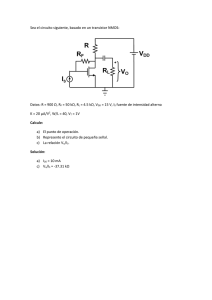
identificacion de mutaciones en el gen iduronato 2 sulfatasa
Anuncio

IDENTIFICACION DE MUTACIONES EN EL GEN IDURONATO 2 SULFATASA (IDS) EN PACIENTES COLOMBIANOS CON SINDROME DE HUNTER DEISY JOHANNA GALVIS RODRÍGUEZ Universidad Nacional de Colombia Facultad de Medicina, Departamento de Morfología Bogotá, Colombia 2013 I IDENTIFICACION DE MUTACIONES EN EL GEN IDURONATO 2 SULFATASA (IDS) EN PACIENTES COLOMBIANOS CON SINDROME DE HUNTER DEISY JOHANNA GALVIS RODRÍGUEZ Tesis o trabajo de investigación presentada(o) como requisito parcial para optar al título de: Magister en Genética Humana Director: MD,MSc, ESP.HARVY MAURICIO VELASCO PARRA Línea de Investigación: Genética Clínica Universidad Nacional de Colombia Facultad de Medicina, Departamento de Morfología Bogotá, Colombia 2013 2 A mi familia 3 AGRADECIMIENTOS A los pacientes y sus familias A la Asociación Colombiana de Pacientes con Enfermedades de Depósito Lisosomal Al personal asistencial de las instituciones visitadas para la inclusión de los pacientes y toma de muestras A la compañía MRC-Holland por aportar el kit para la realización de MLPA y brindar asesoría en el análisis de los resultados A los diferentes especialistas tratantes de los pacientes, que enviaron muestras y/o información clínica relevante para este trabajo Al departamento de Nutrición y Bioquímica de la Pontificia Universidad Javeriana, profesores Janneth González, George Barreto y el estudiante Daniel Torrente, por su orientación en el tema computacional Al Centro de Investigaciones Bioquímicas de la Universidad de Los Andes por el aporte de resultados enzimáticos y muestras Al laboratorio de Errores Innatos del Metabolismo de la Pontificia Universidad Javeriana por el aporte de resultados enzimáticos A mi compañero David Serrano, por su acompañamiento en el inicio de este trabajo A la asistente de la Coordinación de Maestría, por su diligencia en materia de lo administrativo Al Director de esta investigación, por sus valiosas enseñanzas y su creciente comprensión a lo largo del desarrollo de nuestro trabajo A Tatiana Vinasco por su asesoramiento y apoyo en el laboratorio. 4 TABLA DE CONTENIDO TABLA DE CONTENIDO ..................................................................................................................... 5 Resumen ...................................................................................................................................... 10 Abstract ........................................................................................................................................ 11 1. INTRODUCCION ............................................................................................................................. 12 2. JUSTIFICACION ............................................................................................................................... 13 3. OBJETIVOS ..................................................................................................................................... 15 4. 3.1. Objetivo general .......................................................................................................... 15 3.2. Objetivos específicos ................................................................................................... 15 MARCO TEÓRICO ........................................................................................................................... 16 4.1. Definición ..................................................................................................................... 16 4.2. Reseña histórica de la enfermedad ............................................................................. 17 4.3. Epidemiología .............................................................................................................. 19 4.4. Manifestaciones Clínicas de la Mucopolisacaridosis tipo II ........................................ 21 4.5. Fenotipos de MPS II .................................................................................................... 26 4.5.1. Fenotipo Severo o Neuronopático (MPSII A) ............................................................. 26 4.5.2. Fenotipo Atenuado o No neuronopático (MPS II B) .................................................. 27 4.6. Diagnóstico .................................................................................................................. 28 4.7. Manejo de la MPS II .................................................................................................... 28 4.7.1. Soporte y manejo interdisciplinario .............................................................................. 32 4.7.2. Trasplante de médula ósea .......................................................................................... 33 4.7.3. Terapia de reemplazo enzimático ................................................................................ 35 4.7.4. Administración intratecal de Idursulfasa ..................................................................... 40 4.7.5. Terapia génica ................................................................................................................ 40 4.7.6. Herramientas de prevención y diagnóstico temprano .............................................. 42 4.8. Genética de la MPS II .................................................................................................. 43 5 4.8.1. Epidemiología genética ................................................................................................. 46 4.8.2. Correlación genotipo – fenotipo ................................................................................... 47 4.8.3. Herramientas de diagnóstico molecular ...................................................................... 50 4.9. Proteómica y biología celular de la IDS ...................................................................... 52 4.9.1. Estructura y función de la Iduronato 2 sulfatasa (IDS) ......................................... 52 4.9.2. Procesamiento diferencial de IDS ............................................................................... 55 4.9.3. Fisiopatología de la MPS II. .......................................................................................... 56 5. METODOLOGÍA .............................................................................................................................. 64 5.1. Tipo de estudio ............................................................................................................ 64 5.2. Tamaño de muestra .................................................................................................... 64 5.3. Ingreso al estudio ........................................................................................................ 64 5.4. Fase pre analítica ........................................................................................................ 64 5.4.1. Toma de muestra ........................................................................................................ 64 5.4.2. Extracción de DNA ......................................................................................................... 65 5.5. Fase analítica .............................................................................................................. 65 5.5.1. Detección de grandes deleciones y duplicaciones del gen por MLPA ............... 65 5.5.2. Secuenciación ............................................................................................................. 71 5.4.3. Análisis de recombinantes por RFLPs ........................................................................ 74 5.6. Fase Post Analítica ...................................................................................................... 77 5.6.1. Descripción de las variables y análisis estadístico ................................................... 77 5.6.2. Análisis bioinformático de la repercusión de las mutaciones en la estructura tridimensional de la IDS ............................................................................................................. 77 5.6.2.1. Modelamiento y Docking de las IDS mutantes halladas en el estudio ............... 79 5.7. 6. Flujograma ................................................................................................................... 80 RESULTADOS ................................................................................................................................. 81 6.1. Características clínicas de los pacientes .................................................................... 81 6.2. Detección de grandes pérdidas o ganancia de material genético (rearreglos complejos) 91 6.3. Identificación de mutaciones por secuenciación directa ............................................. 94 6 6.4. Análisis de recombinantes por RFLPs ........................................................................ 98 6.5. Clasificación de las mutaciones encontradas ........................................................... 100 6.6. Frecuencia de las mutaciones encontradas en los pacientes MPS II ....................... 103 6.7. Estudio de portadoras ............................................................................................... 105 6.8. Predicción de la repercusión de las mutaciones en la estructura tridimensional de la IDS 109 6.8.1. Modelamiento de la hIDS tipo silvestre ..................................................................... 109 6.8.2. Docking Molecular de la hIDS tipo silvestre ............................................................. 112 6.8.3. Modelamiento y docking de las IDS mutantes ......................................................... 114 7. DISCUSIÓN ................................................................................................................................... 123 7.1. Características clínicas.................................................................................................. 123 7.2. Perfil mutacional y comparación con la literatura .......................................................... 129 7.3. Análisis bioinformático y repercusión de las mutaciones en la estructura tridimensional de la IDS 133 8. CONCLUSIONES ........................................................................................................................... 141 9. LIMITACIONES Y PERSPECTIVAS .................................................................................................. 143 10. BIBLIOGRAFÍA ............................................................................... ¡Error! Marcador no definido. 7 Lista de figuras Figura 4-1 Posicion de los exones del gen IDS. Presencia del Peudogen IDSP1………………………45 Figura 4-2 Pasos en la degradación de GAGs…………………………………………….………………..55 Figura 4-3 Cambios secundarios en las neuronas encontrados en las enfermedades de depósito lisosomal.. ………………………………………………………………………………………..…………….59 Figura 5-4 Amplificación de sondas dependeinte de ligamiento en Múltiplex……………...……………66 Figura 5-5 Representación esquemática de los rearreglos complejos ……………………......……...…76 Figura 6-6 Deleción completa del exón 7 en el paciente MPSII012……………………..……………....93 Figura 6-7 Mutaciones privadas (nuevas) encontradas por secuenciación y MLPA.95 Figura 6-8 Digestión con HinfI (PCR1)……………………………… ……………………………………...97 Figura 6-9 Digestión con digestión con SacI, AcuI………………………………………………………....98 Figura 6-10 Fotografías pacientes MPSII012 y MPSII014………………………………………………..99 Figura 6-11 Distribución de frecuencias mutaciones privadas y reportadas………………………......103 Figura 6-12 Pedigree del paciente MPSII012 ………………………………………………………….....104 Figura 6-13 Portadoras parientes del paciente MPSII12……………………………………………..…..105 Figura 6-14 Exclusión de estado de portadora………………………………………………………..…..106 Figura 6-15 Estudio de portadoras parientes paciente MPSII002……………………………………....108 Figura 6-16 Modelo tridimensional de IDS…………………………………………………………….….109 Figura 6-17 Plot de Ramachandran. …………………………………………………………………..….110 Figura 6-18 Docking de hIDS tipo silvestre……………………………………………………………….112 Figura 6-19 a 6-25 Docking de las distintas IDS mutantes………………………………………………116 Figura 6-26 Modelamiento mutantes truncadas………………………………………………………..…121 Figura 7-27 Modelo propuesto para el mecanismo de recombinación en el intrón 3…………….…..130 8 Lista de tablas Tabla 4-1 Actividd de la IDS en leucicitos, plasma y fibroblastos……………………………………...….33 Tabla 4-2 Resumen del diseño de estudio para los ensayos clínicos de fase I/II y II/III de Idursulfase……………………………………………………………………………………..……………..…37 Tabla 4-3 Transcriptos del gen IDS……………………………………………………………..……………45 Tabla 5-4 Condiciones para la Amplificación de fragmentos génicos…………………………………….71 Tabla 5-5 Set de primers para detección de rearreglos complejos ……………………………….…..….76 Tabla 5-6 RFLPs de los productos de PCR…………………………………………….…………………...76 Tabla 6-7 Características fenotípias de la muestra en estudio…………………………………….…..….84 Tabla 6-8 Edad al diagnóstico, niveles de actividad enzimática de los pacientes y administración de Terapia de Reemplazo Enzimático …………………………………………………………………………..85 Tabla 6-9 Características clínicas de los pacientes………………………………………………..……….89 Tabla 6-10 Valores normalizados de la longitud de fragmentos de las diferentes regiones del gen IDS analizadas por MLPA. Pacientes MPS 001 y 012………………………………………………………….92 Tabla 6-11 Resultados de genotipificación y MLPA…………………………………………….…………93 Tabla 6-12 Mutaciones identificadas por MLPA, secuenciación y RFLPs………………….………….100 Tabla 6-13 Clasificación de Mutaciones pacientes colombianos con MPSII……………….………….101 Tabla 6-14 RMSD y ASA calculados para cada modelo de las IDS mutantes……………..………….114 9 Resumen El Síndrome de Hunter o Mucopolisacaridosis II es una enfermedad ligada X heredada, causada por mutaciones en iduronato 2 sulfatasa (IDS). Esta enzima cataliza la etapa inicial de la degradación de heparán y dermatán sulfato, por lo que su deficiencia conduce al almacenamiento de estos glicosaminoglicanos. La MPSII tiene heterogeneidad alélica, dificultando la correlación genotipo-fenotipo. Este estudio evaluó las características clínicas y la frecuencia de mutaciones en un grupo de pacientes MPSII colombianos y realizó una aproximación a la correlación fenotipo-genotipo. Dieciocho pacientes fueron incluidos en este estudio, 11% no neuronopáticos y 89% neuronopáticos, los cuales fueron sometidos a tres metodologías moleculares: MLPA, secuenciación directa de exones y RFLPs. De un total de 12 mutaciones identificadas, 6 eran nuevas (c.548_564dup16, c.477insT, c.595_607del12, c. 549_562del13, c.182delC y deleción completa del exón 7). La frecuencia de mutaciones recurrentes (R468Q, Q465X, K347Q, K236N, S71N y un fenómeno de conversión) fue del 58%. En dos pacientes (11%) no fue posible establecer la mutación. La mutación S71N fue recurrente en fenotipo atenuado, mientras que las mutaciones de tipo frameshift privadas y rearreglos complejos estaban presentes en pacientes con fenotipo severo. Se realizó Docking molecular con las estructuras de la IDS silvestre y mutantes, encontrando cambios en las interacciones enzima-sustrato para las mutantes de IDS. Palabras clave: Mucopolisacaridosis II, Sulfoiduronato sulfatasa, Análisis Bioinformático, Docking molecular. 10 Abstract Hunter Syndrome or Mucopolysaccharidosis II is an inherited X-linked disease caused by mutations in the iduronate 2 sulfatase (IDS). This enzyme catalyzes the initial stage of the degradation of heparan and dermatan sulfate, therefore its deficiency leads to the storage of these glycosaminoglycans. MPSII has allelic heterogeneity, challenging genotype-phenotype correlation. This study evaluated the clinical characteristics and the frequency of mutations in a group of MPSII Colombians patients and made an approach to phenotype-genotype correlation. Eighteen patients were included in this study, 11% non-neuropathic and 89% neuropathic, which were subjected to three molecular methods: MLPA, direct sequencing of exons and RFLPs. A total of 12 mutations were identified, 6 of them were new (c.548_564dup16, c.477insT, c.595_607del12, c. 549_562del13, c.182delC and complete deletion of exon 7). The frequency of recurrent mutations (R468Q, Q465X, K347Q, K236N, S71N and a phenomenon of conversion) was 58%. In two patients (11%) the molecular diagnosis was not possible. The S71N mutation was recurrent in attenuated phenotype, whereas new frameshift mutations and complex rearrangements were present in patients with severe phenotype. Molecular Docking was performed with the structures of wild type and mutant IDS, finding substantial changes in enzyme-substrate interactions for IDS mutants. Keywords: Mucopolysaccharidosis II, Sulfoiduronate sulfatase, Bioinformatic Analysis, Molecular Docking. 11 1. INTRODUCCION La Mucopolisacaridosis tipo II (MPS II) es una enfermedad genética de herencia recesiva ligada a X. Está catalogada dentro de las “enfermedades raras”, lo que indica que tiene una incidencia menor a 1 en 100.000. El defecto primario es ausencia o inactividad de la enzima Iduronato 2 Sulfatasa (IDS), responsable de la degradación mayoritaria de los glicosaminoglicanos (GAGs) dermatán sulfato y heparán sulfato (también algo de condroitín sulfato) y su consecuencia patológica es la acumulación de estos compuestos en los tejidos del cuerpo, incluyendo el tejido neuronal. En la actualidad en Colombia se ha diagnosticado un número creciente de casos pero no hay datos exactos sobre la incidencia, prevalencia y la epidemiología molecular de la MPS II. Además nuestro país carece de una metodología molecular para el diagnóstico y abordaje genético de esta patología, hay total desconocimiento sobre el perfil mutacional de la MPS II y sobre el estado de portadores de los familiares que presentan esta patología. Hasta ahora, desconocemos la presencia o ausencia de mutaciones privadas en el gen IDS, la frecuencia de las mutaciones más comunes y no hay certeza de la presencia o ausencia de correlaciones genotipo–fenotipo en nuestra población, por lo que se hace clave el desarrollo de propuestas como la que describiremos para entender el comportamiento molecular de este tipo de enfermedades en nuestras poblaciones. 12 2. JUSTIFICACION La Mucopolisacaridosis tipo II (OMIM 309900) o Síndrome de Hunter, es la única MPS con herencia Recesiva ligada a X(Yatziv, Erickson, & Epstein, 1977). El defecto primario de la patología es la ausencia o inactividad de la enzima lisosomal Iduronato 2 Sulfatasa (IDS; EC 3.1.6.13) por mutaciones en el gen IDS que genera en su fenotipo más severo (Hunter A), una alta mortalidad, cercana a 90% antes de los 15 años, la cual es acompañada por discapacidad cognitiva severa, visceromegalias, cardiopatías y enfermedades del sistema respiratorio (Yatziv, et al., 1977). Los estudios sobre incidencia y prevalencia del grupo de enfermedades de depósito lisosomal incluida la MPS II han mostrado diferencias de una región a otra. Malm et al. en población escandinava encontraron una incidencia combinada de las MPS de 4.24 a 7.06 por 100.000 nacidos vivos (Malm, Lund, Mansson, & Heiberg, 2008). Nelson et al. en población australiana observaron una incidencia 3.43 por 100.000 nacidos vivos (Nelson, Crowhurst, Carey, & Greed, 2003) mientras que Baehner en Alemania citó una incidencia de 3.43 por 100.000 (Baehner et al., 2005) y Lin et al. encontraron una incidencia de 2.04 en 100.000 nacidos vivos de Taiwán (Lin et al., 2009). No hay aún reportes al respecto en Latinoamérica ni en Colombia, lo cual va de la mano con el desconocimiento de la epidemiología genética de las mutaciones en IDS y su repercusión en los fenotipos del Síndrome de Hunter. En la actualidad en Colombia se carece de una metodología molecular para el diagnóstico y abordaje genético (incluyendo estudio de portadores) de esta patología y hay total desconocimiento sobre el perfil mutacional de la MPS II en nuestros 13 pacientes. A la fecha no se encuentran estudios en pacientes colombianos sobre la presencia o ausencia de mutaciones privadas en IDS y la frecuencia de las mutaciones más comunes; tampoco hay certeza de la presencia o ausencia de correlaciones genotipo – fenotipo, por lo que se hace clave el desarrollo de propuestas como la que describiremos para entender el comportamiento molecular de este tipo de enfermedades en nuestras poblaciones. Conocer el perfil mutacional de IDS para MPS II permitiría mejorar el diagnóstico, manejo y abordaje integral de este tipo de pacientes y sus familias, generaría mejores herramientas en los procesos de asesoramiento genético, manejo de la información, pronóstico y alcances terapéuticos individualizados (Wraith, Scarpa, et al., 2008). El presente estudio pretende ser el primero de identificación de las mutaciones en el gen IDS en pacientes colombianos que presentan MPS II. El propósito es conocer nuestro perfil mutacional, ofrecer a la comunidad clínica y a los familiares de pacientes los servicios de genotipificación y estudio de portadores de mutaciones en el gen IDS y aportar a la comunidad científica un mayor conocimiento sobre esta entidad en población tan mezclada como la nuestra. 14 3. OBJETIVOS Objetivo general Identificar las mutaciones en el gen IDS en una muestra de pacientes colombianos con MPSII y analizar las repercusiones de estos cambios en la proteína in silico. Objetivos específicos Caracterizar fenotípicamente los pacientes colombianos con Síndrome de Hunter (MPSII) incluidos en el estudio. Identificar la presencia de grandes deleciones/duplicaciones, mutaciones puntuales y rearreglos complejos del gen IDS en los pacientes con Síndrome de Hunter y establecer sus frecuencias. Determinar la presencia de mutaciones comunes y nuevas en el gen IDS en nuestra población a estudio y sus frecuencias. Sugerir posibles mecanismos de origen de los grandes rearreglos encontrados. Estudiar la repercusión de las mutaciones comunes y privadas, sobre la estructura de la proteína in silico. 15 4. MARCO TEÓRICO Definición La MPS II es una enfermedad genética de herencia Recesiva ligada a X. Está catalogada dentro de las “enfermedades raras”, lo que indica que tiene una incidencia menor a 1 en 10.000, tal como se contempla en la Ley de Enfermedades Huérfanas(Ley 1349)(Cáceres, Otero, Gómez, & Rodríguez, 2010). El defecto primario es la ausencia o inactividad de la enzima Iduronato 2 Sulfatasa (IDS), responsable de la degradación de los GAGs dermatán sulfato (también algo de heparán y condroitín sulfato) y su consecuencia patológica es la acumulación de estos compuestos en los tejidos del cuerpo, incluyendo el Sistema Nervioso Central (K. L. Jones & Smith, 2006). Este síndrome tiene un espectro de presentación clínica que va desde un fenotipo severo (Hunter A) hasta un fenotipo leve o atenuado (Hunter B) y su clasificación clínica se obtiene teniendo en cuenta 3 variables: 1. La edad de inicio de síntomas 2. La presencia o ausencia de compromiso neurológico/cognoscitivo 3. El promedio de sobrevida (Neufeld & Muenzer, 2001). Desde el aislamiento y caracterización del gen IDS por Wilson et al. en 1990 se han podido utilizar herramientas moleculares para el diagnóstico de MPS II(Wilson et al., 1990), de gran utilidad en el estudio de portadoras en familias en riesgo, en diagnóstico prenatal y pacientes con edad inferior a 1 año(C. P. Chen et al., 2007). 