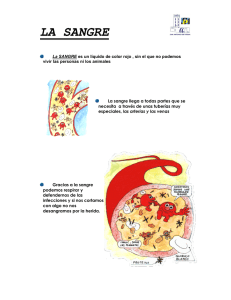
TEMA I MATERIALES DE LABORATORIO El laboratorio es el servicio centralizado de un hospital que generalmente se divide en varios departamentos como son: hematología, bioquímica, inmunología... El laboratorio ha ido adquiriendo con el paso del tiempo un gran protagonismo debido a que se puede obtener una gran información sobre multitud de enfermedades. Ha habido un gran avance en los últimos tiempos utilizándose aparatos cada vez más complicados en sus determinaciones, pero sencillo en su uso. Actualmente la exploración clínica proporciona datos que son completados o confirmados por el laboratorio. Por ejemplo: un paciente que presente síntomas como fatiga, palidez, etc. se puede pensar que padece un proceso anémico, lo cual será confirmado o no mediante un análisis de sangre. Por tanto para sacar una conclusión adecuada debe existir una relación entre los datos obtenidos en clínica y los obtenidos en el laboratorio. Las técnicas analíticas cumplen básicamente 3 objetivos: 1º·- Aportan información, para que un médico diagnostique adecuadamente 2º·- Pueden ser utilizadas como medidas preventivas para conocer el estado de salud de un individuo y detectar precozmente alguna alteración. 3º·- Permiten seguir la evolución de una enfermedad durante el tratamiento. SEGURIDAD EN EL LABORATORIO Par el buen funcionamiento de un laboratorio es necesario seguir unas normas; esto es doblemente importante si se tiene en cuenta que en él hay varias personas trabajando. 1º·- Es necesario antes de empezar cualquier prueba saber que es lo que vamos a hacer para tener preparado el material y reactivos que se necesitan. 2º·- Es esencial la limpieza tanto durante el trabajo como al finalizarlo. 3º·- En el laboratorio existen riesgos potenciales, como por ejemplo: quemaduras por calor o agentes químicos, cortes, pinchazos,... que requieren una atención especial. Es importante evitar los accidentes que en gran manera se deben al descuido y excesiva rapidez en el trabajo, más que a la ignorancia. Las causas más frecuentes de accidentes son: 1º·- Las prisas por llevar a término el trabajo. 2º·- La falta de cuidado y la fatiga. 3º·- La falta de concentración en el trabajo. NORMAS GENERALES EN EL LABORATORIO 1º Debe utilizarse bata mientras se permanece en el laboratorio 2º No se debe comer no beber dentro del laboratorio, ni utilizar recipientes para guardar o conservar elementos. 3º No fumar 4º Antes de utilizar los reactivos leer los rótulos. 5º No dejar destapados los envases no las placas de cultivos. 6º Guardar las medidas de asepsia (lavarse las mano, utilizar guantes,...) 7º Mantener limpias las mesas y lavar el material utilizado al finalizar el trabajo 8º No pipetear por boca líquidos cáusticos o tóxicos o líquidos potencialmente contaminantes, sino utilizar peras de goma. 9º El material de vidrio roto se debe proteger antes de tirarlo para evitar cortes. Si a pesar de las precauciones el accidente se produce el técnico debe conocer algunas nociones de primeros auxilios. Por ejemplo: si hay cortes lo primero es saber si ha quedado algún trozo de un cuerpo extraño dentro; después se dejará fluir la sangre libremente y se hará un lavado con agua oxigenada y luego un antiséptico. NOTA: En el caso de quemaduras producidas generalmente por calor se pondrá sobre la zona una gasa empapada en alcohol. En las quemaduras abiertas se aplicará una pomada antigente y se le pondrá una gasa estéril. MATERIALES DE USO MÁS FRECUENTE En los laboratorios clínicos para la caracterización y cuantificación de sustancias, microorganismos, células y cristales presentes en los fluidos biológicos se utiliza una gran variedad de materiales y dispositivos. Este equipamiento varía según sus características, necesidades, etc., pero en general se disponen de unas mesas en las que se desarrolla en trabajo y sobre las que están colocados los aparatos. En ellas hay conexiones para: gas, luz, etc. Las mesas serán cómodas y de material impermeable y fácilmente lavable. Además habrá taburetes, para evitar la fatiga en unos casos y siendo necesario en otros, como en el caso de las observaciones al microscopio. También habrá armarios en los que se guardarán reactivos y materiales. No se puede dar una norma general en cuanto al material que se va a encontrar en el laboratorio, ya que esto depende de sus necesidades así como de las posibilidades económicas. De forma general el material no instrumenta se puede clasificar en material de vidrio, de plástico, de porcelana, etc. Material de vidrio: el vidrio se caracteriza por una gran resistencia química frente al agua, ácidos, bases, etc. mayor a la de la mayoría de los plásticos, así como por su gran estabilidad resistencia al calor y transparencia. Sin embargo no todo tipo de vidrio es adecuado para su uso en el laboratorio, por el contrario es necesario emplear vidrios que se caracterizan por su resistencia química, mecánica y térmica. Actualmente existen diferentes tipos de vidrios para laboratorios; la mayoría están fabricados en vidrio de boro silicato que se caracteriza por su gran resistencia al calor; por ejemplo: el vidrio pirex... Al trabajar con el vidrio son necesarias unas precauciones como: 1º No someterlo a cambios bruscos de temperatura porque se pueden producir tensiones en él que dan lugar a rupturas, por ejemplo: colocar el material de vidrio en la estufa de secado o esterilización en frío, calentar y dejarlo enfriar antes de sacarlo. 2º No aplicar fuerza sobre llaves o tapones 3º No someterlo a variaciones bruscas de presión 4º No conservar soluciones concentradas de álcalis ó bases en estos vidrios ya que son cáucasis. El vidrio se esteriliza en autoclave generalmente envuelto en papel de filtro. Probetas: son vasos altos graduados de diferentes tamaños. Todos ellos llevan la correspondiente división fraccionada de su capacidad total. Sirven para medir volúmenes sin demasiada precisión. Los volúmenes más usados son: 10, 25, 50, 100, 250, 500, 1000, 2000 y 5000 ml Matraces aforados: son recipientes en forma cónica en la parte inferior y un cuello fino en la parte superior. En el lleva una marca que corresponde al aforo que indica hasta donde hay que llenar el matraz para que el líquido contenido equivalga a la capacidad del mismo. Se utiliza para medir con precisión el volumen que indica el matraz. Son utilizados normalmente para preparar disoluciones de concentración conocida. Los volúmenes más frecuentes son: 5, 10, 25, 50, 100, 250, 500, 1000 y 2000 ml NOTA: En la utilización de material volumétrico, el líquido contenido se debe enrasar en el menisco (curvatura que se encuentra en la superficie de un líquido contenido en un tubo estrecho de modo que el fondo de este menisco se emplea como línea de enrase. Hay que tener en cuenta en el enrase el llamado error de paralelaje, de modo que si el menisco se observa por encima de la línea de enrase el volumen que nos indica es menor del real. Buretas: son tubos cilíndricos estrechos con la parte inferior acabada en punta y la superior como un embudo (exagerado. Son precisos. En la parte inferior tiene una llave de paso. Están graduadas y permiten medir volúmenes pequeños. El cero de la escala se encuentra en la parte superior. Las buretas se mantienen en posición vertical, mediante un soporte. Su tamaño es variable en 50 a 100 cm3. Antes de utilizarlas se deben purgar y para ello se llenan con un poco más de líquido de lo que está medido y se deja caer hasta enrasar en cero, así se elimina el aire. Para manejarla se sujeta la bureta con la mano izquierda y se abre o cierra la llave con el pulgar o índice. El líquido caerá sobre un Erlenmeyer que se sujeta con la derecha. Pipetas: son tubos cilíndricos estrechos cuya parte inferior acaba en punta, también sirven par medir volúmenes con precisiones generalmente volúmenes pequeños (inferiores a 20ml) Son tubos estrechos de poco diámetro abiertos a ambos lados, terminando en punta el inferior, la salida del líquido se regula con el dedo índice que tapa el orificio de la parte superior. Existen 2 tipos de pipetas las graduadas que miden fracciones de líquidos y las aforadas que miden un volumen fijo de líquido y también las hay de doble. Pipetas pasteur: no sirven para medir volúmenes sino echar gotas. Vasos de precipitados: son vasos de vidrio de forma cilíndrica. Miden volúmenes aproximados y los hay graduados y no graduados. Los volúmenes más corrientes son: 50, 100, 500, 1000, 2000 y 5000ml Suelen estar construidos con vidrios que soportan temperaturas elevadas por lo que pueden ponerse en contacto con llamas de mechero o placas de calefacción para calentar sus contenidos. Embudos: se utilizan para realizar operaciones de filtro con ayuda de un papel de fil Tubos de ensayo: son tubos cilíndricos de aproximadamente 2 cm3 de diámetro y con fondo cónico. Sus aplicaciones son múltiples. Por ejemplo: se utilizan para realizar pruebas bioquímicas cualitativas. Tubos de centrífuga: son tubos cilíndricos en los que se colocan el material que va a ser centrifugado; pueden acabar en forma cónica o cilíndrica y pueden estar graduados o sin graduar. Erlenmeyer: es un recipiente de forma cónica utilizada para contener reactivos y soluciones, puede estar graduado o no y en todo caso miden volúmenes con poca exactitud. Es adecuado para evitar pérdidas de materiales efervescencia o en calentamiento prolongado porque la parte alta del matraz actúa como condensador de vapores, por lo tanto, da lugar a que haya menos evaporación. Portaobjetos: es una pequeña lámina de vidrio rectangular donde se coloca la muestra para ser examinada al microscopio. Existen portaobjetos con cavidades semi-esféricas de distintos diámetros de profundidad utilizados para la observación in vivo de microorganismos. Cubreobjetos: es una fina lámina de vidrio con los que se cubren las muestras a examinar a microscopio. Otros: varillas de vidrio, vidrio de reloj, embudos de decantación, kitasato, placas de petri (son recipientes usados para el cultivo de microorganismos). LIMPIEZA Y CONSERVACIÓN DEL MATERIAL DE VIDRIO Hay que insistir en la importancia de la limpieza del material. Puede significar una fuente de error o técnicas que deberán ser repetidas. La limpieza se debe hacer al final de cada técnica sin dejar secar el material utilizado. Normalmente el material de vidrio se limpia con agua y jabón con el uso de escobillas adecuadas. En ocasiones pueden quedar restos siendo entonces necesario un lavado en mayor profundidad mediante la mezcla crómica formada por dicromático potásico 60 gr, agua (primero el agua siempre) 200ml y ácido sulfúrico, suficiente para 1 litro. Con esto se deja unos días u horas. Una vez limpio se debe aclarar varias veces con agua del grifo acabando con un último enjuague con agua destilada. Material de plástico: En la actualidad asistimos en los laboratorios clínicos a una invasión de material puede ser de múltiple uso probetas, vasos de precipitados, etc. o de un solo uso puntas de pipetas de pistón, pipetas pasteur, placas de petri, etc. La mayoría de los plásticos son atacables por disolventes orgánicos y algunos de ellos por lo ácidos fuertes. Además tampoco soportan temperaturas elevadas sin deformarse o descomponerse. Las ventajas frente al vidrio, son que son resistentes a la rotura, tienen poco peso, poco coste por lo que se utiliza como material desechable. Pipetas de pistón o automáticas: existe en el mercado una gran variedad de pipetas de pistón con puntas de plástico desechables. Estas pipetas han surgido al hacerse cada día más evidente el peligro del pipeteo por succión de los fluidos biológicos (suero, sangre, orina,...) con riesgo de contagio de enfermedades. Por otro lado diversos disolventes orgánicos producen vapores tóxicos. Las pipetas de pistón pueden ser de volúmenes fijos o regulables. El émbolo tiene 3 posiciones: normal de reposo, intermedio y de presión a fondo. Las pipetas se cargan apretando el émbolo hasta la posición intermedia y soltándola para llenar la punta y se descargan una vez en la posición normal apretando el émbolo hasta el fondo. Dispensadores: son sistemas que proporcionan repetidamente un volumen seleccionado. Se utilizan normalmente para la adición de reactivos a lotes de muestras reaccionantes. Hay una amplia gama de dispensadores con varios volúmenes de utilización. Cubetas: son recipientes usados como contenedores de muestras generalmente en disolución en las técnicas de espectrometrías. Deben ser transparentes para la longitud de onda en la que se realiza la determinación. Las más usadas son de forma prismática y son de cuarzo o sílice fundido para el ultravioleta y visible; y de cloruro sódico (NaCl) o de fluoruro cálcico (F2Ca) para el infrarrojo. Otros materiales Estufas: es una caja rectangular provista de una serie de calor regulado por un mando situado en el exterior. Se utiliza a diario en el laboratorio de microbiología para incubar cultivos de gérmenes. Mechero: el más utilizado es de tipo Bunsen que consta de un pie sobre el que descansa un tubo por donde sale el gas. En la parte inferior llevan un orificio para la entrada de aire. Sirve para calentar reactivos, flamear asas de siembra,... Gradillas: son soportes para tubos de ensayo, probetas,... TEMA II DISOLUCIONES En numerosas ocasiones los reactivos son adquiridos por el laboratorio en forma de polvo que ha de ser disuelto en la proporción adecuada para su uso, de ahí la importancia de conocer la forma de expresar la concentración variada. Conceptos básicos: la materia está compuesta por unas unidades fundamentales llamadas átomos que se combinan entre sí para formar moléculas. Las masas de esos átomos son tan pequeños que su utilización resulta muy engorrosa para los cálculos por eso se creó una escala relativa en la que se escoge un elemento como tipo y los valores de los demás elementos se refieren a él por combinarse con casi todos los elementos se escogió el O2 al que se asignó de forma arbitraria un peso atómico de 16. Átomos: el átomo es la parte más pequeña e indivisible de la materia. Actualmente podemos definir al átomo como la partícula de un cuerpo simple que es químicamente indivisible y que forma la menor cantidad de un elemento que puede entrar en combinación. Moléculas: al unirse 2 o más átomos se forman las moléculas. La molécula es el límite de la división física, es decir, la parte más pequeña de una sustancia que conserva sus propiedades químicas Se denomina peso molecular de un componente a la suma de los pesos atómicos de los elementos que componen la molécula por cada uno de ellos por un coeficiente igual al número de átomos del mismo que entra a formar parte de ella. Se denomina peso atómico gramo ó átomo gramo de un elemento a la cantidad del mismo equivalente a su peso atómico expresado en gramos. Se denomina mol ó peso molecular ó molécula gramo de un compuesto a la cantidad del mismo que tiene en gramos equivalentes a su peso molecular. Concepto de disolución: se denomina disolución a las mezclas íntimas de 2 ó más cuerpos diferentes que nos ofrecen a simple vista un aspecto homogéneo. El término de disolución normalmente se suele utilizar para describir mezclas homogéneas de 2 ó más líquidos ó de un líquido y 1 ó varios sólidos. En toda disolución hay que distinguir el cuerpo disperso ó soluto que es el que se haya en menor proporción y el cuerpo dispersante o disolvente que es el componente que interviene en mayor cantidad. Tanto el soluto como el disolvente pueden ser sólidos, líquido o gas: soluto (ClNa, azúcar, alcohol) disolvente (agua, leche, agua) La disolución adopta el estado físico del disolvente. En el laboratorio las disoluciones que se manejan con bastante frecuencia son las de sólido en líquido y líquido en líquido generalmente. Especialmente se utilizan las disoluciones acuosas. Una disolución se dice que está saturada cuando no admite más sustancia en disolución de tal modo que si se añade más soluto queda en el fondo sin disolver. Las disoluciones que se hallan lejos de la saturación se llaman disoluciones diluidas y las que están cerca de ella se denominan disoluciones saturadas. La proporción que hay entre soluto y disolvente se conoce como concentración. Hay varias maneras de expresarla. MODOS DE EXPRESAR LA CONCENTRACIÓN La concentración de una disolución nos indica la composición de la misa en función de las cantidades que hay de soluto y disolvente. Unidades físicas: partes por millón, tanto por % en: peso, volumen y p/v. Unidades químicas: molaridad, normalidad y molalidad. Partes por millón: indica el número de gramos por soluto por cada millón de gramos de disolución (el número de miligramos de soluto por cada 1000 gramos de disolución). En disoluciones acuosas es equivalente así mismo a mg de soluto por litros de disolución ya que al tratarse de disolución extremadamente diluida su densidad está muy cerca de la del disolvente, es decir, a la unidad. 5 ppm Fe en agua 5.000 mg de Fe en 1000 ml de agua. % Peso: expresa los gramos de soluto contenidos en 100gr de disolución % Volumen: expresa los ml de soluto contenidos en 100ml de disolución % P/V: expresa los gramos contenidos en 100ml de disolución Molaridad: la M de una disolución acuosa como números de moles de soluto por litros de disolución. Se representa por M Normalidad: la N se define como el número de equivalentes gramos de soluto contenidos en 1 litro de disolución. Se representa por N. Si es valencia 1 la N = M. Por equivalente de un elemento o compuestos entiende el peso de una sustancia que reacciona con 1 gramo de hidrógeno o con un peso que le equivalga. En las prácticas se hallan de la siguiente manera; en los ácidos y bases se dividen su peso molecular por el número de iones H o hidroxilo respectivamente que produce sus moléculas al disociarse.. Peso equivalente: Sosa NaOH Peso equivalente: hidróxido potásico KOH Peso equivalente: Hidróxido cálcico Ca (OH)2 Peso equivalente: Hidróxido de aluminio Al (OH)3 Peso equivalente: Ácido clorhídrico HCl Peso equivalente: Ácido nítrico HNO3 Peso equivalente: SO4H2 Peso equivalente: PO4H3 Para calcular el número de equivalente-gramo en un determinado peso de un ácido o base basta aplicar la siguiente ecuación. N = M Valencia Molalidad: expresa el número el número de moles de soluto disueltos por 1 Kgr de disolvente. Una disolución 2 m de azúcar en agua quiere decir que existe 2 moles de azúcar por cada 1000 gr de disolvente. PREPARACIÓN DE DISOLUCIONES Los disolventes utilizados en la preparación de disoluciones son muy variados aunque el agua es el más utilizado de todos ellos. A la hora de elegir el disolvente se ha de tener en cuenta que tenga propiedades químicas parecidas al soluto. En el laboratorio los productos que se manejan frecuentemente son ácidos y sales utilizando como disolvente el agua. La operación previa será calcular que cantidades son las que vamos a necesitar dependiendo de las concentraciones que queramos obtener y la cantidad que vayamos a preparar. Técnica: se pesa la sustancia en un vidrio de reloj o en un vaso de precipitados (o en un papel de filtro). En el primer caso una vez que se tiene pasada la cantidad deseada se transfiere a un vaso de precipitados lavando con un disolvente el vidrio de reloj y echando estos líquidos de lavado en el vaso. Se disuelve entonces el soluto con pequeñas cantidades de disolvente y se va vertiendo sobre el matraz aforado en el que habremos colocado un embudo. Se realiza varias veces para evitar que queden restos, en el vaso, se va añadiendo disolvente al matraz hasta llegar a la señal del aforo, el enrase es correcto cuando el fondo del menisco es tangente a la línea de aforo. La cantidad de soluto contenido en un volumen dado de solución es igual al producto del volumen por la concentración. Si diluimos una solución, el volumen de la misma va aumentando a la par que su concentración va disminuyendo mientras que la cantidad de soluto presente permanecerá constante, por ello 2 soluciones con diferentes concentraciones, pero que contienen las mismas cantidades de soluto está relacionadas de las siguientes formas (V1 · C1 = C2 · V2) CONCEPTOS GENERALES Cuando nos piden preparar un determinado volumen de una solución de concentración determinada siempre la primera cuestión que se nos plantea es saber que cantidad de sustancia pura deberá contener el volumen solicitado de la misma. Primer supuesto: soluto total/puro- 100%. Si el soluto es una sustancia totalmente pura una vez calculada la cantidad necesaria del mismo no tendríamos más que pensar correctamente, disolver en una proporción de disolvente y enrasar hasta el volumen deseado. Segundo supuesto: si la sustancia de partida es un líquido totalmente puro una vez calculada la cantidad en peso necesaria del mismo, dado que en el caso de líquido es más fácil medirlo en volumen que en peso transformaremos el peso en volumen a través de la densidad que es un dato conocido y luego enrasaremos con agua el volumen solicitado. Tercer supuesto: si la sustancia de partida (soluto) no es totalmente puro (que esta va a ser la situación real) entonces tendríamos que tomar una cantidad mayor de sustancias que si fuera puro. Riqueza: es una expresión que nos indica el grado de pureza de una sustancia. DILUCIONES SERIADAS En diferentes áreas de laboratorio de análisis clínicos se deben diluir las muestras, la sangre pura, líquidos orgánicos, etc. para preparar la concentración medible, por ejemplo: diluciones de recuento de hematies, leucocitos, plaquetas, dilución es de suero para determinar las titulaciones de anticuerpos (ANA), etc. La dilución puede ser definida como una expresión de concentración. La dilución expresa la cantidad ya sea en volumen o en peso de una sustancia en un volumen final específico. Una dilución 1:5 o 1/5 contiene un volumen de una sustancia en 5 volumen del total. Por ejemplo: sangre 1/10 (1 parte de sangre y 9 de agua). Siempre la misma cantidad de agua y de suero (en la proporción). CONCEPTOS DE pH Al preparar una dilución esta va a presentar carácter básico, ácido o neutro. Un ácido es la sustancia capaz de ceder protones (H+) y una base es la sustanica capaz de ceder iones hidroxilo (OH-) o captar protones. El término que nos refleja ese carácter ácido o base es el pH. El pH se puede definir como el logaritmo del inverso de la concentración de protones pH Para los valores entre los que pueden oscilar hay que recurrir a la autoionización del agua. El agua se disocia según la ecuación: H2O [OH-] [H+] La relación que existe entre la parte disociada y la molecular viene dada por la siguiente fórmula Experimentalmente se ha comprobado que Kw · [H2O] = [OH-] · [H+] = 10 Sustituyendo ese valor en la expresióon anterior resulta Al disociarse la molécula de agua nos da igual cantidad de [OH] que de [H] Si predomina la [H+]en una sustancia, esa disolución va a tener carácter ácido 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 La determinación del pH puede hacerse de forma cuantitativa mediante los aparatos peachímetros o de forma cualitativa utilizando los papeles indicadores, estos son de muy fácil manejo. Se impregna el papel con el indicador durante unos segundos y se compara el color con una escala de colores. Ejemplo: si se introduce el papel indicador en una disolución y el color e rojo nos indica que el pH es ácido (bajo) TEMA III EL MICROSCOPIO La palabra microscopio deriva de 2 vocablos griegos `micros' pequeño y `Scopein' ver. En términos generales un microscopio es todo instrumento que permite amplificar la imagen de un objeto o de un ser pequeño. El estudio de los microorganismos, células sanguíneas, cristales en un sedimento orinario,... precisan de instrumentos que amplíen el tamaño de los microorganismos, hasta hacerlos visibles. Según el principio que se funda cada microscopio para ampliar las imágenes se pueden clasificar en 2 tipos: microscopio de luz u ópticos y electrónicos. Entre los primeros se distinguen varios tipos pero todos tienen en común que utilizan lentes ópticas. Tipos: microscopio de campo claro, oscuro, de fluorescencia y de cambio de fase. Los segundos no incluyen lentes ópticas si no que para ampliar las imágenes utilizan un haz de electrones. Podemos hacer otra clasificación: Microscopio simple: son aquellos que utilizan una sola lente (lupas) Microscopio compuesto: son aquellos que están formados por 2 sistemas de lentes una situada cerca del ojo (ocular) y otra cercana al objeto (objetivo). LUPA: es el sistema óptico más sencillo. Permite la observación de un objeto cuando se encuentra a la mínima distancia de visión distinta, esto es, la distancia mínima por la cual el ojo puede percibir un objeto. Cuando se acerca un observador a un objeto crece el ángulo visual y este parece ser mayor. Sin embargo por debajo de una determinada distancia (unos 25 cm) entre el ojo y el objeto, este no se ve con claridad. Este límite se debe a que se supera la capacidad máxima de deformación del cristalino. Si se sitúa entre el ojo y el objeto un sistema óptico capaz de aumentar el ángulo visual se podrá ver el objeto con mayor amplitud y claridad. El aumento habitual de la lupa oscila entre 4 y 60 aumentos (la relación existente entre el tamaño del objeto percibido a simple vista y el apreciado). Por el microscopio, es decir, es el número de veces que se ve el tamaño de un objeto por encima de su valor real. En el microscopio compuesto el aumento se calcula multiplicando el aumento individual del objetivo o del ocular. Se reseña por un número seguido del signo por (x). Existen muchos modelos de lupas pero se utilizan normalmente para la disección de animales y observación de colonias. El PODER DE RESOLUCIÓN: el límite de un microscopio es la capacidad de este para mostrar distintos y separados puntos muy cercanos. El concepto es fácilmente comprensible si imaginamos un coche que viene por la noche desde muy lejos hacia nosotros. En la lejanía sólo se visualiza un haz luminoso pero conforme se va acercando hay un instante a partir del cual se distingue la presencia de 2 faros próximos pero separados. En este momento nuestros ojos tienen un máximo de agudeza visual sobre el objetivo. El límite ó poder de resolución se puede definir también como la distancia mínima entre 2 puntos que pueden distinguirse entre el microscopio. Si queremos que la resolución de un microscopio sea alta el límite o poder de resolución deberá ser lo más pequeño posible. El límite de resolución se calcula mediante la siguiente ecuación: AN = apertura numérica IR = índice de refracción del medio que hay entre la muestra y la lente D = límite de resolución ð = longitud de onda de la luz. De esta ecuación se deduce que la resolución es mayor cuando menor es ð. La resolución es mayor (d es menor) cuanto más grande es la apertura numérica de la lente utilizada (del objetivo). APERTURA NUMÉRICA: es la capacidad de la lente para juntar los grados de luz que se proyectan hacia ella. Depende del índice de refracción del medio que hay entre la lente y la muestra y del sen ð, siendo ð la mitad del ángulo del cono de luz que penetra en la lente. Si se rellena el espacio existente entre la muestra y el objetivo con una sustancia de mayor índice de refracción que el aire (por ejemplo: aceite de cedro `inmersión') se consigue que la mayor parte de los rayos partidos por los fenómenos ópticos ocasionados en el condensador y el porta se refracta y penetra en el objetivo con lo que se incrementa la resolución del microscopio. LA PROFUNDIDAD DE FOCO: es el espesor del objetivo que se aprecia enfocado. El contraste es la diferencia en la absorción de luz entre el objeto estudiado y el medio que le rodea. Se puede aumentar el contraste mediante tinción de la muestra. PARTES DEL MICROSCOPIO ÓPTICO COMPUESTO: Se distingue 2 partes: mecánica y electrónica. PARTE MECÁNICA: dentro de ella tenemos un sistema de soporte y uno de ajuste Sistema de soporte: consta de: Pie: sirve como base al microscopio y tiene el suficiente peso como para sostener el aparato. Antiguamente tenía forma de herradura y ahora rectangular. Brazo: une el pie con el tubo en caso de transporte se debe coger al microscopio por esa pieza. Tubo: es la parte del microscopio que sujeta el objetivo y los oculares; manteniéndolos en la distancia correcta de trabajo. Platina: es una placa horizontal que sostiene las preparaciones para realizar la observación. El porta debe quedar sobre la perforación que hay en el centro de la platina que deja pasar la luz que viene del condensador. Hay una pinza que sujeta el porta. Sistema de ajuste: Manilla de ajuste de los oculares. Tornillo: que permite al aflojarlo girar la pieza del tubo donde están los oculares y observar fácilmente la preparación desde otra posición. Tornillos reguladores de la platina: sirve para desplazar el porta a lo largo y ancho de la platina. Su movimiento queda reajustado en dos escalas móviles lo que permite establecer la posición exacta de cualquier punto de la preparación. 1º·- Se observa el valor más bajo de la escala en mm; está más cercano al 0 que la escala en dm (34) 2º·- Observar el valor de la escala en dm que coincide con uno de los valores de la escala en ml (34'9) Tornillo de elevación del condensador: se utiliza para elevar el condensador y disminuir la iluminación o viceversa. Tornillo de centrado del condensador: se utiliza para centrar el condensador respecto al objetivo. Palanca de cierre del diafragma Tornillo de enfoque: muevan la platina hacia arriba y hacia abajo y son 2 el macromético o de avance rápido y otro es el micrométro o de avance lento. Llevan incorporados un mando del bloqueo que fija la platina a una determinada altura. Regulador de la intensidad de la lámpara. PARTE ÓPTICA: dentro de ella tenemos un sistema de iluminación y lentes. Sistema de iluminación: Fuente de luz: lámpara halógena de intensidad variable. Situada al pie del microscopio. En su superficie externa hay un anillo para colocar filtros que facilitan la visualización de algunas preparaciones. Condensador: concentran la luz generada en la lámpara hacia la preparación. Está entre la fuente luminosa y la platina. Diafragma o iris: sirve para ajustar la apertura numérica. Cuanto más se cierra, más empeora la resolución y mejora el contraste. Está situada en el interior del condensador. Lentes: Objetivos: genera una imagen real invertida y aumentada del objeto. Están colocados en la parte inferior del tubo a nivel de una pieza mecánica que permite cambiarlos fácilmente y que recibe el nombre de revólver. Los de mayor aumento poseen un sistema de amortiguación que dificulta su ruptura al chocar con la preparación. Tiene dibujado un anillo coloreado que indica su número de aumentos: 4X (anillo rojo), 10X (anillo amarillo), 40X (anillo azul), 100X (anillo blanco, inmersión negro). El objetivo de 100 es el de inmersión porque precisa del uso de aceite de cedro sobre la preparación. Oculares: capta la imagen formada por el objetivo y la amplia. Los actuales microscopios son binoculares y están unidos mediante un mecanismo que permite ajustar la distancia interpupilar. Los más usados son de 10X aumentos. MANEJO DEL MICROSCOPIO 1º·- Accionando el revólver seleccionar el objetivo adecuado. Generalmente se empieza por un objetivo de poco aumento y luego se pasa a otro de mayor aumento. Si se hace esto basta con mover el tornillo micrométrico; para enfocar la muestra con este último objetivo. 2º·- Colocar la preparación sobre la platina. 3º·- Encender la fuente de luz y regularla a una intensidad media para evitar que se funda rápidamente la lámpara. 4º·- Situar el condensador: bajo si se utiliza un objetivo de bajo poder de bajo aumento (10X).En la mitad de su recorrido si se utiliza un objetivo de gran poder de ampliación (40X) y alto si se usa un objetivo de inmersión (100X). También conviene ascender el condensador si se observa una muestra teñida y descenderlo si se estudia una muestra en fresco. 5º·- Mirando por fuera de los oculares hacen ascender la platina con el tornillo macromético hasta que el objetivo esté muy cerca de la preparación. Cuando se utiliza un objetivo de poco poder de ampliación el microscopio tiene un tope que impide un acercamiento excesivo. Sin embargo si se emplea un objetivo de inmersión previamente hay que depositar una pequeña gota de aceite sobre la preparación y posteriormente hacer ascender la platina hasta que aquel (objetivo de inmersión) toque el aceite pero no la preparación. 6º·- Ajustar la distancia interpupilar. 7º·- Moviendo el tornillo macrométrico hacer descender lentamente la platina hasta que se vea mirando por los oculares la imagen de la muestra. Con el objetivo de inmersión nunca debe separarse tanto de la preparación como para perder el contacto con el aceite. 8º·- Afinar el enfoque con el tornillo micrométrico. 9º·- Desplazando horizontalmente la platina con movimientos en zig-zag recorrer con el objetivo toda la preparación para realizar una correcta observación de la misma. 10º·- Durante la observación mover continuamente el tornillo micrométrico para enfocar los planos de la muestra. 11º·- Una vez finalizada la observación hacer descender totalmente la platina y se retira la preparación. Si se ha utilizado aceite de inmersión se debe limpiar con una pequeña cantidad de xilol impregnado en una tela suave. Los objetivos secos pueden limpiarse con agua destilada el exterior del aparato con un paño húmedo. AJUSTE DE LOS OCULARES Se realiza una vez enfocada la muestra y se lleva a cabo de la siguiente forma: 1º·- Cerrar el ojo izquierdo y ajustar el enfoque del ojo derecho con el tornillo ðcm 2º·- Cerrar el ojo derecho y ajustar el enfoque del ojo izquierdo con el anillo de ajuste del ocular. TIPOS DE MICROSCOPIOS En el microscopio compuesto el campo está intensamente iluminado y los objetos se ven más oscuros que él. Este microscopio permite el estudio de las estructuras externas de las muestras para la cual esta debe dispuesta en una fina capa que puede ser atravesado por la luz. El microscopio de campo oscuro: es un microscopio óptico ordinario con un condensador especialmente fabricado para dirigir la luz únicamente desde los lados. Con esto se logra que la luz difragtada en un objeto se dirige y penetra en el objetivo. Si en el campo examinado no hay ninguna partícula de distinto índice de refracción se ve todo oscuro. Su máxima aplicación es la de observar la de microorganismos vivos en preparaciones en fresco o en gota pendiente. En ellas se observa el objeto que diafragta la luz brillante y el fondo oscuro. También se ha utilizado mucho para la visualización directa del Treponema Pallioum (sifilis) El microscopio de contraste de fases: consta de un dispositivo situado dentro o debajo del condensador que produce una diferencia de un cuarto de ð en unos rayos luminosos respectos a otros. Esto origina unas variaciones de luminosidad en los elementos estudiados que permiten diferenciarlos del resto de la muestra y observar con mayor detalle su estructura interna. Es muy útil para observar objetos transparentes y no coloreados y así como para preparaciones bacterialógicas. El microscopio de fluorescencia: es un microscopio en el cual la fuente luminosa es luz ultravioleta de modo que la estructura solo se hará visible si es fluorescente. Podemos encontrarnos con sustancias que son fluorescentes por sí mismas o sustancias o estructuras que captan colorantes fluorescentes. Así sucede por ejemplo cuando se utiliza auramina para demostrar la presencia de vacilos de Koch. En esputos o cuando se emplea naranja de acridina se une a la Earduerella Vaginalles en exudados vaginales. Este microscopio se utiliza en inmunofluorescencia de modo que la fijación del colorante fluorescencia al que se le llama también fluorocromo que pueden ser fluoresterina, rodaminas y la ficoeritrina. La fijación de colorante fluorescente a elemento investigado se realiza con un anticuerpo marcado. Por lo tanto el objeto fluorescente se ve como un cuerpo brillante que resalta sobre un fondo oscuro. El microscopio electrónico: está formado por un tubo en cuyo interior se ha hecho el vacío y a través del cuál se propagan los electrones que inciden sobre el objeto. Después estos electrones son refractados y se recogen sobre una pantalla en la cuál se dibuja la imagen del objeto. TEMA IV SANGRE: FISIOLOGÍA, COMPOSICIÓN Y CARACTERÍSTICAS FÍSICO-QUÍMICAS La sangre es un tejido constituido por células (leucocitos, hematies y plaquetas) y plasma (solución proteica en la que se hayan disueltos), iones y otros principios inmediatos Los leucocitos, hematies y plaquetas reciben el nombre de elementos formes y gracias a que se hallan suspendidos en el plasma confieren a la sangre su fluidez característica. Los hematies tienen a su cargo la oxigenación de los tejidos. Los leucocitos la defensa frente a agentes extraños y las plaquetas la hemostasia. El plasma desarrolla diferentes funciones entre las que destaca la inmunitaria (completo, inmunoglobulinas), la hemostática (factores de coagulación), transporte (transferrina), nutritiva (vitamina A y principios inmediatos), el equilibrio iónico (oligoelementos) y la eliminación de sustancias de deshecho (CO2 y productos del metabolismo intermediario). HEMATOPOYESIS Se denomina hematopoyesis al proceso por el que se forma las distintas células de la sangre que tiene lugar en su mayor parte en la médula ósea. Sólo las células maduras pasan a sangre periférica y estas se mantienen en unos límites de normalidad gracias a un equilibrio entre construcción y destrucción. La hematopoyesis comienza en las primeras semanas de vida embrionaria en el saco vitelino. Al tercer mes el principal órgano hematopoyético es el hígado. Hacia la mitad de la vida fetal el bazo y en menor extensión los ganglios linfáticos, el timo y la médula ósea que presentan cierta actividad hematopoyética. En el momento de nacer toda la médula ósea tiene capacidad hematopoyética siendo máxima su actividad hematopoyética. En el individuo adulto la capacidad formadora de las células sanguíneas se limita a la médula ósea de los huesos planos (costillas, esternón, pelvis, cráneo,...) a los huesos cortos (vértebras) y epífisis de los huesos largos. TEJIDO HEMATOPOYÉTICO El sistema hematopoyético engloba a los tejidos y órganos que intervienen en la producción, maduración y destrucción de las células sanguíneas. Este sistema comprende: el sistema mononuclear fagocítico, bazo, hígado, timo, médula ósea y glánglios linfáticos. Sistema Mononuclear Fagocítico: este sistema es un conjunto de monocitos, histocitos y macrófagos; distribuidos en los diferentes espacios intravasculares y extravasculares cuyas principales funciones son fagocíticas e inmunológicas. Bazo: es un órgano muy vascularizado que se localiza bajo el diafragma a la izquierda del estómago. En el bazo se produce una eliminación de los hematies viejos o defectuosos así como de cualquier tipo de células con malformaciones morfológicas o funcionales. El bazo actúa también como reserva de plaquetas. En el feto también participa en la hematopoyesis Hígado: es un órgano que se localiza bajo el diafragma en la parte superior derecha del abdomen. En el feto, el hígado es el lugar principal de la hematopoyesis desde el tercer mes de gestación hasta poco antes del nacimiento. Aunque menos eficaz y selectivo que el bazo, también interviene en la retirada de la circulación de hematies lesionados o defectuosos. Timo: es un órgano linfático-poyético situado en la zona superior del mediastino (entre los dos pulmones) ya se ha desarrollado en el momento del nacimiento y continua creciendo hasta la pubertad. Posteriormente comienza a atrofiarse hasta que en la de servir como un compartimento para la maduración de los linfocitos T. Ganglios Linfáticos: se disponen en grupos a lo largo de los capilares linfáticos más grandes. Contienen una zona interior llamada médula y una exterior llamada corteza. En la corteza se encuentra linfocitos T, macrófagos y linfocitos B. Médula ósea: es el órgano mayor del cuerpo humano. Es el lugar más importante de la formación de las células sanguíneas. Se divide en dos partes: médula ósea roja (donde tiene lugar la hematopoyesis activa) y la médula ósea amarilla (grasa; formada por células adiposas). En los primeros años de vida casi toda la médula ósea es roja. Después se va sustituyendo paulatinamente por médula ósea amarilla. En los adultos la hematopoyesis está limitada a la médula de los huesos planos, a las vértebras y a la epífisis de los huesos largos. La celularidad de la médula ósea puede verse alterado por ciertos estados patológicos. Por lo que es importante conocer las proporciones celulares de las presentes series; así como la morfología de un individuo sano. La médula roja normal es capaz de responder a diferentes estímulos, por un lado puede hacerse hipercelular (incrementando su actividad de proliferación) o por el contrario hipocelular (inactiva como consecuencia de factores ambientales, radiaciones o fármacos). Las células hematopoyéticas una vez maduras pasan a la circulación entre los factores que constituyen la maduración. Salen de una forma ordenada de las células de la médula ósea a la sangre periférica; en la que se encuentran una serie de factores humorales y el crecimiento del parenquima. ORIGEN Y DIFERENCIACIÓN DE LAS CÉLULAS HEMATOPOYÉTICAS Todas las células sanguíneas provienen de una misma célula primitiva denominada célula madre pluripotente (Stem cell o CFU-LM). Capaz de auto-perpetuarse y de diferenciarse hacia cualquier línea madurativa. A partir de la CMP aparecen las células madres comprometidas que van a diferenciarse hacia la línea linfoide que se conoce como CFU-L o hacia la línea mialoide CFU-M y cuyo proceso de diferenciación y maduración finalizará con la aparición de la célula terminal presente en sangre periférica (hematies, leucocitos y plaquetas). El esquema de cómo van a surgir es el siguiente: 1·- Precursor linfoide CFU-L 1.1·- Pre-T A·- Linfocito T 1.2·- Pre-B B·- Linfocito B B.1·- Célula plasmática 2·- Precursor mialoide CFU-GEMM ó CFU-M 2.1·- GFU-GM A·- Mialoblasto Neutrófilo B·- Monoblasto Monocito Macrófago 2.2·- CFU-Bas Promielocito basófilo Basófilo 2.3·- CFU-EO Promielocito eosinófilo Eosinófilos 2.4·- BFU-E Promielocito proeritroblasto Hematie 2.5·- BFU-Meg Megacarioblasto Plaquetas La población celular hematopoyética se clasifica en 3 grupos: Célula Madre ó Stem Cell: se puede definir como la célula que posee una capacidad de automantenimiento que se prolonga durante una gran parte de la vida de un individuo. Todas las células sanguíneas (hematies, plaquetas, granulocitos, monocitos, linfocitos, etc.) proceden. Sólo un 10% de las células Stem se encuentra en fase de síntesis de ADN; pero el 90% restante que se encuentra en fase situación de reposos posee la capacidad de entrar en fase de síntesis en situación de demanda. El reconocimiento de las células madre hematopoyéticas se basa fundamentalmente en los cultivos in vitro de médula ósea. Se ha comprobado que las células maduras de la sangre se desarrollan cultivando médula ósea en diversas condiciones y con la adición de diversas sustancias formando colonias (CFC) o también unidad formadora de colonias (UFC) Células progenitoras comprometidas: (no se observan al microscopio) las células que constituyen este compartimento se caracterizan por tener un gran potencial proliferativo y estar predestinadas a diferenciarse en una línea celular concreta. Se han caracterizado factores de crecimiento, proliferación y diferenciación de estas células a los que se han llamado factores estimulantes de colonias (CSF-S). Estas células progenitoras no pueden identificar morfológicamente al microscopio. Célula de morfología reconocible: son las células con características morfológicas y funcionales específicas y una vida limitada. ERITROPOYESIS Eritropoyesis es la producción de los hematies o eritrocitos. El hematie es el vehículo para el transporte de la hemoglobina. En situaciones anormales se pueden perder o destruir más hematies de los habituales, es entonces, cuando la médula ósea puede aumentar la producción temporalmente para volver a restablecer el equilibrio. Etapas de la eritropoyesis: Proeritroblastos (normoblasto): mide entre 20-25 micras. Es la célula más inmadura reconocible morfológicamente de la serie roja. Es grande tiene un núcleo redondo con varios nucleolos y escaso citoplasma azul. Eritroblasto basófilo (normoblastos iniciales): tiene un tamaño entre 16-20 micras. Procede del proeritroblasto. Es más pequeño. El núcleo ha perdido nucleolos que no se observan. Son de color azul. Eritroblasto policromatófilo: es de menos tamaño que el anterior. El núcleo se va haciendo más pequeño. El citoplasma es policromático (más rosado). Es el último estadio de la serie con capacidad mitótica. Mide entre 8 y 12 micras. Eritroblasto ortocromático: miden entre 7 y 10 micras. Tiene un núcleo pequeño, redondo y muy condensado. El citoplasma es acidófilo (rosado) por su alto contenido en hemoglobina. Una vez finalizado su maduración el núcleo es expulsado de la célula y fagocitado por las células del sistema de la médula ósea (mononuclear fagocítico). No tiene capacidad mitótica. Con la pérdida del núcleo el eritroblasto ortocromático se transforma en reticulocito. Reticulocito: es una célula anucleada con capacidad de síntesis de hemoglobina, proteínas y ARN. Con la tinción vital de azul cresilo brillante puede observarse la sustancia ribosómica reticulada. El reticulocito permanece 48 horas en médula ósea antes de pasar a la sangre periférica donde continua como tal 24 horas hasta convertirse en hematies maduros. El número de reticulocito en niños y adultos entre 0'2 y 2% de hematies. Es de gran valor para conocer la eficacia de la eritropoyesis. Hematies: es el elemento forma más maduro y más pequeño de la serie roja (6-8 micras). Los hematies presentan una vida media de 120 días y su principal misión es la captación de O2 por medio de la hemoglobina y su transporte a los tejidos. Finalmente las funciones del hematie se deterioran y el bazo principalmente hace de filtro retirándolos de la sangre periférica. La transformación de proeritrocitos a hematies dura entre 6 y 9 días. REGULACIÓN DE LA ERITROPOYESIS Depende del grado de oxigenación de los tejidos. Una hormona sintetizada en el riñón y conocida como eritroproyetina estimula la eritropoyesis de la médula ósea. La médula ósea puede aumentar la eritropoyesis hasta 10 veces como respuesta a la eritroproyetina si se dispone de hierro suficiente. Las acciones más importantes de la eritroproyetina son: ~ Estimular la formación y diferenciación celular. ~ Reducir el tiempo de maduración celular ~ Aumentar la concentración de hemoglobina ~ Facilitar la liberación de reticulocitos a la circulación y se va a producir una hipercelularidad eritroblástica en médula ósea. También los andrógenos estimulan la eritropoyesis, las hormonas hipofisarias, tiroideas y suprarrenales. GRANULOPOYESIS Los granulocitos se forman en la médula ósea a partir de la Stem Cell y la primera célula identificable es el Mialoblasto. Las células de la granulopoyesis constituyen el 60-65% de las células de la médula ósea. El CFU-GM es el precursor biopotencial que puede desarrollar CFU-M para producir monocitos. Las células de la serie granulocítica que se desarrollan a partir de CFU-G; son la siguiente. La primera que puede identificarse es el Mialoblasto. Mialoblastos: son células grandes entre 15 y 20 micras. Núcleo redondo, grande y con nucleolo. El citoplasma es escaso y normalmente sin granulaciones y color azul. Promielocito: generalmente algo mayor que el mialoblasto. Presenta gránulos azurófilos. La cromatina nuclear empieza a condensarse y los nucleolos resultan menos evidentes. La relación nucleo-citoplasma es menor (citoplasma es mayor). Mielocito: núcleo redondeado y sin nucleolos. El citoplasma a perdido la basofílea (color rosado) y contiene gran número de granulaciones específicas (neutrófilos, basófilos o eosinófilos) (tintes diferentes). Es la última célula de capacidad mitótica. Metamielocito: ligeramente inferior al mielocito. Se caracteriza por un núcleo arriñonado (forma riñón) con una cromatina más condensador. Cayado ó banda: tamaño ligeramente inferior al anterior. El núcleo en forma de banda. En la base de formación del cayado se produce la constricción parcial del núcleo. Hasta que se forma un fino filamento entre los lóbulos y se forma el leucocito maduro o sementado. Según el tipo de granulación específica se habla de neutrófilos, eosinófilos ó basófilos cada una presenta unas funciones específicas. CFU-LM (Stem-cell) CFU-GM 1·- CFU-G Mieloblasto Promielocito Mielocito Metamielocito Cayado Leucocito maduro (neutrófilo, eosinófilo y/o basófilo) 2·- CFU-M Monocitos. Tras un periodo de 7 horas el neutrófilo emigra por diapedesis (por pseudópodos) a los tejidos donde permanece hasta su destrucción y muerte celular. Seguida de su fagocitosis por los macrófagos existente en todos los tejidos. La granulopoyesis dura entre 10 y 13 días. Los factores estimulantes del crecimiento regulan el desarrollo de los granulocitos y monocitos. También se han identificado sustancias inhibidoras de la granulopoyesis tales como la lactoferrina que es producida por los gránulos secundarios de los neutrófilos. LINFOPOYESIS Los linfocitos se desarrollan principalmente en los tejidos linfoides del cuerpo como: los ganglios linfáticos, los folículos linfoides del bazo, el tracto gastrointestinal (apéndice), las amígdalas y otros sitios. A partir del precursor linfoide (CFU-L) aparece la primera célula primitiva que es el linfoblasto. Linfoblasto: posee un citoplasma no granulado que se tiñe de azul oscuro en la periferia y más claro en el centro. El núcleo es grande. La cromatina está dispuesta en forma reticular y tiende a ser punteada. Por lo general solo contiene 1 ó 2 nucleolos. Prolifocito: esta célula es más pequeña que su precursora y por lo general presenta una banda ancha de citoplasma azul. La cromatina nuclear tiende a aglomerarse y no se detecta un nucleolo definido. Linfocito grande: su citoplasma es bastante abundante y adquiere un color azul celeste. Además pueden existir unos pocos gránulos citoplasmáticos azurófilos pequeños y muy bien definidos. El núcleo es redondo o un poco dentado y la cromatina tiende a estar aglomerada. Linfocito pequeño: de menor tamaño y el citoplasma es escaso. El resto es idéntico al linfocito grande. Desde el año 1962 se sabe que existen 2 tipos de linfocitos fundamentales linfocitos T y linfocitos B. Que a su vez constan de diversas subpoblaciones que se diferencian por una serie de características inmunológicas y funcionales. Los linfocitos T constituyen entre 60-75% de los linfocitos de sangre periférica y los B entre u n10-20%. Los linfocitos B pueden transformarse en células plasmáticas y estas últimas son secretoras de inmunoglobulinas (anticuerpos). Los linfocitos entran y salen varias veces de la circulación sanguínea. Algunos viven entre 10-20 días y otros por espacio de varios años. Algunos linfocitos se transforman por un estímulo antigénico, otros se pierden a través del tracto digestivo y respiratorio y la mayor parte son fagocitados por macrófagos en los ganglios. CFU-L 1·- CFU-L Linfoblatos Prolinfocito Linfocito grande ó pequeño 2·- CFU-M (plaquetas, hematies, neutrófilos,...) TROMBOPOYESIS Es la producción de plaquetas a partir de la Stem-cell que se encuentra en la médula ósea. Las plaquetas se caracterizan por la endomitosis (división nuclear sin citoplasma). La célula médula más primitiva es el megacarioblasto. Megacarioblasto: es la célula más pequeña de la serie. Su núcleo es grande. Tiene nucleolos y el citoplasma es basófilos y sin granulaciones. Promegacariocitos: es mayor que el anterior y se identifica claramente en la médula ósea. El núcleo es multiglobulado sin nucleolos y cromatina densa. Megacariocito: es mayor que los anteriores. Es la célula más grande de la médula ósea. El citoplasma es voluminoso. Cubierto de granulación azurófila. El núcleo es multilobulado. Suele ser pequeño en comparación al citoplasma. La ruptura y desprendimiento del citoplasma dan origen a las plaquetas o trombocitos. Un megacariocito es capaz de producir 16.000 plaquetas. Plaquetas: carecen de núcleo son pequeños fragmentos de citoplasma que se han desprendido de la periferia del megacariocito. Su tamaño suele estar entre 2-4micras. Permanecen en sangre entre 8-12 días. Después las células del sistema fagocítico las destruyen en el bazo. Es muy probable que la maduración y proliferación estén reguladas por dos factores humorales. 