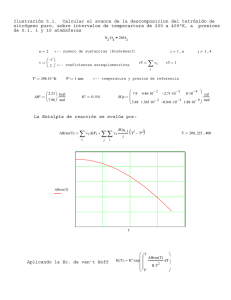
TERMOQUÍMICA Química General Mg. Quím. Mary Flor Cesare Coral [email protected] 1.- Conceptos termodinámicos: Sistemas, calor y trabajo, etc. 2.- Primer principio de la Termodinámica. 2.1.- Energía interna. 2.2.- Primer principio. Entalpía. 3.- Calor de reacción. 3.1.- Entalpía de formación. 3.2.- Otros calores de reacción. 3.3.- Ley de Hess. 3.4.- Calor de reacción y entalpías de formación. 3.5.- Calor de reacción y energías de enlace. 4.- Segundo principio de la Termodinámica. 4.1.- Otras Funciones de estado: Entropía. Energía interna libre. Entalpía libre. 4.2.- Espontaneidad de las reacciones. 4.3 - Segundo principio. 5.- Tercer principio de la Termodinámica. Termodinámica: Es la ciencia que estudia los cambios de energía que tienen lugar en los procesos físicos y químicos. Química: Ciencia que estudia la materia y sus transformaciones Termoquímica: La energía de las reacciones químicas Hay sistemas químicos que evolucionan de reactivos a productos desprendiendo energía. Son las reacciones exotérmicas. Otros sistemas químicos evolucionan de reactivos a productos precisando energía. Son las reacciones endotérmicas. Sistema: región del universo que aislamos para estudiarla ENTORNO Tipos de sistemas Aislados: que no pueden intercambiar ni materia ni energía. Cerrados: son aquellos que pueden intercambiar energía, aunque no materia, con los alrededores. Abiertos: aquellos que pueden intercambiar materia y energía. Clasificación sistemas sistemas Puede intercambiar Abierto Cerrado Aislado Materia Energía Materia Energía Materia Energía Homogéneos: Constan de una sola fase, es decir, se presenta uniforme. Por ejemplo, cuando todas las sustancias están en estado gaseoso o en disolución. Heterogéneos: Constan de varias fases, con superficies de separación entre ellas. Se llama estado a la situación específica en la que se encuentra un sistema. En termodinámica, la descripción del estado de un sistema se realiza mediante los valores de determinadas propiedades macroscópicas denominadas variables termodinámicas, tales como p, V, T, m, ... No todas estas variables son independientes, basta conocer los valores de un pequeño número de ellas para averiguar el valor de otras. Estas variables independientes se denominan variables de estado. Toda función que pueda expresarse con ayuda de las variables de estado se denomina función de estado del sistema. Variables de estado que tienen un valor único para cada estado del sistema. Su variación solo depende del estado inicial y final y no del camino desarrollado. SÍ son:energía, energía interna, entalpía, etc. NO son: calor, trabajo. Funciones de estado 1) Al asignar valores a unas cuantas, los valores de todas las demás quedan automáticamente fijados. 