NTE INEN 0576: Materiales refractarios. Sílico
Anuncio

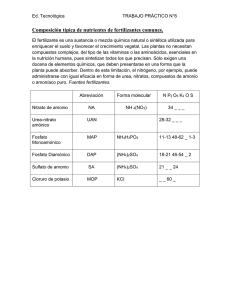
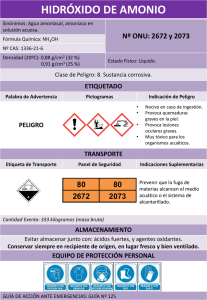
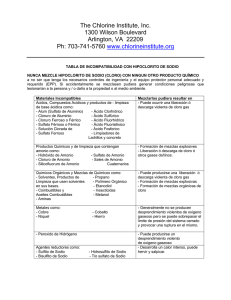
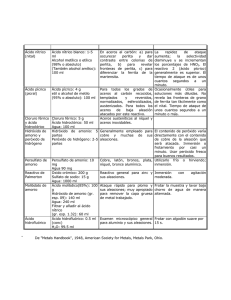
Re p u b l i co fEc u a d o r ≠ EDI CTOFGOVERNMENT± I no r d e rt op r o mo t ep u b l i ce d u c a t i o na n dp u b l i cs a f e t y ,e q u a lj u s t i c ef o ra l l , ab e t t e ri n f o r me dc i t i z e n r y ,t h er u l eo fl a w,wo r l dt r a d ea n dwo r l dp e a c e , t h i sl e g a ld o c u me n ti sh e r e b yma d ea v a i l a b l eo nan o n c o mme r c i a lb a s i s ,a si t i st h er i g h to fa l lh u ma n st ok n o wa n ds p e a kt h el a wst h a tg o v e r nt h e m. NTE INEN 0576 (1981) (Spanish): Materiales refractarios. Sílico aluminosos. Análisis químico CDU: 666.76 Norma Técnica Ecuatoriana MATERIALES REFRACTARIOS-SILICO ALUMINIOS ANALISIS QUIMICO CO 02.07-310 INEN 576 Instituto Ecuatoriano de Normalización, INEN – Casilla 17-01-3999 – Baquerizo Moreno E8-29 y Almagro – Quito-Ecuador – Prohibida la reproducción 1981-01 1. OBJ ETO 1.1 Esta norma establece los análisis químicos para determinar los componentes químicos de los materiales refractarios sílico-aluminosos. 2. ALCANCE 2.1 Esta norma se aplica para la determinación analítica de los siguientes elementos y componentes químicos de los materiales refractarios sílico-aluminosos: humedad, perdida por calcinación, sílice, óxidos de hierro y aluminio, óxido de calcio (cal), óxido de magnesio (magnesia) y álcalis (por el método de J. Jawrence Smith). 2.2 Esta norma se aplica solamente a los materiales refractarios sílico-aluminosos. Para los refractarios de carbono, de magnesita, de cromo, de alta alúmina y de circonio, deberán emplearse otros procedimientos. 3. RESUMEN 3.1 Se tratan las muestras de ensayo con varios disolventes ácido y alcalinos, hasta aislar los cuerpos mencionados en el numeral 2.1. 3.2 La determinación de los cuerpos componentes se llevará a cabo en el orden establecido en el numeral 2.1. 4. DISPOSICIONES GENERALES 4.1 Los análisis químicos da los materiales refractarios son trabajos complicados, que requieren un conocimiento amplio de la Química y un intenso entrenamiento previo de los operadores. 4.2 Pureza de reactivos. En todas las determinaciones deben emplearse reactivos químicos de grado análisis y de calidad comprobada. 4.3 Pureza de agua. A menos que se indique otra cosa, en todas las determinaciones debe emplearse agua de análisis de calidad y pureza comprobadas. 4.4 Concentración de reactivos. 4.4.1 Acidos e hidróxido de amonio concentrados. Se emplearán ácidos hidróxido de amonio concentrados de las siguientes densidades relativas o concentraciones: (Continúa) -1- 1980-0134 NTE INEN 576 1981-01 Acido clorhídrico 1,19 d.r. Acido nítrico 1,40 d.r. Acido sulfúrico 1,84 d.r. Acido fluorhídrico 48% Acido perclórico 60 a 70% Hidróxido de amonio 0,90 d.r. 4.4.2 Acidos o hidróxido de amonio diluidos. Se emplearán ácidos e hidróxido de amonio diluidos, de varios porcentajes en volumen. Se prepararán mediante mezcla de los reactivos concentrados y agua, indicando, entre paréntesis, la relación del volumen concentrado con el volumen de agua, como por ejemplo: ácido sulfúrico (1:9), que tiene 1 parte de ácido sulfúrico y 9 de agua. Las mezclas de ácido sulfúrico deben prepararse vertiendo muy lentamente el ácido