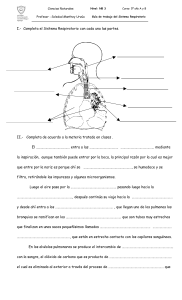
BASES FISIOLOGICAS DE LA FIEBRE Para la adecuada compres de las bases fisiológicas y fisiopatológicas de la fiebre se debe entender la producción de la misma como la respuesta a una serie de eventos que generan interaccion organica y tisular con sustancias capaces de generar una respuesta que generalmente esta medida por la producción de sustancias endógenas conocidas como pirógenos endógenos los cuales se genera por la influencia de sustancias exógenas conocidas como pirógenos endógenos. Esta interacción de pirógenos exógenos con las sustancias pirógenas del cuerpo humano se inicia cuando dichas sustancias pirógenas entran en contacto molecular y químico con células y sustancias endógenas. Un modelo sencillo es sobre las patologías infecciosas las cuales a travez de algunas sustancias propias de su estructura ( pared bacteriana , lípidos de membrana, sustancias secretadas ) interacciona con elementos celulares y químicos a través de receptores moleculares desencadenando la respuesta inflamatoria y la producción de señales internas que van a generar la producción de sustancias endógenas que actúan como pirógenos y causan el desarrollo de la elevación de la temperatura y por consiguiente la fiebre . Modelo de inducción de producción de citoquinas por la via del factor de transcripción nuclear kappa beta y el signalosoma ( se activan a nivel de los linfocitos ) su efecto se dirige hacia el nucleo celular donde genera una serie de interacciones genómicas activando e inhibiendo la expresión génico con la subsecuente producción de señales enfocadas en la producción de interluquinas pro inflamatorias ( IL-1 , IL-6 y IL12 ) y se asocian a la producción de fienre por interacciones posteriores. También está activación del FTNKB se relación con la activación de moléculas de adhesión y con la generación de proteínas de membrana a nivel de las ceulas del sistema retículo endotelal fagocitico que facilitan la migración y diapédesis de estas a diferentes órganos y sistemas ( SNC a través de la barrera hematoencefalica ) Una vez se genera la producción de pirógenos endógenos mediante las diferentes vías de señalización y la interaccion del FTNKB y el signalosoma y otras vías vías , la acción de estas sustancias genera efectos ssitemicos como el aumento de quimiotaxis y diapédesis de células del sistema retículo endotelial fagocitico , aumento de los procesos de la actividad de oxidación celular (metabolica) activación o inhibición de vías metabolicas . porduccion de anticuerpos y citoquinas y potenciación de la capacidad citotóxica de los linfocitos ( LA FIEBRE Y LOS PIROGENOS ENDOGENOS ACTUAN COMO INDUCTORES Y PROPAGADORES DE LA RESPUESTA INMUNE ) Sin importar la via de ña actividad de los pirógenos endógenos estos logran generar el estimulo sobre el SNC la producción final y determinante de la regulación térmica esta medaca por la activación de la cicloxigenasa y la producción de prostaglandina ( E2) y esta general un estímulo a nivel de la región pre óptica localizada en el hipotálamo anterior generando la producción de otra serie de interluquinas que mediaran la producción de la respuesta febril. Entonces lo que se conoce es el efecto de las sustancias pirogenadas y E2 en el punto prefijado de control térmico hipotalámico elevando este lo que genera una adaptación para lograr un nivel más alto de temperatura con la generación de las manifestaciones clínicas como eltemblor , el aumento del matebolismo celular , calambres musculares y etc. FISIOPATOLOGIA DE LA DISNEA En la fisiología de la respiración tenemos que tener en cuenta varios factores anatómicos que son los que van a participar en la respiración, desde la corteza cerebral hasta