- Ninguna Categoria
Interrelación entre elementos en cis
Anuncio
Universidad Nacional de San Martín Instituto de Investigaciones Biotecnológicas Regulación post-transcripcional en Trypanosoma cruzi: Interrelación entre elementos en cis y proteínas de unión a ARN. “Estudio funcional-estructural” Iván D’Orso Tesis presentada para optar por el título de Doctor en Biología Molecular y Biotecnología. Director: Dr. Alberto Carlos Frasch 2003 INDICE Página Resumen 3 Summary 5 Abreviaturas 9 Introducción 11 Objetivos y Antecedentes 45 Resultados 46 Estudio de la regulación de la expresión génica de TcSMUG -Parte I: 47 -Parte II: Identificación de secuencias de ARN en cis capaces de regular la expresión de TcSMUG 53 -Parte III: Identificación de proteínas que reconocen los elementos ARE y GRE 61 -Parte IV: Estructura y Función de proteínas de unión a ARN (TcUBPs 73 -Parte V: Análisis Estructural de las proteínas TcUBPs: Ensayos de Dinámica Molecular 97 Discusión 105 -Conclusiones 115 -Perpectivas futuras 116 Materiales y Métodos 117 -Anexo -Anexo -Anexo -Anexo I II III IV 139 140 141 142 Referencias 143 2 RESUMEN Las mucinas son O-glicoproteínas que participan en procesos de protección, lubricación y adhesión. En T. cruzi, existen dos grupos diferentes de mucinas que presentan patrones de expresión y características especiales (ver revisión en Di Noia, 2000). Una de esas familias, TcSMUG, está codificada por alrededor de 70 genes por genoma haploide y está compuesta por dos grupos denominados S y L, los cuales se expresan preferencialmente en el estadío replicativo epimastigote. Dado que las mucinas son importantes intermediarios en los mecanismos de adhesión celular y protección, el objetivo de esta tesis fue caracterizar el/los mecanismo(s) involucrados en la regulación de la expresión génica de TcSMUG durante el desarrollo del parásito. En primer lugar, se describió que la expresión de TcSMUG se regula a nivel post-transcripcional. Mientras que la tasa de transcripción de TcSMUG no varía entre los diferentes estadíos del parásito, los niveles de ARNms en el estado estacionario difieren entre las formas epimastigote y tripomastigote. Más aún, la vida media del ARNm para los grupos L y S de TcSMUG es > 6hr) en el estadío replicativo epimastigote y baja (t alta (t 1/2 < 0.5hr) en el estadío 1/2 infectivo tripomastigote. A partir de experimentos de transfección de parásitos en forma estable, se describen dos elementos funcionalmente diferentes, que están localizados en la región 3’ no codificante del grupo L y que regulan la estabilidad del ARNm TcSMUG durante el desarrollo del parásito. El primer elemento, denominado GRE por su alto contenido en residuos de Guanosina, está compuesto por 27 nt y confiere estabilidad al ARNm sólo en el estadío epimastigote. El segundo elemento denominado ARE (“AU-rich element”), está compuesto por 44 nt y funciona en la desestabilización del ARNm TcSMUG sólo en el estadío tripomastigote. Ambos elementos, GRE y ARE, son reconocidos específicamente en geles de retardo por proteínas de lisados totales de parásitos. Mientras que las proteínas que reconocen el elemento GRE se expresan en forma constitutiva durante el desarrollo, las proteínas AREBPs (“ARE-binding proteins”) se expresan en forma estadío específica y a partir de ensayos de entrecruzamiento con luz UV se determinó que presentan diferentes masas moleculares. La proteína que interacciona con el ARE en el estadío epimastigote (eAREBP), se purificó parcialmente y analizó por espectrometría de masa. Por comparación con la base de datos PROFound se identificó su posible identidad con la proteína de unión a ARN murina DEF3. Además, se clonó y caracterizó un grupo de proteínas de unión al ARE diferentes a las AREBPs, denominadas TcUBP-1 y TcUBP-2 (por “Uridine-binding protein”). Estas proteínas presentan un único dominio de unión a ARN del tipo RRM (“RNA-recognition motif”) y dominios auxiliares ricos en Glutamina y/o Glicina. Presentan la capacidad de homo-heterodimerizar a partir de residuos de Tirosina ubicados en la región C-terminal rica en Glicina. Las proteínas TcUBPs forman parte de un complejo ribonucleoproteico de ~450kDa, el cual también contiene la proteína de unión a poli(A) (TcPABP1). Experimentos “in vivo” demostraron que la proteína TcUBP-1 está involucrada en la desestabilización selectiva de ARNms conteniendo secuencias AREs, como los grupos L y S de TcSMUG, cuando se sobre-expresa en el estadío epimastigote. 3 A partir de ensayos de Resonancia Magnética Nuclear se estudiaron los mecanismos de reconocimiento del ARN por las proteínas TcUBPs. Por un lado, se utilizaron técnicas de resonancia-triple estándares con el objetivo de asignar residuos de cadena principal. Por otro lado, a partir de experimentos de titulación con ARN, monitoreados por espectros de correlación cuántica simple heteronuclear, se describieron los residuos del motivo RRM que podrían interaccionar con el ARN. No sólo se desplazan químicamente residuos de las hojas β1 y β3, sino que también existen perturbaciones considerables en las hojas β2 y β4. A partir de estos experimentos se compararon los residuos que sufren desplazamientos químicos en las proteínas con un dominio RRM, TcUBPs, y los residuos que interaccionan con ARN en proteínas con múltiples dominios de unión a ARN de otros tipos celulares. Palabras clave Trypanosoma cruzi Expresión génica Regulación post-transcripcional Estabilidad de ARNms Proteínas de unión a ARN Proteína de unión a poli(A) Resonancia Magnética Nuclear 4 Post-transcriptional regulation in Trypanosoma cruzi: Interplay between ciselements and RNA-binding proteins. “Functional and structural study”. SUMMARY. Mucins are O-glicoproteins that can function as a protective barrier, serve for lubrication and act as adhesion molecules. In the protozoan parasite T. cruzi, two different gene groups exists with special features and expression patterns (reviewed in Di Noia, 2000). One group, named TcSMUG, is composed by about 70 genes per haploid genome and is formed by two different groups, named L and S. Group S mRNA is present in the insect related stages. Besides, group L mRNA is present in all the stages, although is much more abundant in the replicative ones. Because of the great importance of mucin molecules in cell-adhesion and the immune system evasion response, the aim of this thesis is to characterize the mechanism(s) involved in the regulatory process of TcSMUG gene expression during parasite development. Firstly, TcSMUG mRNA family was described to be regulated at the post-transcriptional level. While the transcription rate of TcSMUG did not change during parasite development, mRNA steady-state levels change drastically upon differentiation. Moreover, the half-life for the L and S groups of TcSMUG > 6 hr) in the epimastigote stage, whereas they are short-lived in the is higher (t 1/2 trypomastigote stage (t < 0.5 hr). In order to determine the presence of cis-elements 1/2 involved in the stabilization process, stable parasite transfection experiments were performed. Two functionally different elements were identified, localized in the 3’untranslated region (UTR) of TcSMUG-L group. The first element was named GRE, since it is a G-rich element composed by 27 nt and it is involved in confer mRNA stability only in the epimastigote stage. The second element was named ARE, since its resemblance with AU-rich elements of mammalian cells. It is composed by 44 nt and is involved in TcSMUG mRNA decay in the trypomastigote stage. Both elements, GRE and ARE, are specifically recognized in gel shift experiments, by proteins of total parasite lisates and were named GREBPs and AREBPs, respectively. While GREBPs are constitutively expressed during parasite development, AREBPs are expressed with a stage-specific pattern. By UV cross-linking assays it was determined that AREBPs from the different stages present distinct apparent molecular masses. The epimastigote stage specific protein eAREBP was partially purified and analyzed by MALDI-TOF mass-spectrometry. By comparison with the PROFound database, we showed its identity with murin RNA-binding protein DEF3. Two different RNA-binding proteins that recognize the ARE sequence were cloned and characterized and named TcUBP-1 and TcUBP-2, for T. cruzi Uridine-binding protein. TcUBPs proteins present a common single RNA-recognition motif (RRM) and auxiliary domains rich in Gln and/or Gly. They can homo-heterodimerize in vitro and in vivo by Tyr residues present within the C-terminal Gly-rich region. TcUBP-1 and TcUBP-2 proteins form part of a ribonucleoprotein complex of ~450kDa, which also contains the poly(A)binding protein, TcPABP1. TcUBP-1 protein was involved in selective destabilization of ARE-containing mRNAs in vivo, such as TcSMUG L and S, when over-expressed in the epimastigote stage of the parasite. Nuclear Magnetic Resonance experiments were used to study the mechanism of 5 RNA-recognition in TcUBPs proteins, since they are single RRM-type RNA-binding proteins. Triple-resonance standard techniques were used for the backbone assignment. In addition, RNA titration experiments monitored by heteronuclear simple quantum correlation (HSQC) were used to identify the residues in the RRM motif involved in RNA recognition. Residues within β1, β3 but also β2 and β4 sheets showed the highest chemical shifts upon RNA addition. All these information was used to compare the residues perturbed in single RRM-type RNA-binding proteins (TcUBPs) and compared with the ones that interact in multiple RRM-type proteins. Key words Trypanosoma cruzi Gene Expression Post-transcriptional regulation mRNA-stability RNA-binding protein poly(A)-binding protein Nuclear Magnetic Resonance 6 Para los que hacen las cosas simples en la vida, y siempre en positivo, Ileana,.......sin palabras. Mis padres Laura y Carlos, y mi hermana Vera. Gracias..... Carlos Frasch..... por “LA” oportunidad, confianza, apoyo, entusiasmo, su visión de la ciencia.......y por nunca un No!!!!, Daniel Sánchez, también por su apoyo desde los comienzos, allá en 1997, Kalle Gehring, por brindarme tal enorme oportunidad y abrir mi pequeña cabecita molecular a las bases de la espectroscopía......., Javier Di Noia, por la compañia y dirección en los primeros experimentos “pegajosos” con las mucinas, Javier De Gaudenzi, con quien nos “codeamos” durante mi última parte del Doctorado y escuchamos música por las mañanas, a veces del palo, y otras no tanto, Marcelo Guerin, por su buena onda, amistad y bioquímica; aunque las cosas no siempre fueron positivas....Gracias, A las distintas generaciones del lab-3 con quienes compartí muchos mates y tortas: Javier, Carlos, Alejandro, Laura, Gastón, Guido, Vanina, Julieta, Marcela, Roxi, Geo, Adriana, Laura II, Natalia A mi laboratorio en Canadá.....y la enorme compañía y amistad que encontré: Pablito Gutierrez (por “Colombia”), Nadeem Siddiqui (“up Canadien”), Laurent “le grand”, Jean-Francois, Tara y Marta, A Joaquin, Charlie, Rafa y Juampi, por las escaladas a la montaña que nos permitieron olvidarnos de la ciencia....al menos de forma pasajera, A Fabio...por sus hermosas palabras cuando amablemente le pedía inocentes ratones y conejos, A Berta y Lili... por los no tan inocentes parásitos, A la asistencia indiscutida de Andrea Meras en todas las corridas cromatográficas en el HPLC, A la gente de otros labos, por la amistad, bicicleteadas, mates y “locuras” juntos: Cristian “el calabaza”, Augusto “o cabeça”, Ramiro, A Zulmita y Nelly...por esperarme tempranito con los mates “casi” todos los días, A todos los que supieron bancarme en las cortadas “sólo” y en la locura que la ciencia a veces arremete, A Roberto “Bob” Marley.......por su reggae y la atmósfera rastafari siempre en el ambiente positive, KuKi por traer al mundo la “cosita” más hermosa del planeta, Ile...., A la vida....y su creador. “........Jah Rastafari” 8 Abreviaturas ADN ADNc ácido desoxirribonucleico ADN copia ARN ARNm ARNr ácido ribonucléico ARN mensajero ARN ribosomal ARNt cpm dNTP ARN transferencia cuentas por minuto mezcla de dATP, dTTP, dGTP y dCTP EDTA EST g ácido etilen-di-amino-tetra-acético del inglés “Expressed Sequence Tag” aceleración de la gravedad kpb min MOPS kilopares de bases minuto(s) ácido 3-(N-morfolino) propanosulfónico nt ºC K pb kDa PCR PM RT NC SDS PAGE ARNP ActD CHX PABP HnRNP ARE snARN nucleótidos grados centígrados grados Kelvin pares de bases kiloDalton reacción en cadena de la polimerasa peso molecular transcripción reversa No Codificante dodecilsulfato de sodio electroforesis en geles de poliacrilamida ARN-polimerasa Actinomicina D Cicloheximida Proteína de unión a poli(A) del inglés “heteronuclearRiboNucleoprotein” del inglés “AU-rich-element” del inglés “small nuclear ARNs” siARN SL NLS del inglés “small interfering ARNs” del inglés “spliced leader” del inglés “Nuclear Localization Signal” NES RMN HSQC del inglés “Nuclear Export Signal” Resonancia Magnética Nuclear del inglés “Heteronuclear Simple Quantum Correlation” NOE NOESY TOCSY del inglés “Nuclear Overhause Effect” del inglés “NOE SpectroscopY” del inglés “TOtal Correlation SpectroscopY” 9 Los resultados presentados en esta tesis fueron incluídos en los siguientes trabajos: The Trypanosoma cruzi mucin family is transcribed from hundreds of genes having hipervariable regions. (1998). Di Noia J.M., D’Orso I., Aslund L., Sánchez D.O. and Frasch A.C. J. Biol. Chem 273, 10843-10850 AU-rich elements in the 3’ UTR of a new mucin-type gene family of Trypanosoma cruzi confers mRNA instability and modulates translation efficiency. (2000) Di Noia, J.M., D’Orso, I., Sánchez, D.O. and Frasch, A.C. J. Biol. Chem. 275, 10218-10227. Functionally different AU- and G-rich cis-elements confer developmentally regulated mRNA stability in Trypanosoma cruzi by interaction with specific RNA-binding proteins. (2001) Iván D’Orso and Alberto C. Frasch. J. Biol. Chem. 276, 15783-15793. TcUBP-1, a developmentally regulated U-rich RNA-binding protein involved in selective mRNA destabilization in trypanosomes. (2001) Iván D’Orso and Alberto C. Frasch. J. Biol. Chem. 276, 34801-34809. TcUBP-1, a mRNA destabilizing factor from trypanosomes, homodimerizes and interacts with a novel AU-rich element and poly(A)-binding proteins forming a ribonucleoprotein complex. (2002) Iván D’Orso and Alberto C. Frasch. J. Biol. Chem, 277, 50520-50528. The Trypanosoma cruzi mucin coat: Structure, Regulation of the expression and relevance in the host-parasite relationship. (2002) Javier Di Noia, Iván D’Orso and Alberto C. Frasch. In: Molecular Mechanisms in the Pathogenesis of Chagas Disease, Chapter 3. edited by John M. Kelly. Landes Bioscience/Eurekah. RNA-binding proteins and mRNA turnover in trypanosomes. Iván D’Orso, Javier G. De Gaudenzi and Alberto C. Frasch. TRENDS in Parasitology, en prensa. 10 “Todo lo difícil empieza siendo fácil, y toda cosa grande empieza siendo pequeña. El árbol que no pueden rodear los brazos de un hombre crece a partir de un tierno brote. La torre más alta, surge de un puñado de tierra. Un viaje de cien millas empieza a nuestros pies.....” Lao Tse INTRODUCCION Introducción 1- Trypanosoma cruzi y otros parásitos kinetoplástidos. Los protozoos son organismos eucariotas unicelulares primitivos, actualmente considerados un subreino dentro de los protistas. Trypanosoma cruzi se ubica dentro del “phylum” Sarcomastigophora, clase Zoomastigophora, orden Kinetoplastida, familia Trypanosomatidae, sección Estercoraria (Levine et al., 1980). Esta especie no es una población homogénea, sino está compuesta por un conjunto de cepas con diferentes características biológicas, bioquímicas e inmunológicas (Brener, 1973). Las evidencias indican que, a pesar de ser un organismo diploide, la recombinación sexual está por lo menos severamente restringida y se sugiere una estructura multiclonal para las poblaciones de T. cruzi (Tibayrenc et al., 1986). Dada la importante variación intraespecífica que existe en T. cruzi, es de relevancia médica y biológica el análisis de las particularidades de cada población ya que distintas poblaciones difieren en el tropismo por determinados tejidos, virulencia y susceptibilidad a drogas o mecanismos efectores del sistema inmune (Revollo et al., 1998). T. cruzi es un parásito digenético, es decir, posee dos hospedadores naturales: un insecto hematófago, que funciona como vector y un mamífero en el cual se desarrolla la patología. Los insectos pertenecen a la familia Reduvidae, cuya especie Triatoma infestans es la más común en Argentina. El rango de hospedadores mamíferos es amplio, incluyendo al hombre y animales salvajes y domésticos, que actúan como reservorios de la enfermedad (Brener, 1973). El parásito es ingerido por el insecto a partir de un mamífero infectado junto con la sangre que constituye su dieta y pasa por una serie de transformaciones morfológicas irreversibles a lo largo del tubo digestivo. En el intestino medio las formas ingeridas, denominadas tripomastigotes sanguíneos, se diferencian al estadío de epimastigote, que se divide activamente por fisión binaria. Al migrar a la ampolla rectal, los epimastigotes se adhieren al intestino, como condición indispensable para diferenciarse al siguiente estadío llamado tripomastigote metacíclico. La infección es iniciada en el vertebrado por el estadío tripomastigote. Este último, al ser eliminado junto con las heces del insecto durante la ingesta de sangre, penetra en el organismo por las mucosas y/o heridas en la piel. Este estadío no es capaz de dividirse. Por lo tanto, para multiplicarse penetra rápidamente a una célula y se diferencia al estadío replicativo. Para lograr su objetivo, debe atravesar la dermis y matriz extracelular y establecer una infección local. El parásito, escapa del fagolisosoma en que penetra hacia el citoplasma utilizando factores hemolíticos activos a bajo pH (Andrews et al., 1990), en donde se diferencia al estadío de amastigote (Tomlinson et al., 1995). El amastigote se divide intracelularmente por fisión binaria y posteriormente se diferencia a tripomastigote sanguíneo. Este estadío, incapaz de dividirse, es liberado al lisarse la célula huésped, y de esta manera, llega a la sangre donde circula por un período variable antes de invadir otras células, o ser ingerido por el vector, en donde se diferencia al estadío epimastigote, para cerrar el ciclo de vida (Figura 1). La magnitud de la parasitemia diferencia dos etapas de la infección. Durante un período corto posterior a la infección, la parasitemia aumenta hasta llegar a un máximo. La mayoría de los individuos infectados, logran controlar la infección después de 8 a 10 semanas y la parasitemia disminuye hasta hacerse indetectable por análisis microscópicos. Este punto determina el límite entre las fases agudas y crónica de la 12 Introducción infección, que puede prolongarse por años en forma asintomática hasta causar las patologías asociadas a la enfermedad. Durante la fase crónica, el parásito persiste en forma intracelular en regiones determinadas como por ejemplo el bazo, corazón, cólon o nervios periféricos según el tropismo de la cepa, y se cree que en este punto la respuesta inmune humoral del mamífero es capaz de destruir rápidamente a los parásitos liberados al torrente sanguíneo, principalmente por anticuerpos líticos dirigidos contra el epitope Gal(α 1-3) presente en mucinas de la membrana del parásito (Almeida et al., 1994; Gazzinelli et al., 1991). Sin embargo, la respuesta celular no logra montarse en forma adecuada o suficiente para eliminar al parásito y por lo tanto se establece una infección crónica. Figura 1- Ciclo de vida del Trypanosoma cruzi Otras dos especies de parásitos kinetoplástidos relacionados con T. cruzi, son T. brucei y Leishmania spp. Ambos son parásitos digenéticos, pero el primero de ellos no presenta estadío intracelular dentro del hospedador mamífero. En T. brucei, a pesar de que existen varios estadíos reconocidos como intermediarios, podemos mencionar a las formas más diferentes y de mayor importancia. En primer lugar, la forma procíclica presente en la glándula salival del insecto vector tsetse (Glossina sp.) y en segundo lugar, la forma sanguínea presente en el hospedador mamífero y es la que establece la infeccion crónica. En cuanto a Leishmania spp. su ciclo de vida consta de dos estadíos bien diferentes, una forma replicativa no infectiva, el promastigote, y una forma no-replicativa pero infectiva para el hospedador mamífero, el amastigote intracelular (Sacks and Sher, 2002). 1.1-Proteínas tipo-mucinas, O-glicoproteínas de superficie del T. cruzi. Las mucinas, son moléculas altamente glicosiladas, en las cuales los carbohidratos representan alrededor del 80% de la masa molecular. El núcleo proteico está compuesto por residuos de Serina/Treonina y también Prolina. El agente etiológico del mal de Chagas presenta una gran cantidad de genes que codifican para proteínas tipo-mucina. Estas 13 Introducción proteínas han sido muy estudiadas debido a su rol como aceptores de ácido siálico (Schenkman et al., 1993) en la reacción catalizada por la trans-sialidasa (Frasch, 1994; Schenkman et al., 1994). La actividad enzimática de la trans-sialidasa es única y parece haber sido seleccionada durante la evolución para solucionar la falta de la síntesis “de novo” de ácido siálico en T. cruzi (Schauer et al., 1983). En el laboratorio del Dr. Frasch, se describieron tres grupos de genes de mucinas de T. cruzi, los cuales pertenecen a la familia denominada TcMUC, por “T. cruzi Mucins” (Di Noia et al., 1998; Di Noia et al., 1995). La familia de genes TcMUC se parece a los genes que codifican las mucinas de los mamíferos (Di Noia et al., 1995), y está compuesta por 500-700 miembros por genoma haploide (Aguero et al., 2000; Di Noia et al., 1998). Esta familia se divide en tres subgrupos los cuales presentan divergencias en las regiones centrales, aunque se caracterizan por presentar un alto contenido de residuos de Serina/ Treonina (Di Noia et al., 1998). A diferencia de la región central, las regiones N- y Cterminales, las cuales codifican para un péptido de señalización a retículo endoplásmico y una señal de anclaje a la membrana por glicofosfatidilinositol, respectivamente, están muy conservadas. Recientemente se ha clonado y caracterizado una segunda familia de genes de mucina, denominada TcSMUG, por “Small Mucin Genes” (Di Noia et al., 2000). El clonado de los genes de la familia TcSMUG permitió demostrar que existen dos subgrupos denominados L y S, en los cuales la principal diferencia reside en el tamaño de los ARNms por diferencias en el largo de la región 3’-no codificante (NC). Hasta el momento, no existe evidencia alguna, que indique como se regula la expresión de estas dos familias de genes de mucinas durante el desarrollo del parásito. 2-Regulación de la expresion génica. 2.1- Un compendio de mecanismos acoplados en eucariotas superiores. La finalización de la secuencia del genoma humano en el año 2001 (Lander et al., 2001) y como así también de otros organismos tanto procariotas como eucariotas, marcó una nueva era en la biología y medicina moderna. Sin embargo, la identificación de secuencias génicas sin la descripción de la función y regulación de los productos finales generados, es de valor limitado. Sabemos que los distintos tipos celulares que componen un organismo multicelular expresan productos diferentes y presentan, por ende, una identidad propia. De esta manera, la remarcable diversidad encontrada en la especialización celular se logra a través de un estricto control en la expresión y regulación génica. Una gran cantidad de trabajos recientes en el área de biología molecular y celular, permitieron revelar algo nunca evidenciado hasta el momento. Varias etapas o caminos regulatorios de la expresión de genes se encuentran acoplados (Orphanides and Reinberg, 2002). En la Figura 2 se muestra un esquéma donde se resumen los procesos de expresión génica conocidos en células eucariotas, desde la activación de reguladores transcripcionales hasta la síntesis de una proteína funcional. Los niveles de expresión de varios genes son regulados por factores transcripcionales que reconocen secuencias regulatorias de ADN situadas antes del sitio de inicio de la transcripción, excepto para genes controlados por la ARN polimerasa (ARNP) tipo III. El hecho de que más del 5% de 14 Introducción nuestros genes codifican factores de transcripción, demuestra la importancia de esta familia proteica (Tupler et al., 2001). Los factores transcripcionales, una vez activados por diferentes señales intra o extracelulares, se unen a los elementos regulatorios y a través de interacciones con otros componentes de la maquinaria transcripcional, promueven el acceso al ADN y facilitan el reclutamiento de la ARNP en el sitio de inicio de la transcripción. Luego de que la ARNP-II comienza la transcripción de genes que codifican proteínas, el ARN naciente es modificado por la adición de una estructura denominada “cap” en el extremo 5’-terminal (“capping”). Esta modificación sirve para proteger al ARN del ataque de las nucleasas y también como sitio de unión de factores involucrados en el transporte del ARNm del núcleo al citoplasma para su posterior traducción (Proudfoot et al., 2002). El proceso del “capping” también parece coordinar eventos transcripcionales primarios, regulando la transición entre el inicio transcripcional, durante el cual la ARNP-II comienza la síntesis de ARN y la elongación, en el cual la polimerasa se mueve en sentido 5’-3’. Una vez que la ARNP-II alcanza el fin del gen, la transcripción cesa (terminación) por el reconocimiento de secuencias y/o estructuras que impiden el paso de la maquinaria transcripcional. Este pre-ARNm luego se cliva y se poliadenila por agregado de una cola de poli(A) en el extremo 3’ (ver más adelante). Los procesos por los cuales la información génica se transfiere desde el ADN al ARN, transcripción, y desde el ARN a la proteína, traducción, están físicamente separados en las células eucariotas por una membrana que rodea el núcleo, a diferencia de lo que ocurre en procariotas donde ambos procesos están acoplados en forma temporal y espacial. En las células eucariotas los ARNms sintetizados en el núcleo deben ser transportados al citoplasma para su traducción. Gracias a proteínas que se localizan en los poros nucleares, el transporte de macromoléculas es regulado en forma bidireccional (ver revisión en Reed and Hurt, 2002). Este proceso de transporte es mediado por factores que reconocen secuencias en las moléculas del ARNm en el núcleo y lo trasladan al citoplasma. Por su parte, la traducción del ARNm para generar proteínas es llevada a cabo en complejos ribonucleoproteicos llamados ribosomas (Ramakrishnan, 2002) y es mecanísticamente análoga a la transcripción (ver secciones siguientes). Los polipéptidos ahí sintetizados en ribosomas, luego son plegados en forma correcta y modificados químicamente (Fersht and Daggett, 2002) para generar un producto final maduro activo y correctamente localizado (Figura 2). 2.2- Regulación de la expresión génica en tripanosomas. Todos los parásitos helmintos y protistas de importancia médica deben ajustarse a una amplia variedad de cambios en el medio ambiente (condiciones químicas y estrés) durante el pasaje de un hospedador a otro. Para sobrevivir en condiciones ambientales tan variables, estos organismos se protegen utilizando cubiertas proteicas y glico-proteicas. Como parte del proceso adaptativo pueden cambiar su metabolismo y los patrones de expresión de las moléculas esenciales para su supervivencia a partir de la regulación de diferentes etapas del control de la expresión génica. A lo largo de la introducción se presentará una breve comparación entre los procesos regulatorios en tripanosomas y eucariotas superiores. Los tripanosomas presentan rasgos 15 Introducción característicos en sus procesos regulatorios, entre los que cabe mencionar la producción de largas unidades policistrónicas durante la transcripción, el proceso de trans-“splicing” durante el procesamiento del pre-ARNm, su localización y traducción en el citoplasma. Traduccion exosoma ARE PABP e IF A PARN AAAAAAAAAAAA HuR PABP A AAA 4G ARE eIF4E hnR NPD A A PABP eIF m7G-cap 4G eIF4E m7G-cap AAAAAAAAAAAA Estabilizacion ARNm P AUB ARE Clivaje exonucleolitico 3'-5' "Decapping" PABP PABP eI F 4G Desestabilizacion ARNm eIF4E m7G-cap AUBP m7G-cap PABP PABP AAAAAAAAAAAAAAA ARE p15 TAP RAN-GDP XPO1 Citoplasma NPC Nucleo RAN-GTP hnRNPs Transporte de ARNm XPO1 NES AUBP m7G-cap PABP PABP AAAAAAAAAAAAAAA p15 TAP m7G-cap EJC "Splicing" AAAAAAAAAAAAAAA EJC cis-"splicing" procesamiento 3' hnRNPs m7G-cap In In Transcripcion-"splicing" acoplados ADN TBP ARNP-II TBP pre-ARNm "Capping" Decompactacion de la Cromatina Cromosomas condensados Figura 2 - Visión contemporanea de la regulación de la expresión génica nuclear en células eucariotas superiores. Estudios recientes sugieren que cada etapa de la regulación de la expresión génica, desde la transcripción a la traducción, forma parte de un proceso continuo. De acuerdo con esta visión , cada etapa regulatoria, está física y funcionalmente acoplada a la siguiente, asegurando de esta manera una transferencia eficiente de la información entre etapas (ver revisión en Orphanides and Reinberg, 2002). EJC, “Exon Junction complex”; NPC, “Nuclear Pore Complex; NES, “Nuclear export signal”; In, Intrón; hnRNPs, proteínas heteronucleares; AUBP, “AU-rich binding protein”. 16 Introducción 3- Comparación de los procesos regulatorios entre eucariotas superiores y tripanosomas. 3.1- Transcripción y origen de largas unidades policistrónicas en tripanosomas. La estructura genómica y los patrones de transcripción de los parásitos del orden Kinetoplastida son únicos en la evolución. En lugar de presentar un estricto control transcripcional a partir de un promotor por gen, muchos genes son controlados a partir de una misma región promotora; es decir, la transcripción se caracteriza por ser policistrónica (Vanhamme and Pays, 1995). Varios marcos abiertos de lectura separados por largas regiones intergénicas forman parte del mismo ARN todavía no maduro. Además, el arreglo de genes en el genoma de estos parásitos es reminiscente de los operones bacterianos, especialmente por el hecho de que los marcos abiertos de lectura generalmente no están interrumpidos por intrones. Se ha descripto una sola excepción en donde el gen que codifica para la poli(A)-polimerasa de T. cruzi y T. brucei (Mair et al., 2000) presenta un intrón que es eficientemente procesado “in vivo” (ver más adelante). Otro rasgo característico del genoma de estos organismos protozoarios es la distribución y orientación de los genes en el genoma, los cuales no presentan un patrón común. El cromosoma I de Leishmania major, por ejemplo, presenta ~285 kpb de largo. Existe en este cromosoma, una región de 1.6 kpb que no contiene ningún marco abierto de lectura. A su izquierda se encuentran 29 marcos abiertos de lectura todos orientados hacia el mismo telomero, y a la derecha del mismo y orientados hacia el telómero opuesto, hay 50 marcos abiertos de lectura (McDonagh et al., 2000; Myler et al., 1999). Según experimentos de transcripción y sensibilidad a la droga α-amanitina, se describió que esta región de 1.6 kpb contiene un promotor para ARNP-II. Otro ejemplo interesante, es la presencia de unidades transcripcionales sentido y antisentido solapadas en el genoma de T. brucei (Liniger et al., 2001), las cuales pueden servir para mecanismos de regulacion de la expresión a partir de la interferencia por ARN (ver más adelante). Estas unidades transcripcionales provienen de un simple locus cromosómico y la transcripción se extiende hasta más de 10 kpb en forma estadío específica. Más interesante aún, es que en los genomas de los tripanosomas, la frecuencia de unidades transcripcionales antisentido es alta. De esta manera, el hecho de que ARNms estables puedan provenir de las diferentes hebras del ADN, permite hipotetizar que el potencial codificante del genoma puede ser mayor al que se describió previamente (Liniger et al., 2001). Recientemente, se demostró que las tasas transcripcionales de la ARNP-I y ARNP-II, disminuyen drásticamente cuando T. cruzi se diferencia de las formas replicativas no infectivas (epimastigotes y amastigotes) a las formas infectivas no-replicativas (tripomastigotes). Estos cambios transcripcionales parecen estar acoplados a cambios en la estructura nuclear y al remodelamiento de la cromatina (Elias et al., 2001) (ver más adelante). 3.1.1- ARN polimerasas en tripanosomas y su rol en la transcripción. Se han descripto tres tipos de ARNP a partir de la resistencia a la droga α-amanitina, 17 Introducción ARNP-I, -II y -III. La subunidad grande de cada una de las ARNP fue caracterizada por el clonado de su gen (Evers et al., 1989; Jess et al., 1989; Kock et al., 1988). De todas, la ARNP-I fue la más estudiada en tripanosomas. En primer lugar, se sabe que tiene la capacidad de transcribir no sólo genes ribosomales sino también genes que codifican proteínas como los mayores antígenos de superficie del estadío sanguineo infectivo de T. brucei, conocidos como VSG (“Variable Surface Antigen”). En segundo lugar, también sintetizan los ARNms del mayor antígeno de superficie del estadío asociado al insecto vector en T. brucei, conocido como PARP (“Procyclic Acid Repetitive Protein”) y compuesto por dos grupos, EP y GPEET. Una razón por la cual esta ARNP es utilizada por los tripanosomas, puede estar relacionada con su alta eficiencia transcripcional (Vanhamme and Pays, 1995). Finalmente, también se describió que la ARNP-I, es resistente a la droga α-amanitina (Clayton et al., 1990) y su localización intracelular demuestra que está presente alrededor del nucleolo y co-localiza con transcriptos nacientes de la PARP (Lee and Van der Ploeg, 1997). Los promotores para ARNP-I se caracterizaron usando ensayos de “run-on”, ensayos de extensión del cebador y transfecciones con plásmidos conteniendo genes reporteros bajo el control de las secuencias de ADN. A partir de estos ensayos se demostró que la estructura de los promotores para ARNr, VSG y EP se asemejan a los promotores para ARNr descriptos en eucariotas superiores (Clayton, 2002). Además, en ensayos de transcripción “in vitro”, ambos tipos de promotores compiten por los mismos factores celulares (Laufer et al., 1999). Por otro lado, el gen que codifica el factor “TATA-binding protein”, está en la base de datos de Leishmania; por lo tanto, otros factores basales de la transcripción pueden estar presentes en el genoma de tripanosomas (Clayton, 2002). En lo que respecta a la ARNP-III en kinetoplástidos, se sabe que transcribe los U ARNs además de los ARN de transferencia (ARNt). Los promotores para la ARNP-III se mapearon en detalle a partir del uso de sistemas de transcripción “in vitro” y transfecciones transientes de plásmidos diseñados para la producción de ARNs específicos. La transcripción de U ARNs depende de dos regiones A y B ubicadas adelante de los genes para ARNt (Nakaar et al., 1997). Otros ARNs pequeños, los “small nuclear” ARNs o snARNs, son transcriptos por una ARNP-II como parte de una unidad transcripcional única (Xu et al., 2001b). En varias especies de tripanosomas, se han descripto dos genes muy similares que codifican para la ARNP-II. La subunidad grande de esta holoenzima carece de las típicas repeticiones hexapeptídicas ricas en Serina, aunque de todas maneras tiene un alto contenido en Serina y es fosforilada como en otros organismos eucariotas (Smith et al., 1989). En lo que respecta a los promotores tipo II, las pruebas no son concluyentes. Varios reportes describieron la presencia de secuencias promotoras putativas, las cuales han sido mapeadas por ensayos de extensión del cebador y transfecciones transientes (Downey and Donelson, 1999; McAndrew et al., 1998), aunque aún no se han confirmado. El primer promotor identificado para ARNP-II fue descripto recientemente en el gen para el “spliced leader” (SL) de Leptomonas, y presenta un bloque variable con la secuencia CYAC/AYR(+1) (Gilinger and Bellofatto, 2001). Este elemento, así como dos elementos promotores basales adicionales, divergen de los elementos típicos transcripcionales descriptos en eucariotas superiores (ver revisión en Lewin, 1997). El precursor para el 18 Introducción ARN del SL en otros organismos presenta promotores claramente identificados y con una pequeña secuencia iniciadora (Luo et al., 1999). A partir de experimentos de retardo en gel, se caracterizaron varios factores que reconocen el promotor del SL (Gilinger and Bellofatto, 2001; Matkin et al., 2001). Un elemento, denominado PBP-1E, y localizado entre -70 y -60 pbs antes del sitio de inicio de la transcripción es requerido para una eficiente transcripción del ARN del SL. Recientemente se demostró que existe un complejo, compuesto por tres proteínas, que reconoce el elemento PBP-1E (Das and Bellofatto, 2003). Una de estas proteínas es ortóloga a la subunidad (SNAP)50 de las células humanas, involucrada en la regulación de la transcripción de snARNs (Das and Bellofatto, 2003). 3.2- Posicionamiento de la Cromatina. 3.2.1- Su rol en el control transcripcional en eucariotas superiores. El ADN en las células eucariotas se encuentra empaquetado en una estructura nucleoproteica compacta y altamente organizada llamada cromatina. La unidad de organización básica de la cromatina es el nucleosoma, el cual consiste de 146 pbs de ADN envuelta, generalmente 2 veces alrededor de un núcleo proteico que contiene dos copias de histonas del tipo H2A, H2B, H3 y H4 (Luger et al., 1997). Estas pequeñas proteínas presentan una alta carga positiva y están conservadas durante la evolución. Aunque anteriormente se pensaba que la cromatina formaba una estructura estática para el ADN, actualmente se sabe que cumple un rol principal en la regulación de la transcripción génica (Narlikar et al., 2002). Las regiones del genoma que no se transcriben están empaquetadas en una cromatina altamente condensada llamada “heterocromatina”, mientras que la región que presenta unidades transcripcionales se denomina “eucromatina” (Richards and Elgin, 2002). Cada tipo celular empaqueta sus genes con un patrón único de heterocromatina y eucromatina, y este patrón se mantiene también durante la división celular. El patrón de empaquetamiento depende de que genes serán activos en una nueva división celular, asegurando de esta manera las características únicas de cada linaje celular. Para la activación de la expresión génica los factores transcripcionales deben, por lo tanto, abrirse camino en estructuras de cromatina de difícil acceso al ADN. Esto es generado por eventos que conllevan a un aumento en la accesibilidad al ADN (Orphanides and Reinberg, 2002). 3.2.2- Cromatina y transcripción en tripanosomas. En tripanosomas se demostró que existen diferentes tasas de inicio de la transcripción mediadas por ARNP-I y ARNP-II durante las distintas etapas del ciclo de vida del parásito. Además, se observó que estos cambios transcripcionales ocurren en paralelo con modificaciones de la estructura nuclear. Los núcleos de las formas replicativas (epimastigote y amastigote intracelular) son redondeados, contienen pequeñas cantidades de “heterocromatina” periférica y un nucleolo grande. En cambio, los núcleos de las formas infectivas (tripomastigote) son alargados, el nucleolo desaparece y la “heterocromatina” ocupa la mayoría del compartimento nuclear. La disminución de la tasa transcripcional cuando el parásito se diferencia de la forma no-infectiva a la infectiva, se correlaciona con la ruptura del nucleolo (Elias et al., 2001). Sin embargo, hasta el 19 Introducción momento no han sido descriptos factores que regulen el ensamblado y la accesibilidad a la cromatina. No sólo existen diferencias estructurales entre la cromatina de T. cruzi y la de eucariotas superiores, sino también entre los diferentes estadíos del ciclo de vida de este organismo. Dentro del estadío epimastigote, las diferencias existen en las formas de fase exponencial y estacionaria. La cromatina de la forma epimastigote en la fase exponencial está más compactada de acuerdo a ensayos de digestión con ADNasa I (Spadiliero et al., 2002b), mientras que en las formas de fase estacionaria la cromatina se encuentra desplegada extendiéndose hacia el centro del núcleo. Estas observaciones sugieren que, en comparación con células eucariotas superiores, en T. cruzi la cubierta nuclear juega un papel principal e inusual en la interfase y mitosis del ciclo celular (Spadiliero et al., 2002a). Recientemente se describió que en T. cruzi los cromosomas se mueven y, después de entrar en la fase S, se localizan en la periferia nuclear, lugar en donde ocurre la replicación. Una vez finalizada la replicación se observan nuevamente en el interior del núcleo (Elias et al., 2002). En T. brucei se conoce que por mecanismos de variación antigénica (Borst, 2002; Vanhamme et al., 2001), se expresa por vez un miembro de la familia multigénica VSG en un sitio de expresión telomérico. En la forma procíclica la VSG no se expresa, pero sí en la forma infectiva sanguínea. Un estudio de accesibilidad transcripcional con la ARNP del bacteriófago T7, demostró que la transcripción está reprimida solamente (en el sitio de expresión) en el estadío procíclico. Esto sugiere que la cromatina accesible (del sitio de expresión inactivo) en el estadío sanguineo es remodelada después de la diferenciación para producir una estructura que sea accesible a la ARNP. Descubrimientos recientes demuestran que el gen de la proteína VSG a expresar se localiza en un cuerpo extranucleolar el cual contiene la ARNP-I en un estado transcripcionalmente activo (Navarro and Gull, 2001). 3.3- “Splicing” de ARN. Cabe mencionar en esta sección que mientras las células eucariotas utilizan el proceso nuclear de cis-“splicing” como mecanismo de maduración de sus pre-ARNms, los tripanosomas se caracterizan por procesar los transcriptos policistrónicos a través del trans-“splicing” nuclear. 3.3.1- trans-“splicing” y poliadenilación: dos procesos acoplados en tripanosomas. Como se mencionó anteriormente, la transcripción en tripanosomas es policistrónica. Es decir, se generan unidades transcripcionales largas, las cuales deben procesarse para la producción de ARNms maduros. En teoría, este procesamiento puede lograrse por una simple reacción de clivaje en la molécula precursora de ARN. Sin embargo, en la práctica se requieren dos reacciones de procesamiento las cuales se determinó que están acopladas: (1) trans-“splicing” en el extremo 5’ terminal y (2) poliadenilación en el extremo terminal 3’ del ARNm (Figura 3) (Matthews et al., 1994; Ullu et al., 1993). La señal para estos procesos se encuentra adelante del sitio 5’-aceptor de “splicing” (señal AG), el cual normalmente contiene un tracto de polipirimidina (ver Figura 3). Esta 20 Introducción secuencia señaliza el trans-“splicing” de la secuencia del SL al extremo 5’-terminal del ARN, compuesto por el dinucleótido AG. La poliadenilación del pre-ARNm precedente está espacial y temporalmente acoplada al “splicing”, y por lo tanto, se piensa que también está mecanísticamente acoplada. La poliadenilación ocurre, generalmente, entre 100400 nt adelante del sitio 5’ aceptor de “splicing”, dependiendo de las especies (LeBowitz et al., 1993; Matthews et al., 1994). orf-1 ag orf-2 PA ag pPy orf-3 PA ag pPy pre-ARNm policistronico SL nativo orf-1 ag poli(A) SL orf-2 poli(A) SL ag orf-3 poli(A) ag ARNms maduros Figura 3 - Procesamiento de policistrones de ARN en tripanosomas. El acoplamiento de procesos regulatorios también ocurre en tripanosomas y primariamente fue descripto en el procesamiento de policistrones, en donde existe un acoplamiento entre el trans-“splicing” y la poliadenilación como procesos de maduración del pre-ARNm. Tan rápido como son sintetizadas, las unidades policistrónicas son cortadas en sitios específicos, por la supuesta acción de una maquinaria que reconoce tractos de polipirimidina (pPy) en las regiones intergénicas. AG, sitio aceptor 5’ de “splicing”; SL, “spliced leader”; pA, sitio de corte y agregado de la cola de poli(A). orf, marco abierto de lectura. Durante el trans-“splicing”, el ARNm sufre un proceso de “capping”, a partir del cual se transfiere el SL más el “cap” a todos los extremos 5’-terminales de los pre-ARNms. Se sabe que en las células eucariotas la ubicua estructura del “cap” de los ARNms y de los precursores de los snARNs, es el resultado de modificaciones químicas esenciales que ocurren durante la elongación transcripcional. En los tripanosomas, sin embargo, la estructura del “cap” está restringida al ARN del SL y también a los precursores de los snARNs U2, U3 y U4 (Silva et al., 1998). Se sabe que el “cap-SL” es una de las estructuras más elaboradas de la naturaleza y consiste de un residuo 7mGpppG seguido de 4 nucleótidos metilados. En 1998, se caracterizó y purificó la enzima guanililtransferasa de Chritidia fasciculata, encargada de la transferencia de GMP desde el GTP, al extremo difosfato del ARNm. Esta enzima tiene un C-terminal con motivos similares a los descriptos en levaduras y un N-terminal que no presenta homología aparente con otras proteínas involucradas en la generación del “cap” (Silva et al., 1998). Por homología de secuencias se observó que esta enzima también está conservada en otras especies de parásitos kinetoplástidos incluyendo T. brucei (Silva et al., 1998). Por el momento no hay evidencias firmes que indiquen que el trans-“splicing” pueda estar involucrado en la regulación de la expresión de genes durante el desarrollo, salvo el ejemplo de genes acoplados al sitio de expresión de las VSG (Vanhamme et al., 1999). Sin embargo, lo que sí se sabe es que existen sitios 5’-aceptores del “splicing” que son más eficientes que otros. Por ejemplo, los sitios 5’-aceptores de los genes que codifican 21 Introducción las proteínas ribosomales TcP2β son potencialmente fuerte (Vázquez et al., 1994), en comparación con el del ARNm fosfoglicertato-quinasa-a (pgka) en T. brucei (Kapotas and Bellofatto, 1993) y tuzina en T. cruzi (Teixeira et al., 1999). En cuanto a la región 5’-no codificante (NC) de algunos transcriptos de α-tubulina en T. brucei, se describió que contienen elementos que pueden servir como regiones exónicas que modularían la tasa de trans-“splicing” (Lopez-Estrano et al., 1998). En el laboratorio del Dr. Levin, fue de gran interés el estudio de un retrotransposón llamado SIRE por “Short interspersed repetitive element”, el cual puede actuar como sitio 3’-aceptor de “splicing” (Vazquez et al., 1994) y además, como sitio aceptor de poliadenilación (Vazquez et al., 2000). Aproximadamente 1500 copias de SIRE están distribuidas en todos los cromosomas del clon Cl Brener y asociado a una gran variedad de genes (Vazquez et al., 1999). En resúmen, no sólo las regiones intergénicas ubicadas entre los sitios aceptores de trans-“splicing” y de poliadenilación son importantes en la regulación del “splicing”, sino también existen secuencias reguladoras en las regiones no codificantes que pueden actuar como reguladores positivos o negativos. De esta manera, el control de la maduración y el procesamiento del pre-ARNm puede ocurrir en varias etapas diferentes, acopladas y a nivel post-transcripcional. 3.3.2- cis-“splicing” en tripanosomas. A diferencia de lo que ocurre en la mayoría de los organismos eucariotas, el genoma de los tripanosomas carece prácticamente de intrones. Sólo un ejemplo de intrón fue descripto hasta el momento, el cual parece ser la excepción a la regla. El gen que codifica la poli(A)-polimerasa de T. cruzi y T. brucei presenta un intrón de 302 y 653 pbs, respectivamente (Mair et al., 2000). Este intrón presenta las secuencias consenso que funcionan como sitio aceptor 5’ y sitio dador 3’ en la reacción de cis-“splicing” (Mair et al., 2000), y puede ser removido en forma eficiente “in vivo”. Sorprendentemente, este ARNm también puede sufrir trans-“splicing” y el sitio aceptor para cis-“splicing” puede también funcionar como sitio aceptor en trans. No se ha determinado aún si este proceso puede tener una función regulatoria en la actividad polimerasa de la proteína. 3.4- Edición mitocondrial de pre-ARNms en tripanosomas. En el año 1986 se demostró por primera vez que en la mitocondria de los tripanosomas ocurre un proceso post-transcripcional relacionado con la inserción y/o deleción de residuos de Uridina en 12 de 18 transcriptos analizados (Benne et al., 1986). Más adelante, se determinó que el genoma mitocondrial está compuesto por maxicírculos, los cuales codifican ARNms mitocondriales no funcionales, y minicírculos, los cuales codifican pequeños ARNs guías (ARNg), que son quienes proveen la información genética requerida para la edición del ARNm. Las proteínas que forman la maquinaria de la edición del pre-ARNm, están codificadas en el genoma nuclear y son importadas a la mitocondria (ver revisión en Madison-Antenucci et al., 2002). Por lo tanto, esto demuestra que la edición del pre-ARNm necesita tanto de información mitocondrial como nuclear. 22 Introducción 3.5- Transporte de ARNms del núcleo al citoplasma. 3.5.1- Transporte de ARNms en eucariotas superiores. El transporte de ARNms está acoplado a pasos anteriores del control de la expresión génica, como son el “splicing” y eventos posteriores, como es el proceso de NMD (“Nonsense Mediated Decay”), el cual se discute en la sección siguiente. El complejo del poro nuclear (NPC) contiene nucleoporinas que forman un canal central a través del cual el cargo o molécula sustrato es transportada. Este canal tiene un diámetro de ~9 nm y puede expandirse hasta ~26 nm durante el transporte activo de moléculas. Además de las nucleoporinas, contiene un “basket” o canasta nuclear, anillos nucleares y citoplasmáticos y un complejo central llamado “spoke” o radio (Vasu and Forbes, 2001). El transporte del cargo, requiere miembros de una familia de proteínas llamadas karioferinas, que incluye a las exportinas e importinas. Estos receptores interaccionan con proteínas cargo vía señales específicas de localización, conocidas como NLS por “Nuclear Localization Signal” y NES por “Nuclear Export Signal”. Ambas karioferinas requieren para el transporte otra molécula llamada RanGTPasa, que es la que regula la interacción entre el cargo y el receptor. Es así como se forma un complejo ternario Cargo-Karioferina-RanGTPasa, el cual cumple un papel regulador en el transporte de moléculas. Para el importe nuclear, el cargo y la importina se translocan al núcleo y luego se libera el cargo del receptor por unión del RanGTP. En cambio para el exporte nuclear, el cargo y la exportina se unen cooperativamente a RanGTP. Este complejo se trasloca al citoplasma y el cargo se libera con la concomitante hidrólisis de GTP en Ran. De esta manera, la direccionalidad del transporte está mantenida por la presencia de RanGTP en el núcleo y RanGDP en el citoplasma y la acción de una RanGTPasa específica (Reed and Hurt, 2002). El mejor receptor caracterizado hasta el momento es la proteína exportina-1 (CRM1/Xpo1), la cual se requiere para el transporte de proteínas y/o complejos ribonucleoproteicos desde el núcleo hacia el citoplasma. Este receptor se une directamente a secuencias ricas en Leu del tipo NES presentes en las moléculas cargo (Mattaj and Englmeier, 1998). La proteína CRM1/Xpo1 es capaz de transportar ARNms, como así también los precursores de los U snARNs (ver revisión en Macara, 2001). Los U snARNs son transportados como precursores al citoplasma, lugar en donde se ensamblan correctamente en partículas ribonucleoproteicas. El transporte de U snARNs depende de la estructura del “cap”, el complejo de unión al “cap” (CBC), CRM1/Xpo1 y Ran-GTP. Además, se requiere una molécula adaptadora denominada PHAX la cual interacciona con CRM1/Xpo1 y CBC (Ohno et al., 2000). El transporte de otras clases de ARNs, incluyendo ARNt, ARNr y snARN requiere otros miembros de la familia de las karioferinas (Nakielny and Dreyfuss, 1999). Existen proteínas de unión a ARN del tipo hnRNP “heteronuclear RiboNucleoProteins”, que pueden viajar entre el núcleo y el citoplasma. El hecho de que éstas proteínas se muevan entre los dos compartimentos, permitió proponer que además sirven para transportar ARNms. Estudios recientes relacionados con el transporte de ARNms, permitieron identificar una nueva serie de factores no relacionados con las karioferinas que forman una familia de proteínas conservadas, NXF o TAP (Herold et al., 2000). En metazoos, se sabe que TAP heterodimeriza con p15, y el hetero-dímero es 23 Introducción esencial para el transporte de ARNms a través del complejo del poro nuclear (Herold et al., 2001). Es interesante destacar que los componentes de la maquinaria de transporte de ARNm, están física y funcionalmente acoplados al “splicing”. Este descubrimiento permitió entender el acoplamiento que existe entre el “splicing” y NMD, un mecanismo de “vigilancia” utilizado para degradar ARNms que presentan codones prematuros de la traducción (ver sección siguiente). 3.5.2- Transporte nucleo-citoplasmático en tripanosomas. Es intersante destacar que estudios recientes permitieron describir la presencia de una estructura semejante al poro nuclear de eucariotas superiores en células de tripanosomas (Rout and Field, 2001). Además, se describió la presencia de proteínas con secuencias NLS activas, como la histona del tipo H2B y la proteína de unión a ARN La (Marchetti et al., 2000). Recientemente, la proteína exportina-1 (tCRM1/tXpo1) fue identificada en T. cruzi y T. brucei (D’Orso et al., manuscrito en preparación). Existen proteínas en T. cruzi que presentan secuencias NES putativas, las cuales podrían formar parte del camino de transporte de proteínas del núcleo al citoplasma mediado por tCRM1/ tXpo1. 3.6- Degradación de ARNms conteniendo codones prematuros. Acoplamiento entre “splicing”, transporte de ARNm y NMD en eucariotas superiores. Una vez que el pre-ARNm es procesado a través del “capping”, “splicing” y poliadenilación, se genera un ARNm maduro el cual está sujeto a nuevos pasos regulatorios. En primer lugar, existen secuencias específicas del ARNm las cuales son reconocidas por factores proteicos que determinan su destino final en términos de estabilidad, localización y/o modulación de la traducción. Todos estos pasos forman parte de lo que se llama control de la calidad de la función del ARNm (Maquat and Carmichael, 2001). Si el ARN presenta codones prematuros, o los procesos de “splicing” y/o poliadenilación ocurren en forma aberrante, el ARNm puede ser degradado. Parte de este último paso de control, está relacionado con la retención nuclear y con procesos de control antes de que el ARNm sea transportado al citoplasma. En tripanosomas, hasta el momento no hay resultados significativos en esta área de investigación. Sin embargo en otros tipos celulares, incluyendo levaduras y mamíferos, se conocen los mecanismos que regulan el control de la calidad del ARNm sintetizado. Estos se basan en el acoplamiento de los procesos de “splicing” y NMD (Figura 4), cuyas maquinarias están estrictamente interelacionadas (ver revisión en Dreyfuss et al., 2002). Como se mencionó anteriormente, el NMD elimina ARNms que presentan codones prematuros de la traducción (Ishigaki et al., 2001). En metazoos, se sabe que la presencia de codones prematuros, más de 50 nt adelante de la unión exón-exón, desencadena NMD en el citoplasma y está acoplado al “splicing” en el núcleo (Lykke-Andersen et al., 2000). El proceso de NMD se desencadena por la localización de la proteína hUpf3 después del codón prematuro de la traducción (Lykke-Andersen et al., 2000). 