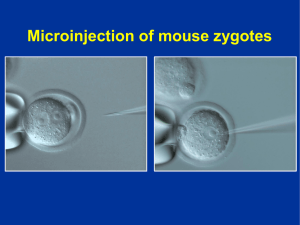
Luque, Guillermina María. 2015 03 12
Anuncio

La disrupción específica del receptor dopaminérgico D2 en lactotropos devela un rol de la prolactina en el control de la ingesta y el metabolismo Luque, Guillermina María 2015 03 12 Tesis Doctoral Facultad de Ciencias Exactas y Naturales Universidad de Buenos Aires www.digital.bl.fcen.uba.ar Contacto: [email protected] Este documento forma parte de la colección de tesis doctorales y de maestría de la Biblioteca Central Dr. Luis Federico Leloir. Su utilización debe ser acompañada por la cita bibliográfica con reconocimiento de la fuente. This document is part of the doctoral theses collection of the Central Library Dr. Luis Federico Leloir. It should be used accompanied by the corresponding citation acknowledging the source. Fuente / source: Biblioteca Digital de la Facultad de Ciencias Exactas y Naturales - Universidad de Buenos Aires UNIVERSIDAD DE BUENOS AIRES Facultad de Ciencias Exactas y Naturales La disrupción específica del receptor dopaminérgico D2 en lactotropos devela un rol de la prolactina en el control de la ingesta y el metabolismo. Tesis presentada para optar al título de Doctor de la Universidad de Buenos Aires en el área: CIENCIAS BIOLOGICAS Lic. Guillermina María Luque Director de tesis: Dra. Damasia Becú de Villalobos Consejero de estudios: Dr. Arturo Romano Laboratorio de Regulación Hipofisaria Instituto de Biología y Medicina Experimental (IBYME-CONICET) Buenos Aires, 2015. LA DISRUPCIÓN ESPECÍFICA DEL RECEPTOR DOPAMINÉRGICO D2 EN LACTOTROPOS DEVELA UN ROL DE LA PROLACTINA EN EL CONTROL DE LA INGESTA Y EL METABOLISMO . La prolactina es una hormona altamente versátil secretada principalmente por los lactotropos de la hipófisis anterior. Su función en la reproducción y fertilidad ha sido muy estudiada, pero poco se sabe de su acción en la regulación del peso corporal y el metabolismo. Altos niveles de prolactina se observan durante la preñez y la lactancia, estados en los que se favorece la hiperfagia. Durante la preñez, los mecanismos normales que regulan el apetito son modificados para generar un balance energético positivo, incrementando la ingesta y los reservorios grasos para abastecer el requerimiento del feto en crecimiento, y ser utilizados posteriormente en la lactancia. Existen numerosos resultados que apoyan la hipótesis de que la prolactina posee un rol significativo en la regulación del peso corporal. Por ejemplo, el ratón deficiente del receptor de prolactina presenta peso corporal y depósitos de grasa disminuidos, la administración de prolactina estimula la ingesta de alimento, y niveles elevados de esta hormona de manera crónica (como ocurre durante la preñez) incrementan la ingesta posiblemente por la inducción de una resistencia a leptina. Sin embargo, ratones macho conteniendo hipófisis ectópicas muestran sólo un mínimo incremento en el peso corporal con un descenso en el tejido adiposo, y ratones hembra que sobreexpresan prolactina no muestran modificaciones en el peso. Por otro lado, en nuestro laboratorio hemos descripto que los ratones hembra carentes del receptor de dopamina D2 en todo su organismo ( Drd2-/ - ) presentan una hiperprolactinemia crónica e hiperplasia hipofisaria, pero con pesos corporales y niveles de ingesta similares a los ratones salvajes. Cabe destacar, que el ratón Drd2-/ - no resultaría un modelo óptimo para el estudio de los efectos de altos niveles de prolactina en el balance energético, dada la importancia fundamental de los receptores de dopamina D2 (RD2s) centrales en las adicciones y los mecanismos de recompensa relacionados al comportamiento alimenticio. La evidencia disponible sugiere que la falta de RD2s centrales en el ratón Drd2-/ - podría estar activando mecanismos compensatorios para limitar finalmente la ingesta de alimento en este modelo hiperprolactinémico. Por lo tanto, nuestra hipótesis sostiene que la deleción específica de los RD2s solamente en lactotropos nos permitiría estudiar los efectos de altos niveles de prolactina en la ingesta y adiposidad, sin los efectos solapados, fisiológicos y patofisiológicos, presentes en el modelo mutante knockout global. Para ello usamos ratones carentes de los RD2s solamente en lactotropos (lacDrd2KO) generados por la tecnología Cre LoxP. Las hembras presentaron hiperprolactinemia crónica e hiperplasia hipofisaria, relacionada a un aumento en la proliferación celular y angiogénesis, resultando un modelo ideal para el estudio de prolactinomas resistentes a agonistas dopaminérgicos. Presentan, por otra parte, un eje de GH conservado. En los ratones hembra lacDrd2KO, pero no los machos, la hiperprolactinemia se asoció a un incremento en el peso corporal relacionado a mayores los niveles de ingesta. En cuanto a la regulación de la ingesta, en el hipotálamo observamos una expresión incrementada del péptido orexigénico Npy , mientras que los niveles de Pomc y Ppo no se vieron alterados (en contraste con los resultados previamente obtenidos en los ratones Drd2-/ - ). Determinamos a su vez, una posible resistencia a la leptina. Es decir que en este nuevo contexto, pudimos determinar un efecto orexigénico de la prolactina, mediado por una acción hipotalámica sobre la expresión del neuropéptido Npy . Se evidenció, además, un incremento significativo en los depósitos de grasa, tamaño de los adipocitos, triglicéridos y ácidos grasos no esterificados séricos, como consecuencia de una disminución en la expresión de enzimas lipolíticas en el tejido adiposo. A nivel hepático, el ratón hembra lacDrd2KO también presentó adiposidad incrementada, con aumento en los triglicéridos hepáticos, que no se relacionó a cambios en la expresión de enzimas lipogénicas/ lipolíticas, pero sí al aumento de factores de transcripción que estimulan la lipogénesis mediada por glucosa. A su vez, los ratones hembra lacDrd2KO presentaron una intolerancia a la glucosa y deficiencia en la liberación de insulina, sin resistencia a la misma. Nuestros resultados revelan un rol fundamental de la prolactina en la ingesta y adiposidad e ilustran el valor de estudiar modelos transgénicos tejido específicos, para evaluar mecanismos patofisiológicos, de otra manera enmascarados en mutantes nulos o animales tratados con agentes farmacológicos. Palabras clave: Receptor de dopamina tipo 2 Prolactina I ngesta Adiposidad Homeostasis de la glucosa SELECTIVE DISRUPTION OF DOPAMINE D2 RECEPTORS IN PITUITARY LACTOTROPES REVEALS A ROLE OF PROLACTIN IN METABOLISM AND FOOD INTAKE REGULATION. Prolactin is a pleiotropic hormone secreted by lactotropes in the anterior pituitary gland. Although its role in reproduction and fertility has been extensively studied, less is known about its action on metabolism and body weight regulation. The high prolactin levels typically observed in pregnant and lactating females contribute to a hyperphagic state. During pregnancy, the normal homeostatic mechanisms regulating appetite are modified to generate a state of positive energy balance, increasing food intake and fat storage to supply the growing fetus with its energy requirements and to be used during lactation. Consistent with the hypothesis that prolactin has a significant role in the regulation of body weight, prolactin receptor deficient mice exhibit lower body weight and reduced fat mass, prolactin administration stimulates food intake, and chronically elevated prolactin levels, such as those seen during pregnancy, increase food intake, probably by inducing a state of leptin resistance. However, male mice bearing ectopic pituitary glands show a small increase in body weight with a decline in fat mass, and female mice overexpressing prolactin do not show greater body weight. Furthermore, we found that female mice lacking dopamine D2 receptors ( Drd2-/ - ) exhibit chronic hyperprolactinemia and pituitary lactotrope hyperplasia but have body weight similar to that of wild-type females in adulthood as well as food intake. However, Drd2-/ - mice are not an optimal model to study the effects of chronic hyperprolactinemia on energy balance, given the fundamental importance of central dopamine D2 receptors (D2Rs) in addiction and reward mechanisms related to feeding behavior. The available evidence suggests that lack of central D2Rs in the Drd2-/ - model might activate compensatory mechanisms that could ultimately limit food intake in this hyperprolactinemic transgenic model. Therefore, we hypothesize that limiting Drd2 ablation specifically in lactotropes would allow us to study the effect of high prolactin secretion on food intake and adiposity, without the confounding physiological and pathophysiological mechanisms present in global null allele Drd2 mutants. We used conditional mutant mice lacking D2Rs in pituitary lactotropes (lacDrd2KO) performed by Cre loxP technology. LacDrd2KO female mice exhibited chronic hyperprolactinemia and pituitary hyperplasia related to an increase in proliferation and angiogenesis, being an excellent animal model for the study of dopamine resistant prolactinomas. They also present a preserved GH axis. I n lacDrd2KO female, but not male mice, hyperprolactinemia was associated with increased body weight and food intake. Hypothalamic Npy mRNA expression was increased, while Pomc and Ppo mRNA levels were unaltered (in contrast to results in global D2R knockout mice). I n addition, a state of leptin resistance was shown. Thus, the orexigenic effect of prolactin and its action on hypothalamic Npy expression were fully evidenced, leading to increased food intake. Marked increments in fat depots, adipocyte size, serum triglycerides, and nonesterified fatty acid levels were seen as a consequence of a decrease in lipolytic enzymes in adipose tissue. On the other hand, the liver of female lacDrd2KO mice had increased lipid content as indicated by oil red staining and triglyceride content, which was not related to changes in lipogenic/ lipolytic enzyme expression, but with an increase in mRNA expression of lipogenic transcription factor regulated by glucose. Furthermore, lacDrd2KO female mice had glucose intolerance, deficient insulin secretion but a preserved response to it. Our results highlight an important role of prolactin in the regulation of food intake and adiposity and illustrate the value of studying cell-specific mutant mice to disentangle the pathophysiological mechanisms otherwise masked in null allele mutants or in animals treated with pervasive pharmacological agents. Key words: Dopamine receptor type 2 Prolactin Food intake Adiposity Glucose homeostasis AGRADECIMIENTOS Y acá estoy, aunque todavía no lo puedo creer, en la etapa final de mi tesis. Porque uno siempre deja para el final final la parte de los agradecimientos, después de haberla chequeado, corregido y releído unas mil veces, y cuando ya está lista para mandar a imprimir. Lo único que tengo bien claro en este momento es que si llegué hasta acá se lo debo a muchísimas personas, a las cuales les estoy enormemente agradecida. A Dama, gracias por haberme enseñado tanto en todo este tiempo, y por haberme acompañado paso a paso en mi crecimiento. Todo lo que soy hoy, como profesional, te lo debo a vos. Gracias por tus consejos, tus guías, tus correcciones, pero sobre todo por la buena onda y apoyo incondicional. Fueron unos hermosos 8 años compartidos que nunca voy a olvidar!!! Gracias a todos los becucuevinos, los de hoy y los de antes! Todos y cada uno de Uds. hicieron que el día a día en el laboratorio sea especial, lleno de risas, charlas, complicidades, anécdotas y alguna que otra frase célebre. Muchos se convirtieron en valiosas amistades que voy a llevar conmigo para siempre. Algunas que son inmunes a las distancias (Mari, Vickota y Claris) y que siempre van a ser buenas excusas y motivos para recorrer nuevas partes del mundo. Y otras que generaron clásicos obligados, como la - o mejor dicho LAS - cervecitaS de Cossab (Ceci y Laris). A Ani, una gran gran amiga, gracias por todooo!!!! Te quiero muchísimo! Sos una persona muy especial para mí. Gracias por tanto apoyo y tanta ayuda incondicional a lo largo de todos estos años. Gracias por los consejos laborales, pero sobre todo por todos los consejos y charlas de la vida, que obviamente seguiremos teniendo… ya te voy a invitar a tomar mate bajo la sombra de mi palo borracho, jaja! Feli, qué te puedo decir? Gracias por haber llegado a mi vida!!!!!! No sólo porque me diste una mano fundamental para poder llegar a tiempo y terminar esta tesis, sino porque en vos encontré una amiga incondicional, que llenó de luz y risas el laboratorio. No me alcanzan las palabras! Gracias por todos y cada uno de los momentos compartidos! Te quiero muchísimo!!! Y te voy a extrañar en cada instante! A Gra y cada una de las integrantes de Graceland, gracias por esos almuerzos y cafecitos reconfortantes, totalmente necesarios para poder seguir adelante! Por escuchar los problemas de cada día y por saber siempre decir la palabra justa. Gracias por compartir la vida!! A los libertunes, gracias compartir mucho más que reactivos y consejos científicos, por la buena onda y por estar siempre dispuestos a dar una mano y a prestar una oreja. Gracias a mi familia, la de siempre y la más reciente. A mis viejos, Moni y Henry, gracias por haberme enseñado y transmitido el amor por la ciencia, y principalmente la pasión por hacerme y responder preguntas. Gracias por demostrarme que uno puede aportar granitos de arena (por más que sean muy muy pequeños) en el descubrimiento de este maravilloso universo. Siempre voy a estar agradecida por los consejos científicos y de la vida, por estar presentes en cada paso que di y por el apoyo incondicional. A mis hermanas, Vicky y Euge, gracias por esas risas interminables de cosas que sólo entendemos nosotras con sólo mirarnos, por la complicidad eterna y por elegir cada día ser parte y compartir nuestras vidas. Por supuesto gracias a mi Nony, Tía Elsa, Abuela Susana, mis tíos, tías, Pablo y cada uno de mis primos, por estar siempre! A todos los integrantes de mi nueva familia: Mirta, Irma, Oscar, Graciela, Claudio, Ale, Rita, Mari, Charly y los niños (Martín, Isa, Tati y Agus). Gracias por haberme aceptado y adoptado como parte de la familia. Por todos los momentos compartidos, los ya clásicos viernes a la noche y los domingos al mediodía. Y especialmente a los chicos, gracias por enseñarme cada día a ser tía (un mundo nuevo del que sigo aprendiendo, jaja). Gracias a mis amigas incondicionales: - a las de siempre, mis santafesinas queridas. Gracias por estar siempre presentes, cada una a su manera, y por crecer a la par. Porque juntas aprendimos que las verdaderas amigas duran más allá de las distancias y los caminos elegidos. Las quiero!!! - al grupete del GYM (o grupo B, jajaja). Gracias por las risas, las anécdotas que quedarán siempre en el recuerdo, los chusmeríos, y por ser un buen motivo para ir al gym, jaja… gracias gracias gracias! - a las chicas cossaberas. Gracias por esas Belgas, Escocesas, I.P.As, etc. etc. etc., totalmente fundamentales y necesarias entre-semana!!! Gracias por las descargas, los consejos, el cable a tierra, las risas y más risas. Porque siempre hay una buena excusa para brindar: para ahogar penas, descargar tensiones, festejar… y por todos los brindis que se vendrán de ahora en adelante!!! - a las caches (vamos que quedamos las mejores!! Jaja). Gracias por todos y cada uno de los momentos compartidos. Por estar presentes SIEMPRE! La vida nos cruzó y nosotras nos encargamos de construir una hermosa amistad verdadera. Las quiero con el alma! - a las vertebratas (más recientemente Tías Vertebratas), por tantos años compartidos, experiencias, crecimientos personales… por estar presentes y acompañarnos en los buenos y malos momentos. Las quiero mis pequeñas haikos! - a Nanu… qué decirte amiga! Nada va a ser suficiente. Gracias, gracias y más gracias! Te adoro con el alma. Sos la mejor amiga que uno puede tener. Gracias por tantas cosas vividas, por estar siempre y crecer juntas! Y finalmente, gracias a Tito, la personita más importante de mi vida, con el que recorrí cada día de este doctorado. Gracias por el apoyo incondicional (sólo vos sabes lo que eso significa), por las palabras alentadoras en el momento indicado, los consejos, las fuerzas transmitidas, la paciencia, las noches en vela. GRACIAS POR TODO! Por ayudarme a ser mejor persona cada día y por la hermosa familia que estamos construyendo… y obviamente, no puedo dejar de mencionar al integrante más pequeño, que llegó para cambiar nuestras vidas, a mi rubio hermoso Ramón. Los amo! Siempre se las ingenian para robarme una sonrisa. Gracias a todos y cada uno de Uds. por ayudarme, cada uno a su manera, a hacer esto posible! ဣMe pregunto si las estrellas se iluminan con el fin de que algún día, cada uno pueda encontrar la suyaဤ El Principito A mi mejor mitad LOS RESULTADOS PRESENTADOS EN ESTA TESIS FORMAN PARTE DE LAS SIGUIENTES PUBLICACIONES: 1. Pituitar a d rai dopa i e D re eptors regulate li er ge e se ual di orphis . María Cecilia Ramírez, Ana María Ornstein, Guillermina María Luque, María Inés Pérez Millán, Isabel García Tornadu, Marcelo Rubinstein, Damasia Becu-Villalobos. Endocrinology (2014) En prensa. 2. ANGIOGENE“I“ IN PITUITARY ADENOMA“. Hu a studies a d e uta t ouse odels . Cristina Carolina, Luque Guillermina María, Demarchi Gianina, López Vicchi Felicitas, Zubeldia Brenner Lautaro, Pérez Millán María Inés, Perrone Sofía, Ornstein Ana María, Lacau-Mengido Isabel, Berner Silvia Inés, Becu-Villalobos Damasia. International Journal of Endocrinology (2014). http://dx.doi.org/10.1155/2014/608497 3. “ele ti e disruptio of dopa i e D re eptors i pituitar la totropes i reases od eight a d adiposit i fe ale i e . Pérez-Millán M. Inés*, Luque Guillermina M.*, Ramírez M. Cecilia, Noain Daniela, Ornstein Ana María, Marcelo Rubinstein*, Damasia Becú-Villalobos*. (* equal contribution). Endocrinology (2014) 155(3):829-39. COMENTADO EN NEWS AND VIEWS DE ENDOCRINOLOGY http://press.endocrine.org/doi/pdf/10.1210/en.2013-2167 SELECCIONADO PARA APARECER EN EL CURRENT ISSUE http://press.endocrine.org/journal/endo 4. Ce tral dopa i e D re eptors o trol od gro th a d ale-to-male social and territorial behavior by a neuroendocrine-e o ri e as ade . Daniela Noaín*, M. Inés Pérez-Millán*, Estefanía P. Bello, Guillermina M. Luque, Rodrigo Casas Cordero, Diego M. Gelman, Marcela Peper, Isabel García Tornadu, Malcolm J. Low, Damasia Becú-Villalobos, Marcelo Rubinstein. (* equal contribution). The Journal of Neuroscience (2013) 33(13):5834-5842. 5. El re eptor dopa i érgi o D : a io es e do ri as o lási as . Isabel García Tornadu, María Victoria Recouvreux, María Cecilia Ramírez, Guillermina María Luque, María Inés Perez-Millan, Rodrigo Lorenzo, María Cristina Camilletti, Ana María Ornstein, Isabel Lacau-Mengido, Carolina Cristina, Graciela Díaz-Torga, Damasia Becu-Villalobos. Acta Bioquímica Clínica Latinoamericana (2011) 45 (4): 599-719. Esta tesis se realizó con el apoyo de CONICET y ANPCyT CONTENIDO ABREVIATURAS UTILIZADAS.............................................................................................. 1 INTRODUCCIÓN GENERAL ................................................................................................. 5 Sistema hipotálamo-hipofisario .................................................................................... 5 Hipotálamo .......................................................................................... 5 Centro regulador de la secreción hipofisaria ............................................................. 7 Centro regulador de la ingesta y metabolismo .......................................................... 8 Núcleos hipotalámicos que participan en la ingesta ........................................... 8 Señales periféricas: Tejido adiposo ................................................................... 11 Hipófisis ............................................................................................. 12 Prolactina ................................................................................................................. 15 Receptores de prolactina .................................................................................. 16 Prolactina: hormona metabólica ....................................................................... 18 Hormona de crecimiento ......................................................................................... 19 Hormona de crecimiento: hormona metabólica ............................................... 20 Alteraciones en la secreción de prolactina y GH: adenomas hipofisarios ................ 21 El receptor de dopamina tipo 2 (RD2) .......................................................................... 22 RD2 en cerebro ................................................................................... 22 RD2 hipofisario ................................................................................... 23 RD2: funciones endócrinas y metabólicas ............................................. 23 Dopamina y regulación de la prolactina .................................................................. 23 Dopamina y regulación de GH-GHRH ....................................................................... 24 Dopamina y péptidos relacionados con la ingesta ................................................... 25 Dopamina y metabolismo de la glucosa .................................................................. 25 HIPÓTESIS ....................................................................................................................... 27 OBJETIVOS GENERALES ................................................................................................... 28 MATERIALES Y MÉTODOS ............................................................................................... 29 Animales ..................................................................................................................... 29 Ratones lacDrd2KO ............................................................................. 29 Genotipificación de los ratones ............................................................................... 30 Extracción de ADN genómico ............................................................................ 30 Reacción en cadena de la polimerasa (PCR)...................................................... 30 Ingesta ........................................................................................................................ 33 Curvas de peso corporal .............................................................................................. 33 Talla corporal .............................................................................................................. 33 Monitoreo del ciclo estral ........................................................................................... 33 Test de tolerancia a la glucosa (GTT) ........................................................................... 34 Secreción de insulina estimulada por glucosa (GSIS) .................................................... 34 Test de tolerancia a la insulina (ITT) ............................................................................ 34 Evaluación del modelo homeostático (HOMA) ............................................................ 35 HOMA-IR ............................................................................................ 35 HOMA-β ell ........................................................................................ 35 Test de sensibilidad a leptina ...................................................................................... 35 Ayuno y realimentación .............................................................................................. 36 Recolección de muestras ............................................................................................. 36 Peso de tejidos ............................................................................................................ 37 Perfil sérico de lípidos y adipoquinas........................................................................... 37 Extracción de ARN y síntesis de ADNc ......................................................................... 37 Diseño de primers ............................................................................... 38 PCR en tiempo real ............................................................................. 40 Histología .................................................................................................................... 41 Procesamiento de muestras ................................................................. 41 Coloraciones ...................................................................................... 42 Inmunohistoquímica (IHQ) ................................................................... 43 Evaluación de parámetros angiogénicos.................................................................. 44 Evaluación de parámetros de proliferación ............................................................. 45 Evaluación de parámetros endócrinos..................................................................... 46 Hipófisis............................................................................................................. 46 Páncreas ............................................................................................................ 47 Evaluación de parámetros de stemness ................................................................... 49 Inmunofluorescencia ........................................................................... 51 Citometría de flujo ...................................................................................................... 53 Protocolo de dispersión de células con colagenasa ................................................. 53 Marcación para CD45/CD31/CD44 .......................................................................... 54 Radioinmunoensayos (RIAs) ........................................................................................ 55 Niveles séricos de hormonas ................................................................ 55 Prolactina .......................................................................................................... 55 IGF-I ................................................................................................................... 55 Progesterona..................................................................................................... 56 Niveles intrahipofisarios de hormonas .................................................. 57 Prolactina .......................................................................................................... 57 GH ..................................................................................................................... 57 Niveles intrapancreáticos de insulina ................................................... 57 ELISA para VEGF .......................................................................................................... 58 Medición de glucógeno hepático ................................................................................. 58 Análisis estadístico ...................................................................................................... 59 CAPÍTULO I – EL RATÓN lacDrd2KO COMO MODELO EXPERIMENTAL DE PROLACTINOMA RESISTENTE A DOPAMINA. ....................................................................................................... 61 Introducción ............................................................................................................... 61 Prolactinomas .................................................................................... 61 Modelos experimentales de prolactinoma .............................................................. 63 Caracterización del ratón knockout total Drd2-/- ............................................... 63 Generación y validación de un nuevo modelo experimental: el ratón lacDrd2KO .................................................................................................................................... 64 Posibles mecanismos tumorigénicos en lactotropos ............................................... 65 Angiogénesis ..................................................................................................... 66 Factor de crecimiento del endotelio vascular (VEGF) ........................... 68 VEGF en hipófisis .......................................................................... 69 Células madre o stem cells ................................................................................ 69 Células madre en hipófisis .................................................................... 70 Células madre tumorales o cancer stem cells (CSC) ............................. 74 Vías de señalización y factores en CSC ........................................ 75 CSC en hipófisis ............................................................................ 75 Objetivos .................................................................................................................... 77 Resultados .................................................................................................................. 78 Validación del modelo ......................................................................... 78 Hiperplasia hipofisaria ............................................................................................. 78 Eje de crecimiento conservado ........................................................................... 79 Ciclo estral ............................................................................................................. 81 Prolactinoma experimental resistente .................................................. 82 Angiogénesis y proliferación ............................................................................... 82 Marcadores de células madre ............................................................................. 83 Discusión .................................................................................................................... 88 CAPÍTULO II – ESTUDIO DE LA INGESTA EN RATONES HEMBRA lacDrd2KO. ..................... 93 Introducción ............................................................................................................... 93 Obesidad ............................................................................................ 93 Señales involucradas en la ingesta ....................................................... 94 Señales centrales ..................................................................................................... 95 Dopamina .......................................................................................................... 95 POMC y α-MSH ................................................................................................. 95 NPY .................................................................................................................... 97 Orexinas - hipocretinas ..................................................................................... 98 Señales periféricas ................................................................................................... 99 Leptina .............................................................................................................. 99 Resistencia a leptina ................................................................................ 100 Caracterización de los mecanismos que regulan la ingesta en el ratón knockout total Drd2 - / - ................................................................................ 102 Objetivos .................................................................................................................. 104 Resultados .............................................................................................................. 105 Ingesta y peso corporal ..................................................................... 105 Leptina .................................................................................................................. 107 Factores orexigénicos y anorexigénicos .......................................................... 110 Machos ................................................................................................................. 111 Discusión .................................................................................................................. 113 CAPÍTULO III – REGULACIÓN DEL METABOLISMO DE LA GLUCOSA EN RATONES HEMBRA lacDrd2KO. ............................................................................................................................. 117 Introducción ............................................................................................................. 117 Homeostasis de la glucosa ................................................................. 117 Mecanismos de regulación .................................................................................... 117 Glucagón ......................................................................................................... 117 Insulina ............................................................................................................ 118 Hormonas hipofisarias .................................................................................... 119 Evaluación del modelo homeostático (HOMA) ...................................................... 120 Obesidad: factor de riesgo para el desarrollo de diabetes .................................... 120 Caracterización delmetabolismo de la glucosa en el ratón knockout total Drd2 - / - ...................................................................................................... 121 Objetivos ................................................................................................................ 122 Resultados .............................................................................................................. 123 Homeostasis de la glucosa alterada .................................................... 123 Machos ................................................................................................................. 131 Discusión .................................................................................................................. 133 CAPÍTULO IV – REGULACIÓN DE LA LIPOGÉNESIS EN RATONES HEMBRA lacDrd2KO. .... 135 Introducción ............................................................................................................. 135 Tejido adiposo blanco ....................................................................... 135 Adipogénesis .......................................................................................................... 135 Lipólisis y lipogénesis ............................................................................................. 136 Prolactina y tejido adiposo .............................................................................. 138 Hígado ............................................................................................. 138 Lipólisis y lipogénesis ............................................................................................. 138 Prolactina e hígado ......................................................................................... 140 Glucocorticoides ............................................................................... 140 Objetivos ................................................................................................................ 142 Resultados .............................................................................................................. 143 Adiposidad incrementada .................................................................. 143 Tejido adiposo gonadal ..................................................................... 145 Lipólisis / Lipogénesis ........................................................................................ 145 Hígado ............................................................................................. 151 Lipólisis / Lipogénesis ........................................................................................ 151 Glucocorticoides ............................................................................... 157 Discusión .................................................................................................................. 159 DISCUSIÓN GENERAL .................................................................................................... 165 BIBLIOGRAFÍA ............................................................................................................... 171 ABREVIATURAS UTILIZADAS ABC Reactivo streptavidina-biotina peroxidasa AC Adenilato ciclasa ACTH Hormona adrenocorticotrófica ADH Hormona antidiurética ADN Ácido desoxirribonucleico AgRP Péptido relacionado a Agouti AMPc Adenosina monofosfato cíclico AMR Anova de medidas repetidas ANOVA Análisis de la varianza APC Aloficocianina Arc Núcleo arcuato ARNm Ácido ribonucleico mensajero ATGL Adipose tissue triacylglycerol lipase (lipasa adiposa de triglicéridos) BMI Body mass index (índice de masa corporal) BSA Albúmina de suero bovino CART Cocaine and Amphetamine-Regulated Transcript (transcripto regulado por cocaína-anfetamina) CD31 (PECAM) Cluster of differentiation molecule Chrebp Proteína de unión al elemento de respuesta a carbohidratos COOH Carboxilo Cpm Cuentas por millón CRH Hormona liberadora de corticotropina CSC Cancer stem cells (células madre tumorales) D2L Receptor de dopamina tipo 2 isoforma larga D2S Receptor de dopamina tipo 2 isoforma corta DA Dopamina DAB Diaminobencidina DAG Diacilglicerol DMN Núcleo dorsomedial DMV Densidad microvascular Drd2 -/- Ratón knockout total para el receptor D2 Drd2 loxP/loxP Ratón con el exón 2 del RD2 flanqueado por secuencias LoxP DTT Ditiotreitol E.S.M. Error estándar de la media 1 EA Albúmina de huevo ELISA Ensayo por inmunoabsorción ligado a enzimas EMT (Epithelial–mesenchymal transition) transición epitelio mesenquimal FACs Fluorescence-activated cell sorting (clasificación de células activadas por fluorescencia) FAS Fatty acids synthase (ácido graso sintasa) FFAs Ácidos grasos libres FGF-2 Factor de crecimiento fibroblástico 2 FITC Isotiocianato de fluoresceína FSH Hormona folículo estimulante GABA Ácido gamma-aminobutírico GH Hormona de crecimiento (somatotropina) GHR Receptor de hormona de crecimiento GHRH Hormona liberadora de somatotropina GnRH Hormona liberadora de gonadotrofina GR Receptor de glucocorticoides GSIS Glucose stimulated insulin secretion (liberación de insulina estimulada por glucosa) GTT Glucose tolerance test (test de tolerancia a la glucosa) Gys2 Glucógeno sintasa 2 HES-1 Hairy and enhancer of split-1 HMG High mobility group (grupo de alta movilidad) HOMA Homeostasis model assessment (modelo homeostático) HSL Hormone-sensitive lipase (lipasa sensible a hormonas) Igfbp3 La proteína 3 de unión al factor de crecimiento insulina-símil IGF-I Factor de crecimiento tipo insulina 1 IgG Inmunoglobulina G IHQ Inmunohistoquímica Ip Intraperitoneal ITT Insulin tolerance test (test de tolerancia a la insulina) Kb Kilobases KDa Kilodaltons lacDrd2KO Ratón knockout para el RD2 solamente en lactotropos LDL Low-density lipoprotein (lipo-proteína de baja densidad) L-DOPA Levodopa (L-3,4 dihidroxifenilalanina) LH Hormona luteinizante LHA Hipotálamo lateral 2 LPL Lipoprotein lipase (lipo-proteína lipasa) MC3R Receptor de melanocortinas tipo 3 MC4R Receptor de melanocortinas tipo 4 MCH Hormona concentradora de melanina mM Milimolar MP Main population (población principal) MZ Marginal zone (zona marginal) NEFA Ácidos grasos no esterificados NGF Factor de crecimiento neural NICD Notch intracellular domain (dominio intracelular de Notch) NIH National Institute of Health, Maryland, Estados Unidos NPY Neuropéptido Y Obrb Receptor de leptina OXA Orexinas tipo A OXB Orexinas tipo B PBS Buffer fosfato PBS-T Buffer fosfato tween PCNA Antígeno nuclear de células en proliferación PCR Reacción en cadena de la polimerasa PE Ficoeritrina PECAM (CD31) Molécula de adhesión endotelial PEPCK Fosfoenolpiruvato carboxi-quinasa Pit-1 Factor de transcripción específico de hipófisis 1 PKA Proteína quinasa A PL Lactógeno placentario PlGF Factor de crecimiento de la placenta POMC Propiomelanocortina PPARγ Peroxisome proliferator-activated receptor gamma (receptor gamma activado por el factor proliferador de peroxisomas) PRL Prolactina PRLR Receptor de prolactina PRLR-L Receptor de prolactina isoforma larga PRLR-S1 Receptor de prolactina isoforma corta 1 PRLR-S2 Receptor de prolactina isoforma corta 2 PRLR-S3 Receptor de prolactina isoforma corta 3 PTTG Pituitary tumor transforming gene (gen transformante del tumor pituitario) PVN Núcleo paraventricular 3 RD1 Receptor de dopamina 1 RD2 Receptor de dopamina 2 RD3 Receptor de dopamina 3 RD4 Receptor de dopamina 4 RD5 Receptor de dopamina 5 rhGH Hormona de crecimiento recombinante humana RIA Radioinmunoensayo Rpm Revoluciones por minuto RT-PCR Reacción en cadena de la polimerasa con transcriptasa inversa SFB Suero fetal bovino SOCS Suppressors of cytokine signaling (proteína supresora de la señalización por citoquinas) SOX-2 Sex determining region Y-box 2 SP Side population (población lateral) Srebf1 Sterol regulatory element-binding transcription factor 1 (factor de transcripción 1 de union al elemento de respuesta a esteroles) STAT Signal transducer and activator of transcription (Transductor de señal y activador de la transcripción) STT Somatostatina TG Triglicéridos TGF-α Factor de crecimiento transformante alfa TIDA Neuronas tuberoinfundibular dopaminérgico TRH Hormona liberadora de tirotropina TSH Tirotropina VEGF Factor de crecimiento de endotelio vascular VEGFR-1 Receptor de VEGF 1 VEGFR-2 Receptor de VEGF 2 VIP Péptido vasoactivo intestinal VLDL Very low-density lipoprotein (lipo-proteína de muy baja densidad) VMN Núcleo ventromedial Vs Versus WT Tipo salvaje α-MSH Hormona melanocito estimulante alfa 4 Introducción INTRODUCCIÓN GENERAL SISTEMA HIPOTÁLAMO-HIPOFISARIO Los organismos pluricelulares complejos requieren sistemas de coordinación que puedan regular e integrar funciones y señales de los diferentes tipos celulares. Existen dos grandes sistemas de coordinación que han evolucionado en gran medida, el sistema nervioso y el sistema endócrino. El primero utiliza señales eléctricas para transmitir muy rápidamente la información entre células; mientras que el segundo utiliza señales químicas, las hormonas. Estas últimas, son producidas por un tipo particular de células y circulan por el torrente sanguíneo hasta alcanzar las células blanco, sobre las que ejercen un efecto regulador a través de su unión a receptores específicos (1). Ambos sistemas, nervioso y endócrino, se encuentran estrechamente relacionados. Muchas neuronas son capaces de secretar hormonas, lo que se conoce como neurosecreción. El sistema neuroendócrino de vertebrados está conformado por el cerebro y la hipófisis, junto con las glándulas endócrinas que controlan: glándula tiroides, paratiroides, corteza adrenal, gónadas e hígado (1). La porción cerebral que está mayormente involucrada en la bioregulación del sistema neuroendócrino es el hipotálamo. Grupos específicos de neuronas hipotalámicas neurosecretoras producen una gran variedad de neurohormonas, las cuales son secretadas a los vasos sanguíneos portales‐hipofisarios especializados. Algunas de estas neurohormonas controlan la secreción de péptidos y hormonas proteicas por parte de la glándula pituitaria (hipófisis), las cuales regulan la actividad de glándulas endócrinas (2), e influyen en aspectos generales del crecimiento, metabolismo y reproducción, afectando muchos tejidos no-endócrinos (1). De esta manera se conforma el eje hipotálamo-hipofisario. HIPOTÁLAMO Durante el desarrollo embrionario en el cerebro de los vertebrados pueden distinguirse cuatro regiones: el telencéfalo (más anterior), diencéfalo, mesencéfalo y romboencéfalo (más posterior). El telencéfalo y diencéfalo comprenden el lóbulo frontal, mientras que el mesencéfalo y romboencéfalo comprenden el cerebro medio y posterior respectivamente. Durante el desarrollo, cada una de estas regiones se diferencia en distintos componentes del cerebro adulto (1) (Figura 1). 5 Introducción Figura 1: Esquema del cerebro de mamíferos. Tomado y modificado de (1). El hipotálamo es de origen diencefálico, y se extiende bajo el tálamo comprendiendo la zona gris que rodea ventralmente al tercer ventrículo. En la porción ventral adquiere una disposición infundibular que se continúa con el tallo hipofisario finalizando en el lóbulo neural (3). Dentro de estos límites se distribuyen las neuronas hipotalámicas, constituyendo núcleos bien definidos con variaciones según la especie, sexo, edad, entre otros factores. El hipotálamo puede dividirse en cuatro regiones principales, organizadas rostro‐caudalmente: área preóptica, hipotálamo anterior, hipotálamo medio e hipotálamo posterior (4). Los núcleos más relevantes de las mismas son: Área preóptica: el núcleo preóptico medial, con neuronas secretoras de la hormona liberadora de gonadotrofinas (GnRH) las cuales proyectan hacia la eminencia media en la mayoría de los mamíferos. Aunque embriológicamente el área preóptica no forma parte del hipotálamo, dado que se origina en el telencéfalo, funcional y topográficamente se considera parte del hipotálamo neuroendócrino. Hipotálamo anterior: el núcleo periventricular (proyecta hacia la eminencia media), el supraóptico y el paraventricular (PVN) (proyectan fundamentalmente hacia el lóbulo neural) y el supraquiasmático (reloj biológico para las regulaciones neuroendócrinas). Hipotálamo medio: en él se reconocen tres partes, el hipotálamo medio basal, el dorsal y el lateral (LHA). Esta región incluye a la eminencia media y a los núcleos arcuato (Arc), ventromedial (VMN) y dorsomedial (DMN) que proyectan hacia la misma. Hipotálamo posterior: comprende los cuerpos mamilares. El núcleo premamilar ventral proyecta hacia la eminencia media. El hipotálamo presenta una compleja regulación de la actividad neuronal de sus núcleos para poder llevar a cabo el procesamiento de la información entrante. En ella intervienen neuronas 6 Introducción que actúan mediante una gran diversidad de neurotransmisores: aminérgicos, colinérgicos, serotoninérgicos, opiáceos, peptidérgicos, GABAérgicos, aminoacídicos, etc (5). Las conexiones neurales del hipotálamo son múltiples y complejas, siendo en su mayoría recíprocas formando circuitos dentro de los núcleos hipotalámicos, entre núcleos y áreas hipotalámicas, como también entre regiones hipotalámicas y diferentes regiones extra- hipotalámicas (4;6). El hipotálamo es el centro neuroendócrino del cerebro, y controla diversos procesos homeostáticos como ser la ingesta de alimentos y bebidas, la reproducción, lactancia, función cardiovascular, control metabólico, termo-regulación, ciclos de sueño y vigilia, como también la secreción hormonal (7). Centro regulador de la secreción hipofisaria El hipotálamo reúne e integra señales de diversos orígenes, estímulos recibidos de los medios externo e interno, y envía señales precisas (neurohormonas) hacia la hipófisis (6;8). Algunas neuronas hipotalámicas responden liberando, de manera pulsátil, diferentes neurohormonas que regulan la síntesis, almacenamiento y secreción de las hormonas tróficas adenohipofisarias. Estas neurohormonas se denominan hormonas liberadoras o inhibitorias según su propiedad de estimular o inhibir la actividad secretoria de la hipófisis respectivamente. La mayoría de estas neurohormonas son oligo- o polipéptidos sintetizados en núcleos hipotalámicos discretos (8;9), algunos de los cuales también actúan como neurotransmisores o neuromoduladores en el sistema nervioso central. En una misma neurona o núcleo pueden sintetizarse y coexistir diferentes tipos de neuropéptidos. Las principales neurohormonas liberadas por el hipotálamo son: la hormona liberadora de corticotropina (CRH), GnRH, la hormona liberadora de hormona de crecimiento (GHRH), la hormona inhibitoria de la liberación de hormona de crecimiento (somatostatina – STT), la hormona liberadora de tirotropina (TRH), la oxitocina, la vasopresina, y la dopamina (DA) inhibidora de la liberación de prolactina. Por otro lado, existe un número creciente de péptidos secretados por el hipotálamo que son liberados junto a las neurohormonas y poseen receptores en la hipófisis capaces de modular la secreción hormonal, tales como la angiotensina I I , la sustancia P, el neuropéptido Y (NPY), el péptido intestinal vasoactivo (VI P), entre otros. Las neurohormonas sintetizadas en los cuerpos neuronales del hipotálamo llegan a las terminales axonales en la eminencia media y son vertidas en el plexo capilar primario formado por la arteria hipofisaria superior (base del hipotálamo). Los capilares del interior de la eminencia media convergen para formar una serie de vasos portales que transportan sangre directamente del tejido neurosecretor del hipotálamo al tejido glandular de la adenohipófisis. Una vez en la glándula estos vasos forman una red de capilares fenestrados en íntimo contacto con las células glandulares donde vuelcan la secreción. Este sistema llamado “sistema porta- hipofisario largo”, permite una 7 Introducción comunicación fluida y directa entre el hipotálamo y la hipófisis, y así, muy bajas concentraciones de los factores hipotalámicos producen efectos sobre la hipófisis, evitando una posible dilución de los mismos por el sistema circulatorio general (8). Por otro lado, las secreciones hormonales de la glándula son volcadas a este sistema de vasos que vuelven a converger y se acoplan al sistema venoso de la hipófisis. Centro regulador de la ingesta y metabolismo El control cerebral sobre la obtención y el gasto energético permite mantener el peso corporal constante dentro de valores normales. Es por ello que los mamíferos con libre acceso al alimento ( ad libitum ) no suelen desarrollar obesidad, y aun alimentándose con dietas hipercalóricas se disparan respuestas metabólicas de modo de evitar el incremento de peso (como ser, disminución en los niveles de ingesta y/ o aumento de gasto energético) (3). Las primeras evidencias que determinaron la importancia de las neuronas hipotalámicas en el balance energético se obtuvieron en 1940 (10). Hetherington & Ranson demostraron que la destrucción de áreas hipotalámicas particulares: VMN y Arc, desencadenaban hiperfagia y obesidad. Mientras que, opuestamente, lesiones en el hipotálamo LHA provocaban disminución en la ingesta y pérdida de peso corporal. A partir de estos resultados, se originó el concepto de centros hipotalámicos duales: el hipotálamo VMN como centro de la saciedad y el LHA como el centro del apetito. Hoy se conoce que estos circuitos resultan de mayor complejidad, siendo un ejemplo el sistema central de melanocortinas (3) como se explicará a continuación. Núcleos hipotalámicos que participan en la ingesta Las áreas hipotalámicas involucradas en la ingesta son los núcleos Arc, PVN, LHA, VMN y DMN. Numerosos componentes del sistema neuroendórino regulan la ingesta de alimentos y el balance energético (11). En el hipotálamo mediobasal, en particular en el núcleo Arc, existen dos poblaciones neuronales que ejercen efectos importantes en la regulación de la ingesta, gasto energético y homeostasis de la glucosa. Las neuronas que coexpresan el péptido relacionado con Agouti ( Agouti-related peptide – AgRP) y el neuropéptido NPY actúan promoviendo la ingesta (neuronas orexigénicas) e inhibiendo el gasto energético; mientras que de manera contraria, las neuronas que coexpresan propiomelanocortina (POMC) y el transcripto regulado por cocaína-anfetamina ( cocaine- and amphetamine-regulated transcript – CART) actúan inhibiendo la ingesta (neuronas anorexigénicas) y estimulando procesos catabólicos (12). Estas dos poblaciones neuronales hipotalámicas ejercen efectos completamente opuestos más allá de estar físicamente próximas entre sí (3). POMC es una proteína precursora de múltiples péptidos, entre ellos las hormonas melanocito estimulantes alfa y beta (α- y -MSH), las cuales actúan sobre receptores de melanocortinas MC3R 8 Introducción y MC4R activando respuestas anorexigénicas (13). Las neuronas de POMC expresan ambos receptores, MC3R y MC4R. En contraposición, AgRP es un antagonista competitivo de α-MSH por el receptor MC3R, lo que reduce su señal promoviendo la ingesta (14). También se ha propuesto a AgRP como agonista inverso de ambos receptores (MC3R y MC4R) independiente de la presencia de α-MSH (12;15). Como consecuencia, a través de los receptores MC3R y MC4R, α-MSH suprime la ingesta y la ganancia de peso corporal, mientras que AgRP promueve el balance energético positivo (3). NPY actúa sobre sus receptores, Y1 a Y5, presentes en el Arc, PVN y otras áreas hipotalámicas para incrementar la ingesta de alimentos (16). Particularmente, NPY inhibe la liberación (17) y síntesis (18) de α-MSH por parte de las neuronas POMC. Por otra parte, ambas poblaciones neuronales expresan el neurotransmisor inhibitorio GABA (ácido gamma-aminobutírico). Mediante el mismo, las neuronas AgRP/ NPY inhiben a las neuronas POMC modulando el gasto energético (12) El rol crucial del sistema de melanocortinas y de las neuronas AgRP/ NPY y POMC en la regulación de la homeostasis energética está bien establecido, y sus vías de regulación y efectos biológicos ampliamente estudiados (Figura 2). 9 Introducción Figura 2: Regulación de la homeostasis energética mediante las neuronas POMC y AgRP/ NPY a través del sistema de melanocortinas. Las neuronas POMC y AgRP/ NPY se encuentran en el Arc, cercanas a la barrera hemato-encefálica, donde tienen acceso a numerosas señales (como la insulina, leptina y nutrientes). Ambas poblaciones neuronales ejercen potentes efectos sobre el balance energético mediante sus neuropéptidos característicos que modulan las neuronas de segundo orden. α- y -MSH, producidos por las neuronas POMC, actúan sobre sus receptores MC3R y MC4R produciendo la disminución de la ingesta e incrementando al gasto energético. NPY y AgRP actúan como antagonistas de estos receptores provocando la respuesta inversa. Por otro lado las neuronas AgRP/ NPY inhibien la neurona POMC, de forma de potenciar su acción orexigénica. Se describió una inhibición GABAérgica por parte de ambas neuronas, POMC y AgRP/ NPY, sobre las neuronas efectoras, así como también por parte de neuronas AgRP/ NPY sobre POMC. Modificado de (12). Estas neuronas del núcleo Arc proyectan hacia otras áreas hipotalámicas como el PVN y el LHA, donde se sintetizan numerosos neuropéptidos que influencian la ingesta y la ganancia de peso. Neuronas de segundo orden presentes en el PVN sintetizan y secretan compuestos con 10 Introducción efectos anorexigénicos como ser CRH, mientras que desde el LHA se liberan neuropéptidos orexigénicos como ser MCH ( melanin-concentrating hormone) y las orexinas A y B (19). Señales periféricas: Tejido adiposo El cerebro necesita obtener información sobre el estado energético del organismo para así poder responder adecuadamente a las necesidades metabólicas, ya sea a largo plazo (mantenimiento del peso corporal), como también modificando decisiones a corto plazo (inicio de la ingesta, o cantidad de la misma). Existen señales periféricas provenientes de hormonas (insulina, ghrelina), adipoquinas (leptina, adiponectina) y nutrientes (glucosa, ácidos grasos), que informan del estado energético al sistema nervioso central, modificando la actividad metabólica de sus neuronas (12). Dejando de lado los nutrientes, estas señales peptídicas son secretadas por células endócrinas especializadas, las cuales se encuentran principalmente en el tejido adiposo, páncreas, estómago e intestino delgado (3). El tejido adiposo es un órgano ampliamente distribuido en el organismo en diferentes depósitos. Puede localizarse subcutáneamente, alrededor de determinados órganos internos, como también estar asociado a otros tejidos (por ejemplo existen adipocitos alrededor de fibras musculares esqueléticas). Está comprendido principalmente por dos tejidos, el tejido adiposo blanco y el pardo. Mientras que el tejido adiposo pardo participa principalmente en la termogénesis, el tejido adiposo blanco resulta el reservorio energético principal en mamíferos (20). En períodos de abundancia calórica, el tejido adiposo blanco acumula ácidos grasos libres (FFAs) en la forma de triglicéridos (TG), a través de su esterificación a glicerol, y los libera a la circulación en los tiempos de deficiencia energética (21). Mientras que el rol del tejido adiposo como fuente central de energía ha sido reconocido hace cientos de años, recién en el año 1953 Kennedy hipotetizó sobre la presencia de señales lipídicas en circulación que actuarían alterando el gasto energético y la ingesta (22). Futuros estudios que involucraron ratones genéticamente obesos ( ob/ ob ) y diabéticos ( db/ db ) confirmaron la presencia de dichos factores (23). Hoy en día se encuentra ampliamente aceptada la idea del tejido adiposo como un órgano endócrino dinámico, fundamental para la regulación del metabolismo. El adipocito expresa receptores asociados con el control del balance energético y produce numerosas adipoquinas, hormonas y péptidos relacionados a la regulación del apetito. Entre ellos se encuentran adiponectina, resistina, leptina, interleuquina-6, los cuales actúan como reguladores del metabolismo lipídico de manera autocrina/ paracrina (21;24). El eje hipotálamo-tejido adiposo contribuye en gran medida al mantenimiento de la homeostasis energética mediante el control nutricional y los niveles de reservas (Figura 3) (24;25). Una desregulación en la secreción de las adipoquinas por parte del tejido adiposo, produce alteraciones en los niveles de ingesta y gasto energético a través de la acción de las mismas sobre el hipotálamo. En particular, las neuronas de POMC y AgRP/ NPY del Arc están opuestamente 11 Introducción reguladas por leptina (Figura 2). Ambos tipos neuronales expresan tanto los receptores para leptina como para insulina, las cuales actúan reduciendo la expresión y liberación de péptidos hipotalámicos orexigénicos (AgRP/ NPY) y activando los anorexigénicos (α-MSH/ CART) (12;24). El Arc coordina a su vez las áreas hipotalámicas de la saciedad (VMN, DMN y PVN) y del apetito (LHA). Por otro lado, estas áreas hipotalámicas, en especial el PVN modularían, vía sistema nervioso simpático, el gasto energético a través de la estimulación de la lipólisis o termogénesis en el tejido adiposo (Figura 3) (24). Figura 3: Esquema representativo del eje hipotálamo-tejido adiposo involucrado en la regulación de la ingesta y gasto energético. El núcleo Arc integra las señales endócrinas periféricas provenientes del flujo sanguíneo (como ser, leptina). Leptina actúa sobre su receptor específico modulando la expresión y liberación de neuropéptidos del Arc. Estos, controlan otras áreas hipotalámicas, como el núcleo VMN, DMN y PVN, considerados el centro de la saciedad, como también el LHA, considerado el centro del apetito. Los circuitos de comunicación entre las distintas poblaciones neuronales se indican como flechas rojas. La señal neuronal (en especial proveniente del PVN) encargada de regular, vía el sistema nervioso autónomo, el gasto energético por parte del tejido adiposo se indica como flecha verde. Modificado de (24). HIPÓFISIS La hipófisis o glándula pituitaria es una glándula endócrina que regula diversas funciones fisiológicas básicas incluyendo crecimiento, reproducción, y homeostasis metabólica. Se encuentra ubicada ventral al cerebro y posterior al quiasma óptico, sobre el bolsillo o fosa del hueso esfenoides denominada silla turca; y permanece unida al hipotálamo por un tallo o pedículo. La hipófisis de mamíferos está compuesta por dos porciones que interaccionan íntimamente entre sí, 12 Introducción y que difieren en su origen embriológico: la hipófisis posterior o neurohipófisis, y la hipófisis anterior o adenohipófisis. La neurohipófisis deriva del neuroectodermo mientras que la adenohipófisis del llamado placode hipofisario. En ambos casos, el origen es ectodérmico (26). La adenohipófisis posee una estructura epitelio-glandular y puede ser subdivida en tres regiones anatómicas: pars distalis, pars tuberalis, y pars intermedia (ausente o vestigial en humanos). Cada región de la adenohipófisis se distingue por sus características histológicas así como también por su relación con la neurohipófisis, en la cual se distinguen tres regiones: eminencia media, pars nervosa y tallo infundibular siendo este último el que conecta ambas estructuras (1) (Figura 4). Figura 4: Estructura de la hipófisis. Tomado de (2). Las funciones endócrinas de la glándula son llevadas a cabo por diferentes tipos celulares ubicados en la adenohipófisis. Estos se encuentran definidos de acuerdo a la hormona que producen y secretan. La pars distalis está compuesta por somatotropos que producen hormona de crecimiento (GH), lactotropos productores de prolactina (PRL), tirotropos que secretan hormona tiroideo estimulante (TSH), corticotropos productores de hormona adrenocorticotrófica (ACTH) y gonadotropos secretores de hormona luteinizante (LH) y hormona folículo estimulante (FSH) (27). La pars intermedia se encuentra altamente inervada por terminales de neuronas hipotalámicas adrenérgicas, dopaminérgicas y serotoninérgicas, que contactan con células denominadas melan otropos. Estas melanocortinas ( ‐MSH, últimas secretan ‐MSH, CLIP, mayoritariamente α‐MSH, como también otras ‐endorfina). La secreción de α‐MSH es inhibida por las fibras dopaminérgicas provenientes del núcleo Arc que forman parte del tallo neural (8). La función principal de este lóbulo se relaciona con la pigmentación. La pars tuberalis está compuesta principalmente por: gonadotropos, corticotropos y tirotropos. Otro tipo celular presente en la glándula son las células folículo estrelladas, no secretoras de hormonas, pero que sintetizan numerosos factores con funciones autocrinas y paracrinas, entre ellos el factor de crecimiento de endotelio vascular (VEGF). 