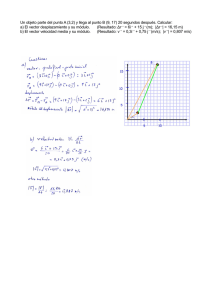
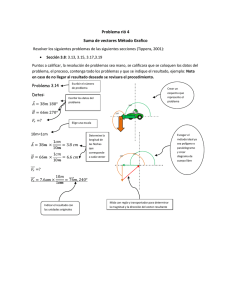
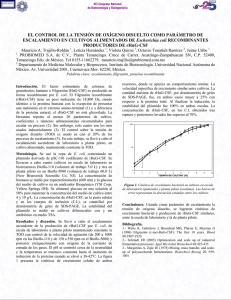
2 materiales y métodos - Tesis
Anuncio

ÍNDICE GENERAL 1 INTRODUCCIÓN………………………………………………………………………………………4 1.1 ANTECEDENTES GENERALES……………………………………………………….…………4 1.2 ANTECEDENTES BIBLIOGRÁFICOS………………………………………………....………...5 1.2.1 PROTEASAS TIPO SUBTILISINA…………………………………………………………5 1.2.2 ENZIMAS PSICROFÍLICAS………………………………………...………………………6 1.2.3 SUBTILASAS T7 Y T8 DE ORIGEN ANTÁRTICO…………………………………….....7 1.2.4 SISTEMAS DE EXPRESIÓN DE PROTEÍNAS RECOMBINANTES……………………..7 1.3. DESCRIPCIÓN DEL PROYECTO Y JUSTIFICACIÓN…………..……………………………..9 1.4. OBJETIVOS…………………………………………………………..……………………………9 2 MATERIALES Y MÉTODOS………………………………………………………………………..10 2.1 MATERIALES…………………………………………………………………………………….10 2.1.1 CÉLULAS HUÉSPED…………………………………………………………...................10 2.1.2 VECTORES…………………………………………………………………………………10 2.1.2.1. Vector pGEM-T Easy…………………………………………………...................10 2.1.2.2. Vectores pHIS1522 y pHIS1525…………………………………………………...10 2.2 MÉTODOS………………………………………………………………………………………...11 2.2.1 PREPARACIÓN DE CÉLULAS COMPETENTES………………………………………..11 2.2.1.1 Preparación de células electrocompetentes de Escherichia coli DH5α…………….11 2.2.1.2. Preparación de protoplastos de Bacillus megaterium WH320……………………..11 2.2.2 TRANSFORMACIÓN DE CÉLULAS DE E. coli DH5α CON pHIS1522 Y pHIS1525....12 2.2.3 CLONAMIENTO DE GENES DE SUBTILASAS T8 Y T7 EN VECTORES DE EXPRESIÓN….………………………………………………………………………..12 2.2.3.1 Adición de sitios de restricción……………………………………………………. 12 2.2.3.2 Purificación de fragmentos amplificados desde gel de agarosa…..………………. 14 2.2.3.3 Clonamiento de insertos T822, T825 y T725 en vector pGEM-T Easy……………14 2.2.3.4 Digestión de plasmidios pGEM-T822, pGEM-T825, pGEM-T725, pHIS1522 y pHIS1525 con enzimas de restricción...………………………………..….……....15 2.2.3.5 Ligación de insertos con sus respectivos vectores de expresión…………..………..15 2.2.3.6 Amplificación in vivo de los vectores pHIS1522-T8, pHIS1525-T8 y pHIS1525-T7……..………………………………………………………………..16 2.2.4 TRANSFORMACIÓN DE PROTOPLASTOS DE Bacillus megaterium CON pHIS1522-T8, pHIS1525-T8 Y pHIS1525-T7..……………………………………………16 2.2.5 PRUEBAS PRELIMINARES DE ACTIVIDAD PROTEASA EXTRACELULAR………17 2.2.6 INDUCCIÓN Y RECUPERACIÓN DE PROTEÍNA RECOMBINANTE DESDE Bacillus megaterium….…………………………………………………………………...17 2.2.7 DETECCIÓN DE PROTEÍNAS RECOMBINANTES………………………………….…17 2.2.8 PRUEBAS DE ACTIVIDAD PROTEASA DE PROTEÍNAS RECOMBINANTES.……..18 2.2.9 ANÁLISIS DE LAS CONSTRUCCIONES pHIS1522-T8, pHIS1525-T8 Y pHIS1525-T7..……………………………………………………………………………...18 3 RESULTADOS Y DISCUSIÓN............................................................................................................20 3.1 AMPLIFICACIÓN DE LOS VECTORES pHIS1522 Y pHIS1525 EN E. coli DH5α…….….. ..21 3.2 EXPRESIÓN RECOMBINANTE DE SUBTILASA T8 EN B. megaterium……………………22 3.2.1 CLONAMIENTO DEL GEN DE SUBTILASA T8 EN pHIS1522 Y pHIS1525…….……22 3.2.2 TRANSFORMACIÓN DE PROTOPLASTOS DE Bacillus megaterium CON pHIS1522-T8 Y pHIS1525-T8..…………………………………….……………………...23 3.2.3 REVISIÓN DE PLASMIDIOS AMPLIFICADOS EN Bacillus megaterium……….……24 3.2.4 PRUEBAS PRELIMINARES DE ACTIVIDAD PROTEASA EXTRACELULAR….…...26 3.2.5 INDUCCIÓN Y RECUPERACIÓN DE PROTEÍNA RECOMBINANTE T8 DESDE B. megaterium…….…………….………………………………………………………....27 3.3 EXPRESIÓN RECOMBINANTE DE SUBTILASA T7 EN Bacillus megaterium…………….29 3.3.1 CLONAMIENTO DEL GEN DE SUBTILASA T7 EN pHIS1525………………………...29 3.3.2 TRANSFORMACIÓN DE PROTOPLASTOS DE Bacillus megaterium CON pHIS1525-T7..……………………………………………………………………………...30 3.3.3 REVISIÓN DE PLASMIDIOS AMPLIFICADOS EN B. megaterium…………………...30 3.3.4 PRUEBAS PRELIMINARES DE ACTIVIDAD PROTEASA EXTRACELULAR………31 3.3.5 INDUCCIÓN Y RECUPERACIÓN DE PROTEÍNA RECOMBINANTE T7 DESDE B. megaterium…...………………………………………………………………………...32 3.4 INDUCCIÓN DE PROTEÍNAS RECOMBINANTES A BAJA TEMPERATURA…….……….33 3.5 ANÁLISIS DE LAS CONSTRUCCIONES pHIS1522-T8, pHIS1525-T8, pHIS1525-T7 Y pHIS1525-T7H…………………………………………………………………………………35 3.5.1 ANÁLISIS DE LAS SECUENCIAS………………………………………..........................35 3.5.2 CORTE CON ENZIMAS DE RESTRICCIÓN…………………….……………………….37 3.6 ANÁLISIS DE pHIS1525 AMPLIFICADO EN E. coli DH5α…………………………………..40 4 CONCLUSIONES……………………………………………………………………………………...42 5 BIBLIOGRAFÍA.....................................................................................................................................44 A. ANEXOS.................................................................................................................................................48 A.1. SECUENCIAS DE T8 Y T7……………………………………………………..………………48 A.2. VECTORES………………………………………………………………………..…………….52 A.2.1 Vector pGEM-T Easy……………….………………………………………..…………….52 A.2.2 Vectores pHIS1522 y pHIS1525……………..………………………………….…………53 A.3. MEDIOS DE CULTIVO…………………………………………………………………………55 A.4. PROTOCOLOS……………………………………….…………………………………….……59 A.5. GELES DE POLIACRILAMIDA PARA PROTEÍNAS……………………………………...…62 A.6. SOLUCIONES STOCK………….………………………………………………………………63 A.7. MAPA DE PARTIDORES PARA SECUENCIACIÓN…………………………………………66 A.8. GELES DE AGAROSA………………………………………………………………………….67 A.9. RESULTADOS DE SECUENCIACIÓN………………………………………………………...69 2 LISTA DE ABREVIATURAS ºC B. megaterium B. subtilis DNA dNTPs OD E. coli EDTA g gr IPTG kΩ kb kDa LB M mM MMBM min mg mL mM mRNA ng PAGE pb PCR pH pmol rpm S-AAPF-pNa SDS SM Tris tRNA µF µL µg V X-gal Grados Celsius Bacillus megaterium Bacillus subtilis Ácido desoxirribonucleico Deoxiribonucleótidos trifosfato Densidad óptica Escherichia coli Etilendiaminotetraacetato ácido Fuerza de gravedad Gramo Isopropil tio-β-D-galactósido Kiloohm Kilobase Kilodalton Medio Luria-Bertani Molar Milimolar Medio mínimo para Bacillus megaterium Minutos Miligramo Mililitros Milimolar Ácido ribonucleico mensajero Nanogramo Electroforesis en gel de poliacrilamida. Pares de bases Reacción en cadena de la polimerasa Potencial hidrógeno Picomol Revoluciones por minuto N-succinil-ala-ala-pro-phe p-nitroanilida Dodecil sulfato de sodio Medio Skim Milk Tris-(hidroximetil)-aminoetano Ácido ribonucleico de transferencia Microfaradios Microlitros Microgramos Voltio 5-bromo-4-cloro-3-indolil-β-D-galactopiranósido 3 1 INTRODUCCIÓN 1.1 ANTECEDENTES GENERALES Durante las últimas décadas se ha generado gran conocimiento sobre las ciencias de la vida y la biología, en base al cual se han desarrollado diversas técnicas y herramientas biotecnológicas, ofreciendo enormes potencialidades en términos de aplicaciones en diferentes ámbitos del desarrollo productivo y social. Una de estas aplicaciones corresponde al uso de enzimas como biocatalizadores en procesos industriales o como componentes de nuevos productos, pues éstas catalizan reacciones específicas, son reutilizables y no generan desechos. De especial importancia es el uso de enzimas en la industria de detergentes, donde actúan removiendo selectivamente manchas de aceites, proteínas o almidón de la ropa. Éstas se vienen usando desde hace más de 40 años con el objetivo de reemplazar a los compuestos sintéticos (blanqueadores), y reducir el consumo de energía producto de la necesidad de altas temperaturas en el proceso. Entre las enzimas utilizadas se encuentran las proteasas, las cuales catalizan la degradación de compuestos de origen proteico presentes, por ejemplo, en las manchas de sangre, huevo, cacao o leche. Sin embargo, esta aplicación aún requiere de altas temperaturas de lavado (alrededor de 60°C), debido a que estas proteasas, usualmente, provienen de organismos que viven en ambientes templados y por lo tanto sus enzimas trabajan óptimamente a estas temperaturas. Es por esto, que en las últimas décadas se han canalizado recursos en el desarrollo de nuevos detergentes que permitan el ahorro energético que implica el lavado con agua fría, y con ello, la necesidad de enzimas con alta actividad proteasa a temperatura ambiental. El descubrimiento y estudio de microorganismos extremófilos, es decir, que viven en condiciones extremas de pH, salinidad o temperatura, ha generado nuevas fuentes de proteínas catalizadoras, activas en condiciones inusuales para los procesos biológicos. Entre ellas se encuentran las enzimas psicrofílicas. Éstas se caracterizan por su adaptación a bajas temperaturas, pues poseen mayor actividad específica que su símil mesófilo a temperaturas entre 0ºC y 30ºC[1], pero se denaturan fácilmente a temperaturas mayores[2]. La aplicación de éste y otros tipos de enzimas en la industria depende de la viabilidad de producirlas a gran escala. Sin embargo, la purificación directa desde el microorganismo de origen suele no ser compatible con las condiciones estándar de producción. Por lo anterior, es preferible producir las enzimas en forma recombinante, es decir, sintetizarla en un organismo distinto al nativo. Los principales microorganismos huésped utilizados con este fin son Escherichia coli y Bacillus subtilis, entre los procariontes, y Saccharomyces cerevisiae, entre los eucariontes. La selección de un sistema adecuado debe considerar el origen biológico de la proteína de interés, el destino celular deseado (intracelular o extracelular), grado de pureza requerido, entre otras características. 4 1.2 ANTECEDENTES BIBLIOGRÁFICOS 1.2.1 PROTEASAS TIPO SUBTILISINA Las proteasas, son enzimas que catalizan la hidrólisis de los enlaces peptídicos de proteínas, produciendo su degradación. Se dividen en exopeptidasas y endopeptidasas o proteinasas, de acuerdo al lugar en el que cortan la cadena polipeptídica[3]. Las exopeptidasas hidrolizan en regiones cercanas a los extremos del péptido, clasificándose en aminopeptidasas o carboxilopeptidasas, de acuerdo al extremo de corte. Las proteinasas se pueden clasificar en base a su mecanismo catalítico como serina proteasas, cisteína proteasas, aspártico proteasas y métalo proteasas[3]. Las serina proteasas, son denominadas también como alcalino proteasas, debido que su actividad es óptima a altos niveles de pH (entre 9.0 y 11.0)[4]. Las subtilisinas, son proteasas que pertenecen a la familia de las serina proteasas. Estas enzimas representan a la subfamilia denominada proteasas tipo subtilisina o subtilasas, donde también se incluyen proteasas como termitasa o proteinasa K[5]. Existen proteasas tipo subtilisina en varios organismos, tales como arqueas, bacterias, hongos, levaduras, y eucariontes mayores, sin embargo, las subtilisinas se han registrado sólo en microorganismos. El sitio catalítico de las subtilasas es conservado, y contiene tres residuos catalíticos: aspartato, histidina y serina, ubicados en ese orden leyendo desde el extremo amino al carboxilo terminal[5]; poseen además un sitio conservado de unión a Ca+2, denominado sitio A, importante para su estabilidad[6]. La mayoría de las proteasas tipo subtilisina son sintetizadas como preproenzimas[5], es decir, poseen en su extremo amino un prepéptido o péptido señal para su exportación al medio extracelular, y un propéptido localizado entre el péptido señal y la parte madura de la proteína. Una vez traslocada la proteína a través de la membrana celular, el prepéptido es eliminado dando lugar a una proenzima, conformada por el propéptido y la enzima madura, desde donde finalmente el propéptido es removido dando lugar a la enzima activa[5]. No obstante, numerosas excepciones son sintetizadas sin la prosecuencia, se le han atribuido varias propiedades, entre ellas la prevención de la actividad de las enzimas proteolíticas durante la secreción, y no se descarta su participación en el proceso. Se cree además que poseen un rol importante durante el replegamiento y activación de las proteasas luego de su traslocación a través de la membrana[7]. Las subtilasas son ampliamente utilizadas en la industria de detergentes. Se consideran las proteasas más adecuadas para este fin, pues poseen baja especificidad[8] y son más compatibles con los compuestos de los detergentes que otros tipos de proteasas: las cisteína proteasas son oxidadas por los agentes blanqueadores y las metaloproteasas pierden sus cofactores metálicos por la acción de los agentes quelantes o de los iones hidróxido. La mayoría de las subtilasas utilizadas como aditivo en detergentes son de la especie Bacillus, y se producen industrialmente en el microorganismo de origen o en forma recombinante[8, 9]. Algunas han sido estudiadas con el fin de mejorar su rendimiento, por ejemplo, tornándolas más estables a los agentes quelantes[10] u oxidantes[11] presentes en los detergentes. Sin embargo, en los últimos años se han desarrollado ampliamente estudios enfocados hacia la 5 incorporación de enzimas activas a bajas temperaturas[ 12 , 13 , 14 ], debido principalmente a la necesidad mundial de una reducción en el consumo energético. 1.2.2 ENZIMAS PSICROFÍLICAS Los ambientes extremos se caracterizan por tener condiciones hostiles para el desarrollo de la vida, tales como extremas condiciones de pH, presión, salinidad y temperatura. Estos factores medioambientales son determinantes en el proceso de adaptación y evolución de los organismos vivos, permitiendo el desarrollo de los organismos extremófilos. La adaptación a temperaturas extremas, en particular a las bajas temperaturas (entre -2 ºC y 20 ºC)[15], da lugar a los organismos psicrofílicos, los cuales han recurrido a diversas estrategias para mantener la fluidez de sus membranas y la actividad metabólica bajo estas condiciones, entre ellas, la capacidad de sintetizar enzimas adaptadas a estas bajas temperaturas. Las bajas temperaturas disminuyen la velocidad de las reacciones químicas en forma exponencial. Este efecto se describe por la ecuación de Arrhenius[16]: k cat = A ⋅ κ ⋅ e − Ea RT donde kcat es la velocidad de la reacción, T la temperatura absoluta, A es el factor preexponencial (o factor de frecuencia, que depende de cada reacción), κ es el coeficiente dinámico de transferencia (relacionado con la viscosidad del medio) que usualmente es 1, E a la energía de activación y R es la constante universal de los gases (8.314 J mol-1 K-1). Sin embargo, las enzimas psicrofílicas poseen una alta actividad específica aún a bajas temperaturas, lo que implica que han desarrollado adaptaciones que alteran la cinética clásica de las enzimas, caracterizadas principalmente como una alta actividad específica y una disminución en la temperatura óptima y en la estabilidad, generando un aumento en la flexibilidad[17]. Las bajas temperaturas tienden a compactar las proteínas generando una disminución de la movilidad necesaria para la actividad catalítica[18]. Esto sugiere que las enzimas psicrofílicas deben aumentar su flexibilidad para poder mantener la acción catalítica bajo estas condiciones. Este aumento en la flexibilidad estaría relacionado con la disminución en la estabilidad térmica de la proteína[1, 14, 17, 18]. Se han determinado varios factores relacionados con el aumento de flexibilidad de las enzimas psicrofílicas con respecto a su contraparte mesófila, entre ellos la agrupación de glicinas, disminución en el número de prolinas en los loops, reducción del número de argininas, disminución de puentes salinos y de interacciones aromáticas e hidrofóbicas, disminución de puentes de hidrógeno y aumento de residuos hidrofóbicos en la superficie; en general, la disminución de factores conocidos como estabilizantes de las enzimas mesófilas[17, 18]. Lo anterior, se traduce también en una baja afinidad por el sustrato, por lo tanto, para facilitar la unión con el sitio activo, en las enzimas psicrofílicas éste se presenta más accesible y de mayor tamaño, para lo cual se reemplazan aminoácidos de mayor tamaño por otros de cadena corta y se aumenta el tamaño de los loops cercanos al sitio activo[17]. 