24 Introducción Y14 Aly Upf3 magoh TAP SRm160 Figura 4 - Acoplamiento entre “splicing” y NMD durante el procesamiento de pre-ARNms en células eucariotas. El complejo EJC (“Exon-Junction Complex”) se ensambla en los ARNms como resultado del “splicing”. La proteína TAP/ p15 se une a los componentes del EJC y media la interacción del complejo ribonucleoproteico con receptores del poro nuclear (NPC). Durante el transporte de ARNms, e inmediatamente más tarde, la composición del complejo se modifica, ya que varios de sus componentes se disocian dejando sólo a Y14, magoh, Upf3 y RNPS1, en la misma posición en la que el complejo se ensambló inicialmente en el núcleo. Upf2 probablemente se une a Upf3 luego del transporte al citoplasma (Dreyfuss et al., 2002). Este complejo controla el destino celular del ARNm. NPC, “Nuclear Pore Complex”; In, Intrón. Upf2 p15 Upf2 RNPS1 Citoplasma NPC Nucleo transporte ARNm TAP p15 EJC EJC Upf3 Aly magoh Y14 RNPS1 SRm160 splicing pre-ARNm In In Sin embargo, el mecanismo que recluta a hUpf3 en el ARNm se desconocía hasta hace un año. Recientemente, dos grupos de trabajo, el del Dr. G. Dreyfuss y el de la Dra. J. Steitz, encontraron en forma simultánea indicios que permiten explicar cómo procede este mecanismo (Figura 5). Existe un complejo llamado EJC por “Exon Junction Complex” que presenta varios componentes conservados entre NMD y la maquinaria de exporte de ARNm (Kim et al., 2001; Lykke-Andersen et al., 2001). Entre ellas, la proteína Y14 (Kataoka et al., 2000), el factor de “splicing” RNPS1 (Mayeda et al., 1999) y hUpf3 (Lykke-Andersen et al., 2000), las cuales pueden movilizarse entre el núcleo y el citoplasma. De esta manera, el complejo EJC funciona en ambos procesos (Figura 5), transmitiendo una marca en la unión exón-exón durante el transporte al citoplasma. Después del transporte del ARNm, otros factores relacionados con el NMD como hUpf1 y hUpf2, se reclutan a través de la interacción con hUpf3. Este complejo, produce la degradación del ARNm si la maquinaria de traducción encuentra el codón prematuro de la traducción. Esta degradación se desencadena por eliminación del “cap” sin deadenilación, seguido por degradación en sentido 5’-3’ mediado por la exoribonucleasa Xrn1. En ARNms normales, es decir sin codones prematuros para la terminación de la traducción, el complejo de NMD es desplazado por el ribosoma durante la traducción, y de esta manera se evita su degradación (Kim et al., 2001; Lykke-Andersen et al., 2001). También se encontró que los factores CBP80-CBP20, ambos componentes del complejo que unen el “cap” en forma co-transcripcional en el núcleo, están involucrados en el proceso de NMD. Ambos realizan una ronda pionera de traducción, después de ser reemplazados por el factor de traducción citoplasmático eIF4E, momento en que las proteínas hUpf2 y hUpf3 se disocian de la unión exón-exón (Ishigaki et al., 2001). De esta manera, el proceso de control nuclear NMD ocurre mientras los ARNms están unidos por las proteínas que reconocen el “cap” 25 Introducción y la proteína de unión a poli(A) (PABP) nuclear o PABP2 (Ishigaki et al., 2001). 3.7- Degradación nuclear y citoplasmática de ARNms y la maquinaria involucrada en células eucariotas superiores. En células de mamíferos se sabe que al menos del 5% de los enlaces fosfodiester nacientes correspondientes a transcriptos sintetizados por ARNP-II finalizan en ARNms citoplasmáticos maduros (Jackson et al., 2000). Resultados similares se observaron para ARNs sintetizados a partir de las ARNP-I y ARNP-III. Estos datos permiten concluir que el principal volúmen de recambio de ARN en las células de mamíferos ocurre en el núcleo en vez del citoplasma. De la misma manera que sucede con el recambio de proteínas, el recambio y la degradación del ARN intracelular debe estar muy regulado para evitar la eliminación aberrante de los transcriptos. Se sabe que la principal maquinaria de degradación intracelular de ARN es exonucleolítica, es decir, a partir de los extremos 5’ o 3’ del ARNm. Por lo tanto, las moléculas de ARN se protegen del ataque promiscuo, evitando exponer sus extremos terminales. El extremo 5’ de los ARNms se protege a partir de la estructura del “cap”, cuya estructura protege al ARNm a partir de proteínas que lo reconocen, como CBP-80 y CBP-20 en el núcleo de la célula (Ishigaki et al., 2001). Un ARNm puede ser degradado por deadenilación, desplazamiento de las proteínas unidas que lo estabilizan, ruptura de su estructura terciaria o clivaje endonucleolítico en sitios de reconocimiento internos ya no protegidos. Una vez que un extremo del ARN está accesible, existen exonucleasas que atacan el ARN en ambos sentidos 3’-5’ y 5’-3’ (Moore, 2002). La maquinaria que degrada el ARNm nuclear ha sido extensivamente caracterizada en levaduras, en donde los caminos de ataque exonucleolítico por deadenilación del transcripto son predominantes (Bousquet-Antonelli et al., 2000). La mayor parte de esta actividad exonucleolítica es provista por el exosoma (Mitchell and Tollervey, 2000; van Hoof and Parker, 1999). El exosoma, descubierto en el laboratorio del Dr. Tollervey, es un complejo de 11 proteínas de las cuales la mayoría son exonucleasas del tipo 3’-5’. Además, existen factores accesorios que incluyen ARN helicasas, una GTPasa y proteínas que trasladan o localizan al exosoma a una secuencia particular del ARN (Mitchell and Tollervey, 2000). Todas las subunidades del exosoma están conservadas en humanos (van Hoof and Parker, 1999). El exosoma también funciona en el citoplasma de la célula, donde es requerido para la degradación en sentido 3’-5’ de los ARNms deadenilados (van Hoof and Parker, 1999). La diferencia entre el complejo exosoma entre ambos compartimentos celulares reside en que el citoplasmático carece de una subunidad, la proteína Rrp6p, una ARNasa de tipo D con actividad 3’ exo-hidrolasa. Esta proteína es la única subunidad del exosoma no requerida para la viabilidad celular (Allmang et al., 1999). También hay evidencias sobre la existencia de un camino nuclear de degradación con sentido 5’-3’. La levadura S. cerevisiae contiene dos exonucleasas 5’-3’ conocidas como Rat1p y Xrn1p, las cuales están conservadas también en humanos. La primer proteína es esencial para el crecimiento. En cambio, Xrn1p aunque no es esencial, forma parte del camino más importante de degradación para ARNms deadenilados y del proceso de recambio NMD como se indicó en la sección anterior. Además se encontró que el 26 Introducción exosoma es atraído a ARNms de vida-media corta por proteínas que se unen a elementos desestabilizantes como los ARE (Chen et al., 2001) (ver más adelante). Esto sugiere un mecanismo paralelo para la atracción del exosoma a transcriptos que están destinados a sufrir NMD (Moore, 2002). Upf3 Upf2 SRm160 magoh Y14 RNPS1 ribosoma ter EJC EJC ter EJC eRFs ARNms normales Upf1 ter CPT ribosoma PROXIMA RONDA DE TRADUCCION EJC EJC EJC ARNms con codones prematuros traduccion INICIACION ter ter CPT EJC EJC eRFs DEGRADACION DEL ARNm "Deccaping" Upf1 ELONGACION Disociacion del EJC TERMINACION Vigilancia Figura 5 - Rol del EJC en la degradación de ARNms que contienen codones prematuros para la terminación de la traducción. En el caso de ARNms normales, los codones de terminación (ter) están generalmente río abajo del extremo 3’, es decir, ~55 nt antes de la última unión exón-exón. De esta manera, los ribosomas remueven el complejo entre Y14-Upf2-Upf3 (EJC, “Exon-Junction Complex”) al término de la traducción. Durante la terminación, Upf1 y el factor de liberación traduccional (eRF3) se reclutan y forman parte del complejo de vigilancia, el cual rastrea por posibles remanentes del complejo Y14-Upf. A menos que el complejo de vigilancia encuentre el complejo Y14-Upf, la traducción procede a una nueva ronda de síntesis. En caso de que el ARNm presente codones prematuros para el término de la traducción (CPT), localizados más de 55 nt antes de la última unión exón-exón, la traducción se frena antes de que el complejo Y14-Upf se remueva. El complejo Y14-Upf remanente interacciona con el complejo de vigilancia y desencadena el “decapping” del ARNm, generando su degradación. 3.8- Degradación de ARNms en tripanosomas. En los parásitos kinetoplástidos, más específicamente en T. brucei, también se encontró la maquinaria del exosoma, tanto en núcleo como en citoplasma, aunque ambos complejos son más pequeños que los de levadura, ya que están compuestos por 8 subunidades (Estévez et al., 2001). Los experimentos relacionados con el estudio de mecanismos de degradación de ARNms en kinetoplástidos han sido principalmente restringidos a observaciones relacionadas con la utilización de inhibidores de la transcripción y traducción (ver más adelante). Básicamente, permitieron determinar que la abundancia de transcriptos particulares es incrementada o disminuida por alguno de estos tratamientos (Brittingham et al., 2001; Graham and Barry, 1996; Teixeira et al., 1995). 27 Introducción 3.9- Interferencia por ARN (RNAi). 3.9.1- RNAi en células eucariotas superiores. Este mecanismo se refiere al silenciamiento génico en forma post-transcripcional a través del cual ARN doble cadena (ARNdc) gen específico, desencadena la degradación del ARNm celular homólogo. La interferencia por ARN es un proceso que está distribuído en todos los organismos eucariotas y está mecanísticamente relacionado al silenciamiento de genes en forma post-transcripcional en plantas (Kooter et al., 1999) y “quelling” en Neurospora (Cogoni and Macino, 1999). Es considerado un mecanismo de defensa contra transcriptos aberrantes los cuales pueden ser producidos por una infección viral o por movilización de transposones (Ketting et al., 1999). Los modelos actuales sugieren que después de la entrada a la célula del ARNdc, una nucleasa produce ARNs pequeños de entre 21-25 nt o “small interfering” ARNs (siARNs), los cuales sirven de secuencias guía para la degradación del ARN blanco (Zamore et al., 2000). Estudios “in vitro” permitieron demostrar que la adición de ARNdc sintético en un extracto de Drosophila, produce el clivaje del ARNm homólogo, generando ARNs pequeños con una periodicidad de corte entre 21-23 nt (Zamore et al., 2000). A partir de estudios de fraccionamiento bioquímico en extractos libre de células de Drosophila, se demostró que existe una actividad que degrada el ARNm blanco y que co-purifica con los siARNs. Recientemente, se identificó un candidato de la actividad ARNasa tipo-III, la cual se llamó Dicer por su habilidad por digerir ARNdc y generar fragmentos homogéneos en tamaño y equivalentes a los siARNs (Bernstein et al., 2001). Algunas proteínas homólogas a Dicer se encontraron en C. elegans y en células humanas (Grishok et al., 2001; Hutvagner et al., 2001). 3.9.2- RNAi en tripanosomas. En tripanosomas, el proceso de interferencia mediada por ARNdc se demostró en el año 1998 (Ngo et al., 1998). El ARN blanco para la degradación es citoplasmático ya que los pre-ARNms no son sustratos de la actividad de degradación. Además, se demostró por primera vez en el laboratorio de la Dra. E. Ullu, que la degradación puede ser desencadenada por ARNdc introducido por electroporación o ARNdc producido “in vivo” a partir de la transfección de un plásmido que contenga promotores opuestos a la secuencia a transcribir (Shi et al., 2000), o también inducido por “hairpins” de ARN transcriptos a partir de promotores inducidos con el sistema de tetraciclina (Bastin et al., 2000; Shi et al., 2000). Más recientemente en T. brucei, se demostró que la activación de la interferencia por ARN resulta en la producción de los siARNs y que el 10% de estos ARNs sedimentan en complejos de alto peso molecular (Djikeng et al., 2001). Se observó que algunos de estos siARNs provienen de la degradación de retro-transposones. Aunque todavía no se ha identificado y caracterizado la(s) proteína(s) que forman parte de este complejo en T. brucei, se especula que la interferencia por ARN en tripanosomas podría servir como un mecanismo de control constitutivo involucrado en el silenciamiento de transposones. 3.10- Control traduccional. 28 Introducción 3.10.1- Traducción en general. El inicio de la traducción en eucariotas comienza con la unión de factores iniciadores a la estructura del “cap”, seguido de la unión de la subunidad ribosomal pequeña y la lectura a partir del primer sitio de inicio de la traducción (ver revisión en Sachs et al., 1997). La presencia de una estructura secundaria larga en la región 5’-NC del ARNm puede impedir la lectura por el ribosoma y además, proteínas que reconocen secuencias en esa región, pueden evitar la traducción en forma efectiva. Está claramente establecido que la mayoría de los ARNms activos para la traducción, se encuentran circularizados debido a la interacción de la proteína PABP1 y el complejo de iniciación eIF4 en el extremo 5’ del ARNm (Dreyfuss et al., 2002). De alguna manera, esto explica porque la traducción es mucho más eficiente y capaz de amplificarse en ARNms que presentan una cola de poli(A) en su extremo 3’-terminal. Sin embargo, no está descripto hasta el momento si el tamaño de la cola del poli(A) regula la eficiencia de traducción. 3.10.2- Regulación a nivel traduccional en tripanosomas. Es probable que el control traduccional juegue un papel importante en la regulación de la expresión génica en tripanosomas; sin embargo, hay pocos resultados en esta área de investigación. La metodología se basa en la comparación de los niveles de ARNms en el estado estacionario, con los niveles o actividades enzimáticas de los productos proteicos finales en diferentes estadíos de desarrollo del parásito. De este modo, se evidenciaron algunas indicaciones de control traduccional. Entre ellas se encuentran como ejemplo las prociclinas EP del tripanosoma africano (Hotz et al., 1998). Por otro lado, los niveles del ARNm de la fructosa bifosfatasa aldolasa son 6 veces mayores en el estadío sanguíneo, sin embargo la proteína está expresada 30 veces más en ese estadío (Hotz et al., 1995). Resultados similares se encontraron durante el estudio de la regulación del gen que codifica el citocromo c de T. brucei, la proteína se expresa 100 veces más en el estadío procíclico, pero el ARNm sólo 3-5 veces más (Priest and Hajduk, 1994; Torri and Hajduk, 1988). De esta manera, los resultados relacionados con el control traduccional en estos organismos, sólo se desprenden de la comparación de los niveles relativos de los productos finales (ARNms y proteínas), durante las diferentes etapas del ciclo de vida del parásito. En tres especies de organismos kinetoplástidos diferentes se determinó que el largo de ambas regiones, 3’-NC y cola de poli(A), no tiene un efecto regulatorio en la traducción (ter Kuile and Salles, 2000). Pocas proteínas se identificaron como posible reguladores de la traducción. La proteína citoplasmática PABP1 en T. cruzi, T. brucei y Leishmania, la cual interacciona con la cola de poli(A) de los ARNms (Bates et al., 2000; Batista et al., 1994; Hotchkiss et al., 1999). Se describió un factor relacionado con la traducción, EF-1α (Billaut-Mulot et al., 1996), aunque ninguna función se asignó “in vivo”. Por último, se clonó un gen de T. cruzi llamado DnaJ que codifica la proteína TcJ6, la cual presenta identidad a la chaperona Sis-1 de S. cerevisiae (Salmon et al., 2001). Esta proteína está localizada en el retículo endoplásmico y se requiere para el inicio de la traducción. 3.10.3 -Función de la proteína PABP1 en la traducción en mamíferos. 29 Introducción La mayoría de los ARNms de eucariotas presentan una cola de poli(A) en su extremo 3’, la cual varía de 50 a 250 pbs en levaduras y eucariotas (Jacobson and Peltz, 1996). La poliadenilación se regula en estos organismos por la presencia de una típica secuencia en la región 3’-NC (5’-AAUAAA-3’), y de un elemento involucrado en la poliadenilación citoplasmática, llamado CPE (“citoplasmic polyadenylation element”). Ambas secuencias interaccionan con diferentes factores celulares. La proteína CPEB interacciona con CPE y recluta al factor de especificidad de poliadenilación y clivaje (CPSF), mientras que la proteína poli(A)-polimerasa, se encarga de la polimerización de la cola de poli(A) de todos los ARNms en el núcleo de la célula (Wahle and Ruegsegger, 1999). También se describió la presencia de una proteína del tipo PAP llamada Gld-2, la cual adiciona tractos de poli(A) a ARNms pero en el citoplasma de la célula (Wang et al., 2002). Una vez que la cola de poli(A) ha sido sintetizada, tanto en núcleo como en el citoplasma, esta porción no codificante del ARNm interacciona con la proteina PABP1. Paip2 eRF3 PABC PABC PABP1 PABP1 AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA PABP1 PABP1 eIF4G ARNm eIF4E 5'-Cap PABC PABC eIF4A Paip1 Figura 6 - Modelo de la interacción entre PABP1 y eIF4 y su rol en la traducción. PABP1 presenta 4 motivos RRM unidos al dominio PABC por una extensa región no plegada. Múltiples moléculas de PABP1 pueden interaccionar con la cola de poli(A) de un ARNm vía los motivos RRM para formar un complejo ARN-proteína. La circularización del ARNm ocurre a través de la unión con eIF4E unido al extremo 5’-terminal “cap” y los dominios RRM de PABP1. El dominio PABC une péptidos de secuencia lineal para reclutar factores proteicos al complejo ARN-proteína. Algunas de las moléculas que interaccionan son Paip1, Paip2 y el factor de liberación traduccional eRF3, que hacen de conectores para atraer a eIF4A y posiblemente otros factores no descriptos hasta el momento. Paip, “PABP-interacting-protein”. La PABP1 es una proteína citoplasmática abundante, involucrada en la regulación de la traducción (Figura 6). Estructuralmente está compuesta por dos porciones, una región N-terminal que contiene 4 motivos RRM (“RNA-recognition motif”) y una región Cterminal que contiene un dominio conservado en todas las proteínas de unión a poli(A), llamado PABC (“poly(A)-binding C-terminal), está compuesto por ~75 residuos (Kozlov et al., 2001). Con una periodicidad de 27 nt de tractos de Adenosina se une una proteína PABP1; por lo tanto, múltiples proteínas pueden unirse en la cola de poli(A) de un ARNm. Este fenómeno de multimerización es requerido para la traducción eficiente del ARNm (Tarun and Sachs, 1995). Además, la PABP1 es capaz de mediar la circularización del ARNm a través de su interacción con el complejo de iniciación de la traducción, eIF4E (Figura 7), el cual a su vez está asociado con la estructura del “cap” en el extremo 5’ (Tarun and Sachs, 1996). Este evento de circularización produce la amplificación de la traducción, ya que facilita el reciclado de los ribosomas (Jacobson and Peltz, 1996). De esta manera, PABP1 funciona como un “andamio” proteico, reclutando varios factores traduccionales en el ARNm. Además de la subunidad eIF4G, existen otras moléculas que interaccionan con PABP1, como Paip1, Paip2, y el factor de terminación traduccional 30 Introducción eRF3 (“eukaryotic Release Factor”). La interacción con estos factores regula positiva o negativamente la traducción (Craig et al., 1998; Khaleghpour et al., 2001). Además, PABP1 puede evitar la degradación del ARNm a partir de la cola de poli(A), inhibiendo indirectamente el ensamblado de una exonucleasa 3’-5’ (Ford et al., 1997) (ver Figura 7). 3.10.4 -Función del dominio C-terminal de PABP1 en la traducción. El extremo C-terminal de la PABP1 (PABC), no fue el centro de atención en comparación con la importancia dada al estudio de los motivos RRM. Sin embargo, durante los últimos años se demostró su función en estructurar la cola de poli(A) del ARNm para la traducción y en regular la localización celular de PABP1. Estas hipótesis fueron confirmadas por estudios que demostraron que PABC es importante para la multimerización proteica y para un correcto transporte núcleo-citoplasmático (Kuhn and Pieler, 1996; Afonina et al., 1998). El dominio PABC presenta una región altamente conservada (residuos 68-72), cuya identidad varía del 40 al 54% entre mamíferos, plantas, levaduras y tripanosomas (Kozlov et al., 2002). La estructura en solución del dominio PABC humano (hPABC), levaduras (yPABC) y T. cruzi (TcPABC), fue resuelta en el laboratorio del Dr. Kalle Gehring (Kozlov et al., 2002; Kozlov et al., 2001; Siddiqui et al., 2003). Las estructuras de hPABC y TcPABC presentan rasgos similares, con una estructura de α-hélice globular, mientras que yPABC carece de la primera de las 5 hélicesα que componen hPABC (Kozlov et al., 2002) y TcPABC (Siddiqui et al., 2003). Existe un gran número de proteínas conocidas que interaccionan con el dominio hPABC. Entre ellas, Pbp1p (Mangus et al., 1998), la cual promueve una correcta poliadenilación de los pre-ARNms; el factor de iniciación de la traducción eIF4G (Tarun et al., 1997); el factor de liberación de la traducción eRF3 (Hoshino et al., 1999); una proteína de mamíferos que muestra identidad con eIF4G, denominada Paip1, y Paip2 (Craig et al., 1998; Khaleghpour et al., 2001). El rango de función de las proteínas que interaccionan con PABP1 es bastante amplio, desde la formación del extremo 3’-terminal del pre-ARNm y poliadenilación, (Pbp1p); regulación de la estabilidad de ARNms, (eRF3); circularización del ARNm y activación traduccional (eIF4G y Paip1) o represión traduccional (Paip2). 4- Elementos en cis y regulación post-transcripcional. 4.1- Elementos en el 3’-NC y su función en la regulación de la estabilidad de ARNms en tripanosomas. El control del inicio de la transcripción es esencial en casi todos los organismos, pero existen ciertas circunstancias en las cuales se requieren niveles adicionales de regulación con el objetivo de obtener un correcto patrón de la expresión. Se sugirió que la regulación a nivel post-transcripcional cumple una función importante en el procesamiento y maduración de los pre-ARNms en tripanosomas (Vanhamme and Pays, 1995). En esta sección se discuten cuáles son las señales en cis- que se caracterizaron y que cumplen un rol fundamental en la regulación de la expresión génica a nivel posttranscripcional. El predominio del control post-transcripcional sobre el transcripcional, está probablemente correlacionado con la organización del genoma de los tripanosomas 31 Introducción en unidades policistrónicas. Los primeros elementos reguladores identificados son de la región 3’-NC del ARNm de los antígenos VSG de los tripanosomas africanos (Berberof et al., 1995). Se demostró que existen elementos en la región 3’-NC de la VSG capaces de regular la expresión de un gen reportero. Más específicamente, una secuencia de ~97 nt, localizada antes de la señal de poliadenilación, es la responsable de regular la estabilidad del ARNm de la VSG. En el estadío sanguíneo, se manifiesta un aumento de la estabilidad del ARNm, mientras que en procíclicos el bajo nivel de proteína madura estaría relacionado con una reducción en la eficiencia de maduración del transcripto (Berberof et al., 1995). Por otro lado, la región 3’-NC de los ARNms que codifican las PARP EP y GPEET de T. brucei, provee el mejor ejemplo de una región regulatoria identificada hasta el momento en tripanosomas. Esta región 3’-NC contiene dos elementos diferentes: un “stem-loop” de 16 nt y una secuencia rica en Uridina de 26 nt. El primer elemento aumenta la traducción de un gen reportero en el estadío procíclico por un mecanismo aún desconocido, pero no juega un rol en la regulación durante el desarrollo del parásito (Furger et al., 1997). El segundo elemento rico en Uridina juega un papel en la regulación de la expresión durante el desarrollo y se demostró que su deleción o mutaciones puntuales reducen o suprimen su función (Hotz et al., 1997; Schurch et al., 1997). Otros dos ejemplos claros se demostraron en el laboratorio de la Dra. C. Clayton. Se observó que la región 3’-NC del ARNm de la fosfoglicerato-quinasa b (pgkb) presenta una secuencia rica en Uridina desestructurada, que también regula la expresión en forma estadío específica. La deleción de este elemento suprime la regulación a nivel de ARNm y de la proteína (Quijada et al., comunicación personal). En segundo lugar, un elemento de 26 nt del ARNm EP1 tiene un rol desestabilizante en el estadío sanguíneo y es el responsable de la destrucción de su 3’-NC por mecanismos de deadenilación o clivaje endonucleolítico (Irmer and Clayton, 2001). Estos dos elementos regulatorios presentan una gran similitud con los elementos desestabilizantes ricos en Adenosina y Uridina, conocidos como “AU-rich elements” (AREs) (Mitchell and Tollervey, 2000). 4.2- Motivos ARE y su función en la degradación de ARNms en mamíferos. El recambio de ARNms es un proceso que está rigurosamente controlado en cualquier célula. Varios elementos pueden regular el destino del recambio de un ARNm en particular, promoviendo su degradación (elementos desestabilizantes) o inhibiendola (elementos estabilizantes). Uno de los mejores elementos regulatorios caracterizados son los ARE, localizados en la región 3’-NC de ARNms de citoquinas y factores de crecimiento, entre otros (Bakheet et al., 2001; Chen and Shyu, 1995). Hay varias clases de AREs que presentan leves variantes de secuencias y que a su vez, son caracterizados por su habilidad de promover la rápida deadenilación y subsiguiente degradación del cuerpo del ARNm (Chen and Shyu, 1995). Los diferentes grupos de AREs identificados hasta el momento en eucariotas están resumidos en la Tabla 1. Básicamente hay tres grupos I, II y III. El grupo I se caracteriza por la presencia de un pentámero AUUUA localizado en una región rica en Uridina como en el ARNm de los proto-oncogenes c-fos y c-myc. El grupo II, puede dividirse en cinco subgrupos dependiendo del contenido de motivos AUUUA solapados. Por último, el grupo III se caracteriza por presentar regiones ricas en Uridina, 32 Introducción pero carecen de los típicos pentámeros AUUUA de los grupos I y II, como por ejemplo los presentes en el ARNm del proto-oncogen c-jun. La variedad de secuencias ricas en Adenosina-Uridina presentes en las regiones 3’-NC de los ARNms, está relacionada con la presencia de distintos grupos de proteínas de unión a ARN que los reconocen y cumplen un papel determinado en el control post-transcripcional de la expresión génica (ver más adelante). Grupo Motivos Ejemplos I WAUUUAW y una región rica en Uridina. c-fos, c-myc IIA AUUUAUUUAUUUAUUUAUUUA GM-CSF, TNF-α IIB AUUUAUUUAUUUAUUUA Interferon-α IIC WAUUUAUUUAUUUAW Cox-2, IL-2, VEGF I ID WWAUUUAUUUAWW FGF2 IIE WWWWAUUUAWWWW receptor u-PA III Región rica en Uridina, sin AUUUA. c-jun Tabla 1 - Tipos de elementos AREs involucrados en la desestabilización de ARNms. Los elementos AREs se definen por su habilidad de promover una rápida degradación del ARNm. Los AREs descriptos pueden agruparse en tres clases según se describió (Xu et al., 1997). Se indican los diferentes grupos I, II y III; el motivo característico de cada grupo y los ejemplos de cada uno. W, significa cualquier ribonucleótido. 4.3- Interrelación entre los procesos de degradación de ARNms y traducción: rol de la cola de poli(A) y elementos en cis en la formación de complejos proteicos en eucariotas superiores. El recambio de ARNms esta mediado tanto por la cola de poli(A), como por secuencias localizadas a lo largo de todo el ARN, las cuales regulan la interacción y formación de complejos proteicos. En el ARNm c-fos, se describió un determinante de inestabilidad (mCRD) localizado en su región codificante, el cual ilustra cómo los procesos de recambio del ARNm y traducción están, en algunos casos, mutuamente interrelacionados (Grosset et al., 2000). La función del mCRD depende de su distancia con respecto a la cola de poli(A), y el mismo interacciona con 5 proteínas formando un complejo multiproteico. La sobre-expresión de cualquiera de estos 5 factores (PABP1, hnRNP D, Paip1, NSAP1 y Unr), aumenta la estabilidad de ARNms que contienen el determinante mCRD, impidiendo su deadenilación (Grosset et al., 2000). En ese trabajo, además se propuso que la formación del complejo multiproteico entre la cola de poli(A) y el mCRD, dificulta el tránsito del ribosoma o el ribosoma puede desarmar el complejo produciendo un rápido decaimiento del ARNm c-fos. Recientemente, se describió que la PABP1 también forma parte de un complejo multiproteico (Figura 7), el cual se forma en la región 3’-NC de ARNms que contienen secuencias ARE (Wilusz et al., 2001). La unión de algunos factores como hnRNP D al ARE, puede alterar la interacción entre PABP1 y las proteínas de unión al “cap” en el extremo 5’-NC, como eIF4G. Este fenómeno facilita el acceso de la poli(A)-ribonucleasa y el ataque exonucleolítico en sentido 3’-5’ (Chen et al., 2001). De esta manera, las proteínas de unión al ARE desencadenan la degradación del transcripto por ruptura de la interacción 33 Introducción del complejo multiproteico del cual forma parte PABP1 (Figura 7). Este evento genera un ARN lineal sin cola de poli(A) por el ataque del exosoma y sin protección en el extremo 5’terminal, por la acción de enzimas que remueven el “cap”. En resúmen, se demostró que existe una conexión funcional entre la actividad del exosoma y el “decapping” del ARNm (Wang and Kiledjian, 2001). DEGRADACION POR DEADENILACION AA PARN NPD ARE A A AAAAA PABP1 hnR PABC eIF 4G 5'-Cap ARNm ESTABILIZACION eIF4E AAAAAAAAAAAA ARE PD N hnR PABP1 eIF 4G AAAAAAAAAAAA ARE R Hu PABC eIF4E ARNm PABP1 eIF 4G PABC eIF4E ARNm 5'-Cap Union hnRNP D 5'-Cap Union HuR Figura 7 - Modelo de regulación de la estabilidad de ARNms mediado por motivos ARE. La interacción del ARE con el factor desestabilizante hnRNPD puede promover una rápida deadenilación del ARNm, ya que esta interacción reduce la afinidad de PABP1 por la cola de poli(A). En cambio, los factores estabilizantes, como proteínas de la familia Hu, HuR, pueden promover el aumento de la interacción de PABP1 con la cola de poli(A). De esta manera, se bloquea la deadenilación y la degradación del ARNm. AAAAAAAAAAAA ARE PABP1 eIF 4G PABC eIF4E ARNm 5'-Cap 4.4- Efecto de inhibidores de la traducción en la estabilidad de ARNms en tripanosomas. La inhibición de la síntesis proteica aumenta la abundancia de varios ARNms inestables en tripanosomas. En general se utilizó cicloheximida, que inhibe la elongación de la traducción por detención de los ribosomas sin liberación de los ARNms (Ross, 1997). De esta manera, la estabilización se puede interpretar de la siguiente forma: (1) la asociación con los polisomas de alguna manera inhibe su degradación, o (2) la degradación de los ARNms, requiere una proteína que es inestable o de vida-media corta; es decir, inhibible por cicloheximida. Esta proteína regulatoria puede ser un componente de la maquinaria de degradación en vez de un factor particular para cada ARNm. Algunos de los ejemplos descriptos en la literatura son: - El ARNm EP1 se estabiliza por inhibición de la traducción en T. brucei (Dorn et al., 1991; Graham and Barry, 1995), en forma independiente del inhibidor que se utilize (cicloheximida, anisomicina, puromicina o emetina). Estos experimentos permitieron concluir que la estabilización no se debe a una asociación del ARNm con los polisomas, ya que los diferentes inhibidores utilizados generaron el mismo efecto (Dorn et al., 1991). Por lo tanto, la ausencia de algún factor proteico debe ser requerida para la degradación del ARNm EP1. - En el estadío amastigote de T. cruzi, la cicloheximida produce una reducción de 34 Introducción casi 3 veces en los niveles de estado-estacionario del ARNm de la amastina y un aumento de 7 veces del ARNm de la tuzina (Teixeira et al., 1995). Sin embargo, la vida-media de ambos ARNms no fue descripta en tal trabajo. Un rasgo interesante es que ambos ARNms provienen de la misma unidad transcripcional (Teixeira et al., 1994) y son regulados en forma diferente durante el desarrollo del parásito. - En L. chagasi, el ARNm mspl es muy estable en la forma virulenta promastigote y de vida-media corta en la fase estacionaria del crecimiento. Experimentos “in vivo” con cicloheximida, demostraron un aumento de la abundancia de entre 4 a 6 veces en la fase estacionaria solamente (Brittingham et al., 2001). - Otros estudios relacionados con el análisis de los niveles de ARNms en ausencia o presencia de inhibidores de la traducción pueden encontrarse en la literatura (Argaman et al., 1994; Quijada et al., 1997). Aunque la mayoría de los resultados obtenidos permiten concluir que los inhibidores utilizados aumentan la estabilidad de ARNms inestables, el(los) mecanismo(s) por el cual/los cuales se regula el metabolismo de estos ARNms no se describió hasta el momento y es sujeto de estudios futuros. 5- Proteínas de unión a ARN: Función y Estructura. Los ARNms están generalmentes asociados con proteínas de unión a ARN, las cuales controlan cada aspecto del metabolismo del mensajero, incluyendo la formación de los extremos 5’- y 3’-terminales maduros, “splicing”, transporte, localización, estabilidad y eficiencia traduccional. Por lo tanto, es de total importancia entender cómo el ARN y las proteínas que lo reconocen interaccionan mutuamente, para finalmente comprender cómo se logran los procesos de regulación de la expresión génica. En general, las proteínas de unión a ARN contienen una estructura modular y presentan dominios de unión a ARN compuestos por 70-150 aminoácidos (Mattaj, 1993). En los últimos tres años se observó un gran progreso en el estudio de la relación estructura-función para los tres grupos más importantes de proteínas en eucariotas. Entre ellas: (1) proteínas con motivos del tipo RRM o “RNA-Recognition motif”; (2) proteínas con motivos KH y (3) proteínas que reconocen ARN doble cadena (“dsRBD”), entre otras. Más aún, estudios computacionales y de dinámica molecular por Resonancia Magnética Nuclear, proveyeron un enorme avance en relación con los aspectos energéticos y dinámicos relacionados con el reconocimiento de moléculas de ARN o complejos ARN-proteína (Perez-Canadillas and Varani, 2001). 5.1- Proteínas con motivos RRM. El motivo RRM o también denominado RNP (“RiboNucleoProtein”) es el dominio de unión a ARN mejor caracterizado y es también el más generalizado en cuanto al número de proteínas que lo contienen (Burd and Dreyfuss, 1994; Varani and Nagai, 1998). Los motivos RRM contienen módulos de entre 80-90 aminoácidos y están compuestos por dos regiones características, un octapéptido ribonucleoproteico-1 (RNP1) y un hexapéptido ribonucleoproteico-2 (RNP2) (Burd and Dreyfuss, 1994). Los motivos RRM interaccionan con una gran variedad de moléculas de ARN y muchos de ellos lo hacen en forma secuencia específica. Todos los motivos RRM estudiados hasta el momento presentan la misma topología y estructura tridimensional (Varani and Nagai, 1998). 35 Introducción Presentan cuatro hojas-β antiparalelas empaquetadas con 2 hélices-α, formando la estructura globular βαββαβ. La única excepción a esta regla se determinó en el tercer RRM de la proteína PTB o “Polypyrimidin-tract Binding Protein”, la cual contiene una quinta hoja-β en su región C-terminal (Conte et al., 2000). Se han resuelto varias estructuras proteicas, incluyendo la proteina humana U1A involucrada en “cis-splicing”, la proteína de unión a tractos de poli(A) (PABP1), hnRNP A1, Sex-lethal (Sxl) y una de las proteínas humana que se une a los ARE (HuC) (Deo et al., 1999; Ding et al., 1999; Handa et al., 1999; Inoue et al., 2000; Nagai et al., 1990). Varias de éstas estructuras también se resolvieron en presencia de ARN y permitieron identificar los contactos específicos ARN-proteína; así como también, evidenciar los cambios conformacionales que ocurren durante la interacción con el ligando o fenómeno de “induced fit” (Williamson, 2000). La mayoría de las proteínas contienen motivos RRM múltiples, los cuales median la unión a ARN en forma cooperativa. Estos dominios, como en Sxl o PABP1, están separados por secuencias conectoras de entre 8-11 residuos (Deo et al., 1999; Handa et al., 1999), las cuales no están estructuradas en la conformación de la proteína libre (Perez-Canadillas and Varani, 2001). El análisis de la covarianza de 300 motivos RRM se describió recientemente (Crowder et al., 2001) y reveló que existe una red de grupos de aminoácidos que covarían, comprendiendo por un lado el núcleo del RRM y por otro lado la superficie del mismo. Este análisis permitió identificar un importante número de residuos, que aunque no están directamente involucrados en el contacto con ARN, pueden influir en la especificidad de la unión (Crowder et al., 2001). 5.2- Cómo dos motivos RRM en una proteína interaccionan con el ARN ? Muchas proteínas de unión a ARN presentan múltiples dominios o superficies de contacto que forman módulos en la organización y estructura final. Recientemente se resolvieron cuatro estructuras del tipo RRM, que permiten entender cómo es la forma en que las proteínas con múltiples dominios reconocen su sustrato. Las proteínas son los motivos RRM1 y RRM2 de Sxl de Drosophila unida a una secuencia rica en Uridina (Handa et al., 1999); la proteína neuronal HuC en presencia de un elemento ARE (Wang and Tanaka Hall, 2001); los RRM1 y RRM2 de la PABP1 humana en presencia de un tracto de poli(A) (Deo et al., 1999) y por último, la proteína nuclear nucleolina en presencia de un “hairpin” de ARN (Ding et al., 1999). Como se mencionó anteriormente, cada una de estas proteínas presentan espaciadores entre los dominios. Se sabe que estas regiones presentan una alta flexibilidad en la proteína libre, permitiendo que los dos dominios sean estructuralmente independientes (Crowder et al., 1999; Inoue et al., 2000). En el complejo ribonucleoproteico, ese espaciador forma una pequeña hélice-α la cual presenta un papel importante en el contacto con el ARN. Los extremos 5’- y 3’-terminales del ARN interaccionan con las regiones N- y C-terminales de las proteínas, respectivamente (PerezCanadillas and Varani, 2001). 5.3- Proteínas con motivos KH. El motivo KH fue identificado por primera vez en la proteína hnRNP K y es otro dominio de unión a ARN bastante común. Consiste de un clásico plegado α-β con una 36 Introducción topología similar a la descripta en varias proteínas ribosomales (Grishin, 2001; Musco et al., 1997). Al igual que los RRM, los motivos KH se identificaron en múltiples copias en más de 100 proteínas y muchas de ellas forman parte de una compleja red de interacciones proteína-proteína y proteína-ARN que regulan la expresión génica. Se ha sugerido también que la unión de los motivos KH al ARN ocurre a través de un mecanismo de cambio conformacional inducido por la interacción (Musco et al., 1996). Una de las proteínas con motivos KH mejor caracterizada hasta el momento es Nova. Por estudios de SELEX (“Systematic Evolution of Ligands by Exponential Enrichment”), se identificó que Nova reconoce secuencias repetitivas del tipo 5’-UCAU-3’ separadas por pequeños tractos de ARN (Buckanovich and Darnell, 1997). Estas secuencias están presentes en ARNms para Nova-1 y Nova-2 y también en un receptor para Glicina. El motivo KH también se lo asoció con una gran variedad de funciones celulares y se lo implicó en diversas enfermedades genéticas, como por ejemplo el síndrome X-frágil, el cual puede ser ocasionado por una reducción en los niveles de expresión de Nova o por un evento de mutación simple, que ocasiona el desplegamiento del dominio KH de la proteína (Siomi et al., 1993). Estudios de espectroscopía por RMN (Musco et al., 1997; Musco et al., 1996) y más recientemente, de Cristalografía de Rayos-X (Lewis et al., 1999), permitieron revelar que los motivos KH comparten la misma topología α-β que los dominios RRM y dsRBD (ver más adelante), los cuales reconocen ARN doble cadena (ver más adelante). La estructura expone segmentos con la secuencia Gli-X-X-Gli (donde X puede ser Lisina, Arginina o Glicina), en los “loops” que conectan las hélices-α1 y α2. A partir de experimentos de RMN, se demostró que pueden participar en la unión con el ARN y son conformacionalmente flexibles (Musco et al., 1997). En contraste con lo esperado, las proteínas con motivos KH, se unen al ARN de una manera completamente diferente a proteínas del tipo RRM y dsRBD. Esto sugiere que la convergencia evolutiva de los dominios de unión a ARN con estructuras comunes del tipo αβ tiene poco que ver con el mecanismo de reconocimiento del ARN (Perez-Canadillas and Varani, 2001). Los contactos entre los “loops” de la proteína y los fosfatos del tetranucleótido 5’-UCAC-3’, juegan un papel importante en el reconocimiento. Sin embargo, no existen interacciones de tipo “stacking” intermolecular como se observa en proteínas con motivos RRM, ni contactos específicos con el OH-2’ como en las proteínas del tipo dsRBD (ver sección siguiente). 5.4- Proteínas de unión a ARN doble cadena (dsRBD). Las proteínas del tipo dsRBD presentan motivos de unión a ARN doble cadena (dsRBM), compuestos por ~70 aminoácidos que forman una estructura terciaria. Ésta puede unir un “dúplex” de ARN y hasta híbridos ARN-ADN, sin ninguna clara especificidad de secuencia (Fierro-Monti and Mathews, 2000). Existen por lo menos 9 sub-grupos de diversas proteínas que contienen uno o más motivos dsRBD el cual participa en interacciones proteína-proteína (Fierro-Monti and Mathews, 2000). Desde el punto de vista estructural, dos estructuras descriptas recientemente permiten clarificar cómo las proteínas del tipo dsRBD reconocen el ARN. Estas son la estructura cristalina del segundo dsRBD de la proteína Xlrbpa de Xenopus laevis (Ryter and Schultz, 1998) y la estructura en solución del tercer dsRBD de la proteína Staufen de Drosophila, unida a un “hairpin” 37 Introducción de ARN (Ramos et al., 2000). A diferencia del cambio conformacional inducido cuando las proteínas con motivos RRM o KH reconocen el ARN, no se identificaron cambios conformacionales significativos en los complejos dsRBD-ARN, en comparación con la estructura de la proteína libre. Sin embargo, a partir de las estructuras dsRBD se determinó la importancia de dos “loops” básicos, los cuáles son necesarios para el contacto y reconocimiento con el ARN doble cadena y también para la discriminación del ADN doble cadena (Ramos et al., 2000). Staufen, el caso más estudiado, reconoce un juego determinado de ARNs en la célula, pero ninguno de ellos tiene un sitio específico de unión. Este tipo de proteínas, contienen múltiples dominios de unión al ARN y se cree que es lo que puede definir su actividad en forma cooperativa (Micklem et al., 2000; Ramos et al., 2000). 5.5- Proteínas de la familia PUF. PUF es otra familia de proteínas reguladoras, que aunque no están estructuralmente relacionadas, se unen a secuencias del 3’-NC de ARNms específicos y pueden modular los niveles de expresión en diferentes tipos celulares. Estas proteínas están involucradas en procesos de degradación de ARNms y en la represión de la traducción, y actúan en combinación con otros factores (Wickens et al., 2002). Se describieron por primera vez en C. elegans y D. Melanogaster y se caracterizan por la presencia de ocho repeticiones consecutivas (repeticiones PUF), cada una de las cuales presenta ~40 aminoácidos. Cada repetición tiene un núcleo consenso que contiene residuos básicos y aromáticos, y se sabe que se requiere el conjunto entero de repeticiones para la unión del ARN (Zamore et al., 1997). La estructura de la proteína Pumilio de Drosophila, miembro de la familia PUF, demostró que se pliega en un dominio con tres hélices-α (Edwards et al., 2001). Desde el punto de vista de la interacción con el ARN, se sabe que dos son los sustratos que se regulan por Pumilio en Drosophila, el ARNm de “hunchback”, el cual participa en la embriogénesis y el ARNm de la ciclina b, cuya represión retarda el ciclo celular. 5.6- Proteínas de unión a ARN con motivos RGG. El motivo o “box” RGG, se identificó inicialmente en la proteína hnRNP U (Kiledjian and Dreyfuss, 1992). Este motivo se define por la presencia de repeticiones RGG (ArgininaGlicina-Glicina) generalmente separadas por residuos aromáticos. Sin embargo, no se conoce el número mínimo de repeticiones requeridas para la unión a ARN. Ejemplos en la literatura muestran que algunas proteínas como hnRNP A1 presentan sólo 6 repeticiones y otras como GAR1 tienen hasta 18 copias (ver revisión en Burd and Dreyfuss, 1994). La alta densidad de residuos de Glicina y las desviaciones dentro del motivo RGG sugieren que éste no es una estructura rígida, aunque ciertos análisis espectroscópicos y de modelado molecular de las repeticiones RGGF de la nucleolina predicen que podría formar una estructura β-espiral (Ghisolfi et al., 1992a). Los motivos RGG también aparecen en proteínas del tipo RRM; por ejemplo en la nucleolina, proteína que reconoce pre-ARNr a partir de sus dos motivos RRM. La región RGG ubicada en el C-terminal del motivo RRM aumenta la afinidad de unión del ARN hasta 10 veces (Ghisolfi et al., 1992b). Esto sugiere 38 Introducción que la unión mediada por el “box” RGG es secuencia específica y facilita el reconocimiento por uno o más dominios, como es el caso de hnRNP U (Kiledjian and Dreyfuss, 1992). Un aspecto interesante del residuo Arginina de los motivos RGG, es el gran potencial de unir hidrógeno, en comparación con otros aminoácidos de carga positiva. Además, el residuo de Arginina puede sufrir modificaciones post-traduccionales como la metilación y la formación de dimetil-Arginina (Liu and Dreyfuss, 1995). Por un lado, la metilación de las proteínas hnRNPs facilita el transporte de hnRNPs del núcleo al citoplasma (Shen et al., 1998) y por otro lado, la fosforilación de residuos de Arginina es una modificación que también regula la actividad de proteínas que contienen motivos RGG (Cobianchi et al., 1993). 5.7- Dominios Auxiliares presentes en las proteínas de unión a ARN. Las proteínas de unión a ARN presentan no sólo los motivos de reconocimiento de sus secuencias blanco, sino también dominios accesorios o auxiliares localizados en los extremos N- y/o C-terminal. Estos dominios auxiliares suelen estar involucrados en procesos de interacción proteína-proteína. Existen dos grandes grupos de dominios auxiliares que se caracterizan por presentar similitudes estructurales y funcionales. En primer lugar, los dominios ricos en Glicina, descriptos principalmente en proteínas hnRNPs básicas, presentan varios residuos de Glicina y aromáticos distribuídos en forma periódica. En segundo lugar, los dominios ricos en Serina-Argininia (SR) (ver revisión en Biamonti and Riva, 1994). El análisis de algunos dominios auxiliares en varios tipos de células eucariotas revela la presencia de rasgos frecuentemente encontrados en los dominios de activación de factores transcripcionales. Algunos de estos dominios son ricos en residuos de Glutamina, Prolina y/o Metionina, capaces de conferir flexibilidad a estas porciones proteicas. Otro tipo de dominios, están compuestos por residuos básicos como Lisina o Arginina, o residuos ácidos, como Aspártico o Glutámico. Se observó en algunos casos, que los dominios auxiliares tienen la capacidad de interaccionar con el ARN o contribuir significativamente en la energía libre de unión con el sustrato. Estos dominios pueden interaccionar con el ARN también en forma inespecífica, anclando la proteína al sustrato ribonucleico y permitiendo que el dominio de unión a ARN pueda adquirir una superficie de contacto en forma específica y directa (Biamonti and Riva, 1994). 6- Proteínas de unión a ARN en tripanosomas. Pocas son las proteínas descriptas con capacidad de unir ARN en tripanosomas. Más aún, a muy pocas se les asignó una función en algún paso del metabolismo y/o procesamiento de ARN. Sin embargo, se pueden definir algunos grupos dependiendo del tipo de dominio y la función asignada a partir de las secuencias que reconocen “in vitro” o “in vivo”. 6.1- Proteínas con motivos RRM. Una de las primeras proteínas con motivos RRM identificadas fue la proteína de unión a poli(A) o TcPABP1 de T. cruzi (Batista et al., 1994), la cual años más tarde se 39 Introducción caracterizó en otras especies de parásitos (Bates et al., 2000; Hotchkiss et al., 1999). En T. brucei, se reportaron dos proteínas (p34 y p37) muy similares en secuencia primaria, y que contienen un sólo dominio del tipo RRM (Zhang et al., 1998), y también se demostró que ambas tienen la capacidad de reconocer “in vivo” al ARNr 5S (Pitula et al., 2002). Esto sugiere un posible papel de estas proteínas en la biogénesis del ARNr. También se identificó la presencia de proteínas con motivos RRM y dominios auxiliares del tipo SR. Estas son capaces de interaccionar con el extremo 5’ del SL de T. brucei y tendrían una función en la regulación del trans-“splicing” (Ismaili et al., 2000; Ismaili et al., 1999). Además, se identificó una proteína de 45kDa (XB1) que reconoce el SL de T. cruzi, y es homóloga al factor PRP31p esencial para el “splicing” en S. cerevisiae, lo que indica que podría estar involucrada en el proceso de trans-“splicing” (Xu et al., 2001a). Además, se reportó la presencia de una secuencia genómica que presenta un marco abierto de lectura de 335 aminoácidos y que codifica para una proteína homóloga a la proteína La con un dominio del tipo RRM y un sitio putativo de unión a ATP en el C-terminal (Westermann and Weber, 2000). 6.2- Proteínas con otros motivos en tripanosomas. Las proteínas de unión a ARN de mayor consideración y estudio desde 1990, son las que forman parte de complejos ribonucleoproteicos involucrados en la edición del ARNm, llamado “editosoma” (ver revisión en Madison-Antenucci et al., 2002), pero no serán descriptas en esta tesis doctoral. Por otro lado, se describió la presencia de proteínas con dedos de zinc o CCCH (TbZFP1 y TbZFP2) en T. brucei y se las implicó en la modulación de la tasa de diferenciación del parásito (Hendriks et al., 2001). Un paso requerido en casi todas las reacciones de procesamiento de ARN es el desplegado de las hebras inter- o intra-moléculas de ARN. De esta manera, y al igual que ocurre en otros tipos celulares existen helicasas de ARN, enzimas claves para las reacciones de catálisis de separación de las hebras del ARN (Schmid and Linder, 1992). Las helicasas forman una superfamilia conocida como proteínas con un “box” DEAD/H, a partir de la presencia de secuencias altamente conservadas del tipo LDEAD/HXXL. Una helicasa de ARN con motivos DEAD-“box” se identificó en T. brucei (Missel et al., 1999). También se identificaron en este microrganismo, proteínas con motivos RGG las cuales forman parte de una familia multigénica con diferentes número de repeticiones (Das et al., 1996). Por úlitmo, se identificó en T. brucei una proteína Pumilio que forma parte de la familia PUF y la cual puede regular la estabilidad de ARNms (Hoek et al., 2002). 7- Comparación de las proteínas de unión a ARN a lo largo de la evolución. En diversos tipos de organismos, desde bacterias, parásitos, levaduras, plantas y células de mamífero se identificaron proteínas con motivos RRM y KH, entre otros. Algunas son específicas de “phylum”, mientras que otras se preservaron en todos los linajes eucariontes durante la evolución: aquellas proteínas requeridas para mecanismos básicos en la regulación post-transcripcional. El genoma de plantas, en particular el de A. thaliana, codifica para 196 proteínas de unión a ARN del tipo RRM y 26 del tipo KH, algo más complejo a lo identificado en C. elegans y D. melanogaster (Lorkovic and Barta, 2002). Un subgrupo interesante dentro 40 Introducción del grupo de proteínas con motivos RRM, es el que presenta una región C-terminal rica en residuos Glicina. Estas proteínas, también de gran diversidad, están involucradas en respuesta a varios tipos de estrés ambiental. Más interesante aún, es la caracterización de distintos grupos de proteínas específicas de organelas como cloroplasto (Lorkovic and Barta, 2002), y mitocondria (Vermel et al., 2002). Por su parte, en cianobacterias hay dos tipos de proteínas con motivos RRM, aquellas que presentan una región C-terminal pequeña rica en Glicina y son reguladas por temperatura, y aquellas que carecen del dominio rico en Glicina y se expresan de forma constitutiva (Sato, 1994). Las proteínas de unión a ARN de cianobacterias y las que presentan regiones ricas en Glicina de eucariotas son similares en estructura y en la forma en que se regulan por temperatura. De esta manera, se sugiere que tal similitud puede ser el resultado de un proceso de evolución convergente (Maruyama et al., 1999). Aunque la función de los dominios ricos en Glicina no está clara hasta el momento, deben ser funcionalmente importantes tanto para cianobacterias como para las proteínas de eucariotas. Es interesante mencionar que muchas especies de bacterias, incluyendo Escherichia coli y Bacilus subtilis, como así también arqueobacterias, carecen de genes que codifiquen proteínas con motivos RRM. Un análisis filogenético (Maruyama et al., 1999) permitió definir un posible origen y camino evolutivo del motivo RRM. Este dominio está presente tanto en procariotas como en eucariotas y todos parecen ser de origen monofilético, es decir, descendientes de un ancestro común. También es interesante resaltar la duplicación intragénica del dominio RRM en un mismo organismo. 8- Formación de complejos ribonucleoproteicos como mecanismo de regulación. Es evidente que todos los mecanismos de regulación de la expresión génica se basan en la formación de complejos compuestos por múltiples componentes proteicos los cuales interaccionan en forma específica y selectiva entre ellos y con moléculas de ARN. Como se mencionó anteriormente, las proteínas de unión a ARN presentan dominios auxiliares. Estas porciones polipeptídicas son las efectoras de la función proteica y modulan la respuesta en forma “downstream” a la unión. Así como los factores transcripcionales presentan dominios de unión a ADN y dominios de activación, las proteínas que unen ARN también presentan similares dominios efectores responsables del reclutamiento de factores adicionales en el ARNm. Aunque se identificaron muchos complejos o partículas ribonucleoproteicas involucradas en los distintos procesos de regulación de la expresión génica (como se mencionó en las secciones anteriores), la conexión funcional entre la formación de estos complejos y la señalización celular está por ser dilucidada. Como se describió, las proteínas y moléculas de ARN están asociadas en forma de complejos, y estas partículas ribonucleoproteícas son las que goviernan el destino de un ARNm particular o un conjunto de ARNms. Entre los complejos importantes descriptos se pueden mencionar los involucrados en la regulación de la transcripción, “splicing”, NMD, transporte del ARN, degradación y/o estabilización del ARNm (como se mencionó con varios ejemplos durante toda la Introducción). También cabe agrupar los motores de localización los cuales pueden estar asociados con el citoesqueleto de la célula y la traducción (Dreyfuss et al., 2002; Orphanides and Reinberg, 2002). 