1º: El factor estimulante de las colonias megacariocíticas que inducen la proliferación de las células progenitoras diferenciadas. 2º: Es la trombopoyetina: también existen constituyentes que inhiben la trombopoyesis algunos de ellos producidos por las propias plaquetas. CFU-LM 1·- CFU-L 2·- CFU-M BFU-Mg Megacarioblasto Promegacarioblasto Megacariocito plaqueta. MADURACIÓN DE LA SERIE MONOCÍTICA Y MACRÓFAGOS Este tipo celular se forma principalmente en el bazo y los tejidos linfoides y en menor medida en la médula ósea. La primera célula madura reconocible al microscopio es el monoblasto. Monoblasto: es muy similar al mieloblasto y por lo general pueden verse nucleolos, excepto que su citoplasma suele ser más claro. Promonocito: algo mayor que el monoblasto. La relación núcleo-citoplasma es moderada. El núcleo es grande y por lo general contorneado y el citoplasma es basófilo. Monocito: (15-20micras) presente tanto en la sangre como en la médula ósea. Con un citoplasma que se colorea de azul grisáceo. A veces pueden reconocerse uno finos gránulos azurófilos y vacuolas. El núcleo suele ser redondo, reniforme o incluso puede ser también globulado. La cromatina está dispuesta en grietas a modo de madeja. RESUMEN CFU-LM 1·- CFU-L (precursor linfoide) linfoblasto prolinfocito linfocito (grande o pequeño). 2·- CFU-GM (precursor mieloide) 2.1·- BFU-E Promieloblasto Eritroblasto basófilo Er. policromatófilo Er. ortocromático reticulocito hematies 2.2·- CFU-G A) CFU-G Mieloblasto Promielocito Mielocito Metamielocito Cayado polimorfonucleares B)CFU-M Monoblasto Promonocito Monocito 2.3·- BFM-Meg Megacarioblasto Promegacariocito Megacariocito Plaquetas. CONCEPTOS La sangre circulante: la sangre es un líquido de color rojo que, circula a través de los vasos del cuerpo a excepción de los vasos linfáticos. Es fácilmente coagulable cuando se detiene. Está constituida por elementos sólidos y un elemento líquido que es el plasma. La sangre es un líquido viscoso y denso que se desplaza lentamente. Si el agua tiene una viscosidad de 1, la sangre tiene una viscosidad media de 5. Ello indica que fluirá a una velocidad 5 veces más lenta que el agua, así mismo, es más pesada que es agua. Tiene una temperatura de 37ºC, un pH entre 7'35-7'45 y una concentración de cloruro sódico de 3'5% similar al agua de mar, dándole un sabor salado. Supone el 8% del peso corporal. Con un volumen de 5 a 6 litros en un hombre de tamaño medio. Elementos que constituyen la sangre: la sangre está constituida por 2 porciones uno es el plasma que es donde se encuentra disuelto las sustancias y otros son los elementos formes (son células y corpúsculos que están suspendidos en el plasma y son las siguientes: 1·Hematies, glóbulos rojos: son células anucleadas que transportan el O2 desde los pulmones a los tejidos. 2·- Leucocitos o glóbulos blancos: Granulares (polinucleares -PMN): Neutrófilos, eosinófilos, basófilos Agranulados (mononucleares): linfocitos, monocitos 3·- Plaquetas Plasma: es un líquido de color amarillento que queda en la parte superior de un tubo de ensayo después de centrifugar el mismo con una determinada cantidad de sangre y un anticoagulante apropiado. Se obtiene por centrifugación de sangre mezclada con anticoagulante, variando el aspecto microscópico del mismo según las condiciones y patologías del individuo. Siendo en condiciones normales un líquido amarillento. La composición es la siguiente: Agua: constituye alrededor del 91% del plasma. Sus funciones son como medio absorbente de calor, actuar como solvente y ser el medio de suspensión de los elementos formes. Proteínas: constituyen el 8% de los solutos del plasma. Básicamente existen las siguientes: ~ Albúminas: suponen más de la mitad de las proteínas plasmáticas. Es producida por el hígado; dan origen a la viscosidad de la sangre; ayuda a conservar el balance hídrico entre los tejidos pues regula el volumen entre la sangre. ~ Globulinas: de estas el grupo más importante son las: • Gammaglobulinas: que intervendrán en la defensa immunitaria • Fibrinógeno: es una proteína de fase aguda; representa una pequeña porción de un papel especial en la coagulación sanguínea. Nitrógeno no proteico: son sustancias que contienen N2 y no son proteínas. Son productos de desecho del metabolismo proteico que se elimina por el riñón, Son el ácido úrico, urea, creatinina y las sales de amonio. Sustancias alimenticias: una vez en el tubo digestivo se descomponen los productos. Estos pasan a la sangre. Para ser distribuidos a todas las células del organismo. Estos productos son los aminoácidos, la glucosa y los lípidos. Sustancias reguladoras: son las enzimas y las hormonas. Los enzimas son producidos por las células del cuerpo y catalizan reacciones químicas. Las hormonas son producidas por las glándulas endocrinas y regulan la defensa de funciones orgánicas. Gases respiratorios: CO2 y O2. Están más relacionadas con los hematies que con el plasma. Electrolitos: son iones que constituyen las sales inorgánicas del plasma. Los cationes son: Na, K, Ca+2, MG+2. Los aniones son: P, SO-4, bicarbonatos, Cl-. Estas salen tienen la función del mantenimiento de una presión osmótica adecuada. La regulación del pH y el mantenimiento de un balance fisiológico adecuado entre la sangre y los tejidos. Suero: el suero es simplemente el plasma menos las proteínas de la coagulación (fibrinógeno). La sangre extraída del organismo, introducida en un tubo de ensayo y centrifugada si se deja pasar un tiempo se observa en el plasma la formación de una masa gelatinosa blanca que no es ni más ni menos un coágulo formado por la acción del fibrinógeno. Si se retira este coágulo tendremos el plasma desprovisto de fibrinógeno y algunos factores de coagulación que es a lo que denominamos suero. El suero se obtiene al centrifugar una muestra de sangre total recogida en un tubo de ensayo sin anticoagulante o bien obtenida en reposo tras la retracción del coagulo sanguíneo. Es de color amarillo limpio y transparente. Cuando se extrae suero después de una comida copiosa puede tener un aspecto lechoso blanquecino por la presencia de innumerables gotitas de grasa (presencia de colesterol o triacilglicéridos elevada). El color del suero puede variar en ciertos estados patológicos: AMARILLO FUERTE: aumento de bilirrubina. COLOR MUY PÁLIDO: en las anemias, en las hemodiluciones. PARDO ROJIZO: hemólisis intensas (ruptura de hematies). ROJIZO: aparece en caso de hemólisis. También en caso de mala manipulación de extracción sanguínea o del proceso de separación de la muestra sanguínea durante la centrifugación de la misma. Anticoagulantes: los 3 anticoagulantes habitualmente utilizados en hematología son: citrato trisódico, EDTA (las sales tripotásica idiosódicas de ácidos etilendiaminotetracético) (los dos primeros previenen la coagulación por extracción del calcio de la sangre mediante precipitación o unión en forma no ionizada.), HEPARINA (actúa formando un complejo con la anti-trombina III del plasma que inhibe la trombosis y otras etapas de la activación de los factores de la coagulación). Citrato sódico: se utiliza para las pruebas del estudio de hemostasia (coagulación) la proporción es 1 parte de citrato en solución acuosa y 9 partes de sangre total. En la actualidad se usa el citrato tamponado porque ayuda a estabilizar el pH del plasma. Para el estudio de VSG 1 de citrato y 4 de sangre. EDTA: se utiliza en una concentración de 1 a 2 mg/ml de sangre y es el anticoagulante preferido para los recuentos celulares y los estudios morfológicos. Heparina: de 0'1 a 0'2 mg/ml de sangre no afecta al tamaño celular o al hematocrito. Es el mejor anticoagulante para la prevención de la hemólisis. No es satisfactorio para recuentos leucocitarios o plaquetarios debido a que produce agregación celular. CAUSAS DE ERROR Las extensiones se deben preparar inmediatamente. Si no se pueden realizar en el término de 2 o 3 horas la sangre debe refrigerarse a 4º. Si se mantiene a temperatura ambiente, el hinchazón de los hematies aumenta entre las 6 y las 24 horas. Los hematies aumenta el hematocrito y el volumen corpuscular medio y disminuye la concentración corpuscular media de hemoglobina y la velocidad de sedimentación. Antes de tomar una muestra de un tubo con sangre venosa para una determinación hematológica es importante mezclar la sangre completamente. Si el tubo ha estado de pie debe invertirse al menos 60 veces o colocarlo durante 2 minutos en un rotador mecánico pues de lo contrario la precisión se vería afectada. TEMA V RECUENTOS DE CÉLULAS HEMÁTICAS Los recuentos de eritrocitos, leucocitos y plaquetas se expresan como concentraciones que serían número de células X unidad de volumen de sangre. Hasta hace poco se expresaban en V de mm3. Actualmente se expresa en litros. RECUENTOS MANUALES Para ellos se utiliza las cámaras de recuento o hemocitómetros; aunque este método se utiliza raramente en el recuento sistemático, sin embargo, se requiere que el técnico está capacitado para aplicarlo en: ~ La comprobación de la validez de los métodos electrónicos con objeto de intentar su calibración. ~ Como método de comprobación de la validez de recuentos electrónicos en caso de leucopenia profunda o por el contrario leucemia. ~ Como método de apoyo: • Cualquier método de recuentos de células incluye 3 fases: 1º Dilución de la sangre. 2º Muestreo de la sangre diluida en un volumen determinado. 3º Recuento de las células en ese volumen. RECUENTOS ERITROCITARIOS Para la realización del recuento manual necesitamos un líquido de dilución, una cámara de recuento o hemocitómetros, una pipeta diluidora y un microscopio. • Líquido de dilución: el más empleado es el líquido de Hayem. Está compuesto de 5g de ClNa, 2'5g de SO4Na2, 2'5g de Cl2Mg y agua destilada cantidad suficiente para 1000ml En general vale cualquier solución isotónica con un conservante que no hemolice, aglutine o deforme a los hematies al menos durante un tiempo (isotónica misma concentración que el suero sanguíneo al menos en este caso). Además lleva un fijador que conserva la forma del eritrocito Las diluciones de la sangre para recuentos celulares y determinaciones de hemoglobina se pueden realizar tanto por métodos manuales como por métodos semiautomáticos (más rápidos y fiables) Los métodos semiautomáticos son instrumentos que aspiran la muestra y la mezcla con el diluyente. Los métodos manuales utilizan pipetas de dilución o pipetas microcapilares o un sistema denominado Unopette que son tubos microcapilares con un vial de plástico que contiene el diluyente. • Pipetas de dilución: son pipetas de cristal que constan de un tubo capilar graduado dividido en 10 partes y marcado con 0'5 en la 5ª señal y con 1 en la 10ª seguido de un ensanchamiento o ampolla que lleva una perlita en su interior para facilitar la mezcla. Por encima de la ampolla lleva un capilar corto por donde se introduce el tubo aspirador y con una señal 101 en la pipeta de hematies y una señal 11 grabado en la pipeta de dilución de los leucocitos. La pipeta de recuento de los hematies es roja y la de blancos es blanca. La pipeta de rojos tiene una capacidad de 100 veces el volumen del capilar y la de los leucocitos 10 veces el volumen del capilar. En la pipeta de eritrocitos cuando la sangre es aspirada, hasta la señal de 0'5 y el líquido de aspiración hasta la señal 101. La dilución resultante es de 1/200 de líquido de dilución. Una vez que se llena la ampolla el capilar no contiene células ya que han sido arrastradas por el líquido de dilución. Para observarlo en la cámara de recuento habrá que vaciar su contenido antes. En la pipeta para recuentos de leucocitos las marcas sobre el tubo capilar son las mismas que la de la pipeta de eritrocitos pero la ampolla es más pequeña con una señal de 11 grabada por encima de ella. Cuando la sangre es aspirada hasta 0'5 y diluida hasta 11 la dilución resultante es de 1/20 • Cámaras de recuentos: el hemocitómetro o cámara de recuento es un grueso portaobjetos de cristal en cuyo tercio medio están fijadas 3 plataformas paralelas que se extiende a lo ancho de su superficie. La plataforma central está subdividida por una ranura transversal en 2 mitades cada una más ancha que las 2 laterales y separadas de ellas y entre sí por fosos. Las plataformas centrales o piezas de fondo están exactamente 0'1ml más bajas que las plataformas laterales y tienen una regla de Neubaver mejorada de 3X3 ml y está subdividida en 9 cuadrados secundarios de 1X1 mm (1mm2). Los 4 cuadrados de las esquinas denominados A, B, C y D se utilizan para el recuento de leucocitos y estos están a su vez subdivididos en 16 partes llamados terciarios. El cuadrado central está dividido en 25 cuadrados terciarios, cada uno de los cuales mide 0'2 X 0'2 mm (0'04mm2) y cada uno de estos se dividen en 16 cuadrados más pequeños. Un grueso cubreobjetos que forma un plano perfecto acompaña a la cámara de reactivos. Cuando el cubre está situado sobre la plataforma de la cámara de recuento hay un espacio de 0'1 mm de altura entre este y la plataforma rayada. Por tanto cada mm2 forma la base que tiene un espacio que mide 0'1 m3. • Limpieza de las cámaras, cubres y pipetas: las cámaras y cubres deben lavarse, inmediatamente después de su utilización, con agua destilada y se dejan secar al aire. Estas superficies no se deben tocar con gasas (trapos,...), puesto que se pueden rayar y confundir con una cuadrícula. La cámara y el cubre no se deben tocar puesto que las huellas son difíciles de sacar. Antes de su utilización las superficies deben estar limpias, secas y sin hilos y marcas de agua. No se deben tocar excepto por los bordes. Las pipetas recién usadas se dejarán con la punta hacia abajo en un recipiente con agua y con una gasa en el fondo. De esta forma evitaremos que se resequen y deterioren las puntas. Para su limpieza definitiva se usará una bomba de succión o un chorro de agua con una jeringuilla y se pasará 1º con agua del grifo, luego con agua destilada, después etanol (alcohol) y finalmente éter. Por último se hace pasar por el interior de las pipetas una corriente de aire hasta que estén secas, lo que se comprueba por la bolita que se mueve libremente sin adherirse a las paredes. Si no se necesitan hasta el día siguiente se coloca con la punta hacia abajo en un recipiente de boca ancha con una gasa en el fondo y se eleva la estufa a 37ºC. TÉCNICA Los hematies son células anucleadas que se observan como discos bicóncavos de 6 a 8 micras de diámetro y 2 micras de espesor. Se puede utilizar sangre capilar o venosa. Si se emplea sangre venosa debe ser recogida sobre EDTA (tapa violeta) debe mezclarse bien con el anticoagulante invirtiendo el tubo varias veces antes de cargar la pipeta. Echaremos una pequeña cantidad de sangre en un vidrio de reloj y procederemos a cargar la pipeta hasta la marca 0'5 con cuidado de que no aparezcan burbujas. Se va llenando la pipeta en posición horizontal. Si nos pasamos de la marca eliminamos el exceso de sangre golpeando suavemente la punta de la pipeta sobre un trapo limpio o papel... Es necesaria mucha exactitud en esta parte ya que un pequeño error lo multiplicaríamos por 200. Limpiamos la punta de la pipeta con un trapo y la introducimos en la disolución de Hayem aspirando a medida que rotamos la pipeta para que se mezcle bien la sangre. Llenamos hasta la marca 0'1 no siendo necesario que este enrase sea tan exacto porque en este caso el error es mínimo. A continuación colocamos el cubre. La adhesión de este a la cámara debe ser perfecta para ello aplicamos una suave presión con los pulgares deslizando este (cubre) hacia arriba y abajo sobre las plataformas laterales hasta que confirmemos su adhesión. Esta adhesión se comprueba por la aparición de los anillos multicolores de Newton que deben persistir cuando dejemos de ejercer la presión (esta maniobra se favorece si previamente se humedece ligeramente la plataforma). A continuación procedemos a cargar la cámara expulsando las 3 ó 4 primeras gotas que ocupan el tubo capilar que no contiene células. Colocamos la pipeta en el borde del cubre controlando que forme unos 30º con la cámara. El líquido se desliza bajo el cubre atraído por capilaridad y debemos controlar esta entrada haciendo presión con la punta de la lengua sobre la boquilla o con el dedo sobre el extremo de la pipeta. Debemos tener cuidado de que el líquido penetre en el espacio debajo del cubre sin que rebose o forme burbujas, ya que el exceso de líquido tiende a elevar el cubre y obtendríamos recuentos erróneamente elevados. Se dejará que las células sedimente unos minutos en la cámara y se llevará al microscopio. Si no vamos a contar enseguida las células se debe proteger el líquido contra la evaporación colocando la cámara en una placa de petri con un papel de filtro humedecido en su interior. Observamos con un objetivo de 10X para comprobar que las células están distribuidas uniformemente sino es así se repetirá el proceso. Se cierra parcialmente el diafragma condensador del microscopio para hacer que los hematies resalten con nitidez al utilizar las lentes de poco aumento. Se cuenta los eritrocitos que hay en 80 cuadrados pequeños con el objetivo de 40X. Para ello se coge un cuadrado mediado central y los cuatro de las esquinas. De aquellos hematies que cabalgan en las líneas de los cuadrados contaremos los que toquen 2 líneas y despreciaremos los que toquen los otros 2. Así podemos contar los que toquen los otros 2. Así podemos contar las que toquen la línea de arriba y la de la derecha y despreciaremos los de abajo y los de la derecha. LAS CARACTERÍSTICAS DE UNA CÁMARA DE RECUENTOS BIEN CARGADA SON: El líquido debe llenar completo o casi completo donde se encuentra el retículo.. No debe existir líquido en el foso. No debe existir burbujas. RECUENTOS DE LEUCOCITOS Para el recuento de leucos se cuentan los 4 cuadrados de las esquinas. Hacemos lo mismo que para los hematies pero empleando la pipeta de blancos, de modo que la solución que obtenemos es de 1/20. Se utiliza el líquidos de Turck. El líquido de Turck es hipotónico de modo que se destruyen los hematies y así no entorpecen en el recuento. El líquido de Turck consta de ácido acético glaciar 2ml, violeta de genciana disolución de 1% 1ml y agua destilada en cantidad suficiente para 1000ml. Deberá filtrarse a menudo para evitar levaduras y hongos. Cálculos En un principio es un poco difícil distinguir los leucocitos de los restos de membranas de los hematies hemolizados. Se debe tener en cuenta que los leucocitos son más refringentes (más brillantes) al moverlo con el microscopio que los restos de hematies. Se debe observar la presencia de la membrana citoplasmática y de la membrana nuclear (a veces más) como líneas finas y brillantes. Esto se observa fácilmente moviendo el microscopio hacia delante y hacia atrás. RECUENTOS DE PLAQUETAS Debe evitarse la aglomeración de plaquetas mediante una correcta y rápida punción venosa. El recuento puede hacerse con la pipeta de rojo o la de blancos cogiendo sangre hasta 0'5 y diluyendo hasta la marca 11 ó de 101 con un líquido de disolución llamado Plaquet-Crom. Está compuesta por oxalato amónico al 1%. Este líquido provoca la hemólisis de los hematies y además contiene un antiagregante plaquetario. Una vez cargada la pipeta la agitamos y esperamos unos minutos para que se hemolicen bien los hematies. Cargamos la cámara y la colocamos en una placa de petri con un papel humedecido para evitar la evaporación del líquido. La dejamos reposar 15 minutos para después hacer el recuento. Este se hace de la misma manera que los hematies de modo que si usamos la pipeta de rojos multiplicaremos el número contado por 10.000 y si ha sido la de blancos por 1.000 MÉTODO DE FONIO PARA RECUENTOS DE PLAQUETAS Se coloca sobre el pulpejo del dedo previamente desinfectado una gota de disolución de sulfato de magnesio (MgSO4) al 14% estéril y a través de ella hacemos una punción dactilar a fin de obtener una rápida dilución de la sangre y evitar la autoaglutinación de las plaquetas. Se procede a hacer una extensión y se tiñe como una fórmula normal pero manteniendo el colorante GIEMSA durante más tiempo. Se lleva al microscopio y se observa con el objetivo de inmersión. Se cuenta el número de plaquetas que hay por cada 1000 hematies. Para ello contamos el número de hematies que hay en un campo en el cual deben estar distribuidos homogéneamente. Calculamos el número de campos que tenemos que ver para que 1000 hematies. Contamos en ese número de campos las plaquetas que se presentarán como pequeños corpúsculos de 2 a 4 micras teñidas de color azul y algunas granulaciones púrpuras. Después sabiendo el número de hematies por mm3 podremos calcular fácilmente el número de plaquetas por mm3 (1000/Xhematies = al número de campos que tenga que mirar) RECUENTOS DE RETICULOCITOS Los reticulocitos o eritrocitos inmaduros existen en la sangre del individuo en proporción de 510 % por cada 1000 hematies en condiciones normales. Debido a que aunque permanecen etc. la médula de 2 a 3 días terminan su maduración en la sangre periférica en 24horas aproximadamente (por eso deben observarse dentro de las 6 primeras horas después de la extracción porque maduran un vitro) Su tamaño es el de los demás hematies pero se caracterizan por contener una pequeña cantidad d ARN formando un retículo granulofilamentoso observable al ser teñido. La cantidad de ARN es tanto menor cuanto más madura es la célula. EL MATERIAL Tubo de hemólisis, porta, porta esmerilado, pipetas pasteur, microscopio, aceite de cedro, solución colorante de azul cresil brillante (fórmula: 1gr de cloruro de sodio al 0'85% cantidad suficiente para 100ml), sangre entera anticoagulada o capilar. TÉCNICA En un tubo de hemólisis se mezclan 3 gotas de sangre bien homogeneizada con 3 gotas de colorante, Mezclar suavemente, dejar reposar 15 minutos de modo que el colorante tiña los reticulocitos. Pasados los 15 minutos se homogeneiza y con la pipeta Pasteur se pone una gota en el porta y se realiza una extensión con el porta esmerilado. NOTA: EXTENSIÓN: se pone una gota a 1cm del borde derecho del porta. En el extremo opuesto se sujeta con el índice y pulgar de la mano izquierda. Con la derecha se toma el esmerilado sujetándolo desde arriba con el índice y pulgar. Se apoya por delante de la gota a 45º de inclinación. Se hace retroceder de modo que el borde coincida con la gota que se extiende por capilaridad por este. Antes de que llegue a los extremos se desliza hacia delante con movimiento firme y rápido por el de la mesa. El movimiento de desplazamiento termina elevando el de la derecha antes de llegar al final. Se debe secar rápidamente por agitación al aire de modo que no se distorsionen las células. Miramos con objetivo de 100X se busca una zona en la extensión en el que el número de eritrocito por campo sea de aproximadamente de 100 y se cuentan los reticulocitos que hay cada 1000 hematies. Para mayor exactitud se harán 2 extensiones. Los eritrocitos y reticulocitos aparecerán con un color verdoso y el retículo de ARN de azul oscuro se expresa en % ó %o de eritrocitos. Los valores son: ~ En recién nacidos (sangre del cordón): 2-6% ~ En adultos y niños: 0'2-2% RECUENTO ELECTRÓNICO DE CÉLULAS La mayoría de los métodos utilizados actualmente para el recuento de célula sanguíneas, están basadas en uno de los principios siguientes: 1º Un rayo de luz atraviesa una corriente de células y estas ocasionan reflexionan en el rayo que se convierten en impulsos eléctricos mediante un tubo que se llama fotomultiplicados. 2º Las células que pasan a través de una abertura provocan cambios en una resistencia eléctrica que quedan registrados como impulsos eléctricos. RECUENTOS CON LUZ DISPERSA: un haz de luz de tipo monofocal es condensado por donde fluye la sangre diluida. Si no hay ninguna partícula la luz va a parar a una pantalla redonda o disco (D) que la retiene. Si este haz condensado encuentra una partícula entonces se refleja la luz que irá a parar a un tipo fotomultiplicador ó foto-detector que lo convierte en impulso eléctrico. Cuando para la sangre diluida los glóbulos entran de uno en uno e irán dando señales al tubo fotomultiplicador que serán posteriormente contadas (las señales). El tubo por el que atraviesan las células son de cristal. En el caso de recuento de hematies el diluyente será una dilución isotópica para evitar la lisis de estos. En el caso de los leucocitos el diluyente llevará un agente hemolítico como por ejemplo la saponina. RECUENTOS BASADOS EN LA RESISTENCIA ELÉCTRICA: se funda en que las células van atravesar una abertura o orificio por la cual está pasando una corriente eléctrica de modo que provocan cambios en la resistencia eléctrica de modo que provocan cambios en la resistencia eléctrica que se registra como impulsos eléctricos. La sangre se diluye en una disolución isotónica conductora de la electricidad. El cilindro de cristal lleva un líquido conductor. También tiene un electrodo en su interior que es el E2 y en su pared un orificio de tamaño adecuado para permitir la entrada de las células. En el recipiente que contiene la dilución celular (sangre diluida) hay otro electrodo que es el E1. El cilindro está conectado a un tubo en forma de U parcialmente lleno de mercurio y que tiene 2 contactos eléctricos. El cilindro está sumergido en la suspensión de células que van a ser contadas y se lleva con la disolución conductora (una bomba de vacío hace llegar el mercurio hasta la parte superior del tubo). Y la suspensión de células fluye a través del orificio hacia el interior del cilindro es menor que en le exterior. Se cuentan los impulsos eléctricos que son proporcionales al volumen de las células. Se empieza el recuento cuando el mercurio establece contacto con CE1 y se detiene cuando toca CE2 contándose un volumen de suspensión de célula igual al volumen que hay entre CE1 y CE2. NORMAS GENERALES DEL MANEJO DEL APARATO DE REACCIÓN AUTOMÁTICO Lo general es seguida los consejos del fabricante. Sin embargo hay una serie de acciones que son comunes a la mayor parte de los aparatos. 1º Al comenzar a hacer los recuentos conectar el aparato a la red y esperar el tiempo necesario para que se caliente. 2º Se limpian los circuitos por donde va a fluir la sangre y líquidos fluyentes con detergentes que nos va a proporcionar el fabricante. 3º Una vez limpio lo circuito se debe calibrar el aparato con las suspensiones patrón 4º Comenzar el recuento 5º Una vez concluido el recuento el aparato debe quedar en conclusiones óptimas de reposo hasta el día siguiente; por lo tanto se deberán lavar los circuitos con el detergente y dejar el aparato según las normas recomendables por el fabricante. LOS SISTEMAS DE RECUENTO SDR No diferencian entre eritroblasto, hematies anucleados y leucocitos por lo que si mediante la observación del frotis se observa una presencia de eritroblastos mayor al 10% se debe hacer una corrección en el recuento de leucocitos. Leucocitos reales = CAUSAS DE ERROR EN LOS RECUENTOS ELÉCTRICOS Al igual en el recuento con la cámara cuenta-glóbulos el empleo de métodos electrónicos puede verse sometido a varios errores, sobre todo si se olvida el control periódico y el calibrado diario de los elementos. El error debido al propio método suele ser menor al 1% pero puede verse notablemente incrementado por las siguientes causas: 1º Uso del diluyente incorrecto 2º Presencia de partículas extrañas en el líquido diluyentes 3º Lisis incompleta de hemólisis en el recuento leucocitario 4º Obstrucción de capilares u orificio de recuento 5º Contaminación de arrastre de una muestra a la siguiente 6º Dilución incorrecta 7º Averías en los componentes eléctricos de los aparatos 8º Mala agitación de la muestra Hoy en día hay aparatos electrónicos que hacen la diferenciación de 5 poblaciones celulares de modo que distinguen linfocito de monocitos y dentro de los basófilos, neutrófilos y eosinófilos. Estos aparatos se fundan en la utilización de tinciones citoquímicas. Además dan alarma cuando hay células anormales, inmaduras. VALORES NORMALES DE LAS CÉLULAS SANGUÍNEAS Los hematies tienen los siguiente parámetros: En niños de 1 año están entre 4'5 + 1'5 · 106 hem/mm3 En niños de 2 años están entre 4'5 + 2 · 106 hem/mm3 En los hombres están entre 5'4 + 1'5 ·106 hem/mm3 (4'5-5) En las mujeres están entre 4'8 + 1'2 · 106 hem/mm3 (4-4'5) En el recién nacido las cifras son más altas próximas a los 6.106 para descender a los pocos días a los valores normales. Estas cifras son constantes no ofreciendo variaciones por causas de ejercicio físico, la noche, etc. Sin embargo puede haber más espesamiento de la sangre, o grandes sudaciones o pérdidas de líquido (deshidrataciones) Los hematies aumentan en personas que viven en lugares altos. Durante el embarazo disminuye por hemodilución (dilución de la sangre). Los leucocitos tienen los siguientes parámetros: En niños de 1 año están entre 10 + 2'5 ·103 hem/mm3 En niños de 2 años están entre 8 + 1'5 ·103 hem/mm3 En hombres están entre 7'4 + 3'5 ·103 hem/mm3 En mujeres están entre 7'2 + 3'5 ·103 hem/mm3 Las cantidades normales son entre 5 · 103 y 10 · 103. Estas cantidades son de amplia variación y las cifras halladas en un mismo individuo con pocas horas de diferencia puede variar en estados normales en hasta en 2.000/mm3; por la mañana existen unos pocos menos que por la noche. Existen ciertos leucocitosis (más de 10.000/mm3) fisiológicas creadas por el ejercicio muscular, la digestión la influencia del calor o del frío, las emociones extensas, embarazo, e incluso por cambios posturales. Al nacer el número de leucocitos es muy elevado y al as 24 horas alcanza incluso las 20.000/mm3. En la primera semana desciende hasta 8.000 a 10.000 permaneciendo en estas cifras y descendiendo paulatinamente hasta la pubertad donde alcanzan las cifras normales; además la cifra de leucocitos en la primera infancia son muy variables individualmente pudiendo encontrarse cifras de hasta 16.000 /mm3 que no se consideran patológicas. Los límites correctos de las plaquetas son entre 100.000 - 400.000 plaq/ mm3. Las cifras normales dependen en gran medida de los métodos empleados en su recuento. Además en el supuesto sano ya existen normalmente variaciones individuales y espontáneas. VARIACIONES EN EL RECUENTO DE LAS CÉLULAS SANGUÍNEAS. Terminología: • ANEMIA: es la disminución de la cantidad de hemoglobina acompañada la mayoría de las veces de disminución de la cantidad de hematies. Puede obedecer a una pérdida por hemorragia, a hemólisis o a una falta de su formación en la médula ósea (insuficiencia medular). • POLICITEMIA: aumento del número de hematies. Puede ser relativa o permanente (policitemia severa). • LEUCOCITOSIS: es el aumento del número de leucocitos por encima de 10.000/mm3. Se presenta en muchas enfermedades infecciosas en procesos inflamatorios, agudos y crónicas y alcanza los valores máximos en las leucemias en las que se pueden sobrepasar los 100.000mm3 (cuando se utilizan sistemas automáticos de recuentos las muestras de sangre con gran número de leucocitos pueden contaminar la siguiente produciendo cifras falsamente elevados). • LEUCOPENIA: disminución de la cantidad de leucocitos por debajo de los 5.000/mm3. Se presentan algún tipo de infecciones. Por ejemplo: fiebres tifoideas, sarampión, brucelosis y algunas enfermedades hemáticas. • TROMBOPENIA: disminuye la cantidad de plaquetas por debajo de las 100.000/mm3. Se presentan por ejemplo: por las púrpuras T (manchas rojas de la piel). • TROMBOCITOSIS: aumento de las plaquetas por encima de 400.000. TEMA VI HEMATÓCRITO La sangre está formada por una parte corpuscular (células sanguíneas) y una parte líquida (plasma). De la parte corpuscular aproximadamente el 90% corresponde a glóbulos rojos. Si la sangre se hace in coagulable (por EDTA en proporción 1-9) y se centrífuga; se obtiene la separación de la parte corpuscular y la plasmática. La primera en la que se observa hematíes, leucocitos y plaquetas es la que tiene mayor densidad por lo que quedará en el fondo del tubo, en cambio el plasma corresponderá a la parte flotante El hematocrito de una muestra de sangre es la relación del volumen de eritrocitos con el de sangre total. Se expresa como un porcentaje P/V o preferiblemente en una fracción decimal en las que las unidades L/L (son longitudes) están implícitas. La heparina seca, el oxalato equilibrado o el EDTA resultan satisfactorios como anticoagulantes. El hematocrito venoso coincide estrechamente con el obtenido con la punción cutánea (yema del dedo). Ambos son mayores que es hematocrito corporal total. El hematocrito se puede medir directamente (por centrifugación con macrométodos o micrométodos) o de forma indirecta (como el producto de CVM -volumen corpuscular mediopor el recuento de hematíes en aparatos automáticos). Analizando la sombra que produce la población eritrocitaria al reflejarse en un campo oscuro. MACROMÉTODO DE WINTROBE Consiste en un tubo de cristal graduado al que se centrífuga para separar las dos fases COMO SE HACE LA LECTURA La lectura se realiza midiendo la columna de eritrocito por ml (L1) y la altura de la muestra total de sangre (L2). Valor hematocrito = L1/L2. Por encima de la capa de glóbulos rojos aparece una capa blanco grisácea que corresponde a leucocitos y plaquetas y que no se debe incluir en L1. En esta técnica el tiempo de centrifugación es alto. Requiere una cantidad relativamente elevada de sangre (5 ml) y además los tubos que se utilizan no son desechables por lo que se ha abandonado a favor del micromatocrito y sobre todo de los métodos automáticos. MÉTODO MICROMATOCRITO Tiene 2 ventajas: la primera es que se utiliza poca cantidad de sangre y la segunda es que se pueden hacer un gran número de pruebas a la vez y en poco tiempo. EQUIPO Se usa un tubo de hematocrito capilar de 7cm de longitud con un orificio uniforme de alrededor de 1mm. Estos tubos pueden estar: • Heparinizados: sirve para tomar sangre capilar directamente. • No heparinizados: sirve para tomar sangre venosa anticoagulada con EDTA. Se debe disponer de centrífugas especiales que producen campos centrífugos que oscila entre 10.000 y 13.000 Ges. MÉTODO El tubo de hematocrito se llena por atracción capilar a partir del punto de punción o de una sangre venosa bien mezclada. Los tubos capilares deben llenarse al menos 5 cm. El extremo vacío se sella con plástico moldeable (plastilina). Los tubos ya llenos se colocan en los canales o servos radiales del aparato de centrifugación con el extremo cerrado dirigido hacia fuera. Se coloca el fondo del tubo contra el relleno de goma para evitar rupturas. La centrifugación durante 5 minutos a 10.000 a 12.000 Ges (11.000 RPM) es satisfactoria. A menos que el hematocrito exceda del 50% en este caso se centrífuga 5 minutos adicionales con objeto de asegurar que se a reducido al mínimos la cantidad del plasma atrapado. Los tubos capilares no están grabados. La longitud de toda la columna y de la columna de eritrocito se debe medir con una regla milimetrada y una lupa o con un lector de hematocrito. Para usar el lector de hematocrito se debe colocar el extremo inferior de la columna de sangre en la línea correspondiente al 0 y el superior al correspondiente al 100. En todo caso seguir las instrucciones del fabricante. La determinación del hematocrito debe utilizarse por duplicado y la diferencia entre los 2 valores no debe ser superior a 0'01. INTERPRETACIÓN DE LOS RESULTADOS El hematocrito normal para valores adultos es de 0'41 a 0'51 y par mujeres 0'36 a 0'45. Un valor por debajo de lo normal en un individuo indica anemia y un valor más alto policitemia. El valor del hematocrito es bajo en la hidriemia del embarazo (proporción desusada de suero en sangre en relación a los corpúsculos sin incremento de la masa total de sangre). El hematocrito puede ser normal o incluso elevado en el shock acompañado por hemoconcentracción debido a la pérdida de sangre aunque la masa total de eritrocitos puede estar considerablemente disminuidas. No es fiable como asignación de anemia y después de una pérdida de sangre aunque sea moderada o después de una transfusión. CAUSAS DE ERROR CENTRIFUGACIÓN: una duración y velocidad de centrifugación adecuadas son esenciales para un hematocrito correcto. En general cuanto más alto es el hematocrito mayor fuerza de centrifugación requiere. En el curso de centrifugación una pequeña cantidad de leucocitos, plaquetas y plasma queda atrapada entre los eritrocitos. El error resultante es por lo general de poca importancia. En general la cantidad de plasma atrapada es mayor en los hematocrito altos que en los bajos. ERROR DEBIDOS A LA MUESTRA: la posición, la actividad muscular y la extasis (estancamiento de la sangre) debida a un torniquete mantenido; provoca la alteración en el hematocrito. Además si hay un exceso de EDTA el hematocrito será falsamente bajo. OTROS ERRORES: Los Errores Técnicos incluyen la mezcla deficiente de la sangre, una lectura inadecuada y la inclusión del estrato leucocitario como parte del volumen eritrocítico. EXAMEN MACROSCÓPICO Siempre que se determina un hematocrito por centrifugación la inspección de la muestra después de la centrifugación puede proporcionar información muy útil. En este sentido deberían anotarse las alturas relativas de la columna de eritrocitos, capa espumosa y columna de plasma. La capa espumosa es el estrato gris-rojizo situado entre los hematíes y el plasma; comprende los leucocitos y plaquetas. Una capa espumosa del 1% del volumen total indica un recuento leucocitario del orden del 10•109 leucocitos/litros. El color naranja o verde del plasma sugiere una gran cantidad de bilirrubina (producto de deshecho de la hemoglobina- da el color amarillo cíatericia) Mientras que el color rosa o rojo sugiere hemoglobilemia. Deben tenerse en cuenta que la causa más frecuente de hemólisis. También le da un color o rosa o rojo al plasma la deficiente recogida de muestras. La presencia de plasma turbio si la muestra no se a recogido después de 1 ó 2 horas de una comida rica en grasas puede señalar necrosis o ciertas hiperglobulinemias anormales especialmente crioglobulinemias (la crioglobulina es una globulina -proteína- que cristaliza a bajas temperaturas). Recién nacidos 0'54 Niños hasta 10 años 0'38 Mujer adulta 0'42 Mujer embarazada 0'39 Hombres 0'45 TEMA VII LA VELOCIDAD DE SEDIMENTACIÓN GLOBULAR Es una propiedad física de la sangre. Si se dispone de un tubo de sangre con anticoagulante y se deja en reposo se observa que después de un cierto tiempo las células sedimentan formándose un paquete de hematíes a nivel del fondo del mismo. Una vez finalizado este proceso quedan 2 fases bien delimitadas que corresponden al plasma y a las células constituidas prácticamente en su totalidad por hematíes. La VSG es equivalente a la longitud del recorrido descendente de la parte superior de la columna de hematíes en un intervalo determinado de tiempo y varios factores contribuyen a este valor. La VSG constituye una medida de agregabilidad de los hematíes y depende fundamentalmente de los siguientes valores: viscosidad plasmática o factores plasmáticos. tamaño de los eritrocitos. diferencia de la distancia entre los hematíes y el plasma. temperatura ambiente. FACTORES PLASMÁTICOS Los niveles elevados de fibrinógeno y en menor medida de las globulinas favorecen una VSG acelerada. Estas moléculas asimétricas de proteínas tienen un efecto mayor que otras proteínas respecto a la disminución de la carga negativa de los eritrocitos que tienden a tener los aparatos. La disminución de este potencial promueve la formación de apilamientos que sedimentan más rápidamente que las células aisladas. La albúmina y la lecitina retrasan la sedimentación y el colesterol la acelera, FACTORES ERITROCITARIOS La anemia es responsable de un incremento de VSG ya que la variación de la relación eritrocitoplasma favorece la formación de apilamientos independientemente de las variaciones en la concentración de proteínas plasmáticas. La VS es directamente proporcional al peso del agregado celular e inversamente proporcional al área superficial. Los microcitos se sedimentan con mayor lentitud que los macrocitos que tienen un área superficial disminuida en relación con el volumen. Las pilas de monedas también tienen un área superficial muy disminuida en relación al volumen y aceleran la VSG. Los hematíes con forma anormal o irregular tales como las células falciformes o los esferocitos dificultan la formación de pilas y diminuyen la VSG. Factores plasmáticos: 1·- Fibrinógeno (ð, ð, ð -globulinas) elevación de la VSG 2·- Colesterol elevación de la VSG 3·- Albúminas y lecitina disminuyen la VSG Factores eritrocitarios 1·- Anemia aumenta la VSG FACTORES QUE HAY EN LA VSG La sedimentación globular se realiza en 3 etapas: 1·- Hemoglutinación: o tendencia de los hematíe4s a formar agregados en forma de “pilas de monedas”. (más importante) (210') 2·- Sedimentación: o desplazamiento de los hematíes hacia el fondo del recipiente a velocidad constante. (340') 3·- Acúmulo de hematíes en el fondo del recipiente (315') La más importante es la hemaglutinación ya que de ella dependerá la velocidad de todo el proceso así cuanto más pequeñas sean los agregados más lentamente se producirá la sedimentación y viceversa. Las proteínas (fibrinógeno y globulinas) facilitan la hemoglutinación y la tendencia a formar pilas de monedas. En un individuo normal la VSG se mantiene constante gracias al equilibrio existente entre el efecto de las albúminas y el de las restantes proteínas del plasma. Aunque se trata de una prueba empírica y altamente inespecífica la determinación de VSG es de gran utilidad en clínica ya que fuera de las posibles variaciones fisiológicas constituye siempre un signo de enfermedad. MÉTODOS 1·- Método de WESTERGREM (método estándar). Este método se utiliza mucho debido a su sencillez. Equipo: el tubo de Westergrem es una pipeta recta de 30cm de largo y un diámetro interno de 2'5mm, está calibrado en mm desde 0 (superior) a 200 (inferior). Tiene un volumen de aproximadamente 1mm y también se utiliza la gradilla de Westergem. Reactivo: Como solución de diluyente anticoagulante se utiliza una solución 0'105M de citrato sódico. Esta solución se filtra y se guarda sin conservantes. Técnica: se añaden 4mm de sangre total a 1mm de citrato de sodio y se mezcla por inmersión. Se llena una pipeta de Westergrem hasta la señal 0 y se coloca verticalmente en el porta pipetas a temperatura ambiente sin vibración ni exposición directa a la luz del sol. Después de 60 minutos exactos se registran en mm la distancia al extremo superior de la columna de hematíes como valor de VSG si la demarcación entre plasma y la columna de hematíes es confusa se toma el nivel donde la distancia total aparece en primer lugar. 2·- Método de WESTERGREM modificado. Una modificación del método de Westergrem produce los mismos resultados pero utiliza sangre anticoagulada con EDTA en lugar de citrato, esto es más conveniente puesto que permite calcular la VSG a partir del tubo de sangre que se utiliza para los demás estudios hematológicos, para ello se diluyen 2 mm de sangre bien mezclada con EDTA en 0'05mm de citrato sódico al 3'8% o en 0'5mm de cloruro sódico al 0'85%. LA VSG aumenta gradualmente con la edad. En la actualidad los límites superiores originados del valor normal de Westergrem (10Km/H para los hombre y 20Km/H para las mujeres) parecen demasiado bajo. Los valores superiores son: HOMBRES MUJERES Menores de 50 años 15 mm/h 20 mm/h Entre 50-80 años 20 mm/h 30 mm/h Mayores de 85 años 30 mm/h 42 mm/h CAUSAS DE ERROR 1·- El valor de la VSG puede estar elevado si la concentración del anticoagulante es mayor de lo recomendado. El citrato sódico o el EDTA no afectan la VSG si se utilizan en la concentración adecuada. Sin embargo, la heparina altera el potencial Z de la membrana y no puede utilizarse como anticoagulante. 2·- La presencia de burbujas en el tubo cuando se llena afectará a la VSG también la hemólisis puede afectarla. 3·- La limpieza del tubo es importante se deben eliminar los restos de alcohol y éter. 4·- Desviación de la verticalidad de la pipeta. Los eritrocitos se agregan a lo largo del lado inferior mientras que el plasma aumenta por el superior (si está inclinada mayor VSG). 5·- La temperatura que debería mantenerse entre 20-25ºC ya que las temperaturas inferiores o superiores en algunos casos alteran la VSG. Si la sangre se ha guardado refrigerada se debería llevar a temperatura ambiente antes de realizar la prueba. 6·- La prueba debería prepararse antes de 2H de haber obtenido la muestra de sangre (0 12H si se utiliza EDTA como anticoagulante y se mantiene la sangre a 4ºC) de lo contrario algunas muestras con VSG elevada darán valores falsamente bajos debido a que los hematíes tienden a adoptar una forma esférica con menor inclinación a formar apilamientos. COCIENTE Z DE SEDIMENTACIÓN Mediante un sistema de centrífuga (zetafuge) se hacen gira los tubos capilares en posición capilar en posición horizontal en 4 ciclos de 45 segundos. Esto provoca una comprensión y dispersión controladas de los hematíes permitiendo la formación y sedimentación de apilamiento en este periodo de 3minutos. El tubo capilar se lee a continuación como si se tratase de un tubo estándar de hematocrito dando un valor denominado zetacrito, el verdadero hematocrito se divide por el zetacrito y el resultado expresado en % es el cociente Z de sedimentación (CZS). Dicho valor no resulta afectado por la anemia por lo que debería ser más fácil de interpretar su sensibilidad al aumentar la VSG por el fibrinógeno es igual que la del método de Westergrem, es tal vez el mejor método de VSG para detectar una enfermedad oculta y una elevación mínima en la VS, sin embargo, se aplana algo entre los niveles moderados y notablemente elevado. Este CZS que sólo requiere 100 microlitros de sangre es considerablemente más rápido. El intervalo de referencia oscila entre 41 y 54 para ambos sexos. El CZS ha demostrado ser una alternativa satisfactoria al VSG en un número reducido de estudios clínicos. MÉTODO MICRO VSG Barrett (1980) describió un método utilizando 0'2 mm de sangre para llenar un tubo de 1 sólo uso de plástico de 230mm de largo y 1mm de luz interna. Los valores de sangre capilar se correlacionaba bien con la sangre venosa en micro VSG por Westergrem. Este método se puede mostrar relativamente útil para enfermos pediátricos. INTERPRETACIÓN DE LA VSG Causas fisiológicas en el que aumenta la VSG es en el embarazo y el envejecimiento. Situaciones patológicas en las que la VSG tiende a elevarse notablemente son en los trastornos de las proteínas plasmáticas como es el mieloma múltiple o la macroglobulinemia. En las hiperglobulinemias policronales graves debidas a enfermedades inflamatorias y en las hiperfibrinogenemias. Los incrementos moderados resultan corrientes en los casos de enfermedad inflamatoria activa, tales como artritis, reumatoides, infecciones crónicas, enfermedad del colágeno y enfermedad neoplásica (cáncer): • La VSG tiene poco valor diagnóstico en estos trastornos pero puede resultar útil par monotorizar la actividad de una enfermedad. Es una prueba más simple que la determinación de las proteínas séricas que tiende a reemplazarla. Aunque a menudo es una prueba normal en pacientes con neoplásicas, tejido conectivo e infecciones, una VSG normal no puede utilizarse para excluir estas actividades diagnósticas. • La VSG sirve en la detención de enfermedades de pacientes asintomáticos como síntomas de alarma. La historia y la exploración física y la utilización de otras pruebas diagnósticas deberán revelar la causa de la elevación de la VSG. • La VSG es útil y está más indicada para establecer el diagnóstico y monotorizar la artritis de la temporal (arteria) y la polimialgia reumática. En la enfermedad de Hodgkin la VSG puede ser una determinación pronóstica de mucha utilidad en ausencia de síntomas sistemáticos. En un estudio 1/3 de los pacientes asintomáticos presentaban una VSG inferior a 10mm/h y una excelente tasa de superviven-cia independientemente de la edad, el estadio y la histopatología. Los paciente asintomáti-cos con una VSG de 60mm/h o más tenían una tasa de supervivencia tan mala como los enfermos con síntomas sistemáticos. En los casos en los que hay una disminución de la VSG tenemos: policitemia vera (verdadera), alteraciones congénicas eritrocitarias (esferocitosis, drepanocitosis), insufi-ciencia cardiaca congestiva (ICC) Esquema Aumento de la VSG: 1·- Fisiológicas A·- Embarazos B·- Envejecimiento 2·- Patológicas A·- Procesos inflamatorios crónicos • Polimialgias reumatódica • Artritis reumatódica • Tuberculosis B·- Inflamación renal C·- ð patías monoclonales (mielomas) D·- Neoplasias y hemopatías malignas E·- Anemia intensa Disminución de la VSG 1·- Policitemia vera 2·- Alteraciones congénitas eritrocitarias (esferocitosis, drepanocitosis) 3·- Insuficiencia cardiaca congestiva 4·- Hipofibrinogemia. TEMA VIII HEMOGLOBINA La hemoglobina es una proteína conjugada que sirve para el transporte de o2 y co2. Cuando está totalmente saturadas, cada gramo contiene alrededor de 1'4ml de O2. La masa total de eritrocitos de un adulto contiene unos 600gr de hemoglobina capaces de transportar 800ml de O2. Una molécula de Hemoglobina consta de 2 pares de cadenas polipeptídicas (unión de aminoácidos) (globina) y 4 grupos prostéicos HEM que contienen cada uno un átomo de Fe en estado ferroso. Las cadenas peptídicas que son iguales 2 a 2 adoptan una posición helicoidal; lo que da a la molécula de Hemoglobina una estructura esteroide. Cada punto HEM se localiza en una zona determinada de una de las cadenas de polipéptidos. Localizado cerca de la superficie de la molécula, el HEM se combina de forma reversible con una molécula de O2 o de CO2. Este grupo HEM es el responsable del color rojo de la Hemoglobina. La parte proteica o globina tiene 4 cadenas polipeptídicas que se denominan con las letras griegas ð, ð, ð, δ y ð. Se diferencian unas de otras en el número o posición (de los aminoácidos de los que están compuestas). Por lo tanto la existen varios tipos de hemoglobinas. En el ser humano se pueden encontrar las siguientes Hemoglobina normales: HEMOGLOBINA A: consta de 2 cadenas ð y 2 cadenas ð. En un adulto normal corresponde a más del 95% del total. HEMOGLOBINA A2: consta de 2 cadenas ð y 2 δ. En un adulto sano está en proporción menor al 3%. HEMOGLOBINA F ó fetal: consta de 2 cadenas ð y 2 ð. Es la Hemoglobina principal en el feto desde el 4º mes de embarazo hasta aproximadamente los 6 meses de edad. La HB F tiene mayor afinidad por el O2 por lo que capta el de la Hemoglobina madre. HEMOGLOBINA Gower: existen 2 tipos de esta Hemoglobina en el embrión. Desaparece casi por completo en el tercer mes de embarazo y empieza a aparecer la HB F. Abreviaturas: Hb Hemoglobina reducida HbO2 OxiHemoglobina COHB CarboxiHemoglobina SHB SulfoHemoglobina SHBCo CarboxisulfoHemoglobina Hi Hemiglobina ó metahemoglobina HCN Hemoglobincianuro ó cianmetahemoglobina La principal función de la hemoglobina es el transporte del O2 de los pulmones donde la tensión es elevada hacia los tejidos donde es baja. A una tensión de O2 de 100mmHg en los capilares pulmonares del 98% de hemoglobina se combina con el O2. En los tejidos donde la tensión de O2 puede descender hasta 20 mmHg el O2 se disocia fácilmente de la hemoglobina; en este caso menos de un 30% del O2 puede permanecer combinado con la hemoglobina. La hemoglobina educida es hemoglobina con su hierro no asociado a no unido al O2. Cuando cada grupo HEM se asocia con una molécula de O2, la hemoglobina correspon-de a la oxihemoglobina. Tanto en la hemoglobina como la oxihemoglobina el hierro permanece en estado ferroso Fe+2. Con el hierro oxidado al estado férrico Fe+3 se forma Hi y la molécula pierde la capacidad de transportar el O2 y/o el CO2. La anemia que e una disolución de la concentración de hemoglobina por debajo de lo normal y casi siempre del número de eritrocitos o del hematocrito constituye una alteración muy corriente y una frecuente complicación de otras enfermedades. El diagnóstico clínico de la anemia basado en la estimación del color de la piel y de las mucosas visibles es de poca garantía. Para complicar más la anemia suele estar enmascarado en muchos procesos por otras manifestaciones. Por estas razones la estimación correcta de la hemoglobina es importante y una de la pruebas habituales que debe llevarse a cabo en casi todos los pacientes. La anemia no constituye una enfermedad en sí misma sino que debe ser considerada como un síndrome derivado de algún trastorno en algún sistema. Quiere esto decir que siempre que aparezca una anemia debe buscarse otra enfermedad o trastorno primario que la motive. No obstante todas las anemias coinciden en la aparición de unos síntomas y signos clínicos que la caracterizan y que se presenta en todo caso. A una concentración de hemoglobina de 16gr por 100ml se a convenido en darle el valor arbitrario de 100%. Los valores normales en un adulto varón son de 13-18gr por 100ml y en una mujer adulta de 11-16gr por 100ml. Si la concentración de hemoglobina es superior a estos valores nos encontramos en un proceso anémico. DERIVADOS DE LA HEMOGLOBINA Las 2 hemoglobinas son la Hb y HbO2 se convierten fácilmente en una serie de compuestos por la acción de ácidos álcalis, sustancias reductoras u oxidantes, el calor u otros agentes. Hi (Hemiglobina ó metahemoglobina): es un derivado de la hemoglobina en la que el hierro en está en estado ferroso se convierte en estado férrico y es incapaz de combinarse de manera reversible con el O2. Sin embargo hasta el 1'5% de la hemoglobina de una persona normal es Hi. Sin embargo los aumentos de Hi causarán cianosis y anemia funcional sí son bastante elevados y simplemente cianosis en concentraciones bajas. En general siempre se forman pequeñas cantidades de Hi pero son producidas dentro del eritrocito. El más importante es el sistema metahemoglobin-reductasa dependiendo de NADH (es lo mismo que NADHcitocromob5reductasa). Otros sistemas que funcionan, sobre todo con sistemas de reserva son el ácido ascórbico; glutación reducción, NADH-metahemoglobín-reductasa. Esta última requiere un cofactor natural o un colorante autoxidante como el azul de metileno para ser activa. La metahemoglobilemia es un aumento de la hemoglobina en los eritrocitos es el resultado de un aumento en la producción de Hi o bien de una disminución de la actividad de la NADH citocromob5 reductasa y puede ser hereditaria o adquirida. 