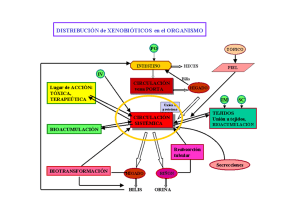
Farmacocinética Conceptos básicos Andrea Tapia Bustos Nicole Sanguinetti Objetivos de la clase • Describir los procesos farmacocinéticos focalizado en: I. Absorción II. Distribución III. Eliminación: - Metabolismo/Biotransformación - Excreción Concepto de farmacocinética Dosis Absorción Nivel plasmático [F] en tejido blanco procesos Distribución Metabolismo Eliminación Excreción Efecto terapéutico Estudio de los procesos que determinan la evolución en el tiempo de las [F] en el organismo Curva Concentración f (tiempo) Esquema de las etapas farmacocinéticas Fase biofarmacéutica Absorción Distribución Eliminación DISOLUCIÓN Rang & Dale Curso temporal de la cantidad de fármaco en el lugar de absorción, del fármaco y su metabolito en el cuerpo, y del fármaco y su metabolito excretados tras la administración de una dosis por vía extravascular. Flórez y col. ABSORCIÓN Outside Presystemic loss Movimiento de un fármaco desde el sitio de administración hasta la circulación sistémica Inside * Bioavailability Mecanismos de transporte Absorción Fig. Influencia de la vía de administración y de la preparación farmacéutica sobre la curva de [F] plasmáticas. Flórez y col. Características de la forma farmacéutica Características del lugar de Absorción (vía de administración) Características FísicoQuímicas del Fármaco Eliminación presistémica Absorción Características de la forma farmacéutica Características del lugar de Absorción Influencia del pKa La mayoría de los F son electrolitos débiles con bajo peso molecular Electrolitos en solución están ionizados de acuerdo a su constante de disociación (pKa) Las membranas celulares sólo son permeables a las formas NO IONIZADAS (depende del pKa del F y del pH del medio donde está disuelto) (vía de administración) Características FísicoQuímicas del Fármaco (masa molecular, liposolubilidad, pKa, solubilidad) Eliminación presistémica Influencia de la solubilidad Capacidad para disolverse completamente en un solvente Depende del coeficiente de reparto (So/w)) Las membranas son permeables a F con ALTO COEFICIENTE DE REPARTO Base débil Ecuación Henderson- Hasselbalch Ácido débil Ecuación Henderson- Hasselbalch Rang & Dale Absorción Características de la forma farmacéutica Características del lugar de Absorción (vía de administración) Características FísicoQuímicas del Fármaco Eliminación presistémica Curva de Niveles plasmáticos en f (t): administración extravascular RAM!!! Fármacos con estrecho margen terapéutico !!! Parámetros Farmacocinéticos Margen terapéutico CME: [ ] mínima efectiva CMT: [ ] mínima tóxica Vel. Absorción aparente = C máx/Tmáx PL: Período de Latencia IE: Intensidad del efecto TE: Tiempo Eficaz-Duración de la acción AUC = Cantidad absorbida = BIODISPONIBILIDAD Flórez y col. Curva de Niveles plasmáticos en f (t): administración intravascular Sin influencia del proceso de absorción Biodisponibilidad (AUC) Biodisponibilidad ABSOLUTA: Fracción (%) de dosis del F administrado por una vía diferente a la i.v. y que llega inalterado a la circulación general (considera la pérdida presistémica) Ej Haloperidol F=60% (40% pérdida presistémica) Biodisponibilidad (AUC) Biodisponibilidad RELATIVA: Medición de la eficacia de la absorción de un mismo F, desde 2 formas farmacéuticas diferentes, para una misma dosis y vía de administración. Ej: 200 mg en comprimidos y cápsulas blandas Fármacos con estrecho margen terapéutico !!! Flórez y col. DISTRIBUCIÓN Es la llegada y disposición de un fármaco a los diferentes tejidos del organismo, condicionando las [F] que se alcanzan en cada tejido. Molécula de un fármaco Transportada en la sangre Disuelta en plasma Fijada a proteínas plasmáticas Variable Unidas a las células sanguíneas Solo las moléculas de fármaco libre pueden abandonar la circulación general y distribuirse a todos los tejidos del organismo. Distribución Liposolubilidad del F Distribución Fármaco Grado de ionización del F Flujo sanguíneo tisular regional Presencia de barreras biológicas Unión a proteínas plasmáticas Fijación (unido)- PP Fármaco (libre) + PP Efecto Terapéutico Distribución Liposolubilidad del F Unión a proteínas plasmáticas: - Albúmina (principal) B-globulina Glicoproteína ácida Lipoproteínas La concentración de fármaco libre Su afinidad por los lugares de unión La concentración de proteína Grado de ionización del F Flujo sanguíneo tisular regional Presencia de barreras biológicas Unión a proteínas plasmáticas Reparto por la grasa corporal (Peso) y otros tejidos: Unión a proteínas tisulares Afinidad por tejidos (acumulación) Fijación Consecuencias de Fijación a PP > 80% I.- Intensidad del efecto farmacológico [F] en la Biofase F libre Restricciones para salir del plasma Vd= Dosis Cp0 Volumen de distribución (Vd) pequeños II.