41 Introducción Actualmente, las técnicas de rastreo de alta-tecnología, conocidas como “highthroughput screenings”, permitieron empezar a revelar parte de las redes de interacción proteica que abarcan la mayoría de las funciones celulares, especialmente en S. cerevisiae (Hirsh and Fraser, 2001). Es evidente que la conectividad de proteínas conservadas en la redes de interacción, está negativamente correlacionada con la tasa de evolución. Aquellas proteínas que presentan mayor cantidad de interactores evolucionan más lentamente, ya que son más importantes para el organismo, y además porque una mayor proporción de proteínas están directamente involucradas en esta función (Fraser et al., 2002). En sitios importantes para la interacción proteína-proteína, los cambios evolutivos deben ocurrir por co-evolución; es decir, la sustitución en una proteína resulta en una presión de selección la cual genera cambios recíprocos en las proteínas que interaccionan. Esto conlleva a que las proteínas que interaccionan y forman parte de un mismo complejo regulatorio, evolucionen con tasas similares. Sin embargo, también es posible que las proteínas que interaccionan, evolucionen en tasas similares porque exhiben homología estructural y por lo tanto, tendrían una distribución similar de los sitios restrictivos. El origen más común de la homología estructural entre proteínas que interaccionan, es la duplicación de un gen que codifica una proteína homodimérica, seguido de la evolución de una de las copias génicas (Fraser et al., 2002). 9- Análisis estructural de proteínas de unión a ARN. Los últimos años fueron de una tremenda explosión en cuanto al número de estructuras de complejos ARN-proteína. Es decir, existen estructuras no sólo para los dominios de unión a ARN en forma aislada sino también acomplejados con sus sustratos ribonucleicos. La principal superfamilia de la que se cuenta con varias estructuras es la del tipo RRM. Esto permitió entender cuáles son los principios básicos del reconocimiento mediado por este dominio de unión. Se utilizaron diferentes enfoques con el objetivo de resolver estructuras atómicas, entre ellos: análisis por RMN y Cristalografía de Rayos X. 9.1-Resonancia Magnética Nuclear y estudios de Dinámica Molecular. RMN es una poderosa técnica espectroscópica que permite realizar un estudio detallado no sólo de estructuras, sino también de dinámica de macromoléculas en solución (Clore and Gronenborn, 1991; Clore and Gronenborn, 1998). Hay varios ejemplos de interacciones proteína-proteína y proteína-ligando los cuales ilustran que la dinámica puede estar relacionada de diversas maneras con la función proteica (Wand, 2001). La RMN puede ser utilizada para monitorear el comportamiento dinámico de una proteína en sitios específicos. Además, se puede estudiar el movimiento de una proteína en un amplio rango de escala de tiempo utilizando diferente clase de experimentos (Ishima and Torchia, 2000). Entre ellos, se encuentra la medida de la tasa de relajación del “spin” nuclear, lo cual permite determinar movimientos en escalas de tiempo rápida (subnanosegundos) o lenta (micro-milisegundos), como así también la tasa de difusión rotacional de una molécula o “tumbling”. Por otro lado, las tasas de transferencia de magnetización entre protones con diferentes desplazamientos químicos permite determinar movimientos de dominios proteicos en escalas de tiempo muy lentas 42 Introducción (milisegundos a días). Esto hace de la Resonancia Magnética Nuclear, una técnica única y poderosa para el estudio de dinámica molecular relacionada con la función proteica. La principal fuente de información geométrica utilizada para la determinación de cualquier estructura 3D por RMN consiste en la medición de las restricciones de distancias menores a ~5Å, derivadas del denominado “Nuclear Overhauser Effect” (NOE). Los datos del NOE se complementan con otro número de restricciones que dependen de la proximidad espacial de los átomos, incluyendo las restricciones torsionales de los ángulos, los cuales a su vez dependen de los acoplamientos escalares de 3 enlaces o “J couplings” (Clore and Schwieters, 2002). Los datos experimentales disponibles a partir de RMN no son suficientes “per se” para determinar la estructura 3D de una molécula. De esta manera, éstos deben ser suplementados por datos de restricciones de geometría covalente, como por ejemplo, el largo de los enlaces, los ángulos, la planaridad y la quilaridad (Clore and Schwieters, 2002). 9.2-Marcado Isotópico de proteínas para estudios de dinámica. El marcado de proteínas en forma selectiva o completa con 13 Cy 15 N provee una oportunidad única para estudiar el movimiento intramolecular localizado en solución (Peng and Wagner, 1994). Esto se debe a que los tiempos de relajación de 13C y 15N, a diferencia de otros núcleos, y la magnitud de los NOEs heteronucleares 13C[1H] o 15N[1H], está casi siempre dominado por un mecanismo, la interacción dipolo-dipolo entre los heteronúcleos y los átomos enlazados directamente. Esta interacción depende de las distancias internucleares y los tiempos de correlación locales (para regiones flexibles) o generales (para una molécula rígida). Una mejor sensibilidad en los espectros se logra midiendo los parámetros de relajación en forma indirecta; es decir, transfiriendo la magnetización de los protones al heteronúcleo, ocasionando que la magnetización heteronuclear evolucione en un tiempo de relajación o manera dependiente de los NOEs, y luego transfiriendo nuevamente la magnetización al protón para observarla. Generalmente, esto se realiza por experimentos bidimensionales y tridimensionales tomando ventaja de la mayor dispersión del espectro (Reid, 1997). 9.3- El uso de RMN para caracterizar la dinámica de proteínas. En la década pasada se desarrollaron cuatro áreas de trabajo en el campo de la espectrosccopía por RMN. Los experimentos de resonancia triple por RMN, las cuales proveen un sólido conjunto de herramientas para la asignación de las resonancias en proteínas de tamaño significante (Clore and Schwieters, 2002). Estos métodos derivan de estrategias de enriqueciminento isotópico, como se comentó, los cuáles se refinaron para permitir el marcado de proteínas. Por otro lado, los experimentos bidimensionales de relajación pueden ser utilizados para analizar los movimientos en escala de tiempo pequeña (sub-nanosegundos) de los vectores de interacción con el cuerpo de la molécula. Los parámetros de relajación por RMN observables, como el “spin-lattice” (T1) y el tiempo de relajación (T2) de un “spin” nuclear, están directamente relacionados con la densidad espectral, la cual a su vez está relacionada en forma directa con la transformada de Fourier. Esta fórmula permite obtener un espectro de frecuencia, es decir, a partir de esta operación se logra extraer las señales de las frecuencias de resonancia y se las 43 Introducción convierte en un espectro de resonancia nuclear. 9.4- Cambios estructurales en el reconocimiento ARN-proteína. La formación de casi todos los complejos ARN-proteína está acompañada de cambios conformacionales en el ARN, la proteína o en ambos (Williamson, 2000). Sin embargo, no hay información específica acerca de los pasos de interacción, los cuales pueden incluir una serie de intermediarios. A pesar de ello se sabe que el ARN y la proteína libres en solución, son flexibles o tienen conformaciones particulares, y que el complejo final ARN-proteína es más rígido o puede presentar diferentes conformaciones (Williamson, 2000). En los complejos ARN-proteína, los contactos intermoleculares entre superficies complementarias de los componentes, proveen un reconocimiento molecular exacto. En la mayoría de los complejos, existen superficies y bolsillos preformados en la proteína, y de este modo el ARN se adapta en forma adecuada para entrar a estos sitios (Gosser et al., 2001). 44 Objetivos y Antecedentes. La línea de investigación comenzó en el laboratorio del Dr. Frasch aproximadamente en 1990, estudiando cuales son las moléculas detectadas por el sistema inmune en un organismo infectado con T. cruzi. Se clonaron varias moléculas, entre ellas la transsialidasa (Frasch, 1994) y moléculas del tipo-mucinas (Reyes et al., 1994), ambos componentes del núcleo glicoproteico de la membrana del parásito. Dentro de todos los genes que codifican para proteínas del tipo-mucinas, se identificaron dos familias multigénicas con rasgos particulares: TcMUC, pertenecientes al estadío tripomastigote (Di Noia et al., 2002; Di Noia et al., 1998) y TcSMUG, pertenecientes al estadío epimastigote (Di Noia et al., 2000). Dada la importancia que las mucinas tienen en el proceso de invasión celular por parte del parásito y además lo particular que son los tripanosomas desde el punto de vista de los mecanismos de regulación de la expresión génica (Vanhamme and Pays, 1995), se propusieron los siguientes objetivos para esta tesis doctoral: • Determinar los niveles de regulación de la expresión de las mucinas del estadío del tripanosoma en el insecto vector (TcSMUG). • Identificar elementos en cis- en el ARNm TcSMUG y factores en trans que formen parte del mismo camino regulatorio. • • Estudiar mecanismos de regulación mediados por estos dos componentes. Estudiar en términos funcionales y estructurales las propiedades de las proteínas de • unión a ARN que regulan la expresión de TcSMUG. Describir por primera vez en tripanosomas, la topología y estructura secundaria por RMN de una proteína de unión a ARN. Fundamento de la Investigación. En organismos eucariotas inferiores, como T. cruzi, en el cual no existe control transcripcional evidente alguno, es de gran importancia estudiar cuales son los mecanismos de regulación de la expresión génica. Se sabe que el principal punto de control de la expresión en estos microorganismos es a nivel post-transcripcional. Sin embargo, no se describieron hasta el momento, secuencias y proteínas involucradas en este proceso. Por lo tanto, el objetivo general de esta tesis doctoral es el estudio de la regulación post-transcripcional, utilizando como modelo las mucinas del tipo TcSMUG. Como objetivos particulares, se pretende identificar y describir elementos en el ARNm y proteínas que los reconozcan específicamente, como así también estudiar el camino regulatorio del cual forman parte. 45 RESULTADOS Resultados PARTE 1 - Estudio de la regulación de la expresión génica de TcSMUG. La familia de genes tipo-mucina TcSMUG. La familia que codifica para genes de mucina TcSMUG fue identificada por el Dr. Javier Di Noia en el laboratorio del Dr. Frasch. Mi trabajo comenzó en 1998 clonando, secuenciando y caracterizando los genes que componen esta familia, junto a Javier, quien centró su tesis doctoral en el estudio de la composición del núcleo proteico de las mucinas (Di Noia, 2000). A partir de la secuencia de 6 clones genómicos y el análisis de otros 28 clones de ADNc, provenientes del projecto de secuenciación del genoma de T. cruzi, se identificaron dos grupos de genes diferentes. La dicotomia identificada fue finalmente correlacionada con las diferencias en la estructura de los genes y ARNms. Estos grupos se denominaron L y S (por “Long” y “Short”), de acuerdo al tamaño de los ADNc. Ambos presentan gran identidad en las regiones N- y C- terminales pero divergen en la región central (Di Noia, 2000). La expresión de la familia de ARNms TcSMUG está regulada durante el desarrollo. Como primer objetivo, se analizó la expresión de TcSMUG a partir de un ensayo de “Northern blot” con ARN total extraído de los 4 estadíos de desarrollo de T. cruzi, y se hibridizó con la sonda denominada N, que reconoce el N-terminal de los ARNms L y S. En la Figura 8A se observa el patrón de expresión de TcSMUG a través de los diferentes estadíos de desarrollo del parásito. Claramente, aparecen dos bandas de 1400 y 1000 pbs (Figura 8A). Como se detalla más adelante, los grupos L y S de ARNms, poseen las diferencias más significativas en sus secuencias 3’-NC (no codificantes). Para corroborar que la banda de 1400 pbs corresponda al ARNm L y la de 1000 pbs al ARNm S, se construyeron dos sondas de ADN denominadas 3’-L y 3’-S las cuales reconocen específicamente las regiones 3’-NC de cada grupo en forma específica. Se realizaron ensayos de “Northern blot” con ARN total extraído del estadío epimastigote. Estas hibridizaciones confirmaron que la banda de mayor tamaño corresponde al grupo L y la de menor tamaño al grupo S (Figura 8B). Como control también se hibridizó el mismo filtro con la sonda N, que revela ambas bandas. De esta manera, concluimos que los dos grupos (L y S) están diferencialmente regulados durante el desarrollo del parásito. Se trabajó inicialmente en los estadíos epimastigote y tripomastigote derivado de células, ya que son estadíos representativos de las formas presentes en el vector insecto y en el mamífero, respectivamente. Además, fue en los estadíos en los cuales se observó la mayor diferencia en los niveles estacionarios de ARNm (Figura 8A). En el estadío epimastigote, se detectan ambas formas y con los mayores niveles de expresión en comparación con las demás etapas del ciclo de vida del parásito. Sin embargo, sólo el grupo L se detectó en el estadío tripomastigote, a partir del tiempo de exposición que se muestra, ya que la sobre-exposición del filtro demuestra que el grupo S también se detecta (resultado no mostrado). Los niveles de ARNms en el estado estacionario se cuantificaron luego de la normalización de las señales observadas por hibridización del mismo filtro con la sonda ARNr 24Sα (Figura 8A, panel inferior). Los valores obtenidos en el estadío epimastigote para el grupo L fueron 6 veces mayores que en tripomastigote y para el grupo S la diferencia fue de 10 veces mayor en epimastigote. 47 Resultados En los otros estadíos del desarrollo también existe una regulación. El grupo L se expresa en todos los estadíos, pero con mayores niveles en los estadíos replicativos (epimastigote y amastigote intracelular); pero, el ARNm del grupo S se expresa preferencialmente en los estadíos asociados con el vector insecto, epimastigote y tripomastigote-metacíclicos (Figura 8A). A B E T Mt A N L S sonda 3'S 3'L L S 24Sα rRNA C 5'-NC L ORF 3'-NC sonda 3'L SL An ARE 90% 87% 82% (SIRE) 93% 67% S An sonda N sonda 3'S 100 bases D L AAUUUUAAAAUUUAUUUAAUGUUGUUUUGAUUUAUUUAUUUUAA S E AUUUUAUUUUAUUUUUACUUUUUUUUU sitio trans-"splicing" CGGGAGAGTTTACACCGCGAGCAACAAG 1 2 3 sitio poliadenilacion GGCGCTGAC(TG) 9 Figura 8 - Patrón de expresión de los ARNms TcSMUG del grupo L y S. A, Ensayo de “Northern blot” realizado sobre ARN total proveniente de los diferentes estadios de desarrollo de T.cruzi: (E) epimastigotes, (T) tripomastigotes derivados de células, (Mt) tripomastigotes metacíclicos y (A) amastigotes intracelulares. El ARN fue extraído y fraccionado en geles desnaturalizantes, transferido a membranas de nylon e hibridizado con la sonda TcSMUG-N. La posición de las bandas L (1.4Kpb) y S (1Kpb), se indica a la derecha del panel. B, Un filtro conteniendo ARN total del estadío epimastigote se hibridizó en forma secuencial con las sondas indicadas: T cSMUG-N, 3’S y 3’L. C, Comparación esquemática de la estructura de los ARNms de los grupos L y S de la familia TcSMUG. El porcentaje de homología entre las regiones conservadas (recuadros grises) se indica en el medio del alineamiento de los ARNms. Las regiones que sólo se encuentran en un sólo tipo de ARNm se muestran como recuadros blancos. La posición de las sondas utilizadas en los paneles A y B, se indica con flechas. La presencia de un bloque conteniendo secuencias ARE se indica con un recuadro negro con la secuencia pertinente en el panel D. D, Secuencias ARE presentes en las regiones 3’NC de los grupos L y S. E, Se indica las secuencias genómicas de los sitios aceptores de trans“splicing” y poliadenilación usados en el 95% de los clones analizados. A partir de las diferencias observadas en los niveles de expresión de las formas L y S durante el desarrollo del parásito, se decidió evaluar y comparar, las secuencias nucleotídicas de las regiones NC e intergénicas de los clones de ADNc y genómicos obtenidos. En la región 5’-intergénica existe un 95% de homología entre L y S (ver Figura 8C) y como se sabe, las regiones 5’-terminales son utilizadas para el trans-“splicing” (Matthews et al., 1994). De los tres sitios posibles aceptores de “splicing” identificados en las secuencias genómicas, todos son utilizados con igual frecuencia, según se obtuvo a partir del análisis de las secuencias de diferentes clones de ADNc (resultado no mostrado). De esta manera, por trans-“splicing” se genera una región 5’-NC que presenta alrededor de 55 pbs, dependiendo del sitio utilizado, la cual es 90% idéntica entre las formas L y S (Figura 8C). Por su parte, la región 3’-NC es la que muestra las diferencias más significativas entre los dos grupos. Esto se ve reflejado principalmente en el largo de la región NC y permite explicar las diferencias de tamaños observadas en el “Northern blot” de la Figura 8A. Esta región está compuesta por bloques altamente conservados entre 48 Resultados ambos grupos y también por bloques específicos de cada grupo (Figura 8C). En la Figura 8C se muestra un esquema comparativo de las regiones NC de los dos grupos de ARNms. Se observa que las primeras 30 pbs después del codón de terminación de la traducción, son 100% idénticas en ambos. Luego, un bloque compuesto por 40 pbs está sólo presente en el grupo L, seguido de un bloque de 90 pbs presente en ambos y con 82% de similitud entre grupos. Luego hay una región de 80 pbs específica del grupo S y 3’ de ésta se observa una región de 100 pbs conservada entre ambos. Hacia el extremo 3’-terminal, existe una región rica en Adenosina y Uridina, denominada ARE (por su similitud con los “AU-rich element” presentes en las regiones 3’-NC de células de mamíferos). El grupo L presenta dos motivos AUUUAUUUA, mientras que en el grupo S, ésta región es más rica en Uridina (ver Figura 8D). Una inserción del elemento descripto como SIRE compuesto por ~450 nt (Vázquez et al., 1994), está presente en el grupo L y es lo que ocasiona la mayor diferencia de tamaño observada. Por último, existe una región de 40 pbs que presenta un 67% de identidad y es en donde se adiciona la cola de poli(A) en el transcripto. Del análisis de los diferentes clones analizados, se concluye que el sitio de poliadenilación está conservado en ambos grupos (Figura 8E); sin embargo, una minoria de los clones de ADNc analizados muestran que la poliadenilación también puede ocurrir dentro del retrotransposón SIRE. Los niveles de transcripción de TcSMUG son similares durante el desarrollo del parásito. Se decidió evaluar si las diferencias en los niveles estacionarios de ARNms entre los estadíos del parásito (Figura 8A) se deben a un efecto transcripcional. Para esto, se realizó un ensayo de transcripción o “run-on”, a partir de núcleos aislados de dos estadíos diferentes del parásito, epimastigote y tripomastigote, los cuáles pueden ser crecidos fácilmente en el laboratorio. En este experimento, los núcleos se incuban en presencia de un “buffer” a 30ºC durante 30 min. y en presencia de [α-32P]-UTP, con el objetivo de producir cadenas nacientes de ARN para determinar las tasas de transcripción de genes diferentes. La producción total de cadenas nacientes fue significativamente diferente en los dos estadíos. En epimastigotes los niveles transcripcionales fueron 10 veces mayores (1.107 cpm totales) y esto podría estar relacionado con resultados recientes que indican que las variaciones en los niveles transcripcionales entre las formas replicativa y noreplicativa se deben a modificaciones en la arquitectura nuclear y remodelamiento de la cromatina (Elias et al., 2001). A partir de las sondas de cadenas nacientes obtenidas, se hibridizaron filtros de Nylon conteniendo puntos de ADNc de un miembro del grupo L, un miembro del grupo S y dos controles, ARNr 24Sα y β-tubulina (Figura 9). Luego de la hibridización, los valores relativos de transcripción se analizaron por densitometría y los datos normalizados se obtuvieron por sustracción de la densitometría proveniente del fondo del experimento (plásmido sin inserto). Los valores finales obtenidos para los miembros de los grupos L y S fueron similares entre estadíos, siendo las razones entre epimastigotes y tripomastigotes de 1.06 para el grupo S y 1.7 para el grupo L (Figura 9). De este modo, la presencia del ARNm del grupo S en el estadío epimastigote, en contraste con su ausencia parcial en tripomastigote, sugiere que podría estar regulado a nivel post-transcripcional, ya que su tasa transcripcional es idéntica en los dos estadíos 49 Resultados analizados (Figura 9). El mismo razonamiento se aplica para el ARNm L, ya que las diferencias mínimas observadas en las tasas transcripcionales no pueden generar las variaciones significativas observadas en los niveles de ARNm en el estado estacionario. De esta manera, si β-tubulina puede ser tomado como un control o indicador de la tasa transcripcional del estadío, y los valores relativos se normalizan a éste, la diferencia entre las tasas de inicio de la transcripción entre estadíos serían mayores. En este caso, el control a nivel post-transcripcional sería aún de mayor importancia, en comparación con los datos no normalizados a un gen de expresión constitutiva, como es el caso de βtubulina. Clon Epimastigote Tripomastigote Mle-1 (Grupo L) 13f5 (Grupo S) 24Sα ARNr β-tubulina 1.9 16.4 21.1 7.5 1.1 15.5 16.2 3.6 Mle-1 13f5 ARNr β-tub Figura 9 - Valores comparativos de las tasas de inicio de la transcripción para los grupos L y S entre los estadíos epimastigote y tripomastigote. Se realizaron experimentos de “run on” a partir de núcleos aislados de los estadíos epimastigote y tripomastigote con el objetivo de marcar cadenas nacientes de ARN con [α 32 P]-UTP. Las señales obtenidas a partir de la hibridización de filtros conteniendo un miembro del grupo S (13f5), grupo L (mle-1) y controles (ARNr y β-tubulina), se cuantificaron por densitometría. Las diferencias en las señales observadas entre 13f5 y mle-1 son debidas a diferencias en el tamaño de los clones aplicados sobre la membrana de nylon y no producto de los valores de transcripción relativos entre ellos. Las diferencias observadas en los niveles de ARNms durante el desarrollo se deben a eventos de regulación post-transcripcional. La comparación de los niveles de ARNms durante el desarrollo del parásito así como también de las tasas transcripcionales, indican que no existe control a nivel transcripcional en la expresión de TcSMUG. Las variaciones evidenciadas en los ARNms podrían deberse a una modificación en los niveles de estado estacionario, a partir de eventos de regulación a nivel post-transcripcional. Por lo tanto, se analizó la vida-media (t ) de los ARNms L y S en los estadíos epimastigote y tripomastigote. Para ello, los 1/2 parásitos se incubaron con Actinomicina D (ActD), una droga que inhibe la transcripción, y después de diferentes tiempos se los colectó por centrifugación para extraer ARN total y realizar un ensayo de “Northern blot” con la sonda N, que reconoce ambos grupos de transcriptos (Figura 10 y 11). En el estadío epimastigote, las vidas-medias de los grupos L y S fueron similares (t > 6 hrs) (Figura 10); en cambio, cuando el mismo experimento 1/2 se realizó en tripomastigotes, la vida-media para el grupo L fue menor, t < 0.5 hrs 1/2 (Figura 11). Para el grupo S no se pudo calcular una vida-media en este estadío ya que su expresión no es detectada por “Northern blot” (Figura 11A). Estos resultados confirman que la familia TcSMUG se regula a nivel post-transcripcional, por mecanismos que controlan los niveles de ARNm durante el ciclo de vida del parásito. Efecto de inhibidores de la traducción en la estabilidad de los ARNms TcSMUG. Una explicación posible para las diferencias observadas en los niveles de estabilidad de los ARNms de la familia TcSMUG entre los estadíos epimastigote y tripomastigote, 50 Resultados puede involucrar la presencia de factores proteicos que afecten su A Tiempo (hrs) Tiempo (hrs) 0 .5 1 2 3 4 6 recambio, ya sea en forma positiva como negativa. Para analizar esta L Tiempo (hrs) 0 .5 1 2 3 4 4 6 Tiempo (hrs) 6 0 .5 1 2 3 4 6 L S L S ARNr CHX+ActD* B C ARNm remanente (%) CHX ARNm remanente (%) la vida-media de TcSMUG a t = 2 hrs 1/2 para el grupo L y t = 2.5 hrs para el 3 ActD cicloheximida (CHX) (Figura 10 y 11). Los parásitos se pre-trataron con CHX doble tratamiento en el estadío epimastigote produjo una reducción de 2 ARNr Control en presencia de un inhibidor de la elongación de la síntesis proteica, como una completa inhibición de la traducción (Maranon et al., 1998). Este 1 L S S posibilidad, se determinó la vida-media de los ARNms L y S en ambos estadíos, durante 4 hrs y luego se agregó ActD, sin lavar los parásitos, para asegurar 0 .5 120 100 80 60 40 20 0 0 1 2 3 4 tiempo (hrs) 5 6 120 100 80 60 40 20 0 0 1 2 3 4 5 6 tiempo (hrs) 1/2 grupo S (Figura 10). Por lo tanto, una disminución de al menos 3 veces se observá en la vida-media de TcSMUG en comparación con parásitos que no fueron tratados con CHX. Por lo contrario, en el estadío tripomastigote no se observan cambios considerables en la vida-media en parásitos pretratados con CHX (Figura 11). Es importante mencionar que el tratamiento con el inhibidor de la traducción CHX sólo, sin agregado de Figura 10 - Estabilidad de los ARNms TcSMUG en el estadío epimastigote. A, Ensayo de “Northern blot” realizado a partir de ARN total extraído de epimastigotes tratados o no con Actinomicina D (ActD) y/o Cicloheximida (CHX), para los tiempos indicados sobre cada panel (0, 0.5, 1, 2, 3, 4 y 6 hrs). Las hibridizaciones fueron secuencialmente realizadas con las sondas TcSMUG-N y ARNr como se indica en cada caso. B, Cuantificación de los niveles de ARNm remanente (%) para el grupo L a partir de la densitometria de las bandas de los ensayos de Northern blot realizados en A. El gráfico muestra la normalización de los datos en comparación con los niveles del control ARNr en cada calle. C, idem al panel B pero para el grupo S. Círculos cerrados, control sin tratamiento; cruces, ActD; círculos abiertos, CHX y cuadrados cerrados, CHX más ActD. ActD luego de 4 hrs, no produce ningún efecto considerable en la vida-media de TcSMUG en ambos estadíos, ya que los niveles de ARNms permanecen constantes (Figura 10A y 11A). De esta manera, la disminución en los niveles de ARNms no es debido a que la CHX, puede estar reduciendo la tasa transcripcional de los genes TcSMUG en forma indirecta, pero sí a que los ARNms son degradados en forma más rápida. Dos posibilidades que permiten explicar estos resultados se detallan en la Discusión. 51 Resultados A Tiempo (hrs) 0 .5 1 2 4 Tiempo (hrs) 6 0 .5 1 L 2 4 Figura 11 - Estabilidad de los ARNms TcSMUG en el estadío tripomastigote. A, Ensayo de “Northern blot” realizado a partir de ARN total extraído de tripomastigotes tratados o no con actinomicina D (ActD) y/o cicloheximida (CHX) para los tiempos indicados sobre cada panel (0, 0.5, 1, 2, 4 y 6 hrs). Las hibridizaciones se realizaron en forma secuencial con las sondas TcSMUG-N y ARNr como se indica en cada caso. B, Cuantificación de los niveles de ARNm remanente (%) para el grupo L a partir de la densitometría de las bandas de los ensayos de “Northern blot” realizados en el panel A. El gráfico muestra la normalización de los datos en comparación con los niveles del control ARNr en cada calle. C, Idem al panel B pero para el grupo S. Círculos cerrados, control sin tratamiento; cruces, ActD; círculos abiertos, CHX y cuadrados cerrados, CHX más ActD. 6 L S S ARNr Control ActD Tiempo (hrs) Tiempo (hrs) 0 .5 1 2 4 6 8 0 .5 1 L L S S 2 4 6 ARNr CHX CHX+ActD* C 120 100 80 60 40 20 0 0 1 2 3 4 tiempo (hrs) 5 6 ARNm remanente (%) ARNm remanente (%) B 140 120 100 80 60 40 20 0 0 1 2 3 4 5 tiempo (hrs) 52 6 Resultados PARTE 2 - Identificación de secuencias de ARN en cis capaces de regular la expresión de TcSMUG. Construcción de mutantes de deleción del 3’-NC del ARNm del grupo TcSMUG-L para analizar la presencia de elementos que regulen su estabilidad. Se ha descripto previamente que TcSMUG se regula a nivel post-transcripcional durante el ciclo de vida del parásito y que la regulación de la estabilidad de los ARNms es un componente importante (Figura 10 y 11). De esta manera, el objetivo siguiente fue identificar elementos que actuen en cis- y regulen los niveles de ARNms después de la transcripción. Se sabe que las regiones no codificantes de los ARNms generalmente están involucradas en procesos regulatorios en otros tipos celulares (Chen and Shyu, 1995; Mignone et al., 2002) y también en tripanosomas, donde controlan los niveles del ARNm (Vanhamme and Pays, 1995). Por lo tanto, se construyó en el vector pTEX (Kelly et al., 1992), un “cassette” para ser utilizado en la transfección de parásitos. En este vector se incluyó el marco abierto del gen reportero cat, el cual se unió a la región 5’-intergénica del grupo L de TcSMUG o a la región 3’-intergénica entera o mutada (ver Figura 12). Se utilizaron las regiones intergénicas enteras, ya que son éstas las que presentan las señales para un correcto procesamiento “in vivo” (Vanhamme and Pays, 1995). Entre ellas se encuentra la región que contiene el sitio 5’ aceptor de trans-“splicing” (ag) y la región 3’intergénica entera, la cual presenta el tracto de polipirimidina (pPy) requerido para la poliadenilación, y sitios aceptores 3’ de trans-“splicing” para un eficiente procesamiento del gen neomicina localizado 3’ de cat. De esta manera, nos aseguramos que “in vivo” ocurriera un procesamiento adecuado no sólo del gen reportero en cuestión, sino del gen de selección neomicina, localizado después de cat en el vector pTEX (Kelly et al., 1992). 5'RI B TcSMUG-L pPy CAT S 5'NC ag 3'RI ARE GRE SIRE 3'ss X pPyag H TcSMUG-LΔGRE TcSMUG-LΔ1 E TcSMUG-LΔ2 E TcSMUG-LΔ3 E TcSMUG-LΔARE E TcSMUG-LΔSIRE E Figura 12 - Construcciones realizadas con el gen reportero cat flanqueado de la región 3’-NC de TcSMUG-L entera o con deleciones parciales. Se muestra una representación esquemática del clon TcSMUG-L completo y mutantes de deleción de su región intergénica 3’ (RI). Todas las construcciones se realizaron por PCR como se indica en Materiales y Métodos, utilizando cebadores con sitios de restricción en sus extremos (B, BamHI; S, SmaI; H, HindIII; E, EcoRI; X, XhoI). Las regiones intergénicas 5’ y 3’ contienen los sitios originales para el trans-“splicing” (AG) y un tracto de polipirimidina (pPy) para el correcto procesamiento del pre-ARNm. 3’ss, sitio 3’-aceptor del “splicing”. 53 Resultados Además de la construcción con las regiones 5’- y 3’-intergénicas completas (denominada TcSMUG-L), se construyeron 6 mutantes adicionales (Figura 12). Cada mutante se delecionó en uno de los bloques en los cuales la región 3’-NC de TcSMUG se organiza, como se mencionó anteriormente (Figura 8C). Las diferentes mutantes se denominaron: TcSMUG-LΔGRE, sin el elemento ubicado después del codón de terminación de la traducción y compuesto por 30 pbs ricas en Guanosina; TcSMUG-LΔ1, el cual carece del segundo elemento compuesto por ~40 pbs; TcSMUG-LΔ2, el cual carece del tercer elemento de ~90 pbs; TcSMUG-LΔ3, el cual está compuesto por ~100 pbs; TcSMUG-LΔARE, sin la región rica en Adenosina-Uridina (ARE), compuesta por ~44 pbs; y por último, TcSMUGLΔSIRE, la cual carece del retrotransposón SIRE de ~450 pbs (Figura 12). Análisis de los sitios aceptores de trans-“splicing” y poliadenilación por RT-PCR en los transcriptos provenientes de las construcciones TcSMUG-L. El primer paso antes del análisis de la estabilidad de los ARNms provenientes de todas las construcciones, fue evaluar los sitios utilizados para el trans-“splicing” y la poliadenilación. Como se indica en Materiales y Métodos, se llevaron a cabo reacciones de RT-PCR con dos juegos diferentes de cebadores para clonar los extremos 5’- y 3’terminales de los ARNms. Luego del clonado de bandas correspondientes a cada una de las construcciones, se secuenciaron los sitios aceptores de 6 clones de cada construcción y se comparó con los sitios utilizados en la construcción control TcSMUG-L. Los resultados de las secuencias permitieron concluir lo siguiente: (1) de los sitios putativos aceptores para trans-“splicing” que se encuentran en la región 5’-intergénica de TcSMUG-L (ver Figura 8E), los clones analizados utilizan preferencialmente el segundo ag, aunque los sitios 1 y 3 también se utilizan para el “splicing”; (2) el sitio de poliadenilación utilizado en los ARNms TcSMUG endógenos (Figura 8E), es el utilizado para el procesamiento del extremo 3’ de los pre-ARNms que derivan de las construcciones TcSMUG-L. De esta manera, las poblaciones de parásitos transfectados evaluadas sirven para realizar los ensayos de determinación de la vida-media de los ARNms generados a partir de las construcciones TcSMUG-L. Determinación de la vida-media de los ARNms provenientes de las mutantes del 3’NC del grupo TcSMUG-L. Las construcciones se transfectaron en epimastigotes de T. cruzi, y se seleccionaron en presencia del antibiótico geneticina. Una vez que las poblaciones transformantes se seleccionaron, se realizaron determinaciones de vida-media de los transcriptos provenientes de las construcciones TcSMUG-L y de las mutantes de deleción de la región 3’-NC (Figura 13). Para ello nos basamos en la presencia de un promotor sensible a ActD en el vector pTEX utilizado (Kelly et al., 1992). Los resultados demuestran que el ARNm proveniente de la construcción completa TcSMUG-L posee una vida media t = 70 min. 1/2 A diferencia, la construcción TcSMUG-LΔGRE, presenta una vida-media inferior (t = 1/2 30 min), lo cual representa una reducción del 58% (Figura 13A y B). Esta secuencia GRE (“G-rich Element”), rica en Guanosina, está compuesta por 27 nt río abajo del codón de terminación de la traducción y presenta dos pentámeros contiguos CGGGG (ver más adelante). Los transcriptos provenientes de otras dos construcciones, TcSMUG-LΔ2 y 54 Resultados TcSMUG-LΔ3, presentan vidas-medias muy similares a TcSMUG-L (t = 75 min. y t = 1/2 1/2 65 min., respectivamente). Finalmente, las construcciones TcSMUG-LΔ1 y TcSMUGLΔSIRE, generan ARNms con mayores vidas-medias (t = 140 min.). Debido a que el 1/2 retrotransposón SIRE es un elemento grande (Vázquez et al., 1994), es necesario realizar deleciones parciales del mismo para definir la región específica que causa tal efecto en la estabilidad del ARNm TcSMUG. En cuanto a la construcción TcSMUG-LΔARE, no se observó ningún efecto significativo en la modificación de la vida-media (t = 68 min.) 1/2 (ver más adelante). Debido a que en la determinación de la vida-media de TcSMUGLΔGRE, menos del 50% de los niveles de ARNm permanecen estables en el primer punto experimental (60 min.) (Figura 13A), el ensayo se repitió tomando muestras entre 0 y 60 min. (Figura 13C y D). Así, se calculó con mayor exactitud la vida-media de TcSMUGLΔGRE (t = 30 min.) y mostró ser idéntica a la determinada en la Figura 13A. 1/2 TcSMUG-L ActD (min) 0 TcSMUG-LΔGRE 60 120 180 0 TcSMUG-LΔ1 60 120 180 0 B 60 120 180 ARNm remanente (%) A CAT NEO ARNr * 100 * 50 * TcSMUG-LΔ1 10 TcSMUG-LΔ3 0 ActD (min) 0 60 120 180 TcSMUG-LΔ3 0 60 TcSMUG-LΔSIRE 60 120 180 0 TcSMUG-L TcSMUG-LΔ2 ** 1 TcSMUG-LΔ2 TcSMUG-LΔSIRE ** * TcSMUG-LΔGRE 120 180 Minutos en ActD 60 120 180 Clon t 1/2 (min) TcSMUG-L TcSMUG-LΔGRE TcSMUG-LΔ1 TcSMUG-LΔ2 TcSMUG-LΔ3 TcSMUG-LΔSIRE CAT NEO 70 30 140 75 65 140 C TcSMUG-L ActD (min) 0 15 30 45 D TcSMUG-LΔGRE 60 0 15 30 45 60 CAT ARNr ARNm remanente (%) ARNr 100 * 50 TcSMUG-L ** **TcSMUG-LΔGRE 10 0 15 30 45 60 Minutos en ActD Figura 13 - Determinación de la vida-media de los ARNms provenientes de las deleciones del 3’-NC de TcSMUG-L. A, Parásitos del estadío epimastigote se transfectaron con los ADN recombinantes indicados en la Figura 12, y se trataron con ActD 10 μg/ml. Se cosecharon parásitos a diferentes tiempos, y se extrajo ARN total para realizar ensayos de “Northern blot”. El mismo filtro se hibridizó en forma secuencial con las sondas de cat, neo y ARNr. La hibridización realizada con la sonda neo sirve como un control interno del experimento ya que éste gen se expresa del mismo vector pTEX que cat (Kelly et al., 1992). B, Cuantificación de las bandas de los ensayos de “Northern blot” del panel A. La vida-media de cada transcripto se indica debajo del gráfico. C, Epimastigotes transfectados con las construcciones TcSMUG-L y TcSMUG-LΔGRE se trataron igual que en el panel A con ActD y se extrajo ARN total en tiempos más cortos, para determinar su vida-media en forma más precisa. D, Cuantificación de las bandas del “Northern blot” del panel C. En los paneles B y D los valores se expresan como la cantidad relativa media de ARNm, +/- el error estándar de la media (n=3) para cada tiempo, luego de la normalización con los valores de ARNr para cada punto. Las diferencias entre TcSMUG-L y las mutantes de deleción fueron significativas cuando se compararon las medias por un test t de Student (*, p < 0.05; **, p < 0.01). 55 Resultados Estos resultados sugieren que el 3’-NC de TcSMUG-L se divide en diferentes regiones funcionales: (1) un elemento positivo llamado GRE, rico en Guanosina y ubicado en los primeros 27 nt; (2) un elemento negativo, llamado E1 por elemento 1, ubicado entre los nucleótidos 28-62 después del codón de terminación; y (3) un elemento rico en AdenosinaUridina (ARE) ubicado entre los nucleótidos 272-318, que es similar en secuencia a los elementos AREs de mamíferos. Las regiones 3’-NC de los ARNms fueron modeladas utilizando el programa Genequest (Lasargene Package, DNAstar Inc.), para predecir si las deleciones del 3’-NC, tendrían algún efecto en la estructura tridimensional predicha del ARN. Estos transcriptos demostraron presentar una estructura modelada semejante, incluyendo todos los “loops” de la región 3’-NC (resultados no mostrados). En resúmen, la composición de la secuencia de los elementos GRE, ARE y E1, son las que confieren el efecto observado sobre la estabilidad del ARNm. El elemento GRE confiere un efecto estadío específico positivo en la estabilidad del ARNm TcSMUG-L, y es funcionalmente diferente al ARE. No se observó ningún efecto en la estabilidad del ARNm TcSMUG-L en el estadío epimastigote al delecionarse el elemento ARE (ver más adelante). Por el contrario, el elemento GRE, sí modula en forma positiva la estabilidad del ARNm en este estadío (Figura 13). Ya que estas dos construcciones son las más interesantes desde el punto de vista funcional, se analizó el efecto de estas deleciones en la estabilidad del ARNm en dos diferentes estadíos: epimastigote y tripomastigote metacíclico. Los resultados se obtuvieron a partir de las construcciones TcSMUG-L (3’-NC completo), TcSMUG-LΔARE (ARE delecionado del 3’-NC) y TcSMUG-LΔGRE (Figura 14A). Los parásitos del estadío epimastigote se diferenciaron a la forma infectiva, tripomastigote metacíclicos, y se incubaron con la droga ActD, para determinar la vida-media de los transcriptos derivados de esas construcciones (Figura 14). Luego del tratamiento con la droga, se colectaron parásitos a diferentes tiempos: 0, 45, 60, 90 y 120 min. para el estadío epimastigote; y 0, 10, 20, 30 min. para el estadío tripomastigote metacíclico. Los tiempos colectados en los dos estadíos fueron diferentes ya que, como se demostró, el ARNm de TcSMUG-L es de vida-media corta (t < 30 min) en el estadío infectivo tripomastigote metacíclico (ver 1/2 Figura 11). Utilizando como sonda de ADN la región codificante del gen reportero cat, se realizó un ensayo de “Northern blot”. En el estadío epimastigote los transcriptos TcSMUGL y TcSMUG-LΔARE, ambos con un el elemento GRE en su región 3’-NC, presentaron vidas-medias similares (t = 70 y 68 min., respectivamente) (Figura 14B y C). En contraste, 1/2 el transcripto TcSMUG-LΔGRE fue menos estable (t = 30 min.), como se mencionó 1/2 anteriormente (Figura 13). Hasta el momento se puede concluir que: (1) el elemento GRE en el estadío epimastigote está involucrado en un proceso de estabilización del ARNm TcSMUG, y (2) el elemento ARE no está involucrado en la estabilización del ARNm TcSMUG-L en epimastigotes, ya que los transcriptos TcSMUG-L y TcSMUG-LΔARE presentan la misma vida-media (ver Figura 14). El análisis de las vidas-medias en el estadío tripomastigote metacíclico, derivados por diferenciación de los epimastigotes transformados, también reveló diferencias en los niveles de ARNm (Figura 14D y E). Los transcriptos TcSMUG-L y TcSMUG-LΔGRE son de vida-media muy corta (t < 10 min.), en comparación con el ARNm TcSMUG-LΔARE, 1/2 56 Resultados el cual presenta una vida-media mayor (t > 30 min.) (Figura 14D y E). De esta manera, 1/2 se puede concluir que la inestabilidad de TcSMUG-L y TcSMUG-LΔGRE en el estadío tripomastigote metacíclico se debe, en parte, a la presencia del elemento ARE en su región 3’-NC. Como control del ensayo de “Northern blot”, el mismo filtro se hibridizó en forma secuencial con una sonda de ADN correspondiente al gen de selección neomicina. Debido a que el gen neomicina está flanqueado por las regiones intergénicas de la gliceraldehído-3-fosfato deshidrogenasa, en el plásmido pTEX (Kelly et al., 1992), éste sirve como control interno para la determinación de la vida-media de diferentes ARNms. Como se muestra, el ARNm neo presenta la misma tasa de recambio en todas las poblaciones de parásitos evaluadas, en forma independiente de la construcción en estudio (Figura 14). A AAUUUUAAAAUUUAUUUAAUGUUGUUUUGAUUUAUUUAUUUUAA S B TcSMUG-L 3'ss H pPy ag GRE X pPy ag ARE TGAGGACGGGGCGGGGCGCGUGCGCC B TcSMUG-LΔGRE 3'ss S X pPy ag pPy ag H B 3'ss S TcSMUG-LΔARE B TcSMUG-L ActD (min) 0 TcSMUG-LΔGRE 45 60 90 120 0 TcSMUG-LΔARE 45 60 90 120 0 pPy ag E H C ARNm remanente (%) pPy ag X 45 60 90 120 CAT NEO 100 50 * ** TcSMUG-L TcSMUG-LΔARE ** 10 0 ARNr D 45 * TcSMUG-LΔGRE 60 90 120 Minutos en ActD ActD (min) 0 10 20 TcSMUG-LΔGRE 30 0 10 20 TcSMUG-LΔARE 30 0 10 20 30 CAT NEO ARNm remanente (%) E TcSMUG-L 100 ** ** ** 10 ARNr 0 F ARN Estadio Epimastigote Tripomastigote TcSMUG-L TcSMUG-LΔARE 50 10 20 30 Minutos en ActD TcSMUG-L TcSMUG-LΔGRE TcSMUG-LΔGRE TcSMUG-LΔARE t 1/2 (min) t 1/2 (min) 70 30 t 1/2 (min) 68 (Estable) (Inestable) (Estable) <10 <10 >30 (Inestable) (Inestable) (Estable) Figura 14 - Dos elementos regulatorios funcionalmente diferentes y localizados en el 3’-NC del ARN TcSMUG-L. A, Representación esquemática de TcSMUG-L (construcción completa), y las mutantes de deleción utilizadas para transfectar parásitos de la forma epimastigote. La secuencia que fue delecionada en los clones TcSMUG-LΔGRE y TcSMUG-LΔARE se indica en el esquema de TcSMUG-L. B, Ensayo de “Northern blot” realizado sobre ARN total extraído de las diferentes poblaciones tratadas con ActD a los tiempos indicados. El mismo filtro se hibridizó en forma secuencial con las sondas cat, neo y ARNr. C, Cuantificación de los niveles de ARNm cat a partir del “Northern blot” del panel B. Los datos se expresaron como la cantidad de ARNm relativa a la media +/- el error estándar de la media (n=3) en cada punto luego de la normalización con los niveles de ARNr. Las diferencias entre TcSMUG-LΔGRE y TcSMUG-L y entre TcSMUGLΔGRE y TcSMUG-LΔARE fueron significativas (*, p < 0.05; **, p < 0.01 cuando se comparó las medias a partir de un test t de Student). D, Tripomastigotes-metacíclicos derivados de los epimastigotes transfectados, se trataron como se indica en B. E, Cuantificación de los niveles de ARNm de cat del panel D. Los datos se expresaron como para el panel C. Las diferencias entre TcSMUG-LΔARE y TcSMUG-L y entre TcSMUGLΔARE y TcSMUG-LΔGRE fueron significativas (**, p < 0.01, cuando se compararon las medias por un test t de Student). En los paneles C y E, la vida-media de cada transcripto se indica al costado del gráfico. F, Tabla que resume los resultados obtenidos en la Figura. 57 Resultados Los elementos de la región 3’-NC también modulan la eficiencia traduccional. Se sabe que en las regiones 3’-NC de un tipo particular de ARNms de tripanosomas existen secuencias que pueden modular la traducción (Furger et al., 1997). De la misma forma, en células de mamíferos la presencia del ARE, por ejemplo en el ARNm del tnf-α, también exhibe un efecto en la tasa traduccional (Kontoyiannis et al., 1999). Con el objetivo de determinar si los elementos de la región 3’-NC de TcSMUG-L controlan la traducción, se midió la actividad enzimática CAT en las diferentes poblaciones transformadas. Los valores de actividad se normalizaron a los niveles de ARNm cat en el estado-estacionario para cada construcción. La medida de actividad enzimática relativa se expresó como el porcentaje de la actividad obtenida con la construcción completa TcSMUG-L normalizada a la cantidad de ARNm cat detectado (Figura 15). vida-media ARNm 5'RI B TcSMUG-L pPy CAT S 5'NC ag 3'RI ARE GRE SIRE 3'ss Eficiencia traduccional minutos (%) (%) de TcSMUG-L 70 (100) 100 30 (42) 117 140 (200) 185 75 (107) 88 65 (93) 112 68 (97) 21 140 (200) 15 X pPy ag H TcSMUG-LΔGRE TcSMUG-LΔ1 E TcSMUG-LΔ2 E TcSMUG-LΔ3 E TcSMUG-LΔARE E TcSMUG-LΔSIRE E Figura 15 – Identificación de elementos negativos y positivos en el 3’-NC de TcSMUG-L involucrados en el control de la estabilidad del ARNm y traducción. Se indica la vida media (en minutos) de cada construcción y el (%) entre paréntesis en comparación con la construcción TcSMUGL. Además, la eficiencia traduccional de cada población, como (%) de la construcción TcSMUG-L. En la población transfectada con TcSMUG-LΔGRE los valores obtenidos de actividad CAT fueron similares a los de la construcción TcSMUG-L (117%). Esto sugiere que si bien este elemento regula los niveles de ARNm, es incapaz de modular la traducción (Figura 15). En cambio, TcSMUG-LΔ1, cuyo transcripto presenta una vida-media mayor que el control TcSMUG-L, presentó niveles mayores de traducción (185%). Este resultado sugiere que el elemento E1 regula ambos procesos en forma negativa en el estadío epimastigote. Las construcciones TcSMUG-LΔ2 y TcSMUG-LΔ3 no producen un efecto considerable en la actividad traduccional (88% y 112% de TcSMUG-L, respectivamente) (Figura 15). El retrotransposón SIRE, sin embargo, produce un efecto positivo, ya que su deleción ocasiona una disminución de la actividad enzimática CAT (15% de TcSMUG-L). Este resultado es interesante, ya que en el laboratorio del Dr. Levin, se sugirió previamente que el elemento SIRE exhibe otras funciones diferentes en el procesamiento y la maduración de los pre-ARNms (Vázquez et al., 1994). El hecho de que SIRE esté presente en distinas posiciones en las regiones intergénicas y/o NC de varios ARNms, sugiere una función determinada en el procesamiento y/o control de la expresión dependiente de su localización. 58 Resultados El elemento ARE está representado en el genoma de T. cruzi. A partir de la funcionalidad del ARE “in vivo” en el control de la estabilidad y la traducción, se analizó la presencia de esta secuencia en otros transcriptos de T. cruzi. Para ello se realizó una búsqueda en la base de datos del NCBI (con un “expect” igual a 1) utilizando el ARE del grupo TcSMUG-L. Esta búsqueda mostró docenas de secuencias similares. En muchos de ellos se encontraron motivos ARE de clase I y II en las regiones 3’-NC. ARNm Secuencia de la región rica en Uridina Grupo plc-1 (AF093565) 20pb-AUAUAUAUAUAUAUAUAUAUAUAUUUAUUUAUUU Proteina unión Ca (L01584.8) 660pb-AUUUUAUUAUUAUUUAUUAUUAUUUAU AUAUAAAU 100pbAUAUUUAUUUAUUUAUAUAUAAA IIA Calmodulina (x52096) 140pb-UU AUUUAAU-25pb-UUUAUUUAUUUUAU IID Transportador de Hexosa (U05588) 413pb-AAAUUAAUUUAUUUAUUUAUUU gdh (AF051892) 350pb-AUUUAUUUAUUUAUUUUUAAA ATPAsa (U40265) 1pb-UUUAUUUUAUUUAUUUUU-40pb-AUAUUUAUU-30pb-UUAUUUAUU I I I IIC Cruzipaina (X54414) 40pb-AUUUUA-90pb-UAUUUAUUUU I Tcce5 (AB003068) 110pb-AUAAUAAUAUAUAUUUAUUUAUU I R27-2 (L04603) 100pb-UUUUUAUUUAUUUUUUUUUU-100bpU UAUUUAUUUAUUUAUUUUAUUAUUAUU gp82 (L14824) 100pb-UUUAUUUUAUGUAUUUUUAUUUUU-160pb-UAUUUAAU I Tssa (AF036421) 85pb-AUUUA-10pb-UUUUUUAUUUAUUUUUAUUUUAU I gp72 (M65021) 350pb-UAUAUAUUUAAUAUUAUAU I Amastina (U04337) 130pb-UUUUAUUUAUUUUAUUUUUUU I IIB Tabla 2 - Los elementos ARE están presentes en las regiones 3’-NC de varios transcriptos de T. cruzi. Las secuencias de T. cruzi presentes en la base de datos del “GenBank” se rastrearon utilizando la secuencia ARE del grupo TcSMUG-L. En esta Tabla se indican algunos ejemplos seleccionados de genes ya caracterizados. El número de acceso al “GenBank” se muestra al lado del nombre de cada ejemplo. El número de pb en las secuencias de los ARE indica la distancia entre el codon de terminación de la traducción y entre el elemento ARE presente en cada ejemplo. Los pentámeros AUUUA están subrayados y también se indica el grupo de cada elemento al cual son similares según su descripción (Xu et al., 1997). En la Tabla 2 se muestran ejemplos de moléculas ya caracterizadas en T. cruzi que contienen AREs; entre ellos una fosfolipasa-c (plc-1), la proteína de unión a calcio, gp-82 (un antígeno de superficie) y amastina entre otros. Lo interesante es que se identificaron motivos ARE que presentan pentámeros AUUUA aislados, además del nonámero UUAUUUAUU o la presencia de pentámeros AUUUAUUUA solapados (Tabla 2). Cuando se rastreó la base de datos de “EST” de T. cruzi , se obtuvieron cientos de secuencias, pero las mismas correspondieron a moléculas no caracterizadas hasta el momento. Comparación de las secuencias ARE de T. cruzi y las presentes en otros tipos celulares. En células de mamífero la degradación de ARNms mediada por elementos ARE, está relacionado con procesos de transformación celular, crecimiento, diferenciación, adhesión celular y también en la regulación de la respuesta inmune (Chen and Shyu, 1995). En la región 3’-NC del grupo L de TcSMUG, el motivo UUAUUUAU(U/A) está 59 Resultados repetido dos veces, como en los ARNms de la interleuquina-3 (Stoecklin et al., 1994) y en el ARNm del interferón-γ (Peppel et al., 1991), donde se asociaron estos motivos con la degradación de ARNms. Además, se reportó que una copia del nonámero UUAUUUAUU produce una leve desestabilización, mientras que dos copias (como es el caso del grupo L) provee una desestabilización de ARNms mayor (Lagnado et al., 1994; Zubiaga et al., 1995). El pentámero AUUUA está repetido 4 veces en el ARE del grupo L y otros grupos de trabajo reportaron que es ésta secuencia, en vez del nonámero, el mínimo elemento funcional del ARE (Xu et al., 1997). El ARNm del grupo S no presenta pentámeros pero si tiene dos motivos AUUUUA, por lo cual es levemente diferente a los ARE descriptos. Sin embargo, existen secuencias ARE que no contienen el motivo AUUUA, que también pueden mediar el decaimiento del ARNm (Chen and Shyu, 1995). 60 Resultados PARTE 3 - Identificación de proteínas que reconocen los elementos ARE y GRE. El elemento GRE es reconocido por proteínas de unión a ARN las cuales forman complejos nucleares y citoplasmáticos. La identificación de elementos involucrados en procesos de estabilización de ARNms, sugirió pensar en la presencia de factores que actúen en trans- y los reconozcan específicamente. Primero se buscaron proteínas que reconozcan al elemento GRE, por su importante papel en la regulación de la estabilidad de TcSMUG-L (Figura 14). La secuencia GRE de 27 nt se sintetizó por transcripción “in vitro” en presencia de [α32P]UTP. Esta ribosonda fue utilizada para ensayos de unión en presencia de extractos proteicos de los diferentes estadíos del parásito. Los complejos formados fueron analizados en geles nativos de poliacrilamida (técnica de retardo en gel). Este ensayo reveló que el GRE es reconocido por 3 complejos proteicos presentes en todos los estadíos del desarrollo de T. cruzi. Como control del experimento, se realizaron las incubaciones en presencia del “buffer” de reacción no evidenciándose bandas de complejos con proteínas (Figura 16A, calle ARN). ARN A Estadio B E Mt A T Figura 16 - El elemento GRE es reconocido por proteínas del parásito que presentan diferentes masas moleculares. A, La secuencia GRE se transcribió “in vitro” y se incubó con extractos proteicos totales provenientes de los cuatro estadíos del parásito: (E) epimastigotes, (T) tripomastigotes derivados de células, (Mt) tripomastigotes metacíclicos y (A) amastigotes intracelulares. Para el análisis de las interacciones ARN-proteína, la reacción de unión “in vitro” se resolvió en un gel nativo. Los complejos GRE identificados (1, 2 y 3) se indican a la izquierda del panel. B, El ARN GRE se incubó con un extracto proteico de epimastigotes y se realizó un gel de retardo preparativo. Los complejos ARN-proteína localizados fueron entrecruzados covalentemente por la irradiación con luz UV y eluídos de la matriz. Luego se corrió un gel desnaturalizante SDS-PAGE 10% y se expuso a placas radiográficas para determinar las masas moleculares aparentes según comparación com marcadores de peso molecular conocidos. GRE kDa 1 2 3 110 97 GRE-1 66 45 GRE-2 29 GRE-3 ARN libre SMUG-L-GRE UGAGGACGGGGCGGGGCGCGUGCGCCG 61 Resultados Para determinar las masas moleculares aparentes de las proteínas que forman parte de los complejos identificados, llamados GRE-1, GRE-2 y GRE-3 (Figura 16A), un extracto proteico total del estadío epimastigote se incubó en presencia de la ribosonda GRE. Las reacciones de unión “in vitro” se corrieron en geles nativos de poliacrilamida en condiciones preparativas; es decir, se sembraron ~20 veces más de muestra que en el gel analítico de la Figura 16A. La posición de los complejos ribonucleoproteicos se identificó por exposición del gel a una placa autorradiográfica durante 2 horas a 4ºC, y luego los complejos se entrecruzaron en forma covalente por irradiación con luz UV y se cortaron para su posterior análisis en geles SDS-PAGE (Figura 16B). Este ensayo demostró que el complejo GRE-1 está compuesto por un sólo polipéptido de ~80kDa, mientras que los complejos GRE-2 y GRE-3 están compuestos por varios polipéptidos de masas moleculares aparentes de ~35, 40 y 66kDa (Figura 16B). Las proteínas que forman parte del complejo GRE-2 presentan diferentes niveles de abundancia en un lisado total del estadío epimastigote. Es así como una banda poco abundante de ~66kDa se detecta junto con dos bandas de ~35 y 55kDa de mayor abundancia (Figura 16B). Ensayos de competición para determinar la especificidad y localización subcelular de los complejos GRE. La especificidad de los complejos que se forman con el GRE se caracterizaron mediante experimentos de competición (Figura 17). En primer lugar, se prepararon reacciones de unión ARN-proteína en ausencia o en presencia de cantidades crecientes de homoribopolímeros sin marcar (poli(A), poli(C), poli(G) y poli(U)) y la ribosonda GRE. Poli(G) es el único ARN que bloquea en forma selectiva la formación de los complejos GRE-1 y GRE-2 (Figura 17A). Este resultado está de acuerdo con la naturaleza del elemento GRE utilizado para los ensayos de retardo en gel, ya que GRE tiene un alto contenido en Guanosina. El complejo GRE-1 se desplaza en forma efectiva con un exceso molar de polímero de 10X, mientras que el complejo GRE-2 se compite parcialmente en presencia de un exceso 1000X de poli(G) (Figura 17A). Esto puede deberse a diferencias en la concentración de las proteínas que forman parte de los complejos en el extracto, lo cual también se sugiere a partir del ensayo de entrecruzamiento de los complejos por UV (Figura 16B). A diferencia de lo que sucede con los complejos GRE-1 y GRE-2, la formación de GRE-3 no se compite en forma eficiente por los polímeros de ARN utilizados (Figura 17A), indicando que su formación es inespecífica. Con el objetivo de determinar cuál es la mínima secuencia reconocida por los complejos en GRE, esta secuencia se dividió en dos partes: a) SMUG-L-GRE-1: 5’GGACGGGGCGGGGC-3’; y b) SMUG-L-GRE-2, este último con un contenido rico en CG y la secuencia 5’-GCGCGUGCGCCG-3’ (ver Figura 17B). Con las dos secuencias marcadas de la misma manera que GRE, se realizaron geles de retardo con fracciones proteicas nucleares y citoplasmáticas, y también con un lisado total como control (Figura 17C). Este ensayo permite concluir que la mínima secuencia requerida para el reconocimiento y la formación de los complejos GRE-1 y GRE-2 es la secuencia SMUG-L-GRE-1, la cual está compuesta por dos pentámeros contiguos CGGGG. Mientras que el complejo GRE1 se localiza en el citoplasma, GRE-2 está distribuído en ambos compartimentos, núcleo y citoplasma (Figura 17C); lo cual indica que estos polipéptidos pueden estar involucrados 62 Resultados en el transporte del ARNm TcSMUG (ver Discusión). poli (A) ARN A - poli (C) poli (G) poli (U) C SMUG-L GRE GRE-1 GRE-2 10 100 1000 10 100 1000 10 100 1000 10 100 1000 T N C T N C T N C GRE-1 GRE-1 GRE-2 * GRE-2 GRE-3 GRE-3 B ARN Secuencia SMUG-L-GRE UGAGGACGGGGCGGGGCGCGUGCGCCG SMUG-L-GRE-1 SMUG-L-GRE-2 GGACGGGGCGGGGC ARN libre GCGCGUGCGCCG Figura 17 - Determinación de la especificidad y localización subcelular de las proteínas que reconocen al GRE. A, Se realizaron reacciones de unión “in vitro” con la sonda GRE sóla (ARN), y extracto de proteínas del estadío epimastigote sin competidor (-) o en presencia de cantidades crecientes (10, 100, 1000X) de polímeros de ARN (poliA, poliC, poliG y poliU), con el objetivo de determinar la especificidad de la interacción ARN-proteína. Las reacciones se resolvieron en un gel nativo 7% como se indica. B, Se muestra la secuencia de los oligoribonucleótidos sintetizados y utilizados en los geles nativos del panel C: SMUG-L-GRE, elemento GRE completo; SMUG-L-GRE-1, primera mitad del elemento GRE; SMUG-L-GRE-2, segunda mitad del elemento GRE. C, Parásitos del estadío epimastigote se lisaron con NP-40 para obtener fracciones nucleares (N) y citoplasmáticas (C). Estas fracciones se utilizaron en reacciones de unión “in vitro” y los complejos se resolvieron por geles nativos. La posición de los complejos GRE-1, GRE-2 y GRE-3 se indica a la izquierda de la Figura. La flecha con el asterisco indica la posición de una banda artefacto debido a la sonda de ARN SMUG-L-GRE-1. 