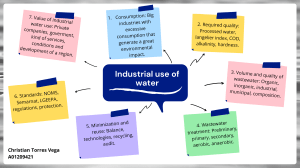
merican public health association. American water works association. Water environment federation - MÉTODOS NORMALIZADOS Para el análisis de aguas potables y residuales-Díaz de Santos (1989)
Anuncio
")
MÉTODOS NORMALIZADOS Para el análisis de aguas potables y residuales Subido por: Libros de Ingeniería Química y más https://www.facebook.com/pages/InterfaseIQ/146073555478947?ref=bookmarks Si te gusta este libro y tienes la posibilidad, cómpralo para apoyar al autor. APHA, AWWA, WPCF MÉTODOS NORMALIZADOS Para el análisis de aguas potables y residuales Preparado y publicado conjuntamente por: AMERICAN PUBLIC HEALTH ASSOCIATION AMERICAN WATER WORKS ASSOCIATION WATER POLLUTION CONTROL FEDERATION Comité Editorial Conjunto LENORE S. CLESCERI, WPCF, PRESIDENTE ARNOLD E. GREENBERG, APHA R. RHODES TRUSSELL, AWWA MARY ANN H. FRANSON Directora de Edición Título original: «Standard Methods» For the Examination of Water and Wastewater. 17 Edition © Copyright, 1989 American Public Health Association American Water Works Association Water Pollution Control Federation © Ediciones Díaz de Santos, S. A., 1992 Juan Bravo, 3-A. 28006 Madrid (España) Reservados todos los derechos. «No está permitida la reproducción total o parcial de este libro, ni su tratamiento informático, ni la transmisión de ninguna forma o por cualquier medio, ya sea electrónico, mecánico, por fotocopia, por registro u otros métodos, sin el permiso previo y por escrito de los titulares del Copyright» ISBN en lengua inglesa: 087553-161-X ISBN en lengua española: 978-84-7978-031-9 Depósito legal: M. 10.447-1992 Diseño de cubierta: Estuart, S. A. (Madrid) Traducción: Diorki, S. A. (Madrid) Fotocomposición: MonoComp, S. A. (Madrid) Impresión: Lavel, S. A. Humanes (Madrid) PRÓLOGO DE LA EDICIÓN EN ESPAÑOL DEL «STANDARD METHODS» ADECAGUA quiere que sus primeras palabras sean para agradecer a Ediciones DÍAZ DE SANTOS el enorme esfuerzo realizado para la publicación en español del «Standard Methods», libro que ha servido de guía a muchas generaciones de técnicos del agua en todo el mundo. Esta publicación facilitará su conocimiento en nuestro país, donde las sucesivas ediciones han sido siempre libro de consulta, y básico, en los temas de contaminación del medio acuático. No creemos, sin embargo, que la publicación se deba valorar únicamente en relación con el mero conocimiento de los métodos más adecuados de análisis. Muchos de sus capítulos son verdaderos compendios de una información básica, sin la cual es muy difícil obtener, con todas las garantías, el conocimiento completo de un problema de contaminación. No podría ser más oportuna esta edición en español. La creciente complejidad de los análisis para determinar microcontaminantes, especialmente orgánicos, y la aplicación de las Directivas Comunitarias, en su versión española, obliga aún más, si cabe, a un rigor en la analítica que sólo podrá conseguirse si se siguen métodos confirmados y seguros. Las mayores exigencias de la Administración, con la posible aplicación de sanciones cada vez más importantes, o incluso la creciente incidencia del delito ecológico, hace necesario disponer de una metodología segura y fiable para definir, hasta límites preestablecidos, una posible fuente contaminante. Muchas son las razones para recomendar vivamente la utilización de Métodos Normalizados para el análisis de aguas potables y residuales, pero parece lógico señalar las siguientes, como más importantes: La primera es la continua revisión a que están sometidos sus métodos de análisis. A la creciente diversidad de contaminantes que aparecen en el medio ambiente, se debe responder con una sistemática revisión de la técnica que permita afrontar los nuevos problemas, con métodos actuales, utilizando los más modernos procesos de análisis y el desarrollo instrumental existente en el mercado. La implantación continua de los procesos de análisis biológicos, como los ensayos de toxicidad, es también del mayor interés, estableciendo la necesaria coordinación entre análisis físico-químicos y biológicos, estos últimos, desgraciadamente, menos utilizados en nuestro país. La mayor atención que la publicación dedica a esta metodología es una prueba más de su interés y de la dedicación de los editores de mantenerla no sólo al día, sino adelantándose a las necesidades, para la protección del medio ambiente. Estamos seguros que, desde su publicación en castellano de la obra Métodos Normalizados para el análisis de aguas potables y residuales será libro de consulta obligado en nuestros laboratorios, empresas de ingeniería y, en general, de todos aquellos que dedicamos nuestra actividad profesional a estos problemas. GAMALIEL MARTÍNEZ DE BASCARÁN Presidente de ADECAGUA PREFACIO A LA DECIMOSÉPTIMA EDICIÓN* Edición decimosexta y anteriores La primera edición de Métodos normalizados se publicó en 1905. Cada una de las subsiguientes ediciones introdujo mejoras significativas en el aspecto metodológico y amplió el alcance de la obra con el fin de incluir técnicas adecuadas para el análisis de la gran cantidad de tipos de muestras encontradas durante la evaluación y control de la calidad del agua y de la contaminación de las aguas. Es interesante revisar brevemente la historia de Métodos normalizados por lo actual de su contenido. Una iniciativa para «garantizar la adopción de métodos de análisis de aguas más eficientes y uniformes» dio lugar en la década de 1880 a la organización de un comité especial en la Sección Química de la American Association for the Advancement of Science. En 1889, este comité publicó un informe titulado: «Método parcial para el análisis sanitario de las aguas y para la presentación de resultados, ofrecido para adopción generalizada»**. En él se trataban cinco temas: 1) amonio «libre» y «albuminoideo»; 2) capacidad de consumo de oxígeno; 3) nitrógeno total en forma de nitratos y nitritos; 4) nitrógeno en forma de nitritos, y 5) presentación de resultados. En 1895, miembros de la American Public Health Association (APHA), conscientes de la necesidad de contar con métodos normalizados para el análisis microbiano de las aguas, patrocinaron una convención de bacteriólogos para estudiar el problema. Como resultado de ello se formó un comité en el seno de la APHA encargado de «diseñar procedimientos para el estudio de bacterias de un modo uniforme y con especial referencia a la diferenciación de especies». A partir de su presentación en 1897†, los procedimientos gozaron de una amplia aceptación. En 1899, la APHA creó el Committee on Standard Methods normalizados para Análisis de Aguas, al que se encargó la extensión de los procedimientos estándar a la totalidad de los métodos relacionados con el análisis de aguas. El informe del Comité, publicado en 1905, constituyó la primera edición de Métodos normalizados, titulado por entonces Métodos normalizados para el análisis de aguas. En él se incluían métodos para el análisis físico, químico, microscópico y bacteriológico de las aguas. En su carta de presentación, el Comité afirmaba: Creemos que los métodos de análisis presentados en este informe como «Métodos normalizados» recogen los mejores procedimientos empleados en la actualidad por los analistas de aguas estadounidenses y pueden aplicarse de forma generalizada en relación con los problemas habituales de purificación de aguas, eliminación de aguas residuales e investigaciones sanitarias. Es evidente que no se pueden emplear métodos de análisis idénticos para problemas ampliamente diferentes, y que los problemas especiales exigen la aplicación de los métodos que mejor se adapten a ellos. No obstante, y sin olvidar este extremo, no deja de ser cierto que el trabajo analítico progresará en relación directa con la adopción general de métodos fiables, uniformes y adecuados. En ocasiones se afirma que la adopción de métodos estándar en el campo de la ciencia aplicada * Esta decimoséptima edición en inglés corresponde a esta edición que se realiza por vez primera en lengua castellana. ** J. Anal. Chem. 3:398 (1889). † Proc. Amer. Pub. Health Assoc. 23:56 (1897). viii MÉTODOS NORMALIZADOS tiende a esclerotizar las investigaciones y a retrasar el auténtico progreso. No hay razón para que ello sea así, sin embargo, si tales métodos se emplean con la mentalidad adecuada. Este Comité desea fervientemente que se continúen todos los esfuerzos encaminados a mejorar las técnicas de análisis de aguas y, especialmente, a comparar los métodos habituales con los aquí recomendados en los casos en que sean diferentes, de tal modo que los resultados obtenidos sean aún más exactos y fiables de lo que lo son en la actualidad. La APHA publicó sucesivas ediciones revisadas y ampliadas de Métodos normalizados para el análisis de aguas en 1912 (segunda edición), 1917 (tercera), 1920 (cuarta) y 1923 (quinta). En 1925, la American Water Works Association se unió a la APHA para la publicación de la sexta edición, a la que se dio el título más amplio de Métodos normalizados para el análisis de aguas limpias y residuales. La publicación conjunta se continuó en la séptima edición, fechada en 1933. En 1935, la Water Pollution Control Federation (WPCF), por entonces llamada Federation of Sewage Works Associations, publicó un informe elaborado por uno de sus comités y titulado «Métodos normalizados para el análisis de aguas residuales»‡. Con pequeñas modificaciones, estos métodos se incorporaron a la octava edición (1936) de Métodos normalizados, la cual ofrecía por primera vez métodos para el análisis de «aguas residuales, vertidos industriales y aguas contaminadas con lodos y fangos». La novena edición, que apareció en 1946, también contenía estos métodos, y al año siguiente la WPCF se asoció plenamente a la publicación. Desde 1947, el trabajo de los comités de Métodos normalizados de las tres asociaciones —APHA, AWWA y WPCF— está coordinado por un consejo editorial conjunto con representación de cada una de ellas. La décima edición (1955) incluía métodos específicos para el análisis de aguas residuales industriales, hecho que se reflejaba en el nuevo título: Métodos normalizados para el análisis de aguas limpias, aguas residuales y residuos industriales. Con el fin de describir con mayor exactitud y concisión su contenido, el título de la undécima edición (1960) se abrevió a Métodos normalizados para el análisis de aguas y aguas residuales, título que se mantuvo sin cambios a lo largo de las ediciones duodécima (1965), decimotercera (1971), decimocuarta (1976) y decimoquinta (1981). En la decimocuarta edición se suprimió la separación entre los procedimientos de análisis para aguas limpias y para aguas residuales, de modo que todos los métodos para la determinación de un componente o una característica dada se agrupaban bajo un único encabezamiento. Con ligeros cambios, la organización de la decimocuarta edición se conservó en la decimoquinta y la decimosexta (1985). En esta última se llevaron a la práctica dos importantes decisiones políticas del consejo editorial conjunto: en primer lugar, se incorporó y adoptó el Sistema Internacional de Unidades (SI), excepto en los casos en que los sistemas o prácticas habituales exigían el empleo de unidades inglesas; en segundo lugar, se redujo al mínimo el empleo de nombres comerciales o referencias a productos de marca, para evitar posibles reclamaciones sobre limitación del comercio o favoritismo comercial. ‡ Sewage Works J. 7:444(1935). PREFACIO A LA DECIMOSÉPTIMA EDICIÓN ix Decimoséptima edición La organización de la decimoséptima edición refleja el compromiso de desarrollar y mantener un sistema de numeración permanente. Se han asignado nuevos números a todas las secciones y se han reservado otros, no empleados en la presente edición, para utilizarlos en el futuro. Se han numerado las distintas partes con múltiplos de 1000, en vez de 100, pero todas ellas conservan su identidad con respecto a la edición anterior, salvo la 6000, dedicada ahora a los métodos para la determinación de compuestos orgánicos específicos; los procedimientos más generales para determinación de compuestos orgánicos aparecen en la parte 5000. La sección introductoria (parte 1000) de la decimoséptima edición ha experimentado una profunda revisión. Las secciones relativas a análisis estadístico, calidad de datos y desarrollo de métodos se han ampliado considerablemente. La sección dedicada al agua como reactivo se ha actualizado para dar cabida al esquema de clasificación actual de los distintos tipos de aguas para reacción. La parte 1000 recoge importante información acerca de la correcta ejecución de los procedimientos y debe ser cuidadosamente estudiada por todos los usuarios de este manual. Las partes restantes se inician con secciones dedicadas a la garantía de calidad y otros temas de aplicación general dentro de cada campo concreto, con el fin de reducir al mínimo las repeticiones en el texto subsiguiente. La escrupulosa observancia de las recomendaciones y precauciones introductorias es fundamental para el éxito del análisis. Antes de emprender un ensayo, el lector debe haber comprendido toda la explicación de cada procedimiento, incluyendo la selección de métodos, los procedimientos de toma de muestras y el almacenamiento de las mismas, así como las posibles fuentes de interferencia. La parte 2000 (propiedades físicas y globales) presenta una nueva sección referente a sobresaturación de gases disueltos; contiene además procedimientos para el análisis de ciertas propiedades globales (acidez, alcalinidad y dureza) que aparecían en otras partes de la obra en ediciones anteriores, así como las pruebas para el gas de digestión de lodos que figuraban en la parte 500 de ediciones anteriores. Se han revisado las secciones referentes a salinidad y saturación de carbonato cálcico para adaptarlas a la práctica actual de análisis de estos parámetros. La parte 3000 (metales) incluye ahora un breve apartado sobre garantía de calidad; se ha actualizado la sección relativa a tratamiento y preparación de las muestras. A continuación se presentan las técnicas instrumentales (espectrometría de absorción atómica y método de plasma de acoplamiento inductivo) útiles para la determinación de numerosos metales; se han revisado los métodos de absorción atómica y se ha incorporado una nueva técnica basada en la generación continua de hidruros. A las secciones dedicadas a análisis para metales específicos se han añadido breves apartados que refieren al lector a las técnicas instrumentales para la determinación de antimonio, arsénico, bismuto, cesio, iridio, molibdeno, osmio, paladio, platino, renio, rodio, rutenio, talio, torio, estaño y titanio. Se ha revisado la sección referente al litio para incluir métodos con umbrales de x MÉTODOS NORMALIZADOS detección más bajos. Se han añadido nuevos métodos para la determinación de compuestos de selenio y aluminio. La parte 4000 (no metales inorgánicos) contiene también una nueva sección dedicada a garantía de calidad. Los métodos analíticos, incluyendo la cromatografía iónica para determinación de varios aniones, así como los métodos para aniones específicos, se mantienen prácticamente sin cambios y se han confirmado por consenso. Para la determinación de cloro residual se han añadido la titulación amperimétrica de bajo nivel y las técnicas de yodometría, eliminándose el método del cristal violeta incoloro. También se han añadido métodos más precisos para la determinación de ozono y dióxido de cloro. Se han suprimido varios métodos para nitratos y se ha añadido un método nuevo para determinación de nitrato mediante cloruro de titanio. La parte 5000 (compuestos orgánicos en general) contiene ahora exclusivamente métodos de determinación de las concentraciones globales de grupos de compuestos orgánicos. Los métodos para determinar compuestos específicos (metano, pesticidas y herbicidas, metanos y etanos halogenados, compuestos causantes de olores y sabores, y contaminantes orgánicos por cromatografía de gases/espectrometría de masas) se han trasladado a la parte 6000 o se han sustituido por procedimientos nuevos. Se han revisado en profundidad los métodos para DOX (anteriormente TOX). Por último, se han incorporado métodos para sustancias procedentes del humus y determinación del potencial formador de trihalometano. La parte 6000 (compuestos orgánicos individuales) es nueva. Contiene información introductoria sobre toma y conservación de muestras para análisis de compuestos orgánicos, así como los principios generales de procedimientos y las fuentes de interferencia para los métodos analíticos instrumentales. Se ha revisado un método de concentración de constituyentes mediante extracción de gases (depuración de bucle cerrado) que se incluía en la parte 500 de la decimosexta edición como método de análisis de compuestos orgánicos causantes de olor o sabor. Cada familia o grupo de compuestos orgánicos se presenta con una introducción sobre su presencia en el medio ambiente y una selección de métodos analíticos para los mismos. La mayoría de los métodos incluidos en la parte 6000 corresponden a procedimientos aprobados por la Agencia del Medio Ambiente, completamente revisados y actualizados, que aparecieron como suplemento a la decimosexta edición. También se incluyen en esta parte varios métodos para la determinación de compuestos específicos que figuraban en la parte 500 en la edición decimosexta. La parte 7000 (radiactividad) no presenta cambios en cuanto a los métodos. Se han revisado las secciones de introducción para aventurar la importancia de la garantía de calidad. La parte 8000 (pruebas toxicológícas) también conserva los métodos de la edición precedente, si bien se ha revisado la introducción y se ha añadido un breve apartado sobre mutagenicidad. La sección de zooplancton de la decimosexta edición ha perdido su carácter unitario, al haberse incluido los métodos PREFACIO A LA DECIMOSÉPTIMA EDICIÓN xi para protozoos ciliados, Daphnia y Acartia en otras secciones de esta misma parte, con el fin de reflejar las relaciones taxonómicas. La parte 9000 (análisis microbiológicos) ha experimentado una revisión y actualización generales. Se ha añadido un nuevo método para recuento directo y se han separado las pruebas de determinación de protozoos patógenos de las referidas a bacterias patógenas. En relación con la determinación de coliformes, la producción de ácido en un medio de cultivo que contiene lactosa se ha incluido nuevamente como reacción positiva. La sección referente a estreptococos, de nueva redacción, hace hincapié en los procedimientos para enterococos. La parte 10000 (análisis biológicos) contiene revisiones sustanciales de los métodos para determinación de plancton, macrófitos y peces. El principal cambio en la sección de plancton es la inclusión de un procedimiento de cromatografía de líquidos de alta resolución (HPLC) para determinación de clorofila. Otros métodos se han sometido igualmente a revisión y a actualización generalizadas. Preparación de reactivos La ejecución estricta de las instrucciones para la preparación de reactivos puede dar lugar a la obtención de cantidades muy superiores a las necesarias. En algunos casos, estos reactivos son tóxicos. Con el fin de favorecer la economía y reducir al mínimo los desperdicios, el analista debe examinar las necesidades y adaptar a ellas las cantidades de solución siempre que resulte apropiado. Esta actitud conservadora debe extenderse igualmente a la política de suministros, de modo que no se acumulen productos químicos sin utilizar de los que haya que deshacerse al expirar su plazo de conservación. Selección y aprobación de métodos En cada nueva edición, tanto los criterios técnicos de selección de métodos como los procedimientos formales de aprobación e inclusión se someten a revisión crítica. Con respecto a los procedimientos de aprobación, se concede especial importancia a garantizar que los métodos presentados hayan sido examinados y se encuentren respaldados por el mayor número posible de personas cualificadas, de modo que constituyan un auténtico consenso entre las opiniones de los expertos. Al preparar la decimocuarta edición, se establecieron grupos de trabajo para cada una de las pruebas, esquema que se mantuvo en cada una de las ediciones subsiguientes. La inclusión de una persona determinada en un grupo de trabajo se basó generalmente en su interés expreso o en el hecho de contar con experiencia reconocida. Fue, en cualquier caso, un esfuerzo encaminado a reunir un grupo con la máxima experiencia posible en cada uno de los métodos de ensayo. A cada grupo de trabajo se le encargó la revisión de los métodos pertinentes de la decimosexta edición, así como de otros procedentes de la literatura, con el xii MÉTODOS NORMALIZADOS fin de recomendar los métodos que deberían incluirse en la decimoséptima edición y presentarlos en forma de manuscrito como propuesta de sección. Posteriormente, cada sección, excepto las de la parte 1000, fue ratificada en rotación por los miembros del Committee on Standard Methods que habían solicitado examinar las secciones de determinada parte. Todos los votos negativos y comentarios surgidos durante la votación fueron examinados posteriormente por el consejo editorial conjunto. Se sometieron a resolución las sugerencias más importantes. En los casos en los que ni el grupo de trabajo ni el consejo editorial conjunto consiguieron resolver los votos negativos de la primera ronda, la sección volvió a someterla a votación entre quienes habían votado afirmativa o negativamente en la primera ronda. Solamente en los pocos casos que no pudieron resolverse así, correspondió al consejo editorial conjunto la decisión final. La información general y la relativa a garantía de calidad presentadas en la parte 1000 recibieron un tratamiento diferente. También en este caso se formaron grupos de trabajo a los que se asignó una tarea y se encomendó la preparación de un borrador de consenso. Este borrador fue examinado por el enlace del consejo editorial conjunto y, finalmente, por el propio consejo. Los borradores de las distintas secciones se enviaron al Committee on Standares Methods, y los comentarios resultantes de esta revisión se incorporaron al borrador final. Al igual que en ediciones precedentes, los métodos presentados en esta obra se considera que son los mejores de los procedimientos disponibles para el análisis de aguas limpias residuales y sustancias relacionadas, y gozan de amplia aceptación; son los recomendados por los especialistas y están ratificados por un elevado número de analistas y profesionales de experiencia más general, por lo que constituyen auténticas referencias estándar de consenso, capaces de ofrecer una base válida y reconocida para control y evaluación. Los criterios técnicos para la selección de los métodos los aplicaron los grupos de trabajo y las personas encargadas de revisar las recomendaciones de éstos, en tanto que el consejo editorial conjunto se limitó a proporcionar directrices generales. Además de los conceptos clásicos de precisión, sesgo y concentración mínima detectable, la selección de un método determinado debe tener en cuenta asimismo aspectos como el tiempo necesario para obtener los resultados, la necesidad de contar con equipos especiales o con analistas que hayan recibido una formación especializada y otros factores referentes al coste del análisis y la viabilidad de un uso generalizado del mismo. Clasificación de los métodos Todos los métodos incluidos en la decimoséptima edición están fechados, con el fin de ayudar a los usuarios a determinar qué métodos han sufrido modificaciones significativas con respecto a la edición precedente. El año en que el Committee on Standard Methods aprobó la sección se indica en una nota a pie de página al comienzo de cada sección. Las secciones o métodos que aparecían en la decimosexta edición y permanecen inalterados en la presente, o han sufrido cambios PREFACIO A LA DECIMOSÉPTIMA EDICIÓN xiii formales pero no de contenido, tienen la fecha de la edición anterior, 1985. Las secciones o métodos que se han modificado significativamente o que se han confirmado mediante consenso general del Committee on Standard Methods se han fechado en 1988, mientras que los demás métodos conservan la fecha de 1985. En la decimoséptima edición, los distintos métodos se han clasificado en cuatro clases fundamentales: PROPUESTOS, ESPECIALIZADOS, NORMALIZADOS y GENERALES. Independientemente de la categoría asignada, todos los métodos deben ser aprobados por el Committee on Standard Methods. Las características de cada clase son las siguientes: 1. PROPUESTOS: Un método PROPUESTO debe someterse a un desarrollo y validación que satisfagan los criterios establecidos en la sección 1040 A de Métodos normalizados. 2. ESPECIALIZADOS: Determinado procedimiento llega a convertirse en un método ESPECIALIZADO por una de las siguientes vías: a) El procedimiento se somete a desarrollo y validación, así como a comprobación en régimen de colaboración, de acuerdo con los requisitos establecidos en la sección 1040 B y C, respectivamente, de Métodos normalizados; o b) El procedimiento en cuestión constituye el «MÉTODO DE ELECCIÓN» de los miembros del Committee on Standard Methods que efectúan el análisis y ha aparecido en DOS EDICIONES PREVIAS de Métodos normalizados. 3. NORMALIZADOS: Un procedimiento se convierte en método NORMALIZADO de una de las dos formas siguientes: a) El procedimiento se somete a desarrollo y validación, así como a comprobación en régimen de colaboración, de acuerdo con los requisitos establecidos en la sección 1040 B y C, respectivamente, de Métodos normalizados, y es «AMPLIAMENTE USADO» por los miembros del Committee on Standard Methods; o b) Se trata de un procedimiento «AMPLIAMENTE USADO» por los miembros del Committee on Standard Methods y ha aparecido en DOS EDICIONES PREVIAS de Métodos normalizados. 4. GENERALES: Un procedimiento se considera método GENERAL cuando ha aparecido en DOS EDICIONES PREVIAS de Métodos normalizados. La asignación de un método a una categoría determinada corresponde al consejo editorial conjunto. Para clasificar los métodos, dicho consejo evalúa los resultados de la encuesta que sobre empleo de métodos por el Committee on Standard Methods se efectúa en el momento de la votación general del método. El consejo editorial conjunto tiene en cuenta asimismo las recomendaciones hechas por los grupos de trabajo y por el coordinador general de la parte de que se trate. En los títulos de los métodos clasificados como «PROPUESTOS», «ESPECIALIZADOS» y «GENERALES» figura la designación correspondiente: los métodos en cuyo título no aparece designación alguna son «NORMALIZADOS». El progreso técnico hace recomendable el establecimiento de un programa xiv MÉTODOS NORMALIZADOS para mantener Métodos normalizados a la vanguardia de los avances en investigación y práctica cotidiana. El consejo editorial conjunto ha desarrollado el siguiente procedimiento para efectuar modificaciones provisionales de los métodos entre ediciones: 1. La categoría asignada a cualquier método en la presente edición puede elevarse por decisión del consejo editorial conjunto, a partir de la publicación de datos que apoyen tal modificación y sean sometidos al comité por el grupo de trabajo correspondiente. La notificación del cambio de categoría se realizará mediante publicación en las revistas oficiales de las tres asociaciones que patrocinan Métodos normalizados. 2. Entre ediciones no puede suprimirse ni rebajarse de categoría ningún método. 3. El consejo editorial conjunto puede decidir entre ediciones la adopción de un nuevo método como propuesto, especializado o estándar, basando su decisión en el procedimiento de consenso habitual. Estos nuevos métodos pueden publicarse en suplementos a las ediciones de Métodos normalizados. Aún más importante para mantener la actual categoría de estos estándares es la intención de los patrocinadores de la publicación y del consejo editorial conjunto de que las próximas ediciones aparezcan con regularidad y a intervalos razonablemente cortos. Los comentarios y preguntas del lector en relación con este manual deben dirigirse a: Standard Methods Manager, American Water Works Association, 6666 West Quincy Avenue, Denver, CO 80235. Agradecimientos El consejo editorial conjunto otorga el mérito de la mayor parte del trabajo de preparación y revisión de los métodos de la decimosexta edición a los Committees on Standard Methods de la American Water Works Association y de la Water Pollution Control Federation, así como al Subcommittee on Standard Methods para el Análisis de Aguas Limpias y Residuales y al Committee on Laboratory Standards and Practices de la American Public Health Association. Miembros de estos comités presiden o forman parte de los grupos de trabajo. Con frecuencia estuvieron ayudados en su labor por consultores que no pertenecían formalmente a los comités ni, en muchos casos, eran miembros de las sociedades patrocinadoras. Deseamos expresar a estos consultores nuestra especial gratitud y reconocimiento por sus esfuerzos. A continuación de estas páginas figura una relación de los miembros y los consultores de los comités. Robert Booth, consultor científico de la Environmental Protection Agency, actuó como enlace entre este organismo y el consejo editorial conjunto; le expresamos nuestro agradecimiento por su interés y ayuda. El consejo editorial conjunto desea manifestar su reconocimiento a PREFACIO A LA DECIMOSÉPTIMA EDICIÓN xv William H. McBeath, MD, director ejecutivo de la American Public Health Association; a John B. Mannion, director ejecutivo de la American Water Works Association, y a Quincalee Brown, director ejecutivo de la Water Pollution Control Federation, por su cooperación y asesoramiento en el desarrollo de esta publicación. Steven J. Posavec, gerente de Métodos normalizados y secretario del consejo editorial conjunto, proporcionó muy numerosos e importantes servicios, esenciales para la preparación de una obra de estas características. Jaclyn Alexander, directora de publicaciones de la American Public Health Association, actuó como editora, y Brigitte Coulton, también de la APHA, lo hizo como directora de producción. Especial reconocimiento merece la labor de Mary Ann H. Franson, directora de edición, quien hizo frente con absoluta eficiencia a las amplias y detalladas responsabilidades que entraña esta publicación. Deseamos expresar nuestro agradecimiento al anterior director ejecutivo de la American Water Works Association, Paul A. Schulte, por el gran apoyo prestado a esta edición y a las anteriores de Métodos normalizados. El fallecido Robert A. Canham realizó inestimables contribuciones al continuo desarrollo y mejora de Métodos normalizados durante su larga trayectoria como director ejecutivo de la Water Pollution Control Federation; conservamos un grato recuerdo de su sabiduría y su dirección. El consejo editorial conjunto agradece los significativos servicios prestados por Frederick W. Pontius, ingeniero de reglamentación de la American Water Works Association y anterior secretario del consejo editorial conjunto, así como por Adrienne Ash, de la American Public Health Association, que actuó como editora durante las fases iniciales de preparación de esta edición. Consejo editorial conjunto Arnold E. Greenberg, American Public Health Association. R. Rhodes Trussell, American Water Works Association. Lenore S. Clesceri, Water Pollution Control Federation, Presidente. En distintos lugares de la presente obra se hace referencia a nombres de fabricantes, a marcas comerciales y a reactivos o compuestos químicos. El empleo de tales nombres no tiene otra finalidad que la de servir de referencia rápida a las características funcionales del producto en cuestión. Dichas referencias por tanto no deben interpretarse como recomendaciones de ninguno de esos productos por parte de los editores; en todos los casos pueden emplearse materiales o reactivos de características equivalentes. CONSEJO EDITORIAL CONJUNTO LENORE S. CLESCERI, Water Pollution Control Federation, Presidente. A RNOLD E. GREENBERG, American Public Health Association. R. RHODES TRUSSELL, American Water Works Association. COORDINADORES DE LAS DISTINTAS PARTES PARA LA DECIMOSÉPTIMA EDICIÓN Las personas siguientes actuaron como coordinadores de la parte indicada. Lenore S. Clesceri, 1000 David J. Rexing, 2000 Andrew D. Eaton, 3000 J. Owen Callaway, 4000 Stephen J. Randtke, 5000 Joseph J. Delfino, 6000 James W. Mullins, 7000 Patrick R. Parrish, 8000 Betty H. Olson, 9000 Hugh D. Putnam, 10000 COMITÉS PARA LA DECIMOSÉPTIMA EDICIÓN Presidentes de los grupos de trabajo Las personas siguientes actuaron como presidentes de los grupos de trabajo que desarrollaron las secciones indicadas. E. Marco Aieta, 4500-CIO2 Harry J. Alexander, 1060 Frank J. Baumann, 6210, 6220, 6230 Daniel F. Bender, 1090 Stuart C. Black, 1010, 1020, 1030, 1040 Robert H. Bordner, 9020 Robert I. Botto, 3120 Lloyd W. Bracewell, 2530 William H. Bruvold, 2160 Gary B. Collins, 10200 John Colt, 2810 Brian J. Condike, 3113 Wm. Bridge Cooke, 9610 Alfred P. Dufour, 9230 David E. Erdmann, 4500-F – Samuel D. Faust, 5530 John M. Ferris, 9810 Edwin E. Geldreich, 9221, 9222 Cari J. George, 10600 Gilbert Gordon, 4500-O 3 Charles N. Haas, 4500-C1 Earl L. Henn, 3111 Anita K. Highsmith, 1080, 9213 Thomas B. Hoover, 4110 Billy G. Isom, 10500 Walter Jakubowski, 9260, 9711 Martha N. Jones, 4500-NO 3 – y NO 2 – Stuart W. Krasner, 6040 Timothy E. Lewis, 350O-A1 William F. McCoy, 9216 Gordon A. McFeters, 9212 Douglas T. Merrill, 2330 Cari J. Miles, 5510 Frank J. Millero, 2520 Leown A. Moore, 5710 James W. Mullins, 7010, 7020, 7110, 7500-Cs, I, Ra, Sr, 3H y U Betty H. Olson, 9010, 9030, 9040, 9050, 9060 Francés Parsons, 9225 xvii xviii Marvin D. Piwoni, 3030, 3110, 3111, 3112, 3113, 3114 Hugh D. Putnam, 10300 Stephen J. Randtke, 5550 Ronald L. Raschke, 10400 Donald J. Reasoner, 9211 David A. Reckhow, 5320 Donald J. Reish, 8510, 10900 Olsen J. Rogers, 4500-Br – Darrell G. Rose, 3112 Robert S. Safferman, 9250 Kenneth Simon, 8010 Robert D. Swisher, 5540 Jack R. Tuschall, 3500-Li Michael J. Vandaveer, 3030 Hugo T. Victoreen, 9215 Oleh Weres, 3500-Se James C. Young, 5210 Committee on Standard Methods y miembros de los grupos de trabajo Las siguientes personas actuaron como miembros del Committee on Standard Methods y como miembros de los grupos de trabajo que desarrollaron las secciones indicadas. John C. Adams, 9213 V. Dean Adams Rose Adams-Whitehead Cacilda Jiunko Aiba E. Marco Aieta, 4500-ClO2 Katherine T. Alben Harry J. Alexander, 1060 James E. Alleman, 4500-NO3 – yNO2Herbert E. Alien Martin J. Alien, 9212, 9215 Osman M. Aly, 5530 Gary L. Amy, 5510 Charles G. Appleby Neal E. Armstrong John A. Arrington, II Robert M. Arthur Donald B. Aulenbach, 4500-NO3– y NO 2 Barry M. Austern Warren F. Averill Guy M. Aydlett Robert M. Bagdigian, 1080 * Personas que no son miembros oficiales del Committee on Standard Methods pero realizaron contribuciones a las secciones indicadas. Rodger B. Baird, 3030, 3110, 3111, 3112, 3113, 3114 L. Malcolm Baker, 6040 Robert A. Baker Gwendolyn L. Ball, 5710, 6040 Roy O. Ball Edmond J. Baratta, 7010, 7020, 7110, 7500-Cs, I, Ra, Sr, 3H, y U Michael J. Barcelona Thomas O. Barnwell, Jr, 5210 Sylvia E. Barrett, 4500-C1 Jerry Bashe*, 6010 Frank J. Baumann, 6210, 6220, 6230 David G. Beckwith, 1080 John W. Beland Daniel F. Bender, 1080, 1090, 4500-C1 y C1O2 Larry D. Benefield Paul S. Berger, 9020, 9215 Thomas Beymer*, 6010 Robert R. Bidigare, 10200 Kenneth E. Biesinger Star F. Birch, 1060, 1090, 9020 Stuart C. Black, 1010, 1020, 1030, 1040 xix H. Curt Blair, 2520, 7010, 7020, 7110, 7500-Cs, I, Ra, Sr, 3H, y U I. Bob Blumenthal Edwin S. Boatman* DebraK. Bolding, 9211, 9230 Robert L. Booth† Robert H. Bordner, 9010, 9020, 9030, 9040, 9050, 9060 Mark E. Bose, 5540 Harry Boston ‡, 10400 Robert I. Botto, 3120 Gerald R. Bouck, 2810 William H. Bouma, 1060 Theresa M. Bousquet Russell E. Bowen George T. Bowman, 5210 William C. Boyle Lloyd W. Bracewell, 2530 Alvin L. Bradshaw, 2520 Brian M. Brady Blaise J. Brazos Geoffrey L. Brock Joe E. Brown Clifford Bruell William H. Bruvold, 2160 Anthony A. Bulich Steven M. Bupp Gary A. Burlingame, 2160, 6040 J. Owen Callaway Craig D. Cameron, 4500-O3 Raúl R. Cárdenas, Jr. Robert E. Carlson Stephen R. Carpenter Anthony R. Castorina Paul A. Chadik, 5510 Peter M. Chapman, 8510, 10500 Eric J. Chatfield Daniel David Chen * Fallecido. † Enlace de la EPA con el consejo editorial conjunto. ‡ Personas que no son miembros oficiales del Commitíee on Standard Methods pero realizaron contribuciones a las secciones indicadas. Mark G. Cherwin, 3030, 3110, 3111, 3112, 3113, 3114 Leonard L. Ciaccio Robert R. Claeys James A. Clark, 9221, 9225 Robert R. Clark William H. Clement, 5530 Dennis A. Clifford Sharon M. Cline John Clugel Colin E. Coggan Robert S. Cohen, 3030, 3110, 3111, 3112, 3113, 3114 Larry D. Cole Gary B. Collins, 10200 Michael Collins, 5510 Tom E. Collins, 6040 John Colt, 2810 Brian J. Condike, 3030, 3110, 3111, 3112, 3113, 3114 Gerald F. Connell Wm. Bridge Cooke, 9010, 9030, 9040, 9050, 9060, 9610, 10900 Sandra D. Cooper, 10300, 10500 Robert C. Cooper, 9225, 9260, 9711 Harold S. Costa C. Richard Cothern, 7010, 7020, 7110, 7500-Cs, I, Ra, Sr, 3H, yU John E. Cowell John V. Crable, 1090 Wendall H. Cross Warren B. Crummett William G. Crumpton, 10200 Nicholas J. Csikai Kenneth W. Cuneo Gregory A. Cutter, 3500-Se Melissa S. Dale, 6040 Richard A. Danforth, 1080 Robert A. Daniels, 10600 Richard-Paul Danner Brian G. D'Aoust, 2810 Patrick H. Davies xx Charles O. Davis, III, 3500-Li Ernst M. Davis, III, 10200 Marshall K. Davis, 3030, 3110, 3111, 3112, 3113, 3114 Laurens M. DeBirk Gary L. De Kock, 3120 Joseph J. Delfino Brian A. Dempsey, 5510 W. Michael Dennis*, 10400 Steven K. Dentel Fred L. DeRoos David Diamond Jack C. Dice, 2160 Denise A. Domínguez John H. Dorsey, 10900 Ronald C. Dressman, 5320 Charles T. Driscoll, 3500-A1 Alfred P. Dufour, 9010, 9030, 9040, 9050, 9060, 9213, 9230, 9260, 9711 Bernard J. Dutka, 9211, 9213, 9215 David G. Easterly, 7010, 7020, 7110, 7500-Cs, I, Ra, Sr, 3H, yU Andrew D. Eaton David L. Edelman, Jr. James K. Edzwald, 5710 Lawrence W. Eichler, 3030, 3110, 3111, 3112, 3113, 3114, 3500Al Gunnar Ekedahl William M. Ellgas, 9222, 9225 G. Keith Elmund, 9020, 9211 Mohamed Elnabarawy Alan W. Elzerman David E. Erdmann, 4110, 4500-F– R. P. Esser, 9810 Paul D. Evans * Personas que no son miembros oficiales del Commiltee on Standard Methods pero realizaron contribuciones a las secciones indicadas. William S. Ewell Samuel D. Faust, 5530 Andrea F. Feagin James C. Feeley, 9213 John M. Ferris, 9010, 9030, 9040, 9050, 9060, 9810 V. R. Ferris, 9810 James V. Feuss, 4500-C1 Duane H. Fickeisen, 2810 Luis Octavio Figueroa Bradford R. Fisher Robert P. Fisher Marvin J. Fishman, 3030, 3110, 3111, 3112, 3113, 3114, 4500-F– Arthur W. Fitchett Ellen P. Flanagan, 9222 Charles Fogle, 2530 Verlin W. Foltz, 4110 Charles T. Ford John A. Fournier Steven P. Fraleigh Martin S. Frant Marlene Frey Clifford Frith, 1080 John L. Fronk Paul L. K. Fu*, 5510 Roger S. Fujioka, 9230 Leo C. Fung, 4500-O3 Joseph T. Fuss, 2810 Anthony M. Gaglierd, 4500-C1 John E. Gannon M. E. Garza, Jr. Anthony F. Gaudy, Jr. Edwin E. Geldreich, 9010, 9030, 9040, 9050, 9060, 9213, 9221, 9222 Stephen R. Gelman, 5210 Cari J. George, 10600 Vincent A. Geraci Michael H. Gerardi Mriganka M. Ghosh Sambhunath Ghosh E. Gilbert, 4500-O3 James M. Gindelberger Thomas S. Gittelman, 6040 xxi William H. Glaze Connie Glover C. Ellen Gonter Reginald K. S. Goo Arley L. Goodenkauf Gilbert Gordon, 45C0-ClO2 y O3 Joseph W. Gorsuch James A. Gouck Joseph P. Gould, 4500-C1 W. O. K. Grabow, 9213 Nancy E. Grams, 5320 Robert L. Graves William B. Gray Philip M. Gschwend Robert J. Gussman, 9020 Rufus K. Guthrie, 9213, 9225 Charles N. Haas, 4500-C1 Earl A. Hadfield, II Stephen W. Hager, 4500-NO3– yNO 2 – Gary E. Hahn Bruce A. Hale, 4110, 4500-CIO2 Nancy H. Hall, 9221, 9260, 9711 David J. Hansen Robert W. Hansen Donald W. Harper David J. Hartman, 5710 Thomas W. Haukebo Steven D. Hawthorne, 10500, 10900 Michael K. Hein, 10300 Charles W. Hendricks, 9225, 9260, 9711 Earl L. Henn, 3030, 3110, 3111, 3112, 3113, 3114 Edwin E. Herricks Delbert Hicks, 10400 Anita K. Highsmith, 1080, 9010, 9030, 9040, 9050, 9060, 9213 * Personas que no son miembros oficiales del Comtnitíee on Standard Methods pero realizaron contribuciones a las secciones indicadas. Albert Hill*, 10400 David R. Hill, 5320 Kenneth M. Hill Phillip A. Hill George D. G. Hilling, 3030, 3110, 3111, 3112, 3113, 3114 George Hoag Laura Mahoney Hodges, 1060 Jimmie A. Hodgeson Robert C Hoehn, 5510, 5710, 9212 Jurg Hoigne, 4500-O3 G. C. Holdren Albert C. Holler, 3030, 3110, 3111, 3112, 3113, 3114 Thomas R. Holm Osmund Holm-Hansen John Homa, Jr, 10600 Thomas B. Hoover, 4110 Peter C. Houle Jack L. Hoyt, 5540 C. P. Huang Wayne B. Huebner, 4500-C1 y C1O2 Donald G. Huggins, 10900 Donald L. Hughes R. DeLon Hull Yung-Tse Hung Joseph V. Hunter Noel M. Hurley Joseph A. Hutchinson, 7010, 7020, 7110, 7500-Cs, I, Ra, Sr, 3H yU Cordelia J. Hwang Verónica Y. Inouye Kurt Irgolic Billy G. Isom, 10500 George Izaguirre, 9250 R. Wayne Jackson, 9222 Walter Jakubowski, 9010, 9030, 9040, 9050, 9060, 9260, 9711 Carol Ruth James Konanur G. Janardan Harían R. Jeche, 9020 S. Rod Jenkins xxii J. Charles Jennett, 3030, 3110, 3111, 3112, 3113, 3114 James N. Jensen J. Michael Jeter Isabel C. Johnson J. Donald Johnson, 4500-C1 Stephen W. Johnson Waynon Johnson Donald L. Johnstone, 9213, 9230 Martha N. Jones, 4500-NO 3 – y NO 2 – Robert A. Jung Swiatoslav W. Kaczmar Sabry M. Kamhawy, 5210 Sandra A. Kaptain Irwin J. Katz, 9250, 9260, 9711 Fred K. Kawahara, 5530 Floyd D. Kefford Michael A. Keirn, 10300 Lawrence H. Keith Nabih P. Kelada William J. Kenney, 5210 Lee G. Kent, 3030, 3110, 3111, 3112, 3113, 3114 Zoltan Kerekes, 9211 Harold W. Kerster Troy Edward King, 1090 Riley N. Kinman Joyce S. Kippin, 9212 Norman A. Kirshen Robert L. Klein, Jr. Donald J. Klemm, 10500, 10900 Margaret M. Knight Bart Koch, 5710 William F. Koch, 4500-F' Frederick C. Kopfler Stuart W. Krasner, 2160, 6040 Richard T. Krause Timothy I. Ladd, 9216 Lawrence E. LaFleur, 5320 Leslie E. Lancy Dennis D. Lañe Russell W. Lañe, 2330 Johan Langewis Alexander Lapteff Richard A. Larson, 5510 Desmond F. Lawler James M. Lazorchak, 10500, 10900 Norman E. LeBlanc Mark LeChevallier, 9212 G. Fred Lee Janet M. Lee Raymond Lee Henry W. Leibee Armond E. Lemke Lawrence Y. C. Leong Raymond D. Letterman Philip A. Lewis Ronald F. Lewis, 9250 Timothy E. Lewis, 3500-A1 Chun-Teh Li Alvin Lieberman Shundar Lin, 9212, 9221 Christopher B. Lind, 9221 Chung-King Liu Larry B. Lobring Stephen R. Lohman, 4500-ClO2 Linda R. Lombardo, 9215 Maxine C. Long, 9225, 9260, 9711, 10200 James E. Longbottom Karl E. Longley, 4500-C1 Marc W. Lorenzen Richard G. Luthy Gerald L. Mahon, 9250 Ronald L. Malcolm, 5510 Joel Mallevialle Bruce E. Manning George Marinenko Leif L. Marking Harold G. Marshall, 10200 Theodore D. Martin, 3120 Margaret Martin-Goldberg Maria T. Martins, 9213 John Y. Masón, 4500-ClO 2 Willy J. Masschelein, 4500-O3 Owen B. Mathre, 3120 xxiii Howard M. May, 3500-A1 Foster L. Mayer Lawrence M. Mayer, 5510 J. Howard McCormick William F. McCoy, 9010, 9030, 9040, 9050, 9060, 9216 Daniel McDaniel Gerald N. McDermott Gordon A. McFeters, 9010, 9030, 9040, 9050, 9060, 9212, 9222 Gerald D. McKee, 5210 James J. McKeown Gerald L. McKinney, 3120 Roy P. McKnight Daniel A. McLean Lilia M. McMillan, 9250 Dale D. McMurtrey Ann Marie McNamara, 1080 Nancy E. McTigue Jay C. Means Robert O. Megard Robert Meglan Morten C. Meilgaard Joseph L. Melnick, 9211 Lori A. Melroy, 1060 Douglas T. Merrill, 2330 Peter L. Meschi Theodore G. Metcalf E. J. Middlebrooks Cari J. Miles, 5510 Arthur M. Miller Frank J. Mulero, 2520 James R. Millette Roger A. Minear, 4500-Br–, 5530 Marc W. Mittelman, 9216 Harold Moehser*, 3500-Se James G. Moncur William J. Montgomery, Jr. Leown A. Moore, 5710 H. Anson Moye * Personas que no son miembros oficiales del Commitíee on Standard Methods pero realizaron contribuciones a las secciones indicadas. Terry I. Mudder James W. Mullins, 7010, 7020, 7110, 7500-Cs, I, Ra, Sr, 3H yU Andrew P. Murray, 10200 Hussein Naimie, 4500-O3 Harry D. Nash, 9020, 9221 Rosemary H. Neal, 3500-Se Stuart Nefft*, 10900 Stanley A. Nichols, 10400 Teresa J. Norberg-King James R. Nugent Gregg Oelker, 3120 Patrick W. O'Keefe Betty H. Olson, 9010, 9030, 9040, 9050, 9060 Cathleen S. O'Neil Joan A. Oppenheimer Edward D. Orme, 4110 John A. Osborne, 10400 Quentin W. Osburn, 5540 Janet G. Osteryoung Edward B. Overton Jeffrey L. Oxenford Arthur T. Palin, 4500-C1 y O3 Sunil P. Pande James M. Pappenhagen Thomas R. Parr, 9225, 9230 Patrick R. Parrish Frances Parsons, 9010, 9030, 9040, 9050, 9060, 9225 Robert A. Paterson, 9610, 10200, 10900 Paul D. Paustian Harry M. Pawlowski, 4500-C1 Richard K. Peddicord Mark E. Peden, 3500-A1 E. Michael Perdue, 5510 Robert K. Pertuit Carol Pesch*, 8510 Deanna L. Peterson, 6040 John Peterson, 9222 William M. Peterson Gary R. Peyton Frederic K. Pfaender JohnD. Pfaff, 4110 Massoud Pirbazari Rodolfo A. Pisigan, Jr., 2330 John T. Pivinski xxiv Marvin D. Piwoni, 3030, 3110, 3111, 3112, 3113, 3114 Russell H. Plumb, Jr. Robert B. Pojasek James M. Polisini William B. Prescott Thomas A. Pressley Fred T. Price Hugh D. Putnam, 10300 James G. Quinn Ánsar A. Qureshi, 9215, 9222, 9230 Neil M. Ram, 5710 Raman K. Raman Steven J. Randtke, 5550 Ronald L. Raschke, 10200, 10400 Inayat A. Rashid Judith A. Rawa Bill T. Ray, 1090 William R. Ray Donald J. Reasoner, 9010, 9030, 9040, 9050, 9060, 9211, 9212 David A. Reckhow, 5320 Kurt A. Reimann, 5540 Martin Reinhard Donald J. Reish, 8510, 10900 Vincent H. Resh, 10900 David J. Rexing Eugene W. Rice, 9221, 9222 George G. Richards, 2330 Harry Ridgway Robert W. Rinehart, Sr. Michele F. Rizet, 3030, 3110, 3111, 3112, 3113, 3114 Morris H. Roberts, Jr., 10500 Andrew Robertson, 10200 Sharon M. Roehm Stephen C. Roesch, 1090 Gary L. Rogers Olsen J. Rogers, 4500-BrPeter F. Rogerson R. R. Romine * Fallecido. † Personas que no son miembros oficiales del Commitiee on Standard Melhods pero realizaron contribuciones a las secciones indicadas. Darrell G. Rose, 3030, 3110, 3111, 3112, 3113, 3114 Bernard Rosenberg, 9225, 9260, 9711 Joel A. Rosenfield William D. Rosenzweig, 9215, 9610 John R. Rossum*, 2330 Richard B. Rubin Peter P. Russell, 2530 Peggy A. Ryker, 9222 William A. Sack, 2520 Robert S. Safferman, 9010, 9030, 9040, 9050, 9060, 9250 Ana M. Sancha Ernest A. Sánchez, 7010, 7020, 7110, 7500-Cs, I, Ra, Sr, 3H yU Rajkamal Sarin Bernard Saunier, 4500-C1 y O, Larry P. Scanlan, 2330 A. David Scarchilli David J. Schaeffer Bernard Schmall John A. Schmitt, 9610 Michael R. Schock, 3500-Li Frank E. Scully, Jr. Bette Seamonds, 1080 Alberta J. Seierstad Kevin G. Sellner†, 10200 Roland L. Seymour, 9610 Joseph Shapiro B. F. Shema, 9260, 9711 Joseph H. Sherrard Marjorie G. Shovlin, 9212, 9225 Peter J. Shuba, 10300, 10500 Mark Shuman Leonard Sideman, 1080 Kenneth Simón, 8010 Philip C. Singer, 5710 J. Edward Singley, 2330 Robert W. Slater, Jr. John P. Slobogin Robert Smith Mark D. Sobsey XXV William C. Sonzogni Charles A. Sorber R. Kent Sorrell David T. Specht Robert L. Spehar Robert G. Spicher Stuart J. Spiegel John B. Sprague Jack R. Stabley, Jr., 3030, 3110, 3111, 3112, 3113, 3114 Vernon T. Stack, 5210 John F. Stafford Alan A. Stevens Raymond M. Stewart Scott Stieg I. H. Suffet, 2160 Makram T. Suidan Bernard F. Sullivan Richard Swartz Robert A. Sweeney, 10200, 10500, 10900 Robert D. Swisher, 5540 James M. Symons John C. Synnott, 4500-NCV Y NO2– Adib F. Tabri Yoshihiro Takahashi, 5320 Lawrence P. Talarico, 5320 Thomas A. Tamayo Mark L. Tamplin, 9260, 9711 Theodore S. Tanaka Donald C. Taylor Raymond H. Taylor, 9215, 9221 Frank Thomas Bruce M. Thomson Earl M. Thurman Robert V. Thurston Edwin C. Tifft, Jr. R. Yucel Tokuz Michael L. Trehy Albert R. Trussell, 6040 * Personas que no son miembros oficiales del Commiitee on Standard Methods pero realizaron contribuciones a las secciones indicadas. León Tsao*, 3500-Se Jack R. Tuschall, 3500-Li, 5510 Ronald E. Twillman, 6040 Mark D. Umphres George S. Uyesugi, 7010, 7020, 7110, 7500-Cs, I, Ra, Sr, 3H yU Michael J. Vandaveer, 3030, 3110, 3111, 3112, 3113, 3114 Schalk W. van der Merwe William H. van der Schalie M. M. Varma Roy M. Ventullo, 9216 P. Aarne Vesilind Hugo T. Victoreen, 9010, 9030, 9040, 9050, 9060, 9215, 9222 Craig O. Vinson Ronald H. Voss Frank F. Wada, 2530 Johannes E. Wajon, 6040 Gerald E. Walsh, 10200 Lawrence K. Wang Mu Hao S. Wang Wuncheng Wang Timothy J. Ward Barron L. Weand David H. Webb, 10400 James H. Webb, 9222 Flora Mae Wellings Oleh Weres, 3500-Se Warren C. Westgarth, 1060, 2160, 9020, 9211 Theodore F. Wetzler, 9221 Willis B. Wheeler Robert E. White James L. Whitlock Greene R. Whitney, 2330 Donald O. Whittemore, 4500-Br– Raymond C. Whittemore, 5210 George P. Whittle Brannon H. Wilder Robert T. Williams Theodore J. Williams, 9020 Kenneth J. Wills, 1060 Frederic J. Winter xxvi John A. Winter, 9020 Charles E. Woelke Roy L. Wolfe Richard E. Wolke, 10600 George T. F. Wong Mark W. Wood Ty R. Woodin Jack Q. Word, 10900 John Wuepper, 2160 * Personas que no son miembros oficiales del Committee on Standard Methods pero realizaron contribuciones a las secciones indicadas. George Yamate In Che Yang, 7010, 7020, 7110, 7500-Cs, I, Ra, Sr, 3H y U Dennis A. Yates Andrew Yee*, 3500-Se Thomas L. Yohe Roger A. Yorton James C. Young, 5210 Michael Young, 9020, 9213 Matthew J. Zabik Stan S. Zaworski Carol A. Ziel, 9211,9213 Melvin C. Zimmerman TABLA C PESOS ATÓMICOS RELATIVOS INTERNACIONALES xxviii TABLA C. PESOS ATÓMICOS RELATIVOS INTERNACIONALES TABLA C. PESOS ATÓMICOS RELATIVOS INTERNACIONALES xxix CONTENIDO PÁGINA Parte 1000 INFORMACIÓN GENERAL 1010 INTRODUCCIÓN ......................................................................... A. Finalidad y aplicación de métodos ............................. B. Estadística....................................................................... C. Glosario .......................................................................... 1020 GARANTÍA DE CALIDAD............................................................ A. Introducción .................................................................. B. Control de calidad......................................................... C. Evaluación de calidad .................................................. 1030 C ALIDAD DE DATOS .................................................................. A. Introducción .................................................................. B. Sesgo ............................................................................ C. Precisión ....................................................................... D. Incertidumbre total ..................................................... E. Límite de detección del método ................................... F. Valoración de la corrección de los análisis .............. 1040 DESARROLLO Y EVALUACIÓN DE MÉTODOS ............................... A. Introducción .................................................................. B. Validación del método .................................................. C. Prueba en colaboración ................................................ 1050 EXPRESIÓN DE RESULTADOS .................................................... A. Unidades ........................................................................ B. Elementos significativos ................................................ 1060 TOMA Y CONSERVACIÓN DE MUESTRAS...................................... A. Introducción .................................................................. B. Toma de muestras ........................................................ C. Conservación de muestras ............................................ 1070 INSTRUMENTAL, REACTIVOS Y TÉCNICAS DE LABORATORIO .. A. Introducción .................................................................. B. Instrumental ................................................................. C. Reactivos ........................................................................ D. Técnicas .......................................................................... 1080 AGUA DE CALIDAD PARA REACTIVOS ........................................ A. Introducción .................................................................. B. Métodos de preparación de agua de calidad para reactivos ..................................................................... C. Calidad del agua para reactivos................................... 1090 SEGURIDAD .............................................................................. A. Introducción .................................................................. B. Equipo de seguridad ................................................... C. Riesgos en el laboratorio ........................................... D. Prácticas de control de riesgos .................................... xxxi 1-1 1-1 1-2 1-4 1-6 1-6 1-7 1-13 1-15 1-15 1-16 1-16 1-18 1-19 1-22 1-24 1-24 1 -24 1-27 1-30 1-30 1-31 1-33 1-33 1-38 1-45 1-47 1-47 1-47 1-49 1-52 1-63 1-63 1-65 1-66 1-68 1-68 1-69 1-73 1-81 CONTENIDO xxxii PÁGINA Parte 2000 2010 2020 2110 2120 2130 2150 2160 2310 2320 2330 2340 2510 2520 PROPIEDADES FÍSICAS Y DE AGREGACIÓN I NTRODUCCIÓN ....................................................................... C ONTROL DE CALIDAD ........................................................ ASPECTO ................................................................................. C OLOR ...................................................................................... A. Introducción ................................................................. B. Método de comparación visual ................................ C. Método espectrofotométrico ....................................... D. Método de filtro triestímulo ....................................... E. Método ADMI de filtro triestimulo (PROPUESTA) T URBIDEZ ............................................................................... A. Introducción ................................................................ B. Método nefelométrico ................................................ OLOR ........................................................................................ A. Introducción ................................................................. B. Prueba de umbral de olor ........................................... G USTO ...................................................................................... A. Introducción ................................................................. B. Prueba de umbral de sabor (PUS) ........................ C. Evaluación de índice de sabor (EIS) ......................... D. Análisis del perfil de sabor (APS) .......................... A CIDEZ ................................................................................... A. Introducción ................................................................. B. Método de titulación .................................. , ................ A LCALINIDAD .......................................................................... A. Introducción ................................................................ B. Método de titulación ................................................... S ATURACIÓN DE CARBONATO CALCICO (PROPUESTA) ... A. Introducción .................................................................. B. índices representativos de la tendencia de un agua a precipitar o disolver el CaCO 3 ........................... C. índices de previsión de la cantidad de CaCO, que puede precipitarse o disolverse .............................. D. Diagramas y códigos de ordenador para los índices de CaCO, .................................................................. D UREZA ................................................................................... A. Introducción .................................................................. B. Cálculo de la dureza ................................................. C. Método titulométrico de EDTA ............................ C ONDUCTIVIDAD ................................................................... A. Introducción ................................................................. B. Método de laboratorio ............................................. S ALINIDAD ............................................................................. A. Introducción ................................................................. 2-1 2-1 2-1 2-2 2-2 2-3 2-5 2-9 2-10 2-12 2-12 2-14 2-18 2-18 2-19 2-26 2-26 2-27 2-30 2-32 2-33 2-33 2-33 2-38 2-38 2-39 2-44 2-44 2-46 2-52 2-53 2-57 2-57 2-57 2-58 2-63 2-63 2-65 2-67 2-67 CONTENIDO xxxiii PÁGINA Parte B. Método de la conductividad eléctrica.......................... C. Método de densidad ................................................... D. Algoritmo de salinidad práctica .................................. 2530 MATERIAS FLOTABLES ................................................................ A. Introducción ................................................................. B. Partículas flotables (GENERAL) ................................ C. Grasas y aceites flotables solubles en triclorotrifluoroetano (GENERAL) ................................................ 2540 SÓLIDOS ................................................................................... A. Introducción ................................................................. B. Sólidos totales secados a 103-105 °C .......................... C. Sólidos totales disueltos secados a 180 °C ................ D. Sólidos totales en suspensión secados a 103-105 °C . E. Sólidos fijos y volátiles incinerados a 550 °C.............. F. Sólidos sedimentables ................................................. G. Sólidos totales, fijos y volátiles en muestras sólidas y semisólidas ............................................................... 2550 TEMPERATURA .......................................................................... A. Introducción ................................................................. B. Métodos de laboratorio y de campo .......................... 2710 PRUEBAS EN LODOS ................................................................. A. Introducción ................................................................. B. Tasa de consumo de oxígeno....................................... C. Volumen de lodo sedimentado .................................... D. índice de volumen de lodo ........................................... E. Tasa de sedimentación de zona .................................. F. Gravedad específica ...................................................... 2720 GAS DIGESTOR DE LODO ......................................................... A. Introducción ................................................................. B. Método volumétrico .................................................. C. Método cromatografía) de gases ................................ 2810 SOBRESATURACIÓN DE GAS DISUELTO .................................... A. Introducción ................................................................. B. Método de difusión de membrana directa ................ 2-68 2-70 2-71 2-72 2-72 2-72 2-87 2-88 2-88 2-89 2-90 2-90 2-90 2-92 2-93 2-94 2-96 2-97 2-97 2-98 2-101 2-104 2-104 2-105 3000 DETERMINACIÓN DE METALES 3010 INTRODUCCIÓN ......................................................................... A. Discusión general ........................................................... B. Toma y conservación de muestras ............................... C. Precauciones generales .................................................. 3020 CONTROL DE CALIDAD .......................................................... 3030 TRATAMIENTO PRELIMINAR DE MUESTRAS ............................... A. Introducción .................................................................. B. Filtración preliminar ..................................................... 3-1 3-1 3-2 3-3 3-4 3-5 3-5 3-6 2-76 2-78 2-78 2-80 2-81 2-83 2-85 2-86 xxxiv CONTENIDO PÁGINA C. Tratamiento preliminar de metales extraíbles por ácido ............................................................................ D. Digestión preliminar de metales ................................... E. Digestión por ácido nítrico ........................................... F. Digestión por ácido nítrico-ácido clorhídrico ......... G. Digestión por ácido nítrico-ácido sulfúrico ............... H. Digestión por ácido nítrico-ácido perclórico .............. I. Digestión por ácido perclórico-ácido fluorhídrico . . . J. Combustión seca ......................................................... 3110 DETERMINACIÓN DE METALES POR ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA ................................................................. 3111 DETERMINACIÓN DE METALES POR ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA DE LLAMA ..................................................... A. Introducción .................................................................. B. Método directo de llama de aire-acetileno .................. C. Método de extracción/llama de aire-acetileno ............ D. Método directo de llama de óxido nitroso-acetileno E. Método de extracción/llama de óxido nitroso-acetileno ......................................................................... 3112 DETERMINACIÓN DE METALES POR ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA DE VAPOR FRÍO.................................................. A. Introducción ................................................................. B. Método espectrometría) de absorción atómica de vapor frío..................................................................... 3113 DETERMINACIÓN DE METALES POR ESPECTROMETRÍA DE ABSORCIÓNATÓMICAELECTROTÉRMICA ................................................ A. Introducción ................................................................... B. Método espectrométrico de absorción atómica electrotérmica .................................................................. 3114 DETERMINACIÓN DE METALES POR GENERACIÓN DE HIDRUROS/ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA .................... A. Introducción ................................................................... B. Método manual de generación de hidruros/espectrometría de absorción atómica ................................. C. Método continuo de generación de hidruros/espectrometría de absorción atómica (PROPUESTA) . ............... 3120 DETERMINACIÓN DE METALES POR ESPECTROSCOPIA DE EMISIÓN DE PLASMA .................................................................................. A. Introducción .................................................................. B. Método de plasma de acoplamiento inductivo (PAI) 3-59 3500-A1 ALUMINIO ............................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ 3-7 3-7 3-9 3-9 3-10 3-11 3-12 3-12 3-13 3-14 3-14 3-21 3-25 3-27 3-30 3-31 3-31 3-32 3-35 3-35 3-39 3-47 3-47 3-48 3-56 3-59 3-59 3-70 3-70 3-70 3-70 xxxv CONTENIDO PÁGINA D. E. Método de eriocromo cianina R ................................ Método automatizado de violeta de pirocatecol (VPC) (PROPUESTA) .............................................. 3500-Sb ANTIMONIO ........................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica.......... C. Método de plasma de acoplamiento inductivo ......... 3500-As ARSÉNICO ............................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica.......... C. Método del dietilditiocarbamato de plata ................ D. Método del tinte de bromuro mercúrico .................. E. Método de plasma de acoplamiento inductivo ......... 3500-Ba BARIO .................................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica.......... C. Método de plasma de acoplamiento inductivo ......... 3500-Be BERILIO .................................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica.......... C. Método de plasma de acoplamiento inductivo ......... D. Método del aluminen ................................................. 3500-Bi BISMUTO ............................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica .......... 3500-Cd CADMIO ............................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica .......... C. Método de plasma de acoplamiento inductivo ......... D. Método de la ditizona................................................... 3500-Ca CALCIO ................................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica .......... C. Método de plasma de acoplamiento inductivo ......... D. Método titulométrico de EDTA ............................... E. Método titulométrico de permanganato ..................... 3500-Cs CESIO ..................................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica .......... 3500-Cr CROMO................................................................................... A. Introducción .................................................................. B. Método de absorción atómica para el cromo total . C. Método de plasma de acoplamiento inductivo ......... D. Método colorimétrico .................................................. 3-71 3-75 3-81 3-81 3-81 3-82 3-82 3-82 3-82 3-83 3-85 3-87 3-87 3-87 3-87 3-87 3-88 3-88 3-88 3-88 3-89 3-91 3-91 3-91 3-91 3-91 3-92 3-92 3-92 3-95 3-95 3-95 3-95 3-96 3-98 3-100 3-100 3-101 3-101 3-101 3-102 3-102 3-102 xxxvi CONTENIDO PÁGINA 3500-Co COBALTO ............................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica.......... C. Método de plasma de acoplamiento inductivo ........ 3500-Cu COBRE .................................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ D. Método de la neocuproína ........................................... E. Método de la batocuproína ........................................ 3500-Au ORO ...................................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... 3500-Ir IRIDIO ...................................................................................... A. Introducción ................................................................. B. Método espectrométrico de absorción atómica ......... 3500-Fe HIERRO.................................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ D. Método de fenantrolina.......................................... 3500-Pb PLOMO .................................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ D. Método de la ditizona ................................................... 3500-Li LITIO ..................................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ D. Método fotométrico de emisión de llama................... 3500-Mg MAGNESIO ........................................................................... A. Introducción ................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ D. Método gravimétrico ..................................................... E. Método de cálculo ......................................................... 3500-Mn MANGANESO ....................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ D. Método del persulfato ................................................... 3500-Hg MERCURIO ............................................................................ A. Introducción .................................................................. 3-105 3-105 3-106 3-106 3-106 3-106 3-107 3-107 3-107 3-110 3-111 3-111 3-112 3-112 3-112 3-112 3-112 3-112 3-114 3-114 3-115 3-120 3-120 3-120 3-120 3-121 3-124 3-124 3-124 3-.124 3-125 3-127 3-127 3-127 3-127 3-128 3-129 3-130 3-130 3-131 3-131 3-131 3-134 3-134 CONTENIDO xxxvii PÁGINA B. Método de absorción atómica de vapor frío .............. C. Método de la ditizona................................................... 3500-Mo MOLIBDENO ........................................................................ A. Introducción .................................................................. B. Método espectrométrico de absorción atómica.......... C. Método de plasma de acoplamiento inductivo ......... 3500-Ni NÍQUEL ................................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica .......... C. Método de fuente de plasma de acoplamiento inductivo ............................................................................. D. Método de la heptoxima (GENERAL) ....................... E. Método de la dimetilglioxima (GENERAL) ........... 3500-Os OSMIO ................................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica.......... 3500-Pd PALADIO .............................................................................. A. Introducción ................................................................. B. Método espectrométrico de absorción atómica.......... 3500-Pt PLATINO .............................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica.......... 3500-K POTASIO ................................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica .......... C. Método de plasma de acoplamiento inductivo ......... D. Método fotométrico de llama....................................... 3500-Re RENIO .................................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica .......... 3500-Rh RODIO ................................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica.......... 3500-Ru RUTENIO ............................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica.......... 3500-Se SELENIO ................................................................................. A. Introducción .................................................................. B. Preparación de la muestra ......................................... C. Método continuo de generación de hidruros/espectrometría de absorción atómica............................... D. Método colorimétrico .................................................. E. Método fluorométrico .................................................. 3-135 3-135 3-137 3-137 3-137 3-137 3-138 3-138 3-138 3-138 3-138 3-140 3-141 3-141 3-141 3-141 3-141 3-141 3-142 3-142 3-142 3-142 3-142 3-143 3-143 3-143 3-144 3-144 3-144 3-145 3-145 3-145 3-145 3-145 3-145 3-146 3-146 3-149 3-153 3-154 3-156 xxxviii CONTENIDO PÁGINA F. G. Determinación del selenio volátil................................. Determinación de compuestos orgánicos de selenio no volátiles ................................................................ H. Método de espectrometría de absorción atómica electrotérmica ................................................................. I. Método de plasma de acoplamiento inductivo ........ 3500-Ag PLATA ................................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ D. Método de la ditizona................................................... 3500-Na SODIO ...................................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ D. Método fotométrico de emisión de llama ................... 3500-Sr ESTRONCIO .............................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ D. Método fotométrico de emisión de llama................... 3500-TI TALIO......................................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ 3500-Th TORIO ...................................................................................... A. Introducción ................................................................. B. Método espectrométrico de absorción atómica ......... 3500-Sn ESTAÑO .................................................................................... A. Introducción ................................................................. B. Método espectrométrico de absorción atómica ......... 3500-Ti TITANIO .................................................................................. A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... 3500-V VANADIO ................................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ D. Método del ácido gálico .............................................. 3500-Zn ZINC .................................................................................... A. Introducción .................................................................. B. Método espectrométrico de absorción atómica ......... C. Método de plasma de acoplamiento inductivo ........ 3-157 3-159 3-161 3-162 3-162 3-162 3-162 3-163 3-163 3-166 3-166 3-167 3-167 3-167 3-171 3-171 3-172 3-172 3-172 3-174 3-174 3-174 3-175 3-175 3-175 3-175 3-175 3-175 3-176 3-176 3-176 3-176 3-177 3-177 3-177 3-177 3-178 3-180 3-180 3-180 3-181 CONTENIDO xxxix PÁGINA D. E. F. Parte Método I de la ditizona .............................................. Método II de la ditizona ........................................... Método del zincón ......................................................... 4000 DETERMINACIÓN DE CONSTITUYENTES INORGÁNICOS NO METÁLICOS 4010 I NTRODUCCIÓN ....................................................................... 4020 CONTROL DE CALIDAD ........................................................... 4110 DETERMINACIÓN DE ANIONES MEDIANTE CROMATOGRAFÍA DE IONES ............................................................................... A. Introducción .................................................................. B. Cromatografía de iones con supresión química de la conductividad del disolvente .................................. 4500-B BORO..................................................................................... A. Introducción .................................................................. B. Método de la curcumina ............................................... C. Método del carmín ........................................................ D. Método de plasma de acoplamiento inductivo .......... 4500-Br– BROMURO ......................................................................... A. Introducción .................................................................. B. Método colorimétrico del rojo de fenol .................... C. Método cromatografía) de iones.................................. 4500-CO2 DIÓXIDO DE CARBONO ..................................................... A. Introducción .................................................................. B. Determinación nomográfica de dióxido de carbono libre y de las tres formas de alcalinidad ................. C. Método titulométrico para el dióxido de carbono libre ........................................................................... D. Dióxido de carbono y formas de alcalinidad mediante cálculo ........................................................................ 4500-CN CIANURO ........................................................................... A. Introducción .................................................................. B. Tratamiento preliminar de muestras ............................ C. Cianuro total después de destilación ........................... D. Método titulométrico..................................................... E. Método colorimétrico .................................................. F. Método del electrodo cianuro-selectivo .................... G. Cianuros susceptibles de cloración después de destilación ........................................................................... H. Cianuros susceptibles de cloración sin destilación (método del atajo) .................................................... I. Cianuro débil y disociable ........................................ J. Cloruro de cianógeno ................................................. 3-181 3-184 3-186 4-1 4-1 4-2 4-2 4-2 4-7 4-7 4-8 4-11 4-12 4-12 4-12 4-13 4-14 4-15 4-15 4-16 4-21 4-22 4-23 4-23 4-28 4-31 4-33 4-35 4-37 4-38 4-40 4-43 4-44 xl CONTENIDO PÁGINA K. Ensayo al toque para selección de muestras ........... L. Cianatos................................................................... M. Tiocianato................................................................ 4500-Cl CLORO (RESIDUAL) ................................................. A. Introducción ............................................................ B. Método yodométrico I .......................................... C. Método yodométrico II ............................................ D. Método amperométrico de titulación ....................... E. Método amperométrico de titulación de bajo nivel . F. Método titulométrico de la DFD ferrosa................. G. Método colorimétrico de la DFD ............................ H. Método de la siringaldacina (PCLD) ...................... I. Técnica yodométrica de electrodo ............................ _ 4500-Cl CLORURO ................................................................... A. Introducción ............................................................ B. Método argentométrico............................................ C. Método del nitrato mercúrico ............................... D. Método potenciométrico........................................... E. Método automatizado del ferrocianuro .................. F. Método de cromatografía de iones ......................... 4500-ClO2 DIÓXIDO DE CLORO ................. ............................... A. Introducción ............................................................ B. Método yodométrico................................................ C. Método amperométrico I ........................................ D. Método de la DFD ................................................. E. Método amperométrico II (PROPUESTA)............... 4500-F– FLUORURO ................................................................ A. Introducción ............................................................ B. Paso de destilación previa ..................................... C. Método de electrodo selectivo de iones .................. D. Método del SFADNS ............................................. E. Método de la complexona ..................................... 4500-H+ VALOR DE PH.............................................................. A. Introducción ............................................................ B. Método electrométrico ............................................. 4500-I– YODO ............................................................................ A. Introducción ............................................................ B. Método del violeta leuco cristal ............................... C. Método amperométrico de titulación ....................... 4500-I– YODURO..................................................................... A. Introducción ............................................................ B. Método del violeta leuco cristal................................ C. Método de reducción catalítica ............................... 4-45 4-46 4-48 4-51 4-51 4-55 4-57 4-61 4-65 4-66 4-70 4-72 4-74 4-76 4-76 4-77 4-78 4-80 4-83 4-85 4-85 4-85 4-86 4-88 4-90 4-91 4-95 4-95 4-97 4-99 4-102 4-104 4-106 4-106 4-107 4-116 4-116 4-117 4-119 4-120 4-120 4-120 4-123 CONTENIDO xli PÁGINA 4500-N NITRÓGENO .............................................................................. 4500-NH3 NITRÓGENO (AMONÍACO) ................................................... A. Introducción .................................................................. B. Paso previo de destilación ......................................... C. Método de la nesslerización (directo y subsiguiente a destilación) ............................................................... D. Método de la sal de fenol ............................................. E. Método titulométrico .................................................... F. Método de electrodo selectivo de amoníaco ............ G. Método de electrodo selectivo de amoníaco con adición conocida ............................................................ H. Método automatizado de la sal de fenol .................. 4-125 4-126 4-126 4-129 4-132 4-136 4-137 4-138 4-140 4-143 4500-NO2– NITRÓGENO (NITRITO) ...................................................... A. Introducción .................................................................. B. Método colorimétrico ................................................. C. Método cromatográfico de iones ................................. 4500-NO3– NITRÓGENO (NITRATO) ...................................................... A. Introducción .................................................................. B. Método espectrométrico ultravioleta selectivo ........... C. Método cromatográfico de iones ................................. D. Método del electrodo de nitrato ................................ E. Método de reducción de cadmio ................................. F. Método automatizado de reducción de cadmio . . . . G. Método de reducción de cloruro titanoso (PROPUESTA) ................................................................. H. Método automatizado de reducción de hidracina (PROPUESTA) ........................................................ 4-145 4-145 4-146 4-148 4-149 4-149 4-149 4-151 4-151 4-153 4-156 4500-Norg NITRÓGENO (ORGÁNICO) ....................................................... A. Introducción ................................................................... B. Método macro-kjeldahl................................................. C. Método semi-micro-kjeldahl ......................................... 4-162 4-162 4-163 4-166 4500-O O XÍGENO (DISUELTO ) ................................................................ A. Introducción .................................................................. B. Métodos yodométricos ................................................ C. Modificación de azida ................................................... D. Modificación de permanganato ................................... E. Modificación de la floculación de alumbre .............. F. Modificación de la floculación de la combinación sulfato de cobre-ácido sulfámico ............................... G. Método de electrodo de membrana .......................... 4-168 4-168 4-169 4-172 4-177 4-178 4500-O3 OZONO (RESIDUAL) (PROPUESTA) .............................. A. Introducción .................................................................. B. Método colorimétrico de índigo ............................... 4-183 4-183 4-183 4-158 4-159 4-178 4-179 xlii CONTENIDO PÁGINA 4500-P FÓSFORO ...................................................................... A. Introducción ............................................................ B. Preparación de muestras .......................................... C. Método colorimétrico del ácido vanadomolibdofosfórico....................................................................... D. Método del cloruro estagnoso ................................. E. Método del ácido ascórbico ..................................... F. Método automatizado de reducción del ácido ascórbico .................................................................... 4500-Si SÍLICE ........................................................................... A. Introducción ............................................................ B. Método espectrométrico de absorción atómica ........ C. Método gravimétrico................................................ D. Método del molibdosilicato .................................... E. Método del azul heteropoli ................................... F. Método automatizado para sílice molibdatorreactiva G. Método de plasma de acoplamiento inductivo ....... 4500-S2– SULFURO .................................................................... A. Introducción ............................................................ B. Separación de sulfuros solubles e insolubles .......... C. Pretratamiento de muestras para eliminar sustancias que puedan interferir o para concentrar el sulfuro D. Método del azul de metileno .................................. E. Método yodométrico................................................ F. Cálculo del sulfuro de hidrógeno desionizado........... 4500 - SO32- SULFITO ................................................................. A. Introducción ............................................................ B. Método yodométrico................................................ C. Método de fenantrolina (PROPUESTA) ................ 4500 - SO 2SULFATO ............................................................... 4 A. Introducción ............................................................ B. Método cromatográfico de iones .............................. C. Método gravimétrico con combustión de residuos . . D. Método gravimétrico con secado de residuos ......... E. Método turbidimétrico .......................................... F. Método automatizado del azul de metiltimol ......... Parte 5000 DETERMINACIÓN DE COMPONENTES ORGÁNICOS 5010 INTRODUCCIÓN .................................................................... A. Discusión general...................................................... B. Toma y conservación de muestras............................ 4-187 4-187 4-191 4-195 4-197 4-199 4-201 4-204 4-204 4-206 4-206 4-208 4-211 4-213 4-215 4-215 4-215 4-218 4-219 4-220 4-222 4-223 4-224 4-224 4-225 4-226 4-229 4-229 4-230 4-230 4-232 4-233 4-235 5-1 5-1 5-1 CONTENIDO xliii PÁGINA 5020 C ONTROL DE CALIDAD ........................................................... 5210 REQUERIMIENTO DE OXÍGENO BIOQUÍMICO ............................... A. Introducción .................................................................. B. Prueba ROB de 5 días .............................................. 5220 REQUERIMIENTO DE OXÍGENO QUÍMICO (ROQ)........................ A. Introducción .................................................................. B. Método de reflujo abierto............................................. C. Reflujo cerrado, método titulométrico ..................... D. Reflujo cerrado, método colorimétrico ...................... 5310 CARBONO ORGÁNICO TOTAL (COT) ........................................ A. Introducción .................................................................. B. Método de combustión-infrarrojo .............................. C. Método de oxidación persulfato-ultravioleta ............. D. Método de oxidación húmeda .................................... 5320 HALÓGENO ORGÁNICO DISUELTO ........................................... A. Introducción ................................................................. B. Método titulométrico de absorción-pirólisis ........... 5510 SUSTANCIAS ACUOSAS HÚMICAS (PROPUESTA) ................... A. Introducción ................................................................. B. Método del dietilaminoetil (DEAE) ............................ C. Método XAD ................................................................. 5520 ACEITE Y GRASA ....................................................................... A. Introducción ................................................................. B. Método de partición-gravimetría ................................ C. Método de partición-infrarrojo ................................ D. Método de extracción de Soxhlet ............................ E. Método de extracción para muestras de lodo............ F. Hidrocarburos................................................................. 5530 FENOLES ................................................................................. A. Introducción .................................................................. B. Procedimiento de limpiado ........................................... C. Método de extracción de cloroformo ........................ D. Método fotométrico directo ......................................... 5540 SURFACTANTES ......................................................................... A. Introducción .................................................................. B. Separación del surfactante por cancelación ................ C. Surfactantes amónicos como SAAM ............................ D. Surfactantes no iónicos como SATC ........................... 5550 TANINO Y LIGNINA .................................................................. A. Introducción .................................................................. B. Método colorimétrico .................................................. 5560 ÁCIDOS ORGÁNICOS Y VOLÁTILES ................................................ A. Introducción .................................................................. 5-2 5-2 5-2 5-4 5-12 5-12 5-14 5-17 5-19 5-20 5-20 5-22 5-26 5-29 5-31 5-31 5-32 5-43 5-43 5-44 5-46 5-48 5-48 5-49 5-51 5-52 5-54 5-55 5-56 5-56 5-58 5-59 5-62 5-63 5-63 5-64 5-69 5-74 5-78 5-78 5-78 5-80 5-80 xliv CONTENIDO PÁGINA B. Método de separación cromatográfica para ácidos orgánicos..................................................................... C. Método de destilación ................................................... 5710 FORMACIÓN DE TRIHALOMETANO (PROPUESTA).................. A. Introducción .................................................................. B. Potencial de formación de trihalometano (PFT) . . . . C. Potencial básico de formación de trihalometano (PBFT) ........................................................................ D. Potencial final de formación de trihalometano (PFFT) ........................................................................ E. Concentración de trihalometano en sistemas de distribución simulados (CTHMSDS)............................ Parte 6000 ANÁLISIS AUTOMATIZADO DE LABORATORIO 6010 INTRODUCCIÓN ......................................................................... A. Discusión general........................................................... B. Toma y conservación de muestras .............................. C. Métodos analíticos ........................................................ 6040 C ONCENTRACIÓN DE COMPONENTES POR EXTRACCIÓN DE 5-81 5-83 5-85 5-85 5-86 5-91 5-93 5-94 6-1 6-1 6-4 6-6 ........................................................................................ 6-11 Introducción .................................................................. Depuración de ciclo cerrado, análisis por cromatografía de gases/espectrometría de masas ................ C. Técnica de purga y atrapamiento .............................. 6210 COMPUESTOS ORGÁNICOS VOLÁTILES ........................................ A. Introducción ................................................................... B. Método I de purga y atrapamiento con cromatografía de gases en columna de relleno/espectrometría de masas .................................................................... C. Método II de purga y atrapamiento con cromatografía de gases en columna de relleno/espectrometría de masas .................................................................... D. Método de purga y atrapamiento con cromatografía de gases en columna capilar/espectrometría de masas ........................................................................... 6211 METANO ................................................................................... A. Introducción ................................................................... B. Método indicador de gas combustible ...................... C. Método volumétrico ................................................... 6220 COMPUESTOS ORGÁNICOS AROMÁTICOS VOLÁTILES ................. A. Introducción .................................................................. B. Método I de purga y atrapamiento con cromatografía de gases ................................................................. 6-11 GAS A. B. 6-12 6-28 6-29 6-29 6-30 6-47 6-56 6-65 6-65 6-66 6-69 6-70 6-70 6-71 CONTENIDO xlv PÁGINA C. 6230 6321 6232 6410 6420 Método II de purga y atrapamiento con cromatografía de gases ................................................................ D. Método de purga y atrapamiento con cromatografía de gases/espectrometría de masas ........................... E. Método de extracción líquido-líquido con cromatografía de gases/espectrometría de masas ................. HALOCARBUROS VOLÁTILES ...................................................... A. Introducción .................................................................. B. Método I de purga y atrapamiento con cromatografía de gases en columna de relleno ........................ C. Método II de purga y atrapamiento con cromatografía de gases en columna de relleno ........................ D. Método de purga y atrapamiento con cromatografía de gases en columna capilar ..................................... E. Método de purga y atrapamiento con cromatografía de gases/espectrometría de masas .......................... 1,2-DIBROMOETANO (DBE) Y 1,2-DIBROMO-3-CLOROPROPANO (DBCP) ...................................................................... A. Introducción .................................................................. B. Método de extracción líquido-líquido con cromatografía de gases ......................................................... C. Método de purga y atrapamiento con cromatografía de gases/espectrometría de masas ........................... D. Método de purga y atrapamiento con cromatografía de gases ............................................................................. TRIHALOMETANOS ..................................................................... A. Introducción .................................................................. B. Método de extracción líquido-líquido con cromatografía de gases .......................................................... C. Método de purga y atrapamiento con cromatografía de gases/espectrometría de masas ........................... D. Método de purga y atrapamiento con cromatografía de gases....................................................................... AGENTES BÁSICOS-NEUTROS Y ÁCIDOS EXTRAÍBLES .................. A. Introducción .................................................................. B. Método de extracción líquido-líquido con cromatografía de gases/espectrometría de masas ................. FENOLES .................................................................................. A. Introducción .................................................................. B. Método de extracción líquido-líquido con cromatografía de gases ......................................................... C. Método de extracción líquido-líquido con cromatografía de gases/espectrometría de masas ................. 6-77 6-85 6-85 6-86 6-86 6-87 6-95 6-101 6-106 6-107 6-107 6-107 6-112 6-113 6-113 6-113 6-114 6-123 6-123 6-123 6-123 6-124 6-147 6-147 6-148 6-158 xlvi CONTENIDO PÁGINA 6431 BIFENILOS POLICLORADOS ............................................................ 6-158 Introducción .................................................................. Método de extracción líquido-líquido con cromatografía de gases ........................................................ C. Método de extracción líquido-líquido con cromatografía de gases/espectrometría de masas ................. 6440 HIDROCARBUROS POLINUCLEARES AROMÁTICOS .................... A. Introducción .................................................................. B. Método de extracción líquido-líquido con cromatografía de gases ......................................................... C. Método de extracción líquido-líquido con cromatografía de gases/espectrometría de masas ................. 6630 PESTICIDAS ORGANOCLORADOS ................................................. A. Introducción .................................................................. B. Método I de extracción líquido-líquido con cromatografía de gases ......................................................... C. Método II de extracción líquido-líquido con cromatografía de gases ........................................................ D. Método de extracción líquido-líquido con cromatografía de gases/espectrometría de masas ................. 6640 HERBICIDAS CLORADOS FENOXI ÁCIDOS ................................... A. Introducción .................................................................. B. Método de extracción líquido-líquido con cromatografía de gases ......................................................... 6-158 A. B. Parte 7000 EXAMEN DE LA RADIACTIVIDAD DE AGUAS LIMPIAS Y RESIDUALES 7010 INTRODUCCIÓN ......................................................................... A. Discusión general............................................................ B. Toma y conservación de muestras ............................... C. Cámara de conteo ........................................................ D. Instrumentos de conteo ................................................. E. Reactivos e instrumental de laboratorio ..................... F. Expresión de resultados ................................................. G. Estadísticas .................................................................... 7020 GARANTÍA DE CALIDAD ............................................................ A. Programa básico de garantía de calidad ................. B. Garantía de calidad para muestras de aguas residuales .......................................................................... 7110 RADIACTIVIDADES BRUTAS ALFA Y BETA (TOTALES, EN SUSPENSIÓN Y EN DISOLUCIÓN ) ............................................... A. Introducción .......................................... ,...................... B. Método de conteo ........................................................ 6-159 6-159 6-159 6-159 6-160 6-171 6-171 6-171 6-171 6-185 6-197 6-198 6-198 6-198 7-1 7-1 7-3 7-5 7-5 7-13 7-14 7-14 7-16 7-16 7-17 7-18 7-18 7-19 CONTENIDO xlvii PÁGINA 7500-Cs CESIO RADIACTIVO .................................................................. A. Introducción ................................................................. B. Método de precipitación .............................................. 7500-I YODO RADIACTICO .................................................................. A. Introducción ................................................................. B. Método de precipitación .............................................. C. Método de intercambio iónico..................................... D. Método de destilación .................................................. 7500-Ra RADIO .................................................................................... A. Introducción .................................................................. B. Método de precipitación............................................... C. Método de emanación .................................................. D. Método secuencial de precipitación (PROPUESTA) . 7500-Sr ESTRONCIO RADIACTIVO TOTAL Y ESTRONCIO 90 ................. A. Introducción .................................................................. B. Método de precipitación............................................... 7500-3H TRITIO ..................................................................................... A. Introducción .................................................................. B. Método espectrométrico de centelleo líquido ............. 7500-U URANIO . .................................................................................. A. Introducción ................................................................. B. Método radioquímico (PROPUESTA) ....................... C. Método fluorométrico (PROPUESTA)....................... Parte 8000 MÉTODOS DE PRUEBA DE TOXICIDAD PARA ORGANISMOS ACUÁTICOS 8010 INTRODUCCIÓN ........................................................................ A. Discusión general........................................................... B. Terminología .................................................................. C. Requisitos básicos para las pruebas de toxicidad . . . D. Desarrollo de pruebas de toxicidad ............................ E. Preparación de los organismos para las pruebas de toxicidad ..................................................................... F. Sistemas, materiales y procedimientos en pruebas de toxicidad .................................................................... G. Cálculo, análisis y comunicación de resultados de pruebas de toxicidad ................................................ H. Interpretación y aplicación de resultados de pruebas de toxicidad ................................................................ 8030 MUTAGENICIDAD ..................................................................... A. Introducción .................................................................. B. Prueba de la Salmonella .............................................. 7-25 7-25 7-25 7-28 7-28 7-28 7-30 7-32 7-34 7.34 7-35 7-40 7-52 7-56 7-56 7-57 7-64 7-64 7-64 7-67 7-67 7-68 7-70 8-1 8-1 8-3 8-5 8-6 8-12 8-27 8-39 8-45 8-48 8-48 8-48 xlviii CONTENIDO PÁGINA 8110 PROCEDIMIENTOS DE PRUEBA PARA ALGAS .............................. 8111 BLOESTIMULACIÓN (PRODUCTIVIDAD ALGAL) ........................... A. Principios generales ...................................................... B. Planificación y evaluación de ensayos en algas .......... C. Instrumental .................................................................. D. Manipulación de muestras .......................................... E. Medio de cultivo sintético para algas .......................... F. Inoculo............................................................................. G. Condiciones y procedimientos de prueba.................... H. Efectos de las adiciones ................................................. I. Análisis e interpretación de datos .............................. 8112 PRUEBAS DE TOXICIDAD EN FITOPLANCTON ........................... A. Introducción ................................................................... B. Inoculo............................................................................. C. Condiciones y procedimientos de prueba.................... 8310 PROCEDIMIENTOS DE PRUEBA DE TOXICIDAD EN PROTOZOOS CILIADOS ................................................................................ A. Introducción ................................................................... B. Selección y preparación de organismos de ensayo . . C. Procedimientos de prueba de toxicidad ..................... D. Evaluación y comunicación de resultados ................. 8410 PROCEDIMIENTOS DE PRUEBA DE TOXICIDAD CON CORAL ESCLERACTINIANO .................................................................. A. Introducción .................................................................. B. Selección y preparación de organismos de ensayo . . C. Procedimientos de prueba de toxicidad ..................... D. Evaluación y comunicación de resultados ................ 8510 PROCEDIMIENTOS DE PRUEBA DE TOXICIDAD PARA ANÉLIDOS A. Introducción .................................................................. B. Selección y preparación de organismos de ensayo .. C. Procedimientos de prueba de toxicidad ..................... D. Evaluación de datos ...................................................... 8610 PROCEDIMIENTOS DE PRUEBA DE TOXICIDAD CON MOLUSCOS A. Introducción .................................................................. B. Selección y preparación de organismos de prueba . . C. Desarrollo de las pruebas de toxicidad....................... D. Comunicación y análisis de resultados ....................... 8710 MICROCRUSTÁCEOS ................................................................... 8711 PROCEDIMIENTOS DE PRUEBA DE TOXICIDAD PARA DAPHNIA A. Introducción .................................................................. B. Selección y preparación de organismos de prueba . . C. Procedimientos de ensayo de toxicidad ................... D. Evaluación y comunicación de resultados ................ 8-49 8-49 8-49 8-50 8-51 8-52 8-53 8-54 8-55 8-59 8-60 8-61 8-61 8-61 8-61 8-63 8-63 8-64 8-65 8-66 8-66 8-66 8-67 8-71 8-74 8-76 8-76 8-76 8-81 8-85 8-85 8-85 8-86 8-89 8-94 8-94 8-94 8-94 8-95 8-97 8-98 CONTENIDO xlix PÁGINA 8712 PROCEDIMIENTOS DE PRUEBA DE TOXICIDAD PARA EL COPÉPODO CALANOIDE ACARTIA TONSA (DANA) ............................. A. Introducción ................................................................. B. Selección y preparación de organismos de ensayo .. C. Procedimientos de prueba de toxicidad .................... D. Evaluación e información sobre resultados .............. 8720 PROCEDIMIENTOS DE ENSAYOS DE TOXICIDAD PARA MACROCRUSTÁCEOS ......................................................................... A. Introducción .................................................................. B. Selección y preparación de especies de prueba ......... C. Desarrollo de pruebas de toxicidad............................. D. Comunicación de resultados......................................... 8750 PROCEDIMIENTOS DE ENSAYOS DE TOXICIDAD PARA INSECTOS ACUÁTICOS .......................................................................... A. Introducción ................................................................. B. Selección y preparación de organismos de prueba .. C. Procedimientos de prueba de toxicidad ..................... D. Evaluación de datos...................................................... 8910 PROCEDIMIENTOS DE PRUEBA DE TOXICIDAD PARA PECES .. A. Introducción .................................................................. B. Selección y preparación de peces ................................. C. Procedimientos de prueba ............................................ Parte 9000 EXAMEN MICROBIOLÓGICO DE LAS AGUAS 9010 INTRODUCCIÓN ........................................................................ A. Discusión general ........................................................... B, Disposiciones de la EPA de Estados Unidos sobre calidad del agua potable........................................... 9020 GARANTÍA DE CALIDAD ........................................................... A. Introducción .................................................................. B. Directrices para el control de calidad intralaboratorio C. Control de calidad interlaboratorios........................... 9030 INSTRUMENTAL DE LABORATORIO ............................................. A. Introducción .................................................................. B. Especificaciones del equipo .......................................... 9040 LAVADO Y ESTERILIZACIÓN ...................................................... 9050 PREPARACIÓN DE MEDIOS DE CULTIVO .................................... A. Procedimientos generales ............................................ B. Agua................................................................................ C. Especificaciones de los medios ..................................... 9060 MUESTRAS ................................................................................ A. Recogida ....................................................................... B. Conservación y almacenamiento ................................. 8-99 8-99 8-99 8-105 8-105 8-106 8-106 8-106 8-120 8-132 8-132 8-132 8-133 8-136 8-139 8-140 8-140 8-140 8-155 9-1 9-1 9-3 9-5 9-5 9-5 9-27 9-29 9-29 9-29 9-34 9-34 9-34 9-36 9-37 9-38 9-38 9-42 I CONTENIDO PÁGINA 9211 9212 9213 9215 9216 MÉTODOS DE DETECCIÓN RÁPIDA .................................................. 9-43 A. B. 9-43 Introducción .................................................................. Prueba de coliformes fecales en siete fases (ESPECIALIZADA) ................................................................... C. Técnicas especiales (ESPECIALIZADAS) ................ D. Detección de colífagos (PROPUESTA) ................... ORGANISMOS BN CONDICIONES ANÓMALAS ............................ A. Introducción .................................................................. B. Incremento de la recuperación ..................................... EXAMEN MICROBIOLÓGICO DE AGUAS EN INSTALACIONES RECREATIVAS ............................................................................ A. Introducción .................................................................. B. Piscinas ......................................................................... C. Baños de hidromasaje .................................................. D. Playas .............................................................................. E. Técnica de filtro de membrana para Pseudomonas aeruginosa .................................................................. F. Técnica de tubos múltiples para Pseudomonas aeruginosa ........................................................................... RECUENTO HETERÓTROFO DE PLACA ...................................... A. Introducción .................................................................. B. Método de placa fluida................................................. C. Método de placa difusa ................................................ D. Método del filtro de membrana ................................... RECUENTO MICROBIANO TOTAL DIRECTO (PROPUESTA) . A. Introducción .................................................................. B. Método microscópico de epifluorescencia ............... 9221 TÉCNICA DE FERMENTACIÓN EN TUBO MÚLTIPLE PARA MIEMBROS DEL GRUPO DE LOS COLIFORMES .......................... A. B. C. D. E. 9222 Introducción .................................................................. Técnicas estandarizadas de fermentación en tubo múltiple (NMP) de coliformes totales..................... Procedimiento de NMP para coliformes fecales . . . . Estimación de la densidad bacteriana ......................... Prueba de presencia-ausencia (P-A) de coliformes . . 9-43 9-44 9-47 9-49 9-49 9-51 9-53 9-53 9-54 9-58 9-59 9-61 9-63 9-64 9-64 9-69 9-72 9-75 9-76 9-76 9-76 9-78 9-78 9-80 9-87 9-89 9-93 TÉCNICA DEL FILTRO DE MEMBRANA PARA MIEMBROS DEL GRUPO DE LOS COLIFORMES................................................................. 9-95 Introducción .................................................................. Procedimiento estándar de filtro de membrana para coliformes totales ...................................................... Procedimiento de incubación retardada para coliformes totales ............................................................... Procedimiento de filtro de membrana para coliformes fecales .......................................................................... 9-95 A. B. C. D. 9-97 9-106 9-109 li CONTENIDO PÁGINA E. Procedimiento de incubación retardada para coliformes fecales .................................................................. F. Procedimiento de filtro de membrana para Klebsiella 9225 DIFERENCIACIÓN DE BACTERIAS COLIFORMES ............................ A. Introducción .................................................................. B. Purificación de cultivos ................................................. C. Diferenciación................................................................. D. Significación de los distintos tipos de coliformes . . . E. Medios, reactivos y procedimientos ............................. 9230 GRUPOS DE ESTREPTOCOCOS Y ENTEROCOCOS FECALES ............. A. Introducción .................................................................. B. Técnica de tubo múltiple ........................................... C. Técnicas de filtro de membrana................................... 9240 IDENTIFICACIÓN DE BACTERIAS DEL HIERRO Y DEL AZUFRE A. Introducción .................................................................. B. Bacterias del hierro ..................................................... C. Bacterias del azufre ..................................................... D. Enumeración, enriquecimiento y aislamiento de las bacterias del hierro y el azufre (PROPUESTA) . . 9250 DETECCIÓN DE ACTINOMICETOS ................................................ A. Introducción .................................................................. B. Recuento de placa de actinomicetos ........................... 9260 DETECCIÓN DE BACTERIAS PATÓGENAS .................................. A. Introducción .................................................................. B. Procedimientos generales cualitativos de aislamiento e identificación de Salmonella ................................ C. Procedimiento de identificación de Salmonella por inmunofluorescencia .................................................. D. Procedimientos cuantitativos para Salmonella............ E. Shigella ............................................................................ F. Escherichia coli patógenos ............................................ G. Campylobacter jejuni .................................................... H. Vibrio cholerae .............................................................. I. Leptospiras patógenas................................................... J. Legionellaceae ................................................................ K. Yersinia enterocolitica ................................................. 9510 DETECCIÓN DE VIRUS ENTÉRICOS ............................................. A. Introducción .................................................................. B. Concentración de virus a partir de pequeños volúmenes de muestra por adsorción y dilución de filtros de mlcroporo (PROPUESTA) ................................ C. Concentración de virus a partir de grandes volúmenes de muestra por adsorción y dilución de filtros de microporo (PROPUESTA) ................................. 9-112 9-113 9-115 9-115 9-116 9-116 9-119 9-120 9-125 9-125 9-127 9-128 9-132 9-132 9-133 9-137 9-139 9-146 9-146 9-148 9-150 9-150 9-151 9-157 9-161 9-162 9-163 9-165 9-167 9-169 9-172 9-177 9-179 9-179 9-183 9-188 CONTENIDO lii PÁGINA D. Concentración de virus por adsorción-precipitación de hidróxido de aluminio (PROPUESTA).............. E. Hidroextracción-diálisis con polietilenglicol (PROPUESTA) .................................................................. F. Recuperación de virus a partir de sólidos en suspensión en aguas limpias y residuales (PROPUESTA) G. Ensayo e identificación de virus en concentrados de muestra (PROPUESTA)............................................ 9610 DETECCIÓN DE HONGOS ......................................................... A. Introducción ................................................................... B. Técnica en placa fluida ................................................. C. Técnica en placa difusa .................................................. D. Técnica de filtro de membrana .................................. E. Técnica para levaduras ................................................. F. Hongos zoospóricos ....................................................... G. Hifomicetos acuáticos ................................................... H. Hongos patógenos para el hombre ............................. 9711 PROTOZOOS PATÓGENOS ............................................................ A. Introducción ................................................................... B. Giardia lamblia ............................................................... C. Entamoeba histolytica .................................................... 9810 EXAMEN NEMATOLÓGICO ........................................................... A. Introducción ................................................................... B. Técnica para nematodos................................................ C. Clave ilustrada para la identificación de nematodos de agua dulce ............................................................. Parte 10000 ANÁLISIS BIOLÓGICO DE LAS AGUAS 10010 INTRODUCCIÓN ......................................................................... 10200 PLANCTON ............................................................................... A. Introducción ................................................................... B. Toma de muestras ......................................................... C. Técnicas de concentración .......................................... D. Preparación ..................................................................... E. Microscopios y calibraciones ....................................... F. Técnicas de recuento para fitoplancton .................... G. Técnicas de recuento para zooplancton .................... H. Clorofila........................................................................... I. Determinación de biomasa (cultivo estacionario) ... J. Medidas de índice metabólico .................................... 10300 PERIFITON .......................................................................... ___ A. Introducción ................................................................... B. Toma de muestras ......................................................... 9-196 9-200 9-202 9-204 9-212 9-212 9-217 9-219 9-221 9-222 9-223 9-225 9-226 9-227 9-227 9-228 9-235 9-236 9-236 9-239 9-241 10-1 10-3 10-3 10-4 10-17 10-19 10-22 10-25 10-31 10-34 10-43 10-47 10-53 10-53 10-54 CONTENIDO liii PÁGINA C. Análisis de muestras ...................................................... D. Productividad ................................................................. E. Interpretación y comunicación de los resultados .... 10400 MACROFITON .......................................................................... A. Introducción .................................................................. B. Estudio preliminar ......................................................... C. Métodos de trazado de mapas de distribución vegetal D. Estimaciones de población ........................................... E. Productividad ................................................................. 10500 MACROINVERTEBRADOS BÉNTICOS ............................................ A. Introducción .................................................................. B. Toma de muestras ........................................................ C. Procesado y análisis de muestras................................. D. Evaluación y presentación de datos .......................... 10600 PECES ........................................................................................ A. Introducción .................................................................. B. Adquisición de datos ..................................................... C. Conservación de la muestra ........................................ D. Análisis de muestras ............................................ , ........ E. Investigación sobre muerte de peces............................ 10900 IDENTIFICACIÓN DE ORGANISMOS ACUÁTICOS ........................... A. Procedimiento en la identificación............................... B. Clave para la identificación de los principales grupos de organismos acuáticos (láminas 1-38) ................. C. Lista de los tipos comunes de organismos acuáticos (láminas 1-38) del nivel trófico ................................. D. Clave para la identificación de las algas de agua dulce comunes en los suministros de agua o en las aguas contaminadas (láminas en color A-F) ..................... E. Cambios recientes en la nomenclatura de las algas.. F. índice de ilustraciones................................................... G. Referencias taxonómicas seleccionadas ....................... Índice analítico....................................................................... 10-57 10-61 10-74 10-76 10-76 10-77 10-79 10-83 10-89 10-106 10-106 10-108 10-122 10-125 10-127 10-127 10-128 10-142 10-145 10-150 10-153 10-153 10-154 10-159 10-200 10-207 10-208 10-211 1-1 PÁG. FIGURAS 1010:1 1020:1 1020:2 1020:3 1020:4 1020:5 1030:1 1030:2 1060:1 1070:1 2120:1 2120:2 2150:1 2150:2 2530:1 2530:2 2530:3 2530:4 2710:1 2710:2 2720:1 2810:1 3112:1 3114:1 3114:2 3500-Al:l Distribuciones normal y desplazada ......................... Gráficos de control para recursos .............................. Análisis duplicados de un estándar ............................ Gráfico de amplitud para concentraciones variables . Gráfico de amplitud para amplitudes variables .......... Gráfico de control de recursos con datos fuera de control (mitad superior) ......................................... Definición de exactitud ............................................ Relación entre límites de detección .......................... Número aproximado de muestras necesarias para calcular una concentración media ............................ Columna de intercambio iónico.................................. 1-2 1-11 1-12 1-12 1-12 Sistema de filtrado para determinaciones de color . . Diagramas de cromaticidad ..................................... Generador de agua inodora ..................................... Conjunto terminal del generador de agua inodora .. Dispositivo de toma de muestras de materias flotables Embudo de flotación de material flotable y soporte de filtro......................................................................... Embudos de flotación y unidad de mezclado............. Tubo de aceite flotable, capacidad 1 l........................ Diagrama esquemático de un vaso de sedimentación para pruebas de volumen de lodo sedimentado .. Diagrama esquemático de un vaso de sedimentación para prueba de tasa de sedimentación de zona .. . Aparato de toma de muestras de gases ...................... Tiempo de respuesta para la prueba de difusión de membrana .............................................................. 2-5 2-8 2-21 2-22 2-73 Esquema de disposición del equipo para medida de mercurio por la técnica de absorción atómica de vapor frío ................................................................ Célula manual de reacción para producir hidruros de As y Se .................................................................... Esquema de un generador continuo de hidruros . . . . Curvas de corrección para estimación de aluminio en presencia de fluoruro .............................................. 1-13 1-15 1-21 1-41 1-53 2-73 2-74 2-76 2-92 2-94 2-97 2-106 3-33 3-49 3-57 3-74 Ivi CONTENIDO PÁG. 3500-Al:2 3500-A1:3 3500-As: 1 3500-As:2 3500-Se:l 3500-Sr:l 4110:1 4110:2 4500-CO2:l 4500:CO2:2 4500:CO2:3 4500:CO2:4 4500-CN:l 4500-Cl– :1 4500-C1– :2 4500-ClO2:I 4500-F –:l 4500-F – :2 4500-H + :l 4500-H + :2 4500-NH3:l 4500- NO3− :l 4500- NO3− :2 4500- NO3− :3 4500-Norg:l 4500-0:1 4500-0:2 Distribuidor de aluminio para Sistema Automatizado I (analizador de inyección de flujo no segmentado) Distribuidor de aluminio para Sistema Automatizado II (analizador de flujo segmentado) ........................ Generador de arsina y ensamblaje del absorbedor .. .. Generador utilizado con el método del tinte de bromuro mercúrico ...................................................... Esquema general para especiación de selenio en agua Método gráfico para el cálculo de la concentración de estroncio ................................................................ Separación típica de aniones inorgánicos utilizando columna de operacional normal .......................... Separación de columna de operación de rápida . . . . Nomogramas para evaluación de la concentración de ion hidróxido ......................................................... Nomogramas para evaluación de la alcalinidad de bicarbonato .............................................................. Nomogramas para evaluación de la alcalinidad de carbonato .............................................................. Nomogramas para evaluación del contenido en dióxido de carbono ......................................................... Destilador de cianuro ................................................... Ejemplo de curva de titulación diferencial (el punto final está 25,5 ml)..................................................... Diagrama de flujo para análisis automatizado de cloruro ......................................................................... Sistema de generación y absorción de dióxido de cloro ........................................................................... Aparato de destilación directa para fluoruro.............. Distribuidor de fluoruro ............................................ Potencial del electrodo frente a pH.............................. Respuesta típica del electrodo de pH como función de la temperatura ...................................................... Distribuidor de amoníaco ........................................... Columna de reducción ................................................. Distribuidor de nitrato-nitrito ................................... Distribuidor de nitrato-nitrito ................................. Aparato de destilación micro-kjeldahl ......................... Montaje para toma de muestras de OD y ROB____ Efecto de la temperatura en la sensibilidad del lectrodo ....................................................................... 3-78 3-79 3-83 3-85 3-148 3-173 4-5 4-5 4-17 4-18 4-19 4-20 4-32 4-82 4-84 4-87 4-98 4-105 4-109 4-109 4-143 4-154 4-156 4-160 4-167 4-171 4-181 Ivii CONTENIDO PÁG. 4500-0:3 4500-0:4 Efecto de salinidad a diferentes temperaturas .......... Tendencia característica del efecto de agitación sobre la respuesta del electrodo ....................................... 4500-P:l Pasos del análisis de las fracciones de fosfato ........... 4500-P:2 Distribuidor de fosfato para cada sistema de análisis automatizado ......................................................... 4500-Si:l Distribuidor de sílice .................................................. 4500-S2¯:l Marcha analítica para determinaciones de sulfuro .. 4500-S2¯:2 Proporción de H2S y HS¯ en sulfuro disuelto ......... 4500- SO32 ¯:1 Aparato para desprendimiento de SO 2 a partir de muestras para análisis colorimétrico...................... 4500- SO 24 ¯:l Distribuidor de sulfato .............................................. 5230:1 5540:1 Sistema de análisis del HOD .................................... Aparato de cancelación .............................................. 6040:1 Esquema del instrumental de depuración de ciclo cerrado............................................................................... Botella alta de depuración de un litro .................... Calentador de gas ....................................................... Extracción de filtro .................................................... Tasa de flujo a través de un filtro de carbono activado de 1,5 mg................................................................. Efecto de la resistencia del filtro, medida como flujo, en la recuperación de sustancias de olor terrosomohoso y estándares internos de C1-C10 ............. Espectros de masas de 2-metilisoborneol bajo diferentes condiciones instrumentales ............................... Espectro de masas de geosmina ................................. Dispositivo de purga ................................................. Relleno y construcción de los dispositivos de atrapamiento para incluir capacidad de deabsorción ... Sistema de purga y atrapamiento (modo purga) . . . . . Sistema de purga y atrapamiento (modo deabsorción) Cromatograma de gases de compuestos orgánicos volátiles ...................................................................... Relleno y construcción de los dispositivos de atrapamiento para incluir capacidad de deabsorción ... Cromatograma de gases de compuestos orgánicos volátiles (columna capilar de diámetro ancho) ...... Cromatograma de gases de compuestos orgánicos volátiles (columna capilar de diámetro mega) .......... Cromatograma de mezcla de prueba (columna capilar de diámetro estrecho) ........................................... 6040:2 6040:3 6040:4 6040:5 6040:6 6040:7 6040:8 6210:1 6210:2 6210:3 6210:4 6210:5 6210:6 6210:7 6210:8 6210:9 4-181 4-182 4-189 4-203 4-214 4-217 4-224 4-227 4-236 5-35 5-66 6-14 6-14 6-15 6-16 6-16 6-17 6-24 6-25 6-32 6-33 6-34 6-34 6-38 6-49 6-58 6-60 6-61 Iviii CONTENIDO PÁG. 6211:1 6220:1 6220:2 6220:3 6220:4 6220:5 6220:6 6220:7 6230:1 6230:2 6230:3 6231:1 6232:1 6232:2 6232:3 6410:1 6410:2 6410:3 6410:4 6410:5 6410:6 6410:7 6410:8 6410:9 6410:10 6410:11 6410:12 6410:13 6420:1 6420:2 6440:1 6440:2 Circuito indicador de gas combustible y diagrama de flujo ........................................................................ 6-67 Relleno y construcción de los dispositivos de atrapamiento para incluir capacidad de deabsorción ... 6-72 Sistema de purga y atrapamiento (modo purga) . . . . . 6-73 Sistema de purga y atrapamiento (modo secado) .... 6-74 Sistema de purga y atrapamiento (modo de absorción) ........................................................................ 6-74 Cromatograma de gases de sustancias aromáticas depuradas .................................................................. 6-75 Cromatograma de mezcla de prueba .......................... 6-78 Cromatograma de mezcla de prueba .......................... 6-80 Cromatograma de gases de halocarburos depurables 6-90 Cromatograma de gases de halocarburos depurables 6-96 Cromatograma dual de sustancias orgánicas volátiles (columna capilar) con detectores de fotoionización (arriba) y conductividad electrolítica (abajo) ......... 6-104 Extracto de agua para reactivos con adición de 0,114 µg/1 de DBE y DBCP ............................................. 6-110 Cromatograma de extracto de agua terminal.............. 6-117 Cromatograma de extracto de estándar ................... 6-118 Cromatograma de extracto de estándar .................... 