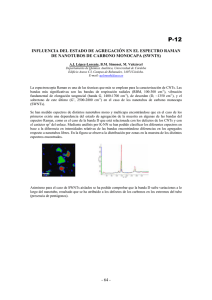
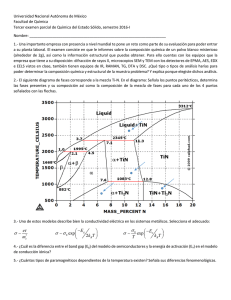
UNIVERSIDAD CATÓLICA DEL NORTE FACULTAD DE
Anuncio

UNIVERSIDAD CATÓLICA DEL NORTE FACULTAD DE CIENCIAS Departamento de Física MEDICIÓN DE PROPIEDADES FERROELÉCTRICAS DE TITANATOS DE BARIO OBTENIDOS POR SÍNTESIS HIDROTERMAL Tesis para optar al grado de Magíster en Ciencias con mención en Física. Lic. Emigdio Chávez Ángel Profesor Guía: Dra. Sandra Fuentes Villalobos Dr. Ramón Zárate Aliaga Antofagasta, Chile 2010 Agradecimientos El presente trabajo de Tesis fue desarrollado en el Departamento de Física de la Universidad Católica del Norte bajo la dirección de la Dra. Sandra Fuentes y el Dr. Ramón Antonio Zarate, dentro del proyecto FONDECYT 1080401 y financiado en su totalidad por la Beca CONICYT para estudios de Magíster Nº 22090805. A todos los diferentes profesores que he conocido en otras universidades y que me han ayudado en el desarrollo de este trabajo, y en forma especial al Dr. Luis Padilla Campos, quien fue el que me integro al mundo de la Química Computacional y le dio sentido a la Mecánica Cuántica, además por su gran ayuda en el uso del programa Gaussian. Al Departamento de Física de la Universidad Católica del Norte, en especial a Dra. Sara Aguilera, Mg. Ramón Fernández y Dr. Miguel Murphy por su ayuda y disposición. A mi familia y forma especial a mi madre Delcira Ángel Ardiles, por todo el apoyo brindado en todos estos años de estudios. ÍNDICE Introducción…………………………………………………………….1 CAPÍTULO 1 CONCEPTOS Y TEORÍA 1.1 Conceptos de ferroelectricidad……………………………………..4 1.2 Teorías fenomenológicas para materiales Ferroeléctricos……………………………………………………..5 1.2.1 Transición de Segundo Orden (Continua)……………….......9 1.2.2 Transición de Primer Orden (Discontinua)…………………11 1.3 Estructuras Cristalinas……………………………………………...15 1.3.1 Titanato de Bario……………………………………………16 1.4 Curva de Histéresis…………………………………………...........18 1.5 Dominios Ferroeléctricos…………………………………………..20 1.6 Métodos de Fabricación de los Titanatos de Bario………………...21 1.6.1 Procesos hidrotermales…………………………………......22 1.7 Motivación………………………………………………………....24 CAPÍTULO 2 MÉTODOS DE CARACTERIZACIÓN Introducción…………………………………………………………....26 2.1 Espectroscopia Raman……………………………………………..26 2.2 Difracción de Rayos X (XRD)……………………………………..29 2.2.1 Formula de Sherrer-Debye y Cálculo de Tamaño de Partícula…………………………………………30 2.2.2 Cálculos de Constantes de Red……………………………..34 2.3 Microscopia de Transmisión de Electrones de Alta Resolución (HRTEM)……………………………………………35 2.3.1 Análisis de Dispersión de Energía de Rayos X (EDX)…………………………………………........36 2.3.2 Difracción de Electrones…………………............................37 2.4 Microscopia de Fuerza Atómica (AFM)…………………………...38 2.5 Circuito Sawyer-Tower…………………………………………….39 CAPÍTULO III SÍNTESIS Y CARACTERIZACIONES ESTRUCTURALES. Introducción……………………………………………………………41 3.1 Métodos de Síntesis Hidrotermal…………………………………..41 3.1.1 Método 1 (Ba)…………………………………………........41 3.1.2 Método 2 (Sf)………………………………………….........42 3.2 Espectroscopia Raman...……………………………………….......44 3.3 Difracción de Rayos X (XRD)…………………………………….49 3.3.1 Calculo de Tamaño de Partícula……………………...……..50 3.3.2 Calculo de constantes de Red……………………………….52 3.4 Microscopia Electrónica de Transmisión de Alta Resolución (HRTEM)…………………………………..…......55 3.4.1 Identificación de Planos Cristalográficos……………….......57 3.5 Microscopia de Fuerza Atómica (AFM)……………………….......59 3.6 Caracterizaciones Ferroeléctricas…………………………………..63 3.7 Discusión de Resultados…………………………………...……….66 CAPÍTULO IV ESTUDIO TEÓRICO Introducción…………………………………………………………..…69 4.1 Metodología de Cluster………………………...…………………..70 4.2 Modelos de Celdas……………………………………...………….71 4.3 Resultados………………………………………………...………..74 CONCLUSIONES…………………………………………………....84 APÉNDICE A.1 Calculo de Primeros Principios…………………………………....86 A.2 Ecuación de Shrödinger……………………………………………87 A.3 Método de Variaciones………………...…………………………..89 A.4 Método de Hartree-Fock (HF)……………………………………..90 A.5 Teoría de la Densidad Funcional (DFT)…………………………...91 A.6 Método de Kohn-Sham (KS)………………………………………92 A.7 LDA, LSDA y GGA……………………………………………….94 A.8 Pseudopotenciales……………………………………………….....96 A.9 Gaussian……………………………………………………………96 A.10 Funcionales Híbridos………………………………………….......98 A.11 Set de Bases…………………………………………………….....99 A.11.1 Set de bases mínimas……………………………………101 A.11.2 Set de base Split-Valence……………………………….102 A.11.3 Set de base Pople………………………………………..102 Listado de Instrumentos. L.1 Microscopía Raman………………………………………………..104 L.2 Tratamientos térmicos……………………………………………...104 L.3 XRD………………………………………………………………..105 L.4 HRTEM………………………………………………………........105 L.5 AFM……………………………………………………………….105 L.6 Osciloscopio……………………………………………………….106 REFERENCIAS………………………………………………………..107 Resumen El interés de esta investigación es el estudio de la síntesis de titanatos de bario nanoestructurados en su fase tetragonal y la posterior caracterización de sus propiedades ferroeléctricas. En esta tesis se propone realizar la preparación de titanatos utilizando el método de Síntesis Hidrotermal en condiciones de solvente sub-críticas con el objeto de obtener Titanatos de Bario en su fase tetragonal. Por este método se obtienen titanatos puros con un alto grado de cristalinidad y con un tamaño de partícula de alrededor de los 50 [nm] de diámetro. La formación de los materiales preparados se caracterizaron mediante medidas de: Espectroscopia Raman, Difracción de rayos X , Microscopía Electrónica de transmisión de alta resolución, Difracción de electrones, Análisis de Energía dispersiva de rayos X y Microscopía de Fuerza Atómica. Debido a divergencias experimentales informadas referente a mediciones de Raman y su relación con la fase tetragonal es que se incorporo a la tesis cálculos teóricos DFT para comprobar cierta vibración, que en la literatura es mencionada como propia de las fase tetragonal, pero no había sido comprobada teóricamente, además también se resolvió cierta inconsistencia entre dos tipos de técnicas de caracterización (Raman y XRD) que asociaban distinta fase cristalina al mismo compuesto. Las características ferroeléctricas fueron medidas a través del circuito r r Sawyer-Tower. Con este circuito se visualiza sus características P − E propias de un ferroeléctrico, es decir, su curva de Histéresis. Abstract The interesting of this research is the study about Synthesis nanostructures of barium titanate in the tetragonal phase, and characterization of their ferroelectric properties. The purpose of this thesis is to prepare titanates using the method Synthesis Hydrothermal in sub-critical conditions of solvent to obtain barium titanate en tetragonal phase. By this method, titanates with a high crystallinity and a particle size about 50 nm are obtained. The formation of the materials was characterized using: Raman Spectroscopy, X Ray Diffraction, High Definition Transmission Electronic Microscopy, Electron Diffraction, Energy Dispersion of X Ray and Atomic Force Microscopy. Due to experimental differences related to Raman’s measures and its relationship with the tetragonal phase is incorporated into the thesis, theoretical calculations DFT to verify a certain vibration that has the tetragonal phase, which is mentioned in the literature, but it had not been theoretically verified. In addition, a certain inconsistency is also solved between two types of skills of characterization (Raman and XRD), which different crystalline phase is associated with the same compound. For this, Cluster method was employed and the calculations were made using the program Gaussian 03W. The ferroelectric characteristics were measured by the circuit SawyerTower; with this circuit we can see the typical characteristics of ferroelectric material. Introducción Con el descubrimiento de la ferroelectricidad en el titanato de bario a mediados de los años 40’, se iniciaron una gran cantidad de investigaciones científicas y tecnológicas para aprovechar sus propiedades ferroeléctricas, tanto en materiales cerámicos como en monocristales. Estos materiales son utilizados para la fabricación de condensadores, sensores, actuadores, dispositivos electromecánicos, dispositivos electroópticos1-3, memorias ferroeléctricas2,3, entre otros. El interés en estos materiales ferroeléctricos y de manera especial el BaTiO3, radica que sus características ferroeléctricas se exhiben a temperatura ambiente4, convirtiéndolo en uno de los mejores materiales para el almacenamiento de cargas eléctricas, ideal para la industria de la electrónica. Otras de las características de estos materiales ferroeléctricos son con respecto a sus propiedades mecánicas, ópticas, piezoeléctricas, piroeléctricas y electro-ópticas1-3. La condición que debe cumplir el Titanato de Bario para exhibir todas estas propiedades, además de su pureza, tamaño de partícula y homogeneidad, es que el sistema cristalino debe encontrarse en su fase ferroeléctrica es decir, una fase no centro simétrica. La importancia de sintetizar esta fase y no otra radica en el hecho que en esta fase existe cierto desplazamiento de los átomos de titanio con respecto a los átomos de oxígeno lo cual produce un dipolo eléctrico. 1 Varios intentos se han realizado para optimizar la obtención de estos titanatos en las condiciones antes descritas, pero no se ha encontrado una síntesis que asegure esta fase. Dentro de los métodos ensayados, se encuentran: La Síntesis de Altas Temperaturas, Calentamiento de Microondas, Evaporación en Vacío, Síntesis Mediante el Método sol-gel y Síntesis Hidrotermales. Y dentro de estos, la síntesis hidrotermal es el proceso experimental más sencillo y económico, debido que sus productos de partida son más baratos que otros procesos y la temperatura y presión de trabajo son bajas y controlables. Por medio de este método se obtendrán titanatos puros con un alto grado de cristalinidad y con un tamaño de partícula de alrededor de los 50 [nm] de diámetro en forma de nanopartículas. Un conjunto de técnicas de caracterización ayudarán al análisis de cada una de las propiedades de las especies preparadas: Espectroscopía Raman, Difracción de Rayos X, Microscopía electrónica de Transmisión de Alta Resolución, Difracción de Electrones, Análisis de Energía Dispersiva de Rayos X y Microscopía de Fuerza Atómica. Por último las características eléctricas se obtendrán mediante el circuito SawyerTower. La tesis se encuentra organizada de la siguiente manera: En el primer capítulo se da una introducción a toda la fenomenología ferroeléctrica, donde se abordan temas tales como: Concepto de Ferroelectricidad, Teoría Ferroeléctrica, Estructuras Cristalinas, Descripción del Titanato de Bario, Curva de Histéresis y Dominios 2 Ferroeléctricos, Métodos de Fabricación de Titanato de Bario y Síntesis Hidrotermal. En el segundo Capítulo se da una descripción de cada una de las técnicas instrumentales utilizadas para caracterizar los materiales además se describe el proceso de síntesis. En el tercer Capítulo se exponen los resultados de las mediciones a través de estas técnicas de caracterización. Además de hacer una discusión de los resultados y plantear los motivos de la inclusión de los cálculos vibracionales. En el cuarto Capítulo se desarrolla todo el estudio teórico que acompaña a este trabajo. Se muestran los resultados de las simulaciones y sus respectivas comparaciones con los antecedentes experimentales. Finalmente, en el apéndice se describe la teoría que acompaña a los cálculos y simulaciones hechas el cuarto Capítulo. 3 CAPÍTULO I: CONCEPTOS Y TEORÍA. 1.1 Conceptos de Ferroelectricidad. Un material ferroeléctrico es aquel que posee un momento dipolar eléctrico diferente de cero, aún en ausencia de un campo eléctrico externo. Además, si se le aplica un campo eléctrico, el sentido de la polarización variara de acuerdo con el sentido del campo. Esta propiedad de la reversibilidad de la polarización no puede ser predicha sólo observando la estructura del material sino que debe ser determinada experimentalmente4. Los materiales ferroeléctricos son dieléctricos no lineales, que cristalográficamente no poseen un centro de inversión5. Estos materiales presentan algunas propiedades tales como: piroelectricidad (variación de la polarización con la temperatura) y piezoelectridad (variación de la polarización con tensiones mecánicas). 4 1.2 Teorías Fenomenológicas para Materiales Ferroeléctricos. La inclusión de términos no lineales en las ecuaciones termodinámicas de estado nos permite formular una descripción de las posibles divergencias en algún tipo de respuesta de los materiales cuando éstos experimentan alguna transición de fase. Estas divergencias pueden ser con respecto a la respuesta elástica (ferroelesticidad) o la respuesta dieléctrica (ferroelectricidad). Para describir el comportamiento ferroeléctrico existen varias teorías y aproximaciones, ahora si sólo nos basamos en consideraciones de simetría, la teoría de Landau puede proveer una adecuada descripción del comportamiento de un sistema en equilibrio en un punto cercano a la transición de fase. La teoría de Landau caracteriza la transición en términos de un parámetro de orden, el cual es una entidad física que es igual a cero cuando un sistema posee alta simetría, y cambia continuamente hasta alcanzar un valor finito cuando se comienza a perder simetría en el sistema, tal como el caso paraeléctrico-ferroeléctrico6,7, donde, a medida que el sistema va perdiendo simetría, la polarización va aumentando. La energía libre ℑ será entonces expandida en serie de potencia en función de la polarización. Cabe hacer notar que la teoría de Landau es estrictamente una aproximación macroscópica y por lo tanto no puede describir el comportamiento microscópico del sistema físico asociado a la transición (por ejemplo desplazamientos atómicos). 5 En general un estado termodinámico de cualquier sistema en equilibrio puede ser descrito completamente especificando ciertos valores. Para un sólido ferroeléctrico estos valores incluyen la Temperatura (T), la Polarización (P), el Campo Eléctrico (E), la deformación (η ) y la tensión ( σ ), pero generalmente la Temperatura, el Campo Eléctrico y la tensión son aplicados externamente, entonces se puede decir que los únicos parámetros o variables internos serán la P y η . Un postulado fundamental de la termodinámica aplicado a materiales ferroeléctricos nos dice que la energía libre ℑ puede ser expresada en función de diez variables: tres componentes de la polarización, seis del tensor σ y una de la Temperatura. El segundo principio fundamental importante que será empleado se refiere a que el valor de las variables dependientes en equilibrio térmico es obtenido en el mínimo de la energía libre, cuando la energía es optimizada, y además teniendo en cuenta que podemos hacer una aproximación en la vecindad de la transición de fase expandiendo ésta en serie de potencias con coeficientes que se pueden determinar experimentalmente. La forma más sencilla de abordar este problema es considerando que el r desplazamiento eléctrico D sólo está dirigido a lo largo del eje polar (ésto restringe al campo eléctrico y a la polarización a la misma dirección), una deformación nula y que su fase paraeléctrica posee centro de inversión (Centro-simétrica). Aunque ninguna de estas restricciones es esencial, al hacer éstas la energía libre puede ser expresada de la forma: 6 ⎛a⎞ ⎛b⎞ ⎛c⎞ ℑP = ⎜ ⎟ P 2 + ⎜ ⎟ P 4 + ⎜ ⎟ P 6 − EP ⎝2⎠ ⎝4⎠ ⎝6⎠ (Ec. 1.1) Aquí sólo se considera los coeficientes del polinomio hasta P6 por simplicidad matemática. Las constantes a , b, c son coeficientes que serán discutidos más adelante. La configuración de equilibrio se determina encontrando el mínimo de ℑP , es decir: ∂ℑP =0 ∂P (Ec. 1.2) De esta operación obtenemos el campo eléctrico en función de la polarización. E = aP + bP 3 + cP 5 (Ec. 1.3) Ahora si volvemos a diferenciar con respecto a P y lo evaluamos en P=0 1 ∂E = a ⇒ E = aP ⇒ a = χ ∂P P = 0 (Ec. 1.4) Donde χ susceptibilidad dieléctrica. La Teoría Landau-Devonshire* asume que en un punto cercano a la temperatura de Curie (T ≈ T0) se cumple que: a =a 0 (T − T0 ) (Ec. 1.5) Y además considera que las constantes b y c son independientes de la temperatura. Combinado las últimas dos ecuaciones, podemos encontrar una expresión para la rigidez dieléctrica. * Devonshire fue el primero en aplicar un la Teoría de Landau a un material ferroeléctrico, basado solamente en la simetría6. 