63 Resultados El elemento ARE interacciona con proteínas de unión a ARN expresadas en forma diferencial durante el desarrollo del parásito. Como se demostró anteriormente, la secuencia rica en Adenosina-Uridina (ARE) de 44 nt es importante en la regulación del ARNm TcSMUG-L, ya que confiere inestabilidad en forma estadío específica (Figura 14). Con el objetivo de determinar si existen proteínas o complejos que reconocen al ARE en los distintos estadíos del parásito, este ARN se marcó por transcripción “in vitro” y se incubó de la misma manera que el GRE con extractos totales de las distintas formas de T. cruzi (Figura 18A). Se identificaron complejos formados entre proteínas del extracto y el elemento ARE (Figura 18A). En el estadío epimastigote se identificó un complejo llamado eAREBP, por epimastigote “ARE-binding protein”, el cual migra más lento que los complejos denominados AREBPs, presentes sólo en los otros tres estadíos del ciclo de vida. Luego se procedió a determinar la composición y las masas moleculares aparentes de los componentes de los complejos que reconocen al ARE a partir de ensayos de entrecruzamiento con luz UV y geles SDSPAGE (Figura 18B). La proteína eAREBP del estadío epimastigote presenta una masa aparente ~100kDa, mientras que las proteínas AREBPs tienen un peso molecular aparente de entre 45-50kDa y están compuestas por al menos dos polipéptidos (Figura 18B). ARN A Estadio B E Mt A T Estadio kDa 110 97 eAREBP E Mt A T eAREBP 66 AREBPs 45 AREBPs 29 ARN libre SMUG-L-ARE AAUUUUAAAAUUUAUUUAAUGUUGUUUUGAUUUAUUUAUUUUAA 64 Figura 18 – La expresión de las proteínas de unión al ARE está regulada durante el desarrollo del parásito. A, El elemento SMUG-L-ARE se generó por transcripción “in vitro” y se incubó con extractos proteícos totales provenientes de los cuatro estadíos del parásito. Para el análisis de las interacciones ARN-proteína, las reacciones de unión “in vitro” se resolvieron en un gel nativo 7%. Las flechas indican la posición de los diferentes complejos ribonucleoproteicos identificados, eAREBP y AREBPs. La posición del ARN incubado en ausencia de proteínas se indica como ARN. B, El ARN se incubó al igual que en el panel A en forma preparativa. Los complejos localizados en el gel nativo, se entrecruzaron covalentemente por irradiación con luz UV y electroforesis en un gel SDS-PAGE 10% para determinar sus masas moleculares aparentes en comparación con marcadores de peso molecular conocidos. Resultados Las proteínas que reconocen al ARE presentan un patrón de regulación durante el desarrollo (Figura 18). Según los resultados obtenidos hasta el momento, habría un proceso de regulación estadío específico durante el ciclo de vida del parásito. Estos factores podrían actuar en forma coordinada y competitiva en trans para influir sobre la estabilidad de los transcriptos TcSMUG, y afectar la expresión final de los productos durante el desarrollo (ver más adelante). Ensayos de competición para determinar la especificidad de las proteínas de unión al ARE. Se realizaron experimentos de competición con homoribopolímeros para confirmar la especificidad de las proteínas que reconocen al ARE (Figura 19). En primer lugar, los experimentos de desplazamiento muestran que eAREBP es selectivamente desplazada en presencia de cantidades crecientes de ARN poli(U), pero no por otros polímeros (Figura 19A). También se realizaron experimentos de competición con las secuencias ARE-sentido y ARE-antisentido transcriptas “in vitro”. El agregado de cantidades crecientes del ARN ARE-sentido en la mezcla de reacción, produce una reducción en la formación del complejo conteniendo eAREBP, el cual es dependiente de la concentración de competidor utilizada (Figura 19B). Por otro lado, el agregado de cantidades crecientes del ARE-antisentido no tiene efecto significativo en la interacción del ARE con eAREBP. En segundo lugar, las proteínas del estadío tripomastigote (tAREBPs) también se compiten en forma selectiva y eficiente con ARN poli(U), pero no con otros polímeros (Figura 19C). Este desplazamiento también se detecta con el poli (A) ARN A - poli (C) poli (G) ARN ARE-sentido, pero no con el ARE-antisentido poli (U) 10 100 1000 10 100 1000 10 100 1000 10 100 1000 (Figura 19D). Estos resultados confirman que las eAREBP B SMUG-L-ARE antisentido ARN poli (U) - 10 50 100 200 10 proteínas tAREBPs, al igual que eAREBP, reconocen en SMUG-L-ARE sentido 50 100 200 10 50 100 200 forma específica al ARE y que su alto contenido en D SMUG-L-ARE SMUG-L-ARE sentido antisentido ARN poli (U) poli (G) - poli (C) ARN C poli (A) eAREBP - 50 100 200 50 100 Uridina es importante para la interacción. 200 tAREBPs Figura 19 – Ensayos de competición de las proteínas de unión al ARE con distintos ARNs. A, Se realizó un gel nativo para resolver las reacciones de unión “in vitro” entre un extracto total de epimastigotes incubado con el elmento ARE sólo (-) o competido con cantidades crecientes de polímeros de ARN sin marcar. ARN, denota la presencia del ARN sin incubar con el extracto. La posición de eAREBP se indica en la izquierda del panel. B, Gel nativo para resolver las interacciones entre eAREBP y cantidades crecientes de los ARN poli(U), ARE-sentido y ARE-antisentido. C, Gel de retardo nativo para resolver las interacciones entre tAREBPs y el ARE en ausencia (-) o presencia de polímeros de ARN sin marcar como competidores. D, Gel de retardo nativo para resolver las interacciones entre tAREBPs y el ARE en ausencia (-) o presencia de cantidades crecientes de los ARNs ARE-sentido o ARE-antisentido. 65 Resultados Las proteínas que reconocen al ARE presentan una localización subcelular diferencial. La localización de eAREBP y tAREBPs se analizó por geles de retardo al igual que para los complejos de unión al GRE. Este experimento mostró que eAREBP es totalmente citoplasmática A o que estaría accesible para reconocer al ARE en el citoplasma y no en el núcleo (Figura 20A), B T C N P PS eAREBP Polisomas - ARNAsa A mientras que las proteínas tAREBPs se encuentran en cantidades similares en ambos + eAREBP compartimientos (Figura 20C). Algunas de las proteínas que reconocen al ARE en otros tipos celulares presentan localización polisomal, y esta particular ubicación estaría relacionada con su capacidad para modular la traducción (Nagy and Rigby, 1995; Winstall et al., 1995; Zhang et al., 1993). Como se describió anteriormente (Figura 15), el elemento ARE regula en forma positiva la ARN libre C T C N tAREBPs traducción en el estadío epimastigote. De esta manera, se intentó analizar si eAREBP, una proteína citoplasmática (Figura 20A), se encuentra asociada con polisomas. Para ello se prepararon polisomas del estadío epimastigote como se describió previamente (Gonzalez and Cazzulo, 1989). Después de la preparación y centrifugación a través de un colchón de sacarosa, el sobrenadante se guardó como fracción postribosomal (PS) y el “pellet” como polisomas (P). Todas las fracciones preparadas, incluyendo citoplasma (C) y núcleo (N), se analizaron en geles de retardo. Como se observa en la Figura 20A, la preparación polisomal tiene algo de actividad de unión, pero es de alrededor del 5% de la actividad de la fracción citoplasmática. De esta manera, para determinar si la falta de una banda de retardo intensa en la fracción polisomal, se debe a la presencia de ARN ricos en Uridina, los cuales pueden actuar como competidores secuestrando parte de eAREBP, el extracto fue pre-tratado con ribonucleasa A (ARNasa A) como se describió previamente (Akhtar et al., 2000). Luego del tratamiento con la nucleasa, esta fue inactivada antes de la reacción de unión “in vitro” al ARE (Figura 20B). Los resultados observados 66 Figura 20 - eAREBP reconoce el elemento ARE en el citoplasma, mientras que las proteínas tAREBPs están localizadas en núcleo y citoplasma. A, Localización subcelular de la proteína eAREBP según se determinó por incubación de extractos proteicos con el ARN marcado ARE y resuelto en un gel de retardo nativo. Parásitos del estadío epimastigote fueron lisados y separados en fracciones: total (T), conteniendo núcleos (N), citoplasma (C), polisomas (P) y sobrenadante postribosomal (PS). Se utilizaron las fracciones del extracto proveniente del mismo número de parásitos (10x106), para los ensayos de interacción ARNproteína. B, El extracto polisomal (P) del estadío epimastigote se pre-trató (+) o no (-) con ARNasa A durante 15 min. a temperatura ambiente e inactivado por el agregado de RNAsin antes de la incubación con el ARN. Los complejos formados fueron resueltos en un gel nativo 7%. C, Localización subcelular de las proteínas tAREBPs determinada a partir de una reacción de unión “in vitro” entre fracciones total (T), nuclear (N) y citoplasmática (C) de un extracto del estadío tripomastigotes derivado de células y el ARN ARE. Resultados demuestran que luego del tratamiento, la unión de eAREBP al ARE en la fracción polisomal incrementa alrededor de 4 veces, en comparación con el control sin tratamiento con nucleasa (Figura 20B). Esto sugiere la existencia de algún ARN competidor en la fracción polisomal, el cual podría estar reclutando eAREBP. Como se observa también en la figura, la sonda de ARN permanece intacta luego de la reacción con el extracto, descartándose así la presencia de alguna nucleasa asociada a polisomas que degrade al ARE. En resúmen, eAREBP es principalmente citoplasmática y está parcialmente asociada a polisomas, mientras que las proteínas tAREBPs están localizadas tanto en núcleo como en citoplasma. Especificidad de unión a ARN de la proteína eAREBP. Para deter minar la mínima porción reconocida por A eAREBP en el ARN SMUG-LARE, se sintetizaron cuatro ARN Secuencia SMUG-L-ARE AAUUUUAAAAUUUAUUUAAUGUUGUUUUGAUUUAUUUAUUUUAA AUUUUAUUUUUAUUUUUA P1 GUUUUGUUUUUGUUUUUG P2 AUGUUGUUGUUGUUGUUG fueron utilizadas en reacciones de unión “in vitro” y análisis por P3 AUGGUGUUGGUGUUGGUG P4 AUAUUAUUAUUAUUAUUA retardo en gel. La interacción de eAREBP con SMUG-L-ARE, se reduce cuando dos motivos AUUUUUA de sus extremos se remueven, como es el caso del oligo SMUG-L-ARE-1 (Figura 21B), demostrando importancia en su el reconocimiento por eAREBP. Además, la unión al nonámero B PLC-ARE muestran las secuencias de ARNs correspondientes que SMUG-S-ARE AUAUAUAUAUAUAUAUAUAUAUAUUUAUUU SMUG-S-ARE SMUG-L-ARE-2 AUUUAUUUAUUUA PLC-ARE HEX-T-ARE UUUUAUUUAUUUU HEX-T-ARE SMUG-L-ARE UUAUUUAUU SMUG-L-ARE-3 ARNs diferentes derivados de éste. En la Figura 21A se SMUG-L-ARE-1 AUUUAUUUAAUGUUGUUUUGAUUUAUUUAUU SMUG-L-ARE-3 SMUG-L-ARE-1 SMUG-L-ARE-2 S P1 P2 P3 P4 eAREBP Figura 21 – Especificidad de unión a ARN de eAREBP. A, Se muestra una lista de los ARNs utilizados en los geles de retardo del panel B. B, Ensayos de retardo en gel entre sustratos de ARN y extractos totales del estadío epimastigote. La posición de la proteína eAREBP se indica con una flecha a la izquierda del panel. La incubación del ARN con el buffer de reacción en ausencia del extracto proteíco, no mostró banda de retardo alguna (resultado no mostrado). UUAUUUUAUU (SMUG-LARE-2) sólo se detecta cuando la película se sobre-expone (resultado no mostrado). La interacción con el sustrato ribonucleico incrementa levemente cuando se adicionan dos Uridinas más en cada uno de sus extremos (SMUG-L-ARE-3). Entonces, el nonámero no es la secuencia mínima requerida para la unión, como sucede con proteínas de unión a elementos ARE de otros tipos celulares. Aunque esta interacción es poco eficiente, puede incrementarse si el mismo está localizado en un contexto rico en Uridina (Figura 21B). Otras secuencias del tipo ARE se encuentran en las regiones 3’-NC del parásito, como el grupo S de la familia TcSMUG (SMUG-S-ARE), el transportador de hexosa (HEX-T-ARE) y la fosfolipasa-c (PLCARE) (ver Tabla 2). Por lo tanto, también se evaluó la capacidad de eAREBP de interaccionar con estos diferentes motivos ARE. Luego de realizar ensayos de retardo en gel, concluímos que de las tres secuencias ARE, eAREBP sólo reconoce el ARN SMUG-S-ARE, presente en la region 3’-NC del grupo de mucinas TcSMUG-S (Figura 21B). 67 Resultados Estos resultados demuestran que eAREBP se une en forma selectiva a alguna de las regiones ARE evaluadas y claramente reconoce secuencias ricas en Adenosina y Uridina de los transcriptos de mucina TcSMUG-L y -S, los cuales contienen motivos AUUUA, AUUUUA y AUUUUUA (Figura 21A). A partir de la secuencia del ARN SMUG-SARE, se sintetizaron 4 ARNs derivados y denominados P1, P2, P3 y P4 (ver Figura 21A) con el objetivo de evaluar más en detalle la afinidad y especificidad de la interacción ARN-proteína. Por un lado, eAREBP reconoce fuertemente P1, así como el ARN SMUG-SARE. Por otro lado, los ARNs P2 y P4 también son reconocidos aunque más levemente, mientras que P3 no es sustrato de eAREBP (Figura 21B). Purificación parcial de la proteína eAREBP. El objetivo siguiente fue caracterizar e identificar la secuencia de la proteína eAREBP. Para ello, se realizaron una serie de pruebas bioquímicas para determinar cual es la mejor combinación de pasos que nos permitieran obtener la banda de ~100kDa relativamente pura para un análisis de secuencia o espectrometría de masa. Sólo nos remitimos a la caracterización de ésta proteína, ya que se pueden obtener grandes cantidades de parásitos sólo del estadío epimastigote en cultivo. A diferencia, la purificación de las proteínas tAREBPs requiere parásitos de los estadíos tripomastigote y/o amastigote intracelulares, difíciles de obtener en grandes cantidades. Varios pasos cromatográficos se ensayaron en colaboración con el Dr. Marcelo Guerin (IIB-UNSAM), quien fue de gran ayuda para la elaboración de un criterio adecuado de purificación. Las columnas evaluadas fueron: A) Cromatografías de Intercambio Iónico: DEAE, Mono Q y Mono S; B) Cromatografía de Filtración en gel: BioSil SEC-250; C) Cromatografía de Hidrofobicidad, Fenil-Sefarosa; D) Cromatografía de afinidad: poli(U) Sefarosa y Heparina. El protocolo preparativo final para la purificación se indica en la Figura 22 y fue el siguiente: 1-Cromatografía de intercambio catiónico, DEAE. Luego de varias pruebas analíticas y preparativas con distintas cantidades de parásitos, se decidió comenzar la purificación a partir de 100gr. peso húmedo de epimastigotes de T. cruzi (Figura 22A). El extracto se preparó de acuerdo a las indicaciones descriptas en Materiales y Métodos. En resumen, los parásitos se resuspendieron en 500 ml de “buffer” de lisis y el extracto resultante se clarificó por ultracentrifugación a 100.000g. El sobrenadante se sembró en una columna DEAE equilibrada con “buffer” Tris-HCl 20 mM pH 7.6. Para la elución de las proteínas retenidas en la matriz, se realizó la elución con un gradiente salino de 0-1 M NaCl en “buffer” Tris-HCl 20 mM pH 7.6. Luego de la elución, se tomaron 10 μl de cada fracción y se midió la capacidad de cada una de reconocer la ribosonda ARE en un ensayo de retardo en gel (Figura 22B). La actividad de eAREBP se detectó entre las fracciones 35 y 47 (Figura 22B), correspondiente a una concentración de NaCl ~250mM. Estas fracciones se juntaron (“pool” ~50 ml), y el mismo se diluyó 1/10 en “buffer” MES 50 mM pH 6.2, de manera tal de alcanzar el pH adecuado y una concentración salina de ~30 mM acorde a los requerimientos del paso siguiente de purificación. 68 Resultados A Extracto epimastigotes (~500 ml) B NaCl Fracciones de elucion Pool Ex P 1517 19 2123 25 27 29 31 33 35 37 3941 4345 47 49 DEAE Sefarosa Elucion: 0-1 M NaCl Heparina HYPER D C NaCl Elucion: 0-1 M NaCl Mono S Ex Fracciones de elucion Pool P 2 4 6 8 10 12 14 1618 20 22 24 26 28 30 32 34 Elucion: 0-0.5 M NaCl Fenil Sefarosa NaCl D Elucion: 1-0 M (NH4) SO 2 Fracciones de elucion 4 Pool Ex P 1 2 3 4 5 6 7 8 9 10 11 12 13 14 151617 1819 SDS-PAGE Tincion Coomasie Blue UV cross-linking E (NH4)2 SO4 Fracciones de elucion Pool Ex 20 21 22 23 24 25 26 27 28 29 30 3132 3334 35 36 37 Figura 22 - Esquema de los pasos bioquímicos utilizados en la purificación de eAREBP. A, Se utilizó un extracto total de proteínas proveniente de 100 gr de parásitos del estadío epimastigote, el cual fue resuspendido en buffer hipotónico conteniendo Chaps 0.5%. Los pasos utilizados para la purificación fueron: B, DEAE Sefarosa, C, Heparina, D, Mono S y E, Fenil Sefarosa. Cada una de las fracciones provenientes de percolado y la elución de las matrices fueron utilizadas para medir la actividad de unión de eAREBP al ARN ARE marcado con [α32P]UTP. Para ello, 10 μl de cada fracción se incubaron con el ARN y las reacciones se resolvieron utilizando geles de retardo nativos de poliacrilamida. Las fracciones que forman parte del “pool” obtenido luego de la elución en cada paso se muestra encima de los paneles. 2-Cromatografía de afinidad, Heparina. Como segundo paso de purificación, se utilizó una columna de Heparina, ya que es un polianión y generalmente su estructura es útil para purificar proteínas de unión a ácidos nucleicos y/o proteínas con una alta carga positiva. El “pool” de elución de la columna DEAE Sefarosa, se sembró en la columna de afinidad Heparina HyperD (BioSepra), previamente equilibrada en “buffer” A (MES 50 mM pH 6.2), en forma manual utilizando una bomba peristáltica y un flujo de 3 ml/min. Luego de lavar y eluir con el “buffer” B (MES 50 mM pH 6.2, 1 M NaCl) haciendo un gradiente salino entre 0-1 M NaCl, se midió la actividad en geles de retardo. Como se observa, eAREBP eluyó entre las fracciones 8-14 (Figura 22C). 69 Resultados 3-Cromatografía de intercambio aniónico, Mono S. El “pool” de la columna Heparina (~10ml) se diluyó nuevamente 1/10 en el mismo “buffer” MES hasta alcanzar una concentración salina baja de alrededor 25 mM NaCl. Este “pool” se cargó en una columna Mono S y otra vez el gradiente se realizó con el “buffer” indicado variando la concentración salina entre 0-0.5 M NaCl. La actividad se identificó a partir de retardo en gel nativo en las fracciones 7-11 (Figura 22D). 4-Cromatografía de hidrofobicidad, Fenil Sefarosa. El cuarto y último paso de purificación fue una columna de hidrofobicidad. Para tal paso, el “pool” de la Mono S (~7ml), se diluyó en “buffer” Tris-HCl 20 mM pH 7.6 y en presencia de Sulfato de Amonio 1 M. La elución se realizó en un gradiente descendiente de concentración de Sulfato de 1 a 0 M, utilizando un “buffer” Tris-HCl 20 mM pH 7.6 5% Sacarosa. La actividad de unión al ARE disminuye un poco en comparación con el paso anterior, porque el Sulfato de Amonio interfiere con la reacción de medición. De todas maneras, el pico de actividad se detectó entre las fracciones 25-30 del gradiente, aproximadamente 0.25 M de sal, pero con mayor actividad en la calle 27 (Figura 22E). 5-Identificación de eAREBP en geles de SDS-PAGE como una banda de ~100kDa. A partir de las fracciones con actividad de la columna de hidrofobicidad se realizó un gel SDS-PAGE 7.5%, el cual se tiñó con “Coomasie-Blue” para identificar bandas proteicas (Figura 23A). En este gel se observó una banda prominente de ~100kDa entre las fracciones 25-30 y especialmente en la calle 27 (Figura 23A). En el gel de retardo de la Figura 22E se observa que la actividad de unión al ARE también aparece en tales fracciones, especialmente en la número 27. Espectrometría de masa MALDI-TOF e identificación de un homólogo putativo en células eucariotas superiores. Luego de la identificación de la banda de ~100kDa correspondiente a eAREBP (Figura 23A), las fracciones con actividad se mezclaron y se las dividió en dos partes. Ambas partes se resolvieron por electroforesis en gel de poliacrilamida desnaturalizante SDS-PAGE 7.5%, en presencia del “buffer” Caps pH 10 (ver Materiales y Métodos). Una parte (50%) se transfirió a una membrana de PVDF. La banda identificada, fue cortada y enviada al Dr. Ulf Hellman (Uppsala University) para el análisis del N-terminal por la reacción de Edman. A partir de éste análisis no se pudo obtener secuencia de aminoácidos, ya que la región N-terminal de la proteína estaba bloqueada. La segunda parte de la muestra (50%) se cortó del gel SDS-PAGE para su digestión con tripsina. Los productos provenientes de la digestión tríptica fueron analizados por espectrometría de masa por MALDI-TOF y la masa de los péptidos protonados obtenidos (Figura 23B) se utilizó para buscar en la base de datos no redundante PROFound. Dentro de la lista de “hits”, el primero correspondió a una proteína de unión a ARN de Mus musculus, denominada DEF3. La única particularidad de esta proteína es que está ausente en un tipo de cáncer de pulmón (Drabkin et al., 1999). Aunque se describió que reconoce poli(G) “in vitro”, no existen evidencias que indiquen su función “in vivo”. En la Tabla 3 se listan los péptidos 70 Resultados compartidos entre las proteínas DEF3 y eAREBP, y sus respectivas masas medias y diferencias en masas moleculares computadas. A kDa 114 Pool 24 25 26 27 28 29 30 31 32 eAREBP 97 66 Figura 23 - Identificación de eAREBP como una proteína de ~100kDa y análisis por espectrometría de masa MALDI-TOF. A, Gel SDS-PAGE teñido con Coomasie Blue e identificación de eAREBP como una proteína de 100kDa en la fracción 27. El asterisco indica la posición de bandas proteicas que copurifican con eAREBP. B, Espectro de masas de la proteína eAREBP digerida con tripsina. Se indican las masas (Daltons) correspondientes a los péptidos utilizados en la búsqueda en la base de datos PROFound (ver en Tabla 3 la lista de los péptidos). * 45 B cuentas 16000 14000 12000 10000 8000 6000 4000 2000 800 1000 1200 1400 1600 1800 m/z Masa Media 790.373 1002.470 1054.362 1103.422 1149.464 1188.480 1214.434 1288.541 1333.564 1364.499 1493.534 1633.625 Masa Computada 790.419 1002.553 1054.446 1103.531 1149.589 1188.588 1214.542 1288.531 1333.583 1364.656 1493.638 1633.758 Residuos desde a 949 955 729 738 273 281 603 611 588 597 167 176 282 291 97 108 149 158 986 996 435 446 936 948 Error (Da) -0.046 -0.083 -0.084 -0.110 -0.124 -0.108 -0.107 0.009 -0.018 -0.157 -0.103 -0.133 Secuencia LACLLCR MVAVNLATGK EVGPCMEFK (1)+0@m KRDEGQESR QPNQPRPADK SRDVPPTDFR DREMPPMDPK GGDFSSSDFQSR ETPHMNYRDR (1)+0@m QSEQELAYLER DAQQDLQDQDYR KENEDDKLTDWNK Tabla 3 - Identificación de péptidos en eAREBP idénticos a la proteína de unión a ARN de mamíferos DEF-3. Se muestra la secuencia de los péptidos compartidos entre eAREBP y la proteína DEF3 de Mus musculus, así como también las masas moleculares de los péptidos de DEF3 (Daltons) y las diferencias (<1 Dalton) con los péptidos de eAREBP. Péptidos medidos: 43; Péptidos iguales: 12; Cobertura mínima de secuencia (12%). Clonado de earebp con el uso de oligos degenerados sintetizados a partir de los péptidos identificados por espectrometría de masa. Con el objetivo de clonar earebp, se sintetizaron varios juegos de oligos degenerados (ver sección Anexo II) a partir de la secuencia de los péptidos compartidos entre eAREBP y el putativo homólogo DEF3 (ver Figura 24). A partir de la posición de los oligonucleótidos en la secuencia de DEF3 (Figura 24), se procedió a combinarlos para realizar ensayos de PCR. De esta manera, se clonaron 71 Resultados varias bandas de amplificación, las cuales presentaban tamaños lógicos según la posición y distancia de los oligonucleótidos en DEF3 (ver Figura 24). Sin embargo, ninguna de las bandas que se secuenció presentó identidad con ésta proteína. Alternativamente, se buscó en diferentes bases de datos con información sobre el proyecto de secuenciación del genoma de diversas especies de tripanosomas, pero tampoco se encontró algún clon con identidad apreciable con la proteína murina DEF3. 1 MWGDSRSANRTGPFRGSQEEGFAPGWNRDYPPPPLKSHARERHSGNFPGRDSLPFDFQGHSGPPFANVEEHSFSYGGR 81 PHGDYQGGEGPGHDFRGGDFSSSDFQSRDSAQLDFRNRDIHSGDFRDREGPPMDYRGGDGTHMDYRGRETPHMNYRD 160 SHTVDFRSRDVPPTDFRGRGTYDLDFRGREGSHSDFRGRDLSDLDFRSRDQSRSDFRNRDVSDLDFRDKDGTQVDSRG 241 ATTDLDFRNRDTPHSDFRGRHRTRTYQDFRGREVGPCMEFKDREMPPMDPKVLDYIQPSPQEREHSGMNMNKREESI 320 MERPAFGVQKGEFEHSETREGETQSGTFEHESQSDFQNSQSPVQEQDKPKLSGGEQQSSDAGLFKEEGDLDFLGQQD 400 RSIEYCDVDHRLPGNQIFGYGQSKSFSQGKMSRDAQQDLQDQDYRTGPSEEKPNRLIRLSGVPENATKEEILNAFRTSDG 477 KDLQLKEYNTGYDYGYVCVEFSLLEDAIGCMEANQGTLMIHDKEVTLEYVPSPDFWYCKRCKASTGGHQSSCSFCKGPK 555 KQELVSYPQPQKTSIPVPSEKQPNQPRPADKEHELRKRDEGQESRLGHQKRDTDRYFPHSRREGLTFRRDREKEPWSG 632 GESKTIMLKRIYRSTPPEVIVEVLEPCVHLTTANVRIIKNRTGPMGHTYGFIDLDSHAEALRVVKILQNLDPPFSIDGKMVAVN 705 GKRRNDSGDHSDHMHYYQGKKYFRDRRGGNRNSDWSSDTNRQGQQSSSDCYIYDSTTGYYYDPLAGTYYDPNTQQEVY 786 PQDPESPDEEEIKEKKSTSQGKSNSKKETSKRDGKEKKDRGMTKFQESTSEGKPPLEDVFKKPLPPTVKKEESPPPPKVV 864 LIGLLGEYGGDSDYEEEEEEEQAPPVQPQPQPREEMTKKENEDDKLTDWNKLACLLCRRQFPNKEVLTKHQQLSDLHK 942 IHRKIKQSEQELAYLERREREGRFKEKGNDRREKLQSFDSPERKRIKYSRETDSDRNPVDKEDTDTSSKGGCAQQATGWR 1020 AGLGYSHPGLGSSEEIEGRMRGPGGGPPGRTSKRQSNETYRDAVRRVMFARYKEL Peptidos con masa molecular identica a DEF3 segun analisis por MALDI-TOF. Cebadores utilizados en reacciones de PCR para clonar earebp. Figura 24 - Secuencia de la proteína DEF3 y los oligonucleótidos utilizados para clonar earebp. Esquema en donde se muestra la secuencia de DEF 3 de Mus musculus y la posición de los oligonucleótidos de ADN utilizados para clonar earebp. Ver Tabla en la Sección Anexo para la secuencia de cada oligonucleótido sintético. 72 Resultados PARTE 4 - Estructura y función de proteínas de unión a ARN (TcUBPs). Con el objetivo de encontrar otros factores que actúen en trans- sobre el ARE del ARNm de TcSMUG-L, se realizó un enfoque alternativo al de la purificación de proteínas. Se buscaron secuencias putativas que codifiquen proteínas de unión a ARN en las bases de datos de EST y GSS del proyecto de secuenciación del genoma de T. cruzi. El rastreo se realizó en la base de datos del NCBI utilizando como entrada la secuencia en aminoácidos de la proteína de unión a ARE de eucariotas superiores HuR (Fan and Steitz, 1998). Este factor funciona en el camino de estabilización de ARNms conteniendo AREs en sus regiones 3’-NC (Fan and Steitz, 1998). Durante la búsqueda de nuevos genes, se identificó una secuencia que presentaba homología con HuR (EST TENG0035). Para clonar en forma completa su ADNc, se realizó una reacción de RT-PCR con ARN total del estadío epimastigote y un cebador que se une a una región conservada en las proteínas de unión a ARN del tipo RRM, llamado RNP-1 (ver más adelante). Para la primer reacción de PCR se utilizó el cebador RNP-2 antisentido, el cual se une río arriba del RNP-1 y la secuencia del SL del extremo 5’-terminal. Para esta reacción se tomó ventaja del hecho de que todos los ARNms de T. cruzi presentan el SL en su región 5’, agregado por una reacción de trans-“splicing” (ver Introducción). A partir de este ensayo se identificaron en geles de agarosa teñidos con bromuro de etidio un conjunto de bandas de entre 200-300 pbs, el tamaño esperado en esa región para el ADN del EST TENG0035. Estos fragmentos se clonaron en el vector pGEM-T Easy. El análisis de la secuencia de 6 clones demostró que 4 eran 100% homólogos a TENG0035, sin embargo otros 2 clones presentaban una región N-terminal levemente diferente y más pequeña. En segundo lugar, se realizó otra reacción de RT-PCR para clonar los extremos 3’de ambas secuencias. Para ello se utilizó ARN total del estadío epimastigote y el cebador oligo(dT) -ancla. Luego, para la reacción de PCR se utilizó el cebador ancla y cebadores 18 que se unen a las regiones N-terminal de los clones identificados (ver Materiales y Métodos). Finalmente, dos bandas de tamaños de ~1000 y ~800 pbs fueron identificadas (resultado no mostrado) y se llamaron Tcubp-1 y Tcubp-2 respectivamente, por T. cruzi “Uridinerich binding protein”, debido a su identidad con proteínas que unen elementos ricos en Uridina de otros tipos celulares. En la Figura 25 se muestra una comparación de los esquemas de ARNms deducido a partir de la secuencia de los ADNc de Tcubp. Comparación entre Tcubp-1 y Tcubp-2. El análisis de las secuencias provenientes de los clones completos Tcubp-1 y Tcubp- 2, demostró lo siguiente: el ADNc de Tcubp-1 está compuesto por 1017 pbs, mientras que Tcubp-2 presenta sólo 854 pbs. El ARNm de Tcubp-1 es más largo que Tcubp-2, tanto en la región codificante como en las regiones no codificantes (Figura 25A). El marco abierto de lectura es la única porción de los ARNms que presenta una gran identidad entre ambos clones (~70%), y de esta región, las porciones con menor identidad son las que codifican los extremos N- y C-terminales. En cuanto a las regiones no codificantes, se observa que son muy diferentes. Sólo 37% de identidad se detecta en la región 3’-NC, mientras que no existe identidad alguna en la región 5’-NC (Figura 25A). 73 Resultados Estructura de los productos deducidos de Tcubp-1 y Tcubp-2. TcUBP-1, está compuesto por 225 aminoácidos y presenta una región N-terminal de 34 aminoácidos la cual es rica en Glutamina (Figura 25). Esta región es seguida por el dominio de unión a ARN o RRM, compuesto por ~92 aminoácidos, el cual presenta los motivos característicos llamados RNP1 y RNP2. RNP2 es un A 5'NC marco abierto lectura 3'NC SL poli-(A) Tcubp-1 hexapéptido presente en la hoja-β1 y RNP1 es un octapéptido localizado ----------- 67 nt (0%) SL (70%) ter minal del motivo RRM, que generalmente presenta funciones regulatorias en términos de afinidad y estabilización de la unión con el ARN (Avis et al., 1996; DeMaria et al., 1997), está compuesta por 14 aminoácidos ubicados a 8 residuos después de la hoja-β4 (ver Figura 25). RNP-2 Tc UBP-1 Tc UBP-2 1 1 RNP-1 en Glicina, por lo cual se la denominó región rica en Glicina. Como característica particular, también se destaca la presencia de residuos de T irosina distribuídos en for ma periódica entre las Glicinas (Figura β2 β3 α2 Tc UBP-1 51 PTTVDEVQLRQLFERYGPIESVKIV C DRET RQSRGYGFVKFQSGSSAQQA Tc UBP-2 37 PTTVDEVQLRQLFERYGPIESVKIV C DRET RQSRGYGFVKFQSGSSAQQA α2 β4 Gli RV Tc UBP-1 101 IAGLNGFNILNKRLKVALAASGHQR --P G IAGAV G DGN GY L G A YGGYGAY Tc UBP-2 87 IAGLNGFNILNKRLKVALAASGHQR NRN G VSTGF G AYG GY - G G YGGYGAY Gln Tc UBP-1 146 AYPS A ANPYAQQ QMM A M YQQ YMM QAPQQTPHQGQQQQMPLPSHPQQQSP P Tc UBP-2 132 AYPS A ANPYAQQ QMM T M QHP YMM T------------------------- - Tc UBP-1 196 QQQ Q QS PS S P Q Q QPQSQQQSQAARPVRR Tc UBP-2 158 --- Q SV PS V P R Q ---------------Dominio Auxiliar C RRM TcUBP-2 de ellos está compuesto por 16 aminoácidos con un alto contenido MSQIPLVSQYDPYGQTA Q L Q QLQQQ QQ QHIP P T QMNPEPD V LRNLMVNYI --------------MSQ Q M Q YYAPR QQ LQQA P A QMNPEPD L LRNLMVNYI α1 identidad alguna con la misma porción en TcUBP-2 (ver más adelante). Hacia el C-terminal, se TcUBP-1, este dominio se puede dividir en dos módulos, el primero ----------- 210 nt β1 TcUBP-1 generalmente sirve para la interacción con otras proteínas. En poli-(A) ----------- 501 nt B Esta región se llamó RV, por Región Variable, ya que no presenta observa el dominio auxiliar de las proteínas de unión a ARN, que (37%) Tcubp-2 143 nt en la hoja-β3, en todas las proteínas con motivos RRM. La región C- 345 nt 638 nt 35 0 0 20 RV RNP2 RNP1 RNP2 RNP1 Gli 126 140 112 121 Gln 157 225 144 167 Figura 25 - Tcubp-1 y Tcubp-2, codifican polipéptidos con identidad a proteínas de unión a ARN. A, Representación esquemática de los ARNms Tcubp, deducida de la secuencia de los ADNc. SL, “spliced leader”. El largo de cada región (marco abierto de lectura y regiones no codificantes 5’ y 3’) se indica en nts debajo de cada esquema. El porcentaje de identidad entre cada una de las regiones para los dos esquemas comparados se muestra entre paréntesis. B, Comparación de las proteínas TcUBPs derivada de la secuencia del ADNc. El alineamiento se realizó utilizando el programa Clustal W (Thompson et al., 1994). Se indican los diferentes dominios: RV, región variable; Gli, región rica en Glicina; Gln, región rica en Glutamina y los octapéptidos RNP-2 y el hexapéptido RNP1 dentro del dominio RRM. La predicción de la estructura secundaria, mostrando las hélices α y hojas β, se determinó utilizando el Software Protein Prediction según: (http:// www.embl-heidelberg.de/). C, Esquema de las proteínas TcUBPs mostrando los diferentes módulos identificados: RRM, dominio de unión a ARN; RV, región variable; Gli y Gln. 25B). El segundo módulo, está compuesto por ~68 aminoácidos y se caracteriza por su alto contenido en Glutamina (~45%). A partir de programas que determinan la estructura secundaria, se predice que es una región sin estructura o “random-coil” (ver más adelante). TcUBP-2 está compuesto por 167 aminoácidos y al igual que TcUBP-1 presenta una región N-terminal corta compuesta por 20 aminoácidos, y rica en Glutamina (45% de los residuos). El dominio RRM (~92 aminoácidos) presenta los motivos conservados RNP1 y RNP2, y es 98% idéntico al de TcUBP-1, con un cambio en una sola posición y 74 Resultados localizado antes de la hoja-β1 (Figura 25B). Nuevamente, el C-terminal del RRM o región RV está compuesta por sólo 8 aminoácidos en comparación con la de TcUBP-1 que presenta 14 aminoácidos. Como se mencionó anteriormente, existe una muy baja identidad en esta región entre ambas proteínas. Finalmente, el extremo C-terminal está también compuesto por una región rica en Glicina, más larga que la de TcUBP-1 y con motivos YGG solapados. Este dominio auxiliar del C-terminal carece de la región rica en Glutamina observada en TcUBP-1 (Figura 25). Tcubp-1 y Tcubp-2 son genes de copia única por genoma haploide y pertenecen a una familia de genes que codifica para proteínas con motivos RRM. Con el objetivo de determinar el número de copias de Tcubp-1 y Tcubp-2 presentes en el genoma de T. cruzi se llevaron a cabo ensayos de “Southern blot” (Figura 26). Se realizaron hibridizaciones en condiciones de alta rigurosidad (65ºC y 20 mM NaH PO ), 2 4 utilizando ADN genómico del clon CL-Brener digerido con varias enzimas de restricción y sondas de ADN correspondientes a las secuencias presentes en la región N-terminal de TcUBP-2 y dominio de unión a ARN (RRM) de A y la hibridización con la sonda común (RRM) muestra el doble de bandas en algunas calles. Estos resultados sugieren que ambos clones están presentes en una copia por genoma haploide (Figura 26B). A partir de un ensayo de “Southern blot” utilizando la sonda RRM en condiciones de baja rigurosidad (55ºC y 200 mM NaH PO ), se 2 4 detectaron varias bandas (resultado no mostrado). Estos resultados indican que si bien Tcubp-1 y Tcubp-2 son genes de copia única, pueden formar parte de una familia de genes cuyos miembros presentan mayor identidad en la porción que codifica para el dominio RRM. La diversidad de esta familia es objeto de estudio del estudiante Javier De Gaudenzi, quien actualmente se encuentra realizando el Doctorado en el laboratorio del Dr. Frasch. El análisis preliminar de la familia que codifica proteínas de unión a ARN del tipo-RRM indica que está compuesta por varios miembros. Hasta el momento, se identificaron y clonaron 6 genes que codifican para proteínas del tipo-RRM en el parásito. 75 Hc 1 Ha NH2 diferente entre ambos clones, mientras que la sonda RRM es capaz de reconocer ambos clones. Como Ha 501 Hc: HincII Ha: HaeIII NH2-RRM Kpb 23.1 9.4 6.6 4.3 9.4 6.6 4.3 2.3 2.0 2.3 2.0 1.4 1.4 NH2 HincII EcoRI PstI Kpb 23.1 DdeI HaeIII B EcoRI PstI DdeI se observa, la hibridización con la región N-terminal de Tcubp-2 demuestra un bajo número de bandas, RRM NH2 TcUBP-2 HaeIII HincII TcUBP-2 (Figura 26A). La primer sonda permite ver sólo a TcUBP-2, porque la región N-terminal es NH2-RRM Figura 26 - Tcubp-1 y Tcubp-2 son genes de copia única. A, Esquema del ARNm de Tcubp-2 y sus distintos dominios: NH 2 y RRM. Se indica la posición en la que cortan las enzimas de restricción HincII (Hc) y HaeIII (Ha), así como también la posición de cada una de las sondas de ADN sintetizadas (NH2 y NH2-RRM). B, Ensayo de “Southern blot” realizado sobre ADN genómico del clon CL-Brener de T. cruzi digerido con las enzimas de restricción que se indican e hibridizados con las sondas: NH2 y NH2-RRM de TcUBP-2. Resultados Tcubp-1 y Tcubp-2 se expresan en un mismo ARN de tamaño mayor que el esperado para el ARNm maduro. Al realizar un ensayo de “Northern blot” con sondas que reconocen específicamente Tcubp-1 y Tcubp-2 se observaron resultados no esperados. En primer lugar, ambas sondas dan señal a una altura mucho mayor (~5.5 kb) que la del tamaño esperado para los ARNms Tcubps (~1 kb). En segundo lugar, ambas sondas colocalizan a la misma altura (Figura 27A). A pesar de que los ARNms maduros para ambos genes existen, ya que se clonaron bandas de RT-PCR realizadas con oligo-(dt) y SL (ver Materiales y Metodos), es posible sugerir que existe un transcripto dicistrónico que contiene Tcubp-1 y Tcubp2. Además, en ensayos de inhibición de la transcripción con ActD se observa que el transcripto es estable (Figura 27B). A partir del rastreo de una biblioteca de cósmidos, el Lic. J. De Gaudenzi identificó (Figura 27C) y secuenció dos cósmidos que presentan ambos genes separados por una región intergénica de ∼3 kpb (ver mapa en Figura 27D). A B NH2 Sonda TcUBP-1 C ActD 2 6 NH2 TcUBP-2 RRM dicistron TcUBP-2/-1 Kb Tiempo (hrs) 0 8 0 CHX / ActD 2 6 8 dicistron 5.5 TcUBP-2/-1 TcUBP-1 TcUBP-2 2.4 2.0 1.5 RRM Tcubp-2 pA ag pA 0.55 Kbp Hc TcUBP-2 NH2 TcUBP-1 Nh2 TcUBP-1 Cooh Sonda ag TcUBP-1 NH2 TcUBP-2 D 2.8 Kbp Hinc II 8 23 Kb TcUBP-1 TcUBP-2 pA Hinc II 8 23 5.5 24S α 24S β 18S 2.4 2.0 1.5 24S α 24S β 18S Hinc II Cosmido 8 23 0.65 Kbp 2.5 Kbp Hc E Hc Tcubp-1 2. Kbp Referencias pPy Hi S Hc tractos GAAAA Figura 27 - Un dicistrón estable contiene Tcubp-1 y Tcubp-2. A, Ensayos de “Northern blot” hibridizados con la sondas RRM, N-terminal de TcUBP-1 y N-terminal de TcUBP-2 sobre filtros de ARN proveniente de parásitos del estadío epimastigote. B, Ensayo de “Northern blot” realizado con la sonda RRM sobre filtros de ARN del estadío epimastigote tratatos con ActD o CHX por 2 hrs y ActD. En el panel A y B se marca la posición de los ARNr de T. cruzi. C, Ensayos de “Southern blot” realizados sobre los cósmidos aislados de una biblioteca ordenada (8 y 23), conteniendo Tcubps, con sondas que reconocen específicamente las porciones RRM, el N-terminal de TcUBP-1 y el N-terminal de TcUBP-2. D, Esquema donde se muestra la posición de Tcubp-1 y Tcubp-2 en el genoma, según la secuencia de un cósmido de ADN de T. cruzi. Identificación de ortólogos de las proteínas TcUBP en otros parásitos kinetoplástidos. Para identificar ortólogos de las proteínas TcUBP en otros parásitos, las bases de datos de EST y GSS de T. cruzi, T. brucei y Leishmania se rastrearon con la secuencia del dominio RRM de TcUBP-1. Se identficaron cuatro secuencias nuevas con los números de acceso al GenBank: AZ050633, AQ638822, AQ637856 y AQ654405. Estos clones presentan una identidad de entre 35-50% en el dominio RRM, siendo los motivos RNP1 y RNP2 las regiones más conservadas entre todos los clones. Si la búsqueda se realiza a partir de la página “web” del Centro Sanger de Secuenciación (http://www.sanger.ac.uk/ Projects) se detectan dos clones con alta identidad a TcUBP-1, (Números de Acceso al 76 Resultados GenBank: AQ846565 de T. brucei y 0.15543 de Leishmania). La comparación de estas dos secuencias con TcUBP-1 muestra una identidad ~70%, y nuevamente el dominio RRM es la porción mas conservada entre las tres proteínas (resultado no mostrado). Estos resultados sugieren que secuencias altamente idénticas a TcUBP-1 están conservadas en diferentes especies de parásitos protozoarios. Comparación entre las proteínas TcUBPs y sus homólogos en otros tipos celulares. Con el objetivo de identificar secuencias idénticas a las proteínas TcUBPs en otros organismos, se realizó una búsqueda en la base de datos del NCBI. A partir de éste enfoque se detectan proteínas de unión ARN del tipo RRM de diversos organismos, incluyendo Drosophila melanogaster, Triticum aestivum, Oryza sativa, Megaselia scalaria, Gallus gallus, Homo sapiens y Arabidopsis thaliana , entre otros. Como se indicó anteriormente, la región de mayor identidad es el dominio RRM. En células de eucariotas superiores, el RRM se asemeja principalmente al descripto para las proteínas de la família Hu de mamífero, U1A y Sxl de Drosophila (Figura 28). RNP-2 RNP-1 TcUBP-1 35 - MNPEPDVLRNLM VN YIP TTVDEVQL R----QLFE R YGPIESVKIVCD RET RQ SRG YGFVKFQSGSSAQQAIA GLN GF NI L NKRLKV ALAASGHQRP Sxl 117 - MNDPRASNTNLI VN YLP QDMTDREL Y----A LFR AIGPINTCRIMRD YKT GYSFG YAFVDFTSEMDSQRAIK VLN GITV R NKRLKV SYARPGGESI HuD 32 --AATD DSKTNLI VN YLP QNMTQEEF R----SLFG SIGEIESCKLVRD KIT GQ SLG YGFVNYIDPKDAEKAIN TLN GLRL Q TKTIKV SYARPSSASI U1A 1 M A VPET R PNHTIYIN NL NEKIKKDEL KKSLYA IFS Q FGQILDILVSRS LKM R - - -G QAFVIFKEVSSATNALRSMQ GF PF Y DKP M R I QYAKTDSDII Figura 28 - El dominio RRM de la proteína TcUBP-1 se asemeja al de las proteínas de unión a ARN de otros tipos celulares. Alineamiento de secuencia de aminoácidos del dominio RRM de TcUBP-1, RRM1 de HuD (mamífero), RRM1 de Sxl (D. melanogaster) y U1A117 (mamífero). Los residuos idénticos entre las 4 proteínas están indicados en negro, y entre 2 o 3 proteínas en gris. Los espacios estan introducidos para un mejor alineamiento. Se indica la posición de los motivos RNP2 y RNP1. Aunque las zonas de mayor identidad son los motivos RNP1 y RNP2, ciertos residuos, especialmente hidrofóbicos, también están conservados entre las proteínas, y en particular son los que componen las hojas-β (Figura 28). Sin embargo, no existe identidad clara en las regiones fuera del dominio RRM entre las proteínas TcUBP-1, HuD, Sxl y U1A. A pesar de esto, algunas porciones de los dominios auxiliares especialmente en la región rica en Glicina de TcUBP-2, como las repeticiones YGG, y también la composición de residuos del C-terminal en TcUBP-1, como por ejemplo el alto contenido en Glutamina, se detectan en proteínas de unión a ARN de otros tipos celulares como hnRNP A1 y hnRNP D (ver Discusión). La porción de las proteínas TcUBPs que más llama la atención es la región rica en Glicina. En TcUBP-1 se observa la secuencia GAYGGYGAY, mientras que en TcUBP-2 esta porción es más extensa y presenta la secuencia GAYGGYGGYGGYGAY. Esto demuestra que en TcUBP-2 el motivo YGG está repetido tres veces mientras que en TcUBP-1 sólo una vez (ver Figura 25). Estos motivos se encuentran en un gran número de proteínas de origen diferente, tal es el caso de la isoforma p42 de la proteína murina hnRNP D (Número de acceso NCBI: BAB03466), la proteína de la pared celular de Chlamydomonas reinhardtii (Número de acceso NCBI: AAA02923) y la región variable de la cadena pesada de la inmunoglobulina de Bos taurus (Número de acceso NCBI: AAA98662), entre otros ejemplos. 77 Resultados TcUBP-1 y TcUBP-2 son proteínas de unión a ARN funcionales y reconocen motivos ricos en Uridina. En esta tesis se demostró que la región ARE, de ~44 nt, localizada en el 3’-NC de A GST L S P1 P2 P3 P4 L GST-TcUBP-1 GST-TcUBP-2 S S P1 P2 P3 P4 P1 P2 P3 P4 L SMUG-L-ARE, se realizó una reacción de unión “in vitro” (Figura 29A). A partir de una reacción de transcripción “in vitro” se sintetizó el ARN SMUG-L-ARE y se lo incubó con las proteínas recombinantes GST y GST-TcUBP1. Las reacciones se resolvieron en un gel nativo, como se observa en la Figura, GSTTcUBP-1 es capaz de reconocer al ARN SMUGL-ARE. Esta unión no se ve afectada por el agregado de heparina (resultado no mostrado), un polianión que sirve como competidor inespecífico. Como era de esperar el experimento control con proteína GST dió negativo (Figura 29A). Luego se intentó demostrar, tanto para TcUBP-1 como para TcUBP-2, cual era el sitio mínimo de reconocimiento en el ARN SMUGL-ARE. Para ello se construyeron otros tres B poli (A) - 1 10 50 500 1 poli (C) poli (G) poli (U) 10 50 500 1 10 50 500 1 10 50 500 TcUBP-1 ARN estadío específica (Figura 14). Con el objetivo de determinar si TcUBP-1 reconoce el ARN ARN TcSMUG-L tiene una función importante en la regulación de la estabilidad en forma - 1 poli (A) poli (C) poli (G) 10 50 500 1 10 50 500 1 10 50 500 1 poli (U) 10 50 500 TcUBP-2 Figura 29- TcUBP-1 y TcUBP-2 interaccionan con ARN poli(U). A, Se realizó un gel de retardo para determinar la posible interacción entre TcUBP-1 o TcUBP-2 y el elemento desestabilizante SMUG-L-ARE (L) y distintas secuencias ricas en Adenosina y Uridina: SMUG-S-ARE (S), P1, P2, P3 y P4. El gel de retardo muestra las interacciones entre los diferentes sustratos y GST, GSTTcUBP-1 o GST-TcUBP-2. B, Ensayo de gel de retardo realizado para resolver las reacciones de unión “in vitro” entre el ARN SMUG-L-ARE sólo (ARN), en presencia de TcUBP-1 o TcUBP-2 y cantidades crecientes de polímeros de ARN (A, C, G, U) como competidores de la reacción de unión a ARN. ARNs conteniendo secuencias parciales de SMUG-L-ARE y se los llamó 1, 2 y 3. La unión de ambas proteínas al ARN de 31 nt SMUG-L-ARE-1, fue un 90% menor comparando con el ARN SMUG-L-ARE, para la misma cantidad de proteína utilizada, ~1μgr (resultado no mostrado). Esto demuestra que los dos motivos AUUUUA de los extremos de SMUG-L-ARE son importantes para el reconocimiento por TcUBP-1 y TcUBP-2. La unión al nonámero UUAUUUAUU característico de los ARE (SMUG-L-ARE-2), es insignificante y se detecta sólo al sobreexponer la película (resultado no mostrado). La interacción se incrementó levemente al utilizarse el nonámero más dos residuos de Uridina en cada uno de sus extremos (SMUGL-ARE-3) (resultado no mostrado). Pero nuevamente se observó una reducción en la interacción de alrededor del 90% como con el ARN SMUG-L-ARE-1. De este modo, se puede concluir que el ARN SMUG-L-ARE de 44 nt, es el mínimo elemento necesario para una eficiente interacción con las proteínas TcUBP, ya que la deleción de las corridas de Uridina afecta su reconocimiento (ver más adelante). Además, se llevaron a cabo experimentos de competición para caracterizar la especificidad de la interacción entre las proteínas TcUBP y el ARN SMUG-L-ARE (Figura 29B). Se prepararon reacciones de unión ARN-proteína en ausencia o presencia de polímeros de ARN poli(A), poli(C), poli(G) y poli(U) sin marcar y la ribosonda indicada. Como se observa, el complejo formado entre 78 Resultados TcUBP-1 o TcUBP-2 y SMUG-L-ARE, se compite en forma eficiente y dependiente de la concentración del ARN poli(U), pero no con poli(A) y poli(C). El ARN poli(G) disminuye la interacción de TcUBP-1 y TcUBP-2 pero sólo cuando se utilizan altas concentraciones (500X) (Figura 29B). Como se describió anteriormente un gran número de transcriptos de tripanosomas presentan regiones ricas en Adenosina-Uridina en sus regiones 3’-NC (ver Tabla 2). Por lo tanto, se evaluó la capacidad de TcUBP-1 y TcUBP-2 de interaccionar con tales ARNs, y se comparó estas interacciones con el reconocimiento de SMUG-L-ARE. Entre los ARNs evaluados están el de la fospolipasa-1 (PLC-ARE), transportador de hexosas (HEX-TARE) y el del grupo TcSMUG-S (SMUG-S-ARE). TcUBP-1 claramente reconoce las regiones ARE de los ARNms TcSMUG-L y TcSMUG-S, los cuales tienen varios motivos del tipo AUUUA, AUUUUA y AUUUUUA (Figura 29A). Por otro lado, TcUBP-1 no es capaz de reconocer, con la cantidad de proteína utilizada en este ensayo (~1μM) las regiones HEXT-ARE y PLC-ARE compuesta por un poli-(AU) más dos pentámeros AUUUA en su 11 extremo 3’-terminal. De esta manera, se concluye que aunque varias secuencias del tipo ARE están presentes en diferentes transcriptos, TcUBP-1 y TcUBP-2 reconocen algunos y no todos esos motivos (resultado no mostrado). TcUBP-1 y TcUBP-2 presentan especificidad y afinidad similares para los ARNs ricos en Uridina testeados. La interacción con el ARN SMUG-L-ARE permite calcular una constante de disociación del complejo ARN-proteína. La constante para la mezcla ARN-proteína, se calculó determinando la concentración de proteína necesaria para unir el 50% del ARN (Smith, 1998). Para ello se realizó un gel de retardo utilizando diferentes concentraciones de la proteína TcUBP-1 desde 0.5 μM hasta 10 μM a una concentración fija de ARN SMUG-L-ARE o SMUG-S-ARE. TcUBP-1 reconoce a SMUG-L-ARE con una K ~ 5 μM d (ver Tabla 4), lo cual representa una interacción de afinidad media. El ARN SMUG-SARE, perteneciente al transcripto TcSMUG-S, es reconocido con más afinidad K ~ 0.5 d μM (Figura 30A y B). De manera tal de analizar en forma más estricta y detallada la interacción entre TcUBP-1 o TcUBP-2 y las secuencias ricas en Uridina, se sintetizaron ARNs modificados basándonos en la secuencia de 18 nt SMUG-S-ARE. Así se construyeron 5 oligoribonucleótidos nuevos (P1, P2, P3, P4 y P1-GGG) (ver Tabla 4). En el ARN P1, los residuos de Adenosina entre las corridas de Uridina, se cambiaron por residuos de Guanosina, generando el oligonucleótido 5’-guuuuguuuuuguuuuug-3’, que también es una secuencia común presente en la región 3’-NC de algunos transcriptos del parásito como EF1α y triparedoxina (Billaut-Mulot et al., 1996; Tetaud et al., 2001). Los ARNs P2 y P3 presentan 1 o 2 Guanosinas adicionales en vez de la segunda o segunda y tercera Uridina(s), respectivamente, en los motivos guuuuug del ARN P1. En el ARN P4, el residuo de Uridina del medio del motivo auuuuua se reemplazó por Adenosina, generando la secuencia (AUU) A presente también en los ARNms de Hsp70 (Dragon et al., 1987). Por 5 último, el ARN P1-GGG presenta tres Guanosinas que reemplazan a Uridinas del último motivo guuuuug, generando la secuencia 5’-guuuuguuuuugugggug-3’ (Tabla 4). La interacción entre TcUBP-1 o TcUBP-2 y los ARNs sintetizados se estudió realizando ensayos de retardo en gel y de esta manera, se calcularon constantes de 79 Resultados disociación aparentes (K ) de los complejos ribonucleoproteicos formados (Figura 30 B y d C). TcUBP-1 reconoce el oligo S con una K ~ 500 nM y el derivado P1 con una mejor d afinidad, K ~ 150nM (Figura 30B). A diferencia, los ARNs P2 y P3 son reconocidos con d afinidades menores (K ~ 750 y 1000 nM, respectivamente). En forma similar al ARN P1, d A L (μM) - 0.5 1 la afinidad por el ARN P1-GGG, es levemente menor (K ~ 250 nM) en comparación con la S 2.5 5 10 - 0.05 0.1 0.25 0.5 d 1 secuencia P1 (Figura 30B). ARN B TcUBP-1 S (nM) P1 P2 P3 P1-GGG - 10 50 250 5001000 10 50 250 500 100010 50 250 500100010 50 250 500100010 50 250 5001000 D M D M ARN C TcUBP-2 S (nM) P1 P2 P3 P1-GGG - 10 50 250 5001000 - 10 50 250 5001000 - 10 50 250 5001000 - 10 50 250 5001000 - 10 50 250 5001000 D M D M Figura 30 - TcUBP-1 y TcUBP-2 reconocen ARNs ricos en Uridina con similar especificidad y afinidad. Ensayo de retardo en gel para determinar las constantes de disociación entre TcUBP-1 (panel A y B) o TcUBP-2 (panel C) y los ARNs indicados. Se incubaron cantidades crecientes de las proteínas con los ARNs y luego se resolvieron en geles nativos. La posición del ARN libre, monómeros (M) y dímeros (D) se indica con flechas. ARN En cuanto a la proteína TcUBP-2, ésta tiene un comportamiento muy similar a TcUBP-1 en términos de actividad de unión a los sustratos ribonucleicos evaluados (Tabla 4). Para los ARNs S y P1 las constantes obtenidas son de alta afinidad y muy similares a las detectadas con TcUBP-1 (K ~ 250 y 100 nM, respectivamente). La d interrupción del último tracto de Uridinas con residuos de Guanosina (ARN P1-GGG), produce un efecto muy leve en la afinidad (K ~ 200 nM). De la misma manera que d sucede con TcUBP-1, los ARNs P2 y P3 son reconocidos con afinidades menores (K ~ d 750 y 850 nM, respectivamente). El ARN P4 presenta constantes de disociación K > 5 d μM para ambas proteínas (resultado no mostrado), debido a la interrupción de los tractos de Uridina con otros nucleótidos. Todos estos resultados se resúmen en la Tabla 4. El elemento rico en GU- es un mejor sustrato para la unión que el elemento AU-. La comparación de las constantes aparentes de afinidad permite concluir que aunque estos diferentes ARNs son reconocidos por TcUBP-1 y TcUBP-2, es claro que no comparten una similitud estructural importante. La única similitud interesante es que algunos de ellos (SMUG-S-ARE, P1 y P1-GGG) contienen más de una repetición del motivo UUUU(U) con residuos de Adenosina o Guanosina intercalados en sus extremos (ver Tabla 4). La sustitución de una de éstas 4 o 5 Uridinas en las repeticiones produce un gran incremento en las constantes aparentes de disociación, así como sucede con los 80 Resultados oligos P2, P3 y P4. De la comparación de las constantes también se puede deducir que probablemente los residuos de Guanosina contacten mejor que las Adenosinas en los complejos ribonucleoproteicos; sin embargo, ambos nucleótidos son purinas. En resumen, ambas proteínas TcUBP-1 y TcUBP-2 presentan afinidades y especificidades muy similares para las secuencias de ARN evaluadas, y se demuestra que los tractos de Uridina son de gran importancia para el reconocimiento (ver Tabla 4). Los valores de las constantes obtenidas, por lo menos para los oligos P1 y P1-GGG, son de gran afinidad en comparación con las afinidades de proteínas de unión a ARN de otros tipos celulares (ver Discusión). A partir de lo observado en este experimento se puede sugerir que tanto TcUBP-1 como TcUBP-2 tienen el potencial de dimerizar en presencia de ARN, cuando en los ensayos de retardo en gel se utilizan concentraciones superiores a ~500 nM de proteína (Figura 30). ARN L Secuencia UAAUUUUAAAAUUUAUUUAAUGUUGUU UUGAUUUAUUUAUUUUU TcUBP-1 (nM) TcUBP-2 (nM) 5000 5000 S AUUUUAUUUUUAUUUUUA 500 ± 25 250 ±10 P1 GUUUUGUUUUUGUUUUUG 150 ± 5 100 ±5 P2 AUGUUGUUGUUGUUGUUG 750 ± 50 750 ± 65 P3 GUGGUGUUGGUGUUGGUG > 1000 850 ± 55 P4 AUAUUAUUAUUAUUAUUA > 5000 > 5000 P1-GGG GUUUUGUUUUUGUGGGUG 250 ± 15 200 ± 15 Tabla 4 - Comparación de las constantes de discociación aparentes obtenidas para TcUBP1 y TcUBP-2 y los diferentes ARNs testeados. Identificación de los productos codificados por Tcubp-1 y Tcubp-2 en extractos de parásitos. Con el objetivo de identificar y caracterizar los productos codificados por los genes Tcubp-1 y Tcubp-2, se prepararon anticuerpos contra diferentes porciones de las proteínas codificadas. En primer lugar, se expresó el dominio RRM junto con el N-terminal de TcUBP-1 como fusión con GST y se inyectaron conejos con esta proteína recombinante. Este anticuerpo policlonal se llamó anti-RRM. Para analizar la expresión de las proteínas, se realizó un ensayo de “Western blot” utilizando un extracto proteico total del estadío epimastigote (Figura 31A). Este anticuerpo reconoce tres bandas de tamaños 18, 27 y 45kDa. La proteína de 18kDa corresponde al tamaño esperado a partir de la región codificante de Tcubp-2 (165 aa, ~18.4kDa) y la proteína de 27kDa presenta el tamaño que se espera para el producto codificado por Tcubp-1 (225 aa, ~27.5kDa). La banda de 45kDa no corresponde al tamaño esperado para ninguna de las proteínas (Figura 31A). Además, se generaron dos anticuerpos adicionales en ratones, contra péptidos de las porciones específicas de TcUBP-1 y TcUBP-2. Para TcUBP-1 se utilizó un péptido de la región N-terminal, que contiene los residuos V7-A17, y para TcUBP-2 un péptido de la región RV, que contiene los residuos G111-Y122 de la proteína. A los péptidos se les agregó en el extremo C-terminal una Cisteína para permitir la unión covalente de los mismos al “carrier” utilizado (KLH). Los anticuerpos policlonales obtenidos se denominaron anti-TcUBP-1 y anti-TcUBP-2, respectivamente. Con el objetivo de confirmar la 81 Resultados especificidad de los anticuerpos generados se realizaron ensayos de “Western blot” utilizando proteínas recombinantes GST-TcUBP-1 y GST-TcUBP-2, y extracto total del estadío epimastigote. En la Figura 31B se observa que el anticuerpo anti-TcUBP-1 reconoce las bandas de 27 y 45kDa en un extracto total, y que sólo la banda de GST-TcUBP-1 reacciona con el anticuerpo. Además, la preadsorción del suero con GST-TcUBP-1 solamente, impide la interacción con las bandas de 27 y 45kDa en un extracto total de epimastigotes (Figura 31B). De la misma manera, se demostró la especificidad del anticuerpo anti-TcUBP-2 (Figura 31C). Este anticuerpo reconoce solamente a GST-TcUBP2 y también reacciona en un extracto total con la banda de 18kDa. Más aún, la preadsorción del suero con GST-TcUBP-2 impide la detección de la banda de 18kDa en un extracto total (Figura 31C). De esta manera, se concluye que la banda de 18kDa está codificada por Tcubp-2 y que las dos bandas de 27 y 45kDa que reaccionan con los anticuerpos anti-RRM y anti-TcUBP-1, están codificadas por Tcubp-1. Se sabe que los dos genes Tcubp-1 y Tcubp-2 están presentes como genes de copia única en el genoma de 66 45 TcUBP-1m 29 TcUBP-1 18 TcUBP-2 kDa 66 45 TcUBP-1m 29 TcUBP-1 18 1 3 4 linea D kDa 66 45 E T A TcUBP-1m 45 29 29 TcUBP-1 18 TcUBP-2 TcUBP-2 18 linea 2 anti-TcUBP-1 Antisuero depletado GST-TcUBP-1 GST-TcUBP-2 kDa 66 Extracto anti-RRM C Antisuero depletado Extracto GST-TcUBP-2 B kDa GST-TcUBP-1 A Extracto T. cruzi y que una sonda dirigida contra el dominio RRM sólo detecta dos bandas en un ensayo de “Southern blot” (Figura 26). Por lo tanto, se puede sugerir que la banda de 45kDa es la proteína TcUBP-1 modificada “in vivo” en forma post-traduccional. De esta manera y para simplificar la nomenclatura, se llamará TcUBP-1 a la banda de 27kDa y TcUBP-1m (m por modificada) a la banda de 45kDa (Figura 31). El patrón de expresión de las proteínas TcUBPs se estudió a partir de ensayos de “Western blot” utilizando lisados proteicos totales de los diferentes estadíos. TcUBP-2 se expresa preferencialmete en epimastigotes; mientras que TcUBP-1 está en todos los estadíos y TcUBP-1m también, aunque su abundancia es mayor en los estadíos asociados con el huésped mamífero, amastigote y tripomastigote (Figura 31D). 