6-118 Cromatograma de gases de una fracción básico/neutra ............................................................................ 6-135 Cromatograma de gases de una fracción acida .......... 6-136 Cromatograma de gases de fracción de pesticida . . . 6-136 Cromatograma de gases del clordán........................... 6-137 Cromatograma de gases del toxafeno ........................ 6-137 Cromatograma de gases del BPC-1016....................... 6-138 Cromatograma de gases del BPC-1221....................... 6-138 Cromatograma de gases del BPC-1232...................... 6-139 Cromatograma de gases del BPC-1242....................... 6-139 Cromatograma de gases del BPC-1248....................... 6-140 Cromatograma de gases del BPC-1254....................... 6-141 Cromatograma de gases del BPC-1260....................... 6-142 Cálculo del factor de decantación ............................. 6-143 Cromatograma de gases de los fenoles ....................... 6-153 Cromatograma de gases de derivados PFB de fenoles 6-154 Cromatograma líquido de hidrocarburos polinucleares aromáticos ......................................................... 6-166 Cromatograma líquido de hidrocarburos polinucleares aromáticos .......................................................... 6-167 CONTENIDO lix PÁG. 6440:3 6630:1 6630:2 6630:3 6630:4 6630:5 6630:6 6630:7 6630:8 6630:9 6630:10 6630:11 6630:12 6630:13 6630:14 6630:15 6640:1 6640:2 7010:1 7500-1:1 7500-Ra:l 7500-Sr:l 8010:1 8010:2 8010:3 8010:4 8010:5 8010:6 Cromatograma de gases de hidrocarbutos polinucleares aromáticos ......................................................... Resultados del procedimiento de cromatografía de gases para pesticidas organoclorados .................... Resultados del procedimiento de cromatografía de gases para pesticidas organoclorados ................... Cromatograma de mezcla de pesticidas ................... Cromatograma de mezcla de pesticidas ................... Cromatograma de mezcla de pesticidas .................. Cromatograma de gases de pesticidas........................ Cromatograma de gases del clordán.......................... Cromatograma de gases del toxafeno ....................... Cromatograma de gases del BPC-1016...................... Cromatograma de gases del BPC-1221...................... Cromatograma de gases del BPC-1232...................... Cromatograma de gases del BPC-1242...................... Cromatograma de gases del BPC-1248...................... Cromatograma de gases del BPC-1254 ..................... Cromatograma de gases del BPC-1260...................... Resultados del procedimiento de cromatografía de gases para herbicidas clorados fenoxi ácidos Cromatogramas de mezcla de herbicidas ................. Forma de las curvas de tasa de conteo, voltaje del ánodo....................................................................... Aparato de destilación para análisis de yodo (no a escala) ...................................................................... Montaje para eliminar emanaciones ........................... Actividad del itrio 90 frente al estroncio 90 en función del tiempo ............................................................... Tanque de mantenimiento para peces y macroinvertebrados .................................................................... Equipo para cultivo de algas .................................... Método de iluminación, bastidor, ubicación de la fuente de medio, bombas de aire y otros aparatos para dispositivos de cultivo algal masivo............... Componentes básicos de un sistema de flujo transversal ............................................................................ Ejemplos de determinación de concentración letal media en dos momentos representativos mediante análisis de probit y línea de ajuste optimizado ... Curva de toxicidad obtenida a partir de las CL50 determinadas en la figura 8010:5 ............................ 6-168 6-175 6-176 6-177 6-178 6-179 6-191 6-191 6-192 6-192 6-193 6-193 6-194 6-194 6-195 6-195 6-202 6-202 7-8 7-33 7-41 7-61 8-16 8-22 8-23 8-29 8-40 8-42 Ix CONTENIDO PÁG. 8610:1 8712:1 8712:2 8712:3 8712:4 8720:1 8720:2 8720:3 9020:1 9215:1 9215:2 9215:3 9221:1 9221:2 9221:3 9240:1 9240:2 9240:3 9240:4 9240:5 9240:6 9240:7 Diagrama del aparato de flujo constante ................. Sistema de cultivo algal ............................................. Aparato para cultivo masivo de copépodos (estático) Aparato para cultivo masivo de copépodos (fluyente) Jaula de generación (según Heinle) ......................... Vaso de cría y exposición y sifón automático para larvas del cangrejo común del Pacífico ............... Tanque para incubado de huevos para langostas . . . Tanque de Hughes para cría de langosta.................. Curva de frecuencia (con distribución desviada positiva) ....................................................................................... Preparación de las diluciones.................................... Pérdida de peso por secado de placas de agar de 15 ml almacenadas individualmente, invertidas y con las cubiertas colocadas ....................................... Pérdida de peso de placas de agar de 25 ml (100 x 15 mm) secadas por separado en una campana de flujo laminar a temperatura ambiente (24 a 36 °C), humedad relativa (30 a 33 por 100) y velocidad del aire 0,6 m/s ............................................................ Esquema de la fase confirmada .............................. Esquema de la prueba completa para la detección de coliformes totales .................................................. Esquema de las confirmaciones opcionales de P-A para distintos organismos indicadores ............... Filamentos de Crenothrix polyspora que muestran variaciones en el tamaño y forma de las células dentro de la vaina ..................................................... Filamentos de Sphaerotilus natans, que muestran células contenidas en los filamentos y algunas células «pululantes» ......................................................... Cultivo de laboratorio de Gallionella ferruginea, que muestra células, tallos y ramificaciones de los tallos en la zona de división de las células..................... Mezcla de fragmentos de tallos de Gallionella ferruginosa y precipitado de hierro-manganeso inorgánico que se encuentra en las muestras naturales tomadas de pozos .......................................................... Célula única de la bacteria del hierro Siderocapsa treubii...................................................................... Bacterias del azufre purpúreas fotosintéticas ......... Bacterias de azufre filamentosas incoloras ............. 8-91 8-101 8-101 8-102 8-103 8-110 8-111 8-113 9-25 9-70 9-73 9-74 9-83 9-84 9-94 9-134 9-135 9-135 9-136 9-136 9-137 9-138 Ixi CONTENIDO PÁG. 9240:8 9240:9 9250:1 9510:1 9510:2 9711:1 9711:2 9711:3 9810:1 10200:1 10200:2 10200:3 10200:4 10200:5 10200:6 10200:7 10200:8 10200:9 10200:10 10200:11 10300:1 10300:2 10300:3 10300:4 10300:5 10400:1 10500:1 Bacterias de azufre filamentosas incoloras ............. Bacterias del azufre incoloras no filamentosas ........ Colonias bacterianas ............................................... Método de adsorción-dilución en dos estadios con filtro microporoso para concentrar virus a partir de un gran volumen de agua ................................ Esquema del aparato utilizado en la concentración inicial ...................................................................... Diagrama de flujo del método para Giardia ........ Esquema del dispositivo para toma de muestras de Giardia .................................................................... Dispositivo para toma de muestras de Giardia ...... Ciclo vital de los nematodos..................................... Características estructurales de los dispositivos de toma de muestras de agua comunes ........................ Trampa para plancton de Schindler-Patalas ......... Ejemplos de redes para toma de muestras de placton utilizadas comúnmente ......................................... Ejemplos de dispositivos de toma de muestras de zooplancton de gran velocidad, usados comúnmente . Embudo de filtro para concentrar zooplancton....... Ajuste de micrómetro ocular..................................... Calibración de la cuadrícula de Whipple ............... Célula de recuento (Sedgwick-Rafter) ................... Dispositivo para concentrar el plancton .................. Dispositivo de corte para plancton de Folsom ....... Cromatograma HPLC de fase inversa para una dilución quíntuple de la muestra EPA .................... Dispositivo de toma de muestras de perifiton ........ Procesos en el metabolismo de oxígeno de una sección de un flujo de agua hipotético durante el transcurso de un día sin nubes .......................... Producción perifitica primaria bruta (PG) determinada con la cámara de O'Connell-Thomas ............ Cálculo de la producción primaria bruta en una estación única .............................................................. Cálculo de la productividad perifitica primaria bruta a partir de las curvas diurnas de flujo ascendentedescendente ............................................................. Curva de Allen para una cohorte de una población de macrofitos acuáticos............................................... Pala de Ponar ............................................................. 9-138 9-139 9-149 9-191 9-192 9-230 9-231 9-231 9-237 10-7 10-10 10-13 10-14 10-19 10-23 10-24 10-27 10-32 10-33 10-41 10-55 10-65 10-69 10-70 10-70 10-94 10-111 Ixii CONTENIDO PÁG. 10500:2 10500:3 10500:4 10500:5 10500:6 10500:7 10500:8 10500:9 10500:10 10500:11 10500:12 10500:13 10500:14 10500:15 10600:1 10600:2 10600:3 10600:4 10600:5 10600:6 10600:7 Pala en gajos de naranja ............................................. Pala de Petersen .......................................................... Pala de Van Veen ........................................................ Pala de Smith-Mclntyre ............................................ Pala de Shipek ........................................................... Pala de Ekman............................................................. Dispositivo de toma de muestras Surber o de piecuadrado ................................................................. Sonda de Phleger ......................................................... Dispositivo de toma de muestras de sondeo de KB . Dispositivo de toma de muestras de Wilding o de tubo de absorción ................................................. Dispositivo de toma de muestras de red de arrastre . Unidad Hester-Dendy de sustrato artificial .............. Dispositivo de toma de muestras de cesta ................. Tomamuestras con banda dentada (visto desde atrás) y banda de cilindro de poliuretano (vista lateral) . Diagrama de una red deslizante sumergida ............... Red barredera utilizada en un arroyo pequeño......... Diagrama de un barco para pesca mediante sistemas electrónicos bien equipado ..................................... Tipos de etiquetas de uso común .............................. Sistema de etiquetado de Transponedor Integrado Pasivo (TIP) ............................................................. Órganos clave y partes corporales externas de peces lisos y espinosos...................................................... Escama de un pez ........................................................ 10-111 10-112 10-112 10-113 10-113 10-114 10-115 10-111 10-116 10-117 10-117 10-119 10-119 10-120 10-132 10-135 10-136 10-139 10-140 10-144 10-147 PÁG. TABLAS 1010:1 1020:1 1020:11 1020:111 1030:1 1030:11 Valores críticos para pruebas de discordancia de 5 por 100 y 1 por 100 para un valor extremo simple en una muestra normal .......................................... Límites de aceptación para muestras duplicadas y adiciones conocidas sobre aguas limpias y residuales ............................................................................ Factores para el cálculo de líneas en gráficos de control de amplitud ..................................................... Verificación de un procedimiento de análisis de suelo Cálculos de precisión mediante el uso de duplicados Cálculos de precisión para adiciones conocidas ........ 1-4 1-9 1-11 1-14 1-17 1-18 Ixiii CONTENIDO PÁG. 1050:1 1060:1 1080:1 1080:11 1090:1 1090:11 2120:1 2120:11 2150:1 2150:11 2160:1 2160:11 2320:1 2330:1 2330:11 2330:111 2340:1 2510:1 2530:1 2710:1 2810:1 2810:11 3030:1 3111:1 3111:11 Factores de conversión ............................................ Resumen de requerimientos especiales para toma de muestras o manipulación ................................... Especificaciones del agua para reactivos................... Procesos de purificación del agua ........................... Valores umbral límite (VUL)*10 para los disolventes especificados en Standard Methods ..................... Valores umbral límite (VUL)* 10 para los reactivos especificados en Standard Methods ..................... 1-30 1-42 1-64 1-64 1-75 1-76 Ordinales seleccionados para determinaciones espectrofotométricas de color* ................................... 2-7 Matices de color para márgenes de longitud de onda dominante ............................................................... 2-7 Números de umbral de olor correspondientes a varias diluciones................................................................ 2-23 Diluciones para varias intensidades de olor............ 2-24 Números de umbral de sabor correspondientes a varias diluciones .............................................................. 2-27 Diluciones para determinación del ÑUS ............... 2-28 Relaciones de alcalinidad ........................................... 2-42 Estimación de constantes de equilibrio y coeficientes de actividad ............................................................ 2-47 Valores precalculados para pK y A a. temperaturas seleccionadas ........................................................... 2-48 Colección de programas gráficos y de ordenador que pueden utilizarse para calcular los índices de saturación* del CaCO3 ................................................. 2-54 Concentraciones máximas de interferencia permitidas con diversos inhibidores ........................................ 2-58 Conductividad de las soluciones de cloruro de potasio a 25 °C* ................................................................... 2-65 Coeficiente de variación y recuperación para prueba de partículas flotables ............................................ 2-75 Factor de corrección de la temperatura ................. 2-96 Coeficiente Bunsen para oxígeno en agua dulce . . . . 2-108 Presión de vapor del agua dulce ............................. 2-110 Ácidos utilizados junto con HNO 3 para preparación de la muestra ....................................................... 3-7 Margenes de concentración de absorción atómica con absorción atómica de aspiración directa .......... 3-17 Datos de precisión y sesgo interlaboratorios para mé- Ixiv CONTENIDO PÁG. 3111:111 3112:1 3113:1 3113:11 3113:111 3113:1V 3113:V 3120:1 3120:11 3500-A1:I 3500-A1:II 3500-Fe:I 3500-V:I 4110:1 4500-ClO2:I 4500-F ¯ :I 4500-N + :I 4500-H + :II 4500-NH3:I todos de absorción atómica. Aspiración directa y metales extraídos ................................................. 3-19 Precisión de un solo operador y niveles de control recomendados para métodos de absorción atómica. Aspiración directa y metales extraídos ........ 3-20 Precisión y sesgo interlaboratorios del método de espectrometría de absorción atómica de vapor frío para el mercurio................................................... 3-34 Modificadores de matrices potenciales para espectrometría de absorción atómica electrotérmica ...... 3-36 Niveles de detección y márgenes de concentración para espectrometría de absorción atómica de atomización electrotérmica........................................ 3-38 Datos de precisión interlaboratorios de un único analista para métodos de atomización electrotérmica 3-44 Datos de precisión global interlaboratorios para métodos de atomización electrotérmica..................... 3-45 Datos de errores relativos interlaboratorios para métodos de atomización electrotérmica..................... 3-46 Longitudes de onda sugeridas, límites de detección estimados, longitudes de onda alternativas, concentraciones de calibración y límites superiores ........ 3-61 Datos de precisión y sesgo de PAI ...................... 3-68 Precisión y sesgo de un solo operador para análisis de aluminio monómero inorgánico.......................... 3-80 Precisión y sesgo de un solo operador en la determinación de alto nivel de aluminio............................ 3-81 Selección de longitud del recorrido de luz para diversas concentraciones de hierro................................. 3-117 Concentración en la que interfieren diversos iones en la determinación de vanadio ............................ 3-178 Precisión y sesgo observados para aniones a distintos niveles de concentración en el agua para reactivos 4-7 Pesos equivalentes para calcular las concentraciones sobre la base de la masa ..................................... 4-94 Concentración de algunas sustancias que producen un error de 0,1 mg/l en 1,0 mg/ f/1 en los métodos de fluoruro ......................................................... 4-96 Preparación de soluciones patrón de pH3 .............. 4-111 Valores patrón del pH3 ........................................................................ 4-112 Datos de precisión y sesgo para los métodos de amoníaco ........................................................................ 4-127 Ixv CONTENIDO PÁG. 4500-NH3:II Precisión y sesgo del electrodo selectivo de amoníaco 4500-NH3:III Preparación de estándares de color permanente para determinación visual de nitrógeno amoniacal . . . . 4500-NH3:IV Valores de Q frente a AE (pendiente de 59 mv) para cambio de volumen del 10 por 100 ....................... 4500-Norg:I Datos de precisión y sesgo para nitrógeno orgánico, método Macrokjeldahl .......................................... 4500-0:1 Solubilidad de oxígeno en agua expuesta a aire saturado de agua a presión atmosférica (101,3 kPa) .. 4500-P:I Datos de precisión y sesgo para los métodos manuales de fósforo........................................................... 4500-P:II Comparación de precisión y sesgo de los métodos del ácido ascórbico ....................................................... 4500-Si:I Selección de la longitud del recorrido de luz para varias concentraciones de sílice ............................ 4500-Si:II Preparación de estándares de color permanente para determinación visual de sílice................................. 4500-S 2¯ :I Valores de pK' logaritmo de la constante de ionización práctica para sulfuro de hidrógeno ............... 5220:1 5310:1 5320:1 5540:1 5710:1 5710:11 5710:111 6010:1 6010:11 6040:1 6040:11 Cantidades de muestra y reactivos para varios vasos de digestión............................................................. Precisión y sesgo para el carbono orgánico total (COT) por oxidación persulfato-ultravioleta ...... Datos de precisión y sesgo, interlaboratorios, un solo operador halógeno orgánico disuelto (procedimiento de microcolumna) ............................................. Recuperación del surfactante por cancelación ......... Precisión y sesgo de un solo operador para determinar el PFT ........................................................... Datos de precisión y sesgo de un solo operador para determinar el PBFT ............................................... Precisión de un solo operador sobre muestras de río diluidas y filtradas, analizadas para determinar el PBFT....................................................................... 4-129 4-135 4-141 4-166 4-174 4-190 4-201 4-209 4-211 4-223 5-18 5-28 5-42 5-68 5-91 5-92 5-93 Métodos de análisis para compuestos orgánicos específicos ...................................................................... 6-1 Conservación recomendada para sustancias volátiles 6-5 Límites de detección instrumental para compuestos de olor terroso-mohoso mediante ADCC-CG/EM 6-13 Límites de detección instrumental para compuestos orgánicos seleccionados mediante ADCC-CG/EM 6-13 Ixvi CONTENIDO PÁG. 6040:111 Condiciones típicas del funcionamiento para análisis CG/EM de extractos ADCC ................................ 6040:IV Datos CG/EM para tres estándares internos y dos compuestos de olor terroso-mohoso....................... 6040:V Sesgo de un único laboratorio para compuestos orgánicos saporíferos y odoríferos seleccionados ........ 6040:VI Datos de precisión para compuestos orgánicos saporíferos y odoríferos seleccionados ........................ 6040:VII Datos de precisión y recuperación para sustancias contaminantes seleccionadas ................................... 6210:1 Condiciones cromatográficas y límites de detección del método (LDM) .................................................. 6210:11 Criterios de abundancia m/z de clave BFB ............. 6210:111 Estándares internos y sustitutivos sugeridos .......... 6210:1 V Criterios de calibración y de admisión al CC ........ 6210:V Masas características para sustancias orgánicas depurables ....................................................................... 6210:VI Sesgo y precisión del método como funciones de la concentración .......................................................... 6210:VII Condiciones cromatográficas para compuestos orgánicos volátiles en columnas de relleno ................. 6210:VIII Masas características (m/z) para compuestos orgánicos depurables.......................................................... 6210:IX Datos de sesgo y precisión de un único laboratorio para compuestos orgánicos volátiles en agua para reactivos (columna de relleno) ............................... 6210:X Condiciones cromatográficas para compuestos orgánicos volátiles en columnas capilares de diámetro ancho ....................................................................... 6210:XI Condiciones cromatográficas para compuestos orgánicos volátiles en columnas capilares de diámetro estrecho .................................................................... 6210:XII Datos de sesgo y precisión de un único laboratorio para compuestos orgánicos volátiles en agua para reactivos (columna capilar de diámetro ancho) … 6210:XIII Datos de sesgo y precisión de un único laboratorio para compuestos orgánicos volátiles en agua para reactivos (columna capilar de diámetro estrecho).... 6220:1 Condiciones cromatográficas y límites de detección del método ............................................................ 6220:11 Criterios de calibración y de admisión al CC ........ 6220:111 Sesgo y precisión del método como funciones de la concentración ......................................................... 6-22 6-24 6-26 6-27 6-28 6-31 6-33 6-37 6-40 6-42 6-45 6-51 6-53 6-55 6-57 6-59 6-62 6-64 6-72 6-76 6-76 Ixvii CONTENIDO PÁG. 6220:1 V 6220:V 6220: VI 6220:VII 6230:1 6230:11 6230:111 6230:IV 6230:V 6230:VI 6230:VII 6230:VIII Tiempos de retención para compuestos aromáticos . Sesgo y precisión de un único laboratorio para sustancias aromáticas no saturadas en aguas potables cloradas y aguas de fuente natural cloradas ....... Precisión de un único analista, precisión global y sesgo para sustancias aromáticas depurables en agua potable………………………………………… Datos de sesgo y precisión para sustancias aromáticas depurables a partir de estudios de evaluación del rendimiento de laboratorios múltiples Condiciones cromatográficas y límites de detección del método (LDM) ................................................. Criterios de calibración y de admisión al CC ......... Sesgo y precisión del método como funciones de la concentración ........................................................ Tiempos de retención para organohaluros................ Sesgo y precisión de un único laboratorio para organohaluros en agua del río Ohio y en agua potable Sesgo y precisión de un único laboratorio para organohaluros en agua del río Ohio y en agua potable Tiempos de retención para compuestos orgánicos volátiles en un detector de fotoionización (DFI) y un detector de conductividad electrolítica (DCE) . . . . Sesgo y precisión de un único laboratorio para compuestos orgánicos volátiles en agua para reactivos Condiciones cromatográficas para 1,2-Dibromoetano (DBE) y l,2-Dibromo-3-Cloropropano (DBCP) .. 6231:11 Sesgo y precisión de un único laboratorio para DBE y DBCP en agua del grifo ................................... 6232:1 Tiempos de retención para trihalometanos............... 6232:11 Precisión y sesgo de un único laboratorio ............... 6410:1 Condiciones cromatográficas, límites de detección del método y masas características para extractos básico/neutros ………………………………………... 6410:11 Condiciones cromatográficas, límites de detección del método y masas características para extractos ácidos .......................................................................... 6410:111 . Criterios de masas de clave de DFTFF y de abundancia ........................................................................... 6410:IV Estándares internos y sustitutivos sugeridos .......... 6410:V Criterios de admisión al CC ...................................... 6410:VI Sesgo y precisión del método como funciones de la concentración ......................................................... 6-81 6-83 6-84 6-85 6-88 6-91 6-93 6-97 6-100 6-101 6-102 6-105 6231:1 6-111 6-112 6-118 6-122 6-126 6-129 6-131 6-132 6-144 6-146 Ixviii CONTENIDO PÁG. 6420:1 6420:11 6420:111 6420:IV 6440:1 6440:11 6440:111 6440:IV 6630:1 6630:11 6630:111 6630:IV 6630:V 6630:VI 6640:1 6640:11 6640:111 Condiciones cromatográficas y límites de detección del método ............................................................ 6-149 Fraccionamiento de gel de sílice y cromatografía de gases con captación de electrones de derivados PFBB ....................................................................... 6-150 Criterios de admisión al CC ....................................... 6-156 Sesgo y precisión del método como funciones de la concentración ......................................................... 6-157 Condiciones cromatográficas líquidas de alto rendimiento y límites de detección del método ........... 6-161 Condiciones cromatográficas de gas y tiempos de retención ..................................................................... 6-163 Criterios de admisión al CC ....................................... 6-169 Sesgo y precisión del método como funciones de la concentración ......................................................... 6-170 Tasas de retención de varios pesticidas organoclorados en relación con el aldrín ................................ 6-182 Datos de precisión y sesgo para pesticidas organoclorados seleccionados ............................................... 6-183 Condiciones cromatográficas y límites de detección del método ............................................................ 6-186 Distribución de pesticidas clorados y BPC en fracciones de columna de gel de sílice-magnesio.............. 6-190 Criterios de admisión al CC ....................................... 6-196 Sesgo y precisión del método como funciones de la concentración ......................................................... 6-197 Tiempos de retención para esteres de metilo de algunos herbicidas clorados fenoxi ácidos relativos al ester de metilo 2,4-D ............................................... 6-203 Precisión de herbicidas fenoxi ácidos a partir de agua de superficie dosificada ........................................... 6-203 Recuperación de herbicidas fenoxi ácidos a partir de agua de superficie dosificada ................................ 6-204 7010:1 Distribución usual de radioelementos comunes entre las fases líquida y sólida de las aguas residuales .. 7500-Ra:I Composición química y radioquímica de muestras usadas para determinar el sesgo y la precisión del método de radio 226 .............................................. 7500-Ra:II Factores de desintegración de radón 222, crecimiento de radón 222 a partir de radio 226 y corrección de la actividad de radón 222 para recuento durante el deterioro ................................................................ 7-4 7-39 7-45 CONTENIDO Ixix PÁG. 8010:1 8010:11 8010:111 8010:IV.A 8010:IV.B 8010:V 8010:VI 8010:VII 8712:1 8910:1 8910:11 9020:1 9020:11 9020:111 9020:IV 9020:V 9020:VI 9020:VII 9020:VIII 9020:IX 9020:X 9211:1 9215:1 9221:1 Composición recomendada para agua dulce reconstituida ........................................................................ Cantidades de agentes químicos de calidad para reactivos que se añaden al agua dulce reconstituida, blanda y aireada para taponar el pH ........................ Procedimiento para la preparación de agua de mar reconstituida ........................................................... Solución madre de macronutrientes ............................ Solución madre de micronutrientes ............................. Nutrientes para medio de cultivo algal en agua de mar........................................................................... Porcentaje de amoníaco desionizado en agua destilada ............................................................................ Datos experimentales extraídos de una prueba de toxicidad hipotética sometida a un análisis probit …. Composición de la dieta algal y concentraciones recomendadas para alimentación de adultos, naupliar y para puesta de huevos………………………… Tratamientos profilácticos y terapéuticos recomendados para peces de agua dulce utilizados con propósitos experimentales..………………….………….. Fotoperíodo de Evansville, Indiana ............................. Calidad del agua purificada utilizada en pruebas microbiológicas ........................................................... Adición de reactivos para pruebas de calidad del agua Tiempo y temperatura para esterilización en autoclave .......................................................................... Tiempo de mantenimiento para medios preparados . Cultivos de control para pruebas microbiológicas .. Cálculo del criterio de precisión .................................. Comprobaciones diarias de la precisión de los recuentos duplicados.......................................................... Recuentos de coliformes y logaritmos correspondientes ........................................................................... Comparación de las frecuencias de los datos de NMP Comparación de las frecuencias de los datos de log NMP ...................................................................... Técnicas rápidas especiales ........................................ Efecto de la temperatura de secado en la pérdida de peso de placas de agar de 15 ml almacenadas individualmente ............................................................... Preparación del medio líquido de lauril triptosa .... 8-18 8-18 8-19 8-20 8-20 8-21 8-34 8-41 8-103 8-145 8-146 9-12 9-13 9-19 9-20 9-21 9-22 9-23 9-26 9-26 9-26 9-45 9-73 9-81 Ixx CONTENIDO PÁG. 9221:11 9221:111 9221:IV 9221:V 9222:1 9222:11 9222:111 9225:1 9225:11 9230:1 9250:1 10200:1 10200:11 10300:1 10300:11 10400:1 Preparación del medio líquido de lactosa.................... Índice de NMP y límites de aceptación del 95 por 100 para distintas combinaciones de resultados positivos y negativos cuando se emplean cinco porciones de 10 ml..................................................................... Índice de NMP y límites de aceptación del 95 por 100 para distintas combinaciones de resultados positivos y negativos cuando se emplean diez porciones de 10 ml ................................................................... Índice de NMP y límites de aceptación del 95 por 100 para distintas combinaciones de resultados positivos y negativos cuando se usan cinco tubos por dilución (10 ml, 1,0 ml, 0,1 ml)............................... Volúmenes de muestra recomendados para la prueba de coliformes totales en filtro de membrana ........ Límites de aceptación del 95 por 100 para los resultados del recuento de coliformes en filtro de membrana utilizando una muestra de 100 ml ...................... Volúmenes de muestra recomendados para la prueba de filtro de membrana en coliformes fecales ........ Nomenclatura de las enterobacteriáceas..................... Diferenciación de los microorganismos indicadores potenciales de las enterobacteriáceas ...................... Algunas características bioquímicas clave de las especies de estreptococos pertenecientes a los grupos de estreptococos fecales y enterococos........................... Propiedades macroscópicas generales de las colonias bacterianas en medio sólido.................................... Características de las redes para toma de muestras de plancton utilizadas comúnmente ........................... Tabla de conversión para la técnica de filtro de membrana (basada en 30 campos puntuados) .................. Libro de cálculo de muestras para cómputo de la tasa corregida del cambio de oxígeno a partir de la curva diurna de una única estación ...................... Libro de cálculo de muestras para cómputo de la tasa corregida del cambio de oxígeno a partir de las curvas diurnas de flujo ascendente-descendente de la concentración de oxígeno y temperatura ........... Métodos usados para determinar la producción macrofitica .................................................................. 9-82 9-90 9-90 9-91 9-102 9-105 9-110 9-117 9-118 9-132 9-149 10-11 10-30 10-71 10-72 10-91 CONTENIDO Ixxi PÁG. LÁMINAS Láminas en blanco y negro de organismos acuáticos 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. Algas azules-verdes: Cocoides (Filum Cyanophyta) ................... 10-162 Algas azules-verdes: Filamentosas (Filum Cyanophyta).............. 10-163 Algas verdes no móviles: Cocoides (Filum Chlorophyta)............ 10-164 Algas verdes no móviles. Filamentosas (Filum Chlorophyta) .. 10-165 Diatomeas. Pennadas (Filum Chrysophyta, clase Bacillariophyceae)................................................................................... 10-166 Diatomeas. Céntricas (Filum Chrysophyta, clase Bacillariophyceae)................................................................................... 10-167 Tipos de algas marinas más grandes (verdes, marrones y rojas) 10-168 Plantas superiores: Plantas flotantes ......................................... 10-169 Plantas superiores: Sumergidas (todas las formas ilustradas son espermatofitas) ........................................................................ 10-170 Plantas superiores: Emergidas (todas las formas ilustradas son espermatofitas)......................................................................... 10-171 Flagelados pigmentados unicelulares (varios filum) ................... 10-172 Flagelados pigmentados tipos coloniales (varios filum) ............. 10-173 Flagelados no pigmentados (Filum Protozoa) ......................... 10-174 Amebas (Filum Protozoa). a) Etapas ameboides; b) etapas flageladas, y c) etapas quísticas .................................................. 10-175 Ciliados (Filum Protozoa) ......................................................... 10-176 Esponjas (Filum Porifera) y Bryozoos (Filum Bryozoa) ........... 10-177 Rotíferos (Filum Rotatoria)......................................................... 10-178 Gusanos (Filum Nemathelminthes) ............................................. 10-179 Tremátodos (Filum Platyhelminthes) y gusanos segmentados (Filum Annelida)...................................................................... 10-180 Crustáceos (Filum Arthropoda, clase crustácea): Tipos de cladóceros (orden Cladocera)........................................................... 10-181 Crustáceos (Filum Arthropoda, clase crustácea): Tipos comunes seleccionados........................................................................... 10-182 Tipos de pupas de insecto .......................................................... 10-183 Plecópteros (orden Plecoptera) .................................................. 10-184 Efímeras (orden Ephemeroptera) ................................................. 10-185 Libélulas (orden Odonata) .......................................................... 10-186 Helgramites y especies afines...................................................... 10-187 Frigáneas (orden Trichoptera)..................................................... 10-188 Moscas de dos alas (orden Díptera)........................................... 10-189 Moscas de dos alas (orden Díptera)........................................... 10-190 Escarabajos (orden Coleóptera) .................................................. 10-191 Chinches (orden Hemiptera, adultos) ......................................... 10-192 Ixxii CONTENIDO PÁG. 32. 33. 34. 35. 36. 37. 38. Moluscos (Filum Molusco): Caracoles (clase Gastropoda).......... Moluscos (Filum Mollusca): Bivalvos (clase Pelecypoda) ........ Tipos de equinodermos (Filum Echinodermata, marinos).......... Algunos invertebrados ............................................................... Algunos tipos de peces (Filum Chordatd) ................................. Tipos de anfibios (Filum Chordata, clase Amphibia) ................ Bacterias y hongos ..................................................................... 10-193 10-194 10-195 10-196 10-197 10-198 10-199 Láminas en color de algas azules-verdes (sección especial en color) . 10-200 A. B. C. D. E. F. Algas saporíferas y odoríferas Algas de obturación de filtros Algas de aguas contaminadas Algas de aguas limpias Algas de plancton y otras algas de aguas superficiales Algas de crecimiento sobre los muros de reservorios PARTE 1000 INTRODUCCIÓN GENERAL 1010 1010 A. INTRODUCCIÓN* Finalidad y aplicación de métodos Los procedimientos descritos en estos estándares están destinados al examen de aguas de calidades comprendidas dentro de amplios márgenes, entre los que se incluyen aguas adecuadas para suministros domésticos e industriales, aguas de superficie, aguas subterráneas, aguas de refrigeración o circulación, aguas de calderas, aguas de alimentación de calderas, aguas residuales urbanas o industriales tratadas o no químicamente, y aguas saladas. La unidad entre los ámbitos del suministro de aguas, del control de su calidad, del tratamiento y la evacuación de aguas residuales se expresa en la presentación de métodos de análisis para cada componente en una sección única para todos los tipos de aguas. Se ha realizado un considerable esfuerzo para presentar los métodos de aplicación más general. En los casos en los que se hacen necesarios métodos alternativos para muestras de diferente composición, los fundamentos para la elección de la opción más adecuada se explican con la mayor claridad posible. Sin embargo, muestras que contengan concentraciones extremas o que presenten composiciones inusuales pueden plantear dificultades e impedir el empleo directo de estos métodos. Por ello, puede ser necesario modificar algún procedimiento en casos específicos. Siempre que se modifique uno de tales procedimientos, el analista debe constatar con claridad la naturaleza de la modificación en el informe de resultados. Algunos procedimientos se destinan a su aplicación en lodos y sedimentos. Una vez más, se ha realizado un importante esfuerzo para presentar los métodos en su más amplio grado de posibles aplicaciones, aunque, cuando se trata de barros o lodos químicos u otras muestras de composición poco habitual, los métodos expuestos en este manual pueden precisar ciertas modificaciones o resultar inapropiados. La mayoría de los métodos tratados han sido refrendados por organismos de control. De hecho, las modificaciones de procedimientos realizados sin aprobación formal pueden resultar inaceptables para un organismo de este tipo. No se incluye en el estudio el análisis de grandes cantidades de productos químicos para el tratamiento de aguas. Un comité de la American Water Works Association prepara y publica estándares al respecto. La información contenida en la parte 1000 tiene utilidad para los laboratorios que desean alcanzar resultados analíticos de una calidad conocida, es decir, de una exactitud conocida y con una incertidumbre conocida dentro de esta exactitud. Para conseguirlo, aplíquense los métodos de garantía de calidad descritos en esta sección a los métodos estándar descritos a lo largo de esta publicación. Otras secciones de la parte 1000 se dedican al equipo de laboratorio, a la seguridad en el laboratorio, a procedimientos de toma de muestras y al desarrollo y la validación de métodos, en todas las cuales se suministra la información necesaria. * La parte 1000 se remitió al Standard Methods Committee a efectos de «revisión y comentarios», y se ofrece únicamente con carácter informativo. No se considera parte de los procedimientos descritos en estos estándares. 1-1 MÉTODOS NORMALIZADOS 1-2 1010 B. 1. Distribución normal Si se repite una medida muchas veces en condiciones esencialmente idénticas, los resultados de cada medida, x, se distribuirán de forma aleatoria en torno a un valor medio (media aritmética) debido a errores incontrolables o experimentales. Si se pudiera acumular un número infinito de estas medidas, sus valores individuales quedarían distribuidos en una curva similar a la que presenta la figura 1010:1. La curva de la izquierda ilustra la distribución normal o gaussiana, descrita de forma precisa por la media, μ, y la desviación estándar, V . La media o promedio de la distribución es sencillamente la suma de todos los valores dividida por el número de valores acumulados, es decir, μ = ¦ i xi / n. Dado que no es posible repetir un número infinito de veces las medidas, se realiza una estimación de la media, mediante el mismo procedimiento de suma aunque con n igual al número finito de medidas repetidas (10 ó 20, o...). Esta estimación de μ se denota como x. La desviación estándar de la distribución normal se define como: De nuevo, el analista no puede sino estimar la desviación estándar, ya que el número de observaciones realizadas es Estadística finito; la estimación de a se denota por , y se calcula del siguiente modo: La desviación estándar fija la anchura, o dispersión, de la distribución normal, e incluye también una fracción fija de los valores que diseñan la curva. Por ejemplo, el 68,27 por 100 de las medidas está comprendido en μ + 1V , el 95,45 por 100 en μ ± 2V , y el 99,70 por 100 en μ ± 3V . Se puede afirmar con suficiente seguridad que el 95 por 100 de los valores está en + 2V , y el 99 por 100 en ± 3V . Cuando se asignan valores a los múltiplos de + V , existen límites de aceptación. Por ejemplo, 10 + 4 indica que los límites de aceptación son 6 y 14, mientras que los valores entre 6 y 14 representan el intervalo de aceptación. Otra útil variable estadística es el error estándar de la media V ȝ , que es la desviación estándar dividida por la raíz cuadrada del número de valores V / n. Ello representa una estimación de la exactitud de la media e implica que otra muestra de la misma población poseería una media comprendida en un intervalo de múltiplos de este error. Los múltiplos de esta variable estadística incluyen la misma fracción de valores que la declarada anteriormente para V . En la práctica, se dispone de un pequeño número de valo- Figura 1010:1. Distribuciones normal y desplazada. INTRODUCCIÓN GENERAL res promedio, por lo que los intervalos de aceptación de la media se expresan como donde t tiene los siguientes valores para intervalos de aceptación del 95 por 100: 1-3 contrará claramente desplazada, tal como se muestra en la parte derecha de la figura 1010:1, en la que la moda, la mediana y la media son muy diferentes. Para obtener una distribución aproximadamente normal, se convierten los resultados en logaritmos y se calcula x y s. Los antilogaritmos de estos dos valores constituyen estimaciones de la media geométrica y la desviación estándar geométrica, xg y sg . 3. En caso de un número pequeño de valores, el uso de t compensa la tendencia a subestimar la incertidumbre. Para n > 15, es normal usar t = 2 en la estimación del intervalo de aceptación del 95 por 100. Otra variable estadística es la desviación estándar relativa, también conocida como coeficiente de variación (CV), que se expresa habitualmente en forma de porcentaje. Esta variable estadística se calcula como 100 ı /μ y su estimación es 100 s/ x . Tal variable estadística normaliza la desviación estándar y en ocasiones facilita la realización de comparaciones directas entre análisis que incluyen una amplia gama de concentraciones. Por ejemplo, si los análisis a bajas concentraciones producen un resultado de 10 ± 1,5 mg/1 y a altas concentraciones de 10 ± 8 mg/1, las desviaciones estándar no parecen comparables. Sin embargo, las desviaciones estándar relativas son 100 (1,5/10) = 15 por 100 y 100 (8/10) = 8 por 100, que indican el menor índice de variación obtenido mediante el uso de este parámetro. 2. Rechazo de datos En una serie de medidas, sucede con bastante frecuencia que uno o más de los resultados difieren en gran medida de los restantes. En teoría, no debe rechazarse ningún resultado, ya que puede indicar un fallo técnico que infunde dudas sobre la validez de todos los resultados o sobre la presencia de una auténtica variante en la distribución. En la práctica, se rechaza el resultado de cualquier análisis en el que se ha producido un error conocido. En estudios medioambientales, las concentraciones extremadamente altas o bajas de sustancias contaminantes pueden ser indicativas de la existencia de áreas con problemas o zonas sin contaminación, por lo que no deben ser rechazadas de forma arbitraria. Existen descripciones de pruebas objetivas para valores extremos1. Si se ordena de modo creciente un conjunto de datos xi, xw...xs, y se calculan el promedio y la desviación estándar, es posible examinar los valores extremos superior e inferior indicativos de posible error mediante el siguiente procedimiento. Se calcula en primer lugar la variable estadística T: Distribución logarítmica normal En muchos casos, los resultados obtenidos a partir de análisis de muestras tomadas del medio ambiente no observan una distribución normal, esto es, la representación gráfica de sus datos se en- T = (xs – x )/s para un valor superior, o T = (x – xi)/s para un valor inferior En segundo término, compárese el valor de T con el valor de la tabla 1010:1 MÉTODOS NORMALIZADOS 1-4 para los niveles de significación de 5 por 100 o 1 por 100. Si el T calculado es mayor que el valor de la tabla para el número de medidas, n, entonces xs o xi es un valor extremo a ese nivel de significación. Consultar en otras fuentes informaciones adicionales sobre las técnicas estadísticas2, 3. 4. 1. 2. 3. Referencias B ARNETT , V. & T. L EWIS. 1984. Outliers in Statistical Data. John Wiley & Sons, Nueva York. NATRELLA, M. G. 1966. Experimental Statistics. National Bur. Standards Handbook 91, Washington, D. C. SNEDECOR, G. W. & W. G. COCHRAN . 1980. Statistical Methods. Iowa State University Press, Ames. TABLA 1010:1. VALORES CRÍTICOS PARA PRUEBAS DE DISCORDANCIA DE 5 POR 100 Y 1 POR 100 PARA UN VALOR EXTREMO SIMPLE EN UNA MUESTRA NORMAL 1010 C. 1. Definición de términos Coeficiente de aceptación: probabilidad, porcentaje, de que el resultado de en una medida esté comprendido en el intervalo de aceptación o entre los límites de aceptación. Glosario Control de calidad: conjunto de medidas que, según una metodología de análisis de muestras, aseguran que el proceso se encuentra bajo control. Duplicado: normalmente, número mínimo de réplicas (dos), aunque en casos específicos se refiere a las muestras du- INTRODUCCIÓN GENERAL plicadas, es decir, dos muestras tomadas en el mismo instante en un lugar concreto. Error aleatorio: desviación en cualquier fase de un procedimiento analítico que puede tratarse mediante técnicas estadísticas estándar. Error de tipo I: también denominado error alfa, es la probabilidad de determinar que un componente está presente cuando en realidad está ausente. Error de tipo II: también denominado error beta, es la probabilidad de no detectar un componente que en realidad está presente. Estándar de comprobación de calibrado: estándar utilizado para determinar el estado de calibrado de un instrumento entre recalibrados periódicos. Estándar de control de laboratorio: estándar, normalmente certificado por un organismo externo, utilizado para medir el sesgo en un procedimiento. Para ciertos componentes y matrices, utilícense los materiales de referencia estándar (Standard Reference Materials) del National Institute of Standards and Technology (NIST)* cuando se encuentren disponibles. Estándar de sustitución: compuesto puro añadido a una muestra en el laboratorio justo antes de su proceso de modo que pueda determinarse la eficacia global de un método. Estándar interno: compuesto puro añadido a un extracto de muestra justo antes del análisis instrumental para permitir la corrección de ineficacias. Evaluación de calidad: procedimiento para la determinación de la calidad de las medidas de laboratorio mediante el empleo de datos a partir de las medidas de control de calidad internas y externas. Exactitud: combinación del sesgo y la precisión de un procedimiento analíti* Anteriormente, National Bureau of Standards (NBS). 1-5 co, que refleja la proximidad de un valor medido a un valor verdadero (véase fig. 1030:1). Garantía de calidad: plan definidor del funcionamiento del laboratorio que especifica las medidas utilizadas para producir datos de una precisión y un sesgo conocidos. Intervalo de aceptación: conjunto de posibles valores en los que el valor verdadero estará comprendido con un grado específico de probabilidad. Límite de aceptación: cada uno de los valores frontera que definen el intervalo de aceptación. Límites de detección: en orden creciente, los diferentes límites son: Límite de cuantificación (LDC): concentración de componente que produce una señal suficientemente mayor que el blanco que puede detectarse en límites específicos por buenos laboratorios durante los acondicionamientos de funcionamiento rutinarios1. Es la concentración típica que produce una señal 10S veces superior a la señal del blanco en agua para reactivos. Límite de detección del método (LDM): concentración de componente que, cuando se procesa a través del método completo, produce una señal con una probabilidad del 99 por 100 de ser diferente del blanco. Para siete réplicas de la muestra, la media debe ser 3,14S superior al blanco, donde s es la desviación estándar de siete muestras. El LDM es mayor que el LID, ya que las repeticiones y las fases de proceso de muestras pueden variar con los componentes y las matrices. Límite de detección instrumental (LDI): concentración de componente que produce una señal superior a cinco veces la relación señal/ruido del instrumento. Similar en muchos aspectos al «nivel crítico» y al «criterio de selección». El último límite se ha establecido en 1,645 veces el valor s de los análisis en blanco. 1-6 MÉTODOS NORMALIZADOS Límite inferior de detección (LID): concentración de componente en agua para reactivos que producen una señal 2(1,645)s por encima de la media de los análisis en blanco, lo que establece los errores de tipo I y II en el 5 por 100. Otra denominación es «límite de detección» (LDD). Precisión: medida del grado de concordancia entre análisis repetidos de una muestra, expresada normalmente como la desviación estándar. Réplica: operación repetida que tiene lugar en un procedimiento de análisis. 1020. Dos o más análisis para el mismo componente en un extracto de una única muestra constituyen análisis de extractos de réplica. Sesgo: desviación consistente de valores medidos a partir del valor verdadero, originada por errores sistemáticos durante un procedimiento. 2. Referencia 1. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1985. National Primary Drinking Water Regulations, 40 CFR Part 141; Federal Register 50: 46936. GARANTÍA DE CALIDAD 1020 A. Introducción La garantía de calidad (GC) es un con-junto de principios de funcionamiento que, si se cumplen estrictamente a lo largo de la toma y el análisis de muestras, generarán datos de calidad reconocida y justificable. En consecuencia, podrá declararse con un alto grado de confianza la exactitud del resultado analítico. En la garantía de calidad se incluyen el control de calidad (sección 1020B) y la evaluación de calidad (sección 1020C). 1. Planificación de la garantía de calidad Establézcase un conjunto de principios de funcionamiento que constituyan un programa de garantía de calidad. Prepárese un plan de GC1 que incluya los siguientes puntos: pliegos de cobertura con Firmas de aprobación del plan, organización y responsabilidades, control de muestras y procedimientos de documentación, procedimiento estándar de funcionamiento (PEF) para cada método analítico, requisitos de formación de analistas, procedimientos de mantenimiento preventivo del equipo, procedimientos de calibrado, acciones correctivas, actividades de control interno de calidad, verificaciones de rendimiento, procedimientos de evaluación de datos para sesgo y precisión, reducción de datos, validación y conjunto de informes. Los pliegos cobertura con las firmas de aprobación indican que el plan ha sido revisado y juzgado corrió adecuado, y que la organización y la división de responsabilidades esbozan la cadena jerárquica y asignan funciones específicas; s cada una de las personas involucradas 1-7 INTRODUCCIÓN GENERAL El control de muestras y los procedimientos de documentación permiten seguir la pista de una muestra y de sus derivados a través de todas las etapas, desde la toma de muestras al análisis y la visualización de resultados. Siempre importante, la documentación lo es especialmente en los casos en que se impone la necesidad de hacer un seguimiento. Un procedimiento estándar de funcionamiento para el método analítico describe el método con tal grado de detalle que un analista experto, incluso no familiarizado con el método, puede obtener resultados aceptables. Los requisitos de formación de analistas deben especificarse. El número de análisis necesarios y la incertidumbre de los resultados variarán en función del tipo de análisis, de las características de la muestra y de la experiencia del analista. Es necesario aplicar procedimientos de mantenimiento preventivo del equipo. Un programa estricto de mantenimiento preventivo reducirá los fallos de funcionamiento de los instrumentos, mantendrá el nivel del calibrado y reducirá los períodos de paralización. Los procedimientos de calibrado, las acciones correctivas, las actividades de control de calidad, las verificaciones de rendimiento y las evaluaciones de datos para sesgo y precisión se exponen en las secciones 1020B y C. La reducción de datos, la validación y el conjunto de informes constituyen los rasgos finales de un programa de GC. La lectura obtenida a partir de un instrumento analítico debe ajustarse a factores tales como la eficacia del instrumento, la eficacia de extracción, el tamaño de la muestra y el valor del fondo antes de llegar a ser un resultado de utilidad. El plan de GC especifica los factores de corrección que han de aplicarse al tiempo que las etapas que se deben seguir en la validación de resultados. Comuníquense los resultados en unidades estándar de masa, volumen o concentración. Utilícese un método prescrito para comunicar resultados por debajo del límite de detección del método. Acompañar cada resultado o conjunto de resultados de la declaración del factor de incertidumbre. 2. Referencia 1. STANLEY, T. W. & S. S. VERNER. 1983. Interim Guidelines and Specifications for Preparing Quality Assurance Project Plans. EPA-600/4-83-004, U. S. Environmental Protection Agency, Washington, D. C. 1020 B. Control de calidad El control de calidad (CC) puede ser interno o externo. El objeto de esta sección es el CC interno; el CC externo, también denominado «avaluación de calidad», se expone en la sección I020C Todos los analistas utilizar, algunos CC en forma de esfuerzos intuitivos para producir rebultados creíbles. Sin embargo, un buen programa de control de calidad se compone, al menos, de siete elementos: certificación de la competencia del operador, recuperación de adiciones conocidas, análisis de estándares suministrados de forme interna, análisis de blancos de reactivos, calibrado mediante estándares, análisis de duplicados y mantenimiento de gráficos de control. Las secciones 1010 y 1030 contienen los cálculos necesarios. MÉTODOS NORMALIZADOS 1-8 1. Certificación de la competencia del operador Antes de permitir que un analista lleve a cabo trabajos susceptibles de ser comunicados, ha de demostrarse su competencia en la realización de los análisis. Los requisitos son variables, aunque para la mayoría de los análisis químicos orgánicos e inorgánicos es suficiente la demostración de sesgos y precisiones aceptables para operación en solitario. Realícese un mínimo de cuatro análisis de réplica de una muestra de comprobación preparada independientemente con una concentración comprendida entre 5 y 50 veces el límite de detección del método (LDM) para el análisis en ese laboratorio. En la figura 1020:1 se muestran los límites generales para trabajos aceptables; algunos métodos pueden especificar límites más rigurosos. 2. Recuperación de adiciones conocidas Utilícese la recuperación de adiciones conocidas como parte de un protocolo analítico regular. Empléense adiciones conocidas para verificar la ausencia de efectos de matriz. Cuando se precise analizar un nuevo tipo de matriz, verifíquese la magnitud de la interferencia. En los casos en que no sea posible aplicar duplicados, por ejemplo cuando el componente de interés se encuentra ausente, realícese una recuperación de adiciones conocidas para el 10 por 100 de las muestras. Si también se encuentran en fase de análisis los duplicados, la suma de los duplicados y las adiciones conocidas debe ser igual como mínimo al 10 por 100 del número de muestras. Realícese la adición conocida entre 5 y 50 veces el LDM o entre 1 y 10 veces el nivel ambiental, por elevados que sean. No deben emplearse adiciones conocidas por encima de la amplitud lineal demostrada del método; utilizar solucio- nes concentradas cuyo cambio de volumen en la muestra sea despreciable. Véanse en la tabla 1020:1 los límites aceptables. 3. Análisis de estándares de suministro externo Los estándares de suministro externo deben analizarse, como mínimo, en los casos en que el análisis de adiciones conocidas no produce recuperaciones aceptables, o una vez al día, según lo que sea más frecuente. Utilícense estándares de control de laboratorio con una concentración entre 5 y 50 veces el LDM o próxima a los niveles ambientales de muestra, en función de cuál sea mayor. Cuando sea posible, utilícense materiales certificados de referencia como estándares de control de laboratorio. Son preferibles, cuando se pueda disponer de ellos, los materiales de referencia estándar (Standard Reference Materials) del National Institute of Standards and Technology (NIST)*. Si se utilizan materiales internos de referencia, prepárense de forma independiente a partir de los estándares usados para el calibrado. Véase tabla 1020:1 sobre límites aceptables para duplicados de alto nivel. 4. Análisis de blancos de reactivos Analícense blancos de reactivos en todos los casos en que se utilicen nuevos reactivos y con la frecuencia que requieran los métodos específicos. Analícese un mínimo del 5 por 100 de la carga de muestra como blancos de reactivos; ello garantiza el seguimiento de la pureza de los reactivos y los blancos de procedimientos globales. Analícese un blanco de reactivos después de cualquier muestra con una concentración superior a la del mayor estándar o que pudiera producir un arrastre de una muestra con la siguiente. * Anteriormente, National Bureau of Standards (NBS). INTRODUCCIÓN GENERAL 1-9 TABLA 1020:1. LÍMITES DE ACEPTACIÓN PARA MUESTRAS DUPLICADAS Y ADICIONES CONOCIDAS SOBRE AGUAS LIMPIAS Y RESIDUALES * Adiciones calculadas como % de la adición conocida recuperada; duplicados calculados como la diferencia en porcentaje de la media. † Bajo nivel se refiere a concentraciones inferiores a 20 veces el LDM. Alto nivel se refiere a concentraciones superiores a 20 veces el LDM. ‡ También límites de aceptación para estándares de control de laboratorios independientes y certificación de competencia del operador. F UENTE : N ATRELLA , M. G. 1966. Experimental Statistics. National Bur. Standards Handbook 91, Washington, D.C. 5. Calibrado con estándares Como mínimo, se miden tres diluciones diferentes del estándar una vez iniciado el análisis. A continuación, verifíquese diariamente la curva estándar mediante el análisis de uno o más estándares en la amplitud lineal, tal como se especifica en el método individual. Los resultados analíticos comunicables son aquellos comprendidos en la escala de las diluciones estándar utilizadas. No deben comunicarse valores por encima del estándar superior a no ser que se haya realizado una comprobación inicial de la mayor amplitud lineal, no se hayan modificado los parámetros y el valor sea inferior a 1,5 veces el estándar superior. El valor inferior comunicable es el LDM, siempre que el estándar de calibrado más bajo sea inferior a 10 veces el LDM. Si se retira el blanco, comunicar el resultado incluso si es negativo. 6. Análisis de duplicados Cuando la mayoría de las muestras poseen índices medibles del componente en fase de determinación, resulta eficaz el análisis de muestras duplicadas para evaluar la precisión. Analícese el 5 por 100 o 1-10 MÉTODOS NORMALIZADOS más de las muestras en duplicados. Analícense duplicados y adiciones conocidas en matrices representativas de las muestras analizadas en el laboratorio. Véase tabla 1020:1 sobre límites aceptables para análisis duplicados. 7. Gráficos de control En los laboratorios se utilizan normalmente tres tipos de gráficos de control1: un gráfico de recursos para estándares, ya sean estándares de control de laboratorio (ECL) o de control de calibrado (ECC); un gráfico de recursos para resultados de fondos o blancos de reactivos; y un gráfico de rango para análisis de réplicas. Los gráficos constituyen instrumentos básicos para el control de calidad. Los diferentes tipos de gráficos se describen a continuación. a) Gráficos de recursos: Los gráficos de recursos para estándares se construyen a partir de la media y la desviación media de un estándar. Esto incluye niveles de alarma (NA) superior e inferior y niveles de control (NC) superior e inferior. Es práctica corriente utilizar los límites ±2s y +3s para el NA y el NC, respectivamente, donde s representa la desviación estándar. Dedúzcanse estos valores a partir de los valores declarados para materiales de referencia estándar, si se utilizan para estándares de control de laboratorio (ECL) o estándares de comprobación de calibrado (ECC), o a partir de análisis de réplica de ECC. El gráfico puede establecerse mediante el uso de valores calculados para la media y la desviación estándar o de porcentajes. Los porcentajes son necesarios cuando varía la concentración. Constrúyase un gráfico para cada instrumento de análisis. Introdúzcanse los resultados sobre el gráfico cada vez que se analice el ECL o el ECC. En la figura 1020:1 se proporcionan ejemplos de gráficos de control para recursos. b) Gráfico de amplitud: Si se conoce la desviación estándar del método, utilícense los factores de la tabla 1020:11 para construir la línea central y los límites de alarma y de control tal como se muestra en la figura 1020:2. Una perfecta concordancia entre los duplicados conduce a una diferencia nula cuando se restan los valores, de modo que la línea de base del gráfico es cero. Se convierte la desviación estándar en la amplitud, de manera que el analista no tenga más que restar los dos resultados para dibujar el valor sobre el gráfico de control. La amplitud media se calcula como: el límite de control como y el límite de alarma como donde: D 2 = factor para convertir s en la amplitud (1,128 para duplicados, como se indica en la tabla 1020:11). s(R) = desviación estándar de la amplitud, y D4 = factor para convertir la amplitud media en 3s(R) 3,267 para duplicados, como se indica en la tabla 1020:11). Un gráfico de amplitud es bastante sencillo cuando se utilizan análisis duplicados de un estándar (fig. 1020:2). Para análisis duplicados de muestras, el dibujo parecerá diferente en virtud de la variación en la concentración de muestras. Si se supone una desviación estándar relativa constante en la amplitud de concentración de interés, los valores R,D4 R, etcétera, pueden calcularse de la forma anteriormente expuesta para varias concentraciones, con una curva suave dibujada a través de los puntos obtenidos, y de esta forma puede determinarse un rango acep- INTRODUCCIÓN GENERAL 1-11 Figura 1020:1. Gráficos de control para recursos. TABLA 1020:11. FACTORES PARA EL CÁLCULO DE LÍNEAS EN GRÁFICOS DE CONTROL DE AMPLITUD FUENTE: ROSENSTEIN, M. & A. S. GOLDEN. 1964. Statistical Techniques for Quality Control of Environmental Radioassays. AQCS Rep. Stat-1. Public Health Serv., Winchester, Massachusetts. table para duplicados para cualquier concentración media. La figura 1020:3 ilustra un gráfico de este tipo. Para el seguimiento de la precisión en el transcurso del tiempo, se hará necesaria una tabla independiente como la sugerida debajo de la figura. Con mayor frecuencia, el rango puede expresarse como una función de la desviación estándar relativa (coeficiente de variación). Normalizar el rango por división por el promedio. Determinar la amplitud media para los pares analizados mediante: y la varianza (cuadrado de la desviación estándar) como Dibújense a continuación las líneas sobre el gráfico en R + 2sR y R + 3sR y, para cada análisis de duplicados, calcúlese la amplitud normalizada e introdúzcase el resultado en el gráfico. La figura 1020:4 es un ejemplo de un gráfico de este tipo. 1-12 MÉTODOS NORMALIZADOS Figura 1020:4. Gráfico de amplitud para amplitudes variables. Figura 1020:2. Análisis duplicados de un estándar. Figura 1020:3. Gráfico de amplitud para concentraciones variables. c) Análisis de gráficos: Si los límites de alarma (LA) se encuentran en un nivel del 95 por 100, un punto de cada 20, como promedio, superará este límite, mientras que únicamente uno de cada 100 superará el límite de control (LC). Empréndanse las siguientes acciones, basadas en estos parámetros estadísticos, que se ilustran en la figura 1020:5: Límite de control: Si una medida supera el límite de control, repítase el análisis inmediatamente. Si la repetición se encuentra dentro del LC, continúese el análisis; si lo supera, hay que interrumpir el análisis y corregir el problema. Límite de alarma: Si dos de cada tres puntos sucesivos superan el LA, analícese otra muestra. Si el siguiente punto es inferior al LA, continúese el análisis; si el punto siguiente supera el LA, hay que interrumpir el análisis y corregir el problema. Desviación estándar: Si cuatro de cinco puntos consecutivos superan 1s, o están en orden creciente o decreciente, analícese otra muestra. Si el siguiente punto es inferior a 1s, o altera el orden, continúese el análisis; en caso contrario, hay que interrumpir el análisis y corregir el problema. Línea central: Si seis muestras sucesivas se encuentran por encima de la línea central, analícese otra muestra. Si el punto siguiente está por debajo de la línea central, continúese el análisis; si el siguiente punto se encuentra en el mismo lado, hay que interrumpir el análisis y corregir el problema. 1-13 INTRODUCCIÓN GENERAL Figura 1020:5. Gráfico de control de recursos con datos fuera de control (mitad superior). Las anteriores consideraciones se aplican cuando las condiciones están por encima o por debajo de la línea central, pero no en ambos lados; por ejemplo, cuatro de cinco valores deben superar o bien + ls o bien – 1S. Después de corregir el problema, se vuelve a analizar la mitad de las muestras analizadas entre la última medida controlada y la última fuera de control. Otra función importante en el gráfico de control es la evaluación de las mejoras en la precisión del método. En los gráficos de amplitud y de recursos, si las medidas nunca o rara vez superan el LA, recalcular el LA y el LC mediante el uso de los 20 puntos de datos más recientes. Las tendencias en la precisión se pueden detectar con mayor prontitud si se investigan promedios corrientes de 20 sobre bases diarias. 8. Referencia 1. GOLDEN, A. S. 1984. Evaluation of internal control measurements in radioassay. Health Phys. 47:361. 1020 C. Evaluación de calidad La evaluación de calidad consiste en el proceso de utilización de medidas de control de calidad externo e interno con objeto de determinar la calidad de los datos obtenidos en el laboratorio. Incluye apartados tales como muestras de evaluación de rendimiento, muestras de comparación entre laboratorios, y verificaciones de rendimiento de forma análoga al CC interno descrito en la sección 1020B, que se aplican para examinar la recuperación, el sesgo, la precisión, el lí- MÉTODOS NORMALIZADOS 1-14 TABLA 1020:111. VERIFICACIÓN DE UN PROCEDIMIENTO DE ANÁLISIS DE SUELO Procedimiento Comentario 1. 2. 3. Muestra introducida en el cuaderno Muestra pesada Procedimiento de secado seguido sí sí no 4a. b. 5. 6. Balanza calibrada Limpia y ajusta en cero Muestra base Molino de bolsas limpio sí sí sí sí Observaciones número de laboratorio asignado peso en seco mantenimiento del horno no realizado una vez al año semanal al pasar 50 retículas debe hacerse después de cada muestra mite de detección y la adhesión a los requisitos de los procedimientos de funcionamiento estándar. 1. Muestras de evaluación del rendimiento Utilícense muestras con cantidades conocidas del componente proporcionadas por un organismo externo o muestras aleatorias preparadas en el propio laboratorio para determinar los resultados obtenidos por el analista. En general, deberá establecerse de antemano la incertidumbre del método; una recuperación aceptable está comprendida en el grado establecido de incertidumbre. Por ejemplo, si la amplitud aceptable de recuperación para una sustancia es del 85 al 115 por 100, se supone que el analista debe conseguir una recuperación dentro de dicha amplitud para todas las muestras de evaluación del rendimiento. 2. Verificaciones del rendimiento Realizar únicamente verificaciones no planificadas mediante un registro de comprobación que permita establecer el modo en que se trata una muestra desde el momento de la recepción hasta la comunicación final de resultados. El objetivo consiste en detectar cualquier desvia- ción del procedimiento estándar de funcionamiento de modo que pueden adoptarse medidas correctivas. En la tabla 1020:111 se muestra un formato recomendado en cuyo registro de comprobación se incluyen algunos apartados iniciales. 3. Muestras de comparación entre laboratorios Los programas comerciales estatales suministran muestras que contienen diversos componentes en diferentes matrices. Para obtener un buen programa de evaluación de calidad es necesario participar en estudios periódicos comparativos de laboratorio. La frecuencia de participación debe ajustarse a la calidad de los resultados obtenidos por los analistas cuyo trabajo está siendo examinado en el estudio. Como procedimiento rutinario es razonable hacer análisis trimestrales. Los organismos oficiales encargados de la dirección de tales estudios se relacionan a continuación. a) Compuestos orgánicos e inorgánicos en agua: Las muestras se hallan disponibles en el Environmental Monitoring Systems Laboratory, EPA, Estados Unidos, 26 West St. Clair, Cincinnati, Ohio 45268. Se pueden utilizar como muestras de evaluación del rendimiento o como estándares para estudios comparativos de laboratorio. 1-15 INTRODUCCIÓN GENERAL b) Radioisótopos en medios diversos: Estas muestras se hallan disponibles en el Environmental Monitoring Systems Laboratory, EPA, Estados Unidos, P. O. Box 93478, Las Vegas, Nevada 891933478. Pueden suministrarse para su uso como muestras de evaluación del rendimiento; el Laboratorio dirige también los estudios comparativos de laboratorio1. Muestras medioambientales con niveles de fondo de radioisótopos se hallan disponibles en División of Laboratories and 1030 4. Referencia 1. JARVIS, A. N. & L. Siu. 1981. Environmental Radioactivity Laboratory. Intercomparison Studies Program. EPA-600/4-81-004, U.S. Environmental Protection Agency, Las Vegas, Nevada. CALIDAD DE DATOS 1030 A. 1. Research, International Atomic Energy Agency, P. O. Box 100, A-1400 Viena, Austria. Introducción Indicadores de calidad Los principales indicadores de la calidad de datos son su sesgo (sección 1030B) y su precisión (sección 1030C), que, combinados, expresan su exactitud. La relación entre estos términos se muestra en la figura 1030:1. De los cuatro resultados posibles, únicamente es exacta la condición de bajo sesgo y alta precisión Indicadores auxiliares de calidad de datos son el límite de detección del método (sección 1030E) y la representatividad. Esta última se puede relacionar tanto con la propia muestra como con la población muestreada. Un método determinado puede tener un alto grado de exactitud, pero, si los resultados no son representativos de la muestra o la muestra no representa a la población, los datos no son de utilidad. La representatividad de la muestra alcanza su mejor evaluación mediante el análisis de diversas muestras en el mismo lugar o a partir de una gran cantidad de muestras de muy diversa procedencia. La representatividad del método se determina de manera óptima mediante la combinación de estudios que utilizan métodos diferentes. 2. Figura 1030:1. Definición de exactitud. Referencia 1. U. S. E NVIRONMENTAL P ROTECTION A GEN CY. 1980. Health Physics Society Committee Report HPSR-1: Upgrading Environmental Radiation Data. EPA-520/1-80-012, U.S. Environmental Protection Agency, Washington, D.C. 1-16 MÉTODOS NORMALIZADOS 1030 B. El sesgo es una medida del error sistemático. Contiene dos componentes: uno se deriva del método, y el otro, del uso del método en el laboratorio. El sesgo de un método se mide adecuadamente mediante la realización de estudios comparativos de laboratorio en los que dicho sesgo se define como la diferencia entre el promedio general y el valor conocido (o verdadero). El sesgo de laboratorio es la diferencia existente entre media de resultados del laboratorio y los valores verda- 1030 C. 1. Definición La precisión es una medida del grado de concordancia entre los análisis múltiples de una muestra dada. Se evalúa mediante análisis de réplicas, análisis repetidos de un estándar estable o análisis de adiciones conocidas sobre las muestras. La precisión se especifica por la desviación estándar de los resultados1. Si se desea obtener la precisión global de un estudio, hay que analizar muestras duplicadas. En esta valoración se incluyen los errores aleatorios implicados en la toma de muestras, así como los errores en la preparación y análisis de muestras. Cuando se emplean sólo algunas repeticiones, por ejemplo, análisis de muestras duplicadas o de extractos duplicados, la amplitud de resultados, R, es aproximadamente tan eficaz como la desviación estándar, ya que las dos medidas difieren en una constante (1,128s = R para duplicados, l,693s = R para triplicados). Sesgo deros, siendo, por consiguiente, una combinación de dos sesgos. Evalúese el sesgo de laboratorio mediante la valoración de los resultados de adiciones conocidas o por medio del análisis de muestras duplicadas registrando el signo de las diferencias al calcular la diferencia promedio. De este valor ha de sustraerse el sesgo del método de un estudio comparativo para determinar el sesgo debido a las prácticas de laboratorio. Precisión 2. Cálculo La tabla 1030:1 contiene los resultados de análisis duplicados y valores de recuperación de los análisis repetitivos de un estándar estable. La tabla 1030:11 muestra el cálculo de precisión que utiliza adiciones conocidas. Para análisis duplicados, la diferencia promedio, o amplitud media, se calcula como la suma de todas las diferencias (valores absolutos) y su división por el número de observaciones: R = ¦ di / n. Esto se transforma en la desviación estándar al dividirse por 1,128. Para análisis múltiples de un estándar estable, calcúlese la desviación estándar del modo acostumbrado. El valor verdadero del estándar es irrelevante para este análisis. Si se utilizan adiciones conocidas para determinar la precisión, como en el ejemplo de la tabla 1030:11, se resta el valor de la recuperación del valor conocido (restar la columna 3 de la columna 4) y se calcula la desviación estándar según el 1-17 INTRODUCCIÓN GENERAL método habitual, tal como se muestra en el pie de la tabla. Si la desviación estándar está en proporción constante con la cantidad presente, puede utilizarse entonces el coeficiente de variación (desviación estándar relativa, s/ x ) en lugar de la desviación estándar. TABLA 1030:1. 3. Referencia 1. AMERICAN SOCIETV FOR TESTING AND MATERIALS. 1977. Standard Practice for Determinaron of Precision and Bias of Methods. Committee D-19 on Water, Designation D2777-77, American Soc. Testing & Materials, Filadelfia, Pennsylvania. CÁLCULOS DE PRECISIÓN MEDIANTE EL USO DE DUPLICADOS 1-18 MÉTODOS NORMALIZADOS TABLA 1030:11. CÁLCULOS DE PRECISIÓN PARA ADICIONES CONOCIDAS 1030 D. 1. Incertidumbre total Definición y cálculo Esta variable estadística constituye una apropiada combinación de las incertidumbres sistemática y aleatoria en un sistema dado de medidas. Las incertidumbres aleatorias se evalúan mediante el cálculo de la precisión; se obtienen de forma estadística. Las incertidumbres sistemáticas se refieren en cambio a los sesgos y a aquellas incertidumbres aleato- rias que no pueden ser evaluadas de forma estadística. Dado que estas últimas pueden estimarse únicamente a partir de un conocimiento profundo de todas las etapas del proceso de medida, según el juicio de un analista experto, existe una considerable vaguedad en la evaluación. Dado que los sesgos pueden ser evaluados, lo habitual es que sólo se consideren éstos en la evaluación de la incertidumbre total. Estos sesgos se pueden produ- 1-19 INTRODUCCIÓN GENERAL cir en varios puntos del sistema, como por ejemplo en el peso de la muestra, en el resultado producido por el instrumento analítico, en cambios en la calidad de los reactivos o en extractos incompletos. Cuando las incertidumbres aleatorias han sido evaluadas con la desviación estándar (sx) y los sesgos se representan por bi, estas cantidades pueden combinarse de forma cuadrática para calcular la incertidumbre1: En general, los sesgos de extracción e instrumental (B) son los de mayor magnitud, con lo que la ecuación se simplifica como: Para expresar la incertidumbre en forma de niveles de aceptación, se supone 1030 E. 1. que el sesgo es conocido con un pequeño error y se añade a los niveles de aceptación superior de la varianza; por ejemplo, para un nivel de aceptación del 95 por 100: Si la concentración del componente se encuentra en una amplitud estrecha, utilícese para B y sx unidades de concentración; en caso contrario, empléese el coeficiente de variación para sx y la forma parcial de B. 2. Referencia 1. U. S. E NVIRONMENTAL P ROTECTION A GEN CY. 1980. Health Physics Society Committee Report HPSR-1: Upgrading Environmental Radiation Data. EPA-520/1-80-012, U.S. Environmental Protection Agency, Washington, D.C. Límite de detección del método Introducción Los límites de detección son objeto de controversia, principalmente por motivo de su inadecuada definición y por cierta confusión de términos. Con frecuencia, el límite de detección instrumental se emplea como límite de detección del método, y al contrario. Independientemente del término que se utilice, la mayoría de los analistas coinciden en que el límite de detección se define a partir de la más pequeña cantidad detectable por encima del ruido en un procedimiento y dentro de un límite declarado de aceptación. Los límites de aceptación se establecen de modo que las probabilidades de que se presenten errores de tipo I y de tipo II sean razonablemente pequeñas. La práctica común identifica varios límites de detección (véase 1010C), cada uno de los cuales posee un propósito definido. Éstos son el límite de detección del instrumento (LDI), el límite inferior de detección (LID), el límite de detección del método (LDM) y el límite de cuantificación (LDC). Ocasionalmente, el límite de detección del instrumento se utiliza como guía para la determinación del LDM. La relación entre todos estos límites es aproximadamente LDI:LID:LDM:LDC = 1:2:4:10. 2. Determinación de los límites de detección Un instrumento de análisis en funcionamiento produce normalmente una señal (ruido) incluso cuando no existe ninguna muestra o se analiza un blanco. Dado que todo programa de GC requie- 1-20 re frecuentes análisis de blancos, la media y la desviación estándar llegan a conocerse bien, la señal de los blancos se hace muy precisa, lo que significa que la curva gaussiana de la distribución de blancos llega a ser muy estrecha. El LDI es la concentración del componente que produce una señal superior a tres desviaciones estándar del nivel de ruido medio o que puede determinarse mediante la inyección de un estándar para producir una señal que sea cinco veces la relación señal-ruido. El LDI resulta muy útil para valorar la concentración de los componentes o la cantidad de un extracto necesaria para producir una señal que permita calcular un límite de detección del método estimado. El LID es la cantidad de componente que produce una señal suficientemente grande como para que pueda detectarse en el 99 por 100 de los ensayos realizados con dicha cantidad. Determínese el LID mediante inyecciones múltiples de un estándar a concentración casi cero (concentración no superior a cinco veces el LDI). Determínese la desviación estándar según el método habitual. Para reducir la probabilidad de un error de tipo I (detección falsa) al 5 por 100, multiplíquese s por 1,645 a partir de una tabla normal acumulativa. Para reducir también la probabilidad de un error de tipo II (no detección falsa) al 5 por 100, duplíquese esta cantidad a 3,290. Por ejemplo, si 20 determinaciones de un estándar de bajo nivel produjeron una desviación estándar de 6 μg/1, el LID sera 3,29 x 6 = = 20 μg/11. El LDM se diferencia del LID en que las muestras que contienen el componente de interés son procesadas a través del método analítico completo. El límite de detección del método es mayor que el LID en virtud de los factores de eficacia de extracción y concentración de los extractos. El LDM puede ser obtenido por analistas experimentados mediante el uso de instrumentos bien calibrados MÉTODOS NORMALIZADOS sobre una base no rutinaria. Por ejemplo, para determinar el LDM se añade un componente al agua para reactivos o a la matriz de interés, al objeto de obtener una concentración próxima al LDM estimado2. Analícense siete partes de esta solución y calcúlese la desviación estándar (s). A partir de una tabla de distribución desigual de t, selecciónese el valor de t para 7 – 1 = 6 grados de libertad y en el nivel 99 por 100; este valor es 3,14. El producto de 3,14 veces s es el LDM deseado. Si bien el LDC resulta de utilidad dentro de un laboratorio, es mayor la utilidad del límite de cuantificación práctica (LCP) definido como el nivel inferior registrable en los límites especificados a lo largo de las operaciones rutinarias de laboratorio3. El LCP tiene una especial importancia por cuanto que laboratorios diferentes producirán LDM distintos incluso si utilizan idénticos procedimientos de análisis, instrumentos y matrices de muestra. El LPC equivale aproximadamente a cinco veces el LDM y representa un límite de detección práctico y alcanzable de forma rutinaria con una certeza relativamente elevada de que los valores comunicados son fiables. 3. Descripción de los límites La figura 1030:2 ilustra los límites de detección expuestos en los apartados anteriores. En esta figura se supone que las señales obtenidas a partir de los instrumentos de análisis poseen una distribución normal y pueden representarse por una curva normal (gaussiana)4. La curva designada por B es representativa de la distribución de señales de blancos o de fondo. Tal como se muestra, la distribución de las señales de blancos es aproximadamente de la misma anchura que las de las restantes distribuciones, es decir, V B V I V L En la medida en que continúe el análisis de blancos, esta curva 1-21 INTRODUCCIÓN GENERAL Figura 1030:2. Relación entre limites de detección se estrechará aún más a causa de los grados crecientes de libertad. La curva designada por I representa el LDI. Su valor medio se sitúa a kV B unidades de distancia de la curva de blancos, y k representa el valor de t (de la distribución desigual de t) que corresponde al límite de aceptación elegido para describir el rendimiento del instrumento. Para un límite del 95 por 100 y n = 14, k = 1,782, y para un límite del 99 por 100, k = 2,68. El solapamiento de las curvas B e I indica la probabilidad de no detección de un componente cuando éste se encuentra presente (error de tipo II). La curva en el extremo derecho de la figura 1030:2 representa el LID. Dado que para el cálculo de LDI y LID se utiliza un número finito de determinaciones, las curvas son más amplias que el blanco, aunque semejantes, lo que hace razonable elegir V I V L . Por consi- guiente, el LID está a una distancia 2kV L de la curva de blancos. 4. 1. Referencias AMERICAN SOCIETY FOR TESTING AND MATERIALS. 1983. Standard Practice for Intralabo- ratory Quality Control Procedures and a Discussion on Reporting Low-Level Data. Designation D4210-83, American Soc. Testing & Materials, Filadelfia, Pensylvania. 2. GLASER , J. A., D. L. F OERST , J. D. M CKEE, S. A. Q UAVE & W. L. B UDDE . 1981. Trace analyses for wastewaters. Environ. Sci. Technol. 15:1426. 3. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1985. National Primary Drinking Water Standards: Synthetic Organics, Inorganics, and Bacteriologicals. 40 CFR Part 141; Federal Register 50: No. 219, 13 de noviembre, 1985. 4. OPPENHEIMER, J. & R. TRUSSELL. 1984. Detection limits in water quality analysis. In Proc. Water Quality Technology Conference (Denver, Colorado, 2-5 de diciembre, 1984). American Water Works Assoc, Denver, Colorado. 1-22 MÉTODOS NORMALIZADOS 1030 F. Valoración de la corrección de los análisis Los procedimientos que se exponen a continuación para la valoración de la corrección de los análisis son aplicables de forma específica a las muestras de agua para las que se han realizado análisis relativamente completos1. Entre ellos se incluyen el pH, la conductividad, los sólidos de disolución total (SDT) y los principales componentes aniónicos y catiónicos, que son indicadores de la calidad general del agua. Las comprobaciones descritas no requieren análisis adicionales de laboratorio. Tres de las valoraciones precisan la realización de cálculos de los sólidos de disolución total y de la conductividad de los componentes medidos. Para calcular los sólidos de disolución total se suman las concentraciones (en miligramos por litro) de los componentes del siguiente modo: Calcúlese la conductividad eléctrica mediante la ecuación: donde: G= conductividad de la solución salina, C= concentración de la solución salina, O = conductancia equivalente de la solución salina en una dilución infinita, K1, k2= constantes para la mitigación de los efectos de nube iónica y electroforético relativos a la movilidad de los iones 1 . litro, deben estar equilibradas, ya que las aguas potables son eléctricamente neutras. El examen se basa en la diferencia porcentual definida del siguiente modo: y los criterios de aceptación se establecen como sigue: 2. La concentración medida de sólidos de disolución total debe ser superior a la calculada, ya que es posible que algún contribuyente significativo no haya sido incluido en el cálculo. Si el valor medido es inferior al calculado, ello puede deberse a algún error en la suma iónica superior y en el valor medido, por lo que debería volverse a analizar la muestra. Si la concentración de sólidos medida es superior en un 20 por 100 a la calculada, la suma inferior de iones no es fiable, por lo que deberían volverse a analizar los componentes seleccionados. La relación aceptable es la siguiente: 3. 1. Equilibrio entre aniones y cationes2 Las sumas de aniones y cationes, cuando se expresan en miliequivalentes por SDT medido = SDT calculado2 CE medida = CE calculada Si la conductividad calculada es superior al valor medido, hay que volver a analizar la suma superior de iones. Si la CE es inferior a la medida, hay que vol- 1-23 INTRODUCCIÓN GENERAL ver a analizar la suma iónica inferior. La relación aceptable es: un valor tan alto como 0,8 veces la CE. El criterio aceptable para esta relación es: SDT calculado/conductividad = 0,55-07 4. CE medida y sumas iónicas Las sumas de aniones y cationes deben ser 1/100 veces el valor medido de la CE. Si alguna de las dos sumas no cumple este criterio, dicha suma no es fiable; analícese de nuevo la muestra. Los criterios aceptables son: 100 x suma anión (o catión), meq/1 = (0,9-11) CE 5. Relación SDT-CE calculada Si la relación calculada SDT-conductividad está por debajo de 0,55, la suma iónica inferior no es fiable; hay que analizarla de nuevo. Si la relación está por encima de 0,7, la suma iónica superior no es fiable; hay que analizarla de nuevo. Si los nuevos análisis no producen cambios en la suma iónica inferior, algún componente no medido, como amoníaco o nitrito, ha de estar presente en una concentración significativa. Si se encuentran presentes iones calcio o sulfato pobremente disociados, el SDT puede alcanzar 6. Relación SDT-CE medida Los criterios aceptables para esta relación comprenden desde 0,55 hasta 0,7. Si la relación SDT-CE se encuentra fuera de estos límites, la SDT o la conductividad medidas no son fiables; hay que analizarlas de nuevo. Existen publicaciones que contienen descripciones más completas sobre las valoraciones del control de calidad 3. 7. 1. Referencias ROSSUM, J. R. 1975. Checking the accuracy of water analyses through the use of conductivity. J. Amer. Water Works Assoc. 67:204. 2. F RIEDMAN , L. C. & D. E. E RDMANN , 1982. Quality Assurance Practices for Analyses of Water and Fluvial Sediments. Tech. Water Resources Inc., libro 5, capítulo A6. U.S. Government Printing Off., Washington, D.C. 3. OPPENHEIMER, J. & A. D. EATON. 1986. Quality control and mineral analysis. in Proc. Water Quality Technology Conference (Houston, Texas, 8-11 de diciembre, 1985). American Water Works Assoc, Denver, Colorado. 1-24 MÉTODOS NORMALIZADOS 1040 DESARROLLO Y EVALUACIÓN DE MÉT0B0S 1040 A. Son muchas las fuentes reconocidas a nivel nacional que pueden proporcionar métodos estándar; no obstante, es posible que en algunas situaciones dichos métodos no sean utilizables, o puedan darse casos en los que no existan métodos estándar para un componente con- 1040 B. Introducción ceto. Por consiguiente, tal vez sea necesario el desarrollo de métodos. El desarrollo de métodos es el conjunto de procedimientos experimentales ideados para medir una cantidad conocida de un componente en diversas matrices. Validación del método Cuando se desarrolla un método completamente nuevo mediante procedimientos aceptables de investigación o se modifica un método existente con el fin de reunir requisitos especiales, se precisa de un proceso de validación en tres etapas: determinación del sesgo y la precisión para un orador único, análisis de muestras desconocidas preparadas de forma independiente y determinación de la rigurosidad del método. 1. Características del operador único Esta parte del procedimiento de validación requiere determinación del límite de detección del método (LDM), de forma análoga a la descrita en la sección 1030; el sesgo del método; y la precisión obtenible por un único operador, es decir, el error aleatorio introdujo en la utilización del método. Para realizar es- tas determinaciones, se analizan como mínimo siete porciones, aunque es preferible que sean 10 o más, de un estándar para cada una de las diversas concentraciones utilizadas en cada matriz. Utilícese una concentración en el LDM, o ligeramente por encima, y una relativamente alta, de modo que pueda especificarse para la que es aplicable el método. El empleo de varias concentraciones para determinar el sesgo y la precisión revelará la naturaleza de las relaciones existentes entre estas características del método y la concentración de la sustancia. Esta relación puede ser constante, lineal o curvilínea, y constituye una característica significativa del método que requiere una explicación clara. En la siguiente tabla de resultados se muestra el cálculo del sesgo y la precisión para una sola concentración en una única matriz a partir de ocho análisis repetidos de un estándar con una concentración conocida de 1,30 mg/l. INTRODUCCIÓN GENERAL 1-25 de rendimiento de EPA-Cincinnati (sección 1020). 3. El sesgo es 0,49/8 = 0,006 mg/1 y la precisión es la raíz cuadrada de 0,2335/ (8-1) = 0 , 03336 , o 0,18 mg/1 (adviértase que se traía de un cálculo similar al de la desviación estándar). 2. Análisis de muestras desconocidas En esta fase del procedimiento de validación del método es necesario realizar el análisis de estándares preparados independientemente cuyo valor es desconocido por el analista. Analícense las muestras desconocidas en réplicas, según el procedimiento de funcionamiento estándar del método. La cantidad media obtenida debe estar comprendida entre las tres desviaciones estándar (s) del valor medio del estándar, aunque preferiblemente en el rango 2 s. Obténganse muestras desconocidas procedentes de otros operadores del laboratorio mediante el empleo de reactivos comerciales para análisis o de estándares proporcionados por el National Institute of Standards and Technology (NIST) *, la EPA, u otras fuentes adecuadas. Si se hallan disponibles para el componente en particular, resultan de especial utilidad las muestras de evaluación * Anteriormente, National Bureau of Standards (NBS). Rigurosidad del método La etapa final de la validación consiste en un examen de rigurosidad, es decir, de la estabilidad del resultado producido cuando se modifican las distintas etapas del método. La determinación de esta característica resulta de especial importancia cuando un método se va a proponer como estándar o método de referencia. Cuando se realiza de forma correcta un examen de rigurosidad, éste señalará las fases del procedimiento en las que resulta de crítica trascendencia respetar un comportamiento riguroso y aquellas en las que se admite cierto grado de flexibilidad. La Association of Official Analytical Chemists1 ha sugerido un método para este examen en el que pueden utilizarse ocho análisis distintos para determinar el efecto de la variación de siete etapas diferentes en un procedimiento analítico. Para ilustrarlo, supóngase que ha de determinarse el efecto de un cambio en los siguientes factores: Para realizar la determinación, se denotan los valores iniciales de los factores por letras mayúsculas de la A a la G, y las variaciones con las letras minúsculas correspondientes. A continuación se establece la siguiente tabla de factores: 1-26 MÉTODOS NORMALIZADOS Si se analiza la combinación 1, el resultado será s. Si se analiza la combinación 2, el resultado sera t, y así sucesivamente hasta que se hayan analizado las ocho combinaciones. Para determinar el efecto de la variación de uno de los factores, se buscan los cuatro resultados en los que el factor se mantuvo como el inicial (mayúsculas) y los cuatro en los que se varió (minúsculas), y se comparan los promedios de los dos grupos. Por ejemplo, para comparar el efecto del cambio de C en c se utilizan los resultados (s + u + w + + y)/4 y (t + v + x + z)/4. Calcúlense los siete pares de resultados para conseguir siete diferencias, las cuales pueden así ordenarse para revelar las que poseen efectos significativos sobre los resultados. Si no se producen diferencias relevantes, calcúlense la media y la desviación estándar de los ocho resultados s a través de z. La desviación estándar es una estimación real de la precisión del método. Este modelo comprueba los efectos principales, no las interacciones. 4. Prueba de equivalencia Después de la validación de un nuevo método por los procedimientos anteriormente mencionados, puede resultar recomendable proceder al examen del método en cuanto a su equivalencia con los métodos estándar, a no ser que no exista ninguno. Ello requiere el análisis de un mínimo de tres concentraciones mediante el método estándar y el alternativo. Si la amplitud de concentraciones es muy grande, se examinarán más concentraciones. Una vez que se ha realizado un conjunto inicial de análisis (cinco o más) para cada concentración elegida, se aplican las siguientes etapas estadísticas2: 1. Examinar la normalidad de la distribución de datos y transformar éstos si es necesario (sección 1010B). 2. Seleccionar un tamaño adecuado de muestra basado en una estimación de la desviación estándar3. 3. Examinar las varianzas de los dos métodos mediante el empleo de la variable estadística de relación F. 4. Examinar los valores promedio de los dos métodos mediante el empleo de una variable estadística / de Student. Existen publicaciones que explican cada una de estas etapas con técnicas adicionales y diversos ejemplos 4. Dado que el número de análisis puede ser bastante alto, los cálculos se vuelven complejos, por lo que es necesario tener algunos conocimientos de estadística básica. Para ello, se encuentra disponible una relación de métodos estándar, de referencia y equivalentes5. 1-27 INTRODUCCIÓN GENERAL 5. 3. Referencias 1. YOUDEN , W. J. & E. H. STEINER. 1975. Statistical Manual of AOAC. Assoc. OfTícial Analytical Chemists, Washington, D.C. 2. WILLIAMS, L. R. 1985. Harmonization of Biological Testing Methodology: A Perform ance Based Approach in Aquatic Toxicology and Hazard Assessment. 8th Symp. ASTM STP 891, R. C. Bahner & D. J. Hansen, eds. American Soc. Testing & Materials, Filadelfia, Pennsylvania. 1040 C. 4. 5. NATRELLA, M. G. 1963. Experimental Statistics. National Bureau of Standards Handbook 91, Washington, D. C. U.S. ENVIRONMENTAL PROTECTION AGEN CY . 1983. Guidelines for Establishing Method Equivalency to Standard Methods. Rep. 600/X-83-037, Environmental Monitoring Systems Lab., Las Vegas, Nevada. U. S. E NVIRONMENTAL P ROTECTION AGEN CY. 1987. Guidelines establishing test procedures for the analysis of pollutants under the Clean Water Act. Interim final rule. 40 CFR Part 136; Federal Register 52:171:33542. Prueba en colaboración Una vez que se ha desarrollado y validado un método nuevo o modificado, resulta apropiado determinar si dicho método puede convertirse en un método estándar. El procedimiento para adecuar un método a la categoría de estándar consiste en realizar la prueba en colaboración. En esta prueba, cierto número de laboratorios utiliza el procedimiento estándar de funcionamiento para analizar un número seleccionado de muestras con el fin de determinar el sesgo y la precisión del método, tal como sucedería en la práctica normal. En la planificación de pruebas en colaboración es preciso considerar los siguientes factores: un procedimiento estándar de funcionamiento escrupulosamente descrito, el número de variables que se han de examinar, el número de niveles que se deben examinar y el número de réplicas necesarias. Como la precisión del método se evalúa mediante la desviación estándar, que es en sí misma el resultado de numerosas fuentes de variación, es preciso examinar las variables que la afectan. Entre ellas, pueden incluirse el laboratorio, el operador, el instrumental y la amplitud de concentración. 1. Variables Examínense como mínimo las siguientes variables: Laboratorio: Deben participar al menos tres laboratorios diferentes, aunque sería de desear un número mayor con el fin de obtener una mejor estimación de la desviación estándar. Instrumental: Dado que las diferencias de modelo y de fabricante pueden constituir fuentes de error, se analizarán como mínimo dos réplicas de cada concentración por laboratorio. Operadores: Para determinar la precisión global, deberán participar como mínimo seis analistas, no siendo más de dos por cada laboratorio. Niveles: Si el desarrollo del método ha señalado que la desviación estándar relativa es constante, se examinarán tres niveles que cubran toda la amplitud del método. Si no es constante, se utilizarán más niveles uniformemente distribuidos MÉTODOS NORMALIZADOS 1-28 a lo largo de la amplitud de funcionamiento. Si se desconfía de los efectos de matriz, deberá llevarse a cabo la prueba en cada uno de los medios para los que se desarrolló el método. Si ello no es factible, utilícense calidades adecuadas de agua para reactivos en el grado que estipule la evaluación resultante de las características del método. 2. Número de réplicas Calcúlese el número de réplicas después de haber completado la determinación del número de variables que se han de examinar mediante el empleo de la fórmula: r > 1 + (30/P) donde: r = número de réplicas, y P = producto de diversas variables. El número mínimo de réplicas es dos. Por ejemplo, cuando un solo operador ha de analizar tres niveles de una sustancia, repitiéndose la operación en seis laboratorios distintos con el mismo tipo de instrumental, P se obtendrá a partir del siguiente cálculo: P = 3 x 1 x 6 x 1 = 18 y el número de réplicas será: r > 1 + (30/18) > 2,7 o r = 3. 3. Ejemplo de prueba en colaboración Envíense a cinco laboratorios cuatro concentraciones de un compuesto (4,3, 11,6, 23,4 y 32,7 mg/1), con instrucciones para que se analicen por triplicado mediante el procedimiento suministrado. Tabular los resultados tal como se muestra en la tabla que se presenta á continuación (se indican los resultados para una única concentración). Dado que no existen valores claramente aberrantes (utilizar el método de la sección 1Q10B para rechazo de los valores extremos), hay que utilizar todos los datos. Calcúlese la media y la desviación estándar para cada laboratorio; utilícense los 15 resultados para calcular una media y una desviación estándar generales. La diferencia existente entre la media de cada laboratorio y la media general revela algunos sesgos significativos, como se aprecia para los laboratorios 1 y 3. La diferencia entre la media general y el valor conocido es el sesgo del método, por ejemplo, 33,0 - 32,7 = 0,3 mg/1 o 0,9 por 100. La desviación estándar relativa de la media general (1,5 mg/1) es 4,5 por 100, que es la precisión del método, y el valor de s para cada laboratorio es la precisión del operador. Tal como se aprecia en la tabla, la suma de las desviaciones desde el valor conocido para los laboratorios fue de 1.3, de modo que la desviación promedio (sesgo) fue 1,3/5 = 0,26, redondeado a 0,3, que es igual a la diferencia entre la media general y el valor conocido. Para las cuatro sustancias desconocidas en este examen, el porcentaje resultante indicó un crecimiento del sesgo y una disminución de la precisión conforme decrece la concentración. Por consiguiente, para describir el método con una enunciación formal, debería proporcionarse la precisión a partir de una línea recta según la fórmula y = mx + b; donde y es la desviación estándar relativa, m la pendiente de la línea, x la concentración y b la desviación estándar relativa para una concentración =J 0. Los valores establecidos a partir de la prueba en colaboración se muestran en la siguiente tabla. INTRODUCCIÓN GENERAL Estos resultados indican que el método es aceptable. Sin embargo, las concentraciones inferiores a 10 mg/1, aproximadamente, requieren especial atención en los análisis. 4. Referencia 1. YOUDEN, W. J. & E. H. STEINER. 1975. Stalistical Manual of the AOAC. Assoc. Official Analytical Chemists, Washington, D.C. 1-29 1-30 MÉTODOS NORMALIZADOS 1050 EXPRESIÓN DE RESULTADOS 1050 A. En el presente texto se emplea el Sistema Internacional de Unidades (SI), y los resultados físicos y químicos se expresan en miligramos por litro (mg/1). Regístrense únicamente las cifras significativas. Cuando las concentraciones son por lo general inferiores a 1 mg/1, puede resultar más conveniente expresar los resultados en microgramos por litro (μg/l). Utilícese μg/1 cuando las concentraciones sean inferiores a 0,1 mg/1. Las concentraciones superiores a 10.000 mg/1 se expresan como porcenta- Unidades jes, donde el 1 por 100 es igual a 10.000 mg/1 para un peso específico de 1,00. En muestras sólidas y residuos líquidos de alto peso específico, hay que realizar una corrección en caso de que los resultados se expresen como partes por millón (ppm) o porcentaje en peso: TABLA 1050:1. FACTORES DE CONVERSIÓN* (Miligramos por litro—Miliequivalentes por litro) * Los factores se basan en la carga iónica y no en las reacciones redox posibles para algunos de estos iones. Los cationes y aniones están clasificados de forma separada por orden alfabético. INTRODUCCIÓN GENERAL 1-31 En estos casos, si el resultado se da en miligramos por litro, ha de declararse el peso específico. La unidad equivalente por millón (epm), o el término idéntico y menos ambiguo de miligramos-equivalentes, o miliequivalentes por litro (me/1), pueden resultar de gran valor en la realización de cálculos de tratamiento de aguas y en la comprobación de análisis por equilibrio entre aniones y cationes. La tabla 1050:1 presenta los factores para la conversión de concentraciones de iones comunes desde miligramos por litro a miliequivalentes por litro, y vicever- 1050 B. sa. El término miliequivalente usado en esta tabla representa la porción 0,001 de un peso equivalente. El peso equivalente se define a su vez como el peso del ion (suma de los pesos atómicos que lo componen) dividido por el número de cargas normalmente asociadas con el ion particular. Los factores de conversión de resultados desde miligramos por litro a miliequivalentes por litro se calcularon mediante la división de la carga del ion por el peso del mismo. De modo inverso, los factores de conversión de resultados desde miliequivalentes por litro a miligramos por litro se calcularon mediante la división del peso del ion por su carga. Elementos significativos 1. Requisitos de comunicación de resultados Para evitar la ambigüedad en la comunicación de los resultados o en la presentación de las directrices de un procedimiento, es corriente la utilización de «elementos significativos». Todos los dígitos transmitidos en una comunicación de resultados han de ser claramente conocidos, con la salvedad del último dígito, que puede ser dudoso. De dicho número se dice que contiene únicamente elementos significativos. Si se aporta más de un dígito dudoso, el dígito adicional (o dígitos) no es significativo. Cuando se comunica un resultado como «75,6 mg/1», el analista debe estar bastante seguro del «75», aunque puede tener ciertas dudas sobre si el «,6» podría ser ,5 o ,7, o incluso ,8, en virtud de la inevitable incertidumbre del procedimiento analítico. Si se conociera una desviación estándar para un trabajo precedente de ± 2 mg/1, el analista podría, o debería, haber redondeado el resultado a «76 mg/1» antes de comunicarlo. Por otro lado, si el método fuera tan bueno que pudiera haberse comunicado con seguridad un resultado de «75,61 mg/1», el analista no debería haberlo redondeado a 75,6. Comuníquense únicamente las cifras que estén justificadas por la exactitud del método. No debe seguirse la extendida práctica de poner el mismo número de cifras a la derecha de la coma decimal en las cantidades que figuran en una columna. 2. Redondeo Para redondear, se eliminan los dígitos que no sean significativos. Si se elimina un dígito de valor 6, 7, 8 ó 9, hay que aumentar el dígito precedente en una unidad; si el dígito eliminado es 0, 1, 2, 3 ó 4, no se altera el dígito precedente. Si se elimina el dígito 5, se redondea el dígito precedente al número par más próximo: así, 2,25 se convierte en 2,2, y 2,35, en 2,4. 1-32 3. MÉTODOS NORMALIZADOS Ceros ambiguos El dígito 0 puede registrar un valor medido cero o servir simplemente como elemento de espaciado para situar la coma decimal. Si se comunica que el resultado de una determinación de sulfato es de 420 mg/1, el receptor del informe puede dudar sobre si el cero posee o no un valor significativo, ya que no se puede borrar. Si un analista calcula un residuo total de 1.146 mg/1, pero se da cuenta de que el 4 resulta dudoso en algunos casos y, por consiguiente, el 6 no tiene valor significativo, debe redondear la respuesta a 1.150 mg/l y comunicarla de esta manera, aunque tampoco en este caso será capaz e1 receptor de determinar si el cero es significativo o no. Aunque el número pueda expresarse como una potencia de 10 (por ejemplo, 11,5 x 102 o 1,15 x 103), esta forma no suele utilizarse, ya que podría no corresponderse con la expresión de resultados y provocar confusión. En la mayoría de los casos restantes no surgirán dudas sobre el sentido en que se usa el dígito 0. Resulta obvio que los ceros son significativos en números como 104 y 40,08. En un número escrito como 5.000, se entiende que todos los ceros son significativos, pues, en caso contrario, el número debería haber sido redondeado a 5,00, 5,0 ó 5, según lo que fuera más apropiado. En los casos en que el cero pueda resultar ambiguo se recomienda acompañar el resultado de una estimación de su incertidumbre. En ocasiones se eliminan ceros significativos sin una razón válida. Si se lee en una bureta «23,60 ml», debe registrarse como tal, y no como «23,6 ml». El primer número indica que el analista se ha tomado el trabajo de valorar la segunda cifra decimal; «23,6 ml» indicaría una lectura más bien descuidada de la bureta. 4. Desviación estándar Si un cálculo produce como resultado «1.476 mg/1» con una desviación están- dar estimada de ±40 mg/1, deberá comunicarse como 1.480 ±40 mg/1. Sin embargo, si la desviación estándar se valora como ±100 mg/1, la respuesta debe redondearse aún más y comunicarle como 1.500± 100 mg/1. Mediante este sistema ye evita la ambigüedad, y el receptor del informe puede interpretar que los ceros son únicamente elementos de espaciado. Incluso en los casos en los casos en los que no se presenta el problema de la ambigüedad del cero, resulta útil mostrar la desviación estándar, pues proporciona una idea de la fiabilidad. 5. Cálculos Como regla práctica de funcionamiento, redondéese el resultado de un cálculo en el que se multipliquen o dividan varios números por el número de cifras significativas que integran el factor que cuenta con un número menor de cifras significativas. Suponer que deben realizarse los siguientes cálculos para obtener el resultado de un análisis: 56 u 0 ,003 462 u 43, 22 1,684 Una calculadora de 10 dígitos suministrará una respuesta de «4,975 740 998». Redondéese este número a «5,0», ya que una de las medidas que intervienen en el cálculo, 56, tiene únicamente dos cifras significativas. Así, fue innecesario medir los tres factores restantes con cuatro cifras significativas, ya que «56» es el «eslabón más débil de la cadena» y limita la exactitud de la respuesta. Si los tres factores restantes se hubieran medido sólo con tres, y no con cuatro cifras significativas, la respuesta no se habría visto afectada y el trabajo habría sido menor. Cuando se suman o restan números, el número que posee la menor cantidad de cifras decimales, y no necesariamente el de menos cifras significativas, establece el 1-33 INTRODUCCIÓN GENERAL límite en el número de dígitos que pueden desplazarse justificadamente en la suma o la diferencia. Así, la suma debe redondearse a «4.928», sin decimales, dado que uno de los sumandos, 4.886, no tiene cifras decimales. Adviértase que otro de los sumandos, 25,9, tiene sólo tres cifras significativas y que ello no limita el número de cifras significativas de la respuesta. La cuestión que se acaba de exponer se ha simplificado en exceso necesariamente. Para estudio más detallado se remite al lector a textos matemáticos especializados. 1060 TOMA Y CONSERVACIÓN DE MUESTRAS 1060 A. Según viejo axioma, el resultado de cualquier determinación analítica no puede ser mejor que la muestra sobre la que se hace. No resulta práctico detallar aquí todos los procedimientos específicos de toma de muestras, dada la gran variedad de propósitos y métodos analíticos, por lo que al lado de cada uno de ellos aparecerá una información más concreta. En el presente capítulo se expone una serie de consideraciones generales aplicables sobre todo a los análisis químicos. El objetivo de la toma de muestras es la obtención de una porción de material cuyo volumen sea lo suficientemente pequeño como para que pueda ser transportado con facilidad y manipulado en el laboratorio sin que por ello deje de representar con exactitud al material de donde procede. Este objetivo implica que la proporción o concentración relativa de todos los componentes serán las mismas en las muestras que en el material de donde proceden, y que dichas muestras serán manejadas de tal forma que no se Introducción produzcan alteraciones significativas en su composición antes de que se hagan las pruebas correspondientes. La persona que recoge una muestra y la lleva a un laboratorio para realizar unas determinaciones específicas es responsable de su validez. Al trabajar con aguas limpias y residuales, el laboratorio suele dirigir u orientar el programa de la toma de muestras, que se determina tras consultar al destinatario de los resultados del análisis. Esta consulta es esencial para asegurar que la elección de las muestras y de los métodos analíticos proporcione una auténtica base para resolver los problemas que plantea la recogida de muestras. 1. Precauciones generales La obtención de una muestra que cumpla los requisitos del programa de toma y manipulación implica que aquélla no debe deteriorarse o contaminarse 1-34 antes de llegar al laboratorio. Antes de llenar el envase con la muestra hay que lavarlo dos o tres veces con el agua que se va a recoger, a menos que el envase contenga un conservante o un declorante. Según los análisis que deban realizarse, hay que llenar el envase por completo (en la mayoría de los análisis orgánicos), o dejar un espacio vacío para aireación, mezclas, etc. (análisis microbiológicos). En el caso de muestras que hayan de ser transportadas, lo mejor es dejar un espacio de alrededor del 1 por 100 de la capacidad del envase para permitir la expansión térmica. En las muestras que contienen compuestos orgánicos y vestigios metálicos hay que tomar precauciones especiales. Teniendo en cuenta que muchos de sus componentes pueden tener unas concentraciones de apenas algunos microgramos por litro, cabe la posibilidad de que se pierdan total o parcialmente si la recogida es defectuosa o no se toman las precauciones necesarias para su conservación. En algunos casos, sólo pueden obtenerse muestras representativas si se hacen mezclas de varias tomas obtenidas a lo largo de un determinado período o en muchos puntos distintos de recogida. Los detalles de la toma de muestras varían mucho según las condiciones locales, y no pueden hacerse recomendaciones específicas que sean de aplicación universal. A veces proporciona más información analizar numerosas muestras en lugar de una sola, ya que de este modo no se pierden los valores máximos y mínimos. La toma debe realizarse con cuidado, al objeto de garantizar que el resultado analítico represente la composición real. Entre los principales factores que influyen sobre los resultados se encuentran la presencia de materia suspendida o de turbidez, el método elegido para la recogida y los cambios físicos y químicos producidos por la conservación o la aireación. Es necesario tomar precaucio- MÉTODOS NORMALIZADOS nes especiales cuando en el procesado de muestras (división, mezcla, separación, filtrado) se han de analizar componentes residuales, sobre todo metálicos y compuestos orgánicos. Algunos análisis, en especial el de plomo, pueden quedar invalidados por alguna contaminación producida durante el procesado. Hay que tratar cada muestra de forma individual según las sustancias a analizar, la cantidad y naturaleza de la turbidez existente y otras condiciones que puedan influir en los resultados. No resulta práctico proporcionar directrices que abarquen todas las situaciones, pudiéndose dejar al juicio del analista la elección de la técnica idónea para conseguir que la muestra recogida sea homogénea. En general, se separa toda cantidad significativa de materia suspendida mediante decantación, centrifugación o filtración adecuada. A menudo es posible tolerar un grado pequeño de turbidez si se sabe por experiencia que ello no interferirá en los análisis gravimétricos o volumétricos y que puede corregirse su efecto sobre los análisis colorimétri-cos sobre los que potencialmente podría ejercer mayores interferencias. Si la turbidez es notable, hay que decidir si se filtra o no la muestra. Para medir la cantidad total de un componente, no hay que eliminar los sólidos suspendidos, sino tratarlos de forma adecuada. Hay que hacer un registro de todas las muestras recogidas e identificar cada envase, preferiblemente pegando una chapa o etiqueta debidamente señalada. Hay que registrar una información suficiente, de manera que se pueda realizar una identificación positiva de la muestra en fechas posteriores, y en esta información debe constar el nombre del que ha hecho la toma, la fecha, la hora y la localización exacta, la temperatura del agua y cualquier otro dato que pueda resultar necesario para establecer una correlación, como son las condiciones meteorológicas, el nivel del agua, la velocidad de la 1-36 2. MÉTODOS NORMALIZADOS Consideraciones sobre seguridad Habida cuenta que los componentes de la muestra pueden ser tóxicos, durante la toma y la manipulación de las mismas hay que adoptar las precauciones adecuadas. Las sustancias tóxicas pueden penetrar a través de la piel y, en el caso de los vapores, a través de los pulmones. Puede producirse una ingestión accidental mediante un contacto directo con los alimentos o por adsorción de vapores por los mismos. Las precauciones pueden limitarse a llevar unos guantes o a proveerse de batas, delantales u otros sistemas de protección. Siempre hay que llevar una protección ocular. Cuando puedan existir vapores tóxicos, la toma de la muestra sólo se realizará en lugares bien ventilados o mediante el uso de un respirador o dispositivos afines. En el laboratorio, los envases se han de abrir en una campana de gases. Nunca deben colocarse alimentos cerca de las muestras o de los lugares de toma; lávense siempre las manos cuidadosamente antes de manipular alimentos1. Si existe la posibilidad de hallar compuestos orgánicos inflamables, se tomarán las adecuadas precauciones. Quedará prohibido fumar cerca de las muestras, de los lugares de toma y en el laboratorio. Manténganse alejadas de las muesiras y de los lugares de recogida las chispas, las llamas y las fuentes de calor excesivo. Evítese la acumulación de vapores inflamables en el refrigerador donde se conserven las muestras, pues los arcos eléctricos que se forman en los contactos del termostato, la luz de la puerta u otros componentes eléctricos pueden desencadenar un fuego o una explosión. Si se sospecha o se sabe que existen compuestos inflamables que han de ser refrigerados, se utilizarán sólo refrigeradores especialmente diseñados a prueba de explosión1. Cuando existan dudas sobre la magnitud de las precauciones a adoptar, se consultará con un especialista en sanidad industrial que posea los conocimientos adecuados. Las muestras con contaminantes radiactivos requieren otras medidas de seguridad; consúltese a un físico especializado en sanidad. 3. Tipos de muestras a) Muestras de sondeo: Estrictamente hablando, una muestra recogida en un lugar y un momento determinados sólo puede representar la composición de la fuente en ese momento y lugar. Sin embargo, cuando se sabe que una fuente es bastante constante en su composición durante un periodo considerable o a lo largo de distancias sustanciales en todas direcciones, puede decirse que una muestra de dicha fuente representará un periodo de tiempo más largo o un volumen mayor o ambas cosas, con respecto al punto específico en el que fue recogida. En estas circunstancias, algunas fuentes pueden estar muy bien representadas por una simple muestra de sondeo. Es el caso de algunos suministros de agua, algunas aguas superficiales y, más raramente, algunas corrientes de aguas residuales. Cuando se sabe que una fuente varía con el tiempo, las muestras de sondeo recogidas a intervalos adecuados y analizadas por separado pueden mostrar la amplitud, la frecuencia y la duración de tales variaciones. Hay que hacer la recogida de las muestras teniendo en cuenta la frecuencia con que se esperan estos cambios, lo que puede variar desde cinco minutos a una hora o más. Las variaciones estacionales de los sistemas naturales pueden exigir la realización de tomas a lo largo de meses. Cuando la composición de la fuente varía en el espacio y no en el tiempo, hay que hacer la toma de las muestras en los lugares adecuados. Hay que tener gran cuidado al hacer tomas de muestras en aguas residuales impuras, orillas lodosas y fangos. No INTRODUCCIÓN GENERAL corriente, la manipulación posterior a la recogida, etc. La etiqueta debe tener espacio suficiente para que puedan ponerse las iniciales de todos los que asumen la custodia de la muestra y para la fecha y el momento del envío al solicitante. Hay que fijar los puntos de recogida mediante una descripción detallada, con mapas o con la utilización de postes, boyas o mojones que permitan su identificación por otras personas sin que éstas tengan que recurrir a la memoria de quién realizó la toma o tengan que ser guiadas al lugar. En los casos en que se prevea la utilización de los resultados de los análisis en litigios, deberán utilizarse procedimientos formales de «cadenas de custodia» (véase, más adelante, el apartado B.l), en los cuales se describirá el historial de la muestra desde su toma hasta el informe final. Las muestras calientes recogidas a presión deben ser enfriadas mientras se mantienen aún a dicha presión. Antes de recoger muestras de un sistema de abastecimiento, hay que dejar que el agua corra por las tuberías al objeto de asegurar que la muestra es representativa del suministro, teniendo en cuenta el diámetro y longitud de la conducción y la velocidad del flujo. La recogida de muestras de un pozo se hará después de haber bombeado una cantidad suficiente como para asegurar que la muestra representa al agua del subsuelo. A veces es necesario bombear a una velocidad determinada para conseguir un descenso de nivel que permita determinar las zonas de donde proviene el aporte al pozo. Se registrarán la velocidad de bombeo y el descenso del nivel. Cuando se analizan muestras recogidas en un rio o arroyo, los resultados pueden variar según la profundidad, la velocidad de la corriente, la distancia de la orilla y la separación entre ambas orillas. Si se dispone del equipo adecuado, se hará una toma «integral» desde la superficie al fondo en la zona media de la 1-35 corriente o de un lado al otro a una profundidad media, de forma que la muestra esté integrada en relación con el flujo. Si sólo puede hacerse una toma pequeña, se hará en el centro de la corriente a una profundidad media. Los lagos y pantanos presentan considerables variaciones debidas a causas normales, como la estratificación estacional, la cantidad de lluvia caída, el desagüe y el viento. La elección del lugar, la profundidad y la frecuencia de las tomas de muestras se hará dependiendo de las condiciones locales y del objetivo del estudio. En cualquier caso, se evitará la espuma superficial. Para determinados componentes es muy importante el lugar en el que se recoge la muestra. Hay que evitar las áreas de turbulencia excesiva, a causa de la posible pérdida de componentes volátiles y presencia de vapores tóxicos. No hay que recoger muestras en vertederos, ya que su localización tiende a favorecer la obtención de compuestos no miscibles más ligeros que el agua. En general, la toma se hará bajo la superficie en áreas tranquilas. Si se necesitan muestras mezcladas, hay que tener cuidado de que al hacer la mezcla no se pierdan los componentes de las mismas a causa de una manipulación inadecuada de las partes que se están combinando. Por ejemplo, el vertido casual de las muestras en lugar de la adición de unas a otras mediante un sifón sumergido puede dar lugar a una volatilización innecesaria. Cuando sea preciso, se refrigerará la mezcla para minimizar la volatilización1. Utilícense sólo muestras representativas (o recogidas según el programa de toma de muestras) para hacer los análisis. La gran variedad de condiciones bajo las cuales han de hacerse las tomas hacen que resulte imposible recomendar un procedimiento único. En general, hay que tener en cuenta las pruebas o análisis que se van a realizar y el fin para el que se requieren los resultados. INTRODUCCIÓN GENERAL pueden recomendarse procedimientos definitivos, pero es preciso adoptar todas las precauciones posibles para conseguir que la muestra sea representativa o se ajuste al programa de toma. b) Muestras compuestas: En la mayoría de los casos, la expresión «muestras compuestas» se refiere a una mezcla de muestras sencillas recogidas en el mismo punto en distintos momentos. A veces se utiliza la expresión «compuesto-tiempo» para distinguir este tipo de muestras de otros. Las muestras compuestas-tiempo son las más útiles para determinar las concentraciones medias que se han de utilizar, por ejemplo, para calcular la carga o la eficiencia de una planta de tratamiento de aguas residuales. Como alternativa al análisis separado de un gran número de muestras seguido de la computadorización de los resultados medios y totales, las muestras compuestas representan un ahorro sustancial de trabajo y gasto de laboratorio. Con este objeto, se considera como estándar para la mayoría de los análisis una muestra compuesta que represente un período de 24 horas. Sin embargo, en determinadas circunstancias puede resultar preferible una muestra compuesta que represente una desviación, un período más corto o el ciclo completo de una operación periódica. Para valorar los efectos de descargas y operaciones especiales, variables o irregulares, han de recogerse muestras compuestas que representen los períodos en los que tienen lugar dichas circunstancias. Para determinar componentes o características sujetas a cambios importantes e inevitables durante la conservación, no deben utilizarse muestras compuestas. Los análisis de este tipo se harán en muestras individuales y lo más rápidamente posible después de su recogida, con preferencia en el mismo lugar de la misma. Ejemplos de este tipo de análisis son los de todos los gases disueltos, el cloro residual, el sulfuro soluble, la tem- 1-37 peratura y el pH. Los cambios en este tipo de componentes, como el oxígeno o el dióxido de carbono disueltos, la temperatura o el pH, pueden producir alteraciones secundarias en otros componentes inorgánicos, como hierro, manganeso, alcalinidad o dureza. Sólo se utilizarán muestras compuestas-tiempo para la valoración de componentes cuya inalterabilidad en las condiciones de toma y conservación de la muestra haya quedado comprobada. Las porciones individuales se recogen en envases de abertura amplia, con un diámetro de al menos 35 mm en la boca y con una capacidad de 120 ml como mínimo. Se recogen estas muestras cada hora (en algunos casos, cada media hora o incluso cada cinco minutos) y se mezclan una vez concluida la toma o se combinan en una sola botella a medida que se van recogiendo. Si se utilizan conservantes, éstos se añadirán inicialmente al envase de la muestra de forma que todas las porciones de la mezcla queden protegidas lo antes posible. A veces puede ser necesario analizar las muestras individuales. Resulta conveniente, y a menudo esencial, combinar las muestras individuales en volúmenes proporcionales al flujo. Un volumen final de muestra de dos a tres litros es suficiente para analizar depuradoras, corrientes y aguas residuales. Existen aparatos automáticos de toma de muestra, pero no deben utilizarse a menos que se conserve la muestra de la forma antes indicada. Hay que limpiar a diario los aparatos de toma, incluidos los envases, para eliminar el crecimiento de organismos biológicos y otros depósitos. c) Muestras integradas: En algunos casos, la información necesaria se obtiene mejor analizando mezclas de muestras individuales, recogidas en distintos puntos al mismo tiempo o con la menor separación temporal que sea posible. A veces, las muestras de este tipo se denominan integradas. Un ejemplo de la nece- 1-38 MÉTODOS NORMALIZADOS sidad de las mismas es el de los ríos o corrientes cuya composición varía según la anchura y la profundidad. Para valorar la composición media o la carga total, hay que recurrir a mezclas de muestras que representen distintos puntos de la sección transversal y que sean proporcionales a los flujos relativos. También puede ser necesario recurrir a muestras integradas cuando se proponen tratamientos combinados para varias corrientes distintas de aguas residuales, cuya interacción puede tener un efecto significativo sobre la tratabilidad o incluso sobre la composición. La predicción matemática de las interacciones puede resultar inadecuada o imposible, de manera que el análisis de una muestra integrada representativa puede proporcionar una información muy útil. Los lagos naturales y artificiales muestran variaciones en su composición, tanto en profundidad como en sentido horizontal. Sin embargo, en muchos casos ni los resultados totales ni los medios resul- 1060 B. tan especialmente significativos; son más importantes las variaciones locales. En estos casos, en lugar de analizar muestras integradas, hay que estudiar muestras individuales. La preparación de muestras integradas suele precisar un equipo especial para hacer la toma a una profundidad conocida sin que ésta se mezcle con el agua de capas más superficiales. Suele ser necesario conocer el volumen, el movimiento y la composición de las distintas partes del agua a estudiar. Por tanto, la toma de muestras integradas es un proceso especializado y complejo que no puede describirse con detalle. 