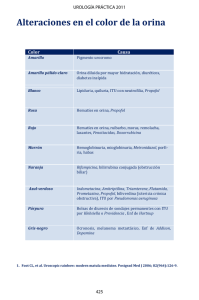
Tesis - Universidad de Colima
Anuncio

UNIVERSIDAD DE COLIMA FACULTAD DE MEDICINA CENTRO UNIVERSITARIO DE INVESTIGACIONES BIOMÉDICAS “FARMACOLOGÍA DE LOS ANESTÉSICOS GENERALES TIOPENTAL SÓDICO Y PROPOFOL” TESIS QUE PRESENTA: MVZ. Miguel Octavio Montoya Domínguez PARA OBTENER EL GRADO DE: MAESTRO EN CIENCIAS FISIOLÓGICAS ASESOR: Dr. José Antonio Sánchez Chapula COLIMA, COLIMA MAYO DEL 2008 AGRADECIMIENTOS Y DEDICATORIA Esta tesis la dedico a la memoria de mi padre el Sr. Miguel Montoya Rodríguez. Agradezco a mi madre la Sra. Ana María Domínguez Morales, por el apoyo, que toda la vida me ha brindado, en lograr las metas de superación académica y profesional, que me he trazado. Un agradecimiento infinito a mi esposa Marissa, por respaldarme y fortalecerme en los momentos difíciles, que enfrente durante la Maestría. A mis maestros, por sus conocimientos aportados, que se verán reflejados en mi actividad como docente. Y por supuesto a mis compañeros de clase Norma, Maribel, Mariana, Iris, Renato y Hebert mil gracias. Índice General I.- Índice de Figuras……………………………………………………………………..1 II.- Abstract ……………………………………………………………………………..2 III.- Resumen ………………………………………………………………………........3 IV.- Introducción ……………………………………………………………………….4 V.- Antecedentes históricos de Anestesia. ……………………………………………..5 -Historia de la anestesia quirúrgica antes de 1846………………………………………..5 -La anestesia después de 1846……………………………………………………………5 VI.- Anestésicos Generales……………………………………………………………....6 -Sitios anatómicos de acción anestésica…………………………………………………..7 - Mecanismos celulares de la anestesia general…………………………………………...8 VII.- Anestésicos intravenosos………………………………………………………….10 VIII.- Farmacología del tiopental sódico………………………………………………12 -Características farmacológicas del tiopental sódico……………………………………..12 -Efectos electrofisiológicos neuronales del tiopental sódico…………………………......15 -Farmacocinética de tiopental sódico…………………………………………………......23 -Farmacocinética del tiopental en presencia de enfermedad……………………………..25 -Aplicaciones clínicas del tiopental sódico…………………………………………….....26 IX.- Farmacología del propofol…………………………………………………………27 -Características farmacológicas del propofol……………………………………………..28 -Efectos electrofisiológicos neuronales del propofol……………………………………..30 -Farmacocinética del propofol………………………………………………………….....33 -Aplicaciones clínicas del propofol…………………………………………………….....34 X.- Efectos cardiovasculares de tiopental sódico………………………………………35 XI.- Efectos cardiovasculares de propofol……………………………………………..37 XII- Efectos electrofisiológicos cardiacos de tiopental y propofol……………………40 -Efectos sobre la duración del potencial de acción……………………………………….40 XIII.-Comparación de tiopental sódico y propofol en procedimientos clínicos……………………………………………………………………………………44 - Intubación endotraquel………………………………………………………………….44 - Cardioversión……………………………………………………………………………44 - Terapia electroconvulsiva………………………………………………………………..44 XIV.- Conclusiones y Perspectivas……………………………………………………. 46 XV.- Bibliografía………………………………………………………………………....47 I.-Índice de Figuras Figura 1. Estructura química del tiopental sódico. (*13) Figura 2. Estructura química del pentobarbital. (*13) Figura 3. Receptor GABAA y sus subunidades. (*16) Figura 4. Estructura del Receptor GABAA. (*17) Figura 5. Sitios de unión de anestésicos al R. GABAA. (*18) Figura 6. Efecto de anestésicos intravenosos sobre unión a radioligandos, para receptor GABAA. (*19) Figura 7. Estructura química del propofol. (*27) Figura 8. Propofol disminuye la excitabilidad neuronal vía conductancia GABAA en interneuronas CA1. (*31) Figura 9. Supresión de la excitabilidad por propofol en células piramidales CA1. (*31) Figura 10. El propofol no altera la morfología del potencial de acción. (*32) Figura 11.Curva concentración-respuesta para el efecto inotrópico negativo de propofol en miocitos ventriculares de rata. (*38) Figura 12. Supresión de IK1 en miocitos ventriculares de rata. (*42) Figura 13. Trazos de corriente en miocito ventricular de rata expuesto a tiopental. (*42) Figura 14. Efecto concentración-depentiente y conductancia fraccional de tiopental sobre IK1. (*43) Figura 15. Relación densidad-voltaje de propofol, control y ketamina sobre IK1. (*43) Figura 16. Efectos de los anestésicos tiopental y propofol en la terapia electroconvulsiva. (*45) * Página donde se localiza la figura. II.- ABSTRACT The ideal anesthetic would be one that has not to biotransform itself, it is not toxic nor irritating, it does not alter the vital functions, furthermore it can induce anesthesia quickly and without difficulties, causing a fast and safe recovery. None of the existing agents at the moment join the mentioned requirements. Two intravenous agents used at the clinical practice are: sodium thiopental and propofol. Thiopental has a fast action and an ultrashort duration, propofol is an hypnotic non barbiturate agent. The mechanism of action through which both drugs exert their anesthetic action is the route of GABAA inhibiting receptors, although other mechanisms could be implied (glycine receptors, NMDA receptors and ionic channels). Cardiovascular clinical effects of thiopental are negative inotropism and tachycardia; on the other hand propofol evokes arterial hypotension and bradycardia; these effects of both drugs are more accentuated in patients having previous cardiovascular disease. III.- RESUMEN El anestésico ideal sería aquel que no necesite biotransformarse, no sea tóxico, irritante, no deprima las funciones vitales, logre inducir rápidamente y sin forcejeos la anestesia, para propiciar una rápida y segura recuperación. Ninguno de los agentes existentes en la actualidad reúne los requisitos mencionados. Dos agentes intravenosos utilizados clínicamente son: tiopental sódico y propofol. El tiopental un barbitúrico de acción rápida y duración ultracorta y el propofol un hipnótico no barbitúrico. El mecanismo de acción por el cual ambos ejercen su acción anestésica es por la vía de los receptores inhibitorios GABAA, aunque otros mecanismos pueden estar implicados (receptores de glicina, receptores NMDA y canales iónicos). Los efectos clínicos cardiovasculares de tiopental son inotropismo negativo y taquicardia, por su parte el propofol produce hipotensión arterial y bradicardia; en ambos casos estos efectos son más acentuados en pacientes con enfermedad cardiovascular previa. IV.-INTRODUCCIÓN Dos de los anestésicos intravenosos más utilizados en la práctica clínica, tanto en medicina humana, como en medicina veterinaria; son el tiopental sódico y el propofol. El tiopental sódico, pertenece al grupo de barbitúricos de acción rápida y duración de acción ultracorta, (5 a 8 minutos de una dosis de inducción en humanos). Por su parte, el propofol o disoprofol es un alquilfenol (2,6-diisopropilfenol) cuya acción anestésica es consecuencia de su interacción con un sitio localizado en el receptor GABAA, produciendo la apertura del canal de cloro. El propofol tiene una potencia 1.8 superior al tiopental, también es de duración ultracorta, y una dosis de inducción dura 4 a 7 minutos. La investigación actual en el área de farmacología de los anestésicos generales está encaminada a dilucidar sus mecanismos de acción, usando técnicas electrofisiológicas y de biología molecular, para comprender de una manera más completa como los anestésicos ejercen su efecto anestésico y los efectos adversos sobre los diferentes órganos y sistemas del organismo. Esta revisión explica las características farmacológicas de estos dos compuestos así como cuales son los efectos y repercusiones más relevantes, particularmente a nivel cardiovascular, esto con el propósito de comprender los efectos clínicos de el tiopental sódico y el propofol, que en su momento pueden ser útiles para afrontar, las complicaciones potenciales durante la anestesia con estos fármacos. V.-Antecedentes históricos de anestesia Historia de la anestesia quirúrgica antes de 1846. Los procedimientos quirúrgicos, antes de esa época, no eran muy frecuentes. Se tenían conocimientos rudimentarios sobre la fisiopatología de las enfermedades, y sobre los fundamentos para tratarlas mediante procedimientos quirúrgicos. Se contaba con algunos medios para aliviar el dolor quirúrgico, que en realidad se habían empleado desde tiempos remotos. Brindaban cierto alivio sustancias como el alcohol, hachís y derivados del opio, tomados por vía oral. En ocasiones se utilizaban métodos físicos para producir analgesia, como cubrir una extremidad con hielo o producir isquemia con un torniquete. La pérdida de conocimiento causada por un golpe en la cabeza o por estrangulación ofrecía cierto alivio del dolor. (Kennedy y Longnecker, en Goodman y Gilman, 1996). La anestesia después de 1846. Aunque rara vez se emplea en la actualidad, el éter fue uno de los primeros anestésicos utilizados. (Kennedy y Longnecker, en Goodman y Gilman, 1996). El siguiente anestésico que se utilizó con amplitud fue el cloroformo; introducido en el campo de la anestesia por el obstetra escocés James Simpson, en 1847. A pesar de la incidencia relativamente alta de defunciones transoperatorias y postoperatorios que acompaño el empleo de este agente, fue el anestésico más utilizado sobre todo en Inglaterra, durante casi 100 años. Tras el entusiasmo inicial, la evolución de la anestesiología en Estados Unidos se caracterizó por un cambio lento y de progreso limitado. En 1868, Edmond Andrews, cirujano de Chicago, describió la administración de óxido nitroso con oxígeno. En 1929 se descubrieron de manera accidental, las propiedades anestésicas del ciclopropano, cuando un grupo de químicos estaba analizando impurezas en un isómero, el propileno. El ciclopropano fue, quizá el agente anestésico general más utilizado durante los siguientes 30 años. Sin embargo, dado el riesgo creciente de explosión en las salas de operaciones por el empleo de equipo electrónico, se incrementó la necesidad de contar con un agente anestésico seguro y no inflamable. Los esfuerzos del British Research Council y de los químicos de Imperial Chemical Industries se vieron retribuidos con el desarrollo del halotano, anestésico no inflamable que se introdujo en el ejercicio clínico en 1956. Por último, en el decenio de 1940, los anestesiólogos usaron el curare para lograr la relajación muscular que antes se lograba sólo con niveles muy profundos de anestesia general. (Kennedy y Longnecker, en Goodman y Gilman, 1996). La situación dio un giro impresionante en 1935, cuando Lundy demostró la utilidad clínica del tiopental, tiobarbitúrico de acción rápida. Originalmente, se le consideró útil como anestésico único, pero las dosis requeridas producían depresión grave de los aparatos circulatorio y respiratorio y del sistema nervioso. Sin embargo, el tiopental se ha aceptado como agente para la inducción rápida de anestesia general. (Kennedy y Longnecker, en Goodman y Gilman, 1996). VI.-Anestésicos generales A pesar de los grandes avances en el área de la anestesiología, no ha podido dilucidarse el mecanismo de acción de los anestésicos generales en su totalidad y más aún los mecanismos por los cuales se presentan los efectos adversos de estos fármacos en los diferentes sistemas y órganos del cuerpo humano y de los animales. Tanto en individuos sanos y enfermos. Los anestésicos generales son un grupo de sustancias estructuralmente diferentes que producen un punto final común, un estado del comportamiento llamado anestesia general. En el sentido más amplio, la anestesia general puede definirse como una depresión global pero reversible de las funciones del sistema nervioso central, lo cual resulta en la pérdida de reacción y percepción de todo estímulo externo. Los barbitúricos por ejemplo, son muy eficaces al generar amnesia y pérdida de conocimiento, pero no sirven como analgésicos. Una manera diferente de definir el estado anestésico es considerarlo como un conjunto de cambios en los “componentes” del comportamiento o percepción. Los componentes del estado anestésico, incluyen amnesia, inmovilidad en la respuesta a la estimulación nociceptiva, atenuación de las reacciones autónomas a la estimulación nociceptiva, analgesia y estado de inconciencia. (Evers y col, en Brunton y col, 2007). Sin embargo, los diferentes anestésicos generales causan grados variables de analgesia, amnesia y relajación muscular, lo común entre todos es que inducen pérdida reversible de la conciencia a bajas concentraciones (a excepción de fenciclidina y ketamina), por lo tanto para evitar confusiones el concepto de anestesia general se reduce a pérdida de conciencia reversible inducida por drogas. (Franks, 2006). Las teorías tradicionales consideraban a los anestésicos como agentes no selectivos que actuaban como consecuencia de las desestructuración de la membrana fosfolipídica de la célula nerviosa. Estas teorías se han abandonado y, al parecer, se demuestra la posibilidad de que la actividad anestésica esté relacionada con sitios de fijación específicos. Así, los anestésicos ejercerían su acción en la transmisión sináptica y no en la conducción axonal y, por otro lado, su diversidad estructural lleva a pensar que no