6 1.2.3 SUBTILASAS T7 Y T8 DE ORIGEN ANTÁRTICO Recientemente en el Centro de Excelencia Académica en Ingeniería Bioquímica y Biotecnología (CIBYB) se han aislado varios microorganismos de origen antártico productores de proteasas extracelulares con alta actividad a baja temperatura. Distintos ensayos han permitido seleccionar las cepas de mayor actividad a pH alcalino (pH=8.0)[19] desde cuyo genoma se han identificado genes de proteasas tipo subtilisina mediante el método de genome walking[20]. Entre ellos se encuentran los genes denominados T8 y T7, encontrados en las cepas 3-17 de Polarisbacter sp. y 2-10 de Pseudoalteromonas sp., respectivamente; ambas son bacterias Gram negativas. La traducción del gen T8 genera una enzima de 536 aminoácidos, con un peso molecular de 57.7 kDa. En el extremo amino presenta una presecuencia o señal de exportación al medio extracelular de 24 aminoácidos. Tiene además una región de 148 aminoácidos, de función no conocida, entre las regiones conservadas que contienen los residuos de aspartato e histidina. Por su parte, el gen T7 codifica para una enzima de peso molecular igual a 72.5 kDa, conformada por 708 residuos aminoacídicos, dentro de los cuales se encuentra un péptido señal de 27 aminoácidos y un propéptido de 117 residuos de longitud. En la Figura 1 se muestra un esquema de la estructura de las enzimas T8 y T7. Su secuencia y alineamiento con las proteasas tipo subtilisina más comunes se puede ver en el Anexo A.1. Figura 1: Esquema de las proteasas tipo subtilisina T8 y T7. Verde: presecuencia; azul: prosecuencia; rojo: región de función no determinada. En blanco se indican los residuos catalíticos. 1.2.4 SISTEMAS DE EXPRESIÓN DE PROTEÍNAS RECOMBINANTES Actualmente, para la producción de proteínas recombinantes existe una gran variedad de organismos hospederos, tales como bacterias, hongos, levaduras, líneas celulares de mamíferos e incluso organismos completos como insectos, plantas o animales. La selección del sistema de expresión dependerá tanto de las características de la proteína objetivo en cuanto a su origen y necesidad de modificaciones postraduccionales, como de las necesidades de productividad y localización celular. Los sistemas más comunes para la expresión de proteínas de origen bacteriano utilizan como microorganismo huésped Escherichia coli y Bacillus subtilis. 7 Escherichia coli es una bacteria Gram negativa, de alta tasa de crecimiento y bajo costo de cultivo; además es el microorganismo mejor caracterizado a nivel genético molecular, por lo que es ampliamente utilizado para la expresión de proteínas heterólogas. Sin embargo, es un microorganismo mal secretor de proteínas debido principalmente a la estructura de su pared celular. La pared celular de las bacterias Gram negativas está conformada por una delgada capa de péptidoglicano, rodeada por una membrana externa de lípidos y proteínas. La presencia de dos membranas (externa y citoplasmática o interna) define un espacio denominado periplásmico[21]. Los mecanismos de secreción de proteínas a través de esta estructura han sido clasificados en Sec-dependientes y Sec-independientes[ 22 ]. Los primeros utilizan un péptido señal para la exportación al periplasma, donde la proteína adquiere su conformación nativa, siendo posteriormente traslocada a través de la membrana externa. Los sistemas Sec-independientes en cambio, traslocan las proteínas directamente desde el citosol al medio extracelular, sin necesidad de un intermediario periplásmico ni péptido señal[22]. Lo anterior se traduce en que la proteína recombinate, en muchos casos, no pueda ser secretada eficientemente y se acumule en el espacio periplásmico, o bien en precipitados insolubles en el citoplasma (cuerpos de inclusión). Por otra parte, Bacillus subtilis, una bacteria Gram positiva, posee una pared celular más simple, conformada sólo por una capa gruesa y homogénea de péptidoglicano[21], por lo que la secreción proteica se hace más fácil, pues requiere el transporte a través de una sola membrana. Lo anterior, hace de B. subtilis un buen secretor de proteínas recombinantes. Sin embargo, produce una gran cantidad de proteasas que afectan la producción, muchas de las cuales aún no han sido identificadas[7]. Los sistemas de expresión recombinante de proteínas requieren además de vectores de expresión específicos para cada microorganismo. Éstos son secuencias circulares de DNA que contienen señales de inicio de transcripción y traducción. También poseen sitios de restricción adyacentes a estas señales para facilitar la inserción del DNA foráneo. Es deseable, además, la presencia de un gen (generalmente resistencia a un antibiótico) que permita seleccionar a las células transformadas, que el vector tenga la capacidad de replicarse en forma autónoma, que sea estable en el huésped, y que posea señales para la regulación de la transcripción del gen foráneo. Algunos vectores han sido diseñados para expresar las proteínas fusionadas por el extremo carboxilo a péptidos cortos que facilitan su posterior purificación y localización, o bien por el extremo amino a péptidos señal para su exportación al medio extracelular, evitando así su degradación por proteasas intracelulares y facilitando su purificación. No obstante existen antecedentes sobre la expresión recombinante de proteasas en Bacillus[8, 9], e incluso de proteasas psicrofílicas en E. coli [23, 24], es conveniente evaluar nuevos sistemas de expresión recombinante. El uso de Bacillus megaterium como microorganismo hospedero posee varias ventajas sobre los sistemas descritos anteriormente. A diferencia de E. coli, es un buen secretor de proteínas, y posee menor actividad proteasa que B. subtilis. Varias proteínas recombinantes han sido exitosamente producidas en Bacillus megaterium, por ejemplo, toxina A de Clostridium difficile[25] (manteniendo la citotoxicidad y actividad glucosiltransferasa in vitro de la toxina nativa) y glucanasa[26] (con actividad 7 veces mayor que en la cepa nativa). No obstante el sistema es relativamente nuevo, existen variados vectores diseñados especialmente con este fin, incluyendo vectores que incorporan señales de 8 exportación y/o péptidos que facilitan la posterior purificación de la enzima recombinante. Entre las proteínas recombinantes producidas con este tipo de vectores se encuentran levansucrasa[27] (con mayor porcentaje de recuperación y mayor actividad específica que la misma enzima expresada en E. coli) y fragmentos de anticuerpos[ 28 ] (con mayor concentración y mayor actividad específica, entendida como unión al antígeno, que el producido en E. coli). 1.3 DESCRIPCIÓN DEL PROYECTO Y JUSTIFICACIÓN El uso de enzimas como aditivos en la industria de detergentes, junto con el interés por desarrollar nuevos productos que permitan disminuir las temperaturas de lavado, genera la necesidad de desarrollar un sistema de producción masiva de proteasas psicrofílicas para esta industria. La ingeniería genética permite expresar enzimas de manera recombinante, con el objeto de facilitar su posterior producción a mayor escala. Es por esto que el estudio de nuevos sistemas de expresión recombinante de proteasas de origen antártico se ve incentivado. Un sistema emergente de expresión recombinante de proteínas es aquel que utiliza células de Bacillus megaterium como microorganismo huésped. Su uso ha permitido la expresión de variadas proteínas heterólogas en forma satisfactoria e incluso, en algunos casos, con mayor actividad específica que la proteína nativa o la expresada en otros sistemas. Se propone, por lo tanto, como proyecto de memoria la “Evaluación de Bacillus megaterium como sistema de expresión recombinante de proteasas tipo subtilisina de origen antártico”. El proyecto consiste en clonar y expresar los genes de las proteasas tipo subtilisina T8 y T7, proveniente de los microorganismos de origen antártico Polarisbacter sp. y Pseudoalteromonas sp., respectivamente, en B. megaterium, y analizar las características del sistema de expresión recombinante. 1.4 OBJETIVOS El objetivo general del Trabajo de Memoria de Título es analizar la expresión recombinante de proteasas de origen antártico en Bacillus megaterium. Con este fin, se fijaron los siguientes objetivos específicos: • • • • Diseñar una estrategia para la expresión recombinante de genes de subtilasas T8 y T7 de origen antártico en B. megaterium. Clonar y expresar los genes de las subtilasas T8 y T7 en B. megaterium. Diseño de un ensayo de actividad proteasa en medio sólido, para el análisis in situ de las subtilasas recombinantes producidas en Bacillus megaterium. Analizar la expresión de proteína recombinante en B. megaterium. 9 2 MATERIALES Y MÉTODOS 2.1 MATERIALES 2.1.1 CÉLULAS HUÉSPED Como huésped de clonación se utilizó células de Escherichia coli cepa DH5α, genotipo: F-, φ80dlacZ∆M15, ∆(lacZYA-argF)U169, deoR, recA1, endA1, hsdR17(rk-, mk+), phoA, supE44, λ-, thi-1, gyrA96, relA1. Para la expresión, se utilizó la cepa de Bacillus megaterium WH320 (MoBiTec), que corresponde a una mutante de la cepa DSM319, lacZ-[ 29]. 2.1.2 VECTORES 2.1.2.1 Vector pGEM- T Easy El vector de clonamiento pGEM-T Easy (Promega) se utilizó para la amplificación in vivo de los productos de PCR desde DNA genómico. Este vector, en su forma lineal, posee un nucleótido de timina en sus extremos 3’, los cuales actúan como extremos cohesivos con los fragmentos de DNA amplificados por PCR, ya que son generados con un nucleótido de adenina en los extremos 3’. La presencia de un nucleótido de timina en los extremos 3’ del vector impide además la recircularización de éste, aumentando la eficiencia del proceso de ligación. Para seleccionar los clones positivos, el sitio de clonamiento múltiple del vector interrumpe la secuencia codificante del péptido α de la enzima β-galactosidasa. Ésta puede hidrolizar el sustrato 5-bromo-4-cloro-3-indolil-β-D-galactósido, X-gal, generando un precipitado de color azul. Así, proporcionando este sustrato al medio de cultivo, las colonias que adquieren este color se identifican como negativas, y las blancas como positivas (Anexo A.2.1 ). 2.1.2.2 Vectores pHIS1522 y pHIS1525 Los vectores pHIS1522 y pHIS1525 (MoBiTec) fueron utilizados para la expresión de proteasas recombinantes en B. megaterium. Estos vectores se caracterizan por tener origen de replicación para E. coli, y para Bacillus, lo que permite realizar el clonamiento y su amplificación in vivo en células de E. coli previo a la transformación de la célula huésped de B. megaterium. En ambos vectores el sitio de clonamiento múltiple deja el gen de interés bajo el control del promotor xylA, PxylA, inducible con xilosa. La presencia de xilosa en el medio produce la 10 liberación del represor (codificado por el gen xylR) del promotor PxylA, permitiendo que se inicie la transcripción. De manera adicional, estos vectores agregan una cola de 6 histidinas al extremo carboxilo de la proteína recombinante, facilitando su posterior purificación. El vector pHIS1525 posee, además, la secuencia de un péptido señal, SPlypA, inmediatamente después del sitio de clonamiento múltiple, fusionando el gen de interés a la señal de exportación. Esto permite que la proteína recombinante sea secretada al medio extracelular por la célula huésped (Anexo A.2.2). 2.2 MÉTODOS 2.2.1 PREPARACIÓN DE CÉLULAS COMPETENTES 2.2.1.1 Preparación de células electrocompetentes de Escherichia coli DH5α Se inoculó 10 mL de medio SOB (Anexo A.3.1) con células stock de Escherichia coli DH5α, conservadas a -80 ºC, y se incubó con agitación a 37 ºC durante la noche. Se midió densidad óptica (O.D.) del cultivo a 600 nm, y se calculó el inóculo necesario para 1 litro de cultivo con O.D600 final igual a 0.05 (considerando que la relación lineal entre O.D y concentración sólo se cumple hasta O.D.=1, de ser necesario se diluyó la muestra 1:10 para medir). A continuación, se creció durante 2 a 4 horas, hasta obtener una O.D600 entre 0.6 y 0.8; entonces se enfrió durante 10 minutos, y luego se colectó las células centrifugando el cultivo a 5000 x g y a 4 ºC, por 10 minutos. El pellet fue lavado 2 veces con igual volumen de glicerol 10% v/v estéril, y resuspendido finalmente en el glicerol residual del último lavado. Luego se alicuotó en volúmenes de 50, 75 y 100 µL, para ser almacenado a -80 ºC. 2.2.1.2 Preparación de protoplastos de Bacillus megaterium WH320 El método de preparación de protoplastos se basó en un protocolo encontrado en bibliografía[30]. Los protoplastos se prepararon a partir de células de B. megaterium regeneradas desde protoplastos comerciales (MoBiTec) según se indica en el protocolo de transformación del Anexo A.4.1, en ausencia de DNA transformante. Con ellas, se inoculó 2 mL de medio SP1 (prolina) (Anexo A.3.2), y se incubó durante la noche a 37 ºC. Luego se diluyó en 200 mL de medio SP1, y se creció a 37 ºC hasta obtener aproximadamente 2x107 cel/mL. El cultivo se lavó 2 veces con 20 mL de medio SMMP (Anexo A.3.3) centrifugando a 1200 x g por 10 minutos, y luego se resuspendió en 10 mL de SMMP. Después se agregó lisozima de clara de huevo de gallina (Sigma) a 80 µg/mL y se incubó a 37 ºC por aproximadamente 30 minutos, observando al microscopio hasta que las células se vieran redondas. Luego se centrifugó a 1200 x g por 10 11 minutos, a 4 °C y se lavó 2 veces con 10 mL de SMMP, manteniendo siempre una baja temperatura. Finalmente, se agregó glicerol hasta dejar el cultivo a glicerol 10% v/v. 2.2.2 TRANSFORMACIÓN DE CÉLULAS DE E. coli DH5α CON pHIS1522 Y pHIS1525 Con el objeto de obtener un mayor número de copias de los vectores pHIS1522 y pHIS1525, se transformó mediante electroporación 20 µL de células electrocompetentes de Escherichia coli con 1 µL de plasmidio 0.2 µg/mL, según protocolo indicado en el Anexo A.4.2. Las células transformadas fueron sembradas en placas con medio LB agar (Anexo A.3.4) con carbenicilina 0.1 mg/mL. Desde las placas se tomó una colonia como inóculo para 4 mL de medio LB (Anexo A.3.5) con carbenicilina 0.1 mg/mL y se incubó a 37 ºC durante la noche para luego extraer el DNA plasmidial mediante minipreparación, con el método QIAprep® Spin Miniprep (ver Anexo A.4.3). 