Diagnostic*) por el laboratorio
Anuncio
 por el laboratorio")
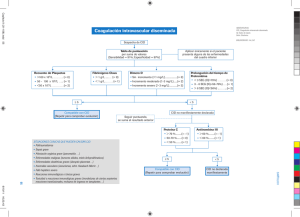
UNIVERSIDAD VERACRUZANA Unidad de Ciencias de la Salud Facultad de Bioanalisis Experiencia Recepcional Profesor de la materia : Dr. Jorge Aquino Carballo Monografia: "Diagnostic*) por el laboratorio clinico de Coagulacion Intravascular Diseminada CID, en Leucemias Promielociticas Agudas" Director de trabajo : M en H. QFB. Enrique de Jesus Gonzalez Cruz Alumna: Sofia Elisa Ramirez Ortega 28-JULIO-2008 preciado, la vida, por rodearme de personas maravillosas, por permitirme concluir una de mis grandes metas. Gracias a mis padres por los sacrificios que realizaron para poder cumplir esta meta, por estar conmigo siempre por su apoyo incondicional, alentarme a seguir adelante por ser los pilares de mi vida. A Fabian, gracia por estar ahi siempre en todo momento, por ser mi mas grande apoyo, por impulsarme para cumplir mis metas y suenos, gracias mi vida por luchar conmigo. Gracias Fabian, por mirar conmigo en la misma direccion. A mi familia por su apoyo incondicional por alentarme a seguir adelante por ensenarme a no redirme jamas, gracias a mi hermana por desvelarse conmigo y por comprender siempre. A mis abuelos por llenarme de amor y por brindarme su incondicional apoyo Gracias a mi director M en H. Q.F.B. Enrique de Jesus Gonzalez Cruz, por asesorarme en la elaboracion de este trabajo y compartir conmigo sus conocimientos, pero sobre todo gracias por compartir conmigo su gran calidad humana, por ensenarme a ver el mundo de diferente manera, por demostrarme que el arma mas importe en este mundo es la preparacidn y el conocimiento. Indice LEUCEMIA PROMIELOCITICA AGUDA Generalidades y Epidemiologia 1 Clasificacion 3 Sintomas 7 Diagnostico 7 Tratamiento 12 COAGULACION INTRAVACULAR DISEMINADA Generalidades 15 Antecedentes 17 Etiologia 19 Patologia 20 Diagnostico 26 DIAGNOSTICO POR EL LABORATORIO CLINICO DE COAGULACION INTRAVACULAR DISMEINDA EN LEUCEMIAS PROMIELOCITICAS 27 CASO CLINICO 31 INTRODUCCION La coagulacion sindrome intravascular frecuente en los diseminada pacientes (CID), con es un leucemias promielociticas agudas (M3), el curso de este sindrome puede llevar a la muerte al paci£nte, por ello de suma importancia diagnosticar oportunamente este sindrome. Para el diagnostico de este existen infinidad de metodos, que con los avances tecnologicos son certeros y rapidos; sin embargo y por desgracia la mayoria de los centros hospitalarios aun no cuentan con tecnicas o equipos tan sofisticados El diagnostico seguro se puede llevar a cabo con los examenes de rutina de un centro hospitalario, como es la citometria hematica complieta y los tiempos de coagulacion, por ello la importancia de dar a conocer los datos relevantes para poder diagnosticar la CID. El laboratorio clinico juega un papel crucial en el diagnostico y monitoreo de este sindrome. El objetivo principal de esta pequeno trabajo es resaltar la importancia del laboratorio clinico en el diagnostico de este padecimiento tan importante, ademas de recalcar aporte que las pruebas de rutina brindan. el gran LEUCEMLA PROMIELOCITICA AOUDA M3 GENERALIDADES Y EPIDEMIOLOGIA La Leucemia Promielocitica Aguda (M3) comprende el 80 % de las leucemias agudas en adultos y del 15-20 % en ninos. Es la leucemia mas frecuente en neonatos. W Tiene concordancia entre gemelos y es mas frecuente en desordenes congenitos (sindrome de Down, sindrome de Klinefelter), con una proportion levemente superior de hombres frente al sexo femenino. (2) Se encuentran distribuidas en todo el mundo, observando variaciones en el tipo de leucemias en diferentes grupos etnicos Su porcentaje en los paises mediterraneos es de 15% y en America Latina de 30%. Esta leucemia se diagnostica sobre todo entre los 15 y 60 anos. <4> El rango de edad de la leucemia promielocitica M3 es de 39 anos de edad y la supervivencia es de 18 meses, el hallazgo mas comun en el examen para el diagnostico inicial es el sangrado. <5) La M3 es una Leucemia Mieloide Aguda, siendo esta una neoplasia clonal del tejido hemopoyetico, caracterizada por la proliferation de celulas blasticas anormales de estirpe mieloide en la medula osea y menor produccion de celulas hematicas normales, condicionando anemia y trombocitopeniaJ 2 ) La leucemia mieloide aguda se produce por danos geneticos adquiridos (no heredados) en el ADN de las celulas en desarrollo dentro de la medula osea. Sus efectos son: 1) el crecimiento incontrolado y exagerado y la acumulacion de celulas llamadas "blastos leucemicos" los que no pueden funcionar como las celulas sanguineas normales 2) el bloqueo de la produccion de celulas normales de la medula, lo que resulta en una deficiencia de globulos rojos (anemia) y plaquetas (trombocitopenia) y de globulos blancos normales (especialmente neutrofilos, es decir, neutropenia) en la sangre. <3> Se algunos conocen factores que aumentan la predisposition al desarrollo de una leucemia promielocitica aguda, como lo son: 1. Radiaciones ionizates. 2. Sustancias quimicas derivadas del benceno o medicamentos citotoxicos 3. Alguna enfermedad hereditaria como sindrome de Bloom, anemia de fanconi, sindrome de Down, anemia de Diamond-Black, sindrome de Kostmann, sindrome de Klinefelter. 4. Virus HTLV 1, Epstein-Barr y tal vez otros <5> Se mencionan alteraciones citogeneticas como son t (15; 17) asociadas con la M3 y son oncogenes relacionados con esta leucemia son los pml-rara, ambas caracteristicas se encuentran en una frecuencia del 100% de este leucemia (y) CLASIFICACION Existen diferentes tipos de clasificaciones para las Leucemias, y especificamente para las leucemias mieloides la OMS (Organization Mundial de la Salud), esta es la forma en que cataloga las leucemias mieloides: o Leucemia Mieloide Aguda con displasias de muchas lineas celulares (precedida o no de SMD) o Leucemia Mieloide Aguda y Sindrome Mielodisplasico asociado a tratamiento previo o Leucemia Mieloide Aguda sin clasificar. (9) Existe tambien otra tipo de clasificacion y esta corresponde a la de la FAB (Grupo Franco-Estadounidense-Britanico), este es el mas amplio sistema de clasificacion morfologica/ histoquimica de la leucemia mieloide aguda fue el desarrollado por el Grupo (FAB, por sus siglas en ingles). Donde la leucemia promielocitica cae dentro de la siguiente clasificacion: Leucemia promielocitica (M3) Las caracteristicas de este trastomo son: a. La gran mayoria de las celulas son promielotitos anormales, con un patron caracteristico de granulation intensa. b. El nucleo varia enormemente en tamano y forma y a menudo es reniforme o bilobulado. <2) Variante de M3 (M3V) Una minoria de los pacientes con leucemia promielocitica aguda presenta granulos multiples. (g) La coagulation intravascular diseminada (CID) y t (15; 17) casi invariablemente estan asociados a la histologia de M3. La M3 es una enfermedad que avanza rapidamente y afecta a la mayoria de las celulas que se estan formando o primitivas (aun no comgletamente desarrolladas o diferenciadas). Estas celulas inmaduras no pueden desempenar suaTiinciones normales. La leucemia cronica avanza lentamente y permite el crecimiento de un mayor numero de celulas mas desarrolladas. (10) La M3 se caracteriza por la presencia de promielocitos patologicos con abundante coloration azurofila. Los nucleos tienen una forma irregular, presentando escotaduras o lobulaciones (11). Es caracteristica la presencia de abundantes cuerpos de Auer formando astillas apiladas. En muchas ocasiones, la simple observation de los mismos con el microscopio optico es suficiente para el diagnostico. Proliferation de blastos y promielocitos anormales. Promielocitos hipergranulares con nucleo reniforme o bilobulado y citoplasma denso con granulos azurofilos. Fig. 1 .