- Duración del efecto farmacológico Eliminación F libre Excreción renal lenta Fracción hepática baja (< 0,3) III.- Interacciones de importancia clínica Si dos o más fármacos se unen al mismo tipo de PP en el mismo sitio de unión. El Vd relaciona la cantidad de F presente en el organismo (Dosis) con la concentración plasmática a tiempo cero del mismo (Cp0) Vd se expresa en L o en L/Kg Es un volumen aparente y NO fisiológico Considera la distribución de F como un proceso instantáneo y homogéneo. Utilidad Clínica: calcular dosis de carga Distribución en regiones especiales El acceso a áreas especiales presenta características particulares al estar limitadas. Por ello, el transporte de fármacos en estas áreas es por difusión pasiva o transporte activo. Factores Fisiológicos y Patológicos que alteran la [Prot. plasmáticas] ejemplo: ALBÚMINA • Puede existir mayor efecto y mayor RAM en aquellos fármacos que se unen en gran proporción a proteínas. Ej: fenitoína, diazepam, anticoagulantes orales. • Si los medicamentos tienen un bajo índice terapéutico y se unen en gran proporción a proteínas plasmáticas, su desplazamiento presentará gran riesgo de toxicidad. Ej: Anticoagulantes orales: crisis hemorrágicas y Sulfonilureas :crisis hipoglucémicas. concentración activa del fármaco en el organismo ELIMINACIÓN La humano disminuye como consecuencia de dos mecanismos. Biotransformación/ Metabolismo: conversión enzimática de una sustancia química en otra diferente en el organismo. Fármacos liposolubles deben ser biotransformados a metabolitos hidrosolubles para ser excretados Excreción: eliminación del cuerpo de un fármaco sin cambios químicos o de sus metabolitos. Metabolismo Citocromo P450 (CYP) Rang & Dale > tamaño molecular > polaridad Ligeramente hidrosoluble Lipófilico Enzimas de Biostransformación Fase 1: “Oxigenasas” Citocromo P450 (CYP450) Monooxigenasas flavinadependientes Epóxido hidrolasas > polaridad Muy hidrosoluble Enzimas de Biostransformación Fase 2: “Transferasas” UDP-glucuronil transferasa (UDPGT) Glutatión Transferasas (GST) Sulfotransferasas N-acetil-transferasas Metiltransferasas Rang & Dale. Consecuencias de la biotransformación • Fase 1: Generar metabolitos inactivos Generar metabolitos activos Generar metabolitos tóxicos • Fase 2: Generar metabolitos inactivos (usualmente) Metabolismo Edad Genético Dietéticos Ambientales Interacciones Farmacológicas Condición clínica o fisiológica Metabolismo Edad Genético Dietéticos Ambientales Interacciones Farmacológicas Condición clínica o fisiológica Metabolismo Edad Genético Dietéticos Ambientales Interacciones Farmacológicas Condición clínica o fisiológica Metabolismo Edad Genético Dietéticos Ambientales Interacciones Farmacológicas Condición clínica o fisiológica Metabolismo Edad Genético Dietéticos Ambientales Interacciones Farmacológicas Condición clínica o fisiológica Inductores hepáticos: Fármacos que pueden aumentar la actividad de las enzimas hepáticas Metabolismo Edad Genético Dietéticos Ambientales Interacciones Farmacológicas Condición clínica o fisiológica Inhibidores hepáticos: Fármacos que pueden inhibir la actividad de las enzimas hepáticas Metabolismo Edad Genético Dietéticos Ambientales Interacciones Farmacológicas Condición clínica o fisiológica EXCRECIÓN Salida de los fármacos o sus metabolitos al exterior del organismo t1/2 = 0,693/Ke -Ke T1/2 = tiempo en el que la [ ] plasmática disminuye a la mitad Flórez y col. Vías de Excreción/Eliminación de fármacos Vía urinaria Vía biliar-Entérica Pulmonar Fanerios (piel-uña-cabellos) Sudor Saliva Leche materna Epitelios descamados Excreción renal de fármacos Fármaco excretado = (Filtración glomerular + secreción tubular) – reabsorción tubular Ultrafiltración máx = 120 mL/min Clearence (Cl) renal máx tiende a 650 mL/min F es filtrado Cl renal F = FG F es filtrado y se reabsorbe Cl renal < FG F es filtrado y secretado Cl renal > FG F es totalmente depurado por secreción Cl renal = Flujo plasmático renal (app 650 mL/min) Excreción Biliar e intestinal: Circulación enterohepática Recirculación Enterohepática Existe una hidrólisis enzimática de los conjugados debido a la flora bacteriana intestinal (Β-glucuronidasa bacteriana): Libera el fármaco en el intestino, el cual puede ser nuevamente absorbido por el intestino. HÍGADO Conjugados Fase II Reabsorción Recirculación enterohepática Bilis INTESTINO Fármaco Hidrólisis Conjugados Heces Flora bacteriana La recirculación enterohepática prolonga los efectos farmacológicos de los fármacos excretados por esta vía. Factores que afectan la excreción de un fármaco Metabolismo Genética ejemplos Características individuales Edad Ácidos Orgánicos Hábitos dietéticos/ Ejercicio físico Enfermedades renales/hepáticas ejemplos Factores Patológicos Obesidad Bases Interacciones Ritmos circadianos Factores ambientalesejemplos Exposición ambiental