16 Reseña histórica de la enfermedad La primera descripción de la enfermedad fue realizada por el mayor Charles Hunter en 1917(Hunter, 1917), que correspondía a dos hermanos de ascendencia británica con macrocránea, fascies tosca, organomegalias, hipoacusia, infecciones faríngeas a repetición, rigidez articular, anormalidades óseas, talla baja e inteligencia normal. Dos años después, Gertrud Hurler reportó sobre dos pacientes femeninas con características similares a las descritas por Hunter, pero además con retardo mental y opacidad corneal (Hurler, 1919). Las dos entidades fueron consideradas una sola, incluso denominadas Síndrome de Hurler-Hunter o Gargolismo hasta la elucidación de la base bioquímica de cada enfermedad, casi 50 años más tarde (Brante, 1952). En 1965 Danes y Bern cultivaron fibroblastos de pacientes con síndrome de Hurler o síndrome de Hunter demostrando con azul de toluidina la presencia de gránulos metacromáticos (Danes & Bearn, 1965), los cuales son el resultado de la acumulación intracelular de GAGs, como se comprobaría en al análisis químico(Danes & Bearn, 1966). Los mismos autores encontraron también en relación con el síndrome de Hunter que los fibroblastos provenientes del padre carecían por completo de gránulos metacromáticos, a diferencia de las condiciones autosómicas recesivas Hurler y Sanfilippo, donde la alteración es evidente en fibroblastos provenientes de ambos progenitores(Pennock & Barnes, 1976). En la búsqueda de la causa de dicha acumulación de GAGs, Fratantoni, Hall y Neufeld demostraron en 1968 la existencia de un “factor de corrección”: al mezclar 17 incidentalmente los cultivos de fibroblastos correspondientes a los síndromes de Hunter y Hurler se produjo la corrección cruzada de los depósitos intracelulares en ambos cultivos(Fratantoni, Hall, & Neufeld, 1968) . Dos años más tarde Cantz et al. lograrían la caracterización inicial de la proteína, y empleando el sustrato radioactivo proteodermatán [35S] sulfato se aclararía su actividad sulfatasa sobre el grupo sulfato del residuo de ácido idurónico, en el primer paso de la degradación de este GAG(Cantz, Chrambach, Bach, & Neufeld, 1972). En 1974 el grupo de Lin, Neufeld y colaboradores, publicó un método radioactivo para medir la actividad enzimática de la IDS empleando un sustrato marcado con tritio (ácido O-a-L-idopiranosilurónico 2 sulfato), puesto que el sustrato con S35 era propenso al error y no era confiable como prueba diagnóstica(Lim, Leder, Bach, & Neufeld, 1974). Más tarde, Liebaers y Neufeld emplearían esta técnica para determinar el estado heterocigoto de la MPSII encontrando que en la mitad de las portadoras obligadas los valores de actividad se encontraban en límites normales, concluyendo así que el ensayo de actividad enzimática no resultaba confiable para identificación de portadoras(Liebaers & Neufeld, 1976). La aproximación inicial a las bases moleculares de la MPS II fue realizada por Upadhyaya y colaboradores mediante análisis de ligamiento con marcadores microsatélites localizados distalmente en el brazo largo del cromosoma X(Upadhyaya et al., 1986). El mismo grupo confirmó que el locus del gen responsable del Síndrome de Hunter se hallaba entre un clúster de marcadores, entre ellos F8C y DXS15, en Xq28. 18 En 1990 Wilson, Hopwood y colaboradores lograron el aislamiento y clonación del cDNA del gen IDS empleando dideoxiterminadores y el fragmento Klenow de la DNA polimerasa I(Wilson, et al., 1990).Con esta información disponible se inició la búsqueda de mutaciones que explicaran el amplio espectro de severidad de la MPSII encontrando en los primeros ensayos grandes deleciones o rearreglos del gen (Wilson et al., 1991), (Wraith et al., 1991). La presencia de un pseudogén corriente abajo del gen IDS explicaría luego la susceptibilidad a eventos complejos de recombinación (Rathmann, Bunge, Steglich, Schwinger, & Gal, 1995). Epidemiología El primer reporte sobre la incidencia del síndrome de Hunter fue realizado en 1971 en Columbia Británica, arrojando un estimado de 0,3 por 100.000 nacidos vivos (Lowry & Renwick, 1971). En 1980 población judía de Israel, Schaap y Bach calcularon 1 por 68.000 nacidos vivos (Schaap & Bach, 1980) y en la misma década en el Reino Unido, Young y Harper reportaron una incidencia de 1 en 132.000 nacidos vivos con una prevalencia de 1 en 3,17 millones de habitantes (Young & Harper, 1982a), (Young & Harper, 1982b). Con el progreso en la identificación y diagnóstico de la MPSII aparece un mayor número de datos epidemiológicos en la literatura. Nelson et al. en Irlanda reportaron 1 en140.000(Nelson, 1997), mientras que en Holanda se calculó en 0,67:100.000 nacidos vivos(Poorthuis et al., 1999). 19 Meikle et al. reportaron una incidencia de 0.62:100.000 nacidos vivos en Australia (Meikle, Hopwood, Clague, & Carey, 1999). Nelson et al. en población de Australia Occidental observaron una incidencia de MPS II de 1 en 165.000 varones nacidos vivos(Nelson, et al., 2003). Baehner en Alemania citó una incidencia de 0,64 por 100.000 (Baehner et al., 2005). Malm et al. recopilaron registros de 25 años en Noruega y 30 años en Suecia y Dinamarca, encontrando una incidencia de 0,13 a 0,27 por 100.000 nacidos vivos y una prevalencia entre el 2 a 6 por millón además de rangos de supervivencia sin tratamiento entre los 2,5 a 58 años(Malm, et al., 2008). En Taiwan, los registros desde 1984 a 2004 muestran la más alta incidencia de MPS II, de 1.07 por 100.000 nacidos vivos (2.05 por 100.000 varones nacidos vivos), representando el 52% de todos los casos de MPS. La incidencia combinada de MPS en esta población fue similar a la de las poblaciones occidentales, pero la proporción de MPSII fue mayor en Taiwan (Lin, et al., 2009). Similar situación se reporta en Japón, donde la incidencia de MPS II se ha estimado en 1:90.000, representando el 50% de todas las MPS (Ochiai et al., 2007). Aún no se dispone de datos epidemiológicos de este tipo en Norteamérica ni América Latina. Un estudio de Coelho et al. en 1997, sobre el diagnóstico de Errores Innatos del Metabolismo (EIM) en Brasil reveló que las Enfermedades de Depósito Lisosomal eran las más frecuentes entre los EIM (59.8% de todos los diagnósticos), y las MPS representaron el 54% de las Enfermedades de Depósito Lisosomal(Coelho, Wajner, Burin, Vargas, & Giugliani, 1997). La Rede MPS Brasilal 20 año 2008 registró 249 pacientes, de los cuales 82 pacientes corresponden a MPSII(Vieira et al., 2008). En Colombia en el año 2012 se registraron 33 pacientes con MPS II, según datos aportados por la organización ACOPEL (Asociación Colombiana de Pacientes con Enfermedades de Depósito Lisosomal). No se han realizado estudios sobre la incidencia y prevalencia de la MPS II en nuestro país, sin embargo, la frecuencia combinada de las MPS calculada en los departamentos de Cundinamarca y Boyacá fue de fue de 1,98 casos por 100.000 nacidos vivos; la mayor frecuencia fue para el tipo IV (0,68), mientras que la MPS II tuvo una frecuencia estimada de 0,45 por 100.000 nacidos vivos(Gómez, García-Robles, & Suárez-Obando, 2012). Manifestaciones Clínicas de la Mucopolisacaridosis tipo II Las características clínicas de la MPS II se deben a la acumulación de GAGs en tejidos y órganos, lo que se traduce en una afección multisistémica con variabilidad fenotípica(Winter & Barainster, 2001).Las principales son: Macrocrania. Epilepsia. Hipoacusia. Facies tosca (puente nasal deprimido, narinas antevertidas, labios y encías gruesas, macroglosia y frente plana). Ausencia de opacidad corneal. Estenosis mitral o de aorta. 21 Hepatoesplenomegalia. Hernia umbilical/inguinal. Talla baja. Múltiples disostosis óseas (hipoplasia de los metacarpianos, acortamiento rizomélico de los dedos y platiespondilia). Rigidez articular. Camptodactilia Retardo en el desarrollo neurológico(Winter & Barainster, 2001), (Neufeld & Muenzer, 2001). Exceptuando el compromiso del Sistema Nervioso Central (SNC), las anormalidades sistémicas son similares independientemente de la severidad del fenotipo. 4.4.1 Características faciales La característica más común que se presenta es un engrosamiento de los rasgos faciales (fascies tosca), que se vuelve aparente entre 2 y 4 años de edad (Young & Harper, 1983). Se observa una base y punta nasal ensanchada, labios gruesos, arcos superciliares y mandíbula prominentes (Young & Harper, 1982), (Neufeld & Muenzer, 2001). 4.4.2. Anomalías esqueléticas El perímetro cefálico se encuentra por lo general sobre +2SD. Los pacientes tienden a ser altos para su edad hasta aproximadamente 4 ó 5 años de edad, cuando empiezan manifestar talla baja(Wraith, Scarpa, et al., 2008). 22 Los hallazgos esqueléticos se conocen colectivamente como disostósis múltiple. El examen radiográfico revela grosor anormal de los huesos, y la osificación epifisaria irregular en las articulaciones de la mano, el hombro y codo(Hunter, 1917). Las manos adquieren forma de garra y en combinación con el síndrome del túnel carpiano puede resultar una pérdida de la función de la mano (Norman et al. 1995), Las costillas y clavículas están engrosadas y tienen una forma inusual. Los cuerpos vertebrales aplanados (platispondilia) con superficies laterales irregularmente dentadas. La movilidad está restringida debido a la rigidez de las articulaciones y contracturas(Link et al., 2010). 4.4.3. Manifestaciones oculares La opacidad corneal no es una característica del síndrome de Hunter, pero una evaluación con lámpara de hendidura puede revelar discretas lesiones de la córnea que no afectan la visión (Ashworth, 2006). Se pueden presentar cambios laterales en la pigmentación de la retina y pérdida de campo de visión en algunos pacientes (Topping et al., 1971). 4.4.4. Manifestaciones otorrinolaringológicas En la mayoría de los pacientes se presentan frecuentes infecciones respiratorias altas (Wraith, Scarpa, et al., 2008).La macroglosia, hipertrofia de adenoides y amígdalas, sumado a cambios en la mandíbula y en el cuello, limitan la apertura de la cavidad oral (Downs, 1995). La mayoría de los pacientes presentan infecciones recurrentes del oído, y casi todos experimentan hipoacusia progresiva (Peck et al., 1984). La pérdida de audición es causada tanto por déficit conductivo como neurosensorial, y la efusión del oído medio es reconocido como un importante 23 contribuyente a la pérdida de audición en los pacientes con síndrome de Hunter(Keilmann, Nakarat, Bruce, Molter, & Malm, 2012). Los dientes son de forma irregular, y el tejido gingival es hiperplásico (Hopkins et al., 1973), (Downs et al., 1995). 4.4.5. Sistema respiratorio La obstrucción de la vía aérea es un hallazgo frecuente en el síndrome de Hunter(Brama, Gay, Feinmesser, & Springer, 1986), (Shih, Lee, Lin, Sheu, & Blickman, 2002). Factores que contribuyen a este fenómeno son: la estrechez y forma anormal de la tráquea y los bronquios (Shih et al., 2002), macroglosia e hipertrofia de amígdalas y adenoides, epiglotis prominente, infecciones frecuentes del tracto respiratorio superior, neumonía recurrente y secreciones nasales y traqueales(Morehead & Parsons, 1993). El movimiento restringido de las articulaciones temporomandibulares, la rigidez de la pared torácica y la distensión abdominal también alteran el patrón respiratorio (Young & Harper, 1982). Una complicación común de la obstrucción de vía aérea en el síndrome de Hunter es la apnea del sueño(Leighton, Papsin, Vellodi, Dinwiddie, & Lane, 2001). 4.4.6. Sistema Cardiovascular La afectación cardiaca está presente en casi todos los pacientes con síndrome de Hunter y es una causa importante de muerte en esta población. Enfermedad valvular mitral y/o aórtica, hipertrofia ventricular y falla cardíaca, son comúnmente reportados(Dangel, 1998). En un estudio de 18 pacientes se reportó insuficiencia mitral en 11 de ellos(M. R. Chen, Lin, Hwang, & Yu, 2005). 24 4.4.7. Sistema Nervioso El compromiso del SNC se sospecha inicialmente por retraso y luego regresión en los hitos del desarrollo, por ejemplo, en la sedestación, marcha y desarrollo del lenguaje; estas manifestaciones se presentan en el fenotipo severo (MPS II A) (Young, Harper, Newcombe, & Archer, 1982). La hidrocefalia comunicante moderada a grave puede acelerar el deterioro neurológico(Yatziv & Epstein, 1977). La presión intracraneal elevada puede causar edema del disco óptico y atrofia del nervio óptico. Las convulsiones se presentan en más de la mitad de los casos con MPS II A que llegan a la edad de 10 años, pero son poco frecuentes en la forma atenuada de la enfermedad (Wraith et al., 2008). El síndrome del túnel carpiano es común en los pacientes con MPS II (NormanTaylor et al., 1995),(Kwon et al., 2011).La compresión de la médula espinal puede ocurrir debido a un estrechamiento del canal vertebral y la inestabilidad de la articulación atlantoodontoidea (Neufeld & Muenzer, 2001). Severas alteraciones del comportamiento, tales como hiperactividad, la obstinación y la agresión, se observan con frecuencia en los pacientes severamente afectados con el síndrome de Hunter, pero no se observa típicamente en las personas con la presentación atenuada (Bax & Colville, 1995). 4.4.8. Afectación gastrointestinal Debido al almacenamiento de GAGs se produce hepatomegalia y esplenomegalia, resultando en distensión abdominal. Se han reportado frecuencias entre el 57% y el 90% de hepato y/o esplenomegalia(Pinto et al., 2006). La hernia umbilical se observa comúnmente, y hernias inguinales se presentan en aproximadamente el 25 60% de los casos. La diarrea crónica es frecuente en pacientes con compromiso neurológico, pero no es común en el fenotipo atenuado (Young et al., 1982). 4.4.9. Otras manifestaciones Los pacientes con síndrome de Hunter pueden tener lesiones en piel consistentes en pápulas blanco marfil que son de 2 a 10 mm de diámetro(Panine & Hashimoto, 1995), (Thappa et al., 1998). Vejiga neurogénica y obstrucción urinaria han sido reportados(Koyama, Moda, Sone, Tanaka, & Hino, 1994). Se estima que aproximadamente la mitad de los pacientes severamente afectados controla esfínteres, pero desarrollan enuresis y encopresis a medida que progresa la enfermedad(Martin et al., 2008). Fenotipos de MPS II La MPS II presenta dos fenotipos: uno severo (MPS II A) o neuronopático y el fenotipo no neuronopático (MPS II B) o atenuado. Sus características e historia natural se exponen a continuación. 4.5.1. Fenotipo Severo o Neuronopático (MPS II A) Se caracteriza por retardo mental y se ha descrito que puede ser hasta 3 veces más frecuente que el fenotipo atenuado (Young, et al., 1982), (Martin, et al., 2008).Los pacientes con fenotipo neuronopático tienen un inicio temprano de la enfermedad, exhiben un curso crónico, progresivo y con severo compromiso multiorgánico y del SNC (Yatziv, et al., 1977), (Jones, et al., 2009). La enfermedad es indetectable en la edad neonatal. La sintomatología más precoz es la otitis media (1,2 años de edad en promedio) y la hernia umbilical (1,3 años) 26 (Wraith, Beck, et al., 2008). Sin embargo en un estudio llevado a cabo en Brasil, las primeras manifestaciones ocurrieron a los 18 meses de edad en promedio (rango intercuartil 6 – 24 meses), siendo más frecuentemente reportadas macrocefalia, retraso en el lenguaje y alteraciones de la conducta, en el fenotipo severo. (Schwartz et al., 2007). HOS (Hunter Outcome Survey), es un instrumento de seguimiento observacional a largo plazo, multicéntrico e internacional. Según los registros la media de edad al diagnóstico es 3 años (rango intercuartil 1.3–8.0 años) (Jones, et al. 2009). La afectación cardiaca, neurológica y respiratoria son los elementos que generan más morbimortalidad (Wraith, Beck, et al., 2008), mientras que la obstrucción de la vía aérea es la causa más frecuente de muerte en pacientes con MPS IIA(J. Muenzer, 1986). Según el análisis de HOS realizado por Jones et al., aquellos pacientes con alteración cognitiva fallecen a edad más temprana, lo cual indica que el compromiso neurológico se asocia a una menor expectativa de vida(S. A. Jones et al., 2009). La media edad de muerte para este grupo es de 11,3 años (rango 11.0–19.0 años)(S. A. Jones, et al., 2009). 4.5.2. Fenotipo Atenuado o No neuronopático (MPS II B) El fenotipo atenuado o no neuronopático cursa con mínimo o ningún deterioro cognitivo, aunque los pacientes experimentan todas las complicaciones somáticas del síndrome (Young & Harper, 1982). Al igual que la MPS II A, es indetectable en la edad neonatal. A diferencia del 27 fenotipo severo, se reporta una edad de inicio de síntomas mayor para la MPS II B,de 3,25 años (39 meses, rango intercuartil 9,7- 60 meses) (Swartz, et al., 2007). Las primeras manifestaciones más frecuentes son anomalías esqueléticas, contracturas articulares e hipoacusia (Schwartz, et al., 2007). Los pacientes con fenotipo no neuronopático son generalmente diagnosticados entre los 4 y 8 años de edad (Jones et al., 2009), inclusive en algunos casos reciben el diagnóstico bioquímico en edades postbuberales por sintomatología poliartropática y deformidades esqueléticas (Neufeld& Muenzer, 2001). Tienen una sobrevida mayor e incluso se han reportado casos con descendencia (DiFerrante & Nichols, 1972). En relación con la morbilidad, es común el síndrome de túnel carpiano (Wraith, Scarpa, et al., 2008). Presentan una alta incidencia de alteraciones cardíacas, que son un importante causa de morbimortalidad (Wraith, Beck, et al., 2008). Según los registros HOS la edad media de muerte es 14,1 años (en contraste con la más temprana para el fenotipo A de 11,3 años) (Jones, et al., 2009), pero se reportan casos de sobrevida hasta la quinta o sexta década (Neufeld & Muenzer, 2001). Diagnóstico Criterios clínicos La presencia de signos y síntomas pueden orientar al diagnóstico, pero en edades inferiores a un año se debe prestar atención a la forma sutil en que suelen estar presentes e indagar por ejemplo sobre otitis media a repetición, hernia umbilical y retraso en el desarrollo del lenguaje(Wraith, Scarpa, et al., 2008). 28 Las MPS representan el 32% de los Errores Innatos del Metabolismo, de ahí la importancia de las pruebas bioquímicas que detectan la excreción de GAGs y que pueden orientar al tipo específico de MPS (Neufeld & Muenzer, 2001). Pruebas cuantitativas para mucopolisacáridos (GAGs) en orina CPC (Cloruro de cetilpiridinio): El CPC es un compuesto de amonio cuaternario que se une a los GAGs negativamente cargados precipitándolos. Sus resultados se expresan en Unidades de CPC/gramo creatinina, cuyo valor máximo es 262 U/g (DiFerrante, 1967), (Pennock, 1973). Alcian blue: Es un colorante polivalente catiónico que bajo el mismo principio del CPC precipita los GAGs. Los resultados se expresan en mgGAG/mol creatinina y el valor máximo es 21,4mg/mol (Scott & Dorling, 1965), (Whiteman, 1973). Albúmina Acida: En esta prueba se precipitan los GAGs en un pH ácido y se realiza una medición por espectrofotometría. Valores superiores a 0.040 se consideran indicativos de excreción de GAGs. Las pruebas cuantitativas no están exentas de error, un estudio de Mahalingam et al. Reporta falsos positivos de 28% (n=26/91) y falsos negativos de 45%(n=60/219) (Mahalingam, et al., 2004). Por esto es necesario realizarlas simultáneamente con las pruebas cualitativas. Pruebas cualitativas: electroforesis de GAGs 29 Establecer con precisión cuáles GAGs han sido excretados en la orina permite clasificar al paciente dentro de los tipos de MPS (Leistner & Giugliani, 1998). El exceso de heparán y dermatán sulfato es indicativo de MPS I, II y VI. Un patrón de heparán sulfato solo sugiere MPS III, queratán sulfato solo sugiere MPS IV y dermatan sulfato solo sugiere MPS VI (Neufeld & Muenzer, 2001). Lo anterior se establece de acuerdo al patrón de migración de GAGs en una electroforesis en gel de agarosa, aunque se prefiere la cromatografía en capa fina (Humbel & Chamoles, 1972). La MPS II se caracteriza por la presencia de bandas de migración correspondientes a dermatán y heparán sulfato, sustratos mayoritarios de la IDS, aunque algunas veces también se observa algo de condroitín sulfato(Kimura, Hayashi, & Tsurumi, 1980). Tamizaje de actividad enzimática en papel de filtro Una vez el paciente ha sido clasificado con posible diagnóstico de MPSII, se procede a la medición de actividad enzimática de la Iduronato-2-sulfatasa en papel de filtro como estudio de pesquisa (Martin, et al., 2008). La técnica se desarrolló a partir de la necesidad de un alto rendimiento, menores costos y disminución de las dificultades en el transporte de la muestra, logrando un tamizaje adecuado y oportuno(Dean, Bockmann, Hopwood, Brooks, & Meikle, 2006). Dean et al., reportaron falsos negativos del 5% (n=1) en una serie de 19 pacientes con MPSII. De 64 controles se encontró 1 individuo con baja actividad pero que aún era el doble del valor de la actividad residual más alta del grupo con MPSII, es decir, no obtuvieron falsos positivos (Dean et al., 2006). 30 Confirmación del diagnóstico de MPS II La medición de la actividad enzimática de la Iduronato-2-sulfatasa en plasma, cultivo de fibroblastos o de leucocitos es la prueba confirmatoria o Gold standard para el diagnóstico de la MPSII(Martin, et al., 2008).Los pacientes con MPS II presentan niveles muy bajos o ausencia de actividad de la enzima contra el sustrato artificial Metilumbeliferil-α-iduronato2-sulfato (Clarke, 2006). El grupo de Voznyi y colegas fue el primero en desarrollar el ensayo fluorimétrico en el año 2001, reemplazando así los métodos radioactivos anteriores(Voznyi, Keulemans, & van Diggelen, 2001). Para ello aislaron leucocitos a partir de muestras de sangre heparinizada y cultivaron fibroblastos de piel obtenidos de pacientes con MPS II y controles sanos. Tras la sonicación de las muestras, tomaron homogenados de 10uL y adicionaron 20uL de 1.25 nmo/L de Metilumbeliferil-α-iduronato2-sulfato en 0,1mol/L de acetato de sodio a pH 4,8-5,0 e incubando por 4 horas a 37ºC (Voznyi et al., 2001). La liberación de 4-Metilumeliferona (4MU) requiere la acción secuencial de IDS y la alpha-iduronidasa, por eso se adiciona en un segundo paso, a un pH 4,5 y se incuba por 24 horas. No realizar esta segunda incubación da lugar a falsos positivos. La reacción se detiene llevando la solución a pH de 10,7 (Voznyi et al., 2001). La reacción final produce sulfato y el fluoróforo 4MU. Los equipos para la medición de la fluorescencia emplean una fuente de luz con intensidad capaz de excitar el fluorocromo o fluoróforo de manera que libera fotones, los cuales chocan contra el sensor. La frecuencia de luz censada es transmitida a la interfase digital que ha sido 31 previam mente calib brada para a obtener a partir de e la longitu ud de ondaa las unida ades de concen ntración en n nanomoles (Lokow wicz, 1999)). Los valo ores en nm mol/4horas s/mg en fibrobla astos y leu ucocitos, y en nmol//4horas/mll en plasm ma obteniddos por Vozyni V y colaborradores se e muestran n en la tabla a 1. Tabla 1 1. Actividad de IDS en e leucocittos, plasma y fibrobla astos, tom ado de Vo oznyi et al., 200 01. Deben medirse además a al menos otrras dos sullfatasas en n el laborattorio, entre e alphadasa, Arilssulfatasa B y Beta-G lucuronida asa, con el fin de desscartar defficiencia iduronid múltiple e de sulfattasas, enfe ermedad ccausada por la muta ación homoocigota en n el gen SUMF2 2, que cod difica para a la Enzim ma Genera adora de Formilgliciina (Dierks s et al. 2001). Man nejo de la MPS II 4 4.7.1. Sop porte y manejo interd disciplinario o Los pa acientes con c MPSII requiere en un abordaje integral com menzando por la evaluacción perriódica neurológica n a, cardííaca, res spiratoria, dica ortopéd y 32 otorrinolaringológica, para detectar a tiempo las anomalías y manifestaciones secundarias con el fin de brindar un tratamiento oportuno (Muenzer et al., 2009) Es necesaria la corrección quirúrgica de hernias abdominales, así como de tejidos orofaríngeos hipertróficos y de alteraciones dentales que por su severidad o riesgo lo ameriten (Muenzer et al., 2009). Durante la anestesia general y la cirugía se debe prestar especial atención en prevenir la luxación atlanto-odontoidea(Walker et al., 1994). En relación con la apnea del sueño se recomienda el uso de dispositivos tipo CPAP (continous positive airway pressure) (Leighton et al., 2001). Si hay obstrucción severa y persistente de la vía aérea se debe realizar traqueostomía permanente(J. Muenzer et al., 2009). En relación con el sistema neurológico, se indican anticonvulsivantes en caso de epilepsia. La descompresión quirúrgica del Tunel carpiano tiene buenos resultados en la mayoría de los pacientes (Norman-Taylor et al., 1995). No menos importantes son las terapias física, ocupacional y del lenguaje (Muenzer et al., 2009). 4.7.2. Trasplante de médula ósea Los experimentos de Neufeld en 1968 y de Kaplan en 1977 sentaron evidencia de que la corrección in vitro de los fibroblastos deficientes se produce por captación de enzimas lisosomales mediada por receptor. (Kaplan et al., 1977). Bajo este principio, se realizaron los primeros transplantes de médula ósea alogénico o de células madre hematopoyéticas (HSCT) en MPS I y VI. (McGovern et al., 1986, Shapiro et al., 1995, Vellodi et al., 1992). 33 Bergstrom et al. reportaron en 1994 el primer trasplante de alogénico de médula ósea en un paciente con MPS IIB quien durante 3 años de seguimiento presentó mejoría en las manifestaciones somáticas de la enfermedad (Bergstrom et al., 1994). Velodi y colegas trataron 10 pacientes con síndrome de Hunter, de los cuales dos requirieron un segundo transplante. Dos murieron por sepsis y otros 3 por enfermedad injerto contra huésped. Sólo tres pacientes sobrevivieron más de 7 años post-procedimiento. En dos pacientes hubo progresión de la discapacidad física y mental, mientras que un paciente mantuvo un desarrollo intelectual normal con leve discapacidad física. Así, el porcentaje de éxito fue de 10%, con un fracaso de 90% y mortalidad por el procedimiento de 50% (Vellodi et al., 1999). El mismo estudio concluyó que no hubo mejoría neurológica para pacientes con fenotipo severo y que el único caso que se benefició del tratamiento correspondía a MPS IIB. Además la alta tasa de efectos secundarios (enfermedad injerto contra huésped y sepsis) y la alta mortalidad podía atribuirse al uso de donantes no relacionados (Vellodi et al., 1999). Guffon et al., reportaron sobre ocho pacientes que recibieron entre los años 19992000 el trasplante de médula ósea de donantes HLA-compatibles. La respuesta fue adecuada, alcanzando 95% de proporción de células de donante después de 1mes del trasplante, con mejoría en las visceromegalias, en las anomalías cardíacas y en la rigidez articular. Sin embargo, los síntomas neurológicos tuvieron evolución variable dependiendo del fenotipo (Guffon et al., 2009). 