elevado de electrodos, se puede procesar una banda espectral más ancha, pero si el número es escaso, debemos priorizar y reducir el rango espectral de forma que se mantenga una resolución espectral razonable en las frecuencias conversacionales54. 1.3.9.2.- Parámetros relacionados con el nivel de estimulación: a) Activación y desactivación de electrodos: problemas tales como la localización extracoclear de uno o varios electrodos o la estimulación del nervio facial o del vestibular por parte de alguno de ellos, tienen su solución en el apagado de los electrodos implicados53. Un problema que suele afectar más a los electrodos de las regiones basales de la cóclea es el estar alojados en zonas con una escasa supervivencia neuronal, lo que hace que tengan muy bajo rendimiento y que finalmente se opte por apagarlos54. b) Ajuste del umbral: Una vez definidos los electrodos que van a mantenerse encendidos, se procede al ajuste del umbral (threshold) que se define como la mínima estimulación que puede ser detectada por el paciente54. c) Ajuste del máximo nivel de comodidad: (nivel C), definido como la máxima estimulación que el paciente tolera sin llegar al umbral doloroso o molesto54. Ambos deben ser establecidos de forma individual para cada electrodo, pudiendo definirse para cada uno de ellos un rango dinámico eléctrico, limitado por el umbral eléctrico de estimulación (nivel T) y el máximo nivel de confort (nivel C). El establecimiento de estos valores puede realizarse mediante el método conductual clásico, que establece los valores en función de las reacciones del paciente implantado, o mediante el método neurofisiológico, que establece los valores en función de los resultados obtenidos mediante la telemetría, y más concretamente en la detección del umbral mediante el e-CAP (potencial de acción compuesto obtenido eléctricamente). En el momento del encendido del procesador, debido a la ausencia de audición previa, los pacientes suelen presentar umbrales muy altos y máximos niveles de comodidad muy bajos, por lo que el rango dinámico es muy reducido. Con el uso del implante, en las sucesivas revisiones, se verá como los umbrales descienden y los niveles C aumentan, incrementándose así el rango dinámico eléctrico y por tanto la resolución en intensidad. Tanto los niveles T como los C son datos subjetivos que nos proporciona el paciente. Existe además otra serie de datos objetivos que podemos medir, y que proporcionan información sobre la funcionalidad de los electrodos, tales como54: a) Telemetría de impedancias, que nos aporta información acerca de la integridad del implante, de las impedancias de los electrodos y la interacción eléctrica entre ellos. Este sistema de telemetría nos informa de la comunicación entre los elementos externos e internos del implante y nos permite detectar problemas tales como cortocircuitos entre electrodos o circuitos en abierto por cables cortados. b) Telemetría de respuesta neural, que permite comprobar el funcionamiento de los electrodos e identificar el potencial de acción compuesto del nervio auditivo. Para ello se utiliza un electrodo del implante que proporciona el estímulo eléctrico mientras que otro electrodo registra el potencial. c) Potenciales evocados auditivos del tronco cerebral evocados eléctricamente (PEATC-e), que exploran la actividad de la vía auditiva en el tronco cerebral en respuesta al implante. Como estímulo eléctrico se utiliza un pulso bifásico con una tasa de estimulación de entre 10 y 100 pulsos por segundo. El electrodo activo se sitúa en el vértex, el de referencia en la mastoides contralateral y el electrodo de tierra en la frente. d) Test de reflejo estapedial, que mide el reflejo del músculo estapedial como respuesta a un estímulo en el nervio auditivo. La existencia de respuesta nos indica el buen funcionamiento del implante coclear y la integridad de la vía auditiva y su conexión con los núcleos motores ipsi y contralateral del VII par, a nivel del tronco cerebral. Se puede ver durante el acto operatorio aunque se puede alterar si el paciente se encuentra muy relajado. 1.4.- IMPLANTE COCLEAR EN PACIENTES CON MALFORMACIONES DE OIDO INTERNO Los primeros criterios para el implante coclear excluían los casos de cócleas malformadas; sin embargo, en 1983 Mangabeira-Albernaz realizó un implante en un paciente cuya malformación coclear no fue descubierta hasta la revisión posquirúrgica55, provocando a partir de entonces el aumento del porcentaje de candidatos a dicha intervención26. En la década de los 80 un limitado número de estos pacientes aparecía en la literatura55, mientras que ya en los 90 algunas series cortas de implantación en cócleas malformadas fueron publicadas55. No todas las malformaciones suponen el mismo grado de contraindicación para el implante. De hecho, las podemos clasificar en contraindicaciones absolutas, malformaciones mayores y menores. 1.4.1.- Contraindicaciones absolutas a) Malformación de Michel o agenesia laberíntica. b) Aplasia coclear. c) Hipoplasia coclear muy grave. d) Anomalías del conducto auditivo interno: las que conlleven una malformación importante del nervio coclear, o incluso su ausencia, y por tanto sea imposible la transmisión del estímulo. Estos pacientes podrían ser candidatos a un implante del tronco del encéfalo, una intervención de mayor riesgo quirúrgico y peores resultados que el implante coclear. 1.4.2.- Malformaciones mayores a) Cavidad común. b) Hipoplasia coclear grave. 1.4.3.- Malformaciones menores Son más prevalentes, y aunque dificultan la cirugía, los riesgos son menores y podemos predecir unos resultados cercanos a los obtenidos en cócleas de morfología normal. En este grupo englobamos: a) Hipoplasia coclear leve. b) Alteraciones de la partición coclear. c) Alteraciones del acueducto del vestíbulo. d) Alteraciones del vestíbulo. En las malformaciones del hueso temporal, llamadas de forma imprecisa displasia de Mondini, existe una reducción del número de células de los ganglios espirales12, en el extremo apical de la cóclea. La estría vascularis suele ser normal pero en ocasiones se presenta atrófica o quística. Estas características histopatológicas de la displasia de Mondini son altamente variables, ya que el Órgano de Corti y sus elementos neurales asociados pueden ser normales o estar parcial o totalmente desorganizados12, lo que explica la gran variabilidad que presentan estos pacientes en los resultados de las pruebas auditivas pre y postimplante. En estos pacientes no hay que olvidar la posibilidad de que presenten un trayecto anómalo del nervio facial, por lo que su monitorización y búsqueda escrupulosa se hacen más importantes que nunca. Por otra parte, es conveniente el uso de antibioterapia de amplio espectro pre e intraoperatoriamente por el riesgo de meningitis, ya que es muy posible la comunicación con el LCR en este tipo de malformaciones14. 1.4.4.- Cirugía según el tipo de malformación La morbilidad global de la intervención se sitúa en torno al 5%56, siendo la mortalidad, tanto por la propia intervención, como por complicaciones postoperatorias, del 0%56. 1.4.4.1.- Malformaciones menores: Toda la intervención se desarrolla según los pasos descritos para una anatomía coclear normal, sin requerir grandes modificaciones, salvo un mayor cuidado en lo que al nervio facial se refiere. 1.4.4.2.- Malformaciones mayores: En estos casos sí será necesaria una modificación del abordaje quirúrgico convencional ya que pueden aparecer fugas de LCR así como un trayecto anómalo del nervio facial. a) Las fugas de LCR durante la cirugía del implante coclear han sido documentadas en numerosas ocasiones26. Según publicaron Hoffman y cols. 55 , las fugas de LCR aparecen en el 40-50% de los pacientes con malformaciones de oído interno. Se producen al realizar la cocleostomía y suelen deberse a una anomalía ósea del extremo lateral del CAI20. Las fugas pueden ser de diferentes calibres, desde un goteo mínimo hasta una fuga importante. El primer caso lo encontramos en las particiones incompletas, donde podemos ver un goteo de perilinfa desde la rampa timpánica en el punto de la cocleostomía pero no debemos encontrar fugas importantes de LCR que nos dificulte la inserción de la guía de electrodos11. Sin embargo, en casos de displasia coclear más grave, la lámina cribiforme que forma la base de la espira media está ausente, y el LCR y los espacios perilinfáticos están comunicados entre sí, por lo que habrá una fuga importante de LCR a través de la cocleostomía. Esto ocurre también en los casos de cavidad común. El oído interno contiene únicamente unos pocos microlitros de endolinfa y perilinfa, por lo que es lógico pensar que el líquido que emana de estas fístulas es líquido cefalorraquídeo que ha atravesado el espacio perilinfático12. Las anomalías del modiolo son claramente detectadas en la TAC en algunos casos, pero otras anomalías más sutiles requieren una exploración más concienzuda, pudiéndose demostrar un ensanchamiento del acueducto del vestíbulo como única malformación12. En cuanto al tipo de cocleostomía más apropiado en estos casos especiales, hay opiniones contradictorias en la bibliografía. Mientras que Weber y cols.20 defienden que la cocleostomía debe ser tan pequeña como se pueda, Graham y cols.20 opinan que debe ser lo mayor posible para que el material reabsorbible con el que se rellena la cavidad quirúrgica al finalizar la intervención, pueda ser introducido más fácilmente sin dañar la guía de electrodos. La salida continua de fluido desde la cocleostomía asegura que ningún líquido potencialmente infectado entra en el espacio intratecal. Cuando se produzca se deberá sellar la cocleostomía con tejidos blandos, siendo necesario en ocasiones taponar la trompa de Eustaquio, aunque existe el riesgo de que una vez que se despierte el paciente aumente la presión de LCR y venza la presión de sellado12. Una vez sellada la cocleostomía, la cabeza se coloca en posición neutra y se observa la cocleostomía durante 10 minutos. Si la fuga no cesa será necesaria la colocación de un drenaje lumbar que se mantendrá de 4 a 5 días. Las fístulas de difícil manejo requerirán una petrosectomía subtotal, cerrar la piel del CAE, taponar la trompa de Eustaquio y rellenar la cavidad mastoidea con grasa abdominal; lo que garantiza el sellado de la fístula12. Otra alternativa médica para tratar y sobre todo prevenir la salida de LCR, es la técnica hiperosmolar, que fue publicada por Natalie Loundon y cols.57, quienes investigaron métodos para disminuir la presión intracerebral durante la cirugía. Para ello se sirven de manitol y suero salino, ya que ambos son efectivos, fáciles de administrar y con escasos efectos secundarios. Los mayores riesgos que pueden presentar son como consecuencia de la deshidratación, tales como cefalea, hiperglucemia transitoria e hipotensión arterial; estando contraindicada esta terapia en pacientes con insuficiencia renal, patología cardíaca, malformaciones cerebrovasculares y hemorragias cerebrales. La monitorización consiste en la medida de la osmolaridad plasmática, que teniendo como referencia los valores normales entre 280 y 290 miliosmoles/litro (mosm/l), en estos pacientes no debe exceder de 300 mosm/l57. b) Un nervio facial con trayecto anómalo aparece en el 0,3% de los oídos con apariencia normal58 y lógicamente, la probabilidad aumenta cuanto mayor es la malformación coclear, pudiendo alcanzar desde el 16%12 hasta el 43%58. En estos casos el nervio facial típicamente discurre por debajo del proceso cocleariforme y cruza el promontorio hacia la ventana redonda, acompañándose a menudo esta dirección anómala de un estribo patológico con cruras ausentes o incompletas12. El desarrollo anómalo de las estructuras derivadas del primer y segundo arcos branquiales, incluyendo la pared ósea del canal del facial, el estribo, la apófisis estiloides y/o el conducto auditivo externo (CAE), se ha asociado a un curso anómalo de los segmentos timpánico y mastoideo del facial8. Siendo lo normal, el discurrir del nervio posterior a las estructuras derivadas del cartílago de Reichert, y dependiendo la posición de las tres porciones del nervio del correcto desarrollo de las estructuras derivadas de dicho cartílago. El receso facial es un espacio quirúrgico que alcanza su morfología definitiva antes del nacimiento, siendo aún desconocido su desarrollo en las personas con malformaciones cocleares26. En algunos niños, el nervio facial puede estar dividido y podemos por tanto, encontrar un segundo nervio localizado más anterior además del que discurre por su trayecto normal posterior al receso facial12, por lo que realizar la cocleostomía anterior a un nervio localizado más anterior de lo habitual es un auténtico reto. La teoría de la migración rostral del nervio facial fue defendida por primera vez en 1967 por Durcan y cols.8, alegando que el anormal desarrollo del estribo y de la pared ósea del canal del facial desde el cartílago de Reichert, permitía al nervio migrar anteriormente siguiendo una tendencia natural hacia su objetivo final, los músculos de la cara. En los casos de cavidad común e hipoplasia coclear, el crecimiento rostral de la cóclea es mínimo o inexistente, permitiendo al nervio migrar de forma anterior; pudiendo producirse este hecho también en los casos en los que el crecimiento coclear se produce correctamente pero retrasado en el tiempo8. Es reseñable la escasa incidencia de posición anómala del nervio facial en las malformaciones de Mondini, pudiendo encontrar la explicación en la función de barrera que desempeña la formación quística que sustituye a las espiras media y apical, que impediría la migración anterior del nervio8. Igualmente, no es frecuente encontrarla en malformaciones cocleares más sutiles como las que afectan al modiolo8. La migración anteromedial del nervio facial se podrá producir siempre y cuando exista un déficit o una anomalía del normal crecimiento rostral de la cóclea entre los 9 y los 13mm del desarrollo embriológico8. La estimulación del nervio facial es una conocida complicación definida por primera vez en 1988 por Cohen y Hoffman59, recogiendo la literatura porcentajes entre el 1 y el 14,9%60 y asumiendo que patologías como la otosclerosis, la otosífilis56, las malformaciones cocleares o la osificación coclear se relacionan con ella en un porcentaje mayor. En estos pacientes es más frecuente que ocurra al iniciar la programación del implante, requiriendo apagar de 1 a 3 electrodos en algunos estudios26 y cuya explicación puede deberse a las dehiscencias que presenta el nervio en su trayecto anómalo. En principio, las malformaciones cocleares no deben impedir alcanzar el receso facial vía transmastoidea para acceder al oído medio durante la intervención del implante coclear26; aún así, si no se puede identificar claramente el facial durante el abordaje transmastoideo tradicional, se debe retirar el yunque para alcanzar a ver mejor la porción timpánica del nervio11. Si la ventana redonda y el nervio facial no pueden ser identificados claramente, una mastoidectomía radical puede ayudar en su localización26, necesitando una reconstrucción posterior con fascia del músculo temporal para evitar que la guía de electrodos salga por el CAE. En los casos de cavidad común, una forma alternativa de entrar en la cóclea es siguiendo al resto cocleovestibular a través del canal semicircular hipoplásico12. Esta técnica evita encontrarse por completo con un trayecto del facial anómalo. McElveen y cols.61 describieron la laberintotomía transmastoidea en los casos de cavidad común para minimizar el riesgo de dañar un nervio facial aberrante y poder tener un mejor control si se produjera una fístula de LCR. Por otro lado, Kronenberg, J y cols.62 proponen un abordaje suprameatal, eliminando así la necesidad de llevar a cabo una mastoidectomía y una timpanotomía posterior. Utilizaron esta técnica en 140 pacientes sin hallar más complicaciones que con la técnica clásica, incluyendo daño del nervio facial o extrusión de la guía al CAE. Este tipo de abordaje lo consideran apropiado tanto para cócleas normales como para aquellas que presenten osificación o alguna malformación. Por lo tanto, en los casos de malformaciones cocleares es más importante que nunca realizar una TAC preoperatoria con el fin de localizar las tres porciones del nervio, así como su monitorización durante toda la intervención para evitar lesionarlo. c) La colocación del implante. Incluso en cócleas normoformadas las complicaciones más frecuentes serían el deterioro inadvertido de la guía de electrodos, el fallo en el sitio de implantación (1,3%) y la migración de los electrodos (2,48%)56. En cuanto a la inserción de la guía, en la hipoplasia coclear, los electrodos pueden quedar comprimidos debido al poco espacio existente, y en la cavidad común, el introducir la guía completamente nos puede llevar a su inserción en el conducto auditivo interno. Tucci y cols.26 describieron la inserción de la guía de electrodos de 12 a 24mm en 3 pacientes con partición incompleta, y Luntz y cols.26 documentaron que el 10% de los pacientes de su estudio tenían insertados 16 de los 22 electrodos que portaba la guía. Existen otras complicaciones leves y graves que resumimos a continuación: A) Leves: - Mareos: poco frecuente. Aparece en las primeras 24-48 horas56. - Acúfenos: pueden aparecer tanto en el oído implantado como en el contralateral56. - Complicaciones de la herida quirúrgica como hematomas, serosas, que en el caso de no resolverse pueden llevar a la retirada del implante56. B) Graves: - Herida quirúrgica: necrosis parcial o total, dehiscencia de la herida e infección postoperatoria del área quirúrgica. La necrosis cutánea debida al imán del implante puede evitarse alejando suficientemente la incisión cutánea de la zona prevista para el receptor45. - Meningitis bacteriana postoperatoria: es una grave complicación con una elevada morbimortalidad. La producida por el streptococo pneumoniae, que es el patógeno más frecuente en la meningitis bacteriana en niños a partir del periodo neonatal63, así como en la edad adulta, tiene una mortalidad del 15 al 60%. En julio de 2002, la US FDA comunicó que había recibido datos acerca de una posible relación entre los implantes cocleares y la meningitis bacteriana63. Los modelos de implantes cocleares que presentan posicionador de electrodos se asociaron a un mayor riesgo de producir meningitis, concretamente 4,5 veces más riesgo64, pensándose que podía deberse entre otros motivos a la cocleostomía de mayor tamaño necesaria para insertar la guía y el posicionador. Sin embargo, Summerfield AQ. Y cols.65, trataron de contrastar esta información en el Reino Unido estudiando a 1.851 niños (66 con posicionador) y a 1.779 adultos implantados (139 con posicionador), sin hallar diferencias estadísticamente significativas dependientes del posicionador al comparar la incidencia de meningitis. - Reimplantación: suele ser necesaria por un fallo total del equipo interno. Diversos estudios revisados por Henson y cols.56 demuestran que los resultados auditivos tras la reimplantación pueden cambiar de manera significativa, aunque suelen depender de los que se tenían con el primer implante. 