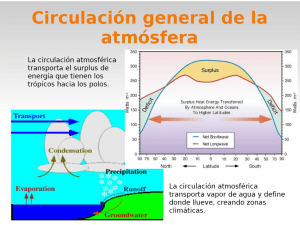
Unidad 3. FENÓMENOS DE OXIDO – REDUCCIÓN 3.1
Anuncio

Unidad 3. FENÓMENOS DE OXIDO – REDUCCIÓN 3.1 Termodinámica electroquímica. Conceptos básicos La primera ley de la termodinámica permite contabilizar los cambios de energía mecánica W y térmica q de un sistema cerrado con la ayuda de la función interna U, energía interna, que representa el estado energético del sistema. La energía U es una función de estado, lo que significa que para pasar de un estado inicial A, a un estado final B, la suma de las cantidades W y q es siempre la misma sin importar cual sea la transformación. Para una transformación infinitesimal, dU = dq ! dW (42) La segunda ley de la termodinámica postula la existencia de la función de estado entropía S, que aparece como la extensión de la energía calorífica asociada a la variable intensiva T, la temperatura termodinámica. La expresión clásica de la segunda ley es: dS = dq rev T (43) donde dqrev corresponde a la variación infinitesimal del calor intercambiado reversiblemente por el sistema con el exterior. En funcionamiento irreversible, la relación anterior es: dS = dq T + dq irrev T (44) donde el término dq/T representa la variación de entropía resultado del intercambio de calor dq con el exterior y dqirrev/T la entropía creada por la irreversibilidad. Para una transformación reversible, dq = T dS, involucrando sólo fuerzas de presión, se obtiene la relación clásica: dU = dq ! dW = TdS ! PdV (45a) Otras funciones de estado, de igual importancia, se obtienen de la misma forma: H = U + PV Entalpía A = U ! TS Energía libre de Helmholtz G = H ! TS Energía libre de Gibbs Para un sistema cerrado, cada función es derivada y sustituida en la expresión (45a), para obtener las siguientes expresiones simples, dH = TdS + VdP (45b) dA = ! PdV ! SdT (45c) dG = VdP ! SdT (45d) Derivando parcialmente estas ecuaciones, podemos deducir varias relaciones. Debido a la importancia de G en la termodinámica electroquímica, podemos obtener de la ecuación (45d) con respecto a la T y a presión P constante: ' (G $ % " = !S & (T # P y con respecto a la P y a T constante: (46) & 'G # $ ! =V % 'P " T (47) Derivando parcialmente, la ecuación (45b) con respecto a la T y a P constante: & 'H # & 'S # $ ! = T$ ! % 'T " P % 'T " P (48) Sustituyendo en la ecuación anterior la definición de capacidad calorífica, Cp, a P constante, Cp " (!H !T )P , se obtiene: Cp & 'S # $ ! = % 'T " P T (49) Derivando otra vez la ecuación (46) con respecto a la T y a P constante, llegamos a la siguiente expresión: ' ( 2G $ Cp ' (S $ % " % (T 2 " = !%& (T "# = ! T P & #P (50) Termodinámica de los sistemas con reacción química o electroquímica Consideremos la reacción electroquímica: v1 A 1 + v 2 A 2 + K ! v3 A 3 + v 4 A 4 + K donde vi representa el coeficiente estequiométrico de la reacción y Ai la fórmula química de la especie i. Por ejemplo, para la electrólisis Cloro – sosa, la reacción global es: 2 NaCl + 2 H 2 O ! 2 NaOH + H 2 + Cl 2 que resulta de la reacción anódica: 2Cl ! " Cl 2 + 2e ! (oxidación del Cl-) y la reacción catódica: 2 H 2 O + 2e ! " H 2 + 2OH ! (reducción del disolvente) los números estequiométricos de la reacción son: v NaCl = !2 v H 2O = !2 v NaOH = 2 v H 2 = 1 vCl2 = 1 El cambio en el número de moles n de cada especie se escribe: dn1 dn 2 dn3 dn 4 = = = = K = d! v1 v2 v3 v4 donde d! representa el avance de la reacción. La expresión anterior se puede escribir, d! = dni dQ = vi nF (51) donde dQ designa la cantidad de carga eléctrica asociada a la variación de dni moles de la especie i, n electrones intercambiados y F=Ne es el número de Faraday. La energía interna es una función de estado del sistema y su variación dU, debe tomar en cuenta, además de S y V, la variación en el número de moles dni. Derivando la energía interna U de la ecuación (45a) con respecto a S, a V y ni constantes, llegamos a: & 'U # =T $ ! % 'S "V ,ni (52) Derivando la energía interna U de la ecuación (45a) con respecto a V, a S y ni constantes, llegamos a: ' (U $ = !P % " & (V # S ,ni (53) Sustituyendo ambas expresiones en la ecuación (45a) y considerando el cambio en la energía interna U con respecto a los moles ni de la especie i, escribimos: ( )U % ( )U % dU = & # dS + & # dS + ' )S $V , ni ' )V $ S , ni ( )U % ## dni i $ S ,V , n j "i ! &&' )n i (54) Las derivadas parciales del tipo ("U "ni )S , V , n j !i se llaman potencial química µi de la especie i. La expresión (54) la reescribimos como, ' (U $ ' (U $ dU = % " dS + % " dS + & (S #V , ni & (V # S , ni ! µ dn i (55) i i Tomando en cuenta la definición del avance de la reacción: dU = TdS " PdS + d! #v µ i (56) i i 3.2 Equilibrio metal – ion metálico en solución en celdas electroquímicas Los procesos de intercambio de carga cuya transferencia tiene lugar entre dos fases, un conductor electrónico (metal) y un conductor iónico (solución), se denominan procesos de electrodo. Los procesos de electrodo agrupan el conjunto de cambios que acompaña a la transferencia, y comprenden: - depósito de metales Cd 2+(solución acuosa ) + 2e" # Cd(sólido) , - desprendimiento de gases 2H +(solución acuosa ) + 2e" # H 2(gas) , - disolución de metales ! Zn(sólido) " Zn 2+(solución acuosa ) + 2e# , ! transformación de PbSO4(sólido) + 2H 2O " PbO2(sólido) + 4H + + SO42# ! - ! sólidos + 2e # Fe 3+ ( soluciónacuosa ) + e ! " Fe 2+ ( soluciónacuosas ) - intercambio electrónico - reacción con intercambio protónico C6 H 5 NO2 + 6H + + 6e" # C6 H 5 NH 2 + 3H 2O Si en el proceso se transfiere carga positiva al electrodo, se habla de un