- Leucemia Aguda Promielocitica M3. En la leucemia M3 microgranular, los promielocitos anormales tienen nucleos muy parecidos a los de los promonocitos, plegados y con cromatina muy fina que permite ver los diferentes lobulos por transparencia (A); estas celulas tienen, aparentemente, pocos granulos azurofilos muy finos. La busqueda cuidadosa muestra que casi siempre coexiste con estas celulas promielociticas con granulos de tamano "estandar" (B). t25) Se conocen dos variantes morfologicas de la leucemia promielocitica aguda, la M3 hipergranular ( n ) que representa la mayor parte de los casos y la variante microgranular denominada M3v que supone es inferior al 30% de los casos inferior. (9) Esta variante presenta las mismas caracteristicas de la forma de los nucleos pero los granulos son muy pequenos, suele presentarse acompanada de hiperleucocitosis. Ambos tipos de Leucemia Promielocitica son fuertemente positivas a las tinciones con mieloperoxidasa, negro de Sudan y cloroacetato-esterasa. Se han desarrollado anticuerpos monoclonales que se fijan especificamente al receptor APL-RAR, que se utilizan para el diagnostico I11) Fig. 2 .- Leucemia Aguda Promielocitica M3. Leucemia M3 teriida citoquimicamente para mieloperoxidasa. Los promielocitos son intensamente positivos. <25> Desde el punto de vista inmunofenotipico los promielocitos en la M3 muestran las caracteristicas siguientes: • tincion positiva para los antigenos miieloides CD 13 y CD33 • tincion negativa para los antigenos CD34, CD7, Cdl lb y CD14 • ocasionalmente positividad para el inarcador CD2 asociado a celulas T (mas frecuente en la variante M3v) En algunos pacientes se ha detectado la expresion del CD-56 (molecula de adhesion neural-celular) <n> SINTOMAS La mayoria de los pacientes comienzan sintiendose mal. Se cansan con mayor facilidad, pueden tener disnea cuando estan fisicamente activos. Pueden verse palidos a ralz de la anemia. Pueden notarse varios signos de hemorragia causados por un recuento muy bajo de plaquetasJ12) Estos signos incluyen hematomas o moretones que ocurren sin razon o debido a una lesion menor, la apariencia de manchas del tamano de la cabeza de un alfiler debajo de la piel, llamadas petequias, o perdida de sangre prolongada en cortes pequenos. Pueden presentarse: fiebre moderada, encias hinchadas, frecuentes infecciones menores, tales como pustulas o llagas perianales, cicatrizacion lenta de las heridas, dolor de huesos o en las articulaciones. <13) DIAGNOSTICO DE LEUCEMIAS M3 Para el diagnostico de la M3 se toman en cuenta diferentes; los primeros hallazgos en los estudios de laboratorio son: anemia y trombopenia en el hemograma, a veces con cifras extremas (anemia normocitica, normocronica e hiporregenerativa). La mitad de los pacientes presentan cifras leucocitarias con neutrofilos < 10 x 109 /L con hipolobulacion, hiposegmentacion u otras alteraciones displasicas. SERIE ROJA Hb Hto Eritro. VGM HbGM CMHb RDW VALORES NORMALES 17.8 g/dl 13.8 41.4 53.40% 4.6 5.6 x 106/|jL 100 Fl 83 28 32 pg 34 g/dl 32 1.20% 12.8 6.6 g/dl 19.40% 2.2 x 106/(jL 88.2 Fl 30 pg 34 g/dl 29.60% TIEMPOS DE COAGULACION Tiempo de Protombina % de actividad INR Tiempo Parcial de Tromboplastina fibrinogeno Sofia Elisa Ramirez Orteea 7 50 0.5 15 250 VALORES NORMALES 14 seg 10 100% 70 2 1 40 seg 20 650 mg/dl 350 -7- SERIE BLANCA LEUCOCITOS TOTALES Netrofilos totales Netrofilos segmentados Neutrdfilos en banda Metamielocitos Promielocitos Blastos Eosinofilos Basofilos Monocitos Linfocitos 28.600 /pL % 61 59 2 0 0 0 18 1 1 SERIE PLAQUETARIA Plaquetas 100 19 4500 Absolute) 17446 16874 572 0 0 0 5148 286 286 5434 11000 Rango Normal /pL 1800 1800 0 0 0 0 20 20 0 1000 Absolute /|jL 7700 7700 700 700 0 0 450 100 800 4800 VALORESNORMALES 150 400x 103/^L Las celulas blasticas comprenden desde el 5 al 95% del total leucocitario y estan presentes en el 80-85% de los pacientes, existiendo formas paucitoblasticas (Leucemia oligoblastica). (14) Fig. 3 Leucemia Aguda Promielocitica M3. C61ula Faggot, la c6lula tiene cambios de pseudos Pelger, con nucleos poco lobulados pero con cromatina muy gruesa. i25) Fig. 4Leucemia Aguda Promielocitica M3. Promielocitos hipergranulares y c61ulas Faggot. C25) Sus caracteristicas morfologicas espetificas como son los bastones de Auer, astillas citoplasmicas, etc., junto con la reactividad citoquimica y la aplicacion de anticuerpos monoclonales, clasifican la LMA dentro de las diferentes variedades de la clasificacion FAB. <15> La biopsia de medula osea es complementaria al aspirado, ofrece una imagen amplia de toda la arquitectura y celularidad medular y esta especialmente indicada en casos de aspirados pobres en grumo o "blancos" (medulas empaquetadas) o con sospecha de fibrosis medular con Citometria de flujo. Existen alteraciones bioquimicas inespetificas consecuencia de la liberation de sustancias intracelulares de las propias celulas leucemicas o de la disfuncion organica por infiltration leucemica (hiperuricemia, hipomagnesemia, hipo o hipercalcemia, aumentos LDH, aumentos de la lisotima, etc.). <2> Fig. 5.- Leucemia Aguda Promielocitica M3. Medula Osea (25-26) En cuanto a la Histoquimica se pueden realizar tinciones histoquimicas especiales en los especimenes de biopsia de medula osea de todos los ninos con leucemia aguda a fin de confirmar el diagnostico. Las tinciones mas empleadas comprenden la mieloperoxidasa, el PAS, el negro B de Sudan y la esterasa. En casi todos los casos el patron de tincion con estas tecnicas histoquimicas permite diferenciar la LMA y de la LLA. (is> Ademas de la morfologia convencional, se dispone de varias tecnicas para confirmar la sospecha de una leucemia promielocitica aguda como la inmunofluorescencia utilizando anticuerpos monoclonales (15> Los marcadores mieloides espetificos son CD 13, CD33, y CD 15 y son coexpresados en la inmensa mayoria de las variantes M0-M5 (con considerable variation en la coexpresion. Desde el punto de vista de los hallazgos citogeneticos: La mayoria de las leucemias promielotiticas (M3) exhiben t (15; 17). El diagnostico incontrovertible de la estirpe es un paso esencial para el manejo de los pacientes y puede requerir de la intervention de especialistas en hematopatologia. <10' El analisis citogenetico