34 4.7.3. Terapia de reemplazo enzimático La Terapia de reemplazo enzimático (TRE ó ERT por sus iniciales en inglés) consiste en la administración intravenosa de la enzima recombinante humana purificada Idursulfasa (Burrow& Leslie, 2008). La Idursulfasa (Elaprase®), desarrollada y patentada por el laboratorio Shire®, es una forma purificada de iduronato-2-sulfatasa producida en la línea celular HT-1080 (células humanas de fibrosarcoma) mediante tecnología de DNA recombinante. La glicoproteína de 525 aminoácidos contiene 8 sitios de N-glicosilación que están ocupados por 2-bis manosa-6-fosfato que contiene cadenas de oligosacáridos (Muenzer et al., 2006).Estos manosa-6-fosfato (M6P) se unen a los receptores de M6P de la superficie celular, permitiendo la subsiguiente internalización y transporte a los lisosomas, donde la enzima es catalíticamente activa. La actividad la de Idursulfasa es dependiente de la modificación postraduccional de la cisteína a Cformilglicina dentro del sitio activo dela enzima. Esta modificación se demostró en 50% de las moléculas de la Idursulfasa (Muenzer et al., 2006). La molécula también contiene glicanos complejos, altamente sialilados, que prolongan la vida media en circulación (Muenzer et al., 2006). García y sus colegas estudiaron los efectos de la Idursulfasa en un modelo de ratón knock out (IDS-KO) (Muenzer et al., 2002). Realizaron infusiones en varias dosis (0,1, 0,25, 0,5, y 1,0 mg / kg) y diferentes intervalos (una vez, semanal, cada dos semanas, y mensual), lo cual resultó en una reducción de los niveles de GAGs en orina y tejidos. Luego, los estudios de seguridad en monos Cynomolgus fueron 35 favorables, permitiendo continuar hacia la fase de ensayos clínicos en pacientes con síndrome de Hunter (Muenzer et al., 2002). La seguridad y eficacia de idursulfasa se evaluó en un ensayo clínico controlado aleatorizado, doble ciego, contra placebo, de fase I / II (Muenzer et al., 2006). En este estudio participaron 12 pacientes, en edades entre 6 y 20 años, sin incluir aquellos severamente afectados. Las características del ensayo clínico controlado se resumen en la tabla 2 (Glamuzina et al., 2011). Tabla 2. Resumen del diseño de estudio y selección de pacientes para los ensayos clínicos fase I/II y II/III Fase I/II Fase II/ III Número 12 pacientes 96 pacientes Criterios de Edad >5 años, talla 119-151cm, Edad 5-31 años, talla 122-130,6 inclusión en capacidad de cooperar. cm. En capacidad de realizar Diagnóstico clínico de MPSII pruebas de función pulmonar, FVC<80% Diagnóstico clínico de MPSII Criterios de Traqueostomía, Transplante de exclusión médula ósea o severo compromiso de SNC Diseño del 6 meses placebo vs 0,15-0,5-1,5 53 semanas con tres brazos: estudio mg/kg/semana de idursulfase Placebo vs idursulfase 0,5 mg/kg Estudio controlado aleatorizado quincenal vs igual dosis semanal doble-ciego contra placebo Estudio controlado aleatorizado doble-ciego contra placebo Puntos de % de cambio de GAG:Cr urinarios Score compuesto: evaluación desde línea de base y a las 5, 13 Test de marcha de 6 minutos primarios y 25 semanas desde línea de base y %FVC predicho. Puntos de Ecocardiograma, distancia en el Cambios en: test de marcha, 36 evaluación test de marcha, función pulmonar, %FVC, volúmenes de hígado y secundarios MRI, arcos de movilidad de bazo por MRI, GAG:Cr, arcos de articulaciones, polisomnografía, movilidad articular. tamaño de hígado y bazo medido por MRI. FVC, capacidad vital forzada Los resultados de la fase I/II se evaluaron a las 48 semanas de tratamiento. La excreción urinaria de GAGs se redujo en 2 semanas de iniciar Idursulfasa, y continuó siendo significativamente reducida después de 48 semanas de tratamiento (p < 0,0001). La mayoría de los pacientes experimentaron reducciones de GAGs en orina cerca de los niveles normales. El volumen de hígado y bazo se redujo significativamente a las 24 y 48 semanas de tratamiento (p<0,01y p <0,001, respectivamente). Sesenta y seis por ciento de los individuos con hepatomegalia y todos los individuos con esplenomegalia experimentaron una normalización de los volúmenes de estos órganos. La distancia de marcha aumentó en un promedio de 48 m(P <0,013) (Burrow & Leslie, 2008). La fase II/III (Tabla 2) fue evaluada a las 53 semanas de tratamiento (Muenzer et al., 2006). Los dos grupos de tratamiento con Idursulfasa exhibieron una mejoría significativa en la variable combinada (marcha y FVC) contra placebo (p = 0,0049 yp = 0,0416, respectivamente). Sin embargo, no hubo diferencias entre los dos grupos de tratamiento. El grupo idursulfasa-semanal experimentó un incremento de 37 m de distancia recorrida en la prueba de caminata de 6 minutos (p = 0,013) en comparación con placebo. Además, el grupo idursulfasa-semanal experimentó un 37 aumento del 2,7% en% del valor de la CVF (p = 0,065) y un aumento de 160 ml en absoluto CVF (p = 0,001) contra placebo (Muenzer et al., 2006) Después de 53 semanas de tratamiento, los niveles de GAGs en orina disminuyeron significativamente en ambos grupos de tratamiento contra placebo (p <0,0001). El 40,6% de los individuos experimentaron normalización de los niveles de GAGs en la orina, mientras que en la mayoría de los pacientes estos niveles se aproximaron el extremo superior de lo normal. El volumen del hígado y del bazo disminuyó significativamente desde el inicio, en comparación con placebo (p <0,0001). Aproximadamente el 80% de individuos con hepatomegalia que recibieron tratamiento con idursulfasa (semanal y cada dos semanas) tenían volúmenes normales del hígado después de 18 y 53 semanas de tratamiento (Muenzer et al., 2006) (Burrow et al., 2008). La FDA aprobó su uso en Julio de 2006. Se recomienda iniciar ERT en el curso temprano de la enfermedad, sin embargo, dado que su paso a través de la barrera hematoencefálica es errático, no se espera mejoría en relación con el compromiso neurológico(Wraith, Scarpa, et al., 2008). Con relación a la Farmacocinética, la concentración sérica de idursulfasa se ha cuantificado usando un ensayo ELISA antígeno-específico. El área bajo la curva de concentración-tiempo (AUC) aumentó de manera proporcional a la dosis cuando esta se incrementó de 0,15 mg / kg a 1,5 mg/kg después de 1 hora de infusión de Elaprase. (Muenzer et al., 2006). La vida media es de 44 +/-19 minutos su 38 eliminación es renal con un aclaramiento de 3 – 3,4 mL/min/kg (DrugBank Database). La dosis inicial recomendada es de 0,5 mg/kg administrados en infusión semanal. La tasa inicial de infusión recomendada es de 8 ml/h durante los primeros 15 minutos, que se puede aumentar en 8ml/hr a intervalos de 15 minutos para restringir la aparición de reacciones adversas, a un máximo de 100 ml / hr. La duración total de infusión es generalmente 1-3 horas, y no debe exceder de 8 horas. Las infusiones pueden administrarse lentas, detenerlas temporalmente o interrumpirlas si se producen reacciones a la infusión. Las reacciones adversas que se pueden presentar son: Cefalea (59-66%) Nasofaringitis (53-59%) Dolor abdominal (50-59%) Artralgias (31-44%) Prurito (19-31%) Rash o Urticaria (16%) Dispepsia (13%) Ansiedad (6-13%) Dolor torácico (13%) Además, Elaprase se pueden causar reacciones anafilactoides en algunos pacientes durante las infusiones. (Muenzer et al., 2006). Anticuerpos IgG contra idursulfasa se detectaron en aproximadamente el 50% de los pacientes después de 27 semanas de tratamiento. Sin embargo, sólo el 31,7% de 39 los pacientes mantiene positivos los anticuerpos después de 53 semanas de tratamiento. No se ha informado sobre asociación alguna entre la presencia de anticuerpos y los eventos adversos o las evaluaciones clínicas. (Muenzer et al., 2006). 4.7.4. Administración intratecal de Idursulfasa La Idursulfasa en su administración intravenosa no atraviesa la barrera hematoencefálica y por ende no corrige las alteraciones del SNC en el fenotipo severo de MPS II (Wraith, Scarpa, et al., 2008). La administración intratecal de la enzima es un abordaje lógico que se ha ensayado con anterioridad en modelos animales de otras Enfermedades de Depósito Lisosomal incluyendo MPSIIIA (Hemsley et al., 2007) y MPS I (Kakkis et al., 2004). La administración intratecal (IT) de Idursulfase en el ratón knock out de MPSII resultó en mejoría del patrón histológico neuronal (Calias et al., 2010) al igual que en perros Beagle (Calias et al., 2010)y por último en monos Cynomolgus (Felice et al., 2011). Una vez demostrada la seguridad de la idursulfase IT en monos Cynomolgus se autorizó el ensayo clínico en humanos (Felice et al., 2011). Desde el año 2012 se dio inicio a la fase de reclutamiento de pacientes con MPSII neuronopático al estudio, con una versión modificada de la enzima recombinante para su administración IT (Joseph Muenzer). 4.7.5. Terapia génica 40 El primer ensayo enfocado en corregir directamente el defecto genético en células MPS II fue realizado Stroncek y colegas en 1999. Emplearon el vector retroviral LS2N con el gen IDS incorporado con el fin de transfectar linfocitos cultivados obtenidos de sangre periférica de pacientes MPS II, pero la proporción de células que al final expresó IDS fue sólo 2.5% (Stroncek et al., 1999). Cardone et al., reportaron el efecto de la terapia génica por medio del vector viral adeno-asociado 2/8 recombinante (AAV2/8), el cual poseía un promotor específico para su expresión en tejido hepático. Emplearon un modelo de ratón knockout con deleción de exones 4 y 5 del gen IDS, y posterior a la administración del vector recombinante observaron completo aclaramiento de los GAGs en los diferentes órganos excepto en cerebro, donde el efecto fue parcial (Cardone et al., 2006). Corradi-Perini y colegas realizaron la corrección genética de Células Madre Mesenquimales (hMSC) obtenidas a partir de pacientes con MPS II empleando un vector retroviral. Dichas células produjeron la corrección cruzada de fibroblastos afectados in vitro. Esta técnica podría representar ventajas para transplante de pacientes, empleando hMSC del mismo paciente y evitando así análisis de histocompatibilidad con donantes (Corradi-Perini et al., 2008). Jung et al. administraron AAV2/8 conteniendo el gen IDS a un nuevo modelo de ratón knockout adulto observando mejoría de la actividad de la IDS. Dicho vector poseía un promotor para expresión del gen en fibroblastos, células linfoides y otros tejidos; sin embargo en cerebro la expresión fue deficiente. Concluyeron que este vector podría ofrecer ventajas para estudios de terapia génica en pacientes con MPS II, debido a su bajo potencial inmunogénico, su capacidad de entrar aún en células 41 que no estén en división e integrarse en un único sitio del genoma de su célula huésped(Jung et al., 2010). Otros ensayos en modelos animales La IDS ha sido rediseñada como una proteína de fusión IgG-sulfatasa para lograr su ingreso al SNC en un modelo de mono Rhesus. Dicha proteína de fusión posee un anticuerpo monoclonal contra el receptor de insulina humana que se comporta como un caballo de Troya molecular para su paso a través de la barrera hematoencefálica (Lu et al., 2011). 4.7.6. Herramientas de prevención y diagnóstico temprano El diagnóstico genético preimplantacional (PGD) permite la identificación de embriones afectados antes de la implantación. Altarescu et al. presentaron el caso de tres familias cuyas mujeres portadoras de mutaciones en el gen IDS fueron sometidas a PGD. Realizaron el análisis genético en 1 ó 2 células de los blastómeros por medio de seis marcadores microsatélites polimórficos. El resultado final en todos los casos fue de neonatos sanos. Adicionalmente, este mismo grupo desarrolló una línea de células madre embrionarias Hunter 46,XX la cual proponen puede ser utilizada para ensayo de fármacos y en el estudio más detallado de la patogénesis de este síndrome(Altarescu et al., 2011). Se debe realizar una tamización en el recién nacido de alto riesgo (con antecedente familiar de MPS II) si se desconoce el diagnóstico molecular de la gestante. Wolfe et al. proponen el uso de espectrometría de masas en tándem (MS/MS) para medir la actividad de IDS en neonatos a partir de manchas secas de sangre, empleando 42 como sustrato umbeliferyl-alfa-L-idurónido-2-sulfato cuya hidrólisis genera un producto no sulfatado que es detectado por MSMS con ionización por electrospray y se cuantifica con un estándar interno marcado con deuterio. Esta metodología sería realizable a gran escala para tamización de MPSII(Wolfe et al., 2011). Se han adelantado otros esfuerzos por detectar la MPS II precozmente a partir de tejido placentario obtenido al nacimiento, hallando una discreta acumulación de GAGs en algunas células endoteliales y pericitos (mas no en fibroblastos). Esto sin embargo requiere confirmación con estudios que involucren un número razonable de pacientes (Baldo et al., 2011). Genética de la MPS II El gen que codifica la IDS, clonado por primera vez por Wilson y colegas (Wilson et al., 1990) está localizado en Xq27.3-q28. Mide cerca de 24Kb y consta de 9 exones (Figura 1). La localización exacta del gen en el cromosoma X es 148, 558, 521-148, 615, 470, en la hebra antiparalela (ensembl.org). Presenta dos variables alélicas y se han reportado 20 SNPs (http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?locusId=3423). La longitud del transcripto primario es de 7619pb. El splicing alternativo del mRNA genera 10 transcriptos más, de los cuales 6se traducen en proteína (ensembl.org), (Malmgren et al., 1995). Las diferencias entre los 11 transcriptos de IDS se observan en la Tabla 3. Tabla 3. Transcriptos del gen IDS. 43 Nombre IDS-001 Exones (codificantes) 9 (9) Longitud (bp) 7619 Longitud proteína (aa) 550 Biotipo IDS-004 IDS-203 IDS-002 IDS-201 IDS-202 IDS-008 IDS-003 8 (8) 5 (4) 3 (2) 7 (4) 2 (1) 7 (1) 9 (5) 1399 2635 1213 1156 1032 572 1529 343 339 179 312 153 86 312 codificante codificante codificante codificante codificante codificante Declinación mediada por mutación nonsense* IDS-005 8 ( 3) 959 106 Declinación mediada por mutación nonsense* IDS-006 IDS-007 7 (0) 3 (0) 945 537 - No codificante No codificante codificante *Se postula que estos transcriptos pasan por un sistema de detección de mutaciones missense que previene expresión de proteínas truncadas o erróneas A. B. 44 Figura 1. A. Posición de los exones en el gen IDS.B. Presencia del Pseudogén (IDSP1) corriente abajo, en orientación contraria al gen. C. Esquema en detalle de la organización de IDSP1, precedido por la región LOC 7277913 con las regiones homólogas a exones 2 y 3 e intrón 2 del IDS; LCR (low copy repeat) homólogo al intrón 7. Regiones CXorf40A y repeticiones L1-Alu-L1 también están implicadas en recombinaciones con el gen (modificado a partir de esquemas de Timms et al., 1997 y de Oshima et al., 2011) Existe un pseudogén (IDSP1) localizado a ~25kb en dirección telomérica al gen, documentado por primera vez por Rathmann y colegas en 1995 (Genbank-EMBL AF011889.1)(Rathmann, et al., 1995).Consta de aproximadamente 1.6kb y su orientación es inversa al gen. Posee regiones homólogas al exón 2, intrón 2 y exón 3 del IDS, además de una región que tiene 96% de homología con el intrón 7. Esta últimaes una repetición de bajo número de copia (LCR)y se encuentra en forma de intrón quimérico 3-7(Figura 1). La presencia delIDSP1favorece rearreglos complejos, que imponen un reto durante el análisis mutacional de los pacientes con Síndrome de Hunter (Bondeson, Malmgren, Dahl, Carlberg, & Pettersson, 1995).Entre estos han descrito inversiones, 45 grandes duplicaciones, grandes deleciones y eventos de conversión (Bondeson et al., 1995) (Lagerstedt et al., 1997) (Bunge et al., 1998), (Lualdi et al., 2005). Un evento de conversión genera un gen IDS que contiene regiones recombinantes con IDSP1 en los intrones 3 y 7. Una timina en la posición 292879 es el punto de ruptura en el intrón 3 que es reemplazado por citosina del intrón quimérico 3-7. De esta manera se yuxtapone también el intrón 7 con su homólogo del IDSP1 y por medio de recombinación no recíproca se producen las regiones recombinantes en los intrones 3 y 7 pero sin alterar los exones en IDS. La secuenciación directa de los exones no puede detectar esta anormalidad (Birot et al., 1996), (Lualdi et al., 2005). Adicionalmente otras regiones en las proximidades del gen y pseudogen como CXorf40A y repeticiones Alu (Figura 1) se han visto implicadas en procesos de recombinación (Oshima et al., 2011). 4.8.1. Epidemiología genética A la fecha han sido descritas más de 350 mutaciones en el IDS (The Human Mutation Gene Database http://www.hgmd.cf.ac.uk/ac/index.php).Hay escasos reportes de mutaciones recurrentes (Froissart et al., 2002; Rathmann et al., 1996; Timms et al, 1997; Wilson et al., 1991; Vafiadaki et al., 1998; Lualdi et al., 2005; Lagerstedt et al., 2000),por lo que se hace indispensable el abordaje de secuenciación total del gen (J. Muenzer et al., 2012). En cuanto a los tipos de mutaciones y su frecuencia global, se encuentra que el 82,6% corresponden a pequeñas alteraciones (Froissart, Da Silva& Maire, 2007). En orden de mayor a menor magnitud dicho grupo comprende mutaciones puntuales – missense y nonsense- (54,9%), pequeñas deleciones/inserciones (17,4%) y 46 mutaciones en sitios de splicing (10,3%). Las grandes alteraciones génicas y rearreglos complejos representan un 17,4% (Froissart, Da Silva, & Maire, 2007). En población Caucásica y de Japón se ha documentado una mayor frecuencia de mutaciones en los exones 9, 3 y 8(Gort, Chabas, & Coll, 1998), (Froissart et al., 1998), (Isogai et al., 1998). Un estudio de China que incluyó 38 pacientes con MPS IIencontró además una importante frecuencia de mutaciones en el exón 2 (Zhang et al., 2011), mientras que en Corea se encontraron con frecuencia mutaciones en los exones 5 y 6 (Sohn et al., 2011). Con relación a América Latina, el estudio de Liessens y colegas encontró en 4 pacientes de Argentina mutaciones en sitios del splicing en los exones 5 y 8, la mutación S132W en el exón 3 y la mutación R468Q en el exón 9 (Liessens et al., 1996). En Brasil, Swartz y colegas reportaron las mutaciones M488I y G498A en un paciente fenotipo neuronopático, mientras que Quaio et al. reportaron la mutación S77D (exón 2) en una familia con casos de fenotipo atenuado (Quaio et al., 2012), y Brusius-Facchin et al. reportaron una gran deleción del gen IDS que involucraba además los genes contiguos FRAXA y FRAXE en tres pacientes con fenotipo severo (Brusius-Facchin et al., 2012). 4.8.2. Correlación genotipo – fenotipo Debido a que el fenotipo algunas veces puede ser confuso, se requiere de metodologías de laboratorio que puedan predecir el desarrollo de la enfermedad, justo antes que las complicaciones aparezcan. El ensayo enzimático para Iduronato 2 Sulfatasa por sí solo no es suficiente para predecir la severidad de la MPS II, por lo 47 que se requiere de otras metodologías para este fin(Parkinson, Muller, Hopwood, & Brooks, 2004). En el caso de algunas MPS, determinadas mutaciones pueden colaborar en la clasificación de los distintos subtipos, sin embargo en MPS II es más complicado. Aunque la mayoría de las mutaciones para Hunter son puntuales, un gran porcentaje de las mismas son privadas y en ellas no es posible la realización de correlaciones genotipo-fenotipo (Moreira da Silva et al., 2001). En términos generales con relación a las correlaciones genotipo fenotipo gran parte de los autores postulan que: Alteraciones genómicas a gran escala, deleciones totales (gen y pseudogen), deleciones parciales, rearreglos gen pseudogen, y pequeñas deleciones o inserciones que alteren el marco de lectura, están asociadas a un fenotipo severo (Froissart, et al., 2007). A continuación se presentan ejemplos destacados: Las deleciones 1220delTT, 1269insCC, 383delAT, 596delAACA, 1148delC, 1063inscambian el marco de lectura y producen codones de parada prematuros, todas identificadas en fenotipo severo (Li et al., 1999). Froissart et al. en 2007 documentaron 21 pacientes con este mismo fenotipo con deleciones R96del, S117del, T118del, R433_G435del, S464_Y466del (Froissart et al., 2007). Inversiones por fenómenos de recombinación fueron reportadas por Lagerstedt et al., 1997 en un paciente, Alves et al. en un paciente y por Lualdi et al. en 5 pacientes; todos presentaban fenotipo neuronopático. 48 En una serie de 155 pacientes europeos, se identificaron inversiones/disrupciones en nueve pacientes (n=9), deleciones parciales del gen (n=6), deleción total del gen (n=6), deleción de exones 4-7 (n=5)y un caso con una gran inserción (kb) involucrando el IDSP1. Todos ellos correspondían a MPS IIA (Froissart et al., 2007) En una serie de 49 pacientes de Corea del Sur, todos aquellos con recombinaciones IDS/IDSP1(n=9) tenían fenotipo severo (Sohn et al., 2012). Pacientes con síntomas atípicos como ptosis palpebral, progresión más rápida semejante a un fenotipo Hurleriano y convulsiones pueden tener deleciones a gran escala incluso afectando el gen FMR2(Probst et al., 2007). Mutaciones missense y en sitios de splicing pueden tener efectos variables en la actividad catalítica de la enzima, lo cual aunado a la baja frecuencia de mutaciones recurrentes complica la correlación fenotípica (Barrager & Cabrera, 2007). La mutación A85T se ha encontrado tanto en fenotipo severo como atenuado, lo cual sugiere variaciones en la transcripción o la influencia de otros factores (Froissart et al., 2007). Las mutaciones de splicing c.104−2A>G, c.241−2A>G, c.241−1G>A, c.418+1G>A, c.880−8A>G dan lugar a proteínas truncadas y se han reportado en relación con fenotipo severo (Alves et al., 2006). La mutación c.1122 C>T (G374G) crea un sitio donador de splicing en el exón 5 dando lugar a pérdida de 60 nucleótidos exónicos pero de manera contradictoria conserva actividad catalítica residual (MPS IIB); similar 49 ocurre con c.542 A>T (N181I), encontrándose en fenotipo atenuado (Froissart et al., 2007). Mutaciones nonsense generalmente se asocian con fenotipo severo: Y234X, E254X, Q298X, Y348X, G365X, Q396X, L482X, W502X, E521X (Froissart et al., 2007) y Q465X (Alves et al., 2006). Como excepciones se encuentran E177X yW475X, asociadas con fenotipo atenuado, y R443X identificada en ambos fenotipos, severo y atenuado (Sukegawa et al., 1995), (Rathmann et al., 1996), (Isogai et al., 1998). 