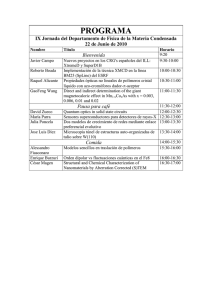
AP.T5.1-MPyC.Tema5.Adtivos.PropiedadesFisicasQuimicas
Anuncio

ASIGNATURA: MATERIALES POLIMÉRICOS Y COMPUESTOS. Tema 5.- ADITIVOS. PROPIEDADES FÍSICAS Y QUÍMICAS. ÍNDICE 1.-Fabricación de monómeros. 2.- Materiales de adición a los polímeros. 2.1.- Introducción. 2.2.- Aditivos 2.2.1.- Introducción 2.2.2.- Lubricantes 2.2.3.- Plastificantes 2.2.4.- Antioxidantes 2.2.5.- Estabilizantes térmicos 2.2.6.- Estabilizantes frente a la radiación ultravioleta 2.2.7.- Retardantes a la llama 2.2.8.- Espumantes 2.2.9.- Colorantes 2.2.10.- Agentes de curado 2.2.11.- Antiestáticos 2.2.12.- Biocidas 2.3.- Cargas 2.4.- Refuerzos. 3.- Propiedades de los materiales poliméricos 3.1.- Introducción 3.2.- Rozamiento y desgate 3.3.- Propiedades eléctricas 3.4.- Propiedades ópticas 3.5.- Propiedades térmicas 3.6.- Permeabilidad a los gases y vapores 3.7.- Estabilidad a altas temperaturas y comportamiento en el fuego 3.8.- Resistencia a los disolventes y reactivos químicos 1.-Fabricación de monómeros. Las materias primas utilizadas en la obtención de los polímeros de síntesis provienen de los recursos naturales (Figura 1.1). Éstos se clasifican en renovables -los procedentes de los seres vivos- y los no renovables, que son los recursos fósiles. Figura 1.1.Clasificación de los recursos naturales. En los seres vivos existen compuestos de carácter macromolecular. Del reino animal destacan las proteínas, el colágeno, la seda, la caseína, etc., y del reino vegetal, el almidón, la celulosa, el látex, etc., como los más conocidos. Con modificaciones químicas adecuadas estos polímeros llegan a ser considerados polímeros semisintéticos: el rayón, el acetato de celulosa, el caucho, etc. En la figura 1.2 se muestra la producción de polímeros de petroquímica respecto a la materia prima en el año 2006. Figura 1.2.- Producción de polímeros de petroquímica respecto a la materia prima En la figura 1.3 se muestra los principales polímeros producidos de biomasa en el año 2007. Figura 1.3.- Principales polímeros producidos de biomasa. Pero para la obtención de los polímeros de síntesis, se utilizan los recursos fósiles. De ellos es el petróleo la materia prima base para la obtención de los plásticos, como consecuencia de la facilidad de extracción del mismo y del desarrollo alcanzado por la tecnología para transformarlo en derivados. Estos dos hechos han supuesto el desplazamiento del carbón por el petróleo con fines sintéticos, ya que en el siglo XIX el carbón era la fuente fundamental de obtención de productos de carácter orgánico y que dio lugar al desarrollo tan importante que alcanzó la industria carboquímica. Con la crisis del petróleo en los años setenta por exigencias económicas de los países productores, el carbón fue considerado nuevamente como alternativa a tener en cuenta para poder mantener a unos niveles aceptables el actual desarrollo económico. Pero mientras no se ponga a punto la tecnología adecuada para transformar este recurso en los productos demandados por la industria, similares a los derivados petrolíferos, no resulta interesante la sustitución del petróleo por el carbón. El gas natural se utiliza fundamentalmente con fines energéticos, ya que dada su composición a base de hidrocarburos de bajo peso molecular, las aplicaciones con fines sintéticos están económicamente limitadas. La primera operación a que se somete el petróleo bruto para su utilización posterior en la industria petroquímica se representa en la figura 1.2 (refinado). El refinado consiste en la separación de los distintos componentes del petróleo por acción del calor. Es una destilación fraccionada en la que se separan a diferentes intervalos de temperatura mezclas de compuestos de tamaño y composición similar. Figura 1.2.- Esquema de la destilación fraccionada del petróleo (refinado). De las fracciones obtenidas, la nafta, que es una mezcla de hidrocarburos de más de cinco átomos de carbono y que tiene un punto de ebullición de hasta 150 º, es la que se utiliza para la fabricación de los plásticos sometiéndola previamente a los procesos de craqueo y reformado, que se reflejan en la figura 1.3. El craqueo es el proceso en el que se produce la ruptura de cadenas hidrocarbonadas dando lugar a moléculas pequeñas de dos a cuatro átomos de carbono con dobles enlaces. Si el proceso se realiza en presencia de vapor de agua, éste se denomina hidrocraqueo. Puede alimentarse el reactor también con fracciones más pesadas -de mayor tamaño-. El craqueo puede realizarse de manera térmica o catalítica. Figura 1.3.- Transformaciones de la nafta hacia olefinas y aromáticos. El reformado es el proceso mediante el cual se modifica la estructura de las moléculas para obtener fundamentalmente compuestos aromáticos, conocidos como fracción BTX (Benceno, Tolueno, Xilenos). Mediante las transformaciones de la nafta (Figura 1.3) se obtienen los dos pilares fundamentales en los que se basa la industria petroquímica: las olefinas y los aromáticos. Ambos son la base de la industria química orgánica actual (plásticos, colorantes, detergentes, pinturas, fármacos, fitosanitarios, etc.). En la figura 1.4 se esquematiza la transformación hacia los materiales poliméricos. Figura 1.4.- Conversión de olefinas y aromáticos a polímeros. 2.- Materiales de adición a los polímeros. 2.1.