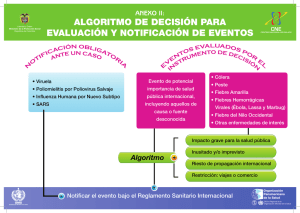
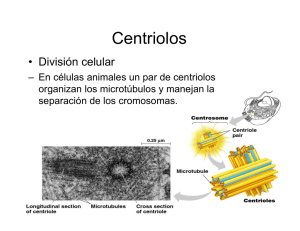
View/Open - Tesis Institucionales
Anuncio

INSTITUTO POLITECNICO NACIONAL ESCUELA NACIONAL DE MEDICINA Y HOMEOPATÍA SECCIÓN DE ESTUDIOS DE POSGRADO E INVESTIGACIÓN PROGRAMA INSTITUCIONAL DE BIOMEDICINA MOLECULAR “Estudio de las Interacciones de la dineína con la región citoplásmica de CD155 y con el virión de poliovirus”. T QUE E PARA MAESTRO S OBTENER EN ESPECIALIDAD P R Q. B. P. EL CIENCIAS DE E I GRADO ELIZABETH E DE: EN BIOMEDICINA S S LA MOLECULAR N GONZÀLEZ T A DURÁN MÉXICO, D.F., 11 DE NOVIEMBRE DEL 2003. INSTITUTO POLITECNICO NACIONAL ESCUELA NACIONAL DE MEDICINA Y HOMEOPATÍA SECCIÓN DE ESTUDIOS DE POSGRADO E INVESTIGACIÓN PROGRAMA INSTITUCIONAL DE BIOMEDICINA MOLECULAR “Estudio de las Interacciones de la dineína con la región citoplásmica de CD155 y con el virión de poliovirus”. TESIS QUE PARA OBTENER EL GRADO DE MAESTRO EN CIENCIAS EN LA ESPECIALIDAD DE BIOMEDICINA MOLECULAR PRESENTA QBP. Elizabeth González Durán DIRECTOR DE TESIS. Dr. Juan Santiago Salas Benito. CODIRECTOR: Dra. Rosa María del Ángel Núñez. ASESORES. Dra. Blanca Lilia Barrón. Escuela Nacional de Ciencias Biológicas. IPN Dra. Ana Lorena Gutierrez E. Patología Experimental. CINVESTAV Dr. Guillermo Pérez Ishiwara. Programa Institucional de Biomedicina Molecular. ENMyHIPN. AGRADECIMIENTOS A mis padres Silvia y Enrique, por su incondicionable apoyo y amor y ser fuerza motora de mi superación. Al Dr. Juan Salas por permitirme realizar uno de mis tantos sueños, por darme sabiduría, tiempo, ideas y una linda amistad; por creer en mí y abrirme todas las puertas... A la Dra. Rosy del Ángel por su amistad y sus acertados consejos; por darme la oportunidad de creer en mí misma, y sobre todo por darme todas las facilidades para realizar este proyecto. A Yina y Fred por estar siempre conmigo, en las buenas y en las malas y por darme la oportunidad de tener una linda familia y conocer a Lila y Ricardo, pero sobre todo por esos dos nenes que amo: Jimena y Ricardito. A mis tíos Mary y Gerardo, por abrirme las puertas cuando mas lo necesitaba y facilitarme el camino a esta meta. A mis Sinodales: Dra. Blanca Lilia Barrón y Dr. Guillermo Pérez I. por el tiempo brindado y los conocimientos que me heredaron. A la Dra. Lorena por su transparente amistad y apoyo durante todo este tiempo. A Fernando Medina por su valiosa ayuda con los cultivos celulares. A Jorge Reyes por su valiosa asesoría con las columnas de afinidad. A Salvador Chavarría por su gran apoyo en el laboratorio con el material y en el bioterio y por su amistad. A Mony y Rebe por estar siempre alentándome pese a la distancia y por su valiosa amistad. A mis compañeros del laboratorio de patología: Mony, Jorge, Jaime, Martha Yoco, Chava, JJ, Román, Melina y Fer, quienes enriquecieron e hicieron posible el trabajo diario de este proyecto. A José Gudiño por su gran amistad y apoyo ilimitado durante todo este tiempo para que hiciera posible uno de mis sueños....por los momentos que hemos compartido y aprendido juntos..... Al mis amigos del “club del Ocotal”: Mony, Cris, Rebe, Juanjo, Ana, la Chinita, JM y Thea por los grandes momentos compartidos dentro y fuera del Cinves.... A mis otros amigos inseparables: Paty, Ady, Chayo, Chivis, Mina, Gaby, Lalo, Iso, Nacho, Gil, Mike y Rodo, por estar siempre conmigo y ser fuerza motora para seguir adelante. A Clau, Susy, lolita y Oc por no dejarme vencer.....y por los buenos momentos juntos. A mis Compañeros y amigas del laboratorio de Genomas de patógenos por apoyarme incondicionalmente todo este tiempo para poder hacer posible este logro. A la Dra. Oliva Clement del laboratorio de poliovirus del InDRE, por su valiosa donación del antisuero contra poliovirus y por su apoyo incondicional... A Laura Enciso por su gran apoyo administrativo y su amistad. A mis amigos de la maestría: Fer, Atenea, Ricardo, Felipe, Judith y Mony. A la UIRH que en algún momento existió y me dio tantas satisfacciones academicas y donde se dio el inicio de este proyecto..... El presente trabajo fue realizado en el Departamento de Patología Experimental del CINVESTAV, bajo la dirección del Dr. Juan Salas Benito y la Dra. Rosa María del Ángel Núñez. Durante el desarrollo del mismo, la autora fue becaria Institucional del IPN y del Programa Institucional para la Formación de Investigadores (PIFI). INDICE Contenido Lista de abreviaturas Lista de figuras y tablas Resumen Abstrac Capítulo 1 Introducción • 1.1 Características de poliovirus • Página I III V VII 1 1 1.2 Ciclo replicativo de poliovirus • 1.3 Diseminación de poliovirus 2 4 • 1.4 Receptor de poliovirus 4 • 1.5 Transporte axonal 7 Capítulo 2 Antecedentes 13 Capítulo 3 Justificación 18 Capítulo 4 Hipótesis 20 Capítulo 5 Objetivos 21 • 5.1 Objetivo general 21 • 5.2 Objetivos particulares 21 Capítulo 6 Estrategia experimental 22 Capítulo 7 Materiales y métodos 23 7.1 Materiales 23 • 7.1.1 Animales de laboratorio 23 • 7.1.2 Cepa Viral 23 • 7.1.3 Líneas celulares 23 • 7.1.4 Medios de cultivo 24 • 7.1.5 Proteínas de fusión 24 7.2 Métodos 26 • 7.2.1 Pureza e integridad de las proteínas de fusión 26 • 7.2.2 Obtención del extracto proteico de médula espinal de 27 ratón BALB/c • 7.2.3 Obtención del extracto S10 de la línea celular SH-SY5Y 28 de neuroblastoma humano • 7.2.4 Detección de la dineína en los extractos proteicos 29 mediante western blot • 7.2.5 Interacciones proteicas con proteínas de fusión y el 32 extracto proteico de médula espinal de ratón BALB/c • 7.2.6 Interacciones de las proteínas de fusión y el extracto S10 33 de neuroblastoma • 7.3 Interacción directa de poliovirus con dineína 34 • 7.3.1 Propagación inicial de poliovirus (stock viral) 35 • 7.3.2 Titulación de poliovirus por el método de formación de 36 placas líticas en células HeLa • 7.3.3 Propagación de poliovirus 38 • 7.3.4 Purificación de poliovirus por gradiente de CsCl 39 • 7.3.5 Acoplamiento de poliovirus purificado a perlas de 42 sefarosa 4B activadas con bromuro de cianógeno para hacer una columna de afinidad • 7.3.6 Interacción de poliovirus con extracto S10 de 43 neuroblastoma • 7.3.7 Detección de poliovirus mediante western blot. 44 Capítulo 8 Resultados 45 Capítulo 9 Discusión 68 Capítulo 10 Conclusiones 79 Capítulo 11 Perspectivas 80 Apéndice 1 82 Apéndice 2 84 Capítulo 12 Referencias bibliográficas 90 Lista de abreviaturas. hPVR. Human poliovirus receptor, por sus siglas en inglés. GST. Glutathione-S-Transferase, por sus siglas en inglés. SNC. Sistema Nervioso Central. PV. Poliovirus. IRES. Sitio de unión interna del ribosoma. RNA. Ácido ribonucleico. VP. Viral protein, por sus siglas en inglés. 3D pol. RNA polimerasa dependiente de RNA de poliovirus. RNAm. Ácido ribonucleico de tipo mensajero. RNT. Región no traducida de poliovirus. CRE. The cis-acting replication element, por sus siglas en inglés. ATPasa. Adenosine tri-phosphate enzyme, por sus siglas en inglés. KIF. Kinesine family, por sus siglas en inglés. KDa. Kilodaltones MAP. Microtubules associate-proteins, por sus siglas en inglés. IC74. Cadena intermedia de la dineína citoplásmica. HC. Cadenas pesadas, IC. Cadenas intermedias. LIC. Cadenas intermedias ligeras. ATP. Adenosine- tri-phosphate. GTPasa. Guanosine- Tri-phosphatase enzyme, por sus siglas en inglés. GTP. Guanosine- tri-phosphate, por sus siglas en inglés. TgPVR1-17. Transgenic poliovirus mouse 1-17, por sus siglas en inglés. HSV. Herpex simples virus, por sus siglas en inglés. DRG. Dorsal root Ganglie, por sus siglas en inglés. TCTEL1. Cadena ligera de la dineína humana. SH-SY5Y. Nombre de la línea celular de neuroblastoma humano. HeLa. Nombre de la línea celular de origen epitelial. DMEM. Dubelco´s modified eagle médium, por sus siglas en inglés. I NTB. Nitroazul y cloruro de tetrazolium. BCIP. 5-bromo-4-cloro-3-indolfosfato, sal de p-tolouidina. ECL. Componente del kit para revelar por quimioluminiscencia. MOI. Multiplicidad de infección. SFB. Suero fetal bovino. TCA. Tri-cloro-acetic acid, por sus siglas en inglés. UFP/ml. Unidades formadoras de placa por ml. IgM. Inmunoglobulina de tipo M. IgG. Inmunoglobulina de tipoG. Ni-NTA. Agarosa-níquel CD155. Nombre del receptor de poliovirus humano. Pα. Nombre de la proteína de fusión que contiene la región citoplásmica de CD155. LFa1. Nombre de la proteína de fusión que no contiene la región citoplásmica de CD155. II Lista de figuras. Figura No. Página Figura No. 1 Estructura de poliovirus humano. 2 Figura No. 2 Isoformas del receptor de poliovirus humano CD155. 5 Figura No. 3 Interacción del receptor CD155 con poliovirus. 6 Figura No. 4 Composición y estructura de la dineína citoplásmica. 10 Figura No. 5 Vías de diseminación de poliovirus. 13 Figura No. 6 Proteínas de fusión Pα y LFa1. 25 Figura No. 7 Curva de precipitación del cloruro de cesio. 41 Figura No. 8 Calidad de la purificación de las proteínas de fusión Pα y 47 LFa1. Figura No. 9 Extracto proteico de médula espinal de ratón. 48 Figura No. 10 Controles del ensayo de la cromatografía de afinidad de 51 Glutatión-S-transferasa para recuperar las interacciones proteicas. Figura No. 11 Extracto citoplasmático S10 de neuroblastoma humano. 53 Figura No.12 Detección de dineína en el extracto S10 de la línea 54 celular SH-SY5Y de neuroblastoma humano. Figura No. 13 Detección de dineína en la interacción de las proteínas 56 de fusión con el extracto S10 de neuroblastoma, recuperadas por columna de afinidad GST-glutatión. Figura No. 14 Determinación del título viral de poliovirus durante el 61 proceso de concentración y purificación por gradiente de cloruro de cesio por el método de formación de placas líticas. Figura No.15 Detección de dineína a diferentes tiempos de crecimiento 62 de la línea celular SH-SY5Y de neuroblastoma humano. Figura No.16 Interacción de poliovirus acoplado a sefarosa 4B con 64 dineína presente en el extracto S10 de neuroblastoma humano. Figura No.17 Detección de poliovirus en la resina de sefarosa 4B 67 mediante western blot y quimioluminiscencia. IIII Figura A. Interacciones proteicas de las proteínas de fusión con el 83 extracto de médula espinal de ratón recuperadas por cromatografía de afinidad con resina Ni-NTA. Lista de tablas. Tabla No. Tabla No. 1 Relación RNA /proteína de cada fracción del gradiente 59 de CsCl. Tabla No. 2 Características de poliovirus purificado por gradiente de 60 CsCl. IV RESUMEN Aunque el mecanismo exacto aún es desconocido, se ha propuesto que uno de los medios por medio del cual poliovirus se disemina al el Sistema Nervioso Central (SNC) es través de los nervios periféricos mediante proteínas motoras involucradas en el transporte axonal retrógrado. Se ha propuesto que poliovirus ingresa a las neuronas desde la sinápsis mediante una endocitosis mediada por el receptor humano de poliovirus (CD155). Una vez que el virus, unido a su receptor, ha sido introducido en el endosoma, es posible que la porción citoplásmica de CD155, localizada en la superficie de la vesícula, interaccione con la dineína citoplásmica, la cual, a su vez, realice el desplazamiento del endosoma, junto con su contenido a lo largo de los microtúbulos, para transportar al virus desde la terminal sináptica hasta el soma neuronal. La finalidad de este trabajo fue determinar si proteínas motoras, como la dineína, interactúan con la región citoplásmica del receptor de poliovirus humano (CD 155) abriendo la posibilidad de que ésta última molécula actuara como intermediario entre el virus y la proteína motora para la diseminación hacia el SNC. Por otro lado, se analizó la posible interacción de la dineína con la cápside de poliovirus con el fin de comprender si el virus es capaz de interactuar directamente con las proteínas motoras para su transporte hacia el cuerpo neuronal. Para confirmar si la región citoplásmica de CD155 interactúa con la dineína citoplasmática, se trabajó con proteínas de fusión GST, de las cuales, una de ellas expresa la región citoplasmática de CD155α. Estás proteínas se hicieron interactuar con el extracto S10 de la línea celular SH-SY5Y de neuroblastoma humano como fuente de dineína. Las interacciones resultantes se recuperaron por cromatografía de afinidad GST-glutatión y se identificaron con anticuerpos monoclonales contra proteínas motoras. Por otra parte, para determinar si poliovirus tiene la capacidad de unirse directamente con la dineína citoplásmica, se realizó una cromatografía de afinidad, donde se realizaron interacciones de V proteínas provenientes de la línea celular de neuroblastoma con poliovirus purificado y acoplado a resina de sefarosa 4B activada con bromuro de cianógeno. Nuestros resultados mostraron que el receptor de poliovirus humano CD 155 no se une a la dineína bajo las condiciones experimentales realizadas, por lo que no podemos aseverar que CD155 actúe como intermediario entre poliovirus y las proteínas motoras en la diseminación del virus hacia el SNC. Sin embargo, la interacción entre poliovirus y la dineína mostró que esta proteína motora puede unirse directamente al virus sin necesidad de emplear su receptor CD155 como intermediario. Estos hallazgos nos sugieren que poliovirus, al igual que otros virus que afectan al SNC, pudiera diseminarse hacia las astas anteriores de la médula espinal sin que se requiera la presencia de su receptor específico. VI ABSTRAC. Even the exact mechanism is unknow, it has been proposed that poliovirus can spreads to central nervous system mainly through the neural pathway by means of motor proteins implicated in the retrograde axonal transport. It has been proposed that the human poliovirus receptor (hPVR) CD155 plays an important role in the neurovirulence of poliovirus inoculated by neuro-muscular injury. If a role of hPVR in the spread of the virus is considered, it is possible that, in the nervous, poliovirus is enclosed by endosomes as a result of hPVR-mediated endocytosis. One possibility, is that while the virus remains attached to the receptor, the cytoplasmic domain of the poliovirus receptor CD155 localizes to the external endosome surface, could interact with the cytoplasmic dynein and then, the endosome complex moves from the synapses to the neural cell body, where uncoating and replication of poliovirus occur. In this work, the objective was determine if motors proteins, like cytoplasmic dynein, could interact with the cytoplasmic domain of the poliovirus receptor CD155, and this event could mediate retrograde transport of poliovirus through the dynein endosome complex to neuron cell. On the other hand, we analysed the possibility that the capside of poliovirus could interact directly with the cytoplasmic dynein and then, the cytoplasmic dynein-poliovius complex was taken up at the neural cell body, without of CD155 participation. To confirm the interaction of CD155 with the cytoplasmic dynein, we using GST fusion proteins. One fusion protein expressed the C-terminal peptide of CD155α (the cytoplasmic domain of the poliovirus receptor CD155α ). Theses proteins was incubated with a cytoplasmic cell lysate of the human neuroblastoma cell line SHSY5Y. The GST fusion proteins, bound to glutathione-agarose beads, were used in a GST-pull down assay to determine affinities to cytoplasmic dynein. The material bound to the glutathione-beads was analysed by immunoblotting with goat monoclonal anti-dynein antibody. VII To prove if poliovirus could bind to cytoplasmic dynein, poliovirus was bound to sepharose-beads 4B activated with cyanogen bromide, and interacted with a cytoplasmic cell lysate of human neuroblastoma. The eluids were analyzed by immunoblotting. Our data indicated that the cytoplasmic domain of poliovirus receptor CD155 did not associate with the dynein under the conditions used in our laboratory. However, we can not in afirm that CD155 could be a bridge between poliovirus and motor protein and then, spreads toward to central nervous system. evidenced the poliovirus and cytoplasmic dynein interaction We without intermediates, like CD155. Our data suggest that poliovirus could spread to CNS without the participation of its cellular receptor CD155. VIII Capítulo I INTRODUCCIÓN. 1.1 Características de Poliovirus. Poliovirus es el agente causal de la poliomielitis, la cual es una enfermedad humana aguda que afecta al Sistema Nervioso Central (SNC) con grados de severidad variable y que, en ocasiones, se complica con parálisis fláccida (Racaniello, 2001; Pallanchs & Ross, 2001). Este virus sólo afecta al ser humano y otros primates y no se han encontrado reservorios en la naturaleza. Poliovirus (PV) es un virus desnudo que pertenece a la familia Picornaviridae y al género Enterovirus. Su cápside es de tipo icosaédrico, compuesta de 60 copias de cada una de las cuatro proteínas estructurales: VP1, VP2, VP3 y VP4 que se encuentran arregladas en 12 pentámeros. En cada uno se localiza una depresión llamada “cañón”, el cual actúa como sitio de unión para el receptor celular del virus (figura 1) (Racaniello, 2001., Tsang et al., 2001; Nomoto et al., 1994). El genoma de PV es una molécula de RNA de una sola cadena de polaridad positiva, cuyo tamaño es alrededor de 7,500 nucleótidos. Se caracteriza por presentar, en el extremo 5´, una región no traducida de 600 a 1200 bases de longitud en la que se encuentra una estructura secundaria conocida como el sitio de unión interna del ribosoma (IRES), la cual es importante para la traducción (Martínez et al.,2001); mientras que en el extremo 3´ se encuentra otra región no traducida de aproximadamente 50 bases, la cual está poliadenilada y es importante para la replicación (Racaniello, 2001; Teterina et al., 2001). El RNA tiene un solo marco de lectura abierto que codifica para una poliproteína, la cual es inicialmente procesada por una proteasa viral para dar origen a tres polipéptidos: P1, P2 y P3 y estos a su vez son procesados para dar origen a las 1 proteínas estructurales VP1(34 KDa),VP2 (29 KDa), VP3 (25 KDa) y VP4 (7 KDa), las proteasas 2A, 2B , 2C , la RNA polimerasa dependiente de RNA 3D pol, la proteasa viral 3C y la proteína 3B o Vpg respectivamente (Racaniello, 2001). 1.2 Ciclo replicativo de poliovirus. La replicación de poliovirus se da en el citoplasma celular. El primer paso consiste en la unión del virus al receptor celular, lo cual genera cambios conformacionales en la cápside, permitiendo el desnudamiento del virus y liberando el RNA genómico al citoplasma (Racaniello, 1996). Una vez ahí, el RNA de polaridad positiva actúa como RNAm, el cual se traduce para producir las proteínas virales esenciales para la replicación y la producción de nuevas partículas (Racaniello, 2001). La replicación del genoma comienza con la síntesis de la cadena negativa de RNA. Para esto se requiere de la formación de los complejos de replicación localizados en vesículas, los cuales están conformados por cadenas de RNA 2 tanto de polaridad positiva como negativa y las ribonucleoproteínas: 3AB, 3CD y 2C ATPasa (Strauss et al., 20003). En la replicación del RNA se emplea la proteína 3B o Vpg, la cual se encuentra unida covalentemente al extremo 5´ del RNA viral (Cheney et al., 2003). Está actúa como cebador para el inicio de la síntesis de la cadena negativa de RNA, la cual es ejecutada por la RNA polimerasa 3D pol a lo largo de la cadena molde. Por otra parte, en el extremo 5´ del RNA se forma otro complejo ribonucleoproteico en el cual se involucra a CRE (elemento activo de replicación en cis, por sus siglas en inglés), este elemento cataliza la uridilación de Vpg (Murray et al., 2003), cuyo producto permite iniciar la síntesis de la cadena positiva de RNA, donde la 3D pol se desplaza a lo largo de la cadena negativa de RNA para terminar la síntesis de la cadena de polaridad positiva. Esto involucra repetidas formaciones de complejos ribonucleoproteíco entre 3CD, 5´ y CRE (Cheney et al., 2003; Murray et al., 2003). La replicación de poliovirus es de tipo asimétrica, es decir, se producen mayor número de copias de RNA de polaridad positiva que de RNA de polaridad negativa; la magnitud de la asimetría de replicación del RNA viral en las células es de aproximadamente de 20 a 50 copias de RNA de polaridad positiva por cada cadena negativa de RNA. La traducción del RNA viral da como resultado la formación de una sola poliproteína, la cual sufre procesamiento tanto co- y postraduccional debido a proteasas virales, permitiendo la síntesis de las proteínas de la cápside viral y de replicación. Enseguida comienza el ensamblaje de los viriones, comenzando con la formación del pentámero, el cual requiere del procesamiento del precursor P1 para dar origen a VPO, VP1 y VP3. Posteriormente VPO es procesado para dar origen a VP2 y VP4, estas dos últimas proteínas de la cápside permiten el ensamblaje final del virus, formando el virión maduro y el ciclo viral culmina con la liberación de los viriones por lisis celular. 