4.8.3. Herramientas de diagnóstico molecular La primera aproximación al diagnóstico molecular de la MPS II fue inmediata a la clonación e identificación del gen. Mediante endonucleasas de restricción, Southern blot y secuenciación directa por dideoxiterminadores (método de Sanger),fue posible identificar grandes deleciones o rearreglos del gen(Wilson, et al., 1991), (SteénBondeson et al., 1992) y en dos pacientes se documentó la deleción completa del IDS(Wraith, et al., 1991). Flomen et al. en Inglaterra, y simultáneamente Bunge et al. en Alemania implementaron la reacción en cadena de la polimerasa (PCR) más secuenciación de Sanger, demostrando así las ventajas de esta metodología para la detección de mutaciones puntuales (Flomen et al., 1992), (Bunge et al., 1992). Oshima et al. emplearon MLPA (Multiplex Ligation-dependent Probe Amplification) en el estudio de dos pacientes, uno de los cuales tenía MPSIIA (del segundo no hay datos clínicos). Esta técnica presenta la ventaja de detectar duplicaciones, las cuales no son detectables por secuenciación. En el primer caso se evidenció una 50 duplicación de ~49kb de la región distal al gen y en el segundo caso una gran deleción que involucraba los exones 1 a 7; estos resultados fueron también reproducibles por Hibridación Genómica Comparativa por Array. A partir de estos hallazgos se plantearon los posibles mecanismos que generaron estos rearreglos complejos, hallando los puntos de ruptura por PCR Primer Walking seguido de secuenciación de los amplicones obtenidos(Oshima et al., 2011). Wei et al.realizaron el estudio molecular de un paciente de origen chino por medio de secuenciación masiva (next-generation sequencing) dirigida a los genes IDS e IDUA (alfa L iduronidasa); estos genes fueron captados específicamente usando la tecnología de chip de hibridación seguido de secuenciación por HiSeq2000 (Illumina®) generando así lecturas de 90pb. El análisis computacional resultado del ensamblaje evidenció una mutación candidata en el gen IDS: una deleción en el exón 9 (c.1270delG) cuya repercusión es un codón de parada prematuro y por consiguiente una proteína truncada (se confirmó dicha mutación por secuenciación de Sanger)(Wei et al., 2011). Si bien hasta el momento hay escasos reportes de mujeres con Síndrome de Hunter, se ha encontrado que pueden desarrollar la enfermedad si presentan una inactivación sesgada del cromosoma X y la mutación se encuentra en el X activo(Zhang et al., 2011). El Ensayo de Inactivación del Receptor de Andrógenos (Kubotta et al., 1999) permite conocer cuál es la proporción de inactivación entre los cromosomas X de origen materno y paterno. Dicho ensayo emplea enzimas de restricción que cortan en sitios del gen activo pero no en el alelo inactivo (metilado), se cuantifica la señal obtenida y se califica como sesgada una relación de 80:20 o 51 mayor. De esta manera se ha dilucidado la causa de la enfermedad en heterocigotas (Kloska, Jakobkiewicz-Banecka, Tylki-Szymanska, Czartoryska, & Wegrzyn, 2011). Proteómica y biología celular de la IDS 3.1.1. Estructura y función de la Iduronato 2 sulfatasa (IDS) La Iduronato-2-sulfatasa humana (IDS) es una enzima lisosomal, monomérica, que hace parte de la superfamilia de las sulfatasas, cuyos dominios son altamente conservados no solo en una misma especie, sino también entre especies diferentes (Diez-Roux & Ballabio, 2005). La secuencia de aminoácidos de IDS corresponde a la traducción conceptual de cDNA, reportada por Wilson et al.(Wilson, Meaney, Hopwood, & Morris, 1993). La secuencia canónica de 550 aminoácidos corresponde a la isoforma1 (Tabla 3). La isoforma 2 producida por splicing alternativo consta de 343 aminoácidos y el cambio WALGEHG → FLMRTNT en el extremo C-terminal. Si bien el splicing alternativo puede generar otras isoformas, falta evidencia experimental de dichas proteínas (Malmgren et al., 1995). Dado que la caracterización completa de la enzima está aún en desarrollo y que su estructura no ha sido aún resuelta por cristalografía de rayos X, gran parte de la información sobre su estructura y función se ha inferido por homología con otras sulfatasas mejor caracterizadas(Miech, Dierks, Selmer, von Figura, & Schmidt, 1998). Los aminoácidos conservados en la IDS son Cys84, Pro86, Ser87, Arg88, Val89, Ser90, Phe91, Leu (Met)92, Thr93, Gly94 y Arg (Lys/Leu)95 (los posibles 52 cambios en otras sulfatasas se indican entre paréntesis)(Dierks, Lecca, Schlotterhose, Schmidt, & von Figura, 1999). Posee un péptido señal de 25 aminoácidos seguido de 8 aminoácidos que son removidos de la proproteína durante su procesamiento postraduccional, y además ocho potenciales sitios de glicosilación (motivos NXS/T) en las posiciones 31, 115, 144, 246, 280, 325, 513 y 537. Estudios de expresión han mostrado que todos estos sitios pueden ser utilizados y que no existe uno en particular que sea esencial para la estabilidad de la IDS, aunque el sitio de N-glicosilación en la posición 280 es importante para el transporte al lisosoma (Millat, Froissart, Maire, & Bozon, 1997). La proteína monomérica está compuesta por dos dominios: un pequeño dominio Cterminal y un gran dominio N-terminal que pertenece a la clase de proteínas α/β el cual contiene el sitio activo. El sitio activo de IDS se caracteriza por poseer un catión divalente (Calcio) y residuos críticos con mecanismos catalíticos que resuelven la estructura intermediaria enzima-éster sulfato. Los aminoácidos presentes en el sitio catalítico se predice son Asp45, Asp46, Cys84, Asp334 e His335, por homología con el sitio activo de las Arilsulfatasas A y B (Dierks, et al., 1999),(Waldow, Schmidt, Dierks, von Bulow, & von Figura, 1999). El residuo Cys84 se encuentra conservado en todas las sulfatasas eucariotas y en la mayoría de sulfatasas procariotas (Dierks et al., 1999). Función de la IDS El primer paso en la degradación de los GAGs dermatán (DS) y heparán sulfato (HS) consiste en la remoción de los motivos 2-O sulfato del ácido urónico que forma 53 parte estos GAGs. Esta reacción es llevada a cabo en los humanos por la Iduronato2-sulfatasa. Al igual que las demás sulfatasas, la IDS requiere de un residuo de Formilglicina (FGly) para ser catalíticamente activa. La formación de FGly es resultado de la oxidación deCys84 por la enzima generadora de formilglicina, en el retículo endoplasmático (Roeser et al., 2006). Los pasos de la reacción llevada a cabo en el sitio activo se han establecido por homología y aún no están del todo esclarecidos. Los aminoácidos del sitio catalítico Asp45, Asp46, Asp334 e His335 poseen cargas positivas que hacen posible la interacción con el sustrato, cargado negativamente. El residuo de FGly es clave en la hidrólisis del O-2-sulfato del ácido urónico, el cual en DS corresponde al Acido Lidurónico (IdoA) y en el caso del HS es Ácido Glucurónico (GlcA). El paso intermediario es la formación de un enlace éster-sulfato, el cual es resuelto con la participación de calcio como cofactor (Bond et al., 1997). Al final de la reacción se libera SO4– y agua (Voznyi et al., 2001). 54 Figura 2. Pasos en la degradación de GAGs. Esquema modificado de Neufeld y Muenzer de acuerdo a los hallazgos de su trabajo y el de Lawrence et al. 3.1.2. Procesamiento diferencial de IDS Distintas formas moleculares de la IDS han sido reportadas en los diferentes tejidos humanos, reflejando un procesamiento diferencial de la enzima. A partir de hígado Bielicki et al. Purificaron la IDS encontrando dos formas de la enzima, de 42 y de 14kDa. Ambas formas mostraron ser activas contra sustratos derivados de heparina 55 y DS. En riñón, placenta y cerebro se detectó la forma de 42kDa (Bielicki et al, 1990). En fibroblastos humanos, un precursor de 76 kDa es convertido en un precursor fosforilado de 90kDa. Este precursor excretado al medio extracelular para luego ser endocitado a través de la vía manosa-6fosfato. Pasa al aparato de Golgi donde, tras la modificación de sus cadenas de oligosacárido, es procesado por escisión proteolítica en pasos sucesivos hasta un intermediario mayor de 55kDa, con la liberación de un polipéptido de 18kDa. Una tiol-proteasa realiza una última proteólisis generando la forma madura de 45kDa, la cual contiene complejas cadenas de oligosacáridos (principalmente glicanos ricos en manosa) (Froissart et al., 1995). 4.9.3. Fisiopatología de la MPS II. Desde los inicios del estudio de la MPSII, quedó claro que la acumulación de GAGs es el principal responsable de las características clínicas del síndrome. Desde 1976 se reportaron los cambios vacuolares citoplasmáticos en hepatocitos, células epiteliales del bazo, células foliculares de la tiroides, células de Sertoli, células cromófobas de la pituitaria y otras células de las meninges, las válvulas cardíacas y el periostio. El principal GAG acumulado en el hígado es heparán sulfato, mientras en el riñón el dermatán sulfato(Nagashima et al., 1976). En las MPS, las vías celulares y bioquímicas secundarias son afectadas por estos cambios: La acumulación del HS extracelular interfiere con la unión de FGF-2 a su receptor y altera la transducción de su señal(Pan et al., 2005). Por otro lado, en modelos animales de MPS se ha reportado la activación de TLR4 (Toll-like 56 Receptor4) y su proteína adaptora MyD88, llevando a una producción aumentada de citoquinas proinflamatorias (Ausseil et al., 2008), (Simonaro et al., 2008). En el tejido neuronal se han estudiado los mecanismos fisiopatológicos involucrados en las Enfermedades de Depósito Lisosomal (LSD) a través de ensayos in vitroy modelos animales(Bellettato & Scarpa, 2010). Estos procesos van más allá del depósito de los productos sensibles al defecto enzimático, pues ocurre simultáneamente la acumulación de diferentes compuestos sin relación con el defecto primario (Figura 3). En efecto, los gangliósidos GM2 yGM3 se acumulan en neuronas afectadas en Niemann-Pick tipo C, MPS I, IIIA, IIIB, VII y Mucolipidosis, pero en vesículas diferentes a las del producto de depósito primario (Walkley et al,. 2004). En las LSD neuropáticas, las manifestaciones en el SNC son pronunciadas aún cuando el almacenamiento es menor en el cerebro que en otros órganos, lo cual se relaciona probablemente con la menor capacidad de regeneración y escasez de vías metabólicas compensatorias en estas células (Futerman&Van Meer, 2004; Tardy et al., 2004). Para la mayoría de LSDs (exceptuando por ejemplo, la Enfermedad de Niemann-Pick) la patología produce más disfunción que pérdida de neuronas (Santavuori et al., 1973; Walkley & Suzuki, 2004). Las neuronas afectadas inicialmente exhiben el depósito del sustrato primario, pudiendo sobrevivir por años mientras continúan secuestrando el material y desarrollando lentamente cambios en la estructura (Walkley 2007). Las neuronas muestran principalmente dos tipos de cambios morfológicos: meganeuritas y esferoides axonales junto con edema somático (Figura 3). 57 Meganeuritas: son enormes ampliaciones del cono axonal que contienen cuerpos de depósito típicos, que a menudo superan el volumen de pericario adyacente. Dos clases de meganeuritas han sido reconocidas: (a) espinosa y cubierto con espinas de tipo dendrítico, expansiones de neuritas y sinapsis, y (b) lisa, sin ninguna evidencia de espinas dendríticas o nuevas formaciones sinápticas. Las meganeuritas están asociadas con un aumento en la expresión del gangliósido GM2 y se ha demostrado que desempeñan un papel fundamental en la regulación de dendritogénesis en las neuronas piramidales corticales (Roisen et al., 1981). Esferoides axonales: son ensanchamientos distales al segmento inicial del axón. Mientras que las meganeuritas contienen el sustrato primario, los esferoides axonales contienen una variedad diferente de materiales con características ultraestructurales que son semejantes a muchos tipos de LSDs (Walkley, 1998). En el cono axonal también pueden surgir nuevas dendritas (dentritogénesis ectópica), generando sinapsis asimétricas potencialmente perjudiciales para la función de la neurona (Walkley y marzo de 1993). Los esferoides pueden causar una interferencia significativa con la eficacia o el tiempo de propagación del potencial de acción (March et al., 1997; Walkley et al., 1991.). Además, se ha demostrado que los esferoides pueden inducir en un bloqueo en la señalización lisosomal que transporta el complejo Factor de Crecimiento-Receptor, crucial para la vida celular –esto explicaría la muerte temprana de las células de Purkinje- (Walkley, 2004). 58 Figura 3. Cambios secundarios en las neuronas encontrados en enfermedades de depósito lisosomal. (Bellettato & Scarpa, 2010). El transporte axonal retrógrado es un importante mecanismo de recaptación de los gangliósidos GM2 y GM3 por medio del endosoma de las terminales axónicas, para ser reciclados (Walkley, 2009). En LSDs se plantea que los cuerpos de depósito en el axón no permiten la fusión lisosoma-endosoma necesaria para la acidificación de este último, alterando el transporte axonal retrógrado (Jeyakumar et al., 2005). La autofagia en las neuronas es esencial para la supervivencia, puesto que proteínas anómalas o agregadas y organelos disfuncionales pueden ser eliminados por este sistema (Tooze & Schiavo, 2008). Se ha demostrado en modelos animales de MPSIV que la autofagia se encuentra comprometida, ocasionando acumulación 59 de proteínas poliubiquitinadas y mitocondrias disfuncionales (Ballabio & Gieselmann, 2009; Tessitore et al., 2009). Están reportadas ciertas anormalidades mitocondriales comunes a distintas LSDs (grandes mitocondrias y acumulación de la subunidad C de la ATP sintasa). Las neuronas son ricas en mitocondrias y son más sensibles que otros tipos celulares, a su funcionamiento anormal (Lucke et al., 2004). Adicionalmente, las ineficientes redes antioxidantes promueven la elevación de especies reactivas de oxígeno (ROS), produciéndose estrés oxidativo y depleción de calcio del RE. Esta elevación del calcio citoplasmático puede activar la UPR (Unfolded Protein Response) y vías apoptóticas mediadas por C-aspasa1 (Breckenridge, 2003; Feng, 2003). A la homeostasis alterada del Calcio, cuya elevación citoplasmática es neurotóxica (Berridge et al., 2003), se suma la del Hierro, similar a lo hallado en enfermedad de Parkinson y Alzheimer. El hierro en el cerebro juega un importante papel (formación de mielina, producción de neurotransmisores como dopamina, norepinefrina, serotonina y en la actividad gabaérgica) (Moos & Morgan, 2004). El material extralisosomal depositado también altera los lípidos de membrana (Futermann & vanMeer, 2004). La inflamación en el SNC es la consecuencia secundaria más prominente y universal del almacenamiento (Jeyakumar et al., 2005), debido a la activación del sistema inmune innato, con células de la microglía y los macrófagos (Wada et al., 2000). La activación de la microglía en la patogénesis LSDs se evidenció en el cerebro de modelos de ratones de MPS I (síndromes de Hurler) y MPS IIIB (síndrome de Sanfilippo tipo B) (Ohmi et al. 2003). 60 Las tauopatías son trastornos neurodegenerativos que tienen en común la acumulación en las neuronas de proteína tau fosforilada, la misma presente en los ovillos neurofibrilares característicos de la Enfermedad de Alzheimer (Duyckaerts & Dickson, 2003). El aumento de proteína tau hiperfosforilada en un modelo de ratón de síndrome de Sanfilippo tipo B (MPS III B), sugieren que la tauopatía es también importante en algunos LSDs (Ohmi et al., 2009). A excepción del SNC, cartílago y hueso, la fisiopatología de las MPS es predominantemente relacionada con aumento en el tamaño de las células, tejidos y órganos (Haskins et al., 2002). Un prominente componente de las válvulas cardíacas es el Dermatán sulfato (Latif et al., 2005). En el corazón la acumulación de GAGs dentro de las células valvulares cambia su forma de fusiforme a redondeada, lo cual se refleja en el engrosamiento de las valvas y cuerdas tendinosas causando estenosis, por lo general con regurgitación. Comúnmente se han encontrado calcificaciones en la región anular de la mitral (Yano et al., 2009; Braunlin et al., 2011). También se ha observado el engrosamiento de la íntima de las arterias coronarias y de grandes vasos (Oudit et al, 2007; Wraith et al., 2008). Una hipótesis de este fenómeno es la activación de la vía del receptor Toll-like 4, que lleva a la sobrerregulación de proteasas e inflamación (Simonaro, 2010) 61 En el hígado, los depósitos de heparán sulfato (HS) son abundantes no sólo en los lisosomas de hepatocitos, sino también de macrófagos perivasculares y células hepáticas sinusoidales, según lo observado en un modelo caprino de MPS IIID (Jones et al., 2004). Por la carga negativa de estos GAGs, móleculas de agua entran a las células causando gran edema. Se ha reportado fibrosis hepática en otros tipos de MPS, pero no en MPS II (Walkley, 2009). Las alteraciones en cartílago son evidentes incluso desde el período embrionario. En un modelo de zebrafish se demostró mediante hibridación in situ la distribución anómala de Sox10, Crestina y una significativa inducción de TGFβ como consecuencia de la subrregulación por IDS, en el cartílago craneofacial en desarrollo (Moro et al., 2010). En los condrocitos, similar a lo ocurrido en SNC, la respuesta inflamatoria desencadenada por el acúmulo de GAGs resulta en apoptosis y liberación de metaloproteinasas, con consecuencias negativas sobre la placa de crecimiento endocondral (Simonaro et al., 2005). La desorganización de la placa de crecimiento ha sido observada en humanos con MPS I y en modelos animales de otras MPS incluyendo MPS VII (Metcalf et al., 2009). Durante el remodelamiento óseo normal, el cartílago es reemplazado por osteoblastos durante el proceso de osificación, proceso en el cual se requiere de osteoclastos para romper el cartílago. La catepsina K es una cisteín-proteasa que juega un importante papel en la resorción ósea llevada a cabo por los osteoclastos (Yasuda et al., 2005). Li et al. demostraron que DS y HS compiten con el condroitín sulfato formando complejos alternos con catepsina K que son colagenolíticamente inactivos (Li et al., 62 2004). Sumado a esto, la vacuolización de los lisosomas en MPS impide el transporte de los fragmentos de colágeno desde las lagunas de resorción hacia la membrana apical, todo lo cual desencadena en un proceso anómalo de osificación (Wilson & Bröme, 2010). En relación con los tejidos de sostén, se demostró alteración en la vía endosomallisosomal y en la fusión autofagosoma-lisosoma en fibroblastos embrionarios de ratón con déficit múltiple de sulfatasas (Fraldi et al., 2010). Recientemente, un estudio in vitro en fibroblastos MPS I, II, IIIA y IVA no evidenció activación de UPR, sin embargo se observó la sobrerregulación de la proteína disulfuro isomerasa A5 (Pdia5), perteneciente a una familia de chaperonas. Esto explica por qué los fibroblastos MPS no fueron más sensibles a apoptosis que los fibroblastos normales 63 5. METODOLOGÍA Tipo de estudio Descriptivo sin prueba de hipótesis. Tamaño de muestra Dieciocho (18) pacientes, colombianos con MPS II. Ingreso al estudio El centro de Investigaciones Bioquímicas de la Universidad de los Andes, ACOPEL y algunos médicos tratantes, brindaron la información socio-demográfica de algunos pacientes colombianos con MPS II. Ellos nos ayudaron a establecer el contacto con cada uno de los pacientes y sus familias para invitarlos a participar de manera voluntaria en este estudio. El paciente o los adultos responsables firmaron el consentimiento informado, el cual contemplo adicionalmente la autorización para la disposición futura de la muestra. El estudio fue evaluado y aprobado por el comité de ética de la Facultad de Medicina de la Universidad Nacional. Fase pre analítica 5.1.1. Toma de muestra Previa aceptación de ingreso al estudio, asentimiento del paciente, asepsia y antisepsia, se procedió a toma única de muestra de sangre periférica de vena 64 braquial, 7 ml de sangre periférica en un tubo vacutainer de color lila (con anticoagulante EDTA). 5.4.2. Extracción de DNA La extracción de DNA genómico se realizó con el kit de extracción UltraClean Blood DNA Isolation Kit® (MOBIO Laboratories, USA) a partir de una muestra de 300 µl de sangre, según las instrucciones del fabricante. El DNA obtenido fue cuantificado por espectrofotometría en un equipo Nanodrop2000®, ThermoScientific®. Fase analítica 5.1.2. Detección de grandes deleciones y duplicaciones del gen por MLPA Se empleó la técnica de MLPA (Multiplex Ligation-dependent Probe Amplification), método que detecta números de copia anormal en una secuencia de DNA dada e incluso distingue entre secuencias que difieren en un solo nucleótido(Schouten et al., 2002). A todos los pacientes se les realizaron MLPA con la finalidad de detectar grandes deleciones o duplicaciones de material genético en el IDS, ya que se reporta en distintas series de pacientes que hasta un 20% presentan rearreglos complejos caracterizados por este tipo de lesiones(Froissart, et al., 2007). Para lo anterior se utilizó el kit SALSA® MLPA® P164 IDS, aportado por la compañía MRC-Holland para esta investigación. Dicho kit consta de 11 sondas para el gen IDS así como otras sondas para el pseudogen IDSP1 y FMR2, además 65 incluye 8 sondas de referencia que detectan diferentes localizaciones en el cromosoma X. MLPA es una forma especial de PCR múltiplex: utiliza dos sondas de oligonucleótidos que hibridizan con la secuencia blanco en un ciclo inicial, después se adiciona a la reacción una ligasa que une estos dos oligonucleótidos, uno de los cuales está marcado con fluorescencia, luego se procede a un nuevo ciclo para la amplificación específica de las mismas sondas (Figura 4) con un único par de primers, ya que los extremos 5’ y 3’ de los oligonucleótidos son iguales. Los productos son separados por electroforesis capilar; las cantidades relativas de los productos de amplificación de las sondas producen picos de diferente altura que se comparan con el DNA de referencia. 66 Figura 4. Amplificación de Sonda dependiente de Ligamiento, en Múltiplex (Shouten, 2002) Esta técnica presenta la ventaja de detectar duplicaciones de material genético, las cuales no son detectables por secuenciación directa. Además permite la cuantificación de hasta 40 secuencias diferentes en una sola reacción. Es altamente sensible (desde 20ng de DNA genómico humano pueden ser empleados) y reproducible. El procedimiento de laboratorio se desarrolló según instrucciones del fabricante siguiendo el nuevo protocolo de 1 tubo, de la siguiente manera: Día 1, reacción de hibridización. Paso de denaturación con 5uL de DNA (100ng por reacción), por 5 minutos a 98ºC, en termociclador. Preparación de master mix: 1,5 ul buffer MLPA y 1,5 uL de probe mix (sondas) por reacción. A la muestra de DNA a 25ºC adición de 3uL de master mix, para volumen final 8uL por reacción. En el termociclador, se continuó incubación 1 minuto 95ºC seguido de 17 horas a 60ºC. Día 2, reacciones de ligamiento y PCR Preparación de master-mix Ligasa: 3uL buffer A, 3 uL buffer B, 25 uL de dH2O y 1 uL de Ligasa-65 por reacción. En el termociclador se llevaron las muestras a 54ºC y adicionó en este punto 32 uL de mix Ligasa a cada tubo para un volumen final de 40uL. 67 El programa se continuó 15 minutos a 54ºC para la reacción de ligamiento, seguido de 5 minutos a 98ºC para inactivación de la enzima, luego mantenimiento de las muestras a 4ºC. Preparación de master mix para la reacción de PCR con 7,5 uL dH2O, 2uL de SALSA PCR primer mix y 0,5uL de SALSA polimerasa. Nota: SALSA PCR primer mix contiene primers marcados con el fluorocromo FAM. A temperatura ambiente, se adicionaron 10 uL de la mix para PCR a cada tubo, para un volumen final de 50uL. Se continuó el programa en termociclador, consistente en 35 ciclos de 30 segundos 95ºC, 30 segundos 60ºC, 1 minuto 72ºC. Finalización 20 minutos a 72ºC y pausa a 15ºC. Se conservaron las muestras a 4ºC en caja protegida de la luz, por un máximo de 5 días para ser llevadas a electroforesis capilar. Se empleó un mínimo de 5 muestras de DNA de pacientes y 3 DNA de controles sanos en cada experimento, y usando la misma cantidad de DNA (100ng) por reacción. También en cada experimento se usó un tubo sin DNA como control de calidad. La electroforesis capilar se realizó en un equipo ABI-Prism 310. La mezcla de inyección consistió en 0,75 uL de producto de PCR (conteniendo flurocromo FAM), 0,5 uL LIZ size standard, 0,75 uL dH2O y 13,5uL de formamida. Se usó un voltaje de inyección de 1.6 kV por 15 segundos, polímero POP4. Incubación a 80ºC por 2 minutos y luego rápido enfriamiento. En el programa Genemapper® se procesaron los datos crudos en formato .FSA provenientes de la electroforesis capilar para asignar los tamaños a los picos que 68 representan los fragmentos. Además se realizó una inspección visual de los electroferogramas buscando los siguientes criterios de calidad de los resultados: a. Presencia de fragmento de 92 nt (dependiente de ligamiento) y productos de amplificación presentes. b. Ausencia de picos de los fragmentos Q en las muestras (los fragmentos Q de 62, 68, 74 y 80 nt son indicativos de baja concentración de DNA y son los únicos que deben aparecer en la muestra sin DNA). c. Señal adecuada de los fragmentos D de 88 y 96 nt (si los productos son mucho más bajos que los de 92 nt, indican denaturación incompleta). d. Altura adecuada de los picos (no menor del 5% de la máxima señal detectada, ni demasiado altos o fuera de escala). e. Ausencia del efecto “ski slope” o declive: picos de las sondas más grandes de MLPA tres veces menores a las sondas más cortas. f. Ausencia de picos extra. g. Ausencia de grandes diferencias en las áreas relativas de los picos que no tienen sentido. h. Las muestras que cumplieron estos criterios fueron llevadas al análisis informático en el software Coffalyser y las que no, se repitieron nuevamente en otro experimento hasta que cumpliesen el control de calidad inicial. El análisis de los fragmentos obtenidos por electroforesis capilar fue realizado en el programa Coffalyser versión 8. Se cargó en el programa la hoja de la mix P164-B1 lote 0110, que contiene las longitudes de los fragmentos clonados por el fabricante.Luego se importaron los datos crudos de los pacientes y los controles, en 69 formato GENEMAPPER.TXT (multiple runs). La verificación de los electroferogramas y archivos .txt de las muestras control permitió identificar y ajustar en el programa la longitud de los fragmentos de X, de Y, de denaturación y de ligamiento para esta población. La opción Autobin on References de Coffalyser ajustó y estableció los fragmentos de los controles como referencia en esta poblaciónse verificó que la desviación estándar para dichos controles fuera menor o igual 0,1.El filtro de los datos arrojó una tabla con los parámetros de control de calidad: concentración de DNA, número de sondas, ligamiento, género (masculino/femenino), fragmentos DD. El programa realiza la normalización dividiendo el área del pico de cada producto por el área del pico de las sondas de referencia. El método de análisis empleado fue population analysis (LS), el cual primero define si hay fragmentos de tamaños extremos sobre todas las muestras y luego somete los demás fragmentos no extremos a un método de regresión, esperando la caída de señal sea linear. Cada sonda se usa separadamente para determinar una razón y la media de todas las razones es la relación final. Cocientes normales (Dosage Quotient) se encuentran entre 0,85 y 1,15. Valores entre 1,35 - 1,55 indican duplicación heterocigota; valores entre 1,7 - 2,2 duplicación homocigota. Entre 0,35 - 0,65 indica deleción heterocigota. Valores fuera de estos intervalos representan número equívoco de copia. 