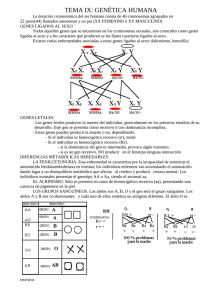
Objetivos Contestados
Anuncio

Autor: José Regino Ríos Gutiérrez. UNIDAD 1. Introducción a la Genética Médica. Conceptos Básicos. Competencia intermedia: Analizar el objeto de estudio de la genética, su clasificación, estado actual y relación con las diferentes especialidades médicas. Criterios de desempeño: 1.1- 1.2- Definir el concepto actual de genética identificando su objeto de estudio. La Genética Médica se ha convertido en una parte del campo más amplio de la medicina genómica, que persigue la aplicación a gran escala del análisis del genoma humano (es la suma de toda la información genética correspondiente a nuestra especie), incluyendo el control de la expresión genética, la variación de los genes humanos y las interacciones entre los genes y el ambiente, con objetivo de mejorar la asistencia médica. La genética médica incluye cualquier aplicación de la genética en la práctica médica. Abarca los estudios de la herencia de las enfermedades familiares, la localización específica de los genes de las enfermedades en los cromosomas, el análisis de los mecanismos moleculares mediante los cuales los genes causan la enfermedad y el diagnóstico y el tratamiento de las enfermedades genéticas. Analizar la evolución histórica de la genética y su clasificación: humana, animal, vegetal y de microorganismos. Los antiguos griegos y hebreos, y después los eruditos medievales, describieron numerosos fenómenos genéticos y propusieron diversas teorías para explicarlos. Gregor Mendel hizo avanzar significativamente la genética al llevar a cabo un conjunto de experimentos, diseñados en organismos vivos (guisantes de jardín) y con esta información formuló una serie de leyes fundamentales de la herencia, que todavía hoy constituyen los fundamentos de la genética. Charles Darwin formuló sus teorías sobre la evolución y su primo Francis Galton desarrolló una amplia serie de estudios en familias (centrado específicamente en gemelos). Las leyes de Mendel fueron redescubiertas en 1900, por tres científicos que trabajaban en tres países distintos. Landstainer descubrió también el sistema de los grupos sanguíneos ABO. En 1902, Archivald Garrod describió la alcaptonuria como el primer “error congénito del metabolismo” y en 1909, Johannsen acuñó el término gen para determinar la unidad elemental de la herencia. 1 H.J. Muller demostró las consecuencias genéticas de la radiación ionizante en la mosca de la fruta. Oswald Avery demostró en 1944 que los genes estaban constituidos por DNA. 1.3- Analizar la clasificación de la genética humana: genética médica, citogenética, genética molecular, genética bioquímica, genética de poblaciones, medicina genómica. La Genética Médica se ha convertido en una parte del campo más amplio de la medicina genómica, que persigue la aplicación a gran escala del análisis del genoma humano (es la suma de toda la información genética correspondiente a nuestra especie), incluyendo el control de la expresión genética, la variación de los genes humanos y las interacciones entre los genes y el ambiente, con objetivo de mejorar la asistencia médica. La Citogenética es el estudio de los cromosomas, su estructura y su herencia aplicado a la práctica de la genética médica. La Genética Molecular se encarga del estudio estructural de los genes, La Genética Bioquímica se enfoca en la función génica, La Genética de Poblaciones investiga las características por grupos o razas. La Asesoría Genética establece el riesgo de recurrencia de alguna enfermedad. La Medicina Genómica se encarga del estudio del genoma su organización y sus funciones. 1.4Clasificar las enfermedades hereditarias cromosomopatías, enfermedades multifactoriales. en: genopatías, Las GENOPATÍAS o defectos monogénicos están causadas por genes mutantes individuales. Como la Fibrosis Quística, la Anemia de células falciformes y el Síndrome de Marfan. Las CROMOSOPATÍAS tienen un exceso o un defecto de los genes contenidos en cromosomas enteros o fragmentos cromosómicos. Como el Síndrome de Down, como grupo afectan a 7 de cada 1000 nacidos vivos. La HERENCIA MULTIFACTORIAL es responsable de la mayor parte de enfermedades, todas las cuales poseen una distribución genética. Como los las malformaciones congénitas, la enfermedad de Hirschsprung, el labio y paladar hendidos, las cardiopatías congénitas y enfermedades como Alzheimer, la diabetes y la hipertensión. 2 5.- Analizar el perfil de los padecimientos que son causa frecuente de referencia a la consulta genética destacando la importancia del asesoramiento genético y de la detección temprana de los desórdenes hereditarios. Las enfermedades genéticas suelen considerarse tan infrecuentes que la mayoría de los profesionales de la salud rara vez se toparán con ellas. Además de su importante contribución a la mortalidad infantil, las enfermedades genéticas son también responsables de gran parte de los ingresos en los hospitales pediátricos. Otra forma de valorar la importancia de este tipo de enfermedades es preguntarse qué proporción de individuos en una población será diagnosticada de alteraciones genéticas, parece una pregunta muy trivial pero no lo es ya que hay varios factores que pueden influir en la respuesta. Teniendo en cuenta las fuentes de variabilidad, merece la pena destacar que en entre el 3-7% de la población será diagnosticada de una enfermedad genética identificable. 6.- Analizar la asesoría genética desde el punto de vista de la bioética. La asesoría genética es un tema muy delicado ya que a la hora de por ejemplo avisar a unos padres sobre un producto que seguramente nacerá con un mal congénito, la decisión de la interrupción del embarazo o de la continuación de éste no solo es cuestión de los padres si no también de los valores morales, religiosos y éticos del médico. 3 Autor: José Regino Ríos Gutiérrez. UNIDAD 2. Aspectos citogenéticos de la herencia. Competencia Intermedia: Analizar los conceptos básicos de la estructura y función de los cromosomas así como su comportamiento durante el ciclo celular para fundamentar la selección de la técnica citogenética en base a la historia clínica del paciente. Criterios de desempeño: 2.1.Analizar los mecanismos que producen las aberraciones cromosómicas: el fenómeno de no disyunción y anafase retardada y su importancia en la formación de alteraciones cromosómicas, así como los mecanismos de alteraciones cromosómicas estructurales: anillos, duplicaciones, deleciones, inversiones, translocaciones recíprocas y robertsonianas. La causa mas frecuente de aneuploidía es la ausencia de disyunción, es decir, la incapacidad de los cromosomas para separarse normalmente durante la meiosis. La ausencia de disyunción puede producirse durante la meiosis I o durante la meiosis II. Un error durante la meiosis I conducirá a que el gameto contenga ambos cromosomas homólogos de un par cromosómico. Por el contrario, la no disyunción en la meiosis II originará un gameto con dos copias de uno de los cromosomas homólogos. El anafase retardado consiste en que uno de los cromosomas, durante el anafase, se retrasa en ir hacia uno de los polos celulares, al terminarse la telofase se pierde. Las aberraciones estructurales pueden ser balanceadas o no balanceadas, en dependencia de que se conserve o no todo el material genético. Las desbalanceadas son: o NO DISYUNCIÓN.- Existe un fallo en la separación de uno de los pares de cromosomas homólogos durante la anafase de la meiosis I materna. Ejemplos: Síndrome Down o trisomía 21; síndrome de Patau o trisomía 13; síndrome de Edwards o trisomía 18. o ANILLOS.- un cromosoma en anillo se forma cuando en cada brazo del cromosoma se produce una rotura que deja dos extremos “cohesivos” en la porción central y que se une formando un anillo. Los dos fragmentos cromosómicos distales se pierden en formas que, si el cromosoma afectado es un autosoma, los efectos son habitualmente graves. Los cromosomas en anillo son a menudo inestables en la mitosis, por lo que es habitual encontrar un cromosoma en anillo en una proporción de células. Las otras células del individuo son habitualmente 4 monosómicas, debido a la ausencia del cromosoma en anillo. o DUPLICACIONES.se pueden producir por entrecruzamiento desigual en la meiosis y también entre los descendientes de portadores de translocaciones recíprocas. o DELECCIONES.- una delección consiste en la pérdida de parte de un cromosoma y se traduce en una monosomía para ese segmento del cromosoma. Una delección muy grave es habitualmente incompatible con la supervivencia a término, en general, cualquier delección que produzca la pérdida de más del 2% del genoma haploide total tendrá un resultado fatal. Las delecciones ocurren a dos niveles: la gran delección cromosómica (síndrome de WolfHirschorn, del maullido del gato, que conlleva la pérdida de material de los brazos cortos de los cromosomas 4 y 5). Y las microdelecciones (síndrome de Prader-Willi y de Angelman). Balanceadas: o INVERSIONES.- es una reconfiguración de dos roturas que afectan a un único cromosoma, y en la que un segmento tiene una posición invertida. Si el segmento invertido afecta al centrómero, se denomina inversión pericéntrica. Si sólo afecta a un brazo del cromosoma, se conoce como inversión paracéntrica. Raramente causan problemas en los portadores, a menos que uno de los puntos de rotura haya alterado un gen importante. Existen inversiones que aunque no causen ningún problema clínico en los portadores equilibrados, pueden producir un desequilibrio cromosómico en la descendencia, con consecuencias clínicas importantes. (HETEROMORFISMO: la inversión pericéntrica afecta al cromosoma número 9, se produce una variante estructural habitual o polimorfismo sin ninguna importancia funcional). o TRANSLOCACIONES RECÍPROCAS.- La transferencia de material genético de un cromosoma a otro se denomina translocación. Una translocación recíproca se forma cuando se produce una rotura en cada uno de los dos cromosomas intercambiándose los segmentos para formar dos nuevos cromosomas derivados. Implica la rotura de al menos dos cromosomas con intercambio de fragmentos. Habitualmente el número de cromosomas se mantiene en 46. En general, las translocaciones recíprocas son únicas de una familia en particular, aunque, por razones que no se conocen, es relativamente habitual una translocación recíproca equilibrada que afecta a los brazos largos de los 5 cromosomas 11 y 22. La incidencia global de la translocación recíproca en la población general es de 1 por 500. o TRANSLOCACIONES ROBERTSONIANAS.- se produce por la rotura de dos cromosomas acrocéntricos (números 13, 14, 15, 21 y 22) en o cerca de sus centrómeros con la posterior fusión de sus brazos largos (se denomina también fusión céntrica). Los brazos cortos de cada cromosoma se pierden sin importancia clínica. El número total de cromosomas se reduce a 45, al no haber pérdida ni ganancia de material genético se considera una reconfiguración funcionalmente equilibrada. La incidencia es de 1 por 1000. Los riesgos es síndrome de Down, abortos espontáneos. 2.2.- Definir mosaico cromosómico estableciendo sus mecanismos de producción el mosaicismo puede definirse como la presencia en un individuo o en un tejido, de dos o más líneas celulares que difieren en su constitución genética pero no derivan de un único cigoto, es decir, no tienen el mismo origen genético. El mosaicismo cromosómico se produce habitualmente por una no disyunción en una división mitótica embrionaria precoz, con persistencia de más de una línea celular. Si, por ejemplo, las dos cromátidas del cromosoma número 21 no se separasen en la segunda división mitótica en un cigoto humano, haría que el cigoto de cuatro células tuviera dos células con 46 cromosomas, una célula con 47 cromosomas (trisomía 21) y una célula con 45 cromosomas (monosomía 21). La siguiente línea celular con 45 cromosomas probablemente no sobreviviría, de forma que es de esperar que el embrión resultante muestre aproximadamente un 33% de mosaicismo para la trisomía 21. El mosaicismo es responsable del 1 al 2 % de todos los casos clínicamente reconocidos de síndrome Down. El mosaicismo puede darse también a escala molecular si una nueva mutación surge a escala en una división celular somática o de la línea germinal precoz. La posibilidad de un mosaicismo de la línea germinal o gonadal es una preocupación importante que surge al aconsejar a los padres de un niño que presente un trastorno, como una distrofia muscular de Duchenne y se trate de un caso asilado. 2.3.- Determinar el objeto de estudio de la citogenética. La citogenética se refiere al estudio de los cromosomas y de la división celular. 2.4.- Analizar las indicaciones e interpretar los resultados de las técnicas para el estudio de los cromosomas con énfasis en técnicas de citogenética convencional y molecular: Hibridación in situ y sus variantes. Interpretar los resultados. 6 BANDEO DE LOS CROMOSOMAS.o BANDEO G (GIEMSA).- método más usado, los cromosomas se tratan con tripsina, que desnaturaliza su contenido proteíco, y se tiñen después con un colorante que se fija al DNA, conocido como Giemsa, que da a cada cromosoma un patrón característico y reproducible de bandas claras y oscuras. o BANDEO Q (quinacrina).- proporciona un patrón de bandeo similar al obtenido con el Giemsa y requiere el examen de los cromosomas con un microscopio de flourescencia ultravioleta. o BANDEO R (reverso).- los cromosomas se desnaturalizan por calor antes de teñirlos con Giemsa, y muestran bandas claras y oscuras que son el inverso de las obtenidas utilizando el bandeo G convencional. o BANDEO C (heterocromatina centromérica).- si los cromosomas son pretratadas con ácido seguido por álcali antes de bandeo G, los centrómeros y otras regiones heterocromáticas que contienen secuencias de DNA altamente repetitivas se tiñen de forma preferecial. o BANDEO POR ALTA RESOLUCIÓN.- el bandeo de alta resolución de los cromosomas en una etapa inicial de la mitosis, como la profase o la prometafase, aporta una mayor sensibilidad, con hasta 800 bandas por conjunto haploide, pero técnicamente es mas exigente. Conlleva en primer lugar la inhibición de la división celular con un agente como el metotrexato o la timidina. Se añade ácido fólico o desoxicitidina al medio de cultivo y se permite a las células que procedan en la mitosis. Se añade entonces la colchicina en un intervalo específico de tiempo, cuando la mayor proporción de células estén en prometafase y los cromosomas no estén completamente contraídos, dando un patrón de bandeo más detallado. o ANÁLISIS DEL CARIOTIPO.- la siguiente etapa en el análisis del cromosoma consiste, en primer lugar, en contar el número de cromosomas presentes en un número especificado de células, algunas veces denominado extensiones metafísicas, seguido por un análisis cuidadoso del patrón de bandeo de cada cromosoma individual en células seleccionadas. Habitualmente el recuento total de cromosomas se hace en 10 a 15 células, pero si se sospecha mosaicismo hay que emprender el recuento de 30 o más células. El análisis detallado del patrón de bandeo de los cromosomas individuales se realiza en ambos miembros de cada pareja de homólogos en aproximadamente tres a cinco extensiones metafísicas, que muestran un bandeo de alta calidad. El patrón de bandeo es específico de cada cromosoma y puede mostrarse en forma de cariotipo estilizado ideal conocido como ideograma. El citogenetista analiza cada pareja de cromosomas homólogos en un microscopio o en una fotografía de la extensión metafísica. El cariotipo es 7 presentado formalmente o cariograma, muestra cada pareja de cromosomas en orden descendente, según su tamaño. o HIBRIDACIÓN IN SITU FLOURESCENTE.- combina la citogenética con la tecnología genética molecular. Se basa en la capacidad única de hibridar una porción de un DNA monocatenario (una sonda) con su secuencia diana complementaria en un cromosoma metafísico, un núcleo en la interfase o una fibra de cromatina extendida. En la hibridación in situ por flourescencia (FISH), la sonda de DNA se marca con un flourocromo que, tras la hibridación con la muestra del paciente, permite visualizar la región donde ha tenido lugar la hibridación usando un microscopio de flourescencia. La FISH se usa ampliamente con fines de diagnóstico clínico y pueden emplearse diferentes tipos de sondas. o TIPOS DE SONDAS FISH. SONDAS CENTROMÉRICAS.- consiste en secuencias de DNA repetitivas encontradas en y alrededor del centrómero de un cromosoma específico. Fueron las sondas originales usadas para el diagnóstico rápido de los síndromes habituales de aneuplodía (trisomía 13, 18, 21), usando células en interfase que no se estaban dividiendo obtenidas de una muestra de diagnóstico prenatal de las vellosidades coriónicas. SONDAS DE SECUENCIA ÚNICA ESPECÍFICAS DE UN CROMOSOMA.- son específicas de un locus en particular. Las sondas específicas de un locus para el cromosoma 13q14 y la región crítica para el síndrome de Down en el cromosoma 21 (21q22.13-21q22.2) pueden usarse conjuntamente con las sondas centroméricas para los cromosomas 18, X e Y para proporcionar un diagnóstico prenatal rápido de algunas de las anomalías cromosómicas numéricas más habituales. Las sondas de secuencia única son particularmente útiles para identificar las minúsculas delecciones y duplicaciones submicroscópicas. Otra aplicación es la utilización de una sonda FISH en interfase para identificar la sobreexpresión de HER2 en los tumores de mama con objeto de detectar a los pacientes con posibilidades de beneficiarse del tratamiento con herceptina. SONDAS TELOMÉRICAS.- Se ha desarrollado un juego completo de sondas teloméricas para los 24 cromosomas (es decir, los autosomas 1-22 más el X y el Y). Para su uso, se ha diseñado un método que permite el análisis simultáneo de la región subtelomérica de cada cromosoma empleando una única extensión microscópica por paciente. Es particularmente útil para identificar a las diminutas anomalías “crípticas” subteloméricas. Como las delecciones y las translocaciones, en una pequeña pero significativa proporción de niños con discapacidad intelectual inexplicada. 8 SONDAS DE PINTADO DEL CROMOSOMA COMPLETO.consiste en un coctel de sondas obtenidas de diversas partes de un cromosoma particular. Cuando esta mezcla de sondas se usa conjuntamente en una hibridación única, todo el cromosoma relevante presenta fluorescencia (es decir, esta “pintado”). El pintado del cromosoma es extremadamente útil para caracterizar las reconfiguraciones complejas, como las translocaciones sutiles, y para identificar el origen del material cromosómico adicional, como los pequeños marcadores o anillos supernumerarios. La última tecnología, descrita como multiplex FISH (M-FISH) o cariotipado espectral (SKY), usa depósitos de sondas de pintado de cromosomas humanos completos para proporcionar un cariotipo humano multicolor, en el que cada pareja de cromosomas homólogos puede identificarse a partir de su color único cuando se estudian mediante análisis de imágenes basados en ordenadores. Estos métodos han demostrado ser extremadamente útiles para detectar las sutiles reconfiguraciones cromosómicas, como las delecciones y las translocaciones, y para identificar a los pequeños marcadores supernumerarios y a los cromosomas en anillo. SONDAS OBTENIDAS A PARTIR DE CROMOSOMAS PURIFICADOS CON EL CITÓMETRO DE FLUJO.- Debido a su diferente tamaño y composición de DNA, los cromosomas se unen a cantidades distintas de colorantes fluorescentes, algunos de los cuales se fijan específicamente a las secuencias GC (“ricas en genes”) y otros a secuencias AT (“pobres en genes”). Esta propiedad de la unión diferencial permite que los cromosomas se separen mediante el proceso de citometría de flujo o separación de las células activadas por fluorescencia (FACS, fluorescente activated cell sorting). Esta técnica consiste en la tinción de los cromosomas en metafase con un colorante fluorescente que se fije al DNA y la proyección de un chorro fino de gotitas mediante un haz de rayos láser enfocado, que excita a los cromosomas hasta que emiten fluorescencia. La intensidad de la fluorescencia se mide con un fotomultiplicador, y los resultados se analizan con un ordenador que dibuja un histograma de distribución del tamaño de los cromosomas. Es lo que se conoce como cariotipo de flujo. HIBRIDACIÓN GENÓMICA COMPARATIVA.(CGH, comparative genomic hybridation) se desarrolló originalmente para solucionar la dificultad de obtener preparaciones metafísicas de buena calidad a partir de tumores sólidos. Esta técnica permite la detección de regiones con pérdida de alelos y con amplificación de genes. El DNA tumoral o de “test” se marca con un fluorocromo verde y el DNA normal de control, con un fluorocromo rojo. Las dos muestras se mezclan y se 9 hibridan competitivamente con cromosomas metafísicos normales y se obtiene una imagen. Si la muestra testada contiene más DNA de una región cromosómica particular que la muestra de control, dicha región se identifica por un incremento de la ratio de fluorescencia verde:rojo. De forma similar, una delección en la muestra se identifica por una reducción en la ratio de flourescencia vede:rojo. La aplicación de la CGH se ha ampliado con le objeto de incluir el análisis de células únicas para el diagnóstico prenatal tras la amplificación de todo el genoma y proporcionar así material suficiente para el análisis. CGH EN MICROMATRICES.- conlleva también también la hibridación del DNA del paciente y del de referencia, pero los cromosomas metafísicos son reemplazados como diana por grandes cantidades de secuencias de DNA unidas a los portas de cristal. Las secuencias de DNA diana pueden ser clones mapeados (cromosoma artificial de levadura (YAC), cromosoma artificial bacterian (BAC), cromosoma artificial derivado de P1 (PAC) o un cósmido) u oligonucleótidos. Se depositan sobre los portas del microscopio usndo un robot mecánico para crear una micromatriz, en la que cada DNA diana tiene una localización única. Después de la hibridación y del lavado para eliminar el DNA no fijado, se miden los valores relativos de fluorescencia usando un programa informático. La aplicación de la CGH en micromatrices se ha extendido desde la citogenética del cáncer hasta la detección de cualquier tipo de ganancia o pérdida, incluyendo la detección de las delecciones subteloméricas en pacientes con una discapacidad intelectual no explicada. 10 Autor: José Regino Ríos Gutiérrez. UNIDAD 3. Síndromes cromosómicos de los autosomas Competencia Intermedia: Clasificar y analizar los síndromes cromosómicos autosómicos más frecuentes, seleccionando e interpretando el cariotipo para hacer el diagnóstico que determina su abordaje, manejo, pronóstico y asesoramiento genético. Criterios de desempeño: 1.- Clasificar la patología estructurales. cromosómica en alteraciones numéricas y ANOMALÍAS NUMÉRICAS.- consisten en la pérdida o la ganancia de uno o más cromosomas, referida como aneuploidía, o la adición de uno o más complementos haploides, conocida como poliploidía. La pérdida de un solo cromosoma produce una monosomía. La ganancia de uno o dos cromosomas homológos se denomina trisomía y tetrasomía, respectivamente. o TRISOMÍA.- (presencia de un cromosoma extra). La mayoría de los casos del síndrome de Down se deben a la presencia de un cromosoma 21 adicional (por ello también se le conoce al sd. De Down como trisomía 21). Otras trisomías autosómicas compatibles con la supervivencia a término son el síndrome de Patau (trisomía 13) y el síndrome de Edwards (trisomía 18). La mayoría de las demás trisomías autosómicas producen la interrupción precoz del embarazo, y la trisomía 16 es un hallazgo habitual en los abortos espontáneos del primer trimestre. La presencia de un cromosoma sexual adicional (X o Y) tienen únicamente efectos fenotípicos leves. La trisomía 21 esta causada habitualmente por un fallo en la separación de uno de los pares de cromosomas homólogos durante la anafase de la meiosis I materna. Este fallo de la separación del bivalente se denomina no disyunción. Con menor frecuencia, la trisomía está causada por una no disyunción que se produce durante la meiosis II, cuando la pareja de cromátides hermanas no pueden separarse. De cualquiera de las formas, el gameto recibe dos cromosomas homólogos (disomía), y si hay una fertilización posterior, se produce un embrión trisómico. o MONOSOMÍA.- (ausencia de un solo cromosoma), la monosomía para un autosoma es casi siempre incompatible con la supervivencia a largo plazo. La falta de contribución de un cromosoma X o Y produce un cariotipo 45, X, que causa el trastorno conocido como síndrome de Turner. Igual que en la trisomía, la monosomía puede producirse por la no disyunción en la meiosis. Si un gameto recibe dos copias de un cromosoma homólogo (disomía), el otro gameto hijo correspondiente no tendrá ninguna copia del mismo cromosoma (nulosomía). La monosomía puede estar también causada por la pérdida de un cromosoma cuando se mueve al polo de la célula durante la anafase, hecho conocido como “retraso de la anafase”. 11 o POLIPLOIDÍA.- las células polipliodes contienen múltiplos del número haploide de cromosomas, como 69 (triploidía) o 92 (tetraploidía). En los seres humanos la triploidía se encuentra relativamente a menudo en el material formado en los abortos espontáneos, y la supervivencia más allá de la mitad del embarazo es rara. o La triploidía puede estar causada por un fallo en la maduración de la división meiótica en un óvulo o en un espermatozoide que conduce, por ejemplo, a la retención de un cuerpo polar o a la formación de un espermatozoide diploide. Alternativamente, se puede producir por la fertilización de un óvulo por dos espermatozoides (dispermia). Cuando la triploidía resulta de la presencia de un juego adicional de cromosomas paternos, la placenta presente habitualmente un tamaño incrementado, lo que se conoce como cambios hidatidiformes. En cambio, cuando la triploidía se produce por un juego adicional de cromosomas maternos, la placenta es habitualmente pequeña. La triploidía ocasiona típicamente un aborto espontáneo precoz. Las diferencias entre la triploidía debida a un juego adicional de cromosomas paternos o de cromosomas maternos proporciona la evidencia de los efectos “epigenéticos” y de “origen del progenitor” importantes en relación con el genoma humano. ANOMALÍAS ESTRUCTURALES.las reconfiguraciones cromosómicas estructurales se producen por rotura del cromosoma con reunificación posterior en una configuración diferente. Pueden ser equilibradas o desequilibradas. En las reconfiguraciones equilibradas el complemento cromosómico está completo, sin pérdidas ni ganancias del material genético. En consecuencia, las reconfiguraciones equilibradas son por lo general inocuas, con la excepción de los casos raros en los que uno de los puntos de rotura daña a un gen funcional importante. No obstante, los portadores de reconfiguraciones equilibradas presentan a menudo el riesgo de generar niños con un complemento cromosómico desequilibrado. Cuando una reconfiguración cromosómica está desequilibrada, el complemento cromosómico contiene una cantidad incorrecta de material cromosómico y los efectos clínicos son habitualmente graves. TIPOS DE ANOMALÍA CROMOSÓMICA.o NUMÉRICA. Aneuploidía. Monosomía Trisomía Tetrasomía Poliploidía. Triploidía 12 o ESTRUCTURAL. o Tetraploidía Translocaciones. Recíprocas Robertsonianas Delecciones Inserciones Inversiones. Paracéntricas Pericéntricas Anillos Isocromosomas DIFERENTES LÍNEAS CELULARES (MIXOPLODÍA). Mosaicismo Quimera 2.- Clasificar las alteraciones numéricas en euploidías y aneuploidías. EUPLOIDÍAS.- Euploidía es el estado celular en el cual la célula tiene uno o más juegos completos de cromosomas (dotaciones monoploides (x)) de su especie; dependiendo de especies, se excluyen los cromosomas sexuales. Los individuos con euploidía pueden presentar o no un número anormal de cromosomas diferente al habitual. Por ejemplo, un humano, tiene 46 cromosomas, que es un número múltiplo de 23, sus número de cromosomas monoploide (x) (que en el caso de los humanos coincide con el número de cromosomas haploide (n); x = n = 23), por lo tanto es euploide (2x = 2n = 46). Un humano hipotético anormal, pero con juegos enteros de cromosomas, por ejemplo, 69, sería también euploide, en tanto que 69 sería múltiplo entero del número monoploide (3x = 69). No se debe confundir con la aneuploidía, que, en contraposición a la euploidía, se refiere a una alteración en la cantidad de uno de los tipos de cromosomas homólogos. Puede Ser de dos tipos: monoploidía y poliploidía. ANEUPLOIDÍAS.- Es la pérdida o ganacia de uno o más cromosomas. Puede ser de tres tipos: monosomía, trisomía y tetrasomía. 3.- Clasificar las alteraciones estructurales en rearreglos balanceados: translocaciones recíprocas y robertsonianas, inversiones paracéntricas y pericéntricas e inserciones; y no balanceados: deleciones, duplicaciones, isocromosomas, anillos, marcadores y dicéntricos. REARREGLOS BALANCEADOS.- 13 TRANSLOCACIONES RECÍPROCAS.- La transferencia de material genético de un cromosoma a otro se denomina translocación. Una translocación recíproca se forma cuando se produce una rotura en cada uno de los dos cromosomas intercambiándose los segmentos para formar dos nuevos cromosomas derivados. Implica la rotura de al menos dos cromosomas con intercambio de fragmentos. Habitualmente el número de cromosomas se mantiene en 46. En general, las translocaciones recíprocas son únicas de una familia en particular, aunque, por razones que no se conocen, es relativamente habitual una translocación recíproca equilibrada que afecta a los brazos largos de los cromosomas 11 y 22. La incidencia global de la translocación recíproca en la población general es de 1 por 500. TRANSLOCACIONES ROBERTSONIANAS.- se produce por la rotura de dos cromosomas acrocéntricos (números 13, 14, 15, 21 y 22) en o cerca de sus centrómeros con la posterior fusión de sus brazos largos (se denomina también fusión céntrica). Los brazos cortos de cada cromosoma se pierden sin importancia clínica. El número total de cromosomas se reduce a 45, al no haber pérdida ni ganancia de material genético se considera una reconfiguración funcionalmente equilibrada. La incidencia es de 1 por 1000. Los riesgos es síndrome de Down, abortos espontáneos. INVERSIONES PARACÉNTRICAS.- Si sólo afecta a un brazo del cromosoma, se conoce como inversión paracéntrica. Raramente causan problemas en los portadores, a menos que uno de los puntos de rotura haya alterado un gen importante. INVERSIONES PERICÉNTRICAS.- es una reconfiguración de dos roturas que afectan a un único cromosoma, y en la que un segmento tiene una posición invertida. Si el segmento invertido afecta al centrómero, se denomina inversión pericéntrica. INSERCIONES.- la inserción se produce cuando se intercala un segmento de un cromosoma en otro cromosoma. Si el material insertado se ha desplazado desde algún otro lugar en otro cromosoma, entonces el cariotipo está equilibrado. De lo contrario, la inserción ocasiona un complemento cromosómico desequilibrado. REARREGLOS NO BALANCEADOS. DELECCIONES.- una delección consiste en la pérdida de parte de un cromosoma y se traduce en una monosomía para ese segmento del cromosoma. Una delección muy grave es habitualmente incompatible con la supervivencia a término, en general, cualquier delección que produzca la pérdida de más del 2% del genoma haploide total tendrá un resultado fatal. Las delecciones ocurren a dos niveles: la gran delección cromosómica (síndrome de Wolf-Hirschorn, del maullido del gato, que conlleva la pérdida de material de los brazos cortos de los cromosomas 4 y 5). Y las microdelecciones (síndrome de Prader-Willi y de Angelman). DUPLICACIONES.- 14 ISOCROMOSOMAS.- un isocromosoma muestra la pérdida de un brazo con la duplicación de otro. La explicación más probable para la formación de un isocromosoma es que el centrómero se haya dividido transversalmente en lugar de longitudinalmente. El isocromosoma más habitualmente encontrado es el formado por dos brazos largos del cromosoma X. Es el responsable de hasta el 15% de todos los casos del síndrome de Turner. ANILLOS.- un cromosoma en anillo se forma cuando en cada brazo del cromosoma se produce una rotura que deja dos extremos “cohesivos” en la porción central y que se une formando un anillo. Los dos fragmentos cromosómicos distales se pierden en formas que, si el cromosoma afectado es un autosoma, los efectos son habitualmente graves. Los cromosomas en anillo son a menudo inestables en la mitosis, por lo que es habitual encontrar un cromosoma en anillo en una proporción de células. Las otras células del individuo son habitualmente monosómicas, debido a la ausencia del cromosoma en anillo. MARCADORES.- polimorfismo de un grupo sanguíneo , bioquímico o del DNA que, si se demuestra que está unido al locus de una enfermedad de interés, puede usar para el diagnóstico presintomático. DICÉNTRICOS.- posee dos centrómeros. 4.- Evaluar las características clínicas, variedades citogenéticas, factores de riesgo y aspectos epidemiológicos para llegar al diagnóstico de las cromosomopatías numéricas autosómicas más frecuentes: síndrome de Down, síndrome de Edwards, síndrome de Patau. SÍNDROME DE DOWN (o trisomía 21).o Características clínicas.- en período neonatal hipotonía grave. En general los rasgos faciales, con las hendiduras palpebrales inclinadas hacia arriba, las orejas pequeñas y lengua sobresaliente. Línea palmar única en 50% de los casos. Cardiopatías congénitas en 40 a 45% de los casos (defectos del canal del canal auriculoventricular, lo del septo ventricular y la persistencia del ducto arterioso). o Base genética.- en los casos debidos a la trisomía 21, el cromosoma hereditario es de origen materno (90%), debido a la no disyunción en la meiosis materna. La translocación robertsoniana es responsable del 4% de los casos. o Patrón hereditario.- anomalías cromosómicas numéricas. SÍNDROME DE EDWARDS (o trisomía 18) .o Características clínicas.- incidencia 1 de 5,000. baja tasa de supervivencia, cardiopatías congénitas. La incidencia sube a medida que aumenta la edad materna, siendo el cromosoma adicional de 15 origen materno. En menos del 10% es causado por mosaicismo o reordenación desequilibrada. o Base genética.- cromosoma adicional es de origen materno. o Patrón hereditario.- anomalías cromosómicas numéricas. SÍNDROME DE PATAU (o trisomía 13).o Características clínicas.- incidencia 1 de 5,000. baja tasa de supervivencia, cardiopatías congénitas. La incidencia sube a medida que aumenta la edad materna. En los raros casos de supervivencia a largo plazo presentan graves dificultades de aprendizaje. o Base genética.- cromosoma adiconal es de origen materno. En 10% de los casos la causa es mosaicismo, o la reordenación desequilibrada. o Patrón hereditario.- anomalías cromosómicas numéricas. NOTA.- como se puede apreciar los síndromes de Edwards y Patau comparten muchas características, se describieron por primera vez en 1960. 5.- Evaluar las características clínicas, variedades citogenéticas, factores de riesgo y aspectos epidemiológicos para llegar al diagnóstico de las cromosomopatías estructurales autosómicas más frecuentes: síndrome de Cri-du-Chat, síndrome de Wolf-Hirschhorn, SÍNDROME DE CRI-DU-CHAT (o síndrome del maullido del gato).o Características clínicas.- el nombre de maullido del gato proviene de lloro característico de los recién nacidos afectados, semejante a un maullido debido al insuficiente desarrollo de la laringe; graves dificultades de aprendizaje, asociada a retraso físico. Incidencia 1 de 50,000. o Base genética.- (5p-) delecciones de las porción terminal del cromosoma 5 o Patrón hereditario.- anomalía cromosómica estructural. SÍNDROME DE WOLF-HIRSCHHORN.o Características clínicas.- dificultades de aprendizaje, unidas a retraso en desarrollo físico; incidencia 1 de 50,000. o Base genética.- (4p-) delección de la porción terminal del cromosoma 4; poca correlación entre el fenotipo y la pérdida precisa de material cromosómico. o Patrón hereditario.- anomalía cromosómicas estructural 16 6.- Evaluar las características clínicas, factores de riesgo y aspectos epidemiológicos para llegar al diagnóstico de los síndromes de genes contiguos: síndrome de Beckwith-Wiedemann, síndrome de Prader-Willi, síndrome de di George. SÍNDROME DE BECKITH-WIEDEMANN.o Características clínicas.- En el cuadro clínico se observan niños afectados con aumento de tamaño corporal en el momento de nacer, con la lengua también muy grande (macroglosia) y a menudo una importante protrusión del ombligo. Algunos pacientes han presentado fibrosis quística y baja estatura. La hipoglucemia grave es una complicación potencialmente mortal, así como la aparición de tumores malignos en riñones, glándulas suprarrenales e hígado. o Base genética.- síndrome ocasionado por la disomía uniparental en una porción del cromosoma 11(11p15), con el consiguiente exceso de genes aportados por el padre, también es posible que sea por una pérdida de los genes aportados por la madre, o a ambas posibilidades. También se encuentra afectado el Gen del factor de crecimiento de tipo insulina 2. o Patrón hereditario.- SÍNDROME DE PRADER-WILLI.o Características clínicas.- 1 de 20,000 nacimientos, estatura corta, obesidad, hipogonadismo y dificultades de aprendizaje o Base genética.- delección intersticial en la porción proximal de brazo largo del cromosoma 15; también puede ser por una delección submicroscópica por hibridación. Casi siempre obtenido del padre o Patrón hereditario.- 7.- Determinar el método de estudio de los síndromes cromosómicos autosómicos más frecuentes: historia clínica, exámenes de laboratorio, gabinete y cariotipo, e interpretar los resultados para establecer diagnóstico, opciones terapéuticas y pronóstico. Estudios moleculares para la detección de mutaciones: Southern blot, PCR y secuenciación. Southern blot: Digestión de ADN mediante enzimas de restricción. Electroforesis de los fragmentos en gel de agarosa. Transferencia del ADN a una membrana de nailon o nitrocelulosa. Hibridación del ADN con una sonda marcada. Sirve para detectar inserciones, deleciones y reordenamientos. 17 PCR: Se necesitan ADN, nucleótidos libres, 2 cebadores adyacentes a la secuencia a analizar y una Taq polimerasa. Se calienta a 95º para desnaturalizar el ADN. Se anillan los cebadores a medida que se enfría a 35-65º. Se calienta a 70-75º y se lo expone a los nucleótidos y la Taq polimerasa para extender la cadena desde el cebador. Secuenciación: Similar a la PCR, se introducen 4 ddNTP’s (A, C, T y G), uno en cada tubo diferente que son los que aparecen en los telomeros. De esta forma se producen cadenas de diferente tamaño de acuerdo con la posición de cada nucleótido. Se hace 4 corridas electroforeticas diferentes y se comparan las posiciones, concluyendo con una “escalera” que se lee de abajo para arriba de acuerdo con el peso de los fragmentos. Detecta inserciones, deleciones, mutaciones puntuales y reordenamientos. Metodología indirecta (método de ligamiento): describe dos loci que se localizan lo bastante próximos en un mismo cromosoma como para que su frecuencia de recombinación sea inferior al 50%. *Cri du Chat: El trabajo terapéutico del niño con cri du chat puede empezar desde los 3 meses, siempre que su estado de salud sea estable (muchos pequeños con esta condición son muy delicados en sus primeras semanas de vida), y se realizará en dos vertientes: terapia física para lograr la mayor independencia de movimientos posible, y de estimulación temprana para desarrollar sus aptitudes y potencial intelectual, se debe abordar el retardo mental y se recomienda la asesoría para los padres. *Sd de Down:La mejoría en los tratamientos de las enfermedades asociadas al SD ha aumentado la esperanza de vida de estas personas, desde los 14 años hace unas décadas, hasta casi la normalidad (60 años, en países desarrollados) en la actualidad. A lo largo de los últimos 150 años se han postulado diferentes tratamientos empíricos (hormona tiroidea, hormona del crecimiento, ácido glutámico, dimetilsulfóxido, complejos vitamínicos y minerales, 5-hidroxitriptófano o piracetam) sin que ninguno haya demostrado en estudios longitudinales a doble ciego que su administración provoque ningún efecto positivo significativo en el desarrollo motor, social, intelectual o de expresión verbal de las personas con SD. No existe hasta la fecha ningún tratamiento farmacológico eficaz para el SD, aunque los estudios puestos en marcha con la secuenciación del genoma humano permiten augurar una posible vía de actuación (enzimática o genética), eso sí, en un futuro todavía algo lejano. *Sd de Edwards: No hay específico y tratamiento sabido para el síndrome de Edward. Los síntomas causados por el síndrome de Edward son también manejables hasta un cierto grado 8.Fundamentar el asesoramiento genético cromosómicas específicas de estos síndromes. en anormalidades 18 Las personas a las que puede resultarle útil incluyen: Aquellas que tienen o piensan que podrían tener un trastorno hereditario o un defecto de nacimiento, mujeres que están embarazadas o tienen pensado tener un hijo después de los 35 años de edad, las parejas que ya han tenido un hijo con retraso mental, un trastorno hereditario o un defecto de nacimiento. Cri du chat: Los padres de un niño con este síndrome deben buscar asesoría genética y someterse a una prueba de cariotipo con el fin de determinar si uno de ellos tiene una reordenación del cromosoma 5. 19 Autor: José Regino Ríos Gutiérrez. Unidad No. 4. Anormalidades de los cromosomas sexuales. Competencia Intermedia: Clasificar y analizar los síndromes más frecuentes de los cromosomas sexuales y los trastornos de la diferenciación sexual, seleccionando e interpretando el cariotipo y pruebas moleculares para hacer el diagnóstico que determine su manejo, pronóstico y asesoramiento genético. Criterios de desempeño: 4.1- Analizar las características estructurales y funcionales del cromosoma X. Cromosoma sexual; los genes transportados en el cromosoma X se denominan ligados a X. El cromosoma X mide más de 153 millones de pares de bases lo que representa un total del 5% de las ADN en células de mujer y un 2,5% en las del hombre. Se cree que el cromosoma X contiene 1336 genes de los cuales 20 quedan por identificar. El cromosoma X tiene una baja proporción de genes si lo comparamos con otros cromosomas. Esta compuesto por muchos segmentos de ADN repetitivo que no codifica para ninguna proteína o su función no es conocida. Solo el 1,7% del cromosoma codifica para proteínas funcionales que curiosamente son de baja longitud, si las comparamos con la media de longitud de un gen humano. El cromosoma X es más grande y tiene mayor cantidad de regiones de eucromatina que su pareja, el cromosoma Y. Se suele aceptar que el cromosoma X procede de una derivación de algún cromosoma autosómico. 4.2- Analizar las características de la inactivación del cromosoma X y la hipótesis de Lyon. La Teoría de la Inactivación señala que en las células somáticas de las mujeres normales (pero no de los hombres normales) se inactiva un cromosoma X, lo que equilibra la expresión de los genes ligados al X en los dos sexos. de la mayor parte de los genes ligados a X en el cromosoma X inactivo. Selección aleatoria de uno de los cromosomas X en las células de la mujer. X inactivo: Heterocromático (corpúsculo de Barr) Duplicación tardía en la fase S Expresa el RNA de XIST Asociado a modificaciones en la histona macroH2A en la cromatida. 20 La Hipótesis de Lyon menciona que el cromosoma X inactivo se identifica citológicamente por la presencia de una masa de heterocromatina denominada corpúsculo de Barr. 4.3- Analizar los genes que se escapan a la inactivación. Aunque la inactivación del cromosoma X es claramente un fenómeno cromosómico, no todos los genes del X están sujetos a inactivación. Al menos 15% de los genes escapa a la inactivación y se expresa tanto en el cromosoma X activo como en el inactivo. Además otro 10% muestra una inactivación X variable; es decir, escapa a la inactivación en algunos individuos de sexo femenino, pero no en todos. Muchos genes escapan a la inactivación en Xp distal, llegando a ser casi un 50%. 4.4- Analizar las características estructurales y funcionales del cromosoma Y. es más pequeño que el cromosoma X, cromosoma sexual. es uno de los cromosomas sexuales (gonosomas o heterocromosomas) de los animales heterogaméticos, sólo presente en los individuos machos. es un elemento acrocéntrico pequeño, que en su mayor parte no se recombina durante la meiosis. Sólo se produce recombinación con el cromosoma X en dos pequeñas regiones pseudoautosómicas denominadas PAR1 y PAR2 [3]. Por otro lado, dado que en él se localizan secuencias polimórficas, éstas serán cedidas de padres a hijos varones en forma obligada. Sólo representa el 2% del complemento cromosómico. Los polimorfismos presentes en este cromosoma proveen de una herramienta adicional en las metodologías de la identificación humana. En particular, los microsatélites hipervariables han contribuído en gran medida en este campo debido al gran número de tales marcadores disponibles, los que han sido ya validados en el ámbito forense internacional. 4.5- Diferenciar las características clínicas, variedades citogenéticas, factores de riesgo y aspectos epidemiológicos de las cromosomopatías numéricas y estructurales gonosómicas mas frecuentes: síndrome de Turner, síndrome de Klinefelter, síndrome de Polisomía X y síndrome de Polisomía Y. SÍNDROME DE TURNER (45,X).o Características clínicas.- 1 de 5,000 a 1 de 10,000; hidropesía (edema generalizado) hasta hinchazón en la nuca (pliegue en la nuca); implantación baja de pelo en la nuca, aumento del ángulo del codo, cuartos metacarpos cortos, pezones muy apartados, coartación de la aorta. Inteligencia normal, baja estatura, insuficiencia ovárica. 21 o Base genética.- el hallazgo más frecuente es el 45,X que a veces se denomina 45, XO. En 80% de los casos surge por la pérdida de un cromosoma sexual (X o Y) durante la meiosis paterna. En algunos casos hay mosaicismo cromosómico. o Patrón hereditario.- SÍNDROME DE KLINEFELTER (47, XXY).o Características clínicas.- 1 de 1,000 varones nacidos vivos. Torpeza, ligeras dificultades de aprendizaje, comportamiento egocéntrico; los adultos ligeramente más altos que la media y piernas largas, moderara ginecomastia, son infértiles (por azoospermia), testículos pequeños y blandos. Incidencia alta de úlceras de los miembros inferiores, osteoporosis y carcinoma de mama. o Base genética.- cromosoma X adicional, misma probabilidad de que el cromosoma extra provenga del padre que de la madre. Los casos que se asocian con la madre es por la edad avanzada de esta. Una pequeña proporción presentan mosaicismo. o Patrón hereditario.- o NOTA.- En raras ocasiones se han encontrado varones con más de dos cromosomas X, como 48,XXXY O 49 XXXXY. Esos individuos suelen tener un grave retraso mental y comparten los rasgos físicos de los varones con Klinefelter, pero con características más marcadas. SÍNDROME DE POLISOMÍA X (MUJERES XXX).o Características clínicas.- no suelen presentar anomalías físicas, pero sí una ligera reducción de 10 a 20 puntos en la capacidad intelectual y a veces presentan comportamiento bastante rebelde. Suelen presentar fertilidad normal y dan a luz hijos con cariotipos normales. Presentan una alta incidencia de dificultad de aprendizaje, cuya intensidad se correlaciona de forma directa con el número de cromosomas X presentes. o Base genética.- el cromosoma X adicional es de origen materno y suele originarse por un error de la meiosis I. o Patrón hereditario.- SÍNDROME DE POLISOMÍA Y.o Características clínicas.- 1 de 1,000. dificultad de aprendizaje, conducta antisocial delictiva, la mayoría de los hombres 47,XYY no presentan ni dificultades de aprendizaje ni un historial delictivo, aunque si inmadurez emocional y comportamiento impulsivo. La fertilidad es normal. Aspecto físico normal y estatura superior a la promedio. La inteligencia presenta una afectación ligera. 22 o Base genética.- el cromosoma Y adicional se origina de la no disyunción durante la meiosis II paterna, o debido a algún episodio ocurrido tras la formación del cigoto. o Patrón hereditario.- 23 Autor: José Regino Ríos Gutiérrez. Unidad No. 5. Enfermedades genéticas de transmisión mendeliana I. Herencia autosómica dominante. Competencia Intermedia: Analizar las características de la herencia autosómica dominante, para diagnosticar las enfermedades genéticas con este patrón de herencia y determinar su abordaje, manejo, pronóstico y asesoramiento genético. Criterios de desempeño: 5.1.- Definir alelo, heterocigoto, doble autosómico, dominante, codominante. heterocigoto, homocigoto, El ALELO es una de las variantes alternativas normales de un gen. El HETEROCIGOTO es un individuo o genotipo con dos alelos diferentes en un locus determinado de un par de cromosomas homólogos. El DOBLE HETEROCIGOTO es un individuo heterocigoto con dos locus diferentes. El HOMOCIGOTO es un individuo con alelos idénticos en un locus determinado de un par de cromosomas. AUTOSÓMICO es lo relativo a cualquier cromosoma nuclear a excepción de los sexuales. Una enfermedad causa por una mutación en un gen o par de genes autosómicos muestra una herencia autosómica. Se dice que un rasgo es DOMINANTE si es expresado fenotípicamente por los heterocigotos (DOMINANTE PURO, SEMIDOMINANTE E INCOMPLETAMENTE DOMINANTE ) CODOMINANTE es si ambos alelos de un par se expresan en un estado heterocigoto, entonces los dos alelos (y los rasgos determinados por ellos) son codominantes 5.2.- Analizar las características de la herencia autosómica dominante. un rasgo autosómico dominante es el que se manifiesta en estado heterocigótico, es decir, en una persona que posee tanto un alelo anómalo o mutante como un alelo normal. 5.3.- Diferenciar los factores que modifican la expresión genética: mutación de novo, expresión variable, pleiotropismo, penetrancia, anticipación, heterogeneidad alélica y heterogeneidad genética o de locus. La MUTACIÓN DE NOVO es cualquier cambio permanente heredable en la secuencia del DNA genómico.(secuencia de nucleótidos o en la disposición del DNA) La EXPRESIÓN VARIABLE.- las características clínicas en los trastornos autosómicos dominantes pueden mostrar una sorprendente variación de una persona a otra, incluso en la misma familia. 24 PLEIOTROPISMO.- Un único gen puede dar lugar a dos o más efectos aparentemente no relacionados. PENETRANCIA.- (“saltarse una generación”) es posible que algunos individuos heterocigóticos para las mutaciones génicas que dan lugar a ciertos trastornos autosómicos dominantes no presenten características clínicas anómalas, representando así la penetrancia reducida. Se cree que la penetrancia reducida es el resultado de los efectos modificadores de otros genes, así como la interacción del gen con los factores ambientales. Un individuo que muestra características de un trastorno a pesar de ser heterocigoto para la mutación de un gen particular, se dice que presenta no penetrancia. ANTICIPACIÓN.- en algunos rasgos o trastornos autosómicos dominantes, como la distrofia miotónica, la aparición de la enfermedad se produce a una edad más joven en los descendientes que en los padres, o la enfermedad aparece con una gravedad creciente en las generaciones sucesivas. HETEROGENEIDAD ALÉLICA.- o heterogeneidad mutacional. Hay individuos que tienen dos mutaciones diferentes en el mismo locus; se conocen como heterocigotos compuestos y constituyen lo que se denomina heterogeneidad mutacional o alélicas. La mayoría de los individuos afectados con un trastorno autosómico recesivo son probablemente heterocigotos compuestos más que homocigotos reales, a menos que sus padres estuvieran relacionados, en cuyo caso es probable que fueran homocigóticos para la misma mutación por descendencia, habiendo heredado dicha mutación de un antepasado común HETEROGENEIDAD GENÉTICA O DE LOCUS.- un trastorno heredado de la misma manera puede deberse a mutaciones en más de un gen, o lo que se conoce como heterogeneidad de locus. 5.4.- Realizar el cálculo de riesgo con el cuadrado de Punnett, analizando la segregación de gametos con las diferentes posibilidades de presentación para los genes autosómicos dominantes A a a a Aa Aa aa aa A a A AA Aa a Aa aa A A a Aa Aa a Aa Aa A A A AA AA a Aa Aa A A A AA AA A AA AA 5.5.- Analizar el concepto de codominancia con los ejemplos de grupos sanguíneos. Es el término usado para indicar la expresión de dos rasgos alélicas en un estado heterocigótico. En las personas del grupo sanguíneo AB se puede demostrar antígenos de los grupos sanguíneos A y B en los eritrocitos, de forma que los grupos sanguíneos A y B son, por lo tanto, codominantes. 25 5.6.- Evaluar las características clínicas, genéticas, factores de riesgo y aspectos epidemiológicos para llegar al diagnóstico de las enfermedades autosómicas dominantes más representativas: Acondroplasia, Osteogénesis imperfecta, síndrome de Marfán, síndrome de Ehlers Danlos, Neurofibromatosis, Charcot Marie Tooth, Hipercolesterolemia familiar. ACONDROPLASIA.o Características clínicas.- 1 de 15,000 a 1 de 40,000 nacidos vivos. Anomalías en la osificación de los cartílagos, cabeza normal o más grande de los normal, brazos y piernas cortos (especialmente muslos y parte proximal de los brazos), tronco normal y marcha bamboleante. Limitación en la extensión del codo, manos en tridente, piernas arqueadas (genum varum), gibosidad toracolumbar en la infancia, protuberancia frontal, hipoplasia de la mitad de la cara. o Base genética.- gen FGFR3 o Patrón hereditario.- autosómica dominante OSTEOGENÉSIS IMPERFECTA.o Características clínicas.- enfermedad de los huesos frágiles o Base genética.