proporciona una de las mas solidas evidencias pronosticas disponibles, prediciendo asi resultados tanto de la induction de remision como la terapia posremision. Las anormalidades citogeneticas que indican un pronostico bueno son la t (8; 21), inv (16) y t (15; 17). La citogenetica normal presagia una LMA de riesgo regular. Los pacientes con LMA que se caracteriza por deleciones de los brazos largos o monosormas de cromosomas 5 6 7; por desplazamientos o inversiones de cromosoma 3, t(6;9), t(9;22); o por anormalidades de cromosoma llq23 tienen pronosticos particularmente precarios con quimioterapia. Estos subgrupos citogeneticos vatitinan el resultado clinico en pacientes de edad avanzada con LMA al igual en pacientes jovenes. Los genes de fusion formados en t (8; 21) e inv <16> pueden ser detectados por reaction en cadena de polimerasa transcriptasa reversa (RT-PCR, por sus siglas en ingles), la cual indicara la presencia de estas alteraciones geneticas en algunos pacientes en quienes la citogenetica estandar era tecnicamente inadecuada. RT-PCR no parece identificar un numero significativo de pacientes con genes de fusion de riesgo favorables con citogenetica normal. Las anormalidades del gen MLL (cromosoma llq23) tambien se puede detectar usando RT-PCR y se pueden detectar en algunos casos de leucemia con citogenetica normal. Estas tecnicas moleculares de diagnostico no estan disponibles facilmente. <15> en oncologia pediatrica y recibir el tratamiento en centros oncologicos especializados o en hospitales dotados con las Instalaciones adecuadas de soporte. En paralelo con las crecientes tasas de supervivencia en los ninos tratados por LMA ha venido creciendo tambien la conciencia sobre las secuelas de largo plazo de los diversos tratamientos. En el caso de los ninos sometidos a quimioterapia intensiva, incluido el uso de las antraciclinas, es fundamental la vigilancia continua de la funcion cardiaca. Tambien se sugiere practicar examenes periodicos de las funciones renal y auditiva. Ademas, la irradiation corporal total que precede al trasplante de medula osea incrementa el riesgo de falla del crecimiento, disfuncion gonadal y tiroidea y aparicion de cataratas. Actualmente se consiguen tasas de remision cercanas al 80-90% en ninos, 70% en adultos jovenes, 50% en personas de edad intermedia y 25% en los ancianos. La media de supervivencia es de aproximadamente 12 m. Si se logra remision, 25% viviran alrededor de 2 anos y un 10% alrededor de 5 anos. Actualmente con el TMO en primera remision completa se logran porcentajes de supervivencia libre de enfermedad a los 4 anos de eritre 30 y el 50%, segun sea autologo o alogenico (17) En cuanto a la quimioterapia se han llevado a cabo en los ultimos anos varios estudios multicentricos utilizando varias combinaciones de acido trans-retinoico y quimioterapia, y trioxido de arsenico. Los resultados de estos estudios indican que el uso concomitante de acido trans-retinoico (16> e idarrubicina es superior al uso secuencial, tanto en lo que se refiere a los efectos antileucemicos como a un mejor control del sindrome de diferenciacion de la APL (tambien llamado sindrome RA-APL acido retinoico-leucemia promielocitica aguda- o sindrome del acido retinoico o sindrome de diferenciacion en la APL) caracterizado por fiebre, disnea, aumento de peso, infiltrados leucocitosis. Este sindrome puede ser fatal. <17> La administration intravenosa de trioxido de arsenico en dosis de 0.15 mg/kg/dia tanto durante la fase de induction como durante la fase de consolidation en leucemias promielotiticas agudas refractarias al tratamiento estandar o en recaidas de la APL ocasiona resultados bastante satisfactorios: en los estudios realizados, el 87% de los pacientes tratados con el trioxido de La tecnica de RT-PCR es esencial para documentor la presencia de genes de fusion PLM-RARa y tratar a los pacientes con los regimenes menos intensos a base de acido t-retinoico y antraciclinas. Tambien es importante para detectar la minima enfermedad residual. Otros metodos para la confirmation de una APL son el analisis citogenetico, la hibridacion fluorescente in situ y la hibridacion de Southern (16) TRATAMIBNTO Generalmente, el tratamiento se divide en dos fases: 1) Induction (cuyo proposito es alcanzar la remision) y 2) consolidation/intensification posteriores a la remision. El tratamiento posterior a la remision puede constar de un numero variable de cursos de quimioterapia intensiva con o sin trasplante de medula osea. Por ejemplo, en el actual estudio national de los Estados Unidos (CCG-2961) se usa el tratamiento de induction seguido de dos cursos de quimioterapia, uno de los cuales se hace con citarabina en altas dosis. Un regimen pediatrico de LMA desarrollado por el Consejo de Investigation Medica del Reino Unido promulga el uso de tres o cuatro cursos de quimioterapia luego del curso initial de quimioterapia de induction de remision. El tratamiento de mantenimiento no hace parte de la mayoria de los protocolos pediatricos de LMA con la exception de los protocolos del grupo de Berlin-Francfort-Munster (BFM). En ninos con PML que han recibido tratamientos radicales, el tratamiento de mantenimiento no parece tener valor. El tratamiento de la PLM suele asociarse con una mielosupresion pronunciada y prolongada y otras complicaciones. Se ha empleado el tratamiento con factores de crecimiento hematopoyeticos (G-CSF, GM-CSF, por sus siglas en ingles) con la intention de reducir la toxicidad asociada con la mielosupresion acusada pero no tiene influencia sobre el resultado ultimo. Practicamente todos los estudios aleatorios en adultos del uso de factores de crecimiento hematopoyeticos (G-CSF, GM-CSF) han demostrado una reduction significativa del tiempo que transcurre hasta la recuperation de los neutrofilos, pero con grados variables de reduction de la morbilidad y poco o ningun efecto sobre la mortalidad. A causa de la intensidad del tratamiento que se instaura en los ninos con PML, estos deben estar bajo el cuidado de especialistas arsenico alcanzaron la remision completa. Los estimados de supervivencia de Kaplan-Meier a los 18 meses fueron del 66% globalmente y del 56% para la supervivencia sin recaidas- <16> Las nuevos descubrimientos como la de los Investigadores del Programa de Diferenciacion y Cancer del Centro de Regulation Genomica de Barcelona han demostrado que las interconexiones entre los mecanismos de las enzimas que metilan el ADN (metiltransferasas) y el complejo de proteinas. Polycomb en el silenciamiento de genes estan en la base de la leucemia promielocitica aguda. Los dos mecanismos antes citados interconectados bioquimicamente entre si podian regular genes de mahera conjunta. Se pensaba que estos dos mecanismos que tienen un papel importante en el desarrollo embrionario, eran independientes uno del otro, pero han visto que colaboran entre si y que, cuando se regulan erroneamente van apagando genes importantes para el control de la proliferation celular. Los dos