70 Simultáneamente todos los pacientes fueron llevados a secuenciación para la detección de mutaciones puntuales. 5.1.3. Secuenciación Se realizó secuenciación directa de los 9 exones para IDS, teniendo en cuenta su alta heterogeneidad alélicay por los datos arrojados por varios grupos (Froissart et al., 2002; Rathmann et al., 1996; Timms et al, 1997; Wilson et al., 1991; Vafiadaki et al., 1998; Lualdi et al., 2005; Lagerstedt et al., 2000) quienes indican que la gran mayoría de las mutaciones son privadas y hay escasas alteraciones génicas recurrentes. Se emplearon los primers descritos en la tabla 4 modificados a partir de los reportes iníciales de Froissart et al. (Froissart et al., 1998) y Lin et al. (2006) para la amplificación de segmentos génicos y limites intrón - exón de los 9 exones para IDS. En este estudio no se evaluaron los segmentos 5´UTR ni 3´UTR y tampoco las alteraciones epigenéticas (metilación) del gen por limitaciones presupuestales y de cronograma. Tabla 4. Condiciones para la Amplificación de Fragmentos Génicos Exón Primer Secuencia Annealing (ºC) MgCl2 Amplificado (pb) 1 1F GCAAAAAGACGGGTAACTGC 57 1.5 548 1R AGGGAGGAAGGGAGAAGAGA 2F AGGACTCAGGCTTCCTCCTC 56.5 1 429 2R AACAAGATGTCCCGCACAAT 2 Fuente (Alves S. et al, 2006)(Alves, et al., 2006) (Alves S. et al, 2006) (Lau KC y Lam CW, 2008) (Lau & Lam, 2008) (Lau KC y Lam 71 CW, 2008) 3 4 5 6 7 8 9 3F TGGTTTGAGCTCTGCATGAC 3R AGAGGGCTCTGCAAAGACAG 4F GTTCCACTTGCCCATTTGTT 4R ACCAGCTTCACAGAACATGC 5F CGTGAAGGGCTGATTATGTG 5R ATGTAGCCACCTTCCCTGTG 6F ACGTGGGGAATGCTAGTGAG 6R GGTGGAGTTGTGTCTACTGAGA A 7F GATTGGGAGAGATGCACAGG 7R CCACTGGTTCACAAAAGAGAA 8F ACAAGCTGTGGTATGATGAT 8R TAAAGGTGATCTTACTGTCAA 9F AGGTGGTGTTTCTAAACGTCTG 9R CAAAACGACCAGCTCTAACTC 57 1.5 409 57 1.5 275 57 1.5 393 57 1.5 397 57 1.5 335 50.5 1 280 57 1.5 705 (Lau KC y Lam CW, 2008) (Lau KC y Lam CW, 2008) (Alves S. et al, 2006) (Alves S. et al, 2006) (Alves S. et al, 2006) (Alves S. et al, 2006) (Alves S. et al, 2006) (Alves S. et al, 2006) (Alves S. et al, 2006) (Alves S. et al, 2006) (Alves S. et al, 2006) (Alves S. et al, 2006) (Alves S. et al, 2006) (Alves S. et al, 2006) La amplificación de dichas regiones se llevó a cabo en una PCR convencional en un volumen final de 25 uL conteniendo Buffer 1X, MgCl2 1 mM ó 1.5 mM dependiendo del par de primers a utilizar (Tabla 4), 0,2mM de cada uno de los primers, 0,25 mM dNTPs, 1 U de Taq polimerasa (Invitrogen®) y 120 ng de gDNA por reacción. En un termociclador MyClycler®(BIORAD) se emplearon las siguientes condiciones: un primer paso de desnaturalización a 95ºC por 4 minutos, un segundo paso de 35 repeticiones de desnaturalización a 95ºC por 30 segundos, hibridación a diferentes temperaturas de acuerdo al par de primers (ver tabla 4, annealing) por 30 segundos y extensión a 72ºC por 40 segundos. Paso de extensión final a 72ºC por 10 minutos. 72 Los amplicones obtenidos se verificaron en electroforesis en gel de agarosa al 1,7%, 80 V por 40 minutos y utilizando el marcador de peso molecular Mass Ruler® DNA ladder Low Range Se empleó el agente intercalante SYBR Safe® para la visualización de los productos. Con el kit Pure Link® (Invitrogen) se realizó la purificación de los amplicones partir de producto de PCR, empleando para ello 23 uL de producto de reacción, 92 uL de Binding Buffer con isopropanol adicionado (4 volúmenes de producto), se trasladó el volumen total a una columna de sílica del kit y se centrifugó a 10.000g por 1 minuto. En un segundo paso se utilizaron 600 uL de Wash Buffer (+etanol) para lavado de la columna, centrifugando 1 minuto a 10,000 g. Tras evacuación del tubo colector se repitió el centrifugado a 15.000 g por 2 minutos para retirar por completo cualquier residuo de etanol. Finalmente se adicionaron al centro de la columna 35 uL de buffer de elusión (Tris) dejando incubar por 2 minutos a temperatura ambiente para luego centrifugar a máxima velocidad (16.000 g) por 2 minutos, obteniendo finalmente el producto purificado para secuenciar. Para la secuenciación se emplearon por reacción 10uL de producto purificado más 1,2 uL de primer (forward o reverse). Por ende, por cada amplicón se enviaron 2 muestras para realizar secuencia forward y reverse, utilizando el kit de secuenciación Big Dye Terminator 3.1 (Applied Biosystems®), siguiendo instrucciones del fabricante. Los productos de la reacción se secuencia se purificaron nuevamente usando el kit Big Dye® XTerminator™ Purification kit (Applied Biosystems) y fueron analizados en un secuenciador ABI prism 3500 (Applied Biosystems). 73 Con el programa BioEdit Sequence Alignment Editor (Hall, 1999) se revisaron en detalle las secuencias obtenidas verificando así su calidad e idoneidad para los posteriores análisis, procediendo a la edición de cada secuencia y obtención de los respectivos formatos FASTA. Las secuencias FASTA fueron sometidas a comparación contra el genoma de referencia mediante la herramienta BLASTn, para así identificar las mutaciones. Empleando la herramienta Translate de Proteomics tools (ExPASy) se obtuvo la secuencia de aminoácidos de las diferentes mutantes encontradas. 5.4.3. Análisis de recombinantes por RFLPs En aquellos pacientes en que los rsultados de MLPA y secuenciación fueron negativos se aplicó el método rápido de PCR y RFLPs reportado por Lualdi y colaboradores para detección de los cuatro tipos de recombinantes IDS/IDSP1 más frecuentes (Lualdi et al., 2005) (Figura 5). Este método consiste en dos PCR intercaladas por la digestión con enzimas de restricción que reconocen puntos de ruptura donde ocurren dichs recombinaciones (Tabla 5). 74 Figura 5. Representación esquemática de los cuatro rearreglos. La numeración es arbitrariamente designada a partir del 1er nt del intrón 3 del pseudogen IDSP1 (IDS2 en la figura, barras blancas). El gen IDS está representado por barras negras. La caja de líneas verticales es región exclusiva del pseudogen. Las flechas indican los sitios de reconocimiento de las enzimas de restricción, Rec A, B, C o D son los posibles patrones que se identifican con esta técnica (tomado del trabajo de Lualdi et al., 2005) Tabla 5. Set de primers. Tomado del trabajo de Lualdi y colaboradores (Lualdi, et al., 2005). 75 En la tabla 6 se observan las enzimas para la digestión de los productos de la primera y segunda PCR y los probables resultados de acuerdo al tipo de recombinación presente. El producto de la primera PCR es digerido con HinfI para evaluar punto de ruptura y recombinación en el intrón 7. Las recombinaciones en este punto generan fragmentos de 556, 397, 271, 71 y 15 bp. La digestión con SacI también evalúa recombinación en el intrón 7 en diferente punto de ruptura (Tabla 6). Tabla 6. RFLPs de los productos de PCR. Primera PCR Enzima Producto 1315 (WT) Producto 1310 (recombinante) Segunda PCR Producto 1300 (recombinante) Posición HinfI Mismatch IDSP1>IDS GAATC>AGAGG HinfI GAATC>AGAGG 1865-1869 SacI T>C 1344 Eco57I (AcuI) C>T 243 Fragmentos de restricción 451, 397, 271, 110, 71, 15 556, 397, 271, 71, 15. -1310 (Rec tipo A o D) - 679,631(Rec tipo B ó C) 771, 403, 126 (Rec tipo A, B o C) 1174, 126 (Rec tipoD) La digestión de los productos de la segunda PCR permite analizar eventos de conversión en intrón 3. Si el intrón 3 está normal, la digestión con Eco57I (AcuI) genera fragmentos de 771 y 403 bp. La presencia de mismatch IDS2-C>IDS-T causa la pérdida de un sitio de reconocimiento de Eco57I (AcuI), produciendo un fragmento de 1174 bp además de los fragmentos 771 y 403 bp. lo que indica un evento de conversión (ver sección 4.8. Genética de la MPS II y Figura 5) (Birot et al., 1996; Bunge et al., 1998; Lualdi et al., 1995). 76 Fase Post Analítica 5.6.1. Descripción de las variables y análisis estadístico Se planteó una descripción de las principales variables clínicas de los pacientes. En el caso de variables discretas se estimó la frecuencia y el porcentaje entre los pacientes. Para variables continuas se realizó el mismo paralelo pero expresado en medidas de tendencia central y dispersión. Se realizó la prueba no paramétrica de Kolmogorov-Smirnov de una sola muestra para conocer si los valores de actividad enzimática tienen distribución normal (hipótesis nula o Ho). El nivel se significancia para rechazo de la Ho fue 0,05 (CI 95). 2 Una vez cumplida esta condición se realizó test Chi cuadrado (χ ) para evaluar si los datos obtenidos presentan variaciones, aceptando la hipótesis nula (Ho) de la existencia de diferencias estadísticamente significativas cuando p<0,05, con 9 grados de libertad. Para todo lo anterior, se empleó el paquete estadístico SPSS free trial versión 21. 5.1.2. Análisis bioinformático de la repercusión de las mutaciones en la estructura tridimensional de la IDS Dado que no se dispone de una estructura cristalizada de la Iduronato 2 Sulfatasa humana, en primer lugar se realizó el modelamiento tridimensional de la enzima mediante herramientas bioinformáticas. Las plantillas o templetes para este modelamiento se obtuvieron partir de la búsqueda y selección de los mejores homólogos en la base de datos Protein Databank (PDB) mediante la herramienta 77 BLASTp, introduciendo en el campo de búsqueda la secuencia de aminoácidos de la IDS en formato fasta. Para esta búsqueda se empleó la secuencia de la forma madura (residuos de la posición 34 a la 550 inclusive) de la IDS humana (hIDS) depositada en Uniprot bajo el código de acceso P22304. El modelamiento se realizó por threading. Se empleó el paquete de software MOE (Molecular Operating Environment) v.2010("Molecular Operating Environment (MOE), 2010.10; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2010.,") en las instalaciones del laboratorio de biomoléculas de la Pontificia Universidad Javeriana, institución que posee licencia para uso del programa. El grupo de investigación en Macromoléculas de la PUJ brindó el soporte necesario para la realización de todas las simulaciones in silico. El campo de fuerza empleado en MOE fue Amber99, parametrizado para simulaciones en medio acuoso; de aquellos modelos generados por el procedimiento aleatorizado de Boltzmann se seleccionó el mejor para ser sometido a minimización de energía por el programa. La calidad energética del modelo obtenido de la hIDS tipo silvestre se evaluó mediante la herramienta Structure Assessment del servidor Swiss-Model. La calidad geométrica y estereoquímica se examinó por plots de Ramachandran generados por el programa RAMPAGE (Lovell et al., 2003). La medición de la desviación de la raíz media cuadrática (RMSD) se llevó a cabo en MOE frente al templete empleado, para evaluar la precisión del modelo; valores totales de RMSD (promedio de la distancia entre dos átomos de dos proteínas sobrelapadas) de 1,0 o cerca a ese valor representan moléculas con alta similitud estructural. 78 Con la estructura de la hIDS tipo silvestre se realizó el docking molecular contra sus sustratos Heparán y Dermatán Sulfato cuyas estructuras fueron obtenidas de la base de datos PubChem. Para ello se empleó el programa Autodock Vina (Trott & Olson, 2010). Con el fin de simular la modificación post-traduccional del residuo C84 se convirtió a FGly con la herramienta Builder en Pymol. Se evaluó este modelo modificado para asegurar que no se generen alteraciones estéricas o de energía. El resultado del docking se analizó mediante el programa LigPlot+ (Laskowski & Swindells, 2011). 5.6.2.1. Modelamiento y Docking de las IDS mutantes halladas en el estudio Con las secuencias de aminoácidos de todas las mutantes encontradas en este estudio (excepto el fenómeno de conversión) se realizó en MOE el modelamiento de cada una empleando como plantilla el modelo tridimensional de la hIDS obtenido en este estudio. Se midieron los RMSD en cada caso frente a la proteína de tipo silvestre. RMSD totales que con un resultado mayores de 1.5 Å o inferiores a 0.5 Å indicaron un cambio estructural. Además se realizó el cálculo del Área Accesible a Solvente (ASA) empleando el servidor Getarea. El docking molecular se llevó a cabo con Autodock Vina y su evaluación con LigPlot+, con cada uno de los modelos de las enzimas mutantes, y se analizaron las diferencias respecto al docking realizado con el modelo tipo silvestre. La visualización de los resultados se realizó en PyMol™ versión 1.3 de uso académico (Schrodinger, 2010). 79 Flujograma FASE PREANALÍTICA RECLUTAMIENTO DE PACIENTES Y TOMA DE MUESTRA Única muestra de sangre periférica máx. 7mL, en tubo EDTA. EXTRACCIÓN DNA UltraClean Blood DNA Isolation Kit®, según instrucciones del fabricante FASE ANALÍTICA DETECCIÓN DE GRANDES DELECIONES Y/O REARREGLOS GENÓMICOS MLPA a todos los pacientes, con el kit SALSA® MLPA® P164 IDS SECUENCIACIÓN Secuenciación de los 9 exones y límites intrón-exón. Condiciones de primers en Tabla 1. EVALUACIÓN DE RECOMBINANTES FASE POST ANALITICA Descripción de variables sociodemográficas y clínicas de los pacientes. Acercamiento a una correlación Fenotipo-Genotipo. ANÁLISIS BIOINFORMÁTICO Predicción de la repercusión de las mutaciones en la estructura terciaria. 80 6. RESULTADOS 6.1. Características clínicas de los pacientes Un total de 18 pacientes colombianos fueron incluidos en el estudio, con una edad promedio de 10.8 años (SD ±7.8 años), todos de género masculino. Los individuos MPS II 006, 016 y 017 son los únicos pertenecientes a una misma genealogía, todos los demás fueron pacientes no relacionados. En cuanto a sus características fenotípicas, se observó que el Síndrome de Hunter de tipo neuronopático (Hunter A) el más frecuente, correspondiendo al 86.6% de los casos (n=16), mientras el fenotipo no neuronopático se presentó en el 13.3% de los pacientes (n=2). La media de la edad de inicio de síntomas para todos los pacientes fue de 2.5 años (SD±1.55 años), siendo el síntoma más frecuente el retraso del desarrollo del lenguaje (66.7%), seguido de rigidez de grandes articulaciones (26,5%) y neumonía recurrente (20%) (Tabla 7). El paciente MPSII002 falleció durante la fase de reclutamiento por razones inherentes a la enfermedad. Para el fenotipo neuronopático (MPS II A) la edad promedio de aparición de síntomas fue 3.5 años con una SD±1.85 años y los síntomas iníciales más frecuentemente referidos en este grupo fueron retraso en el desarrollo del lenguaje (81%) y rigidez articular (18,8%). Para el fenotipo no neuronopático (MPS II B) fue 81 de 0.88 años (SD±0.17), refiriendo como primeras manifestaciones neumonía, visceromegalias y rigidez articular (Tabla 7). 82 Tabla 7. Características fenotípicas de la muestra en estudio. CÓDIGO Edad (años) ANTECED. FAMILIAR MPSII Edad inicio síntomas PRIMER SÍNTOMA FENOTIPO PROCEDENCIA (años) MPSII001 5 0.83 Retraso del lenguaje Neuronopático Bogotá MPSII002 18 2 Retraso del lenguaje Neuronopático Florencia (Caquetá) MPSII003 8 1 Macrocránea/ Neuronopático San Gil (Santander) Retraso del lenguaje MPSII004 14 MPSII005 2 MPSII006 17 MPSII007 6 MPSII008 10 MPSII009 14 Tío materno Primos, tío materno Hermano 2 Retraso del lenguaje /Fascies tosca Neuronopático La Dorada (Caldas) 0.71 Retraso del lenguaje Neuronopático Duitama (Boyacá) 1.5 Retraso del lenguaje Neuronopático Cúcuta (Norte de Santander) 0.75 Neumonía /Visceromegalias No neuronopático Bogotá 5 Retraso del lenguaje /Fascies tosca Neuronopático Bogotá 3 Retraso del lenguaje Neuronopático Bogotá (padres origen 83 Boyacá) MPSII010 13 3 Neumonía/Rigidez articular Neuronopático Girardot (Cundinamarca) MPSII011 13 Tío materno 3 Retraso del lenguaje /Rigidez articular. Neuronopático Bogotá MPSII012 8 Tío y primo 1.5 Macrocránea/ Retraso del lenguaje Neuronopático Bogotá (madre origen Villavicencio-Meta) MPSII013 7 1.5 Hipoacusia/rigidez articular Neuronopático Bogotá MPSII014 1 0.25 Neumonía Neuronopático Bogotá (madre origen Huila) MPSII015 31 Sobrino 1 Rigidez articular No neuronopático Bogotá (madre origen Huila) MPSII016 16 Primos, tío 2.5 Retraso del lenguaje Neuronopático Cúcuta (misma familia MPS II006) MPSII017 6 Primos, tío 1.5 Retraso del lenguaje Neuronopático Cúcuta (misma familia MPS II006) MPSII018 16 1.1 Retraso del lenguaje /Rigidez articular Neuronopático Manizales (Caldas) 84 85 La media de la edad al diagnóstico fue de 6 años (SD ±4.38 años). El 94,4% (n=17) recibió Terapia de Reemplazo Enzimático (TRE) y actualmente el 83% (n=15) asiste a la administración de TRE en dosis semanal. Tres pacientes no continuaron el tratamiento debido a severo compromiso multisistémico y de la vía aérea en etapas avanzadas de la enfermedad. Tabla 8. Edad al diagnóstico, niveles de actividad enzimática de los pacientes y administración de Terapia de Reemplazo Enzimático (actual o previamente). CÓDIGO EDAD INICIO SÍNTOMAS EDAD AL DIAGNÓSTICO (Años) FENOTIPO ACTIVIDAD ENZIMÁTICA (LEUCOCITOS) TERAPIA DE REEMPLAZO ENZIMÁTICO 1 (años) MPSII001 0.83 1 N 0.71 SÍ MPSII002 2 5 N 0.24 SÍ MPSII003 1 5 N 0.32 SÍ MPSII004 2 13 N 0.19 SÍ MPSII005 0.71 2 N 1.10 SI MPSII006 1.5 13 N 0.40 SÍ MPSII007 0.75 5 No N 0.00 SÍ MPSII008 5 10 N 1.6 *(papel filtro) NO MPSII009 3 11 N 1.8 2 SÍ MPSII010 3 3 N 0.47 SÍ MPSII011 3 6 N 0.85 SÍ MPSII012 1.5 2 N 26** (papel filtro) SI MPSII013 1.5 3 N 1.34 SI 86 MPSII014 0.25 1 N 0.47 SI MPSII015 1 9 No N 9.46***(papel filtro) SI MPSII016 2.5 12 N ND SÍ MPSII017 1.5 2 N ND SÍ MPSII018 1.1 10 N 22.27**(papel filtro) SI Controles: 7.5-55.1 nmol/mg Proteína/hora. 2 48-118 nmol/mg Proteína/hora. 1 Actividad IDS en papel filtro: *ref 12-23, **ref 57 – 149, ***ref71-187 (umoles/l.h). ND: no disponible. Los niveles de actividad enzimática (Tabla 8) fueron analizados por medio del estadístico no paramétrico Kolmogorov-Smirnov, encontrando que siguen una distribución normal con una media de 0,54 y desviación típica 0,41, con un nivel se significancia de 0,674 (no se rechazó Ho=distribución normal de la muestra). La prueba de Chi (χ) cuadrado para una sola muestra arrojó un valor de p = 0.818, con lo cual se rechaza Ho y se concluye que no existen diferencias estadísticamente significativas en los niveles de actividad enzimática en este grupo de pacientes de ambos fenotipos. Los pacientes de este estudio tuvieron una talla sobre +2 SD en los primeros años de vida, que se normalizó hacia los 8 años de edad. Las características comunes a todos los pacientes fueron macrocránea, fascies tosca y hernia umbilical. El 88% presentaba hepatoesplenomegalia. 87 La frecuencia de manifestaciones clínicas relacionadas con mayor morbilidad en los pacientes se distribuyó de la siguiente manera: movilidad articular disminuida 88,8% (n=16/18); hipoacusia 83,3% (n=15/18); infecciones recurrentes de vía aérea superior 72,2% (n=13/18); otitis media aguda 55,6% (n=10/18); convulsiones 44,4% (n=8/18). La frecuencia de anomalías relacionadas con mayor mortalidad fue la siguiente: neumonía a repetición (>2 episodios) 66.7% (n=12/18); hipertrofia de amígdalas y adenoides (relación con obstrucción de la vía aérea superior) 61% (n=11/18); cardiopatía 55,6% (n=10/18). Dentro de las cardiopatías los defectos más frecuentes fueron la afección de la válvula mitral (insuficiencia n=2, mixoma n=1, displasia n=1) que representó el 40% de los defectos cardiacos (n=4/10), seguido de la hipertrofia ventricular izquierda, 20%, (n=1/10) (Tabla 9). 88 Cifosis dorsal Inestabilidad cervical Movilidad articular disminuida SÍ NO SÍ >5 SI Insuficiencia mitral trivial S SI SI NO SI 5 >5 SI displasia mitral y aorta S NO SI SI SI 2 1 2 Engrosamie nto valvas mitrales S SI NO NO SI SI >5 2 3 SI SANO S SI SI NO SI NO SI 2 1 4 DESC SANO S SI NO NO SI NO SI >5 0 0 DES ND S SI NO NO SI +2 50 SÍ SI NO NO NO SI 2 2 1 DESC MPSII 002 28 +3 116 <-3 56,5 +2 62 SÍ SI SI SI SI SI >5 0 1 MPSII 003 22 +1 104 <-3 57 +2 ND SI SI NO NO NO SI >5 0 MPSII 004 36 0 127 <-3 58 +2 ND SI SI SI SI NO SI 0 MPSII 005 16, 9 +2 94 +2 54 +2 58 SI SI NO ND NO SI MPSII 006 30 0 117 -3 57 +2 ND SI SI SI SI NO MPSII 007 16 +1 97 -3 50,5 0 53, 5 NO SI NO NO MPSII 31 0 118 -3 57 +2 78 SI SI NO SI PC (DS) 55 perímetro cefálico 0 Talla (SD) 111 TALLA (cm) +1 DESC Hernia umbilical S SÍ 21 Cardiopatía hipertrofia VI MPSII 001 PESO (Kg) Hepato-esplenomegalia Hipertrofia amígdalas/ adenoides IInfecciones vía aérea inferior Otitis media (EPISODIOS) SI* Anomalías dentales SI Compromiso visual (n óptico) no Hipoacusia NO Convulsiones S SI Facies tosca ND Macrocranea perímetro abdominal Peso –para talla (SD) Infecciones va superior (EPISODIOS) Tabla 9. Características clínicas de los pacientes. 89 008 MPSII 009 31 0 114 <-2 56 +2 66, 7 SI SI SI SI NO SI >5 2 >5 SI* SANO S SI SI NO SI MPSII 010 30 0 112 <-2 57 +2 66 SI SI SI SI NO SI >5 3 >5 SI hipertrofia VI, estenosis aórtica S NO SI NO SI MPSII 011 19, 5 +1 111 <-2 56 +2 65, 8 SI SI SI SI NO SI >5 4 1 SI SANO S SI SI NO SI MPSII 012 28 +2 123 -1 57,5 +2 76 SI SI NO SI ** SI SI >5 5 3 SI Mixoma de v mitral + prolapso S SI SI NO SI MPSII 013 32 +3 124 0 59 +2 67 SI SI NO SI NO SI >5 5 0 SI leve hipertrofia VI S SI NO NO +/ - MPSII 014 14 +2 85 +3 51 +2 50 SI SI NO NO NO SI 4 0 >5 DESC hipertensión pulmonar leve S NO SI NO +/ - MPSII 015 54 0 152 <-2 57 +2 ND SI SI NO SI NO SI 0 1 1 NO Insuficiencia aórtica leve S SI SI NO SI MPSII 016 25 +2 110 <-2 56 +2 ND SI SI SI SI NO SI >5 2 2 SI SANO S SI SI NO SI MPSII 017 24 +2 110 -1 56 +2 ND SI SI NO NO NO SI >5 1 0 DESC ND S SI SI NO SI MPSII 018 32 0 135 <-2 55 +2 73 SI SI SI SI NO SI >5 >5 0 SI Insuf. tricuspídea trivial S SI SI NO SI ND: no disponible. DESC: desconocido. *Requirieron traqueostomía (MPSII 002 y 009). ** confirmada neurosensorial y conductiva. 90 6.2. Detección de grandes pérdidas o ganancia de material genético (rearreglos complejos) Por la técnica de MLPA (Multiplex Ligation-dependent probe amplification) se logró en dos pacientes la detección de dos distintos tipos de mutaciones. En el paciente 001 se identificó ganancia en el exón 4 y en el pseudogen (Tabla 10) En el paciente MPSII012 se identificó ausencia de señal para el exón 7, indicando una deleción total de dicho exón (Tabla 10 y Figura 6). Estos pacientes fueron analizados simultáneamente por secuenciación directa, encontrando en el primer caso la inserción de un nucleótido en el exón 4, que se describe en detalle en la siguiente sección; mientras que en el segundo caso no se produjo amplificación con primers específicos para el exón 7. La verificación en las bases de datos X Chromosome gene database® y Human Gene Mutation Database® de Biobase, aclaró que la deleción del exón 7 es una mutación no reportada. Recuérdese que ambos pacientes son hemicigotos para el cromosoma X. El paciente MPSII 001 presenta valores superiores a 1,3 pero sin representar duplicación (punto de corte para duplicación> 1,7). Obsérvese la deleción homocigota de MPS II 012, casilla en color rojo, valor 0 (cero). Valores normales: 0,85 – 1.15. Tabla 10. Valores normalizados de la longitud de fragmentos de las diferentes regiones del gen IDS analizadas por MLPA. Pacientes MPS 001 y 012. Sample name MV36 X-146.8 FMR1 X-147.6 AFF2 X-147.6 AFF2 X-148.4 IDS Exon 10 X-148.4 IDS Exon 09 X-148.4 IDS Exon 08 X-148.4 IDS Exon 07 X-148.4 IDS Exon 06 X-148.4 IDS Exon 05 X-148.4 IDS Exon 04 X-148.4 IDS Exon 03 X-148.4 IDS Exon 02 X-148.4 IDS Exon 01B X-148.4 IDS Exon 01A X-148.4 IDSP1 Intron 10 X-148.4 IDSP1 Exon 10 MPSII001 Reco Length (nt) mmen ded Ratio Order 1 Normal (0,84) 166 2 Normal (0,95) 337 3 Normal (0,92) 355 4 Normal (1,17) 265 5 Normal (1,03) 238 6 Normal (1,15) 212 7 Normal (1,04) 184 8 Normal (0,93) 328 9 Normal (1,09) 310 10 Gain (1,31) 276 11 Normal (0,92) 256 12 Normal (1,17) 229 13 Normal (1,21) 205 14 Normal (1,02) 148 15 Normal (1,26) 292 16 Gain (1,4) 319 17 Normal (0,99) 301 18 Normal (0,85) 190 19 Normal (0,84) 247 20 Normal (1,05) 136 21 Normal (0,95) 373 22 Normal (1,03) 198 23 Normal (1,11) 220 24 Normal (1,01) MPSII012 Ratio Normal (0,94) Normal (1) Normal (0,99) Normal (0,99) Normal (0,99) Normal (1,11) HO Loss (0,02) Normal (0,97) Normal (1,03) Normal (1,08) Normal (0,99) Normal (1,01) Normal (0,93) Normal (0,98) Normal (0,94) Normal (1) Normal (0,92) Normal (0,92) Normal (1,05) Normal (1) Normal (0,97) Normal (1,08) Normal (1,04) Normal (1) 92 MLPA ‐M MPSII012 1,2 1 Ratio 0,8 0,6 0,4 0,2 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 0 Mapview Figurra 6 .Delección completa del exxón 7 en el paciente MPSII0122. Arriba:diagrama técnicco (Technical Chart), el rombo o azul muy y por debajo de los ppuntos de corte – líneass roja y amarilla, indica la no o amplifica ación de la as sondass para el exón e 7. Abajo o: diagrama de barra as, en el e eje X los fragmentos f s específiccos y en el e eje Y cocientes o índ dices norm malizados, el fragme ento del ex xón 7 (7) sin señal alguna (cero)). 