Laboratorio - Universidad Interamericana de Puerto Rico
Anuncio

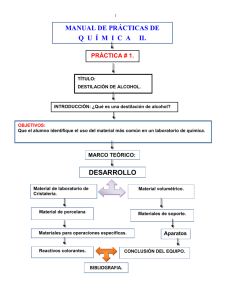
1 UNIVERSIDAD INTERMERICANA DE PUERTO RICO RECINTO DE BAYAMON DEPARTAMENTO DE CIENCIAS NATURALES Y MATEMÁTICAS Guías para el laboratorio Química Orgánica I Química 2221 Semana Práctica 1 Entrega de equipo 2 Punto de fusión 3 Destilación simple 4 Destilación fraccionada 5 Modelos Moleculares Estructuras (información del profesor) 6 Recristalización 7 Extracción 8 Cromatografía 9 Síntesis de 2- cloro – 2 – metilpropano 10 Síntesis de un éster 2 UNIVERSIDAD INTERAMERICANA DE PUERTO RICO CALENDARIO ACADÉMICO – LABORATORIO Curso CHEM 2221 Laboratorio G - 335 Martes 8:30 11:20 am Agosto-Diciembre 2013 PERIODO TÍTULO EXPERIMENTO FECHA ENTREGA DE INFORME * 1, 2, Agosto 20 Agosto 27 3, Sep. 3 Introducción al Laboratorio Entrega de equipo Asignación Tarea en la libreta de Laboratorio Punto de Fusión 4, Sept. 10 Destilación Simple 5, 6, 7, 8, 9, 10, Sept. Sept. Oct. Oct. Oct. Oct. 17 24 1 8 15 22 Destilación Fraccionada Grupos funcionales Tutorial para Modelos Moleculares Extracción N/A Recristalización 11, 12, 13, 14, Oct. Nov. Nov. Nov. 29 5 12 26 Síntesis 2- cloro-2- metilpropano Cromatografía N/A Examen Laboratorio y Entrega de equipo y libreta de laboratorio 15, 16. Dic. 6 *** Ultimo día para baja parcial o total con la anotación de “W” – Diciembre 7 de 2013. 3 UNIVERSIDAD INTERMERICANA DE PUERTO RICO RECINTO DE BAYAMON DEPARTAMENTO DE CIENCIAS NATURALES Y MATEMATICAS Experimento #1 Punto de Fusión QUIM 2221 Introducción El punto de fusión de un compuesto sólido puro se define como la temperatura a la cual el líquido y el sólido coexisten en equilibrio bajo una presión en particular. La presión más utilizada para medir el punto de fusión es la presión atmosférica, bajo estas condiciones el punto de fusión se conoce como el punto de fusión normal. Cambios moderados de presión no afectan significativamente el punto de fusión, sin embargo, una pequeña cantidad de impurezas en el sólido tienden a disminuir su valor. Por lo tanto, el punto de fusión de un compuesto orgánico es una propiedad física que se utiliza comúnmente para establecer la pureza de un compuesto o para determinar su identificación. Experimentalmente se informa el punto de fusión de un compuesto utilizando el intervalo de temperatura en el cual el compuesto se derrite. Es decir, se informa la temperatura en la cual aparece la primera gota de líquido y la temperatura en que la última partícula de sólido desaparece. Las impurezas pueden causar que el intervalo de temperatura en que ocurre la fusión sea más amplio que el intervalo de temperatura del compuesto puro Objetivos 1. Distinguir las diferentes formas para determinar el punto de fusión. 2. Determinar el punto de fusión de una muestra sólida. 3. Discutir la relación entre el punto de fusión y la pureza del compuesto. 4. Identificar un compuesto por el punto de fusión . Procedimiento Los siguientes compuestos están disponibles para determinar el punto de fusión: urea benzamida ácido benzoico ácido esteárico 1-naftol ácido - o- tolúico mezclas (1-naftol y ácido esteárico) 4 Para la determinación del punto de fusión se utilizarán las siguientes técnicas experimentales: 1. Baño de aceite (se utilizarán dos muestras que no sean las mezclas ni sus componentes puros) a. b. c. d. e. 2. Pulverizar la muestra y preparar el tubo capilar siguiendo las instrucciones y las demostraciones del instructor. Unir el tubo capilar al termómetro utilizando un pedazo de goma. Asegurarse que la parte del tubo capilar que contiene la muestra está en contacto con el bulbo del termómetro. Colocar el termómetro con el tubo capilar en un baño de aceite (vaso precipitado de 150 ml con aceite). Asegurarse que el pedazo de goma NO está en contacto con el aceite. Calentar el baño de aceite con mechero o con la plancha de calentamiento. Tomar el punto de fusión experimental. Tubo Thiele (se utilizarán dos muestras que no sean las mezclas ni sus componentes puros). a. b. c. d. e. 3. baño de aceite tubo Thiele melt-temp Pulverizar la muestra y preparar el tubo capilar siguiendo las instrucciones y las demostraciones del instructor. Unir el tubo capilar al termómetro utilizando un pedazo de goma. Asegurarse que la parte del tubo capilar que contiene la muestra está en contacto con el bulbo del termómetro. Colocar el termómetro con el tubo capilar en el tubo Thiele. Asegurarse