Status Epilepticus in Pediatric Emergency: Article Review
Anuncio

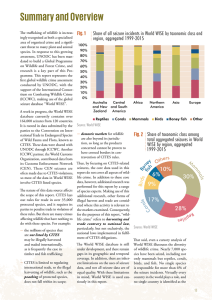
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/281699140 Status epilepticus in the pediatric emergency department Article in Clinical Pediatric Emergency Medicine · March 2015 DOI: 10.1016/j.cpem.2015.01.001 CITATIONS READS 2 1,430 2 authors, including: Jonathan Kurz Northwestern University 31 PUBLICATIONS 303 CITATIONS SEE PROFILE All content following this page was uploaded by Jonathan Kurz on 09 October 2017. The user has requested enhancement of the downloaded file. Abstract: Status epilepticus is a common pediatric neurologic condition that often presents to the emergency department. Prior neurologic abnormalities or preexisting epilepsy are significant risk factors for the development of status epilepticus, although younger children more commonly present with either an acute symptomatic or febrile etiology. Prolonged seizure duration is associated with the development of resistance to anticonvulsants and the potential for permanent neurologic injury. Treatment strategies should focus on rapid administration of appropriate antiepileptic medications, with benzodiazepines used as the first-line agent. Nonconvulsive and subclinical status epilepticus may be difficult to identify. A high index of suspicion should be maintained for nonconvulsive status epilepticus, particularly in encephalopathic and critically ill children. Treatment of refractory status epilepticus often requires intensive care unit admission and the involvement of a neurologist. Initial diagnostic evaluation should include a broad differential and be focused on treatable causes, including central nervous system infections, electrolyte and metabolic disorders, and trauma. Keywords: status epilepticus; subclinical status epilepticus; nonconvulsive status epilepticus; refractory status epilepticus; anticonvulsant Department of Pediatrics, Division of Neurology, Ann & Robert H. Lurie Children's Hospital of Chicago, Northwestern University Feinberg School of Medicine, Chicago, IL; Ruth D. & Ken M. Davee Pediatric Neurocritical Care Program, Ann & Robert H. Lurie Children's Hospital of Chicago, Northwestern University Feinberg School of Medicine, Chicago, IL. Status Epilepticus in the Pediatric Emergency Department S Jonathan E. Kurz, MD, PhD, Joshua Goldstein, MD tatus epilepticus (SE) is a common, life-threatening neurologic emergency, characterized by persistent, continuous seizure activity. It is commonly encountered by pediatric emergency care providers; 6 to 7% of patients presenting to the emergency department (ED) with seizure are in SE, 1,2 and there are approximately 3.1 million pediatric ED visits in the United States for seizure yearly. 3 Prolonged seizures carry a significant risk of mortality, estimated to be 3% among pediatric patients. 4,5 Status epilepticus additionally predisposes patients to short- and long-term neurologic morbidities, including recurrent SE, cognitive deficits, and neurodevelopmental delays. 4,6,7 Animal models suggest that resistance to anticonvulsant therapy develops as the duration of seizure increases. 8 Early recognition of SE and appropriate treatment by prehospital and emergency care providers may therefore improve chances for rapid seizure control and reduce subsequent morbidity and mortality. CLASSIFICATION AND PATHOPHYSIOLOGY Status epilepticus is characterized by continuous clinical and/ or electrographic seizure activity, or episodes of recurrent seizure activity without recovery between seizures. There is considerable variability in the duration of continuous seizures required to meet the threshold for SE in clinical trials and treatment guidelines. Current practices favor a shorter duration of continuous seizures being defined as SE, based on the typical duration of individual seizures and animal data suggesting increased risk for STATUS EPILEPTICUS IN THE PEDIATRIC EMERGENCY DEPARTMENT / KURZ AND GOLDSTEIN • VOL. 16, NO. 1 37 38 VOL. 16, NO. 1 • STATUS EPILEPTICUS IN THE PEDIATRIC EMERGENCY DEPARTMENT / KURZ AND GOLDSTEIN Reprint requests and correspondence: Joshua Goldstein, MD, Ann and Robert H. Lurie Children's Hospital of Chicago, Division of Child Neurology, No. 51, 225 E. Chicago Ave, Chicago, IL 60614. [email protected] (J.E. Kurz), [email protected] (J. Goldstein) 1522-8401 © 2015 Published by Elsevier Inc. pharmacoresistance and neuronal injury with increasing seizure duration. 9,10 Most seizures are self-limited, brief events. In adult epilepsy monitoring units, video and subdural electrode monitoring of patients with secondarily generalized seizures demonstrated a mean clinical and electrographic duration of less than 2 minutes. 11 Another study of adult seizure duration documented mean seizure durations of 130 seconds or less, depending on seizure type. 12 Most seizures in children are similarly brief and self-limited. Among children with new-onset seizure, 76% of seizures were brief, with a mean duration of 3.6 minutes; the remainder lasted more than 30 minutes. 13 Seizures lasting more than 10 to 15 minutes are less likely to stop spontaneously 13 and may signify a failure of the mechanisms that typically terminate a seizure. These longer events are more likely to progress to the self-sustaining and continuous seizures of SE. Data from animal models show that experimentally induced seizures become self-sustaining between 15 and 30 minutes. After this point, a self-perpetuating circuit presumably develops, sustaining seizure activity for hours without further stimulation. 14 Although extrapolation of animal data to clinical practice is imprecise, a continuous seizure in humans is thought to involve both failure of the typical endogenous mechanisms leading to seizure termination and development of self-sustaining seizure activity. Numerous neurophysiologic and biochemical changes are postulated to play a role in the initiation of SE. For example, synaptic expression and subunit-specific trafficking of g-aminobutyric acid type A (GABAA) receptors changes with increasing seizure duration. 15 The resultant change in GABA-nergic inhibitory neurotransmission may help sustain ongoing seizure activity. In addition, seizure-induced changes in GABA receptor subunit composition may decrease response to anticonvulsant therapies by altering receptor sensitivity to benzodiazepines. 