El Tejido Adiposo como Órgano Endocrino
Anuncio

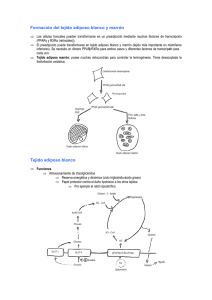
Brandan, Nora C. Profesora Titular. Cátedra de Bioquímica. Facultad de Medicina. UNNE. Llanos, Isabel Cristina. Jefa de Trabajos Prácticos. Cátedra de Bioquímica. Facultad de Medicina. UNNE. Miño, Claudia Alejandra. Jefa de Trabajos Prácticos. Cátedra de Bioquímica. Facultad de Medicina. UNNE. Piccardo, Alejandro Pablo. Ayudante Alumno. Cátedra de Bioquímica. Facultad de Medicina. UNNE. Ragazzoli, Maximiliano A. Ayudante Alumno por Concurso. Cátedra de Bioquímica. Facultad de Medicina. UNNE. Ruiz Díaz, Daniel A. N. Ayudante Alumno por Concurso. Cátedra de Bioquímica. Facultad de Medicina. UNNE. El Tejido Adiposo como Órgano Endocrino Universidad Nacional del Nordeste Facultad de Medicina TEJIDO ADIPOSO. Generalidades.............................................................................. 1 PEROXISOME PROLIFERATOR- ACTIVATED RECEPTORS (PPARs)................ 1 PPAR................................................................................................................ 2 PPAR................................................................................................................ 2 PPAR 2 ADIPOCITOQUINAS ESPECÍFICAS DEL TEJIDO ADIPOSO............................... 3 ADIPONECTINA.................................................................................................... 3 Estructura............................................................................................................ 3 Síntesis................................................................................................................ 3 Secreción.............................................................................................................3 Receptor y mecanismo de acción......................................................................... 3 Acción:................................................................................................................3 RESISTINA............................................................................................................. 4 Estructura............................................................................................................ 4 Síntesis................................................................................................................ 4 Secreción.............................................................................................................4 Mecanismo de acción.......................................................................................... 4 Acción................................................................................................................. 4 LEPTINA................................................................................................................. 4 Estructura............................................................................................................ 4 Síntesis y Secreción............................................................................................. 4 Receptor.............................................................................................................. 4 Mecanismo de Acción......................................................................................... 4 Acción................................................................................................................. 5 ADIPSINA............................................................................................................... 5 ACYLATION STIMULATES PROTEIN (ASP).................................................. 5 ADIPOCITOQUINAS NO ESPECÍFICAS DEL TEJIDO ADIPOSO........................ 5 INTERLEUQUINAS (IL)....................................................................................... 5 IL-1..................................................................................................................... 5 IL-6..................................................................................................................... 5 TNF-................................................................................................................. 6 INHIDOR DEL ACTIVADOR DE PLASMINÓGENO (PAI-1)......................... 6 APLICACIÓN CLINICA: Insulinorresistencia y Adipocitoquinas.............................. 6 BIBLIOGRAFIA............................................................................................................ 