proceso catódico o de reducción, mientras que se trata de un proceso anódico o de oxidación, cuando se transfiere carga !negativa al electrodo. Metal Interfase Metal Seno del metal Gradiente de potencial Seno de la solución Composición constante Zona de la reacción de electrodo Composición constante Figura 11. Sistema electródico En la figura 11 se muestra la interfase metal-solución, caracterizada por un gradiente de potencial que da lugar a una redistribución de carga, responsable de la doble capa. La reacción que se produce en la interfase se llama reacción de electrodo, puede agrupar las siguientes etapas, 1) transporte de las especies electroactivas hacia el electrodo, 2) adsorción de las mismas en el electrodo, 3) transferencia de carga, 4) desorción de los productos de reacción del electrodo, 5) difusión de dichos productos hacia el seno de la solución, 6) reacciones químicas secundarias, y 7) formación de nuevas fases. A veces no tienen lugar las etapas 6 ó 7. Otras veces las etapas 2, 3 y 4 están reunidas en la formación de un complejo de transcisión. En los procesos de depósito y disolución de los metales, las etapas 4 y 5 son el paso inverso de las etapas 1 y 2. El paso 7 consiste en nucleación y crecimiento de cristales (sólidos) o en formación de burbujas de gas. Velocidad de reacción, polarización y sobretensión La transformación electroquímica de 1 mol de especie electroactiva requiere el paso de nF coulombs, donde n es el número de cargas intercambiadas y F es la constante de Faraday (F = 96487 C mol-1 ). Así, el número de moles transformados de la especie electroactiva A, está expresado por, nA = ! It nF (57) donde I y t corresponden a la corriente que circula y al tiempo, respectivamente. Dos tipos de corriente: Corriente faradaica - La corriente implicada en una reacción de electrodo. Corriente no faradaica – La corriente implicada en la carga y descarga de la doble capa electroquímica. Así, la velocidad de una reacción de electrodo se puede definir como el número de moles de la especie electroactiva A, transformados por unidad de tiempo: rA = I nF (58) Puesto que las reacciones de electrodo tienen lugar en la interfase metal – solución, su velocidad depende del área o superficie de electrodo, en consecuencia la velocidad también se puede expresar en función de la densidad ! de corriente j, que se refiera a la corriente por unidad de área. Polarización – Es cuando se pasa una corriente a través de la interfase metal – solución y se desplaza al electrodo de su condición de equilibrio. Si el electrodo mantiene su polarización, es decir, que no tiene lugar un cambio de potencial con el paso de corriente, se habla de un electrodo no-polarizable. En cambio, se tiene un electrodo polarizable cuando un aumento del potencial aplicado no origina paso de corriente apreciable. La polarización da una idea de la diferencia de potencial entre el potencial del electrodo E y el potencial del mismo en condiciones de equilibrio E0: Como medida de la polarización se define el parámetro llamado sobretensión o sobrepotencial η, y viene dado por " = E! E 0 (59) La sobretensión aparece como consecuencia de los fenómenos de polarización, y es debida a la baja velocidad de alguna de las etapas de la reacción de electrodo, que conduce a un retraso del proceso global. Esta etapa lenta controla la reacción de electrodo y constituye la etapa determinante de la velocidad de reacción (figura 12). La etapa lenta puede ser por, Sobretensión por transferencia de carga, si el proceso total viene retrasado por la etapa de transferencia de carga. Sobretensión de difusión, si la variación de la concentración de la especie electroactiva, junto al electrodo respecto a su valor en el seno de la solución, origina un transporte de materia que pueda constituir la etapa lenta del proceso. Sobretensión de reacción, si existe una reacción química acoplada a la reacción electródica y que retrasa el proceso global, de modo que su velocidad viene controlada por la reacción química. Sobretensión de concentración, cuando se tratan juntas la sobretensión de difusión y de reacción, pues ambas están relacionadas con la concentración en la interfase. Sobretensión de cristalización, si en el proceso hay una etapa de formación de una nueva fase puede conducir a una sobretensión de fase. Sobretensión de resistencia, cuando la resistencia de la solución origina, durante el paso de la corriente, una caída óhmica de potencial. Esta se presenta cuando existe una capa poco conductora entre el metal del electrodo y la solución. Transferencia de materia Adsorción A ! e e- Aads Capa reaccional Bads Seno del electrolito Transferencia de materia B Desorción Electrodo Diversas etapas de una reacción electroquímica Figura 12 3.3 Potencial interno, externo y de superficie Potencial eléctrico exterior ! – Es el trabajo realizado para llevar una carga puntual desde un punto en el vacío hasta junto a la superficie de una fase conductora. También se denomina potencial de Volta. Potencial eléctrico de superficie ! – Es el trabajo necesario para llevar una carga desde junto a la superficie conductora, franqueando una capa de cargas y de dipolos orientados, hasta el interior de la fase conductora. Potencial eléctrico interior ! – Es la suma del potencial eléctrico exterior y el potencial eléctrico de superficie, denominado también potencial de Galvani. Así, se puede escribir, # =" + ! (60) El potencial eléctrico interior y el potencial eléctrico de superficie no se pueden medir, mientras que el potencial eléctrico exterior