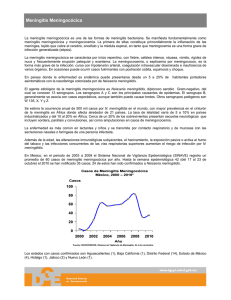
Patología neurológica Crisis convulsiva Convulsión es el fenómeno motor caracterizado por una contracción involuntaria de la musculatura del cuerpo, de apariencia tónica o clónica (según que su naturaleza sea continua o discontinua); puede ser de origen cerebral o espinal y se relaciona con muy diversos mecanismos. Crisis cerebral es el episodio caracterizado por fenómenos anormales súbitos y transitorios de tipo motor, sensitivo-sensorial, vegetativo o de afectación de la conciencia, como resultado de una disfunción cerebral transitoria, parcial o generalizada. Crisis epiléptica es la crisis cerebral resultante de la descarga excesiva de una población neuronal hiperexcitable. Puede ser ocasionalcuando acontece en una persona no epiléptica por efecto de una causa accidental (tóxico, infección, fiebre, trastorno vascular, etc.); se denomina única la crisis epiléptica espontánea no recurrente, son iterativas cuando se repiten a intervalos de tiempo variables y se denominan seriadascuando se presentan ordenadas en series o grupos. Epidemiologia Las crisis cerebrales, en general, y las epilépticas en particular, constituyen una patología frecuente en la infancia, estimándose que hasta un 8-10% de los niños las presentan en alguna ocasión y la prevalencia de la epilepsia en la población escolar de nuestro medio es del 0,5%. Su expresión clínica es alarmante, siendo posible que la familia considere que la vida del niño está en peligro o que puede originar secuelas, por lo que constituyen uno de los más frecuentes motivos de consulta en los servicios de urgencia. Convulsiones del recién nacido Las convulsiones se presentan en alrededor del 1% (entre el 0,2-1,4%) de los RN a término, porcentaje que se incrementa hasta el 20-25% en los prematuros, aunque su incidencia real es difícil de establecer ya que pueden pasar desapercibidas o ser confundidas con otros fenómenos motores, como las tremulaciones, que tienen diferente significado. Las peculiaridades estructurales y bioquímicas ya comentadas condicionan un umbral convulsivo más bajo y la expresión característica de las manifestaciones convulsivas que se relacionan con la etapa madurativa y destacan las crisis erráticas y la facilidad para presentarse estados de mal. Etiología La mayoría de los casos pueden considerarse como convulsiones “sintomáticas”, con una etiología identificable. Diversos factores maternos antenatales (nulíparas añosas, diabetes, deficiencia en vitamina D) y otros intraparto aumentan el riesgo de convulsiones neonatales. Predominan los casos relacionados con: encefalopatía hipóxico-isquémica (que en algunas series es responsable del 45% de los casos) y anomalías metabólicas: hipoglucemia, hipocalcemia, errores del metabolismo de los aminoácidos, deficiencia/dependencia de piridoxina, déficit de biotina o tiamina; trastornos relacionados con la natremia, bicarbonato, amoníaco, bilirrubina (kernícterus, hoy excepcional), trastornos peroxisomales, lisosomales o degenerativos de la sustancia gris. Siguen en importancia las infecciones del SNC, traumatismos del feto y RN, supresión brusca de drogas o fármacos (síndrome de abstinencia), fetopatía alcohólica, madre tratada con hidracidas o cuando se ha utilizado procaína para la anestesia del parto. Hay que considerar las disgenesias cerebrales y síndromes genéticos con afectación neurológica y la hemorragia intraventricular(especialmente en los nacidos pretérmino). Pese a la gran diversidad de posibles factores etiológicos, en porcentajes que oscilan (según las posibilidades de estudio) entre el 3-20% de los casos, no es posible determinar la causa (formas idiopáticas). Formas clínicas La semiología es muy variada, siendo a menudo atípicas y su reconocimiento no siempre es fácil, en especial en el prematuro en que, por razones anatómicas y funcionales, las crisis son menos organizadas. En el cerebro inmaduro del neonato, la actividad convulsiva tiene probablemente un origen subcortical. Por lo que las manifestaciones clínicas más frecuentes son movimientos bucolinguales (chupeteo, masticación) y oculares, así como apnea. Se distinguen los siguientes tipos: Tipos Crisis mínimas, atípicas (sutiles). Difíciles de reconocer por lo que, a menudo, pasan desapercibidas. Se exteriorizan por automatismos motores breves (succiones, clonías palpebrales, movimientos masticatorios, repetición paroxística del pedaleo, movimientos natatorios) o por fenómenos vegetativos (palidez, cianosis, babeo, cambios de ritmo respiratorio, apnea, hipotermia, bradicardia) y son más habituales en los prematuros. Crisis clónicas. Quizá las más características y frecuentes son localizadas (cara, extremidades, cuello, tronco) o difusas, en relación con lesiones focales (por ejemplo, infarto cerebral), aunque también con trastornos metabólicos; en las crisis clónicas difusas o multifocales las sacudidas saltan desordenadamente de unas a otras partes del cuerpo (por lo que también se las llama migratorias o erráticas), pero la migración no es típicamente jacksonniana. Crisis tónicas. Suelen ser generalizadas y se traducen por la extensión de las extremidades superiores e inferiores (recordando la postura de descerebración) o por la flexión tónica de las superiores y extensión de las inferiores (como en la decorticación) y en menos ocasiones son focales con posición mantenida de una extremidad o en una posición asimétrica del tronco, cuello o ambos. Crisis mioclónicas. Se distinguen de las clónicas porque las sacudidas son más rápidas y por su predilección por los músculos flexores. Pueden ser focales, multifocales y generalizadas. Son las menos frecuentes y se observan sobre todo en los prematuros con lesiones graves del SNC. Las crisis tónico-clónicas generalizadas, tan características de edades posteriores, no