1·- Forma Hereditaria: se divide en 2 categorías principales. a) En la primera la metahemoglobile-mia se debe a una disminución de la capacidad del eritrocito para reducir la Hi que se forma constantemente a partir de la hemoglobina. La mayoría de las veces se debe a un déficit de la enzima NADH citocromob5 reductasa y se hereda con carácter autosómico recesivo. El homocigoto tiene unos niveles de Hi del 10 al 50% y su aspecto es cianótico (de color azulado). Sólo de forma ocasional presenta policitemia como mecanismo de compensación. La concentración de hemoglobina del 10-25% puede no mostrar. Los niveles de 35-50% producen síntomas leves como disnea de esfuerzo (ahogo) y cefaleas y las que exceden de 70% son probablemente letales. El tratamiento con ácido ascórbico ó azul de metileno en esta forma de metahemoglobulemia hereditaria reducirá el nivel de Hi aparentes por activación del sistema NADPH metahemoglobin-reductasa. Heterocigoto (menos grave) presentan niveles intermedios de actividad NADH citocromob5reductasa y niveles sanguíneos normales. Sin embargo pueden llegar a ser cianóticos tras la exposición a productos por fármacos oxidantes en cantidades que no afectarían a individuos normales. b) La segunda categoría: en ella los sistemas de reductores dentro de la hemoglobina dentro del eritrocito son normales pero las estructuras de la molécula hemoglobínica es anormal: una determinada alteración genética en la composición de los aminoácidos de las cadenas ð ó ð pueden formar una molécula de hemoglobina que tiene una tendencia aumentada a la oxidación y una capacidad reducida de que la hemiglobina o metahemoglobina se reduce a hemoglobina. Se han identificado sulfohemoglobina aberrantes cuya consecuencia es la cianosis asintomática debida a metahemoglobulemia; se denomina formas diversas de la hemoglobulemia. Se heredan como genes autosómicos dominantes y la terapéutica con azul de metileno no es efectiva. 2·- Metaglobulinas adquiridas: las presentan las mayorías de las metaglobulemias, son debidas principalmente a la exposición a fármacos o productos químicos que producen un aumento de la formación de la metaglobulina. Entre ellos destacar los nitritos, nitratos, cloratos y quironas. Otras sustancias como las anemias cromáticas y los compuestos nitrogenados actúan probablemente de forma indirecta a través de un metabolito dado que in vitro no producen la formación de metahemoglobina. Dichas sustancias incluyen la fenacetina, las sulfamidas,... Los niveles de fármacos o sustancias químicas que no producirían una metaglobulemia significativa en el individuo normal podrían producirla en un individuo con una leve disminución de la actividad NADH citocromob5reductasa estos individuos son los recién nacidos y las personas heterocigóticas para el déficit de esa enzima. La hemoglobina se combina irreversiblemente con varios productos químicos por ejemplo: cianuro, sulfuros, peróxidos, fluoruros, etc. Por su gran afinidad con el cianuro las intoxicaciones cianúricas deben tratarse con nitritos para que la hemoglobina se transforme en metaglobulina que se combinará luego con el cianuro. De esta manera el cianuro, que es extremadamente tóxico para los enzimas respiratorios celulares, se vuelve menos tóxico al transformarse en cianmetahemoglobina. La hemoglobina y la sulfohemoglobina se cuantifica por espectrofotometría. Si la metahemoglobina está elevada se debe eliminar en primer lugar los fármacos o sustancias tóxicas como posible causa. Para determinar si la metahemoglobina es debida al déficit de la NADH citocromob5 reductasa se debe realizar el análisis dela enzima. Una hemoglobina anormal (hemoglobulemia) también puede ser responsable de la metahemoglobulemia observada al nacimiento o en los primeros meses de vida. Sulfohemoglobina (SHb): es una mezcla de formas de hemoglobina oxidadas parcialmente desnaturalizadas que se producen en al hemólisis oxidativa. Durante la oxidación de la hemoglobina el sulfuro que puede proceder de diferentes fuentes se incorpora a los anillos Hem de la hemoglobina con el resultado de un hemocromo verdoso. Una oxidación posterior suele producir como resultado la desnaturalización y precipitación de la hemoglobina en forma de cuerpo de Hem. La sulfohemoglobina no puede transportar oxígeno pero su combinarse con CO para formar carboxihemoglobina no se puede volver a reducir a hemoglobina y permanece los corpúsculos hasta que se disgregan. La sulfohemoglobina se ha observado en pacientes tratados con sulfoamidas o con amidas aromáticas como por fenacetina. Así como en pacientes con estreñimiento grave, en caso de bacteriemia debido a Clostridium Prefringens en un proceso conocido como cianosis enterogénica. La concentración de sulfohemoglobina in vivo suele ser baja alrededor al 1% y raramente sobrepasa el 10% de la hemoglobina total cuando de lugar a cianosis y suele ser asintomática. La carboxihemoglobina (SHBCo): el CO, endrógeno producido en la degradación del Hem a bilirrubina suele constituir alrededor del 0'5% de la carboxihemoglobina en sangre, pero se incrementa en la anemia hemolítica. La hemoglobina suele combinarse con CO en la misma proporción que con O2, sin embargo la afinidad de la molécula de hemoglobina por el CO es 210 veces mayor que por el O2. Esto significa que el CO se fijará a la hemoglobina aunque su concentración en el aire sea sumamente baja por ejemplo desde 0'02%. En estos casos se va formando cada vez más carboxihemoglobina no puede fijar el O2 y por tanto no sirve para transportarlo. A un paciente intoxicado por CO se le debe administrar O2 puro para que la carboxihemoglobina se transforme en oxihemoglobina la carboxihemoglobina es fotosensible tiene un color rojo cereza brillante típico. La intoxicación aguda por CO se conoce bien desde hace mucho tiempo (combustión en presencia de poco O2). Sin embargo la intoxicación crónica debida a una exposición prolongada a pequeñas cantidades de CO se conoce menos pero adquiere cada vez más importancia. Sus causas más corrientes son los motores de gasolina, el gas de iluminación, los calentadores de gas, estufas y hornos defectuosos y el humo de tabaco. La exposición al CO es por tanto uno de los riesgos de la civilización moderna. En las calles más frecuentadas de las grandes ciudades se ha encontrado que el aire tiene una concentración de gas suficiente como para causar síntomas leves en personas que permanezcan durante largos periodos de tiempo en ellas. Por ejemplo: agentes de tráfico. Al exposición crónica al CO a través del humo del tabaco puede provocar una elevación crónica del nivel del carboxihemoglobina para los fumadores, de modo que tienden a presentar hematocrito más altos que los no fumadores y pueden presentar policitemia. Los individuos sanos que han sido expuestos a diversas concentraciones del gas durantes una hora no presentan síntomas definidos (cefalea, vértigo, debilidad muscular y náuseas) a menos que la concentración del gas en la sangre llegue al 20% o al 30%. Mientras que la intoxicación crónica, sobre todo en niños, pueden aparecer síntomas graves con concentración más bajas. La ferroxihemoglobina: se puede cuantificar por espectrofotometría diferencial o por cromatografía de fases. PRUEBAS PARA DERIVADOS DE HEMOGLOBINA. Algunos datos se obtienen examinando a simple vista una muestra de sangre. Un aspecto normal del suero o del plasma revela que el pigmento está en los glóbulos rojos. Si se agita una sangre total normal en el aire durante 15 minutos adquiere un color rojo claro por convertir la hemoglobina en oxihemoglobina. La sangre tiene un color rojo cereza brillante cuando el pigmento es carboxi-hemoglobina en la intoxicación por CO. El color es de chocolate en la metahemoglobu-lemia y la banda malva en al sulfohemoglobulemia. INDENTIFICACIÓN ESPECTROFOTOMÉTRICA Las distintas hemoglobinas tienen espectros de absorción características que se determina en un espectrofotómetro. La identificación de las diferentes formas de la hemoglobina con la determinación de sus espectros de absorción puede hacerse de una manera muy sencilla: se coloca en un tubo de ensayo aproximadamente la mitad de una gota de sangre y se diluye 20ml de agua bidestilada. Se examina la muestra en un espectrómetro empleando agua como blanco y se lee la absorción a intervalos de 5 nm. DETERMINACIÓN DE LA CONCENTRACIÓN DE HEMOGLOBINA. Se considerarán los métodos de cianometahemoglobina (hemiglobincianuro; HiCN), el que mide el contenido de hierro. El método del HiCN, recomendado por el International Comité for Standardizationin Hematology (ICSH, 1978), presenta la ventaja de ser cómodo y de ser una solución estándar estable, fácilmente disponible. Método del hemiglobincianuro (HiCN) Principio: se diluye la sangre en una solución de ferrocianuro potásico y cianuro potásico. El ferrocianuro potásico oxida las hemoglobinas a hemiglobinas (Hi; metahemoglobinas), y el cianuro potásico proporciona los iones cianuro (CN-) para formar hemiglobincianuro (HiCN, cianmetahemoglobina), que tiene una absorción máxima amplia a una longitud de onda de 540 nanómetros. La capacidad de absorción de la solución se mide en un fotómetro o espectrofotómetro a 540 nm y se compara con la de una solución de HiCN estándar. Reactivo: el disolvente de Drabkin modificado con detergente: ferrocianuro potásico 0'2gr, cianuro potásico 0'05gr, fosfato potásico dihidrogenado (anhidro) 0'14gr, detergente no iónico (por ejemplo serox) 1 ml y por último agua destilada 1.000ml. La solución debe ser de color amarillo claro y pálido, tener un pH de 7 a 7'4 y dar una lectura de 0 cuando se miden en fotómetros a 540nm frente a un patrón de agua. La sustitución del KH2PO4, en este reactivo por bicarbonato sódico (NaHCO3) acorta el tiempo necesario en el reactivo original de Drabkin para la conservación total de hemoglobina a CNHi de 10 a 13 minutos. El detergente aumenta la lisis de los eritrocitos y disminuye la turbidez de la precipitación proteica. Se debe tener cuidado con el KCN en la preparación de la solución de Drabkin, ya que las sales o soluciones de cianuro son venosas. El disolvente contiene sólo 50mg/l de KCN, menos que la dosis letal para una persona de 70Kgr. Sin embargo, ya que se libera HCN por acidificación, se debe evitar la exposición del disolvente a ácidos. Es aconsejable deshacerse de los reactivos y muestras a través del agua corriente del fregadero. El disolvente se conserva bien en frasco oscuro a la temperatura del laboratorio, pero se debe renovar cada mes. Método: se añaden 20microlitos de sangre a 5 ml de disolvente (1:251), bien mezclados y se mantienen a temperatura ambiente durante al menos 3 minutos. Se determina la capacidad de absorción, frente al blanco reactivo, en el colorímetro fotoeléctrico a 540nm o con un filtro apropiado. Se abre entonces un vial de HiCN estándar y se mide la capacidad de absorción, a temperatura ambiente, en el mismo aparato de una manera similar. La muestra se debe analizar a las pocas horas la dilución. El estándar se mantendrá en oscuridad cuando no se utilice y se desechará al término del día. Hb (g/dl)= (muestra de prueba A540/estándar A540) X ([estándar(mg/dl)] X 251/1.000(mg/gr) Suele ser conveniente calibrar el fotómetro al utilizarse para hemoglobinometría preparando una curva estándar o una tabla que relaciona la capacidad de absorción a la concentración de Hb en g/dl. La capacidad de absorción del estándar HiCN fresco se mide frente a un blanco reactivo. Se hacen las lecturas de la capacidad de absorción del estándar HiCN fresco y de sus diluciones en el reactivo (1 en 2, 1 en 3, 1 en 4) frente al blanco reactivo. Los valores de Hb en g/dl se calculan para cada solución, como se vio anteriormente. Cuando las lecturas de capacidad de absorción se llevan a un papel gráfico lineal como ordenadas frente a la concentración de Hb en las abscisas, los puntos deben describir una línea recta que pasa por el origen. A partir de esta curva estándar se puede preparar una tabla que da las concentraciones de Hb para las lecturas de capacidad de absorción. * más sencillo: material y reactivos: tubos de ensayo, pipetas de 5ml y 0'02ml, matraz aforado de 1 litro, espectrofotómetro; reactivo de Drabkin, puede encontrarse de forma concentrada o ya diluida. En el 1º caso son viales de 20ml que hay que diluir para obtener 1litro. Se guarda a temperatura ambiente y protegida de la luz; patrón de Hb: viene indicada su concentración, pero suele ser de 15g/100ml de sangre. Los patrones tienen caducidad, se guardan en nevera, pero no se congelan. 3 tubos de ensayo: blanco (5ml reactivo de Drabkin) patrón (5ml reactivo + 0'02ml de sangre patrón) problema (5ml reactivo + 0'02ml sangre) se deja reposar 3 minutos. Y se miden las densidades ópticas a 540nm, de modo que: Hb(g/ml) = (absorbancia problema/ absorbancia patrón) X 15 * La ventaja del método del HiCN es que se miden la mayoría de las formas de hemoglobinas (Hb, HbO2, Hi y HbCO, pero no SHb). La muestra de la prueba se puede comparar directamente con el estándar HiCN, y las lecturas se pueden hacer según la conveniencia del técnico, gracias a la estabilidad del as muestras diluidas. El aumento en la capacidad de absorción no debido a hemoglobina puede estar originado por la turbidez causada por proteínas plasmáticas anormales, hiperlipemias, grandes leucocitos (recuentos superiores a 30x109/l) o gotitas de grasa, factores todos ellos que pueden dar lugar a un incremento en la dispersión de luz y de la capacidad de absorción aparente. Método de la oxihemoglobina (HbO2): ya no se utiliza ampliamente, pero todavía constituye un método satisfactorio la determinación de la hemoglobina como oxihemoglobina. La principal desventaja reside en que no se dispone de un estándar estable para la HbO2. Debido a la sencillez del método, muchas veces se utiliza para comparar niveles de hemoglobina en los casos en los que no se precisa la cantidad absoluta, como, por ejemplo, en las determinaciones de fragilidad osmótica o de HbA2. El método de HbO2 no mide la carboxihemoglobina (HbCO), la metahemoglobina (Hi) o la sulfohemoglobina (SHb), todas las cuales son inactivas para el transporte de oxígeno. Se hace una dilución 1:251 de sangre en NH4OH 0'007N. El agua empleada para preparar la solución amónica debe destilarse en columna, porque cantidades mínimas de cobre en el agua destilada o en otros diluyentes utilizados en las determinaciones de HbO2 pueden transformarla en metahemoglobina y reducir los valores. Agítese bien para asegurar la mezcla y la oxigenación de la hemoglobina. La solución se lee en un fotómetro en que se emplea un filtro verde (540nm) y una solución 0'007N de hidróxido amónico como blanco. La prueba puede leerse transcurridos algunos segundos o, si está en una cubeta tapada, en cualquier momento antes de pasar 3 días. La curva estándar puede trazarse por uno de los procedimientos que se expondrán más adelante. Métodos químicos (contenido en hierro): la hemoglobina puede medirse por determinación de su contenido en hierro. En la sangre, el hierro no hemoglobínico es insignificante en comparación con el hemoglobínico. El hierro debe separarse primero de la hemoglobina, generalmente por medio de un ácido o por incineración, y entonces es titulado con TiCl3 o forma un complejo con un reactivo que desarrolle un color susceptible de medirse fotométricamente. Basado en la estructura molecular, el contenido de hierro de la hemoglobina es de 0'347%. La concentración de hemoglobina en sangre (g/dl) por 3'47. la determinación de la concentración de la hemoglobina por medición del contenido en hierro es demasiado complicada para el trabajo habitual, pero se puede utilizar para verificar otros métodos, y a partir de ella se pueden diseñar curvas de calibración para los métodos de HbO2 y HiCN. Se usa para estandarización en hemoglobinometría si se desea o si no están disponibles estándares de cianometahemoglobina certificados. El método del hierro, por supuesto, mide la hemoglobina total; el método de HbO2 mide solo Hb y HbQ2 y el método de HiCN determina la Hb, HbO2, Hi y HbCO. Errores en hemoglobinometría: los errores pueden depender de la muestra, el método, el equipo o el operador. Algunos han sido comentados al describir los distintos métodos. Errores inherentes a la muestra: una punción venosa inadecuada puede producir hemoconcentración, que aumentará la concentración de hemoglobina y el recuento de células hemáticas. Una técnica inadecuada en la obtención de muestras por punción del dedo o capilares puede producir errores en ambos sentidos. Errores inherentes al método: el método de la oxihemoglobina mide la Hb y la HbO2, pero no la Hi, HbCO o SHb. Por tanto, es el método que mejor determina la hemoglobina fisiológicamente activa, lo que puede tener importancia en algunos enfermos. El método de la HiCN es actualmente el de elección. El empleo del estándar de HiCN para la calibración del instrumento y para la prueba en sí elimina una de las fuentes mayores de error y proporciona una comparabilidad entre todos los laboratorios que lo emplean. La ancha banda de absorción de la HiCN en la región de 540nm permite que se emplee tanto en los fotómetros de filtro como en los espectrofotómetros de banda estrecha. Con excepción de la SHb, todas las variedades de la hemoglobina se convierte en HiCN. Errores inherentes al equipo: la exactitud del equipo no es uniforme. Es conveniente una pipeta bien graduada y con una precisión garantizada de menos de 1% de error. La calibración de pipetas de vidrio reutilizables. Puede producirse un error importante por el empleo de cubetas no seleccionadas. Son preferibles las cubetas intercomunicadas, debido a que eliminan el pequeño error existente, incluso cuando se utilizan cubetas bien calibras. El fotómetro o colorímetro debe calibrarse en el laboratorio antes de su empleo inicial y revisarse con frecuencia. Los dispositivos de longitud de onda, los filtros y las lecturas métricas requieren un control. Si se emplea un fotómetro bien estandarizado y regularmente revisado, el error del método de la cianometahemoglobina puede reducirse hasta +2% (expresado como coeficiente de variación, CV). Errores del operador: la mayoría de los errores por el llamado factor humano son los mismos en todos los procedimientos técnicos. Pueden reducirse mediante un buen adiestramiento, una comprensión profunda de la significación clínica de la prueba y de la necesidad de un método de confianza, el cumplimiento de las instrucciones orales y escritas sobre los principios del método y la familiarización con el equipo y con las causas de error. El técnico ha de estar experimentado en la volumetría de precisión y familiarizado con el rendimiento de su instrumento para reconocer sus fallos. Se ha demostrado que los errores aumentan con la fatiga y tienden a ser mayores al final del día que al principio. Un operador que por naturaleza y por adiestramiento es paciente y crítico, y que se interesa por su trabajo, estará menos expuesto a cometer errores que otros. La descripción anterior se aplica a las técnicas manuales de hemoglobinometría. Hoy se utilizan ampliamente equipos semiautomáticos y automáticos que tienen la propiedad de eliminar los componentes de error en las pipetas y cubetas individuales y gran parte de los errores humanos. TEMA IX ÍNDICES ERITROCITARIOS SECUNDARIOS Los índices eritrocitarios secundarios son los que relacionan el hematocrito con el número de hematíes y la concentración de hemoglobina denominados a su vez índices eritrocitarios primarios. Son fundamentalmente 3: volumen corpuscular medio, hemoglo-bina corpuscular media y concentración corpuscular media de hemoglobina. Su determina-ción es muy útil en la valoración y orientación diagnóstica de las anemias y pueden calcularse a partir de los 3 índices eritrocitarios primarios. En la actualidad la mayoría de los autoanalizadores que se emplean en hematología suministran todos los índices eritrocitarios de forma sistemática. Volumen corpuscular medio: es el valor medio del volumen de cada hematíe se calcula a partir del hematocrito y del número de hematíes expresados en número de hematíes/l. Se expresa en fentolitro. VCM = ----------------------- Hemoglobina corpuscular media: expresa el valor medio del contenido de hemoglo-bina que existe en cada hematíe. Se determina mediante el cociente entre el valor de la concentración de hemoglobina de sangre total (gr/l) y el número de hematíes por litro. Se expresa en picogramo (pg = 10-12gr) HCM = --------------------------- Concentración corpuscular medio de hemoglobina: corresponde a la concentración en un decilitro de hematíes y se calcula a partir de la concentración de hemoglobina partida de litro de sangre y del valor del hematocrito. CMB = ----------------------- VALORES NORMALES VCM CMH CCMH Recién nacidos 1 día 105 35 36'0 Recién nacidos 1 semana 103 36 25'0 Niños de 1 mes 90 30 34'0 6 - 4 años 78 27 - 25 33 - 32 10 años 82 27 24 Adulto 88 + 10 29'5 + 2'5 32'5 + 2'5 Variaciones patológicas en las formas más frecuentes de anemia. VCM CMH CCMH A. normocítica 81 - 98 27 - 33 31 - 34 A. ferropénica 64 - 74 20 - 24 39 - 33 Talasemia 37 - 67 18 - 22 32 - 34 Esferocitosis hereditaria 78 - 90 28 - 32 24 - 36 A. macrocítica > 98 > 33 > 34 Así el VCM permite establecer una primera orientación etimológica; la causa de la anemia al clasificarse en anemia microcítica (<80 fl), la macrocítica (> 100 fl) y la anemia normocítica (82 95). La CCMH permite diferenciar la anemia hipocrómica o férrica. Cuando la CCMH es < 30 pg de la que se acompaña de un contenido hemoglobínico por hematíe normal o incluso elevado. Las anemias microcíticas que se suelen acompañar de hemoglobina y un descenso de la CMH constituyen la alteración más frecuetne en hematología y pueden obedecer a 2 mecanismo principales: El más frecuente es la ferropenia o carencia de hierro. Talasemia menor o enfermedad congénita (hereditaria) caracterizada por el descenso de la síntesis de una de las cadenas de hemoglobina. El carácter inmensamente microcítico de la talasemia y su elevada frecuencia entre los pobladores del área mediterránea han hecho de la determinación sistemática del VCM, mediante autoanalizadores, un buen método para el escrutinio (diagnóstico) de esta hemoglobinopatía. Las anemias macrocíticas: obedecen generalmente a una carencia de alguno de los factores madurativos (vitamina B12 o ácido fólico). No obstante la determinación sistemática del VCM en los grandes hospitales ha permitido conocer otras causas de las hepatopatías y del alcoholismo en la que la determinación del VCM ha sido considerada por algunos autores como un buen índice de ingesta crónica de alcohol. El aumento de reticulocitos puede ser también una causa de macrocitosis debida al mayor tamaño de reticulocitos comparado con el de hematíes maduros. La determinación de la CCMH mediante métodos electrónicos tiene escaso valor práctico en el diagnóstico de las anemias hipocrómicas pero es útil para detectar aumentos del contenido hemoglobínico intraeritrocitario ya que en este caso presenta prácticamente un valor superior al normal. El aumento de la CCMH puede obedecer a una disminución de la relación entre la superficie y el volumen eritrocitario (ej.: esferocitosis) o puede ser debido a una pérdida excesiva de agua por parte del hematíe (ej.: deshidratación eritrocitaria) pero nunca a un aumento de la síntesis hemoglobínica. Morfológicamente se pone con facilidad de manisfiesto por la aparición de hematíes intensamente teñidos (hipercromos) cuando se observa en un frotis sanguíneo teñido mediante coloración panótica convencional. Otros índices eritrocitarios son RDW = ADE es e lancho de la distribución eritrocitaria. Es un índice de la variación del tamaño de los hematíes y se relaciona con la anicocitesis observada en las extensiones al microscopio, que sirve para diferenciar entre la anemia ferropoénica y la talasemia. Volumen plaquetario medio (VPM): es el volumen medio de las plaquetas que se expresa en fentolitros. Plaquetario (PTC): es la relación entre el volumen plaquetario y el volumen total de sangre y se expresa en porcentaje. Ancho de distribución de plaquetas (ADP): es un índice de la variabilidad en el tamaño de las plaquetas. También se expresa en porcentaje. TEMA X REALIZACIÓN DEL FROTIS SANGUÍNEO La práctica de un frotis sanguíneo también llamado extensión es de gran importancia en hematología ya que el diagnóstico de muchas enfermedades hematológicas puede realizarse con solo observar las características morfológicas de las células sanguíneas Debido a ello la técnica de realización del frotis tiene que ser impecable procurando que este no sea excesivamente fino ni excesivamente grueso. Con ello se conseguirá una distribución adecuada de las células en las diferentes áreas del frotis cuyo conocimiento es fundamental tanto para la observación de la morfología eritrocitaria como para la realización de la fórmula leucocitaria o recuento diferencial de leucos. Existen 3 procedimientos para realizar el frotis sanguíneo: Método de los 2 portaobjetos o portas o de la cuña Método de los 2 cubreobjetos Método automático mediante extensor de frotis por centrifugación. En cualquiera de estos métodos se tendrán en cuenta las siguientes precauciones: 1·- Todo el material que hay que utilizar tiene que estar escrupulosamente limpios y desengrasados. 2·- Si se parte de punción digital no se debe utilizar nunca la primera gota. 3·- Si se parte de punción venosa el frotis deberá hacerse con la máxima rapidez posible con sangre no tratada con anticoagulante. En caso de tenerlo que realizar con anticoagulante será el EDTA potásico, debido a su escasa acción sobre la morfología de las células sanguíneas. El frotis debe ser realizado siempre dentro de las 2 primeras horas de practicada la extracción ya que si no es así el efecto del anticoagulante sobre los leucocitos producen hinchamiento nuclear con falso aumento del número de neutrófilos no segmentados, vacuolización citoplasmática y aparición de diversos artefactos. Métodos de los 2 portas o cuñas: para conseguir una buena extensión de sangre es imprescindible utilizar portaobjetos limpios, secos y desengrasados. Estos portas se utilizan cogiéndolos siempre por los bordes. Se deposita sobre el porta que va a recibir el frotis una pequeña gota de sangre (5 microlitros) de no más de 3 mm de diámetro; obtenida por punción cutánea o por extracción venosa. La gota se sitúa sobre la línea central del porta aproximadamente a 1 cm de uno de sus extremos. Este portaobjetos se coloca sobre la mesa de trabajo encima de un papel de filtro con la gota a la derecha y se sujeta por el extremo opuesto con los dedos pulgar e índice de la mano izquierda. Para extender la sangre se utilizar otro portaobjeto esmerilado que sujetándolo con la mano derecha se sitúa delante de la gota de sangre de forma que los 2 portaobjetos formen un ángulo entre 30º y 45º y desplazarlo suavemente hacia atrás hasta que alcance la gota de sangre. Esperar a que por capilaridad se distribuya uniformemente deslizar suavemente y a velocidad moderada un porta sobre otro en sentido longitudinal hasta que la gota de sangre quede bien extendida sobre la superficie del primer portaobjetos. El grosor del frotis sanguíneo se puede variar según sea el ángulo que formen entre sí ambos portas. Así, si es superior de 45º la extensión obtenida será gruesa y corta. Por lo contrario, si el ángulo es muy pequeño será largo y fino. El secado del frotis se hará a temperatura ambiente y en posición horizontal. Una vez que la extensión está seca se enumera o se escribe el nombre del paciente y fecha en la parte más gruesa de la extensión. En las extensiones de sangre sobre portas diferenciamos una cabeza un cuerpo y una cola. La `cabeza' es la línea en la cual hemos apoyado un portaobjetos sobre todo antes de hacer la extensión. Es una zona excesivamente gruesa y en ella se aprecia un aumento de los linfocitos. El `cuerpo' representa la mayor parte de la extensión corresponde a la región intermedia del frotis y en ella existe un reparto equilibrado de las células. Es la zona ideal para hacer el recuento diferencial. La `cola' es una zona excesivamente fina. Corresponde al final de la extensión y termina con un área donde las células adoptan una disposición acordonada que se conoce como flecos o barbas. En esta región existe un exceso granulocitos y monocitos. Así mismo en todos estas zonas lo morfología erotricitaria puede variar ampliamente. En las zonas excesivamente gruesos los hematíes forman aglomerados. Debido a un mayor tiempo de secado. Mientras que en las zonas excesivamente finas los hematíes están casi siempre deformados y presentan una tonalidad uniforme no apreciándose la zona clara central. Debido a ello para apreciar la morfología eritrocitaria debe seleccionarse la zona ideal ya que correspopnde a aquella en que los hematíes se hayan bien separados entre sí y en cada uno de ellos se observa bien diferenciada la zona clara central. Limpieza de material: los portas sucios pueden limpiarse sumergiéndolos durante 24 horas en mezcla crómica [100 gr de dicromato potásico disuelto en 750 ml de agua a la cual se le ha añadido con cuidado 250ml de sulfúrico concentrado] transcurrido este tiempo se lavan los portas con agua corriente y se enjuagan con agua destilada y se guardan en alcohol de 95% o en una mezcla de alcohol-éter. Antes de utilizarlos se secan y se frotan con un paño limpio. Método de los 2 cubreobjetos: se toman 2 cubres de 22mm de lado sin grasa, limpios y secos. Primero se coloca una gota de sangre en el centro del cubre. Para ello se sujeta el cubre por dos ángulos contiguos entre los dedos pulgar é índice de una mano, se toca la gota de sangre sin tocar la piel, se coloca sobre un segundo cubre cruzado con respecto al primero de manera que los ángulos formen una estrella de 8 puntas. La sangre se entiende por capilaridad formando una película delgada y uniforme. En el momento de cesar de extenderse la sangre y antes de que empiece a coagularse separar los cubres rápidos y firmemente, tirando de ellos en el plano paralelo a sus superficies. Los cubres se deben colocar con la extensión hacia arriba sobre papel limpio y dejar que sequen al aire. O también se puede colocar unidos por su cara posterior en hendiduras, echar en una caja una vez secos; se tiñen y se montan, adquiriendo la sangre de la cara del cubre que contiene la sangre teñida a un porta en el que se ha depositado una gota de líquido de montaje. Método automático mediante un extensor de centrifugación o spinner: la extensión automática de la sangre sobre un porta se realiza mediante una centrífuga especial conocida como Spinner que permite obtener una vez finalizada la centrifugación una capa monocelular. Mediante este procedimiento se hace innecesaria la selección de un área ideal del frotis para realizar la fórmula sanguínea o la observación de la morfología eritrocitaria ya que la distribución de las células se realiza de forma irregular sobre toda la superficie del porta. Una pequeña desventaja, es la tendencia de los eritrocitos a presentar una palidez central excéntrica que recuerda los esferocitos. TINCIÓN DEL FROTIS SANGUÍNEOS. El frotis sanguíneo una vez seco se somete a un proceso de fijación y posteriormente a una tinción mediante a un colorante adecuado. En la mayoría de los laboratorios los colorantes más empleados para la tinción hematológica se basan en el de Romanowsky constituido fundamentalmente con la mezcla de eosina (ácida) y azul de metileno (básico). Además se ha incorporado el empleo de derivados por oxidación del azul de metileno que se conoce con el nombre de azures (A, B, C). Son los azures responsables de la coloración púrpura o roja de ciertas estructuras. Tanto la eosina como el azul de metileno son muy sensibles a las variaciones de pH de las diferentes estructuras celulares de forma que las que tienen carácter básico fijan la eosina mientras que las que poseen propiedades ácidas fijan principalmente el azul de metileno. Esto explica que las estructuras basófilas se tiñan de color azul mientras que los competente acidófilas adquieren un color rosado. Igualmente la diferente afinidad de ciertas granulaciones citoplasmáticas por dichos colorantes permite clasificar los leucocitos polimorfos nucleares en 3 grupos: A·- Granulocitos eosinófilos: en los que la granulación específica contiene sustancias de carácter básicos que fijan los colorantes ácidos y se tiñen de color rojo-naranja. B·- Granulacitos basófilos: en los que la granulación específica posee sustancias de carácter ácido que fijan los colorantes básicos y se tiñen de color azul oscuro. C·- Granulocitos neutrófilos: en los que la granulación específica posee compuestos de carácter neutros que fijan ambos colorantes simultáneamente. Debido a ello se tiñen de un color pardo. Los azures (A, B, C) son los responsables de la coloración púrpura o roja de la cromatina de los leucocitos y de ciertas granulaciones citoplasmáticas que por ello se denominan azurófilos. Dentro del grupo de colorante tipo Romanowsky los más utilizados son el de Giemsa, Wright, May- Grünwald- Giemsa: ~ Tinción de Giemsa: • Material: frotis sanguíneo, colorante de Giemsa (constituido por una mezcla de azul de metileno, eosina, y varios azures en dilución acuosa), alcohol metílico absoluto pero libre de acetona (metanol) microscopio con objetivo de inmersión y aceite de inmersión. • Método: 1·- Fijar el frotis en alcohol etílico absoluto de 3 a 4 minutos 2·- Sumergirlo verticalmente en una solución de Giemsa preparada extemporal-mente a partir de 1 volumen de la solución colorante más 9 volúmenes de solución tampón pBs (pH = 6'8) 3·- Esperar durante 10 minutos. 4·- Lavar el frotis con abundante agua destilada y dejarlo secar al aire libre. Una vez seco está listo para ser observada al microscopio. • Resultados: es frecuente encontrar en el método de Giemsa que la granulación esosinófila no presenta la tonalidad rojo-anaranjada característica. Esta dificultad puede prevenirse añadiendo una gota de fusina fenicada por cada 10ml de alcohol metílico utilizado. Las granulaciones neutrófilas y los hematíes se tiñen mal; sin embargo esta coloración permite diferenciar alguna forma de metamielocito que pueden ser confundidos con los monocitos, pues el protoplasma de los monocitos queda de color azulado y el de los metamielocitos de color rosa. ~ Tinción de Wright: • Material: colorante de Wright, tampón fosfato pH = 4, rejilla horizontal o cubeta de tinción o soporte o bien una cubeta de coloración con su cestillo. • Técnica: colocar el frotis secado al aire en una cubeta de tinción con la sangre hacia arriba añadir el colorante gota a gota hasta cubrir bien la preparación. Dejar actuar de 1 a 2 minutos. Añadir igual número de gotas de agua destilada o mejor de una solución tampón igual que las que se añadan después de colorante. Dejar actuar de 4 a 5 minutos. Lavar con agua destilada hasta que la extensión presente un aspecto rosado al examinarlo a simple vista. Limpiar el dorso del potaobjetos con una gasa humedecida en alcohol para eliminar cualquier resto de colorante. Secar al aire y observar con el microscopio con el objetivo de inmersión. • Resultados: los hematíes se observarán de color naranja o rosado, el citoplasma de los monocitos presentarán una tonalidad azul grisácea con gránulo finos rojizos y el de los linfocitos presentará varias tonalidades azules. Los núcleos de los linfocitos y neutrófilos aparecerán de color púrpura oscuro. Los núcleos de los monocitos de color púrpura algo más claros más bien lila. Los gránulos de los eosinófilos son de color rojo anaranjado intenso. Los gránulos de los basófilos púrpura azulado muy oscuro. Y los gránulos de los neutrófilos se aprecian de color lila. Las plaquetas toman color violeta o púrpura. • Observaciones: se en la preparación se observa precipitados de colorante puede ser debido a varias causas: lavado insuficiente al final del proceso, utilización de portas sucios, mala filtración del colorante, mala distribución del colorante sobre el porta por no mantener este en posición horizontal. Durante la tinción el tampón fosfato controla el pH del colorante. Si el colorante es demasiado ácida la extensión resultará demasiado rojiza y si por el contrario es demasiado alcalina la precipitación presentará un aspecto azulado. Si los frotis recién teñidos resultan demasiados pálidos se corrige aumentando el tiempo de tinción o disminuyendo el tiempo de lavado. ~ Tinción de May- Grünwald- Giemsa(panóptica): Es el resultado de combinar la tinción de Giemsa con la de May- Grünwald y se conoce como tinción panóptica. Esta tinción resulta de manera especial las granulaciones leucocitarias y mejora la coloración de los hematíes. • Material: frotis sanguíneo seco, colorante de Giemsa, colorante de May- Grümwald (está constituido por una solución alcohólica de azul de metileno y eosina). • Método: 1·- Fijar el frotis en el porta sumergiéndolo en solución de M-G durante 2 o 3 min. 2·- Transferido a una dilución de May-Grünwald durante 2 o 3 minutos diluida en agua destilada o con pBs preparada extemporáneamente y dejarla uno 2 o 3 minutos. 3·- Sin lavar sumergir el frotis en la solución de Giemsa prepara extemporáneamente durante 20 minutos. 4·- Lavar el frotis con abundante agua destilada y sumergirlo en tampón pBs de pH = 6'8 de unos 2 a 5 minutos. 5·- Secar el frotis al aire y una vez seco está listo para su observación al microscopio. • Resultados: esta preparación proporciona una amplia gama de colores. Los hematíes se tiñen de un color rosa pálido con una zona central más clara. La policromía se advierte con una tendencia de color azulado. La cromatina nuclear se tiñe de violeta oscuro dejando dibujadas las estructuras cromáticas que sirven para ver el grado de madurez de la célula. Las granulaciones de los eosinófilos son rojo-anaranjado casi amarillentas. La de los basófilos son violeta muy oscuro y la de los neutrófilos rojo-púrpura Los linfocitos tienen pequeños granos azurófilos de color rojo. Las plaquetas presentan una porción periférica azulado y gránulos centrales rojos. El citoplasma de los linfocitos es casi siempre azul y el de los monocitos presenta un color ligeramente azulado. CAUSAS DE ERROR EN LA TINCIÓN DEL FROTIS SANGUÍNEO. Una extensión de sangre bien teñida debe caracterizarse por una buena diferenciación de las estructuras subcelulares de los leucocitos una coloración rosada de los hematíes y la ausencia de los precipitados. Los defectos más frecuentes observados en los frotis sanguíneo son: 1·- Una coloración excesivamente azul que puede ser debida a: ~ Excesivamente grueso el frotis ~ Lavado insuficiente ~ Tiempo prolongado ~ Empleo de colorante excesivamente alcalino 2·- Una coloración excesivamente rosada: en este caso el colorante , el tampón o el agua de lavado tiene un carácter demasiado ácido. 3·- Presencia de precipitados. En general obedecen a una acción excesivamente prolongada del colorante May-Grünwald o del colorante fijador; otras veces pueden evitarse mediante la filtración del colorante antes de su empleo. 4·- Apreciación de artefactos morfológicos debido al anticoagulante 5·- Apreciación de artefactos debido a suciedad, deterioro o presencia de grasa en el porta. 6·- Hidratación de los hematíes. - Tinción de May- Grünwald: El colorante de may-grüneald se vende ya preparado para su uso inmediato. - Técnica: la preparación secada al aire, pero sin fijar se tiñe durante 3 minutos con la solución de may-grünwald. Cubrir la preparación con igual volumen de agua destilada neutra que se haya puesto de colorante moviendo el porta para que se seque y dejar de 5 a 10 minutos. Lavar con agua destilada hasta que la preparación tome un color rojo-rosado; secar en posición vertical y observar con objetivo de inmersión. TEMA XI FUNCIONES DE LOS LEUCOCITOS Los glóbulos blancos o leucocitos, son las células sanguíneas con núcleo. Fueron teñidas por primera vez en el siglo XIX, por Ehrlich Pappenheim, clasificándolas por su coloración y características nucleares. Según su comportamiento ante el del colorante se clasifican en: • Acidófilos o eosinófilos: muestran afinidad por la parte ácida del colorante de modo que los gránulos citoplasmáticos aparecen de color rosa-anaranjado. • Basófilos: muestran afinidad por la parte alcalina del colorante con gránulos citoplasmáticos de color azul intenso. • Neutrófilos: los gránulos del citoplasma aceptan los 2 tipos de colorante dando al citoplasma una coloración azul-grisácea. Estos 3 grupos se incluyen dentro de las células granulares o granulocitos o polinucleares, y todos ellos tienen grupos segmentados o polinucleares. Las células no granulares (o agranulocitos o mononucleares) que son los linfocitos y monocitos (son células con un núcleo grande y citoplasma de color azul claro). CLASIFICACIÓN Y VALORES DE REFERENCIA. Granulocitos: Basófilo Neutrófilo Cayado Eosinófilo Agranulocitos: Linfocito Monocito Valores de referencia en sangre periférica: el número total de leucocitos en condiciones normales según la edad, desde el nacimiento hasta los 16 años (edad en la que se estabiliza). CN 1 mes 4 años 6 años Adulto Neutrófilo 39 - 57 % 25 - 35 % 25 - 45 % 45 - 50 % 50 - 65 % Cayado 0-4% 0-4% 0-4% 0-4% 0-4% Eosinófilo 1-5% 1-5% 1-5% 1-5% 1-5% Basófilo 0-2% 0-2% 0-2% 0-2% 0-2% Monocitos 4 - 10 % 4 - 10 % 4 - 10 % 4 - 10 % 4 - 10 % Linfocito 25 - 35 % 45 - 65 % 40 - 65 % 40 - 45 % 25 - 40 % Nº leucos total/mm3 9.000-30.000 5.000-21.000 5.500-15.500 4.000-10.000 NEUTRÓFILOS SEGMENTADOS El polinuclear Neutrófilo es el leucocitos predominante en la sangre periférica del adulto sano. Su tamaño es homogéneo de 12 a 14 micrómetros con un núcleo de cromatina compacta segmentado de 2 a 5 glóbulos conectado con puentes cromatínicos. Las células en cayado o en banda llamadas así por la característica forma de su núcleo son los granulocitos más inmaduros que pueden encontrarse en la sangre periférica de personas sanas. El citoplasma del Neutrófilo contiene 2 tipos de granulaciones: primarias son el 15% y secundarias que son el 85%. Esta granulaciones son de color púrpura y contiene numerosas enzimas como lisoenzimas, fosfatasas ácidas y alcalinas, ribo y desoxirribo-nucleasas, lactoferrina, etc. La membrana celular emite pseudópodos que permite al Neutrófilo moverse para cumplir su función fagocítica. El Neutrófilo se origina en la médula ósea a partir de la célula madre o Stem-Cell y después de diferentes estadios de maduración pasa al torrente circulatorio. La vida media de los neutrófilos desde sus precursores identificables en la médula ósea hasta la muerte del Neutrófilo oscila desde 10 a 14 días de los que apenas en 7 horas pasan a sangre periférica debido a que la función principal de los neutrófilos se realiza en los tejidos; además cuando el Neutrófilo pasa al tejido no vuelve a la circulación sino que cumple se misión y muere, siendo fagocitado por los macrófagos del sistema mononuclear fagocítico (es un conjunto de elementos celulares difundidos por todo el organismo, principalmente en el hígado (esta célula en el hígado se llama célula Kupffer), bazo, órganos linfáticos, médula ósea de función hematopoyética, fagocitaria, metabólica, inmunitaria, pigmentaria, etc. Cuando los monocitos salen de la médula ósea y pasan a la sangre todavía son células muy inmaduras. Al cabo de unas horas penetran en los tejidos donde cambian; se hinchan, desarrollan lisosomas y mitocondrias de forma que parece un saco con gránulos que se denomina macrófagos y son fagocitos mucho más potentes que los neutrófilos. Pueden fagocitar hasta 100 bacterias, el plasmodium, etc. Funciones de los neutrófilos: es principalmente una célula migratoria que está de paso en células periféricas y que efectúa casi todas sus funciones fuera de la circulación. Los neutrófilos que penetran en los tejidos son ya células maduras que pueden empezar inmediatamente la fagocitosis. Al acercarse a una partícula que va a ser fagocitada el Neutrófilo proyecta pseudópodos en todas direcciones a través de la misma. Los pseudópodos se unen entre si por el lado opuesto y se fusionan. Esto crea una cavidad cerrada que contiene la partícula fagocitada. Después esta cavidad se invagina hacia el interior del citoplasma y la porción de membrana celular exterior de la célula para formar una vesícula fagocítica que flota libremente dentro del citoplasma y a la cual atacan todas las enzimas contenidas en las granulaciones citoplasmáticas. Suelen poder fagocitar entre 5 y 20 bacterias antes de ser inactivadas y morir. La fagocitosis tiene varias etapas: 1º·- Proceso de quimiotactismo: (tendencia de las células a moverse en una dirección determinada por influencia de estímulos químicos) Consiste en la atracción de la célula fagocitante a la zona de actuación. Las sustancias quimiotácticas pueden ser: • Fracciones complemento activado: sistema enzimático que consta de 9 componen-tes proteicos de C1 a C9 que se activan en presencia del complejo antígeno-anticuerpo con liberación del sistema biológicamente activa. • Sustancias segregadas por los microorganismo así como interluquinas: sustancias producidas por las células del sistema inmunitario activado 2º·- Proceso de fagocitosis: el neutrófilo puede fagocitar: • Complejos antígenos-anticuerpos para ello utiliza sus receptores de membrana para el fragmento Fc del anticuerpo. • Microorganismo o células recubiertas por una fracción del complemento para ello utiliza su receptor de membrana para el fragmento C3B. • Microorganismo directamente en algunas circunstancias. En este caso es necesaria la presencia de obsoninas que son anticuerpos u otro componente del suelo como por ejemplo la fracción C3B del complemento que cuando se combina con un antígeno por ejemplo una bacterina lo hace fácilmente fagocitable. 