2) Cuando cambia el estado de un sistema, los cambios de dichas funciones sólo dependen de los estados inicial y final del sistema, no de cómo se produjo el cambio. DX = Xfinal –Xinicial Ecuaciones de estado: Relacionan funciones de estado (ej: PV = nRT) Tipos de variables Intensivas • No dependen de la cantidad de materia del sistema • Ej: T, P, Extensivas • Dependen de la cantidad de materia del sistema • Ej: m, V Magnitudes que pueden variar a lo largo de un proceso (por ejemplo, en el transcurso de una reacción química) . Ejemplos: ◦ ◦ ◦ ◦ Presión. Temperatura. Volumen. Concentración. Cuando alguna de las variables de estado cambia con el tiempo se produce un proceso termodinámico. Estos pueden ser Isotermo (T = cte) Isóbaro (P = cte) Isócoro (V = cte) Adiabático (Q = 0) Cíclico (estado final = estado inicial) Sistema en equilibrio: Si las variables que definen el estado del sistema no varían con el tiempo. Proceso irreversible: (casi todos los reales lo son). Si para volver de nuevo al estado inicial hay que provocar alguna variación. Proceso reversible: Se puede pasar de nuevo al estado inicial, sin provocar ninguna CALOR Y TRABAJO: El intercambio de energía entre un sistema y los alrededores puede tener lugar en forma de calor (Q) y de trabajo (W). La energía es función de estado, pero el calor y el trabajo no, ya que los cuerpos no tienen ni calor ni trabajo, éstos tan sólo son energía en tránsito CALOR Un sistema cede E en forma de Q si se transfiere como resultado de una diferencia de T entre el sistema y el entorno. Unidad S.I.: Julio 1 cal = 4,184 J El calor no es función de estado No es una propiedad característica del sistema. No es algo que posea el sistema. Es una forma de intercambio de energía, una “energía en tránsito” Calor específico: Ce, es la cantidad de calor que hay que suministrar a 1 g de sustancia para que aumente su temperatura 1 ºC. T2 T Q abs o ced = m C e . dT 1 sí C e = cte unidades: Q abs o ced = m C e DT a) m = masa (g), T(ºC)= Δ de temperatura, Ce = Calor específico (cal/g .ºC) b) m = masa (kg), T = Δ de temperatura (K), Ce = Calor específico (J/kg.K) Realizar trabajo supone la transmisión de energía debido a la acción de una fuerza que lleva consigo un desplazamiento. En Química interesa el trabajo realizado por un gas sometido a una presión exterior, presión atmosférica, supuesta constante. TRABAJO El trabajo realizado por el sistema (gas) al realizar el desplazamiento Δx , y pasar de ocupar un volumen V1 a ocupar un volumen V2 es, en función de la fuerza ha de vencer: W = Fext . Δx . Cos 180º = Fext . Δx .( -1) = - Fext . Δx ya que el desplazamiento del émbolo, Δx, sentido opuesto a la fuerza F dV S Pint Pext ΔX y teniendo en cuenta que F = P . S , tiene resulta : W = - Pext . S . Δx donde S .Δx es la variación de volumen, Δ V , por tanto: W= -P.ΔV Criterio de signos W>0 W<0 SISTEMA Q>0 Q<0 Equivalencia entre Atm.l y julios: Utilizando el concepto de 1 atm: presión que ejerce una columna de Hg de 76 cm de altura: p=dgh *1 atm = 13600 kgxm-3 x 9,8 mxs-2x 0,76 m = 101300 kgxm-1xs-2=101300 N/m2 *1 l = 10-3 m3. Luego: 1 atm.l = 1atm.1 l = 101.300 N/m2 .10-3 m3 =101,3 N.m = 101,3 J Un gas está encerrado en un recipiente de paredes adiabáticas (no transfiere calor al exterior). Calcula el trabajo que realiza contra el exterior cuando su volumen aumenta 85 cm3 a la presión de 4 atm. Dato: 1 atm = 101.300 Pa. Enunciado Energía Unidades Térmicas de Energía Calor Específico Entalpia Energía Interna y Entalpia Establece que la energía puede ser convertida de una forma a otra, pero no se puede crear o destruir. DE q w DE : expresa el cambio de la