interactuarían en un único lugar específico. (López-Timoneda y Gasco, en Velázquez, y col, 2005). Sitios anatómicos de la acción anestésica Los anestésicos generales podrían, en principio interrumpir funciones del sistema nervioso central en muy diferentes niveles, incluyendo neuronas sensitivas periféricas, médula espinal, tallo y corteza cerebral. La identificación de los sitios anatómicos de acción precisos es difícil, porque muchos anestésicos inhiben de manera difusa la actividad eléctrica en el sistema nervioso central y periférico. No obstante eso, estudios in vitro han demostrado que vías corticales específicas exhiben efectos diferenciales a anestésicos inhalatorios e intravenosos (MacIver y Roth, 1988; Nicoll, 1972). Esto sugiere que diferentes anestésicos pueden producir componentes específicos del estado anestésico vía acciones en sitios específicos del SNC. De esta manera se ha demostrado que los anestésicos inhalados producen inmovilización en respuesta a una incisión quirúrgica por acciones sobre la medula espinal, de tal forma que esta respuesta a la incisión quirúrgica es un parámetro utilizado para determinar la concentración alveolar mínima (CAM) de todos los anestésicos inhalados utilizados. Por ejemplo la CAM del oxido nitroso es de 105 comparada con la del isofluorano que es de 1.2 en humanos; esto quiere decir que se requiere menos isofluorano a nivel alveolar para producir anestesia y por lo tanto la CAM determina la potencia de los anestésicos inhalados (Rampil, 1994; Antognini y Schwartz, 1993). Dado que la amnesia y la inconciencia no pueden resultar de acciones sobre la medula espinal, los diferentes componentes de la anestesia son producidos por efectos en diferentes áreas del SNC. Así, otros resultados han mostrado que los efectos sedantes de barbitúricos como el pentobarbital y el propofol son mediados por receptores GABAA en el núcleo tuberomamilar (Nelson y col. 2002). Los efectos sedantes del anestésico general, dexmedetomidina son producidos vía acciones en el locus coeruleus (Mizobe y col, 1996), acción altamente selectiva sobre receptores alfa-2-adrenérgicos (Franks, 2006). Se ha sugerido que el tálamo puede ser un importante sitio de acción para los efectos producidos por los anestésicos inhalados, ya que la supresión de las neuronas talámicas produce inconciencia (Ries y Puil, 1999). Por otro lado, anestésicos inhalados e anestésicos intravenosos deprimen la transmisión nerviosa en el hipocampo, que es un sitio probable del efecto amnésico (Kendig y col, 1991). La mayoría de los anestésicos inhalados favorecen la acción de GABA y la sinapsis inhibitorias a concentraciones que correlacionan bien con su potencia en vivo. Por ejemplo concentraciones anestésicas de halotano, enfluorano e isofluorano, incrementan marcadamente corrientes inducidas por concentraciones bajas de GABA, pero son inefectivas a concentraciones altas de GABA, esto en estudios realizados en neuronas disociadas de cerebro de rata. (Franks y Lieb, 1994). Actualmente se han identificado residuos de aminoácidos, sobre subunidades de GABAA y Glicina, para anestésicos volátiles y alcoholes, que revelan que estos compuestos actúan por unión a canales iónicos operados por ligando (Wafford y col, 2004). Como es el caso del etomidato, ya que la mutación de un aminoácido de la subunidad β3, reduce la potencia considerablemente y serina reemplazada por asparaginasa en la subunidad β1 del receptor GABAA aumenta la sensibilidad anestésica. (Franks, 2006). Mecanismos celulares de la anestesia general La primera teoría con amplia aceptación propuesta por Meyer y Overton a finales del siglo XIX, postula que los sitios blanco de los anestésicos generales son porciones de lípidos de membranas nerviosas; y la potencia de un anestésico es directamente proporcional al coeficiente de partición agua/aceite. (Evers y Crowder, en Hardman y Limbird, 2003). Modificaciones a la hipótesis lipídica mencionan que pequeños incrementos de temperatura, aumentan la fluidez y permeabilidad lipídica, aumentando la potencia anestésica. Perturbaciones de la bicapa lipídica, pueden producir membranas nerviosas disfuncionales, que puedan alterar la recaptura del neurotransmisor. (Franks, 2006). Así mismo, Franks en 2006, asegura que la presión alta (arriba de 100 atmósferas) revierte la anestesia general, ya que aumenta la excitabilidad del animal, reduciendo quizás la potencia analgésica. Sin embargo, las teorías lipídicas, en la actualidad han sido paulatinamente descartadas o modificadas. (Koblin y col, 1994), debido a que se ha demostrado que los anestésicos inhalados e intravenosos tal vez sean enantioselectivos en su acción como anestésicos. (Etomidato, esteroides, isoflorano). (Tomlin y col, 1998; Lysko y col, 1994; Wittmer y col, 1996); y el hecho de que los enantiómeros tienen acciones únicas con propiedades físicas idénticas indica que otras propiedades, aparte de la liposolubilidad son importantes para determinar la acción anestésica. Esto ha llevado a pensar en la identificación de sitios específicos para la fijación de anestésicos a proteínas. Y además las teorías lipídicas, tienen mucha dificultad para aceptarse en la actualidad, ya que los enantiómeros de anestésicos, usualmente exhiben diferente potencia en animales. (Ej: R(+) etomidato, usado clínicamente es más potente que S(-) etomidato,). (Franks, 2006). A partir de 1980 existen fuertes evidencias experimentales que consolidan las teorías que postulan que los anestésicos generales actúan directamente en blancos proteicos. (Franks y Lieb, 1982). Los anestésicos generales producen dos importantes efectos electrofisiológicos a nivel celular. Primero, los agentes inhalados producen hiperpolarización de las neuronas (Nicoll y Madison, 1982). Este efecto puede ser importante en neuronas con actividad marcapaso o circuitos generadores. Segundo, a concentraciones anestésicas tanto los agentes intravenosos como los inhalados producen efectos principalmente sobre la transmisión sináptica, más que sobre la generación y/o propagación de potenciales de acción (Larrabee y Posternak, 1952). Los anestésicos inhalados inhiben sinapsis excitatorias y aumentan la actividad de sinapsis inhibitorias en varias preparaciones (Perovansky y col, 1995; MacIver y col, 1996). Sin embargo, los receptores de glutamato ionotropicos al parecer son relativamente insensibles a agentes volátiles, ya que corrientes de glutamato activadas en neuronas disociadas del tracto solitario de rata, no son afectadas por 300 μM de halotano y enfluorano. (Franks y Lieb, 1994). Los efectos predominantes de los agentes intravenosos son sobre las sinapsis, donde tienen efectos marcados sobre la respuesta sináptica a los neurotransmisores liberados. La mayoría de estos agentes actúan predominantemente aumentando la acción inhibitoria de neurotransmisores. (Franks y Lieb, 1994). Sorprendentemente existen pocos estudios modernos, del efecto de los anestésicos generales sobre receptores de glutamato ionotropicos, uno de ellos es el realizado en neuronas corticales de rata, donde los barbitúricos inhiben receptores de glutamato ionotropicos del tipo no-NMDA. Particularmente el pentobarbital con una IC50 de 50 μM fue muy efectivo para inhibir corrientes inducidas por kainato; siendo que la EC50 para anestesia general en mamíferos es precisamente 50 μM. (Franks y Lieb, 1994). En este sentido la ketamina, anestésico disociativo actúa predominantemente inhibiendo la transmisión neuroexcitatoria de sinapsis glutamatergicas, inhibiendo el sitio de fenciclidina del receptor NMDA. (Franks y Lieb, 1994). Para los anestésicos generales intravenosos, se ha establecido que el receptor GABAA es el sitio blanco, esto se ha demostrado por medio de estudios electrofisiológicos, ya que por ejemplo 20 μM de pentobarbital incrementa la respuesta a GABA. Existen muchas evidencias más de que los anestésicos generales a concentraciones quirúrgicas, actúan sobre canales iónicos dependiente de ligando (antes que los de dependientes de voltaje), con potenciación postsináptica inhibitoria de la actividad del canal. Aunque el rol de segundos mensajeros permanece incierto es claro que los anestésicos actúan directamente sobre proteínas, más que su efecto sobre lípidos. (Franks y Lieb, 1994). VII.-Anestésicos intravenosos Las necesidades para la anestesia general y la cirugía pueden requerir la administración de diversos fármacos intravenosos con efectos diferentes, para garantizar la hipnosis, analgesia y control de las reacciones reflejas viscerales. Con la introducción del tiopental (PENTOTHAL) por Lundy en 1935, pudo contarse con un barbitúrico de tiempo de acción apropiado para las necesidades de la cirugía. Su uso durante la anestesia general sigue excediendo al de cualquier otro barbitúrico. El propofol (2,6-disopropilfenol; DIPRIVAN) no está relacionado, desde el punto de vista químico, con otros agentes anestésicos intravenosos. El compuesto es un aceite a temperatura ambiente, y se expende en emulsión a una concentración del 1 por ciento. (Kennedy y Longernecker, en Goodman y Gilman, 1996). El tiopental y otros barbitúricos son malos analgésicos, y pueden incluso incrementar la sensibilidad al dolor cuando se administran en dosis subterapéuticas. En estas circunstancias se manifiestan signos de reacción simpática, como taquicardia, pupilas dilatadas, lagrimeo, sudoración, taquipnea, aumento de la presión arterial y movimientos o vocalización por reacción al procedimiento quirúrgico. (Kennedy y Longernecker, en Goodman y Gilman, 1996). Aun cuando se dispone de varios barbitúricos de acción ultracorta, el tiopental es el que más se utiliza para la inducción de la anestesia, a menudo en combinación con anestésicos inhalados (Trevor y Miller, en Katzung, 2002). La inyección intravenosa de propofol (2mg/Kg) induce anestesia con tanta rapidez como el tiopental. Se puede conservar la anestesia mediante administración sostenida de propofol en solución intravenosa de manera conjunta con opioides y óxido nitroso, otros agentes de inhalación o una combinación de ellos (Kennedy y Longernecker, en Goodman y Gilman, 1996). El fármaco (propofol) es también efectivo para producir sedación prolongada en pacientes con cuidados intensivos. Sin embargo, el empleo del propofol para la sedación en niños bajo cuidados intensivos ha dado lugar a acidosis grave en presencia de infecciones respiratorias y posibles secuelas neurológicas cuando se suspende. VIII.-Farmacología del tiopental sódico. Características farmacológicas del tiopental sódico. El tiopental sódico pertenece al grupo de los barbitúricos, y éstos constituyen los hipnóticos más empleados, al punto de haber desplazado a casi todos los otros grupos, por ser muy eficaces y ofrecer más bien pocas reacciones adversas. Los barbitúricos son sustancias de origen sintético y corresponden químicamente a la clase de los ureidos cíclicos o di-ureidos. Los barbitúricos reciben este nombre porque derivan del ácido barbitúrico o malonilurea, que resulta de la condensación de la urea y el ácido malónico para dar lugar al anillo de la tetrahidropirimidina. El ácido barbitúrico es inactivo y adquiere propiedades hipnóticas si se reemplazan los dos átomos de hidrógeno en la posición 5 por grupos alquilos o arilos; en esta forma se han sintetizado una amplia variedad de compuestos, más de 2500, de los cuales se han empleado en medicina unos 50. (Litter, 1969) Los tres barbitúricos que se utilizan para anestesia clínica en humanos son el tiopental sódico (pentotal), tiamilal sódico (surital) y el metohexital sódico (brevital). El tiopental sódico es el barbitúrico más utilizado para inducir la anestesia. Estos tres anestésicos son administrados como mezcla racemica a pesar de la enantioselectividad en su potencia anestésica (Christensen y Lee, 1973; Nguyen y col, 1996). El ácido barbitúrico y sus derivados son poco solubles en agua, siendo en cambio liposolubles, lo que los hace fácilmente absorbibles en el tracto digestivo. La mejor clasificación que puede realizarse de los barbitúricos se refiere a la duración de su acción (Litter, 1969). Barbitúricos de acción prolongada, más de 6 horas como el barbital, fenobarbital sódico y mefobarbital; de acción intermedia, de 3 a 6 horas de duración (butabarbital sódico, amobarbital; de acción corta, de menos de 3 horas (pentobarbital sódico, heptabarbital, secobarbital sódico y hexobarbital) y barbitúricos de acción ultracorta, empleados por vía intravenosa para producir anestesia general siendo la mayoría tiobarbitúricos (tiopental sódico y tialbarbitón sódico). (Litter, 1969). A nivel clínico en la actualidad, en medicina veterinaria se utilizan el tiopental sódico, para inducción de la anestesia inhalada y procedimientos diagnósticos y quirúrgicos de 15 a 20 minutos de duración, el fenobarbital utilizado por vía oral como pro-fármaco (primidona) para control de ciertos tipos de epilepsia en perros y el pentobarbital sódico, empleado también como anticonvulsivo en perros y gatos, sedante a bajas dosis y ampliamente como fármaco para eutanasia. Otro de los tiobarbitúricos no mencionado en esta clasificación y utilizado clínicamente es el tiamilal sódico (Biotal). El tiopental sódico es químicamente muy parecido al pentobarbital sódico, con la diferencia que en la molécula de esta sustancia un átomo de azufre sustituye a un átomo de oxígeno (Fig.1y 2). Figura1. Estructura química del tiopental sódico. Tomado de: www.fass.se/LIF/produktfakta/substance_produc... Figura 2. Estructura química del pentobarbital. Adaptado de: www.answers.com/topic/pentobarbital-sodium. El tiopental sódico se encuentra disponible en forma de polvo, mezclado con carbonato de sodio. Debe almacenarse en lugar fresco y lejos de la luz, el compuesto es inestable en solución, o cuando se expone a la humedad del ambiente, y su grado de deterioro es proporcional a la temperatura de la solución. Por esta razón resulta recomendable que las soluciones de tiopental sódico se preparen exactamente antes de utilizarse, aunque también es posible almacenarlas en un refrigerador, a temperaturas entre 3 y 6 grados centígrados, durante no más de siete días. Estas soluciones no se deben guardar a la temperatura ambiental promedio (18 a 20 grados centígrados) por un lapso superior a tres días. (Sumano y Ocampo, 1997). A nivel clínico, al menos en perros y gatos no se debe utilizar una vez preparado cuando la solución presenta cristales visibles o bien cuando ha sido almacenado por más de 15 días en el refrigerador. Debido a la elevada liposolubilidad y su rápido paso de la barrera hematoencefálica, alcanza concentraciones en el cerebro que inducen una intensa acción depresora y anestesia; ésta aparece a los 10-20 segundos de la inyección de una dosis anestésica y dura 20-30 minutos. (Flórez y Hurlé, en Flórez y col, 1992). La profundidad de la anestesia y la depresión son proporcionales a la dosis; se acompaña de depresión respiratoria que inicialmente puede alcanzar la forma de apnea para después mantenerse en cierto grado de hipoventilación. Si la depresión no es profunda puede aparecer salivación, broncoespasmo y laringoespasmo en especial en respuesta a estímulos químicos o mecánicos. (Flórez y col, en Flórez y col, 1992). Durante la acción del tiopental, el flujo y metabolismo cerebrales están disminuidos, lo que reduce también la presión intracerebral; estos eventos pueden ayudar en situaciones de hipertensión endocraneal, traumatismos craneales, etc. La presión intracraneal lo hace de manera notable y se recurre a este efecto en clínica durante la anestesia para operaciones neuroquirúrgicas, o en otras circunstancias en que se esperan incrementos en la presión intracraneal. (Flórez y col, en Flórez y col, 1992). Debido a que disminuye de manera notoria el metabolismo cerebral, el tiopental se ha evaluado como un fármaco protector contra la isquemia cerebral (Nussmeier y col, 1986). Las dosis de tiopental para producir EEG (Electroencefalograma) isoeléctrico protegen al cerebro durante la isquemia, por ejemplo durante el accidente cerebrovascular (trombosis cerebral y el derrame cerebral), pero no lo protegen si el EEG se ha vuelto isoeléctrico de antemano a causa de paro cardiaco o traumatismo craneoencefálico (Kennedy y Longernecker, en Goodman y Gilman, 2006). El tiopental y otros barbitúricos son malos analgésicos, y pueden incluso incrementar la sensibilidad al dolor cuando se administran en cantidades inadecuadas. En estas circunstancias se manifiestan signos de reacción simpática, como taquicardia, pupila dilatada, lagrimeo, sudoración, taquipnea, aumento de la presión arterial y movimientos o vocalización por reacciones al procedimiento quirúrgico. (Kennedy y Longernecker, en Goodman y Gilman, 2006). El tiopental puede reducir el flujo sanguíneo hepático y la velocidad de filtración glomerular, pero ello no produce efectos posteriores retardados en la función hepática y renal. Los barbitúricos pueden exacerbar la porfiria intermitente aguda y ha precipitado crisis de porfiria cuando se ha utilizado como agente inductor. (Trevor y Miller, en Katzung, 2002). Efectos electrofisiológicos neuronales del tiopental. A la fecha, el mecanismo de acción más reconocido por el cual el tiopental ejerce su efecto anestésico es mediado por los receptores inhibitorios GABAA. (Kash y col, 2004). Los receptores GABAA son substratos moleculares para la regulación de la vigilia, ansiedad, tensión muscular, actividad epileptogénica y funciones de memoria; y el incremento rápido de la inhibición sináptica del GABAA, es la base para la farmacoterapia de varios desordenes neurológicos y psiquiátricos. (Rudolph y Moller, 2003). Diferentes isoformas de unidades del receptor GABAA han sido clonadas, principalmente en el cerebro, las más abundantes formas nativas comprenden: 2α1, 2β2 y 1γ2. (Fig.3) Cada subunidad del receptor es de 50-60 KDa; con su dominio extracelular, dominio transmembrana con 4 segmentos α-hélices (M1-M4) en el dominio citoplasmático. Posee además su filtro de selectividad de 5.6 Ǻ de ancho. Importante es señalar que el dominio transmembrana es la vía permeable, ya que residuos hidrofílicos quizá interactúan con moléculas de agua, formando un caparazón a los alrededores de aniones clorados, permitiendo estabilización energética, para entrar al dominio del poro. (Fig.4) (Kash y col, 2004). Figura 3. Diagrama. A. Receptor GABAA comprende subunidades α1, β2 y γ2. B. Topología de membrana de una subinidad individual del receptor GABAA. Flechas negras indican sitios de unión en la interfase de las subunidades α1 y β2. C. Un modelo de la homología del receptor GABAA, en el cual el DEC (dominio extracelular) está basado en la cristalografía del AChBP y el DTM (dominio transmembranal) es una αHélice junta. Adaptado de Kash y col. 2004. Figura 4. Dominios funcionales del R. GABAA. A. Representación esquemática del sitio de unión a GABA, demostrando la contribución asimétrica de la subinidad α y β, usando la nomenclatura clásica del sitio de unión al asa. B. Diagrama del dominio M2 de las subunidades α1 y β2, del R. GABAA mostrando los residuos que contribuyen para el poro. Los residuos interactúan favorablemente con moléculas de agua. (En azul) C. Acercamiento de los sitios de unión a GABA en un modelo homologo en el dominio de unión a GABA en formato CPK (Corey-Pauling-Kaltun). Dominios en la subunidad β2, involucrados en la unión a agonistas (morado) y en la subunidad α1 (azul).Adaptado de Kash y col. 2004. Para tener una aproximación de los sitios de unión de los fármacos al receptor, se han desarrollado ratones knock-out (que carecen de subunidades del receptor GABAA, como la α1, α5, α6, β2, β3, γ2 y ρ1), así como ratones knockin (que afecta sitios de acción modulatoria por drogas, como:α1(H101R), α2(H101R), α3(H126R), α5(H105R) y β3(N265M). los ratones knock-out, proporcionan información con respecto a la regulación de genes y los ratones knokin, proveen información de la contribución específica de subtipos de receptores individuales para el espectro farmacológico de diazepam y anestésicos generales. (Rudolph y Moller, 2003). (Fig.5) Figura 5. Esquema del receptor GABAA y los principales sitios de unión. 2 de las 5 subunidades son mostradas con líneas punteadas. Cada subunidad posee una cadena polipeptídica con una larga porción Nterminal, 4 regiones transmembranales y una corta porción extracelular C-terminal. Residuos de aminoácidos de las subinidades α y β, contribuyen a los sitios de unión a GABA, además residuos en subunidades α y γ, contribuyen a sitios de unión a benzodiacepinas. Residuos de aminoácidos de subunidades α y β, son críticos para la unión a halotano y enfluorano, con los mismos residuos de aminoácidos en la subunidad β, son críticos para la acción de etomidato y propofol. Los sitios de unión para barbitúricos, involucra, al menos en parte residuos de la subinidad β. Los sitios de unión a neuroesteroides como alfaxolona, no esta molecularmente definido. Adaptado de Rudolph y Moller. (2003). Los sitios de unión al receptor GABAA son: la subunidad α y β, (pero no la γ), son receptores de activación por GABA donde la subunidad γ posee sensibilidad a benzodiacepina en el receptor nativo. (Kash y col, 2004). El tiopental sódico, suprime la excitabilidad intrínseca de interneuronas vía GABA. Adicionalmente el tiopental, incluyendo los barbitúricos metohexital y pentobarbital sódico, anestésicos esteroides (alfaxalona) y otros anestésicos como etomidato, producen corrientes hiperpolarizantes, sensibles a bicuculina (Bieda y MacIver, 2004). Otra de las fuertes evidencias que consolidan que el efecto anestésico de tiopental, es preferencialmente por la interacción con receptores GABAA ha sido demostrada en membranas de corteza de rata, con estudios de binding (unión a radioligandos), donde el tiopental con una IC50 de 37.6µM, inhibe la unión del [35S] TBPS o t-butylbiclorofosforotionato, para picrotoxina (estimulante del sistema nervioso central por bloqueo de receptores GABAA) del receptor GABAA. (Fig.6). (Lingamaneni y Hemmings Jr, 2003). Importante es señalar que la concentración plasmática efectiva para anestesia con tiopental es de 25 µM. (Franks y Lieb, 1994). La IC50 de tiopental para inhibir la unión del radioligando [3H] BTX-B a canales de sodio dependientes de voltaje es mayor a 2000 µM y en el caso del radioligando [3H] PN200-110 (isradipina) a canales de calcio tipo L (sitio de unión a dihidropiridina) fue 355 µM. (Lingamaneni y Hemmings Jr, 2003). Esto indica que para bloquear los canales de sodio y canales de calcio tipo L, al menos en estas condiciones experimentales, se requieren concentraciones excesivas de tiopental. Figura 6. Efecto de anestésicos intravenosos sobre su unión a radioligandos para receptor GABAA en membranas corticales de rata con [35S] TBPS o t-butyl-biclorofosforotionato. Adaptado de Lingamaneni y Hemming Jr. (2003). Sinner y col. (2006) aseguran que las oscilaciones espontáneas de Ca++ son dependientes de glutamato y aparentemente son responsables para la plasticidad neuronal e integración de la información y son influenciadas por el receptor GABAA. Los anestésicos intravenosos midazolam y tiopental suprimen la amplitud y la frecuencia reversiblemente de estas oscilaciones de calcio, de una manera dependiente de la dosis, con una EC50 (concentración efectiva para inhibir la mitad de estas oscilaciones de calcio) en concentraciones clínicas relevantes. Este efecto estuvo mediado vía receptor GABAA y puede ser revertido por el antagonista del receptor GABAA (bicuculina). En contraste la aplicación de ácido barbitúrico (sin propiedades anestésicas) no tiene efectos sobre las oscilaciones espontáneas de Ca++ y éstas pueden representar un modelo interesante para estudiar mecanismos anestésicos sobre el procesamiento de información neuronal. (Sinner y col, 2006). Evidencias experimentales, de los efectos electrofisiológicos de tiopental, también se han reportado, sobre diferentes canales iónicos, aunque las concentraciones utilizadas en estos experimentos están por arriba de las concentraciones que producen el efecto anestésico. Los anestésicos generales inhiben canales de calcio dependientes de voltaje, inhibiendo la transmisión excitatoria en el sistema nervioso central (Kitayama y col, 2002). Kitayama y col. (2002) determinaron la participación de los anestésicos intravenosos, dentro de ellos el tiopental, sobre la liberación de glutamato, evocada por K+ en rebanadas cerebrocorticales de rata. Esta inhibición de la liberación de glutamato aparentemente puede ser debida principalmente a inhibición de canales de calcio dependientes de voltaje del tipo P/Q, aunque a través de la activación del receptor GABAA, que juega un rol en esta respuesta. Los canales de calcio tipo P, aparentemente están más involucrados en la liberación presináptica de neurotransmisores de terminales nerviosas centrales. Los barbitúricos también inhiben los canales de calcio, pero usualmente con una IC50 (Concentración que produce el 50% máximo de inhibición) doblemente mayor de la concentración necesaria para anestesia general y generalmente son insensibles a concentraciones clínicas de los anestésicos generales (Solo pequeña inhibición a concentraciones quirúrgicas) (Franks y Lieb, 1994). Los canales tipo- P, quizá son los más importantes canales de Ca++ dependientes de voltaje involucrados en la transmisión sináptica en cerebro de mamíferos. (Hall y col, 1994). Sin embargo, Hall y col. (1994) que determinaron su sensibilidad a anestésicos a concentraciones clínicas relevantes en neuronas disociadas cerebelares de purkinje de ratas, encontraron que inhibiciones de menos del 10%, fueron obtenidas de halotano, isoflorano, tiopental, pentobarbital, propofol y etanol. Inhibición substancial por anestésicos fue encontrada solamente en concentraciones, mucho más grandes que las que tienen relevancia clínica. Mínima inhibición de canales de Ca++ tipo-P producida por anestésicos volátiles y anestésicos intravenosos para anestesia general en mamíferos, sugiere que estos canales no juegan un rol en la inducción de la anestesia general. (Hall y col, 1994). Criterios opuestos a las conclusiones de Kitayama y col. (2002) que aseguran que varios agentes como tiopental, pentobarbital, fenobarbital, alfaxolona y propofol a concentraciones clínicas inhiben liberación