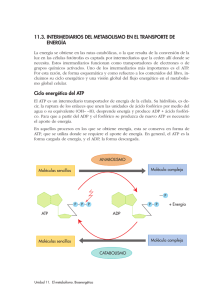
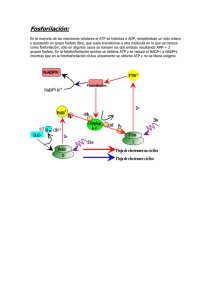
LA BOMBA CARDIACA: CONTRACTILIDAD Y FACTORES
Anuncio

INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 CAPITULO 8 LA BOMBA CARDIACA: CONTRACTILIDAD Y FACTORES METABOLICOS EN LA FISIOPATOLOGÍA DE LA INSUFICIENCIA CARDIACA. La función de la bomba cardíaca depende de[1] : 1) La Precarga; 2) La Poscarga, 3) El estado contráctil que representa las características del desempeño del músculo independiente de las distintas contractilidad condiciones de cargas. 4) La frecuencia cardíaca (FC). En la determinación del volumen sistólico (VS) intervienen la pre- y poscarga, y la contractilidad. El volumen sistólico es directamente proporcional a la PRECARGA ---STARLING VOLUMEN SISTÓLICO POSCARGA precarga e inversamente proporcional a la poscarga. Figura 8-1 Figura 8-1. Factores que determinan el VS Precarga Es la fuerza por unidad de superficie que va a elongar en diástole al músculo cardíaco. Para calcular su valor se han propuesto a distintas variables que podrían ser consideradas como representativas de precarga, tales como la presión de fin de diástole (PFD); presión de llenado ventricular; estrés de fin de diástole(EFD); diámetro de fin de diástole (DFD); o volumen de fin de diástole (VFD). Cada uno de estas estimaciones presenta importantes limitaciones. Para algunos la precarga es el grado de estiramiento del sarcómero existente al final de la diástole (interviene la dimensión o el volumen), mientras que para otros es la fuerza que determina tal estiramiento (concepto de fuerza, Figura 8-2. Efecto de poscarga sobre acortamiento[43]. tensión o stress). Por ejemplo, en el caso de hipertrofia ventricular con disminución de la complianza de cámara la precarga estaría reducida –dado a que hay menor dimensión - aunque la presión de llenado esté aumentada. De allí que la PFD y el EFD pueden usarse como indicadores de precarga sólo si se conoce que la complianza no ha cambiado[2]. 163 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 Por ejemplo si la PFD aumenta de 15 a 20 mms de Hg es probable que la precarga haya aumentado salvo que haya habido reducción de la complianza. El VFD y el DFD dependen en parte del estiramiento del sarcómero: a medida que aumenta el estiramiento también aumentará el VFD y el DFD. Pero en el caso de hipertrofia ventricular excéntrica, un número mayor de sarcómeros estirados en un menor grado producen la misma dimensión diastólica que un número inferior de sarcómeros estirados en mayor grado. O sea que el VFD no es una medida exacta de la precarga, aunque puede ser usado como indicador. Si el DFD se aumenta agudamente es seguro que la precarga ha aumentado. Si ha habido una sobrecarga de volumen y se observa el DFD 3 meses después de la sobrecarga de volumen, la dimensión aumentada puede deberse a precarga aumentada, hipertrofia excéntrica o a ambas. Para Carabello[2] el estiramiento del sarcómero es probablemente la mejor definición de precarga porque es un determinante clave de la función sistólica. De acuerdo con la Ley de Laplace, cuanto mayor sea el radio de la cavidad mayor será la precarga. El volumen puede aumentar al doble de su valor, pero eso implica sólo un aumento del 26% del radio y de la tensión de pared. Kass[3] también considera que la dimensión de fin de diástole indica el grado de precarga (es decir que influye mas el largo del sarcómero que la tensión requerida para obtener tal longitud) y por ello señala que el VFD y no la PFD es la mejor medida de precarga. Es el estiramiento de la cámara o su volumen el que determina primariamente el desempeño cardiaco. Opie[4] estima que la precarga puede definirse como el estrés de pared al final de la diástole (y entonces al máximo largo en reposo del sarcómero). Medidas sustitutivas de precarga serían la PFD o DFD. Para Katz[5] la precarga está determinada por el retorno venoso que llena el ventriculo al final de la diástole y por las propiedades lusitropicas del ventrículo, coincidiendo entonces con Carabello y Kass. En definitiva, la mejor aproximación como medida de la precarga está dada por el VFD, Poscarga Es la carga que el músculo enfrenta en la sístole, que genera un estrés sobre la unidad de superficie; la poscarga generalmente es medida Figura 8-3. A la izquierda IC moderada y a la derecha severa, efectos de estrés presor. Cuando hay reserva de precarga (izquierda) ante un estrés pasa de A a B sin disminuir acortamiento, y solo disminuye este con mayor aumento de estrés. En disfunción severa cualquier estrés presor disminuye el acortamiento. (Tomado de Dell’Italia[43]) al final de la sístole.Puede definirse poscarga como la presión intraventricular suficiente para abrir la válvula aórtica permitiendo la eyección del contenido ventricular; o la carga contra la que el ventrículo se contrae o dicho de otra forma el estrés de pared durante la eyección ventricular[4] El cálculo del estrés de pared se hace por la Ecuación de Laplace que establece: 164 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 σ = P.r / 2 h donde σ es estrés de pared, P es presión intraventricular, r radio de la cavidad ventricular y h el espesor de la pared ventricular. Puede calcularse el estrés en cualquier instante de la eyección, pero habitualmente se toma el fín de sístole. Como puede verse, al considerar poscarga equivalente a estrés sistólico, se pone de manifiesto una interrelación e interdependencia entre precarga y poscarga. Puede verse en la Figura 8-2 y 8-3 como el aumento de poscarga disminuye el acortamiento, en el corazón normal y en el insuficiente La impedancia aórtica podría ser considerada como expresión de la poscarga. Pero consta de dos componentes que son la resistencia periférica y la impedancia sistólica que es la relación instantánea entre la presión y el flujo o caudal durante la eyección. En la impedancia intervienen la elasticidad arterial, la inercia de la sangre y la reflexión de ondas, que se calculan a partir de un análisis de Fourier de sus componentes, separados como armónicas. De los dos componentes mucho más importante es la resistencia periférica, aunque el otro no debe ser olvidado. La resistencia periférica es una aproximación poco exacta de evaluación de la poscarga del ventrículo izquierdo[6] . El estado contráctil. Regulación de la contractilidad Tres mecanismos intervienen para regular la fuerza [1,2,6] contráctil cardíaca Figura 8-4. Ley de Starling (Relación entre Tensión y Longitud) en el sarcómero : 1) Ley de Frank-Starling, que establece que cuando más se estira el músculo mayor será la fuerza contráctil; 2) la fuerza contráctil dependiente de la FC (fenómeno de la escalera o treppe, o de Bowditch) y 3) las propiedades intrínsecas del músculo cardíaco, que además está bajo control neurohumoral (incluyendo SNS, Ang II y endotelína). 1) Ley de Starling. Starling en 1918, basándose en estudios propios y de investigadores que le precedieron, estableció que cuando mas grande es el volumen del corazón, mayor es la energía de su contracción y la cantidad de cambios químicos de cada contracción. Dadas las dificultades para determinar el volumen cardíaco - aún usando ecocardiografía o cardiometría de impedancia – para obtener la presión de llenado ventricular izquierdo se calcula la diferencia entre la presión auricular izquierda y la presión diastólica de ventrículo izquierdo: cuando aumenta la presión de llenado también lo hace la precarga y el desempeño del ventrículo. Frank había establecido en 1895 un principio parecido: cuando el corazón se llena con distintos volúmenes siendo cada uno mayor que el anterior, la estimulación en cada caso generará contracciones (isométricas contra válvulas cerradas experimentalmente) que producen sendas curvas de presión intraventricular creciente con un porción inicial ascendente y luego una descendente. Figura 8-4. 2) La fuerza contráctil dependiente de la FC (fenómeno de la escalera o treppe, o de Bowditch) . Un aumento de la FC incrementa progresivamente la fuerza de la contracción, 165 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 mientras que la disminución produce efecto inverso. Es el llamado Fenómeno de la Escalera + ++ (treppe, en alemán) o de Bowditch. Probablemente se deba a una mayor entrada de Na y Ca en + + las células que al rebasar la capacidad de la bomba Na -K ATPasa, incentivan el intercambio + ++ ++ reverso Na /Ca provocando mayor cantidad de Ca intracelular y mayor contractilidad. Figuras 8-5 y 8-6. 3) El estado contráctil. Depende de las condiciones del músculo en sí, sin la influencia de precarga o poscarga. Si se aumenta la contractilidad manteniendo constante la precarga, la contracción isométrica se altera. Con las catecolaminas hay aumento muy importante de la derivada de la fuerza desarrollada en función del tiempo (df/dt) ++ y de la fuerza pico. Pasa lo mismo agregando Ca al líquido [1] de perfusión . Figura 8-6. El aumento súbito de El proceso de la contracción Recordemos el papel de las catecolaminas y del la frecuencia cardiaca produce un latido de menor amplitud (fuerza) y luego un aumento en escalera de la misma (Tomado de Cingolani H, Fisiología Humana, 7ª.Edición, con mecanismo de transducción en el proceso contráctil, que hemos analizado en el Capítulo 3: El neurotransmisor se liga al receptor adrenérgico y luego se acopla a la adenilciclasa por medio de la proteína G estimulante (Gs), actuando sobre el ATP y llevándolo a AMPc, quien a su vez activa a la proteínquinasa A (PKA), quien va a fosforilar: a) canales de calcio; b) receptores ryanodínicos (RyR2) y proteínas que intervienen en el metabolismo normal (PP1-PPA2, FKBP) ; c) proteínas contráctiles (troponina); d) fosfolamban; y e) enzimas metabólicas. Figura 8-6 Figura 8-5. Aumento de la fuerza contráctil tras el aumento de la frecuencia, en nomal (círculos abiertos) y en IC. (Tomado de Cingolani H, Fisiología Humana, 7ª.Edición, con permiso) Intervención de los receptores beta-adrenérgicos (GPCR) En la IC se produce hiperactividad simpática como mecanismo compensador. Pero el exceso de producción de catecolaminas implica daño miocárdico por toxicidad. El organismo adopta medidas para evitar tal toxicidad, pero ellas producen alteraciones de las vías de señalamiento que van a perturbar a la capacidad contráctil. Aún no se sabe si las anormalidades del señalamiento adrenérgico son un mecanismo de adaptación para prevenir el exceso de estimulación o son cambios que implican una desadaptación que deprime la reserva contráctil e inician la descompensación y contribuyen a la progresión[7]. ACTUALIZACIÓN 12/12/2005 La disfunción miocárdica se debe a insuficiencia de por lo menos uno de tres sistemas: excitación-acoplamiento, mecanismos de reserva contráctil, y maquinería contráctil 166 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 contenida en el sarcómero. Dentro de las anormalidades de excitación-acoplamiento están cambios en la duración del potencial de acción, alteración de la SERCA2a por reducción de niveles de proteinas y de fosfolamban, regulación variables del inercambiador Na+/Ca++, y alteraciones del RyR2 y proteínas asociadas. Hay disminución de reserva contráctil por alteración del señalameinto adrenérgico y apagamiento de la relación fuerza/frecuencia. LeWinter MM, vanBuren P.: Sarcomeric proteins in hypertrophied and failing myocardium: An overview. Heart Fail Rev 2005;10:173-74 Uno de los mecanismos usados en el intento de adaptación de los pacientes con IC es la desensibilización de los receptores adrenérgicos, en el intento de evitar la toxicidad de los excesos de catecolaminas. Recordemos que la estimulación del β1AR es proapoptótica, mientras que el β2AR protege de la apoptosis. La desensibilización sea por disminución de la densidad de receptores o por la internalización de los mismos limita el desgaste energético, la remodelación ventricular y la muerte celular que se asocia a la hiperactividad catecolaminérgica. Intervienen para ello las kinasas de los receptores acoplados a las proteínas G (GPCR) denominadas GRKs, de las cuales la más importante es la βARK1, quien actuando concertadamente con la β-arrestina, fosforila a los receptores beta-adrenérgicos (βAR), inactivándolos. El corazón insuficiente regula hacia arriba a la βARK1, con lo cual se logra desensibilización de los receptores y reducción de la contractilidad. Como contrapartida la expresión del inhibidor de la βARK1, el péptido βARKct (ct = carboxy-terminal), aumenta