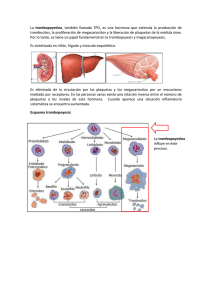
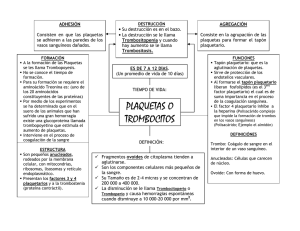
Universidad Latina De Panamá Facultad de Medicina William C. Gorgas Catedra de Hematología Laboratorio Trabajo N°2 Investigación Trastornos Plaquetarios Integrantes: Crisanto Sambrano Jaén: 9- 752-1781 Daniel Rosario: PE-13-2082 Profesora: Miriam C. de Gúette Fecha de entrega: Miércoles 1 de abril de 2020 Introducción: En este trabajo investigativo es importante saber que las plaquetas humanas son células sin núcleo multifuncionales que desempeñan un papel indispensable en la hemostasia. Las plaquetas se originan a partir del citoplasma de los megacariocitos de la médula ósea. Las plaquetas no tienen ADN genómico, pero contienen ARN mensajero (ARNm) derivado de los megacariocitos y la maquinaria translacional necesaria para la síntesis de las proteínas. Su forma y tamaño pequeño permite que las plaquetas sean empujadas hacia los bordes de los vasos sanguíneos, colocándolas en una posición óptima para la vigilancia constante de la integridad de la función plaquetaria vascular. 1. Enfermedad de Von Willebrand (EVW): ¿Qué es? La EVW es causada por un problema con una de las proteínas de la sangre, el factor von Willebrand (FVW). Las personas con EVW o no tienen suficiente factor von Willebrand (FVW) o el que tienen no funciona bien. El FVW tiene dos funciones importantes en la coagulación de la sangre. El FVW es la proteína que hace que las plaquetas se peguen a la pared de un vaso sanguíneo lastimado y que se peguen unas a otras. Sin él, no se puede formar un tapón de plaquetas. El FVW también es una de las proteínas que transporta uno de los factores coagulantes, el factor VIII. Esto significa que ayuda a que haya suficiente factor VIII en la sangre y que ayuda a llevar al factor VIII a donde sea necesario. Sin FVW, el factor VIII será descompuesto en el torrente sanguíneo y posiblemente no haya suficiente para detener un sangrado. El factor von Willebrand es almacenado en las células endoteliales que recubren los vasos sanguíneos. Sólo cerca de una tercera parte del FVW está flotando en el torrente sanguíneo. El resto está almacenado, las plaquetas se mueven al punto de ruptura para tapar el agujero. El factor von Willebrand es lo que permite que las plaquetas se peguen a la pared del vaso sanguíneo y unas a otras. En una persona con EVW, las plaquetas no se pueden pegar lo suficiente para formar un buen tapón de plaquetas. El sangrado durará más. Qué pruebas de Hematología se pueden hacer para aporte en el diagnóstico? Primer nivel: - Factor VIII ( FVIII:C) VWF:RCo ( Cofactor de ristocetina) o pruebas que exploran interacción VWF-GPlb) VWF:Ag ( Antígeno de VW) Aglutinación inducida por ristocetina ( RIPA) Segundo nivel: - Prueba de respuesta a la desmopresina Estudio de multímeros en geles de alta y baja resolución VWFpp(propéptido) VWF:FVIIIB( unión al colágeno) Biología molecular - Se sospecha enfermedad de Von Willebrand en pacientes con sangrado de causa desconocida, en particular en aquellos con antecedentes de una diátesis hemorrágica similar. Los estudios de coagulación revelan recuento plaquetario normal, INR normal y, a veces, TTP ligeramente prolongado. La prueba del tiempo de sangría es poco fiable y ya no se hace. - El diagnóstico requiere determinar el antígeno de FvW plasmático total, la función del FvW determinada por la capacidad del plasma para mantener la aglutinación de plaquetas normales por medio de ristocetina (actividad del cofactor de ristocetina) y la concentración plasmática de factor VIII. Los estímulos que aumentan transitoriamente las concentraciones de FvW pueden causar resultados falsos negativos en la EvW leve; a veces, deben repetirse los estudios. - En la forma común (tipo 1) de EvW, los resultados son concordantes; es decir, se observa una depresión equivalente del antígeno de FvW, la función del FvW y la concentración plasmática de factor VIII. El grado de depresión varía de alrededor del 15 al 60% de lo normal y determina la gravedad de una hemorragia anormal del paciente. Las concentraciones de antígeno de FvW pueden ser de tan solo el 40% de lo normal en individuos