si se puede medir. ~ – Es el trabajo necesario para traer una partícula cargada desde el Potencial electroquímico µ punto donde el potencial es cero hasta el punto situado en el seno de una fase cargada y provista de dipolos en su superficie. Su valor depende de la fase material y de la partícula, por lo que se escribe ~ I , esto es, potencial electroquímico de la partícula B en la fase I (figura 13a). también µ B El potencial electroquímico se puede descomponer en dos contribuciones: − Un potencial químico µ BI , que es el trabajo químico (de corto alcance) necesario para traer la partícula B desde el infinito hasta el punto P de la fase I, desprovista de carga y de dipolos (figura 13b). Este potencial se debe a la interacción química entre la partícula y el material de la fase. − Un potencial electrostático z B F! I , que corresponde al trabajo necesario para traer la partícula B al punto situado en el interior vacío de la envoltura con la misma carga y los dipolos que la fase real (figura 13c). Este trabajo viene dado por el producto de la carga zBF, por el potencial interior de la fase φI. Figura 13. Potencial electroquímico de una partícula cargada en una fase cargada con dipolos en la superficie Por lo tanto, µ~BI = µ BI + z B F! I (61) Las distintas contribuciones del potencial electroquímico (J mol-1) se resumen y se indican a continuación, µ~BI = µ BI + z B F" I + z B F! I donde, ! I el potencial de superficie en volts, no medible, ! I el potencial exterior en volts, medible, # I = " I + ! I el potencial interior en volts, no medible, µ BI el potencial químico en J mol-1, y definimos (62) " BI = µ BI + z B F! I como el potencial real en J mol-1. Cuando se tienen dos fases, la diferencia de potencial entre un punto de la fase material I y un punto de la fase material II define la diferencia de potencial de Volta, "III # = # II $ # I (63) Si la especie B en las dos fases está en equilibrio, µ˜ BI = µ˜ BII , y teniendo en cuenta la ecuación (62), II µ˜ BI = µ˜! " # BI + z B F$ I = # BII + z B F$ II B ! sustituyendo en la ecuación (63) "III # = ! " # BI % # BII = $ II % $ I zB F $ BI % $ BII zB F (64) También se define la diferencia de potencial de Galvani de la fase II con relación a la fase I, según "III # = # II $ # I ! A partir de la ecuación (61), y para las dos fases con la misma composición, µ BI = µ BII , ( µ˜ BII "! µ˜ BI = µ BII + z B F# II " (µ BI + z B F# I ) $ %III µ˜ B = z B F # II " # I (65) ) Así, la diferencia de potencial de Galvani queda en función! de la diferencia de potencial electroquímico, ! "II µ˜ "III # = I B (66) zB F Potencial en los electrodos Al sumergir un metal en una solución iónica, se establece una diferencia de potencial entre el metal ! y el seno de la solución. El valor de esta diferencia de potencial no se puede determinar experimentalmente. Suponer dos fases, Fase I Metal M Fase II Solución M z+ Considerar el proceso entre el metal M, sus iones Mz+ y los z electrones M " M z+ + ze M En el equilibrio, se cumple µ˜ M = µ˜ MS z + + z µ˜ eM ! donde S indica la fase solución. Sustituyendo la ecuación (61) y que el electrón tiene carga negativa, se escribe M M S ! S M M S S M M µ + z F" = µ M z + + z F" + zµ e Reordenando la igualdad, ! # µ = µ M z + + z F" + zµ e $ z F" M S z F" # z F" = µ M S M z+ M # µ + zµ M e M S $ " #" = µ MS z + # µ M + zµ eM zF S donde " # " constituye el potencial eléctrico del electrodo, cuyo valor no es medible experimentalmente. Sustituyendo el potencial químico de la forma iónica en función de la actividad, se tiene ! ! M S " #" = o o en donde "µ = µ M z + # µ ! ! M µ Mo z + + RT ln a M z + # µ M + zµ eM zF + zµ eM , la expresión anterior se escribe "# SM = # M $ # S = 1 "µ o + RT ln a M z + zF ( ) (67) M Si bien, "# S no es medible durectamente, si se puede determinar la diferencia entre dos potenciales de electrodo mediante un potenciostato y donde uno de los potenciales de electrodo sea conocido. El potencial de ! electrodo conocido se le llama potencial de electrodo de referencia y será tratado más adelante. ! La celda electroquímica Una celda electroquímica esta constituida por al menos dos electrodos(metales) y un electrolito. El electrodo donde se lleva a cabo la oxidación de especies se le llama ánodo y el electrodo donde se lleva a cabo la reducción de especies se le llama cátodo. El electrolito esta constituido de una sal disuelta en una solución (agua, por ejemplo) donde la sal puede ser afin a alguno de los electrodos. Esta celda es un dispositivo que puede producir trabajo eléctrico en el entorno, como se muestra en la figura 14 (caso de funcionamiento en pila). El trabajo eléctrico producido Wel, es menor o igual que la disminución en la energía libre de Gibbs, Wel # " !G . Antes de continuar con el desarrollo termodinámico, definiremos algunos conceptos básicos. e Los electrones fluyen de la terminal negativa a la positiva Anodo Motor Trabajo eléctrico Cátodo e Reacción química De la celda Figura 14. Pila electroquímica Potencial eléctrico El potencial eléctrico es el trabajo realizado para traer una unidad de carga positiva desde el infinito (donde el potencial eléctrico es cero), hasta un punto en el espacio. Si φ es el potencial eléctrico en ese punto y W es el trabajo para traer una carga Q desde el infinito al punto, entonces "= W Q (68) Si φ1 y φ2 son los potenciales eléctricos en dos puntos en el espacio y W1 y W2 son las cantidades de trabajo para traer la carga Q a estos puntos, tendremos W1 + W12 = W2 , donde W12 es el trabajo para llevar Q del punto 1 al 2. Despejando