8 El Tejido Adiposo como Órgano Endocrino Página 1 TEJIDO ADIPOSO. Generalidades Tradicionalmente, el tejido adiposo fue visto como el sitio de almacenamiento de energía en forma de triacilglicéridos (TAG) durante la alimentación y liberador de ácidos grasos durante el ayuno para proporcionar combustible a otros tejidos. Sin embargo, hoy es evidente que tiene funciones fisiológicas importantes, secretando numerosas proteínas, la cuales participan en la regulación autócrina y parácrina dentro del propio tejido y además tienen efectos en la función de órganos distantes, tales como el músculo, páncreas, hígado y cerebro. Estas proteínas secretadas, las cuales fueron denominadas bajo el término común de adipocitoquinas o adipocinas se hallan implicadas en: La regulación del peso corporal (leptina, CRP30/adipoQ) La función del sistema inmune (TNFa, IL-1, IL-6) La función vascular (angiotensina e inhibidor del plasminógeno tipo 1), La función reproductiva (estrógenos). Desarrollo de la resistencia a la insulina (resistina) Por lo tanto, se reconoce que el tejido adiposo, especialmente el visceral funciona como un órgano mayor endócrino. Estos nuevos conocimientos tienen implicancias importantes para entender la relación fisiopatológica entre el exceso de grasa del cuerpo y los estados patológicos, tales como la resistencia a la insulina y diabetes mellitus, solo por nombrar algunas. El tejido adiposo está formado por células adiposas (adipocitos) y un componente estromático/vascular en el que residen los preadipocitos. Los adipocitos, con un tamaño de 10 a 200 micras, son células redondeadas que contienen una vacuola lipídica que representa el 95% del peso celular y que desplaza al resto de las organelas hacia la periferia. Existen dos tipos de tejido adiposo, y por lo tanto dos tipos de adipocitos diferentes que los forman: ➢ El tejido adiposo blanco, es el más abundante del organismo humano adulto y por lo tanto el mayor reservorio energético, el cual, como ya se mencionara éste deposito se hace en forma de TAG, proveniente estos de los quilomicrones y VLDL circulantes. Es en éste tejido adiposo blanco donde se pone de manifiesto como órgano productor de sustancias con acción endócrina, parácrina y autócrina. ➢ El tejido adiposo pardo es el encargado de la termogénesis, su color se debe por la gran cantidad de mitocondrias que posee, las cuales expresan altas cantidades de UCP (uncoupling protein); proteínas desacoplantes que producen una fosforilación oxidativa desacopladora, lo que produce disipación de energía en forma de calor. En condiciones normales el 80% del tejido adiposo está localizado en el tejido celular subcutáneo (TCS o hipodermis), mientras que el tejido adiposo visceral (TAV) representa menos del 20%. El TAV está constituido por adipocitos de un tamaño más reducido, con menor capacidad de almacenamiento, más vascularizado, con una mayor inervación simpática y con gran número de receptores 3-adrenérgicos, lo que facilita una mayor actividad metabólica. En el aumento de la cantidad tejido adiposo se hallan implicado dos procesos; por una lado está el aumento de tamaño de los adipocitos (hipertrofia) y por otro, el incremento en el número de adipocitos (hiperplasia), este último se realiza a partir de los preadipocitos mesenquimáticos, lo cual supone un conjunto de pasos de diferenciación en el que participa una cascada de factores de trascripción específicos, uno de los cuales es el receptor activador de la proliferación de los peroxisomas gamma. Debido a la importancia de este tipo de receptor para el desarrollo del tejido adiposo se lo describirá en primer lugar, para luego continuar con las distintas adipocitoquinas producidas por el tejido adiposo. PEROXISOME PROLIFERATOR- ACTIVATED RECEPTORS (PPARs) Los peroxisome proliferator-activated receptors (PPARs) constituyen una subfamilia de receptores nucleares que tras la unión de su ligando funcionan como factores de trascripción. Estos receptores son activados por ácidos grasos poliinsaturados o derivados de estos (ligandos fisiológicos) y tiazolidinedionas o fibratos (ligandos farmacológicos), donde el principal mecanismo de activación involucrado es la fosforilación del PPAR. De esta manera, actuando como hormonas los ácidos grasos activan receptores que gobiernan su propio metabolismo. Estructuralmente, estos receptores están constituidos por unos 400 aminoácidos (aa), y al igual que los demás receptores de esteroides tienen 5 o 6 regiones que conforman 4 dominios y cada uno