sanos con grupo sanguíneo 0. - Se sospechan variantes tipo 2 si los resultados son discordantes; es decir, el antígeno de FvW es más alto que lo esperable para el grado de alteración de la actividad del cofactor de ristocetina. El antígeno de FvW es más alto que el previsto porque, en el tipo 2, el defecto del FvW es cualitativo (pérdida de multímeros del FvW de alto peso molecular) no cuantitativo. El diagnóstico se confirma demostrando un descenso de la concentración de multímeros grandes de FvW en la electroforesis en gel de agarosa. Se reconocen 4 variantes tipo 2 diferentes, que se distinguen por alteraciones funcionales de la molécula de FvW. - Los pacientes con EvW tipo 3 no tienen FvW detectable y presentan marcada deficiencia de factor VIII. 2. Enfermedad de Glanznmann: ¿Qué es? Es un trastorno de la función plaquetaria causado por una anomalía en los genes de las glicoproteínas IIb/IIIa. Estos genes codifican para un grupo de proteínas enlazadas que normalmente se encuentran en la superficie de las plaquetas, el receptor glicoproteína IIb/IIIa (también llamado receptor de fibrinógeno). Las plaquetas participan en la formación de coágulos sanguíneos y en la reparación de vasos sanguíneos dañados. Cuando un vaso sanguíneo se lesiona, las plaquetas se activan y ocurre un proceso de transformación de las plaquetas para convertirse en un tapón plaquetario, este proceso se divide en tres etapas: adhesión, secreción y agregación. En la etapa de agregación, el fibrinógeno (Factor I) se adhiere a lugares específicos en la superficie de las plaquetas (receptores de adhesión). Uno de esos receptores se llama glicoproteína IIb/IIIa (GP IIb/IIIa). Existen tres subtipos de trombastenia de Glanznmann: En el Tipo I, la cantidad de GPIIb/IIIa es menor de 5% de lo normal. En el Tipo II, la cantidad de GPIIb/IIIa es entre 5% y 20% de lo normal. En el Tipo III, hay una cantidad normal de GPIIb/IIIa pero ésta no funciona correctamente. ¿Qué pruebas de hematología se pueden hacer para aporte en el diagnóstico: Laboratorio: Cambio de forma presente Agregación plaquetaria ausente o respuestas anormales de agregación plaquetaria a estímulos fisiológicos, tales como epinefrina, colágeno, ácido araquidónico y ADP. Esto se explica porque las GP IIb/IIIa son necesarias para la unión del fibrinógeno y las plaquetas, durante la agregación Agregación presente con agonistas fuertes (trombina) Reacción de liberación presente Contenido de Fg intraplaquetario disminuido o ausente Prueba de sangre para la deficiencia de GPIIb/IIIa en las plaquetas Anormal retracción del coágulo 3. Síndrome de Bernard Solier: ¿Qué es? El síndrome de Bernard-Soulier (SRS), también conocido como distrofia trombocitopenia hemorrágica, es un trastorno hereditario que afecta a la serie megacariocítica o plaquetaria y que se caracteriza por un síndrome hemorrágico con disminución del número de plaquetas que muestran un gran tamaño Este síndrome es extremadamente raro, ya que se han descrito tan sólo unos 100 casos hasta el momento. Sus manifestaciones clínicas incluyen, por lo general, púrpura, epistaxis, menorragia y sangrado gingival y gastrointestinal. El SRS se transmite con carácter autosómico recesivo y el trastorno subyacente es una deficiencia o disfunción del complejo de la glicoproteína GPIb-V-IX, un receptor formado por múltiples subunidades, restringido a las plaquetas, que es necesario para la hemostasia primaria. El complejo de la glicoproteína GPIb-V-IX se une al factor de von Willebrand (vWF) permitiendo la adherencia de las plaquetas al endotelio y la formación del tapón plaquetario cuando existe una lesión vascular. Los genes que codifican para las cuatro subunidades del receptor, GPIb alfa, GPIb beta, GPV y GPIX, se mapean en los cromosomas 17p12, 22q11.2, 3q29 y 3q21, respectivamente. Se han identificado defectos genéticos en GPIb alfa, GPIb beta y GPIX, pero no en GPV. El diagnóstico se basa en el reducido número de plaquetas (plaquetopenia) de gran tamaño (macrotrombocitopenia), en el alargamiento del tiempo de sangria, en la disminución de la inducción a la agregación plaquetària por la ristocetina y en una baja