Blastoma Pleuropulmonar de la Ingancia. Reporte de un Caso en el
Anuncio

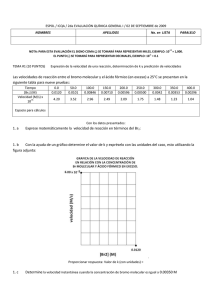
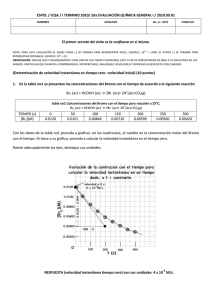
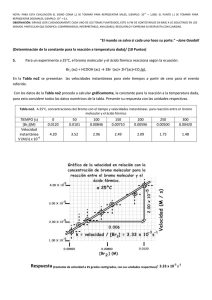
PRESENTACIÓN DE CASOS Blastoma Pleuropulmonar de la Infancia. Reporte de un Caso en el Hospital Universitario San Ignacio y Revisión de la Literatura Luis Fernando Jaramillo G.* Mauricio Peláez A.** Hugo H. Herrera*** RESUMEN Los blastomas pleuropulmonares (BPP) hacen parte de las neoplasias disembriónicas, emparentadas con el tumor de Wilms, el neuroblastoma y el hepatoblastoma. Son neoplasias muy infrecuentes, exclusivas de la infancia y suelen ser confundidas, tanto desde el punto de vista clínico como radiológico con lesiones de naturaleza infecciosa o malformaciones congénitas benignas. En este reporte presentamos un caso recientemente atendido en el Hospital Universitario San Ignacio con el que pretendemos dar una visión actualizada del tema y revisar los principales aspectos clínicos, radiológicos, histológicos y terapéuticos de estos inusuales tumores. PRESENTACIÓN DEL CASO Paciente de sexo femenino, con 23 meses de edad, producto de tercera gestación a término, parto por cesárea debido a preeclampsia materna, peso al nacer de 3.500 g, y talla de 53 cm. Requirió hospitalización por 5 días en la Unidad de Recién Nacidos por cuadro de taquipnea transitoria del recién nacido. Antecedente patológico de importancia: luxación congénita de cadera en tratamiento. Esquema de vacunación completo y desarrollo psicomotor adecuado para su edad. La paciente presentaba cuadro clínico de 10 días de evolución, con fiebre de 39ºC y episodio de convulsión tónico-clónica generalizada con desviación de la mirada, por lo cual fue llevada inicialmente a clínica particular donde se documentó la presencia de neumotórax basal izquierdo, posteriormente la paciente fue dada de alta, citándose para control por la consulta externa. Tres días después presentó nuevo episodio de fiebre y se hospitalizó en otra institución con diagnósticos de: neumotórax tabicado, neumonía izquierda y posible malformación adenomatoide quística pulmonar. De allí se remitió al HUSI para manejo especializado. Al momento de su ingreso se encontró afebril, hidratada, con peso y talla adecuados para su edad, sin signos de insuficiencia respiratoria. Como hallazgos positivos se detectó congestión peritimpánica izquierda y disminución de los ruidos respiratorios en el hemitórax izquierdo, principalmente hacia la base. El resto del examen físico se consideró dentro de límites normales. La evaluación clínica se complementó con TAC que mostró lesión quística multiloculada en pulmón izquierdo compatible con una malformación adenomatoide quística y se evidenció la presencia de neumonía multilobar izquierda y neumotórax tabicado. Cuatro días después, con la impresión clínica anotada la paciente se llevó a cirugía, donde se realizó la resección de la lesión y segmentectomía anterior del lóbulo inferior del pulmón izquierdo. Durante el posoperatorio no se presentaron complicaciones, evolucionando adecuadamente y dándose de alta dos días después en buenas condiciones generales. En el Departamento de Patología se recibió la pieza quirúrgica, que medía 8 x 5 x 3 cm en sus ejes mayores, superficie externa lisa, brillante, de color grisáceo, al corte se observaba una lesión quística, multilocular, con escasa cantidad de material claro viscoso intraluminal, el parénquima pulmonar circundante no mostraba alteraciones macroscópicas. El análisis histológico mostró múltiples formaciones quísticas revestidas por epitelio columnar pseudoestratificado de tipo respiratorio, el estroma era de aspecto denso constituido por células fusiformes, de aspecto inmaduro, con presencia de rabdomioblastos y ocasionales figuras mitóticas (foto 1), uno de los cortes reveló la existencia de áreas sólidas con diferenciación blastematosa y presencia de