13 Introducción A diferencia de la adenohipófisis, la neurohipófisis permanece conectada con el hipotálamo a través de un tracto nervioso (el tracto nervioso hipotálamo ‐ hipofisario). La neurohipófisis secreta dos hormonas, la oxitocina y la hormona antidiurética o vasopresina (ADH). Ambas son sintetizadas dentro de los cuerpos celulares de grandes neuronas que se extienden en los núcleos supraóptico y PVN del hipotálamo. Son transportadas unidas a proteínas específicas, a lo largo de los axones de estas neuronas hasta las terminaciones nerviosas que se extienden dentro de la neurohipófisis. Estas hormonas son secretadas a los capilares que irrigan el lóbulo neural como respuesta a los impulsos nerviosos originados en los núcleos supraóptico y PVN (2) (Figura 5). Figura 5: Relación entre hipotálamo e hipófisis. Tomado de (2). En el presente trabajo, nos centraremos principalmente en las células hipofisarias que sintetizan y liberan prolactina, los lactotropos. 14 Introducción Prolactina La prolactina es una hormona polipeptídica que consiste en 197 aminoácidos en rata y ratón, y 199 en humanos, con un peso molecular de 23 kilodaltons (KDa). Presenta múltiples y diversas funciones biológicas, siendo una de las hormonas más versátiles. Es sintetizada y secretada principalmente por las células lactotropas, las cuales se encuentran presentes en la hipófisis anterior (28). En el ser humano esta hormona es también producida por otros tejidos (como ser: tejido adiposo, diferentes regiones del cerebro, miometrio, placenta, decidua, entre otros) donde es regulada de manera específica y actúa como una citoquina (29;30). Sin embargo, generalmente los niveles producidos por tejidos no hipofisarios no modifican las concentraciones séricas de la hormona. Salvo algunas mínimas excepciones, la producción de prolactina por parte de otros animales se encuentra restringida a la hipófisis; tal es el caso de los ratones (30). En el desarrollo, se detecta presencia de prolactina en la hipófisis fetal en el día 16 en el embrión de ratón, día 17 en la rata y semanas 12 a 15 del desarrollo embrionario humano. En el ser humano, el contenido hipofisario de prolactina en el feto aumenta a partir de la mitad de la gestación hasta el fin de la misma, sin existir una diferencia aparente entre sexos. Consecuentemente, los niveles séricos fetales también aumentan progresivamente hasta alcanzar un pico antes del nacimiento. Luego de la pubertad, las concentraciones séricas de esta hormona resultan el doble en mujeres respecto a los hombres de la misma edad. En el embarazo, la prolactina circulante aumenta progresivamente hasta alcanzar niveles 7-10 veces mayores que los presentes en mujeres no embarazadas. Este aumento es fundamental para el correcto desarrollo mamario necesario para la posterior lactancia (28;30). La síntesis y secreción de la prolactina adenohipofisaria está afectada por numerosos estímulos, tanto externos como internos, los cuales son interpretados y traducidos por el hipotálamo. Las concentraciones plasmáticas de esta hormona en humanos son mayores durante las horas de sueño y menores durante el día, presentando mayor amplitud en las mujeres respecto de los hombres. Esto sugiere que prolactina responde, entre otras cosas, al ritmo circadiano. Por otra parte, la succión durante el amamantamiento es el estímulo externo de mayor importancia, mientras que el estrés y los niveles de esteroides gonadales también aumentan la liberación de esta hormona (28). Los lactotropos, que corresponden al 30-50% de las células hipofisarias, presentan una alta actividad espontánea intrínseca de síntesis y secreción de prolactina, siendo la regulación hipotalámica predominantemente inhibitoria, ejercida principalmente por la dopamina (30). Este neurotransmisor catecolaminérgico llega a la hipófisis a través del sistema porta-hipofisario y se une a receptores dopaminérgicos tipo D2 (RD2s) localizados en el lactotropo, produciendo la inhibición de la síntesis y secreción de la prolactina como así también de la proliferación de las células lactotropas (31;32). Aunque la cantidad de dopamina que llega a la hipófisis es suficiente para inhibir tónicamente al lactotropo, se postulan otros factores inhibitorios cuyos papeles 15 Introducción fisiológicos no están aún bien determinados, como endotelinas, STT, acetilcolina, GABA, entre otros. También la prolactina es capaz de regular su propia liberación al ejercer una retroalimentación positiva sobre el sistema dopaminérgico del hipotálamo. El incremento de prolactina produce un aumento en la actividad de las neuronas dopaminérgicas TI DA (haz dopaminérgico tuberoinfundibular), por lo que disminuye su liberación por parte de los lactotropos (30). Existen también, péptidos estimuladores de la secreción de prolactina, siendo algunos de ellos TRH y VI P o esteroides como los estrógenos. Durante muchos años la prolactina ha sido considerada una hormona encargada de ejercer efectos biológicos limitados a la iniciación y mantenimiento de la reproducción y la lactancia. Sin embargo, los datos de los últimos años permitieron demostrar una gran variedad de funciones de esta hormona, junto con la amplia distribución de su receptor (PRLR) en diferentes tejidos y tipos celulares. Hoy en día, la prolactina es reconocida como una hormona altamente versátil que actúa regulando no solo la reproducción sino también la osmorregulación, metabolismo energético, funciones cerebrales, respuestas inmunológicas, crecimiento y angiogénesis (28;33-35). Sin embargo, la relevancia fisiológica de muchas de estas funciones sigue sin ser respondida, ya que en muchas instancias fueron evidenciadas en condiciones experimentales particulares o en experimentos farmacológicos. Receptores de prolactina El receptor de prolactina (PRLR) pertenece a la superfamilia de receptores de citoquinas de clase I , junto con el receptor de hormona de crecimiento (GHR), compartiendo ambos receptores numerosas características estructurales y funcionales (33). El PRLR se encuentra ampliamente distribuido en los vertebrados (33), y diferentes isoformas del mismo, provenientes de un único gen y producto del splicing alternativo (36) o clivaje post-traduccional (37), han sido detectadas en numerosos tejidos y tipos celulares (33;38). Estas isoformas comparten idéntica secuencia en las dominios extracelulares y transmembrana, pero difieren en la longitud y secuencia de la porción intracelular (38). Hasta el momento se han identificado en el ratón, una isoforma larga (PRLR-L) y tres cortas (PRLR-S1, -S2 y –S3) del receptor (39) (Figura 6). 16 Introducción Figura 6: Representación esquemática (no hecha a escala) de las cuatro isoformas de ARNm del Prlr de ratón ( Prlr-l, Prlr-s1, Prlr-s2 y Prlr-s3): se visualizan el dominio extracelular (blanco), el dominio transmembrana (negro), la porción común del dominio citoplasmático (blanco), y la región única del dominio citoplasmático (gris). Tomado y modificado de (39) . Se ha descripto que la regulación de la expresión de las diferentes isoformas del PRLR varía según los distintos tejidos y los diferentes estados fisiológicos, como ser ciclo estral, preñez y lactancia (39). En hígado de ratón, por ejemplo, las isoformas PRLR-L y PRLR-S3 son las más abundantes mientras que existe una expresión minoritaria de las PRLR-S1 y PRLR-S2 (40) (Figura 7). En tejido adiposo blanco, en cambio, sólo las isoformas PRLR-S2, PRLR-S3 y PRLR-L han sido descriptas, siendo la isoforma predominante la PRLR-L (39). Figura 7: Proporción relativa de los transcriptos de las distintas isoformas del Prlr ( Prlr-l, Prlr-s1, Prlrs2 y Prlr-s3 ) en hígado de ratones control. Tomado y modificado de (40). La activación del PRLR consiste en la unión de prolactina (ligando) la cual genera tanto formas monoméricas como diméricas del PRLR. Esta unión induce cambios estructurales en el dominio extracelular del receptor, conformando el complejo ligando/ receptor. Luego, los cambios estructurales inducidos en este dominio se transmiten al intracelular modulando la actividad del receptor, ya que la porción citosólica controla varias vías de señalización y amplifica la señal (37). Como ya mencionamos, las isoformas larga y cortas se encuentran diferencialmente expresadas en los distintos tejidos (41;42), lo que sugiere que podrían estar activando diferentes 17 Introducción vías de señalización. Se sabe que, prolactina actuando vía PRLR-L activa numerosas quinasas, incluyendo JAK2/ Stat (43), Src quinasa (44), PI 3K/ AKT (45) y las vías de MAPK. Por otro lado, se ha propuesto que las isoformas cortas podrían actuar como dominantes negativos inhibiendo la función del PRLR-L (46;47), ya que carecen del dominio citoplasmático requerido para la asociación con moléculas de señalización tales como Stat. Sin embargo, estudios recientes han demostrado que prolactina a través de PRLR-S regula la actividad transcripcional de múltiples genes, ya sea activando o reprimiéndolos de manera tejido específica (48). Lo cierto es que todavía quedan por determinarse los mecanismos de señalización por los cuales actúa PRLR-S. Prolactina: hormona metabólica Como ya mencionamos, el rol de la prolactina en la reproducción y fertilidad ha sido muy estudiado. Altos niveles de prolactina se observan durante la preñez y la lactancia, en concordancia con un estado de hiperfagia y aumento en la producción láctea (49). En el caso de la preñez, los mecanismos normales que regulan el apetito son modificados de manera tal de generar un balance energético positivo, incrementando la ingesta para abastecer el requerimiento del feto en crecimiento, y aumentando los reservorios lipídicos que van a ser utilizados posteriormente en la lactancia (50). Existen ciertas controversias en la información disponible acerca del rol de la prolactina en la regulación del peso corporal. Algunos estudios llevados a cabo en ratas apoyan la hipótesis de que prolactina posee un rol significativo en la regulación del peso corporal. El incremento crónico de los niveles de prolactina, ya sea inducidos por antagonistas dopaminérgicos, inyecciones diarias con la hormona, o hipófisis ectópicas, han sido asociados con un aumento en los niveles de ingesta y peso corporal (51-54). Consistente con esto, la inhibición de la secreción de prolactina por medio de bromocriptina derivó en el efecto contrario (52). A su vez, inyecciones de prolactina en el PVN incrementan la ingesta de alimentos lo que indica que, además de las acciones periféricas sobre el páncreas y tejido adiposo, prolactina estaría modulando el apetito a nivel hipotalámico (55). Los datos obtenidos en ratones resultan un tanto más contradictorios. Mientras que el ratón deficiente del receptor de prolactina resultó tener un peso corporal y depósitos de grasa disminuidos (56), ratones macho con transplantes de hipófisis ectópicas mostraron sólo un mínimo incremento en el peso corporal con un descenso en la cantidad de tejido adiposo (57), sumado a que ratones hembra que sobreexpresan prolactina no mostraron modificaciones en el peso (39). Por otro lado, en nuestro laboratorio hemos descripto que los ratones hembra que carecen del receptor de dopamina D2 en todo su organismo ( Drd2-/ - ) presentan una hiperprolactinemia crónica e hiperplasia hipofisaria (58;59), pero con pesos corporales y niveles de ingesta similares a los ratones salvajes (WT) (60). Cabe destacar, que el ratón Drd2-/ - no resultaría un modelo óptimo para el estudio de los efectos de altos niveles de prolactina en el balance energético, dada la importancia fundamental de los RD2s centrales en los mecanismos de recompensa relacionados al 18 Introducción comportamiento alimenticio (61;62). La evidencia disponible sugiere que la falta de RD2s centrales en el ratón Drd2-/ - podría estar activando mecanismos compensatorios para limitar finalmente la ingesta de alimento en este modelo hiperprolactinémico. Por otra parte, si bien los efectos de la prolactina sobre la sensibilidad y producción de insulina han sido poco estudiados, los resultados obtenidos hasta el momento apuntan a que la prolactina también estaría involucrada en dichos mecanismos. Se ha demostrado que los estados de hiperprolactinemia se encuentran asociados a una intolerancia a la glucosa y resistencia a insulina en modelos de ratas y humanos (63;64). I ncluso algunos estudios atribuyen la insulinoresistencia causada por estados de hiperprolactinemia en pacientes a efectos de la prolactina sobre la expresión del receptor de insulina (65). Hormona de crecimiento La hormona de crecimiento (GH), es una hormona polipeptídica que presenta homología de secuencia y estructural entre especies y con otras hormonas relacionadas como la prolactina y el lactógeno placentario (PL). Tiene un peso molecular de aproximadamente 22 KDa y está compuesta por una única cadena polipeptídica de 191 aminoácidos con dos puentes disulfuro, que generan un lazo mayor y un lazo menor (66). La mayor parte de la GH circula como monómero no glicosilado, aunque se conocen otras variantes como la desaminada (5% ), acetilada (1-2% ), fosforilada, glicosilada, dimerizada y oligodimerizada, las cuales tienen un mayor peso molecular, son generadas por procesamiento postraduccional, y su función, en general, se desconoce (67). GH es sintetizada, almacenada y secretada por las células somatotropas de la hipófisis anterior, siendo esta hormona la proteína más abundante de la glándula. Los somatotropos la almacenan en gránulos y representan alrededor del 35 al 45 % del total de células de la hipófisis anterior. En humanos, la GH es detectable en el suero fetal a partir del final del primer trimestre y aumenta rápidamente hasta alcanzar un pico en la 20ª semana de gestación. El mayor porcentaje de secreción se observa en la adolescencia y va decreciendo con la edad. En los roedores también hay una disminución en los niveles de GH relacionados con la edad. El aumento de GH en plasma se observa inmediatamente después del nacimiento, siendo máximo en las primeras dos semanas de vida (68). Los valores se mantienen altos hasta los dos meses y luego declinan con la edad hasta valores basales muy bajos. Si bien la influencia del RD2 sobre la prolactina está cabalmente demostrada poco se sabe sobre el control que ejerce este receptor sobre GH. Estudios de nuestro laboratorio han demostrado que en edades tempranas el RD2 es crítico para la estimulación de este eje, y su ausencia en ratones Drd2-/ - causa retardo de crecimiento (68). El eje hipotalámico-hipofisario-somático en mamíferos recibe una multiplicidad de señales a lo largo del desarrollo madurativo de cada individuo lo que aporta un enorme valor adaptativo en 19 Introducción relación a su éxito reproductivo posterior. Dos neuropéptidos hipotalámicos controlan la síntesis y liberación de GH en los somatotropos del lóbulo anterior de la hipófisis: GHRH y STT. Los desbalances en los niveles de GHRH, GH, sus receptores, o mecanismos de traducción de señales, producen alteraciones en fenotipos relacionados al crecimiento somático (69-71) mientras que la falta de STT altera el programa secundario de dimorfismo sexual (72;73). GHRH estimula la proliferación de los somatotropos en la hipófisis así como la síntesis y liberación de GH. Por su parte, STT inhibe tanto la liberación de GHRH como de GH. Una tercera vía de regulación de la síntesis de GH se ha propuesto más recientemente e involucra a ghrelina, una hormona sintetizada por el tracto gastrointestinal, que también está presente en pequeñas cantidades en el hipotálamo e hipófisis e induce la liberación de GH de manera potente (74). Este péptido presenta además un rol fundamental en el control del apetito (75). La liberación de GH sigue un patrón pulsátil que presenta un dimorfismo sexual muy marcado. Mientras que en hembras los niveles plasmáticos de GH son más constantes, de períodos más largos y menor amplitud, en machos la liberación hipofisaria de GH presenta períodos más cortos y de amplitud mucho mayor, por lo que los niveles plasmáticos son más variables a lo largo del día. Estos patrones diferenciales de aumento y descenso en la liberación de GH cumplen un papel clave en la determinación del desarrollo corporal normal en machos y hembras. La GH plasmática tiene muchos blancos de acción, controlando el crecimiento corporal a través de su acción sobre células del hígado, músculo esquelético, tejido óseo y adipocitos. La acción de GH sobre los hepatocitos induce la producción y liberación del factor de crecimiento tipo insulina 1 (I GF-I ) al torrente sanguíneo. I GF-I tiene receptores específicos en varios tejidos del organismo donde participa estimulando el crecimiento somático (76) y la maduración sexual (77). Hormona de crecimiento: hormona metabólica La primera referencia sobre los efectos metabólicos de GH data del año 1948 y sugiere que GH induce el metabolismo lipídico e inhibe la proteólisis en ratones ayunados (78). Ahora está claro que GH afecta el metabolismo tanto de manera directa como indirectamente vía I GF-I o el antagonismo de la acción de insulina (79). La GH es considerada una hormona diabetogénica ya que de manera crónica induce resistencia a insulina e hiperglucemia al oponerse a los efectos de la insulina en el metabolismo de la glucosa, tal como fue demostrado por Houssay (80-83). Frente a una hipoglucemia aguda, GH es secretada, promoviendo la inhibición de la captación de la glucosa por parte del músculo y tejido adiposo, permitiendo de este modo que toda la glucosa disponible sea utilizada por el sistema nervioso central. Se observó a su vez, que promueve la elevación de glucosa en sangre al estimular la gluconeogénesis a partir de lípidos, y fomenta la utilización de ácidos grasos en lugar de la glucosa (84-86). Por otra parte, se sabe que la vía de señalización de GH comparte algunos 20 Introducción efectores con la de insulina (87;88). Sumado a esto, muchos estudios demostraron que GH tiene un efecto trófico directo sobre la célula beta pancreáticas (89-92). En cuanto a los efectos de esta hormona sobre el tejido adiposo, GH ejerce un efecto lipolítico en el tejido adiposo visceral como así también en el subcutáneo, resultando en mayor flujo de FFAs (93;94). A su vez, el ratón transgénico que carece de receptores de GH funcionales (GHRKO) resulta más susceptible a la obesidad inducida por dieta (95). En humanos, existen numerosos reportes donde niños y adultos con deficiencia en GH presentan un incremento en la masa de tejido adiposo, los cuales luego del tratamiento con GH recombinante humana (rhGH) presentan una reducción significativa en los niveles de adiposidad (96-98). Por otra parte, GH también juega un rol importante en la diferenciación de adipocitos (adipogénesis). Numerosos estudios in vitro con líneas de adipocitos han demostrado que GH induce de manera directa la adipogénesis en etapas tempranas del proceso, mediante la activación de Stat5 y consecuentemente de PPAR , un factor adipogénico establecido (99). Además, GH disminuye los niveles de leptina séricos, mientras que los efectos sobre adiponectina resultan contradictorios en humanos y roedores (79). En el hígado, GH estimula la incorporación y almacenamiento de triglicéridos mediante la inhibición de la lipólisis o bien promoviendo la lipogénesis. A su vez, GH posee un efecto estimulatorio en la producción de glucosa hepática como resultado del antagonismo de la acción de insulina, lo que resulta en insulino resistencia hepática o sistémica (79). Alteraciones en la secreción de prolactina y GH: adenomas hipofisarios Los tumores hipofisarios son adenomas monoclonales localizados en la silla turca que representan aproximadamente entre un 10 y 15% de todas las neoplasias intracraneales (100). La mayoría de los tumores hipofisarios crece lentamente a lo largo de los años. Se los considera benignos ya que no son metastásicos y permanecen dentro de la silla turca. Se definen como verdaderos carcinomas sólo en presencia de metástasis craneoespinal y/ o sistémica, y éstas son raras con una incidencia menor al 0,5% de los tumores hipofisarios (101). Los tumores hipofisarios se clasifican según su tamaño en: microadenomas (< 10mm) y macroadenomas (> 10mm). Los pacientes con macroadenomas recurren a la consulta médica debido a efectos de compresión, como dolores de cabeza y/ o falla visual progresiva mientras que los casos de microadenomas son identificados generalmente por la manifestación de síndromes clínicos endócrinos o en forma incidental durante diagnósticos por imagen. Sumado a esto, en un 25% de las autopsias se descubren pequeños adenomas subclínicos. Los tumores hipofisarios se clasifican además según la hormona que producen y se describen como funcionantes si sobreproducen alguna hormona de la hipófisis anterior, o no funcionantes, si no existe síndrome clínico (endócrino) aparente. Los adenomas funcionantes más 21 Introducción frecuentres resultan los secretores de GH (los cuales provocan acromegalia) y los de prolactina (denominados prolactinomas) (102). EL RECEPTOR DE DOPAMINA TIPO 2 (RD2) A lo largo de la evolución el receptor de dopamina tipo 2 (RD2) ha ejercido un rol importante en el arraigo de funciones adaptativas que promueven mejorar la condición física, el éxito reproductivo y la supervivencia. Mecanismos que aumentan la probabilidad del consumo de alimento nutritivo y el apareamiento, utilizan la estimulación por parte de dopamina a través de este subtipo de receptor (103;104). Los RD2s son fundamentales también para la coordinación motora, la actividad locomotora, la motivación o aversión, y la dominancia social (105;106). Asimismo, la participación del RD2 en funciones endócrinas refuerza el rol de la dopamina en el éxito reproductivo y la supervivencia (107). Cinco receptores de dopamina han sido aislados, caracterizados y divididos en dos subfamilias según sus propiedades bioquímicas y farmacológicas, como así también su distribución anatómica: - los receptores tipo D1 que comprenden al RD1 y RD5, y son estimuladores de la adenilato ciclasa (AC) - y los receptores tipo D2 que comprenden los RD2, RD3 y RD4, los cuales inhiben dicha enzima (108). Los miembros de cada familia poseen alta homología de estructura. Todos son receptores con siete pasos transmembrana acoplados a proteína G (109). Los receptores del primer grupo se encuentran acoplados a proteínas G estimulatorias, mientras que los tipo D2 son receptores que se encuentran acoplados a proteína Gi suprimiendo la acumulación de 3´ ,5´ -adenosina monofosfato cíclico (AMPc) mediante la inhibición de la AC, y a proteína G0 regulando los niveles de calcio intracelular. A causa del splicing alternativo existen dos isoformas del RD2, una isoforma larga (D2L) y otra corta (D2S) (110). La isoforma corta posee una deleción de 29 aminoácidos en el tercer dominio citoplasmático, sitio donde ocurre la interacción con las proteínas Gi/ G0. Ambas isoformas del receptor poseen la misma afinidad por dopamina, pero difieren en el acople de segundos mensajeros (111). RD2 EN CEREBRO En tejidos cerebrales el RD2 se expresa predominantemente en el ganglio basal, el núcleo accumbens y la corteza frontal, donde participa en el control extrapiramidal de la actividad locomotora, la recompensa y la organización espacio-temporal de comportamientos orientados, 22 Introducción respectivamente (104;112). En el cerebro, se expresan las dos isoformas del receptor, siendo la D2L la más abundante (112). En el hipotálamo existe una amplia distribución de neuronas que expresan el RD2, como ser en el área preóptica, hipotálamo anterior, HLA y Arc, entre otros (113). RD2 HIPOFISARIO Los RD2s son los receptores predominantes en la hipófisis, presentes en lactotropos y melanotropos, que median las acciones de dopamina en esta glándula (107;109). Los RD2s en lactotropos están involucrados tanto en la inhibición de la liberación y síntesis de prolactina, como en la proliferación de los mismos (107). Del mismo modo, los RD2s inhiben la liberación de α-MSH en los melanotropos (111). Los lactotropos y melanotropos expresan ambas isoformas, D2L y D2S (114). La isoforma D2L es la predominante (115), sin embargo los ratones transgénicos que carecen de D2S presentan hipoplasia de lactotropos, demostrando la importancia de este subtipo de receptor en el control de la prolactina (116). El cociente D2L/ D2S se ve afectado por esteroides sexuales in vitro (115;116). Asimismo, el estradiol disminuye el efecto de dopamina a nivel hipofisario, es decir su actividad inhibitoria sobre prolactina (117), y también afecta el metabolismo de la dopamina hipotalámica (118). RD2: FUNCIONES ENDÓCRINAS Y METABÓLICAS La participación de los RD2s en la regulación de prolactina está bien documentada, mientras que todavía no se logra determinar por completo el rol de los RD2s en el control endócrino de hormonas del crecimiento, la ingesta y el metabolismo de la glucosa (111). Dopamina y regulación de la prolactina La dopamina inhibe la secreción de prolactina por parte de los lactotropos hipofisarios, mediante los RD2s. En respuesta a la dopamina, lo primero que ocurre en el lactotropo es una inactivación de los canales de voltaje dependientes de calcio y por lo tanto una inhibición de la liberación de gránulos con prolactina. Luego se inhibe la enzima AC que produce la inactivación de PKA y una consecuente supresión de la expresión de genes, incluyendo Pit-1 (factor necesario para la expresión tanto del gen de prolactina como de GH). De este modo, no solo inhibe la secreción de prolactina, sino también la proliferación de lactotropos (107). La liberación de prolactina en ratas se encuentra regulada por tres sistemas neuronales dopaminérgicos hipotalámicos: las principales corresponden a las neuronas TI DA cuyos terminales 23 Introducción están en la eminencia media, neuronas tuberohipofisarias con terminales en la hipófisis posterior, y el sistema dopaminérgico periventricular con terminales en la hipófisis intermedia (30). También la prolactina es capaz de regular su propia liberación al ejercer una retroalimentación positiva sobre las neuronas dopaminérgicas TI DA del hipotálamo. El PRLR colocaliza con neuronas que expresan tirosina hidroxilasa, la enzima limitante de la síntesis de dopamina. Durante la preñez tardía y la lactancia, o luego de la formación de un prolactinoma, estas neuronas se vuelven refractarias a la retroalimentación por parte de prolactina provocando una hiperprolactinemia fisiológica o patológica, respectivamente (30). La significancia fisiológica del control inhibitorio por parte de los RD2s en la liberación de prolactina, fue claramente evidenciada en los ratones Drd2–/ –. Los mismos presentan hiperprolactinemia crónica e hiperplasia de lactotropos hipofisarios (58), seguida de la formación de tumores de lactotropos (119). Dopamina y regulación de GH-GHRH El rol de la dopamina en la regulación del GH no ha sido completamente dilucidado. Efectos estimulatorios, así como inhibitorios, del neurotransmisor han sido reportados sobre los niveles plasmáticos de GH in vivo dependiendo de las condiciones experimentales utilizadas (120). Esto puede explicarse por la habilidad que posee la dopamina de liberar tanto GHRH como STT en hipotálamos de ratas (121). I n vitro, tanto respuestas positivas como negativas a la catecolamina han sido descriptas en células hipofisarias (120;122). Los ratones Drd2–/ – resultaron un modelo útil para revelar la participación de los RD2s en el crecimiento y la regulación del eje GH-GHRH. En nuestro laboratorio observamos que estos ratones presentaron un eje de crecimiento alterado, con una baja talla y un menor peso corporal (68). Esto correlacionó con menores niveles de hormona de crecimiento secretados por la hipófisis, y menor I GF-I circulante (68;123). Postulamos que la dopamina central a través del RD2 impediría la correcta liberación de STT o GHRH hipotalámica en la ventana temporal del periodo neonatal, y por ende el desarrollo de la población de somatotropos estaría alterado. En concordancia, en humanos se ha observado que la L-DOPA puede acelerar el crecimiento en niños con deficiencia de GH (124), y que una población de niños con polimorfismos en el RD2 presentó deficiencias en el eje de crecimiento, con talla baja idiopática (125). Finalmente, en adultos bajo tratamiento crónico con antipsicóticos se demostró una respuesta anormal de GH durante el sueño (126). En conjunto todos estos datos apuntan a una participación del RD2 en la regulación del eje GHRH-GH. 24 Introducción Dopamina y péptidos relacionados con la ingesta La dopamina regula el hambre y la saciedad actuando a nivel cerebral. Sin embargo, los efectos de la dopamina sobre la ingesta no son claros ya que interactúa con distintos núcleos hipotalámicos y estimula distintos subtipos de receptores. Se ha descripto que tratamientos sistemáticos con agonistas de los receptores D1/ D2 disminuyen la ingesta (127;128), mientras que también se vio que agonistas selectivos para RD2 incrementan la ingesta de alimentos (129). Por otra parte, el incremento farmacológico de dopamina en el LHA induce anorexia, mientras que la inyección de un antagonista de RD2s en LHA incrementa la ingesta (130). Sumado a esto, diferentes estados energéticos (ayuno, obesidad, anorexia) modulan la expresión del receptor de dopamina (131). En ratones mutantes nulos para el RD2 se describieron dos eventos anorexigénicos: un aumento en la α-MSH (un potente anorexigénico) sérica e hipotalámica, y una disminución del precursor de las orexinas (60). Estos eventos podrían estar siendo compensados por el gran aumento de los niveles de prolactina (estímulo orexigénico). De estos resultados se destaca la compleja regulación del sistema de la ingesta, sistema primordial para la supervivencia. Múltiples factores actuarían en la regulación de la ingesta por el RD2, por un lado estos receptores tienen acciones orexigénicas estimulando las orexinas y α-MSH, y por otro anorexigénicas inhibiendo en forma constante la secreción de prolactina. Por otro lado, en humanos la reducción de los RD2s se asocia con comportamientos adictivos (132), pero la evidencia sobre mutaciones en el RD2 y obesidad es escasa, aunque en general las mutaciones con pérdida de función se asocian a sobrepeso (133;134). Dopamina y metabolismo de la glucosa La relación entre los receptores de dopamina y la homeostasis de la glucosa no ha sido extensamente estudiada, aunque algunos estudios clínicos y experimentales sugieren una conexión entre los mismos. En roedores, inyecciones con L-DOPA resultaron en la acumulación de dopamina en células pancreáticas y en una acción inhibitoria sobre la respuesta a insulina frente a secretagogos (135;136). Por otra parte, ratas macho tratadas con antagonistas de dopamina, presentaron altos niveles tanto de glucosa como de insulina (137). Algunos estudios in vitro, desarrollados en islotes pancreáticos aislados, sugieren la participación local del RD2 en la secreción de insulina (138-141), aunque el efecto sobre la estimulación mediada por el RD2 observada en estos estudios y su importancia funcional, todavía quedan por dilucidarse. Recientemente se describió que existe una estrecha vinculación entre el uso de drogas antipsicóticas y el desarrollo de diabetes tipo I I . Nosotros demostramos que la dopamina también actúa a nivel del islote de Langerhans regulando en forma inhibitoria la secreción de insulina. 25 Introducción Utilizando los modelos animales de mutantes nulos, definimos que la ausencia de la acción dopaminérgica sobre el islote (dada por ejemplo por el uso de drogas que antagonizan al RD2, o en forma constitutiva en los transgénicos), produce una secreción aumentada de insulina, lo que crónicamente conduce a un lento deterioro y agotamiento de las reservas insulínicas del islote (141). 26 HIPÓTESIS Nuestra hipótesis sostiene que la prolactina es un factor hormonal que interviene en el metabolismo de la glucosa y los lípidos, como así también en la ingesta. En este contexto, para evaluar el efecto de altos niveles de prolactina, en presencia de un eje de GHRH-GH conservado, proponemos que la deleción específica de los RD2s solamente en lactotropos nos proporcionaría un modelo experimental óptimo. Nos permitirá estudiar los efectos de altos niveles de prolactina en la ingesta, metabolismo de la glucosa y adiposidad, sin los efectos solapados, fisiológicos y patofisiológicos, presentes en el modelo mutante knockout global de los RD2s, o en animales tratados con agentes farmacológicos en los cuales la interferencia de los cambios sobre los RD2s centrales modificaría las acciones de la prolactina. 27 OBJETIVOS GENERALES Estudiaremos en primer instancia (Capítulo I ) las características de la hiperprolactinemia crónica en los ratones hembra lacDrd2KO en comparación con hembras Drd2loxP/ loxP, el dimorfismo sexual, y la participación de la angiogénesis y las células madre/ progenitoras en la génesis de los prolactinomas que desarrollan estos mutantes. Asimismo determinaremos el perfil endócrino para demostrar la normalidad del eje de GH. En segundo lugar, caracterizaremos el fenotipo de peso corporal e ingesta en presencia de los altos niveles crónicos de prolactina, determinando a nivel hipotalámico la expresión de neuropéptidos relacionados con la ingesta de alimentos. Compararemos los resultados con los previamente obtenidos en ratones Drd2-/ - , para disecar los efectos de los RD2s centrales e hipofisarios (desarrollado en el Capítulo I I ). A continuación (en el Capítulo I I I ) determinaremos los efectos de altos niveles de prolactina sobre la regulación y mantenimiento de la homeostasis de la glucosa. Finalmente, analizaremos el impacto de altos niveles de prolactina sobre la adiposidad y expresión génica a nivel del hígado y tejido adiposo, estableciendo una función de la prolactina en el metabolismo de lipídico (desarrollado en el Capítulo I V). 28 Materiales y métodos MATERIALES Y MÉTODOS ANIMALES Los animales se mantuvieron en el bioterio del I nstituto de Biología y Medicina Experimental (I ByME-CONI CET), en condiciones ambientales reguladas: la temperatura en 22°C, un fotoperíodo de 12 hs de luz (7:00 – 19:00 hs) y 12 hs de oscuridad, y ventilación permanente. Se les permitió libre acceso al agua y alimento balanceado en forma de pellets (Gepsa Feeds, Grupo Pilar), salvo determinados experimentos en los que se especifica lo contrario. Todos los procedimientos experimentales fueron revisados y aprobados por el comité de cuidado y uso de animales del I nstituto de Biología y Medicina Experimental, Buenos Aires. RATONES lacDrd2KO Los ratones que carecen de los RD2s funcionales solamente en lactotropos se obtuvieron por medio de la cruza de ratones Drd2loxP/ loxP (los cuales poseen el exón 2 del RD2 flanqueado por secuencias específicas loxP) (61) con ratones transgénicos que poseen la expresión de la enzima recombinasa Cre bajo el control transcripcional del promotor de prolactina [ Tg(Prl-cre) 1Mrub] (142) (Figura 8). La actividad de la enzima Cre recombinasa fue analizada en hipófisis y demostramos que se encuentra activa en el 96% de las células productoras de prolactina y que esta actividad es altamente específica de lactotropos (142). Los apareos fueron conformados por un ratón hembra Drd2loxP/ loxP junto con un macho Drd2loxP/ loxP.Tg(Prl-cre) 1Mrub (lacDrd2KO), para obtener en una misma camada crías controles ( Drd2loxP/ loxP) y transgénicas (lacDrd2KO), que fueron luego utilizadas para los experimentos subsiguientes. Los ratones lacDrd2KO y sus controles ( Drd2loxP/ loxP) son congénitos con la cepa C57BL/ 6J. 29 Materiales y métodos Figura 8: Generación del ratón transgénico que expresa la recombinasa Cre bajo el control transcripcional del promotor de prolactina (Prl-Cre). Estos ratones fueron cruzados con ratones de otra línea transgénica la cual posee alelos loxP a ambos lados del exón 2 del gen RD2 ( Drd2loxP/ loxP usados como controles). La descendencia, que expresa tanto Prl-Cre como los alelos loxP son denominados lacDrd2KO. Genotipificación de los ratones Extracción de ADN genómico Las crías de ratones se destetaron a los 30 días de su nacimiento y se separaron por sexo hasta que su genotipo fue determinado. Éstas se identificaron con una marcación según un código de perforaciones en las orejas. Para determinar los genotipos se realizaron biopsias de la porción terminal de la cola con un bisturí. Los segmentos de cola u orejas obtenidos se incubaron a 55°C en 20 µl de buffer de digestión (TrisHCl 50 mM pH 8; EDTA 100 mM; SDS 0,5% ; 0,5 mg/ ml de proteinasa K) durante 12 hs. Se precipitó el ADN en frío, durante 40 minutos a -70°C y se centrifugó a 12000 g por 40 minutos a 4°C, para su extracción. Finalmente, se descartó el pellet y el sobrenadante se guardó a -20°C hasta su utilizac ión. La presencia del transgen se detectó por reacción en cadena de la polimerasa (PCR) con cebadores (primers) específicos de las secuencias transgénicas como se detalla a continuación. Reacción en cadena de la polimerasa (PCR) Para la genotipificación de los ratones lacDrd2KO y sus pares Drd2loxP/ loxP deben realizarse dos PCRs diferentes: 30 Materiales y métodos 1) En primer lugar se realiza una para determinar la presencia de sitios loxP. Para la misma, se utilizó un par de primers sentido y antisentido complementarios al intrón 2 del gen RD2 (I nvitrogen Life Technologies), cuyas secuencias se muestran en la Tabla 1. Si existe un alelo mutado el par de primers del intrón 2 amplifica un producto de 452 pares de bases (pb) y en caso de haber un alelo normal amplifican fragmento de 354 pb (Figura 9). Las condiciones de esta PCR se detallan en la Tabla 2. Tabla 1: Primers utilizados para las reacciones de PCR realizadas para la genotipificación de los animales. Secuencia 5’- 3’ Gen Prl-Cre Sentido CCTTCATTTCTGGCCAATG Antisentido AGGCAATTTTGGTGTACGG Intrón 2 Sentido GCTTCACAGTGTGCTGCCTA Antisentido CCATTGCTGCCTCTACCAAG Figura 9: A) Fragmentos amplificados por la reacción de PCR para la genotipificación de los animales con un alelo normal: Drd2+ (fragmento de 354 pb) y otro alelo mutado: Drd2loxP (fragmento de 452 pb). La letra A representa el primer sentido del intrón 2, y la letra B el primer antisentido del intrón 2. Los rombos azules corresponden a los sitios loxP, mientras que el rectángulo verde representa el exón 2 del gen del RD2. B) Gel de agarosa con productos de reacción de PCR de genotipificación para determinar presencia de sitios loxP. La calle 1 corresponde al control positivo Drd2loxP/ loxP, la calle 2 es el control positivo Drd2loxP/ + , la calle 3 es el control positivo Drd2+ / + , y la calle 4 es el blanco (control negativo, sin ADN). El patrón de bandas de las calles 5 y 7 corresponden a animales Drd2loxP/ loxP, mientras que la calle 6 representa un patrón de bandas de un animal heterocigota Drd2loxP/ + . 31 Materiales y métodos Tabla 2: Reacción de PCR para la genotipificación de los animales transgénicos control. MIX PCR Concentración Primers sentido y antisentido 2 µM MgCl2 ADN dntp´s Buffer Taq polimerasa H2O 1,5 mM 200 ng 0,2 mM TrisHCl 20mM; KCl 50mM 1 UE Vf= 30ul Programa 1 ciclo de desnaturalización inicial 34 ciclos: de desnaturalizaciónapareamiento y extensión 1 ciclo de extensión final 2) Temperatura (°C) Tiempo 95 5’ 95 0’’ 62 0’’ 72 0’’ 72 5’ En segundo lugar, se lleva a cabo una PCR para la detección del transgen Prl-Cre, en la cual se utilizó un par de primers conformado por una secuencia complementaria a una porción del promotor de prolactina ( primer sentido), y una secuencia complementaria a la recombinasa Cre ( primer antisentido). Las secuencias se muestran en la Tabla 1 (I nvitrogen Life Technologies) y las condiciones de esta PCR se detallan en la Tabla 3. En este caso se evidencia simplemente presencia o ausencia del transgen, es decir, presencia o ausencia de producto amplificado. Tabla 3: Reacción de PCR para la genotipificación de los animales transgénicos Prl-Cre. MIX PCR Concentración Primers sentido y antisentido 1 µM MgCl2 ADN dntp´s Buffer Taq polimerasa H2O 1,5 mM 200 ng 0,2 mM TrisHCl 20mM; KCl 50mM 1 UE Vf= 30ul Programa 1 ciclo de desnaturalización inicial 35 ciclos: de desnaturalizaciónapareamiento y extensión 1 ciclo de extensión final Temperatura (°C) Tiempo (min) 95 3 94 1 56 1 72 1 72 5 En todos los casos, los productos de PCR fueron corridos en un gel de agarosa al 1,5% y se visualizaron en un transiluminador (Figura 9 y 10). Se sembró un volumen final de 15 µl por calle conteniendo 11 µl del producto de PCR, 2 µl de buffer de siembra (conteniendo azul de bromofenol y glicerol) y 2 µl de SYBR-Green (I nvitrogen Life Technologies) diluido 1:1000 en DMSO, a partir de la concentración del stock comercial. 32 Materiales y métodos Figura 10: Gel de agarosa con productos obtenidos en una reacción de PCR realizada para la determinación del transgen Prl-Cre. La calle 1 corresponde al control positivo para la banda Prl-Cre y la calle 2 es el blanco (control negativo, sin ADN). En la calle 3 se puede identificar un ratón Prl-Cre negativo, mientras que la calle 4 presenta el producto de PCR de un ratón que posee el transgen Prl-Cre (Prl-Cre positivo). INGESTA Los niveles de ingesta fueron determinados en ratones hembra y macho de ambos genotipos ( Drd2loxP/ loxP y lacDrd2KO), a los 3 y 5 meses de edad. Para ello, se colocaron los ratones en jaulas de manera individual y se les proveyó una cantidad conocida (100 g. apróx) de pellets de alimento balanceado regular. La información nutricional del alimento es: 5% de grasas, 19% de proteínas, y 5% de fibras; con un valor calórico de 2,4 kcal/ g. Durante 8 días consecutivos, el alimento residual fue pesado a la misma hora (15 hs). CURVAS DE PESO CORPORAL Una cohorte de ratones fue pesada a lo largo de su vida, desde el primer mes de edad hasta los 11 meses, una vez por mes. Se utilizó siempre la misma balanza, sin estar los animales anestesiados. Este procedimiento fue realizado en el bioterio, siempre en horas de la tarde (15 hs). TALLA CORPORAL La talla corporal fue determinada mediante la medición con una regla de la longitud narizano en ratones totalmente anestesiados con 7 mg/ Kg de xilacina y 80 mg/ Kg de ketamina (inyección intraperitoneal, ip). Los ratones anestesiados son colocados sobre la regla y al estirarlos se determina la distancia comprendida entre la punta de la nariz y el ano. Por otra parte, también se determinó la longitud del fémur, para lo cual se midió el correspondiente a la pata derecha con una regla, en animales eutanasiados. MONITOREO DEL CICLO ESTRAL La identificación de las distintas fases del ciclo estral se realizó mediante la obtención de extendidos vaginales en hembras de 5 meses de edad. Los extendidos fueron observados en un microscopio de campo claro. Los estadíos del ciclo estral se clasificaron como: diestro (presencia 33 Materiales y métodos principalmente de leucocitos), estro (células cornificadas en su mayoría) y proestro (predominancia de células nucleadas). Los resultados fueron expresados como porcentaje (% ) de días en diestro y días en estro-proestro. TEST DE TOLERANCIA A LA GLUCOSA (GTT) El test de tolerancia a la glucosa fue realizado en ratones hembra y macho lacDrd2KO y Drd2loxP/ loxP de 6 y 10 meses de edad. Los animales fueron ayunados durante 8 hs, luego de las cuales fueron inyectados con glucosa ip 2 g/ kg. Los niveles de glucosa se examinaron previo a la inyección de glucosa y a los 15, 30, 60 y 120 minutos post-inyección (Figura 11). Para ello se tomaron 2 µl de sangre por medio de un corte con bisturí en el extremo de la cola, y el valor de glucosa se obtuvo con un medidor electrónico (Dex-I I , Bayer). Figura 11: Esquema representativo de un GTT. SECRECIÓN DE INSULINA ESTIMULADA POR GLUCOSA (GSIS) Ratones hembra y macho, lacDrd2KO y Drd2loxP/ loxP, de 6 y 10 meses de edad fueron ayunados durante 8 hs, luego de las cuales fueron inyectados con glucosa ip 3 g/ kg. Se recolectaron muestras de sangre, por medio de un corte con bisturí en el extremo de la cola, antes (tiempo 0) y 30 minutos post-inyección de glucosa. Las muestras de suero fueron inmediatamente obtenidas por medio de una centrifugación a 300g por 5 minutos, y fueron guardadas a -20°C hasta el momento de su utilización. Los niveles secretados de insulina fueron evaluados mediante un kit de ensayo de inmunoabsorción ligado a enzimas (ELI SA) específico, en el cual se siguieron las instrucciones del manufacturador (sensitive mouse insulin ELI SA kit, Crystal Chem). TEST DE TOLERANCIA A LA INSULINA (ITT) Los ratones fueron ayunados durante 2 hs, luego de las cuales fueron inyectados con insulina humana (Humulin, 1 U/ kg de peso corporal; Eli Lilly). Los niveles de glucosa en sangre fueron monitoreados utilizando un medidor electrónico (Dex-I I , Bayer). Se examinaron previos a la 34 Materiales y métodos inyección de insulina y a los 15, 30, 60, 90 y 120 minutos post-inyección (Figura 12). Para ello se tomaron 2 µl de sangre por medio de un corte con bisturí en el extremo de la cola. Figura 12: Esquema representativo de un I TT. EVALUACIÓN DEL MODELO HOMEOSTÁTICO (HOMA) HOMA-IR Determinamos el modelo homeostático para la evaluación de la resistencia a la insulina (HOMA-I R), indicador de insulino resistencia de los tejidos periféricos frecuentemente utilizado. Se calcula como el producto entre la glucemia basal y la insulina basal, ambas en ayunas, tal como detalla la fórmula siguiente: HOMA-ΒCELL Determinamos el modelo homeostático para la evaluación de la funcionalidad de células beta pancreáticas (HOMA- cell). El mismo se calcula mediante la relación entre los valores de insulina y glucemia basales, ambas en ayunas, tal como detalla la fórmula siguiente: TEST DE SENSIBILIDAD A LEPTINA Ratones hembra lacDrd2KO y Drd2loxP/ loxP de 5 meses de edad fueron colocados en jaulas individuales y ayunados durante 16 hs, luego de las cuales fueron inyectados de manera ip con 35 Materiales y métodos leptina (mLeptin AFP362C NHPP-NI DDK Parlow) en una dosis de 2,5 mg/ kg. Se les presentó una cantidad conocida de alimento (50 g aprox), y se midieron el peso corporal y la cantidad de alimento residual 1, 2, 4 y 8 hs post-inyección (Figura 13). Luego de una semana de reposo, se reiteró el protocolo experimental en la misma cohorte de animales, pero en lugar de inyectar leptina, se les inyectó el vehículo (PBS). Este procedimiento fue realizado en el bioterio, y se utilizó siempre la misma balanza. Figura 13: Esquema representativo del test de sensibilidad a leptina. AYUNO Y REALIMENTACIÓN Ratones hembra lacDrd2KO y Drd2loxP/ loxP, de 5 y 10 meses de edad, fueron colocados en jaulas individuales y ayunados durante 12 hs, registrando su peso corporal inmediatamente antes y después del ayuno para determinar la pérdida de peso por ayuno prolongado. Posteriormente, a un grupo de ratones se les presentó una cantidad conocida de alimento (50 g aprox) durante 1hr (Grupo Realimentado), mientras que el otro grupo continuó en ayuno (Grupo Ayuno). Pasada la hora se registró la cantidad de alimento residual y el peso corporal (ganancia de peso por 1hr de realimentación) en el Grupo Realimentado. Finalmente, ambos grupos fueron eutanasiados para la recolección de muestras. RECOLECCIÓN DE MUESTRAS La colección de muestras de sangre se realizó para determinar los valores de prolactina en suero en ratones hembra de ambos genotipos a lo largo del desarrollo (a partir del primer mes de edad hasta los 11 meses). La extracción se realizó entre las 15 y 17 hs y la muestra se obtuvo por punción de la vena facial sin previa exposición a droga alguna. Con aproximadamente 100 l de sangre se obtuvo suficiente plasma para la medición de la hormona, por duplicado, por radioinmunoensayo (RI A). 36 Materiales y métodos Los ratones de ambos genotipos fueron eutanasiados por decapitación a los 5 y 10 meses de edad según correspondía. Muestras de suero fueron recolectadas para mediciones de prolactina, GH, I GF-I , progesterona, VEGF, leptina, adiponectina, insulina, colesterol total, ácidos grasos no esterificados (NEFAs) y triglicéridos. A su vez, se tomaron muestras de tejidos (hipófisis, hipotálamo, hígado, adrenales, tejido adiposo y páncreas) y se procesaron para PCR en tiempo real, inmunohistoquímica (I HQ) o RI A, como se detalla más adelante. PESO DE TEJIDOS A los 5 y 10 meses de edad, los animales fueron eutanasiados y se determinaron los pesos de las hipófisis, los tejidos adiposos (gonadal y retroperitoneal), adrenales (ambas), páncreas, así como también de los hígados. Para ello se disecaron cuidadosamente, quedando libres de tejido circundante. PERFIL SÉRICO DE LÍPIDOS Y ADIPOQUINAS Por medio del ensayo colorimétrico de Trinder se analizarlos los niveles séricos de colesterol total y triglicéridos. Los NEFAs fueron determinados por el ensayo colorimétrico de Duncombe. En todos los casos, las mediciones se hicieron a partir de 30 µl de suero diluido ½ (dilución realizada con solución salina). Los niveles de adiponectina se analizaron por medio de un kit de ELI SA específico, en el cual se siguieron las instrucciones del manufacturador (mouse adiponectin ELI SA kit, Crystal Chem). Alícuotas de 100 µl de suero diluído (dilución 1/ 10000 realizada con solución salina) fueron utilizadas por duplicado. El límite inferior de detección del ensayo fue de 0,025 ng/ ml. Las concentraciones séricas de leptina fueron analizadas utilizando un ELI SA específico en el cual se siguieron las instrucciones del manufacturador (mouse leptin ELI SA kit, Crystal Chem. Catalog# : 90030). Alícuotas de 5 µl de suero fueron utilizadas por duplicado. El límite inferior de detección del ensayo fue de 0,2 ng/ ml. EXTRACCIÓN DE ARN Y SÍNTESIS DE ADNc El ARN total de los tejidos (tejido adiposo gonadal, hígado e hipotálamo) se aisló usando el reactivo TRI zol® (I nvitrogen Life Technologies). El hipotálamo entero se recolectó en 100 µl de TRI zol® , mientras que se obtuvieron porciones de tejido adiposo e hígado de un peso aproximado de 30 mg para ser colocados en 300 µl de TRI zol® . Se homogenizaron las muestras con un homogeneizador eléctrico (Polytron, Kinematica, Suiza), para luego continuar los pasos necesarios para la separación de fases acuosa y orgánica (adición de 60 µl de cloroformo y centrifugación a 12000 g por 15 minutos a 4°C). El ARN total present e en la fase acuosa se precipitó mediante el 37 Materiales y métodos agregado de 150 µl de isopropanol, incubando luego por 10 minutos a temperatura ambiente y centrifugando a 12000 g por 10 minutos a 4°C. Luego de un lavado con etanol 70% , el pellet de ARN se resuspendió en 20 µl de agua mediante la incubación durante 10 minutos a 60º C en baño seco. En todos los pasos del procedimiento se utilizó agua libre de ARNasas. Se cuantificó utilizando el Nanodrop, el cual mide la absorbancia a 260 nm y evalúa su pureza por la relación de absorbancias a 260/ 280 nm (> 1,8). Se lo guardó a -70°C hasta su posterior utilización en la reacción en cadena de la polimerasa con transcriptasa inversa (RT-PCR). Se obtuvo luego el ADNc por RT-PCR a partir de 1 μg de ARN total en presencia de: 10 mM de MgCl2; 50 mM de buffer Tris-HCl (pH 8,6); 0,5 mM de deoxy- NTPs (dntp’s); 4 mM de DTT; 0,25 μg de primer oligo-(deoxythymidine) (Biodynamics), y 20 unidades de transcriptasa reversa MMLV (Epicentre). Las muestras de RNA junto con el primer oligo-(deoxythymidine) fueron pre-incubadas durante 2 minutos a 65º C. Luego, se incorporó el resto de los reactivos y la reacción se llevó a cabo a 37º C durante 1 hora en el termociclador con un paso final de 5 minutos a 85º C. Se realizaron en paralelo dos controles negativos: - Para verificar la ausencia de ADN genómico, en una muestra control se omitió la transcriptasa reversa. - En otro caso, se omitió el agregado de ARN. La ausencia de fragmentos de ADN amplificados por PCR en estos controles indicó el aislamiento de ARN libre de ADN genómico, como también corroboró la especificidad de la enzima. DISEÑO DE PRIMERS Los primers específicos para cada protocolo de amplificación empleado en este proyecto fueron diseñados con el software “Primer- BLAST” (http:/ / www.ncbi.nlm.nih.gov/ tools/ primerblast/ ), utilizando registros de secuencias de referencia como templado del análisis (http:/ / www.ncbi.nlm.nih.gov/ RefSeq/ ). En el diseño se tuvo en cuenta que éstos amplificaran fragmentos que contuviesen al menos un intrón entre dos exones. De esta manera, se puede determinar la amplificación específica del transcripto y distinguir, en caso que la hubiese, contaminación con ADN genómico, ya que en dicho caso, el fragmento amplificado es de mayor tamaño dado que incluye la secuencia intrónica. Para el análisis de las características de los primers diseñados se empleó el programa “OligoAnalyzer3.1”, (http:/ / www.idtdna.com/ analyzer/ Applications/ OligoAnalyzer/ ). Se tuvo en cuenta que los primers no formen dímeros ni estructuras secundarias de alta estabilidad, y que no presenten repeticiones invertidas ni repeticiones de un mismo nucleótido. Se eligieron primers cuya temperatura de " annealing " estuviera entre 58 y 60°C. El tamaño de los 38 Materiales y métodos fragmentos amplificados por el par de primers diseñados para la PCR en tiempo real no superó los 180 pb. En la Tabla 4 se detallan los primers utilizados. Tabla 4: Primers de ratón utilizados en este trabajo para los ensayos de PCR en tiempo real. Gen Adiponectina Atgl Chrebp Ciclofilina Cyp2b9 Cyp2d9 Fas Ghr Glucokinasa Gr Gys2 Hsl Secuencia (5' - 3') Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido ATCCTGGCCACAATGGCACA CAAGAAGACCTGCATCTCCT AGGACAGCTCCACCAACATC TGGTTCAGTAGGCCATTCCT TCGATCCGACACTCACCCA CCAGGCTCTCCAGATGGCGT TTCTTCATAACCACAGTCAAGACC ACCTTCCGTACCACATCCAT CTGAGACCACAAGCGCCAC CTTGAGCATGAGCAGGACTCC AGTCTCTGGCTTAATTCCTGAT CGCAAGAGTATCGGGAATGC AAGTTGCCCGAGTCAGAGAA CGTCGAACTTGGAGAGATCC CCAACTCGCCTCTACACCG GGGAAAGGACTACACCACCTG CCGTGATCCGGGAAGAGAA GGGAAACCTGACAGGGATGAG CGGGACCACCTCCCAAA CCCCATAATGGCATCCCGAA CCAGCTTGACAAGTTCGA ATCAGGCTTCCTCTTCAG TCTGCTGGCCCCTGACA Fuente Diseñado con Primer Blast Diseñado con Primer Blast Diseñado con Primer Blast Diseñado con Primer Blast (143) (143) Diseñado con Primer Blast Diseñado con Primer Blast Diseñado con Primer Blast Diseñado con Primer Blast Diseñado con Primer Blast Diseñado con Primer Blast Antisentido AGAGCGCAAGCCACAAGGT Igfbp3 Igf-I Lpl Npy Obrb Pomc Ppo Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido ACCCAGAACTTCTCCTCCGA CCGCTTTCTGCCTTTGGAAG TGTAAACGACCCGGACCTAC CACGAACTGAAGAGCATCCA CCCTACAAAGTGTTCCATTACCAA TTGTGTTGCTTGCCATCCTCA GATGCTAGGTAACAAGCGAATG TCAGCCAGAATGCCCAAAC GCATGCAGAATCAGTGATATTTGG CAAGCTGTATCGACACTGATTTCTTC GTGCCAGGACCTCACCACGG CGTTGCCAGGAAACACGGGC GCCTCAGACTCCTTGGGTATTTG Diseñado con Primer Blast (144) Diseñado con Primer Blast (60) Diseñado con Primer Blast Diseñado con Primer Blast (60) 39 Materiales y métodos Prlr* Prlr-l Prlr-s1 Prlr-s2 Prlr-s3 Srebf1 Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido Sentido Antisentido GGCAATCCGGAGAGATGGT CACAGTAAATGCCACGAACG GGCAACCATTTTACCCACAG CTGGGCAGTGGCTTTGAAG CCAAGGCACTCAGCAGTTCT CCTGCATCTTTCCACCAGTTC GGGAAGTCAACTGGAGAATAGAACA CCTGCATCTTTCCACCAGTTC TTTTCAAGTTGCTCTTTGTTGTGAA CCTGCATCTTTCCACCAGTTC GATCCACCTTGTATTTGCTTGGAG CAGCGGCCCTGAGGGTCAAA TGCATGGCAAGAGGCACCGA Diseñado con Primer Blast Diseñado con Primer Blast Diseñado con Primer Blast Diseñado con Primer Blast Diseñado con Primer Blast Diseñado con Primer Blast * Par de primers para el receptor de prolactina (PRLR), pueden amplificar cuatro tipos de ARNm (si se expresan) Prlr-l, Prlr-s1, Prlr-s2 y Prlr-s3. PCR EN TIEMPO REAL La evaluación cuantitativa de los niveles de ARNm se realizó por PCR en tiempo real con una unidad institucional de la firma Biorad, modelo CFX96 Touch TM (Biorad). Las reacciones se llevaron a cabo utilizando una mezcla preformada llamada 5x HOT FI REPol® EvaGreen® qPCR Mix Plus (ROX) (Solis Biodyne) que contiene: 5X EvaGreen® qPCR buffer, ADN polimerasa HOT FI REPol® , dntp’s incluyendo dTTP para mejorar la eficiencia y sensibilidad de reacción respecto de los dUTP, colorantes EvaGreen® y ROX, y 12,5 mM de MgCl 2. A esta mezcla se agregaron los primers (0,48 µM), moldes (50 ng de ADNc) y el agua en un volumen final de 10 µl. Los ensayos se realizaron por duplicado y se incluyeron: el control negativo de la PCR en tiempo real (en el que se omitió el ADNc molde), y los controles negativos de la RT-PCR. Se pusieron a punto programas de amplificación específicos para cada tejido: Hipotálamo: El programa de amplificación estándar utilizado involucró los siguientes pasos: 1) Activación: 95°C por 15 minutos; 2) Desnaturaliz ación: 95°C por 30 segundos; 3) Hibridación: 61°C por 1 minuto; 4) Extensión: 72°C por 30 segund os; 5) Lectura de datos: 88°C por 33 segundos. Los pasos 2 a 5 se repiten 40 veces. Tejido adiposo gonadal: 1) Activación: 95°C por 15 minutos; 2) Desnaturali zación: 95°C por 30 segundos; 3) Hibridación: 61°C por 1 minuto; 4) Extensión: 72°C por 30 segundos; 5) Lectura de datos: 88°C por 33 segundos. Los pasos 2 a 5 se repiten 40 veces. Hígado: 1) Activación: 95°C por 10 minutos; 2) Desnaturali zación: 95°C por 20 segundos; 3) Hibridación/ Extensión: 60°C por 1 minuto; 4) Lec tura de datos: 88°C por 33 segundos. Los pasos 2 a 4 se repiten 40 veces. 