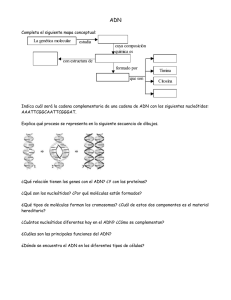
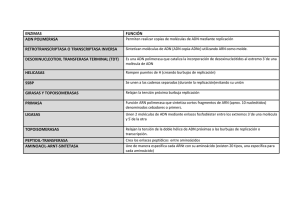
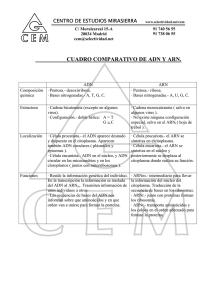
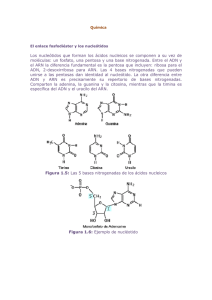
universidad nacional autónoma de méxico - Biblioteca, FES-C
Anuncio

UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO FACULTAD DE ESTUDIOS SUPERIORES CUAUTITLAN Revisión de las técnicas de Retrotranscripción Reacción en Cadena de la Polimerasa (RT-PCR), PCR, secuenciación y sus variantes que se utilizan en la detección de microorganismos que afectan la salu danimal. T R A BA J O P RO F E S I O NA L QUE PARA OBTENER EL TÍTULO DE: LICENCIADA EN MEDICINA VETERINARIA Y ZOOTECNIA P R E S E N TA : CASANDRA TOVAR HERNÁNDEZ ASESOR: DR. HUGO RAMÍREZ ÁLVAREZ CUAUTITLÁN IZCALLI, ESTADO DE MÉXICO 2015 Dedicatorias: A mis padres: Blanca Nieves Hernández Carbajal y Tomas Tovar Salas que sin todo el apoyo incondicional que siempre me han brindado, no hubiera logrado alcanzar esta meta, este sueño que juntos iniciamos y hoy se concluye. gracias por ser mi inspiración en todo momento, mi aliento para dar un paso más cada día, por enseñarme la humildad y forjar en mi valores y por permitirme compartir con ustedes mis logros y mis sueños. Para ustedes con todo mi corazón mil Gracias por tanto y por todo los amo. A mis hermanos: Jesús Alejandro Tovar Hernández y Paulina Tovar Hernández a ustedes que siempre han estado conmigo, que me dan su apoyo y cariño incondicionalmente, y que me aman tal como soy, con todo mi cariño para ustedes hermanos, Gracias por tanto y por todo los amo. A mis abuelos: Josefina Salas Simental y Jesús Nava Reyes a ustedes que me vieron crecer y con su amor me hicieron la mujer que soy, a ustedes que en donde quiera que estén me verán y sonreirán al ver que logre culminar este sueño. Para ustedes y de ustedes con todo mi amor. A ti Mariana Gómez Martínez : mi persona favorita, a ti que nunca dejas de creer en mí ,y que me motivas a ser mejor día a día , a ti que eres parte de este sueño que hoy concluyo , y que sin tu apoyo no se hubiera podido culminarse ,tú que fuiste cómplice para que esto pasara. Con toda mi admiración, mi respeto y mi amor para ti. Mil Gracias. A Guadalupe Tovar Salas y Raquel Guzmán Tovar: por siempre estar ahí para mí y por darme su apoyo y amor en todo momento. Con mi amor para ustedes. Agradecimientos: A dios : que nunca me dejo y que me permitió tener sueños , sueños que me está dejando realizar ,vivir y compartir con toda la gente que amo, a ti que me elegiste para esta profesión hermosa para la cual me diste la pasión y el don , tu mi confidente de vida. Tú que permites que hoy sonría al escribirte estas líneas, por esto y más Gracias por todo lo que me has dado. A mis amigos Berenice Uribe Torres, Juana López Ávila, Edgar Peña Vallejo, Sergio Arturo Trejo, que desde siempre han estado ahí para mí en las buenas y en las malas, por compartir una vida a mi lado, por animarme siempre a realizar mis metas y por formar parte de este sueño que hoy se culmina, por todo esto y más Gracias. A mis amigos y colegas Samuel Ruiz Villalba, Leonardo Pérez Cruz, Ana Karen Peña Fortino, Ana Karen Escobar Arredondo, Paulina García Reyes, Mariana López Serrano, Mitzi Luna nava por compartir conmigo talentos, tiempos, esfuerzos y luchas dentro de esta hermosa profesión que para ustedes y para mí se concreta con un gran esfuerzo y un sueño hecho realidad, a ustedes Gracias. Al Dr. Hugo Ramírez Alvares, por todo su apoyo durante la elaboración de este trabajo profesional, brindándome las herramientas necesarias para que pudiera culminarse exitosamente, por su atención, tiempo y ese apoyo incondicional, Con toda mi admiración y respeto para usted, mil Gracias. A mi querida UNAM y Facultad de Estudios Superiores Cuautitlán, por la oportunidad de formar parte de ella, y hoy orgullosamente ser una egresada de esta la gran casa de estudios mil Gracias. A mis profesores que durante mi carrera me dieron las herramientas necesarias para poder enfrentarme a la vida laboral, y por alentarme a ser cada día mejor Gracias. A SENASICA y CPA por abrirme las puertas de su laboratorio, por la información que me brindaron para poder realizar este trabajo, por el aprendizaje y las facilidades que tanto la institución como los miembros que la integran me brindaron Gracias. A las MVZ Beatriz Elena Moreno Arredondo Y Jenny Dinorah Lozano Alvarado por todo su apoyo y enseñanza que le brindaron a mi persona durante mi estancia en el CPA, por brindarme su tiempo, ética, profesionalismo y compartirme día a día sus conocimientos, por alentarme a concluir este trabajo, a ustedes Gracias. A mis asesores, que se dieron el tiempo y la dedicación para revisar mi trabajo el cual hoy se ha culminado exitosamente, a ustedes Gracias. ÍNDICE PÁGINAS 1. Objetivos. 9 2. Metodología. 9 3. Resumen. 10 4. Antecedentes 10 4.1 Origen e historia de la CPA 10 4.1.1 ¿Qué es la Comisión México Estados Unidos Para la Prevención de la 11 fiebre Aftosa y otras Enfermedades Exóticas (CPA)? 5. Introducción 12 5.1 Orígenes e investigaciones de los ácidos nucleícos 12 5.1.1 Descubrimiento del ácido Desoxirribonucleico (ADN) 12 5.1.2. Estructura de los ácidos nucleicos. 14 5.1.3. Funciones biológicas de los ácidos nucleicos 15 5.1.4. ADN - ARN – Proteína. 16 5.1.5. Papel del Ácido Ribonucleico mensajero (ARNm). 16 5.1.6. Etapas en la síntesis del ARN mensajero. 17 6. Transcripción en células procariotas. 17 6.1. Características de la ARN polimerasa procariota. 18 6.1.1. Iniciación de la síntesis del Ácido Ribonucleico mensajero (ARNm). 18 6.1.2. Elongación de la cadena. 19 6.1.3 Terminación y liberación del Acido Ribonucleico sintetizado (ARNs) 19 7. Transcripción en células eucariontes. 20 7.1 Las ARN polimerasas (Holoenzimas) en eucariontes. 20 7.1.1 El promotor de eucariontes para la ARN polimerasa II. 21 7.1.2. Los exaltadores o "enhacers". 21 7.1.3. El Ácido Ribonucleico mensajero en eucariontes 22 7.1.4. Corte y empalme o splicing. 22 8. Técnicas de la Reacción en Cadena de la Polimerasa (PCR) y “Reverse 25 Transcription” (RT-PCR). 8.1. Componentes de la PCR 26 2 8.1.1. Templado. 26 8.1.2. Características de las muestras. 27 8.1.3. Diseño de los iniciadores 27 8.1.4. La polimerasa. 29 8.1.5. Deoxinucleotidos trifosfato (dNTPs). 29 8.1.6. Sales. 29 8.1.7. Concentración de magnesio. 29 8.1.8. Extracción de Ácido Desoxirribonucleico 30 8.1.9. Fases y parámetros en una Reacción en Cadena de la Polimerasa 32 8.2. Desnaturalización, alineación, elongación o extensión. 32 9. Reacción en Cadena de la Polimerasa en tiempo real “Real Time PCR” 33 9.1. Calidad de las muestras. 36 9.1.1. Controles. 36 9.1.2. Preparación de mezclas de reacción. 37 9.1.3. Solución de problemas. 37 9.1.4. Agentes intercalantes. 38 9.1.5. Sondas. 39 9.1.6. Equipos para la “Real Time PCR”. 40 9.1.7. Opciones de los equipos para la “Real Time PCR”. 40 10. Electroforesis en gel de agarosa para separar productos derivados de la 41 PCR punto final 10.1. Principios físicos de la electroforesis en gel de agarosa. 43 10.1.1. Componentes de la electroforesis en gel de agarosa. 44 10.1.2. Agarosa. 44 10.1.3. Amortiguadores de electroforesis. 44 10.1.4 Concentración de agarosa. 45 10.1.5. Marcador de ADN. 46 10.1.6. Amortiguador de carga. 46 11. Variantes de la PCR 46 12 Secuenciación. 48 3 12.1. El método de degradación química 48 12.1.1. Ventajas y desventajas del método de degradación química 50 12.1.2 El método enzimático Sanger. 51 12.1.3. Limitaciones del método enzimático 52 12.1.4. Secuenciación de ARN 52 12.1.5. Métodos indirectos 53 12.1.6. Métodos directos 53 12.1.7. Método químico 54 13.Descripción de la actividades realizadas en el laboratorio de Bioseguridad 56 nivel 3. 14. Técnicas y diagnósticos durante mi estancia en el laboratorio de 69 Bioseguridad nivel 3. 15. Impacto y relevancia del trabajo profesional 80 16. Perspectiva personal. 81 17. Conclusión 82 18. Apéndice 83 18.1. Formato-Kit Roche 83 18.2. Formato-Kit Trizol 84 18.3. Esquema- Procesamiento de muestras de diagnóstico. 85 19. Bibliografía 86 4 INDICE DE FIGURA PÁGINA Figura 1. Visualización de las instalaciones del laboratorio nivel 3 CPA. 11 Figura 2. Representación esquemática de la estructura de la doble hélice del 14 ADN. Figura 3. Extensión o elongación de las cadenas de ADN. 33 Figura 4. Ejemplificación de una reacción de amplificación de un compuesto 35 específico en un termociclador de tiempo real Figura 5. Amplificación en tiempo real detectando la fluorescencia en 35 diferentes ciclos. Figura 6. Gel de agarosa y productos de Reacción en Cadena de la 43 Polimerasa (PCR) punto final. Figura 7. Secuenciación de ADN con el método de Maxam y Gilbert y 49 separación de los productos. Figura 8. Separación de nucleótidos en gel de acrilamida 50 Figura 9. El método de Sanger. 52 Figura 10. Esquema de un laboratorio de Bioseguridad Nivel 3 56 Figura 11. Fotos de las áreas del laboratorio. 58 Figura 12. Amplificaciones observadas en el fotodocumentador 68 Figura 13. Cámara de electroforesis. 68 Figura 14. Fotoducomentador. 69 Figura 15. Termociclador ABI FAST 7500. 79 Figura 16. Termociclador SMART CICLEY. 79 5 ÍNDICE DE TABLAS PÁGINA Tabla 1. Tipo de polimerasas. 20 Tabla 2. Concentración recomendada de agarosa para separar moléculas de ADN 45 lineales. Tabla 3. Ejemplos de enzimas y su función 55 Tabla 4. Lista de grupos de riesgos por la Organización Internacional de Salud 60 Animal (OIE) Tabla 5. Lista de enfermedades para las cuales se cuenta con herramientas de 61 detección implementadas en el laboratorio Tabla 6. Enfermedades que diagnostica el laboratorio de Bioseguridad nivel 3. 63 Tabla 7. Técnicas de diagnóstico 64 6 GLOSARIO Ácidos Nucleicos: nombre genérico que se aplica indistintamente al ADN o ARN de las dos moléculas informacionales de los seres vivos. ADN (ácido desoxirribonucleico): molécula que almacena la información genética. ADN recombinante: término que se usa en la tecnología aplicada para obtener moléculas de ADN híbridas, por ejemplo, provenientes de diversos seres vivos. Aminoácido: unidad monomérica fundamental de las proteínas. Existen 20 tipos diferentes. Antígeno: sustancia extraña a un organismo, capaz de desencadenar la producción de una respuesta inmune. Blanco de los anticuerpos. ARN (ácido ribonucleico): molécula que transmite información genética. Además cumple con funciones estructurales y de acoplamiento en la maquinaria de traducción de la información. ARN mensajero: molécula de ARN, copia de un gene, que lleva la información desde el genoma hasta donde se realiza la traducción. ARNsn: ácido nuclear de pequeño tamaño. Biología Molecular: rama de la biología nacida a raíz de la identificación de la naturaleza química (molecular) del material genético. Hoy día, nos referimos a biología molecular cuando hablamos de estudios o técnicas centradas en los genes y sus productos inmediatos, las proteínas. CPA: Comisión México Estados Unidos para la prevención de la fiebre aftosa y otras enfermedades exóticas CAP (caperuza, casquete, cofia): Breve secuencia de nucleótidos añadidos en el extremo 5’ de un ARNm eucariota mediante enlaces fosfodiéster 5’-5’ después de la transcripción. Se trata, por lo general, de uno a tres guanilatos (GTP). Cada nucleótido añadido suele estar metilado en posiciones características. ddNTPs: didesoxinucleotidos Trifosfatos nucleótidos carentes de un grupo hidroxilo en el extremo 3´. dNTP: desoxinucleotidos nucleótidos carentes de un hidroxilo en el extremo 3´. EDTA: ácido Etilendiaminotetraacetico amortiguador. OIE: Organización Mundial de Sanidad Animal, organización intergubernamental que transmite la información recibida a todos los demás países, para que puedan protegerse de un posible contagio. 7 PCR: reacción en cadena de la polimerasa. RNPsn: rinonucleoproteinas nucleares. RT-PCR: reversa transcriptasa de la reacción en cadena de la Polimerasa. TRIS: hidroximetil aminometano. Taq Polimerasa: esta es la polimerasa de ADN empleada en PCR. Es derivada de la bacteria “Thermus aquaticus” que vive en riachuelos de agua caliente; la termo-estabilidad de la polimerasa hace posible la PCR. 8 1. Objetivos. Objetivo general. Describir las técnicas que aprendí y desarrollé dentro del laboratorio de bioseguridad nivel 3, durante mis prácticas profesionales en las instalaciones de la Comisión México Estados Unidos para la prevención de la fiebre aftosa y otras enfermedades exóticas (CPA). Objetivos particulares. Revisar los fundamentos de las técnicas de biología molecular más frecuentemente utilizadas en el análisis del Ácido Desoxirribonucleico (ADN) para la identificación de microorganismos que afectan la salud animal. Describir los fundamentos de las técnicas de Reacción en Cadena de la Polimerasa (PCR), Retro transcripción de la Reacción en Cadena de la Polimerasa (RT-PCR), secuenciación y sus variantes que se utilizan en la detección de microorganismos que afectan la salud animal. Mencionar algunas enfermedades de importancia en la sanidad animal en las cuales se ha implementado el uso de técnicas de biología molecular para su detección dentro del laboratorio de bioseguridad nivel 3. 2. Metodología. Se realizó una búsqueda en bases de datos científicas a través de internet (PubMed, revistas electrónicas y ScienceDirect), consultando literatura especializada en bioquímica, biología celular y molecular. 9 3. Resumen. En este trabajo se describen los fundamentos de las técnicas diagnósticas que se realizaron durante una estancia de 6 meses de prácticas profesionales en el área de Biología Molecular en el laboratorio de Bioseguridad nivel 3. Este laboratorio forma parte de la Comisión México Estados Unidos para la prevención de la fiebre aftosa y otras enfermedades exóticas (CPA) y que a su vez depende del Servicio Nacional de Sanidad, Inocuidad y Calidad Agroalimentaria (SENASICA).El desarrollo de prácticas profesionales es una opción que permite optar a obtener el título de Médica Veterinaria y Zootecnista. Para tener los fundamentos teóricos necesarios que me permitieran reforzar mi conocimiento en el área de la Biología Molecular se hizo una búsqueda de información en libros, artículos, revistas e internet. De esta forma adquirí el conocimiento y preparación suficiente para entender con mayor eficacia los procesos que se desarrollan dentro del laboratorio, lo que me permitió sustancialmente tener un mejor desempeño, dado que el enfoque formativo de la carrera de Medicina Veterinaria y Zootecnia no es el desarrollo de habilidades en el campo de la biología molecular, por lo cual, se consideró altamente relevante el desarrollo y descripción de información teórica que se recopiló en una introducción previa a la descripción en sí de las prácticas profesionales propiamente realizadas y la cual es descrita a continuación. 4. Antecedentes 4.1. Origen e historia de la CPA. A consecuencia de un brote de Fiebre Aftosa (FA) en abril de 1947 fue creada la Comisión México-Americana para la Erradicación de la Fiebre Aftosa; organismo binacional cuyo objetivo principal fue la erradicación de esta enfermedad de tipo vesicular que afecta de manera significativa la producción y comercialización pecuaria de los países que la padecen. En 1949 se funda el laboratorio de la misma comisión, con la finalidad de comprobar la presencia de la FA y aplicar las medidas sanitarias de manera más eficiente para su control y erradicación; en 1977 al ampliar la vigilancia epidemiológica surge la necesidad de modificar el laboratorio y convertirlo en uno de bioseguridad nivel 3 (Alta seguridad), el único en su tipo destinado a la sanidad animal del país (Figura 1). Al lograrse el control y la no presencia de focos de FA, en noviembre de 1952 la comisión cambió de actividades y de nombre, a Comisión México-Americana para la 10 Prevención de la Fiebre Aftosa y no es hasta 1954 que se declaró a México libre de la enfermedad. En 1988 por acuerdo de los gobiernos de Estados Unidos y México, se ampliaron sus actividades a otras enfermedades exóticas para nuestro país, por lo que recibió el nombre de Comisión MéxicoEstados Unidos para la Prevención de la Fiebre Aftosa y otras Enfermedades Exóticas de los Animales (CPA). Figura 1.- Visualización del laboratorio nivel 3 CPA. (Fotografía tomado del laboratorio de bioseguridad nivel 3 CPA. Tovar, 2014). 4.1.1. ¿Que es la Comisión México Estados Unidos Para la Prevención de la fiebre Aftosa y otras Enfermedades Exóticas (CPA)? Comisión México-Estados Unidos para la Prevención de la Fiebre Aftosa y otras Enfermedades Exóticas de los Animales (CPA). Es una Dirección de Área dependiente de la Dirección General de Salud Animal (DGSA), del Servicio Nacional de Sanidad, Inocuidad y Calidad Agroalimentaria (SENASICA). Tiene como misión principal la atención de reportes sospechosos de enfermedades exóticas que registran la ciudadanía o el personal de campo, con la finalidad de dar un diagnóstico oportuno a través de la elaboración y actualización de los planes de emergencia. 11 Si se identifica una enfermedad exótica, se lleva a cabo la ejecución de operativos de emergencia a través del Dispositivo Nacional de Emergencia de Sanidad Animal (DINESA). Comisión México-Estados Unidos para la Prevención de la Fiebre Aftosa y otras Enfermedades Exóticas de los Animales (CPA) tiene a su cargo el laboratorio de Bioseguridad Nivel 3, 7 laboratorios regionales y 13 laboratorios de biología molecular estratégicamente ubicados a nivel nacional, que dan una respuesta expedita a las necesidades diagnósticas del patrimonio pecuario y acuícola del país. Además, para la simplificación administrativa, la agilización de procesos y la modernidad técnica en las actividades de vigilancia, durante el 2008 y 2009, la DGSA-CPA desarrolló el Sistema de Información Nacional de Enfermedades Exóticas y Emergentes (SINEXE), una plataforma informática para la notificación y vigilancia epidemiológica de las enfermedades exóticas, emergentes o reemergentes, así como para el registro y análisis de toda información relacionada al seguimiento de las investigaciones llevadas a cabo en el campo. Las funciones de dichos laboratorios dependientes del CPA se enfocan en determinar características genéticas a nivel molecular de agentes causantes de enfermedades, por lo que el personal que labora en estos sitios desarrollan diversas herramientas para la detección de dichas enfermedades; basadas principalmente en la identificación de ácidos nucleícos, las cuales son utilizadas como muestras para la realización de las técnica en biología molecular. 5. Introducción 5.1. Orígenes e investigaciones de los ácidos nucleícos. 