de glutamato, debido principalmente a la acción directa de canales de calcio dependientes de voltaje tipo P/Q. Por otra parte los receptores NMDA (N-metil-D-aspartato) en la corteza prefrontal (PFC) están estrechamente relacionados con la excitabilidad de neuronas piramidales y la función de la PFC. Liu, Dai y Yao en 2006 analizaron el efecto de tiopental con diferentes concentraciones sobre las corrientes NMDA en neuronas piramidales de corteza prefrontal de ratas. Y encontraron que el NMDA induce corrientes de entrada en una manera concentración-dependiente y esta corriente estuvo inhibida por tiopental sódico. Importante es señalar que la IC50 para inhibir la corriente NMDA por tiopental fue de 33.6±6.1 µM. Resultado muy cercano de la concentración (25 µM) farmacológicamente relevante para producir anestesia general en mamíferos. (Franks y Lieb, 1994). Sin embargo, las corrientes NMDA son muy insensibles a pentobarbital (Franks y Lieb, 1994). Otro potencial blanco del tiopental es el receptor de glicina (receptor inhibitorio), que juega un papel a nivel de la medula espinal y tallo cerebral bajo. (Franks y Lieb, 1994). Yang y Xu, en 2006, ensayaron el efecto modulatorio de tiopental sobre este receptor en neuronas del asta dorsal espinal de ratas mecánicamente disociadas. El tiopental inhibe la amplitud, acelera la desensibilización y prolonga la desactivación de corrientes inducidas por glicina (IGly) en una manera dependiente de la concentración. La supresión de la función del receptor de glicina sugiere que las acciones anestésicas de tiopental pueden no ser mediadas por receptores de glicina; esto consolida que el efecto hipnótico del tiopental, se debe a su acción estimulante sobre el receptor inhibitorio GABAA. Yang y Xu (2006) especulan que la débil relajación muscular y el limitado efecto analgésico observado durante la anestesia con tiopental pueden ser atribuidos a efectos inhibitorios sobre receptores de glicina. Tampoco con pentobarbital se encontró potenciación de estas corrientes en neuronas espinales. (Franks y Lieb, 1994). Por otra parte las corrientes de potasio dependientes de voltaje neuronales, son cruciales para varias funciones celulares, como la integración de la información temporal en el sistema nervioso central. (Friederich y Urban, 1999). Friederich y Urban (1999) en neuronas de origen humano y células SH-SY5Y utilizando técnicas electrofisiológicas, evaluaron los efectos relacionados con la concentración sobre corrientes de K+ dependientes de voltaje de tres opiáceos (fentanil, alfentanil y sulfentanil) y siete no opióides (tiopental, pentobarbital, metohexital, propofol, ketamina, midazolam y droperidol), usados en anestesia clínica y observaron que todas las drogas inhiben las corrientes de potasio de una manera reversible, dependiente de la concentración. Supresión de corrientes de potasio por opiáceos ocurre con una IC50 10 veces menor que para barbitúricos. Por ejemplo para el caso de sulfentanil la IC50 fue de 7 µM. La acción de drogas anestésicas sobre corrientes de potasio dependientes de voltaje, quizá contribuyan a los efectos clínicos o efectos colaterales de anestésicos intravenosos. (Friederich y Urban, 1999). Lo que nos permite asegurar que los barbitúricos como el tiopental requieren aproximadamente 3 veces más de la concentración necesaria para la acción anestésica, para bloquear la acción de estas corrientes de potasio. A su vez los canales de potasio sensibles a ATP, están ampliamente expresados en membranas citoplasmáticas de neuronas, y están acoplados al metabolismo celular para excitabilidad. Estos canales también están involucrados en neuroprotección contra daño celular por hipoxia, isquemia y excitoxicidad. Los barbitúricos frecuentemente son usados en pacientes con isquemia cerebral y solamente a altas concentraciones, pero no concentraciones de relevancia clínica inhiben los canales de potasio sensibles a ATP, activados por depleción de ATP intracelular en la sustancia nigra. (Ohtsuka y col, 2006). Farmacocinética del tiopental sódico. Desde la introducción del tiopental a la anestesia clínica en 1936 hasta 1950, la corta duración del efecto anestésico de este agente se atribuyó a su rápida eliminación del organismo. Usando un ensayo espectrofotométrico específico, Brodie y col, en 1950 encontraron que la vida media de eliminación después de un bolo intravenoso era de 4.6 horas, demasiado larga para explicar la brevedad del efecto. Estos mismos investigadores demostraron posteriormente que el elevado flujo sanguíneo cerebral y la liposolubilidad contribuyen a que el tiopental penetre con rapidez al sistema nervioso central. La recuperación rápida de los efectos se atribuyó a redistribución del cerebro a la grasa corporal, que tiene una gran capacidad para almacenar tiopental, aunque posee una irrigación sanguínea mucho menor que el sistema nervioso central. El tiopental fue uno de los primeros fármacos para el que se demostró que la redistribución era el mecanismo de terminación de su efecto farmacológico. El análisis farmacocinético fue determinante para la comprensión de la acción clínica de esta sustancia. Estudios farmacocinéticos posteriores con métodos más sensibles y muestreos más frecuentes han permitido comprender en forma más completa la cinética del tiopental. El hecho que se induzca anestesia unos segundos después de aplicar un bolo intravenoso de tiopental sugiere que el cerebro está probablemente dentro o muy cerca del compartimiento central. Esto es de esperarse porque este compartimiento está formado por tejidos con irrigación abundante. (Stanski, en Prys-Roberts y Hung, 1986). Las concentraciones plasmáticas máximas de tiopental se alcanzan en un tiempo de circulación después de un bolo intravenoso, después de lo cual se inician en forma simultánea dos fases de distribución. La vida media de distribución rápida varía entre 2 y 4 minutos, mientras que la correspondiente a la fase de distribución lenta es de 45 a 60 minutos. La primera probablemente representa el equilibrio en los tejidos bien vascularizados del compartimiento central y la segunda, el equilibrio en el compartimiento superficial (posiblemente músculo esquelético o tejido no graso). Es en esta fase cuando el paciente despierta después de una dosis moderada del barbitúrico. (Stanski, en PrysRoberts y Hung, 1986). Después de 4 a 5 vidas medias de distribución rápida (12 a 17 min) adquiere predominancia la fase de distribución lenta. Esta fase representa el equilibrio entre los compartimientos central y superficial y otro compartimiento más profundo de cinética más lenta (posiblemente grasa). El equilibrio entre este compartimiento y el plasma requiere 2 a 4 horas antes de que sea evidente la fase terminal de eliminación. La vida media de eliminación terminal es de 10-12 horas y solo aplica a dosis de tiopental de hasta 2 g. La vida media del tiopental es mayor que las del metohexital y menor que la del pentobarbital. (Stanski, en Prys-Roberts y Hung, 1986). El tiopental experimenta una biotransformación importante en el hígado. La ruta metabólica predominante en el hombre es oxidación en posición omega, que da origen al ácido carboxílico correspondiente, carente de actividad farmacológica. En menor grado, el tiopental sufre desulfuración para transformarse en el metabolito activo pentobarbital, el cual a su vez se metaboliza a productos inactivos. Se pudieron detectar sólo concentraciones plasmáticas bajas de pentobarbital, en pacientes, 15 minutos después de la administración de dosis totales de tiopental de 450 a 1275mg. (Stanski, en Prys-Roberts y Hung, 1986). En cambio, estudios más recientes muestran que la infusión de dosis altas de tiopental, usadas en la clínica para protección cerebral (300 a 500 mg /Kg en 2 a 3 días) da lugar a concentraciones plasmáticas del metabolito con actividad farmacológica (3 a 7 μg ml-1). Esto sugiere que la velocidad de formación de pentobarbital es baja y que el proceso no contribuye al efecto anestésico de una dosis única de tiopental. (Stanski, en Prys-Roberts y Hung, 1986). La depuración de tiopental (debida a metabolismo) es menor que la de metohexital y mayor que la de pentobarbital. Puesto que el volumen de distribución en estado estable para estos tres barbitúricos es semejante, las diferencias en la vida media de eliminación terminal son debidas por completo a diferencias de depuración hepática (es decir, biotransformación) y debido a la relativamente baja depuración del tiopental, se había postulado que el metabolismo contribuye muy poco a la terminación del efecto anestésico de un bolo intravenoso único. (Stanski, en Prys-Roberts y Hung, 1986). La otra variable farmacocinética que determina la vida media de eliminación terminal del tiopental es el volumen de distribución en estado estable. Debido a su gran liposolubilidad, el fármaco se distribuye extensamente en los tejidos, en especial en la grasa. La baja depuración y gran volumen de distribución del tiopental explican su moderadamente larga vida media de eliminación terminal. (Stanski, en Prys-Roberts y Hung, 1986). El grado de fijación del tiopental a las proteínas plasmáticas, específicamente a la albúmina, es moderado. En pacientes sanos, la fracción libre es de 15 a 25 % si este valor aumenta por enfermedad o por la presencia de otro fármaco, una mayor porción del tiopental existente en la circulación cerebral podrá penetrar al cerebro. Así pues, se presentará un mayor efecto farmacológico con una dosis determinada cuando existe menor fijación a proteínas plasmáticas, en este caso también aumentará la frecuencia de efectos adversos, en particular la depresión cardiovascular. Una mayor concentración de tiopental en el plasma podría incrementar los valores en el corazón y los vasos y predisponer a una mayor depresión miocárdica e hipotensión. La fracción libre de tiopental puede aumentar con la presencia de un segundo fármaco con mayor afinidad por sitios de fijación en la albúmina. Estudios in vitro demuestran que la aspirina, indometacina, ácido mefenámico, fenilbutazona y naproxén pueden producir este efecto. (Stanski, en Prys-Roberts y Hung, 1986). Farmacocinética del tiopental en presencia de enfermedad. En la insuficiencia renal crónica, la dosis anestésica del tiopental se reduce a la mitad, lo cual puede estar relacionado con los cambios en la fijación de proteínas. La fracción libre del barbitúrico aumentó del 22% en sujetos normales a 53% en pacientes con insuficiencia renal crónica. Este aumento incrementa las concentraciones cerebrales para cualquier dosis y puede explicar los requerimientos menores. (Stanski, en Prys-Roberts y Hung, 1986). Una explicación alternativa puede ser que aumente la sensibilidad del cerebro al tiopental en esta enfermedad (Insuficiencia renal crónica). Una administración más lenta en presencia de una menor fijación de proteínas puede evitar la excesiva concentración (y toxicidad) de tiopental en el sistema nervioso central y cardiovascular. La dosis total necesaria para la inducción y mantenimiento de la anestesia no se modificará, suponiendo una sensibilidad normal del sistema nervioso central al tiopental. En presencia de enfermedad hepática crónica, Shideman y col, en 1949 observaron una disminución en los requerimientos de dosis de tiopental, así como una prolongación del efecto, se desconoce el destino farmacocinético del tiopental, aunque se ha demostrado un aumento en la fracción libre. Estudios dosis-respuesta muestran que los requerimientos de tiopental disminuyen con la edad. Jung y col, en 1982 determinaron la farmacocinética de este anestésico en ancianos, la vida media de eliminación aumentó de 5 a 9 horas en pacientes de 20 a 30 años de edad, a 13 a 20 horas en sujetos de 60 a 80 años. Esto se debió a un aumento relacionado con la edad en el volumen de distribución en estado estable; la depuración no se modificó. El mayor volumen de distribución es debido a una reducción de la masa muscular y aumento en la proporción de grasa corporal que ocurre con la edad. Este estudio farmacocinético no explica el menor requerimiento de tiopental en los individuos de mayor edad. La redistribución a músculo y grasa podría ser más lenta en pacientes de edad, o bien el sistema nervioso central más sensible. (Stanski, en Prys-Roberts y Hung, 1986). Cuando se emplean dosis muy altas por tiempos prolongados, la cinética de eliminación del tiopental cambia de una de primer orden a una de orden cero (no lineal). La saturación de las enzimas hepáticas que biotransforman el tiopental es el mecanismo probable de esta cinética no lineal. La eliminación no lineal de dosis altas de tiopental tiene algunas consecuencias clínicas importantes durante una infusión continua de este agente, se necesita determinar la concentración plasmática cada 4 a 6 horas. A medida que los valores se aproximan al punto de saturación (30 a 50 µg ml-1), se hace necesario reducir la velocidad de infusión para evitar concentraciones excesivamente altas. En caso de saturación, la prolongada vida media de eliminación que deriva de la cinética de orden cero aumentará el tiempo entre la suspensión de la infusión y la posibilidad de valorar la