1 2 3 4 anti-TcUBP-2 anti-RRM 82 Figura 31 - Identificación de los productos codificados por Tcubp-1 y Tcubp-2 en el parásito. A, “Western blot” realizado sobre un extracto de proteínas total de epimastigotes de T.cruzi con el anticuerpo policlonal anti-RRM. La posición de las tres bandas se indica con flechas (18, 27 y 45kDa). B, “Western blot” realizado con el anticuerpo policlonal antiTcUBP-1. Las líneas sembradas son: (1) Extracto total de epimastigotes, (2) GSTTcUBP-1, (3) GST-TcUBP-2 y (4) extracto proteico del estadío epimastigote, reaccionado con el suero anti-TcUBP-1 depletado con GST -TcUBP-1. C, “Western blot” realizado con el anticuerpo policlonal anti-TcUBP-2. Las líneas sembradas son: (1) Extracto total de epimastigotes, (2) GST -TcUBP-2, (3) GST -TcUBP-1 y (4) extracto proteico del estadío epimastigote, reaccionado con el suero anti-TcUBP-2 depletado con GST-TcUBP-2. D, “Western blot” realizado sobre extractos proteicos totales de los diferentes estadíos del desarrollo del parásito: (E) epimastigotes, (T) tripomastigotes derivados de células y (A) amastigotes intracelulares, y reaccionado con el anticuerpo anti-RRM. Resultados Las proteínas nativas TcUBP-1 y TcUBP-2 reconocen ARN poli(U) . Para verificar si las proteínas nativas presentan actividad de unión a ARN, se desarrolló un ensayo de unión “in vitro” a partir de extractos totales de parásito seguido por ensayos de “Western blot” (Figura 32A). Polímeros de ARN (A, C, G y U) se unieron covalentemente a bolitas de agarosa activadas con dihidrazida. Luego de equilibrarlas en el “buffer” apropiado, se las incubó con un extracto proteico total del estadío epimastigote durante 1 hr para posibilitar la unión. Las proteínas retenidas fueron eluídas en “buffer” Laemmli 2X y se corrieron en un gel SDS-PAGE 12.5% seguido de un ensayo de “Western blot” con el anticuerpo anti-RRM. Como se observa, todas las proteínas nativas TcUBP-1m, TcUBP-1 y TcUBP-2 son capaces de reconocer en forma específica el ARN poli(U), pero no otros polímeros de ARN (Figura 32A). Para verificar la localización subcelular de las proteínas nativas, se realizó un fraccionamiento bioquímico (Figura 32B). Se cargó en un gel SDS-PAGE, la misma cantidad de proteína correspondiente a cada fracción y luego se realizó un ensayo de “Western blot” con el anticuerpo anti-RRM. TcUBP-2 es la única proteína que localiza en el P1000g, el cual se sabe que contiene proteínas nucleares y proteínas asociadas a la membrana del retículo endoplásmico (Frey et al., 2001) y además, en microsomas (P100.000g). A diferencia, TcUBP-1 y TcUBP-1m no se detectan en el P1000g, aunque sí se localizan en microsomas (P100.000g). TcUBP-1, también está presente en forma soluble en el citoplasma (S100.000g). TcUBP-1m es detectada preferencialmente en las B poli-(ARN) A C G kDa U Extracto kDa Matriz A Extracto fracciones P10.000g y P100.000g (Figura 32B). 1.000g 10.000g 100.000g P S P S P S 97 66 45 TcUBP-1m 29 TcUBP-1 * 66 45 TcUBP-1m 29 18 TcUBP-1 TcUBP-2 18 TcUBP-2 anti-RRM anti-RRM Figura 32 - Las proteínas TcUBP nativas reconocen ARN poli(U) y localizan en diferentes fracciones subcelulares. A, Se preparó un extracto de proteínas del estadío epimastigote y se lo incubó con la matriz agarosa-dihidrazida sóla (Matriz) o conteniendo polímeros de ARN. Luego de los lavados, las proteínas se eluyeron con buffer Laemmli 2X y se realizó un “Western blot” con el anticuerpo policlonal antiRRM. B, Fraccionamiento subcelular seguido de un ensayo de “Western blot” con el anticuerpo anti-RRM de cada fracción. Se muestra la siembra del 10% del extracto preparado (Extracto). P, pellet; S, sobrenadante. Las fracciones utilizadas fueron S y P de 1000g, 10.000 y 100.000g . El asterisco indica la posición de una banda inespecífica debido a la agregación proteíca en el P100.000g. 83 Resultados TcUBP-1 y TcUBP-2 forman parte de un complejo ribonucleoproteico. Se llevaron a cabo experimentos de filtración en gel con el objetivo de determinar si las proteínas TcUBP forman parte de complejos ribonucleoproteicos “in vivo”. Para ello se prepararon extractos libres de células del estadío epimastigote y se pre-trataron con dos “buffers” diferentes (A: 150 mM NaCl y B: 150 mM NaCl y 0.1 μgr/μl ARNasa A). Luego del pre-tratamiento de los extractos por 30 min, se separaron en condiciones nativas en una columna Bio-Sil SEC 250 y las proteínas eluídas se analizaron en geles SDS-PAGE 12.5% seguido por un “Western blot” con el anticuerpo anti-RRM (Figura 33). En el tratamiento del extracto con “buffer” A, las isoformas TcUBP-1 y TcUBP-2 se detectan en un complejo de alto peso molecular de ~450kDa (Figura 33A, línea 9). Por un lado, TcUBP-1 también se detecta en la fracción 17 correspondiente a una masa molecular aparente de ~27kDa y esta banda puede corresponder a la forma monomérica de TcUBP1. Por otro lado, TcUBP-2 se detecta entre las fracciones correspondientes a 17-45kDa (Figura 33A, líneas 16-19), las cuales pueden corresponder a las formas monomérica y dimérica. En presencia del “buffer” B, A 670 kDa kDa 66 Ext 8 180 kDa 45 kDa todas las bandas proteícas correspondientes al complejo de 17 kDa 9 10 11 12 13 14 15 16 17 18 19 20 45 TcUBP-1m 29 TcUBP-1 18 TcUBP-2 B 670 kDa kDa Ext 8 180 kDa 45 kDa 17 kDa TcUBP-1 y TcUBP-2 entre las fracciones 16-19 no cambia significativamente. Como el “buffer” B contiene ARNasa A, se sugiere que un componente ribonucleico es 9 10 11 12 13 14 15 16 17 18 19 20 66 45 TcUBP-1m 29 TcUBP-1 18 ~450kDa desaparecen (Figura 33B, línea 9), mientras que la posición de TcUBP-2 Figura 33 - TcUBP-1 y TcUBP-2 están presentes en un complejo ribonucleoproteico de ~450kDa. Se preparó un extracto proteico total de epimastigotes y se lo pre-trató por 30 min. como se indica: panel A, 150 mM NaCl; panel B, 150 mM NaCl + 0.1 μg/μl de ARNasa A; en un buffer conteniendo 20 mM Tris-HCl pH 6.8. Los diferentes extractos se sembraron en la matriz de cromatografía en gel BioSil SEC 250 y se recolectaron fracciones de 0.5 ml cada una. Las muestras se resolvieron en geles desnaturalizantes SDS-PAGE 12.5%, seguido de un ensayo de “Western blot” con el anticuerpo anti-RRM. La posición de las proteínas TcUBP-1, TcUBP-1m y TcUBP-2, se indica con flechas en la parte derecha del panel. Ext, 10% del extracto total utilizado para el fraccionamiento. En las fracciones 1-7 no se detectó cross-reacción con el suero (no mostrado). Los marcadores de peso molecular utilizados son: 670kDa, T iroglobulina; 180kDa, α2-Macroglobulina; 45kDa, Ovalbumina y 17kDa, Mioglobina. 84 importante para la formación del complejo y la interacción proteínaproteína. El mismo experimento se repitió utilizando un extracto total del estadío tripomastigote derivado de células. Sin embargo, debido a la poca cantidad de material inicial utilizado en dos experimentos independientes, no se detectaron bandas correspondientes a las proteínas TcUBP en ensayos de “Western blot” (resultado no mostrado). Resultados El complejo ribonucleoproteico contiene el heterodímero TcUBP-1-TcUBP-2 y la proteína TcPABP1. De acuerdo a lo observado en el perfil de elución de la cromatografía en gel (Figura 33) y para determinar si las proteínas TcUBP-1 y TcUBP-2 interaccionan “in vivo” en el parásito se realizaron experimentos de inmunoprecipitación. Para esto, se realizaron inmunoprecipitaciones con anticuerpos pre-inmune y anti-TcUBP-1 en extractos sin (-) o con (+) ARNasa A y se analizaron por medio de un ensayo de “Western blot” con el anticuerpo anti-RRM (Figura 34A). Las proteínas TcUBP-1 y TcUBP-2 se detectaron cuando se inmunoprecipitó con anti-TcUBP-1, lo cual sugiere que están asociadas “in vivo” en la forma epimastigote del parásito aún en ausencia de ARN, mientras que la forma TcUBP-1m no co-precipita (Figura 34A). kDa anti-TcPABP1 ARNAsa A anti-TcUBP-2 + anti-TcUBP-1 - PreInmune B anti-TcUBP-1 :IP Extracto kDa Extracto A PreInmune Se sabe que en las células eucariotas superiores, PABP1 es parte de un complejo multiproteico el cual se forma en la región 3’- :IP 66 IgG cp 45 TcPABP1 66 29 TcUBP-1 18 TcUBP-2 IgG cp en Wilusz et al ., 2001). Debido a que TcUBP-1 se 45 anti-TcPABP1 D anti-TcUBP-1 E :IP T A kDa 66 TcPABP1 Extracto kDa Extracto anti-RRM C anti-TcUBP-1 E une a las secuencias ARE “in vitro” (Figura 30), se decidió :IP T - + - + 66 ARNAsa A TcPABP1 45 45 NC de ARNms que contienen elementos ARE (ver revisión anti-TcPABP1 29 anti-TcPABP1 Figura 34 - TcUBP-1 y TcUBP-2 interaccionan y están acomplejadas con la proteína de unión a poli(A). A, Se incubó un extracto proteico total del estadío epimastigote pretratado (+) o no () con ARNasa A, con suero pre-inmune, y anti-TcUBP-1. Los inmunocomplejos se precipitaron con proteína-A-sefarosa y se resolvieron por electroforesis en geles desnaturalizantes SDS-PAGE 12.5%, seguido de un ensayo de “Western blot” con el anticuerpo anti-RRM. IgGcp, indica la posición de la cadena pesada de las inmunoglobulinas. IP, indica el anticuerpo utilizado en el ensayo de inmunoprecipitación. B, Un extracto total de proteínas del estadío epimastigote se incubó con suero pre-inmune, anti-TcUBP-1, antiTcUBP-2 o anti-TcPABP1. Los inmunocomplejos se precipitaron con proteína-A-sefarosa y se resolvieron en geles SDS-PAGE, seguido de un “Western blot” con el anticuerpo anti-TcPABP1. La presencia de TcPABP1 (banda de 66kDa) y la IgGcp, se indican con flechas. C, Extractos totales de epimastigotes (E), tripomastigotes derivados de células (T) y amastigotes (A) se incubaron con anti-TcUBP-1. Los inmunocomplejos precipitados se ensayaron en un “Western blot” con anti-TcPABP1. D, Un extracto proteico total del estadío epimastigote (E) o tripomastigote (T) se pre-trató (+) o no (-) con ARNasa A durante 30 min. y luego se realizó una inmunoprecipitación con el anticuerpo anti-TcUBP-1. Los co-precipitados se resolvieron y ensayaron en un “Western blot” con el suero anti-TcPABP1. 85 evaluar si TcPABP1 está asociada a TcUBP-1 o TcUBP-2 for mando un complejo en el 3’-NC del ARNm de TcSMUG. Para responder esta pregunta, se realizaron reacciones de inmunoprecipitación con los anticuerpos anti-TcUBP-1, anti-TcUBP-2 y antiTcPABP1, seguido de un ensayo de “Western blot” con anti-TcPABP1. Como se muestra en la Figura 34B, en el complejo ARN-proteína que contiene el heterodímero TcUBP-1/TcUBP-2, también se detecta TcPABP1. Dos bandas de 55 y 66kDa se observan en un extracto total de epimastigotes de T. cruzi con el anticuerpo generado Resultados contra el dominio C-terminal de TcPABP1 (ver Materiales y Métodos) (Figura 34B). Sin embargo, se describió que sólo la banda de 66kDa corresponde a la proteína TcPABP1 (Batista et al., 1994). Además, es la única de las dos bandas que co-precipita. La interacción entre TcUBP-1 y TcPABP1 se detecta durante el desarrollo del parásito. Se analizó la interacción entre TcUBP-1 y TcPABP1 durante el desarrollo del parásito (Figura 34C). Para ello se prepararon extractos proteicos totales de las diferentes formas del parásito y se realizaron inmunoprecipitaciones con el anticuerpo anti-TcUBP1 seguido de un ensayo de “Western blot” con anti-TcPABP1. Este experimento demuestra que la interacción entre estas dos proteínas se detecta solamente en los estadíos epimastigote y tripomastigote, pero no en amastigote (Figura 34C). De lo contrario, TcUBP2 que se expresa preferencialmente en el estadío epimastigote (Figura 31D), co-precipita con TcPABP1 en este estadío (Figura 34B) y la interacción no se detecta en las otras formas del parásito. Como se demostró anteriormente, el complejo ARN-proteína de ~450kDa el cual contiene las proteínas TcUBPs se desarma en presencia de ARNasa A (Figura 33). Por lo tanto, se evaluó si la interacción entre TcUBP-1 y TcPABP1 en el estadió epimastigote es dependiente de ARN (Figura 34D). Para tal objetivo, se realizó un ensayo de inmunoprecipitación con extractos de epimastigotes libre de células pre-tratados o no con ARNasa A, demostrando que la interacción entre estos dos factores no se detecta en presencia de la nucleasa. Sin embargo, en el estadío tripomastigote la interacción es independiente del ARN (Figura 34D). TcUBP-1 y TcUBP-2 interaccionan en un ensayo de doble híbrido en levaduras. Se realizó un ensayo de doble híbrido en levaduras para corroborar la interacción entre las proteínas TcUBP-1 y TcUBP-2 en un sistema heterólogo (Figura 35). Para tal efecto, el marco abierto de lectura de TcUBP-1 se clonó en el vector pBTM-116 generando una fusión con el dominio de unión a ADN (DBD) del factor de transcripción LexA (LexA-TcUBP-1) (Fields and Song, 1989). Este vector se transformó en la cepa de levaduras L40 (ver Materiales y Métodos) y se estudió si “per se” tiene la capacidad de activar el sistema transcripcional; es decir, sin interaccionar con el dominio de activación (AD) clonado en el vector pVP16. Este ensayo dio negativo, y aunque un leve crecimiento basal se observó en placas His- no se detectaron niveles de actividad de GAL4 (Figura 35). Por lo tanto, los ADNc de TcUBP-1 y TcUBP-2 se clonaron en el vector presa pVP-16, el cual presenta el AD del factor VP16. En la Figura 35A se lista la combinación de proteínas utilizadas para el 86 Figura 35 - TcUBP-1 y TcUBP-2 interaccionan en un sistema heterólogo. A, Se lista la combinación de proteínas híbridas utilizadas para los ensayos de doble-híbrido. El plásmido pBTM116 se usó para clonar en fusión con el dominio de unión a ADN (DBD) de la proteína LexA. El plásmido pVP16 se usó para clonar las “presas” en fase con el dominio de activación (AD) del factor de transcripción VP16. B, Determinación relativa de las interacciones según se muestra en el panel C (-, sin interacción; ++, interacción fuerte, a partir de su comparación con controles estándares de interacción). C, Ensayo de actividad β-galactosidasa de las levaduras L40 conteniendo ambos plásmidos. Resultados ensayo de doble híbrido y los correspondientes resultados se muestran en la Figura 35B y C. La co-transformación de la cepa L40 con LexA-TcUBP-1 y VP16-TcUBP-1 o VP16TcUBP-2 permiten el crecimiento de las levaduras transformantes, como era de esperar, en medio His- y también mostraron actividad β-galactosidasa positiva (Figura 35C). Se obtuvieron resultados negativos con levaduras transformadas con LexA-TcUBP-1 y VP16 sólo, lo cual indica que la interacción entre TcUBP-1/TcUBP-1 y TcUBP-1/TcUBP-2 es específica. Esto permite concluir que no sólo las proteínas tienen la capacidad de heterodimerizar, sino también pueden homodimerizar, al menos en el caso de TcUBP-1 (Figura 35C). Mapeo de la región mínima requerida en TcUBPs para la unión de ARN y la formación de dímeros. Se demostró que TcUBP-1 y TcUBP-2 son capaces de reconocer ARN-ricos en Uridina con constantes de disociación aparentes casi idénticas (ver Tabla 4). Este resultado sugiere que las diferencias primarias entre ambas proteínas, localizadas en la regiones N- y C-terminales, no contribuyen en el reconocimiento del ARN y/o en procesos de modulación de la actividad de unión al ARN. Además, TcUBP-1 y TcUBP-2 dimerizan; y por lo tanto, se hipotetizó que los dominios auxiliares pueden ser cruciales para la interacción proteína-proteína. Para verificar esta hipótesis, varias mutantes de deleción se construyeron por PCR (ver sección Anexo III). Las diferentes proteínas generadas se llamaron TcUBP-1ΔN, la cual carece de la región N-terminal; TcUBP-1ΔQ, la cual carece de la región rica en Glutamina del C-terminal; TcUBP-1ΔNQ, que carece tanto de las regiones N-terminal y rica en Glutamina; y dos otras construcciones más llamadas TcUBP1ΔQG1 y TcUBP-1ΔQG2 (Figura 36A). La diferencia entre estas dos construcciones reside en la región RV. TcUBP-1ΔQG1 presenta la región RV, compuesta por 14 aminoácidos (PGIAGAVGDGNGYL), después de la hoja-β4 (Figura 36A). Sin embargo, ambas proteínas carecen del motivo GAYGGYGAY presente en la región rica en Glicina. Asimismo, las proteínas de deleción construídas a partir de TcUBP-2 fueron: TcUBP-2ΔN, la cual carece de la región N-terminal; TcUBP-2ΔC, que carece del extremo C-terminal después de la región rica en Glicina y dos construcciones más llamadas TcUBP-2ΔCG1 y TcUBP-2ΔCG2 (Figura 36C). Nuevamente, la diferencia entre estas dos proteínas es que sólo la primera presenta la región RV en su región C-terminal compuesta por 9 aminoácidos (NRNGVSTGF). La predicción de la estructura y los espectros NOE-heteronucleares registrados de las proteínas TcUBP-1 y TcUBP-2 (ver más adelante), sugieren que los dominios auxiliares se comportan como “random-coil”. Por lo tanto, ninguna de las deleciones realizadas tendría un efecto final deletereo en el plegamiento de las proteínas. Las constantes de disociación de la formación de los complejos entre las proteínas construídas y el ARN P1 se determinaron por geles de retardo, repetidos al menos dos veces. La mayoría de las proteínas presentaron constantes similares K ~100-200 nM (Tabla 5). ds Sin embargo, la proteína TcUBP-1ΔQG2 mostró una K > 1000 nM. Esto demuestra que d la afinidad de esta proteína por el ARN disminuyó ~5 veces. A diferencia de ello, TcUBP1ΔQG1 que contiene la región RV en el extremo C-terminal del dominio RRM, muestra la misma K que la proteína completa TcUBP-1 (Tabla 5). De manera similar a lo obtenido d 87 Resultados con las construcciones TcUBP-1, todas las mutantes de TcUBP-2 excepto TcUBP-2ΔCG2, interaccionan con el ARN con la misma afinidad que la proteína entera TcUBP-2 (Tabla 5). Estos experimentos demuestran claramente que el dominio RRM (~92 aminoácidos) es la mínima región requerida para la unión del ARN en las proteínas TcUBPs. La función del C-terminal del dominio RRM (dominio RV) no está clara hasta el momento y requiere, por lo tanto, más investigación. Sin embargo, se especula que puede funcionar en los procesos de interacción con el ARN y/o en la estabilización del complejo ribonucleoproteico (ver Discusión). Figura 36 - Mapeo de los dominios requeridos para la unión a ARN en las proteínas TcUBPs. A, Se muestra los esquemas de TcUBP-1 y las proteínas mutantes utilizadas en el gel de retardo del panel B. Se indica la posición y la extensión del RRM en número de residuos; RV, región variable; Gli, región rica en Glicina y Gln, región rica en Glutamina. B, Gel de retardo para determinar las constantes de disociación (Kd) entre TcUBP-1 o las mutantes de deleción y el ARN P1. Se calculó una constante aparente a partir de las diferentes curvas de ARN unido/ ARN libre vs la concentración de proteína (nM) (ver Tabla 5). C, Esquema de TcUBP-2 y las mutantes de deleción utilizadas en el gel de retardo del panel D. La posición y extensión en residuos de los módulos (RRM, RV, Gli y Cterminal), se indica sobre el esquema de TcUBP-2. D, Las constantes de disociación aparentes se calcularon de la misma manera que en el panel B. Proteína TcUBP-1 TcUBP-1ΔN TcUBP-1ΔQ TcUBP-1ΔNQ TcUBP-1ΔQG1 TcUBP-1ΔQG2 TcUBP-2 TcUBP-2ΔC TcUBP-2ΔCG1 TcUBP-2ΔCG2 Kd (nM) 200 180 150 170 200 >1000 100 100 75 >500 Tabla 5 - Comparación de los valores absolutos de las constantes de disociación aparentes entre las mutantes de deleción de TcUBP-1 y TcUBP-2 y el ARN P1. 88 Resultados La región rica en Glicina del dominio auxiliar es crítica para la dimerización de las proteínas TcUBPs. En los geles de retardo realizados, se observó una disminución en la movilidad de los complejos ribonucleoproteicos en presencia de concentraciones superiores a 500 nM en todas las proteínas mutantes de TcUBP-1, excepto TcUBP-1ΔQG1 y TcUBP-1ΔQG2 (Figura 36). Ambas proteínas carecen de la región rica en Glicina; por lo tanto, podría estar involucrada en procesos de dimerización. Con el objetivo de estudiar la formación de dímeros “in vitro”, se realizaron ensayos de “cross-linking” en presencia de glutaraldehído a una concentración final de 0.01% (Figura 37). Este es un reactivo que permite la formación de una unión covalente entre proteínas que están en contacto directo (Wan et al., 2001). Para este análisis, se utilizaron diferentes proteínas mutantes las cuales se listan en la Figura 37A. En primer lugar, la formación de dímeros se estudió con diferentes cantidades (250, 500 y 1000 nM) de las proteínas TcUBP-1ΔQ y TcUBP1ΔQG2 (Figura 37B). Como se observa, los dímeros se detectaron con concentraciones superiores a 500 nM sólo en la proteína TcUBP-1ΔQ (Figura 37B) y el porcentaje de “cross-linking” detectado es de alrededor del 10-15%, de acuerdo a lo determinado por densitometría de las bandas. Se obtuvieron valores similares de “cross-linking” al observado con proteínas de otros tipos celulares (Wan et al., 2001). Luego de detectar la concentración de proteína requerida para utilizar en las reacciones de “cross-linking” (~500 nM), se estudió la dimerización con todas las proteínas que se listan en la Figura. La proteína TcUBP-1ΔQ, que presenta las regiones RV y rica en Glicina como extensión terminal del dominio RRM, tiene la capacidad de dimerizar (Figura 37C, línea 6); mientras que, la proteína mutante TcUBP-1ΔQG1, que presenta sólo la región RV pero no la región rica en Glicina, forma dímeros de una manera muy ineficiente en comparación con TcUBP-1ΔQ (Figura 37C, línea 8). Por su parte, la proteína TcUBP-1ΔQG2, que carece de ambas regiones, no tiene la capacidad de reaccionar con el glutaraldehído (Figura 37C, línea 10). En cuanto a la proteína TcUBP-1, la formación de dímeros fue casi indetectable con la cantidad de proteína utilizada en este ensayo (Figura 37C, línea 4). En base a estos resultados, es posible que la presencia de la región rica en Glutamina en el extremo C-terminal de TcUBP-1, regule en forma negativa los contactos proteína-proteína debido a su naturaleza “random-coil” o que el ARN o algún factor se requiera “in vivo” para facilitar el proceso de dimerización (ver Discusión). Con respecto a la proteína TcUBP-2, se obtuvieron resultados similares en reacciones de “cross-linking” (resultado no mostrado), en donde también se observa que la región rica en Glicina es el principal determinante para la formación de dímeros. Además, se realizó el análisis por cromatografía de filtración en gel para confirmar los resultados de dimerización obtenidos por “cross-linking”. Para ello las proteínas TcUBP1ΔQ, TcUBP-1ΔQG2, TcUBP-2ΔC y TcUBP-2ΔCG2 se sembraron en una columna Superdex 75 con las condiciones detalladas en Materiales y Métodos. En resúmen, ~1 μM de cada proteína se sembró en la columna y el perfil de elución se comparó con marcadores de peso molecular conocidos. Se observó un perfil de elución similar para las proteínas TcUBP-1ΔQ y TcUBP-2ΔC, las cuales eluyen en dos picos correspondientes al dímero y monómero (resultado no mostrado), en donde el área de cada pico fue aproximadamente el mismo. Sin embargo, las proteínas TcUBP-1ΔQG2 y TcUBP-2ΔCG2 89 Resultados sólo eluyen como un pico el cual corresponde a la posición del monómero. Estos datos corroboran los resultados obtenidos a partir de los geles de retardo (Figura 30 y 36) y los experimentos de “cross-linking” (Figura 37). Figura 37 - La región rica en Glicina es crítica para la dimerización de las proteínas TcUBPs. A, Esquema de TcUBP-1 y las mutantes de deleción usadas para los ensayos de “cross-linking”. En las proteínas TcUBP-1(Y-A) y TcUBP-1(Y-A)ΔQ, todas los residuos Tir de las posiciones 142, 145 y 148 de la región rica en Glicina se reemplazaron por Ala. La posición de la secuencia original GAYGGYGAY y la mutada GAAGGAGAA, se indica sobre ambos esquemas. B, Las proteínas TcUBP-1ΔQ y TcUBP-1ΔQG2 fueron tratadas (+) o no (-) con una concentración final de 0.01% glutaraldehído. Las diferentes concentraciones de proteína utilizadas (250, 500, 1000 nM) se indican debajo del gráfico. Las muestras se trataron por 30 min. y se resolvieron por electroforesis en gel desnaturalizante SDS-PAGE 12.5% y revelado por tinción con Coomasie Blue. La posición de los monómeros y dímeros se indica con flechas. C, Todas las proteínas indicadas en el panel A (concentración de ~ 1 μM) se incubaron (+) en presencia de glutaraldehído 0.01%, y los productos se resolvieron en un gel SDS-PAGE 12.5%. Los residuos de Tirosina de la región rica en Glicina son importantes para la dimerización. Un rasgo interesante que caracteriza la región rica en Glicina es la presencia de residuos aromáticos (preferencialmente Tirosina), distribuídos en forma más o menos espaciada (ver Figura 25B). Este fenómeno se describió para otras proteínas de unión a ARN en células de eucariotas superiores, tales como hnRNP A0 (Myer and Steitz, 1995) y hnRNP A1 (Cartegni et al., 1996). Por lo tanto, para estudiar si los residuos de Tirosina de la región rica en Glicina de TcUBP-1 son importantes en el proceso de dimerización, todos las Tirosinas de las posiciones 142, 145 y 148 de la proteína, se mutaron por residuos de Alanina. Esta proteína mutante se denominó TcUBP-1(Y-A) (ver Figura 37A). A partir de esta construcción, se amplificó todo el marco abierto de lectura excepto la región “random-coil” rica en Glutamina, generando la proteína TcUBP-1(Y-A)ΔQ. Se preparó esta construcción para comparar con el patrón de dimerización de TcUBP-1ΔQ, ya que la proteína entera TcUBP-1 no dimeriza eficientemente (Figura 37C). Los resultados 90 Resultados demuestran claramente que mientras TcUBP-1ΔQ dimeriza “in vitro” (Figura 37C, línea 6), TcUBP-1(Y-A)ΔQ es incapaz de hacerlo (Figura 37C, línea 12). Por lo tanto, el cambio de los residuos de Tirosina por Alanina en la región rica en Glicina, impide la formación de dímeros. También se analizó si la dimerización de las proteínas puede ocurrir a partir de la formación de di-Tirosinas por oxidación de los residuos de Tirosina, en presencia de la enzima “Horse Radish Peroxidase” y H O (Aeschbach et al., 1976). Esta reacción genera 2 2 la producción de un acoplamiento de los anillos fenólicos de residuos de Tirosina intermoleculares próximos, generando el “cross-linking” covalente de las proteínas en cuestión. De esta manera, las proteínas mutantes TcUBP-1ΔQ, TcUBP-1(Y-A)ΔQ y TcUBP1ΔQG2 se trataron como se indica. Sólo la proteína TcUBP-1ΔQ es capaz de formar dímeros y oligómeros, y aún más interesante, la formación de dímeros se desplaza por cantidades crecientes de L-Tirosina (resultados no mostrados). Estos resultados permiten concluir que los residuos de Tirosina de la región rica en Glicina son importantes para producir contactos proteína-proteína en el dímero. Los residuos de Tirosina de la región rica en Glicina se fosforilan in vitro. A partir de los resultados obtenidos, y con la idea de que la dimerización debería ser un proceso regulado “in vivo”, se analizó si los residuos de Tirosina podrían modificarse en forma post-traduccional por fosforilación “in vitro” (Figura 38). Se comparó la capacidad de las proteínas GST como control, GST -TcUBP-1ΔQ y GST TcUBP-1ΔQG1, de fosforilarse “in vitro” en presencia de [γ32P]-ATP y la Quinasa de Tirosina c-Src (ver Materiales y Métodos). Mientras que GST no se fosforila, las proteínas GST-TcUBP-1ΔQ y GST-TcUBP-1ΔQG1 son sustrato de la Quinasa. Sin embargo, GST-TcUBP-1ΔQ se fosforila 4 veces más que GST-TcUBP-1ΔQG1, para la misma cantidad de proteína utilizada en este ensayo (Figura 38). Esto demuestra, aunque en forma indirecta, que las 3 Tirosinas presentes en la región rica en Glicina (GAYGGYGAY) de GST -TcUBP-1ΔQ son sustrato de la Quinasa c-Src “in vitro”. De manera tal de corroborar si TcUBP-1/TcUBP-2 están fosforiladas “in vivo”, se llevaron a cabo reacciones de inmunoprecipitación con el anticuerpo anti-RRM seguido de “Western blot” con un anticuerpo comercial antifosfo-Tirosina. Sin embargo, este ensayo dio negativo debido a la baja reactividad que presentaba el anticuerpo evaluado (resultado no mostrado). 91 Figura 38 - Los residuos Tirosina de la región rica en Glicina de TcUBP-1 se fosforilan in vitro. Se realizaron reacciones de fosforilación “in vitro” utilizando la kinasa c-Src y las proteínas: GST, GST-TcUBP-1ΔQ y GSTTcUBP-1ΔQG1. Los productos de fosforilación se corrieron en geles SDS-PAGE, el cual se expuso a una placa radiográfica (panel A), despúes de realizarse una tinción con Coomasie-Blue (panel B). Resultados La formación de homodímeros de TcUBP-1, pero no TcUBP-2, es bloqueada por la interacción con TcPABP1. Se demostró anteriormente que TcPABP1 co-inmunoprecipita con TcUBPs “in vivo” (Figura 34). Por lo tanto, se trató de estudiar la interacción TcUBPs-TcPABP1 “in vitro”. Para tal objetivo, se clonó el gen Tcpabp1 a partir de ADN genómico de Cl-Brener, en base a la secuencia reportada previamente (Batista et al., 1994). El clon se expresó como fusión con GST, generando la proteína GST-TcPABP1. En primer lugar, se determinó su capacidad de unir ARN “in vitro”, ya que en aquella publicación no se reportó su actividad. Para realizar este ensayo, se incubó TcPABP1 con los distintos homopolímeros de ARN, y como se observa en la Figura 39A, TcPABP1 es capaz de reconocer sólo poli(A). En segundo lugar y para determinar si TcUBP-1 y TcUBP-2, sin el “tag” GST, interaccionan “in vitro” con GST-TcPABP1, se realizó un ensayo de GST-“pull down”. Utilizando las mismas condiciones experimentales, GST-TcPABP1 interacciona con TcUBP-1 pero es incapaz de hacerlo con TcUBP-2 (Figura 39B). Esto sugiere que la unión de TcUBP-1 a TcPABP1 es mediada por contactos proteína-proteína, aunque el ARN pueda ser necesario para reclutar y acercar espacialmente ambas moléculas. Por otro lado, se analizó por gel de retardo el efecto del agregado de cantidades crecientes de TcPABP1 en la interacción ARN-TcUBP-1 y ARN-TcUBP2. Para asegurar que las proteínas reconozcan el ARN en forma dimérica, se utilizó ~2 μM de cada una de las proteínas. Este ensayo demostró que el agregado de TcPABP1 no tiene efecto en la interacción ARN-TcUBP-2, mientras que la interacción ARN-TcUBP-1 se modificó con el agregado de cantidades crecientes de TcPABP1 (Figura 39C). Este resultado demuestra que TcPABP1 no desplaza a TcUBP-1 del ARN, porque no se detecta sonda de ARN libre; sino que, TcPABP1 debe romper la formación de homodímeros de TcUBP-1, generando un patrón de reconocimiento monomérico en solución (Figura 39C). Es probable que la interacción entre estas dos proteínas involucre los extremos N- o Cterminales de TcUBP-1, ya que estas regiones son las más diferentes entre TcUBP-1 y su homólogo TcUBP-2. Figura 39 - Interacción proteína-proteína entre TcUBP-1 y TcPABP1. A, Determinación de la actividad de unión a ARN de la proteína TcPABP1 usando homopolímeros. B, Ensayo de “pull-down” con GST entre GST o GST-TcPABP1 y las proteínas TcUBP-1 o TcUBP-2, sin tag GST. Control, siembra del 10% de la proteína TcUBP-1 o TcUBP-2 antes de hacer el ensayo de interacción proteína-proteína. La integridad de las proteínas no unidas se monitoreó por gel desnaturalizante SDS-PAGE luego que la reacción finalizó (no mostrado). C, Gel de retardo entre TcUBP1 o TcUBP-2 (~ 2 μM) y el ARN P1 en ausencia o en presencia de cantidades crecientes de TcPABP1, sin tag GST. La posición de las formas monómero (M) y dímero (D) se indican con flechas. 92 Resultados La sobre-expresión de TcUBP-1 en epimastigotes afecta en forma selectiva la estabilidad de ARNms TcSMUG. Se analizó la sobre-expresión de TcUBP-1 y TcUBP-2 en la forma epimastigote de T. cruzi, con el objetivo de determinar el efecto de estas proteínas en los niveles de ARNms de TcSMUG y otros ARNms del parásito. Luego de la transfección de las construcciones pRIBOTEX (pR, vector sin inserto) y pRIBOTEX-TcUBP-1 (pR-TcUBP-1), se seleccionaron poblaciones transformantes y se trataron con el inhibidor de la transcripción ActD durante diferentes tiempos (0, 2, 4 y 6 hrs) (ver Figura 40). De esta manera, se analizó por “Northern blot” la vida-media de dos ARNms que contienen regiones ricas en Uridina en sus regiones 3’-NC, como TcSMUG-L y S, y transcriptos que carecen de tales regiones como ductina y Hsp70 (Figura 40A). Los datos de densitometría del “Northern blot” se graficaron luego de la normalización con el control interno de hibridización proveniente de la sonda ARNr (Figura 40B). Se realizaron dos experimentos usando líneas celulares independientes, las cuales se transfectaron y procesaron en forma separada. Los gráficos de la Figura 40B, se realizaron utilizando la media del valor de todos los experimentos, los cuales difieren menos del 5% en cada uno de los puntos indicados. El tiempo inicial que muestra la mayor señal, se tomó como el 100% en todas las curvas, para permitir la comparación de las vidas medias de los diferentes ARNs. En el control Tc negativo, la población transfectada con el vector pR, muestra que las tasas de vida-media para TcSMUG-L y -S, son 5.5 y 5.75 hrs, respectivamente (Figura 40B). Estos valores de vidamedia son muy similares a los obtenidos anteriormente para los ARNms endógenos, sin ninguna transfección (ver Figura 10). Por lo contrario, la tasa de decaimiento de TcSMUG en las poblaciones que expresan TcUBP-1 (pR-TcUBP-1), fue mayor. Para TcSMUG-S la vida-media es de 3 hrs, lo cual representa un 54% de la vida-media en la población pR, y para TcSMUG-L, la vida-media es de 3.75 hrs lo cual representa el 65% de la tasa en la población pR. También se analizó la vida-media de los transcriptos para ductina y Hsp70, los cuales no presentan ni elementos AUni GU- en sus regiones 3’-NC. Ninguno de estos dos transcriptos se afectó por la sobre-expresión de TcUBP-1 (Figura Figura 40 - La sobre-expresión de TcUBP-1 en epimastigotes desestabiliza en forma selectiva, ARNms conteniendo elementos ARE. A, Ensayo de “Northern blot” sobre ARN total proveniente de poblaciones de epimastigotes transfectadas con vector (pR) o sobre-expresando TcUBP-1 (pR-TcUBP-1), tratados con ActD 10 μg/ml. El mismo filtro fue secuencialmente hibridizado con las sondas correspondientes a TcSMUG, ductina, Hsp70 y ARNr. B, Cuantificación de los niveles de ARNm de los diferentes ensayos de Northern blot del panel A, normalizados a los niveles de ARNr. Los datos se expresaron como la cantidad de ARNm relativa a la media, +/- el error estándar de la media (n=2), en cada punto experimental luego de la corrección con la señal proveniente del ARNr. Las diferencias entre pR y pRTcUBP-1 son significativas cuando se compara la media a partir de un test t de Student (*, p < 0.05;**, p < 0.01). 93 Resultados 40B). La vida-media del ARNm para ductina es de 1.5 y 1.65 hrs en parásitos transfectados con pR y pR-TcUBP-1, respectivamente; mientras que la vida-media del ARNm para Hsp70 es de 5.2 y 4.8 hrs en las poblaciones pR y pR-TcUBP-1, respectivamente. Las transfecciones con el vector pR-TcUBP-2 desgraciadamente fueron negativas, en el sentido de que ninguna de las poblaciones se recuperó luego de la selección con el antibiótico utilizado. Estas transfecciones se realizaron en paralelo con otras y además en repetidas ocasiones, por lo que puede descartarse un error técnico. Es posible que la sobre-expresión de TcUBP-2 en epimastigotes de T. cruzi, tenga un efecto deletereo en el crecimiento de los parásitos. Modelo de las interacciones proteína-proteína en el ARNm TcSMUG durante el desarrollo del parásito. Los resultados obtenidos durante esta tesis se pueden resumir en un modelo, que en forma preliminar, puede ser la base para la caracterización de una vasta red de interacciones proteína-proteína en el ARNm de TcSMUG, en los distintos compartimentos subcelulares y durante el desarrollo del parásito (Figura 41). Como se describió anteriormente, existen diferentes proteínas que reconocen a los elementos ARE y GRE, y que regulan la estabilidad del ARNm TcSMUG. En primer lugar, la unión de eAREBP al ARNm es mediada por el elemento ARE localizado en el 3’-NC. Esta unión puede prevenir la asociación de factores desestabilizantes al ARNm, posiblemente a través de la competencia por la unión a elementos en cis- similares. Además, eAREBP puede ser una de las proteínas involucradas en la modulación de la traducción, probablemente a través de su interacción con otros factores pertenecientes al aparato de síntesis de proteínas de la célula. Los resultados obtenidos a partir del fraccionamiento subcelular de la forma epimastigote indican que eAREBP está localizada en el citoplasma o sólo reconoce el ARN en este compartimento celular, donde ocurren la degradación del ARN y/o su traducción. Experimentos futuros de “Western blot”, permitirán determinar si eAREBP está también presente en el núcleo; y de esta manera, es reclutada por algúna(s) proteína(s) que formen parte de un complejo que impida la interacción con el ARE. Se sabe que eAREBP está presente en forma soluble en el citoplasma, aunque quizás también pueda formar parte de algún complejo ribonucleoproteico que no se detecte su actividad a partir de los ensayos de retardo en gel. Además, la proteína eAREBP no se detecta en los demás estadíos, aunque sí existen otras proteínas de unión al ARE (tAREBPs) en tripomastigotes. Se describió que tAREBPs son al menos dos polipéptidos de entre 4550kDa y que a diferencia de eAREBP se detectan en núcleo y citoplasma. El elemento ARE también interacciona “in vitro” e “in vivo” por dos miembros de una familia de proteínas de unión a ARN (TcUBP-1 y TcUBP-2). Se realizaron varios intentos para determinar si existe asociación entre estos dos factores y las proteínas AREBPs; sin embargo, parecen formar parte de complejos proteícos independientes dentro de la célula. Por su parte, Tcubp-1 se expresa como 2 posibles isoformas, TcUBP-1 de 27kDa y TcUBP-1m de 45kDa. Su sobre-expresión ocasiona la desestabilización de ARNms con secuencias ARE en su región 3’-NC, como es el caso de TcSMUG-L y TcSMUG-S. Sin embargo, no se sabe cuál de estas dos isoformas o si las dos están involucradas en el proceso de desestabilización de ARNms. Se sabe que las proteínas están presentes en 94 Resultados compartimentos similares, incluyendo el citoplasma y los polisomas más específicamente, aunque no son proteínas nucleares. TcUBP-1, a diferencia de su isoforma modificada no sólo está en polisomas sino que también localiza en forma soluble en el citosol. Por su parte, TcUBP-2 localiza en el citoplasma, pero también en la fracción nuclear. TcUBP-1, es capaz de formar complejos ribonucleoproteicos con TcPABP1 y también con TcUBP-2, lo cual sugiere un papel en la regulación de la estabilidad de ARNms. Figura 41 - Modelo de las interacciones ARN-proteína y proteína-proteína, reportadas en el ARNm TcSMUG durante el desarrollo del parásito. Ver texto para detalles. Dentro del complejo nativo identificado, TcUBP-1 dimeriza con TcUBP-2 en el estadío epimastigote solamente, y esta asociación diferencial puede regular el destino de los ARNms en forma estadío específico. Más aún, TcUBP-1 es capaz de interaccionar con TcPABP1 en forma dependiente del ARN y en ausencia de TcUBP-2 puede producirse su desplazamiento del ARN. Esto sugiere que TcUBP-2 presenta un rol regulador en la estabilidad y formación del complejo. Las proteínas AREBPs se detectan fácilmente en geles de retardo, mientras que las proteínas TcUBPs no, lo cual sugiere que estas proteínas están interaccionando fuertemente con el ARN “in vivo”; por lo tanto, no se pueden detectar “in vitro” con esta metodología. Además, no se sabe si existe coordinación en la interacción con el ARN o si las mismas interaccionan directamente en el parásito. Es evidente que es necesario clonar el gen que codifica eAREBP, lo cual permitirá obtener anticuerpos necesarios para resolver alguna de estas cuestiones. Las proteínas que reconocen al elemento GRE se expresan en forma constitutiva durante el desarrollo del parásito. Como se determinó, estas proteínas están presentes en ambos compartimentos subcelulares, núcleo y citoplasma. Por lo tanto, es probable 95 Resultados que puedan proteger al ARNm durante su transporte al citoplasma. En el futuro se debe investigar la posibilidad de que exista un complejo proteíco mediado por proteínas que reconozcan al ARE y al GRE y también a la cola de poli(A), u otros factores celulares que impidan el ataque del ARNm por componentes del exosoma, endo- o exo-nucleasas. 96 Resultados PARTE 5 - Análisis Estructural de las Proteínas TcUBPs. Ensayos de Dinámica Molecular. Uno de los objetivos generados durante el desarrollo de esta tesis fue intentar resolver la estructura en solución de las proteínas TcUBPs, realizar ensayos de dinámica molecular y comparar la forma en que proteínas con dominios RRM simples y múltiples interaccionan con el ARN. Para ello, se estableció una colaboración con el Dr. Kalle Gehring (Dept. of Biochemistry, McGill University, Montreal, Canadá), y realicé una pasantía de tres meses en su laboratorio durante el primer semestre del 2002. Marcado isotópico y análisis de los espectros de correlación heteronuclear simple H-15N HSQC. 1 Las proteínas utilizadas para los marcados isotópicos fueron TcUBP-1ΔQ, TcUBP1ΔQG1 y TcUBP-2ΔC, ya que las proteínas enteras TcUBP-1 y TcUBP-2 presentan regiones sin plegamiento aparente en el C-terminal, lo cual significaría la producción de espectros de baja resolución y complicados de analizar. Para tal fin, se crecieron bacterias en presencia del medio mínimo M9 suplementado con 15N-NH Cl. Para las proteínas GST4 TcUBP-1ΔQ y GST-TcUBP-1ΔQG1 fue suficiente con preparar 1,5 litros, mientras que para GST-TcUBP-2 se prepararon ~5 litros de medio de cultivo, debido a los bajos niveles de rendimiento en la inducción obtenidos con esta proteína. El protocolo de inducción y de preparación de las proteínas sin el “tag” GST se detalla en la sección Materiales y Métodos. En la Figura 42 se muestran los espectros 1H-15N HSQC tomados a 500 MHz sobre las tres proteínas uniformemente marcadas con 15N. En la proteína TcUBP-1ΔQG1 se pueden identificar ~126 picos de los 130 esperados porque 9 aminoácidos son Prolina. En TcUBP-1ΔQ se observan ~128 de los 145 residuos esperados, porque 11 son Prolina. Por su parte, en TcUBP-2ΔC se observan ~120 de 131, porque 11 corresponden a residuos de Prolina. Esto significa que algunos picos correspondientes a ciertos residuos, no aparecen en las condiciones que se muestran en este experimento; ni tampoco en muchas otras condiciones evaluadas en las cuales se varió el pH de la muestra, la temperatutra en la que se tomaron los espectros, la fuerza iónica, entre otros (resultados no mostrados). En resumen, en los tres espectros se observa un núcleo central correspondiente a los residuos pertenecientes a porciones no plegadas de la proteína (ver más adelante), pero también regiones plegadas a la izquierda y rodeando el núcleo central. En la parte superior derecha de los espectros, se observan las cadenas laterales de las Glutaminas (~110 ppm 15N y ~7 ppm 1H), las cuales muestran dos picos a la misma altura correspondientes a ambos protones de la amida. Estos presentan resonancias diferentes para el mismo desplazamiento del N. Por último, se muestran las asignaciones de las resonancias de la proteína TcUBP-1ΔQG1 (Figura 42A), según se determinó en los experimentos descriptos más adelante (ver Figura 43). Asignación de las resonancias de cadena principal en la proteína TcUBP-1ΔQG1. La mayoría de las resonancias 1H-15N de las amidas de cadena principal de la proteína TcUBP-1ΔQG1 se obtuvieron usando métodos de RMN bidimiensionales y 97 Resultados tridimensionales. Estas resonancias se asignaron a la secuencia primaria de aminoácidos, a partir de métodos que correlacionan las resonancias de residuos adyacentes usando acoplamientos J (“J couplings”) a través de enlaces en la cadena polipeptídica. Estas secuencias de pulsos requieren que la muestra se marque isotópicamente en forma doble (15N, 13C), como así también la posibilidad de pulsar tres canales de radiofrecuencia a la vez (H, N y C). Para tal fin, la estrategia inicial fue usar los experimentos descriptos: CBCA(CO)NH y HNCACB (Constantine et al., 1993; Grzesiek et al., 1992); ya que la combinación de ambos experimentos se sabe que funciona en forma muy efectiva. Algunas de las correlaciones disponibles a partir de estos experimentos se indican en la Figura 43. Como se observa, no se deter minaron las correlaciones 1H-15N para los residuos M1, Q124, R125, ni las Prolinas (ver Figura 42A). La posición de las resonancias en el espectro 1H-15N HSQC de la proteína TcUBP-1ΔQ, es idéntico al de TcUBP1ΔQG1, con la excepción de que 17 residuos adicionales se observan ya que esta proteína es levemente más larga (Figura 42B). Como se observa, el espectro HSQC es complejo y hay solapamiento de algunos picos debido al gran número de residuos y al área que ocupan los mismos. En la Figura 43A, se muestra los acoplamientos a través de enlaces obtenidos a partir de los Figura 42 - Espectro 1H-15N HSQC de las proteínas TcUBPs libres en solución. Se muestran los espectros de correlación cuántica simple heteronuclear de las proteínas isotópicamente enriquecidas con 15N. A, TcUBP-1ΔQG1. B, TcUBP-1ΔQ. C, TcUBP-2ΔC. Se indica la posición de la mayoría de las resonancias asignadas para las amidas de la proteína TcUBP1ΔQG1. En el panel A se muestra una ampliación de la región central del espectro con las asignaciones respectivas. Las resonancias de las amidas de las proteínas TcUBP-1ΔQ y TcUBP-2ΔC no fueron asignadas. 98 experimentos de correlación tridimensional. En la Figura 43B se muestra el grupo de tiras provenientes del experimento HNCACB de las regiones comprendidas entre los residuos Y13Q18 y G127-Y138. Se observa claramente el alto contenido de Glicinas, con una resonancia característica para el Cα de Resultados 45 ppm y sin Cβ, lo cual facilitó la identificación de los residuos correspondientes. Otro rasgo característico es la presencia de Alaninas, cuyo Cα tiene una resonancia característica de ~18 ppm (Figura 43B). En la misma Figura se muestran las tiras correspondientes al experimento CBCA(CO)NH de los mismos residuos del panel B, el cual permite corroborar las asignaciones realizadas a partir del HNCACB (Figura 43). Otros de los experimentos realizados para establecer asignaciones de las resonancias fueron: HNCO, HNCA, HN(CO)CA, (H)CCH-COSY, TOCSY-HSQC, NOESY-HSQC, HTOCSY-CONH y C-TOCSY-CONH (ver Tabla 7). Todos estos experimentos emplean un número mínimo de pasos de transferencia de la magnetización y permiten obtener constantes de acoplamientos J altas, es decir de gran utilidad. Como era de esperar, el experimento HNCO fue el de mayor sensibilidad, con ~90% de las correlaciones de los grupos carbonilos observadas. Las correlaciones para 1H -1H se obtienen a partir del N α experimento 15N-TOCSY HSQC, aunque la sensibilidad obtenida con el mismo no fue muy buena debido a la constante de acoplamiento J baja. Figura 43 - Experimentos de correlación 3D HNCACB sobre la muestra 15 N- 13 C TcUBP1ΔQG1. La proteína TcUBP1ΔQG1 se enriqueció isotópicamente con 15N y 13C en medio mínimo M9. Las resonancias de cadena principal se asignaron usando métodos que correlacionan resonancias a través de residuos adyacentes usando “J couplings”. A, Las correlaciones disponibles de los experimentos 3D se muestran en for ma esquemática. B, Juego de tiras de los residuos Y13-Q18 y G127-Y138 asignados a partir de la comparación de las resonancias de los experimentos HNCACB y CBCA(CO)NH. Se muestra la posición característica del Cα de las Glicinas con una resonancia de ~45 ppm y la ausencia de Cβ. Los residuos de Alanina presentan desplazamientos característicos para el Cα (~ 18 ppm). Predicción de la estructura secundaria. La estructura secundaria de TcUBP-1ΔQG1 se dedujo a partir del patrón de 99 Resultados desplazamientos químicos correspondientes a los Cα y Cβ ya descriptos (Wishart and Sykes, 1994). La posibilidad de identificar estructuras secundarias a partir de los patrones de las diferencias entre los desplazamientos químicos observados y esperados para un “random-coil”, se documentó previamente (Wishart et al., 1995a; Wishart et al., 1995b). Las diferencias identificadas para 13Cα y 13Cβ se usaron para calcular un índice de desplazamiento químico (CSI o “Chemical Shift Index”), el cual se muestra en la Figura 44A. Debido a que no se observaron todos los desplazamientos químicos para 13Cα , el CSI calculado no es estrictamente el mismo que se presentó originalmente (Wishart and Sykes, 1994). Las hélices-α y las hojas-β se indican a partir de un continuo de valores consenso +1 y -1 para Cα, respectivamente. Lo contrario, es decir desviaciones positivas, son atribuídas a hojas-β en el caso de desplazamientos químicos para Hα y Cβ (resultado no mostrado). De esta manera, la topología observada para esta proteína, βαββαβ, es característica de los dominios de unión a ARN del tipo RRM. Aparentemente, las regiones N- y C-terminales del dominio RRM no tienen un plegamiento esperado a partir del CSI (Figura 44A). A partir de ello, se decidió realizar un experimento NOE-heteronuclear sobre la proteína 15N-TcUBP-1ΔQG1 con el objetivo de confirmar cuáles regiones están plegadas (Figura 44B). En esa figura, claramente se observa que el plegamiento existe entre los residuos ~40 (hoja-β1) hasta ~120 (final hoja-β4). Esto indica que las regiones correspondientes a los extremos del dominio RRM incluyendo la región RV y porción Nterminal, no presentan plegamiento (Figura 44B). Figura 44 - Indice de desplazamiento químico (CSI) y ensayo NOE-Heteronuclear. A, Los desplazamientos químicos para 13Cα y 13Cβ de cada aminoácido, se compararon con los desplazamientos químicos de cada aminoácido en particular en un “random-coil”. Como se define según Wishart et al., (1992), la diferencia entre los valores para “random-coil” y de cada aminoácido en la proteína puede indicar el medio ambiente estructural para cada aminoácido (hélice-α, hoja-β). El signo de la diferencia observada, es función del elemento estructural y la resonancia observada. Esto permite hacer la mejor predicción de la estructura secundaria a partir de los desplazamientos químicos. B, El área de los picos de cada residuo en un espectro 15N-13C HSQC se calculó y comparó con el área esperada, lo cual permite definir el NOEHeteronuclear para cada residuo de la proteína TcUBP-1ΔQG1 a partir del cálculo (valor obtenido/valor esperado). El signo, + o -, representa que ese residuo está en un ambiente plegado o “random-coil” respectivamente. Detección de cambios en los desplazamientos químicos por la interacción con ARN. Se realizaron experimentos 1H-15N HSQC para monitorear y determinar cambios en los desplazamientos químicos en la proteína TcUBP-1ΔQG1 durante la interacción con el ARN. Este método es muy sensible y permite detectar perturbaciones en el medio ambiente de las amidas de cadena principal debido a la unión con el ARN. Los desplazamientos químicos son generalmente sensibles a las perturbaciones de las cadenas laterales conectadas, pero también son afectados por cambios conformacionales en la 100 Resultados proteína, inducidos por la unión del ligando. Para tal objetivo se realizaron dos clases de experimentos. En primer lugar, titulaciones con el ARN rico en Uridina P1, por el cual la proteína TcUBP-1ΔQG1 tiene alta afinidad (ver Tabla 5) y en segundo lugar , titulaciones con ARN poli(U) (Figura 45 y 46). Ambos resultados fueron comparados a partir de los residuos que sufren perturbaciones por la unión del ARN. Para el experimento de titulación con el ARN P1, se utilizaron diferentes relaciones molares proteína-ARN (1:0, 1:0.1, 1:0.2, 1:0.35 y 1:0.7). En la Figura 45, se observan los experimentos de 1H-15N HSQC de las condiciones proteína-ARN 1:0 y 1:0.7, como casos extremos para ver los picos del espectro que pueden moverse en presencia de ARN. Debemos mencionar que se pueden observar cambios importantes en un residuo dado y eso puede ser interpretado de dos maneras diferentes: (1) un cambio local en la conformación proteica que altera la magnetización detectada en un núcleo en particular debido a nuevas modificaciones por los electrones que lo rodean; y (2) que el residuo está involucrado en una interacción directa con el ARN. Es interesante mencionar que varios residuos se desplazan en foma gradual a partir de la titulación con el ARN P1, los cuales se indican en color rojo (Figura 45B). Además, se muestra el gráfico de los valores absolutos de las diferencias de los desplazamientos químicos de las formas libre o unida a ARN para las resonancias 1H y 15 N de las amidas de cada residuo (Figura 45C). Estas diferencias (indicadas como (Δ 15N- Figura 45 - Espectro 1H-15N HSQC de la proteína TcUBP-1ΔQG1 libre o acomplejada con ARN. Los espectros fueron tomados con la proteína libre en solución (A) o en presencia del ARN P1 (B). Sólo se muestra en B el punto de mayor concentración de ARN (relación 1:0.7, proteína-ARN) como parte de un proceso de titulación con el ligando (ver Texto). C, Gráfico en el cual se indican las perturbaciones de los desplazamientos químicos para cada residuo (definido como el valor absoluto de las diferencias entre proteína libre y proteína acomplejada). Las perturbaciones de los desplazamientos químicos para: 1H (blanco) y 15N (negro) de las amidas de cadena principal se muestran en una misma barra para cada residuo como (Δ 15N-1H δ (ppm)). La ubicación de los elementos de estructura secundaria se indica encima del gráfico. 