16 In animal models, as seizure duration increases, resistance to anticonvulsant medications develops. 8 Similarly, in a study of pediatric patients, delay between seizure onset and treatment in the ED was associated with an increase in the risk of seizures lasting more than 60 minutes. 17 These data support aggressive treatment of seizures at earlier timepoints, before the development of self-sustaining seizures and pharmacoresistance impairs the efficacy of abortive anticonvulsive therapy. Seizure-induced neuronal injury may also occur during SE. In an animal model of pilocarpine-induced SE, dead neurons begin to appear in the hippocampus after 20 minutes of continuous seizure activity, and mild damage is present in multiple brain regions by 40 minutes. 18 Similarly, in primate studies, evidence of brain injury was present after 45 to 60 minutes of continuous seizure. 19 Neuronal injury may be due to both ischemic 19 and excitotoxic 20 mechanisms. Evidence for seizure-induced brain injury in human studies is less detailed, and the exact time-point at which injury begins to occur is not well defined. 21 There is limited magnetic resonance imaging (MRI) evidence that suggests that prolonged seizures cause cerebral atrophy. In addition, some authors have postulated that decreases in measured mortality in pediatric SE over the past 4 decades are due to earlier and more effective treatment of SE, implying that morbidity and mortality in prolonged seizure may be duration dependent. 22 In consideration of the short typical duration of self-limited seizures, the potential for pharmacoresistance with longer events, and the potential for seizure-induced neuronal injury that may occur with a longer duration of continuous seizures, the threshold duration of seizure defined as SE in clinical trials and treatment guidelines has been repeatedly revised. Earlier definitions specified 1-hour or 30-minute time-points; 23,24 the 30-minute definition continues to be used extensively in clinical research. More recently, other authors, 25 as well as recent guidelines from the Neurocritical Care Society, 9 have proposed defining SE as 5 minutes of continuous seizure. In addition, some authors propose a category of “early” or “impending” SE at 5 minutes of continuous seizure, for the purpose of initiating early treatment, while maintaining the 30 minute time-point as a threshold for “established” SE for the purpose of epidemiologic studies. 26 Regardless of the definition used, it is reasonable to begin pharmacologic treatment at 5 minutes of continuous seizure, in an attempt to abort the seizure prior to the development of physiologic changes that limit response to treatment or promote neuronal injury. In addition to avoiding brain injury, prompt treatment of continuous seizures may shorten recovery time, reduce morbidity, and reduce the need for intensive care admission. 7,27 STATUS EPILEPTICUS IN THE PEDIATRIC EMERGENCY DEPARTMENT / KURZ AND GOLDSTEIN • VOL. 16, NO. 1 39 Status epilepticus can be classified by semiology into convulsive (CSE) and nonconvulsive SE (NCSE). Convulsive SE typically is characterized by generalized tonic-clonic convulsions and mental status impairment, although myoclonic SE and tonic SE also can present in certain clinical situations. Focal motor SE (epilepsia partialis continua) is generally considered to be a separate entity from CSE and is not covered by this review. Nonconvulsive SE has been defined as a range of conditions in which electrographic seizure activity is prolonged and results in nonconvulsive clinical symptoms, 28 coupled with a proposed minimal seizure duration of 30 minutes. Nonconvulsive SE can be associated with subtle motor manifestations or can be entirely subclinical, presenting only with a clouding of alertness, confusion, or a change in mental status. EPIDEMIOLOGY Status epilepticus is one of the most common pediatric neurologic emergencies, with an incidence of 17 to 38 per 100 000 in children. 4,29 The incidence of pediatric SE is age dependent; reported incidences are as high as 51 to 156 per 100 000 in the first year of life 4,29 and lowest among adolescents. Most epidemiologic studies of SE have used a 30-minute time-point as the definition of SE; including children that present with prolonged seizures in the 5- to 29-minute range would likely result in higher incidences. The mean age of children presenting with SE is 4.4 years. Most of these cases are convulsive SE. 30 Prior neurologic conditions are a risk factor for developing SE. Fifty-six to 60% of children presenting with SE are neurologically normal beforehand, whereas the remainder have a preexisting neurologic abnormality. 4,30 These remote symptomatic causes of SE are less common in younger children (who more commonly present with febrile or acute symptomatic SE) and more common in older children and adolescents. In one study, 60% of children older than 5 years with SE had a prior neurologic abnormality, compared with only 21% of children younger than 2 years. 30 Preexisting epilepsy is a risk factor for developing SE. Approximately 10% of children with childhood-onset epilepsy will experience at least one episode of SE. 31 In an analysis of 2 large cohorts of children with SE, 45% had a history of one or more unprovoked afebrile seizures. 30 Among children with chronic epilepsy, a major risk factor for developing SE is having experienced prior episodes of SE; 32 occurrence of SE is more than 20-fold higher in children with a history of SE than in those TABLE 1. Etiologies of status epilepticus. Febrile SE Presents between 6 mo and 6 y of age Should rule out infectious causes Acute symptomatic Infectious - Bacterial meningitis - Viral encephalitis (including herpes simplex) Metabolic - Hypoglycemia and hyperglycemia - Hyponatremia and hypernatremia - Hypocalcemia - Hypomagnesemia Toxicologic Trauma - Epidural, subdural, or subarachnoid hemorrhage - Intraparenchymal hemorrhage Vascular - Arterial ischemic stroke - Central sinus venous thrombosis Preexisting epilepsy Medication withdrawal or noncompliance Concurrent infection Remote symptomatic Prior central nervous system (CNS) trauma, stroke, cortical malformation, other preexisting CNS abnormality without. Patients with a history of seizure clustering are also more likely to experience SE. 32 The risk of recurrent episodes of SE is highest in children with remote symptomatic etiologies and those with progressive neurologic disease. 