- Introducción. La transformación de los materiales plásticos se realiza fundamentalmente por técnicas de extrusión y de moldeo. Se han hecho algunos ensayos utilizando las técnicas clásicas empleadas para los materiales metálicos con arranque de viruta utilizando el torno y la fresa, pero no han dado buenos resultados en algunos tipos de polímeros como consecuencia del calentamiento local que se crea en el material y que puede originar deformaciones. Las técnicas de moldeo se utilizan tanto en los materiales termoplásticos como en los termofijos. Cada uno de ellos presenta unas características diferentes como consecuencia de la diferencia estructural que presentan. En los materiales termofijos el moldeo se realiza sobre el prepolímero, al que se ha adicionado un entrecruzante o agente de curado. Se introduce en el molde la mezcla que bien puede ser de forma manual o inyectada, y se les somete a presión y temperatura teniendo lugar la reacción de entrecruzamiento. Se obtiene de esta manera el artículo con la forma geométrica deseada. El producto puede ser desmoldeado aunque el molde se mantenga caliente, ya que el material es dimensionalmente estable. En los materiales termoplásticos el moldeo se realiza pasando el material a estado viscoso y fluido. Esto se consigue elevando la temperatura. En esta situación el material ocupa el molde y al enfriarse adquiere su forma. Pero antes de someter el material al proceso de moldeo, hay que incorporar al polímero distintas sustancias que modifiquen sus propiedades mejorándolas y que faciliten la transformación. Es decir primeramente hay que aditivar la macromolécula. La mayoría de los productos de plástico consisten en un material polimérico que ha sido alterado para modificar o mejorar determinadas propiedades. Para utilizar un polímero en la elaboración de productos, por lo común, hay que mezclarlo con otros ingredientes especiales, los cuales tienen varios fines. Los procesos para mezclar también brindan una oportunidad de modificar la forma física del polímero para que se maneje fácilmente en la etapa de transformación final de preparación, pero el propósito principal es el de la introducción de aditivos. Los aditivos son sustancias dispersas en la masa de polímero que se utilizan ampliamente en todo tipo de polímeros: termoplásticos, termofijos y cauchos. Han de cumplir ciertos requerimientos para poder ser incorporados al material, como no ser volátiles a la temperatura de transformación, ni migrar hacia el exterior para no producir contaminación en los productos que estén en contacto con los mismos. No deben tener ningún efecto nocivo ni durante su manipulación ni durante su uso. La mayoría de las propiedades de los polímeros discutidos anteriormente son intrínsecas, es decir, son características esenciales del polímero específico. Algunas de estas propiedades se relacionan y se controlan con la estructura molecular. Sin embargo, muchas veces es necesario modificar las propiedades mecánicas, químicas y físicas en un grado mucho mayor que el permitido por la simple alteración de la estructura molecular fundamental. Por ello, substancias alógenas, que se pueden denominar materiales de adición, se introducen intencionadamente para modificar muchas de esas propiedades y para aumentar la utilidad del polímero, por ejemplo, haciéndolo más duro o más flexible o más barato. Otra función de los materiales que se adicionan es evitar la degradación del polímero cuando se use o durante su tratamiento o en ambos casos por medio de aditivos apropiados. Los materiales que se adicionan a los polímeros se pueden clasificar en tres grandes grupos: - Cargas (Fillers) Aditivos Refuerzos cuyas características y ejemplos de material pueden verse en la tabla 2.1.1 Tabla 2.1.1.- Características y ejemplos de materiales de adición. Las cargas pueden emplearse en grandes cantidades siempre que no comprometan el resto de propiedades del material. Otra clasificación es dividirlos en dos tipos: modificadores y protectores. En la tabla 2.1.2 se muestran los distintos materiales de adición según la función principal que realizan en el material polimérico. La mayor proporción de aditivos de los polímeros corresponde a las cargas y plastificantes, como muestra la figura 2.1.1. Tabla 2.1.2.-Tipos de aditivos según la función que realizan en el material. Aditivo (Denominación) Funciones Lubricantes Plastificantes Cargas, diluyentes y extendedores Cargas reforzantes Antiestáticos Promotores de la adhesión Colorantes Antioxidantes Estabilizantes contra la radiación ultravioleta Ignifugantes (Retardadores de llama) Agentes espumantes Biocidas Facilitan la transformación Modifican las propiedades mecánicas Abaratar el material Mejorar las propiedades mecánicas Modificadores superficiales Modificadores superficiales Modificadores de las propiedades ópticas Aditivos antienvejecimiento Aditivos antienvejecimiento Figura 2.1.1.- Utilización de los diferentes tipos de aditivos. Algunas de las razones por las que se incluyen aditivos, refuerzos y cargas son: • Mejorar la capacidad de tratamiento. • Reducir los costes del material. • Reducir la contracción. • Permitir temperaturas de curado superiores reduciendo o diluyendo materiales reactivos. • Mejorar el acabado de superficie. • Modificar las propiedades térmicas como, por ejemplo, el coeficiente de expansión, la inflamabilidad y la conductividad. • Mejorar las propiedades eléctricas, incluyendo la conductividad o la resistencia. • Prevenir la degradación durante la fabricación y el servicio. • Conseguir un tinte o color determinado. • Mejorar propiedades mecánicas como, por ejemplo, el módulo, la resistencia, la dureza, la resistencia a la abrasión y la tenacidad. • Reducir el coeficiente de rozamiento. Existen multitud de sustancias químicas útiles para los materiales plásticos que producen los cambios en las propiedades pretendidos. No obstante, algunas de las sustancias químicas más rentables son también peligrosas e incluso tóxicas. Un material polimérico se basa en una estructura orgánica y representa el material básico para obtener productos para aplicaciones industriales, uno de tales materiales poliméricos puede ser el polietileno. No obstante, este polímero base en sus aplicaciones corrientes puede necesitar mejorar sus propiedades mecánicas, protegido frente a la acción de los agentes externos (Frente a la luz, a la oxidación, frente a la combustión a temperaturas elevadas, ..), también puede que sea necesario modificar alguna propiedad (densidad, conductividad eléctrica, propiedades químicas,..) o bien simplemente es necesario reducir los costes del producto final (Figura 2.1.2). Todo ello plantea la necesidad de incorporar, al polímero base, unos determinados materiales de adición con el fin de conseguir dichos objetivos. Para mejorar las propiedades mecánicas es necesario utilizar refuerzos, para proteger a los materiales poliméricos frente a la acción de agentes externos se van a utilizar los aditivos de protección, para modificar selectivamente algunas propiedades se utilizarán los aditivos de modificación y, por último, con el fin de reducir los costes vamos a utilizar cargas o “fillers” (Figura 2.1.2). Figura 2.1.2.