- puede explicarse por un mosaicismo somático y/o de la línea germinal en unos de los padres. o Patrón hereditario.- autosómica dominante SÍNDROME DE MARFAN.o Características clínicas.- se usa criterios de Gent para diagnóstico. Individuos altos en relación con la familia no afectada, articulaciones elásticas, la razón entre la envergadura y la estatura es superior a 1.05, la razón entre el segmento superior del cuerpo y el inferior está disminuida, y se presenta deformidad del tórax y escoliosis. Ectopia lenticular (subluxación del cristalino), dilatación de la aorta ascedente. o Base genética.- trastorno del tejido conectivo por defecto de la fibrilina de tipo 1, glicoproteína codificada por el gen FBN1 en 15q21. o Patrón hereditario.- autosómica dominante SÍNDROME DE EHLERS DANLOS.o Características clínicas.hiperlaxitud, cicatrices anormales, pseudotumores, rotura de órganos, periodontitis. Aposición del pulgar al antebrazo, hiperextensión de los dedos y de la muñeca de tal manera que los dedos quedarán paralelos al antebrazo; hiperextensión del codo de mas de 10 grados; hiperextensión de la rodilla de mas de 10 grados; excesiva dorsiflexión y eversión del pie. o Base genética.- heterogeneicidad genética. 26 o Patrón hereditario.- autosómica dominante. NEUROFIBROMATOSIS.o Características clínicas.- uno de los trastornos genéticos más frecuentes en los seres humanos, existen dos tipos NF1 y NF2 ambas pueden incluirse en los síndromes de canceres familiares, uno de los signos mas característicos son pequeñas lesiones pigmentadas conocidas como manchas café con leche y pequeñas tumoraciones carnosas denominadas neurofibromas. Las manchas café con leche surgen en la primera infancia y siguen creciendo en número y tamaño hasta la pubertad. Se requieren mínimo 6 manchas de al menos 5 mm de diámetro para respaldar el diagnóstico en la infancia, así como la presencia de pecas axilares e inguinales. Los neurofibromas son tumores benignos que surgen en la piel. Suelen presentarse al final de la infancia y durante la vida adulta, y se hacen más numerosos con la edad. Macrocefalia relativa y nódulos de Lisch (pequeñas hartotas pigmentados inofensivos situados en el iris); ligero retraso en el desarrollo que se caracteriza por un trastorno del aprendizaje no verbal. Un pequeño número de pacientes desarrollan alguna de las complicaciones mayores, como epilepsia, tumores del sistema nervioso central o escoliosis. o Base genética.- la expresión es muy variable, variedad de gravedad entre los miembros de una misma familia afectada, gen número 17 portador. o Patrón hereditario.- autosómica dominante CHARCOT MARIE TOOTH.o Características clínicas.- también llamada atrofia muscular peronea. Debilidad y atrofia en los músculos distales de la pierna (peroneos, tibial anterior, gemelos), pie arqueado, pie plano, marcha en steppage, pérdida de sensibilidad, manos también afectadas. o Base genética.- se han identificado tres genes (en los cromosomas 1, 17 y X). todos afectan la mielina, y el tipo del cromosoma X afectan al nervio en sí. o Patrón hereditario.- autosómica dominante HIPERCOLESTEROLEMIA FAMILIAR.o Características clínicas.- las personas con hipercolesterolemia familiar tienen valores de colesterol elevados con un riesgo significativo de desarrollar coronariopatías. Pueden presentarse en infancia o adolescencia xantomas. o Base genética.- cromosoma 4 o Patrón hereditario.- autosómica dominante 27 7.- Fundamentar el asesoramiento genético de este tipo de patologías. Cuando uno o mas hijos han nacido con una enfermedad genética, a los padres se les determina el riesgo de recurrencia, que establece la probabilidad de que los siguientes hijos también presenten la enfermedad. Si los padres todavía no han tenido hijos, pero se sabe que tiene riesgo de tener un hijo con una enfermedad genética, se les puede determinar el riesgo de incidencia. Cuando un progenitor esta afectado por una enfermedad autosómica dominante y el otro es normal, los riesgos de incidencia y de recurrencia para cada hijo son de ½. 28 Autor: José Regino Ríos Gutiérrez. Unidad No. 6. Enfermedades genéticas de transmisión mendeliana II. Trastornos autosómicos recesivos. Competencia Intermedia: Analizar las características de la herencia autosómica recesiva, para diagnosticar las enfermedades genéticas con este patrón de herencia y determinar su manejo, pronóstico y asesoramiento genético. Criterios de desempeño: 1.- Definir recesivo, portador, doble heterocigoto, heterocigoto compuesto. RECESIVO.- rasgo expresado en individuos que son homocigóticos para un alelo particular, pero no en los que son heterocigóticos. PORTADOR.- persona heterocigótica para un gen recesivo: hombre o mujer para los genes autosómicos o una mujer para los genes ligados al cromosomas X. DOBLE HETEROCIGOTO.- individuo que es heterocigoto en dos loci diferentes. HETEROCIGOTO COMPUESTO.- individuo afectado con un trastorno autosómico recesivo que tiene dos mutaciones diferentes en genes homólogos. HETEROCIGOTO = PORTADOR 2.- Analizar las características de la herencia autosómica recesiva. Los rasgos y los trastornos recesivos se manifiestan únicamente cuando el alelo mutante está presente en dosis doble, es decir, en una homocigosidad. El árbol genealógico para los rasgos recesivos difiere notablemente del observado en los rasgos autosómicos dominantes. Un rasgo o un trastorno recesivo o puede rastrearse a través de la familia, ya que todos los individuos afectados en una familia pertenecen habitualmente en una sola hermandad (es decir, hermanos y hermanas). Algunas veces a la hermandad se le denomina trasmisión “horizontal”. Los individuos heterocigóticos para tales alelos mutantes no muestran características del trastorno y están perfectamente sanos; se les describe como “portadores”. 3.- Analizar los factores que modifican la expresión genética: heterogeneidad alélica, heterogeneidad genética o de locus y genes modificadores. HETEROGENEIDAD ALÉLICA.- o heterogeneidad mutacional, en la mayoría de los trastornos monogénicas (p. ej. La beta- talasemia) se ha identificado como responsable a una gran cantidad de mutaciones diferentes. Hay individuos que tienen dos mutaciones diferentes en el mismo 29 locus; se conocen como heterocigotos compuestos y constituyen lo que se denomina heterogeneidad mutacional o alélica. La mayoría de los individuos afectados con un trastorno autosómico recesivo son probablemente heterocigotos compuestos más que homocigoto reales, a menos que sus padres estuvieran relacionados, en cuyo caso es probable que fueran homocigóticos para la misma mutación por descendencia, habiendo heredado dicha mutación de un antepasado común. HETEROGENEIDAD GENÉTICA O DE LOCUS.- es un trastorno heredado de la misma manera puede deberse a mutaciones en más de un gen. Por ejemplo, se admite que la alteración/sordera auditiva neurosensorial presenta con mayor frecuencia una herencia autosómica recesiva. Las personas sordas, debido a su escolarización y participación en la comunidad de sordos, a menudo deciden tener hijos con otra persona sorda. Cabría esperar que si dos personas sordas fueran homocigóticas para el mismo gen recesivo, todos los hijos estarían afectados de forma similar. Se han descrito familias en que todos los hijos nacidos de padres sordos debidos a genes autosómicos recesivos han tenido una audición normal, y son lo que se conoce como heterocigotos dobles. La explicación para este hecho debe ser que los padres eran homocigóticos para alelos mutantes en loci diferente, es decir, que un número de genes diferentes pueden ocasionar una sordera neurosensorial autosómica recesiva. De hecho, en los últimos años se ha demostrado que hay otros 20 genes y 15 loci implicados. Los trastornos con el mismo fenotipo debidos a diferentes loci genéticos se denominan genocopias, mientras que cuando el mismo fenotipo es el resultado de causas ambientales se como fenocopia. GENES MODIFICADORES.- genes que alteran o influyen en la expresión o función de otro gen, como la supresión o reducción de la función normal del gen modificado. 4.- Realizar el cálculo de riesgo con el cuadrado de Punnett, analizando la segregación de gametos con las diferentes posibilidades de presentación para los genes autosómicos recesivos. A a A AA Aa a Aa aa A A A AA AA a Aa Aa 30 A A a Aa Aa a Aa Aa 5.- Evaluar las características clínicas, genéticas, factores de riesgo y aspectos epidemiológicos para llegar al diagnóstico de las enfermedades autosómicas recesivas más representativas: Albinismo óculo-cutáneo, fibrosis quística, ictiosis laminar, sorderas neurosensoriales, epidermolisis bulosas, Tay Sachs. ALBINISMO ÓCULO.CUTÁNEO.o características clínicas.- falta de pigmento en la piel, pelo, iris y fondo de ojo. La falta de pigmento en el ojo ocasiona una mala agudeza visual y un movimiento típico pendular del ojo sin control. La pigmentación reducida parece ocasionar el infradesarrollo de parte de la retina para la visión fina, de la fóvea, y la proyección ánomalande las rutas visuales en la corteza óptica. o base genética.- deficiencia de la enzima tirosinasa necesaria para formar melanina a partir de tirosina. o patrón hereditario.- autosómico recesivo FIBROSIS QUÍTICA.o Características clínicas.- Los órganos afectados con más frecuencia es el pulmón y el páncreas. La enfermedad pulmonar crónica ocasionada por las infecciones recurrentes termina por producir cambios fibróticos en los pulmones e insuficiencia cardiaca secundaria, conocida como cor pulmonale. Cuando aparece esta complicación, la única esperanza de supervivencia a largo plazo radica en un transplante pulmonar exitoso. La función pancreática esta dañada en un 85% de los pacientes con FQ, que poseen una secreción enzimática disminuida debido al bloqueo de los ductos pancreáticos por las espesas secreciones. Esto lleva a malabsorción, con aumento del contenido de grasa de las heces. Esta complicación de la enfermedad responde bien al tratamiento con suplementos de enzimas pancreáticas. Otros problemas que podemos encontrar son pólipos nasales, prolapso rectal, cirrosis y diabetes mellitas. 10% de los neonatales presentan íleo meconial (obstrucción del intestino delgado debido al meconio espeso). Casi todos los varones son estériles por la ausencia congénita bilateral de los conductos deferentes. Existe un reducido número de varones con una forma leve de FQ donde el único problema significativo es la ausencia congénita de los conductos deferentes. Otras presentaciones raras de la FQ incluyen la pancreatitis crónica, las bronquiectasias difusas y la aspergilosis alérgica broncopulmonar. 31 o Base genética.- múltiples loci, una elevada tasa de mutación, la deriva meiótica y la ventaja heterocigótica. El locus de la FQ está ubicado en el cromosoma 7q31 o Patrón hereditario.- autosómica recesiva. ICTIOSIS LAMINAR.o Características clínicas.- en recién nacidos (y permanece hasta la vejez), la piel tiene apariencia escamosa y la capas de ésta se desprenden o mudan. Las lesiones son generalmente en el tronco pero también se puede manifestar en el tronco. 1 de 200,000 o Base genética.- enfermedad de bases bioquímicas, por alteración de 5 cromosomas diferentes (2, 3, 14, 17, 19) o Patrón hereditario.- autosómica recesiva. SORDERAS NUEROSENSORIALES.o Características clínicas.- disminución o pérdida de la audición por daño al nervio auditivo en el oído interno. o Base genética.- 20 genes y 15 loci implicados o Patrón hereditario.- autosómica recesiva. EPIDERMOLISIS BULOSAS.o Características clínicas.- aparición de ampollas y heridas de manera espontánea, pueden estar en cualquier parte del cuerpo y en mucosa orgánica. La piel de los afectados es débil, frágil, sensible, vulnerable. Los niños afectados son llamados “niños con piel de mariposa”. o Base genética.- afecta cromosomas de proteínas de la epidermis y la dermis. o Patrón hereditario.- autosómica dominante. TAY SACHS.o Características clínicas.- afecta en particular a los judíos azquenasi, ocurre cuando el cuerpo carece de hexosaminidasa A, una proteína que ayuda a descomponer un químico que se encuentra en el tejido nervioso, llamado gangliósidos. Sin esta proteína, los gangliósidos, en particular el gangliósido GM2 se acumula en las células, especialmente las neuronas del cerebro. El deterioro neurológico comienza intrauterino y se manifiesta a los 6 meses de nacido, generalmente mueren a los 4 o 5 años de edad. Los síntomas más comunes es temblor de manos, defectos del habla, debilidad muscular y pérdida del equilibrio, así como sordera, pérdida de la capacidad visual pudiendo llegar hasta la ceguera, crisis epilépticas, 32 retraso del crecimiento, irritabilidad, capacidades sociales y mentales. apatía o Base genética.- gen defectuoso del cromosoma 15 o Patrón hereditario.- autosómica recesiva y retraso en las 33 Autor: José Regino Ríos Gutiérrez. Unidad No. 7. Enfermedades genéticas de transmisión mendeliana III. Trastornos ligados a los cromosomas sexuales. Competencia intermedia: Analizar las características de la herencia dominante y recesiva ligada al cromosoma X para diagnosticar las enfermedades más frecuentes con este patrón de herencia y determinar su manejo, pronóstico y asesoramiento genético. Criterios de desempeño: 7.1.- Definir hemocigoto, portadora posible. portadora obligada, portadora probable y HEMOCIGOTO.- genotipo de un varón en relación con un rasgo ligado al cromosoma X, dado que los hombres sólo tienen un conjunto de genes ligados al cromosoma X. PORTADORA OBLIGADA.- Madre de un afectado, y con probabilidad de tener otro hijo afectado. PORTADORA PROBABLE.- hija de una portadora obligada. PORTADORA POSIBLE.- madre o hermana de un caso aislado. 7.2.- Analizar las características de la herencia dominante ligada al X, recesivo ligada al X y ligada al Y (herencia holándrica). HERENCIA DOMINANTE LIGADA AL X.- aunque no es habitual, hay trastornos que se manifiestan en la mujer heterocigótica tanto como en el hombre que tienen el alelo mutante en su único cromosoma X. Es lo que se conoce como herencia dominante ligada al cromosoma X. La herencia dominante ligada al cromosoma X recuerda superficialmente a los del rasgo dominante autosómico porque tanto las hijas como los hijos de una mujer afectada tienen una probabilidad de 1 de cada 2 (50%) de verse afectados. Sin embargo, hay una diferencia importante. El hombre afectado con un rasgo dominante ligado al cromosoma X transmite dicho rasgo a todas las sus hijas pero a ninguno de sus hijos. Por tanto, en las familias con un trastorno dominante ligado al cromosoma X hay un exceso de mujeres afectadas y no se produce una transmisión directa hombre-hombre. Un ejemplo de un rasgo dominante ligado al cromosoma X es el raquitismo resistente a la vitamina D. El raquitismo puede deberse a un déficit de vitamina D en la dieta, pero en el raquitismo resistente a la vitamina D el trastorno se produce incluso cuando hay una ingesta dietética adecuada de la vitamina D. La forma dominante ligada al cromosoma X del raquitismo resistente a la vitamina D afecta a los hombres como a las mujeres, aunque en las mujeres los cambios esqueléticos suelen ser menos graves que en los hombres. La forma ligada al cromosoma X de la enfermedad de CharcotMarie-Tooth (neuropatía hereditaria sensorial y motora) es otro ejemplo. En las mujeres heterocigóticas se pude demostrar un patrón en mosaico de la afectación en algunos trastornos dominantes ligados al cromosoma X. un ejemplo es el patrón en mosaico de la pigmentación anómala de la piel que 34 sigue las líneas de desarrollo observadas en las mujeres heterocigóticas para el trastorno dominante ligado al cromosoma X denominado incontinencia pigmentaria. Este es también un ejemplo de trastorno habitualmente letal para los embriones masculinos que heredan el alelo mutado. Otros son los trastornos neurológicos del síndrome de Rett y la heterotopia nodular periventricular. HERENCIA RECISIVA LIGADA AL X.- un rasgo recesito ligado al cromosoma X es el determinado por un gen trasportado en el cromosoma X y que se manifiesta habitualmente en los hombres. Un hombre con un alelo mutante en su único cromosoma X se trasmite por las mujeres portadoras heterocigóticas sanas a los hombres afectados, así como los hombres afectados a sus hijas obligatoriamente portadoras, con el riesgo consiguiente para los nietos masculinos a través de esas hijas. A veces se dice que este tipo de genealogía muestra un patrón de transmisión “diagonal” o de “movimiento de cabello de ajedrez”. El modo de herencia en el que únicamente los hombres resultaban afectados por una enfermedad que era transmitida por mujeres normales fue reconocido por los judíos hace casi 2,000 años. Excusaban de la circuncisión a todos los hijos de todas las hermanas de una madre que tenía hijos con la “enfermedad sangrante”, o sea, hemofilia. No se excusaba a los hijos de los hermanos del padre. HERENCIA LIGADA AL Y (HERENCIA HOLÁNDRICA).- la herencia holándrica o ligada al cromosoma Y implica que sólo los hombres se ven afectados. Un hombre afectado transmite los rasgos ligados al cromosoma Y a todos los hijos y a ninguna de las hijas. En el pasado se sugirió que los trastornos raros como la de piel de puercoespín, las orejas peludas y los dedos de los pies unidos entre sí eran rasgos ligados al cromosoma Y. Con la posible excepción de las orejas peludas, las demás pretensiones de herencia holándrica no se han sostenido en estudios más cuidadosos. Sin embargo, las pruebas indican claramente que el antígeno de la histocompatibilidad H-Y y los genes implicados en la espermatogénesis se transporta en el cromosoma Y, y por tanto manifiesta una herencia holándrica. En el último caso, si están suprimidos, se produce la infertilidad por azoospermia (ausencia de espermatozoides en el semen) en los hombres. La reciente llegada de las técnicas de reproducción asistida, sobre todo la inyección de espermatozoides intracitoplásmica (ICSI), significa que si se produce un embarazo con un feto masculino tras el uso de esta técnica, el niño será también infértil obligatoriamente. 7.3.- Analizar los factores que modifican la expresión genética: influenciados por el sexo, limitados por el sexo, heterogeneidad alélica, heterogeneidad genética o de locus. LIGAMIENTO PARCIAL AL SEXO.- el ligamiento parcial al sexo se ha utilizado en el pasado para explicar ciertos trastornos que parecen exhibir una herencia autosómica dominante en algunas familias y una herencia ligada al cromosoma X en otras. Actualmente se cree que esto es probable que sea así, debido a unos genes transportados en esa porción del cromosoma X que comparten homología con el cromosoma Y y que escapan a la inactivación del cromosoma X. Durante la meiosis se produce un 35 apareamiento entre las partes homólogas distales de los brazos cortos de los cromosomas X e Y, la así denominada región seudoautosómica. Como resultado del entrecruzamiento, se puede transferir un gen desde el cromosoma X al Y o viceversa, lo que permite la posibilidad de una trasmisión hombre-hombre. Los últimos casos serían consistentes con una herencia autosómica dominante. Se ha notificado que una displasia esquelética rara, la discondrosteosis de Leri-Weil, en la que los individuos afectados tienen una estatura corta y una deformidad característica en la muñeca (deformidad de Madelung), presenta tanto una herencia autosómica dominante como ligada al cromosoma X. Se ha demostrado que le trastorno se debe a delecciones (o a una mutaciónen) el gen homeobox de la estatura corta (SHOX), que se localiza en la región seudoautosómica. INFLUENCIA DEL SEXO.- Algunos rasgos autosómicos se expresan más frecuentemente en un sexo que en el otro, lo que se denomina influencia del sexo. La gota y la calvicie presenil son ejemplos de rasgos dominantes autosómicos influidos por el sexo, y los hombres son los predominantemente afectados en ambos casos. La influencia del sexo en esto dos ejemplos se debe probablemente al efecto de las hormonas masculinas. La gota, por ejemplo, es muy rara en las mujeres antes de la menopausia, pero la frecuencia aumenta en el período posterior. La calvicie no aparece en los hombres castrados. En la hemocromatosis, el trastorno autosómico recesivo más habitual en la sociedad occidental, las mujeres homocigóticas tienen menos probabilidades que los hombres homocigóticos para desarrollar una sobrecarga de hierro con los síntomas asociados; la explicación que se da habitualmente es que las mujeres tienen una forma de pérdida sanguínea natural mediante la menstruación. LIMITACIÓN POR EL SEXO.- la limitación por el sexo se refiere a la aparición de ciertas características únicamente en individuos de un sexo en particular. Un ejemplo es la virilización de los lactantes femeninos afectados con el trastorno endocrino autosómico recesivo denominado hiperplasia suprarrenal congénita. 7.4.- Realizar el cálculo de riesgo con el cuadrado de Punnett, analizando la segregación de gametos con las diferentes posibilidades de presentación para los genes dominantes y recesivos ligados al X. X2 Y X1 X1 X2 X1 Y X0 X0 X2 X0 Y X2 Y X1 X1 X2 X1 Y X0 X0 X2 X0 Y 36 7.5.- Evalúa las características clínicas, genéticas, factores de riesgo y aspectos epidemiológicos para llegar al diagnóstico de las enfermedades dominantes y recesivas ligadas al X más representativas: Hemofilia A y B, Distrofia muscular de Duchenne y Becker, Incontinentia Pigmenti, Raquitismo hipofosfatémico familiar. HEMOFILIA A y B.o Características clínicas.- el cuadro clínico es similar en las dos formas de hemofilia, y varía de un sangrado ligero después de un traumatismo importante o una cirugía, a las hemorragias espontáneas en músculos y articulaciones. La gravedad muestra una estrecha correlación con la reducción de la actividad de los factores VIII y IX. Los valores inferiores a 1% suelen asociarse a la tendencia a hemorragias graves desde el nacimiento. Las hemorragias articulares producen fuerte dolor e hinchazón y, cuando son recurrentes, causan una artropatía progresiva con grave incapacidad. En general, los miembros afectados de una familia tienen el mismo estado de gravedad. o Base genética.- la hemofilia A o hemofilia clásica está causada por una deficiencia del factor VIII que desempeña un papel fundamental en la vía intrínseca de activación de las transformación de protrombina en trombina. La hemofilia B está causada por el factor IX, es también conocida como enfermedad de Christmas. o Patrón herencia.- Ligado a X DISTROFIA MUSCULAR DE DUCHENNE.o Características clínicas.- los varones con este problema suelen empezar la sintomatología entre los 3 y los 5 años, con debilidad muscular progresiva que da lugar a una marcha torpe, incapacidad para correr deprisa y dificultad para levantarse del suelo, lo que sólo pueden lograr tirando o “subir trepando” las piernas, y los muslos (signo de Gowers). La mayoría de los niños afectados tienen que usar una silla de ruedas a la edad de 11 años, debido a la grave debilidad muscular proximal de las piernas. El deterioro posterior lleva a lordosis lumbar, contracturas articulares e insuficiencia cardiorrespiratoria que conduce a la muerte a los 18 años, como promedio. Al examen, los niños con DMD muestran un incremento aparente de los músculos de las pantorrillas, que en realidad se debe a la sustitución de las fibras musculares por grasa y tejido conectivo. Es la denominada seudohipertrofia, y por ello en ocasiones la DMD se denomina distrofia muscular seudohipertrófica. Además un tercio de los niños con DMD muestran un déficit intelectual entre ligero y moderado, con un CI promedio de todos los pacientes de 83. 37 o Base genética.- es causada por el gen distrofina en Xp21 o Patrón de herencia.- herencia recesiva ligada a X DISTROFIA MUSCULAR DE BECKER.o Caracteríscas clínicas.- trastorno similar a la distrofia muscular de Duchenne pero más ligero, causado por el mismo gen. El cuadro clínico es muy parecido, pero presenta un curso mucho menos agresivo. La edad promedio de inicio es de 11 años, y muchos pacientes pueden caminar hasta bien entrada la edad adulta. La esperanza de vida global sólo experimenta una ligera reducción. o Base genética.- gen distrofina en Xp21 o Patrón de herencia.- herencia recesiva ligada a X INCONTINENTIA PIGMENTI.o Características clínicas.- las pacientes tienen un patrón en mosaico de pigmentación cutánea, las áreas pigmentadas indican la presencia de tejido en el que se ha inactivado al cromosoma X norma. (líneas de Blaschko). o Base genética.- mutación en un gen de los cromosomas X. o Patrón de herencia.- trastorno dominante ligado a X. RAQUITISMO HIPOFOSFATÉMICO FAMILIAR.o Características clínicas.- deformidad de hueso y cartílago, dolor óseo, hipotonía y debilidad muscular proximal, cardiomiopatía + insuficiencia cardiaca + hipocalcemia, retraso en la deambulación, retardo de crecimiento, tetania, convulsiones, infección respiratoria, mielofibrosis (pancitopenia ó anemia macrocítica), craneotabes, ensanchamiento de suturas craneales, prominencia frontal, aplanamiento posterior del cráneo, rosario raquítico, ensanchamiento del tórax, deformidad de tórax, Genu varo o valgo, marcha de pato, erupción dentaria retardada, hipoplasia del esmalte. o Base genética.- o Patrón de herencia.- dominante ligado al cromosoma X 38 Autor: José Regino Ríos Gutiérrez. Unidad No. 8. Mecanismos no clásicos de transmisión hereditaria I. Competencia intermedia: Comparar las características de la herencia no clásica con las de la herencia mendeliana, determinando la base genética de las mismas y sustentando las diferencias en su expresión y patrón hereditario en patologías mitocondriales, causadas por expansión de microsatélites, por disomía uniperental, impronta genómica, mosaicismo (germinal y somático), determinando su manejo, pronóstico y asesoramiento genético. Criterios de desempeño.1.- Analizar las características génicas y las condiciones hereditarias de la patología genética mitocondrial y por expansión de microsatélites. PATOLOGÍA GENÉTICA MITOCONDRIAL.- el DNA mitocondrial tiene una tasa mayor de mutaciones espontáneas que el DNA nuclear, y se ha propuesto que la acumulación de dichas mutaciones en el DNA mitocondrial es responsable de algunos de los efectos somáticos observados en el envejecimiento. En los seres humanos se ha planteado la herencia mitocondrial o citoplásmica como una explicación posible para el patrón de herencia observado en algunos trastornos raros que afectan tanto a hombres como a mujeres, pero que trasmiten únicamente a través de las mujeres, la así denominada herencia matrilineal o maternal. Se considera que diversos trastornos raros con combinaciones poco habituales de características neurológicas y miocárdicas, que a veces se presentan en asociación con otros trastornos como miocardiopatías y defectos en la conducción, diabetes o sordera, se deben a mutaciones en los genes mitocondriales. Como las mitocondrias tienen un papel importante en el metabolismo celular mediante la fosforilación oxidativa, no es sorprendente que los órganos más susceptibles a las mutaciones mitocondriales sean el sistema nervioso central, el músculo esquelético y el corazón. En la mayoría de las personas el DNA mitocondrial de las diferentes mitocondrias es idéntico, o muestra lo que se denomina homoplasmia. Si se produce una mutación en el DNA mitocondrial de un individuo, inicialmente habrá dos poblaciones DNA mitocondrial, lo que se conoce como heteroplasmia. El porcentaje de mitocondrias con una mutación en su DNA varía entre células y tejidos, y esto, junto con la heterogeneidad mutacional, es una explicación posible para el rango de gravedad fenotípica observado en personas afectadas con trastornos hereditarios mitocondriales. Al tiempo que a herencia maternal se aplica a los trastornos que se deben directamente a las mutaciones en el DNA mitocondrial, es importante ser consciente también de que las proteínas mitocondriales están codificadas principalmente por genes nucleares. Las mutaciones en estos genes pueden tener un impacto devastador sobre las funciones de la cadena respiratoria en el interior de la célula. EXPANSIÓN DE MICROSATÉLITES.- el DNA microsatélite consiste en secuencias de pares de bases repetitivas en tándem de mono, bi, tri y tetranucleótidos localizados por todo el genoma. Es muy raro que las 39 repeticiones microsatélite se produzcan dentro de las secuencias codificantes, pero las repeticiones de trinucleótidos cerca de los genes o en su interior se asocian con ciertos trastornos hereditarios. El DNA microsatélite es altamente polimórfico, es decir, el número de repeticiones de CA varía entre las personas y puede utilizarse en los estudios de ligamiento para la realización del mapa genético. Se cree que esta variación en el número de repeticiones surge por el apareamiento incorrecto de las repeticiones tándem de las dos hebras de DNA, o lo que se conoce como mal apareamiento por deslizamiento de la hebra. Se cree que las duplicaciones o las delecciones de secuencias más largas del DNA repetido en tándem surgen por el cruce desigual de secuencias de DNA no alélicas en las cromátidas de cromosomas homológos o cromátidas hermanas. 2.- Analizar la implicación del fenómeno de anticipación, la hetero y homoplasia, como condicionantes de la expresión fenotípica de algunas patologías con patrón hereditario no clásico. ANTICIPACIÓN.- en algunos rasgos o trastornos autosómicos dominantes, como la distrofia miotónica, la aparición de la enfermedad se produce a una edad más joven en los descendientes que en los padres, o la enfermedad aparece con más gravedad en las generaciones sucesivas. Este fenómeno se denomina anticipación. Solía creerse que este efecto era el resultado de un sesgo de la averiguación, debido a la forma en que se recogían los casos familiares. Se argumentaba que esto aparecía porque las personas en las que la enfermedad comienza a una edad más temprana o es más grave tienen más probabilidad de ser detectadas, y que únicamente los individuos menos gravemente afectados tienden a tener hijos. Además, se creía que, dado que el observador pertenece a la misma generación que los probandos presentes afectados, muchos individuos que en el momento presente no estaban afectados desarrollarían necesariamente la enfermedad en una etapa posterior de la vida. Sin embargo, los estudios recientes han demostrado que en ciertos trastornos, entre los que se incluyen la enfermedad de Huntington y la distrofia miotónica, la anticipación es, de hecho, un fenómeno biológico real que se da como resultado de la expansión de secuencias por repetición de tripletes inestable. HETEROPLASMIA.- mitocondrias de un individuo compuestas por más de una población. HOMOPLASMIA.- mitocondrias de un individuo compuestas por una sola población. 3.- Evaluar las características clínicas, genéticas, factores de riesgo y aspectos epidemiológicos para llegar al diagnóstico de patologías con este tipo de patrón hereditario: atrofia hereditaria de Leber, Merff, Melas, síndrome de X-frágil, enfermedad de Huntington y distrofia miotónica. ATROFIA HEREDITARIA DE LEBER.o Características clínicas.- comienzo agudo de pérdida de visón bilateral y simétrica. Esto evoluciona a una atrofia óptica muy severa 40 y una disminución permanente de agudeza visual. En la fase aguda, que dura algunas semanas, el ojo afectado muestra una apariencia edematosa de la capa de fibras nerviosas y vasos peripapilares inflamados (microangiopatía). Estas características principales se ven en un examen de fondo de ojo justo antes o después de comenzar la pérdida de visión. Un examen revela agudeza visual reducida, pérdida de visión del color, escotoma cecocentral en una prueba de campo visual. o Base genética.- cambios o mutaciones en el DNA mitocondrial en los genes MT-ND1, MT-ND4, MT-ND4L, MT-ND6 o Patrón hereditario.- mitocondrial (ligado a X) ATROFIA HEREDITARIA DE MERFF.o Características clínicas.- también llamado síndrome de epilepsia mioclónica con fibras rojo-rasgadas. Se caracteriza por epilepsia, debilidad muscular, ataxia, convulsiones generalizadas y miopatía mitocondrial con presencia de fibras rojo-rasgadas. Otros síntomas comunes son sordera, demencia, neuropatía, atrofia óptica, fallo respiratorio y cardiomiopatía. Los síntomas pueden aparecer en cualquier momento de la vida y son progresivos. o Base genética.- el 80 -. 90% de los casos de MERFF están asociados a la mutación adenina guanina en la posición 8.344 del gen del ARN de la mitocondria. (mutaciones puntales) o Patrón hereditario.- mitocondrial (ligado a X) ATROFIA HEREDITARIA DE MELAS.o Características clínicas.- migraña, vómitos, demencia, acidosis láctica, encefalopatía mitocondrial, episodios parecidos a un accidente vascular encefálico, epilepsia, sordera, demencia, ataxia, retinosis pigmentaria, cardiomiopatía, disfunción tubular renal proximal. Se manifiesta a cualquier edad. o Base genética.- se asocia con la mutación adenosina guanina del ARNt. (mutaciones puntales) o Patrón hereditario.- mitocondrial (ligado a X) SÍNDROME DE X-FRÁGIL.o Características clínicas.- los chicos mayores y adultos suelen tener rasgos faciales característicos, con frente alta, orejas grandes, cara larga y barbilla prominente. Tras la pubertad, la mayoría tiene testículos grandes (macroorquidia). También hay debilidad del tejido conjuntivo, con articulaciones hiperextensibles, marcas de estiramiento de la piel (estrías) y prolapso de la válvula mitral. Las dificultades de aprendizaje varían de moderadas a graves, y muchos niños afectados muestran rasgos autistas o un comportamiento 41 hiperactivo. El discurso suele ser titubeante y repetitivo. Las mujeres portadoras pueden tener algunos de estos rasgos faciales y alrededor del 50% de las mujeres con la mutación completa muestran dificultades de aprendizaje de ligeras a moderadas. o Base genética.- la mutación FRAXA consiste en un incremento del tamaño de una región en la región 5´no traducida del gen de la dificultad de apredizaje del X frágil. Muestra un sitio “frágil” cercano al telómero del final del brazo largo en Xq27.3 o Patrón hereditario.- mitocondrial (ligado a X) ENFERMEDAD DE HUNTIGTON.o Características clínicas.- de presentación tardía (4ta o 5ta década de vida) y se caracteriza por movimientos coreicos (movimientos espasmódicos e involuntarios, amplios y bruscos de las extremidades, que dificultan incluso la marcha), síntomas psiquiátricos (problemas afectivos y cambio de personalidad, irritabilidad, agresividad, brotes psicóticos, deseo de suicidio) y una degeneración neurológica progresiva que llega a conducir a la demencia; también se caracteriza por parkisonismo, pérdida de expresión facial. En niños menores de 10 años: crisis convulsivas, disfunción oral motora, rigidez, trastornos de marcha, o Base genética.- expansión de la secuencia de repetición poliglutamina CAG situada en la región 5´del gen de la EH. o Patrón hereditario.- mitocondrial de DISTROFIA MIOTÓNICA.o Características clínicas.- a diferencia de la mayoría de las distrofias musculares, el cuadro clínico de la MD no se limita en exclusiva al sistema neuromuscular. Los individuos con esta enfermedad empiezan a tener síntomas de adultos, con debilidad muscular progresiva y miotonía (espasmos musculares tónicos con una relajación prolongada, que puede manifestarse como una tardanza en relajar el apretón de manos al saludar). Otras anomalías clínicas son cataratas, defectos de la conducción cardíaca, alteraciones de la peristalsis gastrointestinal (disfagia, constipación, diarrea), esfínteres débiles, aumento del riesgo de diabetes mellitus y de cálculos biliares, somnolencia, calvicie frontal, atrofia testicular. o Base genética.- cromosoma 19 con inestabilidad en una secuencia de repetición de CTG, que esta presente en la región 3´no traducida dej gen de una proteíncinasa denominada PROTEINCINASA DE LA MD (DMPK). o Patrón hereditario.- autosómica dominante. 42 Autor: José Regino Ríos Gutiérrez. Unidad 9. Mecanismos no clásicos de transmisión hereditaria II. Competencia intermedia.- comparar las características de ka herencia no clásica con las de las herencia mendeliana, determinando la base genética de las mismas y sustentando las diferencias en su expresión y patrón hereditario en patologías mitocondriales, causadas por expansión de microsatélites, por disomía unilateral, impronta genética, mosaicismo (germinal y somático) y herencia oligogénica, determinando su manejo, pronóstico y asesoramiento genético. Criterios de desempeño.1.- Analizar las características génicas y las condicionantes hereditarias de la patología por disomía uniparental, impronta génica, mosaicismo (germinal y somático) y herencia oligogénica. DISOMÍA UNIPARENTAL.- situación en la que un individuo hereda ambos cromosomas de una pareja de homológos de un solo progenitor. IMPRONTA GÉNICA.- expresión diferente del dependiendo del sexo del progenitor que lo transmite. MOSAICISMO GERMINAL.- presencia en la línea germinal o en el tejido gonadal de dos poblaciones de células que difieren genéticamente. MOSAICISMO SOMÁTICO.- aparición de dos líneas celulares diferentes en un tejido o tejidos particulares que difieren genéticamente. HERENCIA OLIGOGÉNICA.- herencia de pocos genes material genético 2.- Analizar la expresión poliálélica en las patologías oligogénicas. HERENCIA POLIALÉLICA.- se debe a la acción de gen que presenta más de dos alelos. Sucede así con los grupos sanguíneos humanos que están determinados por un gen con tres alelos. 3.- Evaluar las características clínicas, genéticas, factores de riesgo y aspectos epidemiológicos para llegar al diagnóstico de patologías con este tipo de patrón hereditario: síndrome de Prader-Willi, síndrome de Angelman, Síndrome de Bardet-Bledl, Retinosis pigmentaria. SÍNDROME DE PRADER-WILLI.o Características clínicas.- estatura corta, obesidad, hipogonadismo y dificultad de aprendizaje, 1 de 20, 000, o Base genética.- delección intersticial en la porción proximal del brazo largo del cromosoma 15, generalmente obtenido del padre. Hay otro porcentaje que tiene una disomía uniparental materna (funcionalmente esto equivale a una delección del cromosoma 15 del padre). o Patrón hereditario.- autosómico dominante. 43 SÍNDROME DE ANGELMAN.o Características clínicas.- 1 de 15,000, epilepsia, dificultades graves para el aprendizaje, marcha atáxica o inestable y aspecto feliz. o Base genética.- delección intersticial de la mismo región 15q11-q13, obtenido de la madre, surge por la pérdida del gen UBE3A o Patrón hereditario.- SÍNDROME DE BARDET-BLEDL.o Características clínicas.- obesidad, retinitis pigmentaria (ceguera nocturna y pérdida progresiva de la visión periférica), retardo mental, hipogonadismo, daño renal, polidactilia (dedos extras en los pies). o Base genética.- cromosoma 16 afectado. o Patrón hereditario.portadores) autosómica recesiva (ambos padres son RETINOSIS PIGMENTARIA.o Características clínicas.- ceguera nocturna (lenta adaptación a la oscuridad), pérdida progresiva del campo visual hasta formar visión en túnel. Se produce por degeneración y apoptosis de los fotorreceptores (bastones-campo periférico) aunque en las fases finales afecta a los conos (visón central), provocando ceguera en un grupo importante de los casos. o Base genética.- o Patrón hereditario.- forma familiar ligada a X 44 Autor: José Regino Ríos Gutiérrez. UNIDAD 10.- HERENCIA MILTIFACTORIAL O COMPLEJA. Competencia intermedia.- evaluar las características de la herencia multifactorial o compleja analizando los determinantes de riesgo genético y ambiental que predisponen a enfermedades comunes. Criterios de desempeño.1.- Explicar los conceptos de multifactorial, poligénico, modelo de distribución normal, modelo del umbral, heredabilidad, gen mayor, coeficiente de relación entre parientes, susceptibilidad genética. MULTIFACTORIAL.- Dada la posibilidad de que muchos factores, tanto genéticos como ambientales, estén implicados en la causa de trastornos, suele referirse a ellos diciendo que presentan una herencia multifactorial. POLIGÉNICO.- también llamada cuantitativa, ésta conlleva la herencia y la expresión de un fenotipo que viene determinado por muchos genes en diferentes loci, y cada gen ejerce un pequeño efecto aditivo (“aditivo” implica que los efectos de los genes son acumulativos, ningún gen es dominante o recesivo sobre otro). MODELO DE DISTRIBUCIÓN NORMAL.- diversas características humanas (presión arterial, circunferencia cefálica, altura, inteligencia, color de piel) muestran continua en la población general que se parece mucho a una distribución normal y que toma la forma de una curva simétrica con forma de campana distribuida uniformemente alrededor de la media. La extensión de la distribución alrededor de la media viene determinada por la desviación estándar. Aproximadamente el 68, el 95 y el 99.7% de las observaciones caen dentro de la media más o menos una, dos o tres desviaciones estándar, respectivamente. Es posible demostrar que un fenotipo con una distribución normal en la población general puede ser producido por una herencia poligénica que implicase la acción de muchos genes en diferentes loci, cada uno de los cuales ejerce un efecto aditivo similar. Esto puede ilustrarse considerando un rasgo como la altura. Si la altura viniera determinada por dos alelos con una frecuencia igual, a (alto) y b (bajo), en un único loci, se produciría un fenotipo discontinuo con tres grupos en una proporción de 1 (alto, aa): 2 (medio ab/ba):1 (bajo, bb). Si el mismo rasgo viniera determinado por dos loci, cada uno con dos alelos que interactuara de forma simplemente aditiva, se originaría una distribución fenotípica de cinco grupos en una proporción de 1 (4 genes altos): 4 (3 altos + 1 bajo): 6 (2 altos + 2 bajos): 4 (1 alto + 3 bajos): 1 (4 bajos). En un sistema con tres loci cada uno con dos alelos la proporción fenotípica sería 1:6:15:20:15:6:1. Puede observarse que a medida que el número de loci aumenta, la distribución se parece cada vez más a una curva normal, apoyando por tanto el concepto de que características como la altura vienen determinadas por los efectos aditivos de muchos genes en diferentes loci. Un apoyo adicional para este concepto proviene del estudio de las correlaciones familiares en características como la altura y, en menor medida, la inteligencia. La correlación es una medida estadística del grado de 45 asociación de fenómenos variables o, en términos más sencillos, una medida del grado de semejanza o de relación entre dos parámetros. MODELO DEL UMBRAL.- en modelo de la predisposición/umbral, todos los factores que influyen en el desarrollo de un trastorno multifactorial, sean genéticos o ambientales, pueden ser considerados como una entidad única conocida como predisposición. Las predisposiciones de todos los individuos en una población forman una variable continua que tiene una distribución normal tanto en la población general como en los familiares de los individuos afectados. Sin embargo, las curvas para los familiares estarán desviadas a la derecha, y el grado de desviación está directamente relacionado con la estrechez de su relación con el caso índice afectado. La predisposición incluye todos los factores que contribuyen a la causa del trastorno. De forman sencilla, una predisposición perjudicial, podría contemplarse como una forma por una combinación de varios genes “malos” y factores ambientales adversos. La predisposición no puede medirse, pero se puede determinar a predisposición media de un grupo por la incidencia de la enfermedad en ese grupo utilizando estadísticas de la distribución normal. Las unidades de medida son las desviaciones estándar, y éstas pueden utilizarse para estimar la correlación entre los familiares. HEREDABILIDAD.- puede definirse como la proporción de la varianza fenotípica total de un trastorno que está causado por una varianza genética aditiva. En términos estadísticos, la varianza es igual al cuadrado de la desviación estándar. Aunque no es posible evaluar la predisposición individual para un trastorno particular, sí se puede calcular qué proporción de la etiología puede atribuirse a los factores genéticos en contraposición a los factores ambientales. Los cálculos de la heredabilidad de un trastorno o de un rasgo indican la importancia relativa de los factores genéticos en su causa, de forma que cuanto mayor sea el valor de la heredabilidad, mayor el papel de los factores genéticos. GEN MAYOR.- es aquel gen que tiene un efecto fenotípico discernible. COEFICIENTE DE RELACIÓN ENTRE PARIENTES.- está basado en la covarianza del fenotipo entre ellos (parientes). SUSCEPTIBILIDAD GENÉTICA.- predisposición hereditaria a una enfermedad o a un trastorno que no se debe a una causa monogénica, sino que habitualmente es el resultado de interacciones complejas de los efectos de múltiples genes diferentes, es decir, de una herencia poligénica. 2.- Explicar las características de la herencia multifactorial y riesgo de recurrencia, estudios de agregación familiar, estudios en gemelos, concordancia, discordancia, estudios de adopción, de asociación y ligamiento. HERENCIA MULTIFACTORIAL.- herencia controlada por muchos genes con pequeños efectos aditivos (poligénicos) más los efectos del entorno. 46 RIESGO DE RECURRENCIA.- probabilidad de que una enfermedad encontrada en un miembro de una progenie aparezca en otros miembros de la misma ascendencia. ESTUDIOS DE AGREGACIÓN FAMILIAR.- el hallazgo de una frecuencia más elevada de determinada enfermedad en la historia médica de una familia que en la población general sugiere una susceptibilidad genética a esa enfermedad. La proporción de familiares afectados según el grado de parentesco (primer grado, segundo grado, etc.) puede ofrecer información acerca del riesgo empírico de recurrencia al realizarse el consejo genético, así como proporcionar pruebas que apoyen una contribución genética. Sin embargo, la agregación familiar no prueba una susceptibilidad genética, ya que las familias comparten un ambiente común. La frecuencia de la enfermedad en cónyuges que viven en el mismo ambiente pero que suelen tener origen genético distinto pueden usarse como control, en especial para posibles factores ambientales que influyen durante la edad adulta.mmm ESTUDIOS EN GEMELOS.- podría pensarse que si los dos componentes de una pareja de gemelos idénticos poseen un mismo rasgo, eso prueba que el rasgo es hereditario. Pero no es necesariamente así. Como los gemelos tienden a compartir el mismo ambiente, es posible que ambos hayan estado expuestos a los mismos factores ambientales. Por ejemplo: cuando uno de los gemelos contrae una enfermedad contagiosa como el impétigo, es probable que el otro gemelo también se vea afectado. Este problema puede solucionarse en parte al comparar las diferencias en la frecuencia de una enfermedad o un trastorno de parejas de gemelos no idénticos o dicigóticos y en gemelos idénticos o monocigóticos. Una forma de sortear esa dificultad es estudiar las diferencias entre parejas de gemelos idénticos que, por circunstancias familiares inusuales, han sido criados de forma separada desde pequeños. Si una determinada enfermedad es enteramente determinada por la genética, cuando un gemelo idéntico esta afectado, el otro también lo está, incluso cuando han crecido en diferentes ambientes. No obstante, es raro que los gemelos idénticos sean separados en la primera infancia, por lo que hay pocos estudios sobre cualquier enfermedad. En una investigación de gemelos idénticos criados por separado, los datos mostraron con claridad que cada pareja de gemelos difería poco en la talla, pero sí lo hacían de manera considerable en el peso corporal. Esas diferencias sugieren que la heredabilidad puede desempeñar un papel mayor en la determinación de la estatura que en la del peso corporal. CONCORDANCIA.- cuando dos miembros de una pareja de gemelos presentan el mismo rasgo se dice que son concordantes. DISCORDANCIA.- si sólo uno de los gemelos tiene el rasgo se dice que son discordantes. ESTUDIOS DE ADOPCIÓN.- otro método que contribuye a diferenciar los factores genéticos y ambientales es la comparación de la frecuencia de una enfermedad entre los individuos que permanecen con sus padres biológicos y los que son adoptados fuera de sus familias biológicas. Los individuos adoptados se llevan sus genes al nuevo ambiente. Si la frecuencia de una 47 enfermedad en los adoptados fuera de su familia biológica es semejante a la verificada en los que permanecen con sus padres biológicos, es probable que los factores genéticos tengan una importancia mayor. Por el contrario, si la frecuencia de la enfermedad en los individuos adoptados es semejante a la de sus padres adoptivos, es probable que los factores ambientales sean más importantes. LIGAMIENTO.- dos loci situados uno al lado del otro en el mismo cromosoma, cuyos alelos se transmiten habitualmente juntos en la meiosis para la formación del gameto. 3.- Explicar las estrategias para el estudio de las enfermedades multifactoriales y para identificación de los genes responsables. Definir los conceptos de clonación posicional y clonación funcional. ANÁLISIS DE LIGAMENTO.- El análisis de ligamiento ha demostrado ser extremadamente valioso para el mapeo de los trastornos monogénicas mediante el estudio de la cosegregación de los marcadores genéticos con la enfermedad. Sin embargo, este tipo de estrategia tiene muchas más dificultades en los trastornos multifactoriales. Puede ser por análisis de las parejas de hermanos (sibling-pair) afectados o mapeo por desequilibrio de ligamiento. ESTUDIOS DE ASOCIACIÓN.- se lleva a cabo comparando la incidencia de una variante particular en los pacientes afectados con la incidencia en un grupo de control cuidadosamente ajustado. Este método se describe a menudo como un estudio de “casos y controles”. Si la incidencia en los grupos difiere de forma significativa, nos da la prueba de una asociación positiva o negativa. ESTUDIOS DE ASOCIACIÓN DEL GENOMA COMPLETO.- los investigadores comparan la totalidad del genoma en un estudio de caso y controles, en lugar de mirar una sola variante cada vez. De esta forma, se puede utilizar este potente nuevo método para identificar nuevos genes de susceptibilidad a la enfermedad. CLONACIÓN POSICIONAL.- mapeo de un trastorno a una región particular de un cromosoma que lleva a la identificación del gen responsable. CLONACIÓN FUNCIONAL.- Identificación de un gen por su función, por ejemplo; aislamiento con los cDNA expresados en un tejido particular en le que hay una enfermedad y un trastorno manifiesto. 4.- Describir los factores genéticos en las enfermedades multifactoriales comunes: equizofrenia, autismo, alcoholismo, artritis, enfermedad de Parkinson, trombosis venosa, enfermedad coronaria, úlcera péptica, Alzheimer, diabetes, hipertensión y obesidad. 48 EQUIZOFRENIA.o Características clínicas.- grave psicosis que suele iniciarse al final de la adolescencia o en la juventud. Se caracteriza por graves alteraciones del pensamiento y el comportamiento, unidas a un notable deterioro social y ocupacional, y puede acompañarse de alucinaciones o delirios. Riesgo de 1% de que una persona lo desarrolle a lo largo de su vida, el .2% de la población mundial se encuentra afectada en un momento dado. La equizofrenia es más frecuente en individuos de clase social mas desfavorecidas y tiene una edad de inicio y peor pronóstico en varones. Esquizoide es el individuo con rasgos semejantes a la equizofrenia encontrados a menudo en familiares de esquizofrénicos. El problema surge porque no hay criterios clínicos para diferenciar la personalidad esquizoide de la normal. Para simplificar, puede considerarse que el término esquizoide se refiere a una persona con los síntomas fundamentales de la esquizofrenia, pero más suaves. Se ha estimado que alrededor del 4% de la población general tiene esquizofrenia o un trastorno esquizoide de la personalidad. o Base genética.- se constata un exceso de nacimientos de individuos equizofrénicos en invierno, lo que sugiere que factores ambientales, como determinadas infecciones víricas o factores nutricionales, podrían contribuir a esa patología. La verificación de diferencias en la concentración de neurotrasmisores (como la dopamina o la 5hidrocitriptamina –serotonina-) en varias zonas del cerebro de los esquizofrénicos en la autopsia, así como la acción farmacológica de los medicamentos antipsicóticos, sugieren que los genes de las sustancias nuerotransmisoras del SNC y de sus receptores pueden ser posibles genes candidatos para los estudios de ligamento. No se ha encontrado un posible ligamiento, pero algunos estudios han producido datos a favor de una asociación con determinadas variantes alélicas, como el receptor de la serotonina tipo 2ª. o Patrón hereditario.- multifactorial AUTISMO.o Características clínicas.- el autismo es un grave trastorno del nuerodesarrollo que afecta entre 4 y 10 personas de cada 10, 000 y que se inicia en los primeros 3 años de vida. La incidencia es de tres a cuatro veces más elevada en niños que en niñas, y las características clínicas son la grave afectación del desarrollo de la respuesta social, una comunicación verbal y no verbal muy pobres, y un compartimiento y unos intereses estereotipados. Muchos niños afectados muestran un retraso significativo en el desarrollo, así como muchos niños con retraso en el desarrollo muestran características autistas. Por ejemplo, el comportamiento autista es muy conocido en niños con esclerosis tuberosa y con varias formas de desequilibrio cromosómico, al igual que en varones con el síndrome del X frágil. El autismo forma parte de un espectro que incluye el síndrome de 49 Asperger, y en conjunto, los trastornos del espectro autista tiene una prevalencia mucho más elevada, de hasta 60 de cada 10,000 individuos; el riesgo para los hermanos de una persona afectada es aproximadamente del 5%. o Base genética.