1
PUBLICACIONES
Artículos
El Dr. Núñez Vázquez y la Dra. Pérez Garrido son licenciados en medicina y cirugía. Ambos comparten
esfuerzos y entorno de trabajo en el Servicio de Hematología y Hemoterapia de la Unidad de Hemofilia y
Unidad de Gestión de Hematología y Hemoterapia
en los HH.UU. Virgen del Rocío de Sevilla. El Dr. Núñez
como Adjunto y la Dra. Pérez como Coordinadora.
¿Sangra menos la
hemofilia B que la A?
Introducción
La hemofilia A y B son desordenes hemorrágicos causados por una deficiencia del factor VIII (FVIII) y factor IX (FIX) de la coagulación, respectivamente, de herencia ligada al cromosoma X. Las primeras referencias
sobre la hemofilia se atribuyen a los escritos judíos del siglo II DC (Talmud),
en los que se eximía de la realización de la circuncisión al tercer hijo de
mujeres, cuyos dos primeros hijos hubieran fallecido por problemas hemorrágicos en relación con dicho procedimiento. Las primeras descripciones
más detalladas las encontramos a finales del siglo XVIII en relación con
familias en las que los varones sufrían episodios hemorrágicos prolongados
después de traumatismos. En 1911 Bulloch y Fildes definieron el concepto
de hemofilia en base a los síntomas, incidencia por sexo y heredabilidad.
Conocida también como la enfermedad de la realeza, la reina Victoria
de Inglaterra, portadora de hemofilia extendió esta enfermedad por las
dinastías europeas incluidas la española, la alemana y la rusa (1).
4
2
3
mente ligado al desarrollo de la
vista genético, ambos genes se
terapia transfusional y se limitó ini-
localizan en el cromosoma X. El gen
cialmente al empleo de sangre total
del FVIII está localizado en la parte
y plasma fresco. Los trabajos de
más distal del brazo largo del cro-
Cohn y posteriormente de Pool en
mosoma X (Xq28), con una estructu-
1965 con la obtención del cripopre-
ra compleja que abarca 186 Kb y
cipitado, culminaron con la disponi-
que está constituido por 26 exones.
bilidad de concentrados de FVIII a
El gen del FIX se encuentra en la
partir de la siguiente década y con
región Xq27, y es considerablemen-
ello la posibilidad de afrontar con
te mas pequeño y menos complejo
garantías intervenciones quirúrgicas,
abarcando solo 33,5 Kb y única-
e instaurar programas de profilaxis y
mente constituido por 8 exones.
autotratamiento domiciliario, con el
Es interesante destacar que en las
objetivo de evitar los sangrados
formas graves, mientras que en he-
espontáneos y de esta manera con-
mofilia A el 40-55% se deben a inver-
trolar o retrasar la aparición de la
siones del intrón 1 y del intrón 22 y
artropatía hemofílica. La evolución
solo un 15% a mutaciones missense,
de dichos concentrados con la
en hemofilia B las mutaciones mis-
introducción de métodos de inacti-
sense representan un 60% de los
En 1952 Aggeler et al. en Estados
vación viral y la fabricación de pro-
casos, lo que puede tener influencia
Unidos y Biggs et al. en Oxford des-
ductos recombinantes después de
en la expresión fenotípica de la
cribieron una entidad, conocida
la clonación de los genes de los
enfermedad en relación con la au-
como enfermedad de Christmas por
factores VIII y IX en 1982 y 1984 res-
sencia completa de factor (5).
el apellido del primer paciente in-
pectivamente, han supuesto gran-
La
vestigado, similar desde el punto de
des avances en el arsenal terapéu-
diferente. Así, la vida media del FVIII
vista clínico y de herencia a la he-
tico de la hemofilia (3,4).
es de unas 8-12 horas y en cuanto a
La hemofilia A es cuatro veces más frecuente que la hemofilia B, de forma que la
prevalencia de la primera es de uno por
cada 5.000-10.000 varones frente a uno por
cada 30.000 varones
en el caso de la segunda.”
que posteriormente se conoció como FIX (2).