33 ETIOLOGIES AND DIAGNOSTIC WORKUP On initial assessment of patients with SE, emergency providers should maintain a broad differential diagnosis. Causes of SE can be separated into febrile SE, acute symptomatic, remote symptomatic, and idiopathic or cryptogenic (Table 1). During acute evaluation and treatment, providers should have a particular focus on identifying reversible causes of SE, such as electrolyte disturbances, hypoglycemia, and for patients already on antiepileptic drugs (AED), subtherapeutic medication levels. Acute evaluations should also be focused on identifying potential underlying etiologies that are life-threatening or will require a change in acute management, such as central nervous system (CNS) infection, trauma, or stroke (Table 2). Febrile seizures are the most common form of pediatric seizure, affecting 2 to 4% of children in the United States and Western Europe. 34 Presenting between 6 months and 6 years of age, most of these 40 VOL. 16, NO. 1 • STATUS EPILEPTICUS IN THE PEDIATRIC EMERGENCY DEPARTMENT / KURZ AND GOLDSTEIN TABLE 2. Initial studies for status epilepticus without identified etiology. Serum electrolytes (sodium, magnesium, calcium) and glucose Computed tomography of head Complete blood count Antiepileptic drug levels (if applicable) Lumbar puncture (if febrile or concern for CNS infection) Consider urine or serum toxicology (if ingestion history or cause not identified) events are simple febrile seizures, or, single, nonfocal events lasting less than 10 minutes occurring in otherwise healthy children. However, febrile SE (defined as duration N 30 minutes) represents 5% of all febrile seizures, and febrile SE is the underlying etiology for approximately one quarter of all episodes of childhood SE. Febrile SE accounts for more than two thirds of SE occurring in children between 1 and 2 years of age. 30 These seizures can be very prolonged; in one study, 24% of episodes of febrile SE lasted longer than 2 hours. 35 Ninety percent of patients in febrile SE required an AED to terminate the seizures, with patients requiring an average of 2 medications. 36 Most of febrile SE is convulsive, and most seizures have some focal features. 35 As with other etiologies of SE, prompt treatment should be initiated to limit neurologic morbidity in febrile SE. Prolonged febrile seizure can lead to acute hippocampal injury detectable on MRI, with increased risk for the development of mesial temporal sclerosis. 37 Although common, febrile SE is a diagnosis of exclusion, and other causes of prolonged seizure with fever, including CNS infection, should be excluded. Cerebrospinal fluid (CSF) analysis in children after febrile SE is typically normal, 38 and CSF pleocytosis should raise a concern for CNS infection or another medical explanation. Central nervous system infection is a common etiology of SE in children. Meningoencephalitis or encephalitis, either bacterial or viral, can lead to SE. A documented CNS infection was reported on average in 12.8% of children with SE in one meta-analysis. 39 In a series of 24 children with SE and fever, bacterial meningitis was detected in 17% of the febrile group, none of whom exhibited typical signs of meningismus. 40 Another report documented acute bacterial meningitis in 11% of children presenting with first ever febrile SE. 4 Given these data, lumbar puncture (LP) and CNS imaging should be obtained in the setting of SE and fever at any age, unless an LP is contraindicated or another etiology is clearly identified. Empiric treatment with antibiotics and acyclovir should be considered until CSF analysis excludes herpes encephalitis or bacterial meningitis as a possibility. Among children with preexisting epilepsy, medication noncompliance or withdrawal is a frequent cause of SE. Even children with well-controlled epilepsy can experience prolonged seizures after missing medication doses. Among children taking AEDs who experienced SE, 32% had low serum levels in one analysis. 39 In children with epilepsy who are taking AEDs, obtaining serum levels should be considered if available. A careful medication history should be obtained to determine if there are missed doses or recent medication changes. Electrolyte abnormalities such as hyponatremia, or metabolic abnormalities such as hyperglycemia or hypoglycemia, can play a role in pediatric SE. Hypocalcemia may present as SE in neonates. In studies of the diagnostic yield of these studies, electrolyte or glucose abnormalities have been reported in 6% of children with SE on average. 39 Status epilepticus induced by electrolyte abnormalities may be refractory to treatment until the underlying metabolic disturbance is corrected. Evaluation of electrolytes and glucose should be obtained on all children presenting with SE. Toxic ingestion may be suggested by the history and should also be considered in cases with unknown etiology. Toxic ingestion is documented in 3.6% of reported cases of SE. 39 Serum and urine toxicology may be helpful in establishing a diagnosis in these cases. Urine toxicology screening tests typically evaluate only for drugs of abuse; if ingestion of a specific agent is suggested by the history, specific serum toxicologic testing for that agent may be more useful. Seizures and SE can be the presenting symptom of traumatic brain injury. Although trauma may be suggested by the history or examination, in some cases, the history may be unclear. In particular, in cases of nonaccidental trauma, the initial history may be incomplete or inaccurate. Nonaccidental injury in infants is strongly associated with the development of prolonged seizure activity. 41 Clinicians should be observant for any external findings that are suggestive of traumatic injury. In cases where a clear etiology of SE cannot be identified, neuroimaging should be obtained once the child is stabilized and the seizures are controlled. These studies are reasonably high yield in this setting; in one study of patients with new-onset seizure presenting as SE, neuroimaging (computed tomography [CT] or MRI) was diagnostic of an underlying etiology in 30% of patients and directed management STATUS EPILEPTICUS IN THE PEDIATRIC EMERGENCY DEPARTMENT / KURZ AND GOLDSTEIN • VOL. 16, NO. 1 41 in 24%. 