función cerebral. (Stanski, en Prys-Roberts y Hung, 1986). La terminación de los efectos del tiopental se atribuyó en un principio a redistribución del cerebro a depósitos grasos. En 1966, Price y col, desarrollaron un modelo fisiológico que simulaba la redistribución del tiopental en todo el organismo. El modelo predijo que los tejidos no grasos (músculo, piel) son más importantes que la grasa corporal en la redistribución de la sustancia desde el sistema nervioso central. Aunque el coeficiente de partición del tiopental entre el plasma y músculo es menor que el de la grasa, la gran masa de tejido corporal (la mitad de la masa corporal, en comparación con la quinta parte en el caso de la grasa) aumenta la contribución de este tejido no graso en la terminación inmediata del efecto anestésico. (Stanski, en Prys-Roberts y Hung, 1986). Aplicaciones clínicas del tiopental sódico. Para inducción y mantenimiento de anestesia se administra un bolo intravenoso inicial de 50 mg, seguido de 100 a 200 mg, pero pueden necesitarse hasta 500 mg en individuos obesos o con gran masa muscular. Posteriormente y según el tipo de intervención quirúrgica se puede seguir con opiáceos o con agentes inhalados o con dosis intermitentes del propio tiopental. En el coma barbitúrico el tiopental se administra en infusión intravenosa., a la dosis de 100mg/Kg/día para conseguir niveles plasmáticos de 2,5-5 mg/100 ml. La duración del efecto, en este caso, depende de los procesos de eliminación (no de redistribución, como en el caso de la anestesia); estos procesos pueden estar condicionados por la capacidad metabólica del individuo, la fijación a tejidos, factores hemodinámicos, etc. En ausencia de otros fármacos potencialmente depresores, puede considerarse que el estado neurológico del paciente es independiente del tiopental cuando se hayan alcanzado niveles por debajo de 0,5 mg/100 ml sin que se haya observado ninguna mejoría en la escala del coma. (Flórez y Hurlé, en Flórez, 1992) En el perro, la dosis anestésica de tiopental sódico es aproximadamente de 15 a 17 mg/Kg de peso y en el gato, de 9 a 11 mg/Kg de peso. Sin embargo, estas cantidades pueden ajustarse de acuerdo con la profundidad de anestesia deseada. El hecho de que el pentotal (nombre comercial del tiopental sódico) ejerza efectos anestésicos de corta duración permite su uso en diversas situaciones clínicas, como la reducción de fracturas, los exámenes ginecológicos o radiológicos, las intervenciones quirúrgicas o la inducción de anestesia durante el empleo de anestésicos inhalados. (Sumano y Ocampo, 1997). IX.-Farmacología del propofol El propofol (2,6-disopropilfenol; DIPRIVAN) no está relacionado desde el punto de vista químico con otros agentes intravenosos. El compuesto es un aceite a temperatura ambiente, y se expende en emulsión a una concentración del 1 %. (Kennedy y Longernecker, en Goodman y Gilman, 1996). La presencia de dos radicales isopropilo en las posiciones 2 y 6 de una molécula de fenol causa dos importantes cambios en las propiedades de este compuesto. (Fig.7) En primer lugar, la molécula se vuelve inerte desde el punto de vista químico y, en segundo término, adquiere actividad anestésica. (Stanski, en Prys-Roberts y Hung, 1986). Figura 7. Estructura química del propofol. Tomado de: www.reactivereports.com/46/46_3.html. En medicina veterinaria, en ocasiones existe la imposibilidad económica de poder utilizar ventilación asistida y seguimiento electrónico de presión sanguínea, saturación de oxígeno e integridad cardiaca, lo que obliga al veterinario a buscar sistemas de anestesia ligera con respiración espontánea para la mayoría de las cirugías. Una técnica alternativa en anestesia clínica es la utilización de propofol por venoclisis; manejado como una opción de la anestesia inhalada. Esto se aplica en particular en pacientes de alto riesgo, por ejemplo: aquéllos con problemas cardiopulmonares (arritmias, taquicardia supraventricular paroxística), insuficiencia renal, intervenciones urológicas con tiempos de cirugía mayores de 480 min, pacientes geriátricos, pediátricos o politraumatizados. (Sumano y Ocampo, 1997). Características farmacológicas del propofol. El propofol es un derivado alquil-fenólico de baja solubilidad en agua por lo que se le suspende en una solución de aceite de soya, fosfolípidos purificados y lecitina de huevo (1% peso/volumen), con esto se logra una emulsión fina que se aplica por vía intravenosa. Se le considera a la solución utilizada para diluir el propofol como uno de los elementos lipídicos más fácilmente aceptados para la alimentación parenteral total. Por tanto, no constituye un riesgo para el paciente. (Sumano y Ocampo, 1997). Este anestésico se debe almacenar a 25 grados centígrados y no se debe congelar. Se agita antes de utilizarse para restablecer la emulsión; si no se logra un aspecto emulsificado uniforme, no debe administrarse el producto. Bajo condiciones ideales puede almacenarse por tres años. El propofol incluido en su vehículo se puede diluir para lograr un volumen mayor de aplicación, únicamente con solución glucosada al 5 %, facilitando su tratamiento para venoclisis continua o con regulador de flujo. Esta maniobra es innecesaria cuando se usa una bomba de infusión programable. (Sumano y Ocampo, 1997). La administración intravenosa de propofol, a la dosis de 2-2,5 mg/Kg, (en humanos) induce pérdida de la conciencia con la misma rapidez que el tiopental. La duración del efecto es muy breve y la recuperación después de una dosis única o tras infusión continua es muy rápida, suave y con confusión postoperatoria mínima. (Flórez y Hurlé, citado en Flórez y col, 1992). La respiración es profundamente deprimida, en particular durante la inducción, efecto que es potenciado por los opiáceos. (Flórez y Hurlé, en Flórez y col, 1992). El propofol no altera la función hepática ni la renal. Disminuye la presión intracraneal y la presión intraocular. No interactúa con bloqueadores neuromusculares. Se describieron inicialmente reacciones alérgicas que fueron atribuibles al disolvente cremofor, pero la nueva formulación en forma de emulsión carece de efectos secundarios anafilactoides. No produce liberación de histamina. Los efectos secundarios más frecuentes son dolor en el sitio de inyección con riesgo de tromboflebitis, náuseas, vómitos y cefalea postoperatoria. (Flórez y Hurlé, en Flórez y col, 1992). Durante la acción de propofol, parecen reducirse flujo sanguíneo cerebral, metabolismo del cerebro y presión intracraneal. Se han publicado algunos informes de convulsiones o movimientos involuntarios durante la inducción o la salida de la anestesia inducida con propofol. La salida de la anestesia con propofol es más rápida que la salida de la administrada con tiopental, incluso después de la administración prolongada. (Kennedy y Longernecker, en Goodman y Gilman, 1996). Este anestésico no parece ocasionar efectos acumulativos ni el fenómeno de despertar prolongado después de su administración prolongada. A estas propiedades favorables se debe el uso extenso del propofol como componente de la anestesia equilibrada, así como su gran popularidad como anestésico para utilización en las “cirugías de un día”. El fármaco es también efectivo para producir sedación prolongada en pacientes con cuidados intensivos. Sin embargo, el propofol para la sedación de niños bajo cuidados intensivos ha dado lugar a acidosis grave en presencia de infecciones respiratorias y posibles secuelas neurológicas cuando se suspende. (Trevor y Miller, en Katzung, 2002). Al propofol se le calcula una potencia 1.8 veces superior al de tiopental. Propofol produce mayor grado de depresión de los reflejos osteotendinosos y oculares con respecto al tiopental. (Sumano y Ocampo, 1997). A pesar de su baja toxicidad se ha relacionado en seres humanos con secuelas convulsivas y preconvulsivas en pacientes sensibles, sin embargo, la relación no es clara y aún se requieren estudios complementarios para definirla. Más todavía, el propofol ha demostrado eficacia en el tratamiento de una emergencia neurológica como en el estado convulsivo cuando ha fallado la administración de diazepam y difenilhidantoína. (Sumano y Ocampo, 1997). Propofol tiene a la vez, una acción anticonvulsiva y neuroexcitadora. Se cree que la actividad anticonvulsiva del propofol está mediada por los receptores del ácido γ-aminobutírico, mientras que el origen de su actividad neuroexcitadora es desconocido. (López-Timoneda, en Velásquez y col, 2005). Aunque anteriormente Dong y Xu en 2002, aseguran que el propofol a concentraciones altas posee efecto pre-convulsivo. El propofol disminuye la tensión del esfínter esofágico posterior por lo que debe hacerse énfasis en un ayuno de ocho horas o menor y la medicación con metoclopramida o cisaprida. (Sumano y Ocampo, 1997). Efectos electrofisiológicos neuronales del propofol En neuronas del asta dorsal disociada de rata, el propofol demostró que potencia la transmisión sináptica inhibitoria, ya que incrementa corrientes inducidas por receptor GABAA y corrientes inducidas por el receptor de glicina a bajas concentraciones; pero también propofol inhibe a ambos receptores a altas concentraciones. Entonces propofol facilita la acción de GABAA y glicina a nivel espinal y puede contribuir significativamente a la anestesia y analgesia inducida por este anestésico general. (Dong y Xu, 2002). Al igual que el tiopental, propofol induce una corriente tónica (sostenida) mediada por el recepto GABAA, incrementando fuertemente corrientes postsinápticas inhibitorias (IPSCs), resultando en depresión del potencial de acción, por depresión de la excitabilidad intrínseca en neuronas hipocampales de rata y finalmente neurodepresión; esta depresión es inhibida por bicuculina y picrotoxina, pero no por estricnina, (Bieda y MacIver, 2004). Así mismo, el propofol incrementa las conductancias GABAA tónicas (sostenidas) a concentraciones menores o iguales a 1 µM en células piramidales CA1 disociadas. (Bieda y MacIver, 2004). Estudios más recientes señalan que tanto propofol como etomidato afectan la subunidad β del receptor GABAA, más claro en el caso del etomidato, ya que ratones knockout que contienen mutaciones en las subuniades β2 y β3, son insensibles a estos dos anestésicos. (Franks, 2006). Mutaciones en la subunidad β3 del receptor GABAA, particularmente N265M, reduce la sensibilidad a propofol. (Jurd y col, 2003). Mediciones directas de las concentraciones libres de propofol en el cerebro durante varios estados de anestesia, no son posibles, por sus características farmacocinéticas. A nivel experimental las concentraciones utilizadas van en rangos desde 0.2µM hasta 500µM, pero los efectos de propofol para inducir anestesia se han calculado de 5 a 10 µM. (Fig.8) (Bieda y MacIver, 2004). Bieda y MacIver en 2004, han demostrado que Propofol (10µM) suprime la excitabilidad en células piramidales CA1. (Fig.9) y no altera la forma del potencial de acción. (Fig.10). Figura 8. Propofol induce corrientes a varias concentraciones y disminuye fuertemente la excitabilidad neuronal vía conductancia GABAA en interneuronas CA1. Adaptado de Bieda y MacIver. (2004). Figura 9. Propofol (10µM) suprime la excitabilidad en células piramidales CA1 en presencia de bloqueadores de receptores de glutamato (17.2 µM CNQX y 100 µM APV) receptores GABAB (5µM CGP55845A) y conductancias GABAA fácicas (conductancias de tiempo de duración relativamente corto, directo y rápido de GABA como por ejemplo potenciales postsinapticos inhibitorios; diferentes a las tónicas, que son conductancias sostenidas de GABA) (10µM SR95531). Adaptado de Bieda y MacIver. (2004). Los bloqueadores están presentes en la solución, y son utilizados para registrar solamente actividad de conductancias GABAA tónicas. Figura 10. La morfología del potencial de acción no es alterada por propofol (10µM).Control. 5 registros superpuestos en neuronas. .Propofol. 