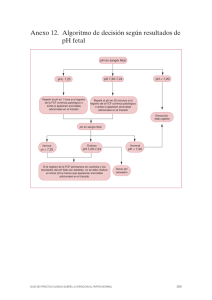
Untitled
Anuncio

RESUMENES DE PRESENTACIONES ORALES O-01. FETOSCOPÍA LÁSER EN EL TRATAMIENTO DEL SÍNDROME TRANSFUNDIDO-TRANSFUSOR: RESULTADOS Y EVALUACIÓN DE DESEMPEÑO EN LOS PRIMEROS 40 CASOS CON LC-CUSUM. Olivia Cambiaso, Lucía Vázquez, César H Meller, Gustavo Izbizky, Horacio A Aiello, Lucas Otaño. Unidad de Medicina Fetal, Servicio de Obstetricia, Hospital Italiano de Buenos Aires Introducción: La ablación láser de anastomosis placentarias por fetoscopía (ALF) es el tratamiento de elección del síndrome transfundido transfusor (STT). Una revisión sistemática muestra que la ALF resulta en 83% de al menos un gemelo sobreviviente (AUGS) y 54% de sobrevida de ambos (SA). En ausencia de programas de entrenamiento, tradicionalmente se ha implementado la ALF a partir de visitas de observación en centros experimentados. Se estima que la curva de aprendizaje para alcanzar la competencia necesaria requiere alrededor de 60 fetoscopías. El método LC-CUSUM es una herramienta estadística que permite evaluar la curva de aprendizaje y determinar objetivamente cuando se alcanzó un nivel de competencia predeterminado. Objetivo: Describir los resultados de los embarazos con STT tratados por ALF en nuestra Unidad y evaluar el nivel de competencia o desempeño alcanzado con la técnica. Materiales y métodos: Estudio retrospectivo de una cohorte consecutiva de embarazos dobles complicados con STT tratados con ALF, entre septiembre de 2009 y febrero de 2014. La ALF fue realizada bajo anestesia regional con técnica de ablación selectiva. Se registraron prospectivamente: estadío del STT, edad gestacional (EG) a la fetoscopía, localización placentaria, complicaciones maternas y sobrevida de los gemelos. Los resultados primarios fueron SA y AUGS. Se evaluó la curva de aprendizaje y nivel de competencia con LCCUSUM en base a la tasa de AUGS, con objetivos de desempeño predefinidos como “aceptable” de 82% e “inaceptable” de 70%. Resultados: Se realizaron ALF en 40 embarazos a una EG media de 20,4 semanas (r=16,3-26,6). Se clasificaron como STT estadío II o III al 93%. Placenta predominantemente anterior en 40%. Hubo 2 er complicaciones maternas severas. Hay 3 casos cursando el 3 trimestre. La mediana de EG al nacimiento de los embarazos con AUGS fue de 33,4 (IQ: 31,4-35). La tasa de AUGS fue de 75% y de SA de 47%. La evaluación con LC-CUSUM muestra que todavía no se ha alcanzado el objetivo de desempeño predefinido como “aceptable”. Conclusión: La implementación del programa de ALF muestra una tasa de AUGS de 75% y de SA de 47%. Si bien no existe diferencia significativa con lo publicado en la literatura (83% y 54% respectivamente), todavía no hemos alcanzado el objetivo de desempeño predefinido. La objetivación del desempeño en cirugía fetal es relevante para el desarrollo de un programa, para su mantenimiento y para el entrenamiento de nuevos operadores. O-02. ¿CUÁNTAS ANOMALÍAS CROMOSÓMICAS SE PIERDEN HACIENDO UN TEST DE ADN FETAL EN SANGRE MATERNA EN LUGAR DE UN CARIOTIPO FETAL? 1 2 1 2 1 Laura Igarzábal , Horacio Aiello , Florencia Petracchi , Lucas Otaño , Enrique Gadow 1. Sección Genética, Dpto. Ginecología y Obstetricia, CEMIC. 2. Servicio de Obstetricia, Unidad de Medicina Fetal, Hospital Italiano de Buenos Aires. Introducción: El test de “ADN fetal libre en sangre materna” (NIPT), introducido recientemente, está limitado a la detección de las anomalías de cromosomas (AC) más frecuentes, a diferencia del cariotipo habitualmente obtenido en análisis en vellosidades coriónicas (VC) o en líquido amniótico (LA). Objetivo: Determinar la proporción de AC en una gran cohorte de cariotipos en VC y en LA para estimar el posible riesgo residual de AC de repercusión clínica luego de un NIPT negativo. Material y Métodos: Evaluación retrospectiva de cariotipos prenatales en VC y LA, realizados en embarazos únicos en 3 centros de Buenos Aires (Grupo de Genética Médica, CEMIC, Hospital Italiano de Buenos Aires), entre 1995-2013. Se clasificaron las indicaciones de estudio en: edad materna avanzada EMA); sin indicación er er médica (SIM); hallazgos ecográficos o “screening” positivo en 1 trimestre (1 Trimestre); hallazgos ecográficos do 2do o “screening” positivo en 2 trimestre ( Trimestre); padres con AC estructurales (AC parental); y Otros. Las AC se dividieron en probablemente detectables por NIPT (trisomías 21, 18, 13 y AC sexuales) y en AC no detectables (otras trisomías, anomalías estructurales, cromosomas marcadores, triploidías). Dentro de la AC no detectables se identificaron aquellas con repercusión clínica. A los efectos del la presente comunicación se excluyeron del análisis los mosaicismos. Se estimó el riesgo residual de AC no detectadas luego de NIPT negativo en el total de la población y en cada categoría de indicación del estudio. Resultados: Se evaluaron 17.253 cariotipos, 16.064 de VC y 1.189 de LA. Excluyendo los mosaicismos (n=109), se diagnosticaron 1082 AC (6,3%), de las cuales el 19,5 % (212/1082) no serían detectadas por NIDT, siendo 7,5 % (82/1082) de repercusión clínica. De acuerdo a la indicación del estudio, la tasa de AC con er repercusión clínica no detectables fueron: 3,2% (11/316) en “EMA”; 8,1% (49/607) en “1 Trimestre”; 20% do (14/70) “2 Trimestre”; 9,8% (5/51) en “AC parental”; 6,3% (1/16) en “Otros”, y ninguna en “SIM”. Conclusión: En un escenario real de diagnóstico prenatal, la aplicación de NIPT en lugar de cariotipo fetal perdería por lo menos 1 de cada 12 AC con repercusión clínica, variando de acuerdo a la indicación del estudio. A esto habría que sumarle los interrogantes en cuanto a la capacidad diagnóstica del NIPT en mosaicismos, cromosomas marcadores, falsos negativos de anomalías detectables (T18, T13 y triplodía), etc. O-03. GEMELAR MONOCORIAL: LA DISCORDANCIA DE LA INSERCIÓN DE LOS CORDONES SE ASOCIA CON DISCORDANCIA EN LOS TERRITORIOS PLACENTARIOS Olivia Cambiaso, Lucía Vázquez, Ignacio Abasolo, César H Meller, Fabián Albornoz, Horacio A Aiello, Lucas Otaño Servicio de Obstetricia, Unidad de Medicina Materno Fetal, Hospital Italiano de Buenos Aires Introducción: La distribución desigual de la placenta (DDP) es el mayor determinante del crecimiento discordante en los embarazos gemelares monocoriales (MC). Se ha descripto la asociación entre la inserción anormal de al menos uno de los cordones umbilicales con la DDP y/o retardo de crecimiento selectivo. Sin embargo, existe poca información en relación a la concordancia o discordancia (simetría o asimetría) entre ambos cordones umbilicales. Objetivo: Evaluar la asociación entre la combinación de las inserciones de los cordones (IC) con la DDP en una cohorte de placentas MC. Materiales y Métodos: Analizamos todas las placentas monocoriales teñidas en el Hospital Italiano de Buenos Aires entre 2010 y 2014. La IC fue registrada en cada gemelo como normal o anormal (marginal o velamentosa). Las placentas fueron categorizadas como con IC concordantes (o simétricas) cuando las dos inserciones eran normales o anormales y discordantes (o asimétricas) cuando una era normal y la otra anormal. El porcentaje de los territorios placentarios individuales fue calculado por un análisis computarizado. La DDP fue definida como una discordancia en la distribución placentaria≥30%. Se analizó la asociación entre la combinación de las IC con la DDP. Resultados: Se analizaron 49 placentas. En el 43% (21/49) se observó una combinación de IC concordante y en el 57% (28/49) una discordante. La tasa de DDP fue del 29% (14/49) en la muestra analizada, en las placentas con IC concordantes fue del 10% mientras que en las con IC discordantes del 43% (p=0.01). OR: 7,1 (IC95%: 1.4-36.6). Conclusión: En la placenta monocorial, las IC discordantes o asimétricas (una inserción normal y otra anormal) se asocian con una distribución desigual de la placenta, en tanto las IC concordantes o simétricas (ambas inserciones normales o ambas anormales) se asocian con territorios placentarios más balanceados. En consecuencia, la observación de una IC anormal sería un factor de riesgo mayor para distribución desigual placentaria y posiblemente para discrepancia de tamaños fetales, cuando la otra IC es normal (OR 7,1). La evaluación de la concordancia de IC podría ser un marcador ecográfico útil de distribución placentaria, tanto en primero como en segundo trimestre, que deberá ser evaluado en series prospectivas. O-04. INTEGRACIÓN DEL SCREENING DE DISFUNCIÓN PLACENTARIA DEL PRIMER Y SEGUNDO TRIMESTRE: RESULTADOS DE UNA COHORTE DE 506 PACIENTES. Etchegaray A, Moren JM, Ciammella RM, Esteban MG, Beruti, E. Unidad de Medicina Fetal, Hospital Universitario Austral Introducción:Las consecuencias de la disfunción placentaria -manifestadas como preeclampsia (PE) y/o restricción de crecimiento (RCIU)- son causas importantes de morbimortalidad maternofetal. En las últimas décadas se han propuesto dos estrategias de cribado o screening con la intención de identificar precozmente a las pacientes con alto riesgo de estas complicaciones. En los años 80 se describió el cribado con doppler uterino en el segundo trimestre (18-24 semanas, S2). Más recientemente se ha propuesto realizar un cribado combinado en el primer trimestre (S1), utilizando características maternas, tensión arterial media, doppler uterino y PAPP-A. Distintos grupos han publicado resultados de las dos modalidades por separado, pero son pocos los trabajos que informan los resultados de integrar ambos cribados. Material y métodos:Se realizó S1 en 952 pacientes entre las 11 y 13 semanas y se lasrecitó para el cribado segundo trimestre. Once pacientes tuvieron un aborto espontáneo y 295 pacientes que no asistieron al segundo cribado. Entre las 18 y 24 semanas se reevaluaron 646 pacientes con doppler uterino. Se excluyeron del análisis 140 pacientes por presentar malformaciones o aneuploidias fetales (29) o no contar con datos perinatales completos (111). El criterio para screening positivo en el primer trimestre (S1+) fue un riesgo ajustado de PE precoz >2% y en el segundo trimestre (S2+),un IP uterino medio mayor al percentilo 95. Resultados:Se establecieron 4 grupos de pacientes de acuerdo a los resultados de los screenings: Grupo 1 (G1): S1+ y S2+, Grupo 2 (G2): S1+ y S2-, Grupo 3 (G3): S1- y S2+; Grupo 4 (G4): S1- y S2-. La sensibilidad y tasa de falsos positivos (TFP) paraS1y S2fueron78,6% para 10% y 85,7% para 10,6%, respectivamente. La sensibilidad combinada deS1 y S2 fue 64,3% para una TFP del 3,6%. Las tasas de PE, y PEG (bajo peso al nacer) fueron: GI: 24%, 24%, G2: 6,1%, 9,1%, G3: 8,6%, 14,3% y G4: 0%, 3,9%. Conclusiones: La combinación de ambos cribados no aumenta la sensibilidad pero reduce significativamente la tasa de falsos positivos. Como era de esperarse, las pacientes con S1 y S2 positivos fueron las de mayor riesgo de PE y RCIU, mientras que ninguna de las que tuvieron S1 y S2 negativos desarrollaron posteriormente PE. Un doppler uterino anormal en el segundo trimestre, es el principal marcador de riesgo de PE y RCIU, aun en pacientes que hayan tenido un cribado negativo anterior. Esto , junto con la reducción de la TFP, justifica repetir el doppler entre las 18-22 semanas independientemente del resultado del cribado del 1er trimestre y establecer un seguimiento personalizadoque incluyabiometrías fetales seriadas y control de la TA materna en aquellas pacientes con índices anormales. O-05. EXPERIENCIA DE CENTRO INDIVIDUAL EN EL USO DE NIPT PARA EL SCREENING DE ANEUPLOIDÍAS. Jorge Hamer, Andrea Quinteiro Retamar, Patricia Kaminker, Sergio Papier, Gabriel Fiszbajn, Cristian Alvarez Sedó, Claudio Chillik CEGYR (Centro de Estudios en Genética y Reproducción), CABA Introducción/objetivos: La tecnología de secuenciación y el análisis de SNPs a partir del ADN en sangre materna, ha permitido desarrollar el screening prenatal no invasivo (NIPT), con el fin de detectar aneuploidías de forma temprana (13,18, 21, X e Y). El propósito de nuestro estudio fue realizar una revisión de nuestra experiencia con el uso de NIPT para el screening de aneuploidías. Materiales y Métodos: Se reclutaron 113 pacientes que realizaron NIPT (agosto 2013 a marzo 2014). El NIPT fue realizado en un único laboratorio. Se analizaron los datos demográficos maternos, resultados del NIPT, así como las variables que afectan la fracción fetal (FF). El análisis estadístico: Chi-cuadrado, t-test y de correlación. Resultados: Datos demográficos maternos (promedio): Edad materna 37.6 (28-45), edad gestacional al momento del test 11.1 semanas (9-18,6), peso 58,8Kg (42,5-85), talla 1,6m (1,53-1,80), BMI 21,9kg/m2 (1730,1). El promedio de FF fue de 10. No se obtuvo resultado del estudio en 5 pacientes (4.4%), de los cuales el 60% obtuvo resultado en una segunda muestra. De las 111 pacientes donde finalmente se obtuvo resultado, el 49.5% correspondieron a sexo XX y 50.5% XY. El 97.3% presento un bajo riesgo para las anomalías cromosómicas. En tres casos (2,7%) se identificó un alto riesgo para trisomía 21; en estas pacientes, dos tenían indicación de NIPT por edad avanzada (>36 años), y la otra por aumento de riesgo detectado mediante ecografía (31 años). En uno de los casos de alto riesgo para T21 por NIPT, se procedió a realizar una punción de vellosidades, encontrándose un cariotipo T21 confirmatorio. La FF presentó una correlación(r) negativa con la edad materna (-0.2), talla (-0.2), peso (-0.3), BMI (-0.23). En las pacientes con FF<4, el 100% tuvo que realizar un segundo estudio debido a un NO resultado del primero, y de las pacientes con una FF>4.1, sólo el 1.8% tuvo que realizar un segundo estudio debido a un NO resultado (p<0.05). Las pacientes con FF>4.1 presentaron un BMI significativamente menor en relación a FF<4 (p<0.05). En la novena semana de gestación, el promedio de FF fue 10.1, siendo no significativamente diferente a las FF de décima (11.2) o >11 (9.9) semanas. En semana nueve ningún necesito de repetición de muestra. Discusión/Conclusiones: Nuestra experiencia evidencia que la obtención de un resultado positivo de NIPT es alta en la primera muestra, y el NO resultado se ve incrementado por una FF<4. La FF se ve influenciada negativamente por el peso, edad y el BMI. El estudio temprano (novena semana) no incrementa la posibilidad de un NO resultado. Estos datos son concordantes con la literatura respecto a este tema. O-06. EXPERIENCIA CON BIOPSIA DE BLASTOCISTO EN EL PROGRAMA DE PGD DE FECUNDITAS. María E. Ducatelli, Andressa G. Mondadori, Fabián Coco, Judith Mincman, Sebastián Neuspiller, Belén Irigoyen, Fernando L. Gismondi, Nicolás Neuspiller, Roberto Coco Fecunditas - Instituto de Medicina Reproductiva -afiliado a la