42 Although MRI identified abnormalities not detected by CT, these were mostly lesions associated with a remote, rather than acute symptomatic cause. Computed tomography is generally appropriate as an acute imaging modality for patients presenting with SE. Limited-sequence rapid MRI protocols are available in some centers and may also be considered for initial imaging. Vascular lesions, such as arterial ischemic stroke or central sinus venous thrombosis, may present as seizure or SE. 43 A persistent new focal neurologic finding should raise a high suspicion for stroke, although prolonged seizure with a subsequent Todd paralysis may mimic stroke-like symptoms. Patients with new focal neurologic findings all require neuroimaging as part of their evaluation. Computed tomographic imaging of the brain may be helpful in identifying acute ischemic stroke and intracranial hemorrhage, although subtle or hyperacute ischemic lesions may require repeat imaging to identify. MRI with diffusion-weighted imaging provides superior evaluation for ischemia, particularly small infarcts or lesions in the posterior fossa. MRI should be considered as part of a patient's evaluation for SE once the patient is stable enough for the longer imaging procedure, particularly if there is a high suspicion for stroke or structural lesion not identified on CT. Remote symptomatic causes, such as occult prior CNS injury, cortical dysplasia, or vascular malformations (in the absence of acute hemorrhage), also may present as SE. Identification of these causes may require imaging with MRI or more detailed laboratory evaluation. Because these remote causes are generally not acutely reversible, this evaluation may continue outside the emergent setting. Similarly, other acute symptomatic etiologies, such as CNS autoimmune conditions, may require extensive serologic testing to identify as part of an ongoing workup after the patient has left the ED. ACUTE TREATMENT Although guidelines and practice parameters regarding the acute management of SE are available, 9 there is no universally accepted treatment paradigm for pediatric SE, and protocols can vary between institutions. 44 Despite this, there is agreement on common principles of treatment of SE, including the need for rapid and appropriate therapy to terminate seizures (Figure 1). As with any critically ill patient, the first steps in management should be assessment of vital signs, airway, breathing, and circulation. The airway should be secured, first with noninvasive maneuvers (midline positioning, jaw thrust, or chin tilt) and with more invasive measures if necessary. Oxygen should be provided, intravenous (IV) access obtained, and any hemodynamic instability addressed. Reassessment of airway, breathing, and circulation and vital signs should continue throughout the acute treatment of SE, and providers should remain alert for changes induced both by prolonged seizure and by adverse effects of therapy. Many AEDs are sedating and may lead to an inability to maintain a safe airway, potentially requiring further intervention; prolonged seizure may induce an encephalopathy with a similar potential effect. Benzodiazepines and barbiturates can suppress respiratory drive. Several medications, particularly barbiturates, can induce hypotension. As discussed above, diagnostic workup should begin concurrently with acute treatment. A glucose check should be obtained during the initial assessment. Most treatment protocols advocate a stepwise approach to anticonvulsant therapy in CSE. Anticonvulsant therapy should begin after 5 minutes of continuous seizure activity. Benzodiazepines are generally given as the first-line agent, followed by a loading dose of an IV AED, most commonly fosphenytoin. It is important to avoid excessive time intervals between medications, underdosing of medications, or provision of medications via an inappropriate route. First-Line Treatment Intravenous lorazepam is the preferred agent for initial therapy of SE. Data from adult studies suggest that IV lorazepam is superior to IV diazepam and IV phenytoin alone for control of SE, 45 although one pediatric randomized clinical trial suggests that IV diazepam and lorazepam may be equivalent in children. 46 Lorazepam should be given at a dose of 0.1 mg/kg IV (at a rate of up to 2 mg/min) to a maximum dose of 2 to 4 mg. If IV access is not available or will require a significant delay in therapy, numerous other benzodiazepine dosage routes are available. Midazolam can be administered via intramuscular, buccal, or intranasal routes; buccal or intranasal dosing is 0.2 to 0.5 mg/kg up to a maximal dose of 10 mg. 47,48 Diazepam is the preferred medication for rectal administration. Rectal dosing ranges from 0.2 to 0.5 mg/kg, depending on age. 49 Benzodiazepine therapy should be provided promptly and at an adequate dose. Importantly, frequent small or subtherapeutic doses of benzodiazepines should be avoided, as this will delay time to adequate serum concentrations of anticonvulsant. Instead, 1 or 2 doses of benzodiazepine should be provided at a therapeutic dose, to allow for more timely administration of a second agent if needed. 42 VOL. 16, NO. 1 • STATUS EPILEPTICUS IN THE PEDIATRIC EMERGENCY DEPARTMENT / KURZ AND GOLDSTEIN Figure 1. Algorithm for initial therapy of status epilepticus. Second-Line Treatments After initial therapy with benzodiazepines, the next phase of management is an IV loading dose of an AED. There are 2 goals of this phase of therapy: terminating ongoing seizure activity and rapidly producing therapeutic serum concentrations of an AED for ongoing seizure control after SE is terminated. Intravenous fosphenytoin is most commonly used as the agent of choice for second-line therapy in pediatric SE. Fosphenytoin is a water-soluble prodrug that is converted to phenytoin by serum and tissue alkaline phosphatase within 10 minutes of administration. Phenytoin acts at neuronal voltage-gated sodium channels to limit repetitive firing of action potentials. In both adult and pediatric randomized controlled trials, a phenytoin-diazepam combination was equivalent to lorazepam in terminating SE as a first-line agent. 45,50 In a study of 3 second-line agents in SE, the efficacy of phenytoin was not statistically different from valproic acid or levetiracetam. 