- mutación en 2q, 7q, 17q, plimorfismo en el gen SLC25A12 en el cromosoma 2q31.1; la causa del autismo aislado (no sindrómico) no está clara, pero cada vez hay más pruebas de que los factores genéticos desempeñan un importante papel en ese trastorno. Esta creencia se basa en los resultados de los estudios de gemelos, que dan una tasa de concordancia para los trastornos del espectro autista de hasta el 80% en gemelos monocigóticos y el 20% en dicigóticos, dependiendo de los criterios de diagnóstico utilizados. Por tanto, el autismo y los trastornos del espectro autista son heredables en alto grado. o Patrón hereditario.- multifactorial ALCOHOLISMO.o Características clínicas.- Creciente tolerancia a los efectos del alcohol, pérdida de control, “lagunas en la memoria”, períodos de amnesia, episodios injustificados de mal humor, nauseas, vómitos, calambres, pérdida de apetito. o Base genética.- el cerebro de algunas personas pueden convertir el alcohol en una sustancia muy adictiva. El cuerpo humano transforma el alcohol en acetaldehído sustancia muy tóxica que, luego de un proceso químico se elimina como dióxido de carbono y agua. En personas genéticamente predispuestas, en cambio, una pequeña cantidad de acetaldehído no es eliminada y se va al cerebro donde se convierte en THIQ. Esta sustancia, la misma que produce el cerebro con la heroína es un sedante de fuerte grado de adicción, mayor que el de la morfina por ejemplo. DRD2. o Patrón hereditario.- multifactorial. ARTRITIS.o Características clínicas.- enfermedad crónica y degenerativa que se caracteriza por provocar inflamación en la membrana sinovial de las articulaciones. La inflamación de las sinoviales provoca dolor, aparecen nódulos en zonas de roce con la piel como los codos, dorso de los dedos de la mano y de los pies. Fiebre, resequedad de la piel y mucosas. Vasculitis. o Base genética.- cromosoma 6q que influye en el comportamiento del gen TNFAIP3. o Patrón hereditario.- multifactorial, herencia mendeliana. 50 ENFERMEDAD DE PARKINSON.o Características clínicas.- temblores, lentitud al escribir, perder la pista de una palabra o pensamiento, falta de expresión en la cara. o Base genética.- mutación en el gen de la alfa-sinocleuina. Parkina, DJ-1, PINK1, LRRK2. o Patrón hereditario.- Autosómico dominante. TROMBOSIS VENOSAS.o Características clínicas.- inflamación en pierna, dolor o sensibilidad en la pierna (unilateral) puede ser al pararse o al caminar. Sensación de calor aumentado en el área que esta inflamada o adolorida. Piel roja o sin color en el área de inflamación o dolor. La trombosis venosa representa un importante problema de salud en todo el mundo, y su incidencia aumenta de 1 de 100,000 en la infancia a 1 de 100 en la vejez. El tromboembolismo venoso, que incluye la trombosis venosa profunda y la embolia pulmonar, es una enfermedad compleja que se produce por múltiples interacciones entre factores de riesgo heredados y adquiridos. Las trombofilias heredadas también aumentan el riesgo de pérdida fetal, tanto en forma de mortiñitos como de abortos tempranos. o Base genética.- la identificación de dos mutaciones prevalecientes en la población caucásica europea ha allanado el camino para numerosos grandes estudios cohortes, que han incrementado el conocimiento de la trombosis venosa. La mutación del factor V de Leiden (R506Q) hace que la proteína del factor V se vuelva resistente a la escisión por la proteína C activada, aumentando así la producción de trombina. La variante de protrombina 20210G>A se sitúa en la región 3´no traducida (UTR) y se asocia con valores de protrombina aumentados. Esas variantes ocasionan un aumento de 4 a 5 veces del riesgo de trombosis en los heterocigotos, pero los individuos homocigotos para una variante o heterocigotos para las dos tienen un riesgo significativamente elevado (hasta 80 veces). Causas heredadas y adquiridas de trombosis venosa: Heredadas.o o COMUNES. Factor de Leiden Variante de la protrombina G20210A Mutación homocigotica C677T en el gen de la metilenotetrahidrofolato reductasa (MTHFR) RARAS.- 51 o Deficiencia de antitrombina Deficiencia de proteína C Deficiencia de proteína S Adquiridas.o Cirugía y traumatismo o Inmovilización prolongada o Trombosis anterior o Embarazo o Anticonceptivos orales y terapia de sustitución hormonal o Edad avanzada Patrón hereditario.- Autosómico recesivo. ENFERMEDAD CORONARIA.o Características clínicas.se deriva de la ateroesclerosis, un proceso que tiene lugar a lo largo de muchos años y que se caracteriza por el depósito de lípidos en el espacio subendotelial (íntima) de las arterias, con el consiguiente estrechamiento de su luz. La primera etapa comprende el depósito de lípidos en la pared arterial a consecuencia de los factores hemodinámicas. Los monocitos se adhieren a las regiones de la superficie endotelial de las paredes arteriales con depósitos lipídicos, penetran en la pared arterial, proliferan y se diferencian en macrófagos. Estos últimos recogen los lípidos, lo que da lugar a las clásicas estrías de grasa, y a través de de la acción de las citocinas, los factores de crecimiento y las moléculas de adhesión, inducen la proliferación de músculo liso y la formación de una matriz extracelular, produciendo el desarrollo de las placas ateroescleróticas fibrosas. El estrechamiento de las arterias coronarias compromete las necesidades metabólicas del músculo cardíaco, lo que conduce a la isquemia miocárdica, que si es grave desemboca en un infarto de miocardio. o Base genética.- Mutaciones en el cromosoma 9. El riesgo de coronapatía es inicialmente multifactorial o poligénico para la mayoría de las personas, se han identificado varios factores de riesgo genéticos y ambientales diferentes, que predisponen al inicio temprano del proceso aterosclerótico. Entre los factores de riesgo ambientales bien conocidos se encuentran la falta de ejercicio físico, el colesterol de la dieta y el tabaquismo. Resulta evidente el potencial de prevención del desarrollo de la coronapatías mediante el control de estos factores de riesgo. o Patrón hereditario.- autosómica recesiva. 52 ÚLCERA PÉPTICA.o Características clínicas.- dolor abdominal; epigástrico, urente, nocturno, alivio con la comida, periódico. Síndrome ulceroso. Solución de continuidad de la mucosa que alcanza hasta la submucosa. o Base genética.- los siguientes caracteres condicionados genéticos se asocian; tendencia familiar a la úlcera péptica, tendencia familiar al vaciamiento gástrico acelerado, respuesta anormal de las células G, hiperpepsinogenemia, ausencia de secreción de antígenos de grupos sanguíneos en la saliva y en el jugo gástrico. o Patrón hereditario.- autosómico dominante. ALZHEIMER.o Características clínicas.la demencia se caracteriza por un deterioro global, irreversible y progresivo del intelecto, la memoria, las habilidades sociales y el control de las reacciones emocionales, en presencia de un estado de conciencia normal. Es heterogénea desde el punto de vista etiológico, y ocurre de forma secundaria a causas no genéticas, como la enfermedad vascular y las infecciones, como el SIDA, al igual que a causas genéticas. La enfermedad de Alzheimer (EA) es la causa mas frecuente de demencia en personas tanto con demencia de inicio temprano (menos de 60 años, o presenil) como inicio tardío (más de 60 años, o senil). El hallazgo nueropatológico clásico en personas con EA es la presencia en el examen post mortem de depósitos amiloides en ovillos neurofibrilares y en placas neuronales o seniles. Además, los individuos con el síndrome Down tienen un riesgo aumentado de desarrollar demencia, que en la autopsia presenta idénticos hallazgos en el SNC que los encontrados en personas con EA típica. o Base genética.- la identificación de la proteína precursora del amiloide beta, A4 (APP) en los depósitos amiloides de las placas neuronales y su ubicación en la región crítica de la parte distal del cromosoma 21q o próxima a ella, asociado a las características fonetípicas del síndrome Down y el incremento del riesgo de EA en personas con el síndrome Down, condujo a la hipótesis de que la duplicación del gen APP podría ser la causa de la EA. o Patrón hereditario.- autosómica dominante. DIABETES.o Características clínicas.- hay dos tipos de diabetes mellitus (DM), clínicamente distintos. El tipo 1 (DM1) es la forma más rara: se inicia en la juventud, es dependiente de la insulina (antes se usaban las siglas DMID diabetes mellitus insulinodependiente), afecta al .4% de la población y muestran una elevada incidencia de complicaciones renales, retinianas y vasculares potencialmente graves. La edad de 53 inicio de la DM1 presenta un pico en la adolescencia y sólo puede controlarse mediante inyecciones regulares de insulina. La diabetes mellitus tipo 2 (DM2) es la forma más frecuente: es de inicio tardío, no es dependiente de la insulina y afecta hasta al 10% de la población. Suele presentarse en personas de más edad y puede responder a la simple restricción dietética de la ingesta de hidratos de carbono, aunque muchos individuos con DM2 necesitan medicamentos hipoglucemiantes orales y algunos precisan insulina. Por último, entre el 1 y 2 % de los diabéticos tienen formas monogénicas (de un solo gen) de diabetes. Entre 1 y 3% de las embarazadas desarrollan intolerancia a la glucosa: es la llamada diabetes gestacional. Esa tolerancia anormal a la glucosa suele normalizarse después del embarazo, aunque del 50 al 75% de esas mujeres desarrollarán diabetes más tarde a lo largo de su vida. La diabetes también puede ser secundaria a una variedad de otros síndromes genéticos raros y a trastornos no genéticos, entre los que figuran el síndrome de Prader- Willi, el síndrome de Bardet-Biedl, el síndrome de Wolfram y la ataxia de Fridreich. Por tanto, la DM es heterogénea desde el punto de vista etiológico. o Base genética.- se conocen casi 20 formas monogénicas de diabetes. La diabetes de la madurez de inicio en la juventud (MODY), se caracteriza por la disfunción de la células beta (HNF1A), las mutaciones en el gen de la glucocinasa causan una hiperglucemia leve. Se han encontrado mutaciones en otros 5 genes, que codifican factores de transcripción necesarios para el desarrollo de las células beta. o Patrón hereditario.dominante. multifactorial, MODY es autosómica HIPERTENSIÓN.o Características clínicas.- la presión arterial puede contribuir hasta con el 50% del total de los problemas cardiovasculares. Las personas con hipertensión se encuadran en dos grupos. En uno de ellos, el inicio de la presión alta suele darse en la juventud y es una consecuencia de otro trastorno, como una enfermedad renal o anomalías en determinadas glándulas endocrinas. Es lo que se denomina hipertensión secundaria. En el otro, más frecuente, la hipertensión suele empezar en la madurez y no tiene una causa identificable, se le denomina hipertensión esencial; se sabe que factores ambientales (como la elevada ingesta de sodio en la dieta, la obesidad, la ingesta de alcohol y la falta de ejercicio) se asocian con un riesgo aumentado de hipertensión. Ese trastorno también tiene una prevalencia más elevada en personas pertenecientes a grupos socioeconómicos más desfavorecidos. Los estudios de personas adoptadas muestran una correlación más baja de la presión arterial con sus padres biológicos que con los hermanos que permanecieron con estos últimos. Además, los estudios de migraciones concernientes a personas que se han desplazado de una población 54 con una baja prevalencia de hipertensión a otra con una prevalencia elevada han mostrado que el grupo inmigrante adquiere la frecuencia de su nuevo grupo población a lo largo de una o dos generaciones. Eso sugiere que los factores ambientales son de gran importancia en la etiología de la hipertensión. o Base genética.- los estudios de familias y de gemelos han mostrado que la hipertensión es familiar y que la presión arterial se correlaciona con el grado de parentesco. Esos hallazgos sugieren la importancia de los factores genéticos en la etiología de la hipertensión. Además, la prevalencia de ese trastorno varía según la población: es más frecuente en las personas de origen afrocaribeño y menos frecuente en los esquimales, los aborígenes australianos y los indios de América Central y del Sur. Los loci que contribuyen a la predisposición de la hipertensión es 17q (HYT1), otro el cromosoma 16 y el cromosoma 15q (HYT2) o Patrón hereditario.- multifactorial. OBESIDAD.o Características clínicas.- Se define como un índice de grasa superior a 30. Un 33% de los norteamericanos tienen obesidad y un 35% sobrepeso. La obesidad no es una enfermedad en si pero contribuye en otras. Existe una fuerte correlacion entre la obesidad de padres e hijos por efectos ambientales. Genes también influyen en la obesidad como el de la leptina, cuando existe niveles disminuidos aumenta el apetito. Ya que la leptina se relación con el MC4R cuando este se ve mutado se produce una obesidad grave. o Base genética.- la falla de un gen que causa resistencia a la insulina podría ser la causa de alrededor de la mitad de las familias obesas. o Patrón hereditario.- multifactorial, poligénico. 55 Autor: José Regino Ríos Gutiérrez. Unidad No. 11.- Defectos congénitos Competencia intermedia.- diagnosticar y clasificar los defectos congénitos para determinar el manejo, prevención y asesoramiento genético así como su impacto social y económico. Criterios de desempeño.1.- Definir defecto congénito y clasificarlos de acuerdo a su impacto sobre la salud del individuo (mayores y menores) y mecanismo de formación (malformación, disrupción, deformación, displasia) DEFECTO CONGÉNITO.- (anomalía congénita o defecto de nacimiento) anomalías estructurales que pueden ocurrir en un embrión, feto o niño recién nacido. IMPACTOS MAYORES SOBRE LA SALUD.- o anomalía mayor, se define como la que tiene consecuencias adversas para la función o la aceptación social del individuo. IMPACTOS MENORES SOBRE LA SALUD.- o anomalías menores, no tienen importancia médica ni cosmética. MALFORMACIÓN.- es un defecto estructural primario de un órgano (o una parte de un órgano) que se deriva de una anomalía inherente al desarrollo. Se solía denominar malformación primaria o intrínseca. La presencia de una malformación implica que el desarrollo temprano de un tejido u órgano determinado se ha detenido o ha seguido una dirección errónea. Ejemplos comunes de malformaciones son las anomalías cardíacas congénitas, como los defectos del septo ventricular o del atrial, el labio leporino con o sin paladar hendido, y los defectos del tubo neural, como la anencefalia o la mielomeningocele lumbosacra. La mayoría de las malformaciones que se limitan a un único órgano muestran una herencia multifactorial, lo que implica la interacción de muchos genes con otros factores. Las malformaciones múltiples tienen mayor probabilidad de deberse a anomalías cromosómicas. DISRUPCIÓN.- se refiere a una estructura anormal de un órgano o un tejido como resultado de factores externos que han interrumpido el proceso de desarrollo normal. Esto solía denominarse malformación secundaria o extrínseca. Entre los factores extrínsecos que pueden afectar al desarrollo normal se incluye la isquemia, las infecciones y el trauma. Un ejemplo de disrupción es el efecto ejercido sobre el desarrollo de un miembro cuando una banda de tejido amniótico se enrolla alrededor de los dedos o del antebrazo de un bebé. Por definición, una disrupción no es genética, aunque en ocasiones los factores genéticos puedan predisponer a hechos de este tipo. Por ejemplo, una pequeña proporción de las bandas amnióticas se debe a defectos del colágeno genéticamente determinados, que debilitan el amnios y aumentan su propensión a rasgarse o romperse de forma espontánea. 56 DEFORMACIÓN.- es un defecto originado por una fuerza mecánica anormal que distorsiona una estructura por lo demás normal. Entre los ejemplos bien conocidos se encuentra la luxación de la cadera y el pie zambo “postural” ligero clubfoot o talipes. Los dos pueden deberse tanto a la falta de líquido amniótico (oligohidramnios) como por la reducción del espacio intrauterino a causa de un embarazo gemelar o a una estructura uterina anormal. Las deformaciones suelen ocurrir cuando la gestación está avanzada e implican un bien pronóstico con el tratamiento adecuado, como por ejemplo el entablillado en el caso del pie zambo, ya que el órgano tiene una estructura básicamente normal. DISPLASIA.- es una organización anormal de las células en un tejido. Los efectos suelen verse en cualquier parte del cuerpo en la que esté presente el tejido en cuestión. Por ejemplo, en una displasia esquelética como la tanatofórica, causada por una mutación en el FGFR3, están afectadas casi todas las partes del esqueleto. De forma análoga, en una displasia ectodérmica se afectan se afectan tejidos muy dispersos de origen ectodérmico, como el pelo, los dientes, la piel y las uñas. La mayoría de las displasias se deben a defectos de un único gen y se asocian a un elevado riesgo de recurrencia en los hermanos y los hijos. 2.-Diferenciar entre patrones de malformaciones múltiples: síndrome, asociación, secuencia, defecto en campo de desarrollo, espectro. SÍNDROME.- en la práctica, la palabra síndrome se usa de forma muy genérica (p. ejm; el “síndrome” de la banda amniótica), pero en teoría se debería reservar para los patrones consistentes y reconocibles de anomalías que, a menudo, tiene una causa subyacente conocida. Estas causas pueden incluir anomalías cromosómicas, como ocurre en el síndrome de Down, o defectos monogénicas, como en el síndrome de Van der Woude, en el que el labio leporino, con o sin fisura palatina, se asocia a depresiones en el labio inferior. Hoy se conocen varios miles de síndromes de malformaciones. Este campo de estudio se denomina dismorfología. El diagnóstico de síndromes concretos ha experimentado un gran adelanto gracias al desarrollo de las bases de datos computarizadas. Es posible obtener una lista de diagnósticos diferenciales proporcionando al programa de búsqueda de la base de datos los detalles de las características anormales claves. Sin embargo, incluso con la ayuda de esta herramienta diagnóstica de gran valor, por desgracia no es posible encontrar un diagnóstico para muchos de los niños dismórficos, por lo que puede resultar extremadamente difícil proporcionar a los progenitores una información precisa acerca del probable pronóstico y del riesgo de recurrencia. ASOCIACIÓN.- el término asociación se ha introducido al reconocerse el hecho de que ciertas malformaciones tienden a ocurrir juntas con una mayor frecuencia de la que sería de esperar por mera probabilidad, pese a que no puedan ser explicadas basándose en una secuencia o síndrome. La asociación se diferencia de un síndrome por la falta de consistencia de las anomalías entre un individuo afectado y otro, y por la ausencia de una explicación subyacente satisfactoria. A menudo los nombres de las asociaciones son acrónimos creados a partir de la primera letra de los 57 órganos o sistemas implicados con más frecuencia. Por ejemplo, la asociación VATER se compone de anomalías Vertebrales, Anales, TraqueoEsofágicas y Renales. Las asociaciones conllevan un riesgo bajo de recurrencia y en general se cree que no tienen un origen genético, pese a que la causa subyacente suele ser reconocida. Hay que convenir que esta clasificación de los defectos de nacimiento no es perfecta. Resulta evidente que dista mucho de ser completa o mutuamente exclusiva, por ejemplo, la obstrucción del flujo urinario causada por una malformación primaria como la de una válvula uretral conduce al oligohidramnios o a la secuencia de Potter, lo que a su vez lleva a malformaciones secundarias, como la luxación de la cadera y el talipes. Para complicar todavía más el tema, la ausencia de ambos riñones, que también suele denominarse erróneamente síndrome de Potter. A pesar de esta confusión semántica, es importante mantener esta clasificación, pues sirve de ayuda para la comprensión de las malformaciones y para los cálculos del riesgo de recurrencia. SECUENCIA.- describe los hallazgos que se producen a consecuencia de una cascada de acontecimientos iniciados por un único factor primario. A menudo éste puede ser la malformación de un solo órgano. En la secuencia de Potter, la pérdida crónica de líquido amniótico o la producción defectuosa de la orina resulta en oligohidramnios. Esto conduce, a su vez, a la compresión fetal, que ocasiona la apariencia facial aplastada, la luxación de la cadera, el talipes y la hipoplasia pulmonar, que suele causar la muerte neonatal por fallo respiratorio. DEFECTO EN CAMPO DE DESARROLLO.- es la alteración de estructuras embrionarias complejas que están sincronizadas y jerarquizadas. ESPECTRO.- grupo poblacional afectado por algún problema genético. 3.- Fundamentar la aplicación del algoritmo de estudio para establecer el diagnóstico del paciente con defectos congénitos, para establecer un plan de manejo y asesoramiento genético. La pregunta mas importante que se formula al evaluar a un niño con una malformación congénita es si el defecto es aislado o si forma parte del conjunto de un síndrome. Es importante diferenciar la secuencia del síndrome. 4.- Analizar los aspectos epidemiológicos de los defectos congénitos en varios estadios de la morfogénesis desde el momento de la concepción (abortos, pérdida gestacional tardía, recién nacidos vivos y muertos) 5.- Justificar las estrategias de la prevención de los defectos congénitos preconcepcional, prenatal, y neonatal: evaluación médica y genética, ingesta de ácido fólico, control prenatal, control de enfermedades maternas, evitar consumo de alcohol, drogas y medicamentos no indicados. Tamiz neonatal, control de niño sano, prevención secundaria de las complicaciones y asesoría para evitar nuevos casos. una buena evaluación médica de los padres antes de la decisión de tener hijos dará un buen control sobre el desarrollo del hijo próximo, el acido 58 fólico junto con la ingesta de vitaminas ayudara a no tener defectos congénitos como los del tubo neural. El consumo de drogas, alcohol y otros nocivos darán como resultado defectos congénitos a cualquier edad gestacional el control de un niño sano es importantísimo ya que prevé cualquier enfermedad por ambiente y ayudara a detectar a tiempo enfermedades genéticas 59 Autor: José Regino Ríos Gutiérrez. Unidad no. 12 Errores innatos del metabolismo Competencia intermedia.Establecer el diagnóstico de los errores innatos del metabolismo fundamentando los exámenes de laboratorio utilizados así como las diferentes opciones terapéuticas. Criterios de desempeño.1.- Definir errores innatos del metabolismo.Se conocen más de 200 errores congénitos del metabolismo que pueden agruparse según el metabolito, la ruta metabólica, la función de la enzima o el orgánulo celular afectado. La mayoría de los errores innatos del metabolismo se heredan de forma autosómica recesiva o ligados al cromosoma X, y sólo unos cuantos lo hacen de forma autosómica dominante. Esto se debe a que la proteína defectuosa en la mayoría de los errores congénitos es una enzima que es difusible, y habitualmente hay suficiente actividad residual en su estado heterocigoto (es decir, mutación de pérdida de función) para que la enzima funcione normalmente en la mayoría de las situaciones. Sin embargo, si la reacción catalizada por la enzima es limitante de la producción (es decir, mutación por haploinsuficiencia) o el producto del gen forma parte de un complejo mutilmérico (es decir, mutación negativa-dominante), el trastorno puede manifestarse en estado heterocigoto, es decir, heredarse de forma dominante. 2.- Analizar las vías metabólicas que producen los errores innatos del metabolismo: ciclo de la urea, catabolismo de los aminoácidos de cadena ramificada, trastornos de la beta oxidación, trastornos de la cadena respiratoria mitocondrial y glucólisis. CICLO DE LA UREA.- El ciclo de la urea es una ruta metabólica de cinco pasos que tiene lugar principalmente en los hepatocitos para la eliminación de los desechos de nitrógeno de los grupos amino de los aminoácidos, que se produce por el recambio normal de las proteínas. Convierte dos moléculas de amoníaco y una de bicarbonato en urea. Las deficiencias de las enzimas del ciclo de la urea producen intolerancia a la proteína debida a la acumulación de amoníaco en el organismo, lo que conduce a una hiperamonemia. Los valores aumentados de amoníaco son tóxicos para el sistema nervioso central y pueden producir el coma y, al igual que algunos de los trastornos del ciclo de la urea, si se dejan sin tratar, la muerte. Los diversos trastornos del ciclo de la Urea son colectivamente raros e individualmente muy raros. Todos ellos se heredan como trastornos autosómicos recesivos, excepto la deficiencia de la ornitina transcarbamilasa, que está ligada al cromosoma X. CATABOLISMO DE LOS AMINOÁCIDOS DE CADENA RAMIFICADA.- los aminoácidos ramificados esenciales leucina, isoleucina y valina tienen parte de sus rutas metabólicas en común. La deficiencia de la enzima implicada da como resultado el enfermedad de la orina de jarabe de arce (los recién 60 nacidos con este trastorno autosómico recesivo presentan en la primera semana de vida vómitos, seguidos de una hipotonía alternante, seguida de la muerte en unas pocas semanas si no se trata. La orina tiene un olor característico parecido al del jarabe de arce. El trastorno está causado por la deficiencia de la descarboxilasa de los cetoácidos de cadena ramificada, que producen un aumento de la excreción de los aminoácidos ramificados valina, leucina e isoleucina en la orina, cuya presencia sugiere el diagnóstico, que se confirma por la demostración de los tres aminoácidos ramificados esenciales en la sangre. El tratamiento consiste en una dieta que limite la ingesta de los tres aminoácidos ramificados a las cantidades necesarias para el crecimiento. Los individuos afectados son particularmente sensibles al deterioro, junto con enfermedades intercurrentes resultantes de la degradación de las proteínas). TRASTORNOS DE LA BETA OXIDACIÓN.- el ciclo de la carnitina es una ruta bioquímica necesaria para el transporte de los ácidos grasos de cadena larga a la matriz mitocondrial, y entonces se activan los que tienen menos de 10 carbonos de longitud, para formar ésteres acil-CoA. El ciclo de la carnitina es una parte de la ruta de la Beta-Oxidación mitocondrial que desempeña un papel principal en la producción de energía, especialmente durante los períodos de ayuno. La deficiencia de carnitina es una característica secundaria de los trastornos de la beta oxidación, con la excepción del defecto en el transporte de la carnitina, en la que es primaria, y este raro trastorno responde espectacularmente a la sustitución de la carnitina. Trastornos en la oxidación de los ácidos grasos más habituales: o Deficiencia de la acil-CoA deshidrogenasa de cadena media.(MCAD) es la más habitual de este grupo de trastornos, y que se presenta con mayor frecuencia como una hipoglucemia hipocetósica episódica provocada por el ayuno. La aparición suele producirse en los primeros 2 años de vida, y en ocasiones es fatal, simulando el síndrome de muerte súbita infantil. El tratamiento se basa en mantener la ingesta calórica adecuada y en evitar el ayuno, que puede ser un desafío en los niños pequeños con enfermedades intercurrentes. Heredado como un trastorno autosómico recesivo, el 90% de los alelos se producen por una mutación puntual única, lo que ha llevado a la discusión de que ésta podría ser una enfermedad candidata para la detección en la población neonatal. o Deficiencia de la acil-Coa deshidrogenasa de cadena larga (LCAD) y de cadena corta (SCAD) y de la 3-hidroxiacil-CoA deshidrogenasa de cadena larga (LCHAD).- todos estos raros trastornos muestran una herencia autosómica recesiva y se presentan precozmente en la vida con una combinación variable de miopatías esquelética y cardíaca, disfunción hepatocelular con hepatomegalia y encefalopatía. El tratamiento gira alrededor del mantenimiento nutriocional y la evitación del ayuno, pero no es muy grato en la SCAD. o Aciduria glutárica.- las acidurias glutáricas tipos I (deficiencia de la glutamil-CoA deshidrogenasa) y II (deficiencia múltiple de la acil-CoA 61 deshidrogenasa) se incluyen como ejemplos de acidurias orgánicas que son intermediarias en la oxidación de los ácidos grasos; ambas muestran una herencia autosómica recesiva. En el tipo I la macrocefalia está presente en el nacimiento y los lactantes presentan episodios de encefalopatía con espasticida, distonía, convulsiones y retraso en el desarrollo. El tratamiento consiste en la restricción dietética de los aminoácidos glutarigénicos: lisina, triptófano y hidroxilisina. Habitual entre los amish de Pensilvania, en su zona se ha introducido a detección neonatal. La aciduria tipo II es variable, y hay dos formas graves que aparecen en el período neonatal; una de ellas incluye anomalías urogenitales. En ambos tipos graves hay hipotonía, hepatomegalia, acidosis metabólica e hipoglucemia hipocetósica. La forma de aparición tardía puede presentarse al principio de la infancia, más que en el período neonatal, con deterioro progresivo, acidosis metabólica, hipoglucemia y encefalopatía. el tratamiento de las formas graves es únicamente de apoyo, pero en las formas más leves la riboflavina, la carnitina y las dietas bajas en proteínas y en grasas han tenido más éxito. TRASTORNOS AFECTAN A LA FUNCIÓN DE LAS MITOCONDRIAS.- en 1962 se identificó por primera vez una enfermedad de las mitocondrias en un paciente cuyas mitocondrias mostraban anomalías estructurales y la pérdida del acoplamiento entre la oxidación y la fosforilación, aunque no fue hasta fines de la década de 1970 y principio de la de 1980 que empezó a apreciarse la importancia del DNA mitocondrial mutado (mtDNA) para la enfermedad humana. El pequeño mtDNA circular de doble cadena contiene genes que codifican la producción del RNA ribosómico (rRNA) y diversos RNA de transferencia (tRNA), necesarios para la biosíntesis de las proteínas mitocondriales, así como de algunas de las proteínas implicadas en el transporte de electrones. Hay 5,523 codones y un total de 37 productos génicos. Los nucleótidos de guanina y de citosina están asimétricamente distribuidos entre las dos cadenas de mtDNA: la cadena rica en guanina, cadena pesada (H, heavy), y la cadena rica en citosina, o cadena ligera (L, light). La replicación y la transcripción están controladas por una secuencia de 1,122 pb del mtDNA conocida como bucle de desplazamiento (bucle-D). La fosforilación oxidativa (OXPHOS) es el proceso bioquímico responsable de la generación de la mayor parte del ATP necesario para la energía celular. El proceso está mediado por cinco complejos I-V, y el mtDNA codifica 13 subunidades OXPHOS, 22 RNAt y 2 RNAr. Los complejos están adecuadamente nombrados. El análisis del complejo I, por ejemplo, ha revelado aproximadamente 41 subunidades diferentes, de las cuales siete son polipéptidos codificados por genes del mtDNA conocidos como ND1, ND2, ND3, ND4, ND5 y ND6, y las restantes 34 subunidades están codificadas por genes del DNA nuclear. El complejo V comprende 12 o 13 subunidades, de las cuales dos (las ATPasas 6 y 8) están codificadas por el mtDNA. La máxima actividad del complejo V parece requerir una unión estrecha de la cardiolipina, codificada por el DNA nuclear. Como la mayoría de las proteínas mitocondriales, incluyendo las subunidades implicadas en el transporte de los electrones, están codificadas por genes nucleares, siguen con mayor frecuencia una herencia autosómica recesiva. Como en otras 62 enfermedades metabólicas recesivas, los trastornos resultantes de las mutaciones en estos genes tienden a reproducirse realmente. Sin embargo, los trastornos debidos a las mutaciones en el mtDNA son extremadamente variables debido al fenómeno de la heteroplasmia. Las características clínicas son principalmente una combinación de signos neurológicos (encefalopatía, demencia, ataxia, distonía, neuropatía y convulsiones) y signos miopáticos (hipotonía, debilidad y miocardiopatía con defectos de conducción). Otros síntomas y signos pueden incluir sordera, diabetes mellitus y pigmentación retiniana, y la acidosis puede ser característica. Las manifestaciones clínicas son tan variables que la citopatía mitocondrial debe considerarse como una posibilidad a cualquier edad, cuando la enfermedad de presentación tenga un componente neurológico o miopático. Se han definido diversas entidades clínicas distintas, y aunque algunas de ellas se solapan considerablemente, hay cierta correlación genotipo-fenotipo. GLUCÓLISIS.- el glucógeno es la forma en la que el azúcar glucosa se almacena en el músculo y en el hígado en forma de polímero, para actuar como una fuente de energía de reserva. En las enfermedades del almacenamiento del glucógeno (GSD), el glucógeno se acumula en cantidades excesivas en el músculo esquelético, el músculo cardíaco y/o ell hígado debido a diversos errores congénitos del metabolismo de las enzimas implicadas en la síntesis y la degradación del glucógeno. Además, debido al bloqueo metabólico, el glucógeno no está disponible como una fuente normal de glucosa. Esto puede ocasionar hipoglucemia, alteración de la función hepática y anomalías neurológicas. En cada uno de los seis tipos principales de GSD hay un defecto enzimático específico que afecta a uno de los pasos de las rutas metabólicas de la síntesis o de la degradación del glucógeno. Los diversos tipos pueden agruparse dependiendo de si afectan principalmente al hígado o al músculo. Los seis tipos se heredan como trastornos autosómicos recesivos, aunque hay variantes de la fosforilasa hepáticas ligadas al cromosoma X. o Enfermedades del almacenamiento del glucógeno que afectan principalmente al hígado. Enfermedad de von Gierke (GSD-I).- fue el primer trastorno descrito del metabolismo del glucógeno y se debe a la deficiencia de la enzima glucosa-6-fosfatasa, responsable de la degradación del glucógeno hepático para liberar glucosa. Los lactantes afectados presentan un hígado agrandado (hepatomegalia) y/o sudoración y taquicardia debida a la hipoglucemia, que puede aparecer después de un ayuno de tan sólo 3-4 horas. El tratamiento es sencillo alimentación frecuente y evitar el ayuno para mantener la concentración de azúcar en la sangre. Enfermedad de Cori (GSD-III).- se debe al déficit de la enzima amilo-1,6-glucosidasa, que se conoce también como la enzima desramificadora. El déficit de la enzima da como resultado la acumulación del glucógeno en el hígado y en otros tejidos debido a la incapacidad para cortar los puntos de 63 “ramificación” del polímero de glucógeno. Los lactantes afectados pueden presentar hepatomegalia por la acumulación del glucógeno y/o debilidad muscular. El tratamiento consiste en prevenir la hipoglucemia mediante la alimentación frecuente y evitar los períodos prolongados de ayuno. o Enfermedad de Anderson (GSD-IV).- se debe a la deficiencia de la enzima ramificadora del glucógeno, que ocasiona la formación de un glucógeno anómalo, consistente en largas cadenas de pocas ramas que las enzimas normalmente responsables de la degradación del glucógeno no pueden romper. Los lactantes afectados presentan hipotonía y una función hepática anómala en el primer año de vida, que evoluciona rápidamente a la insuficiencia hepática. No se dispone de ningún tratamiento eficaz, aparte de la posibilidad de un trasplante hepático. Deficiencia de la fosforilasa hepática (GSD-VI).- es un complejo enzimático multimérico con subunidades codificadas tanto por genes autosómicos como ligados a cromosoma X. la deficiencia de la fosforilasa hepática obstaculiza la degradación del glucógeno, lo que se traduce en niños que, en los primeros 2 años de vida, presentan hepatomegalia, hipoglucemia y deterioro progresivo. El tratamiento consiste en suplementos de hidratos de carbono para mejorar el crecimiento. Enfermedades del almacenamiento del glucógeno que afectan principalmente al músculo. Enfermedad de Pompe (GSD-II).- los lactantes con esta enfermedad presentan habitualmente en los primeros meses de vida flojedad (hipotonía) y retraso en las etapas rudimentarias del desarrollo motor debido a la debilidad muscular. A continuación desarrollan hipertrofia cardíaca y mueren por insuficiencia cardíaca en el primer o segundo año. Los músculos cardíaco y estriados acumulan glucógeno debido a la deficiencia de la enzima lisosómica alfa-1,4-glucosidasa, necesaria para romper el glucógeno. El diagnóstico puede confirmarse por el análisis enzimático de los leucocitos o de los fibroblastos. Las notificaciones iniciales de la terapia de sustitución de la enzima parecen prometedoras. Enfermedad de McArdle (GSD-V).- las personas con esta enfermedad presentan calambres musculares cuando hacen ejercicio en el período de adolescencia. El trastorno se debe a la deficiencia de la fosforilasa muscular, necesaria para la degradación del glucógeno muscular. No hay una forma eficaz del tratamiento, aunque en algunos individuos afectados los calambres musculares tienden a reducirse si se continúa el 64 ejercicio, probablemente como resultado de disponer de otras fuentes de energía por rutas metabólicas alternativas. 3.- Clasificar los errores innatos del metabolismo de acuerdo a substancias involucradas en el trastorno metabólico en: trastornos metabolismo de los carbohidratos, trastornos del metabolismo de lípidos, trastorno del metabolismo de las proteínas y trastornos depósitos de moléculas complejas. las del los por TRASTORNOS DEL METABOLISMO DE LOS CARBOHIDRATOS.- Los errores congénitos del metabolismo de los hidratos de carbono pueden reunirse en dos grupos principales: trastornos del metabolismo de los monosacáridos y trastornos del almacenamiento del glucógeno. o Trastornos del metabolismo de los monosacáridos.- la galactosemia y la intolerancia hereditaria a la fructuosa, son dos ejemplos de los trastornos del metabolismo de los monosacáridos. Galactosemia.- es un trastorno autosómico recesivo debido a la deficiencia de la enzima galactosa 1-fosfato uridil transferasa, necesaria para el metabolismo del azúcar dietético galactosa. Los recién nacidos con galactosemia presentan vómitos, letargo, deterioro progresivo e ictericia en la segunda semana de vida. Si no se tratan, desarrollan complicaciones consistentes en retraso mental, cataratas y cirrosis hepática. La galactosemia puede detectarse por la presencia de sustancias reductoras en orina con pruebas específicas para la galactosa. Las complicaciones de la galactosemia se pueden evitar con el diagnóstico precoz y la alimentación de los lactantes afectados con sustitutos de leche que no contengan galactosa ni lactosa, el azúcar que se encuentra en la leche que se descompone en galactosa. El diagnóstico y el tratamiento precoz son esenciales si se quieren evitar las complicaciones graves. Intolerancia hereditaria a la fructuosa.- es un trastorno autosómico recesivo debido a la deficiencia de la enzima fructuosa 1-fosfato aldolasa. La fructuosa de la dieta está presente en la miel, la fruta y en ciertos vegetales y, en combinación con la glucosa, en el disacárido sacarosa en la caña de azúcar. Los individuos con intolerancia hereditaria a la fructuosa la presentan en distintas edades, dependiendo de cuándo se ha introducido la fructuosa en la dieta. Los síntomas pueden ser mínimos pero también tan graves, como los observados en la galactosemia, que incluyen deterioro progresivo, vómitos, ictericia y convulsiones. El diagnóstico se confirma por la presencia del fructuosa en la orina y el análisis enzimático de la mucosa intestinal, o por una muestra de biopsia hepática. La restricción dietética de fructuosa está relacionada con un buen pronóstico a largo plazo. 65 TRASTORNO DEL METABOLISMO DE LOS LÍPIDOS.la hipercolesterolemia familiar es el trastorno monogénico autosómico dominante más habitual en la sociedad occidental y se asocia con tasas elevadas de morbimortalidad por la coronariopatía prematura. La persona con hipercolesterolemia familiar (FH) tienen valores de colesterol elevados con un riesgo significativo de desarrollar coronariopatía. Pueden presentarse en la infancia o en la adolescencia con depósitos subcutáneos de lípidos, conocidos con xantomas. Comenzando con las familias que habían presentado una coronariopatía precoz, Brown y Goldstein desentrañaron la biología del receptor de las lipoproteínas de baja densidad (LDL) y la base patológica de la FH. Las células obtienen normalmente su colesterol por la síntesis endógena de éste o por la captación del colesterol dietético por los receptores de las LDL en la superficie celular. Los valores intracelulares de colesterol se mantienen por un sistema de retroalimentación, y el colesterol libre inhibe la síntesis de los receptores de las LDL a la vez que reduce el valor de la síntesis del novo del colesterol endógeno. Los valores elevados de colesterol en personas con FH se deben a una función deficiente o defectuosa de los receptores de las LDL que conduce a la síntesis de valores aumentados de colesterol endógeno. Se han identificado 4 tipos principales funcionales o clases de mutaciones en el receptor desde el retículo endosplásmico hasta el aparato de Golgi; unión anómala de las LDL por el receptor, e internalización anómala de las LDL por el receptor. En las personas con FH de ciertos grupos étnicos se encuentran más habitualmente determinadas mutaciones específicas, como resultado de los efectos de fundador. La restricción dietética de la ingesta del colesterol y el tratamiento farmacológico de la circulación enterohempática, pueden reducir los valores de colesterol y, en parte, reducir el riesgo de coronariopatía. TRASTORNO POR DEPÓSITO DE MOLÉCULAS COMPLEJAS.o Trastorno del metabolismo de los esteroides.- consisten en un número de errores innatos autosómicos recesivos de las rutas biosintéticas del cortisol. Pueden dar como resultado la virilización de un feto femenino junto con una pérdida de sal tanto en los lactantes masculinos como en los femeninos, debido a la deficiencia de la hormona aldosterona. Además, los defectos del receptor de los andrógenos ocasiona la falta de virilización de los individuos cromosómicamente masculinos. Hiperplasia suprarrenal congénita (síndrome adrenogenital).- el diagnóstico de la hiperplasia suprarrenal congénita (HSC) debe considerarse en cualquier niña recién nacida que presente virilización de los genitales externos, ya que ésta es la causa más habitual de genitales ambiguos en recién nacidos de sexo femenino. La deficiencia de la 21hidroxilasa es responsable del 90% de los casos. Cerca de la cuarta parte de los lactantes afectados tienen una forma perdedora de sal, y en la segunda o tercera semana de vida presentan colapso circulatorio. La HSC se debe, con menor frecuencia, a la deficiencia de las enzimas 11beta-hidroxilasa o 3beta-deshidrogenasa, y aparece muy raramente como 66 resultado de la deficiencia de enzimas 17alfa-hidroxilasa y 17,20-liasa. La deficiencia de la desmolasa es también muy rara, con bloqueo de todas las rutas. El fenotipo esta invertido, con genitales ambiguos en los hombres, y puede haber crisis addisonianas graves. Los hombres con la deficiencia rara de la 5alfa-reductasa están significativamente Inframasculinizados pero no presentan otros problemas metabólicos, y es probable que sean criados como mujeres. Sin embargo, en la pubertad la oleada de producción de andrógenos es suficiente para estimular el crecimiento del falo, lo que hace problemática la identidad y la asignación del sexo. Las mujeres afectadas con HSC virilizante (clásica) tienen genitales internos normales derivados de los conductos müllerianos, y la virilización de los genitales externos se debe a la acumulación de esteroides corticosuprerrenales proximales al bloqueo enzimático en la ruta biosintética esteroidea, muchos de los cuales tienen una actividad similar a la testosterona. Por supuesto, no se debe olvidar la posibilidad de una HSC en los lactantes masculinos con colapso circulatorio en las primeras semanas de vida. Los lactantes afectados, además de requerir urgentemente una asignación correcta de su sexo, se tratan con cortisol de sustitución, junto con fludrocortisona si tienen la forma pierde sal. Las mujeres virilizadas pueden requerir cirugía plástica a su debido tiempo. La sustitución con esteroides es para toda la vida y necesita aumentarse durante las enfermedades intercurrentes o en los momentos de estrés, como la cirugía. La monarquía en las muchachas con HSC pierde sal es tardía, en ellas la menstruación se muestra irregular y son subfértiles. Síndrome de insensibilidad a los andrógenos.- los individuos con el síndrome de insensibilidad a los andrógenos (SIA) tienen genitales externos femeninos y experimentan desarrollo mamario en la pubertad. Se presentan con amenorrea primaria, debida a la falta de aparición de los períodos menstruales, o con una hernia inguinal que contiene una gónada que resulta ser un testículo. La hernia inguinal es infrecuente en las muchachas y, si está presente, especialmente de forma bilateral, se debe considerar la posibilidad de un SIA. A menudo hay escasez de vello sexual secundario y la investigación de los genitales internos revela la ausencia de útero y de las trompas de Falopio, con una vagina con fondo ciego. El análisis cromosómico revela el cariotipo masculino normal, 46,XY. La producción de andrógenos por los testículos es normal en los individuos afectados, pero los andrógenos no se captan normalmente porque tiene un receptor anómalo para ellos; hay una mutación en el gen receptor de los andrógenos en el cromosoma X. Esto puede comprobarse funcionalmente en los 67 fibroblastos cutáneos. Algunos individuos presentan insensibilidad incompleta o parcial a los andrógenos y experimentan una virilización variable. Los individuos afectados tienen generalmente una orientación sexual femenina pero, evidentemente, serán estériles. Es necesario eliminar los testículos, por el aumento del riesgo de desarrollar una neoplasia testicular, y hay que administrar estrógenos para estimular el desarrollo de los caracteres sexuales secundarios y para evitar la osteoporosis a largo plazo. o Trastorno de almacenamiento en los lisosomas.- además de los errores congénitos del metabolismo, en los que un defecto enzimático ocasiona la deficiencia de un metabolito esencial y la acumulación de precursores metabólicos intermediarios, hay ciertos trastornos en los que la deficiencia de una enzima lisosómica implicada en la degradación de macromoléculas complejas origina su acumulación. Esta acumulación se produce porque las macromoléculas están normalmente en un estado de flujo constante, con un delicado equilibrio entre sus tasas de síntesis y de eliminación. Los niños nacidos con enfermedades por almacenamiento en los lisosomas son habitualmente normales en el nacimiento, pero con el paso del tiempo aparece un curso evolutivo en descenso de diferente duración debido a la acumulación de uno o más tipos diversos de macromoléculas. o MUCOPOLISAARIDOSIS.los niños con alguna de las mucopolisacaridosis (MPS) presentan signos esqueléticos, vasculares o del sistema nervioso central junto con tosquedad de los rasgos faciales. Estas características se deben a la acumulación progresiva de los policacáridos sulfatados (también conocidos como glucosaminoglucanos), causada por una degradación defectuosa de la cadena lateral de hidratos de carbono de los mucopolisacáridos ácidos. Se conocen seis distintas MPS, basadas en sus diferencias clínicas y genéticas. Cada tipo específico de MPS tiene un patrón característico de excreción de orina de los glucosaminoglucanos dermatán, heparán, keratán, y coidritín sulfato. La investigación bioquímica posterior ha revelado que los diversos tipos de deben a deficiencias de distintas enzimas individuales. Todos ellos, a excepción del síndrome de Hunter, que está ligado al cromosoma X, se heredan como trastornos autosómicos recesivos. Síndrome de Hurler (MPS-I).- es la más grave de las MPS. Los lactantes afectados presentan en su primer año la córnea nublada, una curva característica de la columna lumbar y un mal crecimiento posterior. En el segundo año desarrollan pérdida de la audición, tosquedad de los rasgos faciales, hepatoesplenomegalia, rigidez articular y cambios vertebrales. Estas características siguen evolucionando junto con un deterioro mental y, finalmente, la muerte en la mitad de la adolescencia por una combinación de insuficiencia cardíaca e 68 infecciones respiratorias. El diagnóstico de síndrome de Hurler se estableció inicialmente al demostrar la presencia de gránulos metacromáticos en las células, es decir, lisosomas distendidos por el material de almacenamiento, compuesto principalmente por dermatán sulfato. La excreción urinaria aumentada de dermatán y de heparán sulfato se utiliza habitualmente como prueba de detección, pero la confirmación del diagnóstico implica la demostración de una actividad reducida de la hidrolasa lisosómica, alfa-Liduronidasa. Las formas alélicas menos graves del síndrome de Hurler, causadas por grados variables de actividad residual de la alfa-l-iduronidasa, se habían clasificado previamente de forma separada como enfermedad de Scheine (MPS-I) y enfermedad de Hurler/Scheine (MPS-I H/S). Síndrome de Hunter (MPS-II).- los hombres con síndrome de Hunter presentan habitualmente entre los 2 y los 5 años de edad una pérdida de audición, antecedentes de infecciones recurrentes, diarrea y retraso en el crecimiento. El examen revela una tosquedad característica de los rasgos faciales, junto con hepatoesplenomegalia y rigidez articular. La radiografía de la columna revela anomalías en la forma de las vértebras. Hay un deterioro progresivo tanto físico como mental, y habitualmente la muerte se produce en la adolescencia. El diagnóstico se confirma por la presencia de cantidades excesivas de dermatán y heparán sulfato de la orina y por una actividad deficiente o disminuida de la enzima iduronato sulfato sulfatasa en el suero o en los leucocitos. Síndrome de Sanfilippo (MPS-III).- es la MPS más habitual. Los individuos afectados presentan en su segundo año de vida una tosquedad leve de los rasgos, cambios esqueléticos y un deterioro intelectual progresivo con problemas conductuales, continuándose con convulsiones y terminando con la muerte al iniciarse el estado adulto. El diagnóstico se confirma por la presencia del aumento de excreción urinaria de heparán y condroitin sulfato y la deficiencia de una de las cuatro enzimas implicadas en la degradación del heparán sulfato y la deficiencia de una de las cuatro enzimas implicadas en la degradación del heparán sulfato: sulfaminidasa (MPS-IIIA), N-acetil-alfa-Dglucosaminidasa (MPS-IIIB), acetil-CoA-alfa-glucosaminidasaN-acetiltransferasa (MPSIIIC) o N-acetil-glucosamina-6-sulfato sulfatasa (MPSIIID). Los individuos con estas diferentes deficiencias enzimáticas no pueden distinguirse clínicamente. Síndrome de Morquio (MPS-IV).- los niños afectados con este síndrome presentan en el segundo o tercer año de vida anomalías esqueléticas que incluyen estatura corta, deformidad torácica y curvatura de la columna (cifoscoliosis). La inteligencia es normal y la supervivencia es a largo plazo, 69 aunque hay un riesgo de compresión de la médula espinal debido a la evolución de la afectación esquelética. El diagnóstico se confirma por la presencia de keratán sulfato en la orina y la deficiencia de galactosamina-6-sulfatasa (MPS-IV A), o de beta-galactosidasa (MPS-IV B). Síndrome de Maroteaux-Lamy (MPS-VI).- presentan características tipo Hurler al principio de su infancia. Consisten en rasgos faciales toscos, corta estatura con deformidad torácica, cifosis y restricción de la movilidad articular. Además, desarrollan opacidad corneal y anomalías valvulares cardíacas, aunque conservan una inteligencia normal. Una forma menos grave aparece posteriormente con supervivencia hasta bien avanzada la vida de adulto, en contraste con la forma más grave en la que la supervivencia llega generalmente sólo hasta la tercera década. El diagnóstico se confirma por la presencia de la excreción urinaria aumentada de dermatán sulfato y la deficiencia de arilsulfatasa B en los leucocitos o en los fibroblastos. Síndrome de Sly (MPS-VII).- puede presentarse con grados de gravedad muy variables, que van desde características esqueléticas que incluyen cifoscoliosis leve y displasia de la cadera, hasta rasgos faciales toscos, hepatoesplenomegalia, opacidad corneal, anomalías cardíacas y retraso mental, con muerte en la infancia o en la adolescencia. La excreción aumentada de glucosaminoglucanos urinarios y la deficiencia de beta-glucoronidasa en el suero, en los leucocitos o en los fibroblastos confirman el diagnóstico. TRATAMIENTO DE LAS MUCOPOLISACARIDOSIS.- se ha probado el tratamiento de las MPS mediante la sustitución enzimática, pero diversas dificultades prácticas han impedido su éxito. Sin embargo, se ha intentado, más recientemente, el tratamiento con trasplantes de médula ósea, con notificaciones de éxito variable, tanto bioquímica como clínicamente, en las características esqueléticas y cerebrales. ESFINGOLIPIDOSIS (ENFERMEDADES POR ALMACENAMIENTO DE LÍPIDOS.- en las esfingolipidosis hay una incapacidad para degradar los esfingolípidos, lo que ocasiona un depósito progresivo de lípidos o de glucolípidos, sobre todo en el encéfalo, el hígado y el bazo. La afectación del sistema nervioso central da como resultado un deterioro mental progresivo, a menudo con convulsiones, que habitualmente conduce a la muerte en la infancia. Hay al menos 10 tipos diferentes, con deficiencias enzimáticas específicas; las más habituales son las enfermedades de Tay-Sachs, de Gaucher y de Niemann-Pick. o Enfermedad de Tay-Sachs.