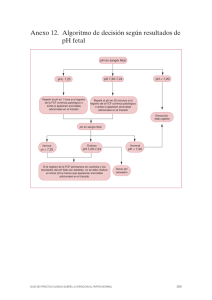
Abrir - ACOG
Anuncio

Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 79 C A P Í TU L O 4 • OBSTETRICIA Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 81 EXAMEN ECOGRÁFICO MORFOLÓGICO FETAL ANORMAL EN EL SEGUNDO TRIMESTRE (18-23 SEMANAS) Dr. Philippe Massoc L. Ginecoobstetra. Departamento de Exploración Fetal. Servicio de Ginecología y Obstetricia. Hospital Guillermo Grant Benavente. Concepción. Chile. La tasa de malformaciones permanece invariable desde inicios del siglo XX. Si se incluyen malformaciones mayores, menores, abortos y mortinatos, las malformaciones afectan al 10% de las gestaciones. La incidencia de anomalías congénitas severas al nacimiento es de 2-3% en la población general, siendo responsables del 20-25% de las muertes perinatales y de porcentajes incluso mayores de morbilidad perinatal. Las malformaciones cerebrales representan en los RN alrededor del 9% de las malformaciones aisladas y el 15,9% en los polimalformados. Lo que hace que el SNC sea un elemento crítico en el Diagnóstico Antenatal(1). El uso del US en obstetricia fue introducido en 1958 por Ian Donald, considerándose uno de los grandes hitos de la medicina. Se considera que hoy entre al 60-100% de las mujeres en EE.UU. y el occidente europeo se les realizan ecografías en prenatal. Los transductores o sondas endovaginales existen desde hace muchos años y permiten un estudio de las estructuras embrionarias de forma bastante precisa. 1 Briard ML; Arch Fr Pediartr 1975; 32: 123-138. EXAMEN ECOGRÁFICO MORFOLÓGICO ANORMAL DEL SISTEMA NERVIOSO CENTRAL El cerebro es uno de los órganos donde su desarrollo es particularmente interesante de estudiar, principalmente en la ecografía. A las 8 SA ya se pueden observar cavidades endocerebrales especialmente visibles en un corte coronal. A las 11 SA el embrión ya se convierte en un feto y reacciona a estímulos externos, donde se observan además movimientos de deglución y de extremidades. EMBRIOLOGÍA Antes del cierre de tubo neural, la extremidad cefálica presenta 3 estructuras bien identificables: prosencéfalo o cerebro anterior, el mesencéfalo o cerebro medio y el romboencéfalo o cerebro posterior. El prosencéfalo y el romboencéfalo se van a dividir. El primero en telencéfalo y el diencéfalo, y el segundo en metencéfalo y en mielencéfalo. El telencéfalo da 2 eminencias laterales que van a constituir los hemisferios cerebrales. Los ventrículos laterales comunican por medio del agujero de Monroe con el tercer ventrículo (V3) y éste con el cuarto ventrículo (V4) por medio del acueducto de Sylvio. A las 8 SA el polo cefálico está flectado, el diámetro biparietal mide 5 mm. En el polo superior, hacia posterior, se observa una cavidad anecogénica que corresponde al romboencéfalo. Entre las 8 y 9 SA es cuando las cavidades embrionarias encefálicas pueden ser mejor estudiadas, el embrión mide 19 mm de longitud, en un corte sagital se reconocen de adelante hacia atrás el diencéfalo, el mesencéfalo y el romboencéfalo. En un corte coronal posterior se puede observar el raquis. A 9 SA se pueden identificar otras estructuras, en corte sagital el diencéfalo, mesenencéfalo y romboencéfalo que se divide en metencéfalo y en mielencéfalo (Figura 1). Figura 1. En un corte sagital estricto podemos observar el prosencéfalo o cerebro anterior que se dividirá en telencéfalo y el diencéfalo (1), el mesencéfalo o cerebro medio que se dividirá en metencéfalo y en mielencéfalo (2) y el romboencéfalo o cerebro posterior (3). 82 Capítulo 4 - Obstetricia A las 10 SA en corte axial, se observan los ventrículos laterales con los plexos coroideos (Figura 2). A 11 SA ya se pueden identificar estructuras de la línea media: en un corte axial, los pedúnculos cerebrales, cerebelo y V4. El cuerpo calloso se empieza a formar a las 15 SA, el que se desarrolla de adelante hacia atrás, terminando a las 22 SA. Particular interés constituye la eco tridimensional que nos permite obtener un volumen del embrión y secundario a esto obtener distintos niveles de cortes en los 3 planos espaciales. Esto nos va a permitir realizar el diagnóstico precoz de anomalías como la acrania, anencefalia y holoprosencefalia. Es entre las 8 y 9 SA que las vesículas cerebrales son mejor visualizadas, y su ausencia puede ser un signo de anomalía cerebral. Ben Pansky. Review of Medical embryology. Macmilliam Publishing Company 1986; 362-427. ser dramáticas. Esta puede ser parcial o completa, aislada o asociada a otras malformaciones, a síndromes genéticos que incluyen anomalías cromosómicas o metabólicas(2-4). El consejo a los padres es difícil. La ACC asociada a una malformación cerebral, a una anomalía cromosómica, a un síndrome malformativo o a una anomalía metabólica, es siempre desfavorable. Pero el pronóstico es incierto cuando se trata de una ACC aislada(5). Es entonces primordial que una ecografía complementada con una resonancia magnética (RM) cerebral, haga o confirme primero el diagnóstico de ACC y en segunda intención realice la búsqueda de otras malformaciones asociadas. El cuerpo calloso (CC) se desarrolla en el periodo post embrionario a partir de la semana 10 de gestación; se completa al sexto mes, después del desarrollo de dos otras conexiones más precoces, las comisuras anteriores y el hipocampo. Esto podría explicar porque su visualización prenatal no puede ser posible hasta las 20 semanas de amenorrea (SA). El CC se desarrolla desde el rostrum hacia la región caudal, con la formación respectiva del genu corporis, el cuerpo y el esplenium. Este proceso de desarrollo es importante: los casos de ACC parcial comprometiendo la porción anterior son principalmente debido a destrucción por patología vascular o infecciosa y aquellos afectados en la porción posterior resultan regularmente de anomalías del desarrollo(5-9) (Figura 3) (Tabla I). En las series actuales de medicina fetal, la ACC representa alrededor de 3-5% de las malformaciones Figura 2. En un corte axial, el telencéfalo da 2 eminencias laterales que van a constituir los hemisferios cerebrales. Atrás se puede observar el romboencéfalo (R). 1. SISTEMA NERVIOSO CENTRAL ANOMALÍAS DE LA LÍNEA MEDIA • AGENESIA DE CUERPO CALLOSO El Cuerpo Calloso es la principal y más larga conexión interhemisférica del cerebro humano, está formado por un reagrupamiento de axones comisurales neocorticales, donde su normal desarrollo es testigo de la adecuada situación de los circuitos interhemisféricos(1). La Agenesia de Cuerpo Calloso (ACC) es una patología rara con consecuencias clínicas que pueden Figura 3. Hipoplasia cuerpo calloso. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 83 TABLA I. SIGNOS EVOCADORES DE AUSENCIA DE CUERPO CALLOSO Corte transversal (axial) • Dilatación moderada de ventrículos laterales. En “aspecto de gota de agua” (Figura 4 a). • Dilatación moderada, bilateral, simétrica de los cuernos occipitales. • Ausencia o aspecto inusual del cavum septum pellucidum. • Aumento del espacio interhemisférico. • Aumento de la distancia entre los cuernos frontales, con aspecto cóncavo (Figura 4b). • Alargamiento del III ventrículo. • Hipoplasia cerebelosa asociada o no a un agrandamiento de la fosa posterior. Corte sagital • Aspecto anormal de las circunvoluciones (Figura 5). • Interrupción o ausencia total de la arteria pericallosa (Figura 6). • Disposición radiar de las cisuras cerebrales de la cara interna de los hemisferios cerebrales (visible a partir de las 32SA). • Imagen hiperecogénica evocando un lipoma de la línea media. Corte frontal • Expansión ascendente del III ventrículo, interpuesto entre los cuernos frontales alcanzando la cisura interhemisférica. Otros signos de sospecha • Microcefalia: medición de perímetro craneano <-3 DS. • Sospecha de hipertelorismo. • Todo contexto polimalformativo, en particular sobre la línea media (cráneo-facial). 4a Figura 5. Agenesia cuerpo calloso, véase la dirección radiar de las circunvoluciones. 4a Figura4 a, b. Ventrículo lateral en forma de “gota de agua” al corte axial, y en “astas de toro” en corte coronal. Figura 6. Al modo energy o Doppler color se puede observar que la arteria cerebral anterior no se continúa con la arteria pericallosa, sino que sigue un trayecto ascendente. 84 Capítulo 4 - Obstetricia del sistema nervioso central (SNC) diagnosticadas por ecografía obstétrica y alrededor de 50% de la anomalías de la línea media(7). La ACC es la malformación comisural más frecuente(10). Su prevalencia es difícil de precisar en la población general, por ser a veces totalmente asintomática y varía en función de las poblaciones estudiadas(5,7,11,12). Se calcula en 3-7/10.000 en la población general. Así es como en grandes series de autopsias el descubrimiento fortuito de una ACC es de 1/19.000(13); y en imagenología en instituciones especializadas en desordenes mentales es de 2-4%(1416) . En función de la gestación, el CC es visto en corte sagital entre las 17-18 SA como una fina imagen lineal hipoecogénica de 1 mm de espesor, destacando el genu corporis y la parte corporal; entre las 22-23 SA el CC es visto en su totalidad en corte sagital, mide 1,8 mm de espesor corporal en promedio. Entre las 32-33 SA el CC mide 2,5 mm de espesor corporal; en corte coronal anterior, permitiendo visualizar muy bien la comisura callosa en toda su extensión. Criterios ecográficos • Diagnósticos Es posible poner en evidencia a través de la ecografía una ACC durante el segundo trimestre del embarazo, a partir de las 17-18 SA.. Una ACC será sospechada por la observación de signos indirectos a partir de las 21-22 SA; en caso de fuerte sospecha un examen complementario a las 25 SA permitirá confirmar el diagnóstico por visualización directa de ausencia parcial o total de CC(7,9) (Tabla II). TABLA II SIGNOS DE SOSPECHA DE ACC Ventriculomegalia • Cuernos frontales en “cuernos de toro” (teardrop sign) • Ausencia cavum septum pellucidum • Ausencia Cuerpo calloso en corte sagital. Signos indirectos de ACC • Aumento de la separación de los hemisferios • • • con una cisura interhemisférica prominente. Ascenso del III ventrículo Lesión de la línea media (lipoma, quiste) Dirección ascendente de la arteria pericallosa El Doppler color tiene un gran aporte ante la sospecha de esta patología. La arteria pericallosa, caracterizada por ser un vaso semicircular en corte sagital, puede estar ausente; en este caso la arteria cerebral anterior no adopta su trayecto curvilíneo en rededor del CC, sino que adopta una trayectoria radial ascendente donde su terminal es la arteria callosomarginal. Este signo puede ser visto precozmente antes de las 22 SA. La distinción entre ACC completa o parcial es difícil de realizar sólo por US y el recurso de la RM se hace necesario para aportar argumentos en favor de una u otra situación. El diagnóstico de ACC parcial se basa en la realización de cortes coronales seriados que tienden a precisar mejor la localización y el espesor del CC. La agenesia parcial es clásicamente de la porción posterior; sin embargo, puede existir una destrucción parcial anterior secundaria de origen clástico (17) . La hipoplasia de CC puede dar falsos positivos de diagnóstico de ACC(8). La RM tiene un importante rol en el diagnóstico de ACC, esta permite una excelente visualización del cerebro fetal y sin consecuencias para el feto, lo que otorga una ayuda complementaria invaluable ante la sospecha de este diagnóstico. Actualmente la RM se sugiere para confirmar o descartar una sospecha de ACC o para la búsqueda de anomalías intracranianas asociadas. Es de mayor sensibilidad para anomalías de la giración, de malformaciones de la fosa posterior y precisa mejor aún la topografía de lesiones quísticas asociadas(18) (Tabla III). En un estudio de Lair-Milan (1997)(19), sobre 29 RM cerebrales antenatales realizadas por dilatación ventricular, 5 ACC fueron detectadas y el diagnóstico de ACC fue propuesto por error en 2 casos (21). Sonigo et al (1995)(17) sobre 48 fetos entre 21 y 38 SA que presentaban una ACC, en 65% de los casos fue sospechado por US; en 35% de los casos fue diagnosticado por la RM. El principal signo de sospecha, así como en otras series, fue una dilatación ventricular(3,5,22,27). El rol de la RM fetal en el estudio antenatal de la ACC es esencialmente en la búsqueda de malformaciones cerebrales asociadas, las que son frecuentes (en alrededor del 75% de los casos) (7,17) . Después de un estudio ecográfico, RM y cariotipo, sólo 15% de ACC se pueden considerar como aisladas; es decir, sin anomalía cerebral o extracerebral, ni anomalía de cariotipo, ni antecedente familiar de ACC con mal desarrollo psicomotor(5,17) (Tabla IV). Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) TABLA III. ANOMALÍAS CEREBRALES EVIDENCIADAS POR RM EN ACC Giración • Paquigiria • Lisencefalia • Esquizencefalia • Anomalía de operculización Silviana Heterotopía subependimaria Nódulo subependimario Atrofia difusa Hipoplasia de lóbulos frontales Microcefalia Fosa posterior • Dandy Walker • Quiste aracnoídeo • Hipoplasia cerebelosa • Mega-gran cisterna Quiste interhemisféricos Hipotelorismo Dismorfia facial Encefalocele occipital Macrocrania con alargamiento de los espacios subaracnoideos Dilatación ventricular Leucodistrofia • Pronósticos La ACC puede ser aislada, ser uno de los elementos claves de un síndrome genético particular o integrarse en un síndrome polimalformativo. La ACC se caracteriza por presentar una gran heterogeneidad: muchos síndromes la asocian. La base de datos de ayuda al diagnóstico LDDB (London Dysmorphology Data Base) que contiene 175 síndromes genéticos que conllevan una ACC. En la mayoría de los casos esta malformación no ha sido reportada más que raramente y la asociación no puede ser retenida como significativa. La recurrencia familiar depende de la etiología de base, si es esporádico o cromosómico (1%), pero hay casos que presentan un patrón autosómico recesivo (25%) o ligado al cromosoma X (50%)(20). Se asocia a veces a patología materno-fetal identificable o no por niveles amnióticos positivos para estas(10,22). • Enfermedades tóxicas: alcohol(23), drogas (valproato, cocaína)(24,25). • Enfermedades inflamatorias: toxoplasmosis, rubéola, virus influenzae(26). • Diabetes materna. • Daño anoxo-isquémico cerebral pre o perinatal (enfermedades metabólicas del feto)(3,27). La distinción entre ACC completa o parcial es 85 TABLA IV. ANOMALÍAS ASOCIADAS MÁS FRECUENTES A ACC Anomalías cerebrales • Dilatación ventricular evolutiva • Hipoplasia parcial del cerebelo • Arnold-Chiari Dandy-Walker • • Microcefalia Anomalía de la giración (lisencefalia, • paquigiria) • Quiste de plexos coroideos • Quiste(s) interhemisférico(s) (aracnoideo) • Lipomas y heterotopías subependimarias Anomalías faciale • Fisura labial uni o bilateral o labiopalatina • Paladar hendido Hipotelorismo o hipertelorismo • • Micrognatia Dismorfias diversas • Anomalías cardiacas • CIV • Canal atrioventricular • Estenosis aórtica (coartación) Anomalías abdominales Hernia diafragmática • Anomalías renales Hidronefrosis • • Ectopía renal Anomalías esqueléticas • Polidactilia Anomalías de cariotipo Trisomía 8, 13, 18 y 21 • • Ploidías (tri, tetra) difícil de realizar sólo por ecografía y la ayuda de la RM es fundamental para aportar argumentos a favor de una o de otra(7). Sin embargo, algunos signos pueden plantear la sospecha de una ACC parcial de la porción posterior: • En corte transversal, la expansión hacia la línea media de los cuernos occipitales modificando la arquitectura normal de los ventrículos laterales. • En corte sagital, la presencia del genu corporis del CC hacia adelante con la presencia de la arteria pericallosa y la disposición radial de las cisuras de la cara interna de los hemisferios cerebrales en la zona donde el CC está ausente. Este diagnóstico aunque difícil, es importante porque la ACC es un factor de riesgo para la presencia de otras malformaciones, ya sean neurológicas o genéticas(7). Pilu et al en 1993 reportan que el US diagnostica 30/35 casos(29) y d’Ercole en 1998 reporta 86 que gracias al US transvaginal la detección es de 4/6 casos(3). Las dificultades pueden ser debidas a la posición fetal, obesidad materna y asociación de anomalías cerebrales(5). Hay que realizar un estudio más acucioso en aquellas pacientes con factores de riesgo: antecedente de síndrome mendeliano con ACC(30), embarazo con alteración de la línea media o antecedente de éste 31 o ante la sospecha de contacto con teratógenos (ya mencionados anteriormente) Es imposible aportar en periodo prenatal elementos pronósticos favorables de certeza. Estudios complementarios con un seguimiento pediátrico estricto son indispensables para poder emitir un adecuado pronóstico fetal(2,7,11); esto no es fácil de realizar, ya que en Europa y en Estados Unidos la ley permite la interrupción del embarazo por razones médicas justificadas (ante cualquier alteración o malformación que conlleve un gran retardo psicomotor y social). El consejo en casos de ACC aislada es difícil de realizar, ya que este puede ser asintomático o revelar déficit durante un examen neurológico como incapacidad manual, discriminación de temperatura y tacto. Pueden tener problemas neurológicos como inestabilidad, déficit intelectual variable y psicosis. Estas alteraciones son más corrientes en casos de asociación de otras anomalías cerebrales; por lo general, los niños con ACC aislada están libres de estos problemas(32). En las series estudiadas de niños seguidos en periodo postnatal, se observa un rango de 32-86% de desarrollo de normalidad; en cambio, en los casos con anomalía asociada se observa siempre algún tipo de alteración neurológica. BIBLIOGRAFÍA 1. Gelot A, Esperandiu O, Pompidou A. Histogénèse du corps calleux. Neurochirurgie, 1998; 44: suppl.1, 61-73. 2. Descamps PH, Lewin G, Body M, Moutard L. et les membres du club francophone de médecine foetale. Neurochirurgie, 1998; 44: suppl.1, 93-95. 3. Parrish ML, Roessmann U, Levinshon MW. Agenesis of the corpus callosum: a study of the frecuency of associated malformations. Ann Neurol 1979; 6: 349-354. 4. Dobyns WB. Agenesis of the corpus callosum and gyral malformations are frequent manifestations of nonketotic hyperglycemia. Neurol 1989; 151: 171-179. 5. D’Ercole C, Girard N, Cravello L, Boubli L, Potier A, Raybaud C, Blanc B. Prenatal Diagnosis of fetal Capítulo 4 - Obstetricia Corpus Callosum Agenesis by Ultrasonography and Magnetic resonance Imaging. Prenat. Diagn. 1998; 18: 247-253. 6. Droulle P, Didier F. L’agénésie calleuse: Problèmes diagnostiques et point de vue de l’echographiste. Medecine foetale et échographie en gynécologie et obstétrique, 1995; 24: 8-10. 7. Maheut-Lourmière J, Paillet Ch. Diagnostique prenatal des anomalies du corps calleux par les ultrasons: le point de vue de l’échographiste. Neurochirurgie, 1998; 44: suppl.1, 85-92. 8. Vergani P, Ghidini A, Strobelt N, Locatelli A, Mariani S, Bertalero C, Cavallone M. Pronostic indicators in the prenatal diagnosis of agenesis of corpus callosum. Am J Obstet Gynecol, 1994; 170: 3, 753-758. 9. Bennett G, Bromley B, Benacerraf B. Agenesis of the corpus callosum: Prenatal detection usually is not possible before 22 weeks of gestation. Radiology 1996; 199: 447-450. 10. Chouchane M, Benouachkou-Debuche V, Giroud M, Durand C, Gouyon JB. Les agénésie du corps calleux: aspects étiologiques, cliniques, moyens de diagnostiques et pronostique. Arch Pediatr 1999; 6: 1306-1311. 11. Talmant C, Yvinec M, Nomballais F, Aubron F, David A, Rival JM, Wetzel M et les membres du Collège Français d’Échographie Foetale. Agénésie du corps calleux: pronostic. Médecine foetale et échographie en gynécologie obstétrique, 1995; 24: 10-18. 12. Lemesle M, Giroud M, Madinier G, Martín D, Baudouin N, Binnert D, Dumas R. Agénésie du corps calleux: Les modes de révélation chez l’adulte. Rev Neurol 1997; 153: 4, 256-61. 13. Grogono JL. Children with agenesis of the corpus callosum. Dev Med Child Neurol 1968; 10: 613-616. 14. Freytag E, Lindenberg R. Neuropathologic findings in patients in a hospital for the mentally deficient: a survey of 359 cases. Jhon Hopkins Med J 1967; 121: 379-392. 15. Jeret JS, Serur D, Wisniewski K et al. Clinicopathological findings associated with agenesis of the corpus callosum. Brain Dev 1987; 9: 25. 16. Jeret JS, Serur D, Wisniewski K, Fisch C (1986). Frecuency of agenesis of the corpus callosum in the developmentally disabled population as determined by computerized tomography. Pediatr Neurosci 12, 101-6. 17. Sonigo P, Rypens F, Carteret M, Brunelle F. L’IRM cérébrale foetale dans le diagnostic antenatal des agénésies du corps calleux. Médecine foetale et échographie en gynécologie obstétrique 1995; 24: 19-22. 18. Brisse H, Sebag G, Fallet C, Elmaleh M, Garel C, Rossler L, Vuillard E, Oury JF, Hassan M. IRM anténatale des agénésies des corps calleux. J Radiol 1998; 79: 659-666. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 19. Lair-Milan F, Gelot A, Baron JM, Lewin F, André Ch, Adamsbaum C. IRM encéphalique anténatale; etude retrospective à propos de 34 examens. J Radiol 1997; 78: 499-505. 20. McKusick VA. Mendelian inheritance in man. 11th Edition. Baltimore: Johns Hopkins University Press, 1994. 21. Deschamps F, Faure JM. Atlas échographique des malformations congénitales du foetus. Ed Sauramps medical 1998: 47-51. 22. Hatem-Gantzer G, Poulain P, Vasseur-Masson D, Ponsot G, Pons JC. Agénésie du corps calleux: un exemple d’incertitude pronostique en médecine foetale. J Gynecol Obstet Biol Reprod 1998; 27: 790-797. 23. Ettlinger G: Agenesis of the corpus callosum (1974). In Vinken P, Bruyn G (Eds): Handbook of Clinical Neurology. Vol. XXX, pp 285-312 (New York: American Elsevier). 24. Lindhout D, Omtzigt JG, Cornel MC (1992): Spectrum of neural tube defects in 34 infants prenatally exposed to antiepileptic drugs. Neurology 42, 111-8. 25. Dominguez R, Villa Coro AA, Slopis JM, Bohan TP (1991): Brain and ocular abnormalities in infants with 87 in utero exposure to cocaine and other street drugs. Am J Dis Child 145, 688-95. 26. Friedman M, Cohen P (1947): Agenesis of the corpus callosum as a possible sequel to maternal rubella during pregnancy. Am J Dis Child 7, 178-85. 27. Janesh G, Lilford R. Assesment and management of fetal agenesis of corpus callosum. Prenatal Diagnosis 1995; Vol.15: 301-312. 28. Bamforth F, Bamforth S, Poskitt K, Applegarth D, Hall J (1988). Abnormalities of the corpus callosum in patients with inherited metabolic diseases. Lancet 2, 451. 