Química Orgánica-III
Anuncio

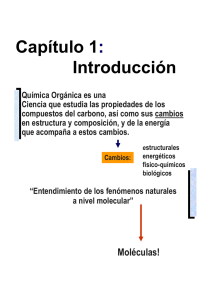
Materia : Química Orgánica-III Texto-guía: Mecanismos de las Reacciones Orgánicas en Esquemas y Tablas (Recapitulación de las conferencias) Prof. Vladímir V. Kouznetsov Escuela de Química UIS 2006 “Aprender sin reflexionar es malgastar la energía” Prefacio Querido lector, lo que Usted tiene en sus manos, no es libro ”clásico” sobre la Química Orgánica, sino un texto-guía que pueda ayudarle a orientarse en ciertos problemas de la Química Orgánica. Este guía es un andamio para conseguir los conocimientos de algunos mecanismos de las reacciones orgánicas. En la literatura química se encuentran muchos libros de excelente calidad; casi todos son obras voluminosas, en donde los estudiantes de carrera de Química pueden “perderse” fácilmente. Por qué? Por la simple razón – la Química Orgánica crea su materia, - moléculas orgánicas, y creándolas, trata de estudiarlas. Cada año se añade la nueva información de las reacciones orgánicas, los métodos de obtención de moléculas orgánicas y sus propiedades. Cómo se puede “orientarse en este universo molecular de información abundante y creciente”? El camino único es clasificar moléculas orgánicas y sus transformaciones. Así, al comienzo del estudio de la Química Orgánica, Usted conoció ciertos tipos de compuestos orgánicos que contienen grupos funcionales, los cuales hacen distinguir las propiedades químicas y biológicas de las moléculas. Al aprender las propiedades químicas de ciertas clases de compuestos, se puede notar que estas propiedades químicas (transformaciones químicas) dependen de la estructura molecular en dónde el grupo funcional tiene un papel influyente para definir estas propiedades. Las transformaciones químicas de compuestos orgánicos se conocen como las reacciones. Como hay tantos compuestos orgánicos (naturales y sintéticos), se tiene otro cartapacio de reacciones orgánicas que dan posibilidad de estudiar moléculas orgánicas químicamente . También hace falta tenerlas clasificadas para poder realizar estos estudios, para predecir los resultados de estas transformaciones, al fin, para entender mejor la Química Orgánica. La quintaesencia del estudio de estas transformaciones químicas es una descripción detallada cómo una molécula se transforma en otra, es decir, el estudio de mecanismos de reacciones orgánicas. En este texto-guía, que será soporte para entender el último curso de Química Orgánica de Carrera de Química en la UIS, se presta mucha atención a los aspectos mecanísticos de de reacciones orgánicas más generales. Buscando el entendimiento mejor de los procesos en la Química Orgánica, este curso se enfoca a las preguntas “Cómo ocurren estas transformaciones ?”, “Cómo se puede confirmar nuestras suposiciones de posibles mecanismos ?” El conocimiento de mecanismos de reacciones orgánicas le ayuda a tener una visión completa de Química Orgánica y sea útil para entender mejor otros procesos, por ejemplo, bioquímicos. A diferencia a los cursos de Química Orgánica-I y II (donde se estudian las propiedades de clases de compuestos orgánicos), el curso de Química Orgánica-III tiene tarea de entender el comportamiento de moléculas orgánicas a través de sus transformaciones químicas, aplicando los criterios del estudio de mecanismos de reacciones orgánicas. Este texto-guía tiene cinco partes: primer capítulo es introductorio dónde se acuerdan los principios químicos más generales que podrían ser útiles en el aprendizaje del material de este curso; segundo capítulo se dedica a las transformaciones químicas más generales de los compuestos alifáticos, prestando atención a las reacciones de sustitución y reacciones de eliminación; en el tercer capítulo se discuten las reacciones de adiciones al doble enlace y las reacciones de cicloadición. El cuarto