101 Resultados H δ (ppm)) son consideradas como perturbaciones en los desplazamientos químicos inducida por la interacción con el ARN. A partir del monitoreo de cada experimento de 1 titulación con ARN y la obtención de los espectros 1H-15N HSQC, los residuos de mayor desplazamiento corresponden a los residuos siguientes: V41 y L45 en la hoja-β1; K73 y V74 en la hoja-β2; R84, G87 y F88 de la hoja-β3; N111, R113 y K115 en la hoja-β4 y A119 y A120 en el “loop” ubicado después de la hoja-β4 (Figura 45C). Sin embargo, de todos estos residuos los de mayor desplazamiento corresponden a K73, V74, N111, K115 y A120. El mismo experimento de titulación se realizó, utilizando ARN poli(U) como sustrato ribonucleico (Figura 46). Las razones de proteína-ARN utilizadas fueron (1:0, 1:0.1, 1:0.2 y 1:0.5). De la misma manera que para la titulación con el ARN P1, aquí se muestran los espectros con las relaciones molares 1:0 y 1:0.5 proteína-ARN, con los residuos que se desplazan en color rojo en el segundo caso (Figura 46). Al igual que en la titulación con el ARN P1, se determinaron los residuos de mayor desplazamiento a partir de las perturbaciones en los desplazamientos químicos inducida por la interacción con poli(U). Entre ellos se encontraron los siguientes residuos: D40, localizado antes de la hoja-β1; V41, N44, L45, V47, N48 y Y49 en la hoja-β1; K73 y V74 en la hoja-β2; S83, R84, Y86 y G87 de la hoja-β3; N111 y K115 en la hoja-β4; y L118, A119 y A120 en el “loop” ubicado después de la hoja−β4 (Figura 46C). Sin embargo, de todos estos residuos los de mayor desplazamiento corresponden a V41, L45, K73, V74 y N111. Figura 46 - Espectro 1H-15N HSQC de la proteína TcUBP-1ΔQG1 libre o acomplejada con ARN poli(U). Los espectros fueron tomados con la proteína libre en solución (A) o en presencia de ARN poli(U) (B). Sólo se muestra en B el punto de mayor concentración de ARN (relación 1:0.5, proteína-ARN) como parte de un proceso de titulación con el ligando. C, Gráfico en el cual se indican las perturbaciones de los desplazamientos químicos para cada residuo (definido como el valor absoluto de las diferencias entre proteína libre y proteína acomplejada). Las perturbaciones de los desplazamientos químicos para: 1H (blanco) y 15N (negro) de las amidas de cadena principal se muestran en una misma barra para cada residuo como (Δ 15N-1H δ (ppm)). La ubicación de los elementos de estructura secundaria se indica encima del gráfico. 102 Resultados Este resultado demuestra claramente, que la mayoría de los aminoácidos que muestran perturbaciones con el ARN P1, también lo hacen con ARN poli(U). La principal diferencia observada, es que con este último sustrato algunos residuos de la hoja-β1 se desplazan en mayor proporción que con el ARN P1 (Figura 46C). Además, con el ARN P1 los residuos de la hoja-β4 o los posteriores, como K115 y A120, se desplazan más que con poli(U). En general, se observa que hay más movimiento de las resonancias en la vecindad de las hojas-β que en las hélices-α, las cuales parecen no verse afectadas por la interacción con el sustrato. Esto es consistente con observaciones previas que indican que las hojas-β antiparalelas sirven como una superficie para la interacción con el ARN (Gorlach et al., 1992; Kanaar et al., 1995). Varios de los residuos que se desplazan en el dominio RRM de TcUBP-1 están compartidos en el primer dominio de unión a ARN de la proteína Sxl de D. melanogaster (Handa et al., 1999) y son los involucrados en el reconocimiento a través de “stacking” de ARN, contactos a partir del esqueleto proteico o cadena lateral. En la Figura 47 se muestra un alineamiento entre el RRM de TcUBP1ΔQG1, U1A, los motivos RRM1 de Sxl y HuD. En esta Figura se compara la localización de los residuos que se sabe hacen contacto con el ARN en las proteínas cuya estructura fue resuelta en presencia del sustrato, con los datos de TcUBP-1ΔQG1 obtenidos a partir de la titulación con ARN. De esta manera, se observa que la N44 en TcUBP-1ΔQG1 presenta la misma posición que la N130 en la hoja-β1 del RRM1 de Sxl. De manera similar, en la hoja-β2 la K73 se reemplaza por R175 en Sxl, y ambos aminoácidos presentan una carga neta positiva. En la hoja-β3, más específicamente en el dominio RNP1, la S83 se reemplaza por S165 y F88 por F170 en Sxl. En la hoja-β4 la N111 se reemplaza por la N193 de Sxl. Aunque este residuo no interacciona con ARN en Sxl (Handa et al., 1999), está conservado en posición en ambas proteínas, y por ende quizás tenga un rol importante en mantener la estructura del dominio RRM. Por su parte, la R113 se reemplaza por la Figura 47 - Alineamiento de secuencias y estructuras secundarias de los motivos RRM de TcUBP-1, Sxl, U1A y HuD. Se indican los motivos RNP-2 y RNP-1 característicos del RRM, que forman parte de las hojas-β1 y -β3 respectivamente. Las interacciones ARN-proteína de TcUBP-1ΔQG1 (obtenidas por titulaciones con ARN), Sxl (Handa et al., 1999), U1A (Oubridge et al., 1994) y HuD (Wang et al., 2001), se indican como sigue. Los aminoácidos involucrados en interacciones “stacking” con el ARN se muestran en colorado. Los aminoácidos que reconocen el ARN con su cadena principal y cadenas laterales se indican por cuadrados abiertos o llenos, respectivamente. El número entre paréntesis luego del nombre de cada proteína significa la posición del aminoácido en la cual comienza el alineamiento. 103 Resultados R195 y la K115 por K197 en el RRM1 de Sxl (Figura 47). Por último, después de la hojaβ4 la A120 se reemplaza por la R220 en Sxl; sin embargo, este cambio no es conservativo (Figura 47). A partir de la estructura tridimensional del RRM1 de Sxl y la estructura modelada de TcUBP-1ΔQG1, se indica la posición de los residuos compartidos entre estas dos proteínas desde el punto de vista de su interacción con el ARN (Figura 47). Si bien se observa que TcUBP-1 y las proteínas comparadas presentan similitudes en cuanto a los aminoácidos que hacen o pueden hacer contacto con el ARN (Figura 47 y 48), existen algunas diferencias en la posición o el tipo de residuo que hace contacto. Por lo tanto, estas diferencias podrían estar relacionadas con distintas formas de reconocimiento en proteínas que presentan uno o más de un dominio del tipo RRM. Figura 48 - Representación estructural de las perturbaciones de los desplazamientos químicos inducidos por la unión del ARN. Comparación entre Sxl-RRM1 y TcUBP-1. La estructura del dominio RRM1 de la proteína Sxl (Handa et al., 1999) y una visión del modelo estructural de TcUBP1ΔQG1, se usan para marcar cada dominio e indicar en rojo las regiones que presentan las mayores perturbaciones en los desplazamientos químicos inducida por la unión del ARN, en el caso de TcUBP-1, o regiones que contacten con el ARN en el caso de Sxl. 104 DISCUSION Discusión Durante esta tesis doctoral se describieron componentes relacionados con la regulación a nivel post-transcripcional de la familia de mucinas TcSMUG. Además, se identificaron y caracterizaron, desde el punto de vista funcional-estructural, una familia de proteínas de unión a ARN involucradas en la regulación de la estabilidad de TcSMUG. Los puntos más interesantes que surgen del análisis de los resultados obtenidos son: (1) La familia de mucinas del grupo TcSMUG se regula a nivel post-transcripcional a partir de mecanismos de control de la vida-media de sus ARNms en las distintas etapas del ciclo de vida del parásito. (2) Existen elementos en cis- localizados en la región 3’-NC de TcSMUG, que están involucrados en procesos de estabilización y desestabilización del ARNm durante el desarrollo del parásito. (3) La identificación de factores en trans- que reconocen los elementos en cis-, y que están involucrados en el mismo camino de regulación de la estabilidad. (4) La purificación bioquímica parcial de la proteína de ∼100kDa eAREBP y su análisis por espectrometría de masas MALDI-TOF. (5) La caracterización de dos miembros de una gran familia de proteínas de unión a ARN, TcUBP-1 y TcUBP-2, en términos de los sustratos ribonucleicos que reconocen “in vitro”. (6) La descripción del rol funcional de la proteína TcUBP-1 en la desestabilización de TcSMUG cuando se sobre-expresa en el estadío epimastigote. (7) Las interacciones proteína-proteína de las cuales forman parte las proteínas TcUBPs en el complejo ribonucleoproteico identificado. (8) La caracterización estructural preliminar por RMN de las proteínas TcUBPs. (9) Estudios de interacción proteína-ARN a partir de ensayos de dinámica molecular. Regulación post-transcripcional de TcSMUG y elementos involucrados. La familia TcSMUG está compuesta por dos grupos de ARNms llamados TcSMUGL y TcSMUG-S, los cuales se regulan a nivel post-transcripcional durante el desarrollo del T. cruzi. Mientras que la tasa de inicio de transcripción de ambos grupos es muy similar entre los estadíos epimastigote y tripomastigote, los niveles estacionarios de ARNm son claramente diferentes (Figura 8). A partir de ensayos de inhibición de la transcripción ~ 6 hrs en el estadío con ActD, se demostró que TcSMUG tiene una vida-media t 1/2 epimastigote (Figura 10), mientras que en tripomastigote TcSMUG es de vida-media corta, t < 0.5 hr (Figura 11). Esto demuestra que la estabilidad de los ARNms TcSMUG se 1/2 regula durante los diferentes estadíos del ciclo de vida del parásito y que por ende, deben existir elementos en cis- involucrados en este proceso regulatorio. Ambos grupos de ARNms (TcSMUG-L y -S) presentan una región 5’-NC conservada entre sí, mientras que el 3’-NC es más variable en secuencia y tamaño, con bloques específicos en cada ARNm, como es el retrotransposón SIRE (Vázquez et al., 1994), el cuál esta presente sólo en el grupo TcSMUG-L. Otros elementos del 3’NC sin embargo, están presentes en ambos ARNms (ver más adelante). Debido a la complejidad del sistema se decidió continuar trabajando sólo con el grupo TcSMUG-L, con el objetivo de identificar elementos 106 Discusión involucrados en la regulación de la estabilidad durante el desarrollo de T. cruzi. A partir de las construcciones de deleción del 3’-NC de TcSMUG-L (Figura 12), y los experimentos de transfección, se identificaron elementos que regulan la estabilidad del ARNm TcSMUGL en los estadíos epimastigote y tripomastigote. En primer lugar, se identificó el elemento GRE, presente en ambos grupos TcSMUG-L y -S. Este elemento está compuesto por 27 nt después del codón de terminación de la traducción y contiene dos pentámeros contiguos del tipo CGGGG. A partir de ensayos de “Northern blot” de las poblaciones transfectantes, se determinó que GRE funciona como un elemento regulador positivo sólo en el estadío epimastigote (Figura 14). En segundo lugar, se demostró que la deleción de otro elemento, denominado E1 y localizado entre los nt 28 y 62 del 3’-NC aumenta la vida-media del ARNm cat utilizado como reportero. Estos resultados sugieren que E1 actúa como un elemento negativo en la estabilidad del ARNm (Figura 13). En tercer lugar, la deleción del elemento ARE tiene un efecto estadío específico (ver más adelante). Finalmente, la deleción de los 450 nt que componen SIRE, produce el mismo efecto negativo que la ausencia de E1 en el 3’-NC, pero debido al gran tamaño de este elemento, es necesario realizar más experimentos de deleción para confirmar y mapear específicamente el dominio involucrado en este proceso regulatorio. Previamente, se demostró en otro laboratorio que SIRE es responsable de la regulación en forma negativa de la expresión de los genes que codifican las proteínas ribosomales TcP2β, y actúa alterando la eficiencia del trans-“splicing” (Vázquez et al., 1994). Ya que la localización del SIRE en diversos ARNms es variable (Vázquez et al., 2000), es posible que cumpla funciones diferentes según su posición en la región codificante o no codificante. Además, es posible que la reducción del largo de la región 3’-NC, por deleción del SIRE, tenga un efecto negativo en la traducción (Figura 15). En resumen, durante ésta tesis se identificaron dos elementos funcionalmente opuestos (ARE y GRE) en el ARNm de TcSMUG-L (Figura 14). Por un lado, el elemento ARE está involucrado en la desestabilización del ARNm en el estadío tripomastigote, pero su deleción no afecta el destino del ARNm en el estadío epimastigote. Estos resultados confirman la idea de que las proteínas que reconocen al ARE en epimastigote, pueden proveer resistencia a endo- o exo-nucleasas. Por otro lado, a diferencia del ARE, el elemento GRE genera un efecto en la estabilidad de TcSMUG durante el desarrollo del parásito. Este elemento puede incrementar la abundancia del ARNm en el estadío epimastigote, ya que la deleción del 3’-NC genera un transcripto más lábil en términos de estabilidad (Figura 14). La presencia del ARE en el 3’-NC del ARNm, también es capaz de modular la tasa traduccional en forma positiva, mientras que GRE no tiene un efecto considerable en tal proceso (Figura 15). Esto sugiere que ambos elementos coordinan en forma cooperativa, la regulación de la abundancia del ARNm TcSMUG en epimastigotes, pero no la traducción. La interacción de elementos negativos y positivos en forma coordinada, se observó para elementos localizados en el 3’-NC de ARNms de las prociclinas de los tripanosomas africanos. Estos elementos afectan tanto la abundancia del ARNm como su tasa traduccional (Furger et al., 1997). En nuestro sistema, el elemento E1 tiene un efecto dual negativo en ambos procesos regulatorios (Figura 15). En resumen, se sugiere que un elemento simple, como ARE o E1, presente dos funciones diferentes pero quizás acopladas dentro del camino de regulación de la expresión. De la misma manera, se 107 Discusión demostró que secuencias ARE en el 3’-NC del ARNm tnf-α afectan también ambos procesos regulatorios de estabilidad y traducción del ARNm (Carballo et al., 1998; Lai et al., 1999; Xu et al., 1997). Proteínas que reconocen los elementos regulatorios de TcSMUG. Proteinas de unión al GRE y al ARE. La coordinación de la expresión de TcSMUG a nivel post-transcripcional permite generar un modelo en el que existen factores que interaccionan “in vivo” con ARE y GRE y formen parte del mismo camino regulatorio. A partir de la resolución de complejos ARN-proteína por geles nativos, se demostró la existencia de factores capaces de reconocer ambos elementos (Figura 16 y 18). Por un lado, se identificaron tres complejos de unión al elemento GRE, los cuales se expresan en forma constitutiva a lo largo del desarrollo del parásito. Los dos primeros complejos (GRE-1 y GRE-2) se compiten en forma específica y eficiente con ARN poli(G) (Figura 17). A partir de ensayos de entrecruzamiento, se determinó que el complejo GRE-1 está compuesto por un polipétido de ~80kDa, mientras que varios factores de masas aparentes 35, 39 y 66kDa componen el complejo GRE-2 (Figura 16). Por otro lado, el elemento ARE interacciona con proteínas cuya expresión está regulada durante el desarrollo del parásito (Figura 18). La proteína de ~100kDa eAREBP interacciona con el ARE en epimastigotes solamente, mientras que en los demás estadíos, al menos dos polipéptidos de entre 45-50kDa, denominados tAREBPs, reconocen al ARE (Figura 18). Es evidente que la presencia de complejos ribonucleoproteicos es capaz de regular la expresión de ARNms en forma coordinada dependiendo de las proteínas que lo componen o de las interacciones proteína-proteína generadas durante las distintas etapas del desarrollo. Debido a la rápida desestabilización de TcSMUG-L en tripomastigotes, la cual es ocasionada por la presencia del elemento ARE en el 3’-NC, es posible que una interacción coordinada entre las proteínas de unión al GRE y al ARE esté regulada en el parásito. Los resultados obtenidos a partir del fraccionamiento subcelular demuestran que la proteína eAREBP se localiza en el citoplasma o sólo está accesible para reconocer el ARN en este compartimiento subcelular. A diferencia de ello, las proteínas tAREBPs están presentes en cantidades equivalentes en núcleo y citoplasma (Figura 20), hecho que permite sugerir que las proteínas tAREBPs transportan ARNms entre ambos compartimientos. Varias proteínas en otros tipos celulares, tienen la capacidad de translocarse entre núcleo y citoplasma (Fan and Steitz, 1998; Loflin et al., 1999; Shyu and Wilkinson, 2000). En algunos casos se describió que existen estímulos extracelulares capaces de regular su capacidad intrínseca de movilización como es el estrés por calor (Gallouzi et al., 2000). En tripanosomas, por su parte, se identificó que existen proteínas como la proteína de unión a ARN homóloga a La y la histona del tipo H2B, las cuales presentan una señal de localización nuclear clásica rica en Lisina, involucrada en el transporte de proteínas del citoplasma al núcleo (Marchetti et al., 2000). De la misma manera, es posible pensar que existan proteínas con señales del tipo NES involucradas en el transporte de ARNms del núcleo al citoplasma. Evidencias recientes en nuestro laboratorio indican que el transporte de proteínas del núcleo al citoplasma está presente en tripanosomas. Además, el crecimiento de los parásitos en cultivo axénico es sensible 108 Discusión a la droga leptomicina B, la cual bloquea el transporte de ARNms mediado por el receptor CRM1/Xpo1. Este receptor se identificó y estudió en T. cruzi y T. brucei y además, se demostró su localización nuclear (D’Orso et al., resultados no publicados). Por lo tanto, la caracterización de este receptor, así como también de las proteínas que interaccionan con éste, es de suma importancia para el estudio de mecanismos de control de la estabilidad de ARNms mediado por el transporte al citoplasma. Proteínas TcUBPs e interacción con ARN. El segundo enfoque utilizado para identificar proteínas que interaccionan con el elemento ARE de TcSMUG, fue la búsqueda en la base de datos de EST y GSS de T. cruzi, de homólogos de las proteínas de unión al ARE de eucariotas superiores, como HuR (Fan and Steitz, 1998). Las proteínas identificadas utilizando este enfoque son las que denominamos TcUBP-1 y TcUBP-2. En esta tesis se obtuvieron evidencias que indican que ambas proteínas presentan un motivo idéntico del tipo RRM con regiones N- y Cterminales auxiliares diferentes (Figura 25). La región C-terminal de TcUBP-1 se divide en dos porciones, una región corta rica en Glicina y un dominio “random-coil” rico en Glutamina (Figura 25 y 44). A diferencia, la región C-terminal de TcUBP-2 presenta una región rica en Glicina levemente más larga y con tres motivos YGG solapados y carece del dominio rico en Glutamina (ver Figura 25). A partir de experimentos de retardo en gel se demostró que ambas proteínas tienen la capacidad de interaccionar con el elemento ARE en reacciones de unión ARNproteína. Si bien el ARE de TcSMUG-L es reconocido “in vitro” por TcUBP-1, esta interacción no es de alta afinidad (K ~ 5 μM). Sin embargo, TcUBP-1 es capaz de regular d la estabilidad de TcSMUG-L “in vivo” (ver Figura 41); por lo cual se sugiere que algún otro componente o modificación post-traduccional de TcUBP-1, se requiera para tal fenómeno. Se observó a partir de reacciones de unión “in vitro” que tanto TcUBP-1 como TcUBP-2 reconocen el elemento ARE de TcSMUG-S con alta afinidad (K ~ 500 nM) y d además, reconocen elementos ricos en Guanosina-Uridina (GU), presentes en regiones 3’-NC de varios transcriptos de T. cruzi (K ~100-200 nM) (Tabla 4). Es evidente que las d proteínas eAREBP, TcUBP-1 y TcUBP-2 presentan, al menos cualitativamente, patrones similares de reconocimiento de sustratos de ARN “in vitro” (ver Tabla 6). Por lo tanto, se puede sugerir que las tres proteínas son capaces de competir “in vivo” por la unión al mismo sustrato ribonucleico. Comparación de las proteínas TcUBPs con proteínas con múltiples motivos RRM. Los experimentos de RMN realizados durantes ésta tesis (ver Tabla 7) y la determinación preliminar de la estructura de TcUBP-1 (ver Figura 47 y 48), permiten sugerir que ambas proteínas presentan una topología βαββαβ característica de las proteínas con motivos RRM. De alguna manera, esto puede explicar porqué las dos proteínas presentan constantes de disociación similares para los ARN utilizados en los experimentos de retardo en gel (Tabla 4). Aunque el dominio RRM es idéntico entre TcUBP-1 y TcUBP-2, la extensión C-terminal del RRM, denominada región RV, es diferente entre ambas proteínas (Figura 25) y su deleción produce un incremento en la constante aparente de disociación (Tabla 5). De manera similar, un estudio bioquímico y 109 Discusión espectroscópico del dominio RRM de la proteína U1A, sin su extensión C-terminal, muestra que presenta la misma topología βαββαβ que la proteína entera. Sin embargo, es incapaz de interaccionar con ARN (Lu and Hall, 1995). Esto demuestra que los residuos del Cterminal del RRM están involucrados en la estabilidad de la estructura de la proteína en su forma libre (Avis et al., 1996). Otro ejemplo se observa en la proteína hnRNP D, en donde la región C-terminal del dominio RRM2 confiere alta afinidad para la unión con el ARE (DeMaria et al., 1997). Este fenómeno sugiere que aquellos residuos localizados en la región RV pueden ser importantes para conferir estabilidad al complejo ARN-proteína a partir de una interacción con los residuos de las hojas β1β3 que reconocen el ARN. ARN Secuencia eAREBP TcUBP-1 TcUBP-2 SMUG-L-ARE-as UAAAAAUAAAUAAAUCAAAACAACAUUAAAUAAAUUUUAAAAUUA - - - SMUG-L-ARE-se U AAUUUUAAAAUUUAUUUAAUGUUGUUUU GAUUUAUUUAUUUUU + + + SMUG-L-ARE-1 AUUUAUUUAAUGUUGUUUUGAUUUAUUUAUU + + + SMUG-L-ARE-2 UUAUUUAUU - - - SMUG-L-ARE-3 U UUUAUUUAUUUU + + + HEX-T-ARE AUUUAUUUAUUUA - - - PLC-ARE AUAUAUAUAUAUAUAUAUAUAUAUUUAUUUAUUU - - - SMUG-S-ARE AUUUUAUUUUUAUUUUUA ++ ++ ++ P1 GUUUUGUUUUUGUUUUUG ++ ++ ++ P2 AUGUUGUUGUUGUUGUUG + + + P3 AUGGUGUUGGUGUUGGUG - - - P4 AUAUUAUUAUUAUUAUUA + - - P1-GGG GUUUUGUUUUUGUGGGUG nd ++ ++ Tabla 6 - Comparación cualitativa de los resultados obtenidos en ensayos de unión “in vitro” entre distintos sustratos ribonucleicos y eAREBP, TcUBP-1 o TcUBP-2. Se muestra cada ARN con su secuencia y el resultado de la interacción en un ensayo de unión “in vitro”, según comparación de los resultados obtenidos. El signo (-, +, ++, representa en forma cualitatva el resultado del ensayo). nd, no determinado. Un rasgo interesante es la estructura modular de los motivos RRM de las proteínas de unión a ARN en células de eucariotas superiores. Estas proteínas presentan múltiples dominios RRM y su composición es la que determina la afinidad final por el ARN. El primer motivo (RRM1), es el más importante en términos de afinidad y especificidad. Sin embargo, los demás motivos RRM tienen funciones accesorias tales como oligomerización y localización (Perez et al., 1997), estabilidad de la estructura en solución (Avis et al., 1996) e interacción proteína-proteína (Khaleghpour et al., 2001). El análisis de mutantes de deleción de la proteína HuR, la cual presenta 3 motivos RRM, permitió sugerir que el RRM1 es crítico para la interacción con la secuencia ARE, pero que por lo menos un motivo RRM adicional es requerido para alcanzar una constante de disociación en el rango nanomolar (Park et al., 2000). Se sabe que cualquier combinación de dos de los tres dominios RRM de la proteína HuR es suficiente para alcanzar una interacción fuerte con el elemento ARE del ARN c-fos “in vitro”. Además, dos dominos RRM, siempre y cuando se incluya el RRM1, pueden estabilizar c-fos “in vivo” de una manera comparable a la proteína entera (Chen et al., 2002). En la proteína hnRNP D, ambos motivos RRM1 y RRM2 son importantes para determinar la afinidad por el ARE, aunque no son suficientes para alcanzar la afinidad de la proteína entera (DeMaria et al., 1997). Es importante 110 Discusión concluir que algunas proteínas de unión a ARN requieren sólo un motivo RRM para alcanzar la afinidad y especificidad necesarias para la interacción, como es el caso de la proteína U1A involucrada en “splicing” (Klein Gunnewiek et al., 2000). Sin embargo, otras proteínas requieren por lo menos dos (Park et al., 2000) u ocasionalmente más dominios o superficies de interacción (Perez-Canadillas and Varani, 2001). Algunos rasgos comunes emergen a partir de la comparación de las estructuras con motivos RRM en “tandem”, tal es el caso de Sxl (Handa et al., 1999) y PABP1 (Deo et al., 1999). Cada una de estas proteínas presentan conectores entre los motivos RRM, de aproximadamente 811 residuos. Estas regiones son de gran mobilidad en la proteína libre, y permiten que los dos dominios sean estructuralmente independientes (Crowder et al., 1999). En esta tesis, se demuestra que las proteínas TcUBPs conteniendo un sólo motivo RRM pueden unirse al ARN con gran afinidad y también pueden dimerizar por la región flexible rica en Glicina (ver más adelante). Este fenómeno, puede estar relacionado con la producción de un dímero en el cual ambas mitades sean independientes. De este modo, una mayor afinidad de reconocimiento del ARN puede lograrse “in vivo” a través de la dimerización, y permitiendo la unión simultánea de dos motivos RRM al ARN. Rol de la región rica en Glicina en la dimerización de las proteínas TcUBPs. La homodimerización, un fenómeno común para los factores transcripcionales de las células procariotas, en eucariotas se extiende hacia la heterodimerización. Esta posibilidad de interacción proteína-proteína, aumenta el rango de secuencias nucleotídicas que pueden ser reconocidas y el grado de especificidad de la interacción. En términos evolutivos, este aumento en la capacidad de unión es necesario cuando los genomas se hacen más grandes y complejos (Branden and Tooze, 1991). Las proteínas TcUBPs pueden homo-heterodimerizar y en ésta tesis se proveen evidencias que demuestran que la región rica en Glicina es importante par tal reacción (Figura 37). Sin embargo, el hecho de que no se detecta en forma efectiva dímeros de TcUBP-1 por “cross-linking” con glutaraldehído, a diferencia de lo que sucede con TcUBP-1ΔQ (Figura 37), sugiere que el dominio Cterminal “random-coil” puede regular la accesibilidad y la interacción proteína-proteína. Se describió que ciertos dominios “coil-coil” de algunos factores de transcripción son capaces de promover o prevenir la formación de homo-heterodímeros (ver revisión en Branden and Tooze, 1991). De esta manera, es lógico pensar que en el parásito ambos procesos de dimerización puedan estar regulados. La proteína recombinante TcUBP1(Y-A)ΔQ, la cual carece de las Tirosinas de la region rica en Glicina, no dimeriza en ensayos de “cross-linking” (Figura 37), a diferencia de lo que sucede con TcUBP-1ΔQ. Estos resultados, nos permiten sugerir que los residuos de Tirosina de esta región sirven para generar contactos en la interfase del dímero. Más interesante aún, es que las Tirosinas de la región rica en Glicina pueden fosforilarse, al menos “in vitro” (Figura 38). Una reacción reversible como esta podría regular el alcance de la dimerización. Dos ejemplos de fosforilación en residuos de Tirosina se describieron como capaces de regular la dimerización de proteínas de unión a ADN y ARN (Amster-Choder and Wright, 1992; Pype et al., 1994). Más aún, los motivos YGG preferencialmente localizados en la región rica en Glicina de TcUBP-2, se identificaron en diferentes proteínas de unión a ARN de Arabidopsis thaliana (Lorkovic and Barta, 2002), la proteína humana hnRNP A0 (Myer 111 Discusión and Steitz, 1995) y el factor de liberación traduccional RF3 de Zygosaccharomyces rouxii (Nakayashiki et al., 2001), entre otros ejemplos. Algunos trabajos interesantes describen la función de la fosforilación en Tirosina en la regulación de la estabilidad y/o traducción del ARNm (Derry et al., 2000; Ostareck-Lederer et al., 2002; Ostrowski et al., 2000). En términos generales, la región rica en Glicina presente en varias proteínas de unión a ARN, no tiene rasgos estructurales comunes más alla de la similitud en la presencia de residuos de Glicina o Prolina que resultan en “coils” flexibles. También se demostró que esta región está involucrada en la interacción proteína-proteína y puede contribuir en la afinidad por la interacción con el ARN (Cartegni et al., 1996; DiNitto and Huber, 2001). En resumen, las proteínas TcUBPs de tripanosomas presentan un único motivo RRM y son capaces de interaccionar con secuencias de ARN ricas en Uridina con gran afinidad. Una segunda porción de la proteína, la región rica en Glicina, podría contribuir a la mobilidad y flexibilidad del motivo RRM en el dímero, como se deduce a partir de los resultados obtenidos y por comparación con las proteínas con motivos RRM de otros tipos celulares. Complejos ARN-proteína y posibles mecanismos de regulación de la expresión génica en tripanosomas. Como se demostró en esta tesis, TcUBP-1 y TcUBP-2 dimerizan y forman parte de un complejo ribonucleoproteico con TcPABP1 (Figura 34). TcUBP-2 co-precipita en el estadío epimastigote, mientras que TcUBP-1 interacciona con TcPABP1 en todos los estadíos de desarrollo del parásito (Figura 34). En experimentos realizados “in vitro”, TcUBP-1 interacciona directamente con TcPABP1 y el efecto de esta interacción está relacionado con la disrupción de la formación de homodímeros de TcUBP-1 “in vitro” (Figura 39). Si bien los dominios involucrados en esta interacción no se mapearon hasta el momento, se especula que el C-terminal de TcUBP-1 puede estar involucrado en este proceso, ya que es la porción más disímil cuando se la compara con TcUBP-2. Aunque TcUBP-1 carece de la secuencia consenso descripta para las proteínas que interaccionan con el dominio C-terminal de PABP1 humana (Kozlov et al., 2001), esta posibilidad debe ser evaluada. Cuando PABP1 se acompleja con la cola de poli(A) en células eucariotas superiores, circulariza moléculas de ARN a partir de la interacción con el complejo de inicio de la traducción eIF4, con la concomitante estabilización del ARNm (Wilusz et al., 2001). Recientemente, se sugirió que la unión de varias proteínas de unión al ARE, como hnRNP D/AUF1, puede alterar la interacción entre PABP1 y la cola de poli(A), facilitando el acceso de una ribonucleasa que actúa deadenilando la cola de poli(A) (Chen et al., 2001). Por lo tanto, la rápida degradación del ARNm involucraría el reconocimiento de los AREs por las proteínas de unión al ARE, varias de las cuales se demostró que son capaces de reclutar el exosoma “in vitro”, como hnRNP D/AUF1, y causar la reducción de la afinidad de la PABP1 por la cola de poli(A) (Chen et al., 2001). En T. brucei, a partir de ensayos de interferencia de ARN realizados contra las diferentes subunidades del exosoma se demostró que el mismo está directamente involucrado en el procesamiento del ARNr 5S (Estévez et al., 2001) y también, al igual que en células eucariotas, en la degradación de ARNms conteniendo secuencias del tipo ARE en su región 3’-NC (Estévez A. et al., 112 Discusión comunicación personal). Más recientemente, se identificó en extractos de tripanosomas una actividad involucrada en el “decapping” de ARNms y otra relacionada con la degradación exonucleolítica 3’-5’, la cual se estimula por secuencias ARE presentes en la región 3’-NC del ARNm (Milone et al., 2002). En nuestro modelo de regulación, la interacción de TcUBP-1 y TcPABP1, rompe la unión entre TcPABP1 y la cola de poli(A) a través de una reducción en la afinidad de su sustrato. Es posible que en el estadío tripomastigote, la presencia de TcUBP-1 sin TcUBP2 en el elemento ARE de TcSMUG pueda reclutar el exosoma (Figura 41). Sin embargo, se sugiere que en el estadío epimastigote, la presencia de TcUBP-2 y otros factores no identificados hasta el momento, pueden estabilizar la interacción entre TcUBP-1 y TcPABP1 en el complejo ribonucleoproteico, generando la estabilización de TcSMUG. Esta interacción podría prevenir, al menos en parte, la pérdida de afinidad de TcPABP1 por la cola de poli(A) generada por la interacción con TcUBP-1. Esta hipótesis además se complementa con los resultados obtenidos a partir de la sobre-expresión de TcUBP1 en epimastigotes, en donde se muestra que niveles de la proteína TcUBP-1 más altos que los celulares desestabilizan al ARNm TcSMUG (Figura 41). Es posible que este fenómeno de desestabilización se alcance, en parte, a través de la depleción de TcPABP1 de la cola de poli(A) por TcUBP-1, y la consecuente degradación del ARNm por deadenilación. Es evidente que los resultados presentados en esta tesis proveen evidencias que demuestran la función de proteínas de unión a ARN en procesos de regulación de la abundancia de ARNms en tripanosomas. Sin embargo, se requieren experimentos adicionales para establecer el/los mecanismo/s por el cual las proteínas TcUBPs estimulan la estabilización y la desestabilización del ARNm TcSMUG. A partir de ensayos de doble híbrido en curso, se pretende encontrar los factores proteicos que interaccionan con TcUBP-1 y/o TcUBP-2 en el complejo ribonucleoproteico identificado; y de esta manera, profundizar los conocimientos sobre los mecanismos de regulación de la expresión de TcSMUG. Pre-ARNms dicistronicos estables Tcubp-1/Tcubp-2 en el citoplasma del parásito. Es evidente que la presencia del dicistrón Tcubp-1/Tcubp-2 identificado, el cual es estable en el citoplasma del parásito, es muy atractivo. Este dicistrón, contiene el SL en su extremo 5’ y un pequeño tracto de poli(A) en su extremo 3’, es transportado al citoplasma y es también relativamente estable (t ∼ 4 hrs) (resultados no publicados). 1/2 De estos resultados surgen distintas preguntas: 1) Cuál es el mecanismo que lleva a éste pre-ARNm no maduro al citoplasma? 2) Cuál es el camino de transporte utilizado por este pre-ARNm? y 3) Debe este pre-ARNm retornar al núcleo de la célula para su procesamiento final y generar los dos transcriptos maduros Tcubp-1 y Tcubp-2? Otros ejemplos de transcriptos que codifican para proteínas regulatorias son también expresados de la misma manera (D’Orso et al., resultados no publicados). Es decir, existe una incongruencia en el tamaño de los ADNc completo y el tamaño del ARNm observado en un ensayo de “Northern blot”. Por lo tanto, es interesante determinar las bases moleculares de este fenómeno que al menos por ahora parece ser un mecanismo de regulación específico para los tripanosomas. Recientemente, se describió que ciertos ARNs celulares presentan regiones 5’-NC relativamente extensas, las cuáles presentan 113 Discusión extensiones del tipo IRES (“Internal Ribosome Entry Site”) (Vagner et al., 2001). En el dicistrón Tcubp-1/Tcubp-2 para las proteínas TcUBPs, se observan regiones ricas en tractos de poli-Purinas, del tipo (A) G(A) GG(A) G(A) GG o G(A) G(A) G(A) . Algunos de 4 2 5 2 3 2 3 estos tractos se describieron como IRES (Chappell et al., 2000; Dorokhov et al., 2002) o son requeridos como “enhancers” del “splicing” en regiones exónicas. Se sabe que la inclusión de algunos exones depende de una secuencia de 81 nt, designada “exonic splicing enhancer”. Esta secuencia contiene un tracto de purina GAAGAAGA denominado elemento A (Caputi et al., 1994; Cramer et al., 1999), que es requerido para la interacción con proteínas involucradas en el “splicing” del tipo SR tales como ASF/SF2 (Tacke and Manley, 1995). Es necesario realizar experimentos para estudiar posibles mecanismos de traducción a partir de secuencias del tipo IRES y revelar cuáles podrían ser los estímulos requeridos para el procesamiento del dicistrón. Interacción TcUBP-ARN y estudios de dinámica molecular. A partir de los estudios de dinámica molecular realizados durante esta tesis se obtuvieron varios resultados. En primer lugar, se corroboró por RMN que las proteínas TcUBP-1 y TcUBP-2, sin su región C-terminal, presentan un espectro 15N-1H HSQC similar. En segundo lugar, se observó que la topología βαββαβ de TcUBP-1ΔQ es la misma que la observada en otras proteínas de unión a ARN con motivos del tipo RRM, como por ejemplo Sxl, HuD y PABP1 (Deo et al., 1999; Handa et al., 1999; Inoue et al., 2000). En tercer lugar, la titulación con ARN permite sugerir que las hojas β son importantes para generar una interfase de reconocimiento del sustrato ribonucleico. Además, se identificaron algunos de los residuos importantes para el reconocimiento del ARN rico en Uridina (Figura 47 y 48), lo cual demuestra similitudes y también diferencias con proteínas de otros tipos celulares. Es importante destacar que ensayos futuros relacionados con la resolución estructural del complejo TcUBP-1-ARN permitirán, en más detalle, analizar las diferencias en el modo de reconocimiento del ARN en proteínas con motivos RRM simples como TcUBP-1 y TcUBP-2 o múltiples, de distintos tipos celulares. Esto permitirá identificar y estudiar nuevos mecanismos y modos de interacción ARN-proteína en organismos evolutivamente tan lejanos como son los eucariotas superiores y los tripanosomas. 114 Discusión Conclusiones. Los aportes más importantes de esta tesis se resumen en los siguientes puntos: (1) T. cruzi presenta una familia de genes de mucina denominada TcSMUG, que se regula en forma post-transcripcional mediante mecanismos de estabilización/ desestabilización del ARNm, durante el desarrollo del parásito. (2) Los elementos ARE y GRE están involucrados en mecanismos de regulación de la estabilidad funcionalmente opuestos, e interaccionan con proteínas presentes en extractos proteicos de los diferentes estadíos del desarrollo del T. cruzi. (3) Las proteínas AREBPs se expresan en forma diferencial durante el desarrollo y las proteínas GREBPs se expresan en forma constitutiva. (4) La expresión de las proteínas TcUBP-1 y TcUBP-2 puede ser regulada a partir de un dicistrón estable que contiene ambos transcriptos. (5) Las proteínas TcUBP-1 y TcUBP-2 forman un complejo ribonucleoproteico con el ARN TcSMUG y con la proteína TcPABP1, y factores adicionales no identificados hasta el momento. (6) Las proteínas TcUBPs homo- y hetero-dimerizan “in vitro” e “in vivo”. Los residuos de Tirosina presentes en la región rica en Glicina del C-terminal están involucrados en la dimerización, y son sustrato de una actividad Quinasa de Tirosina no identificada hasta el momento. Se sugiere que la fosforilación puede regular la formación de dimeros “in vivo”, y de esta manera regular la estabilidad del ARNm de TcSMUG. (7) El motivo RRM de ambas TcUBP es necesario para la interacción con el ARN. Además, aunque la extensión C-terminal (región RV) confiere estabilidad en la formación del complejo ribonucleoproteico. En cuanto al dominio C-terminal rico en Glutamina de TcUBP-1, su función es todavía desconocida aunque posiblemente tenga un rol en la regulación de la formación de homodímeros y/o en la interacción con TcPABP1. (8) A partir de los ensayos de RMN, se demostró que la topología βαββαβ del motivo RRM de las proteínas TcUBP-1 y TcUBP-2 son similares entre sí y al de las proteínas con motivos RRM múltiples de otros tipos celulares. A partir del uso de espectroscopía por RMN se mapearon los residuos importantes para el reconocimiento del ARN por TcUBP-1. 115 Discusión Perspectivas futuras. (1) Purificar los factores que reconocen a los elemntos ARE y GRE, e identificar los genes que codifican estas proteínas sería de gran importancia para el estudio de la regulación de TcSMUG. (2) Estudiar la regulación de las proteínas TcUBP-1/TcUBP-2 a partir del dicistrón de ∼5.5 Kpb identificado. Determinar cuáles son los estímulos ambientales requeridos para el procesamiento de este pre-ARNm. (3) Identificar los factores adicionales que forman parte del complejo ribonucleoproteico del cual TcUBP-1 y TcUBP-2 forman parte, ya sea utilizando experimentos de coinmunoprecipitación y análisis de los productos precipitados por espectrometría de masa MALDI-TOF o a partir de ensayos de doble híbrido en levaduras. (4) Identificar la Quinasa de Tirosina capaz de regular la formación de dimeros entre las proteínas TcUBPs y demostrar la función de la fosforilación/defosforilación en la estabilidad del ARNm TcSMUG durante el desarrollo del parásito. De essta manera, se pretende estudiar la conexión entre procesos de estabilización de ARNms y transducción de señales durante el desarrollo del parásito. (5) Estudiar bioquímica y estructuralmente cómo es la formación de homo- y heterodímeros entre TcUBP-1 y TcUBP-2. (6) Utilizar experimentos de espectroscopía por RMN para definir la estructura de la proteína TcUBP-1 sola en solución o acomplejada con el sustrato ribonucleico, con el objetivo de definir dianas o pequeñas moleculas para bloquear la interaccion ARN-proteína y estudiar su función en el desarrollo del parásito. La identificación de las proteínas TcUBPs en T. cruzi, las cuales interaccionan con el elemento ARE de TcSMUG y pueden interaccionar entre ellas formando un complejo ribonucleoproteico con TcPABP1, sugiere un modelo interesante para estudiar mecanismos de regulación post-transcripcional y control de la formación de homo-heterodímeros “in vivo”. Es necesario la caracterización de los factores adicionales que formen parte del complejo identificado en esta tesis, como así también las proteínas involucradas en la regulación de la formación de dímeros de TcUBP-1, lo cuál permitirá entender cuáles son las señales necesarias para el reclutamiento en ARNms para ser estabilizados o desestabilizados. Este punto de regulación es de relevancia para un organismo que, siendo incapaz de regular la expresión de proteínas a nivel de inicio de la transcripción, requiere de un estricto y rápido control de mecanismos regulatorios. Estos procesos deben ser controlados en forma ordenada una vez que el parásito pasa de un hospedador a otro y se diferencia de estadíos; de lo contrario, las moléculas requeridas para la protección contra el sistema inmune del hospedador no podrían ser expresadas de manera apropiada. 116 MATERIALES y METODOS Materiales y Métodos Manipulación de organismos vivos. Parásitos. La mayoría de los análisis se realizaron sobre la población clonal de Trypanosoma cruzi CL-Brener, utilizada como clon de referencia en el proyecto genoma del parásito (Zingales et al., 1997). Las diferentes formas del parásito, epimastigotes, tripomastigotes derivados de células, tripomastigotes metacíclicos y amastigotes, fueron obtenidas como se describe. Brevemente, el estadío epimastigote de T. cruzi se obtuvo de cultivos axénicos en medio BHT (Life Technologies) con 10% de suero fetal bovino, 100 U/ml penicilina, 100 μg/ml estreptomicina, crecidos a 28ºC con agitación. El estadío de tripomastigote metacíclico se obtuvo por diferenciación a partir de cultivos de epimastigotes en fase estacionaria, incubados en botellas de plástico a 28ºC y sin agitación. Se purificó utilizando columnas de DEAE-celulosa según ya fue descripto (de Sousa, 1983). Los tripomastigotes derivados de células (equivalentes de cultivo al tripomastigote sanguíneo) y amastigotes se obtuvieron a partir de monocapas de células Vero crecidas en medio MEM (Life Technologies) con 4% suero fetal bovino. Los tripomastigotes se cosecharon del medio de las células infectadas, típicamente 5 días post-infección. Los amastigotes se obtuvieron mediante centrifugación diferencial a partir de lisados de células infectadas. Bacterias. Los clonados y la expresión de proteínas recombinantes se realizaron en las cepas DH5αF’ Iq (supE44 Dlac U169 (Ø80 lacZDM15) hsdR17 recA1 endA1 gyr A96 thi-1 relA1 F’ Iq), XL1-Blue (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac (F’ proAB lacI q ZDM15)) o BL21 DE3 pLysS (F–ompT hsdS B (r B–m B–) gal dcm (DE3) pLysS (CmR)). Esta cepa tiene la ventaja de ser deficiente en las proteasas lon y ompT (Phillips et al., 1984). Las bacterias se crecieron de rutina en medio LB (Sambrook et al., 1989). Para la recuperación de bacterias transformadas se usó medio Terrific Broth (Sigma) para obtener mayor eficiencia. Las bacterias conteniendo cósmidos se crecieron en medio Terrific Broth para aumentar el rendimiento de la purificación. Las bacterias competentes se prepararon a partir del método de Cl Ca (Inoue et al., 1990) y se transformaron con la mitad de la 2 reacción de ligación en las condiciones descriptas en el mismo trabajo. Alternativamente se usó el método de electroporación en cubetas de 2 mm utilizando 1/10 de la mezcla de ligación. A esta mezcla se la sometió a un pulso de 2.500 Voltios y 25 μF en un aparato Gene Pulser (BioRad). Purificación y manipulación de ácidos nucleicos. Purificación de ADN total de T. cruzi Se cosecharon cultivos de epimastigotes en fase logarítmica por centrifugación a 7.000g, durante 10 min. a 4°C y se lavaron tres veces con PBS. El pellet de parásitos se resuspendió en solución I (10 mM Tris-HCl pH 7.6, 100 mM NaCl, 100 mM EDTA) a 0,1 gr/ml. Los parásitos en suspensión se lisaron por el agregado de SDS hasta 1%. El lisado se incubó con proteinasa K (Sigma) 100 μg/ml durante 16 hrs a 55°C. Este lisado se extrajo varias veces con un volúmen de fenol:cloroformo (1:1) hasta la desaparición de 118 Materiales y Métodos proteínas en la interfase. El fenol fue equilibrado con Tris-HCl pH 7.6 (Sambrook et al., 1989) y el cloroformo consistió en una mezcla cloroformo:alcohol isoamílico 24:1. Las fases se separaron centrifugando 5 min. a 12.000g. Los restos de fenol se extrajeron dos veces con un volúmen de cloroformo. El ADN se precipitó con 2,5 volúmenes de etanol 100%, el flóculo se extrajo con ansa de vidrio, se lavó en etanol 75%, se dejó secar 5 min. a temperatura ambiente y se resuspendió en buffer TE (10 mM Tris-HCl pH 7.6, 1 mM EDTA). Se agregó ARNasa a 20 μg/ml durante 45 min. a 37°C. Después se llevó a 0,5% SDS y 50 μg/ml proteinasa K y se incubó por 2 hrs a 55°C. Se extrajo una vez con fenol:cloroformo y una vez con cloroformo; se agregaron 0,1 volumen de acetato de sodio 3 M pH 5,2 y 2.5 volúmenes de etanol absoluto para precipitar el ADN y se centrifugó a 12.000g por 15 min. a 4°C. El pellet se lavó con etanol 70% y se resuspendió en buffer TE. La concentración fue estimada comparando con patrones de ADN de cantidad conocida corridos en geles de agarosa. Nota: En estas condiciones se extrae tanto ADN cromosomal como kinetoplástido. Purificación de ARN total de T. cruzi Se utilizó el reactivo comercial TRIzol (Life Technologies), basado en el método de isotiocianato de guanidina y fenol/cloroformo (Simms et al., 1993), según especificaciones del fabricante pero adaptando las cantidades de reactivo para T. cruzi. Se utilizó 1 ml de TRIzol por cada ~5x108 parásitos, no más porque sino la extracción del material no es propicia. La cuantificación se realizó en un espectrofotómetro, sabiendo que 1 OD a 260nm de Absorbancia corresponden aproximadamente a 40 mg/ml de ARN. Purificación de plásmidos y cósmidos de E. coli. Se realizó según el método de lisis alcalina (Sambrook et al., 1989). En el caso de los cósmidos obtenidos de la biblioteca genómica de CL-Brener se realizaron posteriores extracciones con fenol:cloroformo:isoamílico y cloroformo:isoamílico para lograr mayor pureza. Para obtener ADN de calidad superior se utilizaron columnas de intercambio aniónico Tip100 o Tip500 según la cantidad de ADN requerida (QIAGEN Inc.). Purificación de fragmentos de ADN de geles de agarosa Se utilizó el kit QIAEXII (QIAGEN Inc.), el cual se basa en el principio de que el ADN es capaz de adherirse al vidrio en presencia de altas concentraciones salinas y se realizó siguiendo las especificaciones del fabricante. Tratamientos enzimáticos del ADN. Digestiones con endonucleasas de restricción Se utilizaron 5-10 U de enzima por μg de ADN a digerir, con el buffer y temperatura correspondientes a cada enzima según instrucciones del fabricante (New England Biolabs). Para digestiones totales de ADN genómico, se agregaron las unidades de enzima necesarias en dos veces sucesivas, para mejorar la eficiencia de la digestión (Sambrook et al., 1989). La segunda dosis de enzima se agregó en general 2 hrs después de la primera, o tomando 119 Materiales y Métodos en cuenta el tiempo de sobrevida de la actividad de la enzima en cuestión. Después de la segunda dosis se continuó incubando 12-16 hrs. Ligado de fragmentos de ADN Se mezclaron ~50 ng de vector con el inserto de interés, en una relación molar aproximada 1:3, en el buffer de reacción provisto por el fabricante, en un volumen final de 10 μl. Las reacciones se incubaron de 3 hrs a 16°C con 4 U de ADN ligasa del bacteriofago T4 (New England Biolabs) en caso de ser extremos pegajosos o 16 hrs a 16°C con 40 U de ligasa para extremos romos y ligaciones mixtas. Los vectores fueron previamente tratados con fosfatasa alcalina intestinal de ternero (New England Biolabs), 1 U de fosfatasa por pmol de extremos en buffer de restricción, excepto cuando se ligaron oligonucleótidos sintéticos doble cadena. Para el clonado de productos de PCR realizadas con la enzima Taq ADN polimerasa, se utilizó el vector pGEM-T Easy (Promega) en las condiciones sugeridas por el fabricante. Reacción en cadena de la polimerasa (PCR) Las reacciones de PCR se realizaron utilizando la enzima Taq ADN polimerasa purificada en el Instituto a partir de lisados de E. coli, en buffer de reacción 1X (Life Technologies). La concentración de Cl Mg se ajustó al óptimo para cada reacción, siendo 2 por lo general de 1,5 mM y se utilizaron 100 ng de cada oligonucleótido por reacción en un volúmen final de 100 μl. En general, para la amplificación de fragmentos por PCR se utilizaron 35 ciclos de desnaturalización (5 min. 93ºC) - hibridización (30 seg.) - extensión (72ºC) en las mismas condiciones, excepto en el paso de hibridización de los oligonucleótidos, cuya temperatura se fijó en base al oligonucléotido con menor Tm (Tm = 2°C x (A+T) + 4°C x (G+C)); en el paso de extensión el tiempo dependió del largo esperado de la región a amplificar, calculando 1 min. de extensión por kpb. Electroforesis de ácidos nucleicos. Geles no desnaturalizantes para ADN Los fragmentos de ADN fueron fraccionados en geles de agarosa de distintos porcentajes conteniendo 0,5 μg/ml bromuro de etidio, según indicaciones de los manuales de laboratorio (Sambrook et al., 1989). Las condiciones de corrida fueron aproximadamente 5 V/cm durante 1 hr para minigeles de rutina y 0,5-4 V/cm para geles medianos y grandes, corridos durante 12-16 hrs para resolver patrones complejos. A cada muestra se le agregaron 0,2 volúmenes de buffer de muestra (Ficoll 400 15% (p/v), EDTA 1 mM pH 7.6 y naranja-G 2% (p/v)). Se utilizó como buffer de electroforesis de rutina TBE (Tris-borato 90 mM, ácido bórico 90 mM, EDTA 2,5 mM pH 8.3). El ADN fue visualizado por la fluorescencia inducida por luz ultravioleta sobre el bromuro de etidio intercalado. Geles desnaturalizantes para ARN Se utilizó el protocolo descripto en (Fourney et al., 1978). Brevemente, se sembraron 20-30 μg. de ARN total de T. cruzi en cada calle de un gel desnaturalizante de 120 Materiales y Métodos 1,5% agarosa 0,66 M formaldehído. La muestra se mezcló previamente con 5 volúmenes de buffer de muestra (53% formamida, 1X MOPS/EDTA, 6% formaldehído, 0,5% azul de bromofenol, 7% glicerol), y se calentó a 65˚C durante 15 min.. Despues se agregó Bromuro de Etidio (1 μl de una solución 0.2 mg/ml) y se aplicó directamente en geles de agarosa desnaturalizantes. La corrida se realizó a 3 V/cm durante 3-4 hrs. El buffer de armado del gel y de corrida fue MOPS/EDTA (0,02 M MOPS, 0,5 mM acetato de sodio, 0,01 mM EDTA pH 7,0). Inmovilización de ácidos nucleicos en filtros de nylon o nitrocelulosa La transferencia de ácidos nucleicos a filtros de nylon a partir de geles de agarosa se realizó por la técnica de capilaridad según (Sambrook et al., 1989). La transferencia de ADN de colonias bacterianas a filtros para rastreo de recombinantes por hibridización con sondas radiactivas se realizó según (Sambrook et al., 1989). Los ácidos nucleicos se unieron covalentemente al filtro después de su transferencia mediante irradiación con luz ultravioleta (256 nm) durante 1 min. Para geles desnaturalizantes de ARN, la transferencia se realizó por capilaridad durante toda una noche utilizando membranas de nylon Zeta-Probe (Bio-Rad). Marcación radiactiva de fragmentos de ADN La marcación radioactiva se realizó por el método de PCR utilizando la enzima Taq ADN polimerasa sintetizada en nuestro Instituto de Investigación. Se utilizó en el orden de 1 ng de ADN molde, en general a partir de una primer PCR fría o como insertos purificados a partir de geles no desnaturalizantes de agarosa. La PCR se realizó utilizando 100 ng de cada oligonucleótido. Las condiciones utilizadas fueron 1,5 mM Cl Mg, 20 mM 2 Tris-HCl pH 8.4, 50 mM KCl y 1 U de Taq polimerasa. La concentración de deoxinucleótidos (dATP - dGTP - dTTP) se ajustó a 10 μM de c/u y se utilizaron 0,5 mCi/ml dCTP-α32P) (10 mCi/ml, actividad específica 3.000 Ci/mmol, Perkin Elmer Life Sciences), en un volúmen final de 100 μl para lograr sondas de alta actividad específica (Mertz and Rashtchian, 1994). Las sondas se purificaron por precipitación con AcNa 0.3 M y 2.5 volúmenes de etanol 100% o pasando el producto de la PCR por una columna de Sephadex G-50. Las fracciones de 200 μl se cuantificaron en un contador de centelleo. Sondas de ADN utilizadas. NH -TcSMUG: Sintetizada con los cebadores N1, 5’-atgatgctgcgccgcgtcct-3’ y N2, 5’2 gtcgcccccagcagcctca-3’, sobre el clon TcSMUG Mle-1 (Di Noia, 2000). 