Diferencias entre
hemofilia A y B
es
cada unidad infundida por kilogramo de peso se incrementa el nivel
de FVIII en plasma un 2%. En el caso
del FIX su vida media es de 18-24
A pesar de ello, tradicionalmente las
se
En este planteamiento común de la
horas y presenta una recuperación
han descrito sin hacer distinción
hemofilia A y B se establecen tam-
en plasma por cada unidad de
entre ambas hemofilias, general-
bién determinadas diferencias (ta-
factor FIX infundido del 1%. Hay que
mente caracterizadas por sangra-
bla nº1). La hemofilia A es cuatro
añadir que la recuperación del FIX
dos espontáneos o relacionados
veces más frecuente que la hemofi-
recombinante es menor que en le
con traumatismos y siendo los san-
lia B, de forma que la prevalencia
caso de concentrados de FIX deri-
grados articulares, principalmente
de la primera es de uno por cada
vados del plasma, con una amplia
en codos, tobillos y rodillas los even-
5.000-10.000 varones frente a uno
variabilidad individual. La vida me-
tos más característicos. Los hemar-
por cada 30.000 varones en el caso
dia más larga del factor IX repercute
tros de repetición causan una pro-
de la segunda. Las formas graves,
sobre la frecuencia de administra-
gresiva destrucción del cartílago y
definidas por una actividad de fac-
ción de los tratamientos profilácti-
conducen a una artropatía invali-
tor menor del 1%, representan un
cos, de forma que en la hemofilia A
dante característica. El tratamiento
60% en hemofilia A frente al 30-40%
habitualmente el régimen es de tres
de la hemofilia ha estado íntima-
en hemofilia B. Desde el punto de
infusiones en semana mientras que
manifestaciones
2
también
su recuperación se asume que por
mofilia pero ocasionada por un
defecto diferente, la deficiencia del
farmacocinética
hemorrágicas
ASANHEMO. Publicaciones. Artículos
6
7
5
la hemofilia B el factor se administra
mientos de inmunotolerancia, espe-
validado en 100 pacientes adoles-
dos veces por semana. El tratamien-
cialmente en pacientes con reaccio-
centes y adultos con hemofilia
to de los episodios hemorrágicos se
nes previas (7).
mediante los siguientes ítems: inci-
basa en la infusión del factor con-
¿Suponen estas diferencias alguna influencia en el fenotipo hemorrágico de
la hemofilia B con
respecto a la hemofilia A?
dencia de sangrados articulares
centrado deficiente hasta control
del episodio. Sin embargo, existe un
hecho diferencial en la hemofilia A
leve, en la que el empleo de la
desmopresina, un derivado sintético
de la hormona antidiurética, eleva
los niveles basales de factor FVIII de
3 a 5 veces, mientras que en la he-
anuales, estatus ortopédico y uso
anual de FVIII/FIX, incluyendo posibles moduladores de la expresión
fenotípica como los polimorfismos
del
gen
de
la
protrombina
G20210A y del FV Leiden (G1691A).
Los pacientes con hemofilia A presentaron un mayor número de
mofilia B no tiene influencia sobre el
episodios hemorrágicos articulares,
FIX.
un peor estatus ortopédico y un
Respecto a la incidencia de inhibi-
Ya en el año 1959 Quick et al. en una
mayor consumo anual de concen-
dores, encontramos un 30% aproxi-
época previa a la disponibilidad de
trados de FVIII/FIX y consecuente-
madamente para la pacientes con
concentrados de factor y en base a
mente una puntuación más alta
hemofilia A grave y de un 3-5% para
su experiencia con 24 pacientes he-
con significación estadística. Hay
pacientes con hemofilia B, reflejan-
mofílicos B consideraron la posibilidad
que tener en cuenta que el núme-
do probablemente las distintas pro-
de presentación de un fenotipo más
ro de pacientes hemofílicos graves
porciones de mutaciones que dan
suave respecto a la hemofilia “clási-
fue relativamente
lugar a una ausencia de proteína o
ca”. Ramglen en 1962 publicó una
con hemofilia A y 6 con hemofilia B
a una proteína truncada y que se
experiencia similar en 176 pacientes
asocian con un riesgo más elevado
con hemofilia en Suecia (8,9).