capítulo está dedicado a las reacciones más generales de los compuestos aromáticos. En último capitulo se discutan las reacciones de RedOx y las transpociones, El contenido completo de este curso está en el programa que viene a continuación. También se encuentra la lista de libros (obligatorios y optativos) que son “su manantial principal de los conocimientos sobre moléculas orgánicas”. Todo este material representa a unos apuntes del curso en forma de “transparencias” que se exponían durante últimos diez años. A lo largo de este curso se presta atención a las múltiples aplicaciones de moléculas orgánicas, que se puede planear a sintetizar, conociendo los mecanismos generales de reacciones orgánicas, es decir se presta mucha atención a la importancia de la síntesis orgánica. Así, este curso está ligado fuertemente a las actividades en laboratorio Química Orgánica-III. Se espera que al terminar este curso, al lector (que no fue “solo receptor pasivo”) le completan sus conocimientos básicos sobre la Química Orgánica. Entonces, le invito a comenzar reflexionar sobre los procesos químicos que sufren moléculas orgánicas, en general y sobre los mecanismos de las reacciones orgánicas que ayudan a entender generar la diversidad molecular de Química Orgánica, en particular. Prof. Vladímir V. Kouznetsov, PhD, DSc PROGRAMA Capítulo 1: Introducción. Tema 1. Clasificación de las reacciones orgánicas. Sistemas de clasificación: según la ruptura de enlaces (Homólisis y Heterólisis), según la naturaleza de los intermediarios (Reacciones polares y reacciones no polares), según las estructuras de los compuestos participantes en la reacción [Sustitución, Eliminación, Adición (Cicloadición), Transposición]. Reacciones pericíclicas. Tema 2. Criterios en el estudio de un mecanismo y clasificación de los reactivos e intermediarios. Métodos para determinar los mecanismos de reacción orgánica. Demostración de la existencia de productos intermedios. Identificación de los productos intermedios y los productos finales de una reacción. Empleo de isótopos. Nucleófilos y Electrófilos. Fuerza de los ácidos y de las bases. Nucleofilia y basicidad. Radicales libres. Algunos estados intermediarios. Iones carbonio. Carbaniones. Radicales de carbono. Carbenos y nitrenos. Capítulo 2: Química de los compuestos alifáticos. Tema 3. Sustitución Sustitución nucleófila SN. Concepto de Nucleofilia relativa. Constante de Nucleofilia (Swain y Scott). Mecanismos de la SN alifática: Sustitución nucleófila bimolecular SN2. Características cinéticas. Características estéricas. Sustitución nucleófila unimolecular SN1. Características estéricas. Factores que determinan el mecanismo de las reacciones SN: 1) La estructura del substrato; 2) Papel del disolvente; 3) La estructura del nucleófilo. Criterios para distinguir mecanismos SN1 y SN2. Mecanismo SNi. Mecanismo SN2 contra mecanismo SN1. Uso práctico de estas reacciones. Reacciones competitivas en la sustitución nucleófila: eliminaciones y transposiciones. Tema 4. Eliminación. Eliminaciones. Reglas de Saytzeff y Hofmann. Regla de Bredt. Mecanismos de eliminación E1, E2 y E1cB: efectos de temperatura, disolvente y base. Estereoquímica del mecanismo E2. PROGRAMA. continuación Capítulo 3: Química de los compuestos olefínicos y de los compuestos carbonílicos. Tema 5. Adición electrófila y cicloadición. Mecanismos de adición a dobles y triples enlaces carbono-carbono. Regla de Markovnikov. Estereoquímica de la adición. Efecto de Karasch. Hidroboración-oxidación (método de Brown). Adición a sistemas conjugados (1,3-Dienos). Productos de adición 1,2- y 1,4-. Reacción de cicloadición [4+2] (reacción de Diels-Alder) : mecanismo, reglas de Alder, estereoquímica de la cicloadición. Reacción de cicloadición dipolar [3+2]. Aplicación en la síntesis orgánica fina. Noción de metátesis. Su