1.4.5.- Tipos especiales de implantes cocleares Estudios histológicos han demostrado que en las malformaciones mayores, las estructuras neurales no se distribuyen en torno al modiolo, sino en la periferia61, por lo que es conveniente el uso de una guía no precurvada ni centrada en torno al modiolo66. Este dato también lo apoya el estudio de las estructuras nerviosas desde el punto de vista de la embriología. Es decir, estudiar en qué momento del desarrollo embrionario aparecen y qué distribución van adquiriendo conforme avanza el crecimiento del embrión y posterior feto. En las cavidades comunes con comunicación patológica con la porción lateral del CAI, los implantes con guía de electrodos precurvadas permiten controlar que no se introduzca en este último20, 58. Actualmente disponemos en el mercado de varios modelos de implante coclear satisfaciendo las necesidades de cada malformación. a) Para pacientes con cavidades comunes será necesario un modelo de guía recto que no se amolde al modiolo y se adapte a los contornos de la cavidad, donde se encuentran las estructuras nerviosas. Tal y como señalaron McElveen y cols.55, el antiguo diseño Clarion era full-banded y tenía la ventaja de que se alejaba del CAI rotando 180º (o cuando el implante derecho se ponía en el oído izquierdo). Blake C. Papsin, para los casos de cavidad común, utiliza una guía recta que empuja suavemente contra el promontorio antes de insertarla en la cóclea, con el fin de curvar la punta donde están los 3 a 5 electrodos distales, para evitar la inserción de la guía en el CAI o en el canal carotídeo12. McElveen y cols.12 describieron una técnica alternativa para insertar la guía de electrodos en las cavidades comunes sin sufrir las temidas complicaciones, que consiste en insertar la guía a través del canal semicircular lateral hipoplásico. Considerando la posible movilidad de los electrodos en la cavidad, es lógico que se produzca cierta fluctuación de los umbrales audiométricos y los máximos niveles de confort66 en las primeras semanas, por lo que al principio será necesario reprogramar los electrodos en varias ocasiones61. b) En los casos de partición incompleta tipo I (IP-I) en los que no hay septum interescalar y la cóclea es una estructura quística también estarían indicados los modelos señalados anteriormente55. c) En las cócleas hipoplásicas, una guía de electrodos comprimida es a priori la ideal para los casos de marcada hipoplasia a fin de conseguir insertar el mayor número posible de electrodos y evitar su introducción en el CAI66. d) En las particiones incompletas tipo II (IP-II), clásica malformación de Mondini, al estar respetadas la espira basal y la mitad inferior del modiolo, cualquier modelo de implante proporcionará buenos resultados al paciente55. 1.4.6.- Postoperatorio Los resultados de una inserción incompleta de la guía son por lo general menos satisfactorios, pero son lo suficientemente aceptables como para poder afirmar que estos pacientes reciben beneficio del implante coclear26. Puede realizarse un control radiográfico perioperatoriamente, para verificar la posición de la guía de electrodos antes de proceder al sellado de la cocleostomía61. La implantación coclear asistida por fluoroscopia permite visualizar la inserción de la guía de electrodos en tiempo real; correspondiéndose la imagen con la sensación táctil que experimenta el cirujano al introducir la guía67. Una vez finalizada la cirugía la proyección idónea es la radiografía de Stenvers. Esta es una proyección del eje largo del hueso temporal que se estableció para ver la ventana redonda y el canal semicircular posterior así como otras estructuras22. En otorrinolaringología, la proyección de Stenvers es muy empleada para el estudio del oído interno. Sin embargo, a pacientes con vértigo postoperatorio, puede resultarles difícil el adoptar la posición de decúbito prono necesaria para esta proyección. Por esta razón, se ha contemplado la posibilidad de usar una proyección totalmente opuesta a ella, colocándose el paciente en decúbito supino. Maruyama H y cols.68, realizaron un estudio tras el que concluyeron la ausencia de diferencias estadísticamente significativas entre la proyección de Stenvers y la opuesta en cuanto a la dosis de radiación que recibe el cristalino, concluyendo que ésta es una técnica útil. La TAC también puede estar indicada en el postoperatorio del implante. Tiene su mayor indicación en localizar la causa del fallo del implante coclear en aquellos pacientes en los que la radiografía simple en proyecciones de Stenvers y frontoorbitarias sugiere un trayecto anómalo de la guía de electrodos69, siendo las causas más frecuentes de inserción anómala la osificación del laberinto postmeningitis y la otosclerosis. Los cuidados postoperatorios que requieren estos pacientes incluyen cobertura antibiótica por el mayor riesgo de meningitis y vigilancia hospitalaria de 72 horas por la posibilidad de desarrollar una fístula de LCR58. Una infección coclear puede alcanzar el espacio subaracnoideo por múltiples vías, siendo una de ellas el acueducto coclear, que desde la escala timpánica de la espira basal de la cóclea, atraviesa el hueso temporal para desembocar por debajo de la apertura del CAI en el espacio subaracnoideo de la fosa posterior. Otras vías son el extremo lateral del CAI, los poros de las fibras del nervio coclear o una dehiscencia directa en el caso de las malformaciones63. La ruta más frecuente para una fístula de LCR en el oído medio es la ventana oval a través de una dehiscencia en la platina del estribo63. . 2.- ANTECEDENTES Y ESTADO ACTUAL DEL TEMA Numerosos estudios han relacionado los distintos grados malformativos con una determinada edad embriológica. De este modo, Jackler, en 1987 publica la primera clasificación de las malformaciones de oído interno basada en el desarrollo embriológico. Según postula, las malformaciones congénitas de oído interno se desencadenan como resultado de la detención del desarrollo embrionario de las estructuras del oído interno, causadas por una noxa ambiental antes de la octava semana de gestación. Mediante politomografia, clasifica las malformaciones en agenesias cocleares, aplasias (cavidades comunes), hipoplasias y particiones incompletas. De este modo, a menor edad gestacional de aparición de la noxa, mayor grado malformativo. Aceptados los postulados de Jackler, numerosas clasificaciones se suceden hasta la incorporación de la tomografia axial computadorizada y la resonancia magnética nuclear al diagnóstico clínico sistemático. Así, Sennaroglu y cols., Phelps, Schuknecht, y Zheng entre otros desarrollan distintas clasificaciones, basadas eso sí en los postulados iniciales de Jackler. Sin embargo, a pesar de conocerse de forma extensa las diferentes malformaciones del oído interno, tanto óseas como las que interesan a los tejidos blandos, existe aún gran desconocimiento en la distribución y grado funcional de las estructuras nerviosas que conforman el oído interno. Existen escasas citas bibliográficas referidas a la distribución que adoptan las estructuras nerviosas del oído interno en los pacientes afectados de malformación coclear. Parece lógico pensar que el hecho de existir una malformación de oído interno, ya sea ósea o membranosa, condicione la distribución de las estructuras nerviosas emplazadas en esa localización y que adopten una distribución del mismo modo anómala. Además, al igual que la noxa altera la forma coclear y la distribución de las estructuras nerviosas, la funcionalidad y maduración de éstas quedará también afectada. Estos asuntos, relacionados todos ellos con la embriología del oído interno, parecían carecer de importancia hace algunos años por ser irrelevantes en el pronóstico clínico de la hipoacusia severa y profunda, sin realizarse por ello más estudios anatómicos actualizados, dando por sentado que nada nuevo quedaba por conocer. Afortunadamente, hoy día ha vuelto a darse a esta área la importancia que merece y se ha convertido en punto de reflexión y estudio gracias al tratamiento y rehabilitación auditiva de los pacientes hipoacúsicos con implantes cocleares. Las dificultades quirúrgicas que supone una malformación de oído interno a la hora de poner un implante coclear constituyen un verdadero reto tanto para el cirujano como para el anatomista en lo que al desarrollo embriológico del oído se refiere. Conocer en profundidad en qué momento del desarrollo embrionario tiene lugar cada malformación, así como la disposición de las diferentes estructuras nerviosas, vasculares, cartilaginosas, etc., que componen el oído interno, supone una ayuda inestimable de cara a enfocar la cirugía y la rehabilitación de estos pacientes. Por tanto, hoy día la implantación coclear no sólo no está contraindicada en los casos de malformación sino que se encuentra en pleno desarrollo. Eso sí, existen dudas acerca de la mejor estrategia de estimulación a través del implante coclear pues ésta debe ejecutarse conociendo la distribución y funcionalidad de las estructuras nerviosas cocleares. Los clásicos modelos de estimulación con electrodos más o menos perimodiolares y las estrategias de estimulación tonotópica, no parecen ajustarse a los requerimientos estructurales y funcionales de la cóclea malformada, de ahí los escasos resultados funcionales registrados hasta la fecha. El mayor conocimiento de la estructura y funcionalidad neuronal, permitirá desarrollar modelos implantables más ajustados a las necesidades malformativas y obtener mejores resultados funcionales. 3.- OBJETIVOS Los objetivos del presente estudio son: 1.- Estudiar el desarrollo embriológico de las estructuras del oído interno desde el embrión de 5mm hasta el feto de 80mm. 2.- Conocer el desarrollo y la distribución de las estructuras nerviosas del oído interno desde que aparecen tempranamente en el embrión. 3.- Correlacionar el grado de malformación coclear con el grado de maduración del VIII par craneal y de disfunción auditiva. 4.- Conocer el rendimiento obtenido tras la implantación coclear en las diferentes malformaciones de oído interno. 4.- PARTE EXPERIMENTAL 4.1.- ESTUDIO EMBRIOLÓGICO DEL OÍDO INTERNO: 4.1.1.- Objetivos y metodología Con el fin de conocer la evolución de las diferentes estructuras del oído interno, desde su primera aparición hasta la adquisición de su aspecto casi definitivo, hemos realizado una revisión de cortes de embriones y fetos pertenecientes al Departamento de Anatomía y Embriología Humana de la Facultad de Medicina de la Universidad de Granada. Este Departamento dispone de una amplia muestra de los cortes referidos, conseguidos a partir de abortos naturales, tintados con hematoxilinaeosina, y clasificados por tamaños, constituyendo un material de gran valor para el estudio del desarrollo embrionario en general, y del oído interno en nuestro caso. El fin de este estudio embriológico es triple: 1) conocer de primera mano la embriogénesis del oído interno, prestando especial atención a las estructuras nerviosas, con el objetivo de conocer en parte las causas de la mala funcionalidad auditiva de los pacientes con malformaciones cocleares mayores que reciben un implante coclear; 2) tratar de relacionar cada malformación coclear descrita por Jackler con la edad gestacional en la que se detiene el desarrollo; y 3) hacer un estudio comparativo entre las imágenes embriológicas de la cóclea a dichas edades y las obtenidas por TAC en cada caso de malformación. Con un microscopio óptico Leyca, y el programa informático IM50, que permite capturar las imágenes que están siendo vistas por el microscopio, hemos estudiado estos embriones desde los más prematuros en la edad gestacional (5mm) hasta fetos de 80mm, observando los cambios que experimentan las distintas estructuras del oído interno desde su primer esbozo hasta la adquisición de su forma, tamaño y disposición similares a las del adulto, estudiando también las relaciones entre las mismas. Gracias a los diferentes aumentos de que dispone el microscopio óptico, hemos podido obtener una gran variedad de imágenes, tanto panorámicas como de detalles, que muestran de forma clara las diferentes estructuras del oído interno, y que sin duda presentan gran interés. Para el estudio de dichos embriones y fetos nos basamos en los Estadios de Canergie70 para el periodo embrionario precoz (de la primera a la octava semana): Tabla 9.- Estadios de Canergie70. ESTADIO LONGITUD EDAD (días) CANERGIE VERTEX-COCCIX (mm) 1 día 1 0,1mm 2-3 días 2 0,1mm 4 días 3 0,1mm 5-6 días 4 0,1mm 7-12 días 5 0,1-0,2mm 13-15 días 6 0,2-0,3mm 16 días 7 0,4mm 18 días 8 1-1,5mm 20 días 9 1,5-2,5mm 22 días 10 2-3,5mm 24 días 11 2,5-4,5mm 26 días 12 3-5mm 28 días 13 4-6mm 32 días 14 5-7mm 33 días 15 7-9mm 37 días 16 8-11mm 41 días 17 11-14mm 44 días 18 13-17mm 48 días 19 16-18mm Conocer las medidas de los embriones en las primeras etapas del desarrollo resulta relativamente fácil, complicándose la labor cuando los embriones empiezan a mostrar una marcada curvatura del cuerpo. De forma general, la dimensión más corrientemente empleada es la distancia vértex- coccix (C-R), y sirviéndonos de ésta podemos hacer la siguiente clasificación para el periodo fetal71: Tabla 1071.- Periodo fetal. EDAD LONGITUD C-R EDAD LONGITUD C-R (SEMANAS) (mm) (SEMANAS) (mm) 9 39-41mm 19 173mm 10 51-53mm 20 185mm 11 64-66mm 21 197mm 12 77-79mm 22 208mm 13 91-93mm 23 219mm 14 105-107mm 24 230mm 15 119-121mm 28 270mm 16 132-134mm 32 310mm 17 147mm 36 346mm 18 160mm 38 362mm De forma sistemática, comenzando por el embrión de menor tamaño (5mm), hemos visualizado al microscopio la totalidad de los cortes seriados correspondientes a la región cefálica, pudiendo así estudiar las estructuras del oído interno desde su más temprana aparición hasta su configuración casi definitiva. 4.1.2.- Resultados 4.1.2.1.- Embrión de 5mm de longitud: Anterior Posterior 1 4 2 3 5 1 Figura 8.- Embrión de 5mm (2,5x). (1.- Vesículas óticas; 2.- pared rombencefálica; 3.- mesénquima; 4.-rombencéfalo; 5.- médula espinal.) La figura 8 corresponde a un corte de un embrión humano de 5mm de longitud, teñido con hematoxilina-eosina, en el que se pueden observar las vesículas óticas u otocistos, estructuras que aparecen entre los días 23-24 de edad gestacional, en la región más anterior del embrión, a partir de la placoda ótica que aparece sobre el día 22. Como vemos, quedan separadas de la pared rombencefálica, y rodeadas por el mesénquima circundante. La placoda auditiva, y posterior vesícula ótica, es la estructura a partir de la cual se desarrolla el oído interno. Cualquier noxa que interfiera en el desarrollo gestacional antes del día 21, y por tanto impida la aparición de la placoda auditiva, daría lugar a la malformación más grave de oído, que es la ausencia total de oído interno, conocida como síndrome de Michel o aplasia de oído interno (figura 9). Figura 9.- TAC de aplasia oído interno. La figura 9 pertenece a la TAC de una paciente con Síndrome de Michel o aplasia de oído interno. Puede observarse la cadena de huesecillos, y la ausencia de cualquier estructura perteneciente al oído interno. Volviendo a la figura 8, dicho embrión de 5mm se encuentra en un estadio 12-14 según la clasificación de Canergie 83 , lo que supondría una edad gestacional de entre 26 y 34 días (entre 4 y 5 semanas). Si el desarrollo embrionario se detuviera en este momento de la vida embrionaria, el resultado sería una malformación de tipo cavidad común, más precoz, en la que la pérdida total de todo el septo interescalar hace que no exista separación entre la cóclea y el vestíbulo, que forman conjuntamente una única cavidad, o bien a una aplasia coclear, más tardía, en la que hay ausencia de cóclea, pudiendo ser el vestíbulo y los canales semicirculares normales o no. 4.1.2.2.- Embrión de 6mm de longitud Anterior Posterior 4 3 2 1 2 4 1 Figura 10.- Embrión de 6mm. (2,5 x). (1.- vesícula ótica; 2.- ganglio estatoacústico; 3.- ganglio de Gasser; 4.