- los lactantes afectados presentan habitualmente a los 6 meses de edad mala 70 alimentación, letargo, y flojedad. En la segunda mitad del primer año suele hacerse evidente la dificultad para alcanzar las etapas de desarrollo o la involución en él. La alimentación se vuelve cada vez más difícil y el lactante se deteriora progresivamente, con sordera, alteración visual y espasticidad, que evoluciona hasta rigidez. La muerte se produce habitualmente a los 3 años de edad como resultado de una infección respiratoria. También se han notificado formas menos graves juveniles, adultas y crónicas. El diagnóstico de enfermedad de Tay-Sachs se consolida clínicamente por la presencia de una mancha “rojo cereza” en el centro de la mácula del fondo de ojo. La confirmación bioquímica de la enfermedad de Tay-Sachs se hace por la demostración de valores reducidos de hexosaminidasa A en el suero, en los leucocitos o en los fibroblastos cultivados. o Enfermedad de Gaucher.- es la esfingolipidosis más habitual; al igual que la enfermedad de Tay-Sachs, se presenta con mayor frecuencia entre personas con ascendencia judía asquenazi. Hay dos tipos principales, basados en la edad de aparición. En el tipo I, o tipo adulto, es la forma más habitual de la enfermedad de Gaucher; las personas afectadas presentan episodios febriles y dolor en los miembros, las articulaciones o el tronco, con tendencia a presentar fracturas patológicas. El examen clínico revela habitualmente una hepatoesplenomegalia. Las personas afectadas a menudo muestran una anemia leve y cambios radiográficos en los cuerpos vertebrales y en la parte proximal de los fémures. El sistema nervioso central no está afectado. En el tipo II, o enfermedad de Gaucher infantil, una característica principal es la afectación del sistema nervioso central. Los lactantes afectados presentan, entre los 3 y 6 meses de edad, un deterioro progresivo y una hepatoesplenomegalia. A los 6 meses de edad comienzan a mostrar un retroceso en el desarrollo y un deterioro neurológico, con espasticidad y convulsiones, que se continúan con la aparición de infecciones pulmonares recurrentes y con la muerte en el segundo año. El diagnóstico de enfermedad de Gaucher se confirma por la actividad reducida de la enzima glucosiceramida beta-glucosidasa en los leucocitos o en los fibroblastos cultivados. El tratamiento consiste en el alivio sintomático del dolor. Además, a menudo es necesario eliminar el bazo hipertrofiado porque ocasiona una anemia secundaria debida al secuestro prematuro de los eritrocitos, trastorno conocido como hiperesplenismo. o Enfermedad de Niemann-Pick.- los lactantes con esta enfermedad presentan deterioro progresivo y hepatomegalia, y en su mácula puede detectarse una mancha rojo cereza. La involución en el desarrollo evoluciona rápidamente hacia el 71 final del primer año, y la muerte sobreviene a los 4 años. Un signo característico es la presencia de lo que se denominan células en espuma en la médula ósea debido a la acumulación de esfingomielina. La confirmación del diagnóstico se hace por demostración de la deficiencia de la enzima esfingomielinasa. Se ha notificado una forma menos grave sin afectación neurológica. o Trastorno del metabolismo de la purina/pirimidina.- la sensibilidad a la palpación, la inflamación y el dolor articular son el resultado de la respuesta inflamatoria del organismo antes los depósitos de cristal de una sal del ácido úrico. De hecho, sólo a una minoría de las personas que presentan gota se les ha detectado un error congénito del metabolismo. En la mayoría de los casos la causa se produce por una combinación de factores genéticos y ambientales; sin embargo, hay que considerar siempre a los trastornos que pueden producir un aumento del recambio de las purinas (p. ej., una neoplasia como la leucemia) o una secreción reducida de los metabolitos (p. ej., alteración renal) como posibles causas originarias subyacentes. Síndrome de Lesch-Nyhan.- un trastorno particularmente incapacitante del metabolismo de las purinas es el síndrome de Lesch-Nyhan. Este trastorno ligado al cromosoma X se debe a la deficiencia de la enzima hipoxantina guanina fosforribosilpiroferasa, que ocasiona valores aumentados de fosforribosilpirofosfato. Este último es normalmente un compuesto químico limitador de la producción de la síntesis de las purinas. Su presencia en exceso produce una tasa aumentada de la síntesis de las purinas y da lugar a la acumulación de cantidades excesivas de ácido úrico y de algunos de sus precursores metabólicos. El principal efecto lo ejerce sobre el sistema nervioso central, y se manifiesta por movimientos incontrolados, espasticidad, retraso mental y automutilación compulsiva. Aunque los fármacos como el alopurinol, que inhibe la formación de ácido úrico, pueden reducir las concentraciones de este ácido, ninguno ofrece un tratamiento satisfactorio para los efectos debilitantes sobre el sistema nervioso central. Enfermedades por inmunodeficiencia causadas por defectos en el metabolismo de las purinas.- dos trastornos de inmunodeficiencia heredados son, sorprendentemente, errores congénitos del metabolismo de las purinas. Deficiencia de la adenosina desaminasa.- cerca de la mitad de todos los niños con la forma autosómica recesiva de la inmunodeficiencia combinada grave con función alterada de los linfocitos B y T tiene una deficiencia de la enzima adenosina desaminasa. Los 72 niños afectados presentan en su primer año de vida infecciones víricas y bacterianas recurrentes y, si no se tratan, morirán por una infección muy grave en el primer año. El diagnóstico se confirma por una actividad deficiente de la adenosina desaminasa de los eritrocitos. Se ha notificado la corrección de la inmunodeficiencia por transfusión de eritrocitos irradiados. Recientemente se ha llevado a cabo el transplante con éxito de médula ósea -incluso de forma antenatal in útero-. o Deficiencia de la purina nucleósido fosforilasa.se ha demostrado que una parte de los niños susceptibles a las infecciones víricas graves, recurrentes y potencialmente fatales con una alteración aislada de la función de los linfocitos T tienen una deficiencia de la enzima purina nucleósido fosforilasa. Se ha notificado que el tratamiento con eritrocitos irradiados produce una mejoría temporal de la función inmunitaria. Aciduria órotica hereditaria.- los niños con aciduria orótica hereditaria presentan en el primer año de vida anemia megaloblástica, deterioro progresivo y retraso en el desarrollo. Tiene deficiencia de una de estas dos enzimas, orotato fosforribosiltransferasa u orotidina 5´-fosfato descarboxilasa, ambas necesarias para la síntesis de las pirimidinas, lo que se traduce en la excreción de grandes cantidades de ácido orótico en la orina. Se ha notificado que el tratamiento con la pirimidina uridina reduce la excreción de ácido urinario, corrige la anemia y restaura el crecimiento. Trastorno del metabolismo de las porfirinas.- varios trastornos del metabolismo de las porfirinas se deben a una deficiencia de las enzimas en la ruta biosintética del grupo que contiene el hierro en la hemoglobina, el heme. Todas ellas se heredan como trastornos autosómicos dominantes, a excepción de la porfiria eritropoyética congénita, que es un trastorno autosómico recesivo. Esto ocurre porque las enzimas limitadoras de la producción, de forma que las haploinsuficiencia da como resultado la enfermedad clínica. Porfirinas hepáticas. Porfiria intermitente aguda.- (PIA) se caracteriza por ataques de dolor abdominal, debilidad, vómitos y alteraciones mentales en forma de confusión, disgustos emocionales o alucinaciones. Incluso puede ocurrir un coma, y las mujeres se ven más gravemente afectadas que los hombres, con síntomas que a veces se asocian con el ciclo menstrual. Los ataques se pueden desencadenar también por la administración de ciertos 73 fármacos, como los esteroides exógenos, los anticonvulsivantes y los barbitúricos. Se debe a una deficiencia parcial de la enzima uroporfirinógeno I sintasa, que produce un aumento de la excreción de los precursores de las porfirias porbofilinógeno y ácido delta-aminolevulínico en la orina. Coproporfiria hereditaria.- trastorno relacionado heredado también como un rasgo dominante, hay una deficiencia parcial de la enzima coproporfirimógeno oxidasa. El trastonro es clínicamente indistinguible de la porfiria intermitente aguda, aunque aproximadamente un tercio de las personas afectadas tienen también fotosensibilidad cutánea. Porfiria variegata.- tiene un prevalencia particular en los descendientes de los afrikáner de Sudáfrica, con signos neurológicos y viscerales que también pueden desencadenarse por fármacos. Se puede demostrar el aumento de la excreción fecal de los precursores de la porfiria, protoporfiria y coproporfiria, y se ha observado que el trastorno se debe a la deficiencia de la enzima protoporfirinógeno oxidasa. Porfirinas eritropoyéticas. Porfiria eritropoyética congénita.- (PEC) la principal característica es una fotosensibilidad extrema con formación de ampollas en la piel, que dan como resultado una cicatrización amplia, hasta el punto de que la mayoría de las personas afectadas son incapaces de salir al exterior en un día de sol normal. Además, muchas tienen una anemia hemolítica que requiere transfusiones sanguíneas regulares y una esplenectomía frecuente. Los individuos afectados tienen una decoloración rojo-marrón de los dientes, que muestran fluorescencia roja bajo la luz ultravioleta. La porfiria eritropoyética congénita se debe a la deficiencia de la enzima uroporfirinógeno III sintasa. Protoporfiria eritropoyética.- (PPE) de debe a la deficiencia de la enzima ferroquelatasa, responsable de la inserción del ion ferroso en le precursor de la profirina para formar el heme. Las personas afectadas tienen fotosensibilidad y pueden desarrollar a veces una enfermedad hepática crónica. Se ha notificado el tratamiento con éxito de la fotosensibilidad con betacaroteno. 74 o Trastorno de los ácidos orgánicos.- los niños afectados con uno de los trastornos de los ácidos orgánicos presentan episodios periódicos de mala alimentación, vómitos y letargo junto con una acidosis metabólica grave, valores bajos de leucocitos (neutropenia) y de plaquetas (trombocitopenia), valores bajos de azúcar en sangre (hipoglucemia) y altos de amoníaco sanguíneo (hiperamoniemia). Estos episodios a menudo se desencadenan por una enfermedad intercurrente o por un aumento de la ingesta de proteínas y, después de un episodio semejante, los niños afectados pueden perder su capacidad para el desarrollo. El análisis de sangre de los niños en el momento de estos episodios revela valores elevados de glicina (hiperglicinemia). Más tarde se vio que la acidosis en estos episodios se debía a los valores elevados de los ácidos orgánicos, el ácido propiónico o el ácido metilmalónico. Los dos trastornos autosómicos recesivos de los ácidos orgánicos, la academia metilmalónica (AMM) y la academia propiónica (APP), se deben a una deficiencia de las enzimas metilmalonilCoA mutasa y propionil-CoA carboxilasa, respectivamente. La deficiencia enzimática causa la acumulación de los metabolitos tóxicos de los ácidos orgánicos obtenidos por la desaminación de ciertos aminoácidos, de ácidos grasos específicos de cadena larga y de cadenas laterales de colesterol. La terapia para los episodios agudos consistente en el tratamiento de cualquier infección, la sustitución de líquidos, la corrección de la acidosis metabólica y la supresión de la ingesta proteica. El tratamiento profiláctico a largo plazo implica la restricción de la ingesta proteica y el reconocimiento y el tratamiento rápido de cualquier enfermedad intercurrente. Una parte de los individuos afectados con APP son sensibles a la biotina , mientras que algunas personas con AMM lo son a la vitamina B12. o Trastornos del metabolismo del cobre. Enfermedad de Menkes.- es un trastorno ligado al cromosoma X en el que los hombres afectados presentan a los pocos meses de vida dificultades alimentarias, vómitos y poca ganancia de peso. Se continúa con la flojedad correspondiente (hipotonía), convulsiones, y deterioro neurológico progresivo, y la muerte, debida a infecciones respiratorias recurrentes, usualmente a los 3 años. Una característica particular es el pelo, al que le falta pigmento, es ensortijado y quebradizo y se rompe fácilmente. Se observó que se parecía a la lana de la oveja con deficiencia de cobre. Los valores de cobre sérico y de ceruloplasmina son muy bajos. La clonación del gen de la enfermedad de Menkes fue facilitada por una mujer afectada con una translocación autosómica ligada cromosoma X, y reveló que codificada una proteína ATPasa de transporte de cationes de cobre. Los regímenes de tratamiento diferentes fuentes de cobre exógeno han tenido un beneficio limitado hasta la fecha. 75 o Enfermedad de Wilson.- trastorno autosómico recesivo, que se presenta en la infancia o en los primeros años de la adolescencia con convulsiones y signos neurológicos anómalos, que pueden consistir en deterioro de la coordinación, movimientos involuntarios, tono anómalo, disartria (dificultad para hablar), disfagia (dificultad para deglutir) y cambios en la conducta o alteraciones psiquiátricas evidentes. El examen clínico revela la presencia del denominado anillo de Kaiser- Fleischer, un collarete de color marrón dorado o verdoso en el margen corneal. La investigación puede poner de manifiesto la presencia de una función hepática anómala que puede evolucionar hasta una cirrosis. Los altos valores de cobre en el hígado, las concentraciones séricas de ceruloplasmina reducidas, la proteína transportadora de cobre y los resultados anómalos de las pruebas de sobrecarga con cobre son indicativos del diagnóstico. Hay notificaciones llamativas de una mejoría sorprendente de las características neurológicas en las personas con enfermedad de Wilson con el empleo de los agentes quelantes D-penicilamina y trientina, aunque pueden causar efectos secundarios. Trastornos de los peroxisomas.- los peroxisomas son orgánulos subcelulares unidos por una única membrana lipídica de tres capas presente en todas las células; son especialmente abundantes en las células de los parénquimas renal y hepático. La matriz de los orgánulos contiene más de 40 enzimas que realizan diversas reacciones implicadas en la oxidación de los ácidos grasos y en la biosíntesis del colesterol y que interactúan con las rutas metabólicas externas a los peroxisomas. Las enzimas de la matriz de los peroxisomas se sintetizan en los polirribosomas, penetran en el citosol y se transfieren a los peroxisomas. Hay dos tipos de trastornos de los peroxisomas: trastornos de la biogénesis de los peroxisomas, como el síndrome de Zellweger, en el que hay cantidades gravemente reducidas de peroxisomas en todas las células, y deficiencias de enzimas peroxisómicas aisladas, como la adrenoleucodistrofia ligada al cromosoma X. Síndrome de Zellweger.- los lactantes con este síndrome presentan hipotonía y debilidad y tienen características faciales levemente dismórficas, consistentes en frente prominente y una gran fontanela anterior (“mancha blanda”). Pueden tener también cataratas y hepatomegalia. Habitualmente continúan con convulsiones e involución en el desarrolloy suelen morir al año de edad. Las investigaciones pueden poner de manifiesto quistes renales y calcificaciones anómalas en los cartílagos de crecimiento de los huesos largos. El diagnóstico puede confirmarse por los valores elevados en plasma de ácidos grasos de cadena larga. El gen 76 para el sd. De Zellweger codifica una proteína implicada en el ensamblaje de los peroxisomas. Adrenoleucodistrofia.- los hombres con adrenoleucodistria (ALD), trastorno ligado a X, presentan clásicamente al final de la infancia un deterioro del rendimiento escolar, aunque los individuos afectados pueden presentarse a cualquier edad e incluso, en ocasiones, ser asintomáticos. Una parte de los hombres afectados puede aparecer en la vida adulta con características neurológicas menos graves e insuficiencia suprarrenal, lo que se denomina adrenomieloneuropatía. Se ha demostrado que la ALD se asocia con una deficiencia de la enzima de ácidos grasos de cadena muy larga CoA sintasa, pero es secundaria a una deficiencia de la proteína de la membrana peroxisómica. El tratamiento de la ALD consiste en una dieta que como fuente de grasas utiliza un aceite con valores bajos de los ácidos grasos de cadena muy larga. Se ha denominado popularmente “el aceite de Lorenzo”. Sin embargo, la eficacia de esta dieta ha sido decepcionante. 4.- Relacionar las manifestaciones agudas y crónicas de los trastornos metabólicos con la posible alteración metabólica correspondiente: presentación aguda neonatal de tipo intoxicación metabólica, déficit energético, deterioro neurológico agudo presentación pediátrica o tardía con manifestaciones hepáticas, cardíacas, digestivas, presentación crónica y lentamente progresiva o estática. Respiratorios: dificultad para respirar (disnea). Neurológicos: hipotonía (hiperlacticidemia, trastornos de la cadena respiratoria mitocondrial, trastornos del ciclo de la urea), succión débil, rechazo del alimento,. Bradicardia, apnea, hipotermia y coma. Intoxicación alimentaria. Déficit enérgico: hiperglucemia, hipoglucemia, hiperlacticidemia con/sin acidosis metabólica. Hepatoesplenomegalia. Ictericia aguda neonatal: se da generalmente por acumulación de partículas pequeñas, hay letargia, coma y deterioro súbito. Tardía: pasa por un periodo asintomático, hay retraso del desarrollo, infecciones virales, fiebre y estreñimiento. manifestaciones hepáticas: hepatomegalia, hipoglucemia, hiperinsulinemia. Manifestaciones muscular. cardiacas: I.C., cardiomiopatía, hipotonía, debilidad 77 Digestivas: anorexia, vómito crónico y diarrea -- malabsorción, deficiencia de glucosa, deficiencia de lactasa, def. de sacarasa. Neurológicas: deterioro progresivo, se asocia a otros datos. 5.- Analizar la selección de los métodos diagnósticos para detectar los errores innatos del metabolismo. en la mayoría de los errores congénitos del metabolismo en los que pueda identificarse un producto génico deficiente o anómalo puede establecerse el diagnóstico prenatal. Puede efectuarse un análisis bioquímico de los amniocitos cultivados obtenidos por la amniocentesis en el segundo trimestre, pero esta prueba ha cedido abiertamente el paso a otras pruebas más precoces usando las vellosidades coriónicas (VC) directas o cultivada, que permiten establecer un diagnóstico a las 12-14 semanas de gestación. Se realizan dos tipos de pruebas: cualitativas (orina) y cuantitativas (sangre). Olor a ratón mojado: fenilcetonuria. Olor a jarabe de Maple (maggi): deshidrogenasa de cetoacidos alfa. Olor a pies sudorosos: aciduria isovalérica. PRUEBAS DE CAMBIOS DE COLOR: Cloruro férrico: fenilalanina, tirosiuna e histidina. 2,4 dinitrofenilhidracina. Nitroso naftol: tirosina. Obermeyer: triptófano Millon: tirosina Antrona: azúcares Benedict: azúcares reductores p-nitroanilina: ácido metilmalónico. Nitrato de plata: ácido homogentisico. suero Orina Gases arteriales Glucosa Anion gap Proteínas Glucosa Cetonas Amonio ácido úrico y ácido orotico 78 ácido láctico Sustancias reductoras Piruvato Sulfatos ácido úrico nitratos Transaminasas CK 6.- Fundamentar la aplicación de los algoritmos diagnósticos en los errores innatos del metabolismo: hiperamonemia, acidosis metabólica e hiperlactacidemia. HIPERAMONEMIA.- concentraciones elevadas de amonio en sangre, las manifestaciones clínicas son neurológicas: irritabilidad, desorientación, hipotonía alternando con hipertonía, ataxia, convulsiones, vómitos, coma y muerte. ACIDOSIS METABÓLICA.- es debida al hidrógeno que supera las posibilidades de excreción por el organismo que produce una retirada de bicarbonato de los líquidos. La acidosis metabólica se produce por el aumento marcado en la producción endógena de de ácidos como ocurre en la cetoácidosis o en la acidosis láctica, por la pérdida de los depósitos de bicarbonato como ocurre en la diarreas o por la acumulación progresiva de ácidosis endógena. HIPERLACTADEMIA.- es una afección potencialmente mortal causada por el exceso de lactato en la sangre y un Ph sanguíneo bajo. El pH bajo significa que hay demasiado ácido en la sangre, lo que es perjudicial para las células. Se produce cuando hay un fallo en la función de las mitocondrias. 7.- Evaluar características clínicas, genéticas, factores de riesgo y aspectos epidemiológicos para llegar al diagnóstico y fundamentar las opciones terapéuticas de los errores innatos del metabolismo mas frecuentes: FENILCETONURIA.o Cuadro clínico.- retraso mental, piel clara, eccema, epilepsia, artritis. o Base genética.- deficiencia de fenilalanina hidroxilasa. o Patrón hereditario.- autosómica recesiva. ENFERMEDAD DE LA ORINA DE JARABE DE ARCE.o Cuadro clínico.- retraso mental, orina con olor a arce. o Base genética.- deficiencia de descarboxilasa de los beta-cetóacidos ramificados. o Patrón hereditario.- autosómica recesiva. 79 CATABOLISMO DE LA LEUCINA.o Cuadro clínico.- o Base genética.- o Patrón hereditario.- HOMOCISTINURIA.o Cuadro clínico.- retraso mental, luxación del cristalino, trombosis, anomalías esqueléticas. o Base genética.- deficiencia de cistationina beta-sintasa. o Patrón hereditario.- autosómico recesivo. DEFICIENCIA DE ORNITIN TRANSCARBAMILASA.o Cuadro clínico.- hiperemia, muerte al principio de la lactancia. o Base genética.- deficiencia de la ornitina transacarbamilasa. o Patrón hereditario.- dominante ligado al cromosoma X. GALACTOSEMIA.o Cuadro clínico.- cataratas, retraso mental, cirrosis. o Base genética.- deficiencia de galactosa 1-fosfato uridil transferasa. o Patrón hereditario.- autosómica recesiva. MUCOPOLISACARIDOSIS.o Cuadro clínico.retraso mental, hepatoesplenomegalia, opacidad corneal. anomalías esqueléticas, o Base genética.- deficiencia de almacenamiento de lisosomas. o Patrón hereditario.- autosómico recesivo, ligado a cromosoma X. GLUCOGENOSIS.o Cuadro clínico.- hepatomegalia, hipoglicemia, deterioro progresivo. o Base genética.- trastorno por almacenamiento del glucógeno. o Patrón hereditario.- autosómico recesivo, ligado al cromosoma X. ENFERMEDAD DE GAUCHER.o Cuadro clínico.- tipo I: dolores articulares y en los miembros, esplenomegalia. o Cuadro clínico.- tipo II: espasticidad, convulsiones, muerte. 80 o Base genética.- tipo I: deficiencia de glucosilceramida. o Base genética.- tipo II: deficiencia de beta-glucosidasa. o Patrón hereditario.- autosómico recesivo. 8.- Fundamentar el riesgo de recurrencia en los principales errores innatos del metabolismo. La mayoría de los errores innatos del metabolismo son enfermedades autosómicas recesivas exceptuando el síndrome de hunter, deficiencia de OTC y enfermedad de menkes que están ligados al cromosoma X con carácter recesivo. 81 Autor: José Regino Ríos Gutiérrez. Unidad No. 13 Trastornos de la diferenciación sexual Competencia intermedia.- clasificar y analizar los trastorno de la diferenciación sexual más frecuentes, seleccionando e interpretando el cariotipo y pruebas moleculares para hacer el diagnóstico que determine su manejo, pronóstico y asesoramiento genético. Criterios de desempeño.1.- Analizar el proceso de la diferenciación sexual definiendo los conceptos de sexo cromosómico, genético, gonadal, hormonal, fenotípico y de asignación. SEXO CROMOSÓMICO.- Masculino XY, Mujer XX SEXO GENÉTICO.Consiste en la identificación de los cromosomas sexuales (par 23). El par 23 en la forma XX corresponde a una mujer, mientras que en la forma XY define al hombre. Algunos autores lo denominan carácter sexual primario. SEXO GONADAL.- Masculino: Pene, escroto, testículos. Mujer: Labios mayores, labio menores, vagina, útero, ovarios. SEXO HORMONAL.- es la diferenciación que la acción de las hormonas hacen en el individuo. (depende de la acción del tejido gonadal presente en el paciente). Cuando el embrión tiene los cromosomas femeninos XX, las hormonas maternas llamadas estrógenos estimulan el desarrollo de órganos sexuales femeninos en dicho embrión. Pero si el par de cromosomas sexuales es XY, a los dos meses de embarazo comenzará la producción de testosterona, hormona masculina que dará lugar a la formación de los genitales masculinos. SEXO FENOTÍPICO.- Es la clasificación del sexo basada en las características físicas de cada individuo, que incluyen la distribución de la vellosidad, la contextura y tamaño de la musculatura, la forma de los huesos de la pelvis y la distribución de la grasa corporal, entre otros y que en su conjunto se denominan caracteres sexuales secundarios. SEXO DE ASIGNACIÓN.- es la asignación que se hace después de estudios genéticos y cromosómicos a un lactante con genitales ambiguos del sexo con el que se le ha de criar. Es la clasificación del sexo de la persona según el papel que el individuo desempeña en el proceso de reproducción o del coito. El sexo psicológico no depende de los gustos o preferencias de la persona en otras áreas del comportamiento social, como el jugar con carros o muñecas, sino que esta confinado únicamente al área reproductiva o coital. 2.- Analizar las anormalidades en la diferenciación sexual, mediante el estudio clínico, bioquímico, imagenológico, citogenético y molecular. Análisis de cromosomas Niveles hormonales (por ejemplo, nivel de testosterona) Pruebas de estimulación hormonal Pruebas de electrolitos 82 Pruebas moleculares específicas Examen endoscópico (para verificar la ausencia o presencia de la vagina o el cuello uterino) Ultrasonido o IRM para evaluar si los órganos sexuales internos están presentes (por ejemplo, el útero) 3.- Evaluar las manifestaciones clínicas para llegar al diagnóstico de las siguientes alteraciones: Pseudohermafroditismo femenino (hiperplasia congénita por deficiencia de la 21-hidroxilasa).- suprarrenal o Cuadro clínico.- también se le conoce como Síndrome adrenogenital. Se considera este problema en cualquier niña recién nacida que presente virilización de los genitales externos (Es la causa más habitual de genitales ambiguos en recién nacidos femeninos). Cerca de la cuarta parte de los lactantes afectados tienen la forma perdedora de sal, y en la segunda o tercera semana de vida presentan colapso circulatorio, hiponatremia e hiperpotasemia. o Base genética.- trastorno del metabolismo de los esteroides, dando como resultado una deficiencia de la 21-hidroxilasa. o Patrón hereditario.- autosómico recesivo. Pseudohermafroditismo reductasa).- masculino (deficiencia de 5 alfa- o Cuadro clínico.- inframasculinización de los genitales externos. o Base genética.- errores innatos del metabolismo de los esteroides (de las rutas biosintéticas del cortisol). Deficiencia de la 5 alfareductasa. o Patrón hereditario.- autosómico recesivo. Insensibilidad a los andrógenos.o Cuadro clínico.tienen genitales externos femeninos y experimentan desarrollo mamario en la pubertad. Presentan amenorrea primaria, debida a la falta de aparición de los períodos menstruales, o con una hernia inguinal que contiene una gónada que resulta ser testículo. A menudo hay escasez de vello sexual secundario y la investigación de los genitales internos revela ausencia de útero y trompas de Falopio, una vagina con fondo ciego. El análisis cromosómico revela el cariotipo masculino normal, 46,XY. o Base genética.- la producción de andrógenos por los testículos es normal en los individuos afectados, pero los andrógenos no se captan normalmente porque tienen un receptor anómalo en ellos; hay una mutación en el gen receptor de los andrógenos en el cromosoma X. o Patrón hereditario.- autosómico recesivo. 83 Hermafroditismo verdadero.o Cuadro clínico.- el individuo tiene a la vez tejido testicular y tejido ovárico, lo que se asocia a menudo con genitales ambiguos. Cuando se realiza una cirugía exploratoria en estos pacientes es posible encontrar un testículo de un lado y un ovario del otro. Alternativamente puede haber una mezcla de tejido ovárico y testicular en la gónada que se denomina ovotestículo. o Base genética.- la mayoría de los pacientes con hermafroditismo verdadero tiene un cariotipo 46,XX, y en muchos de ellos el cromosoma X derivado del padre es portador de secuencias de DNA específicas del cromosoma Y, resultantes de un entrecruzamiento erróneo entre los cromosomas X e Y durante la meiosis I en la espermatogénesis. o Patrón hereditario.- autosómico recesivo. Varón XX.o Cuadro clínico.- testículos pequeños, ginecomastia, azoospermia, características fenotípicas femeninas. Se le llama también síndrome de la Chapelle. o Base genética.- translocación de brazo corto de Y al brazo corto de uno de los cromosomas X. o Patrón hereditario.- Disgenesias gonadales puras.o Cuadro clínico.- son las malformaciones de las gónadas masculinas o femeninas dando lugar a discrepancia entre los genitales internos y externos. o Base genética.- la persona puede tener tanto tejido ovárico como testicular (ovotestículo) o puede tener un ovario y un testículo. o Patrón hereditario.- 46, XY.o Cuadro clínico.- La persona tiene los cromosomas de un hombre, pero los genitales externos son ambiguos o claramente femeninos. Internamente los testículos pueden ser normales, estar malformados o ausentes. o Base genética.- problemas con los testículos, problemas con la testosterona, deficiencia de la 5-alfa-reductasa, insensibilidad a los andrógenos. o Patrón hereditario.- 84 46, XX.o Cuadro clínico.- La persona tiene los cromosomas de una mujer, los ovarios de una mujer, pero los genitales externos con apariencia de masculinos. Esto es consecuencia en un feto femenino que ha estado expuesto a hormonas masculinas en exceso antes del nacimiento. Los labios mayores se fusionan u el clítoris se agranda para parecer como un pene. Esta persona tiene un útero y trompas de Falopio normales. o Base genética.- hiperplasia suprarrenal congénita, hormonas masculinas consumidas por la madre durante el embarazo, tumores productores de hormonas masculinas en la madre, deficiencia de aromatasa. o Patrón hereditario.- 4.- Establecer problemas. el manejo multidisciplinario y pronóstico de estos Lo ideal es que un equipo de profesionales médicos con experiencia en intersexualidad deba trabajar en conjunto para entender y tratar el niño con intersexualidad, así como comprender, aconsejar y apoyar a toda su familia. En el pasado, la opinión que prevalecía era que generalmente era mejor asignar un sexo lo más rápido posible, a menudo sobre la base de los genitales externos, en vez del sexo de los cromosomas, e instruir a los padres para no tener ambigüedad en sus mentes en cuanto al sexo del niño. A menudo se recomendaba una cirugía rápida en la cual se extirpaba el tejido testicular u ovárico del otro sexo. En general, se consideraba más fácil reconstruir los genitales femeninos que los genitales masculinos funcionales, de tal manera que si la elección “correcta” no era clara, al niño a menudo se le asignaba el sexo femenino. En los últimos tiempos, la opinión de muchos expertos ha cambiado. Un mayor respeto por las complejidades del funcionamiento sexual femenino los ha llevado a concluir que los genitales femeninos insuficientes pueden no ser intrínsecamente mejores que los genitales masculinos insuficientes, incluso si la reconstrucción es “más fácil”. Además, otros factores pueden ser más importantes en la satisfacción del sexo que los genitales externos funcionales. Los factores cromosómicos, neurales, hormonales, psicológicos y conductuales pueden todos influir en la identidad sexual. 85 Autor: José Regino Ríos Gutiérrez. Unidad No. 14. Situaciones comunes de la práctica clínica que requieren valoración genética I. Competencia intermedia.- establecer las bases para hacer el diagnóstico de los problemas genéticos que habitualmente no son detectados en la práctica clínica diaria: talla baja, talla alta. Criterios de desempeño.1.- Analizar los principales motivos de referencia a la consulta genética: talla baja y talla alta. TALLA BAJA.o Condición que afecta el crecimiento lineal en el proceso de desarrollo del individuo ocasionado por múltiples factores pero con un fuerte componente genético. o La falla en el crecimiento es un estado patológico, mientras que la talla baja, a menudo es una variante normal. o Causas de talla baja de origen genético: Condiciones que alteran la producción, secreción y acción de factores hormonales que afectan el crecimiento lineal. Condiciones genéticas que no alteran la producción, secreción y acción de factores hormonales que sí afectan el crecimiento lineal (síndrome Down, mutaciones en el gen SHOX). Talla baja familiar. Retardo constitucional del crecimiento y desarrollo Talla baja idiopática. o La talla baja desproporcionada es el resultado de una displasia esquelética. o En el niño con talla baja proporcionada se deben descartar causas orgánicas, sistémicas y endocrinas. o La presencia de talla baja y sobrepeso en un paciente, tiene una causa orgánica hasta que no se demuestre lo contrario. o La talla baja idiopática se caracteriza por una talla baja significativa (<-25SDS), una velocidad de crecimiento persistentemente lenta para su edad y ningún parámetro bioquímico u otra evidencia específica de una condición que haga retardar el crecimiento. 86 TALLA ALTA.o Se define la talla alta como una estatura de más de dos desviaciones estándar (DE) para la media correspondiente a cada edad cronológica y sexo, o bien por la existencia de una velocidad de crecimiento excesiva (superior al percentil 75). o El hipercrecimiento de comienzo prenatal debe considerarse siempre patológico. o Cuando el hipercrecimiento es una manifestación de comienzo postnatal, deberemos considerar el origen poligénico de la talla definitiva. o Un hipercrecimiento puede originarse por: Exceso de secreción de hormona de crecimiento, como se produce en el gigantismo hipofisiario, en el síndrome de McCune-Albright y en el síndrome MEN-1. Exceso de cierto factores de crecimiento: Exceso de insulina e IGF-I en la obesidad, exceso de insulina en el hijo de madre diabética, en los lactantes gigantes y en la lipodistrofia Sobreexposición de IGF-II en el síndrome de BeckwithWiedemann o modulación de la IGF-II en el síndrome de Simpson-Golabi-Behmel, o por mutilación anormal de H 19. Exceso de receptores para factores de crecimiento (FGFR 3), en el síndrome de Partington. Exceso de un gen de crecimiento SHOX (de la región pseudoautosómica de los cromosomas sexuales humanos), ya sea en el cromosoma X extra (síndrome de Klinefelter y trisomía X) o en el cromosoma Y (varones XYY). Deficiencia de factores relacionados con la detención del crecimiento, como ocurre en la deficiencia de aromatasa para los estrógenos, en la deficiencia de receptores estrogénicos y en el hipogonadismo. Deficiencia de los factores relacionados con la prevención de la elongación ósea y proporciones disarmónicas, como así sucede en el síndrome de Marfan, en el síndrome de Beals y, quizás, en la homocistinuria. Alteración de los genes supresores de tumores, como ocurre en la neurofibromatosis tipo 1, en el síndrome 87 de Bannayan-Riley-Ruvalcaba y en el síndrome de Cowden. Como todo proceso clínico, el hipercrecimiento requiere una valoración ordenada de los datos anamnésicos, de exploración física, tallas familiares y del resultado de las pruebas complementarias. Por ello, es de sumo interés el conocer si la longitud/talla excesiva se manifestó ya en el período neonatal o a lo largo del postnatal. En todos los casos será necesaria la valoración de la edad ósea, que estará adelantada en los hipercrecimientos de causa hormonal, salvo en aquellos que se deben a un déficit o insensibilidad a los esteroides gonadales. En relación con la exploración física, en algunos hipercrecimientos, sus características clínicas sugieren “de entrada” un diagnóstico probable. En otros, la confirmación diagnóstica requiere una reflexión clínica para el empleo de ciertas pruebas complementarias. 2.- Analizar el componente genético en la determinación de la talla. TALLA BAJA.o Mutaciones en el gen SHOX o Mutaciones en el gen del receptor de la hormona de crecimiento (síndrome de Laron) o Mutación en el gen IGF-1 o Mutaciones en el gen del receptor de IGF-1 o Mutaciones en el gen PROP1 o Mutaciones en el gen Pit1 o Mutaciones en el gen GHRH o Mutaciones en el gen GH1 o Mutaciones en el gen del receptor de la insulina (Leprechaunismo) o Mutaciones en el gen DAX-¡ (hipocortisolismo) o Delecciones en la región cromosómica 22q11 TALLA ALTA.o Niveles altos de GH, IGF-I e IGF-3. o Polisomía X (en niñas) o del Y (en varones). o Gonosomopatía X-frágil. o Trisomía 8. 88 o Varones 47 XYY. o Mutación del gen de la fibrilina (síndrome de Marfan). o Mutaciones del gen PTEN (Bannayan-Riley-Ruvalcaba). o Mutación del gen NF-1 (neurofibromatosis tipo 1). o Mutaciones del gen CBS (Homocistinuria). o Sobreexposición de IGF-II (Beckwith-Wiedemann). 3.- Distinguir entre las principales causas genéticas de talla baja y talla alta. Talla alta: neuroendocrinos como acromegalia, síndrome de sotos Esqueleticos: síndrome de marfan, archard, aracnodactilia Metabolicos: hijo de made diabetica, homocistinuria Cormosomico: klinefelter Talla Baja: dismorfico: genopatias, cromosopatias, embriopatias Genetico no dismorfico: hipocrecimiento esencial y uterino Oseos: displasias oseas raquitismo, espondilitis, xifoescoliosis Hormonales: hipotiroidismo, Cushing, déficit Hormona crecimiento Otros: desnutrición, metabolopatias, colagenosis, hipoxico por anemia, cardiopatias o neumopatias o nefropatias 4.- Relacionar las manifestaciones clínicas con los síndromes genéticos con talla alta o talla baja: Síndrome de Silver Russel.o Cuadro clínico.- crecimiento deficiente, peso bajo al nacer, estatura baja, diferencias de tamaño en los dos lados del cuerpo (piernas y brazos de diferente largo), manchas café con leche, curvatura del dedo meñique hacia el dedo anular, retraso en la edad ósea, enfermedad de reflujo gastroesofágico, problemas renales, anchura anormal de cabeza. o Base genética.- Disomía uniparental materna para el cromosoma 7 o Patrón hereditario.- Ligado a X Síndrome de Noonan.o Cuadro clínico.- baja estatura, cuello con pterigium, aumento del ángulo de carga del codo y cardiopatía congénita. Puede encontrarse una deformidad característica del tórax, y la cara puede mostrar hipertelorismo, fisuras palpebrales inclinadas hacia abajo e 89 implantación baja de los pabellones auriculares. Algunos pacientes tiene una ligera diátesis hemorrágica, y la cuarta parte de los afectados presentan dificultades de aprendizaje. o Base genética.- 12q22. mutaciones en el gen de la proteína tirosina fosfatasa, tipo no receptor, 11 (PTPN11). o Patrón hereditario.- Síndrome de Marfán.o Cuadro clínico.- los individuos afectados son altos en comparación con los familiares no afectados, tienen articulaciones elásticas, presentan deformidad del tórax y escoliosis. Ectopia lenticular (subluxación del cristalino); dilatación de la aorta ascendente. o Base genética.- trastorno del tejido conectivo, de la fibrilina de tipo I, glucoproteína codificada por el gen FBN1 en 15q21. o Patrón hereditario.- autosómica dominante. Síndrome de Sotos.o Cuadro clínico.- antes conocido como gigantismo cerebral. Al nacimiento, el peso suele estar aumentado y se percibe macrocefalia. Dificultades tempranas de alimentación e hipotonía, retraso motor y ataxia. La estatura evoluciona alrededor del tope del tope del percentil máximo de crecimiento o lo supera, aunque la estatura final adulta no necesariamente se encuentra aumentada. Puede existir un adelanto de la edad ósea, con manos y pies grandes, y los ventrículos cerebrales pueden mostrarse ligeramente dilatados en la resonancia magnética o en tomografía. El rostro es característico, con frente prominente, hipertelorismo con fisuras palpebrales inclinadas haca abajo, nariz característica en la primera infancia y barbilla larga y puntiaguda. Algunos casos desarrollan escoliosis durante la adolescencia. o Base genética.- traslocaciones cromosómicas equilibradas, dos tenían puntos de ruptura en 5q35. o Patrón hereditario.- autosómico recesivo. 90 Autor: José Regino Ríos Gutiérrez. Unidad No. 15. Situaciones comunes de la práctica clínica que requieren valoración genética II. Competencia intermedia.- Establecer las bases para hacer el diagnóstico de los problemas genéticos que habitualmente no son detectados en la práctica clínica diaria: retraso psicomotor. Criterios de desempeño.1.- Analizar los principales motivos de referencia a la consulta genética: retraso psicomotor. Se empieza preguntando acerca de la situación actual del niño. Inicialmente debemos hacer un interrogatorio libre, inquiriendo sobre qué aspectos preocupan más a los padres. Luego se debe interrogar acerca de todas las áreas de la maduración sin omitir ninguna: social, del lenguaje, emocional, cognitiva, de la visión, de la audición, motora. Preguntar acerca de posibles etiologías, indagando problemas del período prenatal, perinatal y postnatal. Del período prenatal seleccionaremos: antecedentes de posibles infecciones virales, (TORCH), posibles hipoxemias (intención de aborto), hipertensión materna, exposición a agentes teratogénicos como alcohol, drogas, insecticidas, plomo, benceno, radiaciones, etc. Periodo perinatal. Es el período donde existen eventos que contribuyen en forma importante al desarrollo de retraso: hipoxia, hipoglicemia, hiperbilirrubinemia, trauma obstétrico, prematuridad, macro-microcefalia, convulsiones. Período postnatal. Debe indagarse sobre: infecciones del SNC, traumatismos encéfalocraneano severos, trastornos hidroelectrolíticos, intoxicaciones. 2.- Analizar el componente genético de la inteligencia. Se dice que la inteligencia de un individuo esta asociada a la de los padres por lo cual generalmente se tiene un IQ parecido al de los padres, aunque existen casos en que hay padres con IQ alto pero si no se estimula al niño presentará un IQ bajo y viceversa. 3.- Analizar la epidemiología del retraso psicomotor. 95% de los trastornos de RM son por defectos x sd. Cromosomico. 50% de niños con malformaciones tienen retraso mental, 3.5% son defectos por síndromes mendelianos. Es más común el retraso mental en raza negra y en varones. Tiene una prevalencia de 2.3 a 3%. Sd. Down Sd. X frágil 3.7% 4.6% Alteración cromosómica 0.6% PCI, infecciones intrauterinas Trauma postnatal 2.7% 3.2% 91 4.- Fundamentar el algoritmo diagnóstico en casos con sospecha de retraso psicomotor de origen genético con ejemplo clínicos. La asociación con malformaciones sugieren un problema genético, en ausencia de defectos estructurales se debe pensar en errores innatos del metabolismo. DESARROLLO PSICOMOTOR: SINTOMAS Sonrisa social 1 mes Sostén cefálico 1-3 meses Se sienta 6 meses Bipedestación 8 meses marcha 12 meses lenguaje 10 meses Control esfínteres 2 años Funcionamiento de adaptación y de conducta considerable. Leve: IQ 50-70 Moderado: IQ 35-50 Severo: IQ menor a 20 RM leve 8.4 x 1000 RM severo 3.0 x 1000 ABORDAJE: - - hormonas tiroideas estudio citogenético: cariotipo, 1-23% de RM idiopático han sido explicados por deleciones en regiones subteloméricas, disomía uniparental e impronta genómica (cromosomas 7, 15, 16, 20) estudio para síndrome de x frágil TAC. Tamiz metabólico. .-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-.-. RETRASO MENTAL (RM) El retraso mental.- se define como una incapacidad en el desarrollo de las habilidades cognitivas y en la adquisición de un nivel de inteligencia que sería el apropiado para un determinado grupo de edad. 92 El retraso mental puede estar causado por muchos factores, ya sean ambientales como el nacimiento prematuro o infecciones prenatales, o bien genéticos como anomalías cromosómicas o mutaciones en genes concretos. Tipo de retraso metal: o Retraso mental sindrómico (RMLX).- ocurre con penetrancia y expresividad variable como características fenotípicas de numerosos síndromes hereditarios. o Retraso mental no sindrómico o inespecífico (RMX).- el retraso metal se presenta de forma aislada sin ninguna otra característica distintiva. El RM se presenta con más frecuencia en los varones, como consecuencia de mutaciones en genes localizados en el cromosoma X, cuyos efectos evidentes en los varones homocigotos, lo cuales no pueden compensar el efecto de dosis causado por la mutación. Del nacimiento a los 5 años.- retraso psicomotor. De los 5 a los 18 años.- retraso mental. De los 18 años en adelante.- demencia. Se considera RM cuando en pruebas de coeficiente intelectual obtienen 70 o menos puntos. Entre 1 y 3 % de la población mundial presenta RM. Existen tres tipos de retraso mental: o LEVE o MODERADO o SEVERO O GRAVE La causa más común de RM hereditario es: X-Frágil La causa más frecuente de RM genético es: síndrome de Down. Abordaje de un paciente con RM: o Historia clínica. o Exploración física. o Revisión neurológica. o Exámenes de laboratorio. o Consejo genético. 93 Autor: José Regino Ríos Gutiérrez. Unidad No. 16 Patología Genética prenatal y preconcepcional. Competencia intermedia.Evaluar la indicación, limitación y contraindicación de un estudio diagnóstico prenatal, sustentando la selección del más indicado en base a las características del paciente y de los estudios existentes. Criterios de desempeño.1.- Clasificar los métodos de diagnóstico prenatal: No invasivos.o Ultrasonido.- o ecografía, útil para el diagnóstico prenatal. Puede usarse por indicación obstétrica (para detectar la ubicación de la placenta y diagnosticar embarazos múltiples), y también para diagnóstico prenatal de alteraciones estructurales no relacionadas con defectos cromosómicos, bioquímicos o moleculares conocidos. Por ejemplo, se puede buscar polidactilia como manifestación diagnostica de un síndrome de anomalías múltiples, como es el caso de uno de los síndromes de polidactilia con extremidad corta de transmisión autosómica recesiva que acompaña a la hipoplasia pulmonar grave, que es invariablemente letal. Asimismo, puede revelar que el feto tiene una mandíbula pequeña, lo que se relaciona con un paladar hendido posterior y otras alteraciones más importantes en los síndromes que afectan a un solo gen. o Rayos X.- (radiología), el esqueleto fetal puede visualizarse mediante radiografías a partir de las 10 semanas en adelante, esta técnica se usa para diagnosticar displasia esquelética hereditaria. No se usa mucho, en virtud del riesgo que representa para el feto. o Marcadores bioquímicos maternos.- o Alfa fetoproteína (AFP) Estriol no conjugado Gonadotropina coriónica humana (hCG) Inhibina-A Células fetales en sangre materna.- es posible que en un futuro pueda contarse con una media completamente no invasivo de diagnóstico prenatal para las alteraciones cromosómicas y de DNA. Utilizando anticuerpos contra antígenos específicos para el trofoblasto fetal se ha descubierto ahora que durante el primer trimestre se encuentran células de origen fetal en la circulación materna. La validez de estas observaciones se basa en el empleo de la PCR para detectar la presencia de marcadores de DNA derivados de los padres de los padres en la sangre obtenida de mujeres durante las primeras 94 etapas del embarazo. Si bien las técnicas inmunológicas que utilizan anticuerpos contra antígenos de trofoblasto fetal pueden utilizarse para enriquecer las células fetales en la circulación materna, pueden encontrarse problemas para descartar la posibilidad de contaminación importante de las células maternas en la muestra que se está analizando. Esta técnica se ha utilizado en una seria limitada de trastornos, por ejemplo, para predecir el estado Rhesus del feto cuando la madre es Rh negativa y el padre es heterocigoto para tratar de determinar en qué embarazos debe realizarse seguimiento de isoinmunización Rhesus. En tiempos más recientes se ha aprovechado el hecho de que los eritrocitos fetales con nucleados para enriquecer a las células de origen fetal. Invasivos.o Amniocentesis.- implica la aspiración de 10 a 20 mL de líquido amniótico a través de la pared abdominal bajo control ecográfico. Se realiza alrededor de la décimo sexta semana de gestación. La muestra se centrifuga para obtener un sedimento de células y líquido sobrenadante. El líquido puede usarse para el diagnóstico prenatal de defectos del tubo neural mediante el análisis de la alfa-proteína. Las células sedimentadas se usan para cultivo y hacer análisis cromosómico, DNA y bioquímicos. El riesgo de esta técnica es de aborto espontáneo del 0.5 al 1%, y que si el resultado es anormal cabe la posibilidad de que tengan que considerar la interrupción del embarazo en el segundo trimestre, lo cual implica inducción del parto. o Cordocentesis.- se obtiene un pequeña muestra de sangre fetal de uno de los vasos del cordón umbilical. El muestreo de la sangre fetal se utiliza sistemáticamente en el tratamiento de la isoinmunización Rhesus y se puede emplear para el análisis cromosómico a fin de resolver los problemas relacionados con un posible mosaicismo cromosómico en las muestras de Vellosidades Coriónicas o de amniocentesis. o Biopsia de vellosidades coriales.- el muestreo de las vellosidades coriónicas (VC), permite establecer el diagnóstico prenatal durante el primer trimestre del embarazo, puede efectuarse a las 11 o 12 semanas de gestación bajo control ecográfico y consiste en la aspiración transcervical o (más frecuentemente) transabdominal de tejido de las VC (tejido de origen fetal que se deriva de la capa celular externa del blastocisto). El análisis cromosómico puede realizarse en el tejido de las VC en forma directa, analizando frotis de metafase de células en división activa, o después de un cultivo. El procedimiento que se lleva a cabo en etapas más avanzadas del embarazo se denomina biopsia placentaria. La ventaja es el diagnóstico prenatal en el primer trimestre; el inconveniente es el riesgo de 1 a 2% de aborto espontáneo., también hay indicios de que esta técnica puede causar alteraciones en las extremidades del embrión si se lleva a cabo antes de las 9 a 10 semanas de gestación. 95 o Biopsia la piel fetal.- por medio de la fetoscopia se obtienen muestras de tejido del feto que pueden analizarse como un medio para establecer el diagnóstico prenatal de diversos trastornos infrecuentes. Algunos de éstos han sido los trastornos cutáneos hereditarios (como la epidermosis bullosa) y, antes que se contara con análisis de DNA, trastornos metabólicos en los que la enzima se expresa únicamente en determinados tejidos u órganos como el hígado (por ejemplo, la deficiencia de las ornitina transcarbamilasa). o Fetoscopia.- Implica la visualización del feto por medio de un endoscopio; se realiza durante el segundo trimestre para tratar de detectar estructuras sutiles que señalarían un diagnóstico subyacente importante. Lamentablemente la fetoscopia conlleva un riesgo de aborto espontáneo de 3 al 5 %. 2.- Analizar las características e interpretar el resultado de los diferentes métodos de tamizaje materno. Dúo marcador.- edad materna (más de 35 años) y análisis cromosómico. o Triple marcador.- Alfa fetoproteína (AFP), estriol y gonadotropina coriónica humana (hCG). o Durante el embarazo es posible realizar un cribado de alteraciones cromosómicas y sobre todo del síndrome de Down, teniendo en cuenta factores como la edad materna. Durante el embarazo es posible realizar un cribado de alteraciones cromosómicas y sobre todo del síndrome de Down, teniendo en cuenta factores como la edad materna y las concentraciones de tres biomarcadores en el suero materno. Este último enfoque se basa en el descubrimiento de que, a las 16 semanas de gestación, las concentraciones de AFP y del estriol no conjugado en el suero materno tienden a ser menores en los embarazos de síndrome Down que en los embarazos normales, en tanto que las concentraciones de gonadotropina coriónica humana (hCG) en el suero materno suelen estar elevadas. Cuádruple marcador.- AFP, estriol, hCG, inhibina-A. o Recientemente se ha demostrado que otro marcador bioquímico , la Inhibina-A, también está aumentada en el suero materno en los embarazos con síndrome Down. Del ultrasonograma prenatal de segundo nivel. Marcadores ultrasonográficos de primer trimestre.- a las 12 semanas de gestación hay una importante relación entre las alteraciones cromosómicas y la acumulación anormal de líquido en la nuca del feto: un aumento en la translucidez nucal (TN). Esto es aplicable al síndrome Down, a los otros síndromes de trisomía autosómica (trisomía 13 y trisomía 18), al síndrome de Turner y a la triploidía, así como a una amplia gama de otras alteraciones fetales y síndromes infrecuentes. El riesgo de síndrome de 96 Down se correlaciona con valores absolutos de TN lo mismo con la edad materna. Algunos lactantes con síndrome Down tienen atresia duodenal, que se manifiesta como un “signo de burbuja doble” en la ecografía del abdomen fetal. Marcadores ultrasonográficos de segundo trimestre.- a las 18 semanas de gestación, aunque no es posible diagnosticar directamente las anomalías cromosómicas, se sospechará por la detección de una anomalía como en onfalocele o el pie en mecedora. Se encuentra una alteración cromosómica en el 50% de los fetos con onfalocele identificado a las 18 semanas, y el pie de mecedora es un dato muy característico en los lactantes con trisomía 18, que también invariablemente muestran retraso en el crecimiento. 3.- Analizar las características e interpretar el resultado de cada uno de los métodos de diagnósticos prenatal existentes. Diagnóstico preimplantación.