2.2.3 CLONAMIENTO DE GENES DE SUBTILASAS T8 Y T7 EN VECTORES DE EXPRESIÓN 2.2.3.1 Adición de sitios de restricción Para insertar los genes en los vectores pHIS1522 y pHIS1525, se realizó PCR a partir de DNA genómico de las cepas 3-17 de Polarisbacter sp.y 2-10 de Pseudoalteromonas sp., para los genes de las proteínas T8 y T7, respectivamente. En la reacción se utilizaron partidores que agregan sitios de restricción para enzimas del sitio de clonamiento múltiple (MCS) de los plasmidios (ver Figuras A.2.2.a y A.2.2.b, Anexo A.2.2). Para clonar en el vector pHIS1522 se agregaron sitios de restricción para las enzimas SpeI al extremo 5’, y SphI en el 3’, mientras que para pHIS1525 se agregaron los sitios KasI y SphI, respectivamente. Los partidores utilizados se muestran en la Tabla 1. Tabla 1: Partidores Específicos Gen T8 Enzima de restricción SpeI Sitio de restricción Partidor Secuencia del Partidor T8SpeI(s) 5’. . . . ACTAGTGATATTGTTTCAACTCCT . . . 3’ T8 KasI T8KasI(s) 5’. . . . .GGCGCCGATATTGTTTCAACTC . . . . .3’ T8 SphI T8SphI(r) 5’. . . . .GCATGCTTAGATAATTGCTCTG . . . . .3’ T7 KasI T7KasI(s) 5’. . .GGCGCCCAATCAGTTTCAAGTTCAG. . .3’ T7 SphI T7SphI(r) 5’. . . . . GCATGCCGGTTGGTATTGCCC. . . . . .3’ 12 Los fragmentos de amplificación se denominaron T822 y T825, para el gen de T8 con sitios de restricción para pHIS1522 y pHIS1525 respectivamente. El fragmento de amplificación correspondiente al gen de T7 con sitios de restricción para pHIS1525 se denominó T725. La reacción de PCR se llevó a cabo con la enzima Elongasa (Elongase Enzyme), la cual agrega una base de adenina al extremo 3’, permitiendo la posterior ligación al vector de clonamiento pGEM-T. Ésta enzima además posee actividad exonucleasa 3’-5’, permitiendo una amplificación de alta fidelidad. La reacción se realizó de acuerdo a las instrucciones del proveedor y con cada partidor a una concentración final de 0.4 pmol/µL. Los programas de amplificación utilizados se muestran a continuación: Programa 1: Amplificación de T822 desde DNA genómico Paso 1 2 3 4 5 6 7 8 9 10 Temperatura [ºC] 94 94 38 70 Repetir 4 veces los pasos 2 al 4 94 55 70 Repetir 32 veces los pasos 6 al 8 70 Tiempo 1 minuto 32 segundos 32 segundos 2 minutos 30 segundos 32 segundos 32 segundos 2 minutos 30 segundos 6 minutos Programa 2: Amplificación de T825 desde DNA genómico Paso 1 2 3 4 5 6 7 8 9 10 Temperatura [ºC] 94 94 33 70 Repetir 4 veces los pasos 2 al 4 94 55 70 Repetir 34 veces los pasos 6 al 8 70 13 Tiempo 1 minuto 32 segundos 32 segundos 2 minutos 30 segundos 32 segundos 32 segundos 2 minutos 30 segundos 6 minutos Programa 3: Amplificación de T725 desde DNA genómico Paso 1 2 3 4 5 6 7 8 9 10 Temperatura [ºC] 94 94 45 70 Repetir 4 veces los pasos 2 al 4 94 65 70 Repetir 33 veces los pasos 6 al 8 72 Tiempo 1 minuto 32 segundos 40 segundos 2 minutos 30 segundos 32 segundos 40 segundos 2 minutos 30 segundos 6 minutos 2.2.3.2 Purificación de fragmentos amplificados desde gel de agarosa Los productos de PCR de la reacción de amplificación del punto 2.2.3.1, fueron analizados en un gel de agarosa 0.8% o 1% p/v por electroforesis para separar los fragmentos deseados. Luego fueron purificados desde el gel utilizando el sistema QIAEX II Gel Extraction Kit, según las instrucciones del fabricante[31]. 2.2.3.3 Clonamiento de insertos T822, T825 y T725 en vector pGEM-T Easy Con el objeto de obtener amplificación in vivo de los insertos T822, T825 y T725, se utilizó el sistema pGEM-T Easy (Promega), como vector de clonamiento y se transformó E. coli DH5α con los vectores recombinantes. La incorporación de los insertos en un vector facilita además su posterior digestión con enzimas de restricción, pues éstas son poco eficientes cortando en regiones cercanas a los extremos de fragmentos de DNA. La reacción de ligación se realizó con 1 µL de enzima T4 DNA ligasa, 5 µL de buffer Ligasa 2x (ambos incluidos en el sistema de clonamiento), 0.5 µL de vector, y 3.5 µL de inserto, éste último purificado desde gel de agarosa según se indicó en el punto anterior. La reacción se incubó durante 2 horas a 37 ºC. Los plasmidios generados se denominaron pGEM-T822, pGEM-T825 y pGEM-T725. Estas mezclas de ligación fueron utilizadas para transformar células de E. coli DH5α mediante electroporación, según el protocolo indicado en el Anexo A.4.2. Las células fueron plaqueadas en LB agar con carbenicilina 0.1 mg/mL, IPTG 0.5 mM y X-gal 80 ng/mL. De ellas, se hizo PCR de las colonias blancas según se muestra en el protocolo del Anexo A.4.4. Para amplificar el inserto T822 se utilizaron los partidores T8SpeI(s) y T8SphI(r), para el inserto T825 los partidores T8KasI(s) y T8SphI(r), mientras que para amplificar T725 se utilizó el par T7KasI(s) y T7SphI(r) (Ver Tabla 1). Los programas de amplificación se muestran a continuación: 14 Programa 4: PCR de de colonias para T822 y T825 Paso 1 2 3 4 5 6 Temperatura [ºC] 94 94 55 70 Repetir 36 veces los pasos 2 al 4 70 Tiempo 1 minuto 32 segundos 32 segundos 2 minutos 30 segundos 6 minutos Programa 5: PCR de colonias para T725 Paso 1 2 3 4 5 6 Temperatura [ºC] 94 94 65 70 Repetir 36 veces los pasos 2 al 4 72 Tiempo 1 minuto 32 segundos 40 segundos 2 minutos 30 segundos 6 minutos De acuerdo a los resultados del PCR de colonias, se seleccionó una colonia de cada inserto, correspondiente a la de mayor concentración de producto de PCR, y se inoculó en 4 mL de medio LB con carbenicilina 0.1 mg/mL, para luego extraer el DNA plasmidial mediante minipreparación (ver Anexo A.4.3). 2.2.3.4 Digestión de plasmidios pGEM-T822, pGEM-T825, pGEM-T725, pHIS1522 y pHIS1525 con enzimas de restricción Con la finalidad de ligar cada inserto con su respectivo vector, se digirieron ambos con las mismas enzimas de restricción. El inserto T822 ligado al vector de clonamiento pGEM-T Easy, y el vector pHIS1522 se digirieron con las enzimas SpeI (New England Biolabs) y SphI (New England Biolabs). Por otra parte, los plasmidios con insertos T825 y T725, correspondientes al vector pHIS1525, y el mismo vector fueron digeridos con las enzimas de restricción KasI (New England Biolabs) y SphI (NEB). Las digestiones se realizaron en forma doble, de acuerdo a las instrucciones del proveedor, a 37 ºC por un período de 2 horas. Los fragmentos se purificaron de acuerdo al protocolo descrito en el punto 2.2.3.2. 2.2.3.5 Ligación de insertos con sus respectivos vectores de expresión Para ligar los genes de T7 y T8 en los respectivos vectores de expresión para Bacillus, se utilizó la enzima T4 DNA Ligasa (Invitrogen). Se ligó así T822 con pHIS1522, y T825 y T725 con pHIS1525. 15 Para cada reacción se tomó 2 µL de vector y 5 µL de inserto (ambos digeridos y purificados desde gel de agarosa según se indicó en los puntos anteriores), junto con 1 µL de enzima y 2 µL del respectivo buffer y se incubó la mezcla por 2 horas a temperatura ambiente o por 16 horas a 16 ºC. Los plasmidios producto de la reacción de ligación se denominaron pHIS1522-T8, pHIS1525-T8 y pHIS1525-T7. 2.2.3.6 Amplificación in vivo de los vectores pHIS1522-T8, pHIS1525-T8 y pHIS1525-T7 Los vectores pHIS1522-T8, pHIS1525-T8 y pHIS1525-T7 fueron amplificados en células E. coli DH5α. Dichas células fueron transformadas por electroporación siguiendo el protocolo indicado en el Anexo A.4.2, plaqueándose en medio LB agar con carbenicilina 0.1 mg/mL. Se realizó PCR de colonias utilizando los partidores de la Tabla 1. de acuerdo al protocolo indicado en el Anexo A.4.4. Se seleccionó una colonia positiva para el producto de amplificación de cada construcción y se utilizó como inóculo para 6 mL de medio LB con carbenicilina 0.1 mg/mL. Los cultivos se incubaron a 37 ºC durante la noche. Luego se purificó los plasmidios mediante minipreparación de DNA plasmidial según el protocolo del Anexo A.4.3, pero utilizando los 6 mL de cultivo con el fin de obtener una mayor cantidad de DNA. Para chequear el estado y concentración de los plasmidios amplificados, se hizo digestión doble de pHIS1522-T8 con enzimas SpeI (NEB) y SphI (NEB), y de pHIS1525-T8 y pHIS1525T7 con KasI (NEB) y SphI (NEB), durante 2 horas a 37 ºC bajo las condiciones indicadas por el proveedor. 2.2.4 TRANSFORMACIÓN DE PROTOPLASTOS DE Bacillus megaterium CON pHIS1522-T8, pHIS1525-T8 Y pHIS1525-T7 Se transformaron protoplastos de B. megaterium adquiridos a MoBiTec, y protoplastos generados en el laboratorio por el método indicado en el punto 2.2.1.2 de este mismo capítulo. Ambos fueron transformados con los plasmidios pHIS1522-T8, pHIS1525-T8, y con los plasmidios control pHIS1522 y pHIS1525, de acuerdo al protocolo descrito en el Anexo A.4.1. Se transformó además protoplastos generados en laboratorio con pHIS1525-T7 mediante el mismo protocolo. La obtención de DNA plasmidial desde cultivos de B. megaterium, se realizó utilizando el sistema QIAprep® Spin Miniprep según protocolo entregado por el fabricante[32], el cual es en esencia el mostrado en el Anexo A.4.3, con una etapa adicional de lisis con lisozima, y otra de remoción de nucleasas. 16 2.2.5 PRUEBAS PRELIMINARES DE ACTIVIDAD PROTEASA EXTRACELULAR Las colonias transformadas de B. megaterium, fueron transferidas a distintos medios sólidos con el objeto de analizar su actividad proteasa extracelular. En los ensayos se utilizó SM agar (Anexo A.3.6), SM agar con xilosa 0.5% (Anexo A.3.7), MMBM[33] agar (Anexo A.3.8) con S-AAPF-pNa y MMBM agar con sustrato sintético S-AAPF-pNa y xilosa 0.5% (Anexo A.3.9). Las placas se incubaron durante la noche a 37 ºC, para crecer las células, y posteriormente a temperatura ambiente durante el mismo período, para incubar la reacción enzimática. De ser necesario, para facilitar la visualización de los halos de actividad, las placas de SM agar fueron teñidas con azul de Coomassie según se indica en el punto 2.2.7 para el caso de los geles de SDSpoliacrilamida. 2.2.6 INDUCCIÓN Y RECUPERACIÓN DE PROTEÍNA RECOMBINANTE DESDE Bacillus megaterium Se realizó inducción de los cultivos transformados con los plasmidios pHIS1522-T8, pHIS1525-T8, pHIS1525-T7 y pHIS1522 como control. Se tomó muestras de proteína total intracelular, sobrenadante de cultivo, fracción citoplasmática soluble e insoluble, siguiendo el protocolo indicado en el Anexo A.4.5. Posteriormente, siguiendo con el mismo protocolo, las muestras de proteína correspondientes al sobrenadante y a la fracción citoplasmática soluble fueron purificadas en condiciones nativas por cromatografía de afinidad utilizando resina de NiNTA (QIAGEN), que posee afinidad por la cola de polihistidina de las proteínas recombinantes. Finalmente, éstas últimas fueron concentradas mediante centrifugación en tubos Centricon YM10 (Millipore) (Anexo A.4.6). 2.2.7 DETECCIÓN DE PROTEÍNAS RECOMBINANTES Con el objetivo de estudiar la producción de proteína recombinante en las distintas fracciones celulares, se analizó proteína total intracelular, sobrenadante, citoplasmática soluble e insoluble de los cultivos inducidos. Las distintas muestras fueron analizadas en geles de SDS-poliacrilamida (Anexo A.5.1). Se tomó 24 µL de cada muestra con 6 µL de buffer de carga 5x (Anexo A.6.1), y se calentó durante 5 minutos a 100 ºC. Luego se centrifugó 5 minutos a 13000 rpm y el sobrenadante se cargó en el gel. Las muestras fueron corridas a una diferencia de voltaje de 200 V durante 45 minutos. Su visualización se realizó a través de tinción con azul de Coomassie. Para esto, los geles se incubaron en solución de tinción (Anexo A.6.2) un tiempo mínimo de 2 horas agitando suavemente. Luego se realizó destinción del gel incubándolo en solución de destinción (Anexo A.6.3) con agitación suave, hasta apreciar fácilmente las bandas de proteína. 17 2.2.8 PRUEBAS DE ACTIVIDAD PROTEASA DE PROTEÍNAS RECOMBINANTES Se analizó la actividad proteasa de las proteínas recombinantes de las muestras de sobrenadante y fracción citoplasmática soluble, obtenidas de acuerdo al método descrito en el punto 2.2.6, en geles de zimograma (Anexo A.5.2). Se tomó 15 µL de muestra y 15 µL de buffer de carga 2x (Anexo A.6.4). Se centrifugó 5 minutos a 13000 rpm y se cargó el sobrenadante. Las muestras fueron corridas durante 45 minutos a 200 V. Luego, los geles de zimograma fueron incubados en una solución de Tritón 1% durante 20 minutos, con agitación suave. Posteriormente, se transfirió a solución de incubación (Anexo A.6.5) y se incubó durante la noche, a temperatura ambiente, agitando suavemente. Después se incubó en solución de tinción y distinción de igual forma que los geles de SDS-poliacrilamida descrita en el punto anterior. 2.2.9 ANÁLISIS DE pHIS1525-T7 LAS CONSTRUCCIONES pHIS1522-T8, pHIS1525-T8 Y Para analizar la correcta construcción de los vectores pHIS1522-T8, pHIS1525-T8 y pHIS1525-T7, fueron digeridos con enzimas de restricción y secuenciados. Las enzimas de restricción utilizadas se muestran en la Tabla 2. Las reacciones se realizaron bajo las condiciones indicadas por cada proveedor. En los casos de doble digestión no simultánea, el DNA se desaliniza con el método indicado en el manual del QIAEX II Gel Extraction Kit[34], entre digestiones. Los resultados fueron visualizados en geles de agarosa 1%, o en geles de agarosa 0.8% cuando los productos de digestión eran de tamaños mayores a 3 kb. Tabla 2: Enzimas de restricción usadas en el análisis de las construcciones plasmidiales Construcción pHIS1522-T8 pHIS1525-T8 pHIS1525-T7 Enzimas SpeI, SphI, XhoI, EcoRV KasI, SphI, XhoI, EcoRV PstI, BamHI KasI, SphI, XhoI, EcoRV NcoI Proveedor New England Biolabs New England Biolabs Promega New England Biolabs Fermentas La secuenciación de los vectores se realizó en Macrogen Inc. (Corea), con partidores cercanos al sitio de clonamiento. Para la construcción correspondiente a la subtilasa T7 clonada en pHIS1525 se utilizó además partidores pertenecientes al inserto T7 (pT7 y pT7R). El detalle de éstos se muestra en la Tabla 3. En el Anexo A.7 (Figura A.7) se muestra un esquema de la ubicación de los partidores utilizados. 18 Tabla 3: Partidores para secuenciación Partidor pHIS1 pHIS2 pHIS3 pHISR pHISR1 pHISR2 pHISR3 pT7 pT7R Secuencia 5’…GGATAAACAAACTAACTCAATTAAGATAG...3’ 5’……AACCACTCCTTTGTTTATCCACCGAAC…...3’ 5’.……..…AGAGAGTTGCCCTGGAGACAG………..3’ 5’…………GTTCTCCGCAAGAATTGATTG……...