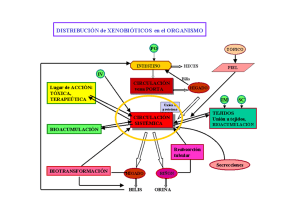
INTRODUCCIÓN La farmacocinética se refiere al movimiento de los fármacos hacia el interior, a través y hacia el exterior del organismo, es decir, de su absorción, distribución, metabolismo y excreción (LADME), aunado a esto podemos hablar de la biodisponibilidad, la cual, podemos definir como el grado y la velocidad con la que el fármaco accede a nuestra circulación, y logra llegar a su lugar de acción, logrando su objetivo, este recorrido suele calcularse mediante la determinación del área bajo la curva concentración plasmática, esta va a ser directamente proporcional a la cantidad total de fármaco que alcanza la circulación, los fármacos pueden ser considerados bioequivalentes en cuanto a la magnitud y a la velocidad de absorción si sus curvas de concentración plasmática son prácticamente iguales. La concentración plasmática es mayor cuanto mayor sea la absorción, la concentración plasmática máxima también llamada pico, se alcanza cuando se igualan la velocidad de eliminación y la de absorción. Este cálculo puede generar errores, ya que la eliminación del fármaco comienza desde el momento en que éste entra a la circulación sistémica, el tiempo pico es el parámetro más utilizado para calcular la velocidad de absorción; cuanto más lenta sea ésta, más tarde se alcanza el pico. Gracias a estos y más cálculos podemos conocer diversos parámetros importantes acerca de los fármacos y su trayecto dentro de nuestro organismo, tal como el volumen de distribución aparente (VDA) el cual podemos utilizar para cuantificar la distribución de los fármacos en todo el cuerpo posterior a la administración vía oral, sublingual, intravenosa, intramuscular etc., este se puede calcular dividiendo la dosis administrada del fármaco entre la concentración plasmática, cuando el paciente toma la tableta o el comprimido, no hay droga en el sistema, por lo que la concentración plasmática es igual a cero; a medida que se van estableciendo los elementos absorbidos van aumentando las concentraciones plasmáticas, siendo esto estrechamente relacionado a la vida media del fármaco al que podemos definir como la velocidad con la que se metaboliza en el hígado pasando a productos fácilmente eliminables por la orina (depurado) y que ya no cuenta con ninguna acción terapéutica siendo este un parámetro útil para determinar los intervalos de dosificación del medicamento OBJETIVOS Simular la administración de un fármaco por vía IV y EV Calcular los parámetros farmacocinéticos (vida media (T1/2 ), Ke, ABC, etc.) intravascular y extravascular. Comparar el comportamiento de los parámetros farmacocinéticos de acuerdo con su vía de administración. MATERIALES 6 Matraces volumétricos de 100mL con tapa (curva) 1 Matraz volumétrico de 50mL con tapa (solución de trabajo) 1 Pipeta volumétrica de 10mL 1 Perilla de succión 1 Balanza analítica 2 Espátulas delgadas 2 Vidrios de reloj o vasos de precipitado de 10mL Espectrofotómetro Celdas para espectrofotómetro Piceta con agua destilada 2 Vasos de precipitado de 200ml Colorante rojo vegetal RESULTADOS: 1) Elaborar una gráfica por cada vía de administración, considerando el logaritmo natural de la concentración (μg/mL) contra tiempo (min). Curva estándar Tubo Blanco 1 2 3 4 5 6 Concentración (mg/mL) 0 0.1031 