la contractilidad al permitir la estimulación del βAR[8]. Otros mecanismos puestos en juego son la regulación hacia arriba de la proteína Gαi, y el aumento de la relación β2/β1, que llevan a un menor acoplamiento con la adenilciclasa y por ende menor formación de AMPc. Aparte de las diferencias funcionales entre los receptores β1 y β2 (Capítulo 3) cuando se usan técnicas de transferencia de genes de cada subtipo y se provoca su sobreexpresión en el corazón de rata se producen fenotipos diferentes. Con una sobreexpresión del receptor β2 de 60 veces más de lo normal se observa aumento de la contractilidad sin consecuencias perjudiciales, necesitando llegar a 350 veces mas para producir modificaciones patológicas, mientras que una sobrexpresión del β1 de 5-30 veces mas produce patología extracelular intracelular [9-11] significativa . Estos datos marcan las importantes βARK diferencias funcionales entre los dos subtipos. Mas aún podría decirse que aumentando la contractilidad por moderada sobreexpresión del β2 puede mejorarse alguna AdenilAdenilα forma experimental de miocardiopatía en el ratón – con β1 ciclasa disfunción e hipertrofia - vinculada a una sobreexpresión β γ [11] βAR de Gαq . Una de las formas de aumentar la contractilidad Proteina G previniendo la disfunción es precisamente administrar el ATP AMPc ya mencionado βARKct en dos formas de miocardiopatías experimentales: en la por disrupción de la proteína lim (MLP), o en la por sobreexpresion de calsecuestrín., lográndose en ambos casos mejoría estructural y [11-13] funcional . Figura 8-6. Estímulo β-1, proteína G y adenilciclasa. AMPc y PKA Papel de los bloqueantes beta-adrenérgicos Los bloqueantes beta-adrenérgicos (metoprolol, bisoprolol, carvedilol, nebivolol) reducen la conducción simpática y de allí la contractilidad, pero el efecto inotrópico negativo es en el corto plazo mientras que a la larga mejoran la función ventricular y la remodelación. Se supone que estas drogas revierten parcialmente la desensibilización y mejoran la contractilidad al aumentar la respuesta de los βAR dentro de ciertos definidos parámetros. Pero también puede suponerse que el tratamiento en sus momentos iniciales descarga al miocardio por un cierto tiempo, promoviendo 167 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 la reversión de la remodelación y mejorando el desempeño sistólico, tal como se ve con los implementos de asistencia ventricular mecánica o con los IECA[11]. Ciclo y canales de calcio. Los canales de Ca++ fosforilados se abren una vez que la corriente despolarizante alcanza el voltaje umbral debido, penetrando Ca++ en la célula en función de un gradiente de concentración. Las concentraciones respectivas de Ca++ son :10-3 mol/lt extracelular; 10-6 mol/lt en la sístole; 10-7 mol/lt en diástole; 10-3 mol/lt en el Retículo Sarcoplásmico. Según Opie y Bers[14] cada canal de Ca++ tiene un muy breve período de apertura que dura ~ 0,2 milisegundos, y entre la apertura de esos canales y el inicio del incremento del Ca++ cistosólico transcurren ~ 4 milisegundos; la concentración pico de Ca++ citosólico se alcanza ~ a los 40-100 mseg con una FC de 60 latidos por minuto, y la contracción pico se produce 200 mseg después; 200 mseg más tarde la relajación es casi completa, y la recuperación del Ca++ citosólico y del ciclo contráctil toma ~ 300 mseg. En total, la mitad del intervalo entre latidos es ocupado por el ciclo contracción-relajación ++ Los canales de Ca tipo L o canales dihidropiridínicos dependientes del voltaje se abren en la despolarización permitiendo la entrada de una pequeña cantidad de Ca++(corriente de entrada de calcio, ICa), que es insuficiente como para iniciar una contracción, pero que provoca la descarga masiva del Ca++ contenido en el Retículo Sarcoplásmico (RS). Esta descarga del Ca++ es posibilitada por la apertura de los receptores ryanodínicos (RyRs), que son miembros de una familia de canales de liberación de Ca++ que se encuentran en el RS, que tienen 3 isoformas, siendo la cardiaca la RyR2. El proceso de liberación del Ca++ del RS por el estímulo producido por el mismo ión al penetrar a través de los canales L ha sido denominado Calcium Induced Calcium Release (CICR). El calcio que penetra en la célula por los canales L de los túbulos T lo hace en forma de “sparks” (chispazos) que van a ser sensadas por los RyR2, que se encuentran en una formación especial del RS. (que establece una relación estrecha con el túbulo T y canales L), llamada pie. Dos genes distintos codifican a los receptores ryanodínicos específicos cardíacos (RyR2) y del músculo [15] + esquelético (RyR1) . Estos canales son aproximadamente 10 veces más grandes que los canales de Na y de ++ ++ [16] Ca , y su apertura aumenta el contenido de Ca del citosol de aproximadamente 100 nmol/lt a 1µmol/lt . El RyR es un complejo macromolecular que incluye a la PKA, a las fosfatasas PP1 y PP2A, la sorcina, el calmodulín y la [15-19] proteína FKBP12 , también conocida como castalbín-2. Las FKBPs son miembros de una familia que ligan a las drogas inmunosupresoras FK506 y rapamicina, y que consta de más de 20 miembros (8 de ellos en los mamíferos). FKBP12 es importante para regular la coordinación de los canales ryanodínicos, debiendo ser fosforilada por la PP1/PPA2. Los RyR2 son múltiples y poseen portones o puertas que deben abrirse simultáneamente (apertura acoplada de portones), sincronizadas por la FKBP12. Para su apertura son fosforilados por la ProteínKinasa A. La hiperfosforilación - como puede suceder en la IC por la hiperactividad simpática con exceso de PKA - disocia a la FKBP12, reduce la ganancia de la excitación/contracción y promueve filtraciones diastólicas de Ca++ y puede ser la causa de pospotenciales tardíos y arritmias fatales[17]. Figura 8-7 168 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 ACTUALIZACIÓN 20/08/05 El receptor ryanodínico es una proteína de 565 kDa con 4.967 aa, codificada por el gen RyR2 del cromosma l(lq42-q43). Es un tetrámero o sea compuesto por 4 monómeros, compuesto cada uno por la terminal carboxilo, que es la porción más importante, dado que controla la localización del RyR2 y forma un canal funcional de liberación de Ca++. La segunda porción constituye 9/10 de la proteina total, y tiene funciones regulatorias. Priori S, Napolitano C.: Cardiac and skeletal muscle disorders caused by mutations in the intracellular Ca2+ release channels. J Clin Invest 2005;115:2033-38 ACTUALIZACION 12/01/2006 Los autores encontraron que los bloqueantes beta-adrenérgicos y un nuevo agente antioxidante llamado edaravone corrigen el defectuoso control de los RyR2 realizado por la FKBP12.6, mejorando la función cardiaca durante el desarrollo de la insuficiencia cardiaca. Se ha determinado que la actividad de canal del RyR está regulada por oxidación o nitrosilación. Yano M, Okuda S, Oda T, et al.: Correction of defective interdomain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation 2005;112:3633-43 La fosforilación de las proteínas regulatorias de los filamentos delgados se hace por medio de las proteinkinasas A (PKA) y C (PKC), para modular el desempeño contráctil miocárdico: la PKA fosforila ATP CITOSOL Gs Æ AC a la troponina I (TnI), disminuyendo su sensibilidad β1 tensional para el calcio, mientras que la PKC Fosfolamb. AMPc deprime la ATPasa y la fuerza de la fibra muscular en la activación máxima del calcio a través de la PKA fosforilación de la TnI y troponina T (TnT). La SERCA Ca++ reducción de la ATPasa miofibrilar en el miocardio S100A1 insuficiente está mediada por la fosforilación ++ Ca CICR dependiente de la PKC de la TnI y la TnT. La CANALES DHP función del filamento delgado se restituye a casi lo ++ Ca++ normal después de asistencia ventricular RyR2 [20] mecánica. troponina FKBP12.6 Ca El Ca++ que ha penetrado en el citosol proveniente del RS, va a activar a la troponina C que a su vez desactiva la inhibición de la troponina I, con lo cual la ACTINA MIOSINA ext PP1-PPA2 RS int Figura 8-7. PKA. “Ciclo” de Ca++. CICR. Receptores ryanodínicos. FKBP12.6 SERCA2. Fosfolamban. Flechas que salen de PKA indican fosforilación. S100A1, modula SERCA2a. PP1-PPA2 modula FKBP troponina T se desacopla de la tropomiosina ubicada en la actina, y la tropomiosina se desplaza dejando libres los puntos de enganche de las cabezas de miosina. La fosforilación de la troponina por la PKA disminuye su sensibilidad al Ca++. El comportamiento de la cabeza de miosina es regulado por una enzima que ha sido activada por el complejo Ca++-calmodulina (CaM). La mayor disponibilidad de Ca++ para las proteínas contráctiles aumenta la fuerza contráctil del miocito y también se facilita la relajación por fosforilación de la troponina I y del fosfolamban. La CaM es una proteína de 16.700 dalton activada, que produce la inactivación de la corriente de entrada de Ca++. También, a través de la proteín kinasa dependiente de la calmodulina llamada calmodulina-kinasa II (CaMK II) fosforila al fosfolambán, aumentando la recaptación de Ca++ por el 169 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 RS. O sea que contribuye a la regulación del flujo del Ca++[14]. Durante el proceso de liberación de Ca++ aumenta el ión en el citosol mientras disminuye en el RS. Hay dos tipos de mecanismos ligados a la terminación de la liberación del ión: 1) La ligadura del Ca++ a sitios de inhibición en los RyR reduce la activación de los canales a través de procesos referidos como “inactivación dependiente del calcio” y 2) disociación del ión de sitios de activación luminal que disminuye la probabilidad de RyR2 abiertos por un proceso que puede ser denominado “desactivación luminal dependente del calcio”. O sea que la terminación del “spark” de Ca++ está gobernada por el Ca++ luminal, más que por la pérdida del ión por el RS[18]. ACTUALIZACIÓN 30/11/05 El filamento grueso que emerge de la línea M está constituido por miosina, titina y proteínaC ligante de miosina. La miosina está constituida por las proteínas miosina de cadena pesada, esencial de cadena liviana y regulatoria de cadena liviana. La elastancia no dependiente de los puentes cruzados está determinada por las porciones más complacientes de titina, la que se extiende entre el filamento grueso y la línea Z. La elastancia que depende de los puentes cruzados está mayormente influenciada por la porciones complacientes de lo molécula de miosina que no están incorporadas en el esqueleto del filamento grueso, o sea la cabeza y las regiones del cuello de la miosina de cadena pesada, incluyendo a las miosinas de cadena liviana esencial y regulatoria. El puente de actina/miosina se forma luego de la hidrólisis del ATP por la miosina y tiene la capacidad de trasladar la energía química del ATP a la energía mecánica del acortamiento del sarcómero. Las dos isoformas de miosina más prevalentes en el miocardio humano son la V-1 y la V-3, constituidas por miosina-α de cadena pesada y miosina-β de cadena pesada, respectivamente. En la IC la miosina-α de cadena pesada es reemplazada por miosina-β de cadena pesada para preservar la producción de fuerza conservando la utilización de ATP aunque en detrimento de la velocidad de acortamiento y la relajación dependiente del miofilamento; hay además regulación hacia arriba de la miosina esencial de cadena liviana auricular que aumenta la velocidad de acortamiento recobrando potencia durante la sístole, pero no compensando la disminución de la relajación. Hay un aumento de la sensibilidad del miofilamento al calcio, que también lleva a disminución de la relajación. Palmer BM. Thick filament proteins and performance in human heart failure. Heart Failure Rev 2005;10:187-97 Otro mecanismo fisiológico es la aceleración de la relajación dependiente de la frecuencia (ARDF, en inglés FDAR), por el cual el llenado diastólico es más rápido a frecuencias más altas. DeSantiago, Maier y Bers han comprobado que el mecanismo es dependiente de la CaMK II, sin intervención del fosfolamban[21]. Luego de la contracción el Ca++ es recaptado por el RS, pero también una fracción significativa es enviada al exterior celular por acción del intercambio Na+/Ca++. La cantidad de Ca++ que sale por acción del intercambiador es igual a la cantidad del catión que penetra por los canales de 170 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 Ca++[19]. Cuatro transportadores sacan al calcio del citosol[22]: (1) SERCA2-ATPasa, (2) NCX del sarcolema, (3) Ca++-ATPasa, y (4) Uniportador de Ca++mitocondrial. La SERCA2 y el NCX son cuantitativamente los más importantes. Bomba de Ca++ (SERCA) del RS. Fosfolamban La bomba de Ca++ del RS, es la Ca++-ATPasa, denominada SERCA (Sarco Endoplasmic Reticulum Calcium) de la cual existen los genes 1, 2 y 3 quienes dan lugar a diversas isoformas. La isoforma SERCA2a es la que se expresa en el tejido cardiaco. El RS forma las cisternas, que son dilataciones en relación de estrecha vecindad con los túbulos T. El Ca++ se almacena interviniendo las en proteínas la cisterna, calreticulín, calsecuestrin, la proteína ligadora rica en histidina y una proteína [23] “junctate”, para retenerlo cardiacas hay denominada . En las células exclusivamente RyR2, mientras que en las musculares lisas vasculares existen además los receptores de los canales de inositol-3-fosfato. Figura 8-8 Entrada/salida de calcio en la célula, por distintas vías: 1)En la membrana celular, con la bomba de sodio, el intercambiador Na+/Ca++ y la Ca++ ATPasa.. 