nos queda W12 = W2 " W1.Sustituyendo en la ecuación (68), ! W "2 # "1 = 12 (69) ! Q ! La diferencia de potencial φ2 - φ1 corresponde al trabajo necesario para llevar la carga positiva del punto 1 al 2. Aplicando para la transferencia de una carga infinitesimal, ! W = "dW = #$ dQ (70) 12 el donde Δφ = φ2 - φ1 expresa la diferencia de potencial eléctrico y dWel el trabajo producido. ! Potencial químico de especies cargadas La tendencia de escape de una partícula cargada, de un ion o un electrón depende del potencial eléctrico aplicado en la fase. Si imprimimos a una placa metálica un alto potencial eléctrico negativo, aumentará la tendencia de escape de las partículas negativas. Pensemos en dos esferas del mismo metal como se muestra en la figura 15, Transferencia de electrones dQ M φ M’ φ' Figura 15. Sistema de dos esferas A partir de la ecuación (70), el trabajo producido será "dWel = (# ' " # ) dQ . Si la transferencia se hace de manera reversible, el trabajo producido es igual a la disminución de la energía libre de Gibbs del sistema, dWel = "dG , de modo que dG = (!!'" !)dQ (71) ~ , si se transfieren dn moles de electrones, En función del potencial químico de los electrones, µ e! ! tenemos ~ ' dn ! µ ~ dn dG = µ (72) e! e! Sustituyendo en la ecuación (71) la carga dQ = -F dn, igualado con la ecuación (72) y dividiendo entre dn, obtenemos ~' " µ ~ = " F (!' " !) µ (73a) e" e" y reordenando µ˜ e " = µ˜ ' e " + F # ' " F # (73b) Sea µ e ! , el potencial químico de los electrones en M cuando φ = 0, entonces µ e " = µ˜ ' e " + F # ' . Restando a la ecuación (73b), ! µ˜ e " = µ e " " F # (74) ~ ! Esta expresión relaciona la tendencia de escape de los electrones, µ , en una fase y el potencial e! ~ eléctrico de la fase, φ . La µ e ! es una función lineal de φ. La ecuación establece que si φ es negativo, ~ es mayor que cuando φ ! µ es positivo. Mediante un argumento análogo, para cualquier tipo de e! especie cargada, la ecuación (74) se escribe, µ˜ i = µ i + z i F " (75) donde zi es la carga de la especie. Para electrones, ze- = -1, de modo que la ecuación (67) se convierte en la ecuación (74). • El primer término µ , es ! la contribución química a la tendencia de escape y es producto del i medio químico en el que se encuentra y es la misma en dos fases de igual composición química, ya que es función de la T, P y composición. • El segundo término, zi F φ, es la contribución eléctrica a la tendencia de escape y depende de la condición eléctrica de la fase expresada por el valor de φ. Así, de la expresión (75) " ! µ˜ i = µ i + z i F # , el potencial electroquímico es µ˜ i , el potencial químico ordinario es µ i , y el potencial eléctrico es zi F φ. ! A continuación se presenta la convención para el potencial químico de especies cargadas: ! • Iones en solución acuosa: φ = 0 en la solución (simplificación), µ˜ i = µ i . • Electrones en metales: Por convención, para cada metal, el potencial químico µ e " = 0. Así µ˜ e " = " F # (76) ! Iones en metales puros: En cualquier metal existe un equilibrio entre los átomos del metal ! M, los iones metálicos Mz+ y los electrones, M " M z + + z e# . La condición de equilibrio es µ M = µ˜ M z + + z µ˜ e"! . Sustituyendo la ecuación (75) para µ˜ M z + y µ˜ e " = " F # para µ˜ e " , se • obtiene µ M = µ M z + + z F " # z F " , o bien µ M = µ M z + . Para un metal puro a 1 atm y 25°C, ! con la convención preliminar de que µ o = 0 para tenemos que µ Mo = µ Mo z + ; de acuerdo ! ! elementos en estas condiciones, obtenemos µ o z! + = 0 . A partir de la ecuación (75), ! M ! µ˜ M z + = z F " (77) ! ! Las ecuaciones (76) y (77) son los valores convencionales del potencial químico de los electrones y ! los iones en cualquier metal puro. ! En resumen: Elementos en su estado de agregación estable: Estado estándar o µ elementos =0 Partículas cargadas: Forma general ! a) Iones en solución acuosa µ˜ i = µ i + z i F " φac = 0 Estado estándar ! µ Ho + = 0 Forma general µ˜ i = µ i = µ io + R T ln a i b) Electrones en cualquier metal ! µ˜ e " (EEH) = 0 o bien #(EEH) = 0 Estado estándar ! µ˜ e " = "F # Forma general c) Iones en metal puro ! Estado estándar ! Forma general ! Diagramas de Celda ! µ Mo z + = 0 µ˜ M z + = z F " La celda electroquímica se representa en un diagrama de celda de la siguiente manera: Los electrodos metálicos se colocan en los extremos del diagrama. Las sustancias insolubles y/o gases se colocan en posiciones interiores adyacentes a los metales. Las especies solubles se colocan en la región media del diagrama. Una frontera de fase se indica con una línea vertical. Una línea vertical discontinua indica la unión entre dos fases líquidas. Dos líneas verticales continuas indican la unión entre dos fases líquidas miscibles donde se ha eliminado el potencial de unión. Se separan con comas las diferentes especies solubles en la misma fase. Ejemplo 1: Completo Pt I (s) Zn(s) Zn 2+ (a Zn 2+ = 0.35) Cu 2+ (aZn 2+ = 0.35) Cu(s) Pt II (s) Abreviado Zn Zn 2+ Cu 2+ Cu Ejemplo ! 