en el agua. 5. PREPARACIÓN DE LAS MUESTRAS 5.1 La muestra, cuidadosamente obtenida por el procedimiento indicado en la Norma INEN 606, debe ser triturada en una pequeña prensa de mandíbulas o de rodillos de acero endurecido, hasta pasar un tamiz de 2,36 mm. 5.2 Después de la primera trituración, la muestra debe triturarse de nuevo hasta pasar un tamiz de 850 m, mezclarse y cuartearse hasta obtener 50 g. 5.3 La muestra de 50 g, obtenida anteriormente, debe ser molida hasta pasar el tamiz de 150 m y luego colocarse en un recipiente cerrado, el cual debe mantenerse libre de contaminación. 5.4 Deben tomarse precauciones para impedir la contaminación de la muestra con partículas de hierro del equipo de muestreo, de trituración o de molienda. La molienda fina debe hacerse en un mortero de ágata, para impedir la introducción de impurezas. 5.5 La determinación de humedad debe hacerse sobre la muestra, en su forma ordinaria, secada al aire ambiente. Las demás determinaciones deben efectuarse sobre la muestra libre de humedad, secada a la temperatura de 105° a 110°C. Si se prefiere, la muestra puede ser secada en una botella de pesada, de la cual se extraerá la cantidad requerida para cada determinación. 6. DISPOSICIONES ESPECIFICAS 6.1 Determinación en blanco. 6.1.1 Deben hacerse determinaciones en blanco sobre los reactivos, para cada constituyente del material refractario, deduciendo en cada caso los resultados correspondientes. 6.1.2 Para la determinación en blanco de dióxido de silicio, deben añadirse a la muestra 0,25 g de óxido de aluminio como cloruro de aluminio. -2- 1980-0134 NTE INEN 576 1981-01 7. DETERMINACION DE LOS COMPONENTES QUIMICOS 7.1 Humedad. 7.1.1 Pesar 2 g de la muestra con aproximación a 0,1 mg y transferir esta cantidad a un crisol tarado de platino o porcelana. Calentar a masa constante de 105° a 110° C. 7.1.2 Cubrir el crisol con una tapa herméticamente ajustada y dejar enfriar a la temperatura del local en un desecador, antes de pesar. 7.1.3 Calcular la pérdida en masa como porcentaje de humedad. 7.2 Pérdida por calcinación. 7.2.1 Procedimiento. 7.2.1.1 Pesar 2 g de la muestra libre de humedad (de 105° a 110°C) con aproximación de 0,1 mg y transferirla a un crisol tarado de platino o porcelana. Calentar a masa constante en aire, en un horno eléctrico de mufla, de 1 000° a 1 100°C. 7.2.1.2 Cubrir el crisol con una tapa herméticamente ajustada y dejar enfriar, a la temperatura del local, en un desecador, antes de pesar. 7.2.1.3 Calcular la pérdida en masa como porcentaje de pérdida por calcinación. 7.3 Sílice. 7.3.1 Procedimiento. 7.3.1.1 Transferir 1,00 g de la muestra libre de humedad (de 105° al 110°C) a un crisol tarado de platino, 3 3 3 de 30 cm . Añadir 10 cm de ácido nítrico (1:1) y 5 cm de ácido fluorhídrico. 7.3.1.2 Cubrir el crisol, colocándolo luego en una placa caliente sobre un bloque de asbesto, y calentarlo, suavemente al principio, aumentando gradualmente la temperatura. La temperatura final debe ser regulada para impedir la ebullición o la salpicadura. 7.3.1.3 La calefacción de esta clase durante una hora es suficiente para descomponer la muestra. Lavar el lado inferior de la tapa del crisol con unas pocas gotas de agua, dejándolas caer dentro del mismo crisol. 3 7.3.1.4 Evaporar el contenido del crisol a sequedad. Enfriar, añadir 10 cm de ácido nítrico (1:1) y volver a evaporar a sequedad. 