que el pedazo de goma NO está en contacto con el aceite. Ver diagrama. Calentar con mechero el tubo Thiele. Tomar el punto de fusión experimental. Melt-temp (se usarán las muestras que corresponden a las mezclas y sus componentes puros.) a. b. Pulverizar la muestra y preparar el tubo capilar siguiendo las instrucciones y las demostraciones del instructor. Colocar el tubo capilar en el melt-temp. 5 c. d. 4. Colocar el termómetro en el melt-temp, instrucciones del instructor. Tomar el punto de fusión experimental. seguir las Empleando cualquiera de las técnicas anteriores determinar el punto de fusión de un desconocido. Para el punto de fusión del desconocido, se recomienda preparar dos muestras en tubos capilares. La primera muestra se utiliza para determinar el punto de fusión aproximado de la muestra. Una vez se conoce esta temperatura se procede a enfriar el sistema por lo menos 10º para entonces correr la segunda muestra a la rapidez recomendada. . Recomendaciones 1. 2. 3. 4. 5. 6. 7. La rapidez de calentamiento debe ser de aproximadamente 2º por minuto. Nunca repita el punto de fusión con el mismo tubo capilar. Se reporta el intervalo de temperatura. Cuando aparece la primera gota de líquido y la temperatura a la cual la última partícula de sólido desaparece. El punto de fusión de las mezclas se pueden hacer simultáneamente para lograr precisión en los resultados. Debe identificar claramente cada tubo capilar. Disponer los tubos capilares y los desperdicios en los envases correspondientes. No lavar el tubo Thiele al terminar el experimento. De ser necesario agitar el baño de aceite para mantener la temperatura uniforme. Preguntas para contestar en el informe. 1. ¿Cuál fue la temperatura eutéctica para la mezcla que utilizó en el laboratorio? 2. ¿Cuál será el efecto observado en el punto de fusión al calentar la muestra rápidamente? 3. Un estudiante determinó que el punto de fusión de un desconocido era 115 ºC. El estudiante dejó que la muestra se enfriara y solidificara, entonces repitió el procedimiento utilizando la misma muestra. En la segunda ocasión el punto de fusión fue 125ºC. Explique el cambio en el punto de fusión. 4. ¿Cuánto calor se requiere para derretir 35.0 gramos de hielo a 0ºC. El calor molar de fusión de hielo es 6.0 KJ/mol. 6 Referencias Eaton, D.C. (1989). Laboratory Investigations in Organic Chemistry. New York.: McGraw Hill. 7 UNIVERSIDAD INTERMERICANA DE PUERTO RICO RECINTO DE BAYAMON DEPARTAMENTO DE CIENCIAS NATURALES Y MATEMATICAS Experimento # 2 y # 3 Destilación Simple y Fraccionada QUIM 2221 Introducción Destilación es la técnica principal utilizada para separar y purificar líquidos orgánicos. En este proceso un líquido en un envase pasa a vapor que posteriormente se condensa y se recibe en otro envase. En la destilación simple cuando el líquido se calienta, su vapor entra en contacto con el termómetro, pasando inmediatamente al condensador donde se enfría y se convierte nuevamente en líquido. Durante la destilación de una mezcla de dos líquidos, el compuesto con el punto de ebullición menor destilará primero a temperatura constante (su temperatura de ebullición). El compuesto con el mayor punto de ebullición destilará cuando la temperatura alcance su punto de ebullición. La destilación fraccionada es una técnica más eficiente para separar líquidos con puntos de ebullición cercanos (+/-50º). En este proceso una columna de fraccionamiento (tubo vertical con material inerte) se intercala entre el matraz de destilación y el condensador (Ver Figura). El material inerte de la columna se utiliza para aumentar el área superficial y por consiguiente los vapores estarán sometidos a varios ciclos de condensación según suben por ella. En otras palabras, cuando el condensado baja por la columna es nuevamente calentado y revaporizado. Cada vez que el condensado se vaporiza, los vapores se concentran en el componente de menor punto de ebullición. El vapor alcanza el termómetro, el condensador y finalmente el envase donde se recoge el destilado Objetivos. 1. Identificar el equipo de la destilación simple. 2. Reconocer el equipo para hacer destilación fraccionada. 3. Diferenciar las dos clases de destilaciones. 4. Aplicar la técnica de destilación para separar líquidos. . 8 Procedimiento El líquido que se usará en la destilación simple y en la destilación fraccionada consta de una mezcla de 2 - propanol / agua (50% v/v). La solución ha sido previamente preparada. 