de ellos tiene una determinada función: ➢ Región aminoterminal (dominio A/B) de función activadora independiente del ligando. ➢ Región de unión al DNA (dominio C) posee la típica estructura en dedos de zinc. ➢ Región denominada bisagra (dominio D), une la región de unión al DNA con la de unión al ligando. ➢ Región de unión al ligando (dominio E/F) de función activadora dependiente del ligando. Página 2 La unión al DNA requiere la formación con un heterodímero de PPAR con el receptor del ácido 9-cisretinoico (RXR), interactuando con un elemento de respuesta proliferador del peroxisoma (ERPP) en el gen blanco (ver Fig. 1). Esta subfamilia de receptores nucleares posee 3 subtipos o isoformas de los PPARs: 1) PPAR: se expresa primariamente en hepatocitos, en menor grado lo hace en cardiocitos, enterocitos, células de corteza adrenal y endotelio. El ligando fisiológico para este con mayor afinidad es un eicosanoide, el acido 8- hidroxieicosatetranoico, sin embargo los ligandos más potente son los fibratos (ligando farmacológico). La activación de este receptor con la consiguiente función de factor de trascripción provoca una serie de modificaciones metabólicas: ➢ Inducen las enzimas mitocondriales de la -oxidación, lo que determina una disminución de los ácidos grasos disponibles para la formación de las lipoproteínas ricas en TAG (PRTG). ➢ Inducen la transcripción de las Apo-AI y Apo-AII, incrementándose así la formación de HDL. ➢ Inhibición de la trascripción de la Apo-CIII, lo cual favorece la reducción en la formación de VLDL. ➢ Induce la trascripción del gen que regula a la lipoprotein-lipasa 1(LPL-1), produciéndose el catabolismo de las PRTG. ➢ Inhibe a la ciclooxigenasa-2 (COX2), disminuyendo la formación de los derivados del ácido araquidónico, lo que trae como consecuencia una disminución de la inflamación endotelial. No ejerce regulación sobre la COX-1. ➢ Inhibe la expresión de las moléculas de adhesión a células vasculares-1 (VCAM-1). 2) PPAR también llamado PPAR , este es ubicuo en cuanto a su localización, su ligando natural (ácido graso) aún no ha sido determinado. De los 3 receptores de esta subfamilia este es el menos conocido, solo puede hacerse algunas conclusiones del todo parciales de las acciones de éste tras su activación: ➢ Inhibiría la proliferación de los queratinocitos. Se ha observado que la carencia del PPAR se asocia con una proliferación aumentada de las células epiteliales, relacionándose con una mayor sensibilidad al cáncer de piel. Esto, si es así, tendría importancia terapéutica, en desórdenes como la psoriasis y el cáncer, ya que podría utilizarse para provocar una diferenciación selectiva e inhibir la proliferación celular. PPAR: este se expresa primariamente en adipocitos, colonocitos (células del colon), macrófagos y células del endotelio vascular. El ligando fisiológico de este receptor se desconoce, a pesar de que la prostaglandina J tiene una alta afinidad. Las tiazolidinedionas son los fármacos que se unen con mayor afinidad al PPAR . Los efectos promovidos por este receptor pueden resumirse de la siguiente manera: (ver Fig. 2) ➢ Promueve la diferenciación celular, en especial de adipocitos. ➢ Codifica proteínas para la captación de ácidos grasos y lipogénesis ➢ Induce la síntesis de GLUT-4. ➢ Induce la expresión de proteínas que intervienen en la termogénesis. ➢ Inhibe la secreción de óxido nítrico sintetasa. ➢ Reduce la secreción de la gelatinasa B (conocida también como metaloproteinasa-9 de matriz), lo que disminuye el daño tisular. ➢ Reduce la expresión de moléculas de adhesión celular, sobre todo VCAM-1. ➢ Regula la captación de LDL oxidadas. ➢ Reduce la secreción de citoquinas del monocito. Fig. 1 Fig. 2 Página 3 ADIPONECTTINA Disminuye flujo AGL Disminuye producción glucosa Disminuye TAG Aumenta Insulinosensibilidad Disminuye Inflamación vascular Aumenta Insulinosensibilidad Aumenta oxidación AGL ADIPOCITOQUINAS ESPECÍFICAS DEL TEJIDO ADIPOSO ADIPONECTINA Estructura: es una proteína de 244 aminoácidos (aa), producto del gen apM1, el que está expresado en el tejido adiposo en forma específica y muy abundante. Pertenece a la superfamilia del colágeno, tiene una estructura homóloga con los colágenos VIII y X y con el factor de complemento C1a-like. En sangre se observan la forma fAdiponectina o forma intacta y la gAdiponectina o globular. Síntesis: sintetizada por el adipocito del tejido visceral, inducida durante la diferenciación del mismo. Se ha demostrado que las variaciones