a desarrollar inhibidor. El índice de
Más recientemente, en un estudio
respuesta a los tratamientos de in-
canadiense los autores comunicaron
munotolerancia
muy
que los pacientes adultos con hemofi-
diferente con un porcentaje de éxito
lia B grave tenían menos hemorragias
del 70% en hemofilia A frente a un
en un 35% que los hemofílicos A gra-
30% en hemofilia B (6). Además, es
ves (10). De igual modo, en una serie
reseñable como dato diferencial
amplia que englobaba a práctica-
que la aparición de un inhibidor
mente toda la población hemofílica
frente al FIX puede asociarse con un
de Canadá (2663 adultos y niños) el
incremento en la frecuencia de
porcentaje de pacientes graves con
reacciones alérgicas, así como a
hemofilia A en profilaxis (69%) fue muy
nefropatía en el contexto de trata-
superior al de hemofilia B (32%). Esta
también
es
pequeño,
36
diferencia fue más acusada en el
" La aparición de un inhibidor frente al FIX puede
asociarse con un incremento en la frecuencia
de reacciones alérgicas,
así como a nefropatía en
el contexto de tratamientos de inmunotolerancia”.
3
grupo de edad de 0-2 años (53% vs
17%), probablemente en relación a
una percepción distinta de la severidad de la sintomatología hemorrágica y menor frecuencia de episodios
articulares (11).
Un sistema de puntuación sobre la
severidad clínica de la hemofilia fue
ASANHEMO. Publicaciones. Artículos
8
9
(12). En otro estudio en el que se recogen los ingresos hospitalarios en dos cen-
Conclusiones
tros de Escocia, los pacientes con hemo-
Aunque tradicionalmente se ha considerado a la hemofilia A y B como
filia A precisaron 2-3 veces más ingresos
entidades similares desde el punto de vista clínico, algunos autores esti-
que los hemofílicos B en todos los grupos
man que la hemofilia B presenta un fenotipo hemorrágico menos grave
de severidad (13,14). Tagariello et al.
en base a variables como son el número de sangrados, pacientes en
recogieron 347 procedimientos de próte-
profilaxis o al número de intervenciones ortopédicas en ambos grupos. La
sis articulares en pacientes con hemofilia
diferencia en el espectro mutacional de ambas hemofilias podría tener
grave de 29 centros de Italia; concluye-
relevancia debido a la mayor proporción de pacientes con ausencia
ron que la frecuencia es 3.38 veces su-
total de factor en hemofilia A comparado con hemofilia B, y por tanto
perior en pacientes con hemofilia A que
sería una posible explicación de la heterogeneidad en el fenotipo de los
con hemofilia B para el mismo nivel de
pacientes considerados como graves (<1%). De confirmarse estas dife-
gravedad y teniendo en cuenta factores
rencias en el fenotipo hemorrágico de ambas hemofilias podría llevar a
como la edad, infección por VIH, hepati-
modificar pautas de tratamiento, como sería la no necesidad de una
tis C y la presencia de inhibidor (15).
profilaxis primaria sistemática en todos los pacientes con hemofilia B gra-
Recientemente, el análisis de 68 pacien-
ve o moderada. Un estudio multicéntrico sería conveniente para estable-
tes con hemofilia A y 20 con hemofilia B
cer la magnitud real de esta diferencia.
de tipo grave y moderado, reveló que el
número medio de sangrados por año fue
de 14.4 para hemofilia A frente a 8.63
para hemofilia B. Del mismo modo, un
14.7% de los hemofílicos A se sometieron
a un procedimiento quirúrgico para corrección de complicaciones musculoesqueléticas comparado con el 4.7% en
hemofílicos B, es decir, una incidencia
3.2 veces superior para el primer grupo
(16).
Por el contrario, otros autores consideran
que la evidencia para considerar la expresión hemorrágica de la hemofilia B
menos grave es insuficiente. En el estudio
de una cohorte holandesa de 282 pacientes con hemofilia grave, 30 con hemofilia B, no se encontraron diferencias
en la edad de aparición del primer episodio hemorrágico articular, uso de profilaxis, consumo de factor anual y procedimientos ortopédicos realizados (17). En
la revisión sobre las causas de mortalidad en pacientes con hemofilia del
Reino Unido entre 1977 y 1998, los episodios debido a hemorragias intracraneales y de cualquier tipo fueron similares en
ambas entidades (18).