1. 2. Destilación Simple a. Antes de comenzar a destilar debe practicar la forma y manera de montar el equipo de destilación simple de forma rápida y segura (Ver diagrama). b. Coloque 50 ml de la mezcla 2 – propanol /agua en el frasco o matraz de destilación de 100 ml y añada 2 ó 3 pedazos de material poroso “glass beads” ,“boiling chips” o “boileezers”. c. Antes de comenzar a destilar debe tener el visto bueno del instructor. Comience la destilación, anote la temperatura inicial. d. Verifique la velocidad de la destilación. e. Recoja el destilado en una probeta y anote la temperatura cada 2 ml del producto recolectado. f. En el informe se presentará una gráfica de temperatura versus volumen de destilado. Destilación Fraccionada a. Preparar una columna de destilación fraccionada para el empaque use “glass beads” (esferas de vidrio) siguiendo las instrucciones del instructor. b. Prepare el equipo de destilación fraccionada (Ver diagrama). c. Coloque 50 ml de la mezcla 2 – propanol /agua en el frasco o matraz de destilación de 100 ml y añada 2 ó 3 pedazos de material poroso “glass beads”, “boiling chips” o “boileezers”. d. Comience la destilación, anote la temperatura inicial. e. Verifique la velocidad de la destilación. 9 . f. Recoja el destilado en una probeta y anote la temperatura cada 2 ml del producto recolectados. g. En el informe se presentará una gráfica de temperatura versus volumen de destilado. Recomendaciones 1. Al unir las partes del equipo de destilación debe colocar un poco de grasa a las partes. ¿Por qué? 2. Las grapas deben sostener firmemente las partes del equipo. Evitar utilizar un exceso de grapas. 3. Los “boiling chips” se utilizan para promover una ebullición uniforme y evitar el sobrecalentamiento. 4. Colocar el bulbo del termómetro en el lugar indicado. Ver diagrama. 5. Disponer de los correspondientes. 6. La rapidez de destilación debe ser de aproximadamente de 1-3 ml por minutos. 7. La destilación termina cuando quedan 2 ml en el matraz de destilación. desperdicios en los envases Preguntas para contestar en el informe. 1. ¿Por qué en la gráfica de Temperatura versus Volumen del Destilado en una sección de la curva la temperatura permanece constante? 2. ¿Cuál es la diferencia entre el punto de ebullición y el punto de ebullición normal? 3. ¿Qué es una mezcla azeotrópica? 4. ¿Cómo se afecta el punto de ebullición de una solución al añadir un soluto no volátil? Explique. 5. ¿Cómo afecta la posición del termómetro en el equipo de destilación la lectura del punto de ebullición? Explique. 10 5. ¿Cuáles son las posibles fuentes de calor usadas en la destilación? 6. ¿Qué sucede si la destilación se hace en forma rápida? Referencias Eaton, D.C. (1989). Laboratory Investigations in Organic Chemistry. New York.: McGraw Hill. 11 UNIVERSIDAD INTERMERICANA DE PUERTO RICO RECINTO DE BAYAMON DEPARTAMENTO DE CIENCIAS NATURALES Y MATEMATICAS Experimento # 4 . Recristalización QUIM 2221 Introducción Recristalización es el proceso que normalmente se utiliza para purificar un sólido impuro (con alrededor de 10% de impurezas). Este proceso está basado en el principio de solubilidad ya que requiere disolver la muestra impura en una cantidad mínima de solvente. Es importante seleccionar el solvente adecuado, ya que el soluto se debe disolver en el solvente caliente y ser insoluble en el solvente frío. Considerando esta característica, si la impureza es menos soluble que el compuesto de interés, entonces no se puede disolver en el solvente caliente y se puede remover en la filtración por gravedad. Si la impureza es más soluble entonces estará presente en el solvente frío y queda en el filtrado después de la recristalización. Si al disolver el soluto en el solvente caliente se observa la presencia de sólidos insolubles (impurezas) se debe filtrar la solución caliente por gravedad. Esta filtración requiere utilizar un embudo de cuello corto o sin él y mantener la solución caliente mientras se está llevando a cabo el proceso de filtración. Si al disolver el soluto se observa que la solución está contaminada con impurezas que muestran color se puede añadir un poco de carbón activado para remover dichas impurezas y llevar a cabo el proceso de filtración en caliente. Después de haber filtrado en caliente se deja enfriar la solución a temperatura de salón se pasa entonces a un baño de hielo. El sólido puro se obtiene finalmente al filtrar la solución fría por la técnica de filtración por succión. Se debe enfriar el sistema para esta filtración por vacío o succión. La masa del material recuperado se determina para poder calcular el por ciento de rendimiento del proceso. La pureza del material se puede examinar tomando el punto de fusión de la muestra recuperada. Objetivos 1. Reconocer las características de un buen solvente 2. Diferenciar la filtración por gravedad de la filtración por succión o vacío. 