a nivel plasmático de adiponectina están influenciadas por el tejido adiposo visceral más que por el tejido adiposo subcutáneo. Secreción: Estimuladores: insulina, IGF-1 (insulin growth factor 1),PPAR (peroxisome proliferator activated nuclear receptor y el LRH-1 (liver receptor homolog-1). Los dos últimos por activación de la trascripción del gen de adiponectina. Inhibidores: TNF- , resistina, leptina, glucocorticoides y ghrelina. Los dos últimos por inhibición de la expresión genética. Receptor y mecanismo de acción: Adiponectina actúa a través de 2 tipos de receptores denominados adipo R, los que se hallan ampliamente distribuidos, incluso en cerebro. Adipo R1 muy abundante en músculo esquelético y adipo R2 expresado mayormente en hígado. Ambos incrementan la fosforilación de la AMP-quinasa (AMPK), una enzima que tiene un rol en las acciones que aumentan la sensibilidad a la insulina y es la responsable directa o indirecta de las acciones de la adiponectina. La AMPK es un sensor de la energía intracelular, que es activada en forma fisiológica por la adiponectina y el ejercicio. Acción: (ver Fig. 3) Músculo esquelético: Aumenta la fosforilación de la tirosina del receptor de insulina y del sustrato del receptor deinsulina-1(IRS-1) lo que favorece la insulinosensibilidad. Aumenta la captación de glucosa, por estimulo de GLUT-4,aumenta la producción de lactato. Se produce la fosforilación de la enzima Acetil-CoA Carboxilasa y con ello suinhibición; lo que favorece la -oxidación de los AGL. Incrementa la actividad del PPAR- induciéndose aun más laoxidación de los AGL. Hígado: Regula dos enzimas clave para la gluconeogénesis como la Fosfoenol-piruvato-carboxi-kinasa y la Glucosa 6-fosfatasa, por lo que produce descenso de los niveles glucémicos. Tejido adiposo: Regula positivamente la acción de la LPL-1, por lo que aumenta el catabolismo de las PRTG. Endotelio vascular: inhibe la expresión de moléculas de adhesión (VCAM e ICAM). Activa la enzima Oxido NítricoSintetasa (NOS), produciendo la formación de óxido nítrico (ON). Inhibe la inducción del factor nuclear kappa beta(NFkB) por parte del factor de necrosis tumoral- . Suprime la expresión de diferentes factores de crecimiento, lo cualimpide la proliferación y migración de células del músculo liso vascular. Además inhibe la expresión del receptorscavenger y consecuentemente la transformación de macrófagos en células espumosas. Regulación de la Síntesis de Adiponectina Estimuladores Inhibidores Concentracionesnormales de insulina Sistemaendocannabinoideinactivado PPAR- activado,tiazolidindionas Altasconcentraciones deinsulina Sistemaendocannabinoideactivado TNF-a Hiperactividad delsistema simpático Fig. 3 Página 4 RESISTINA Estructura: proteína de 114 aa y tiene una estructura similar a proteínas involucradas en procesos inflamatorios. Síntesis: producida en el estroma vascular del tejido adiposo y monocitos principalmente, se la encuentra además en medula ósea, células mononucleares periféricas, pulmón, placenta, células pancreáticas, hipotálamo, hipófisis, glándulas adrenales, miocitos y bazo. Secreción: está fuertemente controlada por condiciones nutricionales y hormonales. Se encuentran bajas concentraciones en el ayuno y su nivel aumenta con la ingesta. La insulina suprimiría el gen de expresión en adipocitos y la hiperglucemia promovería la expresión del gen de la resistina. El IGF (insulin-like growth factor) por ser estimulante del proceso de adipogénesis disminuye la expresión del gen de resistina. Mecanismo de acción: aun no se conocen los mecanismos de señalización. Acción: se sugiere un rol de la resistina en los estados inflamatorios. Se ha reportado una correlación positiva entre resistina sérica y proteína C reactiva (PCR). Tiene efectos antagónicos a la insulina. Reduce el transporte de glucosa dependiente de insulina al músculo esquelético yal tejido adiposo, aumenta la producción hepática de glucosa y la glucemia en ayunas e inhibe la adipogénesis (mediadorade la resistencia insulina). Produce descenso de los niveles séricos de HDL. A nivel vascular, reduce la vasorrelajación, disminuye la expresión de óxido nítrico. Promueve la proliferación yactivación de células musculares lisas y células endoteliales y estimula la expresión de moléculas de adhesión. LEPTINA Estructura: hormona de 146 aa, se produce a partir de un precursor de 167 aa, con una secuencia señal de 21 aminoácidos que se separan antes de que la leptina pase a la sangre; es estructuralmente