los alveolos. En la corteza cerebral lo que se da el estímulo al centro respiratorio que se encuentra en el bulbo raquídeo 1) Esto va ocasión que se produzca una descarga hacia los músculos respiratorio R. SENSITIVOS MECANORECEP TORES 3) por ultimo llega a los alveolos y ahí se da el intercambio gaseoso, sale el CO2 y el O2 entra QUIMIORECEPTORES 2) Y los músculos respiratorios van hacer que podamos expandir nuestra caja torácica y aumentar nuestro volumen pulmonar (disminuye el diafragma y se agranda nuestra caja torácica) Tenemos que tener en cuenta varios aspectos importantes uno es que cada uno de los factores anatómicos mencionados van a tener unos receptores específicos los cuales activaran el centro respiratorio para aumentar la frecuencia o la profundidad de la respiración si es que tenemos la sensación de falta de aire (DISNEA) R. SENSITIVOS: en el momento en que no podamos expandir bien los músculos respetarlos este estimulara la corteza cerebral hacia los centros respiratorios para comenzar a hiperventilar. MECANORECEPTORES: (EN LA EXPACION DE LA CAJA TORACICA CUANDO DISMINUYE) son receptores que si hay una disminución hacen un estímulo para aumentar la frecuencia respiratoria (cuando disminuye nuestro volumen pulmonar tambien tenemos receptores en la vía aérea que se encuentran en el pulmón) cuando hay una distención o una irritación en las fibras celfs neuronales va a producir que también se estimule al centro respiratorio. Quimiorreceptores: (estan dispuestos en 2 aspectos, cuando aumenta la presion arterial de co2 en la sangre princialmente o cuando disminuye la presion de o2 en la sangre) estos 2 aspectos van a ser muy improtantes en lso quimiorrecpetores los cuales van a actuar como estimulo que llegue hacia el centro respiratorio para comenzar aumentar la frecuencia respiratoria. TENER EN CUENTA: EL APARATO RESPIRATORIO ESTA CONFORMADO POR NUESTRO SNC Y SNP Y ESTOS VAN A COORDINAR EL FUNCIONAMIENTOS DE TODAS LAS ESTRUCTURAS MENCIONADAS ( PULMONES Y VIAS AEREAS , VASCULARIZACION PULMONAR , CAJA TORACICA ) SI HAY ALGUNA FALLA O FRACTURA SE PRODUCE UNA ALTERACION DE LA RESPIRACION , LLEVANDO ACONTECER UNA DISNEA . LA COORDINACION CENTRAL TAMBIEN ESTA DADO POR LOS GRUPOS RESIPTAROIOS QUE ESTAN EN EL TALLO CEREBRAL ( G.R DORSAL, G.R VENTRAL , CENTRO NEUMOTAXICO Y CENTRO APNEUSTICO ) G.R.DORSAL = SE ENCARGABA DE ENVIAS AL EFERENCIAS DEL INSPIRACION G. R. VENTRAL : EN CONDICIONES NORMALES NO MANDA EFERENCIAS PERO CUANDO HAY UNA ESPIRACION MAXIMA , ERA EL QUE SE ENCARGABA DE ENVIARLAS . COMPLEJO PRE-BOTZINGER: SE ENCUENTRA EN EL GRUPO RESPIRATORIO VENTRAL Y ES EL ENCARGADO DE REGULAR EL RITMO RESPIRATORIO (MARCAPASO) DICE CUAL ES LA FRENCUENCIA RESPITATORIA Y LO HACE APAORTIR DE CONECCIONES CON EL G.R .DORSAL CON LAS CUALES REGULA LA FRENCUENCIA RESPIRATORIA . ESTAN DESCRITAS LAS 2 POSIBLES SENSACIONES , HAMBRE DE AIRE Y ESFUERZO PARA RESPIRAR U OPRESION EN EL PECHO Hambre de aire: esta mediada por quimiorreceptores periféricos que están en el seno carotideo y en callado de la aorta. Sensación de opresión o esfuerzo para respirar mecano receptores, receptores de estiramiento, receptores j ,ellos cuando censan las alteraciones envían unas aferencias al tallo cerebral y ahí se produce una respuesta que busca o aumentar o disminuir el trabajo de los musculares espiratorios e inspiratorios . COMPENSACION DE LA DISNEA FISIOPATOLOGIA DE PURPURA La púrpura es producida por la extravasación de células sanguíneas a la piel y/o mucosas, dando origen a lesiones de coloración purpúrea que no desaparecen a la vitropresión. A nivel clínico, se manifiesta con hemorragias cutáneomucosas, pudiendo afectar en casos graves a cualquier otro órgano o sistema, condicionando