UBA, CABA Introducción: La biopsia del blastocisto surge gracias a los adelantos logrados en el desarrollo in vitro de los embriones, a los avances en las metodologías de estudio genético, a la eficacia de la vitrificación y a los mejores resultados de la transferencia diferida. Desde el 2011 decidimos cambiar el día de la biopsia, del día 3 al quinto para tener la seguridad del máximo desarrollo de los embriones in vitro que significa la realización del estudio de los potencialmente transferibles. Objetivo: Comunicar la experiencia lograda del nuevo programa PGD de Fecunditas Pacientes y Métodos: Treinta y dos parejas por riesgo para enfermedades monogénicas (15 recesivas, 9 dominantes, 5 ligadas al X, 3 para tipificado de HLA y riesgo genético), 15 por rearreglos balanceados (10 traslocaciones recíprocas, 2 Robertsonianas y 3 inversiones), 5 por edad materna avanzada y 7 por otros motivos (2 para sexado social, 2 por inmunización RhD, 2 para tipificado de HLA y 1 para paternidad de embriones congelados) Todas realizaron ICSI por protocolo del programa. Los ovocitos fecundados fueron cultivados hasta blastocisto y el trofoblasto protruido fue removido y estudiado genéticamente (Minisecuenciación, ligamiento y aCGH para cariotipo molecular). Luego de la biopsia se vitrificaron y se transfirieron de a uno en ciclo diferido. Resultados: De las 59 parejas, 9 no lograron embriones sin afección, 10 no fueron aun transferidas y de las 40 transferidas, 21 lograron el embarazo (52,5%). En el grupo de las génicas, 5 no lograron embriones transferibles, 6 aun no fueron transferidas y de las 21 transferidas, 12 lograron el embarazo (57,1%). En el grupo de los rearreglos equilibrados, 15 parejas lograron embriones transferibles, 3 de ellas aun no fueron transferidas y de las 12 transferidas 4 lograron el embarazo (33,3%). En el grupo de edad materna avanzada, de las 5 parejas, 2 no lograron embriones transferibles y 2 de las 3 transferidas lograron el embarazo (66,6%). Siete parejas del grupo otros motivos, 2 no tuvieron embriones transferibles, 1 aun no se transfirió y de las 4 transferidas, 3 lograron el embarazo (75%). Discusión: Los resultados son mejores que los comunicados en el PGD Consortium de ESHRE, probablemente debido a la mejor sincronización embrio-endometrial en un ciclo natural o preparado fisiológicamente. Su menor costo, el resultado más robusto y la organización de los estudios, lo convierten en la mejor opción para los programas de PGD. O-07. PREDICCIÓN DE HIPOPLASIA PULMONAR LETAL: PERFORMANCE DE DISTINTOS MÉTODOS EN UNA COHORTE DE 23 FETOS CON HERNIA DIAFRAGMÁTICA CONGÉNITA. Etchegaray A, Russo RD, Esteban M, Keller R, Musante G Unidad de Medicina Fetal, Hospital Universitario Austral Introducción: Los avances en el diagnóstico prenatal y la institución de protocolos estandarizados de atención perinatal han conducido a una mayor supervivencia en pacientes con hernia diafragmática (HDC). La oclusión traqueal fetoscópica parece una alternativa promisoria para los pacientes con hipoplasia pulmonar (HP) severa en los que la mortalidad es todavía muy alta, pero es indispensable contar con marcadores prenatales que permitan seleccionar adecuadamente a los candidatos. Se presentan a continuación los resultados de los distintos métodos en una cohorte prospectiva prenatal. Material y métodos:Entre mayo de 2009 y enero de 2014 se realizó el seguimiento prenatal de 23 casos de HDC, 87% del lado izquierdo y 13% del derecho. Todos los casos tuvieron cariotipo normal. En 3 casos (13%) se identificaron otras malformaciones asociadas. Catorce de los 23 casos (61%) nacieron en nuestro hospital. Siete casos (50%) requirieron soporte con ECMO. La sobrevida global fue 50% (7/14) y 64% entre las HDC aisladas. En todos los casos se midió prenatalmente mediante ecografía el índice pulmón cabeza (LHR) y el índice pulmón cabeza observado/esperado (LHRO/E) mediante el método de trazado según lo publicado por Metkus y Jani respectivamente. Se calculó además el índice cuantitativo pulmonar (QLI) según Quintero. En 13 casos se midió además el volumen pulmonar total (VPT) con RMNfetal de acuerdo a la fórmula de Rypens. Resultados: De los cuatro marcadores evaluados, sólo LHR y LHRO/E se correlacionaron significativamente con muerte neonatal (Pearson -0,496, p0,025 y -0,652, p0,001). La herniación hepática y el lado de la lesión no se asociaron significativamente con muerte neonatal (p >0,05). El área bajo la curva de ROC fue0,77, 0,92,0,76 y 0,80 para LHR, LHRO/E, QLI y VPT, respectivamente. Las sensibilidad y especificidad de los distintos marcadores fueron 33,3% y 87,5%para LHR;83,3% y 87,5% para LHRO/E;90,9%y 42,9% para QLI; 62,5% y100%para VPT, respectivamente. Conclusiones:En nuestra poblaciónel mejor marcador para predicción de HP letal fue LHRO/E.QLI fue el marcador con mayor sensibilidad y VPT tuvo la más alta especificidad. La medición de estos marcadores permite asesorar a los padres acerca del pronóstico y puede ser útil en la selección de candidatos para tratamiento fetoscópico prenatal. O-08. NUEVAS TABLAS DE LA BIOMETRÍA FETAL EN POBLACIÓN DE BUENOS AIRES Y COMPARACIÓN CON LAS USADAS ACTUALMENTE EN NUESTRO PAÍS. 1 1 1 1 2 Miguel Ángel Aguirre , Marisa Márquez , Isabel Canosa , Sofía Juarez Peñalba , María de Luján Calcagno ; 2 María Inés Sarchi . 1 Centro Nacional de Genética Médica “Dr. Eduardo Castilla”, ANLIS “Dr. Carlos G. Malbran”. 2 Cátedra de Matemática. Facultad de Farmacia y Bioquímica. UBA. Introducción y objetivos: Las tablas de medidas ecográficas fetales que se usan en nuestro país fueron elaboradas por distintos autores entre 1984 y 1993 en poblaciones de Europa y EE.UU. Existen discrepancias de entre los valores de referencia para cada edad gestacional y en su confección no se usaron criterios metodológicos uniformes. En la última década varios países iniciaron un proceso de revisión y elaboración de tablas propias. Se busca contar con valores de referencia adecuados para cada población y con un tratamiento estadístico más actualizado. Los objetivos del trabajo son: 1) Confeccionar nuevas tablas de la biometría fetal en población propia, y 2) Compararlas con las más usadas en los estudios de rutina en nuestro país. Material y método: Se utilizó un diseño prospectivo de corte transversal. Se seleccionaron 1.440 embarazos únicos en mujeres de la ciudad de Buenos Aires y del Conurbano que concurrieron a la realización de ecografías de rutina entre las 11 y 40 semanas en los años 2009 a 2012. Se excluyeron los casos de fetos malformados, madres diabéticas o fumadores y embarazos de alto riesgo para defectos congénitos. La edad gestacional se estableció por una FUM cierta y siempre que esta fuera coincidente con una ecografía de antes de las 11 semanas. Se tomaron medidas de 12 segmentos corporales fetales y cada embarazo fue tenido en cuenta solo una vez. La curva se ajustó en función de las semanas exactas de gestación, sin aproximación a semana entera, mediante polinomios fraccionales. Se estimaron los valores medios y se modelaron las varianzas para el cálculo de los centilos. En esta presentación se eligieron 4 segmentos fetales: diámetro biparietal (DBP), circunferencia cefálica (CC), circunferencia abdominal (CA) y longitud femoral (LF) y se compararon con tablas de Hadlock, Jeanty, Chitty y de autores sudamericanos. Resultados: Se obtuvieron tablas de la biometría fetal en población de la ciudad de Buenos Aires y el Conurbano por primera vez. Estas mostraron algunas diferencias con las usadas de habitualmente que no son sistemáticas sino dependientes de la edad gestacional, el segmento corporal y el autor. Las medidas de la LF resultaron ser las más coincidentes con las publicadas por Hadlock y otros autores. Las mayores diferencias se concentraron en el DBP y la CA. En nuestra opinión estas diferencias se deberían más a razones metodológicas que a variaciones étnicas. O-09. EVOLUCIÓN DE NIÑOS CON CARDIOPATÍA CONGÉNITA (CC) EN UN HOSPITAL DE REFERENCIA Silvia Andres, Gabriela Bauer, Diego Michelli, Guillermina Soraiz, María Martínez Cacéres, Diana Fariña, Claudia Cannizaro Hospital J.P.Garrahan, CABA Introducción: La incidencia de CC es de 5-8/1000 RN vivos y cerca del 70 % requieren corrección quirúrgica durante el primer año de vida. El Hospital Garrahan es un centro de derivación Nacional de Cardiopatías complejas. En Argentina son la tercera causa de muerte en el período neonatal y la segunda en el post neonatal. La mayoría de las CC se presentan en fetos sin antecedentes o factores de riesgo conocidos de ahí la relevancia del diagnóstico prenatal. Objetivo: describir las características y evolución a los dos años de seguimiento de una cohorte de niños con CC Material y Métodos: estudio descriptivo, observacional y analítico. Población: cohorte prospectiva de todos los pacientes con diagnóstico de CC compleja operados en etapa neonatal y egresados del Servicio de Neonatología y/o Unidad Cardiovascular que son seguidos en el Consultorio de Alto Riesgo del Hospital J.P Garrahan. Variables biológicas y demográficas. Medidas de resultado: diagnóstico prenatal, comorbilidad y crecimiento. Resultados: n: 99 pacientes ( 64 Masc-35 Fem). Características Neonatales (Medianas y Rangos): Diagnósticos más frecuentes: 25% TGV- 14% Co Ao- 12% ATRV- 9% Ventrículo Unico- 6% Tronco Arterioso y 33% otras Cardiopatías. Peso Nacimiento 3.580g (1900-4460), edad gestacional 39 sem (34-41), 60% nacieron por parto vaginal y el 37% eran nulíparas, diagnóstico prenatal 9% el 56% con diagnostico de Hipoplasia Ventricular, edad materna 21 años (15-41), días de vida al ingreso 12 (1-40), 84% requirió cirugía: 67% Correctora- 18% Paliativa- 8% Hemodinamia- 7% no se operó, tiempo de internación 19 días (5-193), ARM 6 días (1-60). Crecimiento Score Z Ẋ±DS ( ) Peso: al nacimiento -0,29 (±1,74), al egreso -2,07 (±1,02), 6m-0,47 (±1,49), 12m -0,40 (±1,33). Talla: al nacimiento -0,89 (±0,60), al egreso -1,30 (±1,10), 6m -0,89 (±0,55), 12m 0,5 (±1,5). Perímetro Cefálico: al nacimiento -0,5 (±0,96),al egreso -0,5 (±1,36), 6m -0,5 (±1,41), 12m -1,0 (±1,16). Comorbilidades en el 1er año: 10 internaciones por cirugía cardiovascular, 15 por infecciones respiratorias y 5 por otras causas. Conclusión: Alerta el bajo porcentaje de diagnóstico prenatal. La comorbilidad de este grupo es alta. No obstante el alto impacto en el crecimiento posnatal inmediato va siendo compensado durante el seguimiento en la mayoría de los niños. RESUMENES DE POSTERS P-1. PREDICCIÓN PRENATAL DE COARTACIÓN DE AORTA Sofía Grinenco, César Meller, Mercedes Saenz Tejeira, Horacio Aiello, Lucas Otaño, Pablo Marantz. Hospital Italiano de Buenos Aires, CABA. Introducción: El diagnóstico prenatal de coartación de aorta, con una alta tasa de falsos negativos y de falsos positivos, representa en la actualidad un verdadero desafío. El objetivo de nuestro trabajo es describir la experiencia en nuestro grupo en detección prenatal de coartación de aorta. Material y métodos: Se incluyeron pacientes con sospecha prenatal y/o diagnóstico postnatal de coartación de aorta nacidos en el Hospital Italiano de Buenos Aires entre marzo 2011 y marzo 2014. Se sospechó coartación de aorta cuando se detectaron al menos 3 de los siguientes hallazgos en el ecocardiograma fetal: dilatación del ventrículo derecho ≥ 30%, dilatación de la arteria pulmonar ≥ 30%, relación istmo -ductal <0.74, presencia de muesca posterior, alteración del flujo en el arco aórtico. Resultados: Se sospechó coartación de aorta en 12 fetos, con una mediana de edad gestacional al diagnóstico de 30.3 (r: 32 a 36) semanas. Tras el nacimiento 8/12 (67%) pacientes presentaron coartación de aorta severa requiriendo cirugía cardíaca neonatal, en tanto que 1/12 (8%) presentó hipoplasia leve del arco transverso sin requerir tratamiento, y 3/12 (25%) presentaron arco aórtico normal (seguimiento postnatal de 3 a 12 meses). Al comparar los pacientes que requirieron cirugía de coartación con quienes no la requirieron no se detectaron diferencias significativas en relación a la presencia de los signos ecocardiográficos fetales referidos. Se constató comunicación interventricular en 4/8 pacientes con coartación de aorta y en el paciente con leve hipoplasia del arco transverso. Por otra parte, tres pacientes sin sospecha prenatal, con ecocardiograma fetal dentro de parámetros de normalidad en el segundo trimestre, presentaron coartación de aorta severa tras el nacimiento. Discusión/Conclusiones: En esta serie 8/11 (73%) casos de coartación de aorta fueron detectados prenatalmente, con un valor predictivo positivo de 67%. Nuevos progresos en investigación son necesarios para mejorar la sensibilidad y especificidad del ecocardiograma fetal en la detección prenatal de coartación de aorta. P-2. EMBARAZO GEMELAR BICORIAL CON MOLA COMPLETA Y FETO VIVO Fabián Albornoz, Maximiliano Alvarez, Justo González Lowy, Lucía Vazquez, Olivia Cambiaso, César H Meller, Horacio A Aiello, Lucas Otaño Servicio de Obstetricia, Unidad de Medicina Fetal, Hospital Italiano de Buenos Aires Introducción: La existencia de un embarazo gemelar que presenta una mola hidatiforme completa (MHC) y un feto vivo es un evento poco frecuente, que se asocia a una alta morbilidad materna y morbi-mortalidad fetal. El diagnóstico prenatal plantea dificultades y la conducta obstétrica continua siendo compleja y controvertida. Objetivo: Describir los resultados de una serie de casos de embarazos gemelares con coexistencia de una MHC y un feto vivo. Materiales y métodos: Análisis retrospectivo de los casos evaluados en la Unidad de Medicina Fetal del Hospital Italiano de Buenos Aires con diagnostico de embarazo gemelar bicorial doble con una MHC y un feto vivo, entre los años 2003 y 2013. Resultados: Se evaluaron 7 casos. La mediana de edad gestacional (EG) al diagnóstico fue de 14,4 semanas (r: 12-27). Los valores de β-hCG se encontraban elevados en todos los casos (mediana: 294.280 mUI/ml, r: 180.000–2.000.000). 