7 κ= 1 χ (Ec. 1.6) = a0 (T − T0 ) La rigidez eléctrica captura el comportamiento de Curie-Weiss observado en la mayoría de los ferroeléctricos para T > T0 . Esto provee un soporte adicional para la dependencia lineal para a . Ahora introduzcamos la Ecuación 1.5 en 1.1, para tener una expresión general para la energía libre: ℑP = 1 1 1 a0 (T − T0 )P 2 + bP 4 + cP 6 − EP 2 4 6 (Ec. 1.7) Donde a0 y c son constantes positivas en todos los ferroeléctricos8,9, la Figura 1.1 muestra la energía libre como una función de la polarización en un paraeléctrico (T>>T0) y la ferroeléctrica (T<<T0). Como transforma esta energía entre esas dos configuraciones será determinada por le signo del coeficiente b; el signo de este parámetro no sólo determinará la naturaleza de la transición sino que también nos indicará cómo evoluciona la polarización a T<T0 si lo hace continua o discontinuamente. F a b P Fig. 1.1 Energía libre como función de la polarización (a) material paraeléctrico (b) material ferroeléctrico 8 1.2.1 Transición de Segundo Orden (Continua). Si b > 0 , entonces ocurre una transición de segundo orden en T=T0, y la energía libre se desarrollará en forma continua como una función que decrece con la temperatura, desde la curva paraeléctrica a la ferroeléctrica (ver Figura 1.1), con mínimos en ± P0 . La polarización espontánea puede ser estimada haciendo el campo eléctrico igual a cero en la Ecuación 1.3 y reteniendo sólo los dos términos de menor grado y considerando que todos los coeficientes son positivos resulta lo siguiente: ⎛a ⎞ Ps = ⎜ 0 (T0 − T )⎟ ⎝b ⎠ 1 2 (Ec. 1.8) De esta expresión se puede observar que a medida que la temperatura aumenta la polarización disminuye hasta hacerse cero en el punto T=T0. Ahora podemos notar que al determinar la rigidez dieléctrica bajo la temperatura de transición ( T < T0 ) encontraremos la siguiente expresión: κ = 2a0 (T0 − T ) (Ec. 1.9) La cual al compararla con la Ecuación 1.5 (expresión para T > T0 ), su valor está sobre T0 ; una mirada rápida sugiere que el valor de κ se hace cero a T = T0 y consecuentemente la susceptibilidad dieléctrica diverge (en la práctica, con materiales reales, la susceptibilidad alcanza valores muy grandes pero finitos, pero aún así esta expresión es apropiada8,9). Ahora podemos resolver también la discontinuidad del calor específico, usando P = 0 para T > T0 y la Ecuación 1.8 para T < T0 , podemos determinar: 9 ( ) ( ∆Cυ = Cυ T = T0+ − Cυ T = T0− Donde Cυ ≡ −T ) (Ec. 1.10) ∂ℑP (ecuación de estado), para obtener ∂T 2 ∆Cυ = a02T0 2b (Ec. 1.11) Esta es la expresión de Landau para el calor específico. Los esquemas asociados con una transición de segundo orden descrito por la teoría de Landau-Devonshire están expresados en la Figura 1.2. 10 (b) χ −1 T0 (a) T>T0 T=T0 T<T0 χ P (C) T0 T Fig.1.2 Transición de fase de segundo orden. (a) Energía libre en función de la Polarización a diferentes temperaturas; (b) La susceptibilidad y su inversa; (c) Polarización espontánea en función de la temperatura 1.2.2 Transición de Primer Orden (Discontinua). Para que ocurra una transición de primer orden la constante b debe ser menor que cero (con a0 , c > 0 ). Con este coeficiente negativo, es evidente que incluso si T > T0 , la energía libre tiene un mínimo en P ≠ 0 . Y, como a ya se le asignó una dependencia de la temperatura, este mínimo será energéticamente menor que un estado no polarizado, y por consiguiente 11 será una configuración termodinámicamente favorable. La temperatura en la cual sucede esto es, por definición, la temperatura de Curie Tc (esta excede a T0). Y la fase no polarizada existirá como un mínimo local de la energía libre, en un rango de temperatura entre Tc y T0 . La característica más importante de esta transición de fase es que el orden del parámetro salta discontinuamente desde cero hasta Tc . Este tipo de transición de fase es usualmente llamada de Primer Orden o discontinua, un ejemplo muy común de ésta son transiciones de sólido a líquido. El procedimiento para encontrar la polarización espontánea y la susceptibilidad es similar a la vista en transiciones de Segundo Orden, pero esta vez tenemos que considerar los términos de grado seis. Cualitativamente, cuando se encuentra la rigidez dieléctrica (la inversa de la susceptibilidad lineal), diverge a T = T0 , correspondiendo a un salto finito de la susceptibilidad y de la polarización espontánea en la transición. En la Figura 1.3 se muestra un esquema de la energía libre, la polarización espontánea, la rigidez dieléctrica y la susceptibilidad lineal. Notar que a T = Tc los tres mínimos son energéticamente degenerados. 12 (b) F T0 Tc T>T0 P T=T0 T<T0 (c) P0 P (a) T T0 Tc Fig.1.3 Transición de fase de primer orden. (a) Energía libre en función de la Polarización a diferentes temperaturas; (b) La susceptibilidad y su inversa; (c) Polarización espontánea en función de la temperatura Como resultado, el comportamiento del sistema en T = Tc dependerá si la aproximación a Tc es desde altas o bajas temperaturas. Más específicamente, el sistema tendrá dos polarizaciones mínimas y finitas ( P ≠ 0) , si es calentada desde una temperatura inicial baja Ti < Tc (estado ferroeléctrico), mientras si la temperatura inicial es alta Ti > Tc tendrá una polarización igual a cero (estado paraeléctrico). Efectivamente el fenómeno de histéresis térmica, donde la transición de temperatura 13 dependerá si la muestra es calentada o enfriada, es dominante en número, para los materiales ferroeléctricos de primer orden incluyendo el BTO8. Cabe hacer notar que el fenómeno de ferroelectricidad es termodinámicamente favorable sólo a temperaturas T0 < Tc . En un material ferroeléctrico bajo la temperatura T0 existen por lo menos dos mínimos en la energía, correspondientes a la polarización espontánea con diferentes orientaciones espaciales. La barrera entre esos dos mínimos indica que un pequeño campo eléctrico no cambiará el sentido de la polarización. Entonces cabe notar que la teoría de LandauDevonshire aquí descrita predice la aparición de una curva de histéresis8,9, en la Figura 1.4 muestra un caso ideal, donde todos los dipolos se invierten en forma conjunta cambiando la orientación de la polarización. (a) (b) Fig. 1.4 Imagen esquemática de la curva de histéresis y dos interpretaciones: (a) variación en la energía libre, (b) deformación de la celda cristalina para el Titanato de Bario. 14 1.3 Estructuras Cristalinas. La estructura cristalina del titanato de bario (BTO) es del tipo pevroskita10. Dicha estructura posee como fórmula general ABO3, donde “A” y “B” son cationes y “O” es un anión. Esta estructura es del tipo cúbica y sus átomos se distribuyen de la siguiente forma: “A” se ubica en las aristas, “B” en el centro y “O” en las caras del cubo (ver Figura 1.5). Para el caso del BTO, la simetría va cambiando a medida que varía la temperatura, ésto se resume en la siguiente tabla. Rango de temperatura T< -90ºC -90ºC < T < 5ºC 5ºC < T < 130ºC 130ºC < T < 1460ºC T > 1460ºC Fase cristalina Romboédrica Ortorrómbica Tetragonal Cúbica Hexagonal Grupo espacial C3v5 - R3m 14 C2v - Amm2 1 C4v - P4mm Oh1 - Pm 3 m 4 D6h - P63/mmc Tabla 1.1 Fases cristalinas del BaTiO3 11 Fig. 1.5 Cristalización tipo pevroskita12 15 1.3.1 Titanato de Bario. El BTO fue la primera pevroskita descubierta que poseía propiedades ferroeléctricas. Su ferroelectricidad se debe a cierto desplazamiento asimétrico dentro del octaedro formado por los átomos de oxígeno y de titanio. Esta deformación genera un dipolo eléctrico y con ésto una polarización neta. A temperatura ambiente el BTO se encuentra en la fase tetragonal, y el desplazamiento de los iones ocurre a lo largo del eje C (ver Figura 1.6), y es de unos 6 a 9 pm aproximadamente. [010] 9 pm [001] 6 pm 6 pm Fig. 1.6 Proyección del plano [100] del BaTiO3. Aquí se muestra el desplazamiento de los iones de oxigeno y de titanio6. En forma aproximada el momento dipolar puede ser obtenido mediante la siguiente expresión: µ = qd (Ec. 1.12) Donde “µ” es el momento dipolar, “q” la carga y “d” es la distancia de separación de las cargas. Para el BTO, y utilizando los datos de la Figura 1.6, el cálculo del momento dipolar neto queda: 16 µ (Ti +4 ) = (1.602 × 10 −19 C )(4)(0.06 × 10 −8 cm) = 3.84 × 10 − 28 C ⋅ cm µ E (O − 2 ) = (1.602 × 10 −19 C )(2)(0.09 × 10−8 cm) (eje z ) = 2.88 × 10 − 28 (Ec. 1.13) C ⋅ cm µC (O − 2 ) = (1.602 × 10−19 C )(2)(0.06 × 10 −8 cm) (caras ) = 1.92 × 10− 28 C ⋅ cm Ahora, si contamos la contribución de cada ión podemos calcular el momento dipolar total de la celda: µ = µ (Ti +4 ) + µ E (O −2 ) + 2 µC (O −2 ) = 1.056 × 10 − 27 C ⋅ cm (Ec. 1.14) Otra importante propiedad que poseen los ferroeléctricos, y en especial el BTO, es la reversibilidad de su polarización debido a la presencia de un campo eléctrico externo. El gráfico de la polarización en función del campo eléctrico externo forma una curva característica de estos materiales, llamada ciclo de histéresis. La Figura 1.7 muestra curvas de histéresis para un dominio ferroeléctrico dentro de un monocristal de BTO y para una cerámica policristalina del mismo material4. Estas curvas fueron tomadas a temperatura ambiente usando una fuente de poder de 50 Hz, mostrando un campo coercitivo o de corte de unos 0.1 MV/m y una polarización espontánea de unos 0.27 C/m2. 17 Fig. 1.7 Histéresis ferroeléctrica de BaTiO3. (a) Un solo dominio ferroeléctrico. (b) Cerámica policristalina 4. 1.4 Curva de Histéresis. …“La histéresis es el fenómeno de inercia por el cual un material, ofreciendo resistencia a un cambio, tiene la tendencia a conservar sus propiedades, haciendo que el proceso de variación sea distinto en un sentido que en el otro”...13. La histéresis se produce debido a un retraso entre la causa externa y el efecto que provoca en él. Para visualizar este fenómeno en un material ferroeléctrico se debe aplicar un campo externo oscilante, lo que genera una modificación de la polarización debido a la presencia de este campo y con ello un comportamiento histérico. 18 Fig. 1.8 Típico ciclo de histéresis14 Para el caso de un material ferroeléctrico la gráfica de campo eléctrico externo versus la polarización, se visualiza como una curva de histéresis, de donde se pueden observar tres variables de interés: • Polarización espontánea o de saturación (Ps): corresponde a la máxima polarización del cristal, cuando todos los dominios ferroeléctricos están apuntando en la misma dirección. • Polarización remanente (Pr): es la polarización residual cuando el campo externo se hace cero. • Campo coercitivo (Ec): Es el campo necesario para poder invertir el sentido de la polarización. 19 1.5 Dominios Ferroeléctricos. Los dominios ferroeléctricos son regiones en donde la polarización espontánea adopta diferentes direcciones, separadas por planos de macla o paredes de dominios. Esta organización en dominios se debe a que los dipolos vecinos se alínean de tal forma que puedan disminuir la energía dentro del sistema. La presencia de dominios vecinos con polarización contraria disminuye el campo creado por las cargas libres y estabiliza el cristal. De lo contrario sucedería que el cristal estaría uniformemente polarizado y las cargas libres en la superficie crearían un campo contrario a la polarización, creando una inestabilidad en el sistema. La existencia de los dominios produce la curva de histéresis característica de los materiales ferroeléctricos, de manera similar como ocurre con los ferromagnéticos. El proceso de histéresis se explica de la siguiente forma: • Se inicia con un cristal ferroeléctrico con un igual número de dominios en direcciones opuestas. • Se aplica un campo eléctrico en alguna dirección de los ejes cristalográficos del cristal. • Los dominios más paralelos al campo comienzan alinearse más rápidamente que los antiparalelos. • Se comienza a incrementar el campo, y la polarización total aumenta rápidamente, hasta el punto de saturación, que es 20 cuando todos los dominios del cristal apunta en la misma dirección creando un sólo gran dominio. • El campo regresa a cero, pero varios dominios mantienen su orientación, de tal forma que la polarización no llega a cero (polarización remanente). • El campo cambia de dirección y la polarización comienza a disminuir hasta hacerse cero (Campo coercitivo). • Comienza nuevamente el ciclo pero esta vez en dirección opuesta. 1.6 Métodos de Fabricación de los Titanatos de Bario. Para la síntesis15,16 de estos materiales en forma de nanopartículas, existen los métodos físicos tales como: oxido mixtos, sol-gel, precursores poliméricos, hidrotermal, microemulsión, mecano-químico, etc.), que han dado como resultados la obtención de material partículado y cristalino con las características ferroeléctricas deseadas. Tradicionalmente e industrialmente, el BTO es preparado por reacción del estado sólido por medio de la calcinación de BaCO3 y TiO2 a altas temperaturas (entre 800 a 1200º C)17,18. Este método es lento e involucra una serie de etapas previas antes de obtener el producto final que son esferas uniformes de tamaños micrométricos (< 5 µm). A pesar del sin números de etapas que involucra este método, su morfología y el tamaño de partículas son difíciles de controlar. 21 Debido a las altas temperaturas con que trabaja este método, hace que el proceso de fabricación genere un gran costo energético y con ello una baja rentabilidad económica. 1.6.1 Procesos Hidrotermales. La técnica hidrotermal ha sido una de las más populares en los últimos 50 años, debido que las temperaturas ocupadas dentro del proceso es baja, comparada con otros métodos de síntesis de materiales. El término hidrotermal es de origen puramente geológico, y fue usado por primera vez por el geólogo británico, Sir Roderick Murchinson (1792-1871), para describir la acción del agua a elevada temperatura y presión en el interior de la tierra, la que causa cambios de la corteza terrestre que conducen a la formación de varios tipos rocas y minerales19. En un laboratorio se utilizan sistemas contenedores capaces de soportar altas temperaturas y presiones llamadas autoclaves. El término hidrotermal por lo general se refiere a cualquier reacción heterogénea en la presencia de solventes acuosos en condiciones de altas temperaturas y presión, que se utiliza para disolver y recristalizar materiales que en condiciones normales son relativamente insolubles. Las condiciones mínimas de temperaturas y de presión no han sido acordadas aún pero la mayoría de los autores coinciden que para que una reacción reciba el nombre de hidrotermal, ésta debe ocurrir con valores de temperatura por encima de los 100º C y presiones iguales o superiores a 1 atm20. 