- vesículas telencefálicas) En la figura 10 podemos observar ambas vesículas auditivas con sus respectivos ganglios estatoacústicos, que ya son visibles en posición ventral. Recordemos que dichos ganglios se originan a partir de células que migran desde la cara anterior de las vesículas óticas. A edades tan tempranas no existe aún diferenciación entre ganglio de Corti y ganglio de Scarpa, por lo que recibe el nombre genérico de ganglio estatoacústico. Sirva de referencia el ganglio de Gasser del V par craneal, de gran tamaño y en posición ventral. 2 3 1 * Figura 11.- Embrión de 6mm (40x). (1.- Vesícula ótica; 2.- tejido ganglionar; 3.tejido nervioso) La figura 11 corresponde al mismo embrión y muestra con detalle el epitelio de la vesícula ótica y su íntima relación tanto con el ganglio estatoacústico como con las fibras nerviosas, características estas últimas por su aspecto ondulado. Podemos ver claramente como las fibras nerviosas, de un color más claro (*), penetran en la vesícula ótica por su cara anteromedial. Es importante resaltar que a edades tan tempranas (6mm) ya son visibles las fibras nerviosas que en un futuro conformarán los nervios del oído interno, tanto el facial como el coclear y el vestibular; y más importante aún es que ya se observan contactando con el epitelio de la vesícula ótica, justificando así la respuesta auditiva, aunque pobre, que los pacientes con importantes malformaciones cocleares tienen al recibir un implante coclear. Sin embargo, el hecho de que esas fibras nerviosas se encuentren tan localizadas, alcanzando únicamente la región anteromedial de la vesícula ótica, y quedando por tanto la mayoría de dicha estructura sin inervación, hace que ese aprovechamiento del implante coclear, aunque existente, sea muy pobre, ya que los electrodos distan mucho de las células nerviosas a las que deben llevar el estímulo eléctrico, requiriendo niveles de carga eléctrica inviables. 4.1.2.3.- Embrión de 10mm de longitud Posterior 1 1 2 * 2 * 3 3 Anterior Figura 12.- Embrión de 10mm (5x) (1.-vesícula ótica; 2.- ganglio estatoacústico; 3.- ganglio de Gasser, *.- fibras nerviosas) En la figura 12 de nuevo podemos observar cómo las fibras nerviosas procedentes del ganglio estatoacústico entran en contacto directo con el epitelio de la región anterior del otocisto, pudiendo diferenciar con facilidad las fibras nerviosas, de un color más claro, entre el tejido ganglionar. En estos estadios existe la posibilidad de que fibras del nervio facial se entremezclen con las procedentes del ganglio estatoacústico. Figura 13.- Embrión de 10mm (20x). La figura 13 muestra con más detalle la única zona de la vesícula ótica que en este estadio está en contacto con las fibras nerviosas, la región más anterior. Figura 14.- Embrión de 10mm (40x) La figura 14 muestra a mayor aumento la proximidad existente entre las células nerviosas y el epitelio de la vesícula ótica, confirmándose lo que ya describíamos en la figura 11 perteneciente a un embrión de 6mm, que ya a edades tan tempranas en el desarrollo embrionario existe contacto entre ambas estructuras. Figura 15.- TAC de cavidad común. Tratando de nuevo de correlacionar la histología y la radiología, mostramos estas dos imágenes de TAC (figura 15) pertenecientes a un paciente con malformación tipo cavidad común, en la que la cóclea y el vestíbulo forman una misma estructura, sin que exista ninguna separación entre ambas. Esta malformación, según la clasificación de Jackler, resulta de la detención del desarrollo embrionario antes de la semana cuarta. Es considerada una malformación mayor y sus importantes diferencias con la anatomía coclear normal, hacen suponer una gran aleatoriedad en la distribución de las estructuras nerviosas, lo que justificaría que en estos casos, los resultados tras el implante coclear sean muy pobres. Vemos la gran similitud de esta estructura con la correspondiente al oído interno vista hasta ahora en los cortes embriológicos. Es una malformación mayor originada por una detención precoz en el desarrollo embrionario. 4.1.2.4.- Embrión de 19mm de longitud 2 4 1 3 Figura 16.- Embrión de 19mm (5x). (1.- Utrículo; 2.- canales semicirculares; 3.ganglio estatoacústico; 4.- ganglio de Gasser.) La figura 16 muestra como las estructuras del oído interno comienzan a diferenciarse. En esta fase y en este corte alto, ya podemos observar el esbozo del utrículo y canales semicirculares cortados. Persiste la vecindad con el ganglio estatoacústico y la referencia anatómica del ganglio de Gasser. Posterior Anterior 8 6 4 1 5 9 3 10 2 7 5 6 4 1 Figura 17.- Embrión de 19mm (2,5x). (1.- Ventrículos laterales; 2.- tercer ventrículo; 3.- adenohipófisis; 4.-G. Gasser; 5.- conducto coclear; 6.- cápsula ótica; 7.- plexos coroideos; 8.- nervio facial; 9.- médula espinal; 10.- atlas.) La figura 17 pertenece al mismo embrión de 19mm pero en un corte más caudal. Es una imagen panorámica en la que podemos observar la práctica totalidad del embrión, destacando según nuestro interés, la presencia de ambos conductos cocleares a ambos lados de la línea media, que corresponderían a sendas espiras basales de la cóclea. Según la clasificación de Canergie83, este embrión sería un estadio 19, con una edad gestacional aproximada de 48 días, entre 6 y 7 semanas, por lo que una detención del desarrollo embrionario en este momento podría dar lugar a una hipoplasia coclear, más grave, o bien a una malformación de tipo partición incompleta, más leve, en la que las espiras media y apical forman una única cavidad quística, sin separación entre ambas. Cóclea hipoplásica Cóclea hipoplásica Figura 18.- TAC de hipoplasia coclear. Tal y como mencionábamos antes, la figura 18 consta de dos imágenes de TAC a distintos aumentos de un paciente con hipoplasia coclear. Podemos ver como la cóclea no presenta sus 2 vueltas y media de espira características sino que es de menor tamaño y forma anómala. Existe gran similitud con la imagen del conducto coclear que veíamos en la figura 17. 4.1.2.5. – Embrión de 20mm de longitud: Figura 19.- Embrión de 20mm (10x). (1.- Utrículo; 2.- canales semicirculares; 3.- ganglio de Gasser; 4.- acúmulo de fibras del nervio y ganglio estatoacústico y del nervio facial). Figura 20.- Embrión de 20mm (20x). * Figura 21.- Embrión de 20mm (20x). Las figuras 20 y 21, pertenecientes al mismo embrión de 20mm, muestran cómo las fibras nerviosas (*) procedentes del ganglio estatoacústico contactan con un engrosamiento del epitelio que dará lugar a la mácula utricular. 4.1.2.6.- Embrión de 28mm de longitud Posterior 3 4 6 Medial Lateral 2 1 7 8 Anterior Figura 22.- Embrión de 28mm (20x). (1.- Nervio facial; 2.- Fibras vestibulares; 3.- CAI; 4.-Conducto endolinfático; 5.- G. Gasser; 6.- Utrículo; 7.- Mácula sacular; 8.- Prolongación sacular) 5 En la figura 22 se aprecian con gran claridad una parte importante de las estructuras nerviosas del oído interno. El conducto endolinfático aparece como una prolongación que parte del utrículo en posición dorsomedial. Es la estructura más medial en los cortes altos del oído interno, lo que la hace fácilmente identificable. Debido a que vemos el conducto endolinfático (4), sabemos que se trata de un corte alto, por lo que recordando la disposición de los nervios en el conducto auditivo interno (CAI), debemos pensar que el nervio que queda más anterior es el nervio facial mientras que el posterior son fibras vestibulares uniéndose en el CAI. Llama también la atención el engrosamiento que presenta el epitelio sensorial en las zonas próximas al contacto con las fibras nerviosas. Una vez más nos servimos del ganglio de Gasser como referencia espacial. Medial Lateral Figura 23.- Embrión de 28mm (10x). 1 Figura 24.- Embrión de 28mm (20x). (Detalle de figura 23) (1.- Fibras del nervio coclear). G.C Figura 25.- Embrión de 28mm (40x). (Detalle de figura 24). (G.C: Ganglio de Corti). La figura 25 muestra la porción del Ganglio de Corti (G.C) que corresponde a la espira basal de la cóclea. 6 7 8 2 1 3 5 4 8 Figura 26.- Embrión de 28mm (5x). (1.-Nervio facial; 2.-martillo; 3.yunque; 4.- estribo; 5.- pabellón auricular; 6.- ganglio de Gasser; 7.- Ganglio de Corti; 8.- cóclea) La figura 26 es una panorámica donde se aprecia la relación de la cóclea con otras estructuras como son el nervio facial, el martillo, el yunque, el estribo, el pabellón auricular y el ganglio de Gasser. Vemos el ganglio de Corti. También podemos apreciar la típica imagen en “helado de cucurucho” de la articulación incudomaleolar del adulto. 4.1.2.7.- Feto de 31mm de longitud 6 7 Figura 27.- Feto de 31mm. (1.- Recesos laterales del cuarto ventrículo; 2.canales semicirculares; *.- desembocadura del conducto endolinfático; 3.- CAI, 4.- fibras vestibulosaculares; 5.- vestíbulo; 6.- cóclea; 7.- Ganglio de Corti). Las cuatro imágenes que conforman la figura 27 corresponden a un feto humano de 31mm de longitud, en cortes sucesivamente más caudales. La número 1 es una panorámica (2,5x) en un corte alto, de la que extraemos la número 2 (5x) y en la que distinguimos claramente la desembocadura del conducto endolinfático (*), los recesos laterales del cuarto ventrículo (1) y los canales semicirculares cortados (2). En la número 3 (5x), que es un corte más bajo, distinguimos el conducto endolinfático (*), el conducto auditivo interno (3), fibras vestibulosaculares (4) y el vestíbulo (5). Y por último, en la número 4 (10x), la más caudal de todas, se ha cortado cóclea apreciándose únicamente dos espiras (6) con sus correspondientes fracciones del ganglio espiral de Corti (7). Esta última imagen, la número 4, correspondería a la cóclea de un paciente con malformación tipo partición incompleta, en la cual las espiras media y apical no han terminado de desarrollarse por lo que el caracol consta únicamente de una vuelta y media de espira. Figura 28.- TAC de malformación tipo partición incompleta. Al igual que hemos hecho con las otras malformaciones, mostramos estas tres imágenes (figura 28) correspondientes a cortes sucesivamente más caudales en la TAC de un paciente con partición incompleta. Véase la división coclear en sólo 2 espiras, la basal (1) y la apical (2), con aspecto ésta última de cavidad quística como resultado de la falta de separación entre la espira media y la apical propiamente dicha. Es la malformación coclear más leve de todas cuantas existen, pudiendo tener algunos de estos pacientes una funcionalidad auditiva muy similar a las personas con cócleas normales. 4.1.2.8. – Feto de 45mm de longitud Figura 29.- Feto de 45mm. (5x). (1.- Nervio facial; 2.- cóclea, 3.estribo; 4.- ganglio geniculado). En la figura 29 se nos muestra el nervio facial (1) en gran parte de su recorrido, viendo sus dos codos con el ganglio geniculado (4) en el primero de ellos y su relación con la cóclea (2) y el estribo (3). Figura 30.- Feto de 45mm. (5x). (1.- espira basal; 2.-espira media; 3.espira apical). La figura 30 es otra sección más caudal del mismo feto de 45mm, en la que pueden apreciarse claramente las espiras basal (1), media (2) y apical (3) de la cóclea. Véase asimismo como la espira basal empieza a presentar una división más o menos clara de sus rampas mientras que las espiras media y apical se encuentran sin segmentar. Cierto es que llegados a este punto, excedemos el periodo embrionario en el que Jackler describió las malformaciones cocleares, por lo que cualquier detención del desarrollo a partir de esta edad gestacional no debería interferir en el correcto desarrollo de la cóclea. Sin embargo, y con el fin de seguir el desarrollo coclear hasta el final, continuamos nuestro estudio con fetos de mayor tamaño. 4.1.2.9. – Feto de 52mm de longitud 4 6 1 2 * 5 3 Figura 31.- Feto de 52mm (5x). (1.- Utrículo; 2.- sáculo; 3.- canal semicircular; 4.-conducto endolinfático; 5.-futuro espacio perilinfático; 6.- cresta ampular). La figura 31 es un corte alto en el que podemos observar la región posterior que corresponde al utrículo (1) a nivel de la desembocadura de dos conductos semicirculares (véase crestas (6) y engrosamiento epitelial), la anterior correspondiente al sáculo (2), un canal semicircular (3) y el conducto endolinfático (4). La zona que rodea al laberinto membranoso es el futuro espacio perilinfático (5) antes de reabsorberse el mesénquima. A B Figura 32.- Feto de 52mm (20x). La imagen A de la figura 32 corresponde a una mácula sacular o utricular, mientras que la imagen B muestra una cresta ampular de un conducto semicircular como la que apreciábamos en la figura 31. Figura 33.- Feto de 52mm (5x) La figura 33 consta de tres imágenes de la cóclea del mismo feto en cortes sucesivamente más caudales. Véase como van perdiéndose las espiras hasta cortar casi únicamente cápsula ótica. Figura 34.- Feto de 52mm (5x). Figura 35.- Feto de 52mm. (10x) (1.- Escala vestibular; 2.- Escala timpánica; 3.- Conducto coclear) 4.1.2.10. – Feto de 70mm de longitud Figura 36.- Feto de 70mm (2,5x). En la figura 36 mostramos una imagen panorámica de un feto de 70mm en el que la cóclea ya está perfectamente formada, con sus dos vueltas y media de espira características y su cápsula ótica cartilaginosa próxima a osificarse. 4.1.2.11. – Feto de 80mm de longitud Figura 37.- Feto de 80mm (5x) En la figura 37 volvemos a ver las tres vueltas de espira de la cóclea, con sus rampas claramente diferenciadas y el tejido cartilaginoso de la cápsula ótica más compacto previo a la osificación. Figura 38.- Feto de 80mm (2,5x) G.G.Corti Corti Figura 39.- Feto de 80mm (10x). (Detalle de figura 38). La figura 39 muestra con un mayor aumento el tejido ganglionar presente en la cara medial del conducto coclear de la espira basal de la cóclea: el ganglio espiral de Corti (G. Corti). Figura 40.- Feto de 80mm (20x) Para finalizar, en la figura 40 volvemos a ver las tres escalas de la coclea: rampa vestibular (R.V), rampa timpánica (R.T) y conducto coclear (C. C), en cuyo suelo se aprecia un engrosamiento celular correspondiente al Órgano de Corti en desarrollo. Véase la membrana tectoria (M. T.) y la lámina espiral (L. E). 4.2.- ESTUDIO ESTADÍSTICO DE PACIENTES IMPLANTADOS: 4.2.1.- Tipo de diseño Hemos diseñado un estudio observacional prospectivo de cohortes con un periodo de seguimiento de 24 meses. 4.2.2.- Muestra 4.2.2.1.- Población de referencia Pacientes hipoacúsicos profundos diagnosticados y tratados en la Unidad de Implantes Cocleares del Hospital Universitario San Cecilio de Granada. 4.2.2.2.- Criterios de inclusión en el estudio Pacientes hipoacúsicos profundos afectos de malformaciones de oído interno y tratados mediante la implantación coclear antes de los 6 años de edad. 4.2.2.3.- Selección de la muestra a estudio La inclusión de pacientes en el estudio se realizó previa comprobación del cumplimiento de los criterios de inclusión. La fecha de inicio en la inclusión del estudio fue enero 2003. 4.2.2.4.- Muestra del estudio La muestra consta de 16 pacientes que cumplen los criterios de inclusión antes descritos y 32 pacientes, igualmente afectos de hipoacusia profunda y tratados mediante implantes cocleares, pero con ausencia de malformación de oído interno. Este segundo grupo se ha denominado grupo de control. 4.2.3.- Método estadístico Todas las variables cuantitativas se expresan como media (x) y desviación estándar (DS). Al tratarse de una muestra con tamaño muestral menor de 20 pacientes y no poder asegurar la normalidad de las variables utilizamos test no paramétricos. Así en la comparación de medias hemos utilizado el test U de Mann Whitney cuando existían variables con 2 categorías y el test de Kruskal-Wallis si existían más de 2 categorías. Las variables cualitativas fueron igualmente tratadas con los mismos tests, siendo previamente sometidas a las recodificaciones oportunas. Se consideró como significación estadística un valor de “p” menor del 5%. 4.2.4.- Variables clínicas del estudio 4.2.4.1.- Edad y sexo El grupo de los casos está formado por 16 pacientes, 11 niños y 5 niñas, con una edad media en el momento del implante de 52,25 meses (D.S 33,63 meses). Tabla 11 El grupo de control está formado por 32 pacientes, 20 niños y 12 niñas, con una edad media en el momento del implante de 38,55 meses (D.S 15,86 meses). Tabla 12. Tabla 11.- Datos epidemiológicos (sexo y edad al implante coclear), grado de hipoacusia (dB), antecedentes de interés, tipo de malformación y modelo de implante utilizado en los pacientes afectos de malformación de oído interno. (V: varón; M: mujer). GRADO EDAD PACIENTE SEXO ANTECEDENTES (MESES) (dB) MODELO MALFORMACION Hipoacusia PERSONALES FAMILIARES IMPLANTE 1 V 16,23 110 SI SI Cavidad común COMBI 2 M 40,77 110 NO NO Cavidad común PULSAR 3 V 72,15 90 NO SI Cavidad común COMBI 4 M 29,72 110 SI NO Hipoplasia PULSAR 5 V 25,55 90 NO NO Partición incompleta COMBI 6 M 51,08 95 NO SI Partición incompleta COMBI 7 V 38, 12 85 SI SI Partición incompleta COMBI 8 V 53,85 100 SI SI Malform. vestíbulo COMBI 9 V 112,03 120 NO NO Malform. vestíbulo COMBI 10 V 143,45 95 SI NO Malform. vestíbulo COMBI 11 V 24,88 90 SI NO Malform. vestíbulo PULSAR 12 M 45,97 100 NO NO Malform. vestíbulo COMBI 13 M 54,37 110 NO NO Hipoplasia PULSAR 14 V 22,28 100 NO NO Malform. vestíbulo SONATA 15 V 49,63 100 NO SI Malform. vestíbulo SONATA 16 V 55,92 110 NO SI Partición incompleta PULSAR Tabla 12.