2: ! Completo Pt H 2 (g, p = 0.8) H 2SO 4 (ac, a = 0.42) Hg 2SO 4 (s) Abreviado Pt H 2 H 2SO 4 (ac) Hg 2SO 4 (s) Hg(L) Hg ! Celda de Daniell ! En la figura 16(a) se muestra una celda electroquímica, la celda de Daniell. Esta celda consiste de dos medias celdas separadas por un puente salino que evita que se mezclen las soluciones pero permite el flujo de corriente entre los dos compartimentos. PtI PtII Figura 16. (a) Celda de Daniell con circuito externo abierto y (b) circuito cerrado y funcionamiento en pila Cada media celda consiste de un metal, una tira de zinc o cobre, inmerso en una solución de una sal muy soluble del metal tal como ZnSO4 o CuSO4. Los electrodos están conectados al exterior mediante dos alambres de platino. El diagrama completo de la celda se escribe, Pt I (s) Zn(s) Zn 2+ (ac ) Cu 2+ (ac ) Cu(s) Pt II (s) En la frontera de la interfase Pt I Zn y Cu Pt II se establece el equilibrio electroquímico por el paso libre de los electrones a través de la interfase. Las condiciones de equilibrio son µ˜ e " (Pt I ) = µ˜ e "! (Zn) y µ˜ e " (Cu) = µ˜ e " (Pt II ) . Utlizando la ecuación (73), se obtiene ! µ˜ e " (Pt I ) = " F # I = µ˜ e " (Zn) = " F # Zn ! $$ # I = # Zn y # Cu = # II (78) donde φI y φII son los potenciales de los dos alambres de platino, φZn y φCu son los potenciales del electrodo de Zn y Cu en contacto con una solución que ! contiene al ion Zn y al ion Cu, respectivamente. Así, la diferencia de potencial de la celda esta definido por, (79) " = # II $ # I = # Cu $ # Zn La diferencia potencial también se puede representar como Δφ o φ, nosotros utilizaremos Ε. La diferencia de potencial φII - φI, se mide a través de un voltímetro y la corriente eléctrica por medio de un amperímetro. Si ! conectamos los dos alambres de platino a través de un amperímetro a un foco (ver figura 15(b)), observamos que: • Se disuelve algo de la tira de Zn (electrodo) y los iones disueltos se van a la solución, • Se depositan algunos iones de Cu sobre la tira de Cu, • En el circuito externo fluyen electrones del electrodo de Zn (cátodo) hacia el de Cu (ánodo), • El movimiento de los electrones generan trabajo o fuerza electromotríz (fem), y por lo tanto, • El foco se ilumina. Los cambios en la celda son: En el cátodo (electrodo izquierdo): Zn(s) " Zn 2+ (ac) + 2e# (Zn) En el circuito externo: 2e" (Zn) # En el ánodo (electrodo derecho) ! Cu 2+ (ac) + 2e" (Cu) # Cu(s) Transformación global: Zn(s) + Cu 2+ (ac) " Zn 2+ (ac) + Cu(s) ! ! ! 2e" (Cu) Cálculo del potencial de celda a partir de la Energía libre de Gibbs de la reacción Esta reacción química es la reacción de celda, donde la energía libre de Gibbs, Δ G, es "G = "G o + RT ln a Zn 2+ aCu 2+ (80) siendo R, T y a la constante de los gases, la temperatura absoluta y la actividad respectivamente. El trabajo realizado sobre el sistema para mover los electrones del electrodo de Zn al electrodo Cu es – Wel, donde "Wel = Q !(# II " # I ) = "2 F $ . Sustituyendo este valor en Wel # " !G , se escribe 2F" # $ %G . Si el proceso es reversible, Wel = " #G , y la expresión (70) se transforma en 2 F " = #$G . Así, la ecuación (80) se convierte en a 2+ "2 F # = " 2F # o + RT ln Zn ! aCu 2+ ! ! ! (81) donde E0 corresponde al potencial de celda en su estado estándar de cada electrodo, es decir, si aZn 2+ =1 y aCu 2+ =1 . En la tabla 1 se muestran los valores de potencial estandar, E0, de algunas ! Despejando el potencial de celda, reacciones de media celda. " = "o # ! RT aZn 2+ ln 2 F aCu 2+ (82) Esta es la Ecuación de Nernst para determinar el potencial de celda de cada electrodo fuera de las condiciones estándar. Esta expresión se puede generalizar para z = n electrones transferidos, ! RT a Zn 2+ (83) " = "o # ln n F aCu 2+ ! Tabla 1. La espontaneidad de una reacción se determina mediante la relación entre el potencial de celda y la energía libre de Gibbs como " = #$G n F . Así, se deduce que si ΔG es negativo, E es positivo y la reacción de celda es espontánea. Por tanto, ! ΔG E La reacción de celda - + Es espontánea + - Es no espontánea 0 0 Esta en equilibrio Forma general de la Ecuación de Nernst Para una reacción general del tipo aA + bB + ... " mM + nN + ... De la ecuación (83) se tiene ! RT a m a n ... ln Ma Na nF a A aB ... " = "o # (84) Sustituyendo la actividad a = [c] γ, donde γ corresponde al coeficiente de fugacidad, tenemos m n ! " = " o # RT ln [ M ] [ N ] ... a b nF [ A] [ B] ... m n ($ M ) ($ N ) ... a b ($ A ) ($ B ) ... (85) En el caso de soluciones diluidas, el coeficiente de fugacidad es " M ! " N ! " A ! " B ! 1 , de modo que la ecuación (85) se reduce a, ! m n M ] [ N ] ... [ RT o "= " # ln (86a) a b nF [ A] [ B] ... En términos del logaritmo natural, m ! n " = "o # [ M ] [ N] ... 2.303 RT log a b nF [ A] [ B] ... " = "o # [ M ] [ N] ... 0.059 log a b n [ A] [ B] ... (86b) y a 298.15 K ó 25°C, se tiene m ! n Así, para toda reacción sencilla de oxidorreducción, se tiene Ox + n e " ecuación (86c), ! [ Red] 0.059 " Ox/Red = " 0Ox/Red # log n [Ox] (86c) ! Red , y a partir de la (87a) " Ox/Red = " 0Ox/Red + [Ox] 0.059 log n [ Red] (87b) " Red/Ox = " 0Red/Ox # [Ox] 0.059 log n [ Red] (87c) ! ! Nótese que la práctica general es de mantener al estado oxidado en el numerador de la expresión logarítmica. Es evidente que para valores fijos de [Ox] y [Red], se tiene " Ox/Red = # " Red/Ox , y para valores unitarios de actividades de las sustancias Ox y Red, o para valores unitarios de ! concentración [Ox] = [Red] = 1 M, se tiene " 0Ox/Red = # " 0Red/Ox ! Convención de signos ! Los textos más modernos se adhieren a las recomendaciones de la Unión Internacional de Química Pura y Aplicada (IUPAC), las cuales estipulan que, por ejemplo, la media celda de cobre, dada por la reacción Cu 2+ (ac) + 2e " ! Cu ( s ) da un valor positivo de potencial de ! 