22 La reacción que ocurre durante el proceso de creación de BTO se describe a continuación21: Ba(OH)2 TiO2·nH2O + 2OHBa2+ + Ti(OH)62CO2+ 2OHBa2+ + CO32- ↔ ↔ ↔ ↔ ↔ Ba2+ + 2OHTi(OH)62- + (n-2)H2O BaTiO3 + 3H2O CO32- + H2O BaCO3 El CO2 proviene del aire que circunda a las reacción, este se disuelve como CO32- ,el cual luego reacciona con Ba2+ formando BaCO3 (Carbonato de Bario). El BaCO3 es una impureza es típica de este proceso, pero puede ser disuelto y eliminado del producto final con Acido Acético. 23 1.7 Motivación. BaTiO3 es uno de los materiales potencialmente más importantes para la tecnología debido a su respuesta ferroeléctrica y su alta constante dieléctrica, lo cual lo hace un importante dieléctrico. Estas propiedades se presentan hasta en partículas de tamaño menores al micrón, en el orden de los cientos de nanómetros, permitiendo grandes aportes en la nanotecnología con cual lo hace extremadamente útil en la industria electrónica, específicamente en electro-cerámica1-3. El BTO se puede utilizar en aplicaciones electro-ópticas, termistores, capacitores y memorias no volatiles (FeRAM)2,3 entre otras. Aprovechando, por un lado las ventajas económicas del método hidrotermal y que la síntesis de materiales ferroeléctricos en fase pura, homogénea, con tamaño y fase cristalina controlada no esta resuelta, es que proponemos en esta tesis la síntesis de óxidos del tipo BaTiO3 en fase tetragonal y la medición de sus características ferroeléctricas. Para lograr este trabajo utilizaremos dos métodos novedosos de síntesis hidrotermales. A medida que se comenzó a desarrollar este trabajo, surgió una segunda motivación importante “Cálculos DFT de los modos vibracionales de distintas fases del BTO”. Éste estudio se centró en la comprobación de ciertos modos vibracionales, que hasta el momento no estaban resueltos teóricamente. Todo esto surgió como necesidad de comprobar cierto modo vibracional que se le asignaba a una fase en específico, pero al analizar nuestros resultados surgían ciertas dudas sobre si esta asignación era la correcta. 24 CAPÍTULO II: MÉTODOS DE CARACTERIZACIÓN. Introducción En el presente capítulo se hace un resumen de las técnicas de caracterización utilizadas para el estudio estructural y ferroeléctrico de los materiales sintetizados. Además se presenta una breve descripción de la fenomenología detrás de cada una de estas técnicas instrumentales. Las técnicas utilizadas para la caracterización estructural de las especies sintetizadas fueron: Espectroscopía Raman, Difracción de Rayos X, Microscopía electrónica de transmisión de Alta Resolución, Análisis de Dispersión de Energía de Rayos X, Difracción de electrones y Microscopía de Fuerza Atómica. Mientras que las caracterizaciones eléctricas fueron hechas a través del circuito de Sawyer Tower. 2.1 Espectroscopia Raman. La espectroscopía surge de la interacción de la radiación electromagnética con la materia. El análisis espectral en el cual se basa esta técnica permite detectar la absorción o emisión de la radiación electromagnética incidente y relacionar éstas con niveles energéticos. De la interacción con la materia pueden existir tres casos posibles: 25 1. Choque elástico: Existe sólo un cambio en el impulso de los fotones sin transferencia energética. Algunos ejemplos son: Difracción de Rayos X, Difracción de Electrones o Anillos de Difracción y Difracción de Neutrones. 2. Choque Inelástico: Existe un cambio en la frecuencia de la luz difractada y por ende una transferencia energética. Por ejemplo Espectroscopía Raman. 3. Absorción o emisión resonante de fotones. La espectroscopía Raman fue detectada por primera vez por el científico indio Chandrasekhara Venkata Raman en el año 1928 quien la describió como una interacción entre un haz de luz incidente y un medio material que genera un nuevo tipo de radiación secundaria. Este tipo de espectroscopía consiste en analizar la radiación dispersada inelásticamente de un haz de luz monocromático, generalmente un rayo láser, que incide en un medio material. El campo oscilante de la radiación electromagnética provoca una oscilación en la densidad electrónica de las moléculas que forman al material estudiado y con ello la aparición de un momento dipolar eléctrico oscilante inducido, el cual actúa como fuente de radiación originando de este modo las dispersiones Raman y Rayleigh. Al irradiar una muestra con un haz de luz, algún fotón incidente hará pasar a la molécula a un estado virtual. Si existe una reemisión inmediata y sin un cambio de la energía se produce la dispersión Rayleigh. En cambio si existe un cambio energético diferente al inicial se provoca la dispersión Raman. Si consideramos que la radiación electromagnética incidente posee una frecuencia ν 0 ésta se dispersará parcialmente al 26 interaccionar con la muestra. La energía de una parte de los fotones dispersados puede disminuir en hν (Líneas Stokes) y ésto debido a una transferencia energética a la muestra en forma de vibración. Por otro lado, si los niveles vibracionales están lo suficientemente poblados, es posible observar también la dispersión de fotones cuya energía ha aumentado en hν (Líneas anti-Stokes)22. Fig. 2.1 Esquema de la actividad Raman El espectro Raman recoge estos datos y representa la intensidad óptica dispersada en función del número de onda normalizado υ al que se produce. El número de onda normalizado es una magnitud proporcional a la frecuencia e inversamente proporcional a la longitud de onda, que es expresada en cm-1. υ= ν c = 1 λ [cm −1 ] (Ec. 2.1) Cada material posee un conjunto de valores característicos de ν debido a su estructura cristalina y a la naturaleza de los enlaces químicos que la forman. Entonces, como estos parámetros son únicos para cada sustancia, es posible utilizar esta técnica como una herramienta de caracterización elemental y estructural de los materiales. 27 2.2 Difracción de Rayos x (XRD). La difracción de rayos x es una poderosa herramienta para la investigación de la estructura cristalina de la materia. Esta técnica se basa en el descubrimiento de von Laue’s, el cual señala que los cristales pueden ser usados como rejillas de difracción para los rayos x, y de esta manera revelar la estructura del cristal. Tiempo después otros usos han sido desarrollados, y hoy en día, no sólo se ocupa para comprobar la estructura de los cristales, sino que también se utiliza para determinar el equilibrio de una fase, determinación de la orientación de un cristal o del ensamble de materiales policristalinos23, el tamaño de partícula24, etc. Toda esta diversidad de usos se debe básicamente a que el espectro electromagnético de los rayos x, que se encuentran entre la luz ultra violeta y los rayos gamma, posee una longitud de onda que va en un rango entre 0.01 y 10 nm, que tiene aproximadamente el mismo orden de magnitud que las distancias interatómicas. Esta técnica consiste en hacer incidir a diferentes ángulos un haz de rayos x a través de una muestra a analizar. Esta interacción produce un patrón de difracción el cual es único para cada sustancia. Fig. 2.2 Ilustración del funcionamiento de la difracción de rayos X 28 De la medición de la posición angular (θ ) de las líneas de difracción se puede calcular las constantes de red o los espacios “d” a través de la Ley de Bragg23: 2d sin(θ ) = nλ (Ec. 2.2) Donde “d” es la distancia interplanar, “ θ ” es el ángulo de incidencia medido desde el plano cristalino que produce la reflexión, “ λ ” es la longitud de onda de la radiación incidente y “ n ” es el orden de la reflexión ( n = 1,2,3.. ). El espaciamiento de los planos interatómicos “d” permite diferenciar las diferentes fases cristalográficas presentes (usando la base de datos del difractómetro). Si la fase es conocida, entonces las reflexiones se pueden rotular con los índices de Miller23. 2.2.1 Formula de Sherrer-Debye y cálculo de tamaño de partícula.24 La Ley de Bragg indica que para que exista un reforzamiento de la difracción las ondas difractadas por un mismo plano los haces deben estar en fase, es decir, que la diferencia de camino tiene que ser un número entero de longitud de onda. Consideremos que tenemos un sistema tal como se muestra en la Figura 2.3. Si la diferencia de caminos de las ondas difractadas por el primer y el segundo plano es un cuarto de longitud de onda, esos dos haces no se cancelarán completamente. Ahora si la onda difractada por la tercera capa también se desfasa un cuarto con 29 respecto a la anterior, tendrá como resultado que se cancelará completamente con la primera. En forma análoga, sucederá lo mismo entre la segunda y cuarta o la primera con la tercera, quinta, etc. Por otro lado, si la diferencia de caminos entre el primer y segundo plano es casi un número entero de longitud de onda, muy cercano al caso de Bragg, el haz difractado en la primera capa estaría complemente fuera de fase sólo con el difractado en la última capa. Entonces, si el cristal es demasiado pequeño no existiría un plano que pueda aniquilar completamente al primer haz difractado y por lo tanto la intensidad no sería cancelada del todo. Una vez hechas estas suposiciones examinemos un cristal de un ancho pequeño “t” que posee planos d-distantes y todos paralelos entre sí, llamaremos Θ B al ángulo que cumple exactamente con la ley de Bragg, empleando la ec. 2.2 para n = 1 nos queda: λ = 2d sin Θ B (Ec. 2.3) En la Figura 2.3 el ángulo de incidencia de los rayos A, D y M forman exactamente este ángulo, entonces el haz D’ está a una longitud de onda desfasado con respecto al haz A’ y en haz M’ lo está “m” longitudes de onda desfasado del primero, de manera que todos los haces están en fase y la intensidad del haz resultante será máxima. Los rayos “B” y “C” forman un ángulo Θ1 y Θ 2 ambos son los ángulos más pequeños alrededor de Θ B que forman un desfase destructivo en las ondas difractadas. El primer ángulo es ligeramente superior y el segundo ligeramente menor que Θ B . Esto provoca que la diferencia de camino del haz B’ con respecto al difractado en el plano m-ésimo esté desfasado 30 m+1 longitudes de onda, y de manera análoga m-1 para el ángulo Θ2 , lo que implica que la mitad superior del cristal se cancela en forma exacta con la parte inferior. A B’ A’ C’ B C D’ D Θ1 Θ2 ΘB 0 1 d ΘB 2 t=md 3 M N N’ L M’ L’ m Fig. 2.3 Difracción de haces de rayos X en infinitos planos d-distantes entre sí. Intensidad I Max. B 2Θ2 2Θ 1/2 (I Max.) 2Θ1 Fig. 2.4 Gráfico del ancho medio para un patrón de difracción De la Figura 2.4 se observa las difracciones para Θ2 y Θ1 son mínimos en el patrón de difracción, la intensidad se va a cero, además el ancho del 31 peak aumenta a medida que el cristal disminuye en grosor, debido a que el rango angular 2Θ1 − 2Θ2 aumenta a medida que m disminuye. La medida B del ancho medio del máximo de difracción se conoce como el ancho a altura media o FWHM por sus siglas en inglés (full widht at half maximum), y una buena aproximación para esta medida esta dada por: B= 1 (2Θ1 − 2Θ2 ) = Θ1 − Θ2 2 (Ec. 2.4) Ahora si se trata de una función tipo Lorenziana la aproximación es exacta. Las ecuaciones de diferencia de caminos para esos dos ángulos relacionados, para un cristal de grosor entero están dadas por: 2t sin Θ1 = (m + 1)λ (Ec. 2.5) 2t sin Θ 2 = (m − 1)λ (Ec. 2.6) Si se restan estas ecuaciones se genera: t (sin Θ1 − sin Θ 2 ) = λ ⎛ Θ + Θ 2 ⎞ ⎛ Θ1 − Θ 2 ⎞ ⇔ 2t cos⎜ 1 ⎟ sin ⎜ ⎟=λ 2 ⎠ ⎝ 2 ⎠ ⎝ (Ec. 2.7) (Ec.2.8) Donde Θ1 y Θ2 son dos ángulos muy cercanos a Θ B , por lo tanto es razonable hacer las siguientes aproximaciones: Θ1 +Θ2 = 2Θ B ⎛ Θ − Θ 2 ⎞ ⎛ Θ1 − Θ 2 ⎞ sin ⎜ 1 ⎟=⎜ ⎟ 2 ⎠ ⎝ 2 ⎠ ⎝ (Ec. 2.9) (Ec. 2.10) 32 Con esas aproximaciones la Ecuación 2.3 se transforma en: ⎛ Θ − Θ2 ⎞ 2t ⎜ 1 ⎟ cos Θ B = λ 2 ⎠ ⎝ (Ec. 2.11) Ahora se introduce la definición de FWHM para llegar finalmente a la ecuación: t= λ B cos Θ B (Ec. 2.12) Si se hace un tratamiento matemático más riguroso de esta formulación resulta la verdadera relación de Debye-Scherrer : t= kλ B cos Θ B (Ec. 2.13) Donde “ k ” es una constante adimensional que varia en un rango entre 0.89 a 1.39, dependiendo de la geometría especifica de las partículas que producen la difracción24. 2.2.2 Cálculos de constantes de red.23 Otra información importante que se puede obtener a partir de los difractogramas de XRD es el cálculo de las constantes de red de la estructura cristalina. Para ello consideremos que, una estructura cristalina en tres dimensiones puede ser caracterizada por un set de vectores, de la forma: 33 r r r r Rn = n1a1 + n2 a2 + n3a3 (Ec. 2.14) r r r Donde n1 , n2 y n3 son enteros, mientras los vectores a1 , a2 y a3 definen las tres direcciones de una celda unitaria del cristal. Para obtener una descripción completa del cristal se necesita construir una base de moléculas o átomos los que deberían ser los mismos para cada y todo sitio dentro de un cristal. Para un tratamiento mas avanzado de la difracción de rayos X, que revele la naturaleza de las intensidades entre los máximos de la difracción, es importante tener una buena descripción de las familias de planos responsables de generar los patrones de interferencia. Por convención para este propósito se utilizan los índices de Miller (h, k , l ) que están definidos como sigue: Para una familia de planos, el plano más cercano al origen tiene como intercepto un punto localizado en ( a1 h r , a 2 k , a3 l ) , con respecto a los ejes a1 , r r a2 , a3 y h, k , l todos los valores enteros que pudiese tener (los intercepto negativos están representados con una barra sobre el número). Entonces al conocer las distancias interplanares podemos determinar la constantes de red del material. Las distancias de espaciamiento entre estos planos están definidos en forma diferente dependiendo de la estructura cristalina. A continuación se muestran algunos ejemplos: 34 Cúbica: d hkl = Tetragonal: d hkl = Hexagonal: d hkl = Ortorrómbica: d hkl = a h + k2 + l2 ac 2 c(h 2 + k 2 ) + al 2 ac 3 4a(h 2 + hk + k 2 ) + 3a 2l 2 abc b 2 c 2 h 2 + a 2 c 2 k 2 + a 2b 2l 2 (Ec. 2.15) (Ec. 2.16) (Ec. 2.17) (Ec. 2.18) Donde a , b y c son las constantes de red. 2.3 Microscopia de Transmisión de Electrones de Alta Resolución (HRTEM). La microscopia electrónica de transmisión (TEM), se ha convertido en una herramienta de suma importancia para la caracterización de los materiales. La óptica electrónica es muy similar a la óptica convencional, sólo que en vez de utilizar fotones o luz visible para formar las imágenes se ocupan electrones. Estos microscopios permiten alcanzar una capacidad de aumento muy superior que la microscopia convencional, y esto debido a que la longitud de onda de los electrones es mucho menor que la de los fotones. Un microscopio electrónico funciona con un haz de electrones generados por un cañón electrónico, acelerados por un alto voltaje y focalizados por una serie de lentes magnéticas, todo ello en el interior de una cámara de ultra alto vacío, debido a que los electrones son fácilmente absorbidos en el aire. Los electrones difractados al pasar a través de la muestra (debidamente tratadas) generan un difractograma que puede ser transformado directamente en una imagen ampliada, mediante lentes magnéticas. 