mecanismos no solo estan interconectados, sino que uno refuerza al otro y, es mas, uno necesita del otro. Esto es relevante para una futura terapia farmacologica, puesto que corrigiendo la actividad de uno de los dos mecanismos regulados erroneamente se revierte la actuation del otro. Para la metilacion ya hay posibles compuestos quimicos y se esta buscando un inhibidor de las proteinas Polycomb <18) COAGULACION INTRAVASCULAR DISEMINADA GENERALIDADES La Coagulation Intravascular Diseminada (CID), es un sindrome con alta prevalecia en Leucemias Promielociticas, donde se encuentra afectada la hemostasis normal del organismo. Distintos factores son capaces de de poner en marcha la formation de trombina intravascular, dando como resultado la presencia de la CID en los pacientes, a continuation se mencionan los factores biologicos en que se presentan con mayor frecuencia la CID (19.3) o Desencadenantes directos: pueden ser endogenos (procedentes de pancreas, celulas malignas o neutrofilos) o exogenos (venenos de serpiente) o Factor tisular (FT): cuando hay rotura de las membranas celulares se liberan a la circulation sanguinea extractos tisulares y se inicia directamente la CID o Varias enzimas proteoliticas: inducen la formation de fibrina o Desencadenantes indirectos: alterando el endotelio vascular inducen la liberation de factor tisular desde el subendotelio, el cual es un desencadenante directo de CID. Los estimulos que lesionan el endotelio tambien pueden activar y agregar las plaquetas o Virus o Microorganismos gramnegativos o Inmunocomplejos. (19) La patologia consiste en la formation incontrolable de fibrina dentro de la microcirculation, produciendo una oclusion trombotica de vasos sanguineos de mediano y pequeno calibre, comprometiendo un adecuado aporte de sangre a los diferentes organos vitales, que unido a alteraciones metabolicas y hemodinamicas contribuye a la falla multiorganica. <2> Se observan episodios hemorragicos, donde llegan a manifestarse en sangrados en tres sitios anatomicos, distintos, al mismo tiempo (*) La presencia de hematuria, hemorragia gastrointestinal, epistaxis, equimosis y petequias, son algunas manifestaciones. La hemorragia puede ser profusa y causar la muerte, por otra parte la obstruction de la microvasculatura por trombos causa anoxia tisular y micro infartos en corazon, rinones, cerebro, higado y pancreas, conduciendo al shock. (4>La finalidad de este trabajo es realizar una recopilacion bibliografica donde se resalte la importancia del Laboratorio Clinico en el diagnostico de este sindrome para lograr una detection oportuna y certera en el menor tiempo posible Fig 6.- Manifestaciones clmicas deCID en una pediatrico con Leucemia M3 ANTECEDENTES El sindrome de coagulacion intravascular diseminada (CID) consiste en la generation extensa de trombina en la sangre circulante con el consiguiente consumo de factores de coagulacion y plaquetas, posible obstruction de la microcirculation y activation secundaria de la fibrinolisis. El consumo de factores de coagulacion y plaquetas conduce a la aparicion de hemorragias y las trombosis obstructivas de la microcirculation a necrosis y disfunciones organicas. (4) Haciendo un poco de historia la primera description de la CID se debe a WH Seegers en su trabajo Factors in the control of bleeding, publicado en 1950. Ratnoff y Prichard en 1955, publicaron un articulo extenso y detallado, que titularon Hemorrhagic State During Pregnancy. Krevans (1957) en el cual reconocio a CID asotiada a reaction transfuntional. Penick (1958) identified la trombicitopenia y los niveles disminuidos del factor VIII en la CID. En 1959 se emplea por primera vez el termino Coagulacion Intravascular Diseminada por Mckay y Hardaway. El mecanismo por el cual el CID produce hemorragia fue aclarado por Lasch (1961), cuando se introdujo el concepto de "coagulopatias de consumo Mackay (1965) correlaciono las presentaciones clinicas con la patologia. En el mismo ano, Rodriguez Erdman publico una extensa revision del tema relacionando las endotoxinas, la reaction de Schwatzman y el CID. Merskey (1996), desarrollo un metodo para medir los productos de la degradation del fibrinogeno y propuso la necesidad de concordancia entre los hallazgos de laboratorio con la del CID con el contexto clinico en el que se desarrolla <5)Los estudios mas recientes como los del HOSPITAL PROVINCIAL CLiNICO-QUIR0RGICO-DOCENTE "JOSE RAMON LOPEZ TABRANE" presenta una publication en el ano 2003 denominada "Coagulacion intravascular diseminada. Diagnostico de laboratorio por los metodos tradicionales. En la cual hace enfasis en las tecnicas que'usualmente se usan en el laboratorio de manera rutinaria para el diagnostico oportuno de la CID. Esta publication tambien explica ademas cuales son las investigaciones de mayor importancia y utilidad para el medico, y Las diferentes opciones con las que puede contar de acuerdo a condiciones concretas del paciente. '20' J. MATEO ARRANZ Y J. FONTCUBERTA BOJ de la Unitat d'Hemostasia i Trombos del Departament dUematologia. Hospital de la Santa Creu i Sant Pau. Barcelona presenta el articulo "Coagulacion intravascular diseminada. Un viejo concepto para un hecho clinico complejo."Donde describe de manera detallada los factores en los cuales se presenta la CID, su fisiopatologia y mecanismo de accion, asi como tambien hace mencion de las pruebas de laboratorio que se realizan para el diagnostico de la CID. W ETIOLOGIA El termino de Coagulacion Intravascular Diseminada (CID), 19 implica coagulacion dentro de los vasos sanguineos ( ). Es un estado en que el equilibrio normal de la hemostasis se ha alterado, permitiendo una formacion inapropiada e incontrolable de fibrina dentro de los vasos sanguineos. La microcirculation es la primera en afectarse, con depositos difusos de fibrina en capilares, arteriolas y venula Durante la formacion de la fibrina, varias proteinas de la coagulacion, en especial el fibrinogeno, se consumen a una velocidad mayor a la que se sintetizan, el resultado es una deficiencia multiple de factores <19). Como la activacion de los factores de la coagulacion desencadena siempre de manera simultanea la activacion del sistema fibrinolitico, se forma tambien plasmina. A continuation aparecen los productos de degradation de la fibrina, conforme aparecen los productos de degradation de la fibrina, interfieren en la funcion plaquetaria y la formacion de mas fibrina. Las manifestaciones clinicas son el resultado de la cadena de acciones inducida por trombina y plasmina al circular en el torrente sanguineo <9)El efecto de los trastornos causados por la coagulacion intravascular diseminada son los e danos a los tejidos, las celulas sanguineas o el endotelio. Se piensa que en la lesion tisular o de celulas sanguineas, sustancias semejantes a la tromboplastina, entran a al circulation y activan el factor extrinseco. (4) De forma fisiologica existe un equilibrio en la hemostasia limitandose la extension de los depositos de fibrina y agregados de plaquetas a las necesidades estrictas para mantener intacta la continuidad del sistema vascular. Cuando un estimulo es demasiado potente y se superan los mecanismos limitantes, -entonces se desarrollara una CID. La intensidad de las alteraciones en la CID dependen de: - La potencia y rapidez con la que actua el desencadenante - Factores modificadores que determinan el que se deposite la fibrina y el lugar en que lo hace. Estos factores son: • Bloqueo del sistema mononuclear fagotitico. • Inhibition de la fibrinolisis. • Potentiation del sistema adrenergico. • Elevacion de los lipidos plasmaticos. • shock (con hipoxia aguda, hipotension, acidosis y tal vez liberation de FT de los organos lesionados).