Energía interna de un sistema q : intercambio de calor entre el sistema y sus alrededores w : trabajo realizado sobre el sistema Capacidad para efectuar un trabajo útil. Una fuerza efectúa un trabajo al actuar sobre un cuerpo durante cierta distancia. Un joule (J) es el trabajo que efectúa una fuerza constante de un Newton cuando el cuerpo sobre el que se ejerce recorre una distancia de un metro en la dirección de la fuerza. La potencia (W) es la rapidez o tasa con que se efectúa el trabajo, o la rapidez de consumo de energía. La energía tiene muchas formas: energía térmica, mecánica, cinética, potencial, Eléctrica, química. Caloría (cal) es la cantidad de energía necesaria para elevar 1°C la temperatura de un gramo de agua, de 14.5 a 15.5°C. En unidades SI, 4.186 J = 1 cal. Cantidad de calor necesaria para aumentar un grado la temperatura de una unidad de masa de la sustancia. Unidades métricas: kcal. kg.-1. K unidades SI, como kJ. kg-1. K -1, (K está en kelvins, y 1 K = 1 °C.) -1, Sustancia Cp (kJ. kg-1. K-1) Aire (293.15 K) 1.00 Aluminio 0.95 Carne 3.22 Cemento Portland 1.13 Concreto 0.93 Cobre 0.39 Maíz 3.35 Suelo seco 0.84 Ser humano 3.47 Hielo 2.11 Hierro colado 0.50 Acero 0.50 Pollo 3.35 Vapor (373.15 K) 2.01 Agua (288.15 K) 4.186 Madera 1.76 Es el intercambio de energía en un recipiente cerrado que no cambia de volumen. Si V= constante, es decir, DV = 0 W=0 Qv = DU La mayoría de los procesos químicos ocurren a presión constante, normalmente la atmosférica. Si p = cte W = – p · D V D U = Qp – p · D V U2 – U1 = Qp – p · (V2 – V1) Q p + U1 + p · V 1 = U 2 + p · V 2 H1 H2 (entalpía) Entalpia (H) H1= U1 + p · V1; H2= U2 + p · V2 Qp + H 1 = H 2 Reac. endotérmica Producto s DH > 0 Q p = H 2 – H1 = D H Reactivos H es una función de estado. Reac. exotérmica Reactivos Entalpia (H) DH < 0 Producto s DH=DU+p·DV Aplicando la ecuación de los gases: p·V=n·R·T y si p y T son constantes la ecuación se cumplirá para los estados inicial y final: p·DV=Dn·R·T DH=DU+Dn·R·T Propiedad termodinámica, depende de la presión, temperatura y composición del material. Se define como: H U PV Donde: H = entalpía (en kJ) U = energía interna (o energía térmica) (en kJ) P = presión (en kPa), V = volumen (en m3). ENERGÍA INTERNA (U) es la energía total del sistema. ◦ Es imposible medir. ◦ Su variación sí se mide. CALOR Q>0 Q<0 TRABAJO TRABAJO W< 0 W>0 DU=Q+W Q y W > 0 si se realizan a favor del sistema. CALOR U es función de estado. Cuando se efectúa un proceso sin cambio de fase y sin cambio de volumen, un cambio de energía interna se define como: DU mCvDT Donde: ∆U = cambio de energía interna. m = masa. Cv = calor específico a volumen constante. ∆ T = cambio de temperatura. La energía interna, U, es una función de estado, es decir, su valor sólo depende de los estados inicial y final del sistema y no del camino seguido para pasar de uno a otro. Por el contrario, el calor y el trabajo no son función de estado, su valor numérico depende tanto de las condiciones iniciales y finales como de los estados intermedios alcanzados para pasar de un estado a otro. Cuando se efectúa un proceso sin cambio de fase sucede sin un cambio de presión, se define un cambio de entalpía como DH mCpDT