- Ejemplo de la necesidad de incorporar materiales de adición al polímero base. Así, dependiendo de la aplicación final de un polímero, se requiere el empleo de diferentes materiales de adición que se incorporan a la formulación del material plástico con diferente finalidad, por ejemplo, protegerlos frentes a agentes externos o dar o mejorar un determinada propiedad. 2.2.- Aditivos. 2.2.1.- Introducción. La finalidad de incorporación de aditivos en un material polimérico puede obedecer a dos razones: 1.- Protección frente a agentes externos Durante la vida en servicio un plástico se encuentra expuesto a agentes ambientales que pueden provocar su degradación (luz ultravioleta y oxidación). En determinadas ocasiones, los plásticos se encuentran sometidos a la acción de temperaturas elevadas en atmosferas oxidantes, que pueden inducir procesos de combustión en contacto con aire. 2.- Modificación de propiedades de forma selectiva El empleo de polímeros en aplicaciones de ingeniería requiere, en muchas ocasiones, la modificación de alguna propiedad (física, química, térmica, eléctrica, bioquímica....) para ampliar el espectro de utilización de estos materiales, ya que se puede adaptar las propiedades según sea la aplicación. El polietileno es un polímero que es la base para la obtención de un plástico con aplicaciones en ingeniería, no obstante este polímero base en condiciones habituales puede estar sometido a temperaturas elevadas, en muchas ocasiones puede estar en contacto con el aire y expuesto a la luz, pero también puede interesar modificar su propiedades como aumentar su ligereza, mejorar su comportamiento eléctrico mediante la disipación de cargas, que sea capaz de soportar impactos o que sea más menos flexible. En tal caso, es necesario incorporar aditivos para conseguir dichos objetivos, tal y como se muestra en la figura 2.2.1.1. Figura 2.2.1.1.- Incorporación de aditivos al polietileno para conseguir determinados objetivos. En relación a los aditivos de protección frente a agentes externos hay que destacar que la luz ultravioleta y la oxidación son dos fenómenos a los cuales se van a enfrentar los materiales poliméricos casi en su totalidad. La luz ultravioleta y la oxidación son los fenómenos responsables de la formación de radicales libres y estos, a su vez, son los responsables del envejecimiento del material, que provoca que se vuelvan amarillos o que el material se oscurezca, fragilidad y, en general, pérdida de propiedades. Con el fin de evitar esta degradación de las propiedades del material polimérico es necesario utilizar estabilizantes a la luz para proteger frente a la luz ultravioleta y estabilizantes antioxidantes para proteger frente a la oxidación, fundamentalmente durante el procesado de los materiales. Por otro lado, un material polimérico es de naturaleza combustible, por lo que es importante tener en cuenta que habitualmente se encuentra en contacto con el aire, la cual aporta O2 que es un comburente, si esto se combina de forma simultánea con elevadas temperaturas, se dispone de los elementos necesarios para tenga lugar un proceso de combustión, que provoca una degradación importante en el material. Para evitar dicha degradación, es necesario incorporar unos aditivos que protegen frente a estos fenómenos de combustión, que son los aditivos retardantes de llama o ignifugantes que trabajan de forma selectiva para disminuir la naturaleza combustible , disminuir el contacto con el comburente y reducir las elevadas temperaturas típicas de un proceso de combustión. Los aditivos se emplean mucho en las aplicaciones de los plásticos en construcción y en todos aquellos casos en los cuales se necesite un buen comportamiento frente al fuego. A continuación, veamos algunas características y posibilidades que ofrecen los aditivos de modificación de propiedades, lo que es necesario para modificar alguna propiedad con el fin de ampliar las posibilidades de aplicación en nuevos sectores industriales, simplemente aportando propiedades que el polímero base inicialmente no poseía. Así, en la tabla 2.2.1.1 se pueden ver distintas posibilidades de modificación de diferentes propiedades. Tabla 2.2.1.1.- Distintas posibilidades de modificación de diferentes propiedades. SOPORTAR IMPACTOS BRUSCOS IMPACTO POR PARTE DE MATERIALES FRÁGILES → AD. MODIFICADORES 2.2.2.- Lubricantes. Es importante que los polímeros no se peguen a la maquinaria de fabricación y procesado y que los objetos moldeados puedan extraerse del molde con facilidad. Los agentes de ayuda al procesado evitan este pegado y pueden disminuir la viscosidad. . Una vez que se extraen los productos del molde, los lubricantes pueden exudar desde el plástico y evitar que los productos se adhieran entre sí, para proporcionar características antiadherentes y de deslizamiento a la superficie plástica. Se incorporan para disminuir las fuerzas de rozamiento entre dos superficies, que en el caso de los plásticos se localizan entre el propio material y las existentes entre el fundido y las superficies metálicas de los equipos de transformación. En el primer caso mejoran el flujo por reducción de la viscosidad del material durante el procesado y en el segundo reducen la adhesión con las paredes metálicas del equipo facilitando el paso del material por la maquinaria que se utiliza en el proceso. Suelen ser productos específicos para cada polímero a transformar. Generalmente los lubricantes son sales sódicas del ácido esteárico, ceras, aceites, etc. Existe una enorme gama de marcas patentadas que se pueden utilizar en tan variados productos, y diversas condiciones de obtención. Se clasifican, por lo común, en dos grupos. 1.- Lubricantes internos, los cuales lubrican los gránulos de polímero, y los de otros aditivos, durante el proceso de transformación. Esto permite una fusión más fácil y más fría, con un riesgo reducido de daño térmico. Estos materiales son, con frecuencia, por lo menos parcialmente miscibles con el polímero fundido. 2.