7.3.1.5 Introducir el ácido lentamente en el crisol, vertiéndolo por los lados. Evaporar tres veces con el ácido nítrico, para asegurar la volatilización completa de los fluoruros. -3- 1980-0134 NTE INEN 576 1981-01 7.3.1.6 Calcinar suavemente al principio y luego a masa constante hasta cerca de 1 200°C. Enfriar en un desecador y pesar. 7.3.1.7 La pérdida en masa, menos la pérdida por calcinación (ver 7.2.1.3), representa todo el posible contenido de sílice en la muestra. 7.4 Óxidos de hierro y aluminio. 7.4.1 Reactivos. 7.4.1.1 Cloruro de amonio pulverizado. 3 7.4.1.2 Solución de cloruro de amonio (20 g/dm ). Disolver 20 g de cloruro de amonio en agua y diluir a 1 dm 3 (1 litro). 3 3 7.4.1.3 Indicador rojo de metilo (1g/dm ). Disolver 0,1 g de rojo de metilo en agua y diluir a 100 cm . 7.4.1.4 Pirosulfato de potasio o de sodio. 7.4.2 Procedimiento. 7.4.2.1 Fundir el residuo de la determinación de sílice en el crisol, calentándolo lentamente con cerca de 2 g de pirosulfato de potasio o de sodio; enfriar, disolver en agua y acidular con tres o cuatro centímetros cúbicos de ácido clorhídrico. 3 7.4.2.2 Diluir de 150 a 200 cm , añadir 5 g de cloruro de amonio y 3 gotas de indicador rojo de metilo. 7.4.2.3 Calentar hasta ebullición, añadir hidróxido de amonio (1:1) lentamente, luego gotear hasta que la solución llegue a ser alcalina para el indicador. 7.4.2.4 Hervir por uno o dos minutos y filtrar a través de un papel filtro de 9 cm. No lavar. Reservar el filtrado. 3 7.4.2.5 Transferir el papel y el precipitado al vaso en el cual se hizo la precipitación, añadir 50 cm de ácido clorhídrico (1:4) y digerir el contenido del vaso cubierto sobre la placa caliente, agitando fuertemente, hasta que el precipitado se disuelva y el papel quede bien macerado. 7.4.2.6 Diluir, añadir rojo de metilo, volver a precipitar con hidróxido de amonio (1:1), hervir y filtrar exactamente como antes. Añadir este segundo filtrado al primero para la subsiguiente determinación de cal (ver 7.8). 7.4.2.7 Limpiar el vaso de precipitación y el agitador, y lavar el papel y el precipitado con cloruro de amonio (20 3 g/dm ). -4- 1980-0134 NTE INEN 576 1981-01 7.4.2.8 Secar y luego calcinar el papel y el precipitado hasta masa constante, en un crisol de platino tarado 3 de 30 cm de capacidad, a una temperatura de 1 000°C con acceso de aire. Enfriar en un desecador y pesar como grupo de óxidos de aluminio y de hierro (R2O3). 7.4.2.9 Para recobrar los restos de dióxido de silicio, que puedan haber permanecido en el grupo R2O3, aña3 dir 2 gotas de ácido sulfúrico (1:4) y 1 cm de ácido fluorhídrico al crisol y evaporar a sequedad. 7.4.2.10 Cubrir el crisol y calcinar a masa constante a 1 100°C. Enfriar y pesar. La pérdida en masa representa el dióxido de silicio adicional, el cual debe ser añadido al que se determinó en 7.3.1. 7.5 Oxido de hierro. 7.5.1 Reactivos. 3 7.5.1.1 Solución de sulfato de magneso. Disolver 70 g de sulfato de magneso cristalino en 500 cm de agua. 3 3 Añadir 140 cm de ácido fosfórico (densidad 1,7) y 130 cm de ácido sulfúrico. Diluir a 1 litro. 7.5.1.2 Solución de cloruro de mercurio. Preparar una solución saturada de cloruro mercúrico. 7.5.1.3 Solución normal (0,04 N) de permanganato de potasio. Disolver 2,5 g de permanganato de potasio en agua y diluir a 2 litros. Dejar en reposo por una semana, filtrar a través de una lámina de asbesto o vidrio poroso, o de un filtro de porcelana, y guardar en un lugar oscuro. 