- Datos epidemiológicos (sexo y edad al implante coclear), grado de hipoacusia, antecedentes de interés y modelo de implante utilizado en los pacientes no afectos de malformación de oído interno. (V: varón; M: mujer). ANTECEDENTES PACIENTE EDAD GRADO (MESES) Hipoacusia MODELO SEXO PERSONALES FAMILIARES IMPLANTE (dB) 1 V 29,87 120 NO NO COMBI 2 V 68,97 120 NO NO COMBI 3 V 43, 63 110 NO NO PULSAR 4 V 29,83 120 NO NO PULSAR 5 V 24,67 120 NO NO PULSAR 6 M 35,60 100 NO NO COMBI 7 V 35,70 110 NO NO PULSAR 8 M 30,28 100 NO NO PULSAR 9 V 20,03 100 NO NO COMBI 10 M 66,37 120 NO NO PULSAR 11 V 45,40 100 NO NO COMBI 12 M 23,67 120 NO NO COMBI 13 M 40,20 100 SI NO PULSAR 14 V 33,83 100 NO SI PULSAR 15 V 22,97 125 NO NO COMBI 16 V 65,77 120 NO NO PULSAR 17 V 31,87 120 NO SI COMBI 18 V 37,83 120 NO NO PULSAR 19 M 45,20 100 NO NO COMBI 20 M 21,03 110 NO NO COMBI 21 V 66,97 125 SI NO PULSAR 22 M 39,63 100 NO NO COMBI 23 V 31,60 100 NO NO COMBI 24 M 21,67 120 NO NO PULSAR 25 M 33,70 120 NO NO PULSAR 26 V 29,28 120 NO NO COMBI 27 V 68,37 100 NO NO PULSAR 28 V 43,40 110 NO NO PULSAR 29 M 24,67 120 NO NO COMBI 30 M 32,83 100 NO NO PULSAR 31 V 29,97 100 NO NO PULSAR 32 V 58,77 120 NO NO PULSAR 4.2.4.2.- Grado de hipoacusia El umbral auditivo estudiado mediante potenciales evocados auditivos del tronco cerebral, muestran un umbral medio de 100,94 dB y una desviación típica de 9,87 para el grupo de malformaciones de oído interno (Mínimo 85 dB y máximo 120 dB). Por otro lado, el umbral medio y la desviación típica fueron de 111,56 dB y 9,95 respectivamente (mínimo 100dB y máximo 125 dB) para el grupo de control. 4.2.4.3.- Antecedentes personales Se recopilaron todos los datos acerca de enfermedades e intervenciones que tuvieran los pacientes independientemente del problema auditivo, así como las posibles incidencias que se desarrollaran en el embarazo y el parto, y que pudieran de una forma u otra, haber colaborado en la instauración de la hipoacusia. De los pacientes afectos de malformación, 6 pacientes (37,5%) presentaron antecedentes de interés, careciendo de ellos los otros 10 (62,5%), tabla 11. El tipo de antecedentes personales de los pacientes con malformación se describen en la tabla 13. En el grupo de control, 30 pacientes carecen de antecedentes personales (93,75%), presentando dichos antecedentes únicamente 2 pacientes (6,25%), tabla 12. Los antecedentes personales de los pacientes sin malformación se detallan en la tabla 14. Tabla 13.- Antecedentes personales de los pacientes afectos de malformación de oído interno. Se muestra la frecuencia y el porcentaje dentro del grupo. Antecedentes Personales Frecuencia Porcentaje CMV al 5º mes de embarazo 1 6,3 Cardiopatía congénita 1 6,3 Hiperglucemia en el embarazo 1 6,3 Infección perinatal 1 6,3 Parto instrumental 1 6,3 Salmonelosis materna embarazo 1 6,3 Sin interés 10 62,5 Tabla 14.- Antecedentes personales de los pacientes sin malformación. Se muestra la frecuencia y el porcentaje dentro del grupo. Antecedentes personales Frecuencia Porcentaje Retraso psicomotor 2 6,25 Sin interés 30 93,75 4.2.4.4.- Antecedentes familiares Siete pacientes (43,75%) de los afectos de malformación presentaron antecedentes familiares de hipoacusia, tabla 11, mientras que en el grupo control únicamente 2 pacientes (6,3%) presentaron antecedentes familiares de hipoacusia, tabla 12. 4.2.4.5.- Tipo de malformación de oído interno Tras la realización de las pruebas de imagen (TAC) se clasificaron los pacientes según el tipo de malformación: 1) cavidad común (3 casos, 18,75%); 2) hipoplasia coclear (2 casos, 12,5%); 3) partición incompleta (4 casos, 25%); 4) dilataciones del acueducto del vestíbulo (3 casos, 18,75%): y 5) vestíbulo ensanchado (4 casos, 25%). A la hora del análisis estadístico, estos dos últimos grupos se han considerado como uno solo bajo el nombre de malformaciones del vestíbulo, al no tratarse de una malformación coclear propiamente dicha. Por tanto, constaría de 7 casos (43,75%). Tabla 11. 4.2.4.6.- Modelo de implante coclear Los implantes utilizados fueron Med-El Combi 40+ (9 casos, 56,25%), Med-El PulsarCI100 (5 casos, 31,25%) y Med-El SonataTI100 (2 casos, 12,5%) en el grupo de pacientes con malformación y 14 casos de Med-El Combi 40+ (43,7%) y 18 (56,3%) de Med-El PulsarCI100 en el grupo de control. 4.2.4.7.- Parámetros de estimulación eléctrica mediante implante coclear. -Umbral (THR): es la mínima intensidad de estímulo eléctrico necesaria para provocar en el paciente una sensación auditiva. Su unidad de medida es el Culombio (Cu). -Máximo nivel de comodidad (MCL): es la máxima intensidad de estímulo eléctrico que el paciente es capaz de percibir sin que le resulte molesto o doloroso. Su unidad de medida es el Culombio (Cu). La diferencia entre el umbral y el máximo nivel de confort eléctricos se denomina rango eléctrico de estimulación. -Duración del pulso: es el tiempo que dura el pulso de estímulo eléctrico. Se mide en microsegundos (µseg.). -Carga eléctrica: es la cantidad total de energía liberada durante el pulso necesaria para desencadenar una respuesta auditiva en el paciente. Resulta del producto entre THR y duración. Su unidad es el Culombio x µseg. -Número de electrodos activos: es el número útil de electrodos para estimular eléctricamente el sistema auditivo del paciente. 4.2.4.8.- Datos de habilidades auditivas -LIP Profile (Listening Progress Profile): creado por Archbold, S en 1993 para el programa de implantes cocleares pediátricos en Nottingham (Inglaterra), con el propósito de valorar la percepción auditiva de los sonidos, del lenguaje y desarrollo de las capacidades auditivas en niños portadores de un implante coclear. Puede aplicarse a niños de todas las edades, y se sirve de unas series de dibujos con dos alternativas, dos hojas de respuestas e instrucciones72. -MTP (Monosyllabic-Trochee-Polysyllabic word test): creado por Norman Rever en 1978 con la finalidad de demostrar la habilidad para identificar diferentes patrones silábicos variando de una sílaba a dos sílabas con diferente acentuación y a más de dos sílabas. Se persigue demostrar la habilidad de identificar palabras con el patrón silábico correcto. Puede aplicarse a pacientes a partir de los dos años de edad, constando el test de 3, 6 ó 12 dibujos subtitulados, 3 objetos y 1 distractor. El niño debe ser capaz de señalizar o repetir claramente el dibujo subtitulado (u objeto). El MTP está diseñado para que el niño señale los objetos, no obstante los niños mayores pueden preferir simplemente repetir la palabra72. Se han recogido los resultados obtenidos al pasar estos tests al paciente en el momento previo a la cirugía, al mes, a los 3 meses, a los 6 meses, a los doce, a los dieciocho y a los veinticuatro. 4.2.5.- Resultados 4.2.5.1.-Estudio de la comparabilidad de las muestras Antes de realizar la descripción de los resultados y de las inferencias se procede al estudio de la comparabilidad de ambos grupos con el fin de detectar las diferencias epidemiológicas inherentes que puedan condicionar las conclusiones del presente estudio. Los grupos de comparación son: 1) pacientes hipoacúsicos profundos afectos de malformación de oído interno y tratados mediante la implantación coclear; y 2) pacientes hipoacúsicos profundos no afectos de malformación de oído interno y tratados mediante la implantación coclear. Los dos grupos a estudio no presentan diferencias estadísticamente significativas en cuanto a la edad de implantación, sexo y tipo de implante utilizado (U de Mann Whitney y Kruskal Wallis; p>0,05). Por otro lado, ambos grupos presentan diferencias estadísticamente significativas en cuanto a los antecedentes personales y familiares (U de Mann Whitney; p<0,05) siendo estos antecedentes más frecuentes en los pacientes con malformación de oído interno. Además, ambos grupos presentan diferencias estadísticamente significativas respecto al grado de hipoacusia, siendo este mayor en los pacientes no afectos de malformación (U de Mann Whitney; p<0,05). 4.2.5.2.- Estudio de diferencias en los parámetros de programación A continuación se detallan los datos obtenidos en la programación del implante coclear en cada paciente con malformación de oído interno (tablas 15 a 30). Con el fin de facilitar la representación de los electrodos inactivos, estos quedan representados con valores de umbral eléctrico (THR) mínimo (2,40 Cu), un Máximo Nivel de Confort (1167 Cu) y una duración del estímulo (65µseg.), de tal manera que la carga sea mínima (156 Cu x µseg.). Estos electrodos inactivos no son contabilizados para el cálculo de los valores promedio del umbral, máximo nivel de confort o carga eléctrica. Tabla 15.- Paciente 1. THR MCL DURACION CARGA (Cu) (Cu) (µseg.) (Cu x µseg) 1 2,40 1167,00 65,00 2 2,40 1167,00 65,00 3 2,40 1167,00 65,00 4 352,80 1276,00 216,70 5 298,50 1235,00 223,30 6 257,80 1208,00 226,70 7 2,40 1167,00 65,00 8 2,40 1167,00 65,00 9 2,40 1167,00 65,00 10 2,40 1167,00 65,00 11 2,40 1167,00 65,00 12 2,40 1167,00 65,00 MEDIA 303,03 1239,66 222,23 DS 47,66 34,23 5,08 Nº electrodo 156,00 156,00 156,00 76.451,76 66.655,05 58.443,26 156,00 156,00 156,00 156,00 156,00 156,00 67.183,36 9.015,87 Tabla 16.- Paciente 2. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 396,90 907,10 54,17 21.500,07 2 415,70 944,90 55,83 23.208,53 3 396,90 944,90 52,50 20.837,25 4 415,70 944,90 54,17 22.518,47 5 415,70 944,90 55,83 23.208,53 6 491,30 944,90 54,17 26.613,72 7 453,50 944,90 52,50 23.808,75 8 415,70 944,90 65,83 27.365,53 9 415,70 944,90 62,50 25.981,25 10 472,40 944,90 55,83 26.374,09 11 2,40 1.167,00 65,00 156,00 12 2,40 1.167,00 65,00 156,00 MEDIA 428,95 941,12 56,33 24.141,62 DS 32,16 11,95 4,37 2.291,44 Nº electrodo Tabla 17.- Paciente 3. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 2,40 1.167,00 65,00 156,00 2 271,40 1.099,00 90,00 24.426,00 3 176,40 1.208,0 136,70 24.113,88 4 407,10 1.208,00 141,70 57.686,07 5 2,40 1.167,00 65,00 156,00 6 2,40 1.167,00 65,00 156,00 7 2,40 1.167,00 65,00 8 2,40 1.167,00 65,00 9 2,40 1.167,00 65,00 10 2,40 1.167,00 65,00 11 2,40 1.167,00 65,00 12 2,40 1.167,00 65,00 MEDIA 284,97 1.171,66 122,8 35.408,65 DS 115,94 62,93 28,51 19.293,44 Nº electrodo 156,00 156,00 156,00 156,00 156,00 156,00 Tabla 18.- Paciente 4. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 56,69 888,20 54,17 21.500,07 2 56,69 944,90 54,17 21.169,64 3 56,69 1.134,00 49,17 19.254,97 4 2,40 1.167,00 65,00 156,00 5 56,69 1.153,00 49,17 19.771,26 6 2,40 1.167,00 65,00 156,00 7 2,40 1.167,00 65,00 156,00 8 2,40 1.167,00 65,00 156,00 9 56,69 1.172,00 49,17 19.854,85 10 2,40 1.167,00 65,00 156,00 11 56,69 944,9 52,50 126,00 12 2,40 1.167,00 65,00 156,00 MEDIA 56,69 1.039,5 51,39 16.946,13 DS 0 126,61 2,50 8.285,72 Nº electrodo Tabla 19.- Paciente 5. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 396,9 1.452,00 53,33 21.166,68 2 415,7 1.425,00 53,33 22.169,28 3 396,9 1.371,00 53,33 21.166,68 4 415,7 1.371,00 51,67 21.479,22 5 415,7 1.371,00 51,67 21.479,22 6 491,3 1.371,00 51,67 25.385,47 7 453,5 1.371,00 53,33 24.185,16 8 415,7 1.289,00 53,33 22.169,28 9 415,7 1.371,00 56,67 23.557,72 10 472,4 1.126,00 53,33 25.193,09 11 2,40 1.167,00 65,00 156,00 12 2,40 1.167,00 65,00 156,00 MEDIA 428,95 1.351,80 53,16 22.795,18 DS 32,16 89,77 1,45 1.650,73 Nº electrodo Tabla 20.- Paciente 6. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 200,5 848,10 36,37 7.352,34 2 208,2 848,10 36,37 7.634,69 3 161,9 848,10 36,37 5.936,87 4 177,3 878,90 36,37 6.501,59 5 200,5 940,60 36,37 7.352,34 6 146,5 909,80 36,37 5.372,16 7 161,9 909,80 36,37 5.936,87 8 154,2 971,50 38,33 5.910,49 9 200,5 909,80 41,67 8.354,84 10 2,40 1.167,00 65,00 156,00 11 2,40 1.167,00 65,00 156,00 12 2,40 1.167,00 65,00 156,00 MEDIA 179,06 896,07 37,41 6.705,8 DS 23,71 43,92 1,80 1.003,34 Nº electrodo Tabla 21.- Paciente 7. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 434,20 1.086,00 26,67 11.580,11 2 461,4 1.208,00 26,67 12.305,54 3 434,2 1.248,00 26,67 11.580,11 4 420,7 1.167,00 26,67 11.220,07 5 488,5 1.425,00 26,67 13.028,3 6 339,3 1.167,00 28,33 9.612,37 7 420,7 1.330,00 26,67 11.220,07 8 434,2 1.045,00 26,67 11.580,11 9 447,8 1.248,00 26,67 11.942,83 10 461,4 1.248,00 26,67 12.305,54 11 366,4 1.208,00 26,67 9.771,89 12 325,7 1.004,00 26,67 8.686,42 MEDIA 419,54 1.198,67 26,81 11.236,11 DS 50,24 117,61 0,47 1.266,19 Nº electrodo Tabla 22.- Paciente 8. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 2,40 1.167,00 65,00 156,00 2 298,50 1.004 33,33 9.949,01 3 239 909,8 80,00 19.120,00 4 200,5 940,6 80,00 16.040,00 5 2,40 1.167,00 65,00 6 2,40 1.167,00 65,00 7 2,40 1.167,00 65,00 8 2,40 1.167,00 65,00 9 2,40 1.167,00 65,00 10 2,40 1.167,00 65,00 11 2,40 1.167,00 65,00 12 2,40 1.167,00 65,00 MEDIA 246 951,46 64,44 15.036,34 DS 49,37 48,03 26,94 4.667,15 Nº electrodo 156,00 156,00 156,00 156,00 156,00 156,00 156,00 156,00 Tabla 23.- Paciente 9. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 271,40 1.262,00 33,33 9.045,76 2 271,40 1.316,00 33,33 9.045,76 3 244,30 1.438,00 33,33 8.142,52 4 271,40 1.425,00 33,33 9.045,76 5 271,40 1.425,00 33,33 9.045,76 6 298,50 1.466,00 33,33 9.949,01 7 298,50 1.466,00 33,33 9.949,01 8 298,50 1.520,00 33,33 9.949,01 9 2,40 1.167,00 65,00 156,00 10 2,40 1.167,00 65,00 156,00 11 2,40 1.167,00 65,00 156,00 12 2,40 1.167,00 65,00 156,00 MEDIA 278,17 1.414,75 33,33 9.271,57 DS 19,16 84,65 0 638,69 Nº electrodo Tabla 24.- Paciente 10. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 246,70 724,70 33,33 8.222,51 2 146,50 771,00 56,67 8.302,16 3 208,20 956,00 38,33 7.980,31 4 190,00 1.004,00 56,67 10.767,30 5 146,50 971,50 56,67 8.302,16 6 176,40 1.058,00 48,33 8.525,41 7 190,00 1.181,00 48,33 9.182,70 8 162,80 1.126,00 48,33 7.868,12 9 135,70 1.004,00 46,67 6.333,12 10 2,40 1.167,00 65,00 156,00 11 2,40 1.167,00 65,00 156,00 12 2,40 1.167,00 65,00 156,00 MEDIA 178,08 977,35 48,14 8.387,09 DS 35,22 149,33 8,18 1.173,28 Nº electrodo Tabla 25.- Paciente 11. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 2,4 1.167 65 156,00 2 340,20 1.181 69,17 23.531,63 3 378,00 1.181 69,17 26.146,26 4 311,80 926 72,50 22.605,50 5 434,60 973,20 72,50 31.508,50 6 434,60 1.011,00 69,17 30.061,28 7 2,4 1.167,00 65 156,00 8 491,30 1.030,00 72,50 35.619,25 9 510,20 1.191,00 69,17 35.290,53 10 2,40 1.167,00 65,00 156,00 11 2,40 1.167,00 65,00 156,00 12 2,40 1.167,00 65,00 156,00 MEDIA 414,38 1.070,45 70,60 29.251,85 DS 74,45 111,40 1,77 11.402,03 Nº electrodo Tabla 26.- Paciente 12. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 257,8 1.533 43,33 11.170,47 2 271,40 1.588 43,33 11.759,76 3 2,4 1.167,00 65 156,00 4 190 1.588 43,33 8.232,70 5 244,30 1.588 43,33 10.585,52 6 230,70 1.533 46,67 10.766,77 7 257,80 1.588 46,67 12.031,53 8 2,40 1.167,00 65,00 156,00 9 2,40 1.167,00 65,00 156,00 10 2,40 1.167,00 65,00 156,00 11 2,40 1.167,00 65,00 156,00 12 2,40 1.167,00 65,00 156,00 MEDIA 242 1.569,66 44,44 9.243,25 DS 28,98 28,40 1,72 1.356,65 Nº electrodo Tabla 27.- Paciente 13. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 258,10 654,1 90,42 23.337,40 2 255,20 655,2 90,42 23.075,18 3 273 654,7 90,42 24.684,66 4 317,70 783,4 75,42 23.960,93 5 299 780,6 75,42 22.550,58 6 324,60 763,4 75,42 24.481,33 7 274,10 763,4 75,42 20.672,62 8 291,30 739,8 75,42 21.969,85 9 383,80 896,6 60,42 23.189,20 10 300,6 718,9 75,42 22.671,25 11 256,2 594,4 90,42 23.165,60 12 279,9 597,5 90,42 25.308,56 MEDIA 292,79 716,83 80,42 23.255,60 DS 36,77 88,68 9,77 1.260,30 Nº electrodo Tabla 28.- Paciente 14. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 363,70 769,5 46,25 16.821,13 2 433 1.001 37,08 16.055,64 3 417,5 975,4 37,08 15.480,90 4 434,20 974,5 37,08 16.100,14 5 218,3 490 73,35 16.012,31 6 448,90 1.005,5 37,08 16.645,21 7 434,20 1.033,8 37,08 16.100,14 8 348,50 854,6 46,25 16.118,13 9 409,30 1.096,8 37,08 15.176,84 10 339,1 908,7 46,25 15.683,38 11 446 1.160,4 37,08 16.537,68 12 258,6 672,9 64,58 16.700,39 MEDIA 379,27 911,93 44,69 16.119,32 DS 76,19 189,29 12,17 503,09 Nº electrodo Tabla 29.- Paciente 15. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 182,6 422,1 68,75 12.553,75 2 166,7 430,4 68,75 11.460,63 3 160,6 430,4 68,75 11.041,25 4 145,2 430,4 68,75 9.982,50 5 155,9 430,4 68,75 10.781,13 6 144,6 430,4 68,75 9.941,25 7 134,4 429,9 68,75 9.240,00 8 121 417,9 68,75 8.318,75 9 116 417,9 68,75 7.975,00 10 142,5 417,9 68,75 9.796,88 11 186,2 417,9 68,75 12.801,25 12 12,3 245,8 137,08 1.686,08 MEDIA 139 40,12 74,44 9.626,29 DS 45,38 52,07 19,72 2.909,19 Nº electrodo Tabla 30.- Paciente 16. THR MCL DURACION CARGA (Cu) (Cu) (µseg) (Cu x µseg) 1 117,4 364,7 64,58 7.581,69 2 2,4 1.167 65 156,00 3 216,9 654,2 37,08 8.042,65 4 230,1 654,2 37,08 8.532,11 5 230,1 673,8 37,08 8.532,11 6 237 673,8 37,08 8.787,96 7 251,5 673,8 37,08 9.325,62 8 266,8 694,1 37,08 9.892,94 9 142,3 359,5 73,75 10.494,63 10 198,9 487,9 55,42 11.023,04 11 321,7 743,7 37,08 11.928,64 12 229,7 510,2 55,42 12.729,97 MEDIA 222,03 589,99 46,24 9.715,58 DS 55,75 135,81 13,59 1.658,25 Nº electrodo Los pacientes afectos de malformación presentan un umbral eléctrico medio en los electrodos activos de 302.08 Cu (DS 98.250 Cu), un máximo nivel de confort promedio de 1028,18 Cu (DS 301,28 Cu) y una carga media de 22.201,63 Cu x µseg (DS 13.758,07 Cu x µseg). La media de electrodos activos fue de 8,25 (DS 3,38). Los resultados promedios y sus desviaciones estándar de los distintos grupos de malformación de oído interno se presentan igualmente en la tabla 31. Los pacientes del grupo control presentaron un umbral eléctrico medio en los electrodos activos de 135,73 Cu (DS 91,98 Cu), un máximo nivel de confort medio de 915,73 Cu (DS 322,94 Cu) y una carga media de 5.384,92 Cu x µseg (DS 4.793,85 Cu x µseg). La media de electrodos activos fue de 10,25 (DS 1,77). Tabla 31. . Tabla 31.