- Los lubricantes externos, que son esencialmente inmiscibles. Lubrican la mezcla para proteger la maquinaria que se usa en el proceso, de esta manera se logra el grado correcto de fricción para que se lleve a cabo el proceso y se evita el exceso de fricción que provocaría también una temperatura local demasiado alta y la degradación. Es muy importante tener la proporción correcta de lubricante en la preparación, ya que un exceso deteriora la mezcla y el proceso posterior. Se emplean muchos lubricantes distintos como ingredientes en los plásticos. Como ejemplos, se pueden mencionar ceras, como ozoquerita, carnauba, parafina y ácido esteárico. Asimismo, se emplean como lubricantes jabones metálicos como estearatos metálicos de plomo, cadmio, bario, calcio y zinc (Tabla 2.2.1). La mayor parte del lubricante se pierde durante el proceso de fabricación de la resina. El exceso de lubricante puede suponer una polimerización más lenta o causar eflorescencia de lubricación, que se manifiesta como un parche irregular y enturbiado en la superficie plástica. Tabla 2.2.2.1.- Lista de los aditivos lubricantes. Algunos plásticos presentan propiedades de antipegajosidad y autolubricantes. Entre los ejemplos se incluyen polifluorocarbonos, poliamidas, polietileno y plásticos de silicona. A veces, se utilizan como lubricantes en otros polímeros. No debe olvidarse que deben seleccionarse cuidadosamente los aditivos por sus posibles efectos tóxicos y según el servicio pretendido. 2.2.3.- Plastificantes. La plasticidad se refiere a la capacidad de un material para fluir o hacerse líquido bajo la influencia de una fuerza. Un plastificante es un agente químico, a ser posible incoloro e inodoro, que se añade al material polimérico con una doble finalidad: (i).- Facilitar el procesado al reducir la temperatura de fundido y la viscosidad. (ii).- Aportar flexibilidad al plástico transformado (formación de estructuras flexibles). Los plastificantes deben ser compatibles con el polímero base, presentar muchos grupos polares, ya que la presencia de grupos polares permite el establecimiento de interacciones con la resina polimérica. Además deben presentar un peso molecular intermedio para evitar problemas de migración y una viscosidad adecuada para poder ser procesados mediante procesos convencionales. De acuerdo con la definición de las normas ASTM (ASTMD883), un plastificante es un material que se incorpora a un material polimérico para facilitar su procesado y mejorar su flexibilidad o "distensibilidad". La adición de un plastificante puede hacer que disminuya la viscosidad en estado fundido, el módulo de elasticidad, y la temperatura de transición vítrea (Tg) de un polímero. Se trata de conocer algunos sistemas de plastificación, así como las condiciones que se deben cumplir para que un determinado compuesto pueda actuar como plastificante de un material polimérico. Se incorporan, pues para facilitar la transformación del material y aumentar la resistencia al impacto, ya que adicionados en pequeñas cantidades aumentan la flexibilidad. La ductilidad y la tenacidad de los polímeros también pueden mejorarse con la ayuda de los plastificantes. Su presencia también reduce la dureza y la fragilidad. Existe una amplia variedad de materiales poliméricos que presentan una estructura que les confiere una elevada rigidez. Es el caso el policloruro de vinilo (PVC) en el cual la presencia del grupo lateral cloro en la estructura da lugar a que el polímero sea rígido. Haciendo una representación esquemática se tendría: donde los anclajes de cada una de las cadenas representa los grupos cloro y se puede observar que en este tipo de estructuras la movilidad está restringida, porque existe un anclaje mecánico entre los grupos cloro de las distintas cadenas poliméricas. Ello da lugar a una baja movilidad de las cadenas poliméricas y a un elevado valor de la temperatura de transición vítrea, Tg (85-90 ºC). A temperatura ambiente su comportamiento será vítreo (duro y frágil), luego en este caso puede ser interesante utilizar agentes plastificantes para aumentar a temperatura ambiente la flexibilidad. El nitrato de celulosa (CN) producido por Schonbein en 1846 fue de uso limitado hasta que Parkes utilizó aceite de ricino como aditivo en 1865. En 1870 Hyatt utilizó alcanfor como plastificante para el CN. En 1910 se utilizó otro plastificante, el fosfato de tricresilo (TCP) como sustituto parcial del alcanfor y para reducir la inflamabilidad del celuloide. En 1933 Waldo Semon patentó el uso de fosfato de tricresilo como plastificante para el PVC. Más tarde este plastificante fue sustituido por uno menos tóxico, el ftalato de di-2-etilhexilo (DOP), que es en la actualidad uno de los plastificantes más ampliamente utilizado. Sin embargo, debido a los posibles problemas relacionados con la migración y la toxicidad de los ftalatos se han sustituido por otros de baja migración y baja toxicidad como los carboxilatos, sebacatos, materiales poliméricos. En los últimos años se está trabajando mucho con los derivados de los aceites vegetales epoxidados (EVO) como el aceite de soja, de ricino y de linaza, que dan muy buenos resultados en el caso del PVC. El efecto de los plastificantes puede explicarse mediante las teorías de la lubricación, de gel y de volumen libre. La primera afirma que los plastificantes se comportan como lubricantes internos y que permiten que las cadenas de polímeros se deslicen entre sí. La teoría del gel, que se aplica a los polímeros amorfos, supone que un polímero como el PVC tiene muchas fuerzas de atracción intermoleculares que se debilitan en presencia de un plastificante como el DOP. Se supone que la adición de un plastificante aumenta el volumen libre de un polímero y que el volumen libre es el mismo para todos los polímeros a la temperatura Tg. En la figura 2.2.3.1 puede verse el mecanismo de plastificación, el cual se produce en diversas etapas, a medida que aumenta la temperatura, hasta alcanzar temperaturas superiores a la de fusión del material. Figura 2.2.3.1.