3º·- Proceso de desgranulación: una vez formado el fagosoma o vesícula fagocítica el contenido de los gránulos del Neutrófilo es vertido a este iniciándose la destrucción del antígeno lo que puede suceder por 2 mecanismos bioquímicos diferentes: mecanismo oxidativos y no oxidativos. En los primeros(los oxidativos) se generan compuestos oxidantes como el agua oxigenada que es el componentes bactericida principal del neutrófilo y en los no oxidativos intervienen el pH ácido del fagosoma, la presencia de lisoenzimas (enzimas inhibidoras por lisis del desarrrollo de numerosas bacterias que se encuentra en las lágrimas muco-nasal y en la mayoría de los tejidos y secreciones) y la lactoferrina que por su propiedad de fijar el hierro priva a las bacterias de este componente principal para su vida. El neutrófilo también interviene en la inflamación; siendo la inflamación un complejo de cambios tisulares (tejidos) que se producen en respuestas a una agresión o lesión en ese tejido causado por bacterias, traumatismos, productos químicos, calor, etc. Las células lesionadas van a liberar histaminas que pasa a los líquidos vecinos de modo que se va a producir un aumento del riego sanguíneo local y de la permeabilidad de los capilares. De este modo se da un escape hacia los tejidos de grandes cantidades de líquido y proteínas, incluyendo fibrinógeno y cuyo resultado es la aparición de un edema extracelular local (acumulación de líquido en un tejido). Los 4 síntomas de una inflamación son: tumor (bulto), rubor (color rojo), calor y dolor. En los neutrófilos por diferentes fenómenos son atraídos hacia la zona lesionada. Será entonces la gran neutrolofília (hasta 20.00 o 30.000 neutrófilos/mm3 de sangre). Junto con los monocitos transformados en macrófagos ejercen su función de fagocitar. Los macrófagos tienen mayor capacidad de fagocitosis que los neutrófilos que van a ser de máxima eficacia en el plazo de 6 a 12 primeras horas. Al cabo de ese tiempo es cuando entran los monocitos procedentes de la sangre, se hinchan, producen lisosomas y fagocitan entre otros a los neutrófilos muertos. Además liberan sustancias ácidas que también afectan a los neutrófilos que después de pasadas las primeras etapas de inflamación no son tan útiles como los macrófagos. Cuando macrófago y neutrófilos captan una gran cantidad de bacterias y tejidos necrótico ellos mismos acaban por morir. Transcurridos varios días suele crearse en los tejidos inflamados una cavidad que contiene tejido necrótico neutrófilos y macrófagos destruidos que se conoce como pus. La formación de pus continúa hasta que la infección ha sido dominada. A veces la cavidad purulenta se abre paso hacia la superficie del cuerpo o hacia una cavidad interna y en esta forma se vacía espontáneamente. Otras veces sigue cerrada incluso después de la destrucción del tejido, entonces, las células muertas y tejido necrótico del pus gradualmente sufre autolisis durantes días; los productos finales de tal autolisis son absorbidos por los tejidos vecinos hasta que desaparecen los signos de infección tisular. EOSINÓFILOS El eosinófilo maduro es una célula de 12 a 17 micrométros de diámetro aproximadamente. Su concentración en sangre periférica en el adulto normal es del 1 al 3 % del total de leucocitos. Posee un núcleo que por lo general presenta forma en banda o bilobulada con una estructura cromatínica densa y adherida. El citoplasma se caracteriza por la presencia de gránulos eosinófilos o Acidófilos. De forma redonda, relativamente grande y uniforme repartidos, excepto sobre el núcleo celular y que cuando se tiñe presenta un color anaranjado brillante con tintes pardos. A diferencia del neutrófilo estos gránulos son de 1 sólo tipo y presenta una matriz homogénea en cuyo interior se dispone un cristal con estructura periódica. En algunas patologías estos cristales de Charcotleyden. Los gránulos posee numerosos enzimas, mielo-peroxidasa, fosfatasa ácida y arilsulfatasa B. Los eosinófilos se forman en la médula ósea a partir de la célula madre y después de diferentes estadios de maduración pasan a sangre periférica donde solo permanece unas 8 horas, ya que la mayor parte de la población corporal de eosinófilos se encuentran bajo la capa epitelial de los tejidos expuestos al agente externo como son los productos nasales y vías urinarias. También están en la mucosa del intestino y en tejidos pulmonares donde normalmente penetran en el organismo sustancias extrañas. Cuando los eosinófilos han cumplido su misión han sido fagocitados por el sistema mononuclear fagocítico. Funciones eosinófilas: el eosinófilo es una célula móvil y con actividad fagocítica aunque en menor grado que el neutrófilo. Los eosinófilos tienen receptores de membrana y responden a las mismas obsoninas que los neutrófilos. Su membrana celular poseee también receptores para la IgE e histaminas aumentándose el número de estos cuando se activa la célula. El papel biológica de estas células es el de modular la reacción anafiláctica al ser capaces de inactivar el mediador liberados por las células cebadas (basófilos en tejidos) (histaminas, serotonina y bradicidina) y las reacciones antígeno-anticuerpo en el proceso alérgico. Conceptos • Anafilaxia: el aumento de la sensibilidad del organismo a un antígeno que administrado previamente provocó una reacción normal. • Choque anafiláctico: la anafilaxia es un estado alérgico en el cual el gasto cardiaco y la presión arterial cae muchas veces de forma drástica. Resulta de una reacción de tipo antígeno-anticuerpo después de que a penetrado en el sistema circulatorio un antígeno al cual la persona es sensible. Afecta de diferentes formas: 1º·- Si la reacción antígeno-anticuerpo ocurre en contacto directo con las paredes de los vasos sanguíneo o del músculo cardiaco habrá una lesión directa en estos tejidos. 2º·- Las células lesionadas de cualquier otra parte del organismo liberan sustancias tóxicas que van a parar a la sangre de las cuales la más importante es la histamina, que es una sustancia intensamente vasodilatadora. 3º·- La histamina: ~ Aumenta la capacidad vascular al dilatar las venas ~ Dilata las arteriolas con lo cual disminuye la presión arterial ~ Provoca un gran incremento de la permeabilidad capilar con rápido escape de líquido, hacia los espacios tisulares y restos de los tejidos. Por lo tanto se produce una intensa reducción del retorno venoso y un choque de tal gravedad que la persona muere en pocos minutos. • Alergias: es un conjunto de fenómenos de carácter respiratorios, nervioso o eruptivo producidos por la absorción de ciertas sustancias (alérgenos) que dan al organismo una sensibilidad especial ante una nueva acción de tales sustancias aún en cantidades mínimas. Los eosinófilos también intervienen en el control de la infestación (organismos más grandes que los microorganismo) por ciertos parásitos cuyo ataque no se hace por fagocitosis sino por adherencia y subsiguiente citotoxicidad al segregar diversas sustancias nocivas, por ejemplo: cuando hay una infestación por Trichinella Spiralis aumenta a un 25 o 50% el total de leucocitos. BASÓFILOS Son los granulocitos más pequeños de 10 a 14 micras. Posee un núcleo que posee generalmente 2 o 3 lóbulos unidos por puentes cromatínicos, en ocasiones difíciles de observar porque está cubierto por numerosas granulaciones. Estas granulaciones son metacromáticas, es decir, adquieren tonalidades rojizas con los colorantes azules (azul de metileno o azul de toluidina) mientras que el resto de las estructuras se tiñen de azul. Con las coloraciones panópticas adquieren un color rojo oscuro. Los gránulos son ricos en mucopolisacáridos, histaminas, heparina, glucógeno, peróxidasas y fosfatasa ácida. Los gránulos basófilos son hidrosolubles y a veces se encuentran en el interior de vacuolas citoplasmáticas; por ello el citoplasma aparece a veces con numerosas vacuolas que no son más que gránulos parcialmente disueltos. El citoplasma puede tener un color rosa o puede no estar teñido. Los basófilos se originan en la médula ósea y después de diferentes procesos de maduración pasa a la sangre periférica donde realiza su función. Los basófilos que se encuentran en tejidos se denomina células cebadas o mastocitos y presentan con respecto a los basófilos sanguíneos ciertas diferencias en la composición de sus gránulos. Función de los basófilos: su función es actuar como mediadores en las respuestas inflamatorias en especial en las de hipersensibilidad. Los basófilos contienen gran cantidad de histaminas que es nivelada bajo estímulos apropiados. Además poseen receptores de membrana capaces de fijar anticuerpos IgE que al adherirse provoca la desgranulación celular liberándose las enzimas vasoactivas, bronconstrictivas y quimiotácticas. Las células cebadas así mismo se localizan inmediatamente por fuera de muchos capilares, de modo que evita la coagulación sanguínea y estimula la desaparición de partículas de grasa en sangre tras una comida rica en lípidos. Los basófilos que tienen la proporción muchas veces menor al 4 por mil, aumenta durante la fase de curación de la inflamación y también durante inflamación crónicas; probablemente debido a que una inflamación prolongada hace que los eritrocitos tienden a aglomerarse, por tanto, es posible que los basófilos en sangre sirva al liberar heparina para compartir la tendencia de los glóbulos rojos a adherirse. MONOCITOS Los monocitos son las células con mayor tamaño en sangre periférica entre 15 y 30 micras. Son de forma variable muchos son redondeados o alargados y otros presentan pseudópodos. El núcleo está en posición central o excéntrica; es de forma oval o en herradura de cromatina laxa y lineal y está desprovisto de nucleolos. El citoplasma es amplio de color azul plomizo y con un número variable de granulaciones azurófilas. Estas granulaciones son de 2 tipos: Primarias que contienen peroxidasas, fosfatasas y arilsulfatasa. Contiene lisosomas. Secundarias que no contienen peroxidasas. Contiene lisosomas. Los monocitos se producen en la médula ósea y en un plazo aproximado de 24 horas pasan a la sangre. Permaneciendo en ella entre 4 y 10 horas para luego emigrar a los tejidos. En donde se convierte en macrófagos donde cumplen su función. El paso de monocitos a macrófagos se caracteriza por un crecimiento celular progresivo; el núcleo recupera su forma redondeada, aumenta su contenido en lisoenzimas, tamaño y número de mitocondrias e incrementando su metabolismo energético. Los nucleolos pueden observarse y el citoplasma se tiñe de azul. Estas células pueden vivir meses en los tejidos y en condiciones normales no retornan a la sangre. Estos macrófagos o histocitos desarrollan características histoquímicas y morfoló-gicas diferentes dependiendo del sitio de maduración y de su hábitat en tejidos; así los macrófagos del hígado se denominan células de Kupffer, los del pulmón como macrófagos alveolares, los de la piel como células de Langerhans y los del cerebro como células microgliales. Funciones de los monocitos: la función principal de los monocitos y macrófagos es fagocitar, que realizan de manera semejante a los neutrófilos, pero tiene mucha más capacidad que estos. Actúan como defensa de los microorganismos en el proceso de la formación antigénica y en la eliminación de las células viejas dañadas o tumorales. Los monocitos sanguíneos limitan el proceso de coagulación ingiriendo factores de coagulación activados, además de proteínas desnaturalizadas y complejos antígeno-anticuerpo. Con respecto a la regulación de la inmunidad el sistema monocito macrófago desempeñan un papel principal en la iniciación y regulación de la respuesta inmunitaria de la siguiente manera: 1º·- Fagocitar y degradar el antígeno para el reconocimiento de este por el linfocito T. De este modo induce a la proliferación de estos linfocitos a través de la secreción de factores solubles como la interlucina I. Esta a su vez estimula a los linfocitos T a secretar interlucina II que también estimula la proliferación de unos linfocitos T. 2º·- Tiene receptores de membrana para la fracción Fc de las Ig E que aumenta la fagocitosis y destrucción de los agentes infecciosos. Segregan gran cantidad de moléculas biológicamente activas que influyen la respuesta inmunitaria, interferón, algunos componentes del complemento, lisoenzima, etc. LINFOCITOS El linfocito maduro es una célula de gran variedad de tamaños. Se clasifica en linfocitos pequeños y linfocitos grandes. • Los linfocitos pequeños: tiene un tamaño entre 7 y 10 micras y constituyen la mayor parte de la población linfocitaria. Son células con un núcleo que ocupa la mayor parte del área celular con cromatina muy condensada que se tiñe de color púrpura intenso con nucleolos. El citoplasma es de color claro pudiendo mostrar gránulos azurófilos que col colorantes actuales presentan coloración rojiza. • Linfocitos grandes: muy heterogéneos de tamaño varían entre 11 y 16 micra. El núcleo puede ser un poco mayor que el de los linfocitos pequeños pero la diferencia de tamaño celular es debida a la mayor cantidad de citoplasma. Este puede ser de color azul claro con basofília periférica (exterior violeta) o más oscuro que el del linfocito peqi (también se puede decir que presentan un halo perinuclear acromófilo sin cromofília). Pueden destacarse también gránulos azurófilos que no posee peroxidasa. La cromatina nuclear es semejante a la de los linfocitos pequeños y el núcleo puede tener una pequeña depresión (se parece a los monocitos). Tanto los linfocitos pequeños como los grandes pertenecen a subpoblaciones funcionales que se dividen en 3 grupos: linfocitos T, linfocitos B y linfocitos no T no B. Estas 3 subpoblaciones tienen características que permiten diferenciarlos ya que poseen diferentes tipos de enzimas receptores de superficie y antígenos de membrana. No se puede diferenciar morfológicamente pero se sabe que los no T, no B pertenecen a la población de linfocitos grandes granulosos. Funciones de los linfocitos: son las principales células de la respuesta inmunitaria. Actúan mediante los receptores que hay en su superficie. Tenemos 2 tipos: ~ Linfocitos B: dependientes de la médula ósea. Son los encargados tras la exterminación del antígeno de la producción de anticuerpos, y de quedar en el organismo como células de memoria para reaccionar ante futuros estímulos. Tienen en su superficie inmunoglobulinas que reconocen al antígeno. ~ Linfocitos T: dependientes del timo. Son los encargados de la inmunidad celular mediada por células. Tienen en su superficie receptores que reconocen el antígeno. Son responsables del desencadenamiento de las reacciones de defensa mediada por células, y son las células que, tras reconocer al antígeno regulan la respuesta inmunitaria celular y humoral (producción de anticuerpos). Hay 2 grandes subpoblaciones de linfocitos T: T4 o Helper y T8; con función de colaboración, estimulando la diferenciación de los linfocitos B a células plasmáticas secretoras de anticuerpos. T8 con función de citoxicidad. PLASMOCITOS Las células plasmáticas o plasmocitos representan un porcentaje muy pequeño aproximadamente 1 % de las células nucleares de la médula ósea pero no se encuentran en sangre periférica de personas sanas; su emplazamiento son los tejidos linfoides (nódulos linfáticos, bazo, placas de Peyer,...). Pueden hallarse en escasa proporción en el torrente sanguíneo en algunos estados patológicos como son por ejemplo infecciones crónicas, enfermedades granulopatosas, alérgicas y en el mieloma múltiple. El tamaño de las células plasmáticas maduras es de 15 a 25 micras siendo generalmente de forma redonda u ovalada con bordes lisos o ligeramente irregulares. El citoplasma no es granuloso y se tiñe de azul oscuro traslúcidos. La zona citoplasmática perinuclear es más clara y se denomina zona clara perinuclear. Pueden tener 1 o varios vacuolas. Los núcleos son pequeños, ovalados o redondos y excéntricos. La cromatina nuclear es gruesa y a menudo adyacente a la membrana nuclear. La función de los plasmocitos es sintetizar y segregar anticuerpos (inmunoglobulinas) PLAQUETAS Llamados también trombocitos; son células desprendidas del citoplasma de los megacariocitos adultos. Son los elementos formes más pequeños entre 1 y 4 micras y están desprovistas de núcleo por lo que no se trata de verdaderas células sino de fragmentos celulares. En los frotis se observan a menudo aglomerados (agregados plaquetarios) debido a su gran capacidad de agregación. En algunas patologías se dan las megaloplaquetas. Vida media de 8 a 11 días. En sangre periférica se presentan como células discoidales (discos), ovalados, redondos o fusiformes (uso) y de superficie lisa. Funciones de las plaquetas: intervienen fundamentalmente, en varios aspectos de la hemostasia o coagulación. Forman los tampones hemostásicos y participan en la hemostasia secundaria. RECUENTO DIFERENCIAL DE LOS GLÓBULOS BLANCOS Puede definirse como el porcentaje de distribución de los diferentes tipos de leucocitos. Sirve además para el estudio cualitativo de la morfología de los hematíes, leucocitos y plaquetas realizados sobre una extensión de una sangre teñida. Fórmula leucocitaria: el estudio porcentual de los distintos elementos de la series blanca se denomina fórmula leucocitaria relativa. Esta fórmula nos da el número de cada número de leucocitos. Si quisiéramos conocer el número real de cada clase de células blancas por mm' de sangre, es decir, la fórmula absoluta. Ejemplo: si tengo un 3% de eosinófilos y 7.500 leuc/mm3 ¿Cuántos eosinófilos por mm3 tengo? 3 ------------ 100 X 7,500 - 3 x ---------- 7.500 100 Para efectuar el recuento leucocitario se coloca la preparación sobre la platina del microscopio y se enfoca con un objetivo de bajo aumento para establecer la calidad de la preparación. Con este aumento se elige una zona del frotis en la cual los hematíes se hallen lo menos supuestos posibles y los leucocitos uniformemente distribuidos. Luego se coloca una gota de aceite y se cambia al objetivo de inmersión; estando el condensador alto y el diafragma abierto. Existen varias maneras de llevar a cabo el recuento diferencial: • Una vez seleccionada la zona se sitúa el objetivo en el borde inferior y se va desplazando gradualmente hacia el borde opuesto, seguidamente se mueve el frotis lateralmente para no contar el mismo área y se desplaza de nuevo hacia el borde exterior. Se cuenta todos los leucocitos que aparezcan clasificándolos al mismo tiempo por sus diferentes características mediante un contador de células, hasta llegar a un total de 100 leucocitos sino se realizará manualmente. • Recuento en almena: consiste en ir siguiendo el borde del frotis en movimiento en grecas o línea quebrada. A diferencia de los anteriores las líneas verticales no atraviesan el frotis sino que llegan a un tercio de su anchura. En ciertos estados patológicos puede darse un aumento absoluto de un tipo específico de leucocitos. Así tenemos: ~ Leucocitosis neutrófila o neutrofília: que se da en muchas infecciones generales y locales extensas. Por ejemplo: neumonía, abscesos, amigdalitis, apendicitis, etc. En las infecciones agudas graves con buenas respuestas, existe una desviación a la izquierda pronunciada, apareciendo neutrófilos, cayados o en bandas. ~ Linfocitosis absoluta: es característica de 3 infecciones; tosferina, mononucleosis infecciosa, linfocitosis granulosa aguada. También aumenta en tuberculosis, sífilis, en rubéola ~ Monocitos: se da en la brucelosis, en la tuberculosis crónica y en la leucemia monocítica. En la mononucleosis infecciosa es dudoso que haya monocitosis, la enfermedad suele comenzar con una fase neutropénica y luego ascienden progresivamente el número de leucocitos hasta cifras elevadas. ~ Eosinofília: el aumento del número de eosinófilos suele ser aislado o sea sin leucocitosis. Se presenta en la escarlatina, alergias y parasitosis. ~ Basofília: es raro encontrar un aumento absoluto de basófilos salvo en la policitemia vera y en la leucemia mielógena crónica Hay otros conceptos como: neutropénia, linfopenia, eosinopenia y pancitopenia (disminución de las 3 series). Observaciones: 1º·- Sólo deben usar buenos frotis, sí son demasiado gruesos es difícil diferenciar linfocitos de monocitos. Si son demasiado finos la mayor parte de los neutrófilos y monos se encuentran en la cola. 2º·- Se debe aprovechar para hacer el estudio morfológico de hematíes y plaq«etas. Habrá que observar entre 6 y 20 plaquetas en campos donde no estén superpuestos los hematíes. 3º·- Es importante anotar la presencia de cualquier elemento anormal. 4º·- Cuando en una fórmula se encuentran más del 10% de eosinófilos, más del 12% de monocitos y mayor número de linfocitos que de neutros (excepto en niños) se deben contar 200 células y dividir por 2 en el resultado; haciéndolo constar en el informe. TEMA XII CARACTERÍSTICAS MORFOLÓGICAS DE LOS ERITROCITOS 0 HEMATIES Los hematíes de una persona sana se presenta en el frotis como corpúsculo redondos homogéneos de tamaño casi uniforme entre 6 y 8 micras de diámetro. Cada uno de ellos tiene el centro más pálido que la periferia y tiende a hendirse si la extensión se ha secado demasiado despacio. Son células sin núcleo. COLOR La intensidad de la tinción indica aproximadamente la cantidad de hemoglobina en los hematíes. Se emplean los términos normocromo, hipocromo e hipercromo. Según la coloración sea normal, esté disminuida, o sea mayor a la normal, respectivamente. La presencia de células hipocromas e normocromas al mismo tiempo en la misma extensión de sangre, se llama anisocromía o en ocasiones se llama anemia diamórfica (pág. 3) [esto es característico, de la sideroblástical. También se observa algunas semanas después del tratamiento de las anemias ferropénicas con hierro [ó en las anemias hipocromas después de una transfusión de células normales]. La existencia de una tinción gris-azulada de los hematíes que se conoce como policromatofília o policromasia se debe a la presencia de reticulocitos que con las tinciones de frotis adquiere esta coloración debido a la presencia de RNA residual y que tiene afinidad por los colorantes básicos. Además son mayores que los hematíes maduros y puede faltar la madurez central (por lo tanto el aumento de polícromía implica reticulocitos la cual es más intensa en la hemólisis y en las hemorragias agudas]. TAMAÑO Puede ser de 2 tipos fundamentales: - Macrocíticos: de hematíes de tamaño mayor al normal ej.: A. megaloblástica. - Microcítica hematíes de tamaño menor al normal, ej.: A. ferropénica, talasemia. En ocasiones pueden coexistir simultáneamente varias alteraciones de tamaño lo que se denomina ansocitosis. FORMA La variación de la fórmula se denomina poiquilocitosis, cualquier eritrocito con forma anormal se denomina poiquilocitos. Entre las variaciones morfológicas pueden citarse: • Eliptocitos: los hematíes son alargados u ovalados, su presencia puede obedecer a una alteración congénita de la membrana eritrocitaria, aparece en eliptocitosis congénita [también puede observarse de forma adquirida en el curso de A. Megaloblástica,,A. Ferropénica,...1 • Esferocitosis: son hematíes de forma esférica, mayor grosor y carecen de la zona pálida central o la tienen más pequeña y excéntrica. Muestran aumento de la fragilidad en las soluciones salinas hipotónicas; la causa más frecuente de esferocitosis hereditaria es debida a una alteración congénita de la membrana eritrocitaria. También en enfermedades adquiridas como algunos casos de lesión celular por el calor. • Equinocitosis o células dentadas con muescas: también se llaman hematíes espiculados o espinosos o crenados. Son hematíes esferoidales cuya superficie se haya repleta de prominencias cortas y distribuidas regularmente. Son células que suelen aparecer como un artefacto durante la preparación de extensiones o también pueden aparecer en la insuficiencia renal [o en el curso de ciertas eritroenzimopatías de la glucólisis como es el déficit de la piruvato quinasa] • Acantocitos: son eritrocitos espiculados con las prominencias superficiales más alargados que los equinocitos y en los extremos de las espiculas unas prominencias rugosas y redondeadas. Aparecen en una enfermedad congénita llamada acantocitosis [también pueden aparecer en el curso de una insuficiencia hepatocelular grave]. • Dianocitos o codocitos o eritrocito en blanco de tiro: son hematíes con el cese de superficie más delgado de la normal, muestran un reborde periférico hemoglobínico con una zona oscura central, la cual contiene hemoglobina. Ambas zonas están separadas entre sí por un anillo pálido no teñido que contiene menos hemoglobina [se observa en la anemia ferropénica, ej. la ictericia obstructiva y en hemoglobinopatías como la talasemia • Drepanocitosis: los hematíes se alargan y adquieren forma de hoz o de media luna, también se denominan hematíes falciformes. Su aparición es propia de la anemia falciforme y su número aumenta si la sangre se expone a condiciones anaerobias. • Dacriocitos: también reciben el nombre de hematíes de forma de lágrima. Suelen observarse en trastornos de la eritropoyesis como la anemia ferropénica, talasemia y anemia megaloblástica ( y finalmente en la fibrosis medular o mielofibrosis]. • Esquistocitos: corresponden a hematíes rotos o fragmentados. Pueden presentar diversas formas; triangulares, forma de casquete. Indican la presencia de hemólisis. Aparecen en anemia de origen mecánico (quemaduras) [en la anemia megaloblástica y la anemia microangioplástica]. • Estomatocitos: son hematíes con una hendidura bicóncava central. Aparecen de forma característica en una enfermedad congénita conocida como estomacitosis. A veces también se pueden observar hematíes pinzados o también excentrocitos. ESTRUCTURAS En ocasiones los hematíes pueden presentar inclusiones de naturaleza diversas de acuerdo con el origen de las mismas. • Punteado Basófilo: se caracteriza por la presencia dentro del hematíe de gránulos basófilos irregulares que varían de finos a gruesos. Un hematíe con punteado basófilo es un hematíe con alto contenido en RNA y puede tratarse de un reticulocito (intoxicación plomo • Granulación azurófila: corresponde a pequeñas granulaciones eritrocitarias de color violetapúrpura que corresponde a restos del núcleo eritroblástico. • Cuerpos de Pappenheimer: corresponde a 1 o a más gránulos de hierro que aparecen en los siderocitos. • Cuerpos de Howell-Jolly: se trata de restos de cromatina nuclear y aparecen como inclusiones azules. Pueden observarse aisladamente en la anemia megaloblástica, en la anemia hemolítica y después de esplenectomía (extirpación del bazo). • Anillos de Cabot: son estructuras en forma de anillo en asa o en ocho (8). Su existencia pone de manifiesto una afectación importante de la eritropoyesis. Se tiñe de color rojo o púrpurarojizo con el colorante de Wright. • Punteado de la malaria: en los eritrocitos que albergan plasmodiun vivas pueden aparecer uno finos gránulos (gránulos de schüffner) y que se tiñe de un color rojo púrpura. Durante los accesos febriles pueden observarse los plasmodiums. • Apilamiento de hematíes: se parecen a pilas de monedas. Se denominan también formación de Rouleaux y se suele observar en estados en que está perturbado el equilibrio entre albúminas y las globulinas séricas como sucede en el mieloma múltiple. • Eritroblastos: son eritrocitos nucleados y estos en condiciones normales sólo se detectan e médula ósea. En sangre periférica sólo se detectan en el feto y en niños muy pequeños. En el adulto su presencia constituye un signo de intensa regeneración eritroblástica o de infiltración medular por células malignas. • Reacción leuco-eritroblástica: se denomina así cuando existen normoblasto y células inmaduras de la serie neutrófila en la sangre. Normalmente indica trastornos invasores de la médula. • Artefactos que pueden aparecer en el frotis sanguíneo: ~ Células rotas: los leucocitos lesionados o rotos constituyen una proporción de las células nucleadas en sangre. [Siempre que existen casos de linfocitosis atípica, leucemia linfática crónica, leucemias agudas estas células tienden a ser numerosas]. ~ Alteraciones degenerativas : el contacto de la sangre con el EDTA en el tubo produce una serie de alteraciones en los leucocitos. Sobre todo si en vez de EDTA se utiliza oxalato. También influye la cantidad de sangre recogida como cuando los tubos no se llenan. ~ Células contraídas: aparecen en la parte más espesa de la extensión, entonces los en leucocitos contraídos es difícil de distinguir el o los núcleos. ~ Células endoteliales: proceden del revestimiento del vaso sanguíneo. Presentan un perfil de cromatina reticular e inmadura. En sangre venosa es raro que aparezcan, pero si pueden aparecer cuando el espécimen es obtenido por punción de un dedo. TEMA XIII PATOLOGÍAS DEL SISTEMA ERITROCITARIO. ANEMIAS Podemos definir la anemia como el descenso por debajo del nivel normal de la hemoglobina funcional total circulante con o sin disminución de hematíes. La OMS y el comité internacional estandarización en hematología (ICSH) establecen unos valores de referencia según la edad y el sexo y por debajo de los cuales debe considerarse la presencia de anemia. Los valores son los siguientes: HEMOGLOBINA HEMATÓCRITO HOMBRE 13gr/dl 40% MUJER 12 gr/dl 38% MUJER EMBARAZADA 11 gr/dl 36% NIÑOS DE 1 AÑO 11 gr/dl 36% RECIÉN NACIDOS 14 gr/dl 45% Estas cifras están basadas en criterios estadísticos y no siempre tienen que concordar con la realidad. En la valoración de una anemia deben conjugar criterios clínicos y analíticos. Por otra parte la valoración de la concentración de hemoglobina en patología debe tener siempre presente las posibles situaciones que cursan con variaciones del volumen plasmático. Así en el embarazo, en la insuficiencia cardiaca, esplenomegalia (aumento del tamaño de bazo), puede acompañarse del aumento del volumen plasmático, capaces de producir un descenso de la concentración de hemoglobina y con ello una falsa anemia por hemodilución. PRUEBAS ANALÍTICAS PARA EL DIAGNÓSTICO DE LAS ANEMIAS. Para realizar en el laboratorio el diagnóstico de las anemias vamos a tener en cuenta: Hemograma: VCM, CCMH, ADE (IDE o RDW) Extensión sanguínea Recuento de reticulocitos Test adicional HEMOGRAMA • VCM: en el se debe basar la primera aproximación diagnóstica y nos permite realizar una clasificación de las anemias en función del tamaño del hematíe siendo esta: ~ Anemia normocítica: la media de las células presenta un tamaño normal entre 82 y 98 fi. Se trata de un grupo complejo ya que supone el paso ineludible e inicial de muchos procesos que posteriormente se decantarán hacia la microcitosis o macrocitosis. ~ Anemia microcítica: se caracteriza por la reducción del tamaño de las células. El VCM es < 80 fi. Suele implicar alteraciones en la síntesis de hemoglobina: * Fallos en la síntesis de la globina: talasemia * Fallos en la síntesis del grupo hemo: anemia sideroblásticas * Fallos en la incorporación del hierro en la molécula: A·- Déficit de hierro: anemias ferropénicas B·- Problemas en al utilización del hierro: anemia en procesos crónicos. ~ Anemias macrocíticas: se caracteriza por un aumento del tamaño de las células donde el VCM es > 98 11. Pueden ser: * Megaloblásticas: se deben a un déficit de ácido fólico y/o vitamina B12 * No megaloblásticas: donde existe un aumento del VCM pero debido a causas que dependen de la formación de la membrana de los hematíes exclusivamente. Esto ocurre en procesos fibrosis hepática, alcoholismo, ... También se puede observar este fenómeno por un aumento de los reticulocitos circulantes tras episodios de sangrado o hemólisis. • CCMH: en la práctica tiene menor interés que el VCM. Nos informa del grado de hemoglobinización y nos permite clasificar las anemias en : ~ Anemias normocíticas: la media de las células contiene una cantidad normal de hemoglobina y se tiñen normalmente. ~ Anemias hipocrómicas: las células aparecen menos teñidas con una palidez central de aproximadamente la tercera parte del diámetro del hematíe. La intensidad de la tinción viene determinada por el espesor y por la concentración de hemoglobina. Algunas causas de microcitosis lo son también de hipocromía; aunque personas con rasgos de (X o P talasemia se presentan con microcitosis pero sin hipocromía. • RDW o IDE o ADE: es el ancho de distribución eiritrocitario. Es una medida de la anisocitosis y alguno autores la consideran útil en el diagnóstico diferencial y la talasemia. FROTIS DE SANGRE PERIFÉRICA: complementa al hemograma y nos permite valorar las anomalías morfológicas no solo de la serie roja sino también de los leucocitos. Así por ejemplo aparece hipersegmentación de los neutrófilos en las anemias megaloblásticas, también nos permite orientar sobre la etiología de muchos procesos, hematíes en pilas de moneda (mielomas), anisocitosis (ferropénica), plasmodium intraeritrocitarios (paludismo), etc,... RECUENTO DE RETICULOCITOS: es básico tanto para: a) Localizar el origen de la anemia b) Valorar la respuesta al tratamiento Aproximadamente el 1% de los hematíes son reemplazados claramente por reticulocitos y su aumento en sangre periférica implica que la médula ósea está respondiendo bien, ante una situación de anemia o lo que es lo mismo el origen de la anemia no es central y que no hay afectación medular. TEST ADICIONALES: cuando la causa de la anemia no es aparente por la historia clínica ni por los anteriores parámetros se recurre a otros test: • Determinaciones en suero de hiero, ferritina y transferrina. • Determinaciones en suero de vitamina B12 Y ácido fólico. • Test renal de la función hepática y tiroidea. Si estas investigaciones no revelan la causa de la anemia está indicando en el aspirado de la médula ósea. En el feto y en el recién nacido la anemia hemolítica puede ser consecuencia del paso trasplacental de anticuerpos o a infecciones intrauterinas por microorganismos que más tarde no serán causa frecuente de anemias (tosoplasmosis, sífilis, citomegalovirus). SINTOMATOLOGÍA Las manifestaciones clínicas de la anemia son una consecuencia de la disminución de la oxigenación hística (hipoxia -oxígeno- tisular -tejido-) y de la puesta en marcha de diversos mecanismos de adaptación del propio @organismo que intenta solventar dicha situación (disnea y taquicardia en un intento de aportar más cantidad de oxígeno a los tejidos, cefalea, vértigos, vasoconstricción periférica, en un intento de preservar una irrigación correcta a órganos vitales; esto justifica la intolerancia del ftío que presentan estos pacientes y la palidez de piel y mucosas). Por otra parte tenemos síntomas y signos propios de la enfermedad causal. La consecuencia de la anemia varían de unos periodos de la vida a otros. Así la anemia en el feto puede provocar hidrops fetalis condición que se caracteriza por un gran edema en el feto y placenta y que a menudo provoca la muerte intrauterina. En el neonato por inmadurez del hígado la hemólisis severa puede provocar una marcada hiperbilirrubile- mia con peligro de daño cerebral. Por esto la identificación de la anemia en el recién nacido y fetos es muy importante. CLASIFICACION DE LAS ANEMIAS En cuanto a la clasificación de las anemias suele atenderse a 2 criterios: • Atendiendo a criterios fisiopatológicos (etiopatogénico): las anemias pueden clasificarse desde el punto de vista fisiopatológico en 2 grandes grupos de acuerdo con la cifra de reticulocitos: ~ Anemias arregenerativas o de origen central con cifra de reticulocitos normal o disminuido. Obedecen a un trastorno situado en la médula ósea, es decir, la médula es incapaz de producir un número adecuado de hematíes para cubrir las necesidades de oxígeno tisulares. Las causas de la anemia arregenerativa son diversas y situadas a diferentes niveles de la línea celular eritropoyética. [1·- Lesión de CFU A) Aplasia medular B) Anemia refractarias Infiltración neoplásica en la médula ósea Mielofibrosis 2·- Lesión a nivel de los precursores eritroblásticos Eritroblastopenia congénita Eritroblastopenia adquirida Autoanticuerpos contra los eritroblastos Medicamentos 3·- Autosuficiencia de producción de erítroproyetina A)Nefropatía crónica B) Hepotiroidismo 4·- Trastornos en la maduración eritroblástica: Alteraciones de la síntesis del DNA por déficit de: ~ Vitarnina B12 ~ Ácido fólico Alteraciones de la hemoglobinogenesis por defecto de la cadena globínica Trastorno de la secuencia de hierro 5·- Diseritropoyesis congénita A) I B) II C) III ] ~ Anemias regenerativas o periféricas: son todas las anemias cuya causa es periféri- ca y en las que se da una desaparición acelerada de hematíes circulantes por pérdida de sangre (hemorragia) o una destrucción excesiva de los hematíes (hemólisis). En estos casos la cifra de reticulocitos es mayor al 2% y la médula ósea trata de compensar la pérdida de hematíes aumenta o acelera la producción de los mismos. 1·- Hemorragias A) Aguda B) Crónica Parasitarias (paludismo) Tóxicas (Cu, Ar, venenos) • Atendiendo a los criterios morfológico (basados en los índices eritrocitarios): la clasificación morfológica de las anemias es la más utilizada en la práctica clínica. Según cuales sean los valores de VCM y de la CCMH las anemias se pueden clasificar en microcítcas, normocíticas y macrocíticas y en hipocromas y normocromas. Combinándose ambos parámetros tenemos: ~ Anemias microcíticas hipocromas (VCM<80fl; CCMH<32grldl; CMH<27pg) * Anemias ferropénícas * Anemias por enfermedades crónicas y neoplasias * Anemias sideroblásticas * Talasemias * Anemias con trastorno del receptor celular a la transferrina con imposibilidad de entrada de hierro a los eritroblastos. ~ A normocíticas normocromas (VCM 80-100fl; CCMH 32-36gr/dl; CMH 27-33pg * Pérdidas agudas de sangre * Anemia hemolítica * Aplasia * Invasión medular ~ Anemia macrocítica (VCM> 1 00fi; CCMH 32-36gr/d1) * Anemias megaloblásticas * Anemias no megaloblásticas ~ Anemia ferropénica y otros trastornos de metabolismo de hierro: la deficiencia de hierro es la causa más frecuente de anemia del mundo. Obedece a una disminución de la síntesis de hemoglobina como consecuencia de la falta de hierro en el organismo. Etiología: la deficiencia de hierro puede deberse a: Dieta inadecuada, que es más frecuente en países subdesarrollados Hemorragias excesivas; como el sangrado digestivo, úlceras, hemorroides, pólipos y pérdidas de sangre menstrual en la mujer. C) Aumento de las necesidades así en niños de entre 1-3 años, en la adolescencia y en el embarazo, sobre todo en el tercer trimestre. Trastornos de absorción de hierro, como es en la cirugía gastrointestinal. Etapas de la deficiencia férrica: ante una deficiencia de hierro el primer compartimiento afectado es el de depósito después el de transporte y en última instancia el funcional. En la deficiencia de hierro se puede establecer 3 etapas: • Ferropenia latente: el hierro de depósito (ferritina y hemosiderina) está disminuido pero no se van afectados ni el compartimiento de transporte ni el funcional. El hematocrito, la hemoglobina y la sideremia (nivel de hierro en sangre) son normales; por tanto no hay anemia. La única prueba para valorar la falta de hierro en los depósitos es la ferritina sérica [también tiene interés para valorar la sobrecarga férrica como en la hemocromatosis]. • Ferropenia larvada: constituye un estadio más avanzado en el cual los depósitos están vacíos. Desciende la sideremia y el índice de saturación de transferrina, puede no haber anemia o ser muy leve. No se alteran prácticamente los índices eritrocitarios. [El índice eritrocitario de saturación de transferrina (IST) = % = 20-50%]. • Anemia ferropénica: en la anemia ferropénica desciende el hierro, el hematocrito, la hemoglobina, el IST. Los hematíes se vuelven microcíticos e hipocrómicas. Sintomatología: a parte de los síntomas propios de las anemias tales como debilidad, cansancio, anorexia, abstemia (falta de apetito). Existen otros 2 que son típicas de la anemia ferropénica: coiloniquia (uña en forma de cuchara) y la ingestión de sustancias como tierra, almidón. La falta de síntomas puede reflejar la lenta instauración del déficit de hierro y la capacidad del organismo para adaptarse a esta carencia de hierro y a la anemia. Datos del laboratorio: en el frotis sanguíneo los hematíes son microcíticos, hipocromos, con anisopoiquilocitosis. En cuanto a los índices eritrocitarios, la hemoglobina y el hematocrito están disminuidos (VCM, CCMH, CMH suelen también estar disminuidos. En cuanto al número de hematíes puede ser normal o puede estar disminuido. La RDW está aumentada. El porcentaje de reticulocitos suele esta aumentado o normal. Los datos bioquímicos muestran hierro disminuido, ferritina disminuida, IST disminuidos. La RDW está aumentada. La existencia de una anemia hipocroma normocítica no implica siempre una deficiencia férrica. El diagnóstico diferencial de las anemias ferropénicas debe hacerse con las anemias de las enfermedades crónicas, las talasemias y las anemias sideroblásticas. ~ Anemia de las enfermedades crónicas: es la más frecuente después de la anemia ferropénica y tiende a instaurarse de forma lenta e insidiosa. Permaneciendo asintomática en muchas ocasiones. Este tipo de anemias se observa en infecciones crónicas, en trastornos inflamatorios no infecciosos (artritis reumatoide), neoplasias. La causa de esta anemia radica en el bloqueo de hierro a los depósitos, lo que impide su paso al compartimiento de transporte y al funcional para la síntesis de hemoglobina. Se presenta como una anemia leve, al principio se caracteriza por ser normocítica normocroma, convirtiéndose después en microcítica hipocroma. Por lo tanto la cantidad de hierro depositada (ferritina) está aumentado y la protoporferina también está aumentada. El hierro, transferrina y capacidad total de saturación de transferrina está disminuido. ~ Anemias sideroblásticas: se engloba dentro de esta denominación aquellas anemias generalmente refractarias (no responden) que se caracterizan por una mala utilización de hierro por los eritroblastos para la síntesis de hemoglobina. Esto conlleva la aparición de sideroblastos en la médula ósea y aumento notable del hierro depositado. Se caracteriza por hematíes hipocromas y generalmente microcíticos (puede aparecer otra población eritrocitaria normocítica). Las formas más frecuente de presentación son adquiridas, cuyo origen es el alcoholismo. La intoxicación por plomo, fármacos como el cloranfenicol y el isoniacida (también existen anemias sideroblásticas hereditarias ligadas al sexo o de herencia desconocida). Los datos del laboratorio más significativas son: * Ferritina elevada * Aumento de sideroblastos en anillo * Incremento de hierro macrofágico en médula ósea ~ Anemias por anticuerpos antireceptores de la transferrina: la causa de este trastornos un proceso autoinmune en el que se desarrolla anticuerpos contra el receptor de la transferrina impidiendo la entrada del hierro en el interior de los eritroblastos. Es un tipo de anemia ferropénica con hipocroma e hipersideremia en al que el hierro prácticamente ausente del sistema mononuclear fagocítico y de los eritroblastos se acumula de modo presente en el parénquimo hepático. ~ Anemias macrocíticas: se acompaña del aumento del tamaño celular (VCM >100) Normalmente la macrocitosis es la traducción periférica de un trastorno madurativo de la línea eritropoyética, pero tu existe un 5% en que existe macrocitosis con médula ósea normoblástica. * Anemias megaloblásticas: su mecanismo es la falta de la vitamina B12 y/o ácido fólico para la síntesis de ADN durante la fase S. Se acompaña de unas alteraciones morfológicas características de los eritroblastos consistentes en gigantismo celular y asincromía madurativa del núcleo citoplasmático. La vitamina B12 y el ácido fólico son sustratos necesarios para la síntesis de nuevas moléculas de ADN de aquí que su insuficiencia origine trastorno en la división celular y anomalías morfológicas y funcionales en las células. Principalmente en aquellas que tiene un alto índice mitético (células eritroideas). La vitamina B12 (cianocobalamina): es una cobalarnina que resulta de la unión asimétrica de 4 anillos pirrólicos en tomo a un átomo de cobalto. Es sintetizada exclusivamente por animales y algunos microorganismos que están presentes en la flora bacteriana y principalmente se va a depositar en el hígado. Las necesidades mínimas de un individuo adulto es de 2 a 6 microgramos/día. Las formas activas de la cobalamina son metilcobatamina y desoxiadenosilcobalamina. La vitamina B12 se libera por digestión de las proteínas de origen animal y luego se fija por el factor intrínseco gástrico (que es una glucoproteína producida en las células epiteliales del ilion donde se absorbe la vitamina B12. Una vez absorbida la vitamina B12 es transportada en el plasma ligada a un grupo de proteínas conocidas como transcobalamina (I, H, III). El 99% de la vitamina B12 se fija a la transcobalarnina 11 que actúa como principal proteína de transporte haciendo llegar rápidamente hígado células hematopoyé-ticas y otras células en división [el intervalo de referencia correspondiente a la vitamina B12 (en suero) es de 200-900mg/l][la carencia de transcobalamina II conduce a una anemia megaloblástica grave en la infancia aunque el nivel sérico de la vitamina B12 se mantiene normal]. La cobalamina en forma de depósito varía entre 2-5mg siendo necesario nucho tiempo para agotar los depósitos y manifestar su carencia. Las 2 funciones de la vitamina B12 en el hombre más importante son: a) Intervenir en la degradación de ciertos ácidos grasos para obtener energía en forma de ATP b) Participar en el metabolismo del folato. El metabolismo del ácido fólico: el ácido fólico es el ácido pteroil glutámico (ácido pteroico + ácido parabinobenzoico -PABA- + una o varias moléculas de ácido glutámico). Su forma activa es el tetrahidrofólico, porque el ácido fólico es por si inerte. Las bacterias de la flora intestinal sintetizan ácido fólico que es eliminado en gran parte por las heces lo que hace necesario su aporte a través de la alimentación. Se encuentran en alimentos de origen animal y vegetal siendo aconsejable la ingestión de 300-400microgramos/día. En la naturaleza se encuentra en forma de poliglutamatos, pero las conjugasas de la bilis y el intestino los hidroliza antes de la absorción en monoglutarnatos y se produce su absorción en el yeyuno proximal. El fólato se elimina rápidamente del plasma y pasa a las células y tejidos para su utilización. El 5-metil-tetrahidrohilofólico representa la principal forma de folato en el suero, en los hematíes y en el hígado. El principal órgano de reserva está en el hígado. El intervalo de referencia para el folato sérico es entre 5-21microgramo/litro y para el folato del hematíe varía entre el 150-600inicrograrnos/litro de hematíe. El folato interviene en numerosos procesos metabólicos, algunos de ellos esenciales en la síntesis de ADN. # Anemia megaloblástica por déficit de vitamina B12: 1·- Dieta inadecuada: es infrecuente en países occidentales, a excepción de los vegetarianos estrictos. 2·- Producción deficiente de factor intrínseco (es la más común) A) Anemia perniciosa: está causada por un fracaso de la mucosa gástrica para secretar el factor intrínseco. Generalmente no se manifiesta hasta la edad avanzada. Suelen presentar gastritis crónicas. Se produce una atrofia de las células parietales (proceso autoinmune). [Se han detectado 2 tipos de autoanticuerpos en el suero de los pacientes con anemia perniciosa: anticuerpos-células-parietales y anticuerpos-antifactor-intrínseco]. También existe un déficit de factor intrínseco congénito. B) Gastrectomía: la extirpación quirúrgica del estómago elimina toda la posible fuente del factor intrínseco lo que determina una fuente megaloblástica. Una vez agotados los depósitos orgánicos de vitamina B12 si no se realiza un tratamiento con vitamina B12. 3·- Absorción deficiente de vitamina B12: A) Síndrome de mala absorción: tales como al enfermedad celiaca, la reacción de intestino delgado, una enfermedad inflarnatoría, cáncer.... B) Falta de disponibilidad de vitamina B12: la infestación por tenia de un pez (dyphilobotrium latum) puede producir deficiencia de vitamina B12 porque la tenia compite por la vitamina B 12 junto al huésped. # Anemia megaloblástica por déficit de ácido fólico: 1·- Dieta inadecuada 2·- Aumento de necesidades de folato como en el embarazo, lactancia, crecimiento, infecciones,... 3·- Absorción deficiente de folato como ocurre en los síndromes de mala absorción. 4·Interferencias metabólicas de los folatos: alcohol, fármacos. Datos de laboratorio: 1·- Hemogramas: hemoglobina, hematocrito y número de hematíes disminuidos. El VCM aumentado. La CCMH y el RDW aumentado y precede al aumento del VCM. 2·- El frotis sanguíneo: la supervivencia de los hematíes es corta. Existe macrocitos ovales. Numerosos cuerpos de Howell-Jolly., Pueden existir anillos de cabot y hematíes nucleados con cariorrexis y punteados basófilos. Características de los leucocitos se encuentran frecuentemente leucopenias y neutrófilos hipersegmentados. 3·- Reticulocitos: el porcentaje de reticulocitos está disminuido. 4·- Test adicionales: • Determinación de vitamina B12 en sueros • Determinación de ácido fólico en suero y en hematíes • Test de absorción de Schilling • Anticuerpos antifactor intrínseco en caso de anemia perniciosa • Anticuerpos anticélulas apriétales normalmente presentes en A. Perniciosa. * Anemias no megaloblásticas: se produce un aumento de VCM pero debido sólo a causas que afectan a la formación de la membrana del hematíe son típicas de enfermedades hepáticas y alcoholemia. Se caracteriza por una leucopenia y trombopenia por efecto del alcohol en médula ósea. También pueden aparecer hiperesplenismo (aumento del bazo) asociada a enfermedades hepáticas. Datos de laboratorio: 1·- Hemograma: hemoglobina, hematocrito y número de hematíes disminuido. VCM aumentada. CCMIII normal y RDW normal. 2·- Frotis sanguíneo: características de los hematíes: los macrocitos son más redondos que ovales. Anisocitosis y poiquilocitosi menos marcada que la A megaloblástica En la enfermedad hepática crónica aparece Rouleaux con aumento de concentración de inmunoglobulinas. En el alcoholismo se puede sufrir una anemia hemolítica con esferocitosis con asociación de hiperlipemia. La característica de los leucocitos es que no aparecen neutrófilos hipersegernentados. 