3 1.3 Diseminación de poliovirus. La infección por poliovirus se adquiere vía oral mediante el consumo de alimentos contaminados con este virus o por contaminación orofecal. Inicialmente es encontrado en la orofaringe y en el intestino delgado, en placas de Peyer y otros órganos linfoides, en donde inicialmente se replica, de ahí atraviesa la barrera intestinal y pasa al torrente linfático y luego a la circulación sanguínea, presentándose la fase de viremia transitoria (figura 5) y sólo en una de cien infecciones el virus llega a invadir el SNC y replicarse en las neuronas causando poliomielitis paralítica (Ouzilou et al., 2002; Ohka & Nomoto, 2001). La entrada de poliovirus a células permisivas es dependiente de la expresión de una glicoproteína de membrana cuya función celular se desconoce. La expresión de este receptor de poliovirus, también conocido como CD155, está limitada a primates, por lo cual la variedad de huéspedes susceptibles está restringida únicamente a humanos y monos (Solecki et al., 2000; He et al., 2000; Solecki et al., 1999; Bernhardt & Bibb et al., 1994). En la actualidad se emplean, como modelo animal para el estudio de la infección por este virus, ratones transgénicos que expresan el receptor de poliovirus humano (hPVR), los cuales son susceptibles a la infección y desarrollan parálisis como en el caso de los humanos (Ren et al., 1990; Bernhardt & Harber et al., 1994; Ren & Racaniello, 1992; Selinka et al., 1992). 1.4 Receptor de poliovirus. El receptor de poliovirus es una glicoproteína celular de superficie que posee 8 sitios de glicosilación y pertenece a la superfamilia de las inmunoglobulinas (Ig). Está constituido de una región amino-terminal, seguida de 3 dominios extracelulares: V-C2-C2, un dominio transmembranal y una región citoplasmática 4 (figura 2) (Mendelsohn et al., 1989; Bernhardt & Bibb et al.,1994; Belnap et al., 2000). Existen 4 variantes generadas por "Splicing alternativo". Los RNAm para hPVRβ y hPVRγ, que carecen de todo o parte del exón que codifica para el dominio de unión a la membrana, generan hPVRs solubles. Las otras dos variantes son hPVRα y hPVRδ, las cuales poseen el dominio de unión a membrana, por lo que actúan como receptores funcionales para poliovirus, difiriendo solamente en la longitud y composición de aminoácidos del dominio intracelular citoplásmico (figura 2) (Bernhardt & Bibb et al.,1994; Bernhardt & Harber et al., 1994; Nomoto et al., 1994). El peso molecular de hPVRα es de 45 kDa y para hPVRδ es de 43 kDa aproximadamente, en estado deglicosilado. 5 Análisis con poliovirus y receptores mutantes muestran que los sitios de unión a poliovirus residen en el dominio 1 de la región amino-terminal (Bernhardt & Harber et al.,1994). PVR tiene una doble función en el proceso de infección de poliovirus: primero, permite la unión de la partícula viral con la superficie celular y segundo, participa en el proceso de desnudamiento del genoma viral, ya que induce cambios conformacionales en el virión (cuya densidad es de 160S), generando la partícula A infecciosa (Joklik & Darnell,1961). Durante este cambio conformacional, la proteína VP4 es liberada de la partícula y la proteína VP1 expone su región amino-terminal (Fricks & Hogle;1990), generando la partícula A, cuya densidad es de 135S y que es altamente hidrofóbica (figura 3), lo que aparentemente le permite interaccionar con la membrana celular y liberar el material genético dentro de la célula huésped (He et al., 2000; Belnap et al., 2000; Racaniello, 1996; Nomoto et al., 1994). 6 1.5 Transporte Axonal. La motoneurona es una de las células blanco de poliovirus y se sabe que su destrucción durante la infección por poliovirus es la causa de la parálisis fláccida. Se trata de un tipo de célula nerviosa que transmite impulsos desde el cerebro o la médula espinal hasta el tejido muscular o glandular. La síntesis de proteínas axonales, neurotransmisores, membranas y el ensamblaje de organelos se lleva a cabo exclusivamente en el cuerpo de la neurona y los componentes destinados para el axón o las terminales sinápticas deben ser transportados a través del axón de una manera eficiente (Cyr & Brady,1992). Para que las funciones del SNC se lleven a cabo se requiere de un sistema que permita el transporte de diversos componentes desde el soma o pericarión a la terminal sináptica y viceversa a través de los axones. Este sistema de transporte es lo que conoce como transporte axonal (Cyr & Brady, 1992; Brady, 1991). El movimiento de los componentes neuronales a través del axón puede ser de dos tipos: transporte axonal lento y el transporte axonal rápido, los cuales sólo difieren en la velocidad de movimiento y el tipo de organelos que transportan. El transporte axonal lento es responsable del movimiento de proteínas del citoesqueleto, como microtúbulos y microfilamentos, y enzimas solubles a la velocidad de 0.1 a 3 mm por día y su importancia radica en que permite el crecimiento de las prolongaciones citoplasmáticas de la neurona así como su mantenimiento. Por otro lado, el transporte axonal rápido permite el movimiento de organelos membranosos, como es el caso de mitocondrias y vesículas citoplasmáticas, a la velocidad de 50 a 200 mm por día (Nobutaka,1997). Dentro del transporte axonal rápido se distinguen otros dos tipos de transporte, que sólo difieren en su direccionalidad: 7 I. Transporte anterógrado, donde la dirección del movimiento es del cuerpo neuronal a las uniones sinápticas. II. Transporte retrógrado, el cual involucra el retorno desde la terminal sináptica hasta el cuerpo neuronal (Cyr & Brady, 1992). Los organelos membranosos que se mueven en dirección anterógrada, incluyendo la mitocondria y estructuras vesiculotubulares, pueden ser precursores de membrana plasmática axonal y vesículas sinápticas. Estas últimas son organelos que normalmente contienen neuropéptidos, neurotransmisores y proteínas asociadas. Por el contrario, los organelos membranosos prelisosomales, cuerpos multivesiculares, mitocondrias, endosomas y receptores del factor neurotrópico se mueven en dirección retrógrada. Se han encontrado sólo dos proteínas motoras unidireccionales llamadas kinesina y dineína cerebral que son responsables para el adecuado transporte de todo tipo de organelos hacia su blanco, a la velocidad requerida (Nobutaka, 1997). Las proteínas motoras que participan en el transporte anterógrado son principalmente del tipo de la kinesinas. Las kinesinas son proteínas motoras con actividad de ATPasa dependiente de microtúbulos. La familia de estas kinesinas se encuentra constituida por diversos tipos de moléculas motoras, las cuales sólo difieren en el tipo de dominio motor que presentan, entendiéndose por dominio motor como el sitio de la molécula donde radica su actividad de ATPasa dependiente de microtúbulos, lo cual hace posible el transporte de tipo anterógrado. Existen distintos tipos de la familia de las kinesinas: Las kinesinas con el dominio motor en la región amino-terminal de la molécula (KIF1A, KIF1B, KIF3A/B, KIF4, KIF5A y KIF5C), la familia de las kinesinas con el dominio motor central (KIF2) y la familia de las kinesinas con el dominio motor en la región carboxilo-terminal (KIFC1, KIFC2 y KIFC3). Otra clasificación de la familia de las 8 kinesinas es en base al dominio motor: si este es de tipo monomérico (KIF1A, KIF1B), homodimérico (KIF2, KIF4, KIF5A, KIF5B, KIF5C, KIFC1, KIFC2 y KIFC3) ó heterodimérico (KIF3A/B) (Nobutaka, 1997). La kinesina neuronal es una proteína tetramérica conformada por dos cadenas pesadas, de 115 y 130 kDa respectivamente, y dos cadenas ligeras de 62 y 70 kDa. En la cadena pesada en el fragmento de 45 KDa se localiza la actividad de ATPasa dependiente de unión a microtúbulos de está molécula (Brady,1991). Estas proteínas participan en el transporte de organelos membranosos por transporte anterógrado a lo largo del axón, pero no participan en el movimiento del citoesqueleto. La kinesina se mueve en dirección hacia el extremo positivo de los microtúbulos y la unión de esta molécula motora a los microtúbulos es muy fuerte. Algunas kinesinas, como la del tipo KIFC2, cuyo dominio motor se encuentra en el extremo carboxilo-terminal y cuya función está asociada en el proceso de división celular, promueve el movimiento en dirección al extremo negativo de los microtúbulos (Nobutaka, 1997). En el transporte axonal retrógrado participan principalmente las proteínas motoras del tipo de MAP1α, dineína y dinactina (Takenata et al., 1998; Cyr & Brady, 1992; Brady, 1991). MAP1α es una proteína motora asociada a microtúbulos (por sus siglas en ingles microtubule-associated proteins), con actividad de ATPasa y caracterizada por mover microtúbulos en sentido opuesto a la kinesina. Esta molécula pertenece a la familia de las dineínas, cuya longitud es de 40 nm, presenta dos dominios citoplasmáticos, dos cadenas pesadas, dos cadenas ligeras y su composición polipeptídica es muy compleja. Se encuentra ampliamente distribuida en la neurona y organelos asociados a la membrana de los axones. Las MAPs presentes en el axón funcionan como reguladores del transporte de organelos membranosos dependiente de fosforilación (Brady, 1991; Nobutaka, 1997; Paschall et al., 1987). 9 Otra proteína motora asociada a microtúbulos es la dineína. Se conocen dos clases de dineína: la dineína axonal y la dineína citoplásmica. La dineína axonal media el movimiento flagelar y esta constituida por brazos externos e internos unidos a un doblete de microtúbulo de axonemas en cilios y flagelos. Algunos parásitos presentan un organelo llamado axonema, el cual es rico en dineína axonal y está encargado de la motilidad de este organismo. La dineína citoplásmica está involucrada en numerosas actividades celulares, entre ellas la translocación de organelos celulares (Pfister∗ et al., 1996). Ambas clases de dineínas son proteínas compuestas de múltiples subunidades polipéptidicas. 10 La dineína citoplásmica es un complejo proteico con múltiples subunidades muy grandes, el cual está compuesto de dos cadenas pesadas idénticas de aproximadamente 410 kilodaltrones (KDa) cada una; dos cadenas intermedias de 74 KDa; cuatro cadenas intermedias de entre 53 y 59 KDa y siete cadenas ligeras cuya función se desconoce (figura 4). Cada cadena pesada contiene cuatro sitios de unión a ATP, incluyendo el sitio catalítico, que provee la energía necesaria para su movimiento a lo largo de los microtúbulos y el sitio de unión a microtúbulos. Las cadenas intermedias de 74 KDa, en conjunto, unen a la dineína a su carga específica, entendiéndose por carga una vesícula del aparato de Golgi, un organelo membranoso, un cinetócoro o los ásteres durante la división celular. La dineína provee la fuerza necesaria para mover su carga hacia lo largo del microtúbulo en dirección al extremo negativo de éste (Cyr & Brady,1992; Vaughan et al.,1996). Kevin Vaughan y Richard Valleé mostraron que las IC74´s son codificadas por dos genes, uno de los cuales genera dos variantes por "splicing" alternativo y el otro tres (Pfister & Vaughan et al.,1996). Steuer y colaboradores demostraron, mediante ensayos de coinmunoprecipitación, que la dineína colocaliza, junto con el "spindle" mitótico y los microtúbulos del cinetócoro durante metafase y anafase de cromosomas aislados, finalmente ellos concluyen que la dineína citoplásmica se localiza en el cinetócoro de cromosomas de células en mitosis (Steuer et al., 1990). En otros estudios de coinmunoprecipitación, se encontró que la dineína se localiza en la parte externa del cinetócoro, en el dominio que representa la corona de este último (Zinkowski et al.,1991). En Drosophila se ha estudiado la función que tiene la dineína durante la oogénesis en el oocito y hay evidencias que demuestran que la dineína es requerida para la localización de determinantes celulares, como la localización del bicoide y los RNA mensajeros, así como la localización y estabilización del núcleo. Además, sorprendentemente, se encontró que la función de la dineína es sensible a la reducción de los niveles de kinesina y se ha observado que 11 mutaciones presentes en kinesinas también alteran el proceso de localización de la dineína citoplásmica y de dinactina, así como el proceso de unión nuclear, los cuales son resultados similares a aquéllos obtenidos en ausencia de dineína. Esto muestra que la actividad de estas dos proteínas motoras es interdependendiente y que la kinesina regula de manera indirecta la localización y reciclaje de la dineína (Duncan & Warrior, 2002). La dineína citoplasmática de tipo cerebral consta de dos cadenas pesadas de aproximadamente 530 KDa, tres cadenas intermedias de 74 KDa y cuatro cadenas ligeras de 50 KDa que, en conjunto, forman un complejo enorme (figura 4). En neuronas se han encontrado seis isoformas de la cadena intermedia de 74 KDa (IC74) cuya expresión cambia durante el proceso de diferenciación neuronal, en particular durante la división celular (Pfister & Vaughan et al., 1996). Aunque la dineína cerebral funciona como proteína motora en el transporte retrógrado de organelos membranosos, se ha encontrado que cierta población de dineínas cerebrales se desplazan de manera anterógrada (Nobutaka,1997; Lye et al.,1987). La dinactina es una de las subunidades de la dinamina, la cual es una molécula motora con actividad de ATP y GTPasa que tiene capacidad de unir GTP inducido por interferón y algunos factores virales (Echeverri et al., 1996; Takenata et al., 1998; Cyr & Brady, 1992; Brady, 1991). 12 Capítulo 2 ANTECEDENTES. Existen dos posibles vías por las cuales poliovirus alcanza el SNC: 1) El virus entra a través de la corriente sanguínea y la barrera hematoencefálica 2) el virus se introduce dentro de los nervios periféricos que inervan al músculo esquelético y se disemina hacia el SNC por transporte axonal retrógrado (Ren & Racaniello,1992; Ohka & Nomoto, 2001). En este último mecanismo se propone que el virus entra al axón de la motoneurona secundaria, después de interaccionar con su receptor en el espacio sináptico. Posteriormente asciende por dicho axón hasta llegar a la médula espinal, donde se replica en el cuerpo de la neurona, causando lisis celular y liberación de la nueva progenie viral, la cual se disemina lateralmente a otras neuronas y a los cuerpos de motoneuronas primarias del encéfalo (figura 5) (Ren & Racaniello, 1992). 13 Existen varias evidencias que apoyan la diseminación neural de poliovirus Nathanson y colaboradores, demostraron que, al inocular poliovirus dentro del nervio isquiático de monos, el virus aparecía inicialmente en el área lumbar de la médula espinal y más tarde en la corteza motora. Al realizar una sección en el nervio isquiático previo a la inoculación, se observó que se bloqueaba la diseminación del virus desde el músculo al SNC, sugiriendo que poliovirus puede diseminarse a lo largo de las fibras nerviosas (Nathanson & Bodian, 1961). Resultados semejantes fueron reportados por Ren y Racaniello al inocular poliovirus dentro del músculo del cojinete plantar de la pata posterior de ratones transgénicos TgPVR1-17, que expresan el receptor de poliovirus humano. Encontraron que el virus entra primero al segmento inferior de la médula espinal y, posteriormente, se disemina a regiones superiores y al cerebro. También demostraron que la expresión del receptor de poliovirus juega un papel muy importante en la diseminación viral por esta ruta (Ren & Racaniello, 1992). Gromeier y Wimmer, al realizar múltiples inoculaciones intramusculares de poliovirus en el músculo gastrocnemio de ratones transgénicos, el cual está inervado por el nervio isquiático, resultó en un incremento del daño neurológico al SNC inducido por poliovirus debido al trauma muscular, demostrando que la parálisis fue causada por el virus que alcanza la médula espinal por el transporte axonal retrógrado a partir de los nervios dañados de los músculos inyectados (Gromeier & Wimmer, 1998). Ponnuraj y colaboradores, demostraron que, tanto la cepa silvestre de poliovirus como la cepa Sabin atenuada, se diseminan al SNC a través de una ruta neural desde su sitio primario de replicación después de la inoculación en el nervio ulnar en monos Bonet, con la diferencia de que sólo la cepa silvestre causa parálisis (Ponnuraj et al., 2001). El transporte a lo largo de grandes distancias es muy crítico, en particular para los virus que infectan neuronas, en donde el sitio de entrada de estos virus puede estar localizado muy lejos del cuerpo neuronal. El transporte retrógrado de dos 14 clases de alfaherpesvirus, de Herpes Simplex 1, del virus de la pseudorabia y adenovirus ocurre de manera rápida y eficiente por el mecanismo mediado por microtúbulos. Hèléne Raux y colaboradores, empleando el sistema de doble híbrido en bibliotecas genómicas de células levadura cepa L40, cuyas células son transformadas con el plásmido pLex-P que codifica para la proteína P del virus de la rabia. Por ensayos de coinmunoprecipitación en células infectadas y en células transfectadas con el plásmido que codifica para la proteína P, confirmaron que la proteína P interactúa con LC8 (cadena ligera, la cual es un componente de la dineína citoplásmica involucrada en el movimiento de organelos a lo largo de microtúbulos en dirección hacia el extremo negativo de estos). Ellos también detectaron la presencia de LC8 en las partículas virales (Raux et al., 2000). El extremo positivo de los microtúbulos está localizado en la sinapsis y el negativo está anclado al el centro de organización de microtúbulos en el cuerpo neuronal. Así, la cápside del virus de la rabia se une a los microtúbulos y usa a la dineína para su transporte hacia el cuerpo neuronal, proceso mediado por la interacción de la proteína P con la cadena ligera de la dineína citoplásmica LC8. La proteína P es una subunidad de la RNA polimerasa viral, cuya función es actuar como cofactor no catalítico de la RNA polimerasa y como molécula chaperona que ayuda a la proteína viral N a unirse correcta y específicamente a la cadena de RNA naciente durante la replicación del genoma. Ye y colaboradores, demostraron que la proteína U (L) 34 de Herpes Simplex 1 se une a la cadena intermedia de la dineína citoplásmica y parece que esta proteína motora está implicada en la localización nuclear del virus (Ye et al., 2000). Herpes Simplex virus (HSV) normalmente infecta dos tipos de células primarias: las células dendríticas y las neuronas del ganglio de la raíz dorsal (DRG). La infección usualmente comienza con la inoculación del virus en la epidermis y posteriormente entra a la terminal axonal y es transportado retrógradamente a lo largo del axón hacia el cuerpo neuronal donde entra en un estado de latencia. Posteriormente puede reactivarse en respuesta al número de 15 estímulos externos (estado de strees de la persona afectada, así como la elevación de la temperatura del organismo) y ser transportado anterogradamente al lo largo del axón desde el cuerpo celular hasta la epidermis (Roizman & Knipe, 2001). Holland y colaboradores, empleando un sistema de doble cámara, donde en la cámara interna se trabaja con un cultivo de neuronas del ganglio dorsal, permitiendo que los axones crezcan hacia la parte externa y penetren la barrera de agarosa que conecta con la cámara externa donde se localiza un cultivo de células dendríticas. Mediante este modelo y microscopía electrónica, se estudió el transporte anterógrado de la nucleocápside viral, proteínas del tegumento y glicoproteínas de HSV. Encontraron en el axón sólo la presencia de nucleocápsides no envueltas , mientras que viriones no envueltos y envueltos estaban presentes en el cuerpo neuronaL. También observaron que las glicoproteínas estuvieron localizadas, separadas de la nucleocápside, en vesículas axonales, por lo que éstas ultimas parecían no transportarse anterogradamente en el axón (Holland et al., 1999). Hay evidencias obtenidas por microscopia electrónica de que adenovirus tipo 5 se asocia con microtúbulos y elementos del citoesqueleto. Se ha propuesto que esta unión representa un mecanismo de transferencia vectorial del virión, desde la membrana celular al núcleo. Lukashok y colaboradores, usando un sistema del doble híbrido, determinaron que una GTPasa celular se une, tanto a TCTEL1(cadena ligera de la dineína humana) como a la proteína viral E3 ( inhibe el factor de necrosis tumoral α) de adenovirus, formando un complejo en el cual la proteína viral se asocia con proteínas motoras unidas a microtúbulos y cuya funcionalidad está asociada al ciclo replicativo del adenovirus (Lukashok et al., 2000). Ohka y Nomoto proponen que poliovirus se incorpora dentro de las neuronas en la terminal sináptica mediante una endocitosis mediada por el receptor humano de poliovirus (CD155), sin que éste genere los conocidos cambios conformacionales en la partícula viral. Una vez formado el endosoma, el cual encierra el virus intacto, la región citoplásmica del receptor CD155, localizado en la superficie externa, puede interaccionar con la dineína citoplásmica y entonces 16 el endosoma es transportado a lo largo de los microtúbulos del axón en sentido retrógrado hacia el soma neuronal. Una vez ahí, ocurriría el desnudamiento del virus y la replicación de éste con la consecuente destrucción de la neurona (Ohka & Nomoto, 2001). 