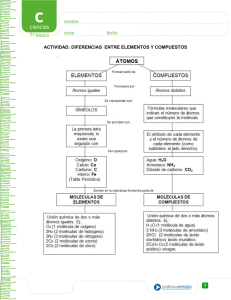
bloque temático i estructura y propiedades de las moléculas orgánicas
Anuncio

BLOQUE TEMÁTICO I ESTRUCTURA Y PROPIEDADES DE LAS MOLÉCULAS ORGÁNICAS Química Orgánica I-www.sinorg.uji.es BLOQUE TEMÁTICO I: ESTRUCTURA Y PROPIEDADES DE LAS MOLÉCULAS ORGÁNICAS Unidad temática I.1: Conceptos fundamentales en Química Orgánica I.1.1. La Química Orgánica como ciencia. I.1.2. El enlace en las moléculas orgánicas. I.1.2.1. El enlace simple Carbono-Carbono I.1.2.2. El doble enlace Carbono-Carbono I.1.2.3. El triple enlace Carbono-Carbono I.1.3. Electronegatividad y polaridad I.1.4. Efecto inductivo Unidad temática I.2 I.2.1. Enlaces deslocalizados: Efecto resonante I.2.1.1. Energía de resonancia I.2.2. Aromaticidad. Unidad temática I.3. Propiedades de los compuestos orgánicos I.3.1. Enlaces más débiles que el enlace covalente I.3.1.1. Las fuerzas, o interacciones, de Van der Waals I.3.1.1.a. Fuerzas dipolo permanente-dipolo permanente (fuerzas de Keesom) I.3.1.1.b. Fuerzas dipolo permanente-dipolo inducido (fuerzas de Debye) I.3.1.1.c. Fuerzas de dispersión de London I.3.1.2. Interacciones intermoleculares por puente de hidrógeno I.3.2. Estructura y propiedades físicas de los compuestos orgánicos I.3.2.1. Puntos de ebullición I.3.2.2. Solubilidad I.3.3. Acidez en las moléculas orgánicas I.3.3.1. Efecto inductivo y fuerza ácida 1.3.3.2. Efectos estéricos y fuerza ácida I.3.3.3. Influencia de otros efectos sobre la fuerza ácida I.3.4. Basicidad en las moléculas orgánicas I.3.4.1. Efecto inductivo y de solvatación en la fuerza básica I.3.4.2. Efecto resonante y fuerza básica 3 4 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) Unidad temática I.1: Conceptos fundamentales en Química Orgánica I.1.1. La Química Orgánica como ciencia La Química Orgánica se ocupa del estudio de las propiedades y transformaciones de los compuestos que contienen el elemento carbono. El elevado número y complejidad de estos compuestos se debe a las características de enlazamiento del carbono, que puede formar enlaces hasta con cuatro átomos más. Además, este elemento se puede unir a otros átomos de carbono para dar lugar a largas cadenas constituidas por cientos e incluso miles de átomos. El carbono puede formar enlaces estables con muchos átomos distintos de la tabla periódica y además, puede formar diferentes tipos de enlaces: simples, dobles o triples. La diversidad de la química que tiene como base el carbono no es sorprendente si se piensa en las diferencias que presentan las formas del carbono elemental: diamante y grafito. El diamante es duro e incoloro mientras que el grafito es suave y negro. Estas diferencias en las propiedades de las distintas formas del carbono son consecuencia de las diferencias estructurales que presentan dichas formas. La Química Orgánica, junto con la Bioquímica, es la ciencia básica que permite explicar los procesos químicos que tienen lugar en los organismos vivos. De hecho, el nombre Química Orgánica proviene de la antigua creencia de que ciertas sustancias sólo podían ser producidas por organismos vivos. Los pueblos prehistóricos hicieron uso de las propiedades de algunos compuestos orgánicos y realizaron algunas reacciones químico-orgánicas. Los antiguos egipcios, los romanos y los fenicios emplearon varios colorantes que eran verdaderos compuesto químicos puros: el índigo, la alizarina y la legendaria púrpura de Tiro. Los dos primeros colorantes se aislaron de las plantas y el último se obtuvo en pequeñas cantidades a partir de una especie rara de molusco. Desde muy antiguo se sabía que la grasa animal se podía convertir en jabón por tratamiento con lejía. Hasta época tan reciente como 1948, los químicos orgánicos no pudieron sintetizar productos que fueran capaces de competir con el jabón (detergentes). La fermentación del almidón y de los azúcares para obtener alcohol se conoce desde tiempos prehistóricos y el método que se aplica en la actualidad no difiere mucho del que se ideó hace cientos de años. La Química Orgánica, tal y como hoy la conocemos, arranca de finales del siglo XVIII cuando se inició el aislamiento de sustancias orgánicas de extractos de origen natural. En este orden de cosas son dignos de mención los estudios que el alemán Carl Scheele llevó a cabo entre los años 1769 a 1786 sobre aislamiento de diversos compuestos orgánicos de fuentes naturales. En 1784, Lavoisier ideó un método, basado en la combustión de la materia orgánica, que permitía determinar los porcentajes de carbono, hidrógeno, oxígeno y nitrógeno que constituían los compuestos orgánicos. En 1807, el químico sueco Berzelius denominó, con el nombre de compuestos orgánicos, a aquellos compuestos derivados de los seres vivos o de la materia viva. Durante todo el siglo XIX, Berzelius y otros químicos creyeron que tales compuestos poseían una fuerza vital y que, por tanto, sería imposible sintetizar un compuesto orgánico a partir de materiales inorgánicos. La teoría de la fuerza vital fue declinando a medida que la aportación creciente de datos analíticos evidenciaba que las leyes químicas que gobernaban el comportamiento de la materia inorgánica eran también válidas para los compuestos orgánicos. 5 Química Orgánica I-www.sinorg.uji.es La teoría de la fuerza vital sufrió un gran revés en 1828, año en el que Wöhler consiguió sintetizar la urea por descomposición térmica del isocianato amónico. Según la clasificación de Berzelius la urea era un compuesto orgánico, poseedor de fuerza vital y, por tanto, imposible de ser sintetizado a partir de compuestos clasificados como inorgánicos: O Δ NH4 NCO isocianato amónico H2N NH2 urea La síntesis de la urea obligó a un replanteamiento de la definición de compuesto orgánico, pasándose a denominar como tal todo compuesto que contuviese carbono en su estructura. Durante el primer tercio de siglo XIX investigadores como Gay-Lussac, Liebig y Berzelius descubrieron y perfeccionaron nuevos métodos analíticos que permitieron determinar la clase de elementos, así como su proporción, que constituían los compuestos orgánicos. Hacia mitad del siglo XIX, el desarrollo incipiente de la síntesis orgánica permitió la preparación de compuestos orgánicos a partir de materiales de partida relativamente simples. Uno de los aspectos de la Química que se resistía a los esfuerzos de las mentes más brillantes del siglo XIX era el relacionado con la estructura de los compuestos orgánicos. Se sabía, por ejemplo, que el alcohol etílico y el dimetiléter tenían la misma fórmula molecular, C2H6O, pero mientras que el primero es un líquido con punto de ebullición 78°C, el segundo es un gas. Los químicos del siglo XIX pensaron que las diferentes propiedades químicas que presentaban compuestos con la misma fórmula molecular se tenían que deber a la forma en la que se ordenaban los átomos en la estructura molecular. I.1.2. El enlace en las moléculas orgánicas En 1858 Kekulé propuso una teoría estructural que permitía asignar la estructura de los compuestos orgánicos más simples. Esta teoría se basaba en la tetravalencia del átomo de carbono y en el concepto de enlace químico, y fue la base de partida para la asignación de las estructuras de moléculas orgánicas sencillas, tales como el metano, el etano o el propano. La teoría estructural de Kekulé permitió explicar el fenómeno de la isomería, es decir la presencia de diferentes propiedades físicas y/o químicas en compuestos con la misma fórmula molecular. En 1916, la introducción del concepto de enlace covalente por el químico estadounidense Lewis proporcionó la base que permitió relacionar las estructuras de las moléculas orgánicas y sus propiedades químicas. Según Lewis una capa llena de electrones es especialmente estable y los átomos transfieren o comparten electrones para tratar de alcanzar una capa llena de electrones y alcanzar, así, la estructura electrónica estable similar a la del gas noble más próximo, que normalmente contiene 8 electrones en su capa más externa. La tendencia de los átomos a adquirir la configuración electrónica externa de 8 electrones se la conoce como regla del octeto. Cuando dos átomos comparten dos electrones entre sí se forma entre ellos un enlace covalente. Los átomos, de acuerdo con su configuración electrónica, pueden cumplir la regla del octeto con pares de electrones compartidos (electrones enlazantes) y pares de electrones sin compartir (electrones no enlazantes). 6 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) Las estructuras de Lewis utilizan un punto para representar a un electrón de valencia, y un par de puntos o una línea para representar a pares de electrones. A continuación, se indica la representación de Lewis de algunas moléculas orgánicas, como el etano, la metilamina, el metanol y el clorometano. Nótese que estas tres últimas contienen átomos que consiguen su octeto electrónico mediante la suma de electrones enlazantes y no enlazantes, como el caso del átomo de nitrógeno de la metilamina, del átomo de oxígeno del metanol, o del átomo de cloro del clorometano. Representación de Lewis de algunos compuestos orgánicos H H H C C H H H Etano H H C N H H H Metilamina H H H C C O H H H Etanol H H C Cl H Clorometano I.1.2.1. El enlace simple Carbono-Carbono El compuesto orgánico más simple es el metano CH4, que como es sabido tiene estructura tetraédrica. La justificación de esta estructura vino de la mano de la teoría mecanocuántica. Así, un átomo de carbono en su estado fundamental tiene dos electrones desapareados. energía creciente 2p n=2 2s n=1 1s estado electrónico fundamental del carbono Se debería esperar que en lugar de formar CH4, el carbono se uniera sólo a dos átomos de hidrógeno y formara CH2, dejando vacío un orbital 2p. El CH2 es una especie química conocida, llamada carbeno, pero es una sustancia muy reactiva y de tiempo de vida media muy corto. Por adición de 96 kcal/mol de energía a un átomo de carbono, uno de los electrones 2s puede promocionarse hasta alcanzar el orbital vacío 2p, dando lugar a la configuración electrónica indicada a continuación: 7 Química Orgánica I-www.sinorg.uji.es energía creciente 2p n=2 2s n=1 1s estado electrónico del átomo de carbono excitado Al promocionar un electrón desde el orbital 2s al 2p el átomo de carbono tiene disponibles cuatro electrones para formar cuatro enlaces covalentes y de esta forma puede conseguir la configuración electrónica de gas noble. La formación de un enlace covalente produce un descenso de energía en el sistema. Por ejemplo, la formación de un enlace C-H produce un descenso de energía de 87 kcal/mol. Por tanto, la formación de dos enlaces covalentes más en el átomo de carbono provoca un descenso de 174 kcal/mol de energía, que compensa sobradamente los 96 kcal/mol que se requieren para promover al átomo de carbono desde el estado fundamental al estado excitado. Este razonamiento explica por qué el átomo de carbono tiende a ser tetravalente en lugar de divalente. Sin embargo, no explica la forma tetraédrica de la molécula de metano. Si admitimos que