sintomatología muy diversa. Como su nombre indica es una situación en la que la piel tienen manchas que ese color, violeta o púrpura, y se debe a que los hematíes han salido de los vasos, de las venas, por debajo de la piel, allí han transformado su hemoglobina tomando ese color violeta. FISIOPATOLOGIA DE LAS PETEQUIAS Las petequias son pequeñas manchas hemorrágicas en la piel, que pueden tener una dimensión variable, y ser tan pequeñas como una cabeza de alfiler o de mayor tamaño. Son lesiones que no desaparecen bajo la presión del dedo, a diferencia de otras lesiones en la piel como por ejemplo los exantemas que tan frecuentemente afectan a los niños. Las petequias se pueden producir por diferentes mecanismos como por ejemplo por rotura de los pequeños capilares que llegan a la piel. El tamaño de las petequias es un factor determinante para sospechar su origen. Igualmente es importante su localización. De esta manera, las de pequeño tamaño (menores de 2 milímetros) y que están por encima de la línea de las mamilas suelen ser orientativas de cuadro más banal. Por ejemplo son las que se observan tras los accesos de tos o los vómitos, especialmente en algunas personas que tienen la piel más blanca y cierta fragilidad capilar. SEPSIS Los microorganismos proliferan en un nido de infección, invaden luego el torrente sanguíneo y liberan una gran cantidad de sustancias, por ejemplo, la toxina 1 del síndrome de shock tóxico (TSST1) o endotoxinas de microorganismos gramnegativos. Estos productos estimulan la liberación de varios mediadores endógenos derivados del huésped y del microorganismo que interactúa y se acelera o inhibe entre sí. 10 Mediadores procedentes de los microorganismos como la toxina A producida por la Pseudomona aeruginosa y la TSST1 producida por los estafilococos o la endotoxina (LPS) lipopolisacárido de la membrana de los gérmenes gramnegativos, son el típico ejemplo inductor de la secuencia patogénica del choque séptico (Anexo 1) 1- Tenemos primero el inicio de una infección, para ello tenemos el ingreso de distintos microrganismos que puede ingresar ya sea bacterias, virus como: Virus sincicial respiratorio (VSR) Mycoplasma Pneumoniae Chlamydia pneumoniae 2- Una vez que ingresan hacen una invasión del torrente sanguíneo y se da la liberación de sustancias como: Endotoxinas (Lipopolisacáridos) Peptidoglicano Ácido lipoproteico 3- Y posteriormente se da la liberación de mediadores de endógenos de huésped que son las citocinas que estar va a desarrollar: Mecanismos de defensa del huésped Efectos nocivos 4- Y ya de los efectos nocivos, posteriormente se va a ver visto actuado por el Factor de Necrosis Tumoral (FNT), que viene hacer la citocinas responsable del inicio de un shock. 5- Posteriormente de esta se van se van a generar otras citocinas proinflamatorias como: IL-1 IL-2 IL-6 6- Asimismo actúa sobre sus receptores en la mayoría de las células. 7- Ambos van a emitir una respuesta inflamatoria. 8- Por otro lado, el FNT también va activar a otros mediadores de células endoteliales y este va a librar un vasodilatador poderoso que viene hacer el OXIDO NITRICO, este va a provocar: Vasodilatación potente Hipotensión 9- Y esto va a activar también una respuesta inflamatoria. 10- Por otra parte, cuando se da en un principio del mecanismo se da la invasión del torrente sanguíneo que posteriormente se va a liberar sustancias, que estas a su vez, van a activar la secuencia de complemento. 11- Y esto va a liberar anafilotoxinas como: C3a C5b 12- Y este a su vez va a provocar lo siguiente: Vasodilatación ↑ permeabilidad vascular Agregación plaquetaria Agregación de neutrófilos 13- Posteriormente se va a ver anormalidades micro vasculares vinculadas con shock séptico. 