17 Capítulo 3 JUSTIFICACIÓN. En los últimos años se ha observado un incremento en el número de casos de poliomielitis paralítica seguida de la administración de la vacuna oral contra polio y este aumento está fuertemente asociado con inyecciones realizadas después de la aplicación de la vacuna oral. La vacuna oral de poliovirus está constituida por virus vivos atenuados, por lo que son capaces de replicarse en el intestino pero no pueden migrar al SNC. Sin embargo, se piensa que el trauma realizado en el músculo y los nervios que lo inervan por las inyecciones, favorecen una puerta de entrada para que el virus se disemine a través de estos nervios dañados hacia el SNC (Ponnuraj et al., 2001). En 1994 la Organización Mundial de la Salud declaró a América como libre de polio, sin embargo, en los últimos dos años se han reportado brotes de poliomielitis paralítica en Haití y República Dominicana, por lo que despierta nuestro interés en profundizar nuestro conocimiento sobre la diseminación de poliovirus a través de la ruta neural. Se ha propuesto que la diseminación de poliovirus hacia el SNC a través de nervios periféricos se realiza mediante proteínas motoras involucradas en el transporte axonal. Dados los antecedentes que apoyan la diseminación de poliovirus silvestre y cepa vacunal al SNC a través de una ruta neural, la meta de este trabajo es conocer que tipo de proteínas motoras están involucradas en el transporte axonal y diseminación del virus desde nervios periféricos hasta el SNC. Lo anterior nos lleva a plantearnos el posible mecanismo, es decir, si las proteínas motoras involucradas interactúan con la región citoplásmica del receptor de poliovirus humano, sirviendo éste como intermediario para la 18 diseminación hacia el SNC, o si las proteínas motoras interactúan directamente con el virus para transportarlo al soma neuronal. Con los resultados obtenidos pretendemos ampliar un poco más la información existente acerca de la fisiopatología de la poliomielitis y que, en un futuro, estos hallazgos puedan contribuir en la erradicación de está enfermedad. 19 Capítulo 4 HIPÓTESIS. En base a los antecedentes acerca de la diseminación de poliovirus al SNC por medio del transporte axonal retrógrado proponemos: 1) Que poliovirus requiere de la interacción de la molécula CD155 con proteínas motoras del transporte axonal retrógrado, principalmente moléculas del tipo dineína. 2)A su vez, dado que en el axón se han encontrado partículas virales sin cambio conformacional, poliovirus requiera de la interacción directa de su cápside con proteínas motoras del transporte axonal retrógrado, principalmente moléculas del tipo dineína. 20 Capítulo 5 5.1 OBJETIVO GENERAL. Identificar las proteínas motoras involucradas en el transporte axonal retrógrado necesarias para la diseminación de poliovirus hacia el SNC. 5.2 OBJETIVOS PARTICULARES. 1) Obtener y valorar la integridad de las proteínas de fusión P y LF1a así como de extractos proteicos de médula espinal de ratón y de la línea celular SH-SY5Y de neuroblastoma humano. 2) Analizar las interacciones de las proteínas de fusión Pα y LF1a con los extractos proteicos de médula espinal de ratón y la línea celular SH-SY5Y de neuroblastoma humano mediante cromatografía de afinidad y western blot. 3) Analizar las interacciones de poliovirus con los extractos proteicos de la línea celular SH-SY5Y de neuroblastoma humano, mediante columnas de afinidad y western blot. 21 Capítulo 6 ESTRATEGIA EXPERIMENTAL. La estrategia experimental de este trabajo consistió en emplear un sistema “in vitro”, donde se trabajó con proteínas de fusión, las cuales están constituidas por la enzima Glutatión-S- Transferasa (GST) y una secuencia que codifica para seis histidinas. La proteína de fusión Pα además tiene clonada la secuencia que codifica para la región citoplásmica del receptor humano de poliovirus, mientras que la proteína de fusión LFa1 carece de ésta . Por una parte, se hicieron interaccionar las proteínas de fusión con el extracto proteico de médula espinal de ratón y con extracto S10 de neuroblastoma como fuente de dineína. Las interacciones resultantes se recuperaron por cromatografía de afinidad GST-glutatión. Por otra parte, se realizó la interacción directa de poliovirus con el extracto S10 de neuroblastoma, como fuente de dineína, y las interacciones proteicas resultantes se recuperaron por columnas de afinidad. Las proteínas involucradas en las interacciones se identificaron con anticuerpos monoclonales mediante western blot. 22 Capítulo 7 MATERIALES Y MÉTODOS. 7.1 MATERIAL 7.1.1 Animales de laboratorio. Se utilizaron 20 ratones BALB/c de aproximadamente 80g de peso corporal. Se trabajaron 10 ratas Winstar de 150 a 200g de peso corporal. Todos los animales se sacrificaron empleando cámara de CO2. 7.1.2 Cepa viral. Se trabajo con la cepa viral Leon-Lansing PRV 6.1de poliovirus. Esta cepa es una quimera que contiene la región 5´ UTR de la cepa viral P3/ Leon y la región que codifica para las proteínas de la cápside de la cepa viral P2/ Lansing (Gutierrez et al.,1997; La Monica et al.,1987). La propagación del virus se realizó en células HeLa y el stock viral se mantuvo a – 20º C. 7.1.3 Líneas celulares. Se obtuvieron extractos citoplasmáticos de los cultivos de la línea celular SH-SY5Y de neuroblastoma humano (de origen neuronal), esta línea celular proviene de cáncer cerebral. La finalidad de emplear esta línea celular fue como fuente de dineína citoplasmática para las interacciones bioquímicas. Por otra parte, se trabajó con la línea celular HeLa proveniente de adenocarcinoma de cervix humano (de origen epitelial). Esta línea celular se empleó para la propagación de poliovirus, así como para la determinación del título viral por el método de formación de placas líticas. 23 7.1.4 Medio de cultivo. Todos los cultivos celulares se trabajaron con el medio DMEM (Medio Dubelco´s Modified Eagle Médium de Gibco-BRL). El medio DMEM se complementa con HEPES a la concentración final de 5.9 g/l, suero fetal bovino al 10%, penicilina a la concentración de 5000 U/ml y estreptomicina a la concentración de 5 µg/ ml y es empleado para el cultivo de células HeLa. Para el cultivo de la línea celular SH-SY5Y se empleó el medio DMEM para células HeLa, el cual se suplemento con aminoácidos no esenciales a una concentración final de 1X (Gibco-BRL) y piruvato de sodio (2mM). El medio se ajustó a un pH final de 7.1 a 7.4, se esterilizó por sistema de filtración positiva a través de una membrana Millipore de 0.22 µM. 7.1.5 Proteínas de fusión. Las proteínas de fusión Pα y LF1a se generaron en la Universidad de Columbia por el Dr. Juan Salas. La construcción se realizó en un vector de expresión llamado pALEX (Panagiotidis & Silverstein, 1995), cuyo tamaño es de 6.15 kb y que contiene los genes que codifican para la enzima glutatión-S-transferasa y para una cola de histidinas. Entre las secuencias de ambos genes se encuentra el sitio múltiple de clonación, en el cual se clonó la secuencia que codifica para la región citoplásmica del receptor de poliovirus humano α y con el que se obtuvo la proteína Pα. Como control del ensayo se construyó el plásmido LF1a donde únicamente se eliminó el sitio de policlonación dejándose íntegras las secuencias para la glutatión-S- transferasa y la cola de histidinas (figura 6). Ambas construcciones fueron verificadas mediante secuenciación automática en la Universidad de Columbia. La expresión de las proteínas de fusión se realizó en bacterias E. coli BL21 transformadas con los plásmidos correspondientes y se purificaron mediante columnas de agarosa-glutatión. Posteriormente las proteínas se dializaron, se resuspendieron en PBS y se almacenaron a –70° C. 24 25 7.2 MÉTODOS. 7.2.1 Pureza e integridad de las proteínas de fusión. La pureza e integridad de las proteínas de fusión se valoró mediante ensayos de western blot. Las proteínas de fusión fueron previamente purificadas por el Dr. Juan Salas mediante columnas de afinidad agarosa-glutation (SIGMA). Como control negativo del ensayo, se pasó por la columna extracto proteico de bacterias BL21 no transformadas. Posteriormente, las proteínas purificadas se sometieron a electroforesis en gel de poliacrilamida al 12% y se transfirieron a una membrana de nitrocelulosa (BIO-RAD) empleando amortiguador de transferencia (tris-base 50mM, glicina 40Mm y metanol al 10%) a 16 volts, 250 miliamperes por 35 minutos empleando la cámara de transferencia semi-seca TRANS-BLOTR de BIO-RAD . Posteriormente se bloqueó la membrana con solución de bloqueo al 0.5% (Boehringer) 1 hora a temperatura ambiente en agitación, seguido de 3 lavados, de 10 minutos cada uno con, PBS 1X- tween 20 al 0.5% ( KH2PO4 1mM, Na2HPO4 10mM, NaCl 136mM, KCl 2.8 mM y tween 20 al 0.5% ). Posteriormente se incubó a 4° C toda la noche con el anticuerpo primario: IgG de conejo anti-histidinas a una dilución 1:1000 o IgG de ratón anti-GST a una dilución 1:2000. Se lavó la membrana tres veces bajo las mismas condiciones descritas anteriormente y se procedió a incubar por 1 h a temperatura ambiente con el anticuerpo secundario: de cabra antiIgG de conejo acoplado a peroxidasa, para los ensayos con el anticuerpo antihistidinas, o el anticuerpo de cabra anti- IgG de ratón acoplado a fosfatasa alcalina para los ensayos con el anticuerpo anti-GST. Las membranas se lavaron dos veces con PBS 20Mm (Na2HPO4 0.5 M, NaH2PO4 0.5 M, NaCl 5M), seguidas de un lavado con PBS 50 mM (Na2HPO4 0.5 M, NaH2PO4 0.5 M) y por último se reveló a temperatura ambiente. Para el caso del anticuerpo anti-histidinas se reveló con 0.001g de diaminobencidina en H202 al 30% en 5 ml de PBS 50 mM, mientras que para el caso del anticuerpo anti-GST se usó 8 ml de amortiguador AP (tris-HCl 100 mM pH. 9.5, NaCl 100 mM y MgCl2 5 mM) y 13.6 µl de BCIP 50 mg/ml (5-bromo-4cloro-3-indolfosfato, sal de p-tolouidina) (GIBCO BRL) y 26.6 µl de NTB 75 mg/ml (nitroazul y cloruro de tetrazolium) (GIBCO-BRL). BCIP y NBT son usados como 26 sustratos cromógenos para la enzima fosfatasa alcalina, donde la reacción colorida se inicia cuando el grupo fosfato de BCIP se rompe por acción de la fosfatasa alcalina. Esta reacción produce un color azul y el NBT se reduce para formar un precipitado insoluble púrpura. 7.2.2 Obtención del extracto proteico de médula espinal de ratón BALB/c. Se realizó la disección y recuperación de la médula espinal de 10 ratones BALB/c de entre 8 y 10 semanas de edad. El tejido se recolectó en un homogenizador de vidrio en presencia de coctel inhibidor de proteasas CompleteTM (extracto de páncreas 0.015mg/ml, pronasa 0.0015 mg/ml, quimiotripsina 0.0015 mg/ml, termolisina 0.0008 mg/ ml, tripsina 0.002 mg/ ml y papaína 1mg/ ml) (Roche) a una concentración final de 1x y 1000 µl de buffer de lisis RSB-NP40 (MgCl2 1.5 Mm, NaCl 10 mM y tris-HCl 10 mM pH. 7.5 y detergente NP40 al 1%) a 4° C. Se realizaron entre 10 y 15 golpes con la finalidad de disgregar el tejido hasta homogenizarlo completamente. Posteriormente, con la finalidad de romper complejos formados por ácidos nucleicos, se sonicó bajo las siguientes condiciones: 2 pulsos de 10 segundos cada uno a 40 watts en un lapso de 1 minuto, 1 pulso de 60 watts por 15 segundos y 1 pulso de 80 watts por 15 segundos. Finalmente se centrifugó a 9500 x g por 10 minutos a 4° C, en centrífuga refrigerada para eppendorf modelo 1-15K de SIGMA, se recuperó el sobrenadante con micropipeta Gilson de 1000 µl y se almacenó a – 70° C. La concentración de proteínas en el sobrenadante se determinó empleando 1 ml del reactivo de Bradford de PIERCE y se mantuvo a temperatura ambiente por 5 minutos, después se leyó a 595 nm en el espectrofotómetro SmartSpec 3000 de BIO-RAD y la concentración se determinó empleando una curva estándar realizada con albúmina sérica bovina. La calidad de los extractos proteicos se evaluó mediante una electroforesis en gel de poliacrilamida al 12% en condiciones reductoras a 150 volts en cámara de electroforesis modelo Mini-PROTEAN de BIORAD. El patrón proteico se observó tiñendo el gel con la solución de azul de 27 Coomassie (azul de Comassie R250 al 2%, metanol al 50% y ácido acético al 7%) por 10 minutos. Finalmente se eliminó el exceso de colorante con solución de desteñido (metanol al 30% y ácido acético al 5%) por 24 horas. 7.2.3 Obtención del extracto proteico S10 de la línea celular SH-SY5Y de neuroblastoma humano. Se trabajaron 60 cajas P100 marca Costar-Corning con células SH-SY5Y de neuroblastoma humano al 40%, 60% y 100 % de confluencia (20 cajas por condición) crecidas en medio DMEM suplementado con aminoácidos a 37º C en incubadora Nuare, con saturación de humedad del 95 % y a una presión de CO2 entre 4.5% y 5.5 %. Se trabajaron cultivos de 46 horas ( 40% de confluencia) y 56 horas (60% de confluencia) y 70 horas (100%) de crecimiento. Las células presentan morfología triangular alargada con prolongaciones finas y alargadas en cada vértice. Las células se observan completamente espaciadas y sin formar bicapa. Cada caja P100 contenía entre 3.5 x 106 y 5 x 106 de células. Se les aspiró el medio de cultivo con pipeta Pasteur y se lavaron con 1ml de PBS 1X estéril. Se retiró el PBS con pipeta Pasteur y se adicionó 1 ml de buffer de despegado para células ( tris-HCl 40 mM pH 7.5, EDTA 1mM y NaCl 150 mM). Las células se desprendieron empleando un gendarme de Costar-Corning y posteriormente se adicionaron 500 µl más de buffer de despegado. Las placas con las células desprendidas se agitaron a temperatura ambiente por 15 minutos en agitador de plancha Compact Rocker de BIO-RAD, las células se cosecharon y se centrifugaron a 380 x g por 5 minutos en centrífuga refrigerada SIGMA. El sobrenadante se decantó y a la pastilla celular se le adicionaron 500 µl de buffer de lisis A para extracto citoplasmático S10 (HEPES 10 mM pH 7.9, MgCl 1.5 mM, KCl 10 mM). Se homogenizó la pastilla celular con ayuda del vórtex Genie 2 durante 10 segundos y se centrifugó a 380 x g por 5 minutos a 4° C . Se eliminó el sobrenadante y a cada tubo se le adicionó amortiguador de lisis A en proporción 1:1 con respecto al tamaño de la pastilla 28 obtenida (en este caso se adicionaron 200 µl en presencia de coctel inhibidor de proteasas Complete TM a la concentración de 1x). La pastilla se homogenizó en vórtex durante 10 segundos y se traspasó a un homogenizador de vidrio tipo Dounce. Se dieron 85 golpes y se observó al microscopio de luz invertida NIKON TMS para comprobar que las células estuvieran lisadas, observando la presencia de núcleos intactos. El homogeneizado se pasó a un tubo eppendorf de 1.5 ml, se centrífugo a 9500 x g por 30 minutos a 4° C y se recuperó el sobrenadante, el cual se almacenó a –70° C. Para ver la integridad del extracto se realizó una electroforesis en gel de poliacrilamida al 12% en condiciones reductoras en cámara de electroforesis modelo Mini PROTEANR de Bio-Rad a 120 volts, 520 mA durante 70 minutos. El gel se transfirió a una membrana de nitrocelulosa en cámara de transferencia semiseca a 16 volts durante 35 minutos y por último se realizó western blot para detectar la presencia de la dineína en el extracto. 