7.5.1.4 Pirosulfato de sodio o de potasio. 3 7.5.1.5 Solución de cloruro estannoso. Disolver 50 g de cloruro estannoso en 100 cm de ácido clorhídrico y diluir a 1 litro. Colocar unos pocos trozos de estaño en la botella. 7.5.2 Procedimiento. 7.5.2.1 Transferir 1,00 g de la muestra libre de humedad (de 105° a 110°C) a un crisol de platino de 30 cm 3 3 3 y humedecerlo con unas pocas gotas de agua. Añadir 3 cm de ácido nítrico y cerca de 15 cm de ácido fluorhídrico. Evaporar a un calor moderado hasta sequedad, teniendo cuidado de evitar cualquier pérdida por salpicadura. 7.5.2.2 Descomponer los nitratros y fluoruros por calcinación a 1 000°C durante 15 min. Añadir al resi- duo del crisol una pequeña cantidad de pirosulfato de sodio o de potasio y fundir. 7.5.2.3 3 Disolver el fundido en 25 cm de ácido clorhídrico (1:1) y calentar hasta la ebullición. Reducir el hierro por adición de Is solución de cloruro estannoso, haciéndola gotear de una pipeta, con agitación constante del vaso de precipitación, hasta que la solución sea incolora. Añadir luego una gota de exceso. 3 7.5.2.4 Enfriar rápidamente en agua corriente y añadir de un golpe 15 cm de solución saturada de cloruro mercúrico. Dejar en reposo por tres minutos y transferir con lavado a un vaso de precipitación de 1 litro, -5- 1980-0134 NTE INEN 576 1981-01 3 3 que contenga 300 cm de agua fría y 25 cm de solución de sulfato de magneso. Titular con permanganato de potasio (0,04 N), añadido muy lentamente con agitación constante, hasta que se obtenga un color rosado permanente como punto final. 7.6 Oxido de titanio (titania). 7.6.1 Reactivos. 7.6.1.1 Peróxido de hidrógeno (30%). Peróxido de hidrógeno concentrado. 7.6.1.2 Pirosulfato de potasio o de sodio. 7.6.1.3 Solución normal de titania. La solución normal de titania se preparará como se indica en el Apéndice Y. 7.6.2 Procedimiento. 7.6.2.1 Fundir el residuo R2O3 calcinado después de la recuperación del dióxido de silicio (ver el numeral 7.4.2), con una pequeña cantidad de pirosulfato de potasio o de sodio. 3 7.6.2.2 Enfriar y disolver el fundido, añadiendo alrededor de 25 cm de agua caliente y uno o dos centímetros cúbicos de ácido sulfúrico al crisol. Es importante mantener la solución en un pequeño volumen. 3 7.6.2.3 Transferir a un frasco volumétrico de 50 cm . Añadir unas pocas gotas de peróxido de hidrógeno y diluir hasta la marca. 3 7.6.2.4 Colocar los 50 cm de solución en un pequeño tubo Nessler. Comparar el color de esta solución con el color de una solución normal conocida. 3 7.6.2.5 Solución satisfactoria es aquella que tiene tal concentración que 1 cm equivale a 0,0001 g de óxido de titanio. 7.6.2.6 Para hacer la comparación, usar cualquier colorímetro normalizado o colocar una cantidad apropiada de solución normal en un segundo tubo Nessler y diluir con agua de una bureta, hasta que el color se equipare. 7.6.2.7 De la cantidad de agua añadida, calcular el porcentaje de óxido de titanio de la muestra. 7.7 Oxido de aluminio (alumina). 7.7.1 Procedimiento. 7.7.1.1 Sustraer la masa calculada de óxido de hierro (ver 7.5), la masa de sílice recuperada (ver 7.3) y la masa calculada de óxido de titanio (ver 7.6) de la masa total de R2O3 (ver 7.4.2.8). La diferencia restante es la masa de óxido de aluminio, con pequeñas cantidades de óxidos de otros elementos que se han precipitado. -6- 1980-0134 NTE INEN 576 1981-01 7.8 Oxido de calcio (cal). 