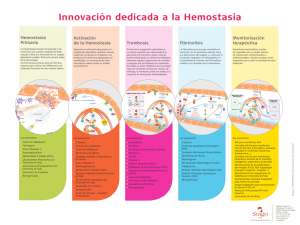
degradacion de fibrina/fibrinogeno y lisis de coagulos
Anuncio

19 OFICINA ESPAÑOLA DE PATENTES Y MARCAS 11 Número de publicación: 2 212 802 51 Int. Cl. : A61K 38/48 7 A61L 33/00 A61P 7/02 ESPAÑA 12 TRADUCCIÓN DE PATENTE EUROPEA T3 86 Número de solicitud europea: 96918996 .8 86 Fecha de presentación: 17.05.1996 87 Número de publicación de la solicitud: 0771146 87 Fecha de publicación de la solicitud: 07.05.1997 54 Título: Degradación de fibrina/fibrinógeno y lisis de coágulos mediante una metaloproteinasa de matriz fibri nolítica. 30 Prioridad: 17.05.1995 US 446887 73 Titular/es: THE NEW YORK BLOOD CENTER, Inc. 310 East 67th Street New York, New York 10021-6295, US 45 Fecha de publicación de la mención BOPI: 72 Inventor/es: Bini, Alessandra 01.08.2004 45 Fecha de la publicación del folleto de la patente: 74 Agente: Justo Vázquez, Jorge Miguel de ES 2 212 802 T3 01.08.2004 Aviso: En el plazo de nueve meses a contar desde la fecha de publicación en el Boletín europeo de patentes, de la mención de concesión de la patente europea, cualquier persona podrá oponerse ante la Oficina Europea de Patentes a la patente concedida. La oposición deberá formularse por escrito y estar motivada; sólo se considerará como formulada una vez que se haya realizado el pago de la tasa de oposición (art. 99.1 del Convenio sobre concesión de Patentes Europeas). Venta de fascículos: Oficina Española de Patentes y Marcas. C/Panamá, 1 – 28036 Madrid ES 2 212 802 T3 DESCRIPCIÓN Degradación de fibrina/fibrinógeno y lisis de coágulos mediante una metaloproteinasa de matriz fibrinolítica. 5 10 15 20 25 30 35 40 Antecedentes de la invención Esta invención se refiere a un método de ruptura de fibrinógeno y fibrina mediado por enzimas. Más particularmente, la invención se refiere a un método para degradar fibrinógeno y producir lisis de coágulos de fibrina mediante la mediación de una metaloproteinasa de matriz fibrinolítica. La invención se refiere además al uso de una metaloproteinasa fibrinolítica como un antitrombótico para reconstruir vasos estenóticos y eliminar depósitos de fibrina. La coagulación sanguínea forma parte de la respuesta natural del organismo ante la lesión o el traumatismo. La formación de coágulos sanguíneos se deriva de una serie de acontecimientos denominados cascada de coagulación, en la que las etapas finales suponen la formación de la enzima trombina. La trombina convierte el fibrinógeno circulante en fibrina, una estructura similar a una red que forma la estructura insoluble del coágulo sanguíneo. Como parte de la hemostasia, la formación de coágulos a menudo es un proceso de salvamento en respuesta a un traumatismo y sirve para detener el flujo de sangre procedente de la vasculatura seccionada. El proceso de salvamento de producción de coágulos en respuesta a una lesión puede convertirse en potencialmente mortal cuando se produce en lugares inapropiados en el organismo. Por ejemplo, un coágulo puede obstruir un vaso sanguíneo y detener el riego sanguíneo a un órgano o a otra parte del organismo. Además, la acumulación de fibrina contribuye a la estenosis parcial o completa de los vasos sanguíneos, dando como resultado la disminución crónica del flujo sanguíneo. Igualmente son potencialmente mortales los coágulos que llegan a desprenderse de sus sitios originales y que fluyen a través del sistema circulatorio produciendo bloqueos en sitios alejados. Tales coágulos se conocen como embolias. En efecto, se ha calculado que las patologías de la coagulación sanguínea, tales como ataques cardiacos, accidentes cerebrovasculares y similares, ascienden a aproximadamente el cincuenta por ciento de todas las muertes hospitalarias. La formación de fibrina durante la inflamación, la reparación tisular o la hemostasia, sólo desempeña un papel temporal y debe eliminarse cuando se restaura la estructura y la función tisular normal. Por tanto, un coágulo de fibrina que se forme rápidamente para parar la hemorragia en un vaso sanguíneo dañado se modifica y después se elimina para restaurar el flujo sanguíneo normal cuando se produce la cicatrización. El sistema responsable de la ruptura de la fibrina y la eliminación del coágulo es el sistema fibrinolítico. La acción del sistema fibrinolítico está estrictamente coordinada mediante la interacción de activadores, zimógenos y enzimas, así como a través de inhibidores de cada uno de estos componentes, para proporcionar activación local concentrada en los sitios de acumulación de fibrina (Francis y col., 1994; Collen 1980; Collen y col., 1991). El principal mediador de la fibrinólisis es la plasmina, una endopeptidasa similar a la tripsina que escinde la fibrina para disolver los coágulos y para permitir que se regeneren los tejidos lesionados. También se ha demostrado que la plasmina desempeña un papel en la degradación de proteínas implicadas en las interacciones célula-célula y célulamatriz, así como en la activación de otras enzimas que reconstruyen tejidos, tales como las metaloproteinasas de matriz (Murphy y col., 1992). A su vez, el control de la actividad de la plasmina, así como estos otros acontecimientos extracelulares, está mediado principalmente por activadores del plasminógeno, que convierten el zimógeno inactivo plasminógeno en la enzima plasmática plasmina. 45 50 55 En la práctica clínica se desea comúnmente activar o potenciar el sistema fibrinolítico. Esto es particularmente necesario en casos de infarto de miocardio en los que las arterias coronarias llegan a ocluirse y requieren recanalización. Se ha demostrado que el cateterismo es, en cierto modo, eficaz en tal recanalización, pero se desean agentes farmacológicos que complementen o sustituyan tales procedimientos invasivos para inhibir la reoclusión. El estudio del intricado sistema de trombólisis y fibrinólisis ha sido un campo en rápido crecimiento, que ha dado como resultado el desarrollo de una nueva generación de agentes trombolíticos. Los tratamientos terapéuticos anteriores para disolver coágulos potencialmente mortales han incluido inyectar en el sistema sanguíneo diversas enzimas que se sabe que rompen la fibrina (Collen, 1996). Los problemas con estos tratamientos han sido que las enzimas no eran específicas de sitio y, por tanto, tendrían más efectos además de producir la disolución del coágulo. Además, estas enzimas destruyen e interfieren con muchas interacciones proteicas vitales que sirven para evitar que el organismo sangre excesivamente debido a las muchas lesiones leves que recibe diariamente. La destrucción de estas salvaguardias mediante tales enzimas puede conducir a hemorragia grave y a otras complicaciones potencialmente mortales. 60 65 En la actualidad, los agentes terapéuticos mejor conocidos para inducir o intensificar la trombólisis son compuestos que producen la activación del plasminógeno, los denominados “activadores del plasminógeno” (Brakman y col., 1992). Estos compuestos producen la hidrólisis del enlace peptídico Arg560-Val651 en el plasminógeno. Esta hidrólisis da lugar a la serín-proteasa activa bicatenaria, la plasmina. Se conocen varios de tales activadores del plasminógeno, incluyendo serín-proteasas tales como el activador del plasminógeno tipo urocinasa (u-PA), el activador del plasminógeno tipo tisular (t-PA), la estreptocinasa (una proteína no enzimática) y la estafilocinasa. De estos, la estreptocinasa es el agente trombolítico terapéutico más ampliamente utilizado. Sin embargo, aunque la estreptocinasa y los demás activadores del plasminógeno han demostrado ser útiles en la recanalización de las arterias coronarias, su capacidad 2 ES 2 212 802 T3 5 10 para mejorar la mortalidad no carece de efectos secundarios y su uso todavía requiere rigurosas condiciones de control para tener éxito en un alto porcentaje de casos (Martin y col., 1994). Además, el uso de tales compuestos puede producir complicaciones hemorrágicas en individuos propensos. Por otra parte, uno de los inconvenientes del uso de t-PA en ensayos clínicos ha sido la rápida formación de nuevo del coágulo después de haberse disuelto, dando como resultado la reoclusión trombótica en algunos pacientes. Numerosos estudios han documentado la capacidad de t-PA para iniciar o potenciar los fenómenos trombolíticos (Sobel y col., 1987). Como resultado, t-PA, específicamente su forma recombinante, rt-PA, se está haciendo más popular como agente farmacéutico trombolítico. No obstante, rt-PA sí que presenta graves limitaciones, incluyendo el coste extremadamente elevado de la dosis y la eficacia variable. Además, se han identificado inhibidores específicos de acción inmediata de t-PA en el plasma humano y en otros fluidos corporales (Collen y col., 1987). Un enfoque adicional para t-PA supone el uso potencial de la transferencia génica de y la expresión del t-PA recombinante en células endoteliales (Lee y col., 1993). Este procedimiento es extremadamente complejo y no es probable que sea práctico como tratamiento trombolítico en el futuro próximo. 15 20 25 También se conocen otras enzimas distintas a la plasmina que pueden degradar fibrina/fibrinógeno en diferentes grados. Por ejemplo, las proteasas leucocitarias endógenas (Bilezikian y col., 1977; Plow y col., 1975), últimamente identificadas como elastasa y catepsina G (Gramse y col., 1978; Plow, 1980; Plow y col., 1982), pueden degradar parcialmente fibrina/fibrinógeno. También se conocen enzimas exógenas que degradan fibrina. Tales enzimas incluyen las enzimas hemolíticas recogidas del veneno de ciertas serpientes, por ejemplo, de las familias crotalidae y viperidae (Purves y col., 1987; Retzios y col., 1992; Sánchez y col., 1991). Las enzimas fibrinolíticas aisladas de las serpientes pueden agruparse en dos clases diferentes (Guan y col., 1991). Estas enzimas (que degradan preferentemente la cadena Aα del fibrinógeno y también las cadenas α y β de la fibrina, son metaloproteasas de zinc (Guan y col., 1991) y todas pueden inhibirse mediante EDTA. Las enzimas de la segunda clase son las serín-proteinasas y muestran especificidad por la cadena β de la fibrina (Guan y col., 1991). Una endopeptidasa procedente del veneno de la víbora bufadora (Bitis arietans) puede escindir en el sitio de reticulación de la cadena γ y de este modo, escindir el Fragmento dímero D en un monómero similar a D (Purves y col., 1987). También se han obtenido enzimas fibrinolíticas de las sanguijuelas (Zavalova y col., 1993; Budzynski, 1991), así como a partir del medio de crecimiento de una bacteria (Aeromonas hydrophilia) que se recogió del tubo digestivo de la sanguijuela (Loewy y col., 1993). 30 35 40 45 50 55 60 65 Las metaloproteinasas de matriz (MMP) endógenas o “matrixinas” (“matrixins”) incluyen tres clases de enzimas: colagenasas, gelatinasas y estromelisinas. Se sabe que las MMP tienen capacidad para degradar varias proteínas y proteoglicanos que están asociados con la matriz extracelular (ECM) del tejido conjuntivo. Se ha demostrado que rompen varias proteínas, incluyendo el colágeno (tipos I-IV, VII y X), la laminina, la fibronectina, la elastina y los proteoglicanos. Las MMP también se han identificado en los leucocitos (Welgus y col., 1990). Se ha demostrado que MMP-2 y MMP-9 tienen actividad elastasa (Señor y col., 1991), a la que podría atribuirse parte de la actividad proteolítica compleja, observada inicialmente en los granulocitos (Sterrenberg y col., 1983). Las MMP participan en la reconstrucción de los tejidos en procesos fisiológicos tales como la morfogénesis y el desarrollo embrionario, así como en la patofisiología de la cicatrización de heridas, la invasión tumoral y la artritis (Matrisian 1992; Nagase y col., 1991; Woessner, 1991; Werb y col., 1992). La expresión de las MMP y de sus inhibidores se produce bajo control molecular y celular amplio y variado (Kleiner y col., 1993; Matrisian, 1992; Woessner, 1991). Factores de regulación conocidos incluyen hormonas, citocinas, protooncogenes, esteroides y factores de crecimiento. Las MMP se bloquean por inhibidores específicos denominados “inhibidores tisulares de metaloproteinasas” (TIMP) que pueden bloquear la actividad de cada elemento de la familia. Se forma un complejo enzima -inhibidor y no tiene lugar ningún recambio del tejido conjuntivo si las MMP están presentes en exceso. El principal enfoque de investigación sobre la ECM ha sido limitar la degradación de la ECM mediante las MMP para interrumpir o interferir en la progresión de los estados patológicos. Varios grupos de investigadores están obteniendo moléculas pequeñas que podrían inhibir las proteinasas para alterar su actividad destructiva en la artritis y como factores antiangiogénicos para inhibir la diseminación tumoral. La metaloproteinasa de matriz 3 (MMP-3) pertenece a la clase de estromelisinas de las metaloproteinasas de matriz. La MMP-3 se expresa en los macrófagos maduros (Campbell y col., 1991), pero también en las células endoteliales, las células del músculo liso y los fibroblastos. Más recientemente, se ha demostrado que la MMP-3 se expresa en células espumosas derivadas de macrófagos procedentes del ateroma de experimentación (Galis y col., 1995). El zimógeno inactivo, pro-MMP-3, se activa mediante la elastasa del neutrófilo, la calicreína plasmática, la plasmina, la quimiotripsina, la tripsina, la catepsina G y la triptasa del mastocito, así como mediante compuestos de mercurio, tales como acetato 4-aminofenilmercúrico (APMA) (Nagase y col., 1992; Kleiner y col., 1993; Nagase y col., 1990; Nagase, 1991). Se han encontrado niveles elevados de MMP-3 en las articulaciones de pacientes que padecen artrosis y artritis reumatoide. En las placas ateroescleróticas hay una gran cantidad de antígeno relacionado con fibrina/fibrinógeno (FRA) que consiste en diferentes formas moleculares (Bini y col., 1987; Bini y col., 1989; Smith y col., 1990; Valenzuela y col., 1992). Dos estudios muy recientes han demostrado la presencia de la metaloproteinasa de matriz 3 en las placas ateroescleróticas (Henney y col., 1991; Galis y col., 1994), pero su función en este contexto sigue sin aclararse. Además, la MMP-3 se ha visto en estos estudios como un factor negativo potencial. Los sustratos conocidos de MMP-3 incluyen proteoglicanos, colágeno tipo IV, fibronectina y laminina. Tales sustratos son habituales de las metaloproteinasas de matriz en general (Doolittle, 1987). Sin embargo, no se ha sugerido 3 ES 2 212 802 T3 que ninguna metaloproteinasa endógena pudiera participar en la degradación de fibrinógeno o fibrina. Ni ha habido ninguna indicación de que las metaloproteinasa pudieran utilizarse para la fibrinólisis o la trombólisis. 