2) Los canales de calcio de la membrana; 3) los canales del RS. También intercambio Na+/Ca++ en la mitocondria. Como el Ca++ libre proveniente del RS va a actuar sobre la troponina y por su intermedio con la actina/miosina. (Esquema tomado de Opie LH, The Heart. Lippincott-Raven.1998, modificado El Ca++ sale del RS y entra en el citosol para iniciar la contracción, pero luego debe ser retirado para que se produzca la relajación. El 7080% del Ca++ es retirado por la SERCA2a, mientras que el resto es enviado al espacio extracelular principalmente por el intercambiador Na+/Ca++ y accesoriamente por sistemas lentos de transporte de Ca++. Figura 8-8 [24,25] ++ Según Netticadan y col. dado que el RS regula la concentración intracelular del Ca , se pueden explicar los cambios en la contractilidad por cambios en la función del RS. La CaMK y la PKA (dependiente del AMPc) están involucradas en la regulación de la función contráctil a través de la fosforilación de distintas proteínas, explicándose así que un defecto en estos mecanismos regulatorios sea de importancia en el desarrollo de disfunción del RS. La fosforilación y desfosforilación juegan un papel crítico en la regulación de variados procesos celulares. Un defecto en la fosforilación de proteínas del SR por la CaMK del RS puede ser parcialmente responsable de la disfunción del RS en el caso de isquemia/reperfusión. Los autores examinaron el estado de la ++ fosforilación - por medio de la CaMK - de las proteínas del ciclado de Ca del RS, asi como del RyR2, SERCA2a, y el fosfolamban en corazones de un grupo control y de pacientes en IC. Los resultados indicaron que la fosforilacion de la proteínas del ciclado por medio del CaMK endógeno está deprimida en la IC debida a IAM. La actividad de SERCA2a en relación a la de CaM se encontró deprimida en casos de IC consecutiva a IAM. La SERCA2a es a su vez modulada por la fosfoproteína fosfolamban o por fosforilación directa por medio de la PK II, dependiente de CaMK II[18,21-25]. El fosfolamban cuando está desfosforilado inhibe a la SERCA2a; cuando la PKA lo fosforila cesa la inhibición Según Koss[26], la proporción entre las cantidades de fosfolamban y SERCA2a es crítica para la regulación de la contractilidad miocárdica, y alteraciones de esa relación pueden contribuir al deterioro funcional observable en la IC. Experimentalmente se ha intentado cambiar la cantidad relativa de fosfolamban en relación a SERCA2a en IC experimental en ratas, con la hipótesis que cambios en la cantidad de fosfolamban (en proporción a la de SERCA2a) puedan ser responsables de las anormalidades de 171 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 manejo del Ca++ observados en el miocardio insuficiente. De esta forma administraron por medio [26] de adenovirus gen de fosfolamban, buscando crear una sobreexpresión del mismo . Los resultados que obtuvieron sugirieron que las alteraciones de la relación entre fosfolamban y ++ SERCA2a pueden considerarse como responsables de las anormalidades del manejo del Ca observable en la IC, y que la sobreexpresión de SERCA2a puede corregir ampliamente esas anormalidades. ++ La recaptación de Ca por el RS está reducida en la IC. Esto se ha visto en distintos modelos [27] experimentales y en la miocardiopatía dilatada humana . Hasenfuss y col. [28] encontraron que los niveles proteicos de la SERCA2a estaban reducidos en un 36% (p = 0,02) en corazones en insuficiencia comparados con otros sin insuficiencia. Los niveles de SERCA2a se correlacionaron ++ con la captación de Ca . Se ha sugerido que las anormalidades de los segundos mensajeros contribuyen a los cambios celulares durante IC. Los segundos mensajeros mas importantes son el AMPc, el IP3 y el DAG. Marks [17,18,29] señala que en la IC terminal se observa regulación hacia abajo de los canales ryanodínicos del RS asi como regulación hacia arriba de los canales de IP3. Quedaría demostrado entonces que en el miocardio insuficiente está prolongado el ciclo de Ca++ y disminuida su recaptación por el RS. La S100A1 es una proteína que ha sido encontrada aumentada en la HVI compensatoria y significativamente [30] ++ regulada hacia abajo en la IC terminal . Aumenta la contractilidad al aumentar el flujo de Ca por modulación de la SERCA2a. Excitación-contracción En la regulación de excitación-contracción y de repolarización-relajación participan fundamentalmente cuatro sitios o zonas celulares, que producen efectos sobre la disponibilidad del ++ Ca o la respuesta celular, cuales son : el sarcolema, el RS, el complejo regulador troponina[19] tropomiosina y los filamentos de actina y miosina . Los dos procesos que regulan la contracción y ++ la relajación de los miocitos son: a) los que alteran la disponibilidad del Ca citoplasmático libre y ++ b) los que alteran la respuesta de los miofilamentos a la activación del Ca . Se sabe, por ejemplo, que la fosforilación de la troponina I por la PKA, el bajo pH, y la reducción de longitud del sarcómero disminuyen la sensibilidad de los miofilamentos al calcio[30]. La fuerza total desarrollada, la tasa de desarrollo de la fuerza y su declinación durante la relajación dependen primero con la cantidad de Ca++ disponible para actuar sobre la troponina y ++ luego con la forma por la cual el Ca es retirado durante la relajación. Las anormalidades funcionales observables en IC responden a una relación F/Fr negativa secundaria a perturbaciones del manejo del Ca++, y están caracterizadas por aumento de la concentración intracelular de Ca++ en diástole, aumento de la duración y disminución de la amplitud del “ciclo” de Ca++ y reducción de su contenido en el RS[31,32]. La evidencia disponible indica que la regulación hacia abajo de SERCA2a subyace en estas anormalidades posiblemente combinadas con aumento de la actividad del intercambio Na+/Ca++. 172 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 ACTUALIZACIÓN 15/03/2005 Diversas alteraciones celulares y moleculares se observan en el miocito hipertrofiado: a) reducción de los almacenes de Ca++ por una tasa disminuida de reabsorción del mismo por la SERCA2a; b) reducción de la cantidad de SERCA2a; c) reducción de la fosforilación del fosfolamban y d) aumento de la filtración o escape de Ca++ del RS por RyR2 hiperfosforilados. Harris DM, Mills GD, Chen X, Kubo H, Berretta RM, Votaw VS, Santana LF, Houser SR. Alterations in early action potential repolarization causes localized failure of sarcoplasmic reticulum Ca2+ release. Circ Res. 2005 Mar 18;96(5):543-50 + ++ Intercambiador Na /Ca (NCX) Studer y col. [33] han encontrado en corazones humanos en IC que la expresión del gen del + intercambiador Na /Ca++ (NCX) está aumentada, compensando en parte la menor recaptación del Ca++ del RS. El NCX es un mecanismo muy importante para el transporte transmembrana de los iones sodio y calcio[19,33-35]. Su modo operacional es alternativo: 1) “hacia adelante” (forward) introduciendo Na+ en el citosol y sacando Ca++, y 2) “reverso” o sea a la inversa el movimiento de iones. Se intercambian 3 iones de Na+ por cada ión de Ca++.. El aumento de la entrada de Ca++ por acción reversa del NCX es una fuente de Ca++ intracellular colaborando con el aporte del RS para lograr la activación de las miofibrillas. Es un tema de actual discusión e investigación en desarrollo. (Lectores interesados pueden consultar una amplia revisión de Blaustein y Lederer[35]) Hasenfuss[28] encuentra función diastólica preservada si está aumentado el NCX, y alterada a la inversa. En el corazón insuficiente se observa que durante la diástole la relajación está retardada y permanece eventualmente incompleta y que durante la sístole la relacion fuerza frecuencia está disminuida. El aporte de Ca++ por el RS puede estar disminuido por[35]: a) Menor cantidad de RyR2; b) Menor sensibilidad del RS al CICR; c) Disminución del contenido de Ca++ del RS. El menor contenido de Ca++ del RS se explica por la disminución de SERCA2a (su expresión y/o su actividad) más el aumento de actividad del NCX En general se piensa que el contenido del Ca++ del RS está disminuido en la IC. También puede ser que tal disminución se deba a un déficit de recaptación o del mecanismo de intercambio Na+/Ca++. Algunos investigadores sugieren que alteraciones de las proteínas encargadas del transporte de Ca++, en especial los RyR2 y su proteína moduladora FKBP12.6 son los principales protagonistas en la fisiopatología de la IC[36]. Jiang y col.[37] investigaron las concentraciones y propiedades funcionales de los RyR2, SERCA2a y fosfolamban, en seres humanos con IC y en perros con IC provocada por marcapaseo. Sus resultados señalaron que interviene el RS en la fisiopatología de la IC, más por captación anormal del Ca++ que por liberación del ión, contribuyendo al ciclo de Ca++ lento y deprimido. 173 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 Se ha dicho que la disminución de la contractilidad en los corazones insuficientes no sería causada únicamente por una disminución de la concentración pico de Ca++ intracelular sino también por una disminución de la sensibilidad del miofilamento. Pero sin embargo la mayoría de ++ los investigadores encuentran que la sensibilidad de las miofibrillas al Ca el estiramiento permanecen sin cambios en la IC. D'Agnolo y col. y su exacerbación ante [38] señalan que en la miocardiopatía dilatada no hay alteraciones de las proteínas contráctiles y regulatorias, y que lo que es anormal es el mecanismo de regulación de apertura de los portones de los RyR2 del RS, estando asi comprometido el acoplamiento éxcito-contráctil. Se ha discutido acerca del papel de los canales de Ca++ dependientes del voltaje, con la idea Chen y col.[39] estiman que que cambios de su expresión pueden contribuir a la aparición de IC: alteraciones (disminución) de la densidad y regulación de los canales L contribuyen a la contractilidad anormal y a la apagada respuesta adrenérgica en el corazón insuficiente. Sin embargo Schröder y col. [40] ++ encuentran que los canales L de Ca muestran aumentos de su apertura y biodisponibilidad en la IC, indicando un nivel mayor en el estado estable de fosforilación, por alteración de la reacción de desfosforilación. Esta aparente contradicción con ++ Chen y col. podría explicarse porque la investigación se refiere a canales L de Ca quienes podrían estar intentando compensar a deficiencias de los canales de Ca ++ superficiales, de los túbulos T. Mecanismos alterados en la IC La potenciación de la contractilidad por medio del aumento de la FC o sea el Fenómeno de Bowditch es un mecanismo importante de la función cardíaca, pero está atenuado o ausente en el corazón insuficiente[41-49]. La estimulación del receptor beta-adrenérgico incrementa la relación Fuerza/Frecuencia (F/Fr) de la contractilidad. O sea que que al aumentar la FC se incrementa la contractilidad cardiaca para cada nivel de estimulación adrenérgica creciente. En la IC experimental se ve alteración de la amplificación adrenérgica de la relación F/Fr durante estrés o ejercicio. Figuras 8-7 y 8-8 El aumento de la FC durante la distensión ventricular mejora la función ventricular en normales pero tiene efectos dañosos en pacientes con miocardiopatía dilatada, como lo han observado Petretta y col.