uso sintético moderno. Tema 6. Adición nucleófila a los compuestos carbonílicos. Adición nucleófila al grupo carbonilo. Mecanismos. AdN de nucleófilos nitrogenados (aminas, oximas etc) y oxigenados (agua, alcoholes), de carbaniones (compuestos organometálicos) e hidruros. Estereoquímica de la adición. Regla de Cram. Reacción de Cannizzaro como ejemplo de la adición del hidruro. Reacciones de condensación aldólica : regioselectividad, catalizadores, complicaciones. Condensación crotónica. Adición a las α,β-enonas. Reacción de Baylis-Hillman. Uso práctico de estas reacciones. Capítulo 4: Química de los compuestos aromáticos. Tema 7. Sustitución nucleófila sobre núcleos aromáticos. Sustitución del hidrógeno. Compuestos de diazonio aromático. Sustitución de halógenos sobre núcleo aromático. Mecanismos SN2Ar y SN1, mecanismo “bencínico”. Estructura y estabilidad de los complejos de Meizenheimer. Uso práctico de estas reacciones. Sustitución nucleófila aromática vía catálisis de metales transitorios: reacción de Heck, reacción de Stille, reacción de Suzuki, reacción de Sonogashira. Tema 8. Sustitución electrófila sobre carbonos aromáticos. Mecanismo SE2Ar. Estructura y estabilidad de los complejos de Wheland. Nitración. Sulfonación. Halogenación. Reacciones de Friedel-Crafts. Formilación. Efecto de los sustituyentes presentes sobre una nueva substitución en el núcleo: grupos del primer genero, grupos del segundo género. Uso práctico de estas reacciones. PROGRAMA. continuación Capítulo 5: Reacciones de RedOx y algunas transposiciones de importancia sintética. Tema 9. Transposiciones. Noción de la reacción electrocíclica. Transposiciones moleculares (Tr). Tr con la participación del ion carbonio: transposiciones de Beckmann, Schmidt, Hofmann, Favorskii, Baeyer-Williger. Tr con la participación de carbaniones: transposiciones de Stevens, Wittig, transposición bencílica. Tr sigmatrópicas: transposición de Claisen, Cope, Frier, transposición bencidínica. Tema 10. Reacciones RedOx. Clasificación. Número de oxidación. Sistemas RedOx-I, sistemas RedOx-II. Oxidaciones comunes: conversión de los alcoholes en aldehídos, cetonas y ácidos carboxílicos. Aperturas oxidativas de los glicoles y alquenos. Reducciones de las cetonas con metales. Reducciones de los derivados de ácidos carboxílicos con los hidruros mixtos. Reacciones RedOx bioquímicas. Nociones generales. Bibliografía: Obligatoria R.T. Morrison, R.N. Boyd “Química orgánica”, 5ª edición, 1998 - ( MB). P. Sykes “Mecanismos de reacción en química orgánica”, 1985 - (Sykes, 1985). K. P.C. Vollhardt, N.E. Schore, ”Organic Chemistry”, 2003 - (VS). F. A. Carey, “Química Orgánica”, 3a edición, 1999. F.A. Carroll “Perspectives on Structure and Mechanism in Organic Chemistry”, 1998. Optativa M.B. Smith, J. March “Advanced Organic Chemistry”, 5a edición, 2001 - (SM). F.A. Carey, R.J. Sunberg “Advanced Organic Chemistry, Parte A”, 3a edición, 1990 - (CS). P. Sykes “Investigación de mecanismos de reacción en química orgánica”, 1982 - (Sykes, 1982). R. Bruckner “Advanced Organic Chemistry. Reaction Mechanisms”, 2002. Capítulo 1: Introducción Química Orgánica es una Ciencia que estudia las propiedades de los compuestos del carbono, así como sus cambios en estructura y composición, y de la energía que acompaña a estos cambios. Cambios: estructurales energéticos físico-químicos biológicos “Entendimiento de los fenómenos naturales a nivel molecular” Moléculas! Introducción Estructura Reacción Mecanismo son tres aspectos fundamentales en la Química Orgánica Química orgánica tiene su idioma (estructura y reacción) y su lógica (mecanismo), basándose en el comportamiento físico-químico de las moléculas. Moléculas, átomos y enlaces... Enlace iónico Enlace covalente (de pares de electrones compartidos) (Lewis, 1816) Enlace covalente polar Ejemplos Cl