40 Materiales y métodos La detección del producto amplificado se monitoreó en el equipo de PCR en tiempo real, el cual mide el aumento de los niveles de fluorescencia en cada ciclo de PCR causados por la unión del SYBR Green al ADN doble cadena. El equipo determina un nivel de fluorescencia umbral dentro de la fase exponencial de la reacción. El número de ciclos que requiere cada muestra para alcanzar dicho umbral se conoce como Ct (Cycle threshold) , el cual está directamente relacionado con la cantidad inicial de molde presente en la PCR. Dado que se obtiene una señal siempre que haya moléculas de ADN doble cadena, a posteriori del ensayo de PCR se realizaron curvas de disociación (Curva de Melting ) para determinar la especificidad de la señal y así eliminar falsos positivos debidos a productos inespecíficos y dímeros de primers, entre otros. La expresión relativa de los transcriptos evaluados se calculó usando Ciclofilina como control endógeno. La expresión matemática utilizada para calcular estas relaciones fue: 2- Ct Dónde: Ct = Ct muestra - Ct referencia. Ct muestra= Ct gen en estudio - Ct gen endógeno. Ct referencia= promedio Ct de las muestras control. Para todos los genes evaluados se realizaron previamente ensayos de estandarización de los protocolos empleados, que involucraron la evaluación de curvas de rango dinámico de amplificación del transcripto de interés y posteriores curvas de disociación. HISTOLOGÍA PROCESAMIENTO DE MUESTRAS Cada tejido tuvo un protocolo de procesamiento específico: Las hipófisis, una vez disecadas, fueron fijadas en formalina (4% ) durante 3 hs, y deshidratadas por pasajes sucesivos en alcoholes de concentración creciente. Etanol 70° durante 1 hr, etanol 96° ON ( over night ) y dos pasajes por etanol 100° de media hora cada uno. Luego se realizaron dos pasajes por xileno (30 min cada uno) para ser posteriormente incluidas en parafina (3 hs a 56°C ). Las adrenales fueron incluidas siguiendo el mismo protocolo recién mencionado. Las porciones de tejido adiposo gonadal fueron fijadas en formalina (4% ) durante toda la noche. Al día siguiente se traspasaron a etanol 70° y 24 hs después a etanol 96° donde quedaron durante 12 hs. Luego se realizaron dos pasajes por etanol 100° de 1 hr cada uno y posteriormente, dos pasajes por xileno (1 hr cada uno). Finalmente fueron incluidas en parafina (6 hs a 56°C ). En el caso del páncreas se llevó a cabo el mismo protocolo de inclusión recién mencionado, con la diferencia de que este tejido se extrae completo (en lugar de una porción) y se incluye estirado sobre un papel para que no se pliegue sobre sí mismo. 41 Materiales y métodos En todos los casos, se cortaron secciones del taco de parafina de 4 µm de espesor en un micrótomo, se montaron en portaobjetos de vidrio cargados positivamente y se dejaron secar ON a 37°C . En el caso del hígado, se guardaron porciones embebidas en OCT a -70°C para ser luego cortadas secciones de 10 µm de espesor en crióstato. De esta manera, los lípidos los cuales son afines a los solventes orgánicos, se mantienen intactos. COLORACIONES En el caso de los hígados se realizó una coloración específica para visualizar contenido lipídico. Los cortes realizados en crióstato se lavaron en primer lugar con agua y luego con isopropanol al 60% ; se colorearon con una solución de Oil Red O (Sigma-Aldrich) durante 15 minutos, y se lavaron nuevamente con isopropanol 60% . Posteriormente, los núcleos fueron coloreados con hematoxilina (durante 20 segundos), y los tejidos fueron montados en un medio acuoso (10 g de gelatina, 60 ml de agua destilada, 70 ml de glicerol y 0,25 g de fenol). De esta manera, los lípidos fueron visualizados en color rojo, mientras que los núcleos en un azul-violáceo. La solución madre de Oil Red O consiste en 0,5 g de C.I . 26125 en 100 ml de isopropanol, la cual luego es diluida con 20 ml de agua destilada cada 30 ml para así preparar la solución de uso. Cortes en parafina de tejido adiposo gonadal fueron coloreados con hematoxilina/ eosina (H&E) para un posterior análisis morfométrico. Los cortes se desparafinaron en xileno durante 20 min y fueron luego, re-hidratados por pasajes sucesivos en alcoholes de concentración decreciente: 100°, 96°, 90° y 70° (10 min cada uno) , hasta que finalmente fueron lavados con agua. Se realizó una coloración con hematoxilina durante 20 seg, y luego de un lavado con agua, se prosiguió a colorear con eosina acuosa durante 1 min. Finalmente los cortes de tejido fueron deshidratados y montados. Análisis morfométrico del tejido adiposo gonadal: Para determinar el tamaño de los adipocitos, se capturaron imágenes (3 imágenes por sección y 2 secciones de tejido por animal) utilizando una cámara digital Canon PowerShot G6 acoplada a un microscopio Zeiss Axiostar plus con un objetivo de 40X. El análisis morfométrico se realizó a partir de 50 células haciendo uso del programa I mage J ( National I nstitutes of Health ). Se crearon rutinas automatizadas para medir áreas celulares, en las que se fijó una escala correspondiente al aumento 40X, para relacionar los pixeles con µm 2. Se delimitó manualmente el perímetro de cada adipocito y luego se calculó el área comprendida por el mismo. Por último, se calculó el área total del tejido analizado. La mediana de las áreas de los adipocitos fue calculada, y se realizó, para cada genotipo, una distribución de porcentaje de adipocitos de un determinado tamaño a intervalos de 200 µm 2. 42 Materiales y métodos Cortes en parafina de las adrenales fueron coloreados con H&E para un posterior análisis cualitativo. Se siguió el mismo protocolo que el utilizado para tejido adiposo gonadal (detallado previamente). INMUNOHISTOQUÍMICA (IHQ) Para la inmunomarcación se calentó la muestra en horno microondas sumergida en Buffer Citrato 0,01 M para la recuperación antigénica y se usó el método de streptavidina-biotina peroxidasa (ABC) como sistema de revelado. El protocolo básico empleado se muestra en la Tabla 5. 43 Materiales y métodos Tabla 5: Protocolo básico de I HQ. S e des parafinó e hidrató: Xilol Alc ohol 100° Alc ohol 96° Alc ohol 90° Alc ohol 70° PBS 2 pas ajes de 10 min c /u 10 min 10 min 10 min 10 min 2 pas ajes de 10 min c /u R ec uperac ión antigénic a: S e c alentó en horno mic roondas el B uffer C itrato 0,01 M, pH:6 PBS S e c alentó el buffer 3 min a 100% de potenc ia, s e agregaron las mues tras y s e c alentó 10 min y 20 min apagado. 2 pas ajes de 10 min c /u Inhibic ión de peroxidas as endógenas : Agua oxigenada 3% en P B S PBS 30 min 2 pas ajes de 10 min c /u B loqueo de uniones ines pec ífic as : L ec he en polvo des c remada 5% en P B S 1h Antic uerpo primario: S e inc ubó c on el antic uerpo es pec ífic o PBS O N (en c ámara húmeda) a 4°C 2 pas ajes de 10 min c /u Antic uerpo s ec undario: Antic uerpo s ec undario biotinilado PBS 1 h (en c ámara húmeda) 2 pas ajes de 10 min c /u S is tema de revelado: AB C (1/50 A + 1/50 B en P B S ) PBS S us trato enz imátic o ( 5 µl de H 2 O 2 ) + c romógeno (2,5 mg de diaminobenc idina: D AB ) dis uelto en 10 ml de P B S Agua 30 min (en c ámara húmeda) 2 pas ajes de 10 min c /u 3 min 2 pas ajes de 10 min c /u S e c ontrac oloreó. S e des hidrataron y montaron las mues tras En todos los casos se realizaron controles negativos de especificidad sustituyendo el anticuerpo primario con PBS. Evaluación de parámetros angiogénicos Se evaluaron parámetros angiogénicos en hipófisis embebidas en parafina, de hembras de 10 meses de edad de ambos genotipos. Mediante la detección por I HQ de un antígeno específico 44 Materiales y métodos de células endoteliales: CD31 ( cluster of differentation molecule) también denominado PECAM ( platelet endothelial cell adhesion molecule-1), se inmunomarcaron tanto vasos maduros como inmaduros. Para ello se siguió el protocolo descripto en la Tabla 5. La Tabla 6 detalla los reactivos utilizados en esta I HQ. Tabla 6: Detalle de los reactivos utilizados. Anticuerpo primario Dilución utilizada Anticuerpo secundario biotinilado Dilución utilizada Cromógeno empleado Contracoloración anti-CD31* 1:200 anticabra 1:100 DAB Hematoxilina *Proveedor: Santa Cruz Biotechnology, Santa Cruz CA. Cuantificación: Se cuantificó el área vascular relativa utilizando cortes histológicos de I HQ para la detección de CD31 combinado con una técnica de análisis de imágenes. Para la obtención de las imágenes se utilizó una cámara digital Canon PowerShot G6 acoplada a un microscopio Zeiss Axiostar plus. Se obtuvieron imágenes de las hipófisis (5 imágenes por sección y 2 secciones de tejido por animal) usando un objetivo de 40X. Luego, utilizando el programa I mage J ( National I nstitutes of Health ) se crearon rutinas automatizadas para medir áreas vasculares. Se fijó una escala correspondiente al aumento 40X, para relacionar los pixeles con µm 2. Se delimitó manualmente el perímetro de cada vaso positivo para CD31 y luego se calculó el área comprendida por el mismo. Por último, se calculó el área total de la foto analizada. El área vascular relativa se determinó dividiendo el área vascular absoluta (definida como la suma de las áreas de todos los vasos correspondientes a una foto) por el área total de cada foto. Finalmente, se calculó el promedio de todas las áreas relativas correspondientes a cada animal. Evaluación de parámetros de proliferación La medida de proliferación celular se ha usado para predecir el comportamiento tumoral. Un antígeno utilizado para evaluar el índice de proliferación es el antígeno nuclear de células en proliferación (PCNA). En 1978 se describió un suero autoinmune aislado de pacientes con lupus eritematoso sistémico, que era capaz de reconocer un antígeno nuclear presente en las células proliferantes, este antígeno fue denominado PCNA (145;146). Es una proteína de 36 KDa, acídica nuclear, no histónica, que actúa como proteína auxiliar de la enzima ADN polimerasa delta, siendo indispensable para la síntesis de ADN. La expresión de PCNA comienza en la fase G1 del ciclo celular, alcanza un máximo en la fase S y comienza a disminuir hacia la fase G2/ M. Tiene una vida media de más de 20 hs. 45 Materiales y métodos Se determinó mediante I HQ el número de núcleos inmunopositivos para PCNA en hipófisis embebidas en parafina, de hembras de 10 meses de edad de ambos genotipos. El protocolo básico empleado se muestra en la Tabla 5. La Tabla 7 detalla los reactivos utilizados. Tabla 7: Reactivos utilizados. Anticuerpo primario Dilución utilizada Anticuerpo secundario biotinilado Dilución utilizada Cromógeno empleado Contracoloración anti-PCNA* 1:200 anticonejo 1:100 DAB Hematoxilina *Proveedor: Santa Cruz Biotechnology, Santa Cruz CA. Cuantificación: Se cuantificó el número de núcleos marcados con PCNA utilizando cortes histológicos sometidos a ensayos de I HQ combinado con una técnica de análisis de imágenes. La obtención de las imágenes se realizó de igual manera que la detallada anteriormente. Se obtuvieron 5 imágenes por sección y 2 secciones de tejido por animal, utilizando un objetivo de 100X de aumento. Con el programa I mage J se marcaron manualmente cada uno de los núcleos inmunopositivos para PCNA presentes en cada imagen (utilizando la opción de Cell Counter ), y luego se cuantificó el número de núcleos totales. Finalmente, se calculó el porcentaje de núcleos PCNA positivos. Evaluación de parámetros endócrinos Hipófisis Se determinó mediante I HQ el número de células inmunopositivas para α-MSH en la intermedia de cortes de hipófisis embebidos en parafina. α-MSH es un péptido de 13 aminoácidos producido en el lóbulo intermedio de la hipófisis, como también en el sistema nervioso central. El protocolo llevado a cabo para la inmunomarcación se detalla en la Tabla 5. La Tabla 8 describe los reactivos utilizados en esta I HQ. Tabla 8: Reactivos utilizados. Anticuerpo primario Dilución utilizada Anticuerpo secundario biotinilado Dilución utilizada Cromógeno empleado Contracoloración anti-αMSH* 1:10000 anticonejo 1:100 DAB Hematoxilina 46 Materiales y métodos *Proveedor: Santa Cruz Biotechnology, Santa Cruz CA. Cuantificación: Se cuantificó el número de células marcadas con α-MSH. Se obtuvieron 3 imágenes por sección y 2 secciones de tejido por animal utilizando un objetivo de 100X, seleccionando fotos que incluyan la zona intermedia de la hipófisis. Se marcaron manualmente, utilizando el programa I mage J, cada una de las células inmunopositivas para αMSH presentes en cada imagen (utilizando la opción de Cell Counter ). Luego se cuantificó el número de células totales (evidenciadas por la coloración de sus núcleos con hematoxilina). Se calculó finalmente el porcentaje de células con α-MSH en función de las totales. Páncreas Se realizaron inmunomarcaciones diferentes en cortes seriados de páncreas enteros. Para cada animal se realizó una I HQ contra insulina en una sección de tejido, y la siguiente sección contra glucagón. El protocolo general se detalla en la Tabla 5 y en las Tabla 9 y 10 se describen los reactivos utilizados en cada caso. Tabla 9: Reactivos utilizados. Anticuerpo primario Dilución utilizada Anticuerpo secundario biotinilado Dilución utilizada Cromógeno empleado Contracoloración anti-Insulina* 1:300 anticobayo 1:200 DAB Hematoxilina *Proveedor: Sigma, Saint Louis, MO. Tabla 10: Reactivos utilizados. Anticuerpo primario Dilución utilizada Anticuerpo secundario biotinilado Dilución utilizada Cromógeno empleado Contracoloración anti-Glucagon* 1:300 anticonejo 1:100 DAB Hematoxilina *Proveedor: Santa Cruz Biotechnology, Santa Cruz CA. Análisis morfométrico: Para la obtención de las imágenes se utilizó una cámara digital Canon PowerShot G6 acoplada a un microscopio Zeiss Axiostar plus. Se obtuvieron tantas imágenes como fueran necesarias para cubrir toda la sección de tejido de cada animal utilizando un objetivo de 5X. 47 Materiales y métodos Luego se fotografiaron cada uno de los islotes identificados en cada animal, utilizando un objetivo 40X. Se determinaron los siguientes parámetros: - Área promedio del islote (µm 2): Utilizando el programa I mage J se crearon rutinas automatizadas para medir áreas de los islotes. Se fijó una escala correspondiente al aumento 40X, para relacionar los pixeles con µm 2. Se delimitó manualmente el perímetro de cada islote y luego se calculó el área comprendida por el mismo. Finalmente, se calculó el promedio de todas las áreas de islotes correspondientes a cada animal. - Porcentaje de islotes chicos-medianos-grandes: Habiendo calculado el área de cada uno de los islotes, se calculó para cada animal, el porcentaje de islotes menores a 5000 µm 2 (considerando a los mismos como “chicos”) y mayores a 5000 µm 2 (considerándolos “medianos y grandes”). - Número de islotes por área (densidad de islotes): Se marcó manualmente cada uno de los islotes presentes en cada imagen (utilizando la opción de Cell Counter del programa I mage J) y luego se calculó el área total del páncreas (sumatoria de áreas totales de todas las fotos tomadas con el objetivo 5X). El número de islotes por unidad de área se determinó dividiendo el número de islotes totales (definido como la suma de todos los islotes correspondientes al total de las fotos) por el área total del tejido analizado. - Área ocupada por islotes: Se calculó el área total del páncreas (sumatoria de áreas de todas las fotos). El área ocupada por islotes (área relativa) se determinó dividiendo el área de islotes absoluta (definida como la suma de las áreas de todos los islotes correspondientes a un animal) por el área total del páncreas. - Área ocupada por células (Insulino positivas): Las imágenes de los cortes histológicos inmunocoloreadas fueron convertidos a escala de grises con la herramienta de máscaras, seleccionando previamente el color correspondiente a la marca específica (de manera de independizarnos de la coloración con hematoxilina). Por segmentación de color se identificaron las células marcadas con el anticuerpo anti-glucagón (células αpositivas) y se cuantificó el área total ocupada por ellas. Por último, se calculó el área total de la foto analizada. El área inmunomarcada se determinó dividiendo el área inmunoreactiva por el área total de cada foto. Considerando despreciable la cantidad de otros tipos celulares en el islote (como ser células δ), se calculó el área ocupada por células , como la diferencia entre el área total del tejido y la ocupada por las células αpositivas. A su vez, calculamos la razón comprendida entre el área ocupada por células y por células α, mediante el cociente de las mismas. - % de células α (Glucagón positivas): Se marcaron manualmente, utilizando el programa I mage J, cada una de las células inmunopositivas para glucagón presentes en cada islote (utilizando la opción de Cell Counter ). Luego se cuantificó el número de células 48 Materiales y métodos totales en el mismo islote (evidenciadas por la coloración de sus núcleos con hematoxilina). Se calculó finalmente el porcentaje de células con Glucagón en función de las totales. - % de células (I nsulino positivas): Considerando despreciable la cantidad de otros tipos celulares en el islote (como ser células δ), se calculó el % de células , como la diferencia entre las células totales presentes en el islote y las células α-positivas. - Área promedio de cada célula : Se delimitó manualmente el perímetro de 30 células insulino positivas de cada animal, y se calculó el área comprendida por cada una de ellas. Finalmente, se calculó el promedio de todas las áreas correspondientes a cada animal. Evaluación de parámetros de stemness Se realizó la detección mediante I HQ de parámetros relacionados a stem cells en cortes de hipófisis incluidas en parafina. En particular, se estudiaron NOTCH-1, NOTCH-3, HES-1 y SOX-2. El protocolo llevado a cabo para la inmunomarcación se detalla en la Tabla 5 y las Tablas 11, 12, 13 y 14 describen los reactivos utilizados en cada caso. Tabla 11: Reactivos utilizados. Anticuerpo primario Dilución utilizada Anticuerpo secundario biotinilado Dilución utilizada Cromógeno empleado Contracoloración anti-NOTCH1* 1:700 anticonejo 1:100 DAB Hematoxilina *Proveedor: Merck Millipore. Tabla 12: Reactivos utilizados. Anticuerpo primario Dilución utilizada Anticuerpo secundario biotinilado Dilución utilizada Cromógeno empleado Contracoloración anti-NOTCH3* 1:200 anticonejo 1:100 DAB Hematoxilina *Proveedor: Santa Cruz Biotechnology, Santa Cruz CA. Tabla 13: Reactivos utilizados. 49 Materiales y métodos Anticuerpo primario Dilución utilizada Anticuerpo secundario biotinilado Dilución utilizada Cromógeno empleado Contracoloración anti-HES1* 1:400 anticonejo 1:100 DAB Hematoxilina *Proveedor: Merck Millipore. Tabla 14: Reactivos utilizados. Anticuerpo primario Dilución utilizada Anticuerpo secundario biotinilado Dilución utilizada Cromógeno empleado Contracoloración anti-SOX2* 1:400 anticonejo 1:100 DAB Hematoxilina *Proveedor: Abcam. Cuantificación: En los casos de NOTCH-1, NOTCH-3 y HES-1, se cuantificó el área positivamente inmunomarcada por los mismos, utilizando cortes histológicos sometidos a ensayos de I HQ combinado con una técnica de análisis de imágenes. Se obtuvieron imágenes de las adenohipófisis inmunomarcadas (3 imágenes por sección y 2 secciones de tejido por animal) usando un objetivo de 40X. Luego, utilizando el programa I mage J se crearon rutinas automatizadas para medir áreas inmunomarcadas. Se fijó una escala correspondiente al aumento utilizado (40X), relacionando los pixeles con µm 2. Las imágenes de los cortes histológicos inmunocoloreadas fueron convertidos a escala de grises con la herramienta de máscaras, seleccionando previamente el color correspondiente a la marca específica (de manera de independizarnos de la coloración con hematoxilina). Por segmentación de color se identificaron las células marcadas con cada uno de los anticuerpos y se cuantificó el área total ocupada por ellas. Por último, se calculó el área total de la foto analizada. El porcentaje de área inmunomarcada se determinó dividiendo el área inmunoreactiva por el área total de cada foto, multiplicado por 100. Finalmente, se calculó el promedio de todos los porcentajes de área correspondientes a cada animal. En el caso de SOX-2, al obtenerse tanto marca nuclear como citoplasmática se cuantificó en forma discriminada el número de células y núcleos marcados positivamente. Se obtuvieron 4 imágenes por sección y 2 secciones de tejido por animal utilizando un objetivo de 40X, seleccionando fotos que solamente incluyan la adenohipófisis (ya que no se obtuvieron representantes suficientes que presentaran la zona del cleft ). Se marcaron manualmente, 50 Materiales y métodos utilizando el programa I mage J, cada una de las células y núcleos inmunopositivos para SOX-2 presentes en cada imagen (utilizando la opción de Cell Counter con canales diferentes para cada uno). Luego se cuantificó el número de células totales (evidenciadas por la coloración de sus núcleos con hematoxilina). Se calculó finalmente el porcentaje de células y núcleos con SOX-2. INMUNOFLUORESCENCIA Se analizó cualitativamente mediante inmunofluorescencia la cantidad de somatotropos mediante una inmunomarcación contra GH. Para ello, se calentó la muestra en horno microondas como método de recuperación antigénica. El protocolo básico empleado se muestra en la Tabla 15 y los detalles de los reactivos en la Tabla 16. 51 Materiales y métodos Tabla 15: Protocolo básico de inmunofluorescencia. S e des parafinó e hidrató: Xilol Alc ohol 100° Alc ohol 96° Alc ohol 90° Alc ohol 70° PBS 2 pas ajes de 10 min c /u 10 min 10 min 10 min 10 min 2 pas ajes de 10 min c /u R ec uperac ión antigénic a: S e c alentó en horno mic roondas el B uffer C itrato 0,01 M, pH:6 S e c alentó el buffer 3 min a 100% de potenc ia, s e agregaron las mues tras y s e c alentó 10 min y 20 min apagado. 2 pas ajes de 10 min c /u PBS B loqueo de uniones ines pec ífic as : L ec he en polvo des c remada 5% en P B S 1h Antic uerpo primario: S e inc ubó c on el antic uerpo es pec ífic o O N (en c ámara húmeda) a 4°C 2 pas ajes de 10 min c /u PBS Antic uerpo s ec undario: Antic uerpo s ec undario 1:30 hs (en c ámara húmeda) en os c uridad 2 pas ajes de 10 min c /u PBS Algunas mues tras s e c ontrac olorearon. En todos los casos se realizaron controles negativos de especificidad sustituyendo el anticuerpo primario con PBS. Tabla 16: Reactivos utilizados. Anticuerpo primario Dilución utilizada Anticuerpo secundario biotinilado Dilución utilizada Contracoloración anti-GH* 1:800 antimono Texas Red** 1:100 Ninguna *Proveedor: Dr. A. F. Parlow, National Hormone and Pituitary Program, Torrance, CA. **Proveedor: Santa Cruz Biotechnology, Santa Cruz CA. 52 Materiales y métodos CITOMETRÍA DE FLUJO Protocolo de dispersión de células con colagenasa Se realizó el protocolo partiendo de dos grupos de hipófisis, uno proveniente de ratones hembra Drd2loxP/ loxP y otro de ratones hembra lacDrd2KO, ambos de 10 meses de edad. Todas fueron disecadas en el momento, provenientes de animales recién decapitados. - Se colocaron las hipófisis de cada grupo en una caja de Petri conteniendo medio A, y utilizando bisturíes se cortó cada hipófisis en 4 o 5 pedazos (cada uno de aproximadamente 2x2 mm). Posterior a un lavado con medio A, se colocaron 8 ml de colagenasa tipo I V (1 mg/ ml), y se incubó durante 40 min a 37°C (en oscuridad). - Se disgregaron las hipófisis utilizando una pipeta de vidrio, resuspendiendo enérgicamente cada 20 min (se resuspendieron al menos 3 veces dejando entre cada una las hipófisis en la estufa a 37°C durante 20 min). - Una vez disgregadas, se procedió al filtrado del sobrenadante a través de un mesh de nylon de 50 µm, quedando finalmente en un falcon de 50 ml. Luego del filtrado se lavó la caja de Petri con 3 ml de PBS + 20% SFB (suero fetal bovino) y se filtró también por si hubieran quedado células. A la solución ya filtrada contenida en el falcon, se le duplicó su volumen con PBS + 20% SFB de manera de inactivar la colagenasa. - Se agregaron 3 ml de BSA 3% al fondo de manera de formar una película protectora. - Se llevó a cabo una centrifugación a 190g por 10 min a temperatura ambiente, y luego se tiró el sobrenadante por volcado. - Se resuspendió el pellet en 1 ml de PBS + 20% SFB, y se contaron las células utilizando trypan blue junto con una cámara de Neubauer. - Conociendo el número de células totales obtenido, se procedió a separar las mismas en distintos subgrupos experimentales como se detalla en la Figura 14. 53 Materiales y métodos Figura 14: Las células totales obtenidas en cada grupo se dividen en 5 subgrupos (en falcons de 15 ml). En simultáneo se realizan cuatro controles que consisten en: el control de isotipo (el cual contiene los tres isotipos) y las tres incubaciones simples (con cada anticuerpo por separado). Finalmente la muestra corresponde a los tres anticuerpos juntos que es lo que nos interesa evaluar. - A las células contenidas en cada falcon de 15 ml se les colocó 5 ml de PBS 1X (exceso de PBS para lavar). - Se centrifugaron a 300g por 10 min a temperatura ambiente, y se eliminó el sobrenadante por volcado. - Se colocó una solución de bloqueo (PBS + 2,5% SFB) de manera tal de obtener 1 millón de células vivas cada 200 µl de solución de bloqueo. Marcación para CD45/CD31/CD44 - Partimos de 1 millón de células vivas cada 200 µl de solución de bloqueo. - Se agregaron los anticuerpos directamente a la solución de bloqueo: “Muestra” = PE-CD45 (stock: 0,2 mg/ ml; concentración final de uso: 10 µg/ ml) junto con FI TC-CD31 (stock: 0,5 mg/ ml; concentración final: 25 µg/ ml) y APC-CD44 (stock: 0,2 mg/ ml; concentración final: 10 µg/ ml). “Control de Isotipo” = control de isotipo PE-ratón (stock: 0,2 mg/ ml; concentración final: 10 µg/ ml) junto con control de isotipo FI TC-rata (stock: 0,5 mg/ ml; concentración final: 25 µg/ ml) y control de isotipo APC-rata (stock: 0,2 mg/ ml; concentración final: 10 µg/ ml). “Incubaciones Simples” = PE-CD45 (stock: 0,2 mg/ ml; concentración final: 10 µg/ ml) 54 Materiales y métodos FI TC-CD31 (stock: 0,5 mg/ ml; concentración final: 25 µg/ ml) APC-CD44 (stock: 0,2 mg/ ml; concentración final: 10 µg/ ml). - Se incubó por 30 min en hielo y en oscuridad, mezclando cuidadosamente cada 10 min. - Se agregaron 2 ml de solución de bloqueo a cada tubo. - Se centrifugaron por 10 min a 190g y 4°C. - Se eliminó el sobrenadante y se resuspendió el pellet con solución de bloqueo para obtener una concentración de 1x106 células/ ml. Se mantuvo en hielo y protegido de la luz hasta el FACs. RADIOINMUNOENSAYOS (RIAs) NIVELES SÉRICOS DE HORMONAS Prolactina Con los sueros obtenidos en cada uno de los casos se procedió a medir los niveles de prolactina mediante RI A, siguiendo el protocolo que se detalla a continuación. - La curva de concentraciones del estándar de referencia y la dilución de las muestras (8 l de suero) fueron realizadas en buffer de ensayo: PBS (buffer fosfato 0,01M, NaCl 0,15M y 0,01% azida sódica, pH final de 7,4) con 1% de albúmina de huevo (EA). Tanto los puntos de la curva (llevados a cabo a partir del estándar de ratón RP3) como las muestras se analizaron por duplicado. - El primer anticuerpo (I gG policlonal de conejo anti-prolactina de ratón, NHPP-NI DDK, dilución de trabajo 1:100000) y la hormona marcada (aproximadamente 18000 cpm/ tubo) fueron agregados a los tubos patrones y de muestra, los cuales fueron posteriormente incubados a temperatura ambiente durante 24 hs. - Al día siguiente, el segundo anticuerpo (suero policlonal de oveja anti-I gG de conejo, dilución de trabajo 1:90) fue agregado al ensayo e incubado a temperatura ambiente durante 2 hs y 30 min a 4°C. Luego de esta incubación se separó la hormona libre de la unida al anticuerpo específico por centrifugación refrigerada (4°C, Bec kman I nstruments) a 2000 rpm durante 30 min. El sobrenadante fue aspirado y se midió la radioactividad asociada al precipitado (correspondiente al complejo hormona-anticuerpo) en un contador Gamma [ Hewlett Packard (eficiencia: 82% )] . Los coeficientes de variación intra- e inter-ensayo fueron 6,3% y 12.5% , respectivamente. IGF-I Las muestras de suero para RI A de I GF-I (10 μl) así como los estándares de I GF-I fueron sujetos al método de crioprecipitación con etanol ácido para extraer el I GF-I de las proteínas de unión a I GF-I (147). Para ello, se tomaron 10 µl de suero y se les agregó 40 µl de solución ácidoetanólica (87 % v/ v etanol + 12,5 v/ v HCl 2 N). Las muestras se mezclaron con vórtex y luego 55 Materiales y métodos fueron incubadas a temperatura ambiente durante 30 min. Posteriormente, se centrifugaron a 3000 g durante 30 min a 4°C, se tomó el sobrenadant e y se agregaron 20 µl de Tris Base 0,855 M. Se mezclaron con vórtex y se dejaron crioprecipitar a -20°C durante 1 hora, luego de la cual, se centrifugaron durante 30 min a 3000 g a 4°C para fi nalmente tomar el sobrenadante. Para el RI A se usaron 10 µl de este extracto de suero por duplicado. El protocolo, una vez obtenidos los extractos, fue semejante al RI A de prolactina previamente descripto. I GF-I se determinó usando un anticuerpo (I gG policlonal de conejo anti-I GF-I de ratón, 1:100000; UB2-495) provisto por los Dres. L. Underwood y J.J. Van Wyk, y distribuido por el “Hormone Distribution Program ” del NIDDK. Se usó I GF-I humano recombinante (rhI GF-I , Chiron Corp., Emeryville, CA) como radio-ligando y como ligando no marcado. La sensibilidad del ensayo fue de 6 pg por tubo. Los coeficientes de variación intra- e inter-ensayo fueron 8,2% y 14,1% , respectivamente. Progesterona A partir de sueros de ratones hembra lacDrd2KO y Drd2 loxP/ loxP se midieron los niveles séricos prog esterona. Se extrajeron alícuotas de 50 μl de suero dos veces con 1 ml de hexano por vez, en tubos de extracción. Los extractos se evaporaron durante 1 hora en un baño a 50°C y se disolvieron, posteriormente, en 2 ml de solución PBS-gelatina (compuesta por solución de fosfato 0,075 M, azida sódica 0,02% , NaCl 154 nM, pH 7,0, conteniendo 0,1% (P/ V) de gelatina. Las muestras resuspendidas permanecieron durante 24 hs a 4°C. De estos extractos se cargaron 500 μl por duplicado. Los estándares se prepararon a partir de una solución de progesterona (Sigma) de 1 μg/ml en etanol, en un rango de 12,5 a 6400 pg por tubo en 500 μl de PBS‐gelatina. Se agregaron 200 μl de anti‐progesterona (Niswender GDN 337), disuelto en PBS‐gelatina en una dilución de 1:2400 a los tubos conteniendo la curva y las muestras. A todos los tubos de ensayo se les agregó la progesterona tritiada (New England Nuclear) de una actividad específica de aproximadamente 100 Ci/ nmol y una pureza radioquímica del 99% , disuelta en PBS‐gelatina (aproxim adamente 10.000 cpm/100 μl). Las muestras y la curva, con el trazador y el anticuerpo, fueron incubadas durante 24 hs a 4°C. Pasado el tiempo de incubación, se agregó a cada tubo 200 μl de una suspensión de Carbón (0,5% ) ‐Dextrano (0,05% ) en PBS, recientemente preparada. De esta forma se logra adsorber la hormona libre, separando la misma del complejo hormona-anticuerpo. Se agitó con un agitador mecánico y se incubó durante 10 minutos a temperatura ambiente. Luego se centrifugó a 3000 rpm durante 10 minutos a 4°C. Finalmente, se tomaron 700 μl del sobrenadante (conteniendo el complejo hormona‐anticuerpo) y se les agregó 1 ml de líquido centellante Optiphase “Hisafe”3. Se registró la actividad de los viales en un contador Beta (Beckmann, EEUU). La sensibilidad del ensayo fue de 500 pg, y los coeficientes de variación intra e inter ‐ ensayo fueron de 7,1% y 12,15% , respectivamente. 56 Materiales y métodos NIVELES INTRAHIPOFISARIOS DE HORMONAS Preparación de las muestras: Las hipófisis de los ratones se homogenizaron en hielo en buffer Tris-HCl 60 mM - EDTA 1 mM, pH 6,8 (TE) con un mezcla de inhibidores de proteasas: fluoruro de fenil metil sulfonilo (PMSF) y otra mezcla preformada de los inhibidores TPCK, TAME, ZPCK y TLCK (Sigma, Saint Louis, MO) utilizando un homogenizador manual. El volumen de TE más inhibidores fue proporcional al peso del tejido (100 µl de buffer por cada 1,5 mg de hipófisis). El homogenato se centrifugó a 3000g durante 5 min a 4°C. Se extrajo cuidadosamente el sobrenadante y se descartó el pellet. Una alícuota del sobrenadante se separó para la medición de proteínas con el kit de cuantificación del Qubit-fluorometer TM (I nvitrogen Life Technologies, Buenos Aires) y el resto se guardó a -20°C para su posteri or utilización. Se diluyó 1 µl de homogenato en buffer EA 1% de modo que hubiera 0,0976 µg de proteínas cada 25 µl (volumen de muestra que se carga para la realización del RI A). De acuerdo a calibraciones previas, esta cantidad de proteínas era la adecuada. Prolactina Se llevó a cabo del mismo modo que lo explicado anteriormente para las mediciones en suero. GH La muestra, previa dilución adecuada, se llevó a 200 μl en PBS-EA 1% , y se incubó con 100 μl de primer anticuerpo diluido en PBS- EDTA y 100 μl de hormona iodinada (diluida en PBS-EA 1% de manera de ten er entre 17000 y 20000 cpm en 100 μl), durante 24 hs a temperatura ambiente. Posteriormente se agregó 50 μl de segundo anticuerpo (anti-mono I gG elaborado en conejo, disuelto en PBS-EDTA en una dilución 1:105000), y se incubó durante 12 hs a temperatura ambiente. Se incubó finalmente con 100 μl de un tercer anticuerpo (anti conejo IgG 1:50 en PBS 0,01M -NRS 0,3% ) durante aproximadamente 20 hs a 4º C. A continuación, se separó la hormona libre por centrifugación a 2000 rpm durante 30 minutos y a 4º C, y se aspiró el sobrenadante. Se contó la hormona unida en un contador Gamma [ Hewlett Packard (eficiencia: 82% )] . Los coeficientes de variación intra e inter ‐ ensayo fueron de 12,8% y 13,2% , respectivamente. NIVELES INTRAPANCREÁTICOS DE INSULINA Preparación de las muestras: Los páncreas fueron pesados y homogeneizados en solución ácido etanólica: 87,5% v/ v etanol + 12,5% v/ v HCl 2N (1 ml de solución cada 0,4 g de tejido), para luego dejarse incubando durante 1 hr a 4º C. 57 Materiales y métodos Se centrifugaron a 800g durante 10 minutos a 4º C y se transfirió el sobrenadante a otro tubo (extracto 1). Se tomaron 100 µl del extracto 1 (el resto se guardó -70º C) y se neutralizó con 60 µl de Tris base 0,855 M. Se mezcló por inversión obteniendo de este modo el extracto 2, el cual se guardó a -70º C hasta su utilización. Se tomaron 2 µl del extracto 2 y se cuantificaron las proteínas con el Qubit TM Quantitation Kit (I nvitrogen Life Technologies, Buenos Aires). Para realizar el RI A se calculó el volumen necesario para cargar 5 ng de proteínas totales por tubo, siendo necesario realizar una dilución 1/ 500 con buffer I GF-I . Protocolo de RIA: La curva de concentraciones del estándar de referencia y la dilución de las muestras fue realizada en buffer de ensayo: 200 mg/ l de sulfato de protamina, fosfato de sodio monobásico 4,14 g/ l, 0,5% de detergente tween, y EDTA 0,1M, pH final 7,5. Se utilizó como estándar para hacer las curvas insulina humana provista por los Laboratorios Beta (Buenos Aires, Argentina). El primer anticuerpo (I gG policlonal de cobayo anti-insulina de ratón, 1:50000, Sigma, St. Louis, Missouri, EEUU) y la hormona marcada (aproximadamente 10000 cpm/ tubo) fueron agregados a los tubos patrones y de muestra, los cuales fueron posteriormente incubados a temperatura ambiente durante 24 hs. Al día siguiente, el segundo anticuerpo (anti-I gG de cobayo hecho en conejo, 1:100000) fue agregado al ensayo e incubado a 4°C por 8 hs. Luego de esta incubación se agregó el tercer anticuerpo (suero policlonal de oveja anti-I gG de conejo, 1:90) y se incubó a 4°C por 12 hs. Se separó la hormona libre de la unida al anticuerpo específico por centrifugación refrigerada (4°C, Beckman I nstrument s) a 3000 rpm durante 30 min. El sobrenadante fue aspirado y se midió la radioactividad asociada al precipitado (correspondiente al complejo hormona-anticuerpo) en un contador Gamma (Perkin Elmer). ELISA PARA VEGF Las concentraciones séricas e intrahipofisarias de VEGF fueron analizadas utilizando un ELI SA específico en el cual se siguieron las instrucciones del manufacturador (Quantikine M Mouse VEGF; R&D Systems). Alícuotas de 15 µl de suero fueron utilizadas por duplicado. En el caso de las mediciones intrahipofisarias se utilizaron homogenatos de hipófisis a partir de los cuales se cargaron 30 µg de proteínas en 15 µl. El límite inferior de detección del ensayo fue de 7,8 pg/ ml, mientras que el coeficiente de variación intra-ensayo de 6,7% . MEDICIÓN DE GLUCÓGENO HEPÁTICO Una porción del tejido hepático fue recolectada en nitrógeno líquido. De cada muestra se tomó entre 20 a 25 mg, registrando el peso exacto. Luego el tejido se transfirió a un recipiente 58 Materiales y métodos conteniendo 400 μl de KOH 30% y se dejó incubar durante 20 minutos en un baño de agua hirviendo. Luego de enfriar cada tubo, se agregó 50 µl del co-precipitante, una solución de sulfato de sodio saturada 5% . Al cabo de un minuto se agregó 1 ml de etanol 50° y mediante una centrifugación (5 min a velocidad máxima) se precipitó el glucógeno extraído. Se eliminó el sobrenadante y se agregó 0,5 ml de agua hexadestilada para resuspender el glucógeno y nuevamente se agregó 1 ml de etanol 50° para re-precipitar el glucógeno (centrifugación de por medio), con la finalidad de purificar la extracción del mismo. El paso siguiente fue eliminar completamente el sobrenadante y resu spender el precipitado en 50 μl de agua. Luego, se agregaron bajo campana 3 ml de la solución de Antrona (Merck Millipore) al 0,15% previamente preparada con solución de agua con ácido sulfúrico (en relación 30:70, agua:ácido). Se dejó en un baño a 90°C por 20 minutos y luego se llevaron los tubos a hielo, donde se dejaron reposar durante 15 min. Luego se sembró cada muestra por duplicado (300 µl) al igual que la curva patrón (detalle más abajo) y se midió la absorbancia a 620 nm en un lector de placas de ELI SA (LECTOR DE MI CROPLACAS MULTI SKAN FC, THERMO SCI ENTI FI C). En paralelo se realizó una curva patrón utilizando distintas concentraciones de glucógeno (Sigma), llevadas a 50 µl (desde 150 µg a 2,34 µg) y se continuó desde el agregado del reactivo Antrona. ANÁLISIS ESTADÍSTICO Las pruebas estadísticas realizadas en los diferentes ensayos se llevaron a cabo utilizando el programa STATI STI CA 7.0 (StatSoft I nc., EEUU), mientras que los supuestos fueron corroborados utilizando el programa Statistix 8.0. En la leyenda de cada figura, de la sección resultados, se indican las pruebas estadísticas utilizadas. En todos los casos los resultados fueron expresados como las medias ± el error estándar de la media (E.S.M). A modo de resumen, se utilizó Prueba de T en aquellas comparaciones entre sólo dos grupos, como ser: la comparación del peso de los tejidos; contenido de prolactina y GH intrahipofisaria medidos por RI A; en las I HQ de PCNA, CD31 y α-MSH, entre otras. Se analizaron mediante ANOVA de dos factores para medidas repetidas (AMR) los datos obtenidos en las curvas de peso y talla corporal, los niveles de prolactina sérica a lo largo del desarrollo, GTT, GSI S e I TT, como también la ganancia de peso en el test de sensibilidad a leptina. Se utilizó ANOVA de dos factores para medidas independientes en las determinaciones de ingesta de alimento, contenido de glucógeno, expresión de ARNm hepático de Cyp , como también todas las expresiones de ARNm en condiciones de ayuno y realimentación. Los porcentajes fueron analizados por Prueba de Х2, como ser el caso de las etapas del ciclo estral, y los tamaños de los islotes pancreáticos (chicos y medianos-grandes). Las medianas de los tamaños de adipocitos fueron 59 Materiales y métodos analizadas por el Test de comparación de medianas (Х2). Para las correlaciones, ya sea entre los niveles de leptina séricos y el peso corporal, o entre los niveles de leptina y prolactina séricos, se utilizó la Prueba de Spearman. En los casos en los que fueron necesarias pruebas post hoc se realizaron comparaciones de Fisher. Un P < 0,05 fue considerado significativo. Comparaciones paramétricas o no paramétricas fueron llevadas a cabo según el cumplimiento o no de supuestos de normalidad y homogeneidad de varianzas, y de ser necesario, se realizaron transformaciones de los datos. 60 Capítulo I - Introducción CAPÍTULO I – EL RATÓN lacDrd2KO COMO MODELO EXPERIMENTAL DE PROLACTINOMA RESISTENTE A DOPAMINA. INTRODUCCIÓN PROLACTINOMAS Los prolactinomas son los tumores hipofisarios más frecuentes, y como el resto de los adenomas hipofisarios tienen un origen monoclonal. Se ha reportado en mujeres que el 35% de los tumores hipofisarios producen prolactina y un 6% producen prolactina junto con otra hormona hipofisaria. La secreción excesiva de prolactina causa oligomenorrea o amenorrea, y galactorrea, y por estos síntomas los prolactinomas son detectados precozmente en las mujeres (Figura 15). En los hombres en cambio, la galactorrea es rara y los síntomas de hipogonadismo tales como impotencia y falta de libido llevan a la consulta médica en forma tardía (32). Figura 15: Regulación hipotálamo-hipofisaria de la secreción de prolactina. La pérdida del tono inhibitorio dopaminérgico puede producir la formación del prolactinoma. Modificado de (32). Los prolactinomas son generalmente divididos en dos categorías: microprolactinomas (< 10 mm de diámetro) y macroprolactinomas (> 10 mm de diámetro). Aunque los prolactinomas invasivos pueden comprimir el quiasma óptico, causar daño hipotalámico y erosionar el tercer ventrículo, rara vez son metastásicos y el origen de verdaderos carcinomas hipofisarios es muy poco frecuente (107). En las mujeres en período fértil son más frecuentes los microprolactinomas 61 Capítulo I - Introducción mientras que en los hombres y en mujeres post-menopáusicas predominan los macroprolactinomas (148). Una hiperprolactinemia patológica es definida como una elevación constante de los niveles de prolactina sérica por encima de 20 ng/ ml en individuos no lactantes y no gestantes. En los casos de hiperprolactinemia es necesario hacer una distinción entre prolactinomas propiamente dichos, que liberan cantidades excesivas de prolactina, y otros tipos de adenomas que, debido a su gran tamaño comprimen el tallo hipofisario, disminuyendo de este modo el flujo de factores que llegan a la hipófisis por el sistema porta, y la deprivación de dopamina ocasiona como consecuencia un aumento en la liberación de prolactina (107). Por otro lado, los niveles de prolactina que son secretados por los prolactinomas son generalmente proporcionales al tamaño del tumor (102) y pueden alcanzar niveles circulantes tan altos como 5000-10000 ng/ ml (107). Adenomas diagnosticados radiológicamente con niveles de prolactina sérica por debajo de 100-150 ng/ ml sugieren hiperprolactinemia secundaria, causada por tumores que no son prolactinomas (107). Como la dopamina es el principal factor inhibitorio de la síntesis y secreción de prolactina, así como de la proliferación de lactotropos (107), cualquier variable química o funcional que interfiera con la acción de este neurotransmisor sobre los receptores dopaminérgicos localizados en la hipófisis producirá variaciones en la secreción de esta hormona. Los prolactinomas conservan generalmente la capacidad de respuesta a la acción inhibitoria de la dopamina, por lo que los agonistas dopaminérgicos producen rápida reducción del tamaño tumoral y de los niveles de prolactina sérica (149), resultando estos prolactinomas sensibles a dicho tratamiento. Las principales drogas que se utilizan son la bromocriptina, y cabergolina (150). Estos derivados del ergot actúan uniéndose y activando directamente el RD2, tanto del lactotropo normal como tumoral, provocando la inhibición de la síntesis y secreción de prolactina y del crecimiento de los lactotropos (reducción de citoplasma junto con disminución de retículo endoplasmático rugoso y aparato de Golgi), con la consiguiente reducción del tamaño tumoral (151;152). Comparando ambos agonistas dopaminérgicos la cabergolina resulta más eficiente en cuanto a la recuperación de los niveles normales de prolactina y de ciclos ovulatorios, y posee efectos secundarios menos frecuentes y severos que bromocriptina (150;153). Por lo tanto, el primer abordaje es el tratamiento médico mediante drogas agonistas dopaminérgicas, sin embargo, aproximadamente un 15% de los pacientes no responden al mismo. Estos se consideran prolactinomas resistentes, los cuales en muchas ocasiones se relacionan con una disminución en la expresión o función del RD2 (menor afinidad del RD2 por los agonistas de dopamina y/ o problemas en la transducción de señales) (154). En estos casos se debe recurrir a un tratamiento quirúrgico mediante una cirugía por vía transepto-esfenoidal o transcraneal (según el tamaño del tumor), en la cual se extirpa selectivamente el adenoma hipofisario conservando la porción de hipófisis normal. En caso de tumores con elevado índice de crecimiento, que no 62 Capítulo I - Introducción responden al tratamiento médico o generan recidivas post-quirúrgicas, se lleva a cabo una radioterapia. Modelos experimentales de prolactinoma Existen varios modelos que han sido utilizados para el estudio de prolactinomas en diversos trabajos, algunos son: administración farmacológica de estrógenos en forma crónica en ratas (155;156), ratas seniles (157), y animales transgénicos, como por ejemplo ratones que sobreexpresan galanina en lactotropos (158), factor de crecimiento transformante alfa (TGF- α) (159), factor de crecimiento neural (NGF) (160), un receptor truncado tipo 4 del factor de crecimiento fibroblástico (FGF) (161) o ratones knockout para PRLR (162). Caracterización del ratón knockout total Drd2 -/En nuestro laboratorio hemos trabajado con ratones Drd2-/ - . Estos ratones fueron generados por el Dr. Malcolm Low (Portland, Oregon, EEUU) y Marcelo Rubinstein (I NGEBI CONI CET, Buenos Aires, Argentina) por deleción completa del exón 7 y parcial del extremo 5´ del exón 8 del gen del Rd2 murino mediante recombinación homóloga (Figura 16) (58). Dichos exones codifican para los últimos 2 dominios transmembrana del RD2, el tercer loop extracelular y la cola COOH-terminal citoplasmática. La zona delecionada fue reemplazada por un cassette que confiere resistencia a neomicina y esta secuencia es a la vez utilizada para la genotipificación de los ratones por PCR a partir de ADN genómico. Figura 16: Generación del ratón deficiente de RD2s mediante recombinación homóloga. Se muestran el locus endógeno del RD2, el vector target con el cassette que confiere resistencia a neomicina y el alelo predicho luego de la recombinación homóloga. Una porción del clon genómico fue retenida para ser utilizada como sonda para la genotipificación. Se señalan también los sitios de corte para EcoR V (RV), Apa1 (A) y Sac1 (S). Modificado de (58). 63 Capítulo I - Introducción Demostramos en el laboratorio que los ratones Drd2-/ - de ambos sexos tienen mayores niveles de prolactina sérica que sus pares WT a partir de la sexta semana de vida, y que en las hembras la hiperprolactinemia siempre es mayor que en los machos (68). A los 8 meses de edad las hembras Drd2-/ - desarrollan hiperplasia hipofisaria de lactotropos con un aumento en el peso de la hipófisis de 2-3 veces respecto a las hembras WT, y una disminución en el número de los demás tipos celulares de la glándula (58;163). A esta edad no se ven signos de transformación neoplásica, manteniéndose intacta la arquitectura determinada por la red de fibras de reticulina. A los 17-18 meses las hembras Drd2-/ - desarrollan verdaderos adenomas hipofisarios evidenciados por la presencia de nódulos multifocales, agrandamiento de la glándula por fuera de la silla turca con aumento del peso de unas 30 veces respecto a las hembras WT y destrucción de las fibras de reticulina, característica patognomónica de adenoma. A esta edad los machos presentan pequeños nódulos multifocales también con ruptura de la red de reticulina indicando desarrollo de múltiples adenomas (119). Este modelo experimental resultó de gran utilidad para estudiar los prolactinomas resistentes a agonistas dopaminérgicos, sin embargo al carecer de los RD2s en todo el organismo, presentan otras múltiples alteraciones que resultan en fenotipos endócrinos tales como un retardo del crecimiento corporal y alteraciones en el eje GH-I GF-I (68;123), modificaciones de los circuitos que controlan la ingesta (60), y fallas en la homeostasis de la glucosa (141). Postulamos que la generación de un ratón tejido específico que carezca del RD2 solamente en lactotropos, resultaría en un modelo más limpio y fiel para el estudio de prolactinomas resistentes. Generación y validación de un nuevo modelo experimental: el ratón lacDrd2KO En nuestro laboratorio en colaboración con el del Dr. Rubinstein, desarrollamos un ratón knockout tejido específico, que carece de RD2s funcionales solamente en lactotropos. Para ello, se utilizó la tecnología de Cre/ loxP, la cual se basa en la expresión de la proteína recombinasa Cre de 38 KDa, enzima que cataliza la recombinación sitio específica del ADN entre secuencias de 34 pb repetidas llamadas loxP (164). Los sitios loxP son palindrómicos donde repeticiones directas de dos sitios loxP provocan la escisión del fragmento contenido entre ellos por medio de la recombinasa (165;166). Estas secuencias loxP pueden ser artificialmente insertadas en el genoma de células embrionarias por recombinación homóloga, de tal forma que flanqueen uno o más exones del gen de interés (llamado “gen floxeado”), lo que producirá una escisión precisa de ADN, sin cortar otras partes del genoma. Se generan ratones transgénicos homocigotas para el gen floxeado por técnicas convencionales, y se cruzan con un segundo ratón transgénico que contiene el transgén Cre bajo el control transcripcional de un promotor tejido o célula específico. En la progenie homocigota para el gen floxeado y que porte el transgén Cre simultáneamente, se producirá la 64 Capítulo I - Introducción recombinación Cre/ loxP, y por lo tanto se delecionará el gen floxeado. Esto sucederá solamente en aquellos tipos celulares en los cuales el promotor asociado a Cre sea activo. En nuestro caso particular los ratones que carecen de RD2s funcionales solamente en lactotropos se obtuvieron por medio de la cruza de dos ratones transgénicos previamente desarrollados: los ratones Drd2loxP/ loxP (los cuales poseen el exón 2 del RD2 flanqueado por secuencias específicas loxP) (61) con ratones transgénicos que poseen la expresión de la enzima recombinasa Cre bajo el control transcripcional del promotor de prolactina [ Tg(Prl-cre) 1Mrub] (142) (Figura 8). Para validar el modelo, determinamos la especificidad de la expresión de esta enzima mediante el análisis de niveles de ARNm de Cre en diferentes tejidos. Observamos altos niveles de Cre hipofisarios, y niveles mínimos o ausentes en hipotálamo, hígado, riñón, ovario y pulmón. A su vez, estudiamos la actividad de la enzima Cre recombinasa en hipófisis. Para ello, se cruzó al ratón [ Tg(Prl-cre) 1Mrub] con un ratón reportero que expresa la proteína fluorescente td-tomato como resultado de la recombinación mediada por Cre (167). Mediante el análisis de células doble positivas (para prolactina y la proteína td-tomato) demostramos que la enzima Cre se encuentra activa en el 96% de las células productoras de prolactina y que esta actividad es altamente específica de lactotropos (142). Posibles mecanismos tumorigénicos en lactotropos Son múltiples los factores que participan en la tumorigénesis de lactotropos, y el fenotipo final del tumor dependerá de la relación entre dichos factores. Dentro de los factores intervinientes se pueden citar: 1) Receptor de dopamina: Como ya mencionamos, los lactotropos a diferencia de los otros tipos celulares endócrinos de la hipófisis, presentan una alta actividad espontánea de secreción de la hormona prolactina. Se encuentran bajo un control tónico predominantemente inhibitorio cuyo principal factor es el neurotransmisor dopamina (168), que está en alta concentración en la eminencia media y sangre portal hipofisaria, y que actúa vía el RD2 en lactotropos. En prolactinomas resistentes a agonistas dopaminérgicos se ha descripto la disminución en la expresión o función del RD2 (154;169;170) confirmando el rol crítico de la dopamina y el RD2 sobre este tipo celular en relación a la secreción de prolactina y a su proliferación (59). Es así como un buen modelo experimental para el estudio de prolactinomas resistentes es el de ratones Drd2-/ - , cuyas hembras desarrollan hiperplasia hipofisaria a lo largo de su vida seguida de la formación de adenomas de lactotropos en forma espontánea (58). 2) Prolactina: La prolactina ejerce un control negativo sobre su propia secreción a través un aumento en el tono dopaminérgico hipotálamo-hipofisario, mediante una retroalimentación sobre las neuronas dopaminérgicas hipotalámicas TI DA, y la alteración de la expresión y actividad de la tirosina hidroxilasa, enzima limitante en la síntesis de dopamina (171). 65 Capítulo I - Introducción Existen PRLR tanto en hipotálamo como en la hipófisis anterior, y se ha demostrado su presencia en lactotropos de ratas y humanos específicamente. En hipófisis, la prolactina tiene acciones inhibitorias directas sobre la proliferación de los lactotropos, pudiendo limitar a su vez, la acción de los factores de crecimiento producidos por células vecinas que en forma paracrina influencian el crecimiento de los mismos. Estudios en ratones que carecen del PRLR demuestran que esta hormona inhibe su propia liberación actuando no solamente a nivel de las neuronas TI DA, sino también a nivel de la hipófisis en forma independiente de dopamina. Estos animales desarrollan hiperprolactinemia, como también hiperplasia hipofisaria y consecuentemente adenomas, los que resultan de mayor tamaño que los observados en ratones Drd2-/ - (162). 3) Estrógenos: Los estrógenos se consideran los factores liberadores de prolactina más importantes, ya que son potentes estimuladores de las funciones del lactotropo. A nivel hipofisario, aumentan la mitosis de lactotropos, favorecen la conversión de somatomamotropos a lactotropos puros, aumentan la síntesis de prolactina por activación génica, provocan hipertrofia de este tipo celular, aumentan los receptores hipofisarios de diferentes factores liberadores hipotalámicos como TRH y disminuyen los receptores de dopamina hipofisarios (172). A nivel hipotalámico, los estrógenos inhiben la actividad de las neuronas TI DA (118). Las acciones estrogénicas son claramente contrarias a las de la dopamina, y del balance entre ambos factores depende en gran parte la regulación de los lactotropos. 4) Otros factores: En la generación de prolactinomas experimentales también se ha descripto la participación de: NGF (160;173;174), galanina (158;175), TGF- α y (159;176;177), y el oncogen transformante tumoral de la hipófisis (PTTG) (178). A continuación detallaremos dos mecanismos posiblemente involucrados en la tumorigénesis hipofisaria. Angiogénesis La glándula pituitaria en los mamíferos es uno de los tejidos más vascularizados y posee doble aporte sanguíneo. Por un lado, recibe oxígeno y nutrientes a partir de las arterias hipofisarias superior e inferior que son ramas directas de la arteria carótida interna; pero también posee un sistema de sangre venosa formado por los vasos portales largos hipofisarios, los cuales proveen un medio de comunicación entre el hipotálamo y la adenohipófisis, llevando neurotransmisores y otros péptidos hipotalámicos estimuladores e inhibidores de la secreción y proliferación de las células de la hipófisis anterior (Figura 17). Los capilares del sistema porta hipofisario son fenestrados lo que favorece el intercambio de sustancias con las células endócrinas de la glándula (179). 66 Capítulo I - Introducción Figura 17: Vasculatura de la hipófisis. Modificado de (179). La angiogénesis es un proceso que requiere la división de las células endoteliales, su migración y diferenciación dando lugar a la formación del lumen de los nuevos vasos y a partir de éstos, la membrana basal. Durante el proceso también se produce la remodelación de la matriz extracelular y el reclutamiento de estructuras vecinas para formar y mantener los nuevos vasos (180). El desarrollo de tumores como los de mama y colon, está asociado a una marcada angiogénesis con profundos cambios en la estructura de los componentes de la matriz extracelular. El papel de la angiogénesis en el desarrollo de tumores hipofisarios, en cambio, ha sido motivo de controversias. Por un lado, se discute el grado de vascularización que poseen los adenomas hipofisarios en sí mismos y en relación a la glándula normal (181). Por otro lado, Elias y Weiner propusieron que la angiogénesis era necesaria para la inducción y el crecimiento de los prolactinomas inducidos por estrógenos en ratas Fisher 344 (182). Asimismo, Schechter y Gorczyca reportaron la formación de nuevas arterias extra-portales en los adenomas hipofisarios (183). Sumado a esto, la tomografía computada y la resonancia magnética mostraron un nuevo aporte de sangre arterial directo en los adenomas humanos (184). En su conjunto, estas evidencias llevaron a hipotetizar que un nuevo aporte directo de sangre arterial facilitaría el desarrollo de tumores hipofisarios ya que esta sangre sistémica contiene escasos niveles de factores hipotalámicos, en particular de dopamina, que inhibe la proliferación de lactotropos (183). 