3·- Test adicionales: • Detenninación de ácido fólico normal • Determinación de vitamina B12 aumenta • [Test de supresión de deoxiuridina en médula ósea normal y nos permite excluir el déficit de vitamina B12 o de ácido fólico en alcohólicas con macrocitosis]. ~ Anemias aplásicas : Es una hipoplasia medular que afecta no solo a la serie roja si no que conlleva a la existencia de trombopenia y leucopenia asociadas. La causa que genera la panacitopenia (disminución de las 3 series) reside en la propia médula ósea. Aproximadamente el 70% de los casos son idiopáticos (origen desconocido) aunque existen estímulos externos como exposición al benceno, xilol, tolueno, determinados fármacos (cloranfenicol -antibiótico) e infecciones o en algunos casos de lupus. El problema básico radica en la disminución de las 3 series celulares de la médula ósea por lo que podemos encontrar alteradas no sólo los procesos de transporte de oxígeno si no también de coagulación y/o de defensa por alteraciones de la serie blanca. Datos de laboratorio: hallamos una disminución de las 3 series en sangre periférica con hemoglobina y hematocrito bajos. Velocidad de sedimentación globular muy elevada. Linfocitosis muy relativa y ausencia de eosinófilos y basófilos con hierro elevado y reticulocitos disminuidos, como dato de afección medular. La anemia es de tipo normocrómico. [En realidad la médula ósea raramente e completamente acelular sí no que puede existir en mayor o menor grado un aumento variable de linfocitos, monocitos y células plasmáticas. Los megacariocitos están disminuidos] El diagnóstico se basa en el cuadro hematológico de anemias, trombopenia y granulopenia. El índice de fosfatasa alcalina granulocítica se encuentra francamente elevada. Con escasa anisocitosis y ausencia de signos de regeneración hemática. ~ Anemia hemolítica: las anemias debidas principalmente a una mayor destrucción de los hematíes son anemias hemolítícas, por tanto, una disminución de la supervivencia del hematíe demuestra la presencia de hemólisis. Cuando la vida media del hematíe se reduce sobreviene el estado hemolítico. La médula ósea puede hacer frente a la masiva desocupación del hematíe acelerando la producción en el mismo grado. Si lo consigue decimos que establece un estado hemolítico compensado. Si no puede, se insatura la anemia hemolítica. El estado hemolítico ya sea compensado o no conlleva a la aparición de signos y síntomas de hiperdestrucción e hiperregeneración que lo caracteriza. Signos de hiperdestrucción: anemias e ictericia, esplenomegalia y hepatomegalia, heces hipercoloreadas, glóbulos rojos de vida corta, hemoglobinemia, hierro sérico elevado, lactoglobina disminuida, hemoglobinuria (hemoglobina en orina), [porfinas elevadas en orina, hemosiderinuria aumentada de la lactodeshidrogenasa (LDH), urobilinógeno fecal elevado, uribilina en orina elevada y el ALA (ácido delta-aminolevulímíco) elevado]. Signos de hiperregeneración: reticulocitosis, [febrículas, alteraciones óseas, metabolismo basal alto, deficiencia de folato,...] Clasificación de las anemias hemolíticas: se pueden clasificar en: - Intracorpuscular o intrínsecas: cuando el defecto radica en el propio hematíes; ya sea en la membrana, las enzimas o la hemoglobina. Son de origen hereditario y generalmente la hemólisis es extravascular. - Extracorpuscular o extrínsecas: se producen cuando la causa de la rotura del hematíe es ajena a ese, así tenemos los anticuerpos plasmáticos dirigidos contra antígenos eritrocitarios, sustancias tóxicas, fármacos, etc. Son generalmente anemias adquiridas y la hemólisis puede ser extravascular (se degrada la hemoglobina) o íntravascular (se libera directamente V.S.) * Anemias hemolítícas intrínsecas: en este grupo vamos a estudiar las anemias producidas por alteración intrínseca del hematíe que son casi en su totalidad de origen congénito. El lugar de hemólisis de las anemias hemolíticas intrínsecas es generalmente extravascular. La anomalía puede estar en la membrana de las moléculas de hemoglobina o en las enzimas del hematíe. Afectaciones de la membrana eritrocitaria: 1·- Esferocitosis hereditaria: es la causa más frecuente de anemia hemolítica crónica. Es una enfermedad que se hereda con carácter autosómica (hombre o mujer) dominante. El defecto radica en las proteínas de membrana espectrina y anquirína que provoca un aumento de permeabilidad para el sodio adquiriendo el hematíe forma esférica lo que da nombre a esta enfermedad. El hematíe esferocítico es muy rígido y al carecer de flexibilidad es atrapado por el bazo. Lo que reduce la supervivencia del hematíe en sangre periférica. La hemólisis crónica provoca anemia, esplenomegalia e ictericia. La presencia de esferocitosis se demuestra en frotis sanguíneos por la presencia de hematíes totalmente redondos y llenos de hemoglobina, lo que provoca un CCMH aumentado. Los estudios de laboratorio demuestra también un aumento de bilirrubina y retículocitosis y una disminución de haptoglobina. 2 pruebas deben realizarse para confirmar la esferocitosis hereditaria. La fragilidad osmótica y la autohemólisis. 2·- Eliptocitosis hereditaria: es una anomalía de la forma del hematíe que se hereda con carácter autosómico dominante y cuyo origen radica en un defecto proteico de membrana (espectrina). Su incidencia es mucho menor que la esferocitosis hereditaria. Se caracteriza por que en el frotis sanguíneo aparecen al menos 25% de eliptocitos. 3·- Acantocitosis hereditaria 4·- Estomatocitosis hereditaria 5·- Piropolquilocitosis hereditaria Trastornos enzimáticas: el hematíe maduro obtiene la energía de la degradación de la glucosa que penetra en el hematíe por un proceso de transporte que no consume energía. El metabolismo de la glucosa sigue 2 caminos: 1·- La glucosa se degrada un 90% por vía anaerobia o vía de Embden-Meyerhof 2·- Vía glucolítica eritrocítaria es la de las pentosas fosfatos o aerobias. Todas las enzimas que intervienen en el metabolismo eritrocitario deben estar presentes en concentraciones fijas. Permanecen activas durante toda la vida del hematíe. El diagnóstico definitivo de la deficiencia requiere del recuento del enzima. Los defectos más frecuentes son: 1·- Deficiencia de la glucosa-6-fosfato deshidrogenasa: dentro del sistema metabóli- co eritrocitario destaca por su frecuencia el déficit de glucosa-6-fosfato deshidrogenasa. Su mecanismo de trasmisión hereditaria se halla ligada al sexo. Los portadores heterocigotos suelen carecer de expresión clínica. Se han identificado más de 300 variantes de la enzima que difiere en estabilidad, actividad y movilidad electroforética. La variante mediterránea es la más frecuente en nuestro país. Su expresión clínica es la aparición de un cuadro hemolítico con fiebre, ictericia, reticulocitosis, cefaleas, etc. La crisis hemolítica suele empezar a los 2 o 3 de iniciada la ingestión de medicamentos (ácido acetil salicílico, sulfonaminas, antipalúdicos, cloranfenicol, ácido ascórbico,...) Otra forma clínica de la deficiencia de glucosa-6-fosfato deshidrogenasa está presente; asociada a infecciones de individuos con déficit de glucosa-6-fosfato deshidroge- nasa. Pueden presentar crisis hemolíticas, coincidiendo con infecciones bacteriológicas o virales. La variante más corriente entre los individuos de raza blanca es la glucosa-6-fosfato deshidrogenasa mediterránea, encontrada en las poblaciones mediterráneas. En subgrupo de sujetos con deficiencia de glucosa-6-fosfato deshidrogenasa de la variante mediterránea algunas horas después de haber consumido habas puede presentar una hemólisis grave llamada fabismo. La cuantificación de la actividad enzimática glucosa-6-fosfato deshidrogenasa en un hemolizado libre de leucocitos es la prueba diagnóstica definitiva. 2·- Deficiencia de la piruvato quinasa: se trasmite de forma recesiva autosómica y sólo los índices homocigotos tienen manifestaciones clínicas. El cuadro clínico es el de una anemia hemolítica de intensidad variable con anemia, ictericia, esplenomegalia y litiasis biliar. Los datos de laboratorio muestran anemia normocítica y normocráma con reticulocitosis. Bilirrubina y LDH sérica aumentada. Haptoglobina disminuida. COOMBS (negativo) y fragilidad osmótica normal. El diagnóstico de deficiencia de priruvato quinasa se realiza mediante la determinación de la actividad enzimática a partir del hemolizado. La deficiencia adquirida del piruvato quinasa se observa ocasionalmente en trastornos mielodisplásicas y en leucemias. 3·- Déficit de piridina-5-nueleotidasa (PN): la deficiencia de piruvato-5- nucleotidasa se hereda con carácter autosémico recesivo, produce una acumulación de piridinas y una degradación alterada de ARN con un marcado punteado basófilo de los eritrocitos en el frotis sanguíneo. El trastorno se caracteriza por una hemolítico crónica, reticulocitosis, intenso punteado basófilo y esplenomegalia. El diagnóstico se confirma demostrando una disminución de la actividad nucleosidásica. La deficiencia adquirida de piruvato-5-nucleotidasa se produce en al intoxicación por plomo y probablemente es responsable del punteado basófilo observado en este cuadro. Hemoglobinopatías o por defectos de la hemoglobina: en este conjunto de enfermedades hereditarias que afectan a la hemoglobina podemos distinguir 2 grupos: 1·- Talasemias: constituyen un grupo de heterogéneos de enfermedad hereditaria originadas por defecto de síntesis parcial o total de una o más de las cadenas polipeptídicas de la globina que forman la hemoglobina. La secuencia de aminoácidos es normal por lo que las talasemias se consideran defectos cuantitativos. La herencia de la talasemia sigue las leyes de Mendel. Clasificación: las talasemias se pueden clasificar atendiendo a 2 criterios: clínicos (según la gravedad la talasemia será menor, intermedia o mayor), genéticos químico (dependiendo de la cadena de globina que deja de sintetizar tendremos talasemia ð, ð, δ o ð). Incidencia y distribución geográfica: la talasemia es el trastorno genético más frecuente en el ser humano encontrado ampliamente distribuida en África, Asia y países mediterráneos. En España la más frecuente es la talasemia heterocigota. Fislopatología: una síntesis defectuosa de la cadena de globina conduce: 1·- Se forma tetrámeros de hemoglobina inadecuados, se produce menos hemoglobina A y como consecuencia anemia microcítica, hipocroma. 2·- Un exceso de cadenas no apareadas que si precipita en eritroblasto dan lugar a una destrucción intramedular y si lo hacen en el hematíe provocan hemólisis. ð Talasemia: está codificada por un solo gen alélico. Talasemia b homocigota (talasemia mayor): la forma más grave conocida es la llamada anemia de Cooley. Se caracteriza este tipo de talasemia mayor por que existe una disminución intensa de la producción de la cadena ð. La producción de las cadenas ð y ð es alta como consecuencia de hemoglobina F es elevada. Los agregados de estas cadena ð y ð son inestables y precipitan en el eritroblasto, se produce una anemia hemolítica grave. Los hallazgos clínicos en la talasemia mayor son: anemia grave con ictericia y esplenomegalia que se hacen evidentes en la primera infancia. Los huesos faciales prominentes y los ojos oblicuos que producen un aspecto mongólico. La pubertad se retrasa y el individuo crece achaparrado. La mayoría de los pacientes requieren transfusiones regulares y experimentan problemas debido a la sobrecarga de hierro. Normalmente se desarrolla hemocromatosis y el fallo cardiaco por siderosis del miocardio es la causa principal de muerte hacia fin de la tercera década. Las características de la sangre es que se produce una anemia hipocroma microcítica, extrema poiquilocitosís, células diana, ovarocitos, anillos de cabot, corpúsculos de Howell-Jolly, fragmentos nucleares, anisocromía, anisocitosis y a menudo normoblastosis extremada. El recuento de reticulocitos es menos elevada que lo previsto. La bilirrubina indirecta está aumentada y también aumenta el hierro sérico y también la resistencia osmótica Talasemia ð heterocigota (talasemia menor) o rasgo de Cooley: los signos son muy variables desde anemia grave moderada a la normalidad clínica total. En muchos individuos se comprueba una leve anemia hipocrorna y microcítica con ligera ictericia hemolítica y esplenomegalia. En sangre' se caracteriza por un aumento del número de hematíes, disminución de hemoglobina y del hematocrito. La CMH, el VCM y el CCNM son bajos. Hay células diana y punteados basófilos, microcitosis y poiquilocitosis. El nivel de hierro es normal o elevado. En médula se caracteriza por una hiperplaxia de médula roja. En cuanto a las hemoglobinas F existe un aumento ligero. Existe una forma denominada talasemia ð tipo 1 con hemoglobina A2 normal y que se presenta un mínimo cambio hematológico. ð Talasemia: está codificada por 2 genes alélicos: 1·- Hydrops fetalis (con hemoglobina Bart): la ausencia absoluta de cadenas a es incompatible con la vida, no existe hemoglobina A, ni F. Se detectan grandes cantidades de hemoglobina de Bart y algo de hemoglobina H (ð4) y ambas se desplazan con mayor rapidez en la electroforesis alcalina que la hemoglobina A. 2·- Enfermedad de la hemoglobina H (ð4): en este caso se caracteriza por ausencia de 3 de los 4 genes ð es frecuente una anemia crónica. La sangre está caracterizada por que: la VCM y la CMH están disminuidas. Hay hipocromía, células diana y anisopoiquilocitosis y reticulocitos entre el 4 y el 5%. Al teñir la sangre con un colorante vital induce a la formación de cuerpos de inclusión de color azul pálido en muchos de los hematíes (debido a precipitados de hemoglobina). Después de la esplenectomía pueden apreciarse grandes corpúsculos aislados. Por electroforesis se puede ver hemoglobina H que emigra más rápido y ligeros indicios de hemoglobina Bart. ð talasemía menor: ausencia de 2 genes que conduce a una anemia muy leve. Existe microcitosis. Los valores de hierro, ferritina y protoferina presentan valores normales. Portador silente de la talasemia a (rasgo talasémico): falta un gen de los 4. En los adultos no existe anormalidad hematológica. En los niños recién nacidos la hemoglobina Bart (ð4) representa entre 1 y 2% de hemoglobina en sangre del cordón umbilical. Existen unas variantes como es la δ talasemia que cursa con hemoglobina A normal o baja y aumento de la hemoglobina F. Métodos para estudiar las hemoglobinas: 1·- Electroforesis de hemoglobina, puede en: - acetato, de celulosa a pH alcalino - agar cítrico en medio ácido 2-- Cuantificación de hemoglobina A2 3-- Desnaturalización alcalina para la hemoglobina F [4-- Prueba de la tira de elusión ácida de la hemoglobina F.] 2·- Hemoglobinapoatías estructurales: son un grupo de enfermedades que se caracteriza por presentar una cadena de globina estructuralmente anómala. Las hemoglobinas anormales difieren de las normales en que un aminoácido de la globina ha sido sustituida por otro. Esto es suficiente para que las hemoglobinas tengan propiedades distintas. Solubilidad, afinidad por el oxigeno, inestabilidad, carga eléctrica, etc. Se hereda con carácter autosómico dominante siguiendo las leyes de Mendel. Estudiaremos las más frecuentes: Hemoglobinopatías S: es la variante estructural más conocida. Se produce por sustitución del aminoácido glutámico por la valina en la posición 6 de la cadena P. Es frecuente en África y muy rara en España. La forma homocigota de lugar a la anemia de las células falciformes (SS). LA forma heterocigota no presenta signos clínicos (AS). La presencia de hemoglobina S provoca una deformación en los hematíes que adoptan forma de hoz y conduce a una anemia hemolítica conocida como anemia de las células falciformes. Esta anemia en su forma homocigota es grave y se traduce en una anemia hemolítica crónica con crisis dolorosa producida por infartos múltiples en diversos órganos. Los pacientes con hemoglobina SS presentan anemia, reticulocitosis e hiperbilirrubinea y en el frotis sanguíneo se observan drepanocitos. La electroforesis pone de manifiesto la banda de hemoglobina S que puede aparecer en una proporción 50-80%. La presencia d hemoglobina S en niveles superiores al 10% impide el fenómeno de falciformación y da un carácter más benigno a la enfermedad. La AS protege a los niños del paludismo. Hemoglobinopatías inestables: se refiere a un grupo de variantes de hemoglobina que provoca inestabilidad en su molécula con formación de cuerpos de Heinz y da lugar a una anemia hemolítica congénita. Se conoce más de 100 variantes con reticulocitosis hiperbilinubilemia y descenso de la haptoglobina. La electroforesis de hemoglobina en acetato de celulosa suele ser normal. En ocasiones puede elevarse la hemoglobina A2 y la hemoglobina F. En la actualidad la confirmación de hemoglobinopatías se realiza con técnica de biología molecular. Hemoglobinopatías coti aumento por la afinidad del oxígeno: se han descrito variantes en las que la hemoglobina por su gran afinidad por el oxígeno no lo libera; lo que provoca hipoxia hística. Se han descrito igualmente variantes de hemoglobina con baja afinidad por el oxígeno, lo que puede provocar cianosis. Otras hemoglobinopatías: metahemoglobilemias congénitas, hemoglobinopatías C, D, E ..... * Anemias hemolíticas adquiridas: a diferencia de las congénitas aparecen en el curso de diferentes enfermedades que al producir una alteración de las propiedades físicas, eritrocitarias disminuye la deformabilidad eritrocitaria o facilitan la fagocitosis de los hematíes por las células de sistema mononuclear fagocitario excepto la hemoglobinuria paraxística nocturna que obedece a un defecto intrínseco de la membrana. Todas las anemias hemolíticas adquiridas restantes tienen origen extracorpuscular. Las anemias hemolíticas adquiridas se dividen en 2 grandes grupos: autoinmunes y no autoinmunes. # Las anemias hemolíticas adquiridas autoinmunes: obedecen a la formación de anticuerpos contra determinados antígenos de la superficie eritrocitaria. # Las anemias hemolíticas adquiridas de mecanismo no autoinmune: responden a causas muy diversas por lo que constituyen un grupo relativamente heterogéneo. # Las A.H.A. autoinmunes: es un síndrome clínico caracterizado por la presencia en el plasma o en los hematíes de autoanticuerpos dirigidos contra determinados antígenos, determinados eritrocitos. Las A.H.A. autoinmunes en el 50% de los casos constituye una manifestación más en el curso de las enfermedades que generan autoanticuerpos contra ciertos antígenos de la membrana eritrocitaria. Otras veces no existe enfermedad de otra causa asociada demostrable por lo que la A.H.A. autoinmune, se denomina idíopática (causa desconocida) debido a que la actividad de los autoanticuerpos dependen de la temperatura. Estos se han clasificado en 2 grandes grupos: 1·- Autoanticuerpos calientes con actividad máxima alrededor de los 371C 2·- Autoanticuerpos fríos en los que esta se sitúa entre 0 y 201C. De acuerdo con ello existen 2 formas de A.H.A. autoinmunes según obedezca a la acción anticuerpos calientes o fríos. Los autoanticuerpos son en general Ig G e Ig M. Siendo excepcionales los de tipo Ig A o Ig D. Los autoanticuerpos Ig G son en la mayoría de los casos de tipo caliente. Tienen carácter policlonal y se unen intensamente a la superficie eritrocitaria. Existe sin embargo un autoanticuerpo Ig G de tipo frío que fija en gran medida el complemento y se denomina hemolisina bífásica; porque su unión a los hematíes sólo se realiza a baja temperatura y la lisis sólo se produce cuando esta recobra su valor normal (se conoce como hemiglobinuria paraxística afrígore). Los autoanticuerpos Ig M son de tipo frío; también se les conoce como crioaglutininas. Cuando aparecen en el curso de una infección son de tipo policlonal. Mientras que su aparición idopática con carácter monoclonal y es lo que se conoce como las crioaglutininas. La presencia de autoanticuerpos (Ig G) o complemento sobre la superficie eritrocitaria puede detectarse en la mayoría de los casos mediante el empleo de una antiglobulina polivalente [obtenida por inmunización de conejos frente a las proteínas de suero (anti-Ig G, anti-C)] que se conoce con el nombre de COONMS. La antiglobulina de COOMBS está formada por anticuerpos completos que producen aglutinación de los hematíes cuando está recubiertos por autoanticuerpos incompletos o el complemento. Ello constituye la base del método diagnóstico fundamentalmente de la anemia hemolítica adquirida autoinmune, que se conoce como prueba de la antiglobulina directa (que se conoce como PAD o prueba de COOMBS). La PAD consiste en poner en contacto los hematíes del paciente con al antiglobulina en general polivalente y observa la aparición de la aglutinina cuando en la superficie eritrocitaria posee autoanticuerpos (en general Ig G) o determinadas fracciones del complemento. Cuando en lugar de hematíe se parte de suero del paciente los autoanticuerpos presentes en el mismo puede también ponerse de manifiesto mediante la llamada prueba de la antiglobulina indirecta (PAI o COOMBS indirecto). Esta consiste en incubar el suero problema (suero paciente) con hematíes lavados con el objeto de producir la fijación de los autoanticuerpos sobre al superficie eritrocitaria y en una segunda fase se realiza la PAD. La positividad de la PAD depende fundamentalmente de la cantidad de la Ig G o del complemento fijado sobre la superficie eritrocitaria ya que si este es insuficiente el resultado será negativo. Aunque por definición la PAD en al anemia hemolítica adquirida autoinmune por autoanticuerpos calientes debe ser positiva en un momento u otro de la evolución. Es posible observar síndromes hemolíticos con características clínicas y hematológicas propias de anemias hemolítica adquirida autoinmune pero con PAD repetidamente negativo. Estos casos se conocen con el nombre de anemia hemolítica adquirida autoinmune, COOMBS negativo y corresponden aproximadamente al 10% del total de anemia hemolítica adquirida autoinmune por autoanticuerpos calientes. Las anemia hemolítica adquirida autoinmune, PAD o COOMBS negativas obedecen probablemente a que la cantidad de autoanticuerpos fijados en al superficie eritrocitaria es insuficiente para que pueda evidenciarse la reacción. Por ello en tales casos deben emplearse otros procedimientos más sensibles que la prueba de COOMS. B-- Anemia hemolítica adquirida autoinmune por anticuerpos fríos: la etiología puede ser idiopática o secundarias a procesos linfa-proliferativos e infecciones. La hemólisis- es discreta, la anemia es crónica y de progresión lenta con alguna crisis aguda con evidente hemoglobinuria. El tipo de anticuerpos es Ig M que se une al hematíes en los vasos periféricos y produce obstrucciones con acrocianosis y adormecimientos. Al volver a la circulación sistemática y con más calor el anticuerpo se suelta, la hemólisis es principalmente intravascular por activación del complemento y en parte por fagocitosis sobre todo hepática de los hematíes recubiertos por fracciones C3. La confirmación diagnóstica se establece por la positividad del PAD o COOMBS y la presencia de un título de crioaglutininas séricas siempre superior a 1/ 152. C·- Anemia hemolítica adquirida autoinmune por anticuerpos bifásicos: la crioliemoglobinuria paraxística es un tipo de anemia muy rara (es lo que se conoce como hemoglobinuria paraxística afrígore). Se presenta en todas las edades y casi siempre secundarias a infecciones. Cursa con hemoglobinuria tras la exposición al frío con escalofríos, dolor de riñones, piernas, abdomen, fiebre y vómitos. Los anticuerpos son de tipo Ig G, la lisis es intravascular por activación del complemento y el complemento antígeno-anticuerpo se separa y por lo tanto no hay fagocitosis. # Las A.H.A. no autoinmunes: normalmente la prueba del COOMS es negativa y al clínica es diferente excepto la hemoglobinuria paraxística nocturna de origen corpuscular. Todas las anemias hemolíticas incluidas aquí obedecen a una agresión extracorpuscular de los hematíes por causas diversas tales como fenómenos inmunes, efectos mecánicos,... 1·- Hemoglobinunía paraxística nocturna: es un síndrome hemolítico secundario, aún a un defecto adquirido de la membrana eritrocitaria por el cual los hematíes presentan un aumento de la sensibilidad a la acción lítica del complemento (C3). Características diagnósticas del laboratorio de la hemoglobina paraxística nocturna: el hemograma muestra un cierto grado de anemia con moderada trombopenia y leucopenia, los reticulocitos suelen haber aumentado durante la fase activa de la enfermedad. 2·- Anemia hemolítica inducida por fármacos: consiste en la aparición de anticuerpos contra determinados medicamentos circulantes o sus metabolitos que lesionan la membrana eritrocitaria y producen hemólisis. La hemólisis puede producirse a través de 4 mecanismos: Adsorción (pegarse) del complemento inmunes a la membrana del hematíe. Adsorción del fármaco a la membrana del hematíe. Inducción de autoanticuerpos por fármacos. Adsorción no inmunológica de la inmunoglobulina a la membrana del hematíe. En algunos de estos casos puede ser el COOMBS positivo. 3·- Anemias hemolíticas mecánicas: [efecto secundario: rompe el hematíe] 4·- Anemias hemolíticas por agentes químicos: la acción de las sustancias químicas depende de la dosis y demás factores. Entre las sustancias químicas que pueden producir anemia hemolítica, tenemos el agua, los nitrototuenos, el plomo, el veneno de víboras. 5·- Anemias por agentes físicos: las quemaduras extensas de tercer grado producen una anemia hemolítica, tal vez a causa de una lesión directa de los hematíes. 6·- Anemia hemolítica por trastornos metabólicos: ciertas enfermedades producen alteraciones sobre el plasma y repercute también en los hematíes, como en el caso de una hepatopatía alcohólica que puede causar hemólisis. 7·- Anemia hemolítica por agentes infecciosos: la anemia del paludismo está causada por la destrucción de los hematíes por plasmodiums. Otros parásitos que pueden causar anemia tenemos la leishmania, babesia y algunas bacterias como pueden ser neumococos, estreptococos,... 8·- Anemia hemolítica del recién nacido: obedece a una agresión inmune de los hematíes fecales pro anticuerpos matemos, normalmente la placentea es impermeable al paso de las células sanguíneas y de macroglobulinas (Ig M) pero no permite el paso de globulinas de bajo peso molecular (Ig G). Si los hematíes fetales poseen un grupo Rh y/o ABO incompatibles con el de la madre esta se inmuniza contra aquellos y reproduce una intensa síntesis de anticuerpos a partir del segundo embarazo. En caso de inmunización la madre produce 2 tipos de anticuerpos Ig M e Ig G. Los anticuerpos antiRh son de tipo Ig G, atraviesan la barrera placentaria y penetran en la sangre fetal causando una hemólisis masiva durante un segundo embarazo incompatible. Las consecuencias aunque varían suelen ser graves: intensa anemia hemolítica e hiperbilinubinemia. El diagnóstico se hace con COOMBS directo e indirecto. ~ Anemia refractaria: son un grupo de anemias que no responde a tratamiento ninguno. Cursan con diseritropoyesis o dishematopoyesis como característica fundamental. POLIGLOBULIAS Y POLICITEMIAS. Poligiobulias: es la elevación del número de hematíes por encima de sus valores normales. Las Poliglobulias pueden producirse a través de diferentes orígenes y mecanismo. Por ejemplo: cuando existe una hipoxia hística va a aumentado la eritroproyetina y al aumentar la eritroproyetína aumenta la poliglobulia. Otro mecanismo muy diferente a este es cuando al tejido hematopoyético propiamente dicho y que desencadena en la poliglobulia afecta concretamente a la célula germinal específica directamente responsable de la eritropoyesis. En este caso la poliglobulia no es secundaria a un exceso de producción de eritropoyetina sino primaria es la policitemia. Policitemia vera: es una enfermedad mieloproliferativa (maligna) crónica caracterizada por un hiper-producción predominante d hematíes que determina el aumento absoluto de la masa de la serie roja debido a la existencia de una patología de la célula madre hematopoyética. En el inicio de este proceso se haya también presentes otras manifestaciones como leucocitosis neutrofília con más o menos eosinofília y basofília, trombocitosis, alteraciones morfológicas dishematopoyéticas, esplenomegalia. La causa de la policitemia vera es desconocida y a diferencia de las poliglobulias se encuentra niveles bajos de eritropoyetina baja. Clínica: afecta más a varones que a mujeres. El comienzo es insidioso (poco a poco) y las primeras manifestaciones pueden ser una trombosis o hemorragia. Pueden aparecer dolores de cabeza, vértigos, acúferos (dolor de oído), trastornos de visión, equimosis (labios) y epistaxis y cianosis vinosa de cara, nariz, orejas, labios. Datos de laboratorio: aumento de la producción de hematíes con una vida media eritrocitaria normal. A medida que el proceso avanza se acorta la VMC por un fenómeno de hiperespienismo. En el examen de sangre periférica el dato más destacado es la gran elevación de la cifra de hematíes (7-106) el contenido hemoglobínico total está muy elevado, pudiendo llegar a cifras de 10-24gr/dl. Los hematíes son microcíticos. Después de varias flebotomías (sangrías) suelen aparecer signos morfológicos regenerativos como cierto grado de anisocitosis, punteado basófilo y policromasia. La elevación eritrocitaria se acompaña de leucocitosis con desviación a la izquierda y presencia ocasional de algunos granulocitos inmaduros, con frecuencia aparecen eosinofília y basofília en grado elevado. Elevación de las fosfatasas alcalinas granulocíticas (leucocitarias) (FAG) mientras que en las poliglobulias son normal. Las plaquetas están numéricamente elevadas y de tamaño gigante. [La policitemia vera es una enfermedad mieloproliferativa cromosoma philadelfia negativa.] Otro dato característico es que los niveles séricos de la vitamina B12 se hayan aumentados. Poliglobulias: se habla de poliglobulias cuando la anomalía misma desencadena la hiperproducción eritrocitaria se halla situada fuera del tejido eritrocitario el cual por otra parte responde normalmente a un estímulo excesivo. Tipo: A-- Poliglobulias secundarias a una hipoxia hística: la hipoxia hística provoca una hipersecreción de eritroproyetina que se traduce en el aumento de la eritroproyetina y eventualmente en una poliglobulia. La eritrocitosis es normocítica y normocroma. B-- Poliglobulias por enfermedades cardiovasculares C-- Poliglobulias de las enfermedades pulmonares y por hipoventilación alveolar. D-Poliglobulias por anomalías en el transporte de oxígeno: las hemoglobinas anormales (metahemoglobinopatías congénitas y la carboxihemoglobina en los grandes fumadores provocan poliglobulias). E-- Poliglobulias por hipoxia tóxica: derivados de anilina, nitrofenoles,... F-- Poliglobulias de las enfermedades renales. G-- Poliglobulias asociadas a diferentes tumores. H-- Pseudopoliglobulias: en este cuadro patológico la masa eritrocitaria es normal pero el volumen plasmático están disminuidos. Esto produce una hemo-concentración por aumento del número relativo de los hematíes por mm3 de sangre. La pseudopoliglobulias suele producirse como consecuencia del uso de diuréticos o en situaciones de deshidratación como por ejemplo las quemaduras graves y al diarrea. También puede generarla el estrés y el consumo abusivo del alcohol. Resumen 1·- Anemias microcíticas hipocromicas: ~ Anemias ferropénica ~ Anemias entrmedades crónicas ~ Anemias sideroblásticas ~ Anemias de receptores de transferrina ~ Talasemias* 2·- Anemias macrocíticas ~ Megaloblásticas: • Por déficit de vitamina B12 • Por déficit de ácido fólico • Por déficit del factor intrínseco. ~ No megaloblásticas (hepatologías) 3·- Anemias aplásicas 4·- Anemias hemolíticas ~ Anemia hemolítica congénita (intrínseca) • Enzimopatías: * Déficit de glucosa-6-fosfato deshidrogenasa * Déficit de piruvato quinasa * Déficit de piridina-5-nucleotidasa • Membranopatías * Esferocitosis hereditaria * Eliptocitosis hereditaria * Acantacitosis hereditaria * Estomatocitosis hereditaria * Piropoiquilocitosis hereditaria • Hemoglobinopatías * Talasemias* 1·- ð talasemia (homocigota y heterocigota) 2·- ð talasemia 3·- Portador silente de la talasemia ð * Hemoglobinopatías estructurales 1·- Hemoglobinopatías 2·- Hemoglobinopatías inestables 3·- Hemoglobinopatías con aumento por la afinidad del oxígeno ~ Anemias hemolíticas adquiridas • Anemias hemolíticas adquirida autoinmune, • Anemias hemolíticas adquirida no autoinmune 5·-Anemias refractarias TEMA XIV ALTERACIONES DE LA SERIE BLANCA 1·- Trastornos no malignos de los leucocitos 1.1·- Cambios cualitativos de los granulocitos Los granulocitos comprenden basófilos, neutrófilos y eosinófilos y su función es participar en la defensa del huésped y en la respuesta inflamatoria. Diversas anomalías en su morfología o en su función pueden ser causa de infecciones bacterianas y fungidas. • Alteraciones morfológicas: ~ Granulaciones tóxicas: consisten en un aumento del tamaño y de la intensidad de la coloración de los gránulos del neutrófilo. Aparecen en infecciones bacterianas y leucocitosis reactivas. ~ Anomalías de Pelger-Huet: es un defecto de la lobulación del núcleo de los granulocitos. Existen 2 formas una hereditaria y otra adquirida. Esta última puede observarse en hemopatías graves. ~ Cuerpos de Döhle: son inclusiones de color azulado que se localiza cerca de los citoplasmas de los neutrófilos. Aparecen en infecciones, quemaduras y tumores. ~ Otras anomalías morfológicas son: Alder-Reilly, May-Hegglin, Chediak,... • Alteraciones funcionales: son trastornos de los granulocitos que dificultan la fagocitosis y posterior destrucción de los microorganismo. Ciertos defectos en la formación de gránulos son: trastornos en el metabolismo del oxígeno, ausencia de enzimas en los gránulos [sobre todo mieloperoxidasas] que pueden ser causa de disfunciones de los neutrófilos que tienen como consecuencia infecciones bacterianas y/o fúngicas. El germen más comúnmente implicado en estas infecciones es el Staphilococus Aureus. 1.2·- Cambios cuantitativos de los granulocitos: • Neutrofília: la leucocitosis neutrofília se refiere a un aumento absoluto de neutrófilos por encima de 7.000leuc/mm3. Causas: ~ Aumento de la producción por la médula ósea. Tiene lugar en infecciones, inflamaciones, neoplasias, tratamiento con litio (maniaco-depresivas) ~ Retención mayor en sangre. Se da cuando se administra glucocorticoides que dificultan las salidas de los neutrófilos desde la sangre a los tejidos. ~ Ejercicio físico, ingestión, digestión, convulsión, ansiedad y cuando hay exceso de adrenalina en la sangre,... La causa más importante es la infección bacteriana, ocurre a las 5 o 6 horas de producirse esta última con paso de neutrófilo de médula ósea a sangre periférica. Cuando la demanda crece excesivamente pueden observarse cayados e incluso otros granulocitos más inmaduros, hablándose entonces de desviación a la izquierda. En infecciones muy graves puede agotarse la reserva de neutrófilos y aparece neutropenia. En las únicas infecciones bacterianas en que se da neutropenia en vez de neutrofília son la fiebre tifoidea y brucelosis. Causas frecuentes de neutrofílias que se acompaña de desviación a la izquierda así mismo están las hemorragias y hemólisis. También la causa son intoxicaciones metabólicas y ciertos tratamientos con medicamentos. • Neutropenia: se considera que existe neutropenia cuando el número absoluto está por debajo de 1500 neutr/mm3. El término granulocitosis se utiliza para las neutropenias graves provocadas por fármacos. Se pueden dividir en tres grado según su gravedad: A·- Neutropenias leves: 1500 - 1000 neutrofilos/mm3. B·- Neutropenia moderadas: 1000 - 500 neutrofilos/mm3. C·- Neutropenia grave: menor de 500 neutrofilos/mm3. Causas: ~ Disminución de la producción por la médula ósea a defectos de la liberación desde la médula ósea a la sangre. ~ Aumento de utilización y paso rápido de la sangre a los tejidos. ~ Aumento de la destrucción celular. Sea cual fuere la causa la tendencia a las infecciones y la fiebre son comunes a la mayoría de las neutropenias. A su vez las infecciones son la causa más frecuente de neutropenia entre ellas tenemos fiebre tifoidea y brucelosis y gripe, hepatitis, rubéola y el sarampión. En todas hay un excesivo consumo de granulocitos. Ciertas sustancias como el benceno o los antineoplásicos lesionan el comportamiento de células primitivas de células óseas provocando una aplasia (incompleto o defectuosos desarrollo) medular y consecuentemente una neutropenia. Se da granulocitosis con medicamentos como granulocitos, cloranfenicol, tiouracilo y/o fenilbutazona. Esta neutropenia es precoz e intensa apareciendo escalofríos, taquicardia, cefalea y fiebre; si se sigue manteniendo el medicamentos se daña el hígado pudiendo producirse septicemia (presencia de bacterias en la sangre) y sobrevenir la muerte generalmente por pulmonía. • Eosinofília: es el aumento de eosinófilos por encima de 500 eos/mm3. Causas: las alergias son la causa más frecuente en los países no tropicales. El asma, la fiebre del heno, las reacciones a fármacos y a ciertos alimentos pueden cursar con cifras superiores a 2000 eos/mm3. La infestación por parásitos como trichinella espiralis que invaden los tejidos tienden a producir eosinofílias superiores a las alérgicas. Los parásitos intestinales suelen causan eosinofília menos pronunciadas. • Eosinopenia: menor a 0'04%, en 400 o más células. 1.3·- Alteraciones linfocitarias no malignas: Se considera que hay linfocitosis cuando hay linfocitos mayores a 4.000 linf/mm3 en el adulto, por encima de 7.000 en niños y por encima de 9.000 en la primera infancia. Se da una falsa linfocitosis cuando aparece neutropenia en infecciones virales puesto que aparecerá una linfocitosis relativa. Las alteraciones linfáticas no malignas pueden ser adquiridas o congénitas y afectan a los linfocitos T ó B ó a ambas poblaciones: • Alteraciones adquiridas: son de naturaleza cuantitativas y se presentan como mecanismos reactivos frente a infecciones (sobre todo virales o estados inflamatorios. En general se afectan tanto los linfocitos T como los B teniendo los frotis un aspecto heterogéneo. • Las afecciones congénitas: se caracterizan por reducción de leucocitos y deterioro de la inmunidad tanto celular (linfocito T) como humoral (linfocito B), teniendo el frotis un aspecto normal y homogéneo. Causas: infecciones como mononucleosis infecciosa, hepatitis, tuberculosis, tos ferina, sífilis y linfocitosis infecciosa. Se da también en las leucemias linfoides aguada y crónicas y en el hipertiroidismo. Enfermedades: ~ Mononucleosis infecciosas: es una enfermedad linfoproliferativa benigna. Causada por la infección del virus de Epstein-Barr (VEB). El diagnóstico exacto de esta enfermedad que generalmente cursa sin complicaciones puede establecerse por el reconocimiento morfológico de sus linfocitos atípicos y por métodos serológicos. Epidemiología: la trasmisión del virus tiene lugar mediante gotas de saliva infectadas. Incide de forma especial en niños, jóvenes siendo poco frecuente después de los 45 años de edad. Las razones es que alrededor del 80 - 90% de los adultos han sido expuestos y presentan inmunidad contra el virus. Síntomas clínicos: tras un periodo de incubación de entre 10 -50 días aparece un periodo prodrómico de algunos días de alteración, con dolor de cabeza y fatiga, seguido de otro periodo de fiebre, dolores en el cuello, linfoadeopatías [adeo- ganglios] Datos de laboratorio: aparece una leucocitosis entre 10.000 - 25.000leu/mm3 con aumento del número de linfocitos (linfocitosis absoluta), monocitos y grandes elementos mononucleares que corresponden a linfocitos T estimulados en respuesta a la colonización de los linfocitos B por el virus de VEB. La velocidad de sedimentación globular es moderada, la anemia es muy rara y aparecen elevadas ligeramente las enzimas hepáticas (GTP, GOT, GGT). Así como una LDH (lipoproteína de altar densidad) Pronóstico: es una virialis benigna que casi siempre se cura sin apoyo terapéutico notable [reposo en cama y medidas locales para la faringitis] ~ Linfocitosis infecciosa: es una enfermedad benigna contagiosa en los niños pequeños cuyo agente causal es probablemente un virus. El periodo de ocupación está entre 11 - 21 días. Los síntomas son leves, diarreas, fiebre e infección respiratoria. No hay faringitis ni linfoadeopatías. Los resultados del laboratorio ayudan a establecer el diagnóstico y los datos son: leucocitosis intensa (de 30.000 a 50.000 leucos) con un 60% al 95% de linfocitos siendo estos pequeños y homogéneos. No hay anemias y la velocidad es normal. El principal diagnóstico diferencial hay que realizarlo con la mononucleosis infecciosa. 1.4·- Monocitosis: Se consideran que existen cuando superan los 1.000 monocitos/mm3. Causas: infecciones como tuberculosis, endopatitis, brucelosis y paludismo y también aumentan en las leucemias monocíticas. En la tuberculosis el monocito desempeña un papel importante ya que los fosfolípidos que recubren el bacilo tuberculosos son degradados dentro del monocito, es decir, la Monocitosis es un signo de extensión del proceso tuberculosos. Los valores más altos de monocitosis se observan en la leucemia monocítica, así como en la enfermedad de Hodgkin y en las agranulocitosis inducidas por drogas. Enfermedad de Hodgkin: es un linfoma maligno (tumor de tejidos linfáticos) caracterizado por la hipertrofia de causa desconocida del tejido linfoadenoideo (ganglio linfático, bazo, médula ósea y amígdalas con fiebre periódica, anemia progresiva, esplenomegalia) picor (purito) y emaciación (enflaquecimiento extremo por causa morbosa). Los monocitos tienen un papel importante en las inflamaciones y en las reacciones inmunitarias de ahí que pueda observarse un aumento de estas células en estado de estado muy diverso. Los monocitos que se presenta en la etapa de recuperación de infecciones agudas se considera de signo favorable. 2·- Trastornos neoplásicos de los leucocitos. Neoplasia: es la neo (nueva) formación de tejido en el que la multiplicación celular no está totalmente controlada por los sistemas reguladores del organismos y que a veces tiene un carácter progresivo (tumor malignoneoplasia, neoplasiatumor maligno). 2.1·- Concepto de leucemia: Es una enfermedad maligna progresiva caracterizada por la proliferación incontrolada de un tipo de leucocito, que infiltra a la médula ósea y otros tejidos con aparición de células inmaduras, a veces morfológicamente anormales en la sangre periférica. Son enfermedades de tipo clonal, lo que quiere decir, que la malignidad afecta a una célula primitiva que viene directamente a través del factor ambiental. De esta forma se crea una progenie de células anormales cuya invasión y proliferación producen la enfermedad. ~ Etiología e incidencia: el origen de las leucemias es desconocido si bien se encuentran implicados factores genéticos, radiaciones, antígenos tóxicos o virus. Se ha visto leucemias en gemelos univitelinos, personas con defectos cromosómicos tienen mayor riesgo de padecer leucemias, por ejemplo: en el síndrome de Dawn en donde la incidencia de la leucemia es mayor en individuos normales. Los individuos expuestos a radiaciones y semejantes (rayos X y/o ð) tienen mayor disposición a padecerlos. Sustancias como el cloranfenicol, el benceno, ciertos colorantes, alquilantes también se relacionan con ella. Hay una impresión cada vez más firme de que la leucemia pueda ser de etiología vírica. Sin embargo es probable que no se deba a un solo factor si no a un conjunto de agentes etiológicos (genéticos, virus, radiaciones, sustancias, etc.) e incluso que la causa varíe de una persona a otra habiendo personas más predispuestas a padecerlo. Se calcula que cada año se presentan 6 nuevos casos de leucemia por cada 100.000habitantes. Del total de leucemias el 60% son agudas y el 40% son crónicas. En niños de hasta 7 años la leucemia constituye el 50% de todas formas de cáncer, después la frecuencia decrece para volver a aumentar a partir de la sexta década de la vida. Las leucemias agudas se manifiestan preferentemente en los primeros años de vida y a partir de los 60 años las crónicas son muy raras en la infancia. ~ Clasificaciones: en el siglo pasado, Virchow, clasificó las leucemias, según su evolución en crónicas y agudas. Las primeras se caracterizan por una mayor supervivencia de los pacientes, con células maduras en sangre periférica y las agudas por células inmaduras y evolución rápida. ~ El pronóstico de las leucemias agudas y crónicas ha sido cambiando: así se ha ido realizando notables adelantos con algunos tratamientos en las leucemias linfoides agudas (LLA) consiguiéndose remisiones hasta el 50% sin embargo no se puede decir lo mismo de la leucemia mieloide crónica (LMC) con un pronóstico y evolución desfavorables. Las leucemias agudas y crónicas se subdividen en mieloides y linfoides de acuerdo con el tipo celular que predomina. La leucemia no se limita a una línea celular hematopoyética sino que es un trastorno de una célula precursora y afecta a toda la progenie celular. La revisión morfológica: la histoquímica, la inmunología y la citogenética (determinación del cromosoma philadelphia,..), etc. Todas ellas constituyen unos medio imprescindibles para una mejor clasificación o tipificación lo que ayuda a obtener un mejor tratamiento del paciente. 2.1·- Leucemia crónicas: ~ Leucemia mieloide crónica: en 1951, Damesher, introdujo la expresión síndrome mieloproliferativo (síndrome que presenta proliferación medular o extra-medular de una o varias series celulares de la médula ósea) para designar a un grupo de alteraciones que tienen una similitud clínica y evolutiva y que puede presentar problemas de diagnóstico diferencial; entre ellas se incluyen la metaplasia mieloide (metaplasia: es la producción por las células de una especie de tejido diferente del que producen normalmente), la policitemia vera, la trombocitemia hemorrágica esencial (síndrome hemorrágico con un alto número de plaquetas mayor a 1.000.000 hiperplasia megacariocitos y hepatoesplenomegalia de curso letal) y la leucemia mieloide crónica. La leucemia mieloide crónica la vamos a estudiar por se interés clínico. Es una enfermedad con un crecimiento neoplásico primario de las células granulocíticas de la médula ósea con elevación excesiva de esta serie en la sangre periférica. Tiene un curso evolutivo bifásico (crónico y aguda). Su origen es desconocido aunque parece relacionarse con algunos medicamentos y algunas radiaciones. Representa aproximadamente el 20% de todos los casos de leucemia en los países occidentales, siendo mayor su incidencia entre los 40-50 años de edad. Curso clínico: el desarrollo de los síntomas y signos de la LMC es insidioso (malicioso con apariencia inofensiva); además son inespecíficos y comunes a otras patologías. En principio aparece una fase crónica que dura entre 3-5años que se caracteriza por: 1º En sangre periférica parecen cifras en 50.000-300.000 leucos/mm3 con presencia de todos los elementos inmaduros de la serie granulocítica (mieloblastos, promielocitos, cayados y segmentados). Es frecuente la presencia de eosinófilos y basófilos, anemia moderada normocítica, normocroma y en ocasiones presencia de eritroblastos. 2º Ácido úrico elevado debido al gran catabolismo celular. Existe un aumento de la proteína fijadora de la vitamina B12 sintetizada normalmente por los granulocitos. 3º La médula ósea destaca el aumento de células con gran predominio de la serie granulocítica. [Un 90% de pacientes de LMC presenta una anomalía cromosómica adquirida, el cromosoma philadelphia, que consiste en la translocación del material genético del cromosoma 22 al 9.] Para su diagnóstico diferencial también se determina la fosfatasa alcalina leucocitaria que alcanza niveles muy bajos en los granulocitos de estos pacientes. La mayoría de los pacientes de LMC sufre una transformación hacia una fase aguda o de crisis blástica en la que la leucemia se hace más agresiva y resistente a los tratamientos. Las células predominantes en esta fase son mieloblastos que invaden ganglios, sistema nervioso y sangre periférica apareciendo otras anomalías cromosómicas en sus células. La mayor aparte de los pacientes de esta fase aguda mueren en un plazo breve siendo un 10% los que mueren en fase crónica. El pronóstico de los pacientes con LMC dependerá de la rapidez con que aparezca la crisis blástica. Tratamiento: en su fase crónica se basa en la quimioterapia convencional. En la fase de crisis blástica sólo un 25% de los pacientes muestran respuestas favorables a la quimioterapia. La crisis blástica de la LMC sigue considerándose como la hemopatía maligna de peor pronóstico. Un grupo de antivirales llamado interferón han sido usados con buenos resultados. La única medida que a logrado la curación en un número considerable de enfermos es el transplante de médula ósea. Sus principales hermano histocompatibilidad. El transplante debe efectuarse en la fase crónica pues en la crisis blástica la tolerancia es muy baja. ~ Leucemia linfoide crónica: es una proliferación mononuclear de linfocitos B con gran capacidad de multiplicación de vida media muy larga que invaden ganglios linfáticos, médula ósea y sangre periférica. Es la leucemia más benigna, de mayor dilución. Tiene su máxima incidencia a partir de los 60 años. Constituyen el 30% de todas las leucopenias y el 75% de las formas crónicas. Su descubrimiento fortuito acontece en un 20% de los casos. Su etiología es desconocida y factores implicados en otras leucemias no parecen desempeñar aquí un papel importante. Clínica: pueden aparecer asintomáticos durante largos periodos de tiempo. Cuando la enfermedad progresa son frecuentes las adenopatías (enfermedad de los ganglios) y la esplenomegalia. Datos de laboratorio: lo más importante es la leucocitosis con cifras entre 50-100•109. siendo el 90% linfocitos de aspecto maduro y homogéneo. A medida que progresa la infiltración medular aparece anemia normocítica normocroma y trombopenia. Un 20% de los pacientes padecen anemia hemolítica adquirida autoinmune. Existen 2 variantes: 1·- Variante clásica: aparecen linfocitos pequeños de núcleos redondos apenas sin citoplasma. Hay descenso de hidrolasas ácidas. Los linfocitos proliferantes son frágiles (sombras Gumprecht). 2·- Variante de células estimuladas: tipo celular grande y citoplasma muy basófilo. Si bien es frecuente la aparición de un linfoblasto, es excepcional. La crisis blástica terminal en la leucemia linfoide crónica. La médula ósea aparece con una gran infiltración linfoide que supera el 40% de las células existentes lo que hace que la eritropoyesis, la granulopoyesis y la trombopoyesis quedan allegadas. No existe un cariotipo específico. Tratamiento: la mayor parte de los pacientes no requieren tratamiento salvo los que se encuentran en un estado avanzado en el momento del diagnóstico por lo que deben ser sometidos a quimioterapia. [Se han descrito otros tipos de trastornos linfoproliferativos de la LLC entre ellos la LLC tipo T que se caracteriza por un número de linfocitos no superior a 40 • 109fl, ausencia de adenopatías, infiltración medular escasa y linfocitos muy abundantes en ð-glucuronidasa y en fosfatasa ácida. Otro trastorno maligno linfoide es la leucemia proliferativa crónica de células B que cursa con esplenomegalia masiva, leucocitosis intensa superior a 300 • 106 mm3 con alta proporción de blastos en sangre y una mala repuesta a la quimioterapia.] 