Cl + Enlace covalente coordinado HCl - Na Cl CCl4 H3C CH3 NH3 H3C Cl (L. Pauling, “The Nature of the Chemical Bond”, 3 ed., Corhell University Press, Uthaca, NY, 1960) Introducción Una reacción orgánica Es la interacción entre unos compuestos orgánicos (sustratos) en ciertas condiciones o también la transformación de un compuesto orgánico en otro(s) (productos finales) en presencia de un compuesto (un reactivo) que puede ser orgánico o inorgánico Una característica importante de las reacciones orgánicas es la ruptura del enlace químico de las moléculas Durante la realización de reacciones químicas, moléculas (sustratos y reactivos) sufren cambios estructurales Una síntesis Construcción intencional de las moléculas orgánicas por medio de reacciones químicas Introducción Clasificación estructural Según las estructuras de sustratos y de productos obtenidos: Sustrato Producto final Sustituciones (S), Adiciones (Ad), Eliminaciones (E), Transposiciones (Tr) Oxidaciones-Reducciones (Red-Ox) Syke (1985), p. 30-32 Ejemplos... A-B Ad: E: Tr: G´ G S: A G, G´ – grupos funcionales B A B - A-B “Herramientas necesarias” para efectuar la reacción: Sustratos, Reactivos, Disolvente, “Ayudante” (Catalizador o Inhibidor) Esta clasificación abarca casi todas las reacciones orgánicas, pero no aclara bien sus aspectos mecanísticos Introducción “Más al fondo...” Clasificación química Estos cambios se pueden clasificar de la siguiente manera: Según el modo de ruptura de los enlaces en una molécula A-B Heterólisis Homólisis A: B A :B Al romper el enlace químico, pueden formarse los intermediarios iónicos (polares) y radicalares (no polares) A- / B+ A+ / B- A./ B. Introducción Sin embrago ! Existe un grupo grande de reacciones que transcurren sin formación de iones, ni radicales y se llaman REACCIONES PERICÍCLICAS. Se puede ahora sumar estas observaciones en la tabla siguiente: REACCIONES POLARES RADICALES PERICÍCLICAS Ad, S, E, RedOx, Tr, Cad Introducción Naturaleza de los reactivos y clasificación de reacciones orgánicas Carey, p. 274 Sykes (1985), p. 28-30 Nucleófilos- :Nu reactivo debe tener por lo menos un par de electrones en los orbitales p- o n- no compartidos o centros con la carga electrónica alta. Electrófilos- E+ reactivo debe tener por lo menos un orbital vacante o centros de poca densidad electrónica Radicales- R. especies que contienen electrones desapareados Tipos de las reacciones según “los participantes” S: SN SE Srad Ad,E: Tr: TrN Trrad Nucleófilas Electrófilas Radicalarias Introducción Mecanismo de reacción en química orgánica El mecanismo es una descripción detallada del camino por el cuál transcurre la reacción. ? ? S ? P Etapas elementales; Intermediarios; Estado de transición Para conocer el mecanismo completo es necesario: Saber la situación exacta de los reactivos en el sistema durante toda la reacción; Conocer la naturaleza de interacción ó de formación de los enlaces entre los átomos que participan en esta interacción; Saber la energía del sistema en todas las etapas de la reacción; Saber la velocidad de los cambios que transcurren en el sistema. Introducción Este volumen de información supera lo que se sabe hasta ahora de cualquier reacción. ??? (es mucho) Simplificación Por eso, nos vemos obligados a limitar el problema de estudio del mecanismo. Vamos a opinar que “sabemos el mecanismo” cuando todos los intermediarios que se forman en la reacción son conocidos y cuando se puede, en términos generales, a indicar cómo ocurre cada etapa separada de la reacción compleja. Esta simplificación es posible sólo cuando se sabe cuales son los átomos que atacan a otros, qué fácil es este ataque y que tipo de enlaces se forman (se rompen) en etapas separadas. Introducción SM, p. 218 Carroll, p. 297 Intermediarios de reacciones orgánicas • • • • • • Carbocationes Carbaniones