el átomo de carbono en la molécula de metano participa con el orbital 2s y los tres orbitales 2p, hay que concluir que se formarían tres enlaces covalentes por solapamiento C2p-H1s, y el cuarto enlace covalente se formaría por solapamiento C2s-H1s. Esto significaría que tres de los ángulos H-C-H serían de 90º, y los otros quedarían indeterminados. H H C H H Estructura que debería presentar el metano si los orbitales enlazantes no están hibidrizados Orbitales 1s y 2p del átomo de carbono El sistema de un orbital 2s y tres orbitales 2p, mutuamente perpendiculares, es una solución satisfactoria aproximada para la ecuación de Schroedinger para la capa n=2, pero pueden formularse combinaciones lineales de estos cuatro orbitales que también sean soluciones satisfactorias para la ecuación de Schroedinger. Matemáticamente está permitido combinar los orbitales 2s y 2p de cualquier modo, con la condición de que en la formación de los cuatro 8 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) orbitales nuevos se empleen exactamente un orbital s y tres p. Una forma de llevar a cabo tal combinación consiste en formar cuatro orbitales nuevos, cada uno de los cuales tiene ¼ de carácter s y ¾ de carácter p. Los cuatro orbitales híbridos son entonces equivalentes entre sí y, teniendo en cuenta que contienen triple carácter p que s, se les denomina híbridos sp3. El contorno de densidad electrónica de un orbital sp3 presenta dos lóbulos, como un orbital p, pero en este caso los lóbulos son bastante desiguales en tamaño. Diagrama de contorno de densidad electrónica de un orbital híbrido sp3 Para formar un enlace fuerte es necesario que los electrones estén situados entre los núcleos de los átomos. Un orbital sp3 puede situar mucha más densidad electrónica, en una dirección determinada, que la que sitúa un orbital s o un orbital p. Por consiguiente, un enlace covalente que se forme con la participación de un orbital sp3 del átomo de carbono será más fuerte que un enlace covalente en el que participe un orbital p o un orbital s. La energía de un enlace covalente que se forma mediante el solapamiento entre el orbital híbrido sp3 del carbono y el orbital 1s del hidrógeno es de 103 kcal/mol, mientras que los enlaces covalentes correspondientes C2p-H1s y C2s-H1s tienen una energía de 60 kcal/mol y 80 kcal/mol. Los cuatro orbitales híbridos sp3 del carbono se sitúan en direcciones tales que forman entre ellos ángulos de 109.5º, como si se dirigieran hacia los vértices de un tetraedro regular. Densidad electrónica alrededor de un átomo de carbono con hibridación sp3. La densidad electrónica total es esférica Los orbitales híbridos sp3 dan la mejor explicación para la formación de enlaces en los hidrocarburos saturados porque el átomo de carbono tiene la misma energía, tanto si está hibridizado como si no lo está, pero la configuración hibridizada puede formar enlaces más fuertes. La geometría tetraédrica tiene una ventaja adicional, puesto que permite que los núcleos de hidrógeno estén lo más alejados posible entre sí para una longitud de enlace C-H. Y puesto que estos núcleos están todos cargados positivamente, cuanto más alejados estén, en igualdad de otras condiciones, menor será la energía del sistema. La molécula del metano es un tetraedro perfecto con ángulos de enlace de 109.5º. La distancia de enlace C-H es de 1.09 Å. La hibridación sp3 en el átomo de carbono explica la estructura del metano. Cada enlace C-H se forma por solapamiento de un orbital sp3 del carbono con un orbital 1s del hidrógeno. 9 Química Orgánica I-www.sinorg.uji.es H o H H C 1.09 A H 109.5º El etano CH3-CH3 está compuesto por dos grupos metilo. Cada átomo de carbono presenta una hibridación sp3 y se une a los átomos de hidrógeno mediante un enlace σ formado por solapamiento del orbital 1s del hidrógeno con un orbital sp3 del carbono. Además, existe un enlace σ C-C formado por el solapamiento de un orbital sp3 de un átomo de carbono con el otro orbital sp3 del otro átomo de carbono. Los dos grupos metilo no están fijos en una posición determinada, sino que pueden girar con relativa libertad alrededor del enlace sigma que mantiene unidos a los dos átomos de carbono. El enlace σ mantiene el solapamiento lineal cuando giran los átomos de carbono (véase el bloque temático II para el concepto de isomería conformacional). H H H giro de 60º H H H H Conformación eclipsada del etano H H H H H Conforrmación alternada del etano conformaciones alternadas del etano I.1.2.2. El doble enlace Carbono-Carbono Como se acaba de ver, cuando se comparte un par de electrones entre dos átomos se forma un enlace simple. Muchas moléculas orgánicas contienen átomos que comparten dos pares electrónicos, como la del etileno, y se dice que estos átomos están unidos mediante un enlace doble. También hay estructuras orgánicas con átomos que comparten tres pares de electrones, como los de la molécula de acetileno, y en este caso se dice que el enlace entre los átomos es un triple enlace. 10 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) Representación de Lewis del etileno y del acetileno H H C C H H Etileno H C C H Acetileno Los alquenos son hidrocarburos con enlaces dobles carbono-carbono. Se les denomina también olefinas. El alqueno más simple es el etileno cuya fórmula molecular es C2H4. El doble enlace se representa, en una estructura de Lewis, mediante dos pares de electrones entre los átomos de carbono. La longitud del enlace C=C en el etileno es de 1.33 Å, mucho más corto que el enlace simple C-C del etano que es de 1.54 Å. La longitud del enlace C-H en el etileno es de 1.08 Å, ligeramente menor que el enlace C-H en el etano que es de 1.09 Å. Los ángulos de enlace de C-C-H y H-C-H son de 121.7° y 116.6° respectivamente. H o o 1.33 A C 1.08 A H 116.6o C 121.7o o 1.54 A H H C H C o H H H H 1.09 A etileno H etano Estas distancias y ángulos de enlace se pueden explicar admitiendo que los dos átomos de carbono que forman el doble enlace presentan una hidridación sp2 y que el doble enlace está constituido por un enlace σ y un enlace π. El enlace σ se forma por solapamiento de los orbitales sp2 de cada átomo de carbono. Cada uno de los enlaces C-H se forma por solapamiento de un orbital híbrido sp2 del carbono con el orbital 1s del hidrógeno. 2p 2p H H C C sp2 H C H H H C H H Enlace σ (Csp2-Csp2) En la región de enlace carbono-carbono deben entrar dos electrones más. Cada átomo de carbono contiene todavía un orbital 2p no hibridizado. El orbital 2p consta de dos lóbulos y a cada uno se le da un signo que representa el signo algebraico de la función de onda en las diferentes regiones. Los signos de la función de onda no representan cargas. Indican que la función de onda de un orbital 2p tiene valor cero en el átomo de carbono. A esto se le denomina un nodo. Los nodos son puntos que marcan un cambio de signo de la función de onda (Nota= en estos apuntes los dos signos de la función de onda, + y -, se representan mediante dos colores diferentes en cada uno de los lóbulos orbitálicos). 11 Química Orgánica I-www.sinorg.uji.es Para que los dos orbitales p se recubran eficazmente, deben estar orientados paralelamente entre sí y perpendicularmente a la estructura del enlace σ, y además el signo de la función de onda tiene que coincidir. Para que esto ocurra, la estructura de los enlaces σ tiene que ser coplanar y los seis núcleos atómicos implicados en el doble enlace tienen que estar en el mismo plano. Si esto ocurre, los dos orbitales paralelos p están lo suficientemente cerca para solaparse en posición lateral y se pueden combinar de dos maneras: a) Cuando se recubren los lóbulos del mismo signo se forma un orbital molecular enlazante π. b) Si los signos de la función de onda no coinciden se genera un orbital molecular antienlazante π*. En el estado fundamental de un alqueno, los dos electrones que forman el enlace π entre los átomos de carbono están en el orbital molecular enlazante π. H C Energía C H no hay solapamiento H H H H C C H H Orbital π∗ antienlazante H H C H C H solapamiento enlazante H H C C H H Orbital π enlazante El solapamiento de los orbitales p es menos eficaz que el solapamiento frontal por el que se forman los orbitales σ. Por consiguiente un enlace π es más débil que un enlace σ. La longitud del enlace C-H es menor en el etileno que en el etano por dos razones: a) El enlace σ del etileno está formado por el solapamiento de dos orbitales sp2 del carbono (33.3% de carácter s), mientras que el enlace σ en el etano está formado por el solapamiento de dos orbitales sp3 (25% de carácter s). b) El solapamiento de los orbitales p que forman el enlace π aproxima a los dos átomos de carbono. Isomería cis-trans La energía de disociación del doble enlace C=C es aproximadamente de 146 kcal/mol y la energía de disociación de un enlace simple C-C es de 83 kcal/mol. Por tanto, la energía de disociación del enlace π debe ser de 63 kcal/mol. Los extremos de la molécula de etileno no pueden torcerse entre sí, porque para ello se debería romper el enlace π. A diferencia de lo que 12 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) ocurre en los enlaces simples, en los enlaces dobles C=C no hay libre rotación. Este es el origen de la isomería cis-trans. Por ejemplo, hay dos alquenos que responden al nombre de 2-buteno: el cis-2-buteno y el trans-2-buteno (véase el bloque temático II para el concepto de isomería configuracional de doble enlace). CH3 H3C C H H3C C C H H C CH3 H cis-2-buteno trans-2-buteno I.1.2.3. El triple enlace Carbono-Carbono Los alquinos son hidrocarburos que contienen un triple enlace C-C. Se les denomina también hidrocarburos acetilénicos porque derivan del alquino más simple que se llama acetileno. La estructura de Lewis del acetileno muestra tres pares de electrones en la región entre los núcleos de carbono. H C C H Acetileno El acetileno tiene una estructura lineal que se explica admitiendo una hidridación sp en cada uno de los átomos de carbono. El solapamiento de dos orbitales sp entre sí genera el enlace σ C-C. Por otra parte, el solapamiento del orbital sp con el orbital 1s del hidrógeno forma el enlace σ C-H. Los dos enlaces π se originan por solapamiento de los dos orbitales p que quedan en cada uno de los dos átomos de carbono. El solapamiento de estos orbitales forma un cilindro de densidad electrónica que circunda al enlace σ C-C. Enlace σ Csp-Csp Enlace σ Csp-H1s C H C H H Sistema de orbitales σ del acetileno C H C Sistema de orbitales π del acetileno La longitud del enlace C-C en el acetileno es de 1.20 Å y cada uno de los enlaces C-H tiene una distancia de 1.06 Å. Los dos enlaces, tanto el C-C como el C-H, son más cortos que los enlaces correspondientes en el etano y en el etileno. 