35 Las imágenes se pueden producir a partir de electrones difractados, entregando imágenes de campo oscuro, o a partir de electrones directos que han atravesado la muestra sin interacción, generando imágenes de campo claro25. Las limitaciones de estos microscopios es que muchos materiales no sobreviven a las condiciones que son sometidos: alto vacío e impacto de los electrones acelerados, además muchas de las muestras se transforman debido a que suelen perder agua de hidratación o las moléculas orgánicas se pueden volatilizar o reaccionar. Sin embargo, esta técnica tiene una ventaja fundamental ya que permite mostrar los defectos a nivel atómico en los materiales que es muy difícil de estudiar con otras técnicas26. 2.3.1 Análisis de Dispersión de Energía de Rayos X (EDX). Esta técnica de análisis entrega la composición química de la muestra analizada, valores de porcentaje en peso por elemento presente y porcentaje de presencia atómica. Durante el análisis la muestra es bombardeada por un haz de electrones, los cuales colisionan con los electrones superficiales de la muestra y en algunos casos los sacan de la estructura dejando un espacio. Este espacio vacante es ocupado por un electrón de alta energía proveniente de un nivel más alto emitiendo un haz de rayos X para minimizar su energía. 36 Este valor de la energía liberada es único para cada tipo de átomo, entonces al medir esta energía se puede asociar a un elemento químico específico27. 2.3.2 Difracción de electrones. Esta técnica de caracterización consiste en hacer incidir un haz de electrones sobre una muestra, lo que genera un patrón de interferencia el cual es analizado para determinar algunos parámetros cristalográficos propios de la muestra analizada, generalmente la identificación de los planos cristalográficos. Esta técnica es similar a la difracción de rayos X o a la difracción de neutrones. Al incidir un haz de electrones sobre un sólido, este actúa como una rejilla de difracción, y genera una serie de anillos concéntricos, que al medir el radio de cada anillo e invertirlo el resultado es una de las distancias de los planos25. d hkl = 1 rn (Ec. 2.19) 37 2.4 Microscopia de Fuerza Atómica (AFM). La microscopia de fuerza atómica (AFM), fue creada microscopio de efecto túnel a partir del (STM), y pertenece a la familia de microscopios llamado Scanning Probe Microscopy (SPM). Esta familia de microscopios se caracterizan por la interacción con la muestra se hace a través de una sonda o punta. El AFM es un equipo capaz de detectar fuerza del orden de los nanoNewton, presentes en la interacción entre la punta y la muestra, esas pequeñas fuerzas son medidas gracias al movimiento de un brazo (Cantilever) muy flexible en el cual está montada la punta de interacción. Cuando el equipo comienza a sondear una muestra el cantilever empieza a flectarse producto de la interacción de la punta con la superficie, este movimiento es detectado por fotodiodo y luego interpretado por un Software como: topografía, fuerza de torción, fuerza eléctrica, fuerza magnética, etc28. Fig. 2.5 Esquema de funcionamiento del microscopio de fuerza atómica AFM29. 38 2.5 Circuito Sawyer-Tower. Las propiedades ferroeléctricas fueron medidas mediante el circuito Sawyer-Tower30. Este circuito consta de dos capacitores puestos en series donde uno de ellos (Cx) es la cerámica ferroeléctrica. De este circuito se puede obtener un ciclo de histéresis y con ello hacer una estimación de la polarización espontánea, la polarización remanente y el campo coercitivo. Para hacer estas estimaciones se puede considerar que la polarización de un material ferroeléctrico cualquiera posee la siguiente forma: r r r P = D − ε0E (Ec. 2.20) Teniendo en cuenta que en un condensador de placas paralelas se cumple que r r D >> ε 0 E si el material ferroeléctrico exhibe una constante r dieléctrica mayor a 20, y P es igual a la carga libre en las placas del condensador dividido por su área, es decir: P≈ Q µ̂ A (Ec. 2.21) Desde el circuito de Sawyer-Tower de la Figura 2.6, el material ferroeléctrico (Cx) está conectado en serie con un condensador lineal de valor C0, lo que indica que la carga en ambos condensadores es igual. Luego, si VC es el voltaje en el condensador lineal, entonces la Ecuación 2.21 toma la forma: r CV P ≈ 0 C µ̂ A (Ec. 2.22) 39 Esto indica que la polarización del material ferroeléctrico es proporcional al voltaje en el condensador lineal. Para obtener la curva de histéresis, se requiere graficar en el eje Y el voltaje en el capacitor de referencia (C0) y en el eje X el voltaje total aplicado al circuito. De esta forma, las dos señales se pueden digitalizarse en un osciloscopio en el modo XY obteniendo un ciclo de histéresis. El voltaje recibido por la cerámica ferroeléctrica se puede calcular restando las dos señales o escogiendo el capacitor de referencia tal que sea unas 1000 veces superior a la cerámica ferroeléctrica de esta forma el voltaje en el circuito será aproximadamente igual al aplicado en el condensador de referencia. En ambas formas utilizadas, se puede visualizar el ciclo de histéresis en el osciloscopio, y de él se pueden estimar parámetros antes mencionados. Fig. 2.6 Circuito esquemático de Sawyer-Tower12 40 CAPÍTULO III: SÍNTESIS ESTRUCTURALES. Y CARACTERIZACIONES Introducción En el presente capítulo se presenta los resultados de la preparación de BTO obtenido a través de la síntesis hidrotermal. Aquí se hace un resumen de los resultados de las caracterizaciones del material partículado, donde primero se presenta los métodos de síntesis ocupados y posteriormente las diversas caracterizaciones estructurales y ferroeléctricas. Al final de cada una de estas técnicas se realiza una breve discusión de cada resultado. Al final de este capítulo se hace una discusión general, y se compara los resultados entre cada técnica, proponiendo un posible modelo de la estructura estabilizada. 3.1 MÉTODOS DE SÍNTESIS HIDROTERMALES. 3.1.1 MÉTODO 1 (Ba). Se prepara una solución que contiene 1 ml de acido nítrico concentrado en 39 ml de agua destilada caliente y separadamente otra solución que contiene 1 ml de tetra-n-butiltitanato (TBT) disuelta en 8 ml etanol 41 absoluto. Esta solución es añadida gota a gota a la solución acida con agitación constante a 60º C y se deja reaccionar por 3 horas. Posteriormente se añade 10 ml de una solución de BaCl2*2H2O (0.25 M) bajo una purga de nitrógeno (N2), seguidamente se agrega aprox., 3ml de NaOH a 6M, hasta alcanzar un pH alcalino (13-14). Posteriormente la solución obtenida se vierte en vaso de teflón y llevado a una autoclave por un lapso de 1 a 3 días a diferentes temperaturas de reacción (70-220º C), al terminar la reacción la autoclave se deja enfriar a temperatura ambiente. 3.1.2 Método 2 (Sf). Se toma 1.1 ml de TiCl4 disuelto en 2.3 ml de HCl a 2M, hasta formar un liquido amarillento, luego 2.4 grs de BaCl2*2H2O se disuelve en 10 ml en agua caliente desionizada. Una vez obtenida ambas soluciones, se mezclaron bajo agitación magnética y un ambiente de N2. Lentamente se comenzó a añadir de NaOH (6M), hasta alcanzar un pH alcalino o básico (14-13). Posteriormente se introduce a un vaso de teflón dentro de una autoclave por tres horas a diferentes temperaturas (70 a 220º C). La reacción se lleva a cabo bajo una atmósfera de oxígeno con una presión parcial de 60 psi. Al terminar la reacción la autoclave se deja enfriar a temperatura ambiente. Para limpiar el sólido resultante de la reacción y disminuir el pH se hacen una serie de lavados con agua destilada, hasta obtener un pH entre 6-7, posteriormente las muestras son secadas al aire bajo luz infrarroja por 24 horas. 42 A continuación se presenta los resultados de las distintas caracterizaciones para muestras obtenidas a través de ambos métodos de fabricación (Ba y Sf). De un conjunto de muestras obtenidas (aprox. 50 por cada método), se escogieron: • Ba 06: obtenida por el método 1, con un tiempo de reacción de 24 h a una temperatura de 220º C. • Ba 07: obtenida por el método 1, con un tiempo de reacción de 24 h a una temperatura de 180º C. • Sf 35: obtenida por el método 2, con un tiempo de reacción de 3 h a una temperatura de 220º C. • Sf 36: obtenida por el método 2, con un tiempo de reacción de 3 h a una temperatura de 180º C. 43 3.2 Espectroscopia Raman. La Figura 3.1 muestra una comparación de los espectros obtenidos para los BTO obtenidos por ambos métodos de síntesis Ba y Sf. Del espectrograma se pueden identificar cinco modos principales: 185, 255, 307, 518, 720 cm-1, todas estas vibraciones son asociadas al BTO en fase tetragonal31,32. La frecuencia ubicada aprox. a 307 cm-1 es la de mayor interés para nuestro estudio debido a que ésta vibración es la que se asocia a la fase tetragonal31,33. Como ya se ha descrito anteriormente, 100 200 300 400 Sf35 Sf36 Ba06 Ba07 720 518 307 255 185 esta es una de las fases que posee propiedades ferroeléctricas. 500 600 700 800 -1 Raman Shift [cm ] Fig 3.1 Espectros Raman asociados a las muestras obtenidas con los dos métodos de síntesis. Del espectro Raman se puede concluir que las especies sintetizadas fue BTO en la fase tetragonal sin presencia de impurezas tales como carbonato de bario (BaCO3) o restos de algún precursor sin reaccionar. Ésto nos indica que tanto la fase y la pureza de las especies sintetizadas son las deseadas. 44 Otra información interesante que se puede obtener de los espectros Raman es el grado de cristalinidad de una muestra, y para esto existen dos formas para determinarlo. El primer método consiste en comparar la actividad Raman con la intensidad de Rayleigh, ya que mientras mas intensa sea la actividad Raman respecto al Rayleigh mayor será su cristalinidad. El segundo método es verificar la agudeza de los peaks Raman, ya que entre más agudos sean estos, también será mayor la cristalinidad en una muestra. Para mejorar la cristalinidad, homogeneidad y pureza de las muestras sintetizadas, estas fueron sometidas a un tratamiento de calcinación. Este tratamiento consiste en elevar la temperatura y con esto logra que se descompongan las impurezas presentes provenientes de la síntesis (Ej:BaCO3), defectos estructurales y vacancias de oxígeno. El tratamiento térmico no solo mejora las características morfológicas, de las muestras, sino que también provoca un aumento del tamaño de partícula, debido a un proceso llamado coalescencia34. Este proceso consiste en que las partículas al sufrir un cambio de temperatura lentamente comienzan a cohesionarse hasta formar granos más grandes. Por otro lado, es conocido que sistemas nanométricos en una dimensión tales como: tubos, alambres o partículas a nivel nanométrico, presentan propiedades únicas y diferentes debido a su tamaño y a la dimensionalidad (relación superficie volumen)35,36. En el caso del BTO se ha informado que al disminuir el tamaño de partícula aparecen propiedades especiales, como por ejemplo propiedades multiferroicas37. Estas nuevas propiedades empiezan a aparecer sólo cuando el tamaño de partícula comienza a disminuir (< 50 nm). 45 Pero a modo de comparación a las muestras se le realizó tratamientos térmicos a diferentes temperaturas. Los espectros Raman de estas se encuentran en la Figura 3.2 donde se fue variando la temperatura de calcinación manteniendo constante el tiempo que duró (12 h). 250º C 300º C 350º C 400º C 500º C Sin Calcinar 250º C 300º C 350º C 400º C 500º C Sin Calcinar 518 307 720 720 255 Tiempo 12 hrs 185 518 307 255 185 Tiempo 12hrs 100 200 300 400 500 600 -1 Raman Shift [cm ] 700 800 (a) 100 200 300 400 500 600 -1 Raman Shift [cm ] 700 800 (b) Fig. 3.2 Espectros Raman de especies calcinadas a diferentes temperaturas:(a) Método 1 (Ba 06) (b)Método 2 (Sf 35) De los espectros obtenidos se puede ver que para el primer método los máximos Raman lentamente comienzan a agudizarse a medida que la temperatura aumenta, si bien la diferencia de los espectros no es muy apreciable, esto nos quiere decir que las especies sintetizadas sin calcinar, son lo bastante homogéneas y cristalinas, ya que la calcinación no modifico en mucho su estructura interna, lo que se ve reflejado en los espectros. Para el segundo método se puede observar que los peaks se comienzan a agudizar a temperaturas bajas, y a declinar con el aumento de la temperatura. Esto nos quiere decir que para este método, habían 46 varias partículas que se encontraban en la fase cúbica o con pequeños defectos estructurales, y que con una pequeña activación térmica estos cristales pasaron rápidamente a la fase tetragonal (aumento en la agudeza de la vibración en 307 cm-1), mientras que si la temperatura va en aumento el proceso de nucleación impide la fase cúbica pase directamente a la tetragonal informada en la literatura, y se genera una fase cúbica de baja simetría. De todas formas, cabe destacar que las especies sintetizadas sin un tratamiento térmico por si solas tienen las características deseadas. Otra forma de inducir la estabilización de la fase tetragonal y su cristalización, es hacer un annealing a alta temperatura31,38. El tratamiento de annealing es un tipo de calcinación de alta temperatura en una atmósfera saturada de oxígeno. El problema de este tratamiento es el aumento brusco del tamaño de partícula además del enorme gasto energético que significa este proceso, y nuestra intensión era poder obtener BTOs pero con temperaturas de trabajo más bajas. En la Figura 3.3, podemos ver como la actividad Raman va aumentando con respecto al Rayleigh y además como los peaks se van agudizando. 47 518 720 307 255 S f3 5 S f3 6 Ba06 Ba07 0 100 200 300 400 500 600 700 800 900 -1 R a m a n S h ift [c m ] Fig. 3.3 Espectros Raman de muestras con tratamiento de annealing de 15h a 950º c bajo un flujo de oxígeno. Finalmente de los espectrogramas de dispersión Raman podemos concluir que las especies sintetizadas fue BTO puro, homogéneo, cristalino y aparentemente estabilizado en una fase tetragonal. Por otro lado, los tratamientos térmicos posteriores a la síntesis no influyeron en una mejora de estas características, por lo tanto podemos afirmar que las especies sintetizadas, por si solas poseen las características buscadas. 48 3.3 Difracción de Rayos X (XRD). La Figura 3.3a muestra el patrón de difracción para las muestras de BTO obtenido por ambos métodos de síntesis y sin calcinar, el análisis del difractograma señala que se ha logrado sintetizar con éxito BTO puros*. 101 Ba07 Ba06 Sf36 sf35 200 111 211 100 202 210 20 25 30 35 40 45 Ba07 Ba06 sf36 sf35 200 50 55 60 65 (a) 2θ [grados] 44,8 45,0 45,2 45,4 45,6 45,8 46,0 (b) 2θ [grados] Fig. 3.3 Difractogramas de XRD a polvos sintetizados sin calcinar: (a) Espectro completo de 20º a 70º, (b) Espectro localizado en 45º Para determinar si la fase en que se estabilizó nuestro compuesto fue la tetragonal hay que analizar más profundamente cada peak del difractograma, y buscar un desdoblamiento de estos. Este desdoblamiento se produce debido a que al tener una menor simetría la fase tetragonal aparecen nuevos peaks asociados a los planos que ya no comparten la misma distancia como lo hacían en la fase cúbica. En la Figura 3.3b se muestra un barrido de alta exposición para un ángulo entre 44 < 2θ < 46 . Dentro de este intervalo existe el desdoblamiento más significativo en el BTO en fase tetragonal y por ende, el más usado en la literatura para verificar esta fase31,39. Para las muestras no se observa * La asignación de los índices de Miller se hizo utilizando la base de datos “Power Diffraction Releate 2001” y también fueron comparadas con simulaciones hechas a través del software Carine Crystallography versión 3.1, ambos pertenecientes a la Facultad de Ciencias Físicas y Matemáticas de la Universidad de Chile. 