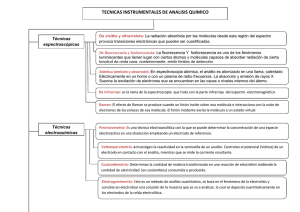
19. crOMaTOGrafía líQUida de alTa resOlUción (HPlc) FranCisCo J. Plou GasCa PaMela torres salas Instituto de Catálisis y Petroleoquímica (CsiC) 19.1. inTrOdUcción La cromatografía líquida de alta resolución (HPLC) es la técnica analítica de separación más utilizada, con ventas millonarias cada año tanto de equipos como de consumibles. La cromatografía, descrita por primera vez en 1906 por el italiano nacido en Rusia Mikhail Tswett, se utilizó inicialmente para separar pigmentos vegetales, clorofilas y xantofilas, mediante el empleo de un tubo de vidrio (en vertical) relleno de carbonato cálcico en polvo (fase estacionaria) y usando éter como eluyente (fase móvil). La solución recorrió el tubo, y los componentes individuales de la mezcla migraron hacia abajo con diferentes velocidades; la columna quedó marcada con bandas horizontales de distintos colores correspondientes a pigmentos diferentes. El resultado fue llamado cromatograma, justificando el nombre que se dio a la técnica: chroma, del griego, que significa «color», y graphein, que significa «escribir». Casi veinte años pasaron sin que esta técnica, que prometía simplificar la separación de mezclas complejas, fuera utilizada. En 1931, Kuhn y Lederer utilizaron la cromatografía líquida para separar muchos productos naturales. En 1952, Martin y Synge introdujeron la cromatografía de reparto, empezando a utilizar el concepto de la distribución y coeficiente de reparto en la separación de mezclas de aminoácidos, lo que les valió el premio Nobel de Química en 1952. En sus comienzos, la cromatografía líquida utilizaba columnas de vidrio de 50-500 cm de longitud y 1-5 cm de diámetro, con rellenos de 150-200 µm para asegurar un caudal adecuado (del orden de microlitros por minuto). Stahl, en 1956, interesado en la separación de los componentes de las células, diseñó un aparato que aplicaba capas de adsorbente en placas, llamándolo cromatografía de capa delgada o fina. En la década de los sesenta, se desarrolló la tecnología que permite utilizar rellenos con diámetro de partícula de 3-10 µm, que generaban altas presiones de trabajo. El avance posterior en fabricación de columnas, rellenos y detectores en continuo permitió el nacimiento de la cromatografía líquida de alta presión (HPLC: high-pressure liquid chromatography), luego denominada de alta reso- Técnicas de análisis y caracterización de materiales 788 lución (HPLC: high-performance liquid chromatography), nombre que se propuso para distinguir esta nueva tecnología de los clásicos métodos de cromatografía líquida a presión atmosférica, que se siguen utilizando con fines preparativos. Desde entonces la cromatografía líquida de alta resolución viene desarrollándose a pasos agigantados, debido principalmente a su versatilidad, alta sensibilidad, fácil adaptabilidad, precisión, la posibilidad de utilizar especies no volátiles o inestables térmicamente, y su gran aplicabilidad a sustancias de interés para la industria, la investigación y en general para la sociedad actual. Ejemplos significativos de aplicaciones de la cromatografía HPLC los constituyen el análisis de aminoácidos, proteínas, fármacos, biocombustibles, drogas, hidratos de carbono, grasas, pesticidas, contaminantes alimenticios, antibióticos, vitaminas o efluentes. En el contexto del libro Técnicas de análisis y caracterización de materiales la cromatografía HPLC es una técnica instrumental que puede utilizarse para, entre otras cosas, analizar determinados componentes presentes en un material, o medir la actividad catalítica de un material analizando el consumo de sustratos o la aparición de los productos de reacción. 19.2. cOMPOnenTes de Un eQUiPO de HPlc Todo equipo de cromatografía líquida de alta resolución debe disponer de, al menos, los siguientes módulos (Figura 19.1): 1. 2. 3. 4. 5. 6. 7. Reservorios o botellas para la fase móvil. Sistema de bombeo. Inyector (manual o automático). Columna. Uno o varios detectores en serie. Sistema de tratamiento de resultados. Botella para residuos. 4 W 3 C 7 5 2 B A 6 1 Figura 19.1. Principales componentes de un equipo de HPLC: (1) reservorios; (2) bomba; (3) inyector automático con diferentes carruseles de muestras; (4) columna cromatográfica; (5) detectores en serie; (6) sistema de tratamiento de datos; (7) botella para desechos. Cromatografía líquida de alta resolución (HPLC) 789 Existen otros módulos complementarios, a veces necesarios para la aplicación, como son el horno de columnas, un desgasificador, un colector de fracciones para cromatografía preparativa, divisores de flujo, etc. así como una serie de pequeños accesorios (soportes de la columna, restrictores de presión a la salida del detector, filtros intermedios, etc.). A continuación se indican las principales características de los componentes de un equipo de HPLC. 19.2.1. reservorios y fases móviles Los reservorios utilizados para las fases móviles van desde el sencillo frasco de disolvente hasta los sistemas más complejos capaces de filtrar, desgasificar, mantener una atmósfera inerte, termostatizar, agitar, etc. Su boca ha de ser lo más estrecha posible para evitar la evaporación del disolvente, especialmente cuando se emplean mezclas en las que no todos los componentes son igualmente volátiles, ya que puede alterarse la composición de la fase móvil. El vidrio suele ser el material más utilizado. El reservorio ha de taparse, para evitar la entrada de polvo. A través del tapón discurre el tubo (generalmente de teflón) que conduce la fase móvil al sistema de bombeo. Este tubo suele tener en su extremo un filtro de titanio de 10 µm de poro, que evita el paso hacia el equipo cromatográfico de partículas sólidas que pueda contener la fase móvil. Los equipos modernos de HPLC suelen incorporar 2-4 reservorios (con una capacidad típica de 1.000 ml cada uno) que permiten la preparación automatizada de mezclas de disolventes como fase móvil o la formación de los correspondientes gradientes. Suelen estar provistos de un sistema para eliminar los gases disueltos (que pueden generar nefastos efectos sobre la bomba, la columna o el detector). Lo más habitual suele ser el bombeo de un gas inerte (típicamente Helio) en forma de pequeñas burbujas para desplazar el aire, y en algunos casos se emplean desgasificadores automatizados en línea. En la Tabla 19.1 se resumen las principales precauciones que deben adoptarse con respecto a la fase móvil para el correcto funcionamiento del sistema de HPLC. Tabla 19.1. acciOnes QUe deben realizarse cOn la fase Móvil acción Filtrar Desgasificar Refrigerar Termostatizar Desoxigenar Agitación 1 cuando Siempre. Utilizar filtros de 0,45 µm1 Indispensable para equipos con gradientes en baja presión Disolventes de bajo punto de ebullición. Fases móviles muy viscosas Fases móviles o muestras oxidables Fases móviles formadas por solventes poco miscibles No deben emplearse filtros de acetato de celulosa cuando el disolvente es acetonitrilo Algunos de los disolventes más utilizados en HPLC son: agua, metanol, acetonitrilo, tetrahidrofurano, isopropanol, diclorometano y hexano. Es muy importante elegir disolventes con una pureza adecuada para cromatografía líquida. Además, cuando se emplean detectores fotométricos (Uv-vIS) es fundamental conocer la longitud de onda a partir de la cual el disolvente no interfiere con la señal del detector (UV-Cutoff). Técnicas de análisis y caracterización de materiales 790 Otras propiedades físico-químicas de los disolventes con una singular importancia en HPLC son: su viscosidad (que afecta de manera notable a la presión de trabajo), su polaridad (que determinará su poder de elución o fuerza elutrópica) y su miscibilidad con otros disolventes (Figura 19.2). n-pentano miscible hexano inmiscible isooctano ciclohexano xileno éter isopropílico cloroformo diclorometano tetrahidrofurano acetona dioxano acetato de etilo acetonitrilo n-propanol etanol metanol agua Figura 19.2. Miscibilidad de los disolventes más comúnmente utilizados en HPLC. 19.2.2. sistemas de bombeo. Gradientes en alta y baja presión Se denomina bomba cromatográfica al dispositivo capaz de proporcionar a la fase móvil la presión necesaria para atravesar, al flujo seleccionado, la columna y el resto del sistema. Las principales características que debe reunir una bomba HPLC convencional (escala analítica) son dos: la obtención de flujos de 0,1 a 5,0 ml/min y la capacidad de trabajar a presiones de hasta 6.000 psi (400 bar). El flujo proporcionado por la bomba debe ser, como cualquier otro valor analítico, lo más exacto y preciso posible (con una desviación estándar inferior al 0,5%). Además, es conveniente que los componentes de la bomba sean resistentes a la corrosión. Las bombas en HPLC son en su mayoría de flujo constante, y operan a la presión que sistema cromatográfico y columna determinan. En particular, se suelen utilizar generalmente bombas de pistón y solo en algunos casos bombas de jeringa. En las bombas de pistón, un dispositivo eléctrico actúa sobre el pistón que entra en una cámara donde se halla el disolvente, para comprimirlo y enviarlo a presión al resto del sistema. El flujo deseado se mide y alcanza en función del número de emboladas por unidad de tiempo. El principal inconveniente de las bombas de pistón simple es la aparición de oscilaciones en la línea base por cada embolada. Para evitarlo se suele recurrir a bombas con dos pistones en paralelo, o bombas con dos pistones en serie. Además, es habitual disponer de un atenuador de pulsos (damper) a la salida de la bomba. Cromatografía líquida de alta resolución (HPLC) 791 Los parámetros fundamentales que caracterizan una bomba HPLC son el rango y la precisión del caudal, su capacidad de mezcla, y la precisión de la mezcla. Por otro lado, los principales problemas que puede presentar una bomba son: – Ruido de flujo: la línea base siempre presenta ruido, que se conoce como rizado. Las causas de un rizado excesivo de la línea base pueden ser eventuales (burbujas, fugas, mal ajuste de los pistones, compresibilidad del disolvente empleado, etc.), constantes (rozamiento mecánico de las partes internas de la bomba, holgura entre las piezas móviles de la bomba) o ajenas a la bomba (sensor de presión, detector, sistema de registro, etc.). – Error de flujo: esto ocurre cuando el flujo real es distinto al estipulado. Puede ser debido a la presencia de burbujas (por no desgasificar correctamente la fase móvil) o a la existencia de fugas en algún punto del sistema. También puede ser consecuencia de la presencia de partículas sólidas en el disolvente (mal filtrado). Cuando la composición de la fase móvil no se modifica durante el análisis, se habla de un sistema de bombeo isocrático. No obstante, suele ser conveniente disponer de bombas capaces de proporcionar gradientes, especialmente para resolver mezclas de sustancias de polaridad dispar. En la Figura 19.3 se muestra la separación de los componentes de una reacción test de síntesis de biodiésel, observándose que el triglicérido (trilaurina) eluye 15 min más tarde que el diglicérido (1,2-dilaurina) y el resto de sustancias más polares de la muestra, bajo condiciones isocráticas. Este hecho supone un tiempo de análisis excesivo con el consiguiente consumo de disolventes. Para la formación de gradientes se disponen varios reservorios con distintas fases 0.10 1 2 3 4 5 Señal del detector (Abs 216 nm) 3 0.08 Glicerol 1-Monolaurina Laurato de metilo 1, 2-Dilaurina Trilaurina 0.06 1 2 4 0.04 0.02 5 0.00 0 5 10 15 20 25 Tiempo de retención (min) Figura 19.3. Separación de los componentes de una reacción test de obtención de biodiésel empleando condiciones isocráticas. Columna Mediterránea C18 Teknokroma (150 × 4,6 mm), 5 µm. Fase móvil: metanol/H2O 95/5. Flujo: 1,2 ml/min. Temperatura: 45 °C. Detección por ELSD. Técnicas