(4) t .S PATOLOGIA La CID se inicia cuando fracasan los mecanismos que limitan la generation de trombina a las zonas donde es necesaria. La via predominante de la activation de la coagulacion en la CID esta mediada por el factor tisular. Existen tejidos que expresan constitutivamente factor tisular que en condiciones normales no estan expuestos a la sangre: adventicia de los vasos sanguineos, mucosas, piel, tejido nervioso, glomerulo renal, liquido amniotico (2). La exposition del factor tisular puede ocurrir por traumatismo (sobre todo encefalico), problemas obstetricos o dano endotelial difuso. El factor tisular puede aparecer en sangre por celulas neoplasicas o por activacion de celulas endoteliales o monocitos. En otras ocasiones son enzimas proteoliticas los causantes de CID ya sean de origen endogeno (enzimas pancreaticos, de celulas neoplasicas, de neutrofilos) o exogeno (venenos de serpiente, etc.). I1) El impacto de CID sobre la via tanto intrinseca como extrinseca afecta directamente en los siguientes aspectos: • 1° Disociar lo fibrinopeptidos A y B del fibrinogeno en forma de monomeros de fibrina. • 2° Activacion de los factores V y VIII que se consumen en la cascada de formation de fibrina. • 3° Contribuir a la trombocitopenia al inducir la agregacion plaquetaria <9> La plasmina se forma dentro de la circulation al activarse el plasminogeno por los activadores tisulares y por los factores de contacto. La plasmina dirige a la fibrina y al fibrinogeno para dar lugar a los productos de degradation de la fibrina, los cuales actuaran como anticoagulantes estas sustancias como la trombina que tiene diversos efectos sobre la hemostasia, todos ellos interviene en la patogenia de la enfermedad, primeramente , causando escision proteolica de los fibrinopeptidos A y B del fibrinogeno , para formar monomeros de fibrina, dando como resultado que dentro del plasma , la concentration de fibrinogeno aumenta.(4) Esto produce que los valores de fibrinogeno se reducen adicionalmente debido a su digestion por la plasmina la cual ataca tambien a los factores V, VIII, IX y XI. (9) La deficiencia inducida del fibrinogeno y de los factores II, V, VIII, IX y XIII, asi como el consumo de plaquetas, conducen a hemorragia y a la formacion de coagulos de manera simultanea. Si La regeneration de trombina excede a la plasmina se forma mas fibrina que la degradada y resulta en una trombosis difusa. La degradation del fibrinogeno, de lo factores V, VIII y IX y de otras proteinas del plasma, por action de la plasmina, contribuye a la hemorragia. <9) Mecanismo de inicio La CID se inicia cuando fracasan los mecanismos que limitan la generation de trombina a las zonas donde es necesaria. La via predominante de la activation de la coagulacion en la CID esta mediada por el factor tisular. Existen tejidos que expresan constitutivamente factor tisular que en conditiones normales no estan expuestos a la sangre: adventicia de los vasos sanguineos, mucosas, piel, tejido nervioso, glomerulo renal, liquido amniotico (5)- La exposition del factor tisular puede ocurrir por traumatismo (sobre todo encefalico), problemas obstetricos o dano endotelial difuso. El factor tisular puede aparecer en sangre por celulas neoplasicas o por activation de celulas endoteliales o monotitos. En otras ocasiones son enzimas proteoliticas los causantes de CID ya sean de origen endogeno (enzimas pancreaticos, de celulas neoplasicas, de neutrofilos) o exogeno (venenos de serpiente, etc.). CID6(2) Lesion endotelial El endotelio vascular regula la formacion del coagulo y su lisis. Se puede alterar por germenes, toxinas, mediadores inflamatorios (endotoxina, complemento, citocinas proinflamatorias o proteasas granulocitarias) o por lesion fisica (6,10) Los agentes infecciosos (bacterias, virus, hongos, protozoos y rickettsias) pueden produtir lesion directa del endotelio vascular (incluso denudation), o perturbation de las celulas endoteliales, lo cual ocasiona un desequilibrio de diferentes sistemas biologicos. En estas situaciones el endotelio se torna en procoagulante ya que expone factor tisular, disminuye la expresion de trombomodulina, libera PAI-1, PAF (factor activador de las plaquetas), endotelina y factor von Willebrand (FVW) y disminuye la sintesis de oxido nltrico Fig. 7. La exposition del subendotelio a la sangre produce adhesion y agregacion de las plaquetas y la puesta en marcha de los mecanismos de la coagulacion. Estas alteraciones, por si solas, pueden desencadenar una CID, o bien, rebajar el umbral para que esta se inicie en respuesta a un estimulo subsiguiente. <n> Incremento de la actimdad procoagulantel Los leucocitos estimulados por la endotoxina, el TNF o la IL-1 (interleucinas) participan en la lesion endotelial. Ademas los monocitos y macrofagos sintetizan factor tisular en respuesta a la endotoxina, expresan factor tisular en su superficie e inician la via de la coagulacion por activacion del factor VII. La production de factor tisular esta mediada por la IL-1 y el TNF. Las celulas endoteliales responden de manera similar. Las plaquetas aportan superficies fosfolipidicas, factor V, lipoproteinas, etc., que participan como cofactores en la generation de factor Xa y en la activacion de la protrombina a trombina- <6> Disminucidn de las actividades anticoagulantes Las actividades anticoagulantes estan representadas por los inhibidores plasmaticos (antitrombina, _2-macroglobulina, TFPI), los inhibidores celulares (PGI2, oxido mtrico) y la via de la proteina C. Las celulas endoteliales sintetizan sustancias heparinoides (proteoglicanos) que unen antitrombina. La antitrombina desciende en la CID por consumo. En condiciones de exposition masiva de factor tisular, el TFPI es insuficiente para inhibirlo. En modelos experimentales, la infusion de endotoxina, IL-1 o TNF reduce la expresion de trombomodulina en las celulas endoteliales y aumenta el factor tisular. Esto produce un desequilibrio procoagulante. La proteina C activada (PCa) ejerce un importante efecto anticoagulante y estimulante de la fibrindlisis. <12-13)Activacion de la fibrindlisis La fibrindlisis esta activada en el curso de la CID, por ello encontramos productos de degradation del fibrinogeno y de la fibrina en estos pacientes <13>- La activation de la fibrinolisis