4. Referencia 1. WATER POLLUTION CONTROL FEDERATION. 1986. Removal of Hazardous Wastes in Wastewater Facilities—Halogenated Organics. Manual of Practice FD-11, Water Pollution Control Fed., Alexandria, Virginia. Toma de muestras 1. Procedimientos de cadena de vigilancia Es esencial asegurar la integridad de la muestra desde su toma hasta la emisión del informe. Ello implica hacer una relación del proceso de posesión y manipulación de la muestra desde el momento en que fue tomada hasta el de su análisis y eliminación final. Este proceso se denomina cadena de vigilancia, y es importante en el caso de que los resultados deban presentarse en un litigio. Si no es éste el caso, el procedimiento de cadena de vigilancia resulta útil como control rutinario de la trayectoria de la muestra. Se considera que una muestra está bajo vigilancia personal si se encuentra en posesión física de una persona, que es la que se encarga de custodiarla y de protegerla de posibles falsificaciones, o si se encuentra en una zona de acceso limitado al personal autorizado. A continuación se resumen los principales aspectos de la cadena de vigilancia. Existen descripciones más detalladas 1, 2. a) Etiquetado de la muestra: Utilícense etiquetas para evitar falsas identificaciones de la muestra. Suelen resultar adecuadas las etiquetas adhesivas o las chapas. En ella debe constar al menos la siguiente información: número de la muestra, nombre del que ha hecho la toma, fecha y momento de la toma y lugar de la misma INTRODUCCIÓN GENERAL Hay que adherir las etiquetas a los envases antes o en el momento de hacer la toma. La etiqueta se rellena con tinta indeleble en el momento de la toma. b) Sellado de la muestra: Se utilizarán sellos para detectar cualquier falsifica ción de la muestra que pueda hacerse antes del análisis. Se recurrirá para ello a sellos adhesivos de papel en los que conste al menos la siguiente información: número de la muestra (idéntico al número de la etiqueta), nombre del que ha hecho la toma y fecha y momento de la misma. También pueden utilizarse sellos de plástico. El sello se colocará de forma tal que sea necesario romperlo para abrir el envase. El sellado ha de realizarse antes de que el envase haya sido apartado de la vigilancia del personal que ha hecho la toma. c) Libro de registro de campo: Toda la información pertinente a un estudio de campo o toma de muestras se registrará en un libro en el que al menos constará lo siguiente: objeto de la toma, localización del punto donde se ha hecho, nombre y dirección del contacto de campo, productor del material del que se ha hecho la toma y dirección de dicho productor, en caso de que sea distinta de la del lugar de obtención de la muestra, y tipo de muestra. Si ésta procede de aguas residuales, hay que identificar el proceso que las produce. También es necesario hacer constar la posible composición de la muestra, incluyendo sus concentracio nes, el número y volumen de las muestras tomadas, la descripción del punto donde se ha hecho la toma y el método de la misma, la fecha y el momento de la toma, el número (o números) de identificación del que ha hecho la toma, referencias del lugar en forma de mapas o fotografías, observaciones y mediciones de campo y firmas del personal responsable de las observaciones. Dado que las situaciones de toma de muestras son muy variadas, no pueden darse reglas generales acerca 1-39 de la información que debe registrarse en el libro, pero en cualquier caso conviene incluir la información suficiente como para que pueda reconstruirse la toma de muestra sin tener que confiar en la memoria del que la ha hecho. El libro de registro debe estar protegido y guardado en lugar seguro. d) Registro de la cadena de vigilancia: Es preciso rellenar el registro de la cadena de vigilancia que acompaña a cada muestra o grupo de muestras. Este registro debe constar de la siguiente informa ción: número de la muestra, firma del que ha hecho la toma, fecha, momento y lugar de la toma, tipo de la muestra, firma de las personas que han participado en la cadena de posesión y fechas de las distintas posesiones. e) Hoja de petición de análisis de la muestra: La muestra irá al laboratorio acompañada por una hoja de petición de análisis. La persona que hace la toma deberá cumplimentar el apartado del impreso referido al trabajo de campo, en el que se incluye gran parte de la informa ción pertinente anotada en el libro de registro. El apartado del impreso que corresponde al laboratorio deberá ser rellenado por el personal de éste, y consta de: nombre de la persona que recibe la muestra, número de la muestra en el la boratorio, fecha de recepción y análisis a realizar. f) Envió de la muestra al laboratorio: La muestra se enviará al laboratorio lo antes posible e irá acompañada del registro de la cadena de vigilancia y de la hoja de petición de análisis. La muestra se entregará a la persona que deba encargarse de su custodia. g) Recepción y almacenamiento de la muestra: En el laboratorio, la persona encargada recibe la muestra e inspecciona su estado y su sello, comprueba la información de la etiqueta y la del sello comparándolas con la del registro de la cadena de vigilancia, le asigna el número de laboratorio, la registra en el libro de 1-40 MÉTODOS NORMALIZADOS entrada al laboratorio y la guarda en una habitación o cabina de almacenamiento hasta que sea asignada a un analista. h) Asignación de la muestra para ser analizada: En general, el supervisor del laboratorio es el que asigna la muestra para que sea analizada. Una vez en el laboratorio, el supervisor o el analista son los responsables del cuidado y la vigilancia de la muestra. 2. Métodos de toma de muestras a) Toma manual: En la torna manual se supone que no se utiliza equipo alguno, pero este procedimiento puede resultar demasiado costoso en tiempo y dinero para programas de toma rutinaria de muestras o a gran escala. b) Toma automática: Mediante la to ma automática se pueden eliminar los errores humanos en la manipulación, se reducen los costes laborales y se propor ciona la posibilidad de hacer tomas con mayor frecuencia 3, por lo que su uso está cada vez más extendido. Es preciso com probar que el aparato automático no contamine la muestra. Por ejemplo, los componentes plásticos pueden ser incompatibles con determinados compues tos orgánicos solubles en los plásticos. Si se sabe aproximadamente cuáles son los componentes de la muestra, el fabricante del aparato automático puede informar sobre las posibles incompatibilidades de los componentes plásticos. En algunos casos, lo mejor es hacer la toma manual con un envase de vidrio según un procedimiento que garantice la adecuada seguridad3. Los aparatos automáticos de toma de muestras se programan de acuerdo con las necesidades específicas de dicha toma. Hay que controlar con precisión la velocidad de bombeo y el tamaño de los tubos según el tipo de muestra que quiera recogerse. 3. Envases de las muestras El tipo de envase que se utilice tiene una importancia capital. En general, los envases están hechos de plástico 0 vidrio, y según los casos puede resultar j preferible uno u otro de estos materia es. Por ejemplo, el sílice y el sodio pueden lixiviarse en el vidrio pero no en el plástico, y los metales pueden dejar residuos absorbidos en las paredes de los envases de vidrio4. Para muestras que contienen compuestos orgánicos, sin embaírlo, conviene evitar los envases de plástico, salvo los fabricados con polímeros fluorados como el politetrafluoretileno (TF3)3. En el caso de muestras que contienen compuestos orgánicos volátiles, algunos de éstos pueden disolverse en las paredes de los envases de plástico o incluso pueden lixiviar sustancias de este «material. Los envases de plástico pueden degradarse y romperse. Algunos compuestos orgánicos son compatibles con el determinados plásticos (véanse las instrucciones de los fabricantes). Sin embargo, aunque se esté seguro de la compatibilidad, hay que tener en cuenta que las paredes dé los envases de plástico pueden resultar porosas para los compuestos orgánicos volátiles. En general, en estos casos es preferible utilizar envases de vidrio3. Los tapones de los envases, que suelen ser de plástico, también pueden plantear problemas al ponerse en contacto con componentes orgánicos. En estos casos se utilizarán de metal o de TFE. Los viales de suero con tabiques de plástico o goma recubierto de TFE pueden resultar útiles. 4. Número de muestras Teniendo en cuenta las variaciones aleatorias, tanto en los procedimientos analíticos como en la presencia de componentes en el lugar de la toma de la muestra, una sola de ellas puede resultar insuficiente para alcanzar el nivel de cer- 1-41 INTRODUCCIÓN GENERAL tidumbre deseado. Si se conoce la desviación estándar global, el número necesario de muestras puede calcularse con la siguiente fórmula4: donde: N = número de muestras, t = t de Student para un nivel de confianza determinado, s = desviación estándar global, y U = nivel de confianza aceptable. Las curvas del tipo de las ilustradas en la figura 1060:1 ayudan a hacer el cálculo. Por ejemplo, si s es 0,5 mg/1, U es ± 0,2 mg/1 y se desea un nivel de confianza del 95 por 100, hay que recoger de 25 a 30 muestras. 5. Para la mayoría de los análisis físicos y químicos se necesitan muestras de 2 1. Para determinadas pruebas pueden requerirse volúmenes mayores. En la tabla 1060:1 se muestran los volúmenes habituales necesarios para análisis. No debe usarse la misma muestra para estudios químicos (orgánicos e inorgánicos), bacteriológicos y microscópicos, pues los métodos de toma y manipulación de las mismas son distintos. 6. 1. Figura 1060:1. Número aproximado de muestras necesarias para calcular una concentración media. Reproducido de: Methods for the Examination of Waters and Associated Materials: General Principles of Sampling and Accuracy of Results. 1980. Her Majesty's Stationery Off., Londres, Inglaterra. Cantidad Referencias U.S. ENVIRONMENTAL PROTECTION AGENCY. 1986. Test Methods for Evaluating Solid Waste: Physical/Chemical Methods, 3.a ed., publ. N.° SW-846, Office of Solid Waste and Emergency Response. Washington, D.C. 2. U.S. ENVIRONMENTAL PROTECTION AGENCY . 1982. NEIC Policies and Procedures. EPA-330/9/78/001/-R (rev. 1982). 3. WATER P OLLUTION C ONTROL F EDERATION . 1986. Removal of Hazardous Wastes in Wastewater Facilities—Halogenated Organics. Manual of Practice FD-11, Water Pollution Control Fed., Alexandria, Virginia. 4. Methods for the Examination of Waters and Associated Materials: General Principles of Sampling and Accuracy of Results. 1980. Her Majesty's Stationery Off, Londres, Inglaterra. 1-42 MÉTODOS NORMALIZADOS INTRODUCCIÓN GENERAL 1-43 1-44 MÉTODOS NORMALIZADOS INTRODUCCIÓN GENERAL 1060 C. 1-45 Conservación de muestras Una completa e inequívoca conservación de las muestras, sean éstas procede aguas residuales domésticas, residuos industriales o residuos naturales, es posible en la práctica. Con independencia de muestras de que se trate, nunca puede conseguirse la estabilidad todos sus componentes. En el menor de los casos, las técnicas de conservación sólo retrasarían los cambios químicos y biológicos que inevitablemente se producen después de la toma. 1. Conservación de la muestra antes del análisis a) Naturaleza de los cambios de la muestra: Algunos análisis pueden verse 55 con mayor facilidad que otros de la conservación de la muestra. Algunos cationes se pierden por adsorción en las paredes de los envases de vidrio o por intercambio iónico con ellas. Entre estos cationes se encuentran el aluminio, el cadmio, el cromo, el cobre, el hierro, el plomo, el manganeso, la plata y el zinc, por lo que, en estos casos, es mejor utilizar un envase diferente y limpio, y acidificar con ácido nítrico hasta un pH inferior a 2,0 para reducir al máximo la precipitación y la adsorción en las paredes del envase. La temperatura cambia rápidamente; el pH puede cambiar de forma significativa en cuestión de minutos; los gases disueltos pueden perderse (oxígeno, dióxido de carbono). Hay que determinar, pues, la temperatura, el Ph y los gases disueltos en el momento de hacer la toma. Al cambiar el equilibrio pH-alcalinidad-dióxido de carbono, el carbonato de calcio puede precipitar y dar lugar a una disminución de los valores del calcio y de la dureza total. El hierro y el manganeso son muy solubles en sus estados de menor oxidación, pero relativamente insolubles en sus estados de mayor oxidación; por tanto, estos cationes pueden precipitar o disolverse si se encuentran en un sedimento, dependiendo del potencial redox de la muestra. La actividad microbiológica puede ser la responsable de los cambios en el contenido de nitrato-nitrito-amoníaco, de la disminución en la concentración de fenol y de BOD, o de una reducción de los sulfatos a sulfitos. El cloro residual es reducido a cloruro. Pueden perderse por oxidación los sulfuros, los sulfitos, los iones ferrosos, el yoduro y el cianuro. Pueden aumentar, disminuir o cambiar de calidad el color, el olor y la turbidez. El sodio, el sílice y el boro pueden lixiviarse a partir del vidrio del envase. El cromo hexavalente puede ser reducido a ion crómico. Los cambios biológicos que se producen en una muestra pueden modificar el estado de oxidación de algunos de sus componentes. Los solubles pueden ser convertidos a materiales unidos orgánicamente en las estructuras celulares, o puede producirse una lisis celular que libere materiales celulares a la solución. Los bien conocidos ciclos del nitrógeno y del fósforo son ejemplos de influencias biológicas en la composición de una muestra. Para la conservación de muestras con orgánicos volátiles es importante que no existan espacios vacíos. Se evitará la pérdida de materiales volátiles si se hace la toma llenando por completo el envase con la muestra, para lo cual se hará rebosar aquél antes de taparlo o sellarlo. Los viales de suero con tapón tabicado son especialmente útiles, ya que a través de dicho tapón puede tomarse una porción de la muestra mediante una jeringa1. 1-46 MÉTODOS NORMALIZADOS b) Intervalo de tiempo entre la toma y el análisis: En general, cuanto menor sea el tiempo que transcurre entre la toma de la muestra y su análisis, más fiable será el resultado del mismo. Para determinados componentes y valores físicos es necesario proceder a un análisis inmediato sobre el terreno. En el caso de muestras compuestas, es habitual considerar el momento en que se acaba de hacer la mezcla como el momento de toma de la muestra. Es imposible establecer con exactitud el tiempo máximo que puede transcurrir entre la toma de la muestra y su análisis; ello depende del carácter de la muestra, del tipo de análisis a realizar y de las condiciones de conservación. Los cambios debidos al crecimiento de microorganismos se retrasan mucho si se mantiene la muestra en la oscuridad y a baja temperatura. Cuando el intervalo entre la toma y el análisis es lo suficientemente largo como para producir cambios en la concentración o en el estado físico de los componentes a medir, se seguirán las instrucciones de conservación que se ofrecen en la tabla 1060:1. Se registrará el tiempo transcurrido entre la toma de la muestra y su análisis y, en el caso de que se haya añadido algún conservante, se identificará éste. 2. Técnicas de conservación Para reducir al máximo la posible volatilización o biodegradación entre el momento de hacer la toma y el de proceder al análisis, se debe mantener la muestra a la menor temperatura posible sin que llegue a congelarse. Lo mejor es empaquetar la muestra antes de enviarla en un recipiente con hielo molido o en cubitos o con sustitutos comerciales del hielo. No debe utilizarse hielo seco, pues éste congelaría la muestrario que podría provocar la rotura del vidrio del envase. El hielo seco puede, además, afectar al pH de la muestra. Mientras se hace la mezcla, las muestras deben mantenerse con hielo o un sistema de refrigeración a 4°C. Las muestras se analizarán lo antes posible una vez llegadas al laboratorio. Si no es posible hacerlo de manera inmediata, se recomienda conservarlas a 4 °C en la mayoría de los casos1. Sólo se utilizarán conservantes químicos cuando se haya demostrado que no van a estropear el análisis. En caso de que se utilicen, deberán añadirse al envase antes de poner la muestra, de manera que todas las partes de ésta entren en contacto con el conservante en el momento en que sean recogidas. No existe ningún método de conservación que sea totalmente satisfactorio. El conservante debe elegirse en función de los análisis a realizar. Un método de conservación útil para un análisis determinado puede ser inadecuado para otros, por lo que a veces es necesario hacer varias tomas y conservarlas por separado para someterlas a análisis múltiples. Todos los métodos de conservación pueden ser inadecuados si se aplican a materias en suspensión. El formaldehído afecta a la mayoría de los análisis, por lo que no debe utilizarse. Los métodos de conservación son relativamente limitados, y están diseñados, en general, para retardar la acción de los microorganismos, retrasar la hidrólisis de los compuestos y complejos químicos y reducir la volatilidad de los componentes. Los métodos de conservación se limitan al control del pH, la adición de productos químicos, el uso de envases ámbar u opacos, la refrigeración, la filtración y la congelación. En la tabla 1060:1 se enumeran los métodos de conservación según los componentes. La exposición anterior no es en absoluto exhaustiva ni completa. Es claramente imposible dar reglas absolutas para evitar todas las alteraciones que puedan producirse. Al abordarse cada 1-47 INTRODUCCIÓN GENERAL uno de los análisis concretos se encontrarán indicaciones adicionales, pero, en gran medida, la precisión de una valoración analítica se fundamenta en la experiencia y el buen juicio de la persona que hace la toma de la muestra. 3. 1. 4. Bibliografía Referencia WATER POLLUTION C ONTROL FEDERATION . 1986. Removal of Hazardous Wastes in 1070 KEITH, L. H., ed. 1988. Principles of Environmental Sampling. ACS Professional Reference Book, American Chemical Soc. INSTRUMENTAL, REACTIVOS Y TÉCNICAS DE LABORATORIO 1070 A. En la presente sección se exponen las necesidades de instrumental, reactivos y técnicas de laboratorio que son habituales en muchos de los análisis descritos en este manual. Las necesidades que son más o menos específicas para una valoración particular se describen junto al método con el que se utilizan. Deben observarse, además, las consideraciones gene- 1070 B. 1. Wastewater Facilities—Halogenated Organics. Manual of Practice FD-11, Water Pollution Control Fed., Alexandria, Virginia. Envases Para uso general en el laboratorio, el material más adecuado es el vidrio de borosilicato*, muy resistente. Existen vidrios especiales con características tales como alta resistencia al ataque por álcalis, bajo contenido en boro u opacidad. Los tapones y tapaderas deben soportar bien el contacto con el material contenido en el envase. Los tapones de metal de rosca constituyen una opción inadecua* Pyrex, fabricado por Corning Glass Works; Kimax, Kimble Glass Co., División of Owens-Illinois, o equivalente. Introducción rales que se presentan. Los requisitos de los métodos radiológicos, bacteriológicos, biológicos y de bioanálisis tienden a diferir en muchos aspectos de los que corresponden a pruebas químicas y físicas. Se presta especial atención a las descripciones de instrumental y procedimientos en las secciones que tratan de esos métodos. Instrumental da para muestras capaces de corroerlos con facilidad. Los tapones de vidrio no resultan satisfactorios para líquidos muy alcalinos, dada su tendencia a adherirse con rapidez. Los tapones de goma resultan excelentes para los líquidos alcalinos pero son inaceptables para los disolventes orgánicos, en los que se desintegran, o para soluciones con metales residuales, a los que pueden contaminar. Utilícese politetrafluoretileno (TFE o PTFE)† para buretas que contengan líquidos fuertemente alcalinos. Cuando se considere † Teflón o equivalente. 1-48 adecuado, pueden utilizarse otros materiales, como porcelana, níquel, hierro, platino, acero inoxidable y vidrio rico en sílice ‡. Las muestras se recogen y se guardan en envases de vidrio de borosilicato, ebonita, plástico u otro material inerte que sea adecuado para cada tipo de análisis (tabla 1060:1). Para períodos de conservación relativamente cortos o cuando los componentes no se alteran por permanecer en envases de vidrio, como es el caso del calcio, el magnesio, el sulfato, el cloruro y quizás otros, resulta satisfactoria la botella para ácidos con cierre de campana de 2,5 1. Este tipo de cierre consiste en un disco de vidrio o polietileno que se ajusta a la superficie del vidrio o en un tapón de polietileno que se inserta en la boca de la botella asegurando una protección adecuada. Cuando una parte de la mezcla se destine a la determinación de la concentración de sílice, sodio u otras sustancias que pudieran resultar afectadas por un almacenamiento prolongado en vidrio blando, se colocará la parte a analizar en una botella pequeña de plástico, dejando el resto de la muestra en la botella de vidrio. Hay que limpiar cuidadosamente los envases de las muestras antes de cada uso. Las botellas de vidrio se lavan, salvo las que vayan a utilizarse para análisis de cromo o manganeso, bien con una mezcla limpiadora compuesta por 1 litro de H2SO2 conc. que se mezcla, lentamente y agitándolo, a 35 ml de solución saturada de dicromato de sodio, o bien con KMnO2 al 2 por 100 en una solución de KOH al 5 por 100 seguida de una solución de ácido oxálico. También se pueden emplear productos comerciales §. ‡ Vycor, fabricado por Corning Glass Works, o equivalente. § Nochromix, Godax Laboratories, Inc., Nueva York, o equivalente. MÉTODOS NORMALIZADOS Para eliminar la materia orgánica, se enjuagará el envase con otros ácidos concentrados. Los detergentes son excelentes limpiadores en muchos casos; pueden utilizarse éstos o HC1 conc. para limpieza de los envases de ebonita y plástico. Una vez limpios, se enjuagan con abundante agua de calidad para reactivos. Para transportarlos, los envases se empaquetan en cajas acolchadas de metal, plástico o fibra prensada, con un compartimento para cada una de las botellas. Las cajas se recubren con material aislante para evitar las roturas. Las muestras en envases de plástico no necesitan protección contra roturas provocadas por golpes o congelación. 2. Material de vidrio para volumetría El material de vidrio ha de ser calibrado, o bien se ha de conseguir un certificado de exactitud de un laboratorio competente. El material de vidrio pan volumetría se calibra bien para «contener» (C) o para «verter» (V). Esto último sólo se conseguirá de una forma exacta cuando su superficie interna está tan está tan escrupulosamente limpia que el agua la humedece inmediatamente y forma una capa uniforme al vaciarlo. Se usará, siempre que sea posible, vidrio de boroslilicato. Para trabajos de precisión se utilizará material de vidrio para volumetría de clase A. Los pesos y volúmenes deben medirse con cuidado cuando se preparen soluciones estándar y curvas de calibración. Estas mismas precauciones se mantendrán cuando se midan los volúmenes de las muestras. En caso de que el volumen se designe en el texto con dos cifras decimales (X,00 ml), se utilizarán pipetas o buretas para volumetría. Cuando se especifique, se emplearán matraces para; volumetría, y lo mismo se hará cuando el volumen se indique como 1.000 ml, y no como 1 l. 1-49 INTRODUCCIÓN GENERAL 3. Tubos de Nessler A menos que se indique lo contrario, se usarán tubos de Nessler de forma «alta» fabricados con vidrio resistente y seleccionados a partir de un proceso de estiramiento uniforme. El vidrio ha de ser claro e incoloro, y el fondo, plano y recto. Cuando el tubo está lleno de líquido y se observa desde arriba con una luz bajo, no deben aparecer puntos oscuros ni distorsiones ópticas. El extremo ha de ser plano, a ser posible pulido al fuego, y suficientemente liso como para que su tapa quede bien pegada al sellarse. Es posible conseguir tubos de Nessler con tapas cónicas de vidrio y con graduación estándar. Las marcas de graduación deben rodear por completo los tubos. Los tubos de 100 ml deben tener una longitud total de alrededor de 375 mm. Su diámetro interno tendrá unos 20 mm, 1070 C. 1. Agua de laboratorio Véase sección 1080. 2. Calidad de los reactivos Sólo se utilizarán reactivos de la mejor calidad de química, aunque ello no se indique cuando se describa un método determinado. La American Chemical Society ha publicado especificaciones «ACS» los productos químicos que deben solicitarse. Los demás productos químicos se pe-dirán como «reactivos para análisis» o como «disolventes orgánicos para análisis espectral». En los libros sobre especificaciones de reactivos citados en la bibliografía (véase más adelante el apartado 4) pueden encontrarse métodos pa- y el externo, 24 mm. La marca de graduación debe estar lo más cerca posible de la medida de 300 mm por encima de la cara superior del fondo. Los tubos que se venden en lotes deben ser muy uniformes, de manera que esta distancia no varíe más de 5 mm. (En algunos lotes comerciales, la diferencia máxima entre los tubos no supera los 2 mm.) Es admisible una marca de graduación a 50 ml. Los tubos de 50 ml deben tener una longitud total de alrededor de 300 mm. Su diámetro interno ha de ser de aproximadamente 17 mm, y el extremo, de 21 mm. La marca de graduación debe estar lo más cerca posible de la medida de 225 mm por encima de la cara superior del fondo. Los tubos vendidos en lotes deben ser muy uniformes, de manera que esta distancia no varíe más de 6 mm. (En algunos lotes comerciales, la diferencia máxima entre los tubos no supera 1,5 mm.) Es admisible una marca de graduación a 25 ml. Reactivos ra comprobar la pureza de los reactivos poco fiables. Por desgracia, muchos colorantes comerciales, para los cuales no se ha establecido el grado ACS, no cumplen exactamente las necesidades analíticas, a causa de variaciones en la respuesta de color de los distintos lotes. En estos casos, deben utilizarse colorantes certificados por la Biological Stain Commission. Cuando no se disponga de un colorante de grado ACS ni certificado por la Biological Stain Commission, se purificará el colorante sólido mediante recristalización. Las siguientes sustancias estándar, cuyas botellas van acompañadas de un certificado de análisis, son suministradas por el National Institute of Standards 1-50 and Technology (NIST)*, Department of Commerce, Washington, D.C., con el objeto de normalizar las soluciones analíticas: En la Special Publication 260 (véase bibliografía) constan otros muchos centenares de estándares suministrados por el NIST. Una prueba satisfactoria de ditizona necesita reactivos de la mayor pureza. Se pueden conseguir cloroformo y tetracloruro de carbono cuyo grado se adapta a los métodos de la ditizona. Para los métodos de ditizona descritos en este manual se seleccionarán reactivos de esta calidad. Las sales de sodio hidrosolubles suelen recomendarse para la preparación de indicadores, debido a que pueden conseguirse con facilidad y su precio es razonable. Cuando la preparación de indicadores del tipo de la fenolftaleína requiera la utilización de alcohol o alcohol etílico, se empleará alcohol etílico al 95 por 100. Cuando se especifique el uso de alcohol isopropílico, la concentración será similar. Algunos reactivos orgánicos son algo inestables si quedan expuestos a la ac* Antes National Bureau of Standards (NBS). MÉTODOS NORMALIZADOS ción atmosférica. Si la estabilidad de un producto químico es limitada o se desconoce, se adquirirán lotes pequeños a intervalos frecuentes. Los reactivos anhidros necesarios para la preparación de soluciones estándar de calibración y tituladores se secan en una estufa a 105-110 °C durante al menos una o dos horas o, preferiblemente, durante una noche. Después de enfriarlos a la temperatura ambiente en un desecador eficaz, se pesa inmediatamente la cantidad necesaria para la solución. Si se requiere una temperatura de secado distinta, se especificará en cada producto químico concreto. El secado de sales hidratadas se realizará con mayor suavidad en un desecador de horno eficaz. 3. Soluciones habituales de ácidos y álcalis a) Unidades de concentración utilizadas: Las concentraciones de los reactivos se expresan en términos de normalidad, molaridad y volúmenes aditivos. Una solución normal (N) contiene un equivalente gramo de peso del soluto por litro de solución. Una solución molar (M) contiene el peso molecular en gramos de soluto por litro de solución. En volúmenes aditivos (a + b), el primer número, a, se refiere al volumen de reactivo concentrado, y el segundo, b, al volumen de agua destilada necesaria para la dilución. Por tanto, « 1 + 9 HC1» significa que se ha de disolver 1 volumen de HC1 concentrado en 9 volúmenes de agua destilada. Para hacer una solución de normalidad exacta de un producto químico que no puede ser medido como estándar primario, se prepara una solución madre relativamente concentrada y después se hace la dilución exacta a la concentración deseada. También puede hacerse una solución con una concentración lige- INTRODUCCIÓN GENERAL ramente mayor que la deseada, estandarizarla y hacer los ajustes necesarios de la concentración mediante dilución; otra posibilidad consiste en utilizar la solución como estandarizada primaria y modificarla por un factor de cálculo. Este último procedimiento es útil, sobre todo, con soluciones que cambian lentamente de concentración y que han de reestandarizarse a menudo, como, por ejemplo, la solución de tiosulfato de sodio. Cuando un laboratorio hace un gran número de análisis con una solución estándar, es recomendable realizar un ajuste a la normalidad exacta especificada. Los análisis se atendrán a las instrucciones de este manual siempre que la normalidad de una solución estándar no dé lugar a una titulación de volumen tan pequeña que impida la exactitud de la medición, o tan grande que provoque una dilución anormal de la mezcla de reacción, y siempre que la solución esté adecuadamente estandarizada y los cálculos sean correctos. b) Preparación y dilución de soluciones: Si se va a preparar una solución de normalidad exacta disolviendo una cantidad pesada de estándar primario o diluyendo una solución más concentrada, mídase el volumen exacto en matraz volumétrico. Prepárense soluciones madre y estándar exactas mediante determinaciones colorimétricas en matraces volumétricos. Cuando no sea necesario que la concentración sea exacta, se mezclará la solución concentrada o el sólido con cantidades medidas de agua, utilizándose cilindros graduados para hacer mediciones. En general, se produce un cambio significativo en el volumen cuando se mezclan soluciones concentradas, lo que hace que el volumen final sea menor que la suma de los utilizados. En el caso de diluciones aproximadas, los cambios de volumen no resultan significativos si las concentraciones son 6N o menores. Las disoluciones se mezclan con cuida- 1-51 do y completamente. Uno de los motivos más frecuentes de error en los análisis en los que se utilizan soluciones estándar diluidas en matraces volumétricos procede de no conseguir una mezcla completa. c) Conservación de las soluciones: Al gunas soluciones estandarizadas se alteran lentamente debido a cambios químicos o biológicos. En estos estándares se indican la vida práctica, la frecuencia de estandarización necesaria y las precauciones de almacenamiento. Otras, como el HC1 diluido, no son reactivas. Sin embargo, también su concentración puede cambiar por causa de la evaporación, que no se evita con un tapón de vidrio. Los cambios de temperatura hacen que las botellas «respiren» y posibilitan cierto grado de evaporación. No debe considerarse válido un estándar de duración superior a un año, a menos que se reestandarice. Sólo será válido para ese período si las condiciones hacen que la evaporación sea mínima y si se ha demostrado previamente que la técnica de conservación es adecuada. Si la botella se abre a menudo o su contenido ocupa mucho menos de la mitad, en pocos meses se produce una evaporación significativa. Hay que comprobar la concentración de las soluciones estándar que han permanecido almacenadas. Utilícense botellas de vidrio de material químicamente resistente, salvo en el caso de que el vidrio sea incompatible (por ejemplo, soluciones de sílice). Para soluciones estándar que no reaccionen con el caucho o el neopreno, se utilizarán tapones de estos materiales; si se ajustan adecuadamente, estos tapones evitan la evaporación durante todo el tiempo que la botella permanezca cerrada. También son eficaces las botellas de tapón de rosca. Si la junta del tapón está hecha de un material suficientemente resistente, puede utilizarse para lo mismo que los tapones de caucho. d) Uso alternativo de los ácidos clorhídrico y sulfúrico: En varias técnicas se 1-52 MÉTODOS NORMALIZADOS utilizan soluciones estandarizadas de H2SO4 y de HC1. A menudo son intercambiables, y, cuando se menciona una, puede utilizarse la otra, siempre que se sepa que ello es posible. e) Preparación: Aunque en general las instrucciones describen la forma de preparar 1 l de solución, pueden prepararse volúmenes mayores o menores según las necesidades. Las instrucciones para la preparación de Í00 ml suelen limitarse a reactivos de corta vida o utilizados en cantidades pequeñas. Una útil regla general consiste en añadir una cantidad adicional de ácido o álcali concentrado al agua, agitando la solución en un vaso que pueda resistir el cambio brusco de temperatura, para diluirla a continuación hasta conseguir el volumen final una vez enfriada a temperatura ambiente. f) Concentraciones uniformes de reactivo: Se ha procurado establecer un determinado número de concentraciones habituales de ácidos y bases uniformes que pudieran servir para ajustar el pH de 1070 D. Existen numerosos trabajos tanto generales como específicos sobre técnicas analíticas (véase apartado 5, Bibliografía). 1. intercambio iónico Las resinas de intercambio iónico tienen un uso muy adecuado y flexible. Utilizadas en columnas de presión atmosférica o de alta presión, efectúan separaciones analíticas de iones orgánicos e inorgánicos. A menudo se presentan en forma de contra-iones de sodio o hidrógeno (unidos a la matriz) para los intercambiadores de cationes, y de contra-iones de cloro, sales fórmicas, acetato e hidróxido para los intercambiadores de aniones. El las muestras antes de desarrollar el color o de la titulación final. Las siguientes concentraciones de ácido son las recomendadas para su empleo general en laboratorios: reactivo comercial concentrado, 6N, IN, 0,1 N y 0,02/V. Véase la contraportada para las direcciones de preparación de estas concentraciones de ácido, así como para las 15N, 6N y 1N de NaOH, y 5N, 3N y 0,2N de NH4OH. 4. Bibliografía R OSIN , J. 1967. Reagent Chemicals and Standards, 5.a ed. D. Van Nostrand Co., Princeton, Nueva Jersey. AMERICAN CHEMICAL SOCIETY. 1974. Reagent Chemicals—American Chemical Society Specifications, 5. a ed. American Chemical Soc., Washington, D. C. The United States Pharmacopoeia. 1975. 19.a rev. U.S. Pharmacopeial Convention Inc., Rockville, Maryland. NATIONAL BUREAU OF STANDARDS. 1988. Catalog of NBS Standard Reference Materials. Nat. Bur. Standards Spec. Publ. 260, 1988-89 ed. Técnicas usuario puede sustituir otros contraiones pasando soluciones regeneradoras a través de la columna de resina según el procedimiento recomendado por los fabricantes. La forma de uso en cada caso específico depende no sólo de la afinidad relativa de la resina por los contra-iones y los iones simples, sino también de los iones que pueden ser aceptados en los casos en los que los iones concentrados de la muestra sean diluidos. Habitualmente se procede a una disolución secuencial de compuestos orgánicos mediante soluciones tampones cuidadosamente elegidas. En el análisis de agua pueden aplicarse los intercambiadores de iones a: a) la eliminación de los iones que producen interferencias, b) la determinación del INTRODUCCIÓN GENERAL contenido iónico total, c) la indicación del volumen aproximado de la muestra para algunas valoraciones gravimétricas, d) la concentración de cantidades residuales de cationes, y e) la separación entre aniones y cationes. En este manual se recomienda el uso de resinas de intercambio iónico para la eliminación de la interferencia en la determinación del sulfato y para la determinación del contenido iónico total. Siempre que pueda aplicarse el intercambio de iones a otras valoraciones, se hará una breve descripción de las operaciones habituales. a) Elección del método: El método de ... mezcla es satisfactorio en muestras de un volumen inferior a 100 ml, aunque no consigue la eliminación o intercambio completo de los iones. El método de la columna puede utilizarse con muestras de cualquier volumen. La técnica de la mezcla consiste en agitar la resina dentro de la muestra durante un tiempo tras el que se retira la resina mediante filtración. El método de la columna es más eficaz, ya que proporciona un contacto conti-nuo entre la muestra y la resina permitiendo una completa reacción de inter-cambio. La solución pasa lentamente por el lecho de resina y los iones son eliminados de la muestra en virtud de pautas cuantitativas, y no cualitativas. La diso-lución de la resina permite recuperar las sustancias intercambiadas. 1-53 Erlenmeyer o un vaso de precipitación de 250 ml y se añade agua destilada suficiente para completar un volumen de 75 ml. Se añaden 2,0 g de resina de intercambio catiónico fuertemente acida y se agita a una velocidad moderada durante 15 minutos. Se filtra a través de un tapón de lana de vidrio colocado en el cuello de un embudo de vidrio de borosilicato de 10 cm. Cuando se ha completado la filtración, se lava la resina con dos porciones de 10 ml de agua destilada y se añade agua destilada hasta completar un volumen total de 100 ml. Para regenerar la resina, se coloca la utilizada en un matraz que contenga 500 ml de HNO3 3N. Cuando se ha acumulado suficiente resina, se lava en la columna (fig. 1070:1) y se regenera pasando HNO3 3N por la columna a una velocidad de 0,1-0,2 ml de ácido/ml de resina/minuto. Se utilizan alrededor de 21 ml de HNO3 3N/ml de resina en la columna. Por último, se lava la resina con suficiente agua destilada hasta que el pH del líquido efluente sea de 5 a 7, utilizan- b) Procedimiento: Se utilizarán resinas especialmente fabricadas para aplica ciones analíticas. Se prepara el intercambiador de iones lavando la resina con varios volúmenes de agua desionizada (agua destilada de buena calidad), de manera que quede eliminada cualquier materia fina o colorante y otros materiales lixiviantes que puedan afectar a los posteriores procedimientos colorimétricos. 1) Método de mezcla para la eliminación de cationes. Se toma una parte de la muestra que contenga 0,1-0,2 miliequivalentes (me) de cationes en un matraz de Figura 1070:1. Columna de intercambio iónico. 1-54 do la misma velocidad de flujo que en el paso de regeneración. Se retira la resina de la columna y se guarda en agua destilada dentro de un envase de boca ancha. Si el agua se colorea durante el almacenamiento, se decanta y se sustituye por agua destilada fresca. Antes de utilizarla, se filtra la resina a través de un tapón de lana de vidrio colocado en el cuello de un embudo, se lava con agua destilada y se deja secar. De esta forma la resina está lista para ser utilizada. 2) Método de la columna para la eliminación de cationes. Se prepara la columna como se ilustra en la figura 1070:1 (longitud del lecho de resina, 21,5 cm; diámetro de la columna, 1,3 cm; representa alrededor de 21 ml o 20 g de resina). También pueden utilizarse otras columnas de intercambio de iones. Una de las más sencillas consiste en una bureta con un tapón de lana de vidrio colocado inmediatamente por encima de la llave. Para fabricar el tubo efluente de una bureta, se curva lentamente un tubo TFE de 3,1 mm de DI y 6,2 mm de DE hasta formar una S alargada; se curva en un ángulo de 45° con respecto a la parte recta, se asegura con tiras de goma o esparadrapo y se deja reposar al menos 48 horas para que pierda la tensión. Se repite la operación hasta conseguir la forma deseada. Los extremos curvados se sujetan con tiras de plástico de unos 2 cm x 100 cm x 2 mm con orificios de 4,7 mm dispuestos adecuadamente. (Cualquiera que sea el tipo de columna que se adopte, nunca se debe dejar que el nivel líquido de la misma descienda por debajo de la superficie superior de la resina, ya que el aire atrapado produce velocidades de flujo irregulares y deteriora la eficacia del intercambio iónico. Se ajustará la muestra y la columna a una misma temperatura.) La columna se carga removiendo la resina en un vaso de precipitados con agua destilada y lavando después cuidadosamente la suspensión según se va pa- MÉTODOS NORMALIZADOS sando a la columna llena de agua destilada a través de un embudo. Si es necesario, se lava la columna contraflujo introduciendo agua destilada por la parte inferior y haciéndola pasar hacia arriba hasta que queden eliminados todos los canales y burbujas de aire. Se conecta un embudo separador a la parte superior de la columna y se utiliza un matraz volumétrico invertido para introducir la muestra, regenerar o lavar las soluciones; hay que asegurarse de que el diámetro del cuello del matraz es bastante grande como para permitir la alimentación automática. Para una mayor eficiencia, se utilizarán pequeñas bombas peristálticas para colocar las soluciones en la columna. Se deja que la muestra fluya hacia el interior de la columna a una velocidad de 0,2 ml de solución/ml de resina/minuto. Una vez colocada la muestra en la columna, se lava la resina con agua destilada hasta que el líquido efluente tenga un pH de 5 a 7. Se utiliza un pH-neutro o tiras de papel indicador del pH para determinar el momento en que la columna ha quedado sin ácido. Para mayor comodidad, al lavar la columna o al adsorber cationes de la muestra de uno o más litros, se comienza esta operación antes de acabar el trabajo del día y se deja que el proceso de intercambio se realice durante la noche. La columna no se seca debido a su salida curva. Una vez retirada el agua destilada, se extraen los cationes adsorbidos haciendo pasar 100 ml de HNO3 3N a través de la columna a una velocidad de 0,2 ml de ácido/ml de resina/minuto. Teniendo en cuenta que un volumen de 100 ml de HNO 3 3N elimina cuantitativamente 3 me de cationes, se utilizarán incrementos adicionales de 100 ml de HNO3 3N para cantidades de iones adsorbidos que sobrepasen los 3 me. Tras la disolución, se lava la columna para extraer el ácido, utilizando agua destilada suficiente como para producir un líquido efluente con un pH de 5 a 7. El lavado se hace a la INTRODUCCIÓN GENERAL misma velocidad de flujo que en el caso de la disolución del ácido. La dilución de éste y el lavado regeneran la columna, dejándola preparada para un uso futuro. Las diluciones de ácidos combinados contienen los cationes que originariamente existían en la muestra. 2. Determinaciones colorimétricas a) Consideraciones generales: Muchas técnicas se basan en la determinación del color con instrumentos colorimétricos. Para obtener los mejores resultados posibles, es imprescindible conocer los principios y limitaciones de estos métodos, sobre todo para elegir el instrumental y la técnica adecuados. Los métodos fotométricos no están exentos de limitaciones específicas. Un analista puede discernir si algo ha ido mal al ver un color o turbidez anómalos cuando hace la comparación visual; es fácil que esta discrepancia no sea detectada al hacer la lectura fotométrica, ya que el instrumento siempre lee un valor, sea éste coherente o no. Hay que comprobar a menudo la sensibilidad y la exactitud mediante el uso de soluciones estándar, de manera que puedan detectarse problemas eléctricos, mecánicos u ópticos en los aparatos. La comprobación, el mantenimiento y la reparación de los mismos pueden requerir conocimientos especializados. Un fotómetro no tiene una exactitud uniforme en toda la extensión de su escala de medida. En casos de absorbancia muy alta, la escala está saturada, de manera que un cambio considerable en la concentración relativa sólo produce cambios pequeños en la posición del indicador o de la aguja. Con una absorbancia muy baja, cualquier pequeña diferencia entre células ópticas, la presencia de humedad condensada, el polvo, las burbujas, las huellas dactilares o una ligera falta de reproducibilidad al colocar las célu- 1-55 las pueden dar lugar a considerables cambios en las lecturas de las concentraciones. Las dificultades se minimizan si se hace que las lecturas caigan en una absorbancia de 0,1 a 1,0 diluyendo o concentrando la muestra o variando el paso de la luz y seleccionando células del tamaño adecuado. En cada uno de los métodos reseñados en el manual se hacen algunas sugerencias sobre las escalas y los pasos de luz adecuados, pero necesariamente debe confiarse mucho en el conocimiento y juicio del analista. Casi todos los fotómetros alcanzan sus mejores resultados en las lecturas de una absorbancia entre 0,1 y 1 con respecto a un blanco ajustado a una absorbancia de 0. Cuanto más se aproxime la lectura a 0 o 0,3, menos exacta será. Si no es posible utilizar una célula con un paso de luz suficientemente largo (lo que sucede con algunos instrumentos comerciales), concentrar más la muestra o elegir una prueba de color más sensible, puede resultar más exacto comparar colores muy tenues en tubos de Nessler que intentar una lectura fotométrica con una transmitancia próxima al 100 por 100. En general, la mejor longitud de onda o filtro es la que produce la mayor amplitud de lecturas entre un estándar y un blanco, lo que suele corresponder a un color visual para el haz de luz complementario al de la solución; por ejemplo, un filtro verde para una solución roja o un filtro violeta para una solución amarilla. Los poderes de absorción (absorcibidades) son útiles para comparar las sensibilidades de los métodos y para calcular la concentración de las soluciones absorbentes, como la ditizona. Puede calcular la absorción según la ley de Beer de la forma siguiente: 1-56 MÉTODOS NORMALIZADOS donde: a = Poder de absorción, l/(g · cm), A = absorbancia de una solución, sin dimensiones, b = concentración, g/1, y c = longitud de paso de la célula, cm. El uso de un instrumento fotoeléctrico hace innecesaria la preparación de lotes completos de estándar para cada lote de muestras a analizar. Sin embargo, debe prepararse un reactivo blanco que contenga agua destilada y todos los reactivos y, al menos, un estándar en el extremo superior de los límites óptimos de concentración para cada grupo de muestras a fin de comprobar la constancia de la curva de calibración. Esta precaución servirá para poner de manifiesto cualquier cambio inesperado en los reactivos, en el instrumento o en la técnica. A intervalos regulares, o cuando aparezca algún resultado dudoso, se preparará un lote completo de estándares (al menos cinco o seis para cubrir los límites óptimos de concentración), al objeto de comprobar la curva de calibración. A este respecto también es útil la información sobre el poder de absorción que se incluye en el presente manual en relación con diversos métodos fotométricos. Hay que comprobar a menudo la exactitud de las curvas o los estándares permanentes comparándola con la de estándares preparados en el laboratorio mediante el uso de los mismos lotes de reactivos, instrumentos y métodos utilizados para el análisis de las muestras. Por muy precisas que sean las curvas de calibración permanente o estándares preparadas por el fabricante, pueden no ser válidas en las condiciones concretas de uso. Los estándares permanentes pueden sufrir un apagamiento o alteración del color, y su validez puede depender también de determinadas condiciones arbitrarias de iluminación. Es posible que los estándares y las curvas de calibración sean incorrectos a causa de pequeñas di- ferencias entre los reactivos, instrumentos o técnicas que ha utilizado el fabricante y los que se emplean en el laboratorio de análisis. El instrumento ha de ponerse a cero mediante un reactivo blanco o, si es necesario, con agua destilada. No poner nunca a cero con un blanco de muestra. Determínese la absorción en comparación con las concentraciones de los estándares para establecer una curva de trabajo y el grado de igualdad con la ley de Beer. Si las lecturas se efectúan en porcentaje de transmitancia, habrán de convertirse en valores de absorbancia antes de consignarse en papel logarítmico. Existen varios métodos para eliminar la eventual turbidez de la muestra. La elección del que se vaya a utilizar depende de la naturaleza de la muestra, el tamaño de las partículas suspendidas y las razones por las que se lleva a cabo el análisis. Puede por ejemplo coagularse añadiendo sulfato de zinc y una base, como se hace en el método directo de nesslerización para el nitrógeno amónico en forma de amoníaco. En muestras de turbidez relativamente manifiesta puede ser suficiente con centrifugar. En algunos casos es posible utilizar filtros de fibra de vidrio, filtros de papel o filtros de vidrio sinterizado de poro fino. Cuando el tamaño de las partículas es muy reducido, los filtros de membrana pueden proporcionar el necesario grado de retención. Utilizados de forma adecuada, cada uno de estos métodos permitirá obtener resultados satisfactorios. Sin embargo, debe recordarse siempre que no existe un único medio ideal para eliminar la turbidez. Además, hay que operar con la máxima atención ya que todos los procedimientos de filtrado o de floculación pueden determinar pérdida de adsorción. Si no es posible eliminar la turbidez sin comprometer la integridad de la muestra, habrá que realizar una compensación fotométrica para corregir la interferencia. Se medirá la muestra sin añadir 1-57 INTRODUCCIÓN GENERAL reactivos (blanco de muestra) comparándola con un blanco de reactivo o con agua destilada para determinar la respuesta del instrumento. La respuesta se debe a la absorción de la muestra o a la turbidez originada por elementos distintos del que se va a determinar. Ténganse en cuenta los cambios de volumen derivados de la no inclusión de reactivos. Si la curva de calibración es lineal, puede sustraerse la absorción del blanco de muestra de la absorción de la muestra antes de consignar la concentración o puede computarse la concentración para el blanco de muestra y sustraerla de la de la muestra analizada. Si se utiliza una calibración no lineal, se computará la concentración del blanco de muestra y se restará de la concentración de la muestra analizada. En este caso no es correcto sustraer el valor de absorbancia. Al desarrollar la técnica, gradúese el cero del instrumento utilizando un blanco de turbidez o, de manera alternativa, determínese la absorbancia de este blanco comparándola con la de un blanco de reactivo o con la del agua destilada, y después réstese de la absorbancia de la muestra. Como en el caso anterior, han de corregirse las diferencias de volumen causadas por la inclusión o no de nuevos reactivos. b) Soluciones de ditizona: En varios métodos colorimétricos para metales (Cd, Pb, Hg, Ag, Zn) se utiliza ditizona (difeniltiocarbazona) como extracto coloreado y formador de complejos metálicos. Los métodos que se expondrán más adelante en este texto se basan en tres soluciones madres de ditizona cuya preparación también se describe. La concentración de ditizona en la solución madre se lleva a cabo a partir de un reactivo de ditizona del 100 por 100 de pureza. Algunos preparados comerciales de ditizona están contaminados por el producto de oxidación difeniltiocabodiazona o por metales. Purifíquese la ditizona de la forma que se indica más adelante. Para so- luciones de ditizona de concentración no superior al 0,001 por 100 (p/v), calcúlese la concentración exacta dividiendo la absorbancia de la solución en una célula de 1,00 cm a 606 nm por 40,6 x 103, la absorcividad o poder de absorción molar. Ajústense las diluciones de las soluciones madre de ditizona hasta conseguir soluciones de trabajo de la concentración deseada, teniendo en cuenta la concentración determinada en la solución madre. 1) Solución madre de ditizona I, 100 mg de ditizona/1.000 ml de CHC13: disuélvanse 100 mg de ditizona en 50 ml de CHC13 en un matraz de 150 ml y fíltrese a través de un papel de 7 cm de diámetro *. Recójase el filtrado en un embudo separador de 500 ml o en un Erlenmeyer de 125 ml a un vacío ligero. Utilícese un dispositivo de filtración que permita manipular el vapor de CHC13. Lávese el matraz con dos porciones de 5 ml de CHCI3 que se filtran. Lávese el papel con tres porciones de 5 ml de CHC13, añadiendo la porción final gota a gota por el borde del papel. Si se ha filtrado a un matraz, pásese el CHC13 a un embudo separador de 500 ml. Añádase 100 ml 1 + 99 de NH4OH al embudo de separación y agítese moderadamente durante un minuto; una agitación excesiva produce emulsiones que tardan en desaparecer. Manténganse separadas las capas moviendo suavemente el embudo para sumergir las gotitas de CHC13 que hayan quedado sobre la superficie de la capa acuosa. Transfiérase la capa de CHC13 a un embudo de separación de 250 ml manteniendo la capa acuosa rojo-anaranjada en el embudo de 500 ml. Repítase la extracción, colocando la capa de CHC13 en otro embudo de separación de 250 ml y pásese la capa acuosa, utilizando NH 4 OH 1 + 99 al * Whatman n.° 42, o equivalente. MÉTODOS NORMALIZADOS 1-58 embudo de 500 ml que contenía el primer extracto. Trasládese la capa acuosa a un embudo de 500 ml después de repetir la operación. Deséchese la capa de CHC13. Para combinar los extractos en el embudo de separación de 500 ml, añádase 1 + 1 de HC1 en porciones de 2 ml, mezclando después de cada adición hasta que la ditizona precipite y la solución pierda su color rojo-anaranjado. Extráigase el precipitado de ditizona con tres porciones de 25 ml de CHC13. Finalmente dilúyanse los extractos combinados hasta 1.000 ml con CHC1 3 ; 1,00 ml = 100 μg de ditizona. 2) Solución madre de ditizona II, 250 mg de ditizona/250 ml de CHC13: disuélvanse 250 mg de ditizona en 50 ml de CHCl 3 en un matraz de 150 ml y fíltrese con un papel de 7 cm de diámetro*. Recójase el filtrado en un embudo de separación de 1.000 ml o en un Erlenmeyer de 125 ml con un vacío ligero; utilícese un dispositivo de filtración que permita manipular el vapor de CHC13. Lávese el matraz con dos porciones de 5 ml de CHC13 y fíltrese. Lávese el papel con tres porciones de 5 ml de CHC13, añadiendo la última gota a gota en el borde del papel. Si se ha filtrado a un matraz, pásese el CHC13 a un embudo de separación de 1.000 ml. Añádanse 200 ml de NH4OH 1 + 99 al embudo de separación y agítese moderadamente durante un minuto; una agitación excesiva produce unas emulsiones que tardan en desaparecer. Manténganse separadas las capas moviendo suavemente el embudo de separación para sumergir las gotitas de CHC13 que hayan quedado sobre la superficie de la capa acuosa. Transfiérase la capa de CHC13 a un embudo de separación de 500 ml, manteniendo la capa acuosa rojo-anaranjada en el embudo de 1.000 ml. Repí* Whatman n.° 42. o equivalente. tase la extracción de la capa de CHC13 con 200 ml de NH4OH 1 + 99 pasando la capa de CHC13 a otro embudo de separación de 500 ml. Pásese la capa acuosa a un embudo de separación de 1.000 mi que contenía el primer extracto. Realícese de nuevo la extracción con una tercera porción de 200 ml de NH 4 OH 1 + 99. Deséchese la capa de CHC13 y pásese la capa acuosa a un embudo de 1.000 ml. Añádanse porciones de 4 ml de HCl 1 + 1 a los extractos combinados, mezclando después de cada adición, hasta que la ditizona precipite y la solución pierda el color rojo-anaranjado. Extráigase el precipitado de ditizona con cuatro porciones de 25 ml de CHC13. Diluyanse los extractos combinados a 250 ml con CHC13; 1,00 ml = 1.000 μg de ditizona. 3) Solución madre de ditizona III, 125 mg de ditizona/500 ml CC14: disuélvanse 125 mg de ditizona en 50 ml de CHC13 y procédase como en el caso de la solución II pero extrayendo el precipitado de ditizona con porciones de 25 ml de CC14. dilúyanse los extractos de CC14 a 500 ml; 1,00 ml = 250 μg de ditizona (PRECAUCIÓN: el CCl4 es tóxico, evitar su inhalación, ingestión y contacto con la piel.) 3. Otros métodos de análisis El uso de otros métodos instrumentales de análisis no descritos específicamente en el presente manual es admisible siempre que los resultados que se obtengan se comprueben de manera periódica, bien en comparación con uno de los métodos estándar expuestos o con una muestra estándar de composición fiable y contrastada. En el informe de laboratorio hay que especificar, junto a los resultados del análisis, cuál ha sido el método instrumental utilizado. a) Espectrometría de absorción atómica: La espectrometría de absorción ató- INTRODUCCIÓN GENERAL mica se ha aplicado a la determinación de metales en el agua, sin necesidad de concentración o tratamientos previos de la muestra. El uso de disolventes orgánicos junto con llamas de oxiacetileno, oxihidrógeno u óxido nitroso-acetileno permite la determinación de metales que forman óxidos refractarios. Entre las técnicas más generalizadas se encuentran métodos de absorción atómica, incluidos algunos sin llama y electrotérmicos (grafito calentado). b) Fotometría de llama: La fotometría de llama se utiliza para determinar sodio, potasio, litio y estroncio. c) Plasma de acoplamiento inductivo (PAI) y sistemas analíticos afines: Se sugiere utilizar PAI para la detección de varios iones metálicos, sobre todo cuando se desconoce si pueden existir posibles interferencias en las técnicas colorimétricas. El PAI ha permitido la determinación de muchos metales a concentraciones de pocos microgramos por litro al reducir a cenizas con HNO3 muestras de 100 ml. También las técnicas de polarografía, como la polarografía por impulsos, la voltametría diferencial por impulsos y la voltametría anódica diferencial de bandas por impulsos, están siendo cada vez más utilizadas para la determinación y especificación de metales pesados en aguas limpias y residuales. Un método muy próximo a la polarografía es la titulación amperométrica, adecuada para determinar cloro residual, dióxido de cloro y yodo, junto con otros métodos yodométricos. d) Titulación potenciométrica: Muchos métodos titulométricos pueden llevarse a la práctica potenciométricamente utilizando milivoltímetros o pH-metros, con electrodos adecuados. e) Electrodos selectivos de iones: Existen electrodos selectivos de iones que permiten una rápida valoración de ciertos componentes del agua. Estos electrodos funcionan de forma óptima en com- 1-59 binación con un pH-metro de escala ampliada o con un milivoltímetro adaptado. En su mayoría, los electrodos operan bajo el principio del intercambio iónico. Existen en la actualidad electrodos selectivos de iones diseñados para medir amoníaco, cadmio, calcio, cobre divalente, dureza del agua, plomo, potasio, plata, sodio, cationes monovalentes y divalentes totales y aniones de bromo, cloro, cianuro, flúor, yodo, nitrato, perclorato y sulfuro, entre otros. Estos aparatos sufren distintos grados de interferencia con otros iones contenidos en la muestra y muchos han de ser aún sometidos a minuciosos estudios antes de ser propuestos y adoptados como métodos estándar. Sin embargo, su valor para el desarrollo de actividades de control es evidente. A fin de eliminar las dudas que puedan existir sobre las variaciones de fiabilidad, debe comprobarse cada electrodo en presencia de interferencias así como con el ion para el que está destinado. En este manual se detallan varios métodos que emplean electrodos. Por su parte, las sondas comerciales de oxígeno disuelto (OD) muestran considerables variaciones de fiabilidad y necesidades de mantenimiento. A pesar de estos inconvenientes, se han aplicado a la monitorización de OD en distintas aguas limpias y residuales. Casi todas ellas llevan un electrodo fijado mediante una membrana permeable al oxígeno. El OD en la solución difunde a través de la membrana y de la capa electrolítica y reacciona con el electrodo, induciendo una corriente proporcional a la actividad y, por tanto, en soluciones de concentración iónica baja o constante, esencialmente proporcional a la concentración de OD. Existen también electrodos de OD sin membrana que ofrecen resultados satisfactorios. En cualquier caso, se debe mantener la superficie del sensor de OD bien agitada y con una compensación de la temperatura que asegure la consecución de datos fiables. 1-60 MÉTODOS NORMALIZADOS f) Cromatografía de gases (CG) y cromatografía de gases/espectrometría de masas (CG/EM): Son muchos los métodos de CG y de CG/EM para el análisis de aguas limpias y residuales. En el presente manual aparecen los destinados a la determinación de los pesticidas de hidrocarburos clorados, los componentes de gases digestores de bolos, fenoles, volátiles, PAH y compuestos causantes de olores y sabores así como análisis CG/EM generalizados para sustancias volátiles y extractos ácidos o básicos/ neutros. g) Análisis de flujo continuo: Se presentan técnicas automatizadas para cloruros, fluoruros, nitrógeno (amoníaco), nitrógeno (nitratos), fosfatos, sílice y sulfatos. h) Cromatografía de iones (CI): La cromatografía de iones es una técnica para la determinación escalonada de aniones o cationes utilizando intercambio iónico y conductividad, detectores amperométricos o colorimétricos. Véase la sección 4110 para la técnica generalizada para aniones. i) Otros métodos de análisis: Continuamente se asiste al desarrollo de nuevas técnicas instrumentales y métodos analíticos. El analista debe estar al corriente de estos progresos. Con regularidad se publican revisiones de cada rama de la química analítica en Analytical Chemistry y una revisión anual de la literatura referida al tema en Journal Water Pollution Control Federation. 4. Control de interferencias Muchos de los métodos analíticos están sujetos a interferencias ocasionadas por sustancias que se encuentran en la muestra. Las más frecuentes son bien conocidas y de ellas se proporciona una detallada información junto a cada uno de los métodos concretos. Es inevitable, sin embargo, que existan interferencias desconocidas e inesperadas. Ello se debe a la diversidad de naturaleza de las aguas y, sobre todo, de las aguas residuales. Por tanto, es necesario prestar atención a los componentes hasta ahora no detectados, los nuevos compuestos de tratamiento (en especial los agentes combinantes) y los nuevos residuos industriales y su potencial amenaza contra la exactitud de los análisis químicos. Cualquier cambio en la aparente composición de un agua que se haya mantenido constante con anterioridad, cualquier color anormal observado en una prueba colorimétrica o durante una titulación, cualquier turbidez, olor u otro hallazgo insólito deben despertar sospechas. Estos cambios pueden ser debidos a variaciones normales en las concentraciones relativas de los componentes habituales, pero también pueden ser consecuencia de la introducción de una sustancia nunca antes observada. Algunas sustancias tales como el cloro y el dióxido de cloro, la alumbre, las sales de hierro, los silicatos, el sulfato de cobre, el sulfato de amonio y los polifosfatos, son utilizadas con tal profusión que merecen especial atención como posibles causas de interferencia. De todas ellas, la más perniciosa probablemente sea el cloro, ya que blanquea o altera los colores de muchos reactivos orgánicos sensibles que se utilizan como indicadores de titulación y como reveladores de color en métodos fotométricos. Entre los métodos que han demostrado su eficacia en la eliminación de los residuos de cloro se encuentran la adición de mínimas cantidades de sulfuro, tiosulfato o arseniato, la exposición a la luz del sol o a la luz ultravioleta artificial y la conservación prolongada. Siempre que se encuentre o se sospeche de una interferencia y no existan recomendaciones específicas para evitarla, es necesario determinar qué técnica —si es que hay alguna— puede eliminarla sin afectar al propio análisis. Cuando existan 1-61 INTRODUCCIÓN GENERAL dos o más opciones metodológicas, habrá que establecer cuál es el procedimiento que afecta en menor medida a los resultados. Si distintos procedimientos dan lugar a resultados considerablemente diferentes, es probable que exista una interferencia. La importancia de algunas de estas interferencias puede atenuarse si se diluye la muestra o se utilizan cantidades más pequeñas; cualquier tendencia de los resultados a aumentar o disminuir de manera constante el diluir la muestra indica que es probable que exista una interferencia. a) Tipos de interferencia: Las interferencias pueden hacer que los resultados analíticos sean demasiado altos o demasiado bajos a consecuencia de alguno de los fenómenos siguientes: 1) Una sustancia de interferencia puede reaccionar como si fuera la sustancia buscada produciendo, por tanto, un resultado elevado; por ejemplo, el bromuro arroja títulos similares a los del cloruro. 2) Puede reaccionar con la sustancia buscada produciendo, en consecuencia, un resultado bajo. 3) Puede asimismo combinarse con el reactivo analítico y evitar que reaccione con la sustancia buscada; por ejemplo, el cloro destruye muchos indicadores y reactivos reveladores del color. Casi todas las interferencias corresponden a uno de estos tipos. Por ejemplo, en un método fotométrico, puede considerarse que la turbidez actúa como la sustancia que ha de determinarse; reduce, pues, la transmisión de la luz. Dos o más sustancias interferidoras pueden asimismo reaccionar de forma no aditiva y cada una de ellas contrarrestar o potenciar los efectos de las demás. b) Anulación de las interferencias: La opción preferencial para minimizar las interferencias es eliminar la sustancia que las produce o convertirla en inocua mediante alguno de los siguientes métodos: 1) Eliminar físicamente la sustancia buscada o la que produce la interferencia. Por ejemplo, destilar el fluoruro o el amoníaco dejando la interferencia. También es posible absorber las interferencias mediante una resina de intercambio iónico. 2) Ajustar el pH de forma que la única sustancia que reaccione sea la buscada; por ejemplo, ajustando el pH a 2, los ácidos volátiles desaparecerán de la solución. 3) Oxidar (digestión) o reducir la muestra hasta convertir a la interferencia en una forma inactiva. Por ejemplo, reducir el cloro o cloruro añadiendo tiosulfato, o digerir muestras para análisis mediante espectrometría de absorción atómica con alguno de los distintos reactivos que destruyen la materia orgánica. 4) Añadir un agente adecuado que se una a la sustancia que produce la interferencia de manera que, aunque persista, sea inocua. Por ejemplo, combinar el hierro con pirofosfato para evitar que interfiera con la determinación de cobre; combinar el cobre con cianuro o con sulfuro para evitar que interfiera con la determinación titulométrica de la dureza. 5) Puede utilizarse una combinación de las cuatro técnicas. Así, por ejemplo, pueden destilarse los fenoles de una solución acida para evitar que se destilen las aminas o puede utilizarse tiosulfato en el método de la ditizona para el zinc a fin de evitar la mayoría de los metales que pueden interferir pasando a la capa de CC14. 6) A veces es posible eliminar el color y la turbidez reduciendo a cenizas por métodos secos o húmedos o utilizando un agente floculante. Algunos tipos de turbidez pueden eliminarse mediante filtración. Sin embargo, estos procedimientos introducen el peligro de que también se elimine el componente objeto del análisis. c) Compensación de la interferencia: Si no puede utilizarse ninguna de estas 1-62 MÉTODOS NORMALIZADOS técnicas, habrá de recurrirse a otros métodos de compensación: 1) Si el color o la turbidez inicialmente presentes interfieren en una determinación fotométrica, es posible utilizar la compensación fotométrica. La técnica se describe en la sección 1070D.2a. 2) Es posible determinar la concentración de las sustancias que producen la interferencia y añadir idénticas cantidades a los estándares de calibración, aunque ello supone una gran cantidad de trabajo. 3) Si la interferencia no sigue aumentando a medida que lo hace la concentración de la sustancia que la produce, sino que tiende a estacionarse en un determinado nivel, se puede añadir una gran cantidad de dicha sustancia a todas las muestras y a todos los estándares. Esta técnica se conoce como «inundación». 4) Puede por fin determinarse la presencia de la sustancia buscada en los reactivos químicos llevando a cabo una determinación de blanco. (reimpresión), Robert E. Krieger Publishing Co., Melbourne, Florida. PECSOK, R. et al. 1976. Modern Methods of Chemical Analysis, 2.a ed. John Wiley & Sons, Nueva York. VOGEL, A. I. 1978. Textbook of Quantitative Inorganic Analysis. Incluye Elementary Instrumental Analysis, 4.a ed. Revisada por J. Basset. Longman, Nueva York. Revisiones y bibliografías analíticas W EIL , B. H et al. 1948. Bibliography on Water and Sewage Analysis, State Engineering Experiment Sta., Georgia Inst. Technology, Atlanta. Annual reviews of analytical chemistry. 19531987. Anal. Chem. 25:2; 26:2; 27:574; 28:559; 29:589; 30:553; 31:776; 32:3R; 33:3R; 34:3R; 35:3R; 36:3R; 37-59:lR. WATER POLLUTION CONTROL FEDERATION RESEARCH COMMITTEE. 1960-1988. Annual literature review, nature and analysis of chemical species J. Water Pollut. Control Fed. 32:443; 33:445; 34:419; 35:553; 36:535; 37:735; 38:869; 39:867; 40:897; 41:873; 42:863; 43:933; 44:903; 45:979; 46:1031; 47:1118; 48:998; 49:901; 50:1000; 51:1108; 52:1083; 53:620; 54:520; 55:555; 56:494; 57:438; 58:419; 59:306; 60:743. Técnicas calorimétricas 5. Bibliografía Técnicas analíticas generales FOULK, C. W., H. V. MOYER & W. M. MACNEVIN. 1952. Quantitative Chemical Analysis. McGraw-Hill Book Co., Nueva York. WlLLARD, H. H., N. H. FURMAN & C. E. BRICKER . 1956. Elements of Quantitative Analysis, 4.a ed. D. Van Nostrand Co., Princeton, Nueva Jersey. HUGHES, J. C. 1959. Testing of Glass Volumetric Apparatus. Nat. Bur. Standards Circ. No. 602. WILSON, C. L. & D. W. WILSON, eds. 1959, 1960, 1962. Comprehensive Analytical Chemistry, Vol. 1A, IB, 1C. Elsevier Publishing Co., Nueva York. MEITES, L., ed. 1963. Handbook of Analytical Chemistry. McGraw-Hill Book Co., Nueva York. KOLTHOFF, I. M., E. J. MEEHAN , E. B. SANDELL & S. BRUCKENSTEIN. 1969. Quantitative Chemical Analysis, 4.a ed. Macmillan Co., Nueva York. WELCHER, F. J., ed. 1975. Standard Methods of Chemical Analysis, 6.a ed. Vol. HA, IIB, IIIA M ELLON , M. G. 1947. Colorimetry and photometry in water analysis. J. Amer. Water Works Assoc. 39:341. NATIONAL BUREAU OF STANDARDS. 1947. Terminology and Symbols for Use in Ultraviolet, Visible, and Infrared Absorptiometry. NBS Letter Circ. LC-857 (19 de mayo). GIBSON, K. S. & M. BALCOM. 1947. Transmission measurements with the Beckman quartz spectrophotometer. J. Res. Nat. Bur. Standards 38:601. SNELL, F. D. & C. T. SNELL. 1948-1971. Colorimetric Methods of Analysis, 3.a ed. Vol. 1, 2, 2A, 3, 3A, 4, 4A, 4AA, 4AAA. Van Nostrand Reinhold, Nueva York. MELLON, M. G., ed. 1950. Analytical Absorption Spectroscopy. John Wiley & Sons, Nueva York. D ISKANT , E. M. 1952. Photometric methods in water analysis. J. Amer. Water Works Assoc. 44:625. S ANDELL , E. B. 1978. Colorimetric Determination of Traces of Metals, 4.a ed. John Wiley & Sons, Nueva York. BOLTZ, D. F., ed. 1978. Colorimetric Determination of Nonmetals, 2.a ed. John Wiley & Sons, Nueva York. 1-63 INTRODUCCIÓN GENERAL Otros métodos de análisis HARLEY, J. H. & S. E. WIBERLEY. 1967. Instrumental Analysis, 2.a ed. John Wiley & Sons, Nueva York. EWING, G. W. 1975. Instrumental Methods of Chemical Analysis, 4.a ed. McGraw-Hill Book Co., Nueva York. H EFTMANN , E., ed., 1975. Chromatography, 3.a ed. Reinhold Publishing Corp., Nueva York. L EDERER , E. & M. L EDERER . 1957. Chromatography, 2.a ed. Elsevier Press, Houston, Texas. LINGANE, J. J. 1958. Electroanalytical Chemistry, 2.a ed. Interscience Publishers, Nueva York. SAMUELSON, O. 1963. Ion Exchangers in Analytical Chemistry. John Wiley & Sons, Nueva York. 1080 AGUA DE CALIDAD PARA REACTIVOS 1080 A. Introducción Uno de los aspectos más importantes del análisis es la preparación del agua de calidad para reactivos que han de utilizarse en la dilución de éstos y para los análisis de blancos. Los niveles de calidad cubren un espectro que oscila entre el tipo I, sin concentraciones detectables de los compuestos o elementos a analizar dentro de los límites de detección del método analítico, y el tipo III, para lavados y análisis cualitativos (véase tabla 1080:1). El agua para análisis no debe contener sustancias que interfieran con los métodos analíticos. La calidad del agua está directamente relacionada con el análisis que vaya a efectuarse. Puede diferir, de hecho, en función de los componentes orgánicos, inorgánicos y microbiológicos y del uso que se vaya a dar a dicha agua. Cualquier método de preparación de agua de calidad para reactivos es acepta- ble siempre que se cumplan los requisitos adecuados, ya que un sistema mal conservado puede dar lugar a la inducción de contaminantes. La osmosis inversa, la destilación y la desionización en distintas combinaciones pueden utilizarse para el mismo propósito si se emplean de forma adecuada. También pueden utilizarse en este proceso la ultrafiltración, el tratamiento con luz ultravioleta o una combinación de ambos. En la sección 1080 se proporcionan normas generales para la preparación de este tipo de aguas. En la tabla 1080:11 se enumeran los procesos más habituales para la purificación del agua y las clases principales de contaminantes que pueden eliminarse mediante esta purificación. Para más detalles sobre la preparación del agua para pruebas microbiológicas, véase la sección 9020B.3c. 1-64 MÉTODOS NORMALIZADOS TABLA 1080:1. ESPECIFICACIONES DEL AGUA PARA REACTIVOS* TABLA 1080:11. PROCESOS DE PURIFICACIÓN DEL AGUA 1-65 INTRODUCCIÓN GENERAL 1080 B. Métodos de preparación de agua de calidad para reactivos 1. Destilación Puede prepararse agua para análisis destilando agua en un alambique de vidrio de borosilicato, cuarzo fundido o titanio. Para eliminar el amoníaco, se destilará a partir de una solución acida. El CO2 puede eliminarse hirviendo el agua durante 15 minutos y enfriándola rápidamente a temperatura ambiente. Por su parte, el CO2 atmosférico se excluye utilizando un tubo que contenga cal sodada o un agente comercial eliminador del CO2*. Téngase en cuenta que al hervir el agua pueden incorporarse otras impurezas que se desprenden del envase. Es necesario realizar tratamiento previo del agua y mantenimiento periódico para evitar la formación de escamas en el alambique. En los casos en que el agua contiene cantidades significativas de calcio, magnesio o bicarbonato puede ser necesario efectuar un pretratamiento de desmineralización mediante osmosis inversa o intercambio iónico. La resistividad del agua destilada (tipo II) debe ser > 1,0 megaohmio-cm a 25 °C, y para la de tipo I el valor deberá ser de 10 megaohmios-cm. Las mediciones son más exactas si se hacen sobre células en serie. 2. Osmosis inversa La osmosis inversa es un proceso en el que se fuerza al agua para que entre a presión a través de una membrana semipermeable, eliminando una parte de los componentes disueltos y de las impurezas suspendidas. La calidad del agua ob- * Ascarite. Fisher Scientifíc Co., o equivalente. tenida depende, obviamente, de la del agua original. Es preciso seleccionar el módulo de membrana de osmosis inversa adecuado a las características del agua a tratar y obtener los datos sobre los contaminantes en el agua original, a la presión a que se vaya a trabajar. Es, asimismo, necesario establecer la producción total de agua para economizar al máximo en su uso sin comprometer la calidad final de la operación. La elección de las configuraciones de las hendiduras depende del grado de suciedad del agua a tratar. Con independencia de la configuración utilizada, puede hacerse necesario un pretratamiento para minimizar la contaminación de la membrana por coloides o partículas, así como la introducción de cloro, hierro y otros compuestos oxidantes que puedan degradar las membranas de osmosis inversa. Es necesario hacer una limpieza periódica de los módulos de membrana. 3. Intercambio iónico Prepárese agua desionizada haciendo pasar el agua a través de un lecho mixto de intercambiador iónico constituido por resinas aniónicas y catiónicas fuertes. Cuando el sistema no está funcionando continuamente, ha de ponerse de nuevo en circulación el agua producida por el lecho de intercambio iónico. La resistencia del agua de tipo I debe ser de 10 megaohmios-cm (en serie) a 25 °C. En los casos en los que sea rentable proceder a una regeneración de la resina, se utilizarán lechos separados de resinas aniónicas y catiónicas. En estos casos, dispóngase el intercambiador aniónico después del catiónico para extraer los residuos que puedan proceder de la resina MÉTODOS NORMALIZADOS 1-66 catiónica. Es esencial que las dimensiones del lecho sean las adecuadas para que las resinas ofrezcan el rendimiento requerido. En especial, hay que establecer la relación entre la longitud y el diámetro del lecho según la máxima velocidad de flujo, para asegurar que no se sobrepasa la velocidad máxima y que el agua permanece durante un tiempo suficiente en contacto con la resina. En situaciones en las que el agua de alimentación contiene cantidades significativas de materia orgánica, es preciso eliminar los microorganismos para disminuir en lo posible la contaminación de las resinas. Los posibles tratamientos previos consisten en prefiltración, destilación, osmosis inversa y adsorción. 4. Adsorción La adsorción suele utilizarse para eliminar el cloro y las impurezas orgánicas. Se suele realizar con carbón activado granular. El nivel de eficacia en la eliminación de organismos depende de la naturaleza de los mismos, de las características físicas del carbón activado y de las condiciones de actuación. La eficacia de 1080 C. la adsorción de organismos es inversamente proporcional a la solubilidad, y, por ello, esta técnica puede resultar inadecuada para la eliminación de compuestos polares de bajo peso molecular. Las diferencias de funcionamiento entre los distintos carbones activados son atribuibles al uso de distintos materiales y a los procedimientos de activación. Selecciónese el carbón activado adecuado teniendo en cuenta estas diferencias. Incluso con un carbón activado óptimo, no se conseguirá un funcionamiento correcto a menos que las dimensiones de la columna permitan obtener velocidad de entrada y tiempo de paso adecuados a la máxima tasa de flujo. El empleo de carbón activado puede producir efectos adversos en la resistividad o resistencia específica. Estos efectos pueden controlarse mediante osmosis inversa, con resinas mixtas o con adsorbentes especiales. Para conseguir el menor nivel posible de organismos contaminantes, se utilizarán mezclas de resinas de pulimentación con carbones especiales en combinación con otros tratamientos adicionales, tales como osmosis inversa, carbones naturales, oxidación ultravioleta o ultrafiltración. Calidad del agua para reactivos 1. Patrones de calidad Existen varios patrones de calidad del agua para análisis, todos ellos basados en los niveles de contaminantes (véase tabla 1080:1)1–3. Los métodos y usos enumerados proceden del National Committee for Clinical Laboratory Standards (NCCLS). Utilícese agua de tipo I en los métodos analíticos que requieran un mínimo de interferencias y desviación y un máximo de precisión. El agua de tipo II está destinada a proporcionar al usuario un agua en la que sea admisible la presencia de bacterias. Se utiliza para la preparación de reactivos, colorantes o tinciones. El agua de tipo III puede utilizarse para la limpieza del material de vidrio o como agua de alimentación para la producción de aguas de mayor nivel de calidad. El agua para análisis de tipo I, con una resistividad mínima de 10 megaohmcm a 25 °C (en serie), se prepara mediante destilación, desionización o tratamiento con osmosis inversa del agua de ali- 1-67 INTRODUCCIÓN GENERAL mentación, seguidos de acabado con un lecho mixto desionizador y paso a través de un filtro de membrana de 0,2 nm de poro. También puede tratarse con osmosis inversa seguida de adsorción con carbón y desionización. Determínese su calidad en el momento de su producción. Los desionizadores de lecho mixto añaden pequeñas cantidades de materia orgánica al agua, sobre todo si el lecho es fresco. La resistividad del agua de tipo I debe ser > 10 megaohm-cm a 25 °C, medida en serie. La determinación de la resistividad no podrá detectar organismos o contaminantes no ionizados ni proporcionará una valoración exacta de los contaminantes iónicos a nivel de microgramo por litro. Por consiguiente, se harán mediciones distintas para contaminantes del tipo de TOC, SiO2 y recuentos bacterianos. El agua de tipo II se produce mediante destilación o desionización. Su resistencia debe ser > 1 megaohm-cm a 25 °C. Hay que adoptar las mismas precauciones al hacer las determinaciones de los contaminantes. El agua de tipo III debe tener una resistividad mínima de 0,1 megaohm-cm. En la tabla 1080:1 se enumeran otros contaminantes del agua para reactivos. El pH del agua de tipo I y II no puede medirse de manera exacta sin contami- narse. Para algunos análisis es necesario medir otros componentes. El agua de tipo I no puede ser almacenada sin que sufra una degradación significativa, por lo que debe producirse de manera continua y consumirse de inmediato. El agua de tipo II puede conservarse, pero hay que procurar que su almacenamiento dure el menor tiempo posible y que mantenga la calidad adecuada al uso que se le va a dar. Consérvese sólo en materiales que la protejan de la contaminación, como el TFE y el vidrio para análisis orgánicos o los plásticos para análisis de metales. El agua de tipo III se conservará en materiales que la protejan de la contaminación. 2. 1. 2. 3. Referencias AMERICAN SOCIETY FOR TESTING AND MATERIALS . 1987. Annual Book of ASTM Standards, Parte 31, Water: Atmospheric Analysis. American Soc. Testing & Materials, Filadelfia, Pennsylvania COMMISSION ON LABORATORY INSPECTION AND A CCREDITATION . 1985. Reagent Water Specifications. College of American Pathologists, Chicago, III. NATIONAL COMMITTEE FOR CLINICAL LABORATORY STANDARDS. 1988. Preparation and Testing of Reagent Water in the Clinical Laboratory - Segunda edición; Tentative Guideline. Publ. C3-T2, National Comm. for Clinical Laboratory Standards, Villanova, Pennsylvania. MÉTODOS NORMALIZADOS 1-68 1090 1090 A. 1. SEGURIDAD Introducción Consideraciones generales Conseguir un ambiente seguro y saludable de trabajo es responsabilidad de la institución, del director del laboratorio, del supervisor del personal y, por último, del propio personal del laboratorio. Cada trabajador debe hacer todo lo posible para protegerse a sí mismo y a sus compañeros. El director del laboratorio debe tener en cuenta que todo accidente tiene una causa y, por tanto, puede evitarse mediante un adecuado programa de seguridad1. Una vez que un empleado se haya acostumbrado a trabajar con técnicas nuevas y seguras, el director puede estar seguro de que dicho empleado propondrá otras ideas acerca de la seguridad. 2. Organización de la seguridad Aunque la responsabilidad del establecimiento y reforzamiento de un programa de seguridad en el laboratorio corresponde, en última instancia, al director, es conveniente, salvo en laboratorios muy pequeños, delegar responsabilidades en un miembro del personal designado como oficial de seguridad2. El responsable de seguridad debe formar parte del equipo de dirección, de forma que tenga capacidad para optimizar el adiestramiento, la inspección y el adoctrinamiento sobre seguridad de los nuevos empleados, y para adquirir una información actualizada sobre el tema. El responsable de seguridad debe inspeccionar periódicamente los equipos de emergencia, como extintores de fuego, sistemas de alar- ma, lavado ocular y duchas de seguridad; debe realizar inspecciones periódicas del laboratorio para descubrir peligros inadvertidos, y debe observar si el personal cumple las reglas de seguridad y recordarle que se mantenga al tanto de las prácticas recomendadas al efecto. La seguridad sobre radiación es una parte especializada del programa general de seguridad. Debe asignarse una persona técnicamente cualificada, que será el Responsable de Seguridad sobre Radiación (RSR). Éste habrá de comprobar que las fuentes y equipos de radiación se usan de manera adecuada y conforme a todas las regulaciones estatales y locales pertinentes, y que se mantienen vigentes las licencias oficiales necesarias. Debe establecerse un comité de seguridad, el cual contará con el apoyo del director del laboratorio. Las recomendaciones de este comité deben ser tomadas en cuenta y aceptadas rápidamente. En los comités de seguridad participan personas con distintos tipos de experiencia en la toma de decisiones en relación con las prácticas de seguridad. Por ejemplo, los químicos deben aportar sus conocimientos en el tratamiento de los peligros químicos y bacteriológicos, y su participación puede ser de gran valor educativo. Es deseable que los miembros roten periódicamente, de forma que la mayoría del personal tenga la oportunidad de participar. Hay que conservar informes bien hechos sobre todos los accidentes, inspecciones, ensayos, etc., siendo preferible utilizar un único impreso estándar para ello. El informe debe contener información suficiente como para que el respon- INTRODUCCIÓN GENERAL 1-69 sable de seguridad, el supervisor y el director puedan determinar quiénes se vieron afectados, qué fue lo que sucedió, cómo y cuándo sucedió y qué lesiones se produjeron. La Occupational Safety and Health Act (OSHA; regulación legal norteamericana) obliga a registrar en un libro los accidentes que producen incapacidades importantes, pero lo más conveniente es registrar todos los accidentes para poder evaluar el programa de seguridad. El registro del momento y la naturaleza de cada accidente puede tener importancia en caso de que se produzcan incapacidades y proceda hacer una reclamación a Worker's Compensation (organismo estadounidense de asistencia laboral). Otra parte del programa de seguridad consiste en mantener un archivo de recomendaciones que hayan hecho el responsable o el comité de seguridad, así como de las acciones que se hayan puesto en práctica. Diversas estipulaciones del OSHA especifican un método para notificar a los empleados los peligros existentes en su puesto de trabajo. El personal expuesto debe estar bajo la supervisión directa y la observación regular de una persona técnicamente cualificada, la cual tendrá conocimiento de los peligros existentes, de sus efectos sobre la salud y de los procedimientos de emergencia pertinentes. El supervisor debe entrenar al personal del laboratorio en las prácticas de seguridad en su trabajo. El personal tiene derecho a conocer cuáles son los materiales peligrosos que maneja, los riesgos específicos que presenta el manejo de esos materia- 1090 B. 1. les y los procedimientos necesarios para protegerse de ellos. El entrenamiento en las técnicas de seguridad exige del director un esfuerzo deliberado. En los laboratorios más grandes es importante realizar seminarios sobre seguridad. Se utilizarán técnicas de enseñanza, incluyendo películas educativas, demostraciones sobre el uso de aparatos tales como extintores de fuego o respiradores, tarjetas donde se indiquen los productos químicos peligrosos y los procedimientos adecuados para manipularlos y manuales de seguridad. Conviene repetir periódicamente los entrenamientos sobre seguridad. Los fabricantes de productos químicos proporcionan hojas de datos sobre seguridad en el manejo de materiales, las cuales tienen una importancia extraordinaria como parte de la información general que puede suministrarse al personal acerca de los peligros de su puesto de trabajo4. Estas hojas de datos deben estar a disposición del personal que utiliza materiales peligrosos. 3. 1. 2. 3. 4. Referencias INHORN , S. L., ed. 1978. Quality Assurance Practices for Health Laboratories. American Public Health Assoc, Washington, D.C. STEERE, N. V., ed. 1971. Handbook of Laboratory Safety, 2.a ed. Chemical Rubber Co., Cleveland, Ohio. Hazard Communication: Final Rule. 1983. Federal Register 48:53280; 1985. 29 CFR Part 1910.1200. Chemical Safety Data Sheets. Manufacturing Chemists' Assoc, Inc. Washington, D.C. Equipo de seguridad Equipo de laboratorio a) Extintores: Existen tres tipos generales de extintores: 1) Extintores de agua, útiles para fue- gos con combustibles ordinarios, como lana, papel o tela. 2) Extintores químicos secos, útiles contra la mayoría de los fuegos, pero sobre todo para apagar los producidos por 1-70 líquidos y metales inflamables y los fuegos eléctricos. 3) Extintores de dióxido de carbono, útiles para fuegos pequeños producidos por líquidos inflamables, y con una eficacia limitada al aplicarse sobre instrumentos y equipos electrónicos. Según los peligros potenciales, un laboratorio puede tener más de un tipo de extintor en cada habitación, en lugar fácilmente accesible y apartado de la zona de máximo peligro1–5. Los extintores de halones son útiles en áreas especiales en donde se encuentren equipos electrónicos o computadoras que puedan dañarse con los extintores convencionales. Tanto los extintores de halones como los de dióxido de carbono reducen la cantidad de oxígeno en el aire. Todos los extintores han de utilizarse con cuidado. A menudo, se recomienda la instalación de extintores multiuso (tipo ABC) en los laboratorios, y de extintores de halones en las campanas que contienen material orgánico. Hay que comprobar que los extintores se revisan y recargan de forma periódica. b) Manta ignífuga: Colóquense mantas ignífugas en lugares fácilmente accesibles en cada laboratorio6. c) Duchas de seguridad: Las duchas de seguridad son una parte integrante del laboratorio, y se utilizan en accidentes por ácidos, cáusticos u otros líquidos peligrosos, cuando se prende fuego a las ropas y en otras situaciones de emergencia. Las duchas deben estar convenientemente situadas, preferiblemente cerca de una puerta y sobre un espacio despejado, y hay que comprobar su estado periódicamente6. d) Lavado de ojos: Cuando sea necesario, se quitarán las lentillas de contacto. Lávense inmediata y cuidadosamente (15 minutos) los ojos para evitar alteraciones visuales o incluso la ceguera en caso de que se produzca una salpicadura de un producto químico. Las salpicaduras en la cara se lavan fácilmente con un MÉTODOS NORMALIZADOS colirio con aplicación en chorro. Algunos expertos opinan que los chorros de colirio introducen partículas en el ojo (de vidrio o metal, polvo, arena), en lugar de retirarlas. Si ocurre un accidente de este tipo, consúltese a un oculista después de haber hecho un suave lavado con agua. Los colirios deben situarse cerca de los fregaderos, en una zona central y muy visible y lejos de las conexiones eléctricas. Hay que controlar periódicamente los recipientes para comprobar su buen funcionamiento; si se utilizan envases portátiles, hay que limpiarlos semanalmente y cambiar el agua para reducir la posibilidad de contaminación. En otro apartado se ha tratado extensamente el uso de los envases de colirio y algunos de sus problemas2. e) Escudos protectores: El tipo de escudos protectores más utilizados es el que protege contra las distintas formas de radiación, como bombas láser y emisiones ultravioletas. Las campanas químicas convencionales deben estar provistas de vidrios de seguridad y paneles móviles. Considérese la conveniencia de utilizar escudos cuando se trabaja con vidrios de vacío o sistemas presurizados. f) Envases de seguridad: Los envases de segundad están diseñados para minimizar las consecuencias de un accidente o para evitar la diseminación de materiales peligrosos. Se utilizan para transportar productos químicos, sobre todo ácidos y álcalis concentrados. Para disolventes inflamables, empléense envases sometidos a controles de calidad por los organismos pertinentes. Otros envases de seguridad más complejos son las cámaras de manipulación con guantes, las campanas de flujo laminar y los compartimentos manejados por control remoto para material radiactivo o patógenos peligrosos. Deben manejarse en condiciones de presión negativa con filtración primaria a la salida de gas o de aire, que se comunique con un sistema de ventilación de orden superior. Hay que INTRODUCCIÓN GENERAL comprobar periódicamente la eficacia del filtro y cambiar y probar los guantes para evitar la liberación accidental de partículas por la acción de bombeo que se produce con su uso. g) Instalaciones de almacenamiento: Para guardar los materiales de laboratorio, hay que conocer sus propiedades, así como las consecuencias de accidentes tales como derramamiento, explosión o fuego. Por regla general, no deben almacenarse grandes cantidades de reactivos en las áreas de trabajo, sino utilizar envases más pequeños que sólo contengan lo suficiente para el trabajo diario o semanal. Evítese la mezcla, en caso de accidente, de productos químicos que puedan combinarse dando lugar a compuestos explosivos, inflamables o peligrosos; para ello deberán almacenarse por separado. Los materiales peligrosos se guardarán en un recinto con recipientes específicos. Los disolventes inflamables se guardarán en cabinas adecuadamente ventiladas y aprobadas por la National Fire Protection Association2 (entidad estadounidense dedicada a la prevención de incendios) o en un frigorífico de seguridad. Utilícense envases especiales para los disolventes inflamables que se guarden en cantidades superiores a 2 1 o cuando la cantidad de disolventes inflamables en una habitación supere los 8 1. (Los disolventes inflamables son líquidos con un punto de ignición inferior a 60 °C a una presión atmosférica superior a 275 kPa y a una temperatura ambiente de 30 °C.) h) Campanas extractoras de emanaciones: Para un trabajo de seguridad en el laboratorio es esencial que existan campanas extractoras de gases y cabinas de seguridad biológica adecuadamente diseñadas7. Utilícense las campanas extractoras para contener y trabajar con materiales peligrosos, pero no como sistema de almacenamiento. 1-71 Se dispone de diferentes tipos de campanas de flujo lento para distintos niveles de peligrosidad. Hay que conocer la capacidad de movimiento de aire que tiene el conjunto del sistema y prestar atención especial al aire que se expela hacia el medio ambiente. Compruébese periódicamente el flujo de aire de las campanas con un anemómetro. Indíquese con una marca de tinta o un esparadrapo la posición necesaria para que el flujo de aire sea de 30 m/min. i) Equipos para el vertido de sustancias: Las zonas de almacenamiento y de trabajo deben disponer de equipos para el vertido de productos químicos; estos equipos pueden comprarse o prepararse en el laboratorio. Utilícense equipos de tamaño adecuado para las cantidades utilizadas de ácidos, bases y disolventes. Es conveniente diseñar y establecer los procedimientos de vertido antes de que se plantee la necesidad. j) Avisos de seguridad en las paredes: Colóquense en un lugar central para informar de las sustancias peligrosas existentes en el laboratorio. 2. Equipo de protección personal El equipo y los materiales de protección personal están constituidos por guantes, zapatos, cascos, gafas, escudos protectores contra radiación y otros artículos de seguridad que debe utilizar cada individuo. Su elección y uso dependen de las distintas tareas que hayan de realizarse. Este equipo, importante para la protección, también sirve como recordatorio constante de la necesidad de adoptar medidas de seguridad. Si se considera necesario, corresponde al director y al supervisor la tarea de comprobar su utilización. a) Indumentaria: La ropa crea una barrera entre el individuo y el peligro. Los trabajadores que manipulan material radiactivo, agentes hipotéticamente 1-72 carcinógenos y material patógeno han de cambiar su indumentaria de calle por ropa de laboratorio al entrar en la zona de trabajo, y cambiarse de nuevo a la salida. Con ello, se evita el transporte de materiales peligrosos fuera del laboratorio, y además se facilita la necesaria manipulación y limpieza de la ropa. Las prendas desechables deben ser ignífugas. b) Guantes: Suelen ser importantes. Consúltense las hojas de datos sobre seguridad en el manejo de materiales para conocer el tipo de guantes que resulta más adecuado para un disolvente o reactivo determinado. Los guantes de goma pueden utilizarse para manipular líquidos peligrosos; los de cuero, para los materiales radiactivos, y los quirúrgicos, para el material patógeno. Los guantes aislantes resultan imprescindibles para manipular objetos calientes o extremadamente fríos, pero no deben utilizarse los de amianto. Para la protección de los instrumentos pueden utilizarse guantes de algodón blancos. c) Calzado protector: Es necesario en los laboratorios en los que se trasladan objetos o instrumental pesados. Cuando se trabaje con máquinas situadas en un nivel elevado, puede ser necesario llevar casco protector. d) Gafas protectoras: Aunque la posibilidad parezca pequeña, las consecuencias de un accidente ocular pueden ser extraordinariamente graves. Se debe solicitar el uso de gafas protectoras a todo el personal del laboratorio. En la mayoría de los laboratorios está prohibido el uso de lentillas de contacto con las gafas protectoras, pero existe una gran controversia al respecto, por lo que conviene considerar dicho uso en cada caso. Las gafas de seguridad protegen de las salpicaduras, del impacto de objetos, del polvo y de la exposición a la luz ultravioleta. No obstante, las gafas que suelen emplearse no proporcionan una seguridad total al ojo. Si un determinado trabajo implica peligros especiales, habrá de pensarse en MÉTODOS NORMALIZADOS una protección adicional. Por ejemplo, para soplar vidrio, soldar, trabajar con láser o realizar tareas que requieran la exposición a otras formas de radiación, se llevarán gafas con filtros especiales. Si se trabaja en una habitación con radiación ultravioleta, se llevarán gafas protectoras con anteojeras o piezas sólidas laterales. Para ciertas actividades será preciso proteger la piel. Si se manipulan ácidos o materiales cáusticos, se llevará un escudo facial para proteger tanto los ojos como el resto de la cara. e) Respiradores: Conviene disponer de respiradores8 para aquellas situaciones de emergencia en las que se formen gases, humos o aerosoles peligrosos y deba entrarse en la habitación antes de que esté totalmente ventilada. En los laboratorios en los que se utilicen gases tóxicos como el trifluoruro de boro, el cloro, la dimetilamina, el óxido de etileno, el flúor y el bromuro de hidrógeno, se dispondrá de respiradores, preferiblemente dispositivos autónomos de respiración (DAR) u otras fuentes de aporte de aire. 3. Referencias 1. NATIONAL INSTITUTE FOR OCCUPATIONAL SAFETY AND HEALTH. 1974. The Industrial Environment—Its Evaluation and Control, 3.a ed. U. S. Dep. Health, Education & Welfare, National Inst. Occupational Safety & Health, Rockville, Maryland. 2. U. S. PUBLIC HEALTH SERVICE. 1975. Lab Safety at the Center for Disease Control. U.S. Dep. Health, Education & Welfare, Publ. núm. CDC 76-8118, National Center Disease Control, Atlanta, Georgia. 3. THE MATHESON COMPANY. 1971. Matheson Gas Data Book, 5.a ed. Matheson Co., East Rutherford, Nueva Jersey. 4. MEIDL, J. H. 1970. Flammable Hazardous Materials. Glenncoe Press, Beverly Hills, California. 5. MCKINNON, G. P. 1976. Fire Protection Handbook, 14.a ed. National Fire Protection Assoc, Boston, Massachusetts. 6. STEERE, N. V., ed. 1971. Handbook of Laboratory Safety, 2.a ed. Chemical Rubber Co., Cleveland, Óhio. INTRODUCCIÓN GENERAL 1-73 7. AMERICAN CONFERENCE OF GOVERNMENTAL INDUSTRIAL HYGIENISTS. 1982. Industrial Ventilation, A Manual of Recommended Practices, 17.a ed. American Conf. of Governmental Industrial Hygienists, Cincinnati, Ohio. 1090 C. Riesgos en el laboratorio Aunque muchos de los riesgos del laboratorio son evidentes, es importante identificar todos los peligros potenciales a la hora de desarrollar un programa efectivo de seguridad. Antes de iniciar cualquier trabajo, establézcase un plan de seguridad, el cual se pondrá en conocimiento de todo el personal que trabaja con materiales peligrosos o cerca de ellos. En este plan deben incluirse las hojas de datos sobre seguridad en el manejo de materiales, y en él se describirán todos los procedimientos rutinarios y de emergencia; todos los empleados deben leer el documento y acreditar con su firma el conocimiento del mismo. Para evitar la ingestión de materiales tóxicos o infecciosos, se prohibirá comer, beber y fumar en todas las zonas del laboratorio. Colóquense señales de advertencia adecuadas en el laboratorio, y compruébese que todos los envases tienen etiquetas claras y fácilmente reconocibles. Hay que mantener despejadas las vías de salida y de acceso a los extintores. Evítese el almacenamiento inadecuado en estantes inseguros, o la colocación de objetos de vidrio o pesados en niveles elevados. 1. 8. N ATIONAL INSTITUTE FOR OCCUPATIONAL SAFETY AND HEALTH. 1976. Guide to Industrial Respiratory Protection. Publ. Núm. 76189, U. S. Dep. Health, Education & Welfare, National Inst. Occupational Safety & Health, Cincinnati. Ohio. Riesgos químicos Las lesiones químicas pueden ser externas o internas1–8. Las primeras se producen como consecuencia de la exposición a sustancias cáusticas o corrosivas, como ácidos, bases o sales reactivas. Adóptense precauciones para evitar acci- dentes como salpicaduras y vertido de los envases. Al mismo tiempo, hay que prestar atención a la corrosión del instrumental, cuyo fallo puede resultar peligroso. Las lesiones internas pueden ser el resultado de los efectos tóxicos o corrosivos de sustancias que haya absorbido el organismo. a) Ácidos y bases inorgánicos: Muchos ácidos y bases inorgánicos se ven sometidos a limitaciones de exposición reguladas por ordenaciones como los Occupational Safety and Health Personal Exposure Limits9 y los valores umbral límite de la American Conference of Governmental Industrial Hygienists10. Estos límites admisibles de exposición y umbrales límites indican la concentración aérea máxima a la que pueden estar expuestos los trabajadores. Los gases de estos ácidos y bases son graves irritantes oculares y respiratorios. Los ácidos y bases líquidos y sólidos pueden provocar graves quemaduras en la piel y en los ojos11. Cuando se calientan los ácidos para incrementar su velocidad de digestión de la materia orgánica, aumenta de forma significativa el peligro de que se produzcan gases, y los ácidos calientes reaccionan de una forma muy rápida con la piel. Los ácidos y las bases se almacenarán por separado en zonas bien ventiladas y fuera del contacto con materiales orgánicos volátiles u oxidables. Se utilizarán envases (de goma o plástico) para transportar tanto los ácidos como las bases. El trabajo con ácidos y bases fuertes sólo debe hacerse en campanas de extracción 1-74 de gases químicos que funcionen adecuadamente. Los ácidos y las bases se añadirán de forma lenta al agua (agitando continuamente), para evitar salpicaduras. En caso de que se produzca un contacto con la piel, lávese cuidadosamente la zona afectada con agua, y consúltese a un médico si persiste la irritación12. No deben volverse a utilizar las ropas hasta que no hayan sido lavadas cuidadosamente. Los objetos de cuero (cinturones y zapatos) retienen el ácido incluso después de enjuagarlos con agua y pueden provocar graves quemaduras si vuelven a usarse. Si se produce un contacto con los ojos, deben lavarse ambos de inmediato durante quince minutos con el colirio de aplicación en chorro, y a continuación se consultará a un médico. El ácido perclórico (véanse secciones 3O3OG y H y 4500-P.D) reacciona violentamente o de forma explosiva en contacto con materia orgánica12. No deben emplearse las campanas extractoras que se hayan utilizado con ácido perclórico para trabajar con reactivos orgánicos, sobre todo si se trata de disolventes volátiles. Además de estos peligros, el ácido perclórico produce graves quemaduras al entrar en contacto con la piel, los ojos y el sistema respiratorio13. Lo mejor es disponer de una campana extractora destinada exclusivamente al trabajo con ácido perclórico. Síganse las instrucciones del fabricante para limpiar adecuadamente los conductos de salida, los cuales quedan taponados, por lo que hay que lavarlos de forma periódica. Las lesiones más frecuentes causadas por el hidróxido de sodio son quemaduras cutáneas y de los ojos. Las soluciones de hidróxido de sodio diluidas hasta 2,5N pueden causar graves lesiones oculares14. En solución, tanto el hidróxido de sodio como otras bases producen una cantidad considerable de calor (a menudo suficiente para que el agua hierva). b) Metales y compuestos inorgánicos: En general, hay que considerar como pe- MÉTODOS NORMALIZADOS ligrosos todos los productos químicos, por lo que deberán utilizarse sólo de la forma prescrita. Manipúlense en una campana extractora utilizando gafas protectoras y bata de laboratorio. En caso de contacto accidental con la piel, lávese cuidadosamente con agua la zona afectada. Si la irritación persiste, es recomendable consultar al médico. Son muchas las fuentes donde conseguirse información sobre toxicidad, precauciones y primeros auxilios 11–15. Algunos de los peligros que presentan algunos metales y compuestos inorgánicos específicos son los siguientes: determinados productos que contienen arsénico o níquel son altamente tóxicos y pueden ser cancerígenos14–15. Evítese la ingestión, la inhalación y el contacto con la piel. La azida de sodio es tóxica, y reacciona con ácidos para producir el aún más tóxico ácido hidrazoico. Cuando se elimina por un sumidero, puede reaccionar con el cobre o el plomo de las tuberías y formar azidas metálicas extraordinariamente explosivas. Pueden destruirse las azidas añadiéndose una solución concentrada de nitrito de sodio, 1,5 g NaNO2/g de ácido de sodio16. La extrema toxicidad del berilio y sus compuestos se refleja por su bajo valor umbral límite, de 2,0 μg/m3,11. Se cree que puede ser carcinógeno para el hombre9–15. Debe manipularse con extrema precaución y siempre en una campana extractora o en una cámara de manipulación con guantes. Los cianuros se utilizan como reactivos y pueden estar presentes en las muestras. El cianuro de hidrógeno es un gas letal. No deben acidificarse soluciones de cianuro salvo en un sistema cerrado o en una campana con ventilación adecuada, ya que puede formarse y liberarse cianuro de hidrógeno. El mercurio tiene la particularidad, con respecto a los demás metales, de que es líquido a temperatura ambiente y posee una apreciable presión de vapor. Un termómetro roto en una habitación mal INTRODUCCIÓN GENERAL ventilada puede dar lugar a que se sobrepase el valor umbral límite del mercurio. Debido a su gran volatilidad y toxicidad, debe manipularse, al igual que sus compuestos, con muchas precauciones, y es preciso disponer de un equipo de limpieza para vertidos. Las sales de perclorato son explosivas si se mezclan con material combustible. También pueden producir irritaciones graves en la piel, los ojos y el sistema respiratorio. Realícense con mucha precaución las tareas de manipulación y almacenamiento de percloratos. El borohidruro de sodio se descompone en el agua liberando hidrógeno, con el consiguiente peligro de explosión. Al igual que otros muchos productos químicos inorgánicos, es un agente fuertemente irritante de la piel y el sistema respiratorio. c) Reactivos y disolventes orgánicos: La mayoría de los disolventes especificados en este manual tienen un valor umbral límite de exposición (véase tabla 1090:1). A diferencia de los disolventes orgánicos, muchos reactivos orgánicos no tienen valor umbral límite (véase la tabla 1090:11 para los que sí lo tienen), pero ello no significa que sean menos peligrosos. Se cree que algunos compuestos son carcinógenos, por lo que deben ser tratados con extrema precaución. Entre ellos se encuentran tanto disolventes como reactivos del tipo del benceno18, el tetracloruro de carbono11, el cloroformo11, el 1,4-dioxano8–11,19, el tetracloroetileno20 y la bencidina21. En los organismos norteamericanos Occupational Safety and Health Administration y National Institute for Occupational Safety and Health proporcionan listas de productos químicos con especiales características de peligrosidad. Las listas de carcinógenos y de productos químicos con evidencia sustancial de ser carcinógenos, son especialmente importantes. La adopción de procedimientos de manipulación a partir de estas listas autorizadas hace 1-75 TABLA 1090:1. VALORES UMBRAL LÍMITE (VUL)*10 PARA LOS DISOLVENTES ESPECIFICADOS EN Standard Methods que disminuyan las probabilidades de error. Los disolventes utilizados pertenecen a distintas categorías: alcoholes, compuestos clorados e hidrocarburos. La exposición a cada una de estas clases de compuestos puede producir diversos efectos sobre la salud 22–24. Los alcoholes, en 1-76 TABLA 1090:11. VALORES UMBRAL LÍMITE (VUL)10 PARA LOS REACTIVOS ESPECIFICADOS EN Standard Methods MÉTODOS NORMALIZADOS ropa protectora. Los compuestos clorados presentan casi los mismos peligros que los disolventes clorados (narcosis y lesión del sistema nervioso central y del hígado). Una etiquetación adecuada, en la que se incluya la fecha para su eliminación según las recomendaciones del fabricante, permite controlar el uso de los productos químicos y eliminar los que hayan caducado. 2. general, son intoxicantes y pueden provocar irritación en las membranas mucosas y adormecimiento. Mientras que los dioles como el etileno glicol son tóxicos, los trioles como la glicerina no lo son en absoluto. Los hidrocarburos clorados provocan narcosis y lesiones en el sistema nervioso central y en el hígado. Los hidrocarburos, al igual que los otros dos grupos, son irritantes de la piel y pueden causar dermatitis tras una exposición prolongada. Debido a la volatilidad de estos compuestos, pueden producirse concentraciones peligrosas de vapor (peligro de fuego o explosión). Es esencial que la ventilación sea adecuada25. Los reactivos orgánicos citados en este manual pertenecen a cuatro categorías principales: ácidos, compuestos halogenados, colorantes e indicadores y pesticidas. La mayoría de los ácidos orgánicos tienen propiedades irritantes22–24. Son predominantemente sólidos, pero a partir de ellos se pueden generar aerosoles. Los colorantes e indicadores también presentan el problema de formación de aerosoles. Los pesticidas han de ser manipulados con precaución, pues son tóxicos22 y no deben entrar en contacto con la piel, para lo cual se llevarán guantes y Riesgos biológicos La seguridad en el laboratorio de análisis de aguas incluye también los riesgos microbiológicos26–30. Los microorganismos patógenos pueden provocar enfermedades por ingestión accidental, inoculación o inyección o por otros medios de penetración a través de la piel. La adopción de unas técnicas adecuadas de seguridad en el laboratorio permitirá controlar estos agentes31. Los peligros fundamentales que entraña la manipulación microbiológica son el contacto manoboca cuando se trabaja con materiales contaminados y la formación de aerosoles al inocular, pipetear, centrifugar o mezclar muestras o cultivos32. No deben mezclarse soluciones soplando a través de una pipeta dentro de un cultivo microbiológico. Cuando se trabaje con muestras muy contaminadas, como residuos o cultivos microbiológicos de alta densidad, utilícese un dispositivo acoplado al extremo bucal de la pipeta para evitar accidentes de ingestión. No pipetear nunca con la boca. Incluso con muestras de agua potable, no es aconsejable hacerlo. Las aguas no tratadas pueden contener patógenos de transmisión hídrica, lo que obliga a colocar todas las pipetas utilizadas en una jarra con una solución desinfectante para que se descontaminen antes de lavar el instrumental de vidrio. Las pipetas utilizadas no deben colocarse sobre mesas, carros de laboratorio o lavabos sin hacer 1-77 INTRODUCCIÓN GENERAL antes una adecuada descontaminación. Como desinfectantes para las pipetas colocadas en la jarra pueden utilizarse satisfactoriamente compuestos de amonio cuaternario que tengan un detergente compatible o soluciones de hipoclorito de sodio. Se utilizará la concentración más elevada entre las recomendadas para estos productos comerciales siempre que dicha concentración no sea tan fuerte como para borrar las señales o enturbiar las pipetas. Cámbiese a diario la solución desinfectante del envase de limpieza. Los materiales contaminados (cultivos, muestras, instrumental de vidrio, desechos de serología, etc.) han de esterilizarse en autoclave antes de ser eliminados o procesados para un nuevo uso. La rotura de los tubos con cultivos durante la centrifugación también produce aerosoles microbiológicos33, 34. Utilícense mezcladores a prueba de escapes, los cuales se mantendrán perfectamente tapados durante la operación. La introducción de un asa recalentada en un matraz de medio de cultivo supone un grave peligro, debido a la dispersión de microorganismos a modo de aerosol35. Para esterilizar asas o agujas de cultivo, es conveniente realizar una incineración en estufa eléctrica, evitando las descargas que podrían producirse por el contacto del asa con el interior de la estufa36. Para controlar las exposiciones al contacto es importante mantener una adecuada higiene personal. Hay que desinfectar con frecuencia las manos y las superficies de trabajo. El agua para beber debe localizarse fuera del laboratorio, preferiblemente en una fuente manejada con el pie. Es recomendable que el personal del laboratorio se vacune contra el tétanos, y quizá contra la fiebre tifoidea u otros agentes infecciosos si se considera conveniente por la naturaleza del trabajo. Elimínense las moscas y otros insectos para evitar la contaminación del instrumental estéril, los medios, las muestras y los cultivos bacterianos, y para evitar el contagio del personal por microorganismos infecciosos transportados por estos vectores. Instálense alambreras en todas las ventanas y puertas que comuniquen con el exterior si no existe aire acondicionado, y pulverícense periódicamente con pesticida las áreas de cabinas de aseo y almacenamiento y las vías de servicio. Teniendo en cuenta que en el laboratorio puede haber también una sección química dedicada al análisis de pesticidas en el agua, la pulverización se aplicará con cuidado en las zonas próximás a los lugares donde se desarrolla la actividad microbiológica. 3. Riesgos de radiación Todo el mundo está expuesto a radiaciones ionizantes. La dosis media de radiación anual que recibe el organismo procedente de fuentes cósmicas, terrestres e internas, de los rayos X en tratamientos médicos y dentales, etc., es de alrededor de 185 mrems/año37. Hay que evitar cualquier exposición laboral continua o intermitente que no sea necesaria, así como los accidentes que puedan dar lugar a una exposición peligrosa. En los laboratorios debe reducirse al mínimo la emisión de rayos X y de luz ultravioleta y la cantidad de material radiactivo que pueda suponer un riesgo. Todas las personas que trabajen con materiales radiactivos o máquinas generadoras de radiación deberán disponer de un manual de seguridad13, 38, en el que se explique la forma de conseguir la autorización necesaria para utilizar, comprar, manipular y almacenar radionúclidos. El manual debe incluir también los métodos para manipular con seguridad el material radiactivo no sellado y los procedimientos a seguir en caso de accidentes radiactivos, para descontaminar, controlar al personal y al laboratorio y para eliminar los materiales radiactivos. MÉTODOS NORMALIZADOS 1-78 a) Materiales radiactivos: En el laboratorio se utilizan radionúclidos para desarrollar y valorar métodos analíticos, para preparar estándares de conteo y para calibrar detectores e instrumentos de conteo (véase parte 7000). Son habituales las fuentes selladas como la célula detectora de níquel 63, utilizada en las unidades de cromatografía de gases para captación de electrones. Hay que instruir a todo el personal que tiene contacto con los materiales radiactivos sobre los posibles riesgos sanitarios. Se establecerán procedimientos adecuados y se pondrán en práctica medidas sobre segundad contra la radiación a fin de reducir al mínimo la posibilidad de exposición y accidentes. b) Máquinas generadoras de radiación: Se minimizarán los peligros derivados de los aparatos, como los de difracción de rayos X o los microscopios electrónicos, si se siguen estrictamente las instrucciones de uso proporcionadas por los fabricantes y los métodos referidos en los manuales sobre seguridad que se citan en la bibliografía. c) Radiación ultravioleta (UV): La radiación UV es de uso frecuente. Cuando los instrumentos están bien fabricados y se manejan adecuadamente, los riesgos son mínimos, pero el empleo de dichos instrumentos puede resultar peligroso cuando se aplican al control de microorganismos en las salas del laboratorio o a la esterilización de objetos. Utilícese una protección adecuada, y recuérdese que las superficies metálicas brillantes reflejan esta clase de energía; apáguense las lámparas UV cuando no se estén utilizando. Se instalarán señales de advertencia e indicadores lumínicos como recordatorios constantes mientras estén encendidas las lámparas UV. Se llevarán gafas o anteojos de protección con anteojeras sólidas siempre que exista la posibilidad de exposición a la radiación UV39. 4. Riesgos físicos a) Eléctricos: La instalación, las conexiones y los aparatos eléctricos se adaptarán a las normas vigentes sobre instalaciones eléctricas. El uso incorrecto de estos aparatos puede dar lugar a graves peligros, como incendios, explosiones, cortes de fluido y descargas eléctricas. Se conectarán a tierra todos los aparatos eléctricos o se utilizará un doble aislamiento. Siempre que sea posible, se usarán tomas de tierra protegidas por interruptor. No deben colocarse receptáculos eléctricos dentro de campanas extractoras, ni se manejarán aparatos con cordones desgastados o aislamientos deteriorados o que produzcan chispas cerca de disolventes inflamables volátiles. Los frigoríficos habrán de tener la aprobación de seguridad. Se desconectarán los aparatos de la red antes de someterlos a trabajos de mantenimiento o reparación, y nunca se harán derivaciones de los interruptores de seguridad. La reparación del equipo por personas que no sean expertas en electricidad puede dar lugar a situaciones especialmente peligrosas40. b) Mecánicos: Dispónganse protecciones o dispositivos de seguridad en las correas de transmisión, las poleas, los transmisores de cadena, los ejes y otros tipos de aparatos mecánicos de transmisión9. Dentro del equipo del laboratorio, requieren este tipo de protección las bombas de vacío, las mezcladoras, las agitadoras y las trituradoras. También se protegerán las herramientas eléctricas utilizadas en el laboratorio9. Las necesarias medidas de seguridad evitarán que determinados aparatos, como las centrifugadoras, que poseen partes que giran a gran velocidad, produzcan salpicaduras. Todos los aparatos que tengan tendencia a vibrar (por ejemplo, centrifugadoras y compresores de aire) se fijarán para impedir que se muevan, y se colocarán lejos de botellas u otros objetos que puedan INTRODUCCIÓN GENERAL caer de estantes o bancos a causa de la vibración13. c) Gases comprimidos: Los cilindros de gas a presión pueden explotar o «salir disparados» si no se manipulan de la forma adecuada. Los escapes de los cilindros pueden dar lugar a explosiones si su contenido es inflamable, presentan riesgos para la salud si el contenido es tóxico y pueden provocar la muerte por sofocación si contienen gases inertes. Las regulaciones OSHA indican la forma de utilizar y almacenar los gases comprimidos9. El transporte se hará en carros, carretillas o plataformas móviles. Durante las tareas de transporte, almacenamiento y manipulación, se asegurarán adecuadamente los cilindros de gas, manteniendo cerrada la válvula de seguridad. Evítese el uso de adaptadores o acopladores. Identifíquese continuamente el contenido de los cilindros. 5. 1. Referencias N ATIONAL I NSTITUTE FOR O CCUPATIONAL SAFETY AND HEALTH. 1976. Registry of Toxic Effects of Chemical Substances. U.S. Dep. Health, Education & Welfare, National Inst. Occupational Safety & Health, Rockville, Maryland. 2. AMERICAN CONFERENCE OF GOVERNMENTAL INDUSTRIAL HYGIENISTS. 1976. Documentation of the Threshold Limit Values for Substances in Workroom Air, 3.a ed. American Conf. of Governmental Industrial Hygienists. Cincinnati, Ohio. 3. WINDHOLZ, M. 1976. The Merck Index, 9.a ed. Merck and Co., Rahway, Nueva Jersey. 4. AMERICAN MUTUAL INSURANCE ALLIANCE. 1966. Handbook of Organic Solvents. Tech. Guide No. 6, American Mutual Insurance Alliance, Chicago, Illinois. 5. B RAKER , W. & A. L. M OSSMAN . 1970. Effects of Exposure to Toxic Gases—First Aid and Medical Treatment. Matheson Gas Products, East Rutherford, Nueva Jersey. 6. S AX , N. I. 1968. Dangerous Properties of Industrial Materials, 3. a ed. Van Nostrand Reinhold Co., Nueva York. 7. BROWNING, E. C. 1965. Toxicity and Metabolism of Industrial Solvents. Elsevier Scientific Publ. Co., Nueva York. 1-79 8. D EICHMANN , W. B. & H. W. G ERARDE . 1969. Toxicology of Drugs and Chemicals. Academic Press, Nueva York. 9. Code of Federal Regulations. 1980. 29 Labor Part 1910. Washington, D.C. 10. AMERICAN CONFERENCE OF GOVERNMENTAL INDUSTRIAL HYGIENISTS. 1986-87. Threshold Limit Values. American Conf. Governmental Industrial Hygienists, Cincinnati, Ohio. 11. N ATIONAL INSTITUTE FOR O CCUPATIONAL SAFETY AND HEALTH/OCCUPATIONAL SAFETY AND H EALTH A DMINISTRARON . 1981. Occupational Health Guidelines for Chemical Hazards. Publ. No. 81-123, U.S. Dep. Health & Human Services, U.S. Dep. Labor, Washington, D.C. 12. NATIONAL RESEARCH COUNCIL. 1981. Prudent Practices for Handling Hazardous Chemicals in Laboratories. National Academy Press, Washington, D.C. 13. STEERE, N. V. 1971. Handbook of Laboratory Safety, 2. a ed. Chemical Rubber Co., Cleveland, Ohio. 14. MUIR, G. C. 1977. Hazards in the Chemical Laboratory, 2.a ed. Chemical Soc, Londres, Inglaterra. 15. C LAYTON , G. D. & F. E. C LAYTON . 1981. Patty's Industrial Hygiene and Toxicology, Vol. II A, 3.a ed. John Wiley & Sons, Nueva York. 16. AMERICAN PUBLIC HEALTH ASSOCIATION, AMERICAN WATER WORKS ASSOCIATION & WATER POLLUTION CONTROL FEDERATION. 1981. Standard Methods for the Examination of Water and Wastewater, 15.a ed. American Public Health Assoc, Washington, D.C. 17. U.S. P UBLIC H EALTH S ERVICE . 1975. Lab Safety at the Center for Disease Control. U.S. Dep. Health, Education & Welfare, Publ. No. CDC 76-8118. National Center Disease Control, Atlanta, Georgia. 18. N ATIONAL INSTITUTE FOR O CCUPATIONAL S AFETY AND H EALTH . 1974. Criteria for a Recommended Standard. Occupational Exposure to Benzene. Publ. No. 74-137, U.S. Dep. Health, Education & Welfare, U.S. Government Printing Off., Washington, D.C. 19. N ATIONAL INSTITUTE FOR O CCUPATIONAL SAFETY AND HEALTH . 1977. Criteria for a Recommended Standard. Occupational Exposure to Dioxane. Publ. No. 77-226, U.S. Dep. Health, Education & Welfare, U.S. Government Printing Office, Washington, D.C. 20. N ATIONAL INSTITUTE FOR O CCUPATIONAL SAFETY AND HEALTH . 1978. Current Intelligence Bulletin 20 Tetrachloroethylene (Perchloroethylene). Publ. No. 78-112, U.S. 1-80 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. Dep. Health, Education & Welfare, U.S. Government Printing Off., Washington, D.C. B OENIGER , M. 1980. Carcinogenicity and Metabolism of Azo Dyes, Especially Those from Benzidine. Publ. No. 80-119, U.S. Dep. Health & Human Services. U.S. Government Printing Off., Washington, D.C. DOULL, J., C. D. KLASSEN & M. O. AMDUR, eds. 1980. Casarett and Doull's Toxicology, 2.a ed. Macmillan Co., Nueva York. Toxic and Hazardous Industrial Chemicals Safety Manual for Handling and Disposal with Toxicity and Hazard Data. 1978. International Technical Information Inst, Tokio, Japón. P ATTY , F. A. 1963. Patty's Industrial Hygiene and Toxicology, 2.a ed. John Wiley & Sons, Nueva York. C LAYTON , G. D. & F. E. C LAYTON . 1978. Patty's Industrial Hygiene and Toxicology, 3. a ed. Vol. 1. John Wiley & Sons, Nueva York. WEDEUN , A. G., E. H ANEL, G. B. PHILLIPS & O. T. M ILLER. 1956. Laboratory Design for Study of Infectious Disease. Amer. J. Pub. Health 46:1102. DARLOW, H. M. 1969. Safety in the Microbiological Laboratory. In J. R. Norris & D. W. Ribbons, eds. Methods in Microbiology. Volumen 1. Academic Press, Nueva York. SCIENCE PRODUCTS DIVISIÓN, MALLINCKRODT CHEMICAL WORKS. 1969. Laboratory Safety Handbook. Mallinckrodt Chemical Works, St. Louis, Missouri. SHAPTON, D. A. & R. J. BOARD. 1972. Safety in Microbiology. Soc. Applied Bacteriology, Tech. Ser. No. 6., Academic Press, Nueva York. U.S. DEPARTMENT OF HEALTH AND HUMAN S ERVICES , P UBLIC HEALTH S ERVICE , C EN TERS FOR D ISEASE C ONTROL & N ATIONAL INSTITUTES OF HEALTH. 1984. Biosafety in Microbiological and Biomedical Laboratories. U.S. Government Printing Off., Washington, D.C. OFFICE OF BIOSAFETY. 1974. Classification of Etiologic Agents on the Basis of Hazard, 4.a ed. Center Disease Control, Atlanta, Georgia. PHILLIPS, G. B. 1961. Microbiological Safety in U.S. and Foreign Laboratories. Tech. Study 35, Biological Laboratories Project 4B11-05-015, U.S. Army Chemical Corps, Fort Detrick, Maryland. REITMAN, M. & G. B. PHILLIPS. 1956. Biological hazards of common laboratory procedures. III. The centrifuge. Amer. J. Med. Technol. 22:100. MÉTODOS NORMALIZADOS 34. HALL, C. V. 1975. A biological safety centrifuge. Health Lab. Sci. 12:104. 35. PHILLIPS, G. B. & M. REITMAN. 1956. Biological hazards of common laboratory procedures. IV. The inoculating loop. Amer. J. Med. Technol. 22:16. 36. GORDON, R. C. & C. V. DAVENPORT. 1973. Simple modification to improve usefulness of the bacti-cinerator. Appl. Microbiol. 26:423. 37. N ATIONAL C OUNCIL ON R ADIATION P RO TECTION AND MEASUREMENTS. 1987. Ionizing Radiation Exposure of the Population of the United States. Rep. No. 93, National Council Radiation Protection, Washington, D.C. 38. CALIFORNIA STATE DEPARTMENT OF HEALTH SERVICES. 1979. Radiation Safety Manual. Berkeley, Calif. (Pueden conseguirse manuales similares en la mayor parte de los departamentos sanitarios, universidades y laboratorios nacionales.) 39. W ALLACE , L. A. 1981. Recent progress in developing and using personal monitors to measure human exposure to air pollutants. Environ. International 5:73. 40. INHORN, S. L., ed. 1978. Quality Assurance Practices for Health Laboratories. American Public Health Assoc, Washington, D.C. 6. Bibliografía NATIONAL COMMITTEE ON RADIATION PROTECTION. 1964. Safe Handling of Radioactive Materials. National Bur. Standards Handbook 92, U.S. Government Printing Off., Washington, D.C. NATIONAL COUNCIL ON RADIATION PROTECTION AND MEASUREMENTS. 1976. Radiation Protection for Medical and Allied Health Personnel. Rep. No. 48, National Council Radiation Protection & Measurements, Washington, D.C. WALTERS, D. B., ed. 1980. Safe Handling of Chemical Carcinogens, Mutagens, Teratogens and Highly Toxic Substances, Vols. 1 and 2. Ann Arbor Science, Ann Arbor, Michigan. FUSCALDO, A. A., B. J. ERLICK & B. HINDMAN. 