se presentan en el periodo neonatal. Convulsiones ocasionales del lactante En el lactante, época en la que todavía no se ha alcanzado la maduración cerebral y persisten la mayoría de los factores predisponentes y desencadenantes, las convulsiones también son frecuentes, con predominio de las crisis unilaterales, parciales (secuelares) y, menos, las generalizadas. El pediatra asistirá, en esta edad, a convulsiones esporádicas, únicas o repetidas, pero son ya más frecuentes las recidivantes, expresión de una verdadera epilepsia. Diagnostico Electroencefalograma. En el RN tiene características peculiares, variando incluso según la edad de gestación, y no siempre existe una correlación entre el EEG, ni siquiera el crítico y la clínica. Los hallazgos intercríticos son diversos: trazados lentos, hipoactivos, periódicos, anomalías focales o multifocales. En el trazado crítico, el origen de las descargas es, casi siempre, focal, pudiendo propagarse a todo el hemisferio del mismo lado o, más rara vez, al contralateral. Diagnóstico de la crisis. Puede plantear problemas en el caso de las mínimas o atípicas, cuyo reconocimiento clínico es difícil, como también lo es en neonatos ventilados mecánicamente, en los que la evidencia de fluctuaciones rítmicas de las constantes vitales y la oxigenación deben alertar acerca de una posible convulsión; al no existir una buena correlación clínico-EEG. Diagnóstico etiológico. Se realiza valorando los datos de la anamnesis, exploración clínica y los estudios complementarios. Las investigaciones de laboratorio a realizar de forma inmediata incluyen: dosificación en sangre de glucosa, calcio, magnesio, bicarbonato y gases, urea, creatinina, bilirrubina y amoniemia; en casos concretos, puede ser preciso efectuar una punción lumbar. Pronostico La mortalidadse ha reducido de forma sensible en los últimos años, pero todavía es significativa, incluso en la actualidad, en que puede recurrirse a los fármacos más recientes y a la asistencia en unidades de cuidados intensivos pediátricos. Se valoran 5 factores: EEG, examen neurológico, etiología, tipo de crisis y peso al nacimiento (que se puntúan 0, 1, 2, excepto el peso, que sólo lo hace 0, 2), se establece el test de Ellison, de utilidad para el pronóstico: cuando la puntuación es igual o inferior a 5, el pronóstico es más favorable; los pacientes con 6 o más puntos constituyen una población de riesgo que es preciso vigilar. La epilepsia posterior se presenta en alrededor del 7-20% de los supervivientes y la mayoría de las veces las crisis se presentan en los 6-9º meses de vida. Tratamiento El objetivo ideal del tratamiento debe ser el cese, no sólo de las convulsiones clínicas, sino también de las descargas en el EEG, ya que es probable que el efecto nocivo de las convulsiones neonatales sobre el cerebro inmaduro se deba a una situación de hiperexcitabilidad neuronal, independientemente de que se acompañen o no de manifestaciones clínicas periféricas. Las medidas terapéuticas deben instaurarse con la máxima urgencia y serán, tanto etiológicas, como sintomáticas. Inicialmente estarán encaminadas a conseguir una ventilación óptima, hidratación adecuada, corrección de los trastornos electrolíticos y del equilibrio ácidobásico y mantenimiento de la función cardiovascular. El edema cerebralse combate con dexametasona y en las convulsiones neonatales idiopáticas se debe administrar siempre piridoxina (100 mg) por vía intravenosa, biotina (20 mg por vía oral o intramuscular) y, si no ceden, ácido folínico (2,5-5 mg dos veces al día), por si se trata de un raro caso en el que las crisis son dependientes de estos factores. La hipoglucemia se trata con administración de 2-4 mL/kg (0,1-1 g/kg) de glucosa al 25%, EV, para mantener la glucemia por encima de 40 mg/100 dL. Tratamiento Fenobarbital (PB) es, probablemente, el más utilizado en el periodo neonatal; en esta edad su vida media es de 45-170 horas, empleándose a dosis de ataque de 20 mg/kg (vía venosa), para continuar con 3-5 mg/kg/día como mantenimiento por VO. La fenitoína(PHT), mejor la fosfenitoína –con menos riesgo de potenciales efectos adversos–, se recomienda como segundo fármaco de elección, empleándose a dosis de ataque de 20 mg/kg y a 3-4 mg/kg como mantenimiento EVen administración muy lenta (por vía IM es irritante, cristaliza, su absorción es irregular y los niveles séricos imprevisibles). Las benzodiazepinasocupan el tercer lugar; el diazepam(DZP), a dosis de 0,5-1 mg/kg, es también efectivo por vía EVo rectal (0,5 mg/kg, repitiendo a los 5 minutos si persisten las crisis, para seguir con PB si no se han controlado); no se recomienda su asociación con PHTya que ésta bloquea los receptores del DZP. Convulsiones febriles Son crisis convulsivas clónicas, tónico-clónicas o atónicas, que acontecen en el lactante o niño, entre los 3 meses y 5 años de edad, con fiebre, sin evidencia de infección intracraneal; se excluyen los casos de convulsiones febriles en niños que han presentado previamente una convulsión afebril. Se presentan en el 2-5% de los niños y constituyen el 30% de todas las convulsiones de la infancia, afectando a ambos sexos, aunque con predominio en el masculino. En su aparición influyen tres factores: edad, elevación térmica (fiebre) y factores genéticos; en cuanto a la edad, la mayoría acontecen entre los 6 meses y los 5 años, en especial entre los 12 y 18 meses. Etiología Los procesos responsables de la fiebre son las infecciones habituales en estas edades: infecciones respiratorias altas, enfermedades exantemáticas, gastroenteritis, así como reacciones postvacunales (el riesgo de convulsiones febriles se incrementa el día de la vacunación DTP–difteria, tétanos, tos ferina– y entre los días 8-14 tras la vacuna del sarampión, paperas y rubéola). Los factores genéticosjuegan un papel destacado