es un mecanismo compensatorio en la CID, secundario a la activacion de la coagulation. Cuando se estimula el endotelio con endotoxina el PAI se libera del endotelio. El incremento de la sintesis y liberation de activadores del plasminogeno en la CID, que puede estar mediado por la trombina, produce activacion de la fibrinolisis con una rapida lisis de los trombos microvasculares. La persistencia de los trombos puede explicarse por tin mayor predominio procoagulante que supere a los activadores del plasminogeno liberados, porque el PAI liberado bloquea los activadores del plasminogeno y por un incremento del inhibidor de la fibrinolisis activable por trombina (TAFI). El uso de farmacos antifibrinoliticos puede favorecer la persistencia de los trombos y la lesion tisular <6>Interacdon de los diversos factores La CID tiene lugar si tres procesos suceden simultanea o consecutivamente: activacion de la coagulacion, reacciones vasomotoras e inhibition de la fibrinolisis. Excepto en algunos estados patologicos (toxinas animales, venenos, dispositivos artificiales, entre otros) las actividades procoagiilantes estan suprarreguladas y las anticoagulantes infrarreguladas. La expresion del factor tisular en los monocitos es importante para desencadenar una coagulacion intravascular. Se producen monomeros de fibrina y la formacion de fibrina soluble. La fibrina soluble puede eliminarse de la circulation o ser lisada por la Action del sistema fibrinolitico activado. Pero cuando se confina en algunas zonas, puede precipitar y polimerizar. Si las actividades fibrinoliticas estan inhibidas, los microtrombos ricos en fibrina pueden persistir, dando como resultado lesion endotelial, lesion tisular y, finalmente, insuficiencia organica. Dependiendo de la intensidad de la reaction del organismo hacia el agente causante, o dependiendo de la prevaletia de los diferentes desencadenantes de la CID, la trombosis microvascular o la hemorragia seran el hecho dominante. La lesion esta causada por la combination del consumo de factores de la coagulacion y plaquetas, la activacion de la fibrinolisis, el efecto de los productos de degradation fibrinoliticos y la lesion vascular. <3) El curso clinico de la coagulacion intravascular diseminada puede ser agudo o cronico, en la forma aguda es en la que se reconoce con mayor facilidad ya que inicia con una hemorragia subita; la forma cronica se desarrolla con el transcurso y puede tener sintomas leves o no. Del 80 al 90% de los pacientes que presentan coagulacion intravascular diseminada de forma aguda y el resto cronica. M Manifestaciones clinicas: Las manifestaciones pueden estar parcialmente eclipsadas por las de la enfermedad desencadenante. La trombosis macrovascular es rara en la CID ya que la formacion de trombos estables esta limitada por la fibrinolisis compensatoria, por el agotamiento del fibrinogeno y por la inhibition de la polimerizacion de la fibrina debida a los PDF (Polimeros de Fibrina). Los defectos de coagulacion que favorecen la hemorragia son la carencia por consumo de los factores de la coagulation y de las plaquetas, la hiperfibrinolisis y la deficiente polimerizacion de la fibrina. La gravedad de la CID es mayor si los mecanismos compensatorios estan disminuidos. En este punto interviene la capacidad sintetizadora del higado para reemplazar los factores de la coagulacion y los inhibidores consumidos, y la capacidad de la medula osea para generar plaquetas. (4) Debido a la trombosis microvascular podemos encontrar disfunciones de organos, a veces tambien causados por otras circunstancias concurrentes propias de la evolution de la enfermedad de base. Puede existir insuficiencia renal y necrosis tubular aguda, insuficiencia hepatica, insuficiencia respiratoria (sindrome de distres respiratorio del adulto) y alteraciones del estado neurologico. En la CID, la hemorragia es la manifestation predominante (70-90% de los pacientes). Los sitios de hemorragia mas frecuentes son: la Purpura cutanea equimotico-petequial, la hemorragia microvascular por puntos de puncion, de insertion de cateteres o drenajes, por superficies cruentas o por heridas quirurgicas. Suele haber microhematuria o, mas raramente, hematuria macroscopica. En pacientes graves pueden aparecer hemorragias digestivas, en general por lesiones agudas, y hemoptisis en pacientes intubados. Las hemorragias intracraneales, suprarrenales, intraparenquimatosas o en cavidades (abdominal, pleural) son menos frecuentes. Los pacientes con Coagulacion intravascular Aguda tienden a inanifestar sintomas hemorragicos, pueden presentar sangrados por en tres sitios, por lo menos, al mismo tiempo, (5) estos tienden a corresponder a los tejidos afectados por el vento desencadenante. Hematuria, Hemorragia gastrointestinal, Epistaxis, escurrimiento en sitios de punciones, equimosis y petequias, son algunas manifestaciones. La hemorragia puede ser profusa y causar la muerte, por otra parte la obstruction de la microvasculatura por trombos causa anoxia tisular y micro infartos en corazon, rinones, cerebro, higado y pancreas, conduciendo al shock. <4>- DIAGNOSTICO Para la detection de CID no existe una prueba de laboratorio especiflca para su diagnostico, ello debido a su fisiopatologia compleja. Sin embargo, existen diferentes hallazgos que nos permiten realizar un diagnostico debemos tener en cuenta cual es la etiologia de CID ya que dependiendo de esta podremos realizar una adecuado diagnostico, en la siguiente tabla se muestra cuales serian los criterios de diagnostico para la CID <14) Clasificacion Biologica CID Clinica CID CID Complicado Definicidn Defecto hemostatico manifestaciones clinicas Criterios Diagnostico D-Dimeros Elevados sin 1 criterio mayor para consumo de plaquetas o factores de coagulacion 2 criterios menores para consumo de plaquetas o factores de coagulacion Defecto hemostatico con Lo mismo que arriba + manifestaciones isquemias o sangrado microvascular y/o trombosis hemorragicas Defecto hemostatico con manifestaciones isquemicas o hemorragicas que pone en Lo mismo que arriba + fallo riesgo la funcion de organos o organico (unico o multiple) el pronostico del paciente « P DIGANOSTICO POR EL LABORATORIO CLINICO DE COAGULACION INTRAVASCULAR DISEMINDA EN LEUCEMIAS PROMIELOCITICAS Las pruebas de rutina para el diagnostico de CID, en el laboratorio clinico son las siguientes: 1. 2. 3. 4. 5. 6. Examen de Extendido Sanguineo Recuento plaquetario Tiempo parcial de Tromboplastina Tiempo de Pro trombina Tiempo de Trombina Productos de degradation de la fibrina y del fibrinogeno Extendido sanguineo: revela la trombocitopenia y puede mostrar el cuadro morfologico de la anemia hemolitica <21) CID aguda. Algunos autores describen tambien la presencia de microesferocitos. Muchos tienen una ligera desviacion izquierda del leucograma. Las alteraciones hematicas se deben al aumento de la fragilidad de los hematies debido al paso por los vasos sanguineos cuya luz esta obstruida por un trombo. En la CID cronica los esquisocitos esquistositos se encuentran presentes en el 90 % de los casos I22) Fig 8 .