7.2.4 Detección de dineína en los extractos proteicos mediante western blot. Una vez que se realizó la transferencia de las proteínas del gel a la membrana de nitrocelulosa, se verificó la eficiencia de la transferencia mediante tinción de la membrana con 15 ml de solución colorante rojo Ponceu (rojo Ponceu al 0.1% en ácido tricloroacético al 7%) por 5 minutos, se retiró el colorante y la membrana se lavó con agua destilada hasta eliminar exceso y poder visualizar las bandas teñidas de rojo. Posteriormente, la membrana teñida se lavó con 5 ml de PBS 1X en agitación para eliminar el colorante hasta que ya no se observaron las bandas. La membrana se bloqueó con 20 ml de leche San Marcos Ligth (0.03 g de proteína / ml) durante 3 horas a temperatura ambiente en agitación suave en agitador de plancha Compac Rocker de BIO-RAD. La membrana se lavó tres veces con 15 ml de PBS-Tween 20 al 0.5% por 10 minutos cada uno. Se adicionó el anticuerpo primario anti-dineína IgM de ratón clona 70.1 de SIGMA a la dilución de 1:2000 en PBS 1X y se incubó a 4° C toda la noche en agitación suave. Se lavó la membrana a 29 temperatura ambiente 4 veces, 15 minutos cada una y con agitación suave, con 15 ml de PBS-Tween 20 0.5% y entonces se incubó con el anticuerpo secundario de conejo anti-IgM de ratón fracción µ acoplado a fosfatasa alcalina 1:2000 a temperatura ambiente en agitación suave por una hora. Se lavó la membrana 3 veces con 15 ml de PBS-tween 20 0.5%, 2 veces con 15 ml de PBS 20 mM, 1 vez con 15 ml de PBS 50 mM; todos los lavados en agitación rápida por 10 minutos. Por último, se lavó la membrana 1 vez con 4 ml de buffer AP por 10 minutos en agitación suave. Se reveló con 4 ml buffer AP en presencia de19.4 µl de NBT y 6.8 µl de BCIP en agitación suave a temperatura ambiente durante 5 minutos. Para aumentar la sensibilidad del ensayo, detectando nanogramos de dineína presente tanto en el extracto S10 de neuroblastoma como en los extractos de médula espinal de ratón y de rata, se realizó un ensayo de western blot empleando el kit de quimioluminiscencia ECLTM Western Blotting Análisis System de Amersham Pharmacia Biotech. Las condiciones fueron las mismas que se describen en el párrafo anterior salvo las siguientes modificaciones: • Dado que el ensayo es altamente sensible se requiere de lavar exhaustivamente la membrana, para esto después de la incubación con el anticuerpo primario anti-dineína IgM de ratón, se realizaron 8 lavados, de 8 minutos cada uno, con 15 ml de PBS-Tween 20 al 0.5% en agitación rápida. • La membrana se incubó con el segundo anticuerpo de cabra anti-IgM de ratón acoplado a peroxidasa de rábano (HPR) a la dilución de 1:8000 en PBStween 20 al 0.5% a temperatura ambiente durante una hora en agitación suave. • La membrana se lavó exhaustivamente con PBS-Tween 20 al 0.5% en 2 proporción 1:2, es decir 2 ml de PBS-Tween 20 al 0.5% por cada cm de membrana, como cada membrana tenía una superficie entre 5 y 20 cm2, se 30 emplearon 10 y 40 ml PBS-Tween 20 al 0.5% respectivamente. Los lavados se efectuaron de la siguiente manera: o 2 lavados rápidos . o 4 lavados de 5 minutos con dos cambios cada uno, en agitación rápida. o 1 lavado de 15 minutos con dos cambios en agitación rápida. o 1 lavado con PBS 50 mM de NaCl en agitación rápida. • Se reveló la membrana en el cuarto obscuro. Para ello se requirió eliminar el exceso de PBS de la membrana con papel absorbente y se colocó en un recipiente. Se mezclaron 500 µl de ECL1 Y 500 µl de ECL2 (reactivos del kit de quimioluminiscencia) y se bañó la membrana con esta mezcla durante dos minutos. Se secó el exceso de reactivo con papel absorbente y se colocó en un cassette de quimioluminiscencia marca SIGMA. Se expuso a una película para diagnóstico X-OMAT de KODAK para autorradiografía por 30 segundos y posteriormente se colocó la membrana en líquido revelador marca Kodak por 1 minuto, se enjuagó y se colocó en fijador marca Kodak por 40 segundos, se lavó la película con agua y se dejó secar a temperatura ambiente. 31 7.2.5 Interacciones proteicas con proteínas de fusión y el extracto proteico de médula espinal de ratón BALB/c. Las interacciones se realizaron incubando las proteínas de fusión LF1a y Pα (100µg) por separado con el extracto de médula espinal (3 mg) en presencia de coctel inhibidor de proteasas CompleteTM a 37° C en incubadora Scientific, en agitación suave por 3 horas. Después de la incubación, las proteínas se recuperaron por cromatografía de afinidad empleando perlas de agarosa–glutatión marca SIGMA. La incubación de las proteínas con las perlas de glutatión se realizó por el método de “Batch”; Se adicionaron 400 µl de resina agarosa-glutatión a cada ensayo, previamente equilibrada con PBS 10 mM pH 7.5 (proporción final 50% v/v), se adsorbió por 3 horas a temperatura ambiente en agitación en vórtex de disco Genie, posteriormente se centrifugó a 323 x g por 1 minuto en la microcentrífuga Sorvall, se recuperó todo el sobrenadante y se colocó en un tubo eppendorf de 1.5 ml constituyendo la fracción de lo “no unido”. La agarosa-glutatión se lavó cinco veces, por 5 minutos, con 500 µl de buffer de lavado (PBS 10 mM, IGPAL 0.2% pH 7.7), centrifugando a 323 x g por 1 minuto cada vez. Las cinco fracciones de lavado se recuperaron en tubos eppendof de 1.5 ml. Por ultimo, las perlas de agarosaglutatión se eluyeron por calor, se adicionaron 100 µl de buffer de muestra Laemli 5X (Tris-base 100 Mm, SDS 10%, β-mercaptoetanol 1%, glicerol 60%, azul de bromofenol 1%) y se calentó a 96º C por 7 minutos. Las muestras se centrifugaron rápidamente para bajar el contenido del tubo y se recuperó el sobrenadante en otros tubos eppendorf de 1.5 ml. Todas las fracciones se les realizó precipitación de proteínas con 1 ml de acetona fría marca MERCK a 20° C por una hora. Se centrifugó a 9500 x g por 10 minutos a 4° C en centrífuga Sorval refrigerada, las pastillas se secaron en centrífuga de vacío marca Vacufuge TM y se resuspendieron en 30 µl de PBS 10 Mm. Posteriormente todas las muestras fueron analizadas mediante electroforesis en gel de poliacrilamida al 12% en condiciones reductoras y el gel se tiñó con azul de Coomassie por 10 minutos. Como controles del ensayo se emplearon las proteínas de fusión Pα y LF1a sin el extracto de médula espinal de ratón, al igual que el extracto de médula espinal sin la 32 presencia de las proteínas de fusión, los cuales se trabajaron bajo las mismas condiciones que las interacciones proteicas. 7.2.6 Interacciones de las proteínas de fusión y el extracto S10 de neuroblastoma. Las interacciones se realizaron incubando las proteínas de fusión LF1a y Pα (100 µg) por separado con el extracto S10 de neuroblastoma (660 µg). Las interacciones se realizaron a 37° C en incubadora Scientific en agitación suave durante 3 horas en presencia de coctel inhibidor de proteasas CompleteTM. Después de la incubación, las proteínas se recuperaron por cromatografía de afinidad empleando perlas de agarosa–glutatión marca Sigma y el método de “Batch”. El procedimiento fue el mismo descrito para las interacciones de las proteínas de fusión con el extracto de médula espinal de ratón BALB/c, salvo las siguientes modificaciones: • Las perlas de agarosa-glutatión se lavaron cinco veces con 1 ml de PBSIGPAL al 0.5%, recuperando cada fracción. • se eluyeron las proteínas unidas a las perlas mediante ebullición a 96° C por 7 minutos en presencia de 100 µl de amortiguador de carga Laemli. • Las fracciones precipitadas con acetona fría se analizaron por electroforesis SDS-PAGE en gel de poliacrilamida al 12%. Posteriormente se transfirió el gel a membrana de nitrocelulosa bajo las condiciones descritas anteriormente y se determinó la presencia de dineína en el ensayo mediante western blot empleando quimioluminiscencia, cuyo método se describió anteriormente. Como controles del ensayo se emplearon las proteínas de fusión Pα y LF1a en ausencia del extracto S10 de neuroblastoma y, del mismo modo, el extracto S10 de neuroblastoma sin la presencia de las proteínas de fusión. 33 7.3 INTERACCION DIRECTA DEL VIRUS CON LA PROTEÍNA MOTORA. 34 7.3.1 Propagación inicial de Poliovirus (Stock viral). Para la propagación de poliovirus se emplearon placas P100 marca Costar-Corning con células HeLa. Cada placa contenía una monocapa de 8.4 x106 células HeLa con el 90% de confluencia. Estas células se crecieron en medio DMEM a 37 º C en incubadora de CO2 de Naure con saturación de humedad del 95% y una presión entre 4.5% y 5.5% de CO2. A cada placa confluente le fue eliminado el medio de cultivo por aspiración con ayuda de una pipeta Pasteur y la monocapa se lavó con 1ml de PBS 1X suplementado con SFB al 0.2%. Posteriormente cada monocapa se infectó con una multiplicidad de infección (MOI) de 100 (100 partículas virales por cada célula). Para esto se adicionaron 3 ml de inóculo viral preparado con PBS–SFB al 0.2% a la dilución de 1:150 (2980 µl de PBS-SFB y 20 µl del stock viral de poliovirus PVR 6.1 cepa Leon–Lansing, el cual expresa las proteínas de la cápside de la cepa Lansing y el genoma de la cepa Leon (Gutierrez et al., 1997). El virus se dejó adsorber a 37° C en incubadora con CO2 Nuare en agitación suave durante una hora. Posteriormente se adicionaron 6 ml a cada placa de medio de cultivo DMEM complementado para células HeLa previamente atemperado a 37° C y se incubó a 37° C por 24 hrs en incubadora húmeda marca Nuare a la concentración de CO2 al 4.5%. El efecto citopático fue valorado a las 15 horas postinfección mediante observación al microscopio de luz invertida marca Nikon TMS. Como control negativo del ensayo se trabajó con una caja de células HeLa sin infectar y en lugar del virus se adicionó PBS-SFB al 0.2%. Se trabajó bajo las mismas condiciones que el virus. En cuanto fue observado el efecto citopático en las células infectadas, se cosecharon junto con el medio de cultivo y se colocaron en un tubo cónico de 15 ml marca Corning. Se clarificó el sobrenadante centrifugando a 11760 x g por 20 minutos a 4° C en centrífuga Sorval RC 26 plus , empleando rotor SLA 3000. Se recuperó el sobrenadante ("stock" viral) y se almacenó a –70° C hasta su titulación. 35 El procedimiento se realizó en condiciones de esterilidad en campana de bioseguridad tipo III marca Scientific. 7.3.2 Titulación de Poliovirus por el método de formación de placas líticas en células HeLa ( plaqueo viral). Todo el procedimiento se realizó en condiciones de esterilidad en campana de bioseguridad tipo III marca Scientific. Se emplearon multiplacas de cultivo de 6 pozos marca Costar-Corning. Cada pozo contenía una monocapa confluente de 1 X 106 células HeLa. A cada pozo se le aspiró el medio de cultivo con pipeta Pasteur y se lavó dos veces con PBS-SFB al 0.2%. Se realizaron las siguientes diluciones del virus empleando PBS-SFB al 0.2% según la siguiente relación: Dilución PBS-SFB 0.2% Poliovirus Volumen final 10-2 1485 µl 15 µl 1500 µl 10-3 1350 µl 150 µl (de 10-2) 1500 µl 10-4 1350 µl 150 µ l (de 10-3) 1500 µl 10-5 1350 µl 150 µl (de 10-4) 1500 µl 10-6 1350 µl 150 µl (de 10-5) 1500 µl 10-7 1350 µl 150 µl (de 10-6) 1500 µl Control negativo 1500 µl ---------- 1500 µl Se agregaron 500 µl de cada dilución en el pozo correspondiente y se dejó adsorber a 37° C en incubadora Scientific por una hora con agitación suave. El ensayo se realizó por duplicado. Durante la incubación se preparó el medio DMEM 2X (se preparó a una concentración 2X y se complementó con SFB al 10%, 5000 U/ ml de penicilina, 5µg/ ml estreptomicina a pH final de 7.1-7.4, esterilizado por sistema de filtración positiva 36 a través de una membrana Millipore de 0.22 m) y se suplemento en SFB al 10%. También se agregó solución de antibióticos penicilina /estreptomicina quedando a una concentración final de 2X. El medio ya preparado se mantuvo en baño maría a 45° C. Por otro lado se fundió bactoagar estéril al 1.8% por calentamiento y se mantuvo a 45° C en baño maría para evitar su solidificación. Después de la adsorción del virus, el sobrenadante se aspiró con pipeta Pasteur y cada pozo fue lavado 2 veces con PBS-SFB al 0.2%. Finalmente se agregaron 2ml por pozo de la mezcla DMEM-bactoagar 1:1 y se dejó solidificar por 15 minutos. Posteriormente se incubó a 37° C en la incubadora Nuare al 4.5% de CO2 por 40 horas. Después del tiempo de incubación, la monocapa se fijó por 10 minutos a temperatura ambiente con 2 ml por pozo de ácido tricloroacético al 10% (TCA). Se retiró el TCA y el bactoagar fue eliminado cuidadosamente con una espátula para no dañar la monocapa. Se agregaron de 2 a 3 ml de cristal violeta ( cristal violeta marca SIGMA al 1%, etanol absoluto al 0.5%, aforar con agua destilada), se tiñó por 15 minutos y se lavó con agua corriente. Se contó el número de placas líticas en cada pozo y se determino el título viral empleando la siguiente formula: 37 UFP /ml = Número de placas líticas x Volumen del inóculo x factor de Dilución Donde: UFP/ ml: Unidades Formadoras de Placa por mililitro. Volumen del inóculo : el volumen final es de 0.5 ml por cada placa. Al despejar la fórmula queda 1/0.5, lo que da un factor de 2. Factor de dilución: se toma en cuenta la dilución en la que son contables las placas líticas. 7.3.3 Propagación de poliovirus. Con la finalidad de incrementar la progenie viral de poliovirus, se propagó el stock viral en células HeLa. Para esto se trabajaron 20 cajas P100 de células Hela con una confluencia del 90%. La infección se realizó a una multiplicidad de infección de 10 MOI. El procedimiento realizado fue idéntico al descrito anteriormente con la diferencia de que al presentarse el efecto citopático, a las 20 horas postinfección, en las células infectadas, las cajas se congelaron -20° C y se descongelaron a temperatura ambiente. El proceso se repitió tres veces con la finalidad de liberar completamente al virus de las células. Posteriormente se cosechó el sobrenadante, el cual se clarificó a 11760 x g por 20 minutos a 4° C en centrífuga Sorvall RC 26 plus, rotor SLA 3000. El sobrenadante clarificado se almacenó a –20º C. Se realizó un segundo pase de poliovirus con el sobrenadante clarificado proveniente de la propagación anterior en 20 cajas P100 de células HeLa con una multiplicidad de infección de 50 MOI. El virus resultante se procesó de la misma manera descrita en el párrafo anterior. Por último, el virus se volvió a propagar a 38 partir del sobrenadante clarificado obtenido con anterioridad en 20 cajas P100 de células HeLa a una MOI de 100. El virus clarificado se recolectó en tubos de ultra centrífuga de polialómero con tapa de 29 x 104 mm de Beckman. Los tubos se equilibraron y se colocaron en el rotor de ángulo fijo tipo S28 Beckman y el virus se concentró y purificó parcialmente por ultracentrifugación en la ultracentrífuga Beckman L7-55 a 46,200 x g durante 4 horas a 4° C. Se decantó el sobrenadante y la pastilla viral se resuspendió en 500 µl de buffer NTE (Tris-HCl 10 Mm pH 7.5, 1Mm EDTA y NaCl 100 mM) dejándolo actuar toda la noche a 4° C. El virus, ya resuspendido, se recolectó en un tubo cónico de 15 ml y se cuantificó la cantidad de proteína por el método de Bradford descrito anteriormente. La viabilidad y el título viral se determinaron por el método de plaqueo como se describió anteriormente. 7.3.4 Purificación de poliovirus por gradiente de cloruro de cesio. Poliovirus se purificó del resto de las proteínas provenientes de las células HeLa en las cuales se propagó, mediante un gradiente de taza zonal. Para esto se colocaron 6.07 g de CsCl sólido en un tubo cónico de 15 ml (para que el CsCl alcancé la densidad de 1.33 g/ ml en un volumen final de 14 ml), se adicionaron 5 ml de buffer NTE y 4ml de poliovirus concentrado por ultracentrifugación obtenido en el paso anterior. Se homogenizó hasta que el cloruro de cesio se disolvió completamente, se traspasó a un tubo de ultracentrífuga ultraclaro de 14 x 89 mm de Beckman, el cual se llenó hasta el borde con buffer NTE. Los tubos se equilibraron y centrifugaron a 120,000 x g (31,000 rpm ) en el rotor de columpio SW41ti Beckman a 4° C por 24 horas en la ultra centrífuga L7-55 de Beckman (Minor, 1981). La velocidad de centrifugación se determinó en base a la curva de saturación del cloruro de cesio para el rotor SW40ti de Beckman, tomando en cuenta la densidad del gradiente de cloruro de cesio y el nivel de llenado del tubo. Así, la densidad del 39 gradiente fue 1.33 g/ ml y este valor se intersectó en la curva de saturación de Cesio en la condición de tubo lleno ("full"). La intersección indica la velocidad de ultracentrifugación a 31,000 rpm (figura 7). En base a métodos de purificación para poliovirus ya reportados en la bibliografía, se determinó que la banda viral migraría a las ¾ partes superiores del gradiente (Minor, 1981; Kajigaya et al., 1985; Joklik & Darnell, 1961; Moscufo et al.,1991; Moscufo & Chow, 1992; Moscufo et al., 1993; Levinton & Darnell, 1960). Después de terminar el proceso de ultracentrifugación, se trabajó en el cuarto oscuro y, con ayuda de una lámpara, se hizo incidir luz blanca directamente sobre el gradiente resultante con el fin de visualizar una banda blanca a nivel de las tres cuartas partes superiores del tubo, cuya banda corresponde al virus. Se recolectó la fracción indicada y se determinó la viabilidad viral mediante titulación por la técnica de plaqueo, como se indicó anteriormente. Por último, se determinó la concentración de proteína por el método de Bradford descrito anteriormente. La fracción resultante se almacenó a –70° C. Se realizó el mismo procedimiento con células HeLa no infectadas como control negativo del ensayo. 40 41 7.3.5 Acoplamiento de poliovirus purificado a perlas de sefarosa 4B activadas con bromuro de cianógeno para hacer una columna de afinidad. Se mezclaron 0.5g de sefarosa 4B activada con bromuro de cianógeno ( SIGMA) con 5 ml de solución de activación (HCl 1mM) en un tubo cónico de 15 ml y se agitó suavemente durante 15 minutos a temperatura ambiente. Se centrífugo a 448 x g por 3 minutos en centrífuga Sorvall TC y el sobrenadante se eliminó por aspiración. La sefarosa hidratada se lavó 3 veces, con 5 ml cada vez, con solución de activación y posteriormente 3 veces con 5 ml cada vez con la solución de acoplamiento (NaCl 0.5M, NaHCO3 0.1 M pH 8.3). El virus purificado por gradiente de CsCl se mezcló con la solución de acoplamiento llevándola a una concentración de 1 mg/ml manteniendo la relación de 4 mg de proteína por cada 5 g de sefarosa 4B activada. Se incubó en agitación suave toda la noche a 4° C y la eficiencia del acoplamiento a la resina fue valorado midiendo la concentración de proteína del sobrenadante por el método de Bradford antes y después del acoplamiento. La sefarosa 4B ya acoplada se lavó 3 veces con solución de bloqueo (Tris base-Glicina 0.1 M pH 8.6), 3 veces con solución de acetatos (ácido acético 0.1 M, NaCl 0.5 M pH 4) y 3 veces con PBS 1X para acoplamiento (KH2PO4 15 mM, k2HPO4 15 mM, NaCl 0.5 M pH.7.4). Finalmente se centrífugo a 448 x g en la centrífuga Sorvall TC por 1 minuto, se eliminó el sobrenadante por aspiración y se adicionaron 2 ml de PBS 1X para acoplamiento en proporción 1:2 con respecto al volumen final de la sefarosa acoplada con poliovirus (1 ml). Se empacaron 0.8 ml de la sefarosa acoplada en una jeringa de 3ml desechable marca PLASTIPAKMR para formar la columna de afinidad, se lavó con 20 volúmenes de PBS 1X para acoplamiento y se almacenó a 4° C hasta su uso. Como control negativo del ensayo se trabajó con 40 cajas P100 con células HeLa no infectadas con poliovirus, de las cuales se clarificó el sobranadante, se concentraron por ultracentrifugación y las proteínas concentradas se pasaron por el gradiente de cloruro de cesio y la fracción resultante del gradiente se acoplo a sefarosa 4B bajo las mismas condiciones que el virus. 42 7.3.6 Interacción de poliovirus con extracto S10 de neuroblastoma. Para determinar si poliovirus tiene afinidad por la dineína presente en el extracto S10 de neuroblastoma, se realizaron las interacciones proteicas entre el extracto S10 de neuroblastoma y poliovirus acoplado a la columna de sefarosa 4B. La interacción se realizó adicionando 900 µl (2700 µg/µl) de extracto S10 de neuroblastoma a la columna de poliovirus, se dejó fluir el extracto por la columna dos veces e interactuar el extracto en la columna a temperatura ambiente por 40 minutos. Posteriormente se dejó fluir y el mismo extracto se pasó 5 veces a través de ésta. Se recuperaron los últimos 500 µl del extracto de la columna (fracción de lo “no unido”) y se lavó la columna con 40 volúmenes de PBS 1X 0.1 M de NaCl (32 ml). Las fracciones de lavado se dividieron en dos partes: la primera o Fw1 (los primeros 15 ml) comprendió 5 fracciones de 500 µl cada una al azar. La segunda o Fw2 estuvo formada por las 5 fracciones, seleccionadas al azar de los restantes 15 ml. Después de que se precipitaron estas fracciones con acetona como se describió anteriormente, se resuspendieron las primeras cinco en un solo tubo y las otras cinco en un segundo tubo. La columna se eluyó con 1 ml de cada uno de los siguientes eluyentes: PBS 0.2 M, PBS 0.5 M, PBS 0.8 M y PBS 1M de NaCl. Las fracciones resultantes se recuperaron en tubos eppendorf de 1.5 ml, cada tubo con 500 µl de cada fracción, y se precipitaron con 1 ml de acetona fría marca MERCK durante 1 hora a -20º C. Posteriormente se centrifugó cada una a 9,500 x g en la centrífuga refrigerada SIGMA por 10 minutos a 4º C, se evaporó el resto de acetona en la centrífuga de vacío VacufugeTM por 15 minutos. Las muestras se resuspendieron en 30 µl de PBS 1X NaCl 10 mM y se realizó electroforesis en gel de poliacrilamida al 12%. Posteriormente las proteínas separadas se transfirieron a membrana de nitrocelulosa y se realizó western blot para determinar la presencia de la dineína por quimioluminiscencia según el método antes descrito. 