- Datos generales de programación. CAVIDAD COMUN THR MCL CARGA Nº ELECT. χ (DS) χ (DS) χ (DS) χ (DS) 338,98 Cu 1.117,49 Cu (78,43 Cu) (156,48 Cu) 42.244,54 Cu x µseg (22.320,28 Cu x µseg) 5,33 (4,04) 20.100,86 Cu x HIPOPLASIA 344,92 Cu 887,63 Cu µseg 8,50 (73,71 Cu) (241,54 Cu) ( 4.461,46 Cu x (4,95) µseg) 12.613,16 Cu x Malformación de oído interno PARTICION INCOMPLETA 312,40 Cu 1.021,16 Cu µseg 10,50 (130,4 Cu) (317,92 Cu) (7.044,2 Cu x (1,29) µseg) 13.847,95 Cu x MALFORM. VESTIBULO 268,13 Cu 1.043,67 Cu µseg 8,14 (99,83 Cu) (375,66 Cu) (7.457 Cu x (3,24) µseg) 22.201,63 Cu x TODAS 302,08 Cu 1.028,18 Cu µseg 8,25 (98,25 Cu) (301,28 Cu) (13.758,07 Cu x (3,38) µseg) 5.384,92 Cu x NO MALFORMACION 135,73 Cu 915,73 Cu µseg 10,25 ( 91,98 Cu) (322,94 Cu) (4.793,85 Cu x (1,77) µseg) Los pacientes afectos de malformación de oído interno presentan en promedio un umbral eléctrico y una carga eléctrica mayor que los pacientes no afectos de malformación de oído interno, existiendo diferencias significativas (U de Mann Whitney; p<0,05). Por otro lado, no presentan diferencias significativas respecto al nivel máximo de confort ni al número de electrodos activos (U de Mann Whitney; p>0,1). Al estratificar el grupo de malformaciones de oído interno en sus distintos grupos malformativos, y comparar los resultados promedio con los obtenidos por el grupo sin malformaciones, obtenemos los siguientes resultados: - Los pacientes afectos de cavidad común presentan un umbral promedio y una carga media mayores que el grupo control, mientras que el número de electrodos activos es menor, presentando diferencias significativas (U de Mann Whitney; p<0,05). No existen diferencias significativas respecto al nivel máximo de confort. - Los pacientes con hipoplasia coclear presentan un umbral promedio mayor que el grupo control, con diferencias estadísticamente significativas (U de Mann Whitney; p< 0,05). No existen diferencias significativas respecto a máximo nivel de confort, carga media ni número de electrodos activos. - Los pacientes con malformación tipo partición incompleta presentan un umbral promedio mayor que el grupo control, con diferencias estadísticamente significativas (U de Mann Whitney; p< 0,05). No existen diferencias significativas respecto a máximo nivel de confort, carga media ni número de electrodos activos. - Los pacientes con malformaciones del vestíbulo presentan un umbral y una carga promedios mayores que el grupo control, con diferencias estadísticamente significativas (U de Mann Whitney; p< 0,05). No existen diferencias significativas respecto a máximo nivel de confort ni número de electrodos activos. Estudiando los diferentes parámetros considerados (THR, MCL, carga y número de electrodos activos) en cada uno de los grupos del estudio, obtenemos los siguientes resultados: -THR: al comparar los umbrales medios de los cinco grupos (cóclea normal, cavidad común, hipoplasia coclear, partición incompleta y malformación del vestíbulo), obtenemos diferencias estadísticamente significativas (KruskalWallis, p<0,05). Si comparamos los pacientes afectos de alguna malformación con los pacientes del grupo de control, las diferencias resultan también estadísticamente significativas (U de Mann-Whitney, p<0,05) Al comparar las malformaciones mayores (cavidad común e hipoplasia) con las menores (partición incompleta y malformaciones del vestíbulo), no encontramos significación estadística (U de Mann-Whitney, p>0,05), sucediendo lo mismo si comparamos las malformaciones mayores entre si o las menores entre si. 400 350 300 250 200 150 100 50 Cu 0 No malform Partición incompleta Hipoplasia Figura 41.- THR medios (Cu). Cavidad común Malf. Vest. -MCL: al comparar los máximos niveles de confort medios en los cinco grupos por separado, no obtenemos diferencias estadísticamente significativas (Kruskal-Wallis, p>0,05). Si comparamos los pacientes con malformación en general con los que no la tienen, las diferencias tampoco resultan estadísticamente significativas (U de Mann-Whitney, p>0,05). Obtenemos el mismo resultado cuando tratamos de comparar las malformaciones mayores con las menores, las mayores entre si y las menores entre si (U de MannWhitney, p>0,05). 1200 1000 800 600 400 200 Cu 0 No malform. Partición incompleta Hipoplasia Figura 42.- MCL medios (Cu). Cavidad común Malf. Vest. -Carga eléctrica: al comparar las cargas eléctricas medias de los cinco grupos de estudio (cavidad común, hipoplasia coclear, partición incompleta, malformación del vestíbulo y cóclea normal) encontramos diferencias estadísticamente significativas (Kruskal-Wallis, p<0,05). Al comparar dichas medias entre el grupo de pacientes con malformación en conjunto y el de cóclea normal, asimismo las diferencias son estadísticamente significativas (U de Mann-Whitney, p<0,05). También existen diferencias significativas en las cargas promedio entre pacientes afectos de malformaciones mayores (cavidad común e hipoplasia coclear) y pacientes afectos de malformaciones menores (malformaciones del vestíbulo y partición incompleta) (U de Mann-Whitney, p<0,05). Entre las malformaciones mayores, hay indicios de significación (U de Mann-Whitney, p=0,083) y entre las malformaciones menores las diferencias no son estadísticamente significativas (U de Mann-Whitney, p=1). 45000 40000 35000 30000 25000 20000 15000 10000 5000 Cu x µseg 0 No malform. Partición incompleta Hipoplasia Cavidad común Malf. Vest. Figura 43.- Cargas eléctricas medias (Cu x µseg). -Número de electrodos activos: al comparar el número medio de electrodos activos en los cinco grupos vemos que no hay diferencias estadísticamente significativas (Kruskal-Wallis, p>0,05). Si comparamos los pacientes con malformación en general con los que no la tienen, obtenemos que hay indicios de significación (U de Mann-Whitney, p=0,095), siendo preciso aumentar la muestra para poder identificar esas diferencias. Al comparar las malformaciones mayores (cavidad común e hipoplasia) con las menores (partición incompleta y malformaciones del vestíbulo), y a su vez las mayores y las menores entre si, no obtenemos significación estadística (U de MannWhitney, p>0,05). 12 10 8 6 4 2 0 No malform. Partición incompleta Hipoplasia Cavidad común Figura 44.- Número medio de electrodos activos. Malf. Vest. 4.2.5.3.- Habilidades auditivas En la tabla 32 se presentan los porcentajes de acierto resultantes de la exploración de las habilidades auditivas de los pacientes (LIP y MTP) antes (0) y 24 meses tras la implantación coclear. Tabla 32.- Porcentajes de acierto de los test logopédicos. LIP LIP MTP3 MTP3 MTP6 MTP6 MTP12 MTP12 PACIENTE 0 24m 0 24m 0 24m 0 24m 1 0% 50% 0% 0% 0% 0% 0% 0% 2 4% 60% 0% 10% 0% 0% 0% 0% 3 0% 69% 0% 67% 0% 0% 0% 0% 4 0% 26% 0% 0% 0% 0% 0% 0% 5 10% 100% 0% 100% 0% 100% 0% 100% 6 0% 82% 0% 10% 0% 8% 0% 0% 7 10% 100% 0% 100% 0% 88% 0% 91% 8 45% 100% 12% 75% 5% 60% 0% 40% 9 90% 100% 45% 100% 40% 100% 30% 96% 10 60% 100% 45% 100% 42% 100% 40% 100% 11 5% 52% 0% 20% 0% 15% 0% 5% 12 60% 100% 20% 100% 40% 100% 34% 100% 13 33% 100% 10% 70% 8% 100% 0% 100% 14 10% 80% 5% 65% 5% 50% 0% 55% 15 20% 100% 10% 90% 10% 100% 10% 80% 16 0% 80% 0% 80% 5% 80% 5% 70% Todos los pacientes afectos de malformación de oído interno mejoraron sus habilidades auditivas tras la implantación coclear existiendo diferencias estadísticamente significativas (test de Wilcoxon, p<0,05). La tabla 33 muestra los resultados promedio (DS) de las habilidades auditivas, previas y 24 meses tras la implantación coclear, de los pacientes afectos de malformación de oído interno, como grupo y estratificado según las distintas malformaciones, y de los pacientes sin malformación coclear. Tabla 33.- Datos de logopedia (χ y DS) en todos los grupos antes (0) y después (24) del implante coclear. LIP 0 LIP 24 MTP3 0 MTP3 24 χ χ χ χ (DS) (DS) (DS) (DS) MTP6 MTP6 MTP12 MTP12 0 24 0 24 χ χ χ χ (DS) (DS) (DS) (DS) CAVIDAD 1,33% 59,67% 0,00% 30,67% 0,00% 0,00% 0,00% 0,00% COMUN (2,30%) (9,50%) (0,00%) (33,85%) (0,00%) (0,00%) (0,00%) (0,00%) HIPOPLASIA 16,50% 63,00% 5,00% 50,00% 40,00% 50,00% 0,00% 50,00% COCLEAR (23,33%) (52,32%) (7,07%) (70,71%) (56,56%) (70,71%) (0,00%) (70,71%) PARTICION 10,00% 95,50% 0,00% 82,50% 0,00% 70,75% 0,00% 62,75% INCOMPLETA (8,16%) (9,00%) (0,00%) (35,00%) (0,00%) (30,80%) (0,00%) (45,20%) Malform. de oído interno MALF. 42,14% 93,14% 17,43% 92,86% 24,57% 78,71% 14,86% 76,86% VESTIBULO (28,55%) (18,14%) (20,28%) (18,89%) (23,23%) (31,48%) (18,75% (24,76%) 21,68% 81,18% 9,18% 61,68% 9,68% 56,31% 7,34% 52,31% (27,84%) (23,52%) (15,20%) (39,54%) (15,70%) (44,56%) (13,89%) (44,46%) 38,56% 92,25% 39,19% 88,81% 38,00% 87,38% 37,44% 84,13% (24,43%) (14,17%) (19,99%) (18,03%) (17,39%) (19,45%) (20,64%) (20,18%) TODAS NO MALFORMACION Los pacientes afectos de malformación de oído interno presentan unos resultados en promedio inferiores a los obtenidos por el grupo de pacientes sin malformación, presentando diferencias estadísticamente significativas (U de Mann Whitney; p<0,05). Únicamente, los resultados en promedio obtenidos en el test de Lip-profile a los 24 meses no muestran diferencias estadísticamente significativas entre ambos grupos de comparación (U de Mann Whitney; p=0.45) Cuando estratificamos el grupo de malformaciones de oído interno en sus distintos grupos malformativos, y comparamos los resultados promedio con los obtenidos por el grupo sin malformaciones, obtenemos que: - Los pacientes afectos de cavidad común presentan resultados inferiores en promedio en todos los tests empleados para valorar las habilidades auditivas, antes y después del implante coclear, frente a los pacientes del grupo control, diferencias estadísticamente significativas (U de Mann-Whitney; p<0,05). - Los pacientes afectos de hipoplasia coclear presentan resultados peores estadísticamente significativos al compararlos con los obtenidos por el grupo control, son el MTP3 y MTP12 ambos previos al implante coclear (U de Mann-Whitney; p<0,05). - Los pacientes afectos de malformación de partición incompleta presentaron peores resultados en LIP, MTP3, MTP6 y MTP12 antes del implante coclear (U de Mann-Whitney; p<0,05). - Los pacientes afectos de malformaciones del vestíbulo presentaron resultados peores en MTP3 y MTP12 previos a la implantación (U de MannWhitney; p<0,05). Analizando cada prueba logopédica independientemente, antes y después de la intervención, con el fin de comparar los resultados obtenidos por cada uno de los grupos de estudio, obtenemos los siguientes resultados: - LIP 0: existen diferencias estadísticamente significativas (KruskalWallis, p<0,05) en los resultados de los 5 grupos de comparación, así como al comparar el grupo con malformación en su conjunto con el grupo de control (U de Mann-Whitney, p<0,05). Al comparar las malformaciones mayores con las menores, y las mayores entre si, no aparece significación estadística (U de Mann-Whitney, p>0,05), habiéndola sin embargo al comparar las malformaciones menores entre si (U de Mann-Whitney, p<0,05). - LIP 24: al comparar la media de aciertos en el LIP en los cinco grupos después de recibir el implante, vemos indicios de significación estadística (Kruskal-Wallis, p=0,063), por lo que aumentando el tamaño muestral podríamos conseguirla. Si comparamos los pacientes con malformación en general con los que no la tienen, las diferencias no resultan estadísticamente significativas (U de Mann-Whitney, p>0,05) Al comparar las malformaciones mayores (cavidad común e hipoplasia) con las menores (partición incompleta y malformaciones del vestíbulo), sí obtenemos diferencias estadísticamente significativas (U de Mann-Whitney, p<0,05). Entre las malformaciones mayores y entre las menores, las diferencias no son estadísticamente significativas (U de Mann-Whitney, p>0,05). 100 % aciertos 90 80 70 60 50 40 30 20 10 0 No malform. Partición incompleta Hipoplasia antes IC Cavidad común Malf. Vest. después IC Figura 45.- LIP (% medio de aciertos). - MTP3 0: hay diferencias estadísticamente significativas al comparar la media de aciertos en el MTP3 previo al implante en los cinco grupos (KruskalWallis, p<0,05), así como al comparar el grupo de los pacientes con malformación en general con los que no la tienen (U de Mann-Whitney, p<0,05). Al comparar las malformaciones mayores (cavidad común e hipoplasia) con las menores (partición incompleta y malformaciones del vestíbulo), y las mayores entre si no observamos diferencias estadísticamente significativas (U de Mann-Whitney, p>0,05). Entre las malformaciones menores en cambio, sí existen (U de Mann-Whitney, p<0,05). - MTP3 24: al comparar la media de aciertos en el MTP3 en los cinco grupos después de recibir el implante, obtenemos diferencias estadísticamente significativas (Kruskall-Wallis, p<0,05). Al igual que si comparamos los pacientes con malformación en general con los que no la tienen, y las malformaciones mayores con las menores (U de Mann-Whitney, p<0,05). Sin embargo no es así si comparamos las malformaciones mayores entre si y las menores entre si (U de Mann-Whitney, p>0,05). 100 % aciertos 90 80 70 60 50 40 30 20 10 0 No malform. Partición incompleta Hipoplasia antes IC Cavidad común Malf. Vest. después IC Figura 46.- MTP3 (% medio de aciertos). -MTP6 0: las diferencias estadísticamente significativas las obtenemos al comparar los cinco grupos de forma independiente (Kruskal-Wallis, p<0,05), el grupo general de malformación con el que no la tiene (U de Mann-Whitney, p<0,05), y las malformaciones menores entre si (U de Mann-Whitney, p<0,05) en cambio no las hay cuando comparamos las malformaciones mayores con las menores y las mayores entre si (U de Mann-Whitney, p>0,05). -MTP6 24: al comparar la media de aciertos en el MTP6 en los cinco grupos 24 meses después de recibir el implante, observamos diferencias estadísticamente significativas (Kruskall-Wallis, p<0,05). Si comparamos los pacientes con malformación en general con los que no la tienen, aparecen indicios de significación (U de Mann-Whitney, p=0,056). Al comparar las malformaciones mayores con las menores, sí obtenemos significación estadística (U de Mann-Whitney, p<0,05), pero no al comparar las malformaciones mayores y las menores entre si (U de Mann-Whitney, p>0,05). 100 % aciertos 90 80 70 60 50 40 30 20 10 0 No malform. Partición incompleta Hipoplasia antes IC Cavidad común Malf. Vest. después IC Figura 47.- MTP6 (% medio de aciertos) -MTP12 0: al comparar los cinco grupos (Kruskal-Wallis, p<0,05) y las malformaciones en conjunto con el grupo sin malformación, aparecen diferencias estadísticamente significativas (U de Mann-Whitney, p<0,05). Esto no ocurre al comparar las malformaciones mayores con las menores, o las mayores y las menores entre si (U de Mann-Whitney, p>0,05). -MTP12 24: al comparar la media de aciertos en el MTP12 en los cinco grupos 24 meses después de recibir el implante, parece haber diferencias (Kruskall-Wallis, p=0,073), debiendo aumentar la muestra para confirmarlo. Si comparamos los pacientes con malformación en general con los que no la tienen, las diferencias sí resultan estadísticamente significativas (U de MannWhitney, p<0,05). Así como al comparar las malformaciones mayores con las menores (U de Mann-Whitney, p<0,05). Estas diferencias no aparecen si comparamos las cavidades comunes con las hipoplasias (malformaciones mayores) o las malformaciones tipo partición incompleta con las malformaciones del vestíbulo (malformaciones menores) (U de Mann-Whitney, p>0,05). 90 80 % aciertos 70 60 50 40 30 20 10 0 No malform. Partición incompleta Hipoplasia antes IC Cavidad común después IC Figura 48.- MTP12 (% medio de aciertos). Malf. Vest. 5.