expresión o ausencia del complejo de la glicoproteína GPIb-V-IX. El consumo de protrombina se encuentra asimismo muy disminuido. Con el seguimiento y la atención adecuados, el pronóstico es generalmente bueno, pero pueden darse episodios graves de sangrado durante la menstruación y en los casos de traumatismo e intervenciones quirúrgicas. El tratamiento o la profilaxis de la hemorragia durante los procedimientos quirúrgicos requieren habitualmente de una transfusión de plaquetas. ¿Qué pruebas de hematología se pueden hacer para aporte en el diagnóstico: Pruebas de agregación plaquetaria Análisis de la glicoproteína de la superficie de las plaquetas Recuento automatizado de plaquetas Citometría de flujo con la utilización de anticuerpos monoclonales en sangre total. En algunas personas con SBS, el conteo de plaquetas es ligeramente menor que lo normal. Una persona con SBS puede tener período de sangrado muy largo. Puede tomar más de 20 minutos para que cese un sangrado de una cortadura pequeña. Los resultados de la prueba PFA 100® también serán anormales. Cuando se observan bajo el microscopio, las plaquetas de las personas con SBS son mucho más grades de lo normal. Una prueba importante para detectar SBS es la agregación de plaquetas. Cuando se mezcla el antibiótico ristocetina con las plaquetas del paciente, éste no hace que se agrupen juntas como lo hace con las plaquetas normales. 4. Trombocitemia Esencial (TE): ¿Qué es? Se define a la TE como una neoplasia mieloproliferativa crónica clonal, que compromete en forma primaria la línea de megacariocitos (MK) de médula ósea (MO), caracterizada por una persistente trombocitosis (mayor a 450.000/µl), hiperplasia megacariocítica, en ausencia de eritrocitosis o leucoeritroblastosis, con un curso clínico relativamente benigno con complicaciones trombóticas arteriales o venosas y hemorrágicas, con un aumento del riesgo de transformación a una neoplasia hematológica más severa . La TE es la entidad más difícil de definir; esto se ve reflejado en la diversidad de criterios propuestos para establecer su diagnóstico. La TE debe ser diferenciada del resto de las NMP crónicas con trombocitosis por la forma de evolución, pronóstico y tratamiento a instituir. ¿Qué pruebas de hematología se pueden hacer para aporte en el diagnóstico: El descubrimiento de la mutación del gen de la tirosinkinasa JAK2V617F en el cromosoma 9, permitió introducir nuevos criterios en el diagnóstico de las NMP crónicas, pero su presencia en otros SMP y su ausencia en muchas TE impide utilizarlo como criterio aislado de diagnóstico; por lo tanto, son necesarios datos clínicos, de laboratorio y anatomopatológicos para poder realizarlo . Es por ese motivo que los criterios de diagnóstico actuales de TE de la Organización Mundial de la Salud (OMS) publicados en el 2008 y que modifican los criterios de la PVSG- requieren de una combinación de datos clínicos, histológicos y citogenéticos, con especial énfasis en los hallazgos morfológicos de los MK de MO. En aquellos pacientes con trombocitosis persistente no reactiva deben excluirse otras patologías mieloproliferativas que pueden simular la TE, como son la LMC, la PV, la MFP en sus distintas fases y las mielodisplasias (MDS). Criterios diagnósticos: Se deben cumplir los 4 criterios para el diagnóstico. Características de los criterios de la WHO: A) Recuento plaquetario: Se reduce el número de plaquetas de 600.000/µl a 450.000/µl, ya que el percentil 95 para el recuento plaquetario normal ajustado para sexo y edad es menor a 400.000. Esto permite reconocer estadios tempranos de la TE. B) Biopsia de MO y Anatomía patológica de la MO: Deben ser excluidas todas las entidades clínicas que cursan con trombocitosis. La celularidad es normal o moderadamente hipercelular para la edad del paciente. Patrón histoarquitectural general conservado. MK. Ubicación centromedular en grupos o dispersos. MK de tamaño grande o gigante. Citoplasma abundante. Núcleos con hiperlobulaciones profundas y contornos irregulares. Emperipolesis frecuente, sin ser un hallazgo especifico. Serie mieloide y eritroide en número normal o incrementado. La serie eritroide