67 Capítulo I - Introducción Varios estudios han demostrado que la angiogénesis en los tumores hipofisarios está relacionada con el comportamiento y la respuesta tumoral, como por ejemplo: -Los microprolactinomas son menos vascularizados que los macroprolactinomas (185). -Los prolactinomas invasivos son significativamente más vasculares que los no invasivos (186). -Los macroprolactinomas y tumores secretores de ACTH con menor densidad vascular tienen mayor probabilidad de remisión (186). -Los carcinomas de hipófisis tienen una mayor vascularización que los adenomas (187-189). Todos estos datos en su conjunto, sugieren que el desarrollo de prolactinomas depende del proceso angiogénico. Factor de crecimiento del endotelio vascular (VEGF) Ferrara y Henzel (en el año 1989) aislaron y purificaron del medio de cultivo de células folículo estrelladas de hipófisis bovina, un factor de crecimiento específico y mitogénico para las células endoteliales que llamaron VEGF (190). El VEGF exhibe dos actividades biológicas principales: el aumento de la permeabilidad vascular mediante la formación de fenestraciones (191) y la capacidad para estimular la proliferación de las células endoteliales de la micro y macro vasculatura, función específica para este tipo celular (190). Además promueve la supervivencia y migración de las células endoteliales. Dvorak y colaboradores propusieron que el aumento en la permeabilidad vascular es un paso crucial para la angiogénesis asociada a tumores y heridas (192). Además, VEGF estimula la migración de las células endoteliales y funciona como un factor antiapoptótico promoviendo la supervivencia de dichas células en los nuevos vasos formados (193). VEGF (o VEGF-A) es una glicoproteína dimérica de 46-48 KDa que forma parte de una familia que incluye al factor de crecimiento de la placenta (PlGF), y también VEGF-B, VEGF-C, VEGF-D. Si bien estas proteínas son productos por distintos genes muestran una alta homología estructural, conservando ocho residuos de cisteína en las mismas posiciones (190;194). El VEGF está principalmente involucrado en la angiogénesis, mientras que el VEGF-C y el VEGF-D participan en la generación de vasos linfáticos (195). El VEGF es producido por las células endoteliales, macrófagos, linfocitos y una variedad de otros tipos celulares (190). Finalmente, tiene un valor potencial en cuanto a la evaluación del pronóstico de pacientes y como blanco terapéutico. 68 Capítulo I - Introducción VEGF en hipófisis Como ya mencionamos, VEGF fue originalmente aislado de células folículo estrelladas provenientes de hipófisis bovina (196;197) y su presencia fue demostrada en hipófisis de varias especies y en la línea celular tumoral GH3 (197-199). Aproximadamente el 90% de los tumores hipofisarios humanos mostraron secreción de VEGF in vitro (200) y varios investigadores detectaron la proteína por inmunohistoquímica e hibridización in situ en adenomas humanos (201;202). Existen controversias en cuanto a la expresión de VEGF y su relación con el comportamiento tumoral. Lloyd y colaboradores (202) hallaron una menor expresión de VEGF en los adenomas hipofisarios respecto a la glándula normal, resultados que se correlacionaban con la menor densidad microvascular encontrada para estos adenomas. A su vez, determinaron una mayor expresión de VEGF en los carcinomas comparados con los adenomas. En cambio, Mc Cabe y colaboradores (203), determinaron una mayor expresión del ARNm de Vegf y su receptor Vegfr-2 en los adenomas respecto a las hipófisis normales. En modelos animales, nuestro laboratorio y otros, demostraron claramente la inducción de VEGF en prolactinomas experimentales (59;204;205). Nosotros demostramos que los factores de crecimiento VEGF, PTTG y el factor de crecimiento fibroblástico 2 (FGF-2), son incrementados por la acción de estrógenos, pero que la ausencia del tono inhibitorio dopaminérgico incrementaría solo la expresión de VEGF (59;204;206;207). Estos resultados sugieren que los prolactinomas experimentales resistentes a agonistas dopaminérgicos podrían tener un incremento en la expresión de VEGF, que estaría involucrado en el desarrollo del tumor. Observamos que el incremento de VEGF hipofisario en ratones Drd2-/ - se correlaciona con los niveles séricos de prolactina (59), y hemos demostrado, a su vez, la expresión de VEGF hipofisario en prolactinomas humanos resistentes a agonistas dopaminérgicos (208;209). Sumado a esto, en el laboratorio hallamos una posible aplicación terapéutica de estrategias anti-VEGF para el tratamiento de prolactinomas resistentes a agonistas dopaminérgicos. Tanto la aplicación local con VEGF-TRAP como el tratamiento sistémico con un anticuerpo monoclonal antiVEGF, resultaron en la inhibición tumoral, con disminución en la vascularización y niveles de proliferación, y caída de los niveles de prolactina en hipófisis hiperplásicas de ratones hembra Drd2-/ - (210). Células madre o stem cells En la mayoría de los tejidos, nuevas células requeridas, ya sea en el recambio celular homeostático, plasticidad celular en respuesta a demandas corporales y/ o regeneración tisular luego de un daño, pueden ser generadas por las llamadas “células madres” (211-213). Una célula madre es aquella que tiene la capacidad de proliferar tanto de manera simétrica como asimétrica. Bajo determinadas circunstancias y estímulos, una célula madre puede dividirse 69 Capítulo I - Introducción produciendo dos nuevas células madre, lo que consiste en una proliferación simétrica (214). El pool de células madre debe ser sostenido en el tiempo, de manera que regularmente se generan nuevas células madre mediante este proceso de auto-renovación, manteniendo así un stock constante (215). Estas células son consideradas inmortales, ya que pueden dividirse indefinidamente hasta que son inducidas a diferenciarse o a sufrir apoptosis. En la división asimétrica en cambio, una célula hija mantiene las características de stemness de la madre (células equivalentes), mientras que la otra resulta en una célula progenitora que dará origen a un linaje de células diferenciadas. Esta célula progenitora no es inmortal, muere ya sea por senescencia o por apoptosis (214). Existen distintos tipos de células madre con diferentes capacidades de diferenciación: una célula madre totipotente es aquella que puede dar origen a un organismo con todas sus células (célula huevo o cigoto). Se llama célula madre pluripotente a la célula embrionaria que puede generar cualquier tipo celular pero resulta incapaz de generar un organismo vivo completo. Las células madre multipotentes tienen la capacidad de diferenciarse en múltiples tipos celulares limitados. Y por último, una célula madre bipolar es aquella que puede generar dos linajes celulares, como por ejemplo, las células ovales del hígado que dan origen a los hepatocitos y a las células ductales biliares (214). Las células madre se encuentran, por lo general, en un microambiente especializado denominado nicho, que regula su mantenimiento, auto-renovación, destino final y reacción a estímulos externos (215). Estos nichos difieren en su composición celular y localización en los diferentes tejidos. La expresión de genes particulares caracteriza a las células madre, tales como Sca1 (antígeno 1 de stem cell), Sox-2, Sox-9, Oct-4, Bmi1, Nanog , Nestina y componentes de las vías de Notch , Wnt ( Wingless-type MMTV integration site family ) y Shh ( Sonic hedgehog ), entre muchos otros. Sumado a esto, se han descripto numerosos marcadores de superficie que permiten su identificación y aislamiento por medio de la técnica de citometría de flujo. Algunos de estos son: CD133 (Prominin-1), CD44, CD34, y CD38. Células madre en hipófisis Los distintos linajes celulares hipofisarios, productores de hormonas específicas, provienen de progenitores comunes presentes en la bolsa de Rathke y se definen durante la embriogénesis bajo la influencia de morfógenos y factores, de crecimiento y transcripción, determinados. Luego del nacimiento, el tamaño y la composición hipofisaria madura. En la adultez, la glándula tiene una lenta, pero continua renovación celular (216). En particular, las células de la adenohipófisis de una rata adulta son reemplazadas cada 6-10 semanas, lo que corresponde a una renovación de cerca de 50000 células por día (217). Del mismo modo, las poblaciones celulares se adaptan dinámicamente a las demandas endócrinas del organismo (crecimiento, pubertad, estrés, 70 Capítulo I - Introducción inflamación y ciclo reproductivo). Por ejemplo, en condiciones de preñez y lactancia, la alta demanda de prolactina es abastecida por un aumento en el número de lactotropos (218). Asimismo, la hipófisis posee la capacidad de regenerarse luego de un daño tisular (219). Existen evidencias de que nuevas células hipofisarias productoras de hormonas son generadas a partir de células no hormonales frente a las diferentes demandas (218). Se sugirió entonces, que la hipófisis alojaría una subpoblación de células madre/ progenitoras específicas que participarían en la generación de nuevas células hipofisarias necesarias para la renovación celular y plasticidad, en la vida post-natal (216) (Figura 18). Se propone como nicho primario a la zona marginal (MZ), ya que contiene células con características madre/ progenitoras en diferentes fases de maduración y del ciclo celular. La MZ se encuentra adyacente al cleft hipofisario, lumen remanente entre el lóbulo anterior y el lóbulo intermedio de la hipófisis (216). Figura 18: Modelo de comportamiento de células madre/ progenitoras hipofisarias. La zona marginal (MZ), propuesta como nicho primario, contiene células stem y progenitoras en diferentes fases de maduración y del ciclo celular. Varios marcadores son expresados en diferentes combinaciones en la zona o fase transicional, y supuestamente en un orden temporal altamente regulado. La zona del wedge (círculo punteado azul) representaría la zona germinativa principal. Se piensa que las células madre de la MZ conectan con las células madre dentro de la glándula (nichos secundarios) para formar una red tri-dimensional (3D) de 71 Capítulo I - Introducción comunicación. Las células madre se transportarían desde los nichos al área glandular por EMT ( Epithelial– mesenchymal transition ), guiadas por células folículo estrelladas y/ o gradientes de factores quemotácticos, como Sfd1. Las células madre hipofisarias son fundamentales para diferentes procesos de remodelado de la glándula. NL: lóbulo neurohipofisario; AL: lóbulo anterior; I L: lóbulo intermedio. Modificado de (215). Dentro de los distintos tipos celulares presentes en la hipófisis, se postulan algunos como candidatos a ser células madre/ progenitoras: Células folículo estrelladas Las células folículo estrelladas son células no productoras de hormonas, que presentan largas proyecciones y se distribuyen en la adenohipófisis, entre las células secretoras de hormonas. En ratas, estas células son generadas en el período post-natal temprano, y se encuentran adyacentes al cleft hipofisario (220). Las células madre/ progenitoras son capaces de generar colonias adherentes in vitro (221). Se utilizó el ensayo de formación de colonias para determinar y caracterizar la posible población de células con características de madre/ progenitoras en la glándula pituitaria. De todas las células de la hipófisis anterior sólo el 0,2% posee la capacidad de formar colonias, y se postula que se trata de las células folículo estrelladas ya que resultan positivas para la expresión de marcadores característicos de las mismas (como ser S100 y GFAP) (220). Células marginales Las células marginales se encuentran en el cleft hipofisario durante la embriogénesis y en la MZ de la hipófisis adulta. Existen algunas evidencias indirectas que sugieren a estas células como posibles candidatos a células madre/ progenitoras como ser, que poseen características estructurales inmaduras (similares a las observadas en las células folículo estrelladas), carecen de gránulos de secreción (220), y presentan una alta actividad proliferativa desde el nacimiento hasta la adultez (222). Células de la Side Population Estudios recientes han demostrado la existencia células madre/ progenitoras en hipófisis, mediante la identificación de una población celular denominada side population (SP) en adenohipófisis adultas de ratón, rata y pollo (223), y más recientemente de humanos (215). Las células de la SP poseen la capacidad de expulsar el colorante vital Hoechst 33342 permitiendo así identificarlas por citometría de flujo como la subpoblación con baja cantidad de este colorante. El transportador de multidrogas ABC ( ATP-binding cassette), en particular el ABCG2, es el responsable de esta actividad. La capacidad de excluir sustancias tóxicas u hostiles es considerada un componente de defensa fundamental de las células madre. Consecuentemente, esta metodología fue utilizada en múltiples tejidos para identificar sus células madre/ progenitoras (224227). 72 Capítulo I - Introducción Se demostró que las células de la SP hipofisaria (las cuales corresponden un 1,5% de las células totales presentes en la adenohipófisis adulta murina) presentan características asociadas a células madre, ya que expresan marcadores de stemness y poseen caminos de señalización activados por encima de los niveles de las células normales ( main population : MP) (218). Como conclusión, la hipófisis contiene una subpoblación celular muy pequeña, la SP, enriquecida de células con fenotipo asociado a células madre y carente de células diferenciadas. Posteriormente, se subdividió a la SP en dos subgrupos según los niveles de expresión del antígeno Sca1: las células con alta expresión de Sca1 resultaron ser aproximadamente el 60% de la SP, mientras que las de baja expresión las restantes, siendo estas últimas sorpresivamente, las que contienen las células madre/ progenitoras hipofisarias, y pasaron a ser denominadas como SCSP ( stem cell-SP). Las mismas, poseen una expresión incrementada de Sox-2, Sox-9, Lgr5, CD44 y Nanog, factores relacionados a las células madre (216). Células nestina positivas Así como ocurrió en otros tejidos, mucha de la evidencia sobre posibles células madre en hipófisis se obtuvo a partir de la identificación de marcadores de stemness en poblaciones de tejido adulto. En particular, en hipófisis de rata, numerosas células nestina positivas fueron identificadas en toda la glándula (228) pero su principal localización resultó ser adyacente al cleft (posible nicho hipofisario) (229). Estas células presentaron tanto fenotipo de folículo estrelladas como de células productoras de hormonas (228). A partir de cultivos celulares enriquecidos en esta población se observó que las células nestina positivas presentaban características mesenquimales (230). En embriones, en cambio, se observa un bajo porcentaje de células nestina positivas, esto sugiere que el pool de células madre en las hipófisis adultas sería diferente de los progenitores embrionarios (230). Células Sox-2+ / Sox-9Se sabe que Sox-2 es fundamental para el normal desarrollo de la hipófisis (231). Este factor se localiza en aproximadamente el 3% de las células de la glándula post-natal, las cuales se localizan principalmente adyacentes al cleft aunque también se encuentran distribuidas en la adenohipófisis (230). Cuando las células Sox-2 se cultivan in vitro son capaces de formar pituesferas, y el 0,03-0,05% de células son capaces de auto-renovarse a esferas secundarias (232). Aquellas que no se auto-renuevan expresan Sox-9, nestina y S100 (220). En la hipófisis adulta el 1% de las células Sox-2 positivas no expresan Sox-9, otro factor de transcripción involucrado en el desarrollo embrionario (230). Se observó que las células Sox2+ / Sox-9- son capaces de diferenciarse en los 5 tipos celulares secretores de hormonas, como también hacia folículo estrelladas (220). 73 Capítulo I - Introducción Células madre tumorales o cancer stem cells (CSC) El modelo de células madre tumorales o, dicho de otro modo, de cancer stem cells (CSC), provee una posible explicación a la heterogeneidad fenotípica y funcional que existe entre células cancerosas en algunos tipos tumorales (233-236). El modelo postula que los tumores están organizados jerárquicamente en subpoblaciones celulares, conformadas en su mayoría, por células no tumorigénicas, y por un pequeño grupo de CSC (237). Una CSC se define como una célula indiferenciada con capacidades de iniciar tumores, autorenovación, y diferenciación en múltiples linajes celulares (238) (Figura 19). Por definición, tanto las CSC como las células madre de tejido normal tienen la capacidad de auto-renovarse y diferenciarse, lo que las distingue entre sí es que las CSC lo hacen de manera desregulada (239). Figura 19: Organización jerárquica de clones tumorales según la teoría de las CSC. Modificado de (240). La presencia de CSC fue primero demostrada en casos de leucemia mieloide aguda (241) y más recientemente en tumores sólidos como tumores de mama (242), próstata (243), colon (244) y cerebro (245;246). Múltiples evidencias sugieren que las CSC juegan un rol fundamental en la iniciación, angiogénesis, mantenimiento, progresión y metástasis tumoral (238;247;248). Las CSC son clínicamente relevantes, ya que varían entre tumores de diferentes pacientes, pueden cambiar constantemente a medida que progresa la enfermedad, y presentan una alta resistencia a drogas (249-251), radioterapia y quimioterapia convencionales (237;238). Por 74 Capítulo I - Introducción consecuencia, para la prevención y tratamiento del cáncer, resulta necesario identificar y caracterizar estas subpoblaciones. Vías de señalización y factores en CSC En la búsqueda por la caracterización e identificación de CSC, se ha descripto la expresión de numerosos factores, característicos de las células madre normales, en las mismas, como ser Oct-4, Nestina, los receptores Notch, Sox-2, entre otros. En mamíferos, la vía de señalización de Notch comprende una familia de 4 receptores: Notch1-4, los cuales son proteínas de un paso transmembrana conformados por tres dominios, un dominio extracelular, uno transmembrana y uno intracelular (NI CD). Sus ligandos, Delta y Jagged (ligandos clásicos), al unirse al receptor lo activan volviéndolo susceptible al clivaje proteolítico, lo que resulta en la liberación del NI CD que transloca al núcleo y actúa como coactivador de la transcripción de genes blanco ( Hes, Hrt y Ciclina D3) (252). Se han descripto más recientemente, ligandos no clásicos de estos receptores como ser Dlk1 ( ligand delta-like 1 homologue), Dlk2 ( ligand delta-like 2 homologue), entre otros. La señalización de Notch es crítica para el desarrollo y mantenimiento de la homeostasis, participando en una variedad de procesos celulares que incluyen el mantenimiento de células madre, la especificación del destino celular, la diferenciación, proliferación, adhesión celular, migración, transición epitelio-mesenquimal (EMT) y apoptosis (253). Se ha reportado que Notch puede promover o reprimir el crecimiento tumoral dependiendo del tipo celular y el contexto (253). La desregulación de la señalización de Notch ha sido vinculada al cáncer de pulmón, próstata, cuello uterino (254) y al neuroblastoma (255). Por otra parte, Sox-2 es una proteína, que pertenece a la familia de factores de transcripción HMG ( high-mobility group ), fundamental para el desarrollo temprano y el mantenimiento de células madre embrionarias indiferenciadas. Es a su vez, uno de los factores de transcripción clave utilizado para inducir una célula madre pluripotente a partir de fibroblastos. Estudios recientes sugieren que la sobreexpresión de Sox-2 se relaciona con el desarrollo de cáncer en múltiples tejidos como ser cáncer de pulmón, esófago y mama. Tal como en las células madre embrionarias, Sox-2 juega un rol en las CSCs, contribuyendo positivamente al mantenimiento de las características de stemness en estas células (256). CSC en hipófisis Algunos de los tumores hipofisarios, de origen monoclonal, presentan múltiples tipos celulares productores de variadas hormonas hipofisarias, lo que apoya la idea de la participación de células madre/ progenitoras en el desarrollo de los mismos (257). Glaiberman y colaboradores describieron la presencia de células con fenotipo de célula madre (con expresión de Nestina, entre 75 Capítulo I - Introducción otras características) en tumores hipofisarios, y plantearon un posible rol de las mismas en la iniciación y crecimiento del tumor (257). Fauquier y colaboradores detectaron una pequeña subpoblación de células que expresan Sox-2 en la glándula hipofisaria anterior en cultivos in vitro. Las mismas resultaron capaces de formar esferas que pueden proliferar, auto-renovarse en esferas secundarias, y diferenciarse a tipos celulares hipofisarios (232). Sin embargo, no relacionaron la expresión de este factor con la génesis y mantenimiento de adenomas hipofisarios. Por otro lado, el ligando no clásico de los receptores Notch: Dlk1, se encuentra aumentado en los tumores hipofisarios humanos (258), mientras que el silenciamiento del locus Dlk1/ MEG se detecta en adenomas hipofisarios no funcionantes (259). Sumado a esto, se ha descripto que las vías de señalización de Notch y Wnt se encuentran activadas y resultan importantes en la progresión de adenomas no funcionantes, ya que se observaron cambios en la expresión de Notch3, Dlk1, entre otros (260). Lo cierto es que los tumores hipofisarios son altamente prevalentes y pueden causar morbilidad severa a causa de las disrupciones hormonales, más allá de que por lo general se trate de tumores benignos. Es por ello que resulta de gran importancia la identificación y caracterización en prof undidad de las “CSC” hipofisarias, para de este modo proveer de nuevas estrategias terapéuticas. Finalmente, es importante aclarar que el término “cancer stem cell” no sería el adecuado en este tejido, ya que como mencionamos los tumores suelen ser benignos. Es por ello que células madre/ progenitoras tumorales sería más acertado. 76 Capítulo I - Objetivos OBJETIVOS Analizar la importancia de los RD2s, en la regulación de la actividad hipofisaria y el proceso de génesis de prolactinomas. La caracterización de este modelo será fundamental para los objetivos posteriores, en los que se determinará el efecto de la hiperprolactinemia sobre el metabolismo. Estudiaremos el perfil endócrino, angiogénico y de proliferación celular en las hipófisis hiperplásicas de ratones hembra lacDrd2KO en comparación con hembras Drd2loxP/ loxP. Y determinaremos la presencia de marcadores de células madre/ progenitoras en hipófisis de ambos genotipos. Los objetivos específicos son estudiar: - Las características del prolactinoma en ratones hembra y macho en cuanto a su evolución, tamaño y peso hipofisario, y niveles de prolactina séricos e intrahipofisarios. - Validaremos y caracterizaremos el modelo de los ratones lacDrd2KO analizando el eje de crecimiento y el ciclo estral. - Parámetros angiogénicos: determinaremos en hipófisis de ambos genotipos el área microvascular mediante la inmunomarcación para PECAM, y los niveles de VEGF tanto séricos como intrahipofisarios. - Niveles de proliferación hipofisaria mediante la inmunomarcación contra PCNA. - La presencia de una población de células con características de células madre/ progenitoras, mediante una técnica de citometría de flujo. Luego compararemos la presencia de marcadores de células madre/ progenitoras en hipófisis de ambos genotipos. Estudiaremos, en particular, las vías de NOTCH y SOX-2. 77 Capítulo I - Resultados RESULTADOS VALIDACIÓN DEL MODELO Hiperplasia hipofisaria Los ratones hembra lacDrd2KO presentaron niveles basales de prolactina superiores a los de las hembras controles ( Drd2loxP/ loxP) desde el primer mes de vida hasta los 11 meses de edad (Figura 20A). Las hipófisis de ratones hembra lacDrd2KO de 10 meses de edad resultaron más pesadas en comparación con sus respectivos controles ( P = 0,0039) (Figura 20C). En forma concordante, la concentración de prolactina intrahipofisaria se encontró incrementada significativamente en los ratones transgénicos (Figura 20B), indicando, ambos eventos en su conjunto, hiperplasia de lactotropos. Figura 20: Hiperplasia hipofisaria. A) Niveles séricos de prolactina en ratones hembra Drd2loxP/ loxP (n = 6) y lacDrd2KO (n = 6), evaluados desde el primer hasta los 11 meses de edad. El análisis por AMR indicó un efecto significativo de genotipo ( P = 0,0028). B) Concentración de prolactina (PRL) intrahipofisaria 78 Capítulo I - Resultados medida por RI A en ratones hembra Drd2loxP/ loxP (n = 6) y lacDrd2KO (n = 9) de 10 meses de edad. C) Peso hipofisario en hembras Drd2loxP/ loxP (n = 6) y lacDrd2KO (n = 9) de 10 meses de edad. A la izquierda una imagen representativa del tamaño hipofisario de una hembra control ( Drd2loxP/ loxP) comparado con una hembra lacDrd2KO. En las figuras B y C los resultados se analizaron mediante una Prueba T, donde * P < 0,05 lacDrd2KO vs Drd2loxP/ loxP de la misma edad. En todas las figuras presentes en este capítulo, las barras indican la media y las barras de error el E.S.M. En los machos, en cambio, no observamos diferencias significativas entre genotipos en los niveles séricos de prolactina, o el peso hipofisario (Figura 21A y B). Figura 21: Machos. A) Peso hipofisario en ratones macho Drd2loxP/ loxP (n = 6) y lacDrd2KO (n = 5) de 10 meses de edad. B) Niveles séricos de prolactina en ratones macho Drd2loxP/ loxP (n = 12) y lacDrd2KO (n = 14) de 10 meses de edad. En ambos casos los resultados se analizaron mediante una Prueba T, lacDrd2KO vs Drd2loxP/ loxP de la misma edad. Eje de crecimiento conservado Se midió la talla corporal en un grupo de hembras lacDrd2KO comparativamente con sus controles ( Drd2loxP/ loxP) durante 9 meses, sin observar diferencias significativas (Figura 22A). Asimismo, tampoco existieron diferencias en la longitud del fémur entre ratones hembra de 10 meses de edad de ambos genotipos (1,64 ± 0,02 y 1,69 ± 0,05 cm para ratones Drd2loxP/ loxP y lacDrd2KO, respectivamente). Consistente con estos resultados, determinamos que la concentración de GH hipofisaria y el porcentaje de somatotropos, así como los niveles séricos y de ARNm hepático de I gf-I , resultaron similares entre ratones lacDrd2KO y Drd2loxP/ loxP (Figura 22B-D). Del mismo modo, la expresión génica de I gfbp3 estaba conservada (Figura 22D). La expresión de algunos genes hepáticos sexualmente dimórficos, en especial, los genes que codifican para citocromo p450 (143;261), son regulados por los perfiles de secreción de la hormona GH. Demostramos que los niveles de expresión de dos genes hepáticos dependientes de 79 Capítulo I - Resultados GH: el gen Cyp2b9 predominante de hembras y Cyp2d9 predominante de machos, resultaron similares en ambos genotipos, indicando que el patrón sexualmente dimórfico de secreción de GH se encuentra conservado en los ratones lacDrd2KO (Figura 22E). En su conjunto estos resultados demuestran que los ratones hembra lacDrd2KO presentan un eje de GH normal. Figura 22: Eje de GH. A) Talla corporal medida como la longitud (en cm) comprendida entre la nariz y el ano. Se estudiaron ratones Drd2loxP/ loxP (n = 6) y lacDrd2KO (n = 5) entre los 2 y 9 meses de edad. Los datos se analizaron estadísticamente mediante un AMR, sin obtener diferencias. B) Concentración de GH intrahipofisaria medida por RI A en ratones hembra Drd2loxP/ loxP (n = 6) y lacDrd2KO (n = 8) de 10 meses de edad. A la derecha, imágenes representativas de una inmunofluorescencia realizada en cortes de 80 Capítulo I - Resultados adenohipófisis de ratones Drd2loxP/ loxP y lacDrd2KO de 10 meses de edad. Se utilizó un anticuerpo primario anti-GH junto con un anticuerpo secundario acoplado a Texas-Red de manera tal que los citoplasmas de los somatotropos se visualizan en rojo. C) Niveles séricos de I GF-I medidos por RI A en ratones Drd2loxP/ loxP y lacDrd2KO (n = 4) de 10 meses de edad. D) Niveles de ARNm de I gf-I e I gfbp3 en hígados de ratones hembra Drd2loxP/ loxP (n = 7) y lacDrd2KO (n = 8) de 10 meses de edad. En las figuras B, C y D los resultados se analizaron mediante una Prueba T, lacDrd2KO vs Drd2loxP/ loxP de la misma edad. E) Niveles de ARNm de genes hepáticos dependientes de GH con patrones de expresión sexualmente dimórficos; a la izquierda un gen predominante de hembras ( Cyp2b9) y a la derecha un gen predominante de machos ( Cyp2d9). Los resultados se analizaron por un ANOVA de dos factores * P ≤ 0,0001 vs hembras de ambos genotipos; n = 5 y 4 para las hembras y n = 4 y 4 para los machos, Drd2loxP/ loxP y lacDrd2KO, respectivamente. Ciclo estral Los ciclos estrales de los ratones hembra lacDrd2KO estaban alterados. Presentaban un incremento en el porcentaje de días en diestro a los 5 meses de edad (Figura 23A), como también un leve aumento en los niveles séricos de progesterona respecto de sus pares Drd2loxP/ loxP ( P = 0,064) a los 10 meses (Figura 23B). Figura 23: Ciclo estral. A) Porcentaje de días en diestro o proestro + estro, observado en hembras Drd2 loxP/ loxP (n = 5) y lacDrd2KO (n = 9) de 5 meses de edad. Los datos se analizaron estadísticamente mediante un test Х2: * P ˂ 0,05 vs Drd2loxP/ loxP en el mismo estadío del ciclo. B) Niveles séricos de progesterona evaluados por RI A en ratones Drd2loxP/ loxP (n = 8) y lacDrd2KO (n = 7) de 10 meses de edad. Se realizó una Prueba T, lacDrd2KO vs Drd2loxP/ loxP de la misma edad. 81 Capítulo I - Resultados PROLACTINOMA EXPERIMENTAL RESISTENTE Angiogénesis y proliferación Estas hipófisis hiperplásicas presentaron un área vascular aumentada, determinada por el análisis de la inmunomarcación contra PECAM ( P ≤ 0,002) (Figura 24A), como así también niveles incrementados de proliferación, evaluados por el porcentaje de núcleos PCNA positivos ( P ˂ 0,05) (Figura 24B). Por otra parte, los niveles de VEGF séricos no mostraron diferencias (Figura 24C), mientras que la concentración intrahipofisaria de este factor presentó una tendencia a estar aumentada, sin resultar estadísticamente significativa ( P = 0,079) (Figura 24D). Figura 24: Angiogénesis y proliferación. A) Porcentaje del área vascular calculada en secciones de tejido hipofisario inmunopositivo para PECAM en ratones hembra Drd2loxP/ loxP (n = 11) y lacDrd2KO (n = 14) de 10 meses de edad. B) Porcentaje de núcleos PCNA positivos en hipófisis de ratones hembra Drd2loxP/ loxP (n = 4) y lacDrd2KO (n = 4) de 10 meses de edad. C) Niveles de VEGF séricos medidos por ELI SA en ratones hembra Drd2loxP/ loxP (n = 6) y lacDrd2KO (n = 6) de 10 meses de edad. D) Contenido de VEGF hipofisario analizado por ELI SA en ratones Drd2loxP/ loxP (n = 5) y lacDrd2KO (n = 6) de 10 meses de edad ( P = 0,079 vs Drd2loxP/ loxP). En todos los casos los resultados se analizaron mediante una Prueba T, * P < 0,05 lacDrd2KO vs Drd2loxP/ loxP de la misma edad. 82 Capítulo I - Resultados Marcadores de células madre Se optimizó una técnica de citometría de flujo para la obtención de una población celular CD31- / CD45- / CD44+ . Esta población estaría enriquecida en células progenitoras (CD44 + ), con exclusión de células progenitoras de endotelio y hematopoyéticas (CD31 - y CD45- , respectivamente). Corroboramos, la presencia de dicha población celular (indicada como P5) en hipófisis de ratones hembra de 10 meses de edad de ambos genotipos (Figura 25). Figura 25: Población celular CD31-/CD45-/CD44+. I magen representativa del resultado obtenido en el análisis por citometría de flujo en un pool de hipófisis de ratones hembra Drd2loxP/ loxP (n = 5) y lacDrd2KO (n = 4). Los anticuerpos utilizados fueron: anti-mouse-CD31-FI TC (0,5 mg/ ml), anti-mouse-CD45PE (0,2 mg/ ml), anti-mouse-CD44-APC (0,2 mg/ ml). A) Resultado obtenido en el pool de hipófisis Drd2loxP/ loxP. B) Resultado obtenido en hembras lacDrd2KO. 83 Capítulo I - Resultados La población P1 se definió según forma y tamaño celular. Dentro de la misma, se encuentran: P2 que es la población celular CD31 - / CD45- ; P6 población CD31 + / CD45- ; y P7 población CD31- / CD45+ . Dentro de la población P2 (CD31- / CD45- ) se definieron dos grupos: P3 células CD44- y P5 células CD44+ , siendo esta última la de nuestro interés. Habiendo determinado la existencia de una población enriquecida en células madre/ progenitoras en hipófisis, decidimos profundizar el estudio de marcadores de stemness en forma comparativa entre genotipos. En cortes de hipófisis de ratones hembra de 10 meses de edad, evaluamos por I HQ la expresión de los receptores NOTCH-1 y NOTCH-3 (Figura 26A y B), al igual que la proteína producto de uno de sus genes blanco, HES-1 (Figura 26C). No observamos diferencias estadísticas en ninguno de dichos marcadores estudiados, aunque en todos los casos se evidenció una tendencia a estar disminuidos en las hipófisis tumorales provenientes de hembras lacDrd2KO (Figura 26D). La localización de ambos receptores, al igual que de HES-1, fue principalmente citoplasmática. 84 Capítulo I - Resultados Figura 26: Vía de Notch. A) Porcentaje de área inmunopositiva para el receptor NOTCH-1 en secciones de tejido adenohipofisario de ratones hembra Drd2loxP/ loxP (n = 4) y lacDrd2KO (n = 4) de 10 meses de edad; P = 0,15 B) Porcentaje de área inmunopositiva para el receptor NOTCH-3 en cortes de 85 Capítulo I - Resultados adenohipófisis de ratones hembra Drd2loxP/ loxP (n = 5) y lacDrd2KO (n = 5) de 10 meses de edad. P = 0,16 C) Porcentaje de área inmunopositiva para HES-1 en adenohipófisis de ratones hembra Drd2loxP/ loxP (n = 5) y lacDrd2KO (n = 5) de 10 meses de edad. P = 0,13. En todos los casos los resultados se analizaron mediante una Prueba T, lacDrd2KO vs Drd2loxP/ loxP de la misma edad. D) I mágenes representativas de las I HQ realizadas en cortes de hipófisis de ratones hembra de ambos genotipos. En todos los casos, las imágenes fueron obtenidas con un objetivo 40X, y corresponden al lóbulo anterior. Los preparados fueron coloreados con hematoxilina, y en marrón se observan los citoplasmas positivos para cada anticuerpo (DAB). Por otra parte estudiamos, también por medio de I HQ, la expresión de SOX-2 en cortes de hipófisis de hembras de 10 meses de edad. Cualitativamente, observamos una localización de las células que expresan SOX-2 en la zona del cleft , y también células SOX-2 positivas ubicadas en forma de clusters en la adenohipófisis (Figura 27A). Cuantitativamente, determinamos que el porcentaje total de células SOX-2 positivas en la adenohipófisis (sin incluir la zona del cleft ), cualquiera sea la localización subcelular del factor de transcripción, resultó el mismo en hipófisis de ambos grupos (7,03 ± 0,27 y 6,07 ± 1,54 % para ratones Drd2loxP/ loxP y lacDrd2KO, respectivamente). A su vez, analizamos en forma discriminada el porcentaje de núcleos y citoplasmas con expresión positiva para SOX-2 en adenohipófisis de ambos genotipos. No existen diferencias estadísticas en estos parámetros estudiados, aunque observamos que los núcleos SOX2 positivos presentaron una clara tendencia a estar disminuidos en las hipófisis tumorales provenientes de hembras lacDrd2KO ( P = 0,088) (Figura 27B y C). Cuando analizamos la relación entre el porcentaje de núcleos y citoplasmas SOX-2 positivos, logramos evidenciar la disminución significativa presente en las hipófisis tumorales provenientes de hembras lacDrd2KO ( P ˂ 0,03) (Figura 27D), lo que claramente indicaba un cambio en la localización subcelular del factor de transcripción. 86 Capítulo I - Resultados Figura 27: SOX-2. A) I magen representativa de una I HQ realizada en un corte de hipófisis de un ratón hembra Drd2loxP/ loxP con un anticuerpo anti-SOX-2. A la izquierda se encuentra la imagen obtenida con un objetivo 10X, y a la derecha con uno 40X. Se recuadra en rojo la porción magnificada. N= neurohipófisis; I= hipófisis intermedia; A= adenohipófisis. Los preparados fueron coloreados con hematoxilina, y en marrón se observa la marca SOX-2 positiva (DAB). B) Porcentaje de núcleos positivos para SOX-2 en adenohipófisis de ratones hembra Drd2loxP/ loxP (n = 5) y lacDrd2KO (n = 5), de 10 meses de edad. P = 0,088 vs. Drd2loxP/ loxP. C) Porcentaje de citoplasmas positivos para SOX-2 en adenohipófisis de ratones hembra Drd2loxP/ loxP (n = 5) y lacDrd2KO (n = 5), de 10 meses. D) Relación comprendida entre el porcentaje de núcleos positivos y el porcentaje de citoplasmas positivos para SOX-2 en adenohipófisis de ratones hembra Drd2loxP/ loxP (n = 4) y lacDrd2KO (n = 5). En todos casos los resultados se analizaron mediante una Prueba T, * P < 0,03 lacDrd2KO vs Drd2loxP/ loxP. Estos resultados en su conjunto demuestran que los ratones hembra lacDrd2KO presentan prolactinomas con aumento en el índice de proliferación y en el patrón angiogénico, y una tendencia a la disminución en la proporción de células madre/ progenitoras. Esto último sugiere que el aumento en los niveles proliferativos no es a expensas de las mismas, o que han participado en estadíos más tempranos de la génesis del prolactinoma. Por otra parte, el modelo experimental tiene RD2s funcionales en el resto del organismo, y por lo tanto el eje de crecimiento conservado. A continuación estudiamos el efecto de la hiperprolactinemia crónica de este modelo sobre la ingesta y el metabolismo usando este modelo. 87 Capítulo I - Discusión DISCUSIÓN En trabajos previos demostramos que en los ratones Drd2-/ - , la ausencia de control dopaminérgico sobre los lactotropos del lóbulo anterior de la hipófisis, promueve la formación de prolactinomas y su consiguiente hiperprolactinemia, resultando en un buen modelo para la búsqueda de terapias alternativas a prolactinomas resistentes a agonistas dopaminérgicos (204). Pero también demostramos, mediante el uso de este modelo experimental, que la dopamina no solamente actuaba sobre la regulación de la prolactina, sino que participaba activamente en la secreción de GHRH y GH (68;123;262), la regulación de la ingesta (60) y la homeostasis de la glucosa (141). La multiplicidad de fenotipos solapados, nos planteó la necesidad de contar con una nueva herramienta genética que permita estudiar solamente los fenotipos asociados a la falta del RD2 hipofisario y consecuente hiperprolactinemia. Es por ello que generamos ratones transgénicos que expresan la recombinasa Cre bajo el control transcripcional del promotor de prolactina (Prl-Cre) en ratones de la cepa C57BL/ 6J. A partir de los mismos, hemos generado una línea de ratones mutantes nulos condicionales en los que la anulación del RD2 está restringida a nivel de los lactotropos (lacDrd2KO), mediante la cruza de los ratones transgénicos Prl-Cre con la línea Drd2loxP/ loxP (142). Se determinaron los niveles de prolactina sérica en ratones hembra a lo largo del crecimiento, y como era de esperar, las hembras lacDrd2KO presentaron una hiperprolactinemia sostenida desde el primer mes hasta los 11 meses de edad como consecuencia de la ausencia de control dopaminérgico. Sumado a esto, las hipófisis de ratones hembra lacDrd2KO resultaron más pesadas, y la concentración de prolactina intrahipofisaria se encontró incrementada, indicando, ambos eventos, hiperplasia de lactotropos. En los machos en cambio, no se observaron diferencias en los niveles séricos de prolactina, como tampoco en los pesos hipofisarios. Es evidente que la falta de tono dopaminérgico es el principal evento que induce la hiperplasia de lactotropos en los ratones hembra lacDrd2KO. Sin embargo esto resulta aparentemente insuficiente como único inductor ya que los machos lacDrd2KO no generan hiperplasia hipofisaria. Se encuentra ampliamente descripto en la literatura que el tono dopaminérgico sobre los lactotropos es mayor en las hembras que en los machos. En 1981 Gudelsky y Porter reportaron que la concentración de dopamina en la eminencia media de ratas hembra en diestro era aproximadamente siete veces mayor que la de machos (263). A su vez, las neuronas TI DA (principal fuente de dopamina hipotalámica) presentan una mayor actividad basal en las hembras respecto de los machos (264) y esto parece deberse a la acción de los estrógenos, dado que la ovariectomía suprime la actividad TI DA, y el reemplazo estrogénico la recupera. En los machos, por el contrario, la castración incrementa la actividad de estas neuronas y la testosterona la inhibe (30). Por lo tanto, probablemente al anular el tono dopaminérgico en la hembra, el cual es mucho más fuerte que en el macho, se desarrolla el prolactinoma. Tales diferencias sexuales en 88 Capítulo I - Discusión la generación de prolactinomas también se evidenciaron en los ratones Drd2-/ - (68) y los Prlr -/ (162). A raíz del resultado de una elevada prolactina sérica en hembras lacDrd2KO, es que decidimos analizar el ciclo estral de las mismas siendo que prolactina tiene un rol fundamental en la regulación del ciclo reproductor. El ciclo estral se encontró alterado, con un aumento en el porcentaje de días en diestro (etapa lútea). Entre las diversas funciones de la prolactina, su rol en la reproducción ha sido uno de los más caracterizados. Ratones hembras knockout para el gen de prolactina y para su receptor son estériles, con marcadas alteraciones del ciclo estral (265;266). Por otro lado, los elevados niveles de prolactina en los ratones Drd2-/ - provocaron, particularmente en las hembras, un fenotipo de infertilidad (204). En roedores, la prolactina es esencial para el sustento de la acción luteotrópica, promueve la producción de progesterona por parte del cuerpo lúteo y el mantenimiento de la gestación (33), sin embargo los niveles de progesterona séricos se encontraron mínimamente incrementados (sin significancia estadística). El control dopaminérgico sobre el eje de GH en ratones ha sido escasamente documentado. Para dilucidar la participación de los RD2s centrales e hipofisarios en el eje de crecimiento, realizamos un estudio exhaustivo de este eje en animales lacDrd2KO comparado con sus controles. No se hallaron diferencias en la talla ni en la longitud del fémur entre ratones lacDrd2KO y Drd2loxP/ loxP. Del mismo modo, no observamos alteraciones en los niveles de I GF-I séricos, concentración de GH intrahipofisaria, y determinamos una pulsatilidad de GH normal, mediante el análisis de genes hepáticos dependientes de GH sexualmente dimórficos. Todas estas evidencias en su conjunto apoyan la idea de que los RD2s hipofisarios no están involucrados en el eje de crecimiento, sino que la ausencia de los RD2s centrales es la que produce un retardo en el crecimiento en los ratones Drd2-/ - . En nuestro laboratorio, se demostró previamente la importancia de la participación de la dopamina a través del RD2 en el crecimiento corporal normal y en la secreción de GH en el período neonatal (68). En conjunto los resultados obtenidos con el ratón lacDrd2KO y con ratones Drd2-/ - asignan a este receptor hipotalámico una acción final estimuladora sobre el eje de GHRH-GH. Habiendo descripto que las hembras lacDrd2KO presentaban hiperplasia de lactotropos, decidimos caracterizar dichos prolactinomas, ya que resultaría un modelo experimental mejorado para el estudio de prolactinomas resistentes a agonistas dopaminérgicos. Encontramos un incremento en los niveles de proliferación celular en las hipófisis lacDrd2KO, lo que condice con el aumento en el peso hipofisario, y analizamos parámetros relacionados con la angiogénesis, proceso por el cual se desarrolla nueva vasculatura a partir de una ya existente, y que tiene un rol crucial en el crecimiento tumoral (267). El rol de la angiogénesis en la generación de adenomas hipofisarios ha sido altamente cuestionado. La mayoría de las neoplasias se caracterizan por presentar un alto nivel de densidad microvascular (DMV: número de vasos por área), considerando que la vasculatura es esencial para el crecimiento del tumor y el desarrollo de metástasis. Los adenomas hipofisarios, contrariamente, son en su mayoría benignos, de 89 Capítulo I - Discusión crecimiento lento con una frecuencia de metástasis extremadamente baja. Sin embargo, su característica de escasa agresividad no excluye la participación de la angiogénesis durante su desarrollo. Apoyando esto, las lesiones benignas o precancerosas de glándula mamaria y cérvix, presentan un incremento de la vascularización respecto a los tejidos normales, lo que demuestra que la necesidad de generación de nuevos vasos no es requisito determinante de malignidad (268). En este caso, los factores relacionados con la angiogénesis evaluados fueron el factor de crecimiendo VEGF y CD31 un antígeno específico de células endoteliales como marcador de vasos totales. Observamos un aumento en el número de vasos CD31 positivos (mayor DMV) en los ratones lacDrd2KO respecto de las hipófisis controles. Esto se correlaciona con los resultados obtenidos previamente por el laboratorio, donde determinamos que las hipófisis de ratones hembra Drd2-/ - también presentaban una mayor DMV, sin diferencias en el tamaño de los vasos con respecto a los presentes en hipófisis de ratones WT, lo que resultó en un aumento en el área vascular relativa (210). En la literatura se ha descripto que los adenomas hipofisarios tienen menor DMV que la glándula normal, y se sugiere que factores inhibitorios de la angiogénesis podrían estar relacionados en la etiopatogenia de los mismos (180;185;268;269). En prolactinomas humanos algunos trabajos apuntan a una disminución en la densidad microvascular respecto a las hipófisis normales (270), sin embargo otros proponen lo contrario. En nuestro laboratorio, a diferencia de los resultados obtenidos con modelos murinos, determinamos una menor área vascular en tumores hipofisarios humanos respecto de glándulas normales, pero destacamos tamaños vasculares diferentes. Observamos, una gran predominancia de vasos de menor tamaño en los adenomas lo que podría estar sugiriendo una mayor angiogénesis, en contraposición con la presencia de vasos de mayor tamaño en las hipófisis normales (271). Por otro lado, analizamos los niveles de VEGF en ambos genotipos, tanto séricos como intrahipofisarios. VEGF es una de las citoquinas más importantes involucradas en la angiogénesis en condiciones fisiológicas y patológicas (272). Es reconocida por su rol fundamental en el crecimiento de tumores que requieren del desarrollo de nueva vasculatura cuando su volumen sobrepasa los pocos milímetros (273). Encontramos un aumento significativo en la concentración de VEGF en las hipófisis hiperplásicas de las hembras lacDrd2KO. Esto concuerda con lo observado en estudios previos del laboratorio donde determinamos un aumento en la expresión de este factor de crecimiento en hipófisis Drd2-/ - y demostramos que el tratamiento de los prolactinomas experimentales, ya sea de forma local o sistémica con drogas anti-VEGF, resultaron en una inhibición sustancial del tamaño tumoral y de los niveles de prolactina (210). Del mismo modo, describimos un aumento en la expresión de VEGF en una cohorte de prolactinomas humanos resistentes a agonistas dopaminérgicos, con una fuerte correlación en la expresión de este factor con CD31, en diferentes adenomas hipofisarios (208). 90 Capítulo I - Discusión Por otra parte, determinamos valores séricos de VEGF normales en las hembras lacDrd2KO respecto de sus pares Drd2loxP/ loxP. Previamente en el modelo de ratón knockout total habíamos observado que los niveles séricos de VEGF se encontraban significativamente aumentados en las hembras Drd2-/ - a los 5 y 9 meses de edad, respecto de sus pares WT de la misma edad (210). Analizando estos datos en su conjunto podríamos postular que la ausencia de RD2s en múltiples tejidos, estaría causando aumentos de VEGF sistémicos, mientras que la ausencia específica de estos receptores a nivel hipofisario provocaría un aumento de VEGF a solo nivel de la glándula pituitaria, y no a nivel sistémico. En trabajos previos del laboratorio relacionados al desarrollo y la regulación de prolactinomas experimentales en distintos modelos animales hemos descripto la participación de factores de crecimiento, tales como VEGF, PTTG y el FGF-2. En particular demostramos que los tres factores son incrementados por la acción de estrógenos, pero la falla del tono inhibitorio dopaminérgico incrementaría solo la expresión de VEGF (59;204;206;207). Está reportado que la dopamina ejerce un efecto antiangiogénico en el endotelio, inhibiendo las acciones permeabilizantes mediadas por VEGF al inducir la endocitosis de su receptor VEGFR-2, actuando vía el RD2, crítico para promover la angiogénesis. Sin embargo, este neurotransmisor, no afecta otros mediadores que influencian la permeabilidad microvascular, la proliferación de células endoteliales y su migración (274). El mayor índice de proliferación observado en las hipófisis lacDrd2KO mediante el antígeno PCNA, al igual que el previamente descripto en prolactinomas de Drd2-/ - (210), es seguramente el resultado de la acción de factores de crecimiento que tienen mayor acceso a las células debido al aumento de permeabilidad vascular y al desarrollo de los nuevos vasos inducidos por factores angiogénicos como el VEGF. Finalmente estudiamos la expresión de marcadores específicos de células madre/ progenitoras en este nuevo modelo de prolactinomas experimentales. Aunque la teoría de las CSCs fue elaborada principalmente para las neoplasias malignas, y las CSCs fueron asiladas principalmente en este tipo de tumores, la presencia de células madre/ progenitoras en hipófisis adultas eleva la posibilidad de que las mismas jueguen un rol la formación de adenomas hipofisarios, frecuentemente benignos (230). Lo cierto es que hasta el momento poco se sabe del rol de las células madre/ progenitoras en el desarrollo de tumores hipofisarios. Existe un estudio en ratones, en el cual se asocia a células nestina positivas en el desarrollo y crecimiento de tumores hipofisarios del lóbulo intermedio. Estas células expresaban además Sox-2 y Lhx3 pero no hormonas hipofisarias (257). Además, en adenomas hipofisarios humanos extraídos de pacientes sometidos a cirugía, se aisló y caracterizó una SP, la cual presentó un incremento (respecto de la MP) en la expresión de transportadores de multidrogas, oncogenes y genes relacionados a células madre/ progenitoras, como ser componentes de las vías Notch y Wnt/ -catenina (230). 91 Capítulo I - Discusión Nosotros encontramos en las hipófisis tumorales de ratones lacDrd2KO una tendencia a la disminución en los factores involucrados en la vía de Notch (dos de sus receptores, Notch-1 y Notch-3 y uno de sus genes blanco Hes-1) en comparación con hipófisis control. En relación a esto, hay evidencias que determinan un aumento en los niveles de Notch-3 en adenomas no funcionantes humanos (275;276), el cual no se observa en el resto de los tumores hipofisarios, entre ellos los productores de GH y prolactina (276). A su vez, no observamos diferencias en la cantidad de células Sox-2 positivas entre genotipos, pero sí cambios en la localización subcelular de este factor. Encontramos una disminución en las células con núcleos Sox-2 positivos en los prolactinomas experimentales, sugiriendo una menor activación de este factor de transcripción en las mismas. Estos resultados en su conjunto sugieren que el aumento en los niveles proliferativos en las hipófisis hiperplásicas de lacDrd2KO no es a expensas de las células con fenotipo de células madre/ progenitoras, o alternativamente, que las mismas han participado en estadíos más tempranos de la génesis del prolactinoma. Nuestros resultados en su conjunto demuestran que los ratones hembra lacDrd2KO resultan un excelente modelo para el estudio de prolactinomas resistentes a dopamina debido a que carecen de RD2s funcionales en lactotropos. A causa de esta ausencia presentan hiperprolactinemia crónica e hiperplasia hipofisaria con un marcado incremento en el índice de proliferación y en el patrón angiogénico, y una tendencia a la disminución en la proporción de células madre. Por otra parte, el modelo experimental tiene RD2s funcionales en el resto del organismo, y por lo tanto el eje de crecimiento conservado. 92 Capítulo II - Introducción CAPÍTULO II – ESTUDIO DE LA INGESTA EN RATONES HEMBRA lacDrd2KO. INTRODUCCIÓN OBESIDAD Los mamíferos se originaron hace más de 100 millones de años, mientras que el primer antepasado del hombre surge alrededor de 7 millones de años atrás. Durante todos estos años hemos evolucionado para asegurar la supervivencia de la especie, lo que implica alimentarse, y una vez asegurado esto, reproducirse. Pero la realidad cambió rotundamente en poco tiempo: existe un desarrollo tecnológico desmesurado y la expectativa de vida incrementó considerablemente, alcanzando un promedio de 76 años en la Argentina. Esto último se debe al gran desarrollo medicinal para el combate de enfermedades, y a la ausencia actual de grandes predadores. Esta nueva realidad trajo consigo nuevas enfermedades y desórdenes que todavía no entendemos del todo. Una epidemia que se ha expandido por todos los continentes es la obesidad (11). En la actualidad existen, según la Organización Mundial de la Salud (OMS), más personas con sobrepeso que con desnutrición, y establece a su vez, que desde 1980 se ha duplicado la tasa de obesidad en todo el mundo. En Argentina los datos de la Organización de Naciones Unidas para la Alimentación y la Agricultura (FAO) muestran que el 29 por ciento de los adultos son obesos. La medida estándar que se utiliza para determinar la presencia de obesidad es el índice de masa corporal ( body-mass index = BMI ), el cual consiste en la relación entre el peso y la altura. En adultos, un BMI mayor a 30 kg/ m 2 está asociado a sobrepeso, lo cual implica un aumento en el riesgo de enfermedades asociadas. La obesidad es un desorden metabólico crónico que predispone a la adquisición de diabetes tipo I I , enfermedades cardiovasculares, cerebrovasculares e hipertensión, siendo éstas algunas de las principales causas de muerte en la actualidad (277). El estudio de la obesidad resulta altamente complejo. A partir del estudio con gemelos se deduce que entre el 40-70% de las variaciones en el peso corporal se deben a factores genéticos (278). Por otro lado, existen estudios que demuestran que la obesidad debe ser tratada como una adicción (adicción a la comida) debido al efecto que produce la comida a nivel del sistema nervioso central en algunos pacientes obesos (279;280). Y por último, no se pueden dejar de lado el balance energético, el metabolismo de los ácidos grasos, ni tampoco las costumbres socioculturales. La realidad es que mucha gente hace esfuerzos cotidianos para bajar de peso con dietas controladas y/ o actividad física, y no lo logra o lo consigue sólo por periodos muy cortos. 93 Capítulo II - Introducción Las razones por las cuales el índice de obesidad ha y sigue incrementando quedan todavía por determinarse. SEÑALES INVOLUCRADAS EN LA INGESTA El hipotálamo integra las señales tanto hormonales como neuronales que regulan el apetito y el metabolismo. Además de los circuitos que responden a señales periféricas, existen circuitos neuronales que se superponen a éstos y que tienen la potencialidad de regular el sistema como un conjunto. Está establecido que el proceso integral de la ingesta también involucra los mecanismos relacionados con el sistema de recompensa a nivel central, el cual incluye aspectos como la motivación para comer, y que responden a numerosas y diversas señales como los aromas, sabor de la comida, entre otras. La regulación central de la ingesta involucra una compleja relación entre señales periféricas hormonales y metabólicas, neuropéptidos, monoaminas y otros mensajeros, los cuales a su vez actúan en varios niveles del sistema nervioso (Figura 28). Detallaremos solo algunos de estos componentes que se relacionan directamente con nuestros objetivos. Figura 28: Esquema representativo de la regulación hipotalámica de la ingesta. Los agonistas dopaminérgicos podrían actuar a varios niveles: 1) a través de la inhibición de prolactina, 2) a nivel de las neuronas de primer orden en el núcleo Arc, estimulando POMC o inhibiendo AgRP/ NPY, y 3) a nivel de las neuronas del HLA en forma indirecta estimulando las orexinas. Las flechas continuas indican estimulación, mientras que las punteadas inhibición. Tomado y modificado de (11). 