- Promielocitos en Leucemia M3. Tincion de Wright <25) Sofia Elisa Ramirez Orteea - 27 - Recuento plaquetario: Disminuidas, a veces marcadamente, constituye la anormalidad mas frecuente de la CID y a menudo un temprano indicador de ella. Suelen encontrarse conteos por debajo de 150,000 o recuentos que disminuyen en forma constante. Comunmente aparecen macro plaquetas representando las plaquetas jovenes que se forman en sustitucion de las utilizadas. (23) Tiempo Tromboplastina de Partial: La tromboplastina en presencia de iones de calcio actua sobre la protombina y produce trombina, la cual actua sobre el fibrinogeno y lo convierte en fibrina, sustancia que es responsable de la formacion del coagulo. Como la cefalina reemplaza las plaquetas, la prueba resulta normal en presencia de deficiencias plaquetarias cualitativas o cuantitativas. Como el extracto de cefalina no coagula el plasma hemofilico tan rapidamente como el normal, se le llamo tromboplastina " Partial" para diferenciarlo de la tromboplatina " Completa", la cual coagula ambos plasmas en el mismo tiempo. <23) Se encuentra prolongado en el 50 % al 60 % de los casos con CID aguda. En la CID cronica se encuentra normal o prolongado. Evalua la via intrinseca (factores XII, XI, X. IX, VIII y parcialmente I, II, y V). (22) Tiempo de Protrombina: La protombina es el precursor inactivo de una enzima proteolitica, la trombina se sintetiza en el higado y la vitamina k resulta esential para este proceso. <23' Se encuentra p prolongado en el 75 % de los casos de CID aguda, el 25 % lo tiene normal o acortado. En la CID cronica suele ser normal o prolongado. Esta prolongado en presencia de graves depresiones de factores de la coagulacion en una CID rapida. Evalua la via extrinseca (factores II, V, VII y parcialmente el I). <22> Tiempo de Trombina: El tiempo necesario para que el plasma coagule despues de la adicion de trombina se describe como tiempo de trombina. Esta prueba no resulta muy sensible a los descensos en el nivel de fibrinogeno ya que por debajo de 100 mg/dl tienen valores prolongados, pero es muy sensible en anormalidades congenitas en la estructura molecular del fibrinogeno, a la presencia o ausencia de calcio, en la detection de inhibidores antitrombina y productos de degradation de fibrinogeno - fibrina y en la administration terapeutica de heparina La trombina actua directamente sobre el fibrinogeno para convertirlo en fibrina. El calcio acelera esta reaction, pero no Se anade en esta prueba. La velocidad de coagulacion con la trombina es una funcion de la concentration de fibrinogeno o de la action de los inhibidores de la action de la trombina sobre el fibrinogeno. (23) Usualmente prolongado, en muy pocos casos es normal o esta acortado. Su prolongation ocurre en tres circunstancias: • Cuando disminuye considerablemente el fibrinogeno. • Cuando existen PDF circulantes que impiden la polimerizacion de los monomeros de fibrina. • Cuando la sangre esta heparinizada. <22) Valores de los productos de degradation del fibrinogeno v de la fibrina (PDF): El fibrinogeno es el precursor soluble de la proteina formadora del coagulo la fibrina. Esta sustancia ha sido designada como factor I. Es un polipeptido de PM 340.000, producido en el higado y el sistema reticulo endotelial, que circula solubilizado en el plasma y que por action de la trombina forma monomera soluble que bajo influencia del factor XIII se polimeriza al formar un coagulo de fibrina en Cid se encuentra elevado <23> Titulo de PDF: Aumentado en el 85 al 100 % de los pacientes. En la CID cronica los PDF se encuentran usualmente elevados. El aumento de los PDF asi como la circulation de monomeros de fibrina se observan en otras situaciones como por ejemplo: mujeres que toman anticonceptivos orales, embolia pulmonar, infarto del miocardio, enfermedades renales, trombosis arteriales o venosas asi como en cualquier otro evento tromboembolico. Cuando la hiperfibrinolisis reaccional o defensiva sobrepasa los limites terapeuticos puede llegar a producirse un estado de hiperfibrinolisis patologica. En esta situation la plasmina circulante puede actuar sobre el fibrinogeno (PDF tempranos o fragmentos X). Estos PDF pueden unirse con los monomeros de fibrina y con el fibrinogeno mismo formando los complejos mf-PDF y F-PDF. Son incoagulables por la trombina y se encuentran en el suero de los pacientes con CID. <22> Existen otras pruebas usadas par el diagnostico de CID en el laboratorio como son: Test de Sulfato de protamina v test de gelation en etanol: Estas dos pruebas son positivas en el 80 al 85 % de los casos con CID aguda o cronica. Miden la presencia de monomeros de fibrina (mf) separandolos de los PDF a los cuales se encuentran unidos formando complejos mf-PDF. Durante la CID pueden formarse intravascularmente complejos solubles de la fibrina dependiente De la action de la trombina o plasmina sobre el fibrinogeno. Cuando el fibrinogeno es degradado, el monomero de fibrina formado puede seguir tres caminos: • Polimerizarse con otros monomeros formando el coagulo de fibrina insoluble. • Polimerizarse con moleculas completas de fibrinogeno formando los complejos solubles mf-F. • Polimerizarse con los PDF que se forman durante el estado de hiperfibrinolisis plasmatica formando los complejos mf-PDF. La detection de los complejos mf-F y mfBDF constituye un hallazgo patognomonico de CID. <22> Tiempo de lisis dp eu^lobulina: la fracpipn euglpbulinica del plasma es precipitajcla ppr dil^cipn y acidjfjp^cion a un pH 5.5, en el precipitado se enp^pjijja los precin^flos dp la lisis: activador del plasminogeno, plai^inogeno, y ||jj^|in6geno, en tanto que los inhibidores sp e^ppipr||:ran erj el ^o^en^dante. Por adicion de cloruFP 4f calcip ftyfnia el coagulo de euglobulinas, el cu^.1 np ^i^elvje q o ^ ^ j p e n t e en menos de 90min. Si esto sucedjjpra eji tipp|pp, mjdicaria la actividad 2 3 fibrinolitica aumentada. < >. en el paso de nijV^xasten pynblernas 11 ! de fibrinolisis. <21). ' " ' '' , Las dos pruebas anteriqj-ps J^nftipq son indicadores- de CtD) pwir su uso en el laboratorio es casi nulo. CASO CUNICO HISTORIA CLINICA Paciente femenino de 27 anos de edad con fiebre, anorexia, perdida de peso, sangrado transvaginal importante, palidez de tegumentos y petequias diseminadas. Se realizo biometria hematica, reticulocitos, TP, TTPa, fibrinogeno y DHL; los resultados se muestran en el cuadro I y las figuras 9.Sobre la base de lo anterior, se establecieron los diagnosticos iniciales de: 1. Anemia normocitica normocromica severa,hiporregenerativa, con anisocitosis. 2. Leucocitosis severa a expensas de serie mieloide, Mieloperoxidasa positiva con eosinofilia e incremento importante de la deshidrogenada lactica. 3. Trombocitopenia severa con plaquetas pequenas. 