…3’ 5’………ATGTTCTGCCAAGGGTTGGTTTGC…...…3’ 5’…..…TCGATTTTTGTGATGCTCGTCAGGG…..…3’ 5’…….ACGATAGTTACCGGATAAGGCGCAG…….3’ 5’……..TTGAAGTGGATCAAATGTTAAAGC……...3’ 5’…..…CATTAAGCATTACGTGATAAGTGC……...3’ 19 3 RESULTADOS Y DISCUSIÓN Para la expresión recombinante de los genes de las subtilasas T7 y T8, se amplificó desde el DNA genómico de Pseudoalteromonas sp. y Polarisbacter sp., respectivamente, la secuencia codificante de dichas proteasas sin su péptido señal, con la intención de que estas sean fusionadas a la señal de exportación del vector de expresión pHIS1525. Lo anterior, se debió principalmente a que los microorganismos de origen son Gram negativos, mientras que el huésped es Gram positivo, y por lo tanto, sus señales de exportación tienen grandes diferencias. Por ejemplo, una mayor cantidad de residuos con carga positiva es encontrada en prepéptidos de bacterias Gram positivas[7]. Se encuentra también diferencias en el tamaño de la secuencia[7]. Además, se tienen antecedentes de que la misma señal produce sitios de corte distintos en cada tipo de microorganismo[7]. La subtilasa T7 fue clonada como propéptido, pues se cree que la prosecuencia puede facilitar la traslocación y ayudar además al adecuado plegamiento de la enzima[7]. La Figura 2 muestra un esquema de las secuencias clonadas y fusionadas al péptido señal del vector. Figura 2: Esquema de las secuencias clonadas. Anarajado: Péptido señal SPlypA del vector pHIS1525; azul: prosecuancia de T7; gris: secuencias de las enzimas. Se utilizó la técnica de PCR con partidores específicos que incorporaron sitios de restricción para enzimas del sitio de clonamiento múltiple de los vectores de expresión para Bacillus megaterium pHIS1522 y pHIS1525 (ver Tabla 1). El primer vector permite estudiar la expresión intracelular de las enzimas recombinantes, mientras que pHIS1525 permite el estudio de su expresión en forma extracelular. Los fragmentos obtenidos fueron clonados en el vector pGEM-T Easy para su amplificación in vivo, desde donde fueron recuperados y ligados a los vectores de expresión. Estos últimos fueron amplificados previamente en células de E. coli DH5α. Se clonó así el gen de la subtilasa T8 en los vectores pHIS1522 y pHIS1525, y el gen de la subtilasa T7 sólo en el vector pHIS1525, debido a que es de especial importancia pues incorpora una señal de exportación a la proteína clonada. Se transformó protoplastos de B. megaterium con las distintas construcciones, y con pHIS1522 y pHIS1525 como control negativo. Las cepas transformadas fueron sembradas en distintos medios sólidos con sustrato, diseñados para detectar in situ la expresión activa de proteína recombinante. Las construcciones plasmidiales fueron analizadas mediante cortes con enzimas de restricción y secuenciadas para verificar el clonamiento del gen en el marco de lectura correcto. Por otra parte, se prepararon fracciones de sobrenadante de cultivo, citoplasmática soluble, citoplasmática insoluble y proteína total intracelular de las cepas transformadas. Las 20 muestras de sobrenadante y fracción citoplasmática soluble fueron cargadas en columnas de NiNTA para purificar las subtilasas recombinantes mediante cromatografía de afinidad. Los análisis de las muestras se realizaron en geles de SDS-poliacrilamida, utilizando como método de detección la tinción con azul de Coomassie. Se realizaron pruebas de actividad proteasa en geles de zimograma de las muestras de proteína recombinante purificada desde sobrenadante y lisis soluble de los cultivos de las cepas transformadas. 3.1 AMPLIFICACIÓN DE LOS VECTORES pHIS1522 Y pHIS1525 EN E. coli DH5α Para obtener un mayor número de copias de los vectores pHIS1522 y pHIS1525, se transformó con ellos células de E. coli DH5α. Los vectores amplificados fueron cortados con enzimas de restricción para linealizarlos y luego sometidos a electroforesis. En paralelo, fueron digeridos plasmidios control (no amplificados en E. coli). Los vectores pHIS1522 fueron digeridos con SpeI, mientras que para digerir los vectores pHIS1525 se utilizó BamHI. Los resultados se muestran en la Figura 3. Figura 3: Producto de amplificación de plasmidios en E. coli DH5α. (1) pHIS1522 control, (2) y (3) pHIS1522 obtenido desde células de E. coli DH5α. (4): pHIS1525 control, (5) y (6): plasmidios pHIS1525 amplificados en E. coli DH5α. (M) Marcador de tamaño 1 kb (Invitrogen). Las bandas obtenidas coinciden con los tamaños esperados para la linealización de ambos vectores control: 7381 pb para pHIS1522 y 7441 pb para pHIS1525 (indicados con una flecha roja en la Figura 3). Los resultados de la amplificación en E. coli parecen ser satisfactorios, pues los tamaños de los vectores amplificados están dentro de lo esperado, coincidiendo con el de los plasmidios originales. 21 3.2 EXPRESIÓN RECOMBINANTE DE SUBTILASA T8 EN B. megaterium 3.2.1 CLONAMIENTO DEL GEN DE SUBTILASA T8 EN pHIS1522 Y pHIS1525 Desde DNA genómico, se amplificó el gen codificante de la subtilasa T8 sin el fragmento que codifica para el péptido señal, con partidores específicos que permitieron la incorporación de sitios de restricción para el posterior clonamiento en los plasmidios de expresión. Se incorporó sitios para KasI y SpeI al extremo 5’ para clonar en pHIS1522 y pHIS1525, respectivamente, y SphI al extremo 3’ en ambos casos. Los productos de amplificación fueron subclonados en el vector pGEM-T Easy, dando lugar a las construcciones pGEM-T-T822 y pGEM-T-T825. Luego se transformó células de Escherichia coli DH5α con la mezcla de ligación. De las colonias transformadas se realizó PCR de colonias blancas para seleccionar aquellas que tenían efectivamente el inserto deseado, y se seleccionó una de ellas para crecer y extraer el DNA plasmidial. Los plasmidios obtenidos fueron digeridos con las enzimas de restricción correspondientes a los sitios de restricción incorporados (SpeI y SphI en el caso de pGEM-T-T822, y KasI y SphI para pGEM-T-T825), para luego ligar los insertos a los vectores de expresión correspondientes digeridos de igual forma. Se digirió pHIS1522 y pHIS1525 amplificados en células de E. coli, y pHIS1525 control. La foto de la Figura 4 muestra el gel para extracción, producto de la digestión de pGEMT-T825 y pHIS1525, con las enzimas KasI y SphI. Figura 4: Gel para extracción de inserto T825 y plasmidio pHIS1525, digeridos con enzimas SphI y KasI. (1)Digestión de pGEM-T-T825, las bandas corresponden al gen de T8 de 1536 pb y al vector pGEM-T Easy de aproximadamente 3 kb. (2) Digestión del vector pHIS1525 amplificado in vivo en E. coli DH5α. (3) Digestión de pHIS1525 control. (M) Marcador de tamaño 1 kb (Invitrogen). Se observó que el vector pHIS1525 amplificado en E. coli (carril nº2) presentó una migración menor que el vector pHIS1525 control (carril nº3), indicando una posible diferencia de tamaños que se discutirá mas adelante. 22 A continuación se ligó cada inserto en el plasmidio correspondiente. Se obtuvieron las construcciones pHIS1522-T8, pHIS1525-T8 y pHIS1525-T8 control. Esta última construcción se realizó con plasmidio control, es decir, no amplificado en E. coli. Las mezclas de ligación fueron utilizadas para transformar, mediante electroporación, células de E. coli DH5α, con el objeto de amplificar in vivo los vectores con inserto. Luego se realizó PCR de las colonias transformadas para corroborar la presencia del inserto en el vector de transformación (Anexo A.8.1, Figura A.8.1). De las colonias positivas, se escogió una de cada transformación, seleccionando aquella con mayor cantidad de producto de amplificación. 3.2.2 TRANSFORMACIÓN DE PROTOPLASTOS DE Bacillus megaterium CON pHIS1522-T8 Y pHIS1525-T8 Para transformar protoplastos de B. megaterium, se realizó una minipreparación de DNA plasmidial de E. coli desde un cultivo de 6 mL. Se obtuvo así los plasmidios pHIS1522, pHIS1522-T8, pHIS1525, pHIS1525-T8 y pHIS1525-T8 control, en una mayor concentración de la habitual debido al mayor volumen de cultivo utilizado. Para verificar el estado de los plasmidios, éstos fueron cortados con enzimas de restricción. Los vectores pHIS1522 y pHIS1522-T8 fueron digeridos con SpeI y SphI, mientras que pHIS1525-T8 y pHIS1525-T8 control, se digirieron con KasI y SphI. Los resultados del análisis se muestran en la Figura 5. Figura 5: Digestión de minipreparación de plasmidios de expresión. (1) pHIS1522, (2) pHIS1522-T8, (3) pHIS1525, (4) pHIS1525-T8 y (5) pHIS1525-T8 control. (M) Marcador de tamaño 1 kb (Invitrogen). Se vio que los vectores tenían las características deseadas. El resultado anterior mostró además, que no hay diferencias apreciables entre los plasmidios amplificados en células transformadas con construcciones control, y los plasmidios de aquellas transformadas con construcciones hechas con plasmidios amplificados previamente en células de E. coli DH5α. Por esto, para la construcción pHIS1525-T8 se continuó el procedimiento sólo con aquella correspondiente al control por tener mayor cantidad de DNA plasmidial (o banda más intensa), eliminándose en adelante este tipo de control. 23 Con los plasmidios pHIS1522, pHIS1522-T8, pHIS1525, y pHIS1525-T8, se transformaron protoplastos generados en el laboratorio y protoplastos control (comerciales), con el objeto de analizar la calidad de aquellos producidos en el laboratorio. Todos los cultivos dieron resultados positivos para la transformación. La diferencia se dio en la distribución de las colonias en las placas con CR5 top agar, la cual fue homogénea en las placas de protoplastos control, pero se limitó a los bordes en las placas de protoplastos de laboratorio. Lo anterior se le atribuye a la presencia de glicerol en los protoplastos producidos en el laboratorio, el cual pudo dificultar la distribución del cultivo en el agar. Se realizó PCR de colonias de 4 colonias de cada transformación con inserto utilizando los partidores T8SpeI(s) y T8SphI(r) en las colonias transformadas con pHIS1522-T8 y T8KasI(s) y T8SphI(r) (ver Tabla 1) en las colonias transformadas con pHIS1525-T8, pero no hubo producto de amplificación. Se sugiere agregar una etapa de lisis con lisozima 1 mg/mL, incubando 10 minutos a temperatura ambiente, al realizar PCR de colonias de B. megaterium, pues se cree que el llevar las células a 100 ºC por 10 minutos no produjo lisis celular, por lo que no se liberó material genético, dejando a la reacción de PCR sin DNA templado. Se escogió al azar una colonia de B. megaterium y una de B. megaterium control de cada transformación con inserto (pHIS1522-T8 y pHIS1525-T8). A partir de estas colonias se preparó cultivos para minipreparación. Se purificó el DNA plasmidial desde estos cultivos, y luego se hizo PCR utilizando como templado los plasmidios obtenidos, en una dilución 1:10. Las cepas transformadas poseen plasmidios con el inserto T8, pues los productos de amplificación coinciden con el tamaño esperado (Anexo A.8.2. Figura A.8.2). 3.2.3 REVISIÓN DE PLASMIDIOS AMPLIFICADOS EN Bacillus megaterium Los plasmidios purificados desde B. megaterium, se cortaron con las enzimas de restricción SpeI y SphI para pHIS1522-T8, y KasI y SphI para pHIS1525-T8. En ambos casos, se esperaba obtener dos bandas en el producto de digestión, correspondientes al vector, de 7381 y 7441 pb, respectivamente, y al inserto T8, de 1536 pb. Sin embargo, para el segundo plasmidio se obtuvo un resultado inesperado, pues si bien se observó la banda correspondiente al inserto, aparecieron al menos otras tres bandas en el gel. Este resultado se muestra en la Figura 6. 24 Figura 6: Digestión de plasmidios pHIS1522-T8 y pHIS1525-T8 purificados desde cultivos de Bacillus megaterium. (1) pHIS1522-T8, (2) pHIS1525-T8 y (3) pHIS1525-T8 de B. megaterium control. (M): Marcador de tamaño 1 kb (Invitrogen). Información del proveedor indica que la enzima KasI es afectada en su desempeño por metilaciones del tipo CpG. Se transformó entonces, células de E. coli con estos plasmidios extraídos desde Bacillus, para verificar si las nuevas células huésped corregían aquellas metilaciones del DNA. Resultó que el DNA amplificado en E. coli presentó un producto de digestión idéntico al esperado, observándose sólo las bandas correspondientes al vector y al inserto (ver Figura 7). Figura 7: Digestión de plasmidios pHIS1525-T8 amplificados en E. coli. (1) pHIS1525-T8 purificado desde B. megaterium y amplificado en E. coli y (2) pHIS1525-T8 amplificado en E. coli como muestra control. (M) Marcador de tamaño 1 kb (Invitrogen). Se decidió entonces cambiar las enzimas de restricción para el análisis de plasmidios pHIS1525-T8 purificados desde B. megaterium, por PstI y SphI, ya que su actividad no es influenciada por la presencia de grupos metilo en el DNA sustrato. Esta digestión debe generar bandas de 1254 pb y 7756 pb. Los resultados de esta digestión se muestran en la Figura 8. 25 Figura 8: Digestión de pHIS1525-T8 de B. megaterium con PstI y SphI. (1) pHIS1525-T8 purificado desde E. coli DH5α, como muestra control y (2) pHIS1525-T8 purificado desde B. megaterium. (M) Marcador de tamaño 1 kb (Invitrogen). Se puede apreciar que la digestión de plasmidios purificados desde E. coli y B. megaterium con PstI genera fragmentos del mismo tamaño, correspondientes al esperado para la construcción pHIS1525-T8. Además, se observa claramente que la eficiencia de la minipreparación desde B. megaterium es mucho menor que desde E. coli. 3.2.4 PRUEBAS PRELIMINARES DE ACTIVIDAD PROTEASA EXTRACELULAR Para determinar en forma preliminar la producción extracelular de proteína recombinante por parte de las cepas transformadas, se realizó pruebas de actividad proteasa en medio sólido de todas las transformaciones. Para esto se utilizó placas de SM agar y SM agar con xilosa 0.5%. Las placas fueron incubadas a 37 ºC durante la noche. Se observó que las transformaciones que utilizaron protoplastos de B. megaterium de origen comercial (control) no crecieron después de la transferencia, a excepción de la colonia 13, mientras que las de protoplastos producidos en el laboratorio sí lo hicieron. Inesperadamente, los resultados fueron indiferentes a la presencia de inductor xilosa en el medio, y asimismo, al contenido de inserto de T8 en el vector. Esto se aprecia claramente en la Figura 9. 26 Figura 9: Actividad proteasa extracelular en Bacillus megaterium. A: SM agar y B: SM agar con xilosa. (1-4): pHIS1522-T8 en B. megaterium; (5-8): pHIS1522-T8 en B. megaterium control; (9-12): pHIS1525-T8 en B. megaterium; (13-16): pHIS1525-T8 en B. megaterium control; (17-18): pHIS1522 en B. megaterium; (19-20): pHIS1522 en B. megaterium control; (21-22): pHIS1525 en B. megaterium; (23-24): pHIS1525 en B. megaterium control. La presencia de halos en cultivos con vectores sin inserto se atribuyó a la presencia de una proteasa endógena producida principalmente en la fase estacionaria de crecimiento en B. megaterium, de acuerdo a información encontrada en bibliografía[35], cuyo peso molecular es de 34.15 kDa[36]. Estos resultados inhabilitan el ensayo de actividad proteasa extracelular por medio de placas con SM agar con inductor, por lo que se debió buscar otro sistema simple y sensible para detectar actividad proteasa extracelular en medio sólido. 