49 desdoblamiento alguno del peak, esto nos quiere decir que, según XRD, la fase estabilizada fue la cúbica. 3.3.1 Calculo del Tamaño de Partícula. Utilizando la Ecuación 2.3 se puede hacer una estimación del tamaño de partícula a partir del FWHM (B). Para los cálculos del tamaño de partícula se utilizaron los peaks mas intensos del difractograma y una aproximación a partículas esféricas, es decir, k=1.3325, los resultados de estos cálculos se muestran resumidos en la siguiente tabla. 50 Muestra Sf 35 Sf 36 Ba 06 Ba 07 2 Θ2 21.70º 30.88º 38.40º 44.52º 55.44º 64.96º 21.68º 30.84º 55.74º 65.32º 21.72º 31.06º 38.52º 44.64º 55.76º 21.66º 30.84º 38.44º 44.66º 55.22º 65.26º 2 Θ1 22.4º 31.82º 39.02º 45.62º 56.58º 65.72º 22.26º 31.84º 56.72º 65.68º 22.58º 31.92º 39.36º 45.74º 56.66º 22.64º 32.10º 39.36º 45.66º 56.80º 66.42º 2 ΘB 22.06º 31.44º 38.72º 45.00º 55.96º 65.66º 22.00º 31.36º 55.90º 65.54º 22.14º 31.52º 38.84º 45.22º 56.12º 22.16º 31.52º 38.86º 45.24º 56.08º 65.78º B* Tamaño[nm] 0.19006º 62.93 0.24618º 49.54 0.21867º 56.90 0.13826º 91.90 0.34467º 38.57 0.28559º 48.92 0.13000º 91.99 0.24871º 49.45 0.21175º 62.76 0.28621º 49.39 0.18709º 63.93 0.24578º 49.63 0.23473º 53.03 0.31579º 40.27 0.34268º 38.82 0.21821º 54.82 0.27899º 43.72 0.29384º 42.37 0.35156º 36.18 0.41009º 32.44 0.39927º 35.01 Tabla 3.1 Análisis de los peaks de los difractogramas y calculo del tamaño de partícula. De la Tabla 3.1 se puede apreciar que el tamaño de partícula varía en un rango entre 30 a 92 nm, con un tamaño preferencial (calculado a partir del peak con mayor intensidad 2θ ~ 31º ) que varía entre 43 a 50 nm. * Para realizar los cálculos el FWHM (B), este debe estar en radianes. 51 3.3.2 Calculo de Constantes de Red. Otro valor de interés que se obtiene a través de los difractogramas de XRD es el cálculo de las constantes de red. Estos cálculos40 fueron hechos utilizando las ecuaciones de distancia interplanar para las dos fases de interés (cúbica y tetragonal). Si las muestras sintetizadas son cúbicas el cálculo de las constantes de red utilizando la distancia para una simetría tetragonal debiese converger a una cúbica, caso contrario habría que analizar mas profundamente los resultados. Los cálculos de las constantes de red y las distancias interplanares se resumen en las siguientes tablas: Planos 1 1 1 2 2 2 2 0 0 1 0 1 1 0 0 1 1 0 0 1 2 Sf 35 Tetragonal [Å] Sf 35 Cúbica [Å] a = b = 4.0260 c = 4.0155 a = b = c = 4.0245 Experimental 4.026 2.843 2.324 2.013 1.799 1.642 1.421 Teórico 4.026 2.843 2.322 2.013 1.800 1.643 1.422 Experimental 4.026 2.843 2.324 2.013 1.799 1.642 1.421 Teórico 4.025 2.846 2.324 2.012 1.800 1.643 1.423 Tabla 3.2 Constantes de red y distancias interplanares (Muestra Sf 35) 52 Planos 1 1 1 2 2 2 2 0 0 1 0 1 1 0 0 1 1 0 0 1 2 Sf 36 Tetragonal [Å] Sf 36 Cúbica [Å] a = b = 4.0290 c = 4.0325 a = b = c = 4.0305 Experimental 4.037 2.850 2.327 2.015 1.801 1.643 1.420 Teórico 4.029 2.850 2.327 2.015 1.802 1.645 1.425 Experimental 4.037 2.850 2.327 2.015 1.801 1.643 1.420 Teórico 4.031 2.850 2.327 2.015 1.802 1.645 1.425 Tabla 3.3 Constantes de red y distancias interplanares (Muestra Sf 36) Planos 1 1 1 2 2 2 2 0 0 1 0 1 1 0 0 1 1 0 0 1 2 Ba 06 Tetragonal [Å] Ba 06 Cúbica [Å] a = b = 4.0095 c = 4.0190 a = b = c = 4.0115 Experimental 4.012 2.836 2.317 2.004 1.793 1.638 1.420 Teórico 4.010 2.838 2.317 2.005 1.793 1.638 1.419 Experimental 4.012 2.836 2.317 2.004 1.793 1.638 1.420 Teórico 4.011 2.837 2.316 2.006 1.794 1.638 1.418 Tabla 3.4 Constantes de red y distancias interplanares (Muestra Ba 06) 53 Planos 1 1 1 2 2 2 2 0 0 1 0 1 1 0 0 1 1 0 0 1 2 Ba 07 Tetragonal [Å] Ba 07 Cúbica [Å] a = b = 4.0085 c = 4.0130 a = b = c = 4.0085 Experimental 4.008 2.836 2.316 2.003 1.793 1.621 1.411 Teórico 4.009 2.836 2.315 2.004 1.793 1.637 1.418 Experimental 4.008 2.836 2.316 2.003 1.793 1.621 1.411 Teórico 4.008 2.834 2.314 2.004 1.793 1.636 1.417 Tabla 3.5 Constantes de red y distancias interplanares (Muestra Ba 07) Los resultados de las tablas 3.2 a 3.5 no son concluyentes para afirmar que la fase sintetizada fue la tetragonal, ya que como es señalado en la literatura11, la razón entre los ejes c y a debe ser igual a 1.0110, y para nuestros cálculos nos arroja un valor entre 0.9974 a 1.0024. Los resultados de las razones de los ejes esta resumido en la siguiente tabla: Muestra Sf 35 Sf 36 Ba 06 Ba 07 a c 4.0260 4.0290 4.0095 4.0085 4.0155 4.0325 4.0190 4.0130 c a 0.9974 1.0008 1.0024 1.0011 Tabla 3.6 Constantes de red de las distintas muestras y su razón entre sí. Como conclusión final podemos afirmar que a partir de los difractogramas de rayos X, se corroboró la pureza, cristalinidad y homogeneidad de las sustancias y se logró hacer una estimación del tamaño de partícula que confirmó el carácter nanométrico de estas. Otra información obtenida por medio del difractograma rayos X nos indicó 54 que la fase que se estabilizó fue la cúbica y no la tetragonal como lo afirmaba los resultados de espectroscopía Raman. Esta aparente controversia, será discutida mas adelante. 3.4 Microscopia Electrónica de Transmisión de Alta Resolución (HRTEM). Las muestras fueron preparadas suspendiendo el sólido en etanol y mezcladas por ultrasonido por unos 20 minutos, luego fueron colectadas y depositadas en grillas de carbono y cobre. Las imágenes que se exponen a continuación fueron tomadas a campo claro, y ellas muestran cúmulos de partículas con diámetros que fluctúan en un rango entre 4090 nm. 100 nm (a) 0.1 µm (b) Fig. 3.4 Microfotografías en campo claro, (a) Sf 35 sin Calcinar (b) Sf 35 Calcinada a 300º C por 12 h. 55 24 24 20 16 16 Frecuecia [%] Frecuencia [%] 20 12 8 4 12 8 4 0 40 45 50 55 60 65 70 75 Tamaño de Particula [nm] 80 85 90 (a) 0 50 55 60 65 70 75 80 85 90 Tamaño de Particula [nm] 95 100 105 (b) Fig.3.5 Histogramas de Tamaño: (a) Sin Calcinar (b) Calcinada a 300º C por 12 h. Al analizar el histograma de tamaño de la muestra Sf 35 sin calcinar (Figura 3.5a) podemos estimar el tamaño de partícula varía en un rango entre 40 a 90 nm, con un tamaño preferencial de unos 57 nm, este valor no se aleja mucho a lo que se había calculado por medio del difractograma de rayos X que nos dio como tamaño preferencial unos 50 nm, y un rango de tamaños entre 39 a 92 nm. Además otro punto a recalcar es el aumento del tamaño de partícula en la muestra calcinada (65 nm), si bien no es mucha la diferencia, se pone de manifiesto que con una activación térmica las partículas comienzan a cohesionarse en un proceso llamado nucleación mencionado anteriormente. 56 3.4.1 Identificación de Planos Cristalográficos. Con las imágenes de alta resolución y de difracción de electrones se pueden identificar planos cristalográficos dentro de un cristal. Estos planos pueden ser atribuibles a cualquiera de las dos fases que se intentan resolver, ya que la diferencia de distancia entre los planos de la fase tetragonal o cúbica cae dentro del error de medida (0.02 Å). (100) 2 nm Fig. 3.6 Imagen de alta Resolución (Sf 35 sin calcinar) La Figura 3.6 muestra la identificación de unos de los planos cristalográficos en la dirección [100]. Por medio de las imágenes de difracción de electrones se logró identificar varios planos tales como: (100), (111), (112), (211), (122). 57 (122) (211) (112) (111) (100) 5 1/nm Fig. 3.7 Anillos de difracción de la muestra Sf 35 sin calcinar Fig. 3.8 Gráfico de dispersión de rayos X (EDX) 58 La Figura. 3.8 muestra el espectro de dispersión de rayos X de la muestra Sf 35 sin calcinar. Este gráfico muestra intensos peaks asociados a los elementos que componen al BTO que son: Bario, Oxígeno y Titanio, esto nos indica la alta pureza de las especies que hemos sintetizado. La aparición de peaks de Cobre (Cu) y Carbono (C) se encuentran presentes producto de la grilla que contiene a la muestra, mientras que el peak de sodio (Na) es una pequeña impureza proveniente del medio de reacción, que puede ser eliminada por medio de lavado. De las mediciones hechas a través de la microscopia electrónica para la muestra Sf 35 se puede verificar que el tamaño de partícula esta presente a nivel nanométrico: de 40 a 90 nm para la muestra sin calcinar y de 50 a 105 nm para la muestra calcinada. Lo que comprueba el aumento en el tamaño de partículas producto de la nucleación. 3.5 Microscopia de Fuerza Atómica (AFM). Las muestras fueron preparadas suspendiendo el sólido en alcohol etílico y mezcladas por ultrasonido por unos 20 minutos. Luego fueron colectadas y depositadas en un porta- muestra de vidrio muy liso especial para AFM. Las imágenes que se exponen a continuación (figuras 3.9 a 3.12) fueron tomadas en el modo AC o Tapping, y estas muestran cúmulo de partículas con un tamaño entre 20 a 120 nm. 59 Fig. 3.9 Micrografía de Topografía de la muestra Sf-35 Fig. 3.10 Micrografía de Fase de la muestra Sf-36 60 (a) (a) (b) Fig. 3.11 Micrografías de la muestra Ba-06 (a) imagen Topográfica (b)imagen de fase (a) (b) Fig. 3.12 Micrografía de la muestra Ba-07: (a) imagen Topográfica (b)imagen de fase 61 25 28 20 Frecuencia [%] Frecuencia [%] 35 21 14 15 10 5 7 0 0 20 30 40 50 60 70 80 90 100 Tamaño de Partícula [nm] 110 20 120 30 40 50 60 70 80 90 Tamaño de Partícula [nm ] (a) 100 (b) Fig. 3.13 Histogramas de tamaño: (a)Sf 35, (b)Sf 36 36 35 32 30 28 24 Frecuencia [%] Frecuencia [%] 25 20 15 20 16 12 10 8 5 4 0 0 40 50 60 70 80 90 T a m a ñ o d e P a rtíc u la [n m ] 100 (a) 110 30 40 50 60 70 80 Tam año de Partícula [nm] 90 (b) 100 Fig. 3.14 Histogramas de Tamaño: (a) Ba 06, (b)Ba 07 Al analizar los histogramas de tamaño de las diferentes muestras (Figura 3.13a, 3.13b, 3.14a y 3.14b) podemos estimar el tamaño de partícula, el cual varía en un rango entre 20 a 120 nm, con un tamaño preferencial entre 50 a 60 nm, este valor no se aleja mucho a lo que se había calculado por medio del difractograma de rayos X que nos dio como tamaño preferencial unos 50 nm, y un rango de tamaños entre 39 a 92 nm. 62 Esto nos confirma los cálculos hechos a través de los difractogramas de XRD que también se ven reforzados por los histogramas obtenidos por HRTEM. 3.6 Caracterizaciones Ferroeléctricas. Una de las características de los materiales ferroeléctricos es que su polarización espontánea es reversible, al menos parcialmente, a través de la aplicación de un campo eléctrico externo, siendo su manifestación experimental la denominada curva de histéresis. El ciclo de histéresis es usado frecuentemente como un una prueba para demostrar que un material es ferroeléctrico41, y una de formas de medir este ciclo es a través del circuito de Sawyer-Tower30 (ver Figura 2.6). Las curvas de histéresis de los materiales sintetizados fueron medidas mediante este circuito. Para ello se comprimió el sólido nanopartículado en pastillas y se le depositó pequeños electrodos de oro mediante sputtering, dando como resultado condensadores ferroeléctricos con un área aproximada de 0.0401 cm2 y un grosor de unos 0.068 cm. Debido a la fragilidad de estos condensadores se debieron fabricar capsulas contenedoras, que consistían en tubos cilíndricos de vidrio donde en su interior tenían dos electrodos, que hacían contacto con la pastilla ferroeléctrica (ver Figura 3.13). 63 Fig. 3.13 Cápsula contenedora: con una pastilla ferroeléctrica en su interior Esta cápsula fue conectada en serie a un condensador cerámico de capacidad conocida C0 = 1µF , a ambos se les aplicó un voltaje variable de unos 500 V a 50 Hz. Los voltajes en cada condensador fueron medidos mediante un osciloscopio. Para visualizar la curva de histéresis en el osciloscopio, este debe estar en el modo XY. Donde en el eje X se adquiere el voltaje del condensador ferroeléctrico Cx y en el eje Y el voltaje del condensador de referencia C0. La medición de la curva de histéresis es fundamental para dilucidar la fase que se sintetizó ya que, si las muestras se estabilizaron en la fase cúbica la curva que se observará en el osciloscopio será una curva dieléctrica16. Este tipo de curva es similar a la de histéresis, pero en esta no existe saturación, ya que al no estar polarizada esta fase no existe una alineación de dominios y por ende no hay contribución de cargas, estos materiales se polarizan, pero al retirarle el campo externo la polarización se hace cero. Mientras que para una curva de histéresis ferroeléctrica, existen zonas donde la curva de histéresis se tiende a aplanar, estas se denominan zonas de saturación y se forman cuando el campo comienza a 64 incrementarse, y la polarización total aumenta rápidamente, hasta el punto de saturación, que es cuando todos los dominios del cristal apunta en la misma dirección creando un sólo gran dominio, lo mismo sucederá cuando el campo cambie de sentido. 2 Polarización [µC/cm ] 4 Zona de Saturación 2 0 -2 Zona de Saturación -4 -75 -50 -25 0 25 50 75 Campo Aplicado [kV/cm] Fig. 3.14 Histéresis Ferroeléctrica de la muestra Ba 06 La Figura 3.14 se observa el ciclo de histéresis ferroeléctrica para la muestra Ba 06 sin calcinar, en este ciclo se observa claramente las zonas de saturación lo que confirma que la especie sintetizada está mayoritariamente en una fase ferroeléctrica. De esta curva se puede hacer una estimación del campo coercitivo EC = 32 kV cm polarización remanente Pr = 2 µC cm2 y de la . La importancia de haber podido medir una curva de histéresis, es que confirma que se logro sintetizar con éxito la fase que era de nuestro interés, si bien no precisa que el sistema se encuentra en una fase 100 % 65 tetragonal podemos decir que en su estructura interna existe cierto desplazamiento que genera un momento dipolar efectivo. 