7.8.1 Reactivos. 3 7.8.1.1 Solución saturada de oxalato de amonio. Disolver 4 g de oxalato de amonio en 100 cm de agua. 7.8.1.2 Solución normal (0,1 N) de permanganato de potasio. Disolver 3,25 g de permanganato de potasio en 1 litro de agua. Dejar en reposo por una semana, filtrar a través de una lámina de asbesto, vidrio poroso o filtro de porcelana y guardar en un lugar oscuro. 7.8.2 Procedimiento. 7.8.2.1 Acidular el filtrado combinado de la precipitación R2O3 con unos pocos centímetros cúbicos de 3 ácido clorhídrico (1:1) (ver 7.4.2.6) y evaporar a un volumen aproximado de 250 cm . 3 7.8.2.2 Calentar hasta que comience a hervir, añadir 25 cm de una solución saturada de oxalato de amonio e hidróxido de amonio hasta una ligera alcalinidad. 7.8.2.3 Agitar completamente y dejar precipitar, manteniendo la solución caliente por tres o cuatro horas, pero sin hervir. Filtrar y lavar cinco veces con agua fría. Reservar el filtrado. 3 7.8.2.4 Disolver el precipitado de oxalato lavado con 50 cm de ácido clorhídrico (1:1), diluir a cerca de 150 3 cm y precipitar como antes. 7.8.2.5 Lavar el vaso de precipitación, el precipitado y el papel con agua fría, hasta que se elimine el oxalato de amonio. Evitar el lavado excesivo. 7.8.2.6 Combinar los nitrados para la determinación de magnesia (ver 7.9.2.1). 3 7.8.2.7 Añadir 150 cm de ácido sulfúrico al vaso usado para la precipitación. Introducir luego el papel que contiene el precipitado. Calentar casi hasta ebullición y titular con permanganato de potasio (0,1 N). 7.8.2.8 Para efectos del uso del papel, puede hacerse previamente una determinación en blanco. 7.9 Oxido de magnesio (magnesia). 7.9.1 Reactivos. 7.9.1.1 Fosfato diamónico. 7.9.2 Procedimiento. 7.9.2.1 Añadir de dos a tres gramos de fosfato diamónico a los filtrados combinados de la determinación de la 3 cal (ver 7.8.2.6), los cuales deben reducirse previamente, por evaporación, a 300 cm . -7- 1980-0134 NTE INEN 576 1981-01 7.9.2.2 Agitar hasta que se disuelva y luego añadir lentamente hidróxido de amonio, hasta conseguir alcalinidad. 3 7.9.2.3 Añadir luego 30 cm de hidróxido de amonio en exceso. Dejar la solución en reposo, por lo menos 3 durante cuatro horas o hasta el día siguiente, si la solución tiene un volumen mayor de 400 cm . 7.9.2.4 Filtrar, lavar con hidróxido de amonio (1:19). Disolver el contenido del papel con ácido clorhídrico caliente (1:1) y volver a precipitar añadiendo 0,1 g de fosfato diamónico, diluyéndolo, volviéndolo amoniacal y dejándolo nuevamente en reposo. 7.9.2.5 Después de filtrar y lavar como antes, carbonizar y quemar el papel a 900°C, y luego calcinar en un horno eléctrico de mufla, de alrededor de 1 100°C, hasta obtener masa constante. El material calcinado consta de priofosfato de magnesio. 7.9.3 Cálculo. 7.9.3.1 Calcular la masa de magnesia multiplicando la masa del pirofosfato de magnesio por 0,3621. 7.10 Álcalis (método de J. Lawrence Smith). 7.10.1 Reactivos. 7.10.1.1 Carbonato de amonio. 7.10.1.2 Cloruro de amonio. 3 7.10.1.3 Oxalato de amonio (solución saturada). Disolver 4 g de oxalato de amonio en agua y diluir a 100 cm . 3 7.10.1.4 Oxalato de amonio (1 g/litro). Disolver 0,1 g de oxalato de amonio en agua y diluir a 100 cm . 7.10.1.5 Carbonato de calcio. 