94 Capítulo II - Introducción Señales centrales Dopamina La dopamina modifica la ingesta de alimentos actuando sobre diversos núcleos hipotalámicos involucrados en la regulación de la misma a través de sus receptores RD1 y RD2 (11), y varios modelos farmacológicos que antagonizan su acción, o la disrupción génica del sistema dopaminérgico en roedores, dan como resultado una hipofagia grave. Por otra parte, el sistema dopaminérgico que proyecta desde el área tegmental ventral al núcleo accumbens del cerebro, tiene una participación activa en este proceso ya que está íntimamente asociado al sistema de recompensa. Esta área es importante porque está relacionada no solo a la ingesta de alimentos, sino también a los mecanismos de adicción a las drogas (132). Los sistemas endógenos de opioides y canabinoides son a su vez importantes, y estarían interactuando con los sistemas dopaminérgicos. Asimismo, la gran mayoría de las drogas que se utilizan en la clínica para el tratamiento de la obesidad a nivel central tienen acciones sobre el sistema monoaminérgico (que involucra a dopamina, serotonina y noradrenalina). Y muchas de ellas se relacionan con la inhibición de la recaptación de los neurotransmisores como serotonina y dopamina. La dopamina regularía el apetito y la saciedad actuando en áreas hipotalámicas específicas. Sin embargo, los efectos de la dopamina en el control del apetito no son claros y resultan conflictivos de ser interpretados. Esto se debe, en parte, a que la dopamina tiene diferentes acciones en los distintos núcleos hipotalámicos, estando además involucrados los diferentes receptores dopaminérgicos. Por otro lado, las respuestas en torno a la ingesta son distintas cuando se administra dopamina de forma local o sistémicamente (281). Algunos autores sugieren que las hormonas involucradas tradicionalmente en el sistema homeostático pueden afectar directamente la vía de recompensa de dopamina. Se observó que insulina, leptina y ghrelina pueden estimular o inhibir respectivamente las señales dopaminérgicas en el núcleo Arc, y que por lo tanto podrían representar un mecanismo adicional de control (132). POMC y α-MSH El gen de la propiomelanocortina (POMC) se expresa tanto a nivel central, principalmente en el núcleo Arc hipotalámico como ya detallamos previamente, como también a nivel periférico. En la periferia POMC se expresa principalmente en la hipófisis, piel y folículos pilosos (282). El gen de POMC codifica para una pre-prohormona de 31-36 KDa de la cual derivan siete péptidos, los cuales corresponden a los neuropéptidos más abundantes del cerebro: ACTH, α- MSH, -MSH, -MSH, péptido símil corticotrop ina del lóbulo intermedio (CLIP), -lipotrofina y -endorfina (Figura 29). A estos péptidos en su conjunto se los denominan melanocortinas, los cuales median sus efectos a 95 Capítulo II - Introducción través de una familia de cinco receptores de melanocortinas acoplados a proteína G (MC1R a MC5R) (282). Figura 29: Procesamiento de POMC en hipotálamo, y sus neuropéptidos derivados. N-POC: fragmento de POMC del extremo amino terminal; JP: péptido de unión; DA-MSH: MSH deacetilada; CLI P: péptido tipo corticotropina del lóbulo intermedio; LPH: lipotrofina y END: endorfina. Tomado y modificado de (283). El procesamiento de POMC es tejido específico, y su clivaje postraduccional depende de varias enzimas convertasas. Así es como en el lóbulo anterior de la hipófisis se genera la producción de ACTH (péptido de 39 aminoácidos conocido por su acción sobre la glándula adrenal) estimulante de la corticoideogénesis, y –lipotrofina (284), mientras que en el lóbulo intermedio de la hipófisis, los melanotropos sintetizan y secretan α- MSH, - MSH, CLIP, - lipotrofina y – endorfina. Las melanocortinas fueron descubiertas en el año 1916, y la primera función atribuida fue la de modular el color de la piel en las ranas (284). Una serie de hallazgos posteriores permitió avanzar sobre el conocimiento de su rol fisiológico en mamíferos. En 1958 se postuló que tanto la ACTH como la α-MSH podrían tener un papel fisiológico en el funcionamiento del sistema nervioso central. La inyección de melanocortinas en el líquido cefalorraquídeo o en áreas específicas del cerebro, producían cambios en el comportamiento en los monos, perros, ratas, conejos y gatos, que consistían en un excesivo cuidado de sí mismos, aseo y reiterados episodios de estiramiento y bostezo (284). Por otra parte, en seres humanos, la inyección d e α-MSH aumenta la coloración en la piel. Durante los años 90, una serie de experimentos farmacológicos y genéticos demostraron concluyentemente que las melanocortinas modulan el peso corporal a través de modificaciones en la ingesta y en el consumo de energía. En el ratón por ejemplo, lesiones en varios puntos del sistema de melanocortinas generan obesidad, lo mismo que la expresión ectópica de un antagonista de MC4R (14), y las deleciones de MC4R, MC3R (285), como también del gen que codifica para POMC (286). En humanos existe el reporte de dos familias con mutaciones nulas del 96 Capítulo II - Introducción gen que codifica para POMC, que presentan como consecuencia lo que se denomina síndrome de obesidad asociada a melanocortinas (287). Además, existen estudios que determinan que la principal causa monogénica de obesidad en humanos es la haplo-insuficiencia de MC4R (más del 5% de los casos) (282). α-MSH, es un péptido de 13 aminoácidos, con funciones anorexigénicas, ya que al actuar sobre el hipotálamo a través de sus receptores (MC3R y MC4R) produce una disminución en la ingesta de alimento y un aumento en el consumo energético (11). Las neuronas de POMC expresan ambos receptores, MC3R y MC4R. La secreción de α-MSH hipofisaria está bajo un control dopaminérgico inhibitorio dado por el hipotálamo (288). Las neuronas dopaminérgicas TI DA son las principales reguladoras (289), como así también las neuronas tuberohipofisaria y el sistema dopaminérgico periventricular, siendo los receptores de dopamina presentes en los melanotropos los RD2s. Tratamientos crónicos en ratas con agonistas dopaminérgicos disminuyen el contenido de POMC, como así también la actividad de la acetiltransferasa, enzima involucrada en el procesamiento de los péptidos derivados de POMC en el lóbulo intermedio. Por otra parte, la hormona CRH aumenta la secreción de α-MSH al igual que agonistas -adrenérgicos (290). Más allá de la regulación negativa de dopamina sobre la liberación de α-MSH desde la hipófisis intermedia, la relación entre dopamina y el sistema de melanocortinas no ha sido muy definida a nivel central. NPY NPY es un péptido de 36 aminoácidos descubierto en el año 1982, y es el neuropéptido hipotalámico más abundante. Los principales sitios hipotalámicos de expresión de NPY son el núcleo Arc y DMN. NPY es el péptido orexigénico más potente y actúa a través de numerosos receptores acoplados a proteínas G. Según sus características, estructurales y farmacológicas, se distinguen cinco receptores: Y1 a Y5 (281), estando todos relacionados con acciones metabólicas salvo el receptor Y3. La administración intracerebroventricular de NPY, en única dosis o de manera continua, produce hiperfagia, ganancia de peso, y los efectos endócrinos asociados a obesidad. NPY también actúa en el PVN donde modula mecanismos que regulan la alimentación, termogénesis y el metabolismo energético. NPY promueve la reserva energética, disminuyendo la termogénesis del tejido adiposo pardo, aumentando la respuesta a insulina por parte del tejido adiposo blanco, y activando el eje hipotálamo-hipófisis-adrenal (281). El ratón knockout para NPY posee niveles de ingesta normales y responde sin alteraciones al ayuno y realimentación, reforzando la idea de que múltiples factores se encuentran involucrados en la regulación del apetito (281). La leptina inhibe la activación de neuronas NPY, mientras que la ghrelina activa a las neuronas AgRP/ NPY del núcleo Arc (291). 97 Capítulo II - Introducción Finalmente, se propone una relación entre el sistema dopaminérgico y NPY, dado que agonistas dopaminérgicos disminuyen la ingesta por una acción antagónica sobre las neuronas hipotalámicas que expresan NPY (292). Se ha demostrado que la administración de agonistas de RD1 y RD2 en ratones ob/ ob reducen los niveles elevados de ARNm y proteicos de NPY (292;293). Orexinas - hipocretinas En el año 1998, dos laboratorios norteamericanos en simultáneo, y utilizando distintas técnicas experimentales, publican y predicen la estructura de dos neuropéptidos hipotalámicos derivados de un precursor común denominado pre-pro-orexina (PPO). Los mismos son llamados orexinas A (OXA) y B (OXB) por el grupo de Sakurai (294), e hipocretinas 1 y 2 por De Lecea y colaboradores (295). Desde entonces, los nombres orexinas e hipocretinas se utilizan alternativa e indistintamente en honor a la historia de su descubrimiento. Las orexinas son producidas en una zona muy circunscripta del cerebro, el área lateral perifornical del hipotálamo. Los axones de estas neuronas proyectan a múltiples zonas del cerebro lo que explica la gran diversidad de acciones biológicas. Los primeros trabajos mostraron que la administración central de OXA producía un aumento en la ingesta de manera dosis dependiente, mientras que OXB aumentaba la frecuencia excitatoria de la sinapsis en neuronas hipotalámicas en cultivo. Hoy se sabe que la inyección de OXA no solamente afecta la ingesta, sino que también aumenta las pulsaciones (296), la presión sanguínea (297) y el metabolismo (298). Algunos trabajos también postulan que la OXA juega un rol en la termorregulación ya que inyecciones de la misma a nivel central producen una disminución de la temperatura corporal (299). También se describió un rol importante de las orexinas en la regulación del sueño y se observó, en perros, ratones y humanos, que la patología de narcolepsia (desorden donde los periodos de vigilia son fragmentados constantemente por episodios de sueño) se relaciona con deficiencias en la neurotransmisión de las orexinas (300). Los ratones que carecen de la expresión de PPO presentan síndromes de desregulación de las fases sueño-vigilia. En forma convergente otro grupo demuestra en perros, que la patología de narcolepsia está asociada a una mutación en el gen que codifica para el receptor de orexinas (301). La importancia de las neuronas de orexinas fue confirmada en humanos al hallarse que un número significativo de casos de narcolepsia estaban asociados a una dramática reducción del número de neuronas hipotalámicas positivas para orexinas, así como también niveles reducidos de orexinas en el cerebro y en el fluido espinal (300). Sin embargo la participación de las orexinas en la ingesta y el metabolismo es también evidente en numerosos trabajos. Los mismos ratones con deleción génica de orexinas, poseen una ingesta menor pero con un peso corporal conservado. Esto se explica, ya que las orexinas estarían también involucradas en la actividad motora y el estado metabólico. Su ausencia provoca entonces una disminución en la ingesta como también en la actividad locomotora (299). 98 Capítulo II - Introducción Un mecasnismo descripto por el cual las orexinas regulan la ingesta es a través de la liberación del neuropéptido NPY. Se cree que OXA es la principal orexina involucrada en el control de la ingesta mientras que no está clara todavía la acción de OXB en el control del apetito (11). Las neuronas de orexinas responden a señales tanto periféricas como centrales que regulan la ingesta, como ser la leptina, α-MSH, la glucosa y ghrelina (302). Las neuronas de orexinas en el LHA reciben la inervación de neuronas POMC y AgRP/ NPY, y expresan receptores de leptina y αMSH. Estas neuronas también proyectan hacia las neuronas POMC y AgRP/ NPY, inhibiéndolas y activándolas, respectivamente (302). Una regulación dopaminérgica en el control de las orexinas ha sido descripta. Agonistas de los receptores RD1 y RD2 son capaces de incrementar la actividad de las neuronas orexigénicas en el LHA (Figura 28). Se demostró que agonistas dopaminérgicos incrementan la expresión de Fos en neuronas de orexinas, y que el tratamiento combinado de antagonistas de RD1 y RD2 bloquean esta activación (303). Este mecanismo no parece ser directo, sino que neuronas y vías intermedias estarían involucradas. En contraposición, algunos estudios in vitro han demostrado que la dopamina reduce o bloquea la activación de las neuronas de orexinas (303). En humanos se demostró que pacientes que padecen de esquizofrenia y son tratados con haloperidol (antagonista del RD2) poseen niveles menores de OXA en el fluido cerebroespinal respecto a pacientes no tratados (304). Señales periféricas Leptina La leptina es una proteína de 146 aminoácidos cuya secuencia se encuentra ampliamente conservada entre las distintas especies de mamíferos (11). Es una hormona producida y secretada por el tejido adiposo blanco, que actúa sobre el hipotálamo afectando la ingesta de alimentos (277). Su regulación es compleja y en ella intervienen insulina, los glucocorticoides y algunas citoquinas (11). El receptor de leptina (OBR) pertenece a la familia de receptores de citoquina de clase I , y se encuentra presente en múltiples tejidos, incluyendo el cerebro, en diferentes isoformas: una larga (OBRb) y cuatro cortas. La isoforma larga, OBRb, es la implicada en la señalización de los efectos sobre la ingesta en el hipotálamo. Comúnmente los niveles de leptina incrementan de manera proporcional a la cantidad de adiposidad, y actúan sobre las neuronas hipotalámicas ejerciendo una retroalimentación negativa para regular el balance energético, a través de su receptor OBRb localizado en el núcleo Arc (305). Se ha demostrado a su vez, que los niveles de leptina incrementan, temporariamente, con la ingesta de alimentos (306;307). 99 Capítulo II - Introducción La leptina actúa sobre dos poblaciones neuronales involucradas en la regulación del balance energético ubicadas en núcleo Arc del hipotálamo (Figura 2 y 28). Una población neuronal sintetiza y libera dos neuropéptidos orexigénicos, los cuales son inhibidos por leptina: AgRP y NPY. La otra población incluye neuronas anorexigénicas productoras de POMC, cuya actividad es estimulada por esta adipoquina. Se ha demostrado que algunos de los efectos de leptina son mediados por el sistema de melanocortinas (11). Los receptores de leptina se expresan en su mayoría en las neuronas POMC del núcleo Arc (308) y el tratamiento con antagonistas del receptor de melanocortinas revierte parcialmente los efectos anorexigénicos de la leptina exógena (309;310). Estos resultados indican que el sistema central de melanocortinas está por debajo de la vía de señalización del OBRb y que jugarían un rol importante en la mediación de los efectos de esta hormona (311). De hecho, leptina regula la expresión de POMC, incrementando la liberación de su derivado α-MSH (péptido anorexigénico). Es decir, leptina reduce la ingesta de alimentos mediante la inhibición de péptidos orexigénicos y la estimulación de aquellos con funciones anorexigénicas. Numerosos estudios sugieren que la leptina modularía las acciones dopaminérgicas, particularmente aquellas asociadas al sistema de recompensa, actuando sobre el sistema mesolímbico. Los receptores de leptina se expresan en el área tegmental ventral y la sustancia nigra pars compacta (312), a través de los cuales regula la expresión de transportadores de dopamina (313). A su vez, inyecciones de leptina en el núcleo Arc disminuyen los niveles basales de dopamina como los estimulados por la ingesta (314). En ratas alimentadas con dietas ricas en grasas (modelo de obesidad en roedores) se asoció un incremento en los niveles de leptina con el aumento en los RD2s y una disminución en la expresión del transportador de dopamina en las áreas tegmental ventral y sustancia nigra pars compacta (313). Finalmente, la información existente sobre la relación entre prolactina y leptina es controvertida. Por ejemplo, los niveles séricos de leptina están disminuidos o inalterados en ratones deficientes del PRLR (56) y están elevados en animales que sobreexpresan la hormona (315). Sin embargo, ratones machos knockout para prolactina presentaron un aumento en los niveles séricos de leptina (316). En humanos no hay una correlación consistente entre prolactina sérica y los niveles de leptina (30). Resistencia a leptina Como ya mencionamos los niveles de leptina incrementan de manera proporcional a la cantidad de tejido adiposo, y actúan inhibiendo la ingesta de alimentos (305). Sin embargo, los individuos obesos no disminuyen la ingesta, más allá del incremento en los niveles de leptina que la mayoría presenta. Se observó en estos pacientes y algunos modelos experimentales, una resistencia a la leptina, que estaría explicando este fenómeno (307). La heterogeneidad en la respuesta a leptina fue evidenciada en primer lugar en roedores. El ratón leptina deficiente ( ob/ ob ) resultó altamente sensible a la acción de la misma, mientras que 100 Capítulo II - Introducción animales con diversas mutaciones en su receptor resultaron resistentes. Por otra parte, animales con obesidad inducida por alimento rico en grasas mostraron respuestas parciales a la administración de leptina (317). Una variabilidad similar fue observada en humanos obesos, donde pacientes con deficiencia en los niveles de leptina resultan extremadamente sensibles a los efectos del tratamiento con la misma, sin embargo es una cohorte muy pequeña la que presenta este fenotipo. La mayoría de los obesos son resistentes a la leptina. Esto indica que la utilidad de leptina como monoterapia es limitada a un pequeño grupo de pacientes (317). La resistencia a leptina es altamente compleja ya que puede resultar selectiva, en tejidos periféricos o a nivel central, y puede ocurrir en diferentes niveles de la cascada de señalización de la misma (277). Ratones obesos a causa de una dieta rica en grasas presentan resistencia periférica a la administración de leptina (318) debido a deficiencias en el transporte de la misma hacia el cerebro. Esto logró revertirse con inyecciones centrales de esta hormona (319;320). Resistencia periférica también fue observada en ratas obesas Otsuka Long-Evans Tokushima, las cuales también respondieron a la leptina administrada a nivel central (321). En humanos existe una correlación entre los niveles de leptina plasmáticos y en fluido cerebroespinal, y la relación entre ambos (cerebroespinal/ plasmático) resulta menor en individuos obesos (322), lo que sugiere como causa de la resistencia a un transporte deficiente hacia el fluido cerebroespinal. Se sabe que el transporte de leptina hacia el cerebro se lleva a cabo a través de un mecanismo saturable que involucra a las isoformas cortas de los receptores de leptina ubicados en la barrera hemato-encefálica (320). Hoy en día se conoce que leptina puede ser además directamente capturada por regiones cerebrales que presentan conexiones directas con la circulación periférica, como el núcleo Arc (323). El núcleo Arc de los ratones con obesidad inducida por dieta permanece inactivo frente a la inyección central de leptina (324), mientras que el resto de los núcleos hipotalámicos sí responden, lo que sugiere una resistencia central parcial. De hecho, la obesidad relacionada con la edad, se encuentra asociada a resistencia central la cual involucra una disminución en los niveles de ARNm y proteína del receptor OBR (325). Los mecanismos de señalización responsables de la resistencia no están del todo dilucidados. Leptina actúa sobre su receptor activando la vía de JAK-Stat, la cual depende en parte de la fosforilación de residuos tirosina, de proteínas específicas como Stat3 (326). Esta señalización es por lo general atenuada por la activación de proteínas SOCS, las cuales inhiben a JAK. Consistente con esto, los efectos de leptina se ven incrementados en células que carecen de SOCS3 o PT1b (una fosfatasa de tirosinas). Ratones que carecen de cualquiera de estos genes, específicamente en neuronas POMC, resultan resistentes a la obesidad y permanecen delgados frente a dietas ricas en grasas, manteniendo la sensibilidad a leptina. La contribución de estas proteínas en el desarrollo de resistencia a leptina es todavía poco claro, pero estas evidencias sugieren que inhibidores químicos de SOCS3 y PT1b podrían resultar potenciales agentes antiobesidad, ya sea solos o combinados con leptina (317). 101 Capítulo II - Introducción CARACTERIZACIÓN DE LOS MECANISMOS QUE INGESTA EN EL RATÓN KNOCKOUT TOTAL Drd2 -/- REGULAN LA En el laboratorio demostramos que el ratón Drd2-/ - presenta modificaciones en los circuitos que controlan la ingesta (60), y fallas en la homeostasis de la glucosa. A pesar de la hiperprolactinemia del modelo, describimos que no existen diferencias en los niveles de ingesta diaria por ratón entre genotipos, y en correlación, la adiposidad de los ratones no estaba aumentada. Determinamos, a su vez, altos niveles de expresión del péptido anorexigénico α-MSH y bajos niveles del precursor de péptidos orexigénicos (PPO) en los Drd2-/ - en comparación con los WT, postulando que los altos niveles de prolactina sérica estarían compensando los efectos anorexigénicos de estos dos eventos (60). Nuestros estudios en ratones Drd2-/ - demostraron que, más allá de que no existen diferencias en los niveles hipotalámicos de MC3R y MC4R, la ausencia de RD2s funcionales causó un incremento en los niveles circulante de α-MSH exclusivamente en las hembras (60). El contenido de α-MSH hipofisario así como el porcentaje de melanotropos en el lóbulo intermedio, se encontraron disminuidos en ambos sexos, indicando una probable disminución en el almacenamiento de α-MSH, y liberación del péptido al torrente sanguíneo, consistente con el rol inhibitorio de los RD2s en su secreción (60;288). Por otra parte, el contenido de α-MSH hipotalámico resultó significativamente incrementado en las hembras Drd2-/ - , por lo que se esperaría una disminución en los niveles de ingesta, pero altos niveles de prolactina están presentes en los ratones hembra Drd2-/ - que podrían estar contrarrestando este efecto. Por otro lado, demostramos, que la ausencia de RD2s no tuvo efecto en los niveles de ARNm de Npy . Los ratones Drd2-/ - poseen altos niveles de prolactina y un eje de GH-I GF-I disminuido (68;262). Mientras que el aumento de prolactina y la disminución de la acción dopaminérgica favorecerían el incremento en la expresión de Npy (292;293), la GH reducida posee el efecto contrario (327). En este modelo también, la expresión hipotalámica de Ppo resultó significativamente disminuida en las hembras y no en los macho Drd2-/ - (60). Los niveles de ARNm del receptor de orexinas estaban conservados entre genotipos, indicando que las orexinas no regulan a su propio receptor. El descenso en los niveles de ARNm de Ppo indica que los RD2s están involucrados, directa o indirectamente, en la regulación de la expresión de orexinas hipotalámicas. La disrupción de los RD2s estaría induciendo una disminución en la expresión de este factor, pero como no se encontraron diferencias en machos, factores adicionales estarían participando. En particular los ratones Drd2-/ - no presentan alteraciones en los niveles séricos de leptina ni en la expresión de OBR hipotalámico (60). Las hembras presentaron niveles de leptina en ayuno mayores que los machos, pero esta diferencia sexual no se vio modificada entre genotipos. Otros 102 Capítulo II - Introducción autores han demostrado que inyecciones centrales de leptina en los ratones Drd2-/ - provocan mayor disminución en el peso corporal e ingesta respecto de los ratones WT (328). Esto sugiere que existe una interacción entre los RD2s y la sensibilidad a leptina en la regulación de la ingesta y el metabolismo, aunque los mecanismos exactos todavía se desconocen. En resumen, demostramos que la falta de función del RD2 en todo el organismo modifica la regulación de la prolactina, del eje de crecimiento como así también la regulación central asociada a la ingesta de alimentos. Es por ello que desarrollamos el raton knockout tejido específico, que carece de RD2s funcionales solamente en lactotropos (lacDrd2KO), para determinar si un modelo de hiperprolactinemia con el sistema dopaminérgico central conservado develaba las acciones de la prolactina sobre la ingesta, adiposidad, y el metabolismo de la glucosa. 103 Capítulo II - Objetivos OBJETIVOS Estudiar el impacto de la ausencia de RD2s en lactotropos sobre el peso corporal y la ingesta profundizando la participación del sistema nervioso central. Los objetivos específicos fueron: - Realizar curvas de peso corporal a lo largo del desarrollo en ratones hembra y macho Drd2loxP/ loxP y lacDrd2KO, y evaluar los niveles de ingesta diaria a los 3 y 5 meses de edad. - Determinar de qué manera responden las hembras control e hiperprolactinémicas al ayuno prolongado (12 hs) y a la realimentación luego de dicho ayuno, analizando las variaciones en el peso corporal y los niveles de ingesta. - Estudiar los niveles de leptina séricos y la sensibilidad a la misma, en ratones hembra de ambos genotipos. - Analizar la expresión hipotalámica de péptidos orexigénicos ( Prlr , Ppo y Npy ) y anorexigénicos ( Pomc), y estudiar la expresión a nivel hipofisario del péptido anorexigénico α-MSH, en ratones hembra de ambos genotipos a los 5 y 10 meses de edad. 104 Capítulo II - Resultados RESULTADOS INGESTA Y PESO CORPORAL Los ratones hembra lacDrd2KO presentaron un incremento significativo en el peso corporal a partir de los 5 meses de edad (Figura 30A y C). Este incremento no se debió a una alteración en el eje de GH, el cual estaba conservado (como detallamos en el Capítulo I ), sino a una ingesta de alimentos aumentada. Este aumento se evidenció en las hembras lacDrd2KO de 3 y 5 meses en comparación con pares controles Drd2loxP/ loxP de la misma edad (Figura 30B). Figura 30: Peso corporal e ingesta. A) Curvas de peso corporal a lo largo del desarrollo en ratones hembra Drd2loxP/ loxP (n = 8) y lacDrd2KO (n = 12). El AMR evidenció una interacción significativa (genotipo x edad), Pinteracción ˂ 0,0001; *P ˂ 0,03 vs Drd2loxP/ loxP de la misma edad. B) Niveles de ingesta promedio (gramos/ día/ ratón) en ratones Drd2loxP/ loxP comparados con lacDrd2KO a los 3 (n = 8 y 4, respectivamente) y 5 (n = 7 y 6, respectivamente) meses de edad. El ANOVA de dos factores resultó en * P ˂ 0,005 para el efecto genotipo. C) I magen representativa de los tamaños de ratones hembra Drd2loxP/ loxP y lacDrd2KO de 10 meses de edad. En todas las figuras del capítulo, se grafica la media y las barras de error corresponden al E.S.M. 105 Capítulo II - Resultados Analizamos luego, qué ocurría con el peso corporal y la ingesta en condiciones de ayuno y realimentación. Los ratones hembra lacDrd2KO de 10 meses de edad presentaron menor pérdida de peso corporal luego de un ayuno de 12 hs en comparación con las hembras Drd2loxP/ loxP (Figura 31A). Por otra parte, mientras que la ingesta de alimentos (durante 1 hr) luego del ayuno no se vio modificada entre genotipos en ambas edades estudiadas (Figura 31B), la ganancia de peso producto de esa hora de realimentación resultó incrementada en las hembras lacDrd2KO de 5 meses en comparación con pares controles Drd2loxP/ loxP de la misma edad (Figura 31C). Es decir que las hembras hiperprolactinémicas mantenían mejor el peso corporal a pesar del ayuno y luego, además, recuperaban más durante la realimentación, indicando una homeostasis del balance de peso corporal alterada. Figura 31: Peso corporal e ingesta en condiciones de ayuno y realimentación. A) Pérdida de peso corporal (g) luego de un ayuno de 12 hs en ratones hembra Drd2loxP/ loxP y lacDrd2KO, a los 5 (n = 7 y 7, respectivamente) y 10 (n = 4 y 6, respectivamente) meses de edad. El ANOVA de dos factores evidenció una interacción significativa (genotipo x edad), Pinteracción ˂ 0,01; donde *P ˂ 0,03 vs Drd2loxP/ loxP de la misma edad; y # P ˂ 0,004 vs lacDrd2KO a los 5 meses de edad. B) I ngesta de alimento (g) en una hora de realimentación posterior a las 12 hs de ayuno, en ratones hembra Drd2loxP/ loxP y lacDrd2KO, a los 5 (n = 10 y 10, respectivamente) y 10 (n = 4 y 5, respectivamente) meses de edad. El ANOVA de dos factores no arrojó diferencias significativas. C) Ganancia de peso corporal (g) luego de una hora de realimentación, posterior a 106 Capítulo II - Resultados las 12 hs de ayuno, en ratones hembra Drd2loxP/ loxP (n = 10) y lacDrd2KO (n = 10), a los 5 meses de edad. Se realizó una Prueba T donde * P ˂ 0,05 vs Drd2loxP/ loxP de la misma edad. Leptina A partir de los resultados obtenidos nos planteamos analizar los niveles de leptina y la sensibilidad a la misma en este modelo animal. Determinamos, en primer lugar, que los niveles séricos de leptina en ratones hembra de 5 meses de edad con libre disponibilidad de alimento ( ad libitum ) no diferían de los controles, mientras que a los 10 meses los ratones hembra lacDrd2KO presentaron niveles de leptina cinco veces mayores que sus pares Drd2loxP/ loxP de la misma edad ( P ˂ 0,00005) (Figura 32A). En segunda instancia analizamos los niveles de leptina luego de un ayuno de 12 hs, sin observar diferencias significativas entre genotipos ni edades (Figura 32B). Encontramos que los niveles de leptina correlacionaron positivamente con el peso corporal de los animales ( P ˂ 0,0000001), mientras que no existió correlación entre los valores de leptina y prolactina séricos (Figura 32C). Finalmente, no observamos diferencias significativas en la expresión de ARNm del receptor de leptina ( Obrb ) en hipotálamos de ratones de 10 meses de edad (Figura 32D). 107 Capítulo II - Resultados Figura 32: Leptina. A) Niveles de leptina séricos medidos por ELI SA en ratones hembra Drd2loxP/ loxP y lacDrd2KO, a los 5 y 10 meses de edad (n = 7), con alimento ad libitum . El ANOVA de dos factores evidenció una interacción significativa (genotipo x edad), Pinteracción ˂ 0,0001; donde *P ˂ 0,00005 vs Drd2loxP/ loxP de la misma edad; y vs ratones de 5 meses de edad. B) Niveles de leptina séricos medidos por ELI SA en ratones hembra Drd2loxP/ loxP y lacDrd2KO, ayunados por 12 hs, a los 5 meses (n = 5 y 6 respectivamente) y 10 meses de edad (n = 5). El ANOVA de dos factores no arrojó diferencias significativas. C) I zquierda: correlación entre los niveles de leptina séricos (ng/ ml) y el peso corporal (g); n = 51. La Prueba de Spearman evidenció una correlación significativa, donde P ˂ 0,0000001 y coeficiente de correlación Rs = 0,69. Derecha: correlación entre los niveles de leptina y los de prolactina séricos (ng/ ml); n = 46. Los datos se analizaron por Prueba de Spearman sin encontrar correlación. D) Niveles de expresión del receptor de leptina hipotalámico ( Obrb ) en ratones hembra de 10 meses de edad Drd2loxP/ loxP (n = 6) y lacDrd2KO (n = 7). Los resultados se analizaron por Prueba T sin obtener diferencias entre genotipos. 108 Capítulo II - Resultados Para determinar la sensibilidad a leptina, realizamos un ensayo in vivo en el cual inyectamos PBS (vehículo) o leptina (dosis de 2,5 mg/ kg), luego de un ayuno de 16 hs, y analizamos los niveles de ingesta y la ganancia de peso corporal en ratones Drd2loxP/ loxP y lacDrd2KO de 5 meses de edad, a la hora, 2, 4 y 8 hs posteriores a presentarles el alimento. No encontramos diferencias en la respuesta a leptina en cuanto a la ingesta entre genotipos, y en ambos casos la leptina provocó una disminución significativa en los niveles de ingesta 1 hora post-inyección y no a los otros horarios (Figura 33A). En cuanto a la ganancia de peso corporal, no encontramos diferencias estadísticas en el efecto droga, sin embargo pudimos observar una mayor respuesta a leptina (evidenciada como una disminución en la ganancia de peso) en los ratones control, en comparación a los ratones lacDrd2KO, indicando una leve resistencia a la misma a esta edad (Figura 33B). Figura 33: Sensibilidad a leptina. A) I ngesta de alimento (g) 1 hora post inyección de PBS (vehículo) o leptina en una dosis de 2,5 mg/ kg. Se utilizaron ratones hembra Drd2loxP/ loxP y lacDrd2KO de 5 meses de edad (n = 4). El ANOVA de dos factores evidenció una interacción no significativa (genotipo x droga); donde * P ˂ 0,005 vs inyección con PBS; y #P ˂ 0,05 lacDrd2KO vs Drd2loxP/ loxP. B) Ganancia de peso corporal (g) 1, 2, 4 y 8 hs posteriores a la inyección de PBS o leptina (dosis de 2,5 mg/ Kg. ip). A la derecha se encuentra el resultado obtenido en ratones hembra Drd2loxP/ loxP (n = 4) y a la izquierda el correspondiente a lacDrd2KO (n = 4) de 5 meses de edad. En ambos casos, el AMR resultó en una interacción no significativa (droga x tiempo). En ratones Drd2loxP/ loxP el Ptiempo ˂ 0,002, mientras que Pdroga = 0,19 en ratones lacDrd2KO el Ptiempo ˂ 0,00005 mientras que Pdroga = 0,53. 109 Capítulo II - Resultados Factores orexigénicos y anorexigénicos Al observar el incremento en los niveles de ingesta decidimos estudiar la expresión de factores orexigénicos ( Prlr , Ppo y Npy ) y anorexigénicos ( α-MSH y Pomc) a nivel central. Los niveles hipotalámicos de ARNm del receptor Prlr (todas sus isoformas) resultaron significativamente aumentados en ratones hembra lacDrd2KO de 10 meses de edad, lo que estaría sugiriendo una posible regulación positiva de prolactina sobre su propio receptor en este tejido ( P ˂ 0,0003) (Figura 34A). Asimismo, la expresión hipotalámica del neuropéptido Npy presentó un incremento significativo en las hembras lacDrd2KO comparado con los controles Drd2loxP/ loxP. Esto se evidenció a los 5 meses, cuando se observó el aumento en la ingesta (Figura 30B), manteniéndose la diferencia a los 10 meses de edad ( P ˂ 0,04) (Figura 34D). Por otra parte, los niveles de ARNm de Ppo resultaron similares entre genotipos (Figura 34D). En cuando a los factores anorexigénicos, determinamos que el porcentaje de melanotropos presentes en la hipófisis intermedia es similar en ratones lacDrd2KO y Drd2 loxP/ loxP (Figura 34B y C), y en forma concordante observamos que los niveles de expresión de su precursor hipotalámico Pomc no difirieron entre ratones hembra de ambos genotipos (Figura 34D). Figura 34: Neuropéptidos orexigénicos y anorexigénicos. A) Expresión hipotalámica del ARNm de receptor Prlr ( primers que reconocen todas las isoformas) en ratones hembra de 10 meses de edad Drd2loxP/ loxP (n = 6) y lacDrd2KO (n = 6). Los resultados se analizaron mediante una Prueba T donde * P ˂ 0,0003 vs Drd2loxP/ loxP de la misma edad. B) I HQ representativa para el análisis de hipófisis intermedias con un 110 Capítulo II - Resultados anticuerpo anti- α-MSH. Las imágenes fueron tomadas con un objetivo 40X. Los preparados fueron coloreados con hematoxilina, y en marrón se observa la marca positiva (DAB). C) El porcentaje de melanotropos inmunomarcados para α-MSH fue similar en hembras de 10 meses de edad de ambos genotipos. AL: hipófisis anterior; I L: intermedia; NL: neurohipófisis. Los resultados se analizaron mediante una Prueba T sin arrojar diferencias significativas. D) Niveles de expresión de los neuropéptidos hipotalámicos Pomc, Npy y Ppo en ratones hembra de 5 y 10 meses de edad Drd2loxP/ loxP y lacDrd2KO. Los resultados se analizaron por ANOVA de dos factores, * P ˂ 0,04 vs Drd2loxP/ loxP de la misma edad (en todos los casos n entre 5 y 7). Machos En machos lacDrd2KO los pesos corporales no variaron respecto de su pares control (Figura 35A), como tampoco presentaron diferencias en los niveles de ingesta diarios (Figura 35B). Figura 35: Peso corporal e ingesta en machos. A) Curvas de peso corporal a lo largo del desarrollo en ratones macho Drd2loxP/ loxP (n = 8) y lacDrd2KO (n = 9). El AMR no mostró diferencias significativas. B) Niveles de ingesta promedio (gramos/ día/ ratón) en ratones Drd2loxP/ loxP comparados con lacDrd2KO a los 3 (n = 3 y 3, respectivamente) y 5 (n = 5 y 5, respectivamente) meses de edad. El ANOVA de dos factores no arrojó diferencias Concluimos que la hiperprolactinemia se asoció a un aumento de peso, relacionado con mayor ingesta. Esta mayor ingesta podría resultar del incremento de factores orexigénicos a nivel central, como ser PRLR y NPY. Por otra parte, postulamos una posible disminución en la sensibilidad a la leptina a los 10 meses de edad ya que los niveles séricos de la misma se encuentran significativamente incrementados, a pesar de la mayor ingesta en esta edad. A los 5 meses, donde comienza a observarse el incremento en los niveles de ingesta y el peso corporal en los ratones hembra lacDrd2KO, no existen todavía diferencias en los niveles de leptina séricos y la 111 Capítulo II - Resultados sensibilidad a la misma se encuentra conservada, aunque con tendencia a una disminución en la en respuesta a la misma (similar ganancia de peso corporal que con PBS). El paso siguiente fue comprobar si el aumento de peso se encontraba relacionado a alteraciones en el metabolismo y la homeostasis de la glucosa. 112 Capítulo II - Discusión DISCUSIÓN Las hembras hiperprolactinémicas lacDrd2KO presentaron, a los 5 meses, un aumento de peso corporal significativo respecto a las controles, y esta diferencia se incrementó aún más con la edad, llegando a ser del 24% a los 11 meses. El aumento de peso correlacionó con un incremento en la ingesta diaria de alimentos. Este resultado es novedoso y original, ya que la relación entre prolactina y la regulación del peso corporal en ratones está en continuo debate. En ratas se describió que una hiperprolactinemia crónica está asociada con un aumento de la ingesta de alimento y de peso corporal, y que este efecto se puede revertir con la administración de un agonista dopaminérgico como la bromocriptina (51;329). I nyecciones de prolactina en el núcleo PVN de rata aumentan la ingesta de alimento, indicando que la prolactina interactúa con centros hipotalámicos que regulan el apetito (55). En ratones, en cambio, los resultados son controvertidos. Experimentos en machos a los que se les trasplantaron hipófisis ectópicas mostraron un pequeño aumento del peso corporal (57). Animales adultos deficientes en el PRLR mostraron un descenso del peso corporal (56), sin embargo este resultado no pudo ser confirmado más tarde en animales jóvenes (330;331). Por otro lado, en nuestro laboratorio hemos demostrado que la hiperprolactinemia crónica en ratones hembra Drd2-/ - no se encuentra acompañada de un incremento en el peso corporal, ni en los niveles de ingesta (68). Nuestro modelo knockout tejido específico permitió dilucidar los efectos de altos niveles de prolactina sin otros fenotipos solapados, y a partir de los resultados obtenidos podemos determinar un efecto de la misma sobre la regulación del peso y la ingesta, así como los mecanismos asociados al fenotipo. Sumado al aumento de peso con la edad, observamos que los ratones lacDrd2KO presentan una homeostasis alterada en relación al mantenimiento del peso corporal en diferentes condiciones. Descienden menos de peso frente a un ayuno prolongado, y lo recuperan más rápido en la realimentación, comparado con los ratones control. Esto sugiere que los altos niveles de prolactina presentes en este modelo provocan tanto alteraciones en la ingesta de alimentos como en el mantenimiento del balance energético. Al analizar la expresión de neuropéptidos hipotalámicos involucrados en la regulación del apetito en los ratones lacDrd2KO, encontramos que existía un incremento significativo en los niveles de ARNm de dos factores orexigénicos: Npy y Prlr , los cuales estarían provocando el aumento en la ingesta en estos ratones. Prolactina estaría causando este aumento ya sea de manera directa o indirecta. Existen evidencias que apoyan la idea de que prolactina favorece la expresión de NPY. La succión promueve en la madre un aumento tanto de prolactina como de NPY. Experimentos en ratas demostraron que el tratamiento con bromocriptina, el cual inhibe la secreción de prolactina, atenúa el aumento en los niveles de ARNm de Npy en respuesta a la succión; mientras que el tratamiento conjunto de bromocriptina y prolactina ovina restaura el incremento de NPY (293). Del 113 Capítulo II - Discusión mismo modo, estudios in vitro demostraron que la estimulación con prolactina provoca un aumento significativo en los niveles de Npy (332). En el laboratorio demostramos que la ausencia total de RD2s no tuvo efecto en los niveles de ARNm de Npy , pero esto puede explicarse por el hecho de que los ratones Drd2-/ - poseen altos niveles de prolactina, pero también un eje de GHI GF-I disminuido (68;262) (Figura 36). Mientras que el aumento de prolactina y la disminución de la acción dopaminérgica favorecerían el incremento en la expresión de NPY (292;293), la GH reducida posee el efecto contrario (327). En cuanto a la regulación de prolactina y su receptor, se observó que la expresión de PRLR en el cerebro se encuentra incrementada en ratas lactantes en comparación con aquellas no preñadas. Particularmente se evidenció un aumento en los niveles de ARNm de Prlr-l (isoforma larga) en diferentes núcleos hipotalámicos, lo que apoya la idea de que prolactina regula las adaptaciones neuroendócrinas y comportamentales de la preñez (333). Otro grupo de investigadores, mediante el uso de un modelo animal de depresión (a través del estrés crónico moderado), describieron un aumento en los niveles plasmáticos de prolactina, con una disminución en la liberación de dopamina por parte del hipotálamo, lo que provocó un incremento en la expresión génica de Prlr-l en el núcleo Arc y un descenso en los niveles de ARNm de Prlr-s y RD2s (334). Nuestros resultados en el ratón lacDrd2KO son consistentes con la idea de que prolactina estaría actuando a nivel hipotalámico, regulando positivamente la expresión de su propio receptor y la del neuropéptido Npy . Por otro lado, la expresión hipotalámica del precursor de orexinas Ppo estaba conservada, a diferencia de lo que observamos previamente en el modelo de ratón knockout total Drd2-/ - (Figura 36) el cual presenta una disminución en este neuropéptido (60). Esto sugiere que prolactina no tendría un efecto sobre los niveles de Ppo sino que los RD2s hipotalámicos estarían involucrados, directa o indirectamente, en la regulación de la expresión de orexinas hipotalámicas. Sin embargo existen estudios hechos en ratas, por parte de García y colaboradores, los cuales demostraron que los niveles de ARNm hipotalámicos de Ppo se encuentran disminuidos en estados de preñez y lactancia, como así también en un modelo de rata hiperprolactinémica inducida por transplante de hipófisis (335). En cuanto a los péptidos anorexigénicos, no encontramos diferencias en Pomc como tampoco en el porcentaje de melanotropos en la hipófisis intermedia. Esto último concuerda con el hecho de que los ratones lacDrd2KO poseen RD2s funcionales en los melanotropos. La ausencia de RD2s en los ratones Drd2-/ - causó un incremento en la liberación de α-MSH exclusivamente en las hembras (60) (Figura 36) , indicando una probable disminución en el almacenamiento de α-MSH consistente con el rol inhibitorio de los RD2s en su secreción (288). 114 Capítulo II - Discusión Figura 36: Comparación entre ambos modelos hiperprolactinémicos en relación a la expresión de los distintos péptidos orexigénicos (estimuladores de la ingesta, verde) y anorexigénicos (inhibidores de la ingesta, rojo). A la derecha se encuentra representado el ratón lacDrd2KO, con deleción específica del RD2 en lactotropos; y a la izquierda el ratón Drd2-/ - con ausencia total de RD2s funcionales. Otra señal fundamental relacionada con la regulación de la ingesta es la leptina. Encontramos que los niveles de leptina séricos se encuentran significativamente incrementados en los ratones hembra lacDrd2KO de 10 meses de edad, lo que concuerda con el aumento significativo en el peso corporal, mientras que no encontramos variaciones a los 5 meses, edad en la cual las alteraciones en el peso corporal recién comienzan a evidenciarse. Tal como era de esperar el peso de los animales correlacionaba con los niveles de leptina en suero. Está bien establecido en la literatura que los niveles de leptina incrementan de manera proporcional a la cantidad de tejido adiposo, y actúan inhibiendo la ingesta de alimentos (305). Pero en la mayoría de los individuos obesos se observa una resistencia a la leptina, es decir altos niveles pero falta de acción (307). En forma similar, en nuestro modelo experimental si bien no detectamos disminución en la respuesta a leptina en los ratones lacDrd2KO de 5 meses, postulamos una posible resistencia a leptina a los 10 meses, momento en el cual en los ratones lacDrd2KO hay un aumento de ingesta en presencia de altos niveles de leptina, en comparación con los ratones controles. Dicha alteración en la sensibilidad a la leptina, no involucraría diferencias en la expresión hipotalámica de su receptor Obrb . Existen evidencias que proponen que la prolactina es la responsable de inducir la resistencia central a leptina en la preñez. Dicho efecto es fundamental para mantener los niveles 115 Capítulo II - Discusión de ingesta y el almacenamiento de energético elevados, para abastecer las demandas metabólicas del feto y la lactancia (336). Por otra parte, los elevados niveles del neuropéptido Npy presentes en las hembras hiperprolactinémicas también son un indicio de resistencia a leptina, ya que las neuronas AgRP/ NPY del Arc están reguladas por esta adipoquina, la cual actúa reduciendo la expresión y liberación de los péptidos hipotalámicos orexigénicos AgRP y NPY (12;24). Finalmente, a partir de los resultados de este capítulo, concluimos que la hiperprolactinemia se asocia a un aumento de peso, como resultado de una mayor ingesta. Esta mayor ingesta podría resultar del incremento de factores orexigénicos a nivel central, como ser PRLR y NPY. Por otra parte, postulamos una posible disminución en la sensibilidad a la leptina (inducida por prolactina) a los 10 meses de edad ya que los niveles séricos de la misma se encuentran significativamente incrementados, a pesar de la mayor ingesta en esta edad. Destacamos la importancia del modelo animal usado, ya que permite dilucidar los efectos de altos niveles de prolactina, de otra manera enmascarados en mutantes nulos o animales tratados con agentes farmacológicos. 116 Capítulo III - Introducción CAPÍTULO III – REGULACIÓN DEL METABOLISMO DE LA GLUCOSA EN RATONES HEMBRA lacDrd2KO. INTRODUCCIÓN HOMEOSTASIS DE LA GLUCOSA La glucosa es la fuente fundamental de energía para todas las células eucariotas. En los mamíferos, si bien todas las células utilizan este azúcar para cubrir sus necesidades energéticas, el principal consumidor en condiciones basales es el cerebro (80% del consumo total). En ausencia de glucosa las neuronas mueren, por lo que la homeostasis de la glucosa y el mantenimiento de una concentración constante en el fluido extracelular resultan de vital importancia. Mecanismos de regulación El páncreas es el órgano que sintetiza y libera las principales hormonas involucradas en la homeostasis de la glucosa: insulina y glucagón. La porción endócrina de este órgano está constituida por los islotes de Langerhans que son estructuras formadas por, al menos, cuatro tipos celulares: Células alfa ( α): secretoras de glucagón Células beta ( ): secretoras de insulina Células delta ( δ): secretoras de STT Células PP: secretoras del polipéptido pancreático 11 Las células beta constituyen alrededor del 80% de las células del islote, y ocupan una posición central quedando rodeadas por células alfa. La sangre fluye a los islotes a través de las arteriolas que se convierten en capilares fenestrados en la porción central del islote (337). Glucagón Glucagón es una hormona polipeptídica sintetizada en las células alfa de los islotes pancreáticos. Es principalmente captada por el hígado y tiene una vida media corta en plasma. Su receptor se expresa mayoritariamente en este tejido, y pertenece a la superfamilia de receptores acoplados a proteínas G, con siete pasos transmembrana (338). Glucagón promueve la movilización de compuestos energéticos, especialmente glucosa, en el hígado. Esto se logra a través de la activación de la proteína quinasa A (PKA), la que favorece la glucogenolisis y gluconeogénesis, e inactiva la glucólisis (339). 117 Capítulo III - Introducción Su síntesis es inhibida por altos niveles de glucosa y estimulada por bajos niveles de la misma. Su inhibición es reforzada por insulina, la cual actúa de manera parácrina, dentro del islote de Langerhans. Las catecolaminas, la GH y los glucocorticoides estimulan su secreción. También los FFAs sirven en el control de esta hormona: altos niveles inhiben su secreción, mientras que bajos niveles incentivan la liberación de glucagón. Insulina La insulina es una proteína sintetizada por las células beta del páncreas. La acción global de la insulina es facilitar el almacenamiento de sustratos e inhibir su liberación (opuestamente a la acción de glucagón), disminuyendo las concentraciones plasmáticas principalmente de glucosa, pero también de FFAs, cetoácidos y aminoácidos. Los principales blancos de acción de la insulina son el hígado, el músculo y el tejido adiposo (339). La insulina promueve el almacenamiento de la glucosa como glucógeno en las fibras musculares, mientras que en los adipocitos la glucosa es convertida a ácidos grasos que son posteriormente almacenados. En ambos tejidos, la insulina promueve el rápido ingreso de la glucosa al reclutar y exponer en membrana a los transportadores GLUT-4. De esta manera las células poseen una mayor concentración de transportadores en la membrana favoreciendo el rápido ingreso de la glucosa a los tejidos. Es por este efecto que estos transportadores se denominan insulino dependientes. El hígado, a excepción de los otros tejidos de almacenamiento, presenta el transportador GLUT-2, insulino independiente y con alta capacidad para transportar glucosa desde la sangre al interior de la célula (340). En el sistema nervioso central la captación de glucosa ocurre a través de transportadores independientes de insulina, al igual que en el páncreas mismo (339). Estos transportadores son GLUT-3 y GLUT-2. La secreción de insulina está regulada principalmente por glucosa, aunque también intervienen hormonas y el sistema nervioso central a través de inervación simpática y parasimpática. En este contexto, estudios recientes demuestran que los islotes pancreáticos expresan RD2s, y sugieren que la dopamina podría ejercer un control en la regulación de la liberación de insulina (139;141). Los niveles elevados de glucosa en sangre, ya sea por una disfunción en la secreción de insulina o en la sensibilidad a la misma, producen la enfermedad conocida como diabetes mellitus. Esta alteración se caracteriza por presentar diversos trastornos metabólicos. La diabetes tipo I , también conocida como diabetes juvenil o diabetes mellitus insulino dependiente, es causada por una destrucción selectiva de las células beta del páncreas por el propio sistema inmune, causando una deficiencia total o parcial de insulina. Generalmente se manifiesta a edades tempranas y la administración de insulina en estos pacientes es esencial. 118 Capítulo III - Introducción En la diabetes tipo I I , ocurre una resistencia a las acciones de insulina. Algunas causas de esta resistencia pueden ser una deficiencia en la expresión y/ o funcionalidad del receptor de insulina o alteraciones en algún punto de la cascada de señalización intracelular de las células blanco de esta hormona. Como mecanismo compensatorio, el organismo secreta inicialmente mayores niveles de insulina, produciendo a largo plazo un deterioro de las células beta pancreáticas y por lo tanto de la secreción hormonal (341). Hormonas hipofisarias La relación entre la hipófisis y la homeostasis de la glucosa fue descripta hace más de 70 años en los trabajos del Dr. Bernardo Houssay y colaboradores cuyos valiosos aportes a la ciencia merecieron su distinción con el Premio Nobel en el año 1947 (342;343). La GH ejerce efectos metabólicos opuestos a los de insulina, dado que estimula la gluconeogénesis a partir de lípidos y bloquea la captación de glucosa por otros tejidos distintos al nervioso. También se describió que la GH produce resistencia a la insulina en tejidos periféricos. A nivel pancreático, GH y prolactina fueron propuestos como factores moduladores de la función pancreática, al observarse que son capaces de estimular la secreción y biosíntesis de insulina in vitro (344-347). Además se observó que GH, prolactina y el lactógeno placentario son capaces de estimular la proliferación de las células beta (344-348). Estos factores estarían involucrados en la regulación de la masa de células beta del páncreas. La población de células beta está en continuo balance entre la formación de células beta nuevas (por neogénesis y proliferación) y muerte por apoptosis, necrosis o senescencia. La función mejor establecida de las hormonas lactogénicas en el páncreas es durante el embarazo. En este periodo se inducen alteraciones en el metabolismo de la madre como respuesta a la demanda energética del feto. Se produce una secreción elevada de insulina, en algunos tejidos se genera resistencia a insulina y aumenta el metabolismo de lípidos. El páncreas tiene un rol muy importante en estas adaptaciones. Durante el embarazo, las células beta sufren cambios estructurales y funcionales que incluyen: 1) aumento en la secreción de insulina estimulada por glucosa; 2) aumento en la síntesis de insulina; 3) aumento en la proliferación de las células beta; y 4) aumento del metabolismo de la glucosa (344;349). En ratas la administración de prolactina produce un descenso en el umbral de respuesta de la insulina a glucosa; y por ende, aumenta la secreción de insulina y el acoplamiento entre células beta. Asimismo, prolactina estimula la liberación de insulina en islotes pancreáticos aislados (57). Estudios clínicos revelan que la prolactina produce efectos diabetogénicos, y que en pacientes la hiperprolactinemia está asociada a hiperinsulinemia y resistencia a insulina (350;351). 119 Capítulo III - Introducción Evaluación del modelo homeostático (HOMA) La evaluación del modelo homeostático o, en inglés, homeostatic model assessment (HOMA), fue descripto por primera vez por Matthews en el año 1985 (352). Es un método para determinar la funcionalidad de células beta ( cell) y la resistencia a insulina (I R) a partir de las concentraciones de glucosa e insulina en ayuno. La relación entre los niveles basales de glucosa e insulina reflejan el balance entre la salida de glucosa hepática y la secreción de insulina, lo que es mantenido por una retroalimentación entre el hígado y las células beta pancreáticas (353). Se ha comparado al método de HOMA con otros ampliamente validados, como el clamp euglucémico, el clamp hiperglucémico o el modelo mínimo, y se vio que resulta un buen equivalente (354). El HOMA es entonces una alternativa no invasiva, rápida, de bajo costo, y altamente confiable (355). El modelo homeostático para la evaluación de la resistencia a la insulina (HOMA-I R), es un indicador de insulino resistencia en los tejidos periféricos. Si bien fue desarrollado para humanos es también utilizado con frecuencia en trabajos con roedores (356). Se calcula como el producto entre los valores de glucemia e insulina basales, ambos en ayunas. Un HOMA-I R aumentado indica que frente a la misma cantidad de insulina en sangre, la glucemia se encuentra más alta, lo cual refleja una capacidad alterada de los tejidos periféricos para la captación de glucosa mediada por insulina. Por otra parte, el índice para la evaluación de la funcionalidad de células beta (HOMA- cell) se calcula como la relación entre los valores de insulina y glucemia basales, en ayunas. Un HOMA- cell disminuido indica una funcionalidad alterada del páncreas. Obesidad: factor de riesgo para el desarrollo de diabetes El desarrollo de la diabetes tipo I , según estudios realizados con gemelos, depende tanto de factores genéticos como ambientales. La importancia de los factores ambientales queda en evidencia con el incremento en los niveles de diabetes tipo I observado en los inmigrantes de regiones con menor hacia aquellas de mayor incidencia. La población emigrante se adapta a la incidencia media del país de acogida en un corto espacio de tiempo (357). La asociación entre el desarrollo de diabetes tipo I y el peso corporal fue estudiada, en principio, por Baum y colaboradores en 1975, quienes sugirieron que existe una relación entre la alimentación excesiva y las alteraciones hormonales (358). La “hipótesis del acelerador” propuesta por Wilkin es considerada la teoría más aceptada que relaciona la masa corporal y la diabetes tipo I . Este autor sugiere que el incremento en el peso corporal acelera la insulino resistencia, aumentando el riesgo de desarrollar diabetes tipo I en los individuos predispuestos genéticamente a la misma (359). 120 Capítulo III - Introducción La asociación entre obesidad y desarrollo de diabetes tipo I I está más extensamente estudiada. La obesidad es considerada el factor más importante en el desarrollo de enfermedades metabólicas. El tejido adiposo afecta al metabolismo mediante la secreción de numerosos factores, los cuales en la obesidad se encuentran incrementados (360). Los ácidos grasos no esterificados (NEFA), liberados por el tejido adiposo, afectan considerablemente la insulino resistencia. Un incremento en los niveles de NEFA se observan en la obesidad y diabetes tipo I I , estando relacionados en ambos casos con resistencia a la insulina. Se vio en humanos, que un incremento agudo en los niveles de NEFA provoca el desarrollo de resistencia a insulina, y la normalización de los niveles revierte el fenotipo (361). La sensibilidad a insulina también se encuentra relacionada a la distribución del tejido adiposo en individuos obesos. Aquellos individuos con mayor acumulación de tejido adiposo en el área del abdomen presentan una resistencia a insulina incrementada (357). CARACTERIZACIÓN DEL METABOLISMO DE LA GLUCOSA EN EL RATÓN KNOCKOUT TOTAL Drd2 -/En estudios previos del laboratorio observamos que los animales Drd2-/ - luego de 8 hs de ayuno presentan valores de glucosa en sangre mayores respecto a los animales WT, sin hallarse diferencias significativas en los niveles de insulina sérica. A los seis meses de edad, los animales Drd2-/ - presentaron una intolerancia moderada a la glucosa en comparación con ratones WT. Estos valores de glucemia elevados correlacionaron negativamente con los niveles de insulina séricos en el mismo test. Se demostró que la intolerancia a la glucosa se asocia a una insuficiente secreción de insulina frente al estímulo gluco-secretor en los animales Drd2-/ - adultos. En estudios previos de nuestro laboratorio se realizó el test de tolerancia a la insulina (I TT) para determinar si los tejidos presentaban una respuesta diferencial a la insulina según el genotipo. En este test se evalúa cómo es la variación de la glucosa a lo largo del tiempo en respuesta a una dosis determinada de insulina intraperitoneal. No se evidenciaron diferencias significativas entre los genotipos, a ninguna de las edades estudiadas, indicando que la sensibilidad a esta hormona no estaría afectada en los animales Drd2-/ - (141). 121 Capítulo III - Objetivos OBJETIVOS Estudiar el impacto de la hiperprolactinemia, por la ausencia de RD2s en lactotropos, sobre la regulación del metabolismo de la glucosa. Los objetivos específicos fueron: - Analizar los niveles de glucosa e insulina basales (en condiciones de ayuno y no ayuno) en hembras y machos de ambos genotipos. - Realizar ensayos de tolerancia a la glucosa, tolerancia a la insulina, y secreción de insulina estimulada por glucosa, en ratones hembra y macho Drd2loxP/ loxP y lacDrd2KO. - Comparar el peso de los páncreas en los ratones hembra de ambos genotipos. Estudiar dicho tejido mediante I HQ, determinando número y tamaño de los islotes, como también el porcentaje de células alfa y beta presentes en cada uno de ellos. Establecer, a su vez, el contenido de insulina intrapancreático en ratones hembra Drd2loxP/ loxP y lacDrd2KO. 122 Capítulo III - Resultados RESULTADOS HOMEOSTASIS DE LA GLUCOSA ALTERADA Considerando la obesidad de los ratones transgénicos decidimos investigar el impacto de la disrupción de los RD2s en lactotropos sobre la homeostasis de la glucosa in vivo. Los niveles de glucosa sérica en ayuno fueron similares en hembras de 6 meses, mientras que a los 10 meses de edad las hembras lacDrd2KO presentaron valores menores (Figura 37A). No hubo diferencias, sin embargo, en los niveles de glucosa en condición de no ayuno entre genotipos. Encontramos, a su vez, diferencias significativas en los niveles basales de insulina a los 6 y 10 meses de edad ( P < 0,04), los cuales resultaron mayores en ratones lacDrd2KO (Figura 37B). Esta insulinemia basal aumentada podría explicar los menores niveles de glucosa en ayuno obtenidos a los 10 meses de edad. A partir de estos datos pudimos calcular los índices HOMA-I R y HOMA- cell que dan cuenta de la resistencia a insulina y la funcionalidad de las células beta, respectivamente. No obtuvimos diferencias estadísticas entre genotipos en ninguno de los índices, y se evidenció en ambos genotipos una diferencia etaria, que indicaría una pérdida de funcionalidad de los islotes con la edad, y un aumento de sensibilidad a la insulina (Figura 37C). 