1980. Laboratory Safety, Theory and Practice. Academic Press, Nueva York. NATIONAL COUNCIL ON RADIATION PROTECTION AND MEASUREMENTS. 1987. Recommendations on Limits for Exposure to Ionizing Radiation. Rep. No. 91, National Council Radiation Protection & Measurements, Washington, D.C. 1-81 INTRODUCCIÓN GENERAL 1090 D. 1. Prácticas de control de riesgos Monitorización El establecimiento de medidas, prácticas, procedimientos y métodos para evitar la exposición del personal del laboratorio a los materiales peligrosos ha de formar parte de un programa eficaz de seguridad. No obstante, para asegurar el funcionamiento real de esta protección es esencial establecer, simultáneamente, un sistema de monitorización o retroalimentación. a) Monitores químicos: Se han descrito dispositivos capaces de realizar mediciones directas de concentración dentro del entorno inmediato de respiración de una persona1. Utilícense instrumentos activos basados en el bombeo de aire o aparatos pasivos que se fundamentan en el fenómeno de la difusión. Estos aparatos pueden medir compuestos orgánicos e inorgánicos con el absorbente adecuado. Las partículas pueden recogerse en un filtro cuando se utilizan aparatos activos. Es posible conseguir una detallada descripción del desarrollo y uso de monitores personales para la medición de la exposición a los contaminantes químicos del ambiente1. b) Monitores de biorriesgos: Son parte esencial del control microbiológico. Inclúyase una exploración física previa acompañada de análisis hematológicos y otros relacionados con la exposición. Realícense exploraciones anuales con análisis serológicos, estudios de funciones bioquímicas y radiografías de tórax. Archívense las muestras de suero para posibles referencias futuras. El programa de seguridad se completa con la vigilancia sobre el continuo cumplimiento de las reglas básicas de seguridad en el laboratorio. c) Monitores radioquímicas: Consisten en limpiadores, instrumentos de control portátiles y estudio de muestras de aire. En general, se requieren monitores múltiples. Mídase la exposición de la radiación externa con dosímetros personales, preferiblemente dosímetros de película, para determinar la radiación acumulada a lo largo de un determinado período. Como complemento de estos dosímetros pueden utilizarse minicámaras de ionización, dosímetros termolumínicos y cámaras de obturación. Los detectores de radiactividad corporal o de detección por espectrometría gamma determinan la existencia de sustancias radiactivas en el organismo, pero se trata de aparatos caros que requieren un aprendizaje específico. También se comprobará la posible presencia de elementos radiactivos en las excreciones. Además de la monitorización personal, se llevará a cabo una monitorización general del laboratorio. Se analizará todo el instrumental junto con el material que haya estado en contacto con las sustancias radiactivas. Para ello se utilizarán pruebas dosimétricas y de frotamiento. Los contadores GM de ventana fina sirven para las muestras de frotamiento y para monitorizar la piel y la ropa. Se necesita un monitor de centelleo alfa para detectar las emisiones alfa. Steere2 hace una excelente exposición de las técnicas de monitorización para radioisótopos. 2. Eliminación de residuos a) Consideraciones generales: Los rigurosos requisitos establecidos para la eliminación de residuos contemplan posibles consecuencias penales y civiles tanto para las organizaciones como para las personas. Estas reglas varían según los estados y lugares y están sujetas a modificaciones. Puede ser necesario obtener licencias, permisos o ambos; consúltese a las autoridades locales o estatales. 1-82 Es importante disponer de un plan para eliminar con seguridad las sustancias químicas y biológicas utilizadas en el laboratorio. Este plan debe discutirse con el supervisor y, si se considera adecuado, con el coordinador de seguridad. Gastón3 ha descrito el cuidado, la manipulación y la eliminación de los productos químicos peligrosos. Se instalarán sistemas adecuados de toma, utilizándose envases adecuadamente etiquetados; el almacenamiento tendrá una protección contra incendios y se dispondrá de una zona separada para los materiales altamente peligrosos o tóxicos. Se utilizarán envases metálicos de segundad para los residuos de disolventes y para los materiales incompatibles. Se considerará la conveniencia de utilizar envases especiales para los residuos extraordinariamente peligrosos o muy tóxicos y se dispondrá de un empaquetado especial que evite la rotura o el deterioro de los envases durante el transporte. Los materiales peligrosos incompatibles se almacenarán por separado4. b) Los métodos de eliminación de residuos son la incineración, el enterramiento, la evaporación, la neutralización, la reacción química, los tratamientos especiales y las técnicas aplicadas por especialistas en la materia. Puede conseguirse una serie de hojas de datos sobre seguridad en el manejo de materiales5, en la que se incluyen los métodos de elección para la eliminación de residuos de compuestos orgánicos e inorgánicos especiales. A veces pueden eliminarse algunos productos químicos peligrosos vertiéndolos por el sumidero si se encuentran a ciertas concentraciones, pero sólo se hará si se dispone de un permiso escrito concedido por las autoridades locales responsables de las aguas residuales y basado en un conocimiento suficiente de dichos productos. 1) Residuos químicos. Una vez utilizados, los disolventes pueden ser destilados y recuperados para nuevo uso. Los MÉTODOS NORMALIZADOS disolventes no combustibles pueden evaporarse siempre que sus vapores no provoquen problemas ambientales. Cantidades pequeñas de disolventes y productos químicos inflamables pueden quemarse en el suelo, en envases metálicos poco profundos o en incineradores, siempre que se cumplan las normas sobre contaminación del aire. Los materiales ácidos y básicos se neutralizarán antes de eliminarse definitivamente. Muchos materiales solubles no tóxicos pueden diluirse con precaución en un sistema de desagüe, pero antes es preciso asegurarse de que no resultarán peligrosos para las cañerías o para el medio ambiente. Siempre que sea posible, los materiales peligrosos se convertirán, mediante reacciones químicas u otros procesos, en compuestos inocuos antes de proceder a su eliminación. Si ello no es factible, lo mejor es contratar a un especialista en técnicas de eliminación de materiales peligrosos o altamente tóxicos. Los cilindros de gas no recuperables se eliminarán sólo con ayuda de personal cualificado. 2) Residuos biológicos. Esterilícense todas las sustancias infecciosas o tóxicas y todos los instrumentos y aparatos contaminados antes de lavarlos, guardarlos o tirarlos6. El método de esterilización más recomendable es el tratamiento en autoclave. En general, se usa el autoclave a una presión de 103 kPa con una temperatura en la cámara de al menos 121 °C y durante un mínimo de 15 minutos. El tiempo se mide una vez que el material a esterilizar ha alcanzado los 121 °C. Cuando la esterilización se haga con el material dentro de bolsas de plástico, añádase agua al contenido para asegurar la obtención de un calor húmedo. Para la esterilización de objetos que no son de plástico, se utiliza también el calor seco y el tratamiento químico. Tras la esterilización pueden manipularse los residuos con seguridad mediante los sistemas habituales de eliminación. Los residuos contaminados combustibles y los 1-83 INTRODUCCIÓN GENERAL cadáveres de animales se recogen en envases impermeables que se eliminan por incineración. 3) Residuos radioquímicos. El National Committee on Radiation Protection and Measurements7 ha desarrollado criterios generales para la eliminación de residuos radiactivos. Estas recomendaciones han sido adoptadas oficialmente en los Estados Unidos mediante su publicación en el Registro Federal8. Dos principios generales constituyen la base de la eliminación de los residuos radiactivos: a) dilución y consiguiente dispersión para reducir la concentración de los radionúclidos mediante dilución de la materia inactiva o dilución en un medio receptor, y b) concentración y confinamiento, lo que suele suponer una reducción en el volumen de los residuos que posteriormente se almacenarán para dejar que se desactiven. Los residuos que se difunden por el aire pueden ser tratados por cualquiera de los dos métodos anteriores. La ventilación supone la salida de la radiación a la atmósfera. Los típicos gases radiactivos son el yodo, el kriptón y el xenón. El yodo puede eliminarse por depuración o haciendo que reaccione con nitrato de plata. Los gases nobles pueden eliminarse por adsorción; para las partículas, pueden utilizarse las técnicas estándar. Los métodos de dilución son adecuados para los líquidos de baja actividad. Los niveles intermedios pueden ser tratados mediante diversos procesos físico-químicos para separar los residuos en una porción no radiactiva y otra de gran actividad que deberá guardarse. Los residuos sólidos pueden consistir en equipos, instrumentos de vidrio y otros materiales. Siempre que sea posible, se descontaminarán para utilizarlos de nuevo. La descontaminación suele dar lugar a residuos líquidos. Los materiales combustibles que no pueden ser descontaminados suelen quemarse con precauciones especiales; puede ser necesario obtener un per- miso para hacer esta operación. Cuando no exista otra alternativa, se almacenarán los residuos hasta que se desactiven o de forma permanente. 4) Otros residuos especiales. En los Estados Unidos, las normas federales sobre bifenilos policlorados (BPC), dioxinas y amianto obligan a adoptar precauciones especiales con los residuos que contengan dichos materiales. 3. 1. 2. 3. 4. 5. 6. 7. 8. Referencias Proceedings of the Symposium on the Development and Usage of Personal Monitors for Exposure and Health Effect Studies, junio 1979. Publ. EPA-600/9-79-032, U.S. Environmental Protection Agency, Washington, D.C. STEERE, N. V., ed. 1971. Handbook of Laboratory Safety, 2.a ed. Chemical Rubber Co., Cleveland, Ohio. GASTON, P. J. 1970. The Care, Handling, and Disposal of Dangerous Chemicals. Inst. of Science Technology, Northern Publishers (Aberdeen) Ltd., Aberdeen, Escocia. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1980. Hazardous waste compatibility chart. Publ. No. EPA-600/2-80-076, Cincinnati, Ohio. Chemical Safety Data Sheets. Manufacturing Chemists' Assoc, Inc., Washington, D.C. NATIONAL INSTITUTES OF HEALTH, 1974. Biohazard Safety Guide. GPO Stock No. 174000383, U.S. Dep. Health, Education & Welfare, Public Health Service, National Inst. Health, Washington, D.C. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1985. Environmental Standards for the Management and Disposal of Spent Nuclear Fuel, High-Level and Transuranic Wastes. 40 CFR Part 191. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1988. Environmental Standards for the Management, Storage and Land Disposal of Low-Level Radioactive Waste. Proposed Rule. 40 CFR Part 193. 4. Bibliografía GHASSEMI, M., S. QUINLIVAN, G. GRUBER & H. CASEY. 1976. Disposing of Small Batches of Hazardous Wastes. Publ. SW 562. Office of 1-84 Solid Waste, U.S. Environmental Protection Agency, Cincinnati, Ohio. BORDNER, R. H., J. A. WlNTER & P. V. SCARPINO, eds. 1978. Microbiological Methods for Monitoring the Environment, Water and Wastes. Publ. EPA-600/8-78-017, Environmental Monitoring and Support Lab., U.S. MÉTODOS NORMALIZADOS Environmental Protection Agency, Cincinnati, Ohio. NATIONAL ACADEMY OF SCIENCES, NATIONAL ACADEMY OF ENGINEERING & INSTITUTE OF MEDICINE . 1983. Prudent Practices for the Disposal of Chemicals from Laboratories. National Academy Press, Washington, D.C. PARTE 2000 PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2010 INTRODUCCIÓN Esta parte del presente manual trata esencialmente de la medición de las propiedades físicas de una muestra, estableciendo las pertinentes distinciones respecto de las concentraciones de componentes químicos o biológicos. Muchas de las determinaciones aquí incluidas, como el color, la conductividad eléctrica y la turbidez, satisfacen inequívocamente esta categoría, pero no siempre pueden separarse totalmente de la composición química. Algunas de las técnicas citadas miden, por otra parte, las propiedades de agregación derivadas de la presencia de algunos componentes. Otras, por ejemplo la saturación de carbonato cálcico, se relacionan o dependen de pruebas químicas. También se incluyen pruebas de 2020 aspecto, olor y sabor que han sido clasificadas tradicionalmente entre las propiedades físicas, criterio que puede ser en ocasiones discutible. En fin, la sección 2710, referida a pruebas en lodos, analiza algunas pruebas bioquímicas; sin embargo, por comodidad, quedan agrupadas con los demás ensayos realizados con lodos. Con estas pequeñas excepciones, el contenido de esta parte del libro guarda una razonable fidelidad a su título. La mayoría de los métodos incluidos son tradicionalmente de naturaleza física a diferencia de los explícitamente químicos, radiológicos, biológicos o bacteriológicos estudiados en otras partes de la obra. CONTROL DE CALIDAD La parte 2000 contiene diversos métodos analíticos, muchos de los cuales no se atienen a las técnicas estándar de control de calidad. En la parte 1000 se proporciona información general sobre tal control y las técnicas específicas se describen en la explicación de cada uno de los métodos. A muchos de los métodos estudiados en este capítulo pueden aplicárseles las siguientes normas generales: Evaluación del rendimiento analítico de cada método. Determinación de la competencia mediante análisis de muestras que contengan concentraciones co- nocidas. Calibración de los instrumentos y confirmación de que las medidas instrumentales no presentarán desviaciones. Evaluación de la precisión de los métodos analíticos, realizados por duplicado al menos en el 10 por 100 de las muestras. Análisis de un mínimo de cada duplicado en cada batería de muestras. Determinación del sesgo del método analítico en cada lote de muestras mediante análisis de blancos, adiciones conocidas con una frecuencia al menos del 5 por 100 de las muestras y, si es posible, un patrón externo. 2110 ASPECTO* Para examinar el aspecto físico general de una muestra, conviene recurrir a términos que describan brevemente sus características más apreciables. Estos tér- * Aprobado por el Standard Methods Committee, 1988. minos pueden referirse, entre otras cosas, a la presencia de color, turbidez, sólidos en suspensión, crustáceos, larvas, gusanos, sedimentos, materias flotables o partículas similares detectables a simple vista. Cuando sea posible, utilícense valores numéricos que valoren color, turbidez o sólidos en suspensión. 2-1 2-2 MÉTODOS NORMALIZADOS 2120 2120 A. El color del agua puede estar condicionado por la presencia de iones metálicos naturales (hierro y manganeso), de humus y turbas, de plancton, de restos vegetales y de residuos industriales. Tal coloración se elimina para adaptar un agua a usos generales e industriales. Las aguas residuales industriales coloreadas suelen requerir la supresión del color antes de su desagüe. 1. Definiciones El término «color» se asocia aquí al concepto de color puro, esto es, el color del agua cuya turbidez ha sido eliminada. El término «color aparente» engloba no sólo el color debido a las sustancias disueltas, sino también a las materias en suspensión. Tal color aparente se determina en la muestra original sin filtrado ni centrifugado. En algunas aguas residuales industriales muy coloreadas, el color se debe principalmente a materiales coloidales o en suspensión. En estos casos deben determinarse ambos colores, el aparente y el real. 2. Pretratamiento para eliminar la turbidez Para determinar el color mediante los métodos actualmente aceptados, es necesario eliminar la turbidez antes de proceder al análisis. Todavía no se ha encontrado un método óptimo para suprimir la turbidez ni eliminar el color. El filtrado proporciona resultados reproducibles de un día para otro y entre laboratorios; sin embargo, algunos procedimientos de * Aprobado por el Standard Methods Committee, 1988. COLOR* Introducción filtrado pueden eliminar parte del color real. El centrifugado evita interacciones del color con los materiales del filtro, pero los resultados varían con la naturaleza de la muestra y el tamaño y velocidad de la centrífuga. Cuando es necesaria la dilución de la muestra, tanto antes como después de la eliminación de la turbidez, ésta puede alterar el valor de color registrado caso de que existan corpúsculos coloreados luminosos. En cada método se señalan procedimientos aceptables de pretratamiento. Cuando se informe sobre resultados, ha de definirse el método de pretratamiento. 3. Selección del método El método de comparación visual es aplicable a casi todas las muestras de agua potable. La polución por algunos residuos industriales suele producir colores poco habituales que no pueden equipararse. En este caso debe utilizarse un método instrumental. Una modificación de los métodos de triestímulo y espectrofotométrico permite el cálculo del valor de un color simple, representando diferencias de cromaticidad uniforme, incluso cuando la muestra exhibe un color significativamente diferente de los patrones de cobalto-platino. Por comparación de valores de color entre laboratorios, calibrar el método visual mediante procedimientos instrumentales. 4. Bibliografía OPTICAL SOCIETY OF AMERICA. 1943. Committee Report. The concept of color. J. Opt. Soc. Amer. 33:544. JONES, H. et al. 1952. The Science of Color. Thomas Y. Crowell Co., Nueva York. 2-3 PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2120 B. 1. Método de comparación visual Discusión general a) Principio: El color se determina mediante comparación visual de la muestra con concentraciones conocidas de soluciones coloreadas. La comparación también puede realizarse con discos especiales de cristal de color, adecuadamente calibrados. El método patrón de medida de color es el de cobalto-platino, siendo la unidad de color el producido por 1 mg de platino/1 en forma de ion cloroplatinato. El índice cobalto-platino puede variarse para equiparar tonalidades en casos especiales; la proporción que se menciona más adelante es satisfactoria por lo general para igualar el color de las aguas naturales. b) Aplicación: El método platino-cobalto es útil para medir el color del agua potable o aquella cuyo color se debe a materiales naturales; no es aplicable en cambio a la mayoría de las aguas residuales industriales de coloración intensa. c) Interferencia: La turbidez, incluso cuando es ligera, hace que el color aparente sea más llamativo que el color real; por tanto, ha de eliminarse la turbidez antes de aproximarse al color real, bien mediante lectura diferencial con filtros de color distintos1, bien por medidas de dispersión diferencial2. Sin embargo, ninguna técnica ha conseguido la calificación de método estándar. Elimínese la turbidez mediante centrifugado o por el método de filtrado descrito como método C. Centrifúguese durante una hora, a menos que se haya demostrado que la centrifugación en otras condiciones pueda dar lugar a una satisfactoria eliminación de la turbidez. El valor del color del agua depende en buena medida y se incrementa invariablemente al aumentar el pH del agua. Cuando se informa sobre un registro numérico referido a color, especifíquese el pH al que fue determinado. Con fines de investigación o cuando se comparen valores de color entre laboratorios, determínese la respuesta de color de un agua dada sobre un amplio margen de cifras de pH3. d) Método de campo: Dado que el método estándar platino-cobalto no resulta adecuado como método de campo, puede establecerse otro basado en la comparación del color del agua con el de los discos de vidrio situados al extremo de tubos metálicos, que contienen a su vez tubos de vidrio de comparación llenos de agua destilada incolora. Iguálese el color de la muestra con el del tubo de agua destilada más el cristal de color calibrado, mirando a través sobre una superficie blanca, y calíbrese cada disco hasta que se corresponda con el color de la escala platino-cobalto. Los discos de cristal proporcionan resultados sustancialmente coincidentes con los obtenidos mediante el método de platino-cobalto y su uso ha sido admitido como un método estándar de campaña. e) Métodos de laboratorio no estándar: El empleo como patrones de discos de vidrio o de líquidos que no sean agua para el trabajo de laboratorio sólo es permisible si éstos han sido calibrados individualmente frente a los patrones platino-cobalto. Las aguas de color poco usual —como las que aparecen a veces en mezclas de algunos residuos industriales— pueden presentar tonalidades tan diferentes de las del estándar platino-cobalto que la comparación es difícil, cuando no imposible. Para tales aguas, empléense los métodos de las secciones 2120 C y D. Hay que señalar, sin embargo, que los resultados obtenidos con ellos no son directamente comparables a los obtenidos con los estándares platinocobalto. f) Toma de muestras: Recójase la muestra en material de vidrio limpio, realizando la determinación colorimétri- MÉTODOS NORMALIZADOS 2-4 ca dentro de un plazo razonable, ya que las alteraciones biológicas y físicas producidas durante la conservación pueden afectar al color. En las aguas coloreadas naturalmente, estas alteraciones conducen siempre a resultados defectuosos. 2. Instrumental a) Tubos de Nessler, iguales, de 50 ml y forma alta. b) Medidor de pH, para determinación del pH de la muestra (véase sección 4500-H + ). 3. Preparación de patrones a) Si no puede conseguirse un suministro fiable de cloroplatinato, utilícese ácido cloroplatínico preparado a partir de platino metal. No conviene utilizar ácido cloroplatínico comercial, porque es muy higroscópico y puede variar en su contenido de platino. El cloroplatinato de potasio no es higroscópico. b) Disuélvanse 1,246 g de cloroplatinato potásico, K2PtCl6 (equivalente a 500 mg de Pt metal), y 1,00 g de cloruro de cobalto cristalizado, CoCl2-6H2O (equivalente a unos 250 mg de Co metal), en agua destilada con 100 ml de C1H concentrado, y dilúyanse hasta 1.000 ml del agua destilada. Este material estándar tiene un color de 500 unidades. c) Si no se dispone de K 2PtCl6, disuélvanse 500 mg de Pt metálico puro en agua regia con calor; elimínese el HNO3 mediante evaporación repetida con fracciones de HC1 conc. reciente. Disolver todo ello con 1,00 g de CoCl2-6H2O cristalizado como se dijo anteriormente. d) Prepárense patrones con los colores 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60 y 70, mediante diluciones de 0,5, 1,0, 1,5, 2,0, 2,5, 3,0, 3,5, 4,0, 4,5, 5,0, 6,0, y 7,0 ml del estándar de color con agua destilada, hasta 50 ml, en tubos de Nessler. Protéjanse estos patrones de la evaporación y la contaminación mientras no se utilicen. 4. Procedimiento a) Estimación de una muestra intacta: Obsérvese el color de la muestra llenando con ésta un tubo de Nessler hasta la marca de 50 ml, y compárese con el estándar. Es conveniente mirar verticalmente hacia abajo a través de los tubos sobre una superficie blanca o especular, situada en un ángulo de forma que la luz se refleje hacia arriba a través de las columnas de líquido. Si existe turbidez y no se ha eliminado, anótese «color aparente». Si el color excede las 70 unidades, dilúyase la muestra en agua destilada a proporciones conocidas hasta que el color se sitúe dentro de los márgenes del estándar. b) Mídase el pH de cada muestra. 5. Cálculo a) Calcular las unidades de color por la siguiente ecuación: donde: A = color estimado de una muestra diluida y B = ml de muestra tomados para la dilución. b) Infórmense los resultados de color en cifras completas y regístrense como sigue: c) Informe del pH de la muestra. PROPIEDADES FÍSICAS Y DE AGREGACIÓN 6. Referencias 1. K NIGHT , A. G. 1951. The photometric estimation of color in turbid waters. J. Inst. Water Eng. 5:623. 2. JULLANDER, I. & K. BRUNE , 1950. Light absorption measurements on turbid solutions. Acta Chem. Scand. 4:870. 3. BLACK, A. P. & R. F. CHRISTMAN. 1963. Characteristics of colored surface waters. J. Amer. Water Works Assoc. 55:753. 7. Bibliografía HAZEN, A. 1892. A new color standard for natural waters. Amer. Chem. J. 14:300. 2120 C. 1. 2-5 HAZEN, A. 1896. The measurement of the colors of natural waters. J. Amer. Chem. Soc. 18:264. Measurement of Color and Turbidity in Water. 1902. U.S. Geol. Surv., Div. Hydrog. Circ. 8, Washington, D.C. RUDOLFS, W. & W. D. HANLON. 1951. Color in industrial wastes. Sewage Ind. Wastes. 23:1125. PALIN, A. T. 1955. Photometric determination of the colour and turbidity of water. Water Water Eng. 59:341. CHRISTMAN, R. F. & M. GHASSEMI. 1966. Chemical nature of organic color in water. J. Amer. Water Works Assoc. 58:723. GHASSEMI, M. & R. F. CHRISTMAN. 1968. Properties of the yellow organic acids of natural waters. Limnol. Oceanogr. 13:583. Método espectrofotométrico Discusión general a) Principio: El color de una muestra filtrada se expresa en términos que describen la sensación percibida al observarla. La tonalidad (rojo, verde, amarillo, etcétera) se designa como «longitud de onda dominante», el grado de brillantez como «luminancias» y la saturación (pálido, pastel, etc.) como «pureza». Como mejor se detectan estos valores es a partir de las características de transmisión de la luz de una muestra filtrada, mediante espectrofotometría. b) Aplicación: Este método es aplicable en aguas potables y de superficie, y en aguas residuales, tanto domésticas como industriales. c) Interferencia: Interfiere la turbidez. Más adelante se describe el proceso de eliminación mediante filtrado. Figura 2120:1. Sistema de filtrado para determinaciones de color. 2-6 2. Instrumental a) El espectómetro tendrá células de absorción de 10 mm, una banda espectral limitada (10 nm o menos) y un margen operativo eficaz comprendido entre 400 y 700 nm. b) Sistema de filtrado, que consta de lo siguiente (véase fig. 2120:1): 1) Matraces de filtrado, 250 ml, con tubos laterales. 2) Soporte de crisol Walter. 3) Crisol de filtro micrometálico, tamaño medio de poro, 40 uní. 4) Auxiliar de filtrado por calcinación*. 5) Sistema de vacío. 3. Procedimiento a) Preparación de la muestra: Tómense dos muestras de 50 ml a temperatura ambiente. Utilícese una a pH original y ajústese el pH de la otra a 7,6 añadiendo ácido sulfúrico (H2SO4) e hidróxido sódico (NaOH), a concentraciones adecuadas para que el cambio de volumen no exceda el 3 por 100. Es necesario un pH estándar debido a la variación de color con el mismo. Elimínese por centrifugado el exceso de materias en suspensión. Cada muestra deberá tratarse por separado según las siguientes indicaciones: Mézclese cuidadosamente 0,1 g de auxiliar de filtrado en una porción de 10 ml de muestra centrifugada y fíltrese hasta formar una precapa en el crisol de filtro. Fíltrese directamente al matraz residual, tal como se indica en la figura 2120:1. Mézclense 40 mg de auxiliar de filtrado en una porción de 35 ml de muestra centrifugada. Todavía en vacío, fíltrese a través de la precapa y pasar el filtrado al matraz de residuos hasta que aparezca limpio; a continuación, diríjase * Celite núm. 505, Manville Corp.. o equivalente. MÉTODOS NORMALIZADOS este filtrado claro al matraz limpio por medio de la espita de tres posiciones, y recoger 25 ml para determinación de la transmitancia. b) Determinación de las características de transmisión de la luz: Límpiense meticulosamente las celdas de absorción de 1 cm con detergente y enjuáguense con agua destilada. Aclárense dos veces con muestra filtrada y límpiense las superficies externas con papel para envolver vidrios ópticos y a continuación llévense la celda con muestra filtrada. Determínense los valores de transmirancia (en %) para cada cifra de longitud de onda visible presentada en la tabla 2120:1, utilizando las 10 ordenadas marcadas con asterisco para obtener una aproximación y las 30 restantes para mayor exactitud. Dispónganse instrumentos para leer el 100 por 100 de transmitancia sobre el blanco de agua destilada y realizar todas las determinaciones con una banda de espectro limitada. 4. Cálculo a) Tabúlense los valores de transmirancia correspondientes a las longitudes de onda mostradas en las columnas X, Y y Z de la tabla 2120:1. Totalícese cada columna de transmitancia y multiplíquense los totales por los factores adecuados (para 10 ó 30 números) que figuran en la parte baja de la tabla, para obtener valores triestímulo X, Y y Z. El valor triestímulo corresponde al porcentaje de luminancia. b) Calcúlense los coeficientes tricromáticos x e y a partir de los valores triestímulo X, Y y Z, mediante las siguientes ecuaciones: PROPIEDADES FÍSICAS Y DE AGREGACIÓN TABLA 2120:1. ORDINALES SELECCIONADOS PARA DETERMINACIONES ESPECTROFOTOMÉTRICAS DE COLOR* 2-7 Localícese el punto (x, y) en uno de los diagramas de cromaticidad de la figura 2120:2 y determínese la longitud de onda predominante (en nanómetros) y la pureza (en porcentaje) directamente a partir del diagrama. Determínese la tonalidad a partir del valor de longitud de onda dominante, de acuerdo con los márgenes de la tabla 2120:11. TABLA 2120:11. MATICES DE COLOR PARA MÁRGENES DE LONGITUD DE ONDA DOMINANTE * Para significancia de «c», véase figura 2120:2. 5. Expresión de resultados Concrétense las características de color (a pH 7,6 y al pH original) en función de la longitud de onda dominante (nanómetros, para la unidad más próxima), la tonalidad (por ejemplo, azul, azul verdoso, etc.), luminancia (porcentaje, respecto a la decena más próxima) y pureza (porcentaje, respecto a la unidad más próxima). Especifíquense el tipo de instrumento (por ejemplo, espectrofotómetro), el número de ordenadas seleccionadas (10 ó 30) y la anchura de banda espectral (nanómetros) utilizada. * insertar en cada columna el valor de transmilancia (%) correspondiente a la longitud de onda mostrada. Cuando basta una exactitud limitada, utilícense solamente los ordinales marcados con asterisco. 6. Bibliografía HARDY , A. C. 1936. Handbook of Colorimetry. Technology Press, Boston, Massachusetts. 2-8 0,40 MÉTODOS NORMALIZADOS 2-9 PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2120 D. Método de filtro triestímulo 1. Discusión general a) Principio: Para obtener datos de color útiles para control rutinario pueden emplearse tres filtros especiales de luz triestímulo, combinados con una fuente de luz y una célula fotoeléctrica de un fotómetro de filtro. El porcentaje de luz triestímulo transmitida por la solución se determina para cada uno de los tres filtros y, a continuación, los valores de transmitancia se convierten en coeficientes tricromáticos y valores característicos de color. b) Aplicación: Este método es aplicable a aguas potables y de superficie y a aguas residuales, tanto domésticas como industriales. Excepto en los trabajos en los que se requieren valores muy exactos, ofrece resultados muy similares a los del método C. c) Interferencia: Debe eliminarse la turbidez. 2. Instrumental a) Fotómetro de filtro*. b) Fuente de luz del fotómetro de filtro: Lámpara de tungsteno a una temperatura de color de 3000 °C†. c) Células fotoeléctricas de fotómetro de filtro, 1 cm‡. d) Filtros triestímulo§. e) Sistema de filtrado: Véanse sección 2120C.26 y figura 2120:1. 3. Procedimiento a) Preparación de la muestra: Véase sección 2120C.3a. * Electrofotómetro Fisher o equivalente. † Lámpara General Electric núm. 1719 (a 6V) o equivalente. ‡ Célula fotovoltaica General Electric, tipo PV-1 o equivalente. § Corning CS-3-IO7 (N.° 1), CS-4-98 (N.° 2) o equivalente. b) Determinación de las características de transmisión de luz: Lávense cuidadosamente (detergente) y aclárense con agua destilada las células de absorción de 1 cm. Ha de enjuagarse dos veces cada célula con muestra filtrada, lavar la superficie con papel para material óptico y llenar las células con muestra filtrada. Colóquese un blanco de agua destilada en otra célula y utilícese ésta para situar el instrumento en un 100 por 100 de transmitancia. Determínese el porcentaje de transmisión de luz a través de la muestra para cada uno de los filtros de luz triestímulo con la intensidad de la lámpara del fotómetro de filtro fijada en una posición equivalente a 4V. 4. Cálculo a) Determínese el valor de luminar»cia directamente como el porcentaje de valor de transmitancia obtenido con el filtro triestímulo núm. 2. b) Calcúlense los valores triestímulo X, Y y Z a partir de la transmitancia porcentual (Tl, T2 y T3) para los filtros núms. 1, 2, 3 como sigue: X = T3 x 0,06 + r, x 0,25 Y = T 2 x 0 ,31 6 Z = r3 x 0,374 Calcúlense y determínense los coeficientes tricromáticos x e y, la longitud de onda dominante, la tonalidad y la pureza según se ha señalado anteriormente en la sección 2120C.4b. 5. Expresión de resultados Se hará según se indica en la sección 2120C.5. MÉTODOS NORMALIZADOS 2-10 2120 E. 1. Método ADMI de filtro triestímulo (PROPUESTA) Discusión general a) Principio: Este método es una ampliación del método triestímulo 2120D, y con él puede obtenerse una medida de la muestra de color, independientemente de la tonalidad. Se basa en el empleo de la fórmala de valor cromático de AdamsNickerson1 para el cálculo de valores numéricos simples de diferencias de color, es decir, diferencias de color uniformes. Por ejemplo, si a simple vista dos colores, A y B, se consideran distintos del tono incoloro en un mismo grado, sus valores ADMI de color serán los mismos. La modificación fue llevada a cabo por miembros del American Dye Manufacturers Institute (ADMI)2. b) Aplicación: Este método se aplica tanto a aguas limpias y residuales coloreadas, que tengan características cromáticas significativamente diferentes de los estándares platino-cobalto, como a las que presentan una tonalidad similar a la de los estándares. c) interferencia: Elimínese la turbidez. 2. Instrumental a) Fotómetro de filtro*, equipado con filtros triestímulo CIE (véase 2120D.2..7). b) Fuente de luz del fotómetro de filtro: Lámpara de tungsteno a una temperatura de color de 3.000 °C (véase 2120D.2b). c) Celdillas de absorción y soportes de celda adecuados: Para valores de color menores de 250 unidades ADMI, empléense células con paso de luz de 5,0 cm; para valores mayores de 250, utilícense celdas con paso de 1,0 cm. a) Sistema de filtrado: Véanse sección 2120C.26 y figura 2120:1; puede * Electrocolorímetro Fisher. modelo 181. o equivalente. emplearse también una centrífuga capaz de conseguir 1.000 x g (véase sección 2120B). 3. Procedimiento a) Calibrado de instrumentos: Establézcase una curva para cada fotómetro; los datos de un instrumento no pueden aplicarse a otro. Prepárese una curva de calibrado distinta para cada longitud de paso de la celdilla de absorción. 1) Prepárense estándares según el procedimiento descrito en 2120B.3. Para una longitud de célula de 5 cm se dispondrán estándares cen valores de color de 25, 50, 100, 200 y 250. mediante dilución de 5,0, 10,0, 20,0, 30,0, 40.0 y 50.0 ml de estándar de color con agua destilada, hasta 100 ml en matraces volumétricos. Para longitudes de paso más cortas, prepárense estándares adecuados con valores de color más altos. 2) Determínese la transmitancia de luz (véase apartado 3c, más adelante) para cada estándar con cada filtro. 3) Utilizando los cálculos descritos en el apartado 3d, calcúlense los valores triestímulo (Xs, Ys, Zs) para cada estándar, determínense los valores Munsell y calcúlese el valor intermedio (DE). 4) Empleando los valores DE para cada estándar, calcúlese un factor de calibrado Fn para cada estándar, a partir de la siguiente ecuación; donde: (APHA)n= valor de color APHA para estándar n, (DF) n = valor intermedio calculado para estándar, y b = paso en centímetros de luz de la celdilla. PROPIEDADES FÍSICAS Y DE AGREGACIÓN Colocando (DE)n en el eje de abscisas y Fn en el de ordenadas, puntéese una curva para las soluciones estándar. Utilícese la curva de calibrado para derivar el valor F a partir de los valores DE obtenidos con las muestras. b) Preparación de muestras: Prepárense dos porciones de muestra de 100 ml (una a pH original y otra a 7,6), como se describe en la sección 2120C.3a, o por centrifugación. (NOTA: el centrifugado sólo es aceptable si permite lograr una eliminación de la turbidez equivalente a la del filtrado.) c) Determinación de las características de transmisión de la luz: Lávense cuidadosamente con detergente las celdillas de absorción y aclárense con agua destilada. Enjuáguese cada celdilla de absorción dos veces con muestra filtrada. Limpíense las superficies externas con papel para limpiar lentes ópticas y llénese la celdilla con muestra. Determínese la transmitancia de luz de la muestra con los tres filtros para obtener los valores T1 del filtro 1, T2 del filtro 2 y T3 del filtro 3. Estandarizar el instrumento con cada filtro, a 100 por 100 de transmitancia, con agua oxigenada. d) Cálculo de valores de color: Los valores triestímulo para muestras son Xs, Ys y Zs; para estándar, Xr, Yr y Zr; y para agua destilada, Xc, Yc y Zc. Los valores Munsell para muestras son Vxs, Vys y Vzs; para estándar, Vxr, Vyr y Vzr; y para agua destilada, Vxc, Vyc y Vzc. Para cada estándar o muestra, calcúlense los valores triestímulo, a partir de las siguientes ecuaciones: 2-11 Xc = 98,09 Yc = 100,0 Z c = 118,35 Para convertir los seis valores triestímulo (Xs, Ys, Zs, Xc, Yc, Zc,) en los valores Munsell correspondientes, utilícense las tablas publicadas 2, 3, 4*, o mediante la ecuación de Bridgeman3. Los valores intermedios de DE se calculan a partir de la ecuación donde: cuando la muestra se compara con agua destilada. Con la curva de calibrado estándar, utilícese el valor DE para determinar el factor F de calibrado. El valor ADMI de color final se calcula como sigue: donde: b = paso de luz de la celda de absorción (cm). Infórmese sobre valores ADMI de color a pH original y a 7,6. 4. Método alternativo El valor ADMI de color puede también determinarse espectrofotométricamente, utilizando un espectómetro de Los valores triestímulo para el blanco de agua destilada utilizado para estandarizar el aparato son siempre: * Instrumental Colour Systems, Ltd., 7 Bucklebury Place, Upper Woolhampton, Berkshire RG7 5UD, Inglaterra. 2-12 MÉTODOS NORMALIZADOS banda espectral limitado (10 nm o menos) y un margen operativo eficaz de 400 a 700 nm. Este método es una ampliación de 2120C. Los valores triestímulo pueden calcularse a partir de las medidas de transmitancia, preferentemente mediante uso del método de ordinales ponderados o de ordinales seleccionados. El método ha sido descrito por Allen y cols.2, quienes incluyen protocolos y ejemplos prácticos. MAN. 1973. Determination of color of water and wastewater by means of ADMI color values. Proc. 28th Ind. Waste Conf., Purdue Univ., Eng. Ext. Ser. N.° 142:661. BRIDGEMAN, T. 1963. Inversion of the Munsell value equation. J. Opt. Soc. Amer. 53:499. 6. 5. 1. 2. Referencias Bibliografía JUDD, D. B. & WYSZECKI. 1963. Color in Business, Science, and Industry, 2.a ed. John Wiley & Sons, Nueva York. (Véanse tablas A, B, y C en Apéndice.) WYSZECKI, G. & W. S. STILES. 1967. Color Science. John Wiley & Sons, Nueva York. (Véanse tablas 6.4, A, B, C, págs. 462-467.) M C L AREN , K. 1970. The Adams-Nickerson colour-difference formula. J. Soc. Dyers Colorists. 86:354. ALLEN , W., W. B. P RESCOTT , R. E. D ERBY , C. E. G ARLAND , J. M. P ERET & M. S ALTZ - 2130 TURBIDEZ* 2130 A. 1. Introducción Fuentes y significación La transparencia del agua es importante para la elaboración de productos destinados a consumo humano y para numerosos usos industriales. Los fabricantes de bebidas, los procesadores de alimentos y el tratamiento de las plantas de extracción sobre agua superficial generalmente confían en la coagulación, la clasificación y el filtrado para garantizar productos aceptables. La transparencia de una masa natural de agua es un factor decisivo para la calidad y productividad de estos sistemas. La turbidez del agua es producida por materias en suspensión, como arcilla, cieno o materias orgánicas e inorgánicas finamente divididas, compuestos orgánicos solubles coloreados, plancton y otros microorganismos. La turbidez es una expresión de la propiedad óptica que origina que la luz se disperse y absorba en vez de transmitirse en línea recta a través de la muestra. La correlación de la turbidez con la concentración en peso de la materia en suspensión es difícil de establecer, ya que en la dispersión luminosa también intervienen el tamaño, la forma y el índice de refracción de las partículas. Partículas ópticamente negras, como las de carbono activado, pueden absorber luz y aumentar significativamente las cifras de turbidez. 2. * Aprobado por el Standard Methods Committee, 1988. Selección del método Históricamente, el método para determinación de la turbidez se basa en el PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-13 turbidímetro de Jackson1; sin embargo, el valor más bajo de turbidez que puede medirse directamente con este instrumento es de 25 unidades. Como la turbidez del agua tratada suele situarse en un intervalo de 0 a 1 unidades, también se desarrollaron métodos indirectos. Por desgracia, ningún aparato puede duplicar los resultados obtenidos para todas las muestras con el turbidímetro de Jackson. Debido a las diferencias fundamentales en el sistema óptico, los resultados obtenidos con distintos tipos de instrumentos secundarios a menudo no concuerdan exactamente, aun cuando los aparatos estén precalibrados frente al turbidímetro de bujía. Muchos de los turbidímetros comerciales disponibles para medida de turbidez baja proporcionan datos comparativamente válidos sobre la intensidad de la luz dispersada en una dirección dada, predominantemente en ángulo recto a la luz incidente. Esos nefelómetros se ven escasamente afectados por las pequeñas variaciones de los parámetros de diseño y, por tanto, resultan especialmente útiles como instrumento estándar para medir turbideces bajas. Los turbidímetros no estándar, como los dispositivos de anterodispersión, son más sensibles para las partículas grandes y útiles para monitorización del proceso. Otra de las causas de discrepancia en el análisis de la turbidez es el empleo de suspensiones de tipos distintos de material en partículas para la preparación de las curvas de calibrado de instrumentos. Como las muestras de agua, las suspensiones preparadas tienen propiedades ópticas distintas que dependen de la distribución del tamaño de las partículas, su forma y su índice de refracción. Para la calibración del nefelómetro se exige una suspensión de referencia estándar que tenga propiedades reproducibles de dispersión luminosa. Dado que no existe relación directa entre la intensidad de la dispersión de luz a un ángulo de 90° y la turbidez de la bujía de Jackson, tampoco existe un fundamento válido para calibración de un nefelómetro en términos de unidades-bujía. Para evitar interpretaciones erróneas, los resultados de las medidas nefetométricas deben expresarse en forma de unidades nefelométricas de turbidez (UNT). Por su precisión, su sensibilidad y su fácil aplicación a un amplio margen de turbideces, el método nefelométrico resulta preferible a los métodos visuales. El método de bujía de Jackson ha sido eliminado de la presente edición de Métodos normalizados. 3. Conservación de la muestra Determínese la turbidez el mismo día en que se toma la muestra. Si es inevitable una conservación más prolongada, almacénense las muestras en ambiente oscuro hasta 24 horas. No almacenar largos períodos por la posible aparición de cambios irreversibles de la turbidez. Agítense vigorosamente todas las muestras antes de su examen. 4. Referencias 1. AMERICAN PUBLIC HEALTH ASSOCIATION, AMERICAN WATER WORKS ASSOCIATION & WATER POLLUTION CONTROL FEDERATION. 1985. Standard Methods for the Examination of Water and Wastewater, 16.a ed. American Public Health Assoc, Washington, D.C. 2-14 MÉTODOS NORMALIZADOS 2130 B. Método nefelométrico 1. Discusión general a) Principio: Este método se basa en la comparación de la intensidad de la luz dispersada por la muestra en condiciones definidas y la dispersada por una solución patrón de referencia en idénticas condiciones. Cuanto mayor es la intensidad de la luz dispersada, más intensa es la turbidez. Como suspensión patrón de turbidez de referencia se emplea el polímero formacina. Es fácil de preparar y, en cuanto a propiedades de dispersión de luz, más reproducible que la arcilla o el agua turbia natural. La turbidez de una concentración especificada de suspensión de formacina se define como el equivalente a 40 unidades nefelométricas. Esta suspensión tiene una turbidez aproximada de 40 unidades Jackson si se mide en el turbidímetro de bujía; por tanto, las unidades nefelométricas basadas en la preparación de formacina se aproximarán a las del turbidímetro de bujía, pero no serán idénticas. b) Interferencia: La turbidez puede determinarse en cualquier muestra de agua libre de residuos y privada de sedimentos gruesos. La suciedad del vidrio, la presencia de burbujas de aire y los efectos de las vibraciones que alteran la visibilidad superficial de la muestra, conducirán a resultados falsos. El «color verdadero», es decir, el color del agua debido a sustancias disueltas que absorben luz, origina que la turbidez sea más baja. Este efecto, por lo general, no resulta significativo en el caso de aguas tratadas. 2. Instrumental a) Turbidímetro, consistente en un nefelómetro en una fuente de luz para iluminar la muestra, y uno o más detectores fotoeléctricos con un dispositivo de lectura exterior para indicar la intensidad de la luz dispersada a 90° de la vía de luz incidente. Utilícese un turbidímetro diseñado de manera que una parte de la luz desviada alcance el detector en ausencia de turbidez y libre de una desviación significativa después de un breve calentamiento. La sensibilidad del instrumento debiera permitir la detección de diferencias de turbidez de 0,02 UNT o menos, en aguas con cifras de menos de 1 UNT, con margen entre 0 y 40 UNT. Para obtener una cobertura adecuada y una sensibilidad suficiente para turbideces bajas son necesarios varios márgenes. Las diferencias en el diseño del turbidímetro producirán diferencias en los valores obtenidos, aunque se use la misma suspensión para el calibrado. A fin de reducir al mínimo estas diferencias, obsérvense los siguientes criterios de diseño: 1) Fuente de luz: Lámpara de filamento de tungsteno dispuesto para una temperatura de color comprendida entre 2.200 y 3.000 °K. 2) Distancia recorrida por la luz incidente y la dispersada dentro del tubo de muestra: Total que no exceda de 10 cm. 3) Ángulo de aceptación de la luz por el receptor: Centrada a 90° de haz de luz incidente y sin exceder ± 30° a partir de 90°. El detector y el sistema de filtro, si se usa, tendrán una respuesta-pico en el espectro entre 400 y 600 nm. b) Tubos de muestra, de cristal incoloro, transparente. Mantener los tubos escrupulosamente limpios, por dentro y por fuera, descartando los rayados y manchados. No manejarlos cuando están bajo la luz. Utilícense de tipo extralargo, con un estuche protector que facilite su manejo. Llénense las muestras y los patrones después de agitación cuidadosa, dejando tiempo para que se eliminen las burbujas. 2-15 PROPIEDADES FÍSICAS Y DE AGREGACIÓN 3. Reactivos a) Agua libre de turbidez: Es difícil de obtener. Para medir una turbidez alrededor de 0,02 UNT es adecuado el siguiente método. Pásese agua destilada a través de una membrana de filtro con orificios de precisión de 0,2 (μm*; no resultan adecuados los filtros bacteriológicos corrientes. Enjuáguese el matraz de recogida al menos dos veces con agua filtrada y deséchense los 200 ml siguientes. Algunas botellas de agua desmiueralizada de uso comercial están casi libres de partículas. Pueden usarse cuando su turbidez sea más baja que la obtenida en el laboratorio. Dilúyanse las muestras con agua destilada hasta una turbidez no menor de 1. b) Suspensión de turbidez de reserva: d) Estándares alternativos: Como alternativa a la preparación y dilución de formacina, utilícense estándares comerciales, como los gránulos de estireno-divinilbenzeno†, si se ha demostrado su equivalencia a la formacina de preparación reciente. e) Estándares diluidos de turbidez: Dilúyanse porciones de suspensiones de turbidez estándar con agua libre de turbidez, según se requiera. Prepárese diariamente. 4. Procedimiento 1) Solución I: Disuélvanse 1,000 g de sulfato de hidracina (PRECAUCIÓN: Cancerígeno, evitar inhalación, ingestión y contacto con la piel), (NH2)2·H2SO4, en agua destilada y dilúyanse hasta 100 ml en un matraz volumétrico. 2) Solución II: Disuélvanse 10,00 g de hexametilenotetraamina, (CH2)6N4, en agua destilada y dilúyanse hasta 100 ml en un matraz volumétrico. 3) En un matraz de 100 ml mézclense 5,0 ml de solución I y 5,0 ml de solución II. Manténgase durante 24 horas a 25 ± 3 °C, dilúyase hasta la marca y mézclese. La turbidez de esta suspensión es de 400 UNT. 4) Prepárense soluciones y suspensiones mensualmente. c) Suspensión de turbidez estándar: Dilúyanse 10,00 ml de suspensión madre de turbidez, hasta 100 ml, en agua libre de turbidez. Prepárese diariamente. La turbidez de esta suspensión se considera de 40 UNT. a) Calibrado de turbidímetro: Síganse las instrucciones del fabricante. A falta de una escala precalibrada, prepárense curvas de calibrado para cada margen del aparato. Utilizando estándares adecuados, compruébese la exactitud de cualquier escala de calibrado de que se disponga sobre un instrumento precalibrado. Verifíquese por lo menos un estándar en cada margen del aparato que se vaya a utilizar. Compruébese que el turbidímetro facilita lecturas estables en todos los márgenes de sensibilidad utilizados. Es probable que las turbideces elevadas determinadas por medida directa difieran apreciablemente de las determinadas por la técnica referida en el apartado 4c. b) Medida de turbideces menores de 40 UNT: Agítese cuidadosamente la muestra. Espérese hasta que desaparezcan las burbujas de aire, y viértase la muestra en el tubo del turbidímetro. Cuando sea posible, viértase la muestra agitada en el tubo y sumérjase en un baño ultrasónico durante 1-2 segundos, obteniendo la eliminación total de las * Nudepore Corporation, 7035 Commerce Circle, Pleasanton, California, o equivalente. * Estándar AMCO-AEPA-1, Advanced Polyrner Systems, 3696 Haven Ave., Redwood City, California. 2-16 MÉTODOS NORMALIZADOS burbujas. Léase directamente la turbidez en la escala del aparato o en la curva del calibrado adecuada. c) Medida de turbideces superiores a 40 UNT: dilúyase la muestra con uno o más volúmenes de agua libre de turbidez hasta que ésta descienda a 30-40 UNT. Calcúlese la turbidez de la muestra original en función de la que tiene la muestra diluida y del factor de dilución. Por ejemplo, si cinco volúmenes de agua libre de turbidez se añaden a un volumen de muestra y la muestra diluida mostró una turbidez de 30 UNT, la turbidez de la muestra original era de 180 UNT. d) Calíbrense soluciones de monitorización continua de turbidez, para cifras bajas de ésta, mediante determinación de la turbidez del agua que entra y sale por ellas, utilizando un turbidímetro modelo de laboratorio. Cuando esto no sea posible, empléese un adecuado estándar diluido (apartado 3e). Para turbideces superiores a 40 UNT, utilícese solución madre no diluida. 5. Cálculo Unidades nefelométricas de turbidez (UNT) donde: A = UNT encontradas en muestra diluida, B = volumen (ml) de agua de dilución, y C = volumen (ml) de la muestra tomada para dilución. 6. Interpretación de los resultados a) Infórmese de las lecturas de turbidez del siguiente modo: b) Para comparar la eficacia del tratamiento de un agua, estímese la turbidez de forma más precisa a como se ha señalado aquí. Las incertidumbres y discrepancias en medidas de turbidez hacen improbable que dos o más laboratorios dupliquen los resultados de una misma muestra con más exactitud que la especificada. 7. Bibliografía WHIPPLE, G. C. & D. D. JACKSON. 1900. A comparativo study of the methods used for the measurement of turbidity of water. Mass. Inst, Technol. Quart. 13:274. AMERICAN PUBLIC HEALTH ASSOCIATION. 1901. Report of Committee on Standard Methods of Water Analysis. Pub. Health Papers & Rep. 27:377. WELLS, P. V. 1922. Turbidimetry of water. J. Amer. Water Works Assoc. 9:488. BAYLIS, J. R. 1926. Turbidimeter for accurate measurement of low turbidities. Ind. Eng. Chem. 18:311. WELLS, P. V. 1927. The present status of turbidity measurements. Chem. Rev. 3:331. BAYLIS, J. R. 1933. Turbidity determinations. Water Works Sewage 80:125. ROSE, H. E. & H. B. LLOYD. 1946. On the measurement of the size characteristics of powders by photo-extinction methods. J. Soc. Chem. Ind. (Londres) 65:52 (Feb.); 65:55 (Mar.). ROSE, H. E. & C. C. J. FRENCH. 1948. On the extinction coefficient: Particle size relationship for fine mineral powders. J. Soc. Chem. Ind. (Londres) 67:283. GILLETT, T. R., P. F. MEADS & A. L. HOLVEN. 1949. Measuring color and turbidity of white sugar solutions. Anal. Chem. 21:1228. PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-17 JULLANDER, I. 1949. A simple method for the measurement of turbidity. Acta Chem. Stand. 3:1309. ROSE, H. E. 1950. Powder-size measurement by a combination of the methods of nephelometry and photo-extinction. J. Soc Chem. Ind. (Londres) 69:266. ROSE, H. E. 1950. The design and use of photoextinction sedimentometers. Engineering 169: 350, 405. BRICE, B. A., M. HALWER & R. SPEISER. 1950. Photoelectric light-scattering photometer for determining high molecular weights. J. Opt. Soc. Amer. 40:768. KNIGHT , A. G. 1950. The measurement of turbidity in water. J. Inst. Waler Eng. 4:449. HANYA, T. 1950. Study of suspended matter in water. Bull. Chem. Soc. Jap. 23:216. JULLANDER. I. 1950. Turbidimetric investigations on viscose. Svensk Papperstidn. 22:1. R OSE , H. E. 1951. A reproducible standard for the calibration of turbidimeters. J. Inst. Water Eng. 5:310. AITKEN, R. W. & D. MERCER. 1951. Comment on «The measurement of turbidity in water.» J. Inst. Water Eng. 5:328. ROSE, H. E. 1951. The analysis of water by the assessment of turbidity. J. Inst. Water Eng. 5:521. KNIGHT, A. G. 1951. The measurement of turbidity in water: A reply. J. Inst. Water Eng. 5:633. STAATS, F. C. 1952. Measurement of color, turbidity, hardness and silica in industrial waters. Preprint 156, American Soc. Testing & Materials, Filadelfia, Pennsylvania. PALIN, A. T. 1955. Photometric determination of the colour and turbidity of water. Water Water Eng. 59:341. SLOAN, C. K. 1955. Angular dependence light scattering studies of the aging of precipitates. J. Phys. Chem. 59:834. CONLEY, W. R. & R. W. PITMAN. 1957. Microphotometer turbidity analysis. J. Amer. Water Works Assoc. 49:63. PACKHAM, R. F. 1962. The preparation of turbidity standards. Proc. Soc. Water Treat. Exam. 11:64. BAALSRUD, K. & A. HENRIKSEN. 1964. Measurement of suspended matter in stream water. J. Amer. Water Works Assoc. 56:1194. HOAIHER, R. C. 1964. Comparison of different methods for measurement of turbidity. Proc: Soc Water Treat. Exam. 13:89. EDEN, G. E. 1965. The measurement of turbidity in water. A progress report on the work of the analytical panel. Proc: Soc Water Treat. Exam. 14:27. BLACK, A. P. & S. A. HANNAH. 1965. Measurement of low turbidities. J. Amer. Water Works Assoc. 57:901. HANNAH, S. A., J. M. COHEN & G. G. ROBECK. 1967. Control techniques for coagulation-filtration. J. Amer. Water Works Assoc 59:1149. REBHUN, M. & H. S. SPERBER. 1967. Optical properties of diluted clay suspensions. J. Colloid inter face Sci. 24:131. DANIELS. S. L. 1969. The utility of optical parameters in evaluation of processes of flocculation and sedimentation. Chem. Eng. Progr. Symp. Ser. N.° 97, 65:171. LIVESEY, P. J. & F. W. BILLMEYER, JR. 1969. Particle-size determination by low-angle light scattering: new instrumentation and a rapid method of interpreting data. J. Colkñd ínter face Sci. 30:447. OSTENDORF, R. G. & J. F. B YRD. 1969. Modern monitoring of a treated industrial effluent. J. Water Pollut. Control Fed. 41:89. EICHNER, D. W. & C. C. HACH. 1971. How clear is clear water? Water Sewage Works 118:299. HACH, C. C. 1972. Understanding turbidity measurement. Ind. Water Eng. 9(2): 18. SIMMS, R. J. 1972. Industrial turbidity measurement. ISA (Instrum. Soc Amer.) Trans. 11(2):146. TALLEY, D. G., J. A. JOHNSON & J. E. P ILZER. 1972. Continuous turbidity monitoring. J. Amer. Water Works Assoc 64:184. MÉTODOS NORMALIZADOS 2-18 2150 2150 A. 1- OLOR* introducción Discusión general El olor, como el gusto, depende del contacto de una sustancia estimulante con la adecuada célula receptora. Los estímulos son de naturaleza química, y por ello se suele decir que el olfato y el gusto son «sentidos químicos». El agua es un medio neutro que siempre se halla presente en o sobre las membranas que perciben la respuesta sensorial. En su forma pura, el agua no produce sensaciones olfativas o gustativas. Hasta ahora no se ha desarrollado ninguna teoría satisfactoria de la olfacción, aunque se hayan formulado muchas. Debido a la respuesta sensorial adversa, el hombre y los animales pueden evitar muchos aumentos potencialmente tóxicos, y, sin esta primitiva forma de protección, muchas especies no habrían sobrevivido. Hoy en día, estos mismos sentidos proporcionan el primer aviso de virtuales riesgos ambientales. El olor se reconoce1 como un factor de calidad que afecta a la aceptabilidad del agua potable (y de los alimentos preparados con ella), que puede corromperse con la presencia de peces y otros organismos acuáticos y anular la estética de Las aguas de instalaciones de recreo. Muchas sustancias orgánicas y algunas inorgánicas influyen en el gusto y el olor. Estas sustancias pueden tener su origen en vertidos de residuos municipales e in- * Aprobado por el Standard Methods Commiittee. 1985. dustriales, en factores naturales, corno la descomposición de materias vegetales, o en una actividad microbiana asociada. La expansión tecnológica verificada en la abundancia y diversidad de materias residuales exige una depuración del agua que permita eliminar contaminantes que antes sólo estaban en el aire, y el crecimiento continuo de la población, con el consiguiente uso repetido del agua disponible, incrementa la posibilidad de deterioro de la calidad sensorial del agua. El consumo doméstico y los procesos industriales, como los de la elaboración de alimentos, bebidas y productos farmacéuticos, requieren el empleo de un agua esencialmente libre de olor y sabor. Algunas sustancias, como ciertas sales inorgánicas, producen sabor sin olor, y se evalúan mediante pruebas de gusto (sección 2160). Muchas otras sensaciones adscritas al sentido del gusto son olores en realidad, aunque la sensación no es percibida hasta que el material no liega a la boca. A pesar de los rápidos avances realizados en lo que respecta a calidades sensoriales para los análisis químicos2, la mayoría de los olores son demasiado complejos y se detectan a concentraciones demasiado bajas como para permitir su definición mediante aislamiento y determinación de las sustancias químicas odoríferas. El dispositivo supremo para la realización de pruebas de olor es la nariz humana. Las pruebas de olor se llevan a cabo para proporcionar descripciones cualitativas y medidas cuantitativas aproximadas de la intensidad del olor. El método de medida de la intensi- 2-19 PROPIEDADES FÍSICAS Y DE AGREGACIÓN dad que aquí se presenta es la prueba de umbral de olor, basada en un método de límites2. No se estudian aquí los métodos supraumbral. La sección 6040B proporciona un procedimiento analítico para cuantificar varios compuestos orgánicos productores de olor, entre los que se incluyen la geosmina y el metilisoborneol. Las pruebas sensoriales son útiles para indagar la calidad de las aguas tratadas y no tratadas, y para controlar el olor a lo largo de los procesos de tratamiento. Con ellas se puede valorar la eficacia de distintos tratamientos y proporcionar un medio de detectar la fuente de contaminación. 2. 1. 2. Referencias U. S. ENVIRONMENTAL PROTECTION AGENCY. 1973. Proposed Criteria for Water Quality. Vol. 1, Washington, D.C. AMERICAN SOCIETY FOR TESTING AND MATERIALS COMMITTEE E-18. 1968. STP 433, Basic principles of sensory evaluation; STP 434, Manual on sensory testing methods; STP 440, Correlation of subjective-objective methods in the study of odors and taste. ASTM, Filadelfia, Pennsylvania. 2150 B. 1. 3. Bibliografía MONCRIEFF, R. W. 1946. The Chemical Senses. John Wiley & Sons, Nueva York. SECHENOV, I. M. 1956 y 1958. Problem of hygenic standards for waters simultaneously polluted with harmful substances [en ruso]. Gig. Sanil. Núms. 10 y 8. Taste and Odor Control in Water Purification, 2.a ed. 1959. West Virginia Pulp & Paper Co. Industrial Chemical Sales División, Nueva York. [Contiene 1.063 referencias clasificadas.] BAKER, R. A. 1961. Problems of tastes and odors. J. Water Pollut. Control Fed. 33:1099. BAKER, R. A. 1963. Odor effects of aqueous mixtures of organic chemicals. J. Water Pollut. Control Fed. 35:728. ROSEN, A. A., R. T. SKEEL & M. B. ETTINGER. 1963. Relationship of river water odor to specific organic contaminants. J. Water Pollut. Control Fed. 35:777. WRIGHT, R. H. 1964. The Science of Smell. Basic Books, Nueva York. AMERINE , M. A., R. M. P ANGBORN & E. B. ROESSLER. 1965. Principles of Sensory Evaluation of Food. Academic Press, Nueva York. ROSEN, A. A. 1970. Report of research committee on tastes and odors. J. Amer. Water Works Assoc. 62:59. GELDARD, F. A. 1972. The Human Senses. John Wiley & Sons, Nueva York. Prueba de umbral de olor Discusión general a) Principio: Determínese el umbral de olor mediante dilución de una muestra con agua inodora hasta eliminar totalmente el olor perceptible. No existe una concentración absoluta de olor umbral debido a la variación inherente a la capacidad olfatoria individual. La sensibilidad de una misma persona varía según el momento, apareciendo diferencias de un día para otro a lo largo del día. Además, las respuestas varían en función de las características y de la concentra- ción del estímulo oloroso. El número de personas seleccionadas para medir el umbral de olor dependerá del objetivo de las pruebas, de factores económicos y del personal disponible. Para las pruebas sensoriales, cuando los resultados tienen que representar al conjunto de la población o se desea una gran precisión, se requiere un amplio número de experimentadores. En estas circunstancias se recomienda un número no menor de cinco personas1, aunque es preferible que sean diez o más. En las plantas de tratamiento de aguas suele resultar necesaria 2-20 la medida de niveles del umbral por una sola persona. La interpretación de los resultados de un solo probador requiere el conocimiento de la agudeza relativa de esa persona. Algunos investigadores han utilizado sustancias olorosas específicas, como el m-cresol o n-butanol, para calibrar la respuesta del probador2. b) Aplicación: Este método de umbral es aplicable a una gama de muestras que incluye desde aguas naturales casi inodoras a residuos industriales con cifras de umbral del orden de varios miles. Estas muestras muy olorosas no presentan dificultades específicas, puesto que su concentración se reduce proporcionalmente antes de ser sometidas a observación. c) Descripciones cualitativas: A pesar de los esfuerzos realizados durante más de un siglo, aún no se ha puesto a punto un sistema satisfactorio para caracterización de un olor. Las ediciones anteriores de este libro contenían una tabla de descripciones de olor, propuesta como guía para expresar la calidad del olor. Las anticuadas abreviaturas de esa tabla pueden resultar muy complicadas para el lector. La 12.a edición ofrece una explicación de esos términos. d) Toma y conservación de muestras: Recójanse las muestras para pruebas de olor en botellas de vidrio, con tapones de cristal o revestidos de TFE. Realícense las pruebas tan pronto como sea posible después de la toma de muestras. Si es necesario conservar las muestras, recójanse por lo menos 500 ml en una botella llenándola hasta el tapón, y refrigérese, comprobando que durante esta refrigeración no puede penetrar en la muestra ningún olor extraño. No deben utilizarse recipientes de plástico. e) Decloración: La mayoría de las aguas de consumo y algunas residuales están cloradas; con frecuencia es deseable determinar el olor de la muestra clorada antes y después de la decloración. Declórese con arsenito o tiosulfato en una can- MÉTODOS NORMALIZADOS tidad estequiométrica exacta, tal como se describe en Nitrógeno (amonio), sección 4500-NH3. PRECAUCIÓN: NO utilizar compuestos de arsénico como productos declorantes en muestras que puedan servir para pruebas de sabor. f) Temperatura: Los valores de umbral de olor varían con la temperatura. Para la mayoría de las aguas depuradas o naturales, una temperatura de muestra de 60 °C permite la detección de olores que de otra manera pasarían inadvertidos; esta temperatura es la estándar para pruebas de umbral de calor. Para algunos fines —cuando el olor sea demasiado evanescente o la sensación de calor sea excesiva—, esta prueba no es aplicable; cuando la experiencia aconseje una temperatura inferior, utilícese una temperatura de prueba estándar de 40 °C. En casos especiales pueden emplearse otras temperaturas. Informar acerca de la temperatura a que se hicieron las observaciones. 2. Instrumental Para garantizar la fiabilidad de las medidas de umbral, utilícese material de vidrio inodoro. Lávese este material, inmediatamente antes de su uso, con jabón inodoro y solución acida de lavado, y enjuáguese con agua inodora. Resérvese este material exclusivamente para pruebas de umbral. No deben utilizarse tapones de goma, corcho o plástico, ni vasos de boca estrecha. a) Botellas de muestra, con tapón de vidrio o tapas revestidas de TFE. b) Baño a temperatura constante: Un baño de agua o una placa caliente eléctrica, capaces de controlar una temperatura de + 1 °C para pruebas de olor a temperaturas elevadas. El baño no aportará olor alguno a los matraces de olor. c) Matraces de olor: Erlenmeyer (ST32) de 500 ml, con tapón de vidrio, PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-21 para mantener diluciones de la muestra durante la prueba. d) Pipetas: 4) Pipetas de transferencia y volumétricas o cilindros graduados: 200, 100, 50 y 25 ml. 4) Pipetas de medida: 10 ml graduadas en décimas. e) Termómetro: De 0 a 110°C, tipo dial con indicador químico o metálico. 3. Agua inodora a) Fuentes: Prepárese agua inodora mediante paso de agua destilada, desionizada o purificada a través de carbón activado, asegurando que el agua solamente entre en contacto con vidrio o TFE. Si no es posible conseguir un instrumental hecho totalmente de vidrio y TFE, utilícese el modelo que se indica más abajo. Si el agua producida no es inodora, reconstrúyase o purifíquese el sistema. En todo caso, contrólese diariamente la calidad del agua obtenida. b) Generador de agua inodora (figuras 2150:1 y 2150:2): 1) Tubo de vidrio borosilicato, diam. 7,5 cm, long. 45 cm. 2) Arandelas de amianto (dos). (PRECAUCIÓN: cancerígeno, manipúlense con cuidado), para tubo de 7,5 cm. 3) Bridas (dos), para tubo de 7,5 cm. 4) Juntas de estanqueidad* (dos), espesor 2,5/10 cm, con orificio de 2,5 cm ranurado a 7,5/20 cm de profundidad, frente a la rejilla, con 3 perforaciones, de 12,5/40 cm de diámetro, para igualarse a los fijadores. 5) Rejillas de acero inoxidable (dos), malla-40, 7,5-7,5/10 cm de diámetro. 6) Placas de latón (dos), 7,5/40 cm de espesor x 15-2,5/10 cm de diámetro. Agujero central roscado para el manguito * Neopreno, como el que puede obtenerse de Netherland Rubber Co., Cincinnati, Ohio. Figura 2150:1. Generador de agua inodora. de 7,5/10 cm. Lábrese un surco circular (2,5/40 cm de profundidad x 2,5/40 cm de anchura y 2,5-2,5/20 cm de diámetro) en la placa para prevenir escapes. Perfórese 3 orificios de 12,5/15 cm de diámetro que coincidan con los fijadores. 7) Manguitos de unión galvanizados (dos), de 3/4-in. x 3-in. Enroscar el manguito en la piaca y soldar en posición. 8) Pernos y tuercas de aluminio (6), 5/16-in. x 2-in., para mantener fijos los ajustes. 9) Carbón activado, de tamaño de grano de tamiz 12 a 40 aproximadamente**. Los accesorios terminales de la unidad de adsorción se insertan en el tubo de cristal. Levántense ligeramente los pernos, adaptando la placa de latón al tubo de cristal hasta conseguir un adecuado ajuste con la junta de estanqueidad. Llénese la unidad de adsorción con carbón activado, golpeando el cilindro suavemente pero sin apretar el carbón. Los accesorios finales se insertan en la unidad y se conectan a la fuente de agua como se indica en la figura 2150:1. ** Nuchar W-VG, Westvaco, Covington, Virginia; Filtrasorb 200, Calgon Corp., Pittsburgh, Pennsylvania; o equivalente. MÉTODOS NORMALIZADOS 2-22 variará con la calidad y cantidad de agua filtrada. A menudo se aprecian ligeros olores de origen biológico cuando los filtros de carbón mojado dejan de utilizarse entre períodos de prueba. La detección de un olor en el agua procedente del carbón indica la necesidad de cambiar éste. 4. Figura 2150:2. Conjunto terminal del generador de agua inodora. Para evitar la introducción de contaminantes orgánicos, se comprueban las juntas de los tubos. Utilícese cinta tipo TFE o una pasta hecha con mezcla de plomo rojo en polvo y agua. Límpiense todos los accesorios nuevos con queroseno y lávense a continuación con detergente. Enjuáguense bien con agua limpia. c) Operación del generador: Se pasa agua corriente o destilada a través del generador de agua inodora, a un ritmo de 100 ml/min. Cuando el generador se pone en marcha, se aplica un chorro de agua para eliminar restos de carbón y proceder a la descarga. Compruébese diariamente antes de su uso la calidad del agua obtenida del generador a 40 y 60 °C. La vida del carbón Procedimiento a) Precauciones: Selecciónense cuidadosamente, mediante pruebas preliminares, las personas que van a hacer las pruebas de sabor y olor. Aunque no se requiere una sensibilidad extremada, excluyanse las personas poco sensibles, concentrando la atención en observadores que muestren un sincero interés por la prueba. Evítense estímulos olorosos extraños, como los que aparecen cuando se ha comido o fumado antes de la prueba, o los producidos por jabones aromáticos, perfumes y lociones de afeitado. Debe comprobarse que el probador no padece resfriado o alergia, lo cual influye en la respuesta olorosa. Limítese la frecuencia de las pruebas a un número inferior al nivel de fatiga, con descansos frecuentes en ambiente inodoro. La sala donde se realicen las pruebas debe carecer de elementos de distracción, corrientes de aire y olores2. Si es necesario, dispóngase un cuarto especial inodoro, ventilado por aire filtrado mediante carbón activado y mantenido a temperatura y humedad confortables y constantes3. Para un trabajo eficaz, el número de experimentadores debe ser de cinco o más. Las personas que realizan medidas de olor no deben preparar mezclas ni conocer las concentraciones de dilución que se evalúan. Conviene que sean instruidas sobre el procedimiento antes de que participen en una prueba de grupo. Preséntese primero la muestra más diluida para evitar la fatiga sensorial que produce la muestra concentrada. Durante la PROPIEDADES FÍSICAS Y DE AGREGACIÓN prueba, manténgase la temperatura de las muestras en un margen de 1 °C por encima o por debajo de la temperatura especificada. Dado que muchas aguas naturales y residuales están coloreadas o tienen una neta turbidez que puede sesgar los resultados, utilícense matraces opacos o de color oscuro, como los Erlenmeyer rojo actínico. b) Caracterización: Como parte de la prueba de umbral o como prueba aislada, se ordena a cada experimentador que describa con sus propias palabras el olor característico de la muestra. Regístrese la posible coincidencia de valoraciones entre los ensayistas, lo cual puede proporcionar una pista sobre el origen del contaminante oloroso. El valor de la prueba de caracterización aumenta a medida que los experimentadores adquieren experiencia sobre una categoría de olor particular, como algas, clorofenol o moho. c) Medidas del umbral*: El «número de umbral de olor», designado con las siglas NUO, es la dilución mayor de la muestra en agua inodora capaz de producir un olor netamente perceptible. Llévese el volumen total de la muestra y el agua inodora hasta 200 ml en cada prueba. Háganse nuevas diluciones y anótese el correspondiente NUO señalado en la tabla 2150:1. Estas cifras se han calculado de la siguiente manera: donde: A = ml de muestra, y B = ml de agua inodora. * Existen numerosos métodos de disponer y presentar las muestras para determinaciones de olor. Los ofrecidos aquí son prácticos y económicos en tiempo y personal y generalmente resultan suficientes. Si se proyectan pruebas más extensas y se requieren análisis estadistidos de los datos, será necesario conocer la prueba del triángulo y los métodos que han empleado ampliamente las industrias de condimentos y similares4. 2-23 TABLA 2150:1. NÚMEROS DE UMBRAL DE OLOR CORRESPONDIENTES A VARIAS DILUCIONES 1) Colóquese el volumen adecuado de agua inodora en el primer matraz, añádase muestra al agua (evitando el contacto de la pipeta o la muestra con el labio o el cuello del matraz), mézclese girando y procédase como sigue: Determínese el rango aproximado del número de umbral mediante adición de 200 ml, 50 ml, 12 ml y 2,8 ml de muestra a Erlenmeyer con tapón de vidrio conteniendo agua inodora hasta un volumen total de 200 ml. Como referencia de comparación utilícese un matraz separado que sólo contenga agua inodora. Caliéntense las diluciones y la referencia hasta la temperatura de prueba deseada. 2) Agítese el matraz que contiene el agua inodora, quítese el tapón y huélanse los vapores, haciendo lo mismo con la muestra de prueba que contiene la cantidad menor de agua portadora de olor. Si puede detectarse olor a esta dilución, prepárense muestras más diluidas, según se describe más adelante (apartado 5). Si no puede detectarse olor en la primera dilución, repítase el procedimiento citado utilizando una muestra que contenga la concentración inmediatamente más alta de agua con olor, y continúese este proceso hasta que el olor se detecte claramente. 2-24 MÉTODOS NORMALIZADOS TABLA 2150:11. DILUCIONES PARA VARIAS INTENSIDADES DE OLOR A partir de los resultados obtenidos en la prueba preliminar, prepárese una batería de diluciones utilizándose como guía la tabla 2150:11. Prepárense las cinco diluciones señaladas en la línea apropiada y las tres más concentradas de la línea siguiente de dicha tabla. Por ejemplo, si el olor se notó primero en el matraz que contenía muestra de 50 ml en la prueba preliminar, prepárense matraces conteniendo muestras de 50, 35, 25, 17, 12, 8,3, 5,7 y 4,0 ml, diluida cada una de ellas hasta 200 ml de agua inodora. Es necesario seguir este procedimiento para estimular el margen de sensibilidades de todo el grupo de experimentadores. Incluyanse dos o más blancos en la serie cercana al umbral esperado, pero evitando cualquier modelo repetido. No hay que dejar que el experimentador sepa cuáles son las diluciones olorosa y en blanco. Instrúyasele para que huela cada matraz según una secuencia, empezando con la muestra menos concentrada hasta que el olor se detecte con certeza. 4) Regístrense las observaciones indicando en qué matraz de prueba se percibe olor. Por ejemplo: cionadas en la tabla 2150:11, prepárese una dilución intermedia consistente en una muestra de 20 ml diluida hasta 200 ml de agua inodora. Utilícese esta dilución para determinar el umbral. Multiplíquese por 100 el NUO obtenido para conseguir la dilución intermedia. Raramente se requerirá un paso de dilución intermedia mayor del décuplo. 5. Cálculo El número de umbral de olor es el índice de dilución al que empiezan a detectarse gusto u olor. En el ejemplo anterior (apartado 4c4), el primer olor detectable apareció cuando una muestra de 25 ml se diluyó en 200 ml. Así, el umbral es 200 dividido por 25, es decir, 8. La tabla 2150:1 muestra los números de umbral correspondientes a las diluciones comunes. El NUO más pequeño que puede observarse es 1, como ocurre cuando el matraz contiene 200 ml de muestra no diluida. Si no se detecta olor a esta concentración, anótese «no se observó olor», en vez de un número de umbral. (En aplicaciones especiales se han calculado números de umbral fraccionarios5.) A veces aparecen respuestas anómalas; una concentración baja puede designarse como positiva, y una mayor de la serie, negativa. En tal caso, desígnese el umbral como el punto a partir del cual no aparecen otras anomalías. Por ejemplo: donde: – significa respuesta negativa, y + respuesta positiva. 5) Si la muestra sometida a prueba exige una dilución mayor que las propor- Ocasionalmente, un matraz contiene algún olor residual o se ha contaminado PROPIEDADES FÍSICAS Y DE AGREGACIÓN inadvertidamente. Si la prueba es exacta, repítase la prueba para determinar si el último matraz marcado con « – » era en realidad un blanco de agua inodora mal etiquetado o si el « + » anterior era una muestra contaminada. Utilícense métodos estadísticos adecuados para calcular el umbral promedio más probable a partir de un número elevado de resultados proporcionados por los experimentadores. En la mayoría de los trabajos, el umbral de un grupo se expresará por la media geométrica de los umbrales individuales. 6. Interpretación de los resultados El número de un umbral no es un valor exacto. En el caso de que sólo haya un experimentador, representa una opinión en el momento de la prueba. Los resultados de grupo son más significativos porque las diferencias individuales influyen menos en el resultado. Cuando se realiza una comparación con grupos amplios para comprobar la sensibilidad de uno o dos experimentadores, los datos proporcionados por éstos pueden resultar muy útiles. No deben realizarse comparaciones entre momentos y lugares distintos a menos que todas las condiciones de prueba se hayan estandarizado detalladamente y exista algún fundamento para comparar las intensidades observadas. 7. Referencias 1. AMERICAN SOCIETY FOR TESTING AND MATERIALS COMMITTEE E.-18. 1968. STP 433, Basic principles of sensory evatuation; STP 434, Manual on sensory lesting methods; STP 440, Correlation of subjective-objective methods in the study of odors and taste. ASTM, Filadelfia, Pennsylvania. 2-25 2. 3. 4. 5. 8. BAKER, R. A. 1962. Critical evaluation of olfactory measurement. J. Water Pollut. Control Fed. 34:582. BAKER, R. A. 1963. Odor testing laboratory. J. Water Pollut. Control Fed. 35:1396. Flavor Research and Food Acceptance. 1958. Reinhold Publishing Corp. Nueva York. ROSEN, A. A., J. B. PETER & F. M. MIDDLETON. 1962. Odor thresholds of mixed organic chemicals. J. Water Pollut. Control Fed. 34:7. Bibliografía HULBERT, R. & D. FEBEN. 1941. Studies on accuracy of threshold odor value. J. Amer. Water Works Assoc. 33:1945. SPAULDING, C. H. 1942. Accuracy and application of threshold odor test. J. Amer. Water Works Assoc. 34:877. THOMAS, H. A.. JR. 1943. Calculation of threshold odor. J. Amer. Water Works Assoc. 35:751. CARTWRIGHT, L. C, C. T. SNELL & P. H. KELLY. 1952. Organoleptic panel testing as a research tool. Anal. Chem. 24:503. LAUGHLIN, H. F. 1954. Palatable level with the threshold odor test. Taste Odor Control J. 20: Núm. 8 (agosto). SCHKLLENBERGER, R. D. 1958. Procedures for determining threshold odor concentrations in aqueous Solutions. Taste Odor Control J. 24: Núm. 5 (mayo). LAUGHLIN, H. F. 1962. Influence of temperature in threshold odor evaluation. Taste Odor Control J. 28: Núm. 10 (octubre). The threshold odor test. 1963. Taste Odor Control J. 29: Núms. 6, 7, 8 (junio, julio, agosto). SUFFET, I. H. & S. SEGALL. 1971. Detecting taste and odor in drinking water. J. Amer. Water Works Assoc. 63:605. S TAHL , W. H., ed. 1973. Compilation of Odor and Taste Threshcld Values Data. Amer. Soc. Testing & Materials Data Ser. DS 48, Filadelfia, Pennsylvania. AMERICAN SOCIETY FOR TESTING AND MATE RIALS , 1973. Annual Book of ASTM Standards. Part 23, D-1292-65, ASTM, Filadelfia, Pennsylvania. 2-26 MÉTODOS NORMALIZADOS 2160 2160 A. 1. GUSTO* Introducción Discusión general El gusto define solamente las sensaciones gustativas que se designan como amargas, saladas, acidas y dulces, resultantes de la estimulación química de las terminaciones nerviosas sensitivas de las papilas de la lengua y del paladar blando. El sabor abarca un complejo de sensaciones olfativas, gustativas y táctiles, originadas por el estímulo de terminaciones nerviosas situadas en la lengua y en las cavidades nasal y bucal1. Las muestras de agua depositadas en la boca para hacer un análisis sensorial siempre producen un sabor, aunque en él puede predominar el gusto, el olor o la sensación bucal, dependiendo del estímulo químico. Los métodos de análisis sensorial que se tratarán a continuación requieren la introducción de la muestra en la boca; es decir, dicha muestra debe someterse al sentido del gusto, pero técnicamente el análisis sensorial exige la evaluación de la sensación compleja llamada sabor. En el sentido que aquí se le da, gusto se refiere a un método de análisis sensorial en el que las muestras son sometidas a valoración bucal, pero las evaluaciones resultantes pertenecen al sabor. Para valoraciones sensoriales de muestras de agua valoradas bucalmente se han desarrollado tres métodos: prueba de umbral de sabor (PUS), evaluación de índice de sabor (EIS) y análisis del perfil de sabor (APS). La PUS es el más antiguo; se ha utilizado con mucha frecuencia y resulta especialmente útil para determinar si el sabor global de una muestra de agua es distinto al de un patrón bien definido. La EIS es particularmente valiosa para determinar si una muestra * Aprobado por el Standard Methods Committee, 1988. de agua es aceptable para el consumo diario3, y el APS sirve, sobre todo, para identificar y caracterizar los distintos sabores específicos de una muestra de agua4. El APS es un método nuevo, pero aún no ha sido estandarizado4. Las pruebas de sabor solamente se realizarán con muestras inocuas para la ingestión, descartándose las que puedan estar contaminadas por bacterias, virus, parásitos o sustancias químicas peligrosas, las que contengan agentes declorantes, como el arsenito de sodio, o las que procedan de una fuente desconocida. No deben llevarse a cabo pruebas con aguas residuales o fluidos similares no tratados. Obsérvense todas las precauciones sanitarias referidas al instrumental y los recipientes que vayan a entrar en contacto con la muestra. Lávense y esterilícense convenientemente los utensilios antes de su uso. Los análisis se realizarán en un laboratorio libre de olor de fondo que pueda interferir y, si es posible, se dispondrá de aire inodoro filtrado por carbón a temperatura y humedad constantes. Utilícese el método descrito en la sección 2150 referido al agua inodora e insípida que se emplea para preparar agua de dilución y muestras de referencia. 2. 1. 2. 3. 4. Referencias G ELDARD , F. A. 1972. The Human, Senses. John Wiley & Sons, Nueva York. BAKER, R. A. 1961. Taste and Odor in Water: A Critical Review. Manufacturing Chemists' Assoc, Washington, D. C. BRUVOLD, W. H. 1968. Scales for rating the taste of water. J. Appl. Psicol. 52:245. MALLEVIALE , J, & I. H. S UFFET , eds. 1987. The identification and Treatment of Tastes and Odors in Drinking Water. American Water Works Association Research Foundation, Denver, Colorado. 2-27 PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2160 B. 1. Prueba de umbral de sabor (PUS) Discusión general TABLA 2160:1. NÚMEROS DE UMBRAL DE SABOR CORRESPONDIENTES A VARIAS DILUCIONES La PUS se utiliza para medir cuantitativamente el sabor detectable. Más exactamente, se emplea para comparar de manera objetiva el sabor de la muestra con el del agua de referencia especificada como diluyente. El número de umbral de sabor (ÑUS) es el valor máximo de dilución de muestra con agua de referencia que obtiene una diferencia significativamente perceptible. El NUS se calcula como sigue: donde: A = volumen de la muestra, ml, y B = volumen de agua de referencia (diluyente), ml. La tabla 2160:1 expresa los ÑUS correspondientes a varias soluciones. 2. Procedimiento a) Selección del grupo de experimen- tación: A partir de ensayos preliminares se selecciona cuidadosamente a las personas interesadas en realizar las pruebas de sabor. Quedarán excluidos los sujetos poco sensibles y los que padezcan alergias o resfriados. Es conveniente que los probadores se familiaricen con el método antes de participar en la prueba, pero no deben preparar muestras ni conocer las concentraciones de dilución que van a evaluarse. Para un resultado eficaz, manéjese un grupo de cinco o más observadores. b) Caracterización del gusto: Cada uno de los experimentadores describe el sabor característico de la muestra más concentrada, y a continuación se registra la posible coincidencia de apreciaciones. El valor de la caracterización aumenta a medida que los experimentadores adquieren más experiencia con una categoría determinada de sabor, como clorofenólico, herbáceo o mohoso. c) Prueba preliminar: Para determinar el intervalo aproximado del ÑUS, añádanse porciones de muestra de 200, 50, 12 y 4 ml a los volúmenes de agua de referencia (véase sección 2150) señalados en la tabla 2160:1, en vasos de precipitados hasta un total de 200 ml en cada vaso, y mézclese suavemente con un agitador limpio. Sepárese un vaso que contenga sólo agua de referencia para comparar. La temperatura de la muestra durante la prueba debe mantenerse dentro de un margen de 1 °C de la temperatura especificada. Preséntense las muestras a cada experimentador de manera uniforme, ofreciendo primero el agua de referencia y a continuación la de la muestra más diluida. Si se detecta algún sabor en esta dilución, prepárese una muestra intermedia diluyendo 20 ml de muestra en 200 ml de agua de referencia. Utilícese 2-28 esta dilución para determinar el valor umbral y multiplíquese el NUS obtenido por 10 para corregir la dilución intermedia. En varios casos se requerirá una dilución intermedia mayor. Si no se detecta ningún sabor en la muestra más diluida, repítase el ensayo con la concentración siguiente, continuando el proceso hasta que se note claramente algún sabor. d) Determinación del ÑUS: En función de los resultados obtenidos en la prueba preliminar, prepárese una batería de diluciones utilizando como modelo la tabla 2160:11. Prepárense las siete diluciones mostradas en la línea correspondiente. Esta ordenación es necesaria para descubrir el margen de sensibilidades de todos los experimentadores del panel. Si la muestra sometida a prueba requiere una dilución mayor de las propuestas en la tabla, realícense diluciones intermedias según se ha indicado en el anterior punto c. Utilícese para cada dilución y muestra de referencia un vaso limpio de precipitados de 50 ml, lleno hasta el nivel de 25 mi, o bien un vaso alto. No debe utilizarse material de vidrio empleado en pruebas sensoriales para otros análisis. Entre las distintas pruebas, limpiar bien los recipientes en un lavavajillas automático con agua a no menos de 60 °C. Manténganse las muestras a 15 i 1 °C. No obstante, si la temperatura del agua del sistema de distribución es mayor de 15 °C, selecciónese una temperatura adecuada, especificando el valor en el informe de resultados. Preséntese a cada experimentador la serie de muestras en orden de concentración creciente, emparejando cada muestra con una referencia conocida. El experimentador saborea un volumen de muestra que resulte cómodo, moviéndolo en la boca durante varios segundos y expulsándolo sin deglutirlo. Deberá comparar la muestra con la referencia, indicando si detecta algún sabor o regusto. MÉTODOS NORMALIZADOS Inclúyanse en la serie dos o más blancos de referencia próximos al umbral esperado, pero evítese la repetición del modelo. El experimentador no debe saber cuál de las muestras tiene sabor y cuál es un blanco. Se le proporcionarán las instrucciones necesarias para que deguste cada muestra según una secuencia, empezando por la menos concentrada, hasta que el sabor se detecte con certeza. TABLA 2160:11. DILUCIONES PARA DETERMINACIÓN DEL ÑUS Recójanse las observaciones indicando cada uno de los vasos de prueba en los que se detectó sabor. Por ejemplo: donde: – significa respuesta negativa, y + respuesta positiva. 3. Cálculo El número de umbral de sabor es el índice de dilución en el que aquél empieza a percibirse. En el ejemplo anterior, el primer sabor detectable apareció cuando 25 ml de muestra se diluyeron en 200 ml: el número de umbral es 8 (tabla 2160:1). Los blancos de referencia no influyen en el cálculo del umbral. 2-29 PROPIEDADES FÍSICAS Y DE AGREGACIÓN El NUS más bajo que puede observarse es 1, cuando el vaso contiene 200 ml de muestra no diluida. Si a esta concentración no se detecta sabor, indíquese «No observado sabor», en vez de un número de umbral. A veces aparecen respuestas anómalas; una concentración baja puede denominarse positiva, y una más alta de la serie puede llamarse negativa. En estos casos, desígnese el umbral como el punto después del cual no aparecen anomalías. A continuación se ilustra una aproximación a una serie anómala (se excluyen las respuestas a los blancos de referencia): Calcúlense la media y las desviaciones estándar de todos los NUS si la distribución es razonablemente simétrica; en otro caso, se expresará el umbral de un grupo como la media o media geométrica de los umbrales individuales. 4. Interpretación de los resultados Un NUS no es un valor exacto. En el caso de un solo observador representa un juicio en el momento de la prueba. Los resultados obtenidos de un grupo son más significativos debido a que las diferencias individuales tienen menos influencia sobre los resultados de la prueba. Uno o dos experimentadores pueden proporcionar datos útiles si se ha establecido una comparación con grupos más amplios para comprobar su sensibilidad. No deben compararse datos de distintos momentos o lugares, a no ser que se hayan estandarizado cuidadosamente las condiciones de prueba y se disponga de algún fundamento para la comparación de los NUS observados. 5. Bibliografía HULBERT, R. & D. FEBEN. 1941. Studies on accuracy of threshold odor value. J. Amer. Water Works Assoc. 33:1945. SPAULDINO, C. H. 1942. Accuracy and application of threshold odor test. J. Amer. Water Works Assoc. 34:877. THOMAS, H. A. 1943. Calculation of threshold odor, J. Amer. Water Works Assoc. 35:751. Cox, G. J. & J. W. N ATHANS . 1952. A study of the taste of fluoridated water. J. Amer. Water Works Assoc. 44:940. LAUGHLIN, H. F. 1954. Palatable level with the threshold odor test. Taste Odor Control J. 20:1. Cox, G. J., J. W. NATHANS & N. VONAU. 1955. Subthreshold-to-taste thresholds of sodium, potassium, calcium and magnesium ions in water. J. Appl. Physiol. 8:283. LOCKHART, E. E., C. L. TUCKER & M. C. MERRITT, 1955. The effect of water impurities on the flavor of brewed coffee. Food Res. 20:598. CAMPBELL, C. L., R. K. DAWES, S. DEOLALKAR & M. C. MERRITT. 1958. Effects of certain chemicals in water on the flavor of brewed coffee. Food Res. 23:575. MIDDLETON, F. M., A. A. ROSEN & R. H. BRUTTSCHELL . 1958. Taste and odor research tools for water Utilities. J. Amer. Water Works Assoc 50:231. SHELLENBERGER, R. D. 1958. Procedures for determining threshold odor concentrations in aqueous solutions. Taste Odor Control J. 24:1. COHEN, J. N., L. J. KAMPHAKE, E. K. HARRIS & R. L. WOODWARD. 1960. Taste threshold concentrations of metals in drinking water. J. Amer. Water Works Assoc. 52:660. BAKER, R. A. 1961. Problems of tastes and odors. J. Water Pollut. Control Fed. 33:1099. ROSEN, A. A., J. B. PETER & F. M. MIDDLETON. 1962. Odor thresholds of mixed organic chemicals. J. Water Pollut. Control Fed. 34:7. BAKER, R. A. 1962. Critical evaluation of olfactory measurement. J. Water Pollut. Control Fed. 34:582. BAKER, R. A. 1963. Threshold odors of organic chemicals. J. Amer. Water Works Assoc. 55:913. B AKER , R. A. 1963. Odor testing laboratory. J. Water Pollut. Control Fed. 35:1396. BAKER, R. A. 1963. Odor effects of aqueous mixtures of organic chemicals. J. Water Pollut. Control Fed. 35:728. MÉTODOS NORMALIZADOS 2-30 ROSEN, A. A., R. T. SKEEL & M. B. ETTINGER. 1963. Relationship of river water odor to specific organic contaminants. J. Water Pollut. Control Fed. 35:777. 2160 C. BRYAN, P. E., L. N. K UZMINSKI , F. M. S AWYER & T. H. FENO. 1973. Taste thresholds of halogens in water. J. Amer. Water Works Assoc. 65:363. Evaluación de índice de sabor (EIS) 1. Discusión general 3. Cuando la finalidad de la prueba sea evaluar la aceptabilidad para el consumo diario, utilícese la evaluación de índice de sabor que se describe a continuación. Este método se ha empleado con muestras de fuentes públicas en investigaciones de laboratorio y estudios de consumo a fin de recomendar estándares para la regulación del contenido mineral del agua potable. Se ofrece a cada experimentador una lista de nueve respuestas sobre el agua, en una escala que va desde muy favorable a muy desfavorable. La tarea del experimentador consiste en seleccionar la respuesta que mejor exprese su opinión. El índice individual es el número de escala de la respuesta seleccionada. El índice del grupo sobre una muestra dada es la medida adecuada de la tendencia principal de los números de escala de todos los experimentadores con respecto a esa muestra. a) Selección y preparación del grupo: A los experimentadores se les proporciona instrucciones completas y sesiones de ensayo y orientación seguidas de preguntas y discusión de los métodos. En las muestras de degustación, los experimentadores actúan por sí mismos. Selecciónese un grupo a partir de los resultados de las sesiones de ensayo. No se permitirá que los observadores conozcan la composición o la fuente de las muestras específicas. b) Prueba de determinación del índice: Para evaluar hasta 10 muestras puede realizarse una sola sesión de determinación del índice, incluyendo las muestras convencionales mencionadas anteriormente en el apartado 2. Concédase un descanso de al menos treinta minutos entre las distintas sesiones de determinación del índice. En cuanto a necesidades de material de vidrio, véase apartado B2d. Preséntense las muestras a la temperatura que según los observadores resulte agradable para beber agua, manteniendo dicha temperatura a lo largo de todo el ensayo. Se recomienda un nivel de 15 °C, pero, en cualquier caso, la prueba rio debe realizarse a temperaturas superiores a las que son habituales en el agua corriente. Esta temperatura se especificará en el informe de resultados. El orden de las muestras se dispone al azar para cada experimentador. De- 2. Muestras Se emplearán muestras de agua lista para consumo humano o de agua tratada experimentalmente, siempre que satisfagan completamente los requerimientos sanitarios expresados en la sección 2160A.1. Utilícese agua inodora e insípida según se describe en la sección 2150, y una solución de 2.000 mg de NaCl/1, preparada con agua inodora e insípida, como muestra convencional. Procedimiento PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-31 ben proporcionarse instrucciones para cumplir los siguientes pasos: 1) degustar aproximadamente la mitad de la muestra manteniendo el agua en la boca durante unos segundos y expulsándola sin tragar; 2) formar un juicio inicial sobre la escala de determinación de índice; 3) realizar de forma similar una segunda degustación; 4) hacer una determinación final y registrar el resultado en el cuestionario adecuado; 5) enjuagar la boca con el agua de referencia; 6) descansar un minuto antes de repetir los pasos 1-5 con la muestra siguiente. c) Caracterización: Si se requiere también la caracterización del sabor, realícese una sesión de determinación final del índice en la que cada experimentador describa el sabor de cada muestra probada. (Véase apartado B2b.) 6) Creo que no aceptaría este agua para beber a diario. 7) No aceptaría este agua para beber a diario. 8) No bebería nunca este agua. 9) No soporto este agua en la boca y no la bebería jamás. 4. Cálculo Para la determinación del índice, utilícese la siguiente escala. Regístrense las determinaciones de índice como números enteros entre el uno y el nueve, dando al uno la calidad de determinación más elevada. Calcúlese la media y la desviación estándar de todas las determinaciones si la distribución es razonablemente simétrica; de otro modo, indíquese la determinación más típica de un grupo como la media o media geométrica de las determinaciones indicadas. Escala de tendencia de acción: 1) Me agradaría mucho aceptar este agua para beber a diario. 2) Me agradaría aceptar este agua para beber a diario. 3) Estoy seguro de que aceptaría este agua para beber a diario. 4) Aceptaría este agua para beber a diario. 5) Quizá aceptaría este agua para beber a diario. 5. Interpretación de los resultados Los valores que representan la tendencia principal y la dispersión de las determinaciones de calidad en un grupo de laboratorio sólo constituyen aproximaciones de esos valores para una población de consumo definido. 6. Bibliografía BRUVOLD, W. H. & R. M. PANGBORN. 1966. Rated acceptability of mineral taste in water. J. Appl. Psychol. 50:22. BRUVOLD, W. H., H. J. ONGERTH & R. C. DILLEHAY. 1967. Consumer attitudes toward mineral taste in domestic water. J. Amer. Water Works Assoc. 59:547. DILLEHAY, R. C, W. H. BRUVOLD & J. P. SIEGEL. 1967. On the assessment of potability. J. Appl. Psychol. 51:89. BRUVOLD. W. H. 1968. Mineral Taste in Domestic Water. Univ. California Water Resources Center, Los Ángeles. BRUVOLD, W. H. & H. J. ONGERTH. 1969. Taste quality of mineralized water. J. Amer. Water Works Assoc. 61:170. BRUVOLD, W. H. & W. R. GAFFEY. 1969. Rated acceptability of mineral taste in water. II: Combinatorial effects of ions on quality and action tendency ratings. J. Appl. Psychoi 53:317. DILLEHAY, R. C, W. H. BRUVOLD & J. P. SIEGEL. 1969. Attitude, object label, and stimulus factors in response to an attitude object. J. Personal. Social Psychol 11:220. BRUVOLD, W. H. 1970. Laboratory panel estimation of consumer assessments of taste and flavor. J. Appl. Psychol 54:326. PANGBORN, R. M., I. M. TRABUE & R. C. BALDWIN. 1970. Sensory examination of mineralized, chlorinated waters. J. Ammer. Water Works Assoc. 62:572 2-32 MÉTODOS NORMALIZADOS PANGBORN, R. M., I. M. TRABUE & A. C. LITTLE. 1971. Analysis of coffee, tea and artifically flavored drinks prepared from mineralized waters. J. Food Sci. 36:355. B RUVOLD , W. H. & P. C. W ARD 1971. Consumer assessment of water quality and the cost of improvements. J. Amer. Water Works Assoc. 63:3 PANGBORN. R. M. & L. L. BERTOLERO. 1972. Influence of temperature on taste intensity and 2160 D. 1. Análisis del perfil de sabor (APS) Discusión general El análisis del perfil de sabor se emplea para identificar y caracterizar sabores desagradables en el ámbito de un conjunto de datos sobre calidades sensoriales que puedan considerarse aceptables. Los experimentadores serán seleccionados y adiestrados cuidadosamente para la ejecución de este método, que puede resultar especialmente útil para instalaciones de agua. Es un método publicado1. 2. Referencias 1. MALLEVIALE , J. & I. H. S UFKET , eds. 1987. The Identification and Treatment of Tastes and Odors in Drinking Water. American Water Works Association Research Foundation, Denver, Colorado. 3. degree of liking of drinking water. .J. Amer. Water Works Assoc. 64:511. B RUVOLD , W. H. 1975. Human perception and evaluation of water quality. Crit. Rev. Environ. Control 5:153. BRUVOLD, W. H. 1976. Consumer Evaluation of the Cost and Quality of Domestic Water. Univ. California Water Resources Center, Davis. Bibliografía SUFFET, I. H. & S. SEGALL. 1971. Detecting taste and odor in drinking water. J. Amer. Water Works Assoc. 63:605. P ERSSON . P. E. 1982. Muddy odour: a problem associated with extreme eutrophication. Hydrobiologia 86:161. MEILGAARD, M. C, D. S. REID & K. A. WYBORSKI . 1982. Reference standards for beer flavor terminology system. Amer. Soc. Brewing Chemists. J. 40:119. DOTY, R. L., P. SHAMAN, S. L. APPLEBAUM, R. GlBERSON, L. SIKSORSKI & L. ROSENBERG. 1984. Smell identification ability: Changes with age. Science 226:1441. KRASNER, S. W., M. J. MCGUIRE & V. B. FERGUSON . 1985. Tastes and odors: The flavor profile method. J. Amer. Water Works Assoc. 77:34. ANSELME, C, K. N’GUYEN. A. BRUCHET & J. MALLEVIALLE. 1985. Can polyethylene pipes impart odors in drinking water? Environ. Technol. Letters 6:477. AMOORE, J. E. 1986. The chemistry and physiology of odor sensitivity. J. Amer. Water Works Assoc. 78:70. BARTELS, J. H. M., G. A. BURLINGAME & I. H. S UFFET . 1986. Flavor profile analysis: Taste and odor control of the future. J. Amer. Water Works Assoc. 78:50. B URLINGAME , G. A., R. M. D ANN & B. L. BROCK. 1986. A case study of geosmin in Philadelphia's water. J. Amer. Water Works Assoc. 78:56. KRASNER, S. W. & E. G. MEANS. 1986. Returning recently covered reservoirs to service: Health and aesthetic considerations. J. Amer. Water Works Assoc. 78:94. MEANS, E. G. & M. J. MCGUIRE. 1986. An early warning system for taste and odor control. J. Amer. Water Works Assoc. 78:77. BARTELS. J. H. M., B. M. BRADY & I. H. SUFFET. 1987. Training panelists for the flavor profile analysis method. J. Amer. Water Works Assoc. 78:50. PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-33 2310 ACIDEZ* 2310 A. Introducción La acidez de un agua es su capacidad cuantitativa para reaccionar con una base fuerte hasta un pH designado. El valor medio puede variar significativamente con el pH final utilizado en la determinación. La acidez constituye la medida de una propiedad sobreañadida del agua y puede interpretarse en términos de sustancias específicas solamente cuando se conoce la composición química de * Aprobado por el Standard Methods Committee, 1985. 2310 B. 1. la muestra. Con arreglo al método de determinación, los ácidos minerales fuertes, los ácidos débiles, como el carbónico y el acético, y las sales hidrolizables, como los sulfatos de hierro y aluminio, pueden incrementar la acidez determinada. Los ácidos incrementan también la corrosividad e interfieren los índices de reactividad química, su especificación y los procesos biológicos. La medida también refleja las variaciones de la calidad de la fuente del agua. Método de titulación Discusión general a) Principio: Los hidrogeniones, presentes en una muestra como resultado de la disociación o hidrólisis de los solutos, reaccionan a la adición de un alcohol estándar. Así pues, la acidez depende del pH o indicador finales que se empleen. La confección de una curva de titulación mediante el registro del pH de las muestras, tras la adición sucesiva de pequeñas cantidades medidas de titulante, permite la identificación de los puntos de inflexión y la capacidad tampón y, en su caso, determinar la acidez con respecto a un pH de interés. En la titulación de una especie acida simple, como es la de estandarización de reactivos, el punto final más exacto se obtiene a partir del punto de inflexión de una curva de titulación. Este punto de inflexión es el pH al que la curva varía de convexa a cóncava o viceversa. Como la identificación exacta de los puntos de inflexión puede ser difícil o imposible en mezclas tamponadas o complejas, la titulación en tales casos se conduce a un pH terminal arbitrario basado en consideraciones prácticas. Para las titulaciones de control habituales o estimaciones preliminares rápidas de acidez puede utilizarse como punto final el cambio de color de un indicador. Las muestras de aguas residuales industriales, drenaje de ácidos minerales u otras soluciones que contienen cantidades apreciables de iones metálicos hidrolizables, como hierro, aluminio y manganeso, se tratan con peróxido de hidrógeno para garantizar la oxidación de cualquier forma reducida de cationes polivalentes, y se hierven para hidrólisis acelerada. Las 2-34 valoraciones de acidez pueden ser muy variables si no se ejecuta el método con exactitud. b) Puntos finales: De una forma ideal, el punto final de la titulación de la acidez corresponde al punto de equivalencia estequiométrica para la neutralización de los ácidos presentes. El pH en el punto equivalente dependerá de la muestra, la elección entre múltiples puntos de inflexión y el uso previsto de los datos. El dióxido de carbono (CO2) disuelto suele ser el principal componente ácido de las aguas de superficie no contaminadas; las muestras procedentes de estas fuentes deben mejorarse con cuidado a fin de reducir al mínimo la pérdida de gases disueltos. En una muestra que contenga solamente CO2-bicarbonatoscarbonatos, la titulación a pH 8,3 a 25 °C corresponde a la neutralización estequiométrica del ácido carbónico a carbonato. Como el cambio de color del indicador de fenolftaleína está próximo al pH 8,3, este valor se acepta en general como un punto final estándar para la titulación de la acidez total, incluyendo el CO2 y ácidos más débiles. El púrpura de metacresol también tiene un punto final a pH 8,3 y proporciona un cambio de color más llamativo. Para mezclas más complejas o soluciones tamponadas, la selección de un punto de inflexión puede ser subjetiva. Por consiguiente, para determinaciones de acidez estándar mediante una titulación potenciométrica de aguas naturales y residuales, en las que no puede aceptarse el simple equilibrio carbonatado que se comentó anteriormente, utilícense puntos finales de pH 3,7 y 8,3. El azul bromofenol presenta un cambio de color muy vivo en su punto final 3,7. Las titulaciones resultantes se identifican tradicionalmente como «acidez naranja metilo» (pH 3,7) y «fenolftaleína» o acidez total (pH 8,3) independientemente del método real de medida. MÉTODOS NORMALIZADOS c) Interferencias: Durante la toma de muestras, la conservación o la titulación, pueden perderse o ganarse gases disueltos que contribuyen a la acidez o la alcalinidad, como CO2, sulfuro de hidrógeno o amoníaco. Redúzcanse al mínimo estos efectos mediante titulación al punto final inmediatamente después de abrir el recipiente de muestra, sin agitarlo o mezclarlo enérgicamente, protegiendo la muestra de la atmósfera durante la titulación y manteniéndola no más caliente de lo que estaba durante la conservación. En la titulación potenciométrica, las materias oleosas, los sólidos suspendidos, los precipitados u otros materiales de desecho pueden cubrir el electrodo de vidrio y producir una respuesta lenta. Es probable que aparezcan dificultades de este tipo en una curva de titulación errática. No deben eliminarse las interferencias de la muestra, pues pueden contribuir a su acidez. Hágase una breve pausa entre las adiciones de titulación para que el electrodo se equilibre, o límpiese éste de vez en cuando. En las muestras que contienen iones oxidables o hidrolizables, como hierro ferroso o férrico, aluminio y manganeso, los índices de reacción a temperatura ambiente pueden ser suficientemente lentos como para causar una desviación de los puntos finales. No deben utilizarse titulaciones con indicador de muestras turbias que pueden oscurecer el cambio de color en el punto final. El cloro libre residual de la muestra suele blanquear el indicador. Elimínese esta fuente de interferencia añadiendo una gota de tiosulfato sódico (Na2S2O3) 0,1M. d) Selección del método: Determínese la acidez de la muestra en función del volumen de álcali estándar requerido para titular una porción a un pH de 8,3 (acidez de fenolftaleína) o de 3,7 (acidez naranja metilo de aguas residuales o muy polucionadas). Titúlese a temperatura ambiente utilizando un medidor de pH 2-35 PROPIEDADES FÍSICAS Y DE AGREGACIÓN convenientemente calibrado, un titulador operado eléctricamente o indicadores de color. Empléese el método de peróxido caliente (apartado 4a) para pretratar las muestras conocidas o que puedan contener iones metálicos hidrolizables o formas reducidas de cationes polivalentes, como líquidos de decapado del hierro, drenaje de ácidos y otros residuos industriales. Los indicadores de color pueden emplearse como método habitual y para titulaciones de control en ausencia de color y turbidez interferentes, y para titulaciones preliminares para seleccionar el tamaño de la muestra y la potencia del titulante (apartado 4b). e) Tamaño de la muestra: La escala de cifras de acidez encontrada en aguas residuales es tan amplia que no puede especificarse un tamaño simple de muestra ni la normalidad de base utilizada como reactivo de valoración. Utilícese un volumen de reactivo suficiente (20 ml o más de una bureta de 50 ml) para obtener una precisión volumétrica relativamente buena, en tanto se mantiene un volumen de muestra suficientemente pequeño como para permitir puntos terminales netos. Para muestras con cifras de acidez menores de 1.000 mg/1, aproximadamente, de carbono cálcico (CaCO3), selecciónese un volumen con acidez equivalente menor de 50 mg de CaCO3, y realícese una titulación con hidróxido sódico (NaOH) 0,02N . Para cifras de acidez mayores, utilícese una porción con una acidez equivalente a menos de 250 mg de CaCO3 y realícese una titulación con NaOH 0,1 N. Si es necesario, realícese una titulación preliminar para determinar el tamaño óptimo de la muestra y/o la normalidad del titulante. f) Toma de muestras y conservación: Recójanse las muestras en botellas de polietileno o vidrio borosilicato y consérvense a baja temperatura. Llénense las botellas por completo y tápense herméti- camente. Dado que las muestras residuales pueden estar sujetas a la acción microbiana y a pérdidas o ganancias de CO2 u otros gases cuando se exponen al aire, las muestras deben analizarse sin demora, preferiblemente el primer día. Si se sospecha la presencia de alguna actividad biológica, analícense dentro de las seis primeras horas. Evítese la agitación de la muestra y su exposición prolongada al aire. 2. Instrumental a) Titulador electrométrico: Utilícese cualquier medidor de pH disponible en el mercado o un titulador eléctrico provisto de un electrodo de cristal y que pueda ser leído hasta unidades de pH 0,05. Estandarícese y equilíbrese con arreglo a las instrucciones del fabricante. Debe prestarse una especial atención a la compensación de la temperatura y al cuidado del electrodo. Si la temperatura no se compensa de forma automática, titúlese a 25 ± 5°C. b) Vaso de titulación: Su tamaño y forma dependerán de los electrodos y del tamaño de la muestra. El espacio que queda libre sobre la muestra debe ser lo más reducido posible, dejando sitio para el reactivo y la inmersión completa de la porción indicadora de los electrodos. Para electrodos de tamaño convencional, llénese un vaso Berzelius sin ranuras, de tipo alto y con una capacidad de 200 ml. Colóquese un tapón con tres orificios, para ajustar los dos electrodos y la bureta. Con un electrodo miniatura de combinación vidrio-referencia, empléese un erlenmeyer de 125 ó 250 ml con un tapón de dos orificios. c) Agitador magnético. d) Pipetas volumétricas. e) Matraces volumétricos, 1.000, 200 y 100 ml. f) Buretas, cristal borosilicato, 50, 25 y 10 ml. g) Botella de poliolefina, 1 l. 2-36 3. Reactivos a) Dióxido de carbono anhidro: Prepárense todas las soluciones de reserva y estándar y el agua de dilución para el método de titulación con agua destilada o desionizada, hervida durante 15 minutos y enfriada a temperatura ambiente. El pH final del agua deberá ser > 6,0 y su conductividad < 2 μmhos/cm. b) Solución de ftalato ácido de potasio, aproximadamente 0,05N: Tritúrense entre 15 y 20 g de estándar primario de KHC8H 4O 4 hasta una malla de 100 y séquese a 120 °C durante dos horas. Enfríese en un desecador. Transfiérase un peso de 10,0 + 0,5 g (miligrámico) a un matraz volumétrico de 1 l, y dilúyase hasta 1.000 ml. c) Reactivo de hidróxido sódico estándar, 0,1 N: Prepárese la solución aproximadamente 0,1 N como se indica en Preparaciones de reactivos de mesa (véase cubierta interior). Estandarícense, mediante titulación, 40,0 ml de solución de KHC8H4O4 (3b), usando una bureta de 25 ml. Titúlese hasta el punto de inflexión (apartado la) que debe estar próximo a un pH 8,7. Calcúlese la normalidad del NaOH: donde: A = g de KHC 8 H 4 O 4 , pesados en matraz de 1 l B = ml de KHC 8 H 4 O 4 , solución tomada para titulación, y C = ml de NaOH, solución utilizada. Empléese la normalidad medida en cálculos posteriores o ajústese a 0,1000N; 1 ml = 5,0 mg CaCO3. d) Reactivo de hidróxido sódico estándar, 0,02N: Dilúyanse 200 ml de NaOH 0,1 N hasta 1.000 ml y consérvense en una botella de poliolefina, protegido del CO2 atmosférico por un tubo de cal sodada o MÉTODOS NORMALIZADOS un cierre hermético. Estandarícese frente a KHC8H4O4 como se indica en el apartado 3c, utilizando 15,0 ml de solución del ftalato y una bureta de 50 ml. Calcúlese la normalidad como se indicó anteriormente (apartado 3c); 1 ml = 1,00 mg CaCO3. e) Peróxido de hidrógeno, H2O2, 30 por 100. f) Solución indicadora de azul de bromofenol, indicador de pH 3,7: Dilúyanse 100 mg de azul de bromofenol, sal sódica, en 100 ml de agua. g) Solución indicadora de púrpura de metacresol, indicador de pH 8,3: Diluyanse 100 mg de púrpura de metacresol en 100 ml de agua. h) Solución alcohólica indicadora de fenolftaleína, indicador de pH 8,3. i) Tiosulfato sódico, 0,lM: Dilúyanse 25 g de NaS2O3-5H2O y disuélvanse hasta 1.000 ml en agua destilada. 4. Procedimiento Si la muestra está limpia de iones metálicos hidrolizables y de formas reducidas de cationes polivalentes, procédase al análisis de acuerdo con b, c o d. Si se sabe o se sospecha que la muestra contiene tales sustancias, sométase a tratamiento previo según se describe en el punto a. a) Tratamiento con peróxido caliente: Pipetéese una muestra idónea (véase apartado lc) en matraces de titulación. Mídase el pH. Si está alrededor de 4, añádanse incrementos de 5 ml de ácido sulfúrico (H2SO4) 0,02N (sección 2320B, 3c) para reducir el pH hasta 4 o menos. Elimínense los electrodos. Añádanse cinco gotas de H2O2 al 5 por 100 y hiérvase de dos a cinco minutos. Enfríese a temperatura ambiente y titúlese con álcali estándar hasta pH 8,3 con arreglo al procedimiento de 4d. b) Cambio de color: Selecciónese el tamaño y la normalidad de la muestra, titulando según los criterios indicados en PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-37 el apartado 1e. Ajústese la muestra a la temperatura ambiente si es necesario, y vacíese con pipeta en un erlenmeyer, manteniendo la punta de la pipeta cerca del fondo del matraz. Si existe cloro residual libre, añádanse 0,05 ml (una gota) de solución de Na2S2O3 0,1M, o destrúyase mediante la aplicación de rayos ultravioleta. Añádanse 0,2 ml (cinco gotas) de solución indicadora y titúlese sobre una superficie blanca hasta conseguir un cambio de color persistente, característico del punto equivalente. Pueden emplearse las soluciones o los sólidos indicadores que se encuentran disponibles en el mercado diseñados para el margen adecuado de pH (3,7 u 8,3). Investíguese el color en el punto final mediante adición de la misma cantidad del indicador utilizado con la muestra a una solución tampón al pH designado. alcanzó el equilibrio entre las adiciones sucesivas de álcali. Determínese a partir de la curva la acidez relativa hasta un pH determinado. d) Titulación potenciométrica hasta pH 3,7 u 8,3: Prepárese conjuntamente la muestra y la titulación como se especifica en el apartado 4cl. Titúlese hasta el pH de punto final preseleccionado (apartado d) sin registrar valores intermedios. A medida que se aproxime el punto final, hay que reducir las adiciones de álcali y comprobar que se alcanza el equilibrio del pH antes de hacer la adición siguiente. c) Curva de titulación potenciométrica: 1) Los electrodos y el vaso de titulación se enjuagan con agua destilada y se drenan. Selecciónese el tamaño de la muestra y la normalidad del reactivo según los criterios expuestos en el apartado le. Si es necesario, ajústese la muestra a la temperatura ambiente y, con pipeta, viértase la muestra en el matraz, manteniendo la punta cerca del fondo de éste. 2) Mídase el pH de la muestra. Añádase álcali estándar en incrementos de 0,5 ml o menos, de forma que se produzca con cada incremento un cambio de menos de 0,2 unidades de pH. Después de cada adición, mézclese cuidadosa y suavemente con un agitador magnético. Evítense las salpicaduras. Regístrese el pH cuando se obtenga una lectura constante. Añádase más reactivo y mídase el pH hasta alcanzar 9. Confecciónese la curva de titulación mediante punteado de los valores de pH observados versus mililitros acumulados de reactivo añadido. Debe obtenerse una curva suave con una o más inflexiones. Una curva discontinua o irregular puede indicar que no se 5. Cálculo donde: A = ml utilizados de NaOH titulador, B = normalidad del NaOH, C = ml utilizados de H2SO4 (apartado 4d), y D = normalidad del H2SO4. Infórmese, como se indica a continuación, del pH de punto final empleado: «La acidez a pH ___ = ___ mg CaCO3/l». Si se obtiene un valor negativo, determínese la alcalinidad según se describe en la sección 2320. 6. Precisión y sesgo Respecto a la precisión, no puede hacerse ninguna afirmación general, dada la gran variedad de características que presentan las muestras. Es probable que la precisión de la titulación sea mayor que las aproximaciones propias de la toma y manipulación de las muestras antes del análisis. 2-38 MÉTODOS NORMALIZADOS Cuarenta analistas de 17 laboratorios analizaron muestras de agua sintética que contenia incrementos de bicarbonato equivalentes a 20 mg de CaCO3/l. La titulación, realizada según el método del apartado 4d, dio una desviación estándar de 1,8 mg del bicarbonato, con un sesgo irrelevante. Cinco laboratorios analizaron dos muestras con ácidos sulfúrico, acético y fórmico y cloruro de aluminio según los procedimientos descritos en los apartados Ab y Ad. La acidez media de una muestra (pH 3,7) fue 487 mg de CaCO3/l, con una desviación estándar de 11 mg/1. La titulación con azul bromofenol de la misma muestra fue mayor en 90 mg/1, con una desviación estándar de 110 mg/1. La otra muestra señaló una titulación potenciométrica de 547 mg/1, con desviación estándar de 54 mg/1, mientras el indicador correspondiente resultó ser 2320 85 mg/1 mayor, con desviación estándar de 56 mg/1. La diferencia esencial entre las muestras fue la sustitución de citrato amónico férrico, en la segunda muestra, por cloruro de aluminio. 7. Bibliografía WINTER, J. A. & M. R. MlDGETT. 1969. FWPCA Method Study 1. Mineral and Physical Analyses. Federal Water Pollution Control Admin., Washington, D. C. B ROWN , E., M. W. S KOUGSTAD & M. J. F ISH MAN. 1970. Methods for collection and analysis of water samples for dissolved minerals and gases. Chapter Al in Book 5, Techniques of Water-Resources Investigations of United States Geological Survey. U.S. Geological Survey, Washington, D.C. SNOEYINK, V. L. & D. JENKINY 1980. Water Chemistry. John Wiley & Sons, Nueva York. ALCALINIDAD* 2320 A. Introducción 1. Discusión general La alcalinidad de un agua es su capacidad para neutralizar ácidos y constituye la suma de todas las bases titulables. El valor medido puede variar significativamente con el pH de punto final utilizado. La alcalinidad es la medida de una propiedad agregada del agua, y solamente puede interpretarse en términos de sustancias específicas cuando se conoce la composición química de la muestra. La alcalinidad es importante en muchos usos y tratamientos de aguas na* Aprobado por el Standard Methods Committee, 1985. turales y residuales. La alcalinidad de muchas aguas de superficie depende primordialmente de su contenido en carbonatos, bicarbonatos e hidróxidos, por lo que suele tomarse como una indicación de la concentración de estos componentes. Los valores determinados pueden incluir también la contribución de boratos, fosfatos, silicatos y otras bases, cuando se hallen presentes. La alcalinidad por exceso de concentración de metales alcalinoférreos tiene importancia para la determinación de la aceptabilidad de un agua para irrigación. Las determinaciones de alcalinidad se utilizan en la interpretación y el control de los procesos de tratamiento de aguas limpias y residuales. Las PROPIEDADES FÍSICAS Y DE AGREGACIÓN aguas residuales domésticas tienen una alcalinidad menor (o sólo ligeramente mayor) que la del suministro. Los digestores anaerobios que actúan adecuadamente presentan alcalinidades sobrenadantes típicas con cifras de 2.000 a 4.000 mg de carbonato cálcico (CaCO3)/11. 2-39 2. Referencia 1. POHLAND, F. G. & D. E. BLOODGOOD. 1963. Laboratory studies on mesophilic and thermophilic anaerobic sludge digestion. J. Water Pollut. Control Fed. 35:11. 2320B. Método de titulación 1. Discusión general a) Principio: Los iones hidróxilo presentes en una muestra como resultado de la disociación o hidrólisis de los solutos reaccionan con las adiciones de ácido estándar. Por tanto, la alcalinidad depende del pH de punto final utilizado. Para conocer los métodos de determinación de puntos de inflexión a partir de curvas de titulación y las normas para titulación a puntos finales de pH fijados, véase sección 2310B.la. Para muestras de alcalinidad baja (menos de 20 mg de CaCO3/l), utilícese una técnica de extrapolación basada en la proporcionalidad cercana de la concentración de hidrogeniones y el exceso de reactivo más allá del punto de equivalencia. Se mide con precisión la cantidad de ácido estándar requerida para reducir el pH exactamente en 0,30 unidades. Como este cambio del pH corresponde a una duplicación exacta de la concentración de hidrogeniones, puede hacerse una extrapolación simple para el punto de equivalencia1, 2. b) Puntos finales: Cuando la alcalinidad se debe enteramente al contenido de carbonato o bicarbonato, el pH en el punto de equivalencia de la titulación se determina en función de la concentración de dióxido de carbono (CO2) en esta fase. Esta concentración depende, a su vez, del tipo de carbonato total nativo existente y de cualquier pérdida que pueda haberse producido durante la titulación. Como puntos de equivalencia de las concentraciones de alcalinidad correspondientes, en mg de CaCO3/l, se sugieren los valores de pH que se expresan a continuación. «Alcalinidad de fenolftaleína» es un término empleado tradicionalmente para designar la cantidad medida mediante titulación a pH 8,3, independientemente del indicador de color utilizado en su caso para la determinación. Las llamativas variaciones de color producidas por el púrpura de metacresol (pH 8,3) y el verde de bromocresol (pH 4,5) conceden utilidad a estos indicadores para la titulación de alcalinidad. 2-40 MÉTODOS NORMALIZADOS c) Interferencias: Los jabones, las materias oleosas y los sólidos en suspensión o precipitados pueden recubrir el electrodo de vidrio y causar una respuesta retardada. Déjese un tiempo adicional entre las adiciones del reactivo para permitir que el electrodo recupere el equilibrio, o límpiese éste en su caso. No se debe filtrar, diluir, concentrar o alterar la muestra. d) Selección del método: Determínese la alcalinidad de la muestra a partir del volumen de ácido estándar requerido para titular una porción a un pH determinado, según el apartado 1b. Titúlese a temperatura ambiente con un medidor de pH adecuadamente calibrado o un titulador eléctrico, o utilizando indicadores de color. Infórmese de una alcalinidad menor de 20 mg de CaCO3/l solamente si ha sido determinada por el método de alcalinidad baja (apartado 4d). Confecciónese una curva de titulación para estandarización de los reactivos. Los indicadores de color pueden utilizarse para titulaciones habituales y de control en ausencia de color y turbidez que puedan interferir, y en titulaciones preliminares para seleccionar el tamaño de la muestra y la potencia del reactivo (véase más adelante). e) Tamaño de la muestra: Véase la sección 2310B.le para seleccionar el tamaño de la muestra que va a titularse y la normalidad del reactivo, sustituyendo ácido sulfúrico (H2SO4) o clorhídrico (HC1) 0,02 o 0,1 N por el álcali estándar de este método. Si se sigue el método de alcalinidad baja, titúlese una muestra de 200 ml con H2SO4 0,02N, a partir de una bureta de 10 ml. f) Toma de muestras y conservación: Véase sección 231OB.1 f. 2. Instrumental Véase la sección 2310B.2. 3. Reactivos a) Solución de carbonato sódico, aproximadamente 0,0SN: Séquense entre 3 y 5 g de Na2CO3 estándar primario a 250 °C durante 4 h y enfríense en desecador. Se pesan 2,5 + 0,2 g (miligrámicos) y se transfieren a un matraz volumétrico de 1 l, llenando hasta la marca con agua destilada y mezclando el reactivo. No debe conservarse más de una semana. b) Ácidos sulfúrico o clorhídrico estándar, 0,1N: Prepárese la solución acida de normalidad aproximada a la indicada en preparación de reactivos de mesa (véase cubierta interior). Estandarícese frente a una solución de 40 ml de Na2CO3 0,05N en probeta, con unos 60 ml de agua, titulando potenciométricamente a un pH aproximado de 5. Elévense los electrodos, enjuáguense en la misma probeta y háganse hervir suavemente durante 3-5 min cubriendo con un vidrio de reloj. Enfríese a temperatura ambiente, enjuáguese el cristal en la probeta y conclúyase la operación titulando en el punto de inflexión de pH. Calcúlese la normalidad. donde: A = g de Na 2 CO 3 , pesados en el matraz de 1 l, B = ml de solución de Na 2 CO 3 tomados para titulación, y C = ml de ácido empleados. Utilícese la normalidad medida en los cálculos o ajústese a 0,1000N; 1 ml de solución 0,1000N = 5,0 mg de CaCO3. c) Ácidos sulfúrico o clorhídrico estándar, 0,02N: Dilúyanse 200,0 ml de ácido estándar 0,1000N hasta 1.000 ml de agua destilada o desionizada. Estandarícese mediante titulación potenciométrica de 15,0 ml de Na2CO3 0,05N, de acuerdo con el procedimiento del apartado 3b; 1 ml = 1,0 mg de CaCO3. 2-41 PROPIEDADES FÍSICAS Y DE AGREGACIÓN d) Solución indicadora de verde de bromocresol, indicador de pH 4,5: Disuélvanse 100 mg de púrpura de verde de bromocresol, sal sódica, en 100 mg de agua destilada. e) Solución indicadora de púrpura de metacresol, indicador de pH 8,3: Disuélvanse 100 mg de púrpura de metacresol en 100 ml de agua. f) Solución alcohólica de fenolftaleína, indicada a pH 8,3. g) Tiosulfato sódico, 0,1N: Véase sección 2310B.3i. vo hasta reducir el pH exactamente a 0,30 unidades, registrando de nuevo el volumen. 5. Cálculos a) Titulación potenciométrica a pH de punto final: donde: 4. A = ml utilizados de ácido estándar, y N = normalidad del ácido estándar Procedimiento a) Cambio de color: Véase sección 2310B.46. b) Curva de titulación potenciométrica: Sígase el método de determinación de la acidez (sección 2310B.4c), sustituyendo la normalidad de la solución acida estándar por NaOH estándar, y continúense las titulaciones hasta un pH 4,5 o más bajo. No se debe filtrar, diluir, concentrar o alterar la muestra. c) Titulación potenciométrica a pH preseleccionado: Determínese el pH de punto final adecuado, según el apartado Ib. Prepárense conjuntamente la muestra y la titulación (sección 2310B.4c). Titúlese a pH de punto final sin registrar valores intermedios y sin provocar retrasos indebidos. A medida que se alcanza el punto final, realícense adiciones de ácido más pequeñas, comprobando que el pH alcance el equilibrio antes de añadir más reactivo. d) Titulación potenciométrica de alcalinidad baja: Para alcalinidades menores de 20 mg/1, titúlense 100-200 ml con arreglo al procedimiento del apartado 4c, antes descrito, utilizando una microbureta de 10 ml y solución acida estándar 0,02N. Deténgase la titulación a un pH del orden de 4,3 a 4,7 y regístrese el volumen y el pH exacto. Añádase más reacti- o donde: t = título del ácido estándar, mg CaCO3/ml. Infórmese de la manera siguiente sobre el pH de punto final utilizado: «La alcalinidad a pH ___ = ____ mg de CaCO3/l» e indíquese claramente si este pH corresponde a un punto de inflexión de la curva de titulación. b) Titulación potenciométrica de alcalinidad baja: Alcalinidad total, mg CaCO3/l donde: B = ml titulante para primer pH registrado, C = total ml de titulante para alcanzar un pH inferior en 0,3 unidades, y N = normalidad del ácido. c) Cálculo de relaciones de alcalinidad: Los resultados obtenidos a partir de las determinaciones de fenolftaleina y al- MÉTODOS NORMALIZADOS 2-42 calinidad total ofrecen un medio de clasificación estequiométrica de las tres formas principales de alcalinidad presentes en muchas aguas. La clasificación adscribe la alcalinidad total a bicarbonato, carbonato e hidróxido, y acepta la ausencia de otros ácidos (débiles) orgánicos e inorgánicos, como el silícico, el fosfórico y el bórico. Presupone, además, la incompatibilidad de las alcalinidades de hidróxido y de bicarbonato. Dado que los cálculos se hacen sobre una base estequiométrica, las concentraciones iónicas en el sentido más estricto no están representadas en los resultados, que pueden diferir significativamente de las concentraciones reales, especialmente a pH > 10. Con arreglo a este esquema: lese la concentración OH como mg de CaCO3 por litro, y calcúlese la concentración de CO32¯ y HCO3¯ en mg de CaCO3 por litro a partir de la concentración de OH¯ y las alcalinidades de fenolftaleína y total mediante las ecuaciones siguientes: De igual forma, si se encuentran dificultades con el punto final de la fenolftaleína, o si se desea hacer una investigación de la titulación de ésta, calcúlese su alcalinidad como CaCO3 a partir de los 1) La alcalinidad de carbonato (CO32¯) se presenta cuando la de la fenolftaleína no es 0, sino menor que la total. 