3.2.5 INDUCCIÓN Y RECUPERACIÓN DE PROTEÍNA RECOMBINANTE T8 DESDE B. megaterium Para estudiar la producción de proteína recombinante, se hizo inducción de la expresión de la proteína en cultivos de B. megaterium transformados con pHIS1522-T8, pHIS1525-T8 y pHIS1522, este último como control negativo. El proceso se realizó a 37 ºC durante 2.5 horas. Se espera encontrar producción de proteína recombinante intracelular de 57.4 kDa en la cepa transformada con pHIS1522-T8. Para el caso de la transformación con pHIS1525-T8, se espera que la proteína se encuentre en el medio extracelular, pues el vector pHIS1525 permite la expresión de la proteína recombinante fusionada a un péptido señal, siendo el tamaño esperado de 59.6 kDa. Se tomaron muestras del sobrenadante, fracción soluble del citoplasma y proteína total intracelular de los tres cultivos. Aquellas correspondientes al sobrenadante y a la fracción citoplasmática soluble fueron purificadas mediante cromatografía de afinidad a cola de histidinas 27 en condiciones nativas. Las muestras de proteína total se utilizaron como control para analizar la presencia de proteína recombinante no purificada. Las muestras fueron cargadas en un gel de SDS-poliacrilamida, y visualizadas mediante tinción con azul de Coomassie. El resultado se muestra en la Figura 10. Figura 10: Inducción y Recuperación de Proteína Recombinante T8 desde B. megaterium. (1-3) Sobrenadante de cultivo: (1) pHIS1522-T8, (2) pHIS1525-T8, (3) pHIS1522; (4-6) Fracción citoplasmática soluble: (4) pHIS1522T8, (5) pHIS1525-T8, (6) pHIS1522; (7-9) Proteína total intracelular: (7) pHIS1522-T8, (8) pHIS1525-T8, (9) pHIS1522, (M) Marcador de masa molecular (Fermentas). Se puede observar que en las muestras de sobrenadante de cultivo no se detectó presencia de proteínas purificadas por el método de cromatografía. En las muestras de la fracción soluble del citoplasma se aprecian varias bandas, entre las que destaca una banda presente en el carril nº4, correspondiente a la transformación con pHIS1522-T8. Considerando que la proteína T8 fusionada a la cola de polihistidina y sin señal de exportación posee un peso molecular de 57.4 kDa, esta banda se identificó como proteína recombinante. Se estableció la presencia de una proteína no recombinante de peso molecular aproximado presente en las muestras correspondientes a pHIS1525-T8 y en el control negativo. Las muestras correspondientes a proteína total intracelular corroboran la producción de proteína recombinante por la cepa transformada con pHIS1522-T8. Se realizó un ensayo de zimograma a las muestras anteriores. No se observó actividad proteasa en ninguna de las muestras analizadas. De lo anterior, se concluyó que hubo expresión intracelular de subtilasa T8 por la cepa transformada con pHIS1522-T8, sin embargo, la proteína producida no posee actividad enzimática. 28 3.3 EXPRESIÓN RECOMBINANTE DE SUBTILASA T7 EN Bacillus megaterium 3.3.1 CLONAMIENTO DEL GEN DE SUBTILASA T7 EN pHIS1525 Se clonó la subtilasa T7 con su prosecuencia en el vector de expresión para B. megaterium pHIS1525. Desde DNA genómico de Pseudoalteromonas sp., se amplificó el gen de la subtilasa T7 sin su presecuencia, pero incluyendo su prosecuencia, para ser clonado en pHIS1525. Se trabajó sólo con este vector debido a que dota a la proteasa recombinante de una señal de exportación, lo que permite su secreción al medio extracelular. En la reacción de PCR, se utilizaron los partidores T7KasI(s) y T7SphI(r), para incorporar sitios de restricción para KasI al extremo 5’, y SphI al extremo 3’, enzimas del sitio de clonamiento múltiple del vector de expresión. El producto de PCR se clonó en pGEM-T Easy para ser amplificado in vivo en E. coli. Luego se realizó minipreparación de DNA plasmidial desde el cultivo en medio líquido de una colonia seleccionada como positiva mediante PCR de colonias. Los plasmidios obtenidos y el vector pHIS1525 fueron digeridos con KasI y SphI, para luego ligar el inserto y el vector de expresión. La Figura 11 muestra el gel de extracción con los productos de digestión de pGEM-TT7 y pHIS1525. En el carril nº1 se aprecian dos bandas con gran cantidad de DNA, una de unos 3000 pb correspondiente al vector de clonamiento y más abajo la correspondiente al gen de T7 de 2043 pb. Figura 11: Gel para extracción de gen de proteína T7 y vector pHIS1525, digeridos con enzimas KasI y SphI. (1) Digestión de pGEM-T-T7; (2) Digestión de pHIS1525. (M) Marcador de tamaño 1 kb (Invitrogen). El producto de ligación del inserto T7 con pHIS1525 fue amplificado en células de E. coli DH5α. De las colonias obtenidas se realizó PCR de colonias para corroborar la presencia del inserto en el vector de expresión de B. megaterium. Se seleccionó una colonia positiva y se preparó cultivo para minipreparación. 29 3.3.2 TRANSFORMACIÓN DE PROTOPLASTOS DE Bacillus megaterium CON pHIS1525-T7 Se realizó una minipreparación de DNA plasmidial de un cultivo de 6 mL de una colonia de E. coli transformada con pHIS1525-T7, con el objeto de obtener mayor cantidad de DNA. Se digirió el DNA obtenido con las enzimas KasI y SphI para verificar la construcción. Luego se transformó protoplastos de B. megaterium generados en el laboratorio. No se transformó protoplastos comerciales pues anteriormente los resultados no fueron satisfactorios con este control. La transformación fue efectiva, obteniéndose varias colonias, las cuales crecieron después de transferirlas a una nueva placa según indica el protocolo. 3.3.3 REVISIÓN DE PLASMIDIOS AMPLIFICADOS EN B. megaterium Se seleccionaron dos colonias de B. megaterium transformadas con pHIS1525-T7 y se realizó minipreparación de DNA plasmidial. Para chequear los plasmidios purificados se realizó digestión con la enzima NcoI. Se escogió esta enzima pues al digerir la construcción pHIS1525T7 genera dos fragmentos fácilmente distinguibles de 1550 pb y 7926 pb, y además no es bloqueada por ningún tipo de metilación. La Figura 12 muestra los resultados de la digestión. Figura 12: Digestión de plasmidios purificados desde B. megaterium con NcoI. (1) y (2) pHIS1525-T7 de distintas colonias de B. megaterium. (M) Marcador de tamaño 1 kb (Invitrogen). La digestión de plasmidios purificados desde B. megaterium generó dos fragmentos de los tamaños esperados, implicando un resultado positivo para la transformación. 30 3.3.4 PRUEBAS PRELIMINARES DE ACTIVIDAD PROTEASA EXTRACELULAR Se realizó pruebas preliminares de actividad proteasa extracelular en medio sólido de cepas tranasformadas con pHIS1525-T7, utilizando como control negativo la transformación con pHIS1522. La información bibliográfica indica que la proteasa endógena de B. megaterium que inhabilita los ensayos en medio SM agar utilizados en el experimento anterior (punto 3.2.4) es una proteasa neutra[35]. Por otra parte, se ha determinado que la proteasa extracelular de la cepa 317 de Polarisbacter sp. (cepa de origen del gen T8) posee pH óptimo entre 7.0 y 9.0[19] y que es muy probable que la subtilasa codificada por el gen T7 sea alcalina debido a que su secuencia presenta gran similitud con las de otras proteasas tipo subtilisina[19]. Lo anterior que genera una importante herramienta de selección, por lo que se utilizó placas de SM agar con xilosa 0.5% a pH alcalino. Se preparó placas de SM agar con xilosa 0.5% a pH 8.0 y a pH 10.0. Se sembró dos colonias de B. megaterium transformadas con pHIS1525-T7 y otras dos transformadas con pHIS1522 como control negativo en cada placa. Los resultados se pueden ver el la Figura 13. Figura 13: Pruebas de actividad proteasa extracelular en placas de SM agar con xilosa a pH alcalino. A: SM agar con xilosa a pH 8.0 y B: SM agar con xilosa a pH 10.0. (1-2) B. megaterium con pHIS1522 (control negativo); (3-4) B. megaterium con pHIS1525-T7. Se estableció que B. megaterium es capaz de crecer en condiciones alcalinas, incluso a pH 10.0. No se obtuvo diferencias entre la actividad proteasa extracelular de los clones transformados con pHIS1525-T7 y de aquellos utilizados como control negativo. Se determinó que la actividad de la proteasa neutra endógena del microorganismo disminuyó con el aumento de la alcalinidad, pero no logró ser inactivada completamente, lo que 31 dificulta la identificación de clones con expresión extracelular de proteasa recombinante mediante este método, pues niveles de expresión pequeños no podrían ser detectados. 3.3.5 INDUCCIÓN Y RECUPERACIÓN DE PROTEÍNA RECOMBINANTE T7 DESDE B. megaterium Para estudiar la producción de proteína recombinante, se indujo su producción en cultivos de B. megaterium transformados con pHIS1525-T7 y pHIS1522 como control negativo. Los cultivos fueron inducidos durante 2.5 horas a 37 ºC. Se espera encontrar una proteína recombinante de 71.14 kDa expresada en forma extracelular pues el vector pHIS1525 fusiona a la proteína clonada a una señal de exportación. Se analizó en un gel de SDS-poliacrilamida con tinción con azul de Coomassie las muestras de sobrenadante de cultivo y fracción soluble citoplasmática purificadas por cromatografía de afinidad en condiciones nativas. Se analizó además muestras de proteína total intracelular de ambos cultivos para detectar la expresión proteína recombinante no purificada, como control para la etapa de purificación. No se observó ninguna banda indicativa de expresión de proteína recombinante en ninguna de las fracciones. Debido a la posibilidad de que existiera expresión de proteína recombinante activa en cantidades no detectables por SDS-PAGE, se realizó ensayo de zimograma de las mismas muestras para detectar la presencia de proteína recombinante a través de su actividad, obteniéndose resultados negativos para todas ellas. En base a éste resultado, se decidió rehacer una de las construcciones realizadas con pHIS1525 que permite la secreción de la proteína recombinante, pues se consideró que la ausencia de expresión de proteína recombinante por las cepas transformadas con las construcciones pHIS1525-T8 y pHIS1525-T7 podría deberse a cambios en el marco de lectura producidos por mutaciones generadas en alguna etapa del proceso. Un análisis de la secuencia de la subtilasa T8 con el programa LipoP 1.0 Server[37] predijo la presencia de un péptido señal para lipoproteínas en el extremo amino, por lo que probablemente corresponda a una lipoproteína. Esto sugiere que la región de 148 aminoácidos entre los residuos catalíticos de aspartato e histidina (ver Figura 1) podría corresponder a la región mediante la cual la subtilasa queda anclada a la membrana citoplasmática. Por lo anterior, se decidió repetir el proceso con la proteína T7. Se amplificó el gen nuevamente desde DNA genómico. La nueva construcción fue realizada de igual forma que pHIS1525-T7, pero utilizando vector pHIS1525 control (no amplificado en E. coli). Se obtuvo entonces la construcción pHIS1525-T7H, con la cual se transformó protoplastos de B. megaterium generados en el laboratorio. Se realizó ensayo de actividad en medio sólido de las colonias transformadas. Debido a los problemas detectados al utilizar placas de SM (ver puntos 3.2.4 y 3.3.4), se decidió utilizar un 32 sustrato específico para serina proteasas denominado S-AAPF-pNa (N-succinil-ala-ala-pro-phe pnitroanilida), el cual al ser hidrolizado genera un producto de color amarillo. Se utilizó como medio nutritivo MMBM (Medio Mínimo para Bacillus megaterium), pues posee un color grisáceo donde el producto de la hidrólisis del sustrato es más fácil de visualizar que en medios más comunes como LB (Luria Broth) de color amarillo. Como control negativo se utilizó B. megaterium transformado con pHIS1522 y como control positivo subtilisina Carlsberg (Sigma). No se observó halos de actividad en ninguna de las colonias de Bacillus megaterium, pero sí sobre las muestras de subtilisina comercial. Lo anterior no permitió concluir sobre la expresión recombinante de la proteína T7, pues no se conoce la sensibilidad del sistema, pero indica que con el uso del medio MMBM agar con S-AAPF-pNa se logra discriminar la actividad proteasa extracelular producto de la expresión de la proteasa neutra de B. megaterium, de la actividad de proteasas del tipo subtilisina, como la que se desea producir en forma recombinante. En lugar de realizar PCR de colonias, se realizó inducción de once colonias de Bacillus megaterium transformadas con pHIS1525-T7H, seleccionadas al azar, usando la transformación con pHIS1522 como control negativo. Se tomó muestras de 2 mL de sobrenadante y se concentró con tubos Centricon® YM-10 (Anexo A.4.6). Luego, se realizó análisis en un gel de SDSpoliacrilamida visualizado con azul de Coomassie, y ensayo de zimograma. No se detectó la presencia de una proteína recombinante por ninguno de los dos métodos. 3.4 INDUCCIÓN DE PROTEÍNAS RECOMBINANTES A BAJA TEMPERATURA La información encontrada en bibliografía indica que la disminución de la temperatura durante el proceso de inducción mejora la expresión de proteínas recombinantes[38], y disminuye la formación de cuerpos de inclusión. Se encontró además que la inducción de algunas proteasas psicrofilicas expresadas en microorganismos mesófilos se realiza a temperaturas menores a 37ºC[23 y 1], posiblemente debido a su termolabilidad. Se decidió por lo tanto, repetir la inducción de pHIS1522-T8 a 25 ºC, pues, bajo estas condiciones podría plegarse adecuadamente y producirse en forma activa. Se usó como control negativo la transformación con pHIS1522. Se realizó además inducción a 25 ºC de las construcciones pHIS1525-T8, pHIS1525-T7 y pHIS1525-T7H, para confirmar los resultados obtenidos con anterioridad. Los cultivos fueron inducidos durante 17 horas, pues a 25 ºC el metabolismo celular es más lento que a 37 ºC. Se analizó muestras de sobrenadante de cultivo y fracción citoplasmática soluble purificadas por cromatografía de afinidad a cola de polihistidinas en condiciones nativas. Se analizó además muestras de fracción citoplasmática insoluble en busca de expresión recombinante de proteína en forma de cuerpos de inclusión y proteína total intracelular para visualizar proteína recombinante no purificada. El análisis se llevó a cabo mediante geles SDS-PAGE (Figura 14 y Figura 15) y zimograma. Estos últimos no mostraron bandas de actividad para ninguna de las muestras. 