123 Capítulo III - Resultados Figura 37: Homeostasis de la glucosa. A) Niveles de glucosa plasmática en ratones hembra Drd2 loxP/ loxP y lacDrd2KO de 6 y 10 meses de edad, ayunados por 8 hs o sin ayunar. Condición de ayuno n = 6 y 6, a los 6 meses; y 5 y 5 a los 10 meses, respectivamente. Condición de no ayuno n = 5 y 7, a los 6 meses; y 5 y 5 a los 10 meses, respectivamente. Se analizaron los datos por ANOVA de dos factores para cada condición estudiada. En ayuno * P < 0,05 vs ratones Drd2loxP/ loxP a los 10 meses = P interacción (genotipo x edad) < 0,05. En condición de no ayuno * P < 0,002 vs ratones de ambos genotipos a los 6 meses. B) Niveles séricos de insulina (ng/ ml) por ELI SA, en ratones hembra Drd2loxP/ loxP (n = 4 a los 6 meses; n = 6 a los 10 meses) y lacDrd2KO (n = 4 a los 6 meses; n = 7 a los 10 meses) luego de un ayuno de 8 hs. Los datos fueron analizados por ANOVA de dos factores, * P < 0,04 vs ratones Drd2loxP/ loxP. C) Í ndices HOMA-I R y HOMA- cell en hembras Drd2loxP/ loxP y lacDrd2KO, en ambas edades estudiadas (6 meses de edad n = 6 y 6; 124 Capítulo III - Resultados 10 meses de edad n = 4 y 6, respectivamente). Se realizaron análisis de ANOVA de dos factores para cada índice calculado. En ambos casos * P < 0,01 vs ratones de ambos genotipos a los 6 meses. En todas las figuras de este capítulo se grafica la media y las barras de error corresponden al E.S.M. Por otro lado, en ambas edades estudiadas, se observó una marcada intolerancia a la glucosa en hembras lacDrd2KO, evidenciada por niveles elevados de glucosa en sangre, en comparación con sus pares Drd2loxP/ loxP, a los 30 y 60 minutos luego de la inyección ip de glucosa (Figura 38A). Para determinar el efecto de la deficiencia de RD2s en la acción de insulina in vivo, analizamos los cambios en las concentraciones plasmáticas de glucosa luego de una única inyección ip con insulina. Como se observa en la Figura 38B, las curvas de glucosa son similares entre genotipos a ambas edades estudiadas, lo que indica que este modelo experimental no presenta un fenotipo de resistencia a la insulina, lo que coincide con lo obtenido en el índice HOMA-I R (Figura 37C). Al estudiar el GSI S a los 6 meses de edad, observamos secreción de insulina estimulada por glucosa en todos los animales Drd2loxP/ loxP estudiados, mientras que, sólo el 50% de las hembras lacDrd2KO tuvo una liberación de insulina frente a glucosa (Figura 38C). A los 10 meses en cambio, los ratones de ambos genotipos respondieron a la inyección de glucosa incrementando los niveles de insulina séricos, sin embargo al analizar la diferencia entre los niveles de insulina antes y post-inyección de glucosa ( Δ insulina), encontramos que los ratones hembra lacDrd2KO de ambas edades tenían valores menores respecto de sus pares control, demostrando una deficiencia en la liberación de insulina estimulada por glucosa ( P < 0,02) (Figura 38D). 125 Capítulo III - Resultados 126 Capítulo III - Resultados Figura 38: Homeostasis de la glucosa. A) GTT (2 g/ kg) en ratones hembra Drd2loxP/ loxP y lacDrd2KO de 6 y 10 meses de edad ayunados por 8 hs (n = 6 y 6, a los 6 meses; y 5 y 5 a los 10 meses, respectivamente). Se realizaron análisis por AMR para cada edad estudiada. A los 6 meses de edad * P < 0,05 vs ratones Drd2loxP/ loxP en el mismo tiempo = P< 0,05 a los 30 min y 0,02 a los 60 min; P interacción (genotipo x tiempo) < 0,00002. A los 10 meses de edad * P < 0,05 vs ratones Drd2loxP/ loxP en el mismo tiempo = P< 0,0001 a los 30 min y 0,0005 a los 60 min; P hembra Drd2 loxP/ loxP interacción (genotipo x tiempo) < 0,0001. B) I TT en ratones y lacDrd2KO de 6 y 10 meses de edad, los cuales fueron inyectados con 1 U/ kg de insulina humana. No se observaron diferencias significativas por AMR para cada edad estudiada (n = 5 y 7, a los 6 meses; y 5 y 5 a los 10 meses, respectivamente). C) GSI S: Secreción de insulina estimulada por glucosa (3 g/ kg) en hembras Drd2loxP/ loxP y lacDrd2KO, a los 6 y 10 meses de edad. No observamos diferencias en el análisis por AMR a los 6 meses de edad (n = 6 y 6). A los 10 meses de edad (n = 4 y 6, respectivamente) se observan diferencias significativas de ambos genotipos vs el Control (sin glucosa). D) Delta insulina (diferencia entre niveles de insulina post- y pre-inyección de glucosa) en hembras Drd2loxP/ loxP y lacDrd2KO, a los 6 y 10 meses de edad. Se analizaron los datos por ANOVA de dos factores, obteniendo diferencias significativas, * P < 0,02 vs ratones Drd2loxP/ loxP. Determinamos a su vez, que los páncreas de los ratones hembra lacDrd2KO de 5 meses de edad tienen una tendencia a ser más pesados que los de sus pares Drd2loxP/ loxP ( P = 0,063) (Tabla 17). La concentración de insulina intrapancreática medida por RI A (Figura 39A) y el área insulino positiva medida por I HQ (Figura 39B) resultó mayor en las hembras lacDrd2KO. El número de islotes se encontró conservado entre grupos (Figura 39C), presentando los ratones hembra lacDrd2KO un aumento en el porcentaje de islotes de gran tamaño, con áreas mayores a 5000 µm 2 (Figura 39D). El tamaño promedio de cada islote no llega a ser significativamente mayor en las hembra lacDrd2KO ( P = 0,081) (Tabla 17), pero sí el área pancreática ocupada por islotes (Tabla 17). 127 Capítulo III - Resultados 128 Capítulo III - Resultados Figura 39: Páncreas. A) Contenido de insulina intrapancreática (µg de insulina/ ng de proteínas) por RI A, en ratones hembra a los 5 meses (n = 5) y 10 meses de edad (n = 12 y 14, Drd2loxP/ loxP y lacDrd2KO). Los resultados se analizaron mediante un ANOVA de dos factores, donde * P < 0,0002 vs Drd2loxP/ loxP. B) Área insulino positiva por I HQ (área ocupada por células beta), en ratones hembra de 5 meses de edad (n = 7 y 6, Drd2loxP/ loxP y lacDrd2KO). Los resultados se analizaron por Prueba T donde * P < 0,01 vs ratones Drd2loxP/ loxP. C) Densidad de islotes (número de islotes por cm 2) por I HQ, en ratones hembra de 5 meses de edad (n = 7 y 6, Drd2loxP/ loxP y lacDrd2KO). Los resultados se analizaron por Prueba T sin obtener diferencias. D) Porcentaje de islotes chicos (área < 5000 µm 2) y medianos-grandes (área > 5000 µm 2) por I HQ, en ratones hembra de 5 meses de edad (n = 7 y 6, Drd2loxP/ loxP y lacDrd2KO). Los datos se analizaron estadísticamente mediante un test Х2: * P ˂ 0,05 vs islotes Drd2loxP/ loxP del mismo tamaño. E) I mágenes representativas de las I HQ realizadas en cortes de páncreas de ratones hembra de ambos genotipos. En todos los casos, las imágenes fueron obtenidas con un objetivo 40X. Los preparados fueron coloreados con hematoxilina, y en marrón se observan los citoplasmas positivos para cada anticuerpo (DAB). Para todas las figuras del panel los ratones fueron alimentados ad libitum . Observamos a su vez, que los ratones hembra lacDrd2KO presentan un incremento en el porcentaje y tamaño promedio de céluas beta (Tabla 17), lo que estaría sugiriendo que el incremento en el área ocupada por islotes (Tabla 17) se debe a una hiperplasia como también hipertrofia de este tipo celular. El incremento en el porcentaje de células beta es a expensas de un detrimento en las células alfa, tal como puede observarse en la Figura 39E. Consitente con esto, la relación entre las áreas insulino y glucagón positivas es significativamente mayor en las hembras lacDrd2KO (Tabla 17). Tabla 17: Páncreas. Se comparan el peso del páncreas, el área promedio de cada islote (en µm 2), el área ocupada por islotes (en µm 2), el porcentaje de células beta, el tamaño promedio de cada célula beta (en µm 2), y la relación entre el área insulino y glucagón positiva, en ratones hembra Drd2loxP/ loxP y lacDrd2KO. Para los datos correspondientes a los pesos n = 4. Para los análisis por I HQ, n = 7 y 6, Drd2loxP/ loxP y lacDrd2KO. En todos los casos se compararon ratones hembra Drd2loxP/ loxP y lacDrd2KO de 5 meses de edad alimentados ad libitum . Los resultados se analizaron por Prueba T donde * P < 0,05 vs ratones Drd2loxP/ loxP. 129 Capítulo III - Resultados Asimismo, encontramos que frente a la realimentación (ingreso de glucosa) los ratones hembra lacDrd2KO tienen una tendencia a liberar menor cantidad de insulina que sus controles, aunque no hay diferencias estadísticas (Figura 40A), en forma similar a lo que ocurrió frente a una sobrecarga de glucosa (ver Figura 38C). Estos resultados, junto con el aumento del contenido de insulina pancreática observado en ratones ad libitum , sugerirían una deficiencia en el proceso de liberación de insulina (Figura 39A y B). Corroboramos a su vez, en el esquema de ayuno y realimentación, el aumento del contenido de insulina pancreática en los ratones lacDrd2KO (Figura 40B). Figura 40: Homeostasis de la glucosa. A) Niveles séricos de insulina (ng/ ml) por ELI SA, en ratones hembra Drd2loxP/ loxP y lacDrd2KO luego de un ayuno de 12 hs, con y sin realimentación por 1 hr. A la izquierda se encuentran los resultados a los 5 meses de edad (n = 4), mientras que a la derecha los correspondientes a 10 meses de edad (en ayuno n = 4; realimentadas n = 3). Los datos fueron analizados por ANOVA de dos factores sin obtener diferencias. B) Contenido de insulina intrapancreática (µg de insulina/ ng de proteínas) por RI A, en ratones hembra a los 5 meses (ayuno n = 6; realimentadas n = 4 y 6, Drd2loxP/ loxP y lacDrd2KO) y 10 meses de edad (ayuno n = 5; realimentadas n = 3 y 5, Drd2loxP/ loxP y lacDrd2KO). Los resultados se analizaron mediante un ANOVA de dos factores, donde * P < 0,0002 vs Drd2loxP/ loxP. 130 Capítulo III - Resultados Machos En machos, en cambio, no observamos diferencias entre genotipos en ninguno de los parámetros estudiados (Figura 41). 131 Capítulo III - Resultados Figura 41: Homeostasis de la glucosa en machos. A) Niveles de glucosa plasmática en ratones macho Drd2loxP/ loxP y lacDrd2KO de 6 meses de edad ayunados por 8 hs o sin ayuno. Condición de ayuno n = 6 y 6, respectivamente. Condición de no ayuno n = 5 y 5, respectivamente. Los resultados se analizaron por una Prueba T. B) GTT (2 g/ kg) en ratones macho Drd2loxP/ loxP y lacDrd2KO de 6 meses de edad ayunados por 8 hs (n = 6 y 6, respectivamente). Se realizaron análisis por AMR sin encontrar diferencias. C) I TT en ratones macho Drd2loxP/ loxP y lacDrd2KO de 6 meses de edad, los cuales fueron inyectados con 1 U/ kg de insulina humana. No se observaron diferencias significativas por AMR (n = 5 y 5, respectivamente). D) Secreción de insulina estimulada por glucosa (3 g/ kg) en machos Drd2loxP/ loxP y lacDrd2KO de 6 meses de edad (n = 3 y 4). El análisis por AMR evidenció un P tiempo = 0,05 sugiriendo diferencias de ambos genotipos vs su propio Control (sin glucosa). E) Contenido de insulina intrapancreática (µg de insulina/ ng de proteínas) por RI A, en ratones macho a los 10 meses ( ad libitum n = 5; ayuno n = 4; realimentados n = 4 y 3, Drd2loxP/ loxP y lacDrd2KO). Los resultados se analizaron mediante un ANOVA de dos factores sin obtener diferencias. F) Í ndices HOMA-I R y HOMA- cell en machos Drd2loxP/ loxP y lacDrd2KO de 6 meses de edad (n = 3 y 5, respectivamente). Los resultados se analizaron por una Prueba T sin obtener diferencias. En su conjunto, todos estos datos estarían sugiriendo una deficiencia en la secreción de insulina a nivel del páncreas en los ratones hembra que poseen disrupción específica de los RD2s en lactotropos en ambas edades. Esto explicaría la intolerancia a la glucosa observada en este modelo animal. El paso siguiente fue analizar si el aumento en el peso corporal, que no correlacionaba con un aumento de tamaño del ratón, estaba relacionado a un aumento de adiposidad; el papel de la prolactina en los procesos de lipogénesis y lipólisis, y su relación con las alteraciones observadas en la homeostasis de la glucosa. 132 Capítulo III - Discusión DISCUSIÓN Sabiendo que la obesidad resulta un factor de riesgo para el desarrollo de diabetes tipo I I , es que decidimos estudiar el efecto de la deleción específica de RD2s en lactotropos sobre la homeostasis de la glucosa, evaluando diferentes parámetros involucrados en este equilibrio como los niveles de glucosa séricos, la sensibilidad periférica a insulina y la funcionalidad de los islotes para secretar insulina in vivo. Los ratones hembra lacDrd2KO presentaron una marcada intolerancia la glucosa y un déficit en la secreción de insulina como respuesta a una sobrecarga de glucosa. Las drogas neurolépticas que bloquean a los receptores de dopamina pueden causar hiperinsulinemia (362) y están asociados con diabetes en los humanos (133;363;364). Estos resultados dependen en parte de la duración del tratamiento, y en muchos casos de la selectividad de las drogas. Al evaluar los parámetros HOMA-I R y HOMA- cell, que dan cuenta de la resistencia a insulina y la funcionalidad de las células beta respectivamente, no obtuvimos diferencias entre genotipos. Sin embargo, se evidenció en ambos grupos una diferencia etaria, la cual indicaría una pérdida de funcionalidad de los islotes y un aumento de sensibilidad a la insulina con la edad. Se sabe que comúnmente existe una pérdida de función de las células beta pancreáticas con la edad, lo que resulta en una disminución en la liberación de insulina y una consecuente tolerancia a la glucosa alterada, lo que en conjunto provoca que haya una mayor incidencia de desarrollo de diabetes tipo I I a edades avanzadas (365). El resultado interesante en nuestro modelo fue la mejora en la sensibilidad a insulina, cuando normalmente se espera lo contrario, ya que la edad avanzada se asocia con un mayor peso corporal y acumulación de adiposidad, factores que favorecen el desarrollo de resistencia a insulina (366). Consistente con el índice HOMA-I R conservado en las hembras lacDrd2KO, la sensibilidad a insulina, evaluada mediante un ensayo in vivo, resultó en curvas normales de glucosa luego de una única inyección insulina. Es decir, este modelo experimental no presenta un fenotipo de resistencia a la insulina, que coincide con lo obtenido en el valor de HOMA-I R. Sin embargo, en ambas edades estudiadas, se observó una marcada intolerancia a la glucosa en hembras lacDrd2KO. Dado que no existe resistencia a insulina, lo que explica esta falla en la regulación de los niveles de glucemia es que estos ratones lacDrd2KO presentan alteraciones en la secreción de insulina estimulada por glucosa. Esto último fue determinado por el ensayo in vivo en el cual se evaluaron los niveles de insulina liberados luego de una inyección de glucosa, como también por la concentración de insulina intra-pancreática incrementada. Este modelo de ratón transgénico posee un aumento significativo en el peso corporal como consecuencia de altos niveles de prolactina de manera crónica, los que inducen un incremento en la ingesta. A partir de los resultados de este capítulo determinamos que presenta además una homeostasis de la glucosa alterada. 133 Capítulo III - Discusión Durante la preñez, existen cambios estructurales y funcionales en los islotes pancreáticos, entre ellos el aumento en la masa de islotes, a causa principalmente de una hipertrofia e hiperplasia de células beta (344-346). La prolactina tiene un rol fundamental en estas adaptaciones, ya que se demostró, en modelos animales y en humanos, que incrementa la proliferación de las células beta, la expresión del gen de insulina y la secreción de insulina inducida por glucosa (344-348). En ratas con hiperprolactinemia crónica se evidenció un aumento en la concentración de glucosa e insulina luego de una sobrecarga de glucosa (63). Sumado a esto, hombres y mujeres con hiperprolactinemia tienen hiperinsulinemia y una respuesta exagerada en la secreción de insulina inducida por glucosa (367-369). Por otro lado, el ratón deficiente del PRLR presenta una reducción en el tamaño y número de islotes pancreáticos, y una respuesta en la liberación de insulina prácticamente nula (370) . Teniendo en cuenta los efectos de prolactina recién mencionados, que varían según la especie en estudio, los resultados en nuestro modelo animal sugieren que la hiperprolactinemia crónica favorece el incremento en el área ocupada por islotes, en la cantidad de islotes de mayor tamaño, como también el incremento en el contenido de insulina intrapancreática. Tal como ocurre en la preñez (344-346), la prolactina estaría induciendo en los ratones lacDrd2KO, una hiperplasia e hipertrofia de células beta. Por otro lado, se ha demostrado que la prolactina induce intolerancia a la glucosa en numerosas especies, entre ellas ratas con hipófisis ectópicas (63). En forma similar en nuestro modelo se observó una marcada intolerancia a la glucosa que podría estar asociada a una falla en la liberación de insulina estimulada por glucosa, un efecto que no necesariamente sería provocado por los altos niveles de prolactina. Otros factores relacionados al aumento en el peso corporal podrían participar en este fenómeno. Por ejemplo, está descripto que el aumento en los niveles de FFAs séricos incrementa el estrés oxidativo en las células beta, lo que provoca fallas en su estructura y función, siendo uno de los resultados la disminución en la secreción de insulina (371). Es por ello que decidimos evaluar a continuación si el aumento en el peso corporal de los ratones lacDrd2KO estaba relacionado a un aumento de adiposidad, y el papel de la prolactina en los procesos de lipogénesis y lipólisis, y su relación con las alteraciones observadas en la homeostasis de la glucosa. 134 Capítulo IV - Introducción CAPÍTULO IV – REGULACIÓN DE LA LIPOGÉNESIS EN RATONES HEMBRA lacDrd2KO. INTRODUCCIÓN TEJIDO ADIPOSO BLANCO El tejido adiposo es un órgano endócrino cuyas hormonas, las adipoquinas, actúan en el cerebro, hígado, páncreas y músculo para regular el balance energético, la resistencia a insulina y la respuesta inflamatoria. El balance entre los procesos de adipogénesis, lipólisis y lipogénesis (síntesis de triglicéridos) es el responsable por la cantidad de tejido adiposo presente en el organismo (372). Adipogénesis El tejido adiposo está compuesto por: adipocitos maduros que contienen lípidos, preadipocitos y células endoteliales e inmunes. La adipogénesis es el proceso de diferenciación celular por el cual los pre-adipocitos se convierten en adipocitos. Numerosos estudios in vitro con líneas celulares pre-adipocitarias permitieron dilucidar los pasos de este proceso (373). Se puede inducir la adipogénesis mediante el tratamiento con agentes tales como glucocorticoides, AMPc, I GF-I y una alta concentración de glucosa. Estos inductores promueven a los pre-adipocitos a llevar a cabo una expansión clonal mitótica para luego iniciarse los procesos de activación transcripcional de genes marcadores del adipocito (374). Numerosos factores de transcripción han sido identificados, los cuales actúan de manera cooperativa y secuencial para gatillar el programa de diferenciación. Estos incluyen a los miembros de las familias C/ EBP (proteína de unión a CCAAT/ enhancer) y PPAR (receptor activado por proliferadores peroxisomales) (374). El proceso de adipogénesis habitualmente se describe como una cascada de eventos genéticos. En primer lugar se induce la expresión de C/ EBP y C/ EBPδ los cuales resultan responsables de la activación, en segundo lugar, de los factores de transcripción PPAR y C/ EBPα. Durante la primera fase, las células fibroblásticas se redondean y se incrementan los niveles de ARNm de marcadores adipogénicos como Lpl (enzima lipoprotein lipase) (375;376). La aparición de PPAR y C/EBPα activa la expresión de la mayoría de los genes que caracterizan al fenotipo adipocitario, como son Fas (enzima fatty acid synthase), la glicerofosfato deshidrogenasa, la acetilCoA carboxilasa (ACO), el receptor de glucosa GLUT-4, el receptor de insulina y la proteína aP2/ FABP (la proteína que liga ácidos grasos, específica del adipocito), entre otros (377). A través 135 Capítulo IV - Introducción de este proceso, en el citoplasma de la célula van apareciendo gotas lipídicas y a lo largo del tiempo, éstas irán incrementando y fusionándose hasta formar una o dos grandes gotas lipídicas que ocuparán gran parte del citoplasma. C/EBP parece ser capaz de inducir la expresión de C/EBPα, incluso en ausencia de PPAR (378), aunque este hecho es controvertido (379) . PPAR y C/EBPα a su vez inducirían la expresión de sí mismos, así como uno la del otro. La expresión forzada de PPAR (en presencia de sus ligandos) y C/EBPα estimula la adipogénesis aún en ausencia de agentes inductores exógenos. Aunque PPAR es suficiente para inducir la expresión de la mayoría de los genes adipocitarios, C/EBPα es necesario para conferir al adipocito sensibilidad a la insulina (380). La expresión de ARNm del factor de transcripción de respuesta a carbohidratos ( Carbohydrate Responsive Element Binding Protein) Chrebp se encuentra incrementada durante la diferenciación de células 3T3-L1 (pre-adipocitos de ratón) como también de pre-adipocitos subcutáneos y omentales de humanos, lo que sugiere su participación durante la adipogénesis (381). El factor de transcripción perteneciente a la familia de proteínas de unión a elementos de regulación por esteroles ( sterol regulatory element binding proteins, SREBP), en particular la isoforma SREBP-1c (codificada por el gen Srebf1 ), está involucrado en la regulación de genes asociados al metabolismo lipídico. Sin embargo existen resultados contradictorios acerca del rol de SREBP en la adipogénesis (382). La cantidad de tejido adiposo blanco no disminuyó significativamente en ratones deficientes en SREBP-1 (383). Asimismo, la cruza entre ratones knockout para SREBP-1 y ratones ob/ ob , determinó que la expresión de SREBP-1 no era necesaria para el desarrollo de la obesidad (384). Por otra parte, se vio que la expresión ectópica del dominante negativo para SREBP-1c disminuye la diferenciación adipocitaria, y que la sobreexpresión de SREBP-1c promueve la actividad adipogénica de PPAR (385). Lipólisis y lipogénesis Las principales actividades metabólicas del tejido adiposo blanco son la lipogénesis (síntesis de ácidos grasos y su almacenamiento) y la lipólisis (movilización o hidrólisis de los triglicéridos). Ambos procesos están regulados coordinadamente por mecanismos neuroendócrinos, que mantienen la cantidad de grasa corporal constante en condiciones normales (372). Los triglicéridos, el componente celular principal del tejido adiposo blanco, son almacenados en una única gota lipídica que ocupa entre el 80-90% del volumen total del adipocito. Para su síntesis tres ácidos grasos son ensamblados con una molécula de glicerol. Los ácidos grasos necesarios pueden provenir de fuentes externas, como FFAs en circulación o triglicéridos transportados por lipoproteínas (quilomicrones, VLDL o LDL) que deben ser previamente hidrolizados por la enzima LPL ( lipoprotein lipase) presente en el endotelio de los capilares del adipocito (372) (Figura 42). 136 Capítulo IV - Introducción Otra fuente de ácidos grasos es la lipogénesis de novo, que consiste en síntesis de ácidos grasos a partir de carbohidratos (372). Este proceso puede ocurrir tanto en el tejido adiposo como en el hígado. Existen estudios que indican que la lipogénesis de novo en humanos es menor en el tejido adiposo respecto del hígado (386). Para que este proceso ocurra es necesaria la activación de la enzima FAS ( fatty acid synthase) la cual permite el ensamblado entre moléculas de malonilCoA y acetil-CoA las cuales van a conformar la cadena de ácidos grasos (Figura 42). Durante periodos de falta de nutrientes, estrés o actividad física, se estimula la lipólisis de los triglicéridos almacenados. Durante la lipólisis los triglicéridos almacenados, ya sea en adipocitos o hepatocitos, son hidrolizados por la enzima ATGL ( adipose triglyceride lipase) para liberar NEFAs y diacilglicerol (DAG). El DAG producido es luego hidrolizado por la enzima HSL ( hormone-sensitive lipase) para liberar más NEFAs y monoacilglicerol (MAG), el cual va a ser finalmente hidrolizado por completo a NEFAs y glicerol (387) (Figura 42). Estos FFAs y glicerol que son liberados al torrente sanguíneo van a ser utilizados como fuente energética por otros tejidos, tales como el corazón y músculo esquelético (372). Figura 42: Esquema de los procesos de lipólisis y lipogénesis. FA: ácidos grasos; CD36: proteína transportadora de FA; FATP: proteína de membrana de unión a FA; ACS: enzima acil-CoA sintasa; TAG: triglicéridos. Tomado y modificado de (388). 137 Capítulo IV - Introducción El factor ChREBP, involucrado en la lipogénesis de novo, se encuentra expresado en tejido adiposo blanco, aunque en menor medida que en hígado. Muchos grupos han reportado que los triglicéridos producidos por el hígado son transferidos al tejido adiposo blanco, siendo menor la lipogénesis de novo en este tejido, lo que explicaría la baja expresión de ChREBP (381;389). Por otra parte, se ha demostrado en tejido adiposo que una disminución en la expresión de ChREBP provoca un descenso en la incorporación de glucosa y resistencia a insulina debido a menores niveles de GLUT-4 (381). Prolactina y tejido adiposo Antes se creía que los PRLR no se expresaban en el tejido adiposo, y se había propuesto que la prolactina no sería un regulador directo de las funciones de los adipocitos (390). Recientemente se han publicado evidencias de lo contrario. Se ha descripto que prolactina es secretada por adipocitos humanos, no así por adipocitos de ratones (391;392) , y se sabe que los PRLR se expresan en el tejido adiposo pardo y blanco de ratones, de rata y de humanos (39). Esta expresión aumenta durante la lactancia y en animales que sobreexpresan prolactina (39). Evidencias recientes muestran que la prolactina estimula la adipogénesis, lo que sugiere un rol de esta hormona en el metabolismo lipídico (99). Sin embargo, los resultados presentes en la literatura son controversiales. Estudios con ratones deficientes en el PRLR muestran una disminución en el peso del tejido adiposo sin alteraciones en el peso corporal ni en la ingesta de alimentos. Esta reducción deriva de un descenso en el número de adipocitos, sin cambios en su volumen (331). En el mismo sentido, se ha demostrado que la prolactina inhibe la lipólisis en ratas y humanos, y no en tejido adiposo de ratones (316). Por otra parte, recientemente se ha descripto que prolactina inhibe la expresión de malonil-coA, lo que sugiere que inhibiría la lipogénesis (393). HÍGADO Lipólisis y lipogénesis El hígado es el principal tejido regulador del balance de glucosa, y la lipogénesis hepática se encuentra íntimamente relacionada con el metabolismo de la glucosa. El hígado convierte el exceso de carbohidratos en reservas lipídicas (394). En momentos de hiperglucemia, la glucosa es almacenada en el hígado como glucógeno; mientras que en estados de hipoglucemia, el glucógeno hepático es degradado, promoviendo la secreción de glucosa. El hígado es además el principal sitio de gluconeogénesis (conversión de aminoácidos, lípidos u otros carbohidratos en glucosa). La insulina ejerce una acción anabólica en el tejido hepático, estimulando la síntesis de glucógeno y la síntesis lipídica. A su vez, tiene una acción inhibitoria sobre la gluconeogénesis 138 Capítulo IV - Introducción hepática y la secreción de glucosa por parte del hígado, por medio de la estimulación de la enzima Glucokinasa (395). Esta enzima fosforila a la glucosa en la posición 6, dando lugar a glucosa-6fosfato que es incapaz de atravesar la membrana celular, quedando retenida en el hígado. Este compuesto tiene una variedad de destinos, dado que el hígado sólo utiliza una pequeña cantidad para satisfacer sus propias necesidades energéticas. Gran parte de la glucosa-6-fosfato es convertida en glucógeno que se almacena en el hígado mediante la acción de la glucógeno sintasa (GYS2), mientras que el resto es metabolizada a acetil-CoA para ser utilizado posteriormente en la síntesis de ácidos grasos, colesterol y sales biliares (396) (Figura 43). El factor SREBP-1c está asociado a la inducción de genes que participan en la glucólisis y la lipogénesis hepática (397). Se sabe que la expresión de este factor de transcripción en el hígado se encuentra estimulada por insulina, y existen trabajos que postulan que la hormona es capaz de activar la expresión y síntesis de SREBP-1c mediante la vía de señalización que involucra a la PI 3K (398). A su vez, SREBP-1c regula negativamente la expresión de la enzima fosfoenolpiruvato carboxiquinasa (PEPCK), involucrada en la gluconeogénesis hepática (399), y positivamente la expresión de Glucokinasa (400). De esto se desprende que SREBP-1c participa como mediador de la acción inhibitoria de la gluconeogénesis y estimulatoria de la glucólisis que ejerce la insulina en el hígado. A su vez, este factor también participa en la vía de lipogénesis hepática a partir de glucosa (Figura 43). El factor ChREBP tiene un rol fundamental en la lipogénesis de novo en el hígado. Un estudio realizado con ratones knockout para ChREBP demostró que el 50% de la lipogénesis hepática se encuentra regulada por este factor de transcripción (394). No se encuentra directamente estimulado por insulina, sino que requiere de glucosa vía Glucokinasa para activar la transcripción de genes glucolíticos, como los transportadores de glucosa GLUT-2 y GLUT-4, y lipogénicos como la enzima FAS (381;401) (Figura 43). 139 Capítulo IV - Introducción Figura 43: Esquema de la relación entre la homeostasis de la glucosa y la lipogénesis hepática. Tomado y modificado de (395). En los hepatocitos, los FFAs son esterificados para generar triglicéridos, los cuales son almacenados en gotas lipídicas. Los NEFAs liberados por el tejido adiposo como consecuencia de la lipolisis son la fuente principal del hígado para la generación de triglicéridos. En condición de ayuno o durante el ejercicio, la glucosa y los triglicéridos hidrolizados son liberados por el hígado hacia la circulación para ser metabolizados por el músculo, tejido adiposo, y otros tejidos extra-hepáticos (402). Los procesos enzimáticos de lipogénesis y lipólisis hepática son similares a los ya descriptos en tejido adiposo blanco (Figura 42). Prolactina e hígado Si bien se ha demostrado la expresión de PRLR en hígado, con alta expresión de las isoformas PRLR-L y PRLR-S3 (40), el papel de la prolactina sobre los hepatocitos en relación al metabolismo lipídico ha sido escasamente estudiado. GLUCOCORTICOIDES Los glucocortiocides son hormonas sintetizadas por la corteza adrenal, bajo el control del eje hipotálamo-hipófisis-adrenal. Los principales glucocorticoides presentes en humanos y roedores son el cortisol y la corticosterona, respectivamente (403). Una acción exacerbada de los glucocorticoides provoca numerosos desórdenes clínicos como ser obesidad, insulino resistencia e intolerancia a la glucosa, tal como se observa en el Síndrome de Cushing (404). 140 Capítulo IV - Introducción Existen numerosas evidencias que sostienen que los glucocorticoides juegan un rol importante en los desórdenes metabólicos (405). Existe una desregulación tejido específica en la expresión de la enzima productora de glucocorticoides, la 11 beta hidroxiesteroide deshidrogenasa (11 -HSD1), frente a la obesidad en roedores (406) y humanos (407). Del mismo modo, deficiencias en 11 -HSD1 (408) o su inhibición (409) protegen contra las disfunciones metabólicas asociadas a la obesidad. Los glucocorticoides promueven el catabolismo de proteínas y lípidos, lo que incrementa la disponibilidad de FFAs y aminoácidos. A su vez, los glucocorticoides incrementan la concentración intracelular de enzimas y sustratos para la gluconeogénesis, lo que resulta en un incremento en la producción de glucosa hepática (403). El receptor de glucocorticoides (GR) se encuentra presente en los hepatocitos y su activación desencadena la expresión de genes gluconeogénicos (410). La deleción específica de GR en hepatocitos provoca el descenso en la expresión de genes gluconeogénicos como también en los niveles de glucosa séricos en ayunas (411). La expresión de GR se ve estimulada por el factor de transcripción Yin Yang 1 (YY1) el cual aumenta durante el ayuno (412). Por otra parte, se ha demostrado que la reducción específica en la expresión de ARNm de Gr en hígado y tejido adiposo blanco resulta suficiente para mejorar los niveles plasmáticos de glucosa y lípidos en un modelo animal de diabetes tipo I I (403). 141 Capítulo IV - Objetivos OBJETIVOS Estudiar el impacto de la hiperprolactinemia, por la ausencia de RD2s en lactotropos, sobre la regulación de la adiposidad en diferentes tejidos. Los objetivos específicos fueron: - Determinar el peso de tejidos con alto contenido lipídico (tejido adiposo gonadal, retroperitoneal e hígado) en ratones hembra de ambos genotipos, a los 5 y 10 meses de edad. Realizar un análisis morfométrico del tamaño de los adipocitos, y estudiar el contenido lipídico hepático. - Detallar el perfil lipídico séricos en ratones hembra Drd2loxP/ loxP y lacDrd2KO mediante la medición de niveles de adiponectina, colesterol, triglicéridos y NEFA. Analizar, a su vez, los mecanismos de lipogénesis y lipólisis en tejido adiposo blanco e hígado, ya sea en ratones con libre acceso al alimento ( ad libitum ), como en condiciones de ayuno por 12 hs, con y sin realimentación. En particular: - Determinar la expresión de ARNm de los receptores de prolactina (todas sus isoformas) y de hormona de crecimiento, en tejido adiposo gonadal e hígado de hembras Drd2loxP/ loxP y lacDrd2KO de 5 y 10 meses de edad. - Estudiar los niveles de expresión de dos enzima lipolíticas ( Hsl y Atgl) y dos lipogénicas ( Fas y Lpl) en tejido adiposo gonadal e hígado de hembras de 5 y 10 meses de edad. - Analizar la expresión génica de factores lipogénicos dependientes del metabolismo de la glucosa ( Chrebp y Srebf1) en ambos tejidos de hembras Drd2loxP/ loxP y lacDrd2KO de 5 y 10 meses de edad. - En hígado puntualmente, precisar el contenido de glucógeno y la expresión de Glucokinasa, una enzima involucrada en la síntesis del mismo, en ratones hembra de ambos genotipos, a los 5 y 10 meses de edad. Finalmente, caracterizaremos el sistema de glucocorticoides en este modelo animal, estudiando: los niveles de expresión génica del receptor de glucocorticoides ( Gr ) en distintos tejidos (hipotálamo, hígado y tejido adiposo), el peso de las adrenales y la morfología histológica de las mismas, en ratones hembra de 5 meses de edad. 142 Capítulo IV - Resultados RESULTADOS ADIPOSIDAD INCREMENTADA A los 5 meses de edad los ratones hembra lacDrd2KO presentaron un aumento en el peso del tejido adiposo retroperitoneal e hígado respecto de sus pares control, y a los 10 meses de edad también un aumento en el peso del tejido adiposo gonadal (Figura 44A). Los adipocitos en las hembras lacDrd2KO resultaron de mayor tamaño, evidenciado por un incremento en la mediana del área (1363 y 2503 µm 2 para los adipocitos de ratones Drd2loxP/ loxP y lacDrd2KO, respectivamente, P ˂ 0,0001) (Figura 44C). La distribución de los mismos de acuerdo su tamaño muestra que el tejido adiposo proveniente de hembras lacDrd2KO tiene una mayor proporción de adipocitos grandes (mayores a 2000 µm 2) (Figura 44B) respecto del tejido adiposo gonadal de hembras Drd2loxP/ loxP de 10 meses de edad. La coloración de los hígados con Oil Red indicó un mayor contenido lipídico en las hembras lacDrd2KO (Figura 44D), que resultó consistente con el mayor contenido de triglicéridos hepáticos encontrado en este genotipo ( P ˂ 0,05) (Figura 44E). 143 Capítulo IV - Resultados Figura 44: Adiposidad. A) Peso (en g) de tejido adiposo blanco gonadal y retroperitoneal, como también del hígado, en hembras Drd2loxP/ loxP y lacDrd2KO de 5 (n = 5) y 10 (n = 5 y 10, respectivamente) meses de edad. Los resultados se analizaron mediante una Prueba T, * P < 0,05 lacDrd2KO vs Drd2loxP/ loxP para cada tejido, a cada edad. B) Distribución de los adipocitos (proveniente de tejido adiposo gonadal) en porcentaje según su tamaño en hembras Drd2 loxP/ loxP y lacDrd2KO de 10 meses de edad (n = 4). C) I mágenes representativas, tomadas con objetivo de 40X, de cortes histológicos de tejido adiposo gonadal, coloreados con H&E. D) I mágenes representativas de la coloración con Oil Red (se visualiza en rojo) realizada en secciones de hígados provenientes de ratones hembra Drd2loxP/ loxP y lacDrd2KO de 10 meses de edad. Los preparados se colorearon con hematoxilina, y las imágenes fueron tomadas con objetivo de 40X. E) Contenido hepático de triglicéridos (TG) en hembras Drd2loxP/ loxP y lacDrd2KO de 10 meses de edad (n = 6 y 7 respectivamente). Los resultados se analizaron mediante una Prueba T, * P < 0,044 vs Drd2loxP/ loxP. En todas las figuras de este capítulo, las barras indican la media y las barras de error el E.S.M. 144 Capítulo IV - Resultados TEJIDO ADIPOSO GONADAL Lipólisis / Lipogénesis Habiendo encontrado adiposidad incrementada en los ratones hembra lacDrd2KO decidimos evaluar la expresión de los receptores de prolactina en tejido adiposo gonadal comparativamente entre genotipos. Determinamos la expresión de ARNm del total de isoformas para Prlr , como también de las distintas isoformas discriminadas. A los 5 meses de edad las hembras lacDrd2KO presentaron una tendencia a mayores niveles de Prlr-l (isoforma larga) respecto de sus pares control ( P = 0,069) (Figura 45A). Por el contrario, la expresión génica de Prlr-l a los 10 meses de edad presentó una tendencia a estar disminuida en los ratones lacDrd2KO comparado con los Drd2loxP/ loxP ( P = 0,078) (Figura 45B). La determinación de ARNm para el conjunto de isoformas, siguió el mismo patrón que la isoforma larga, indicando la preponderancia de la misma. No encontramos diferencias significativas en los niveles de Prlr-s3 a los 5 meses, mientras que a los 10 meses de edad esta isoforma se encontró debajo del límite de detección. En ambas edades estudiadas las isoformas Prlr-s1 y Prlr-s2 se encontraron debajo del límite de detección. Esto resulta en parte consistente con lo ya descripto por otros autores, que determinan que Prlr-s1 no resulta detectable en tejido adiposo blanco (39). Analizamos también la expresión de receptores de GH ( Ghr ) en este tejido, y encontramos la misma tendencia observada en Prlr-l, aunque sin significancia estadística (Figura 45C). 145 Capítulo IV - Resultados Figura 45: Receptores de prolactina y hormona de crecimiento. A-B) Expresión génica de Prlr total, y sus isoformas: Prlr-l, Prlr-s1, Prlr-s2 y Prlr-s3 en tejido adiposo gonadal de hembras Drd2loxP/ loxP y lacDrd2KO; A) a los 5 meses de edad (n = 4). Los resultados fueron analizados por Prueba T, la cual resultó P = 0,069 vs ratones Drd2loxP/ loxP para Prlr-l (isoforma larga). B) A los 10 meses de edad (n = 11). La Prueba T resultó P = 0,078 vs ratones Drd2loxP/ loxP para Prlr-l (isoforma larga). C) Expresión génica de Ghr (receptor de GH) en tejido adiposo gonadal de ratones hembra Drd2loxP/ loxP y lacDrd2KO de 5 (n = 4) y 10 (n = 11) meses de edad. Los resultados se analizaron por Prueba T sin obtener diferencias significativas. En todos los casos los ratones se encontraban con libre acceso a comida ( ad libitum ). Los ratones hembra lacDrd2KO de 10 meses de edad presentaron niveles séricos incrementados tanto de triglicéridos como NEFA comparado con sus pares control ( P < 0,05) (Figura 46A y C), mientras que los niveles de colesterol total y de adiponectina no difirieron entre genotipos (Figura 46B y D). Del mismo modo, cuando analizamos los niveles de expresión génica de adiponectina en tejido adiposo gonadal de ratones hembra de 10 meses de edad, no observamos diferencias entre los grupos (Figura 46D). Figura 46: Perfil lipídico. A) Triglicéridos séricos (n = 6), B) colesterol total (n = 6) y C) NEFA (n = 6), en ratones hembra Drd2loxP/ loxP y lacDrd2KO de 10 meses de edad con libre acceso a comida ( ad libitum ); * P < 0,05 vs ratones Drd2loxP/ loxP. D) Niveles séricos (n = 6) y de ARNm (n = 11) de adiponectina, en tejido adiposo de ratones hembra Drd2loxP/ loxP y lacDrd2KO de 10 meses de edad, con alimento ad libitum . En todos los casos los resultados se analizaron por una Prueba T. 146 Capítulo IV - Resultados La adiposidad incrementada puede ser consecuencia de un incremento en la adipogénesis y/ o una disminución en la lipólisis. Es por ello que decidimos estudiar a continuación los niveles de expresión de dos enzima lipolíticas ( Hsl y Atgl) y dos lipogénicas ( Fas y Lpl) en tejido adiposo gonadal de hembras de 5 y 10 meses de edad con libre acceso a comida ( ad libitum ) y en condiciones de ayuno por 12 hs, con y sin realimentación durante 1 hr. Mientras que a los 5 meses de edad no existieron alteraciones en la expresión de enzimas en condición ad libitum (Figura 47A), ambas enzimas lipolíticas resultaron disminuidas en ratones lacDrd2KO de 10 meses en comparación con Drd2loxP/ loxP (Figura 47B). La expresión de ARNm de Lpl también estaba disminuida a esta edad, sin encontrarse diferencias en Fas (Figura 47B). En cuanto a los ratones ayunados por 12 hs y realimentados (Figura 47C-F), observamos que a los 5 meses los ratones hembra lacDrd2KO, y no los Drd2loxP/ loxP, responden a la realimentación con un incremento significativo en la expresión génica de la enzima lipogénica Fas, respuesta que desaparece a los 10 meses de edad (Figura 47E). Por otra parte, los ratones realimentados de ambos genotipos presentaron una disminución en los niveles de Lpl a los 10 meses con respecto al estado de ayuno, sin observar estas diferencias a los 5 meses de edad (Figura 47F). 147 Capítulo IV - Resultados 148 Capítulo IV - Resultados Figura 47: Enzimas lipolíticas-lipogénicas en tejido adiposo gonadal. A) Expresión génica de Hsl, Atgl, Fas, y Lpl en tejido adiposo gonadal de hembras Drd2loxP/ loxP y lacDrd2KO de 5 meses de edad alimentadas ad libitum (n = 4 y 4, respectivamente). Los datos se analizaron por Prueba T para cada gen sin obtener diferencias entre genotipos. B) Niveles de ARNm de Hsl, Atgl, Fas, y Lpl en tejido adiposo gonadal de hembras Drd2loxP/ loxP y lacDrd2KO de 10 meses de edad con alimento ad libitum . Los resultados se analizaron por Prueba T donde * P < 0,04 vs ratones Drd2loxP/ loxP (n = 11 y 11, respectivamente). C-F) Expresión génica de Hsl, Atgl, Fas, y Lpl en tejido adiposo gonadal de hembras Drd2loxP/ loxP y lacDrd2KO, ayunadas por 12 hs sin realimentación (Ayuno) y con 1 hr de realimentación (Realimentado). Los paneles de la izquierda corresponden a ratones de 5 meses de edad, mientras que los de la derecha a 10 meses. En cada uno de los casos, los resultados se analizaron mediante ANOVA de dos factores donde * P < 0,02 vs ratones del mismo genotipo en Ayuno, y # P < 0,05 vs ratones Drd2loxP/ loxP. Se utilizaron ratones Drd2loxP/ loxP y lacDrd2KO de 5 meses, n = 6 y 6 ayuno; n = 4 y 6 realimentado; respectivamente. Y ratones Drd2loxP/ loxP y lacDrd2KO de 10 meses, n = 5 y 3 ayuno; n = 3 y 5 realimentado; respectivamente. Profundizamos el análisis de los factores lipogénicos en tejido adiposo gonadal, para lo cual determinamos los niveles de expresión de Chrebp y Srebf1, factores de transcripción dependientes también del metabolismo de la glucosa, que sabemos se encuentra alterada en este modelo. No encontramos diferencias significativas entre los grupos con alimento ad libitum a los 5 meses de edad, pero sorprendentemente, observamos una disminución significativa en la expresión de ambos factores en tejido adiposo de ratones lacDrd2KO de 10 meses de edad ( P < 0,04) (Figura 48A). En cuanto al comportamiento de estos factores al ayuno y realimentación, observamos una tendencia a mayor expresión de Chrebp frente a la realimentación en ratones de ambos genotipos a los 5 meses de edad ( Pcondición = 0,069), tendencia que desaparece a los 10 meses (Figura 48B). A los 10 meses de edad, los niveles de Chrebp son significativamente menores en ratones lacDrd2KO respecto de Drd2loxP/ loxP en ambas condiciones ( P < 0,05) (Figura 48B), manteniendo la diferencia observada en ratones alimentados ad libitum . En cuanto a Srebf1, determinamos un incremento significativo en los niveles de este factor como producto de la realimentación a los 5 meses de edad en ambos genotipos ( P < 0,0003) (Figura 48C); y nuevamente dicha respuesta desaparece a los 10 meses de edad. 149 Capítulo IV - Resultados Figura 48: Factores lipogénicos dependientes de glucosa. A) Niveles de ARNm de Srebf1 y Chrebp en tejido adiposo gonadal de ratones hembra Drd2loxP/ loxP y lacDrd2KO con libre acceso al alimento ( ad libitum ). I zquierda: 5 meses de edad (n = 4). Derecha: 10 meses de edad (n = 10 y 9 respectivamente para Chrebp; n = 8 para Srebf1). Los resultados se analizaron por una Prueba T donde * P < 0,04 vs Drd2loxP/ loxP. B) Niveles de ARNm de Chrebp , en tejido adiposo gonadal de ratones hembra Drd2loxP/ loxP y lacDrd2KO de 5 (figura de la izquierda) y 10 (figura de la derecha) meses de edad, ayunados por 12 hs con realimentación (grupo Realimentado) o sin ella (grupo Ayuno). A los 5 meses: n = 6 y 6 ayunado; n = 4 y 6 realimentado, respectivamente. A los 10 meses: n = 5 y 3 ayunado; n = 3 y 4 realimentado, respectivamente. Los resultados se analizaron mediante un ANOVA de dos factores donde * P < 0,05 vs Drd2loxP/ loxP. Cabe destacar que a los 5 meses el Pcondición = 0,069. C) Expresión de Srebf1 en tejido adiposo gonadal de ratones hembra 150 Capítulo IV - Resultados Drd2loxP/ loxP y lacDrd2KO ayunados por 12 hs sin realimentación (Ayuno) y con 1 hr de realimentación (Realimentado). El gráfico de la izquierda corresponde a ratones de 5 meses de edad, mientras que el de la derecha a 10 meses. Los resultados se analizaron por ANOVA de dos factores donde * P < 0,0003 vs grupo ayunado del mismo genotipo. A los 5 meses: n = 6 y 6 ayunado; n = 4 y 6 realimentado, respectivamente. A los 10 meses: n = 5 y 3 ayunado; n = 3 y 4 realimentado, respectivamente. Los ratones hembra lacDrd2KO de 10 meses de edad presentan mayor contenido lipídico en los adipocitos, evidenciado como un mayor tamaño de los mismos, lo que se debe, al menos en parte, a la disminución en la expresión de enzimas lipolíticas Hsl y Atgl. A los 5 meses de edad, la cantidad de tejido adiposo gonadal no se encuentra todavía incrementada y consistente con esto los niveles de las enzimas lipolíticas se encuentran normales. Las alteraciones más profundas en cuanto a la expresión génica en tejido adiposo gonadal son evidentes a los 10 meses de edad, momento en el cual el aumento en el peso corporal y la adiposidad es significativamente mayor. A los 5 meses de edad, en cambio, no se observan diferencias importantes en los niveles de ARNm de factores lipolíticos o lipogénicos respecto de los ratones control. Llamativamente, la respuesta normal a la realimentación de genes dependientes del metabolismo de la glucosa se manifiesta a los 5 meses y se pierde a los 10 meses, a pesar que las alteraciones en la homeostasis de la glucosa fueron observadas en ambas edades (como se detalle en el capítulo anterior), indicando un deterioro creciente del proceso de lipogénesis dependiente de glucosa. HÍGADO Lipólisis / Lipogénesis Evaluamos también la expresión de factores lipogénicos y lipolíticos hepáticos en hembras lacDrd2KO, para explicar el aumento de lípidos de este órgano en nuestro modelo hiperprolactinémico. Estudiamos, en primer lugar, la expresión de los receptores de prolactina, ya sea total ( Prlr ) como de las distintas isoformas ( Prlr-l, Prlr-s1, Prlr-s2 y Prlr-s3) en hígados de ratones Drd2loxP/ loxP y lacDrd2KO, de 5 y 10 meses de edad. En ambas edades encontramos un incremento significativo en los niveles de Prlr en ratones lacDrd2KO respecto de sus pares control, siendo la isoforma Prlr- s2 la significativamente incrementada a los 5 meses, y Prlr-s3 a los 10 meses de edad (Figura 49A). Analizamos también la expresión génica del receptor de GH ( Ghr ), sin encontrar diferencias entre grupos de ambas edades (Figura 49B). 151 Capítulo IV - Resultados Figura 49: Receptores de prolactina y hormona de crecimiento. A) Expresión génica de Prlr total, y sus isoformas: Prlr-l, Prlr-s1, Prlr-s2 y Prlr-s3 en hígado de hembras Drd2loxP/ loxP y lacDrd2KO. I zquierda: Resultados a los 5 meses de edad (n = 5 y 4 respectivamente). Los resultados fueron analizados por Prueba T donde * P < 0,03 vs ratones Drd2loxP/ loxP. Derecha: A los 10 meses de edad (n = 6 y 7 respectivamente). El análisis mediante Prueba T resultó * P < 0,05 vs ratones Drd2loxP/ loxP. B) Expresión génica de Ghr (receptor de GH) en hígado de ratones hembra Drd2loxP/ loxP y lacDrd2KO de 5 (n = 5 y 4, respectivamente) y 10 (n = 7) meses de edad. Los resultados se analizaron por una Prueba T sin obtener diferencias significativas. En todos los casos los ratones se encontraban con libre acceso al alimento ( ad libitum ). A continuación determinamos los niveles de expresión de dos enzimas lipolíticas ( Hsl y Atgl) y dos lipogénicas ( Fas y Lpl) en hígados de hembras de 5 y 10 meses de edad con libre acceso a comida ( ad libitum ). Mientras que a los 5 meses de edad no existieron alteraciones en la expresión de enzimas en condición ad libitum (Figura 50A), la expresión de ARNm de Lpl resultó disminuida en ratones lacDrd2KO de 10 meses comparado con Drd2loxP/ loxP ( P < 0,03), sin encontrar diferencias en el resto de las enzimas (Figura 50B). 152 Capítulo IV - Resultados Figura 50: Enzima lipolíticas y lipogénicas en hígado. A) Niveles de ARNm de las enzimas lipolíticas ( Hsl y Atgl) y lipogénicas ( Fas y Lpl) en hígados de ratones hembra Drd2loxP/ loxP y lacDrd2KO de 5 meses de edad con libre acceso a la comida ( ad libitum ). Los resultados se analizaron por Prueba T sin obtener diferencias entre genotipos. B) Expresión génica de las enzimas Hsl, Atgl, Fas, y Lpl hepáticas en hembras Drd2loxP/ loxP y lacDrd2KO de 10 meses de edad alimentados ad libitum . Los resultados fueron analizados por Prueba T, * P < 0,03 vs ratones Drd2loxP/ loxP (n = 6 y 7, respectivamente). Dado que estos resultados no explicaban el incremento en los niveles de lípidos hepáticos, decidimos evaluar los niveles de expresión de factores involucrados en la lipogénesis y regulados por insulina o glucosa ( Chrebp , Srebf1 , y Glucokinasa) en hígados de hembras de 5 y 10 meses de edad. Estudiamos los niveles de ARNm ya sea en animales con libre acceso a la comida ( ad libitum ) como en condiciones de ayuno por 12 hs, con y sin realimentación durante 1 hr. A los 5 meses de edad encontramos un incremento significativo en la expresión de Chrebp en ratones lacDrd2KO ( P < 0,03), sin existir diferencias entre grupos en Srebf1 y Glucokinasa, con alimento ad libitum (Figura 51A). A los 10 meses, en cambio, Chrebp no presentó variaciones entre genotipos, y observamos un incremento significativo en los niveles de Glucokinasa hepática en los ratones hiperprolactinémicos ( P < 0,03), en condición ad libitum (Figura 51A). Srebf1 tampoco mostró diferencias a esta edad. Por otro lado, observamos que la realimentación, luego de un ayuno de 12 hs provocaba, el incremento de los niveles hepáticos de ARNm de Srebf1 ( P < 0,03), Glucokinasa y Chrebp (en este último gen solo se observa una tendencia) en ratones Drd2loxP/ loxP de 5 meses como era esperable, mientras que a esta edad en los ratones lacDrd2KO el único gen que se modificaba por la realimentación era Glucokinasa, que además estaba basalmente mayor en comparación con los ratones Drd2loxP/ loxP, (en forma similar a lo encontrado en la condición ad libitum ). Sorprendentemente, a los 10 meses de edad tanto Srebf1 como Chrebp no resultaron modificados por la realimentación, y el efecto sobre la Glucokinasa en ambos genotipos era mucho menor que a los 5 meses (Figura 51B-D). Es decir que el efecto de la realimentación, que provoca liberación de insulina endógena, se desensibilizaba con la edad, y además era genotipo dependiente. 153 Capítulo IV - Resultados Figura 51: Factores dependientes de glucosa. A) Niveles de ARNm de factores de transcripción lipogénicos ( Chrebp y Srebf1) y Glucokinasa en hígados de ratones hembra Drd2loxP/ loxP y lacDrd2KO de 5 meses y 10 meses de edad con libre acceso a la comida ( ad libitum ). Los resultados se analizaron por Prueba 154 Capítulo IV - Resultados T donde * P < 0,03 vs ratones Drd2loxP/ loxP (5 meses: n = 5 y 4, respectivamente; 10 meses: n = 6 y 7, respectivamente). B-D) Expresión génica en hígados de hembras Drd2loxP/ loxP y lacDrd2KO de 5 (izquierda) y 10 (derecha) meses de edad, en condición de ayuno por 12 hs con realimentación (Realimentado) o sin ella (Ayuno). B) Chrebp . El ANOVA de dos factores no arrojó diferencias significativas a los 5 meses (n = 6 ayuno; n = 4 y 6 realimentado, respectivamente), como así tampoco a los 10 meses (n = 5 ayuno; n = 3 y 5 realimentado, respectivamente). C) Srebf1. Los resultados se analizaron mediante ANOVA de dos factores. A los 5 meses Pinteracción (genotipo x condición) < 0,02, donde * P < 0,05 vs grupo Ayuno Drd2loxP/ loxP (n = 5 y 6 ayuno; n = 4 y 6 realimentado, respectivamente). A los 10 meses no se observaron diferencias significativas (n = 5 ayuno; n = 3 y 5 realimentado, respectivamente). D) Glucokinasa. Los resultados se analizaron con ANOVA de dos factores. A los 5 meses * P < 0,000006 vs grupo Ayuno del mismo genotipo y # P < 0,01 vs Drd2loxP/ loxP (n = 6 ayuno; n = 4 y 6 realimentado, respectivamente). A los 10 meses * P < 0,02 vs grupo Ayuno del mismo genotipo (n = 5 ayuno; n = 3 y 5 realimentado, respectivamente). Los resultados obtenidos a los 5 meses de edad, con falta de respuesta de dos genes frente a la realimentación en los ratones lacDrd2KO, correlacionarían con una falta de liberación de insulina (tal como fue observada en el capítulo anterior, tanto por la realimentación como en el ensayo de GSI S), en concordancia con la dependencia de su expresión por dicha hormona. La Glucokinasa en cambio, que depende mayormente de la entrada de glucosa, sí aumentó su expresión por realimentación, aunque la respuesta fue menor a los 10 meses (quizás por desensibilización causada por la intolerancia crónica a la glucosa). Evaluamos también la concentración de glucógeno hepático y observamos que a los 10 meses se encontraba significativamente disminuida en los ratones hembra lacDrd2KO ( P < 0,02), sin existir estas diferencias a los 5 meses (Figura 52A). La realimentación provocó un incremento significativo en la concentración de glucógeno en hígados de ratones de ambos grupos y edades analizadas ( P < 0,002) en comparación con sus pares ayunados (Figura 52B), sin presentar diferencias entre genotipos. 155 Capítulo IV - Resultados Figura 52: Niveles de glucógeno. A) Concentración de glucógeno hepático (µg/ mg de tejido) en ratones hembra Drd2loxP/ loxP y lacDrd2KO de 5 meses y 10 meses de edad con libre acceso a la comida ( ad libitum ). Los resultados se analizaron por ANOVA de dos factores donde Pinteracción (genotipo x edad) < 0,05, y * P < 0,02 vs Drd2loxP/ loxP de la misma edad (5 meses: n = 5; 10 meses: n = 6 y 4, respectivamente). B) Niveles de glucógeno hepático (µg/ mg de tejido) en hembras Drd2loxP/ loxP y lacDrd2KO de 5 (izquierda) y 10 (derecha) meses de edad, en condición de ayuno por 12 hs con realimentación (Realimentado) o sin ella (Ayuno). Los resultados se analizaron por ANOVA de dos factores, donde * P < 0,002 vs grupo Ayuno del mismo genotipo, a los 5 meses (n = 6 y 5 ayuno; n = 4 realimentado, respectivamente). A los 10 meses * P < 0,0004 vs grupo Ayuno del mismo genotipo (n = 5 ayuno; n = 3 y 6 realimentado, respectivamente). Encontramos contenido lipídico incrementado en los hígados de hembras lacDrd2KO, que no pudimos explicar mediante la expresión de enzimas lipolíticas y lipogénicas estudiadas. Sin embargo observamos una regulación positiva del receptor de prolactina en hígados de hembra lacDrd2KO, y se sabe que prolactina está involucrada en procesos de lipogénesis. Solo se evidenció un incremento del factor respondedor a carbohidratos, Chrebp , que participa en la lipogénesis asociada a glucosa, en hígados de ratones lacDrd2KO de 5 meses de edad. Por otro lado, la regulación de genes por realimentación se desensibilizó con la edad, y estaba alterada en los ratones transgénicos. La capacidad de almacenamiento de glucógeno también resultó modificada, en especial a los 10 meses, en los cuales su menor concentración en hígados de ratones lacDrd2KO podría estar asociada a niveles más altos de glucosa en sangre. 156 Capítulo IV - Resultados GLUCOCORTICOIDES Como los glucocorticoides presentan un rol fundamental en la regulación del peso corporal, el metabolismo de lípidos y la glucosa actuando a nivel del cerebro, tejido adiposo e hígado, caracterizamos sus receptores en forma comparativa entre genotipos. No observamos diferencias significativas en el peso de las adrenales (Figura 53A) como tampoco en la expresión génica del receptor de glucocorticoides ( Gr ) en todos los tejidos analizados (Figura 53C). Cabe destacar, la existencia de una tendencia a menores niveles de expresión de Gr en hígados de hembras lacDrd2KO de 10 meses de edad ( P = 0,08). Analizamos la morfología de las adrenales mediante una coloración con H&E, sin observar diferencias entre genotipos, en la proporción entre la zona medular y la corteza, ni en la disposición celular (Figura 53B). Observamos sin embargo, que la zona reticular (porción más interna de la corteza adrenal) presentó mayor número de gotas lipídicas en los ratones lacDrd2KO que sus pares control (ver flechas en Figura 53B). 157 Capítulo IV - Resultados Figura 53: Glucocorticoides. A) Peso (en g) de ambas adrenales en hembras Drd2loxP/ loxP y lacDrd2KO de 5 meses de edad (n = 4). Los resultados se analizaron mediante una Prueba T sin obtener diferencias significativas. B) I mágenes representativas de la coloración con H&E realizada en secciones de adrenales provenientes de ratones hembra Drd2loxP/ loxP y lacDrd2KO de 5 meses de edad. Las imágenes fueron tomadas con objetivos de 10X y 40X. M: zona medular, C: corteza adrenal. Las flechas indican gotas lipídicas. C) Niveles de ARNm de Gr (receptor de glucocorticoides) en hipotálamo (HT) de ratones hembra Drd2loxP/ loxP y lacDrd2KO de 10 meses, y en tejido adiposo blanco gonadal (TA) e hígado (HG) de ratones de 5 y 10 meses de edad. En todos los casos los ratones se encontraban con libre acceso a la comida ( ad libitum ). Los resultados se analizaron por Prueba T sin obtener diferencias significativas. HT: n = 5 y 6, respectivamente. TA: 5 meses n = 4, y 10 meses n = 11. HG: 5 meses n = 5 y 4, respectivamente, y 10 meses n = 7 y 8, respectivamente. Cabe destacar que en HG de 10 meses P = 0,08. En su conjunto estos resultados demuestran que los glucocorticoides se encuentran conservados en este modelo transgénico. Esto apoya la idea de que las alteraciones observadas en cuanto a la ingesta de alimentos, peso corporal, incremento en la adiposidad e intolerancia a la glucosa están asociadas a la hiperprolactinemia, descartando una acción de los glucocorticoides. 