51 As compared with phenytoin, fosphenytoin infusion is less likely to induce cardiac arrhythmias and has a lower risk of causing tissue necrosis if extravasation occurs. 52 Fosphenytoin is dosed in phenytoin equivalents (PE). A typical loading dose in SE is 20 mg PE/ kg IV, which should provide a serum total phenytoin level of approximately 20 μg/mL. Some protocols STATUS EPILEPTICUS IN THE PEDIATRIC EMERGENCY DEPARTMENT / KURZ AND GOLDSTEIN • VOL. 16, NO. 1 43 advocate a second dose of 5 to 10 mg PE/kg in children with persistent seizures after the initial loading dose. Other IV AEDs have also been studied as acute therapy for CSE as alternatives to phenytoin. Valproic acid (VPA) is an AED with several proposed mechanisms, including action at T-type calcium channels, voltage-gated sodium channels, and GABA receptors. Valproic acid presents several advantages in the setting of CSE: it can be rapidly administered via IV and is efficacious against a broad variety of seizure types (including primary generalized epilepsy), and loading doses are generally well tolerated. Two randomized controlled trials (primarily with adult patients) have been reported comparing efficacy of VPA to phenytoin in the treatment of acute SE. Both trials demonstrated that VPA was equivalent or superior to phenytoin in achieving seizure control. 53,54 In a retrospective review of VPA loading for SE and repetitive seizures in pediatric patients, VPA was similarly effective in controlling seizures and was not associated with significant adverse effects. 55 Based on these data, IV loading doses of VPA are a reasonable alternative to phenytoin for second-line therapy in acute SE. This may be particularly true in patients with primary generalized epilepsy or in cases where phenytoin is contraindicated (such as patients with sodium channelopathies or documented phenytoin allergy). Loading doses in the range of 20 to 40 mg/kg are typically reported. Limitations to the acute use of VPA include the potential for hepatotoxicity, hyperammonemia, pancreatitis, and teratogenicity. The risk for VPA-induced hepatotoxicity is highest for children younger than 2 years receiving VPA as polytherapy, 56 and caution should be used in this age group. Valproic acid should be avoided in children with preexisting hepatic failure, pancreatitis, or mitochondrial disease. Valproic acid therapy is also associated with development of congenital malformations, particularly if given during the first trimester, 57 and should be avoided in pregnant patients. Phenobarbital is the most commonly used AED in neonatal SE, based on established practice. Studies regarding the use of phenobarbital in neonatal seizures report similar efficacy to phenytoin, although less than 50% of seizures in neonates were controlled with monotherapy of either agent. 58 Bolus doses of IV phenobarbital can also be used for control of SE in older children and adults, with efficacy similar to that of phenytoin or VPA. 45,58 Loading doses in neonates and older children are typically 20 mg/kg IV, which should yield a plasma level of approximately 20 mg/L. A loading dose can be repeated after 15 to 20 minutes if seizures are not controlled. Phenobarbital acts by enhancing GABA-mediated inhibitory neurotransmission. Adverse effects of phenobarbital are possible with loading doses in SE, 59 including excessive sedation, respiratory depression, and hypotension, which may play a factor in the choice of phenobarbital as a second-line medication outside of neonatal SE. Levetiracetam is a newer AED that offers several theoretical advantages in the treatment of SE. It is available in an IV formulation, has minimal interaction with other medications and a benign side-effect profile, and is a broad-spectrum agent that may be effective against both primary generalized and focal seizures. Evidence for its efficacy in the acute treatment of SE is limited, however, one randomized, controlled pilot study evaluated levetiracetam as a first-line agent at 20 mg/kg, enrolling 79 patients between the ages of 1 and 75 years. Levetiracetam was found to be equally efficacious as compared with lorazepam. 60 Further evidence is available from case series: in one prospective series of adult patients with SE, levetiracetam appeared to be most efficacious in focal SE but was ineffective in all cases of secondarily generalized SE, 61 although the number of patients in each group was small. In a retrospective comparison of levetiracetam, phenytoin, and VPA in adult patients, levetiracetam appeared to be less effective than VPA (and not statistically different from phenytoin). 50 Published evidence specifically regarding the use of levetiracetam in pediatric SE is even more limited, with small numbers of patients with SE reported as part of larger series of children with seizures. 62,63 At present, there are insufficient data to recommend the routine use of levetiracetam as an acute second-line therapy in pediatric SE, although this is certainly an area for further study. Second-line therapy should be administered in a timely fashion in patients failing benzodiazepines. A goal for anticonvulsant therapy in SE through the first dose of fosphenytoin (or other second-line IV anticonvulsant) should be approximately 20 minutes. A sample protocol used in our institution for the acute treatment of SE is illustrated in Figure. Refractory Status Epilepticus Children that continue to experience continuous seizure activity despite therapeutic doses of 2 AEDs (benzodiazepine plus second-line therapy) meet the criteria for refractory SE (RSE); there is no minimum duration of seizure required for this diagnosis. Mortality and neurologic morbidity are higher for children experiencing RSE as compared with children who respond to first- or second-line therapy. In a 44 VOL. 16, NO. 1 • STATUS EPILEPTICUS IN THE PEDIATRIC EMERGENCY DEPARTMENT / KURZ AND GOLDSTEIN meta-analysis of pediatric refractory generalized convulsive SE, overall mortality was 16%. Although reporting of neurologic sequelae of SE varied between the studies selected for meta-analysis, a new neurologic morbidity was present after RSE in 57% of the 61 patients where data were available. 64 In another series of children with RSE, 7 of 22 died as a result of the RSE, and no previously healthy child returned to their neurologic baseline. 