0.0515 0.0257 0.0128 0.0064 0.0032 Absorbancia 0 1.798 0.831 0.374 0.164 0.064 0.031 Curva de calibracion para dicromato de potasio 2 y = 17.608x - 0.0439 R² = 0.9979 Absorbancia 1.5 1 0.5 0 0 -0.5 0.02 0.04 0.06 0.08 Concentracion (mg/mL) 0.1 0.12 Tabla de datos y grafica para vía Intravascular VIA INTRAVASCULAR Absorbancia Concentración (mg/mL) 0.747 0.044917083 0.501 0.031938713 0.391 0.025491184 0.302 0.020274548 0.266 0.018164447 0.256 0.017578308 0.228 0.015937119 0.204 0.014530385 0.196 0.014061474 0.227 0.015878505 0.212 0.014999297 Tiempo (Min) 0 5 10 15 20 25 30 35 40 45 60 Grafica via de administracion intravascular 0 -0.5 0 IN CONCENTRACION -1 -1.5 10 20 30 40 y = -0.0171x - 3.4793 R² = 0.7112 -2 -2.5 -3 -3.5 -4 -4.5 -5 TIEMPO 50 60 70 In -3.102937086 -3.443936422 -3.669422594 -3.898388997 -4.008289048 -4.041089628 -4.139104362 -4.231513274 -4.264316543 -4.142788964 -4.19975197 VIA DE ADMINISTRACION EXTRAVASCULAR VASO A Tiempo 0 5 10 15 20 25 30 35 40 45 60 Tiempo 0 5 10 15 20 25 30 35 40 45 60 Absorción 0.681 0.632 0.57 0.548 0.488 0.374 0.331 0.245 0.2 0.19 0.107 Concentración 0.041168787 0.038385961 0.034864834 0.033615402 0.03020786 0.02373353 0.021291458 0.016407315 0.013851658 0.013283735 0.008569968 IN -3.190074909 -3.260063487 -3.356276575 -3.392770922 -3.499653121 -3.740866454 -3.849449299 -4.110028016 -4.279350318 -4.32121495 -4.759491257 Absorción 0.035 0.078 0.1 0.114 0.143 0.207 0.241 0.268 0.25 0.226 0.178 VASO B Concentración 0.004480918 0.00692299 0.008172422 0.008967515 0.010614493 0.014249205 0.016180145 0.017713539 0.016691277 0.01532826 0.012602226 IN -5.407927395 -4.972907587 -4.806990009 -4.714146702 -4.545534909 -4.25105417 -4.123970382 -4.033426 -4.09286905 -4.178057103 -4.373881793 VASO A VASO B 0 20 40 60 -1 -2 -3 y = -0.0278x - 3.0762 R² = 0.9775 -4 -5 TIEMPO 80 0 0 -1 - 4.9858 y = 0.0187x R²-2 = 0.6192 IN CONCENTRACION IN CONCENCTRACION 0 20 40 -3 -4 -5 -6 TIEMPO 60 80 2) Calcular los parámetros farmacocinéticos: constante de eliminación (k), volumen aparente de distribución (Vd), concentración plasmática al tiempo cero (Cpo), depuración Total Clt, el tiempo de vida media (t1⁄2). CONSTATE DE ELIMINACION VASO A (K) K=0.0342401 k eliminación = K/[] Fármaco KE=0.0342401/0.041168787 KE= 0.8317009 CONSTATE DE ELIMINACION VASO B (K) K=0.008569968 k eliminación = K/[] Fármaco KE=0.008569968/0.004480918 KE= 1.9125473842636 Volumen aparente de distribución (Vd) Vd= [] en el organismo/ [] del fármaco sérico 3.748575047litros Para el cálculo del volumen aparente se divide la [] en el minuto 15 del vaso A y b en las gráficas correspondientes al experimento extravascular Concentración plasmática al tiempo cero (Cpo) A tiempo cero, cuando el paciente toma la tableta o el comprimido, no hay droga en el sistema, por lo que la concentración plasmática es igual a cero; a medida que se van estableciendo los elementos absortivos van aumentando las concentraciones plasmáticas. La concentración plasmática del fármaco es mayor cuanto mayor sea la absorción; la concentración plasmática máxima (pico) se alcanza cuando se igualan la velocidad de eliminación y la de absorción. Área bajo la curva (ABC) Es importante determinar el tipo de administración para considerar correctamente el valor de la concentración plasmática inicial. En