de análisis y caracterización de materiales 792 móviles, y la bomba varía la proporción relativa de los distintos componentes con el tiempo. La elución mediante gradientes produce efectos similares a los obtenidos con una programación de una rampa de temperatura en cromatografía de gases. Existen dos posibilidades a la hora de realizar un gradiente en HPLC: mezcla a alta presión y mezcla a baja presión (Figura 19.4). En alta presión, se utiliza una bomba para cada componente de la fase móvil y una cámara de mezcla; esta cámara está situada entre las bombas y el inyector, es decir, en el lado de alta presión de la bomba. En baja presión, una válvula (proportioning valve) controla la composición de la mezcla abriendo la entrada de cada eluyente durante un tiempo proporcional a la composición deseada; la mezcla se realiza en una cámara situada entre la válvula y la bomba, es decir, en la zona donde todavía no hay presión. Mezcla a baja presión Mezcla a alta presión Columna Bomba C D C D Bomba A Columna Inyector Inyector A B A B Cámara de mezcla Bomba B Cámara de mezcla Figura 19.4. Formación de gradientes en alta y baja presión. Los parámetros que hay que considerar cuando se programan gradientes son el tiempo de equilibrado, la miscibilidad de los disolventes y su pureza, la realización de blancos, la reproducibilidad de tiempos/áreas y la compatibilidad con el detector. En la Tabla 19.2 se comparan las propiedades de ambos tipos de gradientes. El volumen de sistema (dwell volume), también denominado volumen de retraso del gradiente, es el volumen medido desde el punto de mezcla de los disolventes hasta la entrada en la columna. Puede oscilar desde un valor típico de 50 µl para gradientes en alta presión hasta 5 ml en el caso de baja presión. El volumen de sistema es muy importante para: (1) calcular el tiempo que tardan los cambios de gradiente en llegar a la columna, (2) calcular el volumen necesario para el equilibrado del sistema entre inyecciones, y (3) ajustar un método de gradiente entre distintos equipos. Tabla 19.2. cOMParación de GradienTes en alTa y baja Presión Parámetro alta presión baja presión Requerimientos técnicos Dos o más bombas Una única bomba Calidad de la mezcla Buena Buena Desgasificación de las fases móviles No necesaria Imprescindible Flujos bajos No adecuado Adecuado Porcentajes bajos de algún disolvente Adecuado No adecuado volumen de sistema Pequeño Grande (> 1 ml) Cromatografía líquida de alta resolución (HPLC) 793 19.2.3. inyectores El inyector es un dispositivo hermético que se encuentra situado a la salida de la bomba y que permite, mediante el empleo de válvulas, incorporar la muestra a la fase móvil antes de la columna, sin pérdidas de presión que alteren el flujo constante proporcionado por la bomba. Los inyectores de válvulas, conocidos como inyectores de bucle (loop), se utilizan prácticamente en el 100% de los cromatógrafos, destacando por la precisión de la inyección y el mantenimiento de la presión de la bomba, siendo sus partes internas fácilmente reemplazables. Resisten presiones de hasta 7.000 psi. Los inyectores pueden ser manuales y automáticos. En los manuales, se puede cambiar fácilmente el bucle (loop), que es el que nos determina el volumen de muestra que entra en la columna. Existen loops desde 5 µl hasta 5 ml (para HPLC semipreparativo). Es conveniente llenar bien el loop cargando un volumen de muestra de al menos 3 veces su volumen. Una vez lleno, se gira la válvula 1/6 de vuelta, lo que permite la introducción de la muestra a la columna. Los inyectores automáticos o automuestreadores trabajan de manera similar a los inyectores de bucles, permitiendo variar el rango de volumen inyectado. Los automuestreadores suelen disponer de termostatización, y presentan una mayor precisión que los manuales, optimizando el rendimiento del equipo HPLC. Algunos parámetros importantes que deben valorarse cuando se adquiere un inyector automático son: mínimo volumen interno, ausencia de contaminación cruzada, corto tiempo de equilibrado, termostatización (factor importante dependiendo de la naturaleza de las muestras a analizar). La posibilidad de utilizar placas multipocillo en lugar de viales es otro de los aspectos a considerar. En cuanto a los viales, existen innumerables posibilidades dependiendo del material de fabricación (plástico, vidrio ámbar o transparente, etc.), tipo de boca (encapsulable, roscada, etc.), composición del septum (silicona, PTFE —teflón—, o mezclas de ambos), volumen (0,3-20 ml), presencia de inserto para muestras de volúmenes reducidos, etc. 19.2.4. Tubos de conexión Los tubos capilares que se utilizan en HPLC para conectar los distintos componentes del cromatógrafo tienen un diámetro externo de 1/16’’ (1,59 mm). Como regla general, se recomienda utilizar tubo no muy capilar (aprox. 1 mm de diámetro interno) hasta el inyector —para que ofrezca la menor resistencia al flujo—, pero después de él, una vez que la muestra entra en contacto con el sistema cromatográfico, el tubo ha de ser del menor diámetro interno posible (por ejemplo, 0,2 mm de diámetro interno) para evitar la difusión de los analitos y el ensanchamiento de los picos. El tubo ha de continuar siendo capilar entre la salida de columna y el detector, para que los picos sigan tan separados como la columna permite. En la Tabla 19.3 se recogen los diámetros y volúmenes de los tubos utilizados en HPLC. Técnicas de análisis y caracterización de materiales 794 Tabla 19.3. caracTerísTicas de lOs TUbOs UTilizadOs en HPlc Posición Antes del inyector Después del inyector diámetro interno pulgadas mm 0,040 1,0 0,009 0,2 volumen 0,8 ml/m 50 µl/m Los tubos utilizados en HPLC pueden estar fabricados de: 1. Metal: suele ser acero inoxidable 316, que es un material estándar, fácil de doblar, no se obtura ni quiebra, y que se puede fabricar prácticamente en cualquier rango de diámetros interno y externo. Otros metales utilizados son: nitronic 50, níquel 200, EFNI (electroformed nickel), hastelloy C-22 (aleación de níquel, cromo y molibdeno), inconel 600, Titanio, etc. 2. Teflón (PTFE, politetrafluoroetileno): es muy resistente químicamente, pero no tanto frente a las altas presiones o temperaturas. Es rígido, difícil de manipular, y poroso. 3. PeeK (poliéter éter cetona): es el polímero inerte y biocompatible por excelencia. Presenta excelente resistencia química, térmica y a las altas presiones (hasta 5.000 psi, 300 bar). Solo es atacado por dimetilsulfóxido, THF, diclorometano y ácidos sulfúrico y nítrico concentrados. Es conveniente utilizar uniones y tubos del mismo material; por ejemplo, al combinar uniones de acero con tubo capilar de PEEK se corre el riesgo de que este último se dañe. Las uniones de acero suelen llevar una férula y un cono por separado, por lo que necesitan herramientas para su fijación. Por el contrario, las uniones de plástico suelen venir con la férula integrada, se enroscan a mano y son además reutilizables. 19.2.5. columnas y fases estacionarias La columna es la parte más importante del sistema cromatográfico, aunque su precio corresponde a no más del 1-5% del coste total del equipo. Las variables importantes a considerar en una columna HPLC, además de la naturaleza química de su fase estacionaria, son su longitud, diámetro, el tamaño de partícula, así como el diámetro de poro y la homogeneidad entre las partículas. La carcasa de las columnas suele ser de acero inoxidable, ya que se trata de un material inerte, resistente a las altas presiones y con el interior liso, aunque también existen columnas de vidrio, PEEK e incluso de polietileno flexible. Es importante utilizar una columna con una longitud suficiente para separar los compuestos de interés. El empleo de una columna más larga de lo necesario implica un mayor consumo de disolvente y de horas de trabajo. Las columnas más habituales para HPLC analítica tienen longitudes entre 10 y 30 cm. A veces se acoplan 2 o más columnas en serie para mejorar la resolución. En cuanto al diámetro de las columnas de HPLC, para columnas analíticas este suele ser de 4-5 mm; este diámetro es un compromiso entre el consumo de disolvente, el tiempo de análisis y la resolución cromatográfica. Cromatografía líquida de alta resolución (HPLC) 795 Para el correcto almacenaje de las columnas, estas deben guardarse con sus propias tuercas en un lugar protegido de la humedad ambiental y de las temperaturas extremas. No deben golpearse, y si se utilizaron tampones o sales en las fases móviles, hay que desplazarlos antes de almacenarlas, empleando los disolventes adecuados. En el caso de que se guarden en mezclas de disolventes que contengan agua, la concentración de esta no debe ser muy alta, evitando así el crecimiento de microorganismos en el interior de la columna. Por otro lado, suele ser necesario controlar la temperatura de la columna. Así, la mayoría de los equipos de HPLC suelen incorporar un horno que permite mantener la temperatura de la columna en el rango 20-100 ºC. En algunos casos, el controlador de temperatura del detector se puede utilizar como controlador de temperatura del horno. Las primeras fases estacionarias estaban formadas por partículas de 35 a 70 µm, porosas, que proporcionaban no más de 3.000 platos teóricos por metro. Hoy se alcanzan hasta 80.000 platos teóricos por metro, con rellenos de 5 µm o menores. Una primera exigencia para la fase estacionaria es su estabilidad y resistencia a las altas presiones. En la década de los ochenta, se sintetizaron sílices con una mejor distribución de grupos silanol-activos y menor cantidad de grupos silanol libres. En los noventa aparecieron columnas específicas para resolver problemas cromatográficos complejos. También se desarrolló la cromatografía de exclusión molecular, para purificar biomoléculas y polímeros. En la Tabla 19.4 se recopilan los principales proveedores de columnas de HPLC. Tabla 19.4. PrinciPales PrOveedOres de cOlUMnas de HPlc Proveedor Agilent Alltech Análisis vínicos BIA Separations Bio-Rad Chiral Technologies Europe Dionex Grace Davison Merck Metrohm Phenomenex Restek Shimadzu Supelco Showa Denko (Shodex) Teknokroma Thermo Scientific Tosoh Bioscience varian Waters dirección web http://www.agilent.com http://www.alltech.com http://analisisvinicos.com http://www.biaseparations.com http://www.bio-rad.com http://www.chiral.fr http://www.dionex.com http://www.discoverysciences.com http://www.merck-chemicals.com/chromatography http://www.metrohm.com http://www.phenomenex.com http://www.restek.com http://www.shimadzu.com http://www.sigmaaldrich.com http://www.shodex.com http://www.teknokroma.es http://www.thermo.com http://www.tosohbioscience.com http://www.varianinc.com http://www.waters.com 796 Técnicas de análisis y caracterización de materiales El empleo de partículas pequeñas da lugar a mayores presiones de trabajo y, en consecuencia, a un menor tiempo de vida de la columna; sin embargo, la eficacia de la separación cromatográfica aumenta considerablemente. En cromatografía analítica suelen emplearse rellenos de 5 µm (aunque la tendencia actual es usar partículas de menor tamaño, generalmente 3 μm), mientras que para cromatografía semipreparativa y preparativa lo más habitual es hacer uso de rellenos de al menos 10 µm. En todos los casos es fundamental que la distribución del tamaño de partícula sea lo más estrecha posible. La cromatografía ultra-rápida (UPLC, ver apartado 19.6) ha irrumpido con fuerza en el campo de la cromatografía HPLC, haciendo uso de columnas más pequeñas que las convencionales, pero con una gran resolución (aproximadamente 200.000 platos/metro). Suelen tener diámetros internos entre 1,0-4,6 mm, con partículas de menos de 2 µm y longitudes de 3,0 a 7,5 cm. Tienen la ventaja de la rapidez de la separación y el menor consumo de disolvente. En función de su composición, las fases estacionarias que se utilizan en HPLC suelen ser de uno de estos tipos: (1) con base de sílice, (2) poliméricas (basadas en estireno-divinilbenceno), e (3) híbridas, formadas por una combinación de las dos anteriores. Las características que debe reunir una sílice para ser utilizada en HPLC son: forma esférica, tamaño de partícula homogéneo, diámetro de poro superior a 5 nm (50 Å) y una alta superficie específica. Recientemente se han desarrollado las columnas empaquetadas con fases monolíticas de sílice, en la cuales no existe un empaquetamiento de partículas, sino un lecho poroso que llena el volumen de la columna. Entre las ventajas principales de estas últimas destacan el rápido transporte entre la fase estacionaria y la fase móvil, el mínimo volumen muerto, así como una menor presión, lo que permite emplear caudales más altos y reducir el tiempo de análisis. Las columnas basadas en polímeros de estireno-divinilbenceno son muy empleadas en HPLC, existiendo rellenos de este tipo para las distintas clases de cromatografía (fase reversa, intercambio iónico, exclusión molecular, etc.). Se trata de columnas muy estables en el rango de pH 1-13. No obstante, una limitación de estos rellenos es que no deben superarse presiones de 1000 psi (69 bar). Finalmente, las columnas híbridas, formadas por una mezcla de sílice con materiales poliméricos (por ejemplo, XTerra® de Waters, Gemini® de Phenomenex, Prevail® de Alltech, etc.), combinan las ventajas de la sílice (resistencia mecánica, reproducibilidad, alta eficacia, etc.) con las de los polímeros (estabilidad frente al pH, baja reactividad química, etc.). La última innovación en rellenos de columnas consiste en la tecnología fusedcore, que consiste en el empleo de partículas de aprox. 