0Cu 2 + /Cu = 0.34 V . Combinación de medias celdas como celdas galvánicas (Caso particular) Considerar una combinación de dos celdas que produce una celda denominada galvánica como se muestra en la figura 17. electrones voltímetro (V) electrodo de zinc ánodo cátodo electrodo de cobre Puente salino CuSO 4 (ac) ZnSO 4 (ac) Figura 17. Medición de la fem de celda para una celda galvánica formada por una combinación de medias celdas de cobre y de zinc. Se utiliza una combinación de celdas Zn2+/Zn y Cu2+/Cu con las soluciones unidas por un puente salino y los electrodos conectados exteriormente. La concentración del Cu2+ y del Zn2+ son [Cu2+] = 10-1 M y [Zn2+] = 10-2 M. El potencial de reducción para el sistema de media celda de Cu está dado por, " Cu 2+ / Cu = " 0Cu 2+ / Cu [ ] Cu 2+ 0.059 + log n [Cu] = 0.34 + 0.059 log 10#1 = 0.31 V a 25°C 2 [ ] Para la reducción en el sistema de media celda de Zn, ! " Zn 2+ / Zn = " 0Zn 2+ / Zn [ ] Zn 2+ 0.059 + log n [ Zn] = # 0.763+ 0.059 log 10#2 = #0.82 V a 25°C 2 [ ] La media celda de Cu posee el valor mayor de potencial de reducción (más positivo), de modo que las medias celdas interconectadas generarán las reacciones y la reacción global, ! Cu 2+ (ac) + 2e " Zn( s ) " ! Cu ( s ) Zn 2+ (ac) + 2e ! Zn( s ) + Cu 2+ (ac) ! Zn 2+ (ac) + Cu ( s ) Reducción ! Cu 2 + / Cu = 0.31 V Oxidación ! Zn / Zn 2 + = 0.82 V Global U 1.13 V = El sistema mostró: • Un flujo de electrones por el circuito exterior, desde el electrodo de Zn hacia el electrodo de Cu. • El electrodo de Zn como polo negativo (cátodo) y el de cobre positivo (ánodo). • Las semirreacciones como se muestran arriba. • Un voltaje o fem entre electrodos de 1.13 V a 25°C. La celda galvánica continuará produciendo trabajo eléctrico externo al sistema durante un tiempo, con una disminución de iones Cu y un aumento de iones Zn. El valor de ! Zn 2+ / Zn aumentará, mientras que el de ! Cu 2+ / Cu disminuirá. Cuando ! Cu 2 + / Cu = ! Zn 2 + / Zn la celda deja de generar una diferencia de potencial y de realizar trabajo eléctrico externo al sistema. En este punto, se ha generado un equilibrio que se escribe como 0.059 log Cu 2+ 2 [ " 0Cu 2+ / Cu + ] eq = " 0Zn 2+ / Zn + 0.059 log Zn 2+ 2 [ ] eq despejando, ! [ [ Zn 2+ 0.059 log 2 Cu 2+ ] ] eq = " 0Cu 2+ / Cu # " 0Zn 2+ / Zn = 0.34 V # (#0.763) V =1.103 V eq de donde, [ Zn ] = [Cu ] " 2(1.103 V) % $$ '' # 0.059 & 2+ ! K eq eq 2+ = 10 =10 37.4 eq donde el valor obtenido representa la constante de equilibrio a 25°C. ! Combinación de medias celdas como celdas galvánicas (Caso general) Considerar la reacción en dos medias celdas y la reacción global, a Ox 2 + n2 e ! c Red1 " b Red 2 " d Ox 1 + n1 e n2 c Red1 + n1 a Ox 2 Reducción (cátodo) ! Oxidación (ánodo) ! n2 d Ox 1 + n1b Red 2 Global La ecuación (86b) se escribe en forma de reducción para las reacciones 1 y 2, d " Ox 1 /Red 1 = " 0Ox 1 /Red 1 + ! [Ox1] 0.059 log c n1 [ Red1] a [Ox 2 ] 0.059 + log b n2 [ Red 2 ] " 0Ox 2 /Red 2 " Ox 2 /Red 2 = Como el valor de ! Ox 2/Red 2 > ! 0Ox1/Red1 , el voltaje de la celda, U, o fem está dado por a ! [Ox 2 ] 0.059 U = " Ox 2 /Red 2 # " Ox 1 /Red 1 = " 0Ox 2 /Red 2 # " 0Ox 1 /Red 1 + log b n2 [ Red2 ] U= " 0Ox 2 /Red 2 # " 0Ox 1 /Red 1 U= 0.059 U + log n1 n 2 ! ! n1 a 1 n1 a [Ox1] 0.059 log c n1 [ Red1] n2 c [Ox 2 ] [ Red1] n b n [ Red2 ] [Ox1] 0.059 + log n1 n 2 2 (88a) d n2 c [Ox 2 ] [ Red1] n b n [ Red 2 ] [Ox1] 0 d # 1 2 (88b) d En el caso particular en que n1 = n2 = n, la expresión (88a) se escribe, ! U= " 0Ox 2 /Red 2 # " 0Ox 1 /Red 1 a c [Ox 2 ] [ Red1] b d [ Red 2 ] [Ox1] 0.059 + log n (89) En el equilibrio, en general se tiene que " Ox 2 /Red 2 = " Ox 1 /Red 1 y a ! " 0Ox 2 /Red 2 + d [Ox 2 ] = 0.059 log b n2 [ Red2 ] ! [Ox1] 0.059 log c n1 [ Red1] " 0Ox 1 /Red 1 + n d n b Ox1 ] 2 [ Red 2 ] 1 [ 0.059 log = " 0Ox 2 /Red 2 # " 0Ox 1 /Red 1 n2 c n1 a n1 n 2 [ Red1] [Ox 2 ] ! n2 d ! ! K eq = [Ox1] [ Red 2 ] n c n a [ Red1] [Ox 2 ] 2 K eq = [Ox1] $" 0 #" 0Ox1/Red1 & Ox 2/Red 2 & 0.059 n1 n 2 % n1 b 1 n2 d [ Red1] =10 [ Red 2 ] n2 c " % U0 $ ' $ 0.059 / n1 n 2 ' # & n1 b [Ox 2 ] n1 a ' ) ) ( =10 (90a) (90b) En el caso particular en que n1 = n2 = n, la expresión (90b) se escribe, ! 0 " % b d [ Red 2 ] [Ox1] K eq = a c [Ox 2 ] [ Red1] =10 U $ ' $ 0.059 n ' # & (91) Factores que influyen sobre los potenciales de los electrodos ! Los factores más importantes que influyen sobre el valor del potencial medido en medias celdas y celdas, son: El efecto de la concentración del ion o iones activos El efecto de la concentración del ion diverso El efecto del pH El efecto de la concentración del ion o iones activos Un aumento en el valor de [Ox]/[Red] produce un aumento en el valor de EOx/Red. Esta expresión nos permite determinar el valor del potencial de la media celda y celda en el caso de soluciones con un valor de concentración diferente a 1M (para líquidos) y 1 atm (para gases). El efecto de la concentración del ion diverso Un ion diverso es aquel ion que se encuentra en la solución, pero no interviene directamente en la reacción de oxidorreducción (participa en la conducción de la corriente eléctrica). La presencia de todo ion diverso en concentración significativa, causará una disminución del valor de la actividad de los estados Ox y Red, donde cada estado representa una especie iónica. La ecuación de Nernst se escribe, " Ox/Red = " 0Ox/Red + [Ox] # Ox 0.059 a 0.059 log Ox = " 0Ox/Red + log n aRed n [ Red] # Red " Ox/Red = " 0Ox/Red + ! [Ox] = " 0 [Ox] 0.059 # 0.059 0.059 log Ox + log log f, Ox/Red + n # Red n n [ Red] [ Red] (92) donde ! 