similar a la interleucina-1. Síntesis y Secreción: es producida principalmente, pero no exclusivamente por el tejido adiposo (en proporción a la grasa corporal). Nuevas evidencias demuestran que la placenta, el músculo esquelético y posiblemente el fundus gástrico son sitios adicionales de síntesis. También puede ser secretada por células inmunocompetentes y endoteliales. La secreción es pulsátil y esta modulada por diversas hormonas; entre quienes aumentan su producción se encuentran los glucocorticoides, la insulina, interleucina-1 y factor de necrosis tumoral- ; en tanto las que atenúan su expresión son la testosterona y las hormonas tiroides. Receptor: son denominados Ob R, pertenece a la familia de los receptores de citoquinas. La unión de la leptina al receptor induce la dimerización del receptor. Existen varias isoformas del receptor de leptina y se encuentran distribuidos por casi todos los tejidos: ➢ Los receptores más largos (OB-Rb) predominan en hipotálamo y sus funciones consisten en mediar las acciones de la leptina a nivel del SNC. Presentan dominios extracelular, transmembranal e intracelular, lo que indica una posible función de transducción de la señal al interior de la célula. ➢ Las formas cortas (OB-Ra, OBRc, OB-Rd y OB-Rf) se localizan además del hipotálamo en tejidos como el cerebro, riñones, pulmones, tejido adiposo, hígado, páncreas, endotelio y corazón. Carecen de dominio intracelular y sus funciones se han relacionado con el transporte, depuración de la leptina y con la regulación del sistema inmune, entre otras. ➢ El receptor más pequeño, la isoforma OB-Re, carece de dominio intracelular. Parece probable que este receptor, al ser una forma soluble, esté relacionado con el transporte de leptina en plasma y a través de la barrera hematoencefálica. Mecanismo de Acción: La activación del receptor produce la transducción de señal a través de la vía Janus kinasa (JAK) /y el activador del camino de la trascripción (STAT), con activación del factor de transcripción nuclear c-fos. La leptina también aumenta la actividad del fosfatidil inositol 3 quinasa (PI- 3K) desencadenando señales que parecen ser importantes en la modulación de los efectos de las leptina sobre la insulina. Se ha propuesto que también activa la vía de la Proteín-Kinasa A de forma directa. Regulación de la Resistina Factores estimuladores Factores inhibidores Catecolaminas Hormona de crecimiento Endotelina-1 Hormonas gonadales Hiperglucemia Dehidroepiandrosterona Sexo masculino Citoquinas (IL-6) Insulina TNF- Página 5 Acción Interviene en la homeostasis energética, evitando un incremento excesivo del porcentaje graso. En el estado de leptinodeficiencia, la activación disminuida del receptor en el hipotálamo, causa una creciente produccióndel neuropéptido Y (NPY), el cual es probablemente responsable de la hiperfagia, la obesidad y los cambios neuroendócrinosvistos en dicho estado. A nivel hepático activa la enzima Acetil-CoA oxidasa y citrato sintetasa e inhibe a la Acetil-CoA carboxilasa (disminuye lalipogénesis en hígado y tejido graso, aumenta la -oxidación, con lo que dirigen los ácidos grasos libres a su catabolismo por elciclo de Krebs y disminuye su concentración intracelular. . A nivel del metabolismo hidrocarbonado estimula la utilización de glucosa por el músculo y promueve su transporte através del intestino delgado. Sobre el metabolismo de los lípidos, estimula la lipólisis en el adipocito. Inhibe la secreción pancreática de insulina. Aumenta la actividad fagocítica de los macrófagos y también la producción de citoquinas proinflamatorias. Proliferación de células hematopoyéticas. Activación de células T. Promueve la angiogénesis, al estimular la proliferación de células endoteliales. Puede mejorar el flujo sanguíneo y facilitar la disipación de calor y la oxidación lipídica. ADIPSINA Proteasa sérica que no es más que el complemento D, enzima iniciadora y velocidad limitadora de la vía alternativa del complemento, la cual es producida por el tejido adiposo. Se encuentra elevada en la obesidad con un sistema regulatorio dependiente del incremento de la insulina y los glucocorticoides. ACYLATION STIMULATES PROTEIN (ASP) Es el resultado de la unión del residuo terminal de arginina del factor C-3a por las carboxipeptidasas. Como C-3a es un producto final del complemento del cual el factor D forma parte, se le llama vía adipsin-ASP. Su función es la captación y esterificación de los ácidos grasos hidrolizados de los