- DISCUSION El estudio de las malformaciones de oído interno constituye un tema de pleno desarrollo en el momento actual, tanto por su prevalencia, como por su relevancia clínica. Las malformaciones cocleares están presentes en el 20% de los pacientes con hipoacusia neurosensorial congénita, produciendo un déficit auditivo inversamente relacionado con el grado de maduración morfológica, desde hipoacusias moderadas-severas hasta hipoacusias profundas y cofosis. La hipoacusia consecuente a la malformación de oído interno es un déficit sensorial que da lugar a todo un conjunto de dificultades y desventajas para el paciente que las sufre. Supone un handicap personal, social y profesional por el aislamiento que conlleva para el sujeto con respecto al resto de la población, tanto por el déficit sensorial como por las consecuencias negativas sobre el lenguaje, siendo el déficit lingüístico directamente proporcional al grado de la hipoacusia. Además, el déficit sensorial supone un gasto social en infraestructuras y profesionales encaminado a reducir las consecuencias personales en el paciente. Desde el ámbito médico, dos estrategias han permitido reducir el handicap de los pacientes afectos de hipoacusia sensorial: 1) El avance de las tecnologías de amplificación del sonido y estimulación de las estructuras auditivas, bien a través del desarrollo de las prótesis acústicas, electromecánicas, electromagnéticas, vibratorias y la aparición con posterior desarrollo de las prótesis auditivas de estimulación eléctrica; y 2) El diagnóstico precoz, el tratamiento protésico y la rehabilitación temprana en pacientes de corta edad. A pesar de estos avances, los pacientes afectos de malformaciones de oído interno se comportan como un grupo heterogéneo en cuanto a resultados finales auditivos y lingüísticos, siendo éstos peores a mayor grado de malformación. Son 3 los factores responsables de estos heterogéneos resultados: 1) La ausencia o presencia de estructuras intracocleares propias de cada malformación; 2) La distribución topográfica de las estructuras nerviosas auditivas, que condiciona una mayor o menor tonotopía y; 3) El grado madurativo de estas estructuras. Existen numerosas clasificaciones morfológicas de las malformaciones de oído interno, que basadas en técnicas de imagen radiológicas intentan establecer un origen embriológico y una secuencia temporal en las noxas responsables de las mismas. La pionera de estas clasificaciones fue la propuesta por Jackler en 1987, clasificación madre que aún perdura en nuestros días, que basándose en las imágenes obtenidas mediante politomografía clasificó las malformaciones cocleares en: 1) aplasia completa o de Michel, descrita por este autor en 1863. Hay una ausencia completa de oído interno y nervio auditivo y se produce por la interrupción de la formación del oído interno antes de la tercera semana de vida intrauterina; 2) cavidad común, cuando el desarrollo embrionario se detiene antes de la cuarta semana de gestación y da como resultado una estructura única que comprende al laberinto anterior y posterior sin separación entre ambos; 3) la agenesia coclear, producto de una noxa que interrumpe la formación de la cóclea antes de la quinta semana. El laberinto posterior puede ser normal o no, pero no llega a formarse la cóclea; 4) la hipoplasia coclear, si la noxa actúa antes de la sexta semana dando como consecuencia una cóclea de menor tamaño, limitada normalmente a la espira basal; y por último 5) la partición incompleta o deformidad de Mondini, producida antes de la séptima semana de gestación y en la que las espiras media y apical de la cóclea están fusionadas, de modo que las dos vueltas y media de espira típicas de la cóclea normal, en estos casos quedan reducidas a una y media. En la actualidad, la aparición de nuevas y más precisas técnicas de imagen (Tomografia Axial Computadorizada y Resonancia Magnética Nuclear) ha contribuido al aumento en el diagnóstico de pacientes con malformación coclear73 y a un mejor conocimiento de ciertos claroscuros de la clasificación de Jackler. Así, Sennaroglu y cols., en el año 2002 proponen dos modificaciones a la primera: 1) La división de la malformación antes descrita como partición incompleta en dos, tipo I y tipo II. Según Sennaroglu, la partición incompleta tipo II corresponde con la clásica descrita por Carlo Mondini, como una cóclea con una vuelta y media, con las espiras medial y apical unidas en una formación quística, acompañada de un vestíbulo mínimamente dilatado y un acueducto vestibular agrandado. Por el contrario, la partición incompleta tipo I fue descrita por Sennaroglu como una cóclea sin área cribiforme ni modiolo acompañada de un vestíbulo quístico grande; 2) La reestadificación de las malformaciones, quedando situada embriológicamente la partición incompleta tipo I tras la cavidad común y antes de la hipoplasia. Otros autores han descrito de forma pormenorizada las alteraciones del acueducto del vestíbulo (Valvassory y Clemis26; Pyle18) y del conducto auditivo interno (Casselman y cols.31, Vilain y cols. 31), tal y como se ha expuesto en la introducción de la presente tesis doctoral. A pesar de los vastos conocimientos acerca de la morfología y estadiaje de las malformaciones, son escasas las referencias acerca de la distribución y maduración de las fibras neurales en cada una de estas malformaciones que son responsables de la heterogeneidad de resultados auditivos. El presente estudio propone y desarrolla los métodos de estudio de la distribución y maduración de las estructuras nerviosas cocleares, describiendo además los resultados obtenidos en nuestra población. 5.1.- Estudio embriológico. Morfología coclear y distribución de las estructuras nerviosas auditivas: Sirviéndonos de la embrioteca del Departamento de Anatomía Humana de la Facultad de Medicina de la Universidad de Granada, hemos estudiado el desarrollo embrionario del oído interno desde sus primeros esbozos hasta su configuración casi definitiva. Hemos podido corroborar lo publicado en toda la bibliografía consultada con respecto a la aparición de estructuras vasculares, nerviosas, musculares, óseas, etc., correlacionando cada una de ellas con una edad gestacional determinada. La observación al microscopio de cortes embriológicos humanos teñidos con hematoxilina-eosina y clasificados por tamaño (longitud vertex-coxis en milímetros) resulta el método más directo de estudiar la embriología del oído interno, siguiendo la evolución de cada estructura desde su aparición y relacionándola con el conjunto del embrión. Ya en el embrión más pequeño estudiado, 5mm de longitud (estadio 1214 de Canergie, 26-32 días de edad gestacional), la placoda auditiva y posteriormente la vesícula auditiva, origen del todo el oído interno, se mostró presente. Según la clasificación de Streeter4 este embrión corresponde al Horizonte XIII cuya estructura más significativa es la vesícula ótica. Dicha vesícula, se localiza a ambos lados del rombencéfalo, imprescindible para la posterior transformación de las vesículas óticas en el oído interno1,2,3. Mucho se está publicando en los últimos años acerca de los factores o elementos que influyen en la embriogénesis del oído interno. Es indiscutible el papel protagonista de los factores de crecimiento fibroblástico (FGFs), demostrando que su acción es fundamental para la inducción del desarrollo del oído interno, sin descartar otros posibles inductores del proceso74,75,76. El segundo embrión en el que nos detenemos es el de 6mm (estadio 1314 de Canergie, 28-32 días de edad gestacional). En él se mantiene la imagen de la vesícula ótica observada en el de 5mm y aparece por primera vez el ganglio estatoacústico en una posición ventral con respecto a aquella. Destaca también la presencia del ganglio de Gasser del V par craneal, en posición ventral con respecto a la vesícula y de las vesículas telencefálicas, también en la región más ventral del embrión. En el embrión de 10mm (estadio 16 de Canergie, 37 días de edad gestacional), la vesícula ótica abandona su forma ovalada y comienza a emitir prolongaciones que irán dando lugar a las diferentes estructuras del oído interno que se desarrollan a partir de ella. La similitud de la imagen en este momento del desarrollo con las cavidades comunes apreciadas en la clínica en individuos adultos, permite señalar este momento como el límite para la acción de la noxa y la aparición de la malformación. La falta de desarrollo neural, estando únicamente presentes el ganglio estatoacústico y ciertas células neurales alrededor de la vesícula ótica, permiten deducir la inmadurez de la distribución neural que se mantendrá hasta la edad adulta. Además, la cavidad no posee en su interior estructura auditiva alguna, quedando como en el adulto como una vesícula común. Las prolongaciones procedentes de la vesícula ótica se desarrollan y en el embrión de 19mm (estadio 19 de Canergie, 48 días de edad gestacional) comienzan a apreciarse. La figura 16 representa un corte alto en el que se aprecian el utrículo, los canales semicirculares y los ganglios estatoacústico y de Gasser. La figura 17 es un corte más bajo del mismo embrión y por tanto nos muestra lo que se ha formado de la cóclea hasta ese momento, que no es más que la espira basal, y que daría lugar por tanto a una hipoplasia coclear si se detuviera en ese momento su desarrollo. El embrión de 28mm es el primero en el que comienza a distinguirse una cóclea más parecida a la definitiva. En él ya se observan fibras del nervio coclear partiendo de la espira basal de la cóclea así como la porción del ganglio de Corti que corresponde a dicha espira. Además, hacia el conducto auditivo interno se dirigen las fibras del nervio facial y fibras procedentes del vestíbulo. El conducto endolinfático que aparece en una posición dorsomedial en los cortes altos, y la prolongación sacular desde el utrículo. El feto de 31mm nos permite ver una imagen de la cóclea que sería el equivalente a la partición incompleta (fig.28-4). En ella vemos la presencia de dos espiras cocleares, estando la espira media y la apical fusionadas y el ganglio de Corti fácilmente identificable. Queremos destacar la bonita imagen del nervio facial (fig.29) que hemos captado del feto de 45mm, viéndose el ganglio geniculado como un engrosamiento de las fibras nerviosas a su nivel. En este mismo feto (fig.30) hemos observado la imagen más precoz de la cóclea membranosa con sus tres espiras, basal, media y apical, en la que se puede apreciar también el comienzo de la segmentación de las espiras en las diferentes escalas, vestibular, timpánica y conducto coclear; fenómeno que no ha sido completo hasta estudiar el feto de 70mm. En el feto de 52mm hemos querido resaltar la imagen de una cresta ampular y de una mácula utricular perfectamente formadas ya (fig.32). Además en este estadio vemos como el mesénquima que rodea al laberinto membranoso comienza a reabsorberse para dar lugar más adelante al espacio perilinfático. El progresivo endurecimiento de la cápsula ótica, también ha podido documentarse, apreciándose con claridad como el tejido cartilaginoso que conforma la cóclea en estos estadios precoces va compactándose previo a la osificación. Las células del tejido cartilaginoso compacto se han podido identificar perfectamente en el feto de 80mm (fig.37). Por último, en el feto de 80mm y con un gran aumento (20x), vimos con relativa claridad el Órgano de Corti (fig.40), como un engrosamiento del epitelio en el suelo del conducto coclear, cubierto además por una capa de células que corresponde a la membrana tectoria. Esta imagen es un fiel reflejo de lo que será la anatomía coclear definitiva. Una vez realizado el estudio embriológico del oído interno, hemos tratado de correlacionar las imágenes histológicas con las de las TAC de los pacientes con malformación coclear que han formado parte de nuestro estudio estadístico, a fin de confirmar las edades gestacionales a las que se produce cada malformación. En cuanto al estudio embriológico de las estructuras nerviosas del oído interno, destacan en la bibliografía los trabajos llevados a cabo por Lorente de Nó y Poljak, entre otros. El primero, realizó una investigación sirviéndose de embriones en diferentes estadios y concluyó que la cóclea se inervaba después de hacerlo las estructuras del laberinto posterior, desde la espira basal hasta la apical, y las células ciliadas internas antes que las externas. Así la primera estructura en recibir su inervación serían las crestas, seguidas de la mácula utricular, después la sacular y finalmente el Órgano de Corti77. A pesar de las limitaciones que ofrece el estudio microscópico de las estructuras neurales sobre tinciones de hematoxilina-eosina, y sin haber podido obtener muestras con la tinción argéntica descrita por Ramón y Cajal, el estudio del desarrollo de las estructuras nerviosas aportó diversas observaciones: Ya en el embrión de 6mm de longitud, correspondiente a un estadio 1314 de Canergie y equivalente a una edad gestacional de 28 a 32 días, se aprecian las fibras nerviosas contactando el ganglio cocleovestibular con el epitelio de la vesícula ótica. Esto confirma lo consultado en la bibliografía, que afirma que la migración de las neuronas del ganglio estatoacústico desde la porción ventral de la vesícula auditiva resulta aparente a la cuarta semana de gestación1. En este embrión y a máximo aumento (fig.11, 40x) hemos podido constatar la íntima relación existente entre las fibras nerviosas y el epitelio de la vesícula ótica, estando en contacto ya desde edades tan tempranas. Ese epitelio se encuentra sólo en la región ventral de la vesícula ótica, en las proximidades del ganglio, quedando el resto de la vesícula sin contacto con las fibras nerviosas. Este hecho determina la escasa funcionalidad auditiva de los pacientes afectos de cavidad común, tanto con estimulación acústica como con estimulación eléctrica intracoclear. Al encontrarse las fibras nerviosas localizadas en la periferia y en una zona muy concreta de la cóclea, los modelos tradicionales de guías de electrodos no estimulan de forma suficiente a estas fibras, que además de tener una distribución anómala, se encuentran en menor cantidad que en las cócleas normales. Existen algunos estudios78,79 acerca de la aparición del ganglio estatoacústico y la distribución de las terminaciones nerviosas tanto en el pollo como en la rata, demostrándose en ambas especies que los axones parten del ganglio estatoacústico y se dirigen a sus objetivos antes de que dicho ganglio comience a diferenciarse en ganglio de Scarpa y ganglio de Corti. Estos estudios han supuesto una fuente de información importante para el estudio en el humano. Por tanto, la inervación del oído interno está presente desde los primeros momentos de la vida intrauterina, evolucionando durante el periodo gestacional, aumentando en número y modificando su distribución, hasta alcanzar el desarrollo definitivo. Ambos aspectos del desarrollo conducirán a una madurez funcional definitiva. Además, tal y como muestra la figura 12, correspondiente a un embrión de 10mm, en estos estadios tan tempranos es muy probable que junto a las fibras del ganglio estatoacústico se entremezclen fibras del nervio facial. De hecho, esto es algo que en algunos casos se mantiene incluso en la edad adulta. Las conexiones entre el nervio intermediario de Wrisberg (rama del nervio facial) y el nervio vestibular fueron documentadas por Paturet en 1951 así como la presencia de algunas fibras que desde el primer codo del nervio facial contactaban con el ganglio de Scarpa80. Estas conexiones tienen relevancia clínica ya que serían la causa de alteraciones a nivel vestibular en pacientes que sufren una parálisis facial80. Asimismo, fibras de los nervios coclear y vestibular pueden entrar en contacto durante su recorrido por el CAI, pudiendo ser el origen, entre otras, de la persistencia de acúfenos en pacientes sometidos a neurectomía coclear, o la no desaparición de los vértigos en quién se ha practicado una neurectomía vestibular. En la figura 16, perteneciente a un embrión de 19mm, y correspondiente con el estadio de detención del desarrollo embrionario resultante de la hipoplasia coclear, destaca el engrosamiento del epitelio utricular en la zona de vecindad con las fibras nerviosas procedentes del ganglio estatoacústico. Los datos embriológicos no han permitido desentrañar las conexiones neurales en la vecindad coclear, quedando por tanto este punto como objetivo para próximos trabajos con nuevos especímenes de estudio. En el trabajo publicado por C. C. D. Shute, en el que estudia diferentes embriones teñidos con tinción argéntica, se afirma que en este estadio, el único epitelio que se encuentra diferenciado en neuroepitelio es el de las crestas77. En el feto de 31mm (fig.28-4), imagen equivalente a la partición incompleta, a pesar de la malformación concerniente a las espiras cocleares, las estructuras nerviosas reproducen de forma fiel las de la cóclea normoformada, siendo esta la razón de la buena funcionalidad auditiva de numerosos pacientes afectos por este tipo de malformación así como de los buenos resultados obtenidos tras someterse a una implantación coclear. Según C. C. D. Shute, en este embrión ya encontraríamos áreas de neuroepitelio en utrículo y sáculo77. 5.2.- Estudio de la funcionalidad neuronal: El inicio del desarrollo embriológico del oído interno queda marcado a los 28-32 días, momento en que se aprecian las fibras nerviosas contactando el ganglio cocleovestibular con el epitelio de la vesícula ótica. Desde ese momento, las fibras nerviosas madurarán aumentando de número, especializándose y distribuyéndose de forma ordenada en el helicotrema de todas las espiras cocleares. La madurez definitiva condicionará una madurez funcional al oído interno1. Las malformaciones congénitas de oído interno, surgidas como consecuencia de una detención o alteración en el desarrollo embrionario, suponen una alteración en la distribución, especialización y número de las fibras nerviosas del oído interno. Como consecuencia serán causa del déficit auditivo del paciente que la padece. La funcionalidad auditiva puede ser valorada en la actualidad gracias a la estimulación eléctrica intracoclear y a los resultados auditivos que con ella se consiguen. 5.2.1.