Donde: ∆H = cambio de entalpía. Cp = calor específico a presión constante. El calor específico es Cte. dentro del intervalo de temperatura (∆T). Los sólidos y líquidos casi son incompresibles y en consecuencia casi no efectúan trabajo. La ΔPV = cero y los cambios de H y U son idénticos. Así, para los sólidos y líquidos se puede suponer: Cv Cp DU DH C3H8 (g) + 5 O2 (g) 3 CO2 (g) + 4 H2O (l) D H = –2219,8 Kj nreactivos = 1+5 = 6 ; nproductos = 3 D n = – 3 Despejando en D U = D H – D n·R ·T = – 2219 kJ + 3 mol ·(8,3 J/mol.K) .298 K = –2214 kJ D U = – 2212 kJ Es el incremento entálpico de una reacción en la cual, tanto reactivos como productos están en condiciones estándar: p = 1 atm; T = 298 K = 25 ºC; conc. = 1 M Se expresa como DH0 y como se mide en J o kJ depende de cómo se ajuste la reacción. Así, DH0 de la reacción “2 H2 + O2 2 H2O” es el doble del de “H2 + ½ O2 H2O”. DH0 = H0productos – H0reactivos Expresan tanto los reactivos como los productos indicando entre paréntesis su estado físico, y a continuación la variación energética expresada como DH (habitualmente como DH0). Ejemplos: CH4(g) + 2 O2(g) CO2(g) + 2 H2O(l); DH0 = –890 kJ H2(g) + ½ O2(g) H2O(g); DH0 = –241’4 kJ ¡CUIDADO!: DH depende del número de moles que se forman o producen. Por tanto, si se ajusta poniendo coeficientes dobles, habrá que multiplicar DH0 por 2: 2 H2(g) + O2(g) 2 H2O(g) ; DH0 = 2· (–241,4 kJ) Con frecuencia, suelen usarse coeficientes fraccionarios para ajustar las ecuaciones: H2(g) + ½ O2(g) H2O(g) ; DH0 = –241,4 kJ Expresan tanto los reactivos como los productos indicando entre paréntesis su estado físico, y a continuación la variación energética expresada como DH (habitualmente como DH0). Ejemplos: CH4(g) + 2 O2(g) CO2(g) + 2 H2O(l); DH0 = –890 kJ H2(g) + ½ O2(g) H2O(g); DH0 = –241’4 kJ Entalpia (H) Reac. endotérmica Productos Entalpia (H) Reactivos DH > 0 Reac. exotérmica Reactivos DH < 0 Productos 2.-El calor de reacción viene referido al estado físico y al número de moles de los componentes indicados en la correspondiente ecuación estequiométrica, por tanto, si una ecuación se multiplica por “n” su calor de reacción se multiplica por “n” y sí la reacción se invierte, el calor de reacción cambia de signo. Ejemplos: HCl (g) H (g) + Cl (g); D H = 431 kJ o 431 kJ/mol 2 HCl (g) 2 H (g) + 2Cl (g); D H = 2. 431 = 862 kJ H (g) + Cl (g) HCl (g); D H = - 431 kJ Las ecuaciones anteriores también se pueden expresar de la siguiente forma: HCl (g) + 431 kJ H (g) + Cl (g) HCl (g) H (g) + Cl (g) - 431 kJ H (g) + Cl (g) HCl (g) + 431 kJ D H también se puede expresar en cal o kcal. ( 1 cal = 4,18 Julios ) 3.El calor de reacción depende de las condiciones de presión y temperatura a las que tiene lugar el proceso: Se llaman “condiciones estándar” de reacción: 25ºC y 1 atm. Se llama estado normal o estándar de un elemento o compuesto, a la forma física más estable del mismo en las condiciones estándar. Ejemplos: Estado estándar del carbono: C (grafito) Estado estándar del agua: H2O (líquida) Estado estándar del hidrógeno: H2 (gas) 4.