- Mecanismo de plastificación Los efectos de la plastificación son: la absorción de moléculas de plastificante por parte de la resina da lugar a una total interacción entre ambos componentes de la formulación, esta interacción intensa permite que el plastificante se interponga entre las cadenas poliméricas ejerciendo una función de lubricación interna y ello da lugar a una reducción de la temperatura de transición vítrea Tg. Los plastificantes suelen tener baja presión de vapor y bajo peso molecular. Las diminutas moléculas de los plastificantes ocupan posiciones entre las grandes cadenas poliméricas, incrementando la distancia entre cadenas y reduciendo los enlaces secundarios intermoleculares. Generalmente se utilizan plastificantes en la elaboración de polímeros frágiles a temperatura ambiente, tales como cloruro de polivinilo y algún copolímero del acetato. Los plastificantes disminuyen la temperatura de transición vítrea y de este modo los polímeros se pueden utilizar a temperatura ambiente en aplicaciones que requieren algún grado de flexibilidad y de ductilidad. Estas aplicaciones incluyen láminas delgadas o películas, tubos, impermeables y cortinas. Puesto que los plastificantes son fundamentalmente disolventes no volátiles, la compatibilidad hace que sea necesario que la diferencia de parámetros de solubilidad del plastificante y del polímero (δ) sea inferior a 1.8H (Tabla 2.2.3.1). Es interesante resaltar que el valor de δ para el PVC es 9.66 H y para el DOP es 8.85 H, de modo que, δ = 0.81H para este sistema de resina-plastificante de uso tan extendido. Cuanto más similar sea el parámetro de solubilidad mayor facilidad para que un plastificante pueda plastificar a un material poliméricos. Cuando los plastificantes se hallan presentes en cantidades pequeñas, normalmente se comportan como antiplastificantes, es decir, aumentan la dureza y disminuyen el alargamiento de los polímeros. Las propiedades varían notablemente según sea la cantidad de plastificante incorporado, pasando de ser un material vítreo hasta un material gomoso. El PVC, por ejemplo, es uno de los materiales representativos de la utilización de plastificantes en su formulación. Así existe en forma de plástico rígido (tuberías sanitarias, persianas) o plástico flexible (recubrimiento de cables de la luz, sustituto del cuero, etc.). Los plastificantes poco eficaces requieren un uso en cantidades relativamente grandes para superar la antiplastificación inicial. Por el contrario, los plastificantes buenos, como el DOP, cambian su comportamiento de antiplastificante a plastificante cuando se añaden al PVC en cantidades inferiores al 10 %. Tabla 2.2.3.1.- Parámetros de solubilidad de algunos plastificantes típicos. Tabla 2.2.3.1.- Parámetros de solubilidad de algunos plastificantes típicos. El desarrollo de los plastificantes ha estado plagado de problemas de toxicidad. Así, por ejemplo, los bifenilos policlorados de alta toxicidad no se usan ya como plastificantes. Los ésteres de ácido ftálico, como el DOP, pueden extraerse de la sangre almacenada en bolsas y tubos de PVC plastificado. Estos ésteres aromáticos pueden desprenderse también de las tapicerías de PVC para automóviles cuando hace calor. Estos problemas han sido resueltos utilizando poliésteres oligoméricos como plastificantes no migradores en lugar del DOP. Muchos copolímeros, como el poli(acetato de vinilococloruro de vinilo) se plastifican internamente debido a la flexibilización ocasionada por el cambio de estructura de la cadena del polímero. Por el contrario, el DOP y el TCP se denominan plastificantes externos. La presencia de grupos voluminosos en la cadena del polímero aumenta el movimiento segmental. Por tanto, la flexibilidad se incrementará con el tamaño de los grupos pendientes. Sin embargo, los grupos voluminosos lineales de más de diez átomos de carbono reducen la flexibilidad debido a la cristalización de cadenas laterales cuando los grupos se hallan espaciados regularmente. La conservación del plastificante todavía es un problema serio, especialmente durante períodos de uso largos. Por ejemplo, la mayoría de las baldosas de plástico se hacen más frágiles después de un uso prolongado, debido principalmente al desprendimiento del plastificante. Esta dificultad está siendo superada mediante una serie de métodos que incluyen el tratamiento superficial del producto dando lugar a superficies menos porosas y el uso de polímeros ramificados que pueden autoplastificarse. A1 ser polímeros ellos mismos, los polímeros muy ramificados son de desprendimiento lento debido al enmarañamiento físico con la matriz del polímero. Se han formulado más de 500 plastificantes diferentes para modificar polímeros. Los plastificantes son ingredientes cruciales en recubrimientos, extrusión, moldeo, adhesivos y películas de plástico. Los plastificantes suelen ser ésteres de alta temperatura de ebullición tales como los ftalatos de largas cadenas alifáticas (ftalato de dioctilo DOP cuyo uso está muy extendido) o ésteres alifáticos (adipato de dioctilo, DOA). Algunos son peligrosos. La EPA concluyó que los plastificantes de ftalato de di-2-etilhexilo son cancerígenos para los animales en ensayos de laboratorio. Actualmente, este producto se considera un cancerígeno potencial. En la tabla 2.2.3.2 se enumeran algunos plastificantes, mostrando la compatibilidad con el polímero. El agua es un plastificante de uso muy extendido en la naturaleza que permite la flexibilidad de gran parte del cuerpo humano además de la elasticidad de las flores, las hojas, las ramas de los árboles, etc. Las grasas y muchas proteínas también se comportan como plastificantes en el mundo animal. La producción mundial anual de plastificantes es de 3.2 millones de toneladas, siendo la de los EE.UU. superior al millón de toneladas. De hecho, los plastificantes son componentes principales de una serie de productos poliméricos. Por ejemplo, el vidrio de seguridad para automóviles está compuesto normalmente de polibutiral de vinilo y de un 30 % de plastificante. Tabla 2.2.3.2.- Compatibilidad de determinados polímeros y resinas. 2.2.4.