5.1.1. Descubrimiento del ácido Desoxirribonucleico (ADN). A mediados del siglo pasado los investigadores no pudieron avanzar más en la elucidación de la estructura primaria del Ácido Desoxirribonucleico (ADN). Ninguno de los requerimientos claves para la determinación de la secuencia estaba a la mano: no habían métodos para obtener muestras puras de ADN con una secuencia de bases homogénea y tampoco estaban disponibles métodos para el corte de cadenas en una base específica. Consecuentemente, toda la atención se centró en la estructura secundaria. En 1953, James Watson y Francis Crick descifraron la estructura del ADN. Watson propuso que el número de nucleótidos en la célula unitaria 12 cristalográfica favorecía una hélice de doble cadena. Crick dedujo de los datos del patrón de difracción que la estructura era una díada, es decir, que tiene una asimetría tal que las cadenas equivalentes son antiparalelas, lo que implica que corren en direcciones opuestas a lo largo del eje longitudinal. Sólo quedaba por resolver un problema: cómo construir el núcleo de la hélice, empacando las bases juntas en una estructura regular. A partir de las conclusiones de Gulland, Watson sabía que los puentes de hidrógeno unían las bases del ADN. Esto lo convenció de que la esencia de la cuestión tenía que ser una regla que gobernara los puentes de hidrógeno entre las bases aconsejado por Jerry Donohue, Watson manipuló modelos de las bases, apareando la Adenina (A) con la Timina (T) y la Guanina (G) con la Citosina (C). Encontró una relación simple y convincente que involucraba dos puentes de hidrógeno para una pareja Adenina·Timina y tres puentes de hidrógeno para una pareja Guanina·Citosina. La característica especial de este esquema de apareamiento de bases es que la geometría relativa de los enlaces uniendo las bases a las pentosas es virtualmente idéntico para los pares Adenina·Timina y Guanina·Citosina. Resultó que si una purina siempre se aparea con una pirimidina, entonces una secuencia irregular de bases en una cadena sencilla de ADN podría estar apareada regularmente en el centro de una doble hélice sin pérdida de simetría. Las reglas de Chargaff fueron reveladas directamente como una consecuencia obligatoria de una estructura de doble hélice para el ADN. Sobre todo, ya que la secuencia de bases de una cadena automáticamente determina la de su pareja, Crick y Watson pudieron visualizar fácilmente cómo una cadena sencilla podría ser el templado para la síntesis de una segunda cadena de secuencia de bases complementaria (Necochea et al, 2004). El modelo de Watson y Crick de la estructura del ADN fue aceptado rápidamente porque lograba dos cuestiones importantes (Figura 2). Primero, daba cuenta de toda la evidencia química y física disponible. Segundo, abría el camino para explicar de manera más detallada como lleva a cabo el ADN las funciones necesarias para ser el portador de la información hereditaria. A partir de este momento, fue aparente que toda la información requerida para especificar la diversidad de las moléculas biológicas necesaria para llevar a cabo las funciones de la célula, había que buscarla en la secuencia irregular de las bases nucleotídicas. Alexander Dounce, en 1950 postuló que el Ácido Ribonucleico (ARN) era el templado que dirigía la síntesis de proteínas celulares y que una secuencia de tres nucleótidos especificaba solo un aminoácido. El reconocimiento de varios tipos de ARN por Robert Holley no tardó en llegar (Necochea et al, 2004). 13 Figura.-2 Representación esquemática de la estructura de la doble hélice del ADN (Figura 2.- que ejemplifica los componentes de un nucleótido (Necochea et al, 2004). 5.1.2. Estructura de los ácidos nucleicos. El ADN es una molécula formada por una doble hélice. Cada una de las hélices es un polímero integrado por millones de nucleótidos que son los monómeros del polímero. Cada nucleótido está formado por una desoxirribosa, una base púrica o pirimidica y un grupo fosfato (Figura 3). Las dos cadenas de ADN son anti paralelas y se unen entre sí a través de puentes de hidrógeno que se forman entre las bases complementarias (Adenina-Timina y Guanina-Citosina) de las dos hebras del ADN. De esta manera, se obtiene una estructura tipo doble hélice, donde las bases de los nucleótidos se encuentran orientadas hacia el interior, mientras que los grupos fosfato y las desoxirribosas lo hacen hacia el exterior, formando los esqueletos fosfodiéster de cada hélice. La diferencia fundamental entre todas las moléculas de ADN que forman el material genético de los seres vivos es la secuencia de los millones de estos cuatro tipos de nucleótidos con sus bases Adenina, Timina, Guanina y Citosina en cada molécula de ADN. Al igual que en el ADN, los estudios del ARN empezaron con su estructura primaria. Esta búsqueda se logró en paralelo con la del ADN, pero tuvo la complejidad extra del grupo hidroxilo 2´ de los ribonucleótidos. A diferencia del ADN, las moléculas de ARN constan generalmente de cadenas únicas de poli-nucleótidos, debido a que se forman copiando la secuencia de bases de una cadena de ADN. Hoy se sabe que el ARN tiene una mayor versatilidad estructural que el ADN en la variedad de sus especies, en la diversidad de sus conformaciones y en su reactividad química. 14 Los ARNs naturales pueden formar estructuras de doble cadena o adoptar una forma globular compuesta por pequeños dominios dobles conectados por segmentos de cadena sencilla. Las dobles hélices del ARN sólo pueden adoptar la forma A, ya que el hidroxilo 2´ de la ribosa constituye un impedimento estérico (Necochea et al, 2004). 5.1.3. Funciones biológicas de los ácidos nucleicos. Las funciones de los ácidos nucleicos son de almacenamiento, expresión y replicación de la información biológica. En términos generales, todas las moléculas de ADN tienen una configuración similar. Sin embargo, el ADN de una determinada especie de organismos tiene una secuencia de bases propia: su estructura primaria está agrupada en unidades funcionales llamadas genes. La información que contiene esta secuencia desempeña diversas funciones. Los genes estructurales codifican para enzimas, proteínas estructurales y proteínas reguladoras. Otros tipos de genes codifican moléculas de Ácido Ribonucleicos (ARN) que no especifican la estructura primaria de un poli-péptido (Necochea et al, 2004). El Ácido Ribonucleico (ARN), en tres de sus variedades, Ácido Ribonucleico mensajero (RNAm), Ácido Ribonucleico ribosómico (ARNr) y Ácido Ribonucleico de transferencia (ARNt), dan como resultado una pieza fundamental dentro del proceso (Curtís et al, 2008). El primer paso en la síntesis de proteínas, es la síntesis de una molécula de Ácido Ribonucleico (ARN) usando como molde un segmento de una de las cadenas del Ácido Desoxirribonucleico (ADN). En la transcripción, el orden de los desoxirribonucleótidos de uno o varios genes se transfiere uno por uno a una secuencia de ribonucleótidos complementaria. Este proceso está mediado por la enzima ARN polimerasa y al igual que la replicación del Ácido Desoxirribonucleico (ADN), siempre ocurre en dirección 5' a 3' (Holland et al, 1991). Como los procariontes no tienen membrana nuclear, las moléculas de ARN que se transcriben de los genes son inmediatamente traducidas a nivel de los ribosomas para sintetizar las proteínas. En el caso de los eucariontes, los ARN transcritos son transportados del núcleo al citoplasma, a través de la membrana nuclear. Además, los genes de los eucariontes contienen intrones, estructuras de ADN que interrumpen la región del gen que codifica para la proteína (exón). Al transcribirse un gen en los núcleos de las células de los eucariontes, el ARN resultante 15 incluye tanto las regiones de los intrones como las de los exones. Esta molécula de ARN se “procesa” para dar lugar al Ácido Ribonucleico Mensajero (ARNm) Maduro que se exporta del núcleo de la célula al citoplasma, donde luego se traduce en proteína (Holland et al, 1991). La síntesis de proteínas inicia al momento que sucede la traducción de la secuencia de nucleótidos presentes en el ARNm maduro, se lleva a cabo mediante la polimerización de aminoácidos en proteínas, a nivel de los ribosomas en dirección 5' a 3'. La secuencia del ARNm realiza la codificación en forma de tripletes de bases (codones) de acuerdo con el código genético, incorporando en cada paso de lectura un aminoácido a la proteína. Cada Ácido Ribonucleico de Transferencia (ARNt) tiene una secuencia complementaria o anti codón para el codón del aminoácido, que le permite reconocer el codón correcto sobre el ARNm (Necochea et al, 2004). 5.1.4. ADN - ARN – Proteína. De acuerdo con el concepto ADN – ARN - Proteína, las molécula de ARN se sintetizan a partir de moldes de ADN originando un proceso denominado “transcripción” y las proteínas se sintetizan a partir de moldes del ARN originando un proceso denominado traducción (Cooper et al, 2011). 5.1.5. Papel del Ácido Ribonucleico Mensajero (ARNm). Aunque la secuencia de nucleótidos en el ADN parecía especificar el orden de los aminoácidos en las proteínas, eso no significaba que el ADN dirigiera por sí mismo la síntesis de proteínas. De hecho, no parece ser el caso, dado que dicha molécula se localiza en el núcleo de las células eucariotas, mientras que la síntesis proteínica se lleva a cabo en el citoplasma. Es necesaria otra molécula para llevar la información genética del ADN a los sitios donde se realiza la síntesis de proteínas específicamente la cual es en los ribosomas (Cooper et al, 2011). El ARNm es un buen candidato para ser dicho intermediario por que la similitud de su estructura con la del ADN sugería que él ARNm podría ser sintetizado a partir de un molde de ADN. El Ácido Ribonucleico Mensajero difiere del ADN porque está compuesto por una sola cadena en vez de ser de doble cadena, sus azúcares son ribosas en vez de desoxirribosas y contiene la base pirimidínica uracilo (U) en vez de timina (T). Sin embargo, el cambio de azúcar ni la sustitución de uracilo por timina altera el apareamiento de las bases, por lo que la síntesis de ARN 16 puede ser realizada de manera correcta sobre un molde de ADN. Además, dado que el ARN se localiza primamente en el citoplasma, parecía un intermediario lógico para transferir información del ADN a los ribosomas (Cooper et al, 2011). 5.1.6. Etapas en la síntesis del ARN mensajero. Con la finalidad de ordenar el estudio de la transcripción esta se puede dividir en cuatro etapas fundamentales: 1° etapa- UNIÓN de la ARN polimerasa al ADN. 2° etapa- INICIACIÓN de la síntesis. 3° etapa- ELONGACIÓN de la cadena de ARN mensajero. 4° etapa- TERMINACIÓN de la síntesis y liberación de la cadena de ARNm (Goyanes, 2010). 6. Transcripción en células procariotas. La expresión génica se concreta por la transformación de la información genética a partir de moléculas de ADN a moléculas de ARN y desde estas hasta los polipéptidos correspondientes. Las moléculas de Ácido Ribonucleico son sintetizadas usando como molde segmentos específicos de ADN, en la reacción de polimerización que es catalizada por la enzima conocida como ARN polimerasa. Debemos tener en cuenta los siguientes puntos en el proceso de transcripción: A) Los precursores en la síntesis de ARN que se consideran como unidades estructurales son cuatro precursores Ribonucleótidos Trifosfatados: (rATP; rCTP; rGTP; rUTP). B) La secuencia de bases del ARN a polimerizar está determinada por medio de la secuencia de bases de la molécula de ADN utilizada como molde para su síntesis. De ahí su denominación como cadena molde de ADN. C) La cadena de ARN en dirección 5’ – 3’. La cual coincide en la dirección en la que se da la síntesis de ADN. 17 D) La ARN polimerasa, a diferencia de la ADN polimerasa es capaz de iniciar su síntesis sin la presencia de ningún tipo de molécula cebadora (Goyanes, 2010). 6.1. Características de la ARN polimerasa procariota. La ARN polimerasa es una de las enzimas más grandes que se conocen. Consta de cinco sub-unidades: dos alfa (α), beta (β), beta prima (β’) y sigma (σ). La enzima completa es denominada holoenzima y se divide en dos componentes principales: • La enzima central, denominada núcleo formada por las sub unidades 2a, B’B • El factor sigma (el polipéptido σ). La holoenzima tiene la capacidad de unirse a la cadena molde de ADN e iniciar la transcripción; pero una vez realizada esta, el factor σ es liberado dejando que el core continúe con la elongación. En definitiva, es el factor el que le da la capacidad a la ARN polimerasa para poder iniciar la síntesis del ARNm en el sitio apropiado. La holoenzima reconoce un sitio de unión específico en el ADN, este sitio es denominado promotor. Como consecuencia de lo dicho, la ADN polimerasa puede encontrarse en dos estados: • Un estado apropiado para la unión con el promotor “estado de iniciación” u holoenzima. • Un estado apropiado para la elongación, núcleo de la enzima. La ARN polimerasa es lo bastante grande como para tomar contacto con muchas bases del ADN, así la enzima cubre aproximadamente unas 60 pb (pares de bases), aunque el segmento de ADN desenrollado que sirve como molde comprende únicamente unos 17 pb (Goyanes, 2010). 6.1.1. Iniciación de la síntesis del Ácido Ribonucleico mensajero (ARNm). La iniciación se produce mediante la unión de la ARN polimerasa (Holoenzima) al ADN de doble cadena. Para que la cadena molde esté disponible para el apareamiento de bases con los ribonucleótidos, las dos cadenas de ADN deben separarse. El desenrollamiento es local y se produce cuando la holoenzima tiene contacto con la caja (TATA). Una vez que el complejo promotor se encuentra abierto, la ARN polimerasa (Holoenzima) comienza la síntesis. La fase de 18 iniciación no se completa hasta que se hallan polimerizado los primeros seis ribonucleótidos (Goyanes, 2010). 6.1.2. Elongación de la cadena. Después que se han colocado los primeros seis ribonucleótidos, la ARN polimerasa (Holoenzima) sufre un cambio en su conformación, perdiendo la sub unidad &. Se continúa la síntesis en el estado de núcleo, anteriormente descrito. Se mueve a lo largo del ADN colocando los ribonucleótidos complementarios a la cadena de ADN molde, abriendo la hélice a medida que se desplaza. Los ribonucleótidos se unen al extremo 3’ de la cadena de ARN en crecimiento, formándose un híbrido de ARN - ADN en la región desenrollada, el cual se mantiene estabilizado mediante enlaces de puentes de hidrógeno. El híbrido tiene una longitud aproximada de 12 pb y el nuevo ARN es liberado de estas uniones cuando el ADN recupera su estado de doble hélice por desplazamiento del núcleo (Goyanes, 2010). 6.1.3. Terminación y liberación del Ácido Ribonucleico sintetizado (ARNs). La terminación ocurre en una secuencia determinada del ADN, denominada secuencia de terminación. En este punto, la enzima deja de añadir los ribonucleótidos a la cadena de ARN en crecimiento, liberando el producto terminado y disociándose del ADN. La terminación requiere que todos los enlaces de puentes de hidrógeno que mantenían al híbrido de ARN – ADN, se rompan volviéndose a reconstituir el ADN doble. Las secuencias terminadoras muestran las siguientes similitudes: • Todas contienen secuencias palindrómicas justo antes del punto de terminación. El palíndromo está caracterizado por poseer repeticiones inversas, conteniendo un segmento central no repetido (ej. ATCATCGACTA). • La secuencia palindrómica continúa con una secuencia de pares Adenina-Timina, las cuales producen en el ARN mensajero una secuencia de 6 a 8 uracilos en el extremo transcripto. La secuencia de uracilos suministra la señal que permite a la ARN polimerasa disociarse del ADN molde (Goyanes, 2010). 19 7. Transcripción en células eucariontes. La estructura del ARNm y el proceso de transcripción en las células eucariotas son similares a lo expresado anteriormente para las células procariotas. Sin embargo, debemos considerar las siguientes diferencias: • Las células de eucariontes poseen tres clases de ARN polimerasas (I, II y III) las cuales se utilizan para sintetizar los distintos tipos de ARN existentes. • Los extremos 3’ y 5´ de los ARNms están modificados. En el caso del extremo 5’ se encuentra una estructura denominada Caperuza (CAP) y en el correspondiente extremo 3’ se encuentra adherido una larga secuencia de nucleótidos cuya base nitrogenada es adenina (Cola Poly A). • Las moléculas de ARNm luego de ser sintetizas son modificadas. La transcripción primaria “transcriptos primarios” en las eucariotas sufren un proceso por el cual determinadas secuencias son eliminadas (Intrones). Los ARNm en los eucariontes son monocistrónicos mientras que en los procariontes se le denomina policistronicos (Goyanes, 2010). 7.1. Las ARN polimerasas (Holoenzimas) en eucariontes. En las células del tipo eucariota encontramos ARN polimerasa I, ARN polimerasa II y ARN polimerasa III. Sus composiciones en sub unidades son complejas y aun no se conoce con exactitud la función de cada una de estas sub unidades. Lo que sí se conoce con exactitud es la localización y el producto de cada una de ellas, como se muestra en la Tabla 1 (Goyanes, 2010). Tabla 1.- Tipo de polimerasas. Enzima Localización Producto ARN Polimerasa I Nucléolo Pre-ARNr ARN Polimerasa II Nucleoplasma Pre-ARNm ARN Polimerasa III Nucleoplasma Pre-ARN t y ARNr 5S 20 7.1.1. El promotor de eucariontes para la ARN polimerasa II. Los promotores de la ARN polimerasa II muestran una mayor variación en secuencia y la enzima es incapaz de iniciar la transcripción sola, por lo que requiere de una serie de factores de transcripción los cuales son los encargados del reconocimiento de un promotor particular. Como se describió en las células procariotas, el factor sigma es el que le confiere a la ARN polimerasa la capacidad de seleccionar a cada uno de los promotores. Los eucariontes al igual que en las procariotas, las homologías en las regiones próximas al punto de inicio están restringidas a secuencias relativamente cortas. La mayoría de los promotores tienen una secuencia denominada caja TATA o Caja “Hogness”, centrada 25 pb del punto de inicio. Esta caja TATA es idéntica a la mencionada en la transcripción de las células procariotas, variando solamente en su localización (Goyanes, 2010). 7.1.2. Los exaltadores o “enhacers”. Los promotores de eucariontes no trabajan solos para asegurar una transcripción eficaz. En algunos genes o tipos celulares la actividad de un promotor está aumentada por la presencia de otra secuencia conocida como Exaltador o “Enhacer”. Las características de los exaltadores son las siguientes: • Están formados por cientos de bases. • Generalmente incluyen secuencias repetidas. • Actúan a distancia, a miles de bases del promotor. • Son activos en cualquier orientación respecto al promotor, incluso suelen encontrárseles dentro del mismo gen. Los exaltadores, entonces actúan como reguladores de la expresión génica. No está claro aún el mecanismo de acción de los exaltadores, más aún cuando estos se encuentran localizados a cualquier distancia (1000 a 2000 pb) y dirección del mencionado promotor (Goyanes, 2010). 21 7.1.3. El Ácido Ribonucleico Mensajero en eucariontes. Si bien la síntesis y estructura del ARNm en eucarionte es similar a las procariotas, deben mencionarse las siguientes diferencias: • Ambos extremos están modificados, el 5’ presenta una estructura denominada CAP (Caperuza) y el 3’ contiene una secuencia de ácido poliadenílico denominada cola Poly A. • La molécula que sirve de molde para la síntesis de proteína (Ácido Ribonucleico Maduro) es más corta que el ARNm recién sintetizado (Pre – ARN mensajero). Esto se debe a que los últimos sufren un proceso de eliminación de secuencias no codificantes, dicho proceso se denomina splicing (Goyanes, 2010). El extremo 5’ contiene dos nucleótidos conectados que siempre están metilados (-CH3). La reacción de la incorporación del CAP ocurre inmediatamente después de iniciada la transcripción y siempre precede a cualquier modificación que se le realice al ARNm precursor. El CAP tiene la función de proteger al ARNm de la degradación, con lo cual aumenta su vida media, En el extremo 3’ según se mencionó antes los ARNm de eucariontas contienen una secuencia de 20 a 200 nucleótidos que poseen como base nitrogenada a la adenina. La secuencia Poly A no está codificada en el ADN, sino que es añadida al ARN después de finalizada la transcripción. La función de la cola Poly A es la de aumentar la estabilidad de los ARNm (Goyanes, 2010). 