29. Pilu G, Sandri F, Perolo A et al. Sonography of fetal agenesis of the corpus callosum: a survey of 35 cases. Ultrasound Obstet Gynecol 1993; 3: 318. 30. Pilu G. Agenesis of the corpus callosum. www.thefetus.net, 1999. List of syndromes featuring agenesis of the corpus callosum (modified from Blum et al). 31. Cohen MM, sedano HO, Gorlin RJ (1971): Frontonasal dysplasia (median cleft face sindrome). Comments on etiology and patogénesis. Birth Defects 7, 117-24. 32. Byrd SE, Radkowski MA, Flannery A (1990): The clinical and radiological evaluation of abscence of the corpus callosum. Eur J Radiol 10: 65-73. 88 Capítulo 4 - Obstetricia HOLOPROSENCEFALIA La holoprosencefalia reagrupa un conjunto de malformaciones complejas del cerebro y de la cara, que van desde la ciclopia con vesícula presencefálica monoventricular, pasando por formas intermediarias como la holoprosencefalia semilobar, hasta anomalías mucho más sutiles de tipo holoprosencefalia lobar. Se trata de alteraciones precoces de la organogénesis que provocan un defecto de clivaje del prosencéfalo(1). Es importante reconocerlas porque las formas más graves, en particular la de tipo alobar conlleva un pronóstico vital y psicomotor muy peyorativo; el diagnóstico es fácil cuando se asocian malformaciones faciales. Las formas diferenciadas, como la lobar, son de diagnóstico mucho más difícil; es sin embargo, muy importante diagnosticarlas, ya que el pronóstico vital no está en juego y las posibilidades terapéuticas quirúrgicas o médicas, como el tratamiento hormonal enteral son muy importantes. La holoprosencefalia asocia malformaciones de -la linea media y del cerebro (Figuras 7-9). Alrededor de la tercera semana de vida, el prosencéfalo se divide en telencéfalo y en diencéfalo. El diencéfalo da origen a los tálamos, al hipotálamo, al quiasma, a las vesículas ópticas, a la posthipófisis y a la comisura gris. Las estructuras faciales nacen de 3 orígenes embriológicos: el tubo neural, la eminencia frontonasal (frente, región interorbitaria, nariz, tercio medio del labio superior, esqueleto medio de la cara: etmoides, vómer, nariz y tabique nasal, hueso premaxilar e incisivos), los arcos branquiales (orejas, pómulos, tercio lateral del labio superior, el resto del maxilar superior y la totalidad del maxilar inferior). En la holoprosencefalia las alteraciones se encuentran en el prosencéfalo y en el esqueleto medio de la cara. Las estructuras del botón frontonasal son hipoplásicos o aplásicos(2). En el cerebro, el clivaje y diverticulización de la vesícula prosencefálica se detienen y el prosencéfalo forma una cavidad ventricular única, en vez de dividirse en 2 ventrículos laterales (Figura 10), por lo que se debería a una alteración en su inducción; por lo que toda anomalía de inducción, de origen físico, tóxico, metabólica (diabetes)(3), genética (trisomía 13 y 18) o inflamatoria (rubeola, toxoplasmosis), son susceptibles de provocar una holoprosencefalia. Figura 7. Corte sagital en embrión de 10+4 SA, donde se puede observar una proboscis. Figura 8. Corte transversal de cerebro en feto a 10+4 SA, donde se observa una cavidad ventricular única: Holoprosencefalia alobar. Figura 9 . Vista frontal del mismo caso anterior. Nótese un hipotelorismo severo (ciclopia). Obsérvese malposición de los dedos. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 10a 10b Clasificación Existen diferentes clasificaciones de esta malformación, pero las más conocidas son las descritas por Demyer y Zeman(13): • Holoprosencefalia alobar: Es la forma más grave. Los hemisferios cerebrales no se separan, el cerebro es de pequeño volumen y contiene una sola cavidad ventricular en lugar de los ventrículos laterales y del tercer ventrículo. Los tálamos y los cuerpos estriados se encuentran fusionados, así como los bulbos y nervios olfativos, el cuerpo calloso y la linea interhemisférica están ausentes. Una imagen quística, “el saco dorsal” de tamaño a veces considerable, se extiende hacia atrás a partir del techo ventricular. Las malformaciones faciales, si bien no son constantes, pero frecuentes, están representadas por una aplasia o hipoplasia de huesos medios de la cara (Figura 11a, b). 11a Figura 10 a, b. Holoprosencefalia Alobar: obsérvese la cavidad monoventricular en forma de herradura. Definición Defecto de la línea media que conlleva a una sola cavidad ventricular, afectando a todas las estructuras de la linea media y el sistema ventricular lateral y tercer ventrículo. Epidemiología Su incidencia es de 0,18 a 1,2 % de los embarazos. En nacidos vivos es de 1x 5000-16000. Es síndrome holoprosencefálico se presenta en 77% de las trisomías 13(4) y en 15% de las trisomías 18(5, 6, 7, 8). El riesgo de recurrencia ante un niño previamente afectado varía entre un 2 a 30%(9, 10), siendo de 25% en los casos autosómicos recesivos y de un 50% en los dominantes(11, 12), siendo de especial interés para un consejo genético posterior. 89 11b Figura11a,b. Hipotelorismo con proboscis en 3D. 90 • Holoprosencefalia semilobar: Es una forma intermediaria con cerebro pequeño y una sola cavidad ventricular, donde la división interhemisférica es incompleta. El cuerpo calloso está ausente, así como generalmente los bulbos olfativos. Los tálamos y los núcleos grises están fusionados. El macizo facial puede estar afectado o absolutamente normal. • Holoprosencefalia lobar: Es la forma menos grave. El cerebro es de tamaño normal y la separación en 2 hemisferios es completa, salvo en su parte anterior. Frecuentemente existe una larga comunicación entre los ventrículos laterales por ausencia del septum pellucidum, pero los cuernos occipitales y temporales son bien individualizados. El cuerpo calloso puede estar ausente, hipoplásico o presente. Los bulbos olfativos están ausentes. Los tálamos y los núcleos grises están rara vez fusionados. Las malformaciones faciales son excepcionales. Probst(14) critica esta clasificación reprochándole que busca establecer una correlación entre las malformaciones faciales y cerebrales. Para él, existe siempre una agenesia de cuerpo calloso, ya que para esta estructura, se requiere una correcta división del telencéfalo y la formación normal de la placa comisural. Por esto propone una nueva clasificación basada en el grado de diferenciación del cerebro: • Tipo I: Prosencefalias muy poco diferenciadas: ausencia de división de ganglios basales y de los tálamos (tipo A) o división parcial de los tálamos e individualización de un pequeño tercer ventrículo (tipo B o C). • Tipo III: Prosencefalias muy diferenciadas o seudohemisféricas, donde existe una cisura interhemisférica incluso a nivel frontal, pero la corteza de éstos está en continuidad. Las estructuras de la base son completamente separadas. • Tipo II: Forma intermediaria entre las anteriores. Persiste un solo lóbulo frontal, y la corteza es de espesor variable. Criterios ecográficos Es una patología del 2º trimestre. • Diagnósticos La presencia de malformaciones faciales es esencial de reconocer, ya que facilita enormemente el diagnóstico antenatal: anomalías del diámetro biorbitario (hipotelorismo, ciclopia)(15), proboscis, fisura lanionarinaria o palatina. Un 17% de holoprosencefalia se manifiesta sin anomalías faciales evidentes(10, 13). Capítulo 4 - Obstetricia •Holoprosencefalia alobar: El elemento esencial es una amplia cavidad ecolúcida que representa al ventrículo único y que se continúa hacia atrás por el saco dorsal bajo la forma de una voluminosa imagen anecógena. El parénquima cerebral se reduce a una delgada lámina en la periferia de la cavidad ventricular, esencialmente a nivel de las regiones anterior y lateral. En un corte coronal, los tálamos se ven fusionados, la cisura interhemisférica está ausente. En el corte sagital paramediano, la cavidad ventricular y el saco dorsal están directamente en contacto con la bóveda ósea(16,17,18). • Holoprosencefalia semilobar: Existe una sola cavidad ventricular en forma de herradura, de tamaño generalmente menos grande que la holoprosencefalia alobar, ya que el parénquima cerebral es habitualmente más importante. La presencia del saco dorsal es menos frecuente y la característica esencial es la presencia de una separación de los cuernos occipitales. Si los tálamos están fusionados, existe a veces una pequeña separación de éstos(18). • Holoprosencefalia lobar: El sistema ventricular está dilatado, pero morfológicamente se aproxima a la normalidad, con una individualización de los ventrículos laterales, los cuernos occipitales y temporales. Sólo los cuernos frontales están fusionados y el septum pellucidum está ausente. • Pronósticos El pronóstico es muy malo, ya que estos niños sufren importante deterioro intelectual, además de numerosas alteraciones neuroendocrinas. Diagnósticos diferenciales y errores ecográficos Hidrocefalia severa, pero la presencia del tercer ventrículo hace la diferencia. Malformaciones asociadas Malformaciones extracraneofaciales asociadas: polidactilia, displasia renal, onfalocele, cardiopatías. Esto se explicaría por la asociación frecuente de una holoprosencefalia y una anomalía cromosómica(19). BIBLIOGRAFÍA 1. Yakoley PI. Pathoarchitectonicstudies of cerebral malformations. III arhinencephalies (holoprosencephalies) J Neuropathol Exp Neurol 1959; 18: 22-55. 2. Demyer W., Zeman W. Alobar holoprosencephaly (arhinencephaly) with median cleft lip and palate: Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) clinical electroencephalographic and nosologic considerations. Confin Neurol 1963; 23: 1-36. 3. Barr M., Hanson JW., Currey K., Sharp S., Toriello H., Schmickel R., Wilson R. Holoprosencephaly in infants of diabetic mothers. J Pedriatr 1983; 102 (2): 565-568. 4. Donnenfield AE, Menutti MT., Sonographic findings in fetuses with commom chromosome abnormalities. Clin Obstet Gynecol 1988; 31: 80-96. 5. Agrata LA., Kovi J., Parshad R. Holoprosencephaly and trisomy 13 in a cyclops. JAMA 1979; 241: 1109. 6. Bundy AL., Saltzman DH., Pober B., Fine C., Ermerson D., Doubilet PM. Anatenatal sonographic findings in a trisomy 18. J Ultrasound Med 1986; 5: 361-364. 7. Lorch V., Fojalo R., Bauer CR. Cebocephalus associated with trisomy 13-15 mosaicism. Arch Neurol 1978; 35: 163-165. 8. Saunders ES., Shortland D., Dunn PM. What is the incidence of holoprosencephaly? J Med Genet 1984; 21: 21-26. 9. Begleiter MI., Harris DJ. Holoprosencephaly and endocrine dysgenesis in brothers. Am J Med Genet 1980; 7: 315-318. 10. D e m y e r W. H o l o p r o s e n c e p h a l y ( c y c l o p i a arhinencephaly). In: Handbook of Neurology. Vol 30. Ed P.V. VINKEN and G.W. BRUYN. Amsterdam 1977; 18: 431-478. 11. Benke PJ., Cohen MM. Recurrence of holoprosencephaly in families with a positive history. Clin Genet 1983; 91 24: 324-328. 12. Pauli RM., Pettersen JC., Arya S. Familial agnathiaholoprosencephaly. Am J Med Genet 1983; 14: 677698. 13. Demyer W., Zeman W., Palmer CG. The face predits the brain: diagnostic significance of nedian facial anomalies for holoprosencephaly (arhinencephaly). Pediatrics 1964; 34: 256. 14. Probst F. The prosencephalies: morphology, neuroradiological appearances and differential diagnosis. Springer Verlag Berlin Heidelberg, New York 1979. 15. Toth Z., Csecsei K., Szeifert G., Torok O., Papp Z. Early prenatal diagnosis of cyclopia associated with holoprosencephaly. J Clin Ultrasound 1986; 14: 550553. 16. Deraskhshan I., Sabouri-Deylami M., Lofti J. Holoprosencephaly. Computarized tomographic and pneumographic findings with anatomic correlation. Arch Neurol 1980; 37: 55-57. 17. Filly RA., Chinn DH., Callen PW. Alobar holoprosencephaly: ultrasonography prenatal diagnosis. Radiology 1984; 151: 455-459. 18. Nyberg DA., Mack LA., Bronstein A., Hirsch J., Pagon RA. Holoprosencephaly: prenatal sonographic diagnosis. A J N R 1987; 8: 871-878. 19. McGahan JP., Nyberg DA., Mack LA. Sonographic of facial features of alobar and semilobar holoprosencephaly. A J R 1990; 154: 143-148. 92 Capítulo 4 - Obstetricia LAS DILATACIONES VENTRICULARES VENTRICULOMEGALIAS Definición El límite de los ventrículos a nivel del atrium o cuerno posterior de los ventrículos laterales está bien establecido; considerándose como normal un valor < 10 mm, borderline entre 10 y 15 mm, y dilatación ventricular >15 mm. Epidemiología La ventriculomegalia presenta un problema relativamente frecuente en US Obstétrico, ya que su prevalencia es de 0,1 a 0,7%(1, 2). Probablemente se detecta más frecuentemente gracias a los avances tecnológicos y la mejor preparación de los ecografistas (Figura 12). Dentro de las etiologías, Valat considera una asociación de malformaciones entre un 70 a un 83 %; siendo éstas 40 % intra-cranianas y 60 % extra-cranianas (principalmente cardiacas, 20 %). Las aneuploidías se presentarían en una incidencia variable, 3 % trisomía 21, trisomía 13 y trisomía 18 entre un 4,2 a 28,6 %(1). 12a 12b Criterios ecográficos Es una patología del 2º y 3er trimestre. • Diagnósticos Se mide a nivel del cuerno posterior del ventrículo lateral, en forma perpendicular al plexo coroideo: < 10 mm 10-15 mm >15 mm Normal Borderline Dilatación Las ventriculomegalias moderadas son estables en 58% de los casos y regresan un 10% antes del nacimiento(2). Cuando son transitorias <12-13 mm; pero pueden ocasionalmente llegar a 18 mm(3,4). Son generalmente bilaterales, pero pueden ser unilaterales o asimétricas; las unilaterales se observan en un 0,007%(5) y afectan predominantemente el atrium izq (1/5); éstas son generalmente estables y regresivas en el 98% casos. 12c 12c Figura 12 a,b,c. Evaluación de una ventriculomegalia en los 3 cortes espaciales: axial, parasagital y coronal. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) • Pronósticos El pronóstico es variable, ya que puede variar en su evolución; estar asociada a otra patología o una malformación grave; de ser así, su pronóstico es por lo general desfavorable. En cambio, si ésta es aislada, moderada o regresiva, presenta un problema en su enfrentamiento; ya que puede corresponder a una variante de la normalidad(3,6,7). Por lo tanto, el descubrimiento de una ventriculomegalia induce la realización de un estudio y seguimiento hacia una etiología, anomalías asociadas o a una evolución desfavorable. Debe siempre realizarse: • • • • • • Ecografía de referencia Exámenes serológicos maternos Amniocentesis para cariotipo Resonancia Magnética Seguimiento evolutivo ecográfico Consulta a genetista Una revisión de la literatura de 234 ventriculomegalias entre 10 y 15 mm, muestra una morbilidad de amplio espectro: aneuploidías 3,8%, malformaciones asociadas 8,6%, deceso perinatal 3,7%, anomalía del desarrollo neurológico 11,5%, presentándose en total un 22,2% de afección(2, 6-8). Si atrium <12 mm sin otras anomalías descubiertas al US, los RN eran normales; y el retardo mental potencial difería poco del de la población general(2, 6). Etiologías(9) Mielomeningocele: 35.5%, agenesia de cuerpo calloso: 11.4%, infecciones: 4.2% 1, anomalías cromosómicas: 7.1%, malformaciones asociadas: 17%, malformaciones cerebrales: 90%(10), malformaciones extracerebrales: 11.3%, presumidas aisladas: 37.5%, pero tras evaluación en el postnatal, sólo el 24% fueron realmente aisladas. Otras causas: isquémicohemorrágica(11). Mayoritariamente, los niños que presentaban una ventriculomegalia moderada y aislada en antenatal, son normales al nacimiento; pero el pronóstico a largo plazo es incierto: presentan entre un 9 a un 21% de retardo mental y de 3 a 4% si atrium <12 mm(2,6,8) siendo de 2,5% en la población general(12). Se asociaría a mayor predisposición a patología neuropsíquicas: autismo y esquizofrenia(12,13). 93 BIBLIOGRAFÍA 1. Valat AS, Dehouck MB, Dufour P, Dubos JP, Djebara AE, Dewismes L,Robert Y, Puech F. Fetal cerebral ventriculomegaly. Etiology and outcome, report of 141 cases. J Gynecol Obstet Biol Reprod 1998; 27: 782-789. 2. Vergani P, Locatelli A, Strobelt N, Cavallone M, Ceruti P, Paterlini G, Ghidini A. Clinical outcome of mild fetal ventriculomegaly. Am J Obstet Gynecol. 1998; 178(2): 218-22. 3. Arora A, Bannister CM, Russell S, Rimmer S. Outcome and clinical course of prenatally diagnosed cerebral ventriculomegaly. Eur J Pediatr Surg. 1998; 8 Suppl 1: 63-4. 4. Weiner Z, Bronshtein M. Transient unilateral ventriculomegaly: sonographic diagnosis during the second trimester of pregnancy. J Clin Ultrasound. 1994; 22(1): 59-61. 5. Kinzler WL, Smulian JC, McLean DA, Guzman ER, Vintzileos AM. Outcome of prenatally diagnosed mild unilateral cerebral ventriculomegaly. J Ultrasound Med. 2001; 20(3): 257-62. 6. Pilu G, Falco P, Gabrielli S, Perolo A, Sandri F, Bovicelli L. The clinical significance of fetal isolated cerebral borderline ventriculomegaly: report of 31 cases and review of the literature. Ultrasound Obstet Gynecol. 1999; 14(5): 320-6. Review. 7. Bromley B, Frigoletto FD Jr, Benacerraf BR. Mild fetal lateral cerebral ventriculomegaly: clinical course and outcome. Am J Obstet Gynecol. 1991; 164(3): 863-7. 8. Patel MD, Filly AL, Hersh DR, Goldstein RB. Isolated mild fetal cerebral ventriculomegaly: clinical course and outcome. Radiology. 1994; 192(3): 759-64. 9. Valat AS, Dehouck MB, Dufour P, Dubos JP, Djebara AE, Dewismes L, Robert Y, Puech F. Fetal cerebral ventriculomegaly. Etiology and outcome, [report of 141 cases] J Gynecol Obstet Biol Reprod (Paris). 1998; 27(8): 782-9. French. 10. Filly RA, Cardoza JD, Goldstein RB, Barkovich AJ. Detection of fetal central nervous system anomalies: a practical level of effort for a routine sonogram. Radiology 1989; 172(2): 403-8. 11. Levine D, Barnes PD, Madsen JR, Abbott J, Mehta T, Edelman RR. Central nervous system abnormalities assessed with prenatal magnetic resonance imaging. Obstet Gynecol. 1999; 94(6): 1011-9. 12. Kinsbourne M. The intralaminar thalamic nuclei: subjectivity pumps or attention-action coordinators? Conscious Cogn. 1995; 4(2): 167-71. 13. Wright DB, Loftus EF. Measuring dissociation: comparison of alternative forms of the dissociative experiences scale. Am J Psychol. 1999; 112 (4): 497519. 94 Capítulo 4 - Obstetricia HIDROCEFALIAS La hidrocefalia es una patología frecuente y compatible con la vida. Se asocia con frecuencia a una espina bífida quística, aunque la hidrocefalia no sea evidente al nacimiento. Hay 3 causas principales: • Exceso de producción de líquido cefalorraquideo (hidrocefalia comunicante). Generalmente se debe a una anomalía genética recesiva ligada al sexo, la que se manifiesta en el 50% de los varones, siendo la mujer portadora de esta anomalía. • Obstáculo en la circulación (hidrocefalia no comunicante). Generalmente se debe a una obstrucción a nivel del acueducto de Sylvio debido a una anomalía, una inflamación o a una tumoración. Se asocia en algunos casos a espina bífida, siendo la hidrocefalia secundaria a una obstrucción del 4º ventrículo por la tracción hacia el agujero occipital (sd de Arnold Chiari tipo II). • Defecto en la reabsorción (hidrocefalia comunicante). Definición Se caracteriza por la acumulación anormal de líquido cefalorraquideo en la cavidad ventricular o, en caso de hidrocefalia externa, en los espacios subaracnoideos (Figura 13, 14). Epidemiología Su incidencia en recién nacidos es de 0.5-3 x 1000 en USA y de 0.21 x 1000 en España de los embarazos. Criterios ecográficos Es una patología del 2º trimestre. Figura 13. En corte axial, se observa una dilatación de los cuernos laterales, así como del tercer ventrículo. Diagnósticos Se producen diferentes alteraciones como consecuencia del aumento de volumen del sistema ventricular dentro de la bóveda craneana. • Aumento del perímetro craneano. • Alargamiento de las estructuras craneanas. • Adelgazamiento progresivo de los huesos del cráneo. • Laminación de la corteza cerebral, en las formas importantes. Pronósticos Se producen diferentes alteraciones de tipo psíquico, que van desde un retardo psicomotor hasta un retardo profundo, manifestaciones convulsivas. Se manifiestan en el 70-80% de los casos. Malformaciones asociadas Se asocia en 70% de los casos a espina bífida con meningocele, asociada a malformación de ArnoldChiari. Se asocia también a agenesia de cuerpo calloso y quistes aracnoideos. Anomalías cromosómicas en 11% de los casos (trisomía 18 y 21). • HEMORRAGIAS INTRAVENTRICULARES Algunos aspectos ecográficos observados en el recién nacido hacen pensar en la existencia de hemorragias en antenatal; es el caso de las imágenes seudoquísticas subependimarias, las hidrocefalias Figura 14. En corte coronal se observa importante dilatación de los cuernos laterales. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) con contenido ecógeno variable intraventricular y engrosamienro de sus paredes, y de las dilataciones con cavitaciones del parénquima (Figuras 15, 16, 17). 95 16a Figura 15. En corte parasagital. Imágenes hiperecogénicas intraventriculares sin dilatación ventricular. 16b Figura 17. En corte parasagital, se observa gran dilatación ventricular con una imagen hiperecogénica que ocupa casi todo el ventrículo. Grado II. Figura 16 a, b. Dilatación ventricular leve a moderada con imagen ecogénica que ocupa el lúmen ventricular. (Grado I-II). El aspecto ecográfico de una hemorragia ha sido reportado tan precozmente como a las 26 SA de gestación(1). El seguimiento ecográfico ha demostrado las mismas imágenes evolutivas del recién nacido: aparición y acentuación progresiva de la dilatación ventricular, limitación, fragmentación y lisis del coágulo intra luminal, evolución cavitaria de una lesión parenquimatosa. El control por medio del Doppler de la hipertensión endocraneana secundaria a una hidrocefalia post hemorrágica, también puede ser utilizado en el feto; esto podría conducir a la decisión de una interrupción prematura de la gestación. Los mecanismos de origen de la hemorragia fetal son desconocidos, pero probablemente sean similares o los mismos que conducen a una hemorragia en el recién nacido: sufrimiento hipóxico isquémico, con agresión de los vasos de la matriz germinal por medio de alteraciones del flujo sanguíneo cerebral, fenómenos de hipertensión venosa, y una afección del endotelio capilar. La descripción de una hemorragia intra-periventricular en un feto y de una leucomalacia periventricular en el otro feto de una gestación gemelar monocorial, apuntan a estos mecanismos patogénicos(2). BIBLIOGRAFÍA 1. Kim MS, elyaderani MK. Sonographic diagnosis of cerebrovascular hemorrhage in utero. Radiology 1982; 142: 479-480. 