33 Figura 14: Efecto de la temperatura en la expresión recombinante de proteasas por B. megaterium. (1-5) Sobrenadante de cultivo purificado: (1) pHIS1522, (2) pHIS1522-T8, (3) pHIS1525-T8, (4) pHIS1525-T7, (5) pHIS1525-T7H; (6-7) Fracción citoplasmática soluble purificada: (6) pHIS1522, (7) pHIS1522-T8, (8) pHIS1525-T8, (9) pHIS1525-T7 y (10) pHIS1525-T7H. (M) Marcador de masa molecular (Fermentas). Figura 15: Efecto de la temperatura en la expresión recombinante de proteasas por B. megaterium. (1-5): Fracción citoplasmática insoluble: (1) pHIS1522, (2) pHIS1522-T8, (3) pHIS1525-T8, (4) pHIS1525-T7, (5) pHIS1525-T7H; (6-7) Proteína total intracelular: (6) pHIS1522, (7) pHIS1522-T8, (8) pHIS1525-T8, (9) pHIS1525-T7 y (10) pHIS1525-T7H. (M) Marcador de masa molecular (Fermentas). Se detectó sólo la expresión de proteína recombinante en las muestras correspondientes a la fracción citoplasmática soluble y proteína total del cultivo transformado con pHIS1522-T8 (Figura 14, carril nº7 y Figura 15, carril nº7, respectivamente), confirmando los resultados anteriores (ver página 28). El análisis de la proteína intracelular insoluble mostró la existencia de cuerpos de inclusión en las células transformadas con pHIS1522-T8 (Figura 15, carril nº2). La acumulación de proteína recombinante dentro de la célula concuerda con los resultados esperados, pues ésta no fue diseñada para su exportación al medio extracelular. Ésta se presenta principalmente mediante 34 la formación de cuerpos de inclusión. El resto de las muestras no presentó diferencias con respecto al control negativo, descartando la producción de proteína recombinante T7 o T8. Se comparó nivel de expresión de T8 de la transformación con pHIS1522-T8 con el obtenido por inducción a 37 ºC por 2.5 horas (punto 3.2.5). Ambos cultivos eran de un volumen de 100 mL y fueron concentrados a 250 µL (como indican los protocolos A.4.5 y A.4.6 del Anexo A.4), por lo que la simple visualización de la intensidad de la banda puede ser utilizada para comparar los niveles de expresión de proteína recombinante. Se comparó entonces las bandas de proteína total obtenida en ambos casos (compare Figura 10, carril nº7 y Figura 15, carril nº7). Se determinó así, que la inducción a 25 ºC no mejoró la expresión de la proteína T8 en Bacillus megaterium, pues las bandas obtenidas son similares en ambas condiciones, y se ve incluso una menor cantidad de proteína citoplasmática soluble purificada en las muestras obtenidas por inducción a baja temperatura (compare Figura 10, carril nº4 y Figura 14, carril nº7). Por otra parte, del ensayo de zimograma se concluyó que la disminución de la temperatura a 25 ºC durante el proceso de inducción no logró que la proteína recombinante soluble T8, producida por la cepa transformada con pHIS1522-T8, se plegara adecuadamente a su forma activa. Lo anterior no descarta que el descenso de la temperatura de inducción pueda mejorar los resultados en sistemas de expresión extracelular de proteínas recombinantes en B. megaterium, pues no se pudo comparar resultados para este caso. 3.5 ANÁLISIS DE LAS CONSTRUCCIONES pHIS1522-T8, pHIS1525-T8, pHIS1525T7 Y pHIS1525-T7H Las posibles causas de la falta de expresión de proteína recombinante usualmente corresponden a corrimientos en el marco de lectura de la proteína clonada y al término anticipado de la traducción debido a la generación de codones de término por mutaciones generadas durante el proceso. Causas menos comunes son la formación de estructura secundaria del mRNA, lo cual impediría el inicio de la traducción, y el desbalance entre aminoácidos requeridos y tRNAs, lo que generaría una traducción inadecuada o el término anticipado de ésta. Por esto, se decidió analizar las secuencias de las construcciones realizadas. 3.5.1 ANÁLISIS DE LAS SECUENCIAS Para analizar el correcto marco de lectura de las construcciones, éstas fueron secuenciadas con partidores distribuidos en las regiones cercanas al inserto y/o dentro del mismo. La construcción pHIS1522-T8 se secuenció con los partidores pHIS2, pHIS3 y pHISR, que alinean en el vector pHIS1525 en la región adyacete al sitio de clonamiento. Para secuenciar pHIS1525-T7 se utilizó además los partidores pT7 y pT7R, los cuales alinean sobre el gen T7. Las construcciones pHIS1525-T8 y pHIS1525-T7H fueron secuenciadas con los partidores pHIS1, pHIS2, pHIS3, pHISR, pHISR1, pHISR2 y pHISR3, los que alinean en las 35 regiones del vector cercanas al gen clonado. Los partidores pT7 y pT7R también se usaron en el caso de pHIS1525-T7H. (Ver Anexo A.7) Las muestras secuenciadas fueron purificadas mediante minipreparación desde cultivos de células de E. coli transformadas con vectores purificados desde B. megaterium. No se utilizó vectores purificados directamente desde B. megaterium debido a la baja eficiencia del método de minipreparación de DNA plasmidial adaptado a células de Bacillus, descrita con anterioridad. Los resultados de secuenciación se muestran en el Anexo A.9 Se encontró que la construcción pHIS1522-T8 presentaba un marco de lectura correcto de la proteína clonada. Con esta información, se sugiere que la inactividad de la proteína recombinante T8 expresada en forma intracelular se debe posiblemente a un mal plegamiento de ésta durante el proceso, y no a una traducción inadecuada producto de errores en la construcción. Para las construcciones pHIS1525-T8 y pHIS1525-T7 se determinó que el marco de lectura se encontraba alterado debido a la inserción de una secuencia de inserción (IS) de E. coli denominada IS1A[39] en la región del péptido señal, generando un codón de término en la lectura del quinto aminoácido. La secuencia de inserción fue identificada mediante el alineamiento en línea, de la secuencia con aquellas de la base de datos del Nacional Center for Biotechnology Information (NCBI)[40]. Posee 768 pb y está insertada en antisentido con respecto al gen clonado de las subtilasas (ver Figura 16, A y B). Figura 16: Resultados de secuenciación. A: pHIS1525-T8; B: pHIS1525-T7 y C: pHIS1525-T7H. Amarillo: región correspondiente al vector pHIS1525; Verde: inserto; Azul: Secuencia de inserción IS1A; Gris: región no secuenciada. SP: péptido señal. Los sitios de restricción para SphI, XhoI y EcoRVse muestran con letras azules. En el caso de pHIS1525-T7H, del análisis de la construcción se determinó que el marco de lectura de la proteína se encontraba correcto e incorporó satisfactoriamente la señal de exportación. Sin embargo, un error en el diseño del partidor encargado de agregar el sitio de restricción de SphI en el extremo 3’ del gen corrió el marco de lectura justo antes de la fusión con la cola de histidinas, impidiendo su adecuada incorporación y término de traducción. Como resultado, la proteína quedó fusionada a un péptido de 61 aminoácidos, lo que en teoría resultaría 36 en la expresión de una proteína de 79.2 kDa, en lugar de la proteasa T7 con cola de histidinas de 71.14 kDa. Se cree que la ausencia de proteína recombinante T7 en las muestras analizadas para esta construcción, se debería entonces a la producción de una proteína de 79.2 kDa, la cual sería secretada al medio extracelular y, en teoría, no sería purificada por el método de cromatografía de afinidad (pues no se traduciría la cola de histidinas), lo que explica que tampoco haya sido detectada en las muestras analizadas. Por otra parte, esta proteína no poseería actividad enzimática, pues probablemente la presencia del péptido fusionado afectaría la estructura de la proteasa. Aunque se analizó muestras de sobrenadante de cultivo no purificado de la inducción de varias colonias transformadas con pHIS1525-T7H, (ver punto 3.3.5), se cree que la proteína recombinante de 79.2 kDa no fue detectada debido a que probablemente la cantidad de la muestra se encontraba bajo el límite de detección, pues se concentró sólo 8 veces (desde 2 mL a 250 µL), mientras que usualmente se visualizó proteína concentrada 400 veces (de 100 mL a 250 µL) (Anexo A.4.5). Se propone la verificación de la expresión de la proteína de 79.2 kDa, mediante el análisis en SDS-PAGE de sobrenadante de cultivo concentrado en un factor mayor. Por otra parte, en las construcciones con pHIS1525 con inserción de la secuencia IS1A se encontró gran cantidad de mutaciones puntuales sobre el gen clonado. Se estimó una frecuencia de mutación de 1.05 x 10-3, mediante el cuociente del total de las mutaciones y el área total secuenciada. Las mutaciones puntuales presentes en la secuenciación en más de una construcción no se consideraron para este cálculo, y se atribuyeron a errores en la secuenciación original. 3.5.2 CORTE CON ENZIMAS DE RESTRICCIÓN Debido a que no fue posible, secuenciar completamente las construcciones, y a la posibilidad de inserciones parciales o múltiples de la secuencia IS1A detectada en la secuenciación de los vectores pHIS1525-T8 y pHIS1525-T7 en cualquiera de las construcciones plasmidiales, éstas fueron analizadas mediante cortes con enzimas de restricción, con el objeto de determinar las características de la inserción y descartar eventos similares en otras regiones. Se analizó además los patrones de digestión de los vectores pHIS1522 y pHIS1525 amplificados en células de E. coli DH5α, pues la Figura 4 (que muestra el gel de extracción para las construcciones pHIS1525-T8 y pHIS1525-T8 control) indica un aumento de tamaño del vector pHIS1525 amplificado en E. coli con respecto al original. Para el caso de este vector se analizan muestras provenientes de diferentes colonias y transformaciones (pHIS1525A, pHIS1525B y pHIS1525C). En el proceso, se utilizó como control a los vectores pHIS1522 y pHIS1525 de origen comercial. Los vectores pHIS1522-T8, pHIS1525-T8, pHIS1525-T7 y pHIS1525-T7H, purificados desde B. megterium, fueron desmetilados mediante amplificación en E. coli, antes de ser digeridos. Se realizó doble digestión con SphI y XhoI y digestión simple con EcoRV para poder analizar las distintas regiones no secuenciadas de los plasmidios. La digestión con SphI y XhoI 37 permite analizar la región adyacente al gen clonado hacia el extremo 3’ incluyendo la región correspondiente al gen mismo, y genera un segundo fragmento correspodiente la región conservada del vector en todos los plasmidios. La digestión con EcoRV permite analizar el estado de gran parte del vector al generar varios cortes que generan también un producto de digestión teóricamente conservado en todas las muestras. Los sitios de corte para las construcciones pHIS1525-T8, pHIS1525-T7 y pHIS1525-T7H, de acuerdo a los resultados de la secuenciación, se muestran gráficamente en la Figura 16. La predicción teórica de los productos de digestión se muestra en la Tabla 4. Los resultados se presentan en la Figura 17 para la digestión doble, y la Figura 18 para la digestión con EcoRV. Tabla 4: Predicción teórica de productos de digestión Vector pHIS1522 pHIS1522-T8 pHIS1525 pHIS1525-T8 pHIS1525-T7 pHIS1525-T7H Enzima(s) SphI y XhoI EcoRV SphI y XhoI EcoRV SphI y XhoI EcoRV SphI y XhoI EcoRV SphI y XhoI EcoRV SphI y XhoI EcoRV Tamaños teóricos de productos de digestión 611 y 6791 2406 y 4996 2106 y 6793 2406 y 6493 681 y 6793 2406 y 5068 2943 y 6793 2406 y 7330 3452 y 6793 244, 2406, 3484 y 4111 2683 y 6793 2406, 2715 y 4355 Figura 17: Digestión con SphI y XhoI. (1) pHIS1522 control; (2) pHIS1522; (3) pHIS1522-T8; (4) pHIS1525 control; (5) pHIS1525A; (6) pHIS1525B; (7) pHIS1525-T8; (8) pHIS1525-T7 y (9) pHIS1525-T7H. (M) Marcador de tamaño 1 kb (Invitrogen). 38 En la digestión con SphI y XhoI (Figura 17), el mapa de restricción predice un producto de 6790 pb aproximadamente, para todas las muestras digeridas, el cual se encontró presente en el patrón de corte obtenido (excepto en pHIS1525A, carril nº5) y se indica con una flecha azul. Este fragmento abarca casi toda la región no secuenciada del vector, lo que se interpretó como la ausencia de inserciones similares en otras regiones de los vectores analizados. El vector pHIS1522 amplificado en E. coli no mostró diferencias con la digestión del vector control (compare carril nº1 y carril nº2). Por otro lado, el producto de digestión de pHIS1522-T8 comparado con el del vector sin inserto, mostró que se incorporó satisfactoriamente la secuencia de la subtilasa T8 de 1536 pb, lo que se aprecia en la diferencia entre las bandas inferiores (compare carril nº1 y carril nº3). Los resultados para el vector pHIS1525 indicaron que su amplificación en E.coli DH5α (carriles nº5 y nº6) produce cambios en el patrón de corte con respecto al control (carril nº4, indicado con flecha roja), generando fragmentos de mayor tamaño, indicativos de eventos de inserción. Los productos de digestión de pHIS1525-T8 y pHIS1525-T7H (carriles nº7 y nº9, respectivamente), mostraron bandas coincidentes con lo esperado (ver Tabla 4), mientras que la digestión de pHIS1525-T7 (carril nº8) generó tres bandas: la banda conservada de 6790 pb aproximadamente, y dos bandas cercanas a 2500 pb y 600 pb. Probablemente se generó una mutación que incorporó un sitio de restricción para alguna de las enzimas dividiendo el fragmento esperado de 3452 pb. Lo anterior confirmó la presencia de la secuencia de inserción IS1A completa en la región correspondiente a la señal de exportación, en las construcciones pHIS1525-T8 y pHIS1525-T7, y por otra parte, descartó un evento similar en alguna región de la construcción pHIS1525-T7H. Figura 18: Digestión con EcoRV. (1) pHIS1522 control; (2) pHIS1522; (3) pHIS1522-T8; (4) pHIS1525 control; (5) pHIS1525A; (6) pHIS1525C; (7) pHIS1525-T8; (8) pHIS1525-T7 y (9) pHIS1525-T7H. (M) Marcador de tamaño 1 kb (Invitrogen). En el caso del análisis por digestión con EcoRV (Figura 18), las muestras conservan, en teoría, una banda de 2406 pb (ver Tabla 4) correspondiente a una región del vector pHIS1525 (ver Figura 16), la cuál efectivamente se produjo. 