- Antioxidantes. Los antioxidantes se usan para proteger a los polímeros contra la oxidación atmosférica, ya que muchos tienen en sus cadenas moleculares sitios que son susceptibles al ataque del oxígeno del aire. Se trata de identificar los mecanismos de actuación de la oxidación y las etapas de que consta, y así establecer los mecanismos de protección frente a la oxidación. Los fenómenos de oxidación de los materiales poliméricos son de naturaleza muy distinta a la "oxidación" corrosión en materiales metálicos. La oxidación se produce por el arranque de átomos de hidrógeno, dando lugar a la formación de radicales libres que provocan un envejecimiento creciente en el material polimérico. Los efectos de la oxidación pueden ser los siguientes: Fragilización del material polimérico. Oscurecimiento y amarilleamiento del material, resultado de la oxidación. Disminución de la estabilidad térmica del material polimérico. Con la utilización de los aditivos antioxidantes se pueden paliar de forma efectiva los efectos negativos anteriores. El empleo de antioxidantes es necesario en diversas etapas del ciclo de vida del material polimérico: Procesado del material con empleo de calor y esfuerzos de cizalla. Vida en servicio de la pieza de material polimérico. Los polímeros como el polipropileno (PP) no pueden utilizarse a la intemperie sin estabilizadores apropiados debido a la presencia de átomos de hidrógeno de desprendimiento fácil en los átomos de carbono terciario. La configuración electrónica de este lugar facilita la pérdida inmediata de este hidrógeno y, entonces, se produce la formación de radicales libres que promueven la oxidación, dando lugar al envejecimiento de la estructura polimérica. Luego sigue una serie de reacciones que provocan finalmente la escisión de la cadena. Cada vez que se produce la escisión, la cadena se acorta, y empeoran las propiedades. La formación de radicales libres está favorecida por los esfuerzos de cizalla, que tensionan la estructura, y por las temperaturas elevadas, que son fenómenos que se dan durante el procesado de los materiales, de ahí que sea tan importante proteger un material poliméricos durante su procesado con el fin de evitar los efectos negativos de la oxidación. En ausencia de estabilizadores, el polipropileno al igual que muchos otros polímeros (RH) es atacado durante su procesado o su uso en el exterior debido a una reacción de degradación en cadena que se produce en diversas etapas: 1.- Iniciación. Se forma una gran cantidad de radicales libres, favorecida por la acción del calor y la tensión mecánica. 2.- Propagación/Ramificación. Se induce la formación de nuevos radicales libres, que se acelera con la temperatura. Da lugar a la reticulación de la estructura polimérica, volviéndola frágil empeorando el comportamiento mecánico. 3.- Terminación. Se descomponen los radicales libres y el polímero queda muerto.Las etapas anteriores se pueden representan mediante las ecuaciones siguientes: La velocidad de las reacciones en cadena de radicales libres que se acaban de mostrar se ve acelerada en presencia de metales pesados como los iones de cobalto(II), Por el contrario, la velocidad de la reacción en cadena de degradación disminuye en presencia de cantidades pequeñas de antioxidantes. Uno de los mecanismos más importantes para proteger a un material polimérico frente a la oxidación es el empleo de captadores de radicales libres, que son compuestos que bloquean los puntos activos presentes en los radicales libres e impiden que continúe el proceso de envejecimiento. Se emplean para evitar la oxidación durante el proceso de fabricación. En la figura 2.2.4.1 puede verse la forma en la que actúan los aditivos antioxidantes. Estos se mueven hacia los puntos donde se encuentran los radicales libres y lo que hacen es anclarse sobre la estructura bloqueando el crecimiento o los efectos negativos o reactividad de los radicales libres, evitando su propagación y, por tanto, la degradación del material. Figura 2.2.4.1.- Mecanismo de actuación de los aditivos antioxidantes. Existen muchos antioxidantes naturales que se encuentran en las plantas, incluidos los árboles de caucho como la hevea. Los primeros antioxidantes sintéticos fueron obtenidos independientemente por Cadwell y por Winkelman y Gray por condensación de aminas aromáticas con aldehídos alifáticos. Productos comerciales sin purificar como la fenil-3-naftilamina todavía se utilizan como antioxidantes para los neumáticos de caucho, a pesar de su toxicidad. Muchos de los antioxidantes naturales son derivados del fenol y fenoles impedidos estéricamente, como el ditebutilpcresol. Como puede verse en la ecuación siguiente, el antioxidante se comporta como un agente de transferencia de cadena para producir un polímero muerto y un radical libre estable que no inicia la reacción de degradación en cadena. Sin embargo, el radical libre de fenoxi puede reaccionar con otros radicales libres para producir un derivado de la quinona. Dado que el negro de carbón dispone de muchos electrones libres, puede añadirse a polímeros como las poliolefinas para retrasar la degradación de radicales libres. Es costumbre añadir cantidades pequeñas de otros antioxidantes, como los tioles alifáticos o los disulfuros, para aumentar la estabilización por efecto sinérgico. Este último término se usa para referirse a la estabilización más eficaz que se obtiene con una mezcla de antioxidantes. La industria de los polímeros americana consume más de 3000 toneladas de antioxidantes al año. La función de los aditivos antioxidantes es contrarrestar el ataque oxidante, impidiendo químicamente la serie de reacciones que conducen a la escisión. Los antioxidantes son necesarios para evitar la oxidación durante el proceso de fabricación y, también, para proteger la estructura del polímero durante la vida útil del producto. En los procesos de fabricación, los polímeros a menudo están expuestos a condiciones bastante severas de temperatura y corte, lo cual favorece el ataque del oxígeno. Sin protección sería imposible obtener satisfactoriamente el polipropileno. por ejemplo, ya que sería inaceptable la pérdida de ciertas características. Los antiozonantes son un tipo de antioxidantes especiales que se usan particularmente en los cauchos. Los dobles enlaces insaturados de las moléculas de caucho son muy sensibles al ataque del ozono, aun a la baja concentración que hay en el aire. El ataque se vuelve más riguroso cuando el caucho está sometido a esfuerzos. El efecto puede verse en las grietas superficiales que se forman en los artículos de caucho que están al aire libre. Sin antiozonantes, este tipo de ataque puede provocar el deterioro rápido de los artículos de caucho. 2.2.5.