- en el procedimiento, se administran hormonas a la mujer para inducir una hiperovulación y luego se obtienen los ovocito mediante por vía transcervical, bajo sedación, con control ecografico. Se añaden los espermatozoides móviles de una muestra de semen a los ovocitos en cultivo (fecundación in Vitro) y se incuban para permitir la fecundación. Si se lleva va a llevar a cabo el análisis genético en DNA de una sola célula (blastómero) del embrión inicial (blastocisto) en la etapa de ocho células en el tercer día, se logra la fecundación utilizando la inyección intracitoplasmática de espermatozoides (ICSI) de un solo espermatozoide para evitar la presencia de otros espermatozoides contaminados. En la etapa de 8 células se obtiene una biopsia del embrión inicial y se retira para análisis de una o a veces dos células. Cualquiera que sea el análisis genético que se realice, es indispensable que ésta sea una posibilidad práctica en el material genómico de una sola célula. De los embriones sometidos a la prueba, se reintroducen en el útero de la madre dos que estén sanos y no afectados por el trastorno para el cual se tiene riesgo. Una variación de la técnica es la extracción del primero y a menudo el segundo corpúsculo polar del ovocito no fecundado, que se encuentra bajo la zona pelúcida. Dado que el primero corpúsculo polar se degenera con bastante rapidez, es necesario proceder al análisis en las primeras 6 horas tras la obtención. El análisis de los corpúsculos polares es un método indirecto para identificar el genotipo en virtud de que el ovocito y el primer corpúsculo polar se dividen entre sí durante la meiosis I y, por tanto, contienen diferentes miembros de cada par de cromosomas homológos. Biopsia de vellosidades coriónicas.- el muestreo de las vellosidades coriónicas (VC), permite establecer el diagnóstico prenatal durante el primer trimestre del embarazo, puede efectuarse a las 11 o 12 semanas de gestación bajo control ecográfico y consiste en la aspiración transcervical o (más frecuentemente) transabdominal de tejido de las VC (tejido de origen fetal que se deriva de la capa celular externa del blastocisto). El análisis cromosómico puede realizarse en el tejido de las VC en forma directa, analizando frotis de metafase de células en división activa, o después de un cultivo. El procedimiento que se lleva a cabo en etapas más avanzadas del 97 embarazo se denomina biopsia placentaria. La ventaja es el diagnóstico prenatal en el primer trimestre; el inconveniente es el riesgo de 1 a 2% de aborto espontáneo., también hay indicios de que esta técnica puede causar alteraciones en las extremidades del embrión si se lleva a cabo antes de las 9 a 10 semanas de gestación Amniocentesis.- implica la aspiración de 10 a 20 mL de líquido amniótico a través de la pared abdominal bajo control ecográfico. Se realiza alrededor de la décimo sexta semana de gestación. La muestra se centrifuga para obtener un sedimento de células y líquido sobrenadante. El líquido puede usarse para el diagnóstico prenatal de defectos del tubo neural mediante el análisis de la alfa-proteína. Las células sedimentadas se usan para cultivo y hacer análisis cromosómico, DNA y bioquímicos. El riesgo de esta técnica es de aborto espontáneo del 0.5 al 1%, y que si el resultado es anormal cabe la posibilidad de que tengan que considerar la interrupción del embarazo en el segundo trimestre, lo cual implica inducción del parto. Cordocentesis.- se obtiene un pequeña muestra de sangre fetal de uno de los vasos del cordón umbilical. El muestreo de la sangre fetal se utiliza sistemáticamente en el tratamiento de la isoinmunización Rhesus y se puede emplear para el análisis cromosómico a fin de resolver los problemas relacionados con un posible mosaicismo cromosómico en las muestras de Vellosidades Coriónicas o de amniocentesis. Biopsia de piel fetal.- por medio de la fetoscopia se obtienen muestras de tejido del feto ue pueden analizarse como un medio para establecer el diagnóstico prenatal de diversos trastornos infrecuentes. Algunos de éstos han sido los trastornos cutáneos hereditarios (como la epidermosis bullosa) y, antes que se contara con análisis de DNA, trastornos metabólicos en los que la enzima se expresa únicamente en determinados tejidos u órganos como el hígado (por ejemplo, la deficiencia de las ornitina transcarbamilasa). 4.- Fundamentar las patologías genéticas que son susceptibles de ser diagnosticadas de forma prenatal. Defectos del tubo neural Síndrome de Down Alteraciones estructurales (SNC, corazón, riñones y extremidades) Alteraciones cromosómicas Trastornos metabólicos (deficiencia de ornitina transcarbamilasa) Defectos moleculares Trastornos hematológicos Infección congénita Trastornos hereditarios de la piel (epidermólisis ampollosa) 98 5.- Justificar las estrategias diagnósticas de los cromosopatías más comunes (síndrome de Turner, trisomía 18, trisomía 13) y otras alteraciones con manifestaciones prenatales como las displasias óseas y malformaciones congénitas asiladas y múltiples. Trisomia 21 Aumenta la incidencia por las altas cantidades de estradiol no conjugado y gonadotropina coriónica humana. Aneuploidias Detectadas por FISH por medio del DX preimplantacional asi como por la amniocentesis. Trisomia 21, 18 y 13 FISH.. aislamiento de celulas fetales en sangre materna Monosomía PCR en células fetales aisladas de sangre materna fetal 6.- Fundamentar el algoritmo de estudio prenatal para un sujeto con riesgo reproductivo de patología genética. Existen múltiples indicaciones para ofrecer el diagnóstico prenatal. en condiciones ideales se identifica y se evalúa a las parejas con un alto riesgo previo de tener una alteración antes de que la mujer quede embarazada, de manera que, sin prisas, se les pueda asesorar y puedan tomar una decisión sobre las opciones que desea elegir. Una alternativa es que se identifique a estas parejas en las primeras etapas del embarazo, de manera que todavía tengan la oportunidad de considerar todas las opciones de diagnóstico prenatal disponibles lo más pronto posible durante la gestación. La edad materna avanzada es la indicación más común para ofrecer el diagnóstico prenatal. Hay una relación bien reconocida de la edad materna avanzada con un mayor riesgo de tener un niño con síndrome Down y los otros síndromes de trisomía autosómica. No hay edad estándar para indicar el estudio pero se sugiere a partir de los 35 años. Niño previo con una alteración cromosómica, si bien se dispone de varias series con cifras de riesgo de recidiva levemente diferentes, para las parejas que han tenido un niño con síndrome de Down a consecuencia de una falta de no disyunción, o de una translocación robertsoniana desequilibrada de novo, el riesgo en un embarazo subsiguiente suele referirse como el riesgo relacionado con la edad materna más aproximadamente el 0.5%. si se ha observado que uno de los padres es potador de una reordenación cromosómica equilibrada (por ejemplo, translocación cromosómica o inversión paracéntrica) que ha ocasionado que el niño previo nazca con problemas importantes a consecuencia de una alteración cromosómica desequilibrada, es probable que el riesgo de recidiva fluctúe entre 1-2 y el 15-20%. El riesgo definitivo dependerá de las características del reordenamiento presente en los padres y de los segmentos específicos de los cromosomas individuales implicados. Antecedente familiar de una alteración cromosómica; a las parejas se las suele remitir para el diagnóstico prenatal cuando hay un antecedente familiar de alteración cromosómica, más comúnmente síndrome de Down. 99 La mayoría de las parejas no tendrán un incremento en el riesgo en comparación con lo observado en la población general, ya que casi todos los casos de trisomía 21 y otros trastornos cromosómicos habrán surgido como resultado de una no disyunción más que como consecuencia de una translocación cromosómica familiar u otro reordenamiento. Antecedente familiar de un trastorno monogénico; si los padres prospectivos y han tenido un niño afectado, o si uno de los padres está afectado y tiene un antecedente familiar positivo de un trastorno monogénico que comporta un riesgo significativo para la descendencia, entonces se comenta con ellos la opción del diagnóstico prenatal. Antecedente familiar de un defecto del tubo neural; es necesario evaluar cuidadosamente el árbol genealógico para determinar el riesgo aplicable a cada embarazo. Los riesgos pueden determinarse basándose en datos empríricos. Antecedentes familiares de otras alteraciones estructurales congénitas; la evaluación del árbol genealógico familiar habrá de comprender la provisión de un riesgo derivado de los resultados de estudios empíricos. Si el riesgo para un embarazo está aumentado, se puede ofrecer un examen ecográfico detallado para analizar la alteración estructural específica alrededor de de las 16 a 18 semanas de gestación. La ecografía en el segundo trimestre detecta las malformaciones más graves craneales, cardíacas, renales y de las extremidades. Alteraciones identificadas durante el embarazo; la introducción general de los procedimientos de detección diagnóstica prenatal (como las pruebas triples y la ecografía de anomalías fetales) ha implicado que inesperadamente muchas parejas presenten incertidumbre diagnóstica durante el embarazo, que sólo puede aclararse mediante un procedimiento invasivo como la amniocentesis o la biopsia de VC. Otros factores (como el crecimiento fetal deficiente) también pueden ser una indicación para el análisis cromosómico prenatal, ya que la confirmación de una alteración cromosómica grave y no viable como la trisomía 18 y la triploidía puede influir en el tratamiento subsiguiente del embarazo y en el método del parto. Otros factores de riesgo elevado; incluyen la consaguinidad de los padres, un antecedente obstétrico desfavorable y determinadas enfermedades maternas. Un antecedente de tres o más abortos espontáneos inexplicables ha de investigarse mediante estudios cromosómicos en los padres para descartar una reordenación cromosómica como una translocación o una inversión. 100 Autor: José Regino Ríos Gutiérrez. Unidad No. 17 Genética del Cáncer. Competencia intermedia.- Analiza el componente genético del cáncer para fundamentar la asesoría genética. Criterios de desempeño.1.- Analizar la biología del cáncer y la teoría clonal, así como los principales carcinógenos ambientales.Todos los cánceres son una enfermedad genética de las células somáticas debida a una división celular aberrante o a la pérdida de la muerte celular programada normal, pero una pequeña parte de ellos está muy condicionada por mutaciones heredadas en la línea germinal que se comportan como rasgos mendelianos. No obstante, esto no contradice nuestro conocimiento tradicional de que, en muchos cánceres, los factores ambientales tienen una importancia etiológica primaria, mientras que la herencia parece desempeñar un escaso o nulo papel. Esto último es absolutamente cierto para los “cánceres industriales”, que se producen por exposición prolongada a los compuestos químicos carcinógenos. Entre los ejemplos están. Cáncer de piel en los trabajadores del alquitrán. Cáncer de vejiga en los que trabajan con colorante anilina. El angiosarcoma de hígado en los trabajadores que procesan el cloruro de polivinilo (PVC). Cáncer de pulmón en los trabajadores del amianto. Los cánceres surgen como resultado final de la acumulación de mutaciones, tanto heredadas como somáticas, en los protooncogenes y en los genes supresores tumorales. También es importante una tercera clase de genes (los genes de reparadores de errores del emparejamiento de DNA), porque se cree que su inactivación contribuye a la génesis de mutaciones en otros genes que afectan directamente a la supervivencia y a la proliferación de las células. Teoría Clonal: En un tumor todas las células que lo constituyen provienen de una célula común. Monoclonales. El cáncer es un conjunto de trastornos con características de un crecimiento celular incontrolado. 2.- Distinguir entre cáncer esporádico, familiar y hereditario y analizar su epidemiología. CÁNCER ESPORÁDICO.- es aquel en el que no hay una historia familiar previa. CÁNCER FAMILIAR.- Cáncer que se manifiesta en familias con mayor frecuencia de la prevista por casualidad. Estos tipos de cáncer se manifiestan a una edad temprana y pueden indicar la presencia de la mutación de un gen que aumenta el riesgo de padecer de cáncer. También pueden constituir un signo de factores medioambientales o de estilo de vida 101 compartidos. o Síndrome del cáncer hereditario.- término utilizado para describir la agrupación de ciertas familias de tipos particulares de cánceres, en los que se ha propuesto que los distintos tipos de neoplasia maligna pueden deberse a un único gen dominante, específicamente Lynch tipo II. CÁNCER HEREDITARIO.- La mayoría de los síndromes que predisponen al cáncer familiar hereditario raros actualmente reconocidos se heredan de forma dominante, y la descendencia de los individuos afectados tiene un 50% de posibilidad de heredar el gen y, por tanto, un riesgo aumentado de desarrollar el cáncer. Hay también diversos síndromes, heredados habitualmente como trastornos autosómicos recesivos, con un riesgo aumentado de desarrollar cáncer asociado con un mayor número de anomalías en los cromosomas cuando son cultivados, lo que se conoce como los síndromes de roturas cromosómicas. Las personas con un síndrome de predisposición al cáncer familiar heredado tienen el riesgo de desarrollar un segundo tumor (multifocal o bilateral en el cáncer de mama), un mayor riesgo de desarrollar un cáncer a una edad relativamente más temprana que las personas con una forma espóradica, y pueden presentar tumores en diferentes sitios del organismo, aunque habitualmente hay predominio de un tipo de cáncer. 3.- Clasificar los genes responsables del cáncer: oncogenes, genes de reparación, genes supresores tumorales y analizar sus mecanismos de inactivación. ONCOGENES.- Los oncogenes son las formas alteradas de los genes normales (protooncogenes) que tienen papeles claves en el crecimiento celular y en las rutas de diferenciación. En las células normales de los mamíferos hay secuencias de DNA que son homólogas a los oncogenes víricos, y son éstas las que se denominan protooncogenes u oncogenes celulares. Aunque los términos protoncogén y oncogén se usan a menudo de forma indistinta, estrictamente hablando protooncogén está reservado para el gen normal, mientras que oncogén celular, o c-onc, se refiere a un protooncogén mutado que tiene propiedades oncogénicas, como los oncogenes víricos, o v-onc. Se han identificado unos 30 oncogenes. o Los oncogenes celulares están altamente conservados en la evolución, lo que sugiere que desempeñan papeles importantes como reguladores del crecimiento celular, manteniendo la progresión ordenada a través del ciclo celular, la división y la diferenciación celular. o Identificación de oncogenes en los puntos de rotura de la translocación cromosómica.- las aberraciones cromosómicas son habituales en las células malignas, que a menudo muestran una notable variación en el número y en la estructura de los cromosomas. Se ha visto que las translocaciones cromosómicas pueden dar lugar a 102 genes quiméricos nuevos con una función bioquímica o un nivel de actividad protooncogénica alterados. o Amplificación del oncogén.- los protooncogenes pueden también activarse por la producción de copias múltiples del gen, lo que se denomina amplificación génica, mecanismo conocido por ser de gran valor para la supervivencia cuando las células se encuentran ante un estrés ambiental. o Función de los oncogenes.- la transcripción de la señal es una ruta compleja de múltiples pasos desde la membrana celular, a través del citoplasma, hasta llegar al núcleo, que afecta a diversos tipos de productos del protooncogén implicados en bucles de retroalimentación positiva y negativa necesarios para una proliferación y diferenciación celular precisa. o Los protooncogenes actúan de tres formas principales en el proceso de la transducción de la señal: o La primera es a través de la fosforilación de los residuos de las proteínas serina, treonina y tirosina, mediante la transferencia de grupos fosfato procedentes del ATP. Esto lleva a la alteración de la configuración que estimula la actividad cinasa de las proteínas diana, dando lugar a la transducción de la señal. La familia de protooncogenes RAS es un ejemplo del segundo tipo, que son GTPasas y que funcionan como interruptores moleculares a través del ciclo de la guanosina difosfataguanosina trifosfato (GDP-GTP) como intermediarios, transmitiendo la señal de transducción desde la tirosina cinasa asociadas a la membrana a las serina treonina cinasas. El tercer tipo implica a proteínas localizadas en el núcleo que controlan el progreso durante el ciclo celular, la replicación del DNA y la expresión de los genes. Tipos de oncogenes. Factores de crecimiento.- el oncogén mejor conocido que actúa como factor de crecimiento es el oncogén v-SIS, que codifica parte de la subunidad B del factor de crecimiento derivado de las plaquetas (PDGF) biológicamente activo. Los productos de oncogenes que muestran homología con los factores de crecimiento de los fibroblastos (FGF) son HST e INT-2, que están amplificados en los cánceres de estómago y en los melanomas malignos, respectivamente. Receptores del factor de crecimiento.- muchos oncogenes codifican proteínas que forman receptores para el factor de crecimiento, con dominios tirosina cinasa que poseen actividad tirosina cinasa, lo que permite a las células eludir los 103 mecanismos normales de control. Se han identificado más de 40 tirosina cinasa diferentes que pueden dividirse en dos tipos principales: las que abarcan la membrana celular (receptores tirosina cinasas para el factor de crecimiento) y las que se localizan en el citoplasma (tirosina cinasa no receptores). Factores intracelulares de la transducción de la señal.- se han identificado dos tipos de factores intracelulares para la transducción de la señal. Proteínas con actividad GTPasa.- las mutaciones en los genes RAS dan como resultado una actividad GTPasa aumentada o sostenida, lo que ocasiona un crecimiento sin restricción. Serina treonina cinasas citoplasmáticas.las mutaciones en el gen pueden dar como resultado una transmisión aumentada o mantenida de una señal impulsora del crecimiento hacia el núcleo. Proteínas nucleares que ligan el DNA.- los oncogenes FOS, JUN y ERB-A codifican proteínas que son factores de transcripción específicos que regulan la expresión del gen activado o suprimiendo las secuencias de DNA cercanas. Las oncoproteínas MYC y MYB estimulan a las células para que evolucionen desde G1 hasta la fase S del ciclo celular. Su sobreproducción impide que las células entren en una fase de descanso prolongada, dando lugar a una proliferación celular persistente. Factores del ciclo celular.- las células cancerosas pueden aumentar su número por un mayor crecimiento y división, o acumularse por una disminución de la muerte celular. Las anomalías en la regulación del ciclo celular por factores de crecimiento, receptores de los factores inhibidores, llevan a la activación de las cinasas dependientes de la ciclina, como la ciclina D1, ocasionando una transformación celular con una división celular descontrolada. Por otra parte, la pérdida de los factores que ocasionan una muerte celular programada normal –proceso conocido como apoptosis- puede ocasionar la acumulación de células, mediante una prolongación de la supervivencia celular, como mecanismo de desarrollo de algunos tumores. La activación del oncogén bcl-2 mediante reconfiguraciones cromosómicas se asocia con una inhibición de la apoptosis, ocasionando ciertos tipos de linfoma. Transducción de la señal y facomatosis.- facomatosis deriva del griego phakos, que significa “lenteja” (en este contexto, “objeto con forma de lenteja”), y originalmente se refería a tres enfermedades que incluían lesiones benignas dispersas: neurofibromatosis, esclerosis tuberosa, y enfermedad de von 104 Hippel-Lindau. Actualmente se han añadido a esta lista el síndrome del carcinoma nevoide de células basales (Gorlin), la enfermedad de Cowden, la poliposis adematosa familiar, el síndrome de Peutz-Jengher y la poliposis juvenil. En el momento presente conocemos los genes de todos estos trastornos y están normalmente activos en la transducción de la señal intracelular, y sus productos proteína son supresores de tumores. GENES DE REPARACIÓN.- son los responsables de coordinar la corrección de nucleótidos mal incorporados. GENES SUPRESORES TUMORALES.- En contraste con los oncogenes, los genes supresores de los tumores son una clase de genes celulares cuya función normal es suprimir la proliferación celular inadecuada, es decir, el desarrollo de una neoplasia maligna se debe a una mutación de pérdida de función. 4.- Analizar los aspectos citogenéticos del cáncer y su papel en el diagnóstico, pronóstico, tratamiento y seguimiento de las neoplasias hematológicas: leucemia granulocítica crónica y cromosoma Filadelfia. LEUCEMIA GRANULOCÍTICA CRÓNICA.- llamada también leucemia mieloide crónica es un padecimiento en el cual aumenta la producción de glóbulos blancos en la médula ósea. En las primeras fases suele no presentar síntomas, cuando aparecen suelen mostrar cansancio intenso, fiebre elevada, falta de apetito, sudoración nocturna, esplenomegalia. Tiene cuatro fases: o Fase crónica.- en esta fase, que puede durar meses o años, se encuentran pocos blastos tanto en la sangre como en la médula ósea. o Fase acelerada.- aparecen blastos en la sangre y en la médula con desaparición de células normales. o Fase blastica.- por lo menos 1/3 de las células de la sangre y médula son blastos, pueden aparecer acúmulos de estos de estas células en forma de tumores a nivel de huesos o ganglio. También puede haber un cuadro de infiltración meníngeo, con infiltración de blastos en el líquido cefalorraquídeo. o Fase refractaria.- cuando los blastos no disminuyen a pesar del tratamiento. CROMOSOMA FILADELFIA.- En 1960, unos investigadores en Filadelfia fueron los primeros en descubrir un cromosoma anómalo en los leucocitos de pacientes con leucemia mieloide crónica. El cromosoma anómalo, denominado cromosoma Filadelfia o cromosoma Ph1, es una anomalía adquirida que se encuentra en la sangre o en las células de la médula ósea pero no en otros tejidos de estos pacientes. El Ph1 es cromosoma pequeño (cromosoma 22) en el que el material de los brazos largos se ha traslocado recíprocamente hacia y desde el brazo largo del cromosoma 9, es decir, 105 t(9;22)(q34;q11). Se ha observado que dicha translocación transfiere el oncogén celular ABL (Abelson) desde el cromosoma 9 a una región del cromosoma 22 conocida como la región del grupo del punto de rotura (BCR, breakpoint cluster region), dando como resultado un transcrito quimérico obtenido de ambos genes, el c-ABL y el BCR. Esto ocasiona un gen quimérico que expresa una proteína de fusión consistente en la proteína BCR en el extremo amino y en la proteína ABL en el extremo carboxilo, asociada con una actividad transformadora. 5.- Analizar las características clínicas de alto riesgo para formas hereditarias de cáncer: Li Fraumeni, Retinoblastoma, cáncer colorrectal, cáncer de mama-ovario, tumor de Wilms, neoplasia endócrina múltiple. LI FRAUMENI.o Cuadro clínico.- los miembros de familias afectadas, tienen una alta susceptibilidad para desarrollar diversas neoplasias malignas a una edad relativamente temprana (sarcomas, carcinomas suprarrenales y cáncer de mama) o Base genética.- mutaciones en el TP53 (codones 254-258) o Patrón hereditario.- autosómico dominante. RETINOBLASTOMA.o Cuadro clínico.- cáncer de las células de la retina ocular en desarrollo, altamente maligno, que aparece en la infancia, habitualmente antes de los 5 años. Puede ocurrir esporádicamente (la denominada forma “no hereditaria”) o familiar (la forma “hereditaria”). Las causas no hereditarias afectan habitualmente a un solo ojo, mientras que los casos hereditarios pueden ser unilaterales, pero por lo general son bilaterales u ocurren en más de un sitio en un ojo (multifocales). La forma familiar tiende también a presentarse a una edad más temprana que la forma no hereditaria o esporádica. o Base genética.- 13q14 o Patrón hereditario.- autosómica dominante. CÁNCER COLORRECTAL.o Cuadro clínico.- se cree que la mayoría de los cánceres colorrectales se desarrollan a partir de adenomas “benignos”. Histológicamente, los pólipos adematosos menores de 1 cm de diámetro contienen raramente áreas de cambios carcinomatosos, mientras que el riesgo de cambio carcinamotoso aumenta de un 5 a un 10% cuando el adenoma alcanza los 2 cms de diámetro. La transición de un pequeño pólipo adematoso a un cáncer invasivo se cree que tiene lugar entre los 5 y los 10 años. Los pólipos adematosos de menos de 1 cm de diámetro tiene mutaciones en el 106 gen RAS en menos del 10% de los casos. A medida que el tamaño del pólipo aumenta entre 1 y 2 cm, la prevalencia de las mutaciones en el gen RAS está alrededor del 40%, y se incrementa a aproximadamente el 50% en los cánceres colorrectales completos. o Base genética.- pérdida del alelo en el cromosoma 18q. o Patrón hereditario.- autosómico dominante. CÁNCER DE MAMA-OVARIO.o Cuadro clínico. Cáncer de mama.- aproximadamente 1 de cada 12 mujeres en las sociedades occidentales presentará un cáncer de mama, y éste es el cáncer más habitual en la mujer entre los 40 y los 55 años de edad; aproximadamente 1 de cada 3 mujeres afectadas desarrolla una enfermedad metastásica. Entre el 15 y el 20 % de las mujeres con cáncer de mama tienen antecedentes familiares del trastorno. Los estudios familiares han demostrado que el riesgo de que una mujer desarrolle cáncer de mama es mayor cuando en los antecedentes familiares figura uno o más de los siguientes factores: Un grupo de casos entre los familiares femeninos más cercanos. Edad temprana de presentación (<50 años). Presentación de la enfermedad bilateralmente. Aparición adicional de cáncer de ovario. Cáncer de ovario.- aproximadamente 1 de cada 70 mujeres desarrolla un cáncer de ovario, y la frecuencia aumenta con la edad. La mayoría se produce como resultado de alteraciones genéticas en el epitelio de la superficie ovárica y se denomina, por tanto, cáncer de ovario epitelial. En las familias que cuentan con múltiples mujeres afectadas de cáncer de ovario, la edad de presentación es 10 -15 años antes que el cáncer de ovario no hereditario en la población general. o Base genética.- BRCA1;17q11.2 o Patrón hereditario.- autosómico dominante. TUMOR DE WILMS.o Cuadro clínico.- raro neoplasma embrionario renal, que cursa también con aniridia, anomalías genitourinarias y retraso del crecimiento y desarrollo, combinación conocida con síndrome de WARG. 107 o Base genética.- WT1; 11p13. o Patrón hereditario.- NEOPLASIA ENDÓCRINA MÚLTIPLE.o Cuadro clínico.- es una neoplasia en la que una o mas glándulas endocrinas son hiperactivas o forman un tumor. Los síntomas varían de una persona a otra y dependen de la glándula involucrada pueden abarcar dolor abdominal, ansiedad, heces negras y pegajosas, sensación de distensión después de las comidas, ardor, dolor o sensación de hambre en la parte superior del abdomen o en la parte baja del tórax que se alivia con antiácidos o comida. Disminución en el interés sexual, fatiga, dolor de cabeza, ausencia de menstruación, infertilidad, inapetencia, pérdida de vello corporal (en hombres), dolor muscular, náuseas, vómitos, sensibilidad al frío, pérdida involuntaria de peso, problemas de visión, debilidad. o Base genética.- RET; 10q11.2; es un defecto en el gen que porta el código para una proteína que se llama menina o Patrón hereditario.- autosómico dominante. 108 Autor: José Regino Ríos Gutiérrez. Unidad No. 18 Proyecto del genoma Humano. Medicina genómica. Competencia intermedia.- analizar los alcances de la medicina genómica y su aplicación en la práctica clínica. Criterios de desempeño.1.- Analizar los alcances del proyecto del genoma humano.- El concepto de un mapa del genoma humano fue propuesto en 1969 por Víctor McKusick, uno de los padres fundadores de la genética médica. Desde 1973 se realizaron regularmente seminarios sobre el mapa genético humano para recopilar los datos de los mapas. La idea de un proyecto sobre genoma humano específico surgió en una reunión organizada por US Departamento f Energy (DOE) en Santa Fe (Nuevo México) en 1986. En Estados Unidos el Proyecto Genoma Humano comenzó en 1991 con un presupuesto estimado en 200 millones de dólares por año. Otras naciones, sobre todo Francia, Reino Unido y Japón, comenzaron inmediatamente con sus propios programas nacionales de genoma humano, y fueron posteriormente seguidos por otros países. Estos proyectos nacionales individuales fueron coordinados por la organización del genoma humano (HUGO), que tiene tres centros, uno para América, basado en Bethesda (Maryland), otro para Europa, localizado en Londres, y otro para el Pacífico, situado en Tokio. Uno de los objetivos del Proyecto Genoma Humano era la secuencia del genoma humano. La secuenciación, finalizó en 2003 y ha facilitado enormemente la identificación de los genes de las enfermedades humanas. 2.- Definir genoma y genómica e identificar su campo de estudio. GENOMA.- es la totalidad de la información genética que posee un organismo particular. GENÓMICA.- conjunto de ciencias y técnicas dedicadas al estudio integral del funcionamiento, el contenido, la evolución y el origen de los genomas. 3.- Definir y clasificar polimorfismo y analizar la importancia de los polimorfismos en la variabilidad génica. POLIMORFISMO.- Aparición en una población de dos o más formas determinadas genéticamente con una frecuencia tal que la más rara de ellas no pueda justificarse únicamente por una mutación. POLIMORFISMO DE CONFORMACIÓN MONOCATENARO (SSCP).Sistema de detección de mutaciones en el que las diferencias en la estructura tridimensional de un DNA monocatenario dan como resultado una movilidad electroforética en gel diferente en condiciones especiales. POLIMORFISMO EQUILIBRADO.- Dos variantes genéticas distintas presentes de forma estable en una población; las ventajas y las desventajas selectivas se cancelan entre sí. 109 POLIMORFISMO TRANSITORIO.- Dos variantes alélicas diferentes presentes en una población cuyas frecuencias relativas varían debido a una ventaja selectiva o a una desventaja de una o de otra. POLIMORFISMO DE DNA.- variación heredada en la secuencia de nucleótidos no codificadores, del DNA. POLIMORFISMO DE LONGITUD DE LOS FRAGMENTOS RESTRICCIÓN (RFLP).- polimorfismos debidos a la presencia oza<z a la ausencia de un lugar de restricción particular. POLIMORFISMO DE LONGITUD DEL DNA HIPERVARIABLES.- número de tipos diferentes de variaciones en la secuencia del DNA que son altamente polimórficos, por ejemplo; número variable de repeticiones en tándem, mini y microsatélites. POLIMORFISMOS DE NUCLEÓTIDO ÚNICO (SNP).- Variación en la secuencia del DNA de un solo nucleótido que es polimórfica; se presenta cada 1/500 A 1/2000 pares de bases. DE 4.- Definir proteómica y metabolómica y analizar su relación con a medicina molecular. PROTEÓMICA.- es el estudio a gran escala de las proteínas, en particular de su estructura y función. METABOLÓMICA.- es el conjunto de ciencias y técnicas dedicadas al estudio completo del sistema constituido por el conjunto de moléculas que formas los intermediarios metabólicos. 5.- Analizar en forma general el flujo de la información genómica, transcriptómica, proteómica y metabolómica dentro de la célula. 6.- Analizar el impacto del (los) componente(s) genético(s) en la susceptibilidad y resistencia a enfermedades y severidad de los síntomas.una ampliación de la ecogenética que tendrá una importancia particular es la identificación de los individuos con riesgo elevado de desarrollar enfermedades consecutivas a exposiciones ambientales, por ejemplo, cánceres después de la exposición a mutágenos o a carcinógenos. Hay informes de un riesgo aumentado de cáncer vesical en personas que con acetiladores lentos y que han tenido una exposición ocupacional a las aminas aromáticas, que se utilizan como colorantes industriales. También existe la posibilidad de un riesgo aumentado de cáncer vesical para los acetiladores lentos en la población general sin que se haya identificado una exposición peligrosa específica. En sentido contrario, trabajos recientes sugieren la posibilidad de un riesgo aumentado para el cáncer colorrectal en los acetiladores rápidos. Los estudios recientes sugieren que el estado de mal metabolizador de la debrisoquina es menos habitual de lo que cabría esperar en personas con cáncer de pulmón. Esto contrasta con otro polimorfismo para la enzima glutatión S-transferasa (GSTM1), que muestra un aumento en la incidencia del fenotipo nulo (es decir, sin actividad) en personas con adenocarcinoma del pulmón cuando se compara con la población en general. Esta enzima está implicada en la conjugación del glutatión con los compuestos electrofílicos, incluyendo a 110 carcinógenos como el benzopireno, y podría tener un papel protector contra el desarrollo del cáncer. Esta susceptibilidad a la enfermedad no se limita únicamente al cáncer. Muchas de las enfermedades comunes en los seres humanos podrían deberse a diferencia genéticamente determinadas en la respuesta a los agentes ambientales u susceptibilidades. Hay notificaciones de un posible aumento del riesgo de desarrollar enfermedad de Parkinson debido a diferencias en la desintoxicación de las neurotoxinas potenciales junto con un fenotipo de mal metabolizador en el gen CYP2D6 del citocromo hepático P450. La capacidad de detectar sistemáticamente los SPN en grandes cantidades de personas debería permitir la identificación de los genes implicados en determinar la contribución hereditaria de muchas enfermedades comunes. Además de identificar a los individuos con un riesgo elevado de desarrollar una enfermedad común, esto permitiría un mejor conocimiento de las rutas implicadas en la enfermedad, sustentando las promesas de unirlas directamente a posibles intervenciones terapéuticas para estos individuos. No es extraño que las principales compañías internacionales/multinacionales farmacéuticas y biotecnológicas estén invirtiendo fuertemente en estos proyectos. Aunque existe la perspectiva de reducir la probabilidad de que los individuos desarrollen varias de las enfermedades comunes, se plantean muchos problemas sociales y éticos cuando el conocimiento de la variabilidad y de la susceptibilidad genética se traslada al orden público con intereses comerciales creados. El perfil genético es un paso hacia la medicina personalizada. Es una información que puede utilizarse para seleccionar el tratamiento adecuado a la dosis correcta y para evitar reacciones adversas a los fármacos. 7.- Definir farmacogenómica medicamentos. y su aplicación en la respuesta a FARMACOGENÓMICA.- Describe la interacción entre los fármacos y el genoma (es decir, la interacción entre la configuración genética de un individuo y su respuesta a un fármaco). Es importante porque las reacciones farmacológicas adversas son una causa principal de morbimortalidad. Es probable que tenga también una importancia creciente en el futuro, sobre todo como resultado del desarrollo de nuevos fármacos por la información disponible a partir del Proyecto Genoma Humano. Además, estos desarrollos ampliarán nuestro conocimiento sobre las diferencias heredadas que ocasionan la susceptibilidad para las enfermedades comunes, que son consecuencia de la interacción con las exposiciones ambientales u ocupacionales, que se denomina ecogenética. FARMACOGENÉTICA.- Estudio de las variaciones genéticamente determinadas que se ponen de manifiesto únicamente por los efectos de los fármacos. Se utiliza actualmente para describir la influencia de los genes en la eficacia y en los efectos secundarios de los fármacos. 8.- Definir expresión génica. Es el proceso por medio del cual todos los organismos procariotas y eucariotas transforman la información codificada en los ácidos nucleicos en las proteínas necesarias para su desarrollo y funcionamiento. 111 9.- Distinguir las pruebas genómicas y sus aplicaciones: secuenciación genómica, clonación, PCR, microarreglos, en la detección de mutaciones, análisis de ligamento, diagnóstico clínico. SECUENCIACIÓN GENÓMICA.- El genoma humano contiene grandes secciones de DNA repetitivo que a menudo son técnicamente difíciles de clonar y de secuenciar. Una pequeña parte está formada por secuencias o genes expresados. CLONACIÓN.- mitosis repetidas de una célula y que comparten una misma información genética. PCR.- (transcriptasa inversa, RT-PCR), uso de un cebador especial que contiene un promotor y un iniciador de la traducción del mRNA (para la PCR) para fabricar un cDNA. MICROARREGLOS.- Una nueva manera de estudiar cómo interactúan entre sí un gran número de genes y como las redes regulatorias de la célula controlan enormes baterías de genes simultáneamente. Esta técnica crea las micromatrices utilizando un computador para aplicar con alta precisión, gotas minúsculas que contienen ADN de genes sobre un portaobjetos. Luego las placas se hibridizan con ADN complementario marcado en forma fluorescente o radiactiva y un computador mide la intensidad de cada punto fluorescente o radioactivo. Con esto se puede saber que tanto de un fragmento de ADN se encuentra presente y en ciertos tipos de micromatrices también es un indicador de la actividad de un gen especifico. 10.- Fundamentar la importancia de las bases de datos genómicas y las herramientas de la bioinformática. La bioinformática ha sido esencial para el éxito global del Proyecto Genoma Humano. Consiste en establecer dispositivos para la recogida, el almacenamiento, la organización, la interpretación, el análisis y la comunicación de los datos del proyecto que puedan ser ampliamente compartidos por la comunidad científica en su totalidad. Para cualquier implicado en algún aspecto del proyecto del genoma humano es esencial tener un acceso rápido y sencillo a los datos/información que contiene. Esta diseminación de la información se logró con el establecimiento de un gran número de bases de datos electrónicas disponibles en Internet (apéndice). Entre ellas están las bases de datos de secuencias de DNA y de proteínas (por ejemplo: GenBank y EMBL), las bases de datos de los mapas genéticos de los seres humanos (GBD, Genethon, CEPH, CHLC y Whitehead Insitute) y de otras especies (Mouse Genoma Database y base de datos C. elegans), los programas de análisis de ligamiento (por ejemplo; la página de Internet de la Rockefeller University), los datos comentados del genoma (Ensembl and UCSC Genome Bioinformatics) y el catálogo de las enfermedades hereditarias en los seres humanos (Online Mendelian Inheritance in Man, u OMIM). Estos avances en la bioinformática permiten actualmente la 112 posibilidad de identificar secuencias codificadoras y de determinar sus posibles funciones por homología con los genes conocidos, llevando a la perspectiva de identificar un nuevo gen sin la necesidad de ningún trabajo experimental de laboratorio, lo que se ha denominado “clonación in silico”. 11.- Analizar las bases genómicas de enfermedades comunes: asma, obesidad, hipertensión, diabetes, enfermedad cerebro-vascular. ASMA.- El asma es una enfermedad compleja que está asociada con hiperreactividad bronquial y atopía, cuyo fenotipo ha sido difícil de definir clínicamente. A pesar de los grandes avances en el tratamiento, el asma tiene una alta prevalencia, la cual ha ido incrementándose en los últimos años en México. Una de las razones principales del aumento en la prevalencia y morbilidad es la falta de entendimiento de los mecanismos fundamentales que predisponen a un individuo a desarrollar asma. La agregación de la patología en grupos familiares ha hecho suponer la existencia de factores genéticos involucrados. Sin embargo, identificar factores genéticos involucrados en la predisposición del asma es una tarea sumamente difícil en una enfermedad compleja, ya que el diagnóstico clínico es difícil, la expresión clínica es modulada por factores ambientales y además no sigue un patrón de herencia mendeliano. Actualmente, criterios basados en la historia clínica y estudios de laboratorio permiten hacer un diagnóstico confiable de asma y diseñar estudios genéticos más precisos. Los diversos estudios realizados en gemelos y en familias con la patología han sugerido un patrón de herencia multifactorial, cuya expresión puede ser modificada por múltiples factores ambientales e individuales. Los estudios de "pares de hermanos" y los análisis de ligamiento en familias asmáticas han permitido identificar varias regiones cromosómicas ligadas con el fenotipo clínico, incluyendo a los cromosomas 5, 6, 7, 11 y 14. La búsqueda de genes mayores en estas regiones se ha enfocado en los loci que están involucrados con la respuesta alérgica. Entre estos genes destacan los que están ubicados en el cromosoma 5 y que codifican para la IL-9 y la IL-13. El conocimiento de la interacción entre el medio ambiente y los genes implicados en el desarrollo del asma permitirá la identificación de individuos en riesgo de desarrollar asma y la implementación de medidas de prevención hacia el asma. OBESIDAD.- La obesidad es una enfermedad crónica multifactorial de gran trascendencia socio-sanitaria y económica, la cual constituye un serio problema de salud pública a nivel mundial. En México, el aumento en la prevalencia de la obesidad en los últimos siete años ha sido alarmante, ya que más del 70% de la población adulta padece sobrepeso u obesidad. En la mayoría de los casos, la obesidad es el resultado de la interacción tanto de factores genéticos como ambientales. Existen evidencias que sugieren que la acumulación de grasa corporal tiene una base genética, no sólo para las formas monogénicas de obesidad, sino también para la obesidad poligénica común. En esta revisión se describen los principales genes asociados a la obesidad humana, con mayor énfasis en aquellos genes recientemente identificados a través del escrutinio completo del genoma, y cuya participación en la obesidad está aún en discusión. De igual manera, se muestran hallazgos recientes sobre la genética de la obesidad en la población mexicana. El conocimiento de las bases genéticas y moleculares 113 que predisponen al desarrollo de esta patología, podrían contribuir al impulso de acciones preventivas y terapéuticas aplicables en los programas de salud pública para disminuir la prevalencia de la obesidad. HIPERTENSIÓN.- La hipertensión arterial es poligénica y multifactorial ya que resulta de la interacción del medio ambiente con un conjunto de genes que confieren riesgo y/o protección. A pesar de lo expuesto, hoy se sabe que en la hipertensión arterial un 1 a 2% de los casos se explican por formas de transmisión mendeliana simple, como el aldosteronismo remediable por glucocorticoides, el síndrome de exceso aparente de mineralocorticoides y el síndrome de Liddle, en los cuales el mecanismo común es un aumento de la reabsorción de sodio renal. Por lo tanto se podría especular que en la vasta mayoría de los casos, variantes muy prevalentes pero poco penetrantes de ciertos genes podrían conferir susceptibilidad o protección para la afección y que en este grupo de genes se encontrarían los genes "candidatos" que codifican para sustancias muy relacionadas a la función cardiovascular y el balance electrolítico. Entre ellos podemos ubicar aquellos que codifican a los componentes del sistema renina-angiotensina. Actualmente se conocen varios polimorfismos muy prevalentes de estos genes. En el caso del gen de la ACE, el polimorfismo más estudiado corresponde a la inserción/deleción (I/D) de un fragmento de 287 bp en el intrón 16. Aunque no esté asociado a hipertensión, el alelo D conferiría riesgo para sufrir insuficiencia coronaria o hipertrofia ventricular izquierda. En el gen del angiotensinógeno, se conocen muchas variantes pero fundamentalmente dos han sido asociadas a hipertensión arterial, aquellas que presentan metionina o treonina (M/T) en los codones 174 o 235. Las variantes 235T y 174M serían más prevalentes en hipertensos que en normotensos y, como demostramos en población argentina correlacionarían positivamente con los niveles de presión arterial tanto sistólica como diastólica de adultos y adolescentes, ya sea tomadas en consultorio o mediante monitoreo ambulatorio de 24 hs. Por lo tanto, los individuos que portan estos alelos poseen un riesgo relativo mayor de sufrir hipertensión que aquellos que no los portan. La posible asociación de la hipertensión con las variantes moleculares del receptor de la angiotensina II del tipo I que media casi todas los efectos del polipéptido aún está por definirse. En conclusión, el análisis de los defectos genéticos que conllevan a ciertas enfermedades cardiovasculares hoy es posible aunque muy complejo, ya que los genes y el número de variantes de cada gen asociado a cada una de estas afecciones crece diariamente. De hecho pueden contabilizarse más de 150 genes candidatos con 5 a 10 variantes muy frecuentes (polimorfismos de nucleótido único o SNPs) cada uno. Sin embargo, se espera que el desarrollo de técnicas automáticas de secuenciación, la búsqueda de mutaciones mediante matrices de ADN y nuevas formas de análisis nos den parte de la solución a este problema. La caracterización de la base genética que produce o predispone a estas enfermedades podría tener claros beneficios en términos de definir medidas preventivas en individuos en riesgo y/o una terapéutica más racional una vez que la enfermedad está establecida. DIABETES.-Diabetes tipo 1 En la mayor parte de casos de diabetes tipo 1, el paciente debe heredar factores de riesgo de ambos padres. Se cree que estos factores de riesgo deben ser más comunes en la raza blanca, ya que los caucásicos tienen la tasa más elevada de diabetes tipo 1. Puesto que la mayoría de personas que están en riesgo de padecer diabetes, no la padece, los investigadores estudian qué factores del medio ambiente la desencadenan en unas personas y en otras no. Uno de los desencadenantes parece estar relacionado con el tiempo frío. La diabetes tipo 1 aparece más frecuentemente en invierno que en verano y es 114 más frecuente en lugares con climas fríos. Otro desencadenante parece ser los virus. Quizá un virus que tiene solo efectos leves en la mayoría de personas, desencadena una diabetes tipo 1 en otras. La dieta en los primeros meses de vida también parece desempeñar un papel. La diabetes tipo 1 es menos frecuente en personas que han sido amamantadas con lecha materna y en los que los alimentos sólidos han sido introducidos a una edad mayor. En muchas personas, el desarrollo de diabetes tipo 1 parece tardar varios años. Estudios que han seguido a familiares de personas con diabetes tipo 1 han mostrado que la mayoría de estos, que posteriormente padecieron diabetes, tenían ciertos autoanticuerpos en su sangre varios años antes. Los autoanticuerpos son proteínas del sistema inmunitario de defensa que en lugar de destruir bacterias y virus, atacan por error a tejidos del propio organismo. Diabetes tipo 2 La diabetes tipo 2 tiene una base genética más potente que la diabetes tipo 1, pero también depende más de los factores ambientales. La historia familiar de diabetes tipo 2 es uno de los principales factores de riesgo para desarrollar la enfermedad, pero solo entre los que viven en los países occidentales desarrollados. Los norteamericanos y los europeos comen mucha grasa y pocos hidratos de carbono y fibra, y hacen poco ejercicio. La diabetes tipo 2 es más frecuente en personas con estos hábitos. Hay grupos étnicos con mayor riesgo de: africano-americanos, mexicano-americanos e indios Pima. Por el contrario, las personas que viven en áreas no accidentalizadas no suelen tener diabetes tipo 2, independientemente de su riesgo genético. La obesidad es un potente factor de riesgo para la diabetes tipo2. la obesidad es de mayor riesgo para la gente más joven y para los que son obesos desde hace años. La diabetes gestacional es como un rompecabezas. Las mujeres que han desarrollado diabetes durante su embarazo tienen más probabilidades de tener una historia familiar de diabetes, especialmente en su rama materna. Pero, como en otras formas de diabetes, los factores no genéticos desempañan un papel. Las madres de más edad y las obesas tienen mayor probabilidad de padecer diabetes gestacional. ENFERMEDAD CEREBRO-VASCULAR.- La patología de base de las ECV es la aterosclerosis, una enfermedad inflamatoria que se caracteriza por la acumulación de lípidos, células inflamatorias y tejido fibroso en las arterias. Su etiología es multifactorial y compleja, interviniendo tanto factores ambientales como genéticos. Estos factores de riesgo suelen presentarse asociados entre sí, potenciando el riesgo cardiovascular. Los factores de riesgo ambientales suelen ser modificables, lo cual permite prevenir el desarrollo de la ECV. Entre ellos, los tres más importantes son el colesterol elevado, la hipertensión arterial y el tabaquismo. No obstante, debemos también considerar factores como la obesidad, el sedentarismo y la diabetes mellitus. Asimismo, existen otros factores de riesgo que no son modificables, como la edad, el sexo y la base genética. 115 Autor: José Regino Ríos Gutiérrez. Unidad No. 19 Recursos terapéuticos en genética I. Competencia intermedia.- evaluar las opciones y analizar las perspectivas de tratamiento de las enfermedades genéticas a fin de identificar los recursos terapéuticos que permitan el menor deterioro de los pacientes con padecimientos genéticos. Criterios de desempeño.1.- Definir terapia génica. Justificar las alternativas de tratamiento para enfermedades multifactoriales y monogénicas. TERAPIA GÉNICA.- Método que se usa para introducir DNA exógeno dentro de una célula con la finalidad de producir un cambio transitorio o estable dentro de ellas y de esa manera modificar su función. TRANSFECCIÓN.- Se refiere a la transferencia de material genético in Vitro. La terapia génica sólo se hace en células somáticas. OBJETIVOS DE LA TERAPIA GÉNICA.o Incrementar la producción de un producto. o Estudiar el efecto de un producto particular sobre la función y metabolismo celular. o Regular la producción de la célula. o Curación definitva de las enfermedades asociadas con la carencia de… 2.- Analizar los factores de éxito y fracaso en la terapia de enfermedades genéticas: genes no identificados, diagnóstico prenatal, severidad del fenotipo, necesidad de evaluación a largo plazo, heterogeneidad genética. La inserción por terapia génica no «cura» las enfermedades Mendelianas monogénicas dominantes, pues para producir el fenotipo enfermo basta la existencia de un alelo enfermo que el paciente ya tiene. No hay terapia génica concebible para las enfermedades genéticas poligénicas, multifactoriales o de interacciones complejas. Las enfermedades dominantes sólo podrían curarse eficientemente si se cambiara el «gen dominante» que produce la patología y que habitualmente se da en un genotipo heterocigoto (con el alelo normal). La inserción génica puede tener complicaciones como reacciones alérgicas o autoinmunes, inducción de cáncer e interferencia con otras funciones génicas. 116 La heterogeneidad genética en donde las mutaciones en genes localizados en diferentes cromosomas producen expresión similar es una barrera para el uso de la terapia génica, ya que el objetivo esperado de esta es la sustitución de un gen, el cual al no tener una localización exacta en todos los fenotipos causa el fracaso de la terapia 3.- Analizar las estrategias terapéuticas de acuerdo a los niveles de intervención: mutación genética, mutación de RNAm, proteína mutante. Disfunción metabólica, fenotipo clínico, familia. Mutación genética.- Modificación del fenotipo somático (transplantes, transferencia génica). Mutación de RNAm.- Modulación farmacológica de la expresión génica. Proteína mutante.- Control de la actividad enzimática (incremento o sustitución proteíca). Disfunción metabólica.- Reemplazo del producto final. Limitación de sustratos tóxicos. Fenotipo clínico.- Manejo médico-quirúrgico. Familia.- 4.- Fundamentar los recursos terapéuticos de los trastornos metabólicos: restricción dietética, terapia substitutiva, diversificación o uso de vías alternas, inhibición, depleción, aumento de la función de la proteína mutante, substitución proteíca. Estrategias generales.o Entrega de genes ex vivo. o Entrega de genes in vivo. Físicos.o Electroporación. o Disparo de partículas. o Microinyecciones. Químicos.o Coprecipitación con fosfato de calcio. o Precipitación con dietil-amino-etil-dextran. o Liposomas. Biológicos.o Retrovirus. 117 o Adenovirus. o Herpes virus. o Adenovirus relacionados. o Receptores. o Nucleótidos antisentido. o Cromosomas artificiales. Riesgos de las estrategias.o Mutagénesis. o Infección por virus recombinante. o Choque tóxico por viremia. o Transferencia de material génico exógeno no viral. o Contaminación con otros virus u otros organismos deletéreos. o Contaminación del genoma humano. o Alteraciones ecológicas. 5.- Fundamentar la terapia de reemplazo enzimático en las enfermedades por almacenamiento lisosomal: enfermedad de Gaucher y mucopolisacaridosis. TERAPIA DE REEMPLAZO ENZIMÁTICO.o Su fundamento proviene de la observación que muchas enzimas lisosomales pueden ser secretadas y después reabsorbidas por lisosomas en tejidos a distancia y pueden transferirse a células vecinas por contacto celular directo. o La absorción se realiza fundamentalmente a través del receptor manosa 6 fosfato. o Con sólo un 1-5% de la actividad intracelular normal pueden corregirse los defectos metabólicos en las células con deficiencia enzimática. Enfermedad de Gaucher.o Defecto.