3/6 pacientes presentaron hipertiroidismo, 1/7 quistes tecaluteínicos bilaterales, 2/7 metástasis pulmonares y ninguna evidenció invasión miometrial ni metástasis abdominales. En una paciente se decidió tratamiento inmediato de la mola con la consiguiente interrupción del embarazo con evolución favorable. En el resto se realizó conducta expectante. De estas 6 pacientes, 5 presentaron metrorragia, requiriendo 2 de ellas la finalización del embarazo por compromiso de la salud materna a las 20 y 14 semanas. Una paciente presento una preeclampsia severa que requirió finalización del embarazo a las 30 semanas. Las 3 pacientes restantes presentaron buena evolución con finalización alrededor de las 36 semanas. Dos pacientes requirieron quimioterapia por enfermedad trofoblástica persistente. Conclusión: La presente serie mostro una alta tasa de resultados adversos consistente con otras series publicadas. No obstante, en la mitad de los casos se observo un buen resultado perinatal. Es de notar, que las pacientes que presentaron un perfil hipertiroideo correspondieron a las que tuvieron un resultado adverso tanto perinatal como materno. Dado que la ecografía de las 12 semanas podría detectar todos estos los casos, sería importante encontrar marcadores pronósticos que ayuden al asesoramiento temprano. P-3. TINCIÓN PLACENTARIA COMO PARTE DE LA EVALUACIÓN SISTEMÁTICA DEL EMBARAZO MONOCORIAL César H Meller, Ignacio Abasolo, Olivia Cambiaso, Lucía Vázquez, Fabián Albornoz, Horacio A Aiello, Lucas Otaño. Servicio de Obstetricia, Unidad de Medicina Fetal, Hospital Italiano de Buenos Aires. Introducción: La angioarquitectura placentaria de los embarazos monocoriales permite explicar la mayoría de las complicaciones propias de este tipo de embarazos. La estandarización de la tinción placentaria es una herramienta esencial para la evaluación de la placenta monocorial. Objetivo: Describir la metodología de tinción y análisis de las placentas MC que se realiza en nuestra Unidad. Material y Métodos: Se evaluaron las placentas de los embarazos MC seguidos en el Programa de Embarazo Gemelar de la Unidad de Medicina Fetal del Hospital Italiano de Buenos Aires. La técnica consiste en: alumbramiento cuidadoso para no lesionar los cordones umbilicales y asegurar la integridad placentaria con identificación mediante un reparo del cordón del feto 1. Almacenamiento de la placenta en una bolsa plástica en heladera, sin ningún conservante. Inspección macroscópica y descripción del número de vasos y el tipo de inserción de cada cordón. Sección del cordón a 5 cm de su inserción. Lavado la placenta con agua, separación del amnios de la placa coriónica y remoción de la sangre de los vasos superficiales por expresión hacia el cordón. Canalización de las venas y arterias de ambos cordones, con cateter de 2.5 F, e inyección de colorante: azul para las arterias y rojo para la vena del feto 1, y verde en las arterias y amarillo en la vena del feto 2. Fotografía de la placenta con el cordón del feto a la izquierda y cinta métrica en el margen como referencia. Registro del tipo y número de anastomosis. Análisis de la territorialidad placentaria correspondiente a cada feto con un programa de computación. Finalmente, análisis anatomopatológico. Resultados: De 54 placentas disponibles, se pudo aplicar el protocolo en 49 (90%), siendo causas de la falla del mismo ruptura de los cordones y/o de la placenta durante el alumbramiento (3 casos) y errores en su almacenamiento (2 casos), por ejemplo, conservación en formol. Comentarios: El análisis sistemático de la angioarquitectura placentaria de embarazos gemelares MC es técnicamente factible en la mayoría de los casos. Por otra parte, se recomienda este tipo de evaluación como control de calidad de la ablación vascular laser por fetoscopia. P-5. CORRELACIÓN ECOCARDIOGRÁFICA FETAL Y POSTNATAL EN CARDIOPATÍAS CONGÉNITAS Sofía Grinenco, César Meller, Mercedes Saenz Tejeira, Horacio Aiello, Lucas Otaño, Pablo Marantz. Hospital Italiano de Buenos Aires, CABA. Introducción: En unidades especializadas en Medicina Fetal, el diagnóstico prenatal de cardiopatías congénitas (CC) alcanza hasta un 85-95 %. A su vez, identificar las CC con menor tasa de detección y/o mayor tasa de falsos positivos podría ayudar a enfocar aquellas áreas que requieren mayor atención e investigación. El objetivo de nuestro trabajo es evaluar la correlación entre el diagnóstico ecocardiográfico fetal y postnatal en los pacientes referidos a nuestra Unidad. Material y métodos: Estudio descriptivo retrospectivo. Se correlacionaron los hallazgos de la ecocardiografía fetal con los diagnósticos postnatales en fetos con diagnóstico prenatal de CC seguidos en la Unidad de Medicina Fetal del Hospital Italiano de Buenos Aires en el período 2010-marzo 2014. Resultados: De un total de 1060 pacientes referidas para ecocardiografía fetal se diagnosticaron 180 CC, de los cuales 154/180 (85.6%) casos tienen seguimiento postnatal. En 139/154 (90.3%) pacientes existió concordancia entre el diagnóstico pre y posnatal. En 11/154 (7.1%) la ecocardiografía posnatal modificó el diagnóstico de la CC. Y 4/154 (2.6%) presentaron ecocardiograma postnatal normal (falsos positivos), siendo el diagnóstico prenatal más frecuente el de sospecha de coartación de aorta (n=3). En el período estudiado nacieron 3 pacientes con ecocardiograma fetal normal que desarrollaron coartación de aorta (falsos negativos). Conclusiones: En nuestra serie existió una correlación diagnóstica en la mayoría (90%) de los casos. En 1 de cada 15 casos, la evaluación posnatal modificó el diagnóstico original prenatal. La detección prenatal de coartación de aorta sigue representando el mayor desafío diagnóstico. La revisión sistemática de los hallazgos ecocardiográficos pre y postnatales constituye una herramienta de control de calidad del diagnóstico prenatal de CC y una oportunidad de mejora para las unidades de medicina fetal. P-6. EVALUACIÓN DE LA CISTERNA MAGNA, TRANSLUCENCIA INTRACRANIAL Y TRONCO DEL ENCÉFALO EN LA ECOGRAFÍA DE 11 – 13,6 SEMANAS COMO MARCADORES ECOGRÁFICOS DE ESPINA BÍFIDA. Igarzábal María Laura; Micchia Celeste; Randazzo Marianela; Matt Cristina; Calvo Diego; Krupitzki Hugo; Petracchi Florencia. CEMIC. Centro de Educación Médica e Investigaciones Clínicas. Instituto Universitario. CABA. Introducción: La espina bífida (EB) abierta se asocia con desplazamiento caudal del cerebro, manifestación que estaría presente desde el primer trimestre del embarazo. Este evento produciría cambios secundarios en la anatomía del cerebro posterior que podrían evidenciarse en la ecografía del primer trimestre. Se ha propuesto la evaluación de los diámetros del tronco del encéfalo (TE) y fosa posterior, en el mismo corte que el utilizado para la medición de la translucencia nucal (TN), como “marcadores” tempranos de EB. Objetivo: Establecer el rango normal de medidas del diámetro AP de TE, TI, CM y distancia tronco del encéfalo-hueso occipital (TEHO), en un corte medio sagital del perfil fetal entre las 11-13,6 semanas en fetos sin EB. Secundariamente, evaluar si existen variaciones en estas estructuras en fetos con EB de esta cohorte de pacientes. Material y Métodos: Entre noviembre de 2009 y septiembre de 2013 se evaluaron en forma prospectiva 3.405 fetos por ecografía transabdominal como parte del “screening” de anomalías de cromosomas. En el mismo corte medio sagital del perfil fetal utilizado para medir la TN, se evaluaron los diámetros AP de TE, TI, CM, y TEHO. Se calculó la relación TEHO/TE. Se realizó regresión logística para determinar el significado de la asociación entre el diámetro AP de TE, TI, CM, y la longitud céfalo-caudal (LCC). Se registró el diagnóstico de EB al nacer, o por ecografía del segundo trimestre. Resultados: De un total de3.405 fetos se logró una imagen satisfactoria de la fosa posterior en 3.337 fetos (98%). Los diámetros AP de TE, TI y CM aumentaron linealmente en forma significativa con la edad gestacional (p<0,001). En 3093 fetos sin EB se registró la TI siempre presente, la relación TEHO/TE fue mayor a 1, y la imagen fue clasificada como “normal” en 3.073 de ellos. Se diagnosticaron 3 fetos con EB. Conclusión: En la ecografía de 11-13,6 semanas las estructuras del cerebro posterior pudieron ser evaluadas en la mayoría de los fetos. En fetos sin EB existe un incremento significativo con la edad gestacional en los diámetros AP evaluados. En este grupo de fetos no afectados son muy poco frecuentes las imágenes consideradas anormales. La observación de la compresión de la CM y/o TI, el aumento del TE, una relación TEHO/TE < a 1, o una combinación de ellos, podría justificar una evaluación detallada y temprana de la columna fetal. Determinar en forma aislada la ausencia de TI no parecería ser suficiente. No es posible sacar conclusiones sobre la capacidad diagnóstica de estos marcadores debido al escaso número de fetos con EB evaluados. P-7. DUCTUS ARTERIOSO RESTRICTIVO IDIOPÁTICO: RESULTADOS PERINATALES DE UNA SERIE DE CASOS Ç +Ç +Ç Ç Ç César Meller* , Sofia Grinenco , Mercedez Tejeira Saenz , Vazquez Lucía* , Lage Fernanda* , Wojakowski =Ç +Ç Ç A , Pablo Marantz , Horacio Aiello* + *Servicio de Obstetricia, Servicio de Cardiología Infantil; Fetal. Hospital Italiano de Buenos Aires =Ç Ç Servicio de Diagnóstico por Imágenes, Unidad de Medicina Introducción: El ductus arterioso restrictivo (DAr) es un fenómeno poco frecuente, asociado a medicación o a lesiones estructurales cardíacas y, menos frecuentemente, idiopático. Una posible complicación es el cierre prematuro intrautero, falla cardíaca y muerte perinatal. El objetivo de este trabajo es presentar los resultados perinatales de una serie de casos de fetos con DAr idiopático aislado. Material y métodos: Serie de casos evaluados en la Unidad de Medicina Fetal del Hospital Italiano de Buenos Aires entre 01/2013-01/2014. Los criterios utilizados para definir DAr fueron al menos 2 de los siguientes: velocidad sistólica máxima >1.4 m/seg , velocidad diastólica máxima >0.35 m/seg, IP< 1.9. Resultados: Se registraron 6 casos de Dar, 3 (50%) en mujeres nulíparas y 3 (50%) en primíparas. La mediana de edad materna fue 35.5 (r:33-39) años y la mediana de edad gestacional (EG) al diagnóstico fue 32 (r:31-38) semanas. El motivo de derivación para realización de ecocardiograma fetal fue regurgitación tricuspidea (RT) 2/6, diabetes gestacional 1/6, disbalance ventricular 1/6, foco ecogénico 1/6 y antecedente de hijo previo con cardiopatía 1/6. Los 6 casos presentaron una velocidad sistólica > 1.4 m/seg, con una mediana de 1,7 (r:1.6-2) m/seg, 5/6 (83 %) una velocidad diastólica máxima > 0.35 m/seg, con una mediana de 0.35 (r:0.21-0.38) m/seg, y 2/6 (33 %) un IP en el DA < 1.9, con una mediana de 2 (r:1.45-2.1). Otros hallazgos fueron 4/6 (66 %) un diámetro del DA < p5, 3/6 (50 %) RT, 2/6 (33%) DA en forma de “s”, 2/6 (33 %) hipertrofia y dilatación de ventrículo derecho, y 1/6 (16,6 %) dilatación de la arteria pulmonar. No hubo casos de hidrops ni de ductus venoso alterado. El seguimiento fue inicialmente a intervalos < 1 semana, y en caso de estabilidad en al menos 2 controles, cada 7-10 días. Los 6 embarazos presentaron una mediana de edad gestacional al nacimiento de 39 (r:38-40) semanas, una media de peso de 3082 + 392 gr, y todos mostraron buena evolución postnatal, sin ingresos a neonatología y con ecocardiogramas postnatales normales al mes de vida. Comentarios: Nuestra serie presentó un pronóstico perinatal favorable en todos los casos de DAr y, si bien no existen guías para el seguimiento de esta patología y son necesarios más trabajos para poder establecer cuál es elmanejo apropiado, en caso de estabilidad parece razonable una evaluación periódica y finalización del embrazo a término. P-8: UTILIDAD DEL ESTUDIO GENÉTICO EN FETOS CON VENTRICULOMEGALIA LEVE AISLADA 1 2 2 2 Juan J. Lunghi , Carlota Rodó , Silvia Arévalo , Elena Carreras . 1- Hospital Iturraspe, Santa Fe, Argentina. 2- Hospital UniversitariValld’Hebron – UniversitatAutònoma de Barcelona, Barcelona, España. Objetivo: Determinar el grado de asociación entre ventriculomegalia leve aislada (VMLA) y alteraciones genéticas. Materiales y métodos: Estudio retrospectivo incluyendo 37 casos de VMLA, definida con ventrículos cerebrales laterales de 10-12 mm en ausencia de otras alteraciones ecográficas en las que se realizó estudio genético mediante cariotipo fetal o array CGH. Resultados: En los 37 fetos estudiados se encontró 1 caso (2,7%) de trisomía 21. Se realizó array CGH en 14 fetos, obteniendo resultado normal en todos ellos. Se constató progresión de la ventriculomegalia (aumento > 2mm) durante los controles sucesivos en 3 casos (8%), en dos de los cuales era unilateral y uno bilateral. La paciente cuyo feto era portador de trisomía 21 tenía 36 años y un cribado de primer trimestre de bajo riesgo. La ecografía morfológica no mostró ningún otro signo de trisomía 21 apartede la ventriculomegalia bilateral de 10.5 mm. Conclusión: La asociación entre VMLA y anomalías genéticas es baja. El estudio mediante array CGH no aportó mayor información. Se necesitan estudios con mayor número de casos para determinar si el array CGH puede o no tener utilidad en la valoración de fetos con VMLA, por lo que su práctica resulta prudente. P-9: HERNIA DIAFRAGMÁTICA CONGÉNITA SEVERA Y CIRUGÍA INTRA-ECMO. EXPERIENCIA INICIAL Russo D, Falke G, Puigdevall JC, Berberian L, Pernas A, Keller R, Musante G, Etchegaray A. Hospital Universitario Austral, Pilar, Buenos Aires. Introducción: La hernia diafragmática congénita (HDC) se asocia a diversos grados de hipoplasia e hipertensión pulmonar (HTP) severa en el RN. A pesar de los avances en diagnóstico prenatal y en los cuidados intensivos neonatales, la tasa de mortalidad general alcanza al 50%. Un pequeño grupo se beneficiará con la utilización de la oxigenación con membrana extracorpórea (ECMO). El momento de la cirugía continúa en constante revisión. Presentamos una experiencia inicial de 3 cirugías realizadas durante la asistencia con ECMO. Material y métodos: Entre mayo de 2009 y enero de 2014 se realizó el seguimiento prenatal de 23 casos de HDC, de los cuales 14 (61%) nacieron en nuestro hospital. Siete casos (50%) ingresaron a ECMO. La sobrevida global fue 50% (7/14). Tres pacientes con HDC e hipoplasia pulmonar severa se operaron mientras recibían asistencia con ECMO. En estos 3 casos se utilizó modalidad veno-arterial, debido a signos de claudicación cardíaca secundaria a HTP suprasistémica persistente. Resultados: En los 3 casos se trataba de un HDC posterolateral izquierda con hígado intratorácico. El paciente 1 presentó además un secuestro broncopulmonar asociado. Todos tuvieron cariotipo prenatal normal y nacieron por cesárea programada a término Paciente 1: Relación pulmón cabeza (LHR): 0.88. Relación pulmón cabeza observado/esperado (LHRO/E): 35.4%. Edad gestacional al nacer (EGN) 38.6sem. Peso 3312g (p49). Apgar: 5/7. Recibió asistencia respiratoria mecánica (ARM) convencional durante 24hs. ECMO desde el 1° al 13° día de vida. Cirugía con Split muscular y resección de secuestro pulmonar al día 7 de ECMO. Reoperación por síndrome compartimental día 9. Fallece el día 10en ECMO. Paciente 2:LHR 0,77 LHR O/E: 27,9%. EGN: 39.2 sem. 3200g (p45). Apgar: 7/8. ARM 6hs. ECMO desde las 6 hs a los18 días de edad. Cirugía