2,7 µm formadas por un núcleo no poroso de sílice de aprox. 1,7 µm, rodeado por una capa porosa de sílice de 0,5 µm. De esta manera, el núcleo sólido impide que los analitos difundan hasta el centro de la partícula (fenómeno que ocurre con partículas totalmente porosas); este hecho reduce el ensanchamiento de las bandas y aumenta la eficacia hasta llegar a 160.000 platos teóricos por metro, es decir, una eficacia ligeramente inferior a la que se consigue con el empleo de partículas menores de 2 µm en cromatografía ultra-rápida. Además, otra ventaja radica en que con columnas rellenas de fases estacionarias fused-core pueden utilizarse equipos de HPLC convencionales, a presiones mo- Cromatografía líquida de alta resolución (HPLC) 797 deradas, mientras que con partículas de diámetros menores o iguales de 2 µm es necesario utilizar equipos especiales de cromatografía ultra-rápida (UPLC). Hay una serie de fenómenos que pueden indicar el deterioro de una columna HPLC, como son el aumento de presión, el desdoblamiento de picos, cambios en los tiempos de retención, derivas en la línea base, etc. Es posible su regeneración (parcial o total) siguiendo determinados protocolos. No obstante, el gasto en disolventes y reactivos suele aconsejar en muchas ocasiones reemplazar la columna por una nueva. 19.2.6. Precolumnas La columna es delicada y puede perder eficiencia y resolución, muchas veces irrecuperables, por ejemplo cuando se emplean disolventes indebidos o incompatibles, incluso en proporciones pequeñas. Las columnas HPLC llevan filtros a su entrada y salida, y conviene filtrar disolventes y muestras. A pesar de ello, es muy conveniente incorporar una precolumna en el sistema. Las precolumnas se disponen antes de la columna HPLC, aumentando la vida de la columna cromatográfica. La precolumna retiene las partículas sólidas y otros contaminantes presentes en la fase móvil, así como otros componentes de la muestra que se unen irreversiblemente a la fase estacionaria. Además, la sílice —que constituye el relleno base de la mayoría de las columnas— puede disolverse lentamente en la fase móvil. Wehrli demostró que «la cantidad de sílice disuelta, en partes por millón, era aproximadamente equivalente al tanto por ciento de agua en el eluyente». Al incorporar la precolumna, la fase móvil, queda saturada en sílice y ya no es capaz de disolver la sílice de la columna. Además, es importante considerar que a valores bajos de pH puede hidrolizarse el enlace siloxano en fases enlazadas (ver apartado 19.4.2). Es conveniente que la composición de la precolumna sea igual o lo más parecida posible a la de la columna. Las funciones de la precolumna son, en definitiva, de diversa índole: 1. físicas: al incorporar dos filtros adicionales, atenúa los pulsos de la bomba (damper). 2. Químicas: satura la fase móvil en fase estacionaria antes de que entre en la columna. 3. Cromatográficas: el conjunto precolumna-columna tiene más eficiencia (platos teóricos). 4. económicas: alargan la vida de la columna. Uno de los sistemas más atractivos en precolumnas es el de los cartuchos intercambiables. Este tipo de dispositivos se enroscan fácilmente sobre la columna, sin necesidad de herramientas. Además, la carcasa (holder) puede utilizarse para fases estacionarias de distinta naturaleza simplemente cambiando el cartucho desechable. Técnicas de análisis y caracterización de materiales 798 19.2.7. detectores El detector cromatográfico debe caracterizarse por una gran sensibilidad, precisión y linealidad. Es preferible que el detector no se vea afectado por cambios en la composición de la fase móvil (gradientes), que tenga un tiempo de respuesta inferior a 0,3 segundos y un volumen de celda lo más pequeño posible, de modo que no contribuya al ensanchamiento de los picos. El volumen de celda del detector suele ser de unos pocos microlitros, por lo que toma la forma de un tubo estrecho capilar con un cierto ensanchamiento en la zona de medida, llamada microcámara. Es fundamental no sobrepasar los límites máximos de presión y caudal para cada tipo de detector. Los detectores pueden clasificarse en dos grandes grupos: 1. Detectores universales, que responden a cambios de una propiedad física general de todos los analitos (por ejemplo, dispersión de la luz en los detectores evaporativos de dispersión de luz o light-scattering) o una propiedad de la fase móvil que varíe en presencia de los analitos (por ejemplo, índice de refracción en los detectores refractométricos). 2. Detectores selectivos, que solo son capaces de detectar los compuestos que tienen una determinada propiedad física, por ejemplo la absorción de luz visible o ultravioleta a una determinada longitud de onda. Otra clasificación de los detectores se puede realizar por su fundamento instrumental: 1. Detectores ópticos: de absorbancia UV-VIS, fluorimétricos, refractométricos, polarimétricos, evaporativos de dispersión de luz, etc. 2. Detectores eléctricos: electroquímicos, conductimétricos. 3. Detectores específicos: radiométricos, viscosimétricos. 4. Detectores de técnicas acopladas: espectroscopía de masas (MS), aerosol cargado (CAD), espectroscopía infrarroja transformadora de Fourier (IRTF), absorción atómica (AA), espectroscopía de emisión por plasma (ICP), etc. Un parámetro importante que define la calidad de un detector HPLC es el límite de detección y el número máximo de picos detectados. En la Tabla 19.5 se recopilan los límites de detección de los principales tipos de detectores utilizados en HPLC. En este contexto, algunos de los detectores de masas de última generación para HPLC alcanzan límites de detección del orden de fentogramos. Hay un límite por encima del cual los aumentos de la concentración ya no se ven correspondidos por los de la señal del detector, momento en que se dice que el detector ha quedado «ciego» o que está «saturado». En ocasiones se observa la denominada deriva de la línea base, la cual se torna ascendente, descendente o caótica, mucho más que lo esperado como ruido de flujo. Puede ser debida a varios factores: empleo de gradientes, cambios de temperatura Cromatografía líquida de alta resolución (HPLC) 799 del laboratorio, cambios de temperatura de la columna, cambio de viscosidad de la fase móvil, dilatación térmica, evaporación parcial de algún componente de la fase móvil en el propio reservorio, etc. Tabla 19.5. caracTerísTicas de lOs PrinciPales deTecTOres UTilizadOs en HPlc detector Tipo límite de detección1 Absorbancia Selectivo 100 pg – 1 ng Fluorescencia Selectivo 1 – 10 pg Índice de refracción Universal 100 ng – 1 µg Quimioluminiscencia Selectivo 0,1 – 1 pg Electroquímico Selectivo 10 pg – 1 ng Evaporativo de light-scattering Universal 1 –100 ng Conductividad Selectivo 500 pg – 1 ng Espectrometría de masas Universal 100 pg – 1 ng Aerosol cargado (CAD) Universal 100 pg – 10 ng 1 El límite de detección se define como la masa de compuesto inyectada que da lugar a una relación señal/ruido de 5/1, para un analito de masa molecular de 200 g/mol y un volumen de inyección de 10 μl. Cuando se utilizan varios detectores en serie, se debe situar en primer lugar aquel de menor volumen de celda; no obstante, si uno de ellos es destructivo (electroquímico, evaporativo, etc.) este se pone en último lugar. En casos excepcionales, se puede intercalar un divisor de flujo (splitter) para que los caudales que lleguen a dos detectores en serie sean distintos. A continuación se detallan las características de los principales detectores utilizados en cromatografía líquida. Se han seleccionado detectores tanto de tipo general (índice de refracción, evaporativos, etc.) como selectivos (absorbancia, fluorescencia, electroquímicos, etc.). Algunos de los detectores con mayores perspectivas futuras, como los de masas o los de aerosol cargado, se incluyen lógicamente en dicha recopilación. 19.2.7.1. Detectores de absorbancia o fotométricos (UV-VIS) Los detectores fotométricos están basados en la absorción de luz Uv-vIS, definida por la ley de Lambert-Beer. La linealidad de esta ley es su propiedad más importante y la que justifica su utilización en detectores HPLC. Son muy versátiles, ya que la mayoría de los compuestos orgánicos absorben en alguna zona del espectro. En general, el detector Uv-vIS se suele utilizar con compues- 800 Técnicas de análisis y caracterización de materiales tos que poseen uno o varios dobles enlaces. Estos detectores presentan una alta sensibilidad, y son compatibles con el empleo de gradientes de elución, siempre y cuando los disolventes empleados no absorban radiación de dicha longitud de onda. Para minimizar el ensanchamiento de los picos, el volumen de la celda suele ser muy pequeño (1-10 µl), con longitudes de celda en el rango 0,2-1,0 cm. La presión en la celdilla Uv-vIS no debe ser nunca superior a 600 psi (40 bar) en la mayoría de los detectores de este tipo. La banda espectral no es tan aguda como en un espectrofotómetro, suele ser de 10 nm. Existen dos grandes grupos de detectores Uv-vIS: los que utilizan una o varias longitudes de onda discretas, y los que emplean un rango espectral continuo (detectores de fotodiodos o photodiode-array, PDA). Los primeros pueden incorporar un sistema de filtros o un monocromador. Los detectores de fotodiodos son capaces de realizar un espectro completo del contenido de la celda en aproximadamente medio segundo. Los detectores de fotodiodos permiten estimar la pureza de un pico analizando, a lo largo del mismo, el cociente de las absorbancias a dos longitudes de onda distintas. Permiten, además, identificar algunos de los compuestos eluidos mediante la comparación de sus espectros Uv-vIS con los contenidos en bibliotecas de espectros. En la Figura 19.5 se muestra un cromatograma de una mezcla de derivados fenólicos empleando un detector PDA, que nos proporciona los espectros Uv-vIS de los cuatro compuestos principales. 