en su aparición; entre los familiares de los niños con convulsiones febriles se recoge una frecuencia de crisis febriles superior a la encontrada en la población general: cuando uno de los padres las ha padecido el riesgo de que su hijo las presente pasa del 2-5 al 10-20%. Diagnostico Es eminentemente clínico. Ninguna prueba de laboratorio puede confirmar o descartar que el episodio que se ha producido sea una convulsión. Las pruebas analíticas estarán dirigidas al estudio y diagnóstico del proceso infeccioso responsable de la crisis: el ionograma, en especial la natremia, puede ser de interés para el diagnóstico de una posible secreción inadecuada de hormona antidiurética, situación evidenciada con frecuencia en estos pacientes. La práctica de una punción lumbar depende del criterio y experiencia del médico, así como de la edad del niño: dado que los signos y síntomas de una infección meningoencefálica se presentan con menos probabilidad en los niños por debajo de los 2 años, es en ellos donde debe valorarse su realización. Tratamiento Es similar al de cualquier crisis convulsiva con independencia de su etiología incluyendo, además, las medidas generales para combatir la hipertermia y el tratamiento del proceso causal. La profilaxis reduce la recurrencia de las convulsiones febriles pero no existe evidencia de que prevenga una epilepsia posterior y no se recomienda de manera sistemática, limitándose a pacientes con factores de riesgo: anomalía neurológica previa, convulsión febril atípica o compleja, cuando existen antecedentes familiares de crisis afebriles en uno de los padres o hermanos, así como, aun sin datos de riesgo, cuando hay gran ansiedad en la familia. Existen dos modalidades para la prevención: Profilaxis Profilaxis intermitente. Indicada cuando el niño tenga fiebre, requiere un precoz reconocimiento de los signos iniciales de la crisis administrando DZP, por vía rectal en forma de microenema (5 mg en los menores de 2 años y 10 mg en los mayores de esa edad), para repetir la misma dosis a las 8 horas. Profilaxis continua diaria. Son útiles el PB (4-6 mg/kg/día, VO) y la PRM (15-20 mg/kg/día, VO), que logran prevenir las recidivas en un 8090% de los casos, pero tienen efectos secundarios, por lo que no son aconsejables para la profilaxis de las crisis febriles. El VPA(20-30 mg/kg/día, VO) es eficaz. Meningitis aséptica Es un síndrome meníngeo, de variada etiología, caracterizado por presentar LCR claro, con pleocitosis de predominio linfocitario, aumento moderado de proteínas y cultivo bacteriano negativo; su evolución en la mayoría de los casos es benigna y cura sin dejar secuelas. Este síndrome puede cursar aisladamente o formar parte de cuadros clínicos más complejos de etiología generalmente viral. Otras denominaciones iguales o próximas son: “meningitis aséptica benigna”, “meningitis linfocitaria”, “meningitis linfocitaria aguda benigna” y meningitis viral. Etiología Etiología Los agentes etiológicos son múltiples. En el Cuadro están esquematizados los agentes productores de las meningitis asépticas, agrupándolos según su naturaleza. Meningitis viral Meningitis viral. Es la predominante. Los enterovirus no polio son responsables del mayor número de casos (hasta el 80-90% de los casos etiquetados); pueden producirse casos durante todo el año, aunque suelen acumularse en brotes en primavera y verano. Dentro de este grupo cabe destacar en España a los Echovirus, especialmente los 30, 9, 6 y 4 y, últimamente, el serotipo 13; también el virus CoxsackieB5. Los virus herpéticos, aunque poco frecuentes en su presentación como meningitis aséptica están, en segundo lugar en cuanto a frecuencia, pero tienen importancia diagnóstica por ser los únicos tributarios de tratamiento específico. El virus varicela zoster, sólo presenta complicación meníngea (cerebelitis) en una de cada mil varicelas. También es poco frecuente la meningitis en el curso de la mononucleosis infecciosa. Los mixovirus seguían en orden de importancia hasta la instauración de la vacuna obligatoria (especialmente el virus de la parotiditis, que representaba hasta un 15% de los casos). Meningitis por agentes no infecciosos Son consideradas excepcionales. Pueden ocurrir en la fase muy inicial de la meningitis tuberculosa, en las meningitis supuradas incorrectamente tratadas y en las meningitis concomitantes. Actualmente es raro encontrar meningitis asépticas por leptospiras, espiroquetas y rickettsias, con un interés creciente para la Borrelia burgdorferi y son excepcionales las provocadas por parásitos. Las producidas por hongos han aumentado a partir de la era antibiótica; aparecen, sobre todo, en enfermos sometidos a tratamientos muy prolongados con antibióticos de amplio espectro o corticoides o inmunodeprimidos. Los hongos más frecuentes, como causantes de meningitis, son Candida albicans, Blastomyces, tórulas y Actinomyces. No obstante, su cuadros clínico y licuoral se confunden difícilmente con el de las meningitis asépticas. Meningitis aséptica en otros procesos Una reacción meníngea de LCR claro y aséptico puede aparecer en afecciones diversas, como: lupus, sarcoidosis, leucemia, diseminación carcinomatosa generalizada, esclerosis en placas, tumor intracraneal, hemorragia subdural y trombosis de los senos cavernosos que también cabría considerar en relación con las encefalitis. Pero en estos casos el cuadro clínico difiere considerablemente de lo definido como meningitis aséptica. La vacuna de la tos ferina desencadena ocasionalmente alteraciones meníngeas, así como las intoxicaciones por plomo, arsénico y otras sustancias y, finalmente ciertas técnicas exploratorias (neurorradiológicas) o terapéuticas (vía intratecal). Clínica Las manifestaciones clínicas pueden limitarse a signos de irritación meníngea o asociar síntomas derivados de la