[42] en 8 pacientes. En experimentos con ratones se ha visto que la FC tiene un efecto modesto pero significativo en los índices de contractilidad obtenidos por medio de las curvas de Presión/Volumen [43]. Una explicación de la relación F/Fr alterada es que se debe al manejo inadecuado del Ca++, con disminución en vez de aumento del ciclo de Ca++ cuando la FC aumenta, debido a disminución de SERCA2a o a menor captación de Ca++ por el RS . Como consecuencia de la alta FC hay disminución del tiempo para el ciclo de Ca++ resultando en menor acumulación del ión en el RS y disponibilidad de éste para la activación sistólica de las proteínas contráctiles. El deterioro de la captación de Ca++ dependiente de la frecuencia causará acumulación diastólica del catión con perturbación de la función diastólica. 174 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 Es importante señalar que la presencia de una relación F/Fr negativa se debe a una alteración celular, local, y que no es un fenómeno ligado a una perturbación global del corazón o sistémica[44]. Maier, Bers y Pieske[45] han demostrado que el aumento de carga de Ca++ del RS contribuye a la relación F/Fr positiva en conejos y a la inversa su disminución da una relación negativa. En ratas la carga disminuida de Ca++ del RS tiene una importancia menor en la relación F/Fr negativa, pero si la tiene la refractariedad de los canales de liberación del Ca++ del RS. La falta del fenómeno de Bowditch en la IC se explica por una disminución de la sensibilidad al Ca de las miofibrillas, por alteraciones del potencial de acción o por mal manejo del ión. Se pone ++ de manifiesto la importancia del manejo del Ca++ por el RS cuando se ve el efecto de agentes que incrementan el AMPc y de allí aumentan la fosforilación del fosfolamban, permitiendo la recaptación del catión por el RS; estos agentes, a bajas dosis, revierten el fenómeno de la escalera negativo y mejoran las anormalidades diastólicas. La sobre-expresión de calsecuestrín provoca disminución de la contractilidad y de los ciclos de ++ Ca intracelulares, y por ende una disminución de la relación F/Fr, aunque habría una tendencia a normalización del estrés de pared y de la función ventricular[46]. En la relación F/Fr interviene como fundamental la función de la SERCA2a, y ésta depende a su vez de la fosforilación del fosfolamban[47] (ver más arriba). De acuerdo con ello Bluhm y col.[48] han comprobado que el fosfolamban es un determinante mayor de la relación F/Fr. Kaprielian, del Monte y Hajjar[49] consideran que las alteraciones de la contractilidad de la IC, acompañadas de ausencia o inversión del Fenómeno de Bowditch se deben a disminución de la carga de Ca++ del RS por disminución de la función de SERCA2. Bhargava y col. [41] han encontrado pérdida del control adrenérgico de la relación F/Fr en la IC provocada por miocardiopatía (isquémica o dilatada). Un aumento de la FC provoca mayor entrada de Na+ en la célula (parcialmente compensada por la bomba Na+-K+ATPasa), y a través del intercambio Na+/Ca++ se retiene Ca++, incrementándose la contractilidad. Pero también hay una mayor entrada de Ca++ por los canales L (modulados por el AMPc-PKA). Es decir que la relación F/Fr tiene dependencia de la activación del receptor beta-adrenérgico. En el estudio de Bhargava se observó la respuesta a incrementos progresivos de la FC a 150-160 latidos/m con marcapaseo de aurícula derecha en tres controles normales y en 5 pacientes con miocardiopatía dilatada severa. Se midieron la presión intraventricular con el dP/dt (max) y la relajación por tau (τ). Las pendientes de la relación entre frecuencia y dP/dt (max) en controles fueron positivas en estado basal, pero la pendiente media se incrementó en forma sustancial y significativa durante la infusión de dobutamina. En los pacientes con IC la relación mencionada estuvo deprimida y achatada y careció de una rama descendente, y la infusión de dobutamina desplazó ligeramente la relación hacia arriba pero sin modificar la pendiente. Es decir que en pacientes con IC no se observó aumento de la contractilidad con la estimulación beta-adrenérgica. Esto adquiere importancia en la interpretación de la disminución de la capacidad para ejercicio de los pacientes. En voluntarios normales se ve que con bajo nivel de ejercicio hay un incremento linear del VM por aumento de la FC y el volumen sistólico (VS). Hay además mejor desempeño diastólico. O sea que en las fases precoces el aumento del VS se produce fundamentalmente por un mayor VFD mientras que el VFS no cambia mayormente, o sea que el mecanismo Frank-Starling contribuye significativamente al aumento del VM. Cuando se llega a altos niveles de ejercicio el VM continúa incrementándose linearmente a expensas de un aumento también linear de la FC, dado que ya a esos niveles no hay aumento del VFD. En grados muy altos de ejercicio puede observarse 175 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 disminución del VFD mientras que crece la presión pulmonar de wedge (PW). Probablemente esto se debe a dificultad de llenado ventricular por la menor duración de la diástole vinculada a la taquicardia. + En la relación F/Fr también tiene mucho que ver el intercambio de Na a través del NCX. Cuando la + ++ concentración de Na intracelular está aumentada, el NCX funciona en “modo reverso” introduciendo Ca en la + ++ célula y sacando Na . Ese Ca contribuirá al llenado del RS y también, juntamente con la entrada del ión por los ++ [50] canales L, modulará la liberación de Ca del RS. Según Pieske y col. en la IC aparecen trastornos de la bomba + + Na /K ATPasa, consistentes en disminución de expresión, cambios a isoformas, o función alterada, que + + producirán acumulación de Na intracellular. En sus investigaciones concluyeron con que en la IC el Na intracelular está significativamente elevado contribuyendo a mantener adecuadamente la función contráctil cuando la frecuencia de estimulación es baja, pero cuando la frecuencia es alta se altera el comportamiento de la relación F/Fr y la función diastólica. Hay disminución de la complacencia indicando que son necesarios mayores estímulos y mayores presiones de fin de diástole (PFD) para alcanzar la fuerza contráctil adecuada. Los estudios en corazones intactos muestran que las presiones de fin de sístole (PFS) y PFD aumentan cuando los volúmenes aumentan. Pero ambas PFS y PFD relacionadas a volumen se desplazan a muchos mas altos volúmenes en corazones miocardiopáticos que en normales, mostrando el alto grado de remodelación existente, siendo el efecto del volumen mayor sobre la PFS, como lo revela el hecho de que la presión desarrollada (Pd = PFS - PFD), también varía directamente con el volumen ventricular. Estas son evidencias de que la precarga influencia el funcionamiento cardíaco. Se ha discutido si la Ley de Frank-Starling se sigue cumpliendo en caso de IC severa. Para Holubarsch y col. [51] el mecanismo Frank-Starling está preservado en esas condiciones. Esto implica que en el manejo de esos pacientes debe mantenerse la presión de llenado lo suficiente como para aumentar contractilidad pero evitando la congestión circulatoria; probablemente el VI trabaja en el límite de su reserva diastólica. [52] Estos resultados son manifiestamente opuestos a los de Schwinger y Böhm , quienes sostienen que el corazón insuficiente es incapaz de usar la Ley de Starling; Holubarsch [51] indica que hay diferencias metodológicas que explican la discrepancia. Este último autor señala que la fuerza contráctil es dependiente de la longitud de la fibra principalmente a través de cambios de la ++ sensibilidad de las miofibrillas al Ca , probablemente como resultado de una afinidad alterada de éste a la troponina. También considera razonable asumir que el VI opera cerca o aún mas allá de las longitudes óptimas del sarcómero, indicando una reserva de precarga reducida o inexistente. Además se encuentra complianza diastólica ventricular reducida en preparados aislados de ventrículos humanos en insuficiencia, indicando que la elevación del estrés y de la PFD son necesarios para alcanzar fuerza contráctil óptima en corazones desfallecientes . Las observaciones de van der Velden y col.[53], muestran que el mecanismo de Starling está preservado en la IC terminal. Vahl y col. [54] analizaron el mecanismo Frank-Starling en miocardio ventricular de corazones con miocardiopatía dilatada explantados y corazones de donantes en ocasión de trasplantes y encontraron que el mecanismo aún está presente en los ventrículos miocardiopáticos. 176 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 Contracción y relajación No forma parte de los objetivos de este libro ofrecer una descripción detallada de la contractilidad, que opinamos está reservada a textos de fisiología cardiaca. Recomendamos a este respecto la excelente descipción de A. Mattiazzi, en Fisiología Humana de Houssay[55]. ACTUALIZACIÓN 10/08/2006. Los ventrículos comparten el septum interventricular (Siv), y cambios de volumen, presión o complianza en un ventrículo generan cambios en la complianza del otro ventrículo. Este fenómeno se denomina interacción ventricular diastólica (IVD), en el cual juega un importante papel la restricción pericárdica. También existe interacción ventricular durante la sístole. El pericardio aumenta marcadamente la IVD. La IVD se manifiesta como un aumento de la presión diastólica de un ventrículo a medida que el volumen del otro ventrículo sea aumentado. La posición del Siv al final de la diástole está determinada por el gradiente de presión transeptal de fin de diástole, siendo convexo cuando es visto desde el ventrículo izquierdo. Este gradiente se revierte cuando se produce sobrecarga de volumen de ventrículo derecho pero sobre todo con sobrecarga de presión de ventrículo derecho. Cuando se revierte el gradiente el Siv se achata o se vuelve cóncavo. En los estadios iniciales de ejercicio el aumento del VM se debe a un aumento de la frecuencia cardiaca y del VS, causado principalmente por un aumento del VFD, con ligero aumento de la Fr.Ey.. Pero en aproximadamente 50% del VO2 máximo el aumento del VM se debe incremento de la frecuencia cardiaca. En el estadio pico de ejercicio no se usa el mecanismo de Starling por le existencia de una restricción externa al llenado. En pacientes en IC con PFD crónicamente alta la oclusión con balón de la vena cava inferior retira la constricción que se opone al llenado, resultando en un aumento inicial del VFD pese a caída de la PFD. Williams L, Frenneaux M: Diastolic ventricular interaction: from physiology to clinical practice. Nature Clin Practice Cardiov, 2006;3:368-75 Estudios de Función ventricular La función sistólica adecuada implica producir la presión suficiente intraventricular para eyectar la sangre en la circulación sistémica, debiendo vencer una serie de impedimentos englobados en el concepto de "impedancia" aórtica. La actividad ventricular es cuantificada habitualmente a través de índices de contractilidad, que Dell'Italia, Freeman y Gaasch [56] dividen en isovolúmicos y de la fase de eyección. Indices isovolúmicos de contractilidad Son el dP/dt o tasa máxima de incremento de presión en función del tiempo, la velocidad de acortamiento del elemento contráctil (VEC) y la velocidad de acortamiento a carga cero (Vmax). Los cálculos de la VEC son de dificil realización e interpretación. 