2.1·- Leucemia agudas: representan un 10% de todo tipo de cáncer humano y el 60% de las leucemias. Son proliferaciones malignas de las células hematopoyéticas inmaduras que se acumulan en médula ósea, sangre periférica, sistema retículo endotelial (conjunto de elementos celulares difundido por todo el organismo pero principalmente en el hígado (células Kuffer), bazo, ganglios linfáticos, médula ósea de función (hematopoyética, fagocitaria, metabólica, inmunitaria pigmentaria, etc), sistema nervioso central y otras áreas. Una vez confirmado el diagnóstico se aplica una quimioterapia combinada para conseguir su remisión. Cuando esta fracasa, la muerte sobreviene por hemorragia o infección. Clasificación: en 1976 un grupo de hematólogos ingleses, americanos y franceses propusieron un sistema de clasificación de las leucemias agudas basadas en criterios morfológicos y citoquímicos que se conoce como clasificación FAB y las divide en 2 grupos: A·- Leucemias mieloides agudas: • M1 Leucemia mieloblástica sin maduración • M2 Leucemia mieloblástica con maduración • M3 Leucemia promielocítica • M4 Leucemias mielo-monocítica • M5 Leucemia monocítica • M6 Eritroleucemia B·- Leucemias linfoides agudas: • L1 Leucemia linfoblástica homogénea • L2 Leucemia linfoblástica heterogénea • L3 Leucemia linfoblástica Burkit Aunque la leucemia aguda puede presentar a cualquier edad hay una incidencia máxima en la primera década; después la frecuencia decrece hasta los 30 años, edad en la que empieza a aumentar, elevándose de forma significativa partir de los 60 años. La mayoría de las leucemias infantiles son de tipo linfoide mientras que las del adulto de tipo mieloide. Fisiología: la leucemia aguda es consecuencia de la transformación maligna de las células precursoras hematopoyéticas. La población de células leucémicas neoplásicas debe alcanzar el tamaño de un tumor de 1•1012 células antes de que una leucemia pueda ser diagnosticada mediante el examen de médula ósea. A medida que crece la población de células neoplásicas la concentración de células normales disminuye. Alrededor del 50% de los individuos enfermos muestran un cariotipo normal. Clínica: la insuficiencia en la hematopoyesis normal es la consecuencia más grave de las leucemias agudas. Los síndromes más frecuentes se deben a: anemia, trombocitopenia o neutropenia que traen consigo episodios hemorrágicos e infecciones. La exploración física revela hepta y esplenomegalia y en ocasiones linfadenopatias pueden infiltrarse en cualquier tejido (bazo, hígado y ganglios linfáticos). Datos de laboratorio: generalmente se observa anemia normocítica normocroma (en una leucemia aguda siempre está presente la anemia y trombocitopenia). Los leucocitos pueden estar aumentados, normales o disminuidos. De echo más del 50% de los pacientes no presentan leucocitosis, mientras que en las leucemias crónicas siempre aparecen estas. La médula ósea es hipercelular aunque a veces puede ser normocelular e hipocelular. También hay anomalías de maduración en la serie roja, blanca y plaquetaria. En todos los tipos de leucemias hay un exceso de ácido úrico, producto normal del metabolismo de los ácidos nucleicos. Tratamientos y pronósticos: los índices de remisión de leucemias linfoides agudas y leucemias mieloides agudas a mejorado notablemente en la últimas décadas, entendiendo como remisión al periodo en el que desaparecen los signos hematológicos y clínicos de las enfermedades. El 50% de los niños con LLA pueden entrar en una remisión completa. En los adultos el pronóstico no es tan favorable. El tratamiento de la LMA no ha tenido el mismo éxito que la LLA (el pronóstico de las leucemias agudas en adultos es en general bastante pesimista). La quimioterapia es el tratamientos de elección al problema; el problema es que además de erradicar células malignas de la médula ósea también erradica células normales. Los transplantes de médula ósea son una esperanza para los enfermos leucémicos. ~ Leucemia mieloide aguda o mieloblástica: se clasifican de M1 a M6 que es el grado de maduración celular y el grado de participación de la célula granulocítica, monocítica o eritroblástica. Hay que señalar que es imprescindible el estudio del conjunto del frotis de sangre periférica o de médula ósea. • Variedad M1 o leucemia mieloblástica sin maduración: es lo más común en adultos y en niños menores de 1 año. La célula predominante en sangre periférica es el mieloblasto. Prácticamente existe una ausencia de estadios posteriores. Los blastos son positivos a los mieloperoxidasa en número superior al 3%. Presencia de bastones de Auer (agujas o bastones azurófilos en el citoplasma). Los mieloblastos tienen normalmente el núcleo redondo con cromatina laxa y presencia de nucleolos. En más del 50% de los pacientes se dan aberraciones cromosómicas en las células leucémicas. • Variedad M2 o leucemias mieloblásticas con maduración: las células predominantes en sangre periférica son promielocitos y mieloblastos que pueden presentar más del 50% del total. Son muy frecuentes los bastones de Auer. La Médula ósea es hipercelular. • Variedad M3 o leucemia promielocítica: las células predominantes son promielocitos tanto en sangre periférica como en médula ósea. Presencia de bastones de Auer. La reacción de peroxidasa es positiva. El dato más significativo en el diagnóstico es el sangrado. Es muy rara en niños. • Variedad M4 o leucemia mielocítica: tiene células primitivas de la estirpe monocítica (monoblasto y promonocito) y células primitivas de la serie mieloide (promielocito y mieloblasto). El mieloblasto y monocito superan el 25% de las células. Así mismo la suma de mieloblasto y promonocitos supera el 20% tanto en sangre periférica con en médula ósea; lo cual se comprueba por la tinción histoquímica de esterasa donde la serie monocitoide es positiva. • Variedad M5 es la leucemia aguda monocítica: es una masiva proliferación de monocitos en distintas etapas de madurez sin embargo, el mieloblasto no supera el 10% del total de células. • Variedad M6 o eritroleucemia: se caracteriza por una proliferación leucémica mixta de la serie mieloide y eritroblástica excediendo esta última en más de un 50%. Son frecuentes las diseritropoyesis (dis- imperfecta anormal) tanto nucleares como citoplasmáticas. ~ Leucemia linfoide aguda o leucemia linfoblástica: son enfermedades linfoproliferativas malignas con especial incidencia en niños que se caracterizan por una invasión linfoide neoplásica de médula ósea, sangre periférica y ganglios linfáticos siendo las células predominante el linfoblasto las LLA presentan diferentes subtipos morfológicos e inmunológicos. Sin tratamiento la supervivencia es corta pero con la quimioterapia actual se alcanzan periodo de remisiones muy prolongados y muchos pacientes se dan por curados. Los síntomas se deben a la anemia trombocitopénica y neutropénica. Incluyen fatigas, palidez, fiebre y anorexia y en un 80% de los casos los pacientes presentan un dolor óseo. Datos del laboratorio: aparece leucocitosis siendo frecuente la neutropenia. Hay linfoblastos en sangre periférica y anemia normocítica normocroma, la médula ósea hipercelular con infiltración linfoide. Superando el 65% los linfoblastos. Estos son peroxidasas negativos. Según la clasificación de la FAB se han descrito 3 tipos: L1 o leucemia linfoblástica homogénea: es la LLA más común en los niños y la mejor pronosticada. Se caracteriza por la homogeneidad de los linfoblastos que son pequeños con núcleo redondo con hendiduras, nucleolos no visibles y citoplasma escaso con basofília moderada. Pueden ser de tipo inmunológico B ó T. L2 o leucemia linfoblástica heterogénea: es las más frecuente en adultos. El linfoblasto presente puede variar mucho en su tamaño. Hay presencia de nucleolos y el citoplasma es abundante con basofília variable. Los linfoblastos son peroxidasas negativas. L3 o leucemia linfoblástica de Burkit: es la LLA menos frecuente. Se presentan en niños y adultos. Los linfoblastos son homogéneos, grandes y generalmente vacuolizados. El núcleo es redondo con 1 ó 2 nucleolos. 3·- Mieloma múltiple Es un tumor de células plasmáticas localizado fura de la médula ósea caracterizado por lesiones óseas diseminadas para proteínas en sangre y orina y a veces trombocitopenia y leucopenia. El mieloma múltiple o plasmocitoma es una proliferación neoplásica monoclonal de células plasmática que sintetizan generalmente globulinas más o menos completos. Se encuentra por lo general en personal mayor de 60 años. Se caracteriza por: lesiones óseas diseminadas, proteinurias de Bence Jones (cadenas ligeras de orina), hipercalcemia frecuente, velocidad de sedimentación globular muy elevada y plasmocitos en médula ósea entre 1090%. El hemograma es poco significativo observando en el frotis sanguíneo pilas de monedas eritrocitarias. La inmuno-electroforesis revela en inmunoglobulina de tipo monoclonal Ig G o más raramente Ig A ó Ig D. La producción de la inmunoglobulina es anormal y la síntesis excesiva de cadenas ligeras conduce a su excreción urinaria (proteínas de Bence Jones). El mielograma es el mejor examen para poner de manifiesto las células plasmáticas muchas de ellas multinucleadas y con características atípicas. Una complicación de esta enfermedad es el deterioro de la función renal con al formación de cilindros proteicos que destruyen los túbulos renales. En ocasiones pero no siempre pueden observarse en sangre periférica células plasmáticas o linfocitos plasmocitoides. Tratamiento: es con agentes alquilantes, radiaciones y predisoma. La infección es la causa más frecuente de muerte que generalmente sobreviene en un plazo no superior a 3 años a partir del diagnóstico. 4·- Macro-globulinemia de Waldenström (Ig M) La macro-globulinemia de Waldenström es un tumor de células linfoplasmocitoides que se asocia a linfoadenopatías y a hepato-esplenomegalia y que conlleva a la hiperproducción monoclonal de Ig M. No suele producir lesiones óseas no insuficiencia renal y sus principales manifestaciones clínicas derivan de la intensa hiperviscosidad que generan. En sangre periférica también se observa una anemia moderada, una agrupación de los hematíes en pilas de monedas y un aumento muy importante de la VSG. En la médula ósea hay una proliferación difusa de células linfoides y de células plasmocitarias que pueden tener atipias además suele abundar en ella los mastocitos hísticos basófilos. Su pronóstico es más benigno que el del mieloma múltiple y su tratamiento se basa en reducir la viscosidad de la sangre y en el empleo de citostáticos alquilantes. 5·- Índices leucocitarios Son parámetros para evaluar el grado de madurez de los leucocitos y en concreto de los neutrófilos. En la elaboración de estos índices no se tienen en cunta las formas juveniles inmaduras de los eosinófilos y basófilos debido a su mucha menor presencia en sangre periférica. Hay 2 formas de determinarlos la de Schilling y la de Arneth. • Recuento de Schilling: consiste en cuantificar el número de formas juveniles y el de formas maduras de los neutrófilos presentes en sangre periférica y relacionar ambos valores. ~ Schilling dividió a los neutrófilos en 2 grandes grupos: * Formas juveniles y dentro existen mielocito, metamielocito, neutrófilos en banda o en cayado. * Formas maduras: segmentados. Si al observar 2 células en un frotis se duda a la hora de clasificarlas entre una forma menos madura y otra más madura se ha de incluir esta en la última categoría. Estas células representan normal en sangre periférica los siguientes porcentajes: 1·- Mielocito ------------------------------ 0% 2·- Metamielocito ---------------------- 0-1% 3·- Neutrófilos en banda o en cayado 3-5% 4·- Neutrófilo segmentado ---------- 40-75% ~ Cálculo del índice de Schilling: el índice de desviación de Schilling se calcula de la siguiente forma. % formas juveniles Índice de Schilling = -------------------------- % formas maduras ~ Valoración del índice de Schilling: en sangre periférica existe una forma juvenil por unas 16 formas maduras. Cuando aumenta el % de las formas juveniles en los neutrófilos se dice que hay una desviación a la izquierda; y cuando asciende el % de segmentados se dice que hay una desviación a la derecha. • Recuento de Arneth: consiste en contar el número de lobulaciones que tiene una determinada cantidad de neutrófilos y seguidamente calcular el número de lóbulos que hay por cada neutrófilo. Esto suele hacerse por autoanalizadores. ~ Clasificación de los neutrófilos según Arneth: los agrupa según el numero de lobulaciones de su núcleo en 5 grupos: Grupo 1 neutrófilo con un núcleo no segmentado, es decir, sin lobulaciones Grupo 2 neutrófilo con un núcleo dividido 1 veces, es decir, 2 lóbulos Grupo 3 neutrófilo con un núcleo dividido 2 veces, es decir, 3 lóbulos Grupo 4 neutrófilo con un núcleo dividido 3 veces, es decir, 4 lóbulos Grupo 5 neutrófilo con un núcleo dividido 4 veces, es decir, 5 lóbulos ~ Cálculo del índice de lobularidad: conociendo el número de neutrófilos de cada tipo de Arneth que hay en sangre periférica se puede calcular el número de lóbulos que tiene una determinada cantidad de neutrófilos y por tanto es posible saber el número de lóbulos por neutrófilos. Los autoanalizadores proporciona esta cifra que es conocida como índice de lobularidad (IL). ~ Valoración del índice de lobularidad: el índice de lobularidad normal está comprendido entre 1'9-3. Si es menor a 1'9 se dice que hay una desviación a la izquierda y si es mayor que 3 se dice que hay una desviación a la derecha. ~ Determinar el índice de lobularidad: 1·- Material necesario: microscopio, bolígrafo, papel, aceite de inmersión y como muestra una extensión sanguínea muy fina. 2·- Técnica: Enfocar con objetivo de inmersión, observar neutrófilos y contar sus lobulaciones Considerando que hay un lóbulo cuando una parte del núcleo está unida al resto solamente por un fino puente de cromatina Clasificar los neutrófilos según Arneth al tiempo que se cuentan los lóbulos de cada neutrófilo observado, se termina el recuento con al menos 100 neutrófilos; se calculará el índice de lobularidad con la siguiente fórmula: Número de lóbulos I.L. = ------------------------------ Número de neutrófilos ~ Interpretación clínica de los resultados: La desviación a la izquierda puede acompañarse de neutrofília o neutropenia. La desviación a la izquierda con neutrofília es típica de algunas infecciones agudas por ejemplo: apendicitis, en caso de sepsis (microorganismo en sangre). La desviación a la derecha por neutropenia es característica de infecciones como fiebre tifoidea o brucelosis. Desviación a la derecha: puede darse en varios proceso patológicos entre los que destacan las anemias megaloblásticas curiosamente también se produce en la agonía. TEMA XV RECUENTO DIFERENCIAL AUTOMATIZADO El recuento diferencial leucocitario es una de las determinaciones más solicitadas al laboratorio de análisis; por ello unos resultados correctos y precisos son de vital importancia. Actualmente la automatización permite mejorar la especificidad, la exactitud, disminuir los errores y abaratar los costes de los recuentos diferenciales de leucocitos. En los sistemas actualmente empleados se dispone de distintos procedimientos para la realización del recuentos diferenciales de leucocitos: • Sistema digital de imagen (cuenta 500 leucocitos) • Sistema por flujo continuo (cuenta 10.000 leucocitos): son sistemas que utilizan la medida de diferentes propiedades de los leucocitos en suspensión además informa sobre el recuento celular completo. Se basan en : 1·- En la medida de la impedancia y en la conductividad celular que informarán sobre el tamaño de la partícula y su estructura celular interna. 2·- Medida de la luz láser y halógena. 3·- Utilización de técnica histoquímicas sobre la suspensión leucocitaria y estudio de los distintas poblaciones leucocitarias por métodos ópticos. Según su capacidad de discriminación se pueden clasificar en sistema de 3 o de 5 poblaciones. • Análisis o sistema digital de imagen: se basa en la identificación de los leucocitos mediante análisis con ordenador de las imágenes de frotis sanguíneos obtenidas al microscopio. La extensión de sangre uniformemente teñida se coloca sobre la platina de un microscopio. El movimiento de la platina es controlada mediante ordenador de esta forma se reconoce la superficie del frotis y el ordenador detiene el movimiento cuando encuentra un leucocito en su campo de visión. Las imágenes ópticas son registradas por una cámara de televisión y digitalizadas por el ordenador que compara las características de las células con una memoria en al que se incluye las características correspondientes a los distintos tipos celulares, si las características coinciden con el del tipo celular normal se identifica como tal de los contrario se clasifica como desconocido. Las coordenadas de esta últimas células son archivadas para que el técnico las pueda clasificar. Los leucocitos son clasificados en 5 categorías en neutrófilos (segmentados o no segmentados), linfocitos, monocitos, eosinófilos y basófilos. Algunos sistemas son capaces de identificar otros elementos celulares que eventualmente su pueden encontrar en sangre periférica como mielocitos, metamielocitos, promielocito, células plasmáticas, linfocitos atípicos o blásticas. Estos sistemas identifican a los leucocitos en base a aspectos morfológicos por lo que además de requerir células perfectamente conservadas necesitan que existan una muy pequeña variación tintorial para evitar la clasificación errónea de células. Es por ello de gran importancia el empleo de sistemas de realización de frotis sanguíneos adecuados para la obtención de preparaciones homogéneos y con la mayor similitud tintorial. Se recomienda por ello la utilización d sistemas automáticos de tinción. La fórmula leucocitaria muchos de estos contadores proporcionan además información sobre la morfología sobre hematíes y cuantificación de plaquetas. Presentan como mayor inconvenientes su bajo rendimiento tanto por el número de células analizadas (máximo 500) como por su escaso velocidad en la clasificación de las mismas. • Sistema por flujo continuo: ~ De 3 poblaciones: son los llamados sistemas de análisis del volumen de leucocitos la medida del tamaño leucocitario sobre un gran número de células permite elaborar una curva de distribución diferenciando 3 poblaciones leucocitarias: pequeñas (linfocitos), mediana (monocitos, eosinófilos, basófilos, linfocitos grandes) y grandes (neutrófilos). Los autoanalizantes diferenciales basados en este principio pueden utilizar 2 métodos distintos de obtención de poblaciones leucocitarias: análisis de los volúmenes del núcleo, o bien, análisis de volumen de las células intactas. ~ Sistemas de 5 poblaciones: estos sistemas obtiene el análisis diferencial de la de acuerdo con su tamaño y su comportamiento citoquímico antes determinados colorantes. Estos sistemas tienen la ventaja de analizan rápidamente mayor número de células (10.000) y reducir significativamente el error estadístico de recuento. Todas las categorías no clasificada resulta difícil de clasificar. Cuando se producen resultados anormales se realiza un frotis sanguíneo sobre cada célula individual son aplicables 3 métodos: 1·- Citoquímico: por la acción de un reactivo en distintos canales como es el canal de la peroxidasa alcalina. 2·- Medida del volumen por impedancia 3·- Medida de la transmisión óptica: para obtener información sobre la estructura interna de las células. Estos aspectos son analizados y comparados por la parte informática del aparato para producir unos resultados precisos exactos y fiables. De estos parámetros obtiene resultados referentes 4 poblaciones leucocitarias: linfocitos, monocitos, neutrófilos e eosinófilos y adicionalmente 2 poblaciones más las grandes células inmaduras (LUC) y los linfocitos atípicos. La peroxidasa es una enzima contenida en el citoplasma de todos los leucocitos excepto los linfocitos en cantidad variable y esta característica es utilizada para la obtención de la fórmula leucocitaria clásica. Esta enzima actúa sobre el agua oxigenada con sustrato y el 4-cloro-1-naftol como cromógeno en función de la cantidad de peroxidasa contenida en cada célula se produce una mayor o menor absorción de luz. De acuerdo con la información obtenida en el canal de la peroxidasa se obtiene una representación gráfica (distinta según el aparato) en al que se observa diferentes zonas. Los basófilos son detectados en un canal independiente (canal de lobularidad) mediante un reactivo son destruidos las membranas de todos los leucocitos excepto la de los basófilos. TEMA XVI TÉCNICAS CITOQUÍMICAS DE IDENTIFICACIÓN LEUCOCITARIA Como complemento a los métodos de tinción hematológicos se han desarrollado una serie de técnicas histoquímicas que permite la identificación de ciertas sustancias presentes en la célula. Fundamentalmente es posible detectar y localizar enzimas y sustancias inorgánicas. Con estas técnicas se mejora la diferenciación celular ya que es posible establecer con mayor exactitud el grado de maduración y funcionalidad de las células. Así mismo son válidas para el diagnóstico de las diferentes leucemias y para valorar el tratamiento anti-leucémico. En la realización de una técnica histoquímica tendremos 3 procesos fundamentales: 1·- Fijación de la extensión: su fin es evitar el deterioro celular y impedir la difusión de sustancias al medio extracelular. La fijación se puede realizar con sustancias como el metanol, etanol y acetona y actúan coagulando las proteínas celulares. Así como el formaldehído y glutaraldehído que actúan formando puentes intra e ínter proteicos de manera que se alteran menos la estructura celular. Es importante eliminar todos los restos de fijador para evitar interacciones con el compuesto que deseamos analizar. 2·- Incubación: al poner en contacto las células con el medio de reacción se liberan unos productos que bien por sí mismos o bien combinando con otros dan lugar a un precipitado apreciable al microscopio óptico. En el caso de identificación de enzimas debemos controlar bien el pH y la temperatura para optimizar la reacción. 3·- Tinción de contraste: se realiza para facilitar la identificación morfología de la célula en la que se ha producido la reacción. En caso de observadores muy experimentados se puede suprimir este tercer paso. 1·- Tipos de reacciones 1.1·- Identificación de carbohidratos A·- Tinción del ácido periódico de schifff (PAS): pone de manifiesto la presencia de glucógeno y mucopolisacáridos en el citoplasma celular. Se aprecia un precipitado de color rojo-púrpura intenso en el interior de las células PAS positivas como son los polinucleares neutrófilos, 1020% de los linfocitos normales, los megacariocitos, plaquetas y en algunas ocasiones las células plasmáticas. Esta tinción se utiliza sobre todo para el diagnóstico de leucemia linfática aguda donde se observa una positividad intensa con formación de mazacotes teñidos. Así como la eritroblastos. 1.2·- Identificación de enzimas A·- Tinción de la peroxidasa: es un enzima contenida en cantidad variable en todos los leucocitos a excepción de los linfocitos. En base a una actividad peroxidásica fuertemente positiva en los granulocitos, débil en los monocitos y negativa en los linfocitos,, se puede realizar una fórmula leucocitaria los modernos contadores hematológicos. En este diagrama distinguimos: 1º·- Linfocitos: son las células más pequeñas y carecen de actividad peroxidasa (A) 2º·- Monocitos: son ligeramente mayores y con más peroxidasa (B) 3º·- Neutrófilos: son de mayor tamaño y ricos en peroxidasa (C) 4º·- Eosinófilos: de menor tamaño que los neutrófilos y muy cargado de peroxidasa D 5º·- LUC: son células de gran tamaño no teñidas (large unstained cells) (E) presentes en la sangre en una proporción del 4% aproximadamente. No tiene actividad peroxidasa. Se corresponden con linfocitos grandes hiper-activos, o con células patológicas como linfoblastos, mieloblastos, etc. Con estos datos el autoanalizador obtiene el MPXI o índice de actividad peroxidásica media de la población de neutrófilos. Si este factor está fuera de los valores normales indicará una patología de la serie mieloide. También permite diagnosticar los déficit de peroxidasa congénita o adquiridas, por una imagen característica en el diagrama de análisis de poblaciones. B·- Tinción de esterasas: son enzimas que hidrolizan los ésteres de cadena corta de los ácidos grasos. Existen muchas clases de esterasas que se diferencian en el sustrato en el que actúan y los niveles de pH óptimos necesarios. La mayoría de las técnicas histoquímicas se basan en la hidrólisis de los sustratos para acabar por formar precipitados coloreados visibles al microscopio óptico, tras su unión con el reactivo. Dependiendo de la esterasa que pretendamos localizar, encontraremos una interpretación de resultado diferente; por ejemplo: los neutrófilos normales dan reacción negativa a la tinción de la ðnaftil-butirato-esterasa y son positivos a la tinción de la naftol As-D cloroacetato esterasa. C·- Tinción de fosfatasa: • Fosfatasa ácida leucocitaria (FAL): es un enzima lisosómico que consta de varios isoenzimas. En condiciones normales la mayoría de las células hemáticas son fosfatasa ácida leucocitaria en diversos grados; por eso su utilidad diagnóstica es algo limitada. Su utiliza principalmente en el diagnóstico diferencial de los síndromes linfoproliferativos en la llamada leucemia de las células peludas donde los tricoleucitos (cél. peludas) presentan una intensa positividad fosfatasa ácida junto con su aspecto morfológica peculiar (cél. peludas) Leucemia de las células peludas o tricoleucemias: es un raro trastorno clínicamente variable en su manifestación y de aparición insidiosa. Se da proliferación de las células anormales en los órganos retículo-endoteliales y sangre. Esplenomegalia. Las células peludas tienen un diámetro entre 10-20micras (tamaño medio), núcleo entre redondo y ovalado, aunque algunos tienen forma acampanada o mellada. Cromatina uniformemente reticular, nucleolos pequeños e insignificantes, en algunas células la cromatina está más condensada y asemeja a un linfocito. Cantidad de plasma moderado que presenta numerosas proyecciones parecidas a pelos y bordes deshilachados que adoptan color gris con el colorante de Wright y contiene fosfatasa ácida. • Fosfatasa ácida granulocitaria (FAG): es un enzima que se encuentra en los gránulos secundarios de los leucocitos polimorfonucleares. Sólo se valora las células maduras o los neutrófilos en banda y se realiza una clasificación de la actividad FAG en 3 grados: Grado 0 si no existe precipitado en el interior celular Grado 1 si hay un ligero precipitado Grado 2 si el precipitado es abundante Se calcula el índice de FAG anotando el grado de reacción observado en cada una de 100 células distintas. Después se suma el número de células con grados 1 y 2. En un individuo normal el índice de FAG se sitúa entre 20 y 80. Podemos encontrar valores aumentados en infecciones, embarazo y síndromes mieloproliferativo, así como en el tratamiento con hormonas esteroideas. D·- Tinción de la ð-glucuronidasa: es una hidrolasa ácida de localización lisosómica aunque también aparece en el aparato de golgi o en el retículo-endoplasmático. La positividad se observe como una granulación rojiza y es intensa en macrófagos, células plasmáticas y linfocitos T, mientras que los monocitos y los neutrófilos presentan una positividad difusa o son negativos. Se usan para diferenciar los síndromes linfoproliferativos (tricoleucemias, FAL positiva, ðglucuronidasa negativa). 1.3·- Identificación de lípidos A·- Tinción del negro de Sudán-B: con esta técnica se tiñen las sustancias lipídicas como los ésteres fosfolípidos y grasas neutras. Se aprecia una granulación gris oscura en los polimorfonucleares neutrófilos y en sus precursores. Los monocitos pueden presentar algún gránulo disperso y los linfocitos son negativos. Su interpretación es similar a la de las peroxidasas. TEMA XVII ANÁLISIS CUALITATIVO DE LA HEMOGLOBINA POR LA ELECTROFORESIS La hemoglobina es una proteína constituida por 2 porciones: • Un grupo prostético que recibe generalmente el nombre de hemo o hem que proporciona a la sangre su color rojo característico • Y otro grupo proteico llamado globina. El grupo hemo consta a su vez de ferro-porfirinas idénticas cada una de ellas está compuestas por una proto-porfirina IX y un átomo de hierro en estado ferroso. La proto-porfirina IX está formada por 4 pirroles. El átomo de hierro está situado en el centro de estos y se une a cada uno de ellos a través de un átomo de nitrógeno que estos poseen. La globina consta de 4 cadenas polipeptídicas iguales 2 a 2. Cada cadena polipeptídica de la globina se engarza (une) a través de un aminoácido, histidina, al átomo de hierro de su ferroporfirina correspondiente. Hay varias clases de cadenas de globinas que son las siguientes: ð, ð, ð, δ, ð. TIPOS DE HEMOGLOBINA NORMALES Se distinguen distintos tipos de hemoglobinas atendiendo a las cadenas globínicas que poseen. Las principales son: • Hb A: está formada por 2 cadenas ð y 2 cadenas ð. Es ala mayoritaria en el adulto ya que constituye el 97% de la hemoglobina total. • Hb A2: está formada por 2 cadenas ð y 2 cadenas δ. En el adulto constituye un 2'5% de la hemoglobina total. • Hb F o fetal: está formada por 2 cadenas ð y 2 cadenas ð. Es la más abundante desde el cuarto mes de embarazo hasta los 6 meses de edad. • Hb Portland: está formada por 2 cadenas δ y 2 cadenas ð • Hb gower I: está formada por 2 cadenas δ y 2 cadenas ð • Hb gower II: está formada por 2 cadenas ð y 2 cadenas ð. Estas tres son las mayoritarias en el embrión. TIPOS DE HEMOGLOBINAS ANORMALES Son aquellas que tienen alguna alteración química en su molécula o bien que aparecen en una etapa distinta a la normal. Se pueden clasificar en 3 grupos: a) En ellas se reemplazan un aminoácido de las cadenas de polipéptidos por otro distinto. Entre ellas cabe destacar las hemoglobinas C, D, E, G y S. b) Se caracteriza por la ausencia o menor producción de una o más cadenas de polipéptidos. A este tipo se le denomina Talasemias. c) Continuación de síntesis de hemoglobina fetales o embrionarias en el adulto, sobre todo ocurre con Hb F. En el laboratorio de análisis para distinguir las hemoglobinas tanto normales como anormales se pueden emplear métodos químicos, cromatográficos o electroforesis. En este tema estudiaremos el análisis cualitativo de hemoglobina por electroforesis. TÉCNICAS ELECTROFORÉTICAS Son técnicas utilizadas para la separación de mezclas de distintas moléculas que tienen carga a un pH determinado. Se denomina electroforesis al transporte de partículas en función de sus cargas y peso molecular al aplicar un campo eléctrico. En el transporte electroforético a la fuerza del campo se opone la resistencia del medio produciéndose una velocidad de emigración de las partículas constantes. La separación electroforética se basa en la distinta velocidad y sentido de migración que tienen la molécula según su naturaleza química y carga eléctrica en un campo eléctrico. Equipo de electroforesis: consta de los siguientes componentes: 1·- Cubeta: recipiente cuyo fondo esté dividido en 2 partes. En ella se introduce el tampón (reactivo para que no varíe mucho el pH) utilizado para mantener el pH constantes durante el proceso. Una de las 2 partes va conectada al ánodo (parte positiva) y la otra al cátodo (parte negativa) los cuales se cubren con el tampón. El tampón nunca debe poner en contacto las 2 partes de la cubeta. La cubeta lleva en su interior un puente sobre el que se coloca el soporte inerte con el fin de mantenerlo por encima del tampón. 2·- Fuente de alimentación: el requisito fundamental que debe poseer la fuente de corriente eléctrica que sea capaz de mantener una corriente constante dado que la resistencia variará durante un recorrido electroforético la corriente debe cambiar concordante con esa variación, razón por la cual hay que utilizar una fuente de alimentación dotada de un sistema estabilizador, va conectado con un cable al polo positivo de la cubeta y con otro al negativo. 3·- Aplicador de muestras: consta de un soporte que lleva en un extremo un asa rectangular la cual dosifica un volumen constante. 4·- Soporte inerte ó fase estacionaria: su misión consiste en oponer resistencia al movimiento de la moléculas y evitar la difusión pasiva. Se pueden utilizar varios tipos de soporte: papel de filtro, geles de agar (agarosa), almidón y tiras de acetato de celulosa. 5·- Tampón: su misión es mantener el pH constante en un nivel en que las moléculas se encuentren disociadas. El intervalo de pH más utilizado es el 8'4-8'9 aunque en algunos casos como en la electroforesis con agar-citrato se utiliza un pH entre 6-6'5. Los tampones más utilizados son el barbital y tris-EDTA-borato (TEB). ANÁLISIS DE HEMOGLOBINA POR ELECTROFORESIS EN ACETATO DE CELULOSA Fundamento: se basa en la separación electroforética de hemoglobina obtenidos por hemólisis de la muestra sanguínea sobre un soporte de acetato de celulosa. A pH=8'4 casi todas las hemoglobinas tienen carga negativa por lo que emigrarán hacia el polo positivo. Pequeños cambios en la cadena de globina modifican la carga y en consecuencia la migración. Por lo que al finalizar el proceso las distintas hemoglobinas se encuentran a distinta distancia del punto de aplicación de la muestra la cual comparada con la de diferentes controles nos indica el tipo de hemoglobina de que se trata. Las distintas hemoglobinas separadas se pueden cuantificar posteriormente por densitometría o por espectrofotometría tras la tinción. La aplicación de esta técnica consiste en la detección y/o cuantificación de las hemoglobinas existentes en una muestra sanguínea principalmente son A1, A2, C, D, E, F, S, G. Material: • Cubeta de electroforesis y alimentador • Aplicador de muestras • Tiras de acetato de celulosa • Centrífuga • Tubos de centrífuga • Pipeta Pasteur • Estufa Pasteur • Placas de vidrio • Cubetas • Pinzas • Papel de filtro Reactivos: • Sangre entera anticoagulada • Colorante proteico (azul brillante de coomasie o ponceau S) ó colorante específico de hemoglobina (orto-toluidina) • Solución de colorante • Solución transparentadora • Tampón pH=8'6 • Suero fisiológico • Cloroformo TÉCNICAS PARA LA REALIZACIÓN DE LA ELECTROFORESIS • Preparación del hemolizado: se centrifuga 2 ó 3 ml de sangre anticoagulada a 3.000 rpm durante 5 minutos. • Se elimina el plasma sobrenadante con una pipeta Pasteur. • Lavar los hematíes 3 veces son suero fisiológico. • Tras el lavado se hemolizan colocándolos en una solución hipotónica, para ello mezclamos en un tubo de centrífuga 1 volumen de hematíes (2ml), 1'5 de volumen de agua destilada (3ml) y medio volumen de cloroformo (1ml). • Agitar fuertemente y centrifugar 20 minutos a 3.000rpm tras lo cual la capa clorofórmica quedará en el fondo del tubo, la acuosa en la superficie y los restos de hematíes hemolizados en la interfase. • Separar el sobrenadante con una pipeta Pasteur con lo cual realizaremos la electroforesis. La hemoglobina se encuentra en la capa acuosa mientras que otros productos de la hemólisis como leucocitos,... sedimentan tras la centrifugación. En la capa clorofórmica quedan retenidos las sustancias apolares. REALIZACIÓN DE LA ELECTROFORESIS Preparamos el equipo de electroforesis para lo cual conectamos la cubeta a la fuente de alimentación y esta al fluido eléctrico. Llenar los 2 lados de la cubeta con la solución tampón evitando la formación de burbujas. El tampón debe cubrir los electrodos pero no pondrá en contacto las 2 partes de la cubeta. Sumergir con una pinzas las tiras de acetato de celulosa en la solución tampón durante un mínimo de 5 minutos y posteriormente eliminar el exceso de esta colocándola entre 2 papeles de filtro. Coloca la tira sobre el puente de las cubeta de modo que sus extremos queden sumergidos en la solución tampón de cada una de las 2 cámaras de la cubeta. Las tiras se debe montar con la cara absorbente hacia arriba. Esto se puede constatar mediante la esquina cortada que esta debe quedar con al tira frente al operador hacia la derecha y abajo. Colocar la tapa superior de la cubeta y dejar estabilizar las tiras durante 2 minutos. Marcar con un bolígrafo o lápiz graso la zona de aplicación situado a 1cm del borde catódico. En un portaobjetos se coloca unas gotas de la muestra obtenida en el apartado anterior y cargamos el aplicador poniéndolo en contacto con la muestra. Depositar la solución de hemoglobina contenida en el aplicador en la zona marcada con anterioridad por simple contacto de este con la tira. Poner en marcha el alimentador y regularlo 1 hora a 250 voltios o 15 minutos a 450 voltios. TINCIÓN DE LAS TIRAS Tras finalizar la electroforesis extraer con cuidado las tiras y sumergirlas en una cubeta con colorante durante 10 minutos. A continuación extraerlas y decolorar efectuar 3 ó 4 lavados de 2 minutos bajo agitación en la solución de colorante contenida en una cubeta Tras esta operación el fondo de la tira debe quedar completamente blanco. Finalmente secar las tiras. Sumergir las tiras en metanol absoluto durante a 3-5 minutos para fijarlas. Transparentar las tiras por inmersión en una solución transparentar durante 10 minutos. Depositarla sobre una placa de vidrio y colocarlas en la estufa a 100ºC-110ºC durante 10-15 minutos hasta su total transparentado. RESULTADOS E INTERPRETACIÓN En muchos caso además de la detección cualitativa es necesario cuantificar las distintas hemoglobinas; el análisis cuantitativo se puede realizar por densitometría o por espectrofotometría La cuantificación por densitometría se realiza con los densitómetros los cuales poseen un sistema mediante el que son arrastradas las tiras teñidas y se les hace pasar por un haz de luz que incide sobre un detector. La luz que llega al detector produce una señal eléctrica que se recoge sobre un registrador. Cuanto mayor sea la concentración de hemoglobina menor es la luz que llega al detector y mayor la deflexión del registrador. Los densitómetros poseen integradores que miden el área de cada tipo y calcular el porcentaje de cada fracción. Además conocida la cantidad de muestra que se ha separado es posible conocer la de cada fracción. La cuantificación espectrofotométrica se puede realizar de 2 formas: 1·- Se cortan las distintas bandas teñidas y se eluye el colorante midiéndose a continuación la absorbancia. 2·- Las bandas cortadas se pueden disolver completamente con un disolvente orgánico midiendo seguidamente esta solución en el fotómetro. OBSERVACIONES AL MÉTODO 1·- Preparación del hemolizado: a) Si la muestra es anémica utilizaremos mayor cantidad de sangre y menos de agua. b) Si la muestra es vieja puede agregarse cianuro potásico para convertir en cianometahemoglobina ya que esta interfiere con la hemoglobina H. c) En caso de no utilizarse en el momento de su preparación el hemolizado se guardará a menos 4ºC. d) La producción de metahemoglobina debe reducirse al mínimo abreviando el contacto entre plasma y eritrocitos. 2·- Realización de la electroforesis: a) El aplicador debe lavarse con agua destilada y luego secarse entre las aplicaciones de distintas muestras. b) Las tiras de acetato de celulosa sólo deben tomarse y sostenerse por los bordes. c) No debe quedar aire atrapado entre el tampón y las tiras cuando se embeben estas d) Es conveniente realizar una electroforesis paralela con patrones de hemoglobina para comparar posteriormente los resultados. 3·- Tinción de las tiras: a) El colorante utilizado puede ser indistintamente de proteínas o de hemoglobina. No obstante este último diferencia las proteínas no hemo y ayuda a la visualización de bandas d hemoglobina débiles. b) Esta técnica no diferencia todas las hemoglobinas como ocurre entre las hemoglobina S y la hemoglobina B o bien entre la F y la G por tener la misma emigración electroforética por tanto para diferenciarlas hay que recurrir a otros métodos como electroforesis en agar-citrato, prueba de solubilidad, etc. c) La anhidrasa carbónica contamina todos los hemolizados, da una coloración positiva con colorantes de proteínas pero no se colorea con específicos de hemoglobina. d) La densitometría no debe utilizarse para cuantificar hemoglobina A2, hemoglobina F. TEMA XVIII BANCO DE SANGRE Ó GRUPOS SANGUÍNEOS Los grupos sanguíneos tienen su origen en la existencia de determinados antígenos eritrocitarios, plaquetarios, leucocitarios y séricos. • Sistemas de grupos sanguíneos eritrocitarios: se denomina sistema de grupo a un conjunto de antígenos que conservando su independencia se trasmite como una unidad. Cada sistema lo forman varios antígenos que admiten combinaciones casi infinitas y hacen posible una clasificación de la sangre característico y prácticamente irrepetible en cada individuo. En medicina clínica tiene especial interés, tanto por la prevención como el tratamiento de las reacciones transfusionales como en la enfermedad hemolítica del recién nacido. En antígenos eritrocitario es el producto de un gen situado en la membrana del hematíe y reconocido por un anticuerpo específico. Se entiende por genotipo la totalidad de genes que recibe un individuo de sus progenitores y por fenotipo al conjunto de caracteres que estos expresan. La mayoría de los antígenos se hallan bien expresado en el recién nacido y otros están débilmente y algunos prácticamente ausentes. Su distribución es variables; así los antígenos del sistema ABO se encuentran en todas las células de la sangre, tejidos y células corporales; mientras que los pertenecientes al Rh se localizan exclusivamente en la hematíes. • Los anticuerpos: son inmunoglobulinas producidas específicamente por el sistema inmunitario tras una estimulación antigénica. Pueden reconocer antígenos ausentes del individuo que los posee y que aparecen en otros individuos de su misma especie, se habla en este caso de isoanticuerpos o aloanticuerpos, son los más frecuentes e importantes en inmunohematología eritrocitaria. Se llama autoanticuerpos cuando están dirigidos contra los propios hematíes y se llaman heteroanticuerpos cuando reconocen antígenos de otras especies, ejemplo de heteroanticuerpos : suero de Coombs o anti-hemoglobina humana. Los anticuerpos naturales son aquellos anticuerpos en los que no se pueden demostrar el estímulo previo que ha desencadenado su formación. Son generalmente de tipo Ig M, es decir, moléculas de gran tamaño incapaces de atravesar la placenta. Los anticuerpos naturales más importantes pertenecen a los sistemas ABO y Lewis. En algún sistema la presencia de anticuerpos naturales es común pues aparecen de forma constantes en el suero de todos los individuos a estos anticuerpos se les llama regulares. Por ejemplo: Anti-A, Anti-B. Anticuerpos inmunes o adquiridas: son los que se forman después de un contacto con sangre incompatibles por embarazo, transfusión, etc. Normalmente los anticuerpos inmunes o adquiridos suelen ser inmunoglobulinas Ig G que atraviesan la barrera placentaria y pueden ocasionar la enfermedad hemolítica del recién nacido en caso de incompatibilidad fetomaterno. Ejemplo de anticuerpos adquiridos: el anti-Rh y el anti-Kell. Existen anticuerpos que son poco frecuentes y no es habitual encontrarlos en el suero, se habla de anticuerpos irregulares por ejemplo: anti-D. Las inmunoglobulinas Ig M son anticuerpos completos o aglutinantes, es decir, reúnen a los hematíes en grumos visibles y dan una reacción de hemoaglutinación en medio salino. La mayoría son activos entre 4-20ºC y tiene poca transcendencia clínica pero cuando actúa también a 37ºC pueden ocasionar accidentes transfusionales muy graves por su capacidad de fijar complemento y producir una rápida hemólisis intra-vascular con compromiso de la función renal. Las inmunoglobulinas Ig G actúan generalmente a 37ºC y son anticuerpos incompletos o sensibilizantes que se fijan a la membrana del hematíe sin aglutinarlo. Esta reacción antígenoanticuerpo se pone de manifiesto en el laboratorio mediante técnica de aglutinación artificial. Entre ellas tenemos el suero o prueba de Coombs y otras que utilizan polímeros como es la albúmina bovina, enzimas, protoelípticos (tripsina), soluciones de baja fuerza iónica. La prueba de la antiglobulina humana o de Coombs utiliza como reactivo un suero de antiglobulina polivalente capaz de reconocer moléculas de IG G y C3D fijadas a la membrana de los hematíes y provocar su aglutinación. Puede ser: ~ Directa: cuando detecta hematíes sensibilizados in vivo por anticuerpos incompletos ~ Indirecta: cuando provocamos la sensibilización en el laboratorio a partir de suero o hematíes problema. SISTEMA ABO Kandsternen describió en el año 1901 el sistema de grupo sanguíneo ABO. Reconoció 3 grupos según el antígeno que estuviera presente en los hematíes. El grupo sanguíneo A presentaba el antígeno A, el grupo B el antígeno B y el O no presentaba antígenos A ni B. Más tarde se describió el 4º grupo que es el AB cuyos hematíes presentan a la vez antígenos A y B. La especial característica de este sistema es la presencia constante en el suero de anticuerpos ó aglutininas frente al antígeno ausente. Antígeno Anticuerpo Grupo sanguíneo Antígeno A Anti-B A Antígeno B Anti-A B -------------- Anti-A y Anti-B O Antígeno A y B ------------------ AB Los anticuerpos del sistema ABO aparecen de forma natural sin sensibilización previa se desarrolla en la infancia por contacto con sustancias similar a los antígeno A y B que se encuentran extensamente distribuidas en la naturaleza. • Antígenos del sistema ABO: el grupo sanguíneo ABO se hereda siguiendo las leyes de Mendel [intervienen 3 genes alelomórficos A, B y O que se sitúan en un solo locus, cada individuo hereda 2 de ellos, uno de cada progenitor estos genes determinan que tipo de antígeno ABO estará presente en los hematíes]. Los antígenos ABO están muy distribuidos en los tejidos del organismo y en algunas de sus secreciones. No se encuentran totalmente desarrollados en el nacimiento y los anticuerpos naturales de este sistema, no se detectan en el suero del recién nacido hasta haber transcurrido 2 o 3 meses. Del fenotipo A: se puede distinguir 2 subgrupos principales el A1 y el A2. Los hematíes A1 tienen más antígenos A que los hematíes A2. El comportamiento frente al reactivo Anti-A es similar para los 2 subgrupos. Aproximadamente el 30% de los individuos A son A1. Otros subgrupos son A3, AX, AM, etc. El grupo B también presenta subgrupos pero menos frecuentes que el A [B3, BM, BX, etc]. Fenotipo (visible) Genotipo (gen heredado) Frecuencia (%) A1A1 A1 A1A2 A A1O 45'56 A2 A2A2 A2O B B BB 8'65 BO AB A1B AB 3'57 A2B A2B O O OO 42'10 • Anticuerpos del sistema ABO: el Anti-A y el Anti-B son producidos por individuos que carecen de los respectivos antígenos A y B. Dichos anticuerpos con predominantemente Ig M, también pueden ser Ig G pero son menos frecuentes y generalmente son producidos por individuos del grupo O. Los anticuerpos del sistema ABO pueden reaccionar a la temperatura corporal (37ºC) y activar el complemento causando una rápida destrucción intra-vascular de los hematíes. El título de Anti-A y Anti-B con frecuencia está disminuido en le anciano y en los pacientes con hipogammaglobulinemia. Los niños recién nacidos no producen ni Anti-A ni Anti-B hasta que alcanzan los 3-6meses de edad. El Anti-A1 es un anticuerpo natural de tipo Ig M que producen algunos individuos A2 y A2B. El Anti-A1 generalmente tiene un rango térmico bajo, por cuya razón no suele tener significación clínica. Con individuos A2 cuyo suero contiene Anti-A1 con un rango térmico bastante elevado para tener significación clínica suele ser transfundidos con sangre del grupo O ó A2. • Sistema Hh: para que puedan expresarse los genes A y B debe existir otro gen llamado H. Está situado en un locus a parte de los anteriores y define el sistema de grupo sanguíneo Hh se admite la existencia de una sustancia precursora sobre la que actuaría el gen H. Esta sustancia H en presencia de los genes A y/o B se transforma en sustancia A y/o B. El gen O no transforma la sustancia H y por tanto en los hematíes del grupo O esta sustancia está ampliamente representada. La cantidad de sustancia H presente en los hematíes depende del grupo sanguíneo al que pertenezca; el grupo sanguíneo O es el que presenta mayor cantidad de sustancia H. Cuando los hematíes tienen poca sustancia H como lo9s grupos sanguíneos A y AB se puede detectar en el suero la presencia de anticuerpo Anti-H. Esta situación se produce de forma natural sin que exista estimulación antigénica conocida. Estos anticuerpos (Anti-H) actúan a baja temperatura y no suelen tener significación clínica. El fenotipo hh (h) se denomina Bombay y es extremadamente raro [fue descrito por 1ª vez por Wendel en 1952] los individuos que posee este fenotipo, carecen del gen H y por tanto no pueden producir sustancias A o B aunque hayan heredado dichos genes. Estos anticuerpos Anti-H actúan a 37ºC y activan el complemento , por lo tanto tienen significación clínica. Los individuos con fenotipo Bombay son individuos del grupo O y en su suero se detecta Anti-A, Anti-B y Anti-H mientras que en un individuo normal se detectan sólo Anti-A y Anti-B. Es por esta razón que los individuos Bombay deben ser transfundidos solo con sangre procedente de otro individuo con fenotipo Bombay. • Gen secretor (SE): los antígenos A, B y H se hallan en el plasma, en la saliva y en otros líquidos orgánicos. Su presencia en esos líquidos está controlada por el gen secretor (SE) que es autosómico dominante. El 80% de los individuos caucasoides heredan el gen Se y son llamados secretores, el 20% restantes son no secretores. Un secretor del grupo A tiene sustancias antígenos A y H en sus líquidos orgánicos. Los no secretores no tienen antígeno ABH en sus secreciones. Para determinar si un individuo es o no secretor se procede a buscar los antígenos ABH en la saliva. Al determinación de secretores tiene escasa importancia tanto clínica como del laboratorio pero puede ser útil en la determinación de grupos sanguíneos ABO de un individuo si el grupo de sus hematíes no es concluyente. • Trascendencia clínica del sistema ABO: de todos los sistemas de grupo sanguíneo el ABO es el que tiene una mayor importancia transfusional fundamental-mente debido a la presencia en el suero de los individuos de anticuerpos naturales frente al antígeno ausentes en sus hematíes. Al transfundirse sangre incompatible se produce una reacción transfusional de tipo hemolítico que normalmente es inmediata y muy grave. Los hematíes del donante deben ser compatibles con los anticuerpos del receptor pars evitar que tenga lugar una reacción antígeno-anticuerpo. Similar reacción aunque más moderada se produciría cuando el plasma del donante poseen un título elevado de Anti-A y/o Anti-B frente a los hematíes de receptores A, B o AB. El menor grado de reacción que generalmente acompaña a esta situación se explica por la dilución que sufre el anticuerpo en el torrente circulatorio del receptor. Existe una regla clásica de compatibilidad para el sistema ABO. Toda transfusión isogrupo es compatible y por tanto bien tolerada, sin embargo la transfusión será también compatible cuando los hematíes del donante no contengan antígenos capaces de ser aglutinados por anticuerpos presentes en el suero del receptor. EL SISTEMA RHESUS La trascendencia clínica de este sistema viene determinada por el elevado poder inmunógeno de sus antígenos, especialmente el D. Fisher y Race denominaron a los 6 antígenos principales D (Rh original), C, E, c, e, d. • El antígeno D es el que determina la positividad Rh [rho, Rh1]. Fue el primero en descubrirse y es fuertemente inmunógeno, desarrollan anticuerpos anti-D entre el 50-80% de individuos Rh+ que reciben una unidad de sangre positiva. En la práctica transfusional suele ser suficiente dividir a la población en Rh+ y Rh--. Posteriormente se identificaron anticuerpos que daban una frecuencia de aglutinación diferente y definían nuevos determinantes Antígeno C [rh', Rh2], Antígeno E [rh'', Rh3], Antígeno c [hr', Rh4], Antígeno e [hr'', Rh5]. Su poder inmunógeno es menor pero todas las especificidades han sido responsables en alguna ocasión de enfermedad hemolítica del recién nacido o reacción transfusional. El fenotipo Rh se define por la presencia o ausencia de los 5 antígenos El sistema Rh se caracteriza por el gran número y la variabilidad de sus antígeno • Antígeno Du: es una variante débil del antígeno D. Posee todas las características de este antígeno pero cuantitativamente reducidas. Se han descrito más grados de prepotencia, da lugar a reacciones débiles negativas con algunos sueros Anti-D y precisan para ser detectados técnicas que favorecen la aglutinación (Coombs indirecto ó un medio enzimático). Las unidades de sangre Du positivas deben clasificarse como Rh+ pues su transfusión puede resultar incompatible a individuos Rh-- que posee anticuerpos Anti-D. Los enfermos Du positivos es más seguro considerarlas como receptores de sangre D--. Los antígenos del sistema Rh se encuentran únicamente en el hematíe. • Anticuerpos del sistema Rhesus: aunque se han descrito anticuerpos naturales la mayoría de los alo-anticuerpos del sistema Rh son inmunes se tratan en general de anticuerpos de tipo Ig G siendo estimulado su producción por transfusión o por embarazo. También se han identificados Anti-D de tipo Ig M. Los anticuerpos del sistema Rh pueden causar reacciones transfusionales y enfermedad hemolítica del recién nacido. SISTEMA KELL Se han incorporado a este sistema alrededor de 30 antígenos. Los antígenos del sistema Kell