5 10 15 A partir de la discusión anterior, está claro que existen lagunas significativas en la comprensión de los procesos implicados en la formación y degradación de trombos. Aunque se han identificado ciertos enfoques que permiten una medida de control sobre estos procesos, estos enfoques adolecen de graves deficiencias relacionadas con el coste, la eficacia o la seguridad. También se ha encontrado que falta el diagnóstico y el tratamiento de estados patológicos asociados con los procesos fisiológicos en los que participan el fibrinógeno y la fibrina. Como resultado, existe la necesidad de composiciones eficaces para su uso en la limitación del desarrollo de trombos y en la inducción de trombólisis. Son necesarios métodos para disgregar coágulos sanguíneos y placas ateroescleróticas, tanto in vitro, tales como para fines diagnósticos, como in vivo, tales como para el tratamiento terapéutico de la embolia, la ateroesclerosis y otros trastornos clínicamente importantes. Además, existe una necesidad de materiales y métodos de diagnóstico y experimentación para revelar más información concerniente a los procesos físicos y químicos que participan en la formación y la degradación de trombos. 20 Además, existe una necesidad de tratamiento eficaz para restaurar al menos parte de la integridad de una pared de vaso dañada, para estimular la regresión de las placas ateroescleróticas y para ayudar en la angioplastia y la cirugía de bypass (revascularización quirúrgica) para evitar la reoclusión. Sumario de la invención 25 30 35 40 45 50 55 60 65 La presente invención proporciona un método para degradar fibrina, fibrinógeno y sustancias relacionadas (es decir, “fibrina/fibrinógeno”) mediante una metaloproteinasa fibrinolítica (FMP). La metaloproteinasa fibrinolítica es una estromelisina, preferiblemente, estromelisina endógena. Más preferiblemente, la metaloproteinasa fibrinolítica incluye la metaloproteinasa de matriz 3 (MMP-3). La metaloproteinasa fibrinolítica escinde o degrada preferiblemente fibrina/fibrinógeno en el enlace peptídico designado como γGly404-Ala405. El método inventivo puede realizarse in vitro. In vitro, el método supone poner en contacto una muestra de tejido, tal como sangre o plasma, con al menos una metaloproteinasa de matriz fibrinolítica. En este método, la fibrina puede degradarse como un constituyente de los coágulos y/o las placas ateroescleróticas con el fin de investigar la estructura de tales materiales, así como para la investigación adicional de los mecanismos de la fibrinólisis y la trombólisis. El fibrinógeno puede degradarse para fines de experimentación o diagnóstico relacionados con la formación de fibrina o para evitar el crecimiento potencial de una red de fibrinógeno-fibrina. En un método de diagnóstico preferido, el método incluye poner en contacto una muestra que contiene fibrina/fibrinógeno con al menos una metaloproteinasa fibrinolítica endógena para proporcionar productos de degradación. Preferiblemente, el método incluye analizar los productos de degradación para caracterizar la fibrina/fibrinógeno. Tal análisis incluye normalmente separar diferencialmente los diversos productos de degradación. Los productos de degradación pueden identificarse o medirse mediante varios medios. Por ejemplo, los fragmentos pueden detectarse mediante anticuerpos que se unen o se asocian específicamente con una región(es) particular(es) de fibrina/fibrinógeno o que no se asocian con ellos debido a la pérdida del epítopo inducida por la degradación enzimática. Preferiblemente, tales anticuerpos son monoespecíficos, más preferiblemente monoclonales. Pueden utilizarse anticuerpos sintéticos y/o quiméricos, como también regiones de unión al antígeno, tal como Fab y F(ab’)2 . La medida de la asociación específica entre tales anticuerpos y los fragmentos de degradación puede proporcionar información cualitativa o cuantitativa acerca de la muestra de fibrina/fibrinógeno que se está analizando. Tales anticuerpos pueden marcarse de manera que se puedan detectar para ayudar en la medición de los tipos y las cantidades de los productos de degradación. Alternativamente, pueden emplearse anticuerpos fijados a un sustrato para ayudar en la separación o la purificación de los fragmentos de degradación producidos por una metaloproteinasa fibrinolítica. La invención también proporciona el uso de una estromelisina para la fabricación de un medicamento para realizar un tratamiento trombolítico, embolítico o aterolítico en un sujeto vertebrado, preferiblemente un primate, más preferiblemente un ser humano. Normalmente, la metaloproteinasa fibrinolítica ha de administrarse en una composición terapéutica que comprende la metaloproteinasa y un vehículo o diluyente farmacológicamente aceptable. Opcionalmente, la composición administrada puede incluir además uno o más de otros principios activos, como un complemento a la actividad fibrinolítica de la metaloproteinasa. Compuestos complementarios adecuados incluyen compuestos que tienen actividad trombolítica o fibrinolítica. Por ejemplo, tales compuestos complementarios pueden ser un activador del plasminógeno, la hirudina, un inhibidor enzimático, un anticoagulante, un anticuerpo o un péptido sintético específico del receptor plaquetario gpIIb/IIIa, o una combinación de los mismos. La estromelisina puede utilizarse para evitar o mejorar las complicaciones asociadas con la ateroesclerosis (es decir, para degradar el fibrinógeno y la fibrina en placas antes de que se produzca la oclusión), así como para evitar la reoclusión tras el tratamiento trombolítico, por ejemplo con t-PA, tras el infarto de miocardio. De hecho, t-PA es de acción inmediata, mientras que una metaloproteinasa MMP3 preferida tiene una acción más lenta y progresiva. Por tanto, los compuestos pueden usarse beneficiosamente en combinación, incluyendo el uso secuencial o concurrente. 4 ES 2 212 802 T3 La estromelisina también puede utilizarse como un agente profiláctico para inhibir la reestenosis tras una intervención quirúrgica, tal como cirugía de bypass o angioplastia. Alternativamente, la estromelisina puede usarse en el tratamiento aterolítico, para inhibir la formación inicial, o para estimular la regresión, de las placas ateroescleróticas. 5 10 15 Además, la invención proporciona equipos diagnósticos y terapéuticos que incluyen una metaloproteinasa de matriz fibrinolítica. La metaloproteinasa fibrinolítica es preferiblemente endógena. La metaloproteinasa incluye una estromelisina, más preferiblemente, MMP-3. También se prefiere que la metaloproteinasa escinda o hidrolice fibrina/fibrinógeno en el enlace peptídico γGly404-Ala405. Tales equipos pueden incluir uno o más envases, así como reactivo(s) adicional(es) y/o principio(s) activo(s) y/o inerte(s) para realizar cualquier variación en el método inventivo. Reactivos de ejemplo incluyen, sin limitación, sustratos sintéticos para probar la actividad enzimática, y anticuerpos (preferiblemente monoespecíficos, por ejemplo, monoclonales) para medir el aumento o la disminución del nivel de antígeno. Equipos preferidos incluyen al menos una dosis unitaria terapéuticamente eficaz de una metaloproteinasa fibrinolítica. También se prefieren los equipos que incluyen medios para administrar, preferiblemente por vía parenteral, más preferiblemente por vía intravenosa, una composición que contiene una metaloproteinasa fibrinolítica. Los equipos pueden incluir uno o más de otros principios activos como complementos, tales como activadores del plasminógeno, hirudina o anticoagulantes, tales como heparina o aspirina. Estos otros componentes pueden incluirse en composiciones separadas, en envases de reactivos separados, o pueden incluirse todos juntos y/o con la metaloproteinasa fibrinolítica en un único envase de reactivo. Los equipos también pueden incluir instrucciones para mezclar o combinar los componentes y/o para usar el equipo según la invención. 20 25 30 35 40 La invención proporciona además un método para controlar la formación de trombos producidos por aparatos relacionados con la medicina. En esta realización, el método incluye poner en contacto un aparato relacionado con la medicina con una composición que incluya una metaloproteinasa de matriz fibrinolítica, preferiblemente MMP-3. La metaloproteinasa se adhiere o se une deseablemente a una superficie del aparato. El método puede usarse para modificar las superficies de contacto con la sangre de dispositivos protésicos implantables, tales como cánulas, catéteres, injertos, stents (endoprótesis), filtros, dispositivos intrauterinos, válvulas y similares, para proporcionar superficies que inhiban la formación de coágulos o placas. Alternativamente, el método permite la modificación fibrinolítica de aparatos tales como agujas, tubos de recogida de sangre, matraces de cultivo, placas de prueba, pipetas, envases de reactivos, tubos, membranas y similares, para estimular la fibrinólisis y para inhibir la polimerización del fibrinógeno y la formación de trombos que, en caso contrario, podrían interferir con los protocolos de experimentación. Asimismo, la invención proporciona aparatos relacionados con la medicina, tales como dispositivos implantables, material de laboratorio y otros dispositivos para usos in vivo e in vitro, aparatos que se han modificado para que incluyan metaloproteinasa fibrinolítica adherida. Éstas y otras ventajas de la presente invención se apreciarán a partir de la descripción detallada y de los ejemplos que se explican a continuación en el presente documento. La descripción detallada y los ejemplos mejoran la comprensión de la invención, pero no están destinados a limitar el alcance de la invención. Breve descripción de los dibujos Las realizaciones preferidas de la invención se han escogido a modo de ilustración y descripción, pero no están destinadas en modo alguno a restringir el alcance de la presente invención. Las realizaciones preferidas de ciertos aspectos de la invención se muestran en los dibujos adjuntos, en los que: 45 50 La figura 1 muestra un análisis electroforético de fibrinógeno tratado con MMP-2 o MMP-3, que muestra la degradación diferencial del fibrinógeno por cada una de las enzimas. La figura 2A es un gráfico que muestra la lisis comparativa de un coágulo de fibrina por MMP y plasmina, medida mediante el porcentaje de radiactividad en los sobrenadantes de la muestra. La figura 2B es un gráfico que muestra la lisis comparativa de un coágulo de fibrina por MMP y plasmina, medida mediante el porcentaje de radiactividad en los coágulos residuales. 55 La figura 3 muestra un análisis electroforético de fibrina tratada con MMP-2, MMP-3 y plasmina, que muestra la degradación diferencial del fibrinógeno por cada una de las enzimas. La figura 4A muestra un inmunoblot (inmunotransferencia) de los productos de degradación de la digestión de fibrina por MMP-3 y plasmina, medido con MoAb/4-5 (γ392-406). 60 65 La figura 4B muestra un inmunoblot (inmunotransferencia) de los productos de degradación de la digestión de fibrina por MMP-3 y plasmina, medido con MoAb/45 (γ397-411). La figura 5 muestra un inmunoblot de los productos de degradación de la digestión de fibrina por MMP-3, medido con MoAb/T2G1 (Bβ15-42) y MoAb/1D4 (Aα349-406). La figura 6A muestra un análisis electroforético (condiciones no reductoras) del dímero D tratado con MMP-3, que muestra la degradación dependiente del tiempo y de la concentración del dímero D por MMP-3. 5 ES 2 212 802 T3 La figura 6B muestra un análisis electroforético (condiciones reductoras) del dímero D tratado con MMP-3, que muestra la degradación dependiente del tiempo y de la concentración del dímero D por MMP-3. 5 La figura 7 es un diagrama esquemático que muestra la región de reticulación en la fibrina reticulada, que muestra los sitios de escisión de MMP-3 y CNBr. La figura 8 muestra un análisis electroforético de los productos de degradación del fibrinógeno y la fibrina reticulada por MMP-3. 