3’-L: Sintetizada con los cebadores L-se, 5’-gggggaagcgtcggtgcgcc-3’ y L-as, 5’tgtcgctatcggctccctggg-3’ sobre el clon TcSMUG 7.2 (Di Noia, 2000). 3’-S: Sintetizada con los cebadores S-se, 5’-atgaggacggggcggggcg-3’ y S-as, 5’tcagcgccgaaaacgtac-3’ sobre el clon 13n22 (Di Noia, 2000). UBP-1/N: Sintetizada con los cebadores UBP1-N-se y UBP-1-N-as sobre el clon Tcubp-1. UBP-2/N: Sintetizada con los cebadores UBP2-N-se y UBP-2-N-as sobre el clon Tcubp-2. RRM: Sintetizada con los cebadores UBP1-N-se y RNP1-as sobre el clon Tcubp-1. D-AUX: Sintetizada con los cebadores NES-se y UBP-1/C sobre Tcubp-1. 121 Materiales y Métodos Ductin: Sintetizada sobre un ADN molde gentilmente provisto por el Sr. Ramiro Verdún. ARNr: Sintetizada con los cebadores “forward” y “reverse” de m13 sobre un ADN molde pUC19 gentilmente provisto por el Dr. Esteban Bontempi (Instituto Fatala Chaben). Hsp70: Sintetizada con los cebadores T7 y T3 utilizando como ADN molde un plásmido pT7 conteniendo un EST TENG 0042 (Número de acceso Genbank: AI034982), proveniente del proyecto genoma de T. cruzi. Hibridización de filtros con sondas radiactivas Las sondas se marcaron por medio de la técnica de PCR, en presencia de [α32P]dCTP. La reacción de PCR en presencia de marca radiactiva para un volumen final de 100 μl fue la siguiente: 1 ng de ADN molde, 10 μM de una mezcla (dATP, dGTP y dUTP), 5 μl[α32P]-dCTP (3000Ci/mmol), 1.5 mM Cl Mg, 1X Buffer Taq Polimerasa, 1U Taq 2 Polimerasa. Se utilizó esta técnica para la identificación de secuencias con distinto grado de homología a una sonda determinada, según protocolos comunes de laboratorio (Sambrook et al., 1989). Los filtros fueron bloqueados entre 2-4 hrs a 65°C en (600 mM NaH PO pH 7.4, 7% SDS, 1 mM EDTA y 1% seroalbúmina bovina). Se utilizó 500.000 2 4 cpm de sonda por ml de mezcla de hibridización. Se incubó toda la noche a 65°C con agitación suave. Se utilizó dos condiciones de lavado distintas, (1) las que se definen como rigurosas o estrictas (20 mM Na+, 0,1% SDS, 65°C) y (2) las que se refieren como de baja rigurosidad o relajadas (200 mM Na+, 0,1%. SDS, 55°C) siempre con agitación (en general 3 lavados de 20 min. cada uno). La marca fue detectada por exposición a placas autoradiográficas del tipo X-OMAT AR (Kodak). Secuenciación de ADN Las secuencias se realizaron en forma manual y automática. En el primer caso se utilizó el kit comercial Sequenase 2.0 (Amersham Life Science), basado en el método de terminación de cadenas con dideoxinucleótidos de Sanger. Se utilizaron de ~5 μg de plásmido doble cadena previamente desnaturalizado como molde. Las reacciones de secuencia se realizaron siguiendo las instrucciones del fabricante utilizando 10-20 ng del oligonucleótido necesario y dATP-γ-(35S) (Perkin Elmer Life Sciences) y se revelaron por autoradiografía utilizando placas del tipo BioMax (Kodak). Alternativamente se utilizó el método de terminadores fluorescentes con el kit ABIPRISM (Perkin Elmer) y se resolvieron en el secuenciador automático ABI377 (Perkin Elmer) del Instituto. En estos casos los electroferogramas se analizaron manualmente para confirmar las asignaciones de bases hechas por el programa. Análisis de secuencias de ADN. Las secuencias se ingresaron y manipularon con el paquete Lasergene (DNASTAR Inc.). Las alineaciones entre secuencias se realizaron en el programa MegAlign de ese paquete utilizando el algoritmo Clustal W (Thompson et al., 1994). Normalmente, el porcentaje de identidad se calculó a partir de la relación de bases o aminoácidos idénticos sobre la longitud total de la secuencia. En algunos casos se utilizó como parámetro para comparar la semejanza entre secuencias, un índice de similitud que calcula este programa. 122 Materiales y Métodos El mismo es un valor numérico calculado teniendo en cuenta las bases o residuos idénticos y el número y calidad de espacios que haya sido necesario incluir para realizar la alineación, así como la longitud de las secuencias. Aislamiento de los clones descriptos en esta tesis. Tcubp-1 y Tcubp-2. Con el objetivo de identificar posibles secuencias de proteínas de unión a ARN en T. cruzi, se realizó un t-blast-n contra la base de datos de ESTs de tripanosomátidos del NCBI utilizando como entrada el marco abierto de lectura de la proteina HuR de mamíferos, una proteína involucrada en la regulación de la estabilidad de ciertos transcriptos celulares (Fan and Steitz, 1998). Esta proteína se caracteriza por estar compuesta por 3 motivos RRM y por tener actividad de unión al ARE. A partir de esta búsqueda, se identificó un marco abierto de lectura que codificaba una proteína con un sólo motivo RRM. Para su clonado, se realizó una RT-PCR con el cebador oligo(dT) -ancla 5’-gcgactccgcggccgcg(t) 18 18 3’ el cual sirvió para hacer la síntesis del ADNc. La primer reacción de PCR se realizó usando los oligonucleótidos SL (5’-tacagtttctgtactatattg-3’), el cual reconoce la secuencia del “spliced leader” de T. cruzi y RNP-I (5’-aaacttcacaatccataggcc-3’). Los productos finales se clonaron en el vector pGEM-T Easy (Promega) para su secuenciación automática. Utilizando esta estrategia, no sólo se clonó parte del gen de interés, denominado Tcubp1, sino también un segundo clon caracterizado por ser un poco más pequeño. El mismo se llamó Tcubp-2, debido a su alta identidad con Tcubp-1. Para el clonado de los dos ADNc enteros, se sintetizaron oligonucleótidos que reconocen la región N-terminal de ambos, UBP1-N-se (5’-cgggatccatgagccaaattgttgtttc-3’) y UBP-2-N-se (5’cgggatccatgtctcaacagatgcaatac-3’). Estos oligonucleótidos, se utilizaron para hacer una segunda reacción de PCR junto con el oligonucleótido ancla (5’-gcgactccgcggccgcg-3’). Los productos fueron clonados en el vector pGEM-T Easy (Promega) para su secuenciación. Una vez determinada la secuencia de ambos ADNcs, se realizó una reacción de PCR para clonar los marcos abiertos de lectura completos en el vector pGEX-2T (Amersham), para su posterior expresión en bacterias (ver más adelante). Para Tcubp-1, se utilizó los oligonucleotidos UBP-1-N-se y UBP-1-C. Para Tcubp-2, se utilizó UBP-2-N-se y UBP-2-C. Los sitios de restricción BamHI y EcoRI, para su clonado direccional, figuran subrayados. NOTA: La lista de los oligonucleótidos sintéticos utilizados se muestra en la sección de Anexos. Tcpabp1 y Tcpabc. El clonado de Tcpabp1 se realizó por PCR utilizando ADN genómico del clon CLBrener y los oligonucleótidos PABP-N y PABC-2 según se describió previamente (Batista et al., 1994). De la misma manera, se amplificaron 249 pbs correspondientes al C-terminal de TcPABP1, llamado TcPABC, con los oligonucleótidos PABC-1 y PABC-2. Ambos productos se clonaron en el vector pGEX-2T (Amersham) y se purificaron proteínas de fusión con GST utilizando columnas de glutation-agarosa (Sigma) como se indica más adelante. El dominio TcPABC se utilizó para generar anticuerpos policlonales en ratones como se indica más adelante. 123 Materiales y Métodos Experimentos de Transcripción con núcleos aislados (“Run-on”). Se aislaron núcleos provenientes de los diferentes estadíos del parásito a partir de una modificación del protocolo de lisis con NP-40 (Teixeira et al., 1994). Brevemente, 1x109 células se lavaron con PBS 1X y se resuspendieron en el buffer hipotónico A (10 mM Tris-HCl pH 7.6, 2 mM Cl Mg, 5 mM KCl, 2 mM Cl Ca, 1 mM DTT, 1 mM EDTA, 1 2 2 mM espermidina, 6% PEG ) y se incubó en hielo por 10 min. Después de la adición del 3350 detergente NP-40 1%, las células se lisaron con la ayuda de un homogeneizador en forma manual. El lisado celular se diluyó con 1 volúmen de buffer B 2X (0.64 M sacarosa, 40 mM Tris-HCl pH 7.6, 60 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT y 1 mM espermidina), y los núcleos se centrifugaron 15 min. a 1000g. El pellet nuclear se lavó una sola vez con 10 ml de buffer B 1X y se resuspendió en 100 μl de buffer C (25% glicerol, 50 mM Tris-HCl pH 8, 60 mM KCl, 1 mM DTT, 0.1 mM EDTA, 1 mM espermidina). Los transcriptos sintetizados se marcaron con [α-32P]-UTP a 30ºC durante 30 min., y luego fueron extraídos con fenol:cloroformo:alcohol isoamílico (25:24:1), precipitados con etanol y acetato de sodio y su radiactividad medida en un contador de centelleo β. La actividad específica de la sonda fue ~1x107 cpm para el estadío de epimastigote y ~1x106 cpm para los estadíos de tripomastigote y amastigote. Las sondas se utilizaron para hibridizar puntos o “dots” conteniendo ADNc pertenecientes a diferentes clones como se indica en la sección de Resultados (β-tubulina, ARNr, Mle-1 y 13f5, entre otros). Se inmobilizaron 3 μg de cada ADN plasmídico por punto. Tratamiento de los parásitos con drogas inhibidoras de la transcripción y traducción. Se tomaron cultivos de parásitos del estadío epimastigote a una densidad ∼40x106 células por ml. Los mismos fueron tratados con 10 μg/ml de Actinomicina D (ActD), un potente inhibidor de la transcripcción que se intercala en el ADN. Resultados previos deter minaron que ésta concentración de droga, inhibe la transcripción en tripanosomatidos ~100% (Charest et al., 1996; Furger et al., 1997). Se tomaron alícuotas de parásitos a diferentes tiempos luego del agregado de la droga y se cosecharon a 1000g durante 5 min. Los parásitos del estadío tripomastigote se cosecharon a partir de medio de cultivo conteniendo células Vero infectadas, resuspendidos en medio esencial mínimo (MEM) con 5% de suero fetal bovino (Life Technologies) a la misma concentración y tratados de la misma manera que los epimastigotes. La cicloheximida (Sigma), un potente inhibidor de la elongación de la traducción, fue utilizada a una concentracion final de 50 μg/ml en las mismas condiciones que la ActD. En experimentos en los cuales se utilizó ambas drogas, los parásitos fueron preincubados con cicloheximida por un período de 2 hrs y después la ActD fue agregada sin remoción de la primer droga. Todas la alícuotas de parásitos fueron cosechadas por centrifugación, lavadas con PBS y congeladas a 70ºC hasta la extracción de ARN total como se indicó. Transfección de parásitos. Las transfecciones se realizaron utilizando un electroporador BTX 600, cubetas de 2mm de separación entre electrodos y los siguientes parámetros: 1500μF, 335V y 24Ω. Se cosecharon aproximadamente 1x108 parásitos, se lavaron con medio BHT sin 124 Materiales y Métodos suero fetal bovino, y se resuspendieron en 400 μl de medio BHT más 10-50 μg de ADN plasmídico de alta pureza. Los parásitos una vez transfectados, se recuperaron en 5 ml de medio BHT-10% suero fetal bovino. Dos días post-transfección, se adicionó geneticina a una concentración final de ~250 μg/ml. Los parásitos se mantuvieron durante dos semanas en este medio, y luego la concentración del antibiótico se aumentó hasta 500 μg/ml. El gen de resistencia a neo, presente en los plásmidos pTEX (Kelly et al., 1992) y pRIBOTEX (Martinez-Calvillo et al., 1997), se utilizó como marcador de selección y como un control interno para medir los niveles de transfección, ya que se transcribe de manera policistrónica por el mismo promotor que el gen introducido. Ensayos de RT-PCR para determinar los extremos 5’ y 3’ maduros de los ARNms provenientes de las construcciones TcSMUG-L. Con el objetivo de determinar los extremos 5’ y 3’ utilizados como sitios aceptores de trans-“splicing” y agregado de la cola de poli(A), se llevaron a a cabo dos tipos de RTPCRs. Por un lado, para determinar el primer sitio se realizó la RT-PCR con el cebador CAT-in 5’-cgcaactgactgaaatgc-3’ y la reacción de PCR con el mismo oligo y utilizando la secuencia del SL de T. cruzi. Por otro lado, para determinar el sitio de agregado de la cola de poli(A), se realizó la RT-PCR con el cebador oligo-dt-ancla seguido de la reacción de PCR con los cebadores ancla y CAT -se (5’-gggatggagaaaaaaatcactggatata-3’). Los fragmentos provenientes de ambas PCRs se clonaron en el vector pGEM-T Easy (Promega) y se secuenciaron en un secuenciador ABI Prism 377. Ensayo de Actividad Cloramfenicol Acetil Transferasa (CAT). Los parásitos utilizados, se cosecharon por centrifugación a 1000g por 5 min. y se lavaron una vez con “buffer” Tris-HCl 0.25 M pH 8. Los extractos libre de células se prepararon a partir de 4 ciclos de congelamiento-descongelamiento e inactivacion a 65ºC por 10 min. Los lisados celulares se utilizaron para medir la actividad CAT como se describió previamente (Martinez-Calvillo et al., 1997). En resúmen, las reacciones de actividad se llevaron a cabo durante 1 hr a 37ºC con un lisado proveniente de 108 parásitos. Este tiempo de reacción se determinó experimentalmente de manera tal de que los valores de actividad obtenidos entren dentro del rango lineal del ensayo. La conversión de [14C]Cloramfenicol a la forma acetilada se analizó por cromatografia en capa delgada (TLC) y para ello se usaron placas de sílica delgada (Dupont, NEN). Los productos de reacción se resolvieron por corrida en “buffer” Cloroformo:Metanol (95:5). Ensayo de doble-híbrido en levaduras. Levaduras. Se utilizaron levaduras de la cepa L40 (Genotipo: MATa his3 D200 trp1-901 leu23112 ade2 LYS2::(4lexAop-HIS3 )URA3::(8lexAop-lacZ) GAL4) y Fenotipo His-, Trp-, Leu, Ade- (Shih et al., 1996). 125 Materiales y Métodos Vectores. Este ensayo se utilizó para detectar interacciones proteína-proteína “in vivo” en un sistema heterólogo que permita la confirmación de los resultados obtenidos por otra técnica. El sistema de doble híbrido utilizado es el desarrollado por Stan Fields y colaboradores (Fields and Song, 1989). El mismo se basa en la reconstrucción de un factor de transcripción híbrido donde el sitio de unión a ADN es el factor LexA y el sitio de transactivación es del factor VP16. El dominio de LexA está en el vector pBTM116 que es en el cual se clona la proteina señuelo y el factor VP16 está en el vector pVP16 que es con el cual se clona la proteina presa. Cepas y selecciones. El vector pBTM116 puede ser seleccionado en levaduras porque otorga autotrofismo para triptofano y pVP16 hace lo propio con leucina. La cepa utilzada es L40 (MATa his3D200 trp1-901 leu2-3,112 ade2 LYS2::lexAop4HIS3 URA::lexAop8-lacZ). Esta cepa es auxótrofa para triptofano y leucina, pero también para histidina. La reacción positiva de doble híbrido puede identificarse porque esta cepa posee dos genes bajo el control de promotores mínimos que tienen sitios de unión para LexA: HIS3 y lacZ. Entonces, cuando la proteína señuelo interacciona con la proteína del vector pVP16, los motivos de LexA y VP16 reconstruyen un factor de transcripción capaz de promover la expresión de un gen para β galactosidasa (lacZ). De este modo, la reacción positiva de doble híbrido se reconoce por el autotrofismo para histidina y la reacción colorimetrica de un ensayo para β galactosidasa. Medios y soluciones. YPD: 2% peptona, 1% extracto de levadura, 2% glucosa autoclavada en forma separada como stock 20%. Medios Yc: para 1 litro de medio disolver 1.2 gr base nitrogenada de levaduras (Difco, número catálogo: 291940), 0.1gr. de los aminoácidos ade, arg, cis, (leu), (lys), (trp), ura y 0.05gr. de asp, (his), ile, met, phe, pro, ser, tir, val, en 900 ml de agua, autoclavar y agregar 100 ml de glucosa 20%. Los aminoácidos entre paréntesis son excluidos en medios drop-out (excluyentes), según corresponda. Medios drop-out: Yc-TULys: Yc sin trp, ura y lys. Yc-THULL: Yc sin trp, his, ura, lys y leu. Solución AcLi: 0.1M AcLi, 10 mM Tris-HCl pH 7.6, 1 mM EDTA pH 8. Solución PEG-AcLi: 40% p/v PEG , 0.1M AcLi, 10 mM Tris-HCl pH 7.6, 1 mM EDTA 3350 pH 8. Transformación de levaduras. Aquí sólo se describe el protocolo de transformación a baja escala. Si la expresión de nuestra proteína no es tóxica para las levaduras, podemos hacer una transformación secuencial, es decir, primero transformando con el plásmido señuelo (pBMT116) y luego con el pVP16. 126 Materiales y Métodos 1- Se inoculan 10 ml de medio YPD con una colonia grande de las levaduras a ser transformadas. Se deja crecer a 28-30°C durante toda la noche con agitación. La OD debe alcanzar valores mayores a 2. 600nm 2- Se diluye en 50 ml de YPD hasta una OD ~0.5 y se deja crecer con agitación 600nm durante 2-4 hrs a la misma temperatura. 3- Se cosecha por centrifugación a 1000g a temperatura ambiente (no a 4°C!). El pellet se resuspende en 40 ml TE 1X y se vuelve a centrifugar a 1000g a temperatura ambiente por 10 min. 4- El pellet se resuspende en 2 ml de solucion AcLi y se incuba por 10 min. a temperatura ambiente. 5- Se mezclan 1-2 μg de plásmido señuelo con 50 μl de ADN “carrier” preparado en el momento. Preparación del “carrier”: se hierven por 5-10 min. 200 μl de ADN de esperma de salmón sonicado de concentración 10-20 μg/ml. Se le agregan inmediatamente 800 μl de solución de AcLi, se mezcla vigorosamente y se coloca en hielo hasta alcanzar temperatura ambiente. 6- A la mezcla de ADN (plásmido + ADN “carrier”) se le agrega 100 μl de las levaduras en solución AcLi y se mezcla bien. 7- Luego se le adicionan 700 μl de la solución PEG-AcLi y se mezcla con “vortex”. Se incuba la preparación por 30 min. a 30°C. 8- Agregar 70 μl de DMSO y dar un golpe de calor a 42°C durante 10 min. 9- Centrifugar a 10.000g por 20 segundos. El sobrenadante se descarta y el pellet de levaduras se resuspende en 150 μl de buffer TE 1X y se plaquea enplacas Yc-TULys. NOTA-1: Para corroborar que la construcción en el pBTM116 no activa “per se” al sistema del doble híbrido, se debe plaquear esta transformación también en placas Yc-THULL. En segundo lugar, las colonias que crecen en la placa YcTULys debemos ensayarlas para corroborar que no tengan actividad β-galactosidasa. Si ocurre que una construcción activa al sistema en forma indirecta, debemos plaquear en medio Yc-THULL con concentraciones crecientes de 3-aminotriazol (3-AT, Sigma), entre 0-50mM. El 3AT es un compuesto tóxico para las levaduras y que es degradado por la enzima codificada por HIS3. De este modo, si existe un nivel basal de expresión de HIS3, la proteína se utilizará para degradar 3AT y no para la síntesis de histidina. Haremos entonces el ensayo de doble híbrido con la concentración más baja de 3-AT necesaria para impedir que las levaduras crezcan en Yc-THULL. NOTA-2: Las levaduras transformadas primariamente con el plásmido señuelo pBTM116 se crecen en Yc-TULys y se transforman ahora con el vector presa pVP16 a ensayar. Esta transformación se plaquea en Yc-THULL con o sin 3AT dependiendo del señuelo, y se le mide actividad β-galactosidasa. Medición de actividad β-Galactosidasa. Para confirmar el carácter positivo de las levaduras L40 transformadas con ambos plásmidos y capaces de crecer en medio Yc-THULL, se realizan ensayos de medición de la actividad β-galactosidasa. 1- Se estrían colonias a una placa fresca Yc-THULL. 2- Se transfieren las colonias de levaduras a filtros de nitrocelulosa. 127 Materiales y Métodos 3- Las membranas se colocan con las colonias hacia arriba sobre un soporte de aluminio que flota en nitrogeno líquido. Luego de 30 seg. se retira y se deja llegar a temperatura ambiente. Se repite el proceso 2 veces más. Este proceso permite la lisis celular. 4- Los filtros se colocan, colonias hacia arriba sobre papel Whatman 3MM humedecido con “buffer” Z + 50 μl de 25 mg/ml X-Gal, y el mismo se monta sobre una caja de Petri. La caja se tapa y se coloca a 30°C entre 15 min. y 2 hrs. Buffer Z: 60 mM Na HPO , 40 mM NaH PO , 10 mM KCl, 1 mM MgSO pH 7. Antes de 2 4 2 4 usar se agregan 25 μl de β-mercaptoetanol cada 10 ml de “buffer”. 4 Manipulación del ARN para ensayos de interacción con proteínas. Reacciones de transcripción in vitro. Todos los plásmidos utilizados para la transcripción “in vitro” se construyeron de la siguiente forma. Se sintetizaron oligonucleótidos de ADN (Life Technologies) complementarios a las cadenas sentido y antisentido de los ARNs a obtener (ver sección Anexo), conteniendo sitios de restricción para las enzimas EcoRI y HindIII ya abiertos o digeridos. Los mismos se calentaron 5 min. a 95ºC y se unieron a 37ºC durante 2 hrs. Luego se clonaron directamente en el vector pBS- (Stratagene), digerido por EcoRI y HindIII. De esta manera, todos los ARNs sintetizados a partir del promotor para la ARN polimerasa T7, presentan la secuencia GAATTC en el extremo 5’ y A en el extremo 3’ terminal. Para transcribir la cadena sentido del ARN, se incubaron en un volúmen final de 20 μl los siguientes reactivos durante 1 h a 37ºC: 1 μg de plásmido digerido con HindIII, ARN polimerasa T7 (Promega), 500 μM de rATP, rCTP y rGTP, 1 μM [α-32P]-UTP (800Ci/mmol) y 10 U RNAsin (Promega). La cadena antisentido del ARN se sintetizó de la misma manera, pero utilizando 1 μg de plásmido digerido con EcoRI y ARN polimerasa T3 (Promega). Todos los transcriptos fueron purificados en geles de poliacrilamida conteniendo UREA 8 M y eluídos con agitación durante 3 hrs a 37ºC, en un “buffer” conteniendo 0.3 M AcNa, 10 mM Cl Mg y 1 mM EDTA. Luego de la elución, los ARNs se 2 precipitaron con etanol 100% y se resuspendieron en agua. Para las transcripciones preparativas en ausencia de material radioactivo, se realizaron reacciones en un volúmen final de 200 μl a 37ºC durante toda la noche. Los ARNs se detectaron en geles de poliacrilamida-UREA por irradiación con luz UV. Análisis de la interacción ARN-Proteína Análisis por retardo en geles nativos. Las reacciones de unión ARN-proteína, se realizaron en un volúmen final de 20 μl en un “buffer” conteniendo: 20 mM Tris-HCl pH 7.6, 100 mM KCl, 5 mM Cl Mg, 1 mM 2 DTT, 50 ng/μl Heparina, 50 ng/μl ARNt. Además se utilizó 10000 cpm de ARN marcado y la concentración indicada de proteína recombinante o lisado de parásitos correspondiente, según cada experimento. Las reacciones se llevaron a cabo durante 10 min. a 25ºC para reacciones realizadas con extractos de parásitos o 37ºC en presencia de proteínas recombinantes. Cada reacción se sembró directamente en geles nativos 128 Materiales y Métodos 10% de acrilamida-bisacrilamida (38:2) y 0.5 X buffer TBE, realizándose la conocida técnica de retardo en gel, la cual permite determinar si alguna proteína interacciona con la secuencia de ARN en cuestión en las condiciones utilizadas. Para los ensayos de competición de la unión ARN-proteína, se utilizaron por un lado homopolímeros de ARN no marcados radiactivamente (rA, rC, rG y rU), y por otro lado transcriptos “in vitro” como se describió anteriormente. Los geles de retardo, se secaron durante 1 hr a 70ºC y luego se expusieron sobre placas radiográficas, en especial del tipo X-Omat AR (Kodak). Entrecruzamiento de los complejos ribonucleoproteicos por irradiación con luz UV. El ARN marcado radiactivamente se incubó en presencia de un extracto total de proteínas y se resolvió en un gel nativo como se describió en la sección anterior. Los complejos ribonucleoproteicos detectados por exposición del gel de retardo a 4ºC sobre placas radiográficas, se entrecruzaron covalentemente por irradiación con luz UV (254 nm, 500 mJ/cm2) durante 15 min. Luego se trataron con ARNAsa T1 y se eluyeron del gel en presencia de 0.1% SDS a 37ºC durante 3 hrs y en agitación. Los productos del entrecruzamiento se resolvieron mediante electroforesis en geles SDS-PAGE 12.5%. De esta manera y en presencia de marcadores de peso molecular se determinó la masa molecular aparente de las proteínas detectadas en los geles de retardo. Determinación de constantes de disociación aparentes. Para la determinación de una K aparente se hicieron varios ensayos para d confirmar: (1) la concentración de ARN necesaria a utilizar en cada ensayo, de manera tal de que, a una dada concentración proteica, la mitad del ARN este unido; (2) la cantidad de proteína recombinante activa para cada ensayo. El primer punto se determinó experimentalmente y en general se utilizó entre 5000-10000 cpm de ARN marcado (lo cual corresponde a ~1ng). El segundo punto, se estudió realizando ensayos de retardo en gel con una concentracion fija de proteína y aumentando la cantidad de ARN. De este modo y utilizando dos cantidades de proteína diferentes, se pudo observar una curva de saturación en cada caso, que permite hacer el cálculo de proteína activa según se describe (Smith, 1998). En general, todas las proteínas de fusión construidas tuvieron una actividad ~95% según se determinó por geles de retardo. Luego de puesta a punto la técnica, se utilizó una concentración fija de ARN (~1ng) y cantidades crecientes de proteínas, de entre 20 y 1000 nM. Las reacciones se realizaron como se indicó en la sección anterior y se corrieron geles nativos 10% (Acrilamida:Bisacrilamida, 38:2). Las bandas de los complejos ribonucleproteicos se cuantificaron por densitometría y el valor de K aparente d para cada complejo ARN-Proteína, se calculó a partir de un gráfico de dos ordenadas, % (ARN unido/ARN libre) vs concentración de proteína utilizada. Entrecruzamiento covalente de ARN a matrices de agarosa-dihidrazida y ensayos de unión ARN-Proteína. En primer lugar se oxidó el ARN, de manera tal de obtener extremos 3’ C=O en los C-2’ y C-3’, los cuales de esa manera, pueden reaccionar con la matriz de agarosa activada con dihidrazida (Sigma). Para este procedimiento se resuspenden 500 pmoles de ARN en 129 Materiales y Métodos AcNa 0.1 M y se usa 50 mM NaIO como agente oxidante durante 1 hr a temperatura 4 ambiente y en oscuridad para preservar su capacidad oxidante. Una vez oxidado el ARN, se lo precipita con etanol 100% y se lo lava con etanol 75% para eliminar el oxidante remanente. El ARN se resuspende en 500 μl de AcNa 0.1 M. Luego se prepara la matriz de la siguiente forma. Por cada muestra de ARN a entrecruzar a la matriz, se toman 400 μl de la agarosa 50% hidratada y se la lava 4 veces con 1 ml de AcNa 0.1 M, centrifugando en cada paso 1000g durante 1 min. Una vez equilibrada la matriz, se la mezcla con el ARN durante 12 hrs a 4°C con agitación para obtener el ARN-entrecruzado con la matriz de agarosa para los ensayos. El día siguiente, se procede a eliminar el ARN que no se unió con la matriz. Para ello se realizan 3 lavados con 2M ClNa de 1 ml cada uno. Por último, se equilibra en el buffer de unión a utilizar posteriormente, 3 lavados de 1 ml cada uno es suficiente. El “buffer” utilizado en mi caso, fue el siguiente: 20 mM Tris-HCl pH 7.6, 1 mM DTT, 100 mM KCl, 5 mM Cl Mg, 10% glicerol, 50 μg/ml Heparina. Para 2 realizar la unión de proteínas recombinantes, se utiliza alrededor de 5 μg, y para extractos de parásitos se usan 200 μl de un lisado (~10 mg/ml) por tubo. El ensayo de unión se lleva a cabo durante 1 hr y luego se procede a lavar con el buffer de unión en presencia de 0.5% NP-40. Se realizan 4 lavados de 10 min. cada uno, centrifugando en cada caso 1 min. a 1000g. Las proteínas unidas a la matriz se eluyen calentando a 100°C por 10 min. en presencia de 2X Laemmli buffer. Proteínas Expresión de Proteínas Recombinantes. La preparación de proteínas recombinantes se realizó en dos vectores de expresión. Por un lado, se usó el vector pGEX-2T (Pharmacia), el cual permite generar una fusión con la Glutation-S-Transferasa (GST) de Schistosoma japonicum; y por otro lado se utilizó el vector pTRIS-A (Invitrogen) el cual genera una fusión en el C-terminal con un “tag” de poli-Histidina. En la Sección Anexo se muestra el nombre y la secuencia de los clones TcUBPs utilizados durante la tesis doctoral, como así también los cebadores que se utilizaron en las reacciones de PCR para su construcción. Los clones Tcubp-1 y Tcubp-2 no se expresan en forma correcta utilizando el vector pTRIS-A (Invitrogen), pero si se logró generar fusiones activas con la GST en el vector pGEX-2T. Los clones se transformaron en E. coli, preferencialmente en la cepa BL21 DE3 pLys S (Novagen), la cual cuenta con una deficiencia en la capacidad para degradar proteínas (DE3) y también presenta una lisozima que facilita la lisis celular (pLYS). A partir de placas de LB-antibiótico frescas, se realizaron cultivos durante toda la noche a 37°C. Al próximo día se realizaron diluciones 1/20-1/50 a un medio rico como es el Terrific-broth, en presencia del antibiótico correspondiente. Una vez alcanzada una DO de aproximadamente 0.8, se adicionó 600nm el inductor de la expresión, en este caso Isopropil β-D-tiogalactopiranósido (IPTG) durante 3 hrs a 37°C. La cantidad de IPTG utilizada fue de 0.2 mM cuando se utilizó el vector pGEX-2T y 1 mM para cultivos conteniendo el plásmido pTRIS-A. Las proteínas recombinantes se purificaron en columnas de Glutation-agarosa o en una columna “HiTrap” quelante de metales, respectivamente. Luego de la inducción durante 3 hrs las bacterias se cosecharon por centrifugación a 5000g durante 10 min. El pellet bacteriano 130 Materiales y Métodos se resuspendió en “buffer” TBS (~1/20 del volúmen inducido), en presencia de 1 mM PMSF y 3 mM EDTA como inhibidores de Serin y metalo-proteasas, respectivamente. La lisis se realizó por sonicación, con 4 pulsos de 30 seg. cada uno, después se adicionó 1% Triton-X100 y se incubó durante 5 min. en hielo para terminar con la lisis. Por último, el lisado se separó de los restos celulares por centrifugación a 10000g por 20 min. Purificación de proteinas de fusión a GST y separación de la GST por digestión con Trombina. Se utilizó una columna de glutatión-agarosa de 1 ml de volúmen, y se equilibró de la siguiente manera: (1) lavado con 5 volúmenes de TBS Triton-X100 1%, (2) lavado con 5 volúmenes ClNa 3 M, y (3) lavado con 10 volúmenes de TBS 1% Triton-X100. El lisado proteico obtenido se pasó por la columna de glutatión-agarosa y luego se lavó con 10 volúmenes de TBS. Por último, se eluyó con 5 ml de glutation reducido (Sigma) en alícuotas de 1 ml cada una. La columna se regeneró lavandola en presencia de 5 volúmnes de NaCl 3 M y luego se la almacenó a 4°C hasta su próximo uso. Para la determinacion de actividad, las proteínas de fusión se cortaron con Trombina, ya que el plásmido pGEX2T tiene sitio de corte para esta proteasa (ver más adelante). De este modo, se separó la proteína de interés de la proteína de fusión con GST. Para tal motivo, la proteína de fusión producida y pegada a la columna no se eluyó con glutation-agarosa, sino que se cortó directamente en la columna. Para el protocolo de digestión, se utilizó 10 U Trombina (Sigma) por mg de proteína fusión y el tiempo de digestión se determinó en forma experimental ya que el mismo depende de la accesibilidad de la proteasa al sitio de corte. Para las proteínas de fusión producidas en esta tesis, incluyendo TcUBP-1, TcUBP-2, sus mutantes de deleción y TcPABP1, fue suficiente con digerir 2 hrs a 25°C en presencia de un “buffer” conteniendo 20 mM Tris-HCl pH 7.6, 150 mM NaCl y 2,5 mM Cl Ca. El 2 sitio de reconocimiento para la Trombina puede ser según (Chang, 1985): (1) A4-A3-PR/K-A1-A2, donde A4-A3 son residuos hidrofóbicos (M,Y,I,L,V) y A1-A2 son residuos no acídicos (G,N,K,L,S); (2) A2-R/K-A1, donde A2 o A1 son G. Fraccionamiento subcelular y preparación de extractos proteicos y polisomas. Para la preparación de extractos citosólicos, los parásitos se resuspendieron en “buffer” de lisis a una concentración de ~1x106 células por μl de solución. El “buffer” de lisis utilizado contiene: 20 mM Tris-HCl pH 7.6, 0.5% NP-40, 10 mM Cl2Mg, 1 mM DTT, 1 mM EGTA, 10% glicerol y se suplementa con inhibidores de proteasas: 1 mM PMSF y 10 μM E-64. Esta mezcla se incubó durante 15 min. a 4ºC y luego se centrifugó a 1000g durante 10 min. para separar los núcleos, restos celulares y parásitos no lisados. El sobrenadante (S1000) corresponde a la fracción citosólica. Si se pretende continuar trabajando con los núcleos hay que realizar dos lisis adicionales y centrifugación de cada una para separarlos de los restos celulares y contaminantes citosólicos. De esta manera, contamos con un “pellet” de 1000g (P1000), el cual está compuesto por las proteínas nucleares y de la membrana nuclear. Esta fracción, por sonicación (10 pulsos de 15 seg. cada uno) rinde una fracción nuclear libre de membranas. Por otro lado, el sobrenadante 1000g (S1000) se puede purificar aun más. Por centrifugacion a 10000g 15 min., clarificamos un poco más el sobrenadante. El mismo se puede centrifugar a 131 Materiales y Métodos 100.000g en una ultracentrifuga refrigerada (rotor 70Ti, ~40000rpm) durante 1 hr. El S100.000 contiene proteínas solubles mayoritariamente y el P100.000 es la fracción correspondiente a microsomas y en la cual están presentes los polisomas libres de retículo endoplásmico. Los polisomas también pueden purificarse de forma alternativa. El S10000, se siembra en un colchón de sacarosa de dos fases, 1 y 2 M, como se describió previamente (Gonzalez and Cazzulo, 1989). Este colchón se centrifuga durante 2 hrs a 4ºC a una velocidad de 100.000g (~40.000 rpm en un rotor 70Ti). El “pellet” del centrifugado contiene los poliribosomas separados de las demás fracciones sub-celulares. Producción de anticuerpos policlonales y conjugación de péptidos a KLH. Los anticuerpos se prepararon mezclando partes iguales de péptidos y/o proteínas con adyuvante de Freund (Sigma) como emulsionante. La primera inyección se dió con adjuvante de Freund completo, es decir, conteniendo el mycoplasma para estimular la respuesta inmunológica. En cambio, el primer y segundo desafío se realizaron en presencia de adjuvante incompleto (Sigma). Se inyectaron conejos y/o ratones cada 15 días y después del segundo o tercer desafío, los animales se sangraron a blanco. Para la primera inyección de los ratones se utilizó alrededor de 30 μg proteína en un volúmen de 200 μl final. La primera inyección de los conejos se hizo con 200 μg de proteína en un volúmen final de 1 ml. Para los desafios con la mezcla proteína-adyuvante incompleto se utilizó sólo la mitad de proteína que para la primera inyección. Para generar anticuerpos contra péptidos sintéticos (Sigma GENOSYS), se adicionó una Cisteína en el C-terminal del mismo. Este aminoácido permite su conjugación covalente a la proteína KLH (“maleimide-activated keyhole limpet hemocyanin”). Para favorecer ésta reacción, se incubó cantidades equimolares de péptido y KLH durante 1 hr a temperatura ambiente con agitación suave. La mezcla de reacción contiene péptido acoplado y libre, y sirve para inyectar animales directamente en presencia de la emulsión. Los anticuerpos generados fueron: 1- anti-RRM, generado contra los 92 aminoácidos del dominio de unión a ARN (RRM) de TcUBP-1 fusionado a GST; 2- anti-TcUBP-1, generado contra el péptido VSQYDPYGQTAC de la región Nterminal de TcUBP-1 acoplado a KLH; 3- anti-TcUBP-2, generado contra el péptido RNRNGVSTFGAC de los aminoácidos 111-122 de TcUBP-2 acoplado a KLH; 4- anti-TcPABC, generado contra los 83 aminoácidos del dominio C-terminal de TcPABP1 fusionado a GST. Geles desnaturalizantes SDS-PAGE. Se utilizaron geles de 7.5, 10 y 12.5% de acrilamida:bisacrilamida (29:1) con el sistema de “buffers” discontinuo de Laemmli (Sambrook et al., 1989). Las muestras de proteínas provenientes de lisados de parásitos, bacterias, levaduras y purificaciones de proteínas recombinantes se sembraron en geles desnaturalizantes mezclándolas con “buffer” de muestra (100 mM Tris-HCl pH 6.8, 2% SDS, 200 mM DTT, 20% glicerol, 0,1% azul de bromofenol). Los geles se tiñeron con “Coomasie Blue” R-250 (Sigma) o con Nitrato de plata (Sigma) según se indique en cada caso, o transferidos a membranas de 132 Materiales y Métodos nitrocelulosa para la realización de ensayos de “Western blot”. Ensayos de “Western blot” e inmunorevelados. Las muestras proteicas separadas en geles SDS-PAGE, se transfirieron a filtros de nitrocelulosa utilizando la técnica de electrotransferencia húmeda durante 1 hr en “buffer” Towbin (25 mM Tris, 192 mM Glicina, 0.1% SDS, 10-20% Metanol) a 250 mA constante. La transferencia se monitoreó por tinción de la membrana en solución Ponceau S (0.1% p/v Ponceau S en 5% ácido acético glacial). La membrana se incuba 1 min. con la solución, y luego se realizan sucesivos lavados con agua deionizada para sacar el exceso de colorante. De esta manera, se puede determinar la posición de los marcadores de peso molecular y cada una de las calles sembradas. Luego se decolora la membrana lavando con TBS por unos minutos. Para detectar bandas específicas, los filtros se bloquearon en TBS-5% leche descremada durante 1 h y se incubaron con el suero de inmunización en TBS-5% leche durante 1-2 hr y en general utilizando una dilución ~1/ 200 del suero primario. Se realizan dos lavados de 10 min. cada uno en presencia de TBS-Tween 0.1%, y luego se incubó con un segundo anticuerpo acoplado a peroxidasa en una dilución 1/10.000, seguido por dos lavados de TBS-Tween 0.1%. Finalmente, se reveló por quimioluminiscencia detectada por autorradiografía utilizando el “kit” Supersignal® West Pico Chemiluminescent Substrate (Pierce). La membrana húmeda se expone a placas radiograficas X-Omat AR (Kodak). Ensayos de inmunoprecipitación con proteína-A-sefarosa. Extractos proteicos correspondientes a 108 parásitos se incubaron con suero preinmune, o el correspondiente suero (anti-TcUBP-1, anti-TcUBP-2, anti-TcPABP1), en una dilución ~1/50, durante 2 hrs a 4°C en agitación. En un segundo paso, se adicionó 50 μl de proteína-A-sefarosa (Sigma) y se incubó por 2 hrs más con agitación a 4°C, luego la mezcla se centrifugó a 1000g por 1 min., se descartó el sobrenadante y el immunoprecipitado se lavó 3 veces con 400 μl de TBS-Tween 0.1%. Las proteínas retenidas durante la immunoprecipitación, se eluyeron calentando en presencia de Laemmli buffer 2X. Estas se separaron en geles SDS-PAGE seguido de un ensayo de “Western blot” con el anticuerpo indicado en cada caso. En algunos casos y donde se indica, el extracto libre de células se pretrató durante 30 min. a 25°C con 0.1 μg/μl ARNasa A (USB). Estudios de “Cross-linking” proteína-proteína. Para los ensayos de “cross-linking” se utilizaron diferentes concentraciones de proteína recombinante según se indica (0.25, 0.5 y 1 μM). Las proteínas resuspendidas en “buffer” TBS se trataron durante 30 min. a temperatura ambiente en presencia de glutaraldehido 0.01% final como agente para generar el entrecruzamiento covalente de las proteínas. El glutaraldehido utilizado a ésta concentración final permite unir covalentemente aquellos residuos que están haciendo contacto a casi 0Å de distancia (Wan et al., 2001). Los productos fueron resueltos en geles SDS-PAGE y teñidos con “Coomasie Blue” R250. Ensayos de Fosforilación “in vitro”. 133 Materiales y Métodos Para los ensayos de fosforilación se utilizó alrededor de 1 μg de proteína recombinante en presencia de la Quinasa de Tirosina c-Src humana (Sigma, S5439), en 15 μl de volúmen final y utilizando un buffer con la siguiente composición: 10% glicerol, 50 mM Hepes pH 7.5, 2 mM DTT, 10 mM Cl Mg, 0.25% Triton X-100 y 1 μl de [γ32P]-ATP 2 (6000 Ci/mmol). Las reacciones se llevaron a cabo a 30ºC durante 1 hr y los productos se resolvieron en geles SDS-PAGE 10% y se tiñeron con “Coomasie Blue” R250 antes de exponer a placas radiográficas X-Omat-AR (Kodak). Métodos de Purificación Analítica o Preparativa de Proteínas. 1- Cromatografia de Filtración en Gel. Se prepararon extractos citosólicos correspondientes a 5-108 parásitos del estadío epimastigote, de la manera que se indicó más arriba. A este extracto se le ajustó la concentración salina, 150 o 500 mM NaCl, es decir baja o alta fuerza iónica, respectivamente. De la misma manera el pH se ajustó a 6.8 con “buffer” Tris-HCl 1 M pH 6.8. La muestra se pasó por un filtro 0.2 μM y se inyectaron no más de 300 μl (10 mg/ml) por corrida, volúmen que permite una resolución adecuada en las columnas utilizadas. Se utilizaron dos tipos de columnas. En primer lugar, BioSil SEC-250 (BioRad) que tiene un rango de separación de 10-300 kDa. En segundo lugar, se utilizó la columna Superosa 6 (Amersham), cuyo rango de separación es mayor. En ambos casos, el flujo de la corrida se mantuvo en 1 ml/min. y se colectaron fracciones de 0.5 ml. Las muestras de cada corrida fueron analizadas en geles SDS-PAGE seguido de ensayos de “Western blot.” Y la calibración de las columnas se realizó previamente utilizando los siguientes estándares de peso molecular (Sigma): 670kDa, Tiroglobulina; 180kDa, α2-Macroglobulina; 45kDa, Ovalbumina; 17kDa, Mioglobina. 2- Cromatografía de intercambio iónico. DEAE Sefarosa. La columna de intercambio catiónico DEAE (Amersham), se equilibró de la siguiente manera: (1) 5 volúmenes de “buffer” A (20 mM Tris-HCl pH 7.6); (2) 5 volúmenes de “buffer” B (20 mM Tris-HCl pH 7.6 + 500 mM NaCl), y (3) 10 volúmenes de “buffer” A. Para la purificación de la proteína eAREBP, se utilizaron 500 ml de un lisado correspondiente a 100 gr. de parásitos. El lisado libre de células se cargó en una columna conteniendo un volúmen de matriz de ~500 ml, a un flujo constante de 5 ml/min. Luego se lavó con 5 volúmenes de columna y se eluyó con un gradiente lineal de NaCl (buffer B) utilizando un flujo de 1 ml/min. Se colectaron 50 fracciones de 9 ml cada una y se midió actividad por geles de retardo utilizando 10 μl de cada fracción. El gel de retardo se corrió por 1 hr 30 min. y luego de su secado por vacio se expuso a -70°C a placas radiográficas durante 2 hrs. 3- Cromatografia de afinidad Heparin-HyperD. Esta columna (BioSepra) se equilibró de la siguiente manera: (1) 5 volúmenes de “buffer” A (50 mM MES pH 6.2); (2) 5 volúmenes de “buffer” B (50 mM MES pH 6.2 + 1 M NaCl), y (3) 10 volúmenes de “buffer” A. El pool de la DEAE Sefarosa (~10 ml), se cargó en una columna de Heparina (BioSepra) de 10 ml de volúmen, utilzando un flujo constante de 3 ml/min. El protocolo de elución utilizado fue el siguiente: a) 0-3.33 min., 0-0.25 M 134 Materiales y Métodos NaCl; b) 3.33-16.6 min, 0.25-0.6 M NaCl; c) 16.6-17, 0.6-1 M NaCl; d) 17-20 min., 1 M NaCl. Se recolectaron 25 fracciones de 1.4 ml y se midió actividad por geles de retardo utilizando 10 μl de cada fracción. El gel de retardo se corrió por 1 hr 30 min. y luego de su secado se expuso a -70°C a placas radiográficas durante 2 hrs. 4- Cromatografía de intercambio catiónico Mono S. La columna Mono S (Amersham) se equilibró de la siguiente manera y utilizando un flujo de 1 ml/min.: (1) 5 volúmenes de “buffer” A (50 mM MES pH 6.2); (2) 5 volúmenes de “buffer” B (50 mM MES pH 6.2 + 0.5 M NaCl), y (3) 10 volúmenes de “buffer” A. El pool de la cromatografía de afinidad en Heparina (~7ml) se diluyó 1/5 y se lo aplicó a esta columna con el flujo indicado. La elución se realizó con un gradiente lineal de 0-0.5 M NaCl y se recolectaron 20 fracciones de 1.4 ml. Se utilizó al igual que en el paso anterior, 10 μl para la medición de actividad evaluada por geles de retardo. 5- Cromatografía de hidrofobicidad Fenil Sefarosa. El último paso para la purificación fue de hidrofobicidad, utilizando una columna de Fenil Sefarosa (Amersham), la cual se equilibró de la siguiente manera a un flujo de 0.5 ml/min. (1) 5 volúmenes de “buffer” A (20 mM Tris-HCl pH 7.6 + 1 M SO (NH4) ); (2) 4 2 5 volúmenes de “buffer” B (20 mM Tris-HCl pH 7.6 + 5% sacarosa), y (3) 10 volúmenes de “buffer” A. La elución se realizó con un gradiente lineal decreciente en concentración de SO (NH4) de 1 a 0 M. Se recolectaron 40 fracciones de 0.5 ml cada uno. 4 2 Transferencia a membranas de PVDF (Polyvinylidene Fluoride) para el análisis del N-terminal por Edman. La mitad de la muestra de purificación se separó en un gel SDS-PAGE 7.5%, ya que la proteína eAREBP tiene un tamaño de ~100kDa. La corrida se realizó en “buffer” CAPS (3-[cilohexilamino]-1-ácido propanesulfónico) 1X (Caps 10 mM pH 11) y para la transferencia se procedió utilizando en “buffer” CAPS 1X con 10% Metanol para análisis. La transferencia se realizó por 1 hr. a 50V constantes a temperatura ambiente y la membrana de PVDF se tiñó en 0.1% “Coomasie-Blue” preparado en 40% Metanol/1% Acido acético, y destiñó con 50% Metanol. Luego se lavó con agua deionizada y se cortó la banda identificada para realizar un ensayo de secuencia de la región N-terminal por la reacción de Edman en el laboratorio del Dr. Ulf Hellman (Uppsala University, Sweden). Espectrometria de masa y análisis por MALDI-TOF. La otra mitad de la purificación se corrió en el mismo gel desnaturalizante SDSPAGE. La banda identificada se cortó y se envió también al Dr. Ulf Hellman, quien realizó un digerido tríptico de la proteína y separación de los péptidos por HPLC y análisis por espectrometría de masa MALDI-TOF según se describió (Hellman, 2000). 135 Materiales y Métodos Resonancia Magnética Nuclear y ensayos de dinámica molecular. Expresión de proteínas en medio mínimo M9. Los plásmidos pGEX2T-TcUBP-1ΔQ, pGEX2T-TcUBP-1ΔQG1 o pGEX2T-TcUBP2ΔCt se transformaron en bacterias BL21 Gold/DE3 (Stratagene) y se crecieron en medio mínimo M9 (6 gr Na HPO , 3 gr KH PO , 0.5 gr NaCl, 0.5 gr 15N-NH4Cl (Isotec, Inc), 2 gr 13 2 4 2 4 C-glucosa (Cambridge Isotope Laboratory) por litro de cultivo, suplementado con 100 μg/ml de ampicilina. La expresión y purificación de las proteínas fusionadas a GST se realizó como se indicó. La proteína de fusión no se eluyó con glutatión reducido, sino que se realizó la digestión de la proteína de fusión con Trombina (Amersham) en la columna, durante 2 hrs a temperatura ambiente y con agitación suave. Se utilizó 50 U de Trombina en 4 ml de “buffer” TBS suplementado con 2.5 mM Cl Ca, para digerir la proteína de 2 fusión proveniente de la inducción de 1-2 litros de medio de cultivo. Luego de la digestión se recuperan los 4 ml conteniendo la proteína de interés, sin el “tag” de GST. Luego de la digestión, también se eluye con glutation-reducido (Sigma) 10 mM para testear que no haya proteína de fusión sin digerir y determinar sí la expresión funcionó correctamente. Para los ensayos de RMN, las proteínas se prepararon por desalado en columnas NAP-25 (Amersham) y concentración por liofilización en una centrífuga refrigerada. La concentración se realizó hasta obtener un volúmen final de 200 μl conteniendo 20 mM Tris-HCl pH 7.2, 150 mM NaCl. Por último, se agregó a la muestra 1 mM NaN y 1 mM 3 DTT. Espectroscopías por RMN. La asignación de las resonancias de la proteína TcUBP-1ΔQG1 se determinó usando técnicas de resonancia-triple estandares sobre la muestra marcada con 13C o 15N (Grzesiek and Bax, 1993). Para ello se utilizó los espectrómetros de RMN Bruker DRX500 y Varian UNITYplus 800 MHz. Todos los experimentos se procesaron a una temperatura de 303K. Las resonancias de cadena principal Cα, N, HN, y cadenas laterales Cα fueron asignadas a partir de experimentos HNCACB y CBCA(CO)NH (Grzesiek et al., 1992); Constantine, 1993 #28]. Las asignaciones de las resonancias de los Hα y de las constantes de acoplamiento 3J N α se obtuvieron a partir del experimento HNHA (Kuboniwa et al., 1994). H -H Otras asignaciones de cadena principal y laterales se obtuvieron a partir de experimentos tridimensionales NOESY (“Nuclear Overhauser Effect SpectroscopY”) y TOCSY (“TOtal Correlation SpectroscopY”) a 500 MHz. Los espectros fueron procesados utilizando los programas GIFA (Malliavin et al., 1998) y XWIN-NMR version 2.5 (Bruker Biospin). El análisis final se realizó con el programa XEASY (Bartels et al., 1995). La estructura secundaria de la proteína TcUBP-1ΔQG1 se analizó usando desplazamientos químicos conocidos para H y C (Wishart et al., 1991). Experimentos de Titulación. Titulación con ARN. Las titulaciones con ARN se llevaron a cabo en experimentos separados, con el ARN P1 (5’-guuuuguuuuuguuuuug-3’), generado por transcripción “in vitro” a partir del “kit” Megashort (Ambion) purificado por gel desnaturalizante PAGE y el homoribipolímero 136 Materiales y Métodos poly(U) (Sigma), utilizando un espectro de correlación cuántica simple heteronuclear (HSQC) 15N-1H a 500MHz, a una temperatura de 303K sobre la muestra 15N-TcUBP1ΔQG1 (~0.5 mM). Se calcularon los desplazamientos químicos en ppm, [(Δ 1H shift)2 + (Δ 15N shift x 0.2)2]1/2, de los residuos de la proteína 15N-TcUBP1ΔQG1 sobre el agregado de cantidades crecientes de los ARNs utilizados. Las razones molares de proteína-ARN P1 utilizadas fueron 1:0.1, 1:0.2, 1;0.35; 1:0.7, de manera tal de facilitar el movimiento de los residuos desplazados y asignar las resonancias de los residuos perturbados por la interacción con el ARN. De la misma manera, la titulación de 15N-TcUBP1ΔQG1 con poly(U) se llevó a cabo con las siguientes relaciones molares (1:0, 1;0.1, 1:0.2, 1:0.5). Este experimento es muy sensible, y permite detectar perturbaciones en el medio ambiente de las amidas de cadena principal debido a la unión del ligando. Los desplazamientos químicos de las amidas también son sensibles a las perturbaciones de las cadenas-laterales conectadas, que también interaccionan con el sustrato, pero también deben ser afectadas por cambios conformacionales en el esqueleto de la proteína, y esto a veces puede complicar la detección del sitio de unión. Para facilitar la delineación apropiada de la superficie de unión proteínaligando (sea éste ARN o proteína), se puso énfasis en la determinación de grandes desplazamientos químicos observados por titulación y de las amidas desplazadas que se ubican en superficies posibles expuestas según (Yuan et al., 2001). Titulación con proteínas. Las titulaciones se realizaron con la proteína TcUBP-2ΔCt (residuos 1-143) sin marcar, utilizando un espectro de correlación cuántica simple heteronuclear (HSQC) 15 N-1H a 500 MHz, a una temperatura de 303K sobre la muestra 15N-TcUBP1ΔQG1 (~0.2mM). Se calcularon los desplazamientos químicos en ppm, [(Δ 1H shift)2 + (Δ 15N shift x 0.2)2]1/2, de los residuos de la proteína 15N-TcUBP1ΔQG1 sobre el agregado de cantidades crecientes de TcUBP-2ΔCt (1:0, 1:0.1, 1:0.3 y 1:0.6). 137 Materiales y Métodos Tabla 7 - Experimentos de Resonancia Magnética Nuclear realizados sobre la proteína TcUBP1ΔQG1. nd, no determinado. Experimento Objetivo del experimento HNCACB CBCA(CO)NH Asignar los Cα y Cβ a partir 1H- 15N HSQC. Para saber si existe degeneración, es decir más de un residuo en la misma posición de la matriz. Identificar los Hα y calcular los ángulos dihedricos entre los átomos HN-N-C-Hα. Asignar los protones de acuerdo a las correlaciones C-C de cada residuo. Asignar los protones de H N de cada residuo. Identificar las restricciones de las distancias interprotones para los cálculos de l a estructura, y chequear el asignamiento. Residuo en a-helix (fuerte NOE entre HN i y HN i +1). Residuo en hoja-β ( fuerte NOE entre Ha i y HN i+1). Asignar los protones a partir de los protones de la HN del próximo residuo. Asignar los carbonos a partir de los protones de la HN del próximo residuo. Asignar protones de cadena lateral, siempre y cuando el residuo no este solapado con otros residuos o con grupos metilos que tienen los protones más de splazados (~1 ppm). Asignar los protones de re siduos aromáticos, de acuerdo a los NOEs entre estos y Hβ. HNCO HNHA (H)CCH-Cosy TOCSY-HSQC NOESY-HSQC H-TOCSY CONH C-TOCSY CONH 2D TOCSY homonuclear 2D NOESY homonuclear C-NOESY 13 Identificar las restricciones de las distancias inter-H para cálculos de estructura. HNCA CA(CO)NH Asignar los Cα a partir del 1 H- 15N HSQC. 512(1H)x64(15N)x92(13C) 512(1H)x64(13C)x96(15N) Número de repeticiones 16 8 Tiempo de mezcla / / 90 % (CO) 512(1H)x66(13C)x100(15N) 4 / 63 % (Ha) Falta analizar los ángulos. 512(1H)x110(15N)x110( 1H) 4 / Falta analizar 512(1H)x104(13C)x100( 1H) 4 / Se obtuvo poca información 512(1H)x48( 15N)x180(1H) 8 70 mseg 512(1H)x48( 15N)x180(1H) 8 100 mseg 512(1H)x78(13C)x150(15N) 8 / 512(1H)x60(15N)x62(13C) 8 / Falta analizar 1024(1H)x512(1H) 104 110 mseg Falta analizar 1024(1H)x512(1H) 176 110 mseg nd nd 100 mseg nd nd / nd nd / (%) residuos asignados Tamaño de la matriz 88 % (N, HN) y 96% (Cα, Cβ) Falta analizar Falta analizar Se realizará en el futuro para refinar la estructura Confirmar las asignaciones de los Cα, por HNCACB y CBCA(CO)NH 138 Materiales y Métodos ANEXO I Lista de oligonucleótidos de ADN utilizados para realizar las construcciones de CAT flanqueadas por las regiones intergénicas completa o delecionada de TcSMUG-L. Clon Oligonucleótido Secuencia CAT CAT-se 5’-gggATGGAGAAAAAAATCACTGGATATA-3’ CAT-as 5’-cccaagcttTTACGCCCCGCCCTGCCA-3’ SMUG-LΔGRE 1-SE 5’-cccaagcttTCGGTCTGGAGTCCCGTTGTG-3’ 3-AS 5’-cggaattcGCACGCACAACCAAAGACACC-3’ 1+-SE 5’-cccaagcttTGCGCCGCGTATATTTGAGC-3’ SMUG-LΔ1 SMUG-LΔ2 SMUG-LΔ3 SMUG-LΔARE SMUG-LΔSire “sense” 5’-gcttTGAGGACGGGGCGGGGCGCGTGCGCCGA-3’ “antisense” 5’-agcttCGGCGCACGCGCCCCGCCCCGTCCTCAA-3’ 2-SE 5’-cggaattcTGCCCGTGCATTCTCTGCGAC-3’ 2-AS 5’-cggaattcAAATATACGCGGCGCAGGCGCC-3’ 3-SE 5’-cggaattcGTGGGGGGAAGCGTCGGTGCG-3’ 3-AS 5’-cggaattcGCACGCACAACCAAAGACACC-3’ 3-SE 5’-cggaattcGTGGGGGGAAGCGTCGGTGCG-3’ ΔARE-1 5’-cggaattcTTGTGAAAGGGATGTTCGC-3’ ΔARE-2 5’-cggaattcGGCGCACCGACGCTTCCCCC-3’ 3-AS 5’-cggaattcGCACGCACAACCAAAGACACC-3’ Sire-SE 5’-cggaattcAGGCGAATATTTTTTTTCATCGG-3’ Sire-AS 5’-cggaattcCCCCCGCGAACATCCCTTTCAC-3’ 139 Materiales y Métodos ANEXO II Se indica la secuencia de los oligonucleótidos utilizados, clasificados según el clon sobre el que se diseñó cada uno. En minúsculas se indican las secuencias agregadas que no corresponden al molde y se subrayan los sitios de restricción utilizados. Oligos de ADN sintetizados para el clonado por PCR de un homologo de DEF-3 en T. cruzi.Nota: R, purina; Y, pirimidina; I, deoxi-Inosina. Oligonucleótido Secuencia Sentido QSESELA 5’-AGRTAIGCIAGYTCYTGCTC-3’ Antisentido KENEDDK 5’-GACGACAARCTIACIGAYTTG-3’ Sentido LACLLC 5’-AAACTGGCTTGTCTGCTCTGC-3’ Sentido EVGPCMEFK 5’-GACCTICARGAYCARGAYTAY-3’ Sentido KRDEGQE-1 5’-CTCYTGICCYTCRTCICGYTT-3’ Antisentido KRDEGQE-2 5’-TTRGCIGAYGARCCICAGGAG-3’ Sentido DREMPPMDPK 5’-GAGATGCCICCIATGGAYCC-3’ Sentido DAQQDLQDQDY-1 5’-GACCTICARGAYCARGAYTAY-3’ Sentido DAQQDLQDQDY-2 5’-TGIAGGTCYTGYTGIGCRTC-3’ Antisentido 140 Materiales y Métodos ANEXO III Números de acceso al GenBank de los clones obtenidos durante esta Tesis. Tcubp-1 AF372519 Tcubp-2 AF497746 Clon a TcUBP-1 TcUBP-1ΔN TcUBP-1ΔNQ TcUBP-1ΔQ TcUBP-1ΔQG1 TcUBP-1ΔQG2 TcUBP-1 (Y-A)ΔQ TcUBP-2 TcUBP-2ΔN TcUBP-2ΔC TcUBP-2ΔCG1 TcUBP-2ΔCG2 TcPABC TcPABP1 Cebador Secuencia NH2 -BamHI COOHh-EcoRI NH2 -int-BamHI COOH-EcoRI NH2 -int-BamHI G ln-as-EcoRI NH2 -BamHI G ln-as-EcoRI NH2 -BamHI dgngyl-EcoRI NH2 -BamHI NES-as-EcoRI NH2 -BamHI UBP1-GNGYLGA UBP1(Y-A) COOHh-EcoRI NH2 -BamHI COOH-EcoRI NH2 -int-BamHI COOH-EcoRI NH2 -BamHI G ln-as-EcoRI NH2 -BamHI gvstgf-EcoRI NH2 -BamHI Nes-as-EcoRI PABC-1 PABC-2 PABP-N PABC-2 5’-cgggatccATGAGCCAAATTCCGTTGGTTTC-3’ 5’-cggaattcTTACCTTCGAACAGGACGGGC-3’ 5’-cgggatccATGAACCCCGAGCCCGATGTT-3’ 5’-cggaattcTTACCTTCGAACAGGACGGGC-3’ 5’-cgggatccATGAACCCCGAGCCCGATGTT-3’ 5’-cgaattcTTACGCGTACGGGTTCGCAGCG-3 ’ 5’-cgggatccATGAGCCAAATTCCGTTGGTTTC-3’ 5’-cgaattcTTACGCGTACGGGTTCGCAGCG-3 ’ 5’-cgggatccATGAGCCAAATTCCGTTGGTTTC-3’ 5’-ggaattcTTAAAGATATCCGTTGCCGTCTCC-3’ 5’-cgggatccATGAGCCAAATTCCGTTGGTTTC-3’ 5’-cgaattcTTAGGGGCGCTGGTGACCACTGGC-3’ 5’-cgggatccATGAGCCAAATTCCGTTGGTTTC-3’ 5’-gatatcCGCGCCAAGATATCCGTTGCC-3’ 5’-gatatcGCCGGTGGCGCCGGCGCAGCCCCT-3’ 5’-cggaattcTTACCTTCGAACAGGACGGGC-3’ 5’-cgggatccATGTCTCAACAGATGCAATAC-3’ 5’-cggaattcCTACTGACGCGGCACCGACGG-3’ 5’-cgggatccATGAACCCCGAGCCCGATGTT-3’ 5’-cggaattcCTACTGACGCGGCACCGACGG-3’ 5’-cgggatccATGTCTCAACAGATGCAATAC-3’ 5’-cgaattcTTACGCGTACGGGTTCGCAGCG-3 ’ 5’-cgggatccATGTCTCAACAGATGCAATAC-3’ 5’-cggaattcTTAAAACCCAGTGCTTACGCCG-3’ 5’-cgggatccATGTCTCAACAGATGCAATAC-3’ 5’-cgaattcTTAGGGGCGCTGGTGACCACTGGC-3’ 5’-ggatccTCTTTGGCTTCACAGGGACAG-3’ 5’-gaattcGTAAACGTTCATGTGGCGATTC-3’ 5’-ggatccATGTCGAACTTTCCTGCTGCG-3’ 5’-gaattcGTAAACGTTCATGTGGCGATTC-3’ b a Todas las construcciones fueron clonadas en el vector pGEX-2T (Amersham). b Los sitios de restricción utilizados para los clonados se encuentran subrayados. 