93 6.3. Identificación de mutaciones por secuenciación directa Por medio de primers modificados de de Froissart et al. (1998) y Lin et al. (2006) se realizó la secuenciación de los 9 segmentos génicos (exones y límites intrón-exón). El alineamiento de las secuencias frente al genoma de referencia permitió identificar las mutaciones puntuales, que se resumen en la tabla 11. Estas fueron verificadas en las bases de datos X Chromosome Gene Database® y Human Gene Mutation Database® de Biobase, aclarando de esta forma si se trababa de mutaciones ya reportadas o privadas. En los pacientes MPS II 004, 013 y 014 no se detectó anormalidad en el MLPA y las secuencias de los exones y regiones flanqueantes fueron normales. Por lo tanto estos pacientes fueron llevados a análisis de recombinantes. Tabla 11. Resultados de genotipificación y MLPA. CÓDIGO MUTACIÓN (DNA) Fen. Reporte MPSII001 c.477 insT N PRIVADA MPSII002 c.548_564dupTTGCCCTGTGGATGTG N PRIVADA Whitley (1993); Villani (2000); SukegawaHayasaka (2006) MPSII003 c.1403 G>A N MPSII004 MLPA y secuenciación normales (se llevó a análisis de recombinantes) N MPSII005 c.708G>C N Gucev (2011) MPSII006 c.1393C>T N Li (1996) J Inherit Metab Dis 19, 93 94 No N Froissart (1998) Clin Genet 53, 362 c. 212G>A MPSII007 ( polimorfismo c.438 C>T) MPSII008 c.1403 G>A N Whitley, Villani. MPSII009 c.1039A>C N Lissens (1997) J Inherit Metab Dis 20, 453 MPSII010 c.595_607delACAGAGCACTGA N PRIVADA MPSII011 c. 549_562delTGCCCTGTGGATG N PRIVADA MPSII012 Deleción exón7 N PRIVADA MPSII013 MLPA y secuenciación normales (se llevó a análisis de recombinantes) N Lualdi (2005). Hum Mut 25,491) MPSII014 MLPA y secuenciación normales (se llevó a análisis de recombinantes) N c. 212G>A MPSII015 No N Froissart (1998) Clin Genet 53, 362 (polimorfismo c.438 C>T) MPSII016 c.1393C>T N Li (1996) J Inherit Metab Dis 19, 93 MPSII017 c.1393C>T N Li (1996) J Inherit Metab Dis 19, 93 MPSII018 c.182 delC N PRIVADA 95 Figura 7. Mutaciones privadas (nuevas) encontradas por secuenciación y MLPA. MPSII 010: N; c.596_608delACAGAGCACTGA Del Q200-E203 WT190 Mut VPEGTLPDKQSTEQAIQL 190 VPEGTLPDK----QAIQ 96 - MPSII018; N. c.182 delC. P62QfsX67 Wt:C T G G T G A G G T C C C C A A A T A T T G A C C A A C T G G *publicación de fotografías de los pacientes con autorización por escrito de sus padres. Todos los pacientes de este grupo presentaban fenotipo neuronopático. 97 6.4. Análisis de recombinantes por RFLPs Dado que el análisis con MLPA y secuenciación directa de exones no permitió establecer el diagnóstico molecular en los pacientes MPSII 004, 013 y 014, se llevaron al método de PCR de Lualdi y colegas (Lualdi et al., 2005). El producto de la primera PCR fue digerido con HinfI para evaluar punto de ruptura y recombinación en el intrón 7. Las recombinaciones en este punto generan fragmentos de 556, 397, 271, 71 y 15 bp. Ninguno de los pacientes presentó este tipo de recombinación (Figura 8). La digestión con SacI también evalúa recombinación al mismo nivel y en concordancia con lo obtenido con HinfI, el resultado fue negativo para recombinación en el intrón 7 en estos pacientes (Figura 9). Ladder(bp) L C 004 013 014 018 L 500 350 300 250 200 451 397 271 100 Figura 8. Digestión con HinfI (PCR1). L: ladder. C: control sano.Se usó al paciente 018 como control adicional, pues tiene mutación puntual. Todos fueron negativos para este tipo de rearreglo involucrando el intrón 7. 98 La digestión de los productos de la segunda PCR permitió analizar eventos de conversión en intrón 3. Si el intrón 3 está normal, la digestión con Eco57I (AcuI) genera fragmentos de 771 y 403 bp. La presencia de mismatch IDS2-C>IDS-T causa la pérdida de un sitio de reconocimiento de Eco57I (AcuI), produciendo un fragmento de 1174 bp además de los fragmentos 771 y 403 bp. Para el paciente MPSII013 se logró identificar este tipo de recombinación en el intrón 3, que sugiere un evento de conversión (Figura 9). 1 2 3 4 5 6 1 2 3 4 5 6 Figura 9. Izquierda digestión de PCR 1 con SacI, generando únicamente fragmentos de 1310 bp (ladder de 1000 bp), es decir, negativo para recombinación en intrón 7. Derecha: digestión de productos de PCR 2 con AcuI. 1, control negativo; 2, MPSII004; 3, MPSII013; 4, MPSII014; 5 y 6 controles negativos. Obsérvese el producto de 1174 bp en canal 3, correspondiente a MPSII013. Ladder 1000 pb. Por tanto se establece que el paciente MPSII013 presentó un evento de conversión, previamente reportado por Birot et al. en 1996, Bunge et al. en 1998 y luego por Lualdi y colaboradores (Lualdi et al., 2005). 99 Los pacientes MPSII 004 y 014 4, a pesa ar de que e presentaaban activ vidades enzim máticas de e 0,19 y 0,47 nm ol/mg/hora a respectivamente (control 7.5-55.1 7 nmol//mg Proteina/hora) y de que s us caracte erísticas erran de Sínndrome de Hunter (MPS SII) no pud dieron ser diagnosticcados molecularmen nte con laas técnicas s arriba anota adas (Figurra 10) Figurra 10. Izqu uierda, pac ciente MPS SII012. Derrecha, pac ciente MPS SII014. Obs sérvese el fen notipo com mpatible co on síndrom me de Hun nter. El aná álisis moleecular con MLPA, secue enciación y RFLPs fue negattivo en es stos dos pacientes. p Publicac ción de fotogrrafías con consentim miento por e escrito de los padres s. 6.5. Clasificcación de las mutacio ones encontradas La ttabla 12 reúne r toda as las mu taciones encontrada e as en estee estudio. En 18 paciientes que e participarron se iden ntificó un total t de 12 2 mutacionnes de las s cuales seis son nuevvas y seis s son pre viamente reportadas s. De las seis mutaciones repo ortadas que fueron id dentificada as en este estudio, 5 fueron misssense y 1 fue de 100 tipo recombinación. Se encontraron 6 mutaciones nuevas en este estudio, 5 de tipo frameshift y 1 con deleción de 4 aminoácidos, sin alteración en el marco de lectura. Llama la atención que la mayoría de mutaciones nuevas se localizan en el exón 5. Ninguna de las mutaciones nuevas fue de tipo missense. (Tabla 13). Tabla 12. Mutaciones identificadas por MLPA, secuenciación y RFLPs. CÓDIGO MUTACIÓN (DNA) Fen. Reporte MPSII001 c.477 insT N PRIVADA MPSII002 c.548_564dupTTGCCCTGTGGATGTG N PRIVADA Whitley (1993); Villani (2000); SukegawaHayasaka (2006) MPSII003 c.1403 G>A N MPSII004 Técnicas MLPA+Secuenciación+RFLPs no detectaron lesión N MPSII005 c.708G>C N Gucev (2011) MPSII006 c.1393C>T N Li (1996) J Inherit Metab Dis 19, 93 MPSII007 c. 212G>A No N Froissart (1998) Clin Genet 53, 362 N Whitley, Villani. ( polimorfismo c.438 C>T) MPSII008 c.1403 G>A MPSII009 c.1039A>C N Lissens (1997) J Inherit Metab Dis 20, 453 MPSII010 c.595_607delACAGAGCACTGA N PRIVADA MPSII011 c. 549_562delTGCCCTGTGGATG N PRIVADA MPSII012 del. exón7 N PRIVADA MPSII013 Conversión por recombinación en intrón 3 N Lualdi (2005). 101 Hum Mut 25,491) MPSII014 Técnicas MLPA+Secuenciación+RFLPs no detectaron lesión N c. 212G>A MPSII015 No N Froissart (1998) Clin Genet 53, 362 (polimorfismo c.438 C>T) MPSII016 c.1393C>T N Li (1996) J Inherit Metab Dis 19, 93 MPSII017 c.1393C>T N Li (1996) J Inherit Metab Dis 19, 93 MPSII018 c.182 delC N PRIVADA Tabla 13. Clasificación de Mutaciones pacientes colombianos con MPSII. CÓDIGO PACIENTE MUTACIÓN A NIVEL DE PROTEÍNA Fen. c.1403 G>A R468Q N 9 c.1393C>T Q465X N 9 MPSII009 c.1039 A>C K347Q N 8 MPSII005 c.708G>C K236N N 5 c. 212G>A S71N No N 2 MUTACIÓN (DNA) EXÓN MISSENSE MPSII003 MPSII008 MPSII006 MPSII016 MPSII017 MPSII007 MPSII015 102 FRAMESHIFT MPSII001 c.477 insT P160S fsX4 N 4 MPSII002 c.548_564dup TTGCCCTGTGGATGTG D190PfsX13 N 5 MPSII011 c. 549_562del TGCCCTGTGGATG N 5 P62QfsX67 N 2 R294GfsX2 N 7 MPSII018 MPSII012 c.182 delC Del. Exón 7 P185WfsX23 PERDIDA DE AMINOÁCIDOS SIN CORRIMIENTO DE MARCO DE LECTURA MPSII010 c.595_607del ACAGAGCACTGA Del Q200_E2003(QSTE) N 5 - N - RECOMBINACIÓN IDSP1/IDS MPSII013 Conversión, intrón 3 Nota: todas las mutaciones missense y la recombinante son reportadas. Las demás fueron privadas de este estudio. 6.6. Frecuencia de las mutaciones encontradas en los pacientes MPS II Se identificaron mutaciones recurrentes en el 58% de los 14 alelos que recibieron diagnóstico molecular, mientras que en el 42 % restante se identificaron mutaciones privadas (Figura 11). La mutación R468Q se identificó en dos alelos 103 difere entes (dos pacientes), igual qu e la mutac ción S71N. Las demáás mutacio ones se prese entaron cad da una en un único p paciente. Figurra 11. Disttribución de d frecuen ncias muttaciones privadas p y reportad das. A la izquierda, las mutaciones repo ortadas en conjunto (amarillo). ( Discrimina adas en das en es ste estudioo. Los ind dividuos otros colores las mutaciiones nue vas hallad perten necientes a una misma genea logía fuero on contado os como unn solo alelo o. En el grupo o de las mu utaciones reportadas r s se incluye e el evento o de Conveersión encontrado media ante RFLP Ps. 104 6.7. Estudio de portadoras Familia MPSII 0012 Se realizó estudio de portadoras en tres mujeres de la genealogía del paciente MPSII012 (Figura 12), confirmando el estado de portadoras en III.5 y III.8 (Figura 13). Para la pariente III.4 el resultado de MLPA fue normal, excluyendo el estado de portadora (Figura 14). Por ultrasonografía de tercer trimestre se confirmó que IV.2 es femenina. Figura 12. Pedigree del paciente MPSII012. 105 MLPA ‐III.5 1,2 1 Ratio 0,8 0,6 0,4 0,2 15 17 19 21 23 15 17 19 21 23 13 11 9 7 5 3 1 0 Mapvview M MLPA ‐III.8 1,4 1,2 Ratio 1 0,8 0,6 0,4 0,2 13 11 9 7 5 3 1 0 Mapvview Figurra 13. Porrtadoras: arriba, a III.5 5; abajo III.8 (ver genealogía)). Diagramas de barras, eje X fragmenttos especcíficos, eje e Y ratio o cocie nte normalizado, Obsé érvese para el fragm mento del exón 7 el valor del cocientee entre 0.3 35-0.65, indica ativo de de eleción hete erocigota. 106 MLPA‐ III.4 1 1,2 1 Ratio 0 0,8 0 0,6 0 0,4 0 0,2 23 21 19 17 15 13 11 9 7 5 3 1 0 Mapvview Figurra 14. Excllusión de estado e de portadora de III.4 po or MLPA, ccuyo resulttado fue norma al. Famillia MPSII 002 0 Por m medio de secuenciac s ción se pud do establecer el esta ado de porrtadoras pa ara tres mujerres emparrentadas en e primer grado con el pacie ente MPS II002. El análisis enzim mático en papel p filtro realizado a dos de ellas (herm manas) arrrojó una actividad de 49 9,11 y 23,9 98 umol/l.h. (control 5 57,39-149 umol/l.h). En todas ellas, a partir de e la posició ón c.548 inicia la lectu ura de seccuencia me ezclada, mente suge estivo de la a existencia a de dos alelos a distin ntos (uno ccon la dup plicación altam y otro o tipo silve estre), es decir, d el esstado de heterocigoc h cidad de laa mutación n en los tres ccasos. Se realizó el experimen nto por duplicado (ca ada muesttra en una a sesión difere ente) para corrobora ar que los amplicone es no estu uviesen coontaminados. Los datoss fueron reproducible es y ademá ás la línea de base de d la secueencia demostraba purezza suficientte (Figura 15) 107 Figura 15. Estudio de portadoras (A., madre; B. y C., hermanas del probando). Como la mutación se encuentra corriente arriba del exón 5 se analizaron las lecturas reverse para obtener la mayor longitud posible de secuencia no mezclada, verificando así la confiabilidad del experimento. Nótese que la secuencia 3’CACATCCACAGGGCAA 5’ es la complementaria de la región duplicada y que justo después de ella se observa “sobrelapamiento” de los picos. 108 6.8. Predicción de la repercusión de las mutaciones en la estructura tridimensional de la IDS 6.8.1. Modelamiento de la hIDS tipo silvestre El modelamiento de la IDS se realizó por threading. La búsqueda en PDB arrojó como mejores plantillas las estructuras 3ED4 y 1AUK.A, homólogas de la IDS. Mediante el empleo del software MOE al realizar el modelamiento se encontró que 1AUK.A (cadena A de la Arilsulfatasa humana A) presenta una disposición espacial de alfa hélices y hojas beta que se superpone con mayor precisión a la IDS humana y a pesar de su cobertura de 44% e identidad de 40% los residuos catalíticamente activos se encuentran cubiertos por completo por esta plantilla. La superposición de la arquitectura del sitio activo tuvo una precisión y un RMSD mejor que el obtenido con 3ED4. La figura 16 muestra la estructura resultante superpuesta con su 1AUK.A. El RMSD fue 0.97, indicando una alta precisión del modelo. 109 Figura 16. hIDS (azul) y el molde 1AUK.A (Amarillo). Environment v.2010. Molecular Operating La herramienta Structure Assessment tool (Swiss-Prot/ ExPASy) fue empleada para la evaluación estereoquímica y energética del modelo. El plot de Anolea 110 (packing quality of the models) mostró que 85% de los residuos tienen valores negativos (energías favorables), mientras GROMOS (empirical force field energy) dio un 75%. La energía del modelo por Dfire fue -644, lo que sugiere una conformación nativa. El QMEAN6 Z-score fue -8.46, (valores para estructuras resueltas por difracción entre 0 y 1) lo que podría estar explicado por el porcentaje de cobertura del template. El plot de Ramachandran (Figura 17) muestra que 75% de los residuos están en la región favorable, 16.3% en la región permitida y 8.3% en la región desfavorable. Los resultados indican que los ángulos diedros φ y ψ en el modelo de la hIDS son razonablemente acertados. Figura 17. Plot de Ramachandran. RAMPAGE.75% de los residuos en la región favorable, 16.3% permitida y 8.3% desfavorable. Los resultados indican que los ángulos diedros φ y ψ en el modelo de la hIDS son razonablemente acertados. 111 6.8.2. Docking Molecular de la hIDS tipo silvestre La hIDS fue sometida a docking molecular contra su sustrato Heparán sulfato (HS). El modelo 1 fue seleccionado por su mejor energía global (-8.5 kcal/mol) y su orientación espacial dentro del pocket catalítico. La evaluación con Ligplot+ permitió identificar las interacciones proteína-sustrato, que fueron principalmente de tipo electrostático. Las interacciones significativas involucran el O-2 sulfato del Ácido Glucurónico (GlcA) con Cys84, Tyr165, Leu244 y Arg297. Interacciones electrostáticas fueron también vistas entre el grupo carboxilo de GlcA y Asn106 (Figura 18). En la porción externa del pocket, tres puentes de hidrógeno se observaron entre un monómero diferente de GlcA, Leu189 y Asp187. El resultado del docking realizado contra Dermatan sulfato (DS) fue muy similar a lo descrito para HS, involucrando al Acido idurónico (IdoA) en lugar de GlcA. 112 Figurra 18. Do ocking de la hIDS tip po Silvestre. Arriba, residuos localizado os en el sitio activo de e la enzim ma, incluyyendo Cys s84 (modificada a FGly parra esta bajo, detalle e. Note el grupo O-2 2 sulfato del GlcA, qque dista 3.6 3 Å de simulación). Ab la cistteína 84 (C Cys84). 113 6.8.3. Modelamiento y docking de las IDS mutantes Cada mutante fue modelada independientemente en MOE, empleando como plantilla la estructura obtenida de la hIDS silvestre. En la tabla 14 se resumen los RMSD y ASA para cada uno de los modelos. Tabla 14. RMSD y ASA calculados para cada modelo de las IDS mutantes, empleando los programas MOE y Getarea, respectivamente. MUTACIÓN A NIVEL DE PROTEÍNA Fenotipo Wild type - - Å x 103 2,15 R468Q N 0,18 2,3 Q465X N 0,28 2,0 K347Q N 0,13 2,2 K236N N 0,21 2,1 S71N No N 0,19 2,33 P160S fsX4 N 0,38 2,25 D190PfsX13 N 0,27 3 P185WfsX23 N 0,34 3 P62QfsX67 N 0,17 3,3 R294GfsX2 N 0,24 3 Del Q200_E2003(QSTE) N 0,62 RMSD (Å) ASA 2 2,4 Se describen a continuación las características de los modelos generados y el docking de cada mutante por separado. 114 R468Q La alteración del procesamiento postraduccional de esta mutante ha sido demostrada experimentalmente: en un Western blot del extracto protéico obtenido a partir de células COS7 transfectadas con esta mutante, sólo fue encontrado el precursor de la enzima (75kDa) (Charoenwattanasatien et al., 2012). Por consiguiente, la secuencia del precursor de 550aa fue empleada para construir el modelo. La figura 19 ilustra las interacciones, el grupo O2-sulfato del ácido urónico del sustrato interactúa principalmente con Arg 297 formando con éste un puente de hidrógeno; otras interacciones electrostáticas fueron con Tyr105 y Asn106. Es de resaltar que Cys84 interactúa con el O3, mas no con el sulfato a ser removido. 115 Figurra 19. Docking de R4 468Q. Cys 84 interacttúa con el O3, mas nno con el sulfato s a ser re emovido. Otras O intera acciones ccon el O-2 sulfato se e presentarron con ph he105 y Asn10 06, ademá ás de un pu uente de h hidrógeno con c Arg 29 97. 5X Q465 No se e observó interacción entre el ligando y Cys84 en este dockking. Se fo ormó un puentte de hid drógeno entre e el O-2 sulfa ato y Asn n167. Otrras interacciones involu ucraron Tyyr300, Arg2 297, Leu31 8 y Phe10 05 (Figura 20). 116 Figura 20. Docking de Q465X. Arg 297 es el único residuo que figura también en el docking de hIDS silvestre. No aparece Cys84 en el resultado de esta simulación. K347Q En el docking de K347Q, el O-2 sulfato del ácido forma puentes de hidrógeno con Asp45 y Arg297 (Figura 21). 117 Figurra 21. Muttante K347 7Q. Leu244 4, Asn106 6, Cys84 y Arg 297 ttambién ap parecen en el Wild type, sin embarrgo los pue entes de hidrógeno h con c este úlltimo y con n Asp45 son u un hallazgo o diferente.. K236 6N Se ob bservaron interaccio ones simila ares a las observadas con K3347Q, pero o sin la forma ación de los puentes de hidróge eno. 118 S71N N Se ob bservó un resultado en el doccking muy y similar al wild typee, excepto por los puenttes de hidrrógeno del O-2 sulfatto con Arg2 297 y Asp4 45 (Figura 23). Figurra 23. Doc cking de S71N. Del Q Q200_E203 3 Las interaccion nes electro ostáticas in nvolucraron O-2 sulffato con LLeu240, Ty yr165 y Cys84 4 (Figura 24). 2 119 Figurra 24. Doccking de la a mutación n privada Del Q200 0_E203. Am minoácidos como Asn10 06 Tyr 165 5 y Cys 84 aparecen también en e el wild ty ype pero n o Leu240. R294 4GfsX2 (De el. Exón 7) 84 interactú úa electros státicamen nte con el grupo g carboxilo del IddoA/GlcA en e lugar Cys 8 de co on el O-2su ulfato (Figu ura 25). 120 Figura 25. R294GfsX2 (Del. Exon 7). No se observaron interacciones electrostáticas importantes con el O-2 sulfato del ácido urónico. Con otros grupos de IdoA/GlcA interactúan Cys84, Ala82, Val83 y Asp45. Las nuevas mutaciones tipo frameshift (con corrimiento del marco de lectura) P160SfsX4, D190Pfs13, P185GfsX2 y P62QfsX67 pierden la gran mayoría del sitio catalítico y tienen menos de 203 aminoácidos. El docking molecular en estos casos no tiene utilidad y las estructuras de estas proteínas truncadas se muestran en la Figura 26. 121 Figura 26. Verde, P160SfsX4; azul, P185GfsX2; magenta, D190Pfs13X. Obsérvese la severa distorsión de la geometría de estas estructuras y gran cantidad de coils. Pymol v 1.3. 122 7. DISCUSIÓN 7.1. Características clínicas Este estudio incluyó un total de 18 pacientes colombianos con Síndrome de Hunter para su caracterización fenotípica, genotípica y realización de un análisis bioinformático sobre las repercusiones de las mutaciones sobre la estructura tridimensional de la enzima. Es el primer estudio en América en realizar el análisis molecular de la MPSII con tres metodologías de laboratorio disponibles para esta patología y en llevar a cabo simulaciones in silico con las mutaciones encontradas. Otros tipos de MPS han sido estudiados en América Latina, incluyendo MPS VI Maroteaux-Lamy- en pacientes de Argentina (Garrido et al., 2007), MPS IVA en pacientes latinoamericanos (Tomatsu et al., 2004.,) y pacientes del área andina con MPS I(Matte et al., 2001), (Velasco, Pineda, et al., 2011; en evaluación), aportando al conocimiento del perfil mutacional de las MPSs. El Síndrome de Hunter de tipo neuronopático (Hunter A) fue el más frecuente, correspondiendo al 88% (n= 16) de los casos, mientras el fenotipo no neuronopático (Hunter B) se presentó en el 11% de los pacientes (n=2). Los criterios para establecer el subtipo de enfermedad fueron clínicos, basados en las características del neurodesarrollo y el lenguaje observadas en la historia clínica y durante el examen físico. Los dos pacientes no neuronopáticos presentaron un nivel de escolaridad esperado para la edad (el individuo adulto es profesional 123 universitario), mientras que los 16 pacientes restantes presentan retraso global del desarrollo (<5 años) o retraso mental de moderado a grave (>5 años). Según una revisión europea sobre los aspectos epidemiológicos y clínicos de la MPS II, dos tercios de los pacientes presentan fenotipo neuronopático (75%), lo cual es similar a lo encontrado en nuestra población (Wraith, Scarpa, et al., 2008). En una serie de 77 pacientes de Brasil los investigadores clasificaron a los pacientes sin deterioro cognitivo como no neuronopáticos únicamente si contaban con 10 años de edad o más al momento de la inclusión en el estudio(Schwartz, et al., 2007). Por otro lado, reporte inicial del Hunter Outcome survey (HOS) que incluyó población de Europa, Norteamérica, Latinoamérica y Asia no clasificó la población en los dos fenotipos, severo y atenuado (Wraith, Beck, et al., 2008). Todo lo anterior dificulta la comparación de las frecuencias de fenotipos con dichas poblaciones. Adicionalmente, carencia de instrumentos de clasificación mundialmente avalados para síndrome de Hunter dificulta el diagnóstico preciso entre los dos subtipos de la enfermedad. La hipoacusia y otros trastornos sensoriales podrían dar lugar a interpretaciones falsas de retraso en el desarrollo cognitivo. Por otro lado, la MPS IIB puede cursar con retraso mental leve. El estudio de Schwartz y colegas en Brasil también alude a la necesidad de desarrollar instrumentos de clasificación confiables (Schwartz, et al., 2007). Se hace necesario el seguimiento y evaluación en el tiempo de nuestros pacientes para corroborar si la clasificación aquí mostrada es acertada. 124 La media de la edad de inicio de los síntomas fue de 2.5 años (SD±1.55 años) para todo el grupo, siendo en promedio 2.7 años para los neuronopáticos y 0.88 para los no-neuronopáticos. El síntoma más frecuente fue retraso del desarrollo del lenguaje (66.7%), seguido de rigidez de grandes articulaciones (26,5%) y neumonía recurrente (20%). El estudio de Young y Harper reportó en una serie de 84 pacientes en Gales una edad promedio de inicio de síntomas de 2.47 años para fenotipo severo y 4.3 años para la forma atenuada(Young, et al., 1982). En el reporte HOS se observaron edades de 24 y 39 meses en promedio para el fenotipo severo y atenuado, respectivamente; la otitis media y la hernia abdominal fueron los síntomas más tempranos(Wraith, Beck, et al., 2008). Schwartz et al., en Brasil reportaron que la media de edad de inicio de síntomas es 18 meses (1.5 años) para el fenotipo neuronopático, siendo las primeras manifestaciones retardo en el lenguaje (90%), rigidez articular, macrocránea y fascies tosca (Schwartz, et al., 2007). En este mismo estudio, la edad de inicio de síntomas para el fenotipo no neuronopático fue 39 meses (3.7 años). Los datos arriba anotados en relación con la edad de inicio muestran una relación inversa a lo encontrado en nuestros pacientes, en los cuales el fenotipo neuronopático tuvo una edad de inicio de síntomas más tardía según lo referido por los padres. Esto se debe a la presencia de un dato extremo en uno de nuestros 125 pacientes con una edad de inicio de síntomas de 5 años, lo cual probablemente sea atribuible a un sesgo de recuerdo. La media de la edad al diagnóstico en la población estudiada fue de 6.06 años (SD±4.35). Un resultado similar fue encontrado en población de Brasil (n=77), con una edad promedio al diagnóstico de 6 años para todos los pacientes MPS II. El mismo estudio observó que los pacientes con fenotipo atenuado eran diagnosticados a una edad promedio de 14 años, más tardíamente que los de fenotipo severo (7 años de edad en promedio) con una diferencia estadísticamente significativa (p<0,001) (Schwartz, et al., 2007). El estudio multicéntrico HOS arrojó un promedio de 4.1 (SD±3,2) años de edad al diagnóstico(Wraith, Beck, et al., 2008),(S. A. Jones, et al., 2009). La similitud con los datos arrojados por Schwartz y colegas para la edad al diagnóstico indica que ha mejorado en nuestro país el conocimiento de los profesionales sobre las MPSs y la reciente disponibilidad de herramientas de diagnóstico en laboratorios de referencia en el ámbito bioquímico. Los pacientes de nuestra población tuvieron una talla sobre +2 SD en los primeros años de vida, que se normalizó hacia los 8 años de edad y luego se produjo un descenso en el crecimiento inferior a -3 SD en la adolescencia. Similares resultados se observaron en el reporte HOS, con normalización de la talla entre los 9 y 19 años de edad (Wraith et al., 2008). 