10 15 20 25 30 35 Descripción detallada de la invención Tal como se observó anteriormente, dos estudios muy recientes han identificado la presencia de MMP-3 en placas ateroescleróticas (Henney y col., 1991; Galis y col., 1994). Estos estudios han considerado la presencia de las MMP en las placas como un factor negativo que podría favorecer la formación de fisuras en las placas. Sin embargo, la presente invención concuerda con una interpretación completamente diferente de la función de MMP-3. La invención se refiere al papel inesperado que se ha descubierto que desempeña MMP-3 en la degradación del fibrinógeno y la fibrina. Para una mayor claridad y exactitud en la descripción de la invención en el presente documento, se han adoptado ciertos convenios terminológicos en la discusión siguiente. Estos convenios están destinados a proporcionar un medio práctico para mejorar la descripción de la invención, pero no están destinados a ser limitantes, y el experto en la técnica apreciará que pueden incluirse otras interpretaciones adicionales, aunque no contradictorias. Por ejemplo, la invención se refiere al uso de una metaloproteinasa de matriz para degradar fibrina y fibrinógeno. Las metaloproteinasas de matriz útiles según la invención deben mostrar alguna actividad enzimática frente a fibrina, fibrinógeno y/o proteínas, polipéptidos u oligopéptidos relacionados. Metaloproteinasas de matriz que muestran esta actividad se denominan “metaloproteinasas de matriz fibrinolíticas” o “MMP fibrinolíticas”. La metaloproteinasa de matriz fibrinolítica puede ser exógena o endógena. Se prefiere que la metaloproteinasa fibrinolítica sea endógena. Tal como se usa en el presente documento, el término “endógeno” significa que la metaloproteinasa fibrinolítica tiene como origen la especie en la que ha de realizarse la invención. La especie puede ser cualquier vertebrado, preferiblemente, un mamífero y más preferiblemente un primate, más preferiblemente un sujeto humano. De acuerdo con esto, cuando la invención se emplea en un sujeto humano, se prefiere que la metaloproteinasa activa sea endógena para seres humanos, es decir, de origen humano, ya se emplee como una composición farmacéutica o como resultado del tratamiento diseñado para mejorar el control interno del sistema fibrinolítico del sujeto. Para los procedimientos in vitro, el origen de la metaloproteinasa fibrinolítica es menos crítico, pero se prefiere que la metaloproteinasa tenga un origen lo más próximo posible al origen de la especie de la muestra biológica que se está examinando. En tales métodos, la metaloproteinasa es endógena para la muestra si se deriva de la especie a la que pertenece la muestra probada. 40 Las metaloproteinasas fibrinolíticas endógenas incluyen cualquiera de la clase de estromelisina de las metaloproteinasas. Las estromelisinas preferidas incluyen MMP-3 (estromelisina-1), MMP-10 (estromelisina-2) y MMP-11 (estromelisina-3). Una MMP fibrinolítica endógena sumamente preferida es MMP-3. Se ha encontrado que MMP-3 hidroliza fibrina/fibrinógeno en el enlace peptídico γGly404-Ala405 en la región de reticulación de la cadena gamma. 45 Se sabe que, debido a la fluidez y la complejidad de la fisiología de la formación y la degradación de fibrina, muchas formas de fibrina y fibrinógeno están presentes en la sangre circulante, así como en las lesiones trombóticas y ateroescleróticas. Las muchas formas de estas moléculas resultan del ataque continuo por enzimas proteolíticas que escinden de manera diversa las moléculas. El método de la invención se realiza mediante una MMP fibrinolítica que tiene actividad al menos frente a un compuesto relacionado con fibrinógeno o fibrina. Si una molécula derivada de fibrinógeno se ha escindido o modificado previamente para eliminar todos los sitios de escisión de MMP, entonces esa molécula ya no puede actuar como sustrato para una MMP fibrinolítica. Ahora se ha encontrado inesperadamente que los sustratos para MMP-3 incluyen, entre otros, fibrinógeno y fibrina naturales y también se esperaría que incluyeran formas modificadas, sintéticas y semisintéticas de estos compuestos, así como un gran número de productos de escisión de estos compuestos. La clase de sustancias que se derivan del fibrinógeno y/o de la fibrina o que están relacionadas con ellos puede denominarse “fibrina/fibrinógeno”. Por tanto, el método de la invención puede llevarse a cabo utilizando cualquier MMP que actúe para degradar un resto de fibrina/fibrinógeno. Aunque tales enzimas se denominan generalmente “fibrinolíticas”, también pueden denominarse de manera más precisa MMP “fibrin(ogen) olíticas”. 50 55 60 65 Se sabe que el fibrinógeno (también abreviado en el presente documento como “Fg”) es una proteína homodimérica, en la que cada monómero incluye tres cadenas polipeptídicas sustancialmente homólogas, identificadas como cadenas α (alfa), β (beta) y γ (gamma). Para una revisión, véase Doolittle (1987). Por tanto, el fibrinógeno tiene la estructura (αβλ)2 . Las tres subunidades del fibrinógeno tienen dominios enrollados que permiten que las subunidades se ajusten entre sí para formar una región de “espiral enrollada” en el monómero de fibrinógeno. Además, las cadenas beta y gamma tienen cada una un dominio globular, mientras que la cadena alfa está presente en dos formas; una forma predominante que no tiene dominio globular correspondiente (α) y una forma menos frecuente en la que está presente un dominio globular (αE )(Fu y col., 1994). De acuerdo con esto, debido a que el fibrinógeno es homodimérico y debido a que se han identificado dos formas de la subunidad alfa, se conocen dos formas principales de fibrinógeno: 6 ES 2 212 802 T3 (αβγ)2 y (αE βγ)2 . Ambas formas de fibrinógeno se consideran sustratos de las MMP fibrinolíticas según la invención. Los heterodímeros artificiales del fibrinógeno, así como las formas recombinantes, también están dentro de la clase de los sustratos de la MMP fibrinolítica. 5 10 15 20 25 30 35 40 Tal como se ha mencionado, la fibrina (también abreviada en el presente documento como “Fb”) se genera mediante una polimerización inducida y controlada de fibrinógeno (Fu y col., 1994). Dado que se conocen varias formas de fibrinógeno en la sangre circulante, se sabe que pueden producirse varias estructuras de polimerización para la fibrina. La estructura de la fibrina puede afectar a los procesos de fibrinólisis (Gabriel y col., 1992). Ahora se ha encontrado que una metaloproteinasa fibrinolítica lisa eficazmente la fibrina. Por tanto, parece que las metaloproteinasas fibrinolíticas son activas frente a la fibrina sin estar sustancialmente limitadas por las peculiaridades de reticulación de la fibrina. De acuerdo con esto, se considera que la fibrina es un sustrato de MMP según la invención. Por tanto, la fibrina que se produce naturalmente en un sujeto es adecuada para la degradación según la invención, como lo es la fibrina inducida in vitro. Por tanto, los coágulos que se inducen en la sangre ex vivo, por ejemplo, en una muestra de sangre, pueden degradarse según la invención. En tales aplicaciones in vitro, puede emplearse una metaloproteinasa fibrinolítica como un recubrimiento de un envase, tal como un tubo de recogida de sangre. Además, la fibrina artificial, formada a partir de fibrinógeno natural, sintético, semisintético, recombinante y/o de otros tipos, también puede degradarse mediante el método descrito en el presente documento. En condiciones fisiológicas, la plasmina es la enzima principal que actúa para degradar la fibrina. La acción de la plasmina se limita al sitio de acumulación de la fibrina mediante mecanismos de control plasmáticos que evitan la proteolisis de las proteínas circulantes. Sin embargo, en condiciones patológicas, se sabe que la plasmina degrada las proteínas plasmáticas, especialmente el fibrinógeno. El fibrinógeno degradado puede separarse mediante cromatografía de intercambio iónico en cinco fracciones (A, B, C, D y E), de las que los fragmentos D y E son los productos finales principales de la molécula original. La identificación y la caracterización de los fragmentos X e Y intermedios y transitorios hizo que se llegara a comprender el desarrollo de un esquema asimétrico de degradación del fibrinógeno (Francis y col., 1994). Clásicamente, la estructura del fibrinógeno es bilateralmente simétrica, incluyendo un dominio E globular central que es un “nudo” constituido por las regiones N-terminales de las seis cadenas en la molécula de fibrinógeno. Desde E se extienden dos espirales enrolladas, conteniendo cada una de ellas partes de un conjunto de las cadenas α, β y γ. En los otros extremos de las espirales enrolladas hay dominios D globulares. Extendiéndose desde los dominios D, hay extensiones de la cadena Aα, que, únicamente en la subunidad αE , terminan en otro dominio globular. Con el ataque proteolítico mediante plasmina, las escisiones iniciales liberan el extremo polar carboxilo terminal de la cadena Aα y un péptido de la parte N-terminal de la cadena Bβ (Bβ1-42). El principal fragmento restante es el Fragmento X. Las escisiones de las tres cadenas polipeptídicas a lo largo de una espiral enrollada que conecta el nudo (E) central N-terminal y un dominio (D) terminal del fragmento X lo dividen asimétricamente. El resultado es una molécula de fragmento D, que consiste en partes carboxilo terminales de las tres cadenas y un resto de fragmento Y, que consiste en los dominios central y terminal todavía conectados mediante una espiral enrollada. La escisión posterior de la espiral enrollada del fragmento Y produce un segundo fragmento D y un resto de fragmento E. El fragmento X se puede coagular lentamente mediante trombina, pero los fragmentos Y y D tienen potentes efectos antipolimerizantes, debido principalmente a la disgregación del alineamiento apropiado y a la continuación de la acumulación de protofibrillas de fibrina. 45 50 55 60 65 El conocimiento de la fragmentación convencional del fibrinógeno ayuda a proporcionar un marco conceptual con el que comparar la actividad de otras enzimas fibrinolíticas potenciales. Además, se han desarrollado anticuerpos que son reactivos específicamente o que se unen específicamente sólo a algunos de los fragmentos, permitiéndose de ese modo la identificación molecular de los fragmentos con gran exactitud y precisión (Kudryk y col., 1989a). Usando este conocimiento, ahora se ha identificado inesperadamente la actividad fibrinolítica de una metaloproteinasa endógena, permitiéndose de ese modo el desarrollo del método de la invención. La invención proporciona, entre otros, un método de degradación de fibrina/fibrinógeno. Generalmente, el método requiere el uso de una metaloproteinasa de matriz endógena que tenga actividad enzimática, es decir, que pueda hidrolizar la fibrina y/o el fibrinógeno. El método incluye poner en contacto la fibrina o el fibrinógeno con una cantidad eficaz de una metaloproteinasa de matriz fibrinolítica, preferiblemente MW-3. El método incluye normalmente poner en contacto el fibrinógeno o la fibrina con una metaloproteinasa de matriz. Las composiciones que incluyen una metaloproteinasa fibrinolítica también pueden incluir otros principios activos y/o inertes. Otros principios activos pueden incluir componentes tales como inhibidores de las MMP, activadores del plasminógeno, otras enzimas fibrinolíticas, reactivos de anticoagulación, etc. Si se emplea otro principio activo, se prefiere incluir en la composición un activador del plasminógeno. Se ha encontrado que la metaloproteinasa de matriz 3, en particular, no compite sustancialmente ni inhibe sustancialmente el t-PA ni la plasmina. De acuerdo con esto, el método de la invención puede incluir activadores del plasminógeno, tales como u-PA, t-PA, estafilocinasa, estreptocinasa o formas recombinantes, sintéticas o semisintéticas de los mismos. La invención también permite ahora la investigación de la interacción de MMP-3 con activadores del plasminógeno, inhibidores de activadores del plasminógeno (por ejemplo, PAI-1) y plasminógeno natural. Por ejemplo, aunque 7 ES 2 212 802 T3 se sabe que la plasmina puede activar MMP-3, no se sabe si MMP-3 activa t-PA o u-PA, o si por el contrario, podría activarse por ellos. Tampoco se sabe si MMP-3 podría activar el plasminógeno natural. 