células anaplásicas. El parénquima pulmonar circundante y el borde de sección de la cuña se encontraban libres de compromiso tumoral. Con estos elementos se hizo el diagnóstico de: blastoma pleuropulmonar tipo I con foco microscópico de tipo II. Un año después se detectó recidiva tumoral mediastinal, llevándose de nuevo a cirugía, y lográndose la resección parcial de la masa. El análisis histológico mostró una lesión de patrón sólido, con marcada anaplasia celular (foto 2), alta actividad mitótica (mayor de 10 mitosis en 10 CGA) y franca diferenciación rabdomioblástica, confirmada con técnicas de inmunohistoquímica para actina muscular específica (foto 3). Con esta evolución hacia un blastoma pleuropulmonar grado III, se realizó quimioterapia complementaria, sin embargo, la paciente falleció en el mes de abril de 2001 como consecuencia del compromiso sistémico generalizado. DISCUSIÓN Los blastomas pleuropulmonares son lesiones muy infrecuentes, que hacen parte de una categoría peculiar de neoplasias llamadas disembriónicas o disontogénicas, las cuales son observadas exclusivamente en la infancia, a este grupo pertenecen además el tumor de Wilms, los neuroblastomas y los hepatoblastomas[1,2]. Los blastomas de la infancia difieren notablemente de su contraparte en los adultos. Ellos están constituidos por un tejido mesenquimal maligno, de aspecto embrionario o blastematoso, con poco o ningún componente epitelial, el cual, cuando es encontrado es de características no neoplásicas y corresponde a epitelio atrapado por el tumor, esto hace que desde el punto de vista biológico, estas neoplasias sean específicamente catalogadas como sarcomas y no como tumores bifásicos (mezcla de elementos epiteliales y mesenquimales) como sí ocurre en los adultos[3]. Los blastomas pleuropulmonares de la infancia tienen un amplio espectro morfológico tanto a nivel macro como microscópico. En un extremo están las neoplasias intrapulmonares de aspecto quístico, con paredes delgadas sobre un mesénquima de aspecto embrionario y rabdomiosarcomatoso, el cual ha sido denominado como blastoma quístico. En el pasado, esta variante recibió múltiples denominaciones como: sarcoma embrionario, o rabdomiosarcoma pulmonar originado en una malformación adenomatoide o quiste broncogénico[3,4]. El otro extremo del espectro está constituido por masas sólidas de tejido mesenquimal de aspecto embrionario, las cuales pueden comprometer además del pulmón, la pleura y el mediastino. Esta variante es denominada como: blastoma pleuropulmonar[3,4]. Sin embargo, la tendencia actual es denominarlos a todos como blastomas pleuropulmonares (BPP), y subclasificarlos en tres tipos histológicos, que a su vez tienen claras implicaciones pronósticas así[1]: • BPP tipo I: tumores completamente quísticos separados por septos de tejido fibroso y tapizados por epitelio de tipo respiratorio. • BPP tipo II: tumores de aspecto mixto, con presencia de áreas quísticas y sólidas. • BPP tipo III: tumores predominantemente sólidos. En los BPP tipo I el estroma es inmaduro constituido por células primitivas que semejan el sarcoma botriodes acompañadas por células de aspecto rabdomioblástico. Constituyen la presentación más infrecuente de los blastomas pleuropulmonares[1,3]. Tanto en los BPP tipo II como III hay islotes de células primitivas con mezcla de elementos blastematosos y sarcomatosos, en estas neoplasias las figuras mitóticas son abundantes con frecuente diferenciación rabdomioblástica similar a lo visto en el BPP tipo I. Hasta en el 60% de los casos se detecta la presencia de elementos condroides con diferentes grados de celularidad y atipismo. La anaplasia celular focal, en forma de células gigantes de aspecto pleomórfico y bizarro se detecta hasta en el 67% de los casos de BPP tipo II ó III, estos cambios son vistos en el componente sólido o blastematoso y menos frecuentemente en las áreas rabdomioblásticas. En ninguno