3.7 Discusión de Resultados. Se logró sintetizar con éxito BTOs, puros, homogéneos y con un tamaño de partícula a nivel nanométrico, pero la fase estabilizada no está resuelta aún debido a controversias entre dos técnicas de caracterización (Raman y XRD). Al analizar cada espectro Raman se pueden visualizar modos vibracionales que según la literatura son pertenecientes únicamente a fases de baja simetría cristalina (Tetragonal, ortorrómbica y romboédrica) y no aparecen en la fase cúbica31,32,38. Mientras que al analizar los difractogramas de rayos X, aparentemente la fase que se logró sintetizar fue la fase cúbica, debido a que no se encontraron desdoblamientos de los máximos de difracción31,42. Esto nos hace cuestionar la asignación del peak en 307 cm-1 sólo a las fases ferroeléctricas, ya que hasta el momento no existe cálculo alguno que demostrara que esto era así. Por lo tanto es posible pensar que esta vibración también pudiera estar presente en la fase cúbica. Otras posibles explicaciones de esta diferencia de resultados pueden ser atribuidas a: i. La naturaleza vibracional de la espectroscopía Raman en contraste al gran rango de orden que dan los patrones de Difracción de Rayos X. 66 ii. Debido a que el tamaño de partícula del BTO es muy pequeño y de acuerdo a diferentes autores31,43-46 sólo cuando el tamaño de partícula supera cierto valor critico (>60 nm), comienza a manifestarse la fase tetragonal. iii. La existencia de cierta deformación del octaedro de TiO6 del BTO la cual no es suficientemente importante como para deformar al cristal completamente, generando así estabilización de una fase pseudo-cúbica metaestable a temperatura ambiente31,47. Por otro lado, al calcular las constantes de red a partir de los difractogramas de rayos X, los resultados no son concluyentes para afirmar en un 100% que la fase estabilizada fuera cúbica, ya que cuando se da como modelo para el cálculo una celda tetragonal esta no converge a una cúbica. Si bien la razón de los ejes no es la indicada en literatura11, la aparición de esta pequeña diferencia entre los ejes, nos hace pensar que quizás la fase sintetizada no es ni cúbica ni tetragonal si no más bien una pseudo-fase de baja simetría, que probablemente esté generando la aparición de los modos vibracionales asociados a la fase tetragonal por Raman, mientras que por difracción de rayos X la diferencia es tan mínima que no alcanza a ser detectada en los difractogramas. La aparición de un ciclo de histéresis ferroeléctrica es señal de que la fase en cuestión no debiese ser centro-simétrica ya que la única forma de que un material como el BTO sea ferroeléctrico es que la fase estabilizada no debe tener centro de inversión, para que de este modo en el interior del cristal exista una pequeña polarización en ausencia de un campo eléctrico externo. Caso contrario si el cristal fuese centro 67 simétrico no existiría una polarización y la curva de histéresis no presentaría zonas de saturación. En base a estos resultados podemos afirmar que la fase cristalina que se logró sintetizar es ferroeléctrica, y quizás no sea una fase pura si no mas bien una mezcla de fases o una pseudo-fase. En vista de que a través de las técnicas experimentales no se logró resolver completamente la fase estabilizada, y que además en la literatura no existían cálculos que demuestren que la vibración en 307 cm-1 sólo este presente en la fases ferroeléctricas. Es que se decidió hacer un estudio teórico de las fases del BTO. 68 CAPÍTULO IV: ESTUDIO TEÓRICO Introducción La interpretación de las posiciones e intensidades Raman no están usualmente resueltas y necesitan un soporte teórico. Por otro lado la teoría de la densidad funcional DFT es utilizada para la determinación de las frecuencias vibracionales, y su aplicación al cálculo de intensidades Raman aún es un desafío debido al enorme costo computacional en el cálculo de los tensores de susceptibilidad Raman. Por esta razón, las investigaciones teóricas, para este propósito, están basadas en aproximaciones empíricas o semi-empíricas, tal como se presenta en el modelo de bandas de polarizabilidad48-50. Esos modelos generalmente confían la descripción de la respuesta dieléctrica en términos de contribuciones de bandas individuales y sus parámetros son obtenidos a través de consideraciones experimentales. La principal ventaja de esos modelos por sobre los cálculos de primeros principios es su fácil implementación, bajo costo computacional y su habilidad para resolver estructura complejas. Sin embargo, esos modelos tiene una baja exactitud y sus parámetros esporádicamente son tranferibles48. Además, para compuestos del tipo ABO3 polarizados, no existen resultados confiables. Debido a la necesidad de dar una interpretación de los espectrogramas de dispersión Raman y demostrar ciertas afirmaciones experimentales, es 69 que se comenzó a trabajar con cálculos teóricos de primeros principios, para distintas fases para el BTO. Para ello se modelaron celdas unitarias en tres fases distintas: dos conocidas (cúbica y tetragonal) y otra ficticia que es una celda cúbica pero con un pequeño desplazamiento del octaedro TiO6 a lo largo del eje Z. Es importante señalar que nuestro mayor interés era comprobar que el peak asociado a la fase tetragonal que aparece a los 307 cm-1 sólo pertenece a esta fase y no a una cúbica. Además, ver la influencia que ejerce una fase cúbica deformada en el peak 307 cm-1. Para este estudio teórico se utilizó el programa Gaussian 03W51. Donde cálculos fueron realizados dentro del formalismo de la teoría de la densidad funcional (DFT) y utilizando el método de cluster. Todos los detalles de acerca de la teoría con que están sustentados estos cálculos esta descrita en el apéndice. 4.1 Metodología de Cluster. Debido al bajo costo computacional, comparado con el cálculo de sistemas extendidos, se decidió modelar el BTO por medio del método de cluster. Este método simula un sólido en base a unos pocos y finitos átomos, lo que lo hace muy conveniente desde el punto de vista computacional para la simulación del sólido. Las ventajas y desventajas de este método están relacionadas con este último aspecto52. 70 La principal ventaja de este método está asociada directamente con el hecho de poder modelar un sólido en base a un número finito de átomos, y con ello generamos un número finito de enlaces, electrones y niveles de energía, cuyas propiedades pueden ser obtenidas al emplear métodos de cálculos como de Hartree-Fock (HF) o Teoría de la densidad funcional (DFT), que son mas fáciles de llevar a cabo que los procedimiento de cálculos ab initio del tipo extendido. También hay que considerar que efectos asociados al relajamiento de los parámetros de red incluyendo impurezas o defectos son más fáciles de incluir en un procedimiento de cluster. 4.2 Modelos de Celdas. El BTO fue simulado mediante el método de Cluster para ello en primera aproximación se usó el octaedro TiO6 para la fase cúbica y tetragonal. Luego, una vez obtenidos los primeros resultados con este modelo se crearon las celdas unitarias de las tres fases de interés: Tetragonal, Cúbica y Pseudocúbica. El grupo espacial, las constantes de red y las coordenadas irreducibles de las fases cúbica y tetragonal fueron obtenidos de la literatura11,53. A continuación se presentan todos los modelos de cluster utilizados para los cálculos. 71 (a) (b) Fig. 4.1 Modelo de TiO6 para dos fases distintas: (a)Tetragonal, (b) Cúbica (a) (b) Fig. 4.2 Modelo de celdas unitarias para dos fases distintas: (a)Tetragonal, (b) Cúbica 72 (a) (c) (b) (d) Fig. 4.3 cluster de BTO de dos y cuatro celdas: (a) Dos celdas cubicas, (b) Dos celdas tetragonales, (c) Cuatro celdas cúbicas, (d) Cuatro celdas tetragonales. 73 4.3 Resultados. El estudio de los cluster de BTO, se hicieron ocupando métodos dentro del formalismo de DFT. La energía del sistema fue calculada a través de la resolución de las ecuaciones de Kohn-Sham (KS) en un set bases atómicas formadas por funciones Gaussianas. Para esto se utilizó un funcional híbrido de correlación e intercambio, el cual consiste en una cuidadosa mezcla de funcionales de intercambio de Hartree-Fock (HF), calculado con orbitales de KS, y el funcional de intercambio54 B88 añadido al funcional de correlación LYP55. Para los átomos de oxígeno y de titanio se utilizó un set de bases 6-31G56-65, mientras que los átomos de bario fueron utilizados como un pseudopotencial del grupo de los Alamos, Lanl1mb, con el set de bases asociadas66. Los clusters de TiO6 fueron utilizados como clusters de prueba para encontrar el funcional y el set de bases más adecuado. Una vez encontrado el funcional y el set de bases se comenzaron a hacer cálculos de los modos vibracionales. De los resultados logramos concluir que las vibraciones de alta frecuencia se debían sólo al octaedro de TiO6 y que las vibraciones de baja frecuencia (ν < 170cm −1 ) están más relacionadas con los átomos de bario. Este resultado era esperable debido a la dependencia de los modos vibracionales, pueden aproximar por: ν= 1 2π k µ (Ec. 4.1) 74 Entonces, debido a que los átomos de bario no aportaban al movimiento en frecuencias más altas, es que se decidió tratar a estos átomos con una base menor y menos costosa, o sea, como un pseudopotencial. Los espectros Raman teóricos, de una, dos y cuatro celdas para las distintas fases, no presentaban diferencias entre sí excepto por un pequeño desplazamiento hacia la izquierda en los espectrogramas. Esto último se debe a que al extender el número de átomos en la simulación la masa total del sistema y de acuerdo con la ecuación 4.1 la frecuencia debiese disminuir provocando el desplazamiento, pero los modos vibracionales seguían siendo los mismos. A continuación se muestran los diferentes espectros teóricos obtenidos para los distintos cluster en fase cúbica y tetragonal. Frecuencia [cm-1] Fig. 4.4 Raman teórico de una celda cúbica. 75 Frecuencia [cm-1] Fig. 4.5 Raman teórico de una celda tetragonal. Frecuencia [cm-1] Fig. 4.6 Raman teórico de dos celdas cúbicas. 76 Frecuencia [cm-1] Fig. 4.7 Raman teórico de dos celdas tetragonal. Frecuencia [cm-1] Fig. 4.8 Raman teórico de cuatro celdas cúbicas. 77 Frecuencia [cm-1] Fig. 4.9 Raman teórico de cuatro celdas tetragonal. Al comparar los espectros teóricos de la fase cúbica y tetragonal, estos muestran que los modos vibracionales son los mismos excepto por un sólo peak presente en la fase tetragonal cercano a 405 cm-1. Esta vibración es del tipo bending a lo largo del eje Z. Además, al comparar de los espectros experimentales Raman, para ambas fases, éstos muestran que la única diferencia distinguible entre las fases es una sola vibración que está presente en el espectro tetragonal31 cercano a 307 cm-1. Por lo tanto, al comparar cualitativamente los espectros teóricos y experimentales podemos decir en común acuerdo que para distinguir una fase de la otra existe un único modo vibracional, que experimentalmente esta ubicado en 307 cm-1, que diferencia el espectro Raman de la fase tetragonal respecto de la cúbica. 78 Una vez demostrada la diferencia de los espectros para ambas fases, se comenzó a trabajar con una fase cúbica a la cual se le fue desplazando levemente el átomo de titanio a lo largo del eje Z (0.5 a 5 pm). A continuación se muestra los diferentes espectros teóricos, aumentados alrededor de 405 cm-1, para las celdas cúbicas distorsionadas. (a) (b) Fig. 4.10 Raman teórico de la celda cúbica: (a) espectro aumentado alrededor de 405 cm-1, (b) todo el espectro. 79 (a) (b) Fig. 4.11 Raman teórico de la celda cúbica distorsionada 0.5 pm en el eje Z. (a) espectro aumentado alrededor de 405 cm-1, (b) todo el espectro. (a) (b) Fig. 4.12 Raman teórico de la celda cúbica distorsionada 1 pm en el eje Z. (a) espectro aumentado alrededor de 405 cm-1, (b) todo el espectro. 80 (a) (b) Fig. 4.13 Raman teórico de la celda cúbica distorsionadas 2 pm en el eje Z: (a) espectro aumentado alrededor de 405 cm-1, (b) todo el espectro. (a) (b) Fig. 4.14 Raman teórico de la celda cúbica distorsionadas 3 pm en el eje Z. (a) espectro aumentado alrededor de 405 cm-1, (b) todo el espectro. 81 (a) (b) Fig. 4.15 Raman teórico de la celda cúbica distorsionadas 4 pm en el eje Z. (a) espectro aumentado alrededor de 405 cm-1, (b) todo el espectro. (a) (b) Fig. 4.16 Raman teórico de la celda cúbica distorsionadas 5 pm en el eje Z. (a) espectro aumentado alrededor de 405 cm-1, (b) todo el espectro. 82 De las Figuras 4.10 a 4.16 se puede observar claramente como va creciendo el peak localizado en 405 cm-1 a medida que aumenta el desplazamiento del átomo de titanio, además pone de manifiesto como una pequeña distorsión genera esta vibración, lo que es una clara señal de la sensibilidad de la espectroscopía Raman a la simetría de la celda. Por lo tanto podemos decir que la vibración que diferencia una fase de otra, nace producto del desplazamiento del átomo de titanio a lo largo del eje Z. 83 v. CONCLUSIONES De los resultados experimentales y teóricos podemos afirmar que: i. Aunque el BTO es tetragonal a temperatura ambiente, esta fase no es fácil de sintetizar cuando el tamaño de partícula es del orden de los nanómetros. Estos cristales son tan pequeños que cualquier defecto estructural evita la transición de fase cúbica a tetragonal, produciendo altas tensiones dentro del cristal, lo que hace que la fase cúbica se distorsione pero no lo suficiente para alcanzar la fase tetragonal. Estas tensiones provienen de defectos dentro de los cristales de BTO formado durante el proceso de la síntesis hidrotermal. Se ha informado que la presencia de iones de OH(proveniente del medio de reacción) quedan incorporados en la red cristalina del titanato compensando las vacancias cationicas presente en la red67. Esta presencia es la responsable de generar tensiones en el interior de la red, lo que da como resultado la estabilización de la fase pseudo-cúbica metaestable a temperatura ambiente17,31,68 y no de la fase tetragonal. A pesar de estos inconvenientes la vía hidrotermal sigue siendo el proceso más económico de obtención de este material, debido que las temperaturas de trabajo (85 -240º C) son muy bajas en comparación con otros tipos de síntesis. ii. Por medio del método de síntesis propuesto se ha logrado sintetizar BTO, puro, homogéneo, cristalino, con un tamaño de partícula de alrededor de unos 50 nm y estabilizado en una fase ferroeléctrica. 84 iii. La aparente controversia entre espectroscopía Raman y XRD, se debe a que existe una distorsión del octaedro de TiO6, pero es tan pequeña que no es detectada por XRD. iv. El desplazamiento del ion de titanio dentro del octaedro de TiO6 es el responsable de la aparición de la vibración asociada a la fase tetragonal (307 cm-1), por lo tanto no se puede afirmar que las especies sintetizadas de BTO estén estabilizadas en fase tetragonal pura, ya que puede tratarse a una celda cúbica de menor simetría, que posee cierto desplazamiento del átomo de titanio. Pero, de todas formas este desplazamiento es suficiente para crear un momento dipolar eléctrico, y con ello una polarización espontánea. Lo que se ve reflejado en la medición de la curva de histéresis. v. Se comprueba que la vibración localizada en 307 cm-1, no está presente en la fase cúbica, tal como era afirmado en espectros experimentales31,32,38, y que hasta el momento no había sido comprobado en forma teórica. 85 APÉNDICE52 A.1 Cálculo de Primeros Principios. Durante las ultimas décadas, han ocurrido grandes progresos en el cálculos de propiedades de estructura, química, eléctrica, óptica y magnética de materiales. Estos progresos se deben principalmente al desarrollo de nuevas aproximaciones teóricas basadas en la Mecánica Cuántica, las que han sido complementados con la implementación de nuevos software y un rápido desarrollo del hardware. Para la predicción de las propiedades de los materiales existen dos tipos de metodologías computacionales: i. La aproximación del Potencial Empírico el cual describe la interacción entre los átomos en una forma semiclásica, dando lugar a la llamada Mecánica Molecular. ii. Los métodos de la Mecánica Cuántica los cuales toman en cuenta los movimientos e interacción de los electrones en un material, desde un punto de vista microscópico de la teoría. Si la aproximación está basada únicamente en constantes físicas fundamentales tales como la masa y la carga de un electrón y no en parámetros específicos del átomo, entonces se llama Ab-Initio o de primeros principios. 