1.54 Å H H 1.33 Å H C C H H 1.09Å C H H 1.20 Å H H C H 1.08Å H C C 1.06 Å H 13 Química Orgánica I-www.sinorg.uji.es etano etileno acetileno El triple enlace es relativamente corto debido al solapamiento de los tres pares de electrones y al elevado carácter s de los orbitales hidridos sp (50% de carácter s), lo que aproxima más a los átomos de carbono que forman el enlace σ del acetileno. I.1.3. Electronegatividad y polaridad Cuando dos átomos comparten por igual los dos electrones del enlace covalente se dice que el enlace es no polar, como ocurre en el enlace covalente de la molécula de hidrógeno, en el enlace covalente de la molécula de cloro, o en el enlace covalente carbono-carbono del etano. Sin embargo, la mayor parte de los enlaces covalentes están formados por dos átomos diferentes, de manera que los electrones del enlace son atraídos con mayor intensidad por uno de los dos átomos que forman el enlace. Cuando esto ocurre el enlace covalente se denomina enlace polar. Por ejemplo, cuando el carbono se enlaza al cloro el par de electrones del enlace se encuentra atraído con más intensidad por el átomo de cloro, de manera que sobre el átomo de carbono aparece una pequeña carga parcial positiva y sobre el átomo de cloro aparece una cantidad igual de carga negativa. En la siguiente figura se indica el enlace covalente polar C-Cl de la molécula de clorometano. La polaridad del enlace se indica con una flecha que dirige su punta hacia el extremo negativo del enlace polar y un signo mas (+) en el extremo positivo del enlace. H μ δ− δ+ H C Cl H Clorometano La polaridad del enlace se mide mediante su momento dipolar (μ) que se define como la cantidad de diferencia de carga multiplicada por la longitud del enlace. El símbolo δ+ quiere decir una pequeña cantidad de carga positiva y el símbolo δ- quiere decir una pequeña cantidad de carga negativa. A fin de predecir si un enlace covalente va a ser polar se recurre a la comparación de las electronegatividades de los átomos que forman el enlace. La electronegatividad se define como la tendencia del núcleo atómico a la atracción de electrones. Pauling desarrolló una escala de electronegatividades relativas para la mayoría de los átomos. En el Sistema Periódico la electronegatividad aumenta de izquierda a derecha y disminuye al bajar en una columna, por lo que el flúor es el elemento más electronegativo. A continuación, se da una tabla de electronegatividades para los principales átomos de interés en Química Orgánica. 14 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) H 2.2 Li 1.0 Na 0.9 Be 1.6 Mg 1.3 B 1.8 Al 1.6 C 2.5 Si 1.9 N 3.0 P 2.2 O 3.4 S 2.6 F 4.0 Cl 3.2 Br 3.0 I 2.7 Como se deduce de la tabla anterior, un enlace C-H debería estar muy poco polarizado, puesto que la electronegatividad del hidrógeno y del carbono es similar. Sin embargo, los halógenos, el oxígeno y el nitrógeno, que son heteroátomos que suelen aparecer en las estructuras orgánicas, son más electronegativos que el carbono y, por tanto, los enlaces Chalógeno, C-O y C-N son polares. A continuación, se representan las estructuras de Lewis de las moléculas de fluoruro de hidrógeno (HF), agua (H2O) y amoníaco (NH3) con indicación de la polaridad de los enlaces. La molécula de metano se puede considerar que está constituida por enlaces C-H muy poco polarizados: δ + H δ− F − δ+ δ δ+ H O H Fluoruro de hidrógeno Agua δ+ H H N δ+ δ− δ+ H Amoniaco H H C H H Metano En algunos de los temas de esta asignatura se dará una representación del contorno de densidad electrónica de determinadas moléculas. La asimetría en la distribución de carga se indicará con un sistema de colores que varía de tonalidad según el valor del potencial electrostático: el color rojo indica una zona de la estructura con elevada densidad de carga negativa, debido a presencia de átomos muy electronegativos, mientras que un color azul indica una zona de la estructura con déficit de carga debido a la presencia de átomos poco electronegativos. En la siguiente figura se muestra esta variación del color respecto al signo del potencial: rojo < potencial electrostático más negativo naranja < amarillo < verde < azul potencial electrostático más positivo En la siguiente figura se muestran los contornos de potencial electrostático que presentan las moléculas descritas anteriormente: 15 Química Orgánica I-www.sinorg.uji.es El enlace H-F del fluoruro de hidrógeno está fuertemente polarizado y la densidad de carga a lo largo del enlace entre el flúor y el hidrógeno está desplazada hacia el átomo más electronegativo (flúor) creando un potencial electrostático negativo alrededor de dicho átomo (color rojo) y en consecuencia un potencial electrostático positivo alrededor del hidrógeno (color azul). Lo mismo ocurre en el caso de la molécula de agua, en el que la mayor electronegatividad del oxígeno provoca la polarización de los enlaces O-H. El caso del amoníaco es similar al de la molécula de agua: el nitrógeno es más electronegativo que el hidrógeno y, por tanto, los tres enlaces N-H son polares. La densidad de carga se halla desplazada hacia el nitrógeno, lo cual se aprecia perfectamente en el diagrama de contorno de potencial electrostático debido a la coloración rojiza en la parte superior de la figura (posición del nitrógeno), y a la aparición de una zona de color azul en la parte inferior, en la cual se ubican los tres átomos de hidrógeno. En el metano no existen enlaces polares debido a las electronegatividades similares de los átomos de carbono y de hidrógeno. La distribución simétrica de la densidad de carga conlleva la aparición de un potencial electrostático más bien neutro (verde) alrededor de todos los átomos de la molécula. I.1.4. Efecto inductivo La diferente electronegatividad de los átomos que constituyen las moléculas orgánicas, y las interacciones secundarias entre sus orbitales provocan, la aparición de ciertos efectos de polarización y deslocalización electrónica. Uno de ellos es el denominado efecto inductivo, que se puede definir como la polarización de enlaces provocada por un átomo o un grupo atómico a lo largo de una cadena carbonada σ. En la siguiente figura se indica el efecto inductivo provocado por el átomo de cloro en la molécula de cloroetano: Efecto inductivo en el cloroetano H δ H H C C H H δ Cl δ El enlace C-Cl de la molécula de cloroetano está polarizado debido a la diferencia de electronegatividad entre el carbono y el cloro. Como consecuencia de ello, sobre el átomo de cloro existe una fracción de carga negativa y sobre el átomo de carbono una fracción de carga 16 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) positiva. Este átomo de carbono cargado positivamente atrae hacia sí los tres pares de electrones que le unen a los otros tres átomos y por tanto en el otro átomo de carbono de la molécula aparece una fracción de carga positiva, aunque menor que en el átomo de carbono unido directamente al cloro. El efecto inductivo es permanente y por tanto no es un fenómeno que ocurra en un momento dado en la molécula. Hay que tener en cuenta que el efecto inductivo decrece rápidamente al aumentar la distancia con respecto al origen del desplazamiento electrónico, y en la práctica se puede despreciar a partir del segundo átomo de la cadena. El efecto inductivo puede ser electrón-atrayente, como el del átomo de cloro en el cloroetano, o electrón-dador como el del grupo alquilo en la molécula anterior. El efecto inductivo se representa transformando el enlace en una flecha y se indica como +I o –I según el efecto sea electrón-dador o electrón-atrayente. Tomando como referencia el hidrógeno, cuyo efecto inductivo se considera 0, aquellos átomos, o grupos de átomos, cuya electronegatividad es mayor que la del C se considera que tienen efecto inductivo –I. Este tipo de átomos, o grupos de átomos, se clasifican como electrónatrayentes, ya que atraen hacia sí el par de electrones del enlace, dejando sobre el átomo de carbono al que están unidos una densidad de carga positiva. Por otro lado, aquellos átomos, o grupos de átomos, que tienden a desplazar la densidad electrónica sobre el átomo de carbono al que están unidos se dice que tienen un efecto inductivo +I, y se clasifican como grupos electrón-dadores. En este grupo cabe clasificar a los radicales alquílicos, los metales, y los grupos cargados negativamente. En la tabla que se da a continuación se da una clasificación de los principales grupos orgánicos según sus efectos inductivos electrón-atrayentes o electrón-dadores. Los grupos alquilo, de los cuales se indican en la tabla solo los más usuales como el t-butilo, el isopropilo, el etilo o el metilo, presentan todos efecto inductivo electrón-dador (+I). Efectos inductivos Efecto electrón-atrayente (-I) Efecto electrón-dador (+I) - -NO2 (grupo nitro) -O (grupo alcóxido) -CN (grupo ciano) -COO (grupo carboxilato) -COOH (grupo carboxilo) -C(CH3)3 (grupo t-butilo) -X (halógenos) -CH(CH3)2 (grupo isopropilo -OCH3 (grupo metoxilo) -CH2CH3 (grupo etilo) -OH (grupo hidroxilo) -CH3 (grupo metilo) - En los enlaces polares las cargas parciales sobre los átomos son reales. Sin embargo, cuando se dibujan las estructuras de Lewis de determinadas especies químicas aparecen cargas eléctricas asociadas a algunos átomos. Estas cargas se denominan cargas formales y se calculan según la siguiente ecuación: 17 Química Orgánica I-www.sinorg.uji.es Carga formal = nº electrones capa de valencia - nº electrones no compartidos + nº electrones enlazantes 2 A continuación se dibujan las estructuras de Lewis del anión carbonato y del nitrometano. O O C O O Anión carbonato H 3C N O Nitrometano En dos de los átomos de oxígeno del anión carbonato aparece una carga formal negativa. Por otro lado, en la molécula de nitrometano aparece una carga formal positiva sobre el átomo de nitrógeno y una carga formal negativa sobre uno de los dos átomos de oxígeno. En la siguiente figura se indica el cálculo de la carga formal de cada uno de los átomos que integran el anión carbonato: carga formal = 6 - ( 4 + O O 4 )= 0 2 8 )= 0 2 carga formal = 4 - ( 0 + C O carga formal = 6 - ( 6 + Anión carbonato 2 ) =-1 2 El mismo cálculo se indica a continuación para los átomos de oxígeno y nitrógeno de la molécula de nitrometano: carga formal = 6 - ( 4 + 4 )= 0 2 O H 3C carga formal = 5 - ( 0 + N O Nitrometano carga formal = 6 - ( 6 + 8 ) = +1 2 2 2 ) = -1 Dos átomos diferentes que poseen la misma configuración electrónica en la capa de valencia, aunque posean distinta carga formal, se denominan átomos isoelectrónicos. Por ejemplo, el átomo de fluor del fluorometano y el átomo de oxígeno del anión metóxido son átomos isoelectrónicos, al igual que el átomo de carbono del metano y el átomo de nitrógeno del catión amonio. Átomos isoelectrónicos H H C F H Fluorometano H H C O H Anión metóxido Átomos isoelectrónicos H H C H H Metano H H N H H Catión amonio 18 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) Unidad temática I.2 I.2.1. Enlaces deslocalizados: efecto resonante Se denominan enlaces conjugados a los enlaces múltiples, ya sean dobles o triples enlaces que están separados por un enlace simple. La presencia en una estructura de enlaces conjugados, o la presencia de enlaces múltiples contiguos a átomos con pares electrónicos libres, provoca la aparición del efecto resonante o mesómero, también conocido como