177 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 Para calcular dP/dt se requiere un catéter con un micromanómetro de alta fidelidad. La dP/dtmax es sensible a la FC, a la precarga y a la masa ventricular izquierda y también a la presión arterial. Los cambios que se producen en la dP/dt opuestos a los de la FC, la presión arterial y la precarga son indicadores de cambios en el estado contráctil. Pero hay circunstancias especiales, tales como cuando la FC se mantiene estable o disminuye, y los mismo pasa con la PFD y también con la presión aórtica, en las que el aumento del dP/dtmax indica claramente un aumento del estado contráctil del ventrículo[6]. ACTUALIZACIÓN 30/06/06 El dP/dtmax está influenciado por el tamaño ventricular y el espesor de su pared, por anormalidades de la función ventricular, por insuficiencia mitral funcional, por asincronía de la contracción y por la precarga. El índice dP/dt dividido por la presión ventricular instantánea (dP/dt/P) es menos influido por la precarga pero no es muy sensible a cambios de la contractilidad. La velocidad de acortamiento durante la contracción isovolúmica (VEC) es estimada por mediciones de presión y requiere una constante de rigidez K. Tiene las mismas limitaciones que el dP/dtmax . Katz AM.: Physiology of the heart, 4th. Edition, 2006 Lippincott Williams & Wilkins, Philadelphia, USA Los estudios en músculo aislado sugirieron que la máxima velocidad de acortamiento a carga cero (Vmax) era un índice útil del desempeño contráctil cardiaco, probablemente relativamente independiente de la precarga, que cambiaba de forma adecuada cuando se modificaba la contractilidad. Cuando se mide la velocidad de acortamiento y el Vmax en el músculo aislado, se necesita además obtener velocidades de acortamiento con muy bajas cargas, complicándose la determinación. Para el cálculo se requieren modelos mecánicos que toman en [6] cuenta el período isométrico sistólico para calcular la velocidad de acortamiento del elemento contráctil . Luego de las primeras investigaciones se demostró que el Vmax es influenciado por la longitud del sarcómero y por [3] fuerzas viscosas internas . Es indicador de cambios del estado contráctil, pero tiene cierta dependencia de la precarga, y además para su cálculo es necesario recurrir a la extrapolación de datos, razones por las cuales no es [2] mayormente utilizado . Indices de la fase eyectiva La extensión de los movimientos de la pared ventricular son estudiados por los índices de la fase eyectiva y comprenden la Fracción de Eyección (Fr.Ey.) y la velocidad de acortamiento circunferencial (VAC), fácilmente determinables por ecocardiografía o por estudios con radionúclidos. La Fr.Ey. o fracción expulsada es igual al volumen sistólico (VS) dividido por el volumen de fin de diástole (VFD). El VFD es de 120-140 cm³, y el VS es igual a 70-70 cm³ por lo cual la Fr.Ey. es aproximadamente igual a 0,60 (60%). Es uno de los índices mas usados y uno de los de menor sensibilidad, siendo influenciable por la poscarga. Al estar inversamente relacionado con el VFD este índice está reducido en corazones dilatados. Por esta razón tiene mala correlación con los síntomas porque la mayor causa de su disminución no es una merma del VS, sino un aumento del VFD. Según Carabello[2] tiene las siguientes ventajas: 1) Es un excelente indicador pronóstico y 2) Es fácil su determinación. La desventaja (aparte de no determinar la generación de fuerza) es que es dependiente de la contractilidad pero también de la pre y poscarga. De allí su poco valor como índice de contractilidad, aunque es útil para apreciar el funcionamiento global de la bomba, y para valorar algo groseramente la gravedad de la IC según 178 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 el grado de descenso que muestre. Pese a sus limitaciones, es de uso constante en clínica. La Velocidad de acortamiento circunferencial (VAC) es la fracción de acortamiento (determinada por ecocardiografía) dividida por el tiempo de eyección, y depende menos de la precarga que la Fr.Ey. pero es dependiente de la poscarga (el estrés sistólico la modifica[3]). En [57] pacientes con IC la VAC puede ser un índice adecuado de la contractilidad . Kass [3] considera que un método apropiado y más sencillo es calcular la Potencia Ventricular Máxima ajustada a la precarga. La potencia de cámara surge de multiplicar la Presión por el flujo, mientras que la potencia muscular es el producto de fuerza y velocidad de acortamiento. Potencia máxima ventricular es el producto pico instantáneo de Presión y flujo y es altamente dependíente de la precarga de la cámara. Dividiendo Potencia máxima (PWRmax) por el volumen de fin de sístole (VFS) al cuadrado, o sea PWRmax/VFS² , se obtiene un parámetro mínimamente influenciable por las cargas, que ha sido demostrado útil en IC, sobre todo para evaluar medicación inotrópica. Indices de contractilidad de fin de sístole El volumen o dimensión de fin de sístole (VFS) depende del estado contráctil, de la poscarga y de la masa ventricular izquierda pero no de la precarga, asi que examinando la dimensión de fin de sístole o el volumen de fin de sístole en vez de toda la fase de eyección, se elimina a la precarga como factor confundidor en la determinación del estado contráctil. El volumen de sístole es un conocido índice insuficiencia usado en casos de mitral, como indicador del momento oportuno de indicación quirúrgica de la valvulopatía. Carabello[2] ha propuesto una relación entre presión sistólica pico y VFS como (PSP/VFS), índice de contractilidad resaltando su mayor Figura 8-9. Curva Presión/Volumen. A: Cierre de válvula mitral; B) Apertura sigmoidea; C) Cierre de sigmoidea; D) Apertura de válvula mitral. D→A : Llenado ventricular; A→B: Fase isovolumétrica sistólica; B→C Expulsión sistólica; C→D: Isovolumétrica diastólica valor pronóstico con respecto al VFS considerado aisladamente. Una de las formas para estudiar el desempeño cardiaco es estimando la relación Presión/Volumen en un ciclo cardiaco y en ciclos sucesivos. En la diástole el ventrículo se llena inscribiéndose la relación presión-volumen pasiva exponencial. Al final de la diástole, luego de Figura 8-10. Ver texto 179 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 la contracción auricular, el ventrículo alcanza su VFD y PFD, que a través del primero de esos parámetros nos sirve para estimar la precarga. Luego comienza la contracción isométrica sistólica incrementándose la presión intraventricular sin que se produzcan cambios en el volumen, hasta que la misma alcance la presión aórtica, momento en el cual se abrirá la válvula aórtica y comenzará la eyección sistólica que terminará al cerrarse la válvula aórtica. La presión intraventricular cae durante la relajación hasta que se iguala con la de la aurícula izquierda, produciéndose la apertura de la válvula mitral y comenzando el llenado ventricular. Este asa de inscripción antihoraria encierra el trabajo realizado por el ventrículo, o sea que el área del asa es el trabajo cardíaco. Figura 8-9 [1] En la Figura 8-10 tomada de Cingolani se exponen 4 paneles con distintas circunstancias que afectan la relación presión-volumen. En el panel A se esquematizan tres bucles con distintas precargas, y no hay diferencias en la PFS.. Tomando las PFS de los distintos bucles, y uniéndo esos puntos se dibuja la recta isométrica sistólica (Relación P Elastancia arterial PA-2 Elastancia ventrícular sistólica e A B d PA-1 c V:Ey. Emax ---Ea (Ees ó Emax) es considerada medida de contractilidad. En V poscarga por lo que el ventrículo expulsará menor volumen Volumen telediastólico de ventrículo izq Figura 8-11. Presión Volumen Fin de Sistóle = RPVFS) cuya pendiente Para un volumen eyectado nulo la presión aórtica es nula y la presión aórtica se eleva proporcionalmente al volumen de eyección ventricular. A: es la elastancia ventricular sistólic; B: es la elastancia arterial. Si el VFS es igual al VFD, la >Fr.Ey. es nula y la P.A también es nula (punto c). Para un VFS bajo (punto d) la PA será baja (PA-1). Para un VFS más elevado (punto e) la PA (PA-2) será la más elevada. La pendiente de la Ea es mas empinada que la de la Emax (aunque no dibujada expresamente así en la figura) el panel B hay tres bucles ,cada uno con presión aórtica distinta; al incrementar la presión aórtica se produce mayor (la precarga se mantiene constante).. El panel D muestra latidos con estado inotrópico aumentado y la unión de los PFS de cada bucle dibuja una recta de mayor pendiente, característica de una intervención inotrópica positiva. En el panel C hay un desplazamiento hacia arriba y a la izquierda de la recta, pero sin cambiar mayormente la pendiente indicando también mayor contractilidad. . Con el estudio de la relación presión-volumen se pueden derivar índices de contractilidad de fin de sístole. Se obtienen múltiples coordenadas de presión de fin de sístole cambiando el volumen ventricular o la presión arterial, procedimiento en el que se usa la cardiometría de impedancia. Las coordenadas tienen una relación casi linear, siendo llamada la pendiente de ésta Emax (elastancia máxima), que ha sido propuesta como índice de contractilidad. La relación se desplaza hacia arriba y a la izquierda cuando se estimula la contractilidad y hacia abajo y a la derecha cuando la misma está deprimida. El Emax ha sido considerado como una efectiva medida de contractilidad. Además, en la curva P-V puede usarse la impedancia arterial para tener un índice de la poscarga, midiendo la Elastancia arterial efectiva (EA) , que es la diagonal que conecta la Presión de fin de sístole al punto mas alto del Volumen de Fin de diástole. Figura 8-11 También con las curvas de presión-volumen se usa la relación entre trabajo sistólico y VFD, observando la pendiente de la misma (relación TS-VFD o SW-EDV)[3]. 180 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 También se ha estudiado la Rigidez de Fin de Sístole. Se define rigidez como la relación entre las variaciones de estrés (σ) con las variaciones de strain (ε) o deformación: ∆σ/∆ε. Se obtiene una constante k que relaciona ambas variables. Cuando hay aumento de rigidez implica aumento de contractilidad. Pareciera ser de utilidad y no influenciable por otras variables confundidoras[2]. Nuevos métodos de Ecocardiografía Doppler para evaluación de contractilidad Nuevos procedimientos con ecocardiografía Doppler tales como el Doppler Tisular (dentro de esa familia de procedimientos están el Tissue Tracking y el Strain Rate) tratan de cuantificar la función ventricular midiendo velocidades diastólicas y sistólicas y desplazamientos del miocardio. Si bien aportan importante información sobre comportamientos segmentarios de pared ventricular, aún están en desarrollo investigacional para poder ser empleados en el diagnóstico de función ventricular global. Factores metabólicos en la IC El corazón puede ser considerado como un transductor, dada su habilidad de convertir la energía química que recibe en energía mecánica. Además provee sustratos y oxígeno para el mismo y para el resto del organismo. Necesita del aporte continuo de oxígeno y de nutrientes, sobre todo los empleados para transformación en productos energéticos, como son los ácidos grasos (AG) y la glucosa, considerados como combustibles por analogía con las máquinas. Estos últimos experimentan ruptura cuando llegan al miocito, dando lugar a la acetil-CoA que va luego a entrar en el ciclo del citrato en la mitocondria. El adenosíntrifosfato (ATP) aporta la energía química disponible para la conversión en energía mecánica; cada día el corazón usa entre 3,5 y 5 kg del mismo para mantenerse funcionando. El ATP se desdobla por medio de ATPasas en adenosina-5-difosfato (ADP) y fosfato inorgánico (Pi), liberando así energía química que interviene en el trabajo de la contracción, en los movimientos iónicos y en la síntesis macromolecular[1,4,58-65]. La hidrólisis del ATP a ADP y Pi libera 7,3 kcal/mol o sea que la célula puede disponer de esa energía del ATP acoplando la hidrólisis de éste a reacciones químicas que necesiten energía[1]. En condiciones normales 2/3 del ATP hidrolizado es usado para el trabajo contráctil, y 1/3 para el movimiento iónico[58]. La energía química que puede ser usada para NADH realizar trabajo se denomina energía libre. Mononucleótido de flavina (FMN) y complejos SS-Fe El balance de ATP (producción y empleo) en el corazón es mucho mayor que en otros órganos de la economía. El corazón, entre producción y gasto, muestra un balance de 37 kgm diarios de ATP, cifra que hace valorar la enorme importancia de este mecanismo [59] energético . El miocardio tiene 30-35 µmol/gm de ATP, Coenzima Q ADP + Pi ATP ADP + Pi ATP ADP + Pi ATP FADH2 Citocromo C -reductasa Citocromo CC-oxidasa O2 Figura 8-12. La oxidación de NADH aporta 3 ATP. La de FADH2 aporta 2 ATP cantidad que permite mantener la función de bomba sólo durante 50 latidos. Por esa razón la 181 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 célula miocárdica sintetiza ATP continuamente, sin interrupción[58]. El corazón en la IC es ineficiente desde el punto de vista energético. El trabajo externo realizado por el ventrículo izquierdo (VI) está disminuido, mientras que el consumo de energía es normal[60]. Son frecuentes las alteraciones en el metabolismo cardiaco, y en la HVI se observa isquemia relativa del subendocardio. Hay disminución de fosfocreatina y reducción de la actividad de la creatina kinasa[61]. La reducción de energía aportada por el ATP afecta el comportamiento de SERCA2a e impide el correcto ciclaje de formación de puentes cruzados en los miofilamentos, básico para la contracción. Se observa reducción de la actividad de la ATPasa de las miofibrillas y en la velocidad de contracción por cambios en las isoformas de la miosina