14- Esto va a activar: F. XII de coagulación Calicreína Cininógeno 15- Por lo que A través de la liberación de BRADICIDINA se va a dar: Vasodilatación potente Hipotensión 16- Lo cual también a activar una respuesta inflamatoria. 17- Como vemos por 3 mecanismos se puede llegar a una respuesta inflamatoria cierto, ahora esa respuesta inflamatoria puede ser la fiebre como se muestra en la fisiopatología, o también puede ser la tos seca, entre otros. FISIOPATOLOGIA DE LUPUS FISIOPATOLOGIA DE ARTRITIS REUMATOIDE La predisponían genética a tener AR se da por la activación de una respuesta mediada por células T a un factor desencadenante inmunitario, como algún microorganismo. Además, hay una expresión de ciertos genes del complejo mayor de histocompatibilidad (CMH) de una manera ordenada en personas con AR. Un locus genético importante que predispone a AR se encuentra presente en los locus del antígeno leucocitario humano (ALH) en las moléculas CMH clase II, con un foco específico en el locus de DRB1. Este gen ALH-DRB1, que forma un bolsillo reumatoide en la molécula de ALH, puede influir sobre los tipos de péptidos que pueden enlazarse por las moléculas de ALH-DR relacionadas con AR, lo cual afecta la respuesta inmune. La patogenia de la AR es compleja y en ella intervienen diferentes poblaciones celulares implicadas en la respuesta inmune innata y adquirida. En su patogenia participan células residentes en la membrana sinovial, como los sinoviocitos B de estirpe fibroblástica o los macrófagos de la íntima, y las células inflamatorias provenientes de la sangre como los linfocitos T, los linfocitos B y los monocitos. Todas ellas contribuyen a la transformación agresiva del fenotipo de los sinoviocitos B y al desarrollo de un intenso infiltrado inflamatorio cuyo resultado final es la destrucción del cartílago y del hueso subcondral. La activación de las células T CD4+ es el punto de partida de una cascada de fenómenos proinflamatorios con producción de gran cantidad de citocinas y proliferación celular que, en caso de perpetuarse de forma mantenida, como ocurre en la AR, da lugar a una inflamación crónica muy activa, capaz de destruir los tejidos en los que se desencadena, principalmente en las articulaciones en el caso de la AR. En la membrana sinovial comienzan a proliferar las células infiltrantes provenientes de la sangre, como los propios linfocitos T y sus subtipos, y también los linfocitos B. Los monocitos se diferencian en macrófagos y osteoclastos y, además, activan a los condrocitos articulares. En este medio se producen grandes cantidades de citocinas proinflamatorias como la interleucina (IL)-1, la IL-6 y el factor de necrosis tumoral (TNF) entre otras muchas. Las células B producen también autoanticuerpos como el factor reumatoide o los anticuerpos antipéptidos citrulinados. Todo lo cual conduce a la destrucción no sólo de la membrana sinovial, sino también del hueso subyacente y del cartílago articular. Las personas con esta enfermedad tienen una sustancia: el factor reumatoide (FR), el cual es un anticuerpo autólogo (Ig FR), el cual va a reaccionar con un fragmento de inmunoglobulina G (IgG) para formar complejos inmunes. Los complejos inmunes (Ig FR + IgG) y los componentes del complemento se encuentran en la sinovia, el líquido sinovial y las lesiones extraarticulares de personas con AR. Estos complejos inmunes son fagocitados por los neutrófilos y macrófagos y en el proceso, liberan enzimas lisosómicas capaces de provocar cambios destructivos en el cartílago articular. La respuesta inflamatoria que sigue atrae células inflamatorias adicionales, lo que pone en movimiento una cadena de acontecimientos que perpetúa la condición. Conforme progresa