19.2.7.2. Detectores fluorimétricos La fluorimetría es muy adecuada para el análisis de moléculas grandes, rígidas y con dobles enlaces conjugados (por ejemplo, hidrocarburos aromáticos policíclicos). Son aproximadamente 1.000 veces más sensibles que los detectores fotométricos y, sobre todo, muy selectivos. Los factores que afectan a la señal de fluorescencia y, por tanto, a la sensibilidad de HPLC con detección fluorimétrica, son: efecto de los átomos pesados externos, quenching o amortiguación de fluorescencia, amortiguación por formación de dímeros, efecto del filtro interno habitual en altas concentraciones, interacción con disolventes y la temperatura. Hay dos tipos de equipos: (1) fluorómetros, basados en filtros, que son dispositivos baratos, sencillos y de aplicaciones concretas; (2) espectrofluorómetros, que tienen monocromadores, más caros y sofisticados que los anteriores. Cuando la molécula a analizar no es fluorescente, se puede recurrir a su derivatización con un marcador o reactivo fluorogénico (por ejemplo, cloruro de dansilo o fluoroescamina, que reaccionan con los grupos amino del compuesto). Se trata de un detector excelente para análisis de hidrocarburos aromáticos (contaminación ambiental) o de antibióticos (tetraciclinas). Es muy clásica la determinación de aminoácidos, no fluorescentes por sí mismos, derivatizándolos con fluorescamina, para determinar la composición de aminoácidos de una proteína (Figura 19.6). Cromatografía líquida de alta resolución (HPLC) 801 1 2 OH HO OH HO 200 250 300 350 400 200 250 Longitud de onda (nm) 300 350 400 Longitud de onda (nm) 4 3 OH OH OH 200 250 300 350 400 200 250 Longitud de onda (nm) 300 350 400 Longitud de onda (nm) 2 3 1 1. Hidroquinona 2. Resorsinol 3. Catecol 4. Fenol 0 4 5 10 15 20 Tiempo de retención (min) Figura 19.5. Análisis de una mezcla de compuestos fenólicos empleando un detector de fotodiodos. Técnicas de análisis y caracterización de materiales 802 52,70 Lys 66,95 Arg 47,84 His 40,47 Tyr 41,86 Phe 29,93 Val 4 34,61 Met 36,24 Ile 37,18 Leu 23,31 Ala 8 16,17 Glu+Gln 11,51 Thr 12 12,35 Ser Abs (570 nm) 16 21,69 Gly 9,86 Asp+Asn 20 0 0 10 20 30 40 50 60 70 Tiempo de retención (min) Figura 19.6. Determinación de la composición de una proteína derivatizando los aminoácidos (resultantes de la hidrólisis ácida) con un reactivo fluorogénico. 19.2.7.3. Detectores de índice de refracción Los detectores de índice de refracción, también denominados refractométricos, son universales y se vienen utilizando desde los primeros desarrollos de la cromatografía HPLC. En la celda de detección se mide la variación de la refracción de un haz de luz cuando la fase móvil lleva un compuesto disuelto respecto a la fase móvil pura (celda de referencia), como se ilustra en la Figura 19.7. Tienen la ventaja de responder a la presencia de prácticamente cualquier tipo de soluto. Son, por tanto, equivalentes a los detectores de llama o conductividad térmica empleados en cromatografía de gases. La temperatura, la longitud de onda de la radiación empleada y la presión a la que está sometida la sustancia son variables que afectan a la medida del índice de refracción. Una temperatura constante facilita medidas más precisas; por ello, en equipos HPLC con detector refractométrico es muy conveniente el uso de hornos de columnas. Además, la fase móvil pasa a través de un tubo capilar de acero donde se termostatiza antes de entrar en el detector. Es importante tener en cuenta que la medida del índice de refracción no es homogénea utilizando diferentes fuentes de radiación, lo que impide comparar resultados al pasar de un detector refractométrico a otro. Cromatografía líquida de alta resolución (HPLC) 803 Referencia Muestra Gas nebulizador Tubo evaporador Gotas Gotículas Partículas Fuente de luz Trampa de luz y fotomultiplicador Figura 19.7. Esquema del funcionamiento de un detector refractométrico (izquierda) y de un detector evaporativo de light scattering (derecha). Su sensibilidad es limitada, aproximadamente 1.000 veces menor que la de un detector basado en la absorbancia; no obstante, los detectores de Uv-vIS suelen complementarse con un detector refractométrico puesto en serie. Un aspecto negativo de estos detectores es su incompatibilidad con los gradientes de elución. Aplicaciones típicas de los detectores de índice de refracción son el análisis de carbohidratos, triglicéridos y polímeros. 19.2.7.4. Detectores evaporativos de dispersión luminosa (light scattering) Un aspecto importante en HPLC es poder detectar sustancias que no absorben la radiación Uv. Si además carecen de propiedades conductimétricas o electroquímicas, el análisis queda supeditado a utilizar un detector universal refractométrico, con sensibilidad moderada. Además, los detectores de índice de refracción son incompatibles con los gradientes de elución, y muy dependientes de las variaciones de temperatura externa. La búsqueda de alternativas para analizar sustancias como azúcares, polímeros, ácidos grasos, triglicéridos o aminoácidos, condujo al detector evaporativo de dispersión luminosa (evaporative light-scattering detector, ELSD) basado en los efectos dispersivos que toda sustancia posee sobre un haz de luz. Se trata, por tanto, de un detector universal, descrito por primera vez en 1978. Este detector lleva a cabo un primer paso de nebulización del eluyente seguida de la evaporación de la fase móvil (Figura 19.7). Los solutos no volatilizados forman una niebla de micropartículas, que atraviesa un haz luminoso y produce en él la dispersión de la radiación, cuya intensidad constituye la señal del detector. El detector consta, por tanto, de tres partes: Técnicas de análisis y caracterización de materiales 804 1. Un sistema de nebulización neumática del líquido procedente de la columna, generalmente empleando nitrógeno como gas nebulizador. Suele tener un dispositivo para eliminar las gotículas de tamaño superior al óptimo. 2. Un tubo evaporador caliente, donde se produce la evaporación de la fase móvil. 3. Un detector alejado del eje de la radiación incidente, normalmente un fotomultiplicador emplazado 90-120º de la fuente de radiación. Al ser necesario evaporar el disolvente o mezcla de disolventes, evitando la volatilización del soluto, la temperatura de evaporación juega un papel fundamental. Se prefiere que la temperatura no sea demasiado alta, ya que se puede producir degradación térmica de los analitos o incluso su volatilización. Algunos de los últimos modelos permiten la evaporación de eluyentes a baja temperatura, lo que representa una gran ventaja. Estos detectores tienen un fácil manejo, y son compatibles con el uso de modificadores y tampones sublimables (por ejemplo, ácido acético, ácido fórmico, ácido trifluoroacético, acetato amónico, hidróxido amónico, trietilamina, etc.). Permiten utilizar disolventes que absorban la radiación Uv, y son compatibles con los gradientes de elución. Además, la respuesta másica es aproximadamente la misma para la mayor parte de los solutos no volátiles. Su sensibilidad es superior a la de los detectores de índice de refracción, incluso algunos modelos presentan una sensibilidad comparable a los de absorbancia Uv-vIS. En la Tabla 19.6 se comparan las características de los detectores ELSD con los de índice de refracción. Tabla 19.6. cOMParación de lOs deTecTOres elsd cOn lOs refracTOMéTricOs característica índice de refracción elsd Compatibilidad con gradientes No Sí Análisis de compuestos volátiles Sí No Empleo de tampones no volátiles Sí No Efecto de las impurezas en la fase móvil Serio Mínimo Sensible a cambios de temperatura Sí No Picos «negativos» Sí No Linealidad Sí Respuesta logarítmica Tiempo de estabilización Largo Corto Necesidad de preinstalación No Sí 1 1 Se requiere una línea de nitrógeno y un tubo de evacuación de gases. Una peculiaridad del detector evaporativo de light scattering es que su respuesta no es lineal, viniendo definida por la fórmula: A = amb siendo (A) el área del pico, (m) la masa del analito y (a) y (b) coeficientes que dependen de varios factores, fundamentalmente del tamaño de las partículas, la naturaleza de los analitos y la temperatura de evaporación. Cromatografía líquida de alta resolución (HPLC) 805 Por ello suele utilizarse representación logarítmica para las rectas de calibrado (la mayoría de los softwares para tratamiento de datos en HPLC ya incorporan dicha opción): log A = b log m + log a Los valores de (b) descritos suelen estar en el rango 0,6-2,0. La Figura 19.8 muestra las curvas de calibrado de un éster metílico utilizando un ajuste lineal y logarítmico. 2,00e+6 6,5 A 1,75e+6 B 6,0 log Área del pico Área del pico 1,50e+6 1,25e+6 1,00e+6 7,50e+5 5,00e+5 5,5 5,0 4,5 4,0 2,50e+5 3,5 0.00 0 2 4 6 8 10 12 14 [Oleato de metilo] (nM) 0,2 0,4 0,6 0,8 1,0 1,2 log [Oleato de metilo] (nM) Figura 19.8. Curva de calibrado de un compuesto (oleato de metilo) utilizando un detector evaporativo de light-scattering: (A) ajuste lineal; (B) ajuste logarítmico. Los campos de aplicación más importantes de estos detectores son el análisis de azúcares, triglicéridos, ácidos grasos, lípidos, fosfolípidos, polímeros, esteroides, alcaloides, tensioactivos, lubricantes, etc. 19.2.7.5. Detectores electroquímicos Para el análisis de sustancias oxidables o reducibles pueden emplearse detectores electroquímicos, que a su vez pueden ser amperométricos o coulométricos. La señal ocasionada por un detector electroquímico amperométrico es una medida de intensidad proporcional a la concentración de soluto, cuando en la celda se aplica un potencial que ocasiona su reducción o bien su oxidación. Los detectores amperométricos llevan un electrodo de referencia, normalmente plata-cloruro de plata, estableciéndose una diferencia de potencial con el electrodo de trabajo (que suele ser de carbón o de oro). En los coulométricos se utiliza un electrodo de referencia hidrógeno-iones hidrógeno. Con estos detectores la fase móvil debe conducir la electricidad. No basta con metanol o acetonitrilo al 100%; la fase móvil debe contener un electrolito (puede ser 806 Técnicas de análisis y caracterización de materiales un par iónico o una sal inorgánica). La característica fundamental de los detectores electroquímicos es su selectividad y, derivando de ella, su sensibilidad. El potencial redox es la variable que el usuario establece en el electrodo, y debe ser suficiente para producir la reacción de oxidación o reducción del analito. El rango de sustancias analizadas por detección electroquímica es muy amplio, tanto por oxidación (hidrocarburos, amidas, aminas, fenoles, etc.) como por reducción (olefinas, cetonas, aldehídos, nitroderivados, etc.). La aplicación más emblemática es la de las catecolaminas en plasma u orina. También se pueden detectar fenoles, benzoquinona o algunas vitaminas como el ácido ascórbico (vitamina C). En cromatografía de intercambio iónico es interesante la aplicación de los detectores amperométricos de pulsos para el análisis de carbohidratos. 19.2.7.6. Detectores de radioactividad Se trata de contadores de centelleo en continuo. Se utilizan para detectar compuestos radioactivos marcados, especialmente en el caso de fármacos, pesticidas, donde se requiere una alta sensibilidad que no pueden ofrecer los detectores convencionales. Los isótopos que se analizan son: emisores beta de baja energía (3H, 14C, 35S), emisores beta de alta energía (32P), emisores gamma (125I, 131I) y emisores alfa. 19.2.7.7. Detectores de conductividad Estos detectores se encuentran íntimamente ligados a la cromatografía iónica y miden la conductividad del eluyente, que es proporcional a la fuerza iónica. Su sensibilidad disminuye a medida que aumenta la conductividad de la fase móvil, por lo que suele ser necesario incorporar las denominadas columnas supresoras que reducen el ruido de fondo. Como la conductividad es muy dependiente de la temperatura, estos detectores suelen llevar un sistema de compensación automática de la temperatura. Un inconveniente de los detectores de conductividad es que el rango de linealidad es bastante corto. 