al día 14. Fallece día18 sin tolerar destete de ECMO. Paciente 3:LHR 0,78, LHRO/E 23,9%. EGN: 39.3 sem 2800g (p13.5). Apgar: 4/7.ARM 24 hs, luego ECMO. Se opera al 4º día de ECMO. Fallece día 8. Sangrado pulmonar y cerebral. Discusión: La principal dificultad de la cirugía durante ECMO consiste en evitar complicaciones hemorrágicas. Se han utilizado para esto terapias antifibrinolíticas como el ácido tranexámico. En los 3 casos que informamos no existió sangrado significativo durante la cirugía pero 2 de ellos tuvieron complicaciones hemorrágicas en los siguientes días. No hubo muertes intraoperatorias. A pesar de que la cirugía fue exitosa desde el punto de vista técnico, ninguno de los pacientes pudo ser retirado de ECMO y todos fallecieron dentro de la siguiente semana. Conclusión: La reparación de la HDC intra-ECMO es factible, pero debido a la alta mortalidad postquirúrgica asociada, debería plantearse sólo en aquellos casos en los que se considere que la misma pueda colaborar al retiro del soporte. P-10. DISFUNCIÓN PLACENTARIA PRECOZ COMO POSIBLE CAUSA DE HIPOSPADIAS María Eugenia Carducci, Cecilia Páez, Olivia Cambiaso, Cecilia Botazzi, Mariana Jacubowicz, Leandro Suarez, Horacio A Aiello, Lucas Otaño. Unidad de Medicina Fetal, Hospital Italiano de Buenos Aires Introducción: El hipospadias es la anomalía de la diferenciación sexual masculina más frecuente y su etiología es heterogénea. Entre sus causas, se ha postulado la disfunción placentaria precoz, que podría interferir en los mecanismos hormonales necesarios para la adecuada diferenciación de los genitales externos masculinos. Objetivo: Describir una serie de casos de fetos con RCIU severo por disfunción placentaria e hipospadias. Material y Métodos: Análisis retrospectivo de pacientes con RCIU e hipospadias evaluados en la Unidad de Medicina Fetal del Hospital Italiano de Buenos Aires en el 2013. Resultados: Se evaluaron 4 pacientes con RCIU e hipospadias. La mediana de la edad gestacional (EG) al momento de la derivación fue de 18 semanas (r:12-25), y el motivo de la derivación fue en 2 casos RCIU y en los otros dos screening del primer trimestre positivo. La mediana de la EG al desarrollo del RCIU fue de 21 semanas (r:16-25). Se realizó cariotipo prenatal en 3/4 pacientes: 46,XY. El Doppler placentario fue anormal en los 4 casos y el fetal en 3/4. Dos pacientes desarrollaron preeclampsia y una presentó un desprendimiento placentario. La mediana de la EG al nacimiento fue de 30 semanas (r:25-39) y del peso fetal 787g (r:450-1100). Tres recién nacidos fallecieron en la etapa neonatal y el restante presenta buena evolución. Comentarios: Similar a lo reportado en la literatura, nuestros datos muestran una posible asociación entre RCIU severo secundario a disfunción placentaria precoz e hipospadias. La detección de genitales ambiguos en el contexto de un RCIU severo y precoz podría deberse a una disfunción placentaria más que a una anomalía genética fetal. Por lo tanto, en la evaluación de los RCIU severos, sería recomendable delinear con precisión los genitales externos. Es necesaria mayor información, a los efectos de establecer la naturaleza de dicha asociación. P-11. MASAS ECOGÉNICAS RELACIONADAS A SECUESTRO EXTRALOBAR INFRADIAFRAGMÁTICO: DIAGNÓSTICO PRENATAL Y EVOLUCIÓN. 1 1 2 1 Ochoa JH , Colubriale A , Defago VH , Mangupli P , Lovagnini Frutos MG 1 1. Diagnus. Centro de Diagnóstico, Docencia e Investigación. Córdoba, Argentina. 2. Hospital de Niños de la Santísima Trinidad. Córdoba, Argentina. Introducción: Las masas ecogénicas subdiafragmáticas son anomalías poco frecuentes que se caracterizan por la presencia de tejido ecogénico en contacto o inmediatamente por debajo del diafragma . El origen de estas masas está fundamentalmente relacionado con la presencia de secuestro pulmonar extralobar infradiafragmático (SPEI), pero otras lesiones también pueden mostrar imágenes similares y una diferenciación clara entre las entidades no siempre es posible de realizar, antes de nacer. Objetivo: Informar sobre la evolución de una serie de casos de masas ecogénicas subdiafragmáticas con diagnóstico presuntivo de SPEI. Material y métodos: Estudio retrospectivo en una población de embarazadas controladas ecográficamente entre los años 2002 a 2012. Resultados: Se detectaron 7 casos con diagnóstico presuntivo de SPEI con adecuado seguimiento postnatal. La edad gestacional media al diagnóstico fue de 26 semanas (22-31 sem.). Seis de 7 se localizaron en el lado izquierdo. La razón M:F fue de 1:2,5 . El tamaño promedio fue de 25 mm (16 - 47mm). Cinco fueron heterogéneos en apariencia y 2 eran homogéneos. Se pudo identificar el origen de la irrigación arterial en 5 casos, en 2 casos el vaso provenía directamente desde la aorta y en lo otras 3, de ramas de la aorta. Evolución: Dos de los casos presentaron involución prenatal y desaparecieron en el tercer trimestre, un caso tuvo regresión espontánea durante la vida postnatal, y en 4 casos la masa persistió (en 3 pacientes disminuyó el tamaño en los controles posteriores) . Los marcadores séricos de neuroblastoma fueron negativos en los casos que persistieron después del nacimiento. Sólo un caso se trató con cirugía, con una excelente evolución. El examen anatomopatológico confirmó SPEI en este caso. Conclusiones: Aunque nuestra serie es pequeña para obtener conclusiones definitivas, creemos que la evolución de los casos es compatible con una conducta expectante siempre y cuando se asegure la posibilidad de seguimiento postnatal. P-12. TRISOMÍA 16 EN DIAGNÓSTICO PRENATAL Florencia Petracchi, María L. Crespo, Marianela Randazzo, Laura Igarzabal. Sección Genética Departamento de Ginecologia y Obstetricia . Centro de Educación Médica e Investigaciones Clínicas CEMIC Buenos Aires Introducción: La trisomía 16 es la anomalía más frecuente en abortos espontáneos, y ocurre en 1% de todas las concepciones. Sin embargo, resulta poco frecuente como hallazgo en diagnóstico prenatal. Puede presentarse como línea única o mosaico y ha sido principalmente asociada a RCIU y malformaciones cardíacas, prematurez y bajo peso al nacer. En un reporte previo de nuestros datos, habíamos encontrado asociación con PAPP-A bajo en el screening para aneuploidías. Objetivo: describir las características de las pacientes con diagnóstico prenatal de trisomía 16 Materiales y métodos: Se consideraron todas las pacientes que habían realizado diagnóstico prenatal invasivo entre 1996 – 2013 Se evaluó: indicación para diagnóstico prenatal, hallazgos ecográficos, evaluación de translucencia nucal, hueso nasal, resultado de vellosidades coriónicas o amniocentesis, realización y resultado de screening bioquímico previo. Resultados: Se encontraron 10 pacientes con resultado de trisomía 16 en 16001 (0,06%) (IC 95%:0,03-0,12) estudios de diagnóstico prenatal invasivo ( vellosidades corionicas o amniocentesis). De ellas, 9 casos presentaron línea pura y un caso, un mosaico con una línea celular euploide. La indicación para realizar el estudio fue: por edad materna avanzada: 4/10 , y por screening positivo de primer trimestre : 6/10. Presentaron TN aumentada y malformaciones asociadas: 2/ 10 : un higroma quístico y una anomalía de cierre de pared abdominal. El hueso nasal estuvo presente en todas las pacientes que se evaluó: 7/7. Habían realizado screening bioquímico previo: 4/10 y todas ellas presentaron valores bajos de PAPP A, con una mediana de 0,16 MoM. Hubo un caso de free B HCG elevada y otro con taquicardia fetal persistente. Conclusiones: La trisomía 16 en diagnóstico prenatal resultó un hallazgo poco frecuente, en consistencia con la literatura. A pesar del bajo número de casos, podrían considerarse algunas conclusiones: la translucencia nucal, el hueso nasal y los niveles de free Beta HCG no presentaron variaciones características. Los niveles bajos de PAPP- A impresionan estar asociados a esta aneuploidía, con valores incluso inferiores a la mediana de otras trisomías como 13 y 18. P-13. DIAGNÓSTICO PRENATAL DE SINDROME MIELODISPLÁSICO EN EMBARAZOS CON TRISOMÍA 21 Lucía Vázquez, María Zabalza, Mariano Uzal, M Arias, Gabriela Salvatore, Mariana Bucich, Gustavo Izbizky, Lucas Otaño. Unidad de Medicina Fetal, Hospital Italiano de Buenos Aires. Introducción: Los fetos con trisomía 21 (T-21)presentan más riesgo de desarrollar sindromes mieloproliferativos. La hepatoesplenomegalia y el hidrops fetal no inmunológico, son dos de los principales signos de sospecha prenatal de este sindrome. Objetivo: Describir dos casos de diagnóstico prenatal de sindromes mielodisplásicos en fetos con T-21. Caso 1: Paciente de 37 años, derivada a las 23 semanas, que presentó un screening positivo del primer trimestre y hallazgos del segundo trimestre asociados a T-21. Ante la aparición de hidrops fetal, pico sistólico aumentado y hepatoesplenomegalia se sospechó la presencia de T-21 asociada a un sindrome mielodisplásico. La cordocentesis mostró un cariotipo 47,XX,+21, Hb 14.5 mg/dl, GB 21.000/ml (28 % de blastos), plaquetas 42.600/ml. Se finaliza el embarazo a las 37 semanas: recién nacido femenino, 2205g. El hemograma y el frotis muestran: GB 51.000/ml con 60% de formas inmaduras y plaquetas 34.900/ml, confirmando el diagnóstico prenatal. La evolución postnatal fue favorable en los días inmediatos al nacimiento. Caso 2: Paciente de 21 años, derivada con diagnóstico prenatal de T-21 en vellosidades coriónicas. A las 16 semanas se evidencia edema nucal, quistes linfangiectasicos, hidrotórax bilateral y canal AV. A las 18 semanas retrograda el derrame pleural y a las 27 semanas se observa derrame pericárdico leve, intestino ecogénico y hepatomegalia hipoecoica. A las dos semanas se agrega cardiomegalia con regurgitación tricuspídea y ascitis leve. A las 31 semanas el PS-ACM es 1.87 MoM. Por los hallazgos se sospecha un sindrome mieloproliferativo, pero a las 33 semanas se produce una RPM y se finaliza el embarazo por sospecha de corioamnionitis. Recién nacido masculino, hidrópico, de 3035g que ingresa en ARM. El laboratorio muestra GB con 53% de formas inmaduras y plaquetopenia, confirmando la presencia del sindrome mielodisplásico. La evolución fue desfavorable con muerte neonatal. Conclusiones: Ante el diagnóstico de un embarazo con T-21 y la presencia de hepatomegalia hipoecoica con o sin hidrops, se debería sospechar la presencia de un sindrome mielodisplásico fetal. La cordocentesis permitiría la confirmación prenatal. La sospecha de este diagnóstico debe advertir al equipo neonatal. P-14. LA PESQUISA DEL PRIMER TRIMESTRE PARA ANEUPLOIDÍAS: EXPERIENCIA PILOTO EN EL SECTOR PÚBLICO EN LA CIUDAD DE BUENOS AIRES 1 1 1 1 1 Mariana A. Vilas , Miguel A. Aguirre , Isabel Canosa , María Ester Móllica , Sebastian Menazzi , Analía 2 2 2 3 3 3 4 Messina , Sabrina Patti , Diego A. Crema , Mabel Poncelas , Gastón Senyk , Romina Gil , Ana María Sequera , 4 Hugo Boquete 1 Centro Nacional de Genética Médica “Dr. Eduardo Castilla”. ANLIS “Carlos G. Malbran”. 2 Servicio de Obstetricia del Hospital Teodoro Álvarez 3 Servicio de Obstetricia del Hospital Cosme Argerich. 4 Laboratorio de Endocrinología -Unidad de Endocrinología del Hospital Teodoro Álvarez Introducción: Las anomalías cromosómicas son frecuentes en el embarazo. Los métodos de pesquisa para detectar embarazos de riesgo para las trisomías 21, 18 y 13 han demostrado ser eficaces y fueron incorporados a las políticas públicas de muchos países. En Argentina estos métodos no están disponibles en la mayoría de los hospitales públicos. Por esa razón se inició en 2011 un trabajo colaborativo entre el Centro Nacional de Genética Médica (CENAGEM) y los hospitales Teodoro Álvarez y Cosme Argerich para desarrollar la PESQUISA DEL PRIMER TRIMESTRE COMBINADA (PPT) con recursos propios. El hospital Teodoro Álvarez concentró los dosajes bioquímicos de las tres instituciones y el CENAGEM ha realizado los estudios invasivos de los casos positivos. Materiales y métodos: Entre diciembre de 2011 y diciembre de 2013 se realizaron 1013 estudios de PPT a embarazadas de cualquier edad materna entre las 11 y 14 semanas de gestación. Cada estudio consistió en una ecografía detallada con medición de la translucencia nucal (TN) y los dosajes en suero materno deβHCG y PAPP-A. Todas las pacientes tuvieron una entrevista previa de asesoramiento genético para evaluación de antecedentes familiares e información respecto del estudio. Para el cálculo de riesgo se utilizó el software de la Fetal Medicine Foundation (FMF). Se tomó 1/300 como valor de corte y se ofreció diagnóstico invasivo por biopsia de vellosidades coriales y/o amniocentesis a todos los casos positivos. Resultados: De los 1013 estudios, 89 resultaron positivos (8,8%). Se realizaron 45 estudios invasivos de los cuales resultaron 11 fetos con aneuploidías: 4 con trisomía 21, 5 con trisomía 18, 1 con trisomía 13 y 1 con triploidía. La media de edad materna fue de 32 años. Según la distribución por edad materna se esperaban 5,1 casos de trisomía 21, e igual número de otras aneuploidías. Discusión/Conclusión: La pesquisa de aneuploidías ha demostrado ser una herramienta útil para diagnóstico prenatal precoz de defectos congénitos. Además, la ecografía 11-14 permite detectar malformaciones fetales y otros síndromes genéticos. Nuestro trabajo demuestra que en los hospitales públicos es posible desarrollar esta metodología porque existen los recursos humanos y técnicos y la voluntad necesaria para realizarlo. El diagnóstico prenatal de defectos congénitos es una actividad multidisciplinaria que, mediante una intervención adecuada, nos permitiría disminuir la mortalidad neonatal. P-15. HEMOGLOBINA DE BART COMO CAUSA DE HIDROPS FETAL NO INMUNE 1 1 1 1 2 1 Olivia Cambiaso , Lucía Vázquez , César H Meller , Fabián Albornoz , Mónica Aixala , Horacio A Aiello , Lucas 1 Otaño . 1. Servicio de Obstetricia, Unidad de Medicina Fetal, Hospital Italiano de Buenos Aires. 