2) La alcalinidad de hidróxido (OH¯) se presenta si la de la fenolftaleína supera la mitad de la total. 3) La alcalinidad de bicarbonato (HCO3¯) se presenta si la de la fenolftaleína es menor de la mitad de la total. Estas relaciones pueden calcularse mediante el siguiente esquema, donde P es la alcalinidad de la fenolftaleína y T la total (apartado Ib): Selecciónese el valor más pequeño de P o (T – P). Entonces, la alcalinidad de carbonato es el doble del valor más pequeño. Cuando este valor más pequeño es P, el equilibrio (T – 2P) es bicarbonato. Cuando el valor más pequeño es (T – P), el equilibrio (2P – 7) es hidróxido. Todos los resultados se expresan en CaCO3. La conversión matemática de éstos se muestra en la tabla 2320:1 (Se ha propuesto una modificación de dicha tabla, que es más exacta cuando P 1/2 T3.) La relaciones de alcalinidad también pueden calcularse nomográficamente (véase dióxido de carbono, sección 4500CO2). Mídase exactamente el pH, calcú- TABLA 2320:1. RELACIONES DE ALCALINIDAD* resultados de las determinaciones monográficas de concentraciones de iones carbonato e hidróxido: 6. Precisión y sesgo Respecto a la precisión del método, no puede hacerse ninguna afirmación general, dada la gran variedad de característi- 2-43 PROPIEDADES FÍSICAS Y DE AGREGACIÓN cas que presentan las muestras. Es probable que la exactitud de la titulación sea mayor que las aproximaciones propias de la toma de muestras y su manipulación del análisis. En la amplitud de 10 a 500 mg/1, en la que la alcalinidad se debe enteramente a carbonatos o bicarbonatos, pueden lograrse una desviación estándar de 1 mg de CaCO3/l. Cuarenta analistas analizaron en 17 laboratorios muestras sintéticas que contenían incrementos de bicarbonato equivalente a 120 mg de CaCO3/l. Se utilizó el método de titulación del apartado 4b, con un pH de punto final de 4,5. La desviación estándar fue de 5 mg/l, y el sesgo promedio (más bajo que el valor verdadero), de 9 mg/l4. En 12 laboratorios, y con arreglo al método del apartado 4b, se analizaron soluciones de carbonato sódico equivalentes a 80 y 65 mg de CaCO3/l5. Las desviaciones estándar fueron de 8 y 5 ml/l respectivamente, con un sesgo no significativo5. En otros cuatro laboratorios se analizaron seis muestras con alcalinidades totales de alrededor de 1.000 mg de CaCO3/l y conteniendo varios índices de carbonato/bicarbonato, utilizándose ambos métodos, el del apartado 4a y el del 4c. La desviación estándar acumulada fue de 40 mg/l, con diferencias irrelevantes entre ambos procedimientos. 7. Referencias 1. LARSON, T. E. & L. M. HENLEY. 1955. Determination of low alkalinity or acidity in water. Anal. Chem. 27:851. 2. THOMAS, J. F. J. & J. J. LYNCH. 1960. Determination of carbonate alkalinity in natural waters. J. Amer. Water Works Assoc. 3. 4. 5. 8. 52:259. JENKINS, S. R. & R. C. MOORE. 1977. A proposed modification to the classical method of calculating alkalinity in natural waters. J. Amer. Water Works Assoc. 69:56. WINTER, J. A. & M. R. MIDGETT. 1969. FWPCA Method Study 1. Mineral and Physical Analyses. Federal Water Pollution Control Admin., Washington, D.C. SMITH, R. 1980. Research Rep. No. 379, Council for Scientific and Industrial Research, Sudáfrica. Bibliografía AMERICAN SOCIETY FOR TESTING AND MATE RIALS. 1982. Standard Methods for Acidity or Alkalinity of Water. Publ. D1067-70 (reapproved 1977), American Soc. Testing & Materials, Filadelfia, Pennsylvania. SKOUGSTAD, M. W., M. J. FISHMAN, L. C. FRIEDMAN, D. E. ERDMAN & S. S. DUNCAN. 1979. Methods for determination of inorganic substances in water and fluvial sediraents. En Techniques of Water-Resources Investigation of the United States Geological Survey. U.S. Geological Survey, Book 5, Chapter Al, Washington, D.C. 2-44 MÉTODOS NORMALIZADOS 2330 SATURACIÓN DE CARBONATO CÁLCICO (PROPUESTA)* 2330 A. 1. Discusión general Los índices de saturación de carbonato cálcico (CaCO3) se utilizan habitualmente para evaluar la tendencia del agua a formar o disolver precipitados. Esta evaluación es útil para los programas de control de corrosión y en la prevención de precipitados de CaCO3 en sistemas de tuberías e instalaciones como intercambiadores térmicos industriales o calentadores de agua domésticos. Las aguas sobresaturadas de CaCO3 tienden a precipitar esta sal, mientras que las infrasaturadas tienden a disolverla. Las saturadas, es decir, aquellas en las que el CaCO3 está en equilibrio, no presentan tendencia a disolverlo ni precipitarlo. La saturación representa la línea divisoria entre precipitación «probable» o «improbable». Para calcular los índices de saturación de CaCO3 que aquí se describen es preciso medir varias características de calidad del agua. Los requerimientos mínimos son la alcalinidad total (2320), el calcio total (3500-Ca), el pH (4500-H + ) y la temperatura (2550). También ha de calcularse o estimarse la potencia iónica en función de la conductividad (2510) o la medida del total de los sólidos disueltos (2540C). Mídase el pH a la temperatura del agua del sistema, utilizando un medidor de pH termocompensado. Si se mide a una temperatura distinta, por ejemplo * Aprobado por el Standard Methods Committee, 1989. Introducción en el laboratorio, corríjase la medida obtenida1–3. Durante las tomas de pH y alcalinidad, redúzcase al mínimo el intercambio de CO2 entre la muestra y la atmósfera. Lo ideal es cerrar herméticamente la muestra durante las medidas4 y, al menos, debe evitarse cualquier agitación fuerte de las muestras no cerradas. Existen dos tipos generales de índices de saturación del CaCO3: los que determinan si un agua tiene tendencia a precipitar CaCO3 (es decir, si está sobresaturada) o a disolverlo (o sea, infrasaturada), y los que estiman la cantidad de esta sal que puede precipitarse a partir de un agua sobresaturada y la cuantía que disolverá una infrasaturada. Estos últimos índices suelen proporcionar más información, pero son más difíciles de determinar. 2. Limitaciones Se acepta en general que el CaCO3 precipitará en las aguas sobresaturadas y no podrá hacerlo en las infrasaturadas, pero existen excepciones. Por ejemplo, los depósitos de carbonato de aguas sobresaturadas se inhiben por la presencia de fosfatos (preferentemente polifosfatos), de algunas sustancias orgánicas naturales y de magnesio5–7. Estos materiales pueden actuar como agentes secuestrantes y como tóxicos cristalinos. A la inversa, en sistemas de tuberías que conducían agua infrasaturada se han encontrado depósitos de CaCO3. Esta aparente con- 2-45 PROPIEDADES FÍSICAS Y DE AGREGACIÓN tradicción está causada por un pH elevado (relativo al pH del agua bruta) en la inmediata vecindad de algunas zonas (cátodos) de superficies metálicas corroídas. Aunque la mayor parte del agua se halle infrasaturada, puede presentarse alguna sobresaturación local, pudiéndose depositar una cantidad de CaCO3 pequeña, pero significativa. Los cálculos aquí indicados, e incluso los obtenidos por ordenador, no describen suficientemente esas excepciones. Por ello, los índices de saturación no deben considerarse absolutos. Considérense más bien como orientaciones de la conducta del CaCO3 en sistemas acuosos y, en lo posible, compleméntense con los datos obtenidos experimentalmente. De modo similar, los efectos previstos por los índices no siempre confirman las expectativas. En este caso, resulta ilustrativa la relación entre los índices y las tasas de corrosión. En teoría, el sistema de tuberías está protegido cuando el carbonato cálcico se precipita en su superficie. Se cree que el CaCO3 inhibe la corrosión actuando contra las áreas reactivas y proporcionando una matriz que retiene los productos de la corrosión, lo que proporciona una hermeticidad adicional a esas superficies. Las aguas con índices positivos se han considerado tradicionalmente como protectoras, y las de índice negativo, como no protectoras o corrosivas. La relación esperada se observa a veces 8, 9, pero no siempre 10, 11. Los resultados inesperados pueden deberse en parte a la limitada capacidad para prever la conducta del CaCO3. Asimismo, las características del agua que no se hallan implicadas directamente en el cálculo de los índices (por ejemplo, oxígeno disuelto, intensidad de tamponado, cloruros, sulfatos y velocidad del agua) pueden modificar significativamente las tasas de corrosión 9, 1 2 - 1 6 . Por consiguiente, no deben estimarse las tasas de corrosión solamente en función de los índices de CaCO3. 3. 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. Referencias MERRILL, D. T. & R. L. SANKS. 1978, 1979. Corrosión control by deposition of CaCO3 films: A practical approach for plant operators. J. Amer. Water Works Assoc. 70:592; 70:634 & 71:12. L OEWENTHAL , R. E. & G. v. R. M ARAIS . 1976. Carbonate Chemistry of Aquatic Systems: Theory and Applications, Ann Arbor Science Publishers, Ann Arbor, Michigan. M ERRILL , D. T. 1976. Chemical conditioning for water softening and corrosión control. En R. L. Sanks, ed. Water Treatment Plant Design. Ann Arbor Science Publishers, Ann Arbor, Michigan. SCHOCK, M. R., W. MUELLER & R. W. BUELOW. 1980. Laboratory techniques for measurement of pH for corrosión control studies and water not in equilibrium with the atmosphere. J. Amer. Water Works Assoc. 72:304. PYTKOWICZ, R. M. 1965. Rates of inorganic carbón nucleation. J. Geol. 73:196. F ERGUSON , J. F. & P. L. M CC ARTY . 1969. Precipitaron of Phosphate form Fresh Waters and Waste Waters. Tech. Rep. No. 120, Stanford Univ., Stanford, California. M ERRILL , D. T. & R. M. J ORDEN . 1975. Limeinduced reactions in municipal wastewaters. J. Water Pollut. Control Fed. 47:2783. D E M ARIIM . F. 1938. Corrosión and the Langelier calciumcarbonate saturation index. J. Amer. Water Works Assoc. 30:85. LARSON, T. E. 1975. Corrosión by Domestic Waters. Bull. 59, Illinois State Water Survey. JAMES M. MONTGOMERY, CONSULTING ENGINEERS, INC. 1985. Water Treatment Principles and Design. John Wiley & Sons, Nueva York. STUMM, W. 1960. Investigations of the corrosive behavior of waters. J. San. Eng. Div., Proc. Amer. Soc. Civil Eng. 86:27. PISIGAN, R. A., J R. & J. E. SINGLEY. 1985. Evaluation of water corrosivity using the Langelier index and relative corrosión rate models. Materials Perform. 24:26. LANE , R. W. 1982. Control of corrosión in distribution and building water systems En Proceedings AWWA Water Quality Technology Conf., Nashville, Tennessee, dic. 5-8, 1982. 2-46 14. 15. MÉTODOS NORMALIZADOS SONTHEIMER, H., W. KOLLE & V. L. SNOEYINK. 1982. The siderite model of the formation of corrosion-resistant scales. J. Amer. Water Works Assoc. 73:572. SCHOCK, M. R. & C. H. NEFF. 1982. Chemical aspects of internal corrosión; theory, prediction, and monitoring. En Proceedings AWWA Water Quality Technology Conf, Nashville, Tennessee, dic. 5-8, 1982. 16. A MERICAN WATER WORKS ASSOCIATION . 1986. Corrosión Control for Plant Operators. ISBW-O-89867-350-X, Denver, Colorado. 2330 B. índices representativos de la tendencia de un agua a precipitar o disolver el CaCO3 1. Discusión general Los índices que marcan las tendencias a precipitación o disolución de CaCo3 definen si un agua está sobresaturada, saturada o infrasaturada respecto a esta sal. Los más ampliamente utilizados son el índice de Saturación (IS), la Saturación Relativa (SR), también llamado índice de Fuerza de Conducción (IFC), y el índice Ryznar (IR). El IS es, con mucho, el más comúnmente utilizado y el que va a describirse a continuación. El SR y el IS están relacionados (véase ecuación 6, sección 233OD). El IR1 se ha utilizado durante años, a veces con buenos resultados. Debido a su carácter semi-empírico, puede resultar menos fiable que el IS. 2 Índice de Saturación mediante cálculo El IS se determina a partir de la ecuación 1. (1) donde: pH = pH medido, y pH, = pH del agua si estuviera en equilibrio con el CaCO3 en las concentraciones existentes de ion calcio [Ca2 +] e ion bicarbonato [HCO3- ] . Un IS positivo significa que el agua está sobresaturada de CaCO3, y uno ne- gativo, que está infrasaturada; un IS cero representa al agua en equilibrio con esta sal. a) Solución analítica para pHs: Determínese el pHs como sigue: (2) donde: K2 = constante de segunda disociación para el ácido carbónico a la temperatura del agua, Ks = constante de solubilidad del producto para el CaCO3 a temperatura del agua, 2+ [Ca ] = concentración de iones calcio, moles-g/1, [HCH 3− ] = concentración iones bicarbonato. moles-g/1, y fm = coeficiente de actividad para especies monovalentes a temperatura especificada. En la ecuación 2, la p que precede a una variable designa el –log10 de esa variable. Calcúlense los valores pK2, pKs y pfm requeridos para resolver la ecuación 2 a partir de las ecuaciones de la tabla 2330:I. Para ahorrar tiempo de cálculo, los valores pK2 y pKs se han calculado previamente para temperaturas seleccionadas (véase tabla 2330:II). Dicha tabla proporciona varios valores para pKs. En los sistemas acuosos pueden formarse distintos isomorfos de la CaCO3, inclu- 2-47 PROPIEDADES FÍSICAS Y DE AGREGACIÓN yendo calcita, aragonita y vaterita, cada uno de ellos con propiedades de solubilidad diferentes. Estas diferencias pueden ajustarse calculando el pHs simplemente con el empleo del pKs para el compuesto de formación más probable, que para el agua natural es la calcita. Úsese, pues, el pKs para la calcita, a menos que se sepa que es una forma distinta de CaCO3 la que controla su solubilidad. Estímese la concentración de calcio a partir de la medida del calcio total con la ecuación siguiente: donde: El calcio asociado con iones pares no es útil para formar CaCO3. Valórese el [ HCO3- ] concentración de ion bicarbonato, a partir de la ecuación 4. (3) TABLA 2330:I. ESTIMACIÓN DE CONSTANTES DE EQUILIBRIO Y COEFICIENTES DE ACTIVIDAD (4) 2-48 MÉTODOS NORMALIZADOS TABLA 2330:II. VALORES PRECALCULADOS PARA pK y A, A TEMPERATURAS SELECCIONADAS donde: Alk 1 = alcalinidad total, determinada por titulación del punto final de ácido carbónico, equivalentes-g/1, KK = constante de disociación para el agua, en la temperatura de ésta, y Alk 0 = Alcalinidad a la que contribuyen NH 30 , H 3SiO-4 , ¯ HPO 24 , B(OH)-4 , ¯ CH3COO (acetato), HS , e iones pa+ res como CaHCO3 y MgOH+. Estas contribuciones son pequeñas, por lo general, en comparación con las de los componentes considerados normalmente HCO3- , CO32- , OH - y H + . Los cálculos pueden simplificarse. Por ejemplo, en la ecuación 4, los términos con exponentes (p. ej., 10 pH p f pk m w pueden descartarse en general para aguas aproximadamente neutras (pH 6,0 a 8,5) con alcalinidad mayor de unos 50 mg/l como CaCO3. Los términos Caip de la ecuación 3 y Alko en la ecuación 4 son difíciles de calcular sin ordenador. Por tanto, generalmente se descartan para cálculos normales. La versión simplificada de la ecuación 2 en estas condiciones es: 1) Cálculo simple. Se entiende mejor con un ejemplo. Supóngase que la calcita controla la solubilidad del CaCO3 y determínese el IS para un agua de la siguiente composición: 2-49 PROPIEDADES FÍSICAS Y DE AGREGACIÓN Antes de evaluar pfm en la ecuación 2, determínese la potencia iónica (1) y otra constante (A). La potencia iónica se valora a partir de la primera ecuación de la tabla 2330:1, suponiendo que toda la alcalinidad se debe al ion bicarbonato. Utilícese la concentración de alcalinidad (2,60 x 10-3) y la carga de bicarbonato (–1) para calcular la contribución de la alcalinidad a la fuerza iónica. Supóngase que la sílice es predominantemente H4SiO4. Como éste tiene una carga cero, la sílice no contribuye a la fuerza iónica. En ausencia de un análisis completo del agua, valórese la fuerza iónica en función de la conductividad o de la cuantía de sólidos disueltos (véanse ecuaciones alternativas, tabla 2330:1). Evaluar A a partir de la ecuación de la tabla 2330:1, después de determinar la constante dieléctrica E aplicando la fórmula de la misma tabla. De forma opcional, utilícense los valores de A calculados previamente en la tabla 2030:11. En ella, A = 0,506 a 20 °C. A continuación, valórese pfm, según la ecuación de la tabla 2030:1: Determínese [HCO3 ] a partir de la ecuación 4. Se ignorará Alk0, pero como el pH excede a 8,5, se calcularán los otros términos. Según la tabla 2030:11, pK2 = 10,38 y pKw = 14,16. [HCCV] Por consiguiente, p[Ca2+] = 2,42 A partir de la tabla 2030:II, el pKs para la calcita es 8,45. Determínese el pHs mediante la ecuación 2: 2-50 MÉTODOS NORMALIZADOS Y, finalmente, determínese el IS a partir de la ecuación 1: IS = 9,00 - 7,27 = 1,73 El IS positivo indica que el agua está sobresaturada de calcita. 2) Efecto de descartar Caip y Alk0. Si se descarta Caip el pHs, es subestimado e IS superestimado en una cuantía de U 1 Yca donde Yca es la fracción ip ip del calcio total en pares de iones. Por ejemplo, si Yca = 0,30, la evaluación de ip IS es demasiado alta, en 0,5 unidades. Del mismo modo, si se descarta Alk0, el IS se sobreestima en una cuantía de U (1 YAlk , donde YAlko es la fracción 0 de alcalinidad total proporcionada por elementos distintos a HCO3- , CO32 , OH y H+ . Los efectos de descartar Caip y Alk0 son aditivos. Tanto Caip como Alk0 pueden descartarse si los factores YCa y YAlko son peip queños y no interfieren en la interpretación del IS. En aguas de pH neutro y bajo, estos factores son efectivamente pequeños, pero aumentan a medida que las cifras de pH se aproximan o exceden a 9. A valores altos, sin embargo, el IS es típicamente más amplio que su sobreestimado, por lo que el descarte de Caip y Alko no causa problemas. Volviendo al ejemplo anterior, cuando los cálculos se realizaron con un código computadorizado de química de aguas (SEQUIL) (véase tabla 2030:III), que considera los valores citados, el IS fue 1,48, es decir, 0,25 unidades más bajo que el resultado obtenido en los cálculos manuales. En este caso, el descarte de Caip y Alko no interfirió en la interpretación del resultado. Ambos cálculos mostraron que el agua estaba intensamente sobresaturada. La posibilidad de interpretación errónea es mayor en las aguas cercanas a la saturación, con elevadas concentraciones de sulfato. El agua de los circuitos de refrigeración es un ejemplo. El calcio es secuestrado por el potente par iónico CaSO 04 , y el IS puede ser sobreestimado en 0,3 a 0,5 unidades como mucho, incluso a pH neutro. En esas condiciones, el IS puede considerarse cero (ni precipitable ni corrosivo), cuando de hecho es negativo. Resuélvase este problema mediante determinación de pHs, utilizando códigos computadorizados de química del agua que tengan en cuenta los pares de iones y otras formas de alcalinidad. La sección 2330D proporciona información sobre códigos de química del agua. Los cálculos más exactos se logran cuando se realiza un análisis mineral completo. Un procedimiento alternativo pero algo menos riguroso consiste en la medida del [Ca2+], actividad del ion cálcico, con un electrodo iónico específico9. Utilícese la ecuación 5 para determinar U[Ca 2+ ] y luego aplíquese éste a la ecuación 2. Esta aproximación elimina la necesidad de determinar Caip. Sin embargo, no se dispone de un método equivalente para evitar la determinación de Alk0. b) Soluciones gráficas para pHs: Para determinar éste, pueden utilizarse los diagramas de Calwell-Lawrence10-12, especialmente útiles para evaluar las dosificaciones químicas necesarias para conseguir las condiciones hídricas deseadas. Consúltense las referencias para descripción de cómo utilizar los diagramas; véase sección 2330D para información adicional sobre éstos. 3. Índice de saturación mediante determinación experimental a) Saturometría: Los saturómetros se desarrollan para medir la saturación relativa de CaCO3 del agua del mar. En un matraz cerrado provisto de un electrodo de pH se equilibra un agua de contenido cálcico conocido, controlando la graduación térmica con un baño a temperatura constante. Durante el equilibrado, el pH 2-51 PROPIEDADES FÍSICAS Y DE AGREGACIÓN desciende si el CaCO 3 precipita, y aumenta si se disuelve. Cuando el pH deja de variar, se considera que se ha logrado el equilibrio. Los valores iniciales del pH y del calcio y el valor final de pH sirven para calcular la saturación relativa (SR)13. Entonces puede utilizarse la ecuación 6, sección 2330D, para determinar el IS. Una ventaja notable de este método es la posibilidad de seguir la pista de la aproximación al equilibrio mediante medidas del pH, reduciendo así las vacilaciones en torno del logro del equilibrio. El método presenta una sensibilidad máxima en márgenes de intensidad mínima de tamponado (pH 7,5 a 8,5). Los cálculos no tienen en cuenta los pares de iones y la alcalinidad no carbonada, excepto los boratos. La técnica se ha utilizado en medidas oceanógraficas14 in situ, así como en el laboratorio. Los cálculos de saturometría que se analizan más adelante utilizan el Ks de la fase CaCO3, aceptado para el control de la solubilidad. Surgen algunas dudas si no se conoce la identidad del sólido de control. Resuélvanse esas dudas mediante la medida del Ks de dicho sólido de control, que será el producto de actividad del CaCO3, [Ca2+] x U[CO3 ], en equilibrio. Calcúlese este último a partir del pH en equilibrio y el calcio inicial, la alcalinidad y las medidas de pH15. producto de actividad inicial por Ks. Calcúlese el IS aplicando la ecuación 6. La ventaja de este método radica en que no presupone nada sobre la fase de CaCO3. Sin embargo, con este método, la determinación del momento en que se ha logrado el equilibrio resulta más difícil que con el de saturometría. Cualquiera que sea el procedimiento utilizado, las temperaturas deben ser las mismas que las de la fuente de agua. Como alternativa, corregir los resultados de prueba respecto a la temperatura citada. 4. 1. 2. 3. 4. 2- 5. 6. b) Técnica de diferencia de alcalini- dad16: El IS también puede determinarse equilibrando un agua de pH, calcio y alcalinidad conocidos con CaCO3 en un sistema cerrado y a temperatura constante. El producto de actividad CaCO3 antes del equilibrio se determina en función de los valores iniciales de calcio, pH y alcalinidad (o carbonatos totales). La constante del producto de solubilidad CaCO3 (Ks) se iguala con el producto de actividad CaCO3 después del equilibrio, que se determina utilizando el cambio de alcalinidad producido durante el equilibrado. La SR se encuentra dividiendo el 7. 8. Referencias RYZNAR, J. W. 1944. A new index for determining the amount of calciumcarbonate formed by water. J. Amer. Water Works Assoc. 36:47. SNOEYINK, V. L. & D. JENKINS. 1980. Water Chemistry. John Wiley & Sons, Nueva York. STUMM, W. & J. J. MORGAN. 1981. Aquatic Chemistry, 2.a ed. John Wiley & Sons, Nueva York. RUSSELL , L. L. 1976. Chemical Aspects of Groundwater Recharge with Wastewaters. Ph.D. thesis, Univ. California, Berkeley. LANGELIER, W. F. 1936. The analytical control of anticorrosion water treatment. J. Amer. Water Works Assoc. 28:1500. ROSSUM , J. R. & D. T. MERRILL . 1983. An evaluation of the calciumcarbonate saturation indexes. J. Amer. Water Works Assoc. 75:95. PLUMMER, L. N. & E. BUSENBERG . 1982. The solubilities of calcite, aragonite, and vaterite in CO 2 -H 2 O solutions between 0 and 90 degrees C, and an evaluation of the aqueous model for the system CaCO 3 CO2-H2O. Geochim. Cosmochim. Acta 46:1011. ELECTRIC POWER RESEARCH INSTITUTE. 1982. Design and Operating Guidelines Manual for Cooling Water Treatment: Treatment of Recirculating Cooling Water. Section 4. Process Model Documentation and User's Manual. EPRI CS-2276, Electric Power Research Inst., Palo Alto, California. 2-52 MÉTODOS NORMALIZADOS 9. GARRELS, R. M. & C. L. CHRIST. 1965. Solutions, Minerals, and Equilibria. Harpe & Row, Nueva York. 10. MERRILL, D. 1 cSi R. L. SANKS. 1978, 1979. Corrosión control by deposition of CaCO3 films: A practical approach for plant operators. J. Amer. Water Works Assoc. 70:592; 70:634 & 71:12. 11. LOEWENTHAL, R. E. & G. v. R. MARAIS. 1976. Carbonate Chemistry of Aquatic Systems: Theory and Applications. Ann Arbor Science Publishers, Ann Arbor, Michigan. 12. MERRILL, D. T. 1976. Chemical conditioning for water softening and corrosión control. En R. L. Sanks, ed., Water Treatment 2330 C. Índices de previsión de la cantidad de CaCO3 que puede precipitarse o disolverse El potencial de precipitación de carbonato cálcico (PPCC) prevé ambas tendencias del CaCO3, precipitación y disolución, y la cantidad en que pueden hacerlo. El PPCC también se conoce por otros nombres, por ejemplo, capacidad de precipitación del carbonato cálcico (CPCC). El PPCC se define como la cantidad de CaCO3 que puede precipitarse, en teoría, en las aguas sobresaturadas o disolverse en las infrasaturadas durante el equilibrado1. Es posible que la cuantía que realmente precipita o se disuelve sea menor, debido a que el equilibrio puede no conseguirse. El PPCC es negativo en las aguas infrasaturadas, cero en las saturadas y positivo en las sobresaturadas. 1. Plant Design. Ann Arbor Science Publishers, Ann Arbor, Michigan. 13. BEN-YAAKOV, S. & I. R. KAPLAN. 1969. Determination of carbonate saturation of seawater with a carbonate saturometer. Limnol. Oceanogr. 14:874. 14. BEN-YAAKOV, S. & I. R. KAPLAN. 1971. Deepsea in situ calciumcarbonate saturometry. J. Geophys. Res. 76:72. 15. PLATH, D. C, K. S. J OHNSON & R. M. PYT K OWICZ . 1980. The solubility of calcite— probably containing magnesium—in seawater. Mar. Chem. 10:9. 16. BALZAR, W. 1980. Calciumcarbonate saturometry by alkalinity difference. Oceanol. Acta. 3:237. Cálculo del PPCC Este factor no se presta de por sí a cálculos ordinarios; es preferible calcularlo con modelos computadorizados para química del agua y con diagramas de Caldwell-Lawrence (véase sección 23330). Los cálculos más fiables tienen en cuenta los pares de iones y la contribución a la alcalinidad de otros elementos además de HCO3- , CO32 , OH - y H + . Los modelos que no toman en consideración estos factores sobreestiman la cantidad de CaCO3 que puede precipitarse y subestiman la que puede disolverse. 2. Determinación experimental del PPCC Estímese mediante una o varias técnicas experimentales. a) Saturometría: Véase sección 2330B. El PPCC se determina formando parte del cálculo de la SR. b) Técnica de diferencia de alcalinidad: Véase sección 2330B. El PPCC equivale a la diferencia entre los valores de alcalinidad (o calcio) del agua inicial y de la equilibrada, expresados en CaCO3. c) Prueba del mármol: Esta prueba 1-5 es similar a la técnica de diferencia de alcalinidad. El PPCC equivale al cambio de los valores de alcalinidad (o de PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-53 calcio) durante el equilibrado, cuando éste se expresa en CaCO3. d) Prueba de Enslow: La prueba de Enslow5 es una modalidad continua de las pruebas de diferencia de alcalinidad o del mármol. Se vierte continuamente agua en una cubeta de nivel o embudo de separación parcialmente lleno de CaCO3. El líquido que fluye desde este dispositivo se filtra a través de mármol triturado, de modo que se supone que el filtrado está en equilibrio con el CaCO3. El PPCC equivale al cambio de valor de la alcalinidad (o calcio), que tiene lugar durante el paso a lo largo del aparato. e) Prueba de deposición del carbonato cálcico6. La PDCC es un método electroquímico que mide la corriente eléctrica producida cuando el oxígeno disuelto es reducido en un electrodo rotatorio. Cuando se coloca en el aparato un agua sobresaturada, el CaCO3 se deposita sobre el electrodo. El depósito interfiere el transporte del oxígeno y la corriente disminuye. El índice de deposición del CaCO3 es directamente proporcional al de aminoración de la corriente. La PDCC y el PPCC están relacionados pero no son lo mismo. La PDCC es un índice y el PPCC es una cantidad. Para determinaciones reales de PPCC (o PDCC), manténgase la temperatura de la prueba al mismo nivel que la de la fuente de agua. Otra posibilidad consiste en corregir los resultados de la prueba hasta la temperatura del origen del agua. 2330 D. 3. 1. 2. 3. 4. 5. 6. Referencias MERRILL, D. T. & R. L. SANKS. 1978, 1979. Corrosión control by deposition of CaCO 3 films: A practical approach for plant operators. J. Amer. Water Works Assoc. 70:592, 70:634 & 71:12. MERRILL, D. T. 1976. Chemical conditioning for water softening and corrosión control. En R. L. Sanks, ed. Water Treatment Plant Design. Ann Arbor Science Publishers, Ann Arbor, Michigan. D E M ARTINI , F. 1938. Corrosión and the Langelier calciumcarbonate saturation index. J. Amer. Water Works Assoc. 30:85. H OOVER , C. P. 1938. Practical application of the Langelier method. J. Amer. Water Works Assoc. 30:1802. DYE, J. F. & J. L. TUEPKER. 1971. Chemistry of the Lime-Soda Process. En American Water Works Association. Water Quality and Treatment. McGraw-Hill Book Co., Nueva York. MCCLELLAND, N. I. & K. H. MANCY . 1979. CCDT bests Ryzner index as pipe CaCO 3 film predictor. Water Sewage Works 126:77. Diagramas y códigos de ordenador para los índices de CaCO3 1. Descripción La tabla 2330:111 contiene los diagramas y códigos de ordenador que pueden utilizarse para determinar el IS y el PPCC, incluyendo también una breve descripción de sus características. Muchos códigos de ordenador no calculan directamente el IS, pero en su lugar facilitan la saturación relativa (SR). Cuando se ofrezcan datos de SR, calcúlese el IS a partir de1: donde: SR = relación de producto de actividad CaCO 3 a constante de producto de solubilidad. Los diagramas y algunos de los códigos definen el pHs como el pH que el agua exhibiría si estuviera en equilibrio con el CaCO3 a las concentraciones de calcio y alcalinidad total existentes2. Esta definición de pHs se diferencia de la que sigue a la ecuación 1 porque utiliza la alcalinidad en vez del bicarbonato. 2-54 TABLA 2330:III. MÉTODOS NORMALIZADOS COLECCIÓN DE PROGRAMAS GRÁFICOS Y DE ORDENADOR QUE PUEDEN UTILIZARSE PARA CALCULAR LOS ÍNDICES DE SATURACIÓN * DEL CACO 3 PROPIEDADES FÍSICAS Y DE AGREGACIÓN TABLA 2330:III. Continuación 2-55 2-56 MÉTODOS NORMALIZADOS Dentro de una amplitud de pH de 6 a 9, el pHs basado en la alcalinidad y el basado en el bicarbonato son virtualmente iguales, debido a que la alcalinidad total se debe casi enteramente al ion bicarbonato. Por encima del pH 9 sí se diferencian, y la ecuación 6 ya no se aplica si el IS se calcula con pH basado en la alcalinidad. Sin embargo, si se determina a partir de pHs basado en bicarbonato, la ecuación 6 mantiene su aplicación. Además, el cálculo del IS con pHs basado en la alcalinidad invierte el signo del IS por encima de valores de pH de aproximadamente pK2, es decir, un IS positivo, y no el negativo usual, que significa que el agua está infrasaturada3. Con pHs basado en bicarbonato o con RS no aparece inversión del signo, lo que evita confusiones. Es por esto por lo que se prefieren estos dos cálculos. La tabla 2330:III incluye la definición de pHs utilizada para cada código. Algunos modelos calculan solamente la cantidad de CaCO3 que puede precipitarse, pero no la que puede disolverse, y otros calculan ambas. Los diagramas y códigos pueden utilizarse para determinar muchos más parámetros que los índices de saturación de CaCO3. El software y los gráficos de ordenador merecen un premio. La información de la tabla 2330:III describe los parámetros de cada código utilizados para calcular el IS. Consúltense las referencias citadas en la tabla para actualizar la información. 2. 1. 2. 3. Referencias SNOEYINK, V. L. & D. JENKINS. 1980. Water Chemistry. John Wiley & Sons, Nueva York. LANGELIER, W. F. 1936. The analytical control of anticorrosion water treatment. J. Amer. Water Works Assoc. 28:1500. L OEWENTHAL , R. E. & G. v. R. M ARAIS . 1976. Carbonate Chemistry of Aquatic Systems: Theory and Applications. Ann Arbor Science Publishers, Ann Arbor, Michigan. 4. MERRILL, D. T. & R. L. SANKS . 1978. Corrosión Control by Deposition of CaCO 3 Films: A Handbook of Practical Application and Instruction. American Water Works Assoc, Denver, Colorado. 5. M ERRILL , D. T. 1976. Chemical conditioning for water softening and corrosión control. En R. L. Sanks, ed., Water Treatment Plant Design. Ann Arbor Science Publishers, Ann Arbor, Michigan. 6. ELECTRIC POWER RESEARCH INSTITUTE. 1982. Design and Operating Guidelines Manual for Cooling Water Treatment: Treatment of Recirculating Cooling Water. Section 4. Process Model Documentation and User's Manual. EPRI CS-2276, Electric Power Research Inst., Palo Alto, California. 7. BROWN, D. S. & J. D. ALLISON. 1987. MINTEQA1 Equilibrium Metal Specialion Model: A User's Manual. EPA-600/3-87-012. 8. PARKHURST , D. L., D. C. THORSTENSON & L. N. PLUMMER. 1980. PHREEQE—A Computer Program for Geochemical Calculations. USGS WRI 80-96 NTIS PB 81-167801. 9. C ROWE , A. S. & F. J. L ONGSTAFF . 1987. Extension of Geochemical Modeling Techniques to Brines: Coupling of the Pitzer Equations to PHREEQE. En Proceedings of Solving Groundwater Problems With Models, National Water Well Assoc., Denver, Colorado, feb. 10-12, 1987. 10. F LEMING , G. W. & L. N. P LUMMER . 1983. PHRQINPT—An Interactive Computer Program for Constructing Input Data Sets to the Geochemical Simulation Program PHREEQE. USGS WRI 83-4236. 11. E LECTRIC P OWER R ESEARCH I NSTITUTE . SEQUIL-An Inorganic Aqueous Chemical Equilibrium Code for Personal Computers; volumen 1, User's Manual/Workbook, Versión 1.0, GS-6234-CCML. Electric Power Research Inst., Palo Alto, California. 12. K HARAKA , Y. K., W. D. G UNTER , P. K. A GGARWAL , E. H. P ERKINS & J. D. D E BRAAL. 1988. Solmineq.88—A Computer Program for Geochemical Modeling of Water-Rock Interactions. USGS WRIR 884227, Menlo Park, California. 13. B ALL , J. W., D. K. N ORDSTROM & D. W. Z ACHMANN . 1987. WATEQ4F—A Personal Computer Fortran Translation of the Geochemical Model WATEQ2 With Revised Data Base. USGS OFR 87-50. PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2340 DUREZA* 2340 A. 1. 2-57 Introducción Definición Originalmente, la dureza del agua se entendió como una medida de su capacidad para precipitar el jabón. El jabón es precipitado preferentemente por los iones calcio y magnesio. Otros cationes polivalentes también pueden hacerlo, pero éstos suelen estar presentes en formas complejas, frecuentemente con componentes orgánicos, y su influencia en la dureza del agua puede ser mínima y difícil de determinar. De acuerdo con los criterios actuales, la dureza total se define como la suma de las concentraciones de calcio y magnesio, ambos expresados como carbonato cálcico, en miligramos por litro. Cuando la dureza es numéricamente mayor que la suma de alcalinidades de carbonato y bicarbonato, esta cantidad de dureza equivalente a la alcalinidad total se denomina «dureza de carbonato»; la cantidad de dureza que excede a ésta se llama «dureza no carbonatada». Cuando la dureza es numéricamente igual o menor que la suma de alcalinidades de carbonato y bicarbonato, toda la dureza es de carbonato, estando ausente la de bicarbonato. La dureza oscila entre cero y cientos de miligramos por litro, dependiendo de la fuente y del tratamiento a que el agua haya sido sometida. 2. Existen dos métodos. El método B, cálculo de la dureza, es aplicable a todas las aguas y proporciona una gran exactitud. Si se realiza un análisis mineral, puede informarse del cálculo de dureza. El método C, de titulación de EDTA, mide los iones calcio y magnesio y puede aplicarse, con las debidas modificaciones, a cualquier clase de agua. El procedimiento descrito facilita un medio de análisis rápido. 3. * Aprobado por el Standard Methods Committee, 1985. 2340 B. 1. Selección del método Informe de resultados Al informar sobre la dureza, señálese el método utilizado, por ejemplo «dureza (cálc.)» o bien «dureza (EDTA)». Cálculo de la dureza Discusión general El método preferido para determinar la dureza es calcular ésta a partir de los resultados de las valoraciones aisladas de calcio y magnesio. 2. Cálculo Dureza, mg equivalente CaCO3 /l = 2,497 [Ca, mg/l] + 4,118 [Mg, mg/l] 2-58 MÉTODOS NORMALIZADOS 2340 C. 1. Método titulométrico de EDTA Discusión general a) Principio: El ácido etilendiaminotetraacético y sus sales de sodio (abreviatura EDTA) forman un complejo de quelato soluble al añadirse a las soluciones de algunos cationes metálicos. Si a una solución acuosa que contenga iones calcio y magnesio a un pH de 10 ± 0,1 se añade una pequeña cantidad de colorante, como negro de eriocromo T o calmagita, la solución toma un color rojo vino. Si se añade EDTA como reactivo de titulación, los iones calcio y magnesio formarán un complejo, y, cuando todos estos iones estén incluidos en dicho complejo, la solución cambiará del rojo vino al azul, señalando el punto final de la titulación. Para obtener un punto final satisfactorio han de estar presentes los iones magnesio. Para asegurar esta presencia, se añade al tampón una pequeña cantidad de sal magnésica de EDTA, neutra desde el punto de vista complexométrico; de este modo se introduce automáticamente una cantidad suficiente de magnesio y evita la necesidad de una corrección de blanco. La nitidez del punto final aumenta con los incrementos de pH. Sin embargo, el pH no puede aumentar indefinidamente debido al peligro de precipitación de carbonato cálcico (CaCO3) o hidróxido magnésico, Mg(OH)2, y porque la tinción cambia de color a pH alto. El valor de pH especificado de 10 ± 0,1 constituye una solución satisfactoria. Se fija un límite de cinco minutos de duración para la titulación, a fin de reducir al mínimo la tendencia a la precipitación de CaCO3. b) Interferencia: Algunos iones metálicos interfieren produciendo puntos finales débiles o indiferenciados, o provocando un consumo estequiométrico de EDTA. Redúzcase esta interferencia añadiendo algunos inhibidores antes de la titulación. El Mg-EDTA [véase 263)] secuestra selectivamente a los metales pesados, libera magnesio en la muestra y puede utilizarse como sustituto de inhibidores tóxicos o malolientes. Solamente es útil cuando el magnesio sustituido por los metales pesados no contribuye significativamente a la dureza total. Con metales pesados a concentraciones de polifosfato por debajo de las señaladas en la tabla 2340:1, utilícese el inhibidor I o II. Cuando existen concentraciones más altas de metales pesados, el calcio y magnesio se determinan por un método no EDTA (véanse secciones 3500-Ca y 3500-Mg), y la dureza se obtiene mediante cálculo. Las cifras de la tabla deben interpretarse como una orientación aproximada, y se basan en el empleo de muestras de 25 ml diluidas a 50 ml. TABLA 2340:I. CONCENTRACIONES MÁXIMAS DE INTERFERENCIA PERMITIDAS CON DIVERSOS INHIBIDORES* 2-59 PROPIEDADES FÍSICAS Y DE AGREGACIÓN Las materias orgánicas coloidales o en suspensión también pueden interferir en el punto final. Elimínese la interferencia mediante evaporación de la muestra por secado en baño de vapor y calentamiento en horno de mufla a 550 °C hasta que se produzca la oxidación completa de la materia orgánica. Dilúyase el residuo en 20 ml de ácido clorhídrico (HC1) 1N, neutralícese a pH 7 con hidróxido sódico (NaOH) lN y complétese hasta 50 ml con agua destilada; enfríese a temperatura ambiente y continúese de acuerdo con el procedimiento general. c) Precauciones en la titulación: Practíquese la titulación a la temperatura ambiente. El cambio de color se hace demasiado lento a medida que la muestra se acerca a la temperatura de congelación. La descomposición del indicador llega a constituir un problema cuando se emplea agua caliente. El pH especificado puede producir un ambiente propicio a la precipitación del CaCO3. Aunque el titulante disuelve lentamente estos precipitados, un punto final desviado suele proporcionar resultados pobres. La realización de la titulación en cinco minutos reduce al mínimo la tendencia a precipitar del CaCO3. Los tres métodos siguientes también reducen la pérdida por precipitación. 1) Dilúyase la muestra con agua destilada para reducir la concentración del carbonato; esta sencilla operación se ha incorporado al método. Si aparece precipitación a esta dilución 1 + 2, utilícense las modificaciones 2) o 3). El empleo de una muestra demasiado pequeña aporta un error sistemático, derivado de la lectura equivocada de la bureta. 2) Si se conoce la dureza aproximada o se determina por una titulación preliminar, añádase a la muestra un 90 por 100 o más de titulante antes de ajustar el pH con un tampón. 3) Acidifíquese la muestra y remuévase dos minutos para expulsar el CO2 antes del ajuste de pH. Determínese la alcalinidad para indicar la cantidad de ácido que ha de añadirse. 2. Reactivos a) Solución tampón: 1) Disuélvanse 16,9 g de cloruro amónico (NH4C1) en 143 ml de hidróxido de amonio (NH4OH) conc. Añádase 1,25 g de sal de magnesio de EDTA (disponible en el mercado) y dilúyase hasta 250 ml de agua destilada. 2) Si no se dispone de sal magnésica de EDTA, disuélvase 1,179 g de sal disódica de ácido etilendiaminotetraacético dihidrato (grado de reactivo analítico) y 780 mg de sulfato magnésico (MgSO4·7H2O) o 644 mg de cloruro magnésico (MgCl2 · 6H2O) en 50 ml de agua destilada. Para alcanzar la máxima exactitud, ajústese a equivalente exacto por medio de la adición de una pequeña cantidad de EDTA, MgSO4 o MgCl2. Consérvense las soluciones 1) y 2) en un recipiente plástico o de vidrio borosilicato, durante un período no superior a un mes. Tapónese herméticamente para evitar pérdidas de amoníaco (NH3) o captura de dióxido de carbono (CO2). Manipúlese la solución tampón mediante una pipeta de bulbo. Se prescindirá del tampón cuando, al añadirse 1 ó 2 ml a la muestra, éstos no puedan producir un pH de 10,0 ± 0,1 en el punto final de la titulación. 3) También pueden adquirirse en el mercado «tampones inodoros», los cuales constituyen una alternativa satisfactoria. Contienen sal de magnesio de EDTA y tienen la ventaja de ser relativamente inodoros y más estables que los tampones de NH4C1-NH4OH. Por lo general, los tampones inodoros no proporcionan un punto final tan favorable como los de NH4C1-NH4OH a causa de su reacción más lenta, y pueden resultar inútiles cuando el método está automatizado. Prepárese uno de esos tampones mezclando 55 ml de HCl conc. con 400 ml de 2-60 agua destilada y a continuación añádase, lentamente y agitándolo, 300 ml de 2-aminoetanol (libre de aluminio y metales pesados). Agréguense 5,0 g de sal de magnesio de EDTA y dilúyase hasta 1 l con agua destilada. b) Agentes complejantes: Para la mayoría de las aguas, no son necesarios. En ocasiones, cuando el agua contenga iones de interferencia, se deberá añadir un complejante adecuado para lograr un cambio neto y exacto del color en el punto final. Son satisfactorios los siguientes: 1) Inhibidor I: Ajústense las muestras acidas a pH 6 o más con tampón de NaOH 0,1N. Añádanse 250 mg de cianuro sódico (NaCN) en polvo. A continuación, añádase tampón suficiente para ajustar a pH 10,00 ± (PRECAUCIÓN: El NaCN es extremadamente tóxico. Su empleo requiere la adopción de precauciones extraordinarias. Las soluciones que contengan este inhibidor deben drenarse con un chorro de agua en cantidad suficiente para asegurar que no queda ácido capaz de liberar cianhídrico tóxico volátil.) 2) Inhibidor II: Disuélvanse 5,0 g de sulfuro sódico no anhidro (Na2S · 9H2O) o 3,7 g de Na2S · 5H2O en 100 ml de agua destilada. La entrada de aire se evita con un tapón de goma fijado fuertemente. Este inhibidor se deteriora por oxidación del aire y produce un precipitado sulfuro que oscurece el punto final cuando existen concentraciones apreciables de metales pesados. Empléese 1 ml en el apartado 3b, más adelante. 3) MgCDTA: Sal magnésica del ácido 1, 2-ciclohexanodiaminotetraacético. Añádanse 250 mg por 100 ml de muestra y disuélvase completamente antes de aportar la solución tampón. Utilícese este complejante para evitar el uso de inhibidores tóxicos u olorosos cuando existan sustancias interferentes a concentraciones que afecten al punto final pero no contribuyan significativamente al valor de dureza. MÉTODOS NORMALIZADOS Pueden adquirirse preparados que incorporan un tampón y un complejante; estas mezclas tienen que mantener un pH de 10,0 ± 0,1 durante la titulación y proporcionar un punto final neto y exacto cuando se titula la muestra. c) Indicadores: Se han propuesto muchos tipos de soluciones indicadoras, que pueden utilizarse si el analista demuestra que proporcionan valores exactos. El principal problema que presentan estas soluciones es que se deterioran con el tiempo, produciendo puntos finales poco netos. Por ejemplo, las soluciones alcalinas de negro de eriocromo T son sensibles a los oxidantes, y sus soluciones acuosas o alcohólicas son inestables. En general, utilícese la menor cantidad de indicador capaz de obtener un punto final neto. Es responsabilidad del analista determinar individualmente la concentración óptima del indicador. 1) Negro de eriocromo T: Sal sódica del ácido l-(l-hidroxi-2-naftilazo)-5nitro-2-naftol-4-sulfónico, n.° 203 en el índice de color. Disuélvanse 0,5 g de colorante en 100 g de 2,2’, 2”-nitrilotrietanol (también llamado trietanolamina) o 2-metoximetanol (también llamado etilenglicol-monometiléter). Añádanse 2 gotas por 50 ml de solución a titular. Si es necesario, ajústese el volumen. 2) Calmagita: Ácido l-(l-hidroxi-4metil-2-fenilazo)-2-naftol-4-sulfónico. Es estable en solución acuosa y produce el mismo cambio de color que el negro de eriocromo T, con un punto final neto. Disuélvanse 0,10 g de calmagita en 100 ml de agua destilada. Utilícese 1 ml por 50 ml de solución a titular, ajustando el volumen si es necesario. 3) Los indicadores pueden utilizarse en forma de polvo seco siempre que se tenga cuidado en evitar su exceso. Existen en el mercado mezclas secas de esos indicadores y una sal inerte. Si el cambio de color de punto final de esos indicadores no es neto y diferenciado, por lo general, eso significa que se PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-61 requiere un complejante apropiado. Si el inhibidor NaCN no define bien el punto final, lo más probable es que sea defectuoso. d) Titulante EDTA estándar, 0,01M: Se pesan 3,723 g de etilendiaminotetracetato disódico trihidrato, grado de reactivo analítico, también llamado (etilenodinitrilo) sal disódica del ácido tetraacético (EDTA); a continuación se disuelve en agua destilada hasta 1.000 ml. Estandarícese frente a solución de calcio estándar (apartado 2e) como se describe más adelante (apartado 3b). El titulante extrae cationes productores de dureza de los recipientes de vidrio blando, por lo que debe conservarse en frascos de polietileno (preferible) o vidrio borosilicato. El deterioro gradual se compensa mediante la reestandarización periódica y la utilización de un factor de corrección adecuado. e) Solución de calcio estándar: Se pesan 1,000 g de polvo de CaCO3 anhidro (estándar principal o reactivo especial, bajo en metales pesados, álcalis y magnesio) en un erlenmeyer de 500 ml. Coloqúese un embudo en el cuello del matraz y añádase, poco a poco, 1 + 1 HCl hasta la disolución total del CaCO3. Añádanse 200 ml de agua destilada y hágase hervir durante unos minutos para expeler el CO2. Enfríese, añádanse unas gotas de indicador rojo de metilo y ajústese al color naranja intermedio por adición de NH4OH 3N o 1 + 1 HC1, según se requiera. Transvásese cuantitativamente y dilúyase hasta 1.000 ml con agua destilada; 1 ml = 1,0 mg de CaCO3. f) Hidróxido sódico, NaOH, 0,1N. b) Titulación de muestras: Selecciónese un volumen de muestra que requiera menos de 15 ml de reactivo EDTA y realícese la titulación en cinco minutos, medidos a partir del momento de la adición del tampón. Dilúyanse 25,0 ml de muestra hasta alrededor de 50 ml de agua destilada en una batea de porcelana u otro recipiente adecuado. Añádase entre 1 y 2 ml de solución tampón. Por lo general, 1 ml será suficiente para dar un pH de 10,0 a 10,1. La ausencia de un cambio de color de punto final neto en la titulación suele significar la necesidad de añadir un inhibidor en este punto (apartado 2b y siguientes), o que el indicador se ha deteriorado. Añádanse una o dos gotas de solución indicadora o una cantidad adecuada del reactivo en polvo seco (apartado 2c3). Poco a poco, añádase titulante EDTA estándar, removiendo continuamente, hasta que desaparezcan los últimos matices rojizos. Añádanse las últimas gotas con intervalos de 3-5 segundos. En el punto final, la solución suele ser azul. Se recomienda utilizar luz natural o una lámpara fluorescente de luz día, ya que las lámparas de incandescencia tienden a producir un matiz rojizo en el azul de punto final. Si se dispone de muestra suficiente y no hay interferencias, puede lograrse una mayor exactitud incrementando el tamaño de la muestra, como se describe más adelante (apartado 3c). c) Muestra de dureza baja: Para fluido intercambiador de iones u otras aguas ablandadas y para aguas naturales de dureza baja (menos de 5 mg/l), tómese para titulación una muestra amplia, de 100 a 1.000 ml, y añádanse cantidades proporcionalmente grandes de tampón, inhibidor e indicador. Añádase lentamente titulante EDTA por medio de una microbureta y realícese un blanco, utilizando agua bidestilada, destilada o desionizada del mismo volumen que la 3. Procedimiento a) Tratamiento previo de muestras de aguas contaminadas y residuales: Utilícese la digestión de ácido nítrico-ácido sulfúrico, o bien ácido nítrico-ácido perclórico (sección 3030). 2-62 MÉTODOS NORMALIZADOS muestra, a la que hay que añadir idénticas cantidades de tampón, inhibidor e indicador. Sustráigase el volumen del EDTA utilizado como blanco a partir del volumen empleado en la muestra. 6. 4. Cálculo Dureza (EDTA) como mg de CaC0 3 1 A x B x 1.000 ml de muestra donde: A = ml de titulación para la muestra, y B = mg CaCO 3 equivalente a 1,0 ml de titulante EDTA. 5. 56 laboratorios mediante el método titulométrico de EDTA, con una desviación estándar relativa del 2,9 por 100 y un error relativo del 0,8 por 100. Precisión y sesgo Una muestra sintética con 610 mg/l de dureza total en CaCO3, constituida por 108 mg Ca/l y 82 mg Mg/l, y las siguientes sustancias suplementarias: 3,1 mg de K/l, 19,9 mg de Na/l, 241 mg de Cl¯/l, 0,25 mg de NO-2 - N/1, 1,1 mg de NO3- N/l, 259 mg de SO 2-4 / 1 y 42,5 mg de alcalinidad total/l (aportada por NaHCO3) en agua destilada, fue analizada en Bibliografía CONNORS, J. J. 1950. Advances in chemical and colorimetric methods. J. Amer. Water Works Assoc. 42:33. D IEHL , H., C. A. G OETZ & C. C. H ACH . 1950. The versenate titration for total hardness. J. Amer. Water Works Assoc. 42:40. BETZ, J. D. & C. A. NOLL. 1950. Total hardness determination by direct colorimetric titration. J. Amer. Water Works Assoc. 42:49. GOETZ, C. A., T. C. LOOMIS & H. DIEHL. 1950. Total hardness in water: The stability of standard disodiumdihydrogen ethylenediaminetetraacetate solutions. Anal. Chem. 22:798. DISKANT, E. M. 1952. Stable indicator solutions for complexometric determination of total hardness in water. Anal. Chem. 24:1856. BARNARD, A. J., JR., W. C. BROAD & H. FLASCHKA. 1956 & 1957. The EDTA titration. Chemist Analyst 45:86 & 46:46. GOETZ, C. A. & R. C. SMITH. 1959. Evaluation of various methods and reagents for total hardness and calcium hardness in water. Iowa State J. Sci. 34:81 (agosto 15). SCHWARZENBACH, G. & H. FLASCHKA. 1969. Complexometric Titrations, 2.a ed. Barnes & Noble, Inc., Nueva York. PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2510 2-63 CONDUCTIVIDAD 2510 A. Introducción La conductividad es una expresión nuEl recíproco de la resistencia es la conmérica de la capacidad de una solución ductancia, que mide la capacidad para para transportar una corriente eléctrica. conducir una corriente y se expresa en Esta capacidad depende de la presencia ohmios recíprocos o mhos. En los análide iones y de su concentración total, de sis de agua es más conveniente la unidad su movilidad, valencia y concentraciones micromhos. Cuando se conoce y se aplirelativas, así como de la temperatura de ca la constante celular, la conductancia la medición. Las soluciones de la mayo- medida se convierte en conductancia esría de los ácidos, bases y sales presentan pecífica o conductividad, Ks, recíproco de coeficientes de conductividad relativa- la resistencia específica: mente adecuados. A la inversa, las moléculas de los compuestos orgánicos que no se disocian en soluciones acuosas tienen una conductividad muy escasa o nula. La medición física practicada en una Se prefiere el término «conductividad», determinación de laboratorio suele ser de resistencia, medida en ohmios o megaoh- y por lo general se expresa en micromhos mios. La resistencia de un conductor es por centímetro μmhos/cm). En el Sisteinversamente proporcional a su área de ma Internacional de Unidades (SIU), sección transversal y directamente pro- el recíproco del ohmio es el siemens (S) porcional a su longitud. La magnitud de y la conductividad se expresa en milisiela resistencia medida en una solución mens por metro (mS/m); 1 mS/m = acuosa depende, por tanto, de las carac- = 10 μmhos/cm. Para expresar resultados terísticas de la célula de conductividad en unidades SIU, divídanse μmhos/cm utilizada, y sólo tiene sentido si se cono- por 10. El agua destilada tiene recién prepacen esas características. La resistencia específica es la resistencia de un cubo de rada una conductividad de 0,5 a 2 1 cm de lado. En soluciones acuosas, esta μmhos/cm, que aumenta tras unas semamedida es rara, debido a las dificultades nas de almacenamiento a 2-4 μmhos/cm. de fabricación del electrodo. Los electro- Este aumento está producido fundamendos prácticos miden una fracción dada talmente por absorción de dióxido de de la resistencia específica, siendo esta carbono y, en menor grado, de amoníaco. La conductividad de las aguas potafracción la constante celular C: bles en los Estados Unidos oscila generalmente entre 50 y 1.500 μmhos/cm. La conductividad de las aguas residuales domésticas puede estar próxima a la del suministro hídrico local, aunque algunos residuos industriales exhiben conductividades superiores a 10.000 μmhos/cm. En * Aprobado por el Standard Methods Committee, sistemas de conducción, canales, corrien1988. 2-64 tes fluviales y lagos, se utilizan instrumentos de medición de la conductividad, los cuales pueden incorporarse a estaciones de monitorización multiparamétrica con registradores. La medición de la conductividad en laboratorio es relativamente exacta, pero otros medios de determinación menos precisos encuentran numerosas aplicaciones, como son el mareaje del agotamiento de resinas de intercambio iónico y la determinación rápida de cambios significativos en el contenido inorgánico de las aguas potables y residuales. Bien instalados y mantenidos, los dispositivos de monitorización pueden proporcionar registros continuos de la conductividad. La mayoría de los problemas que plantea la obtención de registros adecuados con equipos de monitorización radica en la suciedad del electrodo y en la circulación insuficiente de la muestra. Las mediciones de conductividad en laboratorios se utilizan para: a) Establecer el grado de mineralización para determinar el efecto de la concentración total de iones sobre equilibrios químicos, efectos fisiológicos en plantas y animales, tasas de corrosión, etcétera. b) Determinar el grado de mineralización del agua destilada y desionizada. c) Evaluar las variaciones de la concentración de minerales disueltos en aguas naturales y residuales. La variación estacional mínima que se encuentra en las aguas embalsadas contrasta notablemente con las fluctuaciones diarias de algunas aguas de río contaminadas. Las aguas residuales que contienen cantidades significativas de desechos industriales muestran también una variación diaria considerable. d) Valorar el tamaño de la muestra que se vaya a utilizar para determinaciones químicas comunes y para investigar los resultados de un análisis químico. e) Determinar la cantidad de reactivo iónico necesario en algunas reacciones de MÉTODOS NORMALIZADOS precipitación y neutralización, señalándose el punto final por un cambio en la inclinación de la curva como consecuencia del punteo de la conductividad sobre las lecturas de bureta. f) Calcular los sólidos totales disueltos en una muestra multiplicando la conductividad (micromhos por centímetro) por un factor empírico; éste puede variar de 0,55 a 0,9 dependiendo de los componentes solubles del agua y de la temperatura de medición. Para aguas salinas o de caldera pueden requerirse factores relativamente altos, mientras que, si existen cantidades considerables de hidróxido o de ácido libre, se necesitarán factores más bajos. Aunque la evaporación de la muestra produce un cambio de bicarbonato a carbonato, el factor empírico se calcula, para un aporte de agua relativamente constante, dividiendo los sólidos disueltos por la conductividad. Los miliequivalentes por litro, tanto de cationes como de aniones en algunas aguas, se evalúan aproximadamente multiplicando la conductividad (en micromhos por centímetro) por 0,01. La conductividad electrolítica (a diferencia de la metálica) aumenta con la temperatura a un índice de 1,9 por 100/°C aproximadamente. De una medición inexacta de la temperatura pueden derivarse errores significativos. Las soluciones de cloruro potásico (KC1) tienen un coeficiente de conductividad a temperatura más baja que el agua potable común y, por su parte, el cloruro sódico (NaCl) posee un coeficiente que se aproxima mucho al encontrado en la mayoría de las aguas de pozo y de superficie. Nótese que cada ion tiene un coeficiente de temperatura distinto; así pues, un trabajo meticuloso requiere la determinación de la conductividad a 25 °C. El hecho de que la corrección de la temperatura, una parte en 500 para 25 ± 0,1 °C, alcance resultados significativos depende del equipo disponible y de la precisión deseada. PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2510 B. 1. Método de laboratorio Discusión general Véase sección 2510A. 2. 2-65 Instrumental a) Instrumental de conductividad autocontenida: Utilícese un dispositivo consistente en una fuente de corriente alterna, un puente de Wheatstone, un indicador de valor nulo y una célula de conductividad u otro instrumento que mida el índice de corriente alterna y su voltaje a través de la célula, teniendo este último la ventaja de proporcionar una lectura lineal de la conductividad. Elíjase un instrumento capaz de medir la conductividad con un error que no exceda el 1 por 100 o 1 μmho/cm. b) Termómetro, capaz de marcar hasta 0,1 °C cubriendo una amplitud de 23 a 27 °C. Por la rapidez de su respuesta, resulta conveniente emplear un termómetro eléctrico provisto de un pequeño sensor de temperatura. c) Célula de conductividad: 1) Tipo electrodo de platino. Este tipo de célula se presenta en forma de pipeta o de inmersión. La elección de la célula depende de la amplitud esperada de conductividad y de la amplitud de resistencia del instrumento. Ajústese experimentalmente la amplitud del conjunto total de aparatos, comparando los resultados instrumentales con las conductividades reales de las soluciones de KC1 enumeradas en la tabla 2510:1. Límpiense las células nuevas con una mezcla acida crómico-sulfúrica y platinícense los electrodos antes de su uso. A continuación se lavan y se platinizan de nuevo, siempre que las lecturas sean irregulares, cuando no pueda obtenerse un punto final neto o cuando la inspección muestre que se han desprendido capas de negro de platino. Para platinizar, prepárese una solución de 1 g de ácido cloroplatínico, H2PtCl6 · 6H2O, y 12 mg de acetato de plomo en 100 ml de agua destilada. Una solución más fuerte reduce el tiempo requerido para la platinización, por lo que puede emplearse cuando el tiempo es un factor decisivo, por ejemplo, cuando la constante celular es de 1,0/cm o más. Sumérjanse los electrodos en esta solución y conéctense ambos al polo negativo de una pila de 1,5 V. Conéctese el polo positivo a un trozo de alambre de platino e introdúzcase en la solución. Utilícese una corriente que sólo desprenda una pequeña cantidad de gas. Continuar la electrólisis hasta que ambos electrodos se recubran con negro de platino. Consérvese la solución para uso ulterior. Enjuagar cuidadosamente los electrodos y, cuando no se usen, mantenerlos inmersos en agua destilada. TABLA 2510:I. CONDUCTIVIDAD DE LAS SOLUCIONES DE CLORURO DE POTASIO A 25 °C * 2-66 MÉTODOS NORMALIZADOS 2) Tipo electrodo no de platino. Utilícense células de conductividad con electrodos hechos de metales comunes duraderos (acero inoxidable entre otros) para monitorización continua y estudios de campo. Calíbrense estas células mediante comparación de la conductividad de la muestra con los resultados obtenidos con un instrumento de laboratorio. Para reducir al mínimo los errores de la constante celular, utilícese una célula y un aparato adecuadamente diseñados y homologados. 3. Reactivos a) Agua de conductividad: Se hace pasar agua destilada a través de un desionizador, descartándose el primer litro. La conductividad debe ser menor de 1 μmho/cm. b) Solución estándar de cloruro potásico, KC1, 0,0100M: Disuélvanse 745,6 mg de KC1 anhidro en agua de conductividad, diluyendo hasta 1.000 ml a 25 °C. Ésta es la solución de referencia estándar, que a 25 °C tiene una conductividad de 1.413 μmho/cm. Resulta satisfactoria para la mayoría de las muestras cuando la célula tiene una constante de 1 a 2. Para otras constantes celulares, utilícense las soluciones más fuertes o más débiles enumeradas en la tabla 2510:1. Consérvese en frascos de vidrio borosilicato con tapón de vidrio. 4. Procedimiento a) Determinación de la constante de la célula: Aclárese la célula de conductividad al menos con tres porciones de solución KC1 0,01 M. Ajústese la temperatura de la cuarta porción a 25,0 ± 0,1 °C, mídase su resistencia y anótese el valor térmico. Calcúlese la constante celular C: C = (0,001 413) (R KCl ) [1 + 0,0191 (t – 25)] donde: RKCl = resistencia medida, ohmios, y t = temperatura observada, °C. b) Medición de la conductividad: Aclárese la célula con una o más porciones de la muestra. Ajústese la temperatura de una última porción a 25,0 ± 0,1 °C. Mídase la resistencia o conductividad de la muestra y anótese la temperatura. 5. Cálculo El coeficiente de temperatura de la mayoría de las aguas es el mismo aproximadamente que el de la solución estándar de KC1; cuando más se desvía la temperatura de 25,0 °C, mayor es la incertidumbre en la aplicación de la corrección de la misma. Comuníquense todas las conductividades a 25,0 °C. a) Cuando se mide la resistencia de la muestra, la conductividad a 25 °C es: donde: K = conductividad, μmhos/cm, C = constante de la célula, cm -1 , Rm = resistencia medida de la célula, ohms, y t = temperatura de medición. b) Cuando se mide la conductividad de la muestra, dicha conductividad a 25 °C es: donde: Km = conductividad medida, mhos a t °C, y otras unidades definidas como se indicó anteriormente. NOTA: Si la lectura de conductividad se hace en micromhos por centímetro, PROPIEDADES FÍSICAS Y DE AGREGACIÓN suprímase el factor 1.000.000 en el numerador. c) Para instrumentos que den sus valores en unidades del SIU, 1 6. 1 mS/m = 10 μmhos/cm, o a la inversa, μmho/cm = 0,1 mS/m. 2-67 7. 1. ROBINSON, R. A. & R. H. STORES. 1959. Electrolyte Solutions, 2. a ed. Academic Press, Nueva York, pág. 466. 2. LIND , J. E., J. J. Z WOLENIK & R. M. Fuoss. 1959. Calibration of conductance cells at 25 °C with aqueous solutions of potassium chloride. J. Amer. Chem. Soc. 81:1557. Precisión y sesgo 8. Se sometieron a prueba tres muestras sintéticas, con los siguientes resultados: 2520 Bibliografía JONES, G. & B. C. BRADSHAW. 1933. The measurement of the conductance of electrolytes. V. A redetermination of the conductance of standard potassium chloride solutions in absolute units. J. Amer. Chem. Soc. 55:1780. SALINIDAD 2520 A. 1. Referencias Discusión general La salinidad, que no tiene unidad de medida, es una importante propiedad de las aguas naturales e industriales. Se concibió inicialmente como la determinación de la masa de sales disueltas en una masa dada de solución. La determinación experimental del contenido de sal mediante desecación y pesada presenta algunas dificultades a causa de las pérdidas de algunos componentes. La única manera fiable de determinar la salinidad real o absoluta de un agua natural es realizar un análisis químico completo. Sin embar* Aprobado por el Standard Methods Committee, 1988. Introducción go, este método es costoso en tiempo y no puede proporcionar la exactitud necesaria para un trabajo detallado. Así pues, para determinar la salinidad se suelen utilizar métodos indirectos que incluyen la medida de una propiedad física como la conductividad, la densidad, la velocidad del sonido o el índice de refracción. Partiendo de una relación empírica entre la salinidad y la propiedad física determinada para una solución estándar, se hace posible calcular aquélla. La salinidad resultante no es más exacta que la relación empírica. La precisión, en la medida de una propiedad física, determinará la exactitud de la salinidad. A continuación se indican las precisiones de varias medidas físicas y la salinidad MÉTODOS NORMALIZADOS 2-68 resultante; estos datos son hoy asequibles mediante el uso de instrumentos comercializados en el mercado: Aunque la conductividad presenta la mayor precisión, solamente es útil para solutos iónicos. La densidad, aún menos precisa, responde a todos los solutos. 2. Selección del método En el pasado, la salinidad del agua del mar se determinaba por métodos hidro- 2520 B. 1. métricos y argentométricos, ambos incluidos en las ediciones anteriores de Standard Methods (véase sección 210B y C, 16.a ed.). En los últimos años se han utilizado los métodos de conductividad (2520B) y densidad (2520C) por su precisión y sensibilidad elevadas. Los dos se recomiendan para trabajos precisos de campo y laboratorio. 3. Garantía de calidad Calíbrese el salinómetro o densímetro frente a estándar de KC1 o de agua de mar. La precisión esperada es de más de ±0,01 unidades de salinidad, operando con sistemas de operación minuciosos y estándares que cubran el margen de los resultados. Método de la conductividad eléctrica Determinación Véase Conductividad, sección 2510. Debido a su sensibilidad elevada y a su fácil medición, el método de conductividad es el más utilizado para determinar la salinidad 1, 2. Para determinaciones del agua de mar, utilícese la Escala de Salinidad Práctica 1978 3 - 5 . Esta escala se realizó a partir de una solución de KC1. Una muestra de agua de mar con una conductividad a 15 °C igual a la de una solución de KC1 que contiene una masa de 32,4356 g en 1 kg de solución, se define como poseedora de una salinidad práctica de 35. Este valor se determinó como un promedio de tres estudios de laboratorio independientes. Para determinar la salinidad, se estudia la dependencia de ésta con respecto a la conductividad, Rt, en función de la temperatura (t °C, Escala de Temperatura Práctica Internacional 1968) de una muestra da- da, a un estándar S de agua de mar = 35. válidas a partir de S = 2 a 42. Para determinar la conductividad, úsese un puente, calibrado con un agua de PROPIEDADES FÍSICAS Y DE AGREGACIÓN mar estándar*, con una conductividad relativa al KC1 conocida, siguiendo las instrucciones del fabricante y los métodos anotados en la sección 2510. Si las medidas han de hacerse en aguas de estuario, realícense calibraciones secundarias de agua de mar diluida en peso de conductividad conocida, para garantizar que el puente está midiendo conductividades reales. La Escala de Salinidad Práctica se ha extendido recientemente a salinidades bajas6, para dar una ecuación válida desde salinidad 0 a 40. La ecuación es: La salinidad práctica rompe con la antigua relación salinidad-clorinidad, S = 1,806 55 Cl. Aunque la escala puede utilizarse para aguas de estuario7-10 y de mar abiertol1-13, no está libre de limitaciones12, 14-22. 2. 1. 2. 3. Referencias LEWIS , E. L. 1978. Salinity: its definition and calculation. J. Geophys. Res. 83:466. L EWIS , E. L. 1980. The practical salinity scale 1978 and its antecedents. IEEE J. Oceanic Eng. OE-5:3. B RADSHAW , A. L. & K. E. S CHLEICHER . 1980. Electrical conductivity of seawater. IEEE J. Oceanic Eng. OE-5:50. * Disponible en Standard Seawater Services, Institute of Oceanographic Services, Warmley, Godalming, Surrey, Inglaterra. 2-69 4. CULKIN, F. & N. D. SMITH. 1980. Determination of the concentration of potassium chloride solution having the same electrical conductivity, at 15 °C and infinite frequency, as standard seawater of salinity 35.000 per 1.000 (Chlorinity 19.37394 per 1.000). IEEE J. Oceanic Eng. OE-5:22. 5. DAUPHINEE, T. M., J. ANCSIN , H. P. KLEIN & M. J. PHILLIPS, 1980. The effect of concentration and temperature on the conductivity ratio of potassium chloride solutions to standard seawater of salinity 35 per 1.000 (Cl.19.3740) at 15 °C and 24°. IEEE J. Oceanic Eng. OE-5:17. 6. H ILL , K. D., T. M. D AUPH INEE & D. J. WOODS . 1986. The extension of the Practical Salinity Scale 1978 to low salinities. IEEE J. Oceanic Eng. OE-11:109. 7. MILLERO, F. J. 1975. The physical chemistry of estuarines. En T. M. Church, ed. Marine Chemistry in the Coastal Environment. American Chemical Soc. Symposium, Ser. 18. 8. MILLERO, F. J. 1978. The physical chemistry of Baltic Sea waters. Thalassia Jugoslavica 14:1. 9. MILLERO, F. J. 1984. The conductivity-salinity-chlorinity relationship for estuarine waters. Limnol. Oceanogr. 29:1318. 10. MILLERO, F. J. & K. KREMLING. 1976. The densities of Baltic Sea waters. Deep-Sea Res. 23:1129. 11. FERNÁNDEZ, F., F. VÁZQUEZ & F. J. MILLERO . 1982. The density and composition of hypersaline waters of a Mexican lagoon. Limnol. Oceanogr. 27:315. 12. M ILLERO , F. J. & P. V. C HETIRKIN . 1980. The density of Caspian Sea waters. DeepSea Res. 27:265. 13. M ILLERO , F. J., A. Mucci, J. Z ULLIG & P. C HETIRKIN . 1982. The density of Red Sea brines. Mar. Chem. 11:477. 14. BREWER, P. G. & A. BRADSHAW. 1975. The effect of non-ideal composition of seawater on salinity and density. J. Mar. Res. 33:155. 15. C ONNORS , D. N. & D. R. K ESTER . 1974. Effect of major ion variations in the marine environment on the specific gravityconductivity-chlorinity-salinity relationship. Mar. Chem. 2:301. 16. POISSON, A. 1980. The concentration of the KC1 solution whose conductivity is that of standard seawater (35 per 1.000) at 15 °C. IEEE J. Oceanic Eng. OE-5:24. 2-70 17. 18. 19. MÉTODOS NORMALIZADOS POISSON, A. 1980. Conductivity/salinity/ temperatura relationship of diluted and concentrated standard seawater. IEEE J. Oceanic Eng. OE-5:17. M ILLERO , F. J., A. G ONZÁLEZ & G. K. WARD . 1976. The density of seawater solutions at one atmosphere as a function of temperature and salinity. J. Mar. Res. 34:61. M ILLERO , F. J., A. G ONZÁLEZ , P. G. B RE WER & A. BRADSHAW. 1976. The density of North Atlantic and North Pacific deep waters. Earth Planet Sci. Lett. 32:468. 2520 C. 20. M ILLERO , F. J., D. L AWSON & A. G ONZÁ LEZ. 1976. The density of artificial river and estuary waters. J. Geophys. Res. 81:1177. 21. MILLERO, F. J., P. CHETIRKIN & F. CULKIN. 1977. The relative conductivity and density of standard seawaters. Deep-Sea Res. 24:315. 22. M ILLERO , F. J., D. F ORSHT , D. M EANS , J. GRIESKES & K. KENYON. 1977. The density of North Pacific ocean waters. J. Geophys. Res. 83:2359. Método de densidad 1. Determinación Con un densitómetro de precisión de flujo vibrátil es posible realizar determinaciones rápidas de la densidad de las aguas naturales. Las determinaciones se llevan a cabo haciendo pasar la muestra a través de un tubo vibrante insertado en un manguito de temperatura constante. La densidad U de la solución es proporcional al cuadrado del período de la vibración W . donde A y B son términos determinados por calibrado, el B mediante un densitómetro con agua de mar estándar. La diferencia entre la densidad de la muestra y la del agua pura viene dada por: donde W y W 0 son los períodos de la muestra y del agua, respectivamente. El sistema se calibra con dos soluciones de densidad conocida, siguiendo para la calibración las recomendaciones del fabricante. Esas dos soluciones pueden ser gas nitrógeno y agua o agua de mar estándar y agua. La salinidad de la muestra puede determinarse a partir de una ecuación internacional de estado a 1 atm para agua de mar. Esta ecuación relaciona U U 0 a la salinidad práctica (S), como una función de la temperatura1. donde: y la densidad del agua es dada por: Ejecútese una repetición simple ajustando S hasta que se obtenga la determinación p – p0 a una temperatura dada. Si las medidas se toman a 25 °C, la salinidad puede determinarse a partir de la ecuación siguiente: 2-71 PROPIEDADES FÍSICAS Y DE AGREGACIÓN que tiene una W = 0,0012 en S. También pueden determinarse salinidades aproximadas a partir de densidades o gravedades específicas obtenidas con hidrómetro a una temperatura dada (sección 210B, 16.a edición). 2520 D. 2. Referencias 1. MILLERO, F. J. & A. POISSON. 1981. International one-atmosphere equation of state of seawater. Deep Sea Res. 28:625. Algoritmo de salinidad práctica Debido a que todas las determinaciones de salinidad práctica se llevan a cabo con referencia a la conductividad de agua de mar estándar (corregida para 5 = 35), para los cálculos de salinidad será R, la cantidad disponible. Normalmente, Rt se obtiene directamente con salinómetros de laboratorio, pero las determinaciones in situ por lo general producen la cantidad R, relación de la conductividad in situ a la estándar en S = 35, t = 15 °C, p = 0 donde p es la presión superior a una atmósfera estándar, y la temperatura figura en la Escala Internacional de Temperaturas 1968). R se divide en tres partes, Rp y r, pueden expresarse como funciones de los valores numéricos de los parámetros in situ, R, t y p, cuando t es expresado en °C y p en columnas (105Pa), como sigue: donde: donde: Rp = relación de conductividad in situ a conductividad de la misma muestra a la misma temperatura, pero a p = 0 y rt = relación de conductividad del agua de mar de referencia, con salinidad práctica de 35 y temperatura t, a su conductividad a t = 15 °C. A partir de Rp y rt, calcúlese R, utilizando los resultados in situ, por ejemplo: donde: 2-72 MÉTODOS NORMALIZADOS 2530. MATERIAS FLOTABLES* 2530 A. Introducción Un criterio importante para la evaluación del posible efecto de la presencia de residuos en las aguas de superficie, es la cantidad de material flotable que hay en dichos residuos. Se encuentran dos tipos generales de material notable: material en partículas, que incluye «bolas de grasa», y componentes líquidos, que pueden dispersarse como una película fina y muy visible sobre áreas extensas. El material flotable en las aguas residuales es importante porque se acumula en la superficie, suele ser muy visible, es susceptible de ser transportado por el viento, puede conte- * Aprobado por el Standard Methods Committee, 1988. 2530 B. 1. Partículas flotables (GENERAL) Discusión general a) Principio: Este método se basa en la diferencia de gravedad de las partículas que tienen una densidad menor que la del agua circundante. Las partículas que se recogen en la superficie y pueden filtrarse y calentarse a 103-105 °C son definidas por esta prueba como partículas flotables. b) Aplicación: El método puede aplicarse a las aguas residuales, corrientes sometidas a tratamiento primario y secundario, y aguas industriales. Debido a su sensibilidad limitada, no es aplicable a aguas de corrientes con tratamiento ter- * Teflón o equivalente. ner bacterias patógenas y/o virus asociados con partículas aisladas y puede concentrar cifras elevadas de metales e hidrocarburos clorados, como pesticidas y PCBs. El aceite y las grasas dispersadas en forma coloidal se comportan como otras materias orgánicas dispersadas y se incluyen en el material medido por las pruebas COD, BOD y TOC. La prueba de aceite flotable indica la fracción fácilmente separable. Los resultados sirven para designar separadores de aceite y grasa, al revelar la eficacia de los separadores operativos, y para monitorizar corrientes de aguas residuales tratadas o sin tratar. Muchas ciudades y distritos cuentan con límites especificados de grasa y aceites flotables para las aguas residuales vertidas por las alcantarillas. ciario o de depósito, tanto dulces como de mar. c) Precauciones: Variaciones, incluso pequeñas, en la toma de muestras y en su manipulación, durante y después de la recogida, pueden producir grandes diferencias en la cantidad medida de material flotable. Además, para obtener resultados fiables es imprescindible la uniformidad de la capa de TFE* del embudo de separación. Para un análisis reproducible, trátense todas las muestras de manera uniforme, preferentemente mezclándolas según un procedimiento estándar, antes de la flotación, y utilizando embudos de separación lo más uniformemente preparados que sea posible. Como el método se basa en la diferencia de gravedad específica entre el líquido y las partículas PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-73 flotables, las variaciones de temperatura pueden afectar a los resultados. Realícese la prueba a temperatura constante, igual a la de la masa de agua recibida, y consígnese esa cifra en los resultados. tese un pequeño tubo a la parte exterior del desagüe, aplicando una abrazadera, de manera que pueda fluir libremente el líquido. Recúbrase el interior del contenedor con un aerosol de TFE, tan uniformemente como sea posible, para prevenir la adherencia de aceite y grasa a la superficie. b) Embudo de flotación: Se utiliza un cono Imhoff con una espita de TFE en el fondo, de un volumen total de 3,5 1 (figura 2530:2). Recúbrase por dentro con TFE de manera tan uniforme como sea posible, para evitar la acumulación de partículas de grasa flotables despegadas de la pared. Los embudos se montan de Figura 2530:1. Dispositivo de toma de muestras Figura 2530:2. Embudo de flotación de material flotable y soporte de filtro. de materias flotables. d) Concentración detectable mínima: La concentración detectable mínima reproducible es 1 mg/1, aproximadamente. Aunque puedan medirse niveles mínimos inferiores, los resultados no son significativos dentro de la actual eficacia establecida para la prueba. 2. Instrumental a) Preparador de muestras con mezclador de materiales flotables: Utilícese, en un soporte independiente, un recipiente metálico de 5 1 de capacidad como máximo, equipado con un mezclador propelente (figura 2530:1) y con un tubo de desagüe de 20 mm de diámetro interno insertado en un ángulo de 45 ° en la pared del contenedor en la dirección del flujo. El ángulo de 45 ° asegura que incluso las partículas gruesas fluyan desde el contenedor hacia los embudos de flotación, donde se recoge la muestra. Ajús- MÉTODOS NORMALIZADOS 2-74 la forma indicada en la figura 2530:3, con una luz en el fondo para ayudar a leer los niveles. c) Soporte de filtro: Se cubre la cara interna del extremo superior con TFE, tomando de nuevo todas las precauciones posibles para obtener un revestimiento uniforme. d) Filtros, de fibra de cristal, porosidad fina*. e) Matraz de vacío, 500 ml. f) Revestimiento de TFE: Síganse las instrucciones incluidas en las cajas de revestimiento disponibles en el mercado. Un revestimiento uniforme es la clave de la fiabilidad de los resultados de la prueba, pero en la práctica es difícil de conseguir. 3. Procedimiento a) Preparación de los filtros de fibra de vidrio: Véase sección 2340D.3a. * Whatman GF/C o equivalente. Figura 2530:3. Embudos de flotación y unidad de mezclado. b) Recogida y tratamiento de la muestra: Recójase la muestra en un dispositivo de obtención de flotables, en un punto de mezclado total; transpórtese al laboratorio y deposítense 3,0 1 en el embudo de flotación, dentro de las dos horas siguientes a la toma, para reducir al mínimo las alteraciones del material flotante. Mientras se llena el embudo, mézclese el contenido del colector con el mezclador propelente. Ajústese la velocidad de mezclado para lograr una distribución uniforme de las partículas flotantes en el líquido, pero evitando la formación de un remolino demasiado grande que pueda provocar la captura de gran cantidad de aire. c) Corrección para densidad y efectos de concentración: Cuando el agua recibida tiene densidad y concentración iónica diferentes de la residual, ajústense esas cifras de la muestra a las del agua recibida. Por ejemplo, si el agua recibida es de mar abierto, se vierte 1,5 1 en el embudo de flotación y se añade 1,5 1 de agua de mar filtrada desde la zona de recepción, junto con una mezcla de 39,8 g de NaCl, 8,0 g de MgCl2-6H2O y 2,3 g de CaQ2-2H2O. La mezcla final contiene la cantidad de material flotable en una muestra de 1,5 1, en un medio de densidad y concentración iónica aproximadamente iguales a las del agua del mar. d) Flotación: Mézclese el contenido del embudo de flotación a 40 rpm durante quince minutos, utilizando un mezclador de paletas (figura 2530:3). Déjese reposar cinco minutos, mézclese a 100 rpm durante un minuto y déjese reposar de nuevo treinta minutos. Descárguense 2,8 1 a través de la espita inferior, a un ritmo de 500 ml/min. Durante el desagüe, no debe alterarse la superficie de la muestra en el embudo. Con un bote lavador lleno de agua destilada, límpiese cualquier material flotante adherido a las paletas y al embudo. Déjense reposar los restantes 200 ml durante quince minutos y vacíense los sólidos sedimentados y el líquido 2-75 PROPIEDADES FÍSICAS Y DE AGREGACIÓN hasta la marca 40 ml del cono de Imhoff. Se deja nuevamente en reposo durante diez minutos y se desagua hasta que sólo queden en el embudo 10 ml de líquido y partículas flotables. Añádanse 500 ml de agua destilada y remuévase manualmente para separar las partículas sedimentables de las flotables. Déjese reposar quince minutos y vacíese hasta la marca 40 ml. Tras un nuevo reposo de diez minutos, vacíese gota a gota hasta la señal 10 ml. A continuación se filtran estos 10 ml y las partículas flotables restantes a través de un filtro de fibra de vidrio previamente pesado. Lávese con agua destilada la superficie del embudo de flotación para reunir en el filtro todo el material flotable. e) Pesada: El filtro de fibra de vidrio se seca y se pesa a 103-105 °C durante dos horas exactamente (véase sección 2540D.3c). 4. Cálculo donde: A = peso del filtro + mg material flotable, B = peso del filtro, mg, y C = volumen de la muestra, 1. (No incluir el volumen utilizado para corrección de la densidad y la concentración.) 5. Precisión y sesgo La precisión varía con la concentración de material suspendido en la muestra. No hay ningún método completamente satisfactorio para determinar el sesgo del método en muestras de aguas residuales, pero puede establecerse una recuperación aproximada realizando una segunda prueba para material flotable en toda el agua vertida durante la aplicación del método, con excepción de los últimos 10 ml. La precisión y el sesgo se resumen en la tabla 2530:1. La experimentación del método en una planta municipal de tratamiento indica que el límite inferior práctico de detección es 1 mg/1, aproximadamente. 6. Bibliografía HEUKELEKIAN, H. & J. BALMAT. 1956. Chemical composition of the particulate fractions of domestic sewage. Sewage Ind. Wastes 31:413. ENGINEERING-SCIENCE, INC. 1965. Determination and Removal of Floatable Material from Waste Water. Rep. for U. S. Public Health Serv. contracts WPD 12-01 (Rl)-63 and WPD 12-02-64, Engineering-Science, Inc., Arcadia & Oakland, California. HUNTER , J. V. & H. HEUKELEKIAN . 1965. Composition of domestic sewage fractions. J. Water Pollut. Control Fed. 37:1142. NUSBAUMI. & L. BURTMAN. 1965. Determination of floatable matter in waste discharges. J. Water Pollut. Control Fed. 37:577. TABLA 2530:I. COEFICIENTE DE VARIACIÓN Y RECUPERACIÓN PARA PRUEBA DE PARTÍCULAS FLOTABLES * Material flotante adicional añadido a partir de las escorias de una cubeta de sedimentación primaria. 2-76 MÉTODOS NORMALIZADOS SCHERFIG, J. & H. F. LUDWIG. 1967. Determination of floatables and hexane extractables in sewage. En Advances in Water Pollution Research, Vol. 3, pág. 217. Water Pollution Control Federation, Washington, D.C, SELLECK, R. E., L. W. BRACEWELL & R. CARTER. 1974. The Significance and Control of Wastewater Floatables in Coastal Waters. Rep. for U. S. Environmental Protection Agency contract R-800373, SERL Rep. Núm. 74-1, Sa- nitary Engineering Research Lab., Univ. California, Berkeley. BRACEWELL, L. W. 1976. Contribution of Wastewater Discharges to Surface Films and Other Floatables on the Ocean Surface. Thesis, Univ. California, Berkeley. BRACEWELL, L. W., R. E. SELLECK & R. CARTER. 1980. Contribution of wastewater discharges to ocean surface particulates. J. Water Pollut. Control Fed. 52:2230. 2530 C. Grasas y aceites flotables solubles en triclorotrifluoroetano (GENERAL) 1. Discusión general La prueba de grasas y aceites flotantes no mide un tipo exacto de estas sustancias; más bien los resultados vienen determinados por la prueba. La fracción medida incluye aceite y grasa, ambos flotantes y adheribles a las paredes del vaso de prueba. Las porciones adherible y flotante revisten una importancia práctica similar, ya que se presupone que la mayor parte de la porción adherente flotaría en otras condiciones del agua recibida. Se ha comprobado que los resultados representan adecuadamente la cantidad de aceite retirado en separadores con índices de flotabilidad equivalentes a las condiciones de prueba. 2. Instrumental a) Tubo de aceite flotable (figura 2530:4): Antes de utilizarse, límpiese cuidadosamente el tubo cepillándolo con un detergente ligero. El agua debe formar una película suave sobre la superficie interior del vidrio lavado. No debe utilizarse lubricante en la espita. b) Matraz cónico, 300 ml. Figura 2530:4. Tubo de aceite flotable, capacidad 1I. PROPIEDADES FÍSICAS Y DE AGREGACIÓN 3. Reactivos a) 1,1,2-tricloro-1,2,2-trifluoroetano *: Véase sección 5520B.36. b) Ácido clorhídrico, HCl, 6N. c) Papel de filtro**. 4. Procedimiento a) Toma de muestras: Recójanse las muestras en una zona donde haya fuerte turbulencia y el material flotante no esté atrapado en la superficie. Llénese el tubo de aceite flotante para marcar sumergiéndolo en el agua. No deben utilizarse muestras tomadas del laboratorio en un frasco, pues el aceite y la grasa no pueden redispersarse de nuevo en su condición original. b) Flotación: Sosténgase el tubo en posición vertical e iníciese el período de flotación en el lugar de la toma de muestra después de llenar el tubo. El tiempo estándar de flotación es de 30 minutos; si se utiliza un tiempo distinto, indíquese la variación en el informe de resultados. Al final del período de flotación, vacíense con cuidado los primeros 900 ml de agua a través de la espita, parando antes de que escape el aceite u otra sustancia de la superficie. Gírese ligeramente el tubo hacia atrás y adelante alrededor de su eje vertical para despegar el lodo de los lados, y déjese reposar cinco minutos. Descárguese completamente el lodo que se haya depositado en el fondo o que baje por los lados con el líquido. La capa sobrenadante en la parte superior del líquido puede mezclarse con el agua a medida qUe desciende por el tubo. Si sé produce esta mezcla, deténgase la salida del agua antes de haber perdido material flotable. Déjese en reposo durante cinco minutos antes de desechar el agua restante. Después de eliminar el agua, devuélvase * Freón o equivalente. ** Whatman núm. 40 o equivalente. 2-77 el tubo al laboratorio para completar la prueba. c) Extracción: Acidifíquese a pH 2 o menos con unas gotas de HCl 6N, añádanse 50-100 ml de triclorotrifluoroetano y agítese vigorosamente. Déjese en reposo y drénese el solvente en un vaso seco y limpio. Fíltrese a través de un papel de filtro seco en un matraz cónico de tara registrada de 300 ml, teniendo cuidado de que no haya agua sobre el filtro. Añádase una segunda porción de 50 ml de triclorotrifluoroetano y repítase la extracción, el sedimentado y la filtración en el mismo matraz de 300 ml. Si la cantidad de materiales flotantes de la muestra excede 4 mg/1, puede ser necesaria una tercera extracción. Lávese cuidadosamente el papel de filtro con solvente fresco vertido de una botella de lavado á través de una pipeta fina. Evapórese el solvente de la botella como se describe en la sección 5520B.4. Para cada lote de solvente, determínese el peso del residuo que queda tras evaporación a partir del mismo volumen que se utilizó en el análisis. 5. Cálculos Desígnense los resultados como «grasas y aceites solubles flotables, 30 minutos (u otro valor especificado) de tiempo de sedimentación, mg/1». Grasa y aceite flotables, solubles en triclorotrifluoroetano, tiempo de sedimentación 30 minutos, mg/1 donde: A = ganancia de peso total del matraz tarado, mg, y B = residuo calculado a partir del blanco de solvente del mismo volumen qué el utilizado en la prueba, mg. MÉTODOS NORMALIZADOS 2-78 6. Precisión y sesgo No existe un estándar con el que pueda compararse esta prueba. La variabilidad de las repeticiones está influenciada por la heterogeneidad de la muestra. Si aparecen partículas gruesas de grasa, el elemento de cambio en la toma de muestras puede ser un factor decisivo. Se ha realizado un análisis triplicado de un desagüe de vertidos residuales municipales y dos desagües de plantas envasadoras de carne, conteniendo ambos partículas de grasa significativas. Los promedios para las tres aguas residuales fueron de 2540 48, 57 y 25 mg/1; la desviación estándar promedio fue del 1 l por 100. Una refinería de aceites hizo determinaciones por duplicado de un drenaje en 15 días consecutivos, obteniendo resultados que oscilaron entre 5,1 y 11,2 mg/1. La diferencia promedio entre pares de muestras fue de 0,37 mg/1. 7. Bibliografía P OMEROY , R. D. 1953 Floatability of oil and grease in wastewaters. Sewage Ind. Wastes 25:1304. SÓLIDOS* 2540 A. Introducción Los términos «sólidos», «suspendido» y «disuelto», como se utilizarán de ahora en adelante, reemplazan a los vocablos «residuo», «no filtrable» y «filtrable» de los textos anteriores a la 16.a edición. Sólidos son los materiales suspendidos o disueltos en aguas limpias y aguas residuales. Los sólidos pueden afectar negativamente a la calidad del agua o a su suministro de varias maneras. Las aguas con abundantes sólidos disueltos suelen ser de inferior palatabilidad y pueden inducir una reacción fisiológica desfavorable en el consumidor ocasional. Por estas razones, para las aguas potables es deseable un límite de 500 mg/1 de sólidos disueltos. Las aguas altamente mineralizadas tampoco son adecuadas para muchas aplicaciones industriales o incluso resultan estéticamente insatisfactorias * Aprobado por el Standard Methods Committee, 1985. para bañarse. Los análisis de sólidos son importantes en el control de procesos de tratamiento biológico y físico de aguas residuales, y para evaluar el cumplimiento de las limitaciones que regulan su vertido. 1. Definiciones «Sólidos totales» es la expresión que se aplica a los residuos de material que quedan en un recipiente después de la evaporación de una muestra y su consecutivo secado en estufa a temperatura definida. Los sólidos totales incluyen los «sólidos totales suspendidos», o porción de sólidos totales retenida por un filtro, y los «sólidos disueltos totales» o porción que atraviesa el filtro. El tipo de soporte del filtro, el tamaño del poro, la porosidad, el área y el espesor del filtro, así como la naturaleza física y el tamaño de las partículas y la PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-79 cantidad de material depositado en el filtro son los factores principales que afectan a la separación de los sólidos suspendidos y los disueltos. «Sólidos fijados» es la expresión aplicada al residuo de sólidos totales, suspendidos o disueltos después de someterse a ignición durante un tiempo determinado y a una temperatura especificada. La pérdida de peso por ignición se debe a los «sólidos volátiles». Las determinaciones de sólidos fijados y volátiles no distinguen exactamente entre materias orgánica e inorgánica porque la pérdida de peso por ignición no se limita al material orgánico, sino que incluye también pérdida por descomposición o volatilización de algunas sales minerales. Una óptima caracterización de la materia orgánica puede llevarse a cabo mediante pruebas como la del carbono orgánico total (sección 5310), BOD (sección 5210) y COD (sección 5220). «Líquidos sedimentables» es la expresión aplicada al material que se desprende de la suspensión en un período determinado. Puede incluir material flotable, dependiendo de la técnica (2540F.3b). general muy ligera. Dado que la eliminación de agua ocluida es marginal a esta temperatura, la obtención de peso constante puede ser muy baja. Los residuos secados a 180 ± 2°C perderán casi toda el agua ocluida. Puede permanecer un poco de agua de cristalización, especialmente cuando hay sulfatos. La materia orgánica puede perderse por volatilización, pero no desaparece por completo. La conversión de bicarbonatos en carbonatos produce pérdida de CO2, y los carbonatos pueden descomponerse parcialmente en óxidos o sales básicas. Pueden perderse algunos cloruros y sales nitradas. En general, la evaporación y el secado de muestras de agua a 180 °C proporciona valores sobre sólidos disueltos que están más próximos a los obtenidos mediante suma de las especies minerales determinadas individualmente que a los valores de los sólidos disueltos, logrados mediante secado a la temperatura más baja. Los resultados para residuos ricos en aceite y grasa pueden ser cuestionables debido a la dificultad que supone el secado a peso constante en un tiempo razonable. Los análisis realizados con algún propósito especial pueden exigir una desviación de los procedimientos establecidos para incluir un componente no habitual en los sólidos medidos. Cualesquiera que sean las variaciones técnicas que se introduzcan, éstas deben registrarse y presentarse con los resultados. 2. Fuentes de error y variabilidad La temperatura a la que se seca el residuo incide en gran medida en los resultados, debido a que las pérdidas de peso derivadas de la volatilización de materia orgánica, el agua ocluida, el agua de cristalización y los gases a partir de la descomposición inducida por el calor, así como las ganancias producidas por la oxidación, dependen de la temperatura y tiempo de calentamiento. Los residuos secados a 103-105 °C pueden retener no solamente agua de cristalización, sino también algo de agua ocluida. Como resultado de la conversión del bicarbonato en carbonato, habrá una pérdida de CO2. La pérdida de material orgánico por volatilización será por lo 3. Manipulación y preservación de la muestra Utilícense botellas de plástico o vidrio refractario, teniendo siempre en cuenta que el material en suspensión no debe adherirse a las paredes del recipiente. Iníciese el análisis lo antes posible, pues resulta poco útil preservar la muestra. Refrigérese a 4 °C hasta realizar el análisis, MÉTODOS NORMALIZADOS 2-80 para reducir al mínimo la descomposición microbiológica de los sólidos. también para materiales sólidos y semisólidos producidos durante el tratamiento de aguas limpias y residuales. 4. Selección del método 5. Los métodos B al F son adecuados para la determinación de sólidos en aguas potables, de superficie y salinas, así como para aguas residuales domésticas e industriales, en una amplitud de hasta 20.000 mg/1. El método G es idóneo para la determinación de sólidos en sedimentos y THERIAULT, E. J. & H. H. WAGENHALS. 1923. Studies of representative sewage plants. Pub. Health Bull. No. 132. U. S. ENVIRONMENTAL PROTECTION AGENCY. 1979. Methods for Chemical Analysis of Water and Wates. Publ. 600/4-79-020, Environmental Monitoring and Support Lab., U.S. Environmental Protection Agency, Cincinnati, Ohio. 2540 B. Bibliografía Sólidos totales secados a 103-105 °C 1. Discusión general a) Principio: Se evapora una muestra correctamente mezclada en una placa pesada y secada a peso constante en un horno a 103-105 °C. El aumento de peso sobre el de la placa vacía representa los sólidos totales. Es posible que en muestras de aguas residuales los resultados no representen el peso real de los sólidos disueltos y suspendidos (véase parte anterior). b) Interferencias: El agua fuertemente mineralizada con una concentración significativa de calcio, magnesio, cloruro y/o sulfato puede ser higroscópica y requerir un secado prolongado, una desecación adecuada y un pesado rápido. Elimínense las partículas gruesas flotables o los aglomerados sumergidos de materiales no homogéneos si se decide que su inclusión no es deseable en el resultado final. Dispérsese con un mezclador la grasa y el aceite flotantes antes de separar una porción de muestra para análisis. Puesto que un residuo excesivo en la placa puede formar una costra hidrófila, limítese el tamaño de la muestra para que proporcione un residuo no mayor de 200 miligramos. 2. Instrumental a) Placas de evaporación: Placas de 100 ml de capacidad, fabricadas con uno de los materiales siguientes: 1) Porcelana, 90 mm diámetro. 2) Platino, generalmente satisfactorio para tales fines. 3) Vaso alto de sílice*. b) Horno de mufla para operar a 550 ± 50 °C. c) Baño de vapor. d) Desecador, provisto de un desecante que contiene un indicador colorimétrico de concentración de humedad. e) Horno de secado, para operaciones a 103-105 °C. f) Balanza de análisis, capaz de pesar hasta 0,1 mg. * Vycor, producto de Corning Class Works, Corning, Nueva York, o equivalente. PROPIEDADES FÍSICAS Y DE AGREGACIÓN 3. Procedimiento a) Preparación de la placa de evaporación: Si se va a medir sólidos volátiles, incinérese una placa de evaporación limpia a 550 ± 50 °C durante una hora en un horno de mufla. Si solamente se intentan medir sólidos totales, caliéntese la placa limpia a 103-105 °C durante una hora. Consérvese la placa en el desecador hasta que se necesite. Pesar inmediatamente antes de usar. b) Análisis de la muestra: Elíjase un volumen de muestra que proporcione un residuo entre 2,5 y 200 mg. Transfiérase un volumen medido de muestra bien mezclada a la placa pesada previamente y evapórese hasta que se seque en un baño de vapor o un horno de secado. En caso necesario, añádanse a la misma placa, después de la evaporación, nuevas porciones de muestra. Si la evaporación se lleva a cabo en un horno de secado, reducir la temperatura hasta 2 °C aproximadamente por debajo del punto de ebullición, a fin de evitar salpicaduras. Secar la muestra evaporada al menos durante una hora en horno a 103-105 °C, enfriar la placa en desecador para equilibrar la temperatura y pesar. Repítase el ciclo de secado, enfriado, desecación y pesado hasta obtener un peso constante, o hasta 2540 C. 1. 2-81 que la pérdida de peso sea menor del 4 por 100 del peso previo o menor de 0,5 mg (escoger la menor de ambas). 4. Cálculo donde: A = peso de residuo seco + placa, mg, y B = peso de la placa, mg. 5. Precisión Se realizaron análisis por duplicado, en un solo laboratorio, de 41 muestras de aguas limpias y residuales, con una desviación estándar de diferencias de 6,0 mg/1. 6. Bibliografía SYMONS, G. E. & B. MOREV. 1941. The effect of drying time on the determination of solids in sewage and sewage sludges. Sewage Works J. 13:936. Sólidos totales disueltos secados a 180 °C Discusión general a) Principio: Se filtra una muestra bien mezclada por un filtro estándar de fibra de vidrio; posteriormente, el filtrado se evapora hasta que se seque en una placa pesada y secada a peso constante a 180°C. El aumento del peso de la placa representa los sólidos totales disueltos. Es posible que los resultados no coin- cidan con el valor teórico para sólidos calculado a partir del análisis químico de la muestra (véase parte anterior). Se han puesto a punto métodos aproximativos para correlacionar los análisis químicos con sólidos disueltos1. Para determinar los sólidos totales disueltos, puede utilizarse el filtrado a partir de la determinación de sólidos totales en suspensión (sección 2540D). MÉTODOS NORMALIZADOS 2-82 b) Interferencias: Las aguas excesivamente mineralizadas con un contenido considerable de calcio, magnesio, cloruros y/o sulfatos, pueden ser higroscópicas y exigir un secado prolongado, un grado de desecación adecuado y un pesado rápido. Las muestras ricas en bicarbonato requieren un secado cuidadoso y, probablemente, prolongado, a 180 °C, para asegurar la conversión completa de bicarbonato a carbonato. Puesto que un residuo excesivo en la placa puede formar una costra hidrófila, limítese el tamaño de la muestra para que proporcione un residuo no mayor de 200 mg. 2. Instrumental Además de los aparatos enumerados en la sección 2540B.2a-d, son necesarios: a) Discos de filtrado de fibra de vidrio *, sin aglutinante orgánico. b) Aparato de filtrado: Elíjase de entre los dispositivos siguientes el más adecuado para el disco de filtrado seleccionado: 1) Embudo de filtro de membrana. 2) Crisol de Gooch, 25 a 40 ml de capacidad, con adaptador. 3) Dispositivo de filtrado, con reservono y disco de arandela gruesa (40-60 μm) como soporte del filtro. c) Matraz de succión, de capacidad suficiente para el tamaño de muestra seleccionado. d) Horno de secado, para operaciones a 180 ± 2°C. 3. Procedimiento a) Preparación del disco de filtrado de fibra de vidrio: Insértese el disco con la * Grado Whatman 934AH; tipo Gelman A/E; tipo Millipore AP40; E-D Scientific Specialties, grado 161; o equivalente. Disponible en diámetros de 2,2 a 4,7 cm. cara rugosa hacia arriba en el aparato de filtrado. Hágase el vacío y lávese el disco con tres volúmenes sucesivos de 20 ml de agua destilada. Continuar la succión hasta eliminar todo vestigio de agua. Deséchese el agua de lavado. b) Preparación de la placa de evaporación: Si se van a medir sólidos volátiles, incinérese la placa de evaporación limpia a 550 ± 50 °C durante una hora en un horno de mufla. Si únicamente se desea medir sólidos totales disueltos, caliéntese la placa limpia a 180 ± 2 °C durante una hora en horno. Consérvese en el desecador hasta que se utilice. Pesar inmediatamente antes de usar. c) Selección del filtro y tamaños de la muestra: Elíjase un volumen de muestra que proporcione entre 2,5 y 200 mg de residuo seco. Si se requiere más de 10 minutos para completar el filtrado, se deberá aumentar el tamaño del filtro o disminuir el tamaño de la muestra, pero en cualquier caso no se debe producir menos de 2,5 mg de residuo. d) Análisis de la muestra: Fíltrese el volumen medido de la muestra bien mezclada mediante un filtro de fibra de vidrio, lávese con tres volúmenes sucesivos de 10 ml de agua destilada, permitiendo el drenaje completo del filtro entre los lavados, y continúese succionando durante unos 3 minutos después de terminar el filtrado. Transfiérase el producto a una placa de evaporación pesada y evapórese hasta que se seque en un baño de vapor. Si el volumen de filtrado excediera la capacidad de la placa, añádase a la misma, después de la evaporación, nuevas porciones de muestra. Séquese al menos durante una hora en horno a 180 ± 2 °C, enfríese en un desecador para equilibrar la temperatura y procédase a pesar. Repítase el ciclo de secado, enfriamiento, desecación y pesado hasta obtener un peso constante o hasta que la pérdida de peso sea menor del 4 por 100 del peso previo o menos de 0,5 mg (escoger la menor de ambas). 2-83 PROPIEDADES FÍSICAS Y DE AGREGACIÓN 4. Cálculo 6. Referencia SOKOLOFF, V. P. 1933. Water of crystallization in total solids of water analysis. Ind. Eng. Chem., Anal. Ed. 5:336. donde: A = peso de residuo seco + placa, mg, y B = peso de la placa, mg. 5. Precisión Se llevó a cabo en un solo laboratorio el análisis de 77 muestras de un peso de 293 mg/1, con una desviación estándar de diferencias de 21,20 mg/1. 2540 D. 1. 7. Bibliografía HOWARD, C. S. 1933. Determination of total disolved solids in water analysis. Ind. Eng. Chem. Anal. Ed. 5:4. U. S. GEOLOGICAL SURVEY. 1974. Methods for Collection and Analysis of Water Samples for Dissolved Minerals and Gases. Techniques of Water-Resources Investigations, Book 5, Chap. A 1. U. S. Geological Surv., Washington, D.C. Sólidos totales en suspensión secados a 103-105 °C Discusión general a) Principio: Se filtra una muestra bien mezclada por un filtro estándar de fibra de vidrio, y el residuo retenido en el mismo se seca a un peso constante a 103105 °C. El aumento de peso del filtro representa los sólidos totales en suspensión. Si este material obtura el filtro y prolonga la operación de filtrado, la diferencia entre el total de sólidos y el total de sólidos disueltos puede proporcionar un cálculo aproximado de los sólidos totales en suspensión. b) Interferencias: Elimínense de la muestra las partículas gruesas flotables o los aglomerados sumergidos de materiales no homogéneos, si se decide que su inclusión no es deseable en el resultado final. Puesto que un residuo excesivo sobre el filtro puede formar una costra hidrófila, limítese el tamaño de la muestra para que proporcione un residuo no mayor de 200 mg. Para las muestras ricas en sólidos disueltos, lávese meticulosamente el filtro para asegurar la eliminación del material disuelto. Los tiempos de filtración prolongados, consecuencia de la obturación del filtro, pueden originar resultados altos debido a una cantidad excesiva de sólidos capturados en el filtro obturado. 2. Instrumental Además de los aparatos enumerados en las secciones 2540B.2 y 2540C.2, con excepción de las placas de evaporación, el baño de vapor y el horno de 180 °C, se requiere una: Plancheta*, acero inoxidable o aluminio, 65 mm de diámetro. 3. Procedimiento a) Preparación del disco de filtrado de fibra de vidrio: Insértese el disco con la * Disponible en New England Nuclear, Boston, Massachusetts, o equivalente. MÉTODOS NORMALIZADOS 2-84 cara rugosa hacia arriba en el aparato de filtrado. Hágase el vacío y lávese el disco con tres volúmenes sucesivos de 20 ml de agua destilada. Continúese succionando hasta1 eliminar todo vestigio de agua, y retírese el agua de lavado. Quítese el filtro del aparato de filtrado y trasládese a una plancheta de aluminio o acero inoxidable. Alternativamente, procédase a separar el crisol y la combinación de filtro si se está utilizando un crisol de Gooch. Séquese en horno a 103-105 °C durante una hora. Si se van a medir sólidos volátiles, incinérese a 550 ± 50 °C en horno de mufla, enfríese en desecador para equilibrar la temperatura y procédase a pesar. Repítase el ciclo de secado o incineración, enfriamiento, desecación y pesado hasta obtener un peso constante o hasta que la pérdida de peso sea menor de 0,5 mg entre pesadas sucesivas. Consérvese en desecador hasta que se necesite. Pesar inmediatamente antes de usar. b) Selección del filtro y tamaños de la muestra: Véase sección 2540C.3c. Para muestras no homogéneas como agua residual no tratada, utilícese un filtro ancho para permitir el filtrado de una muestra representativa. c) Análisis de la muestra: Móntese el aparato de filtrado y el filtro e iníciese la succión. Para ajustar el filtro, humedézcase éste con una pequeña cantidad de agua destilada. Fíltrese un volumen medido de muestra bien mezclada por el filtro de fibra dé vidrio. Lávese con tres volúmenes sucesivos dé 10 mí de agua destilada, permitiendo el drenaje completó del filtro entre los lavados, y continúese succionando durante unos tres minutos después de terminar el filtrado. Sepárese cuidadosamente el filtro del aparato y trasládese a una plancheta de aluminio o acero inoxidable. Alternativamente, procédase a separar el crisol y la combinación de filtro del adaptador del crisol, si se está utilizando un crisol de Gooch. Séquese en horno a 103-105 °C durante una hora al menos, enfríese en un deseca- dor para equilibrar la temperatura y pésese. Repetir el ciclo de secado, enfriamiento, desecación y pesado hasta obtener un peso constante o hasta que la pérdida de peso sea menor del 4 por 100 del peso previo o menor de 0,5 mg (escoger la menor de ambas). 4. Cálculo donde: A = peso del filtro + residuo seco, mg B = peso del filtro, mg. 5. Precisión En los estudios efectuados por dos analistas sobre cuatro series de 10 determinaciones cada una, la desviación estándar fue de 5,2 mg/1 (coeficiente de variación 33 por 100) a 15 mg/1; 24 mg/1 (10 por 100) a 242 mg/1, y 13 mg/1 (0,76 por 100) a 1.707 mg/1. Se realizaron análisis por duplicado, en un solo laboratorio, de muestra de aguas naturales y residuales, con una desviación estándar de diferencias de 2,8 mg/1. 6. Bibliografía DEGEN, J & F. E. NUSSBERGER. 1956. Notes on the determination of suspended solids. Sewage Ind. Wastes 28:237. C HANIN , G., E. H. C HOW , R. B' A LEXANDER & J. PoWERS. 1958. Use of glass fiber filter mediumin the suspended solids determination. Sewage Ind. Wastes 30:1062. NUSBAUM, I. 1958. New method for determination of suspended solids. Sewage Ind. Wastes 30:1066. PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-85 SMITH, A. L. & A. E. GREENBERG. 1963. Evaluation of methods for determining suspended solids in wastewater. J. Water Pollut. Control Fed. 35:940. WYCKOFF, B. M. 1964. Rapid solids determination using glass fiber filters. Water Sewage Works 111:277. NATIONAL COUNCIL OF THE PAPER INDUSTRY FOR AIR AND STREAM IMPROVEMENT. 1975. A Preliminary Review of Analytical Methods for the Determination of Suspended Solids in Paper Industry Effluents for Compliance with EPA-NPDES Permit Terms. Spec. Rep. No. 75-01. National Council of the Paper Industry for Air & Stream Improvement, Nueva York. NATIONAL COUNCIL OF THE PAPER INDUSTRY FOR AIR AND STREAM IMPROVEMENT. 1977. A Study of the Effect of Alternate Procedures on Effluent Suspended Solids Measurement. Stream Improvement Tech. Bull. No. 291, National Council of the Paper Industry for Air & Stream Improvement, Nueva York. TREES, C. C. 1978. Analytical analysis of the effect of dissolved solids on suspended solids determination. J. Water Pollut. Control Fed. 50:2370. 2540 E. 1. Sólidos fijos y volátiles incinerados a 550 °C Discusión general a) Principio: El residuo obtenido con los métodos explicados en las secciones B, C o D se incinera, a peso constante, a una temperatura de 550 ± 50 °C. Los sólidos remanentes representan los sólidos totales fijos, disueltos o en suspensión, mientras que la pérdida de peso por ignición representa los sólidos volátiles. La determinación es útil para el control de las operaciones en plantas de tratamiento de aguas residuales, porque ofrece un cálculo aproximado de la cantidad de materia orgánica presente en la fracción sólida del agua residual, lodos activados y residuos industriales. b) Interferencias: Durante el proceso de secado pueden producirse errores negativos en los sólidos volátiles por pérdida de materia evaporable. La determinación de bajas concentraciones de sólidos volátiles en presencia de concentraciones elevadas de sólidos fijos puede estar sujeta a errores considerables. En tal caso, deberán medirse los compuestos volátiles mediante otra prueba, por ejemplo, la del carbono orgánico total (sección 5310). 2. Instrumental Véanse secciones 2540B.2, 2540C.2 y 2540D.2. 3. Procedimiento Incinérese el residuo producido por los métodos explicados en las secciones B, C o D, a peso constante, en un horno de mufla a temperatura de 550 ± 50 °C. Se deberá elevar el horno a esta temperatura antes de introducir la muestra. Por lo general, la incineración sólo precisa de 15 a 20 minutos. Enfríese la placa o el disco de filtro al aire hasta que se haya disminuido el calor y transfiéranse a un desecador para proceder a su enfriamiento final en una atmósfera seca, cuidando de no sobrecargar el desecador. Pésense la placa o el disco tan pronto como se hayan enfriado para equilibrar la temperatura. Repetir el ciclo de incineración, enfriado, desecación y pesado hasta obtener un peso constante o hasta que la pérdida de peso sea menor del 4 por 100 del peso previo. 4. Cálculo 2-86 MÉTODOS NORMALIZADOS 5. Precisión donde: A = peso de residuo + placa antes de incineración, mg, B = peso de residuo + placa o filtro después de la incineración, mg, y C = peso de la placa o filtro, mg. 2540 F. 1. Sólidos sedimentables Discusión general Los sólidos sedimentables de las aguas de superficie y salinas, así como de los residuos domésticos e industriales, pueden ser determinados y expresados en función de un volumen (ml/l) o de un peso (mg/1). 2. Instrumental La prueba volumétrica requiere solamente un cono de Imhoff. En la gravimétrica, son necesarios todos los instrumentos enumerados en la sección 2540D.2 y un vaso de vidrio con un diámetro mínimo de 9 cm. 3. En los estudios realizados por tres laboratorios sobre 4 muestras y 10 replicados, la desviación estándar fue de 11 mg/1 a 170 mg/1 de sólidos totales volátiles. No se pudieron obtener datos de sesgo de muestras reales. Procedimiento a) Volumétrico: Llénese un cono de Imhoff hasta la marca 1-1 con una muestra bien mezclada. Déjese sedimentar durante 45 minutos, removiendo a continuación suavemente las paredes del cono con una varilla o mediante rotación; manténgase en reposo 15 minutos más y regístrese el volumen de sólidos sedimentables del cono como milímetros por litro. Si la materia sedimentada contiene bolsas de líquido entre partículas gruesas, evalúese el volumen de aquéllas y réstese del volumen de sólidos sedimen- tados. El límite inferior práctico de la medición depende de la composición de la muestra y, en general, es del orden de 0,1 a 1,0 ml/l. En caso de producirse una separación de materiales sedimentables y notables, no deben valorarse estos últimos como material sedimentable. b) Gravimétrico: 1) Determínense los sólidos totales en suspensión de una muestra bien mezclada (sección 2540D). 2) Viértase una muestra bien mezclada en un vaso de vidrio de no menos de 9 cm de diámetro, con capacidad para 1 l y con una profundidad de 20 cm. Alternativamente, utilizar un vaso de vidrio de diámetro superior y mayor volumen de muestra. Déjese en reposo durante una hora y, sin remover el material sedimentado o flotable, extráiganse 250 ml desde el centro del recipiente en un punto a medio camino entre las superficies del material sedimentado y del líquido. Determínense los sólidos totales suspendidos (miligramos por litro) de este líquido sobrenadante (sección 2540D). Ésos son los sólidos no sedimentables. 4. Cálculo mg de sólidos sedimentables/l = mg de sólidos totales en suspensión/l – mg de sólidos no sedimentables/l PROPIEDADES FÍSICAS Y DE AGREGACIÓN 5. Precisión y sesgo No se dispone actualmente de datos de precisión y sesgo. 2540 G. 1. 2-87 6. Bibliografía FISCHER, A. J. & G. E. SYMONS. 1944. The determination of settleable sewage solids by weight. Water Sewage Works 91:37. Sólidos totales, fijos y volátiles en muestras sólidas y semisólidas Discusión general a) Aplicabilidad: Este método se aplica en la determinación de sólidos totales y sus fracciones fijas y volátiles en muestras sólidas y semisólidas como sedimentos de río o lago, lodos aislados en procesos de tratamiento de aguas limpias y residuales y aglomeraciones de lodo de filtrado al vacío, de centrifugación u otros procesos de deshidratación de lodos. b) Interferencias: La determinación de sólidos, tanto volátiles como totales, en dichos materiales está sujeta a error negativo, debido a la pérdida de carbonato amónico y materia orgánica volátil experimentada durante la desecación. Aunque otro tanto sucede con las aguas residuales, el efecto tiende a ser más pronunciado con los sedimentos y, especialmente, con los lodos y aglomeraciones de lodos. La masa de materia orgánica recuperada del lodo y el sedimento requiere un tiempo de ignición más prolongado que el especificado para aguas residuales. Obsérvese cuidadosamente el tiempo y la temperatura de ignición especificados para controlar las pérdidas de sales inorgánicas volátiles. Realícese el pesado inmediatamente, ya que las muestras húmedas tienden a perder peso por evaporación. Después del secado o de la ignición, los residuos son a menudo muy higroscópicos y absorben rápidamente humedad del aire. 2. Instrumental Se requieren todos los instrumentos enumerados en la sección 2540B.2; también puede utilizarse una balanza capaz de pesar hasta 10 mg. 3. Procedimiento a) Sólidos totales: 1) Preparación de la placa de evaporación: Si van a medirse sólidos volátiles, incinérese una placa de evaporación limpia en un horno de mufla a 550 ± 50 °C durante una hora. Si solamente se desea medir sólidos totales, caliéntese la placa en un horno a 103-105 °C durante una hora. Enfríese en desecador, pésese y consérvese en el desecador hasta que haya de usarse. 2) Análisis de la muestra: a) Muestras líquidas: Si la muestra contiene suficiente humedad para fluir con mayor o menor facilidad, agítese para homogeneizarla; a continuación colóquese de 25 a 50 g en una placa de evaporación y pésese. Evapórese hasta desecación al baño María, séquese a 103105 °C durante una hora, enfríese para equilibrar la temperatura en un desecador individual con desecante activo, y pésese. b) Muestras sólidas: Si la muestra consta de partículas discretas de material 2-88 MÉTODOS NORMALIZADOS sólido (lodo deshidratado, por ejemplo), extráiganse los fragmentos con un sacabocados del n.° 7 o pulverícese toda la muestra sobre una superficie limpia con los dedos, utilizando guantes de goma. Desposítese de 25 a 30 g en una placa de evaporación y pésese. Déjese en un horno a 103-105 °C durante toda la noche. Enfríese para equilibrar la temperatura en un desecador individual con desecante activo y pésese. b) Sólidos fijos y volátiles: Transfiérase la muestra a un horno de mufla frío, caliéntese éste hasta 550 ± 50 °C e incinérese durante una hora. [Si el residuo obtenido en el apartado 2) contiene grandes cantidades de materia orgánica, incinérese primero el residuo en un quemador de gas protegido por una campana de extracción, en presencia de aire suficiente para disminuir pérdidas por reducción y evitar olores en el laboratorio.] Enfríese en desecador para equilibrar la temperatura y pésese. 4. Cálculo 2550 donde: A = peso del residuo seco + placa, mg, B = peso de la placa, C = peso de la muestra húmeda + placa, mg, y D = peso del residuo + placa después de ignición, mg. 5. Precisión y sesgo No se dispone actualmente de datos de precisión y sesgo. 6. Bibliografía GOODMAN , B. L. 1964. Processing thickened sludge with chemical conditioners. Pags. 78 y sig. En Sludge Concentration, Filtration and Incineration. Univ. Michigan Continued Education Ser. No. 113, Ann Arbor. GRATTEAU, J. C. & R. I. DICK. 1968. Activated sludge suspended solids determinations. Water Sewage Works 115:468. TEMPERATURA* 2550 A. La lectura de cifras de temperatura se utiliza en el cálculo de diversas formas de alcalinidad, en estudios de saturación y estabilidad respecto al carbonato de calcio, en el cálculo de la salinidad y en las operaciones generales de laboratorio. En los estudios limnológicos, con frecuencia se requieren temperaturas de agua en * Aprobado por el Standard Methods Committee, 1988. Introducción función de la profundidad. Las temperaturas elevadas, consecuencia de descargas de agua calentada, pueden tener un impacto ecológico significativo. A menudo, la identificación de la fuente de aporte hídrico, como en los manantiales profundos, sólo es posible efectuando medidas de temperatura. Las plantas industriales suelen pedir datos de temperatura del agua para uso sistemático o cálculos de transmisión de calor. PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2550 B. 2-89 Métodos de laboratorio y de campo 1. Medidas de temperatura de aguas no profundas en laboratorios y otras valoraciones de temperatura de aguas no profundas Normalmente, las medidas de temperatura pueden realizarse con cualquier termómetro Celsius de mercurio, que, como mínimo, deberá tener una escala con marcas cada 0,1 °C sobre el tubo capilar y una capacidad térmica mínima que permita un equilibrado rápido. Compruébese periódicamente con un termómetro de precisión certificado por el National Institute of Standards and Technology (NIST, antes National Bureau of Standards)*, que se utiliza con su certificado y cédula de corrección. Para operaciones de campo, utilícese un termómetro con estuche metálico a fin de evitar roturas. de muestras, de modo que puede obtenerse simultáneamente una muestra de agua. Corríjanse las lecturas de los termómetros reversibles respecto a los cambios debidos a diferencias entre temperaturas en la reversión y temperatura en el momento de la lectura. Calcúlese del modo siguiente: donde: 'T = corrección para sumar algebraicamente a la lectura no corregida, T l = lectura no corregida en la reversión, t = temperatura a la que se lee el termómetro, V o = volumen de la ampolleta final del capilar hasta graduación de 0 °C, K = constante que depende de la expansión térmica relativa del mercurio y el vidrio (valor usual de K = 6.100), y L = corrección del calibrado del termómetro dependiendo de T l . 2. Medidas de temperaturas de aguas profundas Las temperaturas de aguas profundas requeridas por los estudios limnológicos pueden medirse con un termómetro reversible, un termófono o un termistor. El termistor es el más conveniente y exacto, pero su elevado precio dificulta su uso. Antes de utilizarse en mediciones de campo, calíbrese cualquiera de los mencionados dispositivos de medida de temperatura con un termómetro certificado del NIST. Efectúense las lecturas con el termómetro o dispositivo similar sumergido en el agua el tiempo suficiente para permitir un equilibrado total. Anótense los valores que más se aproximen a 0,1 o 1,0 °C. El termómetro utilizado generalmente para medidas de profundidad es el termómetro de tipo reversible. A menudo está montado en el aparato de recogida * Algunos termómetros comerciales pueden inducir errores de hasta 3 °C. Si se efectúan observaciones senadas, es conveniente preparar gráficas termométricas, para obtener 'T en función de los valores de T1 y t. 3. Bibliografía WARREN, H. F. & G. C. WHIPPLE. 1895. The thermophone—A new instrument for determining temperatures. Mass. Inst. Technol. Quart. 8:125. SVERDRUP, H. V., M. W. JOHNSON & R. H. FLEMING, 1942. The Oceans. Prentice- Hall, Inc., Englewood Cliffs, Nueva Jersey. AMERICAN SOCIETY FOR TESTING AND MATERIALS. 1949. Standard Specifícations for ASTM Thermometers. Núm. El-58, ASTM, Filadelfia, Pennsylvania. REE, W. R. 1953. Thermistors for depth thermometry. J. Amer. Water Works Assoc. 45:259. 2-90 MÉTODOS NORMALIZADOS 2710 PRUEBAS EN LODOS* 2710 A. Introducción En esta sección se presenta una serie de pruebas únicamente aplicables a lodos * Aprobado por el Standard Methods Committee, 1985. 2710 B. 1. Tasa de consumo de oxígeno Discusión general Esta prueba se usa para determinar la tasa de consumo de oxígeno de una muestra de suspensión biológica, como un lodo activado. Resulta útil en estudios de laboratorio y de planta-piloto, así como en operaciones de plantas de tratamiento a gran escala. Cuando se emplee como prueba rutinaria de operación de planta, a menudo indicará los cambios de las condiciones operativas en etapas iniciales. Sin embargo, dado que las condiciones de prueba no son necesariamente idénticas a las del lugar de toma de la muestra, es posible que las medidas observadas no sean iguales a la tasa real de consumo de oxígeno. 2. o fangos. Los datos de prueba son útiles para diseñar instalaciones destinadas a la separación y concentración de sólidos y para evaluar su conducta operativa, en especial la de los procesos de activación de lodos. Instrumental a) Dispositivos de tasa de consumo de oxígeno: Utilícese cualquiera de los siguientes: 1) Sonda con un electrodo sensible al oxígeno (polarográfico o galvánico), o bien 2) dispositivo manométrico o respirométrico, con escala de lectura adecuada y capacidad para muestra de al menos 300 ml. El dispositivo deberá contar con una capacidad de aporte de oxígeno mayor que la tasa de consumo de oxígeno de la suspensión biológica, o como mínimo de 150 mg/l·h. b) Cronómetro, o cualquier otro dispositivo de medida de tiempo apropiado. c) Termómetro, para lecturas de ±0,5 °C. 3. Procedimiento a) Calibración del dispositivo de tasa del consumo de oxígeno: Sígase cualquiera de los siguientes procedimientos: 1) Calibrar la sonda y el medidor de oxígeno con arreglo al método expuesto en la sección 4500-O.G, o bien 2) Calibrar el dispositivo manométrico o respirométrico de acuerdo con las instrucciones del fabricante. b) Determinación de sólidos volátiles en suspensión: Véase sección 2540. c) Preparación de la muestra: Ajústese la temperatura de una porción ade- PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-91 cuada de la muestra a la del recipiente del que fue recogida o a la temperatura de evaluación requerida, y manténgase constante durante el análisis. Regístrese la temperatura. Auméntese la concentración de OD de la muestra agitándola en una botella parcialmente llena, o inyectando aire u oxígeno. d) Medición de la tasa de consumo de oxígeno: más bajos limitantes del OD. Un OD bajo (< 2 mg/1 al comienzo de la prueba) puede limitar la captación de oxígeno por la suspensión biológica y se manifestará en una tasa decreciente de consumo de oxígeno a medida que la prueba progresa. Rechácense estos datos ya que no son representativos de la tasa de consumo y repítase la prueba empezando por niveles iniciales más altos de OD. Los resultados de esta determinación son bastante sensibles a las variaciones de temperatura y su exactitud será mínima a menos que se hagan determinaciones por duplicado a la misma temperatura. Cuando se emplee el consumo de oxígeno como prueba de control de planta, realícense determinaciones duplicadas periódicas (al menos mensuales) para establecer la precisión de la técnica. Esta determinación también es sensible al lapso de tiempo transcurrido entre la toma de la muestra y la iniciación de la prueba. 1) Llénese el recipiente hasta rebosar con un volumen adecuado de muestra representativa de la suspensión biológica que se va a someter a prueba. 2) Si se usa una sonda sensible al oxígeno, introdúzcase inmediatamente en una botella de ROB que contenga una varilla de agitación magnética y la suspensión. Obténgase con la sonda una suspensión suficiente hasta llenar el borde de la botella y protéjase su contenido de la atmósfera. Actívese el mecanismo de agitación de la onda y el agitador magnético. (NOTA: ES precisa una homogeneización suficiente. Para suspensiones con concentraciones elevadas de sólidos en suspensión, esto es, > 5.000 mg/1, puede ser necesaria una agitación más enérgica que la proporcionada por el mecanismo de agitación de la sonda y por el agitador magnético.) Si se utiliza un dispositivo manométrico o respirométrico, síganse las instrucciones de puesta en marcha del fabricante. 3) Una vez estabilizada la lectura, anótense el nivel de OD inicial y la lectura manométrica o respirométrica y comiéncese a cronometrar. Regístrense, con intervalos de menos de un minuto, dependiendo de la tasa de consumo, los niveles adecuados de OD y los datos respiro-manométricos, durante 15 minutos o hasta que el OD llegue al límite. La extracción de oxígeno puede arrojar valores inexactos por debajo de 1 mg de OD/1. Si se utiliza un dispositivo respirométrico o manométrico, síganse las instrucciones del fabricante para valores 4. Cálculos Si se usa una sonda de oxígeno, puntéense las lecturas registradas (OD, mg/1) frente a tiempo (minutos) en papel mllimetrado, determinando la inclinación de la curva de ajuste óptimo. Dicha inclinación representa la tasa de consumo de oxígeno en mlligramos por litro por mlnuto. Si se utiliza un dispositivo manométrico o respirométrico, síganse las instrucciones del fabricante para calcular la tasa de consumo de oxígeno. Calcúlese la tasa específica de consumo de oxígeno en mlligramos por gramo por hora de la manera siguiente: Tasa específica de consumo de oxígeno (mg/g)/h tasa de consumo de oxígeno mg/l min 60min = u sólidos volátiles en suspensión, g/l h 2-92 MÉTODOS NORMALIZADOS 5. Precisión y sesgo 6. El sesgo no es aplicable. No se ha determinado la precisión de esta prueba. UMBREIT, W. W., R. H. BURRIS & J. F. STAUFFER. 1964. Manometric Techniques. Burgess Publishing Co., Minneapolis, Minnesota. 2710 C. 1. Bibliografía Volumen de lodo sedimentado Discusión general El volumen de lodo sedimentado de una suspensión biológica es útil para la monitorización rutinaria de procesos biológicos. Para el control de una planta de lodos activados, con el fin de determinar la tasa de flujo de retorno del lodo y cuándo debe desecharse éste, se ha utilizado el volumen del lodo sedimentado en 30 minutos o el índice del volumen sedimentado entre 15 y 30 minutos. El volumen de 30 minutos también se ha empleado para determinar el índice de volumen de lodo1 (sección 2710D). 