65 After failure of a second-line agent, complete suppression of seizure activity with a continuous infusion of an anticonvulsant is recommended. In some clinical situations, providers may elect to trial a bolus dose of another anticonvulsant (phenobarbital or VPA) prior to use of a continuous infusion. A neurologist should be consulted if available, and intensive care unit (ICU) admission should be strongly considered. Respiratory suppression may need to be tolerated to achieve seizure control; intubation may be required to secure the airway. Hypotension is a potential adverse effect of continuous infusion of anesthetics and should be avoided, particularly because cerebral autoregulation may be impaired by the continuous seizure activity. Video-electroencephalogram (EEG) monitoring should be considered for all patients in RSE, both to guide management and to monitor for subclinical seizures. Up to one third of these patients experience electrographic seizures after the convulsive SE is controlled. 66 These seizures may only be detectable with EEG. Midazolam or pentobarbital are commonly used as continuous infusions for the treatment of RSE. In a retrospective study of 358 children, midazolam infusion resulted in eventual seizure control in 64.5%. 67 Adverse effects included primarily respiratory suppression; no causal relationship between midazolam and cardiovascular depression was observed. In one meta-analysis, midazolam was associated with a more rapid return to consciousness and lower intubation rates than barbiturates or inhalational anesthetics. 68 Midazolam infusion is started at a rate of 50 to 100 ug/kg/h and titrated upward to a range of 600 to 1200 ug/kg/h. At our institution, we increase by increments of 50 to 100 ug/kg/h every 15 to 20 minutes. As discussed previously, continuous EEG monitoring should be obtained while using continuous infusions of anticonvulsants. In one adult study of midazolam use in RSE, breakthrough seizures were present in 56% of patients, and these were subtle or entirely subclinical in 89%. 69 Similarly, pentobarbital can be administered as a continuous infusion for the control of RSE. 70,71 Pentobarbital infusion can be initiated with a loading dose of 5 to 8 mg/kg, followed by a continuous infusion at a rate of 0.5 to 1 mg/kg/h. This infusion may be increased as needed to 3 to 5 mg/kg/h. Pentobarbital is associated with a significant degree of CNS and respiratory suppression. Hypotension is more common than that seen with infusions of midazolam, 68 and patients on pentobarbital infusion require cardiac and respiratory monitoring in an ICU. The long-half life of pentobarbital can limit the ability of providers to obtain a neurologic examination, even after the infusion has been discontinued. Nonconvulsive Status Epilepticus Nonconvulsive SE in pediatric patients encompasses a broad range of conditions, with varying clinical presentations, prognosis, and management. Common subtypes that may present to emergency providers include absence SE, complex partial SE (focal seizures with alteration of consciousness), and NCSE in children in coma, which may be seen in critically ill children and may follow CSE. 72 Absence SE occurs almost exclusively in children with idiopathic primary generalized epilepsy and may present with confusion and drowsiness. Children may be alert enough to automatically perform some routine activities. Automatisms and jerking movements of the face and limbs may be present. Complex partial SE may present in a similar fashion, with confusion and potentially a lack of focal features; these children may cycle between unresponsive and partially responsive phases. 72 Both of these conditions require EEG for definitive diagnosis and should be considered in the differential diagnosis of children presenting with an unexplained encephalopathy, particularly in children with a known diagnosis of absence or focal epilepsy. The potential for NCSE in coma has been increasingly recognized in recent years for both children and adults. In a study of 236 comatose children and adults, NCSE was identified in 8%. 73 Nonconvulsive SE may represent the primary cause of an encephalopathy, or may be secondary to an underlying CNS lesion leading to both seizures and unresponsiveness. The potential for NCSE is of particular concern in encephalopathic critically ill children. In a series of 178 unresponsive children in ICU monitored with EEG, NCSE was identified in 33%. 74 In another study, EEG monitoring was obtained on all encephalopathic patients admitted to a pediatric ICU, and NCSE was identified in 18%. 75 Nonconvulsive SE may often follow CSE, even if the clinical seizures have responded to AED therapy. In a study of adults and children with CSE, 48% of patients demonstrated persistent electrographic seizures after treatment of SE, whereas 14% met the criteria for NCSE. 76 In a retrospective series of 19 children with NCSE who were comatose or stuporous at the time STATUS EPILEPTICUS IN THE PEDIATRIC EMERGENCY DEPARTMENT / KURZ AND GOLDSTEIN • VOL. 16, NO. 1 45 of diagnosis, 5 initially presented with CSE, whereas the remainder had no seizures prior to diagnosis of NCSE. 77 Children being treated for RSE or children who do not have improvement of their mental status after treatment of CSE should have continuous EEG monitoring to assess for NCSE. Similarly, EEG should be considered in the evaluation of unresponsive children without a clear etiology, particularly in the setting of critical illness. SUMMARY Status epilepticus is a common neurologic emergency that frequently presents to pediatric emergency providers. Although criteria for the definition of SE vary, there is agreement that treatment should begin early in the course of continuous seizures. Delay in treatment increases the risk of prolonged seizure, medication unresponsiveness, and potential neuronal injury. Treatment protocols may differ across institutions, but should share common principles of rapid treatment with appropriate doses of AEDs. Initial therapy with a benzodiazepine, followed by a second-line agent (typically fosphenytoin), should be achieved within the first 20 minutes of SE. The initial diagnostic workup should occur concurrently and should be focused on identifying acute symptomatic causes of SE that are either reversible or impact acute management. Assessment of glucose and electrolytes should be obtained, an LP obtained in febrile patients, and neuroimaging performed in all cases where the underlying etiology is not immediately apparent. Patients failing appropriate doses of 2 AEDs have RSE; further treatment can include additional IV AEDs and continuous infusions of midazolam or pentobarbital. Patients with RSE will often require admission to a pediatric ICU for ongoing treatment. A high index of suspicion should be maintained regarding NCSE, particularly in patients that do not have an improvement in mental status after treatment of CSE. Acknowledgments This work was supported by The Ruth D. & Ken M. Davee Pediatric Neurocritical Care Program. Conflict of interest: The authors have no conflicts of interest to disclose. REFERENCES 1. Huff JS, Morris DL, Kothari RU, et al. Emergency department management of patients with seizures: a multicenter study. Acad Emerg Med 2001;8:622–8. 