este caso, el problema enuncia que el fármaco fue administrado de manera extravascular, por lo tanto, Cp inicial es igual a cero. El cálculo de ABC de cero a t se realiza de la sig. manera : ABC (0 − 1) = ∑ 𝐶𝑝 △ 𝑡 = vaso A 𝐶𝑝0 + Cp1 Cp Cp Cp(n − 1 Cp ) + (n) (𝑡𝑛 − 𝑡(𝑛 − 𝑖) ∗ (𝑡1 − 𝑡0) + 1 + 2 ∗ (𝑡1 − 𝑡0 ) + ⋯ + 2 2 2 Vaso B 3) Analizar la importancia de los modelos farmacocinéticos in vitro y de los parámetros farmacocinéticas calculados Es necesario y vitalmente importante el estudio de los efectos de los fármacos, para pronosticar con veracidad y precisión el comportamiento in vivo de los medicamentos a partir de observaciones in vitro, así brindar información amplia del mismo. Esto asegura la calidad y buen funcionamiento del fármaco, además de permitir calcular la dosificación óptima para el tratamiento sin comprometer al futuro paciente, y de esa manera escoger la vía de administración correcta. 4) Concluya analizando las ventajas y desventajas de cada vía después de realizar los cálculos correspondientes. INTRAVASCULAR Ventajas: 100% biodisponibilidad. Rápido y controlado. Desventajas: El efecto del medicamento permanece poco tiempo en el organismo. EXTRAVASCULAR Ventajas: El efecto del medicamento permanece más tiempo en el organismo. No presenta fenómeno de primer paso. OBSERVACIONES Desventajas: Menor biodisponibilidad. Absorción lenta. Al realizar la práctica nos encontramos con algunas interferencias, esto debido a que nuestros parámetros farmacocinéticos se comportaban de manera contrario; puesto que estos aumentaban su concentración en lugar de disminuir con forme transcurría la práctica. Nuestra teoría nos dice que esto se debe a mezclar agua corriente con agua destilada, mientras se realizaba la obtención de las muestras. CONCLUSIÓN Con base a lo anterior se concluye que los parámetros obtenidos nos dan una representación de cómo varía la concentración de un fármaco en el organismo según la vía de administración que se elige; por consiguiente diremos que los parámetros farmacocinéticos (vida media, Ke, ABC, T1/2 , etc.) serán mayores en la vía intravenosa, esto debido a que la vía EV implica la absorción del medicamento (LADME), mientras que la vía IV será más rápida debido a que el fármaco pasa de forma directa a circulación, obteniendo un rápido comienzo en la acción del fármaco. Haciendo énfasis en que la vía administración que se elige juega un papel muy importante en el tratamiento; ya que puede tener un marcado efecto sobre la velocidad y la eficacia con los cuales actúa el fármaco. BIBLIOGRAFIA Le, J. (s/f). Biodisponibilidad de los fármacos. Manual MSD versión para profesionales. Recuperado el 10 de septiembre de 2023, de https://www.msdmanuals.com/esmx/professional/farmacolog%C3%ADacl%C3%ADnica/farmacocin%C3%A9tica/bi odisponibilidad-de-losf%C3%A1rmacos Farmacocinética: absorción y distribución. (s/f). Medwave.cl. Recuperado el 10 de septiembre de 2023, de https://www.medwave.cl/puestadia/cursos/3449.html Vida media de un fármaco. (2020, septiembre 4). Eugenomic. https://eugenomic.com/recursos/glosario/vida-media-de-un-farmaco/ La circulación, M. del F. A. (s/f). CINETICOS Y BIOFAR-. Sefh.es. Recuperado el 10 de septiembre de 2023, https://gruposdetrabajo.sefh.es/pkgen/images/stories/MANUAL_FIR/Dicc ionario_PK.pdf de