el proceso inflamatorio, las células sinoviales y los tejidos subsinoviales presentan hiperplasia reactiva. La vasodilatación y el flujo sanguíneo incrementado ocasionan enrojecimiento y calor. La tumefacción articular que se encuentra se debe a una mayor permeabilidad capilar que acompaña al proceso inflamatorio. El rasgo característico de la AR es el desarrollo de una red extensa de nuevos vasos sanguíneos en la membrana sinovial que contribuye al avance de la sinovitis reumatoide. Este tejido de granulación vascular destructivo, denominado pannus, se extiende desde la sinovia para afectar una región de hueso desprotegido en la unión entre el cartílago y el hueso subcondral. El pannus es una característica de la AR que la diferencia de otras formas de artritis inflamatoria. Las células inflamatorias encontradas en el pannus tienen un efecto destructivo sobre el cartílago y el hueso adyacentes. Con el tiempo se desarrolla pannus entre los bordes articulares, lo que provoca movimiento articular reducido y la posibilidad de anquilosis. La evidencia señala a una de las metaloproteinasas de matriz (MPM) que degradan colágeno, la MPM tipo 1, como esencial para que el pannus invada y destruya articulaciones en la AR. Con la progresión de la enfermedad, la inflamación articular y los cambios estructurales resultantes ocasionan inestabilidad articular, atrofia muscular por desuso, estiramiento de los ligamentos y afección de los tendones y músculos. El efecto de los cambios patológicos en la estructura y función articulares se relaciona con el grado de actividad patológica, la cual puede cambiar en cualquier momento. Es desafortunado que los cambios destructivos sean irreversibles. FISIOPATOLOGIA DE LA ANEMIA La expresión clínica de la anemia es el resultado de la hipoxia tisular, y sus síntomas y signos específicos representan respuestas cardiovasculares compensadoras según la gravedad y la duración de esta hipoxia. Una anemia grave puede asociarse a debilidad, vértigo, cefaleas, acufenos, manchas en el campo visual, fatiga fácil, mareos, irritabilidad e, incluso, conducta extraña. Puede aparecer amenorrea, pérdida de la libido, molestias gastrointestinales y, en ocasiones, ictericia y esplenomegalia. Finalmente puede presentarse insuficiencia cardíaca o shock. La anemia es el resultado de una o más combinaciones de tres mecanismos básicos: pérdida de sangre, disminución de la producción o aumento de la destrucción de hematíes (hemólisis). La pérdida de sangre debe ser el primer factor a considerar. Una vez que éste se descarta sólo quedan los otros dos mecanismos. Como la supervivencia de los hematíes es de 120 días, el mantenimiento de una población estable requiere la renovación de 1/120 de las células diariamente. El cese completo de la producción de hematíes resulta en una disminución de aproximadamente el 10% semanal del valor control. Los defectos de producción tienen como resultado una reticulocitopenia relativa o absoluta. Cuando los valores de hematíes disminuyen a una velocidad mayor al 10% (es decir, 500.000/(l) sin evidencia de hemorragia, se establece que el factor causal es una hemólisis. Un abordaje apropiado en la mayoría de las anemias debidas a un defecto de producción es examinar los cambios celulares. Así, la presencia de hematíes microcíticos e hipocrómicos indica que el defecto de producción se debe a un trastorno en la síntesis del hemo o de la globina (déficit de hierro, talasemia y defectos de la síntesis de hemoglobina relacionados) o bien a la anemia de las enfermedades crónicas. Por el contrario, las anemias normocrómicas normocíticas por defectos de la producción sugieren un mecanismo hipoproliferativo o hipoplásico. Finalmente, algunas