5 10 15 20 25 30 35 También puede ser deseable incluir un agente de anticoagulación, tal como heparina, para inhibir o evitar una nueva coagulación. Tales medidas serían menos críticas en las aplicaciones in vitro, pero podrían ser significativamente útiles en aplicaciones in vivo. La combinación de t-PA con heparina para definir una composición trombolítica de doble función se muestra en la patente de los EE.UU. número 5.130.143. En una realización preferida, la invención permite el tratamiento fibrinolítico de un mamífero, preferiblemente de un sujeto humano. En esta realización, el método incluye la administración a un sujeto de una cantidad terapéuticamente eficaz de una metaloproteinasa de matriz que degrada fibrina/fibrinógeno. La metaloproteinasa incluye preferiblemente una estromelisina endógena y, más preferiblemente MMP-3. El uso terapéutico puede emplearse para la trombólisis, o para evitar la progresión y para facilitar de la regresión de las placas ateroescleróticas. Por tanto, el método puede llevarse a cabo para el tratamiento de dosis única o de urgencia o para el tratamiento de mantenimiento o prolongado para reducir la posibilidad o para inhibir el desarrollo de placas ateroescleróticas, embolia o trombos anómalos. Los modos de administración de tal composición se conocen en la técnica y están relacionados con aquellas técnicas empleadas en la administración de agentes trombolíticos convencionales. Tales métodos incluyen, sin limitación, métodos parenterales, preferiblemente métodos intravasculares, tales como la inyección intravenosa, inyección intraarterial y administración mediante catéter. La determinación de la cantidad eficaz de una composición de la invención depende del criterio del médico experto. Las dosis terapéuticas o profilácticas específicas y las horas de administración pueden seleccionarse dependiendo de las condiciones imperantes para lograr el tratamiento clínicamente aceptable. El médico experto tendrá en cuenta factores tales como la edad, el sexo, el peso y el estado del sujeto, así como la vía de administración. El médico experto también reconocerá que la actividad fibrinolítica de las MMP puede intensificarse mediante la administración colateral (es decir, la coadministración o la administración secuencial) de otros principios activos y/o inertes. Por ejemplo, puede ser deseable administrar agentes complementarios que tengan actividad trombolítica. Estos agentes pueden tener actividad fibrinolítica directa o pueden ser reguladores o moduladores de la fibrinólisis en el sistema en el que se emplean. Por ejemplo, agentes que tienen actividad trombolítica incluyen activadores del plasminógeno, hirudina, enzimas (por ejemplo, proteasas derivadas del veneno de serpiente), inhibidores enzimáticos, anticoagulantes (por ejemplo, heparina, aspirina), anticuerpos (preferiblemente anticuerpos monoclonales) o péptidos sintéticos específicos para el receptor plaquetario gpIIb/IIIa, o una combinación de los mismos. Varios de tales agentes trombolíticos se describen en Collen (1996). Estos agentes pueden administrarse junto con, o de manera auxiliar a la administración de una composición que contenga MMP. Por tanto, tal otro agente o agentes pueden incluirse en la composición que contiene metaloproteinasa o pueden administrarse como parte de otra composición. 45 En otra realización, la invención incluye metaloproteinasas fibrinolíticas dirigidas, es decir, metaloproteinasas que se unen a restos que tienen especificidad por una molécula biológica diana. Por ejemplo, una metaloproteinasa puede unirse a un anticuerpo mediante métodos conocidos en la técnica para la unión de proteínas a anticuerpos. De esta forma, una metaloproteinasa puede dirigirse preferentemente a un sustrato de fibrina (fibrinógeno) para mejorar la eficacia de fibrin(ogen)olítica. Por tanto, una metaloproteinasa fibrinolítica tal como MMP-3 puede unirse a anticuerpos que tienen especificidad por fibrina o por un producto de degradación de la misma, a plaquetas, específicamente a la P-selectina, a lipoproteínas oxidadas, etc. 50 La invención también proporciona un método de diagnóstico para la caracterización del fibrinógeno. En este método, la fibrina/fibrinógeno se pone en contacto con una metaloproteinasa de matriz endógena, preferiblemente MMP-3, para producir productos de degradación. Los productos de degradación se analizan después para determinar los tipos y las cantidades de los productos de escisión generados por la actividad de la MMP. 40 55 60 65 Normalmente, el método supone la separación diferencial de los productos de degradación, tal como la separación de los productos mediante electroforesis en gel. Los productos se miden después tal como mediante tinción no específica para revelar las cantidades de productos de tamaños diferentes. Alternativamente, los productos pueden identificarse poniendo en contacto los productos con anticuerpos que son específicamente reactivos o que se asocian específicamente con uno o más dominios de la fibrina/fibrinógeno (Kudryk y col., 1989a). Preferiblemente, tales anticuerpos son específicamente reactivos con un único producto de degradación, permitiendo así la caracterización del producto en relación con otros productos. Nuevos anticuerpos, útiles según el método de diagnóstico de la invención, pueden desarrollarse y marcarse de manera que se puedan detectar con cualquier grupo marcador detectable. Tales grupos marcadores adecuados incluyen, por ejemplo, marcadores fluorescentes, marcadores enzimáticos y marcadores radiactivos. Los grupos detectores útiles según la invención incluyen, por ejemplo, fluoresceína como marcador fluorescente, peroxidasa de rábano picante como marcador enzimático y yodo-125 (125 I) como marcador radiactivo. Marcadores fluorescentes adicionales que pueden utilizarse en la invención incluyen, pero no se limitan a, rodamina, ficoeritrina y compuestos adicionales que emitan energía fluorescente. Marcadores enzimáticos adicionales que pueden utilizarse en esta invención incluyen, pero no se limitan a, glucosa oxidasa y fosfatasa alcalina. Marcadores radiactivos adicionales que pueden utilizarse en esta invención incluyen, pero no se limitan a, yodo-131 (131 I) e indio-111 (111 In). 8 ES 2 212 802 T3 5 10 15 20 25 30 Los marcadores detectables adecuados pueden seleccionarse de entre los conocidos en la técnica, tal como marcadores radiactivos, enzimas, componentes de pares de unión específica, sustancias colorantes coloidales, fluorocromos, sustancias reductoras, látex, digoxigenina, metales, materiales particulados, dansil-lisina, anticuerpos, proteína A, proteína G, materiales densos en electrones, cromóforos y similares. Puede emplearse eficazmente cualquier marcador adecuado, ya pueda detectarse directa o indirectamente. Un experto en la técnica reconocerá claramente que estos marcadores expuestos anteriormente son meramente ilustrativos de los diferentes marcadores que podrían utilizarse en el método de diagnóstico de la invención. Los anticuerpos reactivos con las subunidades del fibrinógeno también pueden derivatizarse mediante conjugación con biotina y usarse, con la adición de especies de avidinas que se han convertido en detectables mediante la conjugación con marcadores fluorescentes, marcadores enzimáticos, marcadores radiactivos, marcadores densos en electrones, etc., en una multiplicidad de aplicaciones inmunoquímicas e inmunohistológicas. Alternativamente, el método de la invención puede llevarse a cabo utilizando anticuerpos que se han ligado o unido a materiales sustrato según métodos conocidos por los expertos en la técnica. Tales materiales son en general sustancialmente sólidos y relativamente insolubles, confiriendo estabilidad a la disgregación física y química de los anticuerpos y permitiendo que los anticuerpos se dispongan en distribuciones espaciales específicas. Entre los materiales sustrato, los materiales pueden escogerse según los fines deseados por el experto e incluir materiales tales como geles, hidrogeles, resinas, perlas, nitrocelulosa, filtros de nylon, placas de microtítulo, matraces de cultivo, materiales poliméricos y similares, sin limitación. El método de la presente invención puede incluir ensayos inmunológicos para determinar la presencia de productos de ruptura de fibrina/fibrinógeno en muestras de tejidos procedentes de sujetos humanos o animales. Muestras de biopsia y autopsia de sujetos, así como muestras de librerías de tejidos o bancos de sangre, pueden evaluarse para determinar la presencia de fragmentos de ruptura mediante MMP de fibrina/fibrinógeno utilizando anticuerpos antifibrinógeno. Además, pueden idearse preparaciones adecuadas para su uso in vivo, tales como para la visualización de fibrinógeno o de sustancias y estructuras que contengan fibrinógeno en un sujeto vivo. De esta forma puede evaluarse in situ la progresión de la fibrinólisis inducida por las MMP. En una realización tal, una MMP fibrinolítica endógena, preferiblemente MMP-3, se une a un material sustrato tal como una membrana, tubo de recogida de sangre, placa de microtítulo, matraz de cultivo o similar. De esta forma, el método de la invención puede llevarse a cabo en ausencia de MMP soluble, para inducir fibrin(ogen)ólisis en una muestra de fluido corporal. Alternativamente, este enfoque es útil en membranas de recubrimiento y en dispositivos protésicos. 35 40 45 50 55 60 65 En efecto, en otra realización, la invención proporciona un método para controlar la formación de coágulos o placas producidos o inducidos por aparatos relacionados con la medicina. En esta realización, el método incluye poner en contacto un aparato relacionado con la medicina con una composición que incluya una metaloproteinasa de matriz fibrinolítica, preferiblemente MMP-3. Este método puede usarse para hacer que una metaloproteinasa se una o se adhiera a una superficie. Se cree que cualquier aparato que pueda entrar en contacto con la sangre puede modificarse así mediante métodos conocidos en la técnica, lo que permite la unión de proteínas a materiales sustrato. Por ejemplo, el método puede usarse para modificar las superficies de contacto con la sangre de dispositivos protésicos implantables, tales como cánulas, catéteres, injertos, stents, filtros, dispositivos intrauterinos y válvulas, para proporcionar superficies que inhiban la formación de trombos. Alternativamente, el método permite la modificación fibrinolítica de aparatos tales como tubos de recogida de sangre, matraces de cultivo, placas de prueba, pipetas, envases de reactivos, tubos y membranas, para estimular la fibrinólisis y para inhibir la formación de trombos que, en caso contrario, podrían interferir con los protocolos de experimentación. Asimismo, la invención proporciona aparatos relacionados con la medicina, tales como dispositivos implantables, material de laboratorio y otros dispositivos para usos in vivo e in vitro, que tienen la capacidad de inhibir la formación de trombos estimulando la degradación de fibrina/fibrinógeno. La metaloproteinasa puede adherirse a un aparato permanente o reversiblemente, de manera que se administre la metaloproteinasa de un aparato a una disolución. La invención también proporciona un método para intensificar la regulación de la fibrinólisis en un sujeto con necesidad de tal tratamiento. En esta realización, el método de la invención incluye inducir la intensificación de la regulación de una metaloproteinasa de matriz fibrinolítica endógena en un sujeto. Preferiblemente, el método incluye aumentar o disminuir la actividad o la expresión de una metaloproteinasa de matriz fibrinolítica endógena tratando al sujeto con tratamiento de transferencia génica de células somáticas. Puede emplearse cualquier enfoque de tratamiento génico según esta realización. Por tanto, puede llevarse a cabo la regulación por incremento de la expresión de MMP mediante la introducción de un gen para una MMP fibrinolítica mediante técnicas de trasferencia génica ex vivo o in vivo. Alternativamente, puede llevarse a cabo la regulación por incremento de una MMP mediante la inhibición de la expresión de un inhibidor de la MMP mediante tecnología antisentido. Puede llevarse a cabo la regulación por disminución de la actividad de la MMP mediante estas técnicas, cuyo diseño y puesta en práctica están dentro de las aptitudes de los expertos en la técnica. En Glick y col. (1994) se facilita una breve visión general de varios métodos de tratamiento génico. Pueden emplearse otros enfoques terapéuticos que incluyan composiciones farmacéuticas para estimular o inhibir la actividad o la expresión de una metaloproteinasa de matriz fibrinolítica endógena. Dada la complejidad del sistema de regulación fibrinolítico y dado el inesperado papel de las MMP en este esquema de regulación, a los expertos en la técnica les parecería que existen muchas posibilidades potenciales para el ajuste del estado de regulación fibrinolítica de un sujeto. 9 ES 2 212 802 T3 Los ejemplos siguientes están dirigidos a ayudar en una mayor comprensión de la invención. Los materiales y condiciones particulares empleados están dirigidos a ilustrar adicionalmente la invención y no son limitantes del alcance razonable de la misma. 