19.2.7.8. Detectores viscosimétricos El parámetro de medida básico de estos equipos es la viscosidad relativa, definida como el cociente de la viscosidad de la solución entre la viscosidad del disolvente. Son especialmente útiles para el análisis de polímeros mediante cromatografía de exclusión molecular (GPC). Existen varias posibilidades: detector viscosimétrico de cuatro capilares (que permite la medida de factores estructurales del polímero, como la ramificación y conformación, además del peso molecular), de dos capilares (de menor sensibilidad que el anterior) y de un solo capilar (que requiere de numerosos atenuadores de pulsos en el sistema de bombeo). Cromatografía líquida de alta resolución (HPLC) 807 19.2.7.9. Detectores de quimioluminiscencia La mayor ventaja de la quimioluminiscencia es que no requiere haz de excitación, elemento esencial en fluorimetría (puede provocar un importante ruido de fondo). Son detectores de muy alta sensibilidad, y permiten la detección de trazas de lípidos, nucleótidos, óxidos nitrosos y catecolaminas. La instrumentación es bastante sencilla; no obstante, una desventaja importante es que necesitan una bomba peristáltica para hacer la reacción post-columna. 19.2.7.10. Detectores de dispersión luminosa (light scattering) no evaporativos También es muy interesante la aplicación de detectores de light-scattering no evaporativos para la determinación de masas moleculares en polímeros y macromoléculas biológicas (GPC/SEC). Existen tres tipos de detectores de light scattering en flujo continuo para GPC/SEC: LALLS (low angle laser light scattering system), MALLS (multiple angle laser light scattering system) y RALLS (right angle laser light scattering system). Estos detectores permiten la determinación absoluta de masa molecular, ramificaciones, etc. y suelen estar acoplados a un detector de índice de refracción y a otro viscosimétrico. 19.2.7.11. Detectores de masas La combinación de la cromatografía líquida con espectrometría de masas (LCMS, liquid chromatography-mass spectrometry) tiene gran potencial, y se aplica cada vez a más campos, con el objetivo de separar, identificar y cuantificar componentes de bajos pesos moleculares, y caracterizar productos de altos pesos moleculares (péptidos, proteínas, etc.). Las técnicas de ionización a presión atmosférica han revolucionado la detección en LC-MS. Un espectrómetro de masas para HPLC cuenta con varios componentes instrumentales: (1) una interfase con el sistema cromatográfico mediante la que el eluyente entra en el detector, (2) una cámara de ionización, (3) una cámara de aceleración de iones, (4) un selector que analiza ordenadamente los iones (cuadrupolo, trampa iónica, tiempo de vuelo TOF, etc.), (5) un detector de los iones y, por último, (6) un software de adquisición y procesado de datos. Existen diferentes tipos de ionización: impacto electrónico (Therma-Beam), electrospray (ESI, electrospray ionisation), ionización química a presión atmosférica (APCI, atmospheric pressure chemical ionisation), FAB (fast atom bombardment), MALDI (matrix-assisted laser desorption). Es importante conocer qué tipos de analitos se van a encontrar y qué fase móvil se va a utilizar para poder elegir la mejor configuración. La ionización por electrospray es más adecuada para compuestos desconocidos o que requieran confimación. Existen diversos sistemas para «filtrar» los iones formados respecto a su relación masa/carga. Los más empleados son los de triple cuadrupolo (TQ) y los de 808 Técnicas de análisis y caracterización de materiales tiempo de vuelo (time of flight, TOF). El sistema de cuadrupolos es el más indicado para cuantificación, mientras que el TOF es el preferido para la identificación de compuestos desconocidos. Este último tiene mayor resolución de masa molecular (en el orden de diezmilésismas de Dalton para el TOF frente a un Dalton para el TQ). 19.2.7.12. Detectores de aerosol cargado Los detectores de aerosol cargado (Charged Aerosol Detection, CAD, también denominados Corona) se han desarrollado recientemente, y combinan elementos de los detectores evaporativos de light scattering con otros de los detectores de masas. En primer lugar, se produce la nebulización con nitrógeno de la fase móvil y la selección de las gotas más pequeñas. Se lleva a cabo la evaporación del disolvente; a continuación, el N2 es ionizado en una cámara de ionización y se adsorbe sobre las partículas de analito. Las partículas cargadas llegan a una caja Faraday, donde se produce la transferencia de cargas y la detección de estas, siendo su intensidad proporcional a la concentración de analito en la muestra. Estos detectores son universales, con una sensibilidad similar a la de un detector de absorbancia Uv-vIS, compatibles con los gradientes de elución, muy reproducibles y de fácil manejo. 19.3 ParáMeTrOs crOMaTOGráficOs Los principales parámetros de una separación cromatográfica, así como su significado, se resumen en la Tabla 19.7. 19.4 TiPOs de crOMaTOGrafía HPlc En el proceso cromatográfico intervienen un conjunto de fuerzas que compiten de manera selectiva por un compuesto (analito o soluto) para: (1) o bien fijarlo al relleno de la columna o fase estacionaria; (2) o bien llevarlo disuelto en el eluyente de la columna (fase móvil). Los distintos tipos de fuerzas entre fase estacionaria y solutos definen los distintos tipos de cromatografía HPLC (Tabla 19.8). Como se indicó en el apartado 19.2.5, la mayor parte de los rellenos en HPLC están basados en sílice, seguidos por los que utilizan la resina sintética estirenodivinilbenceno. A diferencia de la cromatografía de gases, en HPLC la elección de la fase móvil es determinante en la separación obtenida; en cambio, el gas portador en cromatografía de gases no contribuye de manera importante al proceso de separación, siendo su principal función transportar los componentes de la mezcla a través de la fase estacionaria. Cromatografía líquida de alta resolución (HPLC) 809 Tabla 19.7. siGnificadO de lOs PrinciPales ParáMeTrOs crOMaTOGráficOs Parámetro Tiempo de retención fórmula Significado Suma del tiempo que el analito permanece en la fase móvil y el tiempo que interacciona con la fase estacionaria Cociente del tiempo en que el analito está en interacción con la fase estacionaria (tR – to) y el que permanece en la fase móvil (to) tR Factor de capacidad t R − to k = to Selectividad (factor de separación) α= velocidad lineal de la fase móvil u= Resolución R= Número de platos teóricos Cociente del factor de capacidad de dos picos adyacentes. Es una medida del potencial del sistema cromatográfico para separar dos compuestos. Se calcula dividiendo la longitud de la columna (L) entre el tiempo que necesita la fase móvil para pasar a través de la columna (to) k2 k1 L t0 t2 − t1 1 2 ( w1 + w2 ) t N = 16 ⋅ R w Altura equivalente de platos teóricos HETP = Ecuación de van Deemter HETP = A + 2 L N B +C ⋅u u La resolución de dos picos adyacentes es el cociente entre la distancia que hay entre los máximos de ambos picos y la media aritmética de sus anchuras respectivas (w). Determina la calidad de la separación de dos picos Este nombre refleja el origen del concepto de la teoría de destilación: una cierta longitud de la columna es ocupada por un plato teórico. Se calcula, para un determinado pico, considerando el tiempo de retención y su anchura. Se calcula dividiendo la longitud de la columna entre el número de platos teóricos. Indica que para cada sistema cromatográfico existe un flujo (definido por la velocidad lineal) para el cual la altura equivalente de un plato teórico es mínima. En la ecuación, A es la contribución de la difusión de Eddy, B corresponde a la difusión longitudinal y C a la transferencia de masa Tabla 19.8. TiPOs de crOMaTOGrafía HPlc Proceso cromatográfico Adsorción Tipos – Partición Fase normal Fase reversa Cromatografía quiral Intercambio aniónico Intercambio catiónico SEC (size exclusion chromatography) GPC (gel permeation chromatography) Intercambio iónico Exclusión por tamaño 810 Técnicas de análisis y caracterización de materiales 19.4.1. cromatografía HPlc de adsorción La cromatografía de adsorción o cromatografía líquido-sólido (LSC, liquid-solid chromatography) es el método cromatográfico más antiguo. Está basada en el mismo principio que la cromatografía en capa fina (TLC, thin-layer chromatography) y la cromatografía de columna en gel de sílice, utilizada con fines preparativos. Se emplean fases estacionarias polares (sílice, alúmina, hidroxiapatito) y fases móviles apolares (mezclas de hexano, ciclohexano, isooctano, tetracloruro de carbono, cloroformo, acetato de etilo, etc.). Resulta especialmente útil para la separación de compuestos apolares que presenten alguna insaturación o algún grupo funcional. El origen de la retención es la interacción de los grupos polares de los analitos con los grupos polares de la fase estacionaria. En dicha adsorción participan enlaces de hidrógeno, enlaces π e interacciones dipolo-dipolo. El orden de elución es, por tanto, de menos polar a más polar. La sílice es el material más utilizado; los grupos OH de su superficie son los responsables de su interacción con la parte polar del analito, que compite con la interacción de la fase móvil con los grupos funcionales de la sílice. Para reducir la retención de los compuestos es conveniente aumentar la polaridad de la fase móvil (p. ej. cambiar hexano por cloroformo). Las principales ventajas y desventajas de este tipo de cromatografía se recogen en la Tabla 19.9. Tabla 19.9. venTajas e incOnvenienTes de la crOMaTOGrafía HPlc de adsOrción ventajas desventajas – Muy útil para compuestos de polaridad intermedia. – Limitada a compuestos solubles en disolventes apolares. – Se obtienen cambios en selectividad (α) a partir de cambios primarios en la fase móvil. – Es difícil regenerar las columnas. – Fácil recuperación de compuestos en HPLC preparativa. – Amplia gama de eluyentes disponibles para ajustar la selectividad de una separación. – El agua adsorbida puede afectar a la cromatografía (utilizar disolventes secos). – La fase estacionaria no distingue entre moléculas de distinta longitud de cadena con el mismo grupo funcional. La cromatografía de adsorción está especialmente indicada para compuestos de masa molecular por debajo de 5 kDa, que sean solubles en medios de baja polaridad. Algunos de los compuestos que pueden ser analizados mediante cromatografía HPLC de adsorción (enumerados en orden creciente de interacción con la fase estacionaria y, por tanto, en orden creciente de tiempo de retención) son: olefinas < hidrocarburos aromáticos < haluros, sulfuros < éteres < nitroderivados < ésteres, aldehídos, cetonas < alcoholes, aminas < sulfóxidos < amidas < ácidos carboxílicos. En la Figura 19.9 se muestra la separación de una mezcla de isómeros de la vitamina E (tocoferoles) mediante cromatografía de adsorción. Cromatografía líquida de alta resolución (HPLC) 811 3 4 1 2 0 2 4 6 8 10 12 14 min. Figura 19.9. Separación de tocoferoles presentes en aceite de soja (1: α-tocoferol; 2: β-tocoferol; 3: γ-tocoferol; 4: δ-tocoferol) mediante cromatografía HPLC de adsorción. Columna Pinnacle II Silica (150 × 4,6 mm), 5 µm, 110 Å. Fase móvil: hexano/isopropanol (99,5/0,5). Flujo: 0,6 ml/min. Temperatura: 30 °C. Detección Uv (295 nm). Fuente: catálogo de Restek/Teknokroma 2008/2009. 