39 Los productos de digestión con EcoRV de los vectores pHIS1522 control, pHIS1522 y pHIS1522-T8 coincidieron con lo esperado (ver Tabla 4) La digestión de vectores pHIS1525 de distinto origen generó productos de amplificación mayores a los del control para pHIS1525A (carril nº5), pero para pHIS1525C (carril nº6) las bandas fueron coincidentes. Lo anterior sugiere que probablemente la inserción no ocurre en todas las colonias. La digestión de pHIS1525-T7 (carril nº8) produjo sólo dos bandas de cuatro predichas teóricamente. Lo anterior se puede atribuir al hecho que la muestra secuenciada, en base a cuyos resultados se realizó la predicción de corte por las enzimas, corresponde a una minipreparación diferente de DNA plasmidial, sumado a la alta frecuencia de mutaciones puntuales encontrada sobre la región del gen clonado. Las bandas generadas en la digestión de pHIS1525-T8 y pHIS1525-T7H coincidieron con lo esperado, confirmando los resultados encontrados al digerir con SphI y XhoI. 3.6 ANÁLISIS DE pHIS1525 AMPLIFICADO EN E. coli DH5α Con la finalidad de estudiar la mutación por inserción de IS1A, se analizó productos de amplificación in vivo de distintas colonias de E. coli. Se transformó por electroporación células de E. coli DH5α con pHIS1525 de origen comercial. Se purificó DNA plasmidial desde 5 colonias seleccionadas al azar, el cual fue digerido con BamHI para linealizarlo. Se digirió además vector comercial como control. Los resultados se pueden ver en la Figura 19. Figura 19: Digestión con BamHI de pHIS1525 de distintas colonias. (1-5) pHIS1525 purificado desde distintas colonias de E. coli DH5α. (6) pHIS1525 control. (M) Marcador de tamaño 1 kb (Invitrogen). Los vectores cargados en los carriles nº1, nº3 y nº5 generaron bandas del mismo tamaño, pero más grandes que la del vector original (indicado con flecha roja), mientras que los vectores cargados en los carriles nº2 y nº4, al parecer, no fueron digeridos, por lo que se cree que perdieron el sitio de restricción para la enzima BamHI durante la amplificación. 40 Esto sugiere que el proceso de amplificación in vivo en E. coli DH5α genera cambios en el vector pHIS1525, que alteran el tamaño del vector en cantidades discretas. Se atribuye este cambio de tamaño a la inserción, dentro de la región correspondiente a la señal de exportación, de la secuencia de inserción IS1A encontrada en las construcciones pHIS1525-T8 y pHIS1525-T7 mediante secuenciación. En pHIS1525-T7, se cree que la inserción ocurrió durante la amplificación del vector pHIS1525 previa a la clonación del inserto. Sin embargo, en el caso de pHIS1525-T8 se cree que la inserción ocurrió en el proceso de amplificación previo a la transformación de B. megaterium, pues esta última se llevó a cabo con la construcción control que consistió en la clonación del inserto directamente en el vector original (ver punto 3.2.2). Esto indica que el evento de inserción de IS1A puede ocurrir tanto en el vector sólo, como en presencia del inserto. Las secuencias de inserción (IS) son secuencias de DNA transponibles de cadena corta (de 750 a 1600 pb) que contiene sólo los genes que codifican para las enzimas necesarias para su transposición, y que está limitada a ambos lados por secuencia idénticas o muy similares de nucleótidos que presentan una orientación invertida de 15 a 25 pb, conocidas como secuencias inversamente repetidas (IR). Cada elemento IS tiene sus propias IR[21]. Normalmente, las IS1 suelen insertarse en sitios de inserción con determinadas secuencias, en un proceso que incluye la duplicación de un sitio de 9 pb de la secuencia blanco entre los cuales queda flanqueada la IS, denominados secuencias directamente repetidas (DR). Se analizó la secuencia de pHIS1525 en busca de secuencias de inserción identificadas para IS1A[39] en la región del evento. No se encontró que ninguna de ellas alineara en esta región del plasmidio. Sin embargo, se encontró información acerca de las características generales de estos sitios de inserción indicando que usualmente corresponden a regiones de alto porcentaje de pares AT[41]. Con esta información, considerando que las secuencias de inserción analizadas anteriormente eran de 29 pb, y suponiendo que la secuencia DR correspondía a los 9 nucleótidos secuenciados al extremo 5’ del punto de inserción, se determinó una probable secuencia blanco 3’…GCCATAAGTACTTTTTTCATTGTACATTT…5’. Se calculó que tiene una composición AT de 72.4%, indicando que posiblemente corresponde a un sitio de inserción para IS1A. De los resultados anteriores, se sugiere el aumento de controles durante el trabajo con pHIS1525, por ejemplo, mediante análisis de patrones de digestión, así como estudiar el proceso de amplificación in vivo en células de E. coli de distinto genotipo. De esta forma, se podría reintentar la expresión de las subtilasas T7 y T8 u otras proteínas con este sistema. Debido a que no hubo problemas de inserción durante el trabajo con pHIS1522, y se obtuvo expresión de proteína recombinante, se propone la clonación de la subtilasa T7 o T8 con su propio péptido señal en este vector para ser exportadas hacia el medio extracelular. Se propone además intentar la renaturación de la proteína producida en forma de cuerpos de inclusión por las células transformadas con pHIS1522-T8. 41 4 CONCLUSIONES De los resultados obtenidos durante el trabajo de Memoria de Título, se concluye lo siguiente: • Fue posible clonar los genes de las subtilasas T8 y T7, en su forma madura, en el vector de expresión pHIS1525, y la misma región de T8 en pHIS1522. • La expresión de proteína recombinante T8 en el medio intracelular, mediante el uso del vector pHIS1522, produce una proteína sin actividad proteasa, principalmente en forma de cuerpos de inclusión. El marco de lectura de la construcción fue corroborado mediante secuenciación, por lo que se cree que la inactividad se debe a que la subtilasa no se pliega en su forma nativa, como es frecuente que ocurra en proteínas producidas como cuerpos de inclusión. • Para las construcciones realizadas con pHIS1525, la expresión del gen clonado se ve saboteada por la interrupción de la señal de exportación mediante la inserción de una secuencia de inserción, IS1A, la cual fue detectada en el análisis por secuenciación. El mismo análisis mostró la existencia de una alta frecuencia de mutagénesis puntual en estas construcciónes. • Una segunda construcción de la subtilasa T7 en pHIS1525, presenta un corrimiento en el marco de lectura que fusiona a la proteína T7 a un péptido de 61 aminoácidos por su extremo carboxilo. Esto se debe a un error en el diseño del partidor que incorpora el sitio de restricción para clonar el gen en el vector por el extremo 3’. • La secuenciación de la construcción del punto anterior indica que debiera producirse la expresión de la proteína T7 fusionada al péptido en el medio extracelular, sin embargo, no fue posible corroborar experimentalmente esta observación. • La detección de una proteasa endógena de Bacillus megaterium, expresada en la fase exponencial de crecimiento, inhabilita el proceso de ensayo in situ de actividad proteasa extracelular por medio de placas con SM agar con inductor, aunque el aumento del pH en las placas reduce la actividad de la proteasa endógena del microorganismo huésped. Se diseñó un método alternativo, consistente en el uso de placas de Medio Mínimo para Bacillus megaterium agar con S-AAPF-pNa e inductor, el cual permite detectar en forma específica la actividad de serina proteasas. Aunque no se ha determinado su límite de detección, es un medio potencial para ensayos preliminares de actividad de subtilasas recombinantes producidas en B. megaterium. • El proceso de inducción de proteasa recombinante a baja temperatura, no genera mayor cantidad de proteína recombinante soluble en el sistema de expresión intracelular de la subtilasa T8, ni mejora su plegamiento. Sin embargo, esto no descarta que pueda mejora los niveles de expresión en sistemas diseñados para la producción extracelular de proteínas en Bacillus megaterium, pues no fue posible analizar este caso. 42 • Al utilizar Bacillus megaterium como sistema de expresión recombinante de proteínas mediante el uso del vector de expresión pHIS1525 es recomendable realizar controles adicionales a lo largo del proceso. Se sugiere usar un genotipo distinto a E. coli DH5α como huésped de clonamiento. 43 5 BIBLIOGRAFÍA [1 ] Gerday, C., Aittaleb. M., Bentahir, M., Chessa, J. P., Claverie, P., Collins, T., D’Amico, S., Dumont, J., Garsoux, G., Georlette, D., Hoyoux, A., Lonhienne, T., Meuwis, M. A. and Feller. G. (2000) “Cold.adapted enzymes: from fundamentals to biotechnology”, Trends in Biotechnology 18: 103-107. [2] Miyazaki, K., Wintrode, P. L.,. Grayling, R. A., Rubingh, D. N., Arnold, F. H. (2000) “Directed Evolution Study of Temperatue Adaptation in Psichrophilic Enzyme”, Journal of Molecular Biology 297:1015-1026. [3] http://delphi.phys.univ-tours.fr/Prolysis/introprotease.html. Les Protéases et leurs inhibiteurs (PROLYSIS). Université François Rabelais [Consulta: Diciembre, 2006]. [4] Priest, F. G. (1977) “Extracellular Enzyme Synthesis in the Genus Bacillus”. Bacteriological Reviews 41 (3): 711-753. [5] Siezen, R. J. and Leunissen, J. (1997) “Subtilases: The superfamily of subtilisin-like serine proteases”,Protein Science 6: 501-523. [6] Bryan, Philip N. (2000), Review: “Protein engineering of subtilisin”, Biochemica et Biophysica Acta 1543:203-222. [7] Simonen, M.; Palva, I. (1993) “Protein Secretion in Bacillus Species” Microbiological Reviews 57 (1): 109-137. [8] Maurer, KH. (2004) “Detergent Porteases”, Current Opinion in Biotechnology 15:330–334. [9] Wells, J. A., Ferrari, E., Henner, D. J., Estell, D. A. and Chen, E. Y. (1983) “Cloning, sequencing, and secretion of Bacillus amyloliquefaciens subtillisin in Bacillus subtilis”, Nucleic Acids Research 11: 7911-7925. [10] Strausberg, S. L., Ruan, B., Fisher, K. E., Alexander, P. A. and Bryan, P. N. (2004) “Directed Coevolution of Stability and Catalytic Activity in Calcium-free Subtilisin” Biochemistry 44: 3272-3279. [11] Estell, D. A., Graycar, T. P. and Wells, J. A. (1985) “Engineering an Enzyme by Sitedirected Mutagenesis to Be Resistant to Chemical Oxidation”, The Journal of Biological Chemestry 260 (11): 6518-6521. [12] Wintrode, P. L., Miyazaky, K. and Arnold, F. H. (2000) “Cold Adaptation of a Mesofilic Subtilisin-like Protease by Laboratory Evolution” , The Journal of Biological Chemestry 275 (41): 31635-31640. [13] Tindbaek, N., Svendsen, A., Oestergaard, P. R. and Draborg, H. (2004) “Engineering a substrate-specific cold adapted subtilisin” Protein Engineering, Design and Selection 17:149-156. 44 [14] Davail, S., Feller, G. Narinx, E. and Gerday, C. (1994) “Cold Adaptation of Proteins. Purification, Characterization, and Sequence of the heat labile subtilisin from the antartic psycrophile Bacillus TA41”, The Journal of Biological Chemestry 269 (26): 17448-17453. [15] Hough, D. W. and Danson, M. J. (1999) “Extremozymes”, Current Opinion in Chemical Biology 3:39-46. [16] Lonhienne, T., Gerday, C., Feller, G. (2000) “Psychrophilic enzymes: revisiting the thermodynamic parameters of activation may explain local flexibility”, Biochim. Biophys. Acta 1543:1-10. [17] Siddiqui, K. S. and Cavicchioli, R. (2006) “Cold Adapted Enzymes”, Annu. Rev. Biochem. 75:403-433 [18] D’Amico, S., Claverie, P., Collins, T., Georlette, D., Gratia, E., Hoyoux, A., Meuwis, M. A. Feller, G. and Gerday, C. (2002) “Molecular basis of cold adaptation”, Phil. Trans. R. Soc. Lond. 357:917-925. [19] Acevedo, J.P. (en desarrollo) “Aislamiento y Producción recombinante de nuevas proteasas bacterianas adaptadas a bajas temperaturas con potencial biotecnológico”. Informe de Calificación para el Programa de Doctorado en Ciencias de la Ingeniería, Mención Química. Universidad de Chile. [20] Acevedo, J. P., Reyes, F., Parra, L., Salazar, O., Andrews, B. A. and Asenjo, J. A. (en revision) “Improved and easy genome walking method tested on Antarctic bacterial genes”, Biotechniques. [21] Prescott, L., Harley, J., Klein, D. (1999) “Microbiología”, McGraw-Hill, Cuarta Edición, Madrid. [22] González-Pedrajo, B., Dreyfus, D. (2003) “Sistemas de secreción de proteínas en las bacterias Gram negativas: Biogénesis flagelar y translocación de factores de virulencia”, Mensaje Bioquímico XXVII: 45-62. [23] Kulakova,L., Galkin, A., Kurihara, T., Yoshimura, T. and Esaki, N. (1999) “Cold-Active Serine Alkaline Protease from the Psychrotrophic Bacterium Shewanella Strain Ac10: Gene Cloning and Enzyme Purification and Characterization”, Applied and Environmental Microbiology 65: 611-617. [24] Arnórsdóttir, J., Smáradóttir, R. B., Magnússon, Ó. Th., Thorbjarnardóttir, S. H., Eggertsson, G. and Kristjánsson, M. M. (2002) “Characterization of a cloned subtilisin-like serine proteinase from a psychrotrophic Vibrio species”, Eur. J. Biochem. 269: 5536-5546. [25] Burger, S., Tatge, H., Hofmann, F., Genth, H., Just, I. and Gerhard, R. (2003) “Expression of recombinant Clostridium difficile toxin A using the Bacillus megaterium system”, BBRC 307: 584-588. [26] Kim, J. Y. (2003) “Overproduction and secretion of Bacillus circulans endo-beta-1,3-1,4glucanase gene (bglBCI) in B. subtilis and B. megaterium” Biotechnol Lett 25: 1445-1449. 45 [27] Malten, M., Biedendieck, R. Gamer, M., Drews, AC., Stammen, S. Buchholz, K., Dijkhuizen, L. and Jahn, D. (2006) “A Bacillus megaterium Plasmid System for the production, Export, and One-Step Purification of Affinity-Tagged Heterologous Levansucrase from Growth Medium”, Applied and Enviromental Microbiology 72(2):1667-1679. [28] Jordan, E., Hust, M., Roth, A., Biedendieck, R., Schirrmann, T., Jahn, D. and Dübel, S. (2007) “Production of recombinant antibody fragments in Bacillus megaterium”, Microbial Cell Factories 6: 2. [29] Yang, Y., Biedendiek, R., Wank, W., Gamer, M., Malten, M., Jahn, D. and Deckewer, W. D. (2006) “High yield recombinant penicillin G amidase production and export into the growth medium using Bacillus megaterium”, Microbial Cell Factories 5:36. [30] Brey, R., Banner, C., Wolf, J. (1986) “Cloning of Multiple Genes Involved with Cobalamin (Vitamin B12) Biosynthesis in Bacillus megaterium”, Journal of Bacteriology 167 (2): 623-630. [31] http://www1.qiagen.com/literature/handbooks/PDF/DNACleanupAndConcentration/QIAEX_II/1 011033_qiaexii.pdf. Manual para QIAEX II Gel Extraction Kit. Pp. 12-13. [Consulta: Abril, 2006]. [32] http://www1.qiagen.com/literature/protocols/pdf/PR01.pdf. Protocolo para purificación de DNA plasmidial desde Bacillus utilizando el Qiaprep Spin Miniprep Kit. [Consulta: Mayo 2006]. [33] http://textbookofbacteriology.net/Bacillus.html: Todar's Online Textbook of Bacteriology. Department of Bacteriology, University of Wisconsin. [Consulta: Junio 2006]. [34] http://www1.qiagen.com/literature/handbooks/PDF/DNACleanupAndConcentration/QIAEX_II/1 011033_qiaexii.pdf . Manual para QIAEX II Gel Extraction Kit. Pp. 16-17. [Consulta: Mayo, 2006]. [35] Hönerlage, W., Hahn, D., Zeyer, J. (1995) “Detection of mRNA of nprM in Bacillus megaterium ATCC 14581 grown in soil by whole-cell hybridation”, Archives of Microbiology 163: 235-241. [36] http://www.expasy.org/uniprot/Q00891#name. Expert Protein Analysis System (Expasy). [Consulta: Mayo, 2006]. [37] Juncker, A. S., Willenbrock, H., von Heijne, G., Nielsen, H., Brunak, S.,and Krogh, A. (2003)"Prediction of lipoprotein signal peptides in Gram-negative bacteria”. Protein Sci. 12(8):1652-62. [38] http://rscott.myweb.uga.edu/protocols/CEP_222.doc. Robert A. Scott Group, Protocols. University of Georgia. [Consulta: Diciembre, 2006]. [39] http://www-is.biotoul.fr. IS Finder. Le Laboratoire de Microbiologie et Génétique Moléculaires. [Consulta: Diciembre, 2006]. 