7.1.4. Corte y empalme o “splicing” Durante la transcripción, el ARNm sufre un proceso de corte y eliminación de secuencias llamadas intrones y el posterior empalme de las secuencias restantes, los exones en un primer paso se unen al RNAm inmaduro unas pequeñas partículas del ARN nucleares asociadas con proteínas denominadas snRNP “small nuclear ribonucleo protein particles” (Curtis, 2008). El análisis de los productos de la reacción y de los intermediarios formados in vitro reveló que el corte y empalmado del pre-ARNm tiene lugar en dos etapas. En primer lugar el preARNm es cortado en el sitio de corte y empalme 5', y el extremo 5' del intrón se une a un nucleótido de adenina del intrón (cerca de su extremo 3'). En este paso se forma un vínculo inusual entre el extremo 5' del intrón y el grupo hidroxilo 2' de la adenina. El intermediario resultante es 22 una estructura en lazo, en la que el intrón forma un bucle. El segundo paso tiene lugar con el corte en 3' y la unión simultánea de los dos exones. El intrón es una estructura en forma de lazo que es posteriormente degradada en el núcleo de las células intactas. Estas reacciones definen tres secuencias críticas en los ARNm: la secuencia en el sitio de corte 5', la secuencia en el sitio de corte 3' y las secuencias en el punto o sitio de ramificación (el punto en el que se une el extremo 5' del intrón para formar la estructura en forma de lazo). Los pre-ARNm contienen secuencias consensos similares en cada una de estas tres posiciones, permitiendo al complejo de corte y empalme reconocer a los pre-ARNm y llevar a cabo las reacciones de corte y unión implicadas en el proceso. El análisis bioquímico de extractos nucleares ha revelado que el splicing tiene lugar en grandes complejos denominados espliceosomas, compuestos de proteínas y ARN (Cooper et al, 2011). Los ARN que forman parte de los espliceosomas, son cinco tipos de Ácido Ribonucleico nuclear de pequeño tamaño (ARNsn) denominados Ul, U2, U4, U5 y U6. Estos ARN que tienen un tamaño de entre 50 y 200 nucleótidos, forman un complejo con entre seis y diez moléculas proteínicas para formar partículas pequeñas de rinonucleoproteínas nucleares I (RNPsn), que tienen un papel central en el proceso de splicing. Las rinonucleoproteínas nucleares (RNPsn) Ul, U2 y U5 contienen una molécula única de ARNsn, mientras que las RNPsn U4 y U6 están unidas entre sí formando una única RNPsn. El primer paso en la formación del espliceosoma es la unión de la RNPsn Ul al sitio de corte 5' del pre-ARNm. Este reconocimiento de los sitios de corte 5' se realiza por medio del apareamiento de bases entre la secuencia del consenso del sitio de corte 5' y una secuencia complementaria en el extremo 5' del ARNsn Ul. Posteriormente la RNPsn U2 se une al punto de ramificación por apareamiento complementario de bases. Un complejo preformado compuesto de las U4/U6 y U5 se incorpora al espliceosoma, estando la U5 unida a secuencias corriente arriba del sitio de corte 5'. La reacción de corte y empalme se acompaña de reordenamientos en los ARNsn. Las RNPsn U4 /U6 y U5 se disocian del complejo y U6 sustituye a Ul en el sitio de corte 5'. A continuación, U6 se compleja con U2, de modo que se ponen en contacto el punto de ramificación y el sitio de corte 5' para llevar a cabo la primera reacción del paso de la reacción de corte y empalme (formación del intermediario con estructura de lazo). U5 se une a secuencias del sitio de corte 3' y se mantiene la alineación entre los exones 5' y 3' para 23 que puedan empalmarse tras la escisión del intrón. Los ARNsn no sólo reconocen las secuencias consenso en los sitios de ramificación y los sitios de corte en los pre-ARN, sino que también catalizan la reacción de corte y empalme de forma directa (Cooper et al, 2011). Las RNPsn se unen a secuencias cortas ubicadas entre los intrones y los exones luego se añaden más proteínas y forman un gran complejo con el ARN que se denomina espliceosoma. Además de desempeñar funciones de reconocimiento de esas secuencias, las RNPsn llevan a cabo funciones catalíticas. El proceso de “splicing” catalizado por espliceosomas ocurre solo en organismos eucariontes. Las secuencias que se eliminan son los intrones, mientras que las secuencias que Permanecen y forman parte del ARNm maduro son los exones. En muchos casos, un mismo transcrito primario puede ser procesado por “splicing” en más de una forma. Este empalme alternativo permite obtener moléculas de Ácido Ribonucleico maduro diferentes a partir de moléculas de ARNm inmaduro originalmente idénticas, lo cual da por resultado polipéptidos con distintas funciones en estos casos, uno o varios exones son eliminados junto con los intrones (Curtis et al, 2008). El investigador Thomas R. Cech y sus colaboradores en los Estados Unidos, estudiando el splicing del Ácido Ribonucleico Ribosomal (RNAr) en un ciliado de agua dulce llamado Tetrahymena, encontraron que en estos organismos unicelulares eucariontes, del propio intron del RNAr inmaduro actúa como catalizador de la escisión y el empalme, es decir, se produce un empalme auto catalítico. Esta secuencia de ARN se pliega formando un empalme auto catalítico que funciona como enzima, que se ha denominado ribozima. Se han encontrado otros ejemplos de empalme auto catalítico en varios organismos como en ARN codificado por genes mitocondriales o de cloroplastos, en algunos genes nucleares de eucariontes unicelulares y en algunos genes de bacteriófagos, pero no en eucariontes multicelulares (Cooper et al, 2011). La información genética contenida en cada molécula de ARNm se traduce en proteínas a través de un proceso enzimático que se realiza en los ribosomas. En la traducción participan principalmente tres tipos distintos de ARN: el Ácido Ribonucleico ribosomal, que junto con varias proteínas forman los ribosomas; el Ácido Ribonucleico mensajero, que acarrea la información genética contenida en genes específicos del ADN y los Ácidos Ribonucleicos de transferencia (ARNt), que sirven como adaptadores específicos para cada aminoácido durante el ordenamiento 24 lineal de éstos en la síntesis de proteínas, conforme la secuencia del ARNm. La síntesis de proteínas, que de facto es la traducción de la secuencia de nucleótidos presentes en el ARNm, se lleva a cabo mediante la polimerización de aminoácidos en proteínas, a nivel de los ribosomas en dirección 5' a 3'. La secuencia del ARNm realiza la codificación en forma de bases organizadas en tripletes de bases (codones) de acuerdo con el código genético, incorporando en cada paso de lectura un aminoácido de la proteína. Cada ARNt tiene una secuencia Complementaria o anti codón para el codón del aminoácido que le permite reconocer el codón correcto sobre el ARNm (Martínez et al, 2004). 8. Técnicas de la Reacción en Cadena de la Polimerasa (PCR) y “Reverse Transcription” (RT-PCR). La Reacción en Cadena de la Polimerasa (PCR) es una reacción enzimática in vitro que amplifica millones de veces una secuencia específica de ADN durante varios ciclos repetidos en los que la secuencia blanco es copiada. Para ello, la reacción aprovecha la actividad de la enzima ADN polimerasa que tiene la capacidad de sintetizar naturalmente el ADN en las células. En la reacción, si usamos como sustrato ADN genómico, entonces típicamente hablamos de la Reacción en Cadena de la Polimerasa, pero si usamos Ácido Desoxirribonucleico Complementario (ADNc) proveniente del ARNm se le conoce como “Reverse Transcription”-PCR (RT-PCR). Esta conversión se logra mediante una reacción conocida como transcripción reversa y controlada por la enzima transcriptasa reversa, capaz de convertir el ARNm en moléculas de ADNc. Este método fue copiado de los retrovirus que usan una transcriptasa reversa para convertir su genoma de ARN en ADN e integrarse en el genoma de las células, para posteriormente replicarse y generar millones de partículas virales. El ADNc se utiliza cuando analizamos la expresión de algún gen de interés a través de los transcritos de ARNm (Dios et al, 2013). Uno de los aportes fundamentales de esta metodología a la investigación básica en Biología Molecular consiste precisamente en la rapidez y eficiencia mediante las cuales se realiza una tarea que antes requería largas y tediosas jornadas. Para replicar el ADN in vitro la técnica de PCR hizo uso en un principio de la ADN polimerasa de la bacteria “Escherichia coli.” Pero esta enzima perdía actividad debido a la alta temperatura requerida para desnaturalizar la doble cadena del ADN, por lo cual debía agregarse enzima fresca al comenzar el tercer paso de cada ciclo. 25 Con esto fue necesario crear automatizaciones y esto dio al origen a la primera máquina termocicladora "Mr. Cycle" fue creada para tratar esa necesidad de agregar la enzima fresca a cada tubo de prueba después del calentamiento y del proceso de enfriamiento (Herschhorn, 2010). Este inconveniente fue solucionado de manera ingeniosa cuando la polimerasa fue remplazada por su equivalente de la bacteria "termófila" “Thermus aquaticus” (Taq). La ADN polimerasa de este microorganismo denominada Taq polimerasa, actúa eficientemente entre los 75°C y los 80°C y resiste más de dos horas a 93°C (Satz et al, 2011). En 1985, la corporación Perkin-Elmer introdujo el Ácido Desoxirribonucleico (DNA) Termal Cycler. De esta manera, es posible mezclar todos los componentes de la PCR al comenzar el primer ciclo y la reacción en cadena puede llevarse a cabo mediante equipos automatizados que realizarán los ciclos térmicos programados. En 1989, se anunció un acuerdo de colaboración con Hoffman-La Roche en el Desarrollo y la comercialización de productos de diagnóstico humanos in vitro y de servicios basados en tecnología de la Reacción en Cadena de la Polimerasa (PCR). En 1990, los científicos alcanzan la primera amplificación y detección simultánea de las secuencias específicas de Acido Desoxirribonucleico usando un colorante fluorescente, sentando el fundamento para la PCR en tiempo real o la PCR "cinética" con sondas TaqMan. En 1991 Hoffmann-La Roche Inc. adquiere los derechos y las patentes mundiales de la PCR. El Dr. Kary Mullis comparte el premio Nobel de Química en 1993 por inventar la tecnología de la PCR. (Satz et al, 2011). 8.1. Componentes de la PCR. 8.1.1. Templado. Una de las características más atractivas de la PCR es que la cantidad y calidad de la muestra de ADN sujeta a amplificación no necesita ser alta. Una sola célula o especímenes con unos pocos pares de bases son usualmente adecuadas para una amplificación exitosa. El criterio esencial es que la muestra contenga al menos una cadena de ADN intacta que abarque la región que va a ser amplificada y que las impurezas sean suficientemente diluidas como para no inhibir la polimerización (Martínez et al, 2004). 26 8.1.2. Características de las muestras. Existen una serie de reglas sencillas para que el ADN molde no sea un problema en la reacción: • Origen de la muestra y proceso de extracción: la muestra no debe llevar agentes quelantes como el Ácido Etilendiaminotetraacetico (EDTA) que reducen la concentración de iones de Mg. en la disolución. Tampoco debe haber determinados factores sanguíneos, fenol, detergentes, que inhibirán la actividad de la polimerasa. • Integridad del ADN • Cantidad de la muestra: debe ser suficiente cantidad para la amplificación de ADN genómico de copia única se usan cantidades de 100-500ng. En el caso de las zonas repetidas se puede reducir esta cantidad a 10-50ng. El mínimo oscila entre 10-100ng y el máximo entre 400-500ng (Mas et al, 2001). 8.1.3. Diseño de iniciadores. Los oligonucleótidos, iniciadores, cebadores o primers son fragmentos complementarios que se van a unir a cada una de las dos cadenas separadas del templado de ADN. La selección de iniciadores es muy importante en la PCR, de este paso depende el éxito en el laboratorio (Martínez et al, 2004). Para la elección de los iniciadores existen programas que nos facilitan esta tarea (DNAsis, Primer3, etc.). Algunas características a considerar para el diseño de los iniciadores son: • Contenido en Guanina+Citosina debe ser aproximadamente del 50%. La relación máxima de purinas/pirimidinas será 60%/40%. • Deben evitarse zonas largas de secuencias de una sola base. • Se recomienda que en los extremos 3´ las últimas bases sean Guanina o Citosina. • Se debe evitar la complementariedad entre la pareja de primers. Si ésta existe entre los extremos 3´, se aumenta la posibilidad de que se creen dímeros de cebadores. • Normalmente deben tener un tamaño de 18-30 bases (b). • La temperatura de hibridación de los cebadores ha de ser similar en ambos y será variable en función de la secuencia de los mismos. Generalmente oscila entre 45 y 65°C. 27 • Si el primer es menor a 20 b, la temperatura de fusión (Tm), se calcula en base a la siguiente fórmula: Tm: 4(Guanina+Citosina) + 2(Adenina+Timina). • Siendo Guanina, Citosina, Timina y Adenina el número de cada una de las bases que forman cada uno de los oligos. La temperatura de hibridación debe ser aproximadamente 5°C menor que la temperatura calculada (Mas et al, 2001). Cuando el objetivo a amplificar es un locus, la posición de los iniciadores debe de ubicarse en los codones de inicio y terminación. Es recomendable que el iniciador "forward o sentido" se encuentre más o menos a 35 pb del inicio de la secuencia que identifica el comienzo del marco de lectura abierto (ORF), de la misma forma el cebador "reverse o antisentido" debe localizarse 35 pb después del codón de terminación. Las concentraciones óptimas de los iniciadores son generalmente entre 0.1 y 0.5 μM. Altas concentraciones de iniciadores pueden promover “mispriming” y acumulación de producto no específico, y puede incrementar la probabilidad de generar un templado independiente llamado dímero de iniciadores. Los productos no específicos y los dímeros de iniciadores son por si mismos sustratos para la PCR y compiten con el producto deseado por la enzima, los Desoxirribonucleótidos trifosfato (dNTPs) e iniciadores, resultando en un bajo rendimiento del producto deseado. Los iniciadores pueden contener extensiones en el extremo 5’ o “mismatches” para incorporar sitios de restricción enzimática, un codón de inicio ATG o secuencias promotoras en la secuencia blanco. Pueden ser usados iniciadores degenerados para extraer genes nuevos en base a su similitud o su secuencia de nucleótidos de genes similares (Martínez et al, 2004). Una razón menos obvia por la que los iniciadores fallan, es la presencia de estructuras secundarias en el templado de ADN. En este caso la substitución de dGTP por 7-deazo-2’deoxiGTP ha sido muy usada. Uno de los componentes importantes para tener éxito en la técnica es la especificidad de los iniciadores, estos deben ser elegidos de modo que tengan una secuencia única dentro del ADN que será amplificado. Un iniciador diseñado en una zona donde la secuencia es altamente repetida dará lugar a productos no deseados. Sin embargo, el mismo iniciador puede dar una sola banda si se amplifica una sola clona de una biblioteca genómica (Martínez et al, 2004). 28 8.1.4. La polimerasa. Los requerimientos de enzima pueden variar con respecto a la secuencia blanco o los iniciadores. Cuando se optimiza una PCR, se recomienda probar rangos de concentración de enzima de 0.5 a 5 unidades/100 μl. Si la concentración de la enzima es muy alta se acumulan productos no específicos y si es muy baja se formara una cantidad insuficiente del producto deseado. Existe una clasificación de estas polimerasas y son las siguientes, Termolábiles: las cuales oscilan en temperatura óptimas de 37-42°C y en donde se desnaturaliza con el calor. Las Termoestables: las cuales oscilan en temperaturas óptimas de 74°C y resiste durante 40-50´a 96°C y actualmente esta polimerasa es la que se utiliza para las técnicas de PCR. 8.1.5. Deoxinucleotidos trifosfato (dNTPs). Son cuatro: dATP, dGTP, dCTP y dTTP. Como se mencionó anteriormente se deben añadir en la solución de la reacción en concentraciones iguales que normalmente oscilan entre los 20 y los 200 µmol. Los dNTPs pueden captar Mg++, por lo que las concentraciones de ambos componentes deben guardar siempre la misma relación. No debemos variar ninguno de ellos de manera independiente. Se aconseja que la concentración de Mg++ sea 0,5 - 1 veces superior a la concentración de dNTPs (Mas et al, 2001). 8.1.6. Sales. Es de gran importancia la concentración de dos cationes que son añadidos en forma de sales cloruro potásico (KCl) y el cloruro de magnesio (MgCl2). En donde las variaciones de sus concentraciones pueden optimizar o perjudicar la reacción como se menciona a continuación. • Bajas y elevadas concentraciones de K+ ayudan a la desnaturalización de secuencias largas de ADN. • Cloruro de magnesio (MgCl2). Aumenta la temperatura de hibridación del ADN. La concentración de este ion resulta fundamental para la optimización de la reacción. 8.1.7. Concentración de magnesio. Es benéfico optimizar la concentración del ion magnesio ya que afecta el alineamiento de los iniciadores, la temperatura de disociación de las cadenas, tanto del templado como del producto de la PCR, la especificidad del producto, la formación de dímeros de primer y la 29 actividad y fidelidad de la enzima. La Taq polimerasa requiere magnesio libre en la unión con el templado, los iniciadores y los dNTPs. La PCR deben contener 0.5 a 2.5 mM de magnesio sobre el total de la concentración de dNTPs. La presencia de EDTA u otros quelantes en el stock del iniciador o del templado puede alterar la concentración óptima aparente de magnesio (Martínez et al, 2004). En resumen de lo anteriormente mencionado, Si nos referimos al Mg++ hay que considerar que altas concentraciones disminuyen la especificidad de la PCR mientras que bajas concentraciones aumentan la especificidad de la reacción (Mas et al, 2001). 8.1.8 Extracción del Ácido Desoxirribonucleico. La aplicación de las diferentes técnicas empleadas en Biología Molecular para el análisis del genoma, depende en gran medida de la habilidad para extraer el ADN. Para lo cual se utilizan diversos métodos de extracción dependiendo del tipo de tejido y ácidos nucleicos a emplear; la mezcla de tejido así obtenida se agrega a una solución amortiguadora de extracción en presencia de agentes quelantes, soluciones de baja o alta fuerza iónica para que sea soluble el ADN y de altas concentraciones de Cloruro de Sodio (NaCl). Los agentes quelantes como el ácido Etildiamino Tetraacético (EDTA) se emplean para proteger el ADN de la acción de enzimas nucleasas; ellos capturan los iones magnesio y no permiten que actúen como cofactores de las nucleasas. Las altas concentraciones de NaCl se emplean para prevenir la contaminación de la muestra con polisacáridos que afectan la pureza del ADN (Martínez et al, 2004). Pudiendo inhibir la actividad de algunas enzimas como polimerasas, ligasas y endonucleasas de restricción. La base para la separación de los polisacáridos de los ácidos nucleicos es su solubilidad diferencial en presencia de las altas concentraciones de NaCl, los polisacáridos precipitan bajo la acción de fuerzas centrifugas. A la mezcla anterior se agrega un detergente aniónico como el duodecil sulfato de sodio (SDS) que solubiliza proteínas, tejidos y membranas, evitando que una cantidad significativa de ADN quede atrapado en los desechos o restos celulares. Para prevenir que el ADN extraído presente coloración gris o parda, la cual se debe a la actividad de las polifenol oxidasas presentes en algunos tejidos, se incluye en el amortiguador de extracción polivinil pirrolidona (P.V.P) (Martínez et al, 2004). 