158 Capítulo IV - Discusión DISCUSIÓN En el presente capítulo demostramos que los ratones hembra lacDrd2KO presentan una adiposidad incrementada que correlaciona con el aumento en su peso corporal. Tanto el tejido adiposo como el hígado eran más pesados al compararlos con ratones control, incrementando esta diferencia con la edad. El aumento en el tejido adiposo fue acompañado por un incremento en el número de adipocitos de mayor tamaño, lo que sugiere un mayor reservorio lipídico. Del mismo modo, corroboramos que el aumento en el peso hepático se debía a mayor contenido de lípidos, por medio de una tinción específica y de la medición de triglicéridos. En suero altos niveles de NEFA y triglicéridos séricos en ratones hembra lacDrd2KO, demostraban junto con los resultados anteriores un fenotipo de alteración en el metabolismo lipídico. Consistente con estos resultados, pacientes tratados con drogas antipsicóticas que antagonizan los RD2s y elevan prolactina, presentan obesidad, hiperglucemia y dislipidemia (alteraciones en las concentraciones séricas de lípidos y lipoproteínas) (413;414), si bien aun no sé sabe a ciencia cierta los mecanismos que producen estos efectos. Se observó en el modelo transgénico murino de obesidad (ratón ob/ ob ) como también en los seres humanos que el tratamiento con bromocriptina, un agonista dopaminérgico que inhibe la secreción de prolactina conlleva a una pérdida de peso corporal (415) y una mejora en la dislipidemia (416;417). Esta información disponible, junto con la obtenida en nuestro trabajo, indica que prolactina es un potente regulador de la adiposidad. Sin embargo, en hiperprolactinémicas Drd2 trabajos -/ - previos hemos demostrado que los ratones hembra presentan en la adultez, pesos corporales similares a sus pares WT (68), y que no existían diferencias en los niveles de adiposidad ni en el peso hepático entre ambos genotipos, en contraposición con lo encontrado en el modelo lacDrd2KO. Tal como detallamos previamente, esto puede deberse a otros fenotipos presentes en este modelo a causa de la ausencia total de RD2s, como ser el eje de GH disminuido o mecanismos de regulación de la ingesta a nivel central (68). Hay estudios que demuestran la expresión de receptores de dopamina en tejido adiposo y adipocitos de humanos, y sugieren que el RD2 media el efecto inhibitorio de dopamina sobre la síntesis y secreción de prolactina en adipocitos (418). Sin embargo, en tejido adiposo de rata y ratón no se han descripto fehacientemente la expresión de los RD2s con lo cual no podríamos infererir un efecto directo del RD2 sobre el adipocito. Nuestro objetivo era determinar el papel de la prolactina sobre la adiposidad, y para ello usamos el modelo transgénico con anulación de los RD2s solamente en los lactotropos hipofisarios que presenta hiperprolactinemia crónica, eje de GH conservado, y el RD2 cerebral funcional. Al evaluar la expresión de los receptores de prolactina y GH, ambas hormonas con roles metabólicos (34) (419), detectamos que el Ghr no estaba alterado en hígado o tejido adiposo en concordancia con el eje de GH conservado, lo que sugería un rol más preponderante de los elevados niveles de prolactina en el fenotipo metabólico descripto. 159 Capítulo IV - Discusión El estudio de los PRLR, y sus subtipos, en hígado y tejido adiposo arrojó interesantes resultados característicos para cada tejido, y dependientes de la edad. En tejido adiposo la expresión de la isoforma larga Prlr-l resultó levemente incrementada ( P = 0,069) en ratones lacDrd2KO de 5 meses de edad, mientras que a los 10 meses el resultado fue el opuesto. En este tejido las isoformas cortas resultaron debajo del límite de detección. Se sabe que el tejido adiposo expresa dos de las isoformas cortas (PRLR-S2 y PRLR-S3) y la larga (PRLR-L) del receptor (39), siendo la isoforma PRLR-L aquella que se expresa en mayor abundancia (39;99;420), lo que resulta consistente con nuestros resultados. Prolactina activa a Stat5 en los adipocitos, un evento que involucra a PRLR-L y no a las isoformas cortas (99), y resultados obtenidos a partir del ratón que expresa solamente la isoforma larga del receptor, sugieren que la isoforma PRLR-L sola puede estimular la hipertrofia adipocitaria en ratones (421). En su conjunto estos datos sugieren que en el tejido adiposo blanco el receptor funcional es PRLRL, a través del cual prolactina media sus efectos. El incremento en los niveles de Prlr-l a los 5 meses de edad podría estar asociado a la regulación positiva de este receptor por parte de prolactina, como lo observamos en el presente trabajo en el hipotálamo e hígado. Sin embargo, cuando los niveles de esta hormona se vuelven muy por encima de los fisiológicos existe una regulación negativa, posiblemente como mecanismo compensatorio, que explicaría lo que ocurre a los 10 meses. Brandebourg y colaboradores demostraron que prolactina regula positivamente a su receptor en adipocitos provenientes de ratas macho (420), y mencionan que concentraciones supra-fisiológicas de esta hormona pueden causar la regulación negativa de su propio receptor, obstaculizar la dimerización del mismo, o inducir la activación de proteínas supresoras (420). Esta regulación dual del PRLR por parte de prolactina parece ser tejido específica, ya que tanto en hígado como en hipotálamo solo se observó la inducción del mismo, en forma opuesta a lo que describimos en el tejido adiposo. En el hígado de ratones lacDrd2KO, todas las isoformas del receptor de prolactina estaban presentes, y su expresión era mayor que en los ratones controles en ambas edades. Está descripto que en el hígado de ratón, las isoformas PRLR-L y PRLR-S3 son las más abundantes mientras que existe una expresión minoritaria de las PRLR-S1 y PRLR-S2 (40), lo que concuerda con nuestros resultados. Por otra parte, aunque se sabe que el PRLR está altamente expresado en hígado, centro regulador de la homeostasis metabólica, poco se sabe de las acciones potenciales de prolactina en este tejido. A continuación intentamos definir el rol de altos niveles de prolactina y de la inducción de sus receptores en la expresión de genes hepáticos y de tejido adiposo relacionados al incremento lipídico. En tejido adiposo de los ratones lacDrd2KO el descenso en la expresión de enzimas lipolíticas ( Atgl y Hsl) podría dar cuenta del incremento de adiposidad. Este descenso no se evidenció a los 5 meses en concordancia con la adiposidad incipiente de ese momento, resultando manifiesto a los 10 meses de edad. 160 Capítulo IV - Discusión Estos resultados apoyan la idea de que prolactina estaría inhibiendo la lipólisis en el tejido adiposo blanco, algo que ya se había observado en explantes de tejido adiposo epididimal y adipocitos de ratas macho (420). Por otro lado, es conocido que la lipogénesis es regulada por factores como la alimentación, el ayuno y la composición dietaria (372). Es por ello que decidimos evaluar, cómo variaba la expresión de las enzimas lipolíticas y lipogénicas frente al ayuno prolongado y a la realimentación en ambos genotipos. No encontramos diferencias en la expresión génica de las enzimas Atgl, Hsl y Fas en los ratones control comparando ambas condiciones. Esto puede estar relacionado con las evidencias que indican que el descenso en la lipogénesis como producto del ayuno se debe a una menor capacidad del tejido adiposo de generar acetil-CoA a partir de glucosa, más que a una inhibición en las enzimas lipogénicas involucradas en la síntesis de ácidos grasos (372). En los ratones lacDrd2KO de 5 meses encontramos una inducción positiva de la expresión de la enzima lipogénica Fas con la realimentación. Esto podría explicar la mayor ganancia de peso observada a esta edad con la realimentación (resultado del Capítulo I I ). Sorprendentemente encontramos una disminución significativa en los niveles de Lpl con la realimentación en ambos genotipos a los 10 meses. Existen numerosas evidencias que reportan que el ayuno se encuentra relacionado con una disminución en la actividad de la enzima LPL pero no de la expresión de su ARNm, teniendo la realimentación el efecto contrario (372;422). Esto demuestra la necesidad de evaluar la actividad de estas enzimas y no sólo su expresión génica para poder realizar un análisis más profundo. Nuestros resultados hasta aquí apuntaban a una disminución en los niveles de lipólisis para explicar el aumento de adiposidad, sin una activación de la lipogénesis. Al evaluar la expresión de factores de transcripción involucrados en la lipogénesis de novo en el tejido adiposo, observamos una disminución significativa en la expresión de ARNm tanto de Srebf1 como Chrebp a los 10 y no a los 5 meses de edad en las hembras lacDrd2KO alimentadas ad libitum . En relación a esto, existen resultados contradictorios en la literatura acerca del rol de SREBP-1c (codificado por el gen Srebf1). Muchos autores sugieren un rol de este factor en la diferenciación adipocitaria (423;424), sin embargo el ratón knockout para SREBP-1c tiene cantidades normales de tejido adiposo (425;426); ratones que carecen de ambas isoformas, SREBP-1c y -1a, tienen adipocitos completamente diferenciados, con expresión normal de los marcadores específicos adipocitarios (383); y la deleción de SREBP-1 no incrementa las cantidades de tejido adiposo en el ratón ob/ ob (384;427). Sumado a esto se observó, en un modelo de obesidad inducida por dieta rica en grasas, que los niveles de ARNm de Srebf1 se encontraban disminuidos en tejido adiposo blanco (en forma similar a lo que encontramos nosotros), y más aun que la expresión génica de Srebf1 se encuentra regulada negativamente en adipocitos de pacientes con obesidad mórbida (428). Esto sugiere que la disminución en los niveles de Srebf1 a los 10 meses está asociada a la gran cantidad de adiposidad adquirida a esta edad, lo que no ocurre a los 5 meses. 161 Capítulo IV - Discusión En cuanto a Chrebp , existen evidencias de que una disminución en su expresión causa un descenso en la incorporación de glucosa periférica mediante la disminución en la expresión de los transportadores GLUT-4 en adipocitos (429), esto explicaría, al menos en parte, las alteraciones en el metabolismo de la glucosa observadas en los ratones hembra lacDrd2KO (resultado del Capítulo I I I ). Los niveles de Srebf1 resultaron significativamente incrementados frente a la realimentación posterior a un ayuno en ratones de ambos genotipos, de 5 meses de edad. Se sabe que la realimentación provoca la síntesis de ácidos grasos de novo, incrementando los niveles de expresión de Srebf1 tanto en hígado (430) como en tejido adiposo (431). Sorprendentemente, esta inducción desaparece a los 10 meses, sugiriendo un deterioro en la capacidad de respuesta a mayores edades. Por lo tanto nuestros resultados demostraban que el aumento de la adiposidad del tejido adiposo blanco podría responder a una disminución en la lipólisis, y no a un aumento en la lipogénesis o la síntesis de ácidos grasos de novo. La vía de la síntesis de ácidos grasos de novo, a su vez se vió disminuída con la obesidad marcada de los ratones lacDrd2KO. Dada la poca información sobre la acción de la prolactina en la función hepática intentamos dilucidar los mecanismos lipogénicos y lipolíticos que estaban alterados en nuestro modelo hiperprolactinémico, el cual presentaba un hígado con aumento de contenido lipídico. No encontramos diferencias entre genotipos en los niveles de ARNm de las enzimas Hsl, Atgl y Fas a los 5 o 10 meses edad. A los 10 meses, los niveles de la enzima lipogénica Lpl se encontró significativamente disminuida tanto en hígado como tejido adiposo de hembras lacDrd2KO. Este descenso estaría probablemente asociado a los altos niveles de prolactina, ya que está descripto que prolactina inhibe la actividad de LPL tanto en tejido adiposo blanco de humano, como en hepatocitos de rata (432;433). Por otra parte, una deficiencia en LPL provoca hipertrigliceridemia (434) como se observó en nuestro modelo experimental. Sin embargo estos resultados no explicaban el incremento en la adiposidad en el hígado, y es por ello que evaluamos la expresión de factores lipogénicos regulados por el metabolismo de la glucosa. El hígado presenta un rol central en la homeostasis de la glucosa, y la prolactina sérica podría participar en la misma actuando a este nivel. Los niveles de glucosa circulante son controlados por un sistema regulatorio complejo para abastecer constantemente al metabolismo celular, y el hígado juega un papel fundamental en el mantenimiento de dichos niveles. Durante la etapa de absorción, la glucosa ingerida es captada por los hepatocitos y convertida en glucógeno y lípidos (394). Los factores de transcripción ChREBP y SREBP participan en este proceso en forma dependiente de glucosa e insulina, y observamos que la expresión de ARNm de Chrebp se encontraba significativamente incrementada en las hembras lacDrd2KO de 5 meses con alimento ad libitum , lo que podría explicarar el incremento en los triglicéridos hepáticos. En este sentido, se ha descripto que la sobreexpresión de ChREBP en hígado provoca esteatosis hepática (435), que los niveles de ChREBP se encuentran elevados en ratones obesos (402), y la deleción génica de 162 Capítulo IV - Discusión Chrebp , o la inhibición específica de ChREBP en hígado, disminuye la lipogénesis hepática y la esteatosis en ratones ob/ ob (436;437). Por otra parte, observamos que la expresión de ARNm de Glucokinasa se encontraba incrementada en las hembras lacDrd2KO de 10 meses con libre acceso al alimento. Esto sugeriría una mayor cantidad de glucosa-6-fosfato en los hepatocitos, sin embargo, la concentración de glucógeno hepático en estos ratones resultó significativamente menor respecto de los controles. Es decir que esta glucosa-6-fosfato podría estar siendo utilizada para la síntesis de lípidos hepáticos y no glucógeno. Sin embargo, los niveles de ARNm de Srebf1 y Chrebp resultaron similares en comparación con los ratones Drd2loxP/ loxP. En su conjunto estos resultados nos indican que probablemente la cantidad de glucosa que ingresa al hepatocito sea menor en ratones lacDrd2KO, posiblemente por una menor expresión de GLUT-2, lo que provocaría la regulación positiva de Glucokinasa para contrarrestar este efecto, sin llegar a ser suficiente, y observándose entonces la caída en el contenido de glucógeno. Analizamos la respuesta de estos factores frente al ayuno y posterior realimentación, y observamos que Srebf1 se induce positivamente frente a la realimentación en ratones Drd2loxP/ loxP de 5 meses, y no en los lacDrd2KO. Tal como mencionamos previamente la realimentación provoca la síntesis hepática de ácidos grasos de novo, mediante el incremento de los niveles de ARNm de Srebf1 (430). La falta de respuesta por parte de los ratones lacDrd2KO indica una falla en la regulación de este factor de transcripción. Tal como ocurrió en tejido adiposo, esta inducción observada en los ratones control, desapareció a los 10 meses. En este punto, es importante recordar que la capacidad de secreción de insulina se encuentra disminuida en los animales lacDrd2KO (resultado Capítulo I I I ), por lo que los niveles de insulina que alcanzan los animales transgénicos luego de la realimentación son menores que la de sus pares controles, esto explicaría la falta de respuesta de Srebf1 dado que es un factor de transcripción regulado por esta hormona. Trabajos publicados indican que en el hígado de rata la transcripción de Glucokinasa permanece inactiva en el estado de ayuno y puede ser re-inducida por realimentación con carbohidratos (438). Este patrón de expresión es adaptativamente importante, ya que una baja actividad de Glucokinasa durante el ayuno permite la liberación a la circulación de la glucosa sintetizada por el hígado mediante la gluconeogénesis. De manera inversa, la alta actividad de Glucokinasa en presencia de compuestos carbohidratados permite la incorporación de glucosa por el hígado para su posterior conversión en glucógeno y ácidos grasos. Esta rápida incorporación al tejido hepático permite el decremento de los valores de glucosa en sangre. En los ratones lacDrd2KO y controles observamos esta inducción positiva en la expresión de Glucokinasa inducida por la realimentación, en ambas edades. Esta respuesta también fue menor a los 10 respecto de los 5 meses, a pesar que la inducción de síntesis de glucógeno fue similar a ambas edades. Finalmente demostramos que los glucocorticoides se encuentran conservados en este modelo transgénico, ya que la histología y los pesos de las adrenales, como también la expresión del receptor de glucocorticoides en los distintos tejidos analizados no difirieron entre genotipos. 163 Capítulo IV - Discusión Esto apoya la idea de que las alteraciones observadas en los ratones lacDrd2KO están asociadas a la hiperprolactinemia, y no a la acción de los glucocorticoides. En conclusión demostramos que la hiperprolactinemia crónica aumenta la adiposidad del tejido adiposo blanco y el hígado. En el primer tejido lo hace por diminución de enzimas lipolíticas, y en el hígado por aumento de factores de transcripción de la vía de síntesis de novo de lípidos. 164 Discusión General DISCUSIÓN GENERAL Los circuitos del sistema nervioso central analizan e integran señales periféricas metabólicas, endócrinas y neuronales, para modificar la energía adquirida y el consumo de la misma, de manera de abastecer las demandas energéticas del organismo. Múltiples componentes del sistema neuroendócrino actúan como reguladores de la ingesta y el metabolismo (439;440). En particular, la prolactina sería un factor mediador de la hiperfagia asociada a la preñez y la lactancia (49;335). La presencia de receptores de prolactina en áreas cerebrales asociadas con la regulación del balance energético y la ingesta, así como también su presencia en tejidos adiposo blanco y pardo, hígado y páncreas, apoyan la idea de que la prolactina está involucrada en el balance energético, actuando a diferentes niveles (34). A nivel del cerebro, los receptores de prolactina se han localizado en el cuerpo estriado, así como también en numerosos núcleos hipotalámicos asociados a la ingesta y metabolismo, incluyendo el núcleo Arc, el hipotálamo VMN, el núcleo hipotalámico PVN, y el hipotálamo DMN. Adicionalmente, la administración de prolactina intracerebroventricular incrementa la ingesta en ratas (441); y la ausencia de receptores de prolactina en esta misma especie, se ve acompañada por una reducción progresiva del peso corporal (56). El neurotransmisor dopamina es también un mediador fisiológicamente relevante del comportamiento alimenticio (132). Se ha demostrado que un incremento en la señalización de dopamina promueve la ingesta, mientras que un descenso tiene el efecto contrario (419). Por otra parte, las hormonas implicadas en la regulación del sistema homeostático actúan sobre las neuronas dopaminérgicas; por ejemplo, leptina e insulina directamente inhiben las neuronas de dopamina (132). Estudios sobre la función dopaminérgica han implicado a los caminos dopaminérgicos nigroestriados en la alimentación (442), mientras que las vías dopaminérgicas mesolímbicas (principalmente las neuronas del área ventral tegmental que proyectan hacia el núcleo accumbens) parecen estar involucradas en los aspectos de mayor jerarquía, como ser la motivación y la recompensa (132;443). Los ratones que poseen selectivamente inactivado el gen de la tirosina hidroxilasa, la enzima limitante en la biosíntesis de dopamina, resultan hipofágicos y mueren de inanición a las 3-4 semanas de vida (444). Sumado a esto, se ha descripto que el déficit en la densidad de RD2s en el núcleo estriado aceleran el comportamiento de búsqueda compulsiva de alimentos, en ratas con acceso ilimitado a comida rica en grasas (62), y que podría contribuir a la hipofunción de recompensa en individuos obesos. Estos datos revelan la compleja participación de los circuitos dopaminérgicos en la adquisición de alimentos. Hemos demostrado previamente en el laboratorio que la hiperprolactinemia crónica en ratones hembra Drd2-/ - no se encuentra acompañada de un incremento en el peso corporal, ni en los niveles de ingesta (68). En el presente trabajo utilizamos ratones knockout que carecen de RD2s selectivamente en lactotropos, para así poder disecar los efectos solapados generados por la falta de RD2s a nivel central, en la ingesta de alimentos. Demostramos que los ratones hembra 165 Discusión General con disrupción selectiva de RD2s además de presentar una hiperprolactinemia sostenida, sí poseen una ingesta y adiposidad incrementada, como también alteraciones en la homeostasis de la glucosa. Esto estaría sugiriendo que el efecto hiperfágico de la prolactina puede ser más claramente evidenciado cuando la acción de la dopamina central, vía los RD2s, se encuentra preservada. Demostramos una hiperprolactinemia sostenida con un eje de GH conservado en los ratones lacDrd2KO validando que este mutante tejido específico carece completamente de RD2s funcionales en los lactotropos hipofisarios, mientras que mantiene normales los RD2s del sistema nervioso central. Como demostráramos previamente la ausencia de control dopaminérgico sobre los lactotropos del lóbulo anterior de la hipófisis, promueve la formación de prolactinomas y su consiguiente hiperprolactinemia, resultando en un buen modelo para la búsqueda de terapias alternativas (modelo de raton Drd2-/ - ) (204). Consistente con esto, las hipófisis de ratones hembra lacDrd2KO resultaron más pesadas, y la concentración de prolactina intrahipofisaria se encontró incrementada, indicando, ambos eventos, hiperplasia de lactotropos. En los machos en cambio, no se observaron diferencias en los niveles séricos de prolactina, como tampoco en los pesos hipofisarios. Es evidente que la falta de tono dopaminérgico es el principal evento que induce la hiperplasia de lactotropos en los ratones hembra lacDrd2KO. Sin embargo esto resulta aparentemente insuficiente como único inductor ya que los machos lacDrd2KO no generan hiperplasia hipofisaria. Se encuentra ampliamente descripto en la literatura que el tono dopaminérgico sobre los lactotropos es mayor en las hembras que en los machos (30;263;264), lo que explica el dimorfismo sexual evidenciado en los ratones lacDrd2KO. La caracterización de dichos prolactinomas demostró un incremento en los niveles de proliferación y un incremento en factores relacionados con el proceso de angiogénesis, avalando la hipótesis del laboratorio de que una terapia alternativa o adicional para los prolactinomas resistentes a agonista dopaminérgicos sería la anti-angiogénesis (210). Por otro lado no encontramos diferencias notables en la expresión de marcadores específicos de células madre/ progenitoras en este modelo. En la actualidad poco se sabe del rol de las células madre en el desarrollo de tumores hipofisarios. Las hipófisis tumorales de ratones lacDrd2KO presentaron una tendencia a la disminución en los factores involucrados en la vía de Notch (dos de sus receptores, Notch-1 y Notch-3 y el ligando Hes-1) en comparación con hipófisis control, como también en la activación del factor Sox-2. Estos resultados sugieren que el aumento en los niveles proliferativos en las hipófisis hiperplásicas de lacDrd2KO no es a expensas de las células con fenotipo de células madre/ progenitoras, o que las mismas han participado en estadíos más tempranos de la génesis del prolactinoma. Esto constituye un novedoso aporte para la caracterización de prolactinomas experimentales. A continuación estudiamos el fenotipo de control de la ingesta y el metabolismo en este modelo único, en el cual se desarrolla una hiperprolactinemia gradual, crónica y robusta a partir de 166 Discusión General los 5 meses de edad, con mantemiento de la función de los RD2s en el resto del organismo. En ratones hembra lacDrd2KO la ingesta estuvo aumentada a los 3 y 5 meses de edad, y un aumento en peso corporal se manifiesta y acentúa a partir de los 5 meses en comparación con sus pares control, llegando a un 24% de aumento a los 11 meses. Los niveles de progesterona, hormona que podría estimular la ingesta (445), estuvieron mínimamente aumentados en este modelo transgénico. Los transgénicos no evidenciaban diferencias en la longitud corporal, pero sí un incremento marcado en los depósitos de grasa. Contrariamente, describimos previamente que las hembras adultas Drd2-/ - no presentaban diferencias en el peso corporal o ingesta respecto de sus pares WT (60;68), como tampoco un incremento en la adiposidad. Dado que ambas líneas de ratones transgénicos poseen hiperprolactinemia crónica, las diferencias observadas en la ingesta y adiposidad deben estar relacionadas con la participación de los RD2s centrales, los cuales son funcionales únicamente en el modelo lacDrd2KO. En este sentido, nuestros resultados revelan que la expresión de neuropéptidos involucrados en la ingesta y regulados por prolactina o dopamina, están selectivamente modificados en los ratones lacDrd2KO. NPY, es un neuropéptido orexigénico potente, que se expresa en el Arc y DMN. En este último núcleo colocalizan NPY y PRLR, y tanto la succión (293) como la prolactina (446) activarían la expresión génica de Npy en estos sitios. En concordancia, observamos un incremento en los niveles de ARNm de Npy y Prlr en los hipotálamos de hembras lacDrd2KO, que concuerdan con los niveles de prolactina e ingesta aumentados. Por otro lado, la expresión hipotalámica de Ppo el precursor de orexinas A y B (294), importante factor que regula positivamente el comportamiento alimenticio, además del sueño, no estaba alterada en contraposición con la disminución presente en los ratones globales Drd2-/ - (60). En relación a dos péptidos anorexigénicos como el POMC (286;447) y α-MSH no hallamos diferencias en su expresión en el modelo de lacDrd2KO, a diferencia del aumento que describimos en el Drd2-/ - . En este sentido debemos destacar que la síntesis y secreción de α-MSH en la hipófisis intermedia se encuentra constitutivamente inhibida por los RD2s (288), y es por ello que su secreción está aumentada en el modelo Drd2 -/ - y no en el lacDrd2KO. En su conjunto, estos resultados sugieren que la señalización a través de los RD2s centrales en los ratones lacDrd2KO, mantiene los niveles de expresión génica de Pomc (y su derivado αMSH) y Ppo, y permite evidenciar el efecto orexigénico de la prolactina a nivel central, incluyendo su acción sobre la expresión de Npy y su propio receptor hipotalámico (Figura 54). En el ratón knockout global, la pérdida de los RD2s en el sistema nervioso central provoca el descenso en los niveles de orexinas junto con el incremento de α-MSH, estos dos eventos anorexigénicos estarían opacando el efecto de la prolactina, sobre la ingesta de alimentos. Asimismo, nuestros resultados sugieren que podría haber una resistencia a leptina en este modelo hiperprolactinémico, similar al reportado en los estados de preñez y lactancia para abastecer las demandas metabólicas del feto y cría (448;449). Un aumento en la masa de tejido graso está asociado a elevados niveles de leptina, la cual incrementa los niveles hipotalámicos de 167 Discusión General POMC y disminuye los de NPY (448;450) para suprimir la ingesta. Por lo tanto, la ausencia de cambios en la expresión de Pomc y el incremento observado en los niveles de ARNm de Npy , podrían estar relacionados con los altos niveles de prolactina y ser evidencias de una disminución en la sensibilidad a leptina. En este sentido, en los ratones lacDrd2KO encontramos un incremento significativo en los niveles de leptina sérica, que correlacionó positivamente con el peso corporal de los mismos. El aumento de ingesta correlacionó con una marcada acumulación de adiposidad en los ratones hembra lacDrd2KO. Se ha reportado que la prolactina estimula los depósitos de grasa en ratas y palomas hembra (34), y que la hiperprolactinemia en hombres y mujeres no embarazadas podría estar acompañada de una ganancia de peso (451). Asimismo, en el ratón knockout para el receptor de prolactina se observó una disminución en el peso corporal asociada con una reducción de la grasa abdominal total, y receptores de prolactina han sido descriptos en adipocitos (30). Nuestros resultados avalaban un nuevo rol metabólico de la prolactina que comienza a estudiarse. Un dato interesante en el estudio de los PRLR fue su regulación dual en tejido adiposo blanco en los ratones lacDrd2KO: a los 5 meses de edad la expresión de la isoforma larga Prlr-l resultó incrementada en ratones lacDrd2KO, mientras que a los 10 meses el resultado fue el opuesto. Postulamos que el incremento en los niveles de Prlr-l a los 5 meses de edad estaría asociado a la regulación positiva de este receptor por parte de prolactina, mientras que cuando los niveles de esta hormona se vuelven muy por encima de los fisiológicos existe una regulación negativa, posiblemente como mecanismo compensatorio. Brandebourg y colaboradores demostraron que prolactina regula positivamente a su receptor en adipocitos provenientes de ratas macho (420). Sin embargo, también hacen mención a que concentraciones supra-fisiológicas de esta hormona pueden causar la regulación negativa de su propio receptor, obstaculizar la dimerización del mismo, o inducir la activación de proteínas supresoras (420). En este sentido, en los ratones lacDrd2KO a los 10 meses, los niveles de prolactina resultan cinco veces mayores que sus controles. Por otro lado en este tejido preponderó la isoforma Prlr-l sobre las isoformas cortas que resultaron debajo del límite de detección. Más allá de la hiperprolactinemia sostenida en ambos modelos de ratones knockouts para los RD2s, se ha demostrado que niveles elevados de α-MSH en suero disminuyen la adiposidad (452;453), y por lo tanto en el ratón knockout global, los niveles incrementados de α-MSH séricos estarían contrarrestando los efectos adipogénicos de prolactina. Por otro lado, el eje de GHRH-GH que está disminuido en el modelo Drd2-/ - (68) y no en el lacDrd2KO, podría incrementar la sensibilidad a insulina lo cual previene la adiposidad en el primer modelo, en oposición a lo que ocurre en el segundo. Una adiposidad incrementada se encuentra relacionada a insulino-resistencia (454). Los ratones lacDrd2KO presentaron intolerancia a la glucosa, y una menor secreción de insulina en respuesta a un estímulo con glucosa, pero no se observó una resistencia a insulina a través del I TT (Figura 54). Esto podría estar indicando una secreción defectuosa de insulina a nivel de las células 168 Discusión General beta pancreáticas. En particular, destacamos que el contenido de insulina pancréatica estaba aumentado en el mutante, a pesar de la falla de su respuesta a glucosa. Una intolerancia a la glucosa se observa en ratas hiperprolactinémicas a causa de transplantes de hipófisis (63), pero también en el ratón deficiente de receptores de prolactina (370), lo que demuestra la compleja y enigmática participación de prolactina en el metabolismo de los carbohidratos. I nteresantemente, en el tejido adiposo de hembras lacDrd2KO observamos un incremento en el porcentaje de adipocitos de mayor tamaño. Células adiposas de gran tamaño han sido asociadas a un mayor riesgo de diabetes tipo I I (455), lo que correlaciona con la pérdida de funcionalidad de las células beta pancreáticas en estos ratones transgénicos. El aumento en la reserva de lípidos observada podría ser consecuencia de la menor expresión de enzimas lipolíticas observada en tejidos adiposos de ratones hembra lacDrd2KO, mientras que el inesperado descenso en los niveles de ARNm de Lpl tanto en tejido adiposo como hígado estaría probablemente asociado a los altos niveles de prolactina. Prolactina inhibe la actividad de LPL tanto en tejido adiposo blanco de humano, como en hepatocitos de rata (432;433), sumado a esto, una deficiencia en LPL provoca hipertrigliceridemia (434) como se observó en nuestro modelo experimental. Encontramos además en los ratones lacDrd2KO de 5 meses una inducción positiva de la expresión de la enzima lipogénica Fas con la realimentación, lo que podría explicar la mayor ganancia de peso observada a esta edad con la realimentación. En relación a la expresión de factores de transcripción involucrados en la lipogénesis de novo en el tejido adiposo, y regulados por glucosa e insulina, encontramos a los 10 meses de edad la expresión de ARNm tanto de Srebf1 como Chrebp se encontraban significativamente disminuidas en las hembras lacDrd2KO alimentadas ad libitum . Este resultado no explicaría entonces el aumento de adiposidad, pero se asocia a resultados que determinan que la expresión génica de Srebf1 se encuentra regulada negativamente en adipocitos de pacientes con obesidad mórbida (428), y que una disminución en la expresión de Chrebp causa un descenso en la incorporación de glucosa periférica en adipocitos (429), explicando, al menos en parte, la intolerancia a la glucosa observada en los ratones hembra lacDrd2KO. Por otro lado la adiposidad en los hígados de ratones hembra lacDrd2KO también estaba aumentada, sin diferencias en la expresión de enzimas lipogénicas y lipolíticas, y con un aumento en el factor Chrebp y los niveles del PRLR (Figura 54). Se ha descripto que la sobreexpresión de ChREBP en hígado provoca esteatosis hepática (435), los niveles de ChREBP se encuentran elevados en ratones obesos (402), y la deleción génica de Chrebp , o la inhibición específica de ChREBP en hígado, disminuye la lipogénesis hepática y la esteatosis en ratones ob/ ob (436;437). Todos estos datos indican que la prolactina podría tener como blanco a este factor en el hígado, efecto mediado quizás por su receptor y potenciado por la intolerancia a la glucosa del modelo. 169 Discusión General Figura 54: Resumen de los resultados obtenidos en los ratones hembra lacDrd2KO en los distintos tejidos evaluados. Por otro lado, la regulación de genes por realimentación, en hígado y tejido adiposo, se desensibiliza con la edad, y está alterada en ratones transgénicos, probablemente a causa de la falla en la liberación de insulina que presenta este modelo. Finalmente demostramos que los glucocorticoides se encuentran conservados en este modelo transgénico, lo que apoya la idea de que las alteraciones observadas en cuanto a la ingesta de alimentos, peso corporal, incremento en la adiposidad e intolerancia a la glucosa están asociadas a la hiperprolactinemia, y no a la acción de los glucocorticoides. Como conclusión, este trabajo revela la importancia del rol de la prolactina en la ingesta de alimentos, y la acumulación de adiposidad, lo que resulta fundamental en las adaptaciones metabólicas necesarias en los estados de preñez y lactancia. Asimismo, nuestro estudio ilustra el valor de utilizar modelos de ratones mutantes tejido específico, para poder discernir mecanismos fisiológicos y patofisiológicos, de otra manera enmascarados en los mutantes nulos totales o en los animales tratados con agentes farmacológicos. 170 Bibliografía BIBLIOGRAFÍA 1. Norris DO 2007 Vertebrate Endocrinology.Elsevier Academic Press, 4th Ed. edn 2. Pocock G, Richards CD 2005 Fisiología Humana: La base de la medicina.Masson, 2nd Ed. edn 3. Vianna CR, Coppari R . A treasure trove of hypothalamic neurocircuitries governing body weight homeostasis. Endocrinology 2011; 152:11-18. 4. Palkovits M . Interconnections between the neuroendocrine hypothalamus and the central autonomic system. Geoffrey Harris Memorial Lecture, Kitakyushu, Japan, October 1998. Front Neuroendocrinol 1999; 20:270-295. 5. Conn PM, Freeman ME 1999 The Neuroendocrinology in Physiology and Medicine.The Humana Press, New Jersey 6. Halasz B 2000 The Hypothalamus as an Endocrine Organ: The Science of Neuroendocrinology. In: Conn M, Freeman ME (eds). Neuroendocrinology in Physiology and Medicine.Humana Press, Totowa, New Jersey:3-21 7. Lopez M, Tena-Sempere M, Dieguez C . Cross-talk between orexins (hypocretins) and the neuroendocrine axes (hypothalamic-pituitary axes). Front Neuroendocrinol 2010; 31:113127. 8. Fink G 2000 Neuroendocrine Regulation of Pituitary Function: General Principles. In: Conn PM, Freeman ME (eds). Neuroendocrinology in Physiology and Medicine.Humana Press, Totowa, New Jersey:107-133 9. Setalo G, Flerko B, Arimura A, Schally AV . Brain cells as producers of releasing and inhibiting hormones. Int Rev Cytol Suppl 1978;1-52. 10. Nutrition Classics. The Anatomical Record, Volume 78, 1940: Hypothalamic lesions and adiposity in the rat . Nutr Rev 1983; 41:124-127. 11. García-Tornadú I, Luque GM, Perez-Millan MI, Ramirez MC, Recouvreux MV, Ornstein A, Diaz-Torga GS, Becu-Villalobos D 2012 Dopaminergic regulation of food intake. Insights obtained from the dopamine receptor D2 knockout mouse. In: Morrison J (ed). Food Intake: Regulation, Assessing and Controlling.Nova Science Publishers, NY:59-82 12. Sanchez-Lasheras C, Konner AC, Bruning JC . Integrative neurobiology of energy homeostasis-neurocircuits, signals and mediators. Front Neuroendocrinol 2010; 31:4-15. 13. Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD . Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 1997; 385:165-168. 14. Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS . Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science 1997; 278:135-138. 15. Haskell-Luevano C, Monck EK . Agouti-related protein functions as an inverse agonist at a constitutively active brain melanocortin-4 receptor. Regul Pept 2001; 99:1-7. 16. Beck B . Neuropeptide Y in normal eating and in genetic and dietary-induced obesity. Philos Trans R Soc Lond B Biol Sci 2006; 361:1159-1185. 17. Roseberry AG, Liu H, Jackson AC, Cai X, Friedman JM . Neuropeptide Y-mediated inhibition of proopiomelanocortin neurons in the arcuate nucleus shows enhanced desensitization in ob/ob mice. Neuron 2004; 41:711-722. 171 Bibliografía 18. Cyr NE, Toorie AM, Steger JS, Sochat MM, Hyner S, Perello M, Stuart R, Nillni EA . Mechanisms by which the orexigen NPY regulates anorexigenic alpha-MSH and TRH. Am J Physiol Endocrinol Metab 2013; 304:E640-E650. 19. Benoit SC, Tracy AL, Davis JF, Choi D, Clegg DJ . Novel functions of orexigenic hypothalamic peptides: from genes to behavior. Nutrition 2008; 24:843-847. 20. Trayhurn P, Bing C . Appetite and energy balance signals from adipocytes. Philos Trans R Soc Lond B Biol Sci 2006; 361:1237-1249. 21. Galic S, Oakhill JS, Steinberg GR . Adipose tissue as an endocrine organ. Mol Cell Endocrinol 2010; 316:129-139. 22. KENNEDY GC . The role of depot fat in the hypothalamic control of food intake in the rat. Proc R Soc Lond B Biol Sci 1953; 140:578-596. 23. Coleman DL . Effects of parabiosis of obese with diabetes and normal mice. Diabetologia 1973; 9:294-298. 24. Breton C . The hypothalamus-adipose axis is a key target of developmental programming by maternal nutritional manipulation. J Endocrinol 2013; 216:R19-R31. 25. Poulos SP, Hausman DB, Hausman GJ . The development and endocrine functions of adipose tissue. Mol Cell Endocrinol 2010; 323:20-34. 26. Zhu X, Gleiberman AS, Rosenfeld MG . Molecular physiology of pituitary development: signaling and transcriptional networks. Physiol Rev 2007; 87:933-963. 27. Quereda V, Malumbres M . Cell cycle control of pituitary development and disease. J Mol Endocrinol 2009; 42:75-86. 28. Freeman ME, Kanyicska B, Lerant A, Nagy G . Prolactin: structure, function, and regulation of secretion. Physiol Rev 2000; 80:1523-1631. 29. Ben Jonathan N, Mershon JL, Allen DL, Steinmetz RW . Extrapituitary prolactin: distribution, regulation, functions, and clinical aspects. Endocr Rev 1996; 17:639-669. 30. Ben Jonathan N, LaPensee CR, LaPensee EW . What can we learn from rodents about prolactin in humans? Endocr Rev 2008; 29:1-41. 31. Lamberts SWJ, MacLeod RM . Regulation of prolactin secretion at the level of the lactotroph. Physiol Rev 1990; 70:279-318. 32. Melmed S . Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest 2003; 112:1603-1618. 33. Bole-Feysot C, Goffin V, Edery M, Binart N, Kelly PA . Prolactin (PRL) and its receptor:Actions, signal transduction pathways and phenotypes observed in PRL receptor knockout mice. Endocr Rev 1998; 19:225-268. 34. Ben Jonathan N, Hugo ER, Brandebourg TD, LaPensee CR . Focus on prolactin as a metabolic hormone. Trends Endocrinol Metab 2006; 17:110-116. 35. Tovar S, Dieguez C . Prolactin and energy homeostasis: pathophysiological mechanisms and therapeutic considerations. Endocrinology 2014; 155:659-662. 36. Nagano M, Kelly PA . Tissue distribution and regulation of rat prolactin receptor gene expression. Quantitative analysis by polymerase chain reaction. J Biol Chem 1994; 269:13337-13345. 37. Brooks CL . Molecular mechanisms of prolactin and its receptor. Endocr Rev 2012; 33:504525. 172 Bibliografía 38. Ormandy CJ, Binart N, Helloco C, Kelly PA . Mouse prolactin receptor gene: genomic organization reveals alternative promoter usage and generation of isoforms via alternative 3'-exon splicing. DNA Cell Biol 1998; 17:761-770. 39. Ling C, Hellgren G, Gebre-Medhin M, Dillner K, Wennbo H, Carlsson B, Billig H . Prolactin (PRL) receptor gene expression in mouse adipose tissue: increases during lactation and in PRL-transgenic mice. Endocrinology 2000; 141:3564-3572. 40. Corbacho AM, Valacchi G, Kubala L, Olano-Martin E, Schock BC, Kenny TP, Cross CE . Tissue-specific gene expression of prolactin receptor in the acute-phase response induced by lipopolysaccharides. Am J Physiol Endocrinol Metab 2004; 287:E750-E757. 41. Davis JA, Linzer DI . Expression of multiple forms of the prolactin receptor in mouse liver. Mol Endocrinol 1989; 3:674-680. 42. Buck K, Vanek M, Groner B, Ball RK . Multiple forms of prolactin receptor messenger ribonucleic acid are specifically expressed and regulated in murine tissues and the mammary cell line HC11. Endocrinology 1992; 130:1108-1114. 43. Frasor J, Barkai U, Zhong L, Fazleabas AT, Gibori G . PRL-induced ERalpha gene expression is mediated by Janus kinase 2 (Jak2) while signal transducer and activator of transcription 5b (Stat5b) phosphorylation involves Jak2 and a second tyrosine kinase. Mol Endocrinol 2001; 15:1941-1952. 44. Fresno Vara JA, Caceres MA, Silva A, Martin-Perez J . Src family kinases are required for prolactin induction of cell proliferation. Mol Biol Cell 2001; 12:2171-2183. 45. Tessier C, Prigent-Tessier A, Ferguson-Gottschall S, Gu Y, Gibori G . PRL antiapoptotic effect in the rat decidua involves the PI3K/protein kinase B-mediated inhibition of caspase-3 activity. Endocrinology 2001; 142:4086-4094. 46. Berlanga JJ, Garcia-Ruiz JP, Perrot-Applanat M, Kelly PA, Edery M . The short form of the prolactin (PRL) receptor silences PRL induction of the beta-casein gene promoter. Mol Endocrinol 1997; 11:1449-1457. 47. Perrot-Applanat M, Gualillo O, Pezet A, Vincent V, Edery M, Kelly PA . Dominant negative and cooperative effects of mutant forms of prolactin receptor. Mol Endocrinol 1997; 11:1020-1032. 48. Devi YS, Shehu A, Halperin J, Stocco C, Le J, Seibold AM, Gibori G . Prolactin signaling through the short isoform of the mouse prolactin receptor regulates DNA binding of specific transcription factors, often with opposite effects in different reproductive issues. Reprod Biol Endocrinol 2009; 7:87. 49. Woodside B . Prolactin and the hyperphagia of lactation. Physiol Behav 2007; 91:375-382. 50. Grattan DR . The actions of prolactin in the brain during pregnancy and lactation. Prog Brain Res 2001; 133:153-171. 51. Baptista T, de Baptista EA, Lalonde J, Plamondon J, Kin NM, Beaulieu S, Joober R, Richard D . Comparative effects of the antipsychotics sulpiride and risperidone in female rats on energy balance, body composition, fat morphology and macronutrient selection. Prog Neuropsychopharmacol Biol Psychiatry 2004; 28:1305-1311. 52. Knudtzon J, Johansen PW, Haug E, Gautvik K . Effects of hypersecretion of growth hormone and prolactin on plasma levels of glucagon and insulin in GH3-cell-tumorbearing rats, and the influence of bromocriptine treatment. Life Sci 1986; 39:617-621. 173 Bibliografía 53. Byatt JC, Staten NR, Salsgiver WJ, Kostelc JG, Collier RJ . Stimulation of food intake and weight gain in mature female rats by bovine prolactin and bovine growth hormone. Am J Physiol 1993; 264:E986-E992. 54. Moore BJ, Gerardo-Gettens T, Horwitz BA, Stern JS . Hyperprolactinemia stimulates food intake in the female rat. Brain Res Bull 1986; 17:563-569. 55. Sauve D, Woodside B . Neuroanatomical specificity of prolactin-induced hyperphagia in virgin female rats. Brain Res 2000; 868:306-314. 56. Freemark M, Fleenor D, Driscoll P, Binart N, Kelly PA . Body weight and fat deposition in prolactin receptor-deficient mice. Endocrinology 2001; 142:532-537. 57. Matsuda M, Mori T, Sassa S, Sakamoto S, Park MK, Kawashima S . Chronic effect of hyperprolactinemia on blood glucose and lipid levels in mice. Life Sci 1996; 58:1171-1177. 58. Kelly MA, Rubinstein M, Asa SL, Zhang G, Saez C, Bunzow JR, Allen RG, Hnasko R, BenJonathan N, Grandy DK, Low MJ . Pituitary lactotroph hyperplasia and chronic hyperprolactinemia in dopamine D2 receptor-deficient mice. Neuron 1997; 19:103-113. 59. Cristina C, Diaz-Torga G, Baldi A, Gongora A, Rubinstein M, Low MJ, Becu-Villalobos D . Increased pituitary vascular endothelial growth factor-A in dopaminergic D2 receptor knockout female mice. Endocrinology 2005; 146:2952-2962. 60. Garcia-Tornadu I, Diaz-Torga GS, Risso G, Silveyra P, Cataldi N, Ramirez MC, Low MJ, Libertun C, Becu-Villalobos D . Hypothalamic orexin, OX1, aMSH, NPY and MCRs expression in dopaminergic D2R knockout mice. Neuropeptides 2009; 43:267-274. 61. Bello EP, Mateo Y, Gelman DM, Noain D, Shin JH, Low MJ, Alvarez VA, Lovinger DM, Rubinstein M . Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci 2011; 14:1033-1038. 62. Johnson PM, Kenny PJ . Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci 2010; 13:635-641. 63. Reis FM, Reis AM, Coimbra CC . Effects of hyperprolactinaemia on glucose tolerance and insulin release in male and female rats. J Endocrinol 1997; 153:423-428. 64. Tuzcu A, Bahceci M, Dursun M, Turgut C, Bahceci S . Insulin sensitivity and hyperprolactinemia. J Endocrinol Invest 2003; 26:341-346. 65. Schernthaner G, Prager R, Punzengruber C, Luger A . Severe hyperprolactinaemia is associated with decreased insulin binding in vitro and insulin resistance in vivo. Diabetologia 1985; 28:138-142. 66. Goffin V, Shiverick KT, Kelly PA, Martial JA . Sequence-function relationships within the expanding family of prolactin, growth hormone, placental lactogen, and related proteins in mammals. Endocr Rev 1996; 17:385-410. 67. Lewis UJ . Growth Hormone What is it and what does it do? Trends Endocrinol Metab 1992; 3:117-121. 68. Diaz-Torga G, Feierstein C, Libertun C, Gelman D, Kelly MA, Low MJ, Rubinstein M, BecuVillalobos D . Disruption of the D2 dopamine receptor alters GH and IGF-I secretion and causes dwarfism in male mice. Endocrinology 2002; 143:1270-1279. 69. Flavell DM, Wells T, Wells SE, Carmignac DF, Thomas GB, Robinson IC . Dominant dwarfism in transgenic rats by targeting human growth hormone (GH) expression to hypothalamic GH-releasing factor neurons. EMBO J 1996; 15:3871-3879. 174 Bibliografía 70. Moore JP, Jr., Cai A, Hostettler ME, Arbogast LA, Voogt JL, Hyde JF . Pituitary hormone gene expression and secretion in human growth hormone-releasing hormone transgenic mice: focus on lactotroph function. Endocrinology 2000; 141:81-90. 71. Alba M, Salvatori R . A mouse with targeted ablation of the growth hormone-releasing hormone gene: a new model of isolated growth hormone deficiency. Endocrinology 2004; 145:4134-4143. 72. Low MJ, Otero-Corchon V, Parlow AF, Ramirez JL, Kumar U, Patel YC, Rubinstein M . Somatostatin is required for masculinization of growth hormone-regulated hepatic gene expression but not of somatic growth. J Clin Invest 2001; 107:1571-1580. 73. Luque RM, Kineman RD . Gender-dependent role of endogenous somatostatin in regulating growth hormone-axis function in mice. Endocrinology 2007; 148:5998-6006. 74. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H . Ghrelin is a growth hormone releasing acylated peptide from stomach. Nature 1999; 402:656-660. 75. Kirkhus B, Clausen OP . Cell kinetics in mouse epidermis studied by bivariate DNA/bromodeoxyuridine and DNA/keratin flow cytometry. Cytometry 1990; 11:253-260. 76. Herington AC, Cornell HJ, Kuffer AD . Recent advances in the biochemistry and physiology of the insulin-like growth factor/somatomedin family. Int J Biochem 1983; 15:1201-1210. 77. Hiney JK, Srivastava V, Nyberg CL, Ojeda SR, Wees WL . Insulin-like growth factor I of peripheral origin acts centrally to accelerate the initiation of female puberty. Endocrinology 1996; 137:3717-3728. 78. SZEGO CM, WHITE A . The influence of purified growth hormone on fasting metabolism. J Clin Endocrinol Metab 1948; 8:594. 79. Vijayakumar A, Novosyadlyy R, Wu Y, Yakar S, LeRoith D . Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm IGF Res 2010; 20:1-7. 80. Houssay BA, RODRIGUEZ RR . Diabetogenic action of different preparations of growth hormone. Endocrinology 1953; 53:114-116. 81. Houssay BA, RODRIGUEZ RR, CARDEZA AF . [Diabetogenic action of the pituitary growth hormone]. C R Seances Soc Biol Fil 1954; 148:910-911. 82. Hill DJ, Hogg J . Growth factor control of pancreatic B cell hyperplasia. Baillieres Clin Endocrinol Metab 1991; 5:689-698. 83. Dominici FP, Hauck S, Argentino DP, Bartke A, Turyn D . Increased insulin sensitivity and upregulation of insulin receptor, insulin receptor substrate (IRS)-1 and IRS-2 in liver of Ames dwarf mice. J Endocrinol 2002; 173:81-94. 84. Liu JL, Coschigano KT, Robertson K, Lipsett M, Guo Y, Kopchick JJ, Kumar U, Liu YL . Disruption of growth hormone receptor gene causes diminished pancreatic islet size and increased insulin sensitivity in mice. Am J Physiol Endocrinol Metab 2004; 287:E405-E413. 85. Merimee TJ, Felig P, Marliss E, Fineberg SE, Cahill GG, Jr. Glucose and lipid homeostasis in the absence of human growth hormone. J Clin Invest 1971; 50:574-582. 86. Jorgensen JO, Vestergaard E, Gormsen L, Jessen N, Norrelund H, Christiansen JS, Moller N . Metabolic consequences of GH deficiency. J Endocrinol Invest 2005; 28:47-51. 87. Yamauchi T, Kaburagi Y, Ueki K, Tsuji Y, Stark GR, Kerr IM, Tsushima T, Akanuma Y, Komuro I, Tobe K, Yazaki Y, Kadowaki T . Growth hormone and prolactin stimulate tyrosine phosphorylation of insulin receptor substrate-1, -2, and -3, their association with 175 Bibliografía p85 phosphatidylinositol 3-kinase (PI3-kinase), and concomitantly PI3-kinase activation via JAK2 kinase. J Biol Chem 1998; 273:15719-15726. 88. Thirone AC, Carvalho CR, Saad MJ . Growth hormone stimulates the tyrosine kinase activity of JAK2 and induces tyrosine phosphorylation of insulin receptor substrates and Shc in rat tissues. Endocrinology 1999; 140:55-62. 89. Nielsen JH, Linde S, Welinder BS, Billestrup N, Madsen OD . Growth hormone is a growth factor for the differentiated pancreatic beta-cell. Mol Endocrinol 1989; 3:165-173. 90. Hugl SR, White MF, Rhodes CJ . Insulin-like growth factor I (IGF-I)-stimulated pancreatic beta-cell growth is glucose-dependent. Synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS-1 cells. J Biol Chem 1998; 273:17771-17779. 91. Harrison M, Dunger AM, Berg S, Mabley J, John N, Green MH, Green IC . Growth factor protection against cytokine-induced apoptosis in neonatal rat islets of Langerhans: role of Fas. FEBS Lett 1998; 435:207-210. 92. Guo Y, Lu Y, Houle D, Robertson K, Tang Z, Kopchick JJ, Liu YL, Liu JL . Pancreatic isletspecific expression of an insulin-like growth factor-I transgene compensates islet cell growth in growth hormone receptor gene-deficient mice. Endocrinology 2005; 146:26022609. 93. Berryman DE, List EO, Coschigano KT, Behar K, Kim JK, Kopchick JJ . Comparing adiposity profiles in three mouse models with altered GH signaling. Growth Horm IGF Res 2004; 14:309-318. 94. Pasarica M, Zachwieja JJ, Dejonge L, Redman S, Smith SR . Effect of growth hormone on body composition and visceral adiposity in middle-aged men with visceral obesity. J Clin Endocrinol Metab 2007; 92:4265-4270. 95. Berryman DE, List EO, Kohn DT, Coschigano KT, Seeley RJ, Kopchick JJ . Effect of growth hormone on susceptibility to diet-induced obesity. Endocrinology 2006; 147:2801-2808. 96. Collipp PJ, Thomas J, Curti V, Sharma RK, Maddaiah VT, Cohn SE . Body composition changes in children receiving human growth hormone. Metabolism 1973; 22:589-595. 97. Parra A, Argote RM, Garcia G, Cervantes C, Alatorre S, Perez-Pasten E . Body composition in hypopituitary dwarfs before and during human growth hormone therapy. Metabolism 1979; 28:851-857. 98. Hoffman AR, Kuntze JE, Baptista J, Baum HB, Baumann GP, Biller BM, Clark RV, Cook D, Inzucchi SE, Kleinberg D, Klibanski A, Phillips LS, Ridgway EC, Robbins RJ, Schlechte J, Sharma M, Thorner MO, Vance ML . Growth hormone (GH) replacement therapy in adultonset gh deficiency: effects on body composition in men and women in a double-blind, randomized, placebo-controlled trial. J Clin Endocrinol Metab 2004; 89:2048-2056. 99. Fleenor D, Arumugam R, Freemark M . Growth hormone and prolactin receptors in adipogenesis: STAT-5 activation, suppressors of cytokine signaling, and regulation of insulin-like growth factor I. Horm Res 2006; 66:101-110. 100. Farrell WE . Pituitary tumours: findings from whole genome analyses. Endocr Relat Cancer 2006; 13:707-716. 101. Seilicovich A, Pisera D, Sciascia SA, Candolfi M, Puntel M, Xiong W, Jaita G, Castro MG . Gene therapy for pituitary tumors. Curr Gene Ther 2005; 5:559-572. 102. Osamura RY, Kajiya H, Takei M, Egashira N, Tobita M, Takekoshi S, Teramoto A . Pathology of the human pituitary adenomas. Histochem Cell Biol 2008; 130:495-507. 176 Bibliografía 103. Salamone JD, Correa M, Mingote SM, Weber SM . Beyond the reward hypothesis: alternative functions of nucleus accumbens dopamine. Curr Opin Pharmacol 2005; 5:3441. 104. Palmiter RD . Dopamine signaling in the dorsal striatum is essential for motivated behaviors: lessons from dopamine-deficient mice. Ann N Y Acad Sci 2008; 1129:35-46. 105. Kelly MA, Rubinstein M, Phillips TJ, Lessov CN, Burkhart-Kasch S, Zhang G, Bunzow JR, Fang Y, Gerhardt GA, Grandy DK, Low MJ . Locomotor activity in D2 dopamine receptordeficient mice is determined by gene dosage, genetic background, and developmental adaptations. J Neurosci 1998; 18:3470-3479. 106. Morgan D, Grant KA, Gage HD, Mach RH, Kaplan JR, Prioleau O, Nader SH, Buchheimer N, Ehrenkaufer RL, Nader MA . Social dominance in monkeys: dopamine D2 receptors and cocaine self-administration. Nat Neurosci 2002; 5:169-174. 107. Ben Jonathan N, Hnasko R . Dopamine as a prolactin (PRL) inhibitor. Endocr Rev 2001; 22:724-763. 108. Missale C, Nash SR, Robinson SW, Jaber M, Caron MG . Dopamine receptors: from structure to function. Physiol Rev 1998; 78:189-225. 109. Missale C, Russel Nash S, Robinson S, Jaber M, Caron MG . Dopamine Receptors: From Structure to Function. Physiol Rev 1998; 78:189-225. 110. Guiramand J, Montmayeur JP, Ceraline J, Bhatia M, Borrelli E . Alternative splicing of the dopamine D2 receptor directs specificity of coupling to G-proteins. J Biol Chem 1995; 270:7354-7358. 111. Garcia-Tornadu I, Perez-Millan MI, Recouvreux V, Ramirez MC, Luque G, Risso GS, Ornstein AM, Cristina C, Diaz-Torga G, Becu-Villalobos D . New insights into the endocrine and metabolic roles of dopamine D2 receptors gained from the Drd2 mouse. Neuroendocrinology 2010; 92:207-214. 112. De Mei C, Ramos M, Iitaka C, Borrelli E . Getting specialized: presynaptic and postsynaptic dopamine D2 receptors. Curr Opin Pharmacol 2009; 9:53-58. 113. Fox CA, Mansour A, Thompson RC, Bunzow JR, Civelli O, Watson SJ, Jr. The distribution of dopamine D2 receptor heteronuclear RNA (hnRNA) in the rat brain. J Chem Neuroanat 1993; 6:363-373. 114. Sarkar DK, Chaturvedi K, Oomizu S, Boyadjieva NI, Chen CP . Dopamine, dopamine D2 receptor short isoform, transforming growth factor (TGF)-beta1, and TGF-beta type II receptor interact to inhibit the growth of pituitary lactotropes. Endocrinology 2005; 146:4179-4188. 115. Guivarc'h D, Vernier P, Vincent JD . Sex steroid hormones change the differential distribution of the isoforms of the D2 dopamine receptor messenger RNA in the rat brain. Neuroscience 1995; 69:159-166. 116. Iaccarino C, Samad TA, Mathis C, Kercret H, Picetti R, Borrelli E . Control of lactotrop proliferation by dopamine: essential role of signaling through D2 receptors and ERKs. Proc Natl Acad Sci U S A 2002; 99:14530-14535. 117. Libertun C, Becu D, Arakelian MC, Somoza GM, Lux VAR 1982 Neuroendocrine control of prolactin secretion. In: De Nicola A, Blaquier J, Soto RJ (eds). Physiopathology of hypophysial disturbances and diseases of reproduction.Alan R. Liss, Inc., New York:131152 177 Bibliografía 118. Sarkar DK, Gottschall PE, Meites J . Damage to hypothalamic dopaminergic neurons is associated with development of prolactin-secreting pituitary tumors. Science 1982; 218:684-686. 119. Asa SL, Kelly MA, Grandy DK, Low MJ . Pituitary lactotroph adenomas develop after prolonged lactotroph hyperplasia in dopamine D2 receptor-deficient mice. Endocrinology 1999; 140:5348-5355. 120. Müller E, Locatelli V, Cocchi D . Neuroendocrine control of growth hormone secretion. Physiol Rev 1999; 79:511-607. 121. Kitajima N, Chihara K, Hiromi H, Okimura Y, Fujii Y, Sato M, Shakutsui S, Watanabe M, Fufita T . Effects of dopamine on immunoreactive growth hormone-releasing factor and somatostatin secretion from rat hypothalamic slices perfused in vitro. Endocrinology 1989; 124:69-76. 122. Bluet-Pajot MT, Mounier F, Durand D, Kordon C . Involvement of dopamine D1 receptors in the control of growth hormone secretion in the rat. J Endocrinol 1990; 127:191-196. 123. Garcia-Tornadu I, Rubinstein M, Gaylinn BD, Hill D, Arany E, Low MJ, Diaz-Torga G, BecuVillalobos D . GH in the dwarf dopaminergic D2 receptor knockout mouse: somatotrope population, GH release, and responsiveness to GH-releasing factors and somatostatin. J Endocrinol 2006; 190:611-619. 124. Huseman CA, Hassing JM . Evidence for dopaminergic stimulation of growth velocity in some hypopituitary children. J Clin Endocrinol Metab 1984; 58:419-425. 125. Miyake H, Nagashima K, Onigata K, Nagashima T, Takano Y, Morikawa A . Allelic variations of the D2 dopamine receptor gene in children with idiopathic short stature. J Hum Genet 1999; 44:26-29. 126. Mann K, Rossbach W, Muller MJ, Muller-Siecheneder F, Pott T, Linde I, Dittmann RW, Hiemke C . Nocturnal hormone profiles in patients with schizophrenia treated with olanzapine. Psychoneuroendocrinol 2006; 31:256-264. 127. Leibowitz SF . Brain monoamines and peptides: role in the control of eating behavior. Fed Proc 1986; 45:1396-1403. 128. Terry P, Gilbert DB, Cooper SJ . Dopamine receptor subtype agonists and feeding behavior. Obes Res 1995; 3 Suppl 4:515S-523S. 129. Hillebrand JJ, de Wied D, Adan RA . Neuropeptides, food intake and body weight regulation: a hypothalamic focus. Peptides 2002; 23:2283-2306. 130. Vucetic Z, Reyes TM . Central dopaminergic circuitry controlling food intake and reward: implications for the regulation of obesity. Wiley Interdiscip Rev Syst Biol Med 2010; 2:577593. 131. Fetissov SO, Meguid MM, Sato T, Zhang LH . Expression of dopaminergic receptors in the hypothalamus of lean and obese Zucker rats and food intake. Am J Physiol Regul Integr Comp Physiol 2002; 283:R905-R910. 