afectación de otros órganos, dando un síndrome más complejo. La asociación con mialgias intensas es provocada sobre todo por enterovirus; pleurodinia por Coxsackie y ECHO; dolores periumbilicales pseudoapendiculares por poliovirus; exantemas máculo-papulosos por virus ECHOy Coxsackie; afectación parotídea, pancreática o genital por virus urliano; poliartritis por Borrelia burgdorferi. La meningitis comienza bruscamente o de forma gradual. Aveces va precedida de un episodio febril acompañado de otros síntomas, como faringitis, diarrea, estreñimiento o un exantema; en otras ocasiones el síndrome meníngeo constituye la primera manifestación de la enfermedad. En general, aparecen pronto los síntomas más específicos, como cefaleas, vómitos, fotofobia, rigidez espinal y de nuca, exaltación de reflejos tendinosos, signos de Kernig, Lassègue y Brudzinski. En adolescentes, en ocasiones es característico un cuadro de cambio de conducta; otras veces predomina la afectación cerebelosa con ataxia. En conjunto, el cuadro clínico es más discreto que en las meningitis bacterianas o tuberculosas siendo en ocasiones subclínico. La evolución es siempre benigna. Por término medio la curación se consigue en un plazo de siete a catorce días. Exámenes complementarios El LCR es imprescindible para el diagnóstico: es necesaria la punción lumbar siempre que se sospeche la enfermedad. La tensión es variable; en general moderadamente alta. El aspecto es claro, pero puede ser deslustrado y aun opalescente, incluso puede detectarse un retículo fibrinoso, en especial en la coriomeningitis linfocitaria. La pleocitosis varía, desde sólo 10 hasta 2.000 células por mm3, con predominio mononuclear, aunque habitualmente no supera las 1.000. La proteinorraquia es normal o poco elevada. La glucorraquia se mantiene dentro de los límites normales o ligeramente aumentada (excepto en infecciones por Mycoplasma, infiltración leucémica y meduloblastoma), igual que los cloruros y el ácido láctico. Diagnostico En general es fácil, si se atiende a la clínica y al resultado analítico del LCR. Clásicamente lo difícil era realizar la confirmación microbiológica de la sospecha clínica por el bajo rendimiento de los cultivos virales o de las técnicas indirectas basadas en la respuesta inmunitaria específica. Actualmente las técnicas de amplificación molecular han incrementado significativamente el diagnóstico etiológico, siendo las de elección para los principales agentes. El aislamiento del virus por cultivo o la detección del ácido nucleico por PCR se realizará a partir del LCR, frotis faríngeo, secreción nasal, heces u orina, si bien la interpretación de los resultados será más fiable en la muestra de LCR, por lo que será la de elección. Tratamiento Es ante todo sintomático: antitérmicos y analgésicos para combatir fiebre, cefaleas y algias; la agitación puede controlarse con sedantes como la clorpromazina, las benzodiacepinas a dosis bajas o, en casos más rebeldes, con propofol. Para los vómitos es útil la metoclopramida (evitar en lo posible para no enmascarar la aparición de signos encefalíticos). En caso de convulsiones se indicarán fenobarbital o hidantoínas principalmente. No hay que descuidar corregir la deshidratación y las posibles desviaciones del equilibrio ácido-básico, mediante la administración de 80-100 mL/kg/día de la solución glucosalina habitual, añadiendo potasio o bicarbonato, según los datos clínicos y analíticos. Meningitis bacteriana Las meningitis bacterianas, supuradas, purulentas o piógenas incluyen los procesos inflamatorios de las leptomeninges encefálicas y medulares, que cursan típicamente con líquido cefalorraquídeo turbio o purulento, intensa pleocitosis y claro predominio de células polinucleares. Alcanzan su máxima frecuencia en lactantes y párvulos, siendo más raras después de los 10 años. En la edad neonatal revisten caracteres especiales, tanto desde el punto de vista clínico, como etiológico y terapéutico. Los progresos de la antibioterapia han aumentado el índice de curabilidad por encima del 90%. Etiología Las meningitis supuradas son producidas por una gran variedad de agentes bacterianos, cuya frecuencia relativa es muy variable. Considerados globalmente, los preponderantes son N. meningitidis, S. pneumo niae y H. influenzae, especialmente los dos primeros. En los países desarrollados, las campañas de vacunación masivas frente a H. influenzae y N. meningitidis C y, más recientemente, S. pneumoniae ha cambiado de forma drástica la distribución de estos gérmenes. Patogenia Los microorganismos que invaden las meninges lo hacen procedentes de un foco infeccioso primitivo. La mayor parte de las veces llegan por diseminación hematógena a partir de la presencia de patógeno en la mucosa nasofaríngea. Entre el 5-25% de los niños sanos están colonizados por N. meningitidis y S. pneumoniae. Si se produce una infección viral a ese nivel se puede facilitar el paso del germen al torrente circulatorio. Se puede producir infección en diferentes localizaciones, entre ellas, las meninges. En otras ocasiones alcanzan las meninges por propagación de continuidad a partir de un foco infeccioso próximo como otitis, sinusitis y mastoiditis. Para la penetración de las bacterias en el encéfalo y meninges es necesario que se produzca el fracaso de la barrera hematoencefálica. Esta teórica barrera está formada por el endotelio de las paredes de los capilares del SNC, cuya constitución anatómica determina una permeabilidad menor de la que es habitual en otros territorios orgánicos. El factor anatómico más importante está constituido por los “pies terminales de los astrocitos”. Apoyados en la pared de los capilares determinan la formación de una especie de membrana que recubre la pared capilar en casi toda su extensión Anatomía patológica Llegados los microorganismos al