Radicales libres Carbenos Nitrenos Bencinos Son especies reales: 1. Tiempo de vida: inestabilidad; 2. Deficiencia en e-s para formar un octeto estable. Introducción Carbocationes (IUPAC) MB, p. 192-195 Iones de carbenio trivalentes tricoordinados CH3+: iones clásicos SM, p. 580; Carroll, p. 284 Iones carbonio hipercoordinados CH5+ : iones no-clásicos J. Am. Chem. Soc., 1972, 94, 808; J. Chem. Edu., 1986, 63, 930-933 El concepto de cationes alquilo como intermediarios fue desarrollado por Meerwein (1902), Ingold y Whimore (años 30-50 del siglo XX), Olah (años 60-80 del siglo XX) G.A. Olah, Premio Nobel de Química, 1994. “For his contribution to carbocation chemistry” Olah G.A., “My search for Carbocations and Their Role in Chemistry (Noble Lecture)”, Angew. Chem. Int. Ed. Engl., 1995, 35, 1393-1405; Olah G.A., “100 Years of Carbocations and Their Significance in Chemistry”, J. Org. Chem., 2001, 66, 5943-5957; Olah G.A., J. Org. Chem., 2005, 70, 2413-2420. Introducción Carbocationes: aspectos generales SM, p. 218 Carroll, p.283 A. Iones clásicos: σ-carbocationes π-carbocationes ∃Geometría planar; ∃ Estabilidad relativa; ∃ Generación + B. Iones no-clásicos: iones carbonio iones cíclicos tipo catión 2-norbornil + OSO2C6H4Br-p H H “Tres centros para compartir carga electrónica” Norbornano (Biciclo[2.2.1]heptano) Introducción Problema del ion metonio Estructura ? Trabajo inicial de Olah (1987) CH5+ EM, IR “Modelo de trípode: CH3+ + H2 “ Ver animación: www.theochem.rub.de/go/ch5p.html Superácidos CH4 CH5+ ? Superácido? Ácido mágico? Introducción SM, p. 227 Carbaniones: aspectos generales ∃ Geometría piramidal; ∃ Estabilidad relativa; ∃ Generación .. Todos carbaniones son bases Rbase Ácidos conjugados débiles R-H (Pirámide de Keops) ácido conjugado Base fuerte Estabilidad baja del carbanión Introducción SM, p. 238 Radicales: aspectos generales Radicales “libres” ( de quién?) “Carboradicales” ∃ Geometría “mixta” planar o piramidal; ∃ Estabilidad relativa; ∃ Generación . . Son especies paramagnéticas o o “Heteroradicales” Ejemplos: difenilpicrilhidrazil radical; nitróxido radical; fenoxi radicales Introducción “Carboradicales” Método de Resonancia Espin Electrónico (R.S.E.) (>1945) Orientación del espin de un electrón no apareado en un campo magnético variable externo CH3. Ubicado en un campo magnético y sometido a radiación electromagnética Sensibilidad – hasta 10-12 mol/L Desdoblamiento de las señales (3+1)=4 1: 3 : 3 :1 (igual como en RMN) Introducción “Heteroradicales” Fenoxi radicales CMe3 CMe3 PbO2 Me3C Me3C OH O . CMe3 CMe3 Muy estables, Inhibidores de procesos radicales Difenilpicrilhidrazil radical O2N . N N O2N NO2 Introducción Carbenos : ∃ Geometría SM, p. 247 ∃ Generación El metileno es el carbeno más simple : CH2 ( ) singulete : CH2 ( ) triplete Utilidad sintética: reactivo de Simmons-Smith CH2I2 – Zn/Cu, Et2O -Ciclopropanación Su “pariente” :CCl2 ¿Su estado electrónico? “Carbenos –campeones” CF3 .. Br Br CF3 J. Am. Chem. Soc., 1999, 121, 10213. Ph N N Ph : N Ph Se vende ! Introducción Criterios del estudio de un mecanismo Sykes (1985), p. 43-44 Cuatro criterios: 1 El mecanismo debe explicar la formación de los productos finales de la reacción. Es bien evidente, pero la importancia de este criterio consiste en que el mecanismo debe explicar no sólo la formación de los productos, sino también su estereoquímica y además, los resultados del estudio de la reacción con compuestos que tengan átomos isotópicos (marcaje isotópico). 2 En un mecanismo de varias etapas donde se supone la formación de unos intermediarios, es deseable la comprobación de su existencia por métodos químicos o físico-químicos. Sin embargo, en todos los casos, estos intermedios deben llevar a la formación de los productos finales obtenidos. Si no es así, el mecanismo puede ser falso. 