de cadena pesada. Hay incremento del estrés oxidativo originado en sistemas citoplasmáticos y mitocondriales generadores de radicales libres; entre ellos se destaca la xantino-oxidasa (XO) en su producción de anión superóxido. Los radicales libres afectan a la ONs impidiendo la función regulatoria del ON (atenuación del consumo de oxígeno miocárdico y aumento de la eficiencia mecánica). La falta de eficiencia energética ha sido denominada “desacoplamiento mecanoenergético”[62]. Estos aspectos son tratados en profundidad por Opie[4], Ingwall[58], Taegtmeyer[59], Stanley[63], [64] Depre , a cuya lectura remitimos para una información más completa.. Mecanismos de producción de ATP Cuanto mayor sea el trabajo cardiaco producido, mayor será el balance de ATP. Las reacciones químicas que usan ATP son manejadas por relaciones proporcionales de ATP/ADP altas, mientras que las reacciones para síntesis de ATP son inhibidas por esas mismas relaciones. Uno de los mecanismos para regenerar el ATP es la fosforilación a nivel de sustrato y se realiza transfiriendo el grupo fosfato de compuestos fosforilados intermedios al ADP para formar ATP. Estas fosforilaciones pueden suceder en ausencia de oxígeno, denominándose este mecanismo metabolismo Figura 8-13. ATP producidos en el metabolismo de AG anaeróbico[1,66]. El otro mecanismo existente es el de la fosforilación oxidativa: esta se realiza en el interior de las mitocondrias, organelas que están profusamente distribuidas en los miocitos y que contienen las enzimas necesarias para el proceso. El 98% de la re-síntesis de ATP se hace por fosforilación oxidativa, mientras que sólo el 2% proviene de la glucolisis. La fosforilación oxidativa y las 182 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 reacciones químicas que llevan a ella es lo que se conoce como metabolismo aeróbico. La síntesis de ATP se mantiene en estricta proporción con la tasa de utilización de ATP. La energía originada en el catabolismo de distintas sustancias proviene de una serie de reacciones de oxidorreducción: cuando una molécula pierde un electrón, se oxida, si otra molécula gana ese electrón, se reduce. Es muy importante la participación de las coenzimas nicotinamida adenina dinucleótido (NAD+) y flavina adenina dinucleótido (FAD+), que cuando aceptan electrones, reduciéndose, se convierten en NADH y FADH2. Estas coenzimas pueden ser reoxidadas a nivel mitocondrial por reacciones en cadena que transportan electrones y que en presencia de oxígeno forman agua. Los electrones necesitan de la intermediación de aceptadores de electrones, que dan lugar a la liberación gradual de la energía libre, la que es almacenada en forma de ATP[1,66]. Se denomina Potencial de reducción de un elemento, ión o compuesto a la tendencia de ganar electrones frente a otro elemento, ión o compuesto Los aceptadores de electrones forman una cadena que está constituida por tres grandes complejos enzimáticos: La NADH-Q-reductasa, la citocromo-reductasa y la citocromoxidasa. Estas enzimas poseen grupos que aceptan electrones y que son flavinas, sulfuro-hierro (S-Fe) iones cobre (Cu++) y hem. Cuando hay reacciones de transferencia desde el NADH o FADH2 se produce un flujo de protones hacia fuera de la mitocondria, creándose un gradiente de pH y de potencial eléctrico transmembrana. Cuando los protones regresan a la mitocondria se sintetiza el ATP, con intervención fundamental de la ATPasa mitocondrial (ATP-sintetasa). Como consecuencia de la oxidación de NADH a través de la cadena de electrones, la fosforilación oxidativa forma 3 ATP. La FADH2 ingresa a la cadena a nivel de la coenzima Q, generando 2 ATP[66]. Fig. 8-12 ATP y sustratos en IC Los combustibles del corazón, denominados genericamente sustratos, pueden ser hidratos de carbono (HdC), ácidos grasos (AG), amino ácidos(aa) o cuerpos cetónicos. En condiciones de ayunas el corazón usa esencialmente a los AG para la producción de energía oxidativa, pero influencias nutricionales, metabólicas u hormonales pueden inducir una mayor contribución de los HdC. En ayunas el nivel de ácidos grasos libres (AG) es alto, y su captación es usada para el metabolismo oxidativo, resultando así la mayor fuente de energía[4]. Cuando se oxidan los AG se inhibe la oxidación de glucosa y la glucosa captada es convertida en glucógeno. Cuando el organismo ha sido alimentado con HdC los niveles de glucosa circulante y de insulina son altos, estando suprimida la circulación de AG. En este caso disminuye la captación por el corazón de AG, se libera la glucolisis y aumenta la oxidación de la glucosa. El metabolismo de la glucosa suprime la oxidación de los AG. Luego de comidas con alto contenido graso se produce hipertrigliceridemia posprandial, siendo los triglicéridos convertidos en AG, quienes van a oxidación de AG. En esas circunstancias los triglicéridos se convierten en el mayor combustible miocárdico. En el caso de ejercicio intenso agudo, aumenta la cantidad de lactato sanguíneo, siendo el lactato el combustible miocárdico para esa circunstancia. El lactato inhibe la oxidación de glucosa 183 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 y la captación de los AG , y estos aportan solamente el 15-20% de las necesidades orgánicas durante ejercicio. Cuando hay isquemia el patrón de captación de sustrato cambia: en vez de ser predominante a partir de los AG, pasan los HdC a ser los principales sustratos.. Metabolismo de los ácidos grasos …..El corazón toma los AG libres del plasma y luego los oxida (el 80%) o los lleva a almacenes de triglicéridos (el 20%). Los AG son esterificados formando acil-CoA la cual es transformada a acilcarnitina que es la que pasará la membrana mitocondrial, catalizado por las carnitina-palmitoiltransferasas, para proceder a la β-oxidación que producirá un acetil-CoA, un NADH y un FADH2, en cada vuelta de espiral; el acetil-CoA es oxidado en el ciclo citrato de Krebs, donde se produce aproximadamente 10 moléculas de ATP en cada giro del ciclo. El ciclo de Krebs muestra mayor producción cuando hay aumento de trabajo cardiaco, mientras que se muestra deprimido cuando hay hipoxia o isquemia. El Nuclear Receptor Peroxisome Proliferator-Activated Receptor-α (PPARα) media la respuesta de los AG al gene de la carnitina-plamitoil-transferasa. La oxidación de los AG de cadena larga (Acido oleico y palmítico) está reducida en corazones hipertrofiados por disminución de la proteínas ligantes de AG y de la translocasa de AG. También hay reducción de carnitina, asi como perturbación de la cooperación entre la acil-CoA-sintetasa y la carnitina pamiltoil-transferasa. Se observa regulación hacia debajo de un contenido de importancia de proteínas vinculadas con la oxidación de los AG – como las acil-CoAdeshidrogenasas - en aquellos corazones con signos de descompensación y no en los simplemente hipertrofiados. Figura 8-13 Metabolismo de la glucosa La captación de glucosa está controlada por transportadores de glucosa ubicados en la membrana celular que pertenecen a la familia de los GLUT, quienes constituyen un sistema de transporte y contratransporte. La isoforma Acido láctico Glucosa GLUT LPC HK PH LDH GlGl-6-P lactato GS Glucógeno piruvato DHAP FruFru-6-P FruFru-1,61,6-P PKK1 Aldolasa TPI Glu3 PG Glu-3-P GAPDA PK 2 PG PGM PEP enolasa predominante en los miocitos es el GLUT 4, sensible la insulina; también hay expresión de GLUT 1, pero este sobre todo en el miocardio fetal. GLUT 1 no depende de la insulina Figura 8-14. Glucolisis. Ver texto El GLUT 4 es el transportador sensible a la insulina, mientras que el GLUT 1 es independiente de la insulina. Cuando la glucosa penetra en el miocito, es utilizada para glucolisis, síntesis de glucógeno, o el shunt de pentosa. Este último provee cadenas de carbonos para la formación de nucleótidos de adenina, como AMP y GTP y regenera el cofactor NADP+. En la vía glucolítica la 184 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 glucosa es convertida en unidades de 3 carbonos, dando lugar no solamente a ATP sino otros productos que pueden ser usados para ulterior producción de ATP en la mitocondria[63]. La glucosa es el sustrato mas digno de confianza para la producción de energía en el corazón[63]. La importancia del metabolismo de la glucosa via glucolisis se aprecia bien en el músculo hipertrofiado e isquémico. Pero el metabolismo aerobico de la glucosa para mantener la funcion contráctil ha sido menos estudiado. Principalmente por el hecho conocido que los AG son habitualmente, en condiciones normales, el sustrato que el corazón usa preferentemente para la producción de energía por el corazón. La glucosa para el corazón proviene de la circulación sanguínea o de almacenes intracelulares de glucógeno. El transporte de glucosa hacia el interior del miocito es regulado por transportadores específicos- La glucosa intracelular es rápidamente fosforilada y se convierte en un sustrato para la vía glucolítica, y para la síntesis de glucógeno, y de ribosa. Después de glucolítico la entrar glucosa en el camino finalmente es desdoblada hasta piruvato el cual es a su vez un sustrato para otros caminos metabólicos mas.. La captación de glucosa, definida como transporte y fosforilación de glucosa, es medida como el producto de la extracción de glucosa por la concentración arterial de glucosa Figura 8-15. La formación de piruvato abre la vía para otros mecanismos metabólicos. (Esquema tomado de Depre[50]) multiplicada por el flujo. La medición de la captación de glucosa neta y la liberación de lactato por la diferencia arteriovenosa ha sido extensamente usada en el humano para evaluar el metabolismo de la glucosa pero las mediciones in vivo no son tan precisas como en corazones aislados La glucosa penetra en la célula transportada por GLUT 4, y en presencia de hexokinasa (HK) forma glucosa-6-fosfato. La Gl-6-P puede ir a los almacenes de glucógeno (GS: Glucógeno sintetasa). El glucógeno puede transformarse nuevamente en Gl-6-P ( PH: Glucógeno fosforilasa. El ciclo de glucolisis sigue así: Gl-6-P→Fructosa-6-fosfato (Fru-6-P)→Fru-1,6-P y esta en dihidrofosfato de acetona y Glu-3-P→3-fosfoglicerato(3 PG)→2 PG →fosfoenolpiruvato (PEP)→ Piruvato ←→Lactato. Ver figura 8-14 La mayor parte de la glucosa sigue el camino glucolítico descrito, aunque la glucosa-6-fosfato (GL-6-P) es también sustrato para la síntesis de glucógeno. El glucógeno ocupa el 2% del volumen cellular en el adulto pero el 30% en el corazón fetal y el del recién nacido y se incrementa con el ayuno. El depósito de glucógeno es aumentado por la insulina. También hay síntesis de glucógeno cuando el principal combustible es el lactato. 185 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 El glucógeno es rápidamente desdoblado cuando la glucógeno fosforilasa es estimulada por la adrenalina o el glucagon. La glucógeno fosforilasa es la principal enzima reguladora de la glucógenolisis, siendo activada por la fosforilación (por la PKA o por la fosforilasa kinasa activada por Ca++). La ruptura del glucógeno es también rápidamente estimulada en los aumentos súbitos de trabajo cardiaco. Los componentes glicosilados provenientes de la ruptura de glucógeno son preferencialmente oxidados en vez de convertirse en lactato. En el caso de concentraciones fisiológicas de AG, la administración de adrenalina provoca incrementos extras de necesidades energéticas que son inicialmente atendidas por la glucógenolisis y luego por un aumento sostenido de la oxidación de glucosa. La diferencia entre la glucolisis aeróbica y anaeróbica consiste en que en caso de anerobiosis el piruvato es convertido en lactato en presencia de la deshidrogenasa láctica, mientras que en aerobiosis entra en el ciclo del ácido tricarboxílico. En el primer caso hay restitución del cofactor NAD+ necesario para mantener la reacción de la gliceraldehido-3-fosfato deshidrogenasa, que pernite continuar con la glucolisis aun en ausencia de oxígeno. En el segundo caso el piruvato entra en el ciclo del ácido tricarboxílico como acetil-coenzima A (CoA) y es oxidado (Fig. 8-10). Algún lactato es producido, y la LDH permite que sea rápidamente oxidado siguiéndole recoversión a piruvato. Entoces glucolisis no es solamente la ruptura aneróbica de glucosa sino que es la condición normal en el corazón. Bajo condiciones aeróbicas los productos de la glucolisis, o sea piruvato y NADH son utlizados para apoyar y sostener la producción oxidativa de ATP en la mitocondria[60]. Figura 8-15 La glucolisis se desarrolla en el citosol, mientras que el metabolismo oxidativo tiene lugar en la mitocondria. La reducción de los sustratos de compuestos de carbono es llevada a cabo por los cofactores FAD+ y NAD+. Los AG inhiben más la glucolisis que la captación de glucosa Es claro entonces que para obtener el aporte de energía necesario el corazón recurre al metabolismo de los AG y de la glucosa. En el caso de isquemia es predominante la oxidación de AG, desacoplándose de la glucolisis y oxidación de la glucosa. El alto nivel de oxidación de AG va a generar aumento de producción de protones perjudiciales, o sea que contribuye al daño isquémico al inhibir la oxidación de glucosa. ↓ P alta energía ↑ AMP/ATP y/o ↓ PCr/Cr AMPK un grupo de enzimas de las que las más importantes son la AMP-activated protein kinase (AMPK), la acetil-CoA carboxilasa (ACC) y la malonil-CoA-descarboxilasa (MCD). La AMPK fosforila e inhibe a la ACC la que reduce la producción de MCD. Además se supone que la rellenado La oxidación de los AG está controlada por Energía: > demanda, < aporte Inactivación ACCβ ACCβ ↑ GLUT ↑ CAPTACION GLUCOSA ↓ Malonil.Malonil.