- Estimulación eléctrica intracoclear: Hoy día y gracias al desarrollo de los implantes cocleares se puede estudiar la funcionalidad de las distintas zonas intracocleares gracias a la posibilidad de conocer la localización de cada uno de los electrodos intracocleares, y a los parámetros de estimulación eléctrica. En nuestro estudio, 16 pacientes fueron clasificados según el tipo de malformación y sometidos a estimulación eléctrica intracoclear con el fin de conocer los umbrales eléctricos subjetivos, siendo posteriormente comparados los resultados entre las distintos tipos de malformación y frente a un grupo control. Los parámetros de estimulación eléctrica estudiados fueron 1) el umbral eléctrico (THR o threshold), 2) la carga eléctrica necesaria para evocar la respuesta auditiva, 3) el número de electrodos activos y su localización, 4) el máximo nivel de confort (MCL). Dada la inestabilidad de los parámetros de estimulación eléctrica en el paciente implantado durante las primeras semanas tras el proceso de implantación coclear, encendido, programación-rehabilitación auditiva del mismo, y con el fin de realizar comparaciones reales de los parámetros de estimulación, se han tomado los valores a los 6 meses tras la implantación coclear. Por otro lado, la obtención de respuestas evocadas auditivas mediante estimulación eléctrica, potenciales de acción compuesto y potenciales evocados auditivos del tronco cerebral, ambos no pudieron ser realizadas al no estar disponible, con la suficiente fiabilidad, en los implantes Med-El en el momento del estudio. La correcta colocación de los electrodos durante el acto quirúrgico se determinó gracias a un estudio radiológico convencional mediante la proyección de Stenvers, que permite la correcta visualización de los electrodos dentro de la cóclea bien o malformada y la disposición aproximada de estos respecto a la capsula ótica. En nuestro estudio, los pacientes afectos de malformación de oído interno presentan umbrales eléctricos promedio superiores (x=302,08 Cu; DS 98,25 Cu), con diferencias estadísticamente significativas, a los existentes en pacientes sin malformación (x=135,73 Cu; DS 91,98 Cu). Estas diferencias quedan igualmente patentes, al comparar los umbrales de cada una de las malformaciones de oído interno por separado con los obtenidos por el grupo control. De todas formas, la distribución de los umbrales promedio no es igual para todas las malformaciones y se distribuyen de forma inversamente proporcional al grado de diferenciación coclear. Así a menor diferenciación coclear, mayores valores umbrales promedio. De todas formas, los resultados no demuestran diferencias significativas. Este resultado, en principio paradójico queda resuelto al comparar los resultados de la carga eléctrica en el umbral. La medida del umbral auditivo eléctrico real debe estar apoyada en la medición de la cantidad de energía eléctrica liberada durante todo el pulso eléctrico y no sobre la energía liberada en un único momento. El parámetro de estimulación que nos marca dicha medida es la carga eléctrica, medida en Culombios por microsegundo. Esta medida se aproxima más a la realidad que el umbral eléctrico, pues determina la cantidad de energía eléctrica que durante el pulso es liberado y que estimula las fibras nerviosas. Considerando esta como mejor medida, observamos que igualmente sigue una distribución inversamente proporcional al grado de diferenciación coclear, pero sí existen diferencias significativas. Las diferencias son significativas entre malformaciones mayores (cavidad común e hipoplasias cocleares) y malformaciones menores (partición incompleta y malformaciones del laberinto posterior). No existen diferencias significativas entre las malformaciones menores pero sí existen indicios entre cavidades comunes e hipoplasias, debiendo aumentar la muestra para esclarecer la presencia o no de dichas diferencias. Los motivos de la alta carga eléctrica necesaria para evocar un estímulo sonoro en la vía auditiva son la anómala distribución de las fibras neurales, que en las malformaciones mayores quedan lejos de la fuente de estimulación, la poca madurez alcanzada por estas fibras y el menor número de las mismas. Schmidt y cols. contabilizaron el número de células ganglionares en seis oídos con displasia de Mondini, estando entre 7.677 y 16.110, mientras que el número de células ganglionares de los oídos normales se estima en 33.000. Linthicum y Anderson documentaron una estimulación auditiva satisfactoria con un implante coclear en pacientes con 3.200 células ganglionares12. La carga media de los pacientes con anatomía coclear anómala de nuestro estudio fue de 22.201,63 Cu x µseg (D.S 13.758,07 Cu x µseg), siendo para el grupo control de 5.384,92 Cu x µseg (D.S 4.793,85 Cu x µseg). Respecto al número de electrodos activos, observamos que los pacientes con malformación de oído interno presentan un menor número de electrodos activos que los pacientes sin malformación y que este número es directamente proporcional al grado de diferenciación coclear, aunque no se encuentran diferencias significativas. Los motivos de la inactividad de los electrodos no son la avería o la imposibilidad de estimular el área coclear que de él depende, sino que al alcanzar grandes niveles de estimulación para estimular de forma eficaz el área de ellos dependiente, los requerimientos energéticos quedan comprometidos. Siguiendo la ley de Ohm (voltaje=impedancia*corriente) y al ser la impedancia eléctrica una variable constante para cada uno de los electrodos, el nivel de corriente o intensidad eléctrica del pulso queda limitado por el voltaje de la batería. Cuando el voltaje de la batería no es suficiente para aportar la intensidad de corriente necesaria, la corriente o intensidad del pulso eléctrico queda comprometido. Inicialmente este compromiso puede solventarse aumentando la duración del pulso y por tanto la carga eléctrica del mismo, aunque se reduzca la tasa de estimulación (nº de pulsos por segundo). Cuando los requerimientos en cuestión de corriente eléctrica de un electrodo son tan elevados, la duración del pulso muy duradera y en consecuencia la tasa de estimulación se reduzca de forma importante, los resultados auditivos del paciente81,82 quedarán mermados. Con el fin de mejorar la tasa de estimulación y por tanto el comportamiento estocástico y la resincronización de las células auditivas, los electrodos comprometidos son apagados83. Este es el motivo real de la inactividad de los electrodos. El máximo nivel de confort carece en la actualidad de valor para el estudio de la funcionalidad auditiva y puede estar relacionado con el grado de reclutamiento auditivo. Para nuestros pacientes con malformación, el MCL medio es de 1.028,18 Cu (D.S 301,28 Cu), mientras que los pacientes con anatomía coclear normal de nuestro estudio presentan un máximo nivel de confort medio de 915,73 Cu (D.S 322,94 Cu) 5.2.2.- Habilidades auditivas: tests de discriminación verbal y auditiva. Tras la programación de los parámetros del implante, el buen funcionamiento del mismo fue comprobado por los especialistas en logopedia del servicio de ORL del Hospital. Dada la inestabilidad de los parámetros, la falta de entrenamiento de los pacientes y los déficits lingüísticos de los mismos, se consideraron como resultados postimplante los alcanzados a los 24 meses tras la implantación. Numerosos fueron los tests de percepción y discriminación realizados a cada paciente pero dada la variabilidad de resultados de los mismos y con vistas a la comparabilidad, se escogieron tests de percepción como el LipProfile y el MTP en sus variantes. El LIP-Profile (Listening Progress Profile) fue creado por Archbold en 1993 para el programa de implantes cocleares pediátricos de Nottingham (Inglaterra), con el propósito de valorar la percepción auditiva de los sonidos, del lenguaje y desarrollo de las capacidades auditivas en niños portadores de un implante coclear. Se sirve de unas series de dibujos con dos alternativas, dos hojas de respuestas e instrucciones72. El MTP (Monosyllabic-Trochee-Polysyllabic word test) fue creado por Norman Rever en 1978 con la finalidad de demostrar la habilidad para identificar diferentes patrones silábicos, variando de una sílaba a dos sílabas con diferente acentuación y a más de dos sílabas. Se persigue demostrar la habilidad de identificar palabras con el patrón silábico correcto. Puede aplicarse a pacientes a partir de los dos años de edad, constando el test de 3, 6 ó 12 dibujos subtitulados, 3 objetos y 1 distractor72. Los resultados obtenidos demuestran que 1) todos los pacientes del estudio mejoraron la percepción y la identificación gracias al tratamiento con implantes cocleares y posterior rehabilitación, existiendo diferencias estadísticamente significativas (test de Wilcoxon, p<0,05) entre los resultados preimplante y postimplante coclear; y 2) que a mayor grado de diferenciación coclear, mejores resultados porcentuales se alcanzan en los tests de evaluación perceptiva e identificativa verbal. De este modo los pacientes sin malformación obtienen mejores resultados que los pacientes afectos de malformación, salvo en la percepción medida a través del Lip-profile a los 24 meses tras la implantación. No existen diferencias significativas en cuanto a los resultados en este test puesto que ambas poblaciones alcanzan resultados porcentuales elevados. Entre las malformaciones de oído interno, los resultados no se distribuyen de forma homogénea, siendo los obtenidos por los pacientes con malformaciones vestibulares y aquellos afectos de malformaciones menores (particiones incompletas o malformaciones de Mondini) similares a los obtenidos por pacientes hipoacúsicos sin malformación. Por otro lado los pacientes afectos de malformaciones mayores (cavidad común e hipoplasia coclear) presentan resultados en promedio inferiores, siendo las diferencias estadísticamente significativas, respecto a los obtenidos por el resto de pacientes. Es más, aún estando la percepción altamente comprometida, no alcanzando valores superiores al 65% en los Lip-Profile tests, la identificación es nula en pacientes con cavidad común y muy deficiente en pacientes con hipoplasia coclear. Estos últimos, en promedio, difícilmente podrán establecer conversaciones sin fijación visual al no identificar el 50% de las palabras emitidas por el interlocutor. Estos resultados concuerdan con los publicados por Jackler y cols.84, Blake C. Papsin 12, Kim L.S y cols.85, Loundon N. y cols.86, por A. Ramos y cols.14 e Incesulu16 entre otros. Los motivos de la escasa percepción sonora e identificación verbal son, según los resultados aportados en el presente estudio: 1) la necesidad de altas intensidades eléctricas para evocar un estímulo sonoro, que conllevan, como ya se ha explicado con anterioridad, a un aumento de la duración de los pulsos con el fin de aumentar la carga eléctrica, una reducción de la tasa de estimulación y un descenso del número de electrodos activos; y 2) la anómala distribución de las fibras nerviosas en la cóclea malformada. Esta distribución no representa en modo alguno la tonotopía coclear, quedando la identificación verbal muy comprometida. La deficiente percepción e identificación han llevado a numerosos autores a considerar los implantes eléctricos de tronco cerebral como el tratamiento electivo para estos pacientes87,88. Esta elección, si bien es indiscutible en aquellas malformaciones sin posibilidad alguna de implantación, como ocurre en la aplasia vestíbulo-coclear y aplasia coclear, es discutible en las malformaciones mayores (cavidad común e hipoplasia coclear). No existen aún criterios universales para la implantación en el tronco cerebral aunque entre las indicaciones más aceptadas en la actualidad destacan las osificaciones cocleares completas89, las malformaciones mayores de oído interno90, las neurectomías del VIII par craneal, los shwanomas del VIII par craneal en relación con la neurofibromatosis tipo II91 y las aplasias o hipoplasias del nervio cocleovestibular92. Algunos autores sugieren implementar el diagnóstico de imagen, en pacientes afectos de hipoacusias congénitas severas y profundas, mediante la realización de resonancias magnéticas nucleares con el fin de detectar aplasias e hipoplasias del nervio coclear, con el fin de planificar la cirugía y el tipo de implante a realizar93. De todas formas, la no existencia de conducto auditivo interno o la no visualización correcta del paquete nervioso, conformado por el VII y VIII par craneal, mediante las técnicas de imagen no puede llevar a pensar en la ausencia de nervio coclear. Tal y como demuestran las imágenes radiológicas y los resultados obtenidos en nuestra población, la ausencia de conducto auditivo interno no excluye la existencia de fibras nerviosas aberrantes, tanto del VIII par como del VII par craneal. Al igual que los pacientes con malformación mayor y con aplasia o hipoplasia de CAI no presentan parálisis facial, no se puede deducir la ausencia de fibras acústicas cuando las pruebas de imagen diagnostiquen la ausencia de CAI. Cabe así pensar que fibras del VIII y VII par craneal adoptan otras variantes anatómicas y no condicionan la parálisis facial ni la imposibilidad de estimulación eléctrica. Quizás, el estudio prequirúrgico más eficaz para detectar la aplasia o hipoplasia del nervio acústico debe estar basado en la obtención de potenciales evocados auditivos del tronco cerebral mediante estimulación eléctrica en el promontorio94 ya que permiten realizar estimulaciones más eficaces y a mayor intensidad que la estimulación acústica, con límites alrededor de los 110dBHL. De todas formas, la realización de estos potenciales presenta dificultades en la obtención de registros siendo imperativo el correcto rechazo del artefacto eléctrico. No existen datos que apoyen o desestimen la implantación en el tronco cerebral de pacientes afectos de malformaciones de oído interno y los intentos de comparabilidad de resultados están dificultados por los distintos parámetros que evalúan la eficacia auditiva en los sujetos implantados con implantes cocleares e implantes del tronco cerebral. Mientras los resultados de los implantes cocleares se miden en parámetros de percepción e identificación, los grupos protagonistas de las implantaciones del tronco en pacientes afectos de malformaciones de oído interno miden parámetros cognitivos como valores resultado95. Ambos procedimientos presentan complicaciones quirúrgicas aunque diferentes. La cirugía del implante coclear en malformaciones de oído interno puede presentar diversas complicaciones, destacando: 1) fugas de LCR. Según documentaron Hoffman y cols.55, las fugas de LCR, de diferente cuantía, aparecen en el 40-50% de los pacientes con malformaciones de oído interno. Surgen en el momento de realizar la cocleostomía cuando existe una anomalía ósea del extremo lateral del CAI20. 2) Nervio facial con trayecto anómalo aparece en el 0,3% de los oídos con apariencia normal58 y la probabilidad aumenta cuanto mayor es la malformación coclear, pudiendo alcanzar desde el 16%12 hasta el 43%58. 3) Estimulación del nervio facial: es una conocida complicación del implante coclear en la población general, siendo más frecuente en estos pacientes durante la programación del implante, debida a la alta estimulación eléctrica necesaria y a la anómala distribución de las fibras del nervio facial. Requiere del apagado de 1 a 3 electrodos26. 4) La inserción anómala de la guía de electrodos es otra complicación habitual en estos casos, pudiendo quedar la guía comprimida en el caso de cócleas hipoplásicas, o introducida en el CAI64. Quizás esta complicación sea la que más ha motivado a los fabricantes de implantes cocleares a la hora de diseñar nuevos modelos que solventen estas dificultades. Nuestro hospital prefirió la implantación de una guía de electrodos no preformada ya que la cóclea de estos pacientes no presenta una morfología típica, y se precisa una adaptación a demanda intracoclear. Entre las complicaciones de la implantación del tronco aparecen la rinolicuorrea, la meningitis, la hemorragia, el edema cerebeloso, la parálisis de los pares craneales VII, IX, X y XI94 e incluso la muerte. Mientras la investigación sobre implantes cocleares ha desarrollado en los últimos años las modernas técnicas de telemetría y las guías de implante preformadas para un mejor ajuste intracoclear en cócleas normoformadas, los implantes de tronco cerebral han centrado su evolución en pasos previos como nuevas estrategias de codificación avanzadas y cambios en la profundidad de los electrodos con el fin de obtener el mayor beneficio posible96. Esperamos que el mejor conocimiento de las malformaciones de oído interno, en cuanto a distribución y maduración neural, aportados en la presente tesis doctoral, suponga un estímulo en el desarrollo de estrategias de estimulación y en el desarrollo de guías de electrodos más eficientes y adaptables a cada malformación coclear. 6.-CONCLUSIONES Una vez realizado este estudio, con el fin de conocer la distribución y maduración neural en pacientes afectos de malformación de oído interno, podemos concluir que: 1.- La detención del temprano desarrollo embriológico en diferentes etapas supone la existencia de diferentes malformaciones de oído interno que cronológicamente pueden clasificarse en: agenesia total de oído interno, cavidad común, aplasia coclear, hipoplasia coclear, partición incompleta, malformaciones del vestíbulo. 2.- Existe una relación directamente proporcional entre el grado de diferenciación coclear y la funcionalidad auditiva de los pacientes, determinada por la distribución y el número de fibras neurales, y que queda reflejada en la necesidad de mayor o menor estímulo para obtener una respuesta auditiva. 3.- Los pacientes afectos de cavidad común e hipoplasia coclear (malformaciones mayores) requieren mayor estímulo eléctrico y presentan peores resultados auditivos mediante la implantación coclear que los pacientes afectos de particiones incompletas y malformaciones del vestíbulo (malformaciones menores). Estos últimos presentan resultados auditivos similares a pacientes no afectos de malformaciones de oído interno. 4.- Los peores resultados obtenidos por las malformaciones mayores se deben a la falta de tonotopía coclear, la falta de fibras neurales y a la mayor necesidad de estímulo eléctrico para desencadenar la sensación auditiva. 5.- A pesar de las diferencias encontradas entre las distintas malformaciones, todos los pacientes afectos de malformaciones de oído interno se benefician de la implantación coclear como tratamiento rehabilitador de su hipoacusia, no existiendo por lo tanto contraindicación en la implantación coclear de estos pacientes. Únicamente la agenesia coclear y la ausencia de fibras aferentes cocleares se mantienen como contraindicación absoluta de la implantación coclear, siendo entonces la implantación en el tronco cerebral la alternativa terapéutica. 7.-BIBLIOGRAFIA 1.- Herman P, Van Den Abbeele T, Portier F, Marianowski R, Copin H et Tran Ba Huy P. Embryologie de l’oreille interne. Encycl Méd Chir (Editions Scientifiques et Médicales Elsevier SAS, Paris, tous droits réservés). Oto-rhinolaryngologie, 20-005-A-40,1997,10p. 2.