SNC, se produce la inflamación purulenta de las meninges aracnoideas y piamadre, con aparición de un infiltrado leucocitario y un exudado purulento, que se extiende por la base y superficie de ambos hemisferios. Se liberan componentes de la membrana celular al espacio subaracnoideo que desencadenan la cascada inflamatoria con liberación de citocinas, entre ellas el TNF e interleucina 1. Se aumenta la permeabilidad de la BHE, pudiendo aparecer edema cerebral e hipertensión intracraneal. La infección puede alcanzar los ventrículos, dando lugar a una ventriculitis; las adherencias que se producen llegan a obstruir las vías de circulación o reabsorción del liquor, ocasionando una hidrocefalia. Adherencias y bridas inflamatorias se producen también en otras zonas y dan lugar a la formación de tabicamientos, con constitución de celdas, en las cuales el pus y los gérmenes patógenos quedan aislados, persistiendo en ellas la infección de forma prolongada. Se producen igualmente infiltraciones perivasculares, vasculitis y trombosis, que condicionan la presentación de hemorragias cerebrales, zonas de necrosis y derrames subdurales MENINGITIS MENINGOCÓCICA La meningitis meningocócica o cerebroespinal epidémica es la más frecuente en pediatría, tanto en España como en muchos otros países. Agente etiológico. Es la Neisseria meningitidis. Pertenece, junto con el agente de la gonococia (N. gonorrhoeae), a la familia de las Neisseriaceae, en las que también están incluidos agentes patógenos: Acinetobacter, Branhamellao Moraxella. La N. meningitidises un diplococo intracelular gramnegativo, con forma de granos de café. Es productor de endotoxinas, se comporta como aerobio y es muy sensible a los agentes externos. Su escasa resistencia a la temperatura ambiente se ha de tener en cuenta al realizar los cultivos. Contagio Contagio. Se verifica de manera directa por las gotitas de Pflügge expulsadas por los enfermos o, con mayor frecuencia, por portadores sanos. Parece que existen más portadores en la población adolescente, fumadora y con condiciones de hacinamiento. Habitualmente es la rinofaringe donde los meningococos causan una sintomatología banal de tipo catarral. Estos microorganismos, además de su complejo lipoproteína-lipopolisacárido, responsable de los 15 antígenos antes citados y de la producción de endotoxina, poseen fimbrias, gracias a las cuales consiguen su adherencia al epitelio de la mucosa de vías respiratorias altas. Clínica La sintomatología varía de acuerdo con los datos etiopatogénicos expuestos. Así, habrá cuadros de predominio séptico (signos cutáneos, inestabilidad hemodinámica hasta llegar al choque) y otros fundamentalmente meníngeos; los primeros son de evolución rápida, requieren diagnóstico temprano y tienen peor pronóstico; los segundos son más lentos y benignos. El comienzo del cuadro clínico de predominio meníngeo viene señalado por una brusca elevación febril, que alcanza ordinariamente 3940 °C, pulso rápido, grave sensación de enfermedad y síntomas de afectación meníngea: cefaleas de gran intensidad localizadas especialmente en región occipital y vómitos “a chorro”, sin náuseas, y que conforman la “tríada meningítica”. A la exploraciónse aprecian rigidez de nuca y signo del trípode, que aumentan conforme progresa la enfermedad, apareciendo en los casos más floridos opistótonos y extremidades inferiores flexionadas en posición de gatillo de fusil. Una tercera parte de los pacientes presentarán inicialmente el temido exantema petequialtromboembólico irregular y a veces claramente hemorrágico o necrótico. Síntomas cutáneos Con frecuencia aparece un herpes simple localizado en los labios u otras regiones, aunque suele ser tardío, ya en la fase de estado. Si no marcó el comienzo, podrá apreciarse ahora la erupción roseoliforme, maculopapulosa, tenue y de escasa duración y, más a menudo, un exantema purpúrico, que constituye la típica lesión cutánea de las meningococemias. Está formado por petequias o elementos hemorrágicos de mayor tamaño, como una lenteja y más grandes, irregulares, estrellados, de carácter tromboembólico. Se distribuyen por toda la superficie cutánea, aunque suelen predominar en las extremidades. Los elementos de mayor tamaño suelen transformarse en flictenas y dejar lesiones necróticas (púrpura necrótica); más rara vez los elementos son muy grandes y adoptan la forma de púrpura fulminante. Las lesiones cutáneas tromboembólicas son manifestaciones de una púrpura infecciosa angiopática con CID. Periodo de estado Se caracteriza por un aumento de los síntomas anteriores: la rigidez de nuca se intensifica, el opistótonos es más marcado, las cefaleas y vómitos continúan. Los signos de Lassègue, Kernig y Brudzinski son, generalmente, muy positivos, y el sensorio va deprimiéndose, adquiriendo el enfermo un aspecto encefalítico. El edema cerebral que se origina, por su componente encefalítico o vasculítico, puede llegar a producir herniación cerebral, con repercusión sobre tronco encefálico: trastornos respiratorios, midriasis arreactiva, hipertermia y bradicardia, que pueden conducir a enclavamiento y muerte cerebral. En los lactanteslos signos meníngeos clásicos son discretos o faltan siendo, en cambio de gran valor clínico la hipertensión de la fontanela, que se halla tensa a la palpación e incluso se aprecia a simple vista como abombada sobre el cráneo. Datos de laboratorio En el hemogramaexiste una leucocitosis elevada (excepto aquellos cuadros sépticos graves, que cursan con leucopenia, signo de mal pronóstico) con predominio polinuclear y desviación a la izquierda. Existe una elevación de los reactantes de fase aguda, como la proteína C reactiva y la procalcitonina. El LCR es de aspecto turbio, si bien en estadios muy precoces o en enfermos parcialmente tratados puede ser claro (Cuadro 23.10.2). La