2. Laboratorio Aixala-Blanco Introducción: La Hemoglobina de Bart (Hb Bart) es la forma más severa de alfa talasemia. A pesar de que en nuestro medio es una condición rara, es la causa más frecuente de hidrops fetal no inmune en el Sudeste Asiático. Objetivo: Describir dos casos de hidrops fetal con diagnóstico prenatal de Hb Bart. Caso 1: Pareja oriunda de China, primigesta, derivada a las 17 semanas por ascitis fetal. En la evaluación ecográfica se observa: ascitis fetal, calcificaciones intestinales e intrahepáticas y placentomegalia, con Doppler fetal normal, serologías para TORCH negativas y hepatitis B positiva. Una semana después se observa aumento de la ascitis y pico sistólico de la arteria cerebral media (PS-ACM) elevado de 1.8 MoM. Se realiza cordocentesis: cariotipo 46,XY, Hb 6.9 mg/dl, HTO 29.8%, eritroblastos abundantes en el frotis y electroforesis de la Hb con presencia de una Hb de movilidad desconocida que representa el 90% del total. Ambos padres presentan anemia leve con microcitosis, electroforesis y perfil férrico normal, compatible con rasgo talasémico. El estudio molecular de ambos progenitores muestra que son heterocigotas para la deleción alfa-SEA. En su país de origen se confirma el diagnóstico y se induce el parto a las 29 semanas falleciendo el recién nacido a las pocas horas. Caso 2: Pareja oriunda de China, G2P1, con antecedente de muerte fetal a las 29 semanas con hidrops y anemia fetal, derivada en el presente embarazo a las 22 semanas por hidrops fetal. Se observa hidrops, cardiomegalia, placento y hepatomegalia y PS-ACM: 1.38 MoM. Ambos padres presentan rasgo talasémico. Ante la sospecha de hidrops por Hb Bart se realiza cordocentesis: cariotipo 46,XX, Hb 6.6 mg/dl, HTO 26.2%, electroforesis de la Hb compatible con Hb Bart que representa el 80% del total. El estudio molecular de la muestra fetal tiene un alelo positivo para la deleción alfa-SEA, mientras que el alelo normal no amplifica y se confirma que ambos progenitores son heterocigotas para la deleción. Se constata la muerte perinatal a las 27 semanas. Comentarios: Aunque excepcional en parejas de otro origen, ante la presencia de hidrops en la segunda mitad del embarazo en una pareja del Sudeste Asiático, la Hb de Bart es el primer diagnostico a considerar. En nuestro medio el diagnóstico es factible y accesible, lo cual es importante para establecer el pronóstico del embarazo y realizar un asesoramiento genético adecuado. P-16. MORBIMORTALIDAD PERINATAL ASOCIADA A MALFORMACIÓN ARTERIOVENOSA DE GALENO: ESTUDIO MULTICÉNTRICO RETROSPECTIVO. 1 1 2 2 3 2 1 Marchueta J , Mangupli P , Vázquez L , Wojakowski A , Echegaray A , Aiello H , Ochoa J . 1. 2. 3. Diagnus Hospital Italiano de Buenos Aires Hospital Universitario Austral Objetivo: valorar la morbimortalidad perinatal de la Malformación arteriovenosa (MAV) de Galeno en una cohorte. Materiales y métodos: Estudio retrospectivo de todos los casos de MAV de Galeno diagnosticados en tres centros de referencia durante un período de 7 años. Inicialmente se dividieron a las pacientes en dos grupos según si se encontraron anomalías asociadas o no. Se evaluó la edad gestacional al diagnóstico, la repercusión cardíaca y del sistema nervioso central, y la evolución neurológica postnatal. Resultados: se evaluaron diecinueve casos con diagnóstico prenatal de MAV de Galeno. Dieciocho (94,7%) casos fueron aislados y uno (5,3%) se asoció a otra anomalía independiente de la MAV de Galeno. Hubo tres (15,8%) muertes fetales intraútero, ocho (42%) muertes postnatales. De los sobrevivientes (42%), dos (25%) desarrollaron secuelas neurológicas y seis (75%) poseen seguimiento neurológico dentro de parámetros normales para la edad. Conclusiones: a pesar de que los fetos con diagnóstico prenatal de MAV de Galeno asociada a otra anomalía consecuencia de la alteración hemodinámica de la misma poseen peor pronóstico que aquellos con MAV de Galeno aislada, en nuestra serie de casos, la mitad de los pacientes que sobrevivieron también presentaron anomalías asociadas y su evolución postnatal fue favorable. P-17. ECLAMC: EXPERIENCIA EN LA MATERNIDAD ITALIANA DE LA PLATA, ARGENTINA Mónica Ermini, Alfredo Uranga, Lorena Chappe, Micaela Palavecino, Hebe Campaña, Jorge S. López-Camelo Servicio de Obstetricia del Hospital Italiano de La Plata, Argentina Dirección de Investigación, CEMIC, CABA, Argentina Introducción: El Hospital Italiano de La Plata, Argentina forma parte desde 1999 de la red ECLAMC (Estudio Colaborativo LatinoAmericano de Malformaciones Congénitas). El ECLAMC es un programa de investigación clínica y epidemiológica de anomalías congénitas detectadas en nacimientos hospitalarios latinoamericanos. El sistema de vigilancia epidemiológica requiere de frecuencias de referencia, las que serán usadas en la comparación con las frecuencias observadas en la maternidad. Objetivo: El propósito de este estudio fue evaluar la frecuencia de anomalías congénitas en una muestra de 15.583 nacimientos, obtenidos en el Hospital Italiano de la Plata, durante el periodo 1999-2012. Metodología: Fueron comparadas las tasas de prevalencia al nacimiento según método de Poisson (O/E, IC 95%) en relación a una muestra de 2.223.341 nacimientos del resto de las maternidades participantes del ECLAMC. Resultados: Tres de las siete anomalías evaluadas mostraron una frecuencia aumentada respecto del resto del ECLAMC: cardiopatías, anomalías renales y síndrome de Down. Conclusiones: Las altas frecuencias observadas probablemente sean resultado del diagnóstico prenatal, derivación para la atención del parto y del recién nacido y a las características demográficas propias de la población atendida. P-18. EVOLUCIÓN DE RECIÉN NACIDOS CON MALFORMACIONES BRONCOPULMONARES (MBP) PERTENECIENTES A UN PROGRAMA DE DIAGNÓSTICO Y TRATAMIENTO FETAL (PDTF) M. Cecilia Rubio Longo, Claudia Cannizzaro, Aixa Reusmann, Mónica Siminovich, Diana Fariña. Área Terapia Intensiva Neonatal del Hospital de Pediatría Garrahan, Buenos Aires, Argentina. Introducción: El diagnóstico prenatal de malformaciones congénitas (MC) permite planificar estrategias de cuidado perinatal. Al Hospital Garrahan se refieren RN con MC para tratamiento quirúrgico. Desde el año 2008 se desarrolla el PDTF que coordina el cuidado fetal y optimiza la derivación de RN con MC. Objetivo: Describir la evolución de RN con MBP que ingresaron al PDTF. Método: estudio observacional descriptivo de la evolución de todos los pacientes con MBP que consultaron al PDTF. Resultados: Desde 2008 se realizaron 796 consultas en 533 embarazadas. De ellas, 18 RN (3,37%) ingresaron con diagnóstico de MBP. El promedio de edad gestacional al diagnóstico fue 21,5 sem y al nacimiento de 38 sem. El 50% de los casos fueron de sexo masculino. El diagnóstico definitivo de malformación adenomatoide quística (MAQ) se sostuvo en 13 casos (72%) (4 asociados a secuestro 30,7%). En 3 casos, durante el seguimiento, las lesiones remitieron. Se realizó lobectomía en 12 casos (66,6%). Conclusión: El seguimiento cercano de los pacientes con MBP a través del PDTF permite establecer mejores estrategias de cuidado, pautar su nacimiento en centros de alta complejidad, controlar la evolución clínica en el período posnatal y programar su correcto diagnóstico y tratamiento. P-19. ENFERMEDAD DE HIRSCHSPRUNG EN PACIENTE CON DIAGNÓSTICO PRENATAL DE DELECIÓN 5P Y DUPLICACIÓN 4Q: MÁS EVIDENCIA EN RELACIÓN AL LOCUS DE SUSCEPTIBILIDAD HSCR9. 1,2 1 3,4 1,5 1 Alberto Plaja , Teresa Vendrell , Ivana Canonero , Ana Cueto-González , Mar Borregan , Asunción 1 1 1 1 Fernández , Neus Castells , Miguel del Campo , E. Tizzano 1 Medicina Molecular y Genética. Hospital Vall d’ Hebron. Barcelona. España 2 Departamento de Citogenética. LABCO Iberia. Barcelona. España 3 Medicina Molecular y Genética. DIAGNUS. Córdoba . Argentina 4 Cátedra de Genética Médica. Universidad Católica de Córdoba. Argentina 5 Servicio de Pediatría. Hospital Universitario Mutua de Terrasa. Barcelona. España. Introducción: La enfermedad de Hirschsprung (HSCR) es una aganglionosis intestinal congénita, que se presenta en forma aislada o sindrómica. Existe una herencia mendeliana para formas sindrómicas, pero la forma aislada, presenta un patrón no mendeliano con baja penetrancia y expresión variable que sugiere la participación de uno o más genes con baja penetrancia. Existen varios genes candidatos involucrados en su patogénesis. Aquí presentamos un niño con HSCR que incluye el locus de susceptibilidad HSCR9 en región 4q32.1 y síndrome de Cri -du –chat. Objetivo: presentar un caso de diagnóstico prenatal de un reordenamiento cromosómico complejo, destacando la utilidad del array-CHG en la caracterización citogenética y correlación genotipo fenotipo. Descripción del caso clínico: Primer embarazo de pareja con antecedentes de infertilidad. Diagnóstico de RCIU a las 23 semanas de gestación. Se practica amniocentesis. El cariotipo fetal evidencia una anomalía cromosómica con material extra en un cromosoma del par 5. Para una correcta caracterización citogenética se realiza array-CGH al paciente y se detecta una deleción 5p terminal y una duplicación 4q terminal. Se realiza cariotipo a ambos padres y se detecta en la madre una reestructuración entre un cromosoma del par 4 y uno del par 5, que involucra inversión-translocación, confirmada por FISH subtelomérico 5pter y 5qter. RNT/ PEG con un fenotipo peculiar con microcefalia, orejas de implantación baja, pie zambo y cardiopatía congénita. Se diagnosticó HSCR a los 2 meses de edad y se confirmó la aganglionosis colónica. Discusión: La duplicación en el cromosoma 4 del paciente incluye 144 genes e incluye el locus de susceptibilidad HSCR9 (OMIM 611644) que podría explicar la aganglionosis del colon del niño. La deleción en el cromosoma 5 implica 77 genes e incluye la práctica totalidad de las regiones críticas asociadas con el síndrome de Cri du Chat, lo que explicaría el fenotipo observado en este paciente. En esta presentación se plantea que los genes de la región 4q32.1 pueden jugar un rol en la patogénesis de la HSCR. Conclusión: Resulta complejo definir la contribución fenotípica de la duplicación 4q y la deleción 5p, ya que estas entidades muestran una variabilidad fenotípica y menor severidad individualmente, pero ha sido muy útil utilizar el array-CGH, para una correlación genotipo-fenotipo e identificar los genes candidatos implicados en el fenotipo. P-20: PLGF: (FACTOR DE CRECIMIENTO PLACENTARIO) PARA EL SCREENING DE PREECLAMPSIA TEMPRANA, ACOPLADO AL ESTUDIO PRENATAL DEL PRIMER TRIMESTRE. RESULTADOS PRELIMINARES. Cecilia Zylbersztein, Ana María Sequera, Alejandro Falco, Jimena Rivera, Patricia Galati , Delia Pelletier, Liliana Voto Laboratorio Ceusa-Laeh , CABA. Introducción: Es conocido que valores bajos de PAPP-A no sólo están asociados a cromosomopatías sino también a complicaciones en la gestación como por Ej la preeclampsia (PE). Ésta, afecta hasta el 5 % de los embarazos,causando morbi-mortalidad fetal y materna. La PE temprana estaría relacionada con una placentación defectuosa durante la invasión trofoblástica, presentando alteración en las arterias espiraladas y menor secreción de PAPP-A. El PIGF es una proteína angiogénica responsable del crecimiento normal de la placenta. Estudios proponen al PLGF bajo, como marcador de PE precoz, manifestada en 2° mitad del embarazo. Según algunos autores el estudio ecográfico con doppler del flujo de arterias uterinas (AUt) si bien es de alta sensibilidad, tiene baja especificidad. La integración a la historia materna y con los marcadores bioquímicos, incrementa la posibilidad de detectar disfunción placentaria y endotelial. Objetivo: mostrar nuestra experiencia con el PIGF como marcador bioquímico de PE y su asociación con Free Beta HCG (FB) y PAPP-A (PP-A), marcadores del 1º trimestre para alteraciones cromosómicas. Material y métodos: Se analizaron los datos de PLGF de 158 pacientes (P), seleccionando 115 con MoM PPA < 0.80 y /o antecedentes de HT en embarazo previo. En todas las P se midió FB, PP-A y PIGF por Delfia (Perkin Elmer) entre sem 9 a13 de gestación. Se las subdividió según la MoM de PIGF en Grupo(G) 1-A(n=22): MoM < 0.80; G 1-B(n=33): 0.81 a 1.0 y G-2 (n=60): > 1.01. Se solicitó información sobre la PA, flujo de AUt y evolución del embarazo. Resultados (mediana y rango). Las MoM del PLGF fueron = G1-A: 0.67(0.39-0.80), G1-B:0.89(0.82-1.0) y G2:1.25 (1.01-2.42). Estuvieron asociados a MoM PP-A < 0.80= 8 P del G1-A (36%), 13 P del G1-B(39%) y 18 P del G-2(30%) y a MoM FB > 1.0= 9 P del G1-A(41%),12 P del G1-B(36%) y 19 P del G-2(32%). Conclusiones: Consideramos estos resultados de PIGF como preliminares para el screening de la PE temprana durante el 1º trimestre, sin descartar su potencial utilidad para aumentar el índice de detección del SME de Down. Si bien el PAPP-A bajo está relacionado a posibles alteraciones durante la evolución del embarazo, incluyendo patología hipertensiva, no sólo se lo encontró asociado a PIGF bajo (predictor de PE). El mayor % de P con FB alta se encontró en el grupo de P con PIGF bajos. Poder identificar tempranamente embarazos de alto riesgo permitiría implementar mayor vigilancia para evitar complicaciones maternas y perinatales. P-21. TRISOMIA 18: DIAGNÓSTICO PRENATAL CITOGENÉTICO DE 53 CASOS EN 25 AÑOS DE EXPERIENCIA Juan Roberto Rossetti, Claudia Bogado, Rolando Vildoza, Maria Adriana Echegaray. DIAGNUS – CORDOBA Objetivos: Mostrar la variación en la frecuencia del diagnóstico de trisomía 18 a través de los años. Analizar las indicaciones y el rol de la ecografía en el diagnóstico prenatal. Evaluar el curso de la gestación luego del diagnóstico de trisomía 18 Material y Métodos: Se realizaron 1952 estudios prenatales entre marzo 1987 y diciembre de 2013; 14 cordocentesis, 478 amniocentesis y 1460 biopsias coriónicas. Las indicaciones consideradas fueron edad materna avanzada (EMA), marcadores ecográficos, anomalías ecográficas y otras. El curso de la gestación se valoró a través de la comunicación personal con todas las pacientes y/o sus médicos derivantes. Resultados: Entre 1987 y 2001 (1006 pacientes) diagnosticamos 3 trisomías 18, (0,3%) 2 consultaron por EMA y 1 por anomalías ecográficas. En el período 2002-2013 sobre 946 pacientes analizadas encontramos 50 trisomías 18, (5,29%). Las indicaciónes fueron EMA 1 caso, en 13 casos translucencia nucal (TN) aumentada y/u otros marcadores del primer trimestre, en 7 casos TN aumentada + anomalías fetales y en los 29 casos restantes solo anomalías ecográficas. Las anomalías ecográficas mas frecuentes fueron: cardiopatía congénita (27/36), manos en garra (13/36), pie bot (12/36), RCIU (14/36), hidrops (10/36), onfalocele (10/36), hernia diafragmática (6/36), arteria umbilical única (5/36). 