19.4.2. cromatografía HPlc de partición De los cuatro tipos principales de cromatografía HPLC (Tabla 19.8), la cromatografía de partición es, con diferencia, la más utilizada actualmente. La fase estacionaria se liga o embebe sobre partículas inertes (generalmente de sílice) para que permanezca fija en la columna. Inicialmente, la fase estacionaria líquida era retenida sobre la superficie de las partículas de relleno mediante adsorción. No obstante, y a fin de favorecer la estabilidad de la fase estacionaria, esta metodología fue reemplazada por la denominada cromatografía en fase ligada (BPC, bonded-phase chromatography), en la que los grupos funcionales se fijan mediante enlace químico a las partículas de relleno, lo que permite a la fase estacionaria resistir las altas presiones aplicadas en HPLC (Figura 19.10). La mayor parte de los soportes de cromatografía de partición en fase ligada están preparados a partir de sílice de tamaño de partícula entre 3 y 10 µm. La superficie de la sílice está compuesta de grupos silanol (con una densidad aproximada de 8 µmol/ m2), que son muy reactivos. La mayoría de las fases estacionarias utilizadas en este tipo de cromatografía se preparan mediante reacción de la sílice con un derivado organoclorosilano (Figura 19.10). La naturaleza de la cadena R de dicho derivado determina el tipo de cromatografía: normal, reversa o quiral. Técnicas de análisis y caracterización de materiales 812 Si OH + Cl CH3 R Si Si O CH3 R Si CH3 CH3 Figura 19.10. Derivatización de la sílice para cromatografía de partición. 19.4.2.1. Fase normal. Cromatografía de interacción hidrofílica (HILIC) Cuando el grupo R es de naturaleza polar, se habla de cromatografía en fase normal. Ello es debido a razones históricas, ya que los primeros trabajos utilizaron fases estacionarias polares, soportando trietilenglicol sobre sílice o alúmina. Esta cromatografía, de manera similar a lo que ocurre en la cromatografía de adsorción, se basa en la interacción de los analitos con los grupos funcionales polares de la fase estacionaria ligada, que alcanza su intensidad máxima cuando se utilizan eluyentes de baja polaridad. Sin embargo, muchos analistas han dejado de utilizarla debido a su complejidad y falta de reproducibilidad. Las columnas comerciales en fase normal contienen estructuras en las que R es un grupo ciano (–C2H4CN), diol (–C3H6OCH2CHOHCH2OH), amino (–C3H6NH2) o dimetilamino (–C3H6N(CH3)2). En la cromatografía en fase normal, la fase móvil suele estar formada por uno o varios disolventes apolares, siendo los más frecuentes diclorometano, cloroformo o hexano. En la Figura 19.11 se muestra la separación de una serie de ésteres del ácido ftálico mediante HPLC con una columna con fase ciano. 3 1 0 1 2 3 2 4 4 1 di-n octilftalato 2 bis(2-ethilexil)ftalato 3 butilbencilftalato 4 di-n butilftalato 5 dietilftalato 6 dimetilftalato 6 5 5 6 7 8 9 min Figura 19.11. Separación de ésteres de ftalato mediante cromatografía HPLC en fase normal con columnas ciano. Columna Luna CN (150 × 4,6 mm), 5 µm. Fase móvil: A/B 99/1 siendo A: Hexano; B: cloruro de metileno/metanol (80/20). Flujo: 1,0 ml/min. Temperatura ambiente. Detección Uv (254 nm). Fuente: catálogo de Phenomenex 2008/2009. Una desventaja de los rellenos de fase normal es que son menos resistentes a la hidrólisis que los de fase reversa, por lo que es importante controlar el pH del eluyente y vigilar el sangrado de la columna. Una particularidad de las columnas con fase estacionaria ciano es que pueden comportarse como rellenos tanto de fase normal como fase reversa, en función de las condiciones de análisis. Cromatografía líquida de alta resolución (HPLC) 813 La cromatografía de interacción hidrofílica (HILIC, hydrophilic interaction chromatography) se puede considerar una extensión de la cromatografía de partición en fase normal, cuando se utilizan fases móviles acuosas, concretamente mezclas de agua o solución tampón (<40%) con disolventes orgánicos polares (por ejemplo, acetonitrilo). Aunque este tipo de cromatografía lleva realizándose bastante tiempo, ha sido en los últimos años cuando el término HILIC ha empezado a utilizarse de forma generalizada. La cromatografía HILIC es especialmente útil para compuestos muy polares como carbohidratos o péptidos. Este tipo de sustancias no pueden ser analizadas en cromatografía de adsorción debido a que su interacción con la fase estacionaria es demasiado fuerte; además, se trata de sustancias poco solubles en los disolventes comúnmente utilizados en cromatografía de adsorción o de partición en fase normal, como el hexano o el cloroformo. Por otro lado, los compuestos muy polares no son retenidos en columnas de fase reversa, por lo que suele producirse su coelución con el frente. Afortunadamente, el empleo de columnas de fase normal (fundamentalmente columnas amino) y fases móviles acuosas permite abordar el análisis de este tipo de sustancias, como se ilustra en el cromatograma de la Figura 19.12 en el que se separan una serie de azúcares. En los últimos años se han desarrollado nuevos rellenos para cromatografía HILIC; así, por ejemplo, fases estacionarias de sílice modificada con grupos fenilo, pentafluorofenilo o amida han mostrado una extraordinaria afinidad y selectividad por compuestos fenólicos. 1 2 3 5 6 4 7 0 4 8 12 16 20 24 9 8 28 32 Tiempo de retención (min) Figura 19.12. Separación de una mezcla de carbohidratos (1 mg/ml) mediante cromatografía de interacción hidrofílica (HILIC) con columnas amino. Columna Luna NH2 (250 × 4,6 mm), 5 µm. Fase móvil: acetonitrilo/agua (80/20). Flujo: 1,0 ml/min. volumen de inyección: 5 µl. Temperatura: 40 °C. Detección ELSD (92 °C, caudal N2 3 l/min). Compuestos analizados: 1: glucosa; 2: fructosa; 3: sacarosa; 4: maltosa; 5: 1-kestosa; 6: rafinosa; 7: maltotriosa; 8: panosa; 9: nistosa. Técnicas de análisis y caracterización de materiales 814 Tanto en cromatografía en fase normal como en su variante HILIC, el componente menos polar eluye en primer lugar. El comportamiento es el mismo que tenía lugar en la cromatografía de adsorción. Al aumentar la polaridad de la fase móvil, disminuye el tiempo de retención de los componentes de la mezcla. 19.4.2.2. Fase reversa La cromatografía de fase reversa es, con gran diferencia, la técnica de separación por cromatografía líquida más utilizada actualmente en los laboratorios. Se habla de fase reversa cuando la cadena funcional R tiene carácter apolar (hidrófobo). El grupo R suele ser una cadena C8 (n-octil) o C18 (n-octadecil). Dichas cadenas alifáticas se orientan paralelas unas a otras, perpendicularmente a la superficie de las partículas (a modo de peine). La fase móvil suele tener un marcado carácter polar, siendo habitual utilizar mezclas de agua con distintas concentraciones de metanol, acetonitrilo o tetrahidrofurano. Es importante no utilizar eluyentes con valores de pH superiores a 7,5 para evitar la hidrólisis de los grupos siloxano. La polaridad de los grupos funcionales de los analitos es determinante en el orden de elución de los compuestos. El orden de elución en fase reversa es de más polar a menos polar (Figura 19.13); por tanto, los tiempos de retención de los analitos siguen el orden: alcoholes < aminas < amidas < aldehídos < cetonas < ésteres < éteres < hidrocarburos. Además, para aumentar el tiempo de retención de los analitos, se debe aumentar la polaridad de la fase móvil, es decir, disminuir su fuerza elutrópica. Esto suele conseguirse aumentando el porcentaje de agua en la mezcla. Cromatografía de adsorción y en fase normal a. Fase móvil de polaridad baja C a. Fase móvil de polaridad alta A B Cromatografía en fase reversa A B Tiempo Tiempo b. Fase móvil de polaridad media C B b. Fase móvil de polaridad media A A C B Tiempo C Tiempo Polaridad de los solutos: A> B> C Figura 19.13. Relaciones entre polaridad y tiempo de retención para cromatografía de adsorción, cromatografía de partición en fase normal y cromatografía de partición en fase reversa. Cromatografía líquida de alta resolución (HPLC) 815 En fase reversa, la cantidad de fase hidrocarbonada enlazada al soporte de la sílice se define como «carga de carbono». Se mide como el porcentaje de C en peso respecto a la masa de sílice. Suele oscilar entre el 5% y el 20%. Al aumentar la carga de carbono aumenta la retención de los analitos. No obstante, la densidad del material de relleno es también un factor importante a la hora de predecir la retención de una columna. Así, por ejemplo, la fase Novapak C18, con una densidad de 0,91 g/l y una carga de carbono del 7%, retiene más los compuestos que la fase µBondapak, que tiene mayor carga de carbono (10%) pero una menor densidad (0,45 g/l). Otro término importante es el end-capping, proceso que implica una derivatización adicional de la sílice con trimetilclorosilano para bloquear los grupos hidroxilo libres, haciéndolos inaccesibles a los solutos. De esta manera aumenta la carga de carbono y, por tanto, la retención de los analitos. En los últimos años se ha dedicado gran esfuerzo al desarrollo de nuevas fases estacionarias para fase reversa con el fin de mejorar la eficacia de las columnas y su estabilidad frente al pH, así como proporcionar selectividades distintas. Las columnas de fase reversa actualmente en el mercado se pueden clasificar en tres categorías: – Basadas en sílice, que pueden funcionar en un intervalo de pH 2,0-7,0. En este capítulo, existen sílices de distinto grado de pureza, con o sin tratamiento de end-capping, y libres de metales en mayor o menor medida. – Híbridas, formadas por una combinación de polímero (estireno-divinilbenceno) y sílice. Presentan un rango de pH más amplio (1,0-12,0). – Específicas de la aplicación. Por ejemplo, para la separación de mezclas complejas en las que están presentes compuestos muy polares y apolares, se han desarrollado columnas (Synergi®, Atlantis®) en las que la sílice, además de cadenas hidrocarbonadas, incorpora grupos polares, lo que provoca una mayor retención de moléculas polares y por tanto una mejor separación de estas últimas. En algunos casos, es necesario separar mezclas que contienen uno o varios compuestos ionizables. Mediante la estrategia de supresión iónica es posible separar estos compuestos iónicos, suprimiendo (o reduciendo) su estado iónico en solución a través del control del pH, de manera que quedan más retenidos en rellenos de fase reversa. Funciona bien para ácidos débiles y bases débiles. Como modificantes de la fase móvil se suelen utilizar ácidos como el acético, el fórmico o el fosfórico, o bien bases como alquilaminas o fosfatos alcalinos. Con algunos solutos iónicos, otra posibilidad para analizar en fase reversa consiste en utilizar una fase móvil polar que contenga un contraión de carga contraria a la de los analitos, de modo que entre ambos se forma una especie neutra (par iónico). Concretamente, se suele añadir a la fase móvil un catión o anión hidrofóbico, que interacciona con el analito formando un par iónico (Tabla 19.10). Es una metodología que permite analizar ácidos y bases fuertes (pKa < 2, pKb > 8) en condiciones de fase reversa. variando la longitud del grupo alquilo se puede conseguir mayor o menor retención en la columna. Técnicas de análisis y caracterización de materiales 816 Tabla 19.10. aGenTes MOdificanTes UTilizadOs en crOMaTOGrafía de Pares iónicOs Para bases Para ácidos alQUilsUlfOnaTOs: 1-Pentano sulfonato (B5) 1-Hexano sulfonato (B6) 1-Heptano sulfonato (B7) Dodecil sulfato sódico (SDS) alQUilaMinas: Tetrabutilamina Trietilamina Un ejemplo representativo del empleo de pares iónicos se muestra en la Figura 19.14, en la que se separan cuatro aminas biogénicas con actividad neurotransmisora. Su separación mediante cromatografía en fase reversa es difícil, ya que sus tiempos de retención son muy similares y además se producen interferencias debido a su carácter catiónico. Mediante la adición de 1-heptanosulfonato de sodio a la fase móvil y la consiguiente formación de los pares iónicos, se consigue la separación eficiente de los cuatro compuestos. HO + NH2 HO HO H3N + HO OH OH NH3 + 2 Dopamina 1 Adrenalina H N + H3N 4 Triptamina 3 Tiramina 4 3 2 1 0,0 2,0 4,0 6,0 8,0 10,0 12,0 14,0 16,0 18,0 20,0 22,0 25,0 Tiempo de retención (min) Figura 19.14. Separación de aminas biogénicas mediante cromatografía HPLC en fase reversa con par iónico. Columna Discovery C18 (250 × 4 mm), 5 µm. Fase móvil acetonitrilo: tampón ácido 1-heptanosulfónico pH 2,4. Flujo: 1,5 ml/min. Gradiente de acetonitrilo: t = 0 min, 6%; t = 5 min, 6%; t = 18 min, 25%. Temperatura ambiente. Detección Uv (220 nm). Fuente: boletín Analytix de Sigma-Aldrich. Cromatografía líquida de alta resolución (HPLC) 817 La cromatografía de partición es la más universal de las técnicas de cromatografía líquida. Las fases móviles polares permiten cromatografiar una amplia variedad de compuestos de interés bioquímico, farmacológico o químico (Tabla 19.11). Más del 90% de las aplicaciones de compuestos de bajo peso molecular se hacen en cromatografía de partición de fase reversa. Es una técnica de análisis muy versátil, ya que, por ejemplo, variando tan solo la composición de la fase móvil es posible lograr la separación de mezclas complejas. Así, la Figura 19.15 muestra la separación de una serie de derivados del ácido oleico, y en la Figura 19.16 se presenta un cromatograma de una mezcla de explosivos, ambos realizados con columnas de octadecilo (C18). Tabla 19.11. caMPOs de aPlicación de la crOMaTOGrafía HPlc de ParTición campo de aplicación ejemplos Farmacia Antibióticos, analgésicos, esteroides Medio ambiente Pesticidas, herbicidas, fenoles, PCBs Industria química Detergentes, polímeros Alimentación Edulcorantes, emulgentes, antioxidantes Bioquímica Proteínas, hidratos de carbono, lípidos Clínica y Diagnóstico Análisis de orina y sangre, dopaje, hormonas 1 2 3 4 5 6 0 2 4 6 8 10 12 14 16 18 20 Tiempo de retención (min) Figura 19.15. Separación de derivados del ácido oleico mediante cromatografía HPLC en fase reversa. Columna Nucleosil 100-C18 (250 × 4,6 mm), 5 µm. Fase móvil metanol/acetona/agua. Gradiente empleado: t = 0 min, 95/0/5; t = 6 min, 95/0/5; t = 7 min, 100/0/0; t = 9 min, 100/0/0; t = 11 min, 50/50/0; t = 17 min, 50/50/0. Flujo: 1,2 ml/min. Temperatura: 45 °C. Detección ELSD a 60 °C. Compuestos analizados: 1, ácido ascórbico; 2, oleato de ascorbilo; 3, 2-monooleina; 4, ácido oleico; 5, 1,2-dioleina; 6, trioleina. Técnicas de análisis y caracterización de materiales 818 1. HMX 2. RDX 3. 1, 3, 5-TNB 4. 1, 3-DNB 5. 3, 5-DNA 6. NB 7. Tetryl 8. 2, 4, 6-TNT 9. NG 5, 6 1 3 7 10. 2-A-4, 6-DNT 11. 4-A-4, 6-DNT 12. 2, 4-DNT 13. 2, 6-DNT 14. 2-NT 15. 4-NT 16. 3-NT 17. PETN 8, 9 2 4 10 1112 13 16 14 15 17 2 4 6 8 10 12 14 16 18 20 22 24 Figura 19.16. Análisis de explosivos mediante cromatografía HPLC en fase reversa. Columna Ultra-C18 Teknokroma (250 × 4,6 mm). Fase móvil: metanol/agua 56/44. Temperatura: 30 ºC, Flujo: 1,0 ml/min. Detector Uv (210 nm). Inyección de 10 µl con 50 mg/ml de cada componente. Fuente: catálogo de Restek/Teknokroma 2008/2009. 19.4.2.3. Cromatografía quiral Las separaciones de compuestos quirales son muy importantes en los sectores farmacéutico, biotecnológico y de compuestos naturales. Los isómeros ópticos (enantiómeros) se pueden separar por HPLC, siempre que el sistema cromatográfico (fase móvil y/o fase estacionaria) sea asimétrico (quiral). Para estas separaciones es preferible utilizar HPLC que cromatografía de gases, ya que las altas temperaturas que requiere esta última suelen producir racemización de la fase estacionaria quiral y/o de los analitos. Existen dos posibilidades en cromatografía HPLC quiral: – La fase móvil es quiral pero la fase estacionaria no lo es. Se utilizan fases móviles a las que se les adiciona algún reactivo que proporciona quiralidad; este compuesto forma un complejo, aducto o par iónico con los dos enantiómeros, de manera que los diastereómeros formados interaccionan de forma diferente con la fase estacionaria, lo que permite su separación. – La fase estacionaria es quiral pero la fase móvil no lo es. En este caso existen diferentes columnas en el mercado, formadas por una base de partículas de sílice de 5-10 µm funcionalizadas con: (1) pequeñas moléculas con grupos π-activos, básicamente de naturaleza aromática (véase en Figura 19.17 la separación de los dos enantiómeros del naproxeno); (2) polímeros helicoidales basados en celulosa o amilosa modificada químicamente; (3) ciclodextrinas o éteres corona, que proporcionan cavidades quirales; (4) proteínas, dada su naturaleza quiral al estar formadas exclusivamente por L-aminoácidos. Cromatografía líquida de alta resolución (HPLC) 10,1 12,5 S 819 10 R 20 min Figura 19.17. Separación de los enantiómeros (R y S) del antiinflamatorio naproxeno mediante cromatografía HPLC quiral. Columna Chirex 3005 Teknokroma (250 × 4,0 mm), formada por sílice modificada con (R)-1-naftilglicina y ácido 3,5-dinitrobenzoico. Fase móvil: 30 mM de acetato amónico en metanol. Flujo: 0,8 ml/min. Detector Uv (254 nm). Fuente: catálogo de Phenomenex 2008/2009. 19.4.3. cromatografía HPlc de intercambio iónico La cromatografía de intercambio iónico (IEC, ion exchange chromatography) permite separar compuestos de naturaleza iónica (aniones o cationes). La fase estacionaria está formada por una resina o un gel que contiene grupos cargados, que son neutralizados por los iones de la fase móvil (contraiones, counter ions, por ejemplo, Na+). De esta manera, si el analito contiene grupos de carga opuesta a la fase estacionaria este se retiene en la columna. Si se hace pasar una fase móvil que contenga una sal, el aumento de la fuerza iónica del medio permite eluir los analitos de la columna de forma diferencial, aunque el orden de elución es con frecuencia difícil de predecir. Las columnas de intercambio iónico suelen tener base de sílice (con capacidad máxima de intercambio de 1 meq/g) y estireno-divinil-benceno (3 meq/g). No obstante, como la densidad de la sílice es mayor, la capacidad de intercambio de ambos sistemas es similar. Los intercambiadores pueden ser aniónicos (atraen aniones) o catiónicos (atraen cationes). Existen intercambiadores fuertes (están ionizados a cualquier pH, por ejemplo, los que contienen grupos sulfato o aminas cuaternarias) o débiles (su estado de ionización depende del pH, por ejemplo, los que contienen grupos carboxílicos). La retención se basa en la afinidad de los diferentes iones por los puntos de intercambio. Se utilizan fases móviles tamponadas que proporcionan contraiones, permiten controlar el pH y variar la fuerza iónica del medio. En general, para disminuir la retención de un analito existen varias estrategias: – Cambiar el pH de la fase móvil para formar especies neutras. – Aumentar la fuerza iónica de la fase móvil. – Aumentar la temperatura. Técnicas de análisis y caracterización de materiales 820 La retención de un compuesto en una columna de intercambio catiónico se ve afectada por el catión (contraión) elegido en la fase móvil. La capacidad de desplazamiento de los cationes sigue el orden: Li+ < H+ < Na+ < NH4+ < K+ < Rb+ < Cs+ < Ag+ < Mn2+ < Mg2+ < Zn2+ < Co2+ < Cu2+ < Cd2+ < Ni2+ < Ca2+ < Sr2+ < Pb2+ < Ba2+. En el caso de las columnas de intercambio aniónico, la capacidad de desplazamiento de los aniones sigue la secuencia: fluoruro < OH- < acetato < cloruro < tiocianato < bromuro < cromato < fosfato < nitrato < borato < yoduro < oxalato < sulfato < citrato. Se trata de una técnica muy útil para cromatografiar iones inorgánicos y orgánicos, proteínas, etc. En la figura 19.18 se muestra la separación de una mezcla de aniones mediante cromatografía de intercambio iónico. Ejemplos representativos de este tipo de cromatografía son la determinación de aniones en agua de bebida, de nitratos en vegetales, de fluoruros en pasta de dientes, de amonio y nitratos en fertilizantes, o de sodio y potasio en muestras clínicas (transfusiones). Con iones más grandes, y muy especialmente con tensioactivos iónicos, la cromatografía de par iónico da mejor resultado que la de intercambio, debido fundamentalmente a una mejor difusión interna y a una menor adsorción irreversible de los analitos (que impide su elución). 1. Fluoruro (2 ppm) 2. Acetato (5 ppm) 3. Propionato (5 ppm) 4. Formato (5 ppm) 5. Clorito (5 ppm) 6 6. Bromato (5 ppm) 7. Cloruro (3 ppm) 8. Nitrito (5 ppm) 9. Bromuro (10 ppm) 10. Nitrato (10 ppm) 11. Clorato (10 ppm) 12. Carbonato (20 ppm) 13. Sulfato (5 ppm) 14. Oxalato (5 ppm) 15. Fosfato (10 ppm) 13 10 4 mS 7 1 14 8 15 9 11 5 2 6 12 0 0 5 10 15 Minutes Figura 19.18. Análisis de una mezcla de aniones inorgánicos mediante cromatografía HPLC de intercambio iónico. Columna IonPac AG17-C AS17-C Dionex (250 × 4 mm). Fase móvil: hidróxido potásico (1 mM de 0 a 3 min, 1-12 mM de 3 a 10 min, 12-35 mM de 10 a 14 min). Flujo: 1,5 ml/min. Temperatura: 30 °C. Detección de conductividad con columna supresora. Fuente: catálogo de Dionex. Cromatografía líquida de alta resolución (HPLC) 821 Para eliminar el ruido en el detector que supone el empleo de una fase móvil con una alta fuerza iónica, se recurre al empleo de la denominada supresión química. Se trata de un sistema de eliminación de los iones de la fase móvil entre la columna y el detector mediante la incorporación de una columna «supresora». Los detectores más utilizados para cromatografía de intercambio iónico son los de conductividad, para el análisis de aniones, cationes, ácidos orgánicos, aminas, sulfonatos, etc. También se emplean detectores electroquímicos, generalmente de tipo amperométrico, para el análisis de cianuros, sulfitos, sulfuros, ioduro, etc. En este contexto, la cromatografía iónica acoplada a un detector amperométrico de pulsos (high performance anionic exchange chromatography coupled to pulse amperometric detection, HPAEC-PAD) se utiliza para el análisis de carbohidratos como alternativa a la cromatografía de partición en fase normal (en su variante HILIC, ver apartado 19.4.2.1), con detectores de índice de refracción o ELSD. La cromatografía HPAEC-PAD permite la cuantificación directa de monosacáridos, oligosacáridos y polisacáridos a escala de picomoles con mínima preparación de muestras. Esta técnica se basa en la naturaleza débilmente ácida de los carbohidratos que permite una separación muy selectiva a alto pH (>13) empleando una columna de intercambio aniónico. La Figura 19.19 muestra el análisis por HPAEC-PAD de la inulina de achicoria, en la que se aprecia la separación nítida de fructanos con un grado de polimerización (DP) de hasta 50. F S DP10 G DP20 DP30 Figura 19.19. Análisis de inulina de achicoria mediante cromatografía HPAEC-PAD. Columna PA100 Dionex (250 × 4 mm). Programa de gradiente utilizando las siguientes fases móviles: (A) 160 mM NaOH; (B) 160 mM NaOH + 1 M acetato sódico; (C) 1 M NaOH. Flujo: 1,0 ml/min. volumen de inyección: 25 µl, con una concentración de 0,8 g/l. G: glucosa; F: fructosa; S: sacarosa; DP: grado de polimerización. Reproducción con permiso de Elsevier a partir de referencia (Ronkart et ál. 2007, Anal Chim. Acta 604, 81-87). Técnicas de análisis y caracterización de materiales 822 19.4.4. cromatografía HPlc de exclusión También es posible separar los analitos en función de su tamaño molecular. Se utilizan rellenos porosos para que los solutos penetren más o menos en la matriz según su tamaño molecular. Se emplean fundamentalmente dos tipos de fases estacionarias: materiales poliméricos (poliestireno-divinilbenzeno) y materiales basados en sílice. Se han desarrollado rellenos basados en gel de sílice, con poros desde 60 Å hasta 20.000 Å (6-2.000 nm), adecuados para disolventes polares y apolares. A menor tamaño molecular, más penetración y mayor tiempo de residencia en la columna, es decir, mayor retención. Esta técnica se conoce abreviadamente como SEC (size exclusion chromatography) cuando se aplica a proteínas y otros compuestos biológicos o GPC (gel permeation chromatography) cuando se aplica a polímeros sintéticos (plásticos) con fases móviles orgánicas. El principal inconveniente es que se trata de un método de baja resolución, ya que la separación tiene lugar en el volumen total de líquido de la columna (Vt), por lo que el número de picos que pueden resolverse es bastante limitado (Figura 19.20). No obstante, es muy útil para la separación de proteínas y otras moléculas de alto peso molecular, como los polímeros. Es importante comprobar que no haya interacción (adsorción) entre la muestra y la fase estacionaria, ya que este hecho alteraría el orden de elución. Vo Vt B A 0 5 10 15 20 25 Tiempo de retención (min) Figura 19.20. Separación de dos compuestos (A y B) mediante cromatografía de exclusión molecular. Se indican el volumen de exclusión (vo, void volume) y el volumen total (vt, total volume). Una aplicación típica de esta cromatografía es la separación de sustancias naturales de elevado peso molecular (cuando están contaminadas con sales y especies de bajo peso molecular). Otra aplicación es la separación de series homólogas de oligó- Cromatografía líquida de alta resolución (HPLC) 823 meros (por ejemplo, ácidos grasos, oligosacáridos, etc.). En general es recomendable una diferencia del 10% en el peso molecular de los distintos compuestos de la serie para obtener una buena resolución. En ocasiones la fase móvil elegida puede afectar al tamaño de un analito. También es muy útil para la determinación del peso molecular (o la distribución de pesos moleculares) de polímeros o sustancias naturales (por ejemplo, proteínas), como se muestra en la Figura 19.21. Para ello los volúmenes de elución de la muestra se comparan con los volúmenes de elución de una serie de patrones que tienen la misma naturaleza que los componentes a analizar. 