2. Krumholz A, Grufferman S, Orr ST, et al. Seizures and seizure care in an emergency department. Epilepsia 1989;30: 175–81. 3. Pallin DJ, Goldstein JN, Moussally JS, et al. Seizure visits in US emergency departments: epidemiology and potential disparities in care. Int J Emerg Med 2008;1:97–105. 4. Chin RF, Neville BG, Peckham C, et al. Incidence, cause, and short-term outcome of convulsive status epilepticus in childhood: prospective population-based study. Lancet 2006;368:222–9. 5. Delorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996;46:1029–35. 6. Martinos MM, Yoong M, Patil S, et al. Early developmental outcomes in children following convulsive status epilepticus: a longitudinal study. Epilepsia 2013;54:1012–9. 7. Scholtes FB, Renier WO, Meinardi H. Generalized convulsive status epilepticus: causes, therapy, and outcome in 346 patients. Epilepsia 1994;35:1104–12. 8. Mazarati AM, Baldwin RA, Sankar R, et al. Time-dependent decrease in the effectiveness of antiepileptic drugs during the course of self-sustaining status epilepticus. Brain Res 1998; 814:179–85. 9. Brophy GM, Bell R, Claassen J, et al. Guidelines for the evaluation and management of status epilepticus. Neurocrit Care 2012;17:3–23. 10. Abend NS, Loddenkemper T. Management of pediatric status epilepticus. Curr Treat Options Neurol 2014;16:301. http:// dx.doi.org/10.1007/s11940-014-0301-x. 11. Theodore WH, Porter RJ, Albert P, et al. The secondarily generalized tonic-clonic seizure: a videotape analysis. Neurology 1994;44:1403–7. 12. Jenssen S, Gracely EJ, Sperling MR. How long do most seizures last? A systematic comparison of seizures recorded in the epilepsy monitoring unit. Epilepsia 2006;47:1499–503. 13. Shinnar S, Berg AT, Moshé SL, et al. How long do new-onset seizures in children last? Ann Neurol 2001;49:659–64. 14. Mazarati AM, Wasterlain CG, Sankar R, et al. Self-sustaining status epilepticus after brief electrical stimulation of the perforant path. Brain Res 1998;801:251–3. 15. Goodkin HP, Joshi S, Mtchedlishvili Z, et al. Subunit-specific trafficking of GABAA receptors during status epilepticus. J Neurosci 2008;28:2527–38. 16. Naylor DE, Liu H, Wasterlain CG. Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci 2005;25:7724–33. 17. Chin RF, Neville BG, Peckham C, et al. Treatment of community-onset, childhood convulsive status epilepticus: a prospective, population-based study. Lancet Neurol 2008;7: 696–703. 18. Fujikawa DG. The temporal evolution of neuronal damage from pilocarpine-induced status epilepticus. Brain Res 1996; 725:11–22. 19. Meldrum BS, Brierley JB. Prolonged epileptic seizures in primates. Ischemic cell change and its relation to ictal physiological events. Arch Neurol 1973;28:10–7. 20. Tsuchida TN, Barkovich AJ, Bollen AW, et al. Childhood status epilepticus and excitotoxic neuronal injury. Pediatr Neurol 2007;36:253–7. 21. Shorvon S. Does convulsive status epilepticus (SE) result in cerebral damage or affect the course of epilepsy—the epidemiological and clinical evidence? Prog Brain Res 2002;135:85–93. 22. Aicardi J, Chevrie JJ. Status epilepticus. Pediatrics 1989;84: 939–40. 46 VOL. 16, NO. 1 • STATUS EPILEPTICUS IN THE PEDIATRIC EMERGENCY DEPARTMENT / KURZ AND GOLDSTEIN 23. Commission ILAE. Report. The epidemiology of the epilepsies: future directions. Epilepsia 1997;38:614–8. 24. Working Group on Status Epilepticus. Treatment of convulsive status epilepticus. Recommendations of the Epilepsy Foundation of America's Working Group on Status Epilepticus. JAMA 1993;270:854–9. 25. Lowenstein DH. Status epilepticus: an overview of the clinical problem. Epilepsia 1999;40:S3–8. 26. Chen JW, Wasterlain CG. Status epilepticus: pathophysiology and management in adults. Lancet Neurol 2006;5:246–56. 27. Tirupathi S, McMenamin JB, Webb DW. Analysis of factors influencing admission to intensive care following convulsive status epilepticus in children. Seizure 2009;18:630–3. 28. Walker M, Cross H, Smith S, et al. Nonconvulsive status epilepticus: Epilepsy Research Foundation workshop reports. Epileptic Disord 2005;7:253–96. 29. Delorenzo RJ, Pellock JM, Towne AR, et al. Epidemiology of status epilepticus. J Clin Neurophysiol 1995;12:316–25. 30. Shinnar S, Pellock JM, Moshé SL, et al. In whom does status epilepticus occur: age-related differences in children. Epilepsia 1997;38:907–14. 31. Berg AT, Shinnar S, Testa FM, et al. Status epilepticus after the initial diagnosis of epilepsy in children. Neurology 2004; 63:1027–34. 32. Shinnar S. Who is at risk for prolonged seizures? J Child Neurol 2007;22:S14–20. 33. Shinnar S, Maytal J, Krasnoff L, et al. Recurrent status epilepticus in children. Ann Neurol 1992;31:598–604. 34. Shinnar S, Glauser TA. Febrile seizures. J Child Neurol 2002; 17:S44–52. 35. Shinnar S, Hesdorffer DC, Nordli DR, et al. Phenomenology of prolonged febrile seizures: results of the FEBSTAT study. Neurology 2008;71:170–6. 36. Seinfeld S, Shinnar S, Sun S, et al. Emergency management of febrile status epilepticus: results of the FEBSTAT study. Epilepsia 2014;55:388–95. 37. Lewis DV, Shinnar S, Hesdorffer DC, et al. Hippocampal sclerosis after febrile status epilepticus: the FEBSTAT study. Ann Neurol 2014;75:178–85. 38. Frank LM, Shinnar S, Hesdorffer DC, et al. Cerebrospinal fluid findings in children with fever-associated status epilepticus: results of the consequences of prolonged febrile seizures (FEBSTAT) study. J Pediatr 2012;161: 1169–71. 39. Riviello JJ, Ashwal S, Hirtz D, et al. Practice parameter: diagnostic assessment of the child with status epilepticus (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2006;67:1542–50. 