-Al igual que la energía interna U, es imposible calcular el valor absoluto de H; tan sólo se puede calcular variaciones de la entalpía (H ), aunque arbitrariamente se toma como entalpía cero (origen de entalpías) a la entalpía de cualquier elemento en su estado estándar y en las condiciones estándar. Ejemplos: Entalpía (H) del H2 (g) a 25ºC y 1 atm. = 0 Entalpía (H) del C (grafito) a 25ºC y 1 atm. = 0 Entalpía (H) del C (diamante) a 25ºC y 1 atm 0 ≠ forma más estable Entalpía (H) del Cl2 (l) a 25ºC y 1 atm. ≠0 Entalpía (H) del H2O (l) a 25ºC y 1 atm. ≠ compuesto. 0 por no ser la por ser un Es el incremento entálpico (calor de reacción a presión constante) de una reacción en la cual, tanto reactivos como productos están en condiciones estándar (p = 1 atm; T = 298 K = 25 ºC; conc. = 1 M). Se expresa como DH0 y como se mide en J o kJ depende de cómo se ajuste la reacción. Así, DH0 de la reacción “2 H2 + O2 2 H2O” es el doble del de “H2 + ½ O2 H2O”. El incremento entálpico (DH) que se produce en la rx de formación de un mol de un compuesto a partir de los elementos en estado físico normal (en condiciones estándar). Se expresa como DHf0. Es un “calor molar”, es decir, el cociente entre DH0 y el número de moles formados de producto. (Se mide en kJ/mol). Ejemplos: C(s) + O2(g) CO2(g) DHf 0 = –393,13 kJ/mol H2(g) + ½ O2(g) H2O(l) DHf0 = – 285,8 kJ/mol Calor de combustión. Calor de neutralización. Calor de disolución. DH en una reacción química es constante con independencia de que la reacción se produzca en una o más etapas. “el calor de reacción (variación de entalpía), es independiente del camino seguido, es decir, es el mismo si la reacción se realiza directamente o a través de etapas intermedias”: Ley de Hess DH3 A DH1 B DH2 C DH3 = DH1 + DH2 GH Hess (1802-1850) La reacción de vaporización es... (3) H2O(l) H2O(g) DH03 = ? (3) puede expresarse como (1) – (2), luego DH03 = DH01 – DH02 = – 241’8 kJ – (–285’8 kJ) = 44 kJ DH0vaporización = 44 kJ /mol H2(g) + ½ O2(g) H DH10 = – 241’8 kJ DH20 = – 285’8 kJ H2O(g) DH30 = 44 kJ H2O(l) Basándonos en la ley de Hess podemos calcular la de una reacción cualquiera, a partir de las entalpías de formación de las sustancias que intervienen en la reacción. En efecto, en una reacción cualquiera los elementos que forman los reactivos son los mismos que forman los productos, pero están combinados de distinta forma. ΔH Reactivos Productos ΣΔH f ( reactivos) Elementos ΣΔH f( productos) D H = DHf (productos) – DHf (reactivos) La reacción de combustión del butano es: C4H10(g) +13/2O2(g) 4 CO2(g) + 5H2O(l) DH0comb= ? D H0 = npDHf0(product.) – nrDHf0(reactivos) = 4 mol(– 393’5 kJ/mol) + 5 mol(– 285’8 kJ/mol) –1 mol(– 124’7 kJ/mol) = – 2878’3 kJ Luego la entalpía estándar de combustión será: D H0combustión = – 2878’3 kJ/mol Si utilizamos la ley de Hess, la reacción: (4) C4H10(g) +13/2O2(g) 4 CO2(g) + 5H2O(l) DH0comb=? Puede obtenerse a partir de: (1) H2(g) + ½ O2(g) H2O(l) DH10 = – 285’8 kJ (2) C(s) + O2(g) CO2(g) DH20 = – 393’5 kJ (3) 4 C(s) + 5 H2(g) C4H10(g) DH30 = – 124’7 kJ (4) = 4 · (2) + 5 · (1) – (3) 4 C(s) + 4 O2(g) +5 H2(g) + 5/2 O2(g) + C4H10(g) 4 CO2(g) + 5H2O(l) + 4 C(s) + 5 H2(g) D H04 = 4 mol(–393’5 kJ/mol) + 5 mol(–285’8 kJ/mol) –1 mol(– 124’7 kJ/mol) = – 2878’3 kJ (1) (2) (3) H2(g) + ½ O2(g) H2O(l) DH10 = – 285,8 kJ C(s) + O2(g) CO2(g) DH20 = – 393,13 kJ C2H4(g) + 