presión está aumentada, con gran pleocitosis (2.000-3.000 o más células, predominando los polinucleares neutrófilos). La albúmina y globulinas se encuentran también muy aumentadas. Los cloruros y, sobre todo, la glucosa aparecerán disminuidos y el ácido láctico (lactato) está elevado (al contrario, en las meningitis de origen viral). En la fase de regresión disminuye la pleocitosis, que se hace de tipo linfocitario, y el líquido se vuelve claro. El examen directo del frotis pone a veces de manifiesto la existencia de meningococos, en forma de diplococos intracelulares gramnegativo. Sepsis meningocócica grave En ella es tanta la gravedad de la diseminación hemática meningocócica (la acción endotóxica del meningococo produce una lesión vascular diseminada y un cuadro de CID) con gran expresividad hemodinámica y cutánea, que provoca un diagnóstico precoz, sin dar tiempo a la invasión meníngea y aparición de sintomatología. Los niños enferman de un modo repentino con postración, obnubilación y gravísima afectación del estado general. Existen fiebre alta, palidez; a las pocas horas aparecen las petequias y tromboembolias características del síndrome de coagulación intravascular, pasando rápidamente al estado de colapso y choque. La afección progresa rápidamente y, antes de 24 horas, puede fallecer el niño, sin llegar a presentar signos de meningitis, con choque refractario. El LCR es normal o está muy poco alterado, aunque es posible hallar meningococos en el hemocultivo. Síndrome de Waterhouse-Friderichsen Es la denominación clásica de una forma de presentación de la sepsis meningocócica con choque refractario, a la que se asocia un infarto hemorrágico bilateral de las suprarrenales, que le confiere un pronóstico casi siempre letal. Aparte la mayor intensidad de los síntomas circulatorios, aparecen en la piel unas manchas equimóticas confluentes o livideces cadavéricas intravitam, sobre todo en las partes declives del cuerpo, como las que aparecen después de la muerte y que son muy distintas del exantema roseoliforme inicial de la infección meningocócica y de la erupción petequial tromboembólica de la sepsis, con la que se asocian casi siempre. Otras meningitis Después de N. meningitidis, el agente etiológico más frecuente en nuestro medio es S. pneumoniae, seguido por Streptococcus, gramnegativos y Staphylococcus .Las meningitis causadas por estos microorganismos presentan características clínicas análogas a las de la meningitis meningocócica, pero difieren en la terapéutica. Son características clínicas comunes: comienzo brusco; signos meníngeos manifiestos, si bien suelen ser menos intensos que en las formas meningocócicas, y rareza de cuadro cutáneo; fiebre elevada y afectación del estado general, que suelen preceder a las manifestaciones meníngeas; posible relación con focos sépticos vecinos (otitis, mastoiditis, sinusitis, fístulas de LCR, etc.). Algunas tienen especial tendencia a recidivar y presentar recaídas, lo cual depende, en parte, del germen patógeno y, en parte, de la eficacia del tratamiento. El LCR presenta las alteraciones características de las meningitis supuradas: turbio o francamente purulento, pleocitosis elevada de predominio polinuclear, proteínas elevadas, glucosa y cloruros disminuidos. Es posible el hallazgo del agente causal, por frotis directo, cultivo, antígenos bacterianos y PCR. Meningitis neumocócica La infección neumocócica ha sido revisada en el capítulo 7.6. Ahora interesa señalar que Streptococcus pneumoniaees el segundo germen en frecuencia para la etiología en la actualidad. Su incidencia en la población general es de aproximadamente 1,5 casos/100.000 personas, llegando en el grupo de < 5 años a 7/100.000. Un grupo de riesgo son los enfermos con drepanocitosis y esplenectomizados. En muchos casos se evidencian focos previos, como neumonía e infecciones de vías respiratorias altas, especialmente del oído medio. El cuadro clínico es semejante al de la meningitis meningocócica, si bien los síntomas meníngeos suelen ser menos manifiestos, debido a que el exudado inflamatorio se localiza sobre todo en la base cerebral. Son frecuentes las manifestaciones encefalíticas (coma, parálisis de pares craneales, trastornos vegetativos) y el bloqueo precoz del sistema ventricular, con desarrollo de hidrocefalia aguda. Meningitis por Streptococcus agalactiae Es el primer germen responsable de meningitis bacteriana en el periodo neonatal precoz < 7 días de vida. La incidencia de meningitis en el RN es mayor que en cualquier otra edad pediátrica, llegando a ser 0,2 a 1 casos/1.000 RN vivos. Su mortalidad puede llegar al 30% y el índice de secuelas es alto, siendo inversamente proporcional al peso y la madurez del niño. La prevalencia de este germen ha disminuido gracias a las medidas profilácticas que, de forma rutinaria, se toman en las madres portadoras y en los niños. Apesar de ello, sigue siendo una enfermedad temida por su agresividad, tanto en la forma meningítica como en la sepsis y sigue siendo el germen predominante en esta edad. Existen formas de presentación precoz, antes de los 7 días de vida, y presentación tardía, hasta los 30-45 días. Meningitis por H.influenzae El Haemophilus influenzae tipo b, revisado en el capítulo 8.13, era uno de los agentes que con más frecuencia provocaba meningitis en la primera infancia hasta el año 1998 en que se introdujo la vacuna en España. Desde entonces ha desaparecido prácticamente este germen en numerosas estadísticas. En el año 2006, en España sólo se declararon 4 casos de enfermedad por H. influenzae. Los casos aislados suelen corresponder a niños no vacunados. La máxima frecuencia corresponde al lactante, entre los 6 y los 12 meses de edad; es rara en el primer trimestre de vida y después de cumplir los 5 años. En gran parte de los casos la enfermedad va