5 10 15 En los ejemplos siguientes, se describen los experimentos que han revelado el papel que desempeñan las MMP, particularmente MMP-3, en la degradación del fibrinógeno (Fg) y la fibrina reticulada (XL-Fb). Se han estudiado los siguientes aspectos: 1) degradación del Fg; 2) efecto de la digestión del Fg mediante las MMP en su capacidad posterior de coagulación con trombina; 3) lisis de XL-Fb y Fragmento dímero D purificado (DD); 4) análisis en el extremo NH2 -terminal de los fragmentos de cadena de los digestos seleccionados; 5) reactividad de los fragmentos escindidos con varios anticuerpos monoclonales específicos. Se muestra la capacidad de MMP-3 para degradar el Fg y para lisar coágulos de XL-Fb. Basándose en este trabajo, se concluye que tanto la metaloproteinasa de matriz 1 (MMP-1) como la metaloproteinasa de matriz 2 (MMP-2) pueden degradar parcialmente el Fg y que MMP-2 tiene una capacidad limitada para degradar XL-Fb. También se determina que, en XL-Fb, el enlace γGly404-Ala405 es un sitio de escisión principal de MMP-3, que conduce a la formación de un monómero similar al Fragmento D. Esto indica un mecanismo específico de degradación de la fibrina, diferente del de la plasmina, mediante una enzima endógena. Los siguientes procedimientos experimentales son relevantes para los ejemplos 1-9, más adelante: Proteínas y otros reactivos 20 25 30 35 Se adquirió Fg sin plasminógeno y sin fibronectina (Fg ≥ 95% de capacidad de coagulación) o Fg humano liofilizado (American Diagnostica Inc., Greenwich, CT). Se eliminó el plasminógeno y la fibronectina mediante cromatografía de afinidad en lisina-Sepharosa y gelatina-Sepharosa, esencialmente tal como describen otros autores (Deutsch y col., 1970; Engvall y col., 1977; Procyk y col., 1985). La cantidad del Factor XIII en estas preparaciones es de 0,1-0,2 unidades Loewy/mg de Fg según el fabricante. Las disoluciones madre de Fg (12 mg/mL en tampón TNE (Tris-HCl 0,05 M (pH 7,4)), que contenía NaCl 0,1 M, EDTA 0,001 M y aprotinina 100 KIU/mL)) se almacenaron a -70ºC hasta su utilización. La concentración de Fg se midió espectrofotométricamente en disolución alcalina-urea utilizando el coeficiente de extinción (1%, 1 cm) = 16,5 a 282 nm. El Glu-plasminógeno humano (1 U/0,5 mg) procedía de Imco (Stockholm, Suecia). La estreptocinasa (450 U/mg sólido), la albúmina sérica bovina (BSA, Fracción V, grado RIA), el acetato 4-aminofenilmercúrico (APMA) y EDTA procedían de Sigma Chemical Company (St. Louis, MO). La aprotinina procedía de Mobay Chemical Corp (Nueva York, NY). La α-trombina humana (2300 U/mg) fue un generoso obsequio del Dr. J. Fenton. El 125 I -Fg, marcado con el método del yodógeno (actividad específica 1,5 x 106 cpm/µg proteína) fue un generoso obsequio de los Drs. M. Nag y D. Banerjee, Laboratory of Membrane Biochemistry II, The New York Blood Center. Pro-MMP-1, pro-MMP-2 y pro-MMP-3 se purificaron tal como han descrito anteriormente otros autores (Okada y col., 1986; Okada y col., 1990; Suzuki y col., 1990). El resto de los reactivos fueron de calidad analítica y se adquirieron en Fisher Scientific (Springfield, NJ). Electroforesis en gel/inmunotransferencia 40 45 50 55 Muestras de Fg y XL-Fb degradadas con plasmina o MMP se sometieron a SDS-PAGE usando condiciones reductoras y no reductoras. Las muestras reducidas se prepararon en tampón Tris 62,5 mM, pH 6,8, que contenía SDS al 4%, urea 8 M, DTT al 5%, glicerol al 10% y azul de bromofenol al 1%. Las muestras no reducidas se prepararon en el mismo tampón sin DTT. SDS-PAGE se realizó utilizando un gradiente del 5-15% o geles de poliacrilamida al 12,5% en tampón Tris-glicina (Laemmli 1970) o con minigeles al 5% y al 7,5% en tampón fosfato (McDonagh y col., 1972) siguiendo procedimientos generales. Los patrones de peso molecular previamente teñidos utilizados fueron miosina (200 kDa), fosforilasa B (97,4 kDa), BSA (68 kDa), ovoalbúmina (43 kDa), α-quimiotripsinógeno (25,7 kDa), β-lactoglobulina (18,4 kDa) y lisozima (14,3 kDa) (Bethesda Research Laboratories, Gaithersburg, MD). La transferencia a membranas de nitrocelulosa para los análisis de inmunotransferencia se realizó tal como describen Towbin y col. (1979) con pocas modificaciones (Kudryk y col., 1989b). En algunos experimentos, las membranas se tiñeron con oro coloidal antes de la inmunotransferencia (Colloidal Gold Total Protein Stain, BioRad, Hercules, CA). Las membranas se bloquearon con leche en polvo al 5% (Carnation, Nestle, Glendale, CA) o con BSA al 5%, se incubaron durante toda la noche con un anticuerpo primario seleccionado (tabla I) y después se sondaron con un segundo anticuerpo. La peroxidasa de rábano picante anti-ratón de conejo (RAM-HRPO) se preparó tal como describe Goding (1986) usando RAM adquirido en Dako (Carpinteria, CA) y HRPO (tipo VI) de Sigma. Los complejos de peroxidasa unida se detectaron utilizando sustrato quimioluminiscente Luminol (sistema de detección de Western blotting ECL, Amersham Life Science, Arlington Heights, IL). Se expuso la película facilitada (Kodak χ-Omat RP, Eastman Kodak Company, Rochester, NY) a la luz emitida a partir de la hidrólisis del sustrato Luminol añadido en de 10 a 30 segundos. Ejemplo 1 60 Degradación de fibrinógeno 65 Se realizó un experimento para evaluar si las MMP tienen actividad fibrin(ogen)olítica. Se incubó fibrinógeno (Fg) (120 µg, 3,5 µM) con MMP-2 o MMP-3 (6 µg/mL o razón E:S de 1:20) a 37ºC durante diferentes intervalos de tiempo. Se activaron pro-MMP-2 y pro-MMP3 (en Tris-HCl 50 mM, pH 7,5, NaCl 0,15 M, Brij 35 al 0,05%, NaN3 al 0,05%) con APMA 1 mM a 37ºC durante 45 min y 24 h, respectivamente, antes de la adición de las disoluciones de Fg. Todas las reacciones se realizaron en presencia de CaCl2 10 mM a 37ºC. Las digestiones se terminaron mediante la adición de EDTA (concentración final de 25 mM). Los productos de reacción se mezclaron con tampón reductor o no reductor 10 ES 2 212 802 T3 y se sometieron a separación en SDS-PAGE (12,5%). Los geles se tiñeron para la determinación de proteínas con azul de Coomassie. 5 La figura 1 muestra la digestión dependiente del tiempo del fibrinógeno mediante MMP-2 y NEAP-3 por comparación con el fibrinógeno no digerido. Las enzimas se usaron a E:S = 1:20 (p/p) y la incubación fue durante 1, 2, 4 y 24 h. La leyenda para la figura 1 es la siguiente: 10 15 20 25 30 35 A partir de la figura 1 está claro que, usando MMP-2 y MMP-3 en cantidades comparables (6 µg/120 µg Fg), tanto las cadenas Aα como las Bβ del Fg se degradaron ampliamente en 1 h (bandas 2 y 6). Una incubación más larga (24 h) dio como resultado una escisión adicional de ambas cadenas (bandas 5 y 9). La degradación de las cadenas γ del Fg con MMP-3 fue amplia a las 24 h (figura 8, banda 9) y diferente de la producida con MMP-2 (banda 5). También se obtuvo degradación significativa con MMP-2 y MMP-3 a una concentración inferior de la enzima (0,2-0,6 µg/120 µg Fg, datos no mostrados). También se probó MMP-1 usando este protocolo y, en la concentración superior (6 µg/120 µg Fg), mostró cadenas Bβ y γ aparentemente intactas y sólo degradación parcial de las cadenas Aα (datos no mostrados). Ejemplo 2 40 45 50 Capacidad de coagulación del fibrinógeno degradado con MMP-3 Los digestos (1, 5 y 3 h) del fibrinógeno con MMP-2 y MMP-3 se prepararon tal como se describió anteriormente. Se usó un digesto del Fg obtenido con plasmina (18-20 h) (Gårdlund y col., 1972) como control. El Fg intacto y los digestos del Fg se hicieron coagular con trombina, tal como describen Bini y col., (1994). En resumen, 1,2 mg Fg/mL o cualquiera de los digestos anteriores del fibrinógeno obtenidos mediante MMP-2 y MMP-3, se hicieron coagular con trombina (0,4 U NIH/mL) en presencia de CaCl2 20 mM. El tiempo de coagulación se determinó como un aumento en la turbidez y se leyó a 350 nm (Blombäck y col., 1982). La capacidad de coagulación se determinó a partir de la representación de la turbidez frente al tiempo. Se trazó una tangente en la parte de más pendiente de la curva; su intersección con el eje del tiempo se define como tiempo de coagulación (Blombäck y col., 1994). En todos los casos, se formaron los geles sin que se observara ninguna precipitación. Los sobrenadantes de coágulos se corrieron en HPLC (Kudryk y col., 1989b) con el fin de determinar la liberación de los fibrinopéptidos A (FPA) y B (FPB). Estos resultados se muestran en la tabla I, a continuación. 55 60 65 11 ES 2 212 802 T3 TABLA I 5 10 15 20 25 30 35 40 45 50 55 60 65 El Fg digerido con MMP-2 (1,5 y 3 h) no coaguló en el plazo de 10 min (tomado arbitrariamente como el tiempo máximo), pero todavía pudo formar un gel de fibrina tras la incubación durante toda la noche con trombina, tal como se muestra por los datos de turbidez (tabla I). Por el contrario, tanto los digestos producidos por MMP-3 como por plasmina, en tiempo y concentración comparables, no tuvieron capacidad de coagulación, ni siquiera tras la incubación durante toda la noche. Los datos de turbidez indicaron que la formación del gel se obtuvo sólo a partir de las mezclas de reacción del Fg digerido con MMP-2 y con la concentración de plasmina inferior. Además, los valores de turbidez de las mismas preparaciones medidos al día siguiente mostraron que las mezclas de reacción de Fg y MMP-2 fueron similares al control, mientras que los digestos del Fg generados por MMP-3 o plasmina tuvieron turbidez baja o 12 ES 2 212 802 T3 ausencia de la misma (tabla I). Los perfiles de HPLC de los sobrenadantes de todos los digestos mostraron liberación normal de los fibrinopéptidos A y B con trombina (no mostrados). Ejemplo 3 5 Degradación de la fibrina reticulada 10 15 20 Se obtuvieron coágulos de fibrina a partir de fibrinógeno purificado según el método de Bini y col. (1994) y de las referencias citadas en el mismo. Se obtuvieron coágulos radiomarcados con 0,1 mL de Fg purificado (1,2 mg/mL en tampón TNE) que contenía 125 I-Fg (20.000 cpm) en presencia de CaCl2 20 mM. Se añadió trombina (1,5 U NIH/mL, concentración final) y las muestras se incubaron a 37ºC durante 18-20 h (Bini y col., 1994). Se añadió MMP-1, MMP-2 o MMP-3 activas en diferentes cantidades (2-60 µg/mL, que corresponde a la razón E:S de 1:600-1:20) en presencia de CaCl2 10 mM. Se generó plasmina añadiendo plasminógeno (50 µg/mL) y estreptocinasa (1080 U/mL) a los coágulos de fibrina (aproximadamente 0,02-0,5 U/mL de plasmina, concentración final, que corresponde a E:S de 1:1200-1:48). Los coágulos se separaron suavemente de la pared del tubo de ensayo con una varilla de madera y el contenido se sometió a vórtex suavemente tras la adición de cada enzima. Los tiempos de incubación fueron de 1-48 h a 37ºC. Los digestos con MMP se terminaron mediante la adición de EDTA (concentración final de 25 mM) y los de plasmina se terminaron con aprotinina 5.000 KIU/mL. Los coágulos se separaron de los sobrenadantes mediante centrifugación a 13.000 rpm durante 20 min en una Sorval RCL-B (rotor SS34). La fibrinólisis se midió tanto mediante la liberación de radiactividad en el sobrenadante (A) como mediante la radiactividad residual en cada coágulo (B). Las muestras se contabilizaron en un Contador Gamma Automático Packard Serie γ-5000. Los coágulos de fibrina control, con y sin digestión con las diferentes enzimas, se obtuvieron al mismo tiempo para utilizarse para SDS-PAGE. Los datos representan los valores medios de 2-4 experimentos separados. 25 30 35 40 Los resultados de este experimento se presentan en las figuras 2A y 2B, que muestran el porcentaje de radiactividad en los sobrenadantes (figura 2A) y en los coágulos residuales (figura 2B). Tal como se muestra, tras 24 horas de incubación, tanto las muestras de MMP-3 (6µg/120 µg Fg) como de plasmina (0,02 IU/mL) mostraron liberación de más del 80% de la radiactividad en los sobrenadantes (media de tres experimentos) (figura 2A). La radiactividad residual en los coágulos a las 24 horas fue inferior al 10% (figura 2B) y los coágulos parecieron disolverse tanto mediante MMP-3 como mediante plasmina. Véanse las figuras 2A y 2B. La concentración de plasmina utilizada en los experimentos de lisis se escogió partiendo de la base de la obtención de una lisis lenta (Liu y col., 1986). En los experimentos preliminares, se midió una liberación similar de radiactividad en los sobrenadantes usando plasmina a 1:240 y 1:1200 (E:S, p/p), lo que se usó durante todo el estudio. La degradación con MMP-2 en la concentración superior a 24 h, fue similar a la obtenida con MMP-3 a 1:200. La degradación con MMP-1 a la concentración superior, tras 48 h, fue similar a la obtenida con MMP-3 a 1:1200. También se muestran los controles, sin adición de enzima, así como un digesto de XL-Fb obtenido con plasmina. La incubación con una mezcla de MMP-3 y plasmina en la muestra produjo resultados similares a los producidos por cualquiera de las enzimas solas (datos no mostrados). Esto indica que las dos enzimas no interfieren entre sí. Se realizaron experimentos idénticos usando coágulos obtenidos a partir del plasma y se obtuvieron resultados similares (datos no mostrados). Ejemplo 4 45 Composición de cadena de la XL-Fb degradada mediante MMP Y plasmina 50 55 Se analizó la digestión de la fibrina reticulada mediante plasmina, MMP-2 y MMP-3. Las muestras de la degradación de fibrina mediante cada una de las enzimas se redujeron y se sometieron a SDS-PAGE (gradiente del 5-15%) (Laemmli y col., 1973; McDonagh y col., 1972). Tras la electroforesis, las proteínas resueltas se transfirieron a nitrocelulosa. La proteína se tiñó con oro coloidal en la membrana de nitrocelulosa. Los patrones de la degradación de fibrinógeno y fibrina dependiente de la dosis y dependiente del tiempo mediante MMP-2, MMP-3 y plasmina se muestran en la figura 3. La leyenda de la figura 3 es la siguiente: 60 65 13 ES 2 212 802 T3 5 10 15 20 25 30 35 La figura 3 muestra la degradación mediante plasmina de la cadena dimérica γ de la fibrina reticulada (94 kDa) en la cadena dimérica γ DD (76 kDa) en la razón E:S superior (bandas 1 y 2). No se observó degradación del dímero γ de la fibrina reticulada con MMP-2, en la razón E:S superior (1:20) (banda 4). Se obtuvo degradación significativa de la cadena γ con MMP-3 (E:S = 1:20) a las 24 h (banda 7) y se obtuvo la degradación completa aumentando dos veces la razón E:S (banda 8). El patrón de degradación de la cadena dimérica γ de la fibrina reticulada mediante MMF-3 es diferente del obtenido con la plasmina. De hecho, la plasmina disminuye el peso molecular del dímero γ, pero no lo convierte en monómero (bandas 1, 2) ni siquiera a la razón de E:S superior (1:240). Los coágulos de XL-Fb se degradaron gradualmente mediante MMP-3 y dieron como resultado una lisis casi completa a las 24 h. La cantidad de degradación con MMP-3 (1:20) a las 24 h fue comparable a la producida por plasmina (1:1200). Por tanto, MMP-3 es una enzima fibrinolítica más lenta que la plasmina. Las tasas de solubilización del coágulo con MMP-1 y MMP-2 fueron mucho más lentas: a la misma razón E:S (1:20), sólo se solubilizó el 34% y el 58%, respectivamente, tras 24 h, mientras que se degradó el 84% mediante MMP-3. En los digestos obtenidos con MMP-3 y plasmina, la radiactividad residual del coágulo fue ≤ 10%. Estos resultados indicaron que la digestión de XL-Fb con MMP-3 progresó adicionalmente, posiblemente con un mecanismo diferente y más específico que el obtenido con MMP-1 o MMP-2. Sin embargo, la actividad más débil de MMP-2 puede deberse a la rápida autólisis de la enzima tras la activación por APMA (Okada y col., 1990). Ejemplo 5 40 45 Escisión inducida por MMP-3 del dominio de reticulación de la cadena γ Se realizaron inmunotransferencias llevadas a cabo utilizando técnicas estándar con digestos de fibrina obtenidos con plasmina y metaloproteinasa, con un panel de anticuerpos monoclonales (MoAbs) (véase la tabla II) para identificar cómo se escinden las tres cadenas de fibrina mediante MMP-3, en comparación con la plasmina. Los anticuerpos monoclonales se prepararon según técnicas conocidas en la técnica. 