3O2(g) 2CO2(g) + 2 H2O(l) DH30 = – 1422 kJ Buscamos el DH de la reacción siguiente: (4) (4) 2 C(s) + 2 H2(g) C2H4(g) (4) = 2·(2) + 2·(1) – (3) luego DH40 = 2·DH20 + 2·DH10 – DH30 = = 2 · (–393,13 kJ) + 2 · (– 285,8 kJ) – (– 1422 kJ) = = 64,14 kJ es decir DHf0 (eteno) = 64,14 kJ/mol Se trata, pues, de una reacción endotérmica. Las reacciones de combustión son, respectivamente: (1) C6H12O6 + 6 O2 6 CO2 + 6 H2O ; DH1 = – 2815 kJ (2) C2H5OH + 3 O2 2 CO2 + 3 H2O ; DH2 = – 1372 kJ La reacción de fermentación de la glucosa es: (3) C6H12O6 2 C2H5OH +2 CO2 DH3 = ? (3) puede expresarse como (1) – 2· (2), luego DH3 = DH1 – 2·DH2 = – 2815 kJ – 2· (– 1372 kJ) = – 71 kJ y la reacción es exotérmica. “Es la energía necesaria para romper un mol de enlaces de una sustancia en estado gaseoso” En el caso de moléculas diatómicas es igual que la energía de disociación: A—B(g) A(g) + B(g) DHdis = Eenlace= Ee Ejemplo: H2(g) 2 H(g) DH = 436 kJ Si es + (es necesario aportar energía al sistema) Es difícil de medir. Se suele calcular aplicando la ley de Hess. La reacción de disociación del HCl será: (4) HCl(g) H(g) + Cl(g) DH04= ? (1) ½H2(g) + ½Cl2(g) HCl(g) DH01 = –92,3 kJ (2) H2(g) 2H(g) DH02 = 436,0 kJ (3) Cl2(g) 2Cl(g) DH03 = 243,4 kJ (4) = –(1) + ½(2) + ½(3) DH04 = –(– 92,3 kJ ) + ½(436,0 kJ) + ½(243,4 kJ) = = 432,0 kJ Ee(HCl) = 432,0 kJ/mol Aplicando la ley de Hess en cualquier caso se obtiene la siguiente fórmula: DH0 = ni · Ee(enl. rotos) – nj · Ee(enl. formados) en donde ni representa el número de enlaces rotos y nj los formados de cada tipo. Reacción: CH2=CH2(g) + H2(g) CH3–CH3(g) En el proceso se rompe un enlace C=C y otro H–H y se forman 2 enlaces C–H nuevos (el etano tiene 6 mientras que el eteno tenía sólo 4) y un enlace C–C. D H0 = Ee(enl. rotos) – Ee(enl. formados) = D H0 = 1Ee(C=C) + 1 Ee(H–H) – 1Ee(C–C) – 2 Ee(C–H) D H0 = 1 mol · 611 kJ/mol + 1mol · 436 kJ/mol – (1 mol · 347 kJ/mol + 2 mol · 413 kJ/mol) = –126 kJ C3H8 + 5 O2 3 CO2 + 4 H2O Enlaces rotos: 8 C–H, 2 C–C y 5 O=O Enlaces formados: 6 C=O y 8 O–H DH0 = Ee(e. rotos) – Ee(e. form.) DH0 = 8 Ee(C–H) + 2 Ee(C–C) + 5 Ee(O=O) – [6 Ee(C=O) + 8 Ee(O–H)] DH0 = 8·413 kJ + 2·347 kJ +5·499 kJ – (6·745 kJ + 8·460 kJ) = –1657 kJ DH0comb(C3H8) = –1657 kJ/mol Enlace Ee (kJ/mol) H–H 436 C–C 347 C=C 620 CC 812 O=O 499 Cl–C 243 C–H 413 C–O 315 C=O 745 O–H 460 Cl–H 432 Es una medida del desorden del sistema que sí puede medirse y tabularse. DS = Sfinal – Sinicial Existen tablas de S0 (entropía molar estándar) de diferentes sustancias. En una reacción química: DS0 = np· S0productos – nr· S0reactivos La entropía es una función de estado. Es una función de estado extensiva que mide el desorden del sistema. A mayor desorden mayor entropía. Significado energético de S: El contenido energético de un sistema viene dado por H o por U, según que el sistema o los alrededores realicen o no trabajo con variación de volumen; pues bien, toda esta energía no se puede comunicar al exterior, sino que hay una parte de energía degenerada, no aprovechable, que no se puede liberar del sistema, la cual está relacionada con S. Ejemplos de cambios de entropía: - En los cambios de estado hay que tener presente que los gases tienen mayor entropía