triglicéridos por LPL-1 en el estado posprandial vía diacilglicerol acyltransferasa. Aunque la adipsina está aumentada en el obeso, el producto ASP no sufre un aumento proporcional, lo que ha llevado a pensar que ésta cumple un papel limitante en la velocidad de la reesterificación de ácidos grasos y del crecimiento del adipocito. En forma general, hay una disminución de ASP, de la actividad de la diacilglicerol acyltransferasa, de la síntesis de triglicéridos y de la esterificación, lo que lleva a un aumento de los AGL. ADIPOCITOQUINAS NO ESPECÍFICAS DEL TEJIDO ADIPOSO INTERLEUQUINAS (IL) Las IL forman parte de la familia de las citoquinas. Son péptidos señalizadores, mediadores químicos, que se producen como respuesta a la agresión de un tejido y causan respuesta inflamatoria. IL-1: Tiene dos isoformas mayores biológicamente activas: IL-1 , predominantemente unida a la membrana, y la IL-1 que es la isoforma circulante secretada principalmente por linfocitos, macrófagos, células endoteliales y células del músculo liso vascular. Media la respuesta inflamatoria a través de la activación de monocitos y la expresión de moléculas de adhesión en lascélulas endoteliales, induciendo la secreción de otras citoquinas, quimioquinas y factores de crecimiento y estimulando laproliferación del músculo liso. Proceso que sucede en la pared vascular durante la aterogénesis. También induce la coagulación. A nivel renal, aumentaría la permeabilidad vascular endotelial y provocaría proliferación de las células del mesangio ysíntesis de la matriz, así como anormalidades de la microcirculación intraglomerular. IL-6: Es una citoquina proinflamatoria multifuncional. Regula la respuesta humoral y celular durante el proceso inflamatorio y la injuria tisular. Es producida por macrófagos, linfocitos T, células del músculo liso vascular, células endoteliales y adipocitos. El TNF- es capaz de aumentar su producción hasta 60 veces. En condiciones de reposo, el 15 al 35% de la IL-6 es derivado del tejido adiposo y la mayoría proviene del tejido adiposo visceral. Disminuye la producción de LPL-1 Aumenta la secreción hepática de triglicéridos. Es un mediador inflamatorio Página 6 TNF-: Producido principalmente por monocitos, linfocitos T, NK (natural killer), tejido adiposo, músculo liso, células endoteliales y algunas células tumorales. Los efectos biológicos son producidos tras unirse a receptores de membrana (TNF-R: 1 y 2). Estos fueron hallados en todos los tipos celulares excepto en eritrocitos. El mecanismo de acción del TNF es mediado por: ❖ La inhibición de la autofosforilación del receptor de insulina. ❖ Activación de serin-kinasa que disminuye la fosforilación del IRS-1 a nivel del músculo y tejido adiposo, sustrato pobremente reconocido. ❖ Sus efectos están principalmente relacionados con inflamación tanto sistémica como local. ❖ Regula la síntesis de algunas proteínas de fase aguda, especialmente fibrinógeno y factor VIII. ❖ Estimula la producción de prostaglandinas. ❖ Aumenta la expresión de moléculas de adhesión endoteliales. ❖ A nivel hepático aumenta la expresión de genes relacionados con la síntesis de novo de colesterol y ácidos grasos. Regula la captación de glucosa y la oxidación de ácidos grasos. ❖ Estimula la lipólisis. ❖ Disminuye la expresión de GLUT 4. ❖ Suprime los niveles de LPL-1. ❖ Genera resistencia insulínica. Los TAG y los AGL son inductores de la expresión de su TNF. De manera local aumenta la expresión de los genes del PAI 1 y C3, además disminuye la expresión de adiponectina. INHIBIDOR DEL ACTIVADOR DE PLASMINÓGENO (PAI-1) Es una proteasa de serina, que tiene como principal función la inhibición de la fibrinólisis. Su gen se localiza en el cromosoma 7. Es sintetizado por células endoteliales y hepatocitos; está presente en las plaquetas (90%) y en el plasma (10%) donde circula en forma activa ligado a una proteína estabilizadora denominada vitronectina, su vida media es de 10 minutos El compartimento sanguíneo más importante de PAI-1 lo constituyen las plaquetas y se libera por acción del colágeno y ADP. Numerosas sustancias van a regular su síntesis a nivel endotelial: endotoxina, interleuquina 1, factor de necrosis tumoral, trombina y diversos factores de crecimiento aumentan dicha síntesis, mientras que la insulina sería el principal regulador de la síntesis de PAI-1 a nivel del hepatocito. APLICACIÓN CLINICA: Insulinorresistencia y Adipocitoquinas La obesidad constituye un serio problema de salud mundial, está vinculada