-CoA ↓ Inhibición CPT 1 ↑ oxidación AG Figura 8-16. Adenosine Monophosphate Activated Protein Kinase (AMPK) AMPK fosforila y activa a la MCD, con disminución de los niveles de malonil-CoA. Cuando hay isquemia se activa rápidamente la AMPK y ésta inhibe a la ACC, provocando descenso de malonil- 186 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 CoA y aumentando la tasa de oxidación de AG, y como consecuencia disminución de las tasas de glucolisis. El AMPK disminuye los niveles de malonil-CoA, y también incrementa las tasas de glucolisis, con desacople de ésta de la oxidación de la glucosa y aumento de producción de protones y lactato. Esto disminuye la eficiencia cardiaca y contribuye a la severidad del daño isquémico[51]. Figura 8-16 Las evidencias actuales sugieren que la AMPK regula la oxidación de los AG y la captación de glucosa en el corazón y músculo esquelético en respuesta a alteraciones en el aporte y la demanda[65]. En la HV y en la IC se ha observado aumento de la captación y utilización de glucosa. La captación aumentada de glucosa es independiente de la insulina y está asociada a un aumento del transportador GLUT 1 y a una disminución de la expresión del GLUT 4 (sensible a la insulina). No se conoce el mecanismo involucrado en el incremento de la captación de glucosa. Tian y col.[65] han observado en ratas con HVI inducida por sobrecarga cardiaca de presión, una estrecha relación entre captación de glucosa y disminución de fosfocreatina, que es un regulador clave de la AMPK (Adenosine Monophosphate.activated protein kinase). O sea que hay la posibilidad que la utilización de glucosa en la HV sea regulada por el metabolismo energético miocárdico, siendo AMPK un intermediario clave de señalamiento. Hay en pacientes con IC experiencias clínicas con medicamentos que disminuyen la oxidación de los AG y promueven la utilización de glucosa, mejoran la función cardiaca, y aumentan la capacidad para ejercicio en pacientes con enfermedad isquémica crónica[67-70]. De ahí que se piensa que el aumento de utilización de la glucosa representa un mecanismo adaptativo que hace que los corazones hipertrofiados soporten mejor la sobrecarga hemodinámica. Efectivamente se comprueba protección contra la progression de la IC y mejoría de la sobrevida en ratones con sobrecarga de presión crónica.. Las alteraciones del metabolismo de la glucosa tienen un impacto significativo sobre la función contráctil, especialmente durante isquemia y reperfusión[70]. Liao y col.[71] demostraron, en corazones hipertrofiados por sobrecarga crónica de presión, que aumentando la utilización de glucosa se protege de la disfunción contráctil y de la dilatación de cámara. . Anormalidades del metabolismo energético Han habido opiniones discordantes sobre si la concentración de ATP se encuentra disminuida en la IC, pero hay consenso sobre la presencia de disminución de la capacidad cardiaca de trasformar la energía química en mecánica. Se supone que los miocitos del corazón insuficiente son incapaces de sintetizar suficiente adenina o de prevenir la degradación del ATP (ATP→ADP→AMP→adenosina→inosina→hipoxantina) y la pérdida de purinas[58]. La disminución de la actividad de la ATPasa de actomiosina corre paralela a la disminución de la velocidad de acortamiento, mecanismo que puede ser protector. Con respecto a la fibrosis, 187 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 hemos visto en el Capítulo sobre Hipertrofia ventricular y Remodelado su muy importante intervención en la fisiopatología del proceso, contituyendo una de las modificaciones que gobiernan el remodelado y la disfunción diastólica. En la IC hay además anormalidades del metabolismo energético, observándose una anormal distribución de compuestos de fosfato de alta energía sugiriendo un discordancia entre demanda y aporte de oxígeno (isquemia subendocárdica relativa). [72] Scheuer ha estudiado una serie de factores metabólicos que probablemente intervienen en la fisiopatología de la IC, o que pueden contribuir a la progresión de la enfermedad, proponiendo el cuadro que acompañamos. Señala que hay muchas preguntas aún no contestadas acerca de transporte, liberacíón y almacenamiento de energía en la IC. En modelos experimentales de hipertrofia cardíaca se han señalado alteraciones de las mitocondrias cardíacas aunque su función aparentemente permanece normal. Scheuer propone probables alteraciones metabólicas. Ver Cuadro 8-1. Se ha pensado que el corazón insuficiente tiene según Scheuer[72] disminución del aporte energético. Según Stanley y Chandler[63] el corazón con insuficiencia crónica es metabólicamente anormal, tanto en pacientes como en Cuadro 8-1. Factores metabólicos probables en la IC, animales de Vinculados con el transporte de energía, su liberación y almacenamiento 1. Perfusión miocárdica limitada y respuesta a aumento de demanda Disminución de la capacidad vasodilatadora experimentación. Consideran que no es posible sacar conclusiones definitivas acerca de las Crecimiento capilar inadecuado Sector capiular sobredistendido 2. Insuficiente generación y transferencia de energía preferencias por uno u otro sustrato en los varios Alteraciones mitocondriales Disminución de almacenes de creatina y estadios de la enfermedad. Hay alguna indicación fosfocreatina de que los pacientes de clase III de la NYHA, Alteración de la creatin-fosfo-kinasa Deficiencias de ATP compensados, tienen alterada la oxidación de los Disminución de producción de ATP por glucolisis hidratos de carbono. En estudios de IC experimental en perros provocada por marcapaseo, Shen y col [73] señalan Utilización de la energía: Factores de control de contracción y relajación 1. Factores que afectan a la concentración que hay disminución del contenido miocárdico de intracelular de calcio. ATP, que no es fácilmente detectable salvo que la Alteraciones de receptores de membrana, canales iónicos y bombas, disfunción sea severa. La disminución es del 20% proteínas G, nucleótidos cíclicos. Función SERCA2, fosfolamban aproximadamente, cantidad similar a la observada en humanos. Destacaron además que la disminución del ATP se acompaña de disminución y col.[61] col. [74] proteínas contráctiles Isoenzimas de miosina Troponina y tropomiosina encontraron en sus pacientes que los niveles de creatina estuvieron reducidos Factores que afectan la respuesta al calcio de las Actividad ATPasa miofibrilar del “pool” total de adenina. Nascimben 2. Fosforilación de troponina I o de cadena liviana de miosina en un 51%, , mientras que Starling y encontraron una reducción del ATP del 39%. 188 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 Beer y col.[75] midieron las concentraciones de fosfocreatina y de ATP en corazones normales, hipertróficos y en insuficiencia. Los pacientes con hipertrofia padecían hipertensión arterial o estenosis aórtica, mientras los con IC padecían miocardiopatía dilatada. En los casos con miocardiopatía dilatada con IC, las concentraciones de tanto el ATP como la fosfocreatina estuvieron significativamente reducidas. O'Donnell y col.[76] consideran que una fuerte disminución de fosfocreatina y de la creatina total indican la transición de hipertrofia compensadora a descompensación e IC en la miocardiopatía hipertrófica en la rata. El ADP aumentado contribuye a la disfunción diastólica en la HVI posiblemente por un enlentecimieto del ciclaje de los puentes de actina miosina. La disminuida capacidad de la reacción de la creatina quinasa para re-fosforilar el ADP es un posible mecanismo contribuyente a [77] la falla de mantener un bajo ADP en la HVI. . La reacción de la creatina quinasa juega un importante papel en el mantenimiento de la alta relación de ATP:ADP por la rápida transferencia de un grupo fosforilo entre fosfocreatina y ATP. Los corazones hipertróficos tienen relajación [77,78] prolongada y mayor disfunción diastólica que corazones normales con deprivación energética y se ha sugerido que la relajación alterada se debe a sobrecarga de calcio. Durante largo tiempo se ha discutido si existen anormalidades en el metabolismo energético miocárdico que contribuyan a la disfunción cardíaca. Con respecto al sistema creatina-quinasa el sistema creatina/fosfocreatina está directamente ligado a la fosforilación oxidativa a través de la creatina quinasa mitocondrial. La reserva de energía está marcadamente reducida en la IC, aunque en condiciones estables no contribuye a la misma; pero en condiciones de estrés el mecanismo se pone de manifiesto por una reducción de la reserva contráctil, sin conocerse cual [79] es la razón de ello . La reducida reserva energética en la IC puede ser que contribuya a la progresión de la enfermedad, pero también puede ser un mecanismo que proteja al miocardio de la sobrecarga. Ingwall[58] ha demostrado que la disminución de la relación fuerza-velocidad de acortamiento (Vmax) y la disminución de contenido de fosfocreatina, separadamente o en combinación llevan a una disminución de la actividad mitocondrial de la creatina-quinasa, por lo cual piensa que la disminución de la reserva energética contribuye a la disminución de la reserva contráctil del corazón insuficiente. La capacidad de resíntesis del ATP a través del sistema creatina quinasa está comprometido en el miocardio insuficiente. La fosfocreatina está disminuida en la IC pero el ATP no está alterado.Los almacenes de fosfatos de alta energía no parecen estar afectados en condiciones basales, pero puede ser que existan limitaciones en la generación de energía necesarias para el mantenimiento de la función y la estructura en el caso de hipertrofia y disfunción. Hay disminución entonces de la reserva energética que limita la reserva contráctil del corazón. 189 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 Bibliografía 1) 2) 3) 4) 5) 6) 7) 8) 9) 10) 11) 12) 13) 14) 15) 16) 17) 18) 19) 20) 21) 22) 23) 24) 25) 26) 27) 28) 29) 30) 31) 32) Cingolani HE : Mecánica cardíaca. Fenómenos sistólicos y diastólicos. En Fisiología Humana de Bernardo A. Houssay, Directores Drs. Horacio E. Cingolani y Alberto B. Houssay. 7ª. Edición. Editorial El Ateneo, Buenos Aires, 2000.. Carabello B.: Abnormalities in cardiac contraction: Systolic dysfunction. In Congestive Heart Failure. Edited by Jeffrey D Hosenpud % Barry . H Greenberg. Lippincott Williams & Wilkins. Philadelphia, USA, 2000 Kass DA : Myocardial Mechanics. In Heart Failure, Ed. by PA. Poole-Wilson, WS Colucci, BM Massie, K Chatterjee and AJS Coats. Churchill Livingstone Inc., New York, USA, 1997 Opie LH, The Heart, Physiology, from cell to circulation. Lippincott-Raven, Pjiladelphia, USA, 1998 Katz AM. Heart Failure, Pathophysiology, Molecular Biology, and Clinical management. Lippincott Williams & Wilkins, Philadelphia, USA, 2000. Parmley WW, Wikman-Coffelt J. : Physiology of muscular contraction. In Cardiology, Ed. by Parmley WW and Chatterjee K- Lipincott, Williams & Wilkins, USA, 1998 Houser SR, Margulies KB : Is depressed myocyte contractility centrally involved in heart failure?. Cir Res. 2003;92:350-58 Akhter SA, Eckhart AD, Rockman HA, Shotwell K, Lefkowitz RJ, Koch WJ : In vivo inhibition of elevated myocardial beta-adrenergic receptor kinase activity in hybrid transgenic mice restores normal beta-adrenergi signaling and function. Circulation 1999;100:648-53 Liggett SB, Tepe NM, Lorenz JN, Canning AM, Jantz TD, Mitarai S, Yatani A, Dorn GW 2nd. Early and delayed consequences of beta(2)-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation. 