se halla incrementada mayormente en los casos de hemorragias previas. Las fibras reticulínicas presentan patrón normal o están mínimamente incrementadas en la TE (el incremento significativo de fibras reticulínicas o colágenas aleja el diagnóstico de TE) y hasta un 3% pueden tener fibrosis mínima, recordando que una terapéutica previa pueda inducir la fibrosis. La presencia de hemosiderina suele observarse en un 40-70% de los casos. No se observan blastos ni alteraciones displásicas de la serie granulocítica y la evolución a leucemia aguda es sumamente rara. La hematopoyesis extramedular es rara. C) Mutación del JAK2V617F u otra manifestación clonal . Entre el 50-60 % de los pacientes con TE son positivos para la mutación JAK2V617F generalmente en estado heterocigota-, siendo infrecuente (<5%) la homocigosidad para esta mutación. Un porcentaje menor, que oscila entre 14%, es portador de mutaciones en el receptor de trombopoyetina MPL, las cuales se detectan más frecuentemente en los pacientes JAK2V617F negativos. Casi el 95% de las TE tienen estudio citogenético normal y, cuando las anormalidades cariotípicas están presentes, son variables; con el uso de la técnica de FISH se pueden detectar anomalías numéricas ocultas en los cromosomas 8 y 9. Las anormalidades citogenéticas asociadas a trombocitosis como la del (5q), t(3;3)(q21;q26) e inv. (3) )(q21;q26) son características de los SMD y de la LMA más que en la TE. La del (20q), del (5q) y la traslocación entre 1q y 7p se reportaron en TE típicas. Su presencia, a menos que se acompañen de los cambios histológicos en MO que caracterizan a las MDS, no deberían excluir a la TE. Algunos pacientes con TE pueden tener por RT-PCR transcriptos de BCR/ABL y su prevalencia y relevancia clínica es controvertida, en algunos casos son LMC atípicas, cuya MO muestra la presencia de MK hipolobulados. D) Exclusión de otros cuadros hematológicos y trombocitosis reactivas Deben ser excluidas la PV, la MFP y la LMC por tener evolución, pronóstico y tratamiento diferentes, siendo el diagnóstico diferencial de mayor dificultad el estadio pre-fibrótico o temprano de la MFP. En aquellos casos con un cuadro clínico superpuesto con TE, la morfología de los MK se utilizará para el diagnóstico diferencial; mientras que en la TE los núcleos de los MK son hiperlobulados, en la MFP son hipercromáticos, con bizarrías nucleares y marcadas anomalías madurativas núcleo-citoplasmáticas definidas como atipia morfológica. La clasificación WHO es cuestionada actualmente por su énfasis en aspectos histológicos específicos, pues existe variabilidad inter-observador en la morfología de los MK y, particularmente, en la definición subjetiva de la forma pre-fibrótica de la MFP, donde no hay similitud de criterios. Tampoco existe una correlación clínica precisa que permita justificar la inclusión de este estadio como una entidad diferente. La presencia de diseritropoyesis, macrocitosis, monocitosis, anomalía pseudoPelgerHuet u otros cambios dismielopoyéticos de los neutrófilos y/o predominio de micromegacariocitos con núcleos no lobulados, sugieren la presencia de una MDS. La distinción entre TE y TR es clínicamente relevante porque las complicaciones trombo-hemorrágicas son más comunes en la primera. Se debe sospechar una trombocitosis secundaria cuando se encuentra: Microcitosis o cuerpos de Howell-Jolly o leucoeritroblastosis en el frotis periférico. Cuando la MO es hipercelular con hiperplasia granulocítica y/o morfología MK normal, con células de pequeño y mediano tamaño y núcleos lobulados. Cuando la ESD está acelerada o PCR aumentada o hiperfibrinogenemia, hiperpotasemia, y patrón de hierro de anemia de los trastornos crónicos o ferropenia. Durante la década pasada se propusieron varios estudios diagnósticos adicionales para poder diferenciar los SMP entre sí (como la morfología del MK en biopsias óseas y el cultivo de colonias de MK, en forma similar a las colonias eritroides endógenas en PV), pero hubo dificultades para estandarizar estos estudios y, además, fue demostrada hipersensibilidad a la trombopoyetina (TPO) tanto en TE como en TR 5. Síndrome de Sebastián: ¿Qué es? El