efecto conjugativo (se describe como efecto K, por la inicial de Konjugativ en alemán). El efecto resonante, mesómero o conjugativo, es un efecto estabilizante y se define como la deslocalización de la densidad electrónica π de un átomo, o grupo de átomos, sobre otros átomos de la estructura. La molécula de butadieno se puede representar con la estructura de Lewis I, que contiene dos enlaces dobles C-C conjugados, por tanto separados por un enlace simple C-C: Estructuras resonantes del butadieno H 2C H C I C H CH2 H 2C II H C C H CH2 H 2C H C C H CH2 III El efecto conjugativo, resonante o mesómero permite describir la estructura del butadieno con dos estructuras de Lewis adicionales, las numeradas como II y III en la figura anterior, las cuales contienen un enlace doble y dos cargas de signo opuesto en los extremos de la molécula. Las estructuras resonantes o mesómeras son aquellas que tienen la misma geometría pero difieren en su distribución electrónica. Este es el caso de las estructuras I, II y III de la figura anterior, cuya geometría (posición de los núcleos) es idéntica, pero se diferencian en la forma en la que se distribuye la densidad electrónica π. La molécula orgánica que se describe mediante la contribución de varias estructuras resonantes tiene características asociadas a cada una de aquéllas. En estos casos se dice que la molécula es un híbrido de resonancia de las estructuras resonantes. El método de la resonancia permite saber, de forma cualitativa, la estabilización que puede conseguir una molécula por deslocalización electrónica. Cuanto mayor sea el número de estructuras resonantes mediante las que se pueda describir una especie química mayor será su estabilidad. El concepto de estructuras resonantes se puede aplicar en la descripción del nitrometano, que se puede representar mediante las dos estructuras de Lewis que se indican a continuación: Estructuras resonantes del nitrometano O O H 3C N O I H 3C N O II 19 Química Orgánica I-www.sinorg.uji.es En realidad, el nitrometano no es la estructura resonante I ni tampoco la estructura resonante II, sino la estructura que resultaría al mezclar las características asociadas a la estructura I y a la II, tal y como se indica a continuación: Híbrido de resonancia del nitrometano δ O δ H 3C N O δ El problema de dibujar los compuestos orgánicos como híbridos de resonancia reside en la imposibilidad de contar el número de electrones sobre determinados átomos. Por ejemplo, en la estructura de híbrido de resonancia del nitrometano se hace difícil saber el número de electrones sobre el átomo de nitrógeno o sobre los átomos de oxígeno. Aunque los híbridos de resonancia dan una imagen más real del orden de enlace y de la distribución electrónica de la molécula no se suelen utilizar en los textos de Química Orgánica, debido al problema acabado de comentar. Una forma de escribir el híbrido de resonancia, que sí permite contar los electrones en cada átomo, consiste en encerrar entre corchetes a todas las estructuras resonantes conectándolas entre sí mediante una flecha de doble punta, tal y como se ha descrito más arriba. La mayor o menor contribución de las estructuras resonantes a la descripción de la molécula se puede relacionar con la mayor o menor estabilidad que teóricamente puede atribuirse a cada estructura. De forma cualitativa se puede evaluar esta mayor o menor estabilidad teniendo en cuenta los siguientes puntos: 1. Una estructura resonante será tanto más estable (contribuirá más al hibrido de resonancia) cuanto mayor sea el número de enlaces formales que posea. 2. Las estructuras iónicas con separación de cargas son más inestables (contribuyen menos al hibrido de resonancia) que las no cargadas. 3. Entre dos estructuras resonantes con separación de cargas, y en igualdad de otras condiciones, será más estable (contribuirá más al hibrido de resonancia) la estructura con la carga negativa en el átomo más electronegativo. 4. Las estructuras resonantes con octetos completos en todos los átomos de la segunda fila del Sistema Periódico son particularmente estables (contribuyen más al hibrido de resonancia), aunque ello suponga la presencia de una carga positiva en un átomo electronegativo. Para dibujar correctamente las estructuras resonantes de un compuesto conviene seguir las siguientes reglas: 1. Dibujar una estructura de Lewis para el compuesto en cuestión. 20 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) 2. Tomando como base la estructura de Lewis acabada de dibujar se dibuja otra estructura de Lewis de manera que: a) Todos los núcleos mantengan su posición original. b) Se mantenga el mismo número de electrones apareados. A continuación se aplican estas reglas para el dibujo de las estructuras resonantes de la acetamida: 1. Se dibuja la estructura de Lewis de la acetamida: O H 3C C I NH2 2. Tomando como base la estructura de Lewis acabada de dibujar, se dibuja otra estructura de Lewis que mantenga los núcleos en la misma posición y que contenga el mismo número de electrones apareados que la primera, tal y como se indica a continuación. Estructuras resonantes de la acetamida O O H 3C C I NH 2 H 3C C NH 2 II El método de las estructuras resonantes permite describir a la acetamida como un híbrido de resonancia entre las estructuras resonantes I y II. De las dos estructuras resonantes la que contribuye en mayor proporción en el híbrido de resonancia es la I, porque tiene un mayor número de enlaces y porque no comporta separación de cargas. Sin embargo, la estructura resonante II, aunque contribuye poco a la hora de determinar las propiedades físicas y químicas de la acetamida, pone de manifiesto que el enlace C-N debe tener un cierto carácter de doble enlace, como así ocurre en realidad. I.2.1.1. Energía de resonancia Como se ha indicado anteriormente, los dobles enlaces separados por tan sólo un enlace sencillo se denominan dobles enlaces conjugados, como en el 1,3-butadieno o en el 1,3pentadieno. Cuando los enlaces dobles están separados por dos o más enlaces sencillos no interaccionan entre sí, y se les denomina enlaces dobles aislados, como en el caso del 1,4pentadieno. 1,4-Pentadieno 1,3-Pentadieno (Dieno no conjugado) (Dieno conjugado) 21 Química Orgánica I-www.sinorg.uji.es Debido a la interacción entre los dobles enlaces, los sistemas con dobles enlaces conjugados son más estables que los sistemas con dobles enlaces aislados. Una forma de comparar la estabilidad relativa de los alquenos es medir el calor desprendido en la reacción de hidrogenación. Por ejemplo, la diferencia entre los calores de hidrogenación del 1-penteno (ΔH° = -30 Kcal/mol) y del trans-2-penteno (ΔH° = -27.4 Kcal/mol) indica que este último es 2.6 kcal/mol más estable que el alqueno monosustituido. Comparación entre los calores de hidrogenación del 1-penteno y del trans-2-penteno 1-penteno 2.6 Kcal/mol energía trans-2-penteno Hº = -30.0 kcal/mol Hº = -27.4 kcal/mol pentano Si una molécula tiene mas de un enlace doble y están aislados entre sí, el calor de hidrogenación se acerca a la suma de los calores de hidrogenación de los dobles enlaces individuales. Por ejemplo, el calor de hidrogenación del 1,4-pentadieno (dieno con enlaces dobles aislados) es de -60.2 kcal/mol, que es aproximadamente el doble del calor de hidrogenación del 1-penteno. H 2, Pt Hº = -60.2 Kcal /mol 1,4-Pentadieno Para el caso de un dieno conjugado, como el trans-1,3-pentadieno se puede calcular un calor de hidrogenación teórico que sería el que correspondería a la suma de los dos enlaces dobles. A continuación, se efectúa este cálculo para el trans-1,3-pentadieno. Cálculo del calor de hidrogenación teórico del trans-1,3-pentadieno 1 enlace doble similar al del 1-penteno trans-1,3-pentadieno 1 enlace doble similar al del trans-2-penteno Hº = -30.0 kcal/mol Hº = -27.4 kcal/mol Hº = -57.4 kcal/mol 22 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) Según este cálculo, la hidrogenación del trans-1,3-pentadieno debería desprender, aproximadamente, 57.4 kcal/mol. Sin embargo, cuando se lleva a cabo la hidrogenación del trans-1,3-pentadieno, y se determina en un calorímetro el calor desprendido, se observa que son sólo 53.7 Kcal/mol, y por tanto existe una diferencia de 3.7 kcal/mol entre el valor teórico y el valor experimental. Este valor indica que el dieno conjugado tiene una estabilidad adicional de unas 3.7 kcal/mol, que se denomina energía de resonancia o energía de conjugación. En el siguiente diagrama se comparan gráficamente los calores de hidrogenación teórico y experimental del trans-1,3-pentadieno, apreciándose claramente la estabilización adicional, calculada en 3.7 kcal/mol, de este dieno conjugado con respecto a la hipotética situación no conjugada. Comparación entre los calores de hidrogenación teórico y experimental del trans-1,3-pentadieno 1,3-pentadieno teórico 3.7 Kcal/mol (Energía de resonancia) 1,3-pentadieno energía Hº = -57.4 kcal/mol Hº = -53.7 kcal/mol pentano Esta estabilización adicional de los dienos conjugados es una situación general. Por ejemplo, el 1,3-butadieno es un dieno conjugado y, teniendo en cuenta que el calor de hidrogenación experimental del 1-buteno es 30.2 kcal/mol, su calor de hidrogenación teórico sería de 60.4 kcal/mol, tal y como se calcula a continuación: H2, Pt Hº = -30.2 kcal/mol 1-buteno o H teórico =2 x (-30.2 kcal/mol) = -60.4 kcal/mol 1,3-butadieno Sin embargo el calor de hidrogenación experimental del 1,3-butadieno es de sólo -56.8 kcal/mol. La diferencia entre el valor teórico y el experimental, que es de 3.6 kcal/mol, es la 23 Química Orgánica I-www.sinorg.uji.es energía de resonancia o de estabilización del 1,3-butadieno. Comparación entre los calores de hidrogenación teórico y experimental del 1,3-butadieno 1,3-butadieno teórico 3.6 Kcal/mol (Energía de resonancia) 1,3-butadieno energía Hº = -60.4 kcal/mol Hº = -56.8 kcal/mol butano I.2.2. Aromaticidad El compuesto orgánico que representa de forma paradigmática el concepto de aromaticidad es el benceno. La primera estructura para este compuesto fue propuesta por el químico alemán Friedrich August Kekulé von Stradonitz (1829-1896) en 1865, y consistía en una mezcla en equilibrio de dos ciclohexatrienos, formados con enlaces sencillos y dobles alternados. En la estructura de Kekulé los enlaces sencillos serían más largos (1.47 Å) que los enlaces dobles (1.33 Å). Cuando se desarrollaron los métodos físicos de determinación estructural y se pudo medir la distancia de enlace C-C del benceno se encontró que todas las distancias eran iguales y median 1.39 Å, que es un promedio entre la distancia de un enlace doble (1.33 Å) y un enlace simple (1.47 Å). Estuctura del hipotético 1,3,5-ciclohexatrieno Estructura real del benceno o 1.33A H H C C o H 1.47A C H C C H H C H H H H C C C C C H C H H H H H H H o enlaces idénticos (1.39A) 24 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) Aparte de las características físicas del benceno, que no resultan explicadas por las estructuras de 1,3,5-ciclohexatrieno, existen una serie de propiedades químicas del benceno que tampoco resultan explicadas por las estructuras de enlaces dobles alternados que propuso Kekulé. De hecho, la hidrogenación del benceno es mucho más lenta que la de los alquenos y requiere condiciones mucho más drásticas: alta presión de hidrógeno y empleo de catalizadores muy activos. Antes se ha visto cómo la comparación de los calores de hidrogenación permite cuantificar la estabilidad relativa de los dienos conjugados con respecto a los dienos no conjugados. En el caso del benceno se puede recurrir al mismo método para determinar cuál es la estabilidad adicional asociada a este compuesto en comparación con otros alquenos cíclicos. En la siguiente gráfica se representan los calores de hidrogenación, determinados experimentalmente, del ciclohexeno, del 1,4-ciclohexadieno, del 1,3-ciclohexadieno y del benceno. También se representa, a modo de comparación, el calor de hidrogenación teórico del hipotético 1,3,5-ciclohexatrieno. 