43 7.3.7 Detección de poliovirus mediante western blot. Para corroborar la presencia de poliovirus en la columna se realizó western blot. Para esto, después de eluir con PBS 1M de NaCl, se resuspendió la sefarosa 4B acoplada con poliovirus empaquetada en la columna y se tomaron 0.5 ml de ésta. Le fueron adicionados 300 µl de amortiguador de carga Laemli y se calentó a 96° C por 10 minutos. Se realizó una centrifugación rápida para concentrar el contenido en el fondo del tubo y el sobrenadante se traspasó a otro tubo eppendorf de 1.5 ml, al cual se le adicionó 1 ml de acetona fría para precipitación de proteínas por el método antes descrito. La pastilla se resuspendió en 30 µl de PBS 10 mM. Se procedió a realizar la electroforesis en gel de poliacrilamida al 10% en condiciones reductoras. En el gel se incluyeron también el control positivo, el cual consiste de poliovirus concentrado por ultracentrifugación (100 µl), y el control negativo el cual consiste de sefarosa 4B acoplada con células HeLa no infectadas. El gel resultante se transfirió a membrana de nitrocelulosa como se describió anteriormente. El ensayo de western blot se realizó empleando quimioluminiscencia para aumentar la sensibilidad del ensayo. Se realizó bajo las mismas condiciones que el empleado para detectar dineína, con la diferencia que se utilizó, como primer anticuerpo, un suero policlonal IgG de conejo anti-poliovirus serotipos 1,2 y 3 a la dilución de 1:1000 y se incubó cinco horas a temperatura ambiente en agitación suave. Como segundo anticuerpo se empleó un suero de cabra anti-IgG de conejo acoplado a peróxidasa de rábano del kit e quimioluminicencia ECL de Amersham a una dilución de 1:30,000 y se incubó o una hora a temperatura ambiente en agitación suave. Los lavados de la membrana y el revelado se realizaron bajo las mimas condiciones que para el western blot empleando quimioluminicencia antes descrito. El tiempo de exposición de la membrana a la película para diagnóstico fue de 1 hora. 44 Capítulo 8 RESULTADOS. La primera parte del trabajo consistió en realizar interacciones proteína-proteína con extractos celulares como fuente de proteínas motoras involucradas en el transporte de organelos. Para esto se trabajó con proteínas recombinantes de fusión, las cuales están constituidas por la enzima Glutatión-S-Transferasa (GST) y una secuencia que codifica para seis histidinas. La proteína de fusión Pα además tiene clonada la secuencia que codifica para la región citoplásmica del receptor humano de poliovirus tipo α, mientras que la proteína de fusión LFa1 carece de ésta y se empleó como control del ensayo. Se hicieron interactuar estas proteínas de fusión con el extracto proteico del sistema nervioso y de la línea celular de neuroblastoma. Las interacciones proteicas resultantes se recuperaron por columnas de afinidad y las posibles proteínas motoras involucradas se identificaron mediante el uso de un anticuerpo monoclonal anti-dineína. La segunda parte de este trabajo consistió en analizar la interacción de poliovirus con el extracto S10 de neuroblastoma mediante una columna de afinidad para analizar la posibilidad de que poliovirus pudiera interaccionar directamente con la dineína sin necesidad de emplear a su receptor celular como intermediario. Pureza de las proteínas de fusión. En primer instancia se corroboró la pureza e integridad de las proteínas de fusión Pα y LFa1, las cuales fueron purificadas a partir del extracto bacteriano donde se indujo su expresión, mediante columnas de afinidad agarosa-glutatión. Posteriormente se sometieron a electroforesis en geles de poliacrilamida, se transfirieron a membranas de nitrocelulosa y se realizó ensayo de western blot como se indicó en materiales y métodos. Este ensayo se llevó a cabo con dos anticuerpos primarios diferentes: uno anti-GST y el otro anti-histidinas. Cada 45 anticuerpo reveló dos bandas en el ensayo correspondiente: una de 29 kDa (figuras 8A y 8B, carril 2) y otra de 35 kDa (figuras 8A y 8B, carril 3). El peso molecular de la banda de 29 kDa correspondió al peso esperado para la proteína de fusión LFa1 y la de 35 kDa al peso esperado de Pα. Estas proteínas no fueron detectadas en el extracto proveniente de bacterias no transformadas (figuras 8A y 8B, carril 1), aunque al emplear el anticuerpo primario anti-GST se detectaron bandas de entre 45 y 50 kDa (figura 8A, carril 1), lo que puede ser debido, a una reacción cruzada o inespecífica dada por el anticuerpo primario. Sin embargo, estas bandas no corresponden a ninguna de las dos proteínas de fusión ni tampoco interfieren en la detección de éstas. Cabe aclarar que estas bandas inespecíficas que aparecen en el extracto proveniente de bacterias no transformadas detectadas con el anticuerpo anti-GST no se observan con el anticuerpo anti-histidinas (figura 8B, carril 1), esto muy probablemente se deba a que los anticuerpos reconocen antígenos diferentes. Para corroborar que la detección de las proteínas de fusión se debía al anticuerpo primario específico y no a una reacción inespecífica por parte del segundo anticuerpo, se realizó un ensayo de western blot similar empleando exclusivamente el anticuerpo secundario. Estos ensayos no revelaron ninguna banda en ambos casos (figura 8C y 8D). En el western blot para detectar el tallo de histidinas, el cual se reveló con peroxidasa, se observa que el anticuerpo primario anti-histidinas, además de reconocer la banda correspondiente a la de la proteína de fusión LFa1 de 29 kDa, reconoce otra segunda banda a la altura de 70 kDa cuya intensidad es muy baja (figura 8B, carril 2) y que no interfiere con la visualización de la banda de LFa1. Esta banda de mayor peso molecular puede deberse a la formación de polímeros de la misma proteína o a la presencia de una proteína bacteriana co-purificada con la LFa1 y que comparte epítopos con ella. 46 Obtención del extracto proteico de médula espinal de ratón BALB/c. Se obtuvieron 1000 µl de extracto proteico de médula espinal (17 µg/µl), de los cuales 50 µg se sometieron a electroforesis en gel de poliacrilamida al 12% y el gel se tiñó con azul de Coomassie. Se pudieron observar una serie de bandas proteicas, bien definidas, en un rango de peso molecular de 250,000 Da hasta 47 14,000 Da (figura 9) sugiriendo que la calidad del extracto fue buena y que no hay degradación importante de proteínas por acción de proteasas u otros factores. 48 Interacciones proteicas de las proteínas de fusión con el extracto de médula espinal de ratón BALB/c. Con la finalidad de analizar la posibilidad de que la proteína de fusión Pα, que contiene la región citoplásmica del receptor CD155, tenga afinidad por alguna proteína motora presente en el extracto total de médula espinal, se realizó la interacción de las proteínas de fusión con el extracto de medula espinal y posteriormente las posibles interacciones se recuperaron por cromatografía de afinidad con perlas de agarosa-glutatión. controles como En este ensayo se incluyeron varios las interacciones independientes de la proteína Pα, LFa1 y del extracto de médula espinal con perlas de agarosa-glutatión. La finalidad de realizar estos controles, en especial el de las proteínas de fusión, fue para demostrar que la Glutation-S-Transferasa (GST) que contienen se está expresando y ha adquirido el plegamiento ideal que permite la unión a su ligando natural, que es el glutatión. Así mismo, este control nos permitió asegurar que, por cromatografía de afinidad GSTglutatión, es posible recuperar las interacciones resultantes. La interacción de la agarosa–glutatión con el extracto de médula espinal se realizó con el fin de comprobar que no hubiera unión de proteínas de manera inespecífica por parte de la agarosa. Las proteínas de fusión Pα, LFa1 y el extracto de médula espinal de ratón fueron puestos en contacto con perlas de agarosa glutatión y se realizaron las interacciones, lavados y eluídos como se explicó en materiales y métodos. Las distintas fracciones obtenidas del ensayo fueron sometidas a electroforesis en gel de poliacrilamida al 12% y los geles resultantes fueron teñidos con azul de Coomassie por 10 minutos para evidenciar las proteínas. Como se observa en la figura 10A, carril 9, la proteína Pα se unió eficientemente a las perlas de agarosa glutatión aunque cabe mencionar que una fracción de la proteína se pierde a lo largo de todos los lavados (carriles 3 al 8), sin embargo, en el eluído (carril 9) se recupera más del 50%, lo que indica que pese a que la interacción de la proteína Pα con la resina no es 100% eficiente, lo es lo suficiente como para realizar las interacciones posteriores. 49 En lo que se refiere a la proteína LFa1, la figura 10B muestra que también es retenida y reconocida por las perlas de glutatión (carril 9), aunque también se observa el mismo fenómeno que con Pα (carriles 3 al 8). En ambos casos, una probable explicación de la pérdida de proteína en los lavados es que quizá la conformación del sitio activo de la enzima GST no es el óptimo y por eso la unión de la enzima a su ligando es ligeramente débil, aunque este fenómeno no bloquea por completo el sitio activo de la enzima. Por otra parte, tampoco descartamos la posibilidad de que las condiciones de lavado de la resina estén influyendo en la pérdida de cierta proporción de la proteína de fusión en éstos, aunado a que la unión de la GST al glutatión es débil, sin embargo, no pudimos modificar las condiciones de lavado, ya que el disminuir la velocidad de agitación así como el tiempo, se incrementa la unión inespecífica de proteínas del extracto de médula espinal de ratón a la agarosa glutatión. Por último, descartamos la posibilidad de que las perlas de agarosa glutatión estuviesen saturadas, es decir, dado que la purificación inicial de estas proteínas de fusión se realizó en columnas de agarosa glutatión, se pudiera sospechar que el sitio activo de la GST de éstas estuviese bloqueado con el glutatión empleado para su elución y, por tal motivo, la unión a la agarosa se mostrase débil pero las proteínas fueron dializadas después de su purificación, lo que elimina la mayor parte del glutatión que pudiera estar presente y además, la presencia se la proteína en todos los lavados sugiere que siempre una fracción de ésta es la que se separa. En la interacción del extracto de médula espinal con las perlas de agarosa-glutatión (figura 10C) se observó una banda muy bien definida que corresponde a un peso molecular de 24,000 Da y otras dos bandas muy ligeras entre 50,000 y 60,000 Da (figura 10C, carril 9). La posible causa por la que la resina glutatión capturara estas moléculas es quizá debido a que existen proteínas homólogas a la GST en el extracto de médula espinal, pero debido a que sólo fueron tres, se consideró que podrían realizarse las interacciones tomando este fondo en cuenta. 50 Cuando se realizaron las interacciones proteicas de Pα con el extracto de médula espinal de ratón, cuyas interacciones se recuperaron por cromatografía de afinidad agarosa-glutatión, encontramos que en el eluído se observaba el mismo patrón que con el extracto de médula espinal sólo con la agarosa glutatión, con la diferencia de que, además, se apreciaba la banda de Pα. Por tal motivo se modificaron las condiciones de lavado, se adicionó IGPAL a la solución empleada para tal fin y ni de esta manera se pudo eliminar ese fondo inespecífico que dio la resina (datos no mostrados). Con los resultados obtenidos hasta el momento no pudimos determinar si había diferencia entre el fondo inespecífico de la agarosa glutatión en presencia del extracto de médula espinal y las interacciones proteicas realizadas entre las 51 proteínas de fusión y el extracto de médula espinal. Quizá la o las proteínas motoras estén en muy baja concentración en el extracto de médula espinal de ratón y por tal motivo no podemos visualizarlas bajo las condiciones empleadas. Estudios previos realizados con el virus de la rabia, HSV1 y alfaherpesvirus han mostrado que estos virus realizan transporte retrógrado de manera eficiente y que está relacionado específicamente con proteínas motoras asociadas a microtúbulos (Ye et al.,2000; Saksena et al., 2000; Holland et al.,1999; Raux et al., 2000; Lukashok et al., 2000;). Entre las proteínas motoras se ha evidenciado a la dineína como la más representativa en el transporte retrógrado de estos virus, que son capaces de migrar hasta el sistema nervioso central mediante la interacción con esta proteína, la cual se encarga de transportarlos hasta el cuerpo neuronal, donde posteriormente se replican y proliferan causando daño neurológico. En base a lo anterior decidimos concentrarnos en la dineína, tomando en cuenta que poliovirus también es un virus neurotrópico que migra hacia SNC. Para esto determinamos la presencia de dicha proteína en el extracto de médula espinal de ratón BALB/c mediante western blot, como se describió en materiales y métodos, sin embargo, no se encontró dineína en el extracto, esto probablemente sea porque la cantidad presente en el extracto no puede ser detectada por el método empleado (datos no mostrados). La línea celular SH-SY5Y de neuroblastoma humano es originaria de cáncer cerebral; son células neuronales en constante división y, dado que la dineína participa durante los procesos de división celular, nosotros esperamos evidenciarla en estás células. Se procedió a obtener el extracto citoplasmático S10 de neuroblastoma, siguiendo el protocolo descrito en materiales y métodos. Se obtuvo un volumen final de 2 ml cuya concentración fue de 4 µg/µl. Se realizó la electroforesis en gel de poliacrilamida al 12% y se tiñó con azul de Coomassie por 10 minutos, lo cual nos permitió observar la integridad de las proteínas que conforman el extracto, encontrándose proteínas de alto peso molecular que van desde 400 KDa hasta 6 KDa (figura 11, carril 1). 52 Al igual que en el extracto de médula espinal de ratón, no se observó degradación del material proteico. Una vez que se corroboró la integridad del extracto citoplasmático S10 de neuroblastoma, se procedió a determinar la presencia de la dineína en el extracto mediante western blot empleando quimioluminiscencia, como se describió en materiales y métodos. Se decidió trabajar con el anticuerpo monoclonal anti-dineína de tipo IgM de ratón de SIGMA, el cual es derivado de la clona 70.1 proveniente de la fusión de células de mieloma de ratón con esplenocitos de ratón BALB/c inmunizados con dineína citoplásmica purificada proveniente de células de cerebro de embrión de pollo. Este anticuerpo identifica las cadenas intermedias de la dineína citoplásmica de aproximadamente 70 kDa . El anticuerpo fue capaz de reconocer una proteína de aproximadamente 74 kDa en el extracto S10 de la línea celular SH-SY5Y de neuroblastoma humano mediante la técnica de western blot. Se observó la banda correspondiente a la dineína, indicada 53 con una flecha, a la derecha de la película de diagnóstico (figura 12, carril 1). Por otra parte, se extrajo la médula espinal de ratas Wistar y se procedió a obtener el extracto total de médula espinal bajo las mismas condiciones en que se obtuvo el extracto de medula espinal de ratón, descrito en materiales y métodos, en cual se determinó la presencia de la dineína. Este ensayo sirvió como control de la expresión de dineína en tejido animal (figura 12, carril 2). El peso de esta proteína corresponde al reportado para la cadena intermedia de la dineína citoplásmica ( Nobutaka, 1997). 54 Interacción del extracto S10 de neuroblastoma humano con las proteínas de fusión. Una vez que se determinó la eficiencia de unión de las proteínas de fusión a las perlas de agarosa glutatión y la presencia de la dineína en el extracto S10 de neuroblastoma, se procedió a realizar las interacciones del extracto celular con las proteínas de fusión. Las interacciones proteicas se realizaron colocando, en un tubo eppendorf, extracto S10 de neuroblastoma con proteína de fusión Pα; en otro extracto S10 de neuroblastoma con la proteína de fusión LFa1 y en un tercer tubo se colocó sólo el extracto S10 de neuroblastoma. Como controles del ensayo se realizaron las interacciones de las proteínas Pα y LFa1 en ausencia del extracto proteico. Los 5 tubos se incubaron a 37º C en agitación suave por tres horas y las interacciones proteicas resultantes se recuperaron por cromatografía de afinidad con perlas de agarosa glutatión por el método de “Batch” descrito en materiales y métodos. Posteriormente se realizó SDS-PAGE en gel de poliacrilamida al 12 % de las fracciones recuperadas de cada tubo, se transfirió a membrana de nitrocelulosa y se realizó western blot, empleando el anticuerpo anti-dineína y quimioluminiscencia, como se describió en materiales y métodos para determinar la presencia de la dineína. En la interacción de la proteína de fusión Pα con el extracto S10 de neuroblastoma, el western blot mostró que la dineína no se une con la región citoplásmica del receptor de poliovirus humano, ya que dicha proteína no se recupera en el eluído (figura 13C, carril 5) por el contrario, se observa en la fracción de lo “no unido” (figura 13C, carril 3), indicándonos que la interacción es muy débil o que no se está llevando acabo. Como control positivo de dineína para el ensayo de western blot se empleó extracto citoplasmático S10 (figura 13C, carril 2). 55 En la interacción de la proteína LFa1 con el extracto S10 de neuroblastoma, lo que viene siendo el control negativo, no se observó unión de la dineína ya que la proteína se observa completamente en la fracción de lo “no unido” (figura 13B, carril 2). Cabe mencionar que las proteínas de fusión P y LFa1, cuyos pesos moleculares son de 35 kDa y 29 kDa respectivamente, estuvieron presentes en los eluídos de ambas interacciones y se corroboró su peso molecular comparando con los controles de las proteínas de fusión (figura 13B y 13C, carriles 1 respectivamente). Esto se visualizó mediante la tinción de la membrana con rojo Ponceu después de realizar la transferencia de proteínas, indicándonos que la ausencia de la dineína en 56 estas fracciones no se debió a falta de interacción entre el glutatión de la resina y la GST de las proteínas de fusión (datos no mostrados). También comprobamos que, tanto el anticuerpo primario como el secundario empleados para la detección de la dineína, no tuvieran reacción cruzada con las proteínas de fusión, ya que no se observa ninguna señal en los carriles donde únicamente se corrieron estas proteínas (figura 13B y 13C, carriles 1 respectivamente). Por último, en la figura 13A observamos que la dineína presente en el extracto S10 de neuroblastoma no se une de manera inespecífica a las perlas de agarosa glutatión, ya que la dineína es eliminada completamente en la fracción de lo “no unido” (carril 2). El carril 1 de está figura muestra el control positivo de dineína. 