30 Para separar las proteínas se lleva la mezcla de tejido con el amortiguador a una incubación de 65ºC con lo cual se desnaturalizan las proteínas. Para retirar las proteínas desnaturalizadas y la mayoría de los polisacáridos se agrega una sal como el acetato de potasio frío. Por centrifugación se separan tejidos, membranas, polisacáridos y proteínas de los ácidos nucleicos, los cuales quedan en el sobrenadante. Finalmente, hay que separar el ADN de la fase acuosa para lo cual se debe precipitar el mismo usando un alcohol como el isopropanol, el cual es selectivo en la precipitación del ADN en presencia de ARN. Después de precipitado el ADN se disuelve en una solución amortiguadora (hidroximetil aminometano (Tris)- Ácido Etilendiaminotetraacético (EDTA), la cual da un pH estable de 8.0) o en agua bidestilada, la solución reguladora hidroximetil aminometano (Tris) es sensible a la temperatura y debe ser utilizada en la temperatura a la cual se logró el pH original para evitar inexactitudes. El ARN se remueve adicionando enzima RNAasa a la solución (Velasco, 2005). Dentro de la extracción, la lisis o ruptura de las células, es el primer paso para la extracción del ADN. Esto se logra mediante el uso de una solución amortiguadora que contenga solución amortiguadora con hidroximetil aminometano (Tris)- y EDTA, este último se une a cationes divalentes tales como el calcio y el magnesio. Dado que estos iones ayudan a mantener la integridad de la membrana celular, la eliminación de estos con desestabiliza la membrana. La solución hidroximetil aminometano (Tris) es el principal componente amortiguador; su función principal es la de mantener el pH del buffer en un punto estable, por lo general 8,0. Además, la solución Tris interactúa con el lipopolisacárido (LPS) de la membrana, lo cual sirve para desestabilizar la membrana aún más. En la etapa final de la extracción, se extrae el propio ADN de la solución. En este punto, es soluble en el amortiguador. Para extraer el ADN de la solución, este se hace insoluble mediante la adición de etanol o isopropanol (alcohol isopropílico). Al hacer esto, el ADN se hace visible en la solución como una sustancia blanca filiforme. A pesar de que cuando el ADN es insoluble se lo puede aislar de los componentes celulares restantes, no es "utilizable". Después de aislarlo, se elimina el alcohol y el ADN debe ser devuelto a una solución amortiguadora biológica, tal como lo es la solución Tris o agua libre de ARNasas, para ser utilizado posteriormente (Velasco, 2005). 31 8.1.9 Fases y Parámetros en una Reacción en Cadena de la Polimerasa. 8.2. Desnaturalización, alineación, elongación o extensión. En primer lugar es necesario que el ADN se desnaturalice, es decir, que las dos cadenas de ADN se separen. Esta primera fase se conoce como desnaturalización y se lleva a cabo elevando la temperatura a alrededor de 94ºC. El siguiente paso consiste en un descenso de la temperatura para permitir que los iniciadores se unan por complementariedad al ADN molde. Esta segunda fase se conoce como hibridación o alineación. Temperaturas habituales en esta fase oscilan entre 35 y 60ºC. Por último, en la fase de elongación o extensión como se muestra en la Figura 3, la enzima polimerasa incorpora nucleótidos complementarios a partir del extremo 3’ libre de la región en que han hibridado los cebadores. La temperatura a la que se lleva a cabo esta fase depende de la enzima polimerasa empleada; si se utiliza Taq polimerasa la temperatura de elongación suele ser de 72ºC. Estas tres fases constituyen un ciclo de PCR. En este primer ciclo, los fragmentos generados no tienen el mismo tamaño, la copia de cada una de las cadenas se inicia a partir del extremo 3’, en el punto en el que el iniciador hibrida con el ADN molde y termina en el momento en el que la enzima polimerasa no es capaz de añadir más nucleótidos, es decir, que la nueva cadena formada no tiene un tamaño definido (Pérez, 2011). La repetición de este ciclo unas 40 veces, por ejemplo, permite obtener como resultado de un experimento de amplificación millones de copias del fragmento de interés. El número óptimo de ciclos dependerá principalmente de la concentración inicial del templado cuando los otros parámetros son optimizados. Un error común es el de ejecutar demasiados ciclos, ya que esto puede incrementar la cantidad y complejidad de productos no específicos. Pocos ciclos dan como resultado un bajo rendimiento del producto. La cantidad de ADN se duplica teóricamente con cada ciclo de PCR. Después de cada ciclo, la cantidad de ADN es dos veces la anterior, así que después de dos ciclos tenemos 2 x 2 veces, después de 3 ciclos 2 x 2 x 2 veces u 8 (23) veces la cantidad inicial, después de 4 ciclos 2 x 2 x 2 x 2 veces o 16 (24) etc. (Martínez et al, 2004) 32 Cadena complementaria Nucleótidos Cadena molde Figura 3.- Extensión o elongación de las cadenas de ADN (En la figura 3.- Se muestra la acción de la taq polimerasa insertando los diferentes nucleótidos complementarios en el orden que le va indicando la cadena que actúa como molde). 9. Reacción en cadena de la polimerasa en tiempo real (“Real Time PCR”). Otros términos con que se conoce la tecnología de la PCR en tiempo real (Real Time PCR), son PCR cinética o PCR cuantitativa, ya que la metodología permite cuantificar una secuencia específica de ADN presente en la muestra biológica en estudio. Los cambios que ha sufrido la PCR como plataforma tecnológica han sido muy notables; conforme los paradigmas de estudio de los ácidos nucleicos han ido cambiando, la necesidad de ir mejorando la técnica se convirtió en una prioridad para los investigadores; es por ello que la PCR ha sufrido modificaciones para ofrecer una tecnología más innovadora apegada a los retos inherentes que implica el estudio del ADN. Actualmente, el panorama de la PCR promete estar a la altura de las necesidades de la investigación a nivel mundial, tan es así, que ahora la modalidad de la PCR en tiempo real ofrece la gran ventaja de usar un sistema cuantitativo (Dios et al, 2013). En la PCR en tiempo real, los procesos de amplificación y detección de ADN se producen de manera simultánea en el mismo vial cerrado, sin necesidad de ninguna acción posterior “en tiempo real”, además, mediante la detección por fluorescencia se puede medir durante una amplificación la cantidad de ADN sintetizado en cada momento, ya que la emisión de fluorescencia producida en la reacción es proporcional a la cantidad de ADN formado. Esto permite conocer y registrar en todo momento la reacción de amplificación (Necochea et al, 2004). 33 La combinación del uso de la PCR cuantitativa con la detección de moléculas de ARN, han permitido desarrollar el método de Retrotranscripción (RT-PCR). El ADNc generado bajo condiciones adecuadas a partir de la Retrotranscripción de un templado de ARN, puede ser utilizado también como ADN blanco en la PCR en tiempo real (Real Time PCR), La Retrotranscripcion + Reacción en Cadena de la Polimerasa (RT-PCR) es una de las metodologías de la era genómica y se ha convertido en el método de elección para la detección y cuantificación de ARN. La misma consta de dos etapas, en la primera la enzima transcriptasa reversa (RT) sintetiza ADNc utilizando como molde ARN. En la segunda etapa los procesos de amplificación y detección ocurren de manera simultánea. La enzima Taq ADN polimerasa sintetiza múltiples copias de un fragmento de ADN específico, incluso en presencia de otras moléculas de ADN. Para ello, se necesitan dos iniciadores complementarios en los extremos del fragmento de ADN a amplificar (Furlan, 2009). La detección se realiza por varios métodos que se basan principalmente en la utilización de sondas fluorescentes. Las sondas son fragmentos de ADN complementarios a una región del fragmento que queremos amplificar, que están acoplados a un fluoróforo en el extremo 5' y a una molécula que inhibe la fluorescencia (quencher) en el extremo 3'. Se produce emisión de fluorescencia cuando la sonda es hidrolizada y desplazada de su sitio por acción de la Taq ADN polimerasa, liberando el fluoróforo de la acción del quencher. La emisión de fluorescencia producida en la reacción es proporcional a la cantidad de ADN sintetizado, lo cual permite hacer un seguimiento y cuantificación como se muestra en la figura 4. Por las características enunciadas, el método de RT-PCR es la metodología de elección para la detección y cuantificación de ARN y la PCR para detección y cuantificación de ADN como se muestra en la Figura 5. Además, la introducción de equipos de última generación y protocolos mejorados, ha resultado en la aplicación de estos métodos para distintas disciplinas, como por ejemplo microbiología, virología, medicina molecular, medicina forense y biotecnología (Furlan, 2009). 34 AMPLIFICACIÓN UMBRAL Ct threshold Fluorescencia de base (Ciclo 2-10) SIN AMPLIFICACIÓN Figura 4.- Ejemplificación de una reacción de amplificación de un compuesto específico en un termociclador de tiempo real. (Figura 4.- se observa la reacción durante la amplificación, la rapidez con que la señal fluorescente alcanza el nivel umbral (Threshold level), se correlaciona con la cantidad inicial de ADN blanco, permitiendo de esta manera poder cuantificarlo. El número de ciclos necesarios para que la señal fluorescente alcance el nivel umbral, se conoce como Threshold cycle (CT) y es el parámetro en el cual se fundamenta la cuantificación. A mayor CT, menor será la cantidad de ADN blanco o templado inicial). (Furlan, 2009). (Figura 5a) (Figura 5b Figura 5.- Amplificación en tiempo real detectando la fluorescencia en diferentes ciclos. En la figura 5 se muestran curvas típicas de amplificación, donde se gráfica en el eje de las ordenadas el aumento de fluorescencia producido durante el transcurso de la reacción graficado en escala lineal (5a) y en escala logarítmica (5b) y en el eje de las abscisas se muestra cada ciclo de la PCR. La línea horizontal de color rojo está indicando dicho valor umbral de fluorescencia a partir del cual se definen los CT, siendo este umbral fijado en la fase exponencial de la reacción (Furlan, 2009). 35 9.1. Calidad de las muestras. • Los ácidos nucleicos son utilizados inmediatamente después de su purificación, o congelados a -20 ºC para su conservación. • No congelar/descongelar repetidas veces las muestras ya que esto puede causar degradación del ARN. • Agitar bien las muestras antes de adicionar a un tubo. • Evitar la formación de burbujas y gotas en los tubos de reacción. • Corroborar el correcto cerrado de todos los tubos de reacción (Furlan, 2009). 9.1.1. Controles. Los controles son materiales procesados en paralelo con las muestras para establecer la validez del resultado obtenido, ellos nos permiten detectar contaminación y monitorear el ensayo. En cada ensayo de RT-PCR se incluyen los siguientes controles: • Blanco o control de reactivos: contiene todos los reactivos necesarios para la reacción de amplificación excepto ácido nucleico. • Control negativo: son del mismo origen biológico que las muestras y no revela presencia del material genético que se quiere amplificar. El control negativo se utiliza para comprobar que ninguno de los componentes de la reacción esté contaminada con ADN de otra procedencia y que no se han producido errores de manejo que hayan provocado la contaminación cruzada entre muestras. Cada lote debe ser verificado previo a su uso. • Controles positivos: se emplean dos clases por cada ensayo. Uno de ellos es una muestra del mismo origen biológico que las muestras problemas y que revela presencia de material genético específico del cual se quiere amplificar (Pérez, 2011). • El otro control positivo es un producto de PCR clonado en un vector, purificado y cuantificado. Se utilizan dos diluciones del plásmido, una cercana al límite de detección (determinado previamente) y otra de una concentración 10 veces mayor. En ambos casos cada lote es verificado previo a su uso (Furlan, 2009). 36 • Control interno: iniciadores correspondiente a un gen que se expresa constitutivamente en la muestra (gen endógeno). Este tipo de control se emplea para poner en evidencia los falsos negativos que se originan por inhibición de la reacción, debido a compuestos que podrían encontrarse en la muestra. Para verificar la eficiencia de la amplificación y la calidad de las muestras se realiza un ensayo doble de PCR, detectando simultáneamente el control interno y el ADN problema (Furlan, 2009). 9.1.2 Preparación de mezclas de reacción. Para minimizar la variación inter-ensayo y disminuir el tiempo empleado en el procesamiento de las muestras, las mezclas de reacción son preparadas previamente, luego alicuotadas, codificadas y guardadas a -20 ºC. De esta manera pueden ser conservadas durante al menos 15 días. Las mediciones de fluorescencia son registradas en gráficos en función de los ciclos, que pueden ser visualizados durante el transcurso del proceso de amplificación del ADN. Se dice que una molécula de ADN que proviene de moléculas de ARN y que por la reacción de la transcriptasa reversa, se genera ADNc ha sido amplificada, cuando la fluorescencia en el tubo de reacción alcanza el valor del umbral, que se define como el punto en el cual la intensidad de fluorescencia supera significativamente la fluorescencia de base. (Furlan, 2009). 9.1.3. Solución de problemas comunes. Hay que tener en cuenta que dentro del proceso como anteriormente se menciono hay factores que deben seguirse conforme a lo estipulado, o basarse en los manuales que ya han sido aprobados para su uso. El no hacerlo, interfiere en nuestro diagnóstico, algunos errores mínimos y comunes pueden ser: a) Cuando el control positivo es negativo: • Error en la preparación de la muestra. • Algún reactivo puede estar en mal estado. • La sonda puede estar vencida y la fluorescencia inactivada. • La enzima puede estar inactivada. • El control positivo puede estar dañado. • Elección de un programa incorrecto en el Termociclador. 37 • Visualización de resultados en un formato incorrecto. b) Cuando el control negativo es positivo: • Puede ser por contaminación cruzada entre las muestras. • Puede deberse a una degradación inespecífica de la sonda. c) El nivel del fondo es muy alto o muy bajo (debe ser aproximadamente 100-200 unidades): • La concentración de la sonda puede estar equivocada. La sonda puede estar degradada o ser muy antigua si el nivel del fondo es muy bajo (Vargas, 2007). Los sistemas de detección por fluorescencia empleados en la PCR en tiempo real pueden ser de dos tipos: agentes intercalantes y sondas especificas marcadas con fluorocromos (Costa, 1993). 9.1.4. Agentes intercalantes. Son fluorocromos que aumentan notablemente la emisión de fluorescencia cuando se unen al ADN de doble hélice, El incremento de ADN en cada ciclo se refleja en un aumento proporcional de la fluorescencia emitida. Este sistema de detección tiene la ventaja de que la optimización de las condiciones de la reacción es muy fácil y además, es más barato que las sondas específicas, el más empleado en la PCR en tiempo real es el SYBR Green (Costa, 2004). El principal inconveniente de los agentes intercalantes es su baja especificidad, debido a que se unen de manera indistinta a productos generados inespecíficamente o a dímeros de iniciadores, muy frecuentes en la PCR. Para mejorar la especificidad se deben emplear condiciones de reacción óptimas y una selección cuidadosa de los iniciadores para disminuir el riesgo de la formación de dímeros. Además, es recomendable iniciar la reacción de síntesis de ADN a temperaturas elevadas lo cual disminuye de forma notable el riesgo de amplificaciones inespecíficas. Para ello se pueden usar polimerasas recombinantes modificadas que solo funcionan después de ser activadas a temperaturas elevadas (hot-start PCR) o anticuerpos que bloquean el centro activo de la polimerasa hasta que la reacción alcanza temperaturas altas en las que el anticuerpo se desnaturaliza liberando la polimerasa y permitiendo su actividad. 38 Además, La mayoría de los equipos en tiempo real tienen la posibilidad de determinar la temperatura de fusión de los fragmentos amplificados, temperatura (tm) a la que el 50% del ADN de la molécula, está desnaturalizado (Costa, 2004). Cada fragmento amplificado tiene una tm característica, que depende sobre todo de su longitud y de la composición de sus bases. Esta aplicación permite comprobar, aunque no siempre con absoluta garantía la especificidad de los fragmentos detectados en la PCR. Por otra parte, los agentes intercalantes no permiten la identificación de polimorfismos en la secuencia diana (Costa, 2004). 9.1.5. Sondas. Sondas de hibridación específicas. Son sondas marcadas con dos tipos de fluorocromos, un donador y un aceptor. El proceso se basa en la transferencia de energía fluorescente mediante resonancia (FRET) entre dos moléculas. Las más utilizadas son las sondas de hidrólisis denominadas también sondas TaqMan, las sondas moleculares beacons y las sondas FRET (Costa, 2004). Sondas Fret. El sistema es de dos sondas que se unen a secuencias adyacentes del ADN diana, una de las sondas lleva un donador en el extremo 3´ y la otra un aceptor en el extremo 5´. Cuando las sondas están hibridadas, los dos fluorocromos están próximos. Al ser excitado el donador transfiere su energía al aceptor, que a su vez emite la fluorescencia que detecta el lector del equipo (Costa, 2004). Sondas de hidrolisis. Son oligonucleótidos marcados con un fluorocromo donador en el extremo 5’ que emite fluorescencia al ser excitado y un aceptor en el extremo 3’ que absorbe la fluorescencia liberada por el donador. Para que esto ocurra, las moléculas donadora y aceptora deben estar espacialmente próximas. Además, el espectro de emisión de la primera se ha de solapar con el espectro de absorción de la segunda (Costa, 2004). Mientras la sonda está intacta, la fluorescencia emitida por el donador es absorbida por el aceptor. Sin embargo, durante la amplificación del ADN diana, la sonda se hibrida con las cadena complementaria al desplazarse a lo largo de la cadena en su acción de síntesis, la ADN polimerasa 39 de “Thermus aquaticus” que tiene actividad 5´ exonucleasa, hidroliza el extremo libre 5´de la sonda, produciéndose la liberación del fluorocromo donador. Como donador y aceptor están ahora espacialmente alejados, la fluorescencia emitida por el primero es captada por el lector. En todos estos sistemas el incremento de ADN en cada ciclo se corresponde con un aumento de hibridación de las sondas, lo que conlleva un aumento en la misma proporción de fluorescencia emitida. El empleo de sondas garantiza la especificidad de la detección y permite identificar polimorfismos o mutaciones puntuales, pero su coste es más elevado que el SYBR Green y la optimización de las condiciones de la reacción resulta más difícil (Costa, 2004). 9.1.6. Equipos para la “Real Time PCR”. Están compuestos por un termociclador y por un lector de fluorescencia y diseñados para poder efectuar la lectura de la fluorescencia emitida en cada uno de los viales usados y en cualquier momento de la reacción. Los más utilizados son los equipos fabricados por “Applied Biosystems” y “Roche Diagnostic”, pero en general todos los modelos actuales permiten todas las aplicaciones. El número de filtros de lectura que presentan los equipos también es importante ya que permite detectar la emisión de distintos fluorocromos a la vez, de esa manera, se pueden usar varias sondas marcadas con distintos fluorocromos para identificar diferentes tipos de ADN diana en la misma reacción (PCR múltiple) o incorporar controles internos a la reacción para detectar la presencia de inhibidores (Costa, 2004). 