- Estabilizantes térmicos. Los estabilizadores térmicos evitan la degradación a las altas temperaturas del proceso. Detienen otro tipo de reacciones secundarias, por ejemplo, la tendencia de algunos polímeros a despolimerizarse. Estos aditivos son principalmente importantes en el cloruro de polivinilo, el cual se degrada fácilmente, se obscurece cuando se calienta y desprende cloruro de hidrógeno. Para contrarrestar este efecto se ha creado una serie de estabilizadores térmicos que son muy efectivos. Para aumentar su resistencia a la degradación térmica durante su transformación y para aumentar la resistencia a la radiación ultravioleta para alargar su vida útil, se incorporan estabilizantes que eviten la degradación térmica, oxidativa o fotoquímica del material. Estos estabilizantes actúan directamente sobre las reacciones de degradación que son de tipo radicálico inactivando y reduciendo la concentración de especies activas una vez formadas o bien actuando sobre las causas que producen la degradación. Además de la degradación en cadena de radicales libres descrita para el caso de las olefinas, también existe otro tipo de degradación (dehidrohalogenación) que se presenta en los polímeros que contienen cloro como el PVC. Como puede apreciarse en la reacción (2.2.5.1), al calentar PVC, éste pierde cloruro de hidrógeno para formar una estructura cromofórica conjugada de polieno. (2.2.5.1) Puesto que los cloruros alílicos producidos son muy inestables, la degradación continúa en una reacción en cadena de tipo de cremallera abierta. Este tipo de degradación se acelera en presencia de sales de hierro, oxígeno y cloruro de hidrógeno. Como existe la posibilidad de sufrir pérdida de ácido clorhídrico, es por lo que los estabilizantes han de cumplir otras condiciones, como no solamente evitar las reacciones radicálicas oxidativas, sino también absorber y neutralizar el ácido clorhídrico generado para evitar la corrosión de los equipos de transformación. Por último, deben evitar la formación de enlaces conjugados en la molécula que comunican color. Las sales de plomo y bario (tóxicas) absorben el cloruro de hidrógeno y se pueden utilizar como estabilizadores térmicos en algunas aplicaciones, como recubrimientos de cables. Las mezclas de estearatos de calcio y magnesio son menos tóxicas. A pesar de su toxicidad, también se han utilizado alquil mercaptidas de estaño y derivados alquílicos de tioácidos de estaño. Las sales de dioctil estaño son menos tóxicas y producen películas de PVC transparentes. Los fosfitos orgánicos, como los fosfitos de arilo y alquilo mezclados o el fosfito de trifenilo, forman complejos con iones metálicos libres y evitan la formación de cloruros metálicos insolubles. Los aceites insaturados epoxidados (menos tóxicos) como el aceite de soja se comportan como agentes eliminadores de HCI, como se muestra en (2.2.5.2). (2.2.5.2) Estos estabilizantes suelen ser fenoles estéricamente impedidos, aminas aromáticas y fosfitos orgánicos. Los compuestos suelen tener carácter básico, como sales básicas de plomo, jabones de cadmio y mercaptanos. 2.2.6.- Estabilizantes frente a la luz ultravioleta. Algunos materiales poliméricos, en condiciones ambientales normales, se deterioran rápidamente, generalmente en términos de integridad mecánica. Este deterioro suele ser el resultado de la exposición a la luz, en particular a la absorción de la radiación ultravioleta, y también a la oxidación. La radiación ultravioleta interacciona con los enlaces covalentes y puede romper algunos de ellos a lo largo de la cadena molecular; esto puede generar también un entrecruzamiento de cadenas. Lo anterior conduce a la decoloración, pérdida de brillo, transparencia y fragilidad del material polimérico. Aunque una gran parte de la radiación solar (Tabla 2.2.6.1) de alta energía es absorbida por la atmósfera, una cierta parte de la radiación en el intervalo de 280 a 400 nm alcanza la superficie terrestre (ultravioleta). La radiación ultravioleta es especialmente dañina para los materiales poliméricos, ya que la energía de esta radiación va de 72 a 100 kcal (301 a 418 kJ) es lo suficientemente elevada como para romper enlaces covalentes que mantienen unidas las estructura poliméricas. Ello ocasiona el amarilleo y fragilidad de los polímeros orgánicos. Tabla 2.2.6.1.- Distribución de las longitudes de onda y los tantos por ciento de las distintas radiaciones que llegan a la superficie terrestre. El polietileno, PVC, poli estireno, poliésteres y polipropileno se degradan cuando se someten a longitudes de onda de 300, 310, 319, 325 y 370 nm, respectivamente. La energía de enlace necesaria para romper un enlace carbono hidrógeno terciario en el propileno es de 90 kcal/mol, (376.2 kJ/mol) que corresponde a una longitud de onda de 318 nm. La acción de la radiación ultravioleta sobre los materiales poliméricos puede verse en la figura 2.2.6.1, donde se muestra el caso del polietileno, en el cual existen dos tipos de enlaces, -C-C- y –C-H-. Yendo a un gráfico donde se representan las energías de los enlaces, se puede ver que le energía de los enlaces anteriores es similar a la que se encuentra en el rango de las longitudes de onda correspondientes a la radiación ultravioleta, con lo que esta radiación es perjudicial para el material, porque tiene suficiente energía o más energía que los propios enlaces y, por tanto, puede producir su rotura. Figura 2.2.6.1.- Acción de la radiación ultravioleta sobre el polietileno. En la figura 2.2.6.2 se muestra la acción de la radiación ultravioleta sobre el polimetilmetacrilato, en el cual existen cuatro tipos de enlaces, -C-C-, –C-H-, -C-O- y-C=O-. La energía de los enlaces anteriores se encuentra en el rango de las longitudes de onda correspondientes a la radiación ultravioleta. Figura 2.2.6.2.- Acción de la radiación ultravioleta sobre el polimetilmetacrilato. Para ver como actúa la radiación ultravioleta sobre un material polimérico, vamos a tomar como ejemplo el polipropileno (Figura 2.2.6.3). La acción de la radiación ultravioleta sobre las cadenas poliméricas da lugar a la formación de radicales libres por rotura de algunos de los enlaces (llega una radiación de mayor energía que la de los propios enlaces) o en la propia cadena por abstracción de átomos de hidrógeno, los cuales promueven el envejecimiento de la estructura polimérica. Figura 2.2.6.3.