Conferencia - PIFRECV - Universidad de Talca
Anuncio

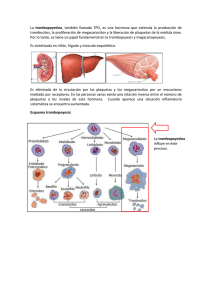
Papel de las Plaquetas en la Patogenia de la Ateroesclerosis Dr. Jaime Pereira G Departamento de Hematología-Oncología Escuela de Medicina Pontificia Universidad Católica de Chile Conferencia organizada por el PIFRECV Facultad de Ciencias de la Salud Universidad de Talca Martes 13 de noviembre de 2007 Ateroesclerosis • Enfermedad inflamatoria sistémica que se caracteriza por la acumulación de células inflamatorias en la íntima de las arterias grandes. Papel “clásico” de las plaquetas en la historia natural de la ateroesclerosis Interacción de las plaquetas con las células endoteliales ¿Lo hacen? ¿Cómo? ¿Consecuencias? Las plaquetas se unen directamente a las células endoteliales in vitro e in vivo. • In vitro Circulation 1997; 96: 1809 Las plaquetas se unen directamente a las células endoteliales in vitro e in vivo. • In vitro Plaquetas en reposo Plaquetas activadas J Exp Med 1998; 187: 329 Las plaquetas se unen directamente a las células endoteliales in vitro e in vivo. • In vivo J Exp Med 2002; 196: 887. Adhesión de plaquetas activadas a HUVEC PLAQUETA ACTIVADA GPIIbIIIa GPIIbIIIa Fbg FVW GPIbα αvβ3 ICAM-1 αvβ3 GPIIbIIIa Fbn αvβ3 CELULA ENDOTELIAL . . . . .. : . . . , . cGMP Plaquetas cAMP ADP PKC Ca++ DAG AC IP3 FvW PLC EctoADPasa : Trombina sGC FT Célula Endotelial PGI2 COX AA Sub-endotelio NO TxA2/PGH2 NOS FvW L-Arg AA FvW Adhesión de plaquetas en reposo a CE dañadas/activadas Blood 1998; 92: 507. Adhesión de plaquetas en reposo a CE dañadas/activadas Blood 1998; 92: 507. Br J Haematol 2006; 134: 426. “Rolling” Adhesión irreversible GPIIb/IIIa PSGL-1 GPIb P-selectina Fibrinógeno GPIIb/IIIa activada αvβ3 Proteínas de los gránulos alfa de las plaquetas Prot. plasmáticas Albúmina IgG Fibrinógeno Factor V FvW Etc. FP4 Prot. plaquetarias Reparación tisular Inflamación β-TG PDGF PDAF PDEGF TGF-alfa TGF-beta H-FGF RANTES ENA-78 SDF-1 IL-1β CD40L Quimioquinas con efecto sobre las células endoteliales Quimioquina Receptor MCP-1 CCR2 IL-8 CXCR2 TARC/MDC CCR4 Fractalquina CX3CR4 RANTES CCR1/CCR5 PF4 ? SDF-1α CXCR4 Isotipo RANTES Circulation 2001; 103: 1772. CD40L plaquetario induce secreción de MCP-1 e IL-8 por las células endoteliales Nature 1998; 391: 591. Thromb Haemost 2007; 98: 798 Plaquetas activadas tienen capacidad de síntesis • Bcl-3 • IL-1β • PAI-1 • Factor tisular Blood 2007;109: 5242 Reclutamiento de células progenitoras J Exp Med 2006; 203: 1221. Reclutamiento de células progenitoras J Exp Med 2006; 203: 1221. CD36 participa en la trombosis in vivo en la dislipidemia Deficiencia de CD36 en hiperlipidemia modula la reactividad plaquetaria Enfermedad Vascular Isquémica y Uso de Cocaína • El uso de cocaína es un reconocido factor de riesgo de complicaciones vasculares isquémicas, que afectan tanto la circulación coronaria como el territorio cerebrovascular. • Clínicamente los usuarios de cocaína tienen un riesgo aumentado de presentar infarto de miocardio, angina de pecho, muerte súbita y accidentes Efectos vasculares del uso de cocaína • Arbol vascular – Ateroesclerosis acelerada* – Trombosis • Corazón – Ateroesclerosis acelerada* – Trombosis coronaria (sindrome coronario agudo, IAM) • Cerebro – Ateroesclerosis acelerada* – Accidente vascular isquémico (RR: 14.0) – Defectos regionales de perfusión cerebral * En ausencia de factores de riesgo clásicos Activación de la hemostasia asociada al uso de cocaína • Pacientes – 18 pacientes de sexo masculino dependientes de cocaína DSM-IV, ingresados a la Clínica Siquiátrica Universitaria. – Edad: 20-49 años; promedio: 35 años – Sin antecedentes de enfermedad cardiovascular – Con último consumo dentro de las 72 horas del ingreso • Controles – 22 hombres sanos – Edad: 22-65 años; promedio: 39 años % Monocyte-platelet aggregates p: <0.0001 100 75 50 25 0 Cocaine Controls P-selectina 10.0 % células positivas P= 0,0376 7.5 * 5.0 2.5 0.0 Cocaína NLE 5000000 p= 0,0003 Microparticles / mL 4000000 3000000 2000000 1000000 0 Cocaine Controls P<0.0001 TF ratio/Isotype 2.0 1.5 1.0 0.5 0.0 Cocaine NLE CEC CEC/mL sangre periferica 1200 1000 Donantes 800 Normal 600 SCA 400 Coca 200 0 Pacientes En resumen…….. • Definitivamente las plaquetas están comprometidas tanto en la iniciación como en aceleración de la ateroesclerosis. • – Las plaquetas inician la ateroesclerosis por adhesión al endotelio intacto (¿inflamado?) – Las plaquetas adheridas al endotelio reclutan monocitos que transmigran al subendotelio transformándose en macrófagos y células espumosas. – Además de los monocitos, las plaquetas reclutan células progenitoras de la MO, que se diferencian en células endoteliales para reparación vascular o células espumosas. Las plaquetas aceleran la AE por incorporación y transporte de ox LDL a los macrófagos/células espumosas. En resumen…….. • Potenciales blancos de intervención farmacológica? • Antiplaquetarios clásicos, sin efecto sobre la progresión de la aterosclerosis • Inhibición de la unión de las plaquetas a la CE? – PSGL-1, anti-P-sel, anti-GPIb • Inhibición de la interacción plaquetas-monocitos – Anti-Mac-1; PSGL-1 • Interacción de las plaquetas con oxLDL – Anti-CD36; CD68-Fc – Estatinas y antagonistas PPAR γ.