1,3,5-ciclohexatrieno -32.9 kcal/mol de energía de resonancia 1.8 kcal/mol energía de resonancia energía -57.4 kcal/mol -55.4 kcal/mol -82.2 kcal/mol - 49.8 kcal/mol -28.6 kcal/mol energía del ciclohexano Cuando el ciclohexeno se hidrogena a ciclohexano se desprenden 28.6 kcal/mol. El 1,4ciclohexadieno, un dieno con conjugado, libera en la hidrogenación 57.4 kcal/mol, aproximadamente el doble del calor de hidrogenación del ciclohexeno. La hidrogenación del 1,3-ciclohexadieno, un dieno conjugado libera 55.4 kcal/mol, 1.8 kcal/mol menos que el doble del valor del ciclohexeno. Una energía de resonancia de 1.8 kcal/mol es típica para un dieno conjugado. Para el hipotético 1,3,5-ciclohexatrieno se puede calcular un calor de hidrogenación de: 0 H = - (3 x 28.6 - 2 x 1.8) kcal/mol = - 82.2 kcal /mol 25 Química Orgánica I-www.sinorg.uji.es Al contrario que los anteriores alquenos, que se hidrogenan a presión atmosférica, la hidrogenación del benceno necesita de elevadas presiones de hidrógeno y de catalizadores muy activos. Cuando se produce la hidrogenación sólo se liberan 49.8 kcal/mol, 32.9 kcal/mol menos que el hipotético calor de hidrogenación del 1,3,5-ciclohexatrieno. A esta diferencia de energía se le conoce como energía de resonancia del benceno. Se dice que el benceno es un hidrocarburo aromático, que es un término que engloba a aquellos compuestos que posen una elevada estabilidad termodinámica y una reactividad química diferente de la de los alquenos y polienos conjugados. La remarcable estabilidad del benceno se puede explicar si se admite la deslocalización de la densidad electrónica asociada a los orbitales p. Las estructuras resonantes se diferencian en la distribución de la densidad electrónica pero no en la posición relativa de los átomos que las integran. En realidad el benceno es un híbrido de resonancia cuyos enlaces π están deslocalizados, con un orden de enlace de aproximadamente 1 ½ entre los átomos de carbono adyacentes. Esto explica que las longitudes de enlace C-C en el benceno sean más cortas que las de los enlaces simples, pero más largas que las de los dobles enlaces. Como los enlaces π están deslocalizados en el anillo a menudo se inscribe un círculo en el hexágono, en lugar de trazar los enlaces dobles localizados. Diferentes representaciones del benceno orden de enlace = 1 1/2 representación de resonancia Por tanto, el benceno consiste en un anillo formado por seis átomos de carbono con hibridación sp2, enlazados entre sí mediante enlaces σ Csp2-Csp2. Cada uno de los átomos de carbono se enlaza además a un átomo de hidrógeno mediante un enlace σ Csp2-H1s. Todos los enlaces C-C tienen la misma longitud y todos los ángulos de enlace son de 120º. Como los átomos de carbono presentan hibridación sp2, cada átomo de carbono tiene un orbital p perpendicular al plano del anillo que se solapa con los orbitales p de los carbonos contiguos para formar un círculo de densidad electrónica π por encima y por debajo del plano molecular. La representación del benceno como un hexágono regular con un círculo en el centro evoca el solapamiento cíclico de los seis orbitales 2p. Representación de la densidad electrónica π del benceno H H H H H H 26 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) Orbitales moleculares del benceno El sistema de orbitales π del benceno se forma en realidad por combinación lineal de los seis orbitales 2p asociados a los átomos de carbono. Esta combinación genera seis orbitales moleculares π de los cuales tres son enlazantes y tres son antienlazantes y son los que se muestran a continuación: Sistema de orbitales moleculares π del benceno 3. O.M. antienlazantes energía 3. O.M. enlazantes La ocupación de los orbitales enlazantes en el benceno es óptima puesto que no se sitúa ningún electrón en los destructivos orbitales antienlazantes. Se puede afirmar, a la vista del diagrama anterior, que la estabilidad del benceno se debe a un grupo de orbitales moleculares de baja energía, que son capaces de acomodar de forma altamente eficiente toda la densidad electrónica asociada a los electrones π. El benceno se engloba dentro de la familia de los compuestos aromáticos. Los componentes de esta familia cumplen las siguientes condiciones: 1) Sus estructuras son cíclicas y contienen enlaces dobles conjugados. 2) Cada átomo de carbono del anillo presenta hidridación sp2, u ocasionalmente sp, con al menos un orbital p no hidridizado. 3) Los orbitales p se solapan formando un anillo continuo de orbitales paralelos. La estructura debe ser plana o casi plana para que el solapamiento de los orbitales p sea efectivo. Química Orgánica I-www.sinorg.uji.es 27 4) Además, los compuestos aromáticos deben cumplir la regla de Hückel cuyo enunciado es el siguiente: Un compuesto aromático contiene 4n+2 electrones π en el sistema cíclico, siendo n un número entero. Si el número de electrones π es 4n, siendo n un número entero, el compuesto es antiaromático. 28 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) Unidad temática I.3. Propiedades de los compuestos orgánicos I.3.1. Enlaces más débiles que el enlace covalente El enlace covalente es la unión que explica el mantenimiento de la unidad estructural de un compuesto orgánico. Además de este enlace intramolecular se pueden dar interacciones entre las moléculas, que son mucho más débiles que los enlaces covalentes, pero que a menudo son las responsables de las propiedades físicas de los compuestos orgánicos. Este tipo de interacciones intermoleculares son de especial importancia en el estado sólido y en el estado líquido, situaciones en las que las moléculas están en íntimo contacto. Los puntos de fusión, de ebullición y las solubilidades de los compuestos orgánicos muestran los efectos de estas fuerzas. Las principales fuerzas intermoleculares son: 1) Las fuerzas de Van der Waals, que se clasifican a su vez en: a) Fuerzas dipolo permanente-dipolo permanente (fuerzas de Keesom) b) Fuerzas dipolo permanente-dipolo inducido (fuerzas de Debye) c) Fuerzas dipolo inducido-dipolo inducido (fuerzas de dispersión de London) 2) Los enlaces por puentes de hidrógeno I.3.1.1. Las fuerzas, o interacciones, de Van der Waals Las fuerzas, o interacciones, de van der Waals son las fuerzas atractivas o repulsivas que se establecen entre las moléculas, ya sean éstas polares o apolares. El valor de las atracciones intermoleculares de tipo van der Waals es bajo, oscilando entre 0.1 y 35 KJ/mol. Sin embargo, estas fuerzas juegan un papel fundamental en campos tan diversos como la química supramolecular, la biología estructural, la ciencia de polímeros, la nanotecnología, la ciencia de superficies, y la física de materia condensada. Existen básicamente tres tipos de interacciones de van der Waals, que se comentan a continuación. I.3.1.1. a. Fuerzas dipolo permanente-dipolo permanente (fuerzas de Keesom) Las fuerzas de atracción que se establecen entre dipolos permanentes se denominan fuerzas de Keesom. El valor de este tipo de interacciones es proporcional a los valores de los momentos dipolares de las moléculas, y se debe a la atracción electrostática entre la zona cargada negativamente de una molécula y la zona cargada positivamente positiva de la otra. Esta atracción electrostática provoca que las moléculas se vayan orientando unas con respecto a otras (véase la figura que se representa a continuación): 29 Química Orgánica I-www.sinorg.uji.es Orientación de moléculas polares debido a las fuerzas de Keesom Las fuerzas de Keesom explican por qué los gases constituidos por moléculas polares, como el SO2 o el HCl, son fácilmente licuables: al disminuir la temperatura, decrece la agitación térmica, los dipolos se orientan entre sí, las moléculas se asocian y se produce un estado más condensado (líquido). I.3.1.1.b. Fuerzas dipolo permanente-dipolo inducido (fuerzas de Debye) Si se aplica un campo eléctrico a una molécula su densidad electrónica responde generando y distribuyendo pequeñas cargas eléctricas en los átomos de la molécula, que en su conjunto se anulan. Por ejemplo, la molécula de cloro carece de momento dipolar permanente, pero si se aplica un campo eléctrico en la dirección del eje molecular, el par de electrones del enlace covalente se polariza, y cada uno de los átomos de cloro que constituyen la molécula adquiere una pequeña carga eléctrica igual, pero de signo opuesto, apareciendo un momento dipolar inducido en la dirección del campo eléctrico. Esta polarización se mide en debyes, en honor a Peter Debye. Cl Cl Aplicación de un campo eléctrico δ δ Cl Cl Las moléculas polares, que contienen un dipolo permanente, actúan como pequeños campos eléctricos, provocando la polarización de las moléculas apolares cercanas. Este fenómeno de polarización inducida es una fuente de atracción entre moléculas y explica la disolución de algunos gases apolares, como el Cl2, en disolventes polares. I.3.1.1.c. Fuerzas de dispersión de London En las moléculas no polares la principal fuerza de atracción es la fuerza de dispersión de London, que surge de la interacción entre dipolos inducidos que se generan temporalmente en las moléculas. Así, la aproximación de las nubes electrónicas de dos moléculas apolares provoca un desplazamiento transitorio de la densidad electrónica de aquéllas, induciéndose un momento dipolar pequeño y temporal que provoca una atracción intermolecular. Estos dipolos temporales solo duran una fracción de segundo y cambian continuamente de orientación. La fuerza de 30 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) atracción entre dipolos inducidos depende del peso molecular y del contacto superficial entre las moléculas y, por tanto, es proporcional al área molecular. El incremento de las fuerzas de dispersión de London, debido al aumento del peso molecular y al de la superficie de contacto entre las moléculas, explica el aumento de la temperatura de ebullición al pasar de metano (temperatura de ebullición = -165ºC), a propano (temperatura de ebullición = -44.5ºC) y a butano (temperatura de ebullición = -0.5ºC). Metano (CH 4 ) Propano (CH 