2. Instrumental a) Columna de sedimentación: Utilícese un cilindro graduado de 1 l, equipado con un mecanismo de agitación que conste de una o más varillas finas, extendidas a lo largo de la columna y situadas a dos diámetros de varilla de la pared del cilindro. Se dispondrá de un agitador capaz de rotar las varillas a no mas de 4 rmp (velocidad punta periférica de 1,3 cm/s aproximadamente). Véase figura 2710:1. b) Cronómetro. c) Termómetro. 3. Procedimiento Colóquese 1,0 1 de la muestra en la columna de sedimentación y distribuyanse los sólidos cubriendo la parte superior Figura 2710:1. Diagrama esquemático de un vaso de sedimentación para pruebas de volumen de lodo sedimentado. e invirtiendo el cilindro tres veces. Insértense las varillas de agitación, actívese el mecanismo agitador y déjese sedimentar la suspensión. Continuar agitando a lo largo de la prueba, manteniendo una temperatura de suspensión igual a la que tenía la muestra en el recipiente de la que fue tomada. Determínese el volumen ocupado por la suspensión a intervalos medidos, por ejemplo, 5, 10, 15, 20, 30, 45 y 60 mlnutos. 2-93 PROPIEDADES FÍSICAS Y DE AGREGACIÓN Regístrese el volumen de lodo sedimentado en milímetros para un intervalo de tiempo dado. Las variaciones que se produzcan en la temperatura de la suspensión, los métodos de toma de muestra y de agitación, el diámetro de la columna de sedimentación y el intervalo de tiempo entre la toma de la muestra y el comienzo de la determinación influyen significativamente en los resultados. 2710 D. 1. 4. El sesgo no es aplicable. No se ha determinado la precisión de este método. 5. 1. D ICK , R. I. & P. A. V ESILIND . 1969. The SVI–What is it? J. Water Pollut. Control Fed. 41:1285. Discusión general 4. 5. Procedimiento Determínese la concentración de sólidos en suspensión de una muestra homogénea de la suspensión (véase sección 2540D). Determínese el volumen de lodo sedimentado en 30 minutos (véase sección 2710C). Cálculo Precisión y sesgo La precisión se determina por la precisión obtenida en la medida de sólidos en suspensión, las características de sedimentación de la suspensión y las variables asociadas a la medida del volumen de lodo sedimentado. No es aplicable el sesgo. 1. 3. Referencia índice de volumen de lodo El índice de volumen de lodo (IVL) es el volumen en mililitros ocupado por 1 g de una suspensión después de 30 minutos de sedimentación. El IVL se utiliza típicamente para monitorizar las características de sedimentación del lodo activado y otras suspensiones biológicas1. Aunque el IVL carece de apoyo teórico2, la experiencia ha demostrado su utilidad para el control de procesos rutinarios. 2. Precisión y sesgo 2. 6. Referencias D ICK , R. I. & P. A. V ESILIND . 1969. The SVI–What is it? J. Water Pollut. Control Fed. 41:1285. F INCH, J. & H. IVES. 1950. Settleability indexes for activated sludge. Sewage lnd. Wastes 22:833. Bibliografía DONALDSON, W. 1932. Some notes on the operation of sewage treatment works. Sewage Works J. 4:48. MOHLMAN, F. W. 1934. The sludge Index. Sewage Works J. 6:119. RUDOLFS, W. & I. O. LACY. 1934. Settling and compacting of activated sludge. Sewage Works J. 6:647. 2-94 MÉTODOS NORMALIZADOS 2710 E. Tasa de sedimentación de zona 1. Discusión general A concentraciones elevadas de sólidos en suspensión, las suspensiones sedimentan según un modelo de sedimentación de zona. Este tipo de sedimentación tiene lugar en condiciones de reposo y se caracteriza por una interfase neta entre el líquido sobrenadante y la zona del lodo. La altura de dicha interfase del lodo se mide en función del tiempo. Los datos de sedimentación zonal para suspensiones que se han sometido a este tipo de sedimentación, por ejemplo, las de lodos activados y las de hidróxidos metálicos, pueden utilizarse en el diseño, operatividad y evaluación de cubetas de sedimentación1, 3. 2. Instrumental a) Vaso de sedimentación: Utilícese un cilindro transparente de 1 m de altura y 10 cm de diámetro como mínimo. Para reducir las discrepancias entre los resultados de laboratorio y los más voluminosos a gran escala, utilícense diámetros mayores y cilindros más altos1, 3. Fíjese una cinta milimétrica calibrada en la parte exterior del cilindro, que deberá equiparse con un mecanismo agitador, por ejemplo, una o más varillas colocadas a dos diámetros de varilla de la pared interna del vaso. Agítese la suspensión cerca de la pared en toda su profundidad, a una velocidad periférica no superior a 1 cm/s. Las velocidades mayores pueden interferir el proceso de espesamiento y arrojar resultados inexactos4. Prepárese el vaso de sedimentación con una abertura en la placa del fondo para llenado y drenaje. Véase figura 2710:2. b) Cronómetro. c) Termómetro. A = 10 cm B = 2 cm mínimo mínimo Figura 2710:2. Diagrama esquemático de un vaso de sedimentación para prueba de tasa de sedimentación de zona. 3. Procedimiento Manténgase la suspensión en un reservorio, en condiciones de mezclado uniforme. Ajústese la temperatura de la suspensión a la del recipiente de donde se tomó o a la requerida para la evaluación, registrando las cifras. Extráigase una muestra homogénea del reservorio y mídase la concentración de sólidos en suspensión (sección 2540D). Actívese el mecanismo de agitación. Llénese el vaso de sedimentación a una 2-95 PROPIEDADES FÍSICAS Y DE AGREGACIÓN altura fija, mediante bombeo de la suspensión o por la fuerza de gravedad. Llénese a un ritmo constante para mantener una concentración uniforme de sólidos en suspensión en toda la extensión del vaso. La suspensión debe aglomerarse, es decir, formar en pocos minutos una estructura espesa con canales de líquido visibles. Si la suspensión no se aglomera, la prueba no es válida y debe repetirse. Regístrese la altura de la interfase sólidos-líquidos a intervalos de un minuto aproximadamente. Recójanse datos durante el tiempo suficiente para garantizar que la suspensión exhibe una velocidad constante de sedimentación de zona y que ha pasado un posible período de refloculación inicial, caracterizado por una velocidad acelerada de sedimentación interfase. La tasa de sedimentación de zona es una función de la concentración de sólidos en suspensión y de la altura de la muestra, así como de los artefactos de laboratorio3. Con el método de llenado descrito anteriormente y un cilindro suficientemente ancho, esos artefactos deben reducirse al mínimo. Sin embargo, incluso con pruebas realizadas meticulosamente, las suspensiones pueden comportarse de forma errática. La conducta imprescindible aumenta para lodos con concentraciones elevadas de sólidos y características pobres de sedimentación, y en los cilindros pequeños. 4. Cálculos Puntéese la altura de interfase en centímetros frente a tiempo en mlnutos1, 3. Trácese una recta a través de los puntos, ingnorando el codo inicial o período de refloculación, y el codo de compresión. Calcúlese la tasa de sedimentación interfase como desviación de la línea en centímetros por minuto. 5. Precisión y sesgo El sesgo no es aplicable. No se ha determinado la precisión de esta prueba. 6. 1. 2. 3. 4. 7. Referencias DICK, R. I. 1972. Sludge treatment. En W. J. Weber, ed, Physicochemical Processes for Water Quality Control. Wiley-Interscience, Nueva York. DICK, R. I. & K. W. YOUNG. 1972. Analysis of thickening performance of final settling tanks. Proc. 27th Ind. Waste Conf., Purdue Univ., Eng. Ext. Ser. Núm. 141, 33. VESILIND, P. A. 1975. Treatment and Disposal of Wastewater Sludges. Ann Arbor Science Publishing Co., Ann Arbor, Michigan. VESILIND, P. A. 1968. Discussion of Evaluation of activated sludge thickening theories. J. San. Eng. Div., Proc. Amer. Soc. Civil Eng. 94:SA1, 185. Bibliografía DICK, R. I. & R. B. EWING. 1967. Evaluation of activated sludge thickening theories. J. San. Eng. Div., Proc. Amer. Soc. Civil Eng. 93:SA4, 9. D ICK , R. I. 1969. Fundamental aspects of sedimentation I & II. Water Wastes Eng. 3:47, 45, & 6:2. DICK, R. I. 1970. Role of activated sludge final settling tanks. J. San. Eng. Div., Proc. Amer. Soc. Civil Eng. 96:SA2, 423. 2-96 MÉTODOS NORMALIZADOS 2710 F. 1. Gravedad específica Discusión general La gravedad específica de un lodo es la relación entre las masas, a volúmenes iguales, del lodo y el agua destilada. Se determina por comparación de la masa de un volumen conocido de una muestra de lodo homogéneo, a temperatura dada, y la masa del mismo volumen de agua destilada a 4 °C. 2. Cálculo Utilícese a o b, adaptando la elección a los métodos descritos anteriormente. a) Calcúlese la gravedad específica, GE, a partir de la fórmula: Instrumental Recipiente: Un matraz marcado o botella para contener durante la pesada un volumen conocido de lodo. 3. 4. Los valores del factor F de corrección de temperatura se dan en la tabla 2710:1. b) Calcúlese la gravedad específica, GE, a partir de la fórmula: Procedimiento Sígase cualquiera de los procedimientos explicados a continuación: a) Regístrese la temperatura de la muestra, T. Pésese el recipiente vacío y regístrese el peso, W. Llénese el recipiente con muestra hasta la marca y regístrese el peso, S. Llénese el recipiente vacío con agua, pésese y regístrese igualmente el peso, R. Mídanse todas las masas con un límite de error máximo de 10 mg. b) Si la muestra no fluye fácilmente, añádase al recipiente tanta cantidad de muestra como sea posible sin ejercer presión, anótese el volumen, pésese y regístrese la masa, P. Llénese el recipiente hasta la señal con agua destilada, teniendo cuidado de no atrapar burbujas de aire en el lodo o en el recipiente. Pésese y anótese la masa, Q. Mídanse todas las masas con un límite de error máximo de 10 mg. Para valores de F, véase tabla 2710:1. TABLA 2710:I. FACTOR DE CORRECCIÓN DE LA TEMPERATURA Temperatura °C Factor de corrección de la temperatura 15 0,9991 20 25 30 35 40 45 0,9982 0,9975 0,9957 0,9941 0,9922 0,9903 2-97 PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2720 GAS DIGESTOR DE LODO* 2720 A. Introducción El gas producido durante la descomposición anaerobia de residuos contiene metano (CH4) y dióxido de carbono (CO2) como componentes principales, con cantidades menores de hidrógeno (H2), sulfuro de hidrógeno (H2S), nitrógeno (N2) y oxígeno (O2). Está saturado con vapor de agua. La práctica común es analizar los gases producidos para estimar su valor de combustión y verificar el proceso de tratamiento. Las proporciones relativas de CO2, CH4 y N2 son normalmente las de mayor interés y las más fáciles de determinar debido a los porcentajes relativamente elevados de estos gases. 1. Selección del método Se han descrito dos métodos de análisis de gas: el volumétrico (B) y el de cromatografía de gases (C). El análisis volumétrico se aplica a la determinación de CO2, H2,CH4 y O2. El nitrógeno se evalúa indirectamente mediante diferencias. Aunque el método requiere largo tiempo, el instrumental es relativamente simple. Puesto que no es necesaria la calibración antes de su uso, este método es especialmente adecuado para análisis que se practican con poca frecuencia. La ventaja principal de la cromatografía de gases es su rapidez. El instrumental comercial se diseña específicamente para el análisis del gas a temperatura ambiente y permite la separación y la medida rutinarias de CO2, N2, O2 y CH4 en menos de 5 minutos. La necesidad de un registro, botellas de gas portador con presión regulada y mezclas certificadas * Aprobado por el Standard Methods Committee. 1988. de gas patrón, encarece el coste hasta tal punto que, cuando los análisis son poco frecuentes, el método resulta antieconómico. Las ventajas de este sistema son la ausencia de errores acumulativos encontrados en las medidas volumétricas secuenciales, la adaptabilidad a análisis de otros gases, la adaptabilidad a análisis y toma de muestras intermitentes y el uso de muestras de 1 ml o menos1. 2. Recogida de muestras Cuando la fuente de gas se encuentre a cierta distancia del instrumental utilizado para el análisis, recójanse las muestras en recipientes sellados y transpórtense al laboratorio. Los colectores de transporte son los recipientes más adecuados. Especialmente útiles resultan los tubos largos de cristal con espita de vidrio de tres vías en ambos extremos, como se muestra en la figura 2720:1. También se Figura 2720:1. Aparato de toma de muestras de gases. 2-98 MÉTODOS NORMALIZADOS preparan con salidas colocadas en el centro y provistas de tabiques para transvase de las muestras con jeringa. Conéctese un extremo del colector a la fuente de gas y ábrase la espita a la atmósfera. Límpiese la línea de aire haciendo pasar 10-15 volúmenes de gas a través del respiradero y ábrase la espita para recibir la muestra. Si se dispone de grandes cantidades de gas, elimínese el aire haciendo pasar de 10 a 15 volúmenes de gas por el tubo. Si el suministro de gas es limitado, llénese el tubo con un líquido que se desplace con el gas, como el mercurio o una solución de sal acidificada. Esta última se utiliza con más facilidad y resulta menos 2720 B. 1. costosa, pero disuelve los gases en cierto grado. Por tanto, llénese el tubo de recogida completamente con el gas y ciérrese herméticamente evitando todo contacto durante su almacenamiento transitorio. Cuando se transfiera el gas al aparato analizador, no deberá transportar ningún líquido. 3. Referencia 1. GRUNE, W. N. & C. F. C HUEH . 1962-63. Sludge gas analysis using gas chromatograph. Water Sewage Works 109:468; 110:43; 77, 102, 127, 171, 220, y 254. Método volumétrico Discusión general a) Principio: Este método puede utilizarse para el análisis del gas digestor o del metano en el agua (véase sección 6211, Metano). Primeramente se pasa un volumen medido de gas a través de una solución de hidróxido de potasio (KOH), para eliminar CO2 y, a continuación, a través de una solución de pirogalol alcalino para eliminar oxígeno, añadiendo luego óxido cúprico sobrecalentado, que elimina el H2 por oxidación del agua. Después de cada uno de los pasos anteriores, mídase el volumen del gas restante; la disminución resultante mide el porcentaje de volumen relativo de cada componente de la mezcla. Finalmente, el CH4 se determina mediante conversión a CO2 y H2O en una pipeta de combustión baja o un conjunto de oxidación catalítica. Se mide el volumen de CO2 formado durante la combustión, para determinar la fracción de metano originalmente presente. El nitrógeno se evalúa aceptando que representa el único gas remanente y equivale a la diferencia del 100 por 100 y la suma de los porcentajes obtenidos para los demás componentes. Cuando se mida solamente el CO2, infórmese solamente de la cifra de éste. No puede hacerse una suposición válida acerca de los gases restantes en ese momento sin hacer un análisis completo. Respecto a los métodos de oxidación, síganse las recomendaciones de los fabricantes del instrumental. PRECAUCIÓN: NO debe efectuarse ningún método de combustión lenta con el gas digestor por la alta probabilidad de que la concentración de CH4 exceda el 5 por 100 de volumen, limite a partir del cual la mezcla es explosiva. 2. Instrumental Aparato de análisis de gas tipo Orsat, que consta, como mínimo, de: 1) una bureta de gas cubierta de agua y ampolla de nivel; 2) una pipeta de absorción de CO2; 3) una pipeta de absorción de O2; 4) un conjunto de oxidación de hidrógeno por óxido cúprico; 5) un conjunto PROPIEDADES FÍSICAS Y DE AGREGACIÓN protegido de oxidación catalítica de CH4 o un conjunto de combustión lenta en pipeta; 6) una ampolla nivelada. Con la pipeta de combustión lenta, utilícese una fuente controlada de corriente para calentar eléctricamente el filamento de platino. Como líquido de desplazamiento se recomienda el mercurio; para recogida de muestras, también se ha empleado con éxito la solución acuosa de Na2SO4H2SO4. Utilícese algún analizador de gas disponible en el comercio que tenga esas unidades. 3. Reactivos a) Solución de hidróxido de potasio: Disuélvanse 500 g de KOH en agua destilada y dilúyase hasta 1 l. b) Reactivo alcalino de pirogalol: Disuélvanse 30 g de pirogalol (también llamado ácido pirogálico) en agua destilada, hasta 100 ml. Añádanse 500 ml de solución de KOH. c) Gas oxígeno: Utilícense aproximadamente 100 ml para cada muestra de gas analizada. d) Líquido de desplazamiento: Utilícese cualquiera de los siguientes: 1) Mercurio, o 2) Solución de sulfato de sodio-ácido sulfúrico. Disuélvanse 200 g de Na2SO4 en 800 ml de agua destilada; añádanse 30 ml de H2SO4 concentrado. 4. Procedimiento a) Introducción de la muestra: Transfiérase de 5 a 10 ml de muestra de gas en una bureta, a través de una conexión de tubo capilar al colector. Expúlsese la muestra a la atmósfera para purgar el sistema. Transfiérase a la bureta hasta 100 ml de muestra de gas. Llevar la muestra de la bureta a presión atmosféri- 2-99 ca o de referencia ajustando la ampolla niveladora. Mídase el volumen exactamente y regístrese como V1 b) Absorción del dióxido de carbono: Elimínese el CO2 de la muestra pasándolo a través de la pipeta de absorción de CO2 cargada con la solución de KOH. Hágase circular el gas hacia delante y hacia atrás hasta que el volumen de muestra permanezca constante. Antes de abrir las espitas entre la bureta y las pipetas de absorción, es preciso asegurarse de que el gas de la bureta está bajo una presión ligeramente positiva para evitar que el reactivo de la pipeta contamine la espita o el distribuidor. Después de la absorción del CO2, transfiérase la muestra a la bureta y mídase el volumen. Regístrese como V2. c) Absorción del oxígeno: Elimínese el O2 haciendo pasar la muestra a través de una pipeta de absorción de O2 cargada con reactivo alcalino de pirogalol hasta que el volumen de la muestra permanezca constante. Mídase el volumen y regístrese como V3. Para muestras de gas digestor, continuar con el procedimiento como se indica en el apartado Ad. Para CH4 en agua, consérvese el gas en la pipeta de CO2 y precédase como se explica más adelante en el apartado 4e. d) Oxidación del hidrógeno: Elimínese el H2 pasando la muestra a través de un conjunto de CuO mantenido a una temperatura del orden de 290 a 300 °C. Cuando se haya obtenido un volumen constante, transfiérase de nuevo la muestra a la bureta, enfríese y mídase el volumen. Registrar éste como K4. Expúlsese todo el gas remanente, a excepción de 20 a 25 ml. Mídase el volumen y consígnese como V5. Consérvese temporalmente en la pipeta de absorción de CO2. e) Oxidación del metano: Purgar las conexiones de entrada a la bureta con O2, introduciendo en la misma de 5 a 10 ml y procediendo luego a su expulsión. Oxidar el CH4 bien por el proceso de 2-100 oxidación catalítica para gas digestor y fase de gas de muestras de agua, o bien por el proceso de combustión lenta para dicha fase. 1) Proceso de oxidación catalítica. Para oxidación catalítica del gas digestor y la fase de gas de muestras hídricas, transfiéranse de 65 a 70 ml de O2 a la bureta registrando este volumen como V6. Pásese el O2 a la pipeta de absorción de CO2 de manera que se mezcle con la muestra conservada en ella. Devuélvase esta mezcla a la bureta y mídase el volumen, que se designará como V7. Dicho volumen debe ser muy aproximado a Vs + V6. Hágase pasar la mezcla de muestra de O2 a través de un conjunto de oxidación catalítica, que se calentará de acuerdo con las instrucciones del fabricante. Manténgase la tasa de paso del gas por debajo de 30 ml/min. Después del primer paso, muévase la mezcla hacia delante y hacia atrás a través del conjunto situado entre la bureta y el depósito, a un ritmo no más rápido de 60 ml/min, hasta obtener un volumen constante. Regístrese como Vs. 2) Proceso de combustión lenta. Para combustión lenta de la fase de gas de las muestras de agua, transfiéranse de 35 a 40 ml de O2 a la bureta y regístrese el volumen como V6. Transfiérase el O2 a la pipeta de combustión lenta y después hágase pasar la muestra de la pipeta de absorción de CO2 a la bureta. Caliéntese hasta incandescencia un asa de platino en la pipeta de combustión, mientras se controla la temperatura ajustando la corriente. Redúzcase la presión de O2 de la pipeta hasta algo menos de la presión atmosférica mediante la ampolla niveladora insertada en la pipeta. Transfiérase la muestra a la pipeta de combustión a un ritmo de 10 ml/min aproximadamente. Después del primer paso, hágase pasar la mezcla de O2 y muestra varias veces de atrás adelante entre bureta y pipeta a ritmo más rápido, permitiendo que el mercurio de la pipeta suba hasta MÉTODOS NORMALIZADOS un punto justamente por debajo del asa calentada. Recójase la muestra de la pipeta de combustión, retírese el asa y enfríese la pipeta y la muestra a temperatura ambiente con un chorro de aire comprimido. Transfiérase la muestra a la bureta y mídase el volumen, que se anotará como Vs. f) Medida del dióxido de carbono producido: Determínese la cantidad de CO2 formado en la reacción mediante paso de la muestra, a través de una pipeta de absorción de CO2, hasta que el volumen permanezca constante. Regístrese el volumen como V9. Compruébese la exactitud de la determinación mediante la absorción del O2 residual de la muestra. Tras esta absorción, regístrese el volumen final como V10. 5. Cálculo a) CH4 y H2 suelen ser los únicos gases combustibles presentes en el gas digestor del lodo. Cuando éste es el caso, determínese el porcentaje por volumen de cada gas como sigue: b) Como alternativa, caliéntese CH4 según una de las ecuaciones siguientes: 2-101 PROPIEDADES FÍSICAS Y DE AGREGACIÓN Los resultados de los cálculos de CH4 por las tres ecuaciones deben ser razonablemente concordantes. Si no es así, repítase el análisis después de indagar en el aparato las fuentes de error, como pueden ser las pérdidas por la espita o por las conexiones. Otros gases combustibles, como etanol, butano o pentano, producirán esa falta de concordancia; sin embargo, la posibilidad de que el gas digestor contenga una cantidad significativa de esos gases es remota. bal para su determinación puede hacerse menor de ± 1 por 100. El de la determinación de O2 y H2, sin embargo, puede ser considerable, debido a las pequeñas concentraciones. Para una concentración tan pequeña como el 1 por 100 puede esperarse un error tan grande como ± 20 por 100. Cuando el N2 se presenta en un bajo porcentaje de volumen, el error en su determinación podría ser incluso mayor, debido a que en su cálculo podrían reflejarse los errores de cada una de las demás determinaciones. 7. 6. Precisión y sesgo Una bureta mide el volumen de gas con una precisión de 0,05 ml y una exactitud probable de 0,1 ml. Con las amplias fracciones de CO2 y CH4 normalmente presentes en el gas digestor, el error glo- 2720 C. 1. Bibliografía YANT, W. F. & L. B. BERGER. 1936. Sampling of Mine Gases and the Use of the Bureau of Mines Portable Orsat Apparatus in Their Analysis. Miner's Circ. Núm. 34, U. S. Bur. Mines, Washington, D.C. MULLEN, P. W. 1955. Modern Gas Analysis. Interscience Publishers, Nueva York. Método cromatográfico de gases Discusión general a) Principio: Véase sección 6630B sobre concentraciones acerca de cromatografía de gases. b) Selección del equipo: Para el análisis de muestras gaseosas se han. propuesto numerosas columnas. Es aceptable cualquiera que proporcione la separación deseada, siempre que se informe sobre todas las condiciones exactas de análisis, con los estándares de calibración. Las directrices que a continuación se exponen son necesariamente generales, y para los instrumentos específicos se seguirán las instrucciones del fabricante. 2. Instrumental a) Cromatografía de gases: Todo aparato que se utilice debe ser equipado con un detector de conductividad térmica. Con algunos rellenos en columna se requieren estufas y controles de temperatura. Utilícese preferentemente una unidad con válvula de muestra de gas. b) Registro: Utilícese un registrador de banda de papel de amplitud total 10-mV con el cromatógrafo de gases. Cuando se detecten componentes menores, como H2 y H2S, utilícese un registro de 1-mV. MÉTODOS NORMALIZADOS 2-102 c) Relleno en columna *: Algunos de los disponibles en el mercado, útiles para separar los componentes gaseosos del lodo, se enumeran a continuación junto con las separaciones habituales, posibles a temperatura ambiente1, 2: 1) Gel de sílice a temperatura ambiente: H2, aire (O2 + N2), CH4, (C02-bajo). 2) Tamiz molecular 13X:H2, O2, N2, CH4. 3) HMPA (hexametilenfosforamida) 30 por 100 en Cromosorb P: CO2 desde (O2, H2, N2, CH4). 4) DEHS (di-2-etilhexilsefacato) 30 por 100 en Cromosorb P: CO2 desde (O2, H2, N2, CH4). Las combinaciones de las columnas 1 con 2, 3 y 2 ó 4 con 2, con un tamaño adecuado y en la secuencia 1.a columnadetector, 2.a columna-detector, separarán fácilmente H2, O2, N2, CH4 y CO2. Se puede adquirir un equipo específico diseñado para estas operaciones2. d) Aparato de introducción de muestras: Se ha diseñado un dispositivo equipado con válvulas de toma de muestras de gas que permite la inspección automática en el cromatógrafo de un volumen de muestra específico. Si no se dispone de este aparato, introdúzcanse las muestras con jeringa y aguja hipodérmica calibre 27. Redúzcase el escape de gas engrasando ligeramente el émbolo con aceite mineral o, preferiblemente, utilizando una jeringa hermética especial. 3. Reactivos a) Gases portadores: Utilícese helio para separar los gases digestores. Si ha de determinarse el H2, empléese argón * Los métodos de cromatografía de gases son extremadamente sensibles al material utilizado. El uso de marcas registradas en el Standard Methods no excluye el de otros productos existentes o todavía no comercializados que proporcionan de forma demostrable resultados equivalentes. como gas portador para aumentar notablemente la sensibilidad. b) Gases de calibrado: Utilícense muestras de CH4, CO2 y N2 de pureza conocida, o mezclas de composición dada. También se emplean muestras de O2, H2 y H2S de pureza conocida si son estos gases los que van a medirse. 4. Procedimiento a) Preparación del cromatógrafo de gases: Ajústese la tasa de flujo de gas portador a 60-80 ml/min. El aparato está listo para su uso cuando el registro marca una línea base estable. El gel de sílice y las columnas del tamiz molecular pierden actividad de forma gradual por la humedad o los materiales permanentemente adsorbidos a temperatura ambiente. Si se producen separaciones insuficientes, reactívese por calentamiento o reempaquetado. b) Calibrado: Para resultados exactos, prepárese una curva de calibrado para cada gas a medir, pues los distintos componentes del gas no dan respuestas equivalentes del detector ni en función del peso ni de la concentración molar. Calíbrese con mezclas sintéticas o con gases puros. 1) Mezclas sintéticas. Cómprense mezclas de gases de composición conocida o prepárense en el laboratorio. Inyéctese un volumen estándar de cada mezcla en el cromatógrafo de gas y anótese la respuesta para cada uno. Calcúlese por ordenador la respuesta del detector, como área bajo la curva o como altura del pico, después de corregir para atenuación. Léanse cuidadosamente las alturas de los picos y relaciónense con las concentraciones de los componentes de la muestra. Reprodúzcanse exactamente los parámetros operativos de un análisis en el siguiente. Si por este método no puede obtenerse una reproducibilidad suficiente, utilícense para calibrado las PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-103 áreas de los picos. Prepárese la curva punteando tanto éstas como las alturas frente a los porcentajes de volumen de cada componente. 2) Gases puros. Introdúzcanse en el cromatógrafo gases puros aislados mediante jeringas. Inyéctense volúmenes de muestra de 0,25, 0,5, 1,0 ml, etc., y realícese el trazado de respuestas del detector, corregido para atenuación, frente a los volúmenes del gas. Cuando el sistema de análisis proporcione una respuesta lineal con concentración creciente del gas desde cero hasta el margen de interés, háganse pasar mezclas estándar con las muestras. Si éstas tienen el mismo tamaño, calcúlese la concentración del gas en proporción directa. c) Análisis de la muestra: Si las muestras van a inyectarse con jeringa, equipar el recipiente de recogida con un respiradero cerrado por un tabique de goma o de silicona. Recójase la muestra para el análisis, vacíese de aire la jeringa presionando el émbolo e introdúzcase la aguja a través del tabique. Retírese el émbolo hasta tomar el volumen de gas deseado, extráigase la aguja del colector e inyéctese rápidamente la muestra en el cromatógrafo. Si las muestras han de inyectarse a través de una válvula de toma de gases, conéctese el recipiente de la muestra al tubo de entrada. Déjese fluir el gas desde el tubo de recolección a través de la válvula para purgar el espacio muerto de aire y llenar el tubo con la muestra. Normalmente son suficientes unos 15 ml para limpiar las líneas y proporcionar de 1 a 2 ml de muestra. Transvásese ésta desde el asa a la corriente de gas portador siguiendo las instrucciones del fabricante. Antes de la inyección, llévense las muestras a la presión atmosférica. Cuando las curvas de calibrado se han preparado con mezclas sintéticas, utilícese el mismo volumen de muestra que el empleado durante dicho calibrado. Cuando se preparan por el método que emplea volúmenes diversos de gases puros, inyéctese un volumen convencional de muestra de gas de unos 2 ml. 5. Cálculo a) Cuando las curvas de calibrado se han preparado con mezclas sintéticas y el volumen de la muestra analizada es igual que el usado en el calibrado, léase directamente el porcentaje de volumen de cada componente en la curva de calibrado, después de someter a ordenador la respuesta del detector para cada componente. b) Cuando las curvas de calibrado se preparan con volúmenes variables de gases puros, calcúlese según la fórmula siguiente el porcentaje de cada gas en la mezcla: donde: A = volumen parcial del componente (leído a partir de la curva de calibrado), y B = volumen de muestra inyectado. c) Cuando se pasan mezclas estándar con las muestras y la respuesta es lineal, desde cero hasta el margen de concentración de interés: donde: C = valor registrador de la muestra, y D = valor registrador del estándar. 6. Precisión y sesgo La precisión y el sesgo dependen del instrumental utilizado y de las técnicas 2-104 MÉTODOS NORMALIZADOS de operación. Si se pone el cuidado necesario, puede lograrse en general una exactitud del 2 por 100. Con gas digestor, la suma de los porcentajes de CH4, CO2 y N2 debe aproximarse al 100 por 100. Si no es así, posiblemente se hayan cometido errores en la toma, la manipulación, la conservación y la inyección del gas, o en los instrumentos y el calibrado. 2810 7. Referencias 1. ANDREWS, J. F. 1968. Chromatographic analysis of gaseous products and reaclants for biological processes. Water Sewage Works 115:54. 2. Column Systems for the Fisher Gas Partitioner. Tech. Bull. TB-154, Fisher Scientific Co., Atlanta, Georgia. Catalog 77, Fisher Scientific Co. SOBRESATURACIÓN DE GAS DISUELTO* 2810 A. El agua llega a estar sobresaturada de gases atmosféricos por varios medios, siendo el más común el calentamiento y arrastre de aire en aguas sometidas a bombeo o vertido. El signo principal de la sobresaturación de gas es la formación de burbujas en las superficies sumergidas o dentro del sistema vascular y los tejidos de los organismos acuáticos. La sobresaturación de gas puede limitar la vida acuática e interferir los procesos de tratamiento del agua. Se han encontrado niveles de sobresaturación letales para los organismos acuáticos en manantiales, ríos, pozos, lagos, estuarios y aguas de mar. La sobresaturación puede producirse en agua procesada o bombeada para preparación de bebidas, suministro a piscifactorías y bioanálisis de laboratorio. También pueden aparecer variaciones temporales, estacionales o de otro tipo en la sobresaturación. Dado que la tasa de equilibrio puede ser baja, la sobresaturación puede persistir en un agua corriente durante días, y de ese modo los * Aprobado por el Standard Methods Committee. 1987. Introducción gases disueltos en exceso persistirán lejos de la fuente de sobresaturación. Las burbujas de gas se forman cuando la presión total de gas disuelto es mayor que la suma de las presiones compensadoras, que incluyen presión del agua, presión barométrica y, para los seres vivos, presión de los tejidos y de la sangre. La presión total de gas disuelto es igual a la suma de las presiones parciales de todos los gases disueltos, incluyendo el vapor de agua. Por lo general, en la mayoría de las aguas naturales sólo se necesita considerar el nitrógeno, el oxígeno, el argón, el dióxido de carbono y las presiones del vapor de agua. La enfermedad de las burbujas de gas de los peces u otros organismos acuáticos es el resultado de una excesiva presión descompensada de gas. Un gas aislado en sobresaturación, como el oxígeno o el nitrógeno, no produce necesariamente la enfermedad de las burbujas, puesto que la formación de éstas depende en gran parte de la presión total de gas disuelto. El grado de saturación de gas debe describirse en función de las presiones más que de la concentración o de las unidades de volumen. 2-105 PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2810 B. 1. Método de difusión de membrana directa Discusión general a) Principio: Para este método se necesita un aparato con un sistema de tubos «permeable al gas» de longitud variable, conectado a un dispositivo de medida de presión. Suelen emplearse tubos de goma de dimetilsilicona, por ser muy permeables a los gases disueltos, incluyendo el vapor de agua. En estado de equilibrio, la presión de calibre dentro de los tubos es igual a la diferencia en la presión de gas ( ' P ) entre la presión total de gas disuelto y la presión barométrica ambiental. Cuando el agua está en equilibrio con la atmósfera, ' P es igual a cero. Si ' P es mayor de cero, el agua está sobresaturada. A la inversa, ' P es negativa si el agua está hiposaturada. b) Margen de trabajo: En este método, el margen de trabajo depende del dispositivo sensor de presión utilizado, pero de forma típica oscilará entre –150 y +600 mm Hg. Los sólidos disueltos en aguas residuales no interfieren en el método. 2. Instrumental Pueden adquirirse en el mercado varios tipos de aparatos de difusión de membrana. Opcionalmente, puede construirse una unidad a partir de componentes disponibles en el mercado. Se han descrito varias unidades, entre ellas un sistema de lectura directa que utiliza transductores de presión y una obtención de datos digital1, una unidad monolínea que puede activar un sistema de alarma2 y un modelo inicial de saturómetro Weiss 3. Cada una de estas unidades posee ventajas y limitaciones específicas; la elección del instrumental dependerá de su aplicación selectiva. Todos estos instrumentos son portátiles, de modo que la recogida de datos puede realizarse en su totalidad en trabajos de campo. Compruébense las posibles fugas del aparato siguiendo las instrucciones del fabricante. Incluso un escape mínimo, difícil de detectar y localizar, producirá datos inútiles. Calíbrese el dispositivo de medida de presión con un manómetro de mercurio o un calibre de presión certificado. Si se utiliza un manómetro, incluyase mercurio nuevo, de manera que pueda fluir con facilidad en el sistema de tubos. Un método alternativo para probar directamente los instrumentos de difusión de membrana consiste en utilizar una cámara pequeña y cerrada, donde los niveles ' P inducidos pueden compararse con los niveles ' P observados2. Los métodos de Van Slyke-Neill4 o de cromatografía de gas son inadecuados para el calibrado, pero pueden utilizarse para comprobar los resultados. Estos métodos miden concentraciones de gases independientes y requieren conversión ulterior a presión ' P o parcial; presentan problemas en la toma y manipulación de muestras 5-7. 3. Procedimiento Al comienzo de cada sesión, compruébense las fugas y recalíbrese el aparato. En el lugar de la monitorización, sumérjase completamente en el agua el elemento sensor, preferiblemente por debajo de la profundidad de compensación hidrostática. A esta profundidad, las presiones hidrostática y total de gas son iguales y, como resultado, no se formarán burbujas en los tubos. La formación de burbujas en el tubo de silicona reduce marcadamente la exactitud. Calcúlese la profundidad de compensación hidrostática5 como sigue: 2-106 y donde: MÉTODOS NORMALIZADOS barómetro portátil. Las presiones barométricas comunicadas por agencias meteorológicas (o aeropuertos) se corrigen a nivel del mar, y suelen ser poco habituales. Z = profundidad, m, y ' P = diferencia de presión, mm Hg. Disgréguense las burbujas formadas en los tubos golpeando suavemente el aparato o moviéndolo rápidamente en el agua. El movimiento de agua a lo largo de los tubos de goma de silicona también facilita el establecimiento de un equilibrio entre las presiones del agua en el gas y en el tubo. Opérese con el aparato «libre de burbujas» hasta observar una ' P estable. Esto puede requerir unos 5 a 30 min, dependiendo de la ' P , la temperatura y el flujo del agua y la geometría del sistema. La respuesta de tiempo del método de difusión de membrana se muestra en la figura 2810:1, para situaciones «libres de burbujas» y «con burbujas». Si se utiliza el aparato en aguas muy contaminadas que contengan aceite u otras sustancias orgánicas, límpiense los tubos de silicona con un detergente suave, de acuerdo con las instrucciones del fabricante. Estos tubos de goma de silicona se han utilizado en aguas naturales no contaminadas en períodos de hasta ocho años, sin que se vieran afectados adversamente por crecimiento de algas2. Los tubos pueden deteriorarse por la acción de arenisca abrasiva, diatomeas, picaduras producidas por organismos acuáticos, algunos compuestos orgánicos y ácidos fuertes2. Obténganse datos de presión barométrica mediante un barómetro de mercurio de laboratorio al menos una vez al día, o a partir de un barómetro portátil calibrado en el lugar de toma de muestras. Si hay más de 5 m de diferencia entre la localización del barómetro y los puntos de toma de muestras, úsese un Figura 2810:1. Tiempo de respuesta para la prueba de difusión de membrana. 4. Cálculo a) Presión de gas total: Preferiblemente, indíquese la presión de gas total como ' P 2, 6, 8 en milímetros de mercurio. También se ha consignado como porcentaje de la presión barométrica local: donde: P b = presión barométrica local verdadera, mmHg No se recomienda definir la presión de gas total como porcentaje. b) Presiones de gases componentes: Cuando se necesita información sobre sobresaturación de componentes gaseosos, indíquense los datos como presiones parciales, presiones diferenciales o porcentaje de saturación5-8. Esto requiere medidas adicionales de oxígeno disuelto, PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-107 temperatura y salinidad * en el lugar de la monitorización. En una mezcla de gases a un volumen dado, la presión parcial de un gas es la que este gas ejercería si fuera el único presente. c) Presiones diferenciales: La presión diferencial de un gas es la diferencia entre las presiones parciales de dicho gas en agua y aire. La presión diferencial del oxígeno puede calcularse como 1) Presión parcial de oxígeno. Calcúlese como sigue: y la del nitrógeno donde: PO2 = presión parcial de oxígeno, E O = coeficiente Bunsen para el oxígeno 2 (tabla 2810:1), y OD = concentración de oxígeno medida, mg/1. Se dispone de coeficientes Bunsen para aguas de mar3. El factor 0,5318 equivale a 760/(1.000K), donde K es la relación entre peso y volumen moleculares para el oxígeno gaseoso5. 2) Presión parcial de nitrógeno. Evalúese restando las presiones parciales de oxígeno y vapor de agua de la presión de gas total. d) Porcentaje de saturación de nitrógeno: En la bibliografía tradicional, los valores de sobresaturación se consignaban como saturación porcentual del gas. Este método se desaconseja, pudiendo calcularse como sigue: Son conversiones útiles las siguientes: donde: PH 2O = presión de vapor de agua a partir de la tabla 2810:11. Esta cifra incluye una pequeña contribución de argón y otros gases presentes, como el dióxido de carbono y el metano. La presión parcial del CO2 es insignificante en aguas de pH > 7,0. 3) índice de presión parcial nitrógeno/oxígeno. Este índice caracteriza la contribución relativa de ambos gases a la presión total de gases disueltos. En agua equilibrada con aire, este índice es de 3,77. * Métodos para estas variables pueden encontrarse en las secciones 4500-O, 2550 y 2520, respectivamente. Préstese atención al comparar estas relaciones con datos antiguos, ya que los PGT(%) y N2(%) se han definido de forma diferente5. 5. Control de calidad La precisión del método de difusión de membrana depende principalmente del dispositivo sensor de la presión. Para un operador con experiencia es aproximadamente de ± 1 a 2 mm Hg, con una exac- 2-108 MÉTODOS NORMALIZADOS PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-109 2-110 MÉTODOS NORMALIZADOS PROPIEDADES FÍSICAS Y DE AGREGACIÓN 2-111 MÉTODOS NORMALIZADOS 2-112 titud de ± 3 a 5 mm Hg 3,6. Los escapes de aire, la formación de burbujas, el equilibrio incompleto o la condensación producen errores en menos, mientras que las pérdidas directas de agua originarán errores en más en las unidades sumergibles. 6. Informe de resultados Inclúyanse los siguientes datos: Profundidad del sensor, m. Presión barométrica, mm Hg. Temperatura del agua, °C. Oxígeno disuelto, mm Hg o mg/1. Salinidad, g/kg. P, mm Hg. nantes significativos. De estas dos situaciones, son los ambientes cerrados los que presentan más probabilidades de que se produzcan casos de morbilidad o mortalidad por enfermedad de las burbujas, los cuales aparecen antes y en niveles menores de ' P . En circunstancias silvestres/naturales, el límite de niveles de seguridad de la sobresaturación depende de la profundidad disponible para la especie y/o la conducta de ésta, pero este límite aparece por lo general a ' P entre 50 y 150 mm Hg. En condiciones de cautividad, la ' P debería estar lo más cerca posible de cero. Para especies y formas de vida sensibles, se han observado efectos letales y subletales a ' P de 10 a 50 mm Hg13. Si se precisa información del gas componente, añádase: Presión parcial de oxígeno, mm Hg. Presión parcial de nitrógeno, mm Hg. índice de presión parcial oxígeno/nitrógeno, o 8. Referencias 1. 2. 3. 7. Interpretación de los resultados Los efectos biológicos de la sobresaturación de gases disueltos dependen de la especie, edad, profundidad de la columna de agua, duración de la exposición, temperatura e índice de presión parcial nitrógeno/oxígeno12. Los límites de seguridad suelen ser distintos en circunstancias silvestres/naturales, en las que la conducta y la presión hidrostática pueden modificar la exposición mediante movimientos horizontales y verticales hacia fuera del lugar de riesgo, y en ambientes cerrados, como acuarios, piscifactorías o laboratorios, en los que la propia instalación impide escapes e incluye otros condicio- 4. 5. 6. 7. D'A OUST , B. G., R. WHITE & H. S IEBOLD . 1975. Direct measurement of total dissolved gas partial pressure. Undersea Biomed. Res. 2:141. BOUCK, G. R. 1982. Gasometer: an inexpensive device for continuous monitoring of dissolved gases and supersaturation. Trans. Amer. Fish. Soc. 111:505. FlCKEISEN, D. H., M. J. SCHNEIDER & J. C. MONTGOMERY. 1975. A comparative evaluation of the Weiss saturometer. Trans. Amer. Fish. Soc. 104:816. BEININGEN, K. T. 1973. A Manual for Measuring Dissolved Oxygen and Nitrogen Gas Concentrations in Water with the Van Slyke-Neill Apparatus. Fish Commission of Oregon, Portland. C OLT , J. 1984. Computation of dissolved gas concentrations in water as functions of temperature, salinity, and pressure. Spec. Publ. 14, American Fisheries Soc, Bethesda, Maryland. D'A OUST , B. G. & M. J. R. C LARK . 1980. Analysis of supersaturated air in natural waters and reservoirs. Trans. Amer. Fish. Soc. 109:708. PIRIE , W. R. & W. A. HUBBERT. 1977. Assumptions in statistical analysis. Trans. Amer. Fish. Soc. 106:646. PROPIEDADES FÍSICAS Y DE AGREGACIÓN 8.8.COLT, J. 1983. The computation and repor- 10. ting of dissolved gas levels. Water Res. 17:841. 9.9. BENSON, B. B. & D. KRAUSE. 1980. The concentration and isolopic fractionation in freshwater in equilibrium with the atmosphere. 1. Oxigen. Limnol. Oceanogr. 25:662. BENSON, B. B. & D. KRAUSE. 1984. The concentration and isotopic fractionation of oxygen dissolved in freshwater and seawater in equilibrium with the atmosphere. Limnol. Oceanogr. 29:620. 2-113 11. GREEN, E. J. & D. E. C ARRITT. 1967. New tables for oxygen saturation of seawater. J. Mar. Res. 25:140. 12. WEITKAMP, D. E. & M. KATZ . 1980. A review of dissolved gas supersaturation literature. Trans. Amer. Fish. Soc. 109-659. 13. C OLT , J. 1986. The impact of gas supersaturation on the design and operation of aquatic culture systems. Aquacult. Eng. 5:49. PARTE 3000 DETERMINACIÓN DE METALES 3010 3010 A. 1. INTRODUCCIÓN Discusión general Significación tos refractarios. En general, los limites de detección de los métodos de PAI son superiores a los de los métodos electrotérmicos. Los métodos colorimétricos son aplicables cuando se conocen las interferencias en los otros métodos. Es frecuente la necesidad de tratamientos preliminares de las muestras. Se describen métodos apropiados de tratamiento preliminar para cada tipo de análisis. Los efectos de los metales en aguas potables y residuales pueden ser beneficiosos, tóxicos o simplemente molestos. Algunos metales resultan esenciales, mientras que otros pueden perjudicar a los consumidores del agua, a los sistemas de tratamiento de aguas residuales y a las aguas de depósitos. En muchos casos el potencial beneficio o riesgo depende de la concentración. 3. 2. Tipos de métodos Los metales se pueden determinar de forma satisfactoria utilizando métodos de absorción atómica, de plasma de acoplamiento inductivo o colorimétricos, aunque estos últimos son de menor precisión y sensibilidad. Los métodos de absorción incluyen técnicas electrotérmicas y de llama. En general, los métodos de llama se aplican en el caso de concentraciones moderadas en sistemas de matrices simples y complejas. Los métodos electrotérmicos tienen mayor sensibilidad, en general, si los efectos de la matriz no son demasiado intensos. Algunos efectos de la matriz pueden compensarse con modificadores de la misma. Las técnicas de plasma de acoplamiento inductivo (PAI) son aplicables dentro de un amplio intervalo lineal y resultan especialmente sensibles en el caso de los elemen- Definición de términos a) Metales disueltos: Son los componentes (metálicos) de una muestra sin acidular que pasan a través de un filtro de membrana de 0,45 μm. b) Metales suspendidos: Son los componentes (metálicos) de una muestra sin acidular que son retenidos por un filtro de membrana de 0,45 μm. c) Metales totales: La concentración de metales determinada en una muestra sin filtrar tras digestión intensa, o la suma de las concentraciones de metales en las fracciones disuelta y suspendida. d) Metales extraíbles con ácido: La concentración de metales en solución tras el tratamiento de una muestra sin filtrar con ácido mineral diluido caliente. Para determinar por separado metales disueltos y suspendidos, hay que filtrar inmediatamente después de recogida la muestra. Se debe conservar en ácido hasta después de filtrar. 3-1 MÉTODOS NORMALIZADOS 3-2 3010 B. Toma y conservación de muestras Antes de recoger una muestra, es necesario decidir cuál es la fracción que se va a analizar (disuelta, suspendida, total o extraíble con ácido). Esta decisión determinará en parte si se acidula la muestra con o sin filtración, así como el tipo de digestión requerido. Durante la toma de muestras y conservación de las mismas pueden producirse graves errores debidos a la contaminación del dispositivo de toma de muestras, a la eliminación defectuosa de los residuos de muestras previas del recipiente de la muestra o a la pérdida de metales por adsorción y/o precipitación en el recipiente de la muestra, como consecuencia de una inapropiada acidulación de la misma. 1. Recipientes para las muestras Los mejores recipientes para las muestras son los fabricados en cuarzo o TFE. Estos recipientes son caros, por lo que se prefieren los de polipropileno o polietileno lineal con tapón de polietileno. También se pueden utilizar recipientes de vidrio de borosilicato, pero hay que evitar el empleo de recipientes de vidrio blando, tratándose de muestras que contienen metales del orden de microgramos por litro. Las muestras para determinación de plata deben guardarse en recipientes que absorben la luz. Sólo se deben utilizar recipientes y filtros que hayan sido enjuagados con ácido. 2. Conservación Consérvense las muestras inmediatamente después de la toma de muestras, acidulando con ácido nítrico concentrado (HNO3) a pH<2. Fíltrense las muestras para metales disueltos antes de guardarlas (véase sección 3030). Normal- mente es suficiente 1,5 ml de HNO3 conc./l de muestra (o 3 ml HNO3 1 + 1/1 de muestra) para una conservación a corto plazo. Para muestras con capacidad tampón elevada hay que aumentar la cantidad de ácido (pueden ser necesarios 5 ml para algunas muestras alcalinas o muy tamponadas). Utilícese ácido comercial de pureza elevada * o prepárese un ácido de gran pureza por destilación del ácido por debajo de la ebullición. Después de acidular la muestra, consérvese preferiblemente en un frigorífico a 4 °C para evitar un cambio de volumen ocasionado por la evaporación. En estas condiciones, las muestras con concentraciones de metal de varios miligramos por litro se mantienen estables a lo largo de un período de hasta seis meses (excepto el mercurio, cuyo límite es de cinco semanas). Tratándose de niveles de metal del orden de microgramos por litro, analícense las muestras lo antes posible después de tomadas. Las muestras para el análisis de mercurio se conservarán añadiendo 2 ml/1 de solución de K2Cr2O7 al 20 por 100 (p/v) (preparada en HNO3 1 + 1). Se conservarán en un frigorífico no contaminado con mercurio. (PRECAUCIÓN: las concentraciones de mercurio pueden aumentar si se guardan en frascos de plástico en laboratorios contaminados con mercurio.) 3. Bibliografía STRUEMPLER, A. W. 1973. Adsorption characteristics of silver, lead, calcium, zinc and nickel on borosilicate glass, polyethylene and polypropylene container surfaces. Anal. Chem. 45:2251. FELDMAN, C. 1974. Preservation of dilute mercury solutions. Anal. Chem. 46:99. * Ultrex, J. T. Baker o equivalente. DETERMINACIÓN DE METALES 3-3 KING , W. G., J. M. R ODRÍGUEZ & C. M. WAI . 1974. Losses of trace concentrations of cadmium from aqueous solution during storage in glass containers. Anal. Chem. 46:771. BATLEY, G. E. & D. GARDNER. 1977. Sampling and storage of natural waters for trace metal analysis. Water Res. 11:745. SUBRAMANIAN, K. S., C. L. CHAKRABARTI, J. E. SUETIAS & I. S. MAINES. 1978. Preservation of 3010 C. 1. Precauciones generales Fuentes de contaminación Evítese la introducción de metales contaminantes que procedan de los recipientes, el agua destilada o los filtros de membrana. Algunos tapones de plástico o forros de tapones pueden contaminar con metales; por ejemplo, se ha encontrado zinc en tapones de rosca de baquelita negra así como en muchos productos de caucho y plástico, e igualmente se ha encontrado cadmio en la punta de pipetas de plástico. Por su parte, el plomo es un contaminante que se halla muy difundido en el aire y el polvo urbanos. 2. some trace metals in samples of natural waters. Anal. Chem. 50:444. BERMAN, S. & P. YEATS. 1985. Sampling of seawater for trace metals. Crit. Rev. Anal. Chem. 16:1. WENDLANDT, E. 1986. Sample containers and analytical accessories made of modern plastics for trace analysis. Gewaess. Wass. Abwass. 86:79. Eliminación de contaminantes Límpiense cuidadosamente los recipientes para muestras con una solución detergente no iónica libre de metales, enjuagar con agua del grifo, sumergir en ácido y enjuagar después con agua libre de metales. Para cuarzo, TFE o material de vidrio, utilícese HNO 3 1 + 1, HC1 1 + 1, o agua regia (3 partes de HC1 conc. + 1 parte de HNO 3 conc.) para la inmersión. En el caso de material plástico, utilícese HNO3 1 + 1 o HC1 1 + 1. Las condiciones adecuadas para la inmersión son 24 h y 70 °C. Se pueden emplear ácido crómico o sustitutivos sin cromo* para eliminar los depósitos orgánicos de los recipientes, pero en el primer caso hay que lavar cuidadosa* Nochromix, Godax Laboratories o equivalente. mente los recipientes con agua con objeto de eliminar las trazas de cromo. No emplear ácido crómico para recipientes de plástico o en el caso de que haya que realizar determinaciones de cromo. Utilícese siempre agua libre de metales en el análisis y en la preparación de reactivos (véase 3111B.2c). En este tipo de métodos, el término «agua» hace siempre referencia a agua libre de metales. 3. Contaminantes que proceden del aire En un análisis de concentraciones de metales con una exactitud del orden de microgramos por litro, los contaminantes procedentes del aire en forma de compuestos volátiles, polvo, hollín y aerosoles del aire del laboratorio, pueden arrojar valores significativos. Para evitar la contaminación hay que operar en condiciones tales como las que proporcionan las vitrinas de aire limpio, de flujo laminar, comerciales, o los lugares de trabajo diseñados al caso y los análisis de blancos que reflejen el procedimiento completo. 4. Bibliografía M ITCHELL , J. W. 1973. Ultrapurity in trace analysis. Anal. Chem. 45:492A. GARDNER, M., D. HUNT & G. TOPPING. 1986. Analytical quality control (AQC) for monitoring trace metals in the coastal and marine environment. Water Sci. Technol. 18:35. 3-4 MÉTODOS NORMALIZADOS 3020 CONTROL DE CALIDAD* En la parte 1000 y en la explicación de cada método se ofrecen recomendaciones detalladas e información general en relación con la garantía y el control de calidad (CC). Consúltese cada método en particular en cuanto a los requisitos de CC específicos. Léase asimismo la parte 1000 para la definición de términos. Recuérdese siempre el propósito general de las medidas: utilícense replicados para establecer la precisión y recuperación de adiciones conocidas para determinar sesgos. Utilícense asimismo patrones, gráficos de control, blancos, calibraciones, y otras medidas necesarias. Dispóngase de documentación adecuada. Las medidas de control de calidad y la justificación de las determinaciones de control de operaciones pueden diferir de las relativas a las determinaciones hechas con propósitos reguladores. Los niveles de metales traza pueden ser de un orden de magnitud inferior a las potenciales fuentes de contaminación. Antes de emplear un método, es necesario determinar su límite de detección (véase sección 1030E). Los métodos instrumentales son más sensibles por lo general que los colorimétricos. Compruébese que el método empleado proporciona la suficiente sensibilidad para el objeto de la medida. Es necesario llevar registros adecuados de todas las operaciones, de manera que personas no familiarizadas con un análisis puedan reconstruir directamente los resultados finales a partir de los datos no procesados y disponer de un juego completo de procedimientos operativos estándar. Al analizar una matriz nueva o con la que no se está familiarizado, utilícese el método de adiciones conocidas para comprobar la ausencia de interferencias * Aprobado por el Standard Methods Committee, 1988. antes del calibrado. Confírmese la ausencia de interferencias analizando las citadas muestras sin diluir y a dilución 1:10; los resultados habrán de ser comparables. Llévese a cabo el análisis de un blanco a lo largo de todo el procedimiento con cada serie de muestras realizando cada etapa con un blanco de agua para reactivos, incluyendo las etapas de digestión. En ácidos y material de vidrio hay muchos metales en cantidades significativas que pueden influir en los resultados. Si las medidas realizadas con el blanco dan concentraciones por encima del límite de detección del método, repítase la preparación de la muestra empleando reactivos o material de vidrio más limpios. Para el análisis de metales hay que comprobar que todo el vidrio y los demás materiales estén especialmente limpios. Para cada marcha analítica, se ha de tener como mínimo una curva de calibración formada por un blanco y dos o más patrones (según los instrumentos), un patrón de referencia externo, un replicado, y una adición conocida para comprobar la ausencia de interferencias de la matriz. Hay que hacer medidas de replicados sobre sub-muestras tomadas de un frasco de muestras único con objeto de establecer la precisión del método (distinta de la precisión de muestras). La precisión y la recuperación de adiciones conocidas deberán estar dentro de los registros conocidos en razón de experimentaciones anteriores. Habrán de mantenerse gráficos de control para control más riguroso de la precisión y el sesgo. Analícese un patrón de control de punto medio y un blanco de calibración al principio, al final y periódicamente (en general, después de cada serie de nueve desconocidas) con cada grupo de muestras, para comprobar que la calibración del instrumento no se ha desviado. Inicialmente, empléese como guía el siguien- DETERMINACIÓN DE METALES te criterio: un valor determinado del patrón de control fuera del 95 a 105 por 100 de la concentración esperada sugiere un problema potencial. Cuando un valor sobrepasa el 90 a 110 por 100 de la concentración esperada, la calibración se suele considerar fuera de control, por lo que es necesario llevar a cabo una corrección. La utilización de límites de control calculados (véase sección 1020) proporcionará mejores indicaciones de las características de ejecución y se recomienda para aquellos laboratorios que realicen análisis frecuentes (por ejemplo, semanales). Ello puede proporcionar límites más ajustados. 3030 3-5 Para determinar si existen efectos de matriz, realícense adiciones conocidas a las muestras antes de una digestión. Recuperaciones entre 85 y 115 por 100 indican que los efectos de matriz no son significativos. Compruébense los patrones de calibración y soluciones de adiciones conocidas frente a una fuente exterior. La concordancia debe ser ±5 por 100. Determinados programas reguladores pueden requerir medidas de control de calidad preceptivas adicionales, por ejemplo, el empleo de un patrón de control al nivel de contaminantes máximo (NCM). TRATAMIENTO PRELIMINAR DE MUESTRAS* 3030 A. Las muestras que contienen partículas o materia orgánica requieren, en general, un tratamiento previo antes del análisis. Los «metales totales» incluyen todos los metales combinados orgánica o inorgánicamente, tanto disueltos como en partículas. Las muestras incoloras, transparentes (principalmente agua potable) con una turbidez de < 1 UNT, inodoras, y de una sola fase, pueden analizarse directamente por espectroscopia de absorción atómica o espectroscopia de plasma de acoplamiento inductivo para metales en total, sin digestión. Para una comprobación posterior o si se encuentran cambios en las matrices existentes, hay que confrontar muestras digeridas o no digeridas para asegurarse de que los resultados son * Aprobado por el Standard Methods Committee, 1989. Introducción comparables. Al tomarlas, se acidulan dichas muestras a pH <2 con ácido nítrico conc. (HNO3) y se analizan directamente. Llévese a cabo una digestión de todas las demás muestras antes de determinar los metales en total. Para analizar los metales disueltos, fíltrese la muestra, acidúlese el filtrado y analícese directamente. Para determinar los metales suspendidos, fíltrese la muestra, realícese una digestión del filtro y del material sobre el filtro y analícese. Para determinar los metales extraíbles con ácido, se extraen los metales tal como se indica después y se analiza el extracto. En esta sección se describe el tratamiento preliminar general de muestras en las que hay que determinar metales, según las secciones 3110a 3500-Zn y con algunas excepciones. Las técnicas especiales de digestión para mercurio se recogen en las secciones 3112B.46 y c, y las de 3-6 MÉTODOS NORMALIZADOS arsénico y selenio en las secciones 3114 y 3500-Se. Cuídese de no introducir metales en las muestras durante el tratamiento preliminar. Durante el pre-tratamiento evítese el contacto con caucho, pinturas a base de metales, humo de cigarrillos, servilletas de papel, así como todos los productos metálicos incluyendo los de acero inoxidable, metal galvanizado y latón. Las campanas de humos convencionales pueden contribuir significativamente a la contaminación de la muestra, sobre todo durante la digestión con ácidos en recipientes abiertos. Las puntas de las pipe- 3030 B. tas de plástico están frecuentemente contaminadas con cobre, hierro, zinc y cadmio; antes de utilizarlas, sumérjanse en HNO3 o HC1 2N durante varios días, enjuagando después con agua desionizada. Compruébese la pureza de los ácidos de calidad para reactivos utilizados para conservación, extracción y digestión. Si se encuentran concentraciones excesivas de metal, hay que purificar los ácidos por destilación o utilizar ácidos ultrapuros. Realícense pruebas con blancos en todas las etapas de digestión y filtración, aplicando las necesarias correcciones a los resultados. Filtración preliminar Si se trata de determinar metales disueltos o suspendidos, fíltrese la muestra en el momento de recogerla, con un dispositivo filtrante de plástico preacondicionado mediante vacío o presión y que lleve un soporte de filtro de plástico o TFE¯ haciéndola pasar por un filtro de membrana (policarbonato o acetato de celulosa) de 0,4 a 0,45 μm de diámetro de poro, prelavado y sin rejilla. Antes de usarlo, fíltrese un blanco de agua de calidad para reactivos, para asegurar la ausencia de contaminación. El preacondicionamiento del filtro y del dispositivo del filtro se realiza enjuagando con 50 ml de agua desionizada. Si el blanco del filtro contiene concentraciones significativas de metales, sumérjanse los filtros de membrana en HC1 aproximadamente 0,5N O HNO3 1 + 1 (recomendado para análisis electrotérmico) y aclárese con agua antes de usarlo. ensáyese el filtro para comprobar la completa recuperación de metales. Si se ha de proceder a la digestión del filtro en el caso de metales suspendidos, regístrese el volumen filtrado e inclúyase un filtro en la determinación del blanco. Antes de filtrar, centrifúguense las muestras muy turbias en tubos de plástico de alta densidad o de TFE lavados con ácido, a fin de reducir la carga sobre los filtros. Las unidades filtrantes a presión con agitación se ensucian menos fácilmente que los filtros con vacío; fíltrese a una presión de 70 a 130 kPa. Después de la filtración, acidúlese el filtrado a pH 2 con HNO3 conc. y analícese directamente. Si se forma un precipitado al acidular, llévese a cabo una digestión del filtrado acidulado antes del análisis, empleando el Método E. Reténgase el filtro y sométase a digestión para determinación directa de los metales suspendidos. NOTA: Filtros diferentes presentan diferentes características de sorción y filtración; en el caso de análisis de trazas, PRECAUCIÓN: NO utilizar ácido perclórico para la digestión de filtros de membrana 3-7 DETERMINACIÓN DE METALES 3030 C. Tratamiento preliminar de metales extraíbles por ácido Los metales extraíbles se adsorben ligeramente sobre material en partículas. Debido a que puede resultar inevitable la digestión de algunas muestras, habrá de operarse en condiciones estrictamente controladas, a fin de obtener resultados significativos y reproducibles. Manténganse constantes el volumen de muestra, el volumen de ácido y el tiempo de contacto. Exprésense los resultados en metales extraíbles y especifíquense las condiciones de extracción. 3030 D. En cuanto se toma la muestra, se acidula con 5 ml de HNO3 conc./l de muestra. Para preparar la muestra, mézclese bien, pásense 100 ml a un vaso o matraz y añádanse 5 ml de HC1 de elevada pureza 1 + 1. Caliéntese durante 15 minutos en baño de vapor. Fíltrese a través de filtro de membrana, ajústese el volumen del filtrado a 100 ml con agua y procédase al análisis. Digestión preliminar de metales Con objeto de reducir la interferencia de la materia orgánica y convertir el metal asociado a las partículas en una forma (normalmente el metal libre) que pueda determinarse por espectrometría de absorción atómica o espectroscopia de plasma de acoplamiento inductivo, utilícese una de las técnicas de digestión señaladas después. Utilícese el método de digestión menos complejo que permita alcanzar una recuperación completa y consistente, compatible con el método analítico y con el metal que se analiza. El ácido nítrico digerirá la mayoría de las muestras en forma adecuada (sección 3O3OE). El nitrato es una matriz aceptable tanto para la llama como para la ab- sorción atómica electrotérmica. Algunas muestras pueden necesitar la adición de ácido perclórico, clorhídrico o sulfúrico para una digestión completa. Estos ácidos pueden interferir en el análisis de algunos metales y todos ellos proporcionan una matriz más pobre para análisis electrotérmico. Confírmese la recuperación del metal para cada digestión y procedimiento analítico utilizado. Utilícese la tabla 3030:1 como una guía para determinar los ácidos que se pueden emplear (además de HNO3) para llegar a una digestión completa. Por regla general, el HNO3 sólo es adecuado en el caso de muestras limpias o materiales que se oxidan con facilidad; la digestión con TABLA 3030:I. ÁCIDOS UTILIZADOS JUNTO CON HNO3 PARA PREPARACIÓN DE LA MUESTRA MÉTODOS NORMALIZADOS 3-8 HNO3-H2SO4 o con HNO3-HC1 es adecuada para la materia orgánica fácilmente oxidable; en el caso de materia orgánica o minerales de difícil oxidación es necesario emplear HNO3-HC1O4 o HNO3-HF para la digestión. Si existen grandes cantidades de materia orgánica, es útil una combustión seca. Para determinar concentraciones de Ag superiores a 0,03 mg/1, diluyase la muestra hasta que su contenido sea inferior a 1 mg/1 de Ag y sométase a digestión siguiendo el método 3O3OF.36. Anótese la técnica de digestión empleada. Las técnicas de digestión con ácidos (sección 3O3OE-I) conducen en general a precisiones y sesgos comparables en la mayoría de tipos de muestras que se digieren en su totalidad con la técnica empleada. Conviene utilizar un volumen mínimo de ácido debido a que los ácidos empleados añadirán metales a las muestras y a los blancos. A continuación se sugieren posibles volúmenes de muestras. Si el volumen recomendado sobrepasa la capacidad de la vasija de digestión, añádase muestra al avanzar la evaporación. Cuando las muestras se concentran durante la digestión (por ejemplo > 100 ml de muestra empleada), determínese la recuperación de metal para cada matriz digerida, con objeto de comprobar la validez del método. El empleo de muestras más grandes hará necesario utilizar más ácido, lo que incrementaría también la concentración de impurezas. La precisión y el sesgo obtenidos con la combustión seca (sección 3O3OJ) son muy variables, dependiendo del tipo de muestra y del metal que se analiza. Empléese la combustión seca únicamente para aquellas muestras que hayan demostrado precisión y sesgo aceptables. Anótense los resultados según la siguiente pauta: B Concentración de metal, mg/1 = A x C siendo: A = concentración del metal en la solución digerida, mg/1, B = volumen final de la solución digerida, ml, y C = tamaño de la muestra, ml. Prepárense muestras sólidas o lodos líquidos con elevados contenidos de sólidos, sobre una base ponderal. Mézclese la muestra y pásese una cantidad adecuada (normalmente 1 g de un lodo con 15 por 100 de sólidos en total) a una vasija de digestión previamente pesada. Vuélvase a pesar y calcúlese el peso de la muestra. Sígase una de las técnicas de digestión que se dan a continuación. Anótense los resultados de peso en húmedo o peso en seco, de la forma siguiente: Concentración de metal, mg/kg (peso húmedo) Concentración de metal, mg/kg (peso seco) siendo: A = concentración de metal en la solución digerida, mg/1, B = volumen final de la solución digerida, ml, y D = sólidos totales, % (véase sección 2540G). DETERMINACIÓN DE METALES 3-9 Prepárense siempre muestras de blancos de ácido para cada tipo de digestión realizada. La experiencia demuestra que un blanco hecho con los mismos ácidos y sometido al mismo procedimiento de di- 3030 E. 1. Digestión por ácido nítrico Instrumental a) Placa caliente. b) Matraces cónicos (erlenmeyer), 125 ml, o vasos Griffin, 150 ml, lavados con ácido y enjuagados con agua. 2. Reactivos Ácido nítrico, HNO3 conc. 3. Procedimiento Mézclese la muestra y pásese un volumen adecuado de la misma (50 a 100 ml) a un matraz erlenmeyer o a un vaso de 125 ml. Añádanse 5 ml de HNO3 y un poco de plato poroso, bolas de vidrio o gránulos Hengar. Llévese a ebullición lenta y evapórese sobre placa caliente 3030 F. 1. hasta el menor volumen posible (aproximadamente 10 a 20 ml) antes de que tenga lugar una precipitación. Continúese calentando y añadiendo el HNO3 conc. necesario para completar la digestión, perceptible porque la solución se hace transparente y ligeramente coloreada. No dejar que la muestra se seque durante la digestión. Recójanse con agua las paredes del matraz o del vaso y fíltrese entonces si es necesario (véase sección 3O3OB). Pásese el filtrado a un matraz volumétrico de 10 ml junto con dos partes de agua de 5 ml, añadiendo este líquido de enjuagado al matraz volumétrico. Enfríese, diluyase hasta la señal y mézclese cuidadosamente. Tómense porciones de esta solución para las determinaciones del metal requeridas. Alternativamente, tómese un volumen mayor de muestra en todo el procedimiento, para concentración (véase 3030D). Digestión por ácido nítrico-ácido clorhídrico Instrumental Véase 3030E.1. 2. gestión que la muestra puede proporcionar la corrección de las impurezas presentes en los ácidos, en el agua para reactivos o en el material de vidrio. Reactivos a) Ácido nítrico, HNO3 conc. b) Ácido clorhídrico, 1 + 1. 3. Procedimiento a) HNO3/HCl en total: Pásese a un matraz o a un vaso un volumen medido de muestra, bien mezclada y mantenida con ácido, apropiado a las concentraciones de metales esperadas (véase 3030E.16). Añádanse 3 ml de HNO3 conc. Colóquese el matraz o vaso sobre 3-10 MÉTODOS NORMALIZADOS una placa caliente y evapórese con precaución hasta menos de 5 ml, asegurándose de que la muestra no hierva ni quede seca ninguna área del fondo del recipiente. Enfríese y añádanse 5 ml de HNO 3 conc. Cúbrase el recipiente con un vidrio de reloj y vuélvase a colocar sobre la placa caliente. Auméntese la temperatura de la placa caliente para dar lugar a un reflujo suave. Continúese calentando, añadiendo más ácido si es necesario, hasta completar la digestión (lo que queda indicado, por lo general, por un ligero color del digestato o ausencia de cambio en su apariencia al continuar el reflujo). Evapórese a menos de 5 ml y enfríese. Añádanse 10 ml de HC1 1 + 1 y 15 ml de agua por 100 ml de volumen final previsto. Caliéntese 15 minutos más para disolver cualquier posible precipitado o residuo. Enfríese, lávense recogiendo con agua las paredes 3030 G. 1. b) HNO3/HCl recuperable: Para este procedimiento de digestión, menos preciso, pásese a un matraz o vaso un volumen medido de muestra bien mezclada y mantenida en ácido. Añádanse 2 ml de HNO3 1 + 1 y 10 ml de HC1 1 + 1 y caliéntese en baño de vapor o sobre placa caliente hasta que el volumen se reduzca a unos 25 ml, asegurándose de que la muestra no hierva. Enfríese y fíltrese para eliminar la materia insoluble o bien centrifúguese o déjese sedimentar durante toda la noche. Ajústese el volumen a 100 ml y mézclese. Digestión por ácido nítrico-ácido sulfúrico Instrumental Véase 3030E.1 2. Reactivos a) Solución de indicador naranja de metilo. b) Acido nítrico, HNO3 conc. c) Ácido sulfúrico, H2SO4 conc. 3. del vaso y el vidrio de reloj y fíltrese para eliminar el material insoluble que podría obstruir el nebulizador. Alternativamente, centrifúguese o déjese sedimentar durante la noche. Ajústese a un volumen predeterminado basándose en las concentraciones de metales esperadas. Procedimiento Mézclese la muestra y llévese con la pipeta un volumen adecuado a un matraz o a un vaso (véase 3030E.1b). Si la muestra no está ya acidulada, acidúlese con H2SO4 conc. hasta punto final con naranja de metilo y añádanse 5 ml de HNO3 concentrado y un poco de plato poroso, bolas de vidrio o gránulos Hengar. Llévese a ebullición lenta sobre pla- ca caliente y evapórese hasta 15 a 20 ml. Añádanse 5 ml de HNO3 conc. y 10 ml de H2SO4 conc. Evapórese sobre placa caliente hasta la aparición de humos blancos de SO3. Si la solución no es transparente, añádanse 10 ml de H2SO4 conc. y repítase la evaporación hasta la aparición de humos de SO3. Caliéntese para eliminar todo el HNO3 antes de continuar el tratamiento. Cuando la solución es transparente y no aparecen humos parduzcos, todo el HNO3 estará eliminado. No dejar que la muestra se seque durante la digestión. Enfríese y diluyase con agua hasta 50 ml aproximadamente. Caliéntese hasta instantes próximos a la ebullición con objeto de disolver las sales que sean casi insolubles. Fíltrese si es necesario, complétese entonces el procedimiento tal como se indica en la sección 3O3OE.3 desde que comienza con «Pásese el filtrado...». DETERMINACIÓN DE METALES 3030 H. 1. Digestión con ácido nítrico-ácido perclórico Instrumental Véase 3030E.1. Se necesitan además los siguientes: a) Pantalla de seguridad. b) Gafas protectoras. 2. Reactivos a) Ácido nítrico, HNO3 conc. b) Ácido perclórico, HC1O4. c) Solución acuosa de indicador naranja de metilo. d) Solución de acetato de amonio: preparada disolviendo 500 g de NH4C2H3O2 en 600 ml de agua. 3. 3-11 Procedimiento PRECAUCIÓN: Las mezclas de HCIOA y materia orgánica calentadas pueden explotar violentamente. Evítese este riesgo tomando las siguientes precauciones: a) no añadir HCIO4 a una solución caliente que contenga materia orgánica: b) tratar siempre previamente con HNO3 las muestras que contengan materia orgánica antes de la adición de HCIO4; c) evitar la repetida formación de humos trabajando en la vitrina (para operaciones de rutina basta conectar la trompa de agua a un extractor de humos de vidrio*. Existen en el comercio campanas extractoras de humos en acero inoxidable con una adecuada instalación de lavado con agua, que son aceptables cuando se utiliza HCIO4.), y d) no evaporar nunca a sequedad las muestras que se están digiriendo con HCIO4. * Tal como el que se puede adquirir en GFS Chemical Co, Columbus, Ohio. Mézclese la muestra y pásese un volumen apropiado de la misma a un matraz o vaso adecuados (véase 3030E.1b). Si la muestra no está ya acidulada, acidúlese con HNO3 conc. hasta punto final con naranja de metilo, añádanse 5 ml más de HNO3 y un poco de plato poroso o bolas de vidrio y evapórese sobre placa caliente hasta 15 a 20 ml. Añádanse 10 ml de HNO3 conc. y 10 ml de HC1O4 conc, enfriando el matraz o el vaso entre las adiciones. Evapórese suavemente sobre placa caliente hasta que aparezcan humos blancos densos de HC1O4. Si la solución no es transparente, se cubre el recipiente con un vidrio de reloj y se mantiene en ebullición hasta que se hace transparente. Si es necesario, añádanse 10 ml de HNO3 conc. para completar la digestión. Enfríese, diluyase hasta 50 ml aproximadamente con agua y llévese a ebullición para expulsar el cloro o los óxidos de nitrógeno. Fíltrese y complétese el procedimiento siguiendo lo señalado en 3O3OE.3, comenzando por «Pásese el filtrado...». En el caso de la determinación de plomo en presencia de cantidades elevadas de sulfato (por ejemplo, determinación de Pb en cenizas volantes de centrales térmicas) hay que disolver el precipitado de PbSO4, según la pauta siguiente: Añádanse 50 ml de solución de acetato amónico al matraz o vaso en el que se ha llevado a cabo la digestión y caliéntese hasta ebullición incipiente. Hágase girar de cuando en cuando el recipiente para mojar todas las superficies interiores y disolver cualquier posible depósito. Colóquese de nuevo el filtro y hágase pasar lentamente la solución a su través. Pásese el filtrado a un matraz volumétrico de 100 ml. enfríese, diluyase hasta la señal, mézclese cuidadosamente y resérvese para la determinación de plomo. 3-12 MÉTODOS NORMALIZADOS 3030 I. 1. Digestión por ácido perclórico-ácido fluorhídrico Instrumental a) Placa caliente. b) Vasos de TFE, 250 ml, lavados con ácido y enjuagados con agua. 2. Reactivos a) Ácido nítrico, HNO3 conc. y 1 + 1. b) Ácido perclórico, HNO4. c) Ácido fluorhídrico, HF, 48 a 51 por 100. 3. Procedimiento PRECAUCIÓN: Véanse las precauciones de 3030H para utilizar el HCIO4; manipúlese 3030 J. con mucho cuidado el HF proporcionando una ventilación adecuada, sobre todo a la solución calentada. Evítese cualquier contacto con la piel. En el caso de quemaduras con HF hay que solicitar ayuda médica. Mézclese la muestra y pásese un volumen adecuado de la misma a un vaso de TFE de 250 ml. Añádase un poco de plato poroso y llévese a ebullición lenta. Evapórese sobre placa caliente hasta 15 a 20 ml. Añádanse 12 ml de HNO, conc. y evapórese hasta casi sequedad. Repítanse la adición de HNO3 y la evaporación. Déjese enfriar la solución, añádanse 20 ml de HC1O4 y 1 ml de HF y elévese a ebullición hasta que la solución se hace transparente y han aparecido humos blancos de HC1O4. Enfríese, añádanse 50 ml de agua aproximadamente y procédase como se indica en 3O3OE.3, comenzando por «Pásese el filtrado...». Combustión seca 1. Instrumental Véanse secciones 2540B.2 y 2540C.2. 2. Procedimiento Mézclese la muestra y pásese un volumen adecuado de la misma a un evaporador de platino o de vidrio de alto contenido en sílice*. Evapórese a sequedad en un baño de vapor. Pásese el evapora- * Vycor, producto de Corning Glass Works, Corning, Nueva York o equivalente. dor a un horno de mufla y caliéntese la muestra hasta obtener una ceniza blanca. Si se van a determinar elementos volátiles, manténgase la temperatura entre 400 y 450 °C. Si sólo se va a determinar sodio, llévese a cabo la combustión seca de la muestra hasta una temperatura de 600 °C. Disuélvase la ceniza en una cantidad mínima de HNO3 conc. y agua caliente. Fíltrese la muestra diluida y ajústese a volumen conocido, preferiblemente, de modo que la concentración final de HNO3 sea aproximadamente de 1 por 100. Tómense porciones de esta solución para la determinación de metales. 3-13 DETERMINACIÓN DE METALES 3110 DETERMINACIÓN DE METALES POR ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA* Los requisitos para determinar metales por espectrometría de absorción atómica varían con el metal y/o con la concentración que se determina, por lo que el método se presenta de la forma siguiente: Sección 3111, Determinación de metales por espectrometría de absorción atómica de llama, que abarca: • Determinación de antimonio, bismuto, cadmio, calcio, cesio, cromo, cobalto, cobre, oro, iridio, hierro, plomo, litio, magnesio, manganeso, níquel, paladio, platino, potasio, rodio, rutenio, plata, sodio, estroncio, talio, estaño y zinc por aspiración directa en una llama de aire-acetileno (311 IB), • Determinación de bajas concentraciones de cadmio, cromo, cobalto, cobre, hierro, plomo, manganeso, níquel, plata y zinc por quelación con pirrolidin ditiocarbamato de amonio (PDCA), extracción con metil isobutil cetona (M1BC) y * Aprobado por el Standard Methods Committee, 1988. aspiración en llama de aire-acetileno (3111C), • Determinación de aluminio, bario, berilio, molibdeno, osmio, renio, silicio, torio, titanio y vanadio por aspiración directa en llama de óxido nitroso-acetileno (3111D), y • Determinación de bajas concentraciones de aluminio y berilio por quelación con 8-hidroxiquinoleína, extracción con MIBC y aspiración en una llama de óxido nitroso-acetileno (3111E). La sección 3112 cubre la determinación de mercurio por la técnica del vapor frío. La sección 3113 se ocupa de la determinación de microcantidades de aluminio, antimonio, arsénico, bario, berilio, cadmio, cromo, cobalto, cobre, hierro, plomo, manganeso, molibdeno, níquel, selenio, plata y estaño por espectrometría de absorción atómica electrotérmica. La sección 3114 se refiere a la determinación de arsénico y selenio por transformación de los mismos en sus hidruros y aspiración en una llama de argón-hidrógeno o de nitrógeno-hidrógeno. 3-14 MÉTODOS NORMALIZADOS 3111 DETERMINACIÓN DE METALES POR ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA DE LLAMA* 3111 A. 1. Principios La espectrometría de absorción atómica se parece a la fotometría de llama de emisión en que la muestra es aspirada en una llama y atomizada. La principal diferencia consiste en que en la fotometría de llama se mide la cantidad de luz emitida, mientras que en la espectrometría de absorción atómica se dirige un rayo luminoso a través de una llama a un monocromador y sobre un detector que mide la cantidad de luz absorbida por el elemento atomizado en la llama. Para determinados metales, la absorción atómica presenta una sensibilidad superior a la emisión de llama. Como cada metal tiene su propia longitud de onda de absorción característica, se utiliza como fuente luminosa una lámpara compuesta de dicho elemento; esto proporciona un método relativamente libre de interferencias espectrales o de radiación. La cantidad de energía absorbida en la llama a una longitud de onda característica es proporcional a la concentración del elemento en la muestra, en un intervalo de concentraciones limitado. La mayor parte de los instrumentos de absorción atómica están equipados para funcionar también en la forma de emisión. 2. Selección del método Véase sección 3110. * Aprobado por el Standard Methods Committee, 1989. Introducción 3. Interferencias a) Interferencia química: Muchos metales se pueden determinar por aspiración directa de la muestra en una llama de aire-acetileno. La interferencia más problemática es la llamada «química», que está originada por la ausencia de absorción de átomos unidos en combinación molecular en la llama. Tal dificultad puede aparecer cuando la llama no es lo bastante caliente para disociar las moléculas o cuando el átomo disociado se oxida de inmediato, dando un compuesto que no se disocia a la temperatura de la llama. Estas interferencias pueden reducirse o eliminarse añadiendo elementos o compuestos específicos a la solución de la muestra. Por ejemplo, la interferencia de fosfato en la determinación de magnesio puede salvarse añadiendo lantano. Asimismo, la introducción de calcio elimina la interferencia de sílice en la determinación de manganeso. Sin embargo, el silicio y metales tales como aluminio, bario, berilio y vanadio requieren la llama de óxido nitroso-acetileno de mayor temperatura para disociar sus moléculas. La llama de óxido nitroso-acetileno también puede ser útil para reducir al mínimo determinados tipos de interferencias químicas observadas con la llama de aire-acetileno. Por ejemplo, la interferencia provocada por concentraciones elevadas de fosfato en la determinación de calcio con llama de aire-acetileno no tiene lugar con la llama de óxido nitrosoacetileno. Las extracciones con MIBC que emplean APDC (3111C) son muy útiles en DETERMINACIÓN DE METALES el caso de interferencias de matrices salinas, por ejemplo, en agua de mar. Este procedimiento también concentra la muestra de manera que se amplían los límites de detección. Las salmueras y el agua del mar se pueden analizar por aspiración directa, pero se recomienda una dilución de la muestra. La aspiración de soluciones que contienen concentraciones elevadas de sólidos disueltos originan formaciones sólidas sobre el cuerpo del mechero. Esto requiere el apagado frecuente de la llama y la limpieza del cuerpo del mechero. Utilícese una corrección del fondo, preferiblemente, cuando se analizan aguas con un contenido en sólidos por encima de un 1 por 100, sobre todo en el caso de que la línea de resonancia primaria del elemento que interesa se encuentre por debajo de 240 nm. Cuando se analizan salmueras y agua de mar es necesario realizar comprobaciones más frecuentes de las recuperaciones para asegurar la precisión de los resultados en estas matrices concentradas y complejas. El bario y otros metales se ionizan en la llama, reduciéndose de esta forma la población (potencialmente absorbente) del estado de base. La adición de un exceso de un catión (sodio, potasio o litio) con ionización similar o inferior resolverá el problema. La longitud de onda de máxima absorción para el arsénico es 193,7 nm y para selenio 196,0 nm, longitudes de onda a las cuales la llama de aire-acetileno absorbe intensamente. La sensibilidad para el arsénico y el selenio puede ser mejorada convirtiendo ambos elementos en sus hidruros gaseosos y analizándolos en una llama de nitrógenohidrógeno o una llama de argón-hidrógeno con tubo de cuarzo (véase sección 3114). b) Corrección de fondo: La absorción molecular y la difusión de la luz causadas por partículas sólidas en la llama pueden conducir a valores de absorción excesivamente altos que dan como resultado 3-15 errores positivos. Cuando tales fenómenos ocurren, es necesaria una corrección de fondo para obtener valores precisos. Utilícese uno de estos tres tipos de corrección de fondo: de fuente continua, de Zeeman o corrección de Smith-Hieftje. 1) Corrección de fondo de fuente continua: Un corrector de fondo de fuente continua utiliza una lámpara de cátodo hueco lleno de hidrógeno con un cátodo metálico o una lámpara de arco de deuterio. Cuando la lámpara de cátodo hueco de fuente de líneas y la fuente continua están colocadas en el mismo trayecto óptico y en el mismo intervalo de tiempo, el fondo de la banda ancha de la señal elemental se sustrae electrónicamente y la señal resultante queda compensada en cuanto al fondo. Tanto la lámpara de cátodo hueco lleno de hidrógeno como la lámpara de arco de deuterio tienen intensidades más bajas que la lámpara de cátodo hueco de fuente de líneas o que la lámpara de descarga sin electrodos. Para obtener una corrección válida, adáptense las intensidades de la fuente continua con la lámpara de cátodo hueco de fuente de líneas o la lámpara de descarga sin electrodos. La adaptación puede causar una disminución de la intensidad de la fuente de líneas o un incremento de la anchura de rendija; estas medidas tienen como inconvenientes la elevación del límite de detección y la posibilidad de ocasionar la no linealidad de la curva de calibración. La corrección del fondo utilizando un corrector de fuente continua es susceptible de interferencias provenientes de otras líneas de absorción en la anchura de banda del espectro. Una absorción atómica significativa de la radiación de fuente continua por elementos distintos al que se determina da lugar a una corrección inadecuada. Cuando se utiliza una lámpara de cátodo hueco de fuente de líneas sin corrección del fondo, la presencia de una línea de absorción de otro elemento en la anchura de banda del es- 3-16 pectro no ocasionará interferencia a menos que solape la línea que interesa. La corrección del fondo de fuente continua no eliminará el solapamiento del espectro de absorción directa, en el caso de que un elemento distinto al que se determina sea capaz de absorber la radiación de línea del elemento que se estudia. 2) Corrección del fondo de Zeeman: Esta corrección se basa en el principio de que un campo magnético divide la línea del espectro en dos haces luminosos polarizados linealmente, en direcciones paralela y perpendicular al campo magnético. Uno es el llamado componente pi (n) y el otro el componente sigma (a). Estos dos haces luminosos tienen exactamente la misma longitud de onda y difieren sólo en el plano de polarización. La línea n será absorbida tanto por los átomos del elemento que interesa como por el fondo originado por la absorción de banda ancha y difusión de la luz de la matriz de la muestra. La línea a será únicamente absorbida por el fondo. La corrección del fondo de Zeeman proporciona una corrección del fondo precisa a niveles de absorción mucho más altos que los conseguidos con sistemas de corrección de fondo de fuente continua. También elimina virtualmente la posibilidad de error proveniente del fondo estructurado. Al no necesitarse fuentes luminosas adicionales, se eliminan las limitaciones de alineamiento e intensidad que se dan al emplear fuentes continuas. Las más notables desventajas del método de Zeeman son su reducida sensibilidad para algunos elementos, su intervalo lineal reducido y un cierto efecto de inversión por el que la absorbancia de algunos elementos comienza a decrecer a concentraciones elevadas, dando lugar a una curva de calibración de dos lados. 3) Corrección del fondo de SmithHieftje: Esta corrección se basa en que la absorbancia medida para un elemento específico se reduce al incrementarse la MÉTODOS NORMALIZADOS corriente que llega a la lámpara de cátodo hueco, mientras que la absorción de sustancias absorbentes no específicas permanece idéntica en todos los niveles de corriente. Cuando se aplica este método, se sustrae la absorbancia con corriente de alto amperaje de la absorbancia con corriente de bajo amperaje. En estas condiciones, se sustrae y corrige la absorbancia debida a fondo no específico. La corrección del fondo de SmithHieftje proporciona una serie de ventajas sobre la corrección de fuente continua. Con ella se puede efectuar una corrección precisa en niveles de absorbancia más altos, eliminándose virtualmente el error de fondo estructurado. En algunos casos también se pueden eliminar las interferencias espectrales. Lo que aún no ha llegado a establecerse es la utilidad de la corrección de fondo de Smith-Hieftje con lámparas de descarga sin electrodos. 4. Sensibilidad, límites de detección e intervalos óptimos de concentración La sensibilidad de la espectrometría de absorción atómica de llama se define como la concentración de metal que produce una absorción de 1 por 100 (una absorbancia de aproximadamente 0,0044). El límite de detección del instrumento se define aquí como la concentración que produce una absorción equivalente al doble de la magnitud de la fluctuación del fondo. La sensibilidad y los límites de detección varían con el instrumento, el elemento determinado, la complejidad de la matriz y la técnica seleccionada. El intervalo de concentración óptimo se extiende normalmente entre la concentración de varias veces la sensibilidad hasta la concentración a la cual la curva de calibración empieza a ser plana. Para conseguir los mejores resultados conviene utilizar concentraciones de muestras y patrones dentro del intervalo óptimo de concentraciones del espectrómetro. Vea- DETERMINACIÓN DE METALES 3-17 TABLA 3111:I. MÁRGENES DE CONCENTRACIÓN DE ABSORCIÓN ATÓMICA CON ABSORCIÓN ATÓMICA DE ASPIRACIÓN DIRECTA se la tabla 3111.I, que indica los intervalos de concentraciones medibles con atomización convencional. En muchos casos, el intervalo de concentración mostrado en la tabla 3111.I puede ser prolongado hacia abajo extendiendo la escala o integrando la señal de absorción un largo tiempo. El intervalo puede prolongarse hacia arriba por dilución, utilizando una longitud de onda menos sensible, haciendo girar el cuerpo del mechero o utilizando un microprocesador para linealizar la curva de calibración a altas concentraciones. MÉTODOS NORMALIZADOS 3-18 5. Preparación de patrones Prepárense soluciones patrón de concentraciones conocidas de metal en agua con una matriz similar a la de la muestra. Utilícense patrones que horquillen la concentración de muestra esperada y que estén dentro del intervalo de trabajo del método. Deben prepararse patrones muy diluidos a partir de las soluciones de reserva con concentraciones superiores a 500 mg/1. Las soluciones patrón de reserva pueden obtenerse de diversas fuentes comerciales. Se pueden preparar también utilizando materiales de referencia del National Institute of Standards and Technology de los Estados Unidos, (NIST, antes National Bureau of Standards) o por los procedimientos descritos en las siguientes secciones. Cuando se trata de muestras que contienen concentraciones variables y elevadas de materiales de la matriz, se deben hacer similares los iones principales en la muestra y en el patrón diluido. Si la matriz de la muestra es compleja y los patrones no pueden adaptarse con precisión a sus componentes, utilícese el método de adiciones del patrón, 3113B.4d2), para corregir los efectos de la matriz. Si se utiliza digestión, los patrones deben someterse al mismo proceso de digestión que las muestras. 6. Instrumental a) El espectrómetro de absorción atómica consiste en una fuente luminosa que emite el espectro de líneas de un elemento (lámpara de cátodo hueco o lámpara de descarga sin electrodos), un dispositivo para vaporizar la muestra (normalmente una llama), medio de aislamiento de la línea de absorción (monocromador o filtro y rejilla ajustable) y un detector fotoeléctrico con su equipo electrónico de amplificación y medida asociado. b) Mechero: El tipo más corriente consiste en una premezcla que introduce el chorro pulverizado en una cámara de condensación para eliminar las gotas grandes. El mechero puede llevar un cuerpo convencional con una sola ranura; un cuerpo Boling que puede preferirse para una aspiración directa con llama de aire-acetileno, o un cuerpo especial para utilizar con óxido nitroso y acetileno. c) Lectura de salida: La mayoría de los instrumentos van equipados con un mecanismo de lectura de salida digital o indicador de cero. Los instrumentos más modernos llevan microprocesadores capaces de integrar las señales de absorción en el tiempo y linealizar la curva de calibración a altas concentraciones. d) Lámparas: Utilícese una lámpara de cátodo hueco o una lámpara de descarga sin electrodos (LDE). Empléese una lámpara para cada elemento que se mide. Las lámparas de cátodo hueco para varios elementos dan por lo general una menor sensibilidad que las lámparas de un solo elemento. Las LDE tardan un cierto tiempo en calentarse y estabilizarse. e) Válvulas reductoras de presión: Manténganse los suministros de combustible y oxidante a presiones algo más altas que la presión controlada de funcionamiento del instrumento empleando válvulas reductoras. Utilícese una válvula reductora separada para cada gas. f) Tubo de ventilación: Colóquese un tubo de ventilación de aproximadamente 15 a 30 cm por encima del mechero para dejar salir los humos y vapores de la llama. Esta precaución protege al personal del laboratorio frente a los vapores tóxicos, protege al instrumento de los vapores corrosivos y evita que la estabilidad de la llama se vea afectada por las corrientes de la habitación. Se recomienda un regulador de tiro o un ventilador de velocidad variable para modular el flujo de aire y evitar perturbaciones de la llama. Selecciónese el tamaño del ventilador con objeto de obtener el flujo de aire recomendado por el fabricante del ins- 3-19 DETERMINACIÓN DE METALES TABLA 3111:II. DATOS DE PRECISIÓN Y SESGO INTERLABORATORIOS PARA MÉTODOS DE ABSORCIÓN ATÓMICA. ASPIRACIÓN DIRECTA Y METALES EXTRAÍDOS truniento. En los laboratorios en los que exista contaminación del aire en forma de partículas pesadas, se deben utilizar instalaciones de laboratorio limpio (sección 3010C). 7. Garantía de calidad/ control de calidad En las tablas 3111:II y III se muestran algunos datos específicos sobre la preci- 3-20 MÉTODOS NORMALIZADOS TABLA 3111:III. PRECISIÓN DE UN SOLO OPERADOR Y NIVELES DE CONTROL RECOMENDADOS PARA MÉTODOS DE ABSORCIÓN ATÓMICA. ASPIRACIÓN DIRECTA Y METALES EXTRAÍDOS sión y el sesgo que pueden obtenerse con los métodos tratados. Analícese un blanco entre las lecturas de la muestra y el patrón para comprobar la estabilidad de la línea de base. Volver al cero cuando sea necesario. Añádase a una muestra de cada diez (o una muestra de cada grupo de muestras si se analizan menos de diez) una cantidad conocida del metal que interesa y vuélvase a analizar para confirmar la recuperación. La cantidad de metal añadida deberá ser aproximadamente igual a la cantidad encontrada. Si hay poco me- tal, añádase una cantidad cercana a la media del intervalo lineal del ensayo. La recuperación del metal añadido deberá estar entre 85 y 115 por 100. Analícese una solución patrón adicional después de cada 10 muestras o con cada grupo de muestras, aunque sean menos, con objeto de confirmar que el ensayo está bajo control. En la tabla 3111:III se muestran las concentraciones recomendadas de patrones para ser utilizados, límites de aceptabilidad y los datos de precisión registrados por un solo operador. 3-21 DETERMINACIÓN DE METALES 8. Referencias 1. 2. 3. 4. 9. AMERICAN SOCIETY FOR TESTING AND MATERIALS . 1986. Annual Book of ASTM Standards, Volume 11.01, Water and Environmental Technology. American Soc. Testing & Materials, Filadelfia, Pennsylvania. U.S. D EPARTMENT H EALTH , E DUCATION AND W ELFARE . 1970. Water Metals No. 6, Study No. 37. U. S. Public Health Serv. Publ. No. 2029, Cincinnati, Ohio. U. S. D EPARTMENT H EALTH , E DUCATION AND W ELFARE . 1968. Water Metals No. 4, Study No. 30. U. S. Public Health Serv. Publ. No. 999-UTH-8, Cincinnati, Ohio. U.S. ENVIRONMENTAL PROTECTION AGENCY. 1983. Methods for Chemical Analysis of Water and Wastes. Cincinnati, Ohio. Bibliografía K AHN , H. L. 1968. Principles and Practice of Atomic Absorption. Advan. Chem. Ser. 3111 B. 1. No. 73, Div. Water, Air & Waste Chemistry, American Chemical Soc, Washington, D.C. RAMÍREZ-MUÑOZ, J. 1968. Atomic Absorption Spectroscopy and Analysis by Atomic Absorption Flame Photometry. American Elsevier Publishing Co., Nueva York. SLAVIN, W. 1968. Atomic Absorption Spectroscopy. John Wiley & Sons, Nueva York. PAUS, P. E. 1971. The application of atomic absorption spectroscopy to the analysis of natural waters. Atomic Absorption Newsletter 10-69. EDIGER, R. D. 1973. A review of water analysis by atomic absorption. Atomic Absorption Newsletter 12:151. PAUS, P. E. 1973. Determination of some heavy metals in seawater by atomic absorption spectroscopy. Fresenius Zeitschr. Anal. Chem. 264:118. BURRELL, D. C. 1975. Atomic Spectrometric Analysis of Heavy-Metal Pollutants in Water, Ann Arbor Science Publishers, Inc., Ann Arbor, Michigan. Método directo de llama de aire-acetileno Discusión general Este método es aplicable a la determinación de antimonio, bismuto, cadmio, calcio, cesio, cromo, cobalto, cobre, oro, iridio, hierro, plomo, litio, magnesio, manganeso, níquel, paladio, platino, potasio, rodio, rutenio, plata, sodio, estroncio, talio, estaño y zinc. 2. Instrumental Espectrómetro de absorción atómica y equipo asociado: Véase sección 3111A.6. Utilícese el quemador recomendado por el fabricante. 3. Reactivos a) Aire, purificado y secado a través de un filtro apropiado que elimina aceite, agua y otras sustancias extrañas. La fuente puede ser un compresor o gas embotellado comercial. b) Acetileno, calidad comercial estándar. Se evitará que la acetona, siempre existente en los cilindros de acetileno, entre y estropee el cuerpo del mechero reemplazando un cilindro cuando su presión de acetileno haya caído a 689 kPa (100 psi). c) Agua libre de metales: Utilícese agua libre de metales para preparar todos los reactivos y como agua de dilución. Prepárese agua libre de metales desionizando el agua del grifo y/o utilizando uno de los siguientes procedimientos, según la concentración de metal en la muestra: destilación simple, bidestilación o sub-ebullición. Compruébese siempre el agua desionizada o destilada para determinar si el elemento que interesa está presente en cantidades traza. (PRECAUCIÓN: Si la fuente de agua contiene Hg u otros metales volátiles, puede no ser suficiente el agua destilada o bidestilada para 3-22 análisis de trazas debido a que estos metales destilan con el agua. En estos casos empléese la sub-ebullición para preparar agua libre de metales.) d) Solución de calcio: Disuélvanse 630 mg de carbonato de calcio, CaCO3, en 50 ml de HC1 1 + 5. Llévese a ebullición suave si es necesario, para obtener una disolución completa. Enfríese y diluyase con agua hasta 1.000 ml. e) Ácido clorhídrico. HC1, al 1 por 100, 10 por 100, 20 por 100, 1 + 5 y 1 + 1 y conc. f) Solución de lantano: Disuélvanse 58,65 g de óxido de lantano, La2O3, en 250 ml de HC1 conc. Añádase el ácido lentamente hasta que el material se ha disuelto, diluyendo después con agua hasta 1.000 ml. g) Peróxido de hidrógeno, al 30 por 100. h) Ácido nítrico, HNO3, al 2 por 100, 1 + 1 y conc. i) Agua regia: Obtenida con tres volúmenes de HC1 conc. y un volumen de HNO3 conc. j) Soluciones patrón de metales: Prepárense una serie de soluciones patrón de metales en un intervalo óptimo de concentraciones por dilución apropiada de las siguientes soluciones de metales de reserva con agua que contiene 1,5 ml de HNO3 conc. Séquense cuidadosamente los reactivos antes de emplearlos. Es necesario utilizar reactivos de la máxima pureza. Si se trata de hidratos, utilícense reactivos recientes. 1) Antimonio: Disuélvanse 0,2669 g de K(SbO)C4H4O6 en agua, añádanse 10 ml de HC1 1 + 1 y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Sb. 2) Bismuto: Disuélvanse 0,100 g de metal bismuto en un volumen mínimo de HNO3 1 + 1. Diluyase a 1.000 ml con HNO3 al 2 por 100 (v/v); 1,00 ml = 100 μg Bi. 3) Cadmio: Disuélvanse 0,100 g de metal cadmio en 4 ml de HNO3 conc. Añádanse 8,0 ml de HNO3 conc. y dilu- MÉTODOS NORMALIZADOS yase con agua hasta 1.000 ml; 1,00 ml = 100 μg Cd. 4) Calcio: Suspéndanse 0,2497 g de CaCO3 (secado a 180° durante 1 hora antes de pesar) en agua y disuélvase con mucha precaución utilizando una cantidad mínima de HNO3 1 + 1. Añádanse 10,0 ml de HNO3, conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Ca. 5) Cesio: Disuélvanse 0,1267 g de cloruro de cesio, CsCl, en 1.000 ml de agua; 1,00 ml = 100 μg Cs. 6) Cobalto: Disuélvanse 0,1000 g de metal cobalto en una cantidad mínima de HNO3 1 + 1. Añádanse 10,0 ml de HC1 1 + 1 y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Co. 7) Cobre: Disuélvanse 0,100 g de metal cobre en 2 ml de HNO3 conc, añádanse 10,0 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Cu. 8) Cromo: Disuélvanse 0,1923 g de CrO3 en agua. Cuando la solución es completa, acidúlese con 10 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Cr. 9) Estaño: Disuélvase 1,000 g de metal estaño en 100 ml de HC1 conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 1,00 mg Sn. 10) Estroncio: Suspéndanse 0,1685 g de SrCO3 en agua y disuélvanse con precaución con una cantidad mínima de HNO3 1 + 1. Añádanse 10,0 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml; 1 ml = 100 μg Sr. 11) Hierro: Disuélvanse 0,100 g de alambre de hierro en una mezcla de 10 ml de HC1 1 + 1 y 3 ml de HNO3 conc. Añádanse 5 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Fe. 12) Iridio: Disuélvanse 0,1147 g de cloroiridato de amonio, (NH4)2IrCl6, en un volumen mínimo de HC1 al 1 por 100 (v/v) y diluyase con HC1 al 1 por 100 (v/v) hasta 100 ml; 1,00 ml = 500 μg Ir. DETERMINACIÓN DE METALES 13) Litio: Disuélvanse 0,5323 g de carbonato de litio, Li2CO3, en un volumen mínimo de HNO3 1 + 1. Añádanse 10,0 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Li. 14) Magnesio: Disuélvanse 0,1658 g de MgO en una cantidad mínima de HNO3 1 + 1. Añádanse 10 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Mg. 15) Manganeso: Disuélvanse 0,1000 g de metal manganeso en 10 ml de HC1 conc. mezclado con 1 ml de HNO3 conc. Diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Mn. 16) Níquel: Disuélvanse 0,1000 g de metal níquel en 10 ml de HNO3 conc. caliente, enfríese y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Ni. 17) Oro: Disuélvanse 0,100 g de metal oro en un volumen mínimo de agua regia. Evapórese a sequedad, disuélvase el residuo en 5 ml de HC1 conc, enfríese y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Au. 18) Paladio: Disuélvanse 0,100 g de alambre de paladio en un volumen mínimo de agua regia y evapórese justo hasta sequedad. Añádanse 5 ml de HC1 conc. y 25 ml de agua y caliéntese hasta completar la disolución. Diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Pd. 19) Plata: Disuélvanse 0,1575 g de nitrato de plata, AgNO3, en 100 ml de agua, añádanse 10 ml de HNO3 conc. y llévese hasta 1.000 ml; 1,00 ml = 100 μg Ag. 20) Platino: Disuélvanse 0,100 g de metal platino en un volumen mínimo de agua regia y evapórese a sequedad. Añádanse 5 ml de HC1 conc. y 0,1 g de NaCl y evapórese de nuevo a sequedad. Disuélvase el residuo en 20 ml de HC1 1 + 1 y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Pt. 21) Plomo: Disuélvanse 0,1598 g de nitrato de plomo Pb(NO3)2, en una cantidad mínima de HNO3 1 + 1, añádanse 3-23 10 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Pb. 22) Potasio: Disuélvanse 0,1907 g de cloruro de potasio, KC1 (secado a 110°C) en agua y llévese hasta 1.000 ml; 1,00 ml = 100 μg K. 23) Rodio: Disuélvanse 0,386 g de hexaclororodato de amonio, (NH4)3RhCl6 . 1,5H2O, en un volumen mínimo de HC1 al 10 por 100 (v/v) y diluyase con HC1 al 10 por 100 (v/v) hasta 1.000 ml; 1,00 ml = 100 μg Rh. 24) Rutenio: Disuélvanse 0,205 g de cloruro de rutenio, RuCl3, en un volumen mínimo de HC1 al 20 por 100 (v/v) y diluyase con HC1 al 20 por 100 (v/v) hasta 1.000 ml; 1,00 ml = 100 μg Ru. 25) Sodio: Disuélvanse 0,2542 g de cloruro de sodio, NaCl, secado a 140 °C, en agua. Añádanse 10 ml de HNO3 conc. y llévese hasta 1.000 ml; 1,00 ml = 100 μg Na. 26) Tallo: Disuélvanse 0,1303 g de nitrato de talio, T1NO3, en agua. Añádanse 10 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg TI. 27) Zinc: Disuélvanse 0,100 g de metal zinc en 20 ml de HC1 1 + 1 y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Zn. 4. Procedimiento a) Preparación de la muestra: La preparación de la muestra requerida depende de la necesidad de medir únicamente los metales disueltos o los metales totales. Si los que se han de determinar son los metales disueltos, véase la sección 3O3OB para preparación de muestras. Si se han de determinar los metales totales o los extraíbles con ácidos, véase sección 3O3OC a J. En todas las muestras hay que cerciorarse de que las concentraciones de ácido y modificadores de la matriz son iguales en muestras y patrones. 3-24 Cuando se determinan Ca o Mg, diluyase y mézclense 100 ml de muestra o patrón con 10 ml de solución de lantano (apartado 3f) antes de la atomización. Cuando se determinan Fe o Mn, mézclense 100 ml con 25 ml de solución de Ca (apartado 3d) antes de la aspiración. Alternativamente, utilícense volúmenes más pequeños. b) Funcionamiento del instrumento: Debido a las diferencias entre las diversas formas y modelos de espectrómetros de absorción atómica, no es posible formular instrucciones aplicables a cada instrumento. Véase al respecto el manual de funcionamiento de cada fabricante. En general, procédase como sigue: Instálese en el instrumento una lámpara de cátodo hueco para el metal deseado y establézcase el dial aproximado de longitudes de onda con arreglo a la tabla 3111:1. Fíjese la anchura de rendija siguiendo las sugerencias del fabricante para el elemento que se mide. Enciéndase el instrumento, aplíquese a la lámpara de cátodo hueco la corriente que aconseja el fabricante y déjese calentar el aparato hasta que se estabiliza la fuente de energía, unos 10 a 20 minutos por lo general. Reajústese la corriente después del calentamiento cuando sea necesario. Optimícese la longitud de onda ajustando el dial de longitudes de onda hasta que se obtiene la ganancia óptima de energía. Alíniese la lámpara siguiendo las instrucciones del fabricante. Instálese una cabeza de quemador adecuada y ajústese su posición. Conéctese el aire y ajústese la velocidad del flujo de aire a lo especificado por el fabricante para obtener la máxima sensibilidad en relación con el metal que se mide. Conéctese el acetileno, ajústese la velocidad de flujo al vapor especificado y enciéndase la llama. Déjese estabilizar la llama unos cuantos minutos. Hágase aspirar un blanco integrado por agua desionizada o una solución acida con la misma concentración de ácido de los pa- MÉTODOS NORMALIZADOS trones y las muestras. Póngase a cero el instrumento. Hágase aspirar una solución patrón y ajústese la velocidad de aspiración del nebulizador para obtener la sensibilidad máxima. Ajústese el mechero vertical y horizontalmente para obtener una respuesta máxima. Hágase aspirar un blanco de nuevo y vuélvase a poner a cero el instrumento. Hágase aspirar un patrón próximo al medio del intervalo lineal. Regístrese la absorbancia de este patrón cuando está recientemente preparado y con una nueva lámpara de cátodo hueco. Hágase referencia a estos datos en subsiguientes determinaciones del mismo elemento para comprobar la consistencia de la instalación del instrumento y el envejecimiento de la lámpara de cátodo hueco y del patrón. El instrumento está ahora preparado para funcionar. Cuando se terminan los análisis, apáguese la llama desconectando primero el acetileno y después el aire. c) Estandarización: Selecciónense al menos tres concentraciones de cada solución patrón de metal (preparada como en el apartado 3j anterior) para establecer límites para la concentración esperada del metal de una muestra. Hágase aspirar un blanco y póngase a cero el instrumento. Hágase aspirar entonces cada patrón sucesivamente en la llama y regístrese la absorbancia. Prepárese una curva de calibración trasladando a un papel de gráfica lineal la absorbancia de los patrones en función de sus concentraciones. Este paso no es necesario en instrumentos equipados con lectura directa de las concentraciones. Existen algunos instrumentos en los que puede necesitarse convertir el tanto por ciento de absorción en absorbancia utilizando una tabla proporcionada generalmente por el fabricante. Trácense curvas de calibración de Ca y Mg con la concentración original de los patrones antes de su dilución con solución de lantano. Trácense curvas de calibración de Fe y Mn con la concentración 3-25 DETERMINACIÓN DE METALES original de los patrones antes de la dilución con solución de calcio. Trácese una curva de calibración de Cr con la concentración original del patrón antes de la adición de H2O2. d) Análisis de muestras: Enjuáguese el nebulizador aspirando agua con 1,5 ml de HNO3 conc./l. Atomícese un blanco y ajústese a cero el instrumento. Atomícese la muestra y determínese su absorbancia. para metales más corrientes, utilizando la curva de calibración apropiada preparada según el apartado 4c. Alternativamente, léase en directo la concentración en el instrumento si éste va equipado de lectura de salida. Si se ha diluido la muestra, multiplíquese por el apropiado factor de dilución. 6. 5. Calcúlese la concentración de cada metal, en microgramos por litro para elementos traza y en miligramos por litro 3111 C. 1. Bibliografía Cálculos WILLIS, J. B. 1962. Determination of lead and other heavy metals in urine by atomic absorption spectrophotomelry. Anal. Chem. 34:614. Véase también la sección 3111A.8 y 9. Método de extracción/llama de aire-acetileno Discusión general Este método es apropiado para la determinación de concentraciones bajas de cadmio, cromo, cobalto, cobre, hierro, plomo, manganeso, níquel, plata y zinc. El método consiste en la quelación con pirrolidin ditiocarbamato de amonio (PDCA) y extracción con metil isobutil cetona (MIBC) seguido de aspiración en una llama de aire-acetileno. 2. Instrumental a) Espectrómetro de absorción atómica y equipo asociado: Véase sección 3111 A.6. b) Cabezas del quemador, convencional. Consúltese el manual de funcionamiento del fabricante para ver el cuerpo de mechero que recomienda. 3. Reactivos a) Aire: Véase 311 lB.3a. b) Acetileno: Véase 3111B.3b. c) Agua libre de metales: Véase 3111B.3c. d) Metil isobutil cetona (MIBC), de calidad reactivo. Para análisis de trazas, purifíquese la MIBC por redestilación o destilación por debajo de la ebullición. e) Solución de pirrolidin ditiocarbamato de amonio (PDCA): Disuélvanse 4 g de PDCA en 100 ml de agua. Si es necesario, purifíquese PDCA con igual volumen de MIBC. Agítese 30 segundos en un embudo de separación, déjese separar y recójase la porción inferior. Descártese la capa de MIBC. f ) Ácido nítrico, HNO3 conc, ultrapuro. g) Soluciones de metal patrones: Véase 3111B.3 j. h) Solución de permanganato de potasio, KMnO4, acuosa al 5 por 100. i) Sulfato de sodio, Na2SO4, anhidro. j) Agua saturada de MIBC: Mézclese una parte de MIBC purificada con una parte de agua en un embudo de separación. Agítese durante 30 segundos y déjese separar. Descártese la capa acuosa. Recójase la capa de MIBC. 3-26 k) Solución de clorhidrato de hidroxilamina, al 10 por 100. 4. Procedimiento a) Funcionamiento del instrumento: Véase la sección 3111B.4b. Después del ajuste final de la posición del quemador, aspírese agua saturada de MIBC en la llama y redúzcase gradualmente el flujo de combustible hasta que la llama sea similar a la de antes de aspirar el disolvente. b) Estandarización: Selecciónense al menos tres concentraciones de soluciones patrones del metal (preparadas como en 3111B.3 j) para fijar los límites de la concentración esperada de metal de la muestra y para que estén, después de la extracción, en el intervalo óptimo de concentraciones del instrumento. Ajústense 100 ml de cada patrón y 100 ml de un blanco de agua libre de metales a pH 3 por adición de HNO 3 1N, o NaOH 1N. Para la extracción de un elemento en concreto, utilícense los siguientes intervalos de pH para conseguir una eficacia de extracción óptima: NOTA: Para la extracción de Ag y Pb el valor óptimo de pH es 2,3 ± 0,2. El complejo de Mn se deteriora rápidamente a la temperatura ambiente dando lugar a una respuesta inferior del instru- MÉTODOS NORMALIZADOS mento. Enfriando el extracto a 0 °C se puede conservar el complejo durante unas cuantas horas. Si ello no resulta posible y no se puede tampoco analizar el Mn inmediatamente, utilícese otro procedimiento analítico. Pásese cada solución patrón y cada blanco a matraces volumétricos individuales de 200 ml, añádase 1 ml de solución de PDCA, sacudiendo para que se mezcle. Añádanse 10 ml de MIBC y sacúdase fuertemente durante 30 segundos. (La relación máxima de volumen de muestra a MIBC es 40.) Déjense separar los contenidos de cada matraz en capas acuosa y orgánica, añádase entonces agua cuidadosamente (ajustada al mismo pH al que se realiza la extracción) haciéndola caer por el costado de cada matraz para que la capa orgánica quede en el cuello y resulte accesible al tubo de aspiración. Aspírense los extractos orgánicos directamente en la llama (llevando el instrumento al cero con un blanco de MIBC saturado de agua) y regístrese la absorbancia. Prepárese una curva de calibración llevando a un papel de gráficos lineales las absorbancias de los patrones extraídos en función de sus concentraciones antes de la extracción. c) Análisis de muestras: Prepárense las muestras de la misma forma que los patrones. Enjuáguese el atomizador por aspiración de MIBC saturado de agua. Aspírese directamente en la llama los extractos orgánicos tratados como se ha indicado antes y regístrense las absorbancias. Con el anterior procedimiento de extracción únicamente se mide el cromo hexavalente. Para determinar el total de cromo, oxídese el cromo trivalente a cromo hexavalente llevando la muestra a ebullición y añadiendo gota a gota la suficiente solución de KMnO4 para dar a la solución un color rosa persistente mientras se hierve la solución durante 10 mi- 3-27 DETERMINACIÓN DE METALES ñutos. Elimínese el exceso de KMnO4 añadiendo una o dos gotas de solución de clorhidrato de hidroxilamina a la solución a ebullición, dejando que la reacción continúe 2 minutos. Si persiste el color rosa, añádase dos gotas más de solución de clorhidrato de hidroxilamina y espérese 2 minutos. Caliéntese durante 5 minutos más. Enfríese, extráigase con MIBC y aspírese. Si se forma una emulsión en la interfase agua-MIBC durante la extracción, añádase Na2SO4 anhidro para obtener una fase orgánica homogénea. En este caso, añádase Na2SO4 también a todos los patrones y muestras en blanco. Para evitar los problemas asociados a la inestabilidad de los complejos metálicos extraídos, determínense los metales inmediatamente después de la extracción. 3111 D. 1. 5. Calcúlese la concentración de cada ion metálico en microgramos por litro acudiendo a la curva de calibración apropiada. 6. Bibliografía ALLAN, J. E. 1961. The use of organics solvents in atomic absorption spectrophotometry. Spectrochim. Acta 17:467. SACHDEV, S. L. & P. W. WEST. 1970. Concentration of trace metals by solvent extraction and their determination by atomic absorption spectrophotometry. Environ. Sci. Technol. 4:749. Método directo de llama de óxido nitroso-acetileno Discusión general Este método se puede aplicar a la determinación de aluminio, bario, berilio, molibdeno, osmio, renio, silicio, torio, titanio y vanadio. óxido nitroso a aire, de manera que la llama pueda encenderse o apagarse con aire como oxidante para evitar un retorno de llama. 3. 2. Cálculos Instrumental a) Espectrómetro de absorción atómica y equipo asociado: Véase sección 3111A.6. b) Cabeza del quemador de óxido nitroso: Utilícese el cuerpo de mechero especial recomendado por el manual del fabricante. Cada 20 minutos en funcionamiento puede ser necesario quitar la capa de carbón formada en la superficie de la ranura empleando una varilla de carbono o similar. c) Válvula de unión en T u otra válvula de conexión para un cambio rápido de Reactivos a) Aire: Véase 3111B.3a. b) Acetileno: Véase 3111 B3b. c) Agua libre de metales: Véase 3111B.3c. d) Ácido clorhídrico, HC1 1N, 1 + 1 y conc. e) Ácido nítrico, HNO3 conc. f) Ácido sulfúrico, H 2SO 4, al 1 por 100. g) Ácido fluorhídrico, HF 1N. h) Óxido nitroso, cilindros comerciales. Instálese en el cilindro de óxido nitroso un regulador especial no congelable o arróllese un serpentín de calefacción alrededor de un regulador ordinario para 3-28 evitar el retorno de llama en el mechero, originado por reducción del flujo de óxido nitroso a través de un regulador congelado (algunos instrumentos de absorción atómica poseen sistemas automáticos de control de gas que interrumpen la llama de óxido nitroso-acetileno, por seguridad, en el caso de una reducción en la velocidad del flujo de óxido nitroso). i) Solución de cloruro de potasio: Disuélvanse 250 g de KC1 en agua y diluyase hasta 1.000 ml. j) Solución de nitrato de aluminio: Disuélvanse 139 g de A1(NO3)3 · 9H2O en 150 ml de agua. Acidúlese ligeramente con HNO3 conc. para evitar una posible hidrólisis y precipitación. Caliéntese para que se disuelva por completo. Enfríese y diluyase hasta 200 ml. k) Soluciones de metales patrón: Prepárese una serie de soluciones patrón de metales con intervalos óptimos de concentración diluyendo apropiadamente las siguientes soluciones de metales de reserva con agua que contiene 1,5 ml de HNO3 conc./l: 1) Aluminio: Disuélvase 1,00 g de metal aluminio en una mezcla acida de 4 ml de HC1 1 + 1 y 1 ml de HNO3 conc. en un vaso. Caliéntese suavemente para efectuar la disolución. Pásese a un matraz de 1 l, añádanse 10 ml de HC1 1 + 1 y diluyase con agua hasta 1.000 ml; 1,00 ml = 100μg Al. 2) Bario: Disuélvanse 0,1516 g de BaCl2 (secado a 250° durante 2 horas), en aproximadamente 10 ml de agua con 1 ml de HC1 1 + 1. Añádanse 10,0 ml de HC1 1 + 1 y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Ba. 3) Berilio: No secar. Disuélvase 1,966 g de BeSO4-4H2O en agua. Añádanse 10,0 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Be. 4) Molibdeno: Disuélvanse 0,2043 g de (NH4)2MoO4 en agua y diluyase hasta 1.000 ml; 1,00 = 100 μg Mo. MÉTODOS NORMALIZADOS 5) Osmio: Obténgase un patrón de solución de tetróxido de osmio* 0,1 M y consérvese en frasco de vidrio; 1,00 ml = = 19,2 mg Os. Háganse diluciones diariamente a medida que se necesitan utilizando H2SO4 al 1 por 100 (v/v). PRECAUCIÓN: el OsO4 es extremadamente tóxico y muy volátil. 6) Renio: Disuélvanse 0,1554 g de perrenato de potasio, KReO4, en 200 ml de agua. Diluyase hasta 1.000 ml utilizando H2SO4 al 1 por 100 (v/v); 1,00 ml = 100 μg Re. 7) Sílice: No secar. Disuélvanse 0,4730 g de Na2SiO3·9H2O en agua. Añádanse 10 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml. 1,00 ml = 100 μg Si. Consérvese en polietileno. 8) Torio: Disuélvanse 0,238 g de nitrato de torio, Th(NO3)4 · 4H2O, en 1.000 ml de agua; 1,00 ml = 100 ng Th. 9) Titanio: Disuélvanse 0,3960 g de cloruro de titanio, TiCl4†, puro (99,8 o 99,9 por 100) en una mezcla de volúmenes iguales de HC1 lN y HF 1n. Llévese hasta 1.000 ml empleando la mezcla acida; 1,00 ml = 100 μg Ti. 10) Vanadio: Disuélvanse 0,2297 g de metavanadato de amonio, NH4VO3, en una cantidad mínima de HNO3 conc. Caliéntese para que se disuelva. Añádanse 10 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 100 μg V. 4. Procedimiento a) Preparación de la muestra: Véase sección 3111B.4a. b) Funcionamiento del instrumento: Véase sección 3111B.4b. Después de ajustar la longitud de onda, instálese una cabeza de quemador de óxido nitroso. Conéctese el acetileno (sin encender la llama) y ajústese la velocidad de flujo al * GFS Chemical Co., P. O. Box 23214 Columbus, Ohio 43223, Cat. No. 64, o equivalente. † Alpha Ventron, P. O. Box 299, 152 Andover St., Danvers, Mass. 01923, o equivalente. 3-29 DETERMINACIÓN DE METALES valor especificado por el fabricante para una llama de óxido nitroso-acetileno. Desconéctese el acetileno. Con ambos suministros conectados, el de aire y el de óxido nitroso, gírese la válvula de conexión hacia el óxido nitroso y ajústese la velocidad de flujo siguiendo las especificaciones del fabricante. Conéctese la válvula a la posición de aire y compruébese que la velocidad del flujo es la misma. Conéctese el acetileno y enciéndase para obtener una llama amarilla brillante. Gírese la válvula con un movimiento rápido al óxido nitroso. La llama deberá tener un cono rojo encima del mechero. Si no fuera así, ajústese el flujo de combustible para conseguir el cono rojo. Una vez encendida la llama de óxido nitroso, déjese que el mechero alcance su equilibrio térmico antes de empezar el análisis. Pulverícese agua que contiene 1,5 ml de HNO3 conc./l y compruébese la velocidad de flujo. Si es necesario, ajústese a una velocidad entre 3 y 5 ml/minuto. Atomícese un patrón del metal deseado con una concentración próxima al punto medio del intervalo óptimo de concentraciones y ajústese el mechero (tanto horizontal como verticalmente) al trayecto luminoso para obtener la máxima respuesta. El instrumento está preparado ahora para trabajar con patrones y muestras. Para apagar la llama, gírese la válvula de conexión desde el óxido nitroso al aire y desconéctese el acetileno. Este procedimiento elimina el peligro del retorno de llama que puede ocurrir en el encendido directo o en el cierre del óxido nitroso y acetileno. c) Estandarización: Selecciónense al menos tres concentraciones de solución patrón del metal (preparadas como en el apartado 3k) con objeto de fijar límites para la concentración esperada de metal en una muestra. Aspírese cada una sucesivamente en la llama. Regístrense las absorbancias. Cuando se trata de Al, Ba y Ti, añádanse 2 ml de solución de KC1 a 100 ml del patrón antes de la aspiración. Para Mo y V añádanse 2 ml de solución de A1(NO3)3 · 9H2O a 100 ml del patrón antes de la aspiración. La mayoría de los instrumentos modernos van equipados con microprocesadores y lectura de salida digital que permite la calibración en términos directos de concentración. Si el instrumento no va equipado de esta forma, prepárese una curva de calibración llevando a un papel de gráficas lineales la absorbancia de los patrones en función de la concentración. Trácense curvas de calibración para Al, Ba y Ti basadas en la concentración original del patrón antes de añadir solución de KC1. Para Mo y V, trácense curvas de calibración basadas en la concentración original de los patrones antes de añadir la solución de A1(NO3)3. d) Análisis de muestras: Enjuáguese el atomizador aspirando agua que contiene 1,5 ml de HNO3 conc./l y póngase a cero el instrumento. Atomícese una muestra y determínese su absorbancia. Para las determinaciones de Al, Ba y Ti, añádanse 2 ml de solución de KC1 a 100 ml de la muestra antes de la atomización. Para Mo y V, añádanse 2 ml de solución de A1(NO3)3 · 9H2O a 100 ml de muestra antes de la atomización. 5. Cálculos Calcúlese la concentración de cada ion metálico en microgramos por litro acudiendo a la curva apropiada de calibración preparada según se indica en el apartado 4c. De forma alternativa, léase directamente la concentración de la lectura de salida del instrumento si se dispone de la instalación adecuada. Si la muestra ha sido diluida, multiplíquese por el apropiado factor de dilución. 3-30 6. MÉTODOS NORMALIZADOS Bibliografía WILLIS, J. B. 1965. Nitrous oxide-acetylene flame in atomic absorption spectroscopy. Nature 207:715. Véase también la sección 3111 A.8 y 9. 3111 E. Método de extracción/llama de óxido nitroso-acetileno 1. Discusión general a) Aplicación: Este método es apropiado para determinación de aluminio a concentraciones inferiores a 900 μg/1 y berilio a concentraciones inferiores a 30 μg /1. El método consiste en la quelación con 8-hidroxiquinoleína, extracción con metil isobutil cetona (MIBC) y aspiración en una llama de óxido nitroso-acetileno. b) Interferencias: Las concentraciones de Fe superiores a 10 mg/1 interfieren suprimiendo la absorción de Al. La interferencia del Fe puede ser enmascarada añadiendo clorhidrato de hidroxilamina/l,10-fenantrolina. Las concentraciones de Mn hasta 80 mg/1 no interfieren si la turbidez del extracto se deja sedimentar. El Mg forma un quelato insoluble con 8-hidroxiquinoleína a pH 8,0 y tiende a eliminar el complejo de Al formando un coprecipitado. Sin embargo, el complejo de Mg se forma lentamente, entre 4 y 6 minutos; su interferencia puede evitarse si la solución se extrae inmediatamente después de añadir tampón. c) Hidróxido amónico, NH4OH conc. d) Tampón: Disuélvanse 300 g de acetato de amonio, NH4C2H3O2, en agua. Añádanse 105 ml de NH4OH conc. y diluyase hasta 1 l. e) Agua libre de metales: Véase 3111B.2c. f) Ácido clorhídrico, HC1 conc. g) Solución de 8-hidroxiquinoleína: Disuélvanse 20 g de 8-hidroxiquinoleína en aproximadamente 200 ml de agua. Añádanse 60 ml de ácido acético glacial y diluyase con agua hasta 1 l. h) Metil isobutil cetona: Véase 3111C.3d i) Ácido nítrico, HNO3 conc. j) Óxido nitroso: Véase 3111D.3h. k) Soluciones de metal patrones: Prepárese una serie de soluciones de metal patrones con 5 a 1.000 μg/1, por dilución apropiada de las soluciones de metal de reserva preparadas según 3111D.3k. f) Solución enmascaradora del hierro: Disuélvanse 1,3 g de clorhidrato de hidroxilamina y 6,58 g de monohidrato de 1,10-fenantrolina en aproximadamente 500 ml de agua y diluyase con agua hasta 1 l. 2. Instrumental Espectrómetro de absorción atómica y equipo asociado: Véase 3111 A.6. 3. Reactivos a) Aire: Véase 3111B.3a. b) Acetileno: Véase 3111.36. 4. Procedimiento a) Funcionamiento del instrumento: Véanse secciones 3111B.4b, C.4a y D.4b. Después del ajuste final de la posición del mechero, aspírese MIBC en la llama y redúzcase gradualmente el flujo de 3-31 DETERMINACIÓN DE METALES combustible hasta que la llama sea similar a la anterior a la aspiración del disolvente. Ajústense las longitudes de onda según la tabla 3111:I. b) Estandarización: Selecciónense al menos tres concentraciones de las soluciones de metal patrones (preparadas como en el apartado 3k) para establecer límites en la concentración esperada del metal de una muestra y pásense 100 ml de cada una de ellas (y 100 ml de un blanco de agua) a cuatro matraces volumétricos de 200 ml. Añádanse 2 ml de solución de 8-hidroxiquinoleína, 2 ml de solución enmascaradora (si se necesita) y 10 ml de tampón a un matraz; añádanse inmediatamente 10 ml de MIBC y agítese vigorosamente. La duración de la agitación influye en la forma del complejo de aluminio. Una agitación rápida, de 10 segundos, favorece el Al monómero, mientras que una agitación de 5 a 10 minutos dará también especies polímeras. El ajuste de la relación 8-hidroxiquinoleína a muestra puede mejorar las recuperaciones de concentraciones extremadamente altas o bajas de aluminio. Trátese de manera similar cada blanco, patrón y muestra. Continúese como en la sección 3111C.4b. c) Análisis de muestras: Enjuáguese el atomizador aspirando agua saturada de MIBC. Aspírense los extractos de muestras tratadas como se ha indicado antes y regístrense las absorbancias. 5. Cálculos Calcúlese la concentración de cada metal en microgramos por litro empleando la curva de calibración apropiada que se ha preparado según el apartado 4b. 3112 DETERMINACIÓN DE METALES POR ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA DE VAPOR FRÍO* 3112 A. Véase sección 3111A para la introducción de métodos. * Aprobado por el Standard Methods Committee, 1988. Introducción 3-32 MÉTODOS NORMALIZADOS 3112 B. Método espectrométrico de absorción atómica de vapor frío 1. Discusión general Este método es aplicable a la determinación de mercurio. 