3. Purificar un sólido impuro. 4. Determinar la pureza del compuesto. 12 Procedimiento 1. Colocar aproximadamente 2 gramos de ácido benzoico impuro en un matraz cónico. Añada el solvente caliente (agua) en pequeñas cantidades hasta que se observe que no se disuelve más material. Añada un exceso de solvente, hasta un 5% del porciento del volumen añadido para prevenir la cristalización prematura. 2. Mantenga caliente la solución del paso #1 y prepare el equipo de filtración por gravedad. Ajuste al soporte un aro de hierro para colocar el embudo de cuello corto que quedará sobre el matraz cónico. Asegure el matraz cónico con una grapa y añada al matraz 5 ml del solvente (agua). Caliente el agua del matraz con una plancha de calentamiento para que el vapor caliente el embudo. Coloque el papel de filtro previamente doblado sobre el embudo. Cuando el embudo esté caliente, retire la plancha y comience la filtración por gravedad. Ver diagrama. 3. Usando una grapa transfiera la solución caliente del paso #1 a través del embudo. Al colocar la solución en el embudo, el nivel no debe pasar de 3-5 mm debajo del borde de papel. Cuando el líquido haya bajado a una tercera parte del volumen original en el embudo, añada más solución caliente. Para evitar evaporación, coloque un cristal de reloj sobre el embudo durante la filtración. 4. Deje que el filtrado se enfríe a temperatura de salón. Luego se coloca en baño de hielo durante 5 minutos para maximizar la cristalización y se procede a filtrar por succión utilizando un embudo Buchner y un aspirador de agua para crear vacío. Ver diagrama. 7. Los cristales se lavan con aproximadamente 5 ml de solvente frío (agua). Se puede utilizar una espátula para separar el precipitado de los bordes del papel de filtro y transferirlo a un cristal de reloj previamente pesado. Recomendaciones 1. 2. 3. 4. Buena coordinación durante la filtración por gravedad, manteniendo la solución caliente. El carbón activado que saque con la punta de la espátula es el apropiado para remover las impurezas. Será discreción del instructor el utilizar una trampa de vacío para proteger el filtrado (licor madre). No añadir carbón activado a una solución hirviendo. 13 5. El añadir mucha solución caliente al filtrar por gravedad de manera que se desborde por atrás del papel de filtro puede resultar en pérdida de producto. Preguntas para contestar en el informe. 1. 2. 3. 4. 5. 6. Mencione tres características más importantes que debe tener el solvente en una recristalización. ¿Por qué se debe enfriar primero la solución a temperatura de salón y luego colocar en baño de hielo? Hacer un esquema de una trampa de vacío, explique su función. ¿Por qué se usa un embudo sin cuello para filtrar en caliente? ¿Qué porciento de ácido benzóico hay en su muestra? ¿Cuál es el punto de fusión del ácido benzóico recuperado? Referencias Eaton, D.C. (1989). Laboratory Investigations in Organic Chemistry. New York.: McGraw Hill. 14 UNIVERSIDAD INTERMERICANA DE PUERTO RICO RECINTO DE BAYAMON DEPARTAMENTO DE CIENCIAS NATURALES Y MATEMATICAS Experimento # 5 Extracción QUIM 2221 Introducción Extracción es la técnica de separación más frecuentemente usada para aislar o separar uno o más componentes en una mezcla. En las extracciones líquido-líquido el proceso consiste en poner dos líquidos (fases) immiscibles en contacto. La separación de un componente en específico de una mezcla se logra por la diferencia en solubilidad de la especie y los solventes utilizados. La regla general que gobierna la separación de los componentes es: igual disuelve igual. Por ejemplo, en una extracción líquido-líquido que consiste de una mezcla de dos compuestos disueltos en un solvente, la solución se coloca en un embudo de separación. Al embudo se le añade un segundo solvente que es immiscible con el primero, ambos son agitados y se espera que las fases se separen. Idealmente, uno de los componentes en la mezcla se extrae por el nuevo solvente