50 (Tabla pasa a página siguiente) 55 60 65 14 ES 2 212 802 T3 TABLA II 5 10 15 20 25 30 Las muestras se incubaron con o sin enzima durante 24 ó 48 h. Las muestras incubadas se sometieron después a SDS-PAGE (geles al 7%) en condiciones reductoras. Tras la electroforesis, las muestras se transfirieron a membranas de nitrocelulosa y las membranas se sometieron a inmunotransferencia con anticuerpos seleccionados. Las cadenas de fibrina/fibrinógeno unidas a anticuerpos específicos se detectaron usando RAM-HRPO y el sustrato quimioluminiscente, tal como se describió anteriormente. Los resultados se ilustran en las figuras 4-5. 35 La figura 4A muestra las inmunotransferencias de los productos de degradación de la digestión de fibrina mediante MMP-3 y plasmina con MoAb/4-2. Este anticuerpo es específico para un epítopo en la cadena γ: γ392-406. Este anticuerpo reacciona con fibrinógeno y los fragmentos D y dímero D sólo tras la desnaturalización. 40 La fibrina reticulada y el fibrinógeno se incubaron cada uno sin enzima a 37ºC, durante 24 h. Además, XL-Fb se incubó con plasmina (24 h) y con MMP-3 (24 h y 48 h). Los digestos se examinaron en condiciones reductoras. La leyenda para la figura 4A es la siguiente: 45 50 55 60 65 Tal como se observa en la figura 4A, MoAb/4-2 (anti-γ392-406) reaccionó tanto con el dímero γ de XL-Fb no digerido (94 kDa, banda 1) como con la cadena γ de Fg no digerida (47 kDa, banda 2). El monómero residual de la cadena γ en la preparación de fibrina reticulada también reaccionó con MoAb/4-2 (banda 1). En el digesto obtenido mediante plasmina de XL-Fb, el dímero γ DD (76 kDa) y el monómero de la cadena γ Fragmento D (que se sabe que resulta de los digestos obtenidos mediante plasmina de Fg y XL-Fb (Siebenlist y col., 1992)) también reaccionaron igualmente con MoAb/4-2 (banda 3). Un digesto a las 24 h de XL-Fb generado por MMP-3 mostró una cadena de dímero γ DD reducida en el coágulo residual, pero cantidades significativas de la cadena γ similar al monómero Fragmento D (36 kDa, banda 4). Una incubación más larga (48 h) sólo mostró la cadena γ similar al monómero 15 ES 2 212 802 T3 Fragmento D en el digesto total. Los digestos obtenidos con ambas enzimas se unieron a MoAb/4-2. 5 También se examinó la fibrina reticulada mediante inmunotransferencia con MoAb/4A5, que es específico para la cadena γ en γ397-411, un epítopo cercano al que es reactivo con MoAb/4-2. Como punto de referencia, el coágulo de XL-Fb se incubó sin enzima a 37ºC durante 48 h. XL-Fb también se incubó con MMP-3 o plasmina durante 24 h. Los resultados de esta inmunotransferencia se muestran en la figura 4B. La leyenda para la figura 4B es la siguiente: 10 15 20 25 30 35 40 45 50 55 Por tanto, tal como se observa en la figura 4B, MoAb/4A5 (anti-γ397-411) mostró reactividad sólo con la banda del dímero γ tanto a partir de XL-Fb intacto como digerido con plasmina (bandas 1, 5) y con el dímero γ DD residual obtenido a partir de los digestos generados por MMP-3 (bandas 2, 4). La cadena γ similar al monómero Fragmento D (36 kDa), completamente reactivo con MoAb/4-2 (figura 4A, bandas 4, 5), no se unió a MoAb/4A5 (figura 4B, bandas 2, 4). El análisis de inmunotransferencia de las mismas muestras en condiciones no reductoras mostró una pérdida similar de inmunorreactividad con MoAb/4A5 (datos no mostrados). Estos experimentos muestran que el patrón de degradación de XL-Fb con MMP-3 es diferente del obtenido con plasmina. En los digestos obtenidos con MMP-3 (1:20), sólo quedan cantidades muy pequeñas del dímero γ. A niveles superiores de esta misma enzima, no pueden detectarse dímeros. MMP-2 no parece afectar a la degradación del dímero γ significativamente. Se usaron dos anticuerpos monoclonales, MoAb/4-2 y MoAb/4A5, reactivos con diferentes epítopos en la secuencia γ392-411, para definir las regiones de escisión de XL-Fb mediante MMP-3, en comparación con plasmina. Este segmento de la cadena contiene residuos (γGln398 y γLys406) que participan en las reticulaciones covalentes (formando enlaces isopeptídicos ε-(γGlu)-Lys) en las moléculas vecinas, lo que conduce a la estabilización de la fibrina mediada por el Factor XIIIa (Chen y col., 1971). MoAb/4A5 reconoce un epítopo en la región COOHterminal de este péptido (γ397-411), mientras que MoAb/4-2 reacciona con su extremo NH2 -terminal (γ392-406). Ambos anticuerpos se unen a placas de microtítulo recubiertas con productos de digestión derivados de la plasmina, los Fragmentos D y DD. Sólo MoAb/4A5 compite con tales fragmentos mientras están cada uno en disolución (Kudryk y col., 1991). En una digestión de 24 h de XL-Fb con MMP-3, tanto la cadena de dímero γ DD residual como la cadena γ similar al monómero Fragmento D fueron reactivos con MoAb/4-2. Digestiones más largas (48 h) dieron como resultado únicamente la cadena γ similar al monómero Fragmento D, que todavía fue reactiva con MoAb/4-2. El análisis de estos mismos digestos con MoAb/4A5 (anti-γ397-411) mostró que sólo la banda del dímero γ DD era reactiva. La cadena γ similar al monómero Fragmento D no se unió a MoAb/4A5. Estos resultados indicaron que había un sitio de escisión principal para MMP-3 dentro del dominio de reticulación de la cadena γ, dando como resultado la degradación del dímero γ. El Fragmento DD purificado también se escindió con MMP-3 hasta obtener un fragmento similar al monómero D. También se realizó la inmunotransferencia de los productos de degradación de la digestión de fibrina mediante MMP-3 y plasmina utilizando MoAb/T2G1 y MoAb/1D4 en condiciones reductoras, tal como se muestra en la figura 5. El protocolo de experimentación fue tal como se describió anteriormente para los otros anticuerpos. La leyenda para la figura 5 es la siguiente: 60 65 16 ES 2 212 802 T3 5 10 15 20 25 30 35 Tal como se indica en la figura 5, las bandas 1-3 se sometieron a inmunotrasferencia con MoAb/T2G1, mientras que las bandas 4-7 se sometieron a inmunotransferencia con MoAb/1D4. MoAb/T2G1 es específico para Bβ15-42, pero sólo de fibrina II, no de fibrinógeno/fibrina I. MoAb/1D4 es específico para Aα349-406 en fibrinógeno y fibrina, así como en sus digestos por plasmina. Tal como se muestra en la figura 5, la inmunorreactividad de MoAb/T2G1 se perdió en los digestos de fibrina obtenidos mediante MMP-3, tanto en la concentración inferior (1:200) como en la superior (1:20) (bandas 4, 5), mientras que está presente en la fibrina intacta (banda 6). La inmunorreactividad de MoAb/1D4 todavía se conserva parcialmente en el digesto de fibrina en la concentración inferior de MMP-3 (banda 1), pero se pierde completamente en los digestos en la concentración superior de MMP-3, tanto a las 24 h (banda 2) como a las 48 horas (no mostrado), mientras que está claramente presente en la muestra control (banda 3). En consecuencia, los productos de degradación de XL-Fb obtenidos mediante MMP-3 se sondaron con MoAbs, T2G1 (anti- Bβ15-42) y 1D4 (anti-Aα349-406), respectivamente. Ambos epítopos están presentes en XL-Fb. En los digestos de XL-Fb obtenidos mediante plasmina, muchas bandas de tamaños diferentes (≥20 kDa) reaccionan con MoAb/1D4, mientras que se pierde toda la reactividad de MoAb/T2G1. Incluso concentraciones relativamente bajas de MMP-3 (1:200) dieron como resultado digestos que no reaccionaron con estos anticuerpos en la inmunotransferencia. Este resultado significa que Aα349-406 se ha escindido de la fibrina mediante MMP-3. No pudo detectarse inmunorreactividad con MoAb/1D4 en los digestos de XL-Fb obtenidos con MMP-3 mediante ELISA de competencia. Este anticuerpo reacciona de manera idéntica con Aα349-406 y con Hi2-DSK (Aα241-476) antes y después de la digestión completa con tripsina. Ejemplo 6 40 45 Digestión del fragmento dímero D (DD) mediante MMP-3 Se llevaron a cabo experimentos para comprobar si MMP-3 escindiría el dímero D purificado (DD) convirtiéndolo en el monómero D. El fragmento D purificado y el dímero D se incubaron a 37ºC durante 4 y 24 h con la concentración inferior (1:20) y superior (1:200) de MMP-3. Las reacciones se terminaron con EDTA 25 mM. Los digestos se resolvieron en PAGE (fosfato al 7%). Las muestras se transfirieron a membranas de nitrocelulosa y se tiñeron con azul de Coomassie. Los resultados se muestran en la figura 6. Las muestras se corrieron en condiciones no reductoras (figura 6A) y reductoras (figura 6B). La leyenda para las figuras 6A y 6B es la siguiente: 50 55 60 65 Tal como se muestra en las figuras 6A y 6B, la degradación progresiva (dependiente del tiempo) del dímero D (186 kDa) da lugar a un fragmento similar al monómero D (bandas 4,5). El tamaño del monómero resultante fue 17 ES 2 212 802 T3 5 ligeramente mayor que el del fragmento D (93 kDa), obtenido durante la degradación con plasmina del Fg (banda 1). Además, dado que este gel se obtuvo 2 meses después de la preparación de estas muestras, esto prueba que la reacción se ha detenido eficazmente y que ya no ha progresado más. Los resultados indican que la monomerización se produce incluso a las 4 horas con la cantidad inferior de enzima (1:200) (banda 3). El incremento de la cantidad de enzima diez veces aumenta la monomerización a las 4 horas (banda 4) y convierte casi completamente el sustrato a las 24 horas (banda 5). Esto se apoya en la figura 6B, que muestra la conversión del dímero γ (78 kDa) en la cadena γ similar al monómero (38 kDa). Ejemplo 7 10 Análisis de productos de la digestión mediante MMP por ELISA 15 20 25 30 Se llevaron a cabo ensayos de unión ELISA utilizando métodos generalmente aceptados. Se cubrieron placas de microtítulo de polivinilo con diluciones apropiadas de diferentes digestos y se sometieron a ensayo para determinar la unión a anticuerpos (Kudryk y col., 1984). De acuerdo con los resultados mostrados en la figura 4, los digestos con concentraciones crecientes de MMP-3 no se unieron a MoAb/4A5 (anti-γ397-411). Esto significa que el sitio de escisión de MMP-3 para el fibrinógeno está en la región de este epítopo (la reticulación de la fibrina se produce en la región γGly397-Lys406). Este tipo de escisión no se produce con plasmina. Sin embargo, se encontró que los digestos eran reactivos con MoAb/4-2 (γ349-406). No pudo detectarse inmunorreactividad con MoAb/1D4 en los digestos de XL-Fb obtenidos con MMP mediante ELISA de competencia, lo que sugiere que el epítopo reactivo de 1D4 se destruye en tales digestos. MoAb/1D4 se une por igual a los digestos de Fg y XL-Fb obtenidos mediante plasmina. Por tanto, el ELISA (unión directa) realizado en los digestos de Fg y XL-Fb con MMP-3 demostró que el epítopo γ392-406 de secuencia lineal (reactivo con MoAb/4-2), pero no γ397-411 (reactivo con MoAb/4A5), podía detectarse mediante este método. Esto confirmó los resultados obtenidos con inmunotransferencia y además sugirió que MMP3 escinde dentro de la secuencia γ397-411. El análisis de la secuencia NH2 -terminal del dipéptido (aislado de la degradación mediante CNBr de XL-fibrina digerida con MMP-3) indicó que γAla405 es el primer residuo de la segunda secuencia (véanse los ejemplos 8-9, a continuación). Se observa que MoAb/4-2 también se une a un péptido sintético cuya secuencia corresponde a γ392-400. MoAb/4A5 reacciona con péptidos sintéticos que corresponden a γ392-411 y γ397-411, que