3 CH 2CH3 ) Peso molecular = 16.0 g/mol Temp. ebullición = -165ºC Peso molecular = 44.1 g/mol Temp. ebullición = -44.5ºC Butano (CH 3CH2 CH 2 CH 3) Peso molecular = 58.1 g/mol Temp. ebullición = -0.5ºC En la siguiente figura se representan los puntos de ebullición y las estructuras de los tres isómeros del pentano (el n-pentano, el iso-pentano (2-metilbutano) y el neo-pentano (2,2dimetilpropano)). n- Pentano i so-Pentano CH3 CH 2 CH 2CH 2CH3 CH 3 CH3 CHCH2 CH 3 Peso molecular = 72.2 g/mol Temp. ebullición = 36.0ºC Peso molecular = 72.2 g/mol Temp. ebullición = 28.0ºC neo-Pentano CH 3 H3 C C CH 3 CH 3 Peso molecular = 72.2 g/mol Temp. ebullición = 9.5ºC La diferencia de temperatura de ebullición entre el isómero lineal n-pentano y el ramificado neo-pentano (2,2-dimetilpropano) es de 27 °C. Esta diferencia se explica por la forma lineal de las moléculas de n-pentano, que provoca un aumento de las fuerzas intermolecularas de dispersión de London. Por el contrario, las fuerzas de dispersión de London entre las moléculas cuasi-esféricas del neo-pentano son mucho más débiles, lo que se traduce en una disminución de su temperatura de ebullición. Química Orgánica I-www.sinorg.uji.es 31 I.3.1.2. Interacciones intermoleculares por puente de hidrógeno Los enlaces de hidrógeno se encuentran en toda la naturaleza. Por ejemplo, son los responsables de las particulares propiedades del agua, las cuales permiten el desarrollo de la vida en la Tierra. Los enlaces de hidrógeno proveen también la fuerza intermolecular que mantiene unidas ambas hebras en una molécula de ADN. El enlace por puente de hidrógeno es un tipo especial de interacción dipolo-dipolo y ocurre cuando un átomo de hidrógeno se encuentra enlazado a un átomo fuertemente electronegativo como el nitrógeno, el oxígeno o el flúor. En estos casos el átomo de hidrógeno posee una carga parcial positiva y puede interactuar con otros átomos electronegativos de una molécula adyacente. Cuanto mayor sea la diferencia de electronegatividad entre el átomo de hidrógeno y el átomo interactuante, más fuerte será el enlace por puente de hidrógeno. Así, el enlace de hidrógeno formado con el F será de mayor intensidad que el formado con el O, y éste a su vez será más intenso que el formado con el N. A continuación se representan, a modo de comparación, las interacciones por puente de hidrógeno entre moléculas de agua, entre moléculas de amoníaco (NH3) y ente moléculas de fluoruro de hidrógeno (HF): El enlace por puente de hidrógeno es mucho más débil que un enlace covalente normal OH, N-H y F-H. La energía de un enlace por puente de hidrógeno puede oscilar entre 8 y 40 KJ/mol. En la siguiente figura se describe la interacción entre dos moléculas de agua mediante un puente de hidrógeno y la formación de agregados intermoleculares de agua por el efecto de los puentes de hidrógeno: 32 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) El etanol y del dimetil éter son dos compuestos isoméricos de fórmula molecular C2H6O. Sin embargo, el etanol hierve a 78ºC mientras que el punto de ebullición del dimetiléter es de 25ºC. En la siguiente figura se representan esquemáticamente las interacciones por puente de hidrógeno entre las moléculas de etanol y las interacciones dipolo-dipolo, mucho más débiles, entre las moléculas de dimetil éter. Interacciones por puente de hidrógeno en el etanol δ− δ− O δ− H3C-H2C O δ− H+ δ CH2-CH3 H δ+ δ− δ− O δ− H3C-H2C O δ− H+ δ CH2-CH3 H δ+ δ− δ− H3C-H2C O δ H δ+ Interacciones dipolo-dipolo en el dimetil éter − H O δ− δ+ CH2-CH3 H3C δ+ H3C δ− O O H3C δ− δ+ CH3 H3C δ+ δ − O H3C H3C O H3C CH3 − δ+ δ O CH3 CH3 δ+ O H3C δ− δ+ H3C δ− δ+ H3C O H3C δ− δ+ H3C O δ− CH3 δ+ CH3 O δ− La diferencia de temperatura de 103° entre los puntos de ebullición del etanol y del dimetil éter se explica por la formación de puentes de hidrógeno entre las moléculas de etanol, y la ausencia de este tipo de interacciones en el dimetil éter, en el cual sólo actúan las fuerzas intermoleculares de tipo dipolo-dipolo (fuerzas de Keesom). I.3.2. Estructura y propiedades de los compuestos orgánicos Las fuerzas intermoleculares influyen de la siguiente manera en las propiedades físicas de los compuestos orgánicos: I.3.2.1. Puntos de ebullición El punto de ebullición se define como la temperatura a la cual la presión de vapor del líquido iguala a la presión del medio que rodea al líquido. Para que un compuesto se vaporice, las fuerzas que mantienen las moléculas unidas unas a otras deben romperse. Esto significa que el punto de ebullición de un compuesto depende de la atracción entre las moléculas. Si las moléculas se mantienen unidas por interacciones fuertes se necesitará mucha energía para separar las moléculas unas de otras y el compuesto tendrá un elevado punto de ebullición. Por otra parte, si las fuerzas intermoleculares son débiles, se necesitará una cantidad de energía relativamente baja para separar las moléculas unas de otras, y el compuesto tendrá un punto de ebullición bajo. Los alcanos, constituidos por moléculas apolares, tienen puntos de ebullición relativamente bajos porque las atracciones intermoleculares se deben a la interacción entre dipolos inducidos (fuerzas de dispersión de London), y este tipo de interacciones son de carácter débil. Las fuerzas de dispersión de London son proporcionales a la superficie de contacto ente las moléculas lo que explica que las alcanos formados por moléculas lineales (mayor superficie 33 Química Orgánica I-www.sinorg.uji.es de contacto) tengan mayores puntos de ebullición que sus isómeros formados por moléculas ramificadas (menor superficie de contacto) Los puntos de ebullición de los éteres, los haluros de alquilo, y en general de las moléculas con heteroátomos, son más altos que los de los hidrocarburos de similar peso molecular debido a la presencia de interacciones intermoleculares dipolo-dipolo, que son más intensas que las interacciones de London. En el caso de los alcoholes y las aminas, además de las interacciones entre dipolos, intervienen las fuerzas por puente de hidrógeno, mucho más fuertes que las primeras. Por ello, los puntos de ebullición de los alcoholes son más altos que los puntos de ebullición de los éteres de igual peso molecular. Los alcoholes también presentan mayor punto de ebullición que las aminas de igual peso molecular. La explicación a este hecho reside en la mayor electronegatividad del átomo de oxígeno en comparación con el átomo de nitrógeno, que hace que los puentes de hidrógeno O-H sean más fuertes que los puentes de hidrógeno N-H. I.3.2.2. Solubilidad Las fuerzas intermoleculares determinan la solubilidad de los compuestos orgánicos. La regla general es que lo semejante disuelve a lo semejante: las sustancias polares se disuelven en disolventes polares y las no polares en disolventes no polares. Hay cuatro casos distintos a la hora de considerar los efectos de la polaridad sobre la solubilidad. a) Un soluto polar con un disolvente polar, por ejemplo la disolución del cloruro sódico (NaCl, compuesto iónico, soluto muy polar) en agua (H2O, disolvente polar). Para conseguir la disolución de un cristal es necesario romper la atracción electrostática que lo iones de signo opuesto se ejercen mutuamente en una red cristalina. Para separar a los iones se necesita una gran cantidad de energía pero el agua, un disolvente muy polar, es capaz de disolver a muchos sólidos iónicos mediante un proceso de solvatación. En la disolución del NaCl, los cationes sodio (Na+) quedan rodeados por una esfera de moléculas de agua que dirigen su extremo negativo (el átomo de oxígeno) hacia los cationes. Solvatación de los aniones Cl- y los cationes Na+ por las moléculas de agua δ+ δ+ δ δO δ+ H + δ+ O δ H H H O H H δ+ δ- O δ+ Na Hδ + O H δ- δ+ δ+ δ+ H O δ- - δ- δ +δ + H H O δHδ+ δ δ+ O + H δ H H O δδ+ δ+H H H Hδ +δ + δ- O H Cl δH δ O δ H + + δ+ δ+ H H O δ- Al mismo tiempo, los aniones cloruro (Cl-) se rodean de una esfera de moléculas de agua que dirigen su extremo positivo (los átomos de hidrógeno) hacia los iones negativos. La energía 34 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) liberada en el proceso de solvatación, junto con el aumento de la entropía, compensa la energía necesaria para romper la red cristalina y el resultado neto es la disolución de los cristales de NaCl. Otro ejemplo de interacción entre un soluto polar y un disolvente polar es el de la disolución del HCl en agua. En este caso se produce la ordenación de las moléculas de soluto y disolvente de manera que las moléculas de agua rodean al cloruro de hidrógeno acercando los oxígenos a la parte positiva (H) del dipolo que forma la molécula de HCl y acercando los hidrógenos hacia la parte negativa (Cl) del dipolo que forma la molécula de HCl. En la siguiente figura se ha representado esta interacción para el caso general de una molécula polar simbolizada como Y-Z. Interacciones electrostáticas entre el agua y un soluto Y-Z b) Un soluto polar con un disolvente no polar, por ejemplo la adición de cloruro sódico (NaCl, soluto polar) a hexano (disolvente no polar). En este caso no se produce la disolución del soluto porque las moléculas no polares del hidrocarburo no solvatan a los iones y no pueden superar la gran energía que se necesita para romper la red cristalina. Cl Na Cl Na Cl Na Soluto polar: NaCl (red iónica) Na Cl Na Cl Na CH3 (CH 2) 4CH3 CH3 (CH 2) 4CH3 CH3 (CH 2) 4CH3 CH3 (CH 2) 4CH3 CH3 (CH 2) 4CH3 CH3 (CH 2) 4CH3 Cl CH3 (CH 2) 4CH3 Disolvente apolar: hexano (hidrocarburo) CH3 (CH 2) 4CH3 c) Un soluto no polar con un disolvente no polar, por ejemplo la disolución de la cera de parafina (soluto no polar) en hexano (disolvente no polar). La cera de parafina está constituida por largas moléculas de hidrocarburo no polares que se atraen débilmente por interacciones de 35 Química Orgánica I-www.sinorg.uji.es London. Estas atracciones también se establecen con el disolvente hexano, pero aunque el intercambio energético es mínimo, la cera se disuelve en el hexano debido al gran aumento de entropía que se produce en el proceso de disolución. En la siguiente figura se ha simbolizado la disolución de la cera de parafina (representada con una figura ovalada de color rojo) en el disolvente apolar hexano. Disolución de un soluto no polar en hexano CH3(CH2)4CH3 CH3(CH2)4CH3 CH3(CH2)4CH3 CH3(CH2)4CH3 CH3(CH2)4CH3 CH3(CH2)4CH3 CH3(CH2)4CH3 CH3(CH2)4CH3 CH3(CH2)4CH3 d) Un soluto no polar con un disolvente polar, como la interacción entre la cera de parafina (soluto no polar) y el agua (disolvente polar). La energía necesaria para separar a las moléculas apolares es mínima puesto que las interacciones que actúan entre este tipo de moléculas son las débiles fuerzas de London. Por el contrario, las interacciones entre las moléculas de agua son mucho más fuertes porque estas moléculas se mantienen unidas por enlaces de hidrógeno. Para que el soluto no polar se disuelva en agua es necesario que se intercale entre las moléculas del disolvente polar, lo que implica un proceso de separación de las moléculas de