de acuerdo a su solubilidad y el otro permanecerá en el solvente original. Los dos solventes son separados y luego de su remoción se obtienen los compuestos de interés. En muchas ocasiones una sola extracción no es suficiente para extraer un compuesto en el solvente utilizado. Para lograr una mejor separación se utiliza el proceso de extracciones múltiples. En este proceso se extrae una o dos veces más usando solvente fresco y luego se combinan los extractos. El agua es frecuentemente usada en esta técnica como uno de los solventes porque tiene la ventaja de ser immiscible con muchos solventes orgánicos. En el caso de usar agua y un solvente orgánico, al separarse las fases, se obtiene lo que se conoce como la capa acuosa (agua) y la capa orgánica. Los solutos se distribuirán entonces entre estas fases de acuerdo a sus solubilidades relativas. Se debe mencionar que las soluciones salinas saturadas se utilizan para extraer exceso de agua en un solvente orgánico y también para que los compuestos orgánicos solubles en agua sean de más fácil extracción por el solvente orgánico. Al separarse una fase orgánica de una acuosa se necesita secar el agua que se quedó aún en la primera. Es por esa razón que se utilizan agentes secantes, tales como: sulfato de sodio anhidro, cloruro de calcio anhidro o sulfato de magnesio. Antes de remover el solvente estos 15 agentes secantes se deben sacar por decantación o filtración por gravedad. Objetivos 1. Identificar las características de un buen disolvente para la extracción. 2. Utilizar el embudo de separación. 3. Identificar las capas orgánica y acuosa. 4. Determinar la pureza de cada sólido recuperado. Procedimiento 1. Colocar aproximadamente 3 gramos de una mezcla que consiste de aspirina y naftaleno en un matraz cónico de 125 ml. Añadir 30 ml de éter, agitar con el agitador hasta disolver. 2. Colocar la mezcla del paso #1 en un embudo de separación. Añadir 30.0 ml de bicarbonato de sodio al 5%. Siguiendo las instrucciones del instructor, lleve a cabo la extracción. Ver diagrama. 3. Coloque el embudo de separación en un aro y remueva la capa acuosa en un vaso precipitado. Caliente en el extractor para eliminar el éter presente. Enfríe y añada cuidadosamente 3M HCl hasta que se precipite la aspirina. 4. Obtenga el producto mediante filtración por succión. 5. A la solución orgánica en el embudo de separación añadir 15 ml de una solución salina saturada. Separar las fases, transfiera la fase orgánica a un vaso precipitado, añada una pequeña cantidad del agente secante (esperar por 15 minutos). 6. Decante el líquido, evapore en un baño de María y obtenga el producto. 7. A cada producto obtenido se le tomará la masa y el punto de fusión en el próximo período del laboratorio. Calcular el porciento de recuperación de cada producto. 16 Recomendaciones 1. Es importante identificar la fase acuosa y la fase orgánica en el embudo de separación antes de separar las fases, para realizar este paso se puede usar la prueba de densidad. Añada una o dos gotas de agua, observe en cual de las fases se distribuye el agua. 2. Seguir las instrucciones en el manejo y operación del embudo de separación. Nunca abra la válvula dirigiendo hacia otra persona o a usted mismo. Siempre sostenga el embudo con ambas manos y de forma segura. 3. Al trabajar con éter, utilice el extractor y mantenga el envase cerrado mientras trabaja. Preguntas para contestar en el informe. 1. Explique por qué se utilizaron en el proceso el bicarbonato de sodio, la solución salina y el agente secante. 2. ¿Qué características debe tener un buen solvente en una extracción? Explique. 3. ¿Usaría usted agua y etanol como solventes para llevar a cabo una extracción? Referencias Eaton, D.C. (1989). Laboratory Investigations in Organic Chemistry. New York.: McGraw Hill. 17 UNIVERSIDAD INTERMERICANA DE PUERTO RICO RECINTO DE BAYAMON DEPARTAMENTO DE CIENCIAS NATURALES Y MATEMATICAS Experimento # 6 Cromatografía QUIM 2221 Introducción Cromatografía es un método que se utiliza para separar uno o más componentes en una mezcla. Hay tres tipos de cromatografías que son principalmente usados en los laboratorios de química: columna, capa final (TLC) y gas (GC). Aunque también se pueden encontrar otros tipos: papel, líquida (HPLC), intercambio iónico y otros. La cromatografía es un método físico en el que los componentes de una muestra se distribuyen entre dos fases: fase estacionaria (adsorbente) y la fase móvil. La fase móvil es un fluido que se mueve a través de la fase estacionaria. En la técnica de cromatografía la muestra pasa a través de la fase estacionaria vía la fase móvil (líquido o gas). Los componentes se separan de acuerdo al grado de adsorción presente en ellos y la fase estacionaria. En otras palabras, en la fase estacionaria los componentes son adsorbidos y la fase móvil los “des-adsorbe” y arrastra a lo largo de la fase estacionaria. La habilidad de un compuesto para ser adsorbido fuertemente por el adsorbente está relacionada con las fuerzas intermoleculares entre ambos. El solvente debe ser capaz de disolver los compuestos que se van a separar, sin embargo, su capacidad de “desadsorción” dependerá de la naturaleza del adsorbente y de los compuestos que son movidos. La cromatografía de papel es el tipo de cromatografía más sencilla. Esta técnica puede ser considerada una forma de cromatografía de capa fina. En ella la fase estacionaria es celulosa. Una muestra de una solución se aplica al papel de cromatografía cerca del borde, se coloca en un envase con solvente en el fondo y se deja que el solvente suba por el papel debido a la acción capilar del mismo. Cuando el solvente llega a la muestra, los componentes más solubles en él comienzan a disolverse (des-adsorción) y a desplazarse hacia arriba. Es decir, mientras el solvente avanza por el papel, los componentes más solubles en el solvente y menos adsorbidos por la fase estacionaria tienden a moverse más rápido por el papel, los componentes menos solubles y más fuertemente adsorbidos tienden a moverse más lentamente. Por lo tanto, como cada soluto se adsorbe y “des-adsorbe” en forma diferente, cada uno de ellos se moverá a diferente velocidad y se conseguirá gradualmente la separación deseada. 18 Para cada soluto se puede determinar el Rf que corresponde a una medida de cuanto se ha movido un componente relativo al frente del solvente. Rf Distancia recorrida por el componente = -----------------------------------------------------Distancia recorrida por el frente del solvente Rf es constante para una sustancia en particular, su valor depende de las fases estacionaria y móvil y de otros factores experimentales. Este valor puede servir para caracterizar los componentes de una mezcla si las condiciones experimentales se controlan cuidadosamente. Objetivos 1. Identificar las clases de cromatografía 2. Aplicar la técnica de cromatografía de papel para separar los componentes de una mezcla. Procedimiento 1. Conseguir cuatro hojas de hojas de papel para cromatografía de aproximadamente 10 cm x 20 cm. Utilizar guantes para no contaminar el papel. Maneje el papel sólo por las orillas (o con pinzas) y colóquela sobre otra hoja de papel que esté limpio. Con mucho cuidado, dibuje en las hojas una línea clara con lápiz alrededor de 10 mm del borde inferior que será uno de los lados largos de la hoja. Bajo la línea anote los números del uno al tres para identificar las muestras. Estos deben estar a una distancia de 2.5 cm de distancia. 2. Para preparar las tres cámaras de revelado, utilice tres vasos de precipitado de 600 ml, en el cual se colocará el solvente que va a utilizar. Una cámara se preparará con volúmenes iguales de la mezcla de NH4OH 2M, etanol 95% y butanol, la segunda cámara con una mezcla (ya preparada) de 60% butanol y 40% de agua y la última cámara contiene butanol. En cada una de las cámaras se añadirá solvente hasta un nivel de aproximadamente 5mm. Una vez colocado el solvente, las cámaras serán cubiertas con un cristal de reloj para saturar el sistema. 3. Utilizando una micropipeta para cada muestra de colores vegetales, coloque una “microgota” en el número asignado en el papel. Permitir que la microgota se seque. Doblar el papel en forma de cilindro evitando solapar los bordes y sujete con grapas 19 los bordes del papel. Este paso se realizará tres veces, una hoja de papel para cada cámara. 4. Colocar con mucho cuidado las hojas de papel en cada cámara de revelado de forma que el borde inferior quede sumergido en el solvente. No permita que el papel toque las paredes del vaso o el fondo y tape el vaso. Deje que el solvente suba por el papel hasta cerca de 1 cm del borde superior. 5. Saque, deje secar el papel con cuidado, y marque una línea con lápiz hasta donde subió el solvente. 