2000;101:1707-14. Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999;96:7059-64. Dorn GW, Molkentin JD : Manipulating cardiac contractility in heart failure. Data from mice and men. Circulation 2004;109:150-58 Rockman HA, Chien KR, Choi DJ, Iaccarino G, Hunter JJ, Ross J Jr, Lefkowitz RJ, Koch WJ. Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci U S A. 1998;95:7000-5 Harding VB, Jones LR, Lefkowitz RJ, Koch WJ, Rockman HA. Cardiac beta ARK1 inhibition prolongs survival and augments beta blocker therapy in a mouse model of severe heart failure. Proc Natl Acad Sci U S A. 2001;98:5809-14. Opie LH, Bers DM.: Excitation-contraction coupling and calcium. In Heart Physiology- From Heart to circulation. Lippincott Williams&Wilkins Pjiladelphia, USA, 2004, chapter 6. Chelu MG, Danila CI, Gilman CP, Hamilton SL: Regulation of ryanodine receptors by FK506 binding proteins. Trends Cardiovasc med 2004;14:227-34 Hasenfuss G, Seidler T. Treatment of heart failure through stabilization of the cardiac ryanodine receptor. Circulation 2003;107:378-80 Marks AR: Clinical implications of cardiac ryanodine receptor/calcium release channel mutations linked to sudden cardiac death. Circulation 2002;106:8-10 Marks AR, Marx SO, Reiken S. Regulation of ryanodine receptors via macromolecular complexes: a novel role for leucine/isoleucine zippers. Trends Cardiovasc Med 2002;12:166-70 Morgan JP.: Cellular physiology of myocite contraction. In Heart Failure. Ed. by P.A. Poole-Wilson, WS Colucci, BM Massie, K Chatterjee and AJS Coats. Churchill Livingstone, USA, 1997, p1. Noguchi T, Hünlich M, Camp PC, et al.: Thin filament-based modulation of contractile performance in human heart failure. Circulation 2004;110:982-87 DeSantiago J, Maier LS, Bers DM. Frequency-dependent acceleration of relaxation in the heart depends on CaMKII, but not phospholamban. J Mol Cell Cardiol 2002;34:975-84 Bers DM : Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res 2000;87:275-81 Frank KF, Bölck B, Erdmann E, Schwinger RHG: Sarcoplasmic reticulum Ca++-ATPase modulates cardiac contraction and relaxation. Cardiovasc Res 2003;57:20-7 Netticadan T, Temsah RM, Kawabata K, Dhalla NS. Sarcoplasmic reticulum Ca(2+)/Calmodulin-dependent protein kinase is altered in heart failure. Circ Res 2000;86:596-605 Netticadan T, Temsah RM, Kawabata K, Dhalla NS. Ca2+-overload inhibits the cardiac SR Ca2+-calmodulin protein kinase activity. Biochem Biophys Res Commun 2002;293:727-32 Koss KL, Grupp IL. Kranias EG : The relative phospholamban and SERCA2 ratio: a critical determinant of myocardial contractility. Basic Res Cardiol 1997;92(Suppl 1):17-24. Pieske B, Kretschman B, Meyer M, Holubarsch C, Weirich J, Posival H, Minami K, Just H, Hasenfuss G.: Alterations in intracelular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation 1995;92:1169-78 Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H : Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ Res 1994;75:434-42 Marks AR.: Expression and regulation of ryanodine receptor/calcium release channels. Trends Cardiovasc Med 1996;6:130-5 Remppis A, Most P, Loffler E, Ehlermann P, Bernotat J, Pleger S, Borries M, Reppel M, Fischer J, Koch WJ, Smith G, Katus HA. The small EF-hand Ca2+ binding protein S100A1 increases contractility and Ca2+ cycling in rat cardiac myocytes. Basic Res Cardiol 2002;97 Suppl 1:I56-62 Mittmann C, Eschenhagen T, Scholz H. Cellular and molecular aspects of contractile dysfunction in heart failure. Cardiovasc Res 1998;39:267-75 Pieske B, Maier LS, Schmidt-Schweda S. Sarcoplasmic reticulum Ca2+ load in human heart failure. Basic Res Cardiol 2002;97 Suppl I:I63-71 190 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 33) Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, Just H, Holtz J, Drexler H : Gene expression of the cardiac Na(+)-Ca2+ exchanger in end-stage human heart failure. Circ 1994;75:443-53 34) Weber CR, Piacentino V 3rd, Margulies KB, Bers DM, Houser SR. Calcium Influx via I(NCX) Is Favored in Failing Human Ventricular Myocytes. Ann N Y Acad Sci 2002;976:478-479 35) Blaustein MP, Lederer WJ: Sodium/calcium exchange: Its physiological implications. Physiolog Rew 1999;79:764854 36) Eisner DA, Trafford AW. Heart failure and the ryanodine receptor: does Occam's razor rule? Circ Res 2002;91:979-81 37) Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res 2002;91:1015-22 38) D'Agnolo A, Luciani GB, Mazzucco A, Gallucci V, Salviati G : Contractile properties and Ca2+ release activity of the sarcoplasmic reticulum in dilated cardiomyopathy. Circulation 1992;85:518-25 39) Chen X, Piacentino V 3rd, Furukawa S, Goldman B, Margulies KB, Houser SR. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res 2002;91:517-24 40) Schröder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, Schwinger RHG, Weil J, Herzig S. : Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation 1998;98:969-76 41) Bhargava V, Shabetai R, Mathiasen RA, Dalton N, Hunter JJ, Ross J Jr : Loss of adrenergic control of the forcefrequency relation in heart failure secondary to idiopathic or ischemic cardiomyopathy. Am J Cardiol 1998;81:1130-37 42) Petretta M, Vicario ML, Spinelli L, Ferro A, Cuocolo A, Condorelli M, Bonaduce D. Combined effect of the forcefrequency and length-tension mechanisms on left ventricular function in patients with dilated cardiomyopathy. Eur J Heart Fail 2002;4:727-35 43) Nemoto S, DeFreitas G, Mann DL, Carabello BA. Effects of changes in left ventricular contractility on indexes of contractility in mice. Am J Physiol Heart Circ Physiol 2002;283:H2504-10 44) Brixius K, Hoischen S, Reuter H, Lasek K, Schwinger RH. Force/shortening-frequency relationship in multicellular muscle strips and single cardiomyocytes of human failing and nonfailing hearts. J Card Fail 2001;7:335-41 45) Maier LS, Bers DM, Pieske B. Differences in Ca(2+)-handling and sarcoplasmic reticulum Ca(2+)-content in isolated rat and rabbit myocardium. J Mol Cell Cardiol 2000;32:2249-58 46) Schmidt AG, Kadambi VJ, Ball N, Sato Y, Walsh RA, Kranias EG, Hoit BD. Cardiac-specific overexpression of calsequestrin results in left ventricular hypertrophy, depressed force-frequency relation and pulsus alternans in vivo. J Mol Cell Cardiol 2000;32:1735-44 47) Munch G, Bolck B, Brixius K, Reuter H, Mehlhorn U, Bloch W, Schwinger RH. SERCA2a activity correlates with the force-frequency relationship in human myocardium. Am J Physiol Heart Circ Physiol 2000 Jun;278(6):H192432 48) Bluhm WF, Kranias EG, Dillmann WH, Meyer M. Phospholamban: a major determinant of the cardiac forcefrequency relationship. Am J Physiol Heart Circ Physiol 2000 Jan;278(1):H249-55 49) Kaprielian R, del Monte F, Hajjar RJ. Targeting Ca2+ cycling proteins and the action potential in heart failure by gene transfer. Basic Res Cardiol 2002;97 Suppl 1:I136-45 50) Pieske B, Maier LS, Piacentino V 3rd, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation 2002 Jul 23;106(4):447-53 51) Holubarsch C, Ruf T, Goldstein DJ, Ashton RC, Nickl W, Pieske B, Pioch K, Lüdeman J, Wiesner S, Hasenfuss G, Posival H, Just H, Burkhoff D.: Existence of the Frank-Starling mechanism in the failing human heart. Circulation 1996;94:683-89 52) Schwinger RHG, Böhm M, Koch A, Schmidt U, Morano J, Eissner H-J, Überfuhr R, Reichart B, Erdmann E.: The failing human heart is unable to use the Frank-Starling mechanism. Circ Res 1994;74:959-69 53) van der Velden J, Boontje NM, Papp Z, Klein LJ, Visser FC, de Jong JW, Owen VJ, Burton PB, Stienen GJ. Calcium sensitivity of force in human ventricular cardiomyocytes from donor and failing hearts. Basic Res Cardiol 2002;97 Suppl 1:I118-26 54) Vahl CF, Timek T, Bonz A, Kochsiek N, Fuchs H, Schaffer L, Rosenberg M, Dillmann R, Hagl S : Myocardial length-force relationship in end stage dilated cardiomyopathy and normal human myocardium: analysis of intact and skinned left ventricular trabeculae obtained during 11 heart transplantations. Basic Res Cardiol 1997;92:26170 55) Mattiazzi AR. Músculo esquelético, cardiaco y liso. En Fisiología Humana de Houssay. Editado por Horacio R. Cingolani y Alberto Houssay. 7ma. Edición. Editorial El Ateneo. Buenos Aires. 2000, Cap. 4. 56) Dell'Italia LJ, Freeman GL, Gaasch WH : Cardiac function and functional capacity: implications for the failing heart. Curr Problems Cardiol. 1883;18:705-60 57) Chatterjee K, Sciammarella MG : Anormalidades en la contractilidad miocárdica: disfunción sistólica. En IC, editado por R.Oliveri. Editorial Médica Panamericana, Buenos Aires, 1999. 58) Ingwall JS : ATP synthesis in the normal and failing heart. In Heart Failure, Ed. by PA. Poole-Wilson, WS Colucci, BM Massie, K Chatterjee and AJS Coats. Churchill Livingstone Inc., New York, USA, 1997 59) Taegtmeyer H.: Energy metabolism of the heart: From basic concepts to clinical applications. Curr Prob in Cardiology. 1994;19:57-116 60) Braunwald E, Bristow MR : Congestive heart failure; fifty years of progress, Circulation 2000;102:IV-14-IV-23. 61) Nascimben L, Ingwall JS, Pauletto P, Friedrich J, Gwathmey JK, Saks V, Pessina AC, Allen PD: Creatine kinase system in mailing and nonfailing human myocardium. Circulation 1996;94:1894-1901 62) Saavedra WF, Paolocci N, St John ME, Skaf MW et al: Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ Res 2002;90:297304 63) Stanley WC, Chandler MP. Energy metabolism in the normal and failing heart: potential for therapeutic interventions. Heart Fail Rev 2002;7:115-30 64) Depre C, Vanoverschelde JL, Taegtmeyer H. Glucose for the heart. Circulation 1999 Feb 2;99(4):578-88 65) Tian R, Musi N, D’Agostino J, Hirshman MF, Googyear LJ : Increased adenosine monofosfato-activated protein 191 INSUFICIENCIA CARDIACA CRÓNICA. Fernando de la Serna. Capítulo 8 :Contractilidad. Metabolismo cardiaco Actualización 1er semestre/2006 kinase activity in rat hearts with pressure overload hypertrophy. Circulation 2001;104:1664 66) Chiappe de Cingolani, G: Generalidades sobre metabolismo. En Fisiología Humana de Bernardo A. Houssay, Directores Drs. Horacio E. Cingolani y Alberto B. Houssay. 7ª. Edición. Editorial El Ateneo, Buenos Aires, 2000.. 67) Rupp H, Zarain-Herzberg A, Maisch B. The use of partial Fatty Acid oxidation inhibitors for metabolic therapy of angina pectoris and heart failure. Herz 2002;27:621-36 68) Opie LH, Sack MN. Metabolic plasticity and the promotion of cardiac protection in ischemia and ischemic preconditioning. J Mol Cell Cardiol 2002;34:1077-89 69) Wolff AA, Rotmensch HH, Stanley WC, Ferrari R. Metabolic approaches to the treatment of ischemic heart disease: the clinicians' perspective. Heart Fail Rev 2002;7:187-203 70) Sambandam N, Lopaschuk GD, Brownsey RW, Allard MF. Energy metabolism in the hypertrophied heart. Heart Fail Rev 2002;7:161-73 71) Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, Ngoy S, Mortensen RM, Tian R. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 2002;106:2125-31 72) Scheuer J.: Metabolic factors in myocardial failure. Circulation 1993;87(supl VII):VII54-57 73) Shen W. , Asai K. and Uechi M. et al. Progressive loss of myocardial ATP due to a loss of total purines during the development of heart failure in dogs: a compensatory role for the parallel loss of creatine. Circulation 1999;100:2113-18. 74) Starling R.C. , Hammer D.F. and Altschuld R.A. Human myocardial ATP content and in vivo contractile function. Mol Cell Biochem 1998;180:171-177. 75) Beer M, Seyfarth T, Sandstede J, Landschutz W, Lipke C, Kostler H, von Kienlin M, Harre K, Hahn D, Neubauer S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol 2002;40:1267-74 76) O'Donnell MJ, Narayan P, Bailey MQ, Abduljalil AM, Altschuld RA, McCune SA, Robitaille PM : 31P-NMR analysis of congestive heart failure in the SHHF/Mcc-facp rat heart. J Mol Cell Cardiol 1998;30:235-41 77) Tian R, Nascimben L, Ingwall JS, Lorell BH : Failure to maintain a low ADP concentration impairs diastolic function in hypertrophied rat hearts. Circulation 1997;96:1313-19. 78) Eberli FR, Apstein CS, Ngoy S, Lorell BH : Exacerbation of left ventricular ischemic diastolic dysfunction by pressure overload hypertrophy: modification by specific inhibition on cardiac angiotensin converting enzyme. Circ res 1992;70:931-43 79) Vogt AM,; Kubler W : Heart failure: is there an energy deficit contributing to contractile dysfunction? Basic Res Cardiol 1998;93:1-10 192