132. Palmiter RD . Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci 2007; 30:375-381. 133. Pijl H . Reduced dopaminergic tone in hypothalamic neural circuits: expression of a "thrifty" genotype underlying the metabolic syndrome? Eur J Pharmacol 2003; 480:125131. 178 Bibliografía 134. Fetissov SO, Meguid MM . On dopamine, D2 receptor, and Taq1A polymorphism in obesity and anorexia. Nutrition 2009; 25:132-133. 135. Ericson LE, Hakanson R, Lundquist I . Accumulation of dopamine in mouse pancreatic Bcells following injection of L-DOPA. Localization to secretory granules and inhibition of insulin secretion. Diabetologia 1977; 13:117-124. 136. Zern RT, Bird JL, Feldman JM . Effect of increased pancreatic islet norepinephrine, dopamine and serotonin concentration on insulin secretion in the golden hamster. Diabetologia 1980; 18:341-346. 137. Baptista T, Lacruz A, Paez X, Hernandez L, Beaulieu S . The antipsychotic drug sulpiride does not affect bodyweight in male rats. Is insulin resistance involved? Eur J Pharmacol 2002; 447:91-98. 138. Nogueira CR, Machado UF, Curi R, Carpinelli AR . Modulation of insulin secretion and 45Ca2+ efflux by dopamine in glucose-stimulated pancreatic islets. Gen Pharmacol 1994; 25:909-916. 139. Rubi B, Ljubicic S, Pournourmohammadi S, Carobbio S, Armanet M, Bartley C, Maechler P . Dopamine D2-like receptors are expressed in pancreatic beta cells and mediate inhibition of insulin secretion. J Biol Chem 2005; 280:36824-36832. 140. Shankar E, Santhosh KT, Paulose CS . Dopaminergic regulation of glucose-induced insulin secretion through dopamine D2 receptors in the pancreatic islets in vitro. IUBMB Life 2006; 58:157-163. 141. Garcia-Tornadu I, OrnsteinA.M., Chamson-Reig A, Wheeler MB, Hill DJ, Arany E, Rubinstein M, Becu-Villalobos D . Disruption of the Dopamine D2 Receptor impairs insulin secretion and causes glucose intolerance. Endocrinology 2010; 151:1441-1450. 142. Noain D, Perez-Millan MI, Bello EP, Luque GM, Casas CR, Gelman DM, Peper M, Tornadu IG, Low MJ, Becu-Villalobos D, Rubinstein M . Central dopamine D2 receptors regulate growth-hormone-dependent body growth and pheromone signaling to conspecific males. J Neurosci 2013; 33:5834-5842. 143. Ramirez MC, Luque GM, Ornstein AM, Becu-Villalobos D . Differential neonatal testosterone imprinting of GH-dependent liver proteins and genes in female mice. J Endocrinol 2010; 207:301-308. 144. Eleswarapu S, Gu Z, Jiang H . Growth hormone regulation of insulin-like growth factor-I gene expression may be mediated by multiple distal signal transducer and activator of transcription 5 binding sites. Endocrinology 2008; 149:2230-2240. 145. Miyachi K, Fritzler MJ, Tan EM . Autoantibody to a nuclear antigen in proliferating cells. J Immunol 1978; 121:2228-2234. 146. Takasaki Y, Robinson WA, Tan EM . Proliferating cell nuclear antigen in blast crisis cells of patients with chronic myeloid leukemia. J Natl Cancer Inst 1984; 73:655-661. 147. Lacau-Mengido IM, Mejía M, Diaz-Torga G, Gonzalez Iglesias A, Formía N, Libertun C, Becu-Villalobos D . Endocrine studies in ivermectin-treated heifers from birth to puberty. J Anim Sci 2000; 78:1-8. 148. Colao A, Sarno AD, Cappabianca P, Briganti F, Pivonello R, Somma CD, Faggiano A, Biondi B, Lombardi G . Gender differences in the prevalence, clinical features and response to cabergoline in hyperprolactinemia. Eur J Endocrinol 2003; 148:325-331. 149. Bevan JS, Webster J, Burke CW, Scanlon MF . Dopamine agonists and pituitary tumor shrinkage. Endocr Rev 1992; 13:220-240. 179 Bibliografía 150. Gillam MP, Molitch ME, Lombardi G, Colao A . Advances in the treatment of prolactinomas. Endocr Rev 2006; 27:485-534. 151. Saeger W, Ludecke DK, Buchfelder M, Fahlbusch R, Quabbe HJ, Petersenn S . Pathohistological classification of pituitary tumors: 10 years of experience with the German Pituitary Tumor Registry. Eur J Endocrinol 2007; 156:203-216. 152. Molitch ME . Dopamine resistance of prolactinomas. Pituitary 2003; 6:19-27. 153. Webster J, Piscitelli G, Polli A, Ferrari CI, Ismail I, Scanlon MF . A comparison of cabergoline and bromocriptine in the treatment of hyperprolactinemic amenorrhea. Cabergoline Comparative Study Group. N Engl J Med 1994; 331:904-909. 154. Caccavelli L, Feron F, Morange I, Rouer E, Benarous R, Dewailly D, Jaquet P, Kordon C, Enjalbert A . Decreased expression of the two D2 dopamine receptor isoforms in bromocriptine-resistant prolactinomas. Neuroendocrinology 1994; 60:314-322. 155. Diaz-Torga G, Gonzalez IA, Achaval-Zaia R, Libertun C, Becu-Villalobos D . Angiotensin IIinduced Ca2+ mobilization and prolactin release in normal and hyperplastic pituitary cells. Am J Physiol 1998; 274:E534-E540. 156. Suarez C, García-Tornadú I, Khalil W, Becu-Villalobos D . Dehydroepiandrosterone treatment attenuates estrogen induced pituitary hyperplasia. J Endocrinol 2002; 174:447454. 157. Goya RG, Lu JK, Meites J . Gonadal function in aging rats and its relation to pituitary and mammary pathology. Mech Ageing Dev 1990; 56:77-88. 158. Cai A, Hayes JD, Patel N, Hyde JF . Targeted overexpression of galanin in lactotrophs of transgenic mice induces hyperprolactinemia and pituitary hyperplasia. Endocrinology 1999; 140:4955-4964. 159. McAndrew J, Paterson AJ, Asa SL, McCarthy KJ, Kudlow JE . Targeting of transforming growth factor-alpha expression to pituitary lactotrophs in transgenic mice results in selective lactotroph proliferation and adenomas. Endocrinology 1995; 136:4479-4488. 160. Borrelli E, Sawchenko PE, Evans RM . Pituitary hyperplasia induced by ectopic expression of nerve growth factor. Proc Natl Acad Sci U S A 1992; 89:2764-2768. 161. Ezzat S, Zheng L, Zhu XF, Wu GE, Asa SL . Targeted expression of a human pituitary tumorderived isoform of FGF receptor-4 recapitulates pituitary tumorigenesis. J Clin Invest 2002; 109:69-78. 162. Schuff KG, Hentges ST, Kelly MA, Binart N, Kelly PA, Iuvone PM, Asa SL, Low MJ . Lack of prolactin receptor signaling in mice results in lactotroph proliferation and prolactinomas by dopamine-dependent and -independent mechanisms. J Clin Invest 2002; 110:973-981. 163. Saiardi A, Bozzi Y, Baik J-H, Borrelli E . Antiproliferative role of dopamine: loss of D2 receptors causes hormonal dysfunction and pituitary hyperplasia. Neuron 1997; 19:115126. 164. Sternberg N, Hamilton D . Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J Mol Biol 1981; 150:467-486. 165. Hoess RH, Wierzbicki A, Abremski K . The role of the loxP spacer region in P1 site-specific recombination. Nucleic Acids Res 1986; 14:2287-2300. 166. Abremski K, Hoess R, Sternberg N . Studies on the properties of P1 site-specific recombination: evidence for topologically unlinked products following recombination. Cell 1983; 32:1301-1311. 180 Bibliografía 167. Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H . A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 2010; 13:133-140. 168. Ben-Jonathan N . Dopamine: a prolactin inhibiting hormone. Endocr Rev 1985; 6:564-589. 169. Pellegrini I, Rasolonjanahary R, Gunz G, Bertrand P, Delivet S, Jedynak CP, Kordon C, Peillon F, Jaquet P, Enjalbert A . Resistance to bromocriptine in prolactinomas. J Clin Endocrinol Metab 1989; 69:500-509. 170. Caccavelli L, Morange-Ramos I, Kordon C, Jaquet P, Enjalbert A . Alteration of G alpha subunits mRNA levels in bromocriptine resistant prolactinomas. J Neuroendocrinol 1996; 8:737-746. 171. Arbogast LA, Voogt JL . Hyperprolactinemia increases and hypoprolactinemia decreases tyrosine hydroxylase messenger ribonucleic acid levels in the arcuate nuclei, but not the substantia nigra or zona incerta. Endocrinology 1991; 128:997-1005. 172. Gudelsky GA, Nansel DD, Porter JC . Role of estrogen in the dopaminergic control of prolactin secretion. Endocrinology 1981; 108:440-444. 173. Fiorentini C, Guerra N, Facchetti M, Finardi A, Tiberio L, Schiaffonati L, Spano P, Missale C . Nerve growth factor regulates dopamine D(2) receptor expression in prolactinoma cell lines via p75(NGFR)-mediated activation of nuclear factor-kappaB. Mol Endocrinol 2002; 16:353-366. 174. Missale C, Boroni R, Sigala S, Buriani A, Fabris M, Leon A, Del Toso R, Spano PF . Nerve growth factor in the anterior pituitary: localization in lactotroph cells and cosecretion with prolactin by a dopamine-regulated mechanism. Proc Natl Acad Sci U S A 1996; 93:42404245. 175. Hyde JF, Moore JPJ, Cai A . Galanin in normal and hyperplastic anterior pituitary cells. From pituitary tumor cell lines to transgenic mice. Ann NY Acad Sci 1998; 863:48-55. 176. Ben-Jonathan N, Liu JW . Pituitary lactotrophs: endocrine, paracrine, juxtacrine and autocrine interactions. Trends Endocrinol Metab 1992; 3:254-258. 177. Pastorcic M, De A, Boyadjieva N, Vale W, Sarkar DK . Reduction in the expression and action of transforming growth factor-b1 on lactotropes during estrogen-induced tumorigenesis. Cancer res 1995; 55:4892-4898. 178. Yu R, Melmed S . Pituitary tumor transforming gene: an update. Front Horm Res 2004; 32:175-185. 179. Melmed S 2002 The pituitary.Blackwell, Second Edition edn 180. Folkman J, Shing Y . Angiogenesis. J Biol Chem 1992; 267:10931-10934. 181. de la Torre NG, Wass JA, Turner HE . Morphologic changes and molecular regulation of angiogenesis in pituitary adenomas. Front Horm Res 2004; 32:133-145. 182. Elias KA, Weiner RI . Direct arterial vascularization of estrogen-induced prolactin-secreting anterior pituitary tumors. Proc Natl Acad Sci U S A 1984; 81:4549-4553. 183. Schechter J, Goldsmith P, Wilson C, Weiner R . Morphological evidence for the presence of arteries in human prolactinomas. J Clin Endocrinol Metab 1988; 67:713-719. 184. Bonneville JF, Cattin F, Gorczyca W, Hardy J . Pituitary microadenomas: early enhancement with dynamic CT--implications of arterial blood supply and potential importance. Radiology 1993; 187:857-861. 181 Bibliografía 185. Turner HE, Nagy Z, Gatter KC, Esiri MM, Harris AL, Wass JA . Angiogenesis in pituitary adenomas and the normal pituitary gland. J Clin Endocrinol Metab 2000; 85:1159-1162. 186. Turner HE, Nagy Z, Gatter KC, Esiri MM, Harris AL, Wass JA . Angiogenesis in pituitary adenomas - relationship to endocrine function, treatment and outcome. J Endocrinol 2000; 165:475-481. 187. Jugenburg M, Kovacs K, Stefaneanu L, Scheithauer BW . Vasculature in Nontumorous Hypophyses, Pituitary Adenomas, and Carcinomas: A Quantitative Morphologic Study. Endocr Pathol 1995; 6:115-124. 188. Turner HE, Nagy Z, Esiri MM, Harris AL, Wass JA . Role of matrix metalloproteinase 9 in pituitary tumor behavior. J Clin Endocrinol Metab 2000; 85:2931-2935. 189. Vidal S, Kovacs K, Horvath E, Scheithauer BW, Kuroki T, Lloyd RV . Microvessel density in pituitary adenomas and carcinomas. Virchows Arch 2001; 438:595-602. 190. Ferrara N, Davis-Smyth T . The biology of vascular endothelial growth factor. Endocr Rev 1997; 18:4-25. 191. Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, Risau W . Vascular endothelial growth factor induces endothelial fenestrations in vitro. J Cell Biol 1998; 140:947-959. 192. Dvorak HF, Harvey VS, Estrella P, Brown LF, McDonagh J, Dvorak AM . Fibrin containing gels induce angiogenesis. Implications for tumor stroma generation and wound healing. Lab Invest 1987; 57:673-686. 193. Alon T, Hemo I, Itin A, Pe'er J, Stone J, Keshet E . Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1995; 1:1024-1028. 194. Halder JB, Zhao X, Soker S, Paria BC, Klagsbrun M, Das SK, Dey SK . Differential expression of VEGF isoforms and VEGF(164)-specific receptor neuropilin-1 in the mouse uterus suggests a role for VEGF(164) in vascular permeability and angiogenesis during implantation. Genesis 2000; 26:213-224. 195. Byrne AM, Bouchier-Hayes DJ, Harmey JH . Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF). J Cell Mol Med 2005; 9:777-794. 196. Gospodarowicz D, Abraham JA, Schilling J . Isolation and characterization of a vascular endothelial mitogen produced by pituitary-derived folliculo stellate cells. Proc Natl Acad Sci U S A 1989; 86:7311-7315. 197. Ferrara N, Henzel W . Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun 1989; 161:851-858. 198. Jabbour HN, Boddy SC, Lincoln GA . Pattern and localisation of expression of vascular endothelial growth factor and its receptor flt-1 in the ovine pituitary gland: expression is independent of hypothalamic control. Mol Cell Endocrinology 1997; 134:91-100. 199. Vidal S, Oliveira MC, Kovacs K, Scheithauer BW, Lloyd R . Immunolocalization of vascular endothelial growth factor in the GH3 cell line. Cell Tissue Res 2000; 300:83-88. 200. Lohrer P, Gloddek J, Hopfner U, Losa M, Uhl E, Pagotto U, Stalla GK, Renner U . Vascular endothelial growth factor production and regulation in rodent and human pituitary tumor cells in vitro. Neuroendocrinology 2001; 74:95-105. 182 Bibliografía 201. Nishikawa R, Cheng SY, Nagashima R, Huang HJ, Cavenee WK, Matsutani M . Expression of vascular endothelial growth factor in human brain tumors. Acta Neuropathol (Berl) 1998; 96:453-462. 202. Lloyd RV, Scheithauer BW, Kuroki T, Vidal S, Kovacs K, Stefaneanu L . Vascular Endothelial Growth Factor (VEGF) Expression in Human Pituitary Adenomas and Carcinomas. Endocr Pathol 1999; 10:229-235. 203. McCabe CJ, Boelaert K, Tannahill LA, Heaney AP, Stratford AL, Khaira JS, Hussain S, Sheppard MC, Franklyn JA, Gittoes NJ . Vascular endothelial growth factor, its receptor KDR/Flk-1, and pituitary tumor transforming gene in pituitary tumors. J Clin Endocrinol Metab 2002; 87:4238-4244. 204. Cristina C, García-Tornadú I, Diaz-Torga G, Rubinstein M, Low MJ, Becu-Villalobos D . The dopaminergic D2 receptor knockout mouse: an animal model of prolactinoma. Arzt E, Bronstein MD, Guitelman M, editors. Front Horm Res 2006; 35:50-63. 205. Banerjee SK, Sarkar DK, Weston AP, De A, Campbell DR . Over expression of vascular endothelial growth factor and its receptor during the development of estrogen-induced rat pituitary tumors may mediate estrogen-initiated tumor angiogenesis. Carcinogenesis 1997; 18:1155-1161. 206. Cristina C, Diaz-Torga G, Gongora A, Guida MC, Perez-Millan MI, Baldi A, Becu-Villalobos D . Fibroblast Growth Factor-2 in hyperplastic pituitaries of D2R knockout female mice. Am J Physiol Endocrinol Metab 2007; 293:E1341-E1351. 207. Cristina C, Diaz-Torga GS, Goya RG, Kakar SS, Perez-Millan MI, Passos VQ, Gianella-Neto D, Bronstein MD, Becu-Villalobos D . PTTG expression in different experimental and human prolactinomas in relation to dopaminergic control of lactotropes. Mol Cancer 2007; 6:4. 208. Cristina C, Perez-Millan MI, Luque G, Dulce RA, Sevlever G, Berner SI, Becu-Villalobos D . VEGF and CD31 association in pituitary adenomas. Endocr Pathol 2010; 21:154-160. 209. Mallea-Gil MS, Cristina C, Perez-Millan MI, Ballarino MC, Rodriguez Villafañe AM, Stalldecker G, Becu-Villalobos D . Invasive giant prolactinoma with loss of therapeutic response to cabergoline: expression of angiogenic markers. Endocr Pathol 2009; 20:35-50. 210. Luque GM, Perez-Millan MI, Ornstein AM, Cristina C, Becu-Villalobos D . Inhibitory effects of antivascular endothelial growth factor strategies in experimental dopamine-resistant prolactinomas. J Pharmacol Exp Ther 2011; 337:766-774. 211. Alvarez-Buylla A, Lim DA . For the long run: maintaining germinal niches in the adult brain. Neuron 2004; 41:683-686. 212. Rando TA . Stem cells, ageing and the quest for immortality. Nature 2006; 441:1080-1086. 213. Slack JM . Origin of stem cells in organogenesis. Science 2008; 322:1498-1501. 214. Trosko JE . Review paper: cancer stem cells and cancer nonstem cells: from adult stem cells or from reprogramming of differentiated somatic cells. Vet Pathol 2009; 46:176-193. 215. Vankelecom H, Chen J . Pituitary stem cells: where do we stand? Mol Cell Endocrinol 2014; 385:2-17. 216. Chen J, Gremeaux L, Fu Q, Liekens D, Van Laere S, Vankelecom H . Pituitary progenitor cells tracked down by side population dissection. Stem Cells 2009; 27:1182-1195. 217. Nolan LA, Kavanagh E, Lightman SL, Levy A . Anterior pituitary cell population control: basal cell turnover and the effects of adrenalectomy and dexamethasone treatment. J Neuroendocrinol 1998; 10:207-215. 183 Bibliografía 218. Vankelecom H . Pituitary stem/progenitor cells: embryonic players in the adult gland? Eur J Neurosci 2010; 32:2063-2081. 219. Landolt AM . Regeneration of the human pituitary. J Neurosurg 1973; 39:35-41. 220. Nassiri F, Cusimano M, Zuccato JA, Mohammed S, Rotondo F, Horvath E, Syro LV, Kovacs K, Lloyd RV . Pituitary stem cells: candidates and implications. Pituitary 2013; 16:413-418. 221. Ramos CA, Venezia TA, Camargo FA, Goodell MA . Techniques for the study of adult stem cells: be fruitful and multiply. Biotechniques 2003; 34:572-4, 586. 222. Vankelecom H . Pituitary stem cells drop their mask. Curr Stem Cell Res Ther 2012; 7:3671. 223. Chen J, Hersmus N, Van D, V, Caesens P, Denef C, Vankelecom H . The adult pituitary contains a cell population displaying stem/progenitor cell and early embryonic characteristics. Endocrinology 2005; 146:3985-3998. 224. Goodell MA, Brose K, Paradis G, Conner AS, Mulligan RC . Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J Exp Med 1996; 183:1797-1806. 225. Hussain SZ, Strom SC, Kirby MR, Burns S, Langemeijer S, Ueda T, Hsieh M, Tisdale JF . Side population cells derived from adult human liver generate hepatocyte-like cells in vitro. Dig Dis Sci 2005; 50:1755-1763. 226. Fontana S, Cohn RD . SP-litting the satellite niche to repopulate muscle. Cell Stem Cell 2009; 4:194-195. 227. Challen GA, Little MH . A side order of stem cells: the SP phenotype. Stem Cells 2006; 24:3-12. 228. Krylyshkina O, Chen J, Mebis L, Denef C, Vankelecom H . Nestin-immunoreactive cells in rat pituitary are neither hormonal nor typical folliculo-stellate cells. Endocrinology 2005; 146:2376-2387. 229. Vankelecom H . Stem cells in the postnatal pituitary? Neuroendocrinology 2007; 85:110130. 230. Florio T . Adult pituitary stem cells: from pituitary plasticity to adenoma development. Neuroendocrinology 2011; 94:265-277. 231. Alatzoglou KS, Kelberman D, Dattani MT . The role of SOX proteins in normal pituitary development. J Endocrinol 2009; 200:245-258. 232. Fauquier T, Rizzoti K, Dattani M, Lovell-Badge R, Robinson IC . SOX2-expressing progenitor cells generate all of the major cell types in the adult mouse pituitary gland. Proc Natl Acad Sci U S A 2008; 105:2907-2912. 233. Dick JE . Stem cell concepts renew cancer research. Blood 2008; 112:4793-4807. 234. Reya T, Morrison SJ, Clarke MF, Weissman IL . Stem cells, cancer, and cancer stem cells. Nature 2001; 414:105-111. 235. Shackleton M, Quintana E, Fearon ER, Morrison SJ . Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell 2009; 138:822-829. 236. Clevers H . The cancer stem cell: premises, promises and challenges. Nat Med 2011; 17:313-319. 237. Wang K, Wu X, Wang J, Huang J . Cancer stem cell theory: therapeutic implications for nanomedicine. Int J Nanomedicine 2013; 8:899-908. 184 Bibliografía 238. Gilbert CA, Ross AH . Cancer stem cells: cell culture, markers, and targets for new therapies. J Cell Biochem 2009; 108:1031-1038. 239. Kreso A, Dick JE . Evolution of the cancer stem cell model. Cell Stem Cell 2014; 14:275291. 240. Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP . Cancer stem cells--old concepts, new insights. Cell Death Differ 2008; 15:947-958. 241. Bonnet D, Dick JE . Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3:730-737. 242. Al Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF . Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003; 100:39833988. 243. Tang DG, Patrawala L, Calhoun T, Bhatia B, Choy G, Schneider-Broussard R, Jeter C . Prostate cancer stem/progenitor cells: identification, characterization, and implications. Mol Carcinog 2007; 46:1-14. 244. O'Brien CA, Pollett A, Gallinger S, Dick JE . A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007; 445:106-110. 245. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB . Identification of a cancer stem cell in human brain tumors. Cancer Res 2003; 63:5821-5828. 246. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB . Identification of human brain tumour initiating cells. Nature 2004; 432:396-401. 247. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA . The epithelialmesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704715. 248. Eyler CE, Rich JN . Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol 2008; 26:2839-2845. 249. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN . Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006; 444:756-760. 250. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL, Clarke MF . Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009; 458:780-783. 251. Oravecz-Wilson KI, Philips ST, Yilmaz OH, Ames HM, Li L, Crawford BD, Gauvin AM, Lucas PC, Sitwala K, Downing JR, Morrison SJ, Ross TS . Persistence of leukemia-initiating cells in a conditional knockin model of an imatinib-responsive myeloproliferative disorder. Cancer Cell 2009; 16:137-148. 252. Fiuza UM, Arias AM . Cell and molecular biology of Notch. J Endocrinol 2007; 194:459-474. 253. Radtke F, Raj K . The role of Notch in tumorigenesis: oncogene or tumour suppressor? Nat Rev Cancer 2003; 3:756-767. 254. Talora C, Sgroi DC, Crum CP, Dotto GP . Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes Dev 2002; 16:2252-2263. 185 Bibliografía 255. Grynfeld A, Pahlman S, Axelson H . Induced neuroblastoma cell differentiation, associated with transient HES-1 activity and reduced HASH-1 expression, is inhibited by Notch1. Int J Cancer 2000; 88:401-410. 256. Liu K, Lin B, Zhao M, Yang X, Chen M, Gao A, Liu F, Que J, Lan X . The multiple roles for Sox2 in stem cell maintenance and tumorigenesis. Cell Signal 2013; 25:1264-1271. 257. Gleiberman AS, Michurina T, Encinas JM, Roig JL, Krasnov P, Balordi F, Fishell G, Rosenfeld MG, Enikolopov G . Genetic approaches identify adult pituitary stem cells. Proc Natl Acad Sci U S A 2008; 105:6332-6337. 258. Altenberger T, Bilban M, Auer M, Knosp E, Wolfsberger S, Gartner W, Mineva I, Zielinski C, Wagner L, Luger A . Identification of DLK1 variants in pituitary- and neuroendocrine tumors. Biochem Biophys Res Commun 2006; 340:995-1005. 259. Cheunsuchon P, Zhou Y, Zhang X, Lee H, Chen W, Nakayama Y, Rice KA, Tessa HedleyWhyte E, Swearingen B, Klibanski A . Silencing of the imprinted DLK1-MEG3 locus in human clinically nonfunctioning pituitary adenomas. Am J Pathol 2011; 179:2120-2130. 260. Moreno CS, Evans CO, Zhan X, Okor M, Desiderio DM, Oyesiku NM . Novel molecular signaling and classification of human clinically nonfunctional pituitary adenomas identified by gene expression profiling and proteomic analyses. Cancer Res 2005; 65:10214-10222. 261. Waxman DJ, Holloway MG . Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol 2009; 76:215-228. 262. Garcia-Tornadu I, Risso G, Perez-Millan MI, Noain D, Diaz-Torga GS, Low MJ, Rubinstein M, Becu-Villalobos D . Neurotransmitter modulation of the GHRH-GH axis. Front Horm Res 2010; 38:59-69. 263. Gudelsky GA, Porter JC . Sex-related difference in the release of dopamine into hypophysial portal blood. Endocrinology 1981; 109:1394-1398. 264. Demarest KT, Moore KE . Sexual differences in the sensitivity of tuberoinfundibular dopamine neurons to the actions of prolactin. Neuroendocrinology 1981; 33:230-234. 265. Lucas BK, Ormandy CJ, Binart N, Bridges RS, Kelly PA . Null mutation of the prolactin receptor gene produces a defect in maternal behavior. Endocrinology 1998; 139:41024107. 266. Horseman ND, Zhao W, Montecino-Rodriguez E, Tanaka M, Nakashima K, Engle SJ, Smith F, Markoff E, Dorshkind K . Defective mammopoiesis, but normal hematopoiesis, in mice with a targeted disruption of the prolactin gene. EMBO J 1997; 16:6926-6935. 267. Crawford Y, Ferrara N . VEGF inhibition: insights from preclinical and clinical studies. Cell Tissue Res 2009; 335:261-269. 268. Niveiro M, Aranda FI, Peiro G, Alenda C, Pico A . Immunohistochemical analysis of tumor angiogenic factors in human pituitary adenomas. Hum Pathol 2005; 36:1090-1095. 269. Turner HE, Harris AL, Melmed S, Wass JA . Angiogenesis in endocrine tumors. Endocr Rev 2003; 24:600-632. 270. Wong AK, Alfert M, Castrillon DH, Shen Q, Holash J, Yancopoulos GD, Chin L . Excessive tumor-elaborated VEGF and its neutralization define a lethal paraneoplastic syndrome. Proc Natl Acad Sci U S A 2001; 98:7481-7486. 186 Bibliografía 271. Perez-Millan MI, Berner SI, Luque GM, De Bonis C, Sevlever G, Becu-Villalobos D, Cristina C . Enhanced nestin expression and small blood vessels in human pituitary adenomas. Pituitary 2012. 272. Ferrara N, Gerber H, LeCouter J . The biology of VEGF and its receptors. Nat Med 2003; 9:669-676. 273. Folkman J . What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst 1990; 82:4-6. 274. Basu S, Nagy JA, Pal S, Vasile E, Eckelhoefer IA, Bliss VS, Manseau EJ, Dasgupta PS, Dvorak HF, Mukhopadhyay D . The neurotransmitter dopamine inhibits angiogenesis induced by vascular permeability factor/vascular endothelial growth factor. Nat Med 2001; 7:569574. 275. Miao Z, Miao Y, Lin Y, Lu X . Overexpression of the Notch3 receptor in non-functioning pituitary tumours. J Clin Neurosci 2012; 19:107-110. 276. Lu R, Gao H, Wang H, Cao L, Bai J, Zhang Y . Overexpression of the Notch3 receptor and its ligand Jagged1 in human clinically non-functioning pituitary adenomas. Oncol Lett 2013; 5:845-851. 277. Karatsoreos IN, Thaler JP, Borgland SL, Champagne FA, Hurd YL, Hill MN . Food for thought: hormonal, experiential, and neural influences on feeding and obesity. J Neurosci 2013; 33:17610-17616. 278. Farooqi IS, O'Rahilly S . Recent advances in the genetics of severe childhood obesity. Arch Dis Child 2000; 83:31-34. 279. Gearhardt AN, Grilo CM, DiLeone RJ, Brownell KD, Potenza MN . Can food be addictive? Public health and policy implications. Addiction 2011; 106:1208-1212. 280. Potenza MN . Obesity, food, and addiction: emerging neuroscience and clinical and public health implications. Neuropsychopharmacology 2014; 39:249-250. 281. Ramos EJ, Meguid MM, Campos AC, Coelho JC . Neuropeptide Y, alpha-melanocytestimulating hormone, and monoamines in food intake regulation. Nutrition 2005; 21:269279. 282. Cone RD . Anatomy and regulation of the central melanocortin system. Nat Neurosci 2005; 8:571-578. 283. Shimizu H, Inoue K, Mori M . The leptin-dependent and -independent melanocortin signaling system: regulation of feeding and energy expenditure. J Endocrinol 2007; 193:19. 284. MacNeil D, Howard A, Guan X, Fong T, Nargumd R, Bednarek M, Goulet M, Weinberg D, Strack A, Marsh D, Chen HY, Shen C, Rosenblum C, MacNeil T, Tota M, MacIntyre E, Van der Ploeg L . The role of melanocortins in body weight regulation: opportunities for the treatment of obesity. Eur J Pharmacol 2002; 440:141-157. 285. Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD . A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology 2000; 141:3518-3521. 286. Yaswen L, Diehl N, Brennan MB, Hochgeschwender U . Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med 1999; 5:1066-1070. 187 Bibliografía 287. Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A . Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet 1998; 19:155-157. 288. Cote TE, Felder R, Kebabian JW, Sekura RD, Reisine T, Affolter HU . D-2 dopamine receptor-mediated inhibition of pro-opiomelanocortin synthesis in rat intermediate lobe. Abolition by pertussis toxin or activators of adenylate cyclase. J Biol Chem 1986; 261:4555-4561. 289. Lindley S, Gunnrt J, Lookingland KJ, Moore K . Effects of alterations in the activity of tuberohypophysial dopaminergic neurons on the secretion of alpha-melanocyte stimulating hormone. Proc Soc Exp Biol Med 1988; 188:282-286. 290. Bower A, Hadley ME, Hruby V . Biogenic Amines and control of melanophore stimulating hormone release. Science 1974; 184:70-72. 291. Zigman JM, Elmquist JK . Minireview: From anorexia to obesity--the yin and yang of body weight control. Endocrinology 2003; 144:3749-3756. 292. Bina KG, Cincotta AH . Dopaminergic agonists normalize elevated hypothalamic neuropeptide Y and corticotropin-releasing hormone, body weight gain, and hyperglycemia in ob/ob mice. Neuroendocrinology 2000; 71:68-78. 293. Chen P, Smith MS . Regulation of hypothalamic neuropeptide Y messenger ribonucleic acid expression during lactation: role of prolactin. Endocrinology 2004; 145:823-829. 294. Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M . Orexins and orexin receptors: a family of hypothalamic neuropeptides and G proteincoupled receptors that regulate feeding behavior. Cell 1998; 92:573-585. 295. de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG . The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A 1998; 95:322-327. 296. Monda M, Viggiano AN, Viggiano A, Viggiano E, Lanza A, De L, V . Hyperthermic reactions induced by orexin A: role of the ventromedial hypothalamus. Eur J Neurosci 2005; 22:1169-1175. 297. Samson WK, Gosnell B, Chang JK, Resch ZT, Murphy TC . Cardiovascular regulatory actions of the hypocretins in brain. Brain Res 1999; 831:248-253. 298. Lubkin M, Stricker-Krongrad A . Independent feeding and metabolic actions of orexins in mice. Biochem Biophys Res Commun 1998; 253:241-245. 299. Ferguson AV, Samson WK . The orexin/hypocretin system: a critical regulator of neuroendocrine and autonomic function. Front Neuroendocrinol 2003; 24:141-150. 300. Taheri S, Zeitzer JM, Mignot E . The role of hypocretins (orexins) in sleep regulation and narcolepsy. Annu Rev Neurosci 2002; 25:283-313. 301. Burdakov D, Alexopoulos H . Metabolic state signalling through central hypocretin/orexin neurons. J Cell Mol Med 2005; 9:795-803. 302. Lopez M, Lage R, Tung YC, Challis BG, Varela L, Virtue S, O'Rahilly S, Vidal-Puig A, Dieguez C, Coll AP . Orexin expression is regulated by alpha-melanocyte-stimulating hormone. J Neuroendocrinology 2007; 19:703-707. 188 Bibliografía 303. Bubser M, Fadel JR, Jackson LL, Meador-Woodruff JH, Jing D, Deutch AY . Dopaminergic regulation of orexin neurons. Eur J Neurosci 2005; 21:2993-3001. 304. Dalal MA, Schuld A, Pollmacher T . Lower CSF orexin A (hypocretin-1) levels in patients with schizophrenia treated with haloperidol compared to unmedicated subjects. Mol Psychiatry 2003; 8:836-837. 305. Ahima RS, Flier JS . Leptin. Annu Rev Physiol 2000; 62:413-437. 306. Baskin DG, Breininger JF, Bonigut S, Miller MA . Leptin binding in the arcuate nucleus is increased during fasting. Brain Res 1999; 828:154-158. 307. Pfaffly J, Michaelides M, Wang GJ, Pessin JE, Volkow ND, Thanos PK . Leptin increases striatal dopamine D2 receptor binding in leptin-deficient obese (ob/ob) mice. Synapse 2010; 64:503-510. 308. Cheung CC, Clifton DK, Steiner RA . Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology 1997; 138:4489-4492. 309. Seeley RJ, Yagaloff KA, Fisher SL, Burn P, Thiele TE, van Dijk G, Baskin DG, Schwartz MW . Melanocortin receptors in leptin effects. Nature 1997; 390:349. 310. da Silva AA, Kuo JJ, Hall JE . Role of hypothalamic melanocortin 3/4-receptors in mediating chronic cardiovascular, renal, and metabolic actions of leptin. Hypertension 2004; 43:1312-1317. 311. Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ . Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001; 411:480-484. 312. Figlewicz DP, Bennett J, Evans SB, Kaiyala K, Sipols AJ, Benoit SC . Intraventricular insulin and leptin reverse place preference conditioned with high-fat diet in rats. Behav Neurosci 2004; 118:479-487. 313. Figlewicz DP, Patterson TA, Johnson LB, Zavosh A, Israel PA, Szot P . Dopamine transporter mRNA is increased in the CNS of Zucker fatty (fa/fa) rats. Brain Res Bull 1998; 46:199-202. 314. Roseberry AG, Painter T, Mark GP, Williams JT . Decreased vesicular somatodendritic dopamine stores in leptin-deficient mice. J Neurosci 2007; 27:7021-7027. 315. Ling C, Billig H . PRL receptor-mediated effects in female mouse adipocytes: PRL induces suppressors of cytokine signaling expression and suppresses insulin-induced leptin production in adipocytes in vitro. Endocrinology 2001; 142:4880-4890. 316. LaPensee CR, Horseman ND, Tso P, Brandebourg TD, Hugo ER, Ben Jonathan N . The prolactin-deficient mouse has an unaltered metabolic phenotype. Endocrinology 2006; 147:4638-4645. 317. Friedman JM . Leptin and the regulation of body weigh. Keio J Med 2011; 60:1-9. 318. Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, Friedman JM . Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci U S A 1997; 94:8878-8883. 319. El Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS . Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest 2000; 105:1827-1832. 320. Hileman SM, Pierroz DD, Masuzaki H, Bjorbaek C, El Haschimi K, Banks WA, Flier JS . Characterizaton of short isoforms of the leptin receptor in rat cerebral microvessels and of brain uptake of leptin in mouse models of obesity. Endocrinology 2002; 143:775-783. 189 Bibliografía 321. Niimi M, Sato M, Yokote R, Tada S, Takahara J . Effects of central and peripheral injection of leptin on food intake and on brain Fos expression in the Otsuka Long-Evans Tokushima Fatty rat with hyperleptinaemia. J Neuroendocrinol 1999; 11:605-611. 322. Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D, Jr. Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med 1996; 2:589593. 323. Faouzi M, Leshan R, Bjornholm M, Hennessey T, Jones J, Munzberg H . Differential accessibility of circulating leptin to individual hypothalamic sites. Endocrinology 2007; 148:5414-5423. 324. Munzberg H, Jobst EE, Bates SH, Jones J, Villanueva E, Leshan R, Bjornholm M, Elmquist J, Sleeman M, Cowley MA, Myers MG, Jr. Appropriate inhibition of orexigenic hypothalamic arcuate nucleus neurons independently of leptin receptor/STAT3 signaling. J Neurosci 2007; 27:69-74. 325. Fernandez-Galaz C, Fernandez-Agullo T, Campoy F, Arribas C, Gallardo N, Andres A, Ros M, Carrascosa JM . Decreased leptin uptake in hypothalamic nuclei with ageing in Wistar rats. J Endocrinol 2001; 171:23-32. 326. Vaisse C, Halaas JL, Horvath CM, Darnell JE, Jr., Stoffel M, Friedman JM . Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet 1996; 14:95-97. 327. Hurley DL, Birch DV, Almond MC, Estrada IJ, Phelps CJ . Reduced hypothalamic neuropeptide Y expression in growth hormone- and prolactin-deficient Ames and Snell dwarf mice. Endocrinology 2003; 144:4783-4789. 328. Kim KS, Yoon YR, Lee HJ, Yoon S, Kim SY, Shin SW, An JJ, Kim MS, Choi SY, Sun W, Baik JH . Enhanced hypothalamic leptin signaling in mice lacking dopamine D2 receptors. J Biol Chem 2010; 285:8905-8917. 329. Gerardo-Gettens T, Moore BJ, Stern JS, Horwitz BA . Prolactin stimulates food intake in a dose-dependent manner. Am J Physiol 1989; 256:R276-R280. 330. Fleenor D, Oden J, Kelly PA, Mohan S, Alliouachene S, Pende M, Wentz S, Kerr J, Freemark M . Roles of the lactogens and somatogens in perinatal and postnatal metabolism and growth: studies of a novel mouse model combining lactogen resistance and growth hormone deficiency. Endocrinology 2005; 146:103-112. 331. Flint DJ, Binart N, Boumard S, Kopchick JJ, Kelly P . Developmental aspects of adipose tissue in GH receptor and prolactin receptor gene disrupted mice: site-specific effects upon proliferation, differentiation and hormone sensitivity. J Endocrinol 2006; 191:101111. 332. Arumugam R, Fleenor D, Freemark M . Lactogenic and somatogenic hormones regulate the expression of neuropeptide Y and cocaine- and amphetamine-regulated transcript in rat insulinoma (INS-1) cells: interactions with glucose and glucocorticoids. Endocrinology 2007; 148:258-267. 333. Augustine RA, Kokay IC, Andrews ZB, Ladyman SR, Grattan DR . Quantitation of prolactin receptor mRNA in the maternal rat brain during pregnancy and lactation. J Mol Endocrinol 2003; 31:221-232. 334. Faron-Gorecka A, Kusmider M, Kolasa M, Zurawek D, Gruca P, Papp M, Szafran K, Solich J, Pabian P, Romanska I, Antkiewicz-Michaluk L, Dziedzicka-Wasylewska M . Prolactin and its receptors in the chronic mild stress rat model of depression. Brain Res 2014; 1555:48-59. 190 Bibliografía 335. Garcia MC, Lopez M, Gualillo O, Seoane LM, Dieguez C, Senaris RM . Hypothalamic levels of NPY, MCH, and prepro-orexin mRNA during pregnancy and lactation in the rat: role of prolactin. FASEB J 2003; 17:1392-1400. 336. Augustine RA, Grattan DR . Induction of central leptin resistance in hyperphagic pseudopregnant rats by chronic prolactin infusion. Endocrinology 2008; 149:1049-1055. 337. Dean L, McEntyre J 2004 The Genetic Landscape of Diabetes.NIDDK, Washington,DC 338. Brubaker PL, Drucker DJ . Structure-function of the glucagon receptor family of G proteincoupled receptors: the glucagon, GIP, GLP-1, and GLP-2 receptors. Receptors Channels 2002; 8:179-188. 339. Berne RM, Levy MN 1993 Physiology.Mosby Year Book, St. Louis, Missouri, 3rd edn 340. Bell GI, Kayano T, Buse JB, Burant CF, Takeda J, Lin D, Fukumoto H, Seino S . Molecular biology of mammalian glucose transporters. Diabetes Care 1990; 13:198-208. 341. Skyler JS 2006 Atlas of Diabetes.Curren Medicine LLC, Philadelphia, 3rd edn 342. Houssay BA, Biassotti A . La diabetes pancreatica de los perros hipofiseoprivos. Rev Soc Argent Biol 1930;251-296. 343. Houssay BA, Biassotti A, Sanmartino R . Modifications functionelles de l'hypophyse apres le lesions infundibulo-tuberriennes chez le crapaud. Comt Rend Soc Biol 1935; 120:725. 344. Brelje TC, Scharp DW, Lacy PE, Ogren L, Talamantes F, Robertson M, Friesen HG, Sorenson RL . Effect of homologous placental lactogens, prolactins, and growth hormones on islet B-cell division and insulin secretion in rat, mouse, and human islets: implication for placental lactogen regulation of islet function during pregnancy. Endocrinology 1993; 132:879-887. 345. Sorenson RL, Brelje TC . Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res 1997; 29:301-307. 346. Sorenson RL, Brelje TC, Roth C . Effects of steroid and lactogenic hormones on islets of Langerhans: a new hypothesis for the role of pregnancy steroids in the adaptation of islets to pregnancy. Endocrinology 1993; 133:2227-2234. 347. Petryk A, Fleenor D, Driscoll P, Freemark M . Prolactin induction of insulin gene expression: the roles of glucose and glucose transporter-2. J Endocrinol 2000; 164:277286. 348. Fleenor DE, Freemark M . Prolactin induction of insulin gene transcription: roles of glucose and signal transducer and activator of transcription 5. Endocrinology 2001; 142:2805-2810. 349. Shao J, Qiao L, Friedman JE . Prolactin, progesterone, and dexamethasone coordinately and adversely regulate glucokinase and cAMP/PDE cascades in MIN6 beta-cells. Am J Physiol Endocrinol Metab 2004; 286:E304-E310. 350. Serri O, Li L, Mamputu JC, Beauchamp MC, Maingrette F, Renier G . The influences of hyperprolactinemia and obesity on cardiovascular risk markers: effects of cabergoline therapy. Clin Endocrinol (Oxf) 2006; 64:366-370. 351. Yavuz D, Deyneli O, Akpinar I, Yildiz E, Gozu H, Sezgin O, Haklar G, Akalin S . Endothelial function, insulin sensitivity and inflammatory markers in hyperprolactinemic premenopausal women. Eur J Endocrinol 2003; 149:187-193. 191 Bibliografía 352. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC . Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28:412-419. 353. Turner RC, Holman RR, Matthews D, Hockaday TD, Peto J . Insulin deficiency and insulin resistance interaction in diabetes: estimation of their relative contribution by feedback analysis from basal plasma insulin and glucose concentrations. Metabolism 1979; 28:1086-1096. 354. Vaccaro O, Masulli M, Cuomo V, Rivellese AA, Uusitupa M, Vessby B, Hermansen K, Tapsell L, Riccardi G . Comparative evaluation of simple indices of insulin resistance. Metabolism 2004; 53:1522-1526. 355. Wallace TM, Levy JC, Matthews DR . Use and abuse of HOMA modeling. Diabetes Care 2004; 27:1487-1495. 356. Vieira VJ, Valentine RJ, Wilund KR, Antao N, Baynard T, Woods JA . Effects of exercise and low-fat diet on adipose tissue inflammation and metabolic complications in obese mice. Am J Physiol Endocrinol Metab 2009; 296:E1164-E1171. 357. Al Goblan AS, Al Alfi MA, Khan MZ . Mechanism linking diabetes mellitus and obesity. Diabetes Metab Syndr Obes 2014; 7:587-591. 358. Baum JD, Ounsted M, Smith MA . Letter: Weight gain in infancy and subsequent development of diabetes mellitus in childhood. Lancet 1975; 2:866. 359. Wilkin TJ . The accelerator hypothesis: weight gain as the missing link between Type I and Type II diabetes. Diabetologia 2001; 44:914-922. 360. Karpe F, Dickmann JR, Frayn KN . Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes 2011; 60:2441-2449. 361. Sarafidis PA, Bakris GL . Non-esterified fatty acids and blood pressure elevation: a mechanism for hypertension in subjects with obesity/insulin resistance? J Hum Hypertens 2007; 21:12-19. 362. Sowell MO, Mukhopadhyay N, Cavazzoni P, Shankar S, Steinberg HO, Breier A, Beasley CM, Jr., Dananberg J . Hyperglycemic clamp assessment of insulin secretory responses in normal subjects treated with olanzapine, risperidone, or placebo. J Clin Endocrinol Metab 2002; 87:2918-2923. 363. Citrome L, Jaffe A, Levine J, Allingham B, Robinson J . Relationship between antipsychotic medication treatment and new cases of diabetes among psychiatric inpatients. Psychiatr Serv 2004; 55:1006-1013. 364. Marder SR, Essock SM, Miller AL, Buchanan RW, Casey DE, Davis JM, Kane JM, Lieberman JA, Schooler NR, Covell N, Stroup S, Weissman EM, Wirshing DA, Hall CS, Pogach L, PiSunyer X, Bigger JT, Jr., Friedman A, Kleinberg D, Yevich SJ, Davis B, Shon S . Physical health monitoring of patients with schizophrenia. Am J Psychiatry 2004; 161:1334-1349. 365. Chang AM, Halter JB . Aging and insulin secretion. Am J Physiol Endocrinol Metab 2003; 284:E7-12. 366. Ryan AS . Insulin resistance with aging: effects of diet and exercise. Sports Med 2000; 30:327-346. 367. Kim SY, Sung YA, Ko KS, Cho BY, Lee HK, Koh CS, Min HK . Direct relationship between elevated free testosterone and insulin resistance in hyperprolactinemic women. Korean J Intern Med 1993; 8:8-14. 192 Bibliografía 368. Pelkonen R, Nikkila EA, Grahne B . Serum lipids, postheparin plasma lipase activities and glucose tolerance in patients with prolactinoma. Clin Endocrinol (Oxf) 1982; 16:383-390. 369. Foss MC, Paula FJ, Paccola GM, Piccinato CE . Peripheral glucose metabolism in human hyperprolactinaemia. Clin Endocrinol (Oxf) 1995; 43:721-726. 370. Freemark M, Avril I, Fleenor D, Driscoll P, Petro A, Opara E, Kendall W, Oden J, Bridges S, Binart N, Breant B, Kelly PA . Targeted deletion of the PRL receptor: effects on islet development, insulin production, and glucose tolerance. Endocrinology 2002; 143:13781385. 371. Cerf ME . Beta cell dysfunction and insulin resistance. Front Endocrinol (Lausanne) 2013; 4:37. 372. Proenca AR, Sertie RA, Oliveira AC, Campana AB, Caminhotto RO, Chimin P, Lima FB . New concepts in white adipose tissue physiology. Braz J Med Biol Res 2014; 47:192-205. 373. Gregoire FM, Smas CM, Sul HS . Understanding adipocyte differentiation. Physiol Rev 1998; 78:783-809. 374. Mandrup S, Lane MD . Regulating adipogenesis. J Biol Chem 1997; 272:5367-5370. 375. MacDougald OA, Lane MD . Transcriptional regulation of gene expression during adipocyte differentiation. Annu Rev Biochem 1995; 64:345-373. 376. Darlington GJ, Ross SE, MacDougald OA . The role of C/EBP genes in adipocyte differentiation. J Biol Chem 1998; 273:30057-30060. 377. Spiegelman BM, Choy L, Hotamisligil GS, Graves RA, Tontonoz P . Regulation of adipocyte gene expression in differentiation and syndromes of obesity/diabetes. J Biol Chem 1993; 268:6823-6826. 378. Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, Satoh S, Nakano R, Ishii C, Sugiyama T, Eto K, Tsubamoto Y, Okuno A, Murakami K, Sekihara H, Hasegawa G, Naito M, Toyoshima Y, Tanaka S, Shiota K, Kitamura T, Fujita T, Ezaki O, Aizawa S, Kadowaki T, . PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell 1999; 4:597-609. 379. Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM . PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell 1999; 4:611-617. 380. Wu Z, Rosen ED, Brun R, Hauser S, Adelmant G, Troy AE, McKeon C, Darlington GJ, Spiegelman BM . Cross-regulation of C/EBP alpha and PPAR gamma controls the transcriptional pathway of adipogenesis and insulin sensitivity. Mol Cell 1999; 3:151-158. 381. Iizuka K . Recent progress on the role of ChREBP in glucose and lipid metabolism. Endocr J 2013; 60:543-555. 382. White UA, Stephens JM . Transcriptional factors that promote formation of white adipose tissue. Mol Cell Endocrinol 2010; 318:10-14. 383. Shimano H, Shimomura I, Hammer RE, Herz J, Goldstein JL, Brown MS, Horton JD . Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene. J Clin Invest 1997; 100:2115-2124. 384. Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, Amemiya-Kudo M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Nagai R, Ishibashi S, Yamada N . Absence of sterol regulatory element-binding protein-1 (SREBP-1) 193 Bibliografía ameliorates fatty livers but not obesity or insulin resistance in Lep(ob)/Lep(ob) mice. J Biol Chem 2002; 277:19353-19357. 385. Kim JB, Wright HM, Wright M, Spiegelman BM . ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc Natl Acad Sci U S A 1998; 95:43334337. 386. Diraison F, Dusserre E, Vidal H, Sothier M, Beylot M . Increased hepatic lipogenesis but decreased expression of lipogenic gene in adipose tissue in human obesity. Am J Physiol Endocrinol Metab 2002; 282:E46-E51. 387. Young SG, Zechner R . Biochemistry and pathophysiology of intravascular and intracellular lipolysis. Genes Dev 2013; 27:459-484. 388. Abel ED . A new twist in the function of the cardiac lipid droplet. Nat Med 2011; 17:10451046. 389. Agius L, Rolls BJ, Rowe EA, Williamson DH . High-energy diets produce different effects on fatty acid synthesis in brown adipose tissue, white adipose tissue and liver in the rat. Biochim Biophys Acta 1983; 750:383-387. 390. Flint DJ, Binart N, Kopchick J, Kelly P . Effects of growth hormone and prolactin on adipose tissue development and function. Pituitary 2003; 6:97-102. 391. Brandebourg T, Hugo E, Ben Jonathan N . Adipocyte prolactin: regulation of release and putative functions. Diabetes Obes Metab 2007; 9:464-476. 392. Hugo ER, Borcherding DC, Gersin KS, Loftus J, Ben Jonathan N . Prolactin release by adipose explants, primary adipocytes, and LS14 adipocytes. J Clin Endocrinol Metab 2008; 93:4006-4012. 393. Nilsson LA, Roepstorff C, Kiens B, Billig H, Ling C . Prolactin suppresses malonyl-CoA concentration in human adipose tissue. Horm Metab Res 2009; 41:747-751. 394. Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K . Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A 2004; 101:7281-7286. 395. Saltiel AR, Kahn CR . Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001; 414:799-806. 396. Stryer L, Berg JM, Tymoczko JL 2002 The Integration of Metabolism. In: W H Freeman (ed). Biochemistry.W H Freeman and Co., New York: 397. Foufelle F, Ferre P . New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem J 2002; 366:377-391. 398. Matsumoto M, Ogawa W, Teshigawara K, Inoue H, Miyake K, Sakaue H, Kasuga M . Role of the insulin receptor substrate 1 and phosphatidylinositol 3-kinase signaling pathway in insulin-induced expression of sterol regulatory element binding protein 1c and glucokinase genes in rat hepatocytes. Diabetes 2002; 51:1672-1680. 399. Chakravarty K, Leahy P, Becard D, Hakimi P, Foretz M, Ferre P, Foufelle F, Hanson RW . Sterol regulatory element-binding protein-1c mimics the negative effect of insulin on phosphoenolpyruvate carboxykinase (GTP) gene transcription. J Biol Chem 2001; 276:34816-34823. 194 Bibliografía 400. Foretz M, Guichard C, Ferre P, Foufelle F . Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci U S A 1999; 96:12737-12742. 401. Dentin R, Pegorier JP, Benhamed F, Foufelle F, Ferre P, Fauveau V, Magnuson MA, Girard J, Postic C . Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem 2004; 279:2031420326. 402. Rui L . Energy metabolism in the liver. Compr Physiol 2014; 4:177-197. 403. Watts LM, Manchem VP, Leedom TA, Rivard AL, McKay RA, Bao D, Neroladakis T, Monia BP, Bodenmiller DM, Cao JX, Zhang HY, Cox AL, Jacobs SJ, Michael MD, Sloop KW, Bhanot S . Reduction of hepatic and adipose tissue glucocorticoid receptor expression with antisense oligonucleotides improves hyperglycemia and hyperlipidemia in diabetic rodents without causing systemic glucocorticoid antagonism. Diabetes 2005; 54:18461853. 404. Andrews RC, Walker BR . Glucocorticoids and insulin resistance: old hormones, new targets. Clin Sci (Lond) 1999; 96:513-523. 405. Livingstone DE, Grassick SL, Currie GL, Walker BR, Andrew R . Dysregulation of glucocorticoid metabolism in murine obesity: comparable effects of leptin resistance and deficiency. J Endocrinol 2009; 201:211-218. 406. Livingstone DE, Jones GC, Smith K, Jamieson PM, Andrew R, Kenyon CJ, Walker BR . Understanding the role of glucocorticoids in obesity: tissue-specific alterations of corticosterone metabolism in obese Zucker rats. Endocrinology 2000; 141:560-563. 407. Rask E, Olsson T, Soderberg S, Andrew R, Livingstone DE, Johnson O, Walker BR . Tissuespecific dysregulation of cortisol metabolism in human obesity. J Clin Endocrinol Metab 2001; 86:1418-1421. 408. Morton NM, Paterson JM, Masuzaki H, Holmes MC, Staels B, Fievet C, Walker BR, Flier JS, Mullins JJ, Seckl JR . Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11 beta-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 2004; 53:931-938. 409. Hermanowski-Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC, Le Grand CB, Li Z, Metzger JM, Mundt SS, Noonan H, Nunes CN, Olson SH, Pikounis B, Ren N, Robertson N, Schaeffer JM, Shah K, Springer MS, Strack AM, Strowski M, Wu K, Wu T, Xiao J, Zhang BB, Wright SD, Thieringer R . 11beta-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med 2005; 202:517527. 410. Winkler R, Benz V, Clemenz M, Bloch M, Foryst-Ludwig A, Wardat S, Witte N, Trappiel M, Namsolleck P, Mai K, Spranger J, Matthias G, Roloff T, Truee O, Kappert K, Schupp M, Matthias P, Kintscher U . Histone deacetylase 6 (HDAC6) is an essential modifier of glucocorticoid-induced hepatic gluconeogenesis. Diabetes 2012; 61:513-523. 411. Opherk C, Tronche F, Kellendonk C, Kohlmuller D, Schulze A, Schmid W, Schutz G . Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol 2004; 18:1346-1353. 412. Lu Y, Xiong X, Wang X, Zhang Z, Li J, Shi G, Yang J, Zhang H, Ning G, Li X . Yin Yang 1 promotes hepatic gluconeogenesis through upregulation of glucocorticoid receptor. Diabetes 2013; 62:1064-1073. 195 Bibliografía 413. Henderson DC, Doraiswamy PM . Prolactin-related and metabolic adverse effects of atypical antipsychotic agents. J Clin Psychiatry 2008; 69 Suppl 1:32-44. 414. Baptista T, Zarate J, Joober R, Colasante C, Beaulieu S, Paez X, Hernandez L . Drug induced weight gain, an impediment to successful pharmacotherapy: focus on antipsychotics. Curr Drug Targets 2004; 5:279-299. 415. Scislowski PW, Tozzo E, Zhang Y, Phaneuf S, Prevelige R, Cincotta AH . Biochemical mechanisms responsible for the attenuation of diabetic and obese conditions in ob/ob mice treated with dopaminergic agonists. Int J Obes Relat Metab Disord 1999; 23:425431. 416. Zhang Y, Scislowski PW, Prevelige R, Phaneuf S, Cincotta AH . Bromocriptine/SKF38393 treatment ameliorates dyslipidemia in ob/ob mice. Metabolism 1999; 48:1033-1040. 417. Cincotta AH, Meier AH . Bromocriptine (Ergoset) reduces body weight and improves glucose tolerance in obese subjects. Diabetes Care 1996; 19:667-670. 418. Borcherding DC, Hugo ER, Idelman G, De Silva A, Richtand NW, Loftus J, Ben Jonathan N . Dopamine receptors in human adipocytes: expression and functions. PLoS ONE 2011; 6:e25537. 419. Narayanan NS, Guarnieri DJ, DiLeone RJ . Metabolic hormones, dopamine circuits, and feeding. Front Neuroendocrinol 2010; 31:104-112. 420. Brandebourg TD, Bown JL, Ben Jonathan N . Prolactin upregulates its receptors and inhibits lipolysis and leptin release in male rat adipose tissue. Biochem Biophys Res Commun 2007; 357:408-413. 421. Le JA, Wilson HM, Shehu A, Devi YS, Aguilar T, Gibori G . Prolactin activation of the long form of its cognate receptor causes increased visceral fat and obesity in males as shown in transgenic mice expressing only this receptor subtype. Horm Metab Res 2011; 43:931937. 422. Bonnet M, Faulconnier Y, Hocquette JF, Bocquier F, Leroux C, Martin P, Chilliard Y . Nutritional status induces divergent variations of GLUT4 protein content, but not lipoprotein lipase activity, between adipose tissues and muscles in adult cattle. Br J Nutr 2004; 92:617-625. 423. Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, Goldstein JL, Brown MS . SREBP1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 1993; 75:187-197. 424. Inoue J, Kumagai H, Terada T, Maeda M, Shimizu M, Sato R . Proteolytic activation of SREBPs during adipocyte differentiation. Biochem Biophys Res Commun 2001; 283:11571161. 425. Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS . Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J Biol Chem 2002; 277:95209528. 426. Shimano H, Horton JD, Shimomura I, Hammer RE, Brown MS, Goldstein JL . Isoform 1c of sterol regulatory element binding protein is less active than isoform 1a in livers of transgenic mice and in cultured cells. J Clin Invest 1997; 99:846-854. 427. Shimomura I, Bashmakov Y, Horton JD . Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem 1999; 274:3002830032. 196 Bibliografía 428. Carobbio S, Hagen RM, Lelliott CJ, Slawik M, Medina-Gomez G, Tan CY, Sicard A, Atherton HJ, Barbarroja N, Bjursell M, Bohlooly Y, Virtue S, Tuthill A, Lefai E, Laville M, Wu T, Considine RV, Vidal H, Langin D, Oresic M, Tinahones FJ, Fernandez-Real JM, Griffin JL, Sethi JK, Lopez M, Vidal-Puig A . Adaptive changes of the Insig1/SREBP1/SCD1 set point help adipose tissue to cope with increased storage demands of obesity. Diabetes 2013; 62:3697-3708. 429. Herman MA, Peroni OD, Villoria J, Schon MR, Abumrad NA, Bluher M, Klein S, Kahn BB . A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012; 484:333-338. 430. Horton JD, Bashmakov Y, Shimomura I, Shimano H . Regulation of sterol regulatory element binding proteins in livers of fasted and refed mice. Proc Natl Acad Sci U S A 1998; 95:5987-5992. 431. Kim JB, Sarraf P, Wright M, Yao KM, Mueller E, Solanes G, Lowell BB, Spiegelman BM . Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest 1998; 101:1-9. 432. Ling C, Svensson L, Oden B, Weijdegard B, Eden B, Eden S, Billig H . Identification of functional prolactin (PRL) receptor gene expression: PRL inhibits lipoprotein lipase activity in human white adipose tissue. J Clin Endocrinol Metab 2003; 88:1804-1808. 433. Julve J, Robert MQ, Llobera M, Peinado-Onsurbe J . Hormonal regulation of lipoprotein lipase activity from 5-day-old rat hepatocytes. Mol Cell Endocrinol 1996; 116:97-104. 434. Weinstock PH, Bisgaier CL, Aalto-Setala K, Radner H, Ramakrishnan R, Levak-Frank S, Essenburg AD, Zechner R, Breslow JL . Severe hypertriglyceridemia, reduced high density lipoprotein, and neonatal death in lipoprotein lipase knockout mice. Mild hypertriglyceridemia with impaired very low density lipoprotein clearance in heterozygotes. J Clin Invest 1995; 96:2555-2568. 435. Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, Ratziu V, Serfaty L, Housset C, Capeau J, Girard J, Guillou H, Postic C . The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest 2012; 122:2176-2194. 436. Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, Girard J, Postic C . Liverspecific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 2006; 55:2159-2170. 437. Iizuka K, Miller B, Uyeda K . Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. Am J Physiol Endocrinol Metab 2006; 291:E358-E364. 438. SALAS M, VINUELA E, SOLS A . INSULIN-DEPENDENT SYNTHESIS OF LIVER GLUCOKINASE IN THE RAT. J Biol Chem 1963; 238:3535-3538. 439. Cone RD . The central melanocortin system and energy homeostasis. Trends Endocrinol Metab 1999; 10:211-216. 440. Cone RD, Cowley MA, Butler AA, Fan W, Marks DL, Low MJ . The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int J Obes Relat Metab Disord 2001; 25 Suppl 5:S63-S67. 441. Sauve D, Woodside B . The effect of central administration of prolactin on food intake in virgin female rats is dose-dependent, occurs in the absence of ovarian hormones and the latency to onset varies with feeding-regimen. Brain Res 1996; 729:75-81. 197 Bibliografía 442. Hnasko TS, Perez FA, Scouras AD, Stoll EA, Gale SD, Luquet S, Phillips PE, Kremer EJ, Palmiter RD . Cre recombinase-mediated restoration of nigrostriatal dopamine in dopamine-deficient mice reverses hypophagia and bradykinesia. Proc Natl Acad Sci U S A 2006; 103:8858-8863. 443. Schwartz MW, Woods SC, Porte D, Jr., Seeley RJ, Baskin DG . Central nervous system control of food intake. Nature 2000; 404:661-671. 444. Zhou QY, Palmiter RD . Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell 1995; 83:1197-1209. 445. Hirschberg AL . Sex hormones, appetite and eating behaviour in women. Maturitas 2012; 71:248-256. 446. Li C, Chen P, Smith MS . Neuropeptide Y and tuberoinfundibular dopamine activities are altered during lactation: role of prolactin. Endocrinology 1999; 140:118-123. 447. Chen AS, Metzger JM, Trumbauer ME, Guan XM, Yu H, Frazier EG, Marsh DJ, Forrest MJ, Gopal-Truter S, Fisher J, Camacho RE, Strack AM, Mellin TN, MacIntyre DE, Chen HY, Van der Ploeg LH . Role of the melanocortin-4 receptor in metabolic rate and food intake in mice. Transgenic Res 2000; 9:145-154. 448. Augustine RA, Ladyman SR, Grattan DR . From feeding one to feeding many: hormoneinduced changes in bodyweight homeostasis during pregnancy. J Physiol 2008; 586:387397. 449. Naef L, Woodside B . Prolactin/Leptin interactions in the control of food intake in rats. Endocrinology 2007; 148:5977-5983. 450. Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG . Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes 1997; 46:2119-2123. 451. Doknic M, Pekic S, Zarkovic M, Medic-Stojanoska M, Dieguez C, Casanueva F, Popovic V . Dopaminergic tone and obesity: an insight from prolactinomas treated with bromocriptine. Eur J Endocrinol 2002; 147:77-84. 452. Kastin AJ, Redding TW, Hall R, Besser GM, Schally AV . Lipid mobilizing hormones of the hypothalamus and pituitary. Pharmacol Biochem Behav 1975; 3:121-126. 453. Wellhoner P, Horster R, Jacobs F, Sayk F, Lehnert H, Dodt C . Intranasal application of the melanocortin 4 receptor agonist MSH/ACTH(4-10) in humans causes lipolysis in white adipose tissue. Int J Obes (Lond) 2011. 454. Kahn SE, Hull RL, Utzschneider KM . Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006; 444:840-846. 455. Laurencikiene J, Skurk T, Kulyte A, Heden P, Astrom G, Sjolin E, Ryden M, Hauner H, Arner P . Regulation of lipolysis in small and large fat cells of the same subject. J Clin Endocrinol Metab 2011; 96:E2045-E2049. 198