2. H u r s t R W , A b b i t t P L . f e t a l i n t r a c r a n i a l hemorrhage and periventricular leukomalacia: complications of twin-twin transfusion. AJNR 1989; 10: 62-63. 96 Capítulo 4 - Obstetricia PATOLOGÍAS QUÍSTICAS DEL SISTEMA NERVIOSO CENTRAL QUISTES DE PLEXOS COROIDEOS Definición Los quistes de los plexos coroideos aparecen cuando la función glucogénica de los plexos coroideos, que conlleva la producción de oxígeno, está aumentada. Deben ser considerados como variantes de lo normal. Su tamaño superior a 10 mm puede traducir una alteración en la maduración funcional encefálica. Epidemiología Su incidencia es de 0,18 a 1,2% de los embarazos. Criterios ecográficos Es una patología del 2º trimestre. Diagnósticos Los quistes de los plexos coroideos aparecen como imágenes ecolúcidas, redondeadas y bien delimitadas en e y l seno de los plexos coroideos (Figuras 18 y 19). Pueden ser únicos o múltiples, uni o bilaterales. Su tamaño es variable, a veces voluminosos, pudiendo llegar a 20 mm de diámetro. La evolución espontánea es marcada por una desaparición completa después de las 24-28 SA, edad gestacional en la que el 80 a 90% de los quistes habrán totalmente desaparecido. • Pronósticos Los quistes de plexos coroideos cuando son aislados deben ser considerados como fisiológicos, cualquiera sea su número, tamaño o localización. Una malformación asociada o su persistencia después de las 24 SA debe hacer pensar en una anomalía de cariotipo. Diagnósticos diferenciales y errores ecográficos Los quistes voluminosos pueden ocupar el lumen del cuerno posterior de los ventrículos laterales y no deben ser confundidos con una dilatación ventricular. Excepcionalmente, existen formas de papiloma de plexos coroideos que pueden ser responsables de una dilatación ventricular unilateral. Figura 19. Malformaciones asociadas Todas las malformaciones mayores o menores, pueden estar asociadas a una anomalía cromosómica (trisomía 18 principalmente, y la 21 rara vez), donde deben ser buscados los signos correspondientes, más aún si persisten tras las 24-26 SA. • QUISTES ARACNOIDEOS Definición Los quistes aracnoideos están constituidos por formaciones ecolúcidas supratentoriales. En el 90% de los casos la naturaleza histológica es aracnoidea. El quiste es periférico y se localiza en los espacios sub aracnoideos (Valle Sylviano, Sillón occipitoparietal, etc). Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) En el 10% de los casos, la naturaleza histológica es de tipo neuroepitelial. El quiste se localiza entonces en los espacios subaracnoideos, en la línea media interhemisférica, en los ventrículos o en el parénquima cerebral. Epidemiología Su incidencia antenatal no está establecida. Se trata de malformaciones raras del sistema nervioso central y no se observan más que en el 1% de la patología tumoral intracraneana sintomática del niño y del adulto. Criterios ecográficos Es una patología del 2º trimestre. • Diagnósticos El quiste aparece como una imagen ecolúcida de contornos regulares y bien delimitados (Figura 20). La topografía del quiste debe ser bien precisada: ♦ Periférico: interesan los espacios sub aracnoideos de los sillones y cisuras hemisféricas. ♦ Medial: es ínter hemisférico. ♦ Supra-selar 97 de una agenesia de cuerpo calloso secundaria a un quiste de la línea media son entre otros factores de mal pronóstico. Los quistes supra selares tienen un riesgo de secuela neuro-endocrino. Pueden ocasionar epilepsia e hidrocefalia secundaria. Diagnósticos diferenciales y errores ecográficos Los principales diagnósticos diferenciales comprenden: w Aneurisma de la vena de Galeno w Cavidades poroencefálicas w Quistes sub ependimarios de germinolisis Las dilataciones quísticas del cavum del septum pellucidum son fisiológicas y desaparecen durante el primer año de vida. Malformaciones asociadas Se trata de anomalías frecuentemente aisladas. • Sistema nervioso central: dilatación ventricular e hidrocefalias, agenesias parcial o completa de cuerpo calloso son secundarias al desarrollo del quiste. • PORENCEFALIA Definición Las cavidades poroencefálicas son lesiones quísticas del parénquima cerebral secundarios a fenómenos isquémicos cerebrales localizados. La encéfalo malacia multiquística es observada a un estado ulterior por la confluencia de múltiples cavidades porencefálicas desestructurando el parénquima cerebral y el sistema ventricular. Figura 20. Se observa imagen que asciende desde la base de cráneo, pasando por la megacisterna, empujando el cerebelo hacia delante, continuando por la tienda del cerebelo hacia la linea media lateral izquierda en este caso. Este caso resultó ser una trisomía 18. • Pronósticos El pronóstico de los quistes aracnoideos diagnosticados en antenatal no está bien establecido. Depende de su localización, de su tamaño y de impacto sobre los tejidos cerebrales adyacentes. La presencia de una dilatación ventricular obstructiva o Epidemiología Anomalías raras, que son secundarias a accidentes isquémicos cerebrales, sea debido a una hipoxia transitoria o debido a bajo débito circulatorio materno (traumatismo craneano con compromiso de conciencia). Estas anomalías pueden observarse tras la muerte de un gemelar en un embarazo con anastomosis vasculares placentarias. Criterios ecográficos Es una patología del 2º trimestre. • Diagnósticos Las cavidades porencefálicas son imágenes ecolúcidas bien delimitadas, pero de contornos irregulares. La topografía es evocadora y concierne el 98 Capítulo 4 - Obstetricia parénquima cerebral fronto-parietal o la sustancia blanca a lo largo del atrium ventricular. Su número es variable. Pueden aparecer en exámenes sucesivos y aumentar el volumen hasta volverse confluentes y desestructurar el parénquima cerebral para constituir una encefalomalacia multiquística. Las cavidades porencefálicas pueden comunicar con los ventrículos laterales por destrucción del parenquimatosa y producen entonces una dilatación de los ventrículos laterales asimétrica, con un tercer ventrículo conservado. No se observan efectos de masa sobre las estructuras adyacentes. • Pronósticos La hidranencefalia presenta además del retardo psicomotor, un alto índice de mortalidad dentro del primer año de vida. • Pronósticos Depende principalmente del número, del tamaño y de la localización parenquimatosa de las lesiones. Definición Es una malformación arterio-venosa que produce una dilatación de la vena de Galeno, donde el sistema arterial aferente proviene del sistema carotídeo o del eje vertebrobasilar. Diagnósticos diferenciales y errores ecográficos Las anomalías quísticas ecolúcidas y principalmente los quistes aracnoideos pueden ser confundidos. La irregularidad de sus contornos, la ausencia de efecto de masa, la topografía y el contexto clínico anoxo-isquémico transitorio están a favor de una porencefalia. • HIDRANENCEFALIA Definición La hidranencefalia caracteriza las lesiones cerebrales mayores del cortex que lo reducen a una fina membrana periférica. Epidemiología La destrucción masiva del cortex es secundaria a un proceso isquémico cerebral difuso que compromete los 3 ejes vasculares arteriales cerebrales: anterior, medio y posterior o secundario a lesiones infecciosas (citomegalovirus y toxoplasmosis congénita). Criterios ecográficos Es una patología del 2º trimestre. • Diagnósticos El contenido de la bóveda craneana está reducida a una cavidad líquida única. La línea interhemisférica puede ser parcialmente visualizada o estar ausente. Los pedúnculos cerebrales, tálamos normales y no fusionados; la fosa posterior y el cerebelo son normales. Diagnósticos diferenciales y errores ecográficos Una hidrocefalia mayor y una holoprosencefalia alobar pueden ser confundidas. La destrucción cortical extrema y la ausencia de la línea media del cerebro orientan poco hacia a una hidrocefalia. La ausencia de fusión de los tálamos y un examen morfológico normal de la facie descartan una holoprosencefalia. ANEURISMA DE LA VENA DE GALENO Epidemiología Es una malformación congénita rara, de incidencia antenatal desconocida. Los aneurismas de Galeno representan menos del 1% de las malformaciones arterio– venosas cerebrales. Criterios ecográficos Es una patología del 2º trimestre. • Diagnósticos El diagnóstico se plantea tras el descubrimiento de una formación de tipo “quístico”, redondeada, ecolúcida pura y de localización interhemisférica. La lesión es supratentorial. El diagnóstico se propone tras el empleo del modo Doppler color que confirma la naturaleza vascular de la lesión (Figura 21 a, b). En corte transversal y en modo-B, el aspecto clásico “en raqueta”, correspondiente a la dilatación aneurismal del seno venoso eferente. • Pronósticos Están determinados por: ♦ La existencia de una insuficiencia cardiaca fetal debido al shunt arterial y al débito aneurismal, conllevando un fenómeno de “robo” vascular, que condiciona el pronóstico neonatal inmediato. El examen ecográfico busca una anasarca fetal y una insuficiencia tricuspídea. ♦ Las aferencias vasculares deben ser precisadas por un examen en modo Doppler color estudiando los Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) ejes carotideos, las arterias cerebrales anteriores, medias y posteriores y del eje vertebro-basilar. La existencia de dilatación arterial asociada en modo Doppler pulsado a una baja de las resistencias vasculares define el territorio aferente. El pronóstico pareciera más ominoso cuando las aferencias comprometen ambos ejes vasculares cerebrales. ♦ Los aneurismas voluminosos pueden asociar una dilatación ventricular por compresión del acueducto de Sylvio. Diagnósticos diferenciales y errores ecográficos El quiste aracnoideo interhemisférico constituye el principal diagnóstico diferencial. Malformaciones asociadas Son anomalías aisladas. Figura 21 a, b. 99 • SÍNDROME DE DANDY WALKER y variantes Definición Los síndromes de Dandy-Walker (DW) y sus variantes reagrupan un conjunto de malformaciones “quísticas” de la fosa posterior relacionadas a una anomalía del desarrollo embrionario del romboencéfalo. Epidemiología Las malformaciones de la fosa posterior representan el 30% de las anomalías del sistema nervioso central. Estas anomalías son mayoritariamente esporádicas, incluyéndose en un contexto genético multifactorial no determinado. Criterios ecográficos diagnósticos • Dandy Walker ♦ E n corte transversal: La fosa posterior se encuentra alargada y su diámetro antero-posterior mide más de 10 mm. El vermis cerebeloso está agenésico, ausente en cortes seriados. Los hemisferios cerebelosos están hipoplásicos y anormalmente separados y desviados hacia fuera. Una dilatación ventricular de tipo supratentorial es frecuente de observar. El 4º ventrículo está dilatado, más bien claramente identificable, y se comunica directamente con la megacisterna magna. ♦ E n corte sagital: La agenesia vermiana es confirmada. La tienda del cerebelo está ascendida. El 4º ventrículo está dilatado, siendo bien visible y se comunica con la cisterna magna sin interposición del vermis. • Dandy Walker variante ♦ E l vermis cerebeloso se encuentra ya sea hipoplásico, o con una agenesia parcial de la porción posteroinferior. El 4º ventrículo está menos dilatado, la dilatación ventricular es poco marcada y la tienda del cerebelo se encuentra en su posición habitual. Diagnóstico diferencial y errores ecográficos El diagnóstico de agenesia vermiana puede ser erróneamente afirmado en un corte transversal, en el caso que el vermis se encuentre en rotación anterosuperior. En este caso la interposición del vermis entre el 4º ventrículo y la cisterna magna no es visible, y la separación de los hemisferios cerebelosos parece anormal. El corte sagital debe corregir este diagnóstico erróneo. 100 Capítulo 4 - Obstetricia Sin embargo, algunas malrotaciones vermianas pueden asociarse a anomalías cromosómicas. Malformaciones asociadas Son frecuentes, 30 a 50% de los casos. • SNC ♦ Agenesia Cuerpo Calloso. ♦ Agenesia del Septum Pellucidum. ♦ Anomalías de la giración y de la migración neuronal (agiria, micropoligiria), las que no son detectables hasta después de las 34 SA. ♦ Microcefalia. ♦ Meningocele occipital. • Otras Anomalías (10 a 30%) ♦ Cara, esqueléticas, cardiopatías congénitas o uropatías. • Sd genéticos ♦ Walker-Warburg ♦ Joubert ♦ Debakan • Anomalías cromosómicas • QUISTES RETROCEREBELOSOS Definición Los quistes retrocerebelosos se deben a una invaginación posterior del piso coroídeo y contiene elementos ependimarios secretores. Epidemiología Estas anomalías malformativas son más raras que los Sd Dandy Walker y aparecen generalmente como aisladas. Criterios ecográficos • Diagnósticos • Quistes retrocerebelosos Quistes de pequeño volumen: formación ecolúcida redondeada bien delimitada y desarrollada en la gran cisterna. Generalmente es excéntrico. Quistes de gran volumen: la gran cisterna magna se encuentra alargada y conformada por la sola presencia de esta imagen ecolúcida. Siempre el vermis cerebeloso, los hemisferios y el 4º ventrículo son normales (Figura 22 a, b). • Megacisterna Magna El diámetro anteroposterior de la gran cisterna, medido en corte transversal, es superior a 10 mm. Los elementos de la fosa posterior son morfológicamente normales. Figura 22 a, b. Quiste retrocerebeloso que ocupa toda la fosa posterior, desplazando el cerebelo hacia anterior y arriba. Diagnóstico diferencial y errores ecográficos Los quistes voluminosos pueden comprimir el vermis cerebeloso, aplastar el 4º ventrículo y no ser diferenciable de un Sd de Dandy Walker. Un Quiste retrocerebeloso medial, de moderado tamaño y subcerebeloso puede no ser distinguido de una megacisterna magna. La medición de la grancisterna debe ser realizada en un corte transversal estricto. Incidencias oblicuas aumentan erróneamente esta medida. • MEGACISTERNA MAGNA Es un capítulo difícil de tratar, donde la mayor prudencia es la regla, en la medida que la etiopatogenia y su valor pronóstico en postnatal sean inciertos. La gran cisterna o “cisterna magna” es un espacio subaracnoideo que se encuentra en la cara posteroinferior del cerebelo(1). Esto puede ser considerado Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) en periodo antenatal como un signo de anomalía estructural o de hipoplasia del cerebelo. 101 cerebelo (vermis y hemisferios), aspecto y volumen del 4º ventrículo y de los espacios pericerebrales(9,10). Definición El diagnóstico de megacisterna es evocado cuando este espacio está alargado, sin anomalía morfológica del cerebelo ni del tronco cerebral, sin quiste indivisualisable, con una fosa posterior de tamaño normal. Hay que tratar de descartar una disgenesia vermiana, las atrofias cerebelosas secundarias, los quistes retrocerebelosos sin comunicación con el 4º ventrículo. Por lo tanto se trata de un diagnóstico de descarte, donde deberá ser considerada como una variante de la normal si no existe ninguna anomalía asociada detectada, ya sea a nivel cerebral como extracerebral(2). Epidemiología En antenatal la incidencia es desconocida. En postnatal su incidencia es muy variable y atañe a pacientes sintomáticos, hay reportes de un 0,1 a 4 x 1000(3,4). El problema de esta incidencia es que los autores no utilizan los mismos criterios de definición y las poblaciones estudiadas no son comparables. Etiopatogénicamente se definen 3 grupos(5): • Origen mecánico(inflamación, tumor): inflamaciones antenatales de tipo infeccioso (CMV) pueden inducir una hipotrofia cerebral y cerebelosa secundaria. • Origen vascular(agudo o crónico que lleva a atrofia): isquemia o hemorragia. • Disgenesia cerebelosa: agenesia parcial o hipoplasia. Una anomalía genética como cromosomopatía (trisomía 18)(6,7). Criterios ecográficos diagnósticos Es un hallazgo más bien del 3er trimestre, ya que en el 2º trimestre se puede observar frecuentemente por la amplitud de los espacios pericerebrales normalmente aumentados. En antenatal la megacisterna se observa en un plano axial oblicuo hacia abajo y atrás, así como en un plano coronal posterior y en uno sagital (Figura 23 a, b). Diagnóstico positivo: este es semicuantitativo, ya que su tamaño varía según la oblicuidad del corte realizado, considerándose anormal un diámetro anteroposterior > a 10 mm en un corte axial(8). Es imperativo descartar otras anomalías, por lo que es necesario realizar cortes coronales y sagitales medios, observándose la posición del vermis cerebeloso, el aspecto morfológico y biométrico del Figura 23 a, b. Megacisterna con integridad de las estructuras de la línea media cerebrales y del vermis cerebeloso. Diagnóstico diferencial y errores ecográficos Habitualmente es posible descartar el diagnóstico de Dandy-Walker, así como el de una agenesia vermiana total o parcial. Es más difícil de realizar el descarte de un quiste aracnoideo, principalmente en su forma medial retrocerebelosa, más aún si su volumen es moderado, ya que es poco probable observar las paredes del quiste. Hipoplasias cerebelosas primitivas o secundarias a un fenómeno infeccioso (CMV) o vascular (isquemia o hemorragia). 102 Antes de las 18 SA es imposible afirmar la integridad de las estructuras cerebelosas, y en particular del vermis, ya que existe una ausencia de cierre del vermis(11,12). En un corte coronal levemente oblicuo, que tome el 4º ventrículo y la megacisterna, justo por delante del vermis, se puede observar una falsa imagen de continuidad entre estos(13). Malformaciones asociadas El estudio del sistema supratentorial es fundamental. Hay que buscar una agenesia de cuerpo calloso, una dilatación ventricular, una anomalía de la giración. Hay que buscar también lesiones extracerebrales, orientadas a variadas causas genéticas, principalmente una trisomía 18, más aún si existe una hipoplasia cerebelosa(14). La realización de un cariotipo no es necesario si parece ser una anomalía aislada, ya que este signo pareciera ser un mal marcador de aneuploidía, aunque se trate de una paciente de riesgo (edad materna elevada)(15). BIBLIOGRAFÍA 1. Liliequist B, Tovi D, Schisano G. Developemental defects of the tentorium and cisterna magna. A report of 2 cases diagnosed byt encephalography and confirmed at operation. Acta Psychiatr. Scand. 1980, 35: 223. 2. Couture A, Droulle P, Didier F. Echographie cérébrale du foetus au nouveau-né. Imagerie et hémodynamique - Chapitre Les malformations cérébrales, pp 267- 370. Ed. Sauramps Médical. 3. Bodensteiner JB, Gay TC, Marks WA, Hamza M, Schaeffer G.G. Macrocisterna magna : a marker for maldevelopement of the brain? Peadiatr. Neurol 1998; 4: 284-286. 4. Adam R, Greenberg JO. The mega cisterna magna J. Neurosurg, Vol 48 : 190-192, 1978. 5. Gonsette R, Potvliege R, Andre-Balisaux F., Stenuit Capítulo 4 - Obstetricia J. La méga grand citerne: étude clinique, radiologique et anatomopathologique. Acta Neurol Belg. 1968; 68: 559-570. 6. Nyberg D, Mahony B, Hegge F, Hickok D, Luthy D, Kapur†R. Enlarged cisterna magna and the DandyWalker malformation : factors associated with chormosome abnormalities. Obstet Gynecol 1991; 77: 436. 7. Thurmond AS. Enlarged cisterna magna in Trisomy 18: Prenatal ultrasound diagnosis. AM J. Obste Gynecol 1989; 161: 83-85. 8. Mahony B, Callen P, Filly R, Hoddick W. The fetal cisterna magna. Radiology 1984; 153: 773-776. 9. Strand RD, Barnes PD, Young Poussaint T, Estroff JA, Burrows PE. Cystic retrocerebellar malformations: unification of the Dandy-Walker complex and the Blake’s pouch cyst. Pediatr Radio 1993; 23: 258260. 10. Duchatel F, Mennesson B, Berseneff H, Oury JF. Mesures échographiques anténatales du cervelet foetal. Intérêt de l’évaluation du développement ftal. J. Gynecol. Obstet. Biol. Reprod. 1989; 18: 879-883. 11. Babcook CJ, Chong BW, Salamat MS, Ellis WG, Goldstein RB. Sonographie Anatomy of the Developing Cerebellum: normal embryology can resemble pathology. AJR 1996;166. 12. Bromley B, Nadel AS, Pauker S, Estroff JA, Benacerraf BR. Closure of the Cerebellar Vermis: Evaluation with Second Trimester US. Obstetrical Ultrasound 1994; 193: 761-763. 13. Lang FC, Frates MCN, Briown DL, Benson CB, Di Salvo DN, Doubilet PM. Sonography of the Fetal Posterior Fossa: False Appearance of Mega-Cisterna Magna and Dandy-Walker Variant. Obstetrical Ultrasound 1994; 192: 247-251. 14. Ghidini A, Fromberg RA, Tiernan J, Wieneke JA, Manz HJ, Sherer DM. Dilated Subarachnoid Cisterna Ambiens : A Potential Sonographic Sign Predicting Cerebellar Hypoplasia. J Ultrasound Med 1996; 15: 413-415. 15. Watson WJ, Katz VL, Chescheir NC, Miller RC, Menard MK, Hansen WF. The Cisterna Magna in Second-Trimester Fetuses With Abnormal Karyotypes. Obstetrics and Gynecology 1992; 79 (59, Part 1). Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 103 DEFECTOS DE CIERRE DEL TUBO NEURAL ANENCEFALIA Definición Defecto completo de cierre del sistema nervioso central en su extremo cefálico. Resulta una ausencia completa de la bóveda craneana y del parénquima cerebral. La presencia de una masa cerebral protruyente, heterogénea, hipertrófica y por sobre el macizo facial caracterizando una exencefalia. Epidemiología Su incidencia es de 1-2 x 1000 nacidos. Habría una incidencia de recurrencia de 3% ante un antecedente directo, asociándose un factor genético multifactorial. Criterios ecográficos Es una patología del 1er trimestre, pudiéndose realizar el diagnóstico a las 10-12 SA. • Diagnósticos ♦ Corte sagital: No se observa bóveda craneana ni tejido cerebral normal. La presencia de una masa parenquimatosa por sobre el macizo facial, de volumen variable, heterogéneo, “que hace de turbante” caracterizando una exencefalia (Figura 24). ♦ Corte transversal: Los reparos anatómicos para la medición del diámetro biparietal no pueden ser hallados. ♦ C orte coronal: Aspecto clásico de “facie de batracio”. Ninguna estructura cerebral es reconocida. Un hidramnios y una mobilidad excesiva son frecuentes. • Pronósticos Es una malformación letal. Diagnósticos diferenciales y errores ecográficos En edad gestacional precoz, un examen rápido puede hacer pasar inadvertida una anencefalia cuando existe la presencia de un tejido angiomatoso proliferante. Malformaciones asociadas • Sistema nervioso central ♦ Raquisquisis cervical y mielomeningocele. • Anexos fetales ♦ Enfermedad de bridas amnióticas. • EXENCEFALIA Definición Se caracteriza por la ausencia completa de la calota craneana y la presencia de tejido cerebral displásico y desorganizado(1). Esto se produce por la falta de protección del sistema nervioso central por la calota, lo que permite que el encéfalo quede expuesto a la agresión permanente del líquido amniótico y a traumatismos repetidos(2,3). Cuando la gestación es prolongada, como en el ser humano, se produce una destrucción, lo que hace las estructuras cerebrales sean destruidas parcial o totalmente para tomar un aspecto morfológico de feto anencéfalo. Una destrucción cerebral completa puede ocurrir en 8 a 10 semanas(2) para muchos la exencefalia es un precursor embriológico de la anencefalia(4, 5, 6). Figura 24. Obsérvese la falta de calota con exteriorización de la masa cerebral. Epidemiología La exencefalia es una patología malformativa extremadamente rara del embrión humano(2, 7). 