precedida o acompañada de infección de vías respiratorias altas. El cuadro clínico es semejante al de las restantes meningitis supuradas, con las salvedades antes comentadas acerca del poco relieve clínico que pueden tener los síntomas meníngeos en el lactante. Meningitis estafilocócica Muy poco frecuente y generalmente debida a una cepa plasmocoagulasa (+), suele producirse por vía hematógena a partir de focos purulentos localizados en el oído, mastoides, senos, piel, cuero cabelludo, huesos, articulaciones; o por contigüidad en fístulas postraumáticas inadvertidas. Anatómicamente existe una intensa inflamación meníngea con gran exudado purulento y abscesos localizados en cerebro y meninges. El cuadro clínico es grave y la frecuencia de secuelas es elevada. En cambio el Staphylococcus plasmocoagulasa(-), muy frecuente en infecciones hospitalarias, RN y en enfermos portadores de derivaciones valvulares (hidrocefalias) o prótesis, es causa de meningitis de evolución subclínica y buen pronóstico. Diagnostico etiológico La anamnesis y clínica pueden ofrecer datos orientadores sobre el agente causal: ambiente epidémico de meningococia, edad, microorganismos entéricos en el RN, H. influenzae en lactante pequeño y antecedentes personales o familiares para eliminar la tuberculosis. El exantema petequial o tromboembólico hará sospechar N. meningitidis, aunque a veces también algunas petequias pueden aparecer en otros procesos: infección por enterovirus, Echovirus, otras virosis, sepsis por H. influenzae, fiebre botonosa mediterránea, púrpura anafilactoide de SchoenleinHenoch, púrpura trombocitopénica idiopática aguda, enfermedad de Kawasaki, alergia medicamentosa y leucemia. El diagnóstico etiológico de certeza se consigue con el examen bacteriológico del LCR, aunque también debe practicarse el hemocultivo. Dada la gravedad de la enfermedad y la importancia del tratamiento precoz, la punción lumbar diagnóstica no debe demorarse, excepto cuando existe inestabilidad hemodinámica o hipertensión endocraneal. Tratamiento Trastornos por déficit de atención e hiperactividad (TDAH) Los trastornos por déficit de atención e hiperactividad (TDAH) vienen definidos por la presencia de tres síntomas fundamentales: 1. Disminución de la atención. 2. Impulsividad. 3. Hiperactividad. En realidad es mucho más que un trastorno. Es un síndrome de dimensiones enormes, que alcanza una gran cantidad de facetas y se debería denominar “Síndrome de Déficit de Atención e Hiperactividad (SDAHA) con mucha más propiedad que TDAH. Prevalencia A medida que ha pasado el tiempo se han ido incrementando los porcentajes de personas que padecen este cuadro, a la vez que ha ido disminuyendo la edad a la que puede ser diagnosticado. Si bien hace unos años se estimaba la prevalencia del TDAH en el 4%-6%, los últimos estudios epidemiológicos dan cifras que rondan el 20% y hasta los más prudentes sitúan la prevalencia por encima del 10%. Es posible que las diferentes pruebas valorativas jueguen un papel importantísimo en los porcentajes de prevalencia. Etiología No hay duda de que el TDAH es un cuadro orgánico, con origen en deficiencias anatómico-biológicas que afectan preferentemente a ciertas estructuras cerebrales y que no es patrimonio exclusivo de los humanos. En seres el reino animal superior (caballos, toros, perros, gatos, monos, etc.) pueden observarse comportamientos similares. La etiología puede ser tanto genética como adquirida, pero en ambas circunstancias con la misma base bioquímica como origen del trastorno. La mayoría de los casos son hereditarios por vía autosómica dominante por parte de ambos progenitores de manera similar. Patogenia El trastorno funcional, es decir, las alteraciones clínicas, se llevan a cabo por problemas bioquímicos en proyecciones de conexión entre los lóbulos frontales y los núcleos basales, que afectan tanto al transporte como a la recaptación de la dopamina y en menor grado, de la serotonina y de la norepinefrina. Ello ocurre tanto en los sujetos en los que el trastorno tiene origen genético como en los de causa adquirida. Clínica Primer año, los niños suelen dormir mal y estar con los ojos muy abiertos y algunos comienzan a andar excesivamente pronto (entre los 6-10 meses), aunque la mayoría marcha sin ayuda algo tarde. Posteriormente estos niños muestran hipotonía, pies planovalgos y miedo a dormir solos por lo que no permiten que se apague la luz – a veces durante toda la noche – y quieren acostarse con los padres. Durante los seis primeros años de vida la característica que mejor define a estos niños es su concepto vital – el mundo se circunscribe a ellos, y los demás son el entorno que les tiene que servir – mostrándose torpes para la motricidad fina, inquietos, caprichosos, entrometidos, acaparadores, egoístas y con poca capacidad de frustración. Durante la edad escolar se muestran dispersos, infantiles, inmaduros, mienten y pueden sustraer dinero en casa, se levantan del pupitre muchas veces, interrumpen a otros niños y a profesores, les cuesta aprender a leer y escribir, tienen dificultades especialmente para las Matemáticas y la Lengua, presentan fracaso escolar con mucha frecuencia, muestran su falta de habilidad motriz tanto en el manejo del lápiz y cuchara como en los deportes de habilidad (ej: el fútbol). Diagnostico Tratamiento El tratamiento de los niños y jóvenes con TDAH conlleva: a) Una información adecuada a los padres de lo que es este síndrome, intentando tranquilizar, relajar y motivar al entorno de los pacientes para facilitar el sacarlos adelante. b) Hacer llegar a los padres y a los profesores el conocimiento de que ni los unos ni los otros son culpables de nada. El problema está en el niño, que lleva el cuadro en su constitución y que tampoco es culpable de su cuadro. c) Que este síndrome persiste siempre, pero que