6. Haga un círculo con lápiz alrededor de cada mancha desarrollada. Mida las distancias requeridas para calcular Rf. Las distancias se deben medir desde el centro de la mancha. 7. En base a los resultados de las pruebas anteriores, decida qué cámara deberá ser utilizada para separar una mezcla desconocida de colores vegetales. Utilizando la cuarta hoja de papel, prepare su desconocido de forma similar al paso #1. 8. Utilizando los valores de Rf de los colores vegetales, identifique los componentes en el desconocido. Recomendaciones 1. No tocar el papel de cromatografía directamente con las manos, usar guantes. Mantener el área de trabajo limpia para mejores resultados. 2. La razón de usar las micropipetas es para colocar una pequeña cantidad de muestra en el papel. Si el tamaño de la muestra es muy grande no se podrá obtener una buena separación. Seguir las instrucciones relacionadas con la separación de las muestras en el papel. 3. No permitir que el solvente alcance el borde superior del papel. Preguntas para contestar en el informe 1. Explique los resultados de los cromatogramas obtenidos de los colores vegetales en la mezcla de solventes y en butanol. 20 2. ¿Qué características debe tener el solvente en una cromatografía? Explique. 3. Alúmina es un adsorbente que se utiliza con frecuencia en cromatografía. ¿Por qué es mayormente usado para mezclas de compuestos no polares? Referencias Eaton, D.C. (1989). Laboratory Investigations in Organic Chemistry. New York.: McGraw Hill. Svoronos, P., Sarlo, E., & Kulawiiec, R.J. (1997). Organic Chemistry: Laboratory Manual.Second Edition.Dubuque, IA.:WCV. Wm. C. Brown Publishers 21 UNIVERSIDAD INTERMERICANA DE PUERTO RICO RECINTO DE BAYAMON DEPARTAMENTO DE CIENCIAS NATURALES Y MATEMATICAS Experimento # 7 Síntesis de 2-cloro-2-metil propano QUIM 2221 Introducción 2-cloro-2-metil propano, un haluro de alquilo terciario, puede ser sintetizado reaccionado un alcohol terciario, 2-metil-2-propanol, con HCl concentrado. El mecanismo de esta reacción es S N1, lo cual implica la formación de un carbocatión terciario. Sin embargo, también es posible la formación de alquenos por el mecanismo E2 Objetivos 1. 2. 3. 4. Preparar un haluro de alquilo a partir de un alcohol. Utilizar las técnicas de destilación simple y extracción. Aplicar los conocimientos de mecanismo de reacción y estequiometria Determinar el rendimiento del producto obtenido, Procedimiento 1. Colocar en un matraz de destilación de 50 ml 5.0 gramos de 2metil-2-propanol (d=0.78 g/ml) y 15 ml de HCl (concentrado). 2. Destilar la mezcla (destilación simple) del paso #1, recogiendo el destilado en hielo. 3. Transfiera el destilado a un embudo de separación y lleve a cabo una extracción con 10 ml de una solución saturada de bicarbonato de sodio. 4. Separar las fases, lavar la fase orgánica en el embudo de separación utilizando 15 ml de agua. 5. Separar nuevamente las fases, colocar la fase orgánica en un matraz cónico y añadir el agente secante (cloruro de calcio), tapar con el cristal de reloj, esperar por 15 minutos. 22 6. Decantar la mezcla del paso #5 a un matraz de destilación de 50 ml. y llevar a cabo una destilación simple. Recoger el destilado (producto destila entre 48ºC - 51ºC)en un envase previamente pesado. 7. Calcular el por ciento de rendimiento. Recomendaciones 1. HCl es corrosivo, trabaje con mucho cuidado en el extractor utilizando la vestimenta adecuada. 2. Llevar a cabo las técnicas de destilación y extracción siguiendo los procedimientos y recomendaciones utilizados previamente. Preguntas para contestar en el informe 1. Dar el mecanismo de la reacción. 2. ¿Cuál es el propósito del bicarbonato de sodio? 3. En esta reacción, ¿es posible un rearreglo del carbocatión? Explique. 4. Escriba la ecuación para la síntesis del 2-cloro-2-metilpropano. Referencias Eaton, D.C. (1989). Laboratory Investigations in Organic Chemistry. New York.: McGraw Hill. Svoronos, P., Sarlo, E., & Kulawiiec, R.J. (1997). Organic Chemistry: Laboratory Manual.Second Edition.Dubuque, IA.:WCV. Wm. C. Brown Publishers 23 Universidad Interamericana de Puerto Rico Recinto de Bayamón Departamento de Ciencias Naturales y Matemáticas Experimento # 8 Sabores artificiales QUIM 2221 Introducción Cuando saboreamos una fruta o muchos otros alimentos el sabor que gustamos se debe a la presencia de compuestos orgánicos que son los ésteres. Estos compuestos químicos proceden no sólo de la naturaleza, se pueden sintetizar para ser usados comercialmente. Los ésteres sirven para