2. Instrumental a) Espectrómetro de absorción atómica y equipo asociado: Véase sección 3111A.6. Los instrumentos y accesorios diseñados específicamente para medir el mercurio por la técnica de vapor frío pueden adquirirse en el mercado y tienen recambios. b) Célula de absorción, un tubo de vidrio o de plástico de aproximadamente 2,5 cm de diámetro. Se considera adecuado un tubo de 11,4 cm de longitud, pero es preferible uno de 15 cm de longitud. Rectifíquense los extremos del tubo perpendiculares al eje longitudinal y encájense las ventanas de cuarzo en su sitio. Acóplense los accesos de entrada y salida del gas (6,4 mm de diámetro) 1,3 cm desde cada extremo. c) Soporte de la célula: Sujétese la célula al cuerpo plano del mechero de óxido nitroso o a otro soporte adecuado y alinéese con el haz luminoso para obtener el máximo de transmitancia. d) Bombas de aire: Utilícese una bomba peristáltica con control electrónico de la velocidad capaz de suministrar 2 1 de aire/min. También se puede utilizar cualquier otro sistema de aire comprimido regulado o cilindro de aire. e) Flujómetro, capaz de medir un flujo de aire de 2 1/min. f) Tubos de aireación, vidrio poroso recto con porosidad gruesa para utilizar en el matraz de reacción. g) Matraz de reacción, matraz erlenmeyer de 250 ml o frasco para KOB, con tapón de caucho para sujetar el tubo de aireación. h) Tubo desecador, de 150 mm x 18 mm de diámetro que contiene 20 g de Mg(ClO4)2. Puede colocarse una bombilla de 60 W con una pantalla adecuada para evitar la condensación de humedad en el interior de la célula de absorción. Colóquese la bombilla de forma que se mantenga la temperatura de la célula 10 °C sobre la ambiental. i) Tubos de conexión, tubos de vidrio para transportar el vapor de mercurio desde el matraz de reacción a la célula de absorción y para interconectar los demás componentes. Los tubos de plástico vinílico * transparente pueden sustituirse por vidrio. 3. Reactivos † a) Agua libre de metales: Véase 3111B.3c. b) Solución de mercurio de reserva: Disuélvanse 1,354 g de cloruro de mercurio, HgCl2, en aproximadamente 700 ml de agua. Añádanse 10 ml de HNO3 conc. y diluyase con agua hasta 1.000 ml; 1,00 ml = 1,00 mg Hg. c) Soluciones patrón de mercurio: Prepárese una serie de soluciones patrón de mercurio con 0 a 5 μg/1 por dilución apropiada de la solución de mercurio de reserva con agua que contenga 10 ml de HNO3 conc./l. Prepárense los patrones diariamente. * Tygon o equivalente. † Utilícense sobre todo reactivos preparados bajos en mercurio. DETERMINACIÓN DE METALES d) Ácido nítrico, HNO3 conc. e) Solución de permanganato potásico: Disuélvanse 50 g de KMnO4 en agua y diluyase hasta 1 l. f) Solución de persulfato potásico: Disuélvanse 50 g de K2S2O8 en agua y diluyase hasta 1 l. g) Solución de cloruro de sodio-sulfato de hidroxilamina: Disuélvanse 120 g de NaCl y 120 g de (NH2OH)2 · H2SO4 en agua y diluyase hasta 1 l. El sulfato de hidroxilamina puede ser sustituido por solución al 10 por 100 de clorhidrato de hidroxilamina. h) Solución de cloruro estannoso: Disuélvanse 100 g de SnCl2 en agua que contenga 200 ml de HC1 conc. y diluyase hasta 1 l. Esta solución se descompone al envejecer. Si forma suspensión, agítese continuamente el reactivo durante su uso. i) Acido sulfúrico, H2SO4 conc. Figura 3112:1. Esquema de disposición del equipo para medida de mercurio por la técnica de absorción atómica de vapor frió. 3-33 4. Procedimiento a) Funcionamiento del instrumento: Véase sección 3111B.4b. Instálese la célula de absorción y alíniese con el trayecto luminoso para obtener la transmisión máxima. Conéctese el equipo asociado a la célula de absorción con los tubos de vidrio o de plástico de vinilo tal como se indica en la figura 3112:1. Conéctese el aire y ajústese la velocidad de flujo a 2 1/min. Déjese que el aire fluya continuamente. Alternativamente, síganse las indicaciones del fabricante para el funcionamiento. NOTA: La iluminación fluorescente puede aumentar los ruidos de la línea de base. b) Estandarización: Pásense 100 ml de cada una de las soluciones patrón de Hg de 1,0, 2,0 y 5,0 μg/1 y un blanco de 100 ml de agua a matraces erlenmeyer de reacción de 250 ml. Añádanse 5 ml de H2SO4 conc. y 2,5 ml de HNO3 conc. a cada matraz. Añádanse 15 ml de solución de KMnO4 a cada matraz y déjese reposar al menos 15 minutos. Añádanse 8 ml de solución de K2S2O8 a cada matraz y caliéntese a 95 °C durante 2 horas en baño maría. Enfríese hasta temperatura ambiente. Se trata cada matraz individualmente, añadiendo suficiente solución de NaClsulfato de hidroxilamina para reducir el exceso de KMnO4, añadiéndose después 5 ml de solución de SnCl2 y acoplando inmediatamente el matraz al dispositivo de aireación. Cuando el Hg se volatiliza y es arrastrado a la célula de absorción, la absorbancia aumenta al máximo en unos cuantos segundos. Tan pronto cómo el registrador vuelva aproximadamente a la línea de base, quítese el tapón que sostiene el tubo poroso del matraz de reacción y reemplácese por un matraz con agua. Límpiese el sistema con una descarga de agua unos cuantos segundos y trabájese con el siguiente patrón de la misma manera. Construyase una curva MÉTODOS NORMALIZADOS 3-34 TABLA 3112:I. PRECISIÓN Y SESGO INTERLABORATORIOS DEL MÉTODO DE ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA DE VAPOR FRÍO PARA EL MERCURIO estándar situando la altura de picos en relación con los microgramos Hg. c) Análisis de muestras: Pásense 100 ml de muestra, o porción diluida hasta 100 ml, con un contenido no superior a 5,0 ng Hg/1, a un matraz de reacción. Trátese como en el apartado 4b. Aguas marinas, salmueras y fluidos de elevado contenido en cloruros requieren hasta 25 ml más de solución de KMnO4. Durante la etapa de oxidación, los cloruros se transforman en cloro libre, que absorbe a 253 nm. Elimínese todo el cloro libre antes de reducir el Hg y efectúese un barrido del interior de la célula utilizando un exceso (25 ml) del reactivo sulfato de hidroxilamina. Elimínese el cloro libre introduciendo aire o nitrógeno tras la adición de la solución reductora de hidroxilamina. Utilícese un tubo aparte y vidrio poroso para evitar la acumulación de remanente estannoso residual, que podría dar lugar a reducción y pérdida de mercurio. 4. Cálculos Determínese la altura de picos de la muestra a partir del gráfico del registrador y léase el valor del mercurio en la curva estándar preparada según el apartado 4b. 5. Precisión y sesgo En la tabla 3112:I se dan los datos de precisión y sesgo interlaboratorios para este método. 6. 1 Referencia 1. K OPP, J. F., M. C. L ONGBOTTOM & L. B. LOBRING. 1972. «Cold vapor» method for determining mercury. J. Amer. Water Works Assoc. 64:20. 7. Bibliografía HATCH , W. R. & W. L. OTT. 1968. Determination of submicrogram quantities of mercury by atomic absorption spectrophotometry. Anal. Chem. 40:2085. UTHE, J. F., F. A. J. ARMSTRONG & M. P. STAINTON. 1970. Mercury determination in fish samples by wet digestion and flameless atomic absorption spectrophotometry. J. Fish. Res. Board Can. 27:805. FELDMAN, C. 1974. Preservation of dilute mercury solutions. Anal. Chem. 46:99. BOTHNER, M. H. & D. E. ROBERTSON. 1975. Mercury contamination of sea water samples stored in polyethylene containers. Anal. Chem. 47:592. HAWLEY, J. E. & J. D. INGLE, JR. 1975. Improvements in cold vapor atomic absorption determination of mercury. Anal. Chem. 41:119. Lo, J. M. & C. M. WAL. 1975. Mercury loss from water during storage: Mechanisms and prevention. Anal. Chem. 47:1869. EL-AWADY, A. A., R. B. MILLER & M. J. CARTER. 1976. Automated method for the determination of total and inorganic mercury in water and wastewater samples. Anal. Chem. 48:110. ODA, C. E. & J. D. INGLE, JR. 1981. Speciation of mercury by cold vapor atomic absorption spectrometry with selective reduction. Anal. Chem. 53:2305. SUDDENDORF, R. F. 1981. Interference by selenium or tellurium in the determination of mercury by cold vapor generation atomic absorption spectrometry. Anal. Chem. 53:2234. 3-35 DETERMINACIÓN DE METALES HEIDEN , R. W. & D. A. A IKENS . 1983. Humic acid as a preservative for trace mercury (II) Solutions stored in polyolefín containers. Anal. Chem. 55:2327. 3113 CHOU, H. N. & C. A. NALEWAY. 1984. Determination of mercury by cold vapor atomic absorption spectrometry. Anal. Chem. 56:1737. DETERMINACIÓN DE METALES POR ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA ELECTROTERMIA* 3113 A. Introducción 1. Aplicaciones La absorción atómica electrotérmica permite la valoración de la mayoría de elementos metálicos con sensibilidades y límites de detección de 20 a 1.000 veces superiores a los de las técnicas de llama convencionales, sin extracción o concentración de la muestra. Este aumento de sensibilidad tiene su origen en el incremento del tiempo de permanencia de los átomos del estado base en el trayecto óptico, que es varias veces mayor que el de la absorción atómica con llama convencional. Numerosos elementos pueden determinarse a concentraciones del orden de 1,0 μg/1. Otra de las ventajas de la absorción atómica electrotérmica es que únicamente se necesita un pequeñísimo volumen de muestra. La técnica electrotérmica está indicada solamente en el caso de niveles de concentración por debajo del intervalo óptimo de absorción atómica directa de llama, ya que está sometido a más interferencias que el procedimiento de llama y requiere un mayor gasto de tiempo en el análisis. Frecuentemente se necesita em* Aprobado por el Standard Methods Committee, 1988. plear el método de adiciones de patrones para asegurarse de la validez de los datos. La elevada sensibilidad de esta técnica obliga a la máxima atención para evitar la contaminación. 2. Principio La espectroscopia de absorción atómica electrotérmica se basa en el mismo principio que la atomización directa a la llama, con la diferencia de que en este caso se emplea un atomizador calentado eléctricamente o un horno de grafito en lugar de una cabeza de quemador estándar. Se introduce un volumen discreto de muestra en el tubo de muestras de grafito (o cubilete). Normalmente, la determinación se realiza por calefacción de la muestra en tres o más etapas. Primero, una corriente de baja intensidad calienta el tubo para secar la muestra. En la segunda etapa, o carbonización, se destruye la materia orgánica y se volatilizan otros componentes de la matriz a una temperatura intermedia. Por último, una corriente de elevada intensidad calienta el tubo hasta la incandescencia y atomiza el elemento cuya concentración se determina en una atmósfera inerte. Se suelen 3-36 MÉTODOS NORMALIZADOS incluir otras etapas auxiliares en el secado y la carbonización y para limpiar y enfriar el tubo entre las muestras. El vapor atómico elemental resultante absorbe la radiación monocromática de la fuente. Un detector fotoeléctrico mide la intensidad de la radiación transmitida, que es inversamente proporcional a la cantidad de átomos elementales en el trayecto óptico en un intervalo limitado. 3. Interferencias Las determinaciones mediante atomización electrotérmica están sometidas a TABLA 3113:I. significativas interferencias derivadas de la absorción molecular y de efectos químicos y de la matriz. Cuando los componentes de la matriz de la muestra se volatilizan durante la atomización puede tener lugar una absorción molecular resultando una absorción de banda ancha. Para compensar esta interferencia existen diversas técnicas de corrección del fondo que se pueden encontrar en el mercado. Una fuente continua (por ejemplo, un arco de deuterio) puede corregir el fondo hasta niveles de absorbancia de 0,8 o similares. Los correctores de fondo de efecto Zeeman pueden manipular absorbancias del fondo de hasta 1,5 a 2,0. La téc- MODIFICADORES DE MATRICES POTENCIALES PARA ESPECTROMETRÍA1 DE ABSORCIÓN ATÓMICA ELECTROTÉRMICA DETERMINACIÓN DE METALES nica de corrección de Smith-Hieftje puede ajustar niveles de absorbancia del fondo hasta de 2,5 a 3,0 (véase sección 3111A.3). Utilícese corrección del fondo cuando se analizan muestras que contienen concentraciones elevadas de ácido o sólidos disueltos y al determinar elementos con los que se utiliza una línea de absorción por debajo de 350 nm. Para reducir al mínimo la interferencia puede acudirse a la modificación de la matriz. Ello se consigue añadiendo diversos productos químicos a la muestra. Alternativamente, se puede programar un dispositivo automático de obtención de muestras que añada modificadores de la matriz directamente a la muestra en la cámara del horno. Algunos modificadores de matriz reducen la volatilidad del elemento que se determina o aumentan su eficacia de atomización por cambio de su composición química. Esto permite emplear temperaturas de carbonización más elevadas para volatilizar las sustancias que interfieren aumentando al mismo tiempo la sensibilidad. Otros modificadores de la matriz aumentan la volatilidad de la misma. En la tabla 3113:I se da una lista de modificadores de matriz. El calentamiento gradual puede servir para disminuir las interferencias del fondo y permite el análisis de muestras con matrices complejas. El calentamiento gradual permite un incremento continuo y controlado de la temperatura del horno en cualquiera de las etapas de la secuencia de temperaturas. Utilícese el secado gradual en el caso de muestras que contengan mezclas de disolventes o muestras con elevado contenido en sal (para evitar salpicaduras). Las muestras que contienen una mezcla compleja de componentes de la matriz requieren a veces una carbonización gradual, para que tenga lugar una descomposición térmica completa y controlada. La atomización gradual puede reducir la absorción de fondo por permitir la volatilización del elemento que se determina antes que la 3-37 matriz. Este método tiene especial aplicación en la determinación de elementos volátiles tales como el cadmio y el plomo. Utilícense también adiciones de patrones para compensar interferencias de la matriz. Cuando se realizan adiciones de patrones para compensar interferencias de matriz, determínese si la especie añadida y el elemento que se determina tienen un comportamiento similar en las condiciones especificadas (véase sección 3113B.4d2). A temperaturas elevadas de carbonización y atomización, tiene lugar una interacción química del tubo de grafito con varios elementos formando carburos refractarios. Los elementos que forman carburos son bario, molibdeno, níquel, titanio, vanadio y silicio. Esta formación de carburos se caracteriza por la aparición de picos de atomización anchos, de amortiguación prolongada y de sensibilidad reducida. La utilización de tubos con recubrimiento pirolítico para estos metales reduce el problema. Para el análisis de aluminio, las plataformas L'vov tratadas con torio dan picos más nítidos a bajas concentraciones y refuerzan la estabilidad de la carbonización. 4. Sensibilidad, límites de detección e intervalo óptimo de concentraciones En la tabla 3113:II se recogen listas de límites de detección estimados e intervalos de concentración óptimos. Estos valores pueden variar con la forma química del elemento que se determina, la composición de la muestra o las condiciones del instrumental. Para una muestra dada, puede conseguirse una mayor sensibilidad empleando un volumen mayor de muestra o reduciendo la velocidad de flujo del gas de purga o con interrupción del gas durante la atomización. Nótese, sin embargo, que estas técnicas también aumentarán los efectos de cualquier posible interferencia. 3-38 MÉTODOS NORMALIZADOS TABLA 3113:II. NIVELES DE DETECCIÓN Y MÁRGENES DE CONCENTRACIÓN PARA ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA DE ATOMIZACIÓN ELECTROTÉRMICA La sensibilidad puede disminuir por dilución de la muestra, por reducción del volumen de muestra, por aumento del flujo de gas de purga o mediante empleo de una longitud de onda menos sensible. El empleo de argón como gas de purga, en lugar de nitrógeno, mejora por lo general la sensibilidad y la reproducibilidad. El hidrógeno mezclado con el gas inerte puede suprimir la interferencia química y aumentar la sensibilidad al actuar como un reductor, ayudando en la producción de más átomos elementales. Utilizando tubos de grafito con recubrimiento pirolítico puede aumentar la sensibilidad para los elementos más refractarios. El accesorio pirómetro óptico/energía máxima de que disponen algunos instrumentos ofrece también una mayor sensibili- dad con temperaturas de atomización inferiores para muchos elementos. Utilizando la técnica de horno de plataforma de temperatura estabilizada (HPTE), que es una combinación de técnicas individuales, se obtiene también una reducción significativa de interferencias al mismo tiempo que una sensibilidad mejorada. La sensibilidad cambia con el envejecimiento del tubo de muestras. Descártense los tubos de grafito cuando se observen variaciones significativas de la sensibilidad o una pobre reproducibilidad. El empleo de concentraciones elevadas de ácido, muestras de salmuera y modificadores de matrices, reducen drásticamente con frecuencia la vida del tubo. En estos casos es preferible utilizar la plataforma de L'vov. 3-39 DETERMINACIÓN DE METALES 5. Referencia 1. SLAVIN, W. 1984. Graphite Furnace AASA Source Book. Perkin-Elmer Corp., Norwalk, Connecticut. 6. Bibliografía FERNÁNDEZ, F. J. & D. C. MANNING. 1971. Atomic absorption analyses of metal pollutants in water using a heated graphite atomizer. Atomic Absorption Newsletter 10:65. SEGAR. D. A. & J. G. GONZÁLEZ. 1972. Evaluation of atomic absorption with a heated graphite atomizer for the direct determination of trace transition metals in seawater. Anal. Chim. Acta 58:7. BARNARD, W. M. & M. J. FISHMAN. 1973. Evaluation of the use of heated graphite atomizer for the routine determination of trace metals in water. Atomic Absorption Newsletter 12:118. KAHN, H. L. 1973. The detection of metallic elements in wastes and waters with the graphite furnace. Int. J. Environ. Anal. Chem. 3:121. WALSH, P. R., J. L. FASCHING & R. A. DUCE. 1976. Matrix effects and their control during the flameless atomic absorption determination of arsenic. Anal. Chem. 48:1014. 3113 B. 1. Método espectrométrico de absorción atómica electrotérmica Discusión general Este método es apropiado para determinar microcantidades de aluminio, antimonio, arsénico, bario, berilio, cadmio, cromo, cobalto, cobre, hierro, plomo, manganeso, molibdeno, níquel, selenio, plata y estaño. 2. HENN, E. L. 1977. Use of Molybdenum in Eliminating Matrix Interferences in Flameless Atomic Absorption. Spec. Tech. Publ. 618, American Soc. Testing & Materials, Filadelfia, Pennsylvania. MARTIN, T. D. & J. F. Kopp. 1978. Methods for Metals in Drinking Water. U. S. Environmental Protection Agency, Environmental Monitoring and Support Lab., Cincinnati, Ohio. HYDES, D. J. 1980. Reduction of matrix effects with a soluble organic acid in the carbon furnace atomic absorption spectrometric determination of cobalt, cooper, and manganese in seawater. Anal. Chem. 52:289. SOTERA, J. J. & H. L. KAHN. 1982. Background correction in AAS. Amer. Lab. 14:100. SMITH, S. B. & G. M. HIEFTJE. 1983. A new background-correction method for atomic absorption spectrometry. Appl. Spectrosc. 37:419. GROSSER, Z. 1985. Techniques in Graphite Furnace Atomic Absorption Spectrophotometry. Perkin-Elmer Corp., Ridgefield, Connecticut. SLAVIN, W. & G. R. CARNICK. 1985. A survey of applications of the stabilized temperature platform furnace and Zeeman correction. Atomic Spectrosc. 6:157. BRUEGGEMEYER, T. & F. FRICKE. 1986. Comparison of furnace & atomization behavior of aluminum from standard & thorium-treated L'vov platforms. Anal. Chem. 58:1143. Instrumental a) Espectrómetro de absorción atómica: Véase sección 3111A.6a. El instrumento debe tener capacidad de corrección de fondo. b) Lámparas fuente: Véase sección 3111A.6d. c) Horno de grafito: Utilícese un dispositivo calentado con circuitos electrónicos de control diseñado para conducir un tubo o cubilete de grafito a través de un programa de calentamiento que proporcione suficiente energía térmica para atomizar los elementos que interesan. Los controles de calor de un horno con sólo tres etapas de calentamiento son adecuados para agua dulce con bajo contenido en sólidos disueltos. En el caso de aguas saladas, salmueras y otras matrices complejas, utilícese un controlador de horno que tenga hasta siete etapas de MÉTODOS NORMALIZADOS 3-40 calentamiento programadas individualmente. Instálese el horno en el compartimento de muestras del espectómetro en lugar de la instalación de mechero convencional. Utilícese argón como gas de purgado para reducir al mínimo la oxidación del tubo de horno y para evitar la formación de óxidos metálicos. Utilícense tubos de grafito con plataformas L'vov para reducir al mínimo las interferencias y mejorar la sensibilidad. d) Lectura de salida: Véase sección 3111A.6C. e) Distribuidores de muestras: Empléense pipetas de microlitros (5 a 100 μl) o un dispositivo automático de toma de muestras diseñado para el instrumento específico. f) Ventilación: Véase sección 3Í11A.6f. g) Suministro de agua de refrigeración: Refrigérese con agua del grifo fluyendo entre 1 y 4 1/minuto o utilícese un dispositivo refrigerador recirculante. h) Dispositivo de filtro de membrana: Utilícese un dispositivo filtrante todo de vidrio y filtros de membrana de 0,45 μm. Para análisis de trazas de aluminio, empléese un dispositivo de polipropileno o de TFE. 3. 4) Nitrato de níquel, 10.000 mg Ni/1: Disuélvanse 49,56 g de Ni(NO3)2·6H2O en agua. Diluyase con agua hasta 1.000 ml. 5) Ácido fosfórico, al 10 por 100 (v/v): Dilúyanse 10 ml de H3PO4 conc. hasta 100 ml utilizando agua. Para la preparación de otros modificadores de matriz, véanse referencias o síganse las instrucciones del fabricante. e) Soluciones de metal de reserva: Véanse secciones 3111B y 3114. f) Resina quelante: de 100 a 200 mallas* purificada por calentamiento a 60 °C en NaOH 10N durante 24 horas. Enfríese la resina y enjuáguese 10 veces con porciones alternativas de HC1 1N, agua libre de metales, NaOH 1N y agua libre de metales, cada vez. g) Agua de mar libre de metales (o salmuera): Llénese una columna de vidrio al borosilicato de 1,4 cm de DI x 20 cm de largo con resina quelante hasta 2 cm del extremo superior. Diluyase la resina con porciones sucesivas de 50 ml de HC1 1N, agua libre de metales, NaOH 1N y agua libre de metales a una velocidad de 5 ml/minuto para extraer los metales traza presentes. Descártense los primeros 10 volúmenes de lecho (300 ml) de soluto. Reactivos a) Agua libre de metales: Véase sección 3111B.3c. b) Ácido clohídrico, HC1 1 + 1 y conc. c) Ácido nítrico, HNO3 1 + 1 y conc. d) Modificadores de matriz: 1) Nitrato de amonio, al 10 por 100 (p/v): Disuélvanse 100 g de NH4NO3 en agua. Diluyase con agua hasta 1.000 ml. 2) Fosfato de amonio, al 40 por 100: Disuélvanse 40 g de (NH4)2HPO4 en agua. Diluyase con agua hasta 100 ml. 3) Nitrato de calcio, 20.000 mg Ca/1: Disuélvanse 11,8 g de Ca(NO3)2·4H2O en agua. Diluyase con agua hasta 100 ml. 4. Procedimientos a) Pretratamiento de muestras: Antes del análisis, trátense previamente las muestras como se indica a continuación. Enjuáguese todo el material de vidrio con HNO3 1 + 1 y con agua. Realícense los procedimientos de digestión en un área del laboratorio limpia y sin polvo para evitar la contaminación de la muestra. Para la digestión de trazas de aluminio, utilícense utensilios de polipropileno * Chelex 100 o equivalente, de Bio-Rad Laboratories, Richmond, California. DETERMINACIÓN DE METALES o TFE para evitar que se arrastre aluminio desde el material de vidrio. 1) Metales disueltos: Véase sección 3030B. Para muestras que requieren análisis de arsénico y/o selenio, añádanse 3 ml de peróxido de hidrógeno al 30 por 100 y una concentración apropiada de níquel antes del análisis. Para los demás metales no se necesitan más tratamientos previos que la adición de un modificador opcional de la matriz (véase tabla 3113:I). 2) Metales recuperables totales (Al, Sb, Ba, Be, Cd, Cr, Co, Cu, Fe, Pb, Mn, Mo, Ni, Ag y Sn): NOTA: Sb y Sn se recuperan siempre que no se utilice HC1 en la digestión. Véase sección 3030D. La muestra digerida se pasa a un matraz volumétrico de 100 ml, se añade una cantidad apropiada de modificador de matriz (si es necesario, véase tabla 3113:I) y se diluye con agua hasta el volumen del matraz. 3) Metales recuperables totales (As, Se): Pásense 100 ml de muestra agitada, 1 ml de HNO3 conc. y 2 ml de H2O2 a un vaso de 250 ml limpio, lavado con ácido. Caliéntese sobre placa caliente, sin dejar que hierva la solución, hasta que el volumen se ha reducido a 50 ml aproximadamente. Quítese de la placa caliente y déjese enfriar hasta temperatura ambiente. Añádase una concentración apropiada de níquel (veáse tabla 3113:I), y diluyase con agua en un matraz volumétrico de 100 ml hasta el volumen del matraz. Prepárese simultáneamente un blanco digerido sustituyendo la muestra por agua y realizando la digestión tal como se ha descrito con anterioridad. b) Funcionamiento del instrumento: Móntese y alíniese el dispositivo del horno siguiendo las instrucciones del fabricante. Conéctese el instrumento y los registradores de la cinta de gráficos. Selecciónese la fuente luminosa apropiada y ajústese a la regulación eléctrica recomendada. Selecciónese la apropiada longitud de onda y establézcanse todas las 3-41 condiciones de acuerdo con las instrucciones del fabricante, incluyendo la corrección de fondo. La corrección de fondo es importante cuando se determinan elementos a longitudes de onda cortas o cuando la muestra tiene un elevado nivel de sólidos disueltos. Por lo general, el proceso no suele ser necesario a longitudes de onda más largas de 350 nm. Por encima de 350 nm, no es útil la corrección con arco de deuterio y hay que emplear otros procedimientos. Selecciónese un flujo apropiado de gas inerte o de protección. En algunos casos conviene interrumpir el flujo de gas inerte durante la atomización. Tal interrupción da lugar a un aumento de sensibilidad al incrementarse el tiempo de residencia del vapor atómico en el trayecto óptico. La interrupción del gas también aumenta la absorción de fondo e intensifica los efectos de interferencia. Considérense las ventajas y desventajas de esta opción para cada matriz cuando se optimizan las condiciones analíticas. Para hacer óptimas las condiciones del horno de grafito, ajústense cuidadosamente las regulaciones de la temperatura del horno para hacer máximas sensibilidad y precisión y mínimas las interferencias. Síganse las instrucciones del fabricante. Utilícense temperaturas de secado ligeramente superiores al punto de ebullición del disolvente, con objeto de facilitar el tiempo y la temperatura suficiente para una evaporación completa sin ebullición o salpicaduras. La temperatura de carbonización debe ser lo suficientemente alta para hacer máxima la volatilización de los componentes de la matriz que interfieren, siendo al mismo tiempo baja para volatilizar el elemento que interesa. Con las temperaturas de secado y atomización fijadas a sus valores óptimos, analícese un patrón a una serie de temperaturas de carbonización con incrementos crecientes de 50 a 100 °C. Cuando se sobrepase la tempe- 3-42 ratura óptima de carbonización, habrá una caída significativa de la sensibilidad. Trácese una curva de temperatura de carbonización frente a absorbancia de muestra: la temperatura de carbonización óptima es la temperatura más alta que no reduce la sensibilidad. Selecciónese la temperatura de atomización determinando la temperatura que proporciona un máximo de sensibilidad sin detrimento de la precisión. Optimícese con una serie de determinaciones sucesivas a diversas temperaturas de atomización utilizando una solución patrón que dé una absorbancia de 0,2 a 0,5. c) Calibración del instrumento: Prepárense patrones para calibración del instrumento diluyendo las soluciones de reserva del metal. Elabórense patrones diariamente. Prepárense un blanco y al menos tres patrones de calibración en el intervalo de concentraciones apropiado (véase tabla 3113:11) correlacionando la concentración del elemento con la respuesta del instrumento. Adáptese la matriz de las soluciones patrón a las de las muestras lo más exactamente posible. En la mayoría de los casos esto se reduce a igualar simplemente el fondo ácido de las muestras. Si se trata de aguas marinas o salmueras, sin embargo, utilícese la matriz libre de metales (apartado 3g) como diluyente de la solución patrón. Añádase también una concentración igual de modificador de la matriz (en caso de que lo requiera el análisis de la muestra) a las soluciones patrón. Inyéctese una porción adecuada de cada solución patrón, con objeto de aumentar la concentración. Analícese cada solución patrón por triplicado para comprobar la precisión del método. Trácese una curva analítica poniendo absorbancias de picos medias o áreas de picos de la solución patrón en función de la concentración en papel de gráficos lineal. Alternativamente, utilícese un instrumento electrónico de calibración en el MÉTODOS NORMALIZADOS caso de que el instrumento tenga esta capacidad. d) Análisis de la muestra: Analícense todas la muestras, excepto las que demuestren estar libres de interferencias de la matriz (basándose en recuperaciones de 85-115 por 100 para adiciones conocidas) utilizando el método de adiciones de patrón. Analícense todas las muestras al menos por duplicado o hasta obtener resultados reproducibles. Una variación de d 10 por 100 se considera una reproducibilidad aceptable. Hállese la media de los valores de los replicados. 1) Determinación directa: Inyéctese en el horno de grafito una porción medida de muestra sometida a tratamiento previo. Empléese el mismo volumen que el utilizado para preparar la curva de calibración. Séquese, carbonícese y atomícese según el programa prefijado. Repítase hasta obtener resultados reproducibles. Compárese el valor medio de la absorbancia o área de pico con la curva de calibración para determinar la concentración del elemento que interesa. Alternativamente, léanse directamente los resultados si la capacidad del instrumento lo permite. Si la absorbancia (o concentración) o área de pico de la muestra más concentrada es superior a la absorbancia (concentración) o área de pico del patrón, diluyase la muestra y vuélvase a analizar. Si se necesitan diluciones muy grandes, puede haber otra técnica (por ejemplo, AA de llama o PAI) más adecuada para esta muestra. Los factores de dilución grandes aumentan los pequeños errores en los cálculos finales. Manténgase un fondo ácido y una concentración de modificador de matriz (cuando se requiera) constante. Si se diluye la muestra con agua, añádanse ácido y modificador de matriz para restablecer la concentración de ambos como en la muestra original. Alternativamente, diluyase la muestra en una solución de blanco de ácido y modificadores de la matriz. DETERMINACIÓN DE METALES Procédase como en el apartado 5a, desarrollado a continuación. 2) Método de adiciones de patrón: Véase el apartado 4e anterior. El método de adiciones de patrón sólo es válido cuando queda dentro de la porción lineal de la curva de calibración. Una vez que la sensibilidad del instrumento ha sido hecha óptima para el elemento que interesa y que se ha establecido el intervalo lineal para el elemento, procédase con el análisis de la muestra. Introdúzcase un volumen medido de muestra en el dispositivo del horno. Séquense, carbonícense o realícese una combustión seca, y atomícense las muestras con arreglo al programa prefijado. Repítase hasta obtener resultados reproducibles. Regístrese la respuesta del instrumento en absorbancia o concentración según lo apropiado. Añádase una concentración conocida del elemento que interesa a una porción separada de muestra de manera que no cambie significativamente el volumen de la muestra. Repítase la determinación. Añádase una concentración conocida (preferiblemente doble de la utilizada en la primera adición) a una porción separada de muestra. Mézclese bien y repítase la determinación. Llévese la absorbancia media o respuesta del instrumento para la muestra y las dos porciones con adiciones conocidas sobre el eje de ordenadas y las concentraciones de elemento añadidas sobre el eje de abscisas de papel de gráficos lineales. Dibújese una línea recta que una los tres puntos y extrapólese la absorbancia a cero. La intersección en el eje horizontal da la concentración de la muestra. El eje de concentraciones a la izquierda del origen habrá de ser la imagen en el espejo del eje a la derecha. 5. Cálculos a) Determinación directa: μg metal/1 = C x F 3-43 siendo: C = concentración de metal leída directamente en el instrumento o en la curva de calibración, en μg/1, y F = factor de dilución. b) Método de adiciones: μg metal/1 = C x F siendo: C = concentración de metal deducida por el método del gráfico de adiciones, en F = factor de dilución. 6. Precisión y sesgo En las tablas 3113:III, IV y V se dan los datos típicos de la precisión y el sesgo obtenibles. 7. Control de calidad Para los procedimientos de control de calidad específicos que han de seguirse durante el análisis, véase sección 3020. Aunque las indicaciones previas muestren unos intervalos óptimos de concentraciones muy bajos para estos metales (véase tabla 3113:II), los datos de la tabla 3113:III que utilizan variaciones de estos protocolos muestran que puede no ser así. Extrémense las precauciones cuando se aplica este método a los intervalos más bajos de concentraciones. Compruébese la precisión del análisis al principio de cada marcha analítica haciendo análisis por triplicado. 8. Referencia 1. COPELAND, T. R. & J. P. MANEY. 1986. EPA Method Study 31: Trace Metals by Atomic Absorption (Furnace Techniques). EPA- 3-44 TABLA 3113:III. MÉTODOS NORMALIZADOS DATOS DE PRECISIÓN INTERLABORATORIOS DE UN ÚNICO ANALISTA PARA MÉTODOS DE ATOMIZACIÓN ELECTROTÉRMICA 1 DETERMINACIÓN DE METALES TABLA 3113:IV. 3-45 DATOS DE PRECISIÓN GLOBAL INTERLABORATORIOS PARA MÉTODOS DE ATOMIZACIÓN ELECTROTÉRMICA 1 3-46 TABLA 3113:V. MÉTODOS NORMALIZADOS DATOS DE ERRORES RELATIVOS INTERLABORATORIOS PARA MÉTODOS DE ATOMIZACIÓN ELECTROTÉRMICA 1 3-47 DETERMINACIÓN DE METALES 600/S4-85-070, U. S. Environmental Protection Agency, Environmental Monitoring and Support Lab., Cincinnati, Ohio. 9. Bibliografía RENSHAW, G. D. 1973. The determination of barium by flameless atomic absorption spectrophotometry using a modified graphite tube atomizer. Atomic Absorption Newsletter 12:158. YANAGISAWA, M., T. TAKEUCHI & M. SUZUKI. 1973. Flameless atomic absorption spectrometry of antimony. Anal. Chim. Acta 64:381. RATTONETTI, A. 1974. Determination of soluble cadmium, lead, silver and indium in rainwater and stream water with the use of flameless atomic absorption Anal. Chem. 46:739. HENN, E. L. 1975. Determination of selenium in water and industrial effluents by flameless atomic absorption. Anal. Chem. 47:428. MARTIN, T. D. & J. F. KOPP. 1975. Determining selenium in water, wastewater, sediment and sludge by flameless atomic absorption spectrometry. Atomic Absorption Newsletter 14:109. MARUTA, T., K. mlNEGISHI & G. SUDOH. 1976. The flameless atomic absorption spectrometric determination of aluminum with a carbon atomization system. Anal. Chim. Acta 81:313. CRANSTON, R. E. & J. W. MURRAY. 1978. The determination of chromium species in natural waters. Anal. Chim. Acta 99:275. HOFFMEISTER, W. 1978. Determination of iron in ultrapure water by atomic absorption spectroscopy, Z. Anal. Chem. 50:289. LAGAS, P. 1978. Determination of beryllium, barium, vanadium and some other elements in water by atomic absorption spectrometry with electrothermal atomization. Anal. Chim. Acta 98:261. CARRONDO, M. J. T., J. N. LESTER & R. PERRY. 1979. Electrothermal atomic absorption determination of total aluminum in waters and waste waters. Anal. Chim. Acta 111:291. NAKAHARA, T. & C. L. CHAKRABARTI. 1979. Direct determination of traces of molybdenum in synthetic sea water by atomic absorption spectrometry with electrothermal atomization and selective volatilization of the salt matrix. Anal. Chim. Acta 104:99. TIMINAGA, M. & Y. UMEZAKI. 1979. Determination of submicrogram amounts of tin by atomic absorption spectrometry with electrothermal atomization. Anal. Chim. Acta 110:55. 3114 DETERMINACIÓN DE METALES POR GENERACIÓN DE HIDRUROS/ESPECTROMETRÍA DE ABSORCIÓN ATÓMICA* 3114 A. Véase sección 3111A para la introducción general sobre métodos espectrométricos de absorción atómica. En esta sección se presentan dos métodos: Un método manual y un método de flujo continuo recomendado especial- * Aprobado por el Standard Methods Committee, 1989. Introducción mente para el selenio. Se prefieren los sistemas automáticos de flujo continuo a los generadores manuales de hidruro debido a que se elimina el efecto de una generación repentina de hidrógeno sobre la transparencia del trayecto luminoso, y cualquier respuesta del blanco de la contaminación del reactivo de HC1 por los elementos que se determinan se incorpora a la línea base del fondo. 3-48 MÉTODOS NORMALIZADOS 3114 B. Método manual de generación de hidruros/espectrometría de absorción atómica 1. Discusión general a) Principio: Este método es aplicable a la determinación de arsénico y selenio por transformación de los mismos en sus hidruros con reactivo borohidruro sódico y aspiración en un atomizador de absorción atómico. El ácido arsenioso y el ácido selenioso, los estados de oxidación As(III) y Se(IV) de arsénico y selenio, respectivamente, se convierten instantáneamente en sus hidruros volátiles con el reactivo borohidruro de sodio en solución acida. Los hidruros se purgan continuamente con argón o nitrógeno en un atomizador apropiado de un espectrómetro de absorción atómica y se convierten en los átomos de la fase gaseosa. El reductor borohidruro de sodio, por una rápida generación de los hidruros de los elementos en una célula de reacción apropiada, hace que sea mínima la dilución de los hidruros por el gas portador y proporciona una determinación rápida y sensible de arsénico y selenio. PRECAUCIÓN: El arsénico, el selenio y sus correspondientes hidruros son tóxicos. Manipúlense con cuidado. A la temperatura ambiente y a valores del pH de la solución de 1 o menores, el ácido arsénico, estado de oxidación As(V) de arsénico, se reduce con relativa lentitud por el borohidruro sódico a As(III), que es transformado instantáneamente en arsina. Los picos de absorción atómica de arsina son normalmente un cuarto o un tercio más bajos para As(V) cuando se compara con As(III). La determinación del arsénico total requiere que todos los compuestos de arsénico inorgánicos estén en el estado de As(III). Las formas orgánicas e inorgánicas de arsénico se oxidan primero a As(V) por digestión con ácidos. El As(V) se reduce entonces cuantitativamente a As(III) con yoduro de sodio o potasio antes de la reacción con borohidruro de sodio. El ácido selénico, el estado de oxidación Se(VI) de selenio, no se reduce perceptiblemente con borohidruro sódico. Para determinar el selenio total por absorción atómica y borohidruro de sodio, se reduce primero el Se(VI) formado durante el procedimiento de digestión a Se(VI), teniendo cuidado de evitar una nueva oxidación por cloro. El rendimiento de la reducción depende de la temperatura, del tiempo de reducción y de la concentración de HC1. Con HC1 4N, caliéntese 1 hora a 100 °C. Con HC1 6N, es suficiente una ebullición durante 10 mlnutos1, 3. Alternativamente, manténganse en autoclave muestra en recipientes cerrados a 121 °C durante 1 hora. NOTA: El tratamiento en autoclave de recipientes cerrados puede dar lugar a una reducción incompleta, debido al parecer a la formación de gas cloro. Para obtener respuestas iguales del instrumento para soluciones de Se(VI) y Se(IV) de iguales concentraciones, regúlense la concentración de HC1 y el tiempo de calentamiento. Para más detalles, véase sección 3500Se. b) Selección del equipamiento: En el mercado existen algunos atomizadores de absorción atómica y células de reacción con hidruros que pueden utilizarse con el reactivo borohidruro de sodio. En la figura 3114:1 se muestra un sistema funcional. Independientemente de cuál sea el sistema atomizador de célula de reacción con hidruro seleccionado, éste debe satisfacer las siguientes consideraciones de control de calidad: a) proporcionar una curva estándar precisa y reproducible entre 0 y 20 μg de As o Se/1 y un límite de detección del instrumental entre 0,1 y 0,5 μg As o Se/1; b) cuando se lleva a cabo el procedimiento completo, los pares de oxidación [As(III)-As(V) DETERMINACIÓN DE METALES 3-49 Figura 3114:1. Célula manual de reacción para producir hidruros de As y Se. o Se(IV)-Se(VI)] deben provocar la misma respuesta del instrumento, y c) la digestión de la muestra debe conducir a un 80 por 100 de recuperación del ácido cacodílico añadido (ácido dimetil arsínico) y una recuperación del 90 por 100 o mayor de As(III), As(V), Se(VI) o Se(IV) añadidos. En la medida de arsénico y selenio se suelen emplear tres tipos de atomizadores de absorción atómica. La mayoría de los fabricantes de instrumentos pueden proporcionar un mechero tipo Boling de llamas de argón (o nitrógeno)-aire con hidrógeno arrastrado. Alternativamente utilícese una célula de cuarzo con calentamiento exterior o una célula de cuarzo con una llama de hidrógeno-oxígeno rico en combustible o de hidrógeno-aire. Las células de atomización de cuarzo proporcionan las determinaciones más sensibles de hidruros de arsénico y selenio y reducen al mínimo el ruido de fondo asociado a la llama de hidrógeno arrastrado por aire-argón. c) Técnicas de digestión: Las aguas naturales y residuales pueden contener cantidades variables de compuestos orgánicos de arsénico y compuestos inorgánicos de As(III), As(V), Se(IV) y Se(VI). Para medir el arsénico y selenio en total de estas muestras se necesita una digestión de la muestra con objeto de solubilizar las formas en partículas y oxidar las formas de arsénico y selenio reducidas, así como para convertir los compuestos orgánicos en inorgánicos. Raramente han sido detectados en el agua compuestos orgánicos de selenio. Se deja al criterio del analista experimentado la necesidad de digestión de la muestra. En el siguiente apartado 4 se ofrecen 3-50 dos procedimientos de digestión. Hay que considerar la digestión con ácido sulfúrico-nítrico-perclórico o la digestión con ácido sulfúrico-nítrico como un paso para la medida del arsénico recuperable total más que arsénico total, ya que no convierte por completo ciertos compuestos orgánicos de arsénico a As(V). La digestión con ácido sulfúrico-nítrico-perclórico destruye eficazmente las sustancias orgánicas y la mayoría de las partículas de las aguas residuales sin tratar o de muestras sólidas. La digestión con persulfato de potasio (apartado Ad) es eficaz para convertir los compuestos orgánicos de arsénico y selenio en As(V) y Se(VI) en las aguas potables superficiales y en la mayoría de aguas residuales4. La reducción con HC1 en autoclave del Se(VI) descrita antes es un procedimiento de digestión eficaz para el Se inorgánico total; pero no ha resultado eficaz para convertir los compuestos de benceno sustituido con selenio a selenio inorgánico. d) Interferencias: Las interferencias son mínimas, ya que los hidruros de As y Se se separan de la solución que contiene la mayoría de las sustancias que pueden interferir. Cuando se hacen variar las matrices de ácidos aparecen ligeras variaciones de la respuesta. Contrólense estas variaciones tratando de la misma manera los patrones y las muestras. Concentraciones bajas de metales nobles (aproximadamente 100 μg/1 de Ag, Au, Pt, Pd, etc.), concentraciones de cobre, plomo y níquel de 1 mg/1 o mayores, y concentraciones entre 0,1 y 1 mg/1 de elementos que forman hidruros (Bi, Sb, Sn y Te) pueden suprimir la respuesta de los hidruros de As y Se. La interferencia por metales de transición depende en buena medida de la concentración de HC1. Las interferencias son menos pronunciadas con HC1 4 a 6N que con concentraciones más bajas5. La presencia de As y Se en las matrices de los otros pueden dar lugar a una supresión similar. Los óxidos MÉTODOS NORMALIZADOS de nitrógeno reducidos provenientes de la digestión con HNO3 y los nitritos también suprimen la respuesta instrumental para ambos elementos. Concentraciones elevadas de yoduro interfieren la determinación de Se por reducción del Se a su forma elemental. No utilizar material de vidrio que haya sido empleado para reducir As(V) con yoduro para la determinación de Se. Para evitar que el gas cloro producido en la reducción del Se(VI) a Se(IV) vuelva a oxidar el Se(IV), genérese el hidruro en el plazo de unas cuantas horas de las etapas de reducción o púrguese el cloro de las muestras por burbujeo6. Las interferencias dependen del diseño del sistema y desafían cualquier descripción cuantitativa debido a sus efectos sinérgicos. Determinadas aguas naturales y residuales pueden presentar sustancias de interferencia en concentración suficiente para suprimir respuestas de As y Se. En el caso de muestras representativas de un determinado laboratorio y tratándose de análisis iniciales de aguas residuales desconocidas, añádanse formas inorgánicas apropiadas de As o Se a las porciones de muestra digerida y mídase la recuperación. Si las recuperaciones medias son inferiores al 90 por 100, considérese la utilización de procedimientos analíticos alternativos. e) Límite de detección e intervalo óptimo de concentraciones: Tanto para el arsénico como para el selenio, analizados por aspiración dentro de una llama de nitrógeno-hidrógeno después de su reducción, el límite de detección del método es de 0,002 mg/1, y el intervalo óptimo de concentraciones, de 0,002 a 0,02 mg/1. 2. Instrumental a) Espectrómetro de absorción atómica: Equipado con flujómetros para argón (o nitrógeno) e hidrógeno, lámparas de descarga sin electrodos de As y Se con DETERMINACIÓN DE METALES suministro de energía, corrección de fondo a las longitudes de onda de medida y registrador de cinta gráfica apropiado. Se necesita un registrador de 10 mV de buena calidad con sensibilidad elevada y un tiempo de respuesta rápido. b) Atomizador: Utilícese uno de los siguientes: 1) Cabeza de quemador de tipo Boling para llama de argón (o nitrógeno)hidrógeno arrastrado por aire. 2) Célula cilíndrica de cuarzo, de 10 a 20 cm de largo, calentada eléctricamente con alambre exterior de aleación «Nichrome» entre 800-900 °C7. 3) Célula cilíndrica de cuarzo con llama interna de hidrógeno rico en combustible-oxígeno (aire)8. La sensibilidad de las células de cuarzo se deteriora a los meses de usarlas. A veces puede restablecerse la sensibilidad tratando con HF al 40 por 100. PRECAUCIÓN: El HF es extraordinariamente corrosivo. Evítese el contacto con la piel. Manipúlese con cuidado. c) Célula de reacción para producir hidruros de As o Se: Véase figura 3114:1. Es aceptable un sistema comercializado siempre que utilice reactivos líquidos de borohidruro de sodio; acepte muestras digeridas según los apartados 4c, d y e; acepte HC1 4 a 6N, y sea agitado eficaz y exactamente por el gas de purga y/o un agitador magnético. d) Cuentagotas o jeringa capaz de suministrar de 0,5 a 3 ml de reactivo borohidruro de sodio. Se necesita una adición exacta y reproducible de manera que la producción de gas hidrógeno no varíe significativamente entre determinaciones. e) Ventilación: Véase sección 3111A.6f 3. Reactivos a) Reactivo de borohidruro de sodio: Disuélvanse 8 g de NaBH4 en 200 ml de 3-51 NaOH 0,1 A7. Prepárese reciente todos los días. b) Solución de agente prerreductor yoduro de sodio: Disuélvanse 50 g de Nal en 500 ml de agua. Prepárese reciente todos los días. Alternativamente empléese una solución de KI equivalente. c) Ácido sulfúrico, 18N . d) Ácido sulfúrico, 2,5N: Añádanse cuidadosamente 35 ml de H2SO4 conc. a aproximadamente 400 ml de agua, déjese enfriar y ajústese el volumen a 500 ml. e) Persulfato de potasio, solución al 5 por 100: Disuélvanse 25 g de K2S2O8 en agua y dilúyase hasta 500 ml. Consérvese en un vaso y refrigérese. Prepárese semanalmente. f) Ácido nítrico, HNO3 conc. g) Ácido per dórico, HC1O4 conc. h) Ácido clorhídrico, HC1 conc. i) Argón (o nitrógeno), comercializado. j) Hidrógeno, comercializado. k) Soluciones de arsénico (III). 1) Solución de As (III) de reserva: Disuélvanse 1,320 g de trióxido de arsénico, As2O3, en agua que contiene 4 g de NaOH. dilúyase hasta 1 l; 1,00 ml = = 1,00 mg As(III). 2) Solución de As(III) intermedia: Dilúyanse 10 ml de solución de As de reserva hasta 1.000 ml utilizando agua que contiene 5 ml de HC1 conc; 1,00 ml = = 10,0 μg As(III). 3) Solución de As (III) patrón: Diluyanse 10 ml de solución de As(III) intermedia hasta 1.000 ml utilizando agua que contiene la misma concentración de ácido utilizada para la conservación de la muestra (2 a 5 ml de HNO3 conc); 1,00 ml = 0,100 fig de As(III). Prepárense soluciones diluidas diariamente. l) Soluciones de arsénico(V): 1) Solución de As(V) de reserva: Disuélvanse 1,534 g de pentóxido de arsénico, As2O5, en agua destilada que contiene 4 g de NaOH. dilúyase hasta 1 l; 1,00 ml = 1,00 mg As(V). 3-52 2) Solución de As(V) intermedia: Prepárese como la anterior de As(III); 1,00 ml = 10,0 μg As(V). 3) Solución de As(V) patrón: Prepárese como la anterior de As(III); 1,00 ml = = 0,100 μg As(V). m) Soluciones de arsénico orgánico: 1) Solución de arsénico orgánico de reserva: Disuélvanse 1,842 g de ácido dimetilarsínico (ácido cacodílico), (CH3)2AsOOH, en agua que contiene 4 g de NaOH. dilúyase hasta 1 l; 1,00 ml = = 1,00 mg As. [NOTA: Compruébese la pureza del reactivo ácido cacodílico frente a un patrón de arsénico intermedio (50 a 100 mg As/1) empleando absorción atómica de llama.] 2) Solución de arsénico orgánico intermedia: Prepárese como la anterior de As(III); 1,00 ml = 10,0 μg As. 3) Solución de arsénico orgánico patrón: Prepárese como la anterior de As(III); 1,00 ml = 0,100 μg As. n) Soluciones de selenio(IV): 1) Solución de Se (IV) de reserva: Disuélvanse 2,190 g de selenito de sodio, Na2SeO3, en agua que contiene 10 ml de HC1 y dilúyase hasta 1 l; 1,00 ml = 1,00 mg Se(IV). 2) Solución de Se(IV) intermedia: Dilúyanse 10 ml de la reserva de Se(VI) hasta 1.000 ml empleando agua que contiene 10 ml de HC1 conc; 1,00 ml = 10,0 μg Se(IV). 3) Solución de Se (IV) patrón: Diluyanse 10 ml de solución intermedia de Se(IV) hasta 1.000 ml con agua que contiene la misma concentración de ácido utilizada para la conservación de la muestra (2 a 5 ml de HNO3 conc). Prepárese diariamente la solución cuando se comprueba la equivalencia de la respuesta del instrumento para Se(IV) y Se(VI); 1,00 ml = 0,100 μg Se(IV). 0) Soluciones de selenio(VI): 1) Solución de Se (VI) de reserva: Disuélvanse 2,393 g de selenato de sodio, MÉTODOS NORMALIZADOS Na2SeO4, en agua que contiene 10 ml de HNO3 conc. dilúyase hasta 1 l; 1,00 ml = = 1,00 mg de Se(VI). 2) Solución de Se (VI) intermedia: Prepárese igual que para la de Se(IV) anterior; 1,00 ml = 10,0 μg Se(VI). 3) Solución de Se (VI) patrón: Prepárese como la de Se(IV) anterior; 1,00 ml = 0,100 μg Se(VI). 4. Procedimiento a) Ajuste del instrumento: Véase figura 3114:1 o síganse las instrucciones del fabricante. Conéctese la entrada de la célula de reacción con el gas de purga auxiliar controlado por un flujómetro. Si se necesita una célula de desecación entre la célula de reacción y el atomizador, utilícese únicamente CaCl2 anhidro, pero no CaSO4, ya que éste podría retener el SeH2. Antes de emplear el sistema de análisis/generación de hidruros, optimícense los parámetros de operación. Aspírense soluciones acuosas diluidas de As y Se directamente en la llama para facilitar el alineamiento del atomizador. Alinéense los atomizadores de cuarzo para obtener una absorbancia máxima. Aspírese un blanco hasta eliminar los efectos de la memoria. Establézcase un flujo de gas de purga, concentración y tasa de adición del reactivo borohidruro de sodio, volumen de solución y velocidad de agitación para una respuesta óptima del instrumento para la especie química que se analiza. Si se emplea un atomizador de cuarzo, optimícese la temperatura de la célula. Si el reactivo de borohidruro de sodio se añade demasiado deprisa, el rápido desprendimiento de hidrógeno desequilibrará el sistema. Si el volumen de solución que se purga es demasiado grande, decrecerá la señal de absorción. Se recomiendan longitudes de onda de 193,7 y 196,0 nm para As y Se, respectivamente. b) Patrones de calibración del instrumento: Pásense 0,00, 1,00, 2,00, 5,00, DETERMINACIÓN DE METALES 10,00, 15,00 y 20,00 ml de soluciones patrón de As(III) o Se(IV) a matraces volumétricos de 100 ml y llévese hasta ese volumen utilizando agua que contiene la misma concentración de ácido empleada para conservación de la muestra (normalmente de 2 a 5 ml de HNO3 conc./l). Así se obtienen blancos y soluciones patrón de 0, 1, 2, 5, 10, 15 y 20 /μg As o Se/1. Prepárense recientes todos los días. c) Preparación de muestras y patrones para arsénico y selenio recuperables en total: Síganse los procedimientos generales de la sección 3030F; alternativamente, añádanse 50 ml de muestra, patrón As(III) o patrón Se(IV), a un vaso de Berzelius de 200 ml. (Alternativamente, prepárense patrones añadiendo 100 μg/l de solución patrón de As o Se directamente en el vaso y diluyendo hasta 50 ml en este vaso.) Añádanse 7 ml de H2SO4 18N y 5 ml de HNO 3 conc. Añádase un trocito de plato poroso o bolas de vidrio si es necesario. Evapórese hasta humos de SO3. Manténganse siempre las condiciones de oxidación añadiendo pequeñas cantidades de HNO3 para evitar que se oscurezca la solución. Manténgase un exceso de HNO3 hasta que se haya destruido toda la materia orgánica. Una solución ligeramente coloreada es normalmente la indicación de que la digestión es completa. Enfríese ligeramente, añádanse 25 ml de agua y 1 ml de HC1O4 conc. y evapórese de nuevo hasta humos de SO3 para expulsar los óxidos de nitrógeno. PRECAUCIÓN: Véase sección 3030H para precauciones en el empleo de HCIO4. Sígase la eficacia del procedimiento de digestión utilizado por adición de 5 ml de solución patrón de arsénico orgánico, o 5 ml de una solución de Se patrón, a una muestra de 50 ml y mídase la recuperación, llevando a cabo el procedimiento completo con los patrones. Para anotar el arsénico recuperable total como arsénico total, las recuperaciones medias de ácido cacodílico deben sobrepasar el 80 por 100. Alterna- 3-53 tivamente, utilícense matraces microkjeldahl de 100 ml para la digestión del arsénico o selenio recuperables en total, con lo que mejora la eficacia de la digestión. Después de terminar la evaporación de humos de SO3, dilúyase hasta 50 ml para las medidas de arsénico o hasta 30 ml para las medidas de selenio. d) Preparación de muestras y patrones para arsénico y selenio en total: Añádanse 50 ml de muestra o patrón a un vaso de Berzelius de 200 ml. Añádanse 1 ml de H2SO4 2,5N y 5 ml de K2S2O8 al 5 por 100. Llevar a ebullición suave sobre placa caliente precalentada durante aproximadamente 30 a 40 minutos o hasta que se alcanza un volumen final de 10 ml. No dejar que se seque la muestra. Alternativamente, caliéntese en autoclave a 121 °C durante 1 hora en recipientes con tapón. Después de la digestión manual, dilúyase hasta 50 ml para subsiguientes medidas de arsénico y hasta 30 ml para las medidas de selenio. Sígase el rendimiento de la digestión midiendo la recuperación de As o Se como antes. Si se obtiene una recuperación pobre del As añadido como ácido cacodílico, vuélvase a analizar utilizando una cantidad doble de K2S2O8. e) Determinación de arsénico con borohidruro de sodio: A 50 ml de patrón o muestra digeridos en un vaso de Berzelius de 200 ml (véase figura 3114:1) añádanse 5 ml de HC1 conc. y mézclese. Añádanse 5 ml de solución prerreductora de Nal, mézclese y espérese al menos 30 minutos. [N OTA: Se ha visto que el reactivo Nal no es necesario para determinados diseños de células de reacción de hidruros si no es importante una pérdida de sensibilidad del instrumento de 20 a 30 por 100 y si pueden controlarse estrictamente las variables de las condiciones acidas de la solución, temperaturas y volúmenes para la producción de As(V) y arsina. Dicho control requiere un sistema automático de suministro; véase sección 3114C] 3-54 Acóplese un vaso de Berzelius a la vez al tapón de goma que lleva el tubo de dispersión de gas para el gas de purga, a la entrada de reactivo borohidruro de sodio y a la salida al atomizador. Conéctese el registrador de cinta de gráficos y espérese hasta que se establece la línea de base por el gas de purga y ha sido expulsado todo el aire de la célula de reacción. Añádanse 0,5 ml de reactivo borohidruro de sodio. Después de que la absorbancia del instrumento ha llegado al máximo y ha vuelto a la línea de base, quítese el vaso, enjuáguese con agua el tubo de dispersión y trabájese con la próxima muestra o patrón. Compárense periódicamente las curvas estándar As(III) y As(V) en relación con la consistencia de la respuesta. Contrólese la presencia de interferencias químicas, que suprimen la respuesta del instrumento para arsina, tratando una muestra digerida con 10 μg/1 de As(III) o As(V) según sea apropiado. Las recuperaciones medias no deberán ser inferiores a 90 por 100. f) Determinación de selenio con borohidruro de sodio: A 30 ml de patrón o muestra digeridos, o a 30 ml de patrón o muestra sin digerir, en un vaso de Berzelius de 200 ml, añádanse 15 ml de HC1 conc. y mézclese. Caliéntese entre 90 y 100 °C durante un período de tiempo predeterminado. Alternativamente, caliéntese en autoclave a 121 °C en recipientes tapados durante 60 minutos, o caliéntese durante un tiempo predeterminado en tubos de ensayo abiertos utilizando un baño maría calentado entre 90 y 100°C o un digestor bloque de aluminio. Compruébese la eficacia de la calefacción elegida por aparición de iguales respuestas del instrumento para las curvas de calibración preparadas con las soluciones patrón de Se(IV) o de Se(VI). Una exposición eficaz al calor para convertir Se(VI) a Se(IV), sin pérdida de Se(IV), varía entre 5 y 60 minutos cuando se emplean vasos o tubos de ensayo abiertos. No someter a digestión las solu- MÉTODOS NORMALIZADOS ciones patrón de Se(IV) y Se(VI) utilizadas para este control de equivalencia. Después de la prerreducción de Se(VI) a Se(IV), acóplense los vasos de Berzelius, uno por uno, al aparato de purga. Conéctese el registrador de cinta de gráficos para cada uno y espérese hasta que se establezca la línea de base. Añádanse 0,50 ml de reactivo de borohidruro de sodio. Después de que la absorbancia del instrumento ha alcanzado un máximo y vuelto a la línea de base, sáquese el vaso, enjuáguese con agua el tubo de dispersión y trabájese con la próxima muestra o patrón. Contrólese la presencia de interferencias químicas que suprimen la respuesta del instrumento de hidruro de selenio tratando una muestra digerida con 10 μg de Se(IV)/l. Las recuperaciones medias no deberán ser inferiores al 90 por 100. 5. Cálculos Trácese una curva estándar anotando las alturas de picos o áreas de picos de los patrones frente a la concentración de los patrones. Mídanse áreas o alturas de picos de las muestras y léanse las concentraciones en la curva. Si la muestra está diluida (o concentrada) antes de la digestión de la muestra, aplíquese el factor apropiado. Léanse las concentraciones directamente después de la calibración estándar, en instrumentos dotados de ese equipo. 6. Precisión y sesgo Se recogieron datos de un laboratorio y de un solo operador para As(III) y arsénico orgánico por métodos manuales y automáticos, y lo mismo para la determinación manual de selenio. A continuación se dan los valores (%) de recuperaciones de siete replicados: 3-55 DETERMINACIÓN DE METALES 7. Referencias 1. VIJAN, P. N. & D. LEUNG. 1980. Reduction of chemical interference and speciation studies in the hydride generation-atomic absorption method for selenium. Anal. Chim. Acta 120:141. 2. VOTH-BEACH, L. M. & D. E. SHRADER. 1985. Reduction of interferences in the determination of arsenic and selenium by hydride generation. Spectroscopy 1:60. 3. 4. 5. 6. 7. 8. JULSHAMN, K., O. RlNGDAL, K.-E. SLINNING & O. R. BRAEKKAN. 1982. Optimization of the determination of selenium in marine samples by atomic absorption spectrometry: Comparison of a flameless graphite furnace atomic absorption system with a hydride generation atomic absorption system. Spectrochim. Acta. 37B:473. NYGAARD, D. D. & J. H. LOWRY. 1982. Sample digestion procedures for simultaneous determination of arsenic, antimony, and selenium by inductively coupled argon plasma emission spectrometry with hydride generation. Anal. Chem. 54:803. WELZ, B. & M. MELCHER. 1984. Mechanisms of transition metal interferences in hydride generation atomic-absorption spectrometry. Part 1. Influence of cobalt, copper, iron and nickel on selenium determination, Analyst 109:569. KRIVAN, V., K. PETRICK, B. WELZ & M. MELCHBR. 1985. Radiotracer error-diagnostic investigation of selenium determination by hydride-generation atomic absorption spectrometry involving treatment with hydrogen peroxide and hydrochloric acid. Anal. Chem. 57:1703. CHU, R. C, G. P. BARRON & P. A. W. BAUMGARNER. 1972. Arsenic determination at submicrogram levels by arsine evolution and flameless atomic absorption spectrophotometric technique. Anal. Chem. 44:1476. SIEMER, D. D., P. KOTEEL & V. JARIWALA. 1976. Optimization of arsine generation in atomic absorption arsenic determinations. Anal. Chem. 48:836. 8. Bibliografía FERNÁNDEZ, F. J. & D. C. MANNING. 1971. The determination of arsenic at submicrogram levels by atomic absorption spectrophotometry. Atomic Absorption Newsletter 10:86. JARREL-ASH CORPORATION. 1971. High Sensitivity Arsenic Determination by Atomic Absorption. Jarrel-Ash Atomic Absorption Applications Laboratory Bull. No. As-3. MANNING, D. C. 1971. A high sensitivity arsenicselenium sampling system for atomic absorption spectroscopy. Atomic Absorption Newsletter 10:123. BRAMAN, R. S. & C. C. FOREBACK. 1973. Methylated forms of arsenic in the environment. Science 182:1247. CALDWELL, J. S., R. J. LISHKA & E. F. MCFARREN. 1973. Evaluation of a low-cost arsenic and selenium determination of microgramper-liter levels. J. Amer. Water Works Assoc, 65:731. AGGETT, J. & A. C. ASPELL. 1976. The determination of arsenic (III) and total arsenic by atomic-absorption spectroscopy. Analyst 101:341. FIORIO, J. A., J. W. JONES & S. G. CAPAR. 1976. Sequential determination of arsenic selenium, antimony, and tellurium in foods via rapid hydride evolution and atomic absorption spectrometry. Anal. Chem. 48:120. CUTTER, G. A. 1978. Species determination of selenium in natural waters. Anal. Chem. Acta. 98:59. GODDEN, R. G. & D. R. THOMERSON. 1980. Generation of covalent hydrides in atomic absorption spectroscopy. Analyst 105:1137. BROWN, R. M., JR., R. C. FRY, J. L. MOYERS, S. J. NORTHWAY, M. B. DENTON & G. S. WILSON. 1981. Interference by volatile nitrogen oxides and transition-metal catalysis in the preconcentration of arsenic and selenium as hydrides. Anal. Chem. 53:1560. SINEMUS, H. W., M. MELCHER & B. WELZ. 1981. Influence of valence state on the determination of antimony, bismuth, selenium, and tel- 3-56 MÉTODOS NORMALIZADOS lurium in lake water using the hydride AA technique. Atomic Spectrosc. 2:81. DEDINA, J. 1982. Interference of volatile hydride forming elements in selenium determination by atomic absorption spectrometry with hydride generation. Anal. Chem. 54:2097. 3114 C. Método continuo de generación de hidruros/ espectrometría de absorción atómica (PROPUESTA) 1. Discusión general El generador continuo de hidruros, recientemente introducido, ofrece las ventajas de sencillez de funcionamiento, reproducibilidad excelente, bajos límites de detección y alta capacidad de volumen de muestra, para el análisis de selenio que sigue a las preparaciones descritas en 3500-Se.B o 3114B.4c y d. a) Principio: Véase sección 3114B. b) Interferencias: El cloro libre en el ácido clorhídrico es una interferencia corriente, pero de difícil diagnóstico (la cantidad de cloro varia según el fabricante y según el lote para un mismo fabricante). El cloro oxida el hidruro y puede contaminar el generador de hidruros, impidiendo así las recuperaciones en cualesquiera de las condiciones. Cuando se detecta la interferencia, o mejor, antes de emplear cada nueva botella de HC1, elimínese el cloro de una botella de 2,3 1 de HC1 conc. haciendo burbujear helio (calidad comercial, 100 ml/minuto) durante 3 horas. Un exceso de oxidante (peróxido, persulfato o permanganato) proveniente de la digestión de selenio total puede oxidar el hidruro. Síganse los procedimientos de 3500-Se.B.2, 3 o 4, para asegurar la eliminación de todos los agentes oxidantes antes de la generación de hidruros. El nitrito es un componente traza corriente en aguas naturales y residuales, y puede reducir la recuperación de seleniuro de hidrógeno del Se(IV) por encima de un 50 por 100 a niveles tan bajos de nitrito como 10 μg/l. Además, durante la reducción de Se(VI) a Se(IV) por digestión con HC1 (3500-Se.B.5), parte del nitrato pasa a nitrito, el cual interfiere sub- siguientemente. Cuando se sospecha esta interferencia, añádase sulfanilamida después de la acidulación de la muestra (o digestión con HC1). La reacción de diazotación entre nitrito y sulfanilamida elimina por completo el efecto de la interferencia (es decir, la pendiente de la adición de patrón es normal). 2. Instrumental a) Generador continuo de hidruros: La unidad básica está compuesta de dos partes: una bomba peristáltica de precisión, utilizada para medir y mezclar reactivos y soluciones de muestras, y el separador gas-líquido. En el separador gaslíquido, un flujo constante de argón arrastra el hidrógeno y los gases de hidruros metálicos formados en la reacción y los lleva a la célula de absorción de cuarzo calentada (3114B.1b y 2b) soportada por una abrazadera metálica montada en la parte de encima de la cabeza del quemador de aire acetileno regular. El líquido gastado sale del separador y fluye a través de un drenaje lateral de nivel constante, a un cubo de residuos. En la figura 3114:2 se muestra el esquema y los parámetros de operación. Contrólense con frecuencia las tasas de flujo para asegurar un flujo estacionario. Un flujo desigual en cualquiera de los tubos originaría una señal errática. Extráiganse los tubos de los rodillos de la bomba cuando ésta no se utilice. Tasas típicas de flujo son: muestra, 7 ml/minuto; ácido, 1 ml/minuto; reactivo borohidruro, 1 ml/minuto. El flujo de argón suele arrojar un registro de 90 ml/minuto. b) Equipo espectrométrico de absorción atómica: Véase sección 3111 A.6. 3-57 DETERMINACIÓN DE METALES Figura 3114:2. Esquema de un generador continuo de hidruros. 3. Reactivos a) Ácido clorhídrico, HC1 5 + 1: Manipúlese el HC1 conc. bajo una campana de humos. Si es necesario, elimínese el Cl2 libre por arrastre con helio desde el HC1 conc, tal como se ha descrito con anterioridad. b) Reactivo de borohidruro: Disuélvanse 0,6 g de NaBH4 y 0,5 g de NaOH en 100 ml de agua. PRECAUCIÓN: El borohidruro de sodio es tóxico, inflamable y corrosivo. c) Solución patrón de referencia de selenio, 1.000 mg/1: Utilícese un patrón comercial; compruébese que el selenio es Se(IV). d) Solución patrón intermedia, 1 mg/1: Dilúyase 1 ml de solución patrón de referencia hasta 1 l en un matraz volumétrico empleando agua destilada. e) Soluciones patrón de trabajo, 5, 10, 20, 30 y 40 μg/1: Dilúyanse 0,5, 1,0, 2,0, 3,0 y 4,0 ml de solución patrón intermedia hasta 100 ml en un matraz volumétrico. f) Solución de sulfanilamida: Prepárese diariamente una solución al 2,5 por 100 (p/v); añádanse varias gotas de HC1 conc. por cada 50 ml de solución para facilitar la disolución. 4. Procedimiento a) Preparación de la muestra: Véanse secciones 3500-Se o 3114B.4c y d para las etapas de preparación de diversas fracciones de Se o Se total. b) Preacondicionamiento del generador de hidruros: Para tubos recién colocados, conéctese la bomba por lo menos 10 a 15 minutos antes de la calibración del instrumento. Tómese una muestra del patrón más alto durante unos pocos minutos para dejar reaccionar el hidruro volátil con los lugares reactivos de las líneas de transferencia y sobre las superficies de la célula de absorción de cuarzo. c) Calibración del instrumento: Dependiendo del volumen vacío total del tubo de muestra, el tiempo de toma de muestras de 15 a 20 segundos es suficiente, por lo general, para obtener una señal estacionaria. Entre una muestra y otra, sumérjase el tubo de captación en agua de enjuague. Calíbrese el instrumento diariamente después de un tiempo de calentado de la lámpara de 45 minutos. Utilícese una lámpara de cátodo hueco o una lámpara de descarga sin electrodos. d) Agentes antiespumantes: Determinadas muestras, sobre todo de aguas residuales que contienen una elevada con- MÉTODOS NORMALIZADOS 3-58 centración de sustancias proteínicas, pueden generar excesiva espuma, que podría transportar líquido directamente a la célula de absorción de cuarzo calentada y dar lugar a salpicaduras de depósitos salinos sobre las ventanas del espectrómetro. Añádase una gota de agente antiespuma* para eliminar este problema. e) Eliminación de nitrito: Después de aciduladas las muestras, o después de su digestión con ácido, añádase 0,1 ml de solución de sulfanilamida por 10 ml de muestra y déjese reaccionar durante 2 minutos. f) Análisis: Para el funcionamiento del equipo analítico, síganse las instrucciones del fabricante. * Dow Corning o equivalente. 7. Bibliografía REAMER, D. C. & C. VEILOON. 1981. Preparation of biological materials for determination of selenium by hydride generation-ASS. Anal. Chem. 53:1192. SINEMUS, H. W., M. MELCHER & B. WELZ. 1981. Influence of valence state on the determination of antimony, bismuth, selenium, and tellurium in lake water using the hydride AA technique. Atomic Spectrosc. 2:81. RODEN, D. R. & D. E. TALLMAN. 1982. Determination of inorganic selenium species in groundwaters containing organic interferences by ion chromatography and hydride generation/atomic absorption spectrometry. Anal. Chem. 54:307. CUTTER , G. 1983. Elimination of nitrite interference in the determination of selenium by hydride generation. Anal. Chim. Acta 149:391. NARASAKI, H. & M. IKEDA. 1984. Automated determination of arsenic and selenium by atomic 5. Cálculos Constrúyase una curva de calibración con la absorbancia en función de la concentración del patrón. Aplíquense factores de dilución a las muestras diluidas. 6. Precisión y sesgo Se analizaron patrones de trabajo junto con las partidas de muestras de agua siguiendo una marcha de rutina. Los patrones se obtuvieron utilizando selenito de sodio y selenato de sodio químicamente puros. Los valores de Se(IV) + + Se(VI) se determinaron convirtiendo Se(VI) en Se(IV) por digestión con HC1. Los resultados se dan en la siguiente tabla: absorption spectrometry with hydride generation. Anal. Chem. 56:2059. WELZ, B. & M. MELCHER. 1985. Decomposition of marine biological tissues for determination of arsenic, selenium, and mercury using hydride-generation and cold-vapor atomic absorption spectrometries. Anal. Chem. 57:427. EBDON, L. & S. T. SPARKES. 1987. Determination of arsenic and selenium in environmental samples by hydride generation-direct current plasma-atomic emission spectrometry. microchem. J. 36:198. EBDON, L. & J. R. WILKINSON. 1987. The determination of arsenic and selenium in coal by continuous flow hydride-generation atomic absorption spectroscopy and atomic fluorescence spectrometry. Anal. Chim. Acta. 194:177. VOTH-BEACH, L. M. & D. E. SHRADER. 1985. Reduction of interferences in the determination of arsenic and selenium by hydride generation. Spectroscopy 1:60. 3-59 DETERMINACIÓN DE METALES 3120 DETERMINACIÓN DE METALES POR ESPECTROSCOPIA DE EMISIÓN DE PLASMA* 3120 A. 1. Discusión general La espectroscopia de emisión, que utiliza plasma de acoplamiento inductivo (PAI), se desarrolló a mediados de los años sesenta1, 2 como un método rápido, sensible y conveniente para la determinación de metales en muestras de aguas limpias y residuales3,6. Los metales disueltos se determinan en muestras filtradas y aciduladas. Los metales totales se determinan tras una apropiada digestión. Debe tenerse cuidado en eliminar las interferencias potenciales, especialmente cuando los sólidos disueltos sobrepasan los 1.500 mg/1. 2. Referencias 1. GREENFIELD, S., I. L. JONES & C. T. BERRY. * Aprobado por el Standard Methods Committee, 1989. 3120 B. 1. Introducción 1964. High-pressure plasma-spectroscopic emission sources. Analyst 89:713. WENDT, R. H. & V. A. FASSEL. 1965. Induction-coupled plasma spectrometric excitation source. Anal. Chem. 37:920. U.S. ENVIRONMENTAL P ROTECTION AGENCY. 1983. Method 200.7. Inductively coupled plasma-atomic emission spectrometric method for trace element analysis of water and wastes. Methods for Chemical Analysis of Water and Wastes. EPA-600/4-79-020, revisado en marzo de 1983. AMERICAN SOCIETY FOR TESTING AND MATERIALS. 1987. Annual Book of ASTM Standards, Vol. 11.01. American Soc. Testing & Materials, Filadelfía, Pennsylvania. F ISHMAN , M. J. & W. L. B RADFORD , eds. 1982. A Supplement to Methods for the Determination of Inorganic Substances in Water and Fluvial Sediments. Rep. No. 82-272, U.S. Geological Survey, Washington, D.C. GARBARINO , J. R. & H. E. TAYLOR. 1985. Trace Analysis. Recent Developments and Applications of Inductively Coupled Plasma Emission Spectroscopy to Trace Elemental Analysis of Water. Vol. 4. Academic Press, Nueva York. Método de plasma de acoplamiento inductivo (PAI) Discusión general a) Principio: Una fuente de PAI consiste en una corriente de flujo de gas argón ionizado por aplicación de un campo de radiofrecuencias típicamente osci- lantes a 27,1 MHz. Este campo está acoplado inductivamente al gas ionizado por una bobina refrigerada con agua que rodea a una «lámpara» de cuarzo que soporta y confina el plasma. En un apropiado nebulizador y cámara de pulveri- 3-60 zación se genera un aerosol de la muestra que se lleva al plasma a través de un tubo inyector colocado dentro de la lámpara. El aerosol de la muestra se inyecta directamente en el PAI, que somete a los átomos que lo componen a temperaturas de aproximadamente 6.000 a 8.000 °K1. Debido a ello, tiene lugar una disociación de moléculas casi completa, consiguiéndose una reducción significativa de interferencias químicas. La elevada temperatura del plasma excita eficazmente la emisión atómica. La ionización de un elevado porcentaje de átomos produce un espectro de emisión iónica. El PAI proporciona una fuente ópticamente «delgada», que no está sujeta a autoabsorción, excepto a concentraciones muy altas. De esta forma, se observan, para muchos elementos2, recorridos dinámicos lineales de orden de magnitud cuatro a seis. La eficaz excitación proporcionada por PAI da lugar a bajos límites de detección para muchos elementos. Esto, unido al extenso recorrido dinámico, permite una determinación multielemental eficaz de metales3. La luz emitida por el PAI se enfoca sobre la rejilla de entrada de un monocromador o policromador que efectúa la dispersión. Se utiliza una rejilla de salida alineada con precisión para aislar una parte del espectro de emisión, para medida de la intensidad empleando un tubo fotomultiplicador. El monocromador utiliza una única rejilla de salida/fotomultiplicador y puede emplear un mecanismo de exploración controlado por ordenador para examinar secuencialmente las longitudes de onda de emisión. El policromador utiliza rejillas de salida fijas múltiples y los correspondientes tubos fotomultiplicadores; supervisa todas las longitudes de onda configuradas utilizando un sistema de lectura de salida controlado por ordenador. El procedimiento secuencial proporciona mayor selección de longitudes de onda, mientras que el procedimiento simultá- MÉTODOS NORMALIZADOS neo puede proporcionar mayor capacidad de muestras. b) Metales a los que se puede aplicar v límites analíticos: La tabla 3120:I recoge una lista de elementos a los que se aplica este método, así como longitudes de onda analíticas recomendadas, y límites de detección del instrumento típicamente estimados utilizando nebulización neumática convencional. Los límites de detección de trabajo reales dependen de la muestra. En la tabla 3120:I también se incluyen los límites superiores para calibración lineal. c) Interferencias: Las interferencias pueden clasificarse como sigue: 1) Interferencias espectrales: Emisiones de luz desde fuentes espectrales distintas a la del elemento que interesa pueden contribuir a una intensidad de una señal neta aparente. Las fuentes de interferencia espectral incluyen solapamientos directos de líneas del espectro, alas prolongadas de líneas intensas del espectro, emisión continua de la recombinación ion-átomo, emisión de bandas moleculares y luz dispersa (difusa) de la emisión de elementos a concentraciones elevadas4. Evítense los solapamientos, seleccionando longitudes de onda analíticas alternas. Evítense asimismo o redúzcanse al mínimo otras interferencias espectrales, mediante adecuada selección de posiciones de corrección del fondo. Una exploración de longitudes de ondas de la región de líneas del elemento es útil para detectar las interferencias espectrales potenciales y para seleccionar posiciones para la corrección del fondo. Háganse correcciones de interferencia espectral residual utilizando factores de corrección determinados empíricamente con el correspondiente programa de ordenador suministrado por el fabricante del espectrómetro o con los cálculos que se detallan a continuación. El método de corrección empírica no se puede utilizar con sistemas de espectrómetros de exploración si las líneas analíticas y las que DETERMINACIÓN DE METALES 3-61 TABLA 3120:I. LONGITUDES DE ONDA SUGERIDAS, LÍMITES DE DETECCIÓN ESTIMADOS, LONGITUDES DE ONDA ALTERNATIVAS, CONCENTRACIONES DE CALIBRACIÓN Y LÍMITES SUPERIORES interfieren no pueden localizarse con precisión y reproducibilidad. Además de esto, si se emplea un policromador, hay que comprobar la ausencia de interferencia espectral de un elemento que podría aparecer en una muestra, pero para el que no hay canal en el dispositivo del detector. Esto se lleva a cabo analizando soluciones de un solo elemento de una concentración de 100 mg/1 y observando para cada canal de elemento la concentración aparente de la sustancia que interfiere, que es superior al límite de detección del instrumento para el elemento. 2) Interferencias no espectrales: a) Las interferencias físicas son efectos asociados a la nebulización de la muestra y a procesos de transporte. Los cambios en las propiedades físicas de las muestras, tales como la viscosidad y la tensión superficial, pueden dar lugar a errores significativos. Esto suele ocurrir cuando muestras que contienen más de 3-62 un 10 por 100 (en volumen) de ácido o más de 1.500 mg/1 de sólidos disueltos se analizan utilizando patrones de calibración que contienen d 5 por 100 de ácido. Siempre que se trata de una matriz de muestra nueva o poco corriente, utilícese el ensayo descrito en el apartado 4g. Si existe interferencia física, compénsese por dilución de la muestra, utilizando patrones de calibración adaptados a la matriz o aplicando el método de adición de patrones (véase el apartado 5d). Un contenido alto en sólidos disueltos puede contribuir también a la desviación del instrumento por formación de sal en el extremo del orificio del gas del nebulizador. Utilizando argón prehumidificado para nebulización de la muestra, se reduce este problema. Un mejor control de la tasa de flujo de argón al nebulizador empleando un controlador de flujo de masa mejora el funcionamiento del instrumento. b) Las interferencias químicas tienen su origen en la formación de un compuesto molecular, efectos de ionización y efectos termoquímicos asociados a la vaporización de la muestra y su atomización en el plasma. Normalmente, estos efectos no son pronunciados y pueden reducirse a un mínimo por una selección cuidadosa de las condiciones de operación (energía incidente, posición de observación del plasma, etc.). Las interferencias químicas dependen en buena medida de la matriz de la muestra y del elemento que interesa. Como en el caso de las interferencias físicas, hay que compensarlas empleando patrones adaptados a la matriz o añadiendo patrones (apartado 5d). Para determinar la existencia de interferencia química, síganse las intrucciones del apartado 4g. 2. Instrumental a) Fuente de PAI: La fuente de PAI consiste en un generador de radiofre- MÉTODOS NORMALIZADOS cuencia (RF) capaz de generar al menos 1,1 KW de energía, lámpara, una bobina de una tesla, una bobina de carga, red de ajuste de impedancia, nebulizador, cámara de pulverización y drenaje. Tanto el argón del nebulizador como el flujo del gas soporte del plasma requieren reguladores de flujo de alta calidad. Para regular el flujo de muestra al nebulizador, se recomienda una bomba peristáltica. El tipo de nebulizador y la cámara de vaporización utilizada pueden depender de las muestras que se van a analizar, así como del fabricante del equipo. En general, se emplean nebulizadores neumáticos del tipo de flujo transversal o concéntrico. En el caso de muestras viscosas, muestras que contengan partículas o con alto contenido en sólidos (> 5.000 mg/1), pueden ser necesarios nebulizadores del tipo Babington. b) Espectrómetro: El espectrómetro puede ser del tipo simultáneo (policromador) o secuencial (monocromador), con óptica de trayecto de aire, purgado con gas inerte o de vacío. Se necesita un paso de banda del espectro de 0,05 nm o menor. El instrumento debe permitir el examen del fondo espectral que rodea las líneas de emisión utilizadas para la determinación de los metales. Si es necesario, se debe poder medir y corregir el fondo espectral a una o más posiciones a uno y otro lado de las líneas analíticas. 3. Reactivos y patrones Utilícense reactivos de calidad de la más alta pureza o equivalente. Son aceptables los ácidos bidestilados. Excepto cuando se advierta, séquense todas las sales a 105 °C durante 1 hora y manténganse en un desecador antes de pesar. Empléese agua desionizada preparada por paso del agua por al menos dos etapas de desionización con resinas de intercambio de lecho mixto catión/anión. Utilícese agua desionizada para preparar to- DETERMINACIÓN DE METALES dos los patrones de calibración, reactivos, y para dilución. a) Ácido clorhídico, HC1 conc. y 1 + 1. b) Ácido nítrico, HNO3 conc. c) Ácido nítrico, HNO3 1 + 1: Añádanse 500 ml de HNO3 conc. a 400 ml de agua y dilúyase hasta un litro. d) Soluciones patrón de reserva: Véanse 3111B, 3111D y 3114B. PRECAUCIÓN: Muchas sales metálicas son extremadamente tóxicas y pueden resultar fatales si se tragan. Lávense cuidadosamente las manos después de manipularlas. 1) Aluminio: Véase 3111D.3kl). 2) Antimonio: Véase 3111B.3j1). 3) Arsénico: Véase 3114B.3k1). 4) Bario: Véase 3111D.3k2). 5) Berilio: Véase 3111D.3k3). 6) Boro: No secar, pero manténgase el frasco bien cerrado y en un desecador. Disuélvanse 0,5716 g de H3BO3 anhidro en agua y dilúyase hasta 1.000 ml; 1 ml = 100 μg B. 7) Cadmio: Véase 3111 B.3j3). 8) Calcio: Véase 3111B.3j4). 9) Cromo: Véase 3111 B.3j6). 10) Cobalto: Véase 3111 B.3j7). 11) Cobre: Véase 3111B.3j8). 12) Hierro: Véase 3111 B.3 j 11). 13) Plomo: Véase 3111B.3 j 12). 14) Litio: Véase 3111 B.3j13). 15) Magnesio: Véase 3111B.3 j 14). 16) Manganeso: Véase 3111B.3 j 15). 17) Molibdeno: Véase 3111D.3k4). 18) Níquel: Véase 3111B.3 j l6). 19) Potasio: Véase 3111 B.3 j 19). 20) Selenio: Véase 3114B.3n1). 21) Sílice: Véase 3111 D.3k7). 22) Plata: Véase 3111 B.3 j 22). 23) Sodio: Véase 3111 B.3 j 23). 24) Estroncio: Véase 3111B.3. j 24). 25) Talio: Véase 3111B.3 j 25). 26) Vanadio: Véase 3111D.3k10). 27) Zinc: Véase 3111B.3j27). e) Patrones de calibración: Prepáren- se patrones mixtos de calibración con las 3-63 concentraciones mostradas en la tabla 3120:I combinando volúmenes apropiados de las soluciones de reserva en matraces volumétricos de 100 ml. Añádanse 2 ml de HNO 3 1 + 1 y 10 ml de HC1 1 + 1 y dilúyase con agua hasta 100 ml. Antes de preparar los patrones mixtos analícese cada solución de reserva por separado para determinar la posible interferencia espectral o la presencia de impurezas. Cuando se preparan patrones mixtos hay que tener en cuenta que los elementos sean compatibles y estables. Consérvense las soluciones patrón mixtas en frascos de fluocarbono FEP o polietileno sin usar. Compruébense inicialmente los patrones de calibración utilizando un patrón de control de calidad; revísese semanalmente la estabilidad. Las siguientes son combinaciones recomendadas que utilizan las líneas analíticas sugeridas en la tabla 3120:I. También son aceptables combinaciones alternativas. 1) Solución patrón mixta I: Manganeso, berilio, cadmio, plomo, selenio y zinc. 2) Solución patrón mixta II: Bario, cobre, hierro, vanadio y cobalto. 3) Solución patrón mixta III: Molibdeno, sílice, arsénico, estroncio y litio. 4) Solución patrón mixta IV: Calcio, sodio, potasio, aluminio, cromo y níquel. 5) Solución patrón mixta V: Antimonio, boro, magnesio, plata y talio. Si la adición de plata da lugar a una precipitación inicial, añádanse 15 ml de agua y caliéntese el matraz hasta que la solución se vuelva transparente. Enfríese y diluyase con agua hasta 100 ml. Para esta combinación de ácidos limítese la concentración de plata a 2 mg/1. En estas condiciones, la plata es estable en una matriz de agua del grifo durante 30 días. Concentraciones más altas de plata requieren HC1 adicional. f) Blanco de calibración: Dilúyanse 2 ml de HNO3 1 + 1 y 10 ml de HC1 1 + 1 hasta 100 ml empleando agua. Pre- MÉTODOS NORMALIZADOS 3-64 párese en cantidad suficiente para inundar el sistema entre patrones y muestras. g) Blanco del método: Realícese el procedimiento completo de preparación de muestras con un blanco de reactivos. Prepárese el blanco del método de forma que contenga los mismos tipos y concentraciones de ácidos que las soluciones de las muestras. h) Patrón para el control del instrumento: Prepárense patrones de control del instrumento combinando elementos compatibles a una concentración de 2 mg/1. i) Muestra de control de calidad del instrumento: Obténgase un patrón acuoso de referencia certificado a partir de una fuente exterior y prepárese según las instrucciones del proveedor. Utilícese la misma matriz de ácidos que en los patrones de calibración. j) Muestra de control de calidad del método: Realícese el procedimiento completo de preparación de muestras con la muestra de control de calidad del instrumento (apartado 3i). k) Argón: Utilícese calidad técnica o de soldador. Si da lugar a problemas, utilícese el prepurificado. 4. Procedimiento a) Preparación de la muestra: Véase sección 3030F. b) Condiciones del funcionamiento: Debido a las diferencias de formas y modelos de los instrumentos, no es posible dar instrucciones del funcionamiento detalladas. Síganse las instrucciones del fabricante. Establézcanse el límite de detección del instrumento, la precisión, las posiciones óptimas de corrección del fondo, los trayectos dinámicos lineales y las interferencias para cada línea analítica. Compruébese que la configuración del instrumento y las condiciones de operación satisfacen las necesidades analíticas y que pueden reproducirse día por día. Se puede emplear una relación de intensidad de emisión de átomo a ion [Cu(I) 324,75 nm/Mn(II) 257,61 nm] con objeto de reproducir las condiciones óptimas para un análisis de múltiples elementos con precisión. La relación de intensidad Cu/Mn puede ser incorporada al procedimiento de calibración, incluyendo especificaciones de sensibilidad y precisión7. Manténganse diaria o semanalmente registros de las intensidades de Cu y Mn y/o las intensidades críticas de líneas de elementos. Regístrense asimismo regulaciones del alineamiento óptico del policromador, tasa de captación de la muestra, lecturas de energía (incidente, reflejada), atenuación del tubo multiplicador, regulaciones del controlador de flujo de masa y mantenimiento del sistema. c) Calibración del instrumento: Prepárese el instrumento como se ha indicado (apartado b). Déjese calentar durante 30 minutos. Alíniense ópticamente los policromadores utilizando la lámpara de perfil o solución. Compruébense el alineamiento de la lámpara de plasma y la rendija de entrada del espectrómetro, sobre todo si se ha mantenido el sistema de introducción de muestra. Hágase un ajuste Cu/Mn o relación de intensidad similar. Calíbrese el instrumento siguiendo el procedimiento recomendado por el fabricante empleando patrones y blancos de calibración. Aspírese cada patrón o blanco durante un mínimo de 15 segundos después de alcanzar el plasma, antes de que comience la integración de la señal. Enjuáguese con blanco de calibración o solución similar durante 60 segundos por lo menos entre cada patrón, para eliminar un posible arrastre del anterior patrón. Utilícese una intensidad media de integraciones múltiples de patrones o muestras con objeto de reducir el error aleatorio. Antes de analizar las muestras, analícese el patrón de comprobación del instrumento. Los valores de la concentra- DETERMINACIÓN DE METALES ción obtenidos no deberán desviarse más de ± 5 por 100 del valor real (o los límites de control establecidos, lo que sea más bajo). d) Análisis de muestras: Comiéncese cada marcha con la muestra haciendo un análisis del blanco de calibración y después analícese el blanco del método. Esto permite una comprobación de los reactivos de preparación de la muestra y procedimientos en cuanto a la contaminación. Analícense las muestras alternando con análisis del blanco de calibración. Enjuáguese con ácido diluido durante 60 segundos por lo menos entre muestras y blancos. Después de introducir cada muestra o blanco, déjese que el sistema se equilibre antes de comenzar la integración de la señal. Examínese cada análisis del blanco de calibración para comprobar que no hay ningún efecto de memoria de arrastre. Si se observa arrastre, repítase el enjuague hasta obtener valores del blanco apropiados. Efectúense adecuadas diluciones y acidulaciones de la muestra para determinar las concentraciones más allá del intervalo lineal de calibración. e) Control de calidad instrumental: Analícese el patrón de control del instrumento una vez cada 10 muestras con objeto de determinar si ha habido una desviación significativa del instrumento. Si la concordancia no queda dentro del ± 5 por 100 de los valores esperados (o dentro de los límites de control establecidos, lo que sea más bajo), póngase fin al análisis de las muestras, corríjase el problema y vuélvase a calibrar el instrumento. Si se utiliza la referencia a la relación de intensidades, la regulación de esta relación puede restablecer la calibración sin necesidad de volver a analizar los patrones de calibración. Analícese el patrón de control del instrumento para confirmar una recalibración apropiada. Vuélvanse a analizar una o más muestras analizadas inmediatamente antes de haber puesto fin a la marcha analítica. 3-65 Los resultados deben estar de acuerdo dentro de un ± 5 por 100; de lo contrario deberían analizarse de nuevo todas las muestras después del último análisis aceptable del patrón de control del instrumento. Analícese una muestra de control de calidad del instrumento con cada marcha. Utilícese este análisis para comprobar la precisión y estabilidad de los patrones de calibración. Si el resultado no está dentro del ± 5 por 100 del valor certificado, prepárese un nuevo patrón de calibración y vuélvase a calibrar el instrumento. Si esto no corrige el problema, prepárese una nueva solución de reserva y un nuevo patrón de calibración y repítase la calibración. f) Control de calidad del método: Analícese la muestra de control de calidad del método dentro de cada marcha. Los resultados deberán quedar comprendidos en un intervalo de ± 5 por 100 de los valores certificados. Desviaciones superiores pueden reflejar pérdidas o contaminación durante la preparación de las muestras. g) Ensayo de interferencia de la matriz: Cuando se analiza una matriz de muestra nueva o inusual, compruébese que no existen efectos positivos o negativos de interferencia no lineal. Si el elemento se presenta en una concentración por encima de 1 mg/1, utilícense diluciones en serie con el blanco de calibración. Los resultados de los análisis de una dilución deberán estar dentro del margen del ± 5 por 100 respecto del resultado original. Alternativamente, o si la concentración está por debajo de 1 mg/1 o resulta indetectable, utilícese una adición posterior a la digestión igual a 1 mg/1. La recuperación de la adición deberá oscilar entre 95 por 100 y 105 por 100 o dentro de los límites de control establecidos de una desviación estándar de ± 2 de la media. Si el efecto de una matriz hace que los resultados del ensayo caigan fuera de los límites críticos, complétese el 3-66 MÉTODOS NORMALIZADOS análisis después de diluir la muestra para eliminar el efecto de la matriz mientras se mantiene una concentración detectable de al menos dos veces el límite de detección o aplicar el método de adiciones de patrón. 5. Cálculos y correcciones a) Corrección del blanco: Sustráigase el resultado de un blanco de calibración adyacente del resultado de cada muestra para la corrección de la desviación de la línea base. (Las concentraciones impresas deberán incluir valores negativos y positivos para compensar la desviación positiva y negativa de la línea de base. Asegúrese de que el blanco de calibración utilizado para la corrección del blanco no ha sido contaminado por arrastres.) Utilícese el resultado del análisis del blanco del método para corregir la contaminación del reactivo. Alternativamente, intercálense blancos del método con las apropiadas muestras. El blanco de reactivo y la corrección de la desviación de la línea base se obtienen por sustracción. b) Corrección por dilución: Si lisis de la muestra. A menos que las condiciones del análisis puedan reproducirse con precisión de un día para otro, o por períodos más largos, vuélvanse a determinar los factores de corrección de interferencia que afecten significativamente los resultados cada vez que se analizan las muestras7, 8. Calcúlense los factores de corrección de interferencia (Kij) a partir de las concentraciones aparentes observadas en el análisis de soluciones de reserva de alta pureza. siendo la concentración aparente del elemento i la diferencia entre la concentración observada en la solución de reserva y la concentración observada en el blanco. Corríjanse las concentraciones de la muestra observadas para el elemento i (ya corregidas para desviación de la línea base) para las interferencias espectrales de los elementos j, k y l; por ejemplo: Concentración del elemento i corregida para interferencia espectral la muestra estaba diluida o concentrada en la preparación, multiplíquense los resultados por un factor de dilución (FD) calculado como sigue: c) Corrección de la interferencia es- pectral: Corríjase la interferencia espectral utilizando el programa de ordenador suministrado por el fabricante del instrumento, o empleando el método del manual basado en factores de corrección de interferencia. Determínense los factores de corrección de interferencia analizando soluciones de reserva de un solo elemento de concentraciones apropiadas adaptando las condiciones lo más cercanamente posible a las empleadas en el aná- Los factores de interferencia pueden ser negativos si se utiliza la corrección de fondo para el elemento i. Puede resultar una Kij negativa en el caso de que una línea de interferencia se encuentre en la longitud de onda de la corrección de fondo antes que en la longitud de onda del pico. Determínense las concentraciones de los elementos que interfieren j, k y l dentro de sus respectivos intervalos lineales. Para el cálculo de las interferencias mutuas (i interfiere con j y j interfiere 3-67 DETERMINACIÓN DE METALES con i) se necesitan métodos repetitivos o de matriz. d) Corrección de interferencias no espectrales: Si se necesita una corrección de interferencia no espectral, utilícese el método de adiciones de patrón. El método se puede aplicar cuando la forma física y química del elemento en la adición de patrón es la misma que la de la muestra, o el PAI convierte al metal de la adición y de la muestra a la misma forma; el efecto de interferencia es independiente de la concentración del metal en el intervalo de concentraciones de las adiciones del patrón, y la curva de calibración analítica es lineal a lo largo del intervalo de concentraciones de las adiciones del patrón. Utilícese una adición no inferior al 50 por 100 ni superior al 100 por 100 de la concentración del elemento en la muestra para no reducir la precisión de la medida y para que las interferencias que dependen de las relaciones elemento/interferente no conduzcan a resultados erróneos. Aplíquese el método a todos los elementos de todo el conjunto de muestras empleando la corrección de fondo a posiciones fuera de línea cuidadosamente seleccionadas. Se puede emplear una adición de patrón de múltiples elementos si se ha comprobado que los elementos añadidos no son interferentes. e) Registro de datos: Regístrense datos analíticos en unidades de concentración de miligramos por litro utilizando hasta tres cifras significativas. Regístrense los resultados por debajo del límite de detección determinado cuando no se han detectado menos que el límite de detección establecido corregido por dilución de la muestra. 6. Precisión y sesgo Como guía de la precisión y sesgo esperados en general, véanse las ecuaciones lineales de regresión de la tabla 3120:119. También existe información adicional interlaboratorios10. 7. Referencias 1. F AIRES , L. M., B. A. P ALMER , R. E NGLE MAN, JR. & T. M. NIEMCZYK. 1984. Temperature determinations in the inductively coupled plasma using a Fourier transform spectrometer. Spectrochim. Acta 39B:819. 2. B ARNES , R. M. 1978. Recent advances in emission spectroscopy: inductively coupled plasma discharges for spectrochemical analysis. CRC Crit. Rev. Anal. Chem. 7:203. 3. P ARSONS , M. L., S. MAJOR & A. R. F ORS TER. 1983. Trace element determination by atomic spectroscopic methods - State of the art. Appl. Spectrosc. 37:411. 4. LARSON, G. F., V. A. FASSEL, R. K. WINGE & R. N. KNISELEY. 1976. Ultratrace analysis by optical emission spectroscopy: The stray light problem. Appl. Spectrosc. 30:384. 5. GARBARINO, J. R. & H. E. TAYLOR. 1979. A Babington-type nebulizer for use in the analysis of natural water samples by inductively coupled plasma spectrometry. Appl. Spectrosc. 34:584. 6. AMERICAN SOCIETY FOR TESTING AND MATERIALS. 1988. Standard specification for reagent water, DI 193-77 (reapproved 1983). Annual Book of ASTM Standards. American Soc. for Testing & Materials, Filadelfia, Pennsylvania. 7. BOTTO, R. I. 1984. Quality assurance in operating a multielement ICP emission spectrometer. Spectrochim. Acta 39B:95. 8. B OTTO , R. I. 1982. Long-term stability of spectral interference calibrations for inductively coupled plasma atomic emission spectrometry. Anal. Chem. 54:1654. 9. M AXFIELD , R. & B. MI NDAK . 1985. EPA Method Study 27, Method 200. 7 (Trace Metals by ICP). EPA-600/S4-85/05. National Technical Information Serv., Springfield, Virginia. 10. GARBARINO, J. R., B. E. JONES, G. P. STEIN, W. T. BELSER & H. E. TAYLOR. 1985. Statistical evaluation of an inductively coupled plasma atomic emission spectrometric method for routine water quality testing. Appl. Spectrosc. 39:53. MÉTODOS NORMALIZADOS 3-68 TABLA 3120:II. DATOS DE PRECISIÓN Y SESGO DE PAI 3-69 DETERMINACIÓN DE METALES TABLA 3120:II. CONTINUACIÓN * X = recuperación media, μg/1, C = valor real, μg /1, S = desviación estándar multi-laboratorio, μg/1, SR = desviación estándar de un único analista, μg/1. MÉTODOS NORMALIZADOS 3-70 3500-AI ALUMINIO* 3500-AI A. 1. Incidencia El aluminio ocupa el tercer lugar en orden de abundancia entre los elementos de la corteza terrestre, formando parte de minerales, rocas y arcillas. Esta amplia distribución es la causa de la presencia del aluminio en casi todas las aguas naturales como sal soluble, coloide o compuesto insoluble. El aluminio soluble, coloidal e insoluble puede encontrarse también en aguas tratadas o en aguas residuales como residuo de la coagulación con material que contiene aluminio. El agua filtrada en una moderna instalación * Aprobado por el Standard Methods Committee, 1988. 3500-AI B. de filtración rápida con arena no tendrá una concentración de aluminio inferior a 50 μg/1. 2. Selección del método Los métodos espectrométrico de absorción atómica y de plasma de acoplamiento inductivo están exentos de interferencias tan corrientes como fluoruro y fosfato, y son los preferidos. El método colorimétrico de eriocromo cianina R es una forma de determinación del aluminio con instrumentación más sencilla. El método automatizado de violeta de pirocatecol constituye una técnica de análisis de gran sensibilidad con inyección de flujo o flujo continuo. Método espectrométrico de absorción atómica Véase el método espectrométrico de absorción atómica de llama, secciones 3111D y E, y el método espectrométrico 3500-AI C. Introducción de absorción atómica electrotérmica, sección 3113. Método de plasma de acoplamiento inductivo Véase sección 3120. 3-71 DETERMINACIÓN DE METALES 3500-AI D. 1. Método de eriocromo cianina R Discusión general a) Principio: Las soluciones diluidas de aluminio tamponadas a pH 6,0 producen con la tinción de eriocromo cianina R un complejo de color rojo a rosado que presenta un máximo de absorción a 535 nm. La intensidad del color desarrollado depende de la concentración del aluminio, el tiempo de reacción, la temperatura, pH, alcalinidad y concentración de otros iones en la muestra. Para compensar el color y la turbidez, se forma un complejo del aluminio de una porción de la muestra con EDTA para obtener un blanco. La interferencia de hierro y manganeso, dos elementos que se encuentran frecuentemente en el agua, se elimina por adición de ácido ascórbico. El intervalo óptimo de aluminio oscila entre 20 y 300 μg/l, pero puede extenderse por exceso mediante dilución de la muestra. b) Interferencia: Tanto el fluoruro como los polifosfatos dan lugar a errores negativos. Cuando la concentración de fluoruro es constante, el porcentaje de error disminuye al aumentar las cantidades de aluminio. Como la concentración de fluoruro se conoce muchas veces o se puede determinar fácilmente, se pueden obtener resultados bastante precisos añadiendo la cantidad conocida de fluoruro a un grupo de patrones. Se puede obtener una corrección más sencilla a partir de la familia de curvas de la figura 3500Al:l. Se da un procedimiento para eliminar la interferencia de fosfatos complejos. El ortofosfato a concentraciones por debajo de 10 mg/1 no interfiere. La interferencia causada por incluso una pequeña alcalinidad se elimina acidulando la muestra justo detrás del punto de neutra- lización con naranja de metilo. El sulfato no interfiere hasta concentraciones de 2.000 mg/1. c) Concentración mínima detectable: La concentración mínima detectable por este método, en ausencia de fluoruros y fosfatos complejos, es de aproximadamente 6 μg/1. d) Manipulación de las muestras: Tómense las muestras en frascos preferiblemente de plástico, limpios, enjuagados con ácido, y examínense tan pronto como sea posible después de recogidas. Si sólo hay que determinar el aluminio soluble, fíltrese una porción de muestra a través de un filtro de membrana de 0,45 μg; descártense los 50 ml primeros de filtrado y utilícese el filtrado obtenido para la determinación. No hay que utilizar papel de filtro, algodón absorbente o lana de vidrio para filtrar una solución en la que se va a ensayar aluminio, ya que éstos eliminarían la mayor parte del aluminio soluble. 2. Instrumental a) Equipo de colorimetría: Se necesita uno de los siguientes: 1) Espectrofotómetro, para ser utilizado a 535 nm, con un trayecto luminoso de 1 cm o más largo. 2) Fotómetro de filtro, con un trayecto luminoso de 1 cm o más largo y equipado con un filtro verde con una transmitancia máxima entre 525 y 535 nm. 3) Tubos de Nessler, 50 ml, forma alargada, igualados. b) Material de vidrio: Trátese todo el material de vidrio con HC1 1 + 1 caliente y enjuagúese con agua destilada libre MÉTODOS NORMALIZADOS 3-72 de aluminio para evitar errores debidos a materiales absorbidos en el vidrio. Enjuagúese suficientemente para eliminar todo el ácido. 3. Reactivos Utilícense reactivos bajos en aluminio y agua destilada libre de aluminio. a) Solución de aluminio de reserva: Utilícese el metal (1) o la sal (2) para preparar una solución de reserva; 1,00 ml = = 500 μg Al: 1) Disuélvanse 500,0 mg de aluminio metal en 10 ml de HC1 conc. calentando suavemente. Dilúyase con agua hasta 1.000 ml, o 2) Disuélvanse 8,791 g de sulfato de aluminio y potasio (llamado también alumbre de potasio), A1K(SO4)212H2O, en agua y dilúyase hasta 1.000 ml. Corríjase este peso dividiendo por la fracción decimal del A1K(SO4)2 12H2O ensayado en el reactivo empleado. b) Solución patrón de aluminio: Diluyanse con agua 10,00 ml de solución de aluminio de reserva hasta 1.000 ml; 1,00 ml = 5,00 μg Al. Prepárese diariamente. c) Ácido sulfúrico, H2SO4, 0,02N y 6N. d) Solución de ácido ascórbico: Disuélvase 0,1 g de ácido ascórbico en agua y llévese hasta un volumen de 100 ml en un matraz volumétrico. Prepárese reciente todos los días. e) Reactivo tampón: Disuélvanse 136 g de acetato de sodio, NaC2H3O2 3H2O, en agua, añádanse 40 ml de ácido acético 1N y dilúyase hasta 1 l. f) Solución de tinción de reserva: Utilícense cualesquiera de los siguientes productos: 1) Solocromo cianina R-200* o erio* Arnold Hoffman & Co., Providence, Rhode Island. cromo cianina†: Disuélvanse 100 mg en agua y dilúyanse hasta 100 ml en un matraz volumétrico. Esta solución habrá de tener un pH de aproximadamente 2,9. 2) Eriocromo cianina R‡: Disuélvanse 300 mg en aproximadamente 50 ml de agua. Ajústese el pH desde aproximadamente 9 a aproximadamente 2,9 con ácido acético 1 + 1 (se necesitarán aproximadamente 3 ml). Dilúyase con agua hasta 100 ml. 3) Eriocromo cianina R§: Disuélvanse 150 mg en aproximadamente 50 ml de agua. Ajústese el pH desde 9 a 2,9 con ácido acético 1 + 1 (se necesitarán aproximadamente 2 ml). Dilúyase con agua hasta 100 ml. Las soluciones de reserva presentan buenas condiciones de estabilidad y pueden conservarse al menos durante 1 año. g) Solución de tinción de trabajo: Diluyanse 10,0 ml de solución de tinción de reserva seleccionada hasta 100 ml en un matraz volumétrico utilizando agua. Las soluciones de trabajo son estables al menos durante 6 meses. h) Solución indicadora de naranja de metilo, o solución indicadora de verde de bromocresol especificada en la determinación de alcalinidad total (sección 2320B.3d). i) EDTA (sal sódica del dihidrato del ácido etilendiaminotetraacético), 0,01M: Disuélvanse 3,7 g en agua y dilúyase hasta 1 l. j) Hidróxido sódico: NaOH, 1N y 0,1 N. 4. Procedimiento a) Preparación de la curva de calibración: † K & K Laboratories, K & K Lab. Div., Life Sciences Group, Plainview, Nueva York. ‡ Pfaltz & Bauer, Inc., Stamford, Connecticut. § EM Science, Gibbstown, Nueva Jersey. DETERMINACIÓN DE METALES 1) Prepárese una serie de patrones de aluminio desde 0 a 7 μg (0 a 280 μg/1 sobre una muestra de 25 ml) midiendo con precisión los volúmenes calculados de solución de aluminio patrón en matraces volumétricos de 50 ml o tubos de Nessler. Añádase agua hasta un volumen total de aproximadamente 25 ml. 2) Añádase a cada patrón 1 ml de H2SO4 0,02N y mézclese. Añádase 1 ml de solución de ácido ascórbico y mézclese. Añádanse 10 ml de solución tampón y mézclese. Añádanse 5,00 ml de reactivo de tinción de trabajo con una pipeta volumétrica y mézclese. Inmediatamente, llévese hasta 50 ml con agua destilada. Mézclese y déjese reposar durante 5 a 10 minutos. El color empieza a desvanecerse después de 15 minutos. 3) Léase la transmitancia o absorbancia en un espectrofotómetro, empleando una longitud de onda de 535 nm o un filtro verde que dé una transmitancia máxima entre 525 y 535 nm. Ajústese el instrumento a absorbancia cero con el patrón libre de aluminio. Llévese a una gráfica la concentración de Al (microgramos de aluminio en 50 ml de volumen final) frente a la absorbancia. b) Tratamiento de la muestra en ausencia de fluoruro y fosfatos complejos: Colóquense 25,0 ml de muestra, o una porción diluida a 25 ml, en una cápsula de porcelana o matraz, añádanse unas cuantas gotas de indicador naranja de metilo y valórese con H2SO4 0,02N hasta un color rosa tenue. Anótese la lectura y descártese la muestra. A dos muestras similares añádase, a temperatura ambiente, la misma cantidad de H2SO4 0,02N empleado en la volumetría y 1 ml en exceso. Añádase 1 ml de solución de EDTA a una muestra. Esto servirá como blanco por formación de complejo de aluminio que pudiera existir y compensación del color y la turbidez. Añádanse, a ambas muestras, 1 ml de ácido ascórbico, 10 ml de reactivo tampón y 5,00 ml de reactivo 3-73 de tinción de trabajo, tal como se ha prescrito en el anterior apartado a2). Póngase el instrumento a absorbancia cero o a transmitancia 100 por 100 empleando un blanco de EDTA. Tras 5 a 10 minutos de tiempo de contacto, léase la transmitancia o absorbancia y determínese la concentración del aluminio empleando la curva de calibración previamente preparada. c) Comparación visual: Si no se dispone de equipo fotométrico, prepárense y trátense muestra y patrones tal como se ha descrito antes, empleando tubos Nessler de 50 ml. Llévese hasta la marca con agua y compárese el color de la muestra con los patrones después de 5 a 10 minutos de tiempo de contacto. Cuando se emplean tubos de Nessler, no se necesita una muestra tratada con EDTA. Si la muestra presenta turbidez o color, el empleo de tubos de Nessler puede dar lugar a un error considerable. d) Eliminación de la interferencia de fosfatos: Añádanse 1,7 ml de H2SO4 6N a 100 ml de muestra en un matraz erlenmeyer de 200 ml. Caliéntese sobre placa caliente durante al menos 90 minutos, manteniendo la temperatura de la solución justo por debajo del punto de ebullición. Al terminar el período de calentamiento, el volumen de la solución deberá ser de aproximadamente 25 ml. Añádase agua si fuera necesario para mantener ese volumen o por encima de él. Después de enfriar, neutralícese con NaOH hasta pH entre 4,3 y 4,5, utilizando NaOH 1N al principio y 0,1N para un ajuste fino final. Sígase la operación con un pHmetro. Llévese hasta 100 ml con agua, mézclese y empléese una porción de 25 ml para el ensayo de aluminio. Trátese un blanco de la misma manera, empleando 100 ml de agua destilada y 1,7 ml de H2SO4 6N. Réstese la lectura del blanco de la lectura de la muestra o utilícese para llevar el instrumento a absorbancia cero antes de hacer la lectura de la muestra. 3-74 MÉTODOS NORMALIZADOS Figura 3500-A1:I. Curvas de corrección para estimación de aluminio en presencia de fluoruro. 3-75 DETERMINACIÓN DE METALES e) Corrección de muestras que contienen fluoruro: Mídase la concentración de fluoruros de la muestra por el SPADNS o método del electrodo. Por una de las dos maneras siguientes: 1) Añádase la misma cantidad de fluoruro de la muestra a cada patrón de aluminio, o 2) Determínese la corrección de fluoruro a partir del juego de curvas de la figura 3500-A1:1. 5. Cálculos 6. Precisión y sesgo Se analizó en 27 laboratorios una muestra sintética con 520 μg Al/1 en agua destilada, sin interferencia, empleando el método de eriocromo cianina R. La desviación relativa estándar fue de 34,4 por 100 y el error relativo de 1,7 por 100. En 35 laboratorios se analizó una segunda muestra sintética con 50 μg Al/1, 500 μg Ba/1 y 5 μg Be/1 en agua destilada. La desviación relativa estándar fue de 38,5 por 100 y el error relativo 22,0 por 100. Una tercera muestra sintética con 500 μg Al/1, 50 μg Cd/1, 110 μg Cr/1, 1.000 μg Cu/1, 300 μg Fe/1, 70 μg Pb/1, 50 μg Mn/1, 150 μg Ag/1 y 650 μg Zn/1 en agua destilada fue analizada en 26 laboratorios. La desviación relativa estándar fue de 28,8 por 100 y el error relativo 6,2 por 100. Una cuarta muestra sintética con 540 μg Al/1 y 2,5 mg de polifosfato/1 en agua destilada fue analizada en 16 laboratorios, que hidrolizaron la muestra según la manera prescrita. La desviación relativa estándar fue de 44,3 por 100 y el error relativo 1,3 por 100. En 12 laboratorios que no aplicaron medidas correctivas, la desviación relativa estándar fue de 49,2 por 100 y el error relativo de 8,9 por 100. Una quinta muestra con 480 μg Al/1 y 750 μg F/l en agua destilada fue analizada en 16 laboratorios, que corrigieron el contenido en fluoruro empleando la curva. La desviación relativa estándar fue de 25,5 por 100 y el error relativo 2,3 por 100. Los 17 laboratorios que añadieron fluoruro a los patrones de aluminio presentaron una desviación relativa estándar de 22,5 por 100 y un error relativo de 7,1 por 100. 7. Bibliografía SHULL, K. E. & G. R. GUTHAN. 1967. Rapid modified Eriochrome cyanine R method for determination of aluminum in water. J. Amer. Water Works Assoc. 59:1456. 3500-AI E. Método automatizado de violeta de pirocatecol (VPC) (PROPUESTA) 1. Discusión general a) Principio: El violeta de pirocatecol (VPC) reacciona con aluminio para formar un complejo coloreado que absorbe a 580 nm. El desarrollo del color depende del pH, que se ajusta a un valor óptimo de 6,1. b) Interferencias: El hierro férrico forma complejo con VPC con un espectro de absorbancias similar al Al-VPC, dando lugar a una interferencia positiva. La interferencia se enmascara reduciendo el Fe(III) a Fe(II) con clorhidrato de hidroxilamina y subsiguiente quelación de Fe(II) con orto-fenantrolina. El hierro 3-76 ferroso no reacciona con VPC. La formación de complejo de Fe(ll)-ortofenantrolina evita la oxidación de Fe(II) a Fe(III). c) Concentración mínima detectable: La concentración mínima detectable del Al monómero total es de 10 y 7 μg Al/1 en análisis de flujo segmentado (AFS) o no segmentado, y análisis de inyección de flujo (AIF), respectivamente. 2. Instrumental a) Equipo analítico automatizado: Utilícese el análisis de flujo segmentado (AFS) o flujo no segmentado o el análisis de inyección de flujo (AIF). Los sistemas utilizan la misma química, pero difieren en la tasa de flujo y en la cantidad de muestra. Ambos constan de los siguientes componentes: 1) Dispositivo de obtención de muestras. 2) Distribuidor. 3) Bomba dosificadora. 4) Colorímetro equipado con célula de flujo tubular de longitud específica. 5) Filtros*. 6) Registrador. 7) Impresora digital (opcional). b) Columna de intercambio catiónico: Si se desea el fraccionamiento de formas orgánicas e inorgánicas de aluminio, utilícese preferiblemente una columna de TFE de 100 mm de larga (10 mm de DI), aunque pueden utilizarse otras medidas. Rellénese la columna con resina † de intercambio catiónico (14 a 50 mallas). c) Utensilios lavados con ácido: Sumérjanse los utensilios de polipropileno o TFE en HNO 3 al 1 por 100 durante toda la noche. Enjuáguense abundantemente con agua destilada o agua desionizada. * Whatman GF/C o equivalente. † Amberlite IR-120, forma de hidrógeno 1 por 100, Rohm & Haas Company o equivalente. MÉTODOS NORMALIZADOS 3. Reactivos a) Sistema automatizado I (AIF): 1) Solución de aluminio de reserva: Véase apartado D.3a. 2) Solución de aluminio patrón: Véase apartado D3b. Prepárese una gama adecuada de patrones diluyendo volúmenes apropiados de solución de aluminio patrón. 3) Ácido clorhídrico, HC1 conc. ‡. 4) Etanol: 95 por 100. 5) Ácido nítrico: HNO3 conc.§. 6) Solución limpiadora: Añádanse lentamente 8,3 ml de HC1 concentrado a 500 ml de agua en una probeta graduada. Añádanse 100 ml de etanol y complétese el volumen con agua. Prepárese bajo la campana de humos. 7) Solución enmascaradora del hierro: Disuélvanse 7,5 g de clorhidrato de hidroxilamina y 0,56 g de ortofenantrolina en 500 ml de agua y dilúyase hasta 1.000 ml. Desmasifíquese el reactivo a través de un filtro de membrana de 0,45 m. Consérvese en frasco con polietileno. Refrigérese hasta su utilización. 8) Solución de PCV: Disuélvanse 0,375 g de ácido 3,3',4'-trihidroxifucsona2"-sulfónico (VPC) en 40 ml de agua. Déjese aproximadamente 5 minutos removiendo de cuando en cuando. dilúyase con agua hasta 1.000 ml. Desmasifíquese a través de un filtro de membrana de 0,45 μm. Consérvese un frasco de vidrio ámbar o de polietileno. Prepárese diariamente. 9) Solución tampón: Disuélvanse 84 g de hexametilentetraamina en 750 ml de agua. Fíltrese a un matraz volumétrico de 1 l. dilúyase hasta 1.000 ml y pásese a un frasco de polietileno. Refrigérese hasta su uso. b) Sistema II automatizado (analizador de flujo segmentado): ‡ Baker Instra-Analyzed o equivalente, d = 0.91 para NH3 conc. y d = 1,2 (38 por 100) para HC1 conc. § Baker Ultrex o equivalente. 3-77 DETERMINACIÓN DE METALES 1) Solución de aluminio de reserva: Véase apartado D.3a. 2) Solución de aluminio patrón: Véase el anterior apartado a2. 3) Solución limpiadora: Véase el anterior apartado a6. 4) Amoníaco, NH3 conc.‡. 5) Ácido clorhídrico, HCL conc. ‡. 6) Solución de enmascaramiento de hierro: Disuélvanse 17,75 g de clorhidrato de hidroxilamina en 200 ml de agua. Añádanse y disuélvanse 0,176 g de monohidrato de orto-fenantrolina. Dilúyase con agua hasta 250 ml. Consérvese refrigerado en frasco de polietileno. 7) Solución de PCV: Disuélvanse 0,150 g de ácido 3,3',4'-trihidroxifucsona2"-sulfónico (VPC) en 40 ml de agua. Déjese reposar durante 5 minutos removiendo de cuando en cuando. dilúyase con agua hasta 400 ml. Consérvese en frasco de polietileno oscuro. Consérvese refrigerada y protegida de la luz del sol. 8) Tampón: Disuélvanse 300 g de hexametilentetraamina en 750 ml de agua. Fíltrese a un matraz volumétrico de 1.000 ml. Añádanse 16,8 ml de amoníaco concentrado y dilúyase con agua hasta 1.000 ml. Utilícese solución tampón sin diluir, pero compruébese, cuando se emplean todos los reactivos a lo largo del sistema, que el pH del fluido/final está entre 6,0 y 6,2. Si el pH de tal fluido no se encuentra dentro de este intervalo, ajústese el tampón añadiendo amoníaco conc. o HC1 2,5N. 9) Reactivo HCl de trabajo: Añádanse 51,1 ml de HCl conc. hasta 200 ml de agua y dilúyase hasta 250 ml de volumen final. Añádanse 2,5 ml de polioxíetilen 23 lauril éter || (solución al 30 por 100) a la solución final. Utilícese agua destilada o desionizada en lugar de reactivo HCl de trabajo si la muestra no altera el pH del efluente de 6,1. ‡ Baker Instra-Analyzed o equivalente, d = 0,91 para NH 3 conc. y d = 1,2 (38 por 100) para HCl conc. || Brij-35 disponible en ICI Americas, Inc., Wilmington. Delaware, o en Technicon Instruments Corp., Tarrytown. Nueva York, o equivalente. 4. Procedimiento a) Aluminio total: Digiérase la muestra tal como se ha indicado en el procedimiento de digestión con ácido nítrico caliente descrito en la sección 3030E. Ajústese la dilución de la muestra y la concentración del tampón para asegurar que el pH del efluente es óptimo (6,1) para desarrollo de color. Quítese la columna de intercambio catiónico del trayecto de flujo. b) Aluminio monómero total: Determínese el aluminio monómero total sin pretratamiento de la muestra. Fracciónese el aluminio monómero orgánico e inorgánico midiendo el Al reactivo al VPC antes y después de su paso a través de la columna de intercambio catiónico. La diferencia representa la forma monómera inorgánica de Al. 1) Columna de intercambio catiónico: Hágase mínima la ruptura de los complejos órgano-alumínicos manteniendo una tasa de flujo a través de la columna de 3,7 a 4,2 ml/min/ml de resina. 2) Sistema automatizado I (análisis de inyección de flujo no segmentado): Instálese un distribuidor tal como se muestra en la figura 3500.Al:2. Colóquese una columna de intercambio catiónico antes del ciclo de la muestra para evitar la dispersión del bolus de la muestra sobre el distribuidor. Comiéncese a bombear los reactivos y espérese a obtener una línea de base estacionaria. No hay que sumergir nunca las líneas de fluido de las células de flujo y válvulas de la muestra en el depósito de residuos, ya que esto daría lugar a una contra-presión y originaría características pobres del flujo. Analícese la muestra después de alcanzar una línea de base estacionaria. 3) Sistema automatizado II (analizador de flujo segmentado): Instálese un distribuidor tal como el que se muestra en la figura 3500-A1:3. La tasa de toma de muestras es de 30 muestras/hora, utili- 3-78 MÉTODOS NORMALIZADOS Figura 3500-A1:2. Distribuidor de aluminio para sistema automatizado I (analizador de inyección de flujo no segmentado)1. (Arriba) Canal 1.—Aluminio reactivo a VPC total. (Abajo) Canal 2.—Aluminio reactivo a VPC no intercambiable. Clave: portador: agua desionizada (o HC1 0,1 N); Rl = solución de enmascaramiento: cloruro de hidroxilamonio; R2 = reactivo de color: violeta de pirocatecol; R3 = solución tampón: hexametilentetraamina; RC1 = bobina de reactancia, 10 cm (0,5 mm DI); RC2 = = bobina de reactancia, 30 cm (0,5 mm DI); RC3 = bobina de reactancia, 60 cm (0,5 mm DI); CEC = columna de intercambio catiónico. zando un minuto para la toma de muestra y para el lavado. Colóquese la columna alineada antes de la segmentación del aire. Como el aire se introduce cada vez que la aguja de la muestra se conecta desde el lavado a la inyección de la muestra, colóquese alineado un eliminador de burbujeo antes de la columna. Determínese la dispersión aparente de muestra causada por la columna rellenando la columna con una resina inerte (por ejemplo, perlas de poliestireno de tamaño de malla comparable). Calcúlese el factor de dispersión y multiplíquense los valores de Al postcolumna por este factor. Esta corrección es necesaria debido a que normalmente no se dispone de un patrón orgánico adecuado. 3-79 DETERMINACIÓN DE METALES Figura 3500-AI:3. Distribuidor de aluminio para sistema automatizado II (analizador de flujo segmentado). Según Rogeberg y Henriksen, Vatten. Reproducido con permiso. c) Elaboración de la curva de calibra- ción: Prepárese una serie de patrones de aluminio cuya concentración varíe entre 0 y 1.000 μg/1. Mídase el aluminio y trácese el gráfico absorbancia en función de la concentración para determinar el intervalo lineal del sistema. Si la linealidad desaparece, utilícense dos curvas de calibración separadas para muestras de concentraciones alta y baja o dilúyanse las muestras para que queden dentro del intervalo lineal. Para la calibración, quítese la columna de intercambio catiónico. 5. Cálculos Determínese la concentración de aluminio, en microgramos por litro, a partir de la curva de calibración directamente MÉTODOS NORMALIZADOS 3-80 empleando una ecuación de regresión lineal apropiada. Corríjase la dilución si es necesario. 6. Precisión y sesgo La precisión obtenida por un solo laboratorio fue de ± 3 μg Al/1 para muestras de aguas naturales superficiales cuyo contenido en Al variaba entre 0 y 300 μg Al/1 analizado por el sistema de AFS. La recuperación obtenida por el mismo laboratorio con el sistema de AFS fue de 99 y 105 por 100 para muestras de aguas superficiales no tratadas a concentraciones de 150 y 200 μg Al/1, respectivamente. Un único analista de un solo laboratorio analizó diversas concentraciones de Al monómero inorgánico preparadas en agua destilada/ionizada con un sistema de inyección de flujo, con los resultados que se dan en la tabla 3500-A1:I. De forma similar se determinaron la precisión y el sesgo, mostrados en la tabla 3500-A1:II, para un intervalo de calibración de alto nivel de Al entre 350 y 1.000 μg /1. Para el aluminio monómero total se analizaron 24 pares de duplicados en un solo laboratorio. Los pares eran muestras de aguas superficiales naturales y se separaron en muestras con concentraciones por encima de 85 μg Al/1 y muestras con concentraciones por debajo de 50 μg Al/1. Los valores de la precisión de los pares duplicados, como desviación estándar, fueron 4,2 y 1,8, respectivamente. De manera análoga, para el Al combinado orgánicamente, los valores de precisión de los pares duplicados fueron 4,1 y 1,1, respectivamente. Para dos muestras de aguas superficiales naturales, sin tratar, con adiciones conocidas de 300 y 100 μg Al/1, la recuperación fue de 99,6 y 102,3 por 100, respectivamente. 7. Referencias 1. H ILLMAN , D. C, T. E. L E WIS , S. H. P IA , F. X. S UÁREZ , E. M. B URKE , D. E. D OBB , J. M. HENSHAW & L. J. B LUME. 1986. Summary Report on the Evaluation of Aquatic Methods. EPA-600/X-86-271, Agencia de Protección de los Estados Unidos. 2. ROGEBERG , E. J. S. & A. H ENRIKSEN . 1985. An automatic method for fractionation and determination of aluminum species in freshwaters. Vatten 41:48. TABLA 3500-AL:I. PRECISIÓN Y SESGO DE UN SOLO OPERADOR PARA ANÁLISIS DE ALUMINIO MONÓMERO INORGÁNICO 3-81 DETERMINACIÓN DE METALES TABLA 3500-AL:II. PRECISIÓN Y SESGO DE UN SOLO OPERADOR EN LA DETERMINACIÓN DE ALTO NIVEL DE ALUMINIO 8. Bibliografía DOUGAN, W. K. & A. L. WILSON. 1974. The absorptiometric determination of aluminum in water: A comparison of some chromogenic reagents and the development of an improved method. Analyst 99:413. TECATOR, A. B. 1985. Determination of aluminum in water and soil extracts by flow injection analysis. Tech, rep., Tecator AB, Hoganas, Suecia. ROYSET, O. 1986. Flow-injection spectrophotometric determination of aluminum in water with pyrocatechol violet. Anal. Chim. Acta 185:75. HENSHAW, J. M., T. E. LEWIS & E. M. HEITHMAR. 1988. A semi-automated colorimetric method for the determination of monomeric aluminum species in natural waters by flow injection analysis. Int. J. Environ. Anal. Chem. 34:119. 3500-Sb ANTIMONIO 3500-Sb A. Introducción 1. Incidencia 2. El antimonio se encuentra en cantidades traza en aguas naturales (normalmente inferiores a 10 μg/1) y puede presentarse en mayores concentraciones en manantiales termales o en aguas que drenan zonas mineralizadas. El antimonio es un contaminante regularizado en diversos programas, tanto federales como estatales. Se elige el método espectrométrico de absorción atómica electrotérmica debido a su sensibilidad. Cuando no es necesaria una alta sensibilidad, empléese el método espectrométrico de absorción atómica de llama o el método de plasma de acoplamiento inductivo. 3500-Sb B. Selección del método Método espectrométrico de absorción atómica Véanse método espectrométrico de absorción atómica de llama, sección 3111B, y método espectrométrico de absorción atómica electrotérmica, sección 3113. 3-82 MÉTODOS NORMALIZADOS 3500-Sb C. Método de plasma de acoplamiento inductivo Véase sección 3120. 3500-As 3500-As A. 1. Incidencia y significación Una cantidad de arsénico tan pequeña como 100 mg puede ocasionar un grave envenenamiento; además, pueden aparecer efectos crónicos por su acumulación en el cuerpo por repetidos niveles bajos de ingesta. También se le atribuyen al arsénico propiedades cancerígenas. La concentración de arsénico en la mayoría de aguas potables raramente excede los 10 μg/1, aunque han sido registrados valores del orden de los 100 μg/1. El arsénico puede encontrarse en el agua como resultado de una disolución de minerales, descargas industriales o aplicación de insecticidas. * Aprobado por el Standard Methods Committee, 1988. 3500-As B. Arsénico* Introducción 2. Selección del método Se selecciona el método (B) de espectrometría de absorción atómica (EAA) de hidruros que transforma el arsénico en su hidruro y emplea una llama de argónhidrógeno, aunque el método EAA electrotérmico directo (B) es más sencillo en caso de que se demuestre la ausencia de interferencia. El método de dietildiriocarbamato de plata (C) es aplicable cuando no hay interferencias. Por su parte, el método de tinte de bromuro mercúrico (D) requiere cuidado y experiencia y únicamente es adecuado para determinaciones cualitativas o semicuantitativas (± 5 μg As). El método de plasma de acoplamiento inductivo (PAI) (E) resulta útil a concentraciones más altas (superiores a 50 μg/1). Método espectrométrico de absorción atómica Véanse método espectrométrico de absorción atómica electrotérmica, sección 3113, y método espectrométrico de ab- sorción atómica de generación de hidruros, sección 3114. DETERMINACIÓN DE METALES 3500-As C. 1. 3-83 Método de dietilditiocarbamato de plata Discusión general a) Principio: El arsénico inorgánico se reduce a arsina, AsH3, utilizando solución acida de zinc en un generador Gutzeit. Se hace pasar entonces la arsina a través de un frasco lavador que contiene lana de vidrio impregnada en solución de acetato de plomo, a un tubo de absorción que contiene dietilditiocarbamato de plata disuelto en piridina o cloroformo. En el tubo de absorción, el arsénico reacciona con la sal de plata, formando un complejo rojo soluble adecuado para medida fotométrica. b) Interferencia: Aunque ciertos metales —cromo, cobalto, cobre, mercurio, molibdeno, níquel, platino y plata— interfieren en la generación de arsina, las concentraciones de estos metales, que normalmente se encuentran en el agua, no interfieren significativamente. Las sales de amonio en la muestra forman estibina, que interfiere con desarrollo de color formando un color rojo con absorbancia máxima en 510 nm. c) Cantidad mínima detectable: 1 μg As. Figura 3500-A s:1. en el intervalo de 530 a 540, con células de 1 cm. 3. 