- Oído. En: Bruce M. Carlson ed. Embriología humana y biología del desarrollo. 3 ed. Ed Elsevier;2005.p.306-316. 3.- Daniel Choo. The role of the hindbrain in patterning of the otocyst. Dev Biol 2007;308:257-265. 4.- Desarrollo del oído. En: Biología del desarrollo. Fundamentos de Embriología por José Mª Genis Gálvez. 1 ed. Ed Espaxs;1970.p.219-226. 5.- Oído. En: Langman ed. Embriología médica con orientación clínica. 10 ed Ed médica Panamericana;2007.p.329-334. 6.- Nathan Jeffery and Fred Sopor. Prenatal growth and development of the modern human labyrinth. J.Anat 2004;204:71-92. 7.- Ojo y oído. En: Moore-Persand ed. Embriología clínica. 5 ed. Ed Interamericana Mc Graw Hill;1995.p.465-474. 8.- Laura Vitale Romo and hugh D. Curtin. Anomalous Facial Nerve Canal with Cochlear Malformations. Am J Neuroradiol 2001;22:838-844. 9.- Caroline D. Robson. Congenital hearing impairment. Pediatr Radiol 2006; 36:309-324. 10.- Adunka, Oliver F. et al. Internal Auditory Canal Morphology in Children with Cochlear Nerve Deficiency. Otol Neurotol 2006;27:793-801. 11.- W.M. Luxford y J.A. Rivas. Implantación coclear en oídos con malformaciones congénitas. En: Manuel Manrique Rodríguez, Alicia Huarte Irujo eds. Implantes cocleares. Barcelona: Ed. Masson;2002.p.229-234. 12.- Blake C. Papsin, MD. Cochlear Implantation in Children With Anomalous Cochleovestibular Anatomy. Laryngoscope 2005;115:1-19. 13.- Jackler R.K, Luxford W.M, House W.F . Congenital malformations of the inner ear: a classification based on embryogenesis. Laryngoscope 1987;97:214. 14.- A. Ramos, J.Cervera, A.Valdivieso, D. Pérez, J.R. Vasallo, J.M. Cuyas. Implante coclear en malformaciones congénitas. Acta otorrinolaringol Esp 2005; 56:343-348. 15.- Kathlyn Marsot-Dupuch, Alessandro Domínguez-Brito, Karim Ghasli and Claude-Henri Chouard. CT and MR findings of Michel Anomaly: Inner ear Aplasia. AJNR Am J Neuroradiol1999;20:281-284. 16.- Cristoph Arnoldner, Wolf Dieter Baumgartner, Wolfgang Gstoettner, Brigitte Egelierler, Christian Czerny, Erich Steiner et al. Audiological Performance after cochlear implantation in children with inner ear malformations. Int J Pediatr Otorhinolaryngol 2004;68:457-467. 17.- J. Cervera Escario, A. Ramos Macías. Malformaciones congénitas de oído interno. Suplementos de actualización en ORL 2005;1:20-26. 18.- Sennaroglu, Levent; Saatci, Isil. Unpartioned Versus Incompletely Partioned Cochleae: Radiological Differentiation. Otol Neurotol 2004;25:520529. 19.- Yiqing Zheng et al. The shortened cochlea: its classification and histopathologic features. Int J Pediatr Otorhinolaryngol 2002;63:29-39. 20.- Sennaroglu, Levent; Saatci, Isil. A new Classification for Cochleovestibular Malformations. Laryngoscope 2002;112:2230-2241. 21.- P. Clarós, A.Clarós Jr. y A.Clarós. Disontogenias en otología. Anomalías en el desarrollo del oído interno. En: Suárez C., Gil-Carcedo L.M., Marco J, Medina J.E., Ortega P., Trinidad J. eds. Tratado de Otorrinolaringología y Cirugía de cabeza y cuello. 2 ed. Buenos Aires; Madrid: Médica Panamericana; 2007.p.1325-40. 22.- John I.Lane, E.Paul Lindell, Robert J.Witte, David R.DeLone and Colin L.W.Driscoll. Middle and Inner Ear: Improved Depiction with Multiplanar Reconstruction of Volumetric CT Data. RadioGraphics 2006;26:115-124. 23.-P. Clarós, J.J. Sanz, M.A. Clavería, C. Costa, A. Clarós. Implante coclear en paciente con dilatación del saco endolinfático y del acueducto del vestíbulo. Acta Otorrinolaringol Esp 2005;56:132-134. 24.- H.Y. Yuen, A.T. Ahuja, K.T. Wong, V.Yue and A.C. van Hasselt. Computed Tomography of Common Congenital Lesions of the Temporal Bone. Clin Radiol 2003;58:687-693. 25.- Lisa H.Lowe; L. Gilbert Vézina. Sensorineural Hearing Loss in Children. RadioGraphics 1997;17:1079-1093. 26.- Zheng, Yiqing; Schachern, Patricia A.; Djalilian, Hamid R.; Paparella, Michael M. Temporal Bone Histopathology Related to Cochlear Implantation in Congenital Malformation of the Bony Cochlea. Otol Neurotol 2002;23:181-186. 27.- Patología malformativa. En: Manuel Trujillo Peco ed. Otología y Otoneurología. Imagen Diagnóstica. 1 ed. Barcelona: Ed Ars Médica;2006.p.4957. 28.- Shannon P. Pryor, MD et al. Investigation of the Role of Congenital Cytomegalovirus Infection in the Etiology of Enlarged Vestibular Aqueducts. Arch Otolaryngol Head Neck Surg 2005;131:388-392. 29.- Wu, Chen-Chi et al. Common Clinical Features of Children With Enlarged Vestibular Aqueduct and Mondini Dysplasia. Laryngoscope 2005;115:132-137. 30.- John E. McClay, et al. Major and Minor Temporal Bone Abnormalities in Children With and Without Congenital Sensorineural Hearing Loss. Arch Otolaryngol Head Neck Surg 2002;128:664-671. 31.- Özgün Ilhan Demir, Handan Cakmakci, Taner Kemal Erdag, Süleyman Men. Narrow duplicated internal auditory canal: radiological findings and review of the literature. Pediatr Radiol 2005;35:1220-1223. 32.- S. Bisdas, M.Lenarz, T. Lenarz, H. Becker. The abnormally dilated internal auditory canal: a non-specific finding or a distinctive pathologic entity. J Neuroradiol 2006;33:275-277. 33.- Alessandro de Alarcón, Robert A. Jahrsdoerfer, and Bradley W. Kesser. Congenital Absence of the Oval Window: Diagnosis, Surgery and Audiometric Outcomes. Otol Neurotol 2007;29:23 -28. 34.- Kenneth L. Tyler and Bernard N. Fields. Biología de los virus. Enfermedades virales. En: Isselbacher, Braunwald, Wilson, Martin, Fauci y Kasper eds. Harrison. Principios de Medicina Interna. 13 ed. Madrid: Ed Mc Graw-Hill;1997.p.893-902. 35.- Moerike S, Pantzar JT, De Sa D. Temporal bone pathology in fetuses exposed to isotretinoin. Pediatr Dev Pathol 2002;5:405-409. 36.- Denoyelle F.,Marlin S. Surdités de perception d’origine génétique.Encycl Méd Chir (Editions Scientifiques et Médicales Elsevier SAS, Paris, tous droits réservés). Oto-rhino-laryngologie, 20-191-A-10, 2005. 37.- Shelley D. Smith and Lee A. Harker. Single gene influences on radiologically-detectable malformations of the inner ear. J Commun Disord 1998;31:391- 410. 38.- Moshe Goldfeld et al. CT of the Ear in Pendred Syndrome. Radiology 2005;235:537-540. 39.- Elena Hernández et al. Síndrome braquio-oto-renal y colesteatoma congénito. ORL-DIPS 2003;30:222-225. Disponible en: URL: www.nexusediciones.com/pdf/orldips2003_4/or-30-4-009.pdf 40.- Blaser, Susan et al. Inner Ear Dysplasia is Common in Children with Down Síndrome (trisomy 21). Laryngoscope 2006;116:2113-2119. 41.- Sotirios Bisdas, Minoo Lenarz, Thomas Lenarz and Hartmut Becker. Inner ear Abnormalities in Patients with Goldenhar Syndrome. Otol Neurotol 2005;26:398-404. 42.- A.K. Morimoto et al. Absent Semicircular Canals in CHARGE Syndrome: Radiologic Spectrum of Findings. Am J Neuroradiol 2006;27:1663-1671. 43.- Craig A. Buchman et al. Cochlear Implantation in Children with Congenital Inner Ear Malformations. Laryngoscope 2004;114:309-316. 44.- Sennaroglu L, Saatci I, Aralasmak A, Gursel B, Turan E. Magnetic resonance imaging versus computed tomography in preoperative evaluation of cochlear implant candidates with congenital hearing loss. J Laryngol Otol 2002;116:804-810. 45- Dauman R, Carbonnière B, Soriano V, Berger-Lautissier S, Bouyé J,Debruge E, Coriat G et Bébéar JP. Implants cochlèaires chez l’adulte et l’enfant. Encycl Méd Chir (Editions Scientifiques et Médicales Elsevier SAS, Paris, tous droits réservés). Oto-rhino-laryngologie, 20-185-D-10, 1998, 12p. 46.- Eisen MD. Djourno, Eyries and the first implanted electrical neural stimulator to restore hearing. Otol Neurotol 2003;24:500-506. 47.- C. Morera, J. Marco, A. Huarte, A. Ramos y M. Manrique. Evaluación y selección de los pacientes y del tipo de dispositivos para el implante coclear. En: Suárez C., Gil-Carcedo L.M., Marco J, Medina J.E., Ortega P., Trinidad J. eds. Tratado de Otorrinolaringología y Cirugía de cabeza y cuello. 2 ed. Buenos Aires; Madrid: Médica Panamericana; 2007.p.1779-1791. 48.- M. J. Manrique Rodríguez y F. Portillo Corado. Definición, clasificación y funcionamiento de un implante coclear. En: Manuel J. Manrique Rodríguez, Ángel Ramos Macías, Pedro López Villarejo y Emilio García-Ibáñez Ferrándiz eds. Prótesis implantables en Otocirugía. Ponencia Oficial del LIV Congreso Nacional de la Sociedad Española de Otorrinolaringología y Patología Cérvicofacial; 2003 Nov 3-7; Madrid, España;2003.p.141-148. 49.- A. Ramos Macías, A. Wiehoff, L. Cabrera, P. Alemán, J. Rivero Suárez y S. Alayón. Evaluación radiológica. En: Manuel J. Manrique Rodríguez, Ángel Ramos Macías, Pedro López Villarejo y Emilio García-Ibáñez Ferrándiz eds. Prótesis implantables en Otocirugía. Ponencia Oficial del LIV Congreso Nacional de la Sociedad Española de Otorrinolaringología y Patología Cérvicofacial; 2003 Nov 3-7; Madrid, España;2003.p.161-166. 50.- M.Trujillo Peco, C. Rodríguez Guirado y J. M. Trujillo Jiménez. Exploración otológica por la imagen. En: Suárez C., Gil-Carcedo L.M., Marco J, Medina J.E., Ortega P., Trinidad J. eds. Tratado de Otorrinolaringología y Cirugía de cabeza y cuello. 2 ed. Buenos Aires; Madrid: Médica Panamericana; 2007.p.1277-1323. 51.- Jeffrey P. Simons, MD; David L. Mandell, MD; Ellis M. Arjmand, MD. Computed tomography and magnetic resonance imaging in paediatric unilateral and asymmetric sensorineural hearing loss. Arch Otolaryngol Head Neck Surg 2006;132:186-192. 52.- Girard N, Magnan J, Caces F, Chays A et Raybaud C. Imagerie de l’angle pontocérébelleux et du conduit auditif interne normal et pathologique. Encycl Méd Chir (Editions Scientifiques et Médicales Elsevier SAS, Paris, tous droits réservés). Oto-rhino-laryngologie, 20-047-A-80, 1997, 10p. 53.- M. Manrique, C. Morera y A. Huarte. Resultados en implantes cocleares. Rehabilitación de los pacientes implantados. En: Suárez C., Gil-Carcedo L.M., Marco J, Medina J.E., Ortega P., Trinidad J. eds. Tratado de Otorrinolaringología y Cirugía de cabeza y cuello. 2 ed. Buenos Aires; Madrid: Médica Panamericana; 2007.p.1807-1820. 54.- M. Sainz Quevedo y A. de la Torre. Programación del implante coclear. En : Manuel J. Manrique Rodríguez, Ángel Ramos Macías, Pedro López Villarejo y Emilio García-Ibáñez Ferrándiz eds. Prótesis implantables en Otocirugía. Ponencia Oficial del LIV Congreso Nacional de la Sociedad Española de Otorrinolaringología y Patología Cérvico-facial; 2003 Nov 3-7; Madrid, España;2003.p.187-194. 55.- Sennaroglu, Levent; Sarac, Sarp; Ergin, Turan. Surgical results of Cochlear Implantation in Malformed Cochlea. Otol Neurotol 2006;27:615-623. 56.- F.J Cervera-Paz y M.J. Manrique Rodríguez. Complicaciones. En: Manuel Manrique Rodríguez, Alicia Huarte Irujo eds. Implantes cocleares. Barcelona: Ed. Masson;2002.p.241-250. 57.- Natalie Loundon, Nicolas Leboulanger, Janine Maillet, Agnès Riggouzzo, Patrick Richard, Sandrine Marlin, Erea Noel Garabedian. Cochlear implant and inner ear malformation. Proposal for an hyperosmolar therapy at surgery. Int J Pediatr Otorhinolaryngol 2008; 72:541-547. 58.- A. Ramos Macías, A. Osorio Acosta, J.R. Vasallo Morillas y D. Pérez Plasencia. Técnica quirúrgica en casos de cócleas alteradas: osificaciones y malformaciones. En: Manuel J. Manrique Rodríguez, Ángel Ramos Macías, Pedro López Villarejo y Emilio García-Ibáñez Ferrándiz eds. Prótesis implantables en Otocirugía. Ponencia Oficial del LIV Congreso Nacional de la Sociedad Española de Otorrinolaringología y Patología Cérvico-facial; 2003 Nov 3-7; Madrid, España;2003.p.175-185. 59.- Cushing, Sharon L. et al. Incidence and Characteristics of Facial Nerve Stimulation in Children With Cochlear Implants. Laryngoscope 2006;116:17871791. 60.- Smullen, Jennifer L. et al. Facial Nerve Stimulation after Cochlear Implantation. Laryngoscope 2005;115:977-982. 61.- Mylanus, Emmanuel A.M.; Rotteveel, Liselotte J.C; Leeuw, Rens L. Congenital Malformation of the Inner Ear and Paediatric Cochlear Implantation. Otol Neurotol 2004;25:308-317. 62.- Kronenberg, J; Baumgartner, W; Migirov, L; Dagan, T; Hildesheimer, M. The suprameatal approach: an alternative surgical approach to cochlear implantation. Otol Neurotol 2004;25:41-44. 63.- Vincent Callanan and Chistopher Poje. Cochlear implantation and meningitis. Int J Pediatr Otorhinolaringol 2004;68:545-550. 64.- Géraldine Pettersen, Philippe Ovetchkine and Bruce Tapiero. Group A Streptococcal Meningitis in a Pediatric Patient following Cochlear Implantation: Report of the First Case and Review of the Literature. J Clin Microbiol 2005;43: 5816-5818. 65.- Summerfield AQ, Cirstea SE, Roberts KL, Barton GR, Graham JM, O´Donoghue GM. Incidence of meningitis and of death from all causes among users of cochlear implants in the United Kingdom. J Public Health (Oxf).2005; 27:55-61. 66.- Eisenman, David J.; Ashbaugh, Carissa; Zwolen, teresa A.; Arta, H. Alexander; Telian, Steven A. Implantation of the Malformed Cochlea. Otol Neurotol 2001;22:834-841. 67.- Fishman AJ, Roland JT Jr, Alexiades G, Mierzwinski J, Cohen NL. Fluoroscopically assisted cochlear implantation. Otol Neurotol 2003;24:882-886. 68.- Maruyama H, Ideguchi T, Ohura H, Azuma T, Orita S, Amano K, Higashida Y. Usefulness of the opposite direction for Stenvers' method. Nippon Hoshasen Gijutsu Gakkai Zasshi 2003;59:872-878. 69.- R. Jain and S.K. Mukherji. Cochlear Implant Failure: Imaging Evaluation of the Electrode Course. Clin Radiol 2003;58:288-293. 70.- Oído. En: Bruce M. Carlson ed. Embriología humana y biología del desarrollo. 3 ed. Ed Elsevier;2005. Tablas del desarrollo / xiii. 71.- Bradley M. Patten ed. Embriología Humana. 2 ed. Buenos Aires: Ed Librería « El Ateneo »;1958.p.198-201. 72.- Test EARS. Evaluation of auditory responses to speech. 73.- Westerhof JP, Rademaker J, Weber BP, Becker H. Congenital malformations of the inner ear and the vestibulocochlear nerve in children with sensorineural hearing loss : evaluation with CT and MRI. J Comput Assist Tomogr 2001;25:719-726. 74.- Selina Noramly, Robert M. Grainger. Determination of the Embryonic Inner Ear. J. Neurobiol 2002;53:100-128. 75.- Hortensia Sánchez-Calderón, Marta Milo, Yolanda León and Isabel Varela-Nieto. A network of growth and transcription factors controls neuronal differentiation and survival in the developing ear. Int J Dev Biol 2007;51:557570. 76.- Jinwoong Bok, Weise Chang and Doris K. Wu. Patterning and morphogenesis of the vertebrate inner ear. Int J Dev Biol 2007;51:521-533. 77.-C.C.D Shute Asymmetrical Growth of neuro-epithelia in the human labyrinth. J Anat 1952;86:246-258. 78.- Donna M. Fekete and Andrea M. Campero. Axon guidance in the inner ear. Int J Biol 2007;51:549-556. 79.- Tanya T. Whitfield and Katherine L. Hammond. Axial Patterning in the developing vertebrate inner ear. Int J Biol 2007;51:507-520. 80.- Ömer Özdogmus, Ozan Sezen, Utku Kubilay, Erdinç Saka, Ugur Duman, Tangül San et al. Connections between the facial, vestibular and cochlear nerve bundles within the internal auditory canal. J. Anat 2004;205:6575. 81.- Loizou, P. C., Poroy, O., Dorman, M. The effect of parametric variations of cochlear implant processors on speech understanding. Acoust Soc Am 2000;108:790-802. 82.- Kiefer J, von Ilberg C, Rupprecht V et al. Optimized speech understanding with the continuous interleaved sampling speech coding strategy in patients with cochlear implants: effect of variations in stimulation rate and number of channels. Ann Otol Rhinol Laryngol 2000;109:1009-1020. 83.- Rubinstein JT, Hong R. Signal coding in cochlear implants: exploiting stochastic effects of electrical stimulation. Ann Otol Rhinol Laryngol Suppl 2003;191:14-19. 84.- Jackler R.K, Luxford W.M, House W.F. Sound detection with the cochlear implant in five ears of four children with congenital malformations of the cochlea. Laryngoscope 1987;97:15-7. 85.- Kim LS, Jeong SW, Huh MJ, Park YD. Cochlear implantation in children with inner ear malformations. Ann Otol Rhinol Laryngol 2006;115:205-214. 86.- Loundon N, Rouillon I, Munier N, Marlin S, Roger G, Garabedian EN Cochlear implantation in children with internal ear malformations. Otol Neurotol 2005;26:668-673. 87.- Colletti V, Carner M, Miorelli V, Guida M, Colletti L, Fiorino F. Auditory brainstem implant (ABI): new frontiers in adults and children. Otolaryngol Head Neck Surg 2005;133:126-138. 88.- Colletti L. Beneficial auditory and cognitive effects of auditory brainstem implantation in children. Acta Otolaryngol 2007;127:943-946. 89.- Bouccara D, Kalamarides M, Bozorg Grayeli A, Ambert-Dahan E, Rey A, Sterkers O. Auditory brainstem implant: indications and results. Ann Otolaryngol Chir Cervicofac 2007;124:148-154. 90.- Colletti V, Carner M, Miorelli V, Guida M, Colletti L, Fiorino F. Cochlear implant failure: is an auditory brainstem implant the answer?. Acta Otolaryngol 2004;124:353-357. 91.- Colletti V, Shannon R, Carner M, Veronese S, Colletti L. Outcomes in Nontumor Adults Fitted With the Auditory Brainstem Implant: 10 Years´ Experience. Otol Neurotol 2009;30:614-618. 92.- Eisenberg LS et al. Comprehensive evaluation of a child with an auditory brainstem implant. Otol Neurotol 2008;29:251-257. 93.- Carner M et al. Imaging in 28 children with cochlear nerve aplasia. Acta Otolaryngol 2009;129:458-461. 94.- Colletti V, Fiorino FG, Carner M, Giarbini N, Sacchetto L, Cumer G. Advantages of the Retrosigmoid Approach in Auditory Brain Stem Implantation. Skull Base Surg 2000;10:165-170. 95.- Colletti L, Zocconte L. Nonverbal cognitive abilities and auditory performance in children fitted with auditory brainstem implants: preliminary report. Laryngoscope 2008;118:1443-1448. 96.-Seki Y. Restoration of hearing—comparing auditory brainstem implant to cochlear implant. Brain nerve 2007;59:323-329. 6 7