0f, Ox/Red recibe el nombre de potencial formal de reducción de la media celda, varía según la ! concentración del ion diverso o según la fuerza iónica de la solución. El efecto del pH En muchos sistemas de oxidorreducción, el [H3 O+] participa efectivamente. Consideremos la siguiente reacción dependiente del pH, a Ox 2 + y H 3 O + + n 2 e ! b Red 2 + w H 2 O Reducción c Red 1 Oxidación ! d Ox 1 + n1 e n 2 c Red 1 + n1 a Ox 2 + n1 y H 3 O + ! n 2 d Ox 1 + n1 b Red 2 + n1 w H 2 O Global El voltaje, U, de la celda será, a U = " Ox 2 /Red 2 # " Ox 1 /Red 1 = " 0Ox 2 /Red 2 # " 0Ox 1 /Red 1 [ ] Siendo U 0 = " 0Ox 2 /Red 2 # " 0Ox 1 /Red 1 , reescribimos ! n a [ ] n1 y n c [Ox 2 ] 1 H 3O + [ Red1] 2 0.059 0 U= U + log n b n w n d n1n2 [ Red2 ] 1 [ H 2O] 1 [Ox1] 2 ! ! y [Ox 2 ] H 3O + 0.059 + log b w n2 [ Red2 ] [ H 2O] d [Ox1] 0.059 # log c n1 [ Red1] ([ ]) [ ] Siendo que la definición de pH = log 1 H 3O + = "log H 3O + , nos queda en términos del pH, n a U= U ! 0 n c [Ox 2 ] 1 [ Red1] 2 0.059 + log n b n w n d n1n2 [ Red2 ] 1 [ H 2O] 1 [Ox1] 2 " 0.059y pH n2 Para soluciones diluidas normales [H2 O] = 1, por lo que la expresión anterior se escribe, ! U= U 0 n1 a n2c [Ox 2 ] [ Red1] n b n d [ Red 2 ] [Ox1] 0.059 + log n1n2 1 " 2 0.059y pH n2 (93) Si n1 = n2 = n, la ecuación (93) se reduce a, a c [Ox 2 ] [ Red1] b d [ Red 2 ] [Ox1] 0.059 U= U + log n ! 0 0.059y pH n " (94) La constante de equilibrio se obtiene ! Ox 2 /Red 2 = ! Ox1/Red1 , ! a " 0Ox 1 /Red 1 [ ] y d [Ox 2 ] H 3O + Ox1 ] [ 0.059 0.059 0 + log = " Ox 2 /Red 2 + log c b w n1 n2 [ Red1] [ Red 2 ] [ H 2O] despejando n b n w n d Red 2 ] 1 [ H 2O] 1 [Ox1 ] 2 [ 0.059 log = " 0Ox 2 /Red 2 # " 0Ox 1 /Red 1 = U 0 n y n1 a n2c + 1 n1n2 [Ox 2 ] H 3O [ Red1] ! [ ] la constante de equilibrio es ! K eq = [ Red 2 ] [Ox 2 ] n1 b n1 a [ [ H 2O] n1 w ] n1 y H 3O + [Ox1] " U0 $ $ 0.059 n1 n 2 # n2d [ Red1] n2c = 10 % ' ' & (95) Si n1 = n2 = n, la ecuación anterior se reduce a, ! b K eq = w d " U0 $ $ 0.059 n # [ Red 2 ] [ H 2O] [Ox1] = 10 y a c [Ox 2 ] [ H 3O + ] [ Red1] % ' ' & (96) SERIE DE PROBLEMAS 6 ! 1. Calcular el potencial de celda, encontrar la reacción de celda y decir si es o no espontánea la reacción de celda para cada uno de los casos: a) Ag(s) Cu 2+ (ac,a = 0.01) Zn 2+ (ac,a = 0.1) Zn(s) Pt II (s) b) Pt(s) Fe 2+ (ac,a =1),Fe 3+ (ac,a = 0.1) Cl" (ac,a = 0.001) AgCl(s) Ag(s) " HgO(s) Hg(L) c) Zn(s) ZnO 22 ( ac,a = 0.1), OH ( ac,a =1) ! 2. Calcular la constante de equilibrio para cada una de las reacciones del problema anterior. ! ! 3. Calcular la constante de equilibrio para cada una de las reacciones: a) Zn + Cu 2+ " Zn 2+ + Cu b) Zn 2+ + 4CN " # Zn(CN) 2" 4 c) 3H 2O + Fe " Fe(OH) 3 (s) + 3 2 H 2 d) Fe + 2Fe 3+ " 3Fe 2+ ! e) 3HSnO2" + Bi 2O3 + 6H 2O + 3OH " # 2Bi + 3Sn(OH) 2" ! 6 ! f) PbSO4 (s) " Pb 2+ + SO42# !4. La celda de almacenamiento de Edison se simboliza como ! Fe(s) FeO(s) KOH (ac,a ) Ni 2O 3 (s) NiO(s) Ni(s) !Las reacciones de media celda son: + H 2O(L) + 2e" # 2NiO(s) + 2OH " $ o = 0.4 V FeO(s) +! H 2O(L) + 2e" # Fe(s) + 2OH " $ o = "0.87 V a) ¿Cuál es la reacción de celda? b) ¿Cómo depende el potencial de celda de la actividad del KOH? c) ¿Cuánta energía eléctrica se obtiene por kilogramo de materiales activos en la celda? Ni 2O 3 (s) ! 5. Considerar la celda de almacenamiento de plomo Pb(s) PbSO 4 (s) H 2SO 4 (ac,a ) PbSO 4 (s) PbO 2 (s) Pb(s) o en la que "SO 2# /PbSO 4 4 /Pb o = #0.356 V y "SO 2# /PbO 4 2 /PbSO 4 /Pb = +1.685 V . a) b) c) ! Si el potencial de celda es 2.016 V, calcular la actividad del ácido sulfúrico. Escribir la reacción de celda y decir si es o no espontánea la reacción. ! Si la celda produce trabajo (descarga), la reacción se lleva a cabo en una dirección, mientras ! (carga), la reacción se realiza en la dirección opuesta. ¿Cuánto que si se destruye trabajo trabajo debe destruirse por mol de PbO2 producido si el potencial promedio durante la carga es 2.15 V? d) Mostrar la expresión de la dependencia del potencial de celda con la actividad del ácido sulfúrico. e) ¿Cuánta energía eléctrica puede obtenerse por kilogramo de materiales activos en la celda? 6. Considerar la celda Hg(L) Hg 2SO 4 (s) FeSO 4 (ac,a = 0.01) Fe(s) a) Escribir la reacción de celda. b) Calcular el potencial de celda, la constante de equilibrio para la reacción de celda y el cambio de energía libre de Gibbs estándar a 250C. ! 7. Considerar la celda Pt(s) H 2 (g, 1 atm) H + (ac,a =1), Fe 3+ (ac), Fe 2+ (ac) Pt(s) ! con Fe 3+ + e" # Fe 2+ , " o = 0.771 V . a) Si el potencial de celda es 0.712 V, ¿cuál es la relación de concentraciones del Fe2+ y el Fe3+? ! b) ¿Cuál es la relación entre estas concentraciones si el potencial de celda es 0.83 V? ! c) Calcular la fracción de hierro total presente como Fe3+ a Ε =0.650 V, 0.700 V, 0.750 V, 0.771 V, 0.800 V, 0.850 V y 0.900 V? Graficar esta fracción como una función de Ε. 8. Los potenciales estándar a 250C son: Pd 2+ (ac) PdCl 24 (ac) 2e" + + 2e" $ o = 0.83 V Pd(s) # $ o = 0.64 V Pd(s) + 4Cl" (ac) # a) Calcular la constante de equilibrio para la reacción Pd 2+ + 4Cl" # PdCl 24 . 0 b) Calcular el Δ G para esta reacción. ! ! 9. Considerar la pareja O + e" # R , siendo la actividad de todas las especies oxidadas y reducidas la unidad. ¿Cuál debe ser el valor de Ε para la pareja si el reductor R ha de liberar hidrógeno a 1 atm a partir de a) Una solución ácida, aH + =1? ! b) Agua a pH = 7? c) ¿En qué es mejor agente reductor el hidrógeno, en una solución ácida o en una básica? 