síndrome de Sebastián es una rara enfermedad genética que se presenta con plaquetas de tamaño mayor al normal y una disminución en el recuento total de plaquetas que ralentizan el proceso de coagulación de las hemorragias. Está clasificada dentro de la familia de los desórdenes de plaquetas gigantes (junto con el Síndrome de Bernard-Soulier, la anomalía de May-Hegglin, el síndrome de plaquetas grisáceas, etc.) y tiene como característica que los leucocitos presentan unas inclusiones características denominadas cuerpos de Döhle. El trastorno está causado por una mutación en el gen MYH9 localizado en el cromosoma 22 (22q12.3) que codifica una enzima conocida como miosina de cadena pesada no muscular 9. El síndrome de Sebastián es una enfermedad hereditaria autosómica dominante por lo que basta con heredar un gen mutado del padre o de la madre para padecer la enfermedad Las personas afectadas con este síndrome presentan sangrados abundantes por problemas en el proceso de coagulación de la sangre. ¿Qué pruebas de hematología se pueden hacer para aporte en el diagnóstico: . El síndrome de Sebastián se relaciona con unas características concretas de las células sanguíneas: Plaquetas gigantes (presencia de plaquetas de tamaño mayor al normal) Trombocitopenia (disminución del número total de plaquetas) Inclusiones leucocitarias (Cuerpos de Döhle) 6 Y 7: Síndrome de Fechtner: y Síndrome de Epstein: ¿Qué es? El grupo de enfermedades causadas por mutación en el gen MYH9 se agrupaban en cuatro síndromes caracterizados por presentar macrotrombocitopenia asociada a otras enfermedades. Síndrome de Fechtner. Síndrome de Epstein. Tanto el síndrome de Epstein como el de Fechtner, en los que la afectación renal está presente, habían sido considerados clásicamente como variantes del síndrome de Alport. Por otra parte, el síndrome de Sebastián y la anomalía de May-Hegglin se presentan sin afectación renal, manifestándose únicamente por la presencia de inclusiones leucocitarias y afectación ocular o auditiva. Se considera que el síndrome de Epstein, el síndrome de Fechtner, la anomalía de May-Hegglin y el síndrome de Sebastián no son más que diferentes expresiones fenotípicas, de un procesó único. En conjunto todos estos síndromes han sido denominados trastornos relacionados con MYH9 (MYH9RD), los cuales presentan como características comunes la existencia de plaquetas grandes y trombocitopenia presente desde el nacimiento. Pruebas hematológicas para el diagnóstico: Síndrome de Epstein: Hemograma Completo, donde se verá si hay trombocitopenia. El frotis de sangre periférica demuestra al microscopio: plaquetas grandes o cuerpos tipo Döhle en el citoplasma de los neutrófilos. La inmunofluorescencia de un frotis de sangre periférica demuestra agregados de proteína MYH9 típicos en el citoplasma de los neutrófilos. Prueba de creatinina en sangre: niveles de creatinina elevados indican la progresión a insuficiencia renal y riesgo de enfermedad renal terminal. Estos trastornos, caracterizados por trombocitopenia con plaquetas gigantes, se clasificaron en función de los aspectos morfológicos de los cuerpos tipo Döhle y las diferentes combinaciones de las otras manifestaciones de mutación en MYH9: pérdida de la audición, nefropatía glomerular y cataratas. Además, los estos síndromes no definen todas las manifestaciones posibles derivadas de las variantes patogénicas heterocigotas en MYH9. Finalmente, los miembros de la misma familia pueden tener diferentes fenotipos y recibir diferentes diagnósticos dentro del espectro de enfermedades asociadas a mutaciones en el gen MYH9. Por estas razones, las patologías asociadas a MYH9 han sido propuestas como una nueva entidad nosológica que incluye a todos los individuos con variantes patogénicas heterocigotas en MYH9 independientemente del aspecto de los neutrófilos y el fenotipo clínico. Estos síndromes históricamente conocidos, son entidades genéticamente distintas al síndrome de Alport que era el principal diagnóstico diferencia. 