4. Coagulopatia hipofibrinogenemia. de consumo con trombocitopenia CUADRO1 L1MITES DE REFERENCIA RETICULOCITOS 15,000.00jiL 50000.00 TP 20seg 10.00 14seg TTPa 65seg 26.00 40 seg Fibrinogeno 80mg/dl 200.00 400 mg/dl DHL 1,500UI/1 313.00 616UI/L Fig. 9. (24) lOOOOO.OOpL CUADRO II xlO/jiL || % LEU 28.59 Neut 69.4 H 19.83 Linf 10.2 L 2.91 Mono 0.6 L 0.1 Bos 18.3 H 5.23 Baso 0.8 H 0.22 CUADRO III LEU HEM HGB HCT VCM HGCM CHCM Rutina REC H L L L 28.59 xl03/yL MCHC 2.15 xl03/viL RDW HDW 6.6 g/dL 19.40% PLQ VPM 90.3 fL PDW 30.5 pg PCT 33.7 g/dL H L L H L 32.7 g/dL 20.60 pg 3.14% 21 xl03/viL 4.80 fL 86.20% 0.01% Por lo que de inmediato se procedio a realizar aspirado de medula osea en el que se pudo observar medula hipercelular, con proliferation de promielocitos hipergranulares, con cuerpos de AUer positivos. Las series roja y megacariocitica se encontraron disminuidas. En la figura 10 se presentan las imagenes observadas con la tincion de Wright (lOOx) Pig. 10 Promielocitos con bastones de Auer. Fig. 10 Promielocitos con bastones de Auer FIG. 10 Promielocitos con abundantes granulos azurofilos. En seguida se procedio a realizar tinciones citoquimicas para mieloperoxidasa y cloracetato esterasa, las cuales se encontraron intensamente positivas, en tanto que las tinciones para alfa naftil - acetato - esterasa fueron negativas figura 11. En la Citometria de flujo se obtuvieron los siguientes CD13(+), CD33(+), CD34(+), CD45(+), cMPO(+). Fig. 11 La tincion citoquimica intensamente Positiva.<24) de resultados: mieloperoxidasa es Diagndstico definitivo Los promielocitos son anormales, tienen abundantes granulos que no permiten ver con claridad las estructuras nucleares y citoplasma; es comun encontrar multiples cuerpos de Auer, los cuales ahora sabemos son granulos de origen lisosomal que contienen cristales de naturaleza proteica, por lo que son patognomonicos de la leucemia mieloide aguda. Los granulos contienen una sustancia semejante al factor tisular, que cuando se libera y al ponerse en contacto con el factor VII del plasma, inicia la coagulopatia de consumo que en el peor de los casos termina en una coagulacion intravascular diseminada, en la que, como en este caso, existe una prolongation del tiempo de protrombina, tiempo de tromboplastina partial activada, con fibrinogeno bajo y trombocitopenia. Generalmente presentan una cuenta leucocitaria baja, fiebre sin evidentia de infection, sin hepatoesplenomegalia; en la variante microgranular los leucocitos pueden alcanzar cifras de hasta 200,000/mL. Las tinciones citoquimicas para mieloperoxidasa y cloroacetato esterasa son intensamente positivas y alfa - naftil - acetato - esterasa es negativa. (24) CONCLUSION La coagulaci6n intravascular diseminada, es un sindrome que la mayoria de las veces es fatal para los pacientes que lo desarrollan. Un diagnostico oportuno aumenta las posibilidades de vida de los pacientes. Las pruebas de rutina del laboratorio son de gran importancia en el diagnostico de CID, la citometria hematica y los tiempos de coagulaci6n son pruebas que se realizan en la gran mayoria de los centros hospitalarios, lo que permite realizar un diagnostico rapido. Una interpretation correcta de las pruebas de rutina permiten diagnosticar padecimientos serios que ponen en peligro la vida de los paciente adem&s, no debemos olvidar que el diagnostico clinico siempre debe de ir acompanado pruebas de laboratorio que permitan realizar diagnosticos que salven vidas. J BIBUOGRAFIA 1.-LEUCEMIA PROMIELOCITICA AGUDA tomada de: http://www.monografias.com/ trabajos 18/leucemia-mieloideaguda/ leucemia-mieloide-aguda. shtml. 2.- LEUCEMIA PROMIELOCITICA AGUDA PUNTES DE MEDICINA INTERNAtomadade:http://www.portalesmedicos.com/publicacion es / articles /663/1 / Leucemia- Aguda- Apuntes-de-HematologiaApuntes-de-Medicina.html. 3.- Honrubia Molines A. leucemias linfoblasticas agudas infantiles. Evolution historica y perspectivas futuras. BSCP Can Ped 2001; 25- n° 2. Servicio de Hematologia y Hemoterapia. Hospital Universitario Materno-Infantil. Las Palmas de Gran Can aria. 4.- Monografla revisada el 28 de Mayo de 2005. Equipo de Redaction de IQB http: / / www.iqb.es/hematologia/monografias/leucemiam3/leuce miam3. 5.- Gutierrez R.M, Sindromes hematologicos , edit prado. 473-474475. 6.- Ruiz Argtielles .G.J. Fundamentos de Hematologia .2da edi. 180: 2000. 7.- Bick,R.L,Disseminated Intravascular Coagulation(DIC)and related sindromes. In perspectives in Hemostasis. Edited by J. Fareed,H.l.Messmore, J.W.Fentonll, and K.M, Brinkhous.New York, pergamon press, 1981. 8.- Gutierrez .Aguirre O .LEUCEMIA PROMIELOCITICA AGUDA [ICD-10: M9866/3) C92.4 Hospital Angel Leano. Guadalajara, Jalisco, Mexico. UNIVERSIDAD VERACRUZANA FACULTAD DE BIOANAUSIS 9.-CLASIFICACION DE LEUCEMIAS tomada de http://apuntesdemedicina.awardspace.com/Leucemias_clasificaci on.titm 10.- http://cancernetwork.com/ (Cancer Management: A Multidisciplinaiy Approach, 8th Edition, 2004) - Mauricio Lema MD 11.- Wiernik P.H. Neoplastic Diseases of the Blood. ChurchillLivingstone. 1996. 140 pag. 12.- www.oncogroup.com.ar/descripcion.html. 13.- Word.M.E, secretos de la Hematologia y Oncologia .2da edi. MacGraw-Hill interamericana. 1334136.2000. 14.- Cotran RS, Kumar V, Collins. PATOLOGIA ESTRUCTURAL Y FUNCIONAL. McGraw-Hill-Interamericana.6° edicion.2000. 15.- Lawrence Tierney et al. DIAGNOSTICO CLINICO Y TRATAMIENTO. Manual Moderno. 35° edicion. 2000 16.- Zhu, J., Okumura H., Ohtake, S., Nakamura, S., Nakao: The Molecular Mechanism of Arsenic-Induced Apoptosis and Oncosis in Leukemia/Lymphoma Cell Lines. Acta Hematologica, 2003, 110 1-10 Wintihobe M. M. et al. Clinical Hematology 8th Edition Philadelphia. Lea and Febiger 1981 17.-Shen, Y, Yan, H., Chen, J, Zeng, J.M.: Studies on the clinical efficacy and pharmacokinetics of low dose arsenic trioxide in the treatment of relapsed acute promyelocytic leukemia: a comparisoon with conventional dosage. Leukemia, (2001) 15, 735741. 18.-DIARIO MfiDICO 13/6/2007 Por aspanion20 @ 12:32 | TIPOS DE CANCE http://aspanion.blogcindario.eom/2007/ll/00303hallan-una-nueva-via-para-controlar-la-leucemia-m3.html. 19.- http://www.iqb.es/varios/asimov/leucemia01.htm creado: 29 de Julio de 2004. 20.- Rozen F.E, GutierrrezR.M, Gonzalez C.R, Linfoblastica. Medicine .1998; 691-5. Sofia Elisa Ramirez Orteea Ensayo Leucemia aguda -37- 21.- Wintihobe M. M. et al. Clinical Hematology 8th Edition Philadelphia. Lea and Febiger 1981. 22.- HOSPITAL PROVINCIAL CLINICO-QUIRURGICO-DOCENTE "JOSE RAMON LOPEZ TABRANE" Coagulacion intravascular diseminada. Diagnostico de laboratorio por los metodos tradicionales. Disseminated intravascular coagulation. Laboratory diagnosis by traditional methods. 23.- Manual de laboratorio de Hematologia. R. Guerrero Garcia.pag. 193. Textos universitarios) 24.-Enrique Solis Cancino, Victoria Esther Balbuena Yanez. Leucemia aguda mieloide M3 (promielocitica). Laboratorio Solcan. Recibido: 23/11/2007 Aceptado: 27/12/2007.tomada de http://www.medigraphic.com/pdfs/patol/pt-2008/pt081e.pdf. 9/07/08. 25.- Carrillo-Farga Joaquin. "Atlas digital de Hematologia". CYBERCELL 26.- universidad de Nagazaki. "atlas virtual de Hematologia "