104 Criterios ecográficos Es una patología del 2º trimestre. • Diagnósticos Si bien la descripción ultrasonográfica en la literatura de esta patología es rara, el diagnóstico malformativo parece fácil y puede ser realizado en forma muy precoz desde las 16 SA(8). El diagnóstico se basa en la ausencia de estructura ósea cefálica y la presencia por sobre las órbitas de una masa heterogénea donde las estructuras ventriculares y parenquimatosas no son reconocibles (Figura 48). La anatomía vascular post mortem habitualmente es normal, con un polígono de Willis íntegro y una distribución arterial periférica relativamente normal. BIBLIOGRAFÍA 1. Bell JE, Green RJL. Studies on the area cerebrovasculosa of anencephalic fetuses. J Pathol 1982; 137: 315- Capítulo 4 - Obstetricia 318. 2. Elwood JM, Elwodd JH. Epidemiology of anencephaly and spina bifida. New-york: Oxford University Press 1980: 15-26. 3. Warkany J. anencephaly. In: congenital malformations: notes and comments. Chicago. Year Book, 1971; 189200. 4. Ganchrow D, Ornoy A. Possible evidence for secondary degeneration of central nervous system in the pathogenesis of anencephaly and brain dysraphia: a study in young man fetuses. Virchows Arch (Pathol Anat) 1979; 384: 285-294. 5. Hendricks SA, Cyr DR, Nyberg DA, Raabe P, Mack LA. Exencephaly: clinical and ultrasonic correlation to anencephaly. Obstet Gynecol 1988; 72: 898-901. 6. Lemire RJ, Beckwith JB, Warkany J. Experimental exencephaly and anencephaly. In: Anencephaly. Newyork. Raven Press 1978; 85: 113. 7. Debakan AS. Anencephaly in early human embryos. J Neuropathol Exp Neurol 1963, 22: 533-548. 8. Cox GG., Rosenthal ST., Holsapple JW. Exencephaly: sonographic finding and radiologic-pathologic correlation. Radiology 1985; 155: 755-756. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 105 ENCEFALOCELE Definición Defecto de cierre localizado de la bóveda craneana secundaria a una ausencia localizada de revestimiento mesodérmico. Resulta una hernia meníngea o meningo-encefalocele. Epidemiología Es el defecto del sistema nervioso central menos frecuente. El encefalocele es aislado o se integra en una asociación malformativa compleja. Criterios ecográficos Es una patología del 1er trimestre. • Diagnósticos Aparece como un tumor redondeado protruyente del polo cefálico. Es de carácter ecolúcido cuando se trata de una hernia meníngea o un meningocele; si es heterogénea de ecogenicidad mixta que contiene elementos del parénquima cerebral, se trata de una hernia meningo-encefálica (Figura 25). La topografía medial es la lesión más típica. La localización más frecuente es medio retro-occipital (75%), medio frontal (13%) que deben ser buscados en corte sagital de la facie, y las inter-parietales (12%). Una dilatación ventricular obstructiva con macrocrania es encontrada frecuentemente en casos de meningoencefalocele. Una microcefalia asociada es de mal pronóstico. Figura 25. Obsérvese gran encefalocele occipital. • Pronósticos El volumen, la localización, la existencia de malformaciones asociadas y esencialmente el contenido de la hernia meníngea condicionan el pronóstico. Generalmente presentan retardo mental por daño secundario. Diagnósticos diferenciales y errores ecográficos Los encefaloceles pequeños pueden pasar inadvertidos a las 12 SA. El defecto óseo puede ser difícil de identificar. La enfermedad de bridas amnióticas es responsable de un encefalocele lateral. Un higroma quístico puede ser confundido con un encefalocele tipo meningocele, el defecto óseo aclara el diagnóstico. Malformaciones asociadas • Sistema Nervioso central: w Agenesia de cuerpo calloso. w Sd de Dandy Walker. w Hidrocefalia. • Sd de Meckel: asocia un encefalocele, una displasia renal y anomalías de extremidades de tipo polidactilia. DEFECTOS DEL CIERRE DEL RAQUIS • ESPINA BÍFIDA Y MIELOMENIN-GOCELE Definición La espina bífida es un defecto de cierre del raquis. El meningocele es una hernia meníngea pura, el mielomeningocele se caracteriza por la presencia de raíces nerviosas en el interior de la hernia. Las anomalías del polo cefálico secundarios a la presencia de un defecto de cierre del raquis definen la malformación de Arnold Chiari tipo II, que consiste en la tracción del cerebelo hacia el orificio occipital. Esta se manifiesta en más del 90% de los casos. Estos son niños muy afectados que presentan numerosos déficit: afección motriz de los miembros inferiores, alteraciones esfinterianas responsables de lesiones renales que pueden llegar a ser graves y lesiones cerebrales dominadas por una hidrocefalia. Epidemiología Su incidencia es de 1 x 1.000 nacidos. 106 Se atribuye un origen genético multifactorial. La diabetes, el ácido valproico y las carencias de folato son factores predisponentes. Capítulo 4 - Obstetricia ♦ En corte coronal: la separación de los arcos vertebrales posteriores es diagnosticado por la pérdida del paralelismo óseo (Figura 27). Criterios ecográficos Es una patología del 2º trimestre, aunque excepcionalmente puede ser sospechada en el 1er trimestre. Diagnósticos • Signos directos ♦ En corte sagital: la interrupción del muro posterior es visible (Figura 26 a). La hernia meníngea o el mielomeningocele son fácilmente identificables y toman el aspecto de un tumor ecolúcido con base en el raquis interrumpido. Las raíces nerviosas pueden ser visualizadas y flotar en el meningocele. ♦ En corte transversal: los arcos vertebrales posteriores son anormalmente separados y divergentes, realizando un aspecto de “v” invertida. El muro posterior del raquis está interrumpido. El revestimiento cutáneo es discontinuo (Figura 26 b). El fondo de saco medular es anormalmente bajo o no visible a nivel de la 4ª vértebra lumbar. Figura 26 a, b. Obsérvese en corte sagital y axial el defecto óseo de la columna. Figura 27. Se observa imagen ecolúcida en región lumbosacra con pequeño defecto óseo a nivel de columna: espina bífida con meningocele. • Signos indirectos Malformación de Arnold Chiari tipo II ♦ Dilatación ventricular más o menos severa. ♦ Anomalías morfológicas de la bóveda craneana que toma el aspecto de “limón” por la depresión de los cuernos frontales(1, 2) (Figura 28). ♦ Una posición anormal de los pedúnculos cerebrales que son traccionados hacia abajo y atrás. ♦ Imposibilidad de ver la cisterna magna. ♦ Una hipoplasia y una posición anormal del cerebelo que se aloja en la cara interna de la base deloccipital, describiendo un aspecto de “banana”(3). En un corte sagital el vermis es anormalmente traccionado abajo(4) (Figura 29). Figura 28. Signo “del limón”: anomalía morfológica de la bóveda craneana por la depresión de los cuernos frontales Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 107 bífida o compromete la porción sacra. Un oligoamnios, la posición del raquis contra las paredes uterinas pueden disminuir la sensibilidad diagnóstica. Malformaciones asociadas Anomalía asociada en 60% de los casos a anomalías renales, sacras, cardiacas, intestinales, cerebrales y de extremidades. Se asocia a síndromes: Vacter, Vater, Síndrome de Klippel Feil. BIBLIOGRAFÍA Figura 29. Signo de “la banana”: ausencia de la cisterna magna por tracción del cerebelo hacia el canal medular (malformación de Arnold – Chiari). Estos signos tienen importancia de ser mencionados por estar presentes en el 90 a 100% de los casos de espina bífida, y debieran ser visualizables desde las 15 SA (5, 6, 7). • Pronósticos En general el 25% de los casos presenta parálisis completa de las extremidades inferiores, otro 25% presenta parálisis parcial, otro 25% requerirá rehabilitación intensiva y el otro 25% no presenta disfunción importante. Diagnósticos diferenciales y errores ecográficos Los signos indirectos pueden ser difícil de ser identificados cuando se trata de una pequeña espina 1. Filly RA. The lemon sign: a clinical perspective. Radiology 1988; 167: 573-575. 2. Campbell J, Gilbert WM, Nicolaides KH, Campbell S. ultrasound screening for spina bifida: cranial and cerebellar signs in a high risk population. Obatet Gynecol 1987; 70: 247-250. 3. Nicolaides K, Campbell CH, Gabbe SG, Guidetti R. Ultrasound screening for spina bifida: cranial and cerebellar signs. Lancet 1986; 12: 72-74. 4. Drouille P, Didier F. actualités dans le dépistage échographique du spina bifida. In: Couture A, Veyrac C, Baud C. Les malformations congénitales: diagnostic anténatal et devenir. Sauramps Med Ed VIGOT 1988; 386-396. 5. Bamberger-Bozo C. Malformation de Chiari II. J Neuroradiol 1982; 9: 47-70. 6. Peach B. Arnold-chiari malformation: anatomic features in 20 cases. Arch Neurol 1965; 12: 613-621. 7. Veyrac C, Couture A, Ferran JL. Aspects échographiques des myéloméningocèles. Ann Radiol 1983; 26: 113122. 108 Capítulo 4 - Obstetricia SÍNDROME REGRESIÓN CAUDAL Es una patología muy grave que afecta a fetos predominantemente de de madres diabéticas. Las mujeres diabéticas presentan un riesgo de 250 veces mayor al de madres sanas. Definición Defecto de cierre del tubo neural localizado en la región sacra, asociándose con anomalía de extremidades, ya sea de formación o de malposición. Epidemiología El 22% de los fetos con agenesia sacra son de madres diabéticas. El 1% de las madres diabéticas va a tener un niño con agenesia sacra. Criterios ecográficos Es una patología del 1er trimestre. • Diagnósticos Hipoplasia de extremidades inferiores Agenesia total o parcial del sacro Defecto lumbosacro cerrado Anomalías varias: genitourinarias, cardiacas, gastrointestinales y sistema nervioso central Malformaciones asociadas ♦ Agenesia renal ♦ Ano imperforado ♦ Fisura labio-palatina ♦ Microcefalia ♦ Meningomielocele • RESONANCIA MAGNÉTICA CEREBRAL FETAL La resonancia magnética (RM) se presenta como el único método complementario cuando la ecografía parece insuficiente. Se trata de una técnica simple, no invasiva y no irradiante, que permite realizar un estudio en todos los planos del espacio; de donde radica todo el interés en el diagnóstico fetal, y por supuesto la posibilidad de proporcionar un pronóstico sobre la patología fetal , está bien establecido(1,2). Actualmente los progresos tecnológicos permiten secuencias rápidas que evitan la curarización fetal, reemplazándola por una simple premedicación maternal oral (comprimido de flunitrazepam de 1mg administrado 1 hora antes del examen)(2,3). El examen es realizado tras un hallazgo de la ecografía del II trimestre, y es preferible no realizarla muy Figura 30. Resonancia magnética que muestra una agenesia de cuerpo calloso en los 3 cortes espaciales (gentileza del Dr. Marc Mohlo, CHI Poissy-Paris, Francia). Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 109 ECOGRAFÍA RESONANCIA MAGNÉTICA Ventajas Ventajas • Bajo costo • Mayor contraste de tejidos, por lo tanto presenta • Alta disponibilidad mejor visualización de estructuras internas • Estudio dinámico (cerebelo) • Permite evaluación de estructuras • Dependencia relativa del operador en movimiento (corazón) • Puede visualizar toda la zona de interés, las partes fetales no afectan las imágenes, y por ende no se producen artefactos por estas: Alta definición anatómica • Independiente de la posición fetal • No presenta efectos adversos demostrados Desventajas Desventajas • Profundidad limitada por la frecuencia • Menor resolución espacial • Operador dependiente • No es en tiempo real: los movimientos fetales • Reproductibilidad no son visualizados • Oligohidroamnios • Artefactos por movimientos fetales • Calota osificada • Limitada en caso de polihidroamnios • Posición fetal • Alto costo • Difícil disponibilidad de equipo • Claustrofobia materna tempranamente en el embarazo (antes de 24-26 SA); si se desea estudiar la giración, esta debe realizarse después de las 30-32 SA. L os me ca n ismos pa r t icipa nt es est a r ía n r el a c io n a d o s c o n c u a d r o s m a l fo r m a t ivo s (hidrocefalia obstructiva, lisencefalia, agenesia del cuerpo calloso, etc) y destructivos (leucomalacia periventricular). Como se puede observar en la tabla subyacente, la anomalía asociada que se detecta con mayor frecuencia en la RM es la agenesia de cuerpo calloso (Figura 30), ya sea parcial o completa (la RM fetal precisa el diagnóstico por signos indirectos, cortes axiales y coronales); así como también la leucomalacia periventricular. Esto nos puede hacer pensar que la dilatación ventricular podría asociarse con cierta frecuencia a fenómenos de tipo isquémico en el segundo trimestre o con menor frecuencia a fenómenos de tipo hemorrágico. En otros países, como en Francia, donde la interrupción médica del embarazo (feticidio) es legal, en situaciones donde las lesiones pueden comprometer inciertamente el futuro estado neurológico del niño y probablemente en forma importante, es sin lugar a dudas un examen de apoyo muy importante y a veces crucial al momento de dar un pronóstico y tomar una decisión al respecto. La RM representa un complemento diagnóstico esencial de la ecografía con ventajas y desventajas, adquiriendo un rol crucial en el pronóstico fetal y de la conducta obstétrica a tomar. Es por esto que requiere un amplio conocimiento de la anatomía cerebral fetal normal a diferentes edades gestacionales, y debe ser interpretada con mucho cuidado, en ocasiones apoyada en un atlas de RM fetal. BIBLIOGRAFÍA 1. Girard N, Raybaud C, Boubli L. Exploration par IRM du cerveau foetal in utero. Rev Im Med 1992; 4: 391-399. 2. Revel MP, Pons JC, Lelaidier C. MRI of the fetus: a study of 20 cases performed without curarization. Prenat Diagn; 1993; 13: 775-99. 3. Weinreb JC, Lowe TW, Santos-Ramos R. Magnetic resonance imaging in obstetric diagnosis. Radiology 1985; 154: 157-161. 110 Capítulo 4 - Obstetricia EXAMEN ECOGRÁFICO MORFOLÓGICO ANORMAL DE LA FACIE FETAL FISURA LABIO – PALATINA EMBRIOLOGÍA La cara se forma a partir de 5 botones embrionarios faciales alrededor de una depresión ectodérmica, el estomodeum o boca primitiva. Los 5 botones embrionarios aparecen alrededor de la 4ª semana, por proliferación mesenquimatosa del primer arco branquial. El botón frontal, de situación media e impar, constituye el límite superior del estomodeum, es una proliferación hacia delante del cerebro, por medio del neuroporo anterior. Los botones maxilares, pares, constituyen los límites laterales del estomodeum. Los botones mandibulares, pares y simétricos, constituyen los bordes inferior del estomodeum. Las placodas olfativas son engrosamientos bilaterales del ectodermo, que se desarrollan a cada lado de la parte inferior del botón frontal y justo por sobre del estomodeum. En los bordes de las placodas, hacia la 5ª semana, se forman los botones nasales internos y externos. El botón externo forma el ala de la nariz, y el botón interno forma la parte media del labio superior, la parte media del maxilar superior y todo el paladar primario. Hacia la 6ª y 7ª semana las fosas nasales están bien formadas. Los botones maxilares se aproximan entre sí y hacia los botones nasales internos, dando origen a la parte media del filtrum y de la mandíbula superior, de las encías y el paladar primario. Los botones maxilares forman las partes laterales del labio superior, de la mandíbula superior y el paladar secundario. La cara del feto se termina de formar hacia la 9-10ª semana. El paladar se forma desde la 5ª a la 12ª semana, constituido por 2 partes: el paladar primario y el paladar secundario. Ben Pansky. Review of Medical embryology. Macmilliam Publishing Company 1986; 138-145. • FISURA LABIO-PALATINA La fisura labial y la fisura palatina son malformaciones comunes de la cara y del paladar. Corrientemente asociadas, son malformaciones embriológicamente y etiológicamente distintas. Aparecen en tiempos distintos del desarrollo y se deben a mecanismos diferentes. Definición La fisura labial es una malformación del labio superior con o sin fisura palatina, se debe a un defecto de fusión embrionaria de los botones nasal interno y maxilar, por delante del canal palatino anterior. Su incidencia es de 1 x 700-900 nacidos. Más frecuentes en varones(1,2). Pueden ser uni o bilaterales, completas o incompletas según comprometan todo el labio superior hasta la fosa nasal ipsilateral o limitada a una parte del rojo del labio. Las fisuras palatinas se deben a la ausencia de fusión de los procesos palatinos por detrás del canal palatino anterior. Su incidencia es de 1 x 2500 nacidos. Es más frecuente en mujeres. Puede comprometer sólo el reborde alveolar o bien extenderse a los paladares blando y óseo. Pueden ser uni o bilaterales. La fisura medial es considerada a parte y debe ser integrada en las malformaciones mediales de las anomalías de división prosencefálicas. Es una característica del síndrome de Mohr. Epidemiología Las fisuras labio-palatinas son aisladas en el 50 a 60% de los casos(3): Se integran en el 30 a 40% de los casos en síndromes polimalformativos genéticos variados (Sd Fryns, Pierre Robin, Apert, Meckel-Gruber, Roberts, etc). El 5 a 10% se asocia a anomalías cromosómicas (trisomías 13 y 18). Las fisuras aisladas presentan un riesgo de recurrencia de un 4% tras un primer antecedente parental directo, evocando un factor genético multifactorial(1). Hay antecedentes medicamentosos que predisponen a la fisura labiopalatina, como son los antiepilépticos, la vitamina A y sus derivados, la intoxicación etílica(4). Criterios ecográficos Es una patología del 2º trimestre, después de las 17-18 SA. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) Diagnósticos El perfil fetal puede parecer normal ante una fisura labial unilateral, en el caso de una fisura bilateral, se observará la protrusión del botón labiomaxilar medio. Ante un defecto facial, el examen ecográfico debe ser sistemático y lo más detallado posible para permitir un diagnóstico anatómico de lo más preciso para entregar al cirujano los elementos que permitan programar el manejo posterior y familiarizar a la pareja con este tipo de defecto. En cortes coronales tangenciales y recurrentes, la interrupción del labio superior aparece como una ruptura de la continuidad labial. El espacio inter labial puede estar ocupado por un botón alveolar premaxilar. El diagnóstico de fisura palatina se realiza cuando el maxilar superior está interrumpido; el arco alveolar es irregular y presenta un defecto variable. La nariz y la asimetría de las narinas son anormales. Las fisuras labiales completas comprometen uno de los orificios narinarios. El tabique nasal se encuentra desviado (Figura 31a). En corte sagital, las fisuras labiales simples de pequeño tamaño pueden pasar inadvertidas. Cuando el defecto es más importante, el perfil es anormal. Los huesos nasales no se encuentran alineados, el botón premaxilar protruye hacia delante en vez del labio superior. El maxilar superior y el paladar óseo, no son visibles en este corte, pero la lengua puede verse ascendida en el defecto palatino durante los movimientos de deglución fetal(5, 6). En corte transversal, la posición anterior de la lengua y su relación con el maxilar superior deben ser precisados en reposo y tras movimientos de deglución. El defecto labial es claramente visto en este corte. En este corte también debe ser evaluado el maxilar superior, así como en lo posible el inferior; e incluso la cavidad faringea (Figura 31 b). Un estudio dinámico es necesario, en modo Doppler color el diagnóstico de fisuras palatinas puede ser precisado por el pasaje continuo y anormal de flujo color desde las fosas nasales hacia la cavidad bucal. • Pronósticos Cuando la fisura es aislada, el pronóstico quirúrgico postnatal depende de: ♦ Uni o bilateral ♦ Existencia de fisura palatina ♦ Existencia de alteraciones de la deglución ♦ Hipoplasia mandibular asociada 111 Figura 54 a, b. En un corte axial a nivel del paladar, se puede observar uana interrupción en la continuidad palatina. En un corte coronal se observa una asimetría bucal y nasal. Diagnósticos diferenciales y errores ecográficos En el 3er trimestre en examen de la cara fetal puede ser difícil, y pasar inadvertida una fisura labiopalatina. Una posición fetal viciosa: flexión, oligoamnios, condiciones parietales maternas adversas, dificultan el examen. Una fisura palatina aislada posterior, es de diagnóstico antenatal casi imposible. Malformaciones asociadas La presencia de anomalías de la cara, especialmente de fisura labio-palatinas deben obligatoriamente hacer 112 Capítulo 4 - Obstetricia un examen morfológico completo, especialmente del sistema esquelético y del sistema nervioso central. Se asocian de 30 a 50% de los casos: • Las asociaciones malformativas más frecuentes son: cardiacas (3-7%), atresia esofágica (5%), anomalías cervico vertebrales (13%)(7). • Anomalías cromosómicas: en las fisuras aisladas las anomalías cromosómicas son de 6% y de 25% si la fisura es asociada a otra anomalía; trisomías 13 (si la fisura es bilateral), 18 (si la fisura es unilateral), y diversas deleciones(8). • Sd genéticos malformativos: Robert, MeckelGruber, Neu-laxova, Oro-facio-digital, Fryns, etc). • Asociación CHARGE. • Holoprosencefalias Aporte de la ecografía 3D La técnica de superficie permite delimitar de mejor forma la altura de la fisura con respecto a las narinas (Figura 32). El estudio del block volumétrico es de un aporte incontestable en las fisuras palatinas, permitiendo objetivarlas y apreciarlas en toda su magnitud y profundidad. BIBLIOGRAFÍA 1. Couly G. Labio-maxillary and velopalatine clefts. Clinical and therapeutics aspects. Rev Prat 1991; 41 (1) 48-50. 2. Melnick M, Bixler D, Boggs WS, Fogh-Andersen P, Conneally PM. Cleft lip +/- cleft palate: an overview of the literature and an analysis of Danish cases born between 1941 and 1968. Am J Genet 1986; 6 (1): 8397. 3. Couly G, Aicardi J. Associated morphological Figura 32. Eco 3D que muestra defecto labial con desviación del ala nasal derecha. 4. 5. 6. 7. 