estrechamente con las principales causas de morbilidad (enfermedad), mortalidad y discapacidad. La insulinorresistencia es quizás su consecuencia más temible, ya que de ella se derivan una serie de alteraciones metabólicas y endoteliales relacionadas con el desarrollo de la enfermedad vascular coronaria, la diabetes mellitus, la hipertensión arterial, las dislipidemias y la enfermedad cerebrovascular. Insulinorresistencia (IR) se define como la incapacidad genética o adquirida de los tejidos blanco (especialmente hígado, tejido adiposo y músculo) de responder normalmente a la acción de la hormona circulante. La alteración de la función de la insulina parece ser consecuencia de un estado de inflamación sistémica de bajo grado, la clave de la IR se encuentra en la función del tejido adiposo, “agrandado e inflamado” como órgano secretor. Cuando el adipocito normal se hipertrofia, aumenta la secreción de hormonas insulinoresistentes y reduce las insulinosensibles. Esta IR se expresa por una disminución del transporte de glucosa y de su metabolismo en el músculo, tejido adiposo e hígado como órganos principales. Estos defectos son el resultado de alteraciones en el sistema de señales de la insulina. El origen del problema es múltiple: por una parte esta el incremento en la obesidad del TNFα, que distorsiona por sí mismo este sistema de señales y que puede alterar la expresión genética del GLUT-4. El aumento de los AGL trastorna el sistema de señales de la insulina y su transporte, al mismo tiempo, los AGL potencia la secreción de insulina estimulada por la glucosa a corto y largo plazo, lo que contribuye a la hiperinsulinemia característica de los estados de resistencia (de los primeros estadios). Por otra parte los AGL compiten con la glucosa como fuente de energía, por lo cual su aumento lleva a la hiperglucemia y esto a su vez disminuye la captación de glucosa dependiente de la insulina, y es aquí donde interviene el mecanismo de toxicidad de la glucosa estimulado por la elevación de los AGL. A partir de esto surgen 2 conceptos que son la glucotoxicidad y la lipotoxicidad (ver Fig 4). Se denomina glucotoxicidad a los efectos adversos producidos por la hiperglucemia crónica sobre las estructuras celulares y sus funciones. Los niveles moderados y altos de glucosa mantenidos en el tiempo inducen IR y disminución progresiva de la secreción de la hormona. Se han postulado tres mecanismos para explicar cómo la hiperglucemia produciría resistencia a la insulina: ➢ Disminución de la síntesis y actividad del transportador de glucosa 4 (GLUT 4) en el músculo.Página ➢ Aumento de la vía de la glucosamina, la glucosamina produce resistencia a la insulina y déficit de secreción, por disminución de los transportadores de glucosa, GLUT 4 en el músculo y GLUT 2 en la célula . ➢ Glucosilación de los transportadores. Esta unión química, cambia la estructura de las moléculas alterando sus funciones, en este caso de los transportadores de la glucosa, con una menor captación de glucosa en los tejidos periféricos. En tanto que para explicar la acción tóxica de la glucosa sobre la secreción de insulina se proponen 4 mecanismos: ➢ La hiperglucemia, por regulación negativa produciría una disminución del transportador GLUT 2, en la célula; éste es el más aceptado. ➢ Menor actividad de la fosfolipasa C, enzima necesaria para la formación de inositidos fosfatos, que participan en la secreción insulínica al aumentar el nivel de calcio intracelular. ➢ La hiperinsulinemia y principalmente la hiperproinsulinemia tendrían un efecto negativo (down regulation), frenando la síntesis de la hormona. ➢ Aumento de radicales libres, la glucosa actúa como un radical libre produciendo citotoxicidad. En 1995 Unger introduce el concepto de lipotoxicidad, definiéndolo como una disminución de la secreción de insulina por el aumento crónico de los AGL. Se postulan los siguientes mecanismos: ➢ Menor actividad de los transportadores GLUT-2. ➢ Cambios en las vías metabólicas normales de los lípidos. La hipótesis es la siguiente: el aumento de los AGL debido a una lipólisis exacerbada y la inhibición de la Acetil-CoA Carboxilasa (por los AGL) tiene como consecuencia una disminución del Malonil-CoA, lo cual lleva a la desinhibición de la Acilcarnitina-Aciltransferasa 1(ACAT-I) por lo que se intensifica la oxidación de ácidos grasos con disminución de los derivados Acil-CoA (metabolitos que estimulan la secreción de insulina), lo que se traduce en una menor liberación de