23 pacientes (43,4%) portadoras de trisomía 18 interrumpieron su embarazo y 30 (56,6%) decidieron continuar, de ellos 18 murieron in útero y 12 nacieron vivos con una sobrevida máxima de 48hs. Comentarios: La trisomía 18 es prácticamente incompatible con la vida, puede y debería ser diagnosticada prenatalmente; la ecografía precoz y sobre todo en el primer trimestre, es el principal método que orienta a su búsqueda citogenética. A partir del 2002, la búsqueda de los marcadores ecográficos del primer trimestre y la adecuada y completa valoración de la anatomía fetal, permitió mejorar notablemente la tasa de detección de trisomía 18. El diagnóstico prenatal permite la toma de decisiones libres y respetables tanto a la pareja como al obstetra y evita intervenciones médicas fútiles. P-22. CARDIOPATÍAS CONGÉNITAS “QUE NO NOS SORPRENDAN” DIAGNOSTICO PRENATAL Lisandro Benmaor Hospital Fernando Barreyro Posadas Misiones Introducción: Las cardiopatías congénitas son las malformaciones fetales más frecuentes y peligrosas que se pueden presentar desde la vida intrauterina. En la provincia de Misiones no contamos con cirugía cardiovascular pediátrica ni tratamientos hemodinámicos, por lo que el diagnostico precoz influye categóricamente al pronóstico de estos pacientes. Misiones es la provincia con más diagnósticos de cardiopatías del país, con un gran impacto en los pacientes que se le detecto su problema cardiológico durante el embarazo. Objetivos: mostrar la experiencia obtenida en la provincia de misiones de diagnósticos intrauterinos y su impacto en el periodo perinatal. Material y métodos: Se diagnosticaron desde marzo 2010 hasta marzo 2014 un número de 78 cardiopatías intrauterinas. Un porcentaje muy similar entre cardiopatías simples y complejas: 52/48; las cardiopatías complejas más diagnosticadas fueron en orden de frecuencia: canal AV, SCHI, coartación ventrículos únicos, enf. de Ebstein CIV severas, atresia tricúspidea, Fallot entre otras. La mortalidad de cardiopatías con diagnostico fetal fue de 25 pacientes (tanto prenatal como post natal). El número de cardiopatías diagnosticadas fue creciente año tras año. Las comorbilidades más frecuentes fueron las trisomías, síndromes polimalformativos, entre factores relacionados a la madre la toma de IM75, DBT materna y el antecedente de cardiopatía materna influyeron en la aparición de cardiopatías. El aporte del diagnóstico prenatal en morbi-mortalidad, derivaciones, tratamientos oportunos, impacto psicológico fue beneficioso, lo que estimula a la continuidad de seguir trabajando en el futuro de la medicina: diagnósticos prenatales. En Misiones se aprobó la ley de realización de ecocardiografía fetal a todas las madres con o sin factores de riesgo a partir de la semana 18 de edad gestacional, siendo la 1º provincia en el país en contar con dicha ley. P-23: DIAGNÓSTICO PRENATAL DE ANOMALÍAS CONGÉNITAS EN EL SISTEMA DE SALUD PÚBLICO DE NEUQUÉN. LOS PRIMEROS DOS AÑOS DE REGISTRO. Avila Silvia, Ponce Zaldúa María, Muntaner Celeste, Femenía Rosa, Alvarez Gabriel, Fernández Miranda Luis, Andrade Silvia Hospital Provincial Neuquén Introduccion: La disminución de la mortalidad infantil por causas prevenibles en la provincia de Neuquén, evidenció la importancia de las anomalías congénitas (AC). El sistema de salud pública se organiza en una red de atención en niveles de complejidad creciente; El Hospital Provincial Neuquén cuenta con servicio de Genética, Neonatología y Obstetricia de mayor complejidad donde son derivadas las embarazadas con gestas con AC y los recién nacidos con AC de todo el sector público. Objetivos: Determinar la eficiencia de las estrategias de diagnóstico prenatal como predictoras del nacimiento de niños con AC Materiales y métodos: Se realizó un análisis descriptivo transversal con los registros de recién nacidos y embarazadas con AC atendidos en el Hospital Provincial Neuquén (2012-2013). Se atendieron 186 nacidos vivos con anomalías congénitas mayores en el sector público (tasa de incidencia de 1.65). No se observó agregación por localidad de residencia materna. Las AC más frecuentes fueron las de etiología multifactorial. Las edades maternas con mayor incidencia de AC correspondieron a las de 20 a 34 años. 30% de los de 2012 y 32% de los de 2013 tuvieron DPN. Las AC de sistema urinario, esquelético y pared abdominal fueron las de mayor detección ecográfica no así las de SNC y cardiovascular. Los casos calificados como letales fueron fundamentalmente los que correspondieron a afección de sistema nervioso, urinario y anomalías cromosómicas con compromiso multisistémico. Se calculó la sensibilidad, como indicador estadístico que evalúa el grado de eficacia a una prueba diagnóstica obteniéndose un True Positive Fraction de 0.26. Conclusiones: La tasa de anomalías congénitas al nacimiento fue similar a la de los países desarrollados, en coincidencia la mortalidad perinatal que tiene la provincia. Se registró un aumento en la detección de AC que fueron diagnosticados antes del nacimiento. La ecografía fue la herramienta esencial para diagnosticar AC del sistema urinario y las displasias esqueléticas, no para las cardíacas ni las del SNC. Las cromosomopatías representaron el 4% las AC. El TPF indica la necesidad de reforzar las estrategias para mejorar las tasas de detección. Las edades de las mujeres con mayor tasa de presentación de anomalías congénitas no son coincidentes con los grupos etéreos definidos clásicamente como población de riesgo. Estas conclusiones muestran la importancia de los registros para reflexionar sobre la propia práctica y plantear estrategias de mejoramiento continuo. P-24: IMPORTANCIA DE LA TRANSLUCENCIA CITOGENÉTICOS DE 276 PACIENTES NUCAL AUMENTADA EN Juan Roberto Rossetti, Claudia Bogado, Rolando Vildoza, Maria Adriana Echegaray. LOS HALLAZGOS . Grupo de Diagnóstico Prenatal Citogenético – Diagnus – Córdoba - Argentina Objetivos: Mostrar los resultados citogenéticos en biopsia corial, según el valor de la translucencia nucal (TN) Pacientes y métodos: Entre febrero 2002 y diciembre de 2013 se realizaron 920 estudios citogenéticos en vellosidades coriales (VCS). Las indicaciones fueron edad materna igual o mayor de 35 años en 428 pacientes (46,53%), Translucencia nucal aumentada 276 (30%), anomalías ecográficas 112 (12,17%), translocación balanceada en uno de los padres 12 (1,30%) y otras 92 (10%). De 276 pacientes con translucencia nucal aumentada, 107 presentaron aneuploidía (38,77%). Se analizo la frecuencia de estas aneuploidías de acuerdo al valor de la TN que arbitrariamente distribuimos en 5 grupos, el primer grupo de 2,60 a 2,99mm, los tres grupos siguientes con intervalo de 1mm y un último grupo con TN de 6 o mas mm. Resultados: El porcentaje de aneuploidias encontradas fue el siguiente, 9,68% (3/ 31) en el primer grupo (=>2,6 - 2,99), 27,10% (29/107) en el grupo con TN entre 3 y 3,99mm; 37,47 (17/45) en el grupo con TN entre 4 y 4,99mm; 61,3% (19/31) en el grupo de 5 a 5,99mm y finalmente 62,90% (39/62) en el grupo con TN =>a 6mm. Conclusión: Se observa franca disminución de la edad materna avanzada como indicación del diagnostico prenatal citogenético, siendo la ecografía fetal la de mayor aporte en las indicaciones del mismo. Se confirma la importancia de la tranlucencia nucal aumentada y el aumento exponencial de las aneuploidias en relación al incremento de la misma. P-25. EXPERIENCIA EN EL MANEJO DE PACIENTES CON SINDROME DE CORAZON IZQUIERDO HIPOPLASICO SaenzTejeira, M; Napoli,N; Villa, A; Barreta, J; Pilan, M; Perez, A; Jorro, F; Marantz, P Hospital Italiano, Fundación Hospitalaria, Buenos Aires Introducción: El avance más dramático en cardiopatías congénitas, en los últimos años, ha sido sin duda el diagnóstico, manejo y resultados del síndrome de corazón izquierdo hipoplásico (SCHI) Objetivo: El objetivo de este trabajo es presentar nuestra experiencia en el manejo los pacientes con SCIH, en 2 centros de tercer nivel, en el período comprendido entre mayo 2010 y mayo 2013 desde el diagnóstico prenatal hasta la cirugía de Norwood-Sano Poblacion y métodos: Estudio descriptivo retrospectivo. La población fue de 17 pacientes. Para su análisis lo dividimos en Pre estadio 1: diagnóstico fetal, factores de riego y manejo neonatal. Estadío 1: Cirugía de Norwood-Sano, manejo perioperatorio. Resultados:Preestadio 1: Se realizó diagnóstico prenatal de SCIH en todos los pacientes. La correlación entre el diagnóstico prenatal - postnatal fue de un 100% excepto para coartación de aorta que fue de un 80%.Estadío 1: Las opciones terapeúticas fueron: cuidados de comfort, cirugía de Norwood o híbrido farmacológico (cerclaje e infusión de prostaglandinas) en el bajo peso (menor de 2,500). La mortalidad fue 5,88%. Falleció un paciente en cada estadio. No se presentaron muertes fetales Conclusión: Analizando las variables consideradas de alto riesgo neonatal para cirugía de Norwood-Sano, en pacientes con SCIH encontramos que el diagnóstico prenatal mejora la supervivencia de estos pacientes; el bajo peso no fue factor de riesgo y la asociación con malformaciones extracardíacas representa mayor gravedad. P-26. DIAGNÓSTICO PRENATAL DE ECTOPIA RENAL CRUZADA: REPORTE DE DOS CASOS Carlos Garnil, Sofía Grinenco, Juan Fazzio, Jorge Pardo, Germán Falke. Proeco, Vicente López, Buenos Aires. Introducción: La ectopia renal constituye una anomalía congénita rara, siendo la variedad en la que el riñón ectópico se encuentra en la pelvis cruzando la línea media y sin fusión con el riñón contralateral una de las menos frecuentes, con una incidencia reportada en 1:75,000 autopsias. El objetivo de este estudio es reportar dos casos de ectopia renal cruzada sin fusión renal diagnosticadosprenatalmente. Descripción: Se presentan dos casos que fueron referidos con sospecha de agenesia renal unilateral a partir de ecografías obstétricas de rutina. En ambos casos en el examen ecográfico se constató biometría fetal conservada y líquido amniótico normal, riñón izquierdo conservado (diámetro en Pc50), fosa renal derecha vacía y un riñón pelviano medial (diámetro en Pc25) cuya pelvis se encontraba dispuesta en sentido anterior, con irrigación proveniente de una rama tributaria de la arteria iliaca derecha. No se constataron malformaciones en otros órganos. El caso 1 fue evaluado inicialmente a las 23 semanas de gestación con hidronefrosis leve que progresó a moderada a partir de la semana 35.Nació por parto vaginal un bebémasculino de 41 semanas, 3600 grs, confirmándose los hallazgos ecográficos prenatales por ecografía. A las 6 semanas de vida presentaba análisis de laboratorio y centellograma renal bilateral que evidenciaban función renal conservada. El caso 2fue referida a las 31 semanas para su valoración, y no presentó hidronefrosis en su evolución.Nació por parto vaginal un bebéfemenino de 41 semanas, 3700grs, confirmándose ecográficamentelos hallazgos prenatales. A los 3 meses de vida presentaba análisis de laboratorio normal, y retardo en la evacuación del riñon ectópico constatado en la gammagrafía con DMSA. Discusión/Conclusiones: La ectopia renal constituye un diagnóstico diferencial a considerar en casos de falta de visualización de un riñón en el feto. Su detección prenatal permite implementar una estrategia de estudio y seguimiento para valoración de la función renal y de la presencia de anomalías genitourinarias o cardíacas que pueden asociarse. La variedad de ectopia renal cruzada sin fusión renal constatada en los casos presentados es poco frecuente pero debe ser considerada cuando no se visualiza la silueta renal en su respectiva fosa renal ni en la pelvis ipsilateral. P-27. INFECCIONES URINARIAS EN EL EMBARAZO. ESTADO DE SITUACIÓN EN UN HOSPITAL PÚBLICO, AÑO 2010. Silvia Balconi, Matías Villacorta, Cecilia Corigliano, Matías Jara, Di Bartolomeo Susana, Ana Corominas Hospital Nacional Prof. Alejandro Posadas, Buenos Aires Introducción: Tanto las Infecciones urinarias (IU) como la bacteriuria asintomática (BA) pueden causar complicaciones durante el embarazo como la pielonefritis, el parto prematuro y el consiguiente bajo peso al nacer, asociados a mortalidad fetal. El urocultivo está incluido en todos los protocolos de seguimiento del embarazo, por lo tanto es necesario profundizar en el conocimiento de la prevalencia de éstas patologías para implementar acciones preventivas. Objetivo: Conocer la prevalencia de IU y de BA en mujeres embarazadas atendidas en el hospital Posadas entre agosto de 2009 y agosto de 2010, y las especies bacterianas asociadas a ambas patologías. Metodología: Estudio retrospectivo. Se incorporaron al estudio todos los urocultivos de mujeres embarazadas ingresados durante el período estudiado. A partir de los registros del laboratorio, se recabaron los datos del sedimento urinario y cultivo. Resultados: Durante el año 2009 se estudiaron 1716 muestras de orina de mujeres embarazadas, de las cuales en 224 (13,05%) se aisló un microorganismo, 738 (43,01%) presentaron flora polimicrobiana y 754 (43,94%) fueron negativos. La bacteria preponderante fue E coli (58,03%), seguida de Estreptococo Grupo B (EGB) (16,96%) y Enterococcus spp (5,82%). El 19% restante se distribuyó entre Proteus spp, Klebsiella spp, Staphilococcus spp y levaduras. De los cultivos positivos, el 60,71% presentó reacción inflamatoria: el 39,29% no presentó sedimento patológico (bacteriuria asintomática). E coli se presentó en una relación 3:1 en infección urinaria: bacteriuria, por el contrario, EGB se presentó en una relación 1:2. Por otra parte, del total de sedimentos con reacción inflamatoria (n=378), se recuperó el gérmen causal en 141 casos (37,30%). Discusión y Conclusiones: La prevalencia y la distribución de gérmenes causales coincide con la reportada por la literatura, se observa una distribución diferente microorganismos entre infecciones urinarias y bacteriurias, lo cual podría revelar distintos patrones epidemiológicos. Resulta notable el gran porcentaje de recuperación de flora polimicrobiana. Ésta situación genera un problema diagnóstico que podría ser controlado con un mayor énfasis en la etapa preanalítica. Es necesario que la paciente comprenda claramente el proceso de preparación, toma y mantenimiento de la muestra hasta remitirla al laboratorio. El conocimiento de éste problema contribuirá a desarrollar una estrategia para su resolución. P-28: QUIRÓFANO LIBRE DE LÁTEX. CESÁREA Y CIRUGÍA NEONATAL MIELOMENINGOCELE EN HOSPITAL PÚBLICO DE NEUQUÉN. EN NIÑOS CON Gabriel Aníbal Alvarez, María Inés Martínez, Silvia Avila, Liliana Vanderhoeven, Luis Fernández Miranda, Hugo Regondi, Laura Martínez. Hospital Castro Rendón, Neuquén Capital. Introducción: La incidencia de alergia al látex en pacientes con espina bífida oscila entre 10 y 73 %. Estos niños se ven sometidos a múltiples intervenciones viéndose expuestos a productos que contienen látex. Se suma que tienen mayor predisposición genética a desarrollar este tipo de alergia. Las manifestaciones clínicas pueden llegar a ser muy graves. Está descripto que la preparación del quirófano libre de látex disminuiría el riesgo de sensibilización. Objetivo: Describir la experiencia de la organización de quirófanos libres de látex con el propósito de disminuir posibles alérgenos para mejorar el crecimiento y desarrollo de los niños con mielomeningocele. Descripcion de