9.1.7. Opción de equipos para la “Real Time PCR”. Se puede cuantificar la concentración inicial de ADN o ARN diana en la muestra de manera muy sencilla, añadiendo simplemente unos controles externos con concentraciones conocidas y crecientes de ADN diana (curva patrón) en la tanda de amplificación. En la PCR tiempo real el programa informático va registrando el incremento de fluorescencia (Proporcional al aumento de ADN) en cada ciclo y esta información se refleja gráficamente en curvas de cinética de la reacción para cada una de las muestras y controles. Por tanto, se puede controlar la amplificación en las fases iniciales cuando la concentración de los reactivos todavía no es limitante y el efecto de la variabilidad en la eficiencia de amplificación es menos importante. Para cada muestra el programa informático calcula el número de ciclo en el que el lector empieza a detectar un incremento de fluorescencia significativo, con respecto a la señal de base. 40 El ciclo en el que se empieza a detectar el aumento de fluorescencia se denomina punto de corte (Cp. de “crossing point”) o ciclo umbral (Ct de “threshold cycle”) y es inversamente proporcional a la concentración inicial de ADN diana presente en la muestra. Con las concentraciones previamente conocidas de los controles externos y sus Cp correspondientes se dibuja una curva patrón interpolando en ella los valores de la Cp de cada muestra se puede inferir su concentración de ADN inicial (Costa, 2004). Las principales ventajas de la Real Time PCR • La rapidez con la que se obtienen los resultados, puesto que no se necesita ningún proceso adicional. • Sistema homogéneo que evita la manipulación del amplificado, evitando el grave problema de contaminaciones con el mismo. • Amplio rango dinámico de cuantificación. • Permite monitorear la eficiencia de la reacción antes de que terminen sus ciclos. • Mínima variabilidad en los resultados, dando una cuantificación confiable y precisa. Una de las ventajas fundamentales de la Real Time PCR en relación a la PCR tradicional de punto final, es que en la primera, las mediciones se realizan en la etapa exponencial de la reacción donde en teoría por cada ciclo de amplificación se acumula el doble de producto respecto al ciclo anterior, asumiendo una eficiencia del 100%. Por el contrario, en la PCR de punto final la detección del producto de amplificación se realiza en la fase final de la reacción, donde la misma ya se detuvo y hay degradación de producto. Por lo tanto, medidas realizadas en esta fase, no ofrecen resultados muy confiables en cuanto a la cantidad inicial de templado de la muestra (Vinuez, 2009). 10. Electroforesis en gel de agarosa para separar productos derivados de la PCR punto final. Todos los componentes para obtener una Real Time PCR y PCR de punto final tienen los mismos componentes excepto por el proceso de electroforesis que a continuación se menciona: La electroforesis en un soporte de agarosa es un método que se emplea para separar macromoléculas en función del tamaño, la carga eléctrica y otras propiedades físicas, esta migración de partículas cargadas bajo la influencia de un campo eléctrico. 41 Dicha técnica aplica corriente eléctrica a las moléculas para que atraviesen una placa de gel, La fuerza motriz de la electroforesis es la tensión eléctrica aplicada a los electrodos en ambos extremos del gel, Las propiedades de una molécula determinan la velocidad con que un campo eléctrico puede desplazarla a través de un medio gelatinoso muchas macromoléculas biológicas por dar un ejemplo, aminoácidos, péptidos, proteínas, nucleótidos y ácidos nucleicos poseen grupos ionizables a un pH determinado, y si están en solución son cargadas eléctricamente, ya sea como cationes (+) o aniones (-). Según la naturaleza de la carga neta, las partículas cargadas migraran hacia el cátodo o hacia el ánodo. Así, por ejemplo, cuando se aplica un campo eléctrico a un gel con pH neutro, los grupos fosfatos del ADN cargados negativamente lo harán migrar hacia el cátodo (Somma et al, 1997). Existen muchos tipos de electroforesis que se engloban en dos categorías fundamentales: • Electroforesis de frente móvil. • Electroforesis de zona. Actualmente, sólo se utiliza la electroforesis de zona en la cual la muestra se desplaza sobre un soporte sólido, como papel filtro, celulosa o gel (agarosa o poliacrilamida) y los componentes de la muestra migran en forma de pequeñas bandas, también llamadas zonas. Los materiales más comunes para separar moléculas de ácidos nucleicos son polímeros como la poliacrilamida o la agarosa. Estos geles se colocan en una camara de electroforesis, sumergidos en un amortiguador de pH ligeramente mayor de 8, de esta forma los ácidos nucleicos son sometidos a electroforesis, se desplazarán al polo positivo ya que a pH superior a 5 poseen carga negativa. y los geles se comportan como un tamiz molecular y permiten separar moléculas cargadas en función de su tamaño y forma. Así, moléculas de ADN de diferente tamaño, van a emigrar de forma distinta en un gel de electroforesis (Padilla et al, 2003). La distancia recorrida por cada fragmento de ADN va a ser inversamente proporcional al logaritmo de su peso molecular. Es importante la utilización de marcadores de tamaño conocido porque nos permitirán calcular el tamaño en pares de bases de las muestras de ADN problema, En el caso de muestras de ADN separadas en geles de agarosa se le añade bromuro de etidio 42 (compuesto altamente mutagénico y teratógenico), sustancia que se intercala entre moléculas de doble cadena y es fluorescente cuando es excitado con luz ultravioleta (UV). Tras la electroforesis, se visualiza el gel con una lámpara de luz ultravioleta (UV) y se verán las bandas correspondientes a las muestras de ADN separadas y los marcadores de pares de bases. Finalmente, la visualización de las amplificaciones se lleva a cabo tomando una foto digital al gel de agarosa expuesto a luz ultravioleta (UV) (Figura 6); adicionalmente un procesador de imágenes se encarga de analizar las bandas observadas (Padilla et al,2003). Escalera (Pb) 500 pb 400 pb Amplificación de un microorganismo especifico 300 pb 200 pb Escalera (Pb) Amplificación de un microorganismo especifico Escalera (Pb) Control + Amplificación de un microorganismo 100 pb Figura 6.- Gel de agarosa y productos derivados de la Reacción en Cadena de la Polimerasa (PCR) punto final. (Figura 6.- la separación de los productos de la PCR en gel de agarosa, están representados mediante bandas de un tamaño específico y se comparan con un marcador de pares de bases conocido para determinar la especificidad de la reacción. pb = número de pares de bases). (Dios et al, 2013). 10.1. Principios físicos de la electroforesis en gel de agarosa. La separación de las macromoléculas depende de dos variables: cargas y masa. Cuando una muestra biológica, como por ejemplo el ARN se mezcla en una solución amortiguadora y se aplica a un gel, esas dos variables actúan conjuntamente. La corriente eléctrica de un electrodo repele las moléculas al tiempo que el otro electrodo las atrae. La fuerza de fricción del material de gel actúa como tamiz molecular, separando las moléculas en función de su tamaño (Somma et al, 1997). Durante la electroforesis las macromoléculas son empujadas a través de los poros, dependiendo su tasa de migración por el campo eléctrico de los siguientes factores: • Fuerza del campo. • Tamaño y forma de las moléculas. 43 • Hidrofobicidad relativa de las muestras. • Fuerza iónica y temperatura del amortiguador en que se desplazan las moléculas. Dado que la mayor parte de la potencia producida en el proceso electroforético se disipa en forma de calor, pueden producirse los siguientes efectos perjudiciales: • Aumento de la tasa de difusión de los iones de la muestra y del amortiguador, con el consiguiente ensanchando de las muestras separadas. • Inestabilidad térmica de las muestras que son sensibles al calor por ejemplo, desnaturalización del ADN. • Disminución de la viscosidad del amortiguador y por ende, reducción de la resistencia del medio (Somma et al, 1997). 10.1.1. Componentes de la electroforesis en gel de agarosa. 10.1.2. Agarosa. La agarosa, coloide natural que se extrae de las algas el cual es un polisacárido lineal formado por unidades básicas repetidas, la agarosa, que incluye unidades alternadas de galactosa y 3,6-anhidrogalactosa. La agarosa es muy frágil y las manipulaciones pueden destruirla con facilidad. Los geles de agarosa poseen grandes poros y se emplean fundamentalmente para separar moléculas grandes con un peso molecular de más de 200 kDa. Los geles de agarosa permiten una electroforesis rápida, pero con una resolución limitada por lo que las bandas que se forman en los geles tienen tendencia a ser difusas y a esparcirse. Ello obedece al tamaño de los poros y no puede controlarse Los geles de agarosa se obtienen por suspensión de agarosa seca en polvo en un amortiguador acuoso, tras lo cual se hace hervir la mezcla hasta que la agarosa se funde y se convierte en una solución transparente. A continuación se vierte esta solución en un molde para gel y se deja enfriar a temperatura ambiente hasta que se forma un gel rígido. Al solidificarse la agarosa forma una matriz cuya densidad viene determinada por su concentración (Somma et al, 1997). 10.1.3. Amortiguadores de electroforesis. La movilidad electroforética del ADN se ve afectada por la composición y la fuerza iónica del amortiguador de electroforesis. En ausencia de iones, la conductancia eléctrica es mínima y el ADN migra lentamente o ni siquiera se desplaza 44 En un amortiguador de elevada fuerza iónica la conducción eléctrica es muy elevada y se genera una importante cantidad de calor. En el peor de los casos el gel se funde y el ADN se desnaturaliza (Somma et al, 1997). Se dispone de varios tipos de amortiguadores para la electroforesis de ADN que contienen ácido etilendiaminotetraacético (EDTA) (pH 8.0) y tris-acetato (TAE), tris-borato (TBE) o tris-fosfato (TPE) a una concentración de aproximadamente 50mM (pH 7.-5 - 7.8). Los amortiguadores de electroforesis suelen prepararse en forma de soluciones concentradas y se conservan a temperatura ambiente. El TBE se utilizaba inicialmente a una concentración de trabajo de 1x en el caso de la electroforesis en gel de agarosa. No obstante, una solución de trabajo de 0.5x proporciona un poder más que suficiente y en la actualidad prácticamente toda las electroforesis en gel de agarosa se practican utilizando estas concentraciones (Somma et al, 1997). 10.1.4. Concentración de agarosa. Un fragmento de ADN de un tamaño determinado migra a diferentes velocidades a través de los geles según la concentración de agarosa. En función de la concentración de agarosa o amortiguador se pueden separar segmentos de ADN que contengan de 20 a 50 000 pb. En los geles horizontales la agarosa se emplea por lo general en concentraciones comprendidas entre un 0,7% y un 3%, ver tabla 2 (Somma et al, 1997). Tabla 2.- Concentración recomendada de agarosa para separar moléculas de ADN lineales. % de agarosa Resolución según el rango de tamaño de ADN (pb) 0,75 10 000-15 000 1,0 500-10 000 1,25 300-5 000 1,5 200- 4000 2,0 100-2 500 2,5 50-1 000 pb = pares de bases (Somma et al, 1997). 45 10.1.5. Marcador de ADN. Según determinadas condiciones de tensión, concentración del gel de agarosa y el amortiguador, la distancia de migración dependerá del peso molecular del material inicial. Así pues, puede cargarse un marcador de ADN de un patrón de tamaño conocido en las ranuras situadas en los extremos derecho e izquierdo del gel. Generalmente un marcador contiene un número determinado de segmentos de ADN conocidos, lo cual facilita la labor de determinar el tamaño de los ADNs desconocidos en caso de que se produjere alguna distorsión sistemática del gel durante la electroforesis. El marcador de peso molecular es fácilmente adquirido en el mercado, ya que se ofrece una gama de marcadores con distintos pesos moleculares para elegir el de nuestro interés (Somma et al, 1997). 10.1.6. Amortiguador de carga. Las muestras de ADN que deben cargarse en el gel de agarosa se mezclan en primer lugar con un amortiguador de carga, que por lo general contiene agua, glicerol y un colorante (por ejemplo, cianol de xileno, azul de bromofenol, verde de bromocresol, etc.). La concentración máxima de ADN que puede detectarse mediante fotografía de los geles teñidos con bromuro de etidio es de aproximadamente 2 ng en una banda de 0,5 cm de ancho. Si una banda de este ancho contiene más de 500ng de ADN la ranura estará sobrecargada y se producirá una saturación (Somma et al, 1997). El tampón de carga se emplea con tres fines: • Aumentar la densidad de las muestras para que las gotas de ADN caigan uniformemente en el pocillo. • Añadir color a la muestra, simplificando de este modo el proceso de carga. • Incorporar un colorante a la muestra que en un campo eléctrico se desplace hacia el ánodo a una velocidad previsible. 11. Variantes de la PCR. • RT-PCR. En este caso se sintetiza el ADN a partir del ARN mensajero de la célula en estudio por medio de la enzima retrotranscriptasa (cADN). Por lo que las secuencias amplificadas corresponden a los ARNm y por tanto, a los genes que se expresan en el microorganismo en el momento de la extracción del material. Esto es muy conveniente 46 para conocer la fisiología de las células y saber cómo cambia la expresión de genes a lo largo de un proceso infeccioso (Mas, 2004). • PCR anidado. En ocasiones, para asegurar que un producto de PCR corresponde al esperado y no a alguno producido por reacción cruzada; al producto de una reacción primaria de PCR se le usa como molde para una segunda amplificación en la cual se usan iniciadores cuya secuencia esta hacia dentro del producto de la secuencia original (anidados). Con este método se genera un producto más pequeño que el de la reacción primaria. Pero de tamaño conocido, el cual sería improbable que fuera generado a partir de una secuencia no especifica mediante las dos amplificaciones sucesivas (Mas, 2004). • PCR multiplex. Este método se uso inicialmente para estudiar genes con varios intrones y se basa en el diseño de iniciadores para cada uno de los exones. Estos se mezclan en una sola PCR originando varios productos de diversos tamaños y que corresponden a los distintos exones. Además, si se conocen los puntos calientes (sitios del ADN donde ocurren más mutaciones de las esperadas solo por azar) en el gen en estudio, los iniciadores se pueden diseñar sobre estos puntos para que, dado el caso de una mutación en ese lugar, se modifique el corrimiento electroforético o desaparezca una banda específica, indicando cual exón esta modificado en ese espécimen. Para diagnóstico de enfermedades infecciosas, este razonamiento se aplicó al análisis de muestras en las que pudieran existir más de un tipo de microorganismos. Con este propósito se añaden a la PCR juegos de iniciadores capaces de reconocer los distintos organismos esperados en la muestra. Los productos de la amplificación indican la presencia de los diversos organismos por medio de las bandas de tamaño específico para cada microorganismo. Con la PCR multiplex se puede en un solo ensayo responder a varias preguntas: que microorganismo es (virus, bacterias o protozoarios) y que cepas están presentes en la muestra, si presentan resistencia a antibióticos o si poseen determinados factores de virulencia (Mas, 2004). 47 12. Secuenciación. Inicialmente, se pensaba que la secuenciación de los ácidos nucleicos era mucho más difícil que la de las proteínas y muy pocos progresos se hicieron hasta 1960. Esto se debió en parte a la falta de substratos puros del tamaño adecuado con los cuales desarrollar los métodos y a la composición de los ácidos nucleicos. Se esperaba que la interpretación de los resultados de la secuenciación de los ácidos nucleicos (cuatro monómeros) fuera más difícil que el de las proteínas (20 aminoácidos) y se tendrían que aislar productos de degradación más grandes para poder traslaparlos y deducir sus secuencias. Al inicio, la dificultad predominante fue la interpretación de los resultados pero a medida que las técnicas se fueron mejorando y que se fueron estudiando moléculas más largas, la cuestión del análisis empezó a ser más importante. Hoy, la secuenciación de ácidos nucleicos es más rápida y simple que la secuenciación de proteínas. La estrategia básica de la secuenciación de ácidos nucleicos es idéntica a la que se utiliza en la secuenciación de proteínas (Necochea et al, 2004). La secuenciación automática del ADN se puede realizar en equipos como los de “Applied Biosystems, los cuales realizan una electroforesis capilar y todas las muestras son purificadas mediante precipitación con el fin de eliminar el ruido de fondo y los iniciadores sobrantes, colaborando en la obtención de unos resultados satisfactorios. Los cebadores utilizados para la reacción de secuenciación pueden ser universales o específicos (Senasica-CPA, 2014). 12.1. El método de degradación química. En este método un fragmento de ADN de cadena doble o sencilla se marca en los extremos 5´ o 3´ de una o ambas hebras con 32P. Después, la muestra de ADN se divide en cuatro alícuotas y se fragmenta en cuatro reacciones químicas distintas. Posteriormente los fragmentos de ADN generados pueden ser separados por electroforesis en cuatro carriles distintos con base en su tamaño (Figura 7). Conociendo el nucleótido en el que se realizaron los cortes, se puede inferir la secuencia de la molécula original. Las reacciones químicas que se utilizan para fragmentar la molécula de ADN son las siguientes: 1. Corte de las purinas. Las purinas adenina y guanina se metilan con dimetilsulfato (DMS). Después la reacción es tratada en condiciones alcalinas; la molécula de ADN se fragmenta en las purinas metiladas. Como resultado, se obtiene una serie de bandas oscuras que corresponden a las 48 guaninas (las cuales se metilan 5 veces más rápido) y bandas claras que corresponden a las adeninas. Para interpretar fácilmente el patrón de bandas generadas se puede comparar contra un tratamiento que favorezca el corte de las adeninas. 2. Corte de adeninas. Esta reacción es una variación de la anterior, las purinas metiladas se tratan inicialmente con un ácido diluido. Esto favorece el corte de las adeninas metiladas. Después de un tratamiento alcalino las guaninas también son cortadas. Este tratamiento genera una serie de bandas oscuras y claras que también corresponden a las adeninas y las guaninas respectivamente. 3. Corte de pirimidinas. Esta reacción utiliza el compuesto hidracina, que corta las bases citosina y timina. Posteriormente se trata con piperidina para completar la reacción. 4. Corte de citosina. La presencia de NaCl 2M inhibe la reacción de hidracina con tiamina y el tratamiento posterior con piperidina produce solamente fragmentos que terminan en citosina. Desde que se reportó este método, no se han encontrado reactivos químicos específicos que corten las bases Adenina o Timina. La estrategia que se sigue es distinguir entre los nucleótidos que se encuentran al final de cada corte y deducir la secuencia de ADN (Maxam et al, 1977). Amplificación Bases nitrogenadas Amplificación en un gel de agarosa de bases nitrogenadas que fueron colocadas en un carril determinado para ser separadas por medio de electroforesis. Figura 7.- Secuenciación de ADN con el método de Maxam y Gilbert y separación de los productos. Figura 7.- Fotografía mostrando la separación de las bases nucleotidicas en gel (Necochea et al, 2004). 