7.10.1.6 Solución ácido cloroplatínica (100 g Pt/litro). 7.10.1.7 Alcohol etílico (80%). Preparar una solución que contenga 80% en volumen de alcohol etílico en agua. 7.10.1.8 Alcohol etílico (absoluto). Ciertas marcas comerciales de alcohol absoluto desnaturalizado con satisfactorias y considerablemente menos costosas que el alcohol absoluto de grado reactivo. 7.10.2 Procedimiento. 7.10.2.1 Pesar 1,00 g de muestra libre de humedad (105° a 110ºC) (molida hasta un polvo impalpable) y 1 g de cloruro de amonio en un mortero de ágata y mezclar bien. -8- 1980-0134 NTE INEN 576 1981-01 7.10.2.2 Añadir de 7 a 8 g de carbonato de calcio y volver a mezclar íntimamente. Colocar una capa de carbonato de calcio de 3 mm de espesor en el fondo de un crisol de platino. Añadir luego la mezcla anterior, golpeando ligeramente el crisol, para obtener una masa compacta. Colocar otra capa de carbonato de calcio de 3 mm de espesor en la parte superior. 7.10.2.3 Calentar el crisol sobre una llama baja, hasta que dejen de emitirse vapores de amonio. Aumentar entonces el calor de tal manera que la mitad inferior del crisol llegue al rojo sombra y mantener esta temperatura por una hora. 7.10.2.4 Enfriar y llenar las tres cuartas partes del crisol con agua. Calentar hasta que el contenido pueda ser extraído en una sola pieza y triturar en un mortero de ágata. 7.10.2.5 Transferir el material a un plato de platino o porcelana por medio de un chorro de agua. Evaporar hasta obtener un bajo volumen, decantar a través de un papel filtro denso y lavar el material en el plato varias veces por decantación con agua tibia. 7.10.2.6 Transferir al papel y lavar varias veces con agua caliente. Acidular con algunos centímetros cúbicos 3 de ácido clorhídrico y evaporar a un volumen de 150 a 200 cm . 7.10.2.7 Añadir unos pocos centímetros cúbicos de hidróxido de amonio y carbonato de amonio suficiente para precipitar la cal, manteniendo el plato cubierto con un vidrio de reloj. Calentar hasta que el precipitado se separe. Filtrar y evaporar con agua tibia. 7.10.2.8 Evaporar la solución a bajo volumen. Añadir luego un pequeño trozo de carbonato de amonio, para determinar si se ha precipitado todo el calcio. Si no se forma más precipitado, evaporar a sequedad; de otro modo, precipitar y filtrar como antes. Eliminar las sales de amonio calentando hasta cerca del rojo sombra. 7.10.2.9 Disolver el residuo en agua y añadir unos pocos centímetros cúbicos de solución saturada de oxalato de amonio y uno o dos centímetros cúbicos de hidróxido de amonio para precipitar el último rastro de calcio. 7.10.2.10 Calentar de 30 a 45 min, filtrar y lavar con agua que contenga oxalato de amonio (1 g/litro). Colocar el filtrado en un plato de platino tarado. Añadir varias gotas de ácido clorhídrico y evaporar a sequedad. Calcinar lentamente como antes y pesar como cloruro de sodio y cloruro de potasio. 7.10.2.11 La separación del sodio y del potasio debe llevarse a cabo en una atmósfera libre de vapores de amoníaco. Añadir a la solución de los cloruros, combinados en un pequeño plato de porcelana, suficiente solución acida cloroplatínica, para reaccionar con la totalidad del sodio y del potasio. 7.10.2.12 La cantidad necesaria para el uso puede calcularse fácilmente a partir de la concentración conocía de la solución de platino y la masa de cloruros mezclados, contados como cloruro de sodio. 