insulina. Pero ya en 1963, Randle planteó la hipótesis de que el aumento de los AGL, productos de la degradación de TAG del tejido adiposo producían IR y fue él quien dio a conocer el ciclo Glucosa-AGL, conocido como Ciclo de Randle. Por lo que el aumento de AGL, eleva su captación y oxidación, usándose éstos como fuente de energía en los distintos tejidos en competencia con la glucosa. Además, los AGL reducen la afinidad insulina-receptor, disminuyendo la acción de la insulina en los tejidos insulinosensibles; favoreciendo así la IR. Se ha encontrado que a nivel de músculo se inhibe la captación y oxidación de glucosa con la consiguiente disminución de la síntesis de glucógeno. En el hígado se produce gluconeogénesis con mayor producción de glucosa. Como consecuencia de todo esto, habría elevación de los niveles de glucemia. El tejido adiposo es imprescindible; el adipocito es la única célula capaz de tolerar los ácidos grasos tóxicos en su seno, y eso siempre que los tenga aislados en la organela especifica la vacuola adiposa, pues de lo contrario sufre también lipotoxicidad. El tejido adiposo sano no muestra signos inflamatorios en su seno. Se compone de adipocitos pequeños que son insulino sensibles, producen sustancias benéficas y conservan suficiente capacidad de seguir captando ácidos grasos. Cuando la vacuola adiposa es de tal tamaño que la célula no puede seguir captando lípidos ni logra reclutar preadipocitos que la auxilien va dejando de emitir adiponectina, y libera la proteína quimioatractiva de monocitos (MCP) que convertidos en macrófagos tratan de continuar limpiando de grasas a la circulación y el tejido adiposo comienza a producir en forma predominante adipocitoquinas deletéreas, entre ellas el TNF- y la interleuquina 6. Esos adipocitos ya no responden a la insulina. Los lípidos que no pueden ser tomados por el tejido adiposo se depositan fuera de él (esteatosis) lo que agrava la insulinoresistencia (teoría de la inundación). TNFIL-6 Leptina Resistina Adiponectina Disfunción Célula Transporte de Glucosa Menor Actividad de GLUT-4 AGL ELEVADOS Aumenta Gluconeogénesis Depósitos grasos Lipólisis Elevada Transporte de Glucosa Menor Actividad de GLUT-4 Fig. 4 Hiperinsulinemia Página 8 Funciones del Endotelio Modula el tono de la inflamación Origen y blanco de hormonas, factores de crecimiento, sustancias vasoconstrictoras y dilatadoras, factores hemostáticos Liga componentes del complemento Expresa receptores de respuesta inmune Fagocita y destruye. Como ya se dijo, la obesidad es el principal factor adquirido responsable de la disminución de la sensibilidad a la insulina, siendo a nivel del endotelio vascular donde se producen la mayoría de los eventos que complican tanto a la obesidad como a la IR, así por ejemplo la ocurrencia de muerte súbita es 3 veces mayor en obesos, mientras que la cardiopatía isquémica, la enfermedad cerebrovascualar e hipertensión arterial son 2 veces mas frecuentes en la población obesa que en la no obesa. Perfil metabólico de la insulinorresistencia: 1. Obesidad abdominal 2. Hipertrigliciridemia 3. Disminución de HDL 4. Hiperuricemia 5. Aumento del inhibidor del activador tisular del plasminógeno 1 6. Hiperagregabilidad plaquetaria 7. Disfunción endotelial BIBLIOGRAFIA 1. Rodriguez Scull, Lidia Esther. La obesidad y sus consecuencias clinicometabólicas. Rev Cubana Endocrinol. [online]. Sep.-dic. 2004, vol.15, no.3 2. Sanchez Muñoz, Fausto y otros. Adipocinas, tejido adiposo y su relacion con células del sistema inmune. Gaceta Medica de México, vol 141, Nº6, 2005. 3. De Pablo Velasco, Pedro Luis y otros. Significado clínico de la obesidad abdominal. Revista Endocrinología y Nutrición. Vol 54, Nº 5, 2007. 4. Moreno M.J y otro. El tejido adiposo: órgano de almacenamiento y órgano secretor. Anales Sis San Navarra, Vol 25, suplemento I, 2002. 5. Zamora Valdes, Daniel y otros. Mecanismos moleculares de resistencia a la insulina. Revista Médica Sur.Vol 11; Nº 3. 2004. 6. Ruiz, Maximino y cols, Factores de Riesgo cardiovascular no tradicionales. Revista de la Sociedad Argentina de Diabetes. Vol 40, Nº4, 2006. 7. Guione Pelayo, Alfredo. El obeso diabético. Revista de la Sociedad Argentina de Diabetes. Vol 41, Nº1, 2007. 8. Bilicich, Silvina. Adipocitoquinas: su relacion con obesidad, insulinorresistencia, diabetes y ateroesclerosis. Revista EPROCAD, Año 11, Nº4; 2006. 9. http://jasn.asnjournals.org/cgi/content/full/16/8/2395 10. http://www.nature.com/cdd/journal/v13/n1/abs/4401713a.html