57 PARTE II Propagación y purificación de poliovirus. La segunda parte del proyecto se enfocó en demostrar si poliovirus tiene la capacidad de unirse directamente con la dineína sin emplear como intermediario el receptor de poliovirus humano, ya que en HSV1 se ha encontrado que este virus se transporta retrógradamente a través del axón hacia el cuerpo neuronal sin que la partícula viral sufra algún cambio; de hecho hay evidencias que muestran que la partícula se encuentra desnuda sin presencia del tegumento (Penfold et al., 1994; Holland et al., 1999). Inicialmente se preparó el stock de poliovirus, el cual se obtuvo propagándolo en células HeLa a una multiplicidad de 100 MOI. Posteriormente se clarificó el sobrenadante, se concentró por ultracentrifugación y se procedió a propagarlo a mayor escala, comenzando con una multiplicidad de infección baja de 10 MOI y luego con una alta de 50 MOI. La progenie viral obtenida se empleó para realizar los experimentos. La infección fue evidenciada por la presencia de efecto citopático: a las 15 horas posinfección cuando la MOI fue alta, y a las 20 horas cuando fue baja. El efecto citopático se caracterizó por la destrucción de la monocapa de células HeLa, presencia de células desprendidas y suspendidas en el medio de cultivo, retraídas y con forma esférica, formando racimos. En el control negativo, es decir, las células no infectadas, se observó la monocapa celular íntegra con presencia de células alargadas tipo fibroblasto (datos no mostrados). Una vez que se obtuvo suficiente población del virus concentrado (5ml de virus en buffer NTE) y cuyo título viral fue de 1x109 UFP/ml, se procedió a purificar el virus por ultracentrifugación en gradiente isopícnico de CsCl durante 24 horas. Las fracciones obtenidas del gradiente de CsCl se leyeron a la longitud de onda de 260/280 nm en el espectrofotómetro para determinar la relación RNA viral/ proteína y asegurar la presencia del virus (tabla 1). 58 Los resultados expresados en la tabla 1 están ordenados en sentido descendente, es decir, de manera en que se forman en el gradiente. En la parte superior se formó una banda gruesa de color amarillo pardo y aspecto translúcido cuya relación ácido nucleico/ proteína es alto, esto es debido a que aunque el virus pasa por un proceso de ultracentrifugación que permite parcialmente eliminar algunos restos celulares, el concentrado contiene todavía una gran cantidad de proteínas y ácido nucleico de origen celular. La fracción viral se concentró en las ¾ parte superiores del tubo del gradiente formando una banda blanquecina translúcida y delgada. La relación de RNA/ proteína nos indica que es proporcional la cantidad de proteína, dada por las partículas virales (cápside), con el RNA viral, indicándonos la presencia de viriones completos. Cabe mencionar que durante todo el proceso de propagación y purificación, la viabilidad de virus se monitoreó determinando el efecto citopático y el título viral por el método de plaqueo en células HeLa. El título viral se determinó después de concentrar el virus, para corroborar que el proceso de ultracentrifugación no afectará 59 la capacidad lítica de éste así como su viabilidad. También se determinó el título viral antes y después de someterse al gradiente de CsCl para corroborar que iones cesio y el proceso en sí no afectaran las propiedades del virus. La figura 7 muestra la formación de placas líticas, la letra C indica el control negativo, es decir, la monocapa de células HeLa no infectada. El título viral antes de la purificación por gradiente de cloruro de cesio fue de 1 x 109 UFP/ml (figura 14B y tabla 2) y después de 1 x 108 UFP/ml (figura 14C y tabla 2). Esta diferencia se debe a que probablemente, durante el proceso de ultracentrifugación, algunas partículas virales son destruidas por la fuerza centrífuga, reflejándose en la disminución del titulo viral. Otra posibilidad es que los iones cesio se adhieran a algunas partículas virales, modificando su densidad, lo cual permitiría que se distribuyeran hacia la banda celular (tabla 2). A la fracción viral se le determinó la concentración de proteínas por el método de Bradford descrito en materiales y métodos. Las características de poliovirus purificado se muestran en la tabla No. 2. Como control negativo del ensayo se trabajó con cultivo de células HeLa no infectadas bajo las mismas condiciones que el virus y se observó que la monocapa de células se mantuvo íntegra y no se observó la formación de placas líticas, lo que 60 indica que las condiciones de trabajo fueron adecuadas y que no hubo contaminación (figura 14A). Una vez purificado el virus se procedió a acoplarlo a perlas de sefarosa 4B activadas con bromuro de cianógeno, como se describió en materiales y métodos. concentración de virus empleada fue de 4 mg/ ml. Una vez La empaquetada la sefarosa acoplada con el virus para formar la columna, se mantuvo en PBS 1X 0.5 M de NaCl a 4º C hasta su uso. 61 Antes de realizar las interacciones proteicas entre el virus y el extracto S10 de neuroblastoma, fue necesario verificar la expresión de la dineína y ver si ésta se mantenía constante en el extracto S10 durante el crecimiento del cultivo celular. Para esto fue necesario trabajar las células a diferentes tiempos de crecimiento, lo cual está relacionado con el porcentaje de confluencia. Se encontró que si los cultivos de neuroblastoma humano presentaban 100% de confluencia celular, la expresión de dineína se abatía por completo y ya no era posible detectarla en el extracto S10 (figura 15, carril 1). Sin embargo, si las células presentaban entre el 40 y 60% de confluencia, lo cual corresponde con los tiempos de incubación de 46 y 56 horas, la dineína se detectaba de manera apreciable (figura 15, carriles 3 y 2 respectivamente). Como control positivo del ensayo se empleó un extracto S10 de neuroblastoma previamente probado (figura 15, carril 4). Las células de neuroblastoma se observaron en el microscopio de luz. A una confluencia del 40 % se encontraban separadas una de otra y presentaban forma triangular y prolongaciones muy largas en cada vértice. A una confluencia del 100% 62 las células perdieron la morfología triangular al igual que las prolongaciones citoplasmáticas y se observaron con forma redonda (datos no mostrados). Dado que la expresión de la dineína en el extracto citoplásmico de neuroblastoma depende del tiempo de crecimiento celular y con la finalidad de ver si este fenómeno tiene efecto sobre la interacción directa entre el virus y la dineína citoplásmica, se realizaron las interacciones proteicas entre poliovirus acoplado a sefarosa 4B con cada uno de los extractos S10 de neuroblastoma obtenidos a las 46 horas y 56 horas de crecimiento. Se recuperó la fracción de lo “no unido”, 8 fracciones de lavado repartidas en dos fracciones totales: las primeras cuatro fracciones, de 500 µl al azar, formaron la fracción de lavado 1 y las cuatro restantes formaron la fracción de lavado 2. Por último se recuperaron 4 fracciones de eluído a diferentes concentraciones de cloruro de sodio. En cada ensayo se obtuvieron las fracciones descritas anteriormente. Las fracciones se precipitaron con acetona fría y se realizó electroforesis en gel de poliacrilamida al 12%, seguida de una electrotransferencia a membrana de nitrocelulosa. La presencia de la dineína en las distintas fracciones se determinó mediante western blot empleando el anticuerpo monoclonal anti dineína IgM de ratón y revelando con el kit ECL de Amersham para quimioluminiscencia, con el fin de aumentar la sensibilidad del ensayo y evidenciar la dineína en las interacciones proteicas (figura 16). 63 Antes de realizar estas interacciones proteicas se determinó la presencia de dineína mediante western blot, empleando quimioluminiscencia en el extracto S10 de neuroblastoma, el cual lo consideramos como control positivo para todas las interacciones (figura 12, carril 1). Como control, se incluyó un ensayo donde la membrana, con las proteínas del extracto S10 de neuroblastoma transferidas, sólo se incubó con el anticuerpo secundario anti-IgM de ratón acoplado a peróxidasa de rábano. Se observó distinguir que el anticuerpo secundario no dio reacción. Se pueden algunas bandas con pesos moleculares inferiores a la banda de la dineína, pero que no interfieren con la detección de ésta (figura 15B). 64 Al realizar la interacción entre poliovirus acoplado a sefarosa 4B con el extracto S10 de neuroblastoma obtenido a las 46 horas, se encontró que la dineína se recuperó en el eluído obtenido con PBS 0.5 M de NaCl (figura 16A, carril 6). Esta banda correspondió al peso molecular de 74 KDa, la cual correlaciona perfectamente con el control positivo de dineína (figura 16A, carril 1). Es importante destacar que la dineína no se pierde en la fracción de lo “no unido” ni tampoco en los lavados (figura 16A, carriles 2, 3 y 4). Cabe mencionar que en carril 6 de la figura 16A, aparte de la banda correspondiente a la dineína, se observan otras bandas de mayor peso molecular que son detectadas por el mismo anticuerpo. También se observa otra banda inespecífica de aproximadamente 100 KDa en la primera fracción de lavado. Este experimento muestra evidencia de que poliovirus se une de manera específica y estable a la dineína, ya que se requiere una concentración de 0.5 M de NaCl para romper la interacción. Cuando se realizó la interacción de poliovirus acoplado a sefarosa 4B con el extracto S10 de neuroblastoma obtenido a las 56 horas, se encontró que la dineína no es retenida como se esperaba, por el contrario, la banda correspondiente se observa en la fracción 2 de lavado (figura 16B, carril 4). La banda de la dineína no se observa en la fracción de lo “no unido” ni tampoco en ninguno de los eluídos (figura 16B, carriles 2,3,5,7,8 y 9). Este experimento demuestra que, pese a que está presente la dineína en el extracto, la unión que se efectúa entre la dineína y poliovirus es demasiado débil o nula, por lo que la interacción se rompe y la dineína es liberada en la fracción de lavado. Con el fin de corroborar que la interacción con la dineína no se debió a la propia sefarosa 4B o a alguna proteína celular presente en la muestra viral purificada, se emplearon, como control del ensayo, células HeLa no infectadas, las cuales se sometieron al mismo procedimiento de purificación que poliovirus y la fracción celular resultante se acopló a sepharosa 4B. Cabe mencionar que no se observó la banda blanquecina en este gradiente debido a la ausencia de virus, sólo se apreció 65 una banda de aspecto amarillo pardo. Una vez preparada la columna, se realizó la interacción con extracto S10 de neuroblastoma obtenido a las 46 horas, como fuente de dineína. Los resultados mostraron que la dineína no se unió a esta columna de manera inespecífica, ya que la banda de la dineína se ve presente en la fracción de lo “no unido” y en la primera fracción de lavado (figura 16C, carriles 2 y 3). Como marcador se empleó extracto S10 de neuroblastoma (figura 16C, carril 1). Por último, para corroborar si el virus estaba acoplado a la sefarosa 4B, toda la sefarosa acoplada que conformaba la columna se sometió a ebullición a 96º C por 10 minutos con amortiguador de carga Laemli. Al sobrenadante resultante se le realizó precipitación de proteínas con acetona fría y posteriormente éstas fueron sometidas a electroforesis en gel de poliacrilamida al 10%, se transfirieron a membrana de nitrocelulosa y se realizó western blot con anticuerpos policlonales anti-polio, revelando finalmente por quimioluminiscencia para detectar la presencia del virus. Como se puede apreciar en la figura 17, se detectó una banda, de aproximadamente 35 kDa, que corresponde al peso molecular de la proteína de la cápside VP1, lo cual evidencía la presencia del virus en la sefarosa 4B (figura 17, carril 4). Como control positivo se empleó la cepa de poliovirus PRV 6.1 concentrado por ultracentrifugación (figura 17, carriles 3 y 5) y se pudo observar la misma banda. 66 Para comprobar la especificidad del anticuerpo anti-poliovirus serotipos 1, 2 y 3, en la membrana de nitrocelulosa también se incluyó extracto de células HeLa no infectadas (figura 17, carril 1) y el eluído por calor de la sefarosa 4B acoplada con células HeLa no infectadas, como control del ensayo de acoplamiento (figura 17, carril 2). En ambos controles negativos no se apreció la presencia de poliovirus, lo cual indica que la especificidad del anticuerpo es adecuada y que la banda que se observa corresponde a una proteína viral . 67 Capítulo 9 DISCUSIÓN. Hasta el momento no se conoce exactamente el mecanismo por el cual poliovirus invade el SNC, específicamente las neuronas motoras, causando su destrucción y, en casos graves, generando parálisis fláccida en personas afectadas . Los antecedentes existentes acerca del transporte axonal retrógrado de poliovirus y otros virus (Nathanson & Bodian, 1961; Ren & Racaniello, 1992; Gromeier & Wimmer, 1998; Jacob et al., 2000; Döhner et al., 2002; Raux et al.,2000; Ye et al.,2000) nos llevó a tratar de elucidar si poliovirus, una vez dentro del organismo y específicamente en la sinapsis neuronal, es capaz de migrar al soma mediante transporte retrógrado, empleando como intermediario a su receptor específico CD155 y a través de su unión con proteínas motoras que participan en dicho transporte, que tienen la capacidad de unirse a microtúbulos y que migran con dirección al extremo negativo de éstos. El presente trabajo se enfocó en estudiar específicamente las interacciones con la dineína citoplásmica. Esta proteína motora tiene la capacidad de transportar organelos de tipo membranoso, mediante el sistema de unión a microtúbulos, con dirección hacia el extremo negativo de éstos y, estudios realizados con otros virus neurotrópicos como el HSV1, el virus de la rabia, lisavirus y el virus de la pseudorabia, demuestran que la cadena ligera de la dineína tiene la capacidad de interactuar directamente con algún componente de la partícula viral y, por tanto, la emplean como parte del transporte retrógrado para invadir el SNC. Por tal motivo esta proteína motora resultó ser el candidato más obvio para ser estudiado en este trabajo (Döhner et al., 2002; Holland et al., 1999; Ye et al., 2000; Raux et al., 2000; Jacob et al., 2000). 68 La interacción entre la dineína y el receptor de poliovirus se trató de evidenciar empleando una proteína de fusión que contiene a la GST, una serie de 6 histidinas y la secuencia de la región citoplásmica de CD155 En los ensayos iniciales se decidió purificar estas interacciones mediante la unión, tanto del tallo de histidinas como de la GST presentes en las proteínas de fusión, con resinas específicas con la finalidad de comparar los resultados obtenidos por ambos métodos. En primer lugar se trató de recuperar las interacciones proteicas empleando la resina de agarosa níquel (NiNTA). El fundamento de esta técnica consiste en la alta afinidad que el níquel, acoplado a perlas de agarosa, tiene por secuencias consecutivas de más de 6 histidinas. Desafortunadamente la agarosa Ni-NTA presentó unión inespecífica con varias proteínas provenientes del extracto total de médula espinal de ratón BALB/c (ver apéndice 1), de tal manera que fue difícil eliminar el fondo obtenido, aún modificando las condiciones de lavado. Esta alta inespecifícidad de la resina Ni-NTA probablemente se debió a que las proteínas presentes en el extracto de médula espinal quizá son ricas en secuencias consecutivas de más de 6 histidinas, por lo que su afinidad a la agarosa es muy alta. Por tal motivo, esta resina no fue de utilidad. Como segunda opción, se realizaron las interacciones proteicas antes mencionadas y se trataron de recuperar por el método de inmunoprecipitación, empleando el anticuerpo monoclonal anti-dineína de tipo IgM de ratón. La finalidad de este ensayo fue de aumentar la especificidad de éste, así como eliminar por completo la unión inespecífica causada por la agarosa Ni-NTA. El fundamento de esta técnica consiste en que el anticuerpo, unido a su antígeno, en este caso la dineína y las proteínas que con ella hayan interaccionado, es capaz de ser precipitada por perlas de sefarosa 4B que tienen unida la proteína A de Staphylococcus aureus. La proteína A tiene la capacidad de unirse a anticuerpos de tipo IgG, y con menor afinidad, a IgM. Desafortunadamente no se pudo precipitar el anticuerpo con ayuda de la proteína A, dado que su unión fue muy débil (datos no mostrados). 69 Ante los problemas anteriormente expuestos, nos enfocamos a recuperar las interacciones proteína-proteína por cromatografía de afinidad a la agarosa glutatión con la enzima GST presente en las proteínas de fusión, donde solamente se observaron tres bandas, provenientes del extracto S10 de neuroblastoma, que se unieron de manera inespecífica a la resina y cuyo peso molecular no interfirió con la detección de la dineína. También se comprobó que la conformación de la GST de la proteína de fusión fue adecuada ya que ambas, P y LFa1, fueron recuperadas, en su mayoría, en el eluído pese a su ligera pérdida en los lavados. Esto último puede deberse a que la conformación del sitio activo de la enzima GST no es el óptimo y por eso la unión de la enzima a su ligando no es tan intensa como la proteína nativa, aunque este fenómeno no interfiere por completo con la unión al sustrato. Tampoco descartamos la posibilidad de que las proteínas de fusión tuviesen el sitio activo de su GST saturado con glutatión, ya que la purificación se realizó por columnas de afinidad con perlas de agarosa glutatión, sin embargo, las proteínas fueron dializadas después del proceso. Se realizó un ensayo de competencia donde las proteínas de fusión se incubaron con un exceso de la enzima GST comercial de Sigma con la finalidad de remover el posible glutatión de la proteína de fusión y así, ya despejado el sitio activo, hubiese mayor afinidad (datos no mostrados). Los resultados no mostraron cambio alguno después de este tratamiento e incluso el empleo de un nuevo lote de agarosa glutation no modificó de manera importante esta situación, por lo que nos inclinamos a pensar en la primera posibilidad como una de las más factibles. Por otra parte, no podemos descartar la idea de que la pérdida de proteína de fusión en los lavados también se pudiese deber a la astringencia de los mismos, sin embargo, al disminuir la velocidad de agitación, así como el número de lavados, se observó un incremento de la unión inespecífica de proteínas provenientes del extracto S10 de neuroblastoma a la agarosa glutatión (datos no mostrados). Por otra parte, la eliminación del detergente tween 20 de la solución de lavado no afectó los resultados (datos no mostrados). Pese a estos inconvenientes se decidió trabajar con la agarosa glutatión, ya que la unión inespecífica de proteínas hacia la resina fue mucho más baja que con Ni-NTA y que en la interacción con las proteínas de fusión se recuperó más del 50 %. 