3 El mecanismo debe explicar la influencia de los cambios de las condiciones de reacción. Más importantes son los cambios del pH, de la temperatura, de la naturaleza del disolvente y la influencia del catalizador sobre el carácter de los productos y sobre la velocidad de reacción. Eso significa que el mecanismo debe explicar: 4 El mecanismo debe explicar la cinética de la reacción. Es el caso particular, pero muy importante del criterio anterior. Uso cinético del marcaje isotópico. Introducción 1 El mecanismo debe explicar la formación de los productos finales de reacción Determinación (confirmación) de la estructura del producto final A. Si es conocida (descrita en la literatura química) a veces basta comparar los datos físicos B. Si es desconocida (nueva en la literatura química) hace falta analizar su estructura por métodos físico-químicos: IR, UV, RMN (1H, 13C, .. ), EM, CG-EM, RSE, DRX Cualquier producto (incluyendo secundario) de una reacción orgánica debe ser analizado (determinado) Introducción 1a Estudio de la estereoquímica del producto final Sykes (1985), p. 50-51 Br2 Br CCl4 Br Trans o Cis?¿ Introducción “Marcaje isotópico” 1b Sykes (1982), p. 38-49 Estudios con sustratos marcados (trazadores) Uso no cinético Uso cinético D, T, 13C, 14C, 15N, 18O, 32P, 35S, 37Cl, 131I George de Hevesy, Premio Nobel de Química, 1943. J. Chem. Edu., 2001, 78, 301 Para recordar: Isótopo es una de dos (o más) formas (especies) de un elemento que tienen el mismo número atómico, pero distintas masas atómicas. La diferencia en la masa se debe a la presencia de uno o más neutrones extra en el núcleo.. Introducción 1b Ejemplos!! Hidrólisis básica de los esteres Trasposición de Claisen ? ? MB., p. 862 Carroll, p. 325 Marcaje isotópico con carbono Utilización valiosa en las investigaciones bioquímicas y biomédicas: 1. 2. 3. Fotosíntesis: empleo de 14CO Biogénesis de los productos naturales Rutas metabólicas de los fármacos Introducción 1b Efecto isotópico primario Uso cinético para determinar la etapa más lenta del proceso. KH/KD ~ 6.9-10.0 KH/KT ~ 16-60 K12C/K13C ~ 1.022-1.250 K12C/K14C ~ 1.5 K14N/K15N ~ 1.14 K16O/K18O ~ 1.19 “Efecto isotópico de deuterio” D-C, D-O, D-N > H-C, H-O, H-N A mayor masa, más fuerte es el enlace Para romper esos enlaces se necesita más energía!! Gráfica! Introducción 1c Carroll, p.323 Experimentos de cruzamiento Reacciones cruzadas (experimentos de cruzamiento) son una herramienta muy útil e importante en el estudio de los mecanismos Ejemplos… Introducción 2 Estudio de los intermediarios de reacción Sykes (1985), p. 48-50 S + R [I] P Tres técnicas comunes: 4 Aislamiento Detección (UV, IR, RMN, RSE, EM) 4 Atrapado (Trampas químicas) Introducción La mayoría de las reacciones orgánicas transcurren a través de uno o más intermediarios, o sea, consisten en varias etapas elementales Un paso elemental 0.01 nm 10-100 fs Sykes (1982), p. 7-8 MB, p. 30-69 CS, p. 192-194 ET “ET” – estado quasi termodinánico con max. G o en él no existe especies moleculares distintas sino una agrupación única E Eact su fuente es la energía cinética de las partículas en movimiento. Cada reacción requiere colisiones de partículas con energía suficiente y de orientación apropiada. S 1 nm 1-10 ns P χ Estudio del ET con el método UED (ultrafast electron diffraction) (“Femtoquímica” 10-15 s - Chem. Rev., 2004, 104, número 4) A.H. Zewail, Premio Nobel en Química, 1999. Introducción Reacción de dos pasos elementales CS, p. 193 Sykes (1985), p. 40 En diferentes gráficas: ET1 I – un intermediario E I ET2 S P χ ¿Cuál diferencias existe entre ambas gráficas? Introducción 3 Influencia de los cambios de las condiciones sobre la velocidad de la reacción Cuatro factores: 4 Temperatura Ecuación de Arrenius: K = A.e -Eact/RT 4 Disolvente 4 pH (Catálisis ácida, catálisis básica) 4 Catalizador Introducción 3b CS, p. 232-236 