86 Estos cálculos buscan encontrar y describir soluciones aproximadas para la ecuación de Schrödinger. A.2 Ecuación de Schrödinger. El objetivo de los cálculos Ab-Initio es resolver la ecuación de Scrödinger53 y encontrar la función de onda asociado al sistema. ih ∂ ψ (q, t ) = Hψ (q, t ) ∂t (Ec. A.1) La función de onda ψ (q, t ) contiene toda la información necesaria para un sistema dado, siendo dependientes tanto de las coordenadas r r r r electrónicas ( r ) como de las nucleares ( R ) ( q = q(r , R) ).Si utilizamos el método de separación de variables podemos expresar la Ecuación A.1 como la multiplicación de dos funciones una dependiente de las coordenadas espaciales y otro dependiente del tiempo. Al realizar esto la ecuación se desacopla y nos queda: (Ec. A.2) Hψ ( q ) = Eψ ( q ) Al considerar un sistema de muchas partículas la Ecuación A.2 se hace intratable, debido al sin número de interacciones entre ellas. Entonces para poder solucionar esta ecuación es necesario recurrir a aproximaciones. Una de las primeras aproximaciones para solucionar este problema es la aproximación adiabática o de Born-Oppenheimer, 87 donde se presenta un modelo en el cual los electrones se mueven mucho más rápidos que los núcleos, pareciendo estoa últimos inmóviles. Esto permite separar la función de onda electrónica (la que tendrá solamente la información de los electrones) y determinar la energía electrónica a través de la Ecuación A.2. Las coordenadas nucleares actúan simplemente como parámetros en el Hamiltoniano (H) que opera sobre la función de onda electrónica. Para el cálculo de la evolución temporal de los elementos nucleares, pueden ser calculados por separado, usualmente considerando un sistema clásico gobernadas por interacciones Coulombianas entre el núcleo y la nube electrónica determinada en cualquier instante por la configuración nuclear dada. Otras simplificaciones adicionales a la Ecuación A.2 están relacionadas con el desprecio de los términos de interacción magnética dentro del Hamiltoniano, como por ser interacciones de momentos angulares magnéticos y de espín en electrones y núcleos. Finalmente el operador Hamiltoniano puede ser expresado por: n ⎛ h 2 2 Ze′2 ⎞ n −1 n e′2 ⎟+∑ ∑ H = ∑ ⎜⎜ − ∇i − 2me ri ⎟⎠ i =1 j = i +1 rij i =1 ⎝ (Ec. A.3) Donde suponemos que el núcleo es una masa puntual infinitamente pesada. La primera sumatoria contiene los operadores de energía cinética para los n electrones y el núcleo de carga Ze’. La segunda suma es la energía potencial para las repulsiones interelectrónicas; la restricción 88 j>i evita contar dos veces la misma repulsión interelectrónicas, y excluye los términos del tipo e′2 rii . A.3 Método de Variaciones. Una manera de resolver la ecuación de Schrödinger con el Hamiltoniano dado en la Ecuación A.3 es usando el Método de las Variaciones. Este método consiste en que dado un sistema cuyo Hamiltoniano Ĥ es independiente del tiempo, y cuyo valor propio de la energía más bajo es E1, si φ es cualquier función dependiente de las coordenadas del sistema, normalizada, que se comporta bien y que satisface las condiciones limite del problema , entonces ∫ φ * Hˆ φdτ = E φ [ ] ≥ E1 , Si φ está normalizada (Ec. A.4) Este teorema nos permite calcular un límite superior a la energía del estado fundamental del sistema. Ahora si el estado no está normalizado la ecuación se transforma en: ∫ φ * Hˆ φdτ ∫ φ * φdτ = φ Hˆ φ = E[φ ] ≥ E1 φφ (Ec. A.5) Como consecuencia de este teorema el valor del límite superior de la energía es mayor al valor verdadero del estado de más baja energía y depende que tan parecida sea la función de prueba φ a la verdadera función del estado fundamental ψ 0 . Para optimizar la función de prueba 89 y minimizar el limite superior, la función E[φ ] debe ser minimizada variacionalmente, por ejemplo, que φ sea resuelta para δE[φ ] = 0 . A.4 Método de Hartree-Fock (HF). Un tipo de función de prueba usada a menudo en el método variacional r es el producto antisimetrizado de espines-orbitales de una partícula φ (r ) , o determinante de Slater: r r r φ1 (r1 ) φ2 (r1 ) ... φn (r1 ) φHF = 1 N ... ... ... ... ... .... ... ... r r r φ1 (rn ) φ2 (r1 ) ... φ1 (rn ) (Ec. A.6) Un determinante de Slater introducido en el método variacional se resuelve posteriormente con las ecuaciones de HF canónicas por los orbitales de HF canónicos. r r Fφi (r )c = ε icφi (r )c (Ec. A.7) Resolviendo estas ecuaciones se obtienen los orbitales ortonormales r φi (r ) en la Ecuación A.6 que minimiza la Ecuación A.5. Este resultado es llamado la aproximación de Hartree-Fock. 90 A.5 Teoría Funcional de la Densidad (DFT). Los métodos ab initio utilizados en los cálculos, están basados en la DFT que tiene algunas ventajas sobre el método de HF. Los cálculos DFT demuestran la existencia de un principio variacional análogo al método de Rayleigh-Ritz con la excepción que utiliza densidades electrónicas en vez de funciones de onda ψ . La densidad electrónica total se define como: r r N ρ (r ) = ∑ ni ∑ ψ i (r , s ) i 2 (Ec. A.8) s Donde: ψ i → Es un espín-orbital. r r → La coordenada espacial. s→ La coordenada de espín. ni → El número de ocupación. N → El número de espín-orbital. El segundo teorema de Hohenberg-Kohn constituye el principio r variacional de la densidad electrónica: para una densidad de prueba ρ (r ) , r r r r tal que ρ ( r ) ≥ 0 y ∫ ρ (r )dr = N (número de electrones), E0 ≤ E[ ρ (r )] donde r 69 E[ ρ ( r )] es el funcional de la energía mostrada en A.9 . r r r E[ ρ ] = T [ ρ ] + ∫ ρ (r )υ (r )dr + Vee [ ρ ] FHK [ ρ ] = T [ ρ ] + Vee [ ρ ] (Ec. A.9) 91 Las únicas entradas en el funcional E[ ρ ] que son únicas para un sistema r dado son υ (r ) , el potencial electroestático creado por el núcleo, y N, el número total de electrones como una restricción en ρ . F [ ρ ] es un funcional universal que es independiente de la carga y el arreglo nuclear. Desafortunadamente esta función es desconocida, de otra forma, todos los estados electrónicos fundamentales para cualquier configuración nuclear podrían ser determinados. La teoría DFT permite la propagación de aproximaciones más precisas que HF debido a que incluye términos de la energía de correlación y menos complicadas debido al uso de tres coordenadas de densidad electrónica, y no 4N coordenadas de la función de onda electrónica. La precisión para resolver las ecuaciones va depender exclusivamente de los valores para la aproximación de F [ ρ ] . Una ventaja adicional de los métodos basados en DFT es el hecho que mejores aproximaciones para F [ ρ ] están siendo desarrolladas, por lo tanto, todos los cálculos DFT mejoran, sin reparar en detalles sistemáticos, ya que F [ ρ ] es universal. A.6 Método de Kohn-Sham (KS). Las ecuaciones de Kohn-Sham (KS) surgieron como un intento para librar el problema de la función desconocida F [ ρ ] , la que al ser rescrita se muestra como: F[ ρ ] = Ts [ ρ ] + J [ ρ ] + Exc [ ρ ] (Ec. A.10) Donde 92 Exc [ ρ ] ≡ T [ ρ ] − Ts [ ρ ] + Vee[ ρ ] − J [ ρ ] Ts [ ρ ] en la Ecuación A.10 es la energía de cinética para un sistema de N electrones no interactúantes. Exc [ ρ ] es conocida como la energía de correlación-intercambio. Esta contiene la diferencia en la energía cinética entre un sistema con N electrones interactuando y uno con N electrones no interactúantes. Esta también contiene la parte no clásica de de Vee[ ρ ] ; ambas, la parte de intercambio capturada en el teorema de HF y la energía de correlación omitida por HF. Reemplazando T [ ρ ] con Ts [ ρ ] e incluyendo la diferencia entre las dos energías cinéticas en Exc [ ρ ] se puede minimizar E[ ρ ] únicamente utilizando N ecuaciones monoelectrónicas de la forma70: r ⎡ 1 r ⎤ r r Hφi (r ) = ⎢− ∇ 2 + υeff (r )⎥φi (r ) = ε iφi (r ) ⎦ ⎣ 2 (Ec. A.11) Con el potencial efectivo de KS dado por: r r υeff (r ) = υ (r ) + δJ [ ρ ] δE xc [ ρ ] r + r δρ (r ) δρ (r ) (Ec. A.12) Una densidad electrónica puede ser derivada a partir de las soluciones de los N orbitales desde la Ecuación A.11 por medio de la ecuación v r N ρ (r ) = ∑ ni ∑ φi (r , s) i =1 2 (Ec. A.13) s La no linealidad de la Ecuación A.11 será resuelta de una manera r autoconsistente. Una densidad electrónica ρ (r ) inicial es supuesta desde r que υeff (r ) es construido por la Ecuación A.12. Los orbitales son calculados con la Ecuación A.11 que produce una nueva densidad 93 electrónica por medio de la Ecuación A.13. Esta nueva densidad electrónica produce un nuevo r υeff (r ) para las ecuaciones monoelectrónicas y así sucesivamente. Las iteraciones son continuadas hasta que la convergencia deseada en la densidad electrónica ρ y la energía asociada E ( ρ ) son alcanzadas. A.7 LDA, LSDA y GGA. El problema en implementar el método de KS consiste en encontrar una buena aproximación para la energía de correlación e intercambio y para el potencial efectivo. Una de las aproximaciones más sencillas es la llamada Aproximación de la Densidad Local (LDA) propuesta por Kohn y Sham (1965). En esta aproximación la energía de correlación e intercambio toma la forma de70,71: ExcLDA[ ρ ] = ∫ ρ[r ]ε xc (ρ )dr (Ec. A.14) ε xc ( ρ ) es igual a la energía de correlación e intercambio por electrón para un gas de electrones, y corresponde a un funcional ordinario de ρ que ha sido parámetrizada desde simulaciones de Monte Carlo. ExcLDA es una buena aproximación para una densidad electrónica que varia lentamente y llega a ser exacta para un gas de electrones homogéneos. Sorprendentemente, esta a veces se comporta bien con densidades electrónicas que varían rápidamente, sin embargo con estados electrónicos altamente localizados casi siempre falla 94 La Aproximación Densidad de Espín Local (LSDA) es equivalente a la LDA excepto que la aproximación está hecha en el contexto de la teoría de DFTS, la cual es una extensión de la teoría DFT normal utilizando densidades electrónicas con polarización de espín. La energía en este caso es un funcional de ambas densidades de espín (α y β) dando el principio variacional: E[ ρ α , ρ β ] ≥ E0 (Ec. A.15) Donde: ρ α ≡ densidad de espín α ( ↑ ); ρ β ≡ densidad de espín β ( ↓ ) En LSDA, ExcLDA[ ρ α , ρ β ] utiliza la energía de correlación e intercambio por electrón α y electrón β derivada desde un gas de electrones homogéneo con polarización de espín y con ρ α y ρ β . Uno de los mayores beneficios de usar LSDA en vez de LDA es que se incorpora espontáneamente el magnetismo, que es especialmente importante en el modelamiento de materiales con metales de transición. Purdue y Yue desarrollaron la Aproximación del Gradiente Generalizado (GGA) en un intento por resolver la mayor fuente de error en LDA, es decir, la energía de intercambio. El método GGA intenta corregir este error incorporando términos polinomiales con el módulo del gradiente de densidad electrónica ( ∇ρ ( r ) ) que captura algunas de las desviaciones de la energía de correlación e intercambio desde el resultado del gas de electrones homogéneo70,71. Como LDA, GGA tiene un equivalente para la polarización de espín apropiado para usarlo con materiales magnéticos. 95 A.8 Pseudopotenciales. El método de los Pseudopotenciales se basa en la división de los electrones de los átomos en core (o electrones internos e inertes) y valencia (o electrones externos y activos) que a su vez implica una división del espacio que rodea a cada átomo en una región de core entorno a los núcleos atómicos, donde están localizados los orbitales inertes de core que no contribuyen directamente al enlace entre átomos ni a las propiedades derivadas del estado sólido, y una región externa al core (intersticial en el caso del sólido) en donde se produce la redistribución de carga que conduce al enlace químico entre los átomos. La forma de los orbitales en la región del core es poco relevante desde el punto de vista de las propiedades del sólido; sin embargo la correcta descripción de los electrones de valencia en la región externa al core es esencial. En un esquema de pseudopotenciales los electrones de core se consideran “congelados” y sólo se trata de forma explicita en el cálculo a los electrones químicamente activos de valencia, representando el efecto del core atómico inerte sobre los electrones de valencia por medio de un pseudopotencial iónico (aproximación de core congelado). A.9 Programa Gaussian. Encontrar las soluciones analíticas de la ecuación de Schrödinger es el gran problema de la Mecánica Cuántica, ya que las soluciones analíticas sólo son conocidas para casos sencillos como por ser: átomos hidrogenoides, partícula en una caja, oscilador armónico y rotor rígido. 96 Mientras que para sistemas más complejos como el átomo de helio solamente se pueden obtener soluciones aproximadas. Sin embargo, la disponibilidad de computadoras más rápidas permite obtener soluciones a la ecuación de Schrödinger para átomos polielectrónicos, moléculas y sólidos con un alto nivel de aproximación utilizando un programa computacional con la metodología de interés. El programa Gaussian, es un paquete de programas versátil, en el que incluyen todos los métodos ab initio más comunes, tales como HartreeFock, Integración de Configuraciones (CI), funcional de la densidad, Teoría de Perturbaciones de Moller-Plesset (MP), Métodos de Cluster Acoplados (CC), y también incluyen muchos métodos semiempíricos. Gaussian optimiza la geometría, calcula frecuencias vibracionales, propiedades termodinámicas y constantes de apantallamiento RMN, busca estados de transición, calcula Camino de Mínima Energía (MEP), e incluye los efectos de disolvente. Gaussian está disponible para supercomputador, estaciones de trabajo y computadores personales que trabajan bajo Windows51. 