70 Los ensayos se realizaron incluyendo los controles necesarios para asegurar la veracidad de los resultados: se usaron extractos proteicos en ausencia de las proteínas de fusión y a la proteína LFa1 (la cual no contiene la región citoplasmática de hPVR). Con el fin de utilizar un extracto proteico rico en dineína, se decidió trabajar con la línea celular SH-SY5Y, la cual es originaria de un carcinoma cerebral con metástasis en médula espinal de origen humano cuyas células se encuentran, en su mayoría, en la fase G1 del ciclo celular, lo cual permite encontrar a la dineína en abundancia en el extracto citoplasmático debido a su participación en la mitosis. Se obtuvo extracto citoplásmico de neuroblastoma, ya que la dineína citoplásmica es una proteína motora soluble que, durante la interfase celular, es liberada al citoplasma (Paschal et al., 1987; Lye et al., 1987; Schroer et al., 1989). Mueller y Wimmer, empleando el sistema del doble híbrido y ensayos de coinmunoprecipitación e interacciones bioquímicas, demostraron que el dominio citoplásmico de CD155 se asocia fuerte y específicamente a la cadena ligera de la dineína, denominada Tctex-1. Con este hallazgo, ellos proponen que dicho dominio de CD155 puede funcionar como blanco para la unión de proteínas motoras, como la dineína. Durante la internalización de polivorius, mediada por su receptor, se formarían vesículas endocíticas que posteriormente serían transportadas a través de la red microtubular hacia el SNC debido a la interacción de CD155 con la dineína (Mueller et al., 2002). Nuestros hallazgos no mostraron ninguna interacción específica entre la región citoplásmica de CD155α y proteínas del sistema nervioso o de neuroblastoma, a diferencia de lo reportado por Mueller y Wimmer. Es importante destacar que en las interacciones bioquímicas que realizaron estos autores, emplearon una proteína de fusión muy semejante a la utilizada en nuestro trabajo. Una posible diferencia que pudiera explicar nuestros resultados es que en sus ensayos “in vivo”, ellos trabajaron con la línea celular de neuroblastoma de ratón, mientras que nosotros trabajamos la 71 línea celular de neuroblastoma de origen humano y probablemente esto pudiese constituir una marcada diferencia en cuanto a la expresión de otras moléculas que pudiesen participar en la unión de la cadena ligera de la dineína citoplásmica a la proteína de fusión. Ahora bien, pudiera ser que la sensibilidad de nuestro método no fue el óptimo, ya que ellos visualizaron la interacción empleando marcaje con radioactividad. Pese a que nosotros empleamos quimioluminiscencia y además observamos la banda de dineína en la fracción de “lo no unido”, no descartamos la posibilidad de que una fracción mínima estuviese interaccionando con la proteína de fusión pero que no visualizamos. Nosotros decidimos trabajar con la proteína motora nativa, es decir, completa en su estructura, para asemejar a las condiciones que ocurriesen in vivo. Realizamos las interacciones bioquímicas entre extractos proteicos totales y la proteína de fusión que expresa la región citoplasmática de CD155, al igual que Mueller & Wimmer, y la recuperamos por cromatografía de afinidad con GST-glutatión. Otra posible explicación que nosotros atribuimos a la diferencia de los resultados obtenidos en comparación con estos investigadores es que, para que se dé la interacción de la dineína citoplásmica con la región citoplásmica de CD155, es necesario que la primera, que es una molécula muy compleja conformada por múltiples péptidos y cuya unión a su carga especifica está regulada por la asociación con otras moléculas complejas, como la dinactina (Duncan & Warrior, 2002) sufra un cambio en su estructura que le permita exponer sus cadenas ligeras, las cuales se encuentran en la parte interna de la molécula cubiertas por las cadenas intermedias. Una vez expuestas las cadenas ligeras, serían capaces de reconocer su secuencia blanco presente en el tallo citoplasmático de CD155, lo que permitiría la unión y el proceso de transporte retrógrado hacia el cuerpo neuronal dentro de una vesícula endocítica. Con lo antes mencionado, es posible que la interacción de la dineína con el receptor de poliovirus no pudo ser evidenciada debido a que, en el extracto citoplasmático, la dineína se encuentra en su forma nativa y las cadenas ligeras no están expuestas para que se lleve a cabo la unión. 72 Desafortunadamente, no hay disponibles anticuerpos comerciales contra las cadenas ligeras de la dineína citoplásmica que nos pudiesen haber evidenciado la presencia de alguna de ellas en las interacciones analizadas. Esto nos inclina a pensar que, en el nervio invadido por poliovirus, la dineína citoplásmica requiera de un procesamiento previo a la interacción con CD155 que le permita exponer sus cadenas ligeras para efectuar el transporte retrográdo del virus hacia el SNC. Por otro lado, no descartamos la posibilidad de que, in vivo, dineína la interacción entre la y la región citoplásmica de hPVR requiera de la participación de otras moléculas que fortalezcan está unión o de la misma región amino de hPVR. Es importante destacar que la determinación de la presencia de la dineína en médula espinal de rata sirvió como control de la expresión de dineína en tejido animal, comprobando que la expresión de la dineína también esta dada en este tipo de tejido y no sólo en la línea celular de neuroblastoma, además de que nos permitió corroborar la expresión de dineína in vivo. Alrededor de cada uno de los cinco ejes de simetría de la cápside icosahédrica del poliovirus, en cada uno de los protómeros, se encuentra una depresión llamada “cañón”, en cuya formación participan las proteínas VP1, VP2 y VP3. Este cañón contiene el sitio de unión al receptor CD155. Como consecuencia de la unión de poliovirus a su receptor, la cápside viral sufre una alteración que reduce su coeficiente de sedimentación de 160S a 135S (Joklik & Darnell, 1961). Las partículas alteradas 135S, también llamadas partículas A, tienen expuesta a la proteína VP4 y la región amino-terminal de VP1 (Fricks et al.,1990). Estas partículas son los principales representantes de la transición conformacional de la cápside inducida por el receptor de poliovirus humano, sin embargo, hay evidencias que muestran que la partícula de poliovirus se mantiene intacta, en forma de partícula 160S, en el nervio isquiático de ratones transgénicos, es decir, sin sufrir el cambio conformacional generado por la unión a su receptor celular ( Ohka et al.,1998). 73 Por otro lado, Ouzilou y colaboradores proponen que poliovirus entra al organismo a través de un proceso de translocación en las células M, las cuales se encuentran esparcidas en la mucosa epitelial que cubre los folículos linfoides de las placas de Peyer. Mediante un sistema in vitro, empleando un cocultivo de células CaCo-2 y linfocitos aislados de placas de Peyer, las cuales se diferencían a células con características morfo y fisiológicas semejantes a las de las células M, analizaron el proceso de transitosis de poliovirus. Al infectar este cultivo con poliovirus se encontró que puede pasar del lado apical al basal de las células constituyendo el posible paso hacia los linfonodos mesentéricos. Es un hecho interesante que en este proceso de transitosis, por el corto tiempo que requiere para llevarse a cabo, aparentemente no ocurre modificación de las partículas virales pese a que estas células M presentan el receptor CD155 en su superficie. Esta es una evidencia que apoya que poliovirus puede ingresar a una célula sin presentar los cambios conformacionales de la partícula (Ouzilou et al., 2002). En base a los antecedentes anteriores, probablemente hPVR no sea la única molécula con la cual poliovirus pueda interaccionar o, que en ciertas circunstancias, la unión de poliovirus a CD155 no induzca el cambio conformacional que conduce al desnudamiento del RNA viral. Probablemente necesite de la interacción con otras moléculas, quizá del tipo motor, que le permitan desplazarse desde el sitio de entrada en la terminal sináptica hasta su sitio blanco de replicación en el SNC, sin necesidad de sufrir el cambio conformacional en la cápside durante este proceso. Por esta razón nos inclinamos a proponer que la partícula de poliovirus puede interactuar, de manera directa, con proteínas motoras asociadas a microtúbulos, sin necesidad de emplear a su receptor celular como intermediario para dicho transporte. En la segunda parte de este trabajo nosotros nos enfocamos en demostrar si poliovirus tiene la capacidad de unirse directamente a la dineína, sin necesidad de emplear a su receptor celular CD155 como intermediario para dicha unión, como se ha propuesto hasta la fecha por otros investigadores (Mueller et al., 2002; Ohka & Nomoto, 2001). 74 Inicialmente se analizó la presencia de la dineína en extractos S10 de células de neuroblastoma obtenidos a distintos grados de confluencia; 100, 40 y 60 % encontrándose la presencia de está en los extractos de células al 40 y 60 % pero no en el de 100 % de confluencia. Generalmente en los cultivos celulares, entre mayor es la confluencia mayor es la expresión de proteínas, sin embargo, en la línea celular de neuroblastoma SH-SY5Y, nosotros encontramos que cuando las células presentaban el 100% de confluencia ya no se detectaba la expresión de la dineína en el extracto citoplásmico pero si cuando tenían entre el 40% y 60% de confluencia. Una probable explicación es que la dineína es una proteína motora que participa en el transporte de organelos membranosos así como en la mitosis celular (Nobutaka, 1997; Steuer et al., 1990;Zinkowski et al., 1991; Duncan & Marrior, 2002). Durante la mitosis celular, la dineína se localiza en la corona del cinétocoro, cuya función principal es participar en el proceso de movimiento de los ásteres hacia los polos para favorecer la división celular, por tal motivo la expresión de esta proteína es alta en esta etapa (Steuer et al.,1990; Wordeman et al., 1991). Nosotros creemos que una vez que las células entran en fase G0 o estacionaria, donde ya no hay división celular, la expresión de la dineína en la célula disminuye o se abate y por tal razón no es posible detectarla en los extractos citoplásmicos. Por otra parte, se ha reportado que durante la interfase del ciclo celular la dineína se libera al citoplasma (Schroer et al., 1989). Una posibilidad, que no podemos descartar, es que una vez que la célula entra en fase estacionaria (100% de confluencia), la dineína concentre su actividad hacia el movimiento de organelos membranosos para el mantenimiento celular y por tal motivo pierda su solubilidad y no se libere al citoplasma. Después de obtener los extractos proteicos se procedió a propagar poliovirus en células HeLa y a purificarlo por gradiente de taza zonal de cloruro de cesio. La viabilidad del virus se monitoreó durante todo el proceso, determinando el título viral por el método de formación de placas líticas, acompañado de la observación del efecto citopático. Para verificar la eficiencia del proceso de purificación, se realizó una electroforesis en gel de poliacrilamida al 10% observándose bandas correspondientes a las proteínas de envoltura del virus: VP0 de 75kDa, VP2 de 29 KDa, y VP3 de 34 KDa (datos no mostrados). 75 Hay evidencias que indican que la purificación de poliovirus por gradiente de CsCl es sumamente eficiente, ya que el rendimiento del virus al igual que la calidad de la purificación son muy altas, pero se ha mostrado que, tanto rhinovirus como poliovirus pueden adsorber los iones cesio en su superficie, modificándose el gradiente de densidad de la partícula viral a 1.4 g/ ml en lugar de 1.34 g/ ml (Medappa & Rueckert, 1974; Minor, 1981). Nosotros demostramos que el ión cesio no modificó la capacidad del virus de unirse a su receptor celular ni tampoco su capacidad lítica, ya que después de la purificación, la fracción viral recuperada fue capaz de infectar células HeLa. Durante el proceso de purificación y concentración de poliovirus, se arrastraron algunas proteínas celulares que tienen una densidad similar al virus y, por tanto, migran a la misma altura del gradiente. Este hecho podría interferir en la veracidad de nuestros resultados por lo que para eliminar la posibilidad de que la unión con la dineína se debiese a la presencia de las proteínas provenientes de las células HeLa donde se propagó el virus, se trabajó con las mismas células no infectadas y se hizo una preparación de proteínas siguiendo el mismo procedimiento de purificación. Se acopló a perlas de sefarosa 4B como control del ensayo y se encontró que, al realizar las interacciones con el extracto S10 de neuroblastoma, no hubo unión a la columna de afinidad, demostrando que la unión con la dineína citoplásmica era debida propiamente al virus. Nuestros resultados de las interacciones bioquímicas entre poliovirus y extracto S10 mostraron que la dineína citoplásmica se une de manera directa a la cápside de poliovirus. La unión directa de poliovirus con esta proteína solamente se observó cuando se realizaron las interacciones con el extracto S10 de neuroblastoma obtenido a las 46 horas de cultivo, mientras que en aquéllas realizadas con el extracto S10 obtenido a las 56 horas no, pese a que la dineína se detectó previamente en dicho extracto. 76 Una probable explicación de la discrepancia de nuestros resultados encontrados en la interacción directa del virus con la dineína, es que probablemente estén presente otras proteínas que se expresan en fase temprana del crecimiento celular que pudiesen colaborar con la dineína para que se una a la cápside de poliovirus. La dinactina es otra proteína motora compuesta de múltiples subunidades, la cual está implicada en modular la capacidad de unión de la dineína citoplásmica a su carga. Hay evidencias que muestran que si se altera la expresión de la dinactina durante la división celular, se altera la función de la dineína, lo cual se refleja en el incorrecto alineamiento de los cromosomas y la organización de los “spindles”, (Echeverri et al.,1996). Al analizar las interacciones con los extractos citoplasmáticos de neuroblastoma obtenidos a diferentes tiempos de crecimiento se encontró diferencia, lo cual nos inclina a pensar en la participación de otras moléculas como la dinactina y la dinamina para que ocurra la interacción de poliovirus con la dineína en diferentes etapas del ciclo celular. Así, una posible explicación de porque poliovirus no se unió a la dineína presente en el extracto citoplasmático de neuroblastoma obtenido a las 56 horas, sea que en esa etapa del ciclo la expresión de dinactina disminuye o se abate, por tal motivo, la capacidad de unión de la dineína al virus se vea afectada y no se lleva a cabo. Finalmente, después de la realización de las interacciones, se demostró la presencia de poliovirus en la sefarosa 4B, con la finalidad de comprobar que el virus se encontraba realmente acoplado a la resina. Para esto se determinó la presencia de poliovirus mediante western blot empleando quimioluminiscencia. Nuestros resultados mostraron que efectivamente poliovirus estaba unido en la sefarosa 4B, ya que se encontró una banda de aproximadamente 35 kilodaltones en el eluído obtenido. El anticuerpo que se empleó fue de tipo policlonal IgG de conejo, el cual es capaz de reconocer la proteína VP1 de los tres serotipos de poliovirus. Mueller y Wimmer proponen que poliovirus disemina hacia SNC mediante endocitosis mediada por su receptor CD155, el cual interacciona con la dineína citoplásmica. Sin embargo, todos nuestros hallazgos plantean la posibilidad de que poliovirus también pueda interaccionar directamente con esta proteína y entonces 77 pueda migrar, a través de la red de microtúbulos, hacia el cuerpo neuronal donde el RNA genómico se desnuda y comienza la replicación. Un posible mecanismo para explicar la participación de la interacción directa del virus con la dineína citoplásmica sería que poliovirus interaccionara con su receptor CD155 sin que se genere cambio conformacional en la partícula viral, debido a la presencia de algún factor celular, encontrándose como partícula 160S. Una Vez que el virus se ha unido a su receptor, esté último favorezca que el virus interaccione directamente con la dineína, sin que el receptor CD155 tenga contacto con está, permitiendo que poliovirus se desplace retrógradamente a través nde las células nerviosas. Otra posibilidad, es que la interacción directa de la dineína con poliovirus se de exclusivamente dentro del cuerpo neuronal. Una probable función de está interacción, es que permita el desplazamiento del virus en el cuerpo neuronal hasta la periferia celular, sin requerir de la interacción de su receptor, con la finalidad de propagarse a células adyacentes. 78 Capítulo 10 CONCLUSIONES. • La calidad y pureza de las proteínas de fusión P y LFa1 fue adecuada para los ensayos y la conformación, tanto de la proteína GST como de la cola de histidinas, fue adecuada para la correcta purificación por cromatografía de afinidad. • Se determinó la presencia de la cadena intermedia de la dineína en extracto citoplasmático S10 de la línea celular SH-Y5Y de neuroblastoma humano, cuyo peso molecular fue de aproximadamente 74 KDa. La dineína citoplásmica sufre modificación en su expresión durante la división celular en la células de neuroblastoma humano observándose la mayor expresión en aquellas células que se encuentran en plena división celular (40-50% de confluencia) y no se detecta su presencia cuando la confluencia es del 90100%. • La región citoplásmica del receptor de poliovirus humano no se une a la dineína citoplásmica presente en el extracto S10 de neuroblastoma humano, bajo las condiciones experimentales realizadas. • La dineína citoplásmica, proveniente del extracto S10 de neuroblastoma obtenido a las 46 horas de crecimiento, se une de manera directa a la cápside de poliovirus. • La dineína citoplasmática proveniente del extracto S10 de neuroblastoma humano obtenido a las 56 horas no se une directamente a la cápside de poliovirus. • Poliovirus no requiere estrictamente de la región citoplásmica del receptor CD155 como intermediario para unirse a la dineína citoplásmica. 79 Capítulo 11 PERSPECTIVAS. En este trabajo nosotros no pudimos demostrar la unión de la dineína a la región citoplásmica de CD155 como lo reportaron Mueller & Wimmer. Como se mencionó en la discusión, ellos sólo trabajaron con la secuencia que codifica para la cadena ligera de la dineína citoplásmica en los ensayos del sistema de doble híbrido y de coinmunoprecipitación, por tal motivo, nosotros no podemos descartar la posibilidad de que esta interacción ocurra realmente. Por tal motivo se requerirá de analizar la posibilidad de que la dineína requiera de modificaciones previas a la interacción que le permitan exponer, tanto sus cadenas ligeras como intermedias, para favorecer la unión con el receptor de poliovirus. Una posible estrategia para demostrar lo anterior sería tratar a la dineína citoplásmica con algún componente químico desnaturalizante como vanadato y ATP, que promueven la desnaturalización de las cadenas pesadas, permitiendo desensamblar este complejo proteico y permitir la exposición tanto de las cadenas intermedias y ligeras de la dineína. Una vez realizado este tratamiento, se procedería a realizar las interacciones proteicas y analizarlas (Schroer et al., 1989). Otra posible estrategia para comprobar lo anterior, sería sintetizando diferentes péptidos representativos de las cadenas intermedias de la dineína citoplásmica, así como de algunas isoformas de la cadena ligera IC74. Se realizarían las interacciones proteicas entre cada uno de los péptidos sintéticos y la proteína de fusión que expresa la región citoplásmica de CD155 con la finalidad de demostrar si efectivamente CD155 puede unirse específicamente a algún componente de la dineína. Para demostrar si en dicha unión se requiere de la coparticipación de otras proteínas, se realizaría la expresión “in vitro” de dinamina y dinactina y se realizarían las interacciones proteicas en presencia de éstas. 