141 Materiales y Métodos ANEXO IV. Oligonucleótidos sintéticos utilizados para preparar los sustratos de ARN por transcripción “in vitro”. Nota: Los sitios de restricción utilizados para los clonados se encuentran subrayados. ARN SMUG-L-ARE SMUG-L-ARE-1 SMUG-L-ARE-2 SMUG-L-ARE-3 SMUG-S-ARE HEX-T-ARE PLC-ARE P1 P2 P3 P4 P1-GGG Oligonucleótidos 5’-aattcAATTTTAAAATTTATTTAATGTTGTTTTGATTTATTTATTTTAAa-3’ 5’-ttcgaATTAAAATAAATAAATCAAAACAACATTAAATAAATTTTAAAATTg-3’ 5’-aattcATTTATTTAATGTTGTTTTGATTTATTTATTa-3’ 5’-ttcgaAATAAATAAATCAAAACAACATTAAATAAATg-3’ 5’-aattcTTATTTATTa -3’ 5’-ttcgaAATAAATAAg-3’ 5’-aattcTTTTATTTATTTT a-3’ 5’-ttcgaAAAATAAATAAAAg-3’ 5’-aattcATTTTATTTTTATTTTTAa-3’ 5’-ttcgaTAAAAATAAAAATAAAATg-3’ 5’-ttcgaTAAATAAATAAATg-3’ 5’-ttcgaTAAATAAATAAATg-3’ 5’-aattcATATATATATATATATATATATATTTATTTa-3’ 5’-ttcgaAAATAAATATATATATATATATATATATATg-3 ’ 5’-aattcGTTTTGTTTTTGTTTTTGa-3’ 5’-ttcgaCAAAAACAAAAACAAAAg-3 ’ 5’-aattcATGTTGTTGTTGTTGTTGa-3’ 5’-ttcgaCAACAACAACAACAACATG-3’ 5’-aattcATGGTGTTGGTGTTGGTGa-3’ 5’-ttcgaCACCAA CACCAACACCATg-3’ 5’-aattcATATTATTATTATTATTAa-3’ 5’-ttcgaTAATAATAATAATAATATg-3’ 5’-aattcGTTTTGTTTTTGTGGGTGa-3’ 5’-ttcgaCACCCACAAAAACAAAAg-3’ 142 REFERENCIAS Referencias Aeschbach, R., Amado, R. and Neukom, H. (1976) Formation of dityrosine cross-links in proteins by oxidation of tyrosine residues. Biochim Biophys Acta, 439, 292-301. Aguero, F., Verdun, R.E., Frasch, A.C. and Sanchez, D.O. (2000) A random sequencing approach for the analysis of the Trypanosoma cruzi genome: general structure, large gene and repetitive DNA families, and gene discovery. Genome Res, 10, 1996-2005. Akhtar, A., Zink, D. and Becker, P.B. (2000) Chromodomains are protein-RNA interaction modules. Nature, 407, 405-409. Allmang, C., Petfalski, E., Podtelejnikov, A., Mann, M., Tollervey, D. and Mitchell, P. (1999) The yeast exosome and human PM-Scl are related complexes of 3' —> 5' exonucleases. Genes Dev, 13, 2148-2158. Almeida, I.C., Ferguson, M.A., Schenkman, S. and Travassos, L.R. (1994) Lytic anti-alpha-galactosyl antibodies from patients with chronic Chagas’ disease recognize novel O-linked oligosaccharides on mucin-like glycosyl-phosphatidylinositol-anchored glycoproteins of Trypanosoma cruzi. Biochem J, 304, 793-802. Amster-Choder, O. and Wright, A. (1992) Modulation of the dimerization of a transcriptional antiterminator protein by phosphorylation.Science, 257, 1395-1398. Andrews, N.W., Abrams, C.K., Slatin, S.L. and Griffiths, G. (1990) A T. cruzi-secreted protein immunologically related to the complement component C9: evidence for membrane pore-forming activity at low pH. Cell, 61, 1277-1287. Aphasizhev, R., Sbicego, S., Peris, M., Jang, S.H., Aphasizheva, I., Simpson, A.M., Rivlin, A. and Simpson, L. (2002) Trypanosome mitochondrial 3' terminal uridylyl transferase (TUTase): the key enzyme in U-insertion/deletion RNA editing. Cell, 108, 637-648. Argaman, M., Aly, R. and Shapira, M. (1994) Expression of heat shock protein 83 in Leishmania is regulated post-transcriptionally. Mol Biochem Parasitol, 64, 95-110. Avis, J.M., Allain, F.H., Howe, P.W., Varani, G., Nagai, K. and Neuhaus, D. (1996) Solution structure of the N-terminal RNP domain of U1A protein: the role of C-terminal residues in structure stability and RNA binding. J Mol Biol, 257, 398-411. Bakheet, T., Frevel, M., Williams, B.R., Greer, W. and Khabar, K.S. (2001) ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res, 29, 246-254. Bartels, C., Xia, T.H., Billeter, M., Güntert, P. and Wüthrich, K. (1995) The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR, 6, 110. Bastin, P., Ellis, K., Kohl, L. and Gull, K. (2000) Flagellum ontogeny in trypanosomes studied via an inherited and regulated RNA interference system. J Cell Sci, 113, 3321-3328. Bates, E.J., Knuepfer, E. and Smith, D.F. (2000) Poly(A)-binding protein I of Leishmania: functional analysis and localisation in trypanosomatid parasites. Nucleic Acids Res, 28, 1211-1220. Batista, J.A., Teixeira, S.M., Donelson, J.E., Kirchhoff, L.V. and de Sa, C.M. (1994) Characterization of a Trypanosoma cruzi poly(A)-binding protein and its genes. Mol Biochem Parasitol, 67, 301312. Benne, R., Van den Burg, J., Brakenhoff, J.P., Sloof, P., Van Boom, J.H. and Tromp, M.C. (1986) Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell, 46, 819-826. Berberof, M., Vanhamme, L., Tebabi, P., Pays, A., Jefferies, D., Welburn, S. and Pays, E. (1995) 144 Referencias The 3'-terminal region of the mRNAs for VSG and procyclin can confer stage specificity to gene expression in Trypanosoma brucei. EMBO J, 14, 2925-2934. Bernstein, E., Caudy, A.A., Hammond, S.M. and Hannon, G.J. (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature, 409, 363-366. Biamonti, G. and Riva, S. (1994) New insights into the auxiliary domains of eukaryotic RNAbinding proteins. FEBS Letters, 340, 1-8. Billaut-Mulot, O., Fernandez-Gomez, R., Loyens, M. and Ouaissi, A. (1996) Trypanosoma cruzi elongation factor 1-alpha: nuclear localization in parasites undergoing apoptosis. Gene, 174, 1926. Borst, P. (2002) Antigenic variation and allelic exclusion. Cell, 109, 5-8. Bousquet-Antonelli, C., Presutti, C. and Tollervey, D. (2000) Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell, 102, 765-775. Branden, C. and Tooze, J. (1991) In: Introduction to Protein Structure. Garland Publishing, Inc., New York. Brener, Z. (1973) Biology of Trypanosoma cruzi. Annu Rev Microbiol, 27, 347-382. Brittingham, A., Miller, M.A., Donelson, J.E. and Wilson, M.E. (2001) Regulation of GP63 mRNA stability in promastigotes of virulent and attenuated Leishmania chagasi. Mol Biochem Parasitol, 112, 51-59. Buckanovich, R.J. and Darnell, R.B. (1997) The neuronal RNA binding protein Nova-1 recognizes specific RNA targets in vitro and in vivo. Mol Cell Biol, 17, 3194-3201. Burd, C.G. and Dreyfuss, G. (1994) Conserved structures and diversity of functions of RNAbinding proteins. Science, 265, 615-621. Caputi, M., Casari, G., Guenzi, S., Tagliabue, R., Sidoli, A., Melo, C.A. and Baralle, F.E. (1994) A novel bipartite splicing enhancer modulates the differential processing of the human fibronectin EDA exon. Nucleic Acids Res, 22, 1018-1022. Carballo, E., Lai, W.S. and Blackshear, P.J. (1998) Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science, 281, 1001-1005. Cartegni, L., Maconi, M., Morandi, E., Cobianchi, F., Riva, S. and Biamonti, G. (1996) hnRNP A1 selectively interacts through its Gly-rich domain with different RNA-binding proteins. J Mol Biol, 259, 337-348. Chang, J.Y. (1985) Thrombin specificity. Requirement for apolar amino acids adjacent to the thrombin cleavage site of polypeptide substrate. Eur J Biochem, 151, 217-224. Chappell, S.A., Edelman, G.M. and Mauro, V.P. (2000) A 9-nt segment of cellular mRNA can function as an internal ribosome entry site (IRES) and when present in linked copies greatly enhances IRES activity. Proc Natl Acad Sci U S A, 97, 1536-1541. Charest, H., Zhang, W.W. and Matlashewski, G. (1996) The developmental expression of Leishmania donovani A2 amastigote- specific genes is post-transcriptionally mediated and involves elements located in the 3'-untranslated region. J Biol Chem, 271, 17081-17090. Chen, C.Y., Gherzi, R., Ong, S.E., Chan, E.L., Raijmakers, R., Pruijn, G.J., Stoecklin, G., Moroni, C., Mann, M. and Karin, M. (2001) AU-binding proteins recruit the exosome to degrade AREcontaining mRNAs. Cell, 107, 451-464. Chen, C.Y. and Shyu, A.B. (1995) AU-rich elements: characterization and importance in mRNA 145 Referencias degradation. Trends Biochem Sci, 20, 465-470. Chen, C.Y., Xu, N. and Shyu, A.B. (2002) Highly selective actions of HuR in antagonizing AU-rich element- mediated mRNA destabilization. Mol Cell Biol, 22, 7268-7278. Clayton, C.E. (2002) Life without transcriptional control? From fly to man and back again. EMBO J, 21, 1881-1888. Clayton, C.E., Fueri, J.P., Itzhaki, J.E., Bellofatto, V., Sherman, D.R., Wisdom, G.S., Vijayasarathy, S. and Mowatt, M.R. (1990) Transcription of the procyclic acidic repetitive protein genes of Trypanosoma brucei. Mol Cell Biol, 10, 3036-3047. Clore, G.M. and Gronenborn, A.M. (1991) Structures of larger proteins in solution: three- and four-dimensional heteronuclear NMR spectroscopy. Science, 252, 1390-1399. Clore, G.M. and Gronenborn, A.M. (1998) New methods of structure refinement for macromolecular structure determination by NMR. Proc Natl Acad Sci U S A, 95, 5891-5898. Clore, G.M. and Schwieters, C.D. (2002) Theoretical and computational advances in biomolecular NMR spectroscopy. Curr Opin Struct Biol, 12, 146-153. Cobianchi, F., Calvio, C., Stoppini, M., Buvoli, M. and Riva, S. (1993) Phosphorylation of human hnRNP protein A1 abrogates in vitro strand annealing activity. Nucleic Acids Res, 21, 949-955. Cogoni, C. and Macino, G. (1999) Homology-dependent gene silencing in plants and fungi: a number of variations on the same theme. Curr Opin Microbiol, 2, 657-662. Constantine, K.L., Goldfarb, V., Wittekind, M., Friedrichs, M.S., Anthony, J., Ng, S.C. and Mueller, L. (1993) Aliphatic 1H and 13C resonance assignments for the 26-10 antibody VL domain derived from heteronuclear multidimensional NMR spectroscopy. J Biomol NMR, 3, 41-54. Conte, M.R., Grune, T., Ghuman, J., Kelly, G., Ladas, A., Matthews, S. and Curry, S. (2000) Structure of tandem RNA recognition motifs from polypyrimidine tract binding protein reveals novel features of the RRM fold. EMBO J, 19, 3132-3141. Craig, A.W., Haghighat, A., Yu, A.T. and Sonenberg, N. (1998) Interaction of polyadenylate-binding protein with the eIF4G homologue PAIP enhances translation. Nature, 392, 520-523. Cramer, P., Caceres, J.F., Cazalla, D., Kadener, S., Muro, A.F., Baralle, F.E. and Kornblihtt, A.R. (1999) Coupling of transcription with alternative splicing: RNA pol II promoters modulate SF2/ ASF and 9G8 effects on an exonic splicing enhancer. Mol Cell, 4, 251-258. Crowder, S., Holton, J. and Alber, T. (2001) Covariance analysis of RNA recognition motifs identifies functionally linked amino acids. J Mol Biol, 310, 793-800. Crowder, S.M., Kanaar, R., Rio, D.C. and Alber, T. (1999) Absence of interdomain contacts in the crystal structure of the RNA recognition motifs of Sex-lethal. Proc Natl Acad Sci U S A, 96, 48924897. Das, A. and Bellofatto, V. (2003) RNA polymerse II-dependent transcription in trypanosomes is associated with a SNAP complex-like transcription factor. Proc Natl Acad Sci U S A, 100, 80-85. Das, A., Peterson, G.C., Kanner, S.B., Frevert, U. and Parsons, M. (1996) A major tyrosinephosphorylated protein of Trypanosoma brucei is a nucleolar RNA-binding protein. J Biol Chem, 271, 15675-15681. de Sousa, M.A. (1983) A simple method to purify biologically and antigenically preserved bloodstream trypomastigotes of Trypanosoma cruzi using DEAE-cellulose columns. Mem Inst Oswaldo Cruz, 78, 317-333. 146 Referencias DeMaria, C.T., Sun, Y., Long, L., Wagner, B.J. and Brewer, G. (1997) Structural determinants in AUF1 required for high affinity binding to A + U-rich elements. J Biol Chem, 272, 27635-27643. Deo, R.C., Bonanno, J.B., Sonenberg, N. and Burley, S.K. (1999) Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell, 98, 835-845. Derry, J.J., Richard, S., Valderrama Carvajal, H., Ye, X., Vasioukhin, V., Cochrane, A.W., Chen, T. and Tyner, A.L. (2000) Sik (BRK) phosphorylates Sam68 in the nucleus and negatively regulates its RNA-binding ability. Mol Cell Biol, 20, 6114-6126. Di Noia, J.M. (2000) Família de genes que codifican proteínas de tipo mucina en Trypanosoma cruzi. Universidad Nacional de Buenos Aires, Argentina, Buenos Aires. Di Noia, J.M., Buscaglia, C.A., De Marchi, C.R., Almeida, I.C. and Frasch, A.C. (2002) A Trypanosoma cruzi small surface molecule provides the first immunological evidence that Chagas’ disease is due to a single parasite lineage. J Exp Med, 195, 401-413. Di Noia, J.M., D’Orso, I., Aslund, L., Sanchez, D.O. and Frasch, A.C. (1998) The Trypanosoma cruzi mucin family is transcribed from hundreds of genes having hypervariable regions. J Biol Chem, 273, 10843-10850. Di Noia, J.M., D’Orso, I., Sanchez, D.O. and Frasch, A.C. (2000) AU-rich elements in the 3'untranslated region of a new mucin-type gene family of Trypanosoma cruzi confers mRNA instability and modulates translation efficiency. J Biol Chem, 275, 10218-10227. Di Noia, J.M., Sanchez, D.O. and Frasch, A.C. (1995) The protozoan Trypanosoma cruzi has a family of genes resembling the mucin genes of mammalian cells. J Biol Chem, 270, 24146-24149. Ding, J., Hayashi, M.K., Zhang, Y., Manche, L., Krainer, A.R. and Xu, R.M. (1999) Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev, 13, 1102-1115. DiNitto, J.P. and Huber, P.W. (2001) A role for aromatic amino acids in the binding of Xenopus ribosomal protein L5 to 5S rRNA. Biochemistry, 40, 12645-12653. Djikeng, A., Shi, H., Tschudi, C. and Ullu, E. (2001) RNA interference in Trypanosoma brucei: cloning of small interfering RNAs provides evidence for retroposon-derived 24-26-nucleotide RNAs. RNA, 7, 1522-1530. Dorn, P.L., Aman, R.A. and Boothroyd, J.C. (1991) Inhibition of protein synthesis results in super-induction of procyclin RNA levels. Mol Biochem Parasitol, 44, 133-139. Dorokhov, Y.L., Skulachev, M.V., Ivanov, P.A., Zvereva, S.D., Tjulkina, L.G., Merits, A., Gleba, Y.Y., Hohn, T. and Atabekov, J.G. (2002) Polypurine (A)-rich sequences promote cross-kingdom conservation of internal ribosome entry site. Proc Natl Acad Sci U S A, 99, 5301-5306. Downey, N. and Donelson, J.E. (1999) Search for promoters for the GARP and rRNA genes of Trypanosoma congolense. Mol Biochem Parasitol, 104, 25-38. Drabkin, H.A., West, J.D., Hotfilder, M., Heng, Y.M., Erickson, P., Calvo, R., Dalmau, J., Gemmill, R.M. and Sablitzky, F. (1999) DEF-3(g16/NY-LU-12), an RNA binding protein from the 3p21.3 homozygous deletion region in SCLC. Oncogene, 18, 2589-2597. Dragon, E.A., Sias, S.R., Kato, E.A. and Gabe, J.D. (1987) The genome of Trypanosoma cruzi contains a constitutively expressed, tandemly arranged multicopy gene homologous to a major heat shock protein. Mol Cell Biol, 7, 1271-1275. Dreyfuss, G., Kim, V.N. and Kataoka, N. (2002) Messenger-RNA-binding proteins and the messages they carry. Nat Rev Mol Cell Biol, 3, 195-205. 147 Referencias Edwards, T.A., Pyle, S.E., Wharton, R.P. and Aggarwal, A.K. (2001) Structure of Pumilio reveals similarity between RNA and peptide binding motifs. Cell, 105, 281-289. Elias, M.C., Faria, M., Mortara, R.A., Motta, M.C., de Souza, W., Thiry, M. and Schenkman, S. (2002) Chromosome localization changes in the Trypanosoma cruzi nucleus. Eukaryot Cell, 1, 944-953. Elias, M.C., Marques-Porto, R., Freymuller, E. and Schenkman, S. (2001) Transcription rate modulation through the Trypanosoma cruzi life cycle occurs in parallel with changes in nuclear organisation. Mol Biochem Parasitol, 112, 79-90. Estevez, A.M., Kempf, T. and Clayton, C. (2001) The exosome of Trypanosoma brucei. EMBO J, 20, 3831-3839. Evers, R., Hammer, A., Kock, J., Jess, W., Borst, P., Memet, S. and Cornelissen, A.W. (1989) Trypanosoma brucei contains two RNA polymerase II largest subunit genes with an altered Cterminal domain. Cell, 56, 585-597. Fan, X.C. and Steitz, J.A. (1998) Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J, 17, 3448-3460. Fersht, A.R. and Daggett, V. (2002) Protein folding and unfolding at atomic resolution. Cell, 108, 573-582. Fields, S. and Song, O. (1989) A novel genetic system to detect protein-protein interactions. Nature, 340, 245-246. Fierro-Monti, I. and Mathews, M.B. (2000) Proteins binding to duplexed RNA: one motif, multiple functions. Trends Biochem Sci, 25, 241-246. Ford, L.P., Bagga, P.S. and Wilusz, J. (1997) The poly(A) tail inhibits the assembly of a 3'-to-5' exonuclease in an in vitro RNA stability system. Mol Cell Biol, 17, 398-406. Fourney, R.M., Miyakoshi, J., Day III, R.S. and Paterson, M. (1978) Northern blotting: efficient RNA staining and transfer. FOCUS, 10, 5-9. Frasch, A.C. (1994) Trans-sialidase, SAPA amino acid repeats and the relationship between Trypanosoma cruzi and the mammalian host. Parasitology, 108, S37-44. Fraser, H.B., Hirsh, A.E., Steinmetz, L.M., Scharfe, C. and Feldman, M.W. (2002) Evolutionary rate in the protein interaction network. Science, 296, 750-752. Frey, S., Pool, M. and Seedorf, M. (2001) Scp160p, an RNA-binding, polysome-associated protein, localizes to the endoplasmic reticulum of Saccharomyces cerevisiae in a microtubule- dependent manner. J Biol Chem, 276, 15905-15912. Furger, A., Schurch, N., Kurath, U. and Roditi, I. (1997) Elements in the 3' untranslated region of procyclin mRNA regulate expression in insect forms of Trypanosoma brucei by modulating RNA stability and translation. Mol Cell Biol, 17, 4372-4380. Gallouzi, I.E., Brennan, C.M., Stenberg, M.G., Swanson, M.S., Eversole, A., Maizels, N. and Steitz, J.A. (2000) HuR binding to cytoplasmic mRNA is perturbed by heat shock. Proc Natl Acad Sci U S A, 97, 3073-3078. Gazzinelli, R.T., Pereira, M.E., Romanha, A., Gazzinelli, G. and Brener, Z. (1991) Direct lysis of Trypanosoma cruzi: a novel effector mechanism of protection mediated by human anti-gal antibodies. Parasite Immunol, 13, 345-356. Ghisolfi, L., Joseph, G., Amalric, F. and Erard, M. (1992a) The glycine-rich domain of nucleolin has an unusual supersecondary structure responsible for its RNA-helix-destabilizing properties. 148 Referencias J Biol Chem, 267, 2955-2959. Ghisolfi, L., Kharrat, A., Joseph, G., Amalric, F. and Erard, M. (1992b) Concerted activities of the RNA recognition and the glycine-rich C- terminal domains of nucleolin are required for efficient complex formation with pre-ribosomal RNA. Eur J Biochem, 209, 541-548. Gilinger, G. and Bellofatto, V. (2001) Trypanosome spliced leader RNA genes contain the first identified RNA polymerase II gene promoter in these organisms. Nucleic Acids Res, 29, 15561564. Gonzalez, N.S. and Cazzulo, J.J. (1989) Effects of trypanocidal drugs on protein biosynthesis in vitro and in vivo by Trypanosoma cruzi. Biochem Pharmacol, 38, 2873-2877. Gorlach, M., Wittekind, M., Beckman, R.A., Mueller, L. and Dreyfuss, G. (1992) Interaction of the RNA-binding domain of the hnRNP C proteins with RNA. EMBO J, 11, 3289-3295. Gosser, Y., Hermann, T., Majumdar, A., Hu, W., Frederick, R., Jiang, F., Xu, W. and Patel, D.J. (2001) Peptide-triggered conformational switch in HIV-1 RRE RNA complexes. Nat Struct Biol, 8, 146-150. Graham, S.V. and Barry, J.D. (1995) Transcriptional regulation of metacyclic variant surface glycoprotein gene expression during the life cycle of Trypanosoma brucei. Mol Cell Biol, 15, 59455956. Graham, S.V. and Barry, J.D. (1996) Polysomal, procyclin mRNAs accumulate in bloodstream forms of monomorphic and pleomorphic trypanosomes treated with protein synthesis inhibitors. Mol Biochem Parasitol, 80, 179-191. Grishin, N. (2001) KH domain: one motif, two folds. Nucleic Acids Res, 29, 638-643. Grishok, A., Pasquinelli, A.E., Conte, D., Li, N., Parrish, S., Ha, I., Baillie, D.L., Fire, A., Ruvkun, G. and Mello, C.C. (2001) Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell, 106, 23-34. Grosset, C., Chen, C.Y., Xu, N., Sonenberg, N., Jacquemin-Sablon, H. and Shyu, A.B. (2000) A mechanism for translationally coupled mRNA turnover: interaction between the poly(A) tail and a c-fos RNA coding determinant via a protein complex. Cell, 103, 29-40. Grzesiek, S. and Bax, A. (1993) Amino acid type determination in the sequential assingment procedure of uniformly 13C/15N-enriched proteins. J Biomol NMR, 3, 185-204. Grzesiek, S., Dobeli, H., Gentz, R., Garotta, G., Labhardt, A.M. and Bax, A. (1992) 1H, 13C, and 15N NMR backbone assignments and secondary structure of human interferon-gamma. Biochemistry, 31, 8180-8190. Handa, N., Nureki, O., Kurimoto, K., Kim, I., Sakamoto, H., Shimura, Y., Muto, Y. and Yokoyama, S. (1999) Structural basis for recognition of the tra mRNA precursor by the Sex- lethal protein. Nature, 398, 579-585. Hellman, U. (2000) In: Sample preparation by SDS-PAGE and in gel-digestion. Proteomics in functional genomics. Protein structure analysis, Birkhauser Verlag AG, Basel, pp. 43-54. Hendriks, E.F., Robinson, D.R., Hinkins, M. and Matthews, K.R. (2001) A novel CCCH protein which modulates differentiation of Trypanosoma brucei to its procyclic form. EMBO J, 20, 67006711. Herold, A., Klymenko, T. and Izaurralde, E. (2001) NXF1/p15 heterodimers are essential for mRNA nuclear export in Drosophila. RNA, 7, 1768-1780. Herold, A., Suyama, M., Rodrigues, J.P., Braun, I.C., Kutay, U., Carmo-Fonseca, M., Bork, P. and 149 Referencias Izaurralde, E. (2000) TAP (NXF1) belongs to a multigene family of putative RNA export factors with a conserved modular architecture. Mol Cell Biol, 20, 8996-9008. Hirsh, A.E. and Fraser, H.B. (2001) Protein dispensability and rate of evolution. Nature, 411, 1046-1049. Hoek, M., Zanders, T. and Cross, G.A. (2002) Trypanosoma brucei expression-site-associatedgene-8 protein interacts with a Pumilio family protein. Mol Biochem Parasitol, 120, 269-283. Hoshino, S., Hosoda, N., Araki, Y., Kobayashi, T., Uchida, N., Funakoshi, Y. and Katada, T. (1999) Novel function of the eukaryotic polypeptide-chain releasing factor 3 (eRF3/GSPT) in the mRNA degradation pathway. Biochemistry (Mosc), 64, 1367-1372. Hotchkiss, T.L., Nerantzakis, G.E., Dills, S.C., Shang, L. and Read, L.K. (1999) Trypanosoma brucei poly(A)-binding protein I cDNA cloning, expression and binding to 5 untranslated region sequence elements. Mol Biochem Parasitol, 98, 117-129. Hotz, H.R., Biebinger, S., Flaspohler, J. and Clayton, C. (1998) PARP gene expression: control at many levels. Mol Biochem Parasitol, 91, 131-143. Hotz, H.R., Hartmann, C., Huober, K., Hug, M. and Clayton, C. (1997) Mechanisms of developmental regulation in Trypanosoma brucei: a polypyrimidine tract in the 3'-untranslated region of a surface protein mRNA affects RNA abundance and translation. Nucleic Acids Res, 25, 3017-3026. Hotz, H.R., Lorenz, P., Fischer, R., Krieger, S. and Clayton, C. (1995) Role of 3'-untranslated regions in the regulation of hexose transporter mRNAs in Trypanosoma brucei. Mol Biochem Parasitol, 75, 1-14. Hutvagner, G., McLachlan, J., Pasquinelli, A.E., Balint, E., Tuschl, T. and Zamore, P.D. (2001) A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science, 293, 834-838. Inoue, H., Nojima, H. and Okayama, H. (1990) High efficiency transformation of Escherichia coli with plasmids. Gene, 96, 23-28. Inoue, M., Muto, Y., Sakamoto, H. and Yokoyama, S. (2000) NMR studies on functional structures of the AU-rich element-binding domains of Hu antigen C. Nucleic Acids Res, 28, 1743-1750. Irmer, H. and Clayton, C. (2001) Degradation of the unstable EP1 mRNA in Trypanosoma brucei involves initial destruction of the 3'-untranslated region. Nucleic Acids Res, 29, 4707-4715. Ishigaki, Y., Li, X., Serin, G. and Maquat, L.E. (2001) Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell, 106, 607-617. Ishima, R. and Torchia, D.A. (2000) Protein dynamics from NMR. Nat Struct Biol, 7, 740-743. Ismaili, N., Perez-Morga, D., Walsh, P., Cadogan, M., Pays, A., Tebabi, P. and Pays, E. (2000) Characterization of a Trypanosoma brucei SR domain-containing protein bearing homology to cis-spliceosomal U1 70 kDa proteins. Mol Biochem Parasitol, 106, 109-120. Ismaili, N., Perez-Morga, D., Walsh, P., Mayeda, A., Pays, A., Tebabi, P., Krainer, A.R. and Pays, E. (1999) Characterization of a SR protein from Trypanosoma brucei with homology to RNA-binding cis-splicing proteins. Mol Biochem Parasitol, 102, 103-115. Jackson, D.A., Pombo, A. and Iborra, F. (2000) The balance sheet for transcription: an analysis of nuclear RNA metabolism in mammalian cells. FASEB J, 14, 242-254. Jacobson, A. and Peltz, S.W. (1996) Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu Rev Biochem, 65, 693-739. 150 Referencias Jess, W., Hammer, A. and Cornelissen, A.W. (1989) Complete sequence of the gene encoding the largest subunit of RNA polymerase I of Trypanosoma brucei. FEBS Lett, 249, 123-128. Kanaar, R., Lee, A.L., Rudner, D.Z., Wemmer, D. and Rio, D.C. (1995) Interaction of the sex-lethal RNA binding domain with RNA. EMBO J, 14, 4530-4539. Kapotas, N. and Bellofatto, V. (1993) Differential response to RNA trans-splicing signals within the phosphoglycerate kinase gene cluster in Trypanosoma brucei. Nucleic Acids Res, 21, 40674072. Kataoka, N., Yong, J., Kim, V.N., Velazquez, F., Perkinson, R.A., Wang, F. and Dreyfuss, G. (2000) Pre-mRNA splicing imprints mRNA in the nucleus with a novel RNA-binding protein that persists in the cytoplasm. Mol Cell, 6, 673-682. Kelly, J.M., Ward, H.M., Miles, M.A. and Kendall, G. (1992) A shuttle vector which facilitates the expression of transfected genes in Trypanosoma cruzi and Leishmania. Nucleic Acids Res, 20, 3963-3969. Ketting, R.F., Haverkamp, T.H., van Luenen, H.G. and Plasterk, R.H. (1999) Mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell, 99, 133-141. Khaleghpour, K., Svitkin, Y.V., Craig, A.W., DeMaria, C.T., Deo, R.C., Burley, S.K. and Sonenberg, N. (2001) Translational repression by a novel partner of human poly(A) binding protein, Paip2. Mol Cell, 7, 205-216. Kiledjian, M. and Dreyfuss, G. (1992) Primary structure and binding activity of the hnRNP U protein: binding RNA through RGG box. EMBO J, 11, 2655-2664. Kim, V.N., Kataoka, N. and Dreyfuss, G. (2001) Role of the nonsense-mediated decay factor hUpf3 in the splicing-dependent exon-exon junction complex. Science, 293, 1832-1836. Klein Gunnewiek, J.M., Hussein, R.I., van Aarssen, Y., Palacios, D., de Jong, R., van Venrooij, W.J. and Gunderson, S.I. (2000) Fourteen residues of the U1 snRNP-specific U1A protein are required for homodimerization, cooperative RNA binding, and inhibition of polyadenylation. Mol Cell Biol, 20, 2209-2217. Kock, J., Evers, R. and Cornelissen, A.W. (1988) Structure and sequence of the gene for the largest subunit of trypanosomal RNA polymerase III. Nucleic Acids Res, 16, 8753-8772. Kontoyiannis, D., Pasparakis, M., Pizarro, T.T., Cominelli, F. and Kollias, G. (1999) Impaired on/ off regulation of TNF biosynthesis in mice lacking TNF AU- rich elements: implications for joint and gut-associated immunopathologies. Immunity, 10, 387-398. Kooter, J.M., Matzke, M.A. and Meyer, P. (1999) Listening to the silent genes: transgene silencing, gene regulation and pathogen control. Trends Plant Sci, 4, 340-347. Kozlov, G., Siddiqui, N., Coillet-Matillon, S., Trempe, J.F., Ekiel, I., Sprules, T. and Gehring, K. (2002) Solution structure of the orphan PABC domain from Saccharomyces cerevisiae poly(A)binding protein. J Biol Chem, 277, 22822-22828. Kozlov, G., Trempe, J.F., Khaleghpour, K., Kahvejian, A., Ekiel, I. and Gehring, K. (2001) Structure and function of the C-terminal PABC domain of human poly(A)- binding protein. Proc Natl Acad Sci U S A, 98, 4409-4413. Kuboniwa, H., Grzesiek, S., Delaglio, F. and Bax, A. (1994) Measurement of HN-H alpha J couplings in calcium-free calmodulin using new 2D and 3D water-flip-back methods. J Biomol NMR, 4, 871878. Lagnado, C.A., Brown, C.Y. and Goodall, G.J. (1994) AUUUA is not sufficient to promote poly(A) 151 Referencias shortening and degradation of an mRNA: the functional sequence within AU-rich elements may be UUAUUUA(U/A)(U/A). Mol Cell Biol, 14, 7984-7995. Lai, W.S., Carballo, E., Strum, J.R., Kennington, E.A., Phillips, R.S. and Blackshear, P.J. (1999) Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol Cell Biol, 19, 4311-4323. Lander, E.S., et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860-921. Laufer, G., Schaaf, G., Bollgonn, S. and Gunzl, A. (1999) In vitro analysis of alpha-amanitinresistant transcription from the rRNA, procyclic acidic repetitive protein, and variant surface glycoprotein gene promoters in Trypanosoma brucei. Mol Cell Biol, 19, 5466-5473. LeBowitz, J.H., Smith, H.Q., Rusche, L. and Beverley, S.M. (1993) Coupling of poly(A) site selection and trans-splicing in Leishmania. Genes Dev, 7, 996-1007. Lee, M.G. and Van der Ploeg, L.H. (1997) Transcription of protein-coding genes in trypanosomes by RNA polymerase I. Annu Rev Microbiol, 51, 463-489. Levine, N.D., Corliss, J.O., Cox, F.E., Deroux, G., Grain, J., Honigberg, B.M., Leedale, G.F., Loeblich, A.R., 3rd, Lom, J., Lynn, D., Merinfeld, E.G., Page, F.C., Poljansky, G., Sprague, V., Vavra, J. and Wallace, F.G. (1980) A newly revised classification of the protozoa. J Protozool, 27, 37-58. Lewin, B. (1997) In: Genes VI. Oxford Uniervsity Press and Cell Press, New York. Lewis, H.A., Chen, H., Edo, C., Buckanovich, R.J., Yang, Y.Y., Musunuru, K., Zhong, R., Darnell, R.B. and Burley, S.K. (1999) Crystal structures of Nova-1 and Nova-2 K-homology RNA-binding domains. Structure Fold Des, 7, 191-203. Liniger, M., Bodenmuller, K., Pays, E., Gallati, S. and Roditi, I. (2001) Overlapping sense and antisense transcription units in Trypanosoma brucei. Mol Microbiol, 40, 869-878. Liu, Q. and Dreyfuss, G. (1995) In vivo and in vitro arginine methylation of RNA-binding proteins. Mol Cell Biol, 15, 2800-2808. Loflin, P., Chen, C.Y. and Shyu, A.B. (1999) Unraveling a cytoplasmic role for hnRNP D in the in vivo mRNA destabilization directed by the AU-rich element. Genes Dev, 13, 1884-1897. Lopez-Estrano, C., Tschudi, C. and Ullu, E. (1998) Exonic sequences in the 5' untranslated region of alpha-tubulin mRNA modulate trans-splicing in Trypanosoma brucei. Mol Cell Biol, 18, 46204628. Lorkovic, Z.J. and Barta, A. (2002) Genome analysis: RNA recognition motif (RRM) and K homology (KH) domain RNA-binding proteins from the flowering plant Arabidopsis thaliana. Nucleic Acids Res, 30, 623-635. Lu, J. and Hall, K.B. (1995) An RBD that does not bind RNA: NMR secondary structure determination and biochemical properties of the C-terminal RNA binding domain from the human U1A protein. J Mol Biol, 247, 739-752. Luger, K., Mader, A.W., Richmond, R.K., Sargent, D.F. and Richmond, T.J. (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature, 389, 251-260. Luo, H., Gilinger, G., Mukherjee, D. and Bellofatto, V. (1999) Transcription initiation at the TATAless spliced leader RNA gene promoter requires at least two DNA-binding proteins and a tripartite architecture that includes an initiator element. J Biol Chem, 274, 31947-31954. Lykke-Andersen, J., Shu, M.D. and Steitz, J.A. (2000) Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell, 103, 1121152 Referencias 1131. Lykke-Andersen, J., Shu, M.D. and Steitz, J.A. (2001) Communication of the position of exonexon junctions to the mRNA surveillance machinery by the protein RNPS1. Science, 293, 18361839. Macara, I.G. (2001) Transport into and out of the nucleus. Microbiol Mol Biol Rev, 65, 570-594. Madison-Antenucci, S., Grams, J. and Hajduk, S.L. (2002) Editing machines: the complexities of trypanosome RNA editing. Cell, 108, 435-438. Madison-Antenucci, S. and Hajduk, S.L. (2001) RNA editing-associated protein 1 is an RNA binding protein with specificity for preedited mRNA. Mol Cell, 7, 879-886. Mair, G., Shi, H., Li, H., Djikeng, A., Aviles, H.O., Bishop, J.R., Falcone, F.H., Gavrilescu, C., Montgomery, J.L., Santori, M.I., Stern, L.S., Wang, Z., Ullu, E. and Tschudi, C. (2000) A new twist in trypanosome RNA metabolism: cis-splicing of pre-mRNA. RNA, 6, 163-169. Malliavin, T.E., Pons, J.L. and Delsuc, M.A. (1998) An NMR assignment module implemented in the Gifa NMR processing program. Bioinformatics, 14, 624-631. Mangus, D.A., Amrani, N. and Jacobson, A. (1998) Pbp1p, a factor interacting with Saccharomyces cerevisiae poly(A)- binding protein, regulates polyadenylation. Mol Cell Biol, 18, 7383-7396. Maquat, L.E. and Carmichael, G.G. (2001) Quality control of mRNA function. Cell, 104, 173-176. Maranon, C., Puerta, C., Alonso, C. and Lopez, M.C. (1998) Control mechanisms of the H2A genes expression in Trypanosoma cruzi. Mol Biochem Parasitol, 92, 313-324. Marchetti, M.A., Tschudi, C., Kwon, H., Wolin, S.L. and Ullu, E. (2000) Import of proteins into the trypanosome nucleus and their distribution at karyokinesis. J Cell Sci, 113, 899-906. Martinez-Calvillo, S., Lopez, I. and Hernandez, R. (1997) pRIBOTEX expression vector: a pTEX derivative for a rapid selection of Trypanosoma cruzi transfectants. Gene, 199, 71-76. Maruyama, K., Sato, N. and Ohta, N. (1999) Conservation of structure and cold-regulation of RNA-binding proteins in cyanobacteria: probable convergent evolution with eukaryotic glycinerich RNA-binding proteins. Nucleic Acids Res, 27, 2029-2036. Matkin, A., Das, A. and Bellofatto, V. (2001) The Leptomonas seymouri spliced leader RNA promoter requires a novel transcription factor. Int J Parasitol, 31, 545-549. Mattaj, I.W. (1993) RNA recognition: a family matter? Cell, 73, 837-840. Mattaj, I.W. and Englmeier, L. (1998) Nucleocytoplasmic transport: the soluble phase. Annu Rev Biochem, 67, 265-306. Matthews, K.R., Tschudi, C. and Ullu, E. (1994) A common pyrimidine-rich motif governs transsplicing and polyadenylation of tubulin polycistronic pre-mRNA in trypanosomes. Genes Dev, 8, 491-501. McAndrew, M., Graham, S., Hartmann, C. and Clayton, C. (1998) Testing promoter activity in the trypanosome genome: isolation of a metacyclic-type VSG promoter, and unexpected insights into RNA polymerase II transcription. Exp Parasitol, 90, 65-76. McDonagh, P.D., Myler, P.J. and Stuart, K. (2000) The unusual gene organization of Leishmania major chromosome 1 may reflect novel transcription processes. Nucleic Acids Res, 28, 28002803. Mertz, L. and Rashtchian, A. (1994) PCR radioactive labeling system: a rapid method for synthesis 153 Referencias of high specific activity DNA probe. FOCUS, 16, 45-48. Micklem, D.R., Adams, J., Grunert, S. and St Johnston, D. (2000) Distinct roles of two conserved Staufen domains in oskar mRNA localization and translation. EMBO J, 19, 1366-1377. Mignone, F., Gissi, C., Liuni, S. and Pesole, G. (2002) Untranslated regions of mRNAs. Genome Biol, 3. Reviews 0004. Milone, J., Wilusz, J. and Bellofatto, V. (2002) Identification of mRNA decapping activities and an ARE-regulated 3' to 5' exonuclease activity in trypanosome extracts. Nucleic Acids Res, 30, 40404050. Missel, A., Lambert, L., Norskau, G. and Goringer, H.U. (1999) DEAD-box protein HEL64 from Trypanosoma brucei: subcellular localization and gene knockout analysis. Parasitol Res, 85, 324330. Mitchell, P. and Tollervey, D. (2000) Musing on the structural organization of the exosome complex. Nat Struct Biol, 7, 843-846. Moore, M.J. (2002) Nuclear RNA turnover. Cell, 108, 431-434. Musco, G., Kharrat, A., Stier, G., Fraternali, F., Gibson, T.J., Nilges, M. and Pastore, A. (1997) The solution structure of the first KH domain of FMR1, the protein responsible for the fragile X syndrome. Nat Struct Biol, 4, 712-716. Musco, G., Stier, G., Joseph, C., Castiglione Morelli, M.A., Nilges, M., Gibson, T.J. and Pastore, A. (1996) Three-dimensional structure and stability of the KH domain: molecular insights into the fragile X syndrome. Cell, 85, 237-245. Myer, V.E. and Steitz, J.A. (1995) Isolation and characterization of a novel, low abundance hnRNP protein: A0. RNA, 1, 171-182. Myler, P.J., Audleman, L., deVos, T., Hixson, G., Kiser, P., Lemley, C., Magness, C., Rickel, E., Sisk, E., Sunkin, S., Swartzell, S., Westlake, T., Bastien, P., Fu, G., Ivens, A. and Stuart, K. (1999) Leishmania major Friedlin chromosome 1 has an unusual distribution of protein-coding genes. Proc Natl Acad Sci U S A, 96, 2902-2906. Nagai, K., Oubridge, C., Jessen, T.H., Li, J. and Evans, P.R. (1990) Crystal structure of the RNAbinding domain of the U1 small nuclear ribonucleoprotein A. Nature, 348, 515-520. Nagy, E. and Rigby, W.F. (1995) Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD(+)-binding region (Rossmann fold). J Biol Chem, 270, 2755-2763. Nakaar, V., Gunzl, A., Ullu, E. and Tschudi, C. (1997) Structure of the Trypanosoma brucei U6 snRNA gene promoter. Mol Biochem Parasitol, 88, 13-23. Nakayashiki, T., Ebihara, K., Bannai, H. and Nakamura, Y. (2001) Yeast [PSI+] “prions” that are crosstransmissible and susceptible beyond a species barrier through a quasi-prion state. Mol Cell, 7, 1121-1130. Nakielny, S. and Dreyfuss, G. (1999) Transport of proteins and RNAs in and out of the nucleus. Cell, 99, 677-690. Narlikar, G.J., Fan, H.Y. and Kingston, R.E. (2002) Cooperation between complexes that regulate chromatin structure and transcription. Cell, 108, 475-487. Navarro, M. and Gull, K. (2001) A pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature, 414, 759-763. Ngo, H., Tschudi, C., Gull, K. and Ullu, E. (1998) Double-stranded RNA induces mRNA degradation 154 Referencias in Trypanosoma brucei. Proc Natl Acad Sci U S A, 95, 14687-14692. Ohno, M., Segref, A., Bachi, A., Wilm, M. and Mattaj, I.W. (2000) PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell, 101, 187-198. Orphanides, G. and Reinberg, D. (2002) A unified theory of gene expression. Cell, 108, 439-451. Ostareck-Lederer, A., Ostareck, D.H., Cans, C., Neubauer, G., Bomsztyk, K., Superti-Furga, G. and Hentze, M.W. (2002) c-Src-mediated phosphorylation of hnRNP K drives translational activation of specifically silenced mRNAs. Mol Cell Biol, 22, 4535-4543. Ostrowski, J., Schullery, D.S., Denisenko, O.N., Higaki, Y., Watts, J., Aebersold, R., Stempka, L., Gschwendt, M. and Bomsztyk, K. (2000) Role of tyrosine phosphorylation in the regulation of the interaction of the heterogenous nuclear ribonucleoprotein K protein with its protein and RNA partners. J Biol Chem, 275, 3619-3628. Park, S., Myszka, D.G., Yu, M., Littler, S.J. and Laird-Offringa, I.A. (2000) HuD RNA recognition motifs play distinct roles in the formation of a stable complex with AU-rich RNA. Mol Cell Biol, 20, 4765-4772. Peng, J.W. and Wagner, G. (1994) Investigation of protein motions via relaxation measurements. Methods Enzymol, 239, 563-596. Peppel, K., Vinci, J.M. and Baglioni, C. (1991) The AU-rich sequences in the 3' untranslated region mediate the increased turnover of interferon mRNA induced by glucocorticoids. J Exp Med, 173, 349-355. Perez, I., McAfee, J.G. and Patton, J.G. (1997) Multiple RRMs contribute to RNA binding specificity and affinity for polypyrimidine tract binding protein. Biochemistry, 36, 11881-11890. Perez-Canadillas, J.M. and Varani, G. (2001) Recent advances in RNA-protein recognition. Curr Opin Struct Biol, 11, 53-58. Phillips, T.A., VanBogelen, R.A. and Neidhardt, F.C. (1984) lon gene product of Escherichia coli is a heat-shock protein. J Bacteriol, 159, 283-287. Pitula, J.S., Park, J., Parsons, M., Ruyechan, W.T. and Williams, N. (2002) Two families of RNA binding proteins from Trypanosoma brucei associate in a direct protein-protein interaction. Mol Biochem Parasitol, 122, 81-89. Priest, J.W. and Hajduk, S.L. (1994) Developmental regulation of Trypanosoma brucei cytochrome c reductase during bloodstream to procyclic differentiation. Mol Biochem Parasitol, 65, 291-304. Proudfoot, N.J., Furger, A. and Dye, M.J. (2002) Integrating mRNA processing with transcription. Cell, 108, 501-512. Pype, S., Slegers, H., Moens, L., Merlevede, W. and Goris, J. (1994) Tyrosine phosphorylation of a M(r) 38,000 A/B-type hnRNP protein selectively modulates its RNA binding. J Biol Chem, 269, 31457-31465. Quijada, L., Soto, M., Alonso, C. and Requena, J.M. (1997) Analysis of post-transcriptional regulation operating on transcription products of the tandemly linked Leishmania infantum hsp70 genes. J Biol Chem, 272, 4493-4499. Ramakrishnan, V. (2002) Ribosome structure and the mechanism of translation. Cell, 108, 557572. Ramos, A., Grunert, S., Adams, J., Micklem, D.R., Proctor, M.R., Freund, S., Bycroft, M., St Johnston, D. and Varani, G. (2000) RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J, 19, 997-1009. 155 Referencias Reed, R. and Hurt, E. (2002) A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell, 108, 523-531. Reid, D.G. (1997) In: Protein NMR Spectroscopy. Humana Press, Totowa, New Jersey, USA. Revollo, S., Oury, B., Laurent, J.P., Barnabe, C., Quesney, V., Carriere, V., Noel, S. and Tibayrenc, M. (1998) Trypanosoma cruzi: impact of clonal evolution of the parasite on its biological and medical properties. Exp Parasitol, 89, 30-39. Reyes, M.B., Pollevick, G.D. and Frasch, A.C. (1994) An unusually small gene encoding a putative mucin-like glycoprotein in Trypanosoma cruzi. Gene, 140, 139-140. Richards, E.J. and Elgin, S.C. (2002) Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell, 108, 489-500. Ross, J. (1997) A hypothesis to explain why translation inhibitors stabilize mRNAs in mammalian cells: mRNA stability and mitosis. Bioessays, 19, 527-529. Rout, M.P. and Field, M.C. (2001) Isolation and characterization of subnuclear compartments from Trypanosoma brucei. Identification of a major repetitive nuclear lamina component. J Biol Chem, 276, 38261-38271. Ryter, J.M. and Schultz, S.C. (1998) Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J, 17, 7505-7513. Sachs, A.B., Sarnow, P. and Hentze, M.W. (1997) Starting at the beginning, middle, and end: translation initiation in eukaryotes. Cell, 89, 831-838. Sacks, D. and Sher, A. (2002) Evasion of innate immunity by parasitic protozoa. Nat Immunol, 3, 1041-1047. Salmon, D., Montero-Lomeli, M. and Goldenberg, S. (2001) A DnaJ-like protein homologous to the yeast co-chaperone Sis1 (TcJ6p) is involved in initiation of translation in Trypanosoma cruzi. J Biol Chem, 276, 43970-43979. Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) In Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. Sato, N. (1994) A cold-regulated cyanobacterial gene cluster encodes RNA-binding protein and ribosomal protein S21. Plant Mol Biol, 24, 819-823. Schauer, R., Reuter, G., Muhlpfordt, H., Andrade, A.F. and Pereira, M.E. (1983) The occurrence of N-acetyl- and N-glycoloylneuraminic acid in Trypanosoma cruzi. Hoppe Seylers Z Physiol Chem, 364, 1053-1057. Schenkman, S., Eichinger, D., Pereira, M.E. and Nussenzweig, V. (1994) Structural and functional properties of trypanosome trans-sialidase. Annu Rev Microbiol, 48, 499-523. Schenkman, S., Ferguson, M.A., Heise, N., de Almeida, M.L., Mortara, R.A. and Yoshida, N. (1993) Mucin-like glycoproteins linked to the membrane by glycosylphosphatidylinositol anchor are the major acceptors of sialic acid in a reaction catalyzed by trans-sialidase in metacyclic forms of Trypanosoma cruzi. Mol Biochem Parasitol, 59, 293-303. Schmid, S.R. and Linder, P. (1992) D-E-A-D protein family of putative RNA helicases. Mol Microbiol, 6, 283-291. Schurch, N., Furger, A., Kurath, U. and Roditi, I. (1997) Contributions of the procyclin 3' untranslated region and coding region to the regulation of expression in bloodstream forms of Trypanosoma brucei. Mol Biochem Parasitol, 89, 109-121. 156 Referencias Shen, E.C., Henry, M.F., Weiss, V.H., Valentini, S.R., Silver, P.A. and Lee, M.S. (1998) Arginine methylation facilitates the nuclear export of hnRNP proteins. Genes Dev, 12, 679-691. Shi, H., Djikeng, A., Mark, T., Wirtz, E., Tschudi, C. and Ullu, E. (2000) Genetic interference in Trypanosoma brucei by heritable and inducible double-stranded RNA. RNA, 6, 1069-1076. Shih, H.M., Goldman, P.S., DeMaggio, A.J., Hollenberg, S.M., Goodman, R.H. and Hoekstra, M.F. (1996) A positive genetic selection for disrupting protein-protein interactions: identification of CREB mutations that prevent association with the coactivator CBP. Proc Natl Acad Sci U S A, 93, 13896-13901. Shyu, A.B. and Wilkinson, M.F. (2000) The double lives of shuttling mRNA binding proteins. Cell, 102, 135-138. Siddiqui, N., Kozlov, G., D’Orso, I., Trempe J.F. and Gehring, K. (2003) Solution Structure of the C-terminal domain from PABP in Trypanosoma cruzi: A vegetal class PABC domain. Enviado a publicar. Silva, E., Ullu, E., Kobayashi, R. and Tschudi, C. (1998) Trypanosome capping enzymes display a novel two-domain structure. Mol Cell Biol, 18, 4612-4619. Simms, D., Cizdziel, P.E. and Chomczynski, P. (1993) TRIZOL™ : A New Reagent for optimal single-step isolation of rna. FOCUS, 15, 99-102. Siomi, H., Siomi, M.C., Nussbaum, R.L. and Dreyfuss, G. (1993) The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell, 74, 291-298. Smith, C. (1998) In RNA:Protein Interactions, A Practical Approach. Oxford University Press, Inc., New York. Smith, J.L., Levin, J.R., Ingles, C.J. and Agabian, N. (1989) In trypanosomes the homolog of the largest subunit of RNA polymerase II is encoded by two genes and has a highly unusual Cterminal domain structure. Cell, 56, 815-827. Spadiliero, B., Nicolini, C., Mascetti, G., Henriquez, D. and Vergani, L. (2002a) Chromatin of Trypanosoma cruzi: In situ analysis revealed its unusual structure and nuclear organization. J Cell Biochem, 85, 798-808. Spadiliero, B., Sanchez, F., Slezynger, T.C. and Henriquez, D.A. (2002b) Differences in the nuclear chromatin among various stages of the life cycle of Trypanosoma cruzi. J Cell Biochem, 84, 832839. Stoecklin, G., Hahn, S. and Moroni, C. (1994) Functional hierarchy of AUUUA motifs in mediating rapid interleukin-3 mRNA decay. J Biol Chem, 269, 28591-28597. Tacke, R. and Manley, J.L. (1995) The human splicing factors ASF/SF2 and SC35 possess distinct, functionally significant RNA binding specificities. EMBO J, 14, 3540-3551. Tarun, S.Z., Jr. and Sachs, A.B. (1995) A common function for mRNA 5' and 3' ends in translation initiation in yeast. Genes Dev, 9, 2997-3007. Tarun, S.Z., Jr. and Sachs, A.B. (1996) Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J, 15, 7168-7177. Tarun, S.Z., Jr., Wells, S.E., Deardorff, J.A. and Sachs, A.B. (1997) Translation initiation factor eIF4G mediates in vitro poly(A) tail- dependent translation. Proc Natl Acad Sci U S A, 94, 90469051. Teixeira, S.M., Kirchhoff, L.V. and Donelson, J.E. (1995) Post-transcriptional elements regulating expression of mRNAs from the amastin/tuzin gene cluster of Trypanosoma cruzi. J Biol Chem, 157 Referencias 270, 22586-22594. Teixeira, S.M., Kirchhoff, L.V. and Donelson, J.E. (1999) Trypanosoma cruzi: suppression of tuzin gene expression by its 5'-UTR and spliced leader addition site. Exp Parasitol, 93, 143-151. Teixeira, S.M., Russell, D.G., Kirchhoff, L.V. and Donelson, J.E. (1994) A differentially expressed gene family encoding “amastin,” a surface protein of Trypanosoma cruzi amastigotes. J Biol Chem, 269, 20509-20516. ter Kuile, B.H. and Salles, F.J. (2000) The length of the combined 3' untranslated region and poly(A) tail does not control rates of glyceraldehyde-3-phosphate dehydrogenase mRNA translation in three species of parasitic protists. J Bacteriol, 182, 3587-3589. Tetaud, E., Giroud, C., Prescott, A.R., Parkin, D.W., Baltz, D., Biteau, N., Baltz, T. and Fairlamb, A.H. (2001) Molecular characterisation of mitochondrial and cytosolic trypanothione- dependent tryparedoxin peroxidases in Trypanosoma brucei. Mol Biochem Parasitol, 116, 171-183. Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res, 22, 4673-4680. Tibayrenc, M., Ward, P., Moya, A. and Ayala, F.J. (1986) Natural populations of Trypanosoma cruzi, the agent of Chagas disease, have a complex multiclonal structure. Proc Natl Acad Sci U S A, 83, 115-119. Tomlinson, S., Vandekerckhove, F., Frevert, U. and Nussenzweig, V. (1995) The induction of Trypanosoma cruzi trypomastigote to amastigote transformation by low pH. Parasitology, 110, 547-554. Torri, A.F. and Hajduk, S.L. (1988) Posttranscriptional regulation of cytochrome c expression during the developmental cycle of Trypanosoma brucei. Mol Cell Biol, 8, 4625-4633. Tupler, R., Perini, G. and Green, M.R. (2001) Expressing the human genome. Nature, 409, 832833. Ullu, E., Matthews, K.R. and Tschudi, C. (1993) Temporal order of RNA-processing reactions in trypanosomes: rapid trans-splicing precedes polyadenylation of newly synthesized tubulin transcripts. Mol Cell Biol, 13, 720-725. Vagner, S., Galy, B. and Pyronnet, S. (2001) Irresistible IRES. Attracting the translation machinery to internal ribosome entry sites. EMBO Reports, 2, 893-898. van Hoof, A. and Parker, R. (1999) The exosome: a proteasome for RNA? Cell, 99, 347-350. Vanhamme, L. and Pays, E. (1995) Control of gene expression in trypanosomes. Microbiol Rev, 59, 223-240. Vanhamme, L., Pays, E., McCulloch, R. and Barry, J.D. (2001) An update on antigenic variation in African trypanosomes. Trends Parasitol, 17, 338-343. Vanhamme, L., Postiaux, S., Poelvoorde, P. and Pays, E. (1999) Differential regulation of ESAG transcripts in Trypanosoma brucei. Mol Biochem Parasitol, 102, 35-42. Varani, G. and Nagai, K. (1998) RNA recognition by RNP proteins during RNA processing. Annu Rev Biophys Biomol Struct, 27, 407-445. Vasu, S.K. and Forbes, D.J. (2001) Nuclear pores and nuclear assembly. Curr Opin Cell Biol, 13, 363-375. Vázquez, M., Ben-Dov, C., Lorenzi, H., Moore, T., Schijman, A. and Levin, M.J. (2000) The short 158 Referencias interspersed repetitive element of Trypanosoma cruzi, SIRE, is part of VIPER, an unusual retroelement related to long terminal repeat retrotransposons. Proc Natl Acad Sci U S A, 97, 21282133. Vázquez, M., Lorenzi, H., Schijman, A.G., Ben-Dov, C. and Levin, M.J. (1999) Analysis of the distribution of SIRE in the nuclear genome of Trypanosoma cruzi. Gene, 239, 207-216. Vázquez, M.P., Schijman, A.G. and Levin, M.J. (1994) A short interspersed repetitive element provides a new 3' acceptor site for trans-splicing in certain ribosomal P2 beta protein genes of Trypanosoma cruzi. Mol Biochem Parasitol, 64, 327-336. Vermel, M., Guermann, B., Delage, L., Grienenberger, J.M., Marechal-Drouard, L. and Gualberto, J.M. (2002) A family of RRM-type RNA-binding proteins specific to plant mitochondria. Proc Natl Acad Sci U S A, 99, 5866-5871. Wahle, E. and Ruegsegger, U. (1999) 3'-End processing of pre-mRNA in eukaryotes. FEMS Microbiol Rev, 23, 277-295. Wan, L., Kim, J.K., Pollard, V.W. and Dreyfuss, G. (2001) Mutational definition of RNA-binding and protein-protein interaction domains of heterogeneous nuclear RNP C1. J Biol Chem, 276, 7681-7688. Wand, A.J. (2001) Dynamic activation of protein function: a view emerging from NMR spectroscopy. Nat Struct Biol, 8, 926-931. Wang, L., Eckmann, C.R., Kadyk, L.C., Wickens, M. and Kimble, J. (2002) A regulatory cytoplasmic poly(A) polymerase in Caenorhabditis elegans. Nature, 419, 312-316. Wang, X. and Tanaka Hall, T.M. (2001) Structural basis for recognition of AU-rich element RNA by the HuD protein. Nat Struct Biol, 8, 141-145. Wang, Z. and Kiledjian, M. (2001) Functional link between the mammalian exosome and mRNA decapping. Cell, 107, 751-762. Westermann, S. and Weber, K. (2000) Cloning and recombinant expression of the La RNA-binding protein from Trypanosoma brucei. Biochim Biophys Acta, 1492, 483-487. Wickens, M., Bernstein, D.S., Kimble, J. and Parker, R. (2002) A PUF family portrait: 3’UTR regulation as a way of life. Trends Genet, 18, 150-157. Williamson, J.R. (2000) Induced fit in RNA-protein recognition. Nat Struct Biol, 7, 834-837. Wilusz, C.J., Wormington, M. and Peltz, S.W. (2001) The cap-to-tail guide to mRNA turnover. Nat Rev Mol Cell Biol, 2, 237-246. Winstall, E., Gamache, M. and Raymond, V. (1995) Rapid mRNA degradation mediated by the cfos 3' AU-rich element and that mediated by the granulocyte-macrophage colony-stimulating factor 3' AU-rich element occur through similar polysome-associated mechanisms. Mol Cell Biol, 15, 3796-3804. Wishart, D.S., Bigam, C.G., Holm, A., Hodges, R.S. and Sykes, B.D. (1995) 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. Investigations of nearest-neighbor effects. J Biomol NMR, 5, 67-81. Wishart, D.S., Bigam, C.G., Yao, J., Abildgaard, F., Dyson, H.J., Oldfield, E., Markley, J.L. and Sykes, B.D. (1995b) 1H, 13C and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR, 6, 135-140. Wishart, D.S. and Sykes, B.D. (1994) The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR, 4, 159 Referencias 171-180. Wishart, D.S., Sykes, B.D. and Richards, F.M. (1991) Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol, 222, 311-333. Xu, N., Chen, C.Y. and Shyu, A.B. (1997) Modulation of the fate of cytoplasmic mRNA by AU-rich elements: key sequence features controlling mRNA deadenylation and decay. Mol Cell Biol, 17, 4611-4621. Xu, P., Wen, L., Benegal, G., Wang, X. and Buck, G.A. (2001a) Identification of a spliced leader RNA binding protein from Trypanosoma cruzi. Mol Biochem Parasitol, 112, 39-49. Xu, Y., Liu, L., Lopez-Estrano, C. and Michaeli, S. (2001b) Expression studies on clustered trypanosomatid box C/D small nucleolar RNAs. J Biol Chem, 276, 14289-14298. Yuan, C., Yongkiettrakul, S., Byeon, I.J., Zhou, S. and Tsai, M.D. (2001) Solution structure of two FHA-1phosphothreonine peptide complexes provide insight into the structural basis of the ligand specificity of FHA1 from yeast. J Mol Biol, 314, 563-575. Zamore, P.D., Tuschl, T., Sharp, P.A. and Bartel, D.P. (2000) RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell, 101, 25-33. Zamore, P.D., Williamson, J.R. and Lehmann, R. (1997) The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA, 3, 1421-1433. Zhang, J., Ruyechan, W. and Williams, N. (1998) Developmental regulation of two nuclear RNA binding proteins, p34 and p37, from Trypanosoma brucei. Mol Biochem Parasitol, 92, 79-88. Zhang, W., Wagner, B.J., Ehrenman, K., Schaefer, A.W., DeMaria, C.T., Crater, D., DeHaven, K., Long, L. and Brewer, G. (1993) Purification, characterization, and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol Cell Biol, 13, 7652-7665. Zingales, B., Pereira, M.E., Almeida, K.A., Umezawa, E.S., Nehme, N.S., Oliveira, R.P., Macedo, A. and Souto, R.P. (1997) Biological parameters and molecular markers of clone CL Brener-the reference organism of the Trypanosoma cruzi genome project. Mem Inst Oswaldo Cruz, 92, 811814. Zubiaga, A.M., Belasco, J.G. and Greenberg, M.E. (1995) The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol, 15, 2219-2230. 160 Notas 161 Notas 162
 0
0
Anuncio
Documentos relacionados








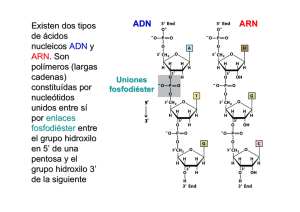
Descargar
Anuncio
Añadir este documento a la recogida (s)
Puede agregar este documento a su colección de estudio (s)
Iniciar sesión Disponible sólo para usuarios autorizadosAñadir a este documento guardado
Puede agregar este documento a su lista guardada
Iniciar sesión Disponible sólo para usuarios autorizados