8. anomalies of the face and brain in infants. Arch Fr Pediatr 1988; 45 (2): 99-104. Couly G. Malformations and anomalies associated with labiomaxopalatal clefts. Chir Pediatr 1983; 24 (4-5): 231-233. Aubry MG, Aubry JP. Diagnostic anténatal des malformations et tumeurs cervico-faciales. Rev Prat (Paris)1991; 4: 16-20. Vasquez MP, BrodatyG. Prise en charge pré et post natal des fentes labiopalatines. J Pedriatr 1992; 5: 258-266. Gorlin R, Cohen M, Levin L. Syndromes of the head and neck. 3ème édition 1990 Oxford University Press. Aymé S. Epidémiologie des malformations orofaciales, L. Med. Génét 1990; 19: 115-25. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 113 PATOLOGÍA OCULAR Y DE ORBITA Las anomalías oculares no son fáciles de determinar. Lo primero es siempre evaluar las órbitas con su tamaño, lo que nos permite diagnosticar o descartar microftalmias (existe ocasionalmente una microftalmia que afecta sólo al globo ocular, sin afectar la órbita; este caso no es posible de diagnosticar en antenatal). Dentro de éstas se evalúan la presencia como la opacidad de los cristalinos así como sus movimientos oculares (Figura 33a). Se han descrito marcadores oculares de aneuploidía, como lo es la persistencia de la arteria hialoidea después de las 24-26 SA (Figura 33b). 34a 34b Figura 34 a, b. Corte parasagital y axial de la órbita que muestra un dacriocistocele. Figura 33 a. Se evalúa la órbita, el cristalino, los movimientos oculares y el sistema nasolacrimal. También se pueden evaluar la indemnidad de estructuras paraoculares, como es el sistema nasolacrimal (Figura 34). PERFIL FETAL El perfil fetal nos puede aportar una enorme información en cuanto a normalidad o anormalidad se refiere. Estas anomalías pueden estar aisladas o formar parte de anomalías cromosómicas o síndromes genéticos. Estas anomalías pueden ser desde sólo una dismorfia facial; es decir, sin una malformación establecida hasta una claramente identificable. Pueden ser tan sutiles como una hipoplasia o agenesia del hueso nasal (Figura 35 a, b). • MICRORETROGNATIA Y SÍNDROME DE PIERRE ROBIN Figura 33 b. La persistencia de la arteria hialoidea (0,8 mm de espesor) se ha descrito como marcador de aneuploidia. La arteria hialoidea aparece en el quiasma óptico y llega hasta la cara posterior del cristalino. Definición Las microretrognatias están definidas por la existencia de una hipoplasia mandibular, responsable 114 Capítulo 4 - Obstetricia Figura 36. Retrognatia en una trisomía 18. • Diagnósticos El retrognatismo es definido por un corte sagital estricto del perfil fetal. La mandíbula normal sobrepasa hacia delante la parte inferior del hueso frontal pasando por el puente nasal. La microrretrognatia se caracteriza por una hipoplasia mandibular donde la retrognatia es la primera traducción. El tamaño y la morfología mandibular son anormales en estos casos. La agnatia es diagnosticada ante la ausencia del arco mandibular óseo en un corte transversal recurrente de la cara. Las anomalías de la deglución deben siempre ser buscadas. La existencia de un hidramnios, la no visualización del estómago y la ausencia de movimientos de deglución y de flujos endobucales en modo Doppler color orientan hacia la asociación de un defecto mayor de la deglución. Figura 35 a, b. Agenesia hueso nasal en un feto de 13 y 23 SA; el primero fue un niño normal y el segundo un sd de Down. de la desaparición de la prominencia mentoniana (Figura 36). Pueden a lo más presentar una agnatia por agenesia mandibular. Epidemiología Malformaciones raras se integran frecuentemente en síndome genéticos polimalformativos complejos de severidad variable. Criterios ecográficos Es una patología del 1er y 2º trimestre, a partir de las 12-14 SA. • Pronósticos Está determinado por la afección causal. Diagnósticos diferenciales y errores ecográficos El diagnóstico puede ser difícil en aquellas formas anatómicas difíciles, donde los signos de anomalía de la deglución deben ser considerados. Existen morfotipos “normales” de retrognatia. El diagnóstico de microrretrognatia es a veces más complejo hacia fines del 2º y en el 3er trimestre. Malformaciones asociadas Las microrretrognatias están presentes en numerosos síndromes genéticos (Pierre Robin, Goldenhar, Nager, etc). Debe hacerse un estudio morfológico completo de la cara y del sistema nervioso central, además de buscar anomalías extra faciocraneales. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) TIROIDE FETAL (Goitre tiroideo) Definición No se trata de una malformación, sino de una disfunción. Su constitución antenatal puede ser secundaria a un hipotiroidismo congénito, una intoxicación o carencia de yodo, o a la ingesta materna de antitiroideos de síntesis. Epidemiología Su incidencia no está establecida. El hipotoridoismo congénito afecta 1 x 5000 nacidos. Criterios ecográficos Es una patología del 2º trimestre. • Diagnósticos El tiroides fetal es visible en un corte transversal del cuello. Aparece bajo la forma de una imagen ecogénica homogénea por delante del raquis. El diagnóstico de Goitre fetal es subjetivo, 115 ya que no existen biometrías de referencia para el tiroides normal. Está definido por una imagen ecogénica, en semicírculo, bilobada y que deforma el cuello. Una deflexión de la cabeza fetal permanente puede observarse cuando el Goitre es voluminoso. • Pronósticos No hay criterios ecográficos que permitan evaluar la calidad de funcional del tiroides. Un Goitre voluminoso puede llegar a comprimir la tráquea o laringe, manifestándose por la aparición de una hiperecogenicidad pulmonar obstructiva o un hidramnios. Diagnósticos diferenciales y errores ecográficos Otros tumores cervicales: • Linfangiomas • Teratomas Malformaciones asociadas Generalmente es una anomalía aislada. 116 Capítulo 4 - Obstetricia EXAMEN ECOGRÁFICO MORFOLÓGICO ANORMAL DE LA CAVIDAD TORÁCICA FETAL MALFORMACIONES PULMONARES EMBRIOLOGÍA Los pulmones presentan 4 estados evolutivos: • Periodo seudoglandular: (5ª a 17ª semana) Las divisiones bronquiales se diferencian y el sistema de conducción aéreo se instala. Hacia la 17ª semana todos los elementos pulmonares están formados, a excepción de aquellos responsables del intercambio gaseoso. • Periodo canalicular: (13ª a 25ª semana) Existe una superposición de periodos, ya que los segmentos superiores se desarrollan más rápido que los inferiores. Hacia la 24ª semana cada bronquiolo terminal da origen al menos a 2 bronquiolos respiratorios, y la respiración es posible a partir de este momento por la presencia de algunos alvéolos primitivos en sus extremidades. • Periodo de saco terminal: (24ª semana al nacimiento) Numerosos sacos terminales se desarrollan; los pulmones pierden su aspecto canalicular y el epitelio de los sacos se hace más delgado (epitelio alveolar o neumocito tipo I). Entre las 23 y 24 SA los neumocitos tipo I se diferencian en células secretoras, denominándose neumocitos tipo II. Hacia las 25 y 28 SA hay suficientes sacos terminales para permitir la sobrevida en caso de nacimiento prematuro. • Periodo alveolar: (fin del periodo fetal hasta los 8 años) El epitelio del saco terminal se hace más escamoso y fino, haciendo que la membrana alveolo-capilar permita el intercambio gaseoso y produzca una cantidad suficiente de surfactante. Entre los 3 y 8 años el número y tamaño de los alvéolos inmaduros aumenta, así como la posibilidad de de formación de nuevos alvéolos primitivos, aumentando de tamaño y maduración al mismo tiempo. Las patologías intratorácicas son muy variadas, pero son relativamente poco frecuentes en relación a la afección de otros órganos como SNC, corazón, genitourinario, tubo digestivo, etc. El diagnóstico ecográfico antenatal de estas patologías intratorácicas es relativamente fácil, aunque más del 50% se manifiestan después del nacimiento (enfisema lobar gigante, secuestro intralobar, quiste broncogénico, traqueomalacia, etc). Los signos ecográficos son de 2 tipos: sean signos indirectos tales como hidramnios, anasarca; sea, lo más frecuente, descubrimiento fortuito de una imagen anormal (anomalía posicional de los reparos anatómicos del corazón, de los diafragmas, del estómago, del hígado o de los ejes vasculares; asociados a anomalías de ecoestructura como colecciones líquidas. Siempre deben buscarse anomalías asociadas de otros órganos. El polihidramnios debe hacer sospechar una compresión esofágica; la anasarca, una compresión cardiaca o una alteración en el drenaje venoso. Es en estos casos en que se puede recurrir a procedimientos terapéuticos de excepción como “el drenaje in útero” o a la extracción prematura en vías de una cirugía neonatal precoz. En general, las anomalías intratorácicas aisladas, no compresivas y sin hipoplasia pulmonar son de excelente pronóstico. Ben Pansky. Review of Medical embryology. Macmilliam Publishing Company 1986; 152-156. • SECUESTRO PULMONAR Definición El secuestro pulmonar se define por la existencia de un tejido pulmonar que no está en comunicación con el tracto aéreo normal. El “pulmón secuestrado” es independiente del parénquima pulmonar normal y es vascularizado por un pedículo arteriovenoso de origen sistémico. La vascularización arterial del secustro pulmonar se hace por medio de una arteria que nace de la aorta, más raramente de una arteria intercostal. El drenaje venoso se realiza por medio del sistema cava – ácigos. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) Existen 2 tipos de secuestro pulmonar: • Secuestro intralobar: (75 a 85%) están incluidas en un lóbulo normal y no poseen pleura propia. Son intratorácicos. • Secuestro extralobar: (15 a 25%) corresponde a un tejido pulmonar independiente del pulmón normal, recubierto por una pleura propia. Son por lo general intratorácicos (85%), pero pueden ser intraabdominales, subdiafragmáticos. Epidemiología Constituyen 5 a 15% de las malformaciones pulmonares congénitas. No se conocen factores etiológicos ni genéticos relacionados. Las anomalías cromosómicas son excepcionales. Criterios ecográficos Es una patología del 2º trimestre. • Diagnósticos El secuestro pulmonar se presenta como una hiperecogenicidad pulmonar. Se trata de una masa homogénea hiperecogénica, bien delimitada, de contornos bien definidos, dentro del parénquima pulmonar. Se ubica generalmente en postero – inferior izquierda en alrededor del 70% de los casos. El tamaño está limitada a un segmento o lóbulo pulmonar y mide raramente más de 50 mm de diámetro. El examen ecográfico en modo Doppler color constituye una ayuda importante al diagnóstico etiológico del secuestro pulmonar. La puesta en evidencia de un pedículo vascular de origen sistémico por medio del modo Doppler color permitiría afirmar este diagnóstico. A veces es difícil evidenciar este pedículo. Su pequeño tamaño, su localización posterior, la vascularización del parénquima pulmonar, así como la proximidad del corazón y los grandes vasos, pueden dificultar el uso del Doppler para el diagnóstico. Pueden haber formas atípicas: bilateral, lobares superiores y quísticas compuestas de formaciones anecógenas, redondeadas, de tamaño variable e insertas en el seno de la hiperecogenicidad. • Pronósticos Son de mal pronóstico las lesiones bilaterales, la existencia de una anasarca fetoplacentaria, y la asociación de malformaciones. La regresión espontánea antenatal, ya sea 117 parcial o más raramente total, es posible. Diagnósticos diferenciales y errores ecográficos Una hiperecogenicidad pulmonar debe hacer pensar en: • Malformación adenomatoidea tipo II • Tapón mucoso intra bronquial • Una estenosis traqueal • Un teratoma mediastínico • Un neuroblastoma mediastinal • Una hernia diafragmática con ascenso hepático intratorácico Malformaciones asociadas Son raras en los secuestros intralobares, 5 a 15%; y son más frecuentes en las extralobares, 50 a 65%. • Torácicas ♦ Hernia diafragmática ♦ Atresia esofágica con o sin fístula con el pulmón secuestrado ♦ Fístula traqueo – esofágica • Pulmonares ♦ Malformación adenomatoídea pulmonar ♦ Quistes broncogénicos • Cardiacas ♦ Canal atrioventricular • Digestivas ♦ Estenosis duodenal ♦ Onfalocele • Urológicas ♦ Valvas de uretra posterior • Esqueléticas ♦ Malformaciones del raquis ♦ Anomalías costales • MALFORMACIONES ADENOMA-TOIDEAS DE PULMÓN Definición Son tumores hamartomatosos de pulmón caracterizados por una hiperplasia bronquiolar seudoglandular y una dilatación quística de los bronquiolos terminales. Clasificación de Stocker • Tipo I (Figura 37 a, b) ♦ Quistes de tamaño mayor a 15-20 mm, únicos o múltiples. ♦ Parénquima interquístico normal. • Tipo II ♦ Quistes de tamaño menor a 15 mm. ♦ Parénquima remanente. 118 Capítulo 4 - Obstetricia Epidemiología Su manifestación es rara. Constituyen un 25 a 50% de la patología pulmonar congénita. Se asocian excepcionalmente a anomalías cromosómicas. No existe factor etiológico conocido. Criterios ecográficos Es una patología del 2º trimestre. • Diagnósticos El aspecto ecográfico varía según la clasificación de Stocker: Figura 37 a, b. Malformación adenomatoidea tipo I: grandes masas quísticas pulmonares. • Tipo III (Figura 38) ♦ Micropoliquístico. Quistes de tamaño menor a 0,5mm, no individualizables. ♦ No existe parénquima sano. • Tipo I ♦ I m a g e n a n e c o g é n i c a i n t r a t o r á c i c a , supradiafragmática, de paredes finas. ♦ Ú nicos o múltiples, de 20 a 60 mm de diámetro. • Tipo II ♦ Imagen anecogénica intratorácica, supradiafragmática. ♦ Son múltiples, de 5 a 15 mm de diámetro. ♦ Parénquima adyacente más ecógeno que el pulmón sano. • Tipo III ♦ Masa hiperecogénica densa. ♦ Microquistes de tamaño menor a 0,5 mm de diámetro, a veces identificables. • Pronósticos Está determinado por la presencia de: ♦ Malformaciones asociadas ♦ Desviación mediastínica y compresión cardiaca ♦ Anasarca fetal ♦ Hidramnios ♦ Lesión bilateral El volumen del tumor, cuando afecta sólo un hemitórax, no pareciera influir en el pronóstico postnatal. La regresión ecográfica espontánea antenatal, parcial o más raramente total. Concierne esencialmente las formas aisladas del tipo II y III de la clasificación de Stocker. Figura 38. Malformación adenomatoidea tipo III: pequeñas masas quísticas pulmonares que le dan el aspecto denso e hiperecogénico. Diagnósticos diferenciales y errores ecográficos • Las formas quísticas hacen pensar en: ♦ Quiste broncogénico ♦ Hernia diafragmática ♦ Secuestro pulmonar quístico Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) ♦ Tumor neuroentérico ♦ Teratoma mediastínico • Hiperecogenicidad pulmonar ♦ Secuestro pulmonar ♦ Tapón mucoso intrabronquial ♦ Estenosis traqueal A favor de una Malformación Adenomatoidea Pulmonar tipo III: el examen en modo Doppler color no encuentra vascularización de origen sistémico de tipo tumoral. Malformaciones asociadas • Torácicas ♦ Secuestro pulmonar ♦ Hernia diafragmática ♦ Atresia bronquial • Cardiacas ♦ CIV ♦ Tetralogía de Fallot • Digestivas ♦ Atresia esófago ♦ Atresia y estenosis de colon ♦ Atresia ano - rectal • Urológicas ♦ Agenesia renal bilateral • QUISTES BRONCOGÉNICOS 119 bien delimitadas, de forma redondeada u ovalada y situada en el centro del parénquima pulmonar. Generalmente son únicos (Figura 39). Su tamaño es rara vez mayor a 40 mm. Se sitúa habitualmente en uno de los pulmones, sobretodo a nivel de los lóbulos inferiores. Los quistes broncogénicos mediatínicos comprometen el mediastino medio en la regiónparaesofágica o la bifurcación traqueobronquial. Hay formas poco comunes: formas multiloculares o quísticas voluminosas que provocan una desviación mediastínica y anasarca fetal. Quistes múltiples. Figura 39. Imagen ecolúcida retrocardiaca izquierda por el interior de la aorta en un corte de 4 cavidades, al Doppler color no reveló flujo interno. Definición Son estructuras disembrionarias revestidas de un epitelio ciliado de tipo bronquial, que puede presentar otro elemento histológico de tipo bronquioalveolar (cartílago, glándula mucosa o músculo bronquial). Estos quistes pueden comunicar con el árbol bronquial. Su topografía varía según la edad embriológica de formación. Si se forman precozmente son mediastínicos y paraesofágicos. Son pulmonares e intraparenquimatosos si se forman más tardíamente. • Pronósticos Son de mal pronóstico ♦ Malformaciones asociadas ♦ Desviación mediastínica y anasarca fetal ♦ Quiste voluminoso con hipoplasia pulmonar Epidemiología Son raros, de incidencia antenatal desconocida. Excepcionalmente se asocian a anomalías cromosómicas (trisomía 21). Diagnósticos diferenciales y errores ecográficos ♦ Malformación adenomatoidea pulmonar tipo I ♦ Duplicación esofágica ♦ Quistes neuroentéricos Criterios ecográficos Es una patología del 2º trimestre. • Diagnósticos Imagen unilocular anecógena, de paredes finas Los quistes broncogénicos son habitualmente aislados y son asintomáticos después del nacimiento. La regresión antenatal es posible. • Mediastino anterior ♦ Quiste broncogénico ♦ Linfangioma ♦ Hemangioma 120 ♦ Quiste pericárdico ♦ Hernia diafragmática • Mediastino medio ♦ Quiste broncogénico ♦ Quiste pericárdico ♦ Hernia diafragmática • Mediastino posterior ♦ Quiste neuro – entérico ♦ Duplicación esofágica Malformaciones asociadas • Pulmonares ♦ Fístula traqueo – esofágica ♦ Secuestración pulmonar • Cardiaca ♦ Cardiopatías congénitas • Digestivas ♦ Duplicación esofágica ♦ Divertículo esofágico • Esqueléticas ♦ Anomalías vertebrales y quistes broncogénicos mediastínicos • QUILOTÓRAX Y DERRAMES PLEURALES El quilotórax es una colección líquida de origen linfático debido a una anomalía en la producción o reabsroción de la linfa, secundaria a una patología del sistema linfático pulmonar. Los derrames pleurales son en la mayoría de las veces quilotórax, con una incidencia de 1 x 10.000 - 12.000 nacidos. El diagnóstico se realiza en el segundo trimestre y no es posible realizar la distinción de su naturaleza quilosa o serosa por ecografía, ya que ambos tienen las mismas características ecogénicas. La etiología es por lo general imprecisa, las anomalías anatómicas del drenaje linfático son difícilmente diagnosticables en antenatal. El diagnóstico diferencial de estos linfotórax debe realizarse con la trisomía 21 (se asocia en forma excepcional), las monosomías X, el síndrome de Noonan, la hernia diafragmática, las secuestraciones pulmonares, las infecciones virales (parvovirus B19 y coxsackie virus) y alteraciones del ritmo cardiaco. En caso de derrames voluminosos compresivos, se puede discutir un drenaje in útero para evitar una hipoplasia pulmonar. Por sobre las 32 – 33 SA, es mejor plantear una extracción prematura del niño tras inducción de maduración pulmonar. Siempre es conveniente descartar patologías asociadas como secuestro pulmonar, linfangectasias pulmonares y fístula traqueo-esofágica. Capítulo 4 - Obstetricia Definición El quilotórax es un derrame pleural de origen linfático debido a una anomalía de la producción o reabsorción de la linfa, secundaria a una patología del sistema linfático pulmonar. Epidemiología Incidencia de 1 x 10.000 nacidos. Criterios ecográficos Es una patología del 2º trimestre. • Diagnósticos ♦ Un derrame torácico es visible, bajo la forma de una imagen ecolúcida intratorácica. Es uni o bilateral. Su volumen es variable, pudiendo ser desde una simple imagen laminar a ser tan importante que comprime todo el parénquima pulmonar (Figura 40 a, b). ♦ N o existen criterios ecográficos para afirmar el origen quiloso de un derrame pleural. La ecogenicidad del derrame es idéntica a la de los de origen seroso. Figura 40 a, b Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) • Pronósticos ♦ Depende de la aparición de una compresión de las cavidades cardiacas, que puede ir desde una simple desviación mediatínica a una anasarca fetal por insuficiencia cardiaca. ♦ El quilotóprax aislado es por lo general de buen pronóstico. Diagnósticos diferenciales y errores ecográficos No existen criterios ecográficos para afirmar el origen quiloso de un derrame pleural. La ecogenicidad es idéntica en los derrames serosos y quilosos. Debe descartarse siempre infecciones cogestacio-nales, principalmente parvovirus B19 y virus coxsackie. 121 Malformaciones asociadas • Pulmonares ♦ Secuestro pulmonar ♦ Linfangectasias pulmonares ♦ Fístula traqueo – esofágicas Esta anomalía ha sido excepcionalmente encontrada en trisomías 21. La existencia de un derrame pleural debe hacer buscar una ascitis y un derrame pericárdico, pudiendo ser incluidos en el cuadro de las anasarcas fetoplacentarias. Derrame pleural bilateral que comprime ambos pulmones, asociado a ascitis secundaria. 122 Capítulo 4 - Obstetricia HERNIA DIAFRAGMÁTICA La hernia diafragmática es una patología que tiene un desarrollo muy precoz, constituyéndose en una malformación por falla del cierre en el conducto pleuroperitoneal entre la 9ª y 10ª semana de gestación, correspondiendo a una embriopatía; por lo tanto la búsqueda de malformaciones asociadas es de extrema importancia(1-3). Esta anomalía es 10 veces más frecuente a izquierda que a derecha. La consecuencia de este defecto diafragmático es el ascenso o desarrollo de las vísceras abdominales en la cavidad torácica; esto dará como resultado una compresión del pulmón del lado afectado, la desviación del mediastino y el corazón hacia contralateral y finalmente la compresión del pulmón contralateral. Es por esto que la hipoplasia pulmonar se presenta en ambos lados(4- 6). Fisiopatológicamente, la hipoplasia pulmonar conlleva una reducción del número alveolar, una hipoplasia global del pulmón, y lesiones vasculares que llevan a una disminución del contingente vascular arterial y venoso, provocando un engrosamiento de las paredes de los vasos. De hecho, las lesiones se encuentran en ambos lados. La consecuencia de estas lesiones no es sólo la reducción en el número alveolar (que podría ser reversible), pero por sobre todo un aumento en las resistencias pulmonares con una persistencia del efecto de shunt. El canal arterial va a atraer todo el flujo y la perfusión pulmonar será insuficiente para asegurar una hematosis correcta. Todas las técnicas de reanimación después del nacimiento tienden a tratar de sobrellevar este problema, ya sea por medio de vasodilatadores pulmonares, sea por la utilización de una circulación (u oxigenación) extracorpórea (venosa: AREC o arterio-venosa: ECMO)(7-13). Definición La hernia diafragmática se debe a la presencia de un hiato posterolateral, o foramen de Bochdalek, del diafragma. Se debe a un defecto del cierre de las membranas pleuroperitoneales con el mesenterio esofágico y el septum transverso. El cierre precoz de este hiato a derecha explicaría la frecuencia más elevada de hernias a este lado (80%). Los pulmones fetales serían anormales, hipoplásicos, sin que esto sea simplemente explicado por la simple compresión visceral debido a la hernia. Diagnósticos Signos indirectos deben hacer evocar el diagnóstico de hernia diafragmática. · Hidramnios: es frecuente (50 a 70%). Debido a la desviación del mediastino y la desaparición de la isometría aurículo-ventricular en el corte de 4 cavidades. · Desviación de reparos vasculares abdominales: la vena umbilical es desviada en corte transversal. El receso umbilical, la vena porta y las venas suprahepáticas están en posición atípica o mal visualizadas. La aorta torácica descendente presenta una curvatura anormal en el corte sagital. · Hernia diafragmática izquierda: El estómago está en posición intratorácica. En un corte transversal se encuentra laterocardiaco y su ascensión puede ser visualizado en corte sagital, sobre todo cuando el diagnóstico es precoz. El corazón se encuentra desviado a derecha. Otras imágenes de talla inferior al estómago, y aspecto heterogéneas son visibles y corresponden al intestino delgado ascendido. El colon transverso e izquierdo pueden ser identificados, es hipoecogénico y más largo que el delgado. El riñón derecho puede estar incluido en la hernia. · Hernia diafragmática derecha: La visualización directa del hígado ascendido es más difícil. La modificación de reparos vasculares (vena umbilical y ductus venoso) que son desviados y ascendidos toman en este caso todo su valor. El corazón es desviado a izquierda. El pulmón derecho es mal visualizado y rechazado hacia el ápex del hemitórax. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) Pronósticos Los criterios pronósticos no son formales. Es sobre todo la afección cardiaca izquierda y las compresiones que provocan hipoplasia ventricular izquierda, las que junto con la hipoplasia pulmonar van a condicionar el pronóstico postnatal. La siguiente etapa es definir los criterios ultrasonográficos de gravedad. Estos signos pueden no existir en un primer examen, a veces un poco precoz; pero pueden establecerse en las 2 a 3 semanas siguientes (Tabla I). Los norteamericanos consideran como elementos peyorativos la presencia de estómago intratorácico asociado a polihidramnios (20) mientras que los europeos consideran que la presencia de hígado intratorácico asociado a un diámetro abdominal transverso inferior al percentil 10, se asocia a una sobreviva menor al 20%. Estos últimos también consideran de mal pronóstico la presencia de una hipoplasia funcional del ventrículo izquierdo asociado a la no visualización del pulmón contralateral, con una sobreviva menor al 20%(21). La relación VI/VD es de 0.90 para los sobrevivientes y de 0.75 para los fallecidos, pero sería significativo sólo entre las 30 y 40 SA(22). La real severidad del pronóstico se debe a la cuantía de hipoplasia pulmonar secundaria a los órganos herniados que comprimen los órganos intratorácicos. Distintos parámetros se han utilizado para determinar la severidad de esta hipoplasia pulmonar (Tabla II). 123 Diagnósticos diferenciales y errores ultrasonográficos Están constituidos por los tumores anecógenos del tórax fetal como malformación adenomatoidea tipo I y II, teratoma y otros tumores mediastínicos. Las hipoplasias ventriculares y el situs inversus pueden ser interpretados como desviaciones mediastinales. MALFORMACIONES ASOCIADAS Anomalías asociadas: 25 - 57%, 95% casos con muerte fetal(29). • Anomalías morfológicas · SNC: Anencefalia, encefalocele, espina bífida y mielo-meningocele, hidrocefalia, agenesia cuerpo calloso · Cara: Fisura labiopalatina · Cardiacas: (100% mortalidad) · CIA, CIV, Transposición de los grandes vasos, tetralogía de Fallot, hipoplasia de corazón izquierdo(30) · Pulmonar: Secuestro extralobar · Urológicas: · Uropatías obstructivas. · Anomalías de forma y número renal. · Hidronefrosis, agenesia renal. · Digestivas · Las malrotaciones y defectos de fijación del meso del tubo digestivo no deben ser considerados, ya que se integran en la génesis de la patología. · Atresia intestinal. TABLA I. CRITERIOS DE GRAVEDAD ULTRASONOGRÁFICOS DE HERNIA DIAFRAGMÁTICA. Semiología ultrasonográfica (Figura 41 a, b) · · · · · · · Diagnóstico antes de las 25 SA(14) Diámetro abdominal transverso menor al percentil 10(15) Presencia del hígado intratorácico(16) Evaluación del pulmón contralateral(17) Hipoplasia funcional del ventrículo izquierdo: reducción del volumen VI: VI / VD < 0.90 para los sobrevivientes y de 0.75 para los decesos; válido entre 30 – 40 sa. Estómago intratorácico(18) Polihidroamnios(19) 124 Capítulo 4 - Obstetricia Figura 41a, b. Ausencia de cámara gástrica (a).Cámara gástrica (E) intratorácica, con desviación mediatínica hacia la derecha; además se observa hígado (H) e intestino (I) intratorácico. Se observa un remanente de parénquima pulmonar en la parte posterior del hemotórax izquierdo (b). (Letras) TABLA II. CRITERIOS ULTRASONOGRÁFICOS PARA DETERMINACIÓN DE HIPOPLASIA PULMONAR. Biometría pulmonar · El pulmón del lado de la Hernia Diafragmática (HD) generalmente no es visible y la severidad de la hipoplasia pulmonar contralateral juega un rol decisivo en la sobreviva. · Harrison(23) muestra que el LHR (lung head ratio) se relaciona con el pronóstico. · Merz(24) calcula la relación diámetro pulmonar / circunferencia torácica (< 0.09 se relaciona con hipoplasia y deceso neonatal). Luxación intratorácica del hígado · El estómago en posición posterior o medial en el tórax, es un buen signo de luxación hepática, pero no es muy frecuente de ver. · Morin(25) describe la desviación del conducto venoso de Arancio. Mientras mayor es la luxación hepática, menor es el ángulo entre el conducto venoso y la vena umbilical (≤ 90º). Flujo perinasal y traqueal fetal (Figura 42) · Harding mostró que la abolición de movimientos respiratorios conlleva a una disminución del desarrollo pulmonar, del volumen líquido pulmonar y de la IGF II. · Fox (26) mostró que el flujo nasal fetal presenta un espectro de 2 componentes: un ritmo rápido y de baja amplitud, y otro dependiente de las contracciones del diafragma, de ritmo más lento y de mayor amplitud. En el caso de HD, el feto estaría más tiempo en ritmo rápido, que independiente del diafragma, relacionándose con una disminución del volumen de líquido pulmonar y en consecuencia, una insuficiencia pulmonar post natal. · Kalache(27) midió el volumen de flujo traqueal en movimientos respiratorios, y encontró que eran muy bajos en las HD de mal pronóstico. Vascularización pulmonar con doppler energy · Roth(28) observó que la vascularización pulmonar estaría reducida en los casos de hipoplasia pulmonar severa. · LHR: sólo para HDC izquierda(41,42) · < 1,0: mortalidad 100%. · 1,0 - 1,4: sobrevida 38%. · > 1,4: sobrevida 100%. Kitano propone un algoritmo simplificado para el manejo de éstos fetos(43) Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) Figura 42. “Respiración fetal”, medición del flujo nasofaringeo por medio del Doppler pulsado. (Letras) • Anomalías cromosómicas: se asocian en un 1030%(31). · Trisomía 21, 18, 13 y 9 en mosaico. · Beckwith-Wiedemann, Pierre-Robin, Fryns. ·Tetrasomía12p(PallisterKillian) · Microdeleciones, etc. · Teratógenos · Talidomida, quinina, antiepilépticos(32) · Tipo familiar(33) · 2% casos, de herencia sp. · Riesgo de recurrencia: 2%. · Esqueléticas ·Anomalías del raquis y sus extremidades. • Anomalías sindromáticas: su incidencia es de hasta un 40%. · Fryns Sd: (AR). HD en 80% de los casos, dismorfia facial, hipoplasia distal de dedos, fisura labio-palatina, Dandy-Walker o agenesia de cuerpo calloso en 50% de casos, malformaciones cardiacas y urinarias. · Pallister Killian Sd: (tetrasomía 12p). Hidramnios, crecimiento normal o macrosomía,dismorfia facial, fémur corto, hipoplasia distal de dedos, malformaciones cardiacas y urinarias (es conveniente utilizar la FISH para la certeza diagnóstica). · Cornelia de Lange Sd: (sp, AR, AD). RCIU, con dismorfia facial característica, anomalía de extremidades (micromelia, focomelia y oligodactilia), malformaciones cardiacas, digestivas y de órganos genitales externos. Otros: Marfan, Ehlers Danlos, Coffin Siris, Disostosis espóndilo-costal, Beckwith Wiedemann, Di George, etc. 125 La severidad del pronóstico, en particular en las formas graves, nos lleva a buscar medios terapéuticos antenatales, es por esto que Harrison y el equipo de la “Fetal treatment program” han intentado alternativas que conciernen la cirugía fetal abierta, que no detallaremos. El principio de esta cirugía se basa en aliviar esta compresión antes de las 28 SA (inicio de la fase de crecimiento alveolar), para permitir un mejor crecimiento del pulmón afectado, ya que se sabe que las lesiones se agravan en el transcurso de la gestación(34-38). Otros equipos europeos están desarrollando alternativas menos invasivas y con resultados alentadores, Deprest y Gratacos por medio de la fetoscopía colocan un balón endotraqueal fetal, con lo que habrían obtenidos resultados realmente alentadores. La cirugía fetal estaría indicada en todo caso de hernia diafragmática aislada; es decir sin anomalías asociadas, diagnosticada antes de las 25-26 SA con criterios de gravedad presentes. En relación al término del embarazo, los anglosajones preconizan una cesárea sistemática poco antes del término de la gestación para programar una reanimación en las mejores condiciones. En Francia, las indicaciones de una cesárea están relacionadas a indicaciones estrictamente obstétricas maternas, principalmente en relación al sufrimiento fetal. Bohn(44) compara la sobrevida entre el uso de ventilador de alta frecuencia (HFOV) y ECMO, entre los cuales no encuentra diferencias, con un 55 y 53 % de sobreviva respectivamente. Para decidir los casos subsidiarios de cirugía se utiliza la hipoxemia preductal e hipercapnia, con un 53% sobrevida (19811994) a 75% (1995-2001): HFOV y control de la SaO2 preductal(45). Además se constata una diferencia de costo significativa: Sin ECMO: USD$98.000, Con ECMO: USD $ 365.000(46). Gerben(47) encuentra una mortalidad global de un 62%, donde un 52% fueron diagnosticados en antenatal; es por esto que el diagnóstico antenatal se asocia a una alta mortalidad, 64% v/s 29% con diagnóstico postnatal. En relación al manejo del recién nacido, no ha habido modificación de la morbimortalidad a pesar de los diferentes tratamientos instaurados, siendo de un 35% para la ECMO, HFOV, Oxido Nítrico y Surfactante. Harrison(48) muestra que en los fetos con signos de mal pronóstico no varía la morbimortalidad al comparar la cirugía endoscópica fetal con la HFOV: encontrando una sobreviva de 73 y 77% respectivamente. Lo único que marcaría una diferencia es el valor del LHR, siendo menor a 0.90 para los de mal pronóstico. 126 BIBLIOGRAFÍA 1. Dechelotte. Med. Foet. & Ech. Gynecol. 1993;13: 4-9. 2. Hamilton. 4th Ed, Mac Millan London 1976 3. Moore. 4th Ed. Saunders Ed. 1988; 482 p. 4. Langham. Clin Pediatr 1996; 23: 671 - 687. 5. Torfs CD. Teratology 1992; 46:555 - 565. 6. Steihorm. Arch Pediatr Adolesc Med 1994; 148: 626631. 7. Barbet. Amer J for Ens. Med. Pathol. 1988; 9: 4044. 8. Emeryl. Arch Dis Chile 1960; 35: 544 -547. 9. Harrison Ed. Basel 1989; p. 130-142. 10. Beals Pediatr Surg 1992; 27: 997-1002. 11. Vacanti. Pediatr. Surg. Int. 1988; 3:1-5. 12. O’Rourke. J Pediatr Surg. 1988; 23(10): 904-7. 13. Brands. Eur J Pediatr Surg 1992; 2: 81-83. 14. Metkus AP. J Pediatr Surg 1996; 31: 148-152. 15. Schwartz SM. J Pediatr Surg 1994; 125: 447-451. 16. Thébaud B. Arch Pediatr 1999; 6 (2): 186-198. 17. Azancot. Intensive Care Med. 1997; 23: 1062-69. 18. Burge DM. J Pediatr Surg 1989; 24: 567-569. 19. Adzick NS. J Pediatr Surg 1989; 24: 654-658. 20. Adzick Ns. J Pediatr Surg 1985; 20: 357-631. 21. Harrison MR. New Engl J Med 1990; 322: 1582-4 22. Thébaud B. 1997 Intensive Care Med. 23: 1062-69. Capítulo 4 - Obstetricia 23. Harrison MR. JAMA 1994; 271: 382-384. 24. Merz E. Prenat Diagn 1999; 19: 614-619. 25. Morin L. Semin Perinatol 1994; 18: 228-253. 26. Fox H.E. Am J Obstet Gynecol 176: 807-13. 27. Kalache K. DUltrasound Obstet Gynecol 1998; 12: 27-32. 28. Roth P. Gynecol Obstet Invest 1998; 46: 153-157. 29. Puri P. J Pediatr Surg 1984; 19: 29-35. 30. Wilson JM. J Pediatr Surg 1994; 29: 815-819. 31. Smith DW; ed 3 Philadelphia, WB Saunders 1982. 32. Hoboth N. Lancet 1962; 2: 1332-1334. 33. Janik JP. Pediatr Surg Int 1996; 11: 169. 34. Adzick Ns. J Pediatr Surg 1985; 20: 357-631. 35. Harrison MR. Surgery 1980; 88: 174-182 36. Harrison MR. J Pediatr Surg 1990; 25: 47-57 37. Iritani I. Anat Embryol 1984; 169: 133-139 38. Soper RT. Creation and repair of diaphragmatic hernia in the fetal lamb . 41. Metkus AP. J Pediatr Surg 1996; 31: 148-152. 42. Lipshutz GS. J Pediatr Surg 1997; 32: 1634-1636. 43. Kitano Y. Semin Perinatol 1999; 23: 448-461. 44. Bohn D. Am J of Respiratory and Critical Care Medecine 2002; 166: 911-915 45. Boloker J. J Pediatr Surg 2002; 37: 357-366. 46. Metkus AP. J Pediatr Surg 1995; 30: 226-230. 47. Gerben S. Pediatrics 2003; 112: 532-535. 48. Harrison M. N Engl J Med 2003; 349: 1916-1924. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 127 PARED ABDOMINAL ONFALOCELE Definición El onfalocele corresponde a una anomalía del cierre del anillo umbilical y a un defecto de reintegración de las asas intestinales. Se produce una hernia de las vísceras abdominales bajo la implantación del cordón umbilical. El contenido y el volumen del onfalocele es variable, pudiendo contener sólo intestino en la minoría de los casos (10 a 25%)(1,2,3), bazo, hígado e incluso páncreas. Se caracteriza porque las vísceras están recubiertas por una membrana peritoneal y amniótica. Esta cubierta a pesar de ser tan delgada no permite el paso de «alfaproteína» como en la gastrosquisis(4). Debe ser diferenciado de las hernias umbilicales, para éstas la pared abdominal es dehiscente con una cubierta cutánea normal. La hernia es siempre de pequeño volumen. Existe un onfalocele fisiológico desde las 9 a 12 semanas amenorreicas (SA), el que regresa espontáneamente; excepcionalmente puede regresar en forma un poco más tardía como a las 13-14 SA. Si este es realmente manifiesto entre las 11 a 14 SA, se cree que la incidencia de anomalías cromosómicas sería de un 61 % (5). Epidemiología La incidencia es variable de 1 en 2000 y 1 en 5000 a 10.000 nacimientos y ésta aumentaría con la edad materna(5, 6). Las anomalías cromosómicas son más frecuentes, dentro de éstas destacan las aneuplodias (trisomías 18, 21 y 13); esta incidencia varía de 10% si es aislado a un 30-50% si se manifiesta dentro de un contexto polimalformativo. Se puede manifestar dentro de síndromes mendelianos como el síndrome de BeckwithWiedeman. pared abdominal por un saco donde las dimensiones pueden ser evaluadas en cortes sagital y transversal (Figura 44). Las vísceras herniadas están delimitadas por una fina membrana peritoneal, a veces difícil de individualizar. Este saco puede contener asas intestinales, hígado, estómago, etc. La presencia de ascitis asociada no tiene valor pronóstico. Según el volumen del onfalocele, se va a afectar el perímetro abdominal; por lo tanto es importante considerar este factor por la eventual confusión con una restricción del crecimiento fetal. Un hecho importante es la consideración de la inserción del cordón umbilical, la que se hace en el vértice del saco (Figura 45). Figura 44. Corte axial abdominal en un feto de 22 SA. Se visualiza una voluminosa masa anterior de aspecto sólido heterogéneo. Diagnóstico Es un signo de fines del 1er trimestre, a partir de las 13 SA. • Criterios ultrasonográficos o Criterios diagnósticos El onfalocele constituye una masa abdominal anterior de desarrollo extrabdominal. Está unida a la Figura 45. Corte axial abdominal en un feto de 16 SA. Se visualiza la inserción distal del cordón umbilical, lo que hace el diagnóstico diferencial con una gastrosquisis. 128 o Criterios pronósticos Las malformaciones asociadas y los síndromes genéticos subyacentes comandan el pronóstico. Ni el tamaño del defecto de la pared, ni el volumen ni contenido del onfalocele parecieran influir en el pronóstico. Incluso aquellos de pequeño volumen que incluyan sólo contenido intestinal pudieran asociarse aún más con anomalías cromosómicas (67%) (Figuras 46 y 47), no así cuando asocia otros órganos como el hígado (16%)(6). Es raro que el saco se rompa in útero, por lo que no existen evidencias que su vía de interrupción deba ser por cesárea(3, 8-11) por lo que debiera ser según indicaciones obstétricas aceptadas. Si el onfalocele es aislado, su pronóstico es bueno, debiendo ser reparado quirúrgicamente; por el contrario si éste se asocia a otras anomalías, el pronóstico se ensombrece con un 20% de sobreviva. Capítulo 4 - Obstetricia • Diagnóstico diferencial y errores ultrasonográficos o El onfalocele es fisiológico hasta las 12 SA. o La laparosquisis constituye el principal diagnóstico diferencial, pero en ésta el cordón umbilical está normalmente inserto, y no existe una membrana que cubra el contenido eviscerado. o Quiste alantoideo: del cordón cuando este está situado a la proximidad de la pared abdominal. Aumenta la incidencia de trisomía 18 (44%)(12). Malformaciones asociadas (30 a 50%)(13) Es casi de regla que se trate de anomalías aisladas en síndromes polimalformativos. • • • • • • • • • Figura 46. Onfalocele de pequeño volumen, en este caso se trató de una trisomía 18. Sistema nervioso central: Mielo-Meningocele. Cara: Fisura labio-palatina. Torácicas: Hernia diafragmática. Cardiacas: (20 a 50%) CIA, CIV, Tetralogía de Fallot, Transposición de los grandes vasos. Digestivas: atresias y estenosis digestivas, Agenesia de vías biliares, Duplicaciones, Celosomías, Enfermedad de bridas amnióticas. Esqueléticas: (30%) Polidactilias, Artrogriposis. Sd de Beckwith-Wiedeman. (10%) Secuencia OEIS: Onfalocele, Extrofia vesical, Imperforación anal, anomalías Sacras. Anomalías cromosómicas: (28 a 36%) trisomías 13 y 18. LAPAROSQUISIS Definición La laparosquisis corresponde a un defecto de cierre de la pared abdominal lateral del ombligo, sin cubierta peritoneal. Es más frecuentemente encontrada a derecha y por lo general lo eviscerado es sólo intestino (Figura 48). Existen diversas teorías con respecto a su origen • Ruptura precoz de un onfalocele • Defecto del desarrollo de la pared abdominal con herniación de contenido digestivo(14) • Involución anormal de la vena umbilical derecha(14) Figura 47. Onfalocele de gran volumen, en este caso no se asoció a otras malformaciones, pero fue una trisomía 18. Al estar el intestino en directo contacto con el líquido amniótico, se incrementa la liberación de alfaproteína(4). Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 129 Figura 49. Corte sagital. Ecografía vía abdominal de un feto de 13+6 SA con una TIN de 1,8 mm y LCC de 68 mm. Obsérvese el intestino exteriorizado en la parte baja del abdomen (flechas). No hay anomalías asociadas. Figura 48. a) Neonato, se observa evisceración a través de defecto situado a izquierda de la inserción umbilical (forma no habitual); b) evisceración a derecha de la inserción umbilical. En estos fetos no es raro encontrar una monitorización no estresante alterada, esto se debería a un reflejo vagal secundario a la acción del líquido amniótico sobre el intestino(15). Es por este hecho que la interrupción vía cesárea tiende a estar incrementada en estos pacientes. Epidemiología La incidencia es de 1 en 5000 a 10.000 nacimientos. La anomalía es generalmente aislada y las anomalías cromosómicas son excepcionales (< 1%). Diagnóstico Es un signo de fines del 1er trimestre (Figura 49), a partir de las 13-14 SA. Presenta una tasa de detección de un 70 a 72%(16, 17). • Criterios ultrasonográficos o Criterios diagnósticos La laparosquisis aparece como una masa abdominal anterior, de desarrollo extraabdominal (Figura 50). El defecto parietal no es siempre visible. Figura 50. Corte transversal de abdomen. Gracias al Doppler color, podemos observar en este caso, claramente, que el defecto (D) se sitúa a la izquierda de la inserción del cordón umbilical (presentación cefálica). Las vísceras nadan en el líquido amniótico. Las asas intestinales son festoneadas y pueden estar dilatadas. Las paredes digestivas pueden estar engrosadas y ecogénicas. La posición del hígado, del estómago y de la vejiga deben ser determinadas. No se deben visualizar membranas periféricas. Un hecho importante, es que el cordón umbilical está normalmente inserto en la pared abdominal. La masa intestinal intraamniótica está lateralizada. 