se pierde cuando el péptido se escinde con tripsina en γLys406-Gln407. Esta característica concuerda con la falta de reactividad observada de MoAb/4A5 observada con el dipéptido. Ejemplo 8 35 40 45 Análisis del tamaño y la secuencia de las cadenas de Fg y XL-Fb digeridas con MMP-3 El Fg y la XL-Fb digeridos con MMP-3 se separaron en condiciones reductoras en un SDS-PAGE al 12,5% (Laemmil, 1970) y se sometieron a electroinmunotransferencia hasta una membrana de poli(difluoruro de vinilideno) (PVDF) (Matsudaira, 1987). La parte de la membrana que contenía el fragmento de la cadena γ se escindió y se sometió a secuenciación automatizada en un secuenciador en fase líquida mediante impulsos modelo 447A de Applied Biosystems Inc con un analizador de aminoácidos de feniltiohidantoína (PTH) on line modelo 120A. Los digestos de Fg y XL-Fb dieron una secuencia Leu-Lys-Ser-Arg-Lys (SEQ ID NO:1) de la cadena γ, lo que indica que MMP-3 escinde tanto Fg como XL-Fb en el enlace γThr83-Leu84. Sin embargo, esta misma banda del digesto de XL-Fb también dio una segunda secuencia Ala-X-Gln-Ala-Gly-Asp (SEQ ID NO:2) de la cadena γ, lo que indica que MMP-3 indujo hidrólisis en el enlace γGly404-Ala405, en la región de reticulación de la cadena γ. Ejemplo 9 50 55 60 Caracterización de un fragmento de la cadena γ derivado por CNBr de los digestos de XL-Fb obtenidos mediante MMP-3 Para confirmar los resultados obtenidos de la secuenciación de la cadena γ de XL-Fb degradada con MMP-3, se trató el mismo digesto con CNBr (Blomback y col., 1968) y se aisló el fragmento reactivo con MoAb/4-2 (anti-γ392406), tal como sigue. Los fragmentos escindidos por MMP-3 se purificaron mediante HPLC en fase inversa usando una columna Vydac C-4 (1,0 x 25 cm, The Sep/a/ra/tions Group, Hesperia, CA). La columna se desarrolló a temperatura ambiente usando el siguiente gradiente construido con ácido trifluoroacético al 0,05% (TFA, disolvente A) y acetonitrilo al 50% en A (disolvente B): con B al 5%/0,5 min; con B a del 5 al 50%/50 min; con B a del 50 al 100%/7 min; con B al 100%/75 min. La velocidad de flujo de la columna fue de 1,0 mL/min y se monitorizaron las fracciones (1 mL) para determinar la reactividad con MoAb/4-2 (anti-γ392-406). La fracción reactiva con el anticuerpo se purificó adicionalmente mediante FPLC utilizando la columna Superdex Peptide HR 10/30 (1,0 x 30-31 cm, Pharmacia Biotech, Piscataway, NJ). La columna se desarrolló a temperatura ambiente utilizando tampón fosfato 20 mM (pH 7,2), que contenía adicionalmente NaCl 0,25 M. La fracción que reaccionó con MoAb/4-2 se reunió, se desalinizó haciéndola pasar sobre Sep-Pak® (Millipore Corp., Milford, MA) y se sometió a análisis de la secuencia. 65 El fragmento secuenciado correspondió a un dipéptido reticulado con dos extremos NH2 -terminales distintos: γLys385, que resulta de la escisión con CNBr, y γAla405 (figura 7). Las dos secuencias se recuperaron en cantidades 18 ES 2 212 802 T3 molares comparables. Los análisis de extremo NH2 -terminal en el ciclo 3 no mostraron residuos de PTH (debido a un error de la máquina), pero la evidencia de Gln e Ile en el ciclo 3 se observó en el ciclo 4 como intervalo. Estos datos apoyan la conclusión de que MMP-3 escinde en γGly404-Ala405 dentro de la región de reticulación de la cadena γ de fibrina. 5 10 La tabla III muestra la secuencia parcial para el dipéptido reticulado aislado de XL-Fb digerido con MMP-3 tras la degradación con CNBr. El dominio de reticulación γ, con los sitios de digestión de MMP-3 y de escisión por CNBr, se muestra esquemáticamente en la figura 7. El dipéptido resultante contiene un único enlace ε-(γGlu)-Lys), con un extremo NH2 -terminal como γLys385, que resulta de la escisión por CNBr en γMet384-Lys385. La presencia del segundo extremo NH2 -terminal, γAla405, apoyó la conclusión de que MMP-3 escinde en γGly404-Ala405 en la región de reticulación de la cadena γ. TABLA III 15 20 25 30 35 40 45 50 Los datos obtenidos del análisis de la secuencia de los digestos de Fg y XL-Fb obtenidos mediante MMP-3 mostraron la proximidad de los sitios de escisión por plasmina en estos mismos sustratos. La plasmina escinde en γLys62Ala63 y más lentamente en γLys85-Ser86 (Collen y col., 1975). Por tanto, el fibrinógeno digerido mediante plasmina da como resultado dos clases de Fragmento D, es decir, γAla63-Val411 y γSer86-Val411. Por el contrario, MMP-3 escinde tanto el Fg como la XL-Fb en γThr83-Leu84. Además, dado que MMP-3 hidroliza la unión peptídica γGly404Ala405, la cadena γ tiene un peso molecular que es sustancialmente similar al de la cadena γ del Fragmento D generado por plasmina. Sin embargo, en los digestos de XL-Fb obtenidos con MMP3, este producto no es un monómero real, dado que el dominio γ405-411 se reticula a la cadena γ adyacente que tiene la secuencia γLeu84-Gly404. Debe observarse que la recuperación de cantidades similares de las dos secuencias de las cadenas γ del dipéptido generado por CNBr indicó que, aunque lenta, la degradación de XL-Fb mediante MMP-3, dio como resultado la formación del fragmento similar al monómero D, que fue casi completa. Ejemplo 10 Caracterización de otros productos de escisión de Fg y XL-Fb mediante MMP-3 55 60 65 MMP-3 también genera otros productos de escisión de Fg y XL-Fb, cortando específicamente, no sólo la cadena γ, sino también las cadenas α y β. El Fg y la XL-Fb se sometieron a digestión usando MMP-3 (E:S = 1:20) durante 24 h, se separaron y se secuenciaron usando los métodos descritos en el ejemplo 9. Tal como se indica en la figura 8, tanto el Fg (banda 1) como la XL-Fb (banda 2) se escindieron específicamente. En la figura 8, se encontró que la banda 1 incluía dos fragmentos: un fragmento de la cadena γ que tenía una secuencia NH2 -terminal de Leu-Lys-SerArg-Lys (SEQ ID NO:1), que corresponde de nuevo a la escisión γ84; y un fragmento de la cadena β (β1 ) que tenía una secuencia NH2 -terminal de Lys-Arg-Gln-Lys-Gln (SEQ ID NO:3) que corresponde a la escisión en β127. Se encontró que la banda 2 contenía otro fragmento de la cadena β (β2 ) que tenía una secuencia NH2 -terminal de Tyr-Ser-Ser-GluLeu (SEQ ID NO:4) que corresponde a la escisión en β142. La banda 3 es la enzima MMP-3 que permanece en la preparación de digesto. Se encontró que la banda 4 contenía un fragmento de la cadena α que tenía una secuencia en el extremo NH2 -terminal de Asn-Arg-Asp-Asn-Thr (SEQ ID NO:5), que corresponde a la escisión en α103. (También se encontró que la banda 4 contenía fragmentos escindidos en γ1 y α414, tanto en los digestos de fibrinógeno como en los fibrina). La banda 5 contenía fragmentos escindidos en β51 y β52, tanto en los digestos de fibrina como en los de fibrinógeno, obtenidos con MMP-3. 19 ES 2 212 802 T3 5 10 En consecuencia, la evidencia presentada en los ejemplos 1-10 anteriores establece que el fibrinógeno se vuelve incapaz de coagulación con trombina cuando se trata con metaloproteinasa de matriz 3 (MMP-3, estromelisina 1), pero no cuando se trata con metaloproteinasa de matriz 2 (MMP-2, gelatinasa A). La incubación de coágulos de XL-Fb (realizada con 125 I-Fg) obtenidos con MMP-3 dio como resultado la lisis completa tras 24 h. Se genera un fragmento similar al monómero D mediante la degradación por MMP-3 de fibrinógeno, XL-Fb y Fragmento DD. La inmunorreactividad con el anticuerpo monoclonal MoAb/4-2 (anti-γ392-406), pero no con MoAb/4A5 (anti-γ397411), sugirió que un sitio principal de escisión estaba dentro de la secuencia que participa en la reticulación de dos cadenas γ. La secuencia NH2 -terminal de la cadena γ del fragmento similar al monómero D y de un dipéptido aislado del digesto de XL-fibrina obtenido mediante MMP-3, identificó la hidrólisis del enlace peptídico γGly404-Ala405. Estos datos indican que la degradación de Fg y XL-Fb mediante MMP-3 es específica y diferente de la de plasmina. Este mecanismo de fibrinólisis es importante en la cicatrización de heridas, la inflamación, la ateroesclerosis, el cáncer, la enfermedad renal y otros procesos patofisiológicos. 20 Puesto que MMP-3 escinde cerca del sitio en el que se produce la reticulación de la fibrina, se puede dar fácilmente un fuerte argumento teleológico de que MMP-3 desempeña un papel singular en la fibrinólisis. Por tanto, la regulación coordinada de las metaloproteinasas y los activadores del plasminógeno podrían dar como resultado efectos sinérgicos y la degradación completa de la matriz extracelular y de la red de fibrina que resulta de la cicatrización de heridas, la inflamación, la trombosis, el cáncer, la enfermedad renal y otros procesos patofisiológicos. En consecuencia, se considera que la coadministración de MMP-3 con otros agentes trombolíticos, tales como t-PA, también lograría un efecto terapéutico complementario, en el que cada componente complemente o potencie al otro. 25 Por tanto, aunque se han descrito las que en la actualidad se cree que son las realizaciones preferidas de la presente invención, los expertos en la técnica comprenderán que pueden realizarse realizaciones adicionales y otras sin apartarse del espíritu de la invención, y se pretende incluir todas estas modificaciones y cambios adicionales dentro del verdadero alcance de las reivindicaciones expuestas en el presente documento. 15 Bibliografía Bilezikian; S.B., y Nossel, H.L., Blood 50:21-28 (1977). 30 Bini, A., Fenoglio, J.J., Jr., Sobel, J., Owen, J., Fejgl, M. y Kaplan K.L., Blood 69:1038-1045 (1987). Bini, A., Fenoglio, J.J., Jr., Mesa-Tejada, R., Kudryk, B. y Kaplan, K.L., Arteriosclerosis 9:109-121 (1989). 35 Bini, A., Calender, S., Pocyk, R., Blombäck, B. y Kudryk, B.J., “Flow and antibody binding properties of hydrated fibrins prepared from plasma, platelet rich plasma and whole blood”, Thrombosis Res 76:145-56 (1994). Blombäck, B., Blombäck, M., Henschen, A., Hessel, B., Iwanaga, S. y Woods, K.R., Nature 218:130-134 (1968). 40 Blombäck, B. y Okada, M., Thromb Res 25:51-70 (1982). Blombäck, B., Carlsson, K., Fatah, K., Hessel, B. y Procyk, R., Thromb Res 75:521-538 (1994). 45 Brakman, P. y Kluft, C., eds., Plasminogen Activation in Fibrinolysis, in Tissue Remodeling, and in Development, Ann NY Acad Sci, vol. 667 (1992). Budzynski, AZ., “Interaction of hementin with fibrinogen and fibrin”, Blood Coagulation and Fibrinolysis 2:14952 (1991). 50 Campbell, E.J., Cury, J.D., Shapiro, S.D., Goldberg, G.I. y Welgus, H.G., J Immunol 146:1286-1293 (1991). Cawston, T., “Metalloproteinase inhibitors-Crystal gazing for a future therapy”, Br J Rheumatol 30:242-44 (1991). 55 Chen, R. y Doolittle, R.F., Biochemistry 10:4486-4491 (1971). Collen, D., Kudryk, B., Hessel, B. y Blombäck, B., J Biol Chem 250:5808-5817 (1975). Collen D., “On the regulation and control of fibrinolysis”, Thromb Haemost 43:77-89 (1980). 60 Collen D., “Biological properties of plasminogen activators”, Chapter 1 in Sobel, B.E., Collen, D. y Grossbard, E.B., eds., Tissue Plasminogen Activator in Thrombolytic Therapy, Marcel Dekker, Inc., New York (1987). Collen, D. y Lijnen, H.R., “Basic and clinical aspects of fibrinolysis and thrombolysis”, Blood 3114-24 (1991). 65 Collen, D., “Fibrin-selective thrombolytic therapy for acute myocardial infarction”, Circulation 93:857-865 (1996). Deutsch, D.G. y Mertz, E.T., Science 170:1095-1096 (1970). 20 ES 2 212 802 T3 Doolittle, R.F., “Fibrinogen and fibrin”, in Bloom, A.L. y Thomas, D.P., eds., Hemostasis and Thrombosis Churchill Livingston, Edinburgh, New York (1987). Engvall, E. y Ruoslahti, E., IntJCancer 201-5 (1977). 5 Francis, C.W. y Marder, V.J., “Physiologic regulation and pathologic disorders of fibrinolysis”, Chapter 54 in Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 3ª ed., Colman, R.W., Hirsh, J., Marder, V.J. y Salzman EW, eds., JB Lippincott Co, Philadelphia (1994). 10 Fu, Y. y Grieninger, G., “Fib420 : A normal human variant of fibrinogen with two extended a chains”, Proc Natl Acad Sci USA 91:2625-28 (1994). Gabriel, D.A., Muga, K. y Boothroyd, E.M., “The effect of fibrin structure on fibrinolysis”, J Biol Chem 267: 24259-63 (1992). 15 Galis, Z.S., Sukhova, G.K., Lark, M.W. y Libby, P., “Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques”, J Clin Invest 94:2493-2503 (1994). Galis, Z.S., Sukhova, G.K., Kranzhofer, R. y Libby, P., Proc Natl Acad Sci USA 92:402-406 (1995). 20 Gárdlund, B., Kowalska-Loth, B., Gröndahl, N.J, y Blombäck, B., Thromb Res 1:371 (1972). Glick, B.R. y Pasternak, J.J., Molecular Biotechnology: Principles and Applications of Recombinant DNA, Chapter 17, págs. 403-20 (1994). 25 Goding, J.W., in Monoclonal Antibodies: Principles and Practice, Academic Press, New York, NY (1986). Gramse, M., Bingenheimer, C. y Schmidt, W., J Clin Invest 61:1027-1033 (1978). 30 Guan, A.L., Retzios, D., Henderson, G.N. y Markland, F.S., Arch Biochem Biophys 289:197-207 (1991). Henney, A.M., Wakeley, P.R., Davies, M.J., Foster, K., Hembry, R., Murphy, G. y Humphries, S., “Localization of stromelysin gene expression in atherosclerotic plaques by in situ hybridization”, Proc Natl Acad Sci USA 88:8154-58 (1991). 35 Kleiner, D.E. Jr. y Stetler-Stevenson WG, “Structural biochemistry and activation of matrix metalloproteinases”, Curr Opin Cell Biol 5:891-97 (1993). Kudryk, B., Rohoza, A., Ahadi, M., Chin, J. y Wiebe, M.E., Mol Immuno 121:89-94 (1984). 