se producen a partir de una sustancia precursora Kx, codificada por el gen XK, se encuentra en el cromosoma X. Posteriormente los genes Kell autosómicos convierten la sustancia Kx en antígenos del sistema Kell. Hay por lo menos 20 antígenos incluidos en dicho sistema. Muchos de ellos de elevada frecuencia. Los más importantes son K (Kell) y k (cellano). El antígeno K es inmunógeno (produce anticuerpos) por tanto muchos individuos que no la poseen K--, producen Anti-K cuando reciben hematíes K+. Por ser baja la frecuencia del antígeno K no es difícil encontrar sangre compatible para los pacientes con Anti-K. El antígeno k tiene una frecuencia del 99%. • Sustancia Kx: la sustancia precursora de los antígenos Kell está presente en los leucocitos y los hematíes de la mayoría de individuos. La mayor parte de la sustancia Kx eritrocitaria es convertida en antígeno del sistema Kell por los genes Kell autosómicos. Si los hematíes de un individuos carece de la sustancia Kx presentan una forma anormal (acantocitos) y un tiempo de vida reducido. De tales individuos se dice que pertenecen al fenotipo McLeod. La ausencia de la sustancia Kx de los leucocitos se ha descrito en individuos con enfermedad granulomatosa crónica. Tales leucocitos pueden fagocitar pero no eliminar las bacterias por ello los pacientes con enfermedad granulomatosa crónica presenta infecciones bacterianas recurrentes. Los individuos que carecen de sustancia Kx en hematíes y leucocitos presentan al mismo tiempo el fenotipo McLeod y la enfermedad granulomatosa crónica. • Anticuerpos del sistema Kell: los anticuerpos del sistema Kell son generalmente Ig G y su producción es estimulada por transfusión o embarazo pueden por tanto ser la causa de reacciones transfusionales y enfermedad hemolítica del recién nacido. Sin Kx en los hematíes Sin Kx en los leucocitos Fenotipo McLeod Enfermedad granulomatosa crónica Acantocitos Con supervivencia Función leucocitaria reducida deficiente SISTEMA DUFFY (Fy) Se han descrito 5 antígenos en el sistema Duffy. Los más importantes son: Fya, Fyb que son producto de 2 genes alélicos. [Los individuos de raza negra pertenecientes al fenotipo Fy (a-b-) no producen anticuerpos Anti-Fya ni Anti Fyb, después de ser transfundidos con sangre Fy (a+) o bien con Fy(b+). Por el contrario los individuos caucasoides Fy(a-b-) se sensibilizan cuando son transfundidos con sangre Fy(a+) ó Fy(b+). Esto sugiere que el fenotipo Fy(a-b-) procede de genes distintos en cada población]. • Anticuerpos del sistema Duffy: la mayor parte de los anticuerpos pertenecientes al sistema Duffy. Son Ig G y pueden activar la secuencia del complemento. Pueden ser causa de reacciones transfusionales y de enfermedad hemolítica del recién nacido. [El antígenos Fya es más inmunógenos que el Fyb]. • Los antígenos del sistema Duffy y malaria: desde hace tiempo se conoce que los individuos de raza negra con fenotipo Fy (a-b-) son resistentes a la infección por plasmodium vivas [este parásito -plasmodium- sólo invade hematíes Fya y Fyb lo que da a entender que estos antígenos o una estructura relacionada con ellos actúan como receptor de membrana para la invasión parasitaria]. SISTEMA LEWIS Los antígenos del sistema Lewis proceden del plasma y se adsorben a los hematíes. Los genes alélicos del sistema Lewis se representan por los símbolos Le o le. El gen Le representa el gen dominante. El gen le es un alelo silencioso. [Se han descrito 3 fenotipos Lewis comunes: Le(a+b-), Le(a-b+), Le(a-b-). Los individuos que carecen del gen Le pertenecen al fenotipo Le (a-b-)]. Es preciso hacer hincapié en que los antígenos del sistema Lewis proceden del plasma y están adsorbidos en la membrana del hematíe. • Anticuerpos anti-Lewis: los anticuerpos anti-Lea y anti-Leb son alo-anticuerpos naturales de clase Ig M. Los anticuerpos del sistema Lewis pueden aglutinar los hematíes y activar el complemento in-vitro, pero in vivo tiene escasa importancia clínica por 2 razones: ~ En primer lugar los antígenos solubles Lea y Leb presentes en el plasma del donante neutralizan los anticuerpos del receptor. ~ En segundo lugar los antígenos Lewis son rápidamente desplazados de los hematíes del donante, los cuales se transforman y adquieren un fenotipo Lewis igual que el del receptor. Los anticuerpos anti-Lewis que solamente reaccionan a temperatura ambiente o mediante técnicas enzimáticas in-vitro no tiene significación clínica. Los pacientes que presentan algún anticuerpo anti-Lewis que reacciona a temperatura superiores a 30ºC o produce hemólisis in-vitro solamente deben recibir sangre compatibles, asegurándose de ellos, mediante pruebas cruzadas. Los anticuerpos del sistema Lewis no producen enfermedad hemolítica del recién nacido ya que los antígenos Lea y Leb no están bien desarrollados en este (recién nacido). Por otra parte los anticuerpos anti-Lea y anti-Leb por ser de clase Ig M no pueden atravesar la placenta. SISTEMA KIDD (JK) Los 3 fenotipos más corriente del sistema Kidd están definidos por 2 genes alélicos Jka y JKb son inmunógenos débiles. • Anticuerpos: los anticuerpos del sistema Kidd son de la clase Ig G y son capaces de fijar el complemento. Se forman como resultado de la recepción de sangre Kidd positiva, ya sea mediante transfusión o embarazo. El título de anticuerpos de este sistema con frecuencia desciende después de su formación hasta niveles no detectables. Por tanto pueden pasar desapercibido en las pruebas rutinarias de compatibilidad, aunque no se detecten en el momento de la transfusión, los pacientes sensibilizados pueden presentar una reacción transfusional retardada. Los anticuerpos Anti-JKb pueden ser causa de la enfermedad hemolítica del recién nacido ya que los antígenos Jka y JKb están bien desarrollados en los hematíes fetales. SISTEMA I (i) Los antígenos I e i se hallan en casi todos los hematíes. El antígeno I se encuentra en grandes cantidades en los hematíes del adulto, pero solo en pequeñas cantidades en los hematíes del recién nacidos. Por el contrario en los hematíes del adulto hay pequeñas cantidades del antígeno i que es muy abundante en los hematíes del recién nacido. • Anticuerpos anti-Ii: los anticuerpos del sistema I son auto-aglutininas Ig M naturales de rango térmico bajo. Cuando reacciona a temperatura ambiente complica las pruebas serológicas. No obstante a menos que reaccione por encima de 30ºC no son clínicamente significativos. Es raro encontrar anti-i en individuos sanos, más bien se produce de modo secundario en enfermedades de origen vírico tales como la mononucleosis infecciosa. Generalmente no tiene importancia clínica aunque puede producir hemólisis si tiene un rango térmico elevado. SISTEMA MNSs Los antígenos M y N difieren únicamente en 2 aminoácidos situados en las posiciones 1 y 5 de la cadena polipeptídica. Los genes alelos S y s están estrechamente unidos al locus MN. La localización exacta y la estructura bioquímica de los antígeno S y s no se conocen. • Anticuerpos MNSs: el anticuerpos M generalmente está constituido por una mezcla de Ig M e Ig G. El anti-M no suele causar reacciones transfusionales debido a su bajo rango térmico; en raras ocasiones el anti-M ha estado implicado en la enfermedad hemolítica del recién nacido. El anti-N es un anticuerpo de tipo Ig M. No produce reacciones transfusionales ni enfermedad hemolítica del recién nacido. El anti-S puede ser Ig M o Ig G mientras que el anti-s generalmente es una Ig G. Tanto el anti-S como el anti-s pueden causar reacciones transfusionales, hemolíticas y enfermedad hemolítica del recién nacido. OTROS SISTEMAS DE GRUPOS SANGUÍNEOS Se han descrito otros sistemas de grupos sanguíneos: sistema P, Lutheran, Diego, pero que raramente los anticuerpos que pueden originar se encuentran en el curso de las investigaciones serológicas. MOMENCLATURA DE LOS GRUPOS SANGUÍNEOS En la descripción de los sistema de grupos sanguíneos se utilizan el mismo símbolo para designar el gen y el antígeno. Para designarlos el símbolo utilizado para designar el gen se describe con carácter itálicos. Por ejemplo: el gen Lewis Le mientras que el antígeno Le. PRUEBAS Y TÉCNICAS EN INMUNOHEMATOLOGIA Factores que afectan a la hemaglutinación: la hemoaglutinación específica es la reacción más importante que tiene lugar en el laboratorio de un banco de sangre por ser un punto final a casi todos los sistemas destinados a detectar los antígenos y anticuerpos eritrocitarios. Existen 4 grupos de factores que influyen sobre el resultado de la reacción: hematíe, suero, medio y circunstancias físicas en que tiene lugar la reacción. 1·- El hematíe o eritrocitos: la importancia del tipo, número y localización de los antígenos sobre los eritrocitos se pone de manifiesto por los antígenos del sistema ABO y Rhesus. El número de puntos antigénicos del sistema ABO puede ser próximo al millón por cada célula y su localización es extra-membranosa. Por lo tanto los hematíes se aglutinan fácilmente bajo la acción de anticuerpos adecuados. Por el contrario los antígenos Rh tienen solamente unos 1.000-30.000 puntos antigénicos células y son además intra-membranoso, con lo que se aglutinan con menos facilidad ante los anticuerpos correspondientes. 2·- El suero (anticuerpos): los anticuerpos que se forman como respuesta a la estimulación eritrocitaria suelen ser de los tipos Ig G ó Ig M. Los Ig M aglutinan a los hematíes suspendidos en solución salina lo que no suelen hacer los anticuerpos Ig G. Esta característica guarda relación con el tamaño de las moléculas y con el número de puntos de unión con el antígeno [así los anticuerpos Ig M que consta de 5 sub-unidades tienen una distancia aproximadamente de 300 Aº, entre dichos puntos de unión. Mientras que las moléculas de Ig G que están formados por una sub-unidad tienen solo 150 Aº entre dichos puntos] Es necesario el empleo de reactivos anti-globulinas para detectar muchos de los anticuerpos Ig G ya que estos provocan la aglutinación directa de los hematíes. 3·- El medio de refuerzo: los medios con pH y potencia iónica bajos como el de glucosa acidificada aceleran la unión de los anticuerpos a los hematíes. Diversas sustancias como las soluciones salinas de baja potencia iónica se utilizan para efectuar la detección de anticuerpos y las pruebas cruzadas, también se utilizan enzimas. 4·- Circunstancias o características físicas: tales como la temperatura y el tiempo de incubación así como la duración y velocidad de centrifugación influye notablemente sobre la reacción de aglutinación. La mayoría de anticuerpos Ig M reaccionan mejor entre 4 y 27ºC, mientras que, el margen óptimo para la mayor parte de los anticuerpos Ig G se encuentra entre 30 y 37 ºC. Las distintas reacciones antígeno-anticuerpo alcanzan el equilibrio en momentos diferentes. Los anticuerpos de los grupos sanguíneos suelen detectarse cuando se emplean tiempo de incubación entre 15-60 minutos. La centrifugación obliga a los hematíes a aproximarse más entre sí lo que facilita la hemoaglutinación. Otros factores que afectan a la hemoaglutinación son: las cargas negativas, la hidratación y el potencial Z. Inducción a la hemoaglutinación: la hemoaglutinación puede inducirse de 2 maneras: una de ellas consiste en reducir la distancia entre hematíes y la otra en proporcionar fuentes entre 2 anticuerpos que son incapaces de unirse simultáneamente entre 2 hematíes, debido a la distancia que separa estos entre sí. Medios para favorecer la hemoaglutinación: • Enzimas proteo-elípticas: cuando se tratan los hematíes con enzimas como tripsina, papaina y bromelina, se reduce irreversiblemente el contenido de ácido siálico y disminuye los valores del potencial Z. De este modo los hematíes tratados aglutinan fácilmente con los anticuerpos adecuados. • Albúmina: se utiliza para poner de manifiesto los anticuerpos del sistema Rh. • Poli-cationes: uno de los cationes que se utiliza es la protamina. Lo que hace es neutralizar las cargas negativas de los hematíes y provoca una agregación inespecífica, esto permite que los pequeños anticuerpos específicos se unan con más de una célula y originan una aglutinación específica. • Soluciones de baja potencia iónica: al emplear una solución de baja potencia iónica y disminuir el numero de iones existentes quedan expuestas las cargas negativas y positivas, lo que aumenta el porcentaje de asociación entre antígenos y anticuerpo e incrementa la cantidad de este último que se fija sobre los hematíes. • Reactivos de antiglobulina humana: para aglutinar los hematíes separados [más de 25 nanómetros] los anticuerpos antiglobulina humana o anti-complemento que se encuentran y a fijados sobre cada hematíes lo que constituye la base de la prueba de antiglobulina. Prueba de la antiglobulina (Coombs directo ó PAG e indirecto ó PAI): esta prueba que se conoce también con el no0mbre de prueba de Coombs. La PAG ó el Coombs está basada en el principio de que los anticuerpos antiglobulina humana induce la aglutinación de los hematíes recubiertos por globulinas. Cuando emplea la PAG para detectar anticuerpos unidos a los hematíes in vivo, se denomina prueba de antiglobulina directa, PAG ó Coombs directo. Si se usa para detectar anticuerpos en el suero mediante la sensibilización de los hematíes in vitro recibe el nombre de prueba de antiglobulina indirecta, PAI ó Coombs indirecto. Los sueros antiglobulina suelen obtenerse por inmunización en el conejo y se emplea animales seleccionados para la inmunización. • Técnica: para el Coombs directo (PAD) hay que recoger las muestras de sangre en EDTA, se prepara una suspensión salina del 3-5% de los hematíes del paciente. Para el Coombs indirecto (PAI) los hematíes se han de incubar con el suero (o plasma) adecuado a 37ºC durante 15-30 minutos. En ambos casos los hematíes han de lavarse al menos 4 veces para eliminar totalmente las DSFASDDFAS libres no fijadas. El tiempo y la velocidad de centrifugación se han de estandarizar mediante controles positivos y negativos. • Pruebas de la antiglobulina falsamente negativas: las reacciones falsamente negativas suelen originarse por procedimientos inadecuados o defectos técnicos. Las causas habituales son: 1·- Lavado insuficiente 2·- Contaminación con proteínas séricas por suciedad 3·- No haber añadido el suero de Coombs 4·- Incubación o centrifugación insuficiente 5·- Lavado excesivo ó temperatura elevada 6·- Cantidad insuficiente de suero • Pruebas de la antiglobulina falsamente positivas: puede ser por centrifugación excesiva, presencia de anticuerpos inesperados en el suero de Coombs y que haya aglutinación antes de añadir suero de Coombs por presencia de diferentes sustancias. • Aplicación del Coombs directo: 1·- Investigación de auto-anticuerpos 2·- Anticuerpos inducidos por medicamentos 3·- Enfermedad hemolítica del recién nacido 4·- Investigación de una reacción transfusional • Aplicación del Coombs indirecto: 1·- En la detección e identificación de los anticuerpos eritrocitarios en suero 2·- Tipificación de los antígenos eritrocitarios 3·- Pruebas de compatibilidad (pruebas cruzadas) • Modificaciones del Coombs indirecto: pueden aplicarse diversas modificaciones del prueba indirecta para favorecer la detección de anticuerpos: 1·- Hematíes tratados con enzimas: los hematíes que se utilizan en la prueba indirecta de la antiglobulina pueden tratarse con enzimas proteo-elípticas tales como papaína, bromelina o tripsina. [Dichas enzimas separan de la membrana del hematíes las proteínas portadoras de ácido xiálico cargado negativamente]. Esto favorece la sensibilización y la aglutinación por algunos anticuerpos clínica significativos por ejemplo el sistema Rhesus. 2·- Albúmina y solución salina de baja potencia iónica: cuando la técnica de la antiglobulina se efectúa con los hematíes del donante suspendidos en solución salina normal es preciso un tiempo de incubación de 30-60 minutos para lograr un máximo de fijación de los anticuerpos a los antígenos de los hematíes. Si los hematíe se suspenden en una solución salina de bajo potencial iónico el periodo puede reducirse de 5-10 minutos. De modo similar la adicción de albúmina a los hematíe permitiendo un tiempo de incubación más corto. Detección de anticuerpos irregulares: denominación anticuerpos irregulares a aquellos que se producen frente a antígenos distintos a los del sistema ABO. Son anticuerpos que generalmente aparecen por una inmunización transfusional feto-materno y pueden producir reacciones post-transfusionales. La identificación de los anticuerpos irregulares es de gran importancia en el diagnóstico y tratamiento de la enfermedad hemolítica del recién nacido y de ciertos trastornos hemáticos así como en la prevención de reacciones transfusionales y estudios durante el embarazo. La detección de los anticuerpos se realiza enfrentando el suero del paciente o receptor a una serie de hematíes tipados de los cuales se conoce con exactitud los antígenos presente en su membrana el resultado positivo es la aglutinación de los hematíes. Se puede llevar a cabo mediante 3 tipos de técnicas: test de Coombs indirecto, técnica enzimática y técnica a 4ºC. A·- Test de Coombs indirecto: consiste en la incubación previa del suero y los hematíes tipados para conseguir la sensibilización de esos hematíes, es decir, que los anticuerpos presentes en el suero se adhieren a la superficie eritrocitaria. Posteriormente añadimos el suero de Coombs (antiglobulina humana) para poner de manifiestos o no de estos anticuerpos mediante la aglutinación de los hematíes. Para acortar el tiempo de incubación y mejorar los resultados podemos emplear un medio de baja fuerza iónica. B·- Técnica enzimática: consiste en tratar previamente los hematíes tipados con un enzima para facilitar la aglutinación de los hematíes se suelen emplear la tripsina., la papaína y la bromelina. C·- Técnica a 4ºC: se utiliza para detectar la crio-aglutinación o anticuerpos que reaccionan mejor a baja temperatura. Consiste en incubar el suero y los hematíes a 4ºC para posteriormente centrifugar y observar la presencia o no e aglutinación. Se recomienda utilizar al menos 2 de estas técnicas para obtener buenos resultados en la detección de anticuerpos irregulares. • Interpretación clínica de los resultados: el resultado positivos es la presencia de aglutinación, debemos anotarlos en la hoja que acompaña al panel de células. Esta hoja indica los antígenos que están presentes en cada una de las células que componen el panel de manera que la aglutinación de uno de los tubos indica la presencia en el suero problema de anticuerpos frente algunos de los antígenos presentes en al membrana del hematíe. Pruebas cruzadas: el término de pruebas de compatibilidad se refiere a una serie de investigaciones pre-transfusionales realizadas para cerciorarse de que se elige la unidad idónea de sangre que se ha de transfundir y de que la supervivencia de los hematíes va a ser aceptable una vez realizada la transfusión. Además de comprobar la unidad donante y de investigar al paciente en busca de anticuerpos eritrocitarios irregulares y de sus datos previos se realizará un aprueba cruzada. • Prueba cruzada mayor: el objetivo de las pruebas pre-transfusionales es detectar las posible reacciones antígeno-anticuerpo entre la sangre del donante y del receptor antes de la transfusión por ellos es imprescindible realizar una serie de determinaciones que se especifican a continuación: 1·- Tipaje correcto del grupo: ABO, Rh del donante y receptor 2·- Escrutinio de los anticuerpo irregulares del receptor 3·- Pruebas cruzada mayor: ~ Fundamento: se prepara una suspensión de los hematíes donantes en solución salina, para lo cual hay que recogerlos en un segmento que forme parte integrante de la unidad de sangre. Habitualmente se lavan los hematíes una vez para liminar el anticoagulante o las proteínas plasmáticas que pudieran interferir en la prueba. A continuación se mezclan estos hematíes donantes con el suero del paciente (es posible realizar diferentes procedimientos para llevar a cabo la prueba cruzada) debido a que el objetivo de esta es detectar los anticuerpos clínicamente significativos DFASDFSDFSD sólo se efectúa la incubación a 37ºC con un medio de refuerzo y luego se realiza la prueba de la antiglobulina. ~ Interpretación clínica de los resultados obtenidos: la ausencia de aglutinación en todos los paso indica que las sangres son compatibles y por tanto la transfusión es posible. Si hay aglutinación en cualquiera de las fases de la prueba indica incompatibilidad. Si se observa hemólisis en cualquiera de los pasos también es signo de incompatibilidad. En ocasiones es conveniente realizar esta prueba en medio enzimñático dado que el empleo de enzimas como la papaína o bromelina refuerzan las reacciones antígeno-anticuerpo. • Prueba cruzada menor: esta prueba consiste en comprobar el plasma o suero del donante con los hematíes del receptor (paciente) con esto se detecta anticuerpos en al unidad donante que podrían reaccionar con los antígenos eritrocitarios del paciente. Esta prueba se halla actualmente obsoleto ya que las unidades donantes se someten a detección de anticuerpos irregulares en el laboratorio donde se recoge la sangre. • Pauta que hay que se debe seguir en la detección de anticuerpos positivos: si la detección de anticuerpos es positiva o se detecta una incompatibilidad en la prueba cruzada hay que identificar el anticuerpo una vez identificado se elegirá para la transfusión aquellas unidades donantes que carezcan del antígeno correspondiente. TEMA XIX FISIOLOGÍA PLAQUETARIA FORMACIÓN DE LAS PLAQUETAS Los trombocitos o plaquetas se originan a partir de un precursor comprometido que es común a la línea eritrocitaria y a la trombocitaria que es el BFU-EM. Este se transforma en otro precursor comprometido exclusivo de la línea trombocitaria y que se denomina CFU-MEG. Este se diferencia hacia megacarioblasto que es la primera célula de morfología reconocible. Este madura a promegacariocito y este a megacariocito (cuyo tamaño es muy grande tiene de 80 a 100 micras de diámetro). En los estados más avanzados de maduración del megacariocito los gránulos azurófilos de su citoplasma se acumulan en su periferia y se agrupan en masas, separadas entre sí por espacios pálidos que se conocen como membranas de demarcación y que serán los límites de las futuras plaquetas. Los megacariocitos en los que se aprecia la presencia de trombocitos en su interior pueden llamarse metamegacariocitos. Al término de su maduración los megacariocitos emiten unas prolongaciones citoplasmáticas de las que se separan las plaquetas. Un megacariocito da lugar a miles de plaquetas. Los núcleos de los megacariocitos tras la fragmentación de su citoplasma son fagocitados. Algunos megacariocitos escapan de la médula ósea y emigran hacia las arterias pulmonares terminales y hacia los capilares alveolares donde realizan su emisión de trombocitos a la sangre . MORFOLOGÍA DE LAS PLAQUETAS Las plaquetas o trombocitos tienen las siguientes características morfológicas: 1·- Su forma es variable aunque suele ser discoidea 2·- Su tamaño es muy pequeño de 1 a 4 micras 3·- Carecen de núcleo 4·- Su citoplasma se tiñe de color azul pálido y contiene un número variable de pequeños gránulos de color púrpura. Estos gránulos no suelen estar presentes en la zona periférica de las plaquetas (zona hialómera) y, suelen acumular en el área central de las mismas (granulómera o cromómera). Además su citoplasma suele proyectarse hacia el exterior dando lugar a una serie de prolongaciones en forma de tentáculos o pseudópodos (falso pus). Muchas plaquetas tienen sin embargo los bordes lisos. Con frecuencia se adhieren unos trombocitos a otros formando conglomerados plaquetarios que se observan más abundantemente al final de la extensión. ESTRUCTURA INTERNA DE LAS PLAQUETAS • Zona periférica o pared celular ~ Capa exterior o glucocálix: es el componente de los trombocitos que está en contacto directo con el plasma circundante. Es la estructura con la que adhiere las plaquetas. Presenta una gran avidez por proteínas externas (por ejemplo: por algunas proteínas plasmáticas) y por tanto intervienen en la recepción de estímulos que ponen en marcha la activación de las plaquetas. ~ Membrana celular: es trilaminar; en ella se encuentra una proteína contráctil que participa en la refracción del coágulo de fibrina (trombostenina de superficie). Además uno de sus fosfolípidos (ácido araquidónico) interviene en la activación de la coagulación y es el factor 3 plaquetario (f3p) o tromboplastina plaquetaria. Sin embargo la principal función de la membrana consiste en mantener la integridad celular de las plaquetas. ~ Área submembranosa: es la parte más interna de la zona periferia. Ayuda a mantener la forma discoidal de las plaquetas y participan en la formación y en la estabilización de los tentáculos de los trombocitos. • Zona del sol-gel o hialoplasma: es la zona periférica del citoplasma de las plaquetas que corresponden al área de las misma que observado con el microscopio óptico se aprecia casi vacía. Por su plasticidad y la rapidez en formar tentáculos se cree que tiene una estructura de sol-gel. Con el microscopio electrónico se observa en su interior 2 tipos de elementos fibrosos (haz marginal de micro túbulos y micro-filamento) que constituyen el sistema contráctil de los trombocitos. ~ Haz marginal de micro-túbulos: consiste en un anillo de micro-túbulos situados debajo de la pared celular. Se comporta como una especie de citoesqueleto que mantiene la forma discoidal en las plaquetas. Se pierde cuando se activan los trombocitos. ~ Micro-filamentos: tienen un diámetro similar a los filamento de que están compuestos los micro-túbulos (filamentos) y se ubican paralelamente a estos. Están formados por proteínas contráctiles (actina, miosina, trombostenina, troponina, tropo-miosina). Intervienen en los cambios de formas de las plaquetas y en la secreción de sustancias por parte de las mismas. • Zona de organelas: ~ Mitocondrias: son poco abundantes y contribuyen al depósito de ATP y de Ca ~ Lisosomas: contienen fundamentalmente enzimas hidroelípticas e intervienen en la lisis celular. ~ Gránulos densos: tienen función secretora y encierran metales (calcio, magnesio y fósforo) nucleótidos (ATP y ADP) y amidas. ~ Gránulos ð o específicos: son fijados por el aparato de Golgi. Son los más numerosos y engloban proteínas específicas (factor de Von Willebrand, factor 4 plaquetario -f4p-..) Vacían su contenido al exterior cuando las plaquetas se activan. ~ Masa de glucógeno: • Sistema membranoso ~ Aparato de golgi: se encuentra en un número muy reducido en las plaquetas y contribuyen en la síntesis de las proteínas. ~ Sistema tubular denso: son una serie irregular de canales que fabrican diversos elementos fibrosos como las prostaglandinas y engloban la reserva principal de calcio empelado en la contracción de alguno componentes de las plaquetas. ~ Sistema canalicular abierto: son invaginaciones de la membrana celular que delimita un extenso sistema de tortuosos canales que comunican el interior de los trombocitos con el plasma. Facilitan el ensamblaje de los factores de coagulación al aumentar la superficie de las plaquetas. También interviene en la secreción de los trombocitos al ser la vía de salida del contenido de sus gránulos citoplasmáticos, tras la fusión de estos últimos en la parte interna del sistema canalicular abierto cuando se activan las plaquetas. FUNCIONES DE LAS PLAQUETAS Del total de la masa plaquetaria presente en el organismo las 2/3 partes circulan por la sangre y la 1/3 parte restante se deposita en el bazo. Hay un intercambio libre y direccional entre los trombocitos sanguíneos y los esplénicos. El número de plaquetas por mm3 de sangre 130.000400.000. La vida media de los trombocitos en sangre periférica varía entre 8-12 días. Tras este periodo d tiempo son destruidas en el hígado y bazo por el sistema mononuclear fagocítico (hemostasia). Los trombocitos intervienen esencialmente en la detención de las hemorragias. Para ello actúa a nivel de la hemostasia primaria mediante la formación del trombo blanco plaquetario y a nivel de la coagulación mediante la producción de factores que participan en algunas de sus etapas. • La formación del trombo blanco plaquetario: este proceso se desarrolla en 3 fases que son: 1·- Interacción de las plaquetas con la pared de los vasos sanguíneos o adhesión plaquetaria. 2·- Activación de los trombocitos 3·- Interacción de las plaquetas entre sí, o agregación plaquetaria. 1·- Adhesión: cuando se da una sección de un vaso sanguíneo las primeras plaquetas que escapan por la lesión se adhiere a las fibras de colágeno por el interior de la pared vascular. La adherencia de las plaquetas al colágeno y a la membrana basal desencadena cambios morfológicos y funcionales en estas (de forma discoidea o esférica con pseudópodos). 2·- Activación: provoca: ~ Cambio de forma en las plaquetas (discoidea, esférica con pseudópodos siendo estos los que permiten el contacto con plaquetas. Los micro-túbulos se contraen reuniendo a los organeros y gránulos en el centro de la plaqueta. ~ Liberación de los gránulos, gránulos densos y lisosomas a través del sistema canalicular abierto. El ácido araquidómico que está en la membrana se convertirá por acción de las enzimas en tromboxano A2 que es un potente vasoconstrictor necesario par ala contracción de los trombocitos y que estimula la agregación plaquetaria. 3·- Agregación: las plaquetas se unen entre sí por acción de sustancias como ADP, adrenalina y serotonina formando un entramado o trombo plaquetario; que en principio es reversible; si el estímulo cesa se produce desactivación y las plaquetas recuperan su forma; se trata de un proceso reversible. Si las sustancias inductoras principalmente la trombina siguen actuando tiene lugar la agregación irreversible, producción de fibrina, y por tanto de un trombo estable, con lo que cesa el sangrado. Este durará más o menos tiempo dependiendo de la profundidad de la lesión, del calibre del vaso y del mecanismo hemostásico. Las plaquetas también intervienen aportando factores de coagulación como son el f3p que forma parte del completo activador de protombina a trombina. De modo que también sintetizan o contiene sustancias que interviene en la coagulación del plasma sanguíneo. En general se les conoce como factor plaquetario de la coagulación: Factor 1 plaquetario: que se corresponde con el factor plasmático V de la coagulación. Factor 2 plaquetario Factor 3 plaquetario: los trombocitos también interviene en la activación de la vía intrínseca de la coagulación. Esto lo realiza mediante la exposición en su superficie de f3p o tromboplastina. El f3p atrae a otros factores de coagulación y facilita su reunión. Como consecuencia del encuentro de estos factores de coagulación se produce la activación de la protombina y su paso a trombina. Por tanto el f3p no es suficiente pero si necesario para iniciar la coagulación por la vía intrínseca. Factor 4 plaquetario Fibrinógeno Factor Von Willebrand Factor plasmático V Factor plasmático XIII Trombostenina Serotonina Catecolaminas ÍNDICES PLAQUETARIOS Los autoanalizadores hematológicos ofrecen una serie de parámetros entre los que cabe destacar: 1·- El plaquetocrito (PTC ó PCT) 2·- El volumen plaquetario medio (VPM ó MPV) 3·- Anchura de distribución plaquetaria (PDW ó ADW) 1·- El plaquetocrito es la relación existente entre el volumen ocupado por los trombos y el ocupado por la sangre total expresado en %. El valor normal está entre 0'12-0'36. 2·- El volumen plaquetario es el valor medio del volumen de las plaquetas. Valores normales entre 7'2-11'1fl. 3·- Anchura de distribución plaquetaria: también se denomina como índice de distribución de las plaquetas o IDP. Es el coeficiente de variación de los volúmenes de las plaquetas [CV - VPLQ = __GSD - VPLQ_ X 100; GSD= desviación estándar geométrica]. VPM Los valores normales entre el 25-65%. Indica el grado de variabilidad existente entre los volúmenes de los trombocitos. TEMA XX HEMOSTASIA INTRODUCIÓN Y CONCEPTOS Aunque vulgarmente se habla de coagulación sanguínea para describir el proceso que impide la salida de la sangre de nuestro aparato circulatorio. Hay que señalar que este proceso es mucho más complejo debiendo incluirse dentro del término hemostasia mucho más amplio que el anterior. Por tanto la coagulación es una parte de la hemostasia. Definimos hemostasia como el conjunto de mecanismos merced a los cuales se consigue detener y cohibir los proceso hemorrágicos. Estos mecanismos son de densidad variable según la importancia de la lesión; así tenemos: • Cuando la lesión se produce en los vaso capilares es suficiente la hemostasia primaria para detener la hemorragia. La hemostasia primaria comprende el conjunto de fenómenos que conlleva a la formación del trombo blanco plaquetario. • Si la lesión se produce en un vaso de mayor calibre la hemostasia primaria es reforzada por la coagulación plasmática, necesaria para la formación de un coagulo de fibrina insoluble y sólido. Se llama coágulo a la red de fibrina que ha inmovilizado en el interior de los vasos a las células de la sangre. Esta transformación tiene lugar tras una serie de reacciones enzimáticas en las que intervienen numerosos factores tanto plasmáticos como plaquetarios, por lo que resulta imposible separar la hemostasia primaria de la coagulación. ETAPAS DE LA HEMOSTASIA El proceso hemostático que parece reservado al momento de solucionar una hemorragia producida por un traumatismo es un proceso casi constante pues debido a pequeños traumatismos de variada índole se producen muy frecuentemente pequeñas hemorragias no aparentes, debido al buen funcionamiento de los procesos hemostáticos. Así pues tomando como ejemplo el caso de una rotura vascular los procesos que suceden para inhibir la hemorragia son: 1·- En primer lugar se da un fenómeno de origen vascular: produciéndose una contracción refleja del vaso afectado mediada por un reflejo nervioso que desencadena el propio traumatismo, dando lugar a un estímulo simpático que contrae el vaso y relentece la circulación. Como consecuencia de ello se estrecha la luz del vaso y el flujo de sangre al exterior se remite. La vasoconstricción ocurre inmediatamente después de la lesión y dura un corto periodo de tiempo. Después se da una vasoconstricción de tipo químico provocada por sustancias como: serotonina, ADP y tromboxano A2, sintetizadas y liberados por las plaquetas. A la vez se producen por las células endoteriales la prostaciclina PGI2 con propiedades vasodilatadoras que contrarrestan el potente efecto vasoconstrictor del tromboxano A2 consiguiéndose un equilibrio entre sustancias de efectos antagónicos. La cantidad de sangre que salga después de la lesión va a depender del tamaño y del tipo de vaso, así como la eficacia del sistema hemostático. 2·- Intervienen las plaquetas: activadas por el colágeno que aparece en la lesión debida la rotura vascular. Esta activación induce a la agresión plaquetaria que tiende a taponar la herida. Así mismo hay una liberación de sustancias plaquetarias que van a favorecer la hemostasia por distintos mecanismos: ADP: que favorece la agregación plaquetaria. Factor 3 plaquetario: que activa la formación del tromboplastina intrínseca principio de la vía intrínseca de la coagulación. Serotonina y esterocolaminas: que favorecen la contracción vascular prolongada. En este momento ya se ha constituido el llamado trombo primario que puede impedir la hemorragia pero no es suficiente sobre este trombo primario debido a un receptor que poseen las membranas plaquetarias para activar la trombina, se producen redes de fibrina dando un armazón a estos trombos primarios, envolviendo, incluso, al vaso lesionado en forma de manguito. 3·- Coagulación: pese al paso anterior, todavía es necesario reforzar el trombo producido realizándose en un tiempo posterior el fenómeno de la coagulación sanguínea. La sangre debido a estímulo que se producen en el punto de la lesión va a coagular. Es un proceso complejo en donde interviene muchos factores. En esencia el proceso de la coagulación se produce mediante la activación de una proteína soluble o factor I (fibrinógeno) que se transforma por la acción de un enzima protoelíptico (trombina en fibrina), proteínas insoluble que forma redes proteicas que son la base o el esqueleto del coágulo. La trombina procede de la protombina activada y, la activación de esta última puede conseguirse por 2 mecanismos: Cuadro Vía intrínseca ó Vía extrínseca ó Vía lenta Vía rápida XII III XI VII IX Ca+2 Ca+2 VIII X Ca+2 f3p V II I Fibrinógeno • Vía intrínseca o vía lenta: comienza con la activación del factor XII. Esta activación in vivo se realiza debido al contacto con el vaso lesionado que a perdido la protección anticoagulante que supone el endotelio intacto. In vitro se produce con el contacto con el vidrio cargado negativamente. Este factor XII activado actúa activando el factor XI que a su vez, en presencia el factor IV (calcio) activa al factor IX. El factor IX unido al factor VIII, al calcio y al f3p actúan activando el factor X. Aquí la vía intrínseca confluye con la vía extrínseca. • Vía extrínseca o vía rápida: comienza cuando la sangre recibe al factor III procedentes de las membranas celulares que se dañan en el traumatismo. Este factor III reacciona con el factor VII dando lugar a un complejo que en presencia de calcio activa el factor X. • Vía común: los 2 procesos anteriores coinciden en el momento de la activación del factor X que junto con el factor V y el calcio da lugar al paso de protombina a trombina que convierte al fibrinógeno a fibrina. 4·- Una vez formado el coágulo con las redes de fibrina el factor XIII en presencia de iones calcio realiza una estabilización de las redes de fibrinas, permitiendo el enlace entre sus moléculas lo que ayuda a estabilizarlo. Además se da una retracción del coágulo: si hay trombocitos en el momento de la constitución del coágulo de fibrina, estos liberan una proteína contráctil llamada trombostenina que hace que el coágulo se retraiga. Este al retraerse engloba células sanguíneas y exuda una cierta cantidad de suero. 5·- Fibrinolisis: consiste en la destrucción del coágulo de fibrina que se ha formado previamente, al término del proceso coagulatorio. La destrucción del coágulo se produce tras la reparación de la pared del vaso dañado y tiene por objeto la reanudación del flujo sanguíneo a través de la luz vascular. Además la Fibrinolisis permite la disolución de los depósitos de fibrina que pueden aparecer espontáneamente al nivel del árbol circulatorio y también hace posible la repermeabilización de los vasos trombosados. Los restos que quedan tras la fibrinolisis son captados por las células macrofágicas. La fibrinolisis destruye la fibrina formada en la coagulación. El proceso se desencadena por la activación de una proteína que circula por la sangre el plasminógeno. Este al entrar en contacto con las redes de fibrina se transforma en plasmina. Esta actúa como sustancias destructoras de fibrina y otras proteínas de la coagulación como los factores I, II, V y VIII. La importancia de este proceso radica en que la perpetuación del coágulo en el organismo seria tan peligrosa como la hemorragia. [Otras sustancias naturales inhibidoras de la coagulación que se dedican a limitar o impedir este proceso serían la colinesterasa (actúa sobre el factor XII a impidiendo su factor coagulante), anti-trombina I (conocemos como factor antitrombina I a la acción del fribrinógeno que, aunque al pasar a fibrina absorbe trombina, esta absorción es seguida de una liberación posterior que da reversibilidad al proceso), anti-trombina IV (son los productos de la degragación del fibrinógeno que actúan como anticoagulantes), anti-trombina III (que es la más importante). Forman complejos con la trombina inactivándola. Este mecanismo es favorecido por la heparina que es parte de la coagulación de la misma. FACTORES DE LA COAGULACIÓN Son glucoproteínas que circulan de forma inactiva en el plasma y, tras un proceso de activación se convierte en enzimas. La característica de los factores activados es su capacidad para romper enlaces en los que el aminoácido presente es la serina de aquí que se denominen serín-proteasas. Factor I fibrinógeno: es una glucoproteína de síntesis hepática cuya propiedad es ser coagulable por la trombina. Sus cifras normales en plasma son entre 200-400mg/100ml. Estos niveles aumentan en el estrés hemorrágico y en procesos inespecíficos tales como el embarazo, inflamción o enfermedades autoinmunes. Pueden disminuir en los trastornos obstétricos (trastorno del embarazo, aborto incompleto), intervenciones por el pulmón, quemaduras extensas, transfunsión de sangre incompatible, etc. Factor II protombina: se sintetiza en el hígado y requiere presencia de vitamina K (antihemorrágica) para su síntesis. La vitamina K se encuentra en los vegetales pero la mayor parte de la vitamina absorbida y utilizada es producida por las bacterias intestinales su insuficiencia primaria es rara, suele ser adquirida y se debe a enfermedades que impiden la absorción de la vitamina K en el intestino o su utilización por el hígado. Factor III tromboplastina tisular (f III p): se puede dar este nombre a cualquier sustancia capaz de transformar la protombina en trombina. Se encuentra en la célula endoterial unido a la membrana y es liberado al plasma cuando se produce una lesión vascular, pasando a formar un complejo con el factor VII en presencia de calcio que activa el factor X. Se encuentra en el endotelio, pulmón, riñón, hígado y en los grandes vasos (vía extrínseca). Factor IV calcio: los iones calcio se necesitan en todas las etapas de coagulación a excepción de la fase de contacto y la activación del factor XIII por la fibrina. Los anti-coagulantes: citrato sódico, oxalato sódico, EDTA neutralizan el ácido. Concentración normal de calcio es de 5-20mg/dl. Se necesita poca cantidad de calcio, por lo que la hipocalcemia no representa problemas hemorrágicos. Factor V factor lábil o acelerina: factor hepático que puede tener valores bajos en pacientes con hepatopatías. Su actividad desaparece rápidamente en la sangre o plasma anti-coagulada, incluso en el plasma congelado su actividad dura unas semanas. Forma un complejo con el factor X activado, el f3p, la tromboplastina tisular y calcio, denominado protrombinasa. Factor VI: no asignado; se considera que es el factor V activado. Factor VII protonvertina o factor estable: hepático dependiente de la vitamina K. Tanto inactivo como activo necesita a la tromboplastina tisular o hística y del calcio para su acción sobre el factor X. No se suele determinar aisladamente sino que se determina junto con el factor X. Algunos venenos de serpientes poseen una actividad semejante al factor XII y pueden sustituirlos en ciertos ensayos. Factor VIII factor anti-hemofílico A: es una glucoproteína termolábil producida por las células endoteriales. Han sido definidas 3 fracciones en la molécula del factor VIII de acción procoagulante, denominada factor VIII-c (coagulante), otra de acción inmunitaria denominada factor VIII-ag (antigénica), fracción responsable de la adhesividad y agregabilidad plaquetaria cuyo defecto está presente en la enfermedad de Von Willebrand y que se llama factor VII-vW (Von Willebrand). Estas 3 fracciones forman un gran complejo macro-molecular cuya estructura no está totalmente definida. Es un factor muy lábil que desaparece muy rápidamente de la sangre o plasma conservados en estado líquido, sin embargo es muy estable el plasma fresco, congelado a -40ºC o bien liofilizado (chupados, sin agua). La deficiencia congénita del factor VIII da lugar a la hemofilia A o hemofilia clásica es la más frecuente en los trastornos hemorrágicos constitucionales. El mecanismo de herencia es un carácter recesivo ligado al sexo. Lo padecen los hombres y lo trasmiten las mujeres. Factor IX christmas anti-hemofílico B o BHF: hepático dependiente de la vitamina K también se llama componente de la tromboplastina plasmática (PTC). La hemofilia B o enfermedad de christmas puede ser congénita o adquirida se parece mucho a la deficiencia del factor VIII en los aspectos del laboratorio. Como no se consume durante la coagulación ni se destruye con el tiempo existe también en el suero. Factor X factor Stuart-Prower: es de síntesis hepática, dependiente de la vitamina K. Ocupa su puesto clave en la activación de la protombina. Tiene actividad enzimática y puede activarse tanto por vía intrínseca como por vía extrínseca. En su insuficiencia aparece el cuadro clínico caracterizado por hematomas, epistaxis (sangrado por la nariz), etc. Que se parece mucho a las insuficiencias del factor VII. Factor XI antecedentes plasmáticos de la tromboplastina ó PTA: de síntesis hepática que es activada por el facto XII activado sin necesidad de la presencia de calcio como cofactor. Después de la deficiencia del factor VII y IX, es la deficiencia congénita más frecuente, pero la diátesis hemorrágica producida es más bien ligera. Se hereda con un carácter dominante simple no autosómico no ligado al sexo, afecta tanto a los hombres como a las mujeres. Factor XII factor Hageman o factor de contacto: su lugar de síntesis es desconocido. Su eficiencia aislada no presenta manifestaciones hemorrágicas y es activado por superficie de carga negativa por ejemplo: el litio y por diversas enzimas. Factor XIII estabilizantes de la fibrina o fibrinasa: tanto el hígado como los megacariocitos parecen estar implicados en la producción de factor XIII. Está presente en el plasma y ausente en el suero. El factor XIII activado hace más estable e insoluble la fibrina y por tanto más resistente a la digestión de la plasmina. Se sospecha una deficiencia cuando todas las pruebas de coagulación son normales pero el coágulo de fibrina sufre lisis. Precaleina o factor fletcher Cininógeno de auto pero molecular o factor fitzgeral Proteína C Proteína S Factor Passovoy Factores presentes en el suero: VII, IX, X, XI, XII Factores que quedan en el plasma después de la adsorción (pegarse): I, V, VIII, XI, XII Factores hepáticos: I, II, V, VII, IX, X Factores vitamina K dependientes: II, VII, IX, X TRASTORNOS DE LA HEMOSTASIA Cuando el mecanismo hemostático falla se produce la salida de sangre al exterior de los vasos sanguíneos, hemorragia. Esta hemorragia puede tener varios orígenes según sea la porción hemostática que falle. Así distinguimos: • Trastornos de la pared vascular: aunque esta pared quede fuera del sistema hemático, debemos considerarla, pues ya se ha visto que la vasoconstricción refleja y la propia estructura del vaso intervienen en la primera fase de la hemostasia. • Trastornos de las plaquetas: tanto en la cantidad de plaquetas como en la calidad de las mismas que altera su funcionamiento. • Trastornos de los factores plamáticos de la coagulación así como la aparición de inhibidores de la misma. Aunque esta clasificación parece clara la realidad clínica es que en muchas ocasiones los procesos son mezcla de varios mecanismos, apareciendo cuadros clínicos complicados. Definiciones de los conceptos sobre trastornos de la hemostasia: las distintas manifestaciones clínicas aparecen con frecuencia en los trastornos de la hemostasia: • Petequias: son pequeñas manchas rojo-púrpura del tamaño de una cabeza de alfiler. Representa sangre extravasada de vasos intactos. Se observan por lo general en las extremidades debido a la alta presión venosa. Aparecen en enfermos con vasculopatías, trombocitopenia y trombopatías. • Púrpuras: cuando las petequias se agrupan se las conoce con el nombre de púrpuras. Son más grandes y aparecen los mismos trastornos que las petequias. • Equimosis: son áreas de sangre extravasadas generalmente de origen hipo-dérmico que aparecen de forma espontánea o por traumatismo. Son dolorosas y sensibles al tacto. Pueden aparecer en enfermos con trastornos vasculares o de plaquetas. • Hematomas: es un tumor (bulto) por acumulación de sangre. Es una gran equimosis que infiltra tejidos subcutáneos o muscular que produce deformidad y la aparición de manchas moradas. Los hematomas aparecen en los trastornos de la coagulación y en la fibrinolisis y en identidades clínicas como leucemias, sobredosis de anticoagulantes orales, etc. • Hermatrosis: es la localización de la hemorragia en la articulación. Aparece en los trastornos graves de la circulación como en la hemofilia. • Hematuria: presencia de sangre en la orina. Puede ser debida a lesiones renales cáncer de otros trastornos graves de la coagulación. Trastornos del sistema vascular: los trastornos vasculares o vasculopatías son un grupo heterogéneo de trastorno muy frecuentes en la clínica práctica que se caracterizan por equimosis y hemorragia espontáneas en los pequeños vasos. La anomalía fundamental parece residir en los propios vasos, por otro lado las manifestaciones hemorrágicas debido a vasculopatías no suelen ser graves. La localización de la hemorragia es principalmente en la piel. Los trastornos vasculares pueden ser congénitos o adquiridos. • Dentro de las congénitas tenemos: ~ Telangiectasia hemorrágica hereditaria: es una dilatación de los capilares de pequeños calibre generalizadas o localizadas. Esta enfermedad se trasmite por herencia autosómica dominante. La lesión básica está constituida por la presencia en piel y mucosas de multitud de telangiectasias producidas por dilatación de capilares. Es frecuente que las hemorragias no aparezcan hasta la juventud y la manifestación más frecuente es la epistaxis. Las pruebas de laboratorio son normales. • Dentro de las adquiridas tenemos: ~ Equimosis simple: es un trastorno frecuente y benigno que aparecen en mujeres sanas en edad fértil comenzando generalmente en la adolescencia. Se caracteriza por recurrencia de equimosis sin causa aparente. La causa de este trastorno se desconoce, por que todas las pruebas de hemostasia son normales. ~ Púrpura senil: vasculatura frecuente en los ancianos. Aparece por lo general en los antebrazos y manos. La piel de las zonas afectadas es delgada y elástica. ~ Púrpura por infección y medicamentos: la púrpura pueden causarla diversas clases de virus, bacterias y sus toxinas. Su origen puede deberse a la formación de micro-trombos y a la acción tóxica sobre el epitelio vascular. Medicamentos como la aspirina o quinina pueden causar equimosis. Su modo de acción se desconoce pero el trastorno mejora cuando se suspende el tratamiento. ~ Escorbuto: falta de vitamina C que cursa con gingivitis hemorrágica, petequias hemorrágicas, etc. ~ Sarcoma hemorrágico de Kaposis: son nódulos múltiples azulados de la piel con hemorragias y caracteres neoplásicos. Es producido por una proliferación de células neoplásicas acompañando a la proliferación de células del sistema mononuclear fagocítico. Es típico en la enfermedad del SIDA. Trastornos de las plaquetas: la función de las plaquetas en la hemostasia es la formación del tapón hemostático para reducir la pérdida de sangre como respuesta a una lesión vascular. Los trastornos plaquetarios se pueden clasificar en: trombopenia, trombocitosis y trombopatías. • Trombopenia: es el recuento plaquetario inferior a 125.000/mm3 si bien las manifestaciones clínicas comienzan cuando las cifras son inferiores a 60.000/mm3. Los signos clínicos más frecuentes son: púrpura, epistaxis, gengivorragias y hemorragias gastrointestinales. Los datos de laboratorio más significativo son: cifra de plaquetas disminuidas, tiempo de sangría prolongado, fragilidad capilar aumentado y coágulo poco retráctil. La trombopenia se puede producir por: 1·- Descenso en la producción de plaquetas: tiene lugar por 2 mecanismos: A·- Disminución en la producción de megacariocitos como consecuencia de lesión medular causada por radiaciones, infecciones, medicamentos, leucemias. B·- Por una trombopoyesis ineficaz, por existir un defecto de maduración que impide el paso de megacariocitos a plaqueta. Se observa en las anemias megaloblásticas 2·- Aumento en la destrucción de plaquetas: esta destrucción puede tener lugar por 2 formas: A·- Por un mecanismo inmunológico: se conocen varias formas clínicas de trombopenia inmunitaria: • Púrpura trombopenia idiopática (PTI): aparece una trombopenia intensa que provoca hemorragia graves. Puede presentarse de forma aguda o crónica siendo más frecuente en mujeres que en hombres. Los datos de laboratorio son: presencia en más del 80% de los casos anticuerpos anti-plaquetarios, así como una reducción de la vida media de las plaquetas. El tratamiento se basa en el empleo de corticoides y en ciertos casos es aconsejable la esplenectomia. • Púrpura inducida por medicamentos: en la mayor parte de los casos el medicamento se combina con las proteínas del plasma. Actuando como antígeno y dando lugar a la formación de anticuerpos contra él. Los medicamentos pueden ser cloranfenicol, eritrombocina, tetraciclinas, sulfamidas, aspirinas, feniltoinas,... B·- Por un exceso de consumo de plaquetas: se presentan en el caso de coagulación intravascular diseminada (CIVD). La causa más conocida es la lesión del endotelio vascular al que se adhieren plaquetas, trombina y bacterias. Se da agregación plaquetaria con secuestro en los capilares y por tanto del número circulante. 