130 Capítulo 4 - Obstetricia o Criterios pronósticos El pronóstico está dado por la existencia de lesiones de necrosis por isquemia del mesenterio a nivel del defecto y de las asas en contacto con el líquido amniótico, responsable de la atresia intestinal (30%)(18, 19) , ya que se produce una peritonitis química 20: Las complicaciones que se pueden observar en la gastrosquisis se describen en la Tabla I. Criterios ultrasonográficos (Figuras 51 y 52). • Disminución del peristaltismo. • Dilatación intestinal. (lumen mayor a 11 - 17 mm)(14, 15, 21) • Engrosamiento y ecogenicidad de las paredes digestivas(15), dificultaría la reparación posterior y aumentaría la aparición de complicaciones. El modo Doppler pulsado y color pueden manifestar la ausencia de vascularización arterial mesentérica a nivel de las asas intestinales extra e intra abdominales, lo que sería de mal pronóstico. ilustrando la isquemia intestinal (Figuras 53 y 54). La presencia extrabdominal hepática sería de mal pronóstico. Figura 52. Asas intestinales libres en la cavidad amniótica, presentan dilatación de su lumen (L) en grado variable. Las paredes intestinales se observan engrosadas e hiperecogénicas (P) al contacto con el líquido amniótico. Ninguna membrana es visible. TABLA I. COMPLICACIONES HABITUALES DE LAPAROSQUISIS. • RCIU: 77% • Prematurez: 55% • Mortalidad: 40% y 80% con cardiopatía congénita • Pequeño para edad gestacional: 29 a 43%(22) • Anomalías no-gastrointestinales asociadas: 7-30% • Anomalías cardiacas: < 8% Figura 53. Corte transversal. Embarazo de 21+3 SA. Con Power Energy observamos la vascularización mesentérica de las asas exteriorizadas. Figura 51. Corte transversal de abdomen. Intestino herniado (I) libre en la cavidad amniótica a través del defecto de la pared, el que es medible (*). Figura 54. Doppler de la arteria mesentérica superior a nivel de las asas exteriorizadas a través del defecto de la pared abdominal. (RI : 0,81 e IP : 1,57) Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 131 La existencia de un oligoamnios (20 a 40% de las laparosquisis) sería también de mal pronóstico. • Diagnóstico diferencial y errores ultrasonográficos El onfalocele constituye el principal diagnóstico diferencial, pero la inserción central del cordón y la visualización de la membrana peritoneal establecen el diagnóstico. Malformaciones asociadas Se trata generalmente de una malformación aislada. Puede asociarse anomalía digestiva como atresia, estenosis o involución intestinal secundaria (15 a 25% de los casos). Figura 55.Evisceración de órganos digestivos. La disrrelación del defecto de la pared con el gran volumen de las vísceras sugiere una manifestación muy temprana en la gestación. LIMB – BODY WALL COMPLEX Es también conocida como «Body Stalk anomaly». Algunos autores la consideran como una variante de una ruptura precoz del amnios. Consiste en un defecto amplio de la pared abdominal con exteriorización de vísceras sin cubierta peritoneal; usualmente se asocia a otras anomalías. Diagnóstico Es un signo del 1er trimestre, pero generalmente se diagnóstica en el 2° trimestre. • Criterios ultrasonográficos o o o o o Figura 56. En este feto se aprecia el importante grado de desviación de columna que presenta. Esto podría deberse a la presencia de un cordón umbilical corto y al peso ejercido por los órganos eviscerados hacia abajo. Criterios diagnósticos El cordón umbilical es corto. Presenta un amplio defecto de la pared abdominal, frecuentemente afectando el lado izuierdo (Figura 55) Asocia defectos de columna, ya sea postural (escoliosis) o disrrafias de ubicación baja (Figura 56) Otros hallazgos son: encefalocele o exencefalia, fisuar labial y/o palatina, y de extremidades (Figura 57) • Criterios pronósticos o Es invariablemente fatal o No se asocia a anomalías cromosómicas o Puede estar asociado a consumo de cocaína(23) ABDOMEN ATRESIA ESÓFAGO Definición Es una falta de comunicación entre 2 segmentos del esófago, se asocia frecuentemente a Figura 57. Se visualiza el defecto de columna (escoliosis) y las anomalías de extremidades. una fístula traqueo-esofágica. Esta anomalía impide la deglución del líquido amniótico hacia el estómago, y su posterior absorción en el intestino, no pudiendo circular hacia la placenta y ser eliminado; por esto se produce un hidramnios. Cabe recordar que la cámara gástrica puede tener un aspecto normal, esto se debe a que el estómago está recubierto por mucosa, la que produce secreción 132 gástrica; además, puede haber una fístula traqueoesofágica distal(24- 28). Clasificación Clasificación anatómica de Ladd: • Tipo A: Atresia con fístula traqueal «distal». (90%) • Tipo B: Atresia pura sin fístula (5 a 10%) • Tipo C: Atresia con doble fístula esófago-traqueal (1 a 2%) • Tipo D: Atresia con fístula «proximal» (1%) • Tipo E: Atresia con fístula bronquial «distal» (4%) Capítulo 4 - Obstetricia ubicación más frecuente se sitúa a nivel del lumen del píloro por hipertrofia de los músculos del esfínter; se sitúa por detrás de la ampolla de Vater. Su causa es desconocida, pero podría tener una explicación genética. Epidemiología La incidencia es de 1 en 4000 a 10.000 nacimientos. Las anomalías cromosómicas son frecuentes alrededor de un 30%, principalmente la trisomía 21. Es la anomalía gástrica más frecuente en los niños. Epidemiología La incidencia es de 1 en 1.500 a 3.000 nacimientos. Diagnóstico Es un signo del 2° y 3er trimestre. Diagnóstico Es un signo del 2° trimestre. • Criterios ultrasonográficos • Criterios ultrasonográficos o Criterios diagnósticos En corte transversal del abdomen, la imagen de la cámara gástrica y de la primera porción duodenal dilatada; manifiesta el aspecto clásico en «doble burbuja» ecolúcida. El corte sagital confirma el diagnóstico poniendo en evidencia la comunicación entre el estómago y duodeno, formando una imagen en «curvatura». Un hidramnios está presente en el 50% de los casos (Figura 58). o Criterios diagnósticos ♦ Atresia sin fístula: • Hidramnios y estómago no visible en más de 1 examen ultrasonográfico seriado • Dilatación permanente del esófago • Movimientos de regurgitación importantes ♦ Atresia con fístula: • Hidramnios y estómago no visible (fístula estenosante). • Hidramnios y estómago visible, pero de pequeño volumen • Dilatación permanente del esófago • Movimientos de regurgitación importantes • Diagnóstico diferencial y errores ecográficos • Diagnósticos de ausencias de visualización de cámara gástrica. • Fístula esófago-traqueal de grueso calibre: el estómago es normalmente visible y la cantidad de líquido amniótico es normal. Malformaciones asociadas Las anomalías cromosómicas son frecuentes entre un 10 a 30%, principalmente la trisomía 18. Secuencia de VACTERL 30 a 70%. ATRESIA Y ESTENOSIS DUODENAL Definición Las estenosis y atresias duodenales son malformaciones obstructivas intestinales donde la Figura 58. Se visualiza a izquierda la imagen «curvatura» en un corte parasagital y a derecha la de «doble burbuja». Movimientos anormales de regurgitación son visibles en modo Doppler color. No es posible determinar en antenatal si la causa se debe a una estenosis o a una atresia del lúmen duodenal. • Diagnóstico diferencial y errores ecográficos Ante una imagen ecolúcida subdiafragmática, evocamos: Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) o Quistes de la mitad superior del abdomen. o Una hidronefrosis y megaureter unilateral. El peristaltismo gastro-duodenal normal puede reproducir una falsa imagen «curvatura», pero ésta es transitoria. Malformaciones asociadas Las anomalías cromosómicas son frecuentes entre un 10 a 30%, principalmente la trisomía 21. El 50% de las atresias duodenales presentan otra anomalía. En la ausencia de otras anomalías, el pronóstico postnatal es excelente(29). Cardiacas: cardiopatías 20%. Digestivas: malrotación intestinal. Secuencia de VACTERL o VATER. 133 • Criterios ultrasonográficos o Criterios diagnósticos En cortes transversales y sagitales del abdomen, las atresias intestinales aparecen bajo la forma de múltiples lagunas ecolúcidas, de paredes adyacentes más o menos hiperecogénicas y que presentan un aspecto de «panal de abejas». Seudoastraciones pueden ser descritas correspondiendo a la yuxtaposición de las asas dilatadas. Las imágenes son móviles en el tiempo a causa del peristaltismo y de la agravación progresiva de las lesiones (Figura 59) Un hidramnios está presente en casi 25% de los casos, más aún a medida que el obstáculo es alto y que la edad gestacional avanza. ATRESIA Y ESTENOSIS YEYUNO-ILEAL Definición La atresia representa 90 a 95% de las obstrucciones yeyuno-ileales. Las estenosis se encuentran sólo en un 5 a 10% de los casos. La topografía de la obstrucción se reparte en forma similar a todo lo largo del intestino delgado, sin un sitio preferencial. Mientras más importante es la estenosis y más distal se ubica, mayor es el grado de dilatación de las asas intestinales, por lo que presenta al ultrasonido mayor número de asas distendidas. Clasificación Clasificación anatómica: • Tipo I: Atresia por simple diafragma, sin discontinuidad intestino-mesentérico (30%). • Tipo II: cordón fibroso que une 2 extremos intestinales distendidos (35%). • Tipo III: interrupción completa intestinal y mesentérico. La atresia es completa en «V» (Tipo IIIa) o de múltiples imágenes ecolúcidas (IIIb) (35%). • Tipo IV: Atresias múltiples (5%). Epidemiología La incidencia es de 1 en 3000 a 5000 nacimientos. Las atresias yeyunales son 2 veces más comunes que las ileales(30). Las anomalías cromosómicas son raras (< 1%). Diagnóstico Es un signo de fines del 2° trimestre, a partir de las 26 a 30 SA. Figura 59 a, b. Corte axial abdominal fetal en un feto de 35 SA. Se visualizan múltiples asas dilatadas e incluso una seudoastración intestinal. 134 Capítulo 4 - Obstetricia o Criterios pronósticos No son asequibles al diagnóstico ultrasonográfico. Ni la topografía, ni el tipo anatómico, ni el largo del intestino delgado funcional restante no son visibles. El estudio de la vascularización mesentérica en modo Doppler no es posible, pero podría aportar una ayuda al pronóstico. • Diagnóstico diferencial y errores ecográficos o Las imágenes ecolúcidas de un megaureter. o Falsas imágenes de dilatación digestiva, heterogéneas y de pequeños tamaños, de tipo intestino delgado, pero a veces de colon, son visibles en la 2° mitad de la gestación y no deben ser interpretadas como una atresia. Malformaciones asociadas Es casi de regla que se trate de anomalías aisladas y no se asocien anomalías extraintestinales. Se asocian anomalías digestivas como malrotación, gastrosquisis, duplicación, ileo meconial. HIPERECOGENICIDAD INTESTINAL Definición La hiperecogenicidad intestinal es un signo ultrasonográfico, no una descripción anatómica. Epidemiología La incidencia es de 1 a 2% de los embarazos, sólo deben considerarse anormales aquellas de «tipo óseo»; es decir, que es similar a la densidad ósea de la pelvis fetal o de las estructuras óseas de la columna. Diagnóstico Es un signo del 1er y del 2° trimestre. • Criterios ultrasonográficos o Criterios diagnósticos La hiperecogenicidad intestinal debe ser considerada como patológica cuando es de densidad ósea (Figura 60). El intestino normal ecogénico es la base de una masa heterogénea sistemáticamente, pero mal delimitada. • Diagnóstico diferencial y errores ultrasonográficos • Las hiperecogenicidades donde la densidad es menor a la del hueso adyacente, deben ser Figura 60. Corte axial abdominal fetal. Se visualiza hiperecogenicidad intestinal en un feto de 39 SA. En este caso se trató de una insuficiencia placentaria con hipoxia fetal. consideradas como una variante de la normal. • Las calcificaciones del abdomen pueden ser discutibles y deben ser diferenciadas entre diversas entidades patológicas: peritonitis meconial, quistes y seudoquistes meconiales, calcificaciones de fetopatías infecciosas (toxoplasmosis, CMV, varicela), enterolitiasis y tumores abdominales heterogénicos. Malformaciones asociadas Generalmente en el 66% de los casos ninguna anomalía fetal es encontrada. En el 35% de los casos existe un contexto patológico: o 13% de muerte fetal inexplicada, donde el 1% presenta restricción del crecimiento fetal severo. o 5% de anomalías cromosómicas (trisomía 21). o 5% de estenosis o atresias digestivas. o 3,8% de infección fetal. o 3 , 3 % d e m u c o v i s c i d o s i s ( e n f e r m e d a d fibroquística). o 3,3% de malformaciones diversas asociadas. PATOLOGIA DE VIAS BILIARES Definición Las anomalías de las vías biliares son excepcionales en la vida intrauterina. La patología quística es descrita en 3 tipos según la clasificación de Alonso-Ley. Los divertículos vesiculares son anomalías benignas de la vesícula biliar. Las litiasis vesiculares son extremadamente raras de describir en el feto. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 135 Epidemiología La patología quística de las vías biliares es frecuente en el Extremo Oriente sin que su etiopatogenia sea claramente definida. Las litiasis biliares son más frecuentemente encontradas en un contexto de enfermedad hemolítica hereditaria. La formación de litiasis podría explicarse por un defecto de reabsorción ileal de las sales biliares, provocado por diversas patologías como la fibrosis pancreática o las infecciones intestinales. • Diagnóstico diferencial y errores ecográficos ♦ Los quistes de colédoco deben hacer sospechar: o Una patología quística del abdomen superior. o Quistes del ovario de desarrollo pélvico. o Quistes solitarios hepáticos, del riñón derecho, de la suprarrenal derecha y quistes del mesenterio. Diagnóstico Es un signo del 2° trimestre. QUISTES ABDOMINALES • Criterios ultrasonográficos o Criterios diagnósticos • Quiste del colédoco: aparece como una formación ecolúcida redondeada en el hipocondrio derecho. El quiste está en contacto con el hígado en la región perihiliar. El diagnóstico de certeza se plantea cuando una comunicación entre el quiste y la vesícula biliar es evidenciada. • Divertículo vesicular: la vesícula biliar presenta una dilatación anormal en alguna parte de su pared. Esta imagen no varía de un examen a otro. • Litiasis vesicular: se manifiesta por imagen hiperecogénica, redondeada, única o múltiples y de pequeño tamaño en el lumen vesicular (Figura 61). La cavidad vesicular está más o menos repleta. Imágenes menos densas pueden ser vistas y atribuidas a un «barro biliar». Figura 61. Corte axial abdominal fetal. Se visualizan múltiples asas dilatadas. Malformaciones asociadas En general se trata de anomalías aisladas. Definición Los quistes abdominales fetales están definidos por imágenes ecolúcidas, a veces con pequeños puntos ecogénicos o tabicaciones. Las imágenes quísticas puras son en general siempre benignas en el feto. Las más frecuentes son los quistes del ovario. Diagnóstico Es un signo del 2° trimestre. • Criterios ultrasonográficos o Criterios diagnósticos • Quiste del ovario: imagen redondeada, única, habitualmente ecolúcida pura, se localiza en la pelvis; pero puede en ocasiones ser voluminosa, desarrollándose hacia la cavidad abdominal. Adyacente a la vejiga, puede a veces dar la impresión de ser binocular (Figura 62). Puede presentar un fino punteado ecogénico o ser tabicada, lo que traduce una probable torsión o hemorragia intraquística. Se trata de un feto de sexo femenino, que aparece después de las 24 SA y tiende a aumentar de volumen a medida que avanza la edad gestacional. Son siempre benignos y se deben a acción hormonal(3133) . En ocasiones si éstos crecen demasiado, más de 5 cm, pueden ser puncionados para ser vaciados in útero, de no ser necesario, generalmente regresan en forma espontánea durante los primeros 6 meses de vida(34, 35). • Quiste del colédoco: imagen redondeada, única y anecogénica. Se localiza en la mitad superior del abdomen en la región retro o subhepática, teniendo una relación estrecha con la vena porta. La vesícula biliar está adyacente, la que puede estar comprimida y mal visualizada. El diagnóstico se realiza tras evidenciar una comunicación entre la vesícula biliar y el quiste. Es más frecuente en mujeres(36). 136 Capítulo 4 - Obstetricia en la fosa renal y sobre todo la no visualización de las estructuras renales deben rectificar el diagnóstico. Figura 62. Corte axial pelviano fetal. Se visualizan en cuadrante a derecha 2 imágenes ecolúcidas, por medio del doppler color podemos identificar la vejiga; la imagen adyacente es un quiste del ovario derecho en un feto de 33+4 SA. • Quiste del mesenterio: imagen redondeada, a veces polilobulada, ecolúcida, de tamaño variable. Se sitúa en la región mesentérica ileal, pero la localización antenatal precisa es dificultosa. Su origen generalmente es linfático y habitualmente requiere de cirugía para su remoción. • Quiste solitario renal, hepático, del bazo o del páncreas: corresponde a una imagen redondeada ecolúcida, de tamaño variable y situada en el seno o periferia del parénquima tisular del cual se forma. • Quiste del uraco: imagen redondeada, ecolúcida, situada en el trayecto del uraco. Son abdominopelvianas de ubicación anterior; pudiendo ser yuxtavesicales o retroumbilicales. Criterios pronósticos Los quistes abdominales son siempre benignos, sea cual sea su origen. o • Diagnóstico diferencial y errores ecográficos El diagnóstico de imágenes quísticas del abdomen obliga a plantear cada hipótesis probable según la topografía, el sexo fetal y la evolución de la lesión considerada. Los principales problemas están dados por la imagen en «doble burbuja» de la atresia duodenal, pero el corte sagital permite corregir el diagnóstico evidenciando la continuidad gastro-duodenal. Las hidronefrosis mayores con dilatación pielocaliciarias que borran el fondo de las cálices y hacen el parénquima renal laminar, pueden ser confundidas con un quiste abdominal. La topografía, Quistes Intra-abdominal o Hígado • Quiste hepático • Quiste del colédoco • Várice de la vena umbilical o Suprarrenal • Quiste simple • Hemoarragia • Neuroblastoma o Intestino • Duplicación quística • Quiste del mesenterio • Linfangioma • Peritonitis meconial o Ovario • Quiste folicular BIBLIOGRAFÍA 1. Bair JH; Russ PD; Pretorius DH; et al. Fetal omphalocele and gastroschisis : A review of 24 cases. Am L Radiol 1986; 147: 1047-1051. 2. Brown BSJ. The prenatal ultrasonographic features of omphalocele: A study of 10 patients. J Can Assoc Radiol 1985; 36: 312 - 316. 3. Redford DH; McNay MB; Whittle MJ. Gastroschisis and exomphalos: Precise diagnosis by mid-pregnancy ultrasound. Br J Obstet Gynecol 1985; 92: 54 - 59. 4. Palomaki GE; Hill LE; Knight GJ; et al. Secondtrimester maternal serum alpha-fetoprotein levels in pregnancies associated with gastroschisis and omphalocele. Obstet Gynecol 1988; 71: 906 - 909. 5. Snijders RJM; sebire NJ; Souka A; Santiago C; Nicolaides KH. Fetal exomphalos and chromosomal defects: relanship to maternal age and gestation. Ultrasound Obstet Gynecol 1995; 6: 250 - 255. 6. Snijders RJM; Nicolaides KH. Ultrasound markers. In: Snijders RJM; Nicolaides KH, eds. Ultrasound markers for chromosomal defects. Frontiers in Fetal Medecine series. London: Parthenon; 1994:95. 7. Hughes MD; Nyberg DA; Mack LA; Pretorius DH. Fetal omphalocele: Prenatal detection of concurrent anomalies and other predictors of outcome. 8. Martin LW; Torres AM. Omphalocele and gastroschisis. Symposium on pediatric surgery. Surg Clin North Am 1985; 65: 1235-1244. 9. Schwaitzenberg SD; Pokorny WJ; McGill CW; et al. Gastroschisis and omphalocele. Am J Surg 1982; 144: 650 - 654. 10. Hasan S; Hermansen. The prenatal dignosis of ventral Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) abdominal wall defects. Am J Obstet Gynecol 1986; 155: 842 - 845. 11. Kirk EP; Wah R. Obstetric management of the fetus with omphalocele or gastroschisis: A review and report of one hundred and twelve cases. Am J Obstet Gynecol 1983; 146: 512 - 518. 12. Chen CP; Jan SW; Liu FF; et al. Prenatal diagnosis of omphalocele associated with umbilical cord cyst. Acta Obstet Gynecol Scand 1995; 74: 832 - 835. 13. Dillon E; Rendick M. The antenatal diagnosis and management of abdominal wall defects: the Northern Region experience. Clin Radiol 1995; 50: 855 - 859. 14. Pryde PG; Bardicef M; Treadwell MC; et al. Gastroschisis: Can antenatal ultrasound predict infant outcomes? Obstet Gynecol 1994; 84: 505 – 510. 15. Adra AM; Landy HJ; Nahmias J; Gomez-Marin O. The fetus with gastroschisis: Impact of route of delivery and prenatal ultrasonography. Am J Obstet Gynecol 1996; 174: 540 – 546. 16. Morrow RJ; Whittle MJ; McNay MB; et al. Prenatal diagnosis and management of anterior abdominal wall dfects in the west of Scotland. Prenat Diagn 1993; 13: 111 – 115. 17. Walkinshaw SA; Rendick M; Hebisch G; Hey EN. How good is ultrasound in the detection and evaluation of anterior abdominal wall defects? Br J Radiol 1993; 65: 298 – 301. 18. Luck SR; Sherman J; Raffensperger JG; et al. Gastroschisis in 106 consecutive newborn infants. Surgery 1985; 98: 677 – 683. 19. Mabogunje OOA; Mahour GH. Omphalocele and gastroschisis: Trends in survival across two decades. Am J Surg 1984; 148: 679 – 686. 20. Klick P; Tibboel D; Van Der Kamp AWM; et al. The effect of fetal urine on the development of bowel in gastroschisis. J Pediatr Surg 1983; 18: 47 – 50. 21. Babcock CJ; Hedrick MH; Glodstein RB; et al. Gastroschisis: Can sonography of the fetal bowel accurately predict postnatal outcome? J Ultrasound Med 1994; 13: 701 – 706. 22. Fries MH; Filly RA; Callen PW; et al. Growyh retardation in prenatally diagnosed cases of gastroschisis. J Ultrasound Med 1993; 12: 583 – 137 588. 23. Viscarello RR; Ferguson DD; Nores J; Hobbins JC. Limb – body wall complex associated with cocaine abuse: Further evidence of cocaine’s teratogenicity. Obstet Gynecol 1992; 80: 523 – 526. 24. Farrant P. The antenatal diagnosis of oesophageal atresia by ultrasound. Br J Radiol 1980; 53: 1202 – 1203. 25. Hobbins JC; Grannum PAT; Berkowitz RI; et al. Ultrasound in the diagnosis of congenital anomalies. Am J Obstet Gynecol 1979; 134: 331 – 334. 26. Pretorius DH; Meier PR; Johnson ML. Diagnosis of esophageal atresia in utero. J Ultrasound Med 1983; 2: 475. 27. Pretorius DH; Drose JA; Dennis MA; et al. Tracheoesophageal fistula in utero. J Ultrasound Med 1987; 6: 509 – 513. 28. Loveday BJ; Barr JA; Aitken J. The intra-uterine demonstration of duodenal atresia by ultrasound. Br J Radiol 1975; 48: 1031 – 1032. 29. Grosfeld JL; Rescorlla FJ. Duodenal atresia and stenosis: Reassessment of treatment and outcome case on antenatal diagnosis, pathological variance and long-term follow up. World J Surg 1993; 17: 301 – 309. 30. De Lorimier AA; Fonkalsrud EW; hays DM. congenital atresia and stenosis of the jejunum and ileum. Surgery 1969; 65: 819 – 827. 31. Bower R; Dehner LP; Tenberg JL. Bilateral ovarian cyst in the newborn. Am J Dis Child 1974; 128: 731–733. 32. Carlson DH; griscom NT. Ovarian cyst in the newborn. AJR 1972; 116: 664 – 672. 33. Desa DJ. Follicular ovarian cysts in stillbirths and neonates. Arch Dis Child 1975; 50: 24 – 50. 34. Sapin E; Bargy F; Lewin F; et al. Management of ovarian cyst detected by prenatal ultrasound. Eur J Pediatr Surg 1994; 4: 137 – 140. 35. Brandt ML; Luks FI; Filiatrault D; et al. Surgical indications in antenatally diagnosed ovarian cyst. J Pediatr Surg 1991; 26: 276 – 281. 36. Lugo-Vicente HL. Preanatally diagnosed choledochal cysts: observation or early surgery? J Pediatr Surg 1995; 30: 1288 – 1290. 