40 Kudryk, B.J., Grossman, Z.D., McAfee, J.G. y Rosebrough, S.F., “Monoclonal antibodies as probes for fibrin (ogen) proteolysis”; Chapter 19 in Monoclonal Antibodies in Immunoscintigraphy, Chatal J-F, ed., CRC Press, Boca Raton (1989a). 45 Kudryk, B., Gidlund, M., Rohoza, A., Ahadi, M., Coiffe, D. y Weitz, J.I., Blood 74:1036-1044 (1989b). Kudryk, B., Rohoza, A., Ahadi, M., Gidlund, M., Procyk, R. y Matsueda, G.R., Thromb Haemostas 65:898 (Abstract 714) (1991). 50 Laemmli, V.K., Nature 227:680-685 (1970). Laemmli, U.K. y Favre, M., “Maturation of the head of bacteriophage T4. I. DNA packaging events”, J Mol Biol 80: 575-99 (1973). 55 Lee, S.W., Kahn, M.L. y Dichek, D.A., “Control of clot lysis by gene transfer”, Trends Cardiovasc Med 3:61-66 (1993). Liu, C.Y., Sobel, J.H., Weitz, J.I., Kaplan, K.L. y Nossel, H.L., Thromb Haemostas 56:100-106 (1986). 60 65 Loewy, A.G., Santer, U.V., Wieczorek, M., Blodgett, J.K., Jones, S.W. y Cheronis, J.C., “Purification and characterization of a novel zinc-proteinase from cultures of Aeromonas hydrophila”, J Biol Chem 268:9071-78 (1993). Martin, G.V., Kennedy, J.W. y Marder, V.J., “Thrombolytic therapy for coronary artery disease”, Chapter 73 in Hemostasis and Thrombosis: Basic Principles and Clinical Practice, 3ª ed., Colman, R.W., Hirsh, J., Marder, V.J. y Salzman, E.W., eds., JB Lippincott Co, Philadelphia (1994). Matrisian, L.M., “The matrix-degrading metalloproteinases”, BioEssays 14:455-63 (1992). 21 ES 2 212 802 T3 Matsudaira, P., J Biol Chem 262:10035-10038 (1987). 5 Matsueda, G.R. y Bernatowicz, M.S., (1988) in Fibrinogen 3-Biochemistry, Biological Functions, Gene Regulation and Expression (Mosesson MW, Amrani D, Siebenlist KR, DiOrio P Eds) pp 133-136 Elsevier Science Publishers BV, Amsterdam. McDonagh, J., Messel, H., McDonagh, R.P., Jr., Murano, G. y Blomback, B., “Molecular weight analysis of fibrinogen and fibrin chains by an improved sodium dodecyl sulfate gel electrophoresis method”, Biochim Biophys Acta 257: 13 5-42 (1972). 10 Murphy, G., Atkinson, S., Ward, R., Gavrilovic, J. y Reynolds, J.J., “The role of plasminogen activators in the regulation of connective tissue metalloproteinases”, Ann NY Acad Sci; 667:1-12 (1992). 15 Nagase, H., Enghild, J.J., Suzuki, K. y Salvesen, G., “Stepwise activation mechanisms of the precursor of matrix metalloproteinase 3 (stromelysin) by proteases and (4-aminophenyl)mercuric acetate”, Biochemistry 29:5783-89 (1990). Nagase, H., Ogota, Y., Suzuki, K., Enghild, J.J. y Salvesen, G., “Substrate specificities and activation mechanisms of matrix metalloproteinases”, Biochem Soc Trans 19:715-18 (1991). 20 Nagase, H., Barrett, A.J. y Woessner, J.F. Jr., “Nomenclature and glossary of the matrix metalloproteinases”, Matrix Supplement 1:421-24 (1992). 25 Okada, Y., Nagase, H. y Harris, E.D., Jr., “A metalloproteinase from human rheumatoid synovial fibroblasts that digests connective tissue matrix components”, J Biol Chem 261:14245-55 (1986). Okada, Y., Morodomi, T., Enghild, J.J., Suzuki, K., Nakanishi, I., Salvesen, G. y Nagase; H, Eur J Biochem 194:721-730 (1990). 30 Plow, E.F. y Edgington, T.S., J Clin Invest 56:30-38 (1975). Plow, E. F., Biochim Biophys Acta 630:47-56 (1980). Plow, E. F. y Edgington, T.S., Semin Thromb Haemostas 8:36-56 (1982). 35 Procyk, R., Adamson, L., Block, M. y Blombäck, B., Thromb Res 40:833-852 (1985). Procyk y col. (1991) Blood 77, 1469-1475. 40 45 Purves, L., Purves, M. y Brandt, W., “Cleavage of fibrin-derived D-dimer into monomers by endopeptidase from puff adder venom (Bitis arietans) acting at cross-linked sites of the γ-chain. Sequence of carboxy-terminal cyanogen bromide γ-chain fragments”, Biochemistry 26:4640-46 (1987). Retzios, A.D. y Markland, F.S. Jr, “Purification, characterization, and fibrinogen cleavage sites of three fibrinolytic enzymes from the venom of Crotalus basiliscus basiliscus”, Biochemistry 31:4547-57 (1992). Sanchez, E.F., Magalhes, A., Mandelbaum, F.R. y Diniz, C.R., “Purification and characterization of the hemorrhagic factor II from the venom of the Bushmaster snake (Lachesis muta muta)”, Biochim Biophys Acta 1074:347-56 (1991). 50 Senior, R.M., Griffin, G.L., Fliszar, J., Shapiro, S.D., Goldberg, G.I. y Welgus, H.G., J Biol Chem 266:78707875 (1991). Siebenlist, K.R. y Mosesson, M.W., Biochemistry 31:936-941 (1992). 55 Smith, E.B., Keen, G.A., Grant, A. y Stirk, C., Arteriosclerosis 10:263-275 (1990). Sobel, B.E., Collen, D. y Grossbard, E.B., eds., Tissue Plasminogen Activator in Thrombolytic Therapy, Marcel Dekker, Inc., New York (1987). 60 Sterrenberg, L., Gravesen, M., Haverkate, F. y Nieuwenhuizen, W., Thromb Res 31:719-728 (1983). Suzuki, K., Nagase, H., Ito, A., Enghild, J.J. y Salvesen, G., Biol Chem Hoppe-Seyler 371:305-310 (1990). 65 Towbin, H., Staehelin, T. y Gordon, J., Proc Natl Acad Sci USA 76:4350-4354 (1979). 22 ES 2 212 802 T3 Welgus, H.G., Campbell, E.J., Cury, J.D., Eisen, A.Z., Senior, R.M., Wilhelm, S.M. y Goldberg, G.I., J Clin Invest 86: 1496-1502 (1990). 5 Valenzuela, R., Shainoff, J.R., DiBello, P.M., Anderson, J.M., Matsueda, G.R., y Kudryk, B.J., Am J Pathol 141:861-880 (1992). Werb, Z., Alexander C.M. y Adler, R.R., Matrix Supplement 1:337-343 (1992). Woessner, J.E., FASEB J 5:2145-2154 (1991). 10 Zavalova, L.L., Kuzina, E.V., Levina, N.B. y Baskova, I.P., “Monomerization of fragment DD by destabilase from the medicinal leech does not alter the N-terminal sequence of the y-chain”, Thrombosis Res 71:241-44 (1993). 15 20 25 30 35 40 45 50 55 60 65 23 ES 2 212 802 T3 REIVINDICACIONES 5 1. Uso de una metaloproteinasa fibrinolítica para la fabricación de un medicamento para su uso en un método de tratamiento fibrinolítico, en el que dicha metaloproteinasa fibrinolítica es una estromelisina. 2. Uso según la reivindicación 1, en el que dicha estromelisina es una estromelisina endógena. 3. Uso según la reivindicación 1, en el que dicha estromelisina es MMP-3. 10 15 20 4. Uso según la reivindicación 1, en el que dicha estromelisina escinde fibrina/fibrinógeno en el enlace peptídico γGly404-Ala405. 5. Uso según la reivindicación 1, en el que dicho método de tratamiento fibrinolítico incluye la administración de estromelisina junto con un compuesto complementario que tiene actividad trombolítica. 6. Uso según la reivindicación 5, en el que dicho compuesto complementario que tiene actividad trombolítica se selecciona del grupo que consiste en un activador del plasminógeno, la hirudina, un inhibidor enzimático, una enzima, un anticoagulante, un anticuerpo o un péptido sintético específico del receptor plaquetario gpIIb/IIIa, o una combinación de los mismos. 7. Uso según la reivindicación 6, en el que dicho activador del plasminógeno se selecciona del grupo que consiste en u-PA, t-PA, estreptocinasa, estafilocinasa y combinaciones de los mismos. 25 8. Uso según la reivindicación 1, en el que dicho método de tratamiento fibrinolítico ha de realizarse tras el tratamiento trombolítico para inhibir la reoclusión vascular. 9. Uso según la reivindicación 1, en el que dicho método de tratamiento fibrinolítico ha de realizarse tras la intervención quirúrgica para inhibir la reestenosis. 30 35 10. Uso según la reivindicación 1, en el que dicho método de tratamiento fibrinolítico ha de realizarse para inhibir la formación inicial, o estimular la regresión, de placas ateroescleróticas. 11. Composición para el tratamiento trombolítico que comprende una metaloproteinasa fibrinolítica que es una estromelisina, un compuesto complementario que tiene actividad trombolítica y un vehículo farmacéuticamente aceptable. 12. Composición según la reivindicación 11, en la que dicha estromelisina es una estromelisina endógena. 40 13. Composición según la reivindicación 11, en la que dicha estromelisina es MMP-3. 14. Composición según la reivindicación 11, en la que dicha estromelisina escinde fibrina/fibrinógeno en el enlace peptídico γGly404-Ala405. 45 50 55 15. Composición según la reivindicación 11, en la que dicho compuesto complementario que tiene actividad trombolítica se selecciona del grupo que consiste en un activador del plasminógeno, la hirudina, un inhibidor enzimático, una enzima, un anticoagulante, un anticuerpo o un péptido sintético específico del receptor plaquetario gpIIb/IIIa, o una combinación de los mismos. 16. Composición según la reivindicación 15, en la que dicho activador del plasminógeno se selecciona del grupo que consiste en u-PA, t-PA, estreptocinasa, estafilocinasa y combinaciones de los mismos. 17. Equipo para llevar a cabo el tratamiento trombolítico, que comprende una composición que comprende una metaloproteinasa fibrinolítica que es una estromelisina, un compuesto complementario que tiene actividad trombolítica y un envase. 18. Equipo según la reivindicación 17, en el que dicha estromelisina es MMP-3. 60 65 19. Equipo según la reivindicación 17, en el que dicho compuesto complementario que tiene actividad trombolítica se selecciona del grupo que consiste en un activador del plasminógeno, la hirudina, un inhibidor enzimático, una enzima, un anticoagulante, un anticuerpo o un péptido sintético específico del receptor plaquetario gpIIb/IIIa, o una combinación de los mismos. 20. Equipo según la reivindicación 17, que comprende además medios para administrar una cantidad terapéuticamente eficaz de dicha composición. 24 ES 2 212 802 T3 21. Equipo según la reivindicación 20, en el que dichos medios de administración incluyen medios para administrar dicha composición por vía parenteral. 5 22. Método de degradación de fibrina/fibrinógeno que comprende poner en contacto fibrina/fibrinógeno in vitro con una cantidad de una metaloproteinasa fibrinolítica eficaz para escindir fibrina/fibrinógeno, en el que dicha metaloproteinasa fibrinolítica es una estromelisina. 23. Método según la reivindicación 22, en el que dicha estromelisina es MMP-3. 10 15 24. Método según la reivindicación 23, en el que dicha estromelisina escinde fibrina/fibrinógeno en el enlace peptídico γGly404-Ala405. 25. Método de diagnóstico para caracterizar fibrina/fibrinógeno que comprende poner en contacto fibrina/fibrinógeno in vitro con una metaloproteinasa fibrinolítica para proporcionar productos de degradación característicos de dicha fibrina/fibrinógeno, en el que dicha metaloproteinasa fibrinolítica es una estromelisina. 26. Método de diagnóstico según la reivindicación 25, en el que dicha estromelisina es MMP-3. 20 25 27. Método de diagnóstico según la reivindicación 25, en el que dicho método comprende además poner en contacto dichos productos de degradación con al menos un anticuerpo que se asocie específicamente con un dominio de la fibrina/fibrinógeno y medir la asociación específica de dicho anticuerpo con dichos productos de degradación separados. 28. Método de diagnóstico según la reivindicación 27, en el que dicho anticuerpo se marca de manera que se pueda detectar con un resto de marcador detectable. 29. Método de diagnóstico según la reivindicación 27, en el que dicho anticuerpo es un anticuerpo monoespecífico. 30 35 30. Método para inhibir la formación de trombos mediante un aparato relacionado con la medicina que comprende poner en contacto un aparato relacionado con la medicina con una composición que comprende una metaloproteinasa fibrinolítica, para proporcionar una superficie de inhibición de trombos sobre dicho aparato relacionado con la medicina, en el que dicha metaloproteinasa fibrinolítica es una estromelisina. 31. Método según la reivindicación 30, en el que dicho aparato relacionado con la medicina se selecciona del grupo que consiste en tubos de recogida de sangre, matraces de cultivo, placas de prueba, pipetas, envases de reactivos, tubos, membranas, agujas, cánulas, catéteres, injertos, stents, filtros, dispositivos intrauterinos, válvulas y similares. 40 32. Aparato relacionado con la medicina que tiene propiedades de inhibición de trombos que comprende un dispositivo relacionado con la medicina al que se ha proporcionado una cantidad para inhibir trombos de una composición que comprende una metaloproteinasa fibrinolítica, en el que dicha metaloproteinasa fibrinolítica es una estromelisina. 45 33. Aparato relacionado con la medicina según la reivindicación 32, en el que dicho aparato relacionado con la medicina se selecciona del grupo que consiste en tubos de recogida de sangre, matraces de cultivo, placas de prueba, pipetas, envases de reactivos, tubos, membranas, agujas, cánulas, catéteres, injertos, stents, filtros, dispositivos intrauterinos, válvulas y similares. 50 55 60 65 NOTA INFORMATIVA: Conforme a la reserva del art. 167.2 del Convenio de Patentes Europeas (CPE) y a la Disposición Transitoria del RD 2424/1986, de 10 de octubre, relativo a la aplicación del Convenio de Patente Europea, las patentes europeas que designen a España y solicitadas antes del 7-10-1992, no producirán ningún efecto en España en la medida en que confieran protección a productos químicos y farmacéuticos como tales. Esta información no prejuzga que la patente esté o no incluida en la mencionada reserva. 25 ES 2 212 802 T3 26 ES 2 212 802 T3 27 ES 2 212 802 T3 28 ES 2 212 802 T3 29 ES 2 212 802 T3 30 ES 2 212 802 T3 31 ES 2 212 802 T3 32 ES 2 212 802 T3 33 ES 2 212 802 T3 34