4
ASANHEMO. Publicaciones. Artículos
10
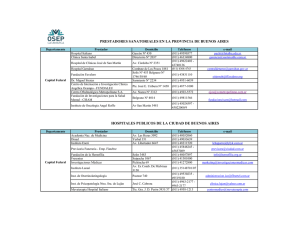
Tabla nº1. Diferencias entre hemofilia A y B.
ITI: inducción de la tolerancia inmune.
Bibliografía
1. Ingram GIC. The history of haemophilia. J. Clin. Path. 1976, 29, 469-479.
2. Biggs R, Douglas As, Macfarlane RG, Dacie JV, Pitney WR, Merskey C. Christmas disease: a condition previously mistaken for haemophilia. Br Med J. 1952
Dec 27;2(4799):1378-82
3. Kurachi K, Davie EW. Isolation and characterization of a cDNA coding for human factor IX. Proc Natl Acad Sci USA 1982; 79:6461-64.
4. Gitschier J, Word WI, Goralka TM, Wion KL, Chen EY, Eaton DH, Vehar GA, Capon DJ, Lawn RM. Characterization of the human factor VIII gene. Nature
1984; 312: 326-30.
5. DiMichele D. Inhibitor development in haemophilia B: an orphan disease in need of attention. Br J Haematol. 2007;138(3):305-15.
6. Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003;9(4):418-35.
7. Franchini M, Lippi G, Montagnana M, et al. Anaphylaxis in patients with congenital bleeding disorders and inhibitors. Blood Coagul Fibrinolysis 2009; 20:
225–29.
8. Quick AJ, Hussey CV. Hemophilia B (PTC deficiency, or Christmas disease). AMA Arch Intern Med. 1959;103: 762-775.
9. Rangrem O. Haemophilia in Sweden. III. Symptomatology, with special reference to differences between haemophilia A and B. Acta Med Scand.
1962;171:237-42.
10. Pai KM, Walker I, Almonte T, et al. Comparing bleed frequency and factor concentrate use between haemophilia A and haemophilia B [abstract]. J
Thromb Haemost. 2005;3 (suppl 1). Abstract P0807.
11. Biss TT, Chan AK, Blanchette VS, et al for the association of hemophilia clinic directors of Canada (AHCDC) and the Canadian association of nurses in
hemophilia care (CANHC). The use of prophylaxis in 2663 children and adults with haemophilia: results of the 2006 Canadian national haemophilia prophylaxis survey. Haemophilia. 2008;14:923-930.
12. Schulman S, Eelde A, Holmstrom M, et al. Validation of a composite score for clinical severity of hemophilia. J Thromb Haemost. 2008;6:1113-1121.
13. Ludlam CA, Lee RJ, Prescott RJ, Andrews J, Kirke E, Thomas AE, Chalmers E, Lowe GDO. Haemophilia care in central Scotland 1980–94. I. Demographic
characteristics, hospital admissions and causes of death. Haemophilia 2000; 6: 494–503.
14. Lowe GD, Ludlam CA. Less severe bleeding in hemophilia B than in hemophilia A. J Thromb Haemost. 2008;6(11):1982-3
15. Tagariello G, Iorio A, Santagostino E, et al. Comparison of the rates of joint arthroplasty in patients with severe factor VIII and IX deficiency: an index of
different clinical severity of the 2 coagulation disorders. Blood. 2009;114: 779-784.
16. Nagel K, Walker I, Decker K, Chan AK, Pai MK. Comparing bleed frequency and factor concentrate use between haemophilia A and B patients.
Haemophilia. 2011 Nov;17(6):872-4.
17. den Uijl IE, Roosendaal G, Fischer K. Insufficient evidence to suggest less stringent therapy in hemophilia B? Blood. 2009, 26;114(23):4907.
18. Darby SC, Kan SW, Spooner RJ, Giangrande PL, Hill FG, Hay CR, Lee CA, Ludlam CA, Williams M. Mortality rates, life expectancy, and causes of death
in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood. 2007, 1;110(3):815-25.
5
ASANHEMO. Publicaciones. Artículos
 0
0