2. Instrumental a) Generador de arsina y tubo de absorción: Véase figura 3500-As:l*. b) Equipo fotométrico: 1) Espectrofotómetro para usarlo a 535 nm con células de 1 cm. 2) Fotómetro de filtro, con filtro verde que tiene una transmitancia máxima * Fisher Scientific Co., n.° 1-405 o instrumental equivalente. Generador de arsina y ensamblaje del absorbedor. Reactivos a) Ácido clorhídrico, HC1 conc. b) Solución de yoduro potásico: Disuélvanse 15 g de KI en 100 ml de agua destilada. Consérvese en frasco topacio. c) Reactivo cloruro estannoso: Disuélvanse 40 g de SnCl2·H2O libre de arsénico en 100 ml de HC1 conc. d) Solución de acetato de plomo: Disuélvanse 10 g de Pb(C2H3O2)2·3H2O en 100 ml de agua destilada. e) Reactivo dietilditiocarbamato de plata: Prepárese este reactivo como se describe en 1) o 2): 3-84 1) Disuélvanse 410 mg de 1-efedrina en 200 ml de cloroformo (CHC13), añádanse 625 mg de AgSCSN(C2H5)2 y ajústese el volumen a 250 ml añadiendo más CHC13. Fíltrese y consérvese en frasco topacio. 2) Disuélvase 1 g de AgSCSN(C2H5)2 en 200 ml de piridina. Consérvese en frasco topacio. f) Zinc, 20 a 30 mallas, libre de arsénico. g) Solución de arsénico de reserva: Disuélvanse 1.320 g de trióxido de arsénico, As2O3, en 10 ml de agua destilada que contiene 4 g de NaOH, y diluyase con agua destilada hasta 1.000 ral; 1,00 ml = 1,00 mg As. (PRECAUCIÓN: Tóxico, evítese la ingestión de soluciones de arsénico.) h) Solución de arsénico intermedia: dilúyanse 5,00 ml de solución de reserva hasta 500 ml empleando agua destilada; 1,00 ml = 10,0 μg As. i) Solución de arsénico patrón: Dilúyanse 10,00 ml de solución intermedia hasta 100 ml empleando agua destilada; 1,00 ml = 1,00 μg As. 4. Procedimiento Para determinar el arsénico total, digiérase la muestra por el procedimiento del apartado 4a. Anótese si la muestra ha sido digerida o no. a) Tratamiento de la muestra: Llévense con la pipeta 35,0 ml de muestra a un frasco generador limpio. Añádanse sucesivamente, cuidando de mezclar después de cada adición, 5 ml de HC1 conc, 2 ml de solución de KI y 8 gotas (0,40 ml) de reactivo SnCl2. Déjese 15 minutos para que se reduzca el arsénico al estado trivalente. b) Preparación del frasco lavador y del absorbedor: Imprégnese la lana de vidrio del frasco lavador con una solución de acetato de plomo. No humedecer demasiado para que el agua no rezume MÉTODOS NORMALIZADOS dentro de la solución de reactivo. Llévense con la pipeta 4,00 ml de reactivo dietilditiocarbamato de plata al tubo absorbedor. c) Generación y medida de arsina: Añádanse 3 g de zinc al generador y conéctese la estructura de lavador-absorbedor inmediatamente. Compruébese que todas las uniones están herméticamente ajustadas. Déjese 30 minutos para un desprendimiento completo de la arsina. Caliéntese ligeramente el generador para asegurarse de que se ha desprendido toda la arsina. Viértase la solución directamente desde el absorbedor a una célula de 1 cm y mídase la absorbancia a 535 nm utilizando el blanco de reactivo como referencia. d) Preparación de la curva estándar: Trátense porciones de solución patrón que contienen 0, 1,0, 2,0, 5,0 y 10,0 μg de As, descritas en los anteriores apartados 4a y c. Trácese el gráfico de absorbancia en función de la concentración de arsénico en el patrón. 5. Cálculos mg As/I = μg As (en 4,00 ml de volumen final) ml de muestra 6. Precisión y sesgo Se analizó en 46 laboratorios una muestra sintética con un contenido de 40 μg As/1, 250 μg Be/1, 240 μg B/l, 20 μg Se/1 y 6 μg V/l en agua destilada utilizando el método de dietilditiocarbamato de plata, con una desviación relativa estándar de 13,8 por 100 y un error relativo de 0 por 100. 7. Bibliografía VASAK, V. & V. SEDIVEC. 1952. Colorimetric determination of arsenic. Chem. Listy 46:341. 3-85 DETERMINACIÓN DE METALES STRATTON, G. & H. C. WHITEHEAD. 1962. Colorimetric determination of arsenic in water with silver diethyldithiocarbamate. J. Amer. Water Works Assoc. 54:861. 3500-As D. 1. BALLINGER, D. C, R. J. LISHKA & M. E. GALES. 1962. Application of silver diethyldithiocarbamate method to determination of arsenic. J. Amer. Water Works Assoc. 54:1424. Método del tinte de bromuro mercúrico Discusión general a) Principio: Después de la concentración de la muestra, el arsénico se libera como arsina, AsH3, por una solución acida de zinc en un generador de Gutzeit. Se hace pasar la arsina generada a través de una columna con algodón arrollado humedecido con solución de acetato de plomo. La arsina generada produce un tinte pardo-amarillo sobre tiras de papel de ensayo impregnado con bromuro mercúrico. La longitud de la parte teñida es aproximadamente proporcional a la cantidad de arsénico presente. b) Interferencia: El antimonio (> 0,10 mg) interfiere al dar un tinte similar. c) Cantidad mínima detectable: 1 μg As. 2. Instrumental Generador de arsina: Véase figura 3500-As:2. 3. Reactivos a) Ácido sulfúrico, H2SO4 1 -I- 1. b) Ácido nítrico, HNO3 conc. c) Algodón arrollado: Córtese un rollo de algodón de dentista en longitudes de 25 mm. d) Solución de acetato de plomo: Prepárese tal como se indica en el método C del apartado 3d. e) Papel de bromuro de mercurio: Utilícense papeles de arsénico comerciales Figura 3500-As:2. Generador utilizado con el método del tinte de bromuro mercúrico. cortados uniformemente en tiras de aproximadamente 12 cm de largo y 2,5 mm de ancho (se pueden conseguir papeles ya cortados y sensibilizados). Sumérjanse las tiras durante al menos 1 hora en una solución filtrada preparada por disolución de 3 a 6 g de HgBr2 en 100 ml de alcohol etílico o isopropílico al 95 por 3-86 100; séquense con ondas de aire. Consérvese en lugar seco y protegido de la luz. Para obtener mejores resultados, háganse los papeles inmediatamente antes de su uso. f ) Solución de yoduro de potasio: Prepárese como se ha indicado en el método C del apartado 3b. g) Reactivo cloruro estannoso: Prepárese como se indica en el método C del apartado 3c. h) Zinc, 20 a 30 mallas, libre de arsénico. i) Solución de arsénico patrón: Prepárese como se ha indicado en el método C del apartado 3i. 4. Procedimiento a) Concentración de la muestra y oxidación de materia orgánica: Introdúzcase en un matraz o en un vaso una muestra adecuada que contenga de 2 a 30 μg As, añádanse 7 ml de H 2 SO 4 1 + 1 y 5 ml de HNO3 conc. Evapórese hasta desprendimiento de humos de SO3. Enfríese, añádase aproximadamente 25 ml de agua destilada y evapórese de nuevo para que los humos de SO3 expulsen los óxidos de nitrógeno. Manténgase un exceso de HNO3 hasta destruir toda la materia orgánica. No dejar oscurecer la solución mientras se destruye la materia orgánica, ya que probablemente el arsénico se reduciría y perdería. Enfríese, añádanse aproximadamente 25 ml de agua destilada y pásese a un frasco generador. b) Preparación de columna de seguridad y tubo de reacción: Sumérjase un extremo del cilindro de algodón de 2,5 cm en solución de acetato de plomo e introdúzcase en una columna de cristal. Póngase entonces el tubo de vidrio estrecho seco en su lugar e introdúzcase el papel de ensayo de HgBr2. Asegúrese de que la tira de papel está recta. MÉTODOS NORMALIZADOS c) Tratamiento del concentrado de muestra: Al concentrado de muestra de 25 ml del generador, añádanse 7 ml de H2SO4 1 + 1 y enfríese. Añádanse 5 ml de solución de K.I, cuatro gotas de reactivo SnCl 2 y 2 a 5 g de zinc. Conéctese inmediatamente el tubo de reacción al generador. Sumérjase el dispositivo hasta 2,5 cm de la parte de encima del tubo estrecho en un baño maría mantenido entre 20 y 25 °C, y déjese que tenga lugar el desprendimiento durante 1,5 horas. Sáquese la tira y calcúlese la longitud media de los tintes en ambos lados. Utilizando una curva de calibración, cuya preparación se describe a continuación, se evalúa la cantidad de arsénico presente. d) Preparación de la curva de calibración: Prepárense un blanco y patrones a intervalos de 3 μg entre 0 y 30 μg de As con 14 ml de H2SO4 1 + 1 y llévese a un volumen total de 25 ml. Colóquese en el generador y trátese como se ha descrito para el conc. de muestra. Extráigase la tira y calcúlese la longitud media en milímetros de los tintes de ambos lados. Trácese un gráfico con longitudes en milímetros en función de los microgramos de arsénico y utilícese como curva estándar. 5. Precisión y sesgo Se ha analizado en cinco laboratorios una muestra sintética con 50 μg As/1, 400 μg Be/1, 180 μg B/l y 50 μg Se/1 en agua destilada por el método de tinte de bromuro mercúrico, con una desviación estándar relativa de 75,0 por 100 y un error relativo de 60,0 por 100. 6. Bibliografía FURMAN , N. H., ed. 1962. Standard Methods of Chemical Analysis, 6.a ed. Vol. I. D. Van Nostrand Co., Princeton, Nueva York, págs. 118124. DETERMINACIÓN DE METALES 3500-As E. 3-87 Método de plasma de acoplamiento inductivo Véase sección 3120. 3500-Ba 3500-Ba A. 1. Incidencia y significación El bario estimula el músculo cardíaco. Sin embargo, una dosis de bario de 550 a 600 mg se considera letal para los seres humanos. Los trastornos que producen su ingestión, inhalación o absorción pueden afectar a corazón, vasos sanguíneos y nervios. A pesar de su relativa abundancia en la naturaleza (es el decimosexto elemento * Aprobado por el Standard Methods Committee, 1985. 3500-Ba B. Introducción en orden de abundancia), en el agua sólo se encuentra en cantidades traza. La concentración de bario en las aguas potables de los Estados Unidos puede oscilar entre 0,7 y 900 μg/l, con una media de 49 μg/1. Las concentraciones más altas en el agua potable son frecuente indicio de contaminación por residuos industriales. 2. Selección del método Realícense los análisis por el método espectrométrico de absorción atómica o el método de plasma de acoplamiento inductivo. Método espectrométrico de absorción atómica Véanse método espectrométrico de absorción atómica de llama y método es- 3500-Ba C. BARIO* pectrométrico de absorción atómica electrotérmica, sección 3113. Método de plasma de acoplamiento inductivo Véase sección 3120. 3-88 MÉTODOS NORMALIZADOS 3500-Be 3500-Be A. 1. Incidencia y significación El berilio y sus compuestos son muy tóxicos y en concentraciones elevadas pueden causar la muerte. La inhalación de polvo de berilio origina una grave enfermedad llamada beriliosis. La enfermedad ocasionada por el berilio puede tomar también la forma de dermatitis, conjuntivitis (enfermedad ocular), neumonía aguda (enfermedad pulmonar) y beriliosis pulmonar crónica. Se utiliza en forma de elemento, compuestos o aleaciones en reactores atómicos, aviación, cohetes y combustibles de * Aprobado por el Standard Methods Committee, 1985. 3500-Be B. Introducción misiles. Su aparición en el agua puede provenir de desechos de las citadas industrias. Los informes sobre el berilio en aguas potables en Estados Unidos señalan un intervalo de 0,01 a 0,7 μg/1, con una media de 0,013 μg /1. 2. Selección del método Los métodos seleccionados son el método espectrométrico de absorción atómica y el de plasma de acoplamiento inductivo. Si no se dispone de los instrumentos necesarios para llevar a cabo el método PAI, se puede utilizar el método colorimétrico, aunque con menores precisión y sesgo. Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, secciones 3111D y E, y método espectrométrico de 3500-Be C. BERILIO* absorción atómica electrotérmica, sección 3113. Método de plasma de acoplamiento inductivo Véase sección 3120. DETERMINACIÓN DE METALES 3500-Be D. 1. 3-89 Método del aluminen Discusión general a) Principio: La adición de una pequeña cantidad de solución complejante de ácido etilendiaminotetraacético (EDTA) evita la interferencia de cantidades moderadas de aluminio, cobalto, cobre, hierro, manganeso, níquel, titanio, zinc y zirconio. Añádase un reactivo tampón de aluminón para formar una laca de berilio midiéndose el color desarrollado a 515 nm. b) Interferencia: En las condiciones especificadas, no pueden tolerarse por encima de 10 mg de cobre. Si hay más, auméntese la cantidad de reactivo EDTA. El cobre complejado absorbe ligeramente a 515 nm; elimínese esta interferencia añadiendo una cantidad equivalente de cobre a los patrones. c) Concentración mínima detectable: 5 μg/1. d) Poder absorbente molar: 900 1 g-1 cm-1. 2. Manipulación de la muestra Acidúlense todas las muestras en el momento de su toma para que los metales se mantengan en solución y se evite su depósito sobre las paredes del recipiente. Tratándose de aguas relativamente limpias que no llevan material en forma de partículas, normalmente resulta suficiente 1,5 ml de ácido nítrico conc. (HNO3)/1 para reducir el pH a 2,0. Las aguas superficiales que pueden contener sedimento, tales como las de corrientes, lagos y fluidos procedentes de plantas de tratamiento de aguas residuales, requieren más ácido. Si la muestra contiene materia en partículas y sólo se desea determinar el contenido de metal «disuelto», fíl- trese la muestra a través de un filtro de membrana de 0,45 μm y acidúlese el filtrado con 1,5 ml de HNO3 conc./l. 3. Instrumental a) Espectrofotómetro: Para ser empleado a 515 nm con un trayecto luminoso de 5 cm. b) Fotómetro con filtro: Que proporciona un trayecto luminoso de 5 cm y va equipado con un filtro verde, que presenta una transmitancia máxima próxima a 515 nm. 4. Reactivos a) Solución de berilio de reserva: Disuélvanse 9,837 g de sulfato de berilio tetrahidratado (BeSO4·4H2O), pureza 0,999, en 100 ml de agua destilada, fíltrese si es necesario y dilúyase hasta 500 ml; 1,00 ml = 1,00 mg Be. b) Solución de berilio patrón: Diluyanse 5,0 ml de solución de berilio de reserva con agua destilada hasta 1.000 ml en un matraz volumétrico; 1,0 ml = 5 μg Be. c) Reactivo EDTA: Añádanse 30 ml de agua destilada y una gota de solución alcohólica de rojo de metilo (50 mg/ 100 ml a 2,5 g de ácido etilendiaminotetraacético. Neutralícese con hidróxido amónico (NH4OH), enfríese y dilúyase hasta 100 ml. d) Reactivo tampón de aluminón: Añádanse 500 g de acetato de amonio, NH4C2H3O2, a 1 l de agua destilada en un vaso de 2 1. Añádanse 80 ml de acético conc. (glacial) y agítese hasta disolución completa. Fíltrese si es necesario. Disuélvase 1 g de sal de triamonio del MÉTODOS NORMALIZADOS 3-90 ácido aurintricarboxílico (aluminón) en 50 ml de agua destilada y añádase a la solución tampón del vaso de 2 1. Disuélvanse 3,0 g de ácido benzoico (C7H6O2) en 20 ml de alcohol metílico y añádanse a la solución tampón agitando al mismo tiempo. dilúyase la mezcla hasta 2 1. Pásense 10 g de gelatina a 250 ml de agua destilada en un vaso de 400 ml. Colóquese el vaso en un baño maría a ebullición y agítese de cuando en cuando hasta que la gelatina se haya disuelto por completo. Viértase la gelatina caliente en un matraz volumétrico de 1.000 ml que contenga 500 ml de agua destilada. Enfríese hasta temperatura ambiente, dilúyase hasta la señal y mézclese. Pásense la solución de gelatina y las soluciones tampón a un frasco de vidrio oscuro de 4 1 resistente a productos químicos. Mézclese y consérvese en lugar frío y oscuro. El reactivo es estable durante un mes al menos. agua destilada y mézclese cuidadosamente. Déjese reposar durante 20 minutos, protegiendo la mezcla de la luz tras la adición del tampón aluminón. Fíltrese si es necesario. Léase la absorbancia de patrón y solución desconocida comparada con el blanco en un espectrofotómetro o fotómetro de filtro a una longitud de onda de 515 nm utilizando células de 5 cm. Trácese una curva de calibración con la absorbancia de los patrones en función de los microgramos de berilio en 100 ml de volumen final. Determínese la cantidad de berilio en la muestra por referencia a la correspondiente absorbancia en la curva de calibración. 6. Cálculos μg Be (en 100 ml de volumen final) mg Be/1 = ml de muestra 5. Procedimiento a) Tratamiento de la muestra: Si hay materia orgánica y se desea determinar el berilio total, digiérase la muestra con HNO3 y H2SO4, tal como se indica en la sección 3O3OG. Si sólo interesa determinar el berilio disuelto, fíltrese la muestra a través de un filtro de membrana de 0,45 μm. b) Reacción con aluminón: Pásense con la pipeta 0, 0,1, 0,5, 1,00, 2,00, 3,00 y 4,00 ml de solución patrón de berilio a una serie de matraces volumétricos de 100 ml. Pásese una muestra de 50 ml, o porción que contenga menos de 200 μg de Be, a un matraz volumétrico de 100 ml. Añádanse 2 ml de reactivo EDTA a cada matraz y dilúyase con agua destilada hasta aproximadamente 75 ml. Añádanse 15 ml de reactivo de tampón aluminón, dilúyase hasta 100 ml con 7. Precisión y sesgo En 32 laboratorios se analizó una muestra sintética de agua destilada con 250 μg Be/1, 40 μg As/1, 240 μg B/l, 20 μg Se/1 y 6 μg V/l, respecto al berilio, con una desviación relativa estándar de 7,13 por 100 y un error relativo de 12 por 100. 8. Bibliografía LUKE, C. L. & M. E. CAMPBELL. 1952. Photometric determination of beryllium in berylliumcopper alloys. Anal. Chem. 24:1056. LUKE, C. L. & K. C. BROWN. 1952. Photometric determination of aluminum in manganese, bronze, zinc die casting alloys, and magnesium alloys. Anal. Chem. 24:1120. 3-91 DETERMINACIÓN DE METALES 3500-Bi 3500-Bi A. El bismuto es muy insoluble en aguas naturales y, por lo general, sólo se encuentra en cantidades traza (inferiores a 3500-Bi B. BISMUTO Introducción 10 μg/1). Puede encontrarse en concentraciones más altas en aguas que drenan áreas mineralizadas. Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, sección 3111B. 3500-Cd 3500-Cd A. 1. Incidencia y significación El cadmio es muy tóxico, y se le han atribuido algunos casos de intoxicación con alimentos. Se cree que muy pequeñas cantidades de cadmio podrían ser la causa de alteraciones adversas en las arterias renales. También produce cánceres generalizados en animales de laboratorio y ha sido relacionado epidemiológicamente con ciertos cánceres humanos. Una concentración de cadmio de 200 μg/1 es tóxica para ciertos peces. El cadmio puede llegar al agua a través de * Aprobado por el Standard Methods Committee, 1985. CADMIO* Introducción vertidos industriales o por deterioro de tuberías galvanizadas. 2. Selección del método Se prefiere el método espectrométrico de absorción atómica electrotérmico (horno de grafito). Los métodos de absorción atómica de llama y de plasma de acoplamiento inductivo proporcionan un aceptable nivel de precisión y sesgo, con límites de detección más altos. El método de la ditizona es adecuado cuando no se dispone de instrumental para la espectrometría de absorción atómica o para el plasma de acoplamiento inductivo y la precisión buscada no es tan grande. 3-92 MÉTODOS NORMALIZADOS 3500-Cd B. Método espectrométrico de absorción atómica Véase método espectrométrico de ab- ción atómica electrotérmica, sección sorción atómica de llama, secciones 3111B 3113. y C, y método espectrométrico de absor- 3500-Cd C. Método de plasma de acoplamiento inductivo Véase sección 3120. 3500-Cd D. 1. Método de la ditizona Discusión general a) Principio: Los iones cadmio reaccionan en condiciones adecuadas con ditizona para formar un color de rosa a rojo que puede extraerse con cloroformo (CHC13). Los extractos clorofórmicos se miden fotométricamente y la concentración de cadmio se obtiene a partir de una curva de calibración preparada con una solución de cadmio patrón tratada de la misma manera que la muestra. b) Interferencia: En las condiciones especificadas no interfieren las concentraciones de iones metálicos encontrados normalmente en el agua. El plomo hasta 6 mg, el zinc hasta 3 mg y el cobre hasta 1 mg en la porción analizada no interfieren. La iluminación normal de la habitación (incandescente o fluorescente) no afecta al color del ditizonato de cadmio. c) Concentración mínima detectable: 0,5 μg Cd en aproximadamente 15 ml de volumen final con un trayecto luminoso de 2 cm. 2. Instrumental a) Equipo de colorimetria: Se necesita uno de los siguientes: 1) Espectrofotómetro, para ser utilizado a 518 nm con un trayecto luminoso mínimo de 1 cm. 2) Fotómetro de filtro, equipado con un filtro verde que tenga una transmitancia luminosa máxima próxima a 518 nm, con un trayecto luminoso mínimo de 1 cm. b) Embudos separadores: 125 ml, preferiblemente con espitas de TFE. c) Material de vidrio: Límpiese todo el material de vidrio, incluidos los frascos para muestras, con HC1 1 + 1 y enjuagúese cuidadosamente con agua del grifo y agua destilada. 3. Reactivos a) Agua libre de cadmio: Vuélvase a destilar el agua destilada en un destila- 3-93 DETERMINACIÓN DE METALES dor hecho completamente de vidrio. Utilícese esta agua para preparar todos los reactivos y soluciones. b) Solución de cadmio de reserva: Pésense 100,0 mg de metal cadmio puro y disuélvanse en una solución compuesta por 20 ml de agua más 5 ml de HC1 conc. Empléese calor para facilitar la disolución del metal. Pásese cuantitativamente a un matraz volumétrico de 1 l y dilúyase hasta 1.000 ml; 1,00 ml = 100 μg Cd. Guárdese en un recipiente de polietileno. c) Solución de cadmio patrón: Llévense con la pipeta 10,00 ml de solución de cadmio de reserva a un matraz volumétrico de 1 l, añádanse 10 ml de HC1 conc. y dilúyase con agua hasta 1.000 ml. Prepárese lo que se necesita utilizar ese mismo día; 1,00 ml = 1,00 μg Cd. d) Solución de tartrato de sodio y potasio: Disuélvanse 250 g de NaKC4H4O6·4H2O en agua y llévese hasta 1 l. e) Soluciones de cianuro de potasiohidróxido sódico: 1) Solución I: Disuélvanse 400 g de NaOH y 10 g de KCN en agua y llévese hasta 1 l. Consérvese en botella de polietileno. Esta solución es estable durante un mes. 2) Solución II: Disuélvanse 400 g de NaOH y 0,5 g de KCN en agua y llévese hasta 1 l. Consérvese en frasco de polietileno. Esta solución es estable durante uno a dos meses. PRECAUCIÓN: El cianuro de potasio es muy tóxico. Hay que tener mucho cuidado al manipularlo. No deben utilizarse pipetas de boca cuando haya que tomar soluciones de cianuro. f) Solución de clorhidrato de hidroxilamina: Disuélvanse 20 g de NH2OH·HC1 en agua y llévese hasta 100 ml. g) Solución de ditizona de reserva I: Véase sección 1070D.22b1). h) Solución de ditizona de trabajo: dilúyase la solución de ditizona de reser- va I con CHC13 para obtener una solución de trabajo de 10 μg/ml. Prepárese diariamente. i) Cloroformo, calidad ACS que sea «adecuada para su uso en el ensayo de ditizona». Para conseguir un CHC13 satisfactorio, añádase una cantidad muy pequeña de ditizona a una porción de CHCI3 en un tubo de ensayo tapado, con lo que se produce un color verde brillante; este color debe ser estable durante un día. j) Solución de ácido tartárico: Disuélvanse 20 g de H2C4H4O6 en agua y llévese hasta 1 l. Consérvese en frigorífico y utilícese cuando aún esté frío. k) Ácido clorhídrico, HC1 conc. l) Solución de indicador azul de timol: Disuélvanse 0,4 g de sal sódica de timolsulfonaftaleína en 100 ml de agua. m) Hidróxido de sodio: NaOH 6N. 4. Procedimiento a) Preparación de la curva estándar: Prepárese un blanco y una serie de patrones a partir de 1 a 10 μg tomando con la pipeta las cantidades apropiadas de solución de Cd patrón y pasándolos a embudos separadores. dilúyase hasta 25 ml y opérese como se indica en el apartado 4c. Trácese una curva de calibración. b) Tratamiento de muestras: Digiérase una muestra tal como se ha señalado en la sección 3030. Tómese con la pipeta un volumen de muestra digerida que contenga 1 a 10 μg Cd y pásese a un embudo separador; dilúyase a 25 ml si es necesario. Añádanse 3 gotas de azul de timol y ajústese el pH a 2,8 con NaOH 6N hasta el primer color amarillo permanente. c) Desarrollo de color, extracción y medida: Añádanse los reactivos en el siguiente orden, mezclando después de cada adición: 1 ml de solución de tartrato de sodio y potasio, 5 ml de solución NaOH-KNC I, 1 ml de solución MÉTODOS NORMALIZADOS 3-94 NH2OHHC1 y 15 ml de solución de ditizona I. Tápense los embudos y agítense durante un minuto, descargando la presión de vapor de los embudos a través de los tapones en vez de hacerlo con la espita. Pásese la capa de CHC13 a un segundo embudo que contenga 25 ml de solución de ácido tartárico fría. Añádanse 10 ml de CHC13 al primer embudo: agítese durante 1 minuto y decántese al segundo embudo. No hay que dejar que la capa acuosa entre en el segundo embudo. Háganse las dos extracciones inmediatamente después de añadir la ditizona, ya que el tiempo de contacto del CHC13 con el álcali fuerte debe reducirse al mínimo (el ditizonato de cadmio se descompone en contacto prolongado con álcali fuerte saturado de CHC13). Sacúdase el segundo embudo durante 2 minutos y descártese la capa de CHCI3. Añádanse 5 ml de CHC13, sacúdase 1 minuto y descártese la capa de CHCI3, haciendo la separación lo más ajustada posible. Añádanse en el siguiente orden 0,25 ml de solución de NH2OH·HC1 y 15,0 ml de solución de ditizona de trabajo. Añádanse 5 ml de solución II de NaOH-KCN. Sacúdase inmediatamente durante 1 minuto y pásese la capa de CHC1 3 a un tubo seco del fotómetro. Léase la absorbancia a 518 nm frente al blanco. Obténgase la concentración de Cd a partir de la curva de calibración. 5. Cálculos 6. Precisión y sesgo Se analizó en 44 laboratorios, empleando el método de ditizona y con una desviación relativa estándar de 24,6 por 100 y un error relativo de 6,0 por 100, una muestra sintética con un contenido de 50 μg Cd/1, 500 μg Al/1, 110 μg Cr/1, 470 μg Cu/1, 300 μg Fe/1, 70 μg Pb/l, 150 μg Ag/1 y 650 μg Zn/1. 7. Bibliografía SALTZMAN, B. E. 1953. Colorimetric micrometermination of cadmium with dithizone method. Anal. Chem. 25:493. GANOTES, J., E. LARSON & R. NAVONE. 1962. Suggested dithizone method for cadmium determination. J. Amer. Water Works Assoc. 54:852. 3-95 DETERMINACIÓN DE METALES 3500-Ca 3500-Ca A. 1. Incidencia y significación La presencia del calcio (el quinto entre los elementos en orden de abundancia) en los suministros de agua proviene de su paso a través o por encima de depósitos de caliza, dolomita, yeso y pizarras yesíferas. El contenido de calcio puede variar entre cero y varios centenares de mlligramos por litro, dependiendo del origen y tratamiento del agua. Las pequeñas concentraciones de carbonato de calcio evitan la corrosión de las tuberías metálicas por depositar una capa protectora. Por otro lado, cantidades apreciables de sales de calcio precipitan al calentar formando incrustaciones perjudiciales en calderas, tuberías y utensilios de cocina. En la sección 2330 se trata la saturación de carbonato de calcio. El calcio contribuye a la dureza total del agua. Para reducir el calcio y la dure* Aprobado por el Standard Methods Committee, 1985. 3500-Ca B. CALCIO* Introducción za asociada a él, se aplica un tratamiento de ablandamiento químico, osmosis inversa, electrodiálisis o intercambio iónico. 2. Selección del método El método de absorción atómica y el método de plasma de acoplamiento inductivo constituyen dos medios precisos para determinar el calcio. Los métodos de titulación con permanganato y EDTA dan buenos resultados en aplicaciones de control y rutinarias. La sencillez y rapidez del procedimiento de titulación con EDTA lo hacen preferible al método del permanganato. 3. Conservación de muestras Son suficientes las precauciones habituales siempre que se procure redisolver el carbonato de calcio que pueda precipitar al estancarse. Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, sección 3111B. 3500-Ca C. Método de plasma de acoplamiento inductivo Véase sección 3120. 3-96 MÉTODOS NORMALIZADOS 3500-Ca D. 1. Método titulométrico de EDTA Discusión general a) Principio: Cuando se añade EDTA (ácido etilendiaminotetracético o sus sales) al agua que contiene calcio y magnesio, aquél se combina primero con el calcio. El calcio se determina directamente con EDTA cuando el pH es lo suficientemente alto para que precipite el magnesio como hidróxido, utilizando un indicador que se combine con el calcio únicamente. Existen varios indicadores que originan un cambio de color cuando todo el calcio ha pasado a formar un complejo con el EDTA a un pH 12 a 13. b) Interferencia: En las condiciones de este ensayo, las siguientes concentraciones de iones no son origen de interferencia cuando se determina la dureza por calcio: Cu2+ 2 mg/1, Fe2+ 20 mg/1, Fe3+ 20 mg/1, Mn2+ 10 mg/1, Zn2 + 5 mg/1, Pb2+ 5 mg/1, Al3+ 5 mg/1 y Sn4 + 5 mg/1. El ortofosfato precipita el calcio al pH del ensayo. El estroncio y el bario dan interferencia positiva, y una alcalinidad por encima de 300 mg/1 puede ser la causa de un punto final indistinguible en las aguas duras. 2. Reactivos a) Hidróxido de sodio, NaOH 1N. b) Indicadores: Para la titulación de calcio se puede disponer de muchos indicadores. Algunos están descritos (véase bibliografía), otros son preparaciones comerciales que también pueden emplearse. El primer indicador de que se dispuso para detectar el punto final de calcio fue la murexida (purpurato de amonio), para cuyo uso se proporcionan indicadores en este procedimiento. Las personas que tengan dificultad para reconocer el punto final con murexida pueden encontrar que es mejor el azul negro de eriocromo R (índice de color número 202) o el azul oscuro de solocromo, ya que el cambio de color es del rojo al azul puro. El azul negro de eriocromo R es ácido sodio-1(2-hidroxi-1 -naftilazo)-2-naftol-4-sulfónico. También se pueden utilizar otros indicadores específicamente diseñados como detectores del punto final en la titulación con EDTA del calcio. 1) Indicador murexida (purpurato de amonio): Este indicador cambia de rosa a púrpura en el punto final. Prepárese disolviendo 150 mg del colorante en 100 g de etilenglicol absoluto. Las soluciones acuosas del colorante no son estables más de 1 día. Una mezcla molida del polvo colorante y cloruro sódico (NaCl) resulta una forma estable del indicador. Prepárese mezclando 200 mg de murexida con 100 g de NaCl sólido y tritúrese la mezcla hasta 40 a 50 mallas. Titúlese inmediatamente de añadir el indicador, ya que éste es inestable en condiciones alcalinas. Facilítese el reconocimiento del punto final preparando un blanco de comparación que contenga 2,0 ml de solución de NaOH, 0,2 g de mezcla sólida de indicador (o de 1 a 2 gotas si se emplea una solución) y suficiente reactivo de titulación EDTA estándar (0,05 a 0,10 ml) para producir un cambio de color. 2) Indicador azul negro de eriocromo R: Prepárese una forma estable del indicador triturando juntos en un mortero 200 mg del colorante pulverizado y 100 g de NaCl sólido hasta 40 a 50 mallas. Guárdese en un frasco herméticamente cerrado. Utilícense 0,2 g de la mezcla molida para la titulación de la misma forma que para el indicador murexida. Durante la titulación, el color cambia de rojo a azul puro, pasando por púrpura y púrpura azulado, sin trazas de tonos rojizos o púrpuras. El pH de algunas aguas (no todas) debe elevarse a 14 (en vez de a 12-13) empleando NaOH 8N para conseguir un buen cambio de color. 3-97 DETERMINACIÓN DE METALES c) Reactivo de titulación estándar EDTA, 0.01M; Prepárese un reactivo de titulación estándar EDTA, tal como se ha descrito para el método de determinación de dureza total con EDTA (sección 2340). El reactivo de titulación estándar EDTA 0,0100M equivale a 400,8 μg Ca/1,00 ml. donde: A = ml de reactivo de titulación para la muestra, y B = mg de CaCO 3 equivalentes a 1,00 ml de reactivo de titulación EDTA en el punto final de indicador para el calcio. 3. Procedimiento a) Tratamiento previo de muestras de agua contaminada y aguas residuales: Sígase el procedimiento descrito en la sección 3030E o I. b) Preparación de muestras: Titúlese inmediatamente después de añadir el álcali y el indicador, debido al elevado pH empleado en este procedimiento. Utilícense 50,0 ml de muestra o una porción más pequeña diluida hasta 50 ml de manera que el contenido en calcio sea, aproximadamente, de 5 a 10 mg. Analícense las aguas duras, con alcalinidad superior a 300 mg CaCO3/l, tomando una pequeña porción y diluyendo hasta 50 ml, o neutralizando la alcalinidad con ácido, hirviendo un minuto y enfriando antes de comenzar la titulación. c) Titulación: Añádanse 2,0 ml de solución de NaOH o un volumen suficiente para producir un pH de 12 a 13. Agítese. Añádanse 0,1 a 0,2 g de la mezcla de indicador seleccionada (o de 1 a 2 gotas si se emplea solución). Añádase poco a poco el reactivo de titulación EDTA, agitando continuamente, hasta el apropiado punto final. Cuando se utiliza murexida, compruébese el punto final por adición de 1 o 2 gotas más de reactivos de titulación para cerciorarse de que no hay más cambio de color. 4. Cálculos 5. Precisión y sesgo En 44 laboratorios se analizó una muestra sintética que contenía 108 mg Ca/1, 82 mg Mg/1, 3,1 mg K/l, 19,9 mg Na/1, 241 mg CP¯/1, 1,1 mg NO3- -N/1, 0,25 mg NO-2 -N/1, 259 mg SO 2-4 /1 y 42,5 mg de alcalinidad total/1 (a la que contribuye el NaHCO3) en agua destilada, empleando el método de titulación con EDTA, con una desviación relativa estándar de 9,2 por 100 y un error relativo de 1,9 por 100. 6. Bibliografía DIEHL, H. & J. L. ELLINGBOE. 1956. Indicator for titration of calcium in the presence of magnesium using disodium dihydrogen ethylenediamine tetraacetate. Anal. Chem. 28:882. PATTON, J. & W. REEDER. 1956. New indicator for titration of calcium with (ethylenedinitrilo) tetraacetate. Anal. Chem. 28:1026. HILDEBRAND, G. P. & C. N. REILLEY. 1957. New indicator for complexometric titration of calcium in the presence of magnesium. Anal. Chem. 29:258. SCHWARZENBACH, G. 1957. Complexometric Titrations. Interscience Publishers, Nueva York. FURMAN, N. H. 1962. Standard Methods of Chemical Analysis, 6.a ed. D. Van Nostrand Co., Inc., Princeton, Nueva Jersey. KATZ, H. & R. NAVONE, 1964. Method for simultaneous determination of calcium and magnesium. J. Amer. Water Works Assoc. 56:121. 3-98 MÉTODOS NORMALIZADOS 3500-Ca E. 1. Método titulométrico de permanganato Discusión general a) Principio: El oxalato de amonio precipita cuantitativamente el calcio en forma de oxalato de calcio. Un exceso de oxalato resuelve los efectos adversos del magnesio. Sólo se obtiene una óptima formación de cristales y una oclusión mínima cuando se lleva lentamente el pH hasta el valor deseado. Esto se realiza en dos etapas, interviniendo la digestión para acelerar la formación de cristales simiente. El oxalato de calcio precipitado se disuelve en ácido y se titula con pérmanganato. La cantidad de permanganato requerida para oxidar el oxalato es proporcional a la cantidad de calcio. b) Interferencia: La muestra deberá estar libre de cantidades interfirientes de estroncio, sílice, aluminio, hierro, manganeso, fosfato y materia suspendida. El estroncio puede precipitar como oxalato y dar lugar a resultados altos. En este caso, determínese el estroncio por fotometría de llama y aplíquese la corrección apropiada. Elimínese la interferencia de la sílice por el clásico procedimiento de deshidratación. Precipítese el aluminio, hierro y manganeso con hidróxido amónico después de tratamiento con persulfato. Precipítese el fosfato como sal férrica. Elimínese la materia suspendida por centrifugación o filtración a través de papel de filtro, vidrio sinterizado o membrana de acetato de celulosa (véase Sólidos, sección 2540). 2. Instrumental a) Bomba de vacío u otra fuente de vacío. b) Matraces filtrantes. c) Crisoles filtrantes: Se recomiendan crisoles de 30 ml de porosidad media. Se pueden utilizar crisoles de vidrio o de porcelana. Los crisoles de estructura completamente porosa son difíciles de lavar cuantitativamente. 3. Reactivos a) Solución de indicador rojo de metilo: Disuélvase 0,1 g de sal sódica de rojo de metilo y dilúyase hasta 100 ml con agua destilada. b) Ácido clorhídrico, HC1 1 + 1. c) Solución de oxalato de amonio: Disuélvanse 10 g de (NH4)2C2O4·H2O en 250 ml de agua destilada. Fíltrese si es necesario. d) Hidróxido de amonio, NH4OH 3N: Añádanse 240 ml de NH4OH conc. a aproximadamente 700 ml de agua destilada y dilúyase hasta 1 l. Fíltrese antes de utilizarlo para eliminar los copos de sílice suspendidos. e) Hidróxido de amonio, 1 + 99. f) Reactivos especiales para eliminar la interferencia de aluminio, hierro y manganeso: 1) Persulfato de amonio, sólido. 2) Solución de cloruro de amonio: Disuélvanse 20 g de NH4C1 en 1 l de agua destilada. Fíltrese si es necesario. g) Oxalato de sodio: Utilícese Na2C2O4 de calidad patrón primario. Póngase a secar a 105 °C durante toda la noche y consérvese en desecador. h) Ácido sulfúrico, H2SO4 1 + 1. i) Reactivo de titulación permanganato potásico estándar, 0,01M: Disuélvanse 1,6 g de KMnO4 en 1 l de agua destilada. Guárdese en un frasco de cristal topacio tapado y déjese por lo menos una semana. Decántese cuidadosamente o tómese con una pipeta el sobrenadante sin agitar el sedimento. Estandarícese frecuentemente esta solución por el siguiente procedimiento (la solución patrón de DETERMINACIÓN DE METALES KMnO4 es exactamente 0,0100M, y equivale a 1,002 mg Ca/1,00 ml): Pésese con precisión 0,1 mg de varias muestras de 100 a 200 mg de Na2C2O4 en vasos de 400 ml. Añádase a cada vaso 100 ml de agua destilada y agítese para que se disuelva. Añádanse 10 ml de H2SO4 1 + 1 y caliéntese rápidamente entre 90 y 95 °C. Titúlese rápidamente con la solución de permanganato que se va a estandarizar, mientras se agita, hasta un punto final de un ligero color rosa que ha de persistir durante 1 minuto, por lo menos. No hay que dejar que la temperatura descienda por debajo de 85 °C. Si es necesario, caliéntese el contenido del vaso durante la titulación; 100 mg consumirán aproximadamente 30 ml de solución. Opérese con un blanco a base de agua destilada y H2SO4. siendo: A = ml de reactivo de titulación para la muestra, y B = ml de reactivo de titulación para el blanco. Sáquese la media de los resultados de varias titulaciones. 4. Procedimiento a) Tratamiento previo de muestras de aguas contaminadas y residuales: Sígase el procedimiento descrito en la sección 3030E o en la 3030I. b) Tratamiento de la muestra: Utilícense 200 ml de muestra con un contenido en Ca no superior a 50 mg (o una porción menor diluida hasta 200 ml). Si existen sustancias que interfieren, procédase como sigue: 1) Eliminación de la interferencia de sílice. Elimínense las cantidades de sílice 3-99 que interfieren por el procedimiento gravimétrico descrito en Sílice, sección 4500Si. Descártese el precipitado de sílice y consérvese el filtrado para eliminar las cantidades que interfieren de los óxidos combinados descritos en el siguiente apartado c. Si no hay óxidos combinados, pásese al apartado d. 2) Eliminación de la interferencia de óxidos combinados. Elimínense las cantidades que interfieren de aluminio, hierro y manganeso, concentrando el filtrado procedente de la determinación gravimétrica de sílice hasta 120 a 150 ml. Añádase el suficiente HC1 de manera que el filtrado procedente de la eliminación de sílice contenga al menos 10 ml de HC1 conc. Añádanse de 2 a 3 gotas de indicador rojo de metilo y NH4OH 3N hasta que el color del indicador cambie al amarillo. Añádase 1 g de persulfato de amonio; cuando comience la ebullición, añádase cuidadosamente NH4OH 3N hasta que la solución se haga ligeramente alcalina y el vapor desprenda un olor a amoníaco claro, aunque no fuerte. Ensáyese con papel tornasol. Hiérvase durante 1 a 2 minutos y déjese reposar 10 minutos hasta que los hidróxidos coagulen, pero no más tiempo. Fíltrese el precipitado y lávese tres o cuatro veces con solución de NH4C1. Trátese el filtrado tal como se describe a continuación. c) Ajuste del pH de la muestra: A 200 ml de muestra, con un contenido en Ca no superior a 50 mg, o a una porción más pequeña diluida hasta 200 ml, añádanse 2 ó 3 gotas de solución de indicador rojo de metilo. Neutralícese con HC1 1 + 1 y hiérvase durante 1 minuto. Añádanse 50 ml de solución de (NH4)2C2O4 · H2O y, si se forma precipitado, añádase justo el suficiente HC1 1 + 1 para redisolverlo. d) Precipitación de oxalato cálcico: Manténgase la solución justo por debajo del punto de ebullición, añádase gota a gota, con una bureta, NH4OH 3N, agi- MÉTODOS NORMALIZADOS 3-100 tando constantemente. Continúese la adición hasta que la solución se enturbie bastante (se requieren aproximadamente 5 ml). Digiérase durante 90 minutos a 90 °C. Fíltrese, preferiblemente a través de crisol filtrante, empleando succión. Lávese inmediatamente con NH 4 OH 1 + 99. Aunque no es necesario pasar todo el precipitado al crisol filtrante, elimínese todo el exceso de (NH4)2C2O4·H2O del vaso. (Si hay que determinar magnesio por gravimetría, colóquense a un lado el filtrado combinado y las aguas de lavado con este propósito.) e) Titulación de oxalato cálcico: Colóquese el crisol filtrante de costado en un vaso y cúbrase con agua destilada. Añádanse 10 ml de H2SO4, agitando al mismo tiempo, y caliéntese rápidamente entre 90 y 95 °C. Titúlese rápidamente con reactivo de titulación KMnO4 hasta un punto final ligeramente rosa que persiste durante por lo menos 1 minuto. No hay que dejar que la temperatura disminuya por debajo de 85 °C; si es necesario, caliéntese el contenido del vaso durante la titulación. Agítese el crisol suficientemente para asegurar la reacción de todo el oxalato. Opérese con un blanco utilizando un vaso y un crisol limpios, 10 ml de H2SO 4 1 + 1, y aproximadamente el mismo volumen de agua destilada utilizado en la titulación de la muestra. 3500-Cs 3500-Cs A. El cesio se encuentra en la mayoría de aguas naturales en proporciones traza (< lμg/1). Las aguas termales pueden 5. Cálculos siendo: A = ml de reactivo de titulación para la muestra, B = ml de reactivo de titulación para el blanco, y M = molaridad de KMnO 4 . 6. Precisión y sesgo En seis laboratorios se analizó una muestra sintética con un contenido de 108 mg Ca/1, 82 mg Mg/1, 3,1 mg K/l, 19,9 mg Na/1, 241 mg Cl¯/1, 1,1 mg NO3- -N/I, 0,25 mg NO-2 -N/1, 259 mg SO 2-4 /1 y 42,5 mg de alcalinidad total/1 (a la que contribuye el NaHCO 3 ) en agua destilada por el método titulométrico de KMnO4, con una desviación relativa estándar de 3,5 por 100 y un error relativo de 2,8 por 100. 7. Bibliografía KOLTHOFF, I. M., E. J. MEEHAN , E. B. S ANDELL & S. BRUCKENSTEIN. 1969. Quantitative Chemical Analysis, 4.a ed. MacMillan Co., Nueva York. CESIO Introducción contener valores mucho más altos (hasta 5 mg/1). 3-101 DETERMINACIÓN DE METALES 3500-Cs B. Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, sección 3111B. 3500-Cr 3500-Cr A. 1. Incidencia Según los informes, las aguas potables de los Estados Unidos, tienen una concentración de cromo hexavalente que oscila entre 3 y 40 μg/l con una media de 3,2 μg/1. En los procesos industriales se utilizan mucho las sales de cromo, y pueden pasar al suministro de agua a través de los desechos industriales. Frecuentemente se añaden cromatos al agua de refrigeración para control de la corrosión. El cromo puede encontrarse en los suministros de agua tanto en estado hexavalente como trivalente, aunque la forma trivalente raramente aparece en el agua potable. 2. Selección del método Utilícese el método colorimétrico para determinar el cromo hexavalente en agua * Aprobado por el Standard Methods Committee, 1985. CROMO* Introducción natural o tratada para su potabilidad. Empléese el método espectrométrico de absorción atómica electrotérmica (horno de grafito) para determinar niveles bajos de cromo total (< 50 μg/1) en aguas naturales y residuales. Utilícese el método espectrométrico de absorción atómica de llama o el método de plasma de acoplamiento inductivo para medir concentraciones del orden de miligramos por litro. 3. Manipulación de las muestras Si únicamente se trata de determinar el metal disuelto, fíltrese la muestra a través de un filtro de membrana de 0,45 μm en el momento de su toma. Después de la filtración, acidúlese el filtrado con ácido nítrico conc. (HNO3) hasta pH < 2. Si lo que se desea es el contenido de cromo total, acidúlese la muestra sin filtrar con HNO3 conc. hasta pH < 2 en el momento de tomarla. MÉTODOS NORMALIZADOS 3-102 3500-Cr B. Método de absorción atómica para cromo total Véase método espectrométrico de absorción atómica de llama, secciones 3111B y C, y método espectrométrico de absor- 3500-Cr C. ción atómica electrotérmica, sección 3113. Método de plasma de acoplamiento inductivo Véase sección 3120. 3500-Cr D. 1. Método colorimétrico Discusión general a) Principio: Este procedimiento mide únicamente el cromo hexavalente (Cr6 + ). Por tanto, para determinar el cromo total hay que convertir todo el cromo a la forma hexavalente oxidando con permanganato potásico. El cromo hexavalente se determina colorimétricamente por una reacción con difenilcarbazida en solución acida. Se produce un color rojovioleta de composición desconocida. La reacción es muy sensible, siendo la capacidad de absorción molar, basada en el cromo, de aproximadamente 40.000 1 g -1 c m - 1 a 540 nm. Para determinar el cromo total, digiérase la muestra con una mezcla acida sulfúrico-nítrico y oxídese después con permanganato potásico antes de la reacción con la difenilcarbazida. b) Interferencias: La reacción con la difenilcarbazida es prácticamente específica para el cromo. Las sales de molibdeno hexavalente y de mercurio reaccionarán para formar color con el reactivo, pero las intensidades son mucho más ba- jas que para el cromo al pH especificado. Se pueden tolerar concentraciones de hasta 200 mg Mo o Hg/1. El vanadio interfiere fuertemente, pero las concentraciones hasta 10 veces las del cromo no causarán problemas. La interferencia potencial de permanganato se elimina por reducción previa con azida. El hierro, a concentraciones superiores a 1 mg/1, puede producir un color amarillo, pero el color de ion férrico (Fe3 + ) no es fuerte, y normalmente no se encuentran dificultades si se mide la absorbancia fotométricamente a la longitud de onda apropiada. Las cantidades de molibdeno, vanadio, hierro y cobre que interfieren pueden eliminarse por extracción de los cupferratos de estos metales con cloroformo (CHC13). Existe un método de extracción, pero no se debe utilizar a menos que sea necesario, pues el cupferrón y CHCI3 residuales en la solución acuosa complican la posterior oxidación. Por tanto, sígase la extracción por tratamiento adicional de ácido fumante para descomponer estos compuestos. 3-103 DETERMINACIÓN DE METALES 2. Instrumental a) Equipo de colorimetría: Se requiere uno de los siguientes: 1) Espectrofotómetro, para utilizarlo a 540 nm con un trayecto luminoso de 1 cm o más largo. 2) Fotómetro de filtro, con un trayecto luminoso de 1 cm o más largo y equipado con un filtro amarillo verdoso que tiene una transmitancia máxima próxima a 540 nm. b) Embudos de separación, 125 ml, forma Squibb, con espita y tapón de vidrio o TFE. 3. Reactivos Utilícese agua redestilada para preparar los reactivos. a) Solución de cromo de reserva: Disuélvanse 141,4 mg de K2Cr2O2 en agua y dilúyase hasta 1.000 ml; 1,00 ml = 50,0 μg Cr. b) Solución de cromo patrón: Diluyanse 10,0 ml de solución de cromo de reserva hasta 100 ml; 1,00 ml = 5,00 μg Cr. c) Ácido nítrico, HNO3 conc. d) Ácido sulfúrico, H2SO4 1 + 1. e) Solución de indicador naranja de metilo. f) Peróxido de hidrógeno, H2O2, al 30 por 100. g) Agua redestilada: Destílese el agua que se vuelve a destilar en un aparato hecho completamente de vidrio. h) Hidróxido amónico, NH4PH conc. i) Solución de permanganato potásico: Disuélvanse 4 g de KMnO4 en 100 ml de agua. j) Solución de azida sódica: Disuélvanse 0,5 g de NaN3 en 100 ml de agua. k) Solución de fenilcarbazida: Disuélvanse 250 mg de 1,5-difenilcarbazida (1,5-difenilcarbohidrazida) en 50 ml de acetona. Consérvese en frasco topacio. Descártese cuando la solución se decolora. l) Cloroformo, CHC13: Evítese, o redestílese, el material que viene en recipientes metálicos o con tapones de forro metálico. m) Solución de cupferrón: Disuélvanse 5 g de C6H5N(NO)ONH4 en 95 ml de agua. n) Ácido fosfórico, H3PO4 conc. o) Ácido sulfúrico, H2SO4 0,2N: Dilúyanse 17 ml de H2SO4 6N hasta 500 ml con agua. 4. Procedimiento a) Preparación de la curva de calibración: Para compensar posibles pérdidas ligeras de cromo durante la digestión u otras operaciones analíticas, trátense los patrones de cromo por el mismo procedimiento que la muestra. Para ello, llévense con la pipeta volúmenes medidos de solución de cromo patrón (5 μg/ml) que varían entre 2,00 y 20,0 ml, para obtener patrones de 10 a 100 μg Cr, a vasos o matraces erlenmeyer de 250 ml. Según el tratamiento previo empleado en el siguiente apartado b, procédase con el subsiguiente tratamiento de patrones como si fueran muestras, realizándose el tratamiento de cupferrón de los patrones si ha sido requerido para las muestras. Desarróllese el color como en el caso de las muestras, pásese una porción adecuada de cada solución coloreada a una célula de absorción de 1 cm y mídase la absorbancia a 540 nm. Utilícese agua destilada como referencia. Corríjanse las lecturas de absorbancia de los patrones restando la absorbancia de un blanco de reactivo que ha sido trabajado con el mismo método. Trácese una curva de calibración llevando valores de absorbancia corregidos frente a microgramos de cromo en un volumen final de 102 ml. b) Tratamiento de la muestra: Si la muestra se ha filtrado y acidulado y sólo 3-104 se desea determinar el cromo hexavalente, sígase el apartado 4e. Si se desea determinar el cromo disuelto total y hay cantidades de molibdeno, vanadio, cobre o hierro que interfieren, sígase el apartado 4c. Si no existen interferencias, sígase el apartado 4d. Si la muestra está sin filtrar y se desea determinar el cromo total, digiérase con HNO3 y H2SO4 como se indica en la sección 3030G. Si existen interferencias, síganse los apartados 4c, 4d y 4e. Si no hay interferencias, procédase como en los apartados 4d y 4e. c) Eliminación de molibdeno, vanadio, hierro y cobre con cupferrón: Llévese con la pipeta una porción de muestra digerida que contenga entre 10 y 100 μg Cr a un embudo de separación de 125 ml. Dilúyase a aproximadamente 40 ml con agua destilada y enfríese en baño de hielo. Añádanse 5 ml de solución de cupferrón enfriada con hielo, sacúdase bien y déjese reposar en el baño de hielo durante 1 minuto. Extráigase en el embudo separador con tres porciones sucesivas de 5 ml de CHC13; sacúdase cuidadosamente cada porción con la solución acuosa, déjense separar las capas y retírese y descártese el extracto de CHC13. Pásese la solución acuosa extraída a un erlenmeyer de 125 ml. Lávese el embudo de separación con una pequeña cantidad de agua destilada y añádase el agua del lavado al matraz. Llévese a ebullición durante aproximadamente 5 minutos para volatizar CHC13 y enfríese. Añádanse 5 ml de HNO3 y el H2SO4 suficiente para que haya aproximadamente 3 ml. Hiérvanse las muestras hasta la aparición de humos de SO3. Enfríese ligeramente, añádanse cuidadosamente 5 ml de HNO3 y llévese de nuevo a ebullición hasta el desprendimiento de humos para completar la descomposición de materia orgánica. Enfríese, lávense las paredes laterales del matraz y llévese otra vez a ebullición hasta desprendimiento de humos de SO3 para eliminar MÉTODOS NORMALIZADOS todo el HNO3. Enfríese y añádanse 25 ml de agua. d) Oxidación de cromo trivalente: Llévese con la pipeta a un erlenmeyer de 125 ml una porción de muestra digerida, con o sin las interferencias eliminadas, y que contenga entre 10 y 100 μg Cr. Empleando indicador naranja de metilo, añádase NH4OH conc. hasta que la solución sea justo básica al naranja de metilo. Añádase H2SO4 1 + 1, gota a gota, hasta que se vuelva acida, más 1 ml (20 gotas) adicional. Ajústese el volumen a aproximadamente 40 ml, añádase un pedacito de plato poroso y caliéntese a ebullición. Añádanse dos gotas de solución de KMnO 4 para obtener un color rojo oscuro. Si se empalidece, añádase KMnO4, gota a gota, hasta mantener un exceso de aproximadamente dos gotas. Hiérvase 2 minutos más. Añádase 1 ml de solución de NaN3 y continúese una suave ebullición. Si el color rojo no desaparece por completo después de una ebullición de aproximadamente 30 segundos, añádase otro ml de solución de NaN3. Continúese la ebullición un minuto más después de desaparecido el color por completo, y enfríese a continuación. Añádanse 0,25 ml (cinco gotas) de H3PO4. e) Desarrollo del color y medida: Utilícese H2SO4 0,2N y un pHmetro para ajustar la solución a pH 1,0 ± 0,3. Pásese la solución a un matraz volumétrico, dilúyase hasta 100 ml y mézclese. Añádanse 2,0 ml de solución de difenicarbazida, mézclese y déjese reposar 5 a 10 minutos para el total desarrollo de color. Pásese una porción apropiada a una célula de absorción de 1 cm y mídase su absorbancia a 540 nm. Utilícese agua destilada como referencia. Corríjase la lectura de la absorbancia restando la absorbancia de un blanco tratado con el mismo método (véase la nota a continuación). Determínense los microgramos de cromo presentes a partir de la absorbancia corregida por referencia a la curva de calibración. 3-105 DETERMINACIÓN DE METALES NOTA: Si la solución está turbia después de dilución a 100 ml en el anterior apartado e, tómese una lectura de absorbancia previa a la adición del reactivo carbazida y corríjase la lectura de absorbancia de la solución coloreada final restando la absorbancia medida previamente. una muestra sintética con 110 μg Cr/1, 500 μg Al/1, 50 μg Cd/1, 470 μg Ca/1, 300 μg Fe/1, 70 μg Pb/1, 120 μg Mn/1, 150 μg Ag/1 y 650 μg Zn/1 en agua destilada. La desviación relativa estándar era de 47,8 por 100 y el error relativo de 16,3 por 100. 7. Bibliografía 5. Cálculos donde: A = ml de muestra original, y B = ml de porción de 100 ml de muestra digerida. 6. Precisión y sesgo Se determinó en 31 laboratorios el cromo disuelto (trivalente y hexavalente) en 3500-Co 3500-Co A. R OWLAND , G. P., J R . 1939. Photoelectric colorimetry—Optical study of permanganate ion and of chromium-diphenylcarbazide system. Anal. Chem. 11:442. SALTZMAN, B. E. 1952. Microdetermination of chromium with diphenylcarbazide by permanganate oxidation. Anal. Chem. 24:1016. URONE, P. F. 1955. Stability of colorimetric reagent for chromium, 5-diphenylcarbazide, in various solvents. Anal. Chem. 27:1354. ALLEN, T. L. 1958. microdetermination of chromium with 1,5-diphenylcarbohydrazide. Anal. Chem. 30:447. SANDELL, E. B. 1959. Colorimetric Determination of Traces of Metals, 3.a ed. Interscience Publishers, Nueva York. COBALTO Introducción 1. Incidencia 2. Selección del método El cobalto se encuentra en las aguas naturales normalmente en una proporción < 10 μg/1. En las aguas residuales puede haber proporciones más altas. Utilícese el método espectrométrico de absorción atómico de llama o de horno, o el método de plasma de acoplamiento inductivo. 3-106 3500-Co B. MÉTODOS NORMALIZADOS Método espectrométrico de absorción atómica Véase el método espectrométrico de absorción atómica de llama, secciones 3111B y C, y el método espectrométrico 3500-Co C. de absorción atómica electrotérmica, sección 3113. Método de plasma de acoplamiento inductivo Véase sección 3120. 3500-Cu 3500-Cu A. 1. Incidencia y significación Las sales de cobre se utilizan en los sistemas de suministro de agua para control de crecimientos biológicos en depósitos y tuberías de distribución y para catalizar la oxidación de manganeso. La corrosión de las aleaciones que contienen cobre en accesorios de tuberías puede introducir cantidades medibles de cobre en el agua de un sistema de conducción. El cobre es esencial para los seres humanos; se calcula en 2,0 mg la necesidad diaria de cobre para una persona adulta. * Aprobado por el Standard Methods Committee, 1988. COBRE* Introducción 2. Selección del método Se recomiendan los métodos espectrométrico de absorción atómica, de plasma de acoplamiento inductivo y de la neocuproína, debido a que no presentan interferencias. El método de la batocuproína puede ser útil para aguas potables. 3. Toma de muestras y almacenamiento Los iones de cobre tienden a adsorberse en la superficie de los recipientes de las muestras. Por esta razón se deben analizar las muestras lo antes posible tras su toma. Si es necesario almacenarlas, utilícense 0,5 ml de HC1 1 + 1/100 ml de muestra para evitar esta adsorción. 3-107 DETERMINACIÓN DE METALES 3500-Cu B. Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, secciones 3111B y C, y método espectrométrico de 3500-Cu C. absorción atómica electrotérmica, sección 3113. Método de plasma de acoplamiento inductivo Véase sección 3120. 3500-Cu D. 1. Método de la neocuproína Discusión general a) Principio: El ion cuproso (Cu+) en solución neutra o ligeramente acida reacciona con 2,9-dimetil-l,10-fenantrolina (neocuproína) para formar un complejo en el que se unen dos moles de neocuproína a un mol de ion Cu + . El complejo se puede extraer por cierto número de disolventes orgánicos entre los que se incluyen una mezcla de cloroformo-metanol (CHCI3-CH3OH), para dar una solución amarilla con una capacidad de absorción molar de aproximadamente 8.000 a 457 nm. La reacción es virtualmente específica para cobre; el color sigue la ley de Beer hasta una concentración de 0,2 mg Cu/25 ml de disolvente; se obtiene un desarrollo completo del color cuando el pH de la solución acuosa está entre 3 y 9; el color es estable en CHC13CH3OH durante varios días. Se trata la muestra con clorhidrato de hidroxilamina para reducir los iones cúpricos a iones cuprosos. Se emplea citrato sódico para complejar los iones metálicos que podían precipitar al elevar- se el pH. Se ajusta el pH entre 4 y 6 con NH4OH, se añade una solución de neocuproína en metanol, y el complejo resultante se extrae con CHC13. Después de diluir el CHC13 hasta volumen exacto con CH3OH, se mide la absorbancia de la solución a 457 nm. b) Interferencia: Pueden interferir cantidades grandes de cromo y de estaño. Evítese la interferencia de cromo añadiendo ácido sulfuroso para reducir el ion cromato y el crómico complejo. En presencia de mucho estaño o de cantidades excesivas de otros iones oxidantes, utilícense hasta 20 ml adicionales de solución de clorhidrato de hidroxilamida. Interfieren también cianuro, sulfuro y materia orgánica, pero pueden ser eliminados por digestión. c) Concentración mínima detectable: La concentración mínima detectable que corresponde a 0,01 de absorbancia o 98 por 100 de transmitancia es de 3 μg Cu cuando se emplea una célula de 1 cm, y de 0,6 μg Cu cuando se emplea una célula de 5 cm. MÉTODOS NORMALIZADOS 3-108 2. Instrumental a) Equipo calorimétrico: Se necesita uno de los siguientes: 1) Espectrofotómetro, para usarlo a 457 nm, que proporcione un trayecto luminoso de 1 cm o más largo. 2) Fotómetro de filtro, que proporcione un trayecto luminoso de 1 cm o más largo y equipado con un filtro violeta de banda estrecha que tiene una transmitancia máxima en el intervalo de 450 a 460 nm. b) Embudos de separación, 125 ml, forma Squibb, con llave y tapón de vidrio o de TFE. 3. Reactivos a) Agua redestilada, libre de cobre: Como la mayor parte del agua destilada ordinaria contiene cantidades detectables de cobre, utilícese agua redestilada, preparada por destilación del agua destilada una vez en un destilador de vidrio resistente, o agua destilada pasada a través de una unidad de intercambio iónico, para preparar todos los reactivos y soluciones. b) Solución de cobre de reserva: Añádanse 10 ml de agua y 5 ml de HNO3 conc. a 200,0 mg de lámina o alambre de cobre electrolítico pulimentado en un erlenmeyer de 250 ml. Cuando la reacción se haya hecho más lenta, caliéntese suavemente para completar la disolución del cobre y llévese a ebullición para expulsar los óxidos de nitrógeno, adoptando precauciones para evitar pérdidas de cobre. Enfríese, añádanse aproximadamente 50 ml de agua, pásese cuantitativamente a un matraz volumétrico de 1 l y dilúyase con agua hasta la señal; 1 ml = 200 μg Cu. c) Solución de cobre patrón: Diluyanse con agua 50,00 ml de solución de cobre de reserva hasta 500 ml; 1,00 ml = 20,0 μg Cu. d) Ácido sulfúrico, H2SO4 conc. e) Solución de clorhidrato de hidroxilamina: Disuélvanse 50 g de NH2OHHC1 en 450 ml de agua. f) Solución de curato de sodio: Disuélvanse 150 g de Na3C6H5O7·2H2O en 400 ml de agua. Añádanse 5 ml de solución de NH2OHHC1 y 10 ml de reactivo neocuproína. Extráigase con 50 ml de CHC13 para eliminar las impurezas de cobre y descártese la capa de CHC13. g) Hidróxido amónico, NH4OH 5N: dilúyanse con agua 330 ml de NH4OH conc. (28-29 por 100) hasta 1.000 ml. Guárdese en frasco de polietileno. h) Papel de rojo Congo u otro papel de ensayo de pH que presente un cambio de color entre pH 4 y 6. i) Reactivo de neocuproína: Disuélvanse 100 mg de hemihidrato de 2,9dimetil-l,10-fenantrolina* en 100 ml de metanol. Esta solución es estable durante un mes o más, en condiciones normales de almacenamiento. j) Cloroformo, CHC13: Evítese o redestílese el material que llega en recipientes con tapa forrada de metal. k) Metanol, CH3OH, calidad reactivo. i) Ácido nítrico, HNO3 conc. m) Ácido clorhídrico, HC1 conc. 4. Procedimiento a) Preparación de la curva de calibración: Llévense con la pipeta 50 ml de agua a un embudo de separación de 125 ml para utilizarlos como blanco de reactivo. Prepárense patrones llevando con la pipeta 1,00 a 10,00 ml (20,0 a 200 μg Cu) de solución de cobre patrón a una serie de embudos de separación de 125 ml y dilúyase con agua hasta 50 ml. Añádase 1 ml de H2SO4 conc. y utilícese el procedimiento de extracción indicado en el apartado siguiente 4b. * GFS Chemical Company, Columbus, Ohio, o equivalente. 3-109 DETERMINACIÓN DE METALES Trácese una curva de calibración llevando la absorbancia frente a microgramos de cobre. Para preparar una curva de calibración para menores cantidades de cobre, dilúyanse 10,0 ml de solución de cobre patrón hasta 100 ml. Trátense volúmenes de 1,00 a 10,00 ml de este patrón diluido por el procedimiento antes descrito, pero utilícense células de 5 cm para medir la absorbancia. b) Tratamiento de la muestra: Pásense 100 ml de muestra a un vaso de 250 ml, añádase 1 ml de H2SO4 conc. y 5 ml de HNO3 conc. Añádase un poco de plato poroso y evapórese cuidadosamente sobre placa caliente hasta desprendimiento de humos blancos densos de SO3. Si la solución queda coloreada, enfríese, añádanse otros 5 ml de HNO3 conc. y evapórese de nuevo hasta que aparezcan humos blancos densos. Repítase, si es necesario, hasta que la solución se vuelva incolora. Enfríese, añádanse aproximadamente 80 ml de agua y llévese a ebullición. Enfríese y fíltrese en un matraz volumétrico de 100 ml. Llévese hasta 100 ml con agua utilizando la mayor parte de los lavados de vaso y filtro. Llévense con la pipeta 50,0 ml u otra porción adecuada que contenga de 4 a 200 μg de Cu, desde la solución obtenida del tratamiento preliminar a un embudo de separación de 125 ml. dilúyase con agua, si es necesario, hasta 50 ml. Añádanse 5 ml de solución de NH2OH·HC1 y 10 ml de solución de citrato sódico y mézclese cuidadosamente. Ajústese el pH a aproximadamente 4, añadiendo incrementos de 1 ml de NH4OH hasta que el papel rojo Congo dé un rojo definido (otros papeles indicadores de pH dan un valor entre 4 y 6). Añádanse 10 ml de reactivo neocuproína y 10 ml de CHC13. Tápese y sacúdase vigorosamente durante 30 segundos o más para extraer el complejo cobreneocuproína con el CHC13. Déjese sepa- rar la mezcla en dos capas y llévese la capa inferior de CHC13 a un matraz volumétrico de 25 ml, teniendo cuidado de no coger nada de la capa acuosa. Repítase la extracción de la capa acuosa con 10 ml de CHC13 adicionales y combínense los extractos. Dilúyanse los extractos combinados hasta 25 ml con CH3OH, ciérrese y mézclese cuidadosamente. Pásese una porción apropiada de extracto a una célula de absorción adecuada (1 cm para 40 a 200 μg Cu; 5 cm para cantidades menores) y mídase la absorbancia a 457 nm o con un filtro de 450 a 460 nm. Utilícese un blanco de muestra preparado por tratamiento de 50 ml de agua con una digestión completa y con el procedimiento analítico. Determínense los microgramos de cobre en la solución final por referencia a la curva de calibración apropiada. 5. Cálculos 6. Bibliografía SMITH, G. F. & W. H. MCCURDY. 1952. 2,9Dimethyl-l,10-phenanthroline: New specific in spectrophotometric determination of copper. Anal. Chem. 24:371. LUKE, C. L. & M. E. CAMPBELL. 1953. Determination of impurities in germanium and silicon. Anal. Chem. 25:1586. GAHLER, A. R. 1954. Colorimetric determination of copper with neocuproine. Anal. Chem. 26:577. FULTON, J. W. & J. H ASTINGS. 1956. Photometric determinations of copper in aluminum and lead-tin solder with neocuproine. Anal. Chem. 28:174. FRANK, A. J., A. B. G OULSTON & A. A. DEACUTIS. 1957. Spectrophotometric determination of copper in titanium. Anal. Chem. 29:750. 3-110 MÉTODOS NORMALIZADOS 3500-Cu E. 1. Método de la batocuproína Discusión general a) Principio: El ion cuproso forma un quelato de color naranja soluble en agua con disulfonato de batocuproína (sal disódica del ácido 2,9-dimetil-4,7-difenil1,10-fenantrolindisulfónico). Aunque el color se forma en el intervalo de pH 3,5 a 11,0, el intervalo de pH recomendado es entre 4 y 5. La muestra se tampona a un pH de aproximadamente 4,3 y se reduce con clorhidrato de hidroxilamina. Se mide la absorbancia 484 nm. El método puede aplicarse de cobre hasta al menos 5 mg/1 con una sensibilidad de 20 μg/l. b) Interferencia: Las siguientes sustancias pueden tolerarse con un error inferior a ±2 por 100: También pueden interferir cianuro, tiocianato, persulfato y EDTA. c) Concentración mínima detectable: 20 μg/1 con una célula de 5 cm. 2. Instrumental a) Equipo colorimétrico: Uno de los siguientes, con un trayecto luminoso de 1 a 5 cm (a menos que se empleen tubos de Nessler): 1) Espectrofotómetro, para su uso a 484 nm. 2) Fotómetro de filtro, equipado con un filtro azul-verde que presenta un máximo de transmisión luminosa próximo a 484 nm. 3) Tubos de Nessler, emparejados, de 100 ml, forma alargada. b) Material de vidrio lavado con ácido: Enjuáguese todo el material de vidrio con HC1 conc. y después con agua libre de cobre. 3. Reactivos a) Agua libre de cobre: Véase método D, apartado 3a. b) Solución de cobre de reserva: Prepárese como se indica en el método D, apartado 3b, pero utilizando 20,00 mg de lámina o alambre de cobre; 1,00 ml = = 20,00 μg Cu. c) Solución de cobre patrón: Diluyanse 250 ml de solución de cobre de reserva hasta 1.000 ml empleando agua; 1,00 ml = = 5,00 μg Cu. Prepárese diariamente. d) Acido clorhídrico, HC1 1 + 1. e) Solución de clorhidrato de hidroxilamina: Véase método D, apartado 3e. 3-111 DETERMINACIÓN DE METALES f) Solución de curato de sodio: Disuélvanse 300 g de Na3C6H5O7·2H2O en agua y llévese hasta 1.000 ml. g) Solución de disulfonato de batocuproína disódico: Disuélvanse 1.000 g de C12H4N2(CH3)2(C6H4)2(SO3Na)2 en agua y llévese hasta 1.000 ml. 4. Procedimiento Llévense con la pipeta 50,0 ml de muestra, o porción adecuada diluida hasta 50,0 ml, a un erlenmeyer de 250 ml. En distintos matraces erlenmeyer de 250 ml, prepárese un blanco de agua de 50,0 ml y una serie de patrones de cobre de 50,0 ml que contengan 5,0, 10,0, 15,0, 20,0 y 25,0 μg Cu. Añádanse a la muestra blanco y patrones, mezclando después de cada adición 1,00 ml de HC1 1 + 1, 5,00 ml de solución de NH2OH·HC1, 5,00 ml de solución de citrato de sodio y 5,00 ml de solución de disulfonato de batocuproína disódico. Pásese a las células y léase la absorbancia de la muestra frente al blanco a 484 nm. Para la curva de calibración, márquese la absorbancia en función de los microgramos de Cu en los patrones. Calcúlese la concentración a partir de la curva de calibración. 3500-Au 3500-Au A. Debido a su extremada insolubilidad, el oro únicamente se encuentra en cantidades traza en las aguas naturales (< 1 μg/1) incluso en áreas mineralizadas. 5. Cálculos 6. Precisión y sesgo Se analizó en 33 laboratorios una muestra sintética con 1.000 μg Cu/1, 500 μg Al/1, 50 μg Cd/l, 110 μg Cr/l, 300 μg Fe/1, 70 μg Pb/1, 50 /μg Mn/1, 150 μg Ag/1 y 650 μg Zn/1, empleando el método de batocuproína, con una desviación relativa estándar de 4,1 por 100 y un error relativo de 0,3 por 100. 7. Bibliografía SMITH, G. F. & D. H. WILKINS. 1953. New colorimetric reagent specific for copper. Anal. Chem. 25:510. BORCHARDT, L. G. & J. P. BUTLER. 1957. Determination of trace amounts of copper. Anal. Chem. 29:414. ZAK, B. 1958. Simple procedure for the single sample determination of serum copper and iron. Clínica Chim. Acta 3:328. BLAIR, D. & H. DIEHL. 1961. Bathophenanthrolinedisulfonic acid and bathocuproinedisulfonic acid, water soluble reagents for iron and copper. Talanta 7:163. ORO Introducción Las aguas residuales de las operaciones de la minería del oro pueden tener concentraciones más altas. 3-112 3500-Au B. MÉTODOS NORMALIZADOS Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, sección 3111B. 3500-lr 3500-lr A. El iridio es un elemento relativamente insoluble y sólo se encuentra normalmente a concentraciones muy bajas y 3500-lr B. IRIDIO Introducción asociado a material en partículas. Las aguas de procesado pueden contener concentraciones más altas. Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, sección 3111B. 3500-Fe HIERRO* 3500-Fe A. 1. Incidencia y significación En las muestras filtradas de aguas superficiales oxigenadas, el hierro raramente alcanza concentraciones de 1 mg/1. Algunas aguas subterráneas y drenajes * Aprobado por el Standard Methods Committee, 1985. Introducción superficiales ácidos pueden contener una cantidad de hierro bastante mayor. El hierro del agua puede ocasionar manchas en la ropa de lavado y en la porcelana. Algunas personas son capaces de detectar el gusto astringente dulce-amargo a niveles por encima de 1 mg/1. En condiciones reductoras, el hierro existe en estado ferroso. En ausencia de iones que forman complejos, el hierro fé- DETERMINACIÓN DE METALES rrico no es significativamente soluble a menos que el pH sea muy bajo. Al exponerlo al aire o al añadir oxidantes, el hierro ferroso se oxida al estado férrico y puede hidrolizarse para formar óxido férrico hidratado insoluble. En muestras de agua, el hierro puede estar en forma de solución auténtica, en estado coloidal que puede ser peptizado por materia orgánica, en complejos inorgánicos u orgánicos de hierro o en partículas suspendidas relativamente gruesas. Puede estar en forma ferrosa o férrica, suspendida o disuelta. El cieno y la arcilla en suspensión pueden contener hierro soluble en ácido. Algunas veces se toman partículas de óxido de hierro con la muestra de agua por el desprendimiento de herrumbre desde las tuberías. El hierro puede provenir, además, del tapón metálico utilizado para cerrar la botella de la muestra. 2. Selección del método Los límites de detección y sensibilidad para el procedimiento espectrométrico de absorción atómica, el método de plasma de acoplamiento inductivo y el procedimiento colorimétrico de fenantrolina, aquí descritos, son similares y en general adecuados para el análisis de aguas naturales o tratadas. Los reactivos complejantes utilizados en los procedimientos colorimétricos son específicos para el hierro ferroso, pero los procedimientos de absorción atómica no lo son. Sin embargo, debido a la inestabilidad del hierro ferroso, que pasa fácilmente a la forma férrica en solución en contacto con el aire, la determinación de hierro ferroso requiere precauciones especiales y puede ser necesario realizarla in situ en el momento de la toma de muestra. El procedimiento de determinación del hierro ferroso con 1,10-fenantrolina (véase 3500-Fe.D.4c) tiene cierta limitación en su aplicabilidad; ha de evitarse un al- 3-113 macenamiento prolongado o la exposición de las muestras a la luz. Con un procedimiento especial, que utiliza la batofenantrolina, se puede obtener una distinción cuantitativa rigurosa entre hierro ferroso y férrico. Los métodos espectrométricos que emplean batofenantrolina1-6 y otros reactivos orgánicos complejantes, tales como ferrozina o TPTZ, son capaces de determinar concentraciones de hierro tan bajas como 1 μg/1. Existe un procedimiento de quimioluminiscencia cuyo límite de detección se ha establecido en 5 ng/1. En otras partes se han descrito otros procedimientos10-13. 3. Toma de muestras y almacenamiento Planifíquense de antemano los métodos de toma, almacenamiento y tratamiento previo de las muestras. Límpiese con ácido el recipiente de la muestra y enjuáguese con agua destilada. Puede ser necesario un equipo de filtración con membrana de las muestras in situ para determinar el hierro en solución (hierro disuelto). Se considera que el hierro disuelto pasa a través de un filtro de membrana de 0,45 μm, pero puede quedar incluido el hierro coloidal. El valor de la determinación depende en gran parte del cuidado puesto en la obtención de una muestra representativa. El hierro de las muestras de agua de pozo o del grifo puede variar en concentración y forma con la duración y el grado de afluencia antes y durante la toma de muestra. Cuando se tome una porción de muestra para determinar el hierro en suspensión, sacúdase la botella de la muestra vigorosamente y con frecuencia para obtener una suspensión uniforme del hierro precipitado. Es necesario poner el máximo cuidado cuando el hierro coloidal se adhiere a la botella de la muestra. Este problema puede agudizarse cuando se utilizan botellas de plástico. MÉTODOS NORMALIZADOS 3-114 Para obtener una determinación precisa del hierro total, utilícese un recipiente separado para la toma de la muestra. Trátese con ácido en el momento de la toma para disolver el hierro y evitar la adsorción o depósito sobre las paredes del recipiente de muestra. Téngase en cuenta el ácido añadido al medir porciones para análisis. La adición de ácido a la muestra puede eliminar la necesidad de añadir ácido antes de la digestión (apartado 3500-Fe.D.4a). 5. 6. 7. 8. 9. 4. 1. 2. 3. 4. Referencias LEE, G. F. & W. STUMM. 1960. Determination of ferrous iron in the presence of ferric iron using bathophenanthroline. J. Amer. Water. Works Assoc. 52:1567. G HOSH , M. M., J. T. O'C ONNOR & R. S. ENGELBRECHT . 1967. Bathophenanthroline method for the determination of ferrous iron. J. Amer. Water Works Assoc. 59:897. B LAIR , D. & H. D IEHL . 1961. Bathophenanthroline-disulfonic acid and bathocuproinedisulfonic acid, water soluble reagents for iron and copper. Talanta 7:163. S HAPIRO , J. 1966. On the measurement of 3500-Fe B. 11. 12. 13. Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, secciones 3111B y C, y método espectrométrico de 3500-Fe C. 10. ferrous iron in natural waters. Limnol. Oceanogr. 11:293. M C M AHON , J. W. 1967. The influence of light and acid on the measurement of ferrous iron in lake water. Limnol. Oceanogr. 12:437. MCMAHON, J. W. 1969. An acid-free bathophenanthroline method for measuring dissolved ferrous iron in lake water. Water Res. 3:743. GIBBS, C. 1976. Characterization and application of ferrozine iron reagent as a ferrous iron indicator. Anal. Chem. 48:1197. D OUGAN , W. K. & A. L. W ILSON . 1973. Absorbtiometric determination of iron with TPTZ. Water Treat. Exam. 22:110. SEITZ, W. R. & D. M. HERCULES. 1972. Determination of trace amounts of iron (II) using chemiluminescence analysis. Anal. Chem. 44:2143. Moss, M. L. & M. G. MELLON. 1942. Colorimetric determination of iron with 2,2'-bipyridine and with 2,2',2"-tripyridine. Ind. Eng. Chem., Anal. Ed. 14:862. W ELCHER , F. J. 1947. Organic Analytical Reagents. D. Van Nostrand Co., Princeton, Nueva Jersey, Vol. 3, págs. 100-104. MORRIS, R. L. 1952. Determination of iron in water in the presence of heavy metals. Anal. Chem. 24:1376. D OIG , M. T., III, & D. F. M ARTIN . 1971. Effect of humic acids on iron analyses in natural water. Water Res. 5:689. absorción atómica termoeléctrica, sección 3113. Método de plasma de acoplamiento inductivo Véase sección 3120. DETERMINACIÓN DE METALES 3500-Fe D. 1. 3-115 Método de fenantrolina Discusión general a) Principio: Se disuelve el hierro, se reduce al estado ferroso por ebullición con ácido e hidroxilamina y se trata con 1,10-fenantrolina a pH de 3,2 a 3,3. El complejo rojo-naranja que se forma es un quelato de tres moléculas de fenantrolina por cada átomo de hierro ferroso. La solución coloreada obedece a la ley de Beer; su intensidad es independiente del pH entre 3 y 9. Un pH entre 2,9 y 3,5 asegura un rápido desarrollo del color en presencia de un exceso de fenantrolina. Los patrones del color son estables durante al menos 6 meses. b) Interferencia: Entre las sustancias que interfieren están los oxidantes fuertes, cianuro, nitrito, y fosfatos (más los polifosfatos que el ortofosfato), cromo, zinc en concentración 10 veces superior a la del hierro, cobalto y cobre por encima de 5 mg/1 y níquel por encima de 2 mg/1. El bismuto, el cadmio, el mercurio, el molibdato y la plata precipitan la fenantrolina. La ebullición inicial con ácido convierte los polifosfatos en ortofosfato y elimina el cianuro y el nitrito, que, por otra parte, podrían interferir. La adición de un exceso de hidroxilamina elimina los errores causados por concentraciones excesivas de reactivos fuertemente oxidantes. En la presencia de iones metálicos que interfieran, utilícese un mayor exceso de fenantrolina para sustituir a la complejada por los metales que interfieren. Cuando existen concentraciones excesivas de iones metálicos que interfieren, puede emplearse el método de extracción. Si existen cantidades apreciables de materia colorante u orgánica, puede ser necesario evaporar la muestra, llevar el residuo a combustión seca suave y volver a disolver en ácido. La combustión seca se puede realizar en crisoles de sílice, por- celana o platino que hayan sido hervidos durante varias horas en HC1 1 + 1. La presencia de cantidades excesivas de materia orgánica puede hacer necesaria la digestión antes de emplear el procedimiento de extracción. c) Concentración mínima detectable: Concentraciones de hierro disuelto o total tan bajas como 10 μg/1 pueden determinarse con un espectrofotómetro provisto de células con un trayecto luminoso de 5 cm o más largo. Trátese un blanco con el procedimiento completo para la corrección. 2. Instrumental a) Equipo calorimétrico: Se necesita uno de los siguientes: 1) Espectrofotómetro, para utilizarlo a 510 nm, que proporciona un trayecto luminoso de 1 cm o más largo. 2) Fotómetro de filtro, que proporciona un trayecto luminoso de 1 cm o más largo y equipado con un filtro verde con una transmitancia máxima próxima a 510 nm. 3) Tubos de Nessler, emparejados, 100 ml, forma alargada. b) Material de vidrio lavado con ácido: Lávese todo el material de vidrio con ácido clorhídrico conc. (HC1) y enjuáguese con agua destilada antes de utilizarlo, con objeto de eliminar depósitos de óxido de hierro. c) Embudos de separación: 125 ml, forma Squibb, con espitas y tapones de vidrio o TFE. 3. Reactivos Utilícense reactivos bajos en hierro. Para preparar las soluciones patrón y de MÉTODOS NORMALIZADOS 3-116 los reactivos, empléese agua destilada libre de hierro. Almacénense los reactivos en botellas con tapón de vidrio. Las soluciones de HC1 y acetato de amonio son estables indefinidamente si están herméticamente tapadas. Las soluciones de hidroxilamina, de fenantrolina y de hierro de reserva son estables durante varios meses. Las soluciones de hierro patrón no son estables; prepárense a diario según se necesiten, por dilución de la solución de reserva. Los patrones visuales en tubos de Nessler son estables durante varios meses si están bien cerrados y protegidos de la luz. a) Ácido clorhídrico, HC1 conc. con menos de 0,00005 por 100 de hierro. b) Solución de hidroxilamina: Disuélvanse 10 g de NH2OH·HC1 en 100 ml de agua. c) Solución tampón de acetato de amonio: Disuélvanse 250 g de NH4C2H3O2 en 150 ml de agua. Añádanse 700 ml de ácido acético conc. (glacial). Como incluso el NH4C2H3O2 de buena calidad contiene una cantidad significativa de hierro, prepárense nuevos patrones de referencia con cada preparación del tampón. d) Solución de acetato de sodio: Disuélvanse 200 g de NaC2H3O2·3H2O en 800 ml de agua. e) Solución de fenantrolina: Disuélvanse 100 mg de monohidrato de 1,10fenantrolina, CX2H8N2·H2O, en 100 ml de agua por agitación y calentamiento a 80 °C. No hervir. Descártese la solución si se oscurece. El calentamiento es innecesario si se añaden al agua dos gotas de HC1 conc. (NOTA: Un mililitro de este reactivo es suficiente para no más de 100 μg Fe.) f) Solución de hierro de reserva: Utilícese metal (1) o sal (2) para preparar la solución de reserva. 1) Utilícese alambre de hierro electrolítico o «alambre de hierro para estandarización» en la preparación de la solu- ción. Si es necesario, límpiese el alambre con papel de lija fino al objeto de eliminar cualquier capa de óxido y producir una superficie brillante. Pésense 200,0 mg de alambre e introdúzcanse en un matraz volumétrico de 1.000 ml. Disuélvanse en 20 ml de ácido sulfúrico (H2SO4) 6N y dilúyase con agua hasta la señal; 1,00 ml = = 200 μg Fe. 2) Si se prefiere sulfato amónico ferroso, añádanse lentamente 20 ml de H2SO4 conc. a 50 ml de agua y disuélvanse 1,404 g de Fe(NH4)2(SO4)2·6H2O. Añádase permanganato potásico (KMnO4) 0,1N gota a gota hasta que persista un color rosa pálido. Diluyase hasta 1.000 ml con agua y mézclese; 1,00 ml = 200 μg Fe. g) Soluciones de hierro patrón: Prepárense diariamente para su uso. 1) Llévense con la pipeta 50,00 ml de solución de reserva a un matraz volumétrico de 1.000 ml y dilúyase con agua hasta la señal; 1,00 ml = 10,0 μg Fe. 2) Llévense con la pipeta 5,00 ml de solución de reserva a un matraz volumétrico de 1.000 ml y dilúyase con agua hasta la señal; 1,00 ml = 1,00 μg Fe. h) Éter diisopropilico o isopropílico. (PRECAUCIÓN: LOS éteres pueden formar compuestos explosivos; pruébense antes del uso.) 4. Procedimiento a) Hierro total: Mézclese cuidadosamente la muestra y mídanse 50,0 ml en un erlenmeyer de 125 ml. Si este volumen de muestra contiene más de 200 μg de hierro, utilícese una porción más pequeña medida con precisión y dilúyase hasta 50,0 ml. Añádanse 2 ml de HC1 conc. y 1 ml de solución de NH2OH · HC1. Añádanse unas cuantas bolas de vidrio y caliéntese a ebullición. Para asegurar la disolución de todo el hierro, continúese la ebullición hasta que el volumen se reduz- DETERMINACIÓN DE METALES ca a 15-20 ml. (Si la muestra ha sido sometida a combustión seca, disuélvase el residuo en 2 ml de HC1 conc. y 5 ml de agua). Enfríese hasta temperatura ambiente y pásese a un matraz volumétrico de 50 o 100 ml o a un tubo de Nessler. Añádanse 10 ml de solución tampón de NH4C2H3O2 y 4 ml de solución de fenantrolina y dilúyase con agua hasta la señal. Mézclese cuidadosamente y déjese al menos durante 10 a 15 minutos para que se desarrolle el color al máximo. b) Hierro disuelto: Inmediatamente después de tomada, fíltrese la muestra a través de un filtro de membrana de 0,45 μg a un matraz de vacío que contenga 1 ml de HC1 conc./100 ml de muestra. Analícese el filtrado en cuanto al hierro total disuelto (apartado 4a) y/o hierro ferroso disuelto (apartado 4c). (Este procedimiento también puede emplearse en el laboratorio sabiendo que la exposición normal de la muestra al aire durante el traslado puede dar lugar a la precipitación del hierro.) Calcúlese el hierro suspendido restando el hierro disuelto del hierro total. c) Hierro ferroso: Determínese el hierro ferroso en el lugar donde se ha tomado la muestra, pues con el tiempo puede cambiar la relación ferroso-férrico en soluciones acidas. Para determinar el hierro ferroso solamente, acidúlese una muestra separada con 2 ml de HC1 conc./ 100 ml de muestra en el momento de su toma. Llénese una botella directamente de la fuente de donde se toman las muestras y tápese. Extráigase a continuación una porción de 50 ml de muestra acidulada y añádanse 20 ml de solución de fenantrolina y 10 ml de solución de NH4C2H3O2 agitando con energía. Diluyase hasta 100 ml y mídase la intensidad del color dentro de los 5 a 10 minutos. No hay que exponerla a la luz del sol. (El desarrollo de color es rápido en presencia de exceso de fenantrolina. El volumen de fenantrolina dado es adecuado para menos de 50 μg de hierro total; 3-117 si existen cantidades mayores, utilícese un volumen de fenantrolina correspondientemente mayor a un reactivo más concentrado. Se necesita un exceso de fenantrolina por la cinética del proceso de formación de complejo.) Calcúlese el hierro férrico por sustracción del hierro ferroso del hierro total. d) Medida del color: Prepárese una serie de patrones llevando con la pipeta volúmenes calculados con precisión de soluciones de hierro patrón (utilícese una solución más débil para medir porciones de 1 a 10 μg) a matraces erlenmeyer de 125 ml, diluyendo hasta 50 ml por adición de volúmenes medidos de agua y realizando los pasos indicados en el apartado 4a. Para comparación visual, prepárese una serie de 10 patrones por lo menos, cuyo contenido varíe de 1 a 100 μg Fe en el volumen final de 100 ml. Compárense los colores en tubos de Nessler de 100 ml de forma alargada. Para la medida fotométrica, utilícese la tabla 3500-Fe:I como una guía aproximada para la selección del apropiado trayecto luminoso a 510 nm. Léanse los patrones frente a agua destilada fijada a absorbancia cero y trácese una curva de calibración, incluyendo un blanco (véase apartado 3c y la Introducción general). TABLA 3500-FE:I. SELECCIÓN DE LONGITUD DEL RECORRIDO DE LUZ PARA DIVERSAS CONCENTRACIONES DE HIERRO 3-118 Si las muestras tienen color o turbidez, realícense todos los pasos del procedimiento con un segundo juego de muestras sin añadir fenantrolina. En lugar de agua destilada, utilícense los blancos preparados para fijar el cero de absorbancia del fotómetro y léase cada muestra revelada con fenantrolina frente al correspondiente blanco sin fenantrolina. Trasládense las lecturas observadas en el fotómetro a valores de hierro empleando una curva de calibración. Este procedimiento no compensa los iones que interfieren. e) Muestras que tienen interferencias orgánicas: Digiéranse las muestras que contienen cantidades sustanciales de materias orgánicas según las indicaciones dadas en las secciones 3030G y H. 1) Si se ha preparado una muestra siguiendo las indicaciones dadas en la sección 3030G o en la 3030H, llévense con la pipeta 10,0 ml u otra porción adecuada que contenga 20 a 500 μg Fe a un embudo de separación de 125 ml. Si el volumen tomado es menor de 10 ml, añádase agua destilada hasta llegar a los 10 ml. Añádanse 15 ml de HC1 conc. al embudo de separación para un volumen acuoso de 10 ml, o si la porción tomada es superior a 10,0 ml, añádanse 1,5 ml de HC1 conc./ml de muestra. Mézclese, enfríese y síganse las indicaciones del apartado 4e3). 2) Para preparar una muestra destinada únicamente a determinación de hierro, mídase un volumen adecuado que contenga 20 a 500 μg Fe y trátese por uno de los procedimientos de digestión descritos en las secciones 3030G y H. Utilícense, no obstante, sólo 5 ml de H2SO4 o HCIO4 y omítase el H2O2. Cuando la digestión es completa, enfríese, dilúyase con 10 ml de agua, caliéntese casi hasta ebullición para disolver las sales de disolución lenta y, si la muestra sigue estando turbia, fíltrese a través de un filtro de fibra de vidrio, de vidrio sin- MÉTODOS NORMALIZADOS terizado o de porcelana, lavando con 2 a 3 ml de agua. Pásese cuantitativamente el filtrado o la solución clara a un matraz volumétrico de 25 ml y llévese hasta 25 ml con agua. Vacíese el matraz en un embudo separador de 125 ml, enjuáguese con 5 ml de HC1 conc. que se añaden al embudo, y añádanse 25 ml de HC1 conc. medidos con el mismo vaso o matraz graduado. Mézclese y enfríese hasta temperatura ambiente. 3) Extráigase el hierro de la solución de HC1 del embudo de separación sacudiendo durante 30 segundos con 25 ml de éter isopropílico (PRECAUCIÓN). Apártese la capa acida inferior en un segundo embudo de separación. Extráigase de nuevo la solución acida con 25 ml de éter isopropílico, drénese la capa acida a un adecuado recipiente limpio y combínense las dos porciones de éter isopropílico. Vuélvase a verter la capa acida en el segundo embudo de separación y extráigase de nuevo con 25 ml de éter isopropílico. Sáquese y descártese la capa acida y añádase la capa etérea al embudo original. La persistencia de un color amarillo en la solución de HC1 después de tres extracciones no significa una separación incompleta del hierro, ya que el cobre, que no se ha extraído, da un color amarillo similar. Sacúdanse los extractos etéreos combinados con 25 ml de agua para hacer volver al hierro a la fase acuosa y pásese la capa acuosa inferior a un matraz volumétrico de 100 ml. Repítase la extracción con una segunda porción de 25 ml de agua, añadiendo ésta a la primera extracción acuosa. Descártese la capa etérea. 4) Añádase 1 ml de solución de NH2OH·HC1, 10 ml de solución de fenentrolina y 10 ml de solución de NaC2H3O2. dilúyase con agua hasta 100 ml, mézclese cuidadosamente y déjese reposar durante 10 minutos. Mídase la absorbancia a 510 nm utilizando una célula de absorción de 5 cm para cantidades de hierro inferiores a 10 μg, o una 3-119 DETERMINACIÓN DE METALES célula de 1 cm para cantidades entre 100 y 500 μg. Como referencia empléese agua destilada o bien una muestra en blanco preparada tratando por el procedimiento analítico completo las cantidades de ácidos especificadas. Si se utiliza el agua destilada como referencia, corríjase la absorbancia de la muestra restándole la absorbancia de una muestra en blanco. Determínense los microgramos de hierro en la muestra a partir de la absorbancia (corregida, si es necesario) por referencia a la curva de calibración preparada utilizando un intervalo apropiado de patrones de hierro que contengan las mismas cantidades de fenantrolina, hidroxilamina y acetato de sodio que la muestra. 5. Cálculos Cuando la muestra ha sido tratada según 4a, b, c, o 4e2): Cuando la muestra ha sido tratada según 4el): Anótense los detalles de la toma de la muestra, almacenamiento y tratamiento previo, si son oportunos para la interpretación de los resultados. 6. Precisión y sesgo La precisión y el sesgo dependen del método de toma de la muestra y su almacenamiento, del método de la medida del color, de la concentración de hierro y de la presencia de color interferente, turbidez e iones extraños. En general, la fiabilidad óptima de la comparación visual en tubos de Nessler no es superior al 5 por 100, y frecuentemente sólo 10 por 100, mientras que, en condiciones óptimas, la medida fotométrica puede ser fiable a 3 por 100 o 3 μg, que constituye una proporción mucho mayor. El límite de sensibilidad para observaciones visuales en tubos de Nessler es de aproximadamente 1 μg Fe. La variabilidad e inestabilidad de la muestra puede afectar a la precisión y al sesgo de esta determinación más que los errores del propio análisis. Se han encontrado serias divergencias en los informes de diferentes laboratorios debidas a las variaciones en los métodos de toma y tratamiento de muestras. En 44 laboratorios se analizó una muestra sintética con 300 μg Fe/1, 500 μg Al/1, 50 μg Cd/1, 110 μg Cr/1, 470 μg Cu/1, 70 μg Pb/1, 120 μg Mn/1, 150 μg Ag/1 y 650 μg Zn/1 en agua destilada, empleando el método de la fenantrolina, con una desviación relativa estándar de 25,5 por 100 y un error relativo de 13,3 por 100. 7. Bibliografía CHRONHEIM, G. & W. WINK. 1942. Determination of divalent iron (by o-nitrosophenol). Ind. Eng. Chem., Anal. Ed. 14:447. MEHLIG, R. P. & R. H. HULETT. 1942. Spectrophotometric determination of iron with o-phenanthroline and with nitro-o-phenanthroline. Ind. Eng. Chem., Anal. Ed. 14:869. CALDWELL, D. H. & R. B. ADAMS. 1946. Colorimetric determination of iron in water with ophenanthroline. J. Amer. Water Works Assoc. 38:727. WELCHER, F. J. 1947. Organic Analytical Reagents. D. Van Nostrand Co., Princeton, Nueva Jersey, Vol. 3, págs. 85-93. KOLTHOFF, I. M., T. S. LEE & D. L. LEUSSING. 1948. Equilibrium and kinetic studies on the formation and dissociation of ferroin and ferrin. Anal. Chem. 20:985. R Y A N , J. A. & G. H. B O T H A M . 1 949 . Iro n in aluminum alloys: Colorimetric determina- 3-120 MÉTODOS NORMALIZADOS tion using 1,10-phenanthroline. Anal. Chem. 21:1521. REITZ, L. K., A. S. O'BRIEN & T. L. DAVIS. 1950. Evaluation of three iron methods using a factorial experiment. Anal. Chem. 22:1470. SANDELL, E. B. 1959. Colorimetric Determination of Traces of Metals, 3.a ed. Interscience Publishers, Nueva York, cap. 22. 3500-Pb 3500-Pb A. 1. El plomo es un importante veneno que se acumula en el organismo. Las aguas naturales rara vez contienen por encima de 5 μg/1, aunque se ha informado sobre valores mucho más altos. El plomo de un suministro de agua puede ser de origen industrial, minero y de descargas de hornos de fundición o de cañerías viejas de plomo. Las aguas de grifo blandas y acidas y que no reciben un tratamiento adecuado contienen plomo como resultado del ataque a las tuberías de servicio. * Aprobado por el Standard Methods Committee, 1985. Introducción Selección del método El método espectrométrico de absorción atómica tiene un límite de detección relativamente alto en la modalidad de llama y requiere un procedimiento de extracción de las bajas concentraciones comunes en el agua potable; el método de absorción atómica electrotérmico es mucho más sensible para las bajas concentraciones y no requiere extracción. El método de plasma de acoplamiento inductivo tiene una sensibilidad similar a la del método de absorción atómica de llama. El método de ditizona es sensible y específico como procedimiento colorimétrico. Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, secciones 3111B y C, y método espectrométrico de 3500-Pb C. PLOMO* 2. Incidencia 3500-Pb B. SKOUGSTAD, M. W., M. J. FISHMAN, L. C. FRIEDMAN, D. E. ERDMANN & S. S. D UNCAN. 1979. Methods for Determination of Inorganic Substances in Water and Fluvial Sediment. Cap. Al en Book 5, Techniques of Water Resources Investigations of the United States Geological Survey. U. S. Geological Surv., Washington, D.C. absorción atómica electrotérmica, sección 3113. Método de plasma de acoplamiento inductivo Véase sección 3120. DETERMINACIÓN DE METALES 3500-Pb D. 1. 3-121 Método de la ditizona Discusión general a) Principio: Se mezcla una muestra acidulada que contenga cantidades del orden de microgramos de plomo con solución reductora amoniacal de citratocianuro y se extrae con ditizona en cloroformo (CHC13) para formar un ditizonato de plomo de color rojo cereza. Se mide fotométricamente el color de la solución de color mlxta1, 2. El volumen de muestra tomada para el análisis será de 21 cuando se emplea digestión. b) Interferencia: La ditizona forma complejos coloreados con bismuto, estaño estannoso y talio monovalente, en solución amoniacal débil de cianuro (pH 8,5 a 9,5). En solución amoniacal fuerte (pH 10 a 11,5) de citrato-cianuro, los ditizonatos de estos iones son inestables y sólo se pueden extraer parcialmente3. Este método utiliza una extracción única de ditizona, de color mixto, y pH elevado. La interferencia de estaño estannoso y talio monovalente se reduce posteriormente cuando estos iones se oxidan durante la digestión preliminar. Una modificación del método permite la detección y eliminación de la interferencia de bismuto. Las cantidades excesivas de bismuto, talio y estaño pueden eliminarse4. La ditizona en CHC1 3 absorbe a 510 nm; contrólese su interferencia mediante el empleo de concentraciones aproximadamente iguales de ditizona en exceso en muestras, patrones y blancos. El método carece de interferencia para la determinación de 0,0 a 30,0 μg Pb en presencia de 20 μg Tl+, 100 μg Sn2+, 200 μg In3 + , así como 1.000 μg de cada uno de los siguientes: Ba2+, Cd2+, Co2+, Cu2+, Mg2+, Mn2+, Hg2+, Sr2+, Zn2+, Al3 + , Sb3+, As3 + , Cr3+, Fe3 + , V3 + , PO3-4 y SO 2-4 . Cantidades del orden de gramos de metales alcalinos no interfieren. Existe una modificación para evi- tar la interferencia de cantidades excesivas de bismuto o estaño. c) Tratamiento preliminar de la muestra: En el momento de la toma acidúlese con HNO 3 conc. hasta pH < 2, pero evítese un exceso de HNO3. Añádanse 5 ml de solución 0,1N de yodo para evitar las pérdidas de compuestos orgánicos de plomo volátiles durante la manipulación y digestión de las muestras. Prepárese un blanco de agua destilada libre de plomo y trátese según el procedimiento completo. d) Digestión de muestras: A menos que la digestión se muestre innecesaria, digiéranse todas las muestras para el plomo disuelto o total, como se ha descrito en 3030G o H. e) Concentración mínima detectable: 1,0 μg Pb/10 ml de solución de ditizona. 2. Instrumental a) Espectrofotómetro: Para utilizarlo a 510 nm, que proporciona un trayecto luminoso de 1 cm o más largo. b) pHmetro. c) Embudos de separación: Tipo Squibb de 250 ml. Límpiese todo el material de vidrio, incluidas las botellas de muestras, empleando HNO3 1 + 1. Enjuáguese cuidadosamente con agua destilada o desionizada. d) Buretas de distribución automática: Utilícense para todos los reactivos con objeto de reducir al mínimo los errores de procedencia indeterminada. 3. Reactivos Prepárense todos los reactivos con agua destilada libre de plomo. MÉTODOS NORMALIZADOS 3-122 a) Solución de plomo de reserva: Disuélvanse 0,1599 g de nitrato de plomo, Pb(NO3)2 (de un mínimo de pureza de 99,5 por 100), en aproximadamente 200 ml de agua. Añádanse 10 ml de HNO3 conc. y dilúyase con agua hasta 1.000 ml. Alternativamente, disuélvanse 0,1000 g de metal plomo puro en 20 ml de HNO3 1 + 1 y dilúyase con agua hasta 1.000 ml; 1,00 ml = 100 μg Pb. b) Solución de plomo de trabajo: Diluyanse 20,0 ml de solución de reserva hasta 1.000 ml empleando agua; 1 ml = = 2,00 μg Pb. c) Ácido nítrico, HNO3 1 + 4: Diluyanse 200 ml de HNO3 conc. hasta 1 l empleando agua. d) Hidróxido amónico, NH4OH1 + 9: dilúyanse con agua 10 ml de NH4OH conc. hasta 100 ml. e) Solución de citrato-cianuro reductora: Disuélvanse 400 g de citrato amónico dibásico, (NH4)2HC6H5O7; 20 g de sulfito de sodio anhidro, Na2SO3; 10 g de clorhidrato de hidroxilamina, NH2OH·HC1, y 40 g de cianuro potásico, KCN (PRECAUCIÓN: es tóxico), en agua y dilúyase hasta 1 l. Mézclese esta solución con 2 1 de NH4OH conc. No debe utilizarse la pipeta con la boca. f) Solución de ditizona de reserva: Véase 1070D.2b1), solución de ditizona de reserva I. g) Solución de ditizona de trabajo: Disuélvanse 100 ml de solución de ditizona de reserva hasta 250 ml empleando CHC13; 1 ml = 40 μg de ditizona. h) Solución de ditizona especial: Disuélvanse 250 mg de ditizona en 250 ml de CHC13. Esta solución no necesita purificación, ya que todos los extractos que la utilizan se descartan. i) Solución de sulfito de sodio: Disuélvanse 5 g de Na2SO3 anhidro en 100 ml de agua. j) Solución de yodo: Disuélvanse 40 g de KI en 25 ml de agua, añádanse 12,7 g de yodo re-sublimado y dilúyase hasta 1.000 ml. 4. Procedimiento a) Con digestión de la muestra: Añádase a una muestra digerida, que no contenga más de 1 ml de ácido conc, 20 ml de HNO3 1 + 4 y fíltrese con papel* de filtro sin plomo y con un embudo filtrante directamente a un embudo de separación de 250 ml. Aclárese el vaso de digestión con 50 ml de agua y añádase al filtro. Añádanse 50 ml de solución amoniacal de citrato-cianuro, mézclese y enfríese a temperatura ambiente. Añádanse 10 ml de solución de ditizona de trabajo, sacúdase vigorosamente durante 30 segundos el embudo tapado y déjese que se separen las capas. Insértese algodón carente de plomo en el vástago del embudo de separación y sáquese la capa inferior. Descártense de 1 a 2 ml de la capa CHC13, y llénese a continuación la célula de absorción. Mídase la absorbancia del extracto a 510 nm, utilizando una solución de ditizona de trabajo, según el aparato 3g, para poner el espectrofotómetro a cero. b) Sin digestión de la muestra: Añádanse a 100 ml de muestra acidulada (pH 2), en un embudo de separación de 250 ml, 20 ml de HNO3 1 + 4 y 50 ml de solución reductora de citrato-cianuro; mézclese. Añádanse 10 ml de solución de ditizona de trabajo y procédase como se indica en el apartado 4a. c) Curva de calibración: Trácese la gráfica de la concentración de al menos cinco patrones y un blanco en función de la absorbancia. Determínese la concentración de plomo en el extracto a partir de la curva. Todas las concentraciones son μg Pb/10 ml de extracto final. d) Eliminación de interferencias de excesos: Los ditizonatos de bismuto, estaño y talio difieren del ditizonato de plomo en la absorbancia máxima. Detéctese su presencia midiendo la absorban- * Whatman n.° 541 o equivalente. 3-123 DETERMINACIÓN DE METALES cia de la muestra a 510 nm y a 465 nm. Calcúlese la absorbancia de la muestra corregida a cada longitud de onda restando la absorbancia del blanco a la misma longitud de onda. Calcúlese la relación de absorbancia corregida a 510 nm a absorbancia corregida a 465 nm. La relación de absorbancias corregidas para ditizonato de plomo es 2,08, y para ditizonato de bismuto es 1,07. Si la relación para la muestra indica interferencia, es decir, si es marcadamente menor que 2,08, procédase como sigue con una nueva muestra de 100 ml: Si la muestra no ha sido digerida, añádanse 5 ml de solución de Na2SO3 para reducir el conservante de yodo. Ajústese el pH de la muestra a 2,5 utilizando un pHmetro y HNO3 1 + 4 o NH4OH 1 + 9, según se necesite. Pásese la muestra a un embudo de separación de 250 ml, extráigase con un mínimo de tres porciones de 10 ml de solución de ditizona especial, o hasta que la capa de CHC13 sea claramente verde. Extráigase con porciones de 20 ml de CHCI3 para eliminar la ditizona (ausencia de verde). Añádanse 20 ml de HNO3 1 + 4, 50 ml de solución reductora de citrato-cianuro y 10 ml de solución de ditizona de trabajo. Extráigase como se indica en el apartado Aa y mídase la absorbancia. 5. Cálculos 6. Precisión y sesgo La precisión de un solo operador al recuperar 0,0104 mg Pb/1 de agua del río Mississippi fue de 6,8 por 100 de desviación relativa estándar y –1,4 por 100 de error relativo. Con la proporción de 0,026 mg Pb/1, la recuperación se hizo con 4,8 por 100 de desviación relativa estándar y 15 por 100 de error relativo. 7. 1. Referencias SNYDER, L. J. 1947. Improved dithizone method for determination of lead—mixed color method at high pH. Anal. Chem. 19:684. 2. SANDELL, E. B. 1959. Coloriraetric Determination of Traces of Metals, 3. a ed. Interscience, Nueva York. 3. WICHMANN , H. J. 1939. Isolation and determination of trace metals—the dithizone system. Ind. Eng. Chem., Anal. Ed. 11:66. 4. AMERICAN SOCIETY FOR TESTINO AND MATERIALS . 1977. Annual Book of ASTM Standards. Part. 26, Method D3112-77, American Soc. Testing & Materials, Filadelfia, Pennsylvania. 3-124 MÉTODOS NORMALIZADOS 3500-Li 3500-Li A. 1. Incidencia El litio, un componente menor de minerales, se encuentra en el agua dulce en concentraciones inferiores a 0,2 mg/1. Las aguas salobres y termales pueden contener proporciones más altas de litio. El empleo de litio o sus sales en unidades de deshumidificación, aguas medicinales, procesos metalúrgicos y manufactura de algunos tipos de vidrio y baterías de acumuladores puede contribuir a su presen* Aprobado por el Standard Methods Committee, 1988. 3500-Li B. LITIO* Introducción cia en aguas residuales. El hipoclorito de litio se encuentra en el mercado como fuente de cloro, pudiéndose utilizar en piscinas. 2. Selección del método Los métodos preferidos son el espectrométrico de absorción atómica y el de plasma de acoplamiento inductivo. También es útil el método fotométrico de emisión de llama en aquellos laboratorios que no están equipados para los métodos preferidos. Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, sección 3111B. 3500-Li C. Método de plasma de acoplamiento inductivo Véase sección 3120. DETERMINACIÓN DE METALES 3500-Li D. 3-125 Método fotométrico de emisión de llama 1. Discusión general 3. Reactivos a) Principio: Lo mismo que los otros metales alcalinos de bajo peso atómico, sodio y potasio, el litio puede determinarse en cantidades traza por métodos fotométricos de llama. Las medidas pueden hacerse a una longitud de onda de 670,8 nm. b) Interferencia: El bario, el estroncio y el calcio interfieren en la determinación espectrométrica de llama del litio, pudiéndose eliminar por adición de una solución de sulfato sódico-carbonato sódico (Na2SO4-Na2CO3) que precipita el sulfato de bario (BaSO4), el carbonato de estroncio (SrCO3) y el carbonato de calcio (CaCO3). El contenido de magnesio no debe sobrepasar los 10 mg en la porción tomada para análisis con objeto de evitar la interferencia de la coprecipitación. c) Concentración mínima detectable: La concentración mínima detectable de litio es de aproximadamente 0,4 μg/1 para el agua de reactivos analizada en un espectrofotómetro de absorción atómica, en la modalidad de emisión. d) Toma de muestras y almacenamiento: Tómese la muestra en un frasco de vidrio de borosilicato o en una botella de polietileno. En el momento de tomarla, ajústese el pH de la muestra a < 2 con ácido nítrico (HNO3). a) Reactivo de sulfato sódico y carbonato sódico: Disuélvanse 5 g de Na2SO4 y 10 g de Na2CO3 en agua destilada y dilúyase hasta 1 l. b) Solución de litio de reserva: Disuélvanse 152,7 mg de cloruro de litio anhidro, LiCl, en agua destilada y dilúyase hasta 250 ml; 1,00 ml = 100 μg Li. Deséquese la sal toda la noche en la estufa a 105 °C. Enfríese en un desecador y pésese inmediatamente después de sacarla del desecador. c) Solución de litio patrón: Diluyanse 10,00 ml de solución de LiCl hasta 500 ml empleando agua destilada; 1,00 ml = 2,0 μg Li. 2. Instrumental Fotómetro de llama: Un instrumento cuya idoneidad para medir las concentraciones de Li haya sido probada por el analista (los fotómetros de llama que suelen encontrarse han sido diseñados para utilizarse en medicina clínica), o un espectrómetro de absorción atómica que funcione en la modalidad de emisión y que emplee una llama de aire-acetileno. 4. Procedimiento a) Tratamiento previo de muestras de agua contaminada y de aguas residuales: Utilícese la digestión con ácido nítrico o la digestión con ácido nítrico-ácido perclórico-ácido fluorhídrico, como se indica en la sección 3030. b) Eliminación de la interferencia de bario, calcio y estroncio: Tómese una muestra de 50,0 ml si se necesita o menor (para concentraciones más altas) con un contenido no superior a 10 mg Mg (para evitar la competencia en la coprecipitación). Añádanse 5,0 ml de reactivo Na2SO4-Na2CO3. Llévese a ebullición para coagular el precipitado de BaSO4, SrCO3, CaCO3 y MgCO3. Continúese calentando para reducir el volumen a un tercio. Retírese del calor y déjese reposar durante al menos 30 minutos para completar la precipitación; de otra forma aparecería un ligero precipitado de BaSO4 después de la filtración. Fíltrese*, láve* Whatman n.° 42 o equivalente. MÉTODOS NORMALIZADOS 3-126 se con agua destilada y dilúyase hasta 50,0 ml después de que la solución haya alcanzado la temperatura ambiente, para medirla por fotometría de llama. c) Tratamiento de soluciones patrón: Prepárese una solución patrón de 2 μg Li/55 ml por dilución de 1 ml de solución de litio patrón hasta 50,00 ml, añadiendo 5 ml de reactivo Na 2SO 4 -Na 2CO3 y mezcla. Prepárense de manera análoga diluciones de la solución patrón de Li para horquillar la concentración de la muestra o para establecer al menos tres puntos sobre una curva de calibración de absorbancia frente a μg Li/55 ml. d) Medida por fotometría de llama: Determínese la concentración de litio por medidas directas de intensidad a una longitud de onda de 670,8 nm. (Con unos instrumentos fotométricos puede emplearse el método de horquillado, mientras que otros exigen la construcción de una curva de calibración.) Trátese la muestra, el agua destilada y el patrón de litio tan simultáneamente como sea posible. Para obtener los mejores resultados, sáquese la media de varias lecturas con cada solución. Síganse las instrucciones del fabricante en cuanto al funcionamiento del instrumento. 5. Cálculos El volumen final real es de 50 ml; la curva de calibración se supone basada en 55 ml de volumen final, como en el apartado 4c, haciéndose el cálculo con el factor 0,9. 6. Control de calidad Trátese un patrón de CC sometiéndolo al protocolo analítico completo como una forma de determinar el sesgo sistemático. Los límites del control de la precisión de determinaciones de duplicados a concentraciones (en agua para reactivos) de 4,0 μg/l y 10,0 μg /1 fueron 4,09 ± ± 0,056 μg/1 y 9,96 ± 0,094 μg/l, respectivamente. La DRE (desviación relativa estándar) fue 1,38 por 100 para un solo operador para una solución de litio con 10 μg/1. 7. Bibliografía KUEMMEL, D. F. & H. L. KARL. 1954. Fíame photometric determination of alkali and alkaline earth elements in cast iron. Anal. Chem. 26:386. BRUMBAUGH, R. J. & W. E. FANUS. 1954. Determination of lithium in spodumene by fíame photometry. Anal. Chem. 26:463. ELLESTAD, R. B. & E. L. HORSTMAN. 1955. Fíame photometric determination of lithium in silicate rocks. Anal. Chem. 27:1229. WHISMAN, M. & B. H. ECCLESTON. 1955. Fíame spectra of twenty metals using a recording name spectrophotometer. Anal. Chem. 27:1861. HORSTMAN, E. L. 1956. Fíame photometric determination of lithium, rubidium, and cesium in silicate rocks. Anal. Chem. 28:1417. URE, A. M. & R. L. MiTCHELL. 1975. Lithium, sodium, potassium, rubidium, and cesium. In J. A. Dean & T. C. Rains, eds. Fíame Emission and Atomic Absorption Spectrometry. Dekker, Nueva York. PICKETT, E. E. & J. L. HAWKINS. 1981. Determination of lithium in small-animal tissues at physiological levels by fíame emission photometry. Anal. Biochem. 112:213. 3-127 DETERMINACIÓN DE METALES 3500-Mg 3500-Mg A. 1. Incidencia El magnesio ocupa el octavo lugar entre los elementos más abundantes y es un componente común de las aguas naturales. Las sales de magnesio, que contribuyen de forma importante a la dureza del agua, se descomponen al calentar formando costras en las calderas. Las concentraciones superiores a 125 mg/1 pueden tener un efecto purificador y diurético. El ablandamiento químico, la osmosis inversa, la electrodiálisis o el intercambio iónico reducen el magnesio y la dureza asociada a él a niveles aceptables. La concentración de magnesio puede variar des- * Aprobado por el Standard Methods Committee, 1985. 3500-Mg B. MAGNESIO* Introducción de cero a varios cientos de miligramos por litro, dependiendo del origen y tratamiento del agua. 2. Selección del método Los cuatro métodos presentados son aplicables a todas las aguas naturales. Con los métodos espectrométrico de absorción atómica y de plasma de acoplamiento inductivo pueden realizarse determinaciones directas. Por el método gravimétrico sólo se puede determinar el magnesio después de la eliminación de las sales de calcio (véase sección 3500Ca). Estos métodos pueden aplicarse a todas las concentraciones seleccionando las porciones de muestra apropiadas. La elección del método es cuestión de preferencia personal y experiencia analítica. Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, sección 3111 B. 3500-Mg C. Método de plasma de acoplamiento inductivo Véase sección 3120. MÉTODOS NORMALIZADOS 3-128 3500-Mg D. 1. Método gravimétrico Discusión general a) Principio: El fosfato hidrógeno diamónico precipita cuantitativamente el magnesio en solución amoniacal en forma de fosfato amónico magnésico. El precipitado se incinera a pirofosfato de magnesio y se pesa como tal. Se puede elegir entre: (a) destrucción del oxalato y las sales de amonio, seguido de precipitación única del fosfato amónico magnésico, y (b) doble precipitación sin pretratamiento. Cuando el tiempo no es un factor, es preferible la doble precipitación porque, aunque el pretratamiento es más rápido, requiere más atención para evitar pérdidas mecánicas. b) Interferencia: La solución deberá estar razonablemente libre de aluminio, calcio, hierro, manganeso, sílice, estroncio y materia suspendida. No deberá contener más de aproximadamente 3,5 g NH4C1. 2. Reactivos a) Ácido nítrico, NHO3 conc. b) Ácido clorhídrico, HC1 conc; también 1 + 1, 1 + 9 y 1 + 99. c) Solución de indicador rojo de metilo: Disuélvanse 100 mg de sal sódica de rojo de metilo en agua destilada y dilúyase hasta 100 ml. d) Solución de fosfato de hidrógeno diamonio: Disuélvanse 30 g de (NH4)2HPO4 en agua destilada y llévese hasta 100 ml. e) Hidróxido amónico, HN4OH conc; también 1 + 19. 3. Procedimiento a) Por eliminación de oxalato y sales de amonio: Al filtrado combinado con los lavados de la determinación de calcio, que contiene no más de 60 mg Mg, o a una porción con menos de esa cantidad, en un vaso de 600 u 800 ml, añádanse 50 ml de HNO3 conc. y evapórese cuidadosamente hasta sequedad sobre una placa caliente. No dejar que la reacción se haga demasido violenta en la última parte de la evaporación; préstese continua atención para evitar pérdidas por salpicado. Humedézcase el residuo con 2 a 3 ml de HC1 conc; añádanse 20 ml de agua destilada, caliéntese, fíltrese y lávese. Añádanse al filtrado 3 ml de HC1 conc, dos a tres gotas de solución de rojo de metilo y 10 ml de solución de (NH4)2HPO4. Enfríese y añádase NH4OH gota a gota, agitando continuamente, hasta que el color vira al amarillo. Agítese durante 5 minutos, añádanse 5 ml de NH4OH conc. y agítese vigorosamente durante 10 minutos más. Déjese reposar toda la noche y fíltrese con papel de filtro. Lávese con NH4OH 1 + 19. Pásese a un crisol incinerado, enfriado y pesado. Séquese cuidadosamente el precipitado, elimínese el papel quemándolo lentamente, dejando que circule el aire. Caliéntese hasta aproximadamente 500 °C hasta que el residuo queda blanco. Incinérese a 1.100 °C durante períodos de 30 minutos, hasta peso constante. b) Por doble precipitación: Al filtrado y los lavados combinados de la determinación de calcio, que contiene no más de 60 mg Mg, o una porción con menos de esa cantidad, añádanse dos a tres gotas de solución rojo de metilo; ajústese el volumen a 150 ml y acidúlese con HC1 1 + 1. Añádanse 10 ml de solución de (NH4)2HPO4. Enfríese. Añádase gota a gota NH4OH, agitando constantemente, hasta que el color vira a amarillo. Agítese durante 5 minutos, añádanse 5 ml de NH4OH conc. y agítese vigorosamente durante 10 minutos más. Déjese 3-129 DETERMINACIÓN DE METALES reposar toda la noche y después fíltrese a través de papel de Filtro*. Lávese con NH4OH 1 + 19. Descártese filtrado y lavados. Disuélvase el precipitado con 50 ml de HC1 1 + 9 templado y lávase bien el papel con HC1 1 + 99 caliente. Añádanse dos a tres gotas de solución de rojo de metilo, ajústese el volumen a 100 a 150 ml, añádanse 1 a 2 ml de solución de (NH4)2HPO4 y precipítese como antes. Déjese reposar en un lugar fresco durante al menos 4 horas o preferiblemente toda la noche. Fíltrese a través de papel de filtro* y lávese con NH 4 OH 1 + 19. Pásese a un crisol incinerado, enfriado y pesado. Séquese cuidadosamente el precipitado y quémese el papel lentamente, dejando que circule el aire. Caliéntese a aproximadamente 500 °C hasta que el residuo sea blanco. Incinérese a 1.100 °C durante períodos de 30 minutos, hasta peso constante. * Cari Schleicher and Schuell Co., S & S n.° 589 White Ribbon, o equivalente. 4. Cálculos 5. Precisión y sesgo En ocho laboratorios fue analizada una muestra sintética con 82 mg Mg/1, 108 mg Ca/1, 3,1 mg K/l, 19,9 mg Na/1, 241 mg Cl¯/1, 1,1 mg NO3- -N/l, 0,250 mg NO-2 -N/l, 259 mg SO 2-4 /l y 42,5 mg de alcalinidad total/1 (a la que contribuye el NaHCO3) utilizando el método gravimétrico, con una desviación relativa estándar de 6,3 por 100 y un error relativo de 4,9 por 100. 6. Bibliografía EPPERSON, A. W. 1928. The pyrophosphate method for the determination of magnesium and phosphoric anhydride. J. Amer. Chem. Soc. 50:321. KOLTHOFF, I. M. & E. B. SANDELL. 1952. Textbook of Quantitative Inorganic Analysis, 3. a ed. MacMillan Co., Nueva York, capítulo 22. HILLEBRAND, W. F. y otros. 1953. Applied Inorganic Analysis, 2.a ed. John Wiley & Sons, Nueva York, capítulo 41 y págs. 133-134. 3500-Mg E. Método de cálculo El magnesio puede calcularse como diferencia entre la dureza y el calcio, como CaCO3, si los metales que interfieren están presentes en concentraciones no interfirientes en la titulación del calcio (sección 3500-Ca.D) y se utilizan inhibi- dores adecuados en la titulación de la dureza (sección 2340C). mg Mg/1 = [dureza total (como mg CaCO3/l) – – dureza de calcio (como mg CaCO 3 /l)] x x 0,243 3-130 MÉTODOS NORMALIZADOS 3500-Mn MANGANESO 3500-Mn A. Introducción 1. Incidencia Aunque el manganeso se encuentra en las aguas subterráneas en la forma iónica divalente soluble, debido a la ausencia de oxígeno, parte o todo el manganeso de una instalación de tratamiento de agua puede aparecer en un estado de valencia superior. La determinación del manganeso total no diferencia entre los diversos estados de valencia. El ion permanganato heptavalente se emplea para oxidar el manganeso y/o la materia orgánica causante del sabor. Debe detectarse con gran sensibilidad el exceso de permanganato, el manganeso trivalente en forma de complejo, o una suspensión de manganeso tetravalente, para el tratamiento de control y para evitar su descarga a un sistema de distribución. Existe la evidencia de que el manganeso se encuentra en las aguas superficiales tanto en suspensión en su forma tetravalente, como en la forma trivalente en un complejo soluble relativamente estable. Aunque raramente sobrepasa 1 mg/1, el manganeso produce manchas tenaces en la ropa lavada y en accesorios de instalaciones sanitarias. Los bajos límites de manganeso impuestos para un agua aceptable derivan de lo anterior más que de consideraciones toxicológicas. Frecuentemente se necesitan medios especiales de eliminación, tales * Aprobado por el Standard Methods Committee, 1988. como precipitación química, ajuste de pH, aireación y empleo de materiales especiales de intercambio iónico. El manganeso se encuentra en las aguas residuales domésticas, efluentes industriales y corrientes receptoras. 2. Selección del método Los métodos espectrométrico de absorción atómica y de plasma de acoplamiento inductivo permiten la determinación directa con aceptable sensibilidad y son los métodos que se eligen. Entre los diversos métodos colorimétricos, el método de persulfato es el preferido, debido a que el empleo de ion mercúrico puede controlar la interferencia de una concentración limitada de ion cloruro. 3. Toma de muestras y almacenamiento El manganeso puede estar en una forma soluble en un agua neutra al principio de tomar la muestra, pero se oxida a un grado de oxidación más alto y precipita o llega a ser adsorbido por las paredes del recipiente. Determínese el manganeso enseguida de tomar la muestra. Cuando es inevitable un cierto retraso, se puede determinar el manganeso total si se acidula la muestra en el momento de su toma empleando HNO 3 hasta un pH < 2. Véase sección 3010B. 3-131 DETERMINACIÓN DE METALES 3500-Mn B. Método espectrométrico de absorción atómica Véase método espectrométrico de absorción atómica de llama, secciones 3111B y C, y método espectrométrico de 3500-Mn C. absorción atómica electrotérmica, sección 3113. Método de plasma de acoplamiento inductivo Véase sección 3120. 3500-Mn D. Método de persulfato 1. Discusión general a) Principio: La oxidación con persulfato de los compuestos mánganosos solubles para formar permanganato se realiza en presencia de nitrato de plata. El color resultante es estable durante 24 horas al menos, si existe un exceso de persulfato y si no hay materia orgánica. b) Interferencia: La interferencia de hasta 0,1 g de cloruro (Cl¯) en una muestra de 50 ml puede evitarse añadiendo 1 g de sulfato mercúrico (HgSO4) para formar complejos ligeramente disociados. Aún interferirán el bromuro y el yoduro y sólo pueden estar presentes en cantidades traza. El procedimiento del persulfato puede ser empleado para agua potable con cantidades traza a pequeñas de materia orgánica si se aumenta el período de calentamiento después de añadir más persulfato. Para las aguas residuales que contienen materia orgánica, utilícese una digestión preliminar con ácidos nítrico y sulfúrico (HNO3 y H2SO4) (véase sección 3O3OG). Si existen grandes cantidades de Cl¯, la ebullición con HNO3 facilita su eliminación. Las trazas de Cl se eliminan con HgSO4 en el reactivo especial. Las soluciones coloreadas de otros iones inorgánicos se compensan en la etapa colorimétrica final. Las muestras que han sido expuestas al aire pueden dar resultados bajos debido a la precipitación de dióxido de manganeso (MnO2). Añádase a la muestra una gota de peróxido de hidrógeno (H2O2) al 30 por 100, después de añadir el reactivo especial, para redisolver el manganeso precipitado. c) Concentración mínima detectable: La capacidad de absorción molar del ion permanganato es aproximadamente 2.300 l g -1 cm -1. Esto corresponde a una concentración mínima detectable (98 por 100 de transmitancia) de 210 μg Mn/1 cuando se utiliza una célula de 1 cm, o de 42 μg Mn/1 cuando se utiliza una célula de 5 cm. 2. Instrumental Equipo colorimétrico: Se necesita uno de los siguientes: MÉTODOS NORMALIZADOS 3-132 a) Espectrofotómetro, para utilizarlo a 525 nm, que proporciona un trayecto luminoso de 1 cm o más largo. b) Fotómetro de filtro, que proporciona un trayecto luminoso de 1 cm o más largo y equipado con un filtro verde que tiene un máximo de transmitancia próximo a 525 nm. c) Tubos Nessler, emparejados, 100 ml, forma alargada. 3. mirán aproximadamente 15 ml de solución de permanganato. Trátese un blanco de agua destilada y H2SO4. donde: A = ml de reactivo de titulación para la muestra, y B = ml de reactivo de titulación para el blanco. Reactivos a) Reactivo especial: Disuélvanse 75 g de H2SO4 en 400 ml de HNO3 conc. y 200 ml de agua destilada. Añádanse 200 ml de ácido fosfórico (H3PO4) al 85 por 100 y 35 mg de nitrato de plata (AgNO3). Dilúyase la solución enfriada hasta 1 l. b) Persulfato amónico, (NH4)2S2O8, sólido. c) Solución de manganeso patrón: Prepárese una solución de permanganato potásico (KMnO4) 0,1N disolviendo 3,2 g de KMnO 4 en agua destilada y llevándolo hasta 1 l. Déjese envejecer durante varias semanas a la luz del sol o calentando durante varias horas cerca del punto de ebullición, fíltrese después a través de un crisol filtrante de vidrio poroso fino y estandarícese frente a oxalato de sodio como sigue: Pésense varias muestras de 100 a 200 mg de Na2C2O4 con precisión de 0,1 mg y pásense a vasos de 400 ml. Añádanse a cada vaso 100 ml de agua destilada y agítese para que se disuelva. Añádanse 10 ml de H2SO4 1 + 1 y caliéntese rápidamente entre 90 y 95 °C. Valórese rápidamente con la solución de KMnO4 que se estandariza, agitando al mismo tiempo, hasta un color rosa ligero de punto final que persiste durante al menos 1 minuto. No dejar que la temperatura caiga por debajo de 85 °C. Si es necesario, caliéntese el contenido del vaso durante la titulación; 100 mg de Na2C2O4 consu- Sáquese la media de los resultados de varias titulaciones. Calcúlese el volumen necesario de esta solución para preparar 1 l de solución en la que 1,00 ml = 50,0 μg Mn, como sigue: Añádanse a este volumen entre 2 y 3 ml de H2SO4 conc. y solución de NaHSO3 gota a gota, agitando al mismo tiempo, hasta que el color del permanganato desaparece. Llévese a ebullición para eliminar el exceso de SO2, enfríese y dilúyase hasta 1.000 ml con agua destilada. Dilúyase más esta solución en el caso de medir pequeñas cantidades de manganeso. d) Solución de manganeso patrón (alternativa): Disuélvanse 1.000 g de metal manganeso (99,8 por 100 mínimo) en 10 ml de HNO 3 redestilado. Dilúyase hasta 1.000 ml con HC1 al 1 por 100(v/v); 1 ml = 1.000 mg Mn. Dilúyanse 10 ml hasta 200 ml con agua destilada; 1 ml = = 0,05 mg Mn. Prepárese la solución diluida diariamente. e) Peróxido de hidrógeno, H2O2, 30 por 100. f) Ácido nítrico, HNO3 conc. g) Ácido sulfúrico, H2SO4 conc. h) Solución de nitrito sódico: Disuélvanse 5,0 g de NaNO2 en 95 ml de agua destilada. DETERMINACIÓN DE METALES i) Oxalato de sodio, Na2C2O4, patrón primario. j) Bisulfito sódico: Disuélvanse 10 g de NaHSÓ3 en 100 ml de agua destilada. 3-133 tabla muestra la longitud apropiada del trayecto luminoso para diversas cantidades de manganeso en 100 ml de volumen final: 4. Procedimiento a) Tratamiento de la muestra: Si se ha preparado una muestra digerida según las indicaciones de reducción de la materia orgánica y/o los cloruros en exceso de la sección 3030G, llévese con la pipeta una porción que contenga 0,05 a 2,0 mg de Mn a un erlenmeyer de 250 ml. Añádase agua destilada hasta 90 ml, si es necesario, y procédase como en el apartado 4b. b) Añádanse 5 ml de reactivo especial y una gota de H2O2 a una porción adecuada de la muestra. Concéntrese hasta 90 ml por ebullición o dilúyase hasta 90 ml. Añádase 1 g de (NH4)2S2O8, llévese a ebullición y déjese hervir durante 1 minuto. No calentar en baño maría. Sepárese de la fuente de calor, déjese reposar 1 minuto y enfríese al grifo (una ebullición demasiado prolongada da lugar a la descomposición del exceso de persulfato y, por consiguiente, a la pérdida de color del permanganato; un enfriamiento demasiado lento tiene el mismo efecto). Dilúyase hasta 100 ml con agua destilada libre de sustancias reductoras y mézclese. Prepárense patrones que contengan 0, 5,00,... 1.500 μg de Mn tratando diversas cantidades de soluciones de Mn patrón de la misma forma. c) Comparación con tubos de Nessler: Utilícense los patrones preparados como en el apartado 4b y que contienen entre 5 y 100 μg Mn/100 ml de volumen final. Compárense muestras y patrones visualmente. d) Determinación fotométrica: Utilícese una serie de patrones de 0 a 1.500 μg de Mn/100 ml de volumen final. Realícense las medidas fotométricas frente a un blanco de agua destilada. La siguiente Prepárese una curva de calibración de concentración de manganeso en función de la absorbancia de los patrones y determínese el Mn en las muestras a partir de la curva. Si existe turbidez o color que interfiere, hágase la corrección como en el apartado 4e. e) Corrección para turbidez o color que interfiere: Evítese la filtración por la posible retención de parte del permanganato sobre el papel de filtro. Si se emplea la comparación visual, sólo puede calcularse el efecto de la turbidez sin que pueda hacerse ninguna corrección para los iones coloreados que interfieren. Cuando se realizan medidas fotométricas, utilícese el siguiente método de «decoloración», que también corrige el color que interfiere. En cuanto se ha hecho la lectura en el fotómetro, añádanse 0,05 ml de solución de H2O2 directamente a la muestra en la célula óptica. Mézclese y, tan pronto como el color de permanganato ha desaparecido por completo y no quedan burbujas, léase de nuevo. Dedúzcase la absorbancia de la solución decolorada de la absorbancia inicial para obtener la absorbancia debida al Mn. 5. Cálculo a) Cuando se toma toda la muestra original para análisis: 3-134 MÉTODOS NORMALIZADOS b) Cuando se toma una porción de la muestra digerida (100 ml) para el análisis: En 17 laboratorios se analizó una segunda muestra sintética análoga en todo, excepto en que tenía 50 μg de Mn/1 y 1.000 μg Cu/1, por el método de persulfato, con una desviación relativa estándar de 50,3 por 100 y un error relativo de 7,2 por 100. 7. 6. Precisión y sesgo Se analizó en 33 laboratorios una muestra sintética con 120 μg Mn/1, 500 μg Al/1, 50 μg Cd/1, 110 μg Cr/1, 470 μg Cu/1, 300 μg Fe/1, 70 μg Pb/1, 150 μg Ag/1 y 650 μg Zn/1 en agua destilada por el método de persulfato, con una desviación relativa estándar de 26,3 por 100 y un error relativo de 0 por 100. 3500 Hg RICHARDS, M. D. 1930. Colorimetric determination of manganese in biological material. Analyst 55:554. NYDAHL , F. 1949. Determination of manganese by the persulfate method. Anal. Chem. Acta. 3:144. MILLS, S. M. 1950. Elusive manganese. Water Sewage Works 97:92. SANDELL, E. B. 1959. Colorimetric Determination of Traces of Metals, 3.a ed. Interscience Publishers, Nueva York, capítulo 26. MERCURIO* 3500-Hg A. 1. Significación Las sales orgánicas e inorgánicas de mercurio son muy tóxicas y su presencia en el ambiente, y en especial en el agua, debe controlarse. Introducción las muestras, aunque el método de ditizona es útil para determinar niveles elevados de mercurio (>2 μg/1) en aguas potables. 3. 2. Selección del método El método de absorción atómica de vapor frío es el seleccionado para todas * Aprobado por el Standard Methods Committee, 1985. Bibliografía Conservación de las muestras Como el mercurio de las muestras puede perderse con facilidad, es necesario tratarlas con HNO3 para reducir el pH a < 2 (véase sección 1060). 3-135 DETERMINACIÓN DE METALES 3500-Hg B. Método de absorción atómica de vapor frío Véase sección 3112. 3500-Hg C. 1. Método de la ditizona Discusión general a) Principio: Los iones mercurio reaccionan con una solución de ditizona en cloroformo para formar un color naranja. Los distintos tonos del color naranja se miden en un espectrofotómetro y las concentraciones desconocidas se calculan a partir de una curva estándar. b) Interferencia: El cobre, oro, paladio, platino divalente y plata reaccionan con ditizona en solución acida. El cobre del extracto con ditizona permanece en la fase orgánica, mientras que el mercurio se disuelve en la fase acuosa. Los otros contaminantes no suelen estar presentes. El ditizonato de