Influencia del disolvente Disolventes apróticos no polares: CCl4 , CS2, ciclohexano etc. Disolventes apróticos bipolares: acetona, acetonitrilo, DMSO, HMPT, DMF, NMP, DMPU, piridina, esteres, CHCl3 etc. Disolventes próticos: H2O, NH3, ROH, RCOOH etc. Las reacciones en solución, incluso con ruptura hemolítica de los enlaces, ocurren más rápido y a temperatura más baja que las mismas en fase gaseosa. La polaridad de los disolventes puede afectar a las rutas de reacciones. Introducción 3d Influencia de la catálisis Sykes (1985), p. 42 En gráficas: E χ Introducción 4 Sykes (1982), p. 11-20 Sykes (1985), p. 36-39 Cinética: La expresión matemática de la relación Velocidad ≅ Concentración xX aA + bB Sustratos + yY Productos finales C C χ χ Etapa determinante.. Velocidad total de una reacción de varias etapas de la velocidad de una etapa más lenta de todas... Introducción Cinética y termodinámica ∆G0 =∆H0-T∆S0 Reacción favorable ∆G0 < 0 (∆H0 < 0, reacción exotérmica) Reacción altamente favorable ∆G0 << 0 (∆H0 < <0, reacción muy exotérmica) Reacción desfavorable ∆G0 > 0 (∆H0 > 0, reacción endotérmica) Gráficas! Introducción Postulado de Hammond CS, p. 211-215 Una teoría de ET que explica la relación entre estructura y reactividad en unas reacciones orgánicas de manera cualitativa (J. Am. Chem. Soc., 1955, 77, 334) Para una reacción de una sola etapa, la geometría de ET se parece al lado que está cerca en energía libre. (Idea similar fue propuesta antes por J.E. Leffler, 1953) Introducción Postulado de Hammond “Si dos estados ocurren consecutivamente durante una reacción y tienen casi el mismo contenido de energía, su interconversión necesita solo una pequeña reorganización molecular” Reacción exotérmica E E ET Reacción endotérmica ET ET P R P P χ R R χ Introducción Control cinético versus Control termodinámico CS, p. 209-210 Sykes (1985), p. 42-43 Sykes (1982), p. 33-37 La composición de los productos pueda determinarse por el equilibrio termodinámico. En este caso, la composición está gobernada por el control termodinámico. Alternativamente, la composición de los productos pueda determinarse por la competencia de velocidades de la formación de los productos. En este caso, esta composición se determina por el control cinético. B E C A A Producto inicial B C χ C – producto que se forma un poco más rápido B – producto termodinámicamente mas estable Si dos reacciones son irreversibles, C se forma más por que él se forma más rápido (Control cinético). Si esas reacciones son reversibles, B será casi siempre único producto cuando se permite alcanzar el equilibrio (Control termodinámico). Introducción Orden y Molecularidad ! La molecularidad de una reacción se define como el número de especies (moléculas, iones, etc) que sufren necesariamente el cambio de covalencia en el paso determinante de la velocidad * reacción monomolecular * reacción bimolecular * reacción trimolecular ! El orden de una reacción es determinado experimentalmente A+B P V = k [A]α [B]β (α + β) - orden * reacción de primer orden * reacción de segundo orden Introducción Ácidos y Bases SM, p.338-351 Sykes (1985), p. 52-74 Teoría de Brønsted Donador de H+ AH ácido Aceptor de H+ Relación de aceptor y donador Aceptor de un par electrónico B base Donador de un par electrónico Teoría de Lewis Los ácidos de Lewis son reactivos electrófilos Los bases de Lewis son reactivos nucleófilos Introducción Ácidos y Bases SM, p.338-351 Sykes (1985), p. 52-74 Teoría de Brønsted: la fuerza de acidez es una tendencia para dar un H+; la fuerza de basicidad es una tendencia para aceptar un H+; se valora a través de pKa (o pKb). Teoría de Lewis: la definición de la fuerza de acidez (basicidad) es difícil, solo se cuenta con los valores relativos Ácidez aumenta HF–SbF5, FSO3H–SbF3–SO3, FSO3H–SbF5, FSO3H pKa << -10 HClO4, HI, H2SO4, HBr, HCl ,ArSO3H pka ~ -10÷ -1 CH(CN)3, HNO3, ……, HCN pKa ~ -2÷+ 9 PhCH3, PhH, CH2=CH2, ……, CH4 pKa ~ 43- 50 La definición de la fuerza de las bases por medio de pKa es más frecuente, así se tiene una única escala continua tanto para los ácidos como para las bases (a través de ácidos conjugados, pKa BH+): Introducción Ácidos y Bases Ácidos conjugados y su fuerza: pKBH (en agua): H3C + 10.73 NH2 H3C 10.66 H3C NH3+ H3C + 9.80 NH CH H3C 3 + 5.23 N H Basicidad decrece: “cuando pKBH es grande, base es fuerte (o ácido conjugado débil) 4.87 NH3+ NH3+ O2 N 1.02 -3.53 Et2O+H H3C C O+ H H3C 5.29 H3CO 4.58 H 3.98 3.91 1.02 O2N NH3+ NH3+ 5.12 H3C -7.50 Basicidad aumenta NH3+ NH3+ Cl NH3+ Br NH3+ pKBH (en agua): (Explicaciones..) Introducción Principio de R. Pearson -HSAB SM, p. 339-341 1963: Teoría cualitativa basada en la transferencia de los e-s para predecir la reactividad química (J. Am. Chem. Soc., 1963, 85, 3533) Ácidos duros Ácidos blandos Bases blandas Bases duras Los ácidos duros prefieren asociarse con las bases duras y Los ácidos blandos prefieren asociarse con las bases blandas (Palabras “duros” o” blandos” no significan fuerte o débil !!) “Dureza” o “blandura” depende de la naturaleza del átomo, sus parámetros: ØTamaño Ø Electronegatividad Ø Polarizabilidad Ø Carga electrónica Introducción HSAB Detalles.. Ácidos duros Ácidos blandos H+, Na+, Ca2+, Mn3+, Al3+, Cr3+, SO3 .. Cu+, Ag+, Hg+, Pd2+, Pt2+, InCl3, BH3, carbenos .. Aceptores: son de átomo pequeño, tienen la carga positiva alta. Son de baja polarizabilidad y alta electronegatividad Bases blandas I-, CN-, SCN-, S2O32-, H-, R-, PPh3, CO, R2S, RSH .. Donadores: son de átomo grande, de alta polarizabilidad y baja electroNegatividad. Son fáciles de oxidarse Aceptores: son de átomo grande, tienen la carga positiva baja. Son de alta polarizabilidad y baja electronegatividad Bases duras H2O, HO-, RO-,F-, AcO-, SO42-, CO32-, NO3-, PO43-, NH3, RNH2, ROH, .. Donadores: son de baja polarizabilidad y alta electronegatividad. Son difíciles de oxidarse Casos de frontera: En ácidos - Fe2+, Co2+, Ni2+, Sn2+, Ru3+, R3C+ En bases - Br-, NO2-, SO32-, PhNH2 üPolarizabilidad es la facilidad con que la distribución electrónica alrededor de un átomo se distorsiona por un campo dentro próximo. Introducción Reactivos Nucleofilos :Nu (del griego, phelein = amar Nucleofilia y Basicidad Basicidad es la capacidad del reactivo no solamente de prestar su par de electrones al carbono deficiente, sino que también, arrancar del Sustrato el protón móvil y formar con él, compuestos menos disociados que el Sustrato. La fuerza nucleofílica (nucleofilia) consiste en la capacidad del reactivo (anión o molécula neutra) de formar un enlace covalente por medio del par de electrones. Los reactivos nucleofílicos suelen tener átomos con un par electrónico y “pueden prestar”. v No siempre el más nucleófilo resulta ser la más fuerte base !! ) Introducción Las fuerzas de nucleofilia y basicidad dependen de la naturaleza electrónica del átomo que contienen los reactivos : C N O F CH4 NH3 H2O HF -CH -NH -OH -F 3 2 Sus aniones Moléculas neutras Átomos en el periodo de la Tabla Química -NH > NH3; (KNH2, NaNH2) 2 Aumenta la electronegatividad (recordar a la escala de Pauling) Disminuye la fuerza nucleofílica y la basicidad! (y acidez aumenta) Introducción Átomos (aniones) dentro del grupo de la Tabla Química Fuerza nucleofílica (Nucleofilia) O- F- 1.72 S- Cl- 6.57 Br- 9.57 I- 14.55 Nucleofilia Masa atómica, - SR > -OR polarizabilidad Basicidad Masa atómica Nucleofilia Polarizabilidad (cm3.1025) FCl- HI >> HF Br- OS- IBasicidad Tamaño del ión I-<< F- Tamaño Basicidad del ión RO- > RS- Introducción Influencia de los ¨vecinos¨ Grupos GDE Grupo Donador de Electrones GAE Grupo Aceptor de Electrones B GDE > B > B GAE Nu GDE > Nu > Nu GAE Factores electrónicos Factores estéricos