49 12.1.1. Ventajas y desventajas del método de degradación química. La baja resolución obtenida cuando se reportó la técnica no se debió a un factor inherente al método de Maxam-Gilbert, sino a una limitante de los geles de acrilamida Para mejorar la resolución del gel se ha reportado el uso de geles de acrilamida muy delgados en conjunto con un voltaje alto de corrimiento, lo que genera bandas más delgadas y mejor separadas como se muestra en la (Figura 8). Otro aspecto del método de Maxam-Gilbert que puede ser un poco laborioso es la necesidad de separar y analizar individualmente las hebras del ADN que se quiere secuenciar. Esto se puede realizar mediante enzimas de restricción que separen los extremos etiquetados para el análisis. Alternativamente, las dos hebras marcadas pueden ser desnaturalizadas y separadas en un gel. Hoy en día, el método más usado para la secuenciación de ácidos nucleicos es el método de Sanger. Sin embargo, es justo decir que el método de MaxamGilbert es el más adecuado para determinar la secuencia de fragmentos cortos de ADN debido a que puede determinar la secuencia desde la primera base; en cambio, el método de Sanger sólo permite la lectura a partir de la base 10-20 (Necochea et al, 2004). Fragmentos de nucleótidos expresando un tamaño diferente en cada banda Figura 8.- Separación de nucleótidos en gel de acrilamida. En la figura 8 gel de acrilamida para diferencia el tamaño de 250 fragmentos y determinar la secuencia. (Necochea et al, 2004). 50 12.1.2. El método enzimático Sanger. El método de secuenciación enzimático salió casi al mismo tiempo que el de Maxam y Gilbert, pero ha sido más utilizado. Esto se debe en gran parte a que se han realizado grandes avances en la automatización de esta técnica. El método de Sanger se basa en el uso de la ADN polimerasa para sintetizar cadenas de ADN con una terminación específica. Con este método se generan fragmentos de ADN de todos los tamaños posibles que se puedan distinguir entre sí, por el tipo de marcaje que llevan o por la incorporación de un terminador específico. Las enzimas del tipo de la ADN polimerasa requieren de un templado de ADN de cadena sencilla y realizan la síntesis de la hebra complementaria extendiéndola a partir de un iniciador en dirección 5’ a 3’. Entre los componentes de la reacción se incluyen nucleótidos que no tienen un grupo hidroxilo en su extremo 3’ llamados Didesoxinucleotidos (ddNTP), para poder obtener una terminación especifica en las cadenas. Una vez que los Didesoxinucleotidos (ddNTP) se incorpora como el residuo terminal evita que la cadena de ADN sintetizada continúe extendiéndose La incorporación de los Didesoxinucleotidos Trifosfatos (ddNTPs) es al azar, de tal forma que se obtienen fragmentos de todos los tamaños posibles que terminan en un residuo específico (Sanger et al, 1977). En el método de Sanger la estrategia es hacer cuatro reacciones diferentes de síntesis de ADN utilizando un Didesoxinucleotidos (ddNTP) distinto en cada tubo. Con la mezcla del nucleótido normal dNTP y su terminador ddNTP, se pueden generar fragmentos complementarios de diferentes tamaños que terminan en el mismo nucleótido. Después estos fragmentos se pueden separar en un gel de electroforesis con cuatro carriles distintos para determinar la secuencia del templado (Figura 9) (Necochea et al, 2004). El método de Sanger tiene varias ventajas sobre el método de Maxam- Gilbert. Las reacciones de secuenciación del método enzimático se pueden realizar en unas horas, en cambio las del método de Maxam-Gilbert tardan al menos un día. Las reacciones del método de Sanger son más “puras”, con menos contaminantes que puedan afectar la resolución del gel (Necochea et al, 2004). 51 Interacción de la ADN polimerasa y los dNTPs Separación y desnaturalización por medio del proceso de electroforesis Figura 9.- El método de Sanger. (Figura 9.- se muestran cuatro reacciones con ddNTPs diferentes las cuales permiten la síntesis de distintos fragmentos con una terminación específica. Estos fragmentos se pueden separar por electroforesis y comparando los tamaños se puede determinar la secuencia del templado). (Necochea et al, 2004). 12.1.3. Limitaciones del método enzimático. Cuando se reportó este método para la secuenciación de ADN se usaba el fragmento Klenow de la polimerasa I y sólo un ciclo de síntesis (incubando a 37 ºC) para obtener fragmentos de distintos tamaños. Todos los fragmentos tenían incorporados en sus cadenas nucleótidos marcados con 32P. El grupo de Sanger reportó que con esta técnica se podía determinar una secuencia de hasta 300 nucleótidos a partir de 15 bases del iniciador aproximadamente. Al momento de publicar esta técnica también reportaron que la mayor dificultad era que los didesoxinucleótidos trifosfatados (ddGTPs) no estaban disponibles comercialmente. Desde entonces se ha experimentado con variaciones del protocolo original y se han realizado grandes avances en la automatización de este método (Necochea et al, 2004). 12.1.4. Secuenciación de ARN. Paralelo al desarrollo de los métodos de secuenciación de ADN, también se reportaron avances en la secuenciación de ARN. Desde que Holley secuenció un ARNt para alanina en 1965 se han desarrollado métodos de secuenciación de ARN similares a los utilizados para secuenciar ADN (Blackburn y Gait, 1996). Básicamente los métodos de secuenciación de ARN se dividen en 2 categorías (Brownlee et al, 1977). 52 12.1.5. Métodos indirectos. En este caso, el ARN se convierte primero a ADNc con la enzima transcriptasa reversa y luego se usa el fragmento obtenido como templado para la reacción de secuenciación. En realidad, este método determina la secuencia de una molécula de ADN a partir de la cual se infiere la secuencia de la molécula de ARN. Este método indirecto es uno de los más comunes para la secuenciación de ARN porque tiene todas las ventajas de la secuenciación de ADN (Necochea et al, 2004). 12.1.6. Métodos directos. Estos métodos se utilizan para secuenciar la molécula de ARN cuando es complicado utilizar el método indirecto. Esto suele suceder con Ácido Ribonucleico pequeño (ARNs) o con estructuras secundarias extensas (ribosomales, transferencia). Todas estas técnicas requieren de que el ARN este en forma pura, para el método enzimático del Ácido Ribonucleico: en los primeros reportes se experimentó con una forma enzimática para secuenciar ARN directamente, en este caso los autores Brownlee y Cartwright (1977) describieron los resultados de la secuenciación de una molécula de ARNm de casi 200 b. Utilizaron un iniciador marcado con 32P y la transcriptasa reversa. Usando reacciones similares a las del método de Sanger, los autores generaron fragmentos de ADNc con una terminación específica dada por didesoxinucleótidos trifosfatados (ddNTPs). (Necochea et al, 2004). Después resolvieron el orden de los fragmentos de ADN generados en un gel de acrilamida. Se ha visto que la concentración del templado de ARN influye mucho en la resolución del gel, no obstante, Carpenter y Simon reportaron que cuanto mayor era la cantidad de ARN viral usado como templado, menor era la resolución obtenida en el gel de acrilamida, debido a que las bandas eran anchas complicando la interpretación del orden. Ellos obtuvieron la mejor resolución utilizando 0.4 μg (0.75 pmol) de ARN como templado (Necochea et al, 2004). En una reacción de secuenciación de Ácido Ribonucleico ribosomal (ARNr), Bakin y Ofengand obtuvieron la mejor resolución empleando 6 veces menos ARN, es decir, solamente 0.13 pmol. A pesar de que se generan fragmentos de ADN, el método enzimático es un método directo porque el templado es una molécula de ARN. La marca se puede incorporar a los 53 fragmentos de ADN de maneras alternativas a la usada por Brownlee y Cartwright (Necochea et al, 2004). El uso de didesoxinucleótidos trifosfatados (ddNTPs) marcados tiene la ventaja de que los fragmentos que sufren una terminación prematura no se detectan ni interfieren con la interpretación de la secuencia. La terminación prematura suele ser un problema más común en la secuenciación de ARN por la formación de estructuras secundarias que interfieren con la actividad de la transcriptasa reversa. Además, la síntesis de fragmentos de ADN a 37 ºC carece de las ventajas de las altas temperaturas que se pueden usar con otras enzimas (Taq polimerasa) (Necochea et al, 2004). 12.1.7. Método químico. En 1977 se presentó un método de ruptura química del ARN similar al de Maxam y Gilbert. La molécula de ARN en este caso ARNr se marca con una molécula de 32P en un extremo. Después se utilizaron nucleasas para hacer digestiones de la molécula de ARN marcado en distintos lugares. La RNAsa T1 corta las guaninas, la RNAsa U2 corta las adeninas y una hidrólisis alcalina rompe todos los enlaces fosfodiéster. Se utiliza un gel de acrilamida para separar los fragmentos de estos tres tipos de ruptura, lo que permite determinar el orden de las guaninas, adeninas y pirimidinas de una molécula de ARN ribosomal. A diferencia del método enzimático en el que se puede usar un iniciador marcado para generar los fragmentos que serán secuenciados, el método químico requiere que la molécula de ARN sea marcada directamente. Esto se puede hacer introduciendo una marca de 32P en el extremo 5’ de la molécula con una cinasa T4 o en el extremo 3’ con una ligasa T4, en la tabla 3 se resumen algunas enzimas y sus funciones (Necochea et al, 2004). 54 Tabla 3.- Ejemplos de enzimas y su función. ENZIMA FUNCIÓN REFERENCIA Fragmento Es una polimerasa de ADN que utilizo Sanger en su Sanger et al., 1977. Klenow. reacción de secuenciación. No es termoestable. T7. Una polimerasa de ADN no termoestable que se Ansorge et al., 1990. utilizaba frecuentemente en las reacciones de secuenciación con el método Sanger. Se utilizaba frecuentemente para incorporar terminadores (ddNTPs) etiquetadas con un fluoroforo. Taq Una polimerasa termoestable aislada de T. aquaticus Innis et al., 1988. Polimerasa. (termófilo). Fue una gran herramienta en el desarrollo de la técnica de PCR. Enzimas de La primera fue aislada de E. coli en 1968 por Maxam Restricción. Matthew Meselson y Robert Yuan. Son nucleasas 1977. y Gilbert, que reconocen y cortan secuencias específicas de ADN (doble cadena). Se utilizan en el método de degradación química para aislar los fragmentos (32P) que serán secuenciados. Transcriptasa Una polimerasa de ADN que sintetiza una cadena de Brownlee y Reversa. ADN utilizando una molécula de ARN como Cartwright, 1977. templado. RNAsa T1. Corta las moléculas de ARN (cadena sencilla) en las Donis-Keller et al., guaninas. RNAsa U2. Corta las moléculas de ARN (cadena sencilla) en las Donis-Keller et al., adeninas. RNA ligasa. 1977. 1977. Se utiliza para unir una marca radioactiva (32P) en el Blackburn y Gait, y Gait, extremo 5’ de la molécula de ARN (cadena sencilla). 1996. Cinasa T4. Se utiliza para unir una marca radioactiva (32P) en el Blackburn extremo 3’ de la molécula de ARN (cadena sencilla). 1996. Tomado de Necochea et al, 2004. 55 13.- Descripción de las actividades realizadas en el laboratorio de Bioseguridad nivel 3. La Comisión México- Estados Unidos para la prevención de la fiebre aftosa y otras enfermedades Exóticas de los animales (CPA-Senasica ), tiene a su cargo uno de los pocos laboratorios Bioseguridad Nivel 3 que se encuentran en México y está dirigido por dicha institución, este laboratorio realiza el diagnóstico de enfermedades que son transmisibles entre los animales y en algunos casos al humano; el laboratorio como se muestraen la figura 10, cuenta con procesos y lineamientos que hay que cumplir por cualquier persona que requiera tener acceso a él, así como, para el personal que labora en dichas instalaciones y para el procesamiento de muestras que son remitidas para el diagnóstico. VENTANAS Y PAREDES CON LAS CONDICIONES ESPECÍFICAS. CAMPANAS DE FLUJO DE AIRE. ENTRADA Y SALIDA POR ESTERILIZACIÓN. ÁREA DE DUCHA. CONTENEDORES DE DESECHOS. ÁREA DE ENTRADA Y CAMBIO DE ROPA. Figura 10.- Esquema de un laboratorio de Bioseguridad nivel 3. Para tener acceso al laboratorio y realizar prácticas profesionales, se hace necesario previamente tomar al menos una plática relacionada con Bioseguridad de un día completo. La información obtenida identifica las funciones, los riesgos y las características que englobaban a un laboratorio de Bioseguridad nivel 3, así como, se firma un documento de confidencialidad y compromiso en donde las personas que estarán ingresando se comprometen a seguir los puntos antes mencionados para el cuidado de su propia salud y resguardo de la información que se genera dentro de dichas instalaciones. También se solicita que el personal se aplique las vacunas de rabia, tétanos y contar con seguro médico, estos son algunos de los requisitos individuales que son indispensables cumplir. 56 Algunas características mínimas requeridas que se deben cumplir para laborar en un laboratorio de bioseguridad nivel 3, aspectos generales y de bioseguridad: Buenas prácticas microbiológicas (prácticas básicas). 1. Higiene y presentación personal. • Higiene personal • Pulcritud • NO maquillaje • NO fragancias • Cabello atado • Manos (Lavado, cepillado, uñas) – Técnica correcta * • Vestimenta exclusiva de laboratorio. Sustitución diaria 2. Conductas básicas. • Respetar lineamientos • Prohibiciones y restricciones • No alimentos y bebidas (Solo sellados, como barras y agua) • No fumar, maquillarse, manipular lentes de contacto. • No pipeteo con la boca Factores de riesgo: prácticas microbiológicas básicas (Buenas prácticas de laboratorio) A los procedimientos: • Técnicas a desarrollar • Equipo y material utilizado • Manipulación de agentes activos • Inoculación de animales • Información de los microorganismos. A las muestras: • Posible presencia de agentes infecciosos • Tipo de muestra (Hisopos, tejidos, fluidos) Procedencia de la muestra. • Especie animal • Zona geográfica 57 Antecedentes clínicos. • Vigilancia de rutina • Reporte de caso clínico • Morbilidad – Mortalidad • Signos A la protección personal: barreras primarias (CDC): protección del personal y del medio interno del laboratorio. En todo momento y al tener contacto con algún microorganismo con el que se vaya a trabajar protegerse con guantes y cubre bocas. • Equipos y prendas de protección personal (Contención primaria) Cada visitante o persona que labora para dicho laboratorio al ingresar a la primera área se desprende de toda ropa que haya tenido contacto con el exterior y se le asigna desde ropa interior, zapatos y pijamas de materiales cómodos e indicados para poder ingresar a la primera puerta. Primera área (Senasica.CPA, 2014). Segunda área (Senasica.CPA, 2014). Pasillo del laboratorio (Senasica.CPA, 2014). Figura 11.- Fotos de las áreas del laboratorio. 58 En la segunda área se hace un aseo personal y después se ingresa al laboratorio. De igual forma para salir se desprende cada persona de la vestimenta utilizada y tiene la obligación de ducharse para eliminar cualquier residuo o presencia de algún microorganismo que pudiera exteriorizar. Cabinas de seguridad biológica. Programas de mantenimiento y desinfección. • Calificación y verificación de parámetros • Desinfección semanal (Sanitización) • Disponibilidad de cabinas de bioseguridad • Uso correcto de ellas • Ropa especial del laboratorio • Protección de manos (Guantes) • Protección respiratoria (Cubre bocas) • Protección ocular y facial (Lentes) Al personal que ingresa a laborar: • Debe ser capacitado • Debe tener experiencia • Habilidades • Tener salud (Física y emocional) • Hacerse un monitoreo periódico • Vacunarse contra rabia y tétanos Resguardo de material biológico: • Almacenamiento seguro dependiendo las condiciones de cada organismo. • Agentes infecciosos (Resguardar etiquetado y separado de otras agentes) en la tabla 4 se menciona una clasificación. • Información importante (Física y digital) • Inventarios actualizados (No quedarse sin material biológico) • Sistema efectivo de identificación de muestras (Cadena de custodia) • Control de acceso al laboratorio (Solo por personal autorizado) • Sistema de vigilancia (Cámaras para controlar el buen manejo de las técnicas y agentes) 59 Barreras secundarias (CDC): protección del medio externo del laboratorio • Características de diseño y construcción (Contención secundaria). • Biocontención. En la siguiente se mencionan los grupos de riesgos establecidos por la OIE Tabla 4.- Lista de grupos de riesgo por la Organización Internación de Salud Animal (OIE) Biocustodia “Biosecurity”: Principios destinados a resguardar, almacenar y vigilar el material biológico y la información relacionada para evitar su pérdida, robo, uso incorrecto, desviación o liberación intencional. Biocontención Tecnologías, medios físicos e infraestructura instalados con el objetivo de evitar la liberación o escape (accidental o intencional) de agentes biológicos. Conocer esta información es de suma importancia cuando se trabaja manipulando microorganismos, ya que estamos expuestos a contagiarnos por una mala manipulación, desinformación y falta de experiencia. La bioseguridad abarca muchos aspectos, solo se mencionan algunos, dado que la actividad principal realizada se enfocó al desarrollo de técnicas diagnósticas. 60 Se enlistan en la Tabla 5 las enfermedades en las que se tiene implementadas herramientas de detección etiológica dentro de los laboratorios, tanto regionales como los de biología molecular que están regidos por Senasica y dirigidos por CPA. Tabla 5.-lista de enfermedades para las cuales se cuenta con herramientas de detección implementadas en el laboratorio. Especie Enfermedad Loque americana (P. larvae) Loque europea (M. plutonius) ABEJAS Nosemosis (N. apis / N. ceranae) Pequeño escarabajo de la colmena (A.tumida) Enfermedad de Newcastle AVES Influenza aviar Salmonelosis aviar (S. gallinarum) Diarrea viral bovina Encefalopatía espongiforme bovina BOVINOS Pleuroneumonía contagiosa bovina Tuberculosis bovina Circovirus porcino II Diarrea epidémica porcina Enfermedad vesicular porcina CERDOS Fiebre porcina clásica Influenza porcina Síndrome disgenésico y respiratorio porcino Enfermedad hemorrágica del conejo CONEJOS Mixomatosis Enfermedad de la cabeza amarilla Enfermedad de la cola blanca Enfermedad de las manchas blancas CRUSTACEOS Mionecrosis Infecciosa Necrosis hipodérmica y hematopoyética infecciosa Síndrome de taura 61 Especie Enfermedad Anemia infecciosa equina Arteritis viral equina Durina (T. equiperdum) Encefalomielitis equina del Este Encefalomielitis equina del Oeste EQUINOS Encefalomielitis equina venezolana Influenza equina Metritis contagiosa equina Muermo (P. mallei) Piroplasmosis equina (B. caballi / B. equi) Herpesvirus del ostión MOLUSCOS Infección por Mateilia refringens Infección por Perkinsus (P. marinus / P. olseni) Artritis encefalitis caprina OVINOS Ectima contagioso Y CAPRINOS Maedi-visna Prurigo lumbar (scrapie) Enfermedad de Aujeszky Estomatitis vesicular Fiebre aftosa VARIAS Fiebre del Nilo Occidental ESPECIES Gusano barrenador del ganado Infestación por garrapatas Lengua azul Rabia VENADOS Enfermedad crónica desgastante del venado VISONES Encefalopatía transmisible del visón (SENASICA- CPA, 2014). De todas las enfermedades enlistadas anteriormente, se mencionan a continuación solo las que se remitieron durante mi estancia dentro del laboratorio y a las que se les realizó procedimientos de diagnóstico (Tabla 6). 62 Tabla 6.- Enfermedades que diagnóstica el laboratorio de Bioseguridad nivel 3. Enfermedad de Newcastle AVES Influenza aviar, H5 Y H7 Newcastle Diarrea viral bovina BOVINOS Encefalopatía espongiforme bovina Fiebre Porcina Clásica PORCINOS Diarrea epidémica porcina Circo virus porcino II PRRS Encefalomielitis equina del Este EQUINOS Encefalomielitis equina del Oeste Encefalomielitis equina venezolana OVINOS Y CAPRINOS Ectima contagioso Enfermedad de Aujeszky Estomatitis vesicular VARIAS ESPECIES Fiebre aftosa Lengua azul Rabia (SENASICA- CPA, 2014). Las siguientes técnicas que se mencionan, son las que realizan en todos los laboratorios dirigidos por CPA. 63 Tabla 7.