- deficiencia de glucocerebrosidasa. o Tratamiento con preparados de enzimas y proteínas de betaglucosidasa. Mucopolisacaridosis.- 118 o Defecto.- en la mucolpolisacaridosis VII deficiencia de betaglucoronidasa. En general en las mucopolisacaridosis el defecto es el acumulo de proteoglicanos. o Tratamiento.- transplante de médula ósea. 119 Autor: José Regino Ríos Gutiérrez. Unidad No. 20 Recursos terapéuticos en genética II Competencia intermedia.- Evaluar las opciones y analizar las perspectivas de tratamiento de las enfermedades genéticas a fin de identificar los recursos terapéuticos que permitan el menor deterioro de los pacientes con padecimientos genéticos. Criterios de desempeño.1.- Comparar modulación de la expresión génica, modificación del genoma somático y terapia génica. MODULACIÓN DE LA EXPRESIÓN GÉNICA.- incrementar la expresión de un gen normal que compense el efecto de la mutación en otro locus MODIFICACIÓN DEL GENOMA SOMÁTICO.- se usa en trasplantes donde la celula transplantada tiene el fenotipo del donante. TERAPIA GÉNICA.- Es la introducción deliberada de material genético en células somáticas humanas con fines terapéuticos, profilácticos o diagnósticos. Incluye técnicas para administrar ácidos nucleicos sintéticos o recombinantes en seres humanos; vectores biológicos genéticamente modificados (como los virus o plásmidos), células madre (stem cells) genéticamente modificadas, virus oncolíticos, ácidos nucléicos asociados a vehículos, ácidos nucleicos desnudos, técnicas antisentido (por ejemplo; vacunas génicas), técnicas de DNA o de RNA como la interferencia en el RNA, y xenotrasplante de células animales (pero no de órganos sólidos). 2.- Fundamentar la aplicación y desuso de los métodos de transferencia de genes en: cáncer, inmunodeficiencia severa combinada y deficiencia de adenosin desaminasa. Cáncer.o Aporte de genes supresores de tumor.- se ha propuesto que la introducción dirigida de genes supresores de tumor reconocidos, como Tp53, en las células cancerosas podría ocasionar el control de su crecimiento. Se precisan conocimientos más detallados de la biología del cáncer para poder llevar a cabo este enfoque de manera fiable y segura. o Introducción de genes que lesionan selectivamente a las células cancerosas.- se ha propuesto como método de terapia génica en el cáncer la introducción del gen del factor necrosis tumoral en los linfocitos que infiltran el tumor, que después pueden restituirse al paciente. Más recientemente se ha propuesto la introducción de lo que sea denominado genes condicionales tóxicos o suicidas en las células cancerosas. Un ejemplo es el gen de la timidincinasa del virus del herpes simple, que permite el metabolismo del fármaco ganciclovir por las cinasas celulares para producir la forma trifosfatada que inhibe a la polimerasa de DNA, lo cual da 120 como resultado la muerte de las células cancerosas y de las células circundantes por un efecto “inespecífico” (bystander). También se ha propuesto que los genes “antiangiógenos” podrían utilizarse para alterar el riesgo sanguíneo de los tumores. Un ejemplo es la inhibición del factor de crecimiento endotelial vascular (VEGF angiógeno). Inmunodeficiencia severa combinada.- es un síndrome raro de diversas causas genéticas en las que existe ausencia combinada de linfocitos T y B. en algunos casos existe también la ausencia de las funciones del linfocito asesino natural (NK, natural Killer). Se han identificado 5 tipos: o Deficiencia de la cadena gamma común de los linfocitos T receptores para factores de crecimiento. o Deficiencia de cinasa Janus 3. o Deficiencia de la cadena alfa del receptor IL-7. o Deficiencia de adenosin desaminasa. o Deficiencia de genes activadores de recombinasa. Deficiencia de adenosin desaminasa.- una de las primeras enfermedades en las que se ha intentado la terapia génica en el ser humano es el trastorno de inmudeficiencia hereditaria causado por una deficiencia de adenosina desaminasa (ADA). El tratamiento convencional más satisfactorio de la deficiencia de ADA es el trasplante de médula ósea, pero cuando o se dispone de un donante compatible, a los pacientes se les puede tratar con ADA conjugada con polietilenglicol. En 1990 el primer ensayo sobre terapia génica incorporó a diez pacientes con SCID por deficiencia de ADA. Si bien no se comunicaron efectos adversos, ninguno de los pacientes se “alivio”, probablemente por la escasa eficiencia de la transferencia del gen por le vector retrovírico. Si bien se ha demostrado que los linfocitos T transducidos persisten más de 10 años y todavía expresan ADA transgénica, el efecto terapéutico de la terapia génica sigue siendo difícil de valorar en virtud del tratamiento concomitante con ADA bovina conjugada con ADA-PEG conlleva una importante ventaja selectiva para los linfocitos T de gen corregido que se acompañaba de la restauración de las funciones de los linfocitos T y las respuestas en la producción de anticuerpos a antígenos, pero de una corrección incompleta del defecto metabólico. Más recientemente, mejoras en el protocolo de transferencia de genes en células CD34+ de la médula ósea en combinación con la quimioterapia a dosis baja dio por resultado el implante estable del injerto, le restablecimiento de las funciones inmunitarias, la corrección del defecto metabólico de ADA y una utilidad clínica demostrada, sin la administración de ADA-PEG. 3.- Definir clonación y diferenciar entre clonación de genes y clonación celular y entre clonación terapéutica y clonación reproductiva. Clonación.- Proceso por el que se consiguen copias idénticas de un organismo, célula o molécula ya desarrollado de forma asexual. 121 Clonación de genes.- Es una técnica mediante la cual se selecciona un gen que nos interesa por alguna razón, generalmente porque codifica alguna proteína de interés para el hombre, se introduce en una célula sencilla, generalmente bacteriana, o de algún protista sencillo como las levaduras, y se hace que esa célula se divida muchas veces y que fabrique la proteína que nos interesa; luego se purifica la proteína y se puede distribuir para su uso. Clonación celular.Clonación celular Clonar una célula consiste en formar un grupo de ellas a partir de una sola. En el caso de organismos unicelulares como bacterias y levaduras, este proceso es muy sencillo, y sólo requiere la inoculación de los productos adecuados. Sin embargo, en el caso de cultivos de células en organismos multicelulares, la clonación de las células es una tarea difícil, ya que estas células necesitan unas condiciones del medio muy específicas. Una técnica útil de cultivo de tejido utilizada para clonar distintos linajes de células es el uso de aros de clonación (cilindros). De acuerdo con esta técnica, una agrupación de células unicelulares que han sido expuestas a un agente mutagénico o a un medicamento utilizado para propiciar la selección se ponen en una alta dilución para crear colonias aisladas; cada una proviniendo de una sola célula potencialmente y clónicamentediferenciada. En una primera etapa de crecimiento, cuando las colonias tienen sólo unas pocas células; se sumergen aros estériles de poliestireno en grasa, y se ponen sobre una colonia individual junto con una pequeña cantidad de tripsina. Las células que se clonan, se recolectan dentro del aro y se llevan a un nuevo contenedor para que continúe su crecimiento. Clonación reproductiva.- Tiene como objetivo obtener individuos adultos. Se presenta como una moderna técnica de reproducción asexuada asistida. 4.- Analizar los principios básicos de la terapia de reemplazo celular: célula totipotencial, pluripotencial, multipotencial, unipotencial, plasticidad, transdiferenciación y fisión celular. Células madre.- células precursoras capaces de renovarse a sí mismas. Células totipoteciales.- células capaces de diferenciarse células posibles, incluso en membranas extraembrionarias. Células pluripotenciales.- masa celular interna. No trofoblasto. Células multipotenciales.- forman capas germinativas, no embrión. Células unipotenciales.- o progenitoras. Células con la habilidad de desarrollar una sola línea de tipo de células. Plasticidad.- es la capacidad de diferenciación que tienen las células madre. Primero se pensaba que una célula podía diferenciarse en otra y no volver a su estado original, sin embargo las evidencias experimentales demuestran que si son capaces de hacerlo. Esto se debe a que la diferenciación no es un proceso irreversible mediado por ejemplo con en todas las 122 elecciones o reordenaciones génicas, sino que es reversible y se produce gracias silenciamientos epigenéticos o activación de genes silenciados. Transdiferenciación.- tiene lugar cuando una célula que no es célula madre se transforma en otro tipo diferente de célula, o cuando una célula madre ya diferenciada crea células fuera de su ruta de diferenciación ya establecida. Es un tipo de metaplasia que incluye todos los cambios en el destino de una célula incluyendo la interconversión de células madre. Fisión celular.- es una de las tres formas de división celular, relativamente simple; se denomina también fisión binaria, los organismos como las bacterias tienen un solo cromosoma. Al inicio del proceso de la fisión binaria, la molécula de ADN del cromosoma de la célula se replica, produciendo dos copias del cromosoma. Un aspecto clave de la reproducción celular de las bacterias es asegurarse de que cada célula hija recibe una copia del cromosoma. Citocinesis es la separación física de las dos células hijas nuevas. 5.- Comparar las fuentes de células madre: médula ósea, cordón umbilical y otros tejidos y las indicaciones de su uso en las diferentes enfermedades genéticas: neurodegenerativas, inmunodeficiencias y hepáticas. Células madre.- células que tienen la capacidad de autorrenovarse mediante divisiones mitóticas o bien de continuar la vía de diferenciación para la que está programada y, por lo tanto, producir células de uno o más tejidos maduros, funcionales y plenamente diferenciados en función de su grado de multipotencialidad. Células madres embrionarias.- son aquellas que forman parte de la masa celular de un embrión de 4-5 días de edad y tienen la capacidad de formar todos los tipos celulares de un organismo adulto. Células madres adulto.- son células no diferenciadas que se encuentran en tejidos y órganos adulto y que poseen la capacidad de diferenciarse para dar lugar a células adultas del tejido en el que se encuentran, por lo tanto se consideran células multipotenciales. En el individuo adulto se conocen hasta ahora alrededor de 20 tipos distintos de células madre, que son las encargadas de regenerar tejidos en continuo desgaste (como la piel y la sangre) o dañados (como el hígado). Su capacidad es mas limitada para generara células especializadas. Célula madre de médula ósea.- las células madre hematopoyéticas de la medula ósea (encargadas de la formación de la sangre) son las más conocidas y empleadas en la clínica desde hace tiempo. Células madres de cordón umbilical.- al ser inmunológicamente inmaduras son más proliferas, no tienen el mismo proceso de envejecimiento, ni exposición a agentes infecciosos externos. Son 100% compatibles con el dueño de ellas y altamente compatibles con los padres y hermanos del dueño. 123 Células madre en otros tejidos.- células madre fetales, aparecen en los órganos y tejidos fetales como hígado, sangre y pulmón y poseen características similares a sus homólogas en tejidos adultos, aunque parecen mostrar mayor capacidad de expansión y diferenciación. Su procedencia no ésta del todo clara. En el tejido adiposo se han encontrado también células madre denominadas mesenquimal que pueden diferenciarse en numerosos tipos de células de los tres derivados embrionarios (musculares, vasculares, nerviosas, etc) Tratamientos con las células madre.o La mayoría se encuentran en estado experimental. o El uso de las células madre adulto, implica menos riesgos de rechazo y también menos problemas éticos y legales. 124 Autor: José Regino Ríos Gutiérrez. Unidad No. 21 Genética de poblaciones. Competencia intermedia.- fundamentar la utilidad de programas de tamizaje poblacional basados en la epidemiología de los padecimientos genéticos. Criterios de desempeño.1.- Analizar el origen de la variabilidad genética. Se refiere a la variación en el material genético de una población o especie, e incluye los genomas nucleares, mitocondrial y ribosomal, además de los genomas de otros orgánelos. La variabilidad genética nueva puede estar causada por mutaciones, recombinaciones y alteraciones en el cariotipo (el número, forma, tamaño, y ordenación interna de los cromosomas). Los procesos que afectan la variabilidad genética son la selección natural y la deriva genética. 2.- Analizar la epidemiología de las enfermedades genéticas en México y en diferentes poblaciones: judíos Ashkenazi, africanos y caucásicos. La epidemiologia de los judíos Ashkenazi indica de acuerdo a su ADN mitocondrial que provienen de solo cuatro mujeres en Europa, por lo tanto y por la consanguineidad los hace mas propensos a sufir de ciertas enfermedades como lo son Síndrome de Bloom, Enfermedad de Canavan, Fibrosis quística, Disautonomía familiar o síndrome de Riley-Day, Anemia de Fanconi, Enfermedad de Gaucher, Distonía de torsión idiopática (DTI) Distonía generalizada, Mucolipidosis IV, Enfermedad de Niemann-Pick y Enfermedad de Tay-Sachs, las enfermedades genéticas en africanos mas comunes son la talasemia, la drepanocitoscis o anemia falciforme, en caucásicos la fibrosis quística 3.- Analizar las causas de mutación. Mutación somática y mutación de la línea germial.- la mutación somática es la que afecta a las células somáticas del individuo. Como consecuencia aparecen individuos mosaico que poseen dos líneas celulares diferentes con distinto genotipo. Una vez que una célula ha sufrido una mutación, todas las células que deriven de ella, heredarán la mutación (herencia celular). Las mutaciones de la línea germinal son las que afectan a las células productoras de gametos apareciendo de este modo, gametos con mutaciones. Estas mutaciones se transmiten a la siguiente generación y tiene mayor importancia desde el punto de vista evolutivo. Tipos de mutación según su consecuencias.- 125 o Mutaciones morfológicas.- afectan a la morfología del individuo, a su distribución corporal. Suele producir malformaciones. o Mutaciones letales y deletéreas.- son las que afectan la supervivencia de los individuos, ocasionándoles la muerte antes de la madurez sexual. Cuando la mutación no produce la muerte, sino una disminución en la capacidad del individuo para sobrevivir y/o reproducirse, se dice que la mutación es deletérea. Este tipo de mutaciones suelen producirse en cambios inesperados en genes que son esenciales o imprescindibles para la supervivencia del individuo. En general las mutaciones letales son recesivas, es decir, solo se manifiestan en homocigosis o bien en hemicigosis para aquellos genes ligados al cromosoma X en humanos. o Mutaciones condicionales.- son aquellas que se presentan en el fenotipo mutante en determinadas condiciones ambientales (condiciones restrictivas), mostrando la característica silvestre en las demás condiciones del medio ambiente (condiciones permisivas). o Mutaciones bioquímicas o nutritivas.- son los cambios que generan una pérdida o un cambio de alguna función bioquímica como, por ejemplo, la actividad de una determinada enzima. o Mutaciones de pérdida de función.- suelen determinar que la función de un gen no lleven a cabo bien su función, por lo que desaparece la función del organismo que la presenta. Suelen ser recesivas. o Mutaciones de ganancia de función.- cuando ocurre un cambio de ADN, lo más normal es que corrompa algún proceso normal del ser vivo. Sin embargo, existen raras excepciones donde una mutación puede producir una nueva función del gen, generando un fenotipo nuevo. Si ese gen mantiene la función original, o si se trata de un gen duplicado, puede dar lugar a un primer paso en la evolución. Tipos de mutación según el mecanismo causal.o Según el mecanismo que haya provocado el cambio en el material genético, se suele hablar de tres tipos de mutaciones, mutaciones cariotípicas o genómicas, mutaciones cromosómicas y mutaciones génicas o moleculares. o Mutaciones cariotípicas o genómicas.- son las mutaciones que afectan al número de cromosomas o todo el complemento cromosómico (todo el genoma). Poliploidía.- aumento del número normal de juegos de cromosomas. Los seres pueden ser autopoliploides si todos los juegos proceden de la misma especie, o alopoliploides si proceden de la hibridación, es decir, del cruce de dos especies. 126 o Haploidía.- provocan la disminución en el número de juegos de cromosomas. Aneuploidía.- mutaciones que afectan solo a un número de ejemplares de un cromosoma o más, pero sin llegar a afectar al juego completo. Pueden ser monosomías, trisomías, tetrasomías, etc. Cuando en lugar de dos ejemplares de cada tipo de cromosomas, que es lo normal, hay o sólo uno, o dos o cuatro, etc. Mutaciones cromosómicas.- son modificaciones en el número total de cromosomas, la duplicación o supresión de genes o de segmentos de un cromosoma y la reordenación del material genético dentro o entre cromosomas. Las alteraciones de la dotación diploide de cromosomas se denominan aberraciones cromosómicas o mutaciones cromosómicas. Hay tres tipo de mutaciones cromosómicas: Reordenamientos cromosómicos.- implican cambios en la estructura de los cromosomas (duplicación, delección, inversión y traslocación) Aneuploidías.- supone un aumento o disminución en el número de cromosomas. Poliploidía.cromosomas. presencia de conjuntos adicionales de o Mutaciones génicas o moleculares.- alteran la secuencia de nucleótidos del ADN. Pueden llevar a la sustitución de aminoácidos en las proteínas resultantes (mutaciones no sinónimas). Un cambio en un solo aminoácido puede no ser importante si es conservatorio y ocurre fuera del sitio activo de la proteína. Así, existen las denominadas mutaciones sinónimas o “mutaciones silenciosas” en las que la mutación altera la base situada en la tercera posición del codón pero no causa sustitución aminoacídica debido a la redundancia del código genético. El aminoácido insertado será el mismo que antes de la mutación. En el caso de las mutaciones neutras, el aminoácido insertado es distinto pero con unas propiedades fisicoquímicas similares. También pueden considerarse neutras aquellas mutaciones que afectan una zona del genoma sin función aparente, como las repeticiones en tándem o dispersas, las zonas intergénicas y los intrones. La mutación génica o puntual, puede tener consecuencias fatales. o Bases moleculares de las mutaciones génicas. Mutación por sustitución de bases.se produce al cambiar de posición un par de bases por otro: Mutaciones transicionales.o simplemente transiciones, cuando un par de bases es sustituido por una alternativa del mismo tipo. 127 Mutaciones transversionales.- o transversiones, cuando un par de bases es sustituida por otra del otro tipo. Mutaciones de corrimiento estructural.- cuando se añaden o se quitan pares de nucleótidos alterándose la longitud de la cadena. Si se añaden o quitan pares en un número que no sea múltiplo de tres (es decir, si no se trata de un número exacto de codones), las consecuencias son especialmente graves, porque a partir de ese punto y no sólo en él, toda la información queda alterada. Hay dos casos: Mutación de pérdida o delección de nucleótidos, en la secuencia de nucleótidos se pierde uno y la cadena se acorta en una unidad. Mutación por inserción de nucleótidos, dentro de la secuencia del ADN se introducen nucleótidos adicionales, interpuestos entre los que ya había alargándose la cadena. Mutaciones en los sitios de corte y empalme (Splicing).las mutaciones de corrimiento del marco de lectura también pueden surgir por mutaciones que interfieren con el Splicing del ARN mensajero. El comienzo y final de cada intrón en un gen están definidos por secuencias conservadoras de ADN. Si un nucleótido muta en una de las posiciones altamente conservada, el sitio no funcionara más, con las consecuencias predecibles para el ARNm maduro y la proteína codificada. 4.- Definir probabilidad y heredabilidad. Probabilidad.- Proporción de veces en que aparece un resultado en una serie amplia de sucesos. Heredabilidad.- Proporción de la variación total de un carácter atribuible a la genética en vez de a los factores ambientales. 5.- Calcular la tasa de mutación en diferentes condiciones genéticas y relacionarlo con probabilidad y heredabilidad.6.- Analizar los supuestos para la ley de Hardy-Weinberg, evolución, mutación, deriva genética, efecto fundador. Ley de Hardy-Weinberg.- La relación entre las frecuencias alélias y las genotípicas fue establecida de forma independientemente por Godfrey Hardy y Wilheim Weinberg y se denomina la ley de Hardy-Weinberg. Podemos emplear esta ley para calcular las frecuencias alélicas y genotípicas cuando no es posible distinguir los homocigotos dominantes de los heterocigotos. Éste suele ser el caso de las enfermedades recesivas como la fibrosis quística. La ley de Hardy-Weinberg establece que la frecuencia de aa debería ser q2. Para la fibrosis quística en la población de raza blanca q2=1/2500 (la prevalecía de la enfermedad entre los recién nacidos). Para calcular q, 128 hallamos la raiz cuadrada a ambos lados de la ecuación q=√1/2500 = 1/50= 0.02. Dado que p + q= 1 , p= 0.98 , podemos calcular la frecuencia de los genotipos de AA y Aa. Este último genotipo, que representa a los heterocigotos portadores del alelo de la enfermedad, tiene especial interés. Dado que p está próximo a 1, podemos simplificar el cálculo redondeando p hasta 1; sin una pérdida significativa de precisión. Encontramos entonces que 2pq = 2q = 2/50 = 1/25 . Esto quiere decir que mientras la incidencia de homocigotos afectados sólo es de 1/2500 , los portadores heterocigotos de los genes de la enfermedad son mucho mas frecuentes, 1 de cada 25 individuos. Equilibrio de Hardy-Weinberg.- mantenimiento de las secuencias de alelos en una población con apareamiento aleatorio y ausencia de selección. Fórmula de Hardy-Weinberg.- ecuación binomial simple de la población genética que puede utilizarse para determinar la frecuencia de los distintos genotipos de uno de los fenotipos. Principio de Hardy-Weinberg.- las proporciones relativas de los distintos genotipos permanecen constantes de una generación a la otra. Evolución.- proceso de cambio continuo y progresivo de un órgano u organismo por el cual se hace cada vez más complejo por diferenciación de sus partes. Mutación.- cambio en el material genético ya sea de un gen único o en el número o estructura de los cromosomas. La mutación que ocurre en los gametos es hereditaria; la mutación en las células somáticas no es hereditaria. Deriva genética.- es una fuerza evolutiva que actúa junto con la selección natural cambiando las características de las especies en el tiempo. Efecto fundador.- ciertos trastornos genéticos pueden ser relativamente habituales en poblaciones particulares a través de todos los individuos que descienden de un número relativamente pequeño de antepasados,o algunos de los cuales tenía un trastorno particular. 7.- Fundamentar el uso de la ley de Hardy-Weinberg para calcular la frecuencia de los genes en la población, teniendo como ejemplo el sistema ABO.8.- Evaluar los principios y utilidad del tamizaje poblacional.El tamizaje poblacional se define como el uso de una prueba sencilla en una población saludable, para identificar a aquellos individuos que tienen alguna patología, pero que todavía no presentan síntomas. Garantiza acceso a toda la población y el seguimiento al paciente. Ayuda a la detección e intervención temprana de las enfermedades, detecta personas en riesgo y se puede confirmar la enfermedad antes de que aparezcan los síntomas para comenzar el tratamiento. Los principios son: Las muestras se pasan por una máquina que las pica y extrae una ruedita de 3,2 milímetros de diámetro para enviarla al laboratorio, después 129 analizan todas las muestras de sangre, un espectrómetro de masas analiza las muestras. Sirve para diagnosticar muchas enfermedades Checar el siguiente link: o http://www.uacj.mx/ICB/RedCIB/MaterialesDidacticos/Monografas/Pr uebas%20de%20Tamiz.pdf 130 Autor: José Regino Ríos Gutiérrez. PREGUNTAS TIPO TEST Capítulo 2.- Bases celular y molecular de la herencia. 1.- Sustituciones de bases.a. Pueden dar lugar a mutaciones terminadoras. Verdadera. Cuando un codón de terminación sustituye a un aminoácido. b. Pueden afectar al proceso de corte y empalme (splicing) Verdadera. Por ejemplo, por mutación de los sitios de corte y empalme (splicing) conservados del donante y del aceptante. c. Son siempre patogénicas. Falsa. Las mutaciones o sustitución silentes en las regiones no codificadoras pueden que no sean patogénicas. d. Pueden afectar a la expresión del gen. Verdadera. Por ejemplo las mutaciones en el promotor pueden afectar a la unión de los factores de trasncripción. e. Ocasionan mutaciones del marco de lectura. Falsa. Los cambios en los marcos de lectura están causados por la inserción o la delección de nucleótidos. 2.- Transcripción.a. Describe la producción de polipéptidos a partir de la plantilla de mRNA. Falsa. Durante la transcripción, el mRNA se produce a partir de la platilla de DNA. b. Se produce en el núcleo. Verdadera. El producto de mRNA se transloca después al citoplasma. c. Produce un mRNA monocatenario utilizando la hebra de DNA antisentido como plantilla. Verdadera. El mRNA es complementario a la hebra antisentido. d. Está regulada por los factores de transcripción que se unen en la UTR en 3´. Falsa. Los factores de transcripción se unen a las secuencias reguladoras en el promotor. e. Precede al encaperuzamiento en 5´y a la poliadenilación. 131 Verdadera. La adición de la caperuza en 5´y la cola poli (A) en 3´facilitan el trasnporte al citoplasma. 3.- Están directamente implicados en la reparación del DNA.a. Glucosilasas. Verdadera. La MYH DNA glucosilasa stá implicada en la reparación por escisión base. b. DNA polimerasas. Verdadera. Incorporan las bases correctas. c. Ligasas. Verdadera. Sellan las brechas después de la escisión de la base anómala y corrigen la inserción de la base. d. Corte y empalme. Falsa. El proceso de corte y empalme (splicing) elimina los intrones durante la producción de la base. e. Ribosomas. Falsa. Los ribosomas están implicados en la traducción. 4.- Durante la replicación del DNA.a. La helicasa del DNA separa al DNA bicantenario. Verdadera. Desenrrolla la hélice del DNA. b. El DNA se sintetiza en una dirección. Falsa. La replicación se produce en ambas direcciones. c. Se sintetizan los fragmentos de Okazaki. Verdadera. Estos fragmentos están unidos por la DNA ligasa para formar la hebra retardada. d. El DNA se sintetiza de forma conservadora. Falsa. La replicación del DNA es semiconservadora ya que sólo una de las hebras se sintetiza de nuevo. e. El uracilo se inserta para emparejarse con la adenina. Falsa. El uracilo se incorpora en el mRNA, la timina en el DNA. Capítulo 3: cromosomas y división celular.1.- La meiosis se diferencia de la mitosis de las siguientes formas.a. Las células hijas son haploides, no diploides. 132 Verdadera. Durante la meiosis humana el número de cromosomas se reduce de 46 a 23. b. La meiosis está restringida a los gametos y la mitosis se produce únicamente en las células somáticas. Falsa. Las divisiones celulares iniciales en la gametogénesis son mitóticas; la meiosis se produce únicamente en la división final. c. En la mitosis hay una única división. Verdadera. En la meiosis las dos divisiones se conocen como se conocen como meiosis I y II. d. La meiosis genera la diversidad genética. Verdadera. Los ambivalentes se separan de forma independiente durante la meiosis I y se producen entrecruzamientos (quiasmas) entre los cromosomas homólogos. e. La profase de la mitosis consta de una sola etapa; en la meiosis I hay cuatro etapas. Falsa. Las cinco etapas de la profase en la meiosis I son: leptoteno, zigoteno, paquiteno, diploteno, diacinesis. 2.- Las anomalías cromosómicas microscopio óptico son: detectadas con fiabilidad por el a. Trisomía. Verdadera. Un cromosoma extra (por ejemplo, el cromosoma 21 en el síndrome de Down) se observa fácilmente. b. Monosimía. Verdadera. Un cromosoma que falta (por ejemplo, el síndrome de Turner en las mujeres con un único cromosoma X) se observa fácilmente. c. Translocación recíproca. Falsa. Puede que no sea visible una translocación sutil. d. Delección intersticial. Falsa. Una pequeña delección puede no ser visible. e. Translocación robertsoniana. Verdadera. La fusión en el centro de los brazos largos de dos cromosomas acrocéntricos se detecta fácilmente. 3.- La hibridación in situ por flourescencia utilizando todo el cromosoma (pintura) o sondas en locus específicos permite la detección rutinaria de: a. Amplificación del gen. 133 Falsa. Los cambios en la dosis génica pueden identificarse por hibridación genómica comparativa (CGH). b. Delección subtelomérica. Verdadera. Las sondas subteloméricas son útiles para la investigación de las dificultades del aprendizaje inespecíficas. c. Trisomía. Verdadera. Las trisomías pueden detectarse en las células en interfase. d. Cromosomas marcadores supernumerarios. Verdadera. El origen de los cromosomas marcadores puede determinarse por la pintura cromosómica. e. Translocación recíproca. Verdadera. Las reconfiguraciones sutiles pueden detectarse por la pintura cromosómica. 4.- Los compuestos químicos utilizados en la preparación de los cromosomas en la metafase para su análisis por microscopia óptica incluyen: a. Colchicina. Verdadera. La colchicina inhibe la formación del huso, deteniendo por tanto a las células en la metafase. b. Fitohemaglutinina. Verdadera. Las fitohematoglutinina estimula la división celular de los linfocitos T. c. Giemsa. Verdadera. El Giemsa s usa para teñir cromosomas con color rosa/púrpura. d. Quinacrina. Falsa. La quinacrina es un colorante fluorescente no visible con el microscopio óptico. e. Salino hipotónico. Verdadera. El suero salino hipotónico inflama las células, causando la lisis celular y la diseminación de los cromosomas. Capítulo 4.- Tecnología y aplicaciones del DNA. 1.- Las siguientes afirmaciones corresponden a las enzimas de restricción.a. Pueden generar fragmentos de DNA con extremos “cohesivos”. 134 Verdadera. El DNA bicantenario se puede someter a digestión para producir unos extremos sobresalientes (COHESIVOS) o extremos romos. b. Son víricos en origen. Falsa. Se han asilado más de 300 enzimas de restricción de diversas bacterias. c. Se utilizan para detectar las mutaciones puntuales. Verdadera. Si la mutación crea o destruye un sitio de reconocimiento. d. Se utilizan en la transferencia Southern. Verdadera. La digestión del DNA por una enzima de restricción es el primer paso para la transferencia Southern. e. Se denomina también exonucleasa de restricción. Falsa. Son endonucleasas porque digieren fragmentos de DNA internamente, en oposición a la digestión por exonucleasas desde los extremos 5´o 3´de los fragmentos de DNA. 2.- Describen la reacción en cadena de la polimerasa (PCR).a. Tipo de clonación sin células. Verdadera. Se pueden producir millones de copias de DNA a partir de una plantilla sin utilizar vectores de clonación. b. Proceso que utiliza una DNA polimerasa termolábil. Falsa. La PCR utiliza a la polimerasa Taq termoestable, ya que se requiere una elevada temperatura desnaturalizante (alrededor de 95ºC) para separar los productos bicentenarios en el comienzo de cada ciclo. c. Un método muy sensible para amplificar el DNA que tiene tendencia a la contaminación. Verdadera. La PCR puede utilizarse para amplificar el DNA de células únicas (por ejemplo, en el diagnóstico genético preimplantación), por lo que las medidas de control apropiadas son importantes para evitar la contaminación. d. Técnica que puede amplificar rutinariamente hasta 100kb de DNA. Falsa. La PCR amplifica rutinariamente objetivos de hasta 1 kb y la PCR de largo alcance está limitada a alrededor de 40 kb. e. Método para amplificar los genes que no requiere un conocimiento previo de la secuencia. Falsa. El conocimiento de la secuencia es necesario para designar a los cebadores que flanquean la región de interés. 3.- Los tipos de hibridación del ácido nucleico incluyen.a. Transferencia Southern. 135 Verdadera. La transferencia Southern describe la hibridación de una sonda marcada radiactivamente con fragmentos de DNA separados por electroforesis. b. Micromatrices (microarrays). Verdadera. La hibridación entre el objetivo y la sonda de DNA tiene lugar en un portaobjetos de vidrio. c. Transferencia Western. Falsa. La transferencia Western se utiliza para analizar la expresión de la proteína utilizando métodos de detección de anticuerpos. d. Transferencia de Northern. Verdadera. La transferencia Northern se utiliza para examinar la expresión del RNA. e. Huella génica o del DNA (fingerprinting). Verdadera. La huella génica del DNA (fingerprinting) utiliza una sonda de DNA minisátelite para hibridarse a fragmentos de DNA hipervariables. 4.- Técnicas que se pueden utilizar para detectar a los genes con mutaciones desconocidas.a. Secuenciación. Verdadera. Se puede utilizar la secuenciación para detectar mutaciones conocidas o desconocidas y caracterizar a una mutación desconocida. b. Polimorfismo de conformación monocatenario (SSCP). Verdadera. El SSCP es un método barato para detección selectiva de las mutaciones aunque su sensibilidad es limitada. c. Cromatografía (DHPLC). líquida desnaturalizante de alto rendimiento Verdadera. La DHPLC es un método eficaz para detectar las mutaciones heterocigóticas. d. Análisis de ligamento del oligonucleótido. Falsa. La OLA se utiliza para detectar mutaciones conocidas ya que el diseño de la sonda es específico de la mutación. e. PCR en tiempo real. Falsa. La PCR en tiempo real se utiliza también para detectar mutaciones conocidas ya que el diseño de la sonda es específico de la mutación. 136 Capitulo 5: Mapeo e identificación de genes en los trastornos monogénicos. 1.- La clonación posicional utiliza: a. Bases de datos genéticas. Verdadera. Ahora que la secuencia del genoma humano está completa, es posible identificar un gen asociado a una enfermedad in silico. b. Conocimiento de los genes ortólogos. Verdadera. Una vez que un gen se ha mapeado en una región puede ser útil comprobar las regiones sintéticas en modelos animales. c. Pacientes con anomalías cromosómicas. Verdadera. Se han identificado muchos genes mediante el mapeo de la translocación o la delección de los puntos de rotura. d. Genes candidatos seleccionados por conocimiento biológico. Falsa. La clonación posicional describe la búsqueda de genes basándose en su localización. e. Marcadores microsatélites. Verdadera. Una exploración de todo el genoma utiliza marcadores microsatélites localizados por todo el genoma para el mapeo de ligamiento. 2.- Es probable que un gen candidato esté asociado a una enfermedad sí: a. Una mutación de pérdida de función causa el fenotipo. Verdadera. Esto implica causalidad. b. Un modelo animal con una mutación en el gen ortólogo tiene el mismo fenotipo. Verdadera. Esto es una evidencia fuerte. c. Múltiples mutaciones diferentes causan el fenotipo. Verdadera. Esto excluye la posibilidad de que una única variante sea un arcador en un desequilibrio de ligamiento más que una mutación patogénica. d. El patrón de la expresión del gen es consecuente con el fenotipo. Verdadera. Por ejemplo, cabría esperar que un gen asociado con la ceguera se expresase en el ojo. e. Es un seudogén. Falsa. Un seudogén no codifica a una proteína funcional y por tanto no es probable que las mutaciones sean patogénicas. 3.- Los logros del Proyecto Genoma Humano son: 137 a. Borrador de la secuencia publicado en 2000. Falsa. El borrador se completó en el 2000, pero su fecha de publicación fue febrero de 2001. b. Secuenciación completada en 2003. Verdadera. La secuencia se terminó 2 años antes de la fecha prevista original. c. Desarrollo de herramientas bioinformáticas. Verdadera. Se desarrollaron herramientas de anotaciones como Ensembl para ayudar a los usuarios. d. Identificación de todos los genes causantes de enfermedad. Falsa. Casi 1500 se han identificado hasta la fecha. e. Estudios de problemas éticos, legales y sociales. Verdadera. Cerca del 5% del presupuesto de Estados Unidos para el Proyecto Genoma Humano se dedicó a estudiar estos problemas. Capítulo 6: Genética del desarrollo: 1.- En el desarrollo, los genes HOX: a. Funcionan como factores de transcripción. Verdadera. Son importantes para la determinación espacial y el modelado. b. Una vez mutados se ha demostrado que están relacionados con muchos síndromes malformativos. Falsa. Actualmente, sólo unos cuantos síndromes malformativos pueden ser directamente atribuidos a mutaciones en el gen HOX, probablemente por una compensación paráloga. c. Muestran estructuras muy divergentes según las distintas especies. Falsa. Contienen una importante homebox conservando de 180 mb. d. Son funcionalmente redundantes en la vida posnatal. Verdadera. Probablemente sólo tienen importancia al inicio del desarrollo. e. Pueden ser importantes individualmente para el desarrollo normal de sistemas corporales muy diferentes. Verdadera. En los sitios en que se han identificado mutaciones causantes de malformaciones, puede que haya diferentes sistemas orgánicos implicados, por ejemplo, el síndrome de mano-pie-genital (HOXA13). 138 2.a. Las gastrulación es el proceso que conduce a la formación de un embrión inicial de 16 células a los 3 días de la fertilización. Falsa. Ocurre más tarde y es el proceso de dejar el eje primario del cuerpo en la segunda y tercer semanas. b. La organogénesis tiene lugar entre las 8 y las 12 semanas de gestación. Falsa. La organogénesis tiene lugar principalmente entre las 4 y las 18 semanas de gestación. c. Las rutas señalizadoras notch y Sonic hedgehog son importantes para garantizar el desarrollo normal en los diversos órganos y tejidos. Verdadera. Lo genes en estas rutas se expresan ampliamente por todo el cuerpo. d. Los somitas se forman mesodermo presomítico. en dirección cuadorrostral desde el Falsa. Los somitas se forman en dirección rostro-caudal. e. Los genes TBX parecen ser cruciales para el desarrollo normal del miembro. Verdadera. Cuando mutan, estos genes producen el síndrome ulna-mamario y el síndrome de Holt-Oram. 3.a. En el desarrollo de los mamíferos la mandíbula se forma del segundo arco faríngeo. Falsa. Se forma del primer arco faríngeo (branquial). b. Las arterias del arco faríngeo se convierten finalmente en los grandes vasos alrededor del corazón. Verdadera. Se produce un remodelado de forma que estos vasos se convierten en grandes arterias. c. TBX1 es un gen clave en los defectos relacionados con el síndrome de Di George. Verdadera. Esto se ha establecido en los animales y se está demostrando que tanga una alta similitud en los seres humanos. d. La acodroplasia puede estar causada por una amplia variedad de mutaciones en el gen FGFR3. 139 Falsa. La acondroplasia se asocia con mutaciones FGFR3 muy específicas que afectan a la parte de unión a la membrana de la proteína. e. Las mutaciones de pérdida de función y de ganancia de función causan habitualmente defectos similares. Falsa. Estos tipos diferentes de mutaciones habitualmente causan fenotipos muy distintos, por ejemplo, el gen RET. 4.a. En la mayoría de los varones con el cariotipo 46, XX el gen SRY está presente y se encuentra en uno de los cromosomas X. Verdadera. A veces el gen SRY está implicado en la recombinación con las regiones pseudoautosómicas de X e Y. b. En la Iyonización, o inactivación del cromosoma X, todos los genes de uno de los cromosomas X están inactivados. Falsa. No todas las regiones del cromosoma X se inactivan, de lo contrario no habría presumiblemente efectos fenotípicos en el síndrome de Turner. c. Como resultado de la Iyonización, mosaicos del cromosoma X. todas las mujeres son Verdadera. Sin embargo, esto sólo tiene consecuencias cuando hay un gen mutado en un cromosoma X. d. El desarrollo fetal masculino depende únicamente de que el gen SRY tenga un funcionamiento normal. Falsa. El SRY tiene una importante función de iniciación, pero hay otros genes muy importantes. e. La inactivación del cromosoma X puede estar ligada de alguna forma al proceso gemelar monocigótico. Verdadera. Algunos fenómenos inusuales que se producen en gemelos llevan a la conclusión de que estos procesos están relacionados. 5.- Factores de transcripción: a. Son secuencias del RNA que interfieren con la traducción en los ribosomas. Falsa. Habitualmente son proteínas que se unen a secuencias de DNA reguladoras específicas. b. Su única función es inactivar a los genes durante el desarrollo. Falsa. También activan genes. c. Cuando se mutan en los segmentos corporales de Drosophila pueden estar completamente reorganizados. 140 Verdadera. Por ejemplo, se puede desarrollar una pierna en vez de una antena. d. No están implicados en los defectos de la lateralidad. Falsa. Los factores de transcripción son cruciales para la lateralidad normal. e. Incluyen genes que tienen un motivo de dedo de cinc (zinc finger). Verdadera. El motivo dedo de cinc (zinc finger) codifica una proyección de aminoácidos tipo dedo que forma un complejo con ion cinc. Capítulo 6, preguntas basadas en casos: Caso 1.- un niño de 2 años se remite a un genetista por presentar una circunferencia cefálica superior al percentil 97, aunque su desarrollo es paralelo a las líneas del centil. A los padres les gustaría tener otro hijo y preguntan por el riesgo de recidiva. Los ventrículos cerebrales están dilatados y han tenido muchas discusiones con neurocirujanos sobre la posible derivación ventriculo-peritoneal. Al hacer la historia de la familia completa, se descubre que la abuela paterna está en revisión con el dermatólogo por lesiones cutáneas, algunas de las cuales se han extirpado, y un tío paterno ha tenido varios quistes dentales eliminados por un dentista hospitalario. 1.- ¿Hay un diagnóstico que abarque las diversas características de los distintos miembros de la familia? La combinación de macrocefalia, queratoquistes odontógenos y carcinomas de células basales aparece en el síndrome de Gorlin (carcinoma nevoide de las células basales). Este trastorno debe incluirse en el diagnóstico diferencial de un niño con macrocefalia, e investigar adecuadamente los antecedentes familiares. Es comprensible que la hidrocefalia sea la principal preocupación, pero la hidrocefalia verdadera no es habitual en el síndrome de Gorlin. 2.- ¿Qué pruebas serían adecuadas para el padre del niño y cuál es la respuesta a la pregunta de la pareja sobre el riesgo de recidiva? El padre del niño es un portador obligado de la mutación del gen PTCH que causa el síndrome de Gorlin en la familia. Debe someterse a exploraciones regulares (al menos anualmente) mediante una radiografía para los queratoquistes odontógenos y estar vigilando regularmente por un dermatólogo para los carcinomas de las células basales. El análisis de la mutación del gen PTCH es posible en cualquier miembro afectado de la familia. Caso 2.- Llevan a una niña de 4 años a la consulta de un pediatra por dificultades de conducta, incluyendo problemas con el control de esfínteres. El pediatra decide haber pruebas de los cromosomas de la niña porque ha visto previamente un caso de un síndrome de 47,XXX (triple X) en que la niña presentaba dificultades de conducta. Para su sorpresa, el resultado cromosómico es 47, XY, es decir, “la niña” es genéticamente “masculina”. 141 1.- ¿Cuáles son las causas más importantes de inversión sexual en un niño de 4 años que es fenotípicamente femenino y por lo demás se encuentra en buena salud física? Las dos causas más probables de inversión del sexo en una “niña” joven son el síndrome de insensibilidad a los andrógenos, que es un trastorno ligado al cromosoma X y que se debe a mutaciones en el gen del receptor de los andrógenos, y las mutaciones en el gen SRY en el cromosoma Y. 2.- ¿Qué debería decir el pediatra a los padres y qué pruebas deberían realizarse? Se puede realizar un análisis de la mutación en los genes, tanto del receptor de los andrógenos como el SRY, par determinar la base genética de la inversión sexual. Es muy importante investigar y localizar, si están presentes, los restos de tejido gonadal, porque tendrán que ser eliminados para evitar el riesgo de un cambio maligno. Po resta causa, hay que dar a los padres una explicación completa, pero el sexo fenotípico del niño debe ser reafirmado como femenino. Capítulo 7: Patrones de Herencia. 1.- Relativos a la herencia autosómica recesiva. a. Las mujeres tienen más probabilidad de estar afectadas que los hombres. Falsa. La ratio sexual es la misma. b. Si ambos padres son portadores, el riesgo en la concepción de que cualquier hijo sea portador es de ¾. Falsa. El riesgo en el momento de la concepción es de ½. c. Las enfermedades que siguen este patrón hereditario son más comunes en las sociedades donde el matrimonio entre primos es normal. Verdadera. Todas las personas portan genes mutados; es más probable que los primos compartan un gen mutado heredado de un abuelo común. d. Habitualmente es una sola generación la que tiene individuos afectados. Verdadera. Los individuos afectados tienen que emparejarse con un portador o con otra persona afectada para que su descendencia resulte también afectada. e. El síndrome de Angelman sigue este patrón. Falsa. Los mecanismos que ocasionan el síndrome de Angelman son variados, pero la herencia autosómica recesiva no es uno de ellos. 2.- Relativos a la herencia ligada al cromosoma X. a. El trastorno no puede transmitirse desde el padre afectado al hijo. 142 Verdadera. Un padre transmite su cromosoma Y a su hijo. b. Cuando es recesivo, un hombre afectado no verá el trastorno en su hijo pero puede aparecer en su nieto. Verdadera. Pueden tener nietos afectados a través de sus hijas que son partadoras. c. Cuando es dominante, las mujeres están gravemente afectadas como los hombres. habitualmente tan Falsa. Aunque el trastorno afecta a las mujeres, los hombres están más gravemente afectados porque la mujer tiene una copia del gen en su otro cromosoma X, y la inactivación del cromosoma X significa que la copia normal se expresa en cerca de la mitad de sus tejidos. d. Cuando es dominante, hay habitualmente más mujeres afectadas que hombres afectados en una familia. Verdadera. Todas las hijas de un hombre afectado estarán afectadas, pero ninguno de los hijos. e. El riesgo de mosaicismo de la línea germinal no necesita ser tenido en cuenta. Falsa. Siempre hay que considerar un mosaicismo de la línea germinal cuando aparezca un caso asilado de un trastorno ligado al cromosoma X. 3.a. La heteroplasmia se refiere a la presencia de más de una mutación en las mitocondrias. Falsa. Esto se refiere a dos poblaciones de DNA mitocondrial, una normal y otra mutada. b. Los genes mitocondriales mudan con menor frecuencia que los genes nucleares. Falsa. Lo opuesto, probablemente porque se replican con mayor frecuencia. c. Los trastornos mitocondriales afectan únicamente al músculo y al tejido nervioso. Falsa. Cualquier tejido con mitocondrias puede estar afectado. d. El riesgo de transmitir un trastorno mitocondrial a la siguiente generación puede ser tan alto como el 100%. Verdadera. Si los ovocitos de la mujer afectada contienen únicamente mitocondrias afectadas. e. Las enfermedades mitocondriales no tiene relación con los genes nucleares. 143 Falsa. Muchas proteínas mitocondriales están codificadas por genes nucleares. 4.a. La heterogeneidad de locus significa que la misma enfermedad puede estar causada por genes en cromosomas diferentes. Falsa. La misma enfermedad causada necesariamente en diferentes cromosomas. por distintos genes- pero no b. Las seudodominancia se refiere al riesgo para la descendencia cuando ambos padres tienen el mismo trastorno heredado de forma dominante. Falsa. El patrón básico de herencia en la seudodominancia es autosómico recesivo. c. Si un trastorno muestra una penetrancia reducida puede saltar generaciones. Verdadera. Una parte de los individuos con el gen mutado no muestran signos ni síntomas. d. La expresión variable caracteriza a las enfermedades que muestran anticipación. Verdadera. Las enfermedades que presentan anticipación muestran una gravedad aumentada, y una edad de aparición cada vez más temprana, en las generaciones sucesivas. e. El pleiotropismo es simplemente una forma más llamativa de expresión variable. Falsa. No una variación en la gravedad (expresión variable) sino dos o más efectos aparentemente no relacionados del mismo gen. 5.a. Un trastorno autosómico recesivo puede en ocasiones surgir de una disomía uniparental. Verdadera. Ambas copias de un gen mutado pueden transmitirse de esta forma a un hijo. b. Los genes con impronta pueden ponerse de manifiesto por una disomía uniparental. Verdadera. Esto explica una parte de los casos de síndromes de Prader-Willi y de Angelman. c. La herencia digénica es simplemente otra forma de referirse a la disomía uniparental. Falsa. La herencia digénica se refiere a la heterocigosidad para dos genes distintos que causan un fenotipo. 144 d. Los factores hormonales pueden ser responsables de los trastornos que muestran una influencia del sexo. Verdadera. Esto explica la calvicie presenil y la gota. e. La mayoría del genoma humano está sujeto a la impronta genómica. Falsa. Sólo una pequeña proporción. Capítulo 7: Preguntas basadas en casos. Caso 1.- Un hombre de 34 años ha desarrollado cierta espasticidad en las piernas durante los últimos años, y su familia ha notado algunos problemas de memoria y de alteración en la conducta. Tiene reflejos periféricos muy aumentados. Es atendido con su madre en la clínica genética y se detecta que ella tiene reflejos periféricos significativamente aumentados en el examen pero sin problemas de salud. Recuerda que su propio padre tuvo probablemente problemas similares a los de su hijo cuando tenía 30 años, pero murió en la guerra. 1.- ¿Cuáles son los patrones de herencia que hay que considerar en este caso? Es posible que los problemas descritos en los miembros de la familia no estén relacionados, pero no es probable. Si el trastorno se ha transmitido desde el abuelo, la herencia mitocondrial es poco pausible. El trastorno puede ser autosómico dominante con variabilidad, o ligado al cromosoma X. 2.- ¿Qué posibilidades diagnósticas hay que considerar? Las ataxias espinocerebelosas son un grupo genéticamente heterogéneo de trastornos que, por lo general, siguen una herencia autosómica dominante y pueden presentarse de esta forma. Es posible que haya una forma de paraparesia espástica hereditaria, también genéticamente heterogénea y que sigue habitualmente un herencia autosómica dominante, aunque se han descrito las formas recesiva y ligada al cromosoma X. Aparte de éstas, hay que considerar la adrenoleucodistrofia ligada al cromosoma X, sobre todo porque el hombre tiene signos de problemas cognitivos y conductuales. Esto es muy importante, no sólo porque puede presentarse precozmente en la vida, sino también por la posibilidad de una insuficiencia suprarrenal. Caso 2.- Una pareja tiene un hijo que ha presentado algunas fracturas óseas al pirncipio de su infancia consecutivas a un traumatismo menor y se le dice que probablemente sea una forma leve de osteogénesis imperfecta. Los padres no tuvieron fracturas durante la infancia, y cuando tiene otro hijo que también presenta fracturas se les dice que la herencia es autosómica recesiva. Se les explica también que el trastorno no tiene porqué aparcer en la descendencia de los niñso afectados en el futuro. 1.- ¿Es correcta la información dada a los padres? La información puede ser correcta, pero probablemente no lo sea y hay que explicarles otras posibilidades. 145 2.- Si no lo es, ¿cuál es el patrón de herencia y la explicación más probable para la recidiva de las fracturas en el hermano? La mayoría de las formas de osteogénesis imperfecta (enfermedad de los huesos frágiles) sigue una herencia autosómica dominante. La recidiva en los hermanos, cuando ninguno de los padres tiene signos o síntomas, puede explicarse por un mosaicismo somático y/o de la línea germinal en uno de los padres. El riesgo para la descendencia de estar afectado sería por lo tanto del 50% (es decir, alto). En este caso se debe considerar la posibilidad de un diagnóstico no genético, principalmente una fractura ósea no accidental. Por tanto, es importante tratar de confirmar el diagnóstico. Capítulo 8: Matemáticas y genética de poblaciones. 1.- Al aplicar el equilibrio de Hardy-Wienberg se hacen las siguientes asunciones: a. La población es pequeña. Falsa. La población debe ser amplia para aumentar la probabilidad del apareamiento no aleatorio. b. No hay consaguinidad. Verdadera. La consaguinidad es una forma de apareamiento no aleatorio. c. No hay nuevas mutaciones. Verdadera. La introducción de nuevos alelos crea nuevas variables. d. No hay niños nacidos por inseminación de donante. Verdadera. En teoría, si la población es pequeña y los donantes se utilizan muchas veces, esto introduciría una forma de apareamiento no aleatorio. e. No hay un movimiento significativo de la población. Verdadera. La migración introduce nuevos alelos. 2.- Si la incidencia en la población de una enfermedad recesiva es de 1 por 10,000, la frecuencia de portador en la población es: a. 1 por cada 100 Falsa. b. 1 por cada 200 Falsa. c. 1 por cada 25 Falsa. d. 1 por cada 50 146 Verdadera. La frecuencia de portador es el doble de la raíz cuadrada de la incidencia. e. 1 por cada 500 Falsa. 3.