46 [40] http://www.ncbi.nlm.nih.gov/BLAST. Basic Local Alignment Search Tool (BLAST). National Center for Biotechnology. [Consulta: Diciembre, 2006]. [41] Mahillon, J., Chandler, M. (1998) “Insertion Sequences”, Microbiology and Molecular Biology Reviews 62 (3): 725-774. 47 A. ANEXOS A.1. SECUENCIAS DE T8 Y T7 48 49 50 Figura A.1: Secuencias de las proteasas T8 y T7 alineadas con proteasas tipo subtilisina típicas: Savinasa, Termitasa y Proteinasa K. Secuencias destacadas: verde: péptido señal; azul: propéptido; rojo: secuencia de función desconocida. Los aminoácidos del sitio catalítico se destacan en amarillo. 51 A.2. VECTORES A.2.1. Vector pGEM-T Easy 52 A.2.2. Vectores pHIS1522 y pHIS1525 Figura A.2.2.a: Mapa de pHIS1522. Vector de clonamiento para E. coli/B. megaterium. TetR(Bac), resitencia a la tetraciclina (Bacillus); TetR1, resistencia a la tetraciclina, incompleta e inactiva; AmpR, resistencia a la ampicilina (E. coli); xylR, represor dependiente de xilosa; orf1, origen de lectura controlado por el promotor inducible por xilosa PxylA; PxylA,, promotor xylA; MCS, sitio de clonamiento múltiple; pBC16 ori, origen de replicación para Bacillus; pBR322, origen de replicación para E. coli; repU, gen esencial para la replicación del plasmidio. 53 Figura A.2.2.b: Mapa de pHIS1525. Vector de clonamiento para E. coli/B. megaterium. TetR(Bac), resitencia a la tetraciclina (Bacillus); TetR1, resistencia a la tetraciclina, incompleta e inactiva; AmpR, resistencia a la ampicilina (E. coli); xylR, represor dependiente de xilosa; orf1, origen de lectura controlado por el promotor inducible por xilosa PxylA; PxylA,, promotor xylA; MCS, sitio de clonamiento múltiple; pBC16 ori, origen de replicación para Bacillus; pBR322, origen de replicación para E. coli; repU, gen esencial para la replicación del plasmidio 54 A.3. MEDIOS DE CULTIVO A.3.1. Medio SOB (1 L) Triptona Extracto de levadura NaCl KCl 20.0 g 5.0 g 0.584 g 0.186 g Llevar a 1 L con H2O (HPLC) A.3.2. Medio SP1 (Prolina) (300 mL) Sulfato de Amonio K2HPO4 KH2PO4 Citrato de sodio Sulfato de magnesio Casaminoácidos Extracto de levadura Prolina 0.6 g 4.2 g 1.8 g 0.3 g 0.06 g 0.06 g 0.3 g 0.03 g Llevar a 292.5 mL con H2O (HPLC) Agregar 7.5 mL de glucosa 20% estéril. A.3.3. Medio SMMP (200 mL) SMM 2x (100 mL) Sacarosa Sal disodio de ácido maleico MgCl2 34.23 g. 0.64016 g. 0.8132 g. Llevar a 100 mL con H2O (HPLC) Ajustar a pH 6.5 Autoclavar 12 minutos AB3 2x Antibiotic Medium Nº3, DIFCO 3.5 g Llevar a 100 mL con H2O (HPLC) Autoclavar 15 minutos Mezclar volúmenes iguales de SMM 2x y AB3 2x 55 A.3.4. Medio LB agar para placas (100 mL) Luria Broth NaCl Agar 1.55 g 0.95 g 1.5 g Llevar a 100 mL con H2O (HPLC) A.3.5. Medio LB (100 mL) Luria Broth NaCl 1.55 g 0.95 g Llevar a 100 mL con H2O (HPLC) A.3.6. Skim Milk (SM) Agar (100 mL) Leche descremada en polvo Extracto de levadura Agar 1g 0.1 g 1.5 g Llevar a 100 mL con H2O (HPLC) A.3.7. Skim Milk (SM) Agar + Xilosa (100 mL) Leche descremada en polvo Extracto de levadura Agar Xilosa 20% 1g 0.1 g 1.5 g 2.5 mL Llevar a 100 mL con H2O (HPLC) 56 A.3.8. Medio Mínimo para Bacillus megaterium (MMBM) + s-AAPF-pNa Agar (100 mL) Sacarosa K2HPO4 KH2PO4 (NH4)2HPO4 Agar MnSO4·7H2O 0.007% H2O (HPLC) 1g 0.25 g 0.25 g 0.1 g 1.5 g 1 mL 97 mL Ajustar pH 7.0 y autoclavar MgSO4·7H2O 2% FeSO4·7H2O 0.01% s-AAPF-pNa 0.125% en DMSO 1 mL 1 mL 0.2 mL A.3.9. Medio Mínimo para Bacillus megaterium (MMBM) + s-AAPF-pNa Agar + Xilosa (100 mL) Sacarosa K2HPO4 KH2PO4 (NH4)2HPO4 Agar MnSO4·7H2O 0.007% H2O (HPLC) 1g 0.25 g 0.25 g 0.1 g 1.5 g 1 mL 97 mL Ajustar pH 7.0 y autoclavar MgSO4·7H2O 2% FeSO4·7H2O 0.01% s-AAPF-pNa 0.125% en DMSO Xilosa 50% 1 mL 1 mL 0.2 mL 1 mL 57 A.3.10. CR5-top agar (500 mL) Componente a: Sacarosa MOPS NaOH H2O (HPLC) 51.5 g 3.25 g 0.33 g 250 mL Ajustar a pH 7.3 y esterilizar por filtración Componente b: Agar Casaminoácidos Extracto de Levadura H2O (HPLC) 2g 0.1 g 5g 142.5 mL Autoclavar por 20 minutos, en botella de 500 mL con agitador Combinar los componentes a y b, a temperatura menor a 50 °C Agregar: Sales CR5 8x 12% prolina (p/v; estéril por filtración) 20% glucosa (p/v; estéril por filtración) 58 57.5 mL 25 mL 25 mL A.4. PROTOCOLOS A.4.1. Protocolo Transformación de Bacillus megaterium. 1. A 500 µL de protoplastos de B. megaterium, agregar 5 µg de DNA (en SMM 2x) (usar tubo de 12 mL) 2. Agregar 3 volúmenes de PEG-P (1.5 mL) e incubar a temperatura ambiente por 2 minutos. 3. Agregar 10 volúmenes (5 mL) de SMMP. Mezclar invirtiendo el tubo cuidadosamente. 4. Centrifugar a 3000 rpm por 10 minutos a temperatura ambiente, y remover inmediatamente el sobrenadante. 5. Resuspender en 500 µL de SMMP. 6. Incubar a 37 ºC por 90 min, con agitación suave. 7. Preparar tubos con 2.5 mL de CR5-top agar estéril, y precalentar (máx. 43 ºC) 8. Agregar 50-200 µL de cultivo a los tubos con CR5-top agar. Mezclar suavemente invirtiendo los tubos con ambas manos. 9. Plaquear en placas con LB con tetraciclina, precalentadas. 10. Incubar a 37 ºC durante la noche. 11. Transferir a placas nuevas dentro 2 días. A.4.2. Protocolo de transformación de células de E. coli por electroporación 1. Se agrega 20 µL de células electrocompetentes y 1µL de mezcla de ligación 2. Las condiciones de electroporación dependen del tipo de célula. (Ver manual del equipo Cell-Porator® Electroporation System (Gibco-BRL Life Technologies, Inc., MD-USA). Para E. coli se realiza a 330 µF, impedancia (DCvolt) baja, tasa de carga rápida, 420 V y 4 kΩ. 3. Las células transformadas se incuban en 1 mL de medio LB a 37 ºC por 1-2 horas 4. Centrifugar a 13000 rpm durante 1 minuto 5. Descartar 900 µL de sobrenadante y resuspender en el remanente 6. Plaquear sobre LB agar adecuado 7. Incubar a 37 ºC durante toda la noche (invertidas para evitar deshidratación) A.4.3. Protocolo de Minipreparación de DNA plasmidial desde células de E. coli DH5α, con el método QIAprep® Spin Miniprep. 1. Inocular una colonia en 4 mL de medio LB con antibiótico y crecer a 37 ºC durante la noche. 2. Centrifugar el cultivo de a 1 mL a la vez, descartando el sobrenadante (máx. 5 mL de cultivo) (1 minuto a 13000 rpm). 3. Resuspender con la pipeta en 250 µL de Buffer P1. 4. Agregar 250 µL de Buffer P2, y mezclar invirtiendo el tubo suavemente 4-6 veces. No mantener esta reacción de lisis por más de 5 minutos. 59 5. Agregar 350 µL de Buffer de N3 e invertir inmediatamente el tubo en forma suave para mezclar. 6. Centrifugar 10 minutos a 13000 rpm. 7. Recuperar el sobrenadante y depositar en una columna de spin. 8. Centrifugar 1minuto a 13000 rpm. 9. Descartar el líquido y lavar el DNA de la columna de spin con 750 µL de Buffer PE. Centrifugar 1minuto a 13000 rpm. 10. Descarta el líquido y centrifugar nuevamente durante 1minuto para remover los residuos de buffer. 11. Poner la columna en un tubo de 1.5 mL, agregar 50 µL de H2O pH 8.5 (precalentada a 50 ºC) en el centro. Dejar reposar 1 minuto y centrifugar 1 minuto a 13000 rpm. A.4.4. Protocolo para PCR de colonias de E. coli DH5α transformadas 1. Transferir colonias en una nueva placa de LB con antibiótico, identificadas con un número, y resuspender el remanente de las células en 30 µL de H2O estéril. 2. Incubar la placa a 37 ºC. 3. Calentar las células resuspendidas en H2O a 100 ºC por 10 minutos, y usar como DNA templado. 4. Reacción de PCR utilizando Taq DNA Polimerasa (Promega) (1 tubo): DNA 5 µL dNTPs [10mM] 0.4 µL Mg Free Buffer 10x 2 µL Primer (s) 1 µL Primer (r) 1 µL MgCl2 1.6 µL Taq DNA Polimerasa 0.35 µL H2O 8.65 µL 5. Programa PCR con aproximadamente 36 ciclos. A.4.5. Protocolo Inducción y recuperación de proteína recombinante en B. megaterium 1. Crecer durante la noche a 37 ºC, un inóculo en 4 mL de LB con tetraciclina 10 µg/mL. 2. Inocular en 100 mL de LB con tetraciclina 10 µg/mL, un volumen de inóculo de manera que quede a una DO600 de 0.05. 3. Crecer durante 2-4 horas a 37 ºC, hasta una DO600 de 0.3. 4. Inducir el promotor de xilosa agregando xilosa 0.5%. 5. Incubar a 37 ºC por 2.5 horas u overnigh a 25 ºC. 6. Detener la inducción dejando las muestras en hielo. 7. Tomar 1 mL de cada muestra. Centrifugar 2 minutos a 13000 rpm y resuspender las células en 100 µL de agua. Tratar con lisozima 1 mg/mL, incubar a temperatura ambiente por 20 minutos y almacenar a 4 ºC. Esta muestra corresponde a la Proteína Total Intracelular (PT). 8. Centrifugar los cultivos 20 minutos a 7000 rpm. 60 9. Separar el sobrenadante y agregar Binding Buffer 10x de manera de que este último quede diluido 10 veces. Esta muestra corresponde al Sobrenadante de cultivo (SN). 10. Resuspender el pellet en Buffer Tris 20mM pH 8.0. Tratar con lisozima (Sigma) 1 mg/mL e incubar 20 minutos a temperatura ambiente. 11. Centrifugar las células lisadas 30 minutos a 15000 rpm. El sobrenadante corresponde a la fracción citoplasmática soluble (LS) y el pellet a la insoluble (LI). 12. Resuspender LI en SDS 2%. Conservar a 4 ºC. 13. Agregar a LS Binding Buffer 10x diluyéndolo 10 veces. 14. A las muestras SN y LS agregar 200 µL de resina Ni-NTA (QIAGEN) por cada 100 mL de muestra, o la misma cantidad para volúmenes menores. 15. Dejar agitando suavemente durante 1 hora a 4 ºC 16. Cargar las muestras con resina en columnas de afinidad. 17. Lavar las columnas 10 veces con 1 mL de Buffer de lavado. 18. Colectar eluyendo 3 veces con 0.5 mL de Buffer de elución. 19. Concentrar las muestras en tubos Centricon® YM-10 (Anexo A.4.6). Conservar a 4 ºC. Nota: La resina y las columnas deben ser previamente ambientadas en Binding Buffer 1x. A.4.6. Protocolo para concentración de proteínas con tubos Centricon YM-10 (Millipore) 1. Centrifugar los tubos Centricon® YM-10 con 100 µL de Tween 20 0.1%, a 6500 rpm por 30 minutos. 2. Cargar las muestras y centrifugar a 6500 rpm y 4 °C, durante aproximadamente 2 horas, o hasta que la muestra haya sido filtrada y concentrada a un volumen de al menos 250 µL. 3. Descartar el filtrado, y recuperar la proteína concentrada con el colector, centrifugando a 3000 rpm y 4 °C, por 5 minutos. 61 A.5. GELES DE POLIACRILAMIDA PARA PROTEÍNAS A.5.1. Gel SDS-poliacrilamida Gel de Resolución: Acrilamida-Bisacrilamida 29:1 Tris 1.5 M pH 8.8 + 0.4% SDS H2O destilada TEMED PSA 10% 1.58 mL 0.95 mL 1.27 mL 2 µL 20 µL Gel Concentrador: Acrilamida-Bisacrilamida 29:1 Tris 0.5 M pH 6.8 + 0.4% SDS H2O destilada TEMED PSA 10% 0.1675 mL 0.25 mL 0.575 mL 1.25 µL 7.5 µL A.5.2. Gel para Zimograma Gel de Resolución: Acrilamida-Bisacrilamida 29:1 Tris 1.5M pH8.8 + 0.4% SDS H2O destilada Gelatina 2% TEMED PSA 10% 1.625 mL 1.4 mL 0.6 mL 0.2 mL 5 µL 15 µL Gel Concentrador: Acrilamida-Bisacrilamida 29:1 Tris 0.5 M pH 6.8 + 0.4% SDS H2O destilada Gelatina 2% TEMED PSA 10% 0.25 mL 0.625 mL 0.125 mL 0.125 mL 3.5 µL 20 µL 62 A.6. SOLUCIONES STOCK A.6.1. Buffer de carga 5x para geles SDS-acrilamida (10 mL) Tris-HCl 1 M (pH 6.8) Glicerol 50% SDS 10% Azul de bromofenol 1% H2O 0.6 mL 5 mL 2 mL 1 mL 0.9 mL A.6.2. Solución de tinción (1 L) Coomassie Metanol Ácido acético H2O (HPLC) 1g 450mL 100mL 450mL A.6.3. Solución de destinción (1 L) Metanol Ácido acético H2O (HPLC) 450mL 100mL 450mL A.6.4. Buffer de carga 5x para zimograma Tris-HCl 0.5 M (pH 6.8) Glicerol SDS 10% Azul de bromofenol 1% 1 mL 0.8 mL 1.6 mL 0.5 mL A.6.5. Buffer de Incubación para zimograma (1 L) Glicina CaCl2 7.5 g 0.3 g Llevar a 1L con H2O (HPLC) Ajustar pH 8.0 A.6.6. Carbenicilina [100 mg/mL] Carbenicilina H2O (HPLC) 0.1 g 1 mL Filtrar para esterilizar 63 A.6.7. Tetraciclina [12.5 mg/mL] Tetraciclina Etanol 80% 0.0125 g 1 mL A.6.8. IPTG [0.5 M] IPTG H2O (HPLC) 0.11915 g 1 mL Filtrar para esterilizar A.6.9. Glucosa 20% (50 mL) Glucosa 10 mg Llevar a 50 mL con H2O (HPLC) Filtrar para esterilizar A.6.10. Xilosa 20% (50 mL) Xilosa H2O (HPLC) 10 g 50 mL Filtrar para esterilizar A.6.11. PEG-P 40%(p/v) PEG6000 en SMM1x Autoclavar por 12 minutos A.6.12. Sales CR5 8x K2SO4 MgCl2·6 H2O KH2PO4 CaCl2·2H2O H2O (HPLC) 1.25 g 50 g 0.25 g 11 g 625 mL Autoclavar 20 minutos. 64 A.6.13. Biding Buffer 10x (1 L) Na2HPO4 Na2HPO4 NaCl Imidazol 82.95 g 4.69 g 175.3 g 6.808 g Llevar a 1 L con H2O (HPLC) Ajustar pH 8.0 A.6.14. Wash Buffer 1x (100 mL.) Biding Buffer 10x Imidazol 10 mL 0.0681 g Llevar a 100 mL con H2O (HPLC) Ajustar pH 8.0 A.6.15. Elute Buffer 1x (100 mL) Biding Buffer 10x Imidazol 10 mL 1.634 g Llevar a 100 mL con H2O (HPLC) Ajustar pH 8.0 65 A.7. MAPA DE PARTIDORES PARA SECUENCIACIÓN Figura A.7: Partidores para secuenciación. Partidores sobre el vector pHIS1525: pHIS1, pHIS2, pHIS3, pHISR, pHISR1, pHISR2 y pHISR3. Partidores sobre el gen T7: pT7 y pT7R. Estos últimos fueron usados sólo en la secuenciación de las construcciones pHIS1525-T7 y pHIS1525-T7H. 66 A.8. GELES DE AGAROSA A.8.1. Gel de Agarosa 1% con resultados de PCR de colonias para construcciones pHIS1525-T8 y pHIS1525-T8 control Figura A.8.1: Resultados PCR de colonias para construcciones con el gen de T8. A.: (1-4): PCR de colonias transformadas con pHIS1525-T8. B.: (1-4): PCR de colonias transformadas con pHIS1525-T8 control. 67 A.8.2. Gel de Agarosa 1% con resultados de PCR de DNA plasmidial de B. megaterium para construcciones pHIS1522-T8 y pHIS1525-T8 Figura A.8.2: PCR de T8 desde DNA plasmidial de B. megaterium: (1) pHIS1522-T8. El control para este plasmidio no se muestra, pues no creció en el cultivo previo a la minipreparación. (2) pHIS1525-T8. (3) pHIS1525T8 de B. megaterium control. 68 A.9. RESULTADOS DE SECUENCIACIÓN A.9.1. pHIS1522-T8 69 Figura A.9.1: Resultados de secuenciación de pHIS1522-T8. Secuencias destacadas: Verde: T8. Las mutaciones puntuales se indican con una flecha de color rojo. A.9.2. pHIS1525-T8 70 Figura A.9.2: Resultados de secuenciación de pHIS1525-T8. T: secuencia teórica. S: Resultado de secuenciación. Secuencias destacadas: Anaranjado: Pétido señal del vector; Azul: IS1A; Verde: T8. Las mutaciones puntuales se indican con una flecha de color rojo. 71 A.9.3. pHIS1525-T7 72 73 Figura A.9.3: Resultados de secuenciación de pHIS1525-T7. T: secuencia teórica. S: Resultado de secuenciación. Secuencias destacadas: Anaranjado: Pétido señal del vector; Azul: IS1A; Verde: T7. Las mutaciones puntuales se indican con una flecha de color rojo. A.9.4. pHIS1525-T7H 74 75 Figura A.9.4: Resultados de secuenciación de pHIS1525-T7H. T: secuencia teórica. S: Resultado de secuenciación. Secuencias destacadas: Anaranjado: Pétido señal del vector; Azul: IS1A; Verde: T7; Rosado: Cola de polihistidinas. Las mutaciones puntuales se indican con una flecha de color rojo. 76