7.10.2.13 La dilución de la solución resultante debe ser tal, que, cuando sea calentada en baño María, cualquier precipitado que se haya formado se disuelva totalmente. De esta manera se puede evitar la inclusión de licor madre en una masa de cristales repentinamente formados. -9- 1980-0134 NTE INEN 576 1981-01 7.10.2.14 Evaporar hasta que la solución llegue a estar suficientemente espesa para solidificarse por enfriamiento. No evaporar a sequedad, por cuanto pueden deshidratarse las sales de sodio y volverse menos solubles en el alcohol. 7.10.2.15 Humedecer el residuo con alcohol etílico (al 80%), filtrar por decantación a través de un papel pequeño, lavar por decantación con más alcohol, triturar con un pequeño majador o una varilla de vidrio. 7.10.2.16 Reservar el filtrado y los lavados, si se va a determinar directamente el sodio. El residuo debe ser amarillo dorado. El color anaranjado-rojizo indica una separación incompleta de la sal de sodio. No es necesario colocar la masa del precipitado sobre el filtro. 7.10.2.17 Secar el plato y el papel por un momento para retirar el alcohol adherido. Disolver el precipitado en el filtro con agua caliente, colocando la solución en un crisol tarado o pequeño plato de platino. 7.10.2.18 Evaporar a sequedad y añadir la sal que esté todavía en el plato de porcelana. Si la sal se ha solidificado totalmente, volver a disolverla en agua y volver a evaporar a sequedad. 7.10.2.19 Calentar por una hora a 130°C en un horno de aire (100°C son suficientes, en caso de una cantidad muy pequeña de precipitado de grano fino). Es necesario cubrir el recipiente desde un principio, por cuanto el precipitado es propenso a crepitar. Cuando se seque, enfriar y pesar como cloroplatinato de potasio. 7.10.3 Cálculo. 7.10.3.1 Calcular la masa de los óxidos de acuerdo a las siguientes fórmulas: KCI = masa de K2 Pt Cl6 x 0,3068 K2 O = masa de K2 Pt Cl6 x 0,1941 NaCI = cloruros totales — K Cl Na2O = Na Cl x 0,5303 -10- 1980-0134 NTE INEN 576 1981-01 APENDICE Y Y.1 PREPARACIÓN DE LA SOLUCIÓN NORMAL DE TITANIA Y.1.1 Pesar 0,05 g de óxido de titanio calcinado. Fundir con 10 g de pirosulfato de potasio en un crisol de platino limpio, manteniendo la temperatura más baja posible para conservar la fluidez. 3 Y.1.2 Enfriar y disolver en cerda de 300 cm de ácido sulfúrico (1:5). Enfriar, transferir a un frasco volumétrico 3 de 500 cm , diluir con agua hasta la marca y mezclar completamente. 3 Y.1.3 Para normalizar la solución, tomar dos porciones de 50 cm , en dos vasos de precipitación de 400 3 cm . Diluir, hervir y precipitar con hidróxido de amonio. 3 Y.1.4 Filtrar y lavar con agua caliente. Colocar los papeles en los vasos originales, añadiendo 15 cm de ácido clorhídrico. Agitar hasta macerar el papel, diluir y precipitar de nuevo con hidróxido de amonio. Y.1.5 Filtrar y lavar con agua caliente, hasta eliminar las sales alcalinas. Calcinar cuidadosamente y pesar. Y.1.6 De acuerdo a la masa determinada, calcular la concentración de la solución. -11- 1980-0134 NTE INEN 576 1981-01 APENDICE Z Z.1 NORMAS A CONSULTAR INEN 606 Materiales refractarios. Muestreo, inspección y recepción. Z.2 BASES DE ESTUDIO Norma ASTM C 575-70. Standard methods for Chemical Analysis of Silica refractories. American Society Testing and Materials. Filadelfia, 1970. -12- for 1980-0134 INFORMACIÓN COMPLEMENTARIA NTE INEN 576