- Ventaja del heterocigoto: a. Puede producir un aumento en la incidencia de los trastornos autosómicos dominantes. Falsa. Se refiere a los trastornos que siguen una herencia autosómica recesiva. b. No significa que la eficacia biológica esté aumentada en el estado homocigótico. Verdadera. El homocigoto puede mostrar una eficacia biológica marcadamente reducida, por ejemplo, la fibrosis quística. c. Puede explicar la distribución mundial de la enfermedad de las células falciformes y del paludismo. Verdadera. Las personas con rasgo de células falciformes son más capaces de eliminar a las células parasitadas de la circulación. d. Puede llevar una distorsión del equilibrio de Hardy-Weinberg. Verdadera. Puede haber un efecto de un proceso de ventaja selectiva. e. Es muy poco probable que pueda ser restreada hasta un efecto fundador. Falsa. La presencia del alelo en una población puede ser de hecho un efecto fundador. 4.- Loci polimórficos: a. Se definen como los loci en los que hay al menos dos alelos, cada uno con frecuencia superior al 10% Falsa. Los alelos necesitan tener frecuencia de 1% solamente. b. Han sido cruciales para los descubrimientos génicos. Verdadera. Son cruciales para el mapeo génico en virtud de su cosegregación con la enfermedad. c. Pueden ser útiles para determinar el estatus genético de cada uno en una familia. Verdadera. El análisis de ligamiento utilizando locus polimórficos puede ser la única forma de determinar el estatus genético en el diagnóstico presintomático y en las pruebas prenatales. d. No tienen nada que ver con el cálculo de las puntuaciones LOD. 147 Falsa. La asociación de locus polimórficos con la segregación de la enfermedad es clave para calcular una puntación LOD. e. Por ellos mismos, no tienen consecuencias para la enfermedad genéticamente determinada. Falsa. Pueden ser importantes, por ejemplo, los grupos sanguíneos. 5.- En la genética de poblaciones: a. Para calcular la tasa de mutación de un trastorno es necesario conocer únicamente la eficacia biológica del trastorno. Falsa. Hay que conocer también la incidencia de la enfermedad. b. Si el tratamiento médico puede mejorar la eficacia biológica, la secuencia de un trastorno autosómico dominante aumentará más rápidamente que la de un trastorno autosómico recesivo. Verdadera. En la enfermedad autosómica recesiva la mayoría de los genes en la población están presentes en la heterocigotos no afectados. c. Incluso cuando se estudia un número grande de familias, la ratio de segregación calculada para un trastorno puede no arrojar las cifras esperadas para un patrón hereditario dado. Verdadera. En los trastornos recesivos no se detectará a los hermanos afectados. d. Para los trastornos ligados al cromosoma X la frecuencia de los hombres afectados iguala a la frecuencia del alelo mutado. Verdadera. Básicamente, sólo los hombres resultan afectados y siempre manifiestan el trastorno. e. El mapeo de la autocigosidad es una estrategia útil para buscar el gen en un trastorno autosómico recesivo. Falsa. Es útil sólo cuando hay un antepasado común en ambos lados de la familia, es decir, una endogamia. Capítulo 8: Preguntas basadas en casos. Caso 1.- La incidencia de cierto trastorno autosómico recesivo en la población. A está bien establecido en aproximadamente 1 de cada 10,000, mientras que, en la población B, la incidencia del mismo trastorno es mucho más elevada, aproximadamente 1 de cada 900. Un hombre del primer grupo de población y una mujer del segundo están planeando casarse y formar una familia. Conscientes de la relativamente alta incidencia del trastorno en la población B, buscan consejo genético. 1.- ¿Cuál es la pregunta esencial que cabe esperar de cada individuo? Está claro que es esencial saber si se ha dado algún caso del trastorno en cuestión en las familias de alguno de los dos consultantes. Si ha sido así, modificaría 148 potencialmente el riesgo de portador para uno de los consultantes independientemente de la frecuencia de la enfermedad en la población. 2.- ¿Cuál es el riesgo de que el trastorno ocurra en el primer embarazo, basándose en la aplicación del equilibrio de Hardy-Weinberg? Asumiendo que el trastorno en cuestión no ha ocurrido previamente en la familia, la frecuencia de portador de la población A es de 1 de cada 50 y de 1 de cada 15 en la población B. El riesgo en el primer embarazo es por tanto 1/50 X 1/15 X 1/14 = 1/3000* Caso 2.- La neurofibromatosis tipo 1 es un trastorno mendeliano relativamente habitual. En una encuesta poblacional de 50,000 personas en una cuidad, se identifican doce casos de los cuales ocho pertenecen todos a una gran familia afectada. 1.- Basándose en estas cifras, ¿cuál es la tasa de mutación en el gen de la neurofibromina? A partir de las cifras dadas, cuatro casos en la cuidad parecen ser nuevas mutaciones, es decir, 4 nuevas mutaciones por 100,000 genes heredados. La tas de mutación es por tanto de 1 por 25,000 gametos. 2.- Citar algunas limitaciones para la validez de los cálculos de la tasa de mutación a partir de una encuesta como ésta. La muestra de la población es pequeña y puede, por tanto, no ser representativa de toda la población. Por ejemplo, si existe un sesgo hacia una población jubilada de mayor edad, la subpoblación reproductora puede ser más pequeña y las cifras estar distorsionadas por la emigración de las personas jóvenes fuera de la cuidad. Además, los cuatro casos de “nuevas mutaciones” debe verificarse mediante el examen adecuado de los padres. Capítulo 9: Herencia poligénica y multifactorial. 1.- En relación con el autismo. a. Está mejor clasificado como un error congénito del metabolismo. Falsa. Es un trastorno del desarrollo nervioso y no se encuentran anomalías metabólicas. b. La tasa de de concordancia aproximadamente del 50%. en los gemelos dicigóticos es Falsa. Esto implicaría una herencia autosómica dominante. La tasa es cercana al 20%. c. El síndrome del cromosoma X frágil es una causa principal. Falsa. Aunque el autismo se produce en el síndrome del cromosoma X frágil, la gran mayoría de los individuos afectados no tienen este trastorno. 149 d. El riesgo para los hermanos aproximadamente del 5%. de una persona afectada es Verdadera. La cifra está cercana al 3% para el autismo completamente desarrollado y otro 3% para características más leves-trastorno en el espectro autista. e. Las niñas se ven afectadas con mayor frecuencia que los niños. Falsa. La ratio hombre:mujer es de aproximadamente 4:1 2.- En el análisis de ligamientos es más difícil en los trastornos multifactoriales que en los trastornos monogénicas porque: a. Es probable que las variantes en más de un gen contribuyan al trastorno. Verdadera. La detección de los poligenes con pequeños efectos es muy difícil. b. Es probable que el número de personas afectadas en una familia sea menor que en un trastorno monogénico. Verdadera. Es un trastorno monogénico con penetrancia total es más fácil encontrar familias con meiosis suficientemente informativas. c. La forma de herencia es habitualmente incierta. Verdadera. El análisis de ligamiento paramétrico requiere que se conozca el modo de herencia. d. Es probable que algunos trastornos multifactoriales tengan más de una etiología. Verdadera. Pueden estar implicados diferetes factores genéticos y ambientales. e. Muchos trastornos aparición. multifactoriales tienen una edad tardía de Verdadera. La edad tardía de aparición significa que el estatus de la afección puede ser incierto. 3.- Estudios de asociación: a. Pueden dar resultados falsos positivos debidos a la estratificación de la población. Verdadera. La enfermedad y la variante probadas pueden ser habituales en un subconjunto de la población pero no hay una relación causal. b. Incluyen la prueba de la transmisión/desequilibrio (TDT). Verdadera. Las pruebas TDT utilizan controles familiares y de esta forma se evitan los efectos de estratificación de la población. c. Los estudios de asociación positivos deben reproducirse. 150 Verdadera. La repetición de estudios con resultados positivos en distintas poblaciones aumentará la evidencia de una asociación. d. Se utilizan para mapear genes en los trastornos multifactoriales. Falsa. Los estudios de asociación se utilizan para comprobar variantes identificadas por técnicas de mapeo génico, incluyendo el análisis de las parejas de hermanos afectados. e. Requieren grupos estrechamente. de pacientes y controles que se ajusten Verdadera. Se pueden perder las variantes con pequeños efectos si los pacientes y los controles no se ajustan perfectamente. 4.- Las variantes en los genes que confieren susceptibilidad a la diabetes tipo 2 (T2DM) se han encontrado: a. Por análisis de ligamiento utilizando parejas de hermanos afectados. Verdadera. El gen calpain-10 se identificó por clonación posicional en parejas de hermanos mexicano-americanos. b. Utilizando modelos animales. Falsa. No se ha identificado ningún gen de susceptibilidad T2DM confirmado con este método. c. Por estudios del gen candidato a partir de subtipos monogénicas de diabetes. Verdadera. Los ejemplos incluyen dos subtipos de diabetes juvenil de aparición en la madurez. d. Por el estudio de los candidatos biológicos. Verdadera. Los genes que codifican las subunidades del canal de potasio en las células beta pancreáticas eran candidatos biológicos. e. En poblaciones aisladas. Verdadera. Por ejemplo, la variante G319S del HNF-1ª se ha notificado únicamente en la población Oji Cree. 5.- Variantes en el gen NOD2/CARD15: a. Están asociadas con la enfermedad de Crohn y la colitis ulcerosa. Falsa. La evidencia hasta la fecha apoya su papel en la enfermedad de Crohn pero no en la colitis ulcerosa. b. Pueden dar lugar a un aumento de 40 veces en el riesgo de la enfermedad. 151 Verdadera. El aumento del riesgo se calcula en 40 veces para los homocigotos y 2.5 veces para los heterocigotos. c. Se identificaron después de que el gen se mapease en el cromosoma 16p12 por clonación posicional. Verdadera. Una exploración de todo el genoma para la enfermedad inflamatoria intestinal identificó inicialmente la región 16p12. d. Ha llevado a nuevas terapias. Falsa. El gen NOD2/CARD15 activa a NF-kappaB, pero este complejo es ya el objetivo de los fármacos más eficaces utilizados para tratar la enfermedad de Crohn. e. Son muy raras en la población general. Falsa. Las tres variantes notificadas se encuentran con una frecuencia del 5% en la población general, en comparación con el 15% en los pacientes con enfermedad de Crohn. Capítulo 9: preguntas basadas en casos: Caso1.- una chica de 16 años pide anticonceptivos orales a su médico de familia. Al interrogar acerca de los antecedentes familiares, se descubre qe su madre sufrió una trombosis venosa profunda a la edad de 40 años y murió tras una embolia pulmonar a los 55 años. No hay otros antecedentes familiares importantes. 1.- ¿Cuál es la prueba genética adecuada? La prueba del factor V de Leyden y de la variante de la protrombina G20210A adecuada. Un resultado positivo proporcionará de forma más exacta su riesgo desarrollar un tromboembolismo y aportará información para su elección anticonceptivos. La heterocigosidad para el factor V de Leyden o para la variante la protrombina G20210A aumentaría su riesgo en cuatro o en cinco veces. homocigosidad o la heterocigosidad compuesta aumentarían su riesgo hasta en veces. es de de de La 80 2.- ¿Cuáles son las limitaciones de las pruebas en esta situación? Los resultados negativos para el factor v de Leyden y la variante de la protrombina G20210A en la consultante debe interpretarse con cautela, ya que hasta el 50% de los casos de trombosis venosa no están asociados con estos factores de riesgo genético. Caso 2.- Una mujer de 35 años es diagnosticada de diabetes y comienza con un tratamiento de insulina. Ella y su hermano de 29 años fueron adoptados y no tuvieron contacto con sus padres biológicos. Su hermano no tiene síntomas de hiperglucemia. Ambos tienen una audición normal, sin otros signos significativos. 1.- ¿Qué posible subtipos de diabetes puede tener y cuáles son las formas de herencia de estos subtipos? 152 El probando puede tener una diabetes tipo 1 (DM1), una diabetes tipo 2 (DM2) o una diabetes juvenil de aparición en el adulto (MODY). Ya que ambos tienen una audición normal, no es probable el diagnóstico de diabetes y sordera heredados por vía materna (MIDD). Las DM1 y DM2 muestran una herencia multifactorial, y hay factores ambientales que desempeñan un papel además de los factores de susceptibilidad genética predisponentes. La MODY presenta una herencia autosómica dominante. 2.- Para cada uno de estos subtipos, ¿cuál es riesgo de que su hermano desarrolle una diabetes? El riesgo del hermano de desarrollar diabetes es de 6, del 35 o del 50%, respectivamente. Si se descubre que su hermana tiene una mutación es uno de los genes que ocasionan una MODY, entonces se le podrían hacer pruebas genéticas predictivas. Una prueba positiva permitiría un control regular para hacer un diagnóstico precoz de la diabetes y evitar las complicaciones diabéticas debidas a la diabetes no diagnósticada de larga duración. Capítulo 10: Hemoglobina y hemoglobinopatías. 1.a. La cadena de hemoglobina fetal, gamma, recuerda a la cadena beta del adulto. Verdadera. La población debe ser amplia para aumentar la probabilidad del apareamiento no aleatorio. b. Las cadenas de hemoglobina alfa, beta y gamma se expresan todas durante la vida fetal. Falsa. Esto es cierto para la cadenas alfa y gamma únicamente; la cadena beta aparece hacia el final de la vida fetal. c. En una talasemia alfa hay demasiadas cadenas alfa. Falsa. Hay demasiadas pocas cadenas alfa, que son sustituidas por cadenas beta. d. La hemoglobina de Barts es una forma de talasemia beta. Falsa. Es una forma de talasemia alfa. e. Los portadores de la talasemia beta padecen frecuentemente anemia sintomática. Falsa. Tienen una anemia leve y los síntomas clínicos con raros. 2.- En relación con la enfermedad de células falciformes: a. El efecto de falciformidad de los eritrocitos es el resultado de una fijación anormal de la hemoglobina con la membrana del eritrocito. Falsa. El efecto se debe a una solubilidad reducida y a la polimeración. 153 b. Puede producirse una trombosis con amenaza para la vida. Verdadera. La obstrucción de las arterias puede ser el resultado de la crisis de falciformidad. c. La hemoglobina S difiere de la hemoglobina A normal por la sustitución de un único aminoácido. Verdadera. Un residuo valina está sustituido por un residuo ácido glutámico. d. Puede haber un infarto esplénico, peto tiene pocas consecuencias clínicas. Falsa. Del infarto esplénico puede resultar una sepsis con amenaza para la vida. e. Las mutaciones puntuales (de cambio de aminoácido) son la causa habitual de una hemoglobina anómala en los trastornos falciformes. Verdadera. Estas mutaciones dan lugar a una sustitución de aminoácidos. 3.a. Muchas variantes de la hemoglobina son inocuas. Verdadera. Esto se aplica a la mayoría de los que se conocen. b. Los tipos de mutación que se producen en las homoglobinopatías son muy limitados. Falsa. Se conocen todos los tipos de mutación. c. En las talasemias hay una hipoplasia de la médula ósea. Falsa. Aparece una hiperplasia de la médula ósea, que lleva a cambios físicos como el engrosamiento de la bóveda craneal. d. En las talasemias la hemoglobina muestra una afinidad anómala por el oxígeno. Verdadera. El oxígeno no se libera tan fácilmente en los tejidos. e. En algunas talasemias se produce un aumento de la hemólisis de los eritrocitos. Verdadera. La hemoglobina H, por ejemplo, es inestable. 4.a. La persistencia de la hemoglobina fetal en la vida de adulto es un trastorno adquirido. Falsa. Es un trastorno hereditario. b. Durante la vida fetal es el hígado el que produce la mayor parte de la hemoglobina corporal. 154 Falsa. Esto es cierto únicamente entre los 2 y los 7 meses de gestación. c. La médula ósea no está implicada en la producción de hemoglobina antes del nacimiento. Falsa. La médula ósea comienza a producir hemoglobina desde los 6-7 meses de vida fetal. d. El hígado continúa produciendo hemoglobina hasta el segundo año de vida posnatal. Falsa. La producción cesa desde los 2 a los 3 meses de vida posnatal. e. La persistencia de la hemoglobina fetal en la vida del adulto es un trastorno benigno. Verdadera. No da lugar a síntomas: las cadenas de hemoglobina producidas son normales. Capítulo 10: Preguntas basadas en casos. Caso1.- Una pareja china residente en Reino Unido ha tenido dos embarazos y en ambos bebés nacieron muertos y edematosos (hidropesía fetal). Estos embarazos ocurrieron cuando vivían en Asia y no tiene hijos vivos. Buscan un consejo genético sobre las posibilidades de que esto suceda de nuevo, pero no disponen de registros médicos de los embarazos. 1.- ¿Qué posibilidad diagnóstica hay que considerar? El origen étnico de la pareja y la poca información deben sugerir la posibilidad de un trastorno hematológico. La talasemia alfa es la causa probable de nacimiento del niño muerto y la hidropesía es secundaria a la insuficiencia cardíaca, que a su vez es secundaria a la anemia. La isoinmunización con Rhesus y la deficiencia de la glucosa-6-fosfato deshidrogenasa son otras posibilidades. Las formas graves de cadiopatía congénita se asocian frecuentemente con la hidropesía, pero las posibilidades de una recurrencia en el hermano (como ocurrió en este caso) son bajas. Sin embargo, hay muchas otras causas de hidropesía que necesitarían ser consideradas. Entre las que son genéticas con posibilidad de recidiva están las formas letales de las raras displasias esqueléticas y unamplio rango de enfermedades metabólicas. 2.- ¿Cuáles son las pruebas adecuadas para esta situación? Se debe hacer a la pareja un hemograma completo, grupos sanguíneos electroferesis de la hemoglobina y valoración de los anticuerpos maternos y de la deficiencia de la glucosa-6-fosfato deshidrogenasa. El análisis de DNA puede detectar la mutación habitual observada en el sudeste de Asia, lo que posibilitaría proponer un diagnóstico genético prenatal mediante muestras de las vellosidades coriónicas. Si no se identifica ningún trastorno con estas pruebas, no es probable que se haga ningún progreso diagnóstico posterior, a menos que la pareja tenga otro embarazo afectado que pueda ser completamente investigado mediante el examen del feto. 155 Caso 2.- Un adolescente cuyos padres son originarios de la India occidental es ingresado desde el servicio de urgencias tras presentar dolor abdominal grave y febrícula. Se sospecha un abdomen agudo y el paciente se somete a laparotomía por una posible apendicitis. Sin embargo, no se identifica ninguna patología quirúrgica. Posteriormente la orina aparece oscura. 1.- ¿Qué otras pruebas pueden ser adecuadas en esta etapa? Esta presentación concuerda con la porfiria intermitente aguda y con el síndrome hemolítico urémico. Sin embargo, el origen étnico debe sugerir la posibilidad de la enfermedad de las células falciformes. El contenido de la orina oscura y las pruebas específicas para la porfiria ayudarán a diferenciarlas, y hay que realizar las pruebas para las células falciformes. 2.- ¿Qué seguimiento es el adecuado? Si el diagnóstico es enfermedad de las células falciformes, se puede intentar utilizar diversos agentes para reducir la frecuencia de las crisis de falciformidad, en particular la hidroxiurea. La penicilina profiláctica es importante para reducir el riesgo de las infecciones neumocócicas graves y hay que proponer a la familia el consejo genético y la detección selectiva en cascada de los familiares. Capítulo 11: Genética Bioquímica. 1.- En la hiperplasia suprarrenal congénita: a. Las mujeres puede presentar virilización y genitales ambiguos. Verdadera. El defecto enzimático más habitual es la deficiencia de la 21hidroxilasa. b. Los hombres ambiguos. pueden mostrar inframasculinización Verdadera. Esto ocurre en las formas raras: deshidrogenasa, 5 alfa-reductasa y desmolasa. deficiencias y genitales de 3beta- c. El déficit de mineralcorticoides puede ser una amenaza para la vida. Verdadera. La hiponatremia y la hiperpotasemia pueden ser graves y producir un colapso circulatorio. d. Se requiere tratamiento durante la infancia pero habitualmente no en la vida de adulto. Falsa. El cortisol y la fludrocortisona se requieren durante toda la vida en la hiperplasia suprarrenal congénita con pérdida salina. e. En las mujeres afectadas la fertilidad está básicamente conservada. Falsa. La fertilidad está reducida en la forma pierde sal. 2.- Fenilcetonuria: 156 a. Es la única causa de un valor de fenilalanina elevado en el período neonatal. Falsa hay una forma benigna así como anomalías en la síntesis del cofactor. b. Requiere tratamiento durante toda la vida. Falsa. La restricción dietética de la fenilalanina es necesaria únicamente durante la infancia y el embarazo. c. Es una causa de epilepsia y de eccema. Verdadera. Éstas son características que aparecen si no se trata. d. Da como resultado valores reducidos de melanina. Verdadera. Los individuos afectados tienen un pigmento reducido y son rubios. e. Forma parte de la misma ruta que la producción de colesterol. Falsa. Una ruta diferente. 3.- La hepatomegalia es una característica importante de: a. Síndrome de Hurler. Verdadera. La hepatomegalia mucopolisacaridosis. es característica de la mayoría de las b. Trastornos del almacenamiento de glicógeno. Verdadera. La hepatomegalia es característica de la mayoría de los trastornos por almacenamiento del glucógeno, aunque no de todos. c. Anomalías del metabolismo de la porfirina. Falsa. No es característica, incluso en las denominadas porfirias hepáticas. d. Enfermedad de Niemann-Pick. Verdadera. Ésta es una almacenamiento de lípidos. de las esfingolipidosis, o enfermedades por e. Galactosemia. Falsa. La cirrosis puede presentarse en los no tratados. 4.- En relación con los trastornos mitocondriales: a. Todos siguen una herencia matrilineal. Falsa. También se aplican los principales patrones de herencia cuando las proteínas mitocondriales están codificadas por los genes nucleares. b. La pigmentación retiniana y la diabetes pueden ser características. Verdadera. Especialmente en NARP y MIDD, respectivamente. 157 c. Hay menos de 50 productos génicos del genoma mitocondrial. Verdadera. Son 37 productos génicos. d. En la enfermedad de Leigh se está siempre causada por la misma mutación puntual. Falsa. La enfermedad de Leigh es genéticamente heterogénea. e. El gen del síndrome de Barth se conoce pero la ruta metabólica es incierta. Verdadera. El gen G4.5 aparece mutado, el ácido 3-metilglutacónico urinario está elevado, pero la relación sigue estando por dilucidar. 5.a. Los ciclos de la carnitina y de los ácidos grasos de cadena larga están relacionados. Verdadera. El ciclo de la carnitina es importante para el transporte de los ácidos grasos de cadena larga en las mitocondrias. b. Una mutación puntual única explica la mayoría de los casos de deficiencia de MCAD (acil CoA deshidrogenasa de cdena media). Verdadera. El 90% de los alelos se deben a la misma mutación, habiéndose sugerido la detección selectiva en la población neonatal. c. Los trastornos de los peroxisomas incluyen a la enfermedad de Menkes y a la enfermedad de Wilson. Falsa. Éstos son errores congénitos del metabolismo del transporte del cobre. d. Los errores congénitos del metabolismo pueden presentarse con hipotonía y acidosis únicamente. Verdadera. Estas características deben iniciar a la investigación de las acidurias orgánicas y de los trastornos mitocondriales, entre otros. e. Los rayos X no tiene utilidad para el diagnóstico de los errores congénitos del metabolismo. Falsa. Las características radiológicas importantes pueden observarse en los trastornos de los peroxisomas y del almacenamiento. Capitulo 11: Preguntas basadas en casos. Caso 1.- Un niño de 2 años, que tiene una hermana de 4 meses, es ingresado al hospital con vómitos y somnolencia. Aunque los vómitos mejoran bastante rápidamente con el aporte de líquidos intravenosos, su glicemia se mantiene baja y los líquidos intravenosos son necesarios durante más tiempo del que cabría esperar normalmente. Los padres dicen que le había pasado ya algo semejante, aunque él se recupero sin llegar a ver al médico. 158 1.- ¿Qué sugiere esta historia? La hipoglucemia pude ser parte de una enfermedad grave en los niños pequeños, pero en este caso el problema intercurrente parece ser relativamente menor, lo que sugiere que la capacidad metabólica del niño para afrontar el estrés ésta comprometida. Este caso debe incitar la investigación de un posible error congénito del metabolismo y, si se establece el diagnóstico, hacer pruebas al hermano más pequeño. 2.- ¿Cuáles son las pruebas adecuadas? La hipoglucemia es una consecuencia habitual de varios errores congénitos del metabolismo de los aminoácidos y de los ácidos orgánicos. La investigación debe comenzar con el análisis de los ácidos orgánicos en la orina y de los ácidos plasmáticos, el amoníaco y las pruebas de función hepática. Caso 2.- Una mujer de 28 años se da cuenta, al cabo de varios años, de que no tiene la misma energía de cuando tenía 20 años. Se cansa con relativa facilidad al hacer ejercicio y los miembros de la familia han observado que tiene los párpados ligeramente caídos, al tiempo que se preguntan si su audición se está deteriorando, lo que ella niega rotundamente. 1.- ¿Cómo ayudarían unos antecedentes familiares detallados en el diagnóstico de este caso? Si hay antecedentes familiares de síntomas similares podrían demostrar una herencia matrilineal, y todos los descendientes de los varones afectados serían normales. Si esta persona es el único individuo afectado, los antecedentes familiares no aportarán ninguna información con respecto al diagnóstico. 2.- ¿Qué pruebas deben realizarse? Hay que considerar todas las causas de miopatía, pero la combinación de características es sugerente de una citopatía mitocondrial. Esto explicaría los síntomas musculares, la ptosis y la alteración auditiva, y podría haber también pruebas de una miocardiopatía con alteraciones neurológicas, retinitis pigmentaria y diabetes mellitus. Es posible que el análisis del DNA mitoondrial de los linfocitos periféricos identificarse una mutación, aunque un resultado negativo no descartaría el diagnóstico. Probablemente una biopsia del músculo mostraría las fibras rojas rasgadas y el análisis del DNA de este tejido aportaría más información que los linfocitos. Capítulo 12: Farmacogenética. 1.- Fármacos con tiopurina para tratar la leucemia: a. Incluyen a la 6-mercaptopurina, la 6-tioguanina y la azatioprina. Verdadera. b. Se utilizan también para suprimir el sistema inmunitario. Verdadera. Se utilizan para tratar trastornos autoinmunitarios y para evitar el rechazo de los trasplantes de órganos. 159 c. Pueden ser tóxicos en el 1 al 2% de los pacientes. Falsa. Pueden ser tóxicos en el 10 a 15% de los pacientes. d. Pueden tener efectos secundarios graves. Verdadera. Entre ellos están leucocitopenia y el daño hepático grave. e. Son metabolizados por la tiopurina metiltransferasa (TPMT). Verdadera. Las variantes en el gen TPMT se asocian con la toxicidad de la tiopurina. 2.- Las enzimas hepáticas que muestran variación genética en su expresión y por tanto influyen en la respuesta a los fármacos incluyen: a. UDP-glucoronosiltransferasa. Verdadera. La deficiencia completa de esta enzima ocasiona la enfermedad de Crigler-Najjar tipo 1. b. O-acetiltransferasa. Falsa. La variación en la N-acetiltransferasa (NAT2) influye en el metabolismo de la isoniazida. c. Alcohol deshidrogenasa. Falsa. La ausencia de ALDH2 (acetaldehído deshidrogenasa) está relacionada con una rubefacción aguda en respuesta al alcohol. d. CYP2D6. Verdadera. Aproximadamente el 5 a 10% de la población europea son malos metabolizadores de la debrisoquina debido a una variante homocigótica en el gen CYP2D6. e. CYP2C9 Verdadera. Las variantes en CYP2C9 se asocian con un metabolismo reducido de la warfarina. 3.- Tienen riesgo aumentado de complicaciones debido a la anestesia: a. Familiares de primer grado de pacientes afectados con hipertermia maligna. Verdadera. La hipertermia maligna se hereda de forma dominante. b. Pacientes con mutaciones en RYR1. Verdadera. Las mutaciones en RYR1 se asocian con la hipertermia maligna. c. Pacientes con deficiencias de la G6DP. 160 Falsa. La deficiencia en G6DP da lugar a la sensibilidad a ciertos fármacos y a las habas. d. Pacientes con mutaciones en CHE1. Verdadera. Las mutaciones homocigóticas en CHE1 ocasionan sensibilidad al suxametonio. e. Pacientes con valores elevados de la seudocolinesterasa. Falsa. La seudocolinesterasa metaboliza al suxametonio, de aquí que los valores reducidos se relacionen con sensibilidad al suxametonio. 4.- Los ejemplos de enfermedades en las que el tratamiento puede estar influido por la farmacogenética son: a. Diabetes juvenil de aparición en la madurez (MODY) subtipo glucocinasa. Falsa. Los pacientes con mutaciones en la glucocinasa se tratan habitualmente sólo con dieta. b. Diabetes juvenil de aparición en la madurez (MODY), subtipo HNF1alfa. Verdadera. Los pacientes con mutaciones en HNF1A son sensibles a la sulfonilureas. c. Infección por VIH. Verdadera. El abacavir es un fármaco eficaz, pero aproximadamente el 5% de los pacientes muestran una hipersensibilidad potencialmente fatal. d. Epilepsia. Verdadera. Algunos pacientes muestran felbamato. reacciones adversas al fármaco e. Tuberculosis. Verdadera. Los inactivadotes lentos de la isoniazida tienen mayor tendencia a presentar efectos secundarios. Capítulo 13. Inmunogenética. 1.a. La cascada del complemento puede activarse únicamente por la unión del anticuerpo y el antígeno. Falsa. La cascada puede activarse también por la ruta alternativa. b. La deficiencia del inhibidor de C1 puede dar lugar a la activación del complemento a través de la ruta clásica. 161 Verdadera. Los valores de C4 están reducidos y la producción de C2b fuera de control. c. Los valores de C3 están reducidos en el edema angioneurótico hereditario. Falsa. Los valores de C3 son normales y los valores de C4 están reducidos. d. El complemento colabora microorganismos. directamente en el ataque a los Verdadera. La C3b se adhiere a la superficie de los microorganismos. e. El complemento intracelular. se encuentra principalmente en la matriz Falsa. El complemento es una serie de al menos 20 proteínas plasmáticas interactivas. 2.a. La molécula de inmunoglobulina está compuesta por seis cadenas de polipéptidos. Falsa. Está compuesta por cuatro cadenas de polipéptidos: dos “ligeras” y dos “pesadas”. b. Los genes de las diversas cadenas ligeras y pesadas de las inmuno globulinas se encuentran juntos en el genoma humano. Falsa. Se distribuyen alrededor de los distintos cromosomas. c. Los familiares próximos son los mejores donantes de órganos porque es probable que compartan los mismos haplotipos del complemento. Falsa. Los donantes tienen tendencia a compartir los haplotipos HLA, que son cruciales para la compatibilidad tisular. d. El DNA que codifica la cadena ligera Kappa contiene cuatro regiones distintas. Verdadera. Son las regiones variable, de diversidad, de unión y constante. e. La diversidad del receptor del antígeno de superficie en el linfocito T puede compararse con el proceso de la diversidad de las inmunoglobulinas. Verdadera. Los receptores de los antígenos contienen dos dominios tipo inmunoglobulina. 3.a. La movilidad transplacentaria materna de los anticuerpos proporciona a los lactantes protección durante unos 12 meses. 162 Falsa. Están protegidos únicamente durante 3-6 meses. b. La inmunodeficiencia combinada grave ligada al cromosoma X (SCID) es responsable de cerca del 5 al 10% del total de SCID. Falsa. La SCID ligada al cromosoma X representa del 50 al 60% del total. c. La SCID, a pesar de su nombre, no siempre es un trastorno grave. Verdadera. La SCID positiva para los linfocitos B debida a una deficiencia en JAK3 puede ser subclínica. d. Siempre hay una anomalía de los linfocitos T en las distintas formas de SCID. Verdadera. Un defecto en la función de los linfocitos T o en el desarrollo. e. La enfermedad granulomatosa crónica (EGC) es un trastorno de la inmunidad humoral. Falsa. La EGC es un trastorno ligado al cromosoma X de la inmunidad mediada por células. 4.a. El síndrome de Di George/Sedlácková es un trastorno primario de la función inmunitaria. Falsa. Se clasifica como inmunodeficiencia secundaria o asociada. b. Las infecciones bacterianas oportunistas graves son infrecuentes en el síndrome de DiGeorge. Verdadera. La inmunodeficiencia es habitualmente leve y el sistema inmunitario mejora con la edad a medida que el timo crece; hay una tendencia a las infecciones víricas en la infancia. c. El diagnóstico genético prenatal es posible en la inmunodeficiencia variable común. Falsa. Las causas de la CVID no se conocen y es a menudo un trastorno de la vida de adulto. d. Los trastornos autoinmunitarios siguen una herencia autosómica dominante. Falsa. El riesgo para los familiares de primer grado está aumentado pero el patrón genealógico es más sugerente de trastornos multifactoriales. e. La investigación de la función inmunitaria debe considerarse en cualquier niño con deterioro progresivo. Verdadera. La FTT inmunodeficiencia. puede ser la única pista de un trastorno por 163 Capítulo 13. Preguntas basados en casos. Caso 1.- Un hombre de 32 años ha tenido lumbago y rigidez durante 2 años y recientemente presenta cierta irritación ocular. Se realiza una radiografía y se establece el diagnóstico de espondilitis anquilosante. Recuerda que su abuelo materno tuvo problemas similares de espalda así como artritis en otras articulaciones. Tiene tres niños pequeños. 1.- ¿Es probable anquilosante? que su abuelo tuviera también espondilitis La naturaleza de los síntomas de su abuelo son bastante inespecíficos; tanto el dolor de espalda como la artritis son muy habituales en la población general. Sin embargo, es ciertamente posible que él también tuviera espondilitis anquilosante, una forma de entesitis (inflamación en el sitio de inserción de un ligamento o tendón en el hueso) con afectación de las articulaciones sinoviales, ya que la heredabilidad es mayor del 90%. 2.- ¿Cuál es el riesgo de transmitir el trastorno a sus tres hijos? Cerca del 95% de los pacientes con espondilitis anquilosante dan positivo al antígeno HLA-B27; sin embargo, en la población general esta prueba tiene únicamente un bajo valor predictivo positivo. Sus hijos tienen un 50% de posibilidades de ser HLA-B27 positivos; si son positivos, el riesgo de desarrollar una espondilitis anquilosante clínica es de alrededor del 9%; si son negativos, el riesgo es inferior al 1%. Caso 2.- Una niña de 4 años presenta frecuentes infecciones de las vías respiratorias altas con afección torácica, y cada episodio se prolonga más tiempo que el de sus compañeros de preescolar. Los médicos han asumido siempre que esto se debe, de alguna manera manera sus primeros meses de vida tormentosos, cuando se sometió a una importante cirugía cardíaca por la tetralogía de Fallot. Presenta también habla nasal y en su ficha neonatal hay una observación de un radiológo sobre el pequeño tamaño de la glándula timo. 1.-¿ Hay otro diagnóstico que pueda explicar sus prolongadas infecciones de las vías respiratorias altas? frecuentes y Esta historia apunta frecuentemente hacia un diagnóstico de síndrome de delección de 22q11 (DiGeorge/Sedlácková), que puede confirmarse muy fácilmente por pruebas de FISH específicas. La inmunidad está alterada pero habitualmente sólo de forma leve, y se produce una mejoría gradual durante la infancia y la adolescencia. 2.-¿ Qué otro tratamiento de la familia está indicado? El síndrome de la delección de 22q11 puede ser familiar y no siempre da lugar a cardiopatía congénita. Si se confirma en la niña, se deben proponer las pruebas con FISH a ambos padres y a otros miembros de la familia según sea necesario. El consejo genético para la niña será importante cuando sea mayor. 164 Capítulo 14: Genética del cáncer. 1.a. Las translocaciones cromosómicas pueden generar un cáncer por modificaciones en la actividad del oncogén. Verdadera. El ejemplo mejor conocido es la leucemia mieloide crónica y el cromosoma Filadelfia. b. Los oncogenes son las formas más habituales de los genes que predisponen a los síndromes del cáncer hereditario. Falsa. Los genes supresores de los tumores son más comunes que los oncogenes. c. La apoptosis defectuosa puede ocasionar tumorogénesis. Verdadera. La apoptosis es la muerte celular programada normal. d. La pérdida de la heterocigosidad (LOH) es otro término para definir el episodio mutacional en un oncogén. Falsa. La PH se refiere a la presencia de dos alelos defectuosos en un gen supresor de tumores. e. Una mutación en el gen APC es suficiente para ocasionar un cáncer colorrectal. Falsa. Aunque son importantes, las mutaciones en APC forman parte de una secuencia de cambios genéticos que llevan al cáncer de colon. 2, a. La hipótesis de las dos mutaciones predice que se desarrollará un tumor cuando ambas copias de un gen crítico se hayan mutado. Verdadera. El paradigma era el retinoblastoma, y posteriormente se demostró que la hipótesis era correcta. b. Las mutaciones en TP53 se encuentran únicamente en el síndrome de Li-Fraumeni. Falsa. Las mutaciones TP53 se encuentran en muchos cánceres, pero el síndrome de Li-Fraumeni están en la línea germinal. c. El protooncogén RET está implicado en todas las formas de neoplasia endocrina múltiple (MEN). Falsa. Está implicada en MEN-2 pero no en MEN-1. d. Los individuos con poliposis adematosa familiar (PAF) deben someterse a una detección selectiva en el tracto gastrointestinal superior. 165 Verdadera. Hay un riesgo significativo de pólipos en el intestino delgado y de cáncer duodenal. e. El cáncer endometrial es característico del cáncer colorrectal sin pólipos hereditario (CCSPH). Verdadera. Las mujeres con este trastorno tienen un riesgo durante toda su vida de hasta del 50%. 3.a. Existe riesgo de cáncer de tiroides en el síndrome de BannayanRiley-Ruvalcaba. Verdadera. Este síndrome es alélico en la enfermedad de Cowden, en el que puede aparecer un carcinoma tiroideo papilar. b. Los varones con una mutación en la línea germinal en BRCA2 tienen un riesgo aumentado de cáncer de próstata. Verdadera. El riesgo durante toda su vida puede estar rondando el 16%. c. La base genética de todos los cánceres de mama familiares está muy bien establecida. Falsa. Los genes BRCA1 y BRCA2 no son responsables de todos los cánceres de mama familiares. d. El cáncer de mama familiar tiene habitualmente una penetrancia completa. Falsa. El riesgo durante toda la vida de cáncer de mama para las portadoras femeninas de BRCA1 y BRCA2 es del 60-85%. e. En los hombres con cáncer de próstata, el 3% de los familiares masculinos de primer grado están afectados de forma similar. Falsa. La cifra es aproximadamente el 15%. 4.a. El meduloblastoma es un tumor común en la enfermedad de vonHippel-Lindau (VHL). Falsa. El hemangioblastoma cerebeloso es un tumor habitual en la enfermedad de VHL. b. El feocromocitoma se observa frecuentemente en el síndrome de Gorlin. Falsa. Este tumor se observa en MEN-2 y en la enfermedad de VHL 166 c. Existe riesgo de cáncer ovárico en el síndrome de Peutz-Jehgers y en el CCSPH. Verdadera. Hay también un riesgo aumentado en el cáncer de mama familiar. d. Las manifestaciones cutáneas aparecen en el síndrome de PuetzJehgers, síndrome de Gorlin y el CCSPH. Verdadera. Manchas de melanina en el síndrome de Puetz-Jehger, carcinomas de células basales en el síndrome de Gorlin y tumores cutáneos en la forma Muir-Torre del CCSPH. e. En dos tercios de los casos de CCSPH se desconoce el gen predisponente. Falsa. La cifra es aproximadamente un tercio. 5.a. Se recomienda la detección selectiva del cáncer renal en el VHL. Verdadera. El carcinoma renal de células claras es un riesgo significativo en la enfermedad de VHL. b. La mamografía detecta el cáncer de mama más fácilmente en las mujeres premenopáusicas que en las posmenopáusicas. Falsa. Es más difícil detectar un cáncer de mama mediante una mamografía en las mujeres posmenopáusicas. c. La detección selectiva del retinoblastoma debe comenzar en el segundo año de vida. Falsa. Debería comenzar en el nacimiento. d. El cáncer colorrectal en dos familiares próximos es suficiente para indicar la necesidad de una detección selectiva por colonoscopia en otros miembros de la familia. Falsa. No cumple los criterios de Amsterdam para el CCSPH. e. La cirugía preventiva está fuertemente indicada en la PAF y las mujeres que den positivo para las mutaciones en BRCA1. Falsa. Está fuertemente indicado en la PAF pero no en las mujeres con mutaciones BRCA1 positivas. Capítulo 14. Preguntas basadas en casos. Caso 1.- Una mujer de 38 años, que se ha sometido recientemente a una mastectomía por cáncer de mama, pide la remisión a un servicio de genética. A su padre le extirparon algunos pólipos intestinales cuando tenía 50 años, y un primo del mismo lado de la familia sufrió un tipo de cáncer de tiroides a los 40. El médico de familia consulta un conjunto de guías que sugieren que no es probable una forma familiar de cáncer de 167 mama, ya que ella es la única afectada, aunque sea bastante joven. Se muestra reacio a remitirla. 1.- ¿Sugieren los antecedentes otro trastorno familiar y, si es así, cuál? En primer lugar hay que confirmar los antecedentes familiares con el consentimiento de los individuos afectados. Si el cáncer de tiroides del primo era de tipo papilar y los pólipos en su padre hamartomatosos, el patrón sería muy sospechoso de la enfermedad de Cowden. Esto también se conoce como síndrome de los hamartomas múltiples, que es autosómico dominante y se debe a menudo a mutaciones de PTEN; el riesgo de cáncer de mama es aproximadamente del 50% en las mujeres. 2.- ¿Qué otras características clínicas diagnóstico? darían una clave para el La macrocefalia, el aspecto adoquinado de la mucosa oral y los lipomas generalizados son otras características que hay que buscar en los pacientes con esta historia poco habitual. Caso 2.- Una mujer de 30 años se remite a consejo genético porque está preocupada sobre el riesgo de desarrollar cáncer de mama. La madre de la consultante ha sido diagnosticada recientemente de cáncer mamario a la edad de 55 años. La hija de su hermano (prima de la consultante) tuvo cáncer de mama bilateral diagnosticada a la edad de 38 años y murió hace 5 por metástasis. La prima había participado en un estudio de investigación que identificó una mutación en el gen BRCA2. El genetista clínico sugiere que hay que hacer pruebas a la madre de la consultante antes de proponer unas pruebas predictivas a la hija. Se sorprenden cuando el laboratorio emite un resultado negativo. 1.-¿Cuáles son las posibles explicaciones de este resultado? Si no se ha confirmado la mutación en BRCA2 en otro miembro de la familia o probando otra muestra de la prima fallecida (por ejemplo, una sección tisular incrustada en parafina), no se puede excluir la posibilidad de una mezcla de muestras en el laboratorio de investigación. Sin embargo, si las muestras del tío son positivas para la mutación, la madre de la consultante es una fenocopia. Por otra parte, se puede haber heredado la mutación de la madre de la prima. 2.- ¿Cuál es el riego del tío de la consultante de desarrollar cáncer mamario? Si el resultado de las pruebas del tío es positivo para la mutación den BRCA2, entonces el riesgo de desarrollar cáncer de mama durante toda su vida es de aproximadamente el 6%, más de 100 veces superior al de la población general. Capítulo 15: factores genéticos en las enfermedades comunes. 1.- 168 a. Se cree que hay un factor genético presente en el 10% de las epilepsias. Falsa. La cifra es aproximadamente el 40% b. Las mutaciones en los genes del canal de potasio predisponen a la epilepsia. Verdadera. Una forma de epilepsia son las convulsiones neonatales familiares, benignas, autosómicas dominantes. c. La epilepsia mioclónica juvenil sigue una herencia autosómica dominante. Falsa. La epilepsia no es mendeliana. d. La epilepsia es habitual en el síndrome de la delección de 1p36. Verdadera. Una gran proporción de pacientes tienen epilepsia de diferentes clases, incluyendo los espasmos infantiles. e. La enfermedad de Unverricht-Lundborg es una forma benigna de epilepsia medeliana. Falsa. Es mendeliana (autosómica recesiva) pero es progresiva con una neurodegeneración. 2.a. Hay un riesgo elevado de enfermedad depresiva bipolar en el síndrome de DiGeorge. Falsa. El riesgo es para la equizofrenia. b. La tasa de concordancia para la equizofrenia en los gemelos monocigóticos es aproximadamente del 80% Falsa. El riesgo está entre el 40 y el 50%. c. Hasta ahora no se ha encontrado ninguna mutación en ningún gen que sea causa de la equizofrenia. Verdadera. Ésta es la situación a pesar de que hay muchos estudios que sugieren una relación significativa. d. Los estudios en gemelos sugieren una heredabilidad similar para la enfermedad uni y bipolar. Verdadera. Las tasas de concordancia en los gemelos monocigóticos y dicigóticos son del 67 y del 20 %, respectivamente. e. Se recomienda el análisis de APOEepislon4 como procedimiento de detección selectiva para la enfermedad de Alzheimer. 169 Falsa. Aunque está presente en cerca del 40% de los pacientes con enfermedad de Alzheimer, no tiene suficiente valor discriminativo para un cribado. 3.a. La susceptibilidad para la enfermedad de Creutzfeldt-Jakob la confiere la homocigosidad para un polimorfismo de la proteína prión (PRNP). Verdadera. Es más probable la conversión de la proteína prión a la isoforma anómala PrPsc. b. Hasta ahora, se conocen tres genes que causan la enfermedad de Alzheimer autosómica dominante. Verdadera. Los genes son APP, Presenilina-1 y Presenilina-2. c. La presenilina-1 es un gen de baja penetrancia enfermedad de Alzheimer de aparición precoz. para el Falsa. La penetrancia es muy alta para todas las enfermedades de Alzheimer mendelianas notificadas hasta la fecha. d. Los estudios que ligamiento son más importantes que los estudios en gemelos para comprender la contribución genética a la enfermedad bipolar. Falsa. Los estudios epidemiológicos en gemelos han demostrado ser más importantes hasta la fecha a la hora de responder a esta pregunta. e. Las personas con síndrome de Down significativo de enfermedad de Alzheimer. tienen un riesgo Verdadera. Sin embargo, las razones no están completamente claras, a pesar de que el gen APP se localiza en el cromosoma 21. 4.- Hemocromatosis: a. Es genéticamente heterogénea. Verdadera. Las mutaciones en al menos cinco genes pueden ocasionar una hematocromatosis. b. Afecta más a las mujeres que a los hombres. Falsa. Los varones tienen cinco veces más probabilidad de resultar afectados. c. Puede tratarse por flebotomía. Verdadera. La flebotomía regular es un tratamiento eficaz. d. Tiene una prenetrancia completa. 170 Falsa. La penetrancia puede ser tan baja como del 1%. e. Habitualmente está causada por una mutación homocigótica en H63D en el gen HFE. Falsa. La causa más habitual es la homocigosidad para la mutación en C282Y del gen HFE. 5.- Las siguientes verdaderas: afirmaciones sobre la trombosis venosa son a. El factor V de Leiden y la variante G20210A de la protrombina son causas habituales de la trombosis venosa. Verdadera. Son responsables de más del 50% de los casos. b. Las pruebas del factor V de Leiden y de la variante de la protrombina cambiarán el tratamiento de un paciente con trombosis venosa. Falsa. Poco probable, pero se pueden ofrecer pruebas y tratamientos profilácticos si es necesario a sus familiares en primer grado. c. La mutación en el factor V de Leiden produce una expresión reducida del gen del factor V. Falsa. La mutación en el factor V de Leiden convierte a la proteína del factor V resistente a la disociación por la proteína C activada. d. La variante de la protrombina ocasiona un aumento de la expresión del gen de la protrombina. Verdadera. Los valores elevados de protrombina sérica se producen por la variante G20210A. e. Los heterocigotos compuestos para el factor V de Leiden y para la variante de la protrombina tienen un aumento de 20 veces mayor riesgo de trombosis venosa. Verdadera. Los riesgos se multiplican. Capítulo 16. Preguntas basadas en casos. Caso 1.- Una niña de 2 años presenta convulsiones parciales. El episodio es breve y no se acompaña de fiebre. Como la niña está bien y no tiene deficiencias neurológicas, se toma la decisión de no tratarla con fármacos anticonvulsionantes. Un año después presenta una convulsión generalizada, de nuevo sin fiebre. En esta ocasión su madre de 30 años pregunta si ellos tiene relación con sus propias convulsiones, que comenzaron a la edad de 15 años, aunque ella sólo ha tenido dos episodios desde entonces. Se sometió a una tomografía computarizada del encéfalo y los médicos mencionaron un trastorno cuyo nombre no puede recordar. La resonancia magnética del encéfalo de la niña muestra nódulos no calcificados en las paredes laterales ventriculares. 171 1.- La madre pregunta si la epilepsia es genética y si podría suceder de nuevo en caso de que tuviera otro hijo. ¿Qué se le puede responder? Generalmente, el riesgo de epilepsia para los familiares de primer grado es de alrededor del 4%. Sin embargo, aquí la madre y la hija están afectadas, lo que sugiere la posibilidad de una forma mendeliana de epilepsia. Además, parece que ambas tienen un signo anómalo en la imagen encefálica, lo que obliga a localizar y revisar las tomografías computarizadas de la madre. En esta etapa se debe dar una explicación sobre la herencia autosómica dominante y la ligada al cromosoma X, así como la posibilidad de que los dos casos de epilepsia sean coincidentes. 2.-¿Cuáles son los diagnósticos que deben considerarse?¿Se le pueden proponer pruebas genéticas? El trastorno que los médicos de la madre mencionaron sería, casi con certeza, una esclerosis tuberosa, que sigue una herencia autosómica dominante. Está indicada tanto para la madre como de la hija en busca de las características clínicas de la esclerosis tuberosa, y disponemos además de las pruebas genéticas. Sin embargo, los nódulos en las paredes del ventrículo lateral pueden ser patognomónicos de una heteropatia nodular periventricular bilateral (BPVNH), y las imágenes han de ser revisadas por alguien que pueda reconocerla. La BPVNH es una anomalía de la migración de las neuronas y se hereda como un trastorno dominante ligado al cromosoma X, ocasionado por mutaciones en el gen filam-1 (FLNA). Se dispone de las pruebas. En general, las formas mendelianas de la epilepsia son raras. Caso 2.- Un niño de 5 años ingresa en el hospital con fiebre inexplicada y se le detecta hiperglucemia. Se recupera bien, pero 2 semanas más tarde su valor de glucemia ha ascendido hasta 7mmol/L. Hay fuertes antecedentes familiares de diabetes por el lado materno, y su madre, tío materno y abuelo materno están afectados. Su padre no tiene síntomas de diabetes, pero su hermana tuvo diabetes gestacional durante su último embarazo. Las pruebas genéticas moleculares identifican una mutación del gen de la glucosinasa heterocigótica en el niño. 1.-Los padres creen que su hijo ha heredado la hiperglucemia del lado materno de la familia. ¿Es correcto?. No necesariamente. Muchas personas con mutaciones en el gen de la glucocinasa son asintomáticas y su hiperglucemia leve se descubre únicamente con la detección selectiva (exámenes médicos rutinarios, durante el embarazo o en enfermedades interecurrentes). La diabetes gestacional en la hermana del padre sugiere que la mutación podría haberse heredado de esta rama de la familia. 2.-¿Cuáles son las consecuencias de encontrar una mutación en el gen de la glucocinasa para esta familia? La identificación de un gen de la glucocinasa es una “buena noticia”, ya que es probablemente que la hiperglucemia leve se mantenga estable durante toda la vida, tratada únicamente con dieta (excepto durante el embarazo), y que no ocasione complicaciones diabéticas. Las pruebas en cascada pueden proporcionarse a los demás familiares. Si la mutación se ha heredado del padre, se deben hacer pruebas 172 a su hermana y su hijo. La hermana podría haberse evitado la ansiedad de tener un hijo pequeño diagnosticado de hiperglucemia inexplicada. Capítulo 16: Anomalías congénitas y síndromes dismórficos. 1.a. Cerca del 5% de todas las muertes de los lactantes se deben a anomalías congénitas. Falsa. La cifra es aproximadamente el 25% b. Al menos la mitad de todos los abortos espontáneos tienen una base genética. Verdadera. Ésta es la cifra de los estudios cromosómicos. Podría ser mucho más elevada si se incluyeran todas las anomalías monogénicas letales. c. Una anomalía congénita mayor afecta aproximadamente a un lactante nacido por cada 100. Falsa. La cifra es de -3% d. La deformidad congénita del pie posicional es un ejemplo de una disrupción del desarrollo intrauterino normal. Falsa. Éste es un ejemplo de deformación. e. Las anomalías múltiples son a veces el resultado de una secuencia. Verdadera. La “secuencia” implica una cascada de sucesos rastreados hasta una anomalía única. 2.a. El síndrome de Down debería ser denominado de forma más precisa “asociación de Down”. Falsa. El nombre de síndrome es correcto debido a la naturaleza altamente reconocible del trastorno. b. El síndrome de Sotos, como el síndrome de Down, se debe a una anomalía cromosómica. Falsa. Se han encontrado que se debe a mutaciones en un único gen. c. La espina bífida afecta a aproximadamente 2 de cada 1,000 nacimientos. Verdadera. La cifra varía entre poblaciones y se reduce con la administración de ácido fólico alrededor de la concepción. d. La enfermedad del riñón poliquístico infantil es un ejemplo de un trastorno con diferentes patrones de herencia. Falsa. Esta entidad bien definida es un trastorno autosómico recesivo. 