de los BPP tipo I se ha encontrado anaplasia[1,3]. Desde el punto de vista clínico la sintomatología es vaga e inespecífica, siendo los signos y síntomas más frecuentes los siguientes: dificultad respiratoria (42%), fiebre (32%), dolor torácico y/o abdominal (26%), tos (26%) y anorexia (12%)[1,3,4,5]. El rango de edad de los pacientes afectados está entre 1 y 9 años, pero la mayoría de pacientes con BPP tipo III están por debajo de los 3 años de edad[3]. La impresión clínica inicial suele ser de neumonía y ocasionalmente de neumotórax, o empiema[1]. Los quistes pulmonares son detectados radiológicamente por la presencia de espacios con aire y/o neumotórax, esta presentación se encuentra hasta en el 38% de los casos[1] y la mayoría de pacientes son intervenidos con diagnóstico de malformación adenomatoide quística o quiste broncogénico[3]. Las lesiones son de tamaño variable, fluctuando entre 2 y 28 cm de diámetro y pesos superiores a los 1.100 g[5]. En algunos casos se detecta compromiso mediastinal, principalmente en los BPP tipo III, este hallazgo se asocia a pobre pronóstico[1,3]. Se han detectado algunos casos familiares o constitucionales de BPP los cuales parecen asociarse a pérdida de la heterocigoticidad en el cromosoma 11p15.5 en la región del gen del tumor de Wilms[1]. Esta región también ha sido implicada en el rabdomiosarcoma, que es un componente importante del BPP[1]. En el estudio de Priest et al.[1] se confirmó que los BPP son neoplasias agresivas, con sobrevida del 42% a 5 años para pacientes con lesiones tipo II y III, la tipo I presenta una sobrevida significativamente mayor, del 83% en el mismo período de tiempo. Además, se encontró alta tendencia a las recurrencias locales (60%), y a la aparición de lesiones metastásicas en el 44% de los casos, principalmente en SNC, huesos del cráneo, hígado y tejidos blandos, ambos eventos más frecuentes en pacientes con tumores tipos II y III. El tratamiento recomendado es multimodal e incluye cirugía y quimioterapia complementarias, las cuales deben ser tan agresivas como puedan ser toleradas por los pacientes, sin embargo, dada la rareza de estos tumores, no existen estudios que muestren la efectividad de un determinado agente quimioterapéutico o una combinación de ellos. La radioterapia muestra limitaciones por la toxicidad cardiopulmonar, sin embargo, es empleada en aquellos casos donde no es posible la excisión quirúrgica completa del tumor[1]. Finalmente, debe considerarse el estudio genético tanto de los pacientes como de sus padres y hermanos con el fin de detectar alteraciones cromosómicas que favorezcan la aparición de otras neoplasias relacionadas[2]. * Profesor asistente, Departamento de Patología, Facultad de Medicina, Pontificia Universidad Javeriana. ** Profesor asistente, Departamento de Cirugía, Unidad de Cirugía de Tórax, Facultad de Medicina, Pontificia Universidad Javeriana. *** Instructor, Departamento de Patología, Facultad de Medicina, Pontificia Universidad Javeriana. LECTURAS RECOMENDADAS 1. Priest J.R., McDermott M.B., Bhatia S., Watterson J., Manivel J.C., Dehner L.P. Pleuropulmonary blastoma: a clinicopathologic study of 50 cases. Cancer, july 1, 1997, vol. 80, no 1. 2. Isaacs H., Jr. Tumors. In: Gilbert-Barnes E., editor. Potter´s pathology of the fetus and infant. St. Louis: Mosby, 1996; 1242-339. 3. Colby T.V., Koss M.N., Travis W.D. Tumors of the lower respiratory tract. In: Atlas of tumor pathology, 1995, Armed Forces Institute of Pathology, fascicle 13: 403-10, Washington, D.C. 4. Koss M.N., Travis W.D., Moran C.A. Pulmonary sarcomas, blastomas, carcinosarcomas y teratomas. In: Hasleton P.S., editor. Spencer´s pathology of the lung. Fifth edition. International edition: McGraw-Hill, 1996; 1065-1109. 5. Koss M.N., Moran C.A., Stocker, J.T. Mixed epithelial-mesenchymal tumors. In: Saldana MJ., editor. Pathology of pulmonary disease. J.B. Lippincott Company, 1994; 617-29.