5 registros superpuestos expuestos a propofol no modifican la forma del potencial de acción, pero si disminuyen la amplitud. Adaptado de Bieda y MacIver. (2004). Al igual que el tiopental, el mecanismo de acción anestésico es mediado principalmente por receptor GABAA, y esto también ha sido demostrado por técnicas de unión a radioligandos (binding), donde la IC50, para inhibir la unión al radioligando [35S] TBPS o tbutyl-biclorofosforotionato, para picrotoxina fue de 9.2µM. Por otro lado la IC50 para inhibir al radioligando [3H] BTX-B de canales de sodio dependientes de voltaje fue de 28.1µM y para canales de calcio tipo L; inhibiendo al radioligando [3H] PN200-110 fue de 175µM. (Lingamaneni y Hemmings Jr, 2003). Donde las concentraciones para inhibir los canales de sodio dependientes de voltaje y canales de calcio tipo L, son por arriba de las concentraciones anestésicas, en particular para canales de calcio tipo L. Otros mecanismos de acción para el efecto anestésico del propofol han sido propuestos; aunque las concentraciones utilizadas, no correlacionan para inducir el efecto anestésico; a continuación se presentan algunas de estas evidencias. El bloqueo de la transmisión sináptica excitatoria, puede ocurrir también a nivel postsináptico por antagonismo de receptores de glutamato del subtipo N-metil-D-aspartato (Mantz, 1992; Bieda y MacIver, 2004). Ying y col, en 2006, determinaron el efecto de propofol sobre la corriente (Ih) in vitro y en vivo y observaron que propofol hace más lento el umbral de rebote de excitación, y también suprime la conductancia de Ih, asociada con una disminución de la regularidad y la frecuencia de oscilaciones delta en neuronas VB (ventrobasales) de ratón. Propofol reduce la Ih de una manera dependiente de la dosis, en neuronas CA1 de hipocampo de rata, la reducción de esta corriente de entrada activada por hiperpolarización, se traduce en reducción de la actividad neuronal. Quizá la efectividad de propofol como anticonvulsivo o antiemético, está asociada al bloqueo del canal, que induce esta corriente. (Funahashi y col, 2001). Por su parte Frenkei y Urban, en 1991, observaron y cuantificaron el efecto de propofol sobre canales de sodio unitarios de tejido de corteza cerebral de humano, en la presencia de batracotoxina. Este anestésico deprime dos grandes funciones del canal de sodio: reducción del tiempo promedio de apertura fraccional, (con un bloqueo máximo del 28%) y la interacción con el estado de activación en equilibrio. Sin embargo, aunque estos canales pueden alterar los patrones de disparo neuronal, no hay evidencia de que estos canales jueguen una función substancial en la producción del estado anestésico. (Frank y Lieb, 1994). Farmacocinética de propofol. En animales de experimentación el disopropil fenol muestra una vida media de distribución muy corta y en esta fase la concentración sanguínea disminuye a un décimo de su valor inicial. La eliminación del fármaco también es rápida en animales, encontrándose vidas medias terminales de 16 a 55 min. Los cálculos preliminares en el hombre indican una vida media de eliminación terminal de alrededor de 55 min y una depuración total de 3000 a 3500 ml min-1 (Stanski, en Prys-Roberts y Hung, 1986). Se biotransforma con eficacia en el hígado y no parece acumularse; se une notablemente a las proteínas plasmáticas y eritrocitos, y puede ser desplazado de éstas por medicamentos opioides como el fentanil o la meperidina por lo que pequeñas dosis de estos medicamentos inducen un efecto anestésico notable debido al propofol libre y al efecto narcótico por sí solo. (Sumano y Ocampo, 1997). En otras palabras, su unión a las proteínas plasmáticas se calcula que es de 97 a 99%. Por lo que si ocurre un desplazamiento de 1 a 3 % se puede aumentar la anestesia 300%, dado que la concentración de propofol libre en sangre se incrementa con la magnitud referida. Las concentraciones sanguíneas útiles para la inhibición del reflejo palpebral varían entre 1.88 y 3.92 µg/mililitro. En estudios de toxicidad hepática se llegó a la conclusión de que el propofol no afecta el funcionamiento de el hígado, evaluándolo mediante la determinación de las enzimas clave como AST o TGO (Aspartato-amino-transferasa), ALT o TGP (Alanino-aminotransferasa), FAS (Fosfatasa alcalina sérica), colesterol y pruebas de excreción de bromosulftaleína. (Sumano y Ocampo, 1997). Aplicaciones clínicas del propofol Después de la autorización para el uso general del propofol en 1989, se ha difundido su utilización para la inducción y la conservación de la anestesia, lo mismo que para producir sedación durante la anestesia regional o en las unidades médicas de cuidados intensivos. (Kennedy y Longernecker, en Goodman y Gilman, 1996). Se ha usado con éxito en niños sometidos a procedimientos de diagnostico o radioterapia. Se recomienda controlar la saturación de oxígeno ya que puede producirse bradipnea y/o hipoxemia. La cirugía oftálmica, la terapia electroconvulsiva, la cardioversión y la intubación o la colocación de una mascarilla laríngea son otros contextos clínicos en los que el propofol puede ser útil como agente anestésico. Una desventaja importante del uso prolongado de propofol es su costo, el cual excede el de las benzodiacepinas y barbitúricos. Las infecciones clínicas debidas a contaminación bacteriana del propofol que se han comunicado, posiblemente se relacionen con rechazo a descartar cantidades relativas de un fármaco costoso. (Trevor y Miller, en Katzung, 2002). En medicina veterinaria; el propofol se puede emplear como anestésico sin premedicación, sin embargo, con el uso de neuroplégicos se reduce su dosis hasta 30%. Habitualmente, este anestésico se ha utilizado sobre todo en procedimientos rápidos y de diagnostico radiológico en perros, gatos y caballos. (Sumano y Ocampo, 1997). X.-Efectos cardiovasculares de tiopental sódico Al aplicar el tiopental sodico por via intravenosa., se puede producir inicialmente una caída de la presión arterial que se recupera pronto. Generalmente, en individuos sanos y en los cuales los mecanismos homeostaticos funcionan adecuadamente, los efectos cardiovasculares no son importantes. Por otro lado, en situaciones de hipovolemia, hemorragia, toxemia, sepsis y shock, la dosis normal de tiopental puede ocasionar colapso circulatorio. (Flórez y col, en Flórez y col, 2002; Kennedy y Longernecker, en Goodman y Gilman, 2006). El gasto cardiaco suele disminuir en cierto grado, pero la resistencia vascular periférica se conserva sin cambios o se incrementa. Disminuye también el flujo sanguíneo a la piel y cerebro, pero en otros órganos se conserva básicamente normal. (Kennedy y Longernecker, en Goodman y Gilman, 2006). El sistema baroreceptor parece no verse afectado por el tiopental, pero la actividad nerviosa simpática se reduce. (Kennedy y Longernecker, en Goodman y Gilman, 2006). Aunque una ligera afectación del reflejo barostático, puediera reducir la contractilidad miocárdica. (Russo y Bressolle, 1998). El efecto sobre la presión venosa central es variable, y puede ocurrir una depresión circulatoria grave después de la administración imprudente de tiopental que produce un deterioro de los mecanismos homeostáticos vasculares. Bolos intravenosos, disminuyen el volumen intratorácico, ya que es transferido a la periferia, resultando en reducción del retorno venoso e incremento de la resistencia vascular sistémica. (Russo y Bressolle, 1998). Dosis de 4 mg/Kg, producen una disminución significativa de la presión arterial media (PAM) de 89 a 74 mmHg y del gasto cardiaco de 6.17 a 4.76 L/minuto; después de la inducción de la anestesia. (Russo y Bressolle, 1998). El efecto directo del tiopental sódico sobre el corazón, se desconoce del todo a la fecha, la contractilidad miocárdica puede verse afectada, principalmente por la reducción del llenado ventricular y por depresión del flujo simpático. Separando efectos vasculares, de efectos inotropicos cardiacos directos, en perros, se encontró que la aplicación de tiopental a dosis anestésicas suficientes para acceder a anestesia quirúrgica, con las cuales se alcanzaron concentraciones en la circulación coronaria de 3 mg/100 ml, no afectaron la fuerza de contracción miocárdica. Solo administrando dosis altas, con las que se alcanzan concentraciones en la circulación coronaria de 12 mg/100 ml produjeron efectos inotrópicos negativos significativos. Por lo tanto la depresión miocárdica, no es un factor importante en los cambios circulatorios asociados a tiopental. (Russo y Bressolle, 1998). Otro efecto no menos importante, que caracteriza a los agentes anestésicos intravenosos, es el potencial arritmogénico. En términos generales, el tiopental, solo bajo ciertas circunstancias clínicas, puede desencadenar taquiarritmias y el propofol por el contrario inducir la aparición de bradiarritmias. Se mencionarán solo algunas evidencias obtenidas en ensayos clínicos. El Tiopental prolonga el intervalo QT (duración del potencial de acción, incluyendo la repolarización) más que propofol en pacientes con hemorragia subaracnoidea y predispone a anormailidades electrocardiográficas y disrritmias cardiacas. (Tankasen y col, 2002). Por su parte Ay y col, en 2003, indican que los anestésicos intravenosos tiopental y etomidato, pueden desencadenar disrritmias cardiacas en pacientes con enfermedad vascular coronaria, por aumento en la dispersión QT, y en pacientes que no padecen este tipo de enfermedades la posibilidad de disrritmias es menor por lo que los pacientes con patología cardiovascular previa presentan más riesgo de padecer arritmias. Factores fisiopatológicos influyen en la dosis de requerimiento de tiopental, por ejemplo: en condiciones de obesidad, los requerimientos de tiopental son menores de 3.9mg/Kg contra 5.1mg/Kg en pacientes sanos. Dosis bajas también son requeridas en pacientes con daño hepatico y uremia. Contrariamente, pacientes con alcoholismo crónico requieren dosis mayores de tiopental, ya que existe tolerancia cruzada entre tiopental sódico y alcohol, no así en los requerimientos para supresión de EEG (electroencefalograma), donde los requerimientos de tiopental, prácticamente son iguales. (Alcoholismo crónico 823 mg y pacientes no alcohólicos 733mg). Y por último pacientes con insuficiencia cardiaca, requieren menos del 60% de la dosis utilizada en un individuo sano. (Russo y Bressolle, 1998). XI.-Efectos cardiovasculares de propofol En el sistema cardiovascular el propofol induce hipotensión por reducción de las resistencias periféricas sin modificar el gasto cardiaco. Deprime la respuesta del reflejo barorreceptor originando bradicardia que puede llegar al paro cardiaco. Disminuye el consumo de oxígeno y el flujo sanguíneo miocárdico. El propofol no parece causar arritmias ni isquemia miocárdica, pero puede incrementar la arritmogenicidad de la adrenalina. (Kennedy y Longernecker, en Goodman y Gilman, 1996; Sumano y Ocampo, 1997). Su efecto sobre el reflejo baroreceptor y su efecto vagotónico directo, es debido a que pequeños incrementos en la frecuencia cardiaca son observados, después de la caída en la presión arterial posterior a una dosis de propofol (Evers y Crowder, en Hardman, Limbird y Gilman, 2001). No obstante, se ha especulado que la disminución global del tono adrenérgico produce una reducción de la resistencia periférica, lo que explica en parte que la inducción con propofol no induce taquicardia refleja (Sumano y Ocampo, 1997). Efectos hemodinámicos moderados, son obtenidos con dosis pequeñas y velocidad de administración lenta, esto combinado con una adecuada terapia de líquidos. (Sneyd, 2004). La disminución de la presión arterial media y la frecuencia cardiaca, con propofol son bien tolerados en pacientes saludables, bien hidratados, pero puede ser peligroso en pacientes geriátricos y en pacientes con una enfermedad clínicamente significativa tanto cardiovascular como cerebrovascular. (Taha, y col, 2005). Esto ha sido bien establecido por Errando y col, en 1998 en un modelo biológico experimental donde se induce hipovolemia en cerdos. Ellos usando varios anestésicos, incluido el propofol, establecen que tiopental y la asociación ketamina-midazolam quizá sean los anestésicos de elección en estas circunstancias ya que propofol causa un grado mayor de inestabilidad hemodinámica. Por lo anterior cuando se requiere someter a un procedimiento anestésico a una persona o a un animal, con un déficit hídrico importante, parece ser que tiopental sódico quizá sea la mejor elección, ya que al no alterar de manera importante la resistencia vascular periférica, o incluso elevarla ligeramente, causa menos hipotensión arterial que el propofol. Aunque resultados de estudios realizados con propofol, a nivel electrofisiológico, revelan cambios importantes en diferentes canales iónicos y corrientes cardiacas de potasio, calcio y sodio, las concentraciones utilizadas, no correlacionan con las dosis eficaces para producir anestesia. Tal es el caso del estudio de Nagashima y col, en 1999, que aseguraron que el efecto cronotropico e inotropico negativo de propofol es mayor que el de tiopental y además el efecto inotrópico y cronotrópico negativo, inducido por propofol, no es inhibido por atropina, demostrando que el propofol no afecta la respuesta cardiaca a acetilcolina, por lo tanto no involucra la activación de receptores muscarínicos. Nagashima y col, en 1999 aseguran que propofol deprime directamente la actividad marcapaso del nodo seno auricular y contractilidad miocárdica; estos estudios fueron realizados, en aurícula canina, pero las concentraciones utilizadas fueron excesivamente altas (30-1000 µg, siendo el efecto concentración dependiente). Por su parte Hamilton y col, en 2000, en miocitos ventriculares intactos aislados de rata observaron que el efecto del propofol produjo una reducción de la contractilidad dependiente de la concentración. (Fig.11); pero la K0.5 para el efecto inotrópico negativo de propofol fue observado en concentraciones más altas (34.5µM) de la EC50 para el valor de la anestesia (0.4µM). Figura11.Curva dosis respuesta para el efecto inotrópico negativo de propofol, durante 1 minuto de exposición a propofol. La K 0.5 para ese efecto fue calculada en 34.5µM. La línea punteada representa en el eje de las abscisas el valor de la EC50 para la anestesia con propofol. La línea sólida representa el mejor punto de los datos y las líneas punteadas representan arriba y abajo del 95% de confiabilidad. Adaptado de: Hamilton y col. (2000). Existen en la literatura resultados contradictorios sobre la arritmogenicidad del propofol, por ejemplo, Kennedy y Longernecker, en Goodman y Gilman, 1996; Sumano y Ocampo, 1997, mencionan que propofol no causa arritmias. Por otro lado, Zeballos y col, (2004) compararon las propiedades electrofisiológicas del tiopental y propofol, así como la vulnerabilidad auricular en un modelo porcino donde se indujo intoxicación alcohólica aguda con etanol, para facilitar la aparición de taquiarritmias auriculares. El propofol fué más arritmogénico que el tiopental, ya que las arritmias inducidas fueron de larga duración, y particularmente el aleteo auricular (ritmo auricular regular y frecuencia auricular mayor a 250 latidos por minuto). Estos resultados tienen relevancia clínica ya que la fibrilación auricular, el aleteo auricular y la taquicardia auricular, son complicaciones frecuentes en el periodo perioperatorio, y son reconocidas como la mayor causa de morbilidad, estancia hospitalaria y costo en los cuidados de la salud. Además, un gran número de pacientes alcohólicos son presentados a procedimientos de emergencia y requieren sedación o anestesia. (Zeballos y col, 2004). Los resultados antes mencionados merecen consideración en términos de futuros estudios que determinen la selección de agentes anestésicos en situaciones clínicas específicas. (Zeballos y col, 2004). XII.