97 A.10 Funcionales Híbridos. Los funcionales híbridos son una clase de aproximaciones a la energía de correlación e intercambio, dentro del formalismo de la teoría DFT, que incorpora una porción exacta de intercambio proveniente de la teoría de HF y porciones de correlación e intercambio provenientes desde otras fuentes (Ab Initio, LDA o empírica). El valor exacto del funcional de la energía de intercambio está expresado en terminos de los orbitales KS. Un funcional hibrido es usualmente construido como una combinación lineal del funcional de intercambio HF ( ExHF ) y un número cualquiera de funcionales de densidad de correlación e intercambio. Para el funcional B3LYP72 la combinación lineal queda*: ( ) ( ) ( ExcB3LYP = ExcLDA + a0 ExHF − ExLDA + ax ExGGA − ExLDA + ac EcGGA − EcLDA ) (Ec. A.16) Donde los tres parámetros a0 = 0.20 , ax = 0.72 y ac = 0.81, están determinados graficando valores predichos para un set de energías de atomización, potenciales de ionización, afinidad protónica y el total de las energías atómicas; ExGGA y EcGGA son Aproximaciones del Gradiente Generalizado: Un funcional Becke 88 de intercambio54 y funcionales de correlación de Lee, Yang y Parr55; y EcLDA es un funcional de correlación proveniente de la Aproximación de la Densidad Local. * El subíndice x indica intercambio, y el subíndice c indica Correlación 98 A.11 Set de Bases. Un set de bases en química es un set de funciones usadas para crear orbitales moleculares, las cuales son expandidas como combinación lineal de funciones con coeficientes a determinar. Usualmente esas funciones son orbitales atómicos centradas en el átomo, y pares de funciones centradas en los lóbulos del orbital “p”. Adicionalmente también a menudo son usadas sets de bases compuestas de ondas planas con longitudes de onda (λ) bajos, estas se aplican especialmente en sistemas con condiciones de borde periódicas. En química cuántica computacional los cálculos se elaboran típicamente con un set finito de bases. En estos casos, las funciones de onda son representadas como vectores, cuyos componentes corresponden a los coeficientes de una combinación lineal de las funciones bases en el set usado. Los operadores están representados como matrices. Al realizar cálculos de moléculas, es común usar bases compuestas de un número finito de orbitales atómicos, centrados en cada núcleo atómico dentro de la molécula (combinación lineal de orbitales atómicos, método LCAO). Inicialmente, esos orbitales atómicos eran orbitales de Slater, los cuales correspondían a un set de funciones que decaen exponencialmente con la distancia. Mas tarde esos orbitales de Slater fueron aproximados por una combinación lineal de orbitales tipo gaussianos, debido a que los cálculos se hacían menos complejos. Hoy en día existen ciento de bases compuestas por orbitales tipo gaussianos (GTOs). El set de bases más pequeño se denomina set de bases mínima, y esta compuesto de un mínimo de funciones bases requeridas para 99 representar todos los electrones de cada átomo, mientras que la más grande puede estar formadas por cientos de funciones bases para cada átomo. Un set de bases mínima es un set al cual para cada átomo de la molécula, una sola función base es usada para cada orbital en el calculo (HF) de cada átomo libre. Sin embargo, para átomos como el litio, funciones bases tipo “p” son añadidas a las funciones bases correspondientes a los orbitales 1s y 2s de átomo libre. Por ejemplo, en cada átomo en la primera fila de la tabla periódica (Li-Ne) tendría un set de base de 5 funciones base (“2” funciones s y 3 funciones “p”). La adición mas común a un set de base mínima son la adición de funciones de polarización, denotada por uno o dos asteriscos (*, **), dos asteriscos indican que las funciones de polarización son también añadidas a los átomos ligeros (H, He). Esas son funciones auxiliares con un nodo adicional. Por ejemplo, la sola función base localizada en el átomo de hidrógeno en un set de bases mínimo sería una función aproximada del orbital 1s, cuando la polarización es añadida a este set de base, una función p también es añadida al set de base, esto añade alguna flexibilidad adicional dentro del set de base, efectivamente permitiendo que los orbitales moleculares del hidrógeno sean más simétricos con respecto al núcleo. Esto es un resultado importante cuando son consideradas representaciones exactas de los átomos vinculados, porque la misma presencia del átomo hace que el ambiente energético de los electrones sea esfericamente asimétrico. Similarmente, las funciones tipo “d” pueden ser añadidas al set de base con orbitales “p”, y funciones “f” a los orbitales “d”, y así sucesivamente. Otra 100 notación mas precisa indica exactamente cuáles y cuántas funciones bases serán añadidas al set de base (ej. p, d). Otra adición común a los set de base son la adición de funciones de difusión, denotadas con un signo mas (+, ++), los dos signos mas indican que también fueron añadidas a los átomos ligeros (H, He). Esas son funciones bases gaussianas muy pequeñas, las cuales representan con más exactitud la parte final (la cola) de los orbitales atómicos mas distantes al núcleo atómico. Esta adición de funciones bases puede ser importante cuando consideramos aniones y otros sistemas moleculares mayores. A.11.1 Set de Bases Mínimas. El set más común de bases mínimas es STO-ng donde “n” es un entero. Este valor de “n” representa el numero de funciones gaussianas primitivas comprimida en una sola función base. En este set de base, el mismo número de gaussianas primitivas comprenden la capa y los orbitales de valencia. Un set de base mínimo típicamente da resultados no muy exactos, pero son menos costosas que sus contrapartes mayores. Comúnmente son usadas las siguientes set de base mínima: • • • • STO-3G STO-4G STO-6G STO-3G* (polarización de STO-3G) 101 A.11.2 Set de Bases Split-Valence. Durante la vinculación molecular, son los electrones de valencia principalmente los que toman parte del vínculo. En reconocimiento a este hecho es común representar los orbitales de valencia por más de una función base, (cada uno puede estar compuesto de una combinación lineal de funciones gaussianas primitivas). Los sets de bases en el cual hay múltiples funciones bases correspondientes a cada orbital de valencia, son llamados valencia doble, triple…. Set de bases, ya que los diferentes orbitales del split tienen diferentes extensiones espaciales. La combinación permite ajustar la densidad electrónica a una extensión espacial adecuada para un ambiente molecular en particular. Un set de bases mínimas es fijado y son incapaces de ajustarse a los diferentes ambientes moleculares. A.11.3 Set de Bases Pople73. La notación para el set de base split-Valence la dio John Pople, y es de la forma X-YZg, donde la X representa el número de gaussianas primitivas comprendidas para cada función base del orbital o de las capas del núcleo, la Y y la Z indican que los orbitales de valencia están compuestos por dos set de funciones bases cada uno, el primero compuesto de una combinación lineal de “Y” funciones gaussianas primitivas, mientras que el otro de una combinación lineal de “Z” funciones gaussianas primitivas. En este caso, la presencia de dos números después del signo menos implica que este set de base es un 102 Split-valence doble zeta, un Split-valence triple o cuadruple usualmente es denotado como X-YZWg, X-YZWVg, y así sustantivamente, a continuación se presenta una lista de las fuentes Split-valence de este tipo. • • • • • • • • • • • 3-21g 3-21g* polarizada 3-21+g funciones de difusión 3-21+g* con polarización y difusión 6-31g 6-31g* 6-31+g* 6-31g(3df, 3pd) 6-311g 6-311g* 6-31+g* 103 Listado de Instrumentos. A continuación se mostrarán los datos técnicos de todos los equipos y programas que se utilizaron para desarrollar este trabajo. L.1 Microscopía Raman. Todos los espectros Raman fueron tomados a temperatura ambiente con un microscopio de espectroscopía Raman y Fuerza Atómica Witec Alpha300R equipado con una cámara CCD. Las muestras fueron excitadas con un Láser de argón de 200 mW Lasos 60 series de longitud de onda 514.5 nm (verde), estas fueron enfocadas con oculares Nikon de 10× / 0.25 y 100× / 0.9 , usando dos grillas diferentes de 600 y 1800 divisiones por milímetro. Este equipo se encuentra en el laboratorio de espectroscopia, perteneciente al departamento de Física de la Universidad Católica del Norte. Los espectros e imágenes Raman fueron analizados mediante el software Witec Project y OriginPro 8 pertenecientes la misma institución. L.2 Tratamientos Térmicos. Las calcinaciones de las muestras se hicieron en un mufla Barnstead Thermolyne, perteneciente al laboratorio termodinámica ubicado en el departamento de Física de la Universidad Católica del Norte. 104 L.3 XRD. Las caracterizaciones de XRD fueron tomadas a temperatura ambiente y se realizaron con un difractómetro de polvos Siemens D5000 con radiación de CuKα no monocromatizada operado a 40 kV y 30 mA. Este equipo pertenece a la Facultad de Ciencias Físicas y Matemáticas de la Universidad de Chile. L.4 HRTEM. Las microfotografías obtenidas por HRTEM fueron tomadas con el microscopio Tencai F20 FEG-TEM equipado con un detector EDS y operado a 200 kV. Este equipo pertenece al laboratorio de HRTEM del departamento de Geología de la Universidad de Chile. Todos los análisis de las imágenes en TEM, HRTEM y difracción de electrones fueron hechos a través del Software Digital Micrograph(TM) versión 3.9.0, perteneciente a la misma casa de estudio. L.5 AFM. Las micrografías de fuerza atómica fueron realizadas en un microscopio de Espectroscopía Raman y Fuerza Atómica Witec Alpha300R del laboratorio de espectroscopía perteneciente al departamento de Física de la Universidad Católica del Norte. 105 L.6 Osciloscopio. Los voltajes para el circuito de histéresis fueron medidos en un osciloscopio Tektronix TK2221A, perteneciente al Laboratorio de Electrónica del Departamento de Física de la Universidad Católica del Norte. Los datos fueron adquiridos con el Software WaveStar V 1.1. 106 REFERENCIAS 1. G. H. Haertling, J. Am. Ceram. Soc. 82 (1999), 797. 2. J. F. Scott, C. A. Paz de Arujo, Sciences 246 (1989), 1400. 3. D. Hernandez Cruz, B. Sahouli, A. Tork, E. J. Knystautas, R. A. Lessard, SPIE Proc. 244 (2001), 4296. 4. C. Barry Carter, M. Grant Norton: “Ceramics Materials Science and Engineering”, Springer, EE.UU., 2007. 5. J. Reitz, F. Milford, R. Christy, “Fundamentos de la teoría electromagnética”, Addison-Wesley Iberoamericana, 2000. 6. A. F. Devonshire, Phil. Mag. 40 (1949), 1040; ibid 42, 1065 (1951); Adv. Phys. 3 (1954), 85. 7. H. Mueller, Phys. Rev. 57(1940), 829. 8. E. Fatuzzo, W. M. Mertz, “Ferroelectricity”, North-Holland, Holanda, 1967. 9. M. E. Lines, A. M. Glass, “Principles and applications of ferroelectrics and related materials”, Oxford University Press, Gran Bretaña, 1977. 10. Y. Xu, “Ferroelectric materials and their applications”, Elsevier, Holanda, 1991. 11. H. Dittrich, N. Karl, S. Kück, H. W. Schock, “Ternary Compounds, organic semiconductors”, Springer-Verlag, EE.UU., 2000. 12. R. A. Zárate, “Fabricación y caracterización de películas delgadas de titanato de bario”, Tesis para optar al grado de Doctor en ciencias exactas con mención en Física, Pontificia Universidad Católica, Chile, 1998. 107 13. G. Manestar, “La histéresis”, <http://techgk.wordpress.com/2006/12/09/la-histeresis/>, 2006. 14. Ruedas de Prensa, S. L., “Histéresis”, <http://www.km77.com/glosario/histe resis.asp>, 1999. 15. C. Pithan, D. Hennings, R. Waser. Int. J. Appl. Ceram. Technol. 2 (2005), 1. 16. S. Luo, Z. Tang, W. Yao, Z. Zhang. Microelectron. Eng. 66 (2003), 147. 17. C. Xia, E. Shi, W. Zhong, J. Gou, J. Cryst. Growth. 166 (1996), 961. 18. L. Qi, B. Lee, Journal of the European Ceramic Society 24 (2004), 3553. 19. P. A. Psaras, H. D. Langford, “Advancing Materials Research”, National Academy Press, EE.UU., 1987. 20. K. Byrappa, M. Yoshimura, “Handbook of Hydrothermal technology A Technology for Crystal Growth and Materials Processing”, Noyes Publications, EE.UU., 2001. 21. L. Guo, Material Letters 60 (2006) 3012. 22. D. Bermejo, “Introducción a la espectroscopía Raman”, Secretaría general de la organización de los Estados Americanos, EE.UU., 1988. 23. B. D. Cullity, “Elements of X-ray Diffraction”, Addison-Wesley Publishing Company, Inc, EE.UU., 1956. 24. A. L. Patterson, PRL, 56 (1939), 978. 25. J. C. Spence, “High-Resolution Electron Microscopy”, Oxford University Press, EE.UU., 2003. 108 26. B. Fultz, J. Howe, “Transmission Electron Microscopy and Diffractometry of Materials”, Springer-Verlag, EE.UU.,2000. 27. G. Gauglitz and T. Vo-Dinh, “Handbook of Spectroscopy”, Wiley-VCH Verlag GmbH & Co. KGaA, Alemenia, 2003. 28. B. Bhushan, “Springer Handbook of Nanotechnology”, Springer Verlag, Alemania, 2003. 29. Witec, “Atomic Decription”, Force Witec Microscopy alpha300A: Wissenschaftliche Technical Instrument and Technologie GmbH, Alemania, 2008. 30. B. Sawyer, C. H. Tower, Phys. Rev., 35 (1930), 269. 31. G. Busca, V. Buscaglia, M. Leoni, P. Nanni, Chem. Mater., 6 (1994), 955. 32. Y. Shiratori, C. Pithan, J. Dornseiffer, R. Waser, J. Raman Spectrosc., 38 (2007), 1300. 33. L. Rimai, J. L. Parsons, J. T. Hickmott, T. Nakamura, Phys. Rev., 168 (1967), 2. 34. J. Schmelzer, “Nucleation Theory”, Wiley-VCH Verlag GmbH & Co. KGaA, Alemania, 2005. 35. M. Bockrath, W. Liang, D. Bozovic, J. Hafner, C. Lieber, M. Tinkham, H. Park, Science 291 (2001), 283. 36. J. Colmes, K. Johnston, R. Doty, B. Korgel, Science 287 (2000), 1471. 37. R. Mangalam, N. Ray, U. Waghmare, A. Sundaresan, C. Rao, Solid State Communications, 149 (2009), 1. 38. Y. Shiratori, C. Pithan, J. Dornseiffer, R. Waser, J. Raman Spectrosc., 38 (2007), 1288. 39. K.Y. Chen, Y. W. Chen, Powder Tech, 41 (2004), 69. 109 40. Programa INTERACCIÓN, para el cálculo de parámetros cristalográficos de muestras policristalinas. Desarrollado por el Dr. Salvador Barbato, Departamento de Química, Facultad de Ciencias, Universidad de Antofagasta. 41. I. Mayergoyz, G. Bertotti, “The science of Hysteresis”, Elsevier, 2005. 42. X. Zhu, J. Wang, Z. Zhang, J. Zhu, S. Zhou, Z. Liu, N. Ming, J. Am. Ceram. Soc., 91 (2008), 2683. 43. E. Shi, C. Xia, W. Zhong, B. Wang, C. Feng, J. Am. Ceram. Soc. 80 (1997), 1567. 44. C. Xia, E. Shi, W. Zhong, J. Gou, J. Cryst. Growth 166 (1996), 961. 45. J. C. Niepce “Surfaces and interfaces of ceramic Materials”, Kluwer Academic Publisher, Holanda, 1989. 46. K. Kajiyoshi, N. Ishizawa, M. Yoshimura, J. Am. Ceram. Soc. 74 (1991), 369. 47. Y. Jung, D. Lim, J. Nho, S. Cho, R. Riman, B. Lee, J. Cryst. Growth 274 (2005), 638. 48. P. Hermet P, N. Izard, A. Rahmani, Ph. Ghosez J. Phys. Chem. B 110 (2006) 24869. 49. H. Chadli, A. Rahmani, K. Sbai, P. Hermet, S. Rols, J. L. Sauvajol, Phys. Rev. B 74 (2006), 205412. 50. S. Guha, J. Menéndez, J. B. Page, G. B. Adams, Phys. Rev.B 53 (1996), 13106. 51. Gaussian 03, Revision E.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. 110 Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, and J. A. Pople, Gaussian, Inc., Wallingford CT, 2004. 52. S. Conejeros, “Estudio Teórico de Estabilidad de las Fases LixK1-xCuFeS2 con Estructura del Tipo ThCr2Si2”, Memoria para optar al grado de Magíster en Ciencias mención Química, Universidad Católica del Norte, Chile, 2009. 53. P. Hermet, M. Veithen, Ph. Ghosez, J. Phys. Condens. Matter. 21 (2009), 215901. 54. A.D. Becke, Phys. Rev. A 38 (1988), 3098 55. C. Lee, W. Yang, R. G. Parr, Phys. Rev. A 37 (1988), 785 56. R. Ditchfield, W. J. Hehre, and J. A. Pople, J. Chem. Phys. 54 (1971), 724. 111 57. W. J. Hehre, R. Ditchfield, J. A. Pople, J. Chem. Phys. 56 (1972), 2257. 58. P. C. Hariharan and J. A. Pople, Theo. Chim. Acta 28 (1973), 213M. 59. P. C. Hariharan and J. A. Pople, Mol. Phys. 27 (1974), 209. 60. M. S. Gordon, Chem. Phys. Lett. 76 (1980), 163 61. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, D. J. DeFrees, J. A. Pople, M. S. Gordon, J. Chem. Phys 77 (1982), 3654. 62. R. C. Binning Jr. and L. A. Curtiss, J. Comp. Chem. 11 (1990), 1206. 63. J. P. Blaudeau, M. P. McGrath, L. A. Curtiss, and L. Radom, J. Chem. Phys. 107 (1997), 5016. 64. V. Rassolov, J. A. Pople, M. A. Ratner, T. L. Windus, J. Chem. Phys. 109 (1998), 1223. 65. V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern, and L. A. Curtiss, J. Comp. Chem. 22 (2001), 976. 66. P.J. Hay, W.R. Wadt, J. Phys. 82 (1985), 270. 67. E. Shi, C. Xia, W. Zhong, B. Wang, C. Feng, J. Am. Ceram. Soc. 80 (1997), 1567. 68. J. Clark, T. Takeuchi, N. Ohtori, D. Sinclair, J. Mater. Chem. 9 (1999), 83. 69. B. H. Brandsten, C. J. Joachin, “Introduction to Quantum Mechanics”, Longman Group UK Limited, Reino Unido, 1989. 70. R. G. Parr, W. Yang “Density Functional Theory of Atoms and Molecules”, Oxford University Press, Reino Unido, 1989. 71. K. Ohn, K. Esfarjani, Y. Kawazoe, “Computational Material Science”, Springer-Verlag Berlin Heidelberg, Alemania, 1999. 112 72. A.D. Becke, J.Chem.Phys. 98 (1993), 5648. 73. K. I. Ramachandran, G. Deepa, K. Namboori, “Computational Chemistry and Molecular Modeling, Principles Applications”, Springer Berlin Heidelberg, Alemania, 2008. and 113