10. Considerar la misma ! pareja a las mismas condiciones del problema anterior. ¿Cuál debe ser el valor de Ε0 de la pareja si el oxidante libera oxígeno a 1 atm utilizando la reacción de media celda, O 2 (g) + 2H 2O(L) + 4e" # 4OH " $ o = 0.401 V a) a partir de una solución básica, aOH " =1? b) a partir de una solución ácida, aH + =1? c) !A partir de agua a pH = 7? d) ¿En qué es mejor agente oxidante el oxígeno, en una solución ácida o en una básica? ! ! Cálculo del potencial de celda Ε a diferente temperatura En una reacción electroquímica, el sistema pasa de un estado inicial 1 a un estado final 2 con un cambio en su energía interna o energía libre de Gibbs ΔG, donde ΔG = G2 – G1. A partir de una Serie de Taylor truncada en la segunda derivada parcial, podemos determinar la energía libre de Gibbs en el estado final G2 = G1 + ΔG en función de la temperatura, ( 2 T ) T1 & '(G # G2 = G1 + (T2 ) T1 )$ ! + 2 2 % 'T " P 2 )&$ ' (G #! 2 $ 'T 2 ! + K % "P (97) donde T1 y T2 son las temperaturas absolutas en el estado 1 y 2, respectivamente. Sustituyendo en la expresión anterior la energía G = - nFΕ, ( 2 T ) T1 & '( # ) nF( 2 = ) nF(1 ) nF (T2 ) T1 )$ ! ) nF 2 2 % 'T " P ( 2 T ) T1 & '( # ( 2 = (1 + (T2 ) T1 )$ ! + 2 2 % 'T " P 2 )&$ ' ( #! 2 )&$ ' ( #! 2 $ 'T 2 ! + K % "P 2 $ 'T 2 ! + K % "P (98) A partir de las expresiones (46) y (50), podemos determinar la primera y la segunda derivada. Para un cambio del estado 1 al estado 2, !G = !H " T!S . Despejando ΔS y sustituyendo en la expresión (46), se obtiene: !G " !H ( )!G % & # = T ' )T $ P (99) y para la segunda derivada, ( ) 2 !G % !Cp & # & )T 2 # = " T ' $P (100) Sustituyendo G = - nFΕ en las expresiones (99) y (100), !H " !G ( )* % & # = nFT ' )T $ P (101) ( ) 2* % 1 !Cp & 2# =" & )T # nF T ' $P (102) Sustituyendo las expresiones de las derivadas parciales (101) y (102) en la expresión (98), ( & 'H ( 'G # T2 2 ( T1 2 !! + ) 2 = )1 + (T2 ( T1 )$$ 2 % nF T " )&$ ( 'Cp #! + K $ nF T ! % " (103) El valor de la primera y de la segunda derivada suele ser del orden de 10-4 ó 10-5 V K-1 y 10-6 ó 10-8 V K-2, respectivamente. Por lo anterior, si la celda no contiene un electrodo de gas, podemos despreciar el segundo término de la Serie de Taylor. Si conocemos los valores del potencial de celda y de (!" !T )P podemos determinar ΔG, ΔH y ΔS para la reacción de la celda ó de la media celda. Esto es posible a partir de la expresión ΔG = - nFΕ y de la expresión (101), donde se despejan las funciones ΔH, ΔS: Para ΔH, & '( # nFT $ ! = *H ) *G % 'T " P + & '( # *H = *G + nFT $ ! % 'T " P + & '( # *H = ) nF( + nFT $ ! % 'T " P & & '( # #! *H = ) nF $$ ( ) T $ ! % 'T " P !" % (104) & '( # )S = n F $ ! % 'T " P (105) y para ΔS, Ejemplo: Para la reacción de celda, Hg 2 Cl 2 (s) + H 2 (1 atm) ! 2Hg(L) + 2 H + (a = 1) + Cl - (a = 1) con el valor de U0(298.15 K) = 0.2676 V y de (!" !T )P = -3.19 x 10-4 V K-1. Calcular: a) el valor de ΔG0, ΔH0 y ΔS0 de la reacción. b) la tensión de celda a 65°C. Solución: a) Como n = 2, ( ) ( ) #G 0 = ! n F " 0 = !2 96 487 C mol !1 (0.2676 V )10 !3 kJ J -1 = !51.64 kJ mol -1 & & '( # #! *H 0 = ) nF $$ ( ) T $ ! % 'T " P !" % ( )( ( ))( ) "H 0 = ! 2 96487 C mol -1 0.2676 V ! 298.15 K ! 3.19 x 10 -4 V K -1 10 !3 kJ J -1 = 69.99 kJ mol -1 ' () $ !1 *S 0 = n F % ! 3.19 x 10 -4 V K !1 = !61.56 J mol !1 K !1 " = 2 96487 C mol ( T & #P ( )( ) b) De la ecuación (17), sustituimos los valores, ' (U $ !4 !1 U 2 = U 1 + (T2 ! T1 )% " = 0.2676 V + (40 K ) ! 3.19 x 10 V K = 0.2548 V & (T # P ( ) Del problema anterior observamos que podemos obtener los valores de las funciones ΔG, ΔH y ΔS a partir del potencial de media celda Ε o de la tensión de celda U. Además, observamos que el valor de la tensión de celda U disminuye con el aumento en la temperatura T. Cálculo del potencial de celda Ε a diferente presión El valor del potencial de celda es función de la presión. Para obtener una expresión de Ε en términos de un estado inicial 1 a un estado final 2, partimos de la ecuación (47), ' (!G $ % " = !V & (P # T (47) Sustituyendo ΔG = -nFE, se obtiene, !V ( )* % & # =" nF ' )P $ T (106) Considerando el cambio del volumen de los líquidos despreciables comparado con el de los gases, y RT suponiendo que los gases obedecen la ley del gas ideal, ΔV es igual a !V = !n gases . P Sustituyendo en la expresión (106) e integrando: $2 % d$ = # "n gases $1 RT nF P2 % P1 dP P ! # 2 = #1 " !ngases R T P2 ln n F P1 (107) Ejemplo: 1 O2 esta caracterizada, a 25°C y 1 atm, 2 por una tensión de descomposición U0 = 1.23 V. Calcular el valor de la tensión de descomposición a 10 atm de presión. La descomposición electrolítica del agua es H 2 O ! H 2 + Solución: Datos U0 = 1.23 V "n gases = #n productos ! #n reactivos = 3 3 !0= 2 2 n = 2 (electrones intercambiados) P1 = 1 atm P2 = 10 atm U 2 = U 1 ! (n gases ( ) !1 !1 R T P2 ' 3 $ 8.314 J mol K (298.15 K ) 10 atm ln = 1.23 V ! % " ln n F P1 1 atm 2 96487 C mol !1 &2# ( ) U 2 = 1.23 V ! (0.04436 V ) = 1.18 V Observamos en el ejemplo anterior que el valor de la tensión de celda disminuye con el incremento en la presión. SERIE DE PROBLEMAS 7 1. Calcular la tensión de celda estándar U0 a partir de tablas de potencial de media celda estándar Ε0 de la electrólisis del cloro con: a) reducción de H2 en el cátodo, b) reducción de O2 en el cátodo c) Decir cual es más espontánea. 2. Calcular la tensión de celda estándar a partir de de los datos termodinámicos en tablas para la electrólisis del cloro con: a) reducción de H2 en el cátodo, b) reducción de O2 en el cátodo c) Decir cual es más espontánea. 3. Retomando la electrólisis del cloro, calcular la tensión de celda U0 a 338 K considerando que la reacción en el cátodo es la reducción del hidrógeno. 4. La reacción de reducción de plata Ag + + 1 H 2 ! Ag + H + está caracterizada a 25°C por los 2 siguientes valores estándar: ΔG0 = -18.43 kcal, ΔS0 = -23.06x10-3 kcal K-1, ΔCp0 = -6.34x10-3 kcal K-1. Determinar la influencia de la temperatura sobre la energía libre de Gibbs a presión constante.