126 En nuestra población de estudio las manifestaciones más frecuentes asociadas a morbilidad fueron movilidad articular disminuida 88,8% (n=16/18); hipoacusia 83,3% (n=15/18); infecciones recurrentes de vía aérea superior 72,2% (n=13/18); otitis media aguda 55,6% (n=10/18) y convulsiones 44,4% (n=8/18). En la serie HOS se encontró frecuentemente rigidez articular (84%) y otitis (74%), la prevalencia de otras manifestaciones se agruparon por órganos o sistemas, complicaciones asociadas al oído/audición disminuida, 90%; sistema neurológico 83%; y vía aérea superior 77% (Wraith et al., 2008). En nuestros pacientes, comorbilidades como la hipertrofia de amígdalas y adenoides 61% (n=11/18) y cardiopatía 55,6% (n=10/18) tienen diferente frecuencia a lo reportado en la serie HOS: hipertrofia amígdalas/adenoides 79% afección cardiovascular 81% (Wraith et al., 2008). La neumonía a repetición se presentó en 66.7% (n=12/18). En los registros HOS se reporta un 80% de prevalencia de alteraciones pulmonares incluida la neumonía, la cual es una causa menos frecuente de muerte (Wraith et al., 2008). Dentro de las cardiopatías el defecto más frecuente en nuestros pacientes colombianos fue la afección–insuficiencia, mixoma o displasia- de la válvula mitral (40%; n=4/10), lo cual concuerda con la serie HOS, donde las anomalías valvulares sumaron 53% (Wraith et al., 2008). En la serie de Brasil, la regurgitación mitral se presentó en el 63% y el engrosamiento de la válvula se documentó en 36.8% (Scwartz, et al., 2007). 127 Hallazgos en las neuroimágenes tales como ventriculomegalia, alteración de la sustancia blanca y del parénquima cerebral y adelgazamiento del cuerpo calloso han sido descritos en las MPSs (Calleja et al., 2012). También se encuentra reportada la presencia de lesiones quísticas principalmente en cercanía o dentro del cuerpo calloso (Shinomiya et al., 1996) pero no hay reportes de lesiones en SNC que sean distintivas de MPSII. 7.1.1. Características enzimáticas La medición de la actividad enzimática en muestra de sangre seca en papel de filtro es un estudio de pesquisa, mientras que el ensayo en cultivo de fibroblastos o leucocitos es la prueba confirmatoria(Martin, et al., 2008). El 82% de los individuos en este estudio fue confirmado por actividad medida en leucocitos. Un 12% tiene hasta la fecha sólo el resultado en papel filtro pero las manifestaciones clínicas y la genotipificación son prueba del diagnóstico de MPSII. La razón principal por la cual estos cuatro pacientes carecen aún de análisis enzimático en cultivo de leucocitos o fibroblastos son las limitaciones económicas y/o geográficas de las familias para trasladarse directamente al laboratorio de referencia, ya que se requiere muestra fresca para dicho análisis. Parkinson et al. reportan que ni la cantidad ni la actividad de la IDS se correlacionan con la severidad del fenotipo(Parkinson, et al., 2004). En este estudio no se observaron diferencias estadísticamente significativas entre los valores de 128 actividad enzimática en el grupo de pacientes incluyendo ambos fenotipos, lo cual es congruente con lo anotado por expertos: la actividad de la IDS es igualmente deficiente en ambas formas de la enfermedad (Scarpa et al., 2011). 7.2. Perfil mutacional y comparación con la literatura Con el empleo de MLPA, genotipificación y RFLPs se logró en este estudio la confirmación del diagnóstico molecular en el 88% de los pacientes (n=16/18). Las mutaciones en los pacientes 002, 010 y 011 involucraron un número importante de nucleótidos (deleciones e inserciones de 12 a 20 nt) pero no fueron detectadas por MLPA. Esto ocurrió porque las sondas X-148.4 específicas para el exón 5 hibridan corriente abajo de la alteración hallada en el exón 5, sin flanquear esa región en todos los tres casos. Al analizar los datos genotípicos observamos una alta frecuencia de mutaciones privadas (42%). En una serie de 155 pacientes europeos provenientes principalmente de Francia, Portugal, Túnez y Suiza se reportó hasta un 64% de mutaciones privadas (Froissart, et al., 2007). Otra serie de 38 pacientes en China tuvo un 45% de mutaciones nuevas (Zhang, et al., 2011). Dentro de las mutaciones recurrentes en individuos no relacionados de fenotipo neuronopático (Hunter A), la más frecuente fue R468Q (pacientes MPSII003 y 008), en la cual hay una transición de Guanina a Adenina en la posición 1403 del 129 cDNA ocasionando en la proteína la sustitución de Arginina, aminoácido cargado positivamente, por una Glutamina, con carga neta cero. La mutación R468Q se ha reportado en series de pacientes de Japón (Sukegawa et al., 1995), (Isogai et al., 1998), Reino Unido (Vafiadaki et al., 1998), Estados Unidos (Li et al., 1999), Taiwan (Lin et al., 2006), Francia (Froissat et al., 2007) y China (Zhang et al., 2011). En cuanto a las demás mutaciones, cada uno de los pacientes tuvo una única mutación individual, y todas fueron puntuales (Q465X, K347Q, K236N).Sus implicaciones funcionales se discutirán más adelante. Tres pacientes en los cuales no se pudo establecer el diagnóstico molecular por MLPA y secuenciación directa fueron llevados al método de RFLPs para detección de rearreglos IDSP1/IDS, demostrando la presencia de un fenómeno de conversión en el paciente MPS II 013. Este evento de conversión que involucró al intrón 3 ha sido reportado previamente en un paciente alemán (Bunge et al., 1998) y en un paciente italiano (Lualdi, et al., 2005). Ambos tenían fenotipo neuronopático. Este evento de conversión genera un gen IDS que contiene regiones recombinantes con IDSP1 en los intrones 3 y 7. Una timina en la posición 292879 es el punto de ruptura en el intrón 3 que es reemplazado por citosina del intrón quimérico 3-7 (Figura 27). De esta manera se yuxtapone también el intrón 7 con su homólogo del IDSP1 y por medio de recombinación no recíproca se producen las regiones recombinantes en los intrones 3 y 7 (Figura 5) (Birot et al., 1996, Bunge et al., 1998, Lualdi et al., 1995). 130 Figura 27. Modelo propuesto para el mecanismo de recombinación en el intrón 3, a partir de los datos de Lualdi et al., 2005. En azul IDSP1 con la hebra invadiendo la región del gen a partir del punto de ruptura T 292879>C y recombinación no alélica por yuxtaposición de regiones homólogas de exón 3 (barra amarilla) e intrón 7 (triángulo). En total se observaron 2 mutaciones consistentes en rearreglos complejos: la conversión en el intrón 3 y la deleción del exón 7 detectada por MLPA la cual no ha sido reportada previamente, representando el 16% de las mutaciones en nuestra población colombiana. Esto es similar a lo reportado en la literatura, donde se refiere un 20% de estos fenómenos de recombinación, en población de Suecia (Steen-Bondeson, et al., 1002), Alemania (Bunge et al., 1998), Italia (Lualdi et al., 2005), Francia (Froissart et al., 2007). Las mutaciones privadas fueron mayoritariamente pequeñas inserciones y deleciones con corrimiento del marco de lectura, sumando éstas el 38% del total de los alelos y todas pertenecientes a pacientes con fenotipo neuronopático. Llama la atención que la mitad de estas (3/6) se presentaron en el exón 5, el cual hace codifica los aminoácidos desde la posición 171 a 239, del dominio N-terminal, adyacentes al core catalítico. 131 Nosotros encontramos 2 mutaciones en el exón 9 (R468Q y Q465X). Una mayor frecuencia de mutaciones en el exón 9, en su mayoría tipo missense y recurrentes ha sido reportada en series de Corea (Sohn et al., 2012), y China (Zhang et al., 2011).En este grupo de pacientes colombianos sólo hubo una mutación en el exón 2 y ninguna en el exón 3. Sin embargo se observa una lata frecuencia de mutaciones en exones 2 y 3 en población de Europa (Froissart et al., 2007), además de Corea (Kim et al., 2003) y China (Zhang et al., 2011). El estudio de Sohn et al. en población coreana encontró un número importante de mutaciones en el exón 5 (Sohn, et al., 2012), similar a lo encontrado en nuestro estudio. Los dos pacientes no relacionados de fenotipo no neuronopático (Hunter B) presentaron la misma mutación S71N, la cual ha sido publicada hallándose en pacientes europeos con este mismo fenotipo(Froissart, et al., 1998). En este estudio no se identificaron mutaciones en sitios del splicing, ni deleción completa del gen IDS o de más de un exón. En un estudio de Portugal se reportó una importante incidencia de mutaciones en sitios del splicing (seis mutaciones intrónicas y dos exónicas de sitios de splicing) , lo cual generaba transcriptos aberrantes, involucrando exones 2 y 3, intrón 2, y rara vez el intrón 3 (Alves et al., 2006). Estas diferencias en la distribución de las mutaciones, probablemente se deban al acervo étnico – genético interpoblacional. En dos casos no se pude establecer el diagnóstico molecular por las técnicas arriba mencionadas. Filocamo et al. reportaron en Italia que hasta un 17% de la muestra puede ser negativa a pesar del diagnóstico clínico y bioquímico 132 confirmado (Filocamo et al., 2001). Una de las posibles causas de este hallazgo es que los pacientes presenten eventos de recombinación no incluidos en los cuatro rearreglos tamizados aquí (Lualdi et al., 2005), o a la presencia de una mutación intrónica o alteraciones en el patrón de metilación. Existe un reporte de una mutación intrónica, distante de la región flanqueante del exón, en un paciente de origen alemán. Esta mutación fue identificada en el intrón 7, y crea un sitio de splicing que ocasiona la inclusión de 78 bp intrónicos en el mRNA (Rathmann et al., 1996). Por otro lado, Tomatsu et al., demostraron que las islas CpG el exón 3 de pacientes varones sanos están hipometiladas mientras que en pacientes con MPSII se encuentra hipermetilada. Los investigadores postularon este estado de hipermetilación como factor predisponente para transiciones y por ende mutaciones (Tomatsu et al., 2006), pero aún no se ha demostrado la existencia de anomalías de metilación aisladas en como defecto genético primario en MPSII. 7.3. Análisis bioinformático y repercusión de las mutaciones en la estructura tridimensional de la IDS Se puede observar, tal como lo han demostrado estudios moleculares en otros países, que la MPS II tiene una alta heterogeneidad alélica y la correlación genotipo/fenotipo es difícil de establecer. Por otro lado, existen pocos reportes sobre ensayos in vitro que evalúen la expresión y grado de afectación de la enzima con estas mutaciones. Nosotros realizamos una aproximación inicial para un mayor 133 entendimiento de la repercusión de las mutaciones identificadas sobre la estructura de la IDS empleando herramientas bioinformáticas. La estructura tridimensional de la hIDS obtenida fue empleada para simular la interacción enzima-sustrato (docking molecular) encontrando que el HS/DS se une a la enzima en un pocket catalítico conservado, como se muestra en la figura 18. Kato et al. realizaron en 2005 el modelamiento tridimensional de la IDS y reportaron que los residuos que hacen parte del sitio catalítico son Asp45, Asn46, Cys84, Arg88, Lys135, His138, Asp334, His335 y Lys347, pero este grupo no realizó simulaciones tipo docking. En contraste, la simulación aquí presentada encontró además de Cys84, los residuos Arg297, leu244, Tyr165 y Asn106 como parte del sito catalítico. Probablemente la unión del metal catiónico requerida para la actividad catalítica induzca cambios en los aminoácidos presentes en el pocket, pero este tipo de simulaciones están fuera de los alcances de nuestro trabajo. El residuo de Cys84 mostró en este análisis una interacción electrostática con el grupo O-2 sulfato del ácido urónico, lo cual está en concordancia con la función biológica de la enzima. El puente de hidrógeno formado a la entrada del pocket podría servir como punto de fijación entre la enzima y el sustrato durante la reacción bioquímica (Negishi, Dong, Darden, Pedersen, & Pedersen, 2003). Respecto a las mutantes de IDS, el docking reveló que incluso una mutación puntual puede alterar toda la estructura tridimensional de la proteína. Todos los valores de RMSD indicaron alteración estructural, ya que presentaban valores muy inferiores a 0.5Å. Esto se explica porque al superponer cada proteína mutante con 134 su plantilla (IDS wild type), la distancia entre los átomos de ambas estructuras varía de tal manera que se obtienen RMSD con valores muy diferentes al esperado, es decir, cercano a 1 Å para proteínas con alta similitud estructural. En contraste, al superponer a IDS wild type con su homólogo 1AUK.A se obtuvo un RMSD de 0,97. Las diferencias en los valores de ASA indican que en cada mutante los cambios estructurales acarrean consigo diferencias en los aminoácidos expuestos a la interfase hidrofílica. En la mutación R468Q un residuo cargado positivamente (Arginina), es sustituidopor uno polar no cargado (Glutamina), lo cual podría resultar en un cambio en la afinidad por el sustrato y no sólo en la estructura terciaria. El fraccionamiento subcelular demostró que la mutante R468Q tiene además un pobre transporte a los lisosomas (Villani, Daniele, Balzano, & Di Natale, 2000) Un trabajo publicado por Kato et al. plantea la hipótesis de que la mutación no conservativa en R468 podría afectar el campo electrostático para la entrada del sustrato en la cavidad del sitio activo, generando una enzima inactiva (Kato et al., 2005). Los resultados del Western blot publicados más tarde por el mismo grupo mostraron sólo precursores primarios, indicio indirecto de cambios estructurales en la proteína (Sukegawa-Hayasaka et al., 2006). La mutación Q465X, como la anterior, se asocia con fenotipo neuronopático. Hay escasos reportes acerca de esta mutación recurrente, y ninguno de estos contiene abordajes computacionales (Li et al., 1999). El docking realizado en este trabajo mostró ausencia de Cys84 en el core catalítico (Figura 22). Aunque los residuos en el extremo carboxilo-terminal no tienen directa participación en el sitio catalítico, los 135 cambios conformacionales que resultan de la interrupción de la secuencia primaria podrían alterar los aminoácidos presentes en él. Adicionalmente, el codón de parada causa la pérdida de dos potenciales sitios de glicosilación –N513 y N537con potenciales consecuencias en el transporte subcelular (Millat, et al., 1997). Sukegawa et al. demostraron in vitro que todas las mutantes por ellos estudiadas y asociadas con fenotipo severo sintetizan únicamente formas precursoras, lo cual sugiere que los cambios estructurales de estas mutaciones tienen consecuencias sobre los eventos postraduccionales (Sukegawa-Hayasaka et al., 2006) En la mutante K347Q un aminoácido cargado positivamente (Lisina) es sustituido por uno polar no cargado (Glutamina). Se encuentra reportado que Lys347 se encuentra adyacente al sitio activo. Esta mutación no conservativa podría modificar severamente el core catalítico. A pesar de que Cys84 aparece en el docking, las diferencias en las interacciones atómicas podrían estar explicando la deficiente actividad enzimática. Por otro lado, K236N es una mutación sólo reportada una vez en un paciente de Bulgaria, y no existen por ahora más estudios sobre ella(Gucev et al., 2011) La muestra de pacientes estudiada incluye solo dos pacientes con fenotipo no neuronopático y en ambos la mutación S71N (recurrente) fue identificada. Tanto en el modelo silvestre como en el mutante, el residuo en la posición 71 se observó distante del core catalítico. Otras mutantes reportadas en asociación con este mismo fenotipo han sido estudiadas in vitro, encontrando que R48P y W337R no siguen un proceso de maduración normal (evidenciado por el precursor de 73 136 75kDa en el Western blot), mientras que A85T sí se procesa a la forma madura de 55-45kDa. No existen hasta la fecha estudios similares sobre la mutante S71N, así que nosotros realizamos el modelamiento y docking de esta mutante con ambas formas (madura y precursor) encontrando los mismos resultados, que se explican en la sección correspondiente. El resultado del docking de S71N fue muy similar a lo encontrado en el wild type excepto por la formación de puentes de hidrógeno con el grupo O-2sulfato del ácido urónico. Propondriams que este cambio dentro del sitio catalítico dificulta la resolución del intermediario éster-sulfato y por lo tanto la actividad catalíca, pero queda sin explicar lo que ocurre con la enzima en el SNC de estos pacientes con inteligencia normal. Hay pocos reportes acerca de la corrección de mutaciones a nivel traduccional, en especial en casos de presentación atenuada de la enfermedad (Bunge et al., 1993) (Lualdi et al., 2010). Se requiere mayor investigación de esta mutante c.212G>A (S71N) buscando posibles eventos de corrección del mRNA o cualquier otro fenómeno traduccional o de procesamiento. Del.Q200_E203 y R294GfsX2 (Del. exón 7) son dos nuevas mutantes encontradas en este grupo de pacientes. El análisis de docking mostró unas interacciones marcadamente distorsionadas dentro del pocket, especialmente en el caso de R294GfsX2, la cual pierde 254 aminoácidos y con ello gran parte del core catalítico. El análisis estructural indicó que las mutantes privadas P160SfsX4, D190Pfs13X y P185GfsX2 pierden la mayoría de la estructura del dominio catalítico, ya que 137 carecen de 386, 347 y 363 aminoácidos respectivamente (más del 50% de la proteína), así que se puede inferior de ello que estos polipéptidos distorsionados no retendrán actividad enzimática y el docking molecular no tiene utilidad en este caso. 7.4. Potenciales implicaciones en el tratamiento de la MPSII La identificación de las mutaciones en la MPSII tiene utilidad en la detección de portadoras con el fin de brindar un adecuado asesoramiento genético. Sin embargo, a pesar de la disponibilidad de estrategias como la fertilización asistida con óvulos de donante y el diagnóstico preimplantacional, la prevención de nuevos casos de MPS II no siempre es posible en nuestro medio. El conocimiento de la mutación en una familia permitiría dar a la paciente gestante la información de la probabilidad de que su hijo, en caso de presentar MPSII, pueda cursar con fenotipo neuronopático o no neuronopático, y esto tiene implicaciones en el tratamiento del paciente. Guffon y colegas realizaron seguimiento a ocho pacientes con MPS II sometidos a transplante de médula ósea, concluyendo que sólo la forma no neuronopática podía beneficiarse con este tratamiento, sin embargo, al momento de la intervención los pacientes tenían entre 3 y 16 años de edad. 138 Un trabajo reciente de Tanaka y colegas en Japón empleó transplante de células madre hematopoyéticas (HSCT) en pacienes con MPSII a edad más temprana observando mejoría en el compromiso del SNC en los pacientes con fenotipo neuronopático (Tanaka et al., 2012). Basados en este último estudio, la intervención temprana con transplante de HSC al paciente en quien se ha identificado una mutación relacionada con fenotipo neuronopático tendría una posibilidad de 85% de mejoría en el aspecto cognitivo, además de los beneficios en las manifestaciones somáticas (Tanaka et al., 2012). En el paciente en el cual se identifique una mutación asociada con fenotipo no neuronopático, la Terapia de Reemplazo Enzimático con Idursulfase sería la opción más acertada (Muenzer et al., 2012). La Terapia con Chaperonas Farmacológicas es un campo emergente en las enfermedades de depósito lisosomal y se encuentran disponibles para enfermedad de Fabry y Gaucher en estudios clínicos fase 3 y 2, respectivamente (Valenzano et al., 2011). En relación con las MPS, los síndromes de Sanfilippo tipo C y Morquio B poseen chaperonas farmacológicas en fase preclínica de investigación (Feldhammer, Durand, & Pshezhetsky, 2009),(Schitter et al., 2010),(Fantur et al., 2010). El conocimiento acerca de la distribución de las mutaciones en la IDS y sus efectos funcionales sobre sus propiedades catalíticas o su estabilidad podrían tener implicaciones en el diseño y desarrollo de nuevos agentes terapéuticos. 139 Las mutaciones periféricas (presentes en la interfase hidrofílica de la proteína y distantes del core catalítico) como R468Q, Q465X y S71N podrían ser susceptibles de terapia con chaperonas farmacológicas, a través del rescate del sistema de control del retículo endoplasmático, corrección del procesamiento y del transporte, para finalmente lograr la translocación de una enzima activa dentro del lisosoma. 140 8. CONCLUSIONES El presente estudio constituye el primer acercamiento colombiano a la genotipificación del gen Iduronato 2 sulfatasa en pacientes con MPSII. En nuestra población se observó una edad de inicio de síntomas de la MPS II con un comportamiento inverso a lo reportado en la literatura para los fenotipos neuronopático y no neuronopático, pero la edad al diagnóstico fue similar a lo reportado en Brasil (6 años). Así mismo, coincide con la literatura la alta frecuencia del retraso del lenguaje como síntoma inicial en el fenotipo neuronopático. Las características fenotípicas tales como rigidez articular, hipertrofia de adenoides/amígdalas y otitis media tuvieron similares frecuencias a las descritas en la literatura, igualmente ocurrió con los factores de morbimortalidad como las cardiopatías y el compromiso del SNC. El estudio de genotipificación presentó una considerable complejidad, ya que requiere la implementación de al menos tres metodologías para lograr una sensibilidad del 88%. Similares hallazgos se han reportado en otras poblaciones. El diagnóstico molecular es de crucial importancia en la identificación de portadoras, ya que, como se observó en dos mujeres portadoras y en la literatura, el análisis enzimático puede resultar en falsos negativos. 141 La frecuencia de mutaciones nuevas (42%) es similar a lo reportado en pacientes de China y la alta frecuencia de mutaciones nuevas en el exón 5 coincide con los datos de un estudio en pacientes coreanos. Por otro lado, la ocurrencia de rearreglos complejos fue similar a lo observado en series de Europa y Asia. El uso de herramientas bioinformáticas fue de gran utilidad para predecir las repercusiones de las distintas mutaciones sobre la estructura tridimensional de la IDS humana, encontrando que mutaciones puntuales como R468Q, Q465X ó S71N alteran su conformación, lo que se reflejó en un cambio en los RMSD totales. El docking molecular permitió conocer los cambios a nivel del sitio catalítico de cada mutante, pudiendo observar interacciones enzima-sustrato con diferencias respecto al tipo silvestre de la enzima. 142 9. LIMITACIONES Y PERSPECTIVAS A pesar de haberse realizado Secuenciación, MLPA y RFLPs, aún quedaron sin diagnóstico molecular dos pacientes (MPSII004 y 0014). Es necesario por lo tanto el análisis de los transcriptos y eventualmente, la búsqueda de alteraciones intrónicas o en los patrones de metilación del gen IDS en estos dos casos, lo cual no fue posible en este estudio por limitaciones presupuestales y del cronograma. Las perspectivas de este trabajo son en primera instancia el ofrecimiento de herramientas para diagnóstico molecular temprano para los pacientes, de manera que se pueda establecer con prontitud la clasificación del fenotipo de MPS II y poder anticipar el manejo adecuado, además de tener a disposición la información para el estudio de portadoras y brindar un asesoramiento genético integral a las familias. Por último, se requiere el diseño de estudios funcionales in vitro para estudiar la repercusión de las mutaciones nuevas sobre la actividad de la enzima. 