ello no es óbice para que la inmensa mayoría de los sujetos que lo presentan sean personas normales. Tratamiento farmacologico El fármaco estimulante de elección es el metilfenidato en cualquiera de sus presentaciones, aunque la de liberación lenta se ha impuesto definitivamente entre los niños que ya pueden tragar la pequeña cápsula, los adolescentes y los adultos debido a la comodidad de una toma al día (por la mañana con el desayuno), ya que se mantienen los niveles de la medicación en sangre sin altibajos a lo largo del día, mientras que la presentación en liberación rápida tiene una vida corta en sangre, se requieren 2-3 tomas al día y presenta fases en que, debido a su bajo nivel en sangre, su eficacia disminuye. Las dosis, en una y otra presentación, están entre 0,3 y 1 mg/Kg/día. Se puede administrar todos los días o bien descansar las fechas en las que no hay actividad escolar. Espectro autista (Autismo) El autismo es un conjunto de alteraciones heterogéneas a nivel del neurodesarrollo que inicia en la infancia y permanece durante toda la vida. Implica alteraciones en la comunicación e interacción social y en los comportamientos, los intereses y las actividades. La prevalencia mundial está alrededor del 1%. Se da más frecuentemente en hombres que en mujeres, en una relación 4:1, aunque se ha observado que las mujeres con autismo tienden a expresar un mayor compromiso cognitivo. Con el DSM-5 (2013), versión más reciente del manual, todos los subtipos del autismo quedaron en una sola categoría: trastornos del espectro autista, que reemplaza el término trastornos generalizados del desarrollo y en la que se fusionan cuatro de los cinco subtipos vigentes en el DSM-4-TR. Etiología A pesar de todos los avances en neurociencias y en genética, aún no se ha podido establecer un modelo que explique la etiología y fisiopatología de los TEA, aunque en diferentes estudios se han evidenciado alteraciones neurobiológicas y genéticas asociadas, así como factores epigenéticos y ambientales involucrados. Precisamente por esto, y por tratarse de una serie de trastornos crónicos, han surgido en ocasiones distintas iniciativas que, sin basarse en métodos científicamente comprobados, prometen efectos positivos. Anormalidades en el trazado electroencefalográfico y trastornos convulsivos se han observado hasta en un 20% a 25% de los pacientes con autismo. Las altas tasas de epilepsia sugieren un papel fundamental de los factores neurobiológicos en la génesis del autismo. Signos de alarma Es importante que todo pediatra conozca las señales de alarma, dado que, sobre todo en las etapas iniciales, los padres pueden tener más una sensación de intranquilidad acerca de su hijo que una necesidad de búsqueda de un diagnóstico. La presencia de estos signos de alarma indicaría la necesidad de realizar estudios más específicos y, si fuese necesario, la derivación a centros de atención temprana, servicios de psiquiatría infantil y neuropediatría u otros especialistas. Clínica Los signos clínicos tempranos pueden incluir desde retraso en algunos patrones del desarrollo a otros síntomas subjetivos, como pueden ser el contacto visual o la reciprocidad emocional con los cuidadores. Los síntomas pueden ser malinterpretados o clasificados incorrectamente como retraso del desarrollo o trastornos inespecíficos del comportamiento o del lenguaje. Las alteraciones que se evidencian en el niño con autismo están centradas en dos focos: • La dificultad en el lenguaje expresivo y comprensivo, que altera el desempeño social. • La presencia de intereses o actividades muy restringidas que afectan su comportamiento. Diagnostico Según la última versión del Manual diagnóstico y estadístico de los trastornos mentales, el DSM-5, se definen los siguientes criterios diagnósticos. • Alteraciones persistentes en la comunicación social y en la interacción social alrededor de múltiples contextos, manifestadas por lo siguiente, actualmente o por los antecedentes • Patrones restrictivos y repetitivos de comportamiento, intereses o actividades, que se manifiestan en dos o más de los siguientes síntomas, actuales o pasados, como movimientos, utilización de objetos o habla estereotipados • Los síntomas deben estar presentes en las primeras fases del período de desarrollo. • Los síntomas causan un deterioro clínicamente significativo en lo social, laboral u otras áreas importantes del funcionamiento habitual. • Estas alteraciones no se explican mejor por la presencia de una discapacidad intelectual (trastorno del desarrollo intelectual) o por el retraso global del desarrollo. Tratamiento En cuanto a las recomendaciones terapéuticas establecidas por la Academia Americana de Psiquiatría Infantil y del Adolescente (AACAP, por sus siglas en inglés), se encuentran las siguientes: • Existen varios modelos terapéuticos pero cualquiera que se escoja deberá ser psicoeducativo, individualizado, transdisciplinario, estructurado y predecible, y deberá incluir el análisis permanente de las conductas problema y las variables del contexto en el que se desenvuelve el paciente, para lograr un impacto positivo en su calidad de vida y la de su familia. • La risperidona y el aripiprazol han sido aprobadas por la FDA para el tratamiento de la irritabilidad, que consiste principalmente en agresión física y rabietas graves asociadas con el autismo. • El clínico debe mantener un papel activo en la planeación del tratamiento a largo plazo, ofreciendo un apoyo constante a las familias y al individuo, pues las necesidades del núcleo familiar cambiarán en las diferentes etapas vitales. Bibliografía Cruz-Hernández, M., & Martínez Valverde, A. (2013). Tratado de Pediatría, Volumen ll. https://scp.com.co/wp-content/uploads/2016/04/2.-Trastorno-espectro.pdf https://www.aeped.es/sites/default/files/documentos/20-tdah.pdf