agua, y por tanto la consiguiente ruptura de puentes de hidrógeno. La energía necesaria que hay que invertir en este proceso no queda compensada por la energía desprendida en el proceso de solvatación, que es prácticamente nulo, y por tanto el soluto no se disuelve porque la red de puentes de hidrógeno de las moléculas de agua excluye a las moléculas de parafina. Soluto no polar Disolvente polar (agua) 36 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) 1.3.3. Acidez en las moléculas orgánicas Según la teoría de Arrhenius, desarrollada a finales del siglo XIX, un ácido es una sustancia que se ioniza en disolución acuosa dando iones H+ y una base es una sustancia que se ioniza en disolución acuosa generando aniones hidróxido (OH-). Según esta definición, el HCl, que se disuelve en agua generando H+ y Cl-, cabe clasificarlo como un compuesto ácido. Por otro lado, el NaOH, que se disuelve en agua generando Na+ y OH-, se clasifica como una base. La definición de ácidos y bases de Arrhenius ayudó en su tiempo a comprender el comportamiento de muchos ácidos y bases, pero no explicaba las características ácidas y básicas de otros compuestos, como el amoniaco NH3, que neutralizaba a los ácidos sin contener una función hidroxilo en su fórmula molecular. En 1923 Brönsted y Löwry definieron un ácido como una sustancia capaz de ceder protones y una base como una sustancia capaz de aceptar protones. Según esta definición, cualquier compuesto que contenga un H en su estructura puede potencialmente actuar como ácido y cualquier compuesto con un par de electrones solitario puede actuar como una base. Las dos especies, ácido y base, se necesitan en cualquier reacción de transferencia de protones, puesto que si una base cede un protón ha de haber una base que lo acepte. Por eso a las reacciones de transferencia de protones se las conoce con el nombre de reacciones ácido-base. Cuando un compuesto pierde un protón, la especie que se genera es su base conjugada. De la misma forma, cuando un compuesto acepta un protón, la especie generada es su ácido conjugado. H-A + A H 2O H 3O Base conjugada Ácido B + + H2 O + B H Ácido conjugado Base OH Cuanto más fuerte es el ácido más débil será su base conjugada y viceversa. Se podría decir que la basicidad es una medida de la capacidad que posee un compuesto para compartir sus electrones con un protón. La fuerza de un ácido depende de la capacidad para donar protones. Cuando un ácido se disuelve en agua se disocia hasta alcanzar un equilibrio en el que coexisten especies iniciales (ácido) y finales (base conjugada). Este equilibrio viene definido por una constante que se denomina constante de disociación ácida (Ka). H-A + H 2O Ka A + Base conjugada Ácido Ka = [ A ] [ H3O ] [HA] H3 O 37 Química Orgánica I-www.sinorg.uji.es La constante de acidez es una medida de la fuerza del ácido, de manera que cuanto mayor sea su valor más fuerte será el ácido (mayor facilidad para donar el protón). La acidez también se puede expresar como pKa, que se define como: pKa = -log Ka Cuanto menor sea el pKa mayor será la acidez del compuesto. Hay que tener presente que la acidez y basicidad son conceptos termodinámicos: no importa la velocidad a la que se alcance el equilibrio sino la posición de éste. Un ácido fuerte es el que tiene una elevada Ka, lo que significa que el equilibrio está muy desplazado a la derecha, lo que a su vez implica una gran estabilización termodinámica de la base conjugada en relación con el ácido que la genera. Determinados compuestos orgánicos poseen características ácidas. Los más importantes son: a) Los ácidos carboxílicos (RCOOH) con pKa del orden de 3 a 5. Estos compuestos son, al igual que el agua, anfotéricos y por tanto capaces de actuar como ácidos o como bases. c) Las sales de amonio, ácidos conjugados de las aminas, con valores de pKa entre 5-10. Son más ácidos que los alcoholes. b) Los alcoholes, mucho menos ácidos que los ácidos carboxílicos, con valores de pKa alrededor de 16. Son también anfóteros. Especies Valores pKa Ácidos carboxílicos (RCOOH) Alrededor de 5 Aminas protonadas (R3NH+) Alrededor de 10 Alcoholes (ROH) Alrededor de 15 La disociación de un ácido carboxílico en agua conlleva la cesión de un protón al disolvente y la formación de un anión carboxilato. Normalmente, los valores de la constante de acidez de los ácidos carboxílicos simples son de alrededor de 10-5 (pKa=5). Por ejemplo, la constante de acidez del ácido acético (CH3COOH) es de 10-4.7 (pKa=4.7). O H3C + H2O O H Ácido acético K a=10-4.7 O H3C + O Anión acetato H O H H 38 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) Aunque los ácidos carboxílicos no son tan ácidos como los ácidos minerales (HCl, H2SO4, HNO3) son mucho más ácidos que los alcoholes. Por ejemplo, el ácido acético es 1011 veces más ácido que los alcoholes más ácidos. De hecho, el ácido acético concentrado puede provocar quemaduras graves en contacto con la piel. Al igual que el protón del hidroxilo del agua, el protón del hidroxilo de un alcohol es débilmente ácido. Una base fuerte puede sustraer el protón del hidroxilo de un alcohol para generar un alcóxido. R O H + Alcohol (ácido) R B O B + H Alcóxido (base conjugada) Base La constante de disociación de un alcohol queda definida por el siguiente equilibrio: Ka R O H + Ka = R H2O R O + H3O H3O O ROH La constante de disociación ácida de los alcoholes varía según su estructura desde, aproximadamente, 10-16 para el metanol hasta, aproximadamente, 10-19 par la mayor parte de los alcoholes terciarios. La acidez de los alcoholes disminuye a medida que aumenta el grado de sustitución en el carbono unido al grupo hidroxilo (OH). Variación de la acidez de los alcoholes con el grado de sustitución H H C OH H metanol > H3C H C OH H etanol > H3C H3C C OH H isopropanol > H3C H3C C OH H3C t-butanol En la siguiente tabla se comparan los valores de pKa de los alcoholes anteriores y el agua. Compuesto Agua Fórmula H2O pKa 15.7 Metanol CH3OH 15.2 Etanol CH3CH2OH 15.9 Isopropanol (CH3)2CHOH 16.5 t-Butanol (CH3)3COH 16.6 39 Química Orgánica I-www.sinorg.uji.es Este orden de acidez en disolución acuosa se explica mediante el diferente grado de solvatación del anión alcóxido, la base conjugada del alcohol. La solvatación tiene un efecto estabilizante sobre el anión alcóxido: cuanto más solvatado esté el alcóxido más desplazado hacia la derecha estará el equilibrio ácido-base. El anión metóxido, la base conjugada del metanol, es relativamente pequeño y se rodea de un número relativamente elevado de moléculas de agua de solvatación. De esta forma la densidad electrónica asociada al átomo de oxígeno se reparte entre las moléculas de agua de solvatación y la especie se estabiliza. S H H B C H OH BH + S C H metanol S S O S H H S S S ión metóxido (fácil de solvatar) El anión t-butóxido es mucho más voluminoso que el anión metóxido y la aproximación de las moléculas de agua del disolvente al átomo de oxígeno que transporta la carga negativa se ve muy impedida. En consecuencia, el anión t-butóxido está menos solvatado que el anión metóxido S H3C B C H3C OH H3C BH S +H C 3 S C H3C H3C t-Butanol O S S ión t-butóxido (difícil de solvatar) Al estar la carga negativa del anión t-butóxido menos repartida por solvatación el equilibrio ácido-base del t-butanol se desplaza menos a la derecha y, como consecuencia, el tbutanol es un ácido más débil que el metanol. 1.3.3.1. Efecto inductivo y fuerza ácida Cuando un ácido carboxílico se disocia se genera un anión carboxilato, de manera que cuanto más estable sea el anión carboxilato más desplazado hacia la derecha estará el equilibrio y más ácido será el ácido carboxílico: R O C O H + B R O C O + B H Si un ácido carboxílico contiene en su estructura átomos electronegativos su acidez aumenta porque el efecto inductivo electrón-atrayente de esta clase de átomos contribuye a deslocalizar la carga negativa del anión carboxilato. Este efecto inductivo puede ser muy grande si están presentes uno o más grupos electrón-atrayentes en el átomo de carbono α. Por ejemplo, el ácido cloroacético (ClCH2COOH) tiene un pKa de 2.86, lo que indica que es un ácido más 40 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) fuerte que el acético (pKa =4.74). El ácido dicloroacético (Cl2CHCOOH) es todavía más fuerte, con un pKa de 1.26. El ácido tricloroacético (Cl3CCOOH) tiene un pKa de 0.64, comparable en fuerza a algunos de los ácidos minerales. O H3C + H2O Ka=10-4.7 O H3C O H O + H2O Ka=10-2.86 + H2O Cl2HC Cl3C H O H + H H O H O anión dicloroacetato ácido dicloroacético + H2O H O O Ka=10-1.26 O H O + anión cloroacetato ácido cloroacético O O O ClH2C O H Cl2HC H O H anión acetato ácido acético ClH2C H + Ka=10-0.64 O Cl3C O H + H O H O H anión tricloroacetato ácido tricloroacético La magnitud del efecto de un sustituyente depende de su distancia al grupo carboxilo. Los sustituyentes electrón-atrayentes en el carbono α son los más eficaces a la hora de aumentar la fuerza de un ácido. Los sustituyentes más alejados tienen efectos mucho más pequeños sobre la acidez, lo que pone de manifiesto que los efectos inductivos decrecen rápidamente con la distancia, como se puede observar en los valores de las constantes de acidez de los ácidos clorados que se indican a continuación: H Cl H3C C C H H O + H2O O H ácido 2-clorobutanoico Cl H H3C C C H H O + H2O O + H2O O H ácido 4-clorobutanoico O + H O O H H anión 2-clorobutanoato Cl H H3C C C H H Ka=10-4.05 O H ácido 3-clorobutanoico H H ClH2C C C H H H Cl H3C C C H H Ka=10-2.86 O + H O O H H anión 3-clorobutanoato Ka=10-4.52 H H ClH2C C C H H O O anión 4-clorobutanoato + H O H H 41 Química Orgánica I-www.sinorg.uji.es El aumento de la acidez provocada por el efecto electrón-atrayente también se pone de manifiesto en los alcoholes. Por ejemplo, el 2,2,2,-trifluoroetanol (CF3CH2OH, pKa=12.8) es mas de mil veces más ácido que el etanol (pKa = 15.9), porque el anión trifluroetóxido está más estabilizado que el etóxido debido al efecto atrayente sobre la carga negativa que ejercen los átomos de halógeno. CH3 CH2 OH + H2O F3C CH2 OH + H2O O + CH3 CH2 F3C CH2 H3O pKa = 15.9 H3O pKa = 12.8 O + En la siguiente tabla se comparan las constantes de disociación ácida de algunos alcoholes. Alcohol Estructura Ka pKa Metanol CH3-OH 3.2 x 10-16 15.5 Etanol CH3CH2-OH 1.3 x 10-16 15.9 2-Cloroetanol Cl-CH2CH2-OH 5.0 x 10-15 14.3 2,2,2-Trifluroetanol F3C-CH2-OH 4.0 x 10-13 12.4 Isopropanol (CH3)2CH-OH 1.0 x 10-18 18.0 t-Butanol (CH3)3C-OH 1.0 x 10-19 19.0 Ciclohexanol C6H11-OH 1.0 x 10-18 18.0 Fenol C6H5-OH 1.0 x 10-10 10.0 Comparación con otros ácidos Agua H2O 1.8 x 10-16 15.7 Ácido acético CH3COOH 1.6 x 10-5 4.8 Ácido clorhídrico HCl 1.6 x 102 -2.2 Los ácidos carboxílicos son mucho más ácidos que los alcoholes análogos. Por ejemplo el ácido acético tiene un pKa=4.7 y el etanol tiene un pKa=15.9. La disociación de un ácido o un alcohol implica, en ambos casos, la ruptura heterolítica de un enlace O-H, pero cuando la disociación se produce sobre el ácido carboxílico se genera un anión carboxilato con la carga negativa repartida por igual sobre dos átomos de oxígeno, mientras que la ionización de un alcohol genera un anión alcóxido, en el cual la carga negativa se encuentra casi en su totalidad sobre un sólo átomo de oxígeno. La deslocalización de la carga en el anión carboxilato hace que éste sea mucho más estable que un anión alcóxido y por tanto, la disociación de un ácido carboxílico es menos endotérmica que la de un alcohol. 