40. Chin RFM, Neville BGR, Scott RC. Meningitis is a common cause of convulsive status epilepticus with fever. Arch Dis Child 2005;90:66–9. 41. Goldstein JL, Leonardt D, Kmytyuk N, et al. Abnormal neuroimaging is associated with early in-hospital seizures in pediatric abusive head trauma. Neurocrit Care 2011;15: 63–9. 42. Singh RK, Stephens S, Berl MM, et al. Prospective study of new-onset seizures presenting as status epilepticus in childhood. Neurology 2010;74:636–42. 43. Singh RK, Zecavati N, Singh J, et al. Seizures in acute childhood stroke. J Pediatr 2012;160:291–6. 44. Cook AM, Castle A, Green A, et al. Practice variations in the management of status epilepticus. Neurocrit Care 2012;17: 24–30. 45. Treiman DM, Meyers PD, Walton NY, et al. A comparison of four treatments for generalized convulsive status epilepticus. N Engl J Med 1998;339:792–8. 46. Chamberlain JM, Okada P, Holsti M, et al. Lorazepam vs diazepam for pediatric status epilepticus. JAMA 2014;311: 1652–60. 47. Holsti M, Dudley N, Schunk J, et al. Intranasal midazolam vs rectal diazepam for the home treatment of acute seizures in pediatric patients with epilepsy. Arch Pediatr Adolesc Med 2010;164:747–53. 48. Chamberlain JM, Altieri MA, Futterman C, et al. Prospective, randomized study comparing intramuscular midazolam with intravenous diazepam for the treatment of seizures in children. Pediatr Emerg Care 1997;13:92–4. 49. Kriel RL, Cloyd JC, Pellock JM, et al. Rectal diazepam gel for treatment of acute repetitive seizures. Pediatr Neurol 1999; 20:282–8. 50. Sreenath TG, Gupta P, Sharma KK, et al. Lorazepam versus diazepam-phenytoin combination in the treatment of convulsive status epilepticus in children: a randomized controlled trial. Eur J Paediatr Neurol 2010;14:162–8. 51. Alvarez V, Januel J-M, Burnand B, et al. Second-line status epilepticus treatment: comparison of phenytoin, valproate, and levetiracetam. Epilepsia 2011;52:1292–6. 52. DeToledo JC, Ramsay RE. Fosphenytoin and phenytoin in patients with status epilepticus: improved tolerability versus increased costs. Drug Saf 2000;22:459–66. 53. Agarwal P, Kumar N, Chandra R, et al. Randomized study of intravenous valproate and phenytoin in status epilepticus. Seizure 2007;16:527–32. 54. Mehta V, Singhi P, Singhi S. Intravenous sodium valproate versus diazepam infusion for the control of refractory status epilepticus in children: a randomized controlled trial. J Child Neurol 2007;22:1191–7. 55. Yu K-T, Mills S, Thompson N, et al. Safety and efficacy of intravenous valproate in pediatric status epilepticus and acute repetitive seizures. Epilepsia 2003;44:724–6. 56. Bryant AE, Dreifuss FE. Valproic acid hepatic fatalities. III. U.S. experience since 1986. Neurology 1996;46:465–9. 57. Jentink J, Loane MA, Dolk H, et al. Valproic acid monotherapy in pregnancy and major congenital malformations. N Engl J Med 2010;362:2185–93. 58. Painter MJ, Scher MS, Stein AD, et al. Phenobarbital compared with phenytoin for the treatment of neonatal seizures. N Engl J Med 1999;341:485–9. 59. Malamiri RA, Ghaempanah M, Khosroshahi N, et al. Efficacy and safety of intravenous sodium valproate versus phenobarbital in controlling convulsive status epilepticus and acute prolonged convulsive seizures in children: a randomised trial. Eur J Paediatr Neurol 2012;16:536–41. 60. Misra UK, Kalita J, Maurya PK. Levetiracetam versus lorazepam in status epilepticus: a randomized, open labeled pilot study. J Neurol 2012;259:645–8. 61. Eue S, Grumbt M, Müller M, et al. Two years of experience in the treatment of status epilepticus with intravenous levetiracetam. Epilepsy Behav 2009;15:467–9. 62. Reiter PD, Huff AD, Knupp KG, et al. Intravenous levetiracetam in the management of acute seizures in children. Pediatr Neurol 2010;43:117–21. 63. McTague A, Kneen R, Kumar R, et al. Intravenous levetiracetam in acute repetitive seizures and status epilepticus in children: experience from a children's hospital. Seizure 2012;21:529–34. 64. Gilbert DL, Gartside PS, Glauser TA. Efficacy and mortality in treatment of refractory generalized convulsive status STATUS EPILEPTICUS IN THE PEDIATRIC EMERGENCY DEPARTMENT / KURZ AND GOLDSTEIN • VOL. 16, NO. 1 47 65. 66. 67. 68. 69. 70. View publication stats epilepticus in children: a meta-analysis. J Child Neurol 1999; 14:602–9. Sahin M, Menache CC, Holmes GL, et al. Outcome of severe refractory status epilepticus in children. Epilepsia 2001;42: 1461–7. Sánchez Fernández I, Abend NS, Arndt DH, et al. Electrographic seizures after convulsive status epilepticus in children and young adults: a retrospective multicenter study. J Pediatr 2014;164:339–46. Hayashi K, Osawa M, Aihara M, et al. Efficacy of intravenous midazolam for status epilepticus in childhood. Pediatr Neurol 2007;36:366–72. Gilbert DL, Glauser TA. Complications and costs of treatment of refractory generalized convulsive status epilepticus in children. J Child Neurol 1999;14:597–601. Claassen J, Hirsch LJ, Emerson RG, et al. Continuous EEG monitoring and midazolam infusion for refractory nonconvulsive status epilepticus. Neurology 2001;57:1036–42. Claassen J, Hirsch LJ, Emerson RG, et al. Treatment of refractory status epilepticus with pentobarbital, propofol, or midazolam: a systematic review. Epilepsia 2002;43:146–53. 71. Kim SJ, Lee DY, Kim JS. Neurologic outcomes of pediatric epileptic patients with pentobarbital coma. Pediatr Neurol 2001;25:217–20. 72. Korff CM, Nordli DR. Diagnosis and management of nonconvulsive status epilepticus in children. Nat Clin Pract Neurol 2007;3:505–16. 73. Towne AR, Waterhouse EJ, Boggs JG, et al. Prevalence of nonconvulsive status epilepticus in comatose patients. Neurology 2000;54:340–5. 74. Hosain SA, Solomon GE, Kobylarz EJ. Electroencephalographic patterns in unresponsive pediatric patients. Pediatr Neurol 2005;32:162–5. 75. Schreiber JM, Zelleke T, Gaillard WD, et al. Continuous video EEG for patients with acute encephalopathy in a pediatric intensive care unit. Neurocrit Care 2012;17:31–8. 76. Delorenzo RJ, Waterhouse EJ, Towne AR, et al. Persistent nonconvulsive status epilepticus after the control of convulsive status epilepticus. Epilepsia 1998;39:833–40. 77. Tay SKH, Hirsch LJ, Leary L, et al. Nonconvulsive status epilepticus in children: clinical and EEG characteristics. Epilepsia 2006;47:1504–9.