- Efectos electrofisiológicos cardiacos de tiopental y propofol. Efectos en la duración del potencial de acción. Estudios realizados por Sakai, y col. en 1996, determinaron los efectos de tiopental y propofol en membranas celulares de miocitos ventriculares de cobayo, utilizando patchclamp en la configuración de whole-cell. Concentraciones de propofol mayores que 0.5 µmol litro-1 acortan la meseta y la duración del potencial de acción y tiopental 10 µmol litro-1 prolonga la duración del potencial de acción, además concentraciones de 50 µmol litro-1 o mayores disminuyen la amplitud de la meseta y el potencial de membrana de reposo. Con fijación de voltaje, propofol 1 µmol litro-1 disminuye la corriente de Ca++ tipo L en 88.4% del valor control, sin afectar la corriente de K+ rectificadora tardía (IK) y el mismo propofol a 10 µmol litro-1 disminuyo la corriente de Ca++ tipo L y la IK en 75.0% y 78.4% respectivamente, sin afectar la corriente de potasio rectificadora de entrada (IK1). Tiopental 10µmol litro-1 disminuye la corriente de Ca++ tipo L en 88.5% y la IK en 78.3%; así mismo tiopental a 100 µmol litro-1 disminuye la corriente de Ca++ tipo L en 88.8% y la IK1 en 67.3%. (Sakai y col, 1996) Estos efectos explican al menos en parte la bradicardia característica del propofol y la taquicardia que puede presentarse con tiopental. Particularmente el efecto en la duración del potencial de acción es definitivamente contrastante entre estos dos anestésicos. Propofol a concentraciones altas es clínicamente relevante, acortando la duración del potencial de acción, principalmente por supresión de la corriente de Ca++ tipo L; y por su parte el tiopental causa efectos bifásicos en la duración del potencial de acción por depresión de IK, Ica.L y IK1 a altas concentraciones. Los distintos efectos cardiodepresivos de propofol y tiopental se atribuyen quizá, a las diferentes acciones en las corrientes de membrana de calcio y potasio (Sakai y col, 1996). Un año después Carnes, Muir y Van en 1997, determinaron el efecto de anestésicos intravenosos, en la corriente de potasio rectificadora de entrada (IK1) en miocitos ventriculares de humano y rata. La inhibición de la corriente rectificadora de entrada, puede causar disrritmias cardiacas por disminución del potencial de membrana o de reposo o por prolongación de la duración del potencial de acción. Carnes y col (1997) encontraron que el tiopental reduce la conductancia de la corriente de potasio rectificadora de entrada (IK1), en una manera dependiente de la concentración en miocitos ventriculares de rata (Fig.12, Fig.13 y Fig.14) y así mismo el efecto de tiopental sobre la conductancia de la IK1 en células ventriculares humanas, también es comparable con los miocitos ventriculares de rata. Por el contrario propofol o ketamina, (Fig.15) no alteran la conductancia de la corriente de potasio rectificadora de entrada (IK1)(Carnes y col, 1997). Lo anterior coincide con lo reportado por Sakai y col en 1996, donde determinan que propofol a 10 µmol litro-1 no afecta la IK1; esto nos indica que al menos en las condiciones experimentales de este estudio el efecto de estos anestésicos intravenosos, sobre estas corrientes repolarizantes es el mismo en estas dos especies y quizá nos lleve a poder asegurar, que lo que sucede en ratas, se pueda transpolar al humano, abriendo un abanico muy amplio de investigación de fármacos no solo agentes anestésicos, sino de otra índole, con repercusiones en las terapias actuales de algunos desordenes cardiacos como las disrritmias y la elección adecuada de los agentes anestésicos en pacientes con antecedentes de enfermedad cardiaca. Dos componentes de IK, han sido separados, son diferentes canales de potasio y quizá puedan ser distinguidos por sus cinéticas de activación, farmacología y regulación. (Heath y Terrar, 1996). Un componente de activación rápido (IKr), es sensible a muchas drogas antiarritmicas clase III, como E4031 y dofetilida; (Sanguinetti y Jurkicwiez, 1990; Carmeliet, 1992)) y un componente de activación lenta (IKs), es regulado por proteínas cinasas y es insensible a los componentes anteriores, pero es bloqueado por anestésicos como propofol y tiopental (Takashoshi y Terrar, 1995; Heath y Terrar, 1996). El componente de activación lenta de la corriente de potasio rectificadora tardía (IKs) en el corazón, es importante durante la repolarización de el potencial de acción cardiaco. (Heath y Terrar, 1997). Propofol y tiopental muestran supresión de la IKs cardiaca, sin efecto en el componente de activación rápida de la corriente de potasio rectificadora tardía (IKr). La inyección de la proteína Kmin, dentro de oocitos de xenopus induce una corriente similar a IKs (IsK). Propofol y tiopental causan reducciones concentración-dependiente de la IsK; por lo que la proteína de K min, contribuye a las bases moleculares de la IKs del canal cardiaco. (Heath y Terrar, 1997). Figura12. Supresión de IK1 en miocitos ventriculares de rata. Tomado de: Carnes y col. (1997). Figura13.Trazos de corriente en miocito ventricular de rata expuesto a 10 microM de tiopental. A. Protocolo de fijación de voltaje. B. Control C. 10 microM de tiopental. D. Diez minutos después de lavado. Tomado de: Carnes y col. (1997). Figura.14. A. Efecto concentración-dependiente de tiopental sobre IK1 y relación densidad-voltaje en miocitos ventriculares de rata. B. Conductancia fraccional de IK1. IC50= 10.5 microM. Adaptado de: Carnes y col. (1997). Figura15. A. Efecto de ketamina concentración-dependiente sobre IK1. Relación densidad-voltaje. B Efecto de 2.5 microM. de propofol y control, sobre IK1. Relación densidad-corriente. Adaptado de: Carnes y col. (1997). XIII.-Comparación de tiopental sódico y propofol en algunos procedimientos clínicos. Intubación traqueal La intubación endotraqueal es un procedimento clínico indispensable, cuando se utiliza anestesia inhalada, y en la mayoría de los casos aunque la inducción se puede realizar con gases anestésicos como el óxido nitroso, la realidad es que la mayoría de las veces se realiza con agentes anestésicos intravenosos, como el tiopental sódico y el propofol. Lo que facilita o dificulta este procedimiento clínico dependiendo entre otros muchos factores, es el agente anestésico utilizado. Y precisamente uno de estos factores atribuibles al agente anestésico es la profundidad anestésica alcanzada con determinada dosis; y considerando que a mayor dosis, es más probable la aparición de efectos adversos, principalmente cardiovasculares. Por ello resulta interesante desde el punto de vista clínico cual de estos dos anestésicos (tiopental sódico y propofol) en determinadas circunstancias logra una mejor calidad de la intubación, sin presentar o presentando los efectos colaterales mínimos. Como por ejemplo; la inducción rápida intravenosa de anestesia general está indicada en infantes, si existe posibilidad de vómito y regurgitación para reducir el riesgo de aspiración de contenido gástrico e intubación de personas adultas para cirugía electiva, demuestran que propofol brinda mejor calidad de anestesia, pero con mayor hipotensión y bradicardia y tiopental menor calidad de la anestesia, sin cambios importantes en la presión arterial y frecuencia cardiaca. (Wodey y col, 1999; Taha y col, 2005). Ensayos en medicina veterinaria han arrojado los mismos resultados (Prassinos y col, 2005). Cardioversión Otro procedimiento clínico en pacientes con arritmias es la cardioversión. Valtonen y col. en 1988, no encontraron cambios en las características de la inducción y la respuesta hemodinámica de tiopental y propofol en pacientes con fibrilación auricular. Y más tarde Gupta y col. en 1990, aseguran que tiopental es el más satisfactorio para la anestesia para cardioversión, ya que propofol significativamente disminuye la presión arterial media después de la inducción. Terapia electroconvulsiva El anestésico ideal usado para ECT (Terapia electroconvulsiva) debe reunir las siguientes características: inducción rápida, corta duración de acción, mínimos efectos colaterales, rápida recuperación y no interferir con la eficacia de la ECT. (Fig.16) En este escenario al parecer el propofol reduce las convulsiones comparado con barbitúricos en aproximadamente 20% y además la presión arterial media y la frecuencia cardiaca, aumentan después del shock eléctrico con tiopental, y estos cambios circulatorios sistémicos son abolidos con propofol, por disminución de la velocidad del flujo sanguíneo en la arteria cerebral media.(Saito y col, 2000) Figura 16. Frecuencia cardiaca durante terapia electroconvulsiva. La frecuencia cardiaca en el grupo de tiopental se incrementa significativamente después del shock eléctrico y el incremento continúa hasta por 15 minutos después del shock eléctrico. En el grupo de propofol la frecuencia cardiaca, no cambia significativamente durante toda la terapia electroconvulsiva. El comportamiento de la frecuencia cardiaca en ambos grupos es diferente y los valores inmediatamente después de la anestesia a los 0.5, 1, 2, 3 y 5 minutos después del shock eléctrico es significativamente más alto en el grupo de tiopental, comparado con el grupo de propofol. (Lo mismo sucede para la presión arterial media y la velocidad de flujo de la arteria cerebral media; no mostrados) Adaptado de Saito y col. (2000). XIV.-CONCLUSIONES 1.- El tiopental sódico y el propofol son dos anestésicos intravenosos de duración ultracorta, su mecanismo de acción anestésico principal y científicamente más sustentado, es por su acción sobre los receptores inhibitorios GABAA. A pesar de que farmacológicamente son de diferente grupo y también son estructuralmente diferentes. 2.- Otras propuestas, sobre el mecanismo de acción de estos anestésicos intravenosos son sobre receptores NMDA, receptores de glicina y canales iónicos de potasio, calcio y sodio. 3.- Los efectos clínicos colaterales sobre el sistema cardiovascular del tiopental sódico son: depresión de la contractilidad miocárdica, incremento en la frecuencia cardiaca y arritmias cardiacas; esto parece ser más frecuente en pacientes con enfermedad cardiovascular previa. 4.- Los efectos clínicos colaterales sobre el sistema cardiovascular del propofol son: bradicardia e hipotensión (frecuentes), arritmias auriculares (ocasionales) y bloqueo auriculoventricular y paro cardiaco (raro). PERSPECTIVAS 1.- La investigación con herramientas electrofisiológicas, en el área de anestesiología y particularmente con estos dos anestésicos ampliamente utilizados, a nivel clínico, debe realizarse en concentraciones equivalentes a las concentraciones de anestesia clínica, para poder realizar conjeturas más precisas del impacto de estos fármacos a nivel de canales iónicos y su correlación con los eventos clínicos, principalmente a nivel del sistema nervioso central y sistema cardiovascular. 2.- La Utilización de los enantiómeros de tiopental y quizá a futuro, fármacos derivados del propofol por modificación de su estructura química, que mantenga o incluso haga más eficaz sus propiedades anestésicas y disminuya sus efectos adversos, principalmente a nivel cardiovascular, deben ser tema de atención por parte de los investigadores, ya que al reducir los riesgos anestésicos, por ende se incrementan los éxitos quirúrgicos. XV.-Bibliografía Antognini, J.F. y Schwartz, K. (1993). Exaggerated anesthetic requeriments on the prefere ntially anesthetized brain. Anesthesiology. 79: 1244-1249. Ay, B., Fak, A.S., Toprak, A., Gogus, Y.F., y Oktay, A. (2003). QT dispersion increases during intubation in patients with coronary disease. Journal of Electrocardiology. 36: 99104. Bieda, M.C., y MacIver, M.B. (2004). Major role for tonic GABAA conductances in anesthetic supresión of intrinsic neuronal excitability. J. Neurophysiol. 