- Técnicas de diagnóstico Técnica Diagnóstica Aislamiento en animales de laboratorio Aislamiento en cultivos celulares Aislamiento en embrión de pollo Ensayo de inmunoabsorción ligado a enzimas Fijación del complemento Identificación morfológica del agente Índice de patogenicidad intracerebral Índice de patogenicidad intravenosa Inhibición de la hemoaglutinación Inmunocromatografía de flujo lateral Inmunodifusión en gel de agar Inmunofluorescencia Inmunohistoquímica Prueba de neutralización por reducción de placas Reacción en cadena de la polimerasa de punto final Reacción en cadena de la polimerasa en tiempo real Secuenciación de ácidos nucleicos Seroneutralización Tiempo Medio de Mortalidad Embrionaria Western blot (SENASICA-CPA, 2014). Las siguientes técnicas que se enlistaran, son las que se realizaron en el laboratorio de bioseguridad nivel 3 mientras mi estancia, se mencionaran dependiendo del área específica en donde se procesan, ya que el laboratorio de Bioseguridad se divide en varias áreas pero solo mencionare las áreas que realizan diagnóstico: 64 AISLAMIENTO VIRAL: En esta área se realizan las siguientes técnicas. Aislamiento en animales de laboratorio Aislamiento en cultivos celulares Aislamiento en embrión de pollo Inmunofluorescencia Inhibición de la hemoaglutinación BIOLOGIA MOLECULAR: En esta área se realizan las siguientes técnicas. Reacción en cadena de la polimerasa de punto final Reacción en cadena de la polimerasa en tiempo real Secuenciación de ácidos nucleicos SEROLOGIA: para estas pruebas los encargados de la técnica puede ser personal tanto de aislamiento viral como de Biología Celular. Seroneutralización Elisa Das Durante los 6 meses de prácticas profesionales mantuve mi estancia en el área de biología molecular aprendiendo las técnicas ya antes mencionadas, antes de comenzar a apoyar a los médicos encargados de estas técnicas se me impartió una capacitación constante que comenzó con ejercicios de pipeteo que diario realice aproximadamente con dos horas diarias para obtener habilidad en ello ya que todo el tiempo se ocupan cantidades pequeñas y eran las de conflicto para mi poca experiencia, conocer cada una de las pipetas que ocupan así como todos los componentes, materiales y medidas de higiene y seguridad que necesitaría para poder iniciar mi Práctica dentro de dicho laboratorio, mi habilidad creció día con día, se me fueron asignando actividades dentro del área como: alicuotar diariamente aguas libres de ARNasas, Trizol, preparar agarosa o cualquier amortiguador que fuera necesario durante el dia, elaborar geles de agarosa y mantener las bitácoras al día. Al mes, se me permitió empezar a realizar técnicas a la par de los 65 médicos con los mismos agentes y al reproducir los mismos resultados, se me permitió directamente apoyarlos en dichos procesos. En promedio el laboratorio recibe aproximadamente 70 muestras a la semana, y esto puede variar dependiendo la estación de año, en verano se realizaba la técnica de ELISA para diagnóstico de enfermedades vesiculares, por la carga de trabajo y el tiempo que se lleva en realizar la técnica, no pude practicarla, pero tuve noción del modo de preparación de los reactivos, placas y del proceso de la técnica. En el apéndice verá ejemplificado el proceso de recepción de muestras y proceso de estas, por lo que mi labor fue el de macerar de 30 a 50 muestras a la semana, todo dependía del tiempo estimado de llegada de las muestras, lleve a cabo de 10 a 30 geles a la semana y como era una actividad asignada del diario mencionare el proceso de elaboración. Electroforesis en gel de agarosa para productos obtenidos por RT-PCR en punto final. Material. • Cámara de electroforesis horizontal con alimentación eléctrica. • Horno de microondas. • Balanza. • Espátulas. • Material de vidrio. • Foto documentadora. Reactivos • Agarosa para electroforesis de ADN. • Tris (hidroximetil) aminometano – EDTA 50X. • Ácido bórico. • Bromuro de etidio. • Sacarosa • Cianol de xileno FF • Marcadores de ADN (escalera a 100 pb). 66 Procedimiento para poder hacer un gel en agarosa • Sellar los bordes de un molde de plástico limpio y seco con cinta adhesiva, colocar el peine adecuado de modo que se formen pocillos completos al añadir la solución de agarosa. • Diluir amortiguador y preparar la cantidad adecuada para llenar la cubeta de electroforesis y preparar el gel. • Pesar la agarosa en polvo hasta lograr 6g en un matraz de Erlenmeyer con tapa. • Calentar la mezcla en un horno de microondas hasta disolver la agarosa. • Verte la solución en el molde y dejar que gelifique. • Una vez completamente formado el gel retirar cuidadosamente el peine y la cinta adhesiva, y colocar el gel en la cámara de electroforesis. • Cargar ADN marcador adecuado en el primer carril y el último. • Agregar el templado con el colorante en cada carril. • Cerrar la tapa de la cámara y conectar adecuadamente los cables eléctricos para que el ADN migre hacia el cátodo por medio de corriente eléctrica. • Aproximadamente se deja correr el gel entre 20 min aproximadamente. • Teñir el gel con bromuro de etidio por 5 min aproximadamente. • Tomar la foto en el fotodocumentador, lo que observaremos serán bandas al intercalarse el colorante en la muestra y de la cual se determinará el tamaño en pares de bases, tomando como referencia las bandas del marcador comercial. Este gel se hace para poder hacer La interpretación del diagnóstico de la técnica de RT- PCR punto final en donde se realiza en base al tamaño en pares de bases de la amplificacion, determinándolo con el marcador de pares de bases comercial (escalera) con el que el laboratorio cuenta. Expresando los Productos positivos que mostrare posteriormente en la figura 12 y en la figura 13 mostrare la cámara de electroforesis en donde se coloca el gel de agarosa, figura 14 se muestra el fotodocumentador de donde se toma la foto del gel de la figura 12. 67 MUESTRAS IDENTIFICADAS MARCADOR DE PESO MOLECULAR AMPLIFICACIÓN DE PRODUCTOS PARA PCR EN GEL DE AGAROSA Figura 12.- Amplificaciones observadas en el fotodocumentador. (Figura12.-MPM= Marcador de pares de bases; H6N8 resultado negativo; los demás carriles muestran resultados positivos de un producto de 300 pb). (Tovar, 2014). Figura 13.- Cámara de electroforesis. (Tovar, 2014). 68 Figura 14.- Fotodocumentador. (Figura 14.-Fotodocumentador para captura de imágenes de productos de PCR separados por electroforesis, el cual es utilizado en el laboratorio de bioseguridad nivel 3). (Tovar, 2014). 14. Técnicas y diagnósticos durante mi estancia en el laboratorio de Bioseguridad nivel 3. Electroforesis en gel de agarosa para productos obtenidos por RT-PCR anidada. La RT-PCR anidada se realiza en el laboratorio específicamente para el diagnóstico de los virus de la encefalitis equina venezolana, encefalitis equina del este y encefalitis equina del oeste. Los cuales pertenecen al género Alphavirus y la familia Togaviridae. Son virus ARN de cadena sencilla y polaridad positiva, con cápside icosaédrica y envoltura. Las encefalitis equinas del este, oeste y venezolana son infecciones transmitidas por mosquitos, que pueden causar encefalitis grave en caballos y humanos. Ocasionalmente, algunos de estos virus también pueden causar enfermedades en otros mamíferos y aves (CFSPH, 2010). Muestras remitidas al laboratorio para el diagnóstico: linfonodos, encéfalo y sangre con EDTA. Situación en México. Es importante mencionar que la última detección del virus de la encefalitis equina venezolana se reportó en 2011 en los estados de Tabasco y Veracruz y en el caso de la otra encefalitis nunca se han detectado en el país. 69 Uso de las técnicas de RT-PCR, PCR en tiempo real y punto final para el diagnostico de enfermedades en el laboratorio. Influenza aviar (IA): para esta enfermedad el templado se somete a una prueba llamada IA MATRIZ. De la cual si hay amplificación se realiza posteriormente una segunda corrida (IA FUSION) para diferenciar entre IA H5 y H7. En el caso de IA H5 se utilizan tres diferentes parejas de primer: norteamericanos, europeos y mexicanos, para establecer el tipo genético viral se realiza la secuenciación de los productos de PCR positivos. Dada su patogenicidad, virulencia y capacidad de mutar de los virus de IA, desde formas levemente patógenas a formas altamente patógenas. De acuerdo a lo establecido por la OIE los virus de influenza aviar se dividen en los de declaración obligatoria altamente patógenas (IA de alta patogenicidad, IAAP) y los de declaración obligatoria levemente patógenas (IA de baja patogenicidad, IABP) (Amaya el at, 2009). El virus de influenza pertenece a la familia Orthomyxoviridae, es un virus ARN segmentado y con envoltura. De acuerdo a sus nucleoproteínas y proteínas de matriz, se clasifican en tres tipos: A, B y C. A su vez, atendiendo a sus dos antígenos de superficie, hemoaglutinina (H) y neuraminidasa (N), se clasifican en subtipos. Los virus de influenza aislados de aves pertenecen sin excepción al tipo A y contienen todos los subtipos hasta ahora conocidos (16 H y 9 N) en las más variadas combinaciones. Entre los virus de influenza de las aves y de los mamíferos existen relaciones de parentesco antigénico (Amaya el at, 2009). Muestras remitidas al laboratorio para el diagnóstico: tráquea, pulmón, bazo, intestino, hisopo cloacal, líquido alantoideo (hisopos conservados en PBS o caldo triptosafosfato). Se trabaja a la par con aislamiento viral en embrión de pollo. Situación en México. Los siguientes estados se encuentran bajo esquema de vacunación para H7N3: Aguascalientes, Chiapas, Coahuila, Durango, Guanajuato, Hidalgo, Jalisco, Michoacán, Nayarit, Nuevo León, Puebla, Querétaro, San Luis Potosí, Tlaxcala, Veracruz y Zacatecas. Los demás se encuentran libres de H7N3. Los siguientes estados se encuentran con baja prevalencia para H5N2: 70 Aguascalientes, Coahuila, D.F, Durango, Guanajuato, Guerrero, Hidalgo, Jalisco, Michoacán, Morelos, Oaxaca, Puebla y Querétaro. Los demás estados están libres de H5N2. El brote más reciente de H7N3 se dio este año en el mes de abril en Puebla y Oaxaca. Es una enfermedad endémica y de notificación obligatoria. Newcastle velogénico y lentogénico. El virus que causa la enfermedad de Newcastle o neumoencefalitis aviar es un miembro de la familia Paramyxoviridae del género Paramixovirus, el cual está integrado por 9 grupos de virus que son serológicamente distintos y que además tienen diferentes hospederos primarios (Moreno, 1994). El virus de Newcastle (VNC) posee un genoma ARN de cadena sencilla y de polaridad negativa; su cápside es de tipo helicoidal con una envoltura lipoproteica. El tamaño de su genoma es de 15,156 nucleótidos (Moreno, 1994). Cepas del virus de Newcastle. Las cepas de virus de Newcastle han sido agrupadas en lentogénicas, mesogénicas y velogénicas Cepas lentogénicas. El grupo de las cepas lentogénicas (casi avirulentas) está integrado por las cepas Hitchner BI, clona 30, La Sota y F, que han sido ampliamente usadas como cepas vacúnales. Cepas mesogénicas. Las cepas de virulencia media llamadas mesogénicas son las Roakin, Komarov, Meekteswar y H, que además han sido usadas ocasionalmente como cepas vacúnales. Cepas velogénicas. Las cepas velogénicas o cepas virulentas de campo que se han identificado son: Milano, Hertz 33, NY, Parrot 70181 y ESSEX 70. Las cuales son viscerotrópicas y la Texas GB neurotrópica y se han utilizado como cepas de desafío. Otras cepas como la Ca 1083 y Largo son aislamientos considerados como velogénicos viscerotrópicos de Newcastle, que se usan también en condiciones de laboratorio con sistemas de alta bioseguridad para hacer pruebas de desafío y 71 comprobar la eficacia de las vacunas. Las cepas mexicanas Querétaro e Iztapalapa se consideran en este grupo (Moreno, 1994). Muestras remitidas al laboratorio para el diagnóstico: encéfalo, tráquea, pulmón, bazo, intestino, hisopo cloacal, líquido alantoideo (hisopos conservados en PBS o caldo triptosafosfato). Se trabaja a la par con el aislamiento viral en embrión de pollo. Situación en México. México está libre de la enfermedad de Newcastle según el SIVE desde 1993. Es una enfermedad exótica y de reporte obligatorio. . Fiebre Porcina Clásica. La Fiebre Porcina Clásica (FPC) es una enfermedad infectocontagiosa viral del cerdo, tanto doméstico como salvaje, está ampliamente distribuida geográficamente y se caracteriza por un cuadro hemorrágico con alta morbilidad y mortalidad. El agente causal es un virus ARN que pertenece al género Pestivirus y a la familia Flaviridae. En este género se incluyen otros virus genética y antigénicamente relacionados como el virus de la Diarrea Viral Bovina tipos 1 (BVDV1) y 2 (BVDV2) y el virus de la Enfermedad de la Frontera (BDV). Los pestivirus pueden ser citopatogénicos o no citopatogénicos en monocapas de cultivo de células (Pérez et al, 2008). El genoma viral es una molécula de ARN de cadena sencilla, polaridad positiva, coeficiente de sedimentación de 40-45s y un tamaño del genoma de aproximadamente 12.284 nucleótidos (Pérez et al, 2008). Muestras remitidas al laboratorio para el diagnóstico: tonsila, linfonodos, riñon, bazo, sangre con EDTA. Para esté virus el laboratorio trabaja a la par dos técnicas de PCR, la RT-PCR tiempo real y punto final, así como con el aislamiento viral. En caso de un resultado positivo a la muestra se le realizará la técnica de PCR anidada y será confirmado el resultado con la secuenciación. 72 Situación en México. México erradico la Fiebre Porcina Clásica el 30 de enero de 2009 y se reconoce como país libre el 14 de agosto de 2012. Es una enfermedad exótica y de notificación obligatoria. Diarrea epidémica porcina. La diarrea epidémica porcina (DEP) a veces designada como “síndrome de la diarrea epidémica porcina”, es una enfermedad viral no zoonótica de los cerdos, causada por un coronavirus y caracterizada por diarrea acuosa y pérdida de peso. Pese a que la enfermedad se identificó y notificó por primera vez en 1971, recientemente se ha diagnosticado en poblaciones porcinas en países no afectados anteriormente entre ellos México. La diarrea epidémica porcina (DEP) no forma parte de la lista de enfermedades de la OIE. No obstante, y de acuerdo con las obligaciones de notificación de los países miembros que figuran en el Código Sanitario para los Animales Terrestres de la OIE, específicamente en el Artículo 1.1.4., relativo a las enfermedades emergentes, se ha registrado un aumento en el número de declaraciones de enfermedad recibidas y transmitidas a través del sistema mundial de información sanitaria de la (OIE) .El virus de la diarrea epidémica porcina (DEP) es un virus con envoltura Ácido Ribonucleico (ARN) clasificado como Alphacoronavirus, de la familia Coronaviridae. No presenta inmunidad cruzada con otros coronavirus entéricos porcinos tales como el virus responsable de la gastroenteritis transmisible (GET) (OIE, 2014). Muestras remitidas al laboratorio para el diagnóstico: intestino, hisopos rectales, a la par con aislamiento viral en cultivo celular. Situación en México. La aparición de la enfermedad en México se dio en julio de 2013 y se reportó a la OIE en mayo de 2014 dando así la declinación en la producción porcina. Actualmente está en control y en enero y febrero del 2015 hubo rebrotes de diarrea epidémica porcina (DEP) en México en los estados de Guanajuato, Michoacán, Querétaro, Puebla y Estado de México Es una enfermedad de notificación obligatoria. (Quintero et al, 2015). 73 Ectima contagioso. El ectima contagioso (EC) es una enfermedad de origen viral que afecta en forma particular a los ovinos y los caprinos, aunque también ha sido encontrada en otros rumiantes domésticos y silvestres, y en condiciones particulares pueden presentarse lesiones característica en el hombre. La enfermedad se presenta de forma enzoótica en todo el mundo con diferentes nombres según el país del que se trate: dermatitis pústular contagiosa (Inglaterra); estomatitis pústular contagiosa (Francia); ORF (Escocia); boca costrosa (Australia y Nueva Zelanda); estomatitis ulcerativa (USA) y boquera (Argentina y Uruguay). El ectima contagioso es producido por un virus ADN de la familia Poxviridae, del género Parapoxvirus, con cápside compleja y envuelto (Tortora, 1987). Muestras remitidas al laboratorio para el diagnóstico: costras conservadas en glicerol al 50 %, se trabaja a la par el aislamiento viral en cultivo celular. Situación en México. Actualmente en control, enfermedad endémica y de notificación. Fiebre aftosa. La fiebre aftosa es una enfermedad muy contagiosa que afecta a los animales de pezuña hendida como bovinos, cerdos, cabras y ovejas, producida por cualquiera de los tipos o subtipos serológicos de virus perteneciente a la familia Picornaviridae, ARN, con cápside icosaédrica y sin envoltura. Pocas veces es letal para dichos animales, pero su importancia radica en las grandes pérdidas económicas como consecuencia de la merma de la productividad, los costosísimos programas de vacunación y enormes gastos en tratamientos para evitar complicaciones y las graves restricciones para la comercialización de la carne y sus derivados. La fiebre aftosa está extendida a casi todo el mundo. La actual situación ecológica de la enfermedad puede alterarse rápidamente y en cualquier momento, de tal manera que un solo país infectado en el mundo es un evidente peligro para todos los demás. En términos generales podría afirmarse que las regiones permanentemente afectadas son aquellas donde hay más movimiento de ganado, mientras que en regiones de explotación mas fija aun dentro o cerca de una zona severamente infectada la enfermedad aparece solo de tanto en tanto como brote aislado. En condiciones naturales el virus es 74 capaz de producir la enfermedad solo en mamíferos de doble pezuña. Pese a la difundida creencia popular que el hombre puede sufrir de fiebre aftosa, no parece ser muy sensible al virus. Es evidente que en la inmensa mayoría de las personas en contacto constante con grandes cantidades de virus, como son los que elaboran vacunas o veterinarios que revisan animales enfermos son los que aparentemente tienen más riesgo. No obstante, al parecer el virus no encuentra terreno para producir lesiones en el hombre y no se han podido encontrar tampoco anticuerpos neutralizantes en estas personas. Los casos diagnosticados como fiebre aftosa en el hombre se deben a otros microorganismos capaces de producir lesiones vesiculares en la mucosa (Inta, 2001). Se conoce la existencia de siete tipos serológicos de virus que no protegen mutuamente uno contra el otro en convalecencia. Los siete tipos de virus conocidos son: Tipo A, Tipo O, Tipo C, Sat 1, Sat 2, Sat 3 y Asia. Ciertas cepas pertenecientes a uno de estos tipos se diferencian formando subtipos, los cuales se designan con un subíndice. La presencia de tipos serológicos del virus obliga a elaborar vacunas que deben contener como antígeno todos los tipos serológicos que puedan eventualmente aparecer en la región donde la vacuna será aplicada. La amenaza de la aparición de subtipos serológicos distintos a los que se usan en la fabricación de vacunas es permanente por ello es importante la rápida identificación del tipo y subtipo que produce cada foco de fiebre aftosa (Inta, 2001). Muestras remitidas al laboratorio para el diagnóstico: epitelio en glicerol al 50%, hisopos de arrastre conservado en PBS, líquido vesicular. En esta enfermedad también se realiza la técnica de ELISA DAS para el diagnóstico. Situación en México. México es libre de fiebre aftosa desde 1955. La enfermedad es exótica y de notificación obligatoria. PRRS (Síndrome reproductivo y respiratorio porcino). El síndrome reproductivo y respiratorio porcino viral (PRRSV) es un proceso infeccioso reconocido en todo el mundo y ha tenido efectos devastadores, diezmando poblaciones porcinas en USA y en Europa. La enfermedad es producida por un virus del género Arterivirus y familia Arteriviridae, de genoma ARN, con cápside icosaédrica y con envoltura, referido como ATTC75 2332 en Norteamérica y como virus Lelystand en Europa (Díaz, 1999).Esquema de vacunación. Cabe mencionar que el cuadro de vacunación dependerá de las características particulares de la piara y la prevalencia de la enfermedad (Ingelvac, 2015). Subclinica: vacunación masiva 2 dosis, piara dos veces al año. Epidemia aguda o reproductiva: vacunación masiva a piara reproductiva, revacunación 6 semanas después y revacunación 3 meses después. Endémica o crónica: vacunación a piara masiva, revacunación a las 6 semanas y revacunación 3 meses después. Vacuna disponible en México Ingelvac PRRS MV vacuna viva atenuada. Muestras remitidas al laboratorio para el diagnóstico: tonsila, linfonodo, bazo, pulmón y sangre. Situación en México. Fase de control por medio de vacunación. Enfermedad endémica y de notificación. Estomatitis vesicular (EV). La EV ha sido reconocida desde 1884. El virus fue descrito por primera vez por Olitsky en 1926. En 1926 y 1927 Cotton identificó los dos serotipos principales del virus (New Jersey e Indiana) como causantes de la enfermedad. Desde entonces fueron identificados y descritos numerosos brotes de EV, particularmente en Estados Unidos de América y en menor grado en México y en América Central y del Sur (Mason, 1978). Recientemente fueron aislados dos nuevos subtipos: Cocal o Indiana 2 y Alagoas o indiana 3. Actualmente se clasifica el virus de la EV dentro de la familia Rhabdoviridae. Se trata de un virus que contiene ARN con cápside en forma cilíndrica de bala (helicoidal) y con presencia de envoltura (Mason et al, 1978). Muestras remitidas al laboratorio para el diagnóstico: epitelio en glicerol al 50%, hisopos de arrastre conservado en PBS y líquido vesicular. 