143 10. BIBLIOGRAFÍA Altarescu, G., Renbaum, P., Eldar-Geva, T., Brooks, B., Varshaver, I., Avitzour, M., et al. (2011). Preventing mucopolysaccharidosis type II (Hunter syndrome): PGD and establishing a Hunter (46, XX) stem cell line. Prenat Diagn. Alves, S., Mangas, M., Prata, M. J., Ribeiro, G., Lopes, L., Ribeiro, H., et al. (2006). Molecular characterization of Portuguese patients with mucopolysaccharidosis type II shows evidence that the IDS gene is prone to splicing mutations. J Inherit Metab Dis, 29(6), 743-754. Baehner, F., Schmiedeskamp, C., Krummenauer, F., Miebach, E., Bajbouj, M., Whybra, C., et al. (2005). Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis, 28(6), 1011-1017. Baldo, G., Matte, U., Artigalas, O., Schwartz, I. V., Burin, M. G., Ribeiro, E., et al. (2011). Placenta analysis of prenatally diagnosed patients reveals early GAG storage in mucopolysaccharidoses II and VI. Mol Genet Metab, 103(2), 197198. Barrager, J., & Cabrera, M. (2007). Mucopolysaccharidosis II Lysosomal storage disorders. (pp. 410-420). Bax, M. C., & Colville, G. A. (1995). Behaviour in mucopolysaccharide disorders. Arch Dis Child, 73(1), 77-81. Bellettato, C. M., & Scarpa, M. (2010). Pathophysiology of neuropathic lysosomal storage disorders. J Inherit Metab Dis, 33(4), 347-362. Bondeson, M. L., Malmgren, H., Dahl, N., Carlberg, B. M., & Pettersson, U. (1995). Presence of an IDS-related locus (IDS2) in Xq28 complicates the mutational analysis of Hunter syndrome. Eur J Hum Genet, 3(4), 219-227. Brama, I., Gay, I., Feinmesser, R., & Springer, C. (1986). Upper airway obstruction in Hunter syndrome. Int J Pediatr Otorhinolaryngol, 11(3), 229-235. Brante, G. (1952). Gargoylism; a mucopolysaccharidosis. Scand J Clin Lab Invest, 4(1), 43-46. Brusius-Facchin AC, De Souza CF, Schwartz IV, Riegel M, Melaragno MI, Correia P, Moraes LM, Llerena J Jr, Giugliani R, Leistner-Segal S.(2012). Severe phenotype in MPS II patients associated with a large deletion including contiguous genes. Am J Med Genet A, 158A(5):1055-1059. Bunge, S., Steglich, C., Zuther, C., Beck, M., Morris, C. P., Schwinger, E., et al. (1993). Iduronate-2-sulfatase gene mutations in 16 patients with mucopolysaccharidosis type II (Hunter syndrome). Hum Mol Genet, 2(11), 1871-1875. Bunge S, Rathmann M, Steglich C, Bondeson ML, Tylki-Szymanska A, Popowska E, Gal A. (1998). Homologous nonallelic recombinations between the iduronate-sulfatase gene and pseudogene cause various intragenic deletions and inversions in patients with mucopolysaccharidosis type II. Eur J Hum Genet. 6(5),492-500. Calleja ML, González Gutiérrez-Solana L, López Marín L, López Pino MA, Fournier Del Castillo C, Duat Rodríguez A.(2012). Neuroimaging findings in patient series with mucopolysaccharidosis. Neurologia, 27(7), 407-413. 144 Cantz, M., Chrambach, A., Bach, G., & Neufeld, E. F. (1972). The Hunter corrective factor. Purification and preliminary characterization. J Biol Chem, 247(17), 5456-5462. Coelho, J. C., Wajner, M., Burin, M. G., Vargas, C. R., & Giugliani, R. (1997). Selective screening of 10,000 high-risk Brazilian patients for the detection of inborn errors of metabolism. Eur J Pediatr, 156(8), 650-654. Charoenwattanasatien, R., Cairns, J. R., Keeratichamroen, S., Sawangareetrakul, P., Tanpaiboon, P., Wattanasirichaigoon, D., et al. (2012). Decreasing activity and altered protein processing of human iduronate-2-sulfatase mutations demonstrated by expression in COS7 cells. Biochem Genet, 50(11-12), 990-997. Chen, C. P., Lin, S. P., Tzen, C. Y., Hwu, W. L., Chern, S. R., Chuang, C. K., et al. (2007). Prenatal diagnosis and genetic counseling of mucopolysaccharidosis type II (Hunter syndrome). Genet Couns, 18(1), 49-56. Chen, M. R., Lin, S. P., Hwang, H. K., & Yu, C. H. (2005). Cardiovascular changes in mucopolysaccharidoses in Taiwan. Acta Cardiol, 60(1), 51-53. Danes, B. S., & Bearn, A. G. (1965). Hurler's syndrome: demonstration of an inherited disorder of connective tissue in cell culture. Science, 149(3687), 987-989. Danes, B. S., & Bearn, A. G. (1966). Hurler's syndrome. A genetic study in cell culture. J Exp Med, 123(1), 1-16. Dangel, J. H. (1998). Cardiovascular changes in children with mucopolysaccharide storage diseases and related disorders--clinical and echocardiographic findings in 64 patients. Eur J Pediatr, 157(7), 534-538. Dean, C. J., Bockmann, M. R., Hopwood, J. J., Brooks, D. A., & Meikle, P. J. (2006). Detection of mucopolysaccharidosis type II by measurement of iduronate-2-sulfatase in dried blood spots and plasma samples. Clin Chem, 52(4), 643-649. Dierks, T., Lecca, M. R., Schlotterhose, P., Schmidt, B., & von Figura, K. (1999). Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J, 18(8), 2084-2091. Diez-Roux, G., & Ballabio, A. (2005). Sulfatases and human disease. Annu Rev Genomics Hum Genet, 6, 355-379. DiFerrante, N., & Nichols, B. L. (1972). A case of the Hunter syndrome with progeny. Johns Hopkins Med J, 130(5), 325-328. Fantur, K., Hofer, D., Schitter, G., Steiner, A. J., Pabst, B. M., Wrodnigg, T. M., et al. (2010). DLHex-DGJ, a novel derivative of 1-deoxygalactonojirimycin with pharmacological chaperone activity in human G(M1)-gangliosidosis fibroblasts. Mol Genet Metab, 100(3), 262-268. Feldhammer, M., Durand, S., & Pshezhetsky, A. V. (2009). Protein misfolding as an underlying molecular defect in mucopolysaccharidosis III type C. PLoS One, 4(10), e7434. Felice, B. R., Wright, T. L., Boyd, R. B., Butt, M. T., Pfeifer, R. W., Pan, J., et al. (2011). Safety evaluation of chronic intrathecal administration of idursulfaseIT in cynomolgus monkeys. Toxicol Pathol, 39(5), 879-892. Filocamo M, Bonuccelli G, Corsolini F, Mazzotti R, Cusano R, Gatti R. (2001). Molecular analysis of 40 Italian patients with mucopolysaccharidosis type II: 145 new mutations in the iduronate- 2-sulfatase(IDS) gene. Hum Mutat, 18,164– 165. Fratantoni, J. C., Hall, C. W., & Neufeld, E. F. (1968). Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science, 162(3853), 570-572. Froissart, R., Da Silva, I. M., & Maire, I. (2007). Mucopolysaccharidosis type II: an update on mutation spectrum. Acta Paediatr Suppl, 96(455), 71-77. Froissart, R., Maire, I., Millat, G., Cudry, S., Birot, A. M., Bonnet, V., et al. (1998). Identification of iduronate sulfatase gene alterations in 70 unrelated Hunter patients. Clin Genet, 53(5), 362-368. Gómez, A., García-Robles, R., & Suárez-Obando, F. (2012). Estimación de las frecuencias de las mucopolisacaridosis y análisis de agrupamiento espacial en los departamentos de Cundinamarca y Boyacá. Biomédica, 32,602-609. Gort, L., Chabas, A., & Coll, M. J. (1998). Hunter disease in the Spanish population: molecular analysis in 31 families. J Inherit Metab Dis, 21(6), 655-661. Gucev, Z. S., Tasic, V., Sinigerska, I., Kremensky, I., Tincheva, R., Pop-Jordanova, N., et al. (2011). Hunter syndrome (Muccopolysaccharridosis Type II) in Macedonia and Bulgaria. Prilozi, 32(2), 187-198. Guffon N, Bertrand Y, Forest I, Fouilhoux A, Froissart R. (2009). Bone marrow transplantation in children with Hunter syndrome: outcome after 7 to 17 years. J Pediatr, 154(5):733-7. Hunter, C. (1917). A Rare Disease in Two Brothers. Proc R Soc Med, 10(Sect Study Dis Child), 104-116. Hurler, G. (1919). A type of multiple degeneration that mainly affects the skeletal system [in German]. Z Kinderheilkd, 24(5-6), 220-234. Isogai K, Sukegawa K, Tomatsu S, Fukao T, Song XQ, Yamada Y, Fukuda S, Orii T, Kondo N. (1998). Mutation analysis in the iduronate-2-sulphatase gene in 43 Japanese patients with mucopolysaccharidosis type II (Hunter disease). J Inherit Metab Dis. 21(1), 60-70. Jones, K. L., & Smith, D. W. (2006). Smith's Recognizable Patterns of Human Malformation (6th ed.): Elsevier Saunders. Jones, S. A., Almassy, Z., Beck, M., Burt, K., Clarke, J. T., Giugliani, R., et al. (2009). Mortality and cause of death in mucopolysaccharidosis type II-a historical review based on data from the Hunter Outcome Survey (HOS). J Inherit Metab Dis, 32(4), 534-543. Jung, S. C., Park, E. S., Choi, E. N., Kim, C. H., Kim, S. J., & Jin, D. K. (2010). Characterization of a novel mucopolysaccharidosis type II mouse model and recombinant AAV2/8 vector-mediated gene therapy. Mol Cells, 30(1), 13-18. Kato, T., Kato, Z., Kuratsubo, I., Tanaka, N., Ishigami, T., Kajihara, J., et al. (2005). Mutational and structural analysis of Japanese patients with mucopolysaccharidosis type II. J Hum Genet, 50(8), 395-402. Keilmann, A., Nakarat, T., Bruce, I. A., Molter, D., & Malm, G. (2012). Hearing loss in patients with mucopolysaccharidosis II: data from HOS - the Hunter Outcome Survey. J Inherit Metab Dis, 35(2), 343-353. Kim CH, Hwang HZ, Song SM, Paik KH, Kwon EK, Moon KB, Yoon JH, Han CK, Jin DK. (2003). Mutational spectrum of the iduronate 2 sulfatase gene in 25 unrelated Korean Hunter syndrome patients: identification of 13 novel mutations. Hum Mutat. 21(4),449-450. 146 Kimura, A., Hayashi, S., & Tsurumi, K. (1980). Characterization of dermatan sulfate and heparan sulfate in the urine of a patient with the Hunter syndrome. Tohoku J Exp Med, 131(3), 227-239. Kloska, A., Jakobkiewicz-Banecka, J., Tylki-Szymanska, A., Czartoryska, B., & Wegrzyn, G. (2011). Female Hunter syndrome caused by a single mutation and familial XCI skewing: implications for other X-linked disorders. Clin Genet, 80(5), 459-465. Koyama, K., Moda, Y., Sone, A., Tanaka, H., & Hino, Y. (1994). Neurogenic bladder in Hunter's syndrome. J Med Genet, 31(3), 257-258. Kwon, J. Y., Ko, K., Sohn, Y. B., Kim, S. J., Park, S. W., Kim, S. H., et al. (2011). High prevalence of carpal tunnel syndrome in children with mucopolysaccharidosis type II (Hunter syndrome). Am J Med Genet A, 155A(6), 1329-1335. Laskowski, R. A., & Swindells, M. B. (2011). LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model, 51(10), 27782786. Lau, K. C., & Lam, C. W. (2008). Molecular investigations of a novel iduronate-2sulfatase mutant in a Chinese patient. Clin Chim Acta, 392(1-2), 8-10. Leighton, S. E., Papsin, B., Vellodi, A., Dinwiddie, R., & Lane, R. (2001). Disordered breathing during sleep in patients with mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol, 58(2), 127-138. Ley 1392(2010). Li P, Bellows AB, Thompson JN. (1999). Molecular basis of iduronate-2-sulphatase gene mutations in patients with mucopolysaccharidosis type II (Hunter syndrome). J Med Genet. 36(1),21-27. Liebaers, I., & Neufeld, E. (1976). Iduronate sulfatase activity in serum, lymphocytes, and fibroblasts--simplified diagnosis of the Hunter syndrome. Pediatr Res, 10(8), 733-736. Lim, T. W., Leder, I. G., Bach, G., & Neufeld, E. F. (1974). An assay for iduronate sulfatase (Hunter corrective factor). Carbohydr Res, 37(1), 103-109. Lin, H. Y., Lin, S. P., Chuang, C. K., Niu, D. M., Chen, M. R., Tsai, F. J., et al. (2009). Incidence of the mucopolysaccharidoses in Taiwan, 1984-2004. Am J Med Genet A, 149A(5), 960-964. Link, B., de Camargo Pinto, L. L., Giugliani, R., Wraith, J. E., Guffon, N., Eich, E., et al. (2010). Orthopedic manifestations in patients with mucopolysaccharidosis type II (Hunter syndrome) enrolled in the Hunter Outcome Survey. Orthop Rev (Pavia), 2(2), e16. Lissens W, Seneca S and Liebaers I. (1997). Molecular analysis in 23 Hunter disease families J. Inher. Metab. Dis, 20, 453– 456 Lovell, S. C., Davis, I. W., Arendall, W. B., 3rd, de Bakker, P. I., Word, J. M., Prisant, M. G., et al. (2003). Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins, 50(3), 437-450. Lowry, R. B., & Renwick, D. H. (1971). Relative frequency of the Hurler and Hunter syndromes. N Engl J Med, 284(4), 221-222. Lualdi, S., Regis, S., Di Rocco, M., Corsolini, F., Stroppiano, M., Antuzzi, D., et al. (2005). Characterization of iduronate-2-sulfatase gene-pseudogene recombinations in eight patients with Mucopolysaccharidosis type II revealed by a rapid PCR-based method. Hum Mutat, 25(5), 491-497. 147 Lualdi, S., Tappino, B., Di Duca, M., Dardis, A., Anderson, C. J., Biassoni, R., et al. (2010). Enigmatic in vivo iduronate-2-sulfatase (IDS) mutant transcript correction to wild-type in Hunter syndrome. Hum Mutat, 31(4), E1261-1285. Malm, G., Lund, A. M., Mansson, J. E., & Heiberg, A. (2008). Mucopolysaccharidoses in the Scandinavian countries: incidence and prevalence. Acta Paediatr, 97(11), 1577-1581. Martin, R., Beck, M., Eng, C., Giugliani, R., Harmatz, P., Munoz, V., et al. (2008). Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome), Pediatrics (2008/02/05 ed., Vol. 121, pp. e377-386). Matte, U., Leistner, S., Schwartz, I., Lima, L., Chamoles, N., Yogalingam, G., et al. (2001). Mutation analysis in South American patients with Mucopolysaccharidosis type I. Paper presented at the Revista Argentina de Antropología Biológic. Meikle, P. J., Hopwood, J. J., Clague, A. E., & Carey, W. F. (1999). Prevalence of lysosomal storage disorders. JAMA, 281(3), 249-254. Miech, C., Dierks, T., Selmer, T., von Figura, K., & Schmidt, B. (1998). Arylsulfatase from Klebsiella pneumoniae carries a formylglycine generated from a serine. J Biol Chem, 273(9), 4835-4837. Millat, G., Froissart, R., Maire, I., & Bozon, D. (1997). Characterization of iduronate sulphatase mutants affecting N-glycosylation sites and the cysteine-84 residue. Biochem J, 326 ( Pt 1), 243-247. Molecular Operating Environment (MOE), 2010.10; Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2010. Morehead, J. M., & Parsons, D. S. (1993). Tracheobronchomalacia in Hunter's syndrome. Int J Pediatr Otorhinolaryngol, 26(3), 255-261. Moreira da Silva, I., Froissart, R., Marques dos Santos, H., Caseiro, C., Maire, I., & Bozon, D. (2001). Molecular basis of mucopolysaccharidosis type II in Portugal: identification of four novel mutations. Clin Genet, 60(4), 316-318. Muenzer, J. A Safety and Dose Ranging Study of Idursulfase (Intrathecal) Administration Via an Intrathecal Drug Delivery Device in Pediatric Patients with Hunter Syndrome Who Have Central Nervous System Involvement and Are Receiving Treatment With Elaprase®: Shire Genetic Therapies. Muenzer, J. (1986). Mucopolysaccharidoses. Adv Pediatr, 33, 269-302. Muenzer, J., Beck, M., Eng, C. M., Escolar, M. L., Giugliani, R., Guffon, N. H., et al. (2009). Multidisciplinary management of Hunter syndrome. Pediatrics, 124(6), e1228-1239. Muenzer, J., Bodamer, O., Burton, B., Clarke, L., Frenking, G. S., Giugliani, R., et al. (2012). The role of enzyme replacement therapy in severe Hunter syndrome-an expert panel consensus. Eur J Pediatr, 171(1), 181-188. Nagashima, K., Endo, H., Sakakibara, K., Konishi, Y., Miyachi, K., Wey, J. J., et al. (1976). Morphological and biochemical studies of a case of mucopolysaccharidosis II (Hunter's syndrome). Acta Pathol Jpn, 26(1), 115132. Negishi, M., Dong, J., Darden, T. A., Pedersen, L. G., & Pedersen, L. C. (2003). Glucosaminylglycan biosynthesis: what we can learn from the X-ray crystal structures of glycosyltransferases GlcAT1 and EXTL2. Biochem Biophys Res Commun, 303(2), 393-398. 148 Nelson, J. (1997). Incidence of the mucopolysaccharidoses in Northern Ireland. Hum Genet, 101(3), 355-358. Nelson, J., Crowhurst, J., Carey, B., & Greed, L. (2003). Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet A, 123A(3), 310-313. Neufeld, E. F., & Muenzer, J. (2001). The mucopolysaccharidoses. In: Scriver CR. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill. Ochiai, T., Suzuki, Y., Kato, T., Shichino, H., Chin, M., Mugishima, H., et al. (2007). Natural history of extensive Mongolian spots in mucopolysaccharidosis type II (Hunter syndrome): a survey among 52 Japanese patients. J Eur Acad Dermatol Venereol, 21(8), 1082-1085. Oshima, J., Lee, J. A., Breman, A. M., Fernandes, P. H., Babovic-Vuksanovic, D., Ward, P. A., et al. (2011). LCR-initiated rearrangements at the IDS locus, completed with Alu-mediated recombination or non-homologous end joining. J Hum Genet, 56(7), 516-523. Pan, C., Nelson, M. S., Reyes, M., Koodie, L., Brazil, J. J., Stephenson, E. J., et al. (2005). Functional abnormalities of heparan sulfate in mucopolysaccharidosis-I are associated with defective biologic activity of FGF-2 on human multipotent progenitor cells. Blood, 106(6), 1956-1964. Panine, V. V., & Hashimoto, K. (1995). What syndrome is this? Hunter syndrome (mucopolysaccharidosis II). Pediatr Dermatol, 12(4), 370-372. Parkinson, E. J., Muller, V., Hopwood, J. J., & Brooks, D. A. (2004). Iduronate-2sulphatase protein detection in plasma from mucopolysaccharidosis type II patients. Mol Genet Metab, 81(1), 58-64. Pennock, C. A., & Barnes, I. C. (1976). The mucopolysaccharidoses. J Med Genet, 13(3), 169-181. Pinto, L. L., Schwartz, I. V., Puga, A. C., Vieira, T. A., Munoz, M. V., & Giugliani, R. (2006). Prospective study of 11 Brazilian patients with mucopolysaccharidosis II. J Pediatr (Rio J), 82(4), 273-278. Poorthuis, B. J., Wevers, R. A., Kleijer, W. J., Groener, J. E., de Jong, J. G., van Weely, S., et al. (1999). The frequency of lysosomal storage diseases in The Netherlands. Hum Genet, 105(1-2), 151-156. Probst, F. J., Roeder, E. R., Enciso, V. B., Ou, Z., Cooper, M. L., Eng, P., et al. (2007). Chromosomal microarray analysis (CMA) detects a large X chromosome deletion including FMR1, FMR2, and IDS in a female patient with mental retardation. Am J Med Genet A, 143A(12), 1358-1365. Quaio CR, Grinberg H, Vieira ML, Paula AC, Leal GN, Gomy I, Leistner-Segal S, Giugliani R, Bertola DR, Kim CA. (2012). Report of a Large Brazilian Family With a Very Attenuated Form of Hunter Syndrome (MPS II). JIMD Rep. 4:125-128. Rathmann, M., Bunge, S., Steglich, C., Schwinger, E., & Gal, A. (1995). Evidence for an iduronate-sulfatase pseudogene near the functional Hunter syndrome gene in Xq27.3-q28. Hum Genet, 95(1), 34-38. Rathmann M, Bunge S, Beck M, Kresse H, Tylki-Szymanska A, Gal A. (1996). Mucopolysaccharidosis type II (Hunter syndrome): mutation "hot spots" in the iduronate-2-sulfatase gene. Am J Hum Gene, 59(6),1202-9. Roeser, D., Preusser-Kunze, A., Schmidt, B., Gasow, K., Wittmann, J. G., Dierks, T., et al. (2006). A general binding mechanism for all human sulfatases by 149 the formylglycine-generating enzyme. Proc Natl Acad Sci U S A, 103(1), 8186. Scarpa, M., Almassy, Z., Beck, M., Bodamer, O., Bruce, I. A., De Meirleir, L., et al. (2011). Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J Rare Dis, 6, 72. Schaap, T., & Bach, G. (1980). Incidence of mucopolysaccharidoses in Israel: is Hunter disease a "Jewish disease"? Hum Genet, 56(2), 221-223. Schitter, G., Scheucher, E., Steiner, A. J., Stutz, A. E., Thonhofer, M., Tarling, C. A., et al. (2010). Synthesis of lipophilic 1-deoxygalactonojirimycin derivatives as D-galactosidase inhibitors. Beilstein J Org Chem, 6, 21. Schouten, J. P., McElgunn, C. J., Waaijer, R., Zwijnenburg, D., Diepvens, F., & Pals, G. (2002). Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res, 30(12), e57. Schwartz, I. V., Ribeiro, M. G., Mota, J. G., Toralles, M. B., Correia, P., Horovitz, D., et al. (2007). A clinical study of 77 patients with mucopolysaccharidosis type II. Acta Paediatr Suppl, 96(455), 63-70. Shih, S. L., Lee, Y. J., Lin, S. P., Sheu, C. Y., & Blickman, J. G. (2002). Airway changes in children with mucopolysaccharidoses. Acta Radiol, 43(1), 40-43. Shinomiya N, Nagayama T, Fujioka Y, Aoki T. (1996). MRI in the mild type of mucopolysaccharidosis II (Hunter's syndrome). Neuroradiology, 38(5):483-5. Sohn, Y. B., Ki, C. S., Kim, C. H., Ko, A. R., Yook, Y. J., Lee, S. J., et al. (2012). Identification of 11 novel mutations in 49 Korean patients with mucopolysaccharidosis type II. Clin Genet, 81(2), 185-190. Steén-Bondeson ML, Dahl N, Tönnesen T, Kleijer WJ, Seidlitz G, Gustavson KH, Wilson PJ, Morris CP, Hopwood JJ, Pettersson U. (1992). Molecular analysis of patients with Hunter syndrome: implication of a region prone to structural alterations within the IDS gene. Hum Mol Genet. 1(3),195-198. Sukegawa-Hayasaka, K., Kato, Z., Nakamura, H., Tomatsu, S., Fukao, T., Kuwata, K., et al. (2006). Effect of Hunter disease (mucopolysaccharidosis type II) mutations on molecular phenotypes of iduronate-2-sulfatase: enzymatic activity, protein processing and structural analysis. J Inherit Metab Dis, 29(6), 755-761. Tanaka A, Okuyama T, Suzuki Y, Sakai N, Takakura H, Sawada T, Kato S, et al. (2012). Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: a nationwide survey in Japan. Mol Genet Metab, 107(3):513-20 Tomatsu S, Sukegawa K, Trandafirescu GG, Gutierrez MA, Nishioka T, Yamaguchi S, Orii T, Froissart R, Maire I, Chabas A, Cooper A, Di Natale P, Gal A, Noguchi A, Sly WS. (2006). Differences in methylation patterns in the methylation boundary region of IDS gene in Hunter syndrome patients: implications for CpG hot spot mutations. Eur J Hum Genet.14(7), 838-845. Trott, O., & Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem, 31(2), 455-461. Upadhyaya, M., Sarfarazi, M., Bamforth, J. S., Thomas, N. S., Oberle, I., Young, I., et al. (1986). Localisation of the gene for Hunter syndrome on the long arm of X chromosome. Hum Genet, 74(4), 391-398. 150 Vafiadaki E, Cooper A, Heptinstall LE, Hatton CE, Thornley M, Wraith JE. (1998). Mutation analysis in 57 unrelated patients with MPS II (Hunter's disease). Arch Dis Child, 79(3),237-41. Velasco, H., Pineda, t., & et al, e. a. (2011). Identificación de mutaciones en el gen IDUA en pacientes de Colombia, Ecuador y Perú con mucopolisacaridosis Tipo I.Unpublished manuscript. Vellodi, A., Young, E., Cooper, A., Lidchi, V., Winchester, B., & Wraith, J. E. (1999). Long-term follow-up following bone marrow transplantation for Hunter disease. J Inherit Metab Dis, 22(5), 638-648. Vieira, T., Schwartz, I., Munoz, V., Pinto, L., Steiner, C., Ribeiro, M., et al. (2008). Mucopolysaccharidoses in Brazil: what happens from birth to biochemical diagnosis? Am J Med Genet A, 146A(13), 1741-1747. Villani, G. R., Daniele, A., Balzano, N., & Di Natale, P. (2000). Expression of five iduronate-2-sulfatase site-directed mutations. Biochim Biophys Acta, 1501(23), 71-80. Voznyi, Y. V., Keulemans, J. L., & van Diggelen, O. P. (2001). A fluorimetric enzyme assay for the diagnosis of MPS II (Hunter disease). J Inherit Metab Dis, 24(6), 675-680. Waldow, A., Schmidt, B., Dierks, T., von Bulow, R., & von Figura, K. (1999). Amino acid residues forming the active site of arylsulfatase A. Role in catalytic activity and substrate binding. J Biol Chem, 274(18), 12284-12288. Wei, X., Jin, F., Ye, Y., Xu, C., Qu, N., Ju, X., et al. (2011). A novel mutation of IDS gene in a Chinese patient with mucopolysaccharidosis II by next-generation sequencing. Clin Chim Acta, 412(23-24), 2340-2342. Wilson, P. J., Meaney, C. A., Hopwood, J. J., & Morris, C. P. (1993). Sequence of the human iduronate 2-sulfatase (IDS) gene. Genomics, 17(3), 773-775. Wilson, P. J., Morris, C. P., Anson, D. S., Occhiodoro, T., Bielicki, J., Clements, P. R., et al. (1990). Hunter syndrome: isolation of an iduronate-2-sulfatase cDNA clone and analysis of patient DNA. Proc Natl Acad Sci U S A, 87(21), 8531-8535. Wilson, P. J., Suthers, G. K., Callen, D. F., Baker, E., Nelson, P. V., Cooper, A., et al. (1991). Frequent deletions at Xq28 indicate genetic heterogeneity in Hunter syndrome. Hum Genet, 86(5), 505-508. Wolfe, B. J., Blanchard, S., Sadilek, M., Scott, C. R., Turecek, F., & Gelb, M. H. (2011). Tandem mass spectrometry for the direct assay of lysosomal enzymes in dried blood spots: application to screening newborns for mucopolysaccharidosis II (Hunter Syndrome). Anal Chem, 83(3), 1152-1156. Wraith, J. E., Beck, M., Giugliani, R., Clarke, J., Martin, R., & Muenzer, J. (2008). Initial report from the Hunter Outcome Survey. Genet Med, 10(7), 508-516. Wraith, J. E., Cooper, A., Thornley, M., Wilson, P. J., Nelson, P. V., Morris, C. P., et al. (1991). The clinical phenotype of two patients with a complete deletion of the iduronate-2-sulphatase gene (mucopolysaccharidosis II--Hunter syndrome). Hum Genet, 87(2), 205-206. Wraith, J. E., Scarpa, M., Beck, M., Bodamer, O. A., De Meirleir, L., Guffon, N., et al. (2008). Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr, 167(3), 267-277. Yatziv, S., & Epstein, C. J. (1977). Hunter syndrome presenting as macrocephaly and hydrocephalus. J Med Genet, 14(6), 445-447. 151 Yatziv, S., Erickson, R. P., & Epstein, C. J. (1977). Mild and severe Hunter syndrome (MPS II) within the same sibships. Clin Genet, 11(5), 319-326. Young, I. D., & Harper, P. S. (1982a). Incidence of Hunter's syndrome. Hum Genet, 60(4), 391-392. Young, I. D., & Harper, P. S. (1982b). Mild form of Hunter's syndrome: clinical delineation based on 31 cases. Arch Dis Child, 57(11), 828-836. Young, I. D., Harper, P. S., Newcombe, R. G., & Archer, I. M. (1982). A clinical and genetic study of Hunter's syndrome. 2. Differences between the mild and severe forms. J Med Genet, 19(6), 408-411. Zhang, H., Li, J., Zhang, X., Wang, Y., Qiu, W., Ye, J., et al. (2011). Analysis of the IDS gene in 38 patients with Hunter syndrome: the c.879G>A (p.Gln293Gln) synonymous variation in a female create exonic splicing. PLoS One, 6(8), e22951. 152 153