92: 1658-1667. Carmeliet, E. (1992). Voltage-and time dependent block of the delayed K+ current in cardiac myocites by dofetilide. Journal of Pharmacology and Experimental Therapeutics. 264: 553-560. Carnes, C.A., Muir, W.W.3rd, y Van, W. (1997). Effect of intravenous anestheticas on inward rectifier potassium current in rat and human ventricular myocites. Anesthesiology. 87: 327-334. Christensen, H.D., y Lee, I.S. (1973). Anesthetic potency and acute toxicity of optically active disubstituted barbituric acids. Toxicol. Appl. Pharmacology. 26: 495-503. Dong, X.P., Xu, T.L. (2002). The actions of propofol on gamma-aminobutyric acid-A and glycine receptors in acutely dissociated spinal dorsal horn neurons of the rat. Anesth. Analg. 95: 907-914. Errando, C.L., Valia, J.C., Sifre, C., Moliner, S., Gil, F., Gimeno, O., y Palanca, J.M. (1998). Cardiocirculatory effects of intravenous anesthetic induction in an experimental model of acute hypovolemia. Revista españoña de anestesiología y reanimación. 45: 333339. Evers, A.S., Crowder, C.M., y Bolser, J.R. (2007). Acciones y Mecanismos de los Anestésicos Generales. Mecanismos de Anestesia. Sitios anatómicos de la acción anestésica. Mecanismos celulares de la anestesia. Acciones moleculares de los anestésicos generales. En Brunton, L.L., Lazo, J.S., y Parker, K.L (Eds), Las bases farmacológicas de la terapéutica (Undécima Edición) (pp. 343-346). Colombia: McGrawHill-Interamericana. Evers, A. S., Crowder, M. (2003). Anestésicos Generales. En Hardman J.G., Limbird, L.E. Las Bases Farmacológicas de la Terapeutica. (Decima Edición)(pp.347). México. McGraw-Hill Interamericana. Flórez, J. y Hurlé, M.A. (1992). Fármacos anestésicos generales. En Flórez, J., Armijo, J.A., y Mediavilla, A. (Eds.). Farmacología Humana. (Segunda Edición)(pp. 407-414). Barcelona: Masson. Franks, N.P. (2006). Molecular targets underlyng general anaesthesia. British Journal of Pharmacology. 147: S72-S81. Franks, N.P., y Lieb, W.R. (1994). Molecular and cellular mechanisms of general anaesthesia. Nature. Review Article. Vol. 367. 607-614. Franks, N.P., y Lieb, W.R. (1982). Molecular mechanisms of general anaesthesia. Nature. 300. 487-493. Frenkel, C., y Urban, B.W. (1991). Human brain sodium channels as one of the molecular target sites for the new intravenous anaesthetic propofol (2,6-disopropylphenol). Eur. J. Pharmacol. 208: 75-79. Friederich, P., y Urban, B.W. (1999). Interaction of intravenous anesthetics with human neuronal potassium currents in relation to clinical concentrations. Anesthesiology. 91: 1853-1860. Funahashi, M., Higuchi, H., Miyawaki, T., Shimada, M., y Matsuo, R. (2001). Propofol suppresses a hyperpolarization-activated inward current in rat hippocampal CA1 neurons. Neurosci. Lett. 311: 177-180. Gupta, A., Lennmarken, C., Veqfors, M., y Tyden, H. (1990). Anaesthesia for cardioversion. A comparison between propofol, thipentone and midazolam. Anaesthesia. 45: 872-875. Hall, A.C., Lieb, W.R., Franks, N.P. (1994). Insensitivity of P-type calcium channels to inhalational and intravenous general anesthetics. Anesthesiology. 81: 117-123. Hamilton, D.L., Boyett, M.R., Harrison, S.M., Davies, L.A., y Hapkins, P.M. (2000). The Concentration- Dependent Effects of Propofol on Rat Ventricular Myocytes. Anesth. Analg. 91: 276-282. Heath, B.M., y Terrar, D.A. (1997). Block by propofol and thiopentone of the min K current (IsK) expressed in Xenopus Oocytes. Naunyn Schmiedebergs Arch. Pharmacol. 356: 404-409. Heath, B.M., y Terrar, D.A. (1996). The deactivation Kinetics of the delayed rectifier components IKr and IKs in guinea-pig isolated ventricular myocites. Experimental Physiology. 81: 605-621. Jurd, R., Arras, M., Lambertm, S., Drexler, B., Sieqwart, R., Crestani, F., Zaugg, M., Vogt, K.E., Ledermann, B., Antkowiak.B., y Rudolph, V. (2003). General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 unit. FASEB J. 17: 250-252. Kash, T.L., Trudell, J.R., y Harrison, N.L. ( 2004). Structural elements involved in activation of the γ-aminobutiric acid type A (GABAA) receptor. Biochemical Society Transactions. Volume 32, part 13: 540-546. Kending y col. (1991). General Anesthetics. Citado por Evers, A.S., y Crowder, C.M. En Hardman, J.G., Limbird, L.E., y Gilman, A.G. (Eds). Goodman & Gilman’s The Pharmacological Basis Of Therapeutics (Tenth Edition) (p.338). New York: McGraw-Hill. (2001). Kennedy, S. K., y Longnecker, D.E. (1996).Farmacología del Sistema Nervioso Central. En Goodman y Gilman. Las Bases Farmacológicas de la Terapéutica. (9a ed.)(pp. 313-315 y 341-349). Ciudad de México: McGrawHill-Interamericana. Kitayama, M., Hirota, K., Kudo, M., Kudo, T., Ishihara, H., y Matsuki, A. (2002). Inhibitory effects of intravenous anaesyhetic agents on K(+)-evoked glutamate release from rat cerebrocortical slices. Involvement of voltage-sensitive Ca(2+) channels and GABA(A) receptors. Naunyn Schmiedebergs Arch. Pharmacol. 366: 246-253. Koblin, D.D., Chortkoff, B.S., Laster, M.J., Eger, E.I. II, Halsey, M.J., y Ionesco, P. (1994). Polyhalogenated compuneds that disobey the Meyer-Overton hypothesis. Anesth. Analg. 79: 1043-1048. Larrabe, M.G., y Postenak, J.M. (1952). Selective actions of anestheticas on synapses and axons in mammalian sympathetic ganglia. J. Neurophysiol. 15: 91-114. Lingamaneni, R., y Hemmings Jr, H.C. (2003). Differential interaction of anaesthetics and antiepileptic drugs with neuronal Na+ channels, Ca2+ channels y GABAA receptors. British Journal of Anaesthesia. 90: 199-211. Litter, M. (1969). Farmacología (4a ed.)(pp. 244-246). Buenos Aires: El Ateneo Editorial. Liu, H., Dai, T., y Yao, S. (2006). Effect of thiopental sodium on N-methyl-D-aspartategated currents. Can. J. Anaesth. 53: 442-448. López-Timoneda, F., y Gasco, M.C. (2004). Fármacos anestésicos generales. En Velásquez, P.L., Moreno, A., Leza, J.C., Lizasoain, I., y Moro, M.A. Farmacología Básica y Clínica.Velázquez (17a ed.) (pp. 233-235 y 240-244). Buenos Aires: Editorial Medica Panamericana. Lysko, G.S., Robinson, J.L., Casto, R., y Ferrone, R.A. (1994). The stereo especific effects of isoflurane isomers in vivo. Eur. J. Pharmacol. 265: 25-29. Mantz, J. (1992). Effects of intravenous anesthetics on neurons of the central nervous system: mechanisms of cellular and molecular action. Ann. Fr. Anesth. Reanim. 11: 540557. McIver, M.B., Mikulec, A.A., Amagasu, S.M., y Monroe, F.A. (1996). Volatile anestheticas depress glutamate transmission via presynaptic actions. Anesthesiology. 85: 823-834. McIver, M.B., y Roth, S.H. (1988). Inhalation anesthetics exhibit pathway-specific and differential actions on hippocampal synaptic responses in vitro. Br. J. Anaesth. 60: 680691. Mizobe, T., Maghsoudi, K., Sitwala, K., Tianzhi, G., Ou, J., y Maze, M. (1996). Antisense technology reveals the alpha2A adrenoreceptor to be the subtype mediating the hypnptic response to the highly selective agonist, dexmetomidine, in the locus coeruleus of the rat. J.Clin. Invest. 98: 1076-1080. Nagashima, Y., Furukawa, Y., Hirose, M., y Chiba, S. (1999). Cardiac effects of propofol and its interaction with autonomic nervous system in isolated cross-circulated canine atria. Journal of Anesthesia. 13: 34-39. Nelson, L.E., Guo, T.Z., Lu, J., Saper, C.B., Franks, N.P., y Maze, M. (2002). The sedative component of anesthesia is mediated by GABAA receptors in an endogenous sleep pathway. Nat. Neurosci. 5. 979-984. Nguyen, K.T., Stephens, D.P., McLeish, M.J., Crankshaw, D.P. y Morgan, D.J. (1996). Pharmakocinetics of thiopental and pentobarbital enantiomers after intravenous administration of recemic thiopental. Anesth. Analg. 83: 552-558. Nicoll, R.A. (1972). The effects od anesthetics on synaptic excitation and inhibition in the olfactory bulb. J.Physiol. 223:803-814. Nicoll, R.A., y Madison, D.V. (1982). General anesthetics hyperpolarize neurons in the vertebrate central nervous system. Science. 217: 1055-1057. Nussmeier , N.A. , Arlund, C., y Slogoff, S. (1986). Neuropsychiatric complications after cardiopulmonary bypass: cerebral protection by a barbiturate. Anesthesiology.64: 165-170. Ohtsuka, T., Ishiwa, D., Kamiya, Y., Itoh, H., Nagata, I., Saito, Y., Yamada, Y., Sumitomo, M., y Andoh, T. (2006). Effects of barbiturates on ATP-sensitive K channels in rat substantia nigra. Neuroscience. 137: 573-581. Perovansky, M., Barnov, D., Salman, M., y Yaari, Y. (1995). Effects of halothane on glutamate receptor-mediated excitatory postsynaptic currents. A match clamp study in adult Mouse hippocampal slices. Anesthesiology. 83: 109-119. Prassinos, N.N., Galatos, A.D., y Raptopoulus, D. (2005). A comparision of propofol, thiopental or ketamine as induction agents in gotas. Veterinary anaesthesia and analgesia. 32: 289-296. Rampil, I.J. (1994). Anesthetic potency is not altered after hypothermic spinal cord transaction in rats. Anesthesiology. 80: 606-610. Ries, C.R., y Puil, E. (1999). Mechanism of anestesia revealed by shunting actions of isoflurane on thalamocortical neurons. J. Neurophysiol. 81: 1795-1801. Rudolph, U., y Moller, H. (2003). Análisis of GABA . Receptor Function and Disection of the pharmacology of benzodiecepines and General Anesthetics Trough Mouse Genetics. Annu. Rev. Pharmacol. Toxicol. 44: 475-498. Russo, H., y Bressolle, F. (1998). Pharmacodynamics and Pharmacokinetics of thiopental. Clin. Pharmacokinetic. 35: 95-134. Saito, S., Kadoi, Y., Nava, T., Sudo, M., Obata, H., Morita, T., y Guto, F. (2000). The comparative effects of propofol versus thiopental on middle cerebral artery blood. Flow velocity during electroconvulsive therapy. Anesth. Analg. 91: 1531-1536. Sakai, F., Hiroaka, M., y Amahak. (1996). Comparative actions of propofol and thipentone on cell membranes of isolated guinea pig ventricular myocytes. British Journal of Anaesthesia. 77: 508-516. Sanguinetti, M.C., y Jurkicwiez, N.K. (1990). Two components of cardiac delayed restifier K+ currents. Diffential sensitivity to block by class III-antiarrhytmic agents. Journal of General Physiology. 96: 195-215. Sinner, B., Friedrich, O., Zink, W., Fink, R.H., y Graf, B.M. (2006). GABAmimetic intravenous anaesthetics inhibit spontaneous Ca2+-oscilations in cultured hippocampal neurons. Acta Anaesthesiol. Scand. 50: 742-748. Sneyd, J.R. (2004), Recent advances in anaesthesia. Br. J. Anaesth. 93: 725-736. Stanski, D.R. (1986). Farmacocinética de los Barbitúricos. En Prys-Roberts, C., y Hung, C.C. Farmacocinética de los anestésicos (pp. 103-111, 126 y 136-137). México, D.F: El Manual Moderno. Sumano, H.S., Ocampo, L. (1997). Farmacología Veterinaria (2a ed.) (pp. 349-358 y 385405) México: McGrawHill-Interamericana. Taha, S., Siddik-Sayyid, S., Alameddine, M., Wakim, C., Dahabra, C., Moussa, A., Khatib, M., y Baraka, A. (2005). Propofol is superior to thiopental for intubation without muscle relaxants. Canadian Journal Anaesthesia. 52: 249-253. Takahashi, H., y Terrar, D.A. (1995). Selectivity of action of propofol and thiopentone on components of the delayed rectifier potassium current in guinea-pig isolated ventricular myicytes. British Journal of Pharmacology. 115: 20p. Tankasen, P.E., Kytta, J.V., y Randell, T.T. (2002). QT interval and QT dispersion during the induction of anaesthesia in patients with subarachnoid haemorrhage: a comparision of thiopental and propofol. European Journal of Anaesthesiology. 19: 749-754. Tomlin, S.L., Jenkins, A., Lieb, W.R., y Franks, N.P. (1998). Stereoselective effects of etomidate optical isomers on gamma-aminobutyric acid type A. receptor and animals. Anesthesiology. 88: 708-717. Trevor, A.J., y Miller, R.D. (2002). Anestésicos Generales. En Katzung, B.G, Farmacolgía básica y clínica (8a ed.)(pp. 488-492). México: El Manual Moderno. Valtonen, M., Kanto, J., y Klossner, J. (1998). Anaesthesia for cardioversion: a comparision of propofol and thiopentona. Can. J. Anaesth. 35: 479-483. Wafford, K.A., Macaulay, A.J., Frodley, R., O’Meara, G.F., Reynolds, D.S., y Rosahl, T.W. (2004). Differentiating the role of γ-aminobutyric acid type-A (GABAA) receptor subtypes. Biochemical Society. Volume 32, part 3: 553-556. Wittmer, L.L., Hu, Y., Kalkbrenner, M., Evers, A.S., Zorumski, C.F., y Covey, D.F. (1996). Enantioselectivity of steroid-induced gamma-aminobutyric acid A receptor modulation and anesthesia. Mol. Pharmacol. 50: 1581-1586. Wodey, E., Chonow, L., Beneux, X., Azzis, O., Bansard, J.Y., y Ecoffey, C. (1999). Haemodynamic effects of propofol vs thiopental in infants: an echocardiografic study. British Journal of Anaesthesia. 82: 516-520. Yang, C.X., y Xu, T.L. (2006). Thiopental inhibits glycine receptor function in acutely dissociated rat spinal dorsal horn neurons. Neurosci. Lett. 397: 196-200. Ying, S.W., Abbas, S.Y., Harrison, N.L., y Goldstein, P.A. (2006). Propofol block of I(h) contributes to the suppression of neuronal excitability and rhythmic burst firing in thalamocortical neurons. Eur. J. Neurosci. 23: 465-480. Zeballos, M., Almendral, J., Anadón, M.J., Gonzalez, P., y Navia, J. (2004). Comparative effects of thiopental and propofol on atrial vulnerability: electrophysiological study in a porcine model including acute alcoholic intoxication. British. Journal of Anaesthesia. 93: 414-421.