173 e. La holoprosencefalia es un de un trastorno con diferentes patrones de herencia Verdadera. Puede ser cromosómica, autosómica dominante y autosómica recesiva. 3.a. La embriopatía por talidomida fue un ejemplo de una disrupción del desarrollo intrauterino normal. Verdadera. Un teratógeno representa una disrupción química o tóxica. b. La deformación congénita del pie puede ser una consecuencia de la agenesia renal. Verdadera. La agenesia renal ocasiona un oligohidramnios, que produce una deformación congénita del pie. c. Los defectos en las extremidades no están causados por exposición fetal al valproato sódico. Falsa. Puede producirse una variedad de defectos en las extremidades. d. Los defectos simétricos tienden a presentarse en una displasia. Verdadera. Hay un efecto generalizado en un tejido particular, como el hueso o la piel. e. Los defectos en el nacimiento son inexplicables en 20% de los casos. Falsa. La cifra es más elevada, alrededor del 50% 4.a. Una infección congénita puede ocasionar que alguien sea ciego y sordo. Verdadera. La sordera y los defectos visuales diversos son característicos. b. El trimestre medio es el momento más peligroso para que un feto se exponga a una infección materna. Falsa. El primer trimestre es mucho más peligroso. c. Los defectos del cuerpo vertebral pueden ser consecuencia de una diabetes mellitus mal tratada en el primer trimestre. Verdadera. Es posible que haya defectos vertebrales a cualquier nivel, incluyendo la agenesia sacra. d. Un polimorfismo en el gen de la metilentetrahidrofolato reductasa (MTHFR) está siempre asociado con un riesgo mayor de defectos en el tubo neural. 174 Falsa. Esto es cierto en algunas poblaciones, no en todas. e. La estenosis pulmonar es una característica del síndrome de Noonan y de la rubéola congénita. Verdadera. La estenosis periférica de la arteria pulmonar en el caso de la rubéola congénita. 5.a. El labio leporino/paladar hendido aparece superior a 1 de cada 1,000 nacimientos. con una frecuencia Verdadera. La incidencia está entre 1 de cada 500 y 1 de cada 1,000 b. Las asociaciones tienen generalmente un alto riesgo de recurrencia. Falsa. El riesgo de recidiva es bajo porque no se cree que sean genéticas. c. El riesgo de recurrencia de un trastorno multifactorial puede habitualmente determinarse mirando la genealogía familiar del paciente. Falsa. Se necesitan grandes estudios de muchas familias. d. Una de las causas de la holoprosencefalia es un defecto metabólico. Verdadera. El síndrome de Smith-Lemli-Opitz es un defecto del metabolismo del colesterol que afecta a la ruta del Sonic hedgehog. e. La cardiopatía congénita afecta a 1 de cada 1,000 nacidos. Falsa. La cifra es mucho más próxima a 1 de cada 100. Capítulo 16: Preguntas basadas en casos. Caso 1.- Una pareja joven acaba de perder su primer embarazo por una anomalía fetal. En la ecografía se diagnóstico un polihidramnios así como un riñón fetal pequeño en un lado. Se realizó amniocentesis y el cariotipo mostró un patrón 46,XY normal. La pareja no estaba segura de qué hacer, pero finalmente decidieron la terminación del embarazo a las 21 semanas. Estaban muy molestos y no quisieron realizar ninguna prueba complementaria, incluyendo la autopsia. Accedieron a que se hiciera una radiografía completa del cuerpo del feto y se encontró que algunas de las vértebras torácicas superiores estaban malformadas. 1.- La pareja pregunta si dicho problema podría reaparecer: no creen que puedan pasar por esta situación de nuevo. ¿Qué se les puede decir? Éste no es un escenario infrecuente. El cariotipo en la amniocentesis era normal y el polihidramnios sugiere la posibilidad de obstrucción gastrointestinal, como una atresia esofágica. Es más probable que las anomalías representen una “asociación” más que síndrome o un trastorno mendeliano. El riesgo de recidiva empírica es bajo, y todo lo que puede proponerse es una ecografía en los embarazos posteriores. 175 2.- ¿Qué pruebas complementarias podrían haber ayudado a informar sobre el riesgo genético? La autopsia fetal es latamente deseable en esta situación para conocer la extensión total de las anomalías de los órganos internos. Un cariotipo repetido de la piel fetal podría haber mostrado algo que no se detectase en la amniocentesis, y ha de conservarse el DNA para un posible uso futuro. Hay que excluir la diabetes materna. Como último recurso se deben hacer pruebas cromosómicas de los padres, incluyendo las detecciones selectivas de los telómeros buscando la posibilidad de una translocación críptica. Caso 2.- En el examen neonatal de rutina del segundo día, se encuentra que un lactante tiene un paladar hendido. El embarazo no tuvo complicaciones ni hubo exposición a teratógenos potenciales y los antecedentes familiares son negativos. El residente de pediatría se pregunta también si los miembros no son ligeramente cortos. El peso del lactante en el nacimiento está en el percentil 25, y la altura en el segundo percentil. 1.- ¿Cuáles son los diagnósticos que se debe considerar? El paladar hendido no sindrómico aislado es estadísticamente el diagnóstico má probable, pero la estatura ligeramente baja puede ser significativa. Las posibilidades sindrómicas incluyen a la displasia congénita espondiloepisifisaria (SEDC), aunque hay muchos síndromes raros con una estatura baja más acusada y con otras características. 2.- ¿Cuáles con los problemas de tratamiento en un caso como éste? La baja estatura parece leve; por tanto, es importante intentar determinar si pudiera ser familiar: hay que evaluar a los padres. Está indicado el seguimiento del lactante, incluyendo una investigación radiológica del esqueleto para ver si hay una displasia esquelética identificable. La SEDC puede acompañarse de miopía y de la alteración auditiva neurosensorial, por lo que es importante valorar la audición y la visión. Sin embargo, el lactante tiene un paladar hendido y ha resultas de ello tiene riesgo de presentar problemas de audición conductiva. Hay que implicar al equipo de paladar hendido desde el comienzo. Capítulo 17: Consejo genético. 1.a. El individuo que busca consejo genético es el probando. Falsa. Éste es el consultante; el probando es el individuo afectado. b. La retinitis pigmentaria no es genéticamente heterogénea. Falsa. La retinitis pigmentaria puede seguir todos los patrones principales de herencia. c. El consejo genético trata de los riesgos de recurrencia. 176 Falsa. Es mucho más de una transferencia de información relevante, presentación de opciones y facilitación de la toma de decisiones frente a elecciones difíciles. d. La opinión propia del asesor sobre la elección difícil es simpre de utilidad. Falsa. El consejo sin guía es el objetivo porque los pacientes/clientes deben tomar sus propias decisiones. e. El buen asesoramiento no debe medirse por la capacidad del paciente/cliente para recordar los riesgos genéticos. Verdadera. Los pacientes no recuerdan la información del riesgo con exactitud y existen otras medidas importantes para valorar la satisfacción del paciente. 2.a. Hay 10 veces más probabilidad de tener niños con anomalías congénitas en la relaciones entre primos hermanos que en la población en general. Falsa. El riesgo es aproximadamente el doble del riesgo general. b. Como media, un abuelo y un nieto comparten ¼ de sus genes. Verdadera. Ésta es una relación de segundo grado. c. Las relaciones incestuosas producen prácticamente dificultades de aprendizaje graves en la descendencia. Falsa. El riesgo es aproximadamente del 25%. d. La consaguinidad debe ser contemplada como extremadamente anómala. Falsa. Es perfectamente normal en muchas sociedades. e. La consaguinidad se refiere matrimonios/relaciones entre primos. exclusivamente a los Falsa. Se refiere a cualquier grado desde, por ejemplo, relaciones entre tíosobrina (segundo grado) a relaciones entre primos terceros (séptimo grado). 3.a. Los trastornos genéticos son accidentes de la naturaleza, por lo que los sentimientos de culpabilidad son raros. Falsa. Los sentimientos de culpabilidad de los padres de los abuelos son habituales cuando se diagnóstica por primera vez una enfermedad genética en un niño. b. El consejo genético claro modifica las decisiones reproductivas de los pacientes en prácticamente todos los casos. 177 Falsa. Muchos pacientes toman la decisión que habría tomado antes del consejo genético, pero después del consejo deberían estar mucho mejor informados. c. La posibilidad de que el primer hijo de unos primos hermanos resulte afectado con un trastorno autosómico recesivo debido a ungen deletéreo heredado de un abuelo es de 1 de casa 32. Verdadera. El riesgo de cada abuelo es de 1 de cada 64. d. Se hacen muchas más pruebas genéticas en los niños para la adopción que en los niños criados por sus padres biológicos. Falsa. Semejante práctica está fuertemente desaconsejada y las indicaciones para las pruebas genéticas deberían ser las mismas. e. Los grupos de apoyo al paciente tiene poco valor dado que la genética médica moderna tiene mucha complejidad. Falsa. Los grupos buenos de apoyo al paciente tienen un inmenso papel, y los mismos pacientes/familias se convierten en expertos en su trastorno. Capítulo 17: Preguntas basadas en casos.Caso 1.- Una pareja tiene un hijo con características dismórficas, estatura corta y retraso en el desarrollo moderadamente grave. En la segunda ocasión en que se analiza su cariotipo, se encuentra que tiene una translocación cromosómica sutil que fue pasada por alto la primera vez. S descubre que el padre es portador de una translocación equilibrada. Su familia siempre ha culpado a la madre del trastorno del niño debido a su pasado de consumo de drogas, dando lugar a que la pareja haya dejado de hablar al resto de la familia. Sin embargo, a través de amigos, él ha sabido que su hermana está tratando de formar una familia. 1.- ¿Cuáles son los problemas genéticos importantes? La pareja esta en riesgo de tener otros niños afectados y se les puede proponer el diagnóstico prenatal. El padre puede haber heredado la translocación equilibrada de uno de sus progenitores y su hermana puede ser también una portadora. Se deben proponer las pruebas de portador a la familia, sobre todo a su hermana que está tratando de quedarse embarazada. 2.- ¿Qué otros problemas plantea este caso? Toda la familia del padre necesita conocer el diagnóstico del hijo, aunque tienen sus propios juicios preconcebidos y puede ser muy difícil aceptar que los problemas del niño tengan su origen en este lado de la familia. Hay un problema de comunicación grave, pero hay que encontrar una manera de informar a toda la familia del padre del riesgo genético. Podría ser necesario implicar a sus médicos de familia. Caso 2.- Una pareja tiene un niño que es diagnosticado de fibrosis quística (FQ) a los 18 meses de edad. El niño es homocigótico para la mutación alfaF508 habitual. Solicitan un diagnóstico prenatal en el siguiente embarazo, pero el análisis del DNA muestra que el padre no es portador de 178 alfaF508. Debe asumirse que él no es el padre biológico con FQ, y esto confirma cuando un análisis posterior muestra que el niño no tiene un haplotipo en común con él. 1.- ¿Qué problema médico plantea esta situación? Actualmente no hay necesidad de que la mujer se someta a una prueba prenatal invasiva en embarazos posteriores; sería desperdiciar los recursos y poner el embarazo en un riesgo pequeño, pero innecesario, de aborto. 2.- ¿Qué problemas de asesoramiento plantean los resultados? Es difícil comunicar el hecho de que la prueba prenatal no es necesaria, pero la revelación de la no paternidad puede tener consecuencias de largo alcance en el matrimonio. Los consejeros no saben si el “padre” sospecha la no paternidad, y la madre puede creer que él es el padre biológico del niño. Capítulo 18: Trastornos cromosómicos. 1.a. El número de cromosomas de los seres humanos se identificó después de la estructura del DNA. Verdadera. El número de cromosomas se identificó en 1956, la estructura del DNA en 1953. b. El cariotipo del síndrome de Turner es la anomalía cromosómica más habitual en los abortos espontáneos. Verdadera. En los abortos espontáneos se produce una gran variedad de cariotipos anómalos, pero el más habitual es el 45,X c. La tasa de abortos en el síndrome de Down es similar a la de los fetos cariotípicamente normales. Falsa. Se calcula que el 80% de todos los fetos con síndrome de Down se pierden espontánemente. d. La mayoría de los niños con síndrome de Down nacen de madres con edades superiores a los 30 años. Verdadera. Aunque el riesgo del síndrome de Down aumenta con la edad materna, la gran proporción de niños nacidos de madres jóvenes indica que la mayoría de los niños con síndrome de Down nace en este grupo de edad. e. Todos los niños con síndrome Down han de ir a escuelas especiales. Falsa. Una pequeña proporción tiene un CI en el extremo inferior del intervalo normal. 2.a. La esperanza de vida de los niños con trisomía del 18 (síndrome de Edwards) es de alrededor de 2 años. 179 Falsa. Tales niños mueren habitualmente a los pocos días o semanas de nacimiento. b. Los varones 47,XYY son fértiles. Verdadera. Los hombres con el síndrome de Klinefelter (47,XXY) no son habitualmente fértiles. c. El origen del síndrome de Turner (45,X) puede estar en al meiosis paterna. Verdadera. Ésta es responsable de una proporción sustancial de casos. d. Todas las personas con síndrome de Angelman tienen una delección citogenéticamente visible en el cromosoma 15q. Falsa. Esto no se observa ni en la disomía uniparental ni en los casos de defecto en el centro de la impronta. e. El síndrome de Di George se produce por una recombinación homóloga mal alineada entre los grupos de genes repetidos flaquenates. Verdadera. La delección en 22q11.2 s una región de 3 Mb flanqueada por secuencias de DNA muy similares. 3.a. En los adultos con síndrome de Williams aparecen problemas vasculares prematuros. Verdadera. Probablemente debida a una haploinsuficiencia para la elastina. b. La cardiopatía congénita es una característica de los síndromes de Prader-Willi y de Smith-Magenis. Falsa. La cardiopatía congénita no es una característica reconocida del síndrome de Prader-Willi. c. El locus del tumor de Wilms está en el cromosoma 13. Falsa. Está en el cromosoma 11p13 y puede ser una característica de los síndromes de WARG y de Beckwith-Wiedemann. d. La aniridia puede estar causada por una mutación genéica o por una microdelección cromosómica. Verdadera. Una mutación en PAX6 o una delección que afecta a este locus 11p15. e. La conducta de un niño puede ayudar a diagnosticar un síndrome malformativo. Verdadera. Los fenotipos de la conducta pueden ser muy informativos, por ejemplo, el síndrome de Smith-Magenis. 180 4.a. El síndrome de Klinefelter afecta aproximadamente a 1 de cada 10,000 hombres nacidos vivos. Falsa. La cifra es aproximadamente de 1 de cada 1,000. b. Las dificultades de aprendizaje son habituales en el síndrome de Klinefelter. Falsa. El CI está reducido en 10 a 20 puntos, pero las dificultades de aprendizaje no son características. c. El mosaicismo cromosómico síndrome de Turner. se observa habitualmente en el Verdadera. La otra línea celular puede ser normal, pero podría también contener material del cromosoma Y. d. Las mujeres con cariotipo 47,XXX no son fértiles. Falsa. Tienen una fertilidad normal. e. Los síndromes de rotura cromosómica pueden ocasionar cáncer. Verdadera. Esto se produce por una inestabilidad del DNA. 5.a. En el síndrome del cromosoma X frágil la repetición de tripletes no cambia significativamente su tamaño cuando se traspasa del padre a la hija. Verdadera. Un hombre transmisor normal traspasa la mutación a sus hijas esencialmente sin cambios. b. El síndrome del cromosoma X frágil es un trastorno único y bien definido. Falsa. Además de FRAXA, hay también FRAXE y FRAXF. c. Se deben realizar pruebas cromosómicas a las niñas con hernia inguinal bilateral. Verdadera. El síndrome de insensibilidad a los andrógenos puede presentarse de esta forma. d. El cariotipo normal es un buen medio para diagnosticar el síndrome del cromosoma X frágil en las niñas. Falsa. Esto no es fiable. El análisis del DNA es necesario. e. El análisis por FISH utilizando sondas multiteloméricas diagnostica cerca del 25% de las dificultades de aprendizaje inespecíficas. Falsa. La cifra es de alrededor del 5% 181 Capítulo 18: Preguntas basadas en casos.Caso 1.- Una niña recién nacida parece poco dismórfica, se le diagnostica una cardiopatía congénita con defecto de tabique auriculoventricular (AVSD) y los pediatras consideran que puede tratarse de un síndrome de Down. Se discute con los padres y se realiza un cariotipo. El resultado es normal: 46, XX. La niña es muy “buena” durante la lactancia y llora muy poco, y no se le realizan más pruebas complementarias. Posteriormente la niña muestra un retraso en el desarrollo global, golpea su cabeza, se despierta cada noche durante unas 3 horas y tiene una braquidactilia leve. Los pediatras la remiten a un genetista para que dé su opinión. 1.- ¿Sugiere la historia un diagnóstico? Los golpes en la cabeza no son raros al inicio de la infancia, sobre todo en los niños con retraso en el desarrollo y, no es necesariamente una característica útil para hacer un diagnóstico. Sin embargo, cuando se combinan con el patrón del sueño persistentemente alterado, se debe considerar el diagnóstico del síndrome de Smith- Magenis. Estos niños pueden ser tranquilos lactantes y tener cardiopatías congénitas. 2.- ¿Qué pruebas deben solicitarse? El síndrome de Smith-Magenis se debe habitualmente a una microdelección en 17p11.2, para lo cual disponemos de una prueba con una sonda FISH. Pueden también presentar una conducta de auto-abrazos y desarrollar escoliosis. La melatonina ha demostrado ser un tratamiento muy eficaz para los trastornos del sueño. Caso 2.- Los padres de una niña de 10 años piden una cita de seguimiento en la clínica de genética. A la edad de 4 años tuvo algunos problemas de conducta y se hizo un análisis de sus cromosomas a partir de una muestra sanguínea. El resultado fue 47,XXX y se les explicó que estas niñas a veces tienen problemas de conducta, son generalmente altas, la fertilidad es normal y que “no habría ningún problema”. Sin embargo, a los 10 años es la niña más pequeña de la clase, y aún tiene el ligero cuello con alas que presentaba en el nacimiento. 1.- ¿Qué diagnóstico debe proponerse actualmente? considerarse y qué pruebas deben El consejo anterior dado de forma natural asumía que la chica era 47,XXX pura. Sin embargo, la evolución posterior plantea la posibilidad de que tuviera un mosacismo cromosómico y, sobre todo, de que fuera un mosaico para el síndrome de Turner (45,X). Se debe proponer la realización de un frotis bucal y/o una biopsia cutánea para ver los cromosomas en un tejido distinto de la sangre. Si fueran normales, habría que considerar otras causas de estatura baja. 2.- ¿En qué forma se modifica el consejo genético y el tratamiento posterior con el nuevo diagnóstico? 182 Si de hecho se encontrase que la chica es un mosaico 45,X/47,XXX, se necesitaría ser investigada por las complicaciones del síndrome de Turner: cardiopatía congénita y riñón en herradura. Además, se cuestiona su fertilidad, por lo que habría que remitirla a un endocrinólogo pediátrico, quien le asesoraría también sobre un posible tratamiento con hormona de crecimiento. Capítulo 19: Trastornos monogénicos. 1.a. En la enfermedad de Huntington es más probable una edad de aparición más temprana en la descendencia si el gen se transmite por una madre afectada en vez de por un padre afectado. Falsa. La inestabilidad de la meiosis es mayor en la espermatogénesis que en la ovogénesis. b. En la enfermedad de Huntington, los que son homocigóticos para la mutación no están más gravemente afectados que los que son heterocigóticos. Verdadera. Esto se ha demostrado en estudios en Venezuela. c. Desde la aparición de la enfermedad de Huntington, la duración media de la enfermedad hasta el episodio terminal es de 25-30 años. Falsa. La duración es de aproximadamente de 10-15 años. d. En la enfermedad de Huntington, la no penetrancia de la enfermedad puede estar relacionada con los alelos anómalos con baja repetición. Verdadera. Esto es así por los alelos de penetrancia reducida de las repeticiones de 36 a 39. e. La anticipación es una característica de la herencia de la distrofia miotónica. Veradera. El trastorno se debe a una expansión muy amplia de repeticiones de tripletes, como en el síndrome del cromosoma X frágil. 2a. El insomnio es una característica de la distrofia miotónica. Falsa. La somnolencia es habitual. b. La distrofia miotónica es una causa de hipertonía neonatal. Falsa. Hipotonía neonatal. c. Los efectos clínicos de la distrofia miotónica están mediados por el RNA. 183 Verdadera. A través de una proteína de unión al RNA, CUG, que interfiere con diversos genes. d. Los defectos de conducción cardíaca son una característica de la distrofia miotónica y de las canalopatías iónicas. Verdadera. Una característica importante de la distrofia miotónica y de la animalía que define a muchas canalopatías. e. El diagnóstico de fibrosis quística (FQ) únicamente en la clínica de infertilidad. puede conocerse Verdadera. Los hombres con FQ leve o subclínica tienen una ausencia congénita bilateral del conducto deferente. 3a. En la FQ, la mutación en F117H es la más habitual en el norte de Europa. Falsa. La mutación deltaF508 es la más habitual. b. En el gen CFTR un polimorfismo intragénico modificador afecta al fenotipo. Verdadera. El tracto politimidina-5T, 7T y 9T- se puede correlacionar rigurosamente con distintos fenotipos de la FQ. c. Las miocardiopatías hipertróficas se deben principalmente a mutaciones en los genes de las canalopatías iónicas. Falsa. Esto es cierto para la mayoría de las arritmias cardíacas heredadas; las miocardiopatías se deben a menudo a defectos en las proteínas del músculo cardíaco. d. Muchas distrofias musculares hereditarias diferentes pueden estar ligadas al complejo que incluye a la distrofina (mutada en las distrofias musculares de Duchenne y de Becker). Verdadera. Este complejo de glicoproteínas en la membrana muscular contiene diversas unidades; los defectos en ellas ocasionan distrofias de cintura. e. Las dificultades de aprendizaje forman parte de la atrofia muscular espinal. Falsa. Estos pacientes tiene una inteligencia normal. 4.a. Es poco probable que la fibrosis quística y la hemofilia sean candidatas para la terapia génica. Falsa. Son buenas candidatas según el pensamiento actual. 184 b. Una relación envergadura: altura anómala es una característica principal del síndrome de Marfan. Falsa. Es únicamente esquelético. un componente de los criterios del sistema c. La neurofibromatosis tipo 1 (NF1) a veces “ salta generaciones” Falsa. Se cree que es un trastorno con penetrancia total. d. La escoliosis puede ser una característica de la NF1 y del síndrome de Marfan. Verdadera. No es habitualmente grave pero es una característica reconocida. e. Las cataratas pueden ser una característica de la NF1 pero no de la NF2. Falsa. La situación es opuesta. 5.a. Los tipos I y II de la neuropatía hereditaria sensoriomotora se refieren a una clasificación genética. Falsa. Ésta es una clasificación neurofisiológica. b. La neuropatía hereditaria sensoriomotora puede seguir todos los patrones principales de herencia. Verdadera. Autosómica cromosoma X. dominante, autosómica recesiva y ligada al c. La vaina del nervio, más que el mismo nervio, es la que está alterada en la forma más habitual de neuropatía hereditaria sensoriomotora. Verdadera. Las mutaciones en la proteína de la mielina periférica afectan a las células de Schwann d. La estimación del valor de la creatina cinasa y del factor VIII es válida para identificar a los portadores de la dstrofia de Duchenne y de la hemofilia respectivamente. Falsa. No son buenas pruebas discriminatorias y se prefiere el análisis del DNA. e. El síndrome de Brugada es una de las variedades de la atrofia muscular espinal. Falsa. Es una arritmia cardíaca hereditaria. Capítulo 19. Preguntas basadas en casos. 185 Caso 1.- una mujer de 31 años querría formar una familia, pero está preocupada porque su hermano de 39 se le diagnóstico una distrofia muscular de Becker hace 30 años y ella recuerda que se le dijo que el trastorno afecta a los varones pero lo transmiten las mujeres. Su hermano vive aún, pero bastante incapacitado por su trastorno. No hay más antecedentes familiares de distrofia muscular. 1.- ¿Es fiable el diagnóstico original? Los antecedentes en el hermano son consistentes con que tenga una distrofia muscular de Becker (DMB), pero también con otras posibilidades diagnósticas, por ejemplo, una distrofia muscular de cinturas (DMC). A veces es difícil diferenciar entre estos dos trastornos y la herencia es distinta, con implicaciones bastante diferentes para la mujer que desea formar familia. 2.- ¿Cuáles son los siguientes pasos para tratar esta situación? Hay que revisar y volver a valorar los registros médicos del hermano afectado. Hace 30 años la pruebas para la DMB eran muy básicas, pero ahora disponemos del análisis de la mutación del gen de la distrofina. Una biopsia muscular sujeta a una tinción específica para la distrofina puede ser diagnóstica, pero si es negativa se dispone de técnicas de tinción para las diferentes formas de la DMC. Si pertenecen al grupo de la DMC, se puede tranquilizar a la mujer porque siguen una herencia autosómica recesiva. Si se trata de una DMB, las pruebas de portador para la consultante serían inmediatas con que sólo se encontrase una mutación específica en el hermano. Caso 2.- Una pareja de mediana edad está destrozada porque su hija de 21 años padece un colapso en un baile y no la pueden resucitar. En el examen postmortem todas las pruebas toxicológicas son negativas y no se encuentra ninguna causa de muerte. La madre recuerda que su padre murió de repente a los 50 creyéndose, en aquel momento, que era de causa cardíaca y su hermana ha tenido episodios cortos de mareo pero no ha considerado necesario consultar con un médico. La pareja tiene otros tres hijos jóvenes y amantes del deporte y se preguntan si esto puede volver a ocurrir. 1.- ¿Cuáles son las pruebas adecuadas? La muerte súbita e inesperada de los adultos jóvenes, sobre todo cuando no se puede identificar un causa, es extremadamente impactante para una familia. La atención se centra en las arritmias y en las miocardiopatías hereditarias; a veces estas últimas no muestran características evidentes en el examen postmortem. Todos los miembros próximos de la familia son candidatos para una valoración cardíaca mediante una ecografía y un ECG con pruebas de esfuerzo, buscando evidencias de los síndromes de QT largo y de Brugada. Se dispone de pruebas genéticas, pero no está garantizado que identifiquen una mutación patógena. Algunas formas de arritmias/miocardiopatías hereditarias son susceptibles de tratamiento profiláctico; para las demás, poco se puede hacer actualmente. 2.- ¿Qué consejo se debe dar a la familia? 186 El tratamiento dependerá del resultado de los exámenes y de las pruebas genéticas. Sin embargo, si no se encuentran signos específicos, es muy difícil saber cómo aconsejar a familias como éstas. Probablemente se deberían evitar los deportes y la natación de alta intensidad, en caso de que tales actividades sean un factor precipitante de una arritmia con amenaza para la vida. Capítulo 20: Cribado de las enfermedades genéticas. 1.a. Los estudios de inactivación del cromosoma X proporcionan un medio útil para identificar a las mujeres portadoras de algunos trastornos ligados al cromosoma X. Verdadera. Buscando las pruebas de dos poblaciones de células. b. Faltan signos clínicos fiables para detectar a la mayoría de los portadores de los trastornos ligados al cromosoma X. Verdadera. Los signos clínicos sólidos son la excepción más que la regla. c. Las variantes en la secuencia de DNA son útiles para el cribado mientras no sean polimórficas. Falsa. Las variantes en la secuencia de DNA deben ser polimórficas para que sean útiles. d. Los marcadores de DNA son útiles para el cribado en la retinitis pigmentaria. Falsa. Hay demasiada heterogeneidad en el locus en este trastorno. e. Con el fin de realizar un cribado de los miembros de la familia, se deben buscar las oportunidades para crear bancos de DNA de los probandos con trastornos letales. Verdadera. Como la regla general esto puede ser vital, pero debe ser emprendido con un consentimiento informado. 2.a. Los pacientes con esclerosis tuberosa presintomática tiene siempre un exantema facial característico. Falsa. El exantema facial del angioqueratoma (adenoma sebáceo) no está a menudo presente. b. Siempre es posible diagnosticar una neurofibromatosis tipo 1 a la edad de 2 años porque es un trastorno de penetrancia completa. Falsa. Puede que no haya un número significativo de manchas de café con leche hasta los 5-6 años. c. Las pruebas bioquímicas no deben considerarse pruebas genéticas diagnósticas. 187 Falsa. Pueden ser completamente informativas del estatus genético de un individuo. d. La resonancia magnética de la columna lumbar puede ser útil para diagnosticar un síndrome de Marfan. Verdadera. La ectasia dural de la columna lumbar es uno de los principales criterios. e. Las pruebas genéticas predictivas debe realizarse siempre por análisis directo del gen. Falsa. Los marcadores de DNA ligados, y a veces las pruebas bioquímicas, pueden ser la mejor modalidad disponible. 3.a. Los programas de detección selectiva de la población deberían estar respaldados legalmente. Falsa. La participación debe ser, en principio, voluntaria. b. Los programas de detección selectiva de la población deberían ofrecerse si se dispusiera de alguna forma de tratamiento o de prevención. Verdadera. El resultado de los programas de detección selectiva de la población debe ser una mejoría en beneficio de la salud. c. La sensibilidad de una prueba se refiere a la amplitud con que la prueba detecta únicamente a los individuos afectados. Falsa. Eso es la especificidad de la prueba. d. El valor predictivo positivo de una prueba de detección selectiva se refiere a la proporción de pruebas positivas que son positivos reales. Verdadera. Es diferente de la sensibilidad, que se refiere a la proporción de casos afectados que se detectan (es decir, puede haber algunos falsos negativos). e. Si no hay un tratamiento eficaz para un trastorno de aparición tardía, las pruebas genéticas predictivas deben realizarse con mucho cuidado. Verdadera. Un consejo experto adecuado debe formar parte del programa de pruebas predictivas. 4.a. Una proporción elevada de personas sometidas a pruebas de portador no pueden recordar sus resultados adecuadamente. Verdadera. El propio recuerdo de los resultados, o su interpretación, es frecuentemente inexacto. 188 b. La detección selectiva de los portadores de la fibrosis quística es el programa más útil entre los chipriotas griegos. Falsa. La incidencia más elevada de una enfermedad grave es la de la talasemia beta: 1 de cada 8 son portadores. c. La posibilidad de que una prueba de detección selectiva lleve a una discriminación en el empleo no es una preocupación importante. Falsa. Esto ha sucedido antes y debe ser una preocupación principal. d. La detección selectiva neonatal de la distrofia muscular de Duchenne mejora la esperanza de vida. Falsa. El beneficio radica en informar ala familia para la toma de decisiones reproductivas posteriores. e. La detección selectiva neonatal de la fibrosis quística es una prueba basada en el DNA. Falsa. El primer análisis es bioquímico, una medida de la tripsina inmurreactiva. 5.a. El cribado en los recién nacidos de la hemocromatosis, el gen más habitualmente mutado en las poblaciones europeas, es un programa que se realiza a escala nacional en el Reino Unido. Falsa. Aunque la frecuencia de portador es cerca de 1 de cada 10, no se ha emprendido ninguna detección selectiva de la población en Reino Unido. b. El cribado presintomático en los niños de enfermedades genéticas de aparición en el adulto es una decisión que toman los padres. Falsa. En general, a menos que se pueda ofrecer una intervención médica beneficiosa, tales pruebas deben ser diferidas hasta que el niño sea lo suficientemente mayor para tomar una decisión. c. El cribado neonatal de la fenicetonuria y el del hipotiroidismo congénito son los programas de detección selectiva más antiguos. Verdadera. Han estado operativos durante cerca de 30 años. d. El cribado de la deficiencia de MCAD (acil-CoA deshidrogenasa de cadena media) está integrado en el cribado de la población neonatal. Falsa. Se encuentra únicamente en la fase de investigación. e. Los registros investigación. genéticos se hacen principalmente para la Falsa. Su función principal es un departamento de servicios es para el tratamiento clínico de los pacientes y de las familias. Capítulo 20: Preguntas basadas en casos. 189 Caso 1.- un hombre de 32 años es alto y delgado y hace 20 años su padre murió de repente a la edad de 50. El médico de familia se pregunta si su paciente tiene un síndrome de Marfan y le refiere a una clínica de genética. Presenta algunas características de síndrome de Marfan pero, estrictamente hablando, sólo cumpliría los criterios aceptados si los antecedentes familiares fueran definitivamente positivos para el trastorno. Tiene un hermano con una altura media y tres niños pequeños que gozan de buena salud. 1.- En términos de pruebas genéticas, ¿cuáles son las limitaciones para la detección selectiva si el diagnóstico es síndrome de Marfan? Es posible realizar un análisis de la mutación en el gen fibrillin-1 para el consultante, pero no está garantizado identificar una mutación incluso si el diagnóstico clínico es seguro. Sería factible un análisis de ligamiento en el locus de fibrillin1en el cromosoma 15 utilizando DNA de la madre del consultante e infiriendo el haplotipo del padre, cuyo estatus de afectado tendría que ser asumido. Sin embargo, en una pequeña familia el ligamiento puede ser consistente con la segregación del trastorno debida al azar. Además, en casos poco habituales el síndrome de Marfan se debe a un gen mutado en el locus separado del cromosoma 13. hay graves dificultades para realizar las pruebas genéticas en esta situación. 2.- ¿Cuáles son los problemas de la detección selectiva para la familia? La complicación importante con amenaza para la vida del síndrome de Marfan es la dilatación progresiva de la raíz aórtica que conlleva un riesgo de disección. Hay quehacer seguimiento a los que tienen un diagnóstico firme hasta que tengan al menos 30 años. Si existen dudas sobre el diagnóstico, la realización de pruebas cardíacas de forma periódica es probablemente una precaución sensata para todos los que están en riesgo hasta la mitad de la segunda veintena. Caso 2.- Se está evaluando una prueba de cribado para la fibrosis quística (FQ) en una población de 100,000 habitantes lactantes recién nacidos. La prueba fue positiva en 805 lactantes, de los cuales se ve que finalmente 45 tienen FQ por una combinación de análisis del DNA y pruebas del sudor. De los lactantes cuya prueba de detección fue negativa, cinco desarrollaron posteriormente síntomas y se les diagnóstico FQ. 1.- ¿Cuál es la sensibilidad y la especificidad de esta prueba de cribado? La sensibilidad es la proporción de verdaderos positivos detectados por la prueba, es decir, 45/45 + 5 = 90%. La especificidad es la proporción de verdaderos negativos detectados por la prueba, es decir, 99.190 (los casos no afectados con resultado negativo)/99.190 + 760(los casos no afectados con resultado positivo)=99.2%. 2.- ¿Cuál es el valor predictivo positivo de la prueba de cribado? El valor predictivo positivo es la proporción de casos con resultado positivo que tienen realmente la enfermedad, es decir, 45/805=5.6%. 190 Capítulo 21: Pruebas prenatales y genética de la reproducción. 1.a. La amniocentesis se practica rutinariamente cada vez más temprano en el embarazo. Falsa. Aún se realiza principalmente alrededor de las 16 semanas de gestación. b. Las células que crecen procedentes de una amniocentesis tiene su origen únicamente en la piel fetal. Falsa. Se obtienen también del amnios y del epitelio del tracto urinario fetal. c. El riesgo de aborto es mayor en la biopsia coriónica que en la amniocentesis. Verdadera. El riesgo es del 1 al 2 %, comparado con el 0.5 al 1%. d. La biopsia coriónica es un procedimiento seguro a las 9 semanas de gestación. Falsa. Hay un pequeño riego de causar anomalías en los miembros; la CVS no debe realizarse antes de las 11 semanas de gestación. e. Los trastornos de la piel fetal pueden diagnosticarse por ecografía. Falsa. Es necesaria la fetoscopia si no se dispone de una prueba de DNA. 2.a. En los embarazos de un síndrome de Down los valores de gonadotropina coriónica humana (hCG) en el suero materno están habitualmente altos. Verdadera. Forma parte de la prueba triple. b. En los embarazos de un síndrome de Down la concentración de alfafetoproteína (Alfa-fetorpoteína) en el suero materno está habitualmente reducida. Verdadera. Forma parte de la prueba triple. c. En los embarazos de una trisomía del 18 los marcadores del suero materno se comportan de la misma forma que en los embarazos de un síndrome de Down. Falsa. La trisomía del 18 todos los marcadores del suero materno están bajos. d. Cerca del 95% de los embarazos de un síndrome de Down se identifican determinando la edad materna, la alfa-fetoproteína y los valores de hCG en el suero y el grosor de la translucencia o pliegue nucal. Falsa. La mejor cifra conseguida es de alrededor del 86% 191 e. El embarazo gemelar es una causa de valores de alfa-fetoproteína materna elevados en el suero. Verdadera. Hay dos fetos en lugar de uno. 3.a. El mosaicismo cromosómico se detecta en cerca del 5% de las muestras de biopsia coriónica. Falsa. la cifra es de alrededor del 1%. b. Los trastornos cromosómicos translucencia nucal anómala. son la causa principal de la Verdadera. Especialmente aneuploidías. c. El intestino fetal ecogénico en la ecografía es un factor de riesgo para la fibrosis quística. Verdadera. Probablemente debido a la presencia de meconio espesado. d. En una pareja que ha tenido un hijo con el síndrome de Down, el riesgo en el siguiente embarazo no está habitualmente muy incrementado. Verdadera. La mayoría de los casaos del síndrome de Down se deben a una no disyunción en la meiosis. e. Los cromosomas marcadores significativos clínicamente. familiares son habitualmente no Verdadera. No es probable que tengan efectos clínicos diferentes en los diversos miembros de la misma familia. 4.a. La inseminación de un donante es un procedimiento que no requiere una licencia de la HFEA. Falsa. Se requiere una licencia de la HFEA. b. El alquiler del vientre para la gestación es ilegal en el Reino Unido. Falsa. No es ilegal pero requiere una licencia de la HFEA. c. Para el diagnóstico genético preimplantación (DGP), la fertilización del huevo se consigue mediante la inyección de un espermatozoide intracitoplasmático (ICSI). Verdadera. Esto se hace para evitar la presencia de esperma foráneo. d. La tasa de éxito de la FIV, en términos de llevarse un niño a casa, es sólo de 50%. Falsa. La cifra es alrededor del 25%. 192 e. El mayor grupo de enfermedades que se comprueban en el DGP son los trastornos monogénicas. Falsa. Los trastornos cromosómicos son el grupo mayor. 5.a. Hay un mayor riesgo concebidos por ICSI. de trastornos genéticos en los niños Verdadera. Las anomalías cromosómicas están presentes en 10 al 12 % de los varones con azoospermia o con una oligospermia grave, algunas de ellos son heredables. b. El esperma de un donante puede utilizarse sólo cinco veces. Falsa. Actualmente, la regla es que no puede haber más de 10 embarazos de un donante. c. Los niños concebidos por inseminación de donante tienen derecho a tanta información como los niños adoptados sobre sus padres biológicos. Falsa. Actualmente no tienen derecho a conocer la identidad de sus padres biológicos. d. Si se pudiera conseguir un diagnóstico prenatal analizando las células fetales en la circulación materna, la pareja aún tendría que considerar la interrupción del embarazo. Verdadera. El análisis de las células fetales en la circulación materna sólo elimina el riesgo de aborto. e. La infertilidad afecta a cerca de 1 de cada 20 parejas. Falsa. La cifra es aproximadamente de 1 de cada 7 Capítulo 21: Preguntas basadas en casos. Caso 1.- Una mujer de 36 años embarazada decide someterse a pruebas prenatales mediante una biopsia de las vellosidades coriónicas tras el hallazgo de la detección de un incremento en el grosor de la translucencia nucal en la ecografía. El resultado inicial, usando las sondas FISH, es bueno (no hay evidencia de trisomía 21) y la mujer se siente enormemente aliviada. Sin embargo, después de 2 semanas, en las células cultivadas aparece mosaicismo para la trisomía del 20. Se somete a una amniocentesis una semana más tarde y a las tres semanas el resultado muestra también algunas células con trisomía del 20. 1.- ¿Porqué se realizó una amniocentesis además de la biopsia de las vellosidades coriónicas? El hallazgo de un mosaicismo para la trisomía del 20 en el tejido de las vellosidades coriónicas podría haber sido un caso de mosaicismo confinado a la placenta. Esto 193 último no es un suceso raro en una gran variedad de aberraciones cromosómicas, pero al estar confinado, no hay consecuencias graves para el embarazo. El problema que existe sen seguir con la realización de la amniocentesis está en la interpretación del resultado. Si no se encuentran células anómalas, no se descarta completamente un mosaicismo cromosómico en el feto. Si se encuentran células anómalas, las implicaciones clínicas son muy difíciles, si no imposibles, de predecir. 2.- ¿Qué otra amniocentesis? cosa puede hacerse después del resultado de la Este caso ilustra las fluctuaciones emocionales y experiencias que algunas mujeres y parejas tiene que afrontar, como resultado de las diferentes formas de pruebas prenatales y de su interpretación. De hecho, no es probable que el mosaicismo de la trisomía del 20 tenga una gran importancia clínica, aunque es muy difícil estar seguro. Se han notificado anomalías renales, y se puede proponer una exploración detallada de las anomalías fetales durante el resto del embarazo. Sin embargo, lo que podría haber sido un embarazo feliz será, probablemente, uno lleno de ansiedad. Caso 2.- Una pareja tiene dos hijos autistas y les gustaría mucho tener otro hijo, están preparados para hacer cualquier cosa con tal de garantizar que el problema no se presente de nuevo. Han recopilado mucha información en Internet y saben que los chicos están afectados con mayor frecuencia (la relación masculino:femenino es de aproximadamente 4:1). Según su punto de vista, la solución más simple para su problema es la selección del sexo mediante diagnóstico genético preimplantación (DGP). 1.- ¿Qué pruebas deben realizarse a los hijos autistas? En la gran mayoría de los casos de autismo no se alcanza ningún diagnóstico específico. Se debe realizar un análisis cromosómico, incluyendo una revisión multitelomérica, el síndrome del cromosoma X frágil, una evaluación metabólica y un examen de los trastornos neurocutáneos. 2.- Si las pruebas a los hijos no permiten hacer un diagnóstico, ¿puede el genetista atender la petición de la pareja en relación con la selección del sexo por DGP? Ésta es una situación muy difícil. Sin embargo, en este caso no hay pruebas de que el autismo esté ligado al cromosoma X, y, por tanto, no hay garantías de que ninguna de las hijas no resulte afectada. En consecuencia, sería muy difícil apoyar esta petición en el Reino Unido, donde el DGP está regulado por la Human Fertilization and Embryolofy Authority y la selección del sexo no está permitida para otras situaciones distintas de los trastornos claramente ligados al cromosoma X. En otros países, donde estas técnicas no están reguladas, la pareja podría encontrar clínicos que accedieran a su petición. Capítulo 22: Cálculo de riesgo. 1.a. Una probabilidad de 0.5% es lo mismo que un riesgo del 50%. 194 Verdadera. Éstas son las dos formas de expresar la misma probabilidad. b. La probabilidad de un suceso nunca excede la unidad. Verdadera. Una probabilidad de 1 significa que el suceso ocurrirá el 100% de las veces. c. En un embarazo de gemelos dicigóticos la probabilidad de que los niños tengan el mismo sexo es 0.5 Verdadera. La probabilidad de que ambos sean niños es ½ x ½ = ¼ para las niñas igual; por lo tanto la probabilidad de ser del mismo sexo es de ¼ + ¼ = ½ (0.5) d. El teorema de Bayes tiene en cuenta tanto la probabilidad previa como la información condicional. Verdadera. Estos dos enfoques del cálculo de la probabilidad son esenciales. e. Es un trastorno autosómico dominante, una penetrancia de 0.7 significa que el 30% de los heterocigotos no manifestará el trastorno. Verdadera. El 70% de los heterocigotos manifestarán el trastorno. 2.- Es un trastorno autosómico recesivo la probabilidad de que el sobrino de un individuo afectado sea un portador es: a. 1 de cada 8 Falsa. b. 1 de cada 2 Falsa. c. 1 de cada 4 Verdadera. Los padres del individuo afectado son portadores obligados, los tíos y las tías tiene un riesgo de 1 de cada 2, los primos un riesgo de 1 de cada 4. d. 1 de cada 10 Falsa. e. 1 de cada 6 Falsa. 3.- En la herencia recesiva ligada al cromosoma X: a. Los hijos de una mujer portadora tienen una probabilidad de 1 de cada 4 de estar afectados. Falsa. El riesgo es 1 de cada 2 si se sabe que el sexo del feto es masculino. 195 b. La madre de un varón afectado es una portadora obligada. Falsa. El varón puede estar afectado porque se haya producido una nueva mutación. c. El riesgo de mosaicismo gonadal en la distrofia muscular de Duchenne puede ser tan alto como el 10% Verdadera. Esto es significativo y ha de ser tenido en cuenta en el cálculo del riesgo y en el consejo. d. En una mujer con un hijo afectado, la posibilidad de ser una portadora se reduce si tiene tres hijos más no afectados. Verdadera. Ésta es una información condicional que puede incluirse en un cálculo de Bayes. e. Se denomina “consultante ficticio” a un individuo en una genealogía que se ignora a la hora de calcular el riesgo. Falsa. Éste es un individuo clave cuyo riesgo debe calcularse antes que el riesgo del consultante. 4.- En una herencia autosómica recesiva el riesgo de que el sobrino de un individuo afectado, nacido del hermano sano del individuo afectado, sea un portador es: a. 1 de cada 2. Falsa. b. 1 de cada 4. Falsa. c. 2 de cada 3. Falsa. d. 1 de cada 3. Verdadera. El hermano sano del individuo afectado tiene una probabilidad de 2 de cada 3 de ser un portador; el hijo de esta persona tiene la mitad de riesgo. e. 1 de cada 6. Falsa. 5.a. Al calcular el riesgo, la información condicional puede incluir datos de DNA negativos. Verdadera. Por ejemplo, los hallazgos de mutaciones negativas cuando se realizan pruebas para la fibrosis quística. 196 b. En la aparición retrasada de un trastorno heredado de forma dominante, el cálculo del riesgo de un heterocigoto requiere los datos de expresión clínica. Verdadera. Los datos de la edad de aparición (expresión clínica) deben obtenerse de grandes estudios familiares. c. El cálculo de cociente de posibilidades no necesita información sobre las probabilidades previas. Falsa. Sin está información se cometerán enormes errores. d. Los riesgos empíricos obtenidos de los estudios epidemiológicos tienen aplicaciones limitadas en una situación particular. Verdadera. Un riesgo empírico es realmente una cifra de compromiso. e. Al utilizar los datos de un marcador de DNA para predecir el riesgo, la fracción de recombinación no importa realmente. Falsa. Puede importar mucho porque es una medida de la probabilidad que se produzca un suceso de recombinación meiótica entre el marcador y la mutación del gen que ocasiona la enfermedad. Capítulo 22. Preguntas basadas en casos. Caso 1.- Dos primos se han casado y quieren formar una familia. Sin embargo, su tío murió hace muchos años de un síndrome de Hurler, una mucopolisacaridosis, un error del metabolismo que sigue una herencia autosómica recesiva. No se dispone de muestras tisulares para los estudios genéticos. 1.- ¿Cuál es el riesgo de que el primer hijo de la pareja resulte afectado por el síndrome de Hurler? Cada uno de los hermanos de la tía afectada tiene la posibilidad de ser un portador; por tanto, cada uno de los primos tiene la posibilidad de ser un portador. La posibilidad de que el primer hijo de la pareja resulte afectado es 1/3 X 1/3 X ¼ = 1/36. 2.- ¿Se le puede proponer a la pareja algo más que la cifra de riesgo? Incluso si no se pueden realizar estudios genéticos, se pueden proponer pruebas bioquímicas prenatales para sus embarazos, aunque las pruebas bioquímicas o determinarán con fiabilidad si son portadores. Si se decidieran por la pruebas prenatales sería también bueno realizar las pruebas para el síndrome de Hunter, que puede confundirse fácilmente con el síndrome de Hurler clínicamente y está ligado al cromosoma X. Caso 2.- Una mujer tiene un hermano y un tío materno afectados de hemofilia A. Ella misma tiene dos hijos no afectados y le gustaría tener más hijos. La remiten a una clínica de genética para analizar el riesgo y las opciones. 197 1.- Basándose exclusivamente en la información dada, ¿cuál es el riego de que la mujer sea portadora de la hemofilia A? Se puede realizar un simple cálculo de Bayes, teniendo en cuenta que ella ha tenido dos hijos normales (tabla 1). Por tanto, ella tiene una posibilidad de 1/5 o del 20% de ser portadora. Tabla 1. PROBABILIDAD ES PORTADORA NO ES PORTADORA PREVIA 1/2 1/2 CONDICIONAL ½x½ 1 CONJUNTA 1/8 1/2 POSTERIOR 1/8 / 1/8 + ½ = 1/5 (2 HIJOS NORMALES) 2.- ¿Se puede hacer algo para modificar su riesgo? Hay una buena posibilidad de identificar la mutación en el gen del factor VIII en su hermano o en su tío si alguno de los dos está aún vivo. De ser así, entonces podría ser posible determinar definitivamente su estatus de portadora. De lo contrario, las pruebas del valor del factor VIII y del antígeno relacionado con el factor VIII pueden dar un resultado que puede modificar su riesgo, pero esto no es necesariamente discriminatorio. El análisis de ligamiento del DNA sería mucho más fiable, siempre y cuando se disponga de muestras de DNA. Capítulo 23: Tratamiento de las enfermedades genéticas. 1.- Los métodos actualmente utilizados para tratar la enfermedad genética incluyen: a. Terapia génica de la célula germinal. Falsa. La terapia génica de las células germinales se considera inaceptable debido al riesgo de transmitir cambios genéticos a las generaciones futuras. b. Trasplante de células progenitoras. Verdadera. Por ejemplo, el transplante de médula ósea se utiliza para tratar diversas inmunodeficiencias heredadas. c. Sustitución de enzimas/proteínas. Verdadera. Los ejemplos incluyen la sustitución del factor VIII o IX en pacientes con hemofilia. d. Restricción dietética. 198 Verdadera. Por fenilcetonuria. ejemplo, la fenilalanina restringida en pacientes con e. Reparaciones in situ de las mutaciones por los mecanismos de reparación del DNA celular. Falsa. Este tratamiento potencial ha sido probado en modelos animales. 2.- La terapia génica puede administrarse por: a. Liposomas. Verdadera. Los liposomas se utilizan mucho porque son seguros y pueden facilitar la transferencia de genes grandes. b. Virus adenoasociados. Verdadera. Los ensayos con la terapia del gen CFTR han utilizado vectores víricos adeno-asociados. c. Oligonucleótidos antisentido. Falsa. Es necesario suministrar oligonucleótidos antisentido a las células diana. d. Lentivirus. Verdadera. Los lentivirus pueden ser útiles para el suministro de genes a las células que no están en división. e. Inyección de un DNA plásmido. Un ejemplo es la inyección del factor IX ligado a un plásmido en los fibroblastos de pacientes con hemofilia B. 3.- La terapia génica se ha utilizado con éxito para tratar a pacietes con las siguientes enfermedades: a. Fibrosis quística. Falsa. Los ensayos han mostrado un aporte seguro del gen CFTR en los conductos nasales, pero el tratamiento eficaz de la fibrosis quística no es posible en el momento actual. b. Inmunodeficiencia combinada grave (XL-SCID). Verdadera. Se ha tratado a un número de pacientes con éxito, aunque apareció un problema cuando dos niños desarrollaron leucemia. c. Enfermedad de las células falciformes. Falsa. Será difícil porque el número de cadenas de globina alfa y beta debe ser el mismo o se puede producir un fenotipo de talasemia. d. Hemofilia. 199 Verdadera. Algunos pacientes han sido capaces de reducir sus factores de coagulación exógenos. e. Deficiencia de la adenosina desaminasa. Verdadera. Aunque los intentos iniciales no tuvieron éxito, actualmente se ha tratado a dos pacientes con éxito por transferencia génica ex vivo. 4.- los métodos potenciales de terapia génica para el cáncer incluyen: a. Inhibición de las proteínas de fusión. Verdadera. Un ejemplo es el inhibidor de la proteincinasa utilizada para tratar la leucemia mieloide crónica. b. Estimulación del sistema inmunitario. Verdadera. Quizá mediante la sobreexpresión de las interleucinas. c. Expresión aumentada de los factores angiogénicos. Falsa. Los factores anti-angiogénicos pueden utilizarse para reducir el aporte sanguíneo a los tumores. d. Interferencia del RNA. Verdadera. La interferencia del RNA es una nueva técnica prometedora que puede utilizarse para identificar a genes sobreexpresados asociados con cánceres. e. Oligonucleótidos antisentido. Verdadera. Hay un número de ensayos en marcha para determinar la utilidad de esta técnica. 200