que los líquidos y éstos que los sólidos, por lo que en la evaporación (líquido gas), S > 0 -En una variación de temperatura sin cambio de estado: si T2 > T1 S (T2 ) > S (T1 ) -En las disoluciones de sólidos en líquidos: S (disolución) > S (soluto) + S (disolvente) -En una mezcla de gases: S (mezcla) > Σ S (gases puros) - En una reacción entre gases: la S aumenta con el número de moles. Existen tablas de S0 (entropía molar estándar) de diferentes sustancias. En una reacción química: DS0 = np· S0productos – nr· S0reactivos DS0 = np· S0productos – nr· S0reactivos a) DS0 = 2 mol · 210,7 J ·mol–1 ·K–1 – (191,5 J·K–1 + 205 J·K–1 ) = 24,9 J·K–1 b) DS0 = 2·192,3 J·K–1 – (3 mol ·130,6 J· mol–1·K–1 + 191,5 J·K–1 ) = –198,7 J·K–1 G = H – T.S H G T.S E degenerada Contenido energético del sistema En principio cabría esperar que una reacción exotérmica fuese espontánea, es decir, que se realice por si misma, ya que pasa a un estado de menor energía, sin embargo, hay reacciones endotérmicas como la expansión de un gas, fusión del hielo, etc., que son espontáneas. También cabría esperar que las reacciones que transcurren con un aumento de entropía fuesen espontáneas, ya que los fenómenos en los que las cosas se desordenan son más probables que aquellos que entrañan una ordenación. La espontaneidad no solo depende de ΔH , sino que se ha visto que también depende de ΔS y en algunos casos de la T. La espontaneidad de una reacción viene determinada realmente por el valor de ΔG. En una reacción en la que se pasa de un estado inicial (1) de los reactivos a uno final (2) de los productos, siendo la temperatura constante, tendremos que G1 = H1 – T.S1 G2 = H2 – T.S2 Δ G = G2 – G1 = H2 – T.S2 – (H1 – T.S1) = (H2 – H1) – T.(S2 – S1) Δ G = Δ H - T ΔS Reac. no espontánea Productos Reactivos DG > 0 T, p = ctes. Reac. espontánea Energía libre (G) Energía libre (G) Reactivos DG < 0 Productos T, p = ctes. No siempre las reacciones exotérmicas son espontáneas. Hay reacciones endotérmicas espontáneas: ◦ Evaporación de líquidos. ◦ Disolución de sales... Ejemplos de reacciones endotérmicas espontáneas: NH4Cl(s) NH4+(aq) + Cl– (aq) D H0 = 14’7 kJ H2O(l) H2O(g) DH0 = 44’0 kJ Una reacción es espontánea cuando DG (DH – T x DS) es negativo. Según sean positivos o negativos los valores de DH y DS (T siempre es positiva) se cumplirá que: D H < 0 y D S > 0 DG < 0 Espontánea D H > 0 y D S < 0 DG > 0 No espontánea D H < 0 y D S < 0 DG < 0 a T bajas DG > 0 a T altas D H > 0 y D S > 0 DG < 0 a T altas DG > 0 a T bajas *La ley de Hess tiene aplicación a todas las funciones de estado : H, U, S y G. *Al igual que el incremento entálpico el incremento de energía libre de una reacción puede obtenerse a partir de DGf0 de reactivos y productos: DG = DGf (productos)– DGf (reactivos) *DGf de un compuesto es indicativo de la estabilidad del mismo respecto a sus elementos. Si DGf < 0 el compuesto es más estable que sus elementos. Si DGf > 0 el compuesto es menos estable que sus elementos. *DGf de los elementos en su estado estándar es cero. D H0 = npDHf0(productos)– nrDHf0(reactivos) = = 2 DHf0(H2O) + DHf0(O2) – 2 DHf0(H2O2) = 2 mol(–285,8 kJ/mol) – 2 mol(–187,8 kJ/mol) = –196,0 kJ DS0 = np· S0productos – nr· S0reactivos = 2 S0(H2O) + S0(O2) – 2 S0(H2O2) = 2 mol(69,9 J/mol·K) + 1 mol(205, J/mol·K) – 2mol(109,6 J/mol·K) = 126,0 J / K = 0,126 kJ / K DG0 = D H0 – T · D S0 = –196,0 kJ – 298 K · 0,126 kJ/ K DG0 = – 233,5 kJ luego será espontánea DH < 0 DS > 0 DS Espontánea a todas las temperaturas DH > 0 DS > 0 Espontánea a temperaturas altas DH DH < 0 DS < 0 DH > 0 DS < 0 Espontánea a temperaturas bajas No Espontánea a cualquier temperaturas DS0 = np· S0productos – nr· S0reactivos a) DS0 = 2 mol · 210,7 J ·mol–1 ·K–1 – (191,5 J·K–1 + 205 J·K–1 ) = 24,9 J·K–1 a) DS0 = 2·192,3 J·K–1 – (3 mol ·130,6 J· mol–1·K–1 + 191,5 J·K–1 ) = –198,7 J·K–1 “En cualquier proceso espontáneo la entropía total del universo tiende a aumentar siempre”. DSuniverso = DSsistema + DSentorno 0 A veces el sistema pierde entropía (se ordena) espontáneamente. En dichos casos el entorno se desordena. “La entropía de cualquier sustancia a 0 K es igual a 0” (máximo orden). Equivale a decir que no se puede bajar de dicha temperatura. ¡CUIDADO! Las S de los elementos en condiciones estándar no son 0 sino que es positiva. En procesos reversibles y a temperatura constante se puede calcular DS de un sistema como: Q DS = — T y si el proceso químico se produce a presión constante: DHsistema – DHsistema DSsistema = ——— ; DSentorno= ———— T T S0 (entropía molar estándar) se mide en J·mol–1·K–1. DSreacción se mide en J·K–1. En procesos a T constante se define como: G=H–T·S DG = D H – T · DS En condiciones estándar: DG0 = DH0 – T· DS0 DSuniverso = DSsistema + DSentorno > 0 (p. espontáneos) Multiplicando por “–T” y como “–T DSentorno = DHsist –T · DSuniverso = – T · DSsist + DHsist = DG < 0 En procesos espontáneos: DG < 0 Si DG. > 0 la reacción no es espontánea Si DG. = 0 el sistema está en equilibrio G es una función de estado. Al igual que el incremento entálpico el incremento de energía libre de una reacción puede obtenerse a partir de DGf0 de reactivos y productos: DG0 = npDGf0(productos)– nrDGf0(reactivos) Reac. no espontánea Productos DG > 0 Reactivos T, p = ctes. Reac. espontánea Energía libre (G) Energía libre (G) Reactivos DG < 0 Productos T, p = ctes. No siempre las reacciones exotérmicas son espontáneas. Hay reacciones endotérmicas espontáneas: ◦ Evaporación de líquidos. ◦ Disolución de sales... Ejemplos de reacciones endotérmicas espontáneas: NH4Cl(s) NH4+(aq) + Cl– (aq) H2O(l) H2O(g) D H0 = 14,7 kJ DH0 = 44,0 kJ D H0 = npDHf0(productos)– nrDHf0(reactivos) = = 2 DHf0(H2O) + DHf0(O2) – 2 DHf0(H2O2) =2 mol(–285,8 kJ/mol) – 2 mol(–187,8 kJ/mol) = –196,0 kJ DS0 = np· S0productos – nr· S0reactivos = 2 S0(H2O) + S0(O2) – 2 S0(H2O2) = 2 mol(69,9 J/mol·K) + 1 mol(205, J/mol·K) – 2mol(109,6 J/mol·K) = 126,0 J / K = 0,126 kJ / K DG0 = D H0 – T · D S0 = –196,0 kJ – 298 K · 0,126 kJ/ K = DG0 = – 233,5 kJ luego será espontánea Establece que la energía fluye de una región de mayor concentración a una de menor y no al revés, y que la calidad de esa energía se degrada al transformase Cuando dos objetos a diferente temperatura se ponen en contacto térmico entre sí, la energía térmica siempre fluye del objeto más caliente al más frío, nunca del más frío al más caliente. Video Refrigerador de Abba - Proyecto 1