anemias se caracterizan por hematíes de gran tamaño, o macrocitos, lo que sugiere un defecto en la síntesis de ADN. Estas anemias se deben generalmente a defectos en el metabolismo de la vitamina B12 o del folato, o a una interferencia en la síntesis de ADN por agentes quimioterápicos citorreductores. Una respuesta medular adecuada a la anemia se evidencia por reticulocitosis o policromatofilia en sangre periférica. FISIOPATOLOGÍA Cuando existe anemia se producen una serie de efectos en el organismo, algunos debidos a la propia situación de hipoxia, pero la mayoría originados por la entrada en acción de distintos mecanismos compensadores. El principal efecto compensador consiste en la mayor capacidad de la hemoglobina para ceder oxígeno a los tejidos, como consecuencia de la desviación hacia la derecha de la curva de disociación de la hemoglobina. Esta disminución de la afinidad de la hemoglobina por el oxígeno se debe a la acción de dos mecanismos: en primer lugar, al producirse la hipoxia y, como consecuencia del metabolismo anaerobio ácido láctico, hay un descenso del pH y, por tanto, una desviación de la curva hacia la derecha (efecto Bohr). Con algo más de retraso se inicia el segundo mecanismo compensador que, aunque tardío, es más efectivo que el efecto Bohr: consiste en el aumento del 2,3difosfoglicerato (2,3-DPG) que actúa sobre la hemoglobina disminuyendo de forma eficaz su afinidad por el oxígeno. Al parecer, el aumento de la desoxihemoglobina produciría, por medio del aumento de la 2,3-DPG-ratomutasa, el incremento del 2,3-DPG. El siguiente mecanismo compensador en importancia consiste en la redistribución del flujo sanguíneo. Dado que en la anemia existe cierto grado de hipoxia tisular y que algunos órganos, como el cerebro y el miocardio, precisan para su funcionamiento una concentración de oxígeno mantenida dentro de límites estrechos, se produce una redistribución del flujo sanguíneo de órganos con menores requerimientos de oxígeno, como la piel y el riñón, hacia aquellos que más lo necesitan. El riñón no sufre efectos apreciables por la redistribución del flujo gracias a que, en condiciones normales, recibe el doble de oxígeno del mínimo necesario. Cuando la hemoglobina desciende por debajo de 7,5 g/dl (4,6 mmol/l), entra en acción otro mecanismo de compensación, el aumento del gasto cardíaco, que en situaciones graves puede incluso cuadruplicarse. El gasto cardíaco aumenta fundamentalmente gracias a la disminución de la poscarga (disminución de las resistencias periféricas y de la viscosidad sanguínea). En casos graves, la disminución de la concentración de oxígeno en la circulación coronaria servirá de estímulo para aumentar más el flujo cardíaco. La presión sistólica suele mantenerse, pero la diastólica tiende a descender, con lo que la tensión diferencial aumenta. Teóricamente, el mecanismo compensador más apropiado es el aumento de la producción de hematíes. En cualquier caso, este mecanismo es lento y sólo es efectivo si la médula ósea es capaz de responder de forma adecuada, como en la anemia posthemorrágica aguda, pero en otros casos no responde de manera apropiada, como ocurre en la anemia ferropénica o en la perniciosa. El aumento de la eritropoyesis, en los casos en que éste es posible, se debe al incremento de eritropoyetina, que se produce como respuesta a la hipoxia renal y posiblemente también extrarrenal. El papel compensador del aparato respiratorio es casi nulo, ya que la oxigenación de los hematíes es excelente a su paso por los pulmones en situación eupneica. La disnea y la taquipnea de esfuerzo que presentan los enfermos se debe a una respuesta inapropiada del centro respiratorio a la hipoxia o a una congestión pulmonar asociada.