1. Poliestireno 34500 2. Poliestireno 9200 3. Poliestireno 3250 4. Poliestireno 580 5. Poliestireno 162 1 5 2 3 4 0 25 Figura 19.21. Análisis de una mezcla de patrones de poliestireno mediante cromatografía HPLC de exclusión molecular. Columna Chrompack Microgel 3 Mix varian (250 × 7,7 mm). Fase móvil: tetrahidrofurano. Flujo: 1,0 ml/min. Detector de índice de refracción. 19.5. escalas de aPlicación de crOMaTOGrafía HPlc Aunque lo más habitual es utilizar la cromatografía HPLC en escala analítica, las posibilidades de aplicación son muy variadas, tanto en escala micro (con flujos muy bajos) como en escala preparativa (para la obtención de productos puros). En la Tabla 19.12 se recogen las distintas escalas a las que se puede trabajar en HPLC, incluyendo los diámetros típicos de las columnas empleadas y los flujos habituales de trabajo. Las técnicas HPLC a escala micro y capilar permiten mayor sensibilidad, eficacia, velocidad de análisis y menor consumo de disolvente que la cromatografía convencional (analítica). Su mayor limitación es no disponer de tanta instrumentación como en HPLC analítica, sobre todo en bombas y formadores de gradientes. En realidad puede considerarse una miniaturización de HPLC analítica en cuanto a flujos, volumen de inyección, celda del detector, tubos, etc. Técnicas de análisis y caracterización de materiales 824 Tabla 19.12. escalas UTilizadas en crOMaTOGrafía HPlc escala diámetro columna flujos Industrial, Planta Piloto 77-150 mm 100-1000 ml/min Preparativa 41-77 mm 20-100 ml/min Semipreparativa 10-21 mm 2-20 ml/min Analítica 3,0-4,6 mm 0,45-2,0 ml/min HPLC microbore 2,0-2,1 mm 150-250 μl/min Micro-LC 1,0 mm 20-100 μl/min LC capilar 0,5 mm 1-20 μl/min Nano-LC 0,1 mm 0-1 μl/min La teoría cromatográfica (ver apartado 19.3) indica que la eficiencia de una separación (definida por el número de platos teóricos, N) es independiente del diámetro interno, viniendo definida básicamente por la longitud de la columna y del tamaño de partícula. Es por ello por lo que a escala micro se logran del orden de 100.000 platos teóricos o más por metro. No obstante, para obtener esa misma eficacia al disminuir el diámetro, es necesario reducir el flujo de manera significativa (Tabla 19.12). La cromatografía HPLC microbore, también denominada cromatografía de baja dispersión, suele utilizar columnas de 2,1 mm de diámetro interno y rellenos de 5 μm, con las ventajas de ahorro de disolvente y más sensibilidad. Concretamente, se suelen lograr aumentos de sensibilidad de aproximadamente 5 veces con las técnicas microbore respecto a las analíticas. La instrumentación es la misma que en HPLC analítica, aunque es muy conveniente minimizar los volúmenes extra-columna (reduciendo las longitudes de los tubos entre inyector y detector) para evitar ensanchamiento de los picos; además, es muy importante ajustar las condiciones del gradiente para tener la misma separación y tiempo de análisis que con la columna analítica. Las técnicas capilares en HPLC son especialmente indicadas en situaciones de poca cantidad de muestra y escasa concentración. La cromatografía capilar y nanoLC se emplean fundamentalmente en biotecnología (especialmente en proteómica), ya que además de la alta eficiencia se obtiene una gran sensibilidad para muestras diluidas. Otra de las aplicaciones típicas de la cromatografía líquida capilar es la separación de estereoisómeros empleando fases móviles quirales (especialmente cuando todavía no se habían desarrollado ampliamente las fases estacionarias quirales), ya que los modificantes quirales de las fases móviles tienen un alto precio y en escala capilar su consumo se reduce de manera significativa. Para convertir un sistema analítico convencional en capilar se puede emplear un divisor de flujo, aunque en este caso el ahorro de disolvente no es tan notable. Una técnica de gran aplicación es la cromatografía semipreparativa y preparativa. Las técnicas preparativas en HPLC son excelentes y más rentables que otras técnicas (cromatografías planares, extracción líquido-líquido, precipitación, etc.). Proporcionan elevada resolución y alta capacidad. Requieren un equipo más sofisticado (y por tanto más caro que uno analítico) que incluye bombas de flujos altos, células de detectores que permitan pasar flujos elevados, colectores de fracciones, Cromatografía líquida de alta resolución (HPLC) 825 divisores de flujo, etc. Las columnas son más voluminosas en todo: mayor tamaño de partícula (10-40 µm), mayores dimensiones (≥ 30 cm), mayor diámetro interno (> 10 mm). Para optimizar la separación y aumentar el rendimiento del proceso se suele adoptar alguna de las siguientes soluciones: reciclar los picos no resueltos, haciéndolos pasar de nuevo por la columna; sincronizar un colector de fracciones con el detector para colectar solo picos; hacer ciclos repetitivos de todo el proceso cromatográfico, etc. Generalmente la fase estacionaria que se utiliza es la misma que la de la columna analítica, ofreciendo por tanto la misma resolución cromatográfica. Esta estrategia permite el ahorro en el desarrollo de métodos, ya que el método analítico puede ser directamente aplicado a escala en fase semipreparativa o preparativa, sin cambio en la resolución cromatográfica. Para escalar una cromatografía analítica a una preparativa usando una columna con el mismo relleno y poder así obtener cromatogramas idénticos es necesario calcular el denominado factor de escalado X. Para ello se aplica la siguiente fórmula: X = (Lpreparativa /Lanalítica) (dpreparativa/danalítica)2 siendo L las longitudes de las columnas utilizadas y d sus respectivos diámetros. Este factor X se multiplica por el flujo y el volumen de muestra, obteniendo así las nuevas condiciones de análisis que deben emplearse en la cromatografía semipreparativa. 19.6. crOMaTOGrafía UlTra-ráPida (UPlc) En los últimos años, con objeto de reducir el tiempo de análisis y así poder realizar un mayor número de ensayos por unidad de tiempo, se ha desarrollado de manera espectacular la cromatografía ultra-rápida (ultra-performance liquid chromatography, UPLC), que permite aumentar la velocidad de análisis manteniendo la resolución. Para ello se han desarrollado columnas de pequeña longitud (< 10 cm) con fases estacionarias cuyo tamaño de partícula es inferior a 2 µm, que permiten separaciones con una gran eficiencia (> 200.000 platos/m). Estas columnas requieren unos equipos de HPLC especiales, capaces de soportar las altas presiones que se generan (> 10.000 psi). Además, los tubos de conexión están minimizados al máximo, para evitar dispersión de los analitos y las celdas de los detectores tienen un volumen mínimo, por el mismo motivo. En la conocida ecuación de van Deemter que rige las separaciones cromatográficas, y que relaciona la eficiencia de la columna con el caudal de trabajo, una de las variables importantes es el tamaño de partícula de la fase estacionaria. Así, al disminuir el tamaño de partícula de 10 µm a 5 µm, y luego a 2,5 µm, se produce un aumento de la resolución cromatográfica; además, esta eficacia se obtiene a flujos superiores en el caso de partículas pequeñas, lo que permite reducir el tiempo de análisis. La Figura 19.22 demuestra cómo al reducir el tamaño de partícula de 5 µm a 3,5 µm disminuye el tiempo de análisis considerablemente manteniendo la resolución cromatográfica, simplemente por un aumento del caudal utilizado. Técnicas de análisis y caracterización de materiales 826 Utilizando partículas todavía más pequeñas (< 2 µm) todavía es posible mejorar la eficacia de la separación, incluso empleando flujos de 5 ml/min. Al flujo óptimo de trabajo, una columna de 15 cm empaquetada con un relleno de 1,7 µm, genera una presión de 15.000 psi. Además, la anchura de pico en estas condiciones suele ser inferior a 1 segundo, lo que implica que el detector debe cumplir unas especificaciones muy determinadas. A) 0,06 1 0,04 AU 3 2 0,02 4 6 5 0,00 0,00 2,00 4,00 6,00 8,00 10,00 12,00 14,00 16,00 18,00 20,00 Minutos B) 1 0,10 0,08 AU 0,06 3 2 0,04 0,02 4 5 6 0,00 0,10 0,00 0,20 0,40 0,60 0,80 1,00 1,20 1,40 1,60 1,80 0,08 AU 2,00 Minutos 0,06 0,04 0,02 0,00 0,00 2,00 4,00 6,00 8,00 10,00 12,00 14,00 16,00 18,00 20,00 Minutos Figura 19.22. Ejemplo de escalado HPLC a cromatografía ultra-rápida: A) separación de 6 analitos en una columna XTerra C18 Waters (150 × 4,6 mm), 5 µm, 1,4 ml/min, ciclo de 25 min; B) columna XTerra C18 Waters (20 × 4,6 mm), 3,5 µm, 3 ml/min, ciclo de 3 min. Compuestos separados: 1, cafeína; 2, anilina; 3, N-metilanilina; 4, 2-etilanilina; 5, 4-nitroanisol; 6, N,N-dimetilanilina. Figura adaptada (con permiso) de Screening 4, 30-31 (2003). Las columnas de UPLC suelen utilizar fases híbridas (polímeros-sílice). Algunos de los parámetros determinantes en cromatografía ultra-rápida son el volumen del sistema, la velocidad del inyector y la respuesta del detector. En particular, el sistema de inyección debe eliminar las caídas de presión, evitar la contaminación cruzada y acortar al máximo el ciclo de inyección. Cromatografía líquida de alta resolución (HPLC) 827 bibliOGrafía 1. Pavia, D. L. et ál. Introduction to organic laboratory techniques, Saunders College Publishing, 1999. 2. SubraManian, G. Preparative and process-scale liquid chromatograph, Ellis Horwood Series in Chemical Engineering, 1991. 3. FonG, G. W.; LaM, S. K. HPLC in the pharmaceutical industry, Marcel Dekker Inc., 1991. 4. Dean, j. A. Analytical Chemistry Handbook, Mc Graw-Hill, New York, 1995. 5. GoodinG, K. M.; ReGnier, F. E. HPLC of Biological macromolecules, Marcel Dekker Inc., 1990. 6. Lloyd, R. et ál. Practical HPLC Method Development, Wiley-Interscience, 2.a ed., 2002. 7. Meyer, v. R. Practical High-Performance Liquid Chromatography, WileyInterscience, 3.a ed., 1999. 8. DonG, M. W. Modern HPLC for practicing scientist, Wiley-Interscience, 2006. 9. MCMaster, M. HPLC: A practical user’s guide, Wiley-Interscience, 2006. 10. Snyder, L. R. Introduccion to Modern Liquid Chromatography, Wiley-Interscience, 2009. 11. LC/GC EUROPE. Europe solutions for separation scientists. Publicada mensualmente por Advanstar, Cheste (UK): http://www.lcgceurope.com.