casos: Se presentan en nuestro servicio dos pacientes cursando embarazos de término con diagnóstico de mielomeningocele (hidrocefalia, defectos en la fosa posterior, hallazgos que encuadran con la malformación de ArnoldChiari ) Previa entrevista con nuestro equipo de diagnóstico prenatal integrado por genetistas, tocoginecólogo y neonatólogo, se realizó ateneo interdisciplinario y se decidió la preparación del quirófano libre de látex para estas cirugías, con la buena predisposición del personal de quirófano. La preparación en ambos casos se realizó con 12 hs de anticipación y luego de la cesárea permanecieron los quirófanos cerrados hasta la cirugía de corrección por neurocirugía Discusión/ conclusión: El trabajo de implementación de estas prácticas combinó esfuerzo de los médicos, enfermeras y personal de quirófano y gran parte de los recursos que a veces son escasos en los hospitales públicos. Se destacó la buena predisposición, la rapidez, y efectividad de los agentes, que cada uno dentro de sus posibilidades permitió hacer lo programado sin inconvenientes. Dada las fuertes recomendaciones en la bibliografía, creemos que la implementación de los quirófanos libres de látex es el primer paso para la prevención de la alergia al latex que sufren estos pacientes a lo largo de su vida. El mérito de realizar este esfuerzo en el hospital público es mejorar la calidad de vida a todos los niños con esta enfermedad. P-29. DIAGNÓSTICO PRENATAL DE UN PACIENTE CON SÍNDROME DE CORNELIA DE LANGE. EL VALOR DEL ESTUDIO DETALLADO DEL FENOTIPO ECOGRÁFICO Pecis Juan, Avila Silvia, Fernández Miranda Luis Hospital Provincial Neuquén, Hospital Bouquet Roldán de Neuquén Introducción: En los últimos años se lograron importantes avances técnicos en la pesquisa de anomalías cromosómicas en la gestación. Sin embargo, se estima que el 95% de las anomalías congénitas de los nacidos vivos cursan con normalidad cromosómica. El diagnóstico certero permite explicar la etiología del cuadro clínico y brindar el asesoramiento genético a la familia sobre el riesgo de recurrencia de la anomalía en un futuro embarazo. Objetivo: Presentamos un caso clínico que ejemplifica cómo los aportes de las imágenes permiten lograr un diagnóstico prenatal a través de la descripción detallada del fenotipo Descripción: En una evaluación de rutina se detecta a las doce semanas edema en velo facio fronto ceéfalo cervical con TN de 5mm. Se trata de una mujer de 26 años que se refiere sana con dos gestas previas la última de las cuales culmina con muerte fetal atribuida a insuficiencia placentaria. En la semana 13 se detecta derrame pleural, micrognatia, hipoplasia cubital izquierda, flexión no reductible del miembro, braqui/oligodactilia bilateral.. A las 20 semanas se constata disminución del edema y se reconfirman los datos anatómicos descriptos anteriormente. Se deriva a la consulta genética y se decide punción de LA para descartar cromosomopatía. Los registros de 3D son compatibles con microcefalia, orientación antimongoloide de las hendiduras palpebrales, hueso nasal deprimido, narinas antevertidas, implantación baja de las orejas, restricción de crecimiento, polihidramnios. La ecografía cardíaca muestra defecto ventrícuoseptal con salida de único vaso sobre el septo conotruncal. El cariotipo fetal es normal. Se postula como diagnóstico diferencial el síndrome de Cornelia De Lange. A las 31 semanas se produce el óbito fetal. El examen físico del mortinato confirma los hallazgos ecográficos: restricción de crecimiento simétrica, facies compatible con síndrome de Cornelia De Lange con hendiduras palpebrales con orientación antimongoloide, pestañas largas y arqueadas, sinofris, hipoplasia radial bilateral, oligodactilia bilateral. Conclusión: La descripción ultrasonográfica minuciosa y detallada permite lograr y a veces confirmar el diagnóstico sindrómico. Esto es de fundamental importancia cuando se considera que no más del 5% de las anomalías congénitas de nacidos vivos y un porcentaje similar de mortinatos se explica por anomalías cromosómicas. P-30. TRISOMÍA 21 EN GEMELOS BIAMNIÓTICOS BICORIÓNICOS: CONTRIBUCIÓN DE ULTRASONOGRAFÍA Y LOS ESTUDIOS GENÉTICOS PARA EL DIAGNÓSTICO Y ASESORAMIENTO. LA Alicia Vaglio. Jorge Pereyra, Pablo Putti, Roberto Quadrelli. Instituto de Genética Médica, Hospital Italiano, Montevideo, URUGUAY Introducción: La detección de la misma anomalía cromosómica en ambos fetos de igual sexo en un embarazo biamniótico bicoriónico plantea dificultades para el asesoramiento genético de riesgo de recurrencia en futuras gestaciones. Material y métodos: Primer embarazo de pareja joven no consanguínea referido por translucencia nucal aumentada y edema en ambos fetos. Se realizó estudio citogenético y de cigosidad en vellosidad corial de ambos sacos. Resultados: El estudio citogenético mostró trisomia 21 libre en ambos fetos. La cigosidad mostró perfiles de ADN idénticos en ambos fetos por lo que se interpretó que se trataba de un embarazo gemelar monocigota con escisión antes del 4o. dia post concepción. Se asesoró de bajo riesgo de recurrencia de anomalía cromosómica en futuras gestaciones. Conclusiones: Se destaca la importancia de la aplicación de la ultrasonografía, los estudios citogenético y de cigosidad para realizar asesoramiento genético en embarazos gemelares. P-31. DIAGNÓSTICO ECOGRÁFICO Y MOLECULAR DE DISPLASIA DIASTRÓFICA A LAS 16 SEMANAS DE GESTACIÓN. Alicia Vaglio, Jorge Pereyra, Pablo Putti, Andrea Quadrelli, Roberto Quadrelli. Instituto de Genética Médica, Hospital Italiano, Montevideo, URUGUAY Introducción. La Displasia Diastrófica (DTD) es una osteocondrodisplasia autosómica recesiva con fenotipo ecográfico característico y puede confirmarse mediante estudio molecular. Material y Métodos: Primer embarazo de pareja no consanguínea referida por translucencia nucal aumentada. Se realizó ecografía y estudio molecular del gen DTDST Resultados: Se detectaron dos mutaciones que confirmaron el diagnóstico. El estudio anatomopatológico fue concordante con el diagnóstico establecido. Conclusiones La ultrasonografía y el estudio molecular permitieron realizar diagnóstico para el embarazo en curso y asesoramiento genético de alto riesgo de recurrencia de igual patología en futuras gestaciones. Con la definición molecular es posible ofrecer estudio prenatal ó preimplantacional para futuros embarazos. P-32. MEGALOURETRA - DIAGNÓSTICO PRENATAL – TRATAMIENTO NEONATAL César Benmaor, Francisco de Badiola, Valeria Frasca, Carlos Esperanza, Lucas Frias, Laura Ranaletti, Horacio Cozzi. Instituto Gema. Unidad de Medicina Fetal. Posadas, Misiones. Introducción: La megalouretra es una anomalía genital poco frecuente caracterizada por la dilatación de la uretra peneana, asociada con la ausencia o hipoplasia de los cuerpos espongioso y/o cavernoso. Las complicaciones postnatales pueden ser tanto disfunciones miccionales, eréctiles, como insuficiencia renal o hipoplasia pulmonar. Resumen: Se presenta un paciente con diagnóstico de megalouretra a las 24 semanas de edad gestacional, imagen ecográfica anecoica de 9mm en cara dorsal de pene, sin patología asociada. Seguimiento ecográfico prenatal. Nacimiento a las 39 SEG. Buen estado general. Sin requerimiento de O2. Ex Fis al nacimiento: dilatación de uretra peneana, 2/3 distales, hasta glande. Diuresis conservada. Se vacía con la compresión uretral. Asintomático. Función renal normal. Sin hidronefrosis. Eco: Hipoplasia de cuerpos espongioso a nivel de uretra mediopeneana hasta glande. Cuerpo cavernoso conservado. Cistoscopía: Uretra glandular conservada. Dilatación a partir de uretra mediopeneana hasta glande. Sin infecciones urinarias. Cirugía: 20 días de vida: Uretrostomía en hora 6. 6 meses: Se realizó Uretroplastía. Conclusión: La megalouretra es la dilatación uretral secundaria a la obstrucción distal de la misma que produce como consecuencia la hipoplasia de los cuerpos espongiosos y del tejido eréctil peneano. Cuando el volumen de líquido amniótico es normal, la supervivencia es posible. Sin embargo, todos los niños tienen algún grado de disfunción sexual, disfunción en la micción o falla renal. La megalouretra debería ser sospechada en fetos del sexo masculino con un falo elongado y o distendido como con anomalías urinarias asociadas. P-33. SEGUIMIENTO POSTNATAL DE PACIENTE CON DIAGNÓSTICO PRENATAL DE HIDRONEFROSIS BILATERAL. PRESENTACIÓN DE UN CASO. Jimena S. Esnaola, María N. Ormaechea, Juan I. Bortagaray, Roberto L. Vagni, Francisco De badiola, Juan M. Moldes. Hospital Italiano de Buenos Aires Paciente masculino con diagnóstico prenatal de hidronefrosis bilateral grado IV mas megauréter con oligohidramnios. Madre con diagnóstico de mielomeningocele. Nacido en otro centro por cesárea a las 36 semanas con 2,650 kg. Presentó diuresis espontánea al nacimiento. Al primer día de vida se realizo cistouretrografía que evidenció válvula uretra posterior y reflujo vesicoureteral grado V izquierdo. Con una creatinina de 8, se le realizó a los 6 días de vida ureterostomía derecha mas vesicostomía. Al mes de vida fue derivado a nuestro hospital para inicio de terapia de reemplazo renal. A los 2 meses de vida inicio diálisis peritoneal. Laboratorio U: 16, Creatinina de 4.92. La Cistovideourodinamia de control informa Vejiga piriforme de contornos regulares. Capacidad: 30 cc. RVU g. III izquierdo, en el mismo momento se realiza pielografía ascendente por ureterostomía derecha donde se visualiza Uréter derecho, tortuoso, dilatado. A los 14 meses se procede mediante una Cistovideoendoscopía terapeútica a la Resección endoscópica de válvula de uretra posterior con Inyección de Vantris ( antireflujo)en meato ureteral izquierdo y cierre de vesicostomía. Ecografias de seguimiento mostraban Ambos riñones de aspecto atrófico, con cambios quísticos. RD: 40 mm, RI: 41 mm. A los 18 meses presentó episodios de infección urinaria mas shock séptico por peritonitis fúngica que requirió extracción de cánula de diálisis peritoneal con colocación de catéter de hemodiálisis mas antibioticoterapia prolongada. Se determino a los 19 meses la realizacion de una nefroureterectomía derecha mas trasplante renal con donante vivo relacionado (padre) con un peso de 10, 26 kg. , en hemodiálisis trisemanal con Ur de 16 y Creatinina de 3.83 mg/dl. No requiriendo Terapia sustitutiva postransplante. Al día de la fecha se encuentra en aceptable estado general, en seguimiento por nefrología pediátrica bajo tratamiento inmunosupresor. Ultimo valor de creatininemia de 0.4 mg/dl P-34. CORIOANGIOMA PLACENTARIO GIGANTE, REPORTE DE CASO José A. Gómez Maidana, Roxana A. Galindo, Andrea M. Segovia, José Rodeyro, Ivete Vargas Hospital Regional “Rio Gallegos”, Rio Gallegos, Santa Cruz Introducción: El corioangioma es el tumor benigno más frecuente de la placenta. Cuando su tamaño es mayor de 5 cm se denomina Corioangioma gigante, pudiendo asociarse a complicaciones maternofetales. El objetivo es la presentación de un caso. Descripción del caso: Paciente de 32 años, G2 C1, con embarazo controlado. Antecedente de feto muerto previo sin causa aparente. En ecografía de 25 semanas se constata formación heterogénea de bordes lobados, subcoriónica, cercana a la inserción del cordón umbilical, de aproximadamente 10 cm. LA normal, resto de morfología fetal sin alteraciones. En doppler pulsado y color se obtiene flujo periférico pero sin flujo central. Se le realiza Resonancia Magnética la cual confirma el hallazgo ecográfico, impresionando como hematoma placentario subcorionico. Posteriormente se decide su internación donde se descarta patologías ligadas a la coagulación con alta posterior. A las 31 semanas vuelve a internarse por amniorexis espontánea y hemoginecorragia de abundante cuantía, con latidos cardiofetales positivos, decidiéndose cesárea de urgencia, obteniendo un RN de sexo masculino de 32 semanas, de 1440 gramos, Apgar 7/9. Posteriormente el estudio anatomopatológico informa: hemangioma arterio-venoso placentario, infarto isquémico vellositario peritumoral, hematoma submembranoso e inserción velamentosa de cordón Conclusión: El corioangioma gigante es un tipo de tumor de la placenta, que se asocia a complicaciones materno-fetales tales como: polihidramnios, anemia fetal, hidrops, crecimiento intrauterino restringido, insuficiencia cardiaca congestiva, y hemorragias anteparto lo cual conlleva a un aumento de la morbimortalidad perinatal, tal como fue el caso descripto. Durante el diagnostico precoz (por imágenes) se puede evidenciar una imagen nodular hipoecoica, bien circunscripta vecina a la placa corial, como también áreas de heterogeneidad debido a procesos degenerativos. El empleo de doppler color y power angio son fundamentales para identificar masas más vascularizadas y por ende más proclives a complicaciones, permitiendo también realizar el diagnóstico diferencial con hematoma subamniotico, trombohematoma subcorial, mola parcial, mioma o teratoma. En ocasiones un corioangioma no suele tener flujo detectable mediante doppler color, pero por esto no debe excluirse la presencia de un tumor vascular. Su diagnóstico oportuno nos permite un control más exhaustivo. P-35. TUMOR ABDOMINAL FETAL Torroba Marcelo, Osores Oscar, Guilburd Guillermo, Ibarreche Guillermo, Nuñez Maria Paz, Sinner Ricardo, Valeria Ulloa Sanatorio San Jorge, Ushuaia, Tierra del Fuego Objetivo: analizar la presentación clínica, las características ecograficas y los resultados anatomo-patológicos Descripcion del caso: Paciente femenina, 32 años de edad, primigesta, FUM: 03/07/1013, FPP: 10/04/2014, peso ant: 62kg -1,65 M, antecedentes personales: crio por HPV-madre CA mama, lumbago . Los controles prenatales comienzan en nuestra clínica a la semana 30. Todas las Ecografias anteriores eran normales. A la semana 32, se observa imagen anecoica simple en hemiabdomen izquierdo que mide 41x37x25 mm, Doppler negativo. Por la topografía y feto fetal se sospecha de tumor simple ovárico. A la semana 35 se controla a la paciente y la imagen continua de las mismas características un poco mas pequeña de 37x34x22 mm.A la semana 37 las características del tumor anexial, cambian haciéndose heterogénea, con tabiques y pasa a ser catalogado como tumor mixto. Se programa operación cesárea a las 38 sem,nace Rn sexo femenino,apgar 9/10'peso 3231g. Se le realiza ecografia abdominal que resulta normal y ecografia ginecológica que informa en topografia anexial izquierda lesión ocupante de espacio, multiloculada ,paredes finas, contenido heterogéneo, Doppler negativo, que mide 26x20x36 mm (10cc) Se sospecha disgerminoma y se biopsia. A la espera de resultado. Discusión: Los quistes ováricos no son tan infrecuentes