8. Síndrome de Alport – Like: ¿Qué es? El síndrome de Alport (SA) es una enfermedad hereditaria de las membranas basales, debida a mutaciones en la colágena tipo IV. Clínicamente se caracteriza por nefropatía hereditaria progresiva, comúnmente asociada a sordera sensorial y/o lesiones oculares y, en ocasiones, leiomiomatosis, con una prevalencia aproximada de 1 caso por cada 50.000 nacidos vivos, aunque probablemente esté infradiagnosticado por la dificultad del diagnóstico ante casos aislados o sin sordera. Es la causa del 1-2% de los pacientes que inician tratamiento renal sustitutivo. El SA presenta una clínica sistémica que afecta a las membranas basales de diferentes tejidos debido a la alteración del colágeno tipo IV. Según el patrón de herencia diferenciamos dos tipos: SA ligado a X, en el 85% de los casos, y SA autosómico recesivo, en el 15% de los casos. ¿Qué pruebas de hematología se pueden hacer para aporte en el diagnóstico: Sangre extraída con EDTA para separación de leucocito sanguíneo. 9. Anomalía de May-Hegglin: La anomalía de May-Hegglin es uno de los síndromes debido a mutaciones en el gen MYH9 (da lugar a muy pocos síntomas o a ninguno. La prevalencia de la anomalía de May-Hegglin es desconocida. La macro trombocitopenia se define por la presencia de plaquetas gigantes, con un diámetro igual o superior al diámetro de un glóbulo rojo. La anomalía de May-Hegglin, así como el síndrome de Sebastián, es una forma puramente hematológica del síndrome MYH9. Se caracteriza por la presencia de inclusiones citoplasmáticas en las células de la línea de los granulocitos. Las inclusiones citoplasmáticas que caracterizan a la anomalía de May-Hegglin presentan forma de huevo, se tiñen de azul en presencia de MayGrünwald-Giemsa (MGG) y se conocen como cuerpos Döhle. La tendencia hemorrágica asociada con este síndrome es generalmente leve, y la mitad de los pacientes son asintomáticos. Sin embargo, el 40 % de los pacientes presentan un sangrado anormal (trombopenia púrpura: epistaxis, menstruaciones profusas, equimosis). La trombocitopenia de May-Hegglin se transmite siguiendo un patrón autosómico dominante, con mayor frecuencia como resultado de mutaciones puntuales en el gen MYH9. Sin embargo, se estima que el 20 % de los casos son esporádicos, asociados a una mutación de novo o a mosaicismo. El gen MYH9, localizado en 22q12-13, codifica para la cadena pesada de la miosina no muscular de tipo IIA (MYHIIA), que se expresa en algunas células sanguíneas (células polinucleares, monocitos y plaquetas), en la cóclea y en los riñones. Estas anomalías moleculares provocan la dimerización anómala de la proteína MYHIIA, que se vuelve inestable y coprecipita con la MYHIIA normal en el citoplasma de los leucocitos, lo que da lugar a cuerpos de inclusión citoplasmáticos. Esta dimerización anómala provoca también un fallo en la organización del citoesqueleto de los megacariocitos, lo que desencadena una trombopenia macrocítica. La mayoría de los pacientes no presentan sangrados anómalos, en cuyo caso no se requiere un tratamiento específico. Sin embargo, puede ser recomendable una transfusión de plaquetas antes de una intervención quirúrgica. La esperanza de vida es normal. ¿Qué pruebas de hematología se pueden hacer para aporte en el diagnóstico: Hemograma completo: donde se verá trombocitopenia y cuerpos de Döhle. El frotis de sangre periférica demuestra al microscopio: plaquetas grandes La inmunofluorescencia de un frotis de sangre periférica demuestra agregados de proteína MYH9. Conclusión Las plaquetas son indispensables para la hemostasia primaria. Los defectos de la función plaquetaria abarcan un grupo grande y heterogéneo de trastornos hemorrágicos cuyas manifestaciones pueden ser de leves a graves. A medida que se dilucidan mejor las complejas vías bioquímicas internas y de transducción de señales, y conforme avancen los análisis estructurales de plaquetas, se comprenderán más mecanismos que provocan defectos de la función plaquetaria. Bibliografía http://www1.wfh.org/publication/files/pdf-1148.pdf https://www.ecured.cu/S%C3%ADndrome_de_Sebasti%C3%A1n https://www.ecured.cu/Enfermedad_de_Glanzmann https://www.cdc.gov/ncbddd/spanish/vwd/facts.html https://marte.enfermedadesraras.org/recursos/cuestionario/patologias/editar/10001524/