42 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) R O H + H2O alcóxido alcohol O R C O H pKa = 16 + H3O R O O R C O + H2O ácido carboxílico O R C O + H3O pKa = 5 anión carboxilato En la siguiente gráfica se representan los niveles energéticos relativos del anión carboxilato y del alcóxido. La reacción de formación del alcóxido es mucho más endotérmica que la reacción de formación del carboxilato, lo que está de acuerdo con la menor constante de equilibrio del proceso de ionización del alcohol. R O + H 3O + Energía O δ− R ROH + H2O RCOOH + H2O + H3 O+ O δ− Coordenada de reacción 1.3.3.2. Efectos estéricos y fuerza ácida Las interacciones desestabilizantes de van der Waals que se establecen entre átomos, o grupos de átomos espacialmente próximos, se denominan efectos estéricos. Los efectos estéricos también pueden jugar un papel importante en la acidez. El impedimento estérico a la solvatación, causado por el tamaño del ácido o del disolvente, puede inhibir la estabilización de la base conjugada por parte del disolvente y, en consecuencia, disminuir la acidez, como se pone de manifiesto en las constantes de acidez del ácido benzoico y del ácido 2,6-di-t-butilbenzoico: O O + H 2O O H pK a= 4.20 H 3O pK a = 6.25 O Ácido benzoico Anión benzoato O CO2 H + H 3O + H 2O + O Ácido 2,6-di-ter-butilbenzoico Anión 2,6-di-ter-butilbenzoato 43 Química Orgánica I-www.sinorg.uji.es El ácido benzoico es un ácido más fuerte que el ácido 2,6-di-t-butilbenzoico porque en aquél el grupo ácido carboxílico (COOH) está rodeado tan sólo de un anillo aromático y las moléculas de agua pueden solvatar sin dificultad al anión benzoato. Sin embargo, cuando se disocia el ácido 2,6-di-t-butilbenzoico se genera el anión 2,6-di-ter-butilbenzoato, en el cual los voluminosos grupos ter-butilo, que se encuentran flanqueando al grupo carboxilato (-COO-), impiden el acercamiento de las moléculas de disolvente y por tanto la estabilización del anión por solvatación, como se indica en la siguiente figura: H3 C CH3 CH3CH3 O Impedimento estérico a la solvatación del anión carboxilato O CH3 H3C I.3.3.3. Influencia de otros efectos sobre la fuerza ácida El ácido o-hidroxibenzoico es unas 40 veces más ácido que el ácido p-hidroxibenzoico. O HO + H 2O O HO O H O Ácido 4-hidroxibenzoico O + H3 O Anión 4-hidroxibenzoato (menor estabilización) O + H 2O O H OH Ácido 2-hidroxibenzoico + H 3O O OH Anión 2-hidroxibenzoato (mayor estabilización) Esto se explica por la estabilización adicional que consigue el anión o-hidroxibenzoato por formación de un puente de hidrógeno intramolecular, que no se puede formar en el anión phidroxibenzoato. Como este anión está menos estabilizado, el equilibrio se desplaza menos hacia la derecha y el ácido p-hidroxibenzoico es menos fuerte que el ácido o-hidroxibenzoico I.3.4. Basicidad en las moléculas orgánicas Según la definición de Brönsted y Löwry una base es una sustancia capaz de aceptar protones, y por tanto cualquier compuestos con un par de electrones solitario puede actuar como una base. Cuando un compuesto acepta un protón, la especie generada es su ácido conjugado. La fuerza básica se puede medir por la constante de basicidad Kb (o pKb = -logKb) de manera que las sustancias que presenten constantes de basicidad elevadas (pKb pequeña) se comportan como 44 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) bases fuertes y si tienen constantes de basicidad pequeñas (pKb grandes) se comportan como bases débiles. B + Kb H 2O B H + Ácido conjugado Base OH [ BH ] [ OH ] Kb = [ B] Las bases más usuales de la Química Orgánica son las aminas. Una amina puede actuar como base de Bronsted-Lowry aceptando el protón de un ácido. Reacción de una amina como base H H R N R N H H X H X H ácido prótico base ácido conjugado Cuando una amina se disuelve en agua se observa un importante aumento del pH, lo que se explica por la sustracción del protón del agua, lo que genera un catión amonio y un anión hidróxido. A la constante de equilibrio de esta reacción se le llama constante de basicidad de la amina y se representa por Kb. R NH2 + H2O Kb = Kb R NH3 R NH3 + OH OH R NH2 I.3.4.1. Efecto inductivo y de solvatación en la fuerza básica Cualquier característica estructural y/o electrónica que estabilice al ión amonio (en relación con la amina libre) desplaza la reacción hacia la derecha, haciendo que la amina sea una base más fuerte. Por el contrario, cualquier característica estructural y/o electrónica que tienda a estabilizar a la amina libre (en relación con el ión amonio) desplaza la reacción hacia la izquierda, haciendo que la amina sea una base más débil. Las alquilaminas son bases más fuertes que el amoniaco. Por ejemplo la metilamina (amina primaria, pKb= 4.74) es más básica que el amoniaco (pKb= 3.36). 45 Química Orgánica I-www.sinorg.uji.es H H H N H O H H H N OH pKb=4.74 H + OH pKb=3.36 H H H + H H H3C H N O H3C H N H La diferencia de basicidad entre la metilamina y el amoníaco se explica por el efecto electrón-dador de los grupos alquilo. En el caso de la metilamina, el grupo metilo ayuda a estabilizar la carga positiva del nitrógeno, lo que provoca una disminución de la energía potencial del catión metilamonio y desplaza el equilibrio hacia la derecha. Siguiendo el anterior razonamiento, se debería esperar que las aminas secundarias fuesen bases más fuertes que las aminas primarias, y las aminas terciarias fuesen bases más fuertes que las aminas secundarias. La situación real es más complicada debido a la solvatación. Como los iones amonio tienen carga positiva, están fuertemente solvatados por el agua y la energía de solvatación contribuye a aumentar su estabilidad. Si el átomo de nitrógeno en el catión amonio presenta muchos grupos alquilo, caso de las aminas secundarias y terciarias, la solvatación se ve dificultada porque las moléculas de agua no se pueden acercar al átomo de nitrógeno que porta la carga positiva. Por tanto, los grupos alquilo sobre los átomos de nitrógeno en las aminas ejercen dos efectos contrapuestos: por una parte estabilizan al catión amonio por efecto inductivo electróndador, pero por otra desestabilizan al catión amonio al impedir una buena solvatación. Como resultado de todo ello, las aminas primarias, secundarias y terciarias muestran valores semejantes de basicidad. En fase gaseosa no puede actuar el efecto de solvatación del catión amonio y en este caso el efecto inductivo electrón-dador hace que una amina terciaria sea más básica que una amina secundaria y ésta más básica que una amina primaria. I.3.4.2. Efecto resonante y fuerza básica Las aminas aromáticas son bases mucho más débiles que las aminas alifáticas. Esta disminución de la basicidad se debe a la deslocalización por resonancia de los electrones no enlazantes de la amina. Por ejemplo, en la anilina, una amina aromática, el par aislado de electrones no enlazantes en el nitrógeno está deslocalizado sobre el sistema π del anillo aromático. Este solapamiento es imposible en el catión anilinio y por ello el reactivo está estabilizado en comparación con el producto. La reacción está desplazada hacia la izquierda y la anilina no es tan básica como las aminas alifáticas. 46 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) H H N N H H H H Anilina (estabilizada por resonancia) Catión anilinio + NH3 estabilización por solapamiento RNH2 + H+ + RNH3 reacción más endotérmica (anilina menos básica) NH2 + H+ Los efectos de hibridación también juegan un papel en la basicidad de las aminas. Por ejemplo, la piridina es una base más débil que las aminas alifáticas, como la piperidina. En la piridina, el par de electrones no enlazantes ocupan un orbital con hibridación sp2 y en la piperidina ocupan un orbital con hibridación sp3. Cuanto mayor es el carácter s de un orbital mayor es la atracción que ejerce el núcleo sobre los electrones. En la piridina, el par electrónico solitario está en un orbital con más carácter s que el orbital que ocupa el par de electrones solitarios de la piperidina y por tanto los electrones están menos disponibles para unirse al protón. hibrido sp2 H N N piridina pKb = 8.75 piperidina pKb = 2.88 hibrido sp3 47 Química Orgánica I-www.sinorg.uji.es Sumario de conceptos 1) Electronegatividad: tendencia del núcleo atómico a la atracción de electrones Tabla de electronegatividades de los átomos más comunes en Química Orgánica H 2.2 Li 1.0 Na 0.9 Be 1.6 Mg 1.3 B 1.8 Al 1.6 C 2.5 Si 1.9 N 3.0 P 2.2 O 3.4 S 2.6 F 4.0 Cl 3.2 Br 3.0 I 2.7 2) Efecto inductivo: polarización de enlaces provocada por un átomo o un grupo atómico a lo largo de una cadena carbonada σ. Efectos inductivos Efecto electrón-atrayente (-I) Efecto electrón-dador (+I) - -NO2 (grupo nitro) -O (grupo alcóxido) -CN (grupo ciano) -COO (grupo carboxilato) -COOH (grupo carboxilo) -C(CH3)3 (grupo t-butilo) -X (halógenos) -CH(CH3)2 (grupo isopropilo -OCH3 (grupo metoxilo) -CH2CH3 (grupo etilo) -OH (grupo hidroxilo) -CH3 (grupo metilo) - 3) Efecto resonante, mesómero o conjugativo: efecto estabilizante provocado por la deslocalización de la densidad electrónica π de un átomo, o grupo de átomos, sobre otros átomos de la estructura. 4) Estructuras resonantes o mesómeras: las que tienen la misma geometría (misma posición de los núcleos) pero difieren en su distribución electrónica 5) Fuerzas, o interacciones, de van der Waals: las fuerzas atractivas o repulsivas que se establecen entre las moléculas, ya sean éstas polares o apolares. 6) Fuerzas de Keesom: las fuerzas de atracción que se establecen entre dipolos permanentes. 48 Bloque Temático I. Estructura de las moléculas orgánicas (www.sinorg.uji.es) 7) Fuerzas de Debye: las fuerzas de atracción que se establecen entre dipolos permanentes y dipolos inducidos. 8) Fuerzas de dispersión de London: las fuerzas que surgen de la interacción entre dipolos inducidos generados temporalmente en las moléculas. 9) Enlace por puente de hidrógeno: interacción dipolo-dipolo que se establece entre el átomo de hidrógeno y otros átomos, cuando aquél se encuentra enlazado a un átomo fuertemente electronegativo como el nitrógeno, el oxígeno o el flúor. 10) Efecto estérico: efecto desestabilizante provocado por las repulsiones de van der Waals entre átomos, o grupos de átomos, espacialmente próximos. 11) Concepto de ácido y base según Arrhenius: un ácido es una sustancia que se ioniza en disolución acuosa dando iones H+. Una base es una sustancia que se ioniza en disolución acuosa generando aniones hidróxido (OH-). 12) Concepto de ácido y base según Brönsted: Un ácido es una sustancia capaz de ceder protones. Una base es una sustancia capaz de aceptar protones.