138 Capítulo 4 - Obstetricia EXAMEN ECOGRÁFICO MORFOLÓGICO ANORMAL DEL APARATO GENITOURINARIO FETAL EMBRIOLOGÍA Hacia el 3er mes, los nefrones están constituidos y son anatómicamente e histológicamente similar a aquellos del riñón definitivo del adulto y comienza a producir orina. Ningún nefrón se desarrolla después del nacimiento, salvo en los prematuros; sólo se produce un aumento del volumen, por lo que el aumento de volumen del riñón se produce por hipertrofia del nefrón y no por un aumento del número de éstos. Esto es importante a considerar por el daño que pudieran sufrir en antenatal, con el fin de determinar conductas a futuro. Las anomalías renales y uretrales se manifiestan en el 3 a 4% de los nacidos. Morfológicamente se distinguen malformaciones de la porción excretora y de la porción colectora, o de ambas. Ben Pansky. Review of Medical embryology. Macmilliam Publishing Company 1986; 248253. Las uropatías diagnosticadas en antenatal tienen una doble importancia: por una parte evitar la infección a corto tiempo, y a largo plazo evitar la degradación de la función renal. Se debe evaluar el pronóstico a largo plazo, informar a los padres y plantear la posibilidad de una intervención si fuera necesario. Las repercusiones sobre el riñón pueden ser funcionales en las uropatías obstructivas donde la hiper presión urinaria puede ser responsable de un cese precoz de la nefrogénesis más o menos temprana durante la vida fetal(1). Los tratamientos son generalmente conservadores, más bien de tipo preventivo, evitando las infecciones y las obstrucciones. Aún deben ser evaluadas a la edad adulta, lo que podría cambiar la conducta en el sentido de ser más conservadora cada vez. Sin embargo, hoy podemos conocer el pronóstico durante la infancia, en particular el riesgo de ver aparecer una insuficiencia renal terminal. Se estima que las uropatías congénitas son el origen del 27% de las insuficiencias renales terminales (IRT) antes de los 5 años, y del 34% de las IRT antes de los 15 años. 1. MALFORMACIONES RENALES O NEFROPATÍAS CONGÉNITAS • AGENESIA RENAL Es una anomalía rara, se presenta en 1 x 30004000 embarazos. Su diagnóstico es muy difícil de establecer antes de las 15 SA. Unilateral: no presenta consecuencias, está marcada por el descubrimiento fortuito en el 2º trimestre de una fosa renal que no contiene estructura renal ni pedículo vascular ipsilateral. Bilateral: (Sd de Potter). Es una afección letal. Se diagnostica generalmente en el 2º trimestre ante un anamnios; las fosas renales no contienen estructuras renales y la vejiga no es visible; los pedículos vasculares renales no son identificables. Se pueden asociar malformaciones genitales, cardiacas y esqueléticas; el retardo de crecimiento intrauterino y la hipoplasia pulmonar son constantes. • ANOMALÍA DE TAMAÑO ♦ ♦ ♦ La hipertrofia compensadora en un riñón único es habitual. Las hipertrofias bilaterales se observan en el cuadro de una visceromegalia: Sd de Beckwith-Wiedeman. Las hipotrofias renales pueden ser uni o bilaterales. • ANOMALÍAS DE NÚMERO, SITUACIÓN Y DE FORMA • Duplicación ♦ Renal y ureteral ♦ Uréteres normales o hidronefrosis con ureterocele • Situación ♦ Ectopía renal baja: riñón pelviano o ileopelviano: uni o bilateral, directo o cruzado • Fusión ♦ Riñón en herradura: ambos riñones están unidos por su polo inferior Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) ♦ ♦ Riñón sigmoideo: el polo inferior de uno de los riñones está fusionado al polo superior del otro Riñón oval: ambos riñones están fusionados completamente ANOMALÍAS DE ESTRUCTURA • Displasia renal multiquística Cuando es bilateral es incompatible con la vida. Antiguamente se le denominaba Sd Potter II. Se caracteriza por una voluminosa masa ecógena con numerosos quistes corticales de tamaño variable, distribuidos en forma anárquica. Generalmente es unilateral. La función renal depende del riñón contralateral, el que debe ser evaluado minuciosamente. En caso de bilateralidad se presenta con anamnios e hipoplasia pulmonar. • Poliquistosis renal infantil o Sd de Potter I Es autosómica recesiva. Ambos riñones están aumentados de volumen, de aspecto uniformemente ecogénico, sin quistes macroscópicos, con o sin diferenciación corticomedular detectable. Generalmente se presenta con anamnios y la vejiga no es visible. • Poliquistosis renal adulta o Sd de Potter III Es autosómica dominante. Ambos riñones son hiperecogénicos, puede presentar quistes macroscópicos. La función renal in útero es normal, por lo que el líquido amniótico es normal. SÍNDROMES • Meckel – Gruber: es autosómico recesivo. Se caracteriza por presentar riñones hiperecogénicos y aumentados de volumen, encefalocele y polidactilia. • Wiedeman-Beckwith: está relacionado a una neomutación en 11p15.5. se caracteriza por onfalocele, visceromegalia (riñones engrosados). Un diagnóstico antenatal es posible, con el fin de evitar los traumas obstétricos y principalmente la hipoglicemia del recién nacido, que sería la sola responsable del retardo mental. POLIQUISTOSIS RENAL UROPATÍAS OBSTRUCTIVAS Constituyen sin duda las anomalías más frecuentes del sistema urinario. El obstáculo conlleva una dilatación del aparato urinario alto. La dilatación de las cavidades es directamente proporcional al obstáculo y al flujo renal. 139 Se considera que a partir de diámetros anteroposterior mayor a 6 mm antes de las 20 SA, > 8 mm entre 20 y 30 SA, y > 10 mm después de las 30 SA, las anomalías persisten siempre en periodo postnatal. La pielectasia e hidronefrosis son sólo un signo que traducen una patología. El diagnóstico de la dilatación del uréter es posible en antenatal, sea espontáneamente, sea exclusivamente cuando está en fase de llenado. Ante un uréter anormalmente visible, la dificultad es reconocer aquél que presenta reflujo. Un uréter se dilata ya sea porque se vacía mal, sea porque se llena de manera retrógrada, o sea porque es constitucionalmente largo. Sea cual sea la etiología, los grados de dilatación son variables, las repercusiones sobre el aparato urinario alto son de difícil apreciación, el aspecto de la ecoestructura no permite hacer un pronóstico funcional fiable(2). ANOMALÍA UNIÓN PIELOURETERAL Constituyen entre el 20 a 50% de las anomalías obstructivas del aparato urinario. Mientras más estrecha sea la estenosis, más precoz será su aparición. El uréter no será visualizado mientras el débito renal no sea mayor al débito de vaciamiento de la estenosis; es por esto que es una patología de aparición tardía, habitualmente del 3er trimestre. En el 2º trimestre la pielectasia es un signo que hay que considerar, justifica un atento control en buscan de estructuras ecolúcidas abdominales en el 3er trimestre. Es de excelente pronóstico funcional si es unilateral, que es el caso más frecuente. Por lo general, el obstáculo conlleva una disminución del flujo sanguíneo, una disminución de la tasa de filtración glomerular, una disminución de la filtración de K+ y una hipertrofia contralateral compensatoria. La degradación de la función renal es parcialmente reversible tras la liberación de la obstrucción. La ausencia de cálices dilatadas es un factor de buen pronóstico. PIELECTASIAS E HIDRONEFROSIS En las ecografías de las 22 y 32 SA debe ser sistemática la evaluación de los riñones en busca de dilataciones piélicas. La importante heterogenicidad de estudios referente a este tema (retrospectivos o prospectivos, metodología diferente en la medición de la pelvis renal, valor de corte del diámetro de la pelvis a 140 considerar, criterios de vigilancia y de pronóstico post natal, etc), hacen difícil un meta análisis comparativo y sistemático. Este es un signo que en el 2º trimestre se asocia a un aumento en el riesgo de estar frente a una aneuploidía, por lo que la importancia de este signo no sólo radica en su presencia, diagnóstico relativamente simple; sino que si se está ante una de tipo aislada o asociada a otra anomalía: malformación locorregional asociada, ausencia de malformación en otro sistema (canal atrio ventricular y estenosis duodenal), anomalía cromosómica como la trisomía 21. En este último punto, la literatura en los últimos 5 años es más o menos concordante en afirmar que este signo aislado no aumenta significativamente el riesgo de aneuploidía, más aún si la edad materna y la traslucencia nucal la sitúan en la población de bajo riesgo. Capítulo 4 - Obstetricia Figura 63. Dilatación pielocaliciaria con discreta hiperecogenicidad parenquimatosa. Definición Dilatación anormal de la pelvis renal. Epidemiología La incidencia de una pielectasia leve se observa en un 2% de los fetos, independiente de la edad gestacional; y de 1% de los embriones a las 12 SA. Su frecuencia es 2 veces mayor en los varones. Puede ser bilateral en el 20% de los casos, e incluso asimétrica. Criterios ecográficos • Diagnósticos La técnica de medición de la pelvis renal más utilizada y aceptada, es la medición del diámetro anteroposterior sobre un plano de corte axial o transversal del abdomen. Valores de corte de las mediciones de la pielectasia varía según las publicaciones y los textos consultados, pero: • 12 SA: > a 3 mm. • 22 SA: 4 a 6 mm pielectasia moderada; > a 7 mm se considera moderada a severa. • 32 SA: 7 a 9 mm pielectasia leve; 10 a 15 mm pielectasia moderada; > 15 mm pielectasia severa o > a 10 mm pero asociada a dilatación caliciaria (Figuras 63, 64). • Pronósticos ♦ Pielectasia visible a 12 SA: El 50% regresa antes de las 22 SA yel 75% antes de las 32 SA; pero la gran mayoría de las pielectasias descubiertas desde las Figura 64. Dilatación de la pelvis renal bilateral, en el primero se observa aún parénquima renal; en cambio en el segundo, no se le logra distinguir. 16 SA eran inexistentes a las 12 SA. Esto implica que probablemente las pielectasias embrionarias y fetales corresponden a poblaciones y a una fisiopatología parcialmente distinta. ♦ Pielectasia visible a 22 SA: entre las pielectasias leves a las 22 SA, entre el 50 y 80% desaparecen en antenatal, y sólo 4 a 10% persisten en post natal, pero el 10% se agravan y por lo mismo hay que controlarlas a las 32 SA. Entre las pielectasias moderadas a las 22 SA, sólo 10% desaparecen en post natal. ♦ Pielectasia visible después de las 32 SA: el valor de corte considerado varía entre 7 y 10 mm. ♦ Importancia de la pielectasia para el diagnóstico de aneuploidías: a las 12 SA el likelihood ratio para la trisomía 21 es de 8, y por lo tanto aumenta el riesgo basal estimado a partir de la edad materna Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 141 y la traslucencia nucal. En el 2º trimestre, por el contrario es de poca importancia con un likelihood ratio de 1.5. ♦ Importancia de la pielectasia como pronóstico de una hidronefrosis: la importancia de una pielectasia y su evolución en el transcurso del embarazo, son importantes factores pronósticos de una uropatía. Cuando una pielectasia se agrava en el transcurso del embarazo o es superior a 10 mm hacia el término de la gestación o está asociada a una dilatación caliciaria, debe obligadamente seguirse de una vigilancia post natal. mide más de 7 mm de diámetro. Se atribuye a los casos de estenosis congénita de la unión vesicoureteral. En general desde el punto de vista pronóstico, el megauréter se caracteriza por una tendencia a la mejoría(4, 5). Si la manifestación es unilateral, el pronóstico es excelente; en cambio si es bilateral, se debe descartar la presencia de una valva de uretra posterior. La propuesta de una operación va en relación a la evolución posterior, basados en criterios clínicos: sintomatología, infecciones urinarias, criterios ecográficos y cintigráficos. Wickstrom propone un diagrama correlacionando el diámetro anteroposterior de la pelvis renal y la edad gestacional para calcular el riesgo teórico de la presencia de una uropatía post natal. Como sea, la visualización de una pielectasia a las 22 SA no tiene importancia a menos que persista después de las 32 SA(3). A las 32 SA, el valor predictivo de una hidronefrosis postnatal presenta, cuando el corte es de 10 mm, una sensibilidad de 82% y de 100% si el corte es de 7 mm, pero al precio de falsos positivos notoriamente mayor. La tasa de intervenciones quirúrgicas postnatales es muy variable, y se sitúa en alrededor de un 15% para las series más importantes, siendo mayor en los casos en que el valor de corte es mayor a 15 mm hacia el término del embarazo. Es por eso que el corte establecido en 10 mm tendría una adecuada relación entre los pacientes subcidarios de exámenes postnatales, evitando una manipulación inútil de estos niños. El megauréter puede ser: • Megauréter secundario a un proceso obstructivo, no excluyendo el reflujo (Figura 65). Bajo: la vejiga se dilata, se vacía mal y sus paredes son más o menos engrosadas. De la unión pielovesical: la vejiga es normal, la micción es completa. Ureterocele: obliga a la búsqueda de un doble sistema colector o duplicidad renal. Los uréteres están a veces muy dilatados, la hidronefrosis es frecuente. • Megauréter constitucional, no obstructivo, no refluyente: La vejiga y las micciones son normales, con uréter dilatado y las cavidades renales no están o muy poco dilatadas. URETEROCELE Es una inserción ectópica del uréter, donde la extremidad inferior dilatada abomba en la vejiga, el uréter suprayacente se dilata; el ureterocele puede ser Malformaciones asociadas La asociación a defectos cromosómicos es del 1,1% para defectos aislados, mientras que con 2-3 ó más anomalías asociadas la prevalencia es de 5,4%, 23% y 64% respectivamente. Se trata de anomalías normalmente aisladas: • Sistema Nervioso Central: espina bífida. • Cardiovascular: canal atrioventricular. • Digestivo: estenosis duodenal (8%), atresia esofágica (13%) MEGA-URETER Se manifiesta secundariamente a una disfunción de todo o de la parte terminal del uréter que se vacía mal. Este término designa los casos donde el uréter Figura 65. Megaureater (U), el que se observa sinuoso por debajo del riñón y por sobre la vejiga (V). 142 compresivo del bajo uréter contralateral. Obliga a la búsqueda de un doble sistema colector o duplicidad renal. REFLUJO URETERAL El reflujo se acompaña de una ausencia de micción total, se manifiesta por un residuo post miccional, alimentado por los uréteres que se llenan por vía retrógrada después del esfuerzo miccional y que se vacían secundariamente en la vejiga, caracterizándose por un aumento moderado de la dilatación de los uréteres al momento de la micción. Si existe pielectasia, es moderada, sin dilatación importante de las cavidades intrarrenales. La ausencia de dilatación de los uréteres no excluye el reflujo, el que habitualmente no presenta manifestaciones ecográficas antenatales. Diagnóstico diferencial y errores ecográficos de las dilataciones uropatías obstructivas El diagnóstico de las diferentes etiologías es difícil en antenatal. La dilatación del uréter puede ser evidente con imágenes tubulares ecolúcidas que serán diferenciadas de las asas intestinales, o sea sutil con episodios de llenado y vaciamiento de los uréteres, siendo el aspecto habitual del reflujo o del megauréter congénito. Algunos elementos ecográficos son más o menos evocadores y pueden orientar hacia algunas patologías: • Pielectasia aislada del 2º trimestre: obliga a un examen atento y meticuloso en el 3 trimestre en busca de dilataciones ureterales. • Hidronefrosis: se asocia rara vez a un megauréter no obstructivo o a reflujo, la dilatación del uréter tiene un rol protector del riñón. • Dilatación precoz del uréter: obliga a la búsqueda de un ureterocele con o sin duplicidad del sistema colector. • Dilatación de uréter asociada a hidronefrosis: evoca un sd obstructivo de la unión vesico-ureteral si la vejiga es normal con micciones completas; si está asociada a una megavejiga o una vejiga con “signos de lucha” (paredes engrosadas e hipertróficas), se trata probablemente de una valva de uretra posterior. • Riñón con parénquima hiperecogénico: evoca una displasia secundaria a un proceso obstructivo, pero este aspecto no permite afirmar un pronóstico funcional. Capítulo 4 - Obstetricia MEGAVEJIGA Generalmente una megavejiga que se manifiesta tempranamente en el embarazo como a las 12 SA, regresa espontáneamente si mide menos de 12 mm de diámetro, pero si se asocia con otras anomalías, se asocia a un 40% de cromosomopatías(1). Si este signo aparece en el 2º trimestre (Figura 66), puede pertenecer a un amplio grupo heterogéneo de patologías, donde el diagnóstico por lo general no es fácil. Siempre hay que realizar el diagnóstico diferencial entre reflujo primario, cloaca, duplicación ureteral y megavejiga – microcolon(6) existen otras anomalías más raras aún, como son síndromes familiares de miopatía visceral, que se caracterizan por seudoobstrucciones digestivas y urinarias a nivel vesical(7). • SÍNDROME DE PRUNNE-BELLY Una obstrucción crónica y la disfunción en el vaciamiento vesical provocan riñones displásicos. La vejiga, los uréteres y la pelvis renal se encuentran muy distendidos. Figura 66 a, b. Se observa megavejiga, la que se confirma al visualizar las arterias umbilicales que rodean esta masa anecoica. Examen ecográfico morfológico fetal anormal en el segundo trimestre (18-23 semanas) 143 Se desconoce su real causa, pero se piensa que pudiera deberse a una obstrucción uretral asociada a una ausencia de musculatura lisa urológica y musculatura abdominal; esto ocasiona una falta de peristaltismo vesical y uretral, impidiendo su vaciamiento. Estas características otorgan en el recién nacido un aspecto muy particular denominándose: “abdomen en ciruela”. Ecográficamente: el cuadro se caracteriza por una megavejiga y un oligoamnios severo a anhidramnios, puede haber criptorquidia. El pronóstico es muy malo, con un 20% de mortalidad en el primer mes de vida. SÍNDROME MEGAVEJIGA–MICROCOLON HIPOPERISTALSIS Se trata de una asociación de malformaciones que demuestran una megavejiga con hidronefrosis, con liquido amniótico normal o aumentado hacia el polihidroamnios(8). Se presenta en el 80% de los casos en fetos femeninos(9). VALVA URETRA POSTERIOR Se trata de la uropatía congénita más grave, que compromete siempre la función renal global. Se les diagnóstica en antenatal en el 50% de los casos, siendo los casos más graves los que se manifiestan con dilatación alta del sistema. Debe ser considerada como una urgencia neonatal. Se caracteriza por un aumento de volumen vesical con paredes engrosadas, dilatación de la uretra proximal, lo que da un aspecto infundibular (Figura 67 a, b). Antes de la aparición del Diagnóstico Antenatal (DAN), la valva de uretra posterior presentaba un 60% de mortalidad. Hoy la mortalidad neonatal es de 0 a 3% relacionada con la insuficiencia renal o respiratoria. La mortalidad en la infancia es todavía de 6% antes de los 12 años(10). Los principales indicadores pronósticos en antenatal son: diagnóstico precoz antes de 24 SA, oligoamnios y una tasa de Beta 2 microglobulina urinaria fetal > 5 mg/ml. La función renal está alterada en periodo neonatal en 70% de los casos, y antes de 3 meses en 80% de los casos. Esta alteración es transitoria en el 60% de los casos. Cuando la tasa de creatinina sérica es constantemente superior a 8 mg/dl durante el primer año de vida, el riesgo de insuficiencia renal terminal es de 67 a 100%. Globalmente el riesgo de IRT es de 20 a 30% entre los 10 y 15 años. Figura 67 a, b. Obsérvese el cuello vesical con las paredes engrosadas de la vejiga. VESICOCENTESIS Crombleholme(11) Jonhson(12) Osmolaridad Na+ Cl – Ca 2+ B2 microglobulina Proteína total < 210 mosm/l < 100 meq/l < 90 meq/l < 2 mg/dl < 2 mg/dl ——— < 200 mosm/l < 100 mg/dl < 90 mg/dl < 8 mg/dl < 6 mg/dl < 20 mg/dl Según Smith(11) el riesgo de IRC es de 34% a 10 años y de 51% a 20 años, y el riesgo de IRT de 10% a 10 años y de 38% a 20 años. En el 75% de los casos existen anomalías urodinámicas. El porcentaje de incontinencia diurna y nocturna es importante: 81% a 5 años, 56% a 10 años y 1% a 20 años(11). 144 EXTROFIA VESICAL Definición Cierre incompleto, a nivel de la linea media, de la pared abdominal inferior sub umbilical y de la pared anterior de la vejiga, produciendo una protrusión de la pared posterior de la vejiga (Figura 68). Figura 68. Se observa un defecto de la pared abdominal inferior, con exposición de la mucosa vesical. Además este paciente presenta una cloaca persistente. Epidemiología Es una patología poco frecuente, se observa 1 x 50.000 nacidos. Criterios ecográficos • Diagnósticos Alteración de la pared inferior abdominal, con imposibilidad de observar la vejiga. Se demuestra la ausencia vesical al observar al Doppler color las arterias umbilicales. • Pronósticos Generalmente estos niños presentan asociada una cloaca persistente, con anomalías genitales y recto-anales. Capítulo 4 - Obstetricia Malformaciones asociadas Se puede asociar a una epispadia, una disyunción sinfisio-pubiana, una división del pene y del clítoris y a una larga separación de los labios o bolsas escrotales. BIBLIOGRAFÍA 1 Gasser B, Lindner V, Bussière L, Faure C, Istier L, Vetter JM, Laborde K. L’appréciation du parenchyme rénal en amont d’une uropathie: Les lésions anatomopathologiques. Les malformations congénitales. Diagnostic anténatal et devenir. Sauramps janv 2002; 17-24. 2 Veyrac C. L’appréciation du parenchyme rénal en amont d’une uropathie: possibilités et insuffisances de l’imagerie. Les malformations congénitales. Diagnostic anténatal et devenir. Sauramps janv 2002: 25-32. 3 Wickstrom E, Maizels M, Sabbagha RE, Cohen LC, Pergament E. Isolated fetal pyelectasis: assesment of risk for postnatal uropathy and Down syndrome. Ultrasound Obstet Gynecol 1996; 8: 236-40. 4 Porto M, McGahan JP. The fetal abdomen and pelvis. In McGahan JP, Porto M (eds): Diagnostic Obstetrical Ultrasound. Philadelphia, JB Lippincott 1994; p343. 5 Romero R, Pilu G, Jeanty P. Prenatal Diagnosis of Congenital Anomalies. East Norwalk, CT, Appleton & Lange 1998. 6 McHugo J, Whittle M. Enlarged fetal bladders: aetiology, management and outcome. Prenat Diagn 2001; 21(11):958-963. 7 Guze CD, Hyman PE, Payne VJ. Family studies of infantile visceral myopathy: myopathic pseudo- obstruction syndrome. Am J Med Genet 1999; 82(2): 114-122. 8 McHugo J, Whittle M. Enlarged fetal bladders: aetiology, management and outcome. Prenat Diagn 2001; 21(11) 958-963. 9 De Vaux-Boitouzet V, Barau G, Blin G, Bulwa S, Engelmann P. Megabladder. Microcolon. P r e n a t a l ultrasonic diagnosis and review of the literature. A propos of a case. J Gynecol Obstet Biol Reprod (Paris) 1990; 19 (3): 327-332. 10 D u c k e t t J W , G a z a k J M . C o m p l i c a t i o n s of ureterosigmoidostomy. Urol Clin North Am. 1983;10(3):473-81. 11 Crombleholme TM, Harrison MR, Golbus MS. Fetal intervention in obstructive uropathy: Prognostic indicators and efficacy of intervention. Am J Obstet Gynecol 1990; 162: 1239. 12 Johnson MP, Bukowski TP, Reitleman C. In utero surgical treatment of fetal obstructive uropathy: A new comprhensive approach to identify appropiate candidates for vesicoamniotic shunt therapy. Am J Obstet Gynecol 1994; 170: 1770.