80 El hecho de que nosotros hayamos encontrado que poliovirus puede unirse directamente a la molécula de dineína citoplásmica sin que requiera de su receptor celular, nos abre otra posibilidad en el mecanismo de transporte axonal hacia el SNC. De ser esto cierto, se tendría que comprobar su unión “in vivo”, ya sea trabajando con ratones transgénicos que expresan el receptor de poliovirus humano y los hace susceptibles a la infección por poliovirus o empleando un sistema de cámara de difusión que permitiera mantener la funcionalidad y viabilidad de células nerviosas que conformaran un nervio. Mediante estas dos herramientas se podría demostrar, por técnicas de colocalización con anticuerpos anti-dineína y anti-polio y monitoreo digital a lo largo del axón hasta el cuerpo neuronal, la interacción directa de poliovirus con la dineína citoplásmica desde la región sináptica del axón hasta el cuerpo neuronal. De hecho, no nos limitaríamos a pensar que la unión directa de poliovirus con la dineína sólo pueda ocurrir en células neuronales, si no que también pudiese ocurrir en otro tipo de células, como es el caso de las células del nódulo linfático. 81 APÉNDICE 1. Interacciones proteicas de las proteínas de fusión y el extracto proteico recuperadas por cromatografía de afinidad con sefarosa Níquel. Con el fin de analizar las interacciones bioquímicas entre las proteínas de fusión y las de médula espinal de ratón, se realizaron ensayos de cromatografía de afinidad empleando la resina Ni-NTA, la cual tiene afinidad por aquellas proteínas que presentan un tallo de 6 histidinas consecutivas. Las interacciones se realizaron incubando las proteínas de fusión LF1a y Pα (100 µg) con el extracto de médula espinal de ratón (3 mg) en presencia de coctel inhibidor de proteasas a 37°C en agitación por 3 horas. Como controles del ensayo se emplearon las proteínas de fusión Pα y LF1a sin el extracto de médula espinal de ratón, al igual que el extracto de médula espinal en ausencia de las proteínas de fusión, las cuales se trabajaron bajo las mismas condiciones que las interacciones proteicas. Después de la incubación, el sobrenadante se trasladó a un tubo cónico de 5 ml, se adicionaron 2 µl de imidazol 10 mM y 300 µl de resina Ni-NTA (Quiagen) previamente equilibrada con PBS al 50% (v/v). Se incubó toda la noche a 4° C y posteriormente se centrifugó a 323 x g por 1 minuto. Se recuperó el sobrenadante, constituyendo la fracción de lo “no unido”, y la resina se lavó cinco veces con buffer de lavado (NaHPO4 50 Mm, NaCl 150 Mm, etanol absoluto al 20% e imidazol 10 Mm pH 8.0) por 15 minutos a temperatura ambiente en agitación. Se recuperaron las fracciones de lavado, las proteínas unidas a la resina se eluyeron tres veces, cada una con 500 µl de buffer de elusión (NaHPO4 50 Mm, NaCl 300 Mm, imidazol 500 mM y tritón X-100 1%, pH 8.0) durante 30 minutos. Las fracciones obtenidas se precipitaron con 1000 µl de acetona a –20° C por una hora, se centrifugaron a 9500 x g en centrífuga Sigma refrigerada a 4° C por 10 minutos, la pastilla se secó en centrífuga de vacío “Vacufuge”por 15 minutos y se resuspendió en 30µl PBS 10 mM. Todas las muestras se calentaron a 96° C en presencia de amortiguador de 82 carga Laemli y se realizó una electroforesis en gel de poliacrilamida al 12%. El gel se tiñó con la técnica de plata. En estos ensayos no se pudo evidenciar ninguna interacción específica entre la proteína Pα y las de médula espinal, especialmente por el número de proteínas celulares que interaccionan de manera inespecífica con la resina de níquel. La resina Ni-NTA une cinco veces más proteína de manera inespecífica en comparación con la resina-glutatión, por lo que decidimos no continuar los experimentos con ésta (figura A). La posible causa de estos resultados es que quizá las proteínas del SNC son ricas en secuencias consecutivas de histidinas, lo que implica que cualquier secuencia con más de 6 histidinas consecutivas es reconocida por la resina Ni-NTA, resultando en interacciones directas con ella y no a través de las proteínas de fusión. 83 APÉNDICE 2. SOLUCIONES. Amortiguador de fosfatos (PBS ) 1X. KH2PO4 1mM Na2HPO4 10 Mm NaCl KCl 136 mM 2.8 Mm Ajustar pH a 7.4 PBS 20 mM. Na2HPO4 0.5 M 10 ml NaH2PO4 0.5 M 10 ml NaCl 15.4 ml 5M Llevar a 400 ml con agua destilada y ajustar pH a 7.4 y aforar a 500 ml. PBS 50 mM. Na2HPO4 0.5 M 25 ml NaH2PO4 0.5 M 25 ml Llevar a 400 ml con agua destilada, ajustar pH a 7.4 y aforar a 500 ml. REACTIVOS PARA WESTERN BLOT. Buffer de corrida para proteínas 10x. Glicina 200 Mm Trizma –Base 24 Mm Duodecil Sulfato de sodio (SDS) 1% Disolver en agua destilada. Se usa a concentración final 1X. 84 Solución de Azul de Coomassie. Azul de Coomassie R250 0.2g Metanol 50% 50 ml Ácido acético 7% 7 ml Adicionar agua destilada hasta aforar a 100 ml. Solución Rojo Ponceu. Rojo Ponceu 0.1% Ácido tricloroacético 7% Solución de desteñido de gel de poliacrilamida. Metanol 30% Ácido acético 7% Buffer de muestra Laemli 2X. Tris-base 100 Mm 1ml SDS 10% 2ml β-mercaptoetanol 1% 0.1 ml Glicerol 60% H20 6 ml 0.6 ml Azul de bromofenol 1% 0.1 ml Proteger de la luz. Buffer de transferencia. Tris-Base 50 Mm Glicina 40 Mm Metanol 10% Amortiguador AP para revelar con fosfatasa alcalina. Tris-HCl 1000 Mm NaCl MgCl2 100 Mm 5 mM, ajustar pH a 9.4 y aforar. 85 REACTIVOS PARA EXTRACTOS CELULARES. Buffer de despegado para células. Tris-HCl 40 mM pH 7.5 EDTA 1mM NaCl 150 mM. Se esteriliza a 120 libras por 20 minutos. Buffer de lisis “A” para extracto citoplásmico S10 HEPES 10 mM pH 7.9 MgCl 1.5 mM KCl 10 Mm Esterilizar a 120 libras por 20 minutos. Buffer RSB-NP40 MgCl2 1.5 Mm, NaCl 10 mM Tris-HCl 10 mM pH. 7.5 NP40 o IGPAL 1% REACTIVOS PARA LA TITULACIÓN DE POLIOVIRUS. Solución Stock de cristal violeta. Cristal violeta Etanol al 20% 1gr. 99 ml. Solución de trabajo de Cristal Violeta. Solución Stock de cristal violeta Etanol al 95% 20 ml 40 ml 86 Agua destilada 150 ml Ácido Tricloroacético al 10%. TCA 10 ml Aforar a 100 ml con agua destilada. Buffer NTE. NaCl 100 mM Tris-HCl pH 7.5 EDTA 10 mM 1 mM Aforar a 1 litro con agua destilada y esterilizar a 121 º C por 20 minutos. SOLUCIONES PARA ACOPLAMIENTO DEL VIRUS A SEFAROSA 4B. Solución de activación (HCl 1 mM). HCl 1N 0.1 ml Aforar a 100 ml con agua. Solución de acoplamiento (NaCl 0.5M/NaHCO3 0.1 M pH. 8.3 NaCl 0.5M NaHCO3 0.1 M Ajustar el pH a 8.3 con NaOH. Solución de bloqueo (Tris-glicina 0.1M pH 8.6). Glicina 0.75g Tris-base 1M 50 ml Ajustar el pH a 8.6 con NaOH y aforar a 100 ml. 87 Solución de acetatos 0.1 M pH 4.0. Ácido acético 1M 25 ml NaCl 7.31g Ajustar el pH a 4 y aforar a 250 ml. PBS (15 mM/NaCl 0.5 M). KH2PO4 1M K2HPO4 1M NaCl 0.5M Se ajusta el pH a 7.4 con HCl o NaOH. SOLUCIONES PARA LAS INTERACCIONES PROTEICAS. Buffer de lavado para las interacciones con la columna de poliovirus (PBS 1X 0.1 M de NaCl) KH2PO4 1 mM Na2HPO4 10 mM NaCl 0.1 M KCl 2.8 mM Ajustar pH a 7.4 Buffer de lavado para resina Ni-NTA. NaH2PO4 NaCl 50 mM 150 mM Imidazol 10 mM Etanol 20 % Se ajusta el pH a 8.0. 88 Buffer de elusión para la resina Ni-NTA. NaH2PO4 50 mM NaCl 300 mM Imidazol 500 Mm Tritón X100 1% Se ajusta el pH a 8.0 Buffer de lavado para resina glutatión (PBS/ NaCl 10 mM, IGPAL 0.2% pH 7.7) KH2PO4 1M 2.92 ml K2HPO4 1M 4.56 ml NaCl IGPAL 0.1M 2.96 g 0.2 % Ajustar pH a 7.7, aforar a 500 ml. 89 Capítulo 12 REFERENCIAS BIBLIOGRÁFICAS. Belnap, M.D., McDermott, M. B., Filman, J.D., Cheng, N., Trus, L. B., Zuccola, J. H., Racaniello, R. V.,Hogle, M. J. and Steven, C.A. 2000. The three-dimensional structure of poliovirus receptor bound to poliovirus. PNAS 97(1):73-78. Bernhardt, G., Bibb, A. J., Bradley, J. and Wimmer, E. 1994. Molecular characterization of the cellular receptor for poliovirus. Virology. 199:105-113. Bernhardt, G., Harber, J., Zibert, A., Crombrugghe, M. and Wimmer E. 1994. The poliovirus receptor: Identification of domains and amino acid residues critical for virus binding. Virology. 203: 344-356. Brady, T. S. 1991. Molecular motors in the Nervous System. Neuron. 7: 521-533. Cheney, I. W., Naim, S., Shim, J.H., Reinhardt, M., Pai, B., Wu, J.Z., Hong, Z. and Zhong, W. 2003. Viability of poliovirus / rhinovirus VPg chimeric viruses and identification of an amino acid residue in the VPg gene critical for viral RNA replication. Journal of virology.77 (13):7434-7443. Cyr, L. J. and Brady, T.S. 1992. Molecular motors in axonal transport: Cellular and molecular biology of kinesin. Molecular Neurobiology. 6: 137-155. Döhner, K., Wolfstein, A., Prank, U., Echeverri, C., Dujardin, D., Valle, R. and Sodeik, B. 2002. Function of dynein and dynactin in herpes simplex virus capsid transport. Molecular Biology of the Cell. 3: 2795-2809. 90 Duncan, E.J. and Marrior, R. 2002. The citoplasmic dynein and kinesin motors have interdependents roles in patterning the Drosophila oocyte. Current Biology. 12 (23): 1982-1991. Echeverri, C. J., Paschal, B. M., Vaughan, K. T. and Valle, R. B.1996. Molecular characterization of the 50-KD subunit of dynactin reveals function for the complex in chromosome alignment and spindle organization during mitosis. Journal of cell biology.132 (4): 617-633. Fricks, E. C. and Hogle, M. J. 1990. Cell-induced conformational change in poliovirus: Externalization of the amino terminus of VP1 is responsible for liposome binding. Journal of virology. 64 (5): 1934-1945. Gromeier, M. and Wimmer, E. 1998. Mechanism of injury-provoked poliomyelitis. Journal of Virology. 72: 5056-5060. Gutierrez, AL., Denova-Ocampo, M., Racaniello, R.V. and Del Angel, R.M. 1997. Attenuating mutations in the poliovirus 5´ untranslated region alter its interaction with polypyrimidine tract-binding protein. Journal of virology. 71(5): 3826-3833. He, Y., Bowman, D. V., Mueller, S., Bator, M. C., Bella, J., Peng, X., Baker, S.T., Wimmer E., Kuhn, J.R. and Rossmann, G. M. 2000. Interaction of the poliovirus receptor with poliovirus. PNAS. 97 (1): 79-84. Holland, J.D; Miranda-Saksena, M., Boadle, A. R., Armati, P. and Cunningham, L.A. 1999. Anterograde transport of Herpes Simplex Virus proteins in axons of peripheral human fetal neurons: an immunoelectron microscopy study. Journal of Virology. 73 (10):8503-8511. Jacob, Y., Badrane, H., Ceccaldi, E-P. and Tordo, N. 2000. Cytoplasmic dynein LC8 interacts with Lyssavirus phosphoprotein. Journal of virology. 74 (21): 1021710222. 91 Joklik, W. K. and Darnell, E.Jr.1961. The adsorption and early fate of purified poliovirus in HeLa cells. Virology. 13: 439-447. Kajigaya, S., Arakaya, H., Kuge, S., Koi, T., Imura, N. and Nomoto, A. 1985. Isolation and characterization of defective-interfering particles of poliovirus Sabin 1 strain. Virology. 142: 307-316. King, M. S., Barbarese, E., Dillman III, F. J., Benashski, E. S., Do, T. K., KingPatel, S. R. and Pfister, K. K. 1998. Cytoplasmic dynein contains a family of differentially expressed light chains. Biochemistry. 37 (43): 15033-15041. La Monica, N., Almond, W.J. and Racaniello, R.V. 1987. A mouse model for poliovirus neurovirulence identifies mutations that attenuate the virus for humans. Journal of virology. 61(9): 2917-2920. Levinton, Leon and Darnell, E.Jr.1960. A simplified procedure for purification of large amounts of poliovirus:characterization and amino acid analysis of type 1 poliovirus. The Journal of Biological Chemistry. 235 (1): 70-73. Lukashok, A.S., Tarassishin, L., Li, Y. and Horwitz, S.M. 2000. An Adenovirus inhibitor of Tumor Necrosis Factor Alpha-induced apoptosis complexes with dynein and small GTPase. Journal of Virology. 74 (10): 4705-4709. Lye, J. R., Porter, E. M., Scholey, J. M. and McIntosh, R. J. 1987. Identification of a microtubule-based cytoplasmic motor in the nematode C. elegans. Cell. 51: 309-318. Martínez, S.E., Ramos, E., Lafuente, E. and López de Quinto S. 2001. Funtional interactions in internal translation initiation directed by viral and cellular IRES elements. Journal of General Virology. 82: 973-984. 92 Mendelsohn, L.C., Wimmer, E. and Racaniello, R. V. 1989. Cellular receptor for poliovirus: Molecular cloning, nucleotide sequence, and expression of a new member of the Immunoglobulin superfamily. Cell. 56: 855-865. Minor, P.D. 1981. Growth, assay and purification of picornaviruses, p. 25-41. In Mahy, J. W. B. (ed). Virology a practical approach. Oirl Pres, Oxford-Washington D.C. Moscufo, N. and Chow, M. 1992. Myristate-protein interactions in poliovirus: interactions of VP4 threonine 28 contribute to the structural conformation of assembly intermediates and the stability of assembled virions. Journal of Virology. 66 (12):6849-6857. Moscufo, N., Simons, J. and Chow, M. 1991. Myristoylationis important at multiple stages in poliovirus assembly. Journal of Virology. 65(5):2372-2380. Moscufo, N., Yafal-Gomez, A., Rogove, A., Hogle, J. and Chow, M. 1993. A mutation in VP4 defines a new step in the late stages of cell entry of poliovirus. Journal of Virology. 67 (8): 5075-5078. Mueller, S., Cao, X., Welker, R. and Wimmer, E. 2002. Interaction of the poliovirus receptor CD 155 with the dynein light chain Tctex-1 and its implication for poliovirus pathogenesis. The Journal of Biological Chemistry. 277 (10): 78977904. Murray, K.E. and Barton, D.J. 2003. Poliovirus CRE-dependent Vpg uridylation is required for positive-strand RNA synthesis but not for negative-strand RNA synthesis. Journal of virology. 77 (8): 4739-4750. Nathanson, N. and Bodian, D. 1961. Experimental poliomyelitis following intramuscular virus injection. The effect of neural block on a neurotropic and a pantropic strain. Bull Johns Hopkins Hosp. 108: 308-319. 93 Nobutaka, Hirokawa. 1997. The mechanisms of fast and low transport in neurons: identification and characterizations of the new kinesin superfamily motors. Current Opinion in Neurobiology. 7: 605-614. Nomoto, A., Koike, S. and Aoki, J. 1994. Tissue tropism and species specificity of poliovirus infection. Trends in Microbiology. 2(2): 47-51. Ohka S., and Nomoto A. 2001. Recent insights into poliovirus pathogenesis. TRENDS in Microbiology. 9(10):501-505. Ohka, S., Yang, X-W., Terada, E., Iwasaki, k. And Nomoto, A. 1998. Retrograde transport of intact poliovirus the axon via the fast transport system. Virology. 250: 67-75. Ouzilou, L., Caliot, E., Pelletier, I., Prévost, MC., Pringault, E. and ColbereGarapin, F. 2002. Poliovirus transcytosis through M-like cells. Journal of General Virology. 83: 2177-2182. Pallansch, A. M. and Ross, P. R. 2001. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses, p. 723-775. In B.N. Fields, D.M. Knipe, P. M. Howley, et al.(ed), Fields virology, forth ed Vol. 1. Lippencott Raven, New York. Panagiotidis, A. CH. And Silverstein, J. S. 1995. pALEX, a dual-tag prokariotyc expression vector for the purification of full-length proteins. Gene. 164 (1):45-47. Paschal, M. B., Shpetner, S. H. and Valle, R. B. 1987. MAP 1C is a microtubuleactivated ATPase which translocates microtubules in vitro and has dynein-like properties. The Journal of Cell Biology. 105: 1273-1282. Penfold, M. E. T., Armati, P. and Cunningham, L. 1994. Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc. Natl. Acad. Sci. USA. 91: 6529-6533. 94 Pfister∗, K. K., Salata, M. W., Dilman, J.F., Torre, E. and Lye, R.G. 1996. Identification and developmental regulation of a neuron-specific subunit of cytoplasmic dynein. Mol. Biol. Cell. 7: 331-343. Pfister, K. K., Salata, M. W., Dilman, J.F., Vaughan, K.T., Valle, R.B., Torre, E. and Lye, R.G. 1996. Differential expression and phosphorylation of the 74-KDa intermediates chains of cytoplasmic dynein in cultured neurons and glia. Journal of Biological Chemistry. 271: 1687-1694. Ponnuraj, M.E., John, J.T., Levin, J.M. and Simoes, F.A.E. 2001. Sabin attenuated LSc/2ab strain of poliovirus spreads to the spinal cord from a peripheral nerve in bonnet monkeys (Macaca radiata). Journal of General Virology. 82: 1329-1338. Racaniello, R. V. 1996. The poliovirus receptor: a hook, or an unzipper. Current Biology. Structure 4 (7):769-773. Racaniello, R.V. 2001. Picornaviridae: The viruses and their replication, p. 685722. In B.N. Fields, D.M. Knipe, P. M. Howley, et al.(ed), Fields virology, forth ed Vol. 1. Lippencott Raven, New York. Raux, H., Flamand, A. and Blondel, D. 2000. Interactions of the Rabies Virus P protein with the LC8 dynen light chain. Journal of Virology. 74 (21): 10212-10216. Ren, R., Costantini, F., Gorgacz, J. E., Lee. J. J. and Racaniello, R. V. 1990. Transgenic mice expressing a human poliovirus receptor: A new model for poliomyelitis. Cell. 63: 353-362. Ren, R. and Racaniello, R.V. 1992. Poliovirus spreads from muscle to the central nervous system by neural pathways. The Journal of Infectious Diseases. 166: 747-752. 95 Roizman, B., and Knipe, M. D. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In B.N. Fields, D.M. Knipe, P. M. Howley, et al.(ed), Fields virology, forth ed Vol. 2. Lippencott Raven, New York. Saksena, M.M., Armati, P., Boadle, A.R., Holland, J.D. and Cunningham, L.A. 2000. Anterograde transport of Herpes Simplex Virus type 1 in cultured, dissociated human and rat dorsal root ganglions neurons. Journal of Virology. 74 (4): 1827-1839. Schroer, A. T., Steuer, R. E. and Sheetz, P. M. 1989. Cytoplasmic dynein is a minus end-directed motor for membranous organelles. Cell. 56: 937-946. Selinka, C-H., Zibert, A. and Wimmer, E. 1992. A chimeric poliovirus /CD4 receptor confers susceptibility to poliovirus on mouse cells. Journal of Virology. 66 (4): 2523-2526. Solecki, D., Bernhardt, G., Lipp, M. and Wimmer, E. 2000. Identification of a nuclear respiratory factor-1 binding site within the core promoter of the human polio virus receptor/CD155 gene. The Journal of Biological Chemistry. 275 (17):12453-12462. Solecki, D., Wimmer, E., Lipp, M. and Bernhardt, G. 1999. Identification and characterization of the cis-Acting elements of the human CD155 gene core promoter. The Journal of Biological Chemistry. 274 (3):1791-1800. Steuer, R.E., Woderman, L., Schroer, A.T. and Sheetz, P. M. 1990. Localization of citoplasmic dynein to mitotic spindles and kinetochores. Nature. 345: 266-268. Strauss, D.M., Glustrom, L.W. and Wuttke, D.S. 2003. Towards an understanding of the poliovirus replication complex: the solution structure of the soluble domain of the poliovirus 3A protein. Journal of Molecular biological. 330:225-234. 96 Takenaka, T., Kawakami, T., Hori, H., Hashimoto, Y., Himura, H. and Kusakabe, T. 1998. Axoplasmic transport and its signal transduction mechanism. Japenese Journal of Phisiology. 48: 413-420. Teterina, L. N., Egger, D., Bienz, K., Brown, M. D., Semler, L. B. and Ehrenfeld, E. 2001. Requirements for Assembly of poliovirus replication complexes and negative-strand RNA synthesis. Journal of Virology. 75 (8): 3841-3850 . Tsang, K. S., McDermott, M. B., Racaniello, R. V. and Hogle, M. J. 2001. Kinetic analysis of the effect of poliovirus receptor on viral uncoating: the receptor as a catalyst. Journal of Virology. 75 (11):4984-4989. Vaughan, K. T., Mikami, A., Paschal, B. M., Holzbaur, E. L., Hughes, S. M., Echeverri, C. J., Moore, K. J., Gilbert, D. J., Copeland, N. G., Jenkins, N. A. and Valle, R. B. 1996. Multiple mouse chromosomal loci for dynein-based motility. Genomics. 36 (1): 29-38. Wordeman, L., Steuer, R.E., Sheetz, P.M. and Mitchison, T. 1991. Chemical subdomains within the kinetochore domain of isolated CHO mitotic chromosomes. The Journal of Cell Biology. 114 (2): 285-294. Ye, G. J., Vaugham, K.T. and Roizman, B. 2000. The Herpes Simplex Virus 1 U(L) 34 proteins interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. Journal of Virology. 74 (3): 1355-1366. Zinkowski, P.R., Meyne, J. and Brinkley, R.B. 1991. The centromere-kinetochore complex: a repeat subunit model. The Journal of Cell Biology. 113(5):1091-1110. Medappa, K. C. And Rueckert, R. R. 1974. Am. Soc. Microbiol. Abstr., 207. 97