como se pensaba. La introducción de la ecografía como método de estudio prenatal y posnatal nos ha permitido conocer realmente su frecuencia, así como obtener más datos referentes a su evolución. Los quistes foliculares ováricos fetales pueden identificarse ecográficamente a partir de la semana 20 de gestación. Su frecuencia aumenta a medida que progresa el embarazo, de manera que la mayoría se detectan durante el tercer trimestre. Se consideran patológicos cuando su tamaño es mayor de 2 cm de diámetro. La incidencia estimada del quiste de ovario es de 1 por cada 2.625 recién nacidos. La mayoría de estos quistes involucionan en el transcurso de los primeros meses de vida, por depleción de hormonas maternas, pero pueden presentar complicaciones durante el periodo fetal o neonatal, entre las cuales la torsión ovárica es la más frecuente . La incidencia de torsión en los quistes ováricos detectados en el periodo fetal es del 38%y la incidencia de torsión en los quistes ováricos detectados en el periodo neonatal es del 50-78% . Su etiología sigue siendo controvertida. La mayoría de los autores los relacionan con una estimulación ovárica por estrógenos maternos y/o feto placentarios, con la gonadotrofina coriónica humana placentaria y con la hormona folículo estimulante fetal. Conclusiones: el diagnostico prenatal ecografico de patología anexial presento alta sensibilidad en detección de patología ovárica y permitió la preparación de los padres con información de las posibles causas y pronostico. Logrando tambien evitar complicaciones fetales como la torsión, con el seguimiento exhautivo y busqueda de patologias asociadas. P-36. REPORTE DE CASO DE SÍNDROME DE CORDÓN CORTO Micaela Palavecino, Maria F. Rouillet, Lorena E. Chappe, Monica L. Ermini Hospital Italiano, La Plata Introducción: El síndrome de cordón corto o Body Stalk Syndrome, pertenece a un grupo de anomalías del cordón umbilical, y se caracteriza por un acortamiento anormal del mismo que generalmente se asocia con anomalías del desarrollo de estructuras mayores; particularmente con defectos de la pared abdominal, defectos del sistema nervioso central y bandas amnióticas. La incidencia de este síndrome es de 0.4 en 1,000 casos Como objetivo nos proponemos reportar un caso clínico nacido en la Maternidad Italiana de La Plata en el mes de diciembre de 2013 y revisar la literatura publicada sobre su etiología, características clínicas y diagnóstico Descripción de caso: Paciente de 21 años, primigesta, sana, sin antecedentes personales ni familiares de relevancia, serologías negativas, sin hábitos tóxicos y embarazo controlado, derivada al tercer nivel de complejidad cursando embarazo de 23 semanas de gestación con diagnostico presuntivo ecográfico de gastrosquisis. Se realiza ecografía obstétrica, en varias oportunidades, en nuestra institución durante el control prenatal, donde se sospecha diagnóstico de Síndrome de cordón corto o Body Stalk Syndrome por la presencia de defectos de la pared abdominal, hipoquinesia y defectos posturales de la columna vertebral; el resto de la anatomía fetal sin particularidades, acompañado de un cordón con longitud aproximada de 10 cm, sin enrollamiento de los vasos. Se decide cesárea abdominal a las 34 semanas de gestación, obteniéndose recién nacido vivo de sexo masculino de 2070 gramos, 45cm de talla y 35 cm de perímetro cefálico, Apgar 1 al minuto y Apgar 4 a los 5 minutos. El feto se encontraba unido a bloque placentario por cordón umbilical de 11cm de longitud, presentando, además, defectos de la pared abdominal observándose hígado, intestinos grueso y delgado fuera de cavidad abdominal, escoliosis marcada, dextrocardia, tórax pequeño y asimétrico y fusión de segundo y tercer dedo de mano derecha. Nace con severas dificultades debidas a su cuadro clínico, produciéndose el óbito pasada una hora del nacimiento. Conclusiones: El síndrome de cordón corto, es una patología del desarrollo del cordón umbilical, de etiología desconocida, de infrecuente presentación. Este caso demuestra la gran gama de defectos estructurales que pueden presentar los fetos afectados por este síndrome, y la importancia de la ecografía en el diagnostico prenatal para realizar el diagnostico a edades tempranas. P-37. GEMELOS UNIDOS: DESCRIPCIÓN DE UN CASO CON DIAGNÓSTICO ECOGRÁFICO PRENATAL 1 1 1 2 2 2 Mariel Ormazábal , Solari Andrea , Mariana Vilas , Marcelo Castresana , Enrique Aguilera , Josué Buliubasich , 1 Miguel Aguirre 1.Centro Nacional de Genética Médica “Dr. Eduardo E. Castilla”. ANLIS “Dr. Carlos G. Malbrán” 2.Hospital General de Agudos “J. A. Penna” Introducción: Los gemelos unidos (GU), también llamados “siameses”, son infrecuentes y su incidencia se estima en 1:50.000 gestaciones y 1:200.000 recién nacidos vivos. Hay un predominio del sexo femenino 2:1. Existen dos hipótesis sobre la causa: la de la fisura, representando una división tardía e incompleta del disco embrionario, y la de la fusión, que es el resultado de la unión de los dos discos embrionarios previamente separados. Los GU se clasifican según el sitio de unión. Los toracópagos constituyen el 40% de los casos publicados y son los de peor pronóstico. Los cefalópagos son menos frecuentes. En todos se ha reportado una alta tasa de prematurez y de muerte intrauterina y perinatal. Las anomalías asociadas más frecuentes son las cardiopatías, los defectos de miembros y de pared abdominal. El pronóstico y la eventual corrección quirúrgica dependen de las estructuras que comparten. La resonancia nuclear magnética (RMN) es de gran utilidad para lograr definirlas. La detección se puede hacer desde el primer trimestre de embarazo, aunque la mayoría se realiza en el segundo trimestre, y el hallazgo tardío se asocia más a complicaciones maternas. Descripción del caso: Se trata del primer embarazo de una pareja no consanguínea, de 19 y 27 años, sin antecedentes familiares de relevancia. Fue derivada a nuestro centro a las 25 semanas de gestación para definir las anomalías ecográficas. Se concluyó que eran dos fetos unidos parcialmente por la cabeza, tórax y abdomen superior. Compartían estructuras craneanas y cerebrales con una cara única. Se observaron dos corazones, cada uno en su posición correspondiente y con actividad independiente. Dos columnas vertebrales en posiciones opuestas y cuatro miembros inferiores. Fue difícil definir el número de miembros superiores. Los genitales externos eran femeninos. El embarazo finalizó en un parto vaginal espontáneo a las 27 semanas por óbito fetal. Discusion: El caso de GU fue de cefalópagos de sexo femenino. El diagnóstico se hizo en el segundo trimestre de embarazo y tuvieron una evolución desfavorable. La ecografía ha demostrado ser útil para detectar gemelos unidos y así poder planear el parto y la posibilidad de corrección quirúrgica, pudiendo alcanzarse el diagnóstico en el primer trimestre de la gestación. P-38. LA EVALUACION DEL CUELLO UTERINO COMO PREDICTOR DEL PARTO PREMATURO Valdivieso, Alicia Maria, Prado, Andrea Centro de Diagnóstico Los Ceibos, Yerba Buena, Tucuman. Introducción: la ecografía transvaginal en el 2° trimestre del embarazo, nos permite evaluar cuello uterino: longitud cervical, forma, y OCI, como elementos predictores del parto prematuro Objetivos:. – Identificar precozmante pacientes de riesgo de PP, permitiendo asi una intervención en el momento adecuado. - Predecir PP. Material y Métodos:- pacientes en 2° trimestre del embarazo. - transductorendocavitario de 6 a 10 MHz. - vejiga vacia. - canal cervical horizontal y totalmente visible en la pantalla. - tomar 3 medidas del canal cervical y promediar. Tomar medidas con compresión abdominal y maniobra de valsalva, promediar y observar modificaciones del mismo. Conclusiones: laUS transvaginal, presenta mayor capacidad predictiva de PP realizada en pacientes sintomáticas en el 2° trimestre. La dilatación del OCI con cuña mayor al 50 % y canal cervical meno a 15 mm, tienen riesgo elevado de PP antes de la semana 35, debiéndose tomar medidas terapéuticas adecuadas. P-39. SÍNDROME DE DANDY WALKER. A PROPÓSITO DE UN CASO Lorena E. Chappe, Micaela Palavecino, María F. Rouillet, Rubén Neme, Alfredo Uranga. Hospital Italiano de La Plata Introducción: El síndrome de Dandy Walker se define como la asociación de hidrocefalia de grado variable con aumento de tamaño de la fosa craneal posterior y defecto del vermis del cerebelo, a través del cual se produce una comunicación con el cuarto ventrículo. De etiología desconocida, aunque se han propuesto varias causas como mendelianas, cromosómicas, ambientales y multifactoriales. La incidencia es de 1:25000 a 1:30000. La malformación de Dandy Walker tiene una alta mortalidad y una alta incidencia de impacto intelectual y neurológico Objetivo: Presentar un caso que presentaba diagnóstico prenatal, su atención al nacimiento y seguimiento neonatal. Descripción del caso: Paciente de 27 años, (g2 a1), de 38 semanas, que ingresa en trabajo de parto con diagnóstico prenatal presuntivo de síndrome de Dandy Walker, ventriculomegalia bilateral, fosa posterior aumentada de tamaño y vermis cerebeloso patológico. El nacimiento se realizó por cesárea, se llevaron a cabo estudios postnatales que llevaron a la confirmación del diagnóstico. Se incluyen imágenes de tomografía axial computada y seguimiento del recién nacido en su etapa neonatal Conclusión: El síndrome de Dandy Walker, es una patología poco frecuente, con una gran gama de alteraciones a nivel estructural en sistema nervioso central. El diagnóstico prenatal presenta una alta sensibilidad, se debe considerar siempre que se encuentre ecográficamente una fosa posterior aumentada de tamaño. P-40. SÍNDROME DE BANDA AMNIÓTICA: PRESENTACIÓN DE UN CASO María C. Zelaya, Ana L. Quaglio, Esteban Heredia, Mónica Rotella Hospital Eva Perón Banda de Rio Salí, Tucumán Objetivo: Presentar caso de Síndrome de Banda Amniótica/Limb body Wall, mostrar las dificultades en el diagnóstico prenatal de patologías de baja incidencia. Caso Clínico: Paciente de 21 años, G3P2, que consulta a las 22 semanas. Ecografía de 13 semanas describe alteraciones de la anatomía fetal: acrania, hernia diafragmática, onfalocele y pie bot. Se realiza ecografía que muestra acrania, defecto grande de pared abdominal, no se identifica diafragma, corazón hacia la parte anterior del tórax, asas intestinales en tórax, no es identifican pulmones, riñones, ni vejiga. Columna vertebral irregular, anomalías de miembros superiores. Fémur acortado, no es identifica cordón umbilical. No es posible hacer un diagnóstico sindromatico. A las 34,1 semanas RPM, dilatación cervical y presentación pelviana. En la ecografía se observa:, anencefalia, onfalocele gigante, ectopia cordis, columna irregular y se plantea un diagnóstico Pentalogia de Cantrell. Se realiza cesárea, extracción cefálica dificultosa con adherencias del polo cefálico a las membranas, RN femenino, 1050 grs, cordón muy corto. Fallece 49 minutos después de nacer. Cariotipo normal 46,XX El examen mostró defecto amplio de pared toracoabdominal, onfalocele roto con hígado e intestinos, el cordón umbilical adherido al lado derecho de la pared abdominal, encefalocele frontal, acrania, amputación de miembro superior izquierdo adherido a una brida que lo une con la fosa nasal izquierda, cifoescoliosis marcada, pie bot izquierdo, sindactilia en mano y pie derecho, pulmón derecho bilobulado. Diagnostico final: Síndrome de Banda amniótica. Discusión/ Conclusiones: La secuencia de Banda amniótica es poco frecuente (1 de cada 11.000 naciemientos en América Latina), de ocurrencia esporádica. Se presenta como un conjunto de malformaciones fetales asociadas a la presencia de bridas o bandas fibrosas que parecen atrapar o estrangular diferentes partes fetales ocasionando deformaciones, amputaciones y disrupciones. Es importante conocerla ya que puede presentar desde una afección nula o leve del feto a graves compromisos orgánicos e incompatibilidad con la vida extrauterina como en el caso presentado. Conocer esta patología ayuda al asesoramiento. La base del diagnóstico prenatal es la ecografía y del diagnóstico postnatal el examen físico. El cariotipo permite excluir patología de origen genético y brindar información más correcta en la planificación de embarazos posteriores. En nuestro caso el diagnostico no pudo completarse en la etapa prenatal pero en la postnatal se completó y se pudo llegar al diagnóstico incluyendo cariotipo por lo que a nuestra paciente se le brindó información completa y asesoramiento adecuado. P- 41. INDICACIONES DE ECOCARDIOGRAFÍA FETAL Héctor G. Gallardo, Sofía Grinenco Planificación Familiar, Centro de Diagnóstico Médico, Merlo, provincia de Buenos Aires Introducción: Las indicaciones de ecocardiografía fetalestán dadas por la detección de alteraciones en la valoración cardíaca en la ecografía obstétrica y, por la identificación de poblaciones de riesgo que presentan una mayor incidencia de cardiopatía congénita. El objetivo de este trabajo es la identificación de las indicaciones de ecocardiografía fetal, su frecuencia y su relación con la detección de cardiopatía congénita en pacientes atendidas en un centro de atención primaria. Material y Métodos: Estudio descriptivo retrospectivo. Se revisaron los registros clínicos de aquellas pacientes en quienes se realizó ecocardiograma doppler color fetal entre abril 2011 y marzo 2014. Se estudiaron las indicaciones de dichos estudios y su relación con la detección de cardiopatía congénita. Resultados: Se realizaron 256 ecocardiogramas fetales, en 48 (18.8%) de los cuales se constató cardiopatía congénita. En 73 casos (28.5%) la indicación fue la detección de alteraciones en la valoración del corazón fetal en la ecografía obstétrica. En 49 (19%) lo fue la edad materna mayor a 35 años, en 33 (12.9%) diabetes mellitus, en 22 arritmia fetal (8.6%), y 9 casos presentaron otros factores de riesgo (3.5%). En 70 casos (27.3%)el estudio fue solicitado “sin indicación”. En 42/48 (87.5%)de los fetos en quienes se diagnosticó cardiopatía congénita la indicación del estudio fue la detección de alteraciones en la valoración del corazón fetal en la ecografía obstétrica. Los 6 pacientes restantes presentaron factores de riesgo para cardiopatía congénita. Conclusiones: La detección de alteraciones en la valoración del corazón fetal en la ecografía obstétrica constituyó la indicación más frecuente y más efectiva en términos de detección de cardiopatía congénita en esta muestra. La solicitud de ecocardiogramas fetales “sin indicación “ constituye una práctica frecuente en nuestro medio, que no ha demostrado mejorar la tasa de detección de los defectos cardíacos.