3·- Incremento de plaquetas en el bazo por esplenomegalia: la esplenomegalia cualquiera que sea su origen lleva consigo un aumento de plaquetas almacenadas y como consecuencia una disminución de plaquetas circulantes. • Trombocitosis: se da cuando las cifras de plaquetas circulantes es superior a 500.000/mm3. Aparece después de intervenciones quirúrgicas y hemorragias, en quemaduras, etc. Si superan el millón de plaquetas se habla de trombocitemia siendo muy amplio el riesgo de trombosis en extremidades. • Trombopatías: engloban todos aquellos trastornos hemostáticos cuya causa principal sea una anomalía en la función de las plaquetas: ~ Trombo-astenia de Glanzmann: es una diátesis hemorrágica que se hereda de forma recesiva autosómica y que se caracteriza por un recuento de plaquetas normal, tiempo de sangría prolongado y ausencia de agregación plaquetaria. ~ Enfermedad de Bernand-Soulier: es una enfermedad grave que se hereda de forma autosómica recesiva y que se caracteriza por hemorragias gastrointestinales, epistaxis y menorragia (menstruación larga y abundante). Los datos de laboratorio son: número de plaquetas normal, tiempo de sangría alargado, alteración en la agregación plaquetaria. ~ Defecto de liberación de plaquetas: el más conocido es el defecto conocido como aspirin-lire. Es un trastorno tanto en la adhesión como en la agregación plaquetaria. Trastornos de la coagulación: las anomalías de los factores plasmáticos de la coagulación causan diátesis hemorrágica ya que se altera la formación de fibrina. En los trastornos de los factores la hemorragia tiende a producirse en tejidos profundos en oposición al sangrado superficial de la vasculopatías y de los trastornos plaquetarios. La deficiencia en la actividad de los factores pueden producirse por diversos mecanismos: 1·- Síntesis disminuida del factor (defecto cuantitativo) 2·- Síntesis anormal del factor (defecto cualitativo) 3·- Pérdida o exceso de destrucción de factores 4·- Inactivación de los factores de coagulación por anticuerpos circulantes. En los trastornos hereditarios de los factores de la coagulación se ha observado que las deficiencias se producen tanto por síntesis de proteínas anormales como disminución de la síntesis. En estos trastornos se afecta casi siempre un solo factor y el sangrado aparece en un solo sitio. En los trastornos adquiridos de la coagulación, muchos más numerosos se afectan en general más factores y las hemorragias suelen aparecen en varios lugares. Estos trastornos están ocasiones principalmente por enfermedades hepáticas o por un exceso de fármaco anticoagulantes cumarínicos (sustancia anticoagulante). En general existe correlación entre la cuantía del defecto de un factor y la gravedad de la clínica hemorrágica. Trastornos congénitos de la coagulación: se han descrito defectos hereditarios de cada uno de los factores de coagulación sin embargo más del 95% de los pacientes con una coagulopatía congénita tienen una alteración del factor VIII (hemofilia A), del factor IX (hemofilia B) o la enfermedad de Von-Willebrand. • Hemofilia A: el término hemofilia se aplica a los trastornos hereditarios de coagulación que se trasmiten de manera recesiva y ligada al sexo (cromosoma X). La frecuencia normal es de 1 por cada 10.000 personas. Dos déficit son responsables de las hemofilias: ~ Déficit del factor VIII, hemofilia A ó clásica que son el 85% de los enfermos. ~ Déficit del factor IX o hemofilia B que son un 15%. La trasmisión y los síntomas son idénticos pero las pruebas en laboratorio y tratamiento son distintos. Las mujeres la trasmiten pero muy rara vez la sufren; al poseer 2 cromosomas X pues, si ambas están afectadas, que sería en el único caso en la que la sufriría no llegaría a ser un ser viable pues muere antes de nacer. El varón al poseer un solo cromosoma X sufre la enfermedad. Todas las hijas de un hemofílico serán portadoras y todos sus hijos será normales ya que llevan forzosamente el cromosoma Y normal del padre. Las hermanas de un hemofílico tendrán un 50% de posibilidades de ser portadores. Las portadoras hemofílicas transmiten la enfermedad a la mitad de sus hijos varones y el carácter de portadoras a la mitad de sus hijas. Aproximadamente el 30% de los hemofílicos no tienen antecedentes familiares de hemorragia lo que sugiere mutaciones continuas del gen productor del factor VIII normal. La deficiencia del factor VIII en la hemofilia es el resultado de la síntesis de una molécula anormal que se traduce en una actividad coagulante reducida (factor VIII-c) con una actividad antígeno normal (VIII-Ag) y una actividad Von Willebrand normal (VIII-vW). En cuanto a la clínica el signo fundamental es la hemorragia. Se produce por pequeños golpes ó por cualquier otro traumatismo que en una persona normal no causaría hemorragia. En el hemofílico esta hemorragia no cede espontáneamente y tiende recidivar. Pueden ser externas, en la piel (mucosa) o internas formando hematomas subcutáneas o infiltrando cavidades. Es característica la formación de hematomas musculares por traumatismo o inyecciones intravasculares. Estos hematomas pueden comprimir vasos y nervios dando la sintomatología propia de la comprensión, incluyendo necrosis y parálisis. La hermatrosis (hemorragias intra-articulaciones) constituyen una manifestación muy importante y típica. Estas hemorragias van poco a poco inutilizando la articulación hasta su total destrucción. Además pueden aparecer espontáneamente. Las frecuencias de las crisis hemorrágicas dependen del nivel sanguíneo del factor VIII. Datos de laboratorio: ~ Tiempo de coagulación alargado ~ Tiempo de cefalina-caolín alargado ~ Pruebas de hemostasia primarias normales ~ Tiempo de quick, fibrinógeno y trombina normales ~ Actividad de factor VIII-c El tratamiento de los hemofílicos: requiere laboratorio y centros científico y especializado. La base del tratamiento es la administración sustitutiva del factor VIII el cual puede obtenerse de varios fuentes: ~ Plasma fresco congelado ~ Concentrados comerciales del factor VIII ~ Crio-precipitados ~ Sangre fresca Esta última solo deberá emplearse en casos muy graves y para reemplazar la volemia (volumen hemático) ya que la cantidad de factor VIII en sangre es baja. En casos de hemorragias intensa o cirugía es necesario mantener los niveles del factor VIII en sangre elevados. Los pacientes con hemofilia tienen un riesgo elevado de padecer enfermedades infecciosas, como resultado de las múltiples transfusiones que reciben durante su vida. Últimamente este riesgo se a reducido como consecuencia del mayor control de calidad de los preparados del factor VIII. Posibilidades genéticas en la descendencia de hombre -mujer: • Hemofilia B: la deficiencia de factor IX o enfermedad de christmast es una coagulopatía hereditaria similar a la hemofilia A. Se diferencia de la hemofilia A en 2 pruebas: 1·- Si la adicción del plasma adsorbido corrige el tiempo de cefalina caolí, se trata de hemofilia A. 2·- Si la adicción de suero corrige el tiempo de cefalina caolí se trata de una hemofilia A. El tratamiento de la hemofilia B se puede realizar con plasma y preferentemente con concentrados del factor IX. • Enfermedad de Von-Willebrand: es una enfermedad hemorrágica hereditaria trasmitida de forma autosómica dominante que se caracteriza en su forma típica por: 1·- Tiempo de sangría alargado. 2·- Niveles factor VIII-c, factor VIII-ag y factor VII-vW. Existen diversas variantes de esta enfermedad con múltiples formas clínicas que serán más intensas cuanto mayor sea el déficit de actividad del factor VII-vW. El tratamiento se basa en medidas hemostáticas locales y en transfusión de plasma fresco ó crio-precipitado lo cual además de corregir la deficiencia del factor VIII normaliza el tiempo de sangría. Trastornos adquiridos de la coagulación: son mucho más frecuentes que los trastornos congénitos. En los adquiridos se afectan varios factores, mientras que en los congénitos suele ser uno. Los que estudiaremos son: trastornos por déficit de vitamina K, trastorno por hepatopatías inhibidores de coagulación y por consumo de factores de coagulación. • Déficit de vitamina K: la vitamina K es necesaria para que el hígado sintetice los factores de coagulación II, VII, IX y X. En ausencia de vitamina K el hígado sintetizan moléculas con estructuras idénticas que tienen inmunidad pero que carecen de capacidad coagulativas. Se las conoce como Pivka (proteínas inducidas por ausencia de vitamina K). Datos de laboratorio: alargamiento del tiempo de quick en grado variables según sea el déficit de factores II, VII y X. El tiempo de tromboplastina parcial activa, ó también puede estar alargada por el descenso del factor IX presente en la vía intrínseca. ~ Alteraciones que causan déficit de vitamina K: 1º·- Enfermedad hemolítica del recién nacido: este trastorno es causado por defecto en la síntesis de los factores dependientes de la vitamina K. Es el resultado de 2 actuaciones: Por un lado de falta de vitamina K Por otra la inmadurez funcional del hígado del recién nacido. El riesgo hemorrágico puede agravarse cuando la madre a tomado anticoagulantes orales al final del embarazo, puesto que atraviesan la barrera placentaria. 2º·- Maladsorción de vitamina K: se da en situaciones en las que por faltar la flora intestinal normal debido a un tratamiento antibiótico prolongado junto con una dieta deficiente en vitamina K podría haber falta de esta en el hígado para la síntesis de factores. 3º·- Anticoagulantes orales: estos anticoagulantes bloquean la vitamina K. • Hepatopatías: el hígado es el órgano de síntesis de proteínas plasmáticas activadoras e inhibidoras de la coagulación y de la fibrinolisis; de aquí que en las enfermedades hepáticas graves se presentes alteraciones de la hemostasia. [Un tiempo de quick alargado que no se consiga con administración de vitamina K revela la existencia de daño hepático. El factor V está frecuentemente por debajo del nivel normal en hepatopatías, cirrosis, etc. El factor de coagulación con una vida media más corta, es el factor VII, y por tanto se disminución en el plasma por enfermedad hepática es la primera alteración que puede detectarse]. El estudio de la coagulación en las enfermedades hepáticas puede permitir valorar la capacidad de síntesis del hígado y facilitar el diagnóstico de hepatopatías graves. • Inhibidores de la coagulación: para un control normal del mecanismo de la hemostasia, existen inhibidores fisiológicos como la anti-trombina III. En situaciones patológicas aparecen inhibidores o anticoagulantes que pueden producir diátesis hemorrágica. Puede ser de 2 tipos: 1º·- Inhibidores dirigidos contra un factor de la coagulación 2º·- Inhibidores de reacción [o contra un grupo de factores] 1·- Entre los inhibidores específicos de factores de la coagulación tenemos: el factor-inhibidor VIII que se encuentra en un 10% de los enfermos con hemofilia A (aunque también en otras patologías). Estos inhibidores se originan como consecuencia de la infusión a estos enfermos de plasma, crio-precipitados o concentrados del factor VIII. Como consecuencia pueden aparecer graves diátesis hemorrágicas. 2·- Los inhibidores de reacción no destruyen específicamente ningún factor de coagulación. El más conocido es el tipo LUPUS, puesto que aparece sobre todo en enfermos con lupus eritematoso diseminado [parece que inhibe el complejo protombina, el tiempo del quick y el tiempo de cefalina-caolí estarán alargadas]. • Consumo de factores de coagulación también se llaman coagulopatías de consumo o coagulación intra-vascular diseminada (CIVD), por coagulación intra-vascular se entiende un proceso de intra-vascular coagulación acelerada. Por medio de ciertos mecanismos como el paso de factores tisulares a la sangre como la presencia en la misma en complejos inmunitarios, bacterias, virus o una flexión endoterial, se puede romper el equilibrio normal de hemostasia, desencadenando: 1·- Deposito intra-vascular de fibrina 2·- Consumo de factores de coagulación y plaquetas 3·- Activación del sistema fibrinolítico Como consecuencia trombosis y hemorragias son manifestaciones clínicas presentes en la CIVD. TEMA XXI TROMBOSIS Y TRATAMIENTO ANTI-TROMBÓTICO FORMACIÓN DEL TROMBO La trombosis figura entre las causas más frecuentes entre las causas de enfermedades y muerte entre los enfermos hospitalizados. El término trombosis se refiere a la formación de una masa intra-vascular de fibrina, plaquetas, eritrocitos y leucocitos que interfieren en el flujo normal de sangre y que se produce por una falta de Inactivación del proceso de coagulación. El trombo o parte de él, puede desprenderse de la pared vascular y circular y ocluir diferentes vasos formando trombos arteriales, venosos o capilares según ocluyen arterias, venas y capilares. No hay pruebas específicas de laboratorio que detecten la presencia de trombos ni la predisposición de su formación. En las arterias caracterizadas por alta presión y alta velocidad de flujo, la interacción de un endotelio vascular alterado con las plaquetas es el factor más importante en la formación de un trombo arterial, o trombo blanco. Los factores que elevan el riesgo de trombos arteriales son: dietas ricas en colesterol, el consumo de cigarrillos, usos de anticonceptivos orales, así como la presencia de hipertensión, diabetes, arterosclerosis, etc. La circulación venosa donde existe baja presión y disminuye la velocidad de flujo, es el estado de hiper-coagubilidad y el estancamiento sanguíneo, lo que conducirá a un trombo venosos o trombo rojo, denominación que recibe por su alto contenido en eritrocitos que quedan atrapados en la malla de fibrina. La trombosis venosa se produce principalmente en las venas profundas de las piernas, de modo que este tipo específico reciben el nombre de trombosis venosa profunda (TVP), que si se acompaña de inflamación con dolor, sensibilidad excesiva y enrojecimiento es conocida como tromboflebitis. Los test de laboratorio son una ayuda en el tratamiento cuando la trombosis ocurre en venas no en arterias. La complicación principal de la trombosis venosa profunda, al igual que los trombos arteriales, es el desprendimiento del émbolo y su desplazamiento al corazón o pulmón, dando lugar a infartos de miocardio y embolias pulmonares. Existen estados que predisponen a la trombosis venosa profunda entre ellos las insuficiencias congénitas de anti-trombina III, proteínas C y proteínas S. Los trombos en los capilares son de tipo mixto y contiene fibrina y plaquetas. Diversas patologías presentan un riesgo mayor de accidentes trombo-embólicos: neoplasias, síndrome mielo-proliferativos, síndrome nefrótico, diabetes, etc. Igualmente existe un alto riesgo de trombo-embolia en pacientes ancianos que han sufrido operaciones quirúrgicas, sobre todo aquellas que han de estar largo tiempo inmovilizados, pues la estasis no facilita la dilución de factores activados por el flujo sanguíneo y por tanto la coagulación. TRATAMIENTO ANTI-TROMBÓTICO De acuerdo con la patogénica se puede realizar una prevención eficaz y un tratamiento adecuado de la trombosis. Los medicamentos utilizados actúan a diferentes niveles: 1·- Medicamentos para prevenir la formación, del coágulo de fibrina (anticoagulante) 2·- Medicamentos que impide la adhesión y agregación plaquetaria (anti-agregantes) 3·- Medicamentos trombo elípticos que disuelven el coágulo de fibrina (fibrinolípticos). 1·- Heparina y anticoagulantes orales: no disuelven el coágulo pero impiden que se forme o que se extienda. Heparina: se utiliza en la prevención de la trombo-embolia venosa y en la embolia pulmonar. La hemorragia es su principal complicación. Se puede administrar por vía venosa o subcutánea. La dosis correcta de heparina varía de un enfermo a otro y siempre a de ser controlada por pruebas de coagulación, siendo la prueba más sensible la TTPA. Su mecanismo de acción es a través de la anti-trombina III, bloquear la trombina y los factores X a, XI a y XII a. Los anticoagulantes orales o anti-vitaminas K se utilizan en la profilaxis y tratamiento de la trombosis tanto arterial como venosa. Se trata de un grupo de sustancias cuya acción es reducir la síntesis hepática de los factores II, VII y X y de las proteínas C y S que son vitaminas K dependientes. Los anticoagulantes orales son de 2 tipos: Derivados cumarínicos: discumarol, acencumaral, warfarina y biscumacetato Derivados indandiona: fenindiona El control de la terapia anticoagulante oral, se realiza mediante el tiempo de quick que es la prueba más sensible • Indicaciones de los coagulantes orales: trombosis venosa, embolia pulmonar y cirugía aortacoronaria. La hemorragia es el efecto adverso más frecuente y se debe generalmente a una sobredosis. El riesgo aumenta por la ingestión de aspirina. 2·- Anti-agregantes: desde que se conoció el papel esencial que desempeña las plaquetas en el origen de la ateroesclerosis y de la trombosis han aparecido numerosos fármacos que inhiben la función plaquetaria y que por tanto intervienen en la prevención de los proceso trombóticos principalmente la formación de trombos arteriales. Los anti-agregantes evitan la adhesión de las plaquetas al tejido sub-endoterial inhibe la agregación de las mismas e impiden la liberación de sustancias de sus ganglios El antiagregante prolonga el tiempo de sangría. Otros fármacos anti-agregantes son el: tipirridamol, sulfín-pirizona. 3·- El objetivo de la terapia fibrinolíptica es activar el plasminógeno contenido en el trombo para su transformación en plasma y su acción sobre la fibrina, provocando la lisis del trombo. Los medicamentos son: extreptoquinasa y uroquinasa que son agentes de primera generación. Actualmente se utiliza un trombolíptico de segunda generación conocido como activación tisular del plasminógeno (t-PA) obtenidos mediante recombinación genética. TEMA XXII HEMOTERAPIA INTRODUCCIÓN Los conocimientos inmuno-hematológicos actuales permiten evitar en la mayor parte de los casos problemas de incompatibilidad y sensibilización. Las técnicas cada día más específicos y sobretodo más sensibles reducen en gran medida la posibilidad de infecciones. Los procedimientos de conservación, fraccionamiento y purificación de los componentes de la sangre permiten aportar con mayor fiabilidad los elementos necesarios en cada caso. Existe el riesgo de una utilización excesiva o inapropiada de la transfusión por lo que el médico debe valorar el beneficio y el riesgo en cada caso realizándolo solo cuando sea verdaderamente necesario. En primer lugar sigue existiendo riesgo de infecciones, inmunizaciones y reacciones adversas. En segundo lugar la escasez de estos componentes hacen que se deban utilizar cuando sea verdaderamente necesario y con un criterio clínico adecuado. SELECCIÓN DEL DONANTE Y TÉCNICA DE EXTRACCIÓN Actualmente en la mayor parte de los países se tiende a que la donación de la sangre no sea remunerada evitándose así una serie de factores de riesgos. Esto tiene una consecuencia negativa debido a la escasez de donantes y la necesidad de imputar hemoderivados. Los requisitos que se exigen en el donante cubren 2 objetivos: 1º·- Preserva de todo riesgo al receptor 2º·- Conseguir todas las garantías para el donante En primer lugar se realiza al posible donante una entrevista en un lugar que permita la discreción seguida de una sencilla exploración clínica. El historial médico incluye: A·- Aspecto saludable B·- Edad comprendida entre 18-65 años C·- Peso estable y superior a 50 Kg. D·- Temperatura inferior a 37'5ºC E·- Pulso que debe ser regular y entre 60-100 latidos por minuto F·- Tensión arterial sistólica inferior a 180 mm de Hg. y la diastólica inferior a 100 mm de Hg. G·- Concentración de hemoglobina en sangre y hematocrito en hombres será superior a 12'5g/dl y 38% de hematocrito. En la mujer será superior a 12gr/dl y H·- Estudio de enfermedades padecidas y hábitos I ·- Una vez finalizado el reconocimiento el donante debe firmar un documento en el que debe constancia clara de que ha comprendido los motivos que excluyen de donar y de que estos no le afecten, así como su conformidad par realizar la donación. Es causa de rechazo permanente permanecer a ciertos grupos como varones homosexuales, drogadictos por vía parenteral, personal sub-saharianas y haitíes hemofílicos, parejas sexuales de los grupos antes citados o infectados por el VIH o bien haber padecido o padecer hepatitis B ó C, paludismo, procesos neoplásicos, proceso crónicos renales, hepáticos, cardio-pulmonares. Son causa de rechazo temporal las enfermedades de la piel, infecciones respiratorias, tuberculosis, sífilis, anemias, afecciones transitorias renales, gastrointestinales y hepáticos no víricos. El intervalo mínimo entre 2 extracciones de sangre total no podrá ser inferior a 2 meses. El número máximo de extracciones anuales no podrá superar 4 los hombres y 3 para las mujeres. La extracción de sangre se hace colocando al donante en una camilla y en un ambiente sosegado, se registra al donante y se rotula correctamente la bolsa y tubos de muestra. Se elige una zona del brazo sin lesiones cutáneas y una vena, se desinfecta la zona, frotando con energía con un algodón impregnado con un antiséptico adecuado de arriba abajo, se repite con otro algodón. Se pinza el sistema de la bolsa para evitar la entrada de gérmenes y se hace la punción canalizando la vena. Se fija la aguja para evitar que se extravase. A continuación se suelta la pinza comprobando que al sangre sale con normalidad. Durante la extracción se mueve la bolsa con suavidad para mezclar la sangre con el anticoagulante. La extracción puede durar de 7-10 minutos. La cantidad de sangre extraída deberá tener en cuenta el peso del donante, no deberá superar el 13% del volumen sanguíneo teórico del donante. Al terminar se pinza el tubo cerca de la aguja y se extrae esta se hace pasar la sangre del tubo a la bolsa para que se mezcla con el anticoagulante se tapona el orificio con una torunda de algodón estéril. Durante la extracción se pueden presentar mareos, sudoración, palidez, náuseas, vómitos, etc. A veces hay que parar la extracción sobretodo si se presenta hipotensión, en este caso se eleva las piernas para que la sangre llegue mejor al cerebro y se administran líquidos por vía oral. Si la punción produce hematoma se comprime en zona elevando el brazo al mismo tiempo. RECOGIDA Y CONERVACIÓN DE SANGRE Los 450 ml de sangre extraídos de un donante se denominan unidad de sangre y recogen bolsas de plástico que contienen aproximadamente 50ml de anticoagulante y conservante. Se necesita también una parte laicota (representación) de sangre para los análisis de tipificación y pruebas de ideoniedad de la sangre extraída. Hay que tener en cuenta que a partir del memento de la extracción se inicia una serie de cambios metabólicos que continúan en el almacenamiento que produce una disminución de células viables (buenas). Se considera que la conservación de la sangre ha sido adecuada cuando a las 24 horas de transfundirla se recupera el 70% de los hematíes. La conservación a baja temperatura disminuye la glucólisis. Los preparados más utilizados son: • Ácido citrato dextrosa: conservación durante 21 día entre 1-6ºC • Citrato fosfato dextrosa: conservación durante 21 día entre 1-6ºC • Citrato fosfato dextrosa con adenina: conservación durante 35 día entre 1-6ºC • Heparina: se conserva refrigerado sólo 48 horas. • Nuevos conservantes: se preparan como soluciones aditivas que contienen en 100ml de suero fisiológico dextrosa, manitol y adenina, incorporando al concentrado de hematíes después de eliminar el plasma. Previamente la sangre se recoge sobre citrato fosfato dextrosa. No se añaden sustancias antisépticas o bacteriostáticas a los productos sanguíneos. PRUEBAS DE TIPIFICACIÓN 1º Determinación del sistema ABO: una vez detectados estos antígenos se comprueban los anticuerpos correspondientes en el plasma, se rotula adecuadamente a que grupo pertenece la sangre. 2º Determinación del título de los anticuerpos anti-A y/o anti-B naturales e inmunológicos si los hubiera 3º Determinación del factor D 4º Determinación de anticuerpos irregulares PRUEBAS DE SELECCIÓN 1·- Determinación de GPT (ALT) y GOT (AST): si son altas se deshecha la sangre y se investigan posibles infecciones víricas. 2·- Determinación antígeno de superficie de la hepatitis B (HBsAg): se hace por técnica de EIA ó RIA. Si alguna es positiva se hacen las confirmaciones pertinentes, rechazando de forma permanente la unidad de sangre con HBsAg positivo e incluso las que sólo sean anti-HBc con objeto de evitar cualquier riesgo aunque lo más posible es que no se infectante. 3·- Determinación de anti-HC (anticuerpo anti-HVC): desde hace unos años esta prueba nos permite detectar son anticuerpos el echo de que un elevado número de ellos se hagan crónicos hace que se rechace la sangre con estos anticuerpos. Las técnicas de EIA que se usan en el banco de sangre pueden dar falsos positivos. Hay que tener en cuenta que las técnicas utilizadas en los bancos de sangre deben ser muy sensibles aunque pierden algo de especificidad, así se evita cualquier riesgo. Nos interesan técnicas que no den falsos negativos. 4·- Determinación de anticuerpos anti-VIH1 y VIH2: se aplica una técnica de EIA. Si da positivo se confirma por Westerh Blat, si la confirmación es positiva se rechaza la sangre. Si los resultados son dudosos se repiten las pruebas no aceptando una sangre hasta que tenga la total seguridad de que no está afectada por VIH. 5·- Determinación de RPR: prueba que detecta anticuerpos inespecíficos anti-treponema (cusa la sífilis). Si da positivo es necesario determinar anticuerpos específicos anti-treponema pallidus por inmuno-fluorescencia o hemoaglutinación. CRITERIOS E INDICACIONES EN LA TRANSFUSIÓN Es necesario que el médico valore un serie de circunstancias como la edad del paciente, el origen, grado y desarrollo de la anemia, la estabilidad hemodinámica y la existencia de factores cardiacos y pulmonares antes de decidir una transfusión. En las anemias hemolítica autoinmunitarias debido a que los auto-anticuerpos se suelen producir contra antígenos eritrocitarios muy comunes y que la tipificación del hematíe alterado es difícil por estar recubierto de anticuerpos la transfusión no es en principio recomendable puede ocurrir que los auto-anticuerpos ataquen los hematíes transfundidos y que además se formen, aloanticuerpos contra estos hematíes por no ser compatibles ante la dificultad del tipado valorado todas las circunstancias y dedicada la transfusión el médico (hematólogo) e el que establece que producto sanguíneo es el adecuado en cada caso. PREPARACIÓN DEL RECEPTOR Los protocolos establecidos tienen que figurar por escrito figurando todos los pasos de forma clara. Deben ser conocidos por todos el personal sanitario con total coordinación. El historial del paciente estará bien detallado. Determinación de los antígenos A, B y D (Rh) y la investigación de anticuerpos irregulares. Registro cuidadosos de todos los resultados. Realización de pruebas cruzadas para ver la compatibilidad de la sangre del donante y la sangre del receptor. Se ponen en contacto los hematíes del donante con el suero del receptor haciendo una prueba de aglutinación directa y un test de Coombs indirecto. Con el fin de detectar anticuerpos completos tipos Ig M y anticuerpos incompletos tipos Ig G que pueden encontrarse en el suero del receptor y alterar los hematíes del donante. Si la prueba cruzada no se realiza por una situación de urgencia se usa sangre del grupo O, Rh-, libre de anticuerpos irregulare y con título bajo de anti-A y anti-B. REALIZACIÓN DE LA TRANSFUSIÓN Los puntos que hay que tener en cuenta en el momento de la transfusión: 1·- Es conveniente que el paciente no tenga fiebre al comenzar la transfusión. 2·- El ritmo de administración habitual es de 500 ml en 1 ó 2 horas. La transfusión no debe durar más de 4 horas ya que la bolsa de sangre a temperatura ambiente pudiera proliferar bacterias. 3·- Durante los primeros 3º minutos, la transfusión se hará lentamente y el responsable de la misma deberá permanecer al lado del enfermo. 4·- Se debe controlar la transfusión comparando los valores del producto transfundido ante y después de dicha transfusión y el aumento no es el adecuado hay que investigar. 5·- Se debe preguntar al enfermo como se siente durante la transfusión y después de la misma. 6·- No debe mezclarse con la sangre otras soluciones intravenosas o medicamentos. 7·- Si la transfusión es de un gran volumen y hay que hacerla a una velocidad mayor de 100ml/minuto es necesario calentar la sangre. Se puede hacer pasando la sangre por un serpentín sumergido en un baño de agua caliente o en bloque de calentamiento en contacto con la sangre a través de una bolsa de plástico. La sangre no puede calentarse por encima de 37ºC ya que podría hemolizarse. 8·- La mayor parte de las transfusiones que suponen un gran volumen de sangre se hacen a través de filtros. TEMA XXIII HEMODERIVADOS, COMPONENTES SANGUÍNEOS Y DERIVADOS DEL PLASMA La sangre se recoge en una bolsa de plástico que contiene anti-coagulantes y soluciones conservadoras. La sangre total puede conservarse a 4ºC durante 5 semanas. De la sangre total pueden separarse varios componentes en el mismo banco de sangre. Los hematíes y las plaquetas se aíslan de la sangre total mediante centrifugación suave, después de la centrifugación los hematíes y las plaquetas son procesados para varios preparados distintos. El plasma residual puede utilizarse directamente o bien ser fraccionado nuevamente par obtener otros componentes. • Sangre total: una unidad de sangre total contiene 450 ml de sangre más 63 ml de solución anticoagulantes y conservantes. La sangre anticoagulada usual contiene citrato, fosfato, dextrosa y adenina [CPD-A]. El citrato fija el ión calcio del plasma evitando así el proceso de la coagulación. El fosfato proporciona el sustrato para ayudar a mantener el nivel 2,3-di-fosfatoglicerato [2,3-DPG] de los hematíes. La dextrosa y la adenina son sustrato para los procesos metabólicos de los componentes celulares. Los hematíes, las plaquetas y los leucocitos y los factores de la coagulación está presentes en una unidad de sangre recién extraída. Durante la conservación a 4ºC, las plaquetas y los leucocitos dejan de ser funcionales al cabo de pocas horas después de la extracción. Hay una reducción gradual de la viabilidad de los hematíes relacionado con el tiempo almacenamiento. Los hematíes conservados durante 3 semanas en CPD-A presentan una recuperación media del 70%, la recuperación mínima aceptable. Con el paso del tiempo desciende los niveles de los factores de coagulación. Aunque es necesario dispones de un pequeño almacén de sangre total raras veces se utiliza. COMPONENTES DE LA SANGRE El término componente sanguíneo generalmente hace referencia a un producto separado de una unidad de sangre total El término derivado del plasma indica un producto separado de un gran volumen de mezclas de plasma mediante un proceso llamado fraccionamiento. • Componentes sanguíneos transportadores de oxígeno: las transfusiones de sangre con frecuencia se administran para restablecer la capacidad de transporte de oxígeno asegurando así la oxigenación de órganos vitales tales como el cerebro, el corazón y los riñones. A·- Concentrados de hematíes: se preparan separando aproximadamente 200 ml de plasma de la unidad de sangre total después de ser centrifugada. Este preparado contiene los hematíes correspondientes a una unidad de sangre total más unos 100 ml de plasma residual. Suele conservarse durante 35 días a 4ºC, debido a su levado valor hematocrito los concentrados de hematíes son viscosos y por ello su velocidad de sedimentación es lenta. La velocidad de infusión puede incrementarse mediante la adicción de suero salino para disminuir la viscosidad. No deben utilizarse nunca soluciones y contengan calcio ni soluciones que contengan glucosa. Las unidades estándar de sangre total de concentrados de hematíes contiene leucocitos, no viables o fragmento de leucocitos. Generalmente la presencia de leucocitos no tiene consecuencias pero en algunos pacientes puede producir una reacción transfusional de tipo febril. En tales pacientes deben recibir sangre desleucocitada. B·- Sangre leucocitada: la mayor parte de los glóbulos blancos pueden separarse descartando la capa leucocitaria mediante una técnica tan simple como la centrifugación invertida. C·- Los hematíes pueden ser congelados utilizando técnicas especiales de crio-congelados. Dichos técnicas permiten periodo de conservación hasta 10 años. Se tratan de técnicas caras por tanto el uso de hematíes congelados solamente en circunstancias especiales. Entre ellas están el suministro de hematíes a individuos pertenecientes a tipos sanguíneos raros o para auto-transfusión. • Productos plaquetarios: A·- El plasma rico en plaquetas (PRP): se obtiene de centrifugación suave de la sangre total. EL sobrenadante es transferido a la segunda de plástico de sistema cerrado. A partir de este plasma rico en plaquetas se obtienen el concentrado plaquetario mediante una segunda centrifugación y la reparación subsiguiente de plasma dejando o el sedimento de plaquetas en suspensión de 50 ml de plasma. B·- Los concentrados plaquetarios contienen aproximadamente entre 60-80% de las plaquetas contenidos en una unidad de sangre total, según la bolsa de plástico utilizada. Las plaquetas son viables durante 5 días ó más si se mantienen a 22ºC sometidas a una agitación horizontal constante. Los concentrados de plaquetas procedentes de donantes Rh+ no deben ser administrados a mujeres Rh-- en edad fértil con objeto de evitar la sensibilización al antígeno D. • Productos procedentes del plasma: A·- Plasma pobre en plaquetas (PPP): de este plasma pueden separarse numerosos productos: plasma fresco-congelado, plasma congelado, crio-precipitado y plasma de recuperación. 1·- Plasma fresco-congelado: se prepara a partir de sangre total recién extraída dentro de las 6 horas siguientes a la extracción. La pronta congelación de este plasma permite la máxima conservación de los factores lábiles de la coagulación. El plasma fresco-congelado no contiene elementos celulares y puede conservarse a -30ºC durante un periodo de hasta 12 meses y se puede utilizar para todos los factores de coagulación. 2·- Plasma congelado: es plasma separado de la sangre total dentro de las 12 horas después de la extracción. Contiene como mínimo el 50% de los factores VIII y V iniciales. El plasma congelado puede utilizarse para tratar insuficiencias de factores de coagulación de leves a moderadas Puede conservarse a -30ºC durante un periodo de 12 meses. 3·- Crio-precipitados: se prepara a partir del plasma congelado dentro de las 6 horas siguientes a la extracción, congelándola a -70ºC y dejándola descongelar a 4ºC. El precipitado que se forma a modo de copos blancos es rico en factor VIII, en fibrinógeno y fibrinectina. El crioprecipitado contiene aproximadamente 250mg de fibrinógeno y aproximadamente 80 unidades de factor VIII. Pueden conservarse a -30ºC durante 12 meses. El plasma sobrenadante del crio-precipitado debe utilizarse dentro de las 5 semanas que siguen a su obtención si se conservase a 4ºC. Pero pueden conservarse durante 2 años a -30ºC. Suele utilizarse para el tratamiento del factor VIII del fibrinógeno y de la fibrinectina. 4·- Plasma de recuperación: es plasma separado de la sangre total después de que esta ha sido conservada a 4ºC durante 24 horas. También puede proceder del sobrenadante de crioprecipitados. Está indicado en los pacientes que necesitan aumento de volemia o un aporte de proteínas plasmáticas y no necesitan el aporte. En factores de coagulación. Los producto procedentes del plasma no requieren pruebas de compatibilidad antes de ser utilizadas pero es conveniente que sean ABO compatibles. DERIVADOS DEL PLASMA Algunos derivados del plasma pueden obtenerse por fraccionamiento del plasma frescocongelado ó del plasma de recuperación. El fraccionamiento permite procesar mayor cantidad de pool (mezclas de diferentes muestras) de plasma. Pero el echo de mezclar muchas cantidades de plasma aumenta el riesgo de trasmitir enfermedades de origen víricos al receptor. 1·-Concentrados de factores de coagulación: estos concentrados se presentan como productos liofilizados. Están indicados en primer lugar para pacientes con deficiencias congénitas de factores de coagulación. Dichos concentrados no deben utilizarse para deficiencias adquiridas benignas por ser elevado el riesgo de transmisión de hepatitis. El concentrado del factor VIII es un preparado liofilizado procedentes del plasma de factores de coagulación contiene gran cantidad de factor VIII junto con pequeñas cantidades de fibrinógeno y otras proteínas. Se utiliza para el tratamiento de hemofilia A. Los concentrados del factor IX se utilizan especialmente para la deficiencia del factor IX ó hemofilia B. EL complejo del factor IX es distribuido en forma de producto liofilizado en viales que contiene 500 unidades de este factor. El uso de concentrados del factor IX, está contraindicada en pacientes con enfermedades hepáticas y se relaciona con trombosis ó coagulación intra-vascular diseminada (CIVD) Los concentrados del complejo del factor IX también se han utilizado para tratar pacientes con inhibidores adquiridos del factor VIII debido a que algunos concentrados del factor IX tienen actividad bypass sobre el factor VIII. 2·- Agentes oncóticos: la albúmina se prepara mediante fraccionamiento de un pool de plasma. Con la albúmina no existe riesgo de trasmisión de hepatitis ya que se esteriliza durante el proceso de preparación. La albúmina mantiene la presión osmótica celular y actúa como proteínas transportadora de fármaco, hormona, enzimas y metabolitos. La función principal es para la reposición de albúmina. Existen soluciones de productos sintéticos macro-moleculares (dextrano, gelatina) que se utilizan como expansores del volumen sanguíneo pero no son tan efectivos como los productos que contienen la albúmina. 3·- Inmunoglobulinas: A·- Séricas: contiene principalmente Ig G y se utiliza para prevenir algunas enfermedades de origen vírico para la terapia de reposición con pacientes con hipo-gamma-globulinemia y en el tratamiento de las inmuno deficiencias congénitas. B·- Inmunoglobulina anti-hepatitis B: es un preparado de inmunoglobulinas séricas obtenida por fraccionamiento de plasma de donantes con un título elevado de anticuerpos contra el antígeno de superficie de la hepatitis B (HBsAg). La administración de inmunoglobulinas antihepatitis B, está indicada después de al exposición a material que contiene el HBsAg, para ser efectiva la administración debe efectuarse dentro de las 48 horas siguientes a la exposición. También está indicada en los recién nacidos, cuyas madres son portadoras del HBsAg. C·- Inmunoglobulina anti-varicela-zoster: se obtiene del plasma de individuos recientemente afectados de herpes zoster. Numerosos estudios han demostrado que la administración pasiva que los anticuerpos específicos dentro de las 72 horas siguientes a la exposición, puede prevenir o atenuar dichas infecciones. D·- Inmunoglobulina anti-Rh: se obtiene a partir del plasma de individuos Rh- que han producido anti-D por inmunización. La administración de la inmunoglobulina Rh reduce sustancialmente la frecuencia de albunización rhesus en mujeres Rh- con hijos Rh+. La administración de inmunoglobulinas Rh previene precisamente la sensibilización al Rh resultante de este paso de hematíes fetales a la circulación materna. E·- Inmunoglobulina anti-tetánica: es la fracción de inmunoglobulina sérica obtenida de individuos que han sido específicamente con tosoide tetánico se aplica par la profilaxis en individuos que presentan riesgo elevado después de una herida. ESQUEMA DE LOS COMPONENTES DE LA SANGRE (hemoderivados) • Componentes de la sangre 1·- Componentes transportadores de oxígeno A·- Concentrados de hematíes (CH) B·- Sangre desleucotizada C·- Hematíes congelados 2·- Productos plaquetarios A·- Plasma pobre en plaquetas B·- Concentrados de plaquetas 3·- Productos plasmáticos A·- Plasma fresco-congelado (todos los factores de la coagulación) B·- Plasma congelado (factor VIII y V) C·- Crio-precipitado (factor VIII, fibrinógeno y fibrinectina) D·- Plasma de recuperación (volemia) • Derivados del plasma 1·- Concentrados de factores de coagulación (deficiencias congénitas) A·- Concentrados del factor VIII (hematíes hemofilia A) B·- Concentrados del complejo del factor IX (hemofilia B y en pacientes inhibidores adquiridos del factor VIII). 2·- Agentes oncóticos: A·- Albúminas B·- Otros (dextrano, gelatina) 3·- Inmunoglobulinas A·- Inmunoglobulinas séricas B·- Inmunoglobulinas anti-hepatitis-B C·- Inmunoglobulinas anti-zoster D·- Inmunoglobulinas anti-Rh E·- Inmunoglobulinas anti-tetánica TEMA XXIV REACCIONES TRANSFUSIONALES Un 3% aproximadamente de los individuos que reciben transfusiones sanguíneas experimentan un efecto adverso llamado reacciones transfusionales. La reacción transfusional puede ser producida por mecanismos inmunológicos y no inmunológicos. Las reacciones transfusionales que se producen durante la transfusión del producto sanguíneo o poco después se llaman reacciones inmediatas. Las que se producen transcurrido algún tiempo se llaman reacciones retardadas. REACCIONES INMUNOLÓGICAS A·- Hemólisis 1·- Reacciones hemolíticas inmediatas: la reacción transfusional hemolítica aguda generalmente tiene lugar después de la administración de sangre ABO incompatible. En estos casos el paciente suele quejarse de fiebre, dolor en el lugar de transfusión, sensaciones de o presión torácica y dolor en la región lumbar. Entre los signos físicos que se observan se incluyen fiebre hipotensión, hemoglobinuria y hemorragia pudiendo evolucionar hacia la insuficiencia renal y por último la muerte. 2·- Reacciones transfusionales hemolíticas agudas: suceden cuando los alo-anticuerpos anti-A y anti-B del plasma del receptor, se unen a los antígenos de los hematíes transfundidos del donante. La interacción del antígeno con el anticuerpo activa la cascada del complemento y tiene como resultado la lisis de los hematíes transfundidos. La hemoglobina libre se fija a la ato-globina y a la albúmina. Estas proteínas fijadoras quedan pronto satura y la proteína libre residual es aclara por el riñón pasando a la orina. La activación del complejo tiene como resultado la liberación del fragmento que son potentes vasos dilatadores, produciéndose también generación de trombina y activación plaquetaria. Estos procesos dan lugar a hipotensión y a CIVD. La CIVD consume plaquetas y factores de coagulación y por ello pueden ser causa de hemorragia Las reacciones transfusionales hemolíticas agudas desembocan con frecuencia en insuficiencia renal. 3·- Reacciones transfusionales hemolíticas retardadas: a veces la sangre aparentemente compatibles pueden producir reacciones transfusionales si el receptor desarrolla anticuerpos frente a antígenos presentes en los hematíes transfundidos durante la transfusión ó después de ella. En la mayoría de los casos el paciente había estado expuesto al antígeno por transfusión ó embarazo pero el nivel de anticuerpos era demasiado bajo para que fueran detectadas en la prueba cruzada. En algunas ocasiones este puede producirse después de 24 horas pero lo más frecuente es que el nivel de anticuerpo aumenta lentamente y que la destrucción de los hematíes empiece a las 2 ó 3 semanas. La única señal de reacción transfusional es el descenso en el nivel de hemoglobina. El diagnóstico generalmente, se efectúa en el banco de sangre al hacer nuevamente pruebas cruzadas, la prueba directa de la antiglobulina suele ser positiva. La prueba directa de la anti-globulina suele ser positiva. Los anticuerpos que con mayor frecuencia están implicados en estas reacciones van dirigidas contra antígenos de los sistemas Kidd, Duffy, Rhesus, Kell, S (MNSs) B·- Reacción debida a los leucocitos: 1·- Reacciones febriles: son las reacciones transfusionales más frecuentes su causa es la reacción entre los leucocitos del donante y los alo-anticuerpos producidos en el receptor por transfusiones previas o por embarazo. Suelen producirse hacia el final de la transfusión y se caracteriza por escalofríos y fiebre. Las reacciones febriles se pueden tratarse con antipirético (termalgín) y evitarse mediante transfusión pobre en leucocitos. 2·- Infiltrados pulmonares: los infiltrados son debidos a agregados leucocitarios en la sangre transfundida que obstruyen la circular pulmonar. Hay las que producen una reacción que interviene el complemento que es la causa del edema pulmonar. 3·- Enfermedad del injerto contra huésped: se caracteriza por una infiltración de linfocitos del donante en la piel, hígado ó tracto intestinal del receptor donde reacciona con las células del huésped causando exantema, diarrea ó hepatitis. C·- Reacciones debido a las plaquetas: 1·- Púrpura post-transfusional: se caracteriza por una trombocitopenia severa por consumo que generalmente tiene lugar en mujeres a los 7-10 días después de la transfusión de un producto sanguíneo. La púrpura se auto-limita y dura entre 2-6 semanas. D·- Reacciones debidas a proteínas plasmáticas. 1·- Anafilaxia: durante la transfusión puede producirse shock anafiláctico grave, con hipotensión y bronco-espasmo. Estas reacciones normalmente tiene lugar en pacientes con deficiencia de Ig A cuyo suero contiene anticuerpos específicos Ig A. Si se produce reacción transfusional debe detenerse inmediatamente y administrar adrenalina y corticoides. 2·- Urticaria: constituye el segundo tipo más frecuente de reacción transfusional. Se caracteriza por prurito y exantema que aparece durante la transfusión o después de esta. La reacción de urticaria tiene su causa en el anticuerpo del receptor que reacciona con antígenos del plasma del donante. Si una reacción de urticaria no va acompañada de otros síntomas o signos puede continuarse la transfusión. La reacción de urticaria puede prevenirse tratando al receptor con antihistamínicos previamente. REACCIONES NO INMUNOLÓGICAS A·- Inmediatas: 1·- Septicemias: aproximadamente el 3% de cada 1.000 unidades de sangre o de componentes sanguíneos están contaminadas con una pequeña cantidad de bacterias. Algunas especies de bacterias entre ellas las pseusomonas creen a temperaturas bajas y pueden estar presentes en gran cantidad en el momento de transfundir la unidad de sangre. Esto puede causar una reacción grave en el receptor. La reacción producidas por los productos sanguíneos infectados incluyen fiebre, escalofríos, hipotensión y muerte. AL transfusión debe detenerse inmediatamente y el componente sanguíneo causante del shock debe ser sometido a examen bacteriológico. 2·- Embolia gaseosa: el empleo de bolsas de plástico en la preparación y administración de productos sanguíneos ha eliminado virtualmente las embolias gaseosas, con objeto de evitar este tipo de accidente nunca debe introducirse aire dentro de los recipientes ni de sistemas de filtros ya que con ello se aumenta la probabilidad de dicho accidentes. B·- Retardadas: 1·- Transmisión de enfermedades: algunos individuos sanos tienen agentes infecciosos en su circulación. La transfusión de su sangre puede dar como resultados la transmisión de la infección al receptor. • Virus de la hepatitis (A, B, C): ~ Hepatitis A: la infección por el virus de la hepatitis A no conduce a un estado de portador crónico. ~ Hepatitis B: está producido por un virus DNA, el virus invade las células hepáticas y se replica en ellas. El HBsAg se investiga en todos los donantes de sangre. Esta medida ha reducido significativamente el riesgo de transmisión de la hepatitis B por transmisión de la hepatitis B por transfusión sanguínea. ~ Hepatitis C: también se investiga en todos los donantes de sangre. • Mononucleosis infecciosa • SIDA • Paludismo • Sífilis • Otras bacterias 2·- Sobrecarga de líquidos: en una transfusión las células, las proteínas, los electrolitos y el agua, tienen tendencia a ser retenido en el espacio intra-vascular. Este aumento en el volumen intra-vascular puede ser causa de insuficiencia cardiaca y edema pulmonar. 3·- Sobrecarga de hierro: cada unidad de sangre contiene 250 mg de hierro, así pues los pacientes que reciben muchas transfusiones de sangre pueden experimentar una sobrecarga de hierro. El hierro se acumula en el hígado en el corazón y en algunas glándulas endocrinas disminuidas su función. Los pacientes que requieren un soporte transfusional periódica tienen un alto riesgo de recibir una sobrecarga de hierro, con objeto de reducir el número de transfusión algunos médicos administran concentrados de hematíes preparados especialmente y enriquecidos con células jóvenes denominados neocitos. ESQUEMA • Reacciones inmunológicas A·- Hemólisis 1·- Reacciones hemolíticas inmediatas 2·- Reacciones transfusionales hemolíticas agudas 3·- Reacciones transfusionales hemolíticas retardadas B·- Reacciones debido a los leucocitos 1·- Reacciones febriles 2·- Infiltrados pulmonares 3·- Enfermedad de injerto contra huésped C·- Reacciones debidas a las plaquetas 1·- Púrpura post-transfusional D·- Reacciones debidas a las proteínas plasmáticas 1·- Anafilaxia 2·- Urticaria • Reacciones no inmunológicas A·- Inmediatas 1·- Septicemia 2·- Embolia gaseosa B·- No inmediatas o retardadas 1·- Transmisión de enfermedades ~ Virus de la hepatitis (A, B, C) ~ Mononucleosis, SIDA, paludismo, sífilis otras bacterias 2·- Sobrecarga de líquido 3·- Sobrecarga de hierro 1 Aviso de Privacidad | Términos y condiciones | Cookies | FAQ | Contacto