76 Situación en México. Actualmente en control, cabe mencionar que en temporadas de lluvia aumenta la prevalencia de dicha enfermedad, es un diferencial de F.A y se considera zoonótica. Por lo que su diagnóstico en el CPA es de rutina por medio de la técnica de ELISA DAS. Enfermedad endémica de notificación obligatoria. Circovirus porcino tipo I y tipo II. A finales de los 70 se encontró un virus como contaminante de algunas líneas celulares de riñón de cerdo (PK15), llamado actualmente Circovirus porcino tipo I (PCV1), el cual demostró no ser patogénico en cerdos infectados experimentalmente. Sin embargo, el Circovirus tipo II (PCV2) fue aislado por primera vez en 1998 en cerdos con Síndrome Multisistémico de Adelgazamiento Post-destete (SMAP). Los Circovirus son virus pequeños (17nm), icosaédricos, no envueltos y contienen un genoma de una cadena sencilla circular de ADN de sentido negativo; de ahí el nombre de Circovirus. Está incluido en una nueva familia denominada Circoviridae, en la que se encuentran además el virus de la anemia infecciosa aviar y el de la enfermedad del pico y la pluma, constituyendo la familia de virus de más reducido tamaño capaz de infectar a vertebrados (Marlon et al, 2007). Muestras remitidas al laboratorio para el diagnóstico: tonsila, linfonodos, bazo, pulmón y sangre. A la par se realiza el aislamiento viral. Situación en México. Actualmente en control. Es una enfermedad endémica y de notificación. Aujeszky. La enfermedad de Aujeszky (EA) o Pseudorrabia es de origen viral producido por el Herpesvirus porcino 1. Está considerada como propia de los porcinos, porque éstos pueden sobrevivir a la enfermedad y permanecer infectados en forma latente, manteniendo el virus en la naturaleza de forma indefinida. Sin embargo, también afecta a otras especies animales aunque en éstos la enfermedad es de curso rápido y normalmente letal. En los cerdos, la EA se manifiesta con trastornos nerviosos, respiratorios o reproductivos según se trate de individuos jóvenes o adultos. 77 La primera descripción de la EA fue en Argentina, con el aislamiento del virus en el año 1979 y a partir de entonces se produjeron varios brotes de la enfermedad. Distintos estudios de prevalencia demostraron la presencia y propagación del virus en las principales zonas de producción porcina del país y el mundo (Iglesias et al, 1987). El virus de la enfermedad de Aujeszky (VEA) o Herpesvirus porcino 1, pertenece a la familia Herpesviridae, subfamilia Alphaherpesvirinae, género Varicellovirus. Tiene un tamaño de 150 a 180 nm. Su estructura consiste en una doble cadena de ácido desoxirribonucleico (ADN) rodeado de una nucleocápside proteica de simetría icosaédrica y una doble envoltura con peplómeros de naturaleza glicoproteíca (Sota et al, 2004). Muestras remitidas al laboratorio para el diagnóstico: tonsila, linfonodos, riñón y bazo, a la par se realiza el aislamiento viral en cultivo celular. Situación en México. Libre de enfermedad de Aujeszky desde 1997 registrado por el SIVE. Actualmente es una enfermedad que ya fue reportada libre en el 2015. Es exótica y de notificación obligatoria. 78 En las siguientes imágenes se mostraran los equipos con los que cuenta el laboratorio de Bioseguridad nivel 3, para realizar sus técnicas antes mencionadas. Figura 15.- Termociclador ABI FAST 7500. (Figura 15.-Equipo para RT-PCR). (Tovar, 2014). Figura 16.- Termociclador SMART CICLEY. (Figura 16.-Equipo para RT-PCR). (Tovar, 2014). 79 15.-Impacto y relevancia del trabajo profesional. La caracterización de pequeñas cantidades de material genético procedente de tales organismos hace posible conocer su procedencia e identificar su dinámica. Por lo cual la técnica de RT-PCR y PCR en tiempo real y punto final, así como la secuenciación han sido una herramienta de impacto para los médicos veterinarios que trabajan en laboratorios del país vinculadas con la sanidad animal, ya que al establecerse estas técnicas dentro de los laboratorios regionales que son apoyados por el gobierno, permiten llevar un diagnóstico molecular que conlleva un alto costo y que difícilmente los productores podrían tener acceso a estas técnicas para poder diagnosticar su explotación e incluso para la prevención de estas enfermedades en sus hatos, por lo que el control y la erradicación de enfermedades emergentes y reemergente que entran y pueden establecerse en nuestro país que pueden poner en riesgo nuestra ganadería y nuestra salud ha sido la clave para que nuevos médicos se involucre en conocer estos diagnósticos y el tipo de enfermedades que también nos afectan en el consumo de alimentos y en la manipulación de estos animales así como el gobiernos de varios estados pone en acción campañas zoosanitarias para llevar acabo control, erradicación y en su momento llevar al país libre de dichas enfermedades y no podría ser esto posible sin el personal que se capacita en realizar esta técnicas. Por lo cual instituciones como la (CPA) que cuentan con médicos en campo, que recopilan muestras de animales muertos y enfermos para analizarlas posteriormente en los laboratorios de diagnóstico molecular que se encuentran distribuidos en todo el país, hacen una función importante en la detección de dichas enfermedades. Los resultados de las técnicas permiten establecer las medidas de control de las enfermedades, desarrollar vacunas contra brotes, y preparar a Médicos Veterinarios que prestan sus servicios dentro de estas instituciones a aprender y seguir investigando para mantener informado al país de nuevos bichos que ataquen nuestra ganadería. Las herramientas moleculares han facilitado la obtención de genomas completos, lo que ha ampliado el conocimiento de los agentes patógenos. Es necesario mantenerse en permanente actualización, muchos de los protocolos, kits, equipos y reactivos se irán reemplazando por otros más nuevos, más económicos, más sensibles y más eficientes a medida que se vayan validando por los laboratorios de referencia u otras instituciones. El mantener una comunicación constante a través de las redes de científicos y personal que labora en torno a temáticas de diagnóstico es de vital importancia para poder intercambiar opiniones, actualizar 80 protocolos e ir estableciendo los cambios que paulatinamente se van ir generando con las nuevas tecnologías. 16.-Perspectiva personal Dentro de los 6 meses de Prácticas Profesionales en el laboratorio de Bioseguridad Nivel 3 se cumplieron las expectativas personales y profesionales esperadas dentro del laboratorio, y por las cuales se tomo esta opción para poder titularme de la carrera de Medicina Veterinaria y Zootecnia. Quedo satisfecha de los logros obtenidos, y de las aportaciones desempeñadas que mi profesión y persona aporto al laboratorio. Me doy cuenta de la gran importancia que tienen esta técnicas para el diagnostico de diferentes enfermedades que por fuera uno desconoce, hago inca pie de la gran responsabilidad que tienes dicho laboratorio y el personal acreditado para llevar a cabo todos los procesos que interfieren Para que el país este informado, en control y alerta a cualquier situación que pudiera presentarse y afectar nuestra ganadería y salud. Estoy consciente que no hay que bajar la guardia y seguir actualizándonos, y no dejar de dar aportaciones a nuestro país por lo que las investigaciones deben de estar a la vanguardia en este tipo de laboratorios, se que como país nos falta mucho para innovarnos en Biología Molecular, México necesita mantener el nivel que tiene, y seguir investigando para poder estar a la vanguardia de otros países. Sé que el país bajo la resguarda de Médicos Veterinarios que se interesen en obtener experiencia en estas área podrán destacar en función de alguno de estos laboratorios o e incluso dar los conocimientos obtenidos en otros países. Después de mi estancia tengo las bases y la práctica para poder incorporarme ante una nueva área de trabajo y competir con estas habilidades aprendidas ante la situación laboral. Mi perspectiva ahora es introducirme en el campo laboral de la Biología Molecular, prepararme, seguir practicando e incluso investigando para mi facultad y mi país. Pero ya como una Medica Veterinaria y Zootecnista. 81 17.-Conclusión. Concluyo mencionando que la Biología Molecular, específicamente técnicas como la RTPCR y la secuenciación ya se están revisando actualmente dentro del plan de estudio de la carrera de MVZ de la FES-Cuautitlán y por la impartición frecuentes de cursos en diferentes instituciones, por lo cual, puede ser un campo de trabajo para el médico veterinario de hoy en día que está interesado en adquirir información y capacitación en estas nuevas tecnologías. El estar a la vanguardia y la necesidad de establecer alternativas de diagnóstico sensibles y específicas para la detección de patógenos que afectan la sanidad animal, hace necesario capacitarse en el uso de estas técnicas para que médicos veterinarios especializados puedan ser considerados en este tipo de laboratorios o simplemente para profundizar en la investigación utilizando la Biología Molecular para el diagnostico de muchas de las enfermedades que afectan la salud animal. 82 18. Apéndice. 18.1. Formato Kit Roche. 83 18.2. Formato Kit Trizol. 84 18.3. Procesamiento de muestras de diagnóstico. AISLAMIENTO VIRAL. HEMOAGLUTINACIÓN, INOCULACIÓN DE EMBRIÓN DE POLLO. RECEPCIÓN DE MUESTRAS, MACERACIÓN Y RETENCIÓN DE MUESTRAS. APROXIMADAMENTE 70 POR DÍA. SEROLOGÍA (ELISA) RT-PCR, r-RT-PCR etc. SECUENCIACIÓN. INTERPRETACIÓN DE RESULTADO Y NOTIFICACIONES (APROX AL SEGUNDO DÍA). 85 19. Bibliografía 1. Amaya C., Casamiquela C., Guillén D., Dillon J. Servicio Nacional de Sanidad y Calidad Agroalimentaria. Manual de procedimientos influenza aviar; SENASA 2009. http://www.senasa.gov.ar/Archivos/File/File2820-influenzaaviar.pdf. (Fecha de consulta en abri2015). 2. Brownlee, G. Carterwright, E. Rapid gel sequencing of RNA by primed synthesis whith reverse transcriptase. J Mol Biol 114. 1997. 3. Curtis, H. Barnes, N,S. Schnek, A. Massarini, A. Biología. Panamericana.7nd Ed. Madrid-México. 2008. 4. Cooper M, G. Hausman, R, E. La célula .Marban.5nd Ed.USA.2011. 5. Costa J. Reacción en cadena de la polimerasa (PCR) a tiempo real. Servicio de Microbiología. Hospital Clínic i Provincial. Barcelona España. 2004. http://external.elsevier.es/espacioformacion/eimc/temas/m2t12.pdf (Fecha de consulta abril 2015). 6. CFSPH. The Center Food Security and Public Health. College of Veterinary Medicine Iowa State University, Iowa. Encefalomielitis equina del este, del oeste y venezolana. 2010. http://www.cfsph.iastate.edu/Factsheets/es/encefalomielitis_equina.pdf. (Fecha de consulta en abril 2015). 7. Dios T.L., Ibarra C., Velasquillo C. Fundamentos de la reacción en cadena de la polimerasa (PCR) y de la PCR en tiempo real, Vol. 2, Núm. 2: Medigraphic 2013. http://www.medigraphic.com/pdfs/invdis/ir-2013/ir132d.pdf. (Fecha de consulta abril 2015). 86 8. Días C.T. Síndrome reproductivo y respiratorio porcino. Detección de antígeno tisular aspectos clínicos, anatomopatologicos y serológicos en los estados de Aragua y Carabobo. Universidad Central de Venezuela. FCV-LUZ. Ladivet. Venezuela. 1999. http://www.saber.ula.ve/bitstream/123456789/27168/2/articulo7.pdf. (Fecha de consulta abril 2015). 9. Furlan G., Zini C., Nuñez M. Procedimiento operativo: análisis cualitativo de ARN viral por RT/qPCR. V congreso virtual Iberoamericano gestión de calidad en laboratorios (Iberolab), 2009. http://www.iberolab.org/opencms/opencms/congreso/IberolabV2009/Comunicaciones/co municaciones1.htm(Fecha de consulta 2015). 10. Goyanes M.F. Transcripción celular en biología molecular. 1-19. 2010. http://www.buenastareas.com/ensayos/Transcripsion-Celular/425962.html. (Fecha de consulta 2015). 11. Herschhorn A, Hizi A. Retroviral reverse transcriptases.Cell Mol Life Sc. 2010. 12. Holland P.M., Abransom R.D., Watson R., Gelfand DH. Detection of specific polymerase chains reaction product by utilizing the 5´-3´ exonuclase activity of thermus aquaticus DNA polymerase; Proceeding of the national academy of sciences. USA. 1991. 13. Iglesias G.S. Infección con el virus de la enfermedad de aujesky en cerdos. FMVZ. UNAM. Ciencia veterinaria. México 1987. 14. Innis, M, D. Myambo, D. Gelfand. Brow, M. DNA sequencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase Chain reaction-amplified DNA. Proc Natl Acad Sci.1988 15. Ingelvac PRRS MLV y Síndrome reproductivo y respiratorio porcino. http://www.bivetmedica.com.mx/content/dam/internet/ah/vetmedica/mx_ES/flash/prrst2. swf. (Fecha de consulta en abril 2015). 87 16. Mason J.D.V. Epidemiologia de la estomatitis vesicular. Comisión México Americana para la prevención de la Fiebre Aftosa: Ciencia Veterinaria. México. 1978. http://www.fmvz.unam.mx/fmvz/cienciavet/revistas/CVvol2/CVv2c4.pdf. (Fecha de consulta en abril 2015). 17. Maxam, A .Gilbert, W. A. New method for sequencing DNA. Proc Natl Acad Sci. 74.1977. 18. Moreno R.C. La enfermedad de Newcastle y algunos avances recientes de diagnóstico. Universidad Nacional Autónoma de México Ciudad Universitaria; Ciencia Veterinaria, 1994. http://www.fmvz.unam.mx/fmvz/cienciavet/revistas/CVvol6/CVv6c3.pdf. (Fecha consultada abril 2015) 19. Martínez C., Rincón A.S., Patricia E. Métodos Físico-Químicos en biotecnología. Universidad Nacional Autónoma de México: Instituto de Biotecnología. 2004. 20. Mas O.J. Diagnóstico por análisis del DNA amplificado por medio de PCR, Diagnóstico molecular en medicina: Manual moderno. 2004. 21. Marlon O. Torres A. Enfermedades asociadas al circovirus porcino tipo 2. XX Reunión ALPA. XXX Reunión APPA. Vol. 15. 2007. http://www.bioline.org.br/pdf?la07047. (Fecha de consulta en abril 2015). 22. Manual de la OIE. Estomatitis vesicular sobre animales terrestres. Institute for Animal Health. Pirbright Laboratory, Ash Road, Pirbright, Woking, Surrey GU24 0NF.Capitulo.2.1.19. Reino Unido 2010. http://www.oie.int/fileadmin/Home/esp/Health_standards/tahm/2.01.19VESICULARST OMATITIS.pdf. (Fecha de consulta 2015). 88 23. Mas E., Poza J., Ciriza J., Zaragoza P., Osta R., Rodellar C. Fundamentos de la reacción en cadena de la polimerasa (PCR). Laboratorio de Genética, Bioquímica y Grupos Sanguíneos; Facultad de Veterinaria. Universidad de Zaragoza. España. 2011. 24. Necochea R.C., Canultec J.C. Secuenciación de ácidos nucleicos. Proyecto de investigación. Instituto de biotecnología. UNAM: Cuernavaca. 2004. 25. Padilla C.A.P., Diez J D., Martínez E.G., Bárcena J.A.R., García C.A. Electroforesis de ácidos nucleicos en geles de agarosa. Aislamiento y caracterización electroforética de DNA plasmídico. Departamento de Bioquímica y Biología Molecular: Campus Universitario de Rabanales. Córdoba. 2003. http://www.uco.es/organiza/departamentos/bioquimicabiolmol/pdfs/17%20ELECTROFO RESIS%20ACS%20NUCLEICOS%20GELES%20AGAROSA.pdf. (Fecha de consulta abril 2015). 26. Pérez R., Lester J., Díaz A.L., Heidy. Peste Porcina Clásica: diagnóstico y control. Grupo de Virología Animal. Dirección de Microbiología. REDVET. Volumen IX: veterinaria organización 2008. http://www.veterinaria.org/revistas/redvet/n111108/111104.pdf. (Fecha de consulta abril 2015). 27. Pérez C.A.M. Reacción en cadena de la polimerasa (Polymerase Chain Reaction, PCR). Universitat Politècnica de València. Escuela Técnica Superior de Ingeniería Agronómica y del Medio Natural - Escola Tècnica Superior d'Enginyeria Agronòmica i del Medi Natural. 2011. https://riunet.upv.es/bitstream/handle/10251/10700/Reacci%F3n%20en%20cadena%20de %20la%20polimerasa.pdf?sequence=1. (Fecha de consulta 2015). 89 28. Quintero R.V Rebrotes de diarrea epidémica porcina (PED) y herramientas para su control.:porcicultura.com pecuarios.2015. http://www.porcicultura.com/porcicultura/home/articulos_int.asp?cve_art=1335. (Fecha de consulta abril 2015). 29. Quintero R.V., Nava. J., Amador J., Trujillo. ME. Efectos económicos de la Diarrea Epidémica (PED) en México 2015. http://www.porcicultura.com/porcicultura/home/articulos_int.asp?cveart=1297. (Fecha de consulta abril 2015). 30. Somma M.M., Querci M. Análisis de la Presencia de Organismos Genéticamente Modificados en Muestras de Alimentos. Sesión nº 5 Electroforesis en gel de agarosa. Organización mundial de la salud oficina regional: Europa.1997. http://gmocrl.jrc.ec.europa.eu/capacitybuilding/manuals/Manual%20ES/Sesi%C3%B3n5. pdf (Fecha de consulta abril 2015). 31. Sota D.M. Manual de procedimientos enfermedad de aujesky; SENASA. 2004. http://www.msd-salud-animal.mx/binaries/Folleto_Porcilis__Begonia_tcm92-66562.pdf. (Fecha de consulta en abril 2015). 32. Satz M.L., Kornblihtt A.R. La reacción en cadena de la polimerasa, el método y sus aplicaciones. Instituto de investigaciones en ingeniería genética y biología molecular (INGEBI-CONICET). Facultad de ciencias exactas y naturales, UBA. 2011. http://www.buenastareas.com/ensayos/La-Reaccion-En-Cadena-De-La/2132700.html. (Fecha de consulta abril 2015). 33. Sistema Mundial de Información Sanitaria de la OIE. Infección por el virus de la diarrea epidémica porcina 2014. http://www.oie.int/fileadmin/Home/esp/Our_scientific_expertise/docs/pdf/E_factsheet_P EDV.pdf. (Fecha de consulta en abril 2015) 90 34. Sanger, F. Nicklen, A, R. Coulson. DNA sequencing whit chain terminating 35. SENASICA-CPA,2015http://www.senasica.gob.mx/?id=4332.Fecha de consulta mayo del 2015. 36. Tortora L.J Ectima contagioso de ovinos y caprinos. Coordinación e investigación y posgrado FESC. UNAM. Departamento de Fisiopatología. INIF AP-SARH: Ciencia Veterinaria. 1987. http://www.fmvz.unam.mx/fmvz/cienciavet/revistas/CVvol4/CVv4c9.pdf. (Fecha de consulta en abril 2015). 37. Velasco R.M. Marcadores moleculares y la extracción de ADN: Asubagroin fca Unicauca. 2005. http://www.unicauca.edu.co/biotecnologia/ediciones/vol3/Art32.pdf. (Fecha de consulta abril 2015). 38. Vargas M.T. Manual de procedimientos para el diagnóstico molecular de la influenza aviar de alta patogenicidad: FAO. 2007. http://www.cridlac.org/digitalizacion/pdf/spa/doc17764/doc17764.htm. (Fecha de consulta abril 2015). 91