resaltar el sabor de los helados, gelatinas, bebidas, bizcochos y muchos otros productos. Los ésteres sintéticos se usan con más frecuencia en la industria de alimentos. Un método comúnmente utilizado para preparar ésteres en mediante la reacción de un ácido carboxílico con un alcohol. La reacción es reversible y requiere un catalizador. En esta práctica de laboratorio el estudiante puede sintetizar el éster de su interés, utilizando el alcohol y el ácido carboxílico correspondiente. Lista de ésteres que pueden ser sintetizados. SABOR ALCOHOL ÁCIDO CARBOXÍLICO Guineo “Peach Pera Piña “Raspberry” “Wintergreen” Yerba buena Isopentanol Älcohl bencílico Propanol Etanol Isobutanol Metanol Acido acético Acido acético Acido acético Acido butírico Acido fórmico Acido salicílico Objetivos 1. Sinetizar un éster. 2. Aplicar las destrezas de laboratorio relacionadas con las técnicas de reflujo,extracción y destilación. 3. Aplicar los conocimientos de mecanismo de reacción y estequiometria. 24 4. Calcular el por ciento de rendimiento de la reacción. Procedimiento 1. Escoger el sabor que desea sintetizar. 2. Use un matraz de destilación de 100 mL y agregar las cantidades de alcohol y ácido carboxílico según lo especifica la NOTA # 1 y ver las NOTAS # 2 y # 3. 3. Añadir cuidadosamente 4 mL de ácido sulfúrico, agitar suavemente. Añadir varios “boiling sone” y refluir por 2 a 3 horas. Ver NOTA # 2. 4. Enfríe la mezcla a temperatura ambiente y ponga el contenido en un embudo de separación. Enjuagar el matraz con varias porciones de una solución fría a de NaCl 15% , y añadir estas porciones al embudo de separación. La fase acuosa en el embudo de separación debe doblar el volumen de la fase orgánica. 5. Agitar y separar las fases. 6. Lavar la fase orgánica con un volumen similar de una solución fría de bicarbonato de sodio al 10%. Ver NOTA # 4. 7. Verificar el pH de la fase orgánica. Si el pH es ácido lava nuevamente con la solución fría de bicarbonato de sodio al 10% hasta pH neutral.. 8. Dejar que la capa orgánica se aclare y transferir el éster (fase orgánica) a un matraz cónico límpio y seco. Añadir sulfato de magnesio anhidro. Agitar y dejar reposar por 10 minutos. 9. Decantar o filtrar hacia en matraz de destilación de 50 mL.. Añadir “boiling chips” y destilar. 10. Recolectar el destilado en una probeta previamente pesada, luego la coloca dentro de en un baño con hielo. 11. Pesar el producto y calcular el rendimiento de la reacción 25 NOTAS 1. Reactivos utilizados SABOR ALCOHOL ÁCIDO CARBOXÍLICO Guineo Isopentanol (0.10 moles) “Peach Pera Älcohl bencílico l (0.10 moles) Propanol (0.20 moles) Piña Etanol (0.15 moles) “Raspberry” Isobutanol (0.15 moles) “Wintergreen” Yerba buena Metanol (1.0 moles) Acido acético (0.30 moles) Acido acético (0.30 moles) Acido acético (0.40 moles) Acido butírico (0.25 moles) Acido fórmico (0.30 moles) Acido salicílico (0.10 moles) Antes de empezar el laboratorio, el estudiante debe tener en la libreta de laboratorio la información correspondiente del alcohol, el ácido carboxílico y el éster como: a. Fórmula estructural b. Peso molecular c. Punto de ebullición d. Densidad e. Masa o volumen de los moles indicados Precauciones y recomendaciones 2. Los ácidos sulfúrico y acético son extremadamente peligrosos y corrosivos. Usar el extractor. 3. Las cantidades de los reactivos que usan en este experimento deben ser medidas con mucha precisión. Use pipetas y probetas apropiadas para medir los volúmenes correspondientes. 4. Destape continuamente el embudo de separación para liberar la presión. 5. Si el sabor (éster) tiene un punto de ebullición menor de 200⁰C use destilación simple. 26 6. Si el sabor (éster) tiene un punto de ebullición mayor de 200⁰C use destilación simple al vacío. Preguntas para contestar en el informe 1. ¿En qué consiste el proceso de esterificación? 2. Escriba la ecuación de equilibrio para la esterificación. 3. ¿Cómo actúa el principio de Le Chatelier en la estificación? 4. ¿Para qué se usan en el experimento las soluciones de bicarbonato de sodio y cloruro de sodio? 5. ¿Por qué la industria de los alimentos no usa los sabores naturales? Referencias Eaton, D.C. (1989). Laboratory Investigations in Organic Chemistry. New York.: McGraw Hill. Svoronos, P., Sarlo, F. & Kulawiec, R. (1996). Organic Chemistry Laboratory Manual. Second Edition. Dubuque. IA.: Wm C. Brown Publishers. Revisado por María Elvira Ballesteros B Agosto 2012