- Mecanismo de actuación de la radiación ultravioleta sobre el polipropileno. Los mecanismos más importantes para proteger a un material polimérico frente a la radiación ultravioleta son: 1.- Utilización de absorbedores de la radiación ultravioleta. Se consumen con el paso del tiempo e impiden o evitan la formación de radicales libre durante el tiempo en el que actúan. 2.- Utilización de captadores de radicales libres. Se consumen con el paso del tiempo. Los de absorbedores de la radiación ultravioleta son compuestos que experimentan reacciones químicas en presencia de la radiación ultravioleta dando lugar a la formación de nuevos compuestos químicos y a una radiación, generalemente infrarroja, que no es perjudicial para la estructura del material polimérico (Figura 2.2.6.4) Figura 2.2.6.4.- Mecanismo de actuación de los absorbedores de la radiación ultravioleta. Los captadores de radicales libres son compuestos que bloquean los puntos activos presentes en los radicales libres e impiden que continúe el proceso de envejecimiento. En la figura 2.2.6.5 puede verse la forma en la que actúan los aditivos estabilizantes frente a la radiación ultravioleta. Los aditivos se mueven hacia los puntos donde se encuentran los radicales libres y lo que hacen es anclarse sobre la estructura bloqueando el crecimiento o los efectos negativos o reactividad de los radicales libres, evitando su propagación y, por tanto, la degradación del material. Figura 2.2.6.5.- Mecanismo de actuación de los captadores de radicales libres. Dado que el efecto de la luz ultravioleta en los polímeros sintéticos es parecido al efecto sobre la piel humana, no es sorprendente que estabilizadores de ultravioleta como el salicilato de fenilo se utilice desde hace años en las cremas bronceadoras como filtro solar. Como se muestra en la reacción (2.2.6.1), el salicilato de fenilo se reordena en presencia de una radiación de alta energía para formar 2,2'-dihidroxibenzofenona. Este último producto al igual que otras 2-hidroxibenzofenonas se comporta como agente de transferencia de energía, es decir, absorbe energía para formar quelatos que liberan energía de longitud de onda superior por formación de derivados de la quinona. (2.2.6.1) Muchos de los estabilizadores de ultravioleta comerciales tienen grupos alcoxilo en el carbono 4 del grupo fenilo. Las 2-alcoxibenzofenonas y los que tienen grupos voluminosos en el carbono 6 no son de utilidad como estabilizadores. Como estabilizadores de ultravioleta se pueden citar además los benzotriazoles, como el 2-(2'-hidroxifenil) benzotriazol. Los acrilonitrilos sustituidos, como el acrilato de etil-2-ciano-3,3'-difenilo. Los complejos metálicos como el dibutilditiocarbamato de níquel; y pigmentos como el negro de carbón. Los complejos metálicos se comportan como agentes de transferencia de energía, eliminadores de radicales libres, y descomponen los hidroperóxidos. Los pigmentos absorben radiación ultravioleta y se comportan como agentes de pantalla. La industria de, los polímeros consume en los EE.UU. 100000 toneladas de estabilizadores de UV al año. La incorporación de estabilizantes tiene la función de reducir el nivel de energía de estas radiaciones absorbiéndolas y emitiéndolas de nuevo a menor longitud de onda sin causar ningún daño sin descomponerse más rápidamente que el polímero -efecto pantalla-, o bien desactivar rápidamente las especies radicálicas formadas desactivadores de estados excitados. El negro de carbono presenta un alto grado de absorción de luz pero cuando se quiere conseguir productos transparentes debe utilizarse otro tipo de aditivos derivados de la benzofenona o del benzotriazol o-hidroxibenzofenona 2-(o-hidroxifenil) benzotriazol. Su capacidad de estabilización radica en la gran conjugación de dobles enlaces que presenta la molécula, y en la formación de puente de hidrógeno intramolecular. 2.2.7.- Retardadores de llama. Los materiales poliméricos presentan una naturaleza orgánica y, generalmente, las altas temperaturas pueden provocar procesos de combustión. Son numerosas las aplicaciones de ingeniería donde se requiere un buen comportamiento frente a altas temperaturas o bien frente al fuego, como pueden ser los materiales plásticos para instalaciones eléctricas (cortocircuitos), componentes de maquinaria o materiales plásticos en el sector de la construcción. Si bien es imposible evitar que el plástico no arda, por su naturaleza orgánica, pero sí que es posible retrasar el proceso de combustión, mediante la utilización de aditivos retardantes de llama o ignifugantes, que mejoran el comportamiento frente al fuego de los materiales poliméricos. La inflamabilidad de los polímeros es una característica del máximo interés, sobre todo en la fabricación de textiles (tiendas de campaña, ropa y tejidos del hogar), de juguetes para niños, etc. Por tanto, es fundamental que tengan una buena resistencia de llama. La mayoría de los polímeros, en estado puro son inflamables, a excepción de los que contienen elevada proporción de cloruros y/o fluoruros, tales como los cloruros de polivinilo y politetrafluoretileno. La resistencia a la inflamabilidad de los polímeros combustibles aumenta adicionando aditivos denominados ignífugos (o retardadores de llama). Estos aditivos funcionan de maneras diferentes bien interfiriendo el proceso de combustión mediante una fase gaseosa, bien alterando el mecanismo normal de degradación térmica, favoreciendo un proceso de baja energía consistente en una reacción química que enfría la región de combustión y cesa el fuego, lo que conduce a la carbonización o formando un recubrimiento protector para aislarlo de la energía térmica. Mientras que algunos polímeros como el PVC no se inflaman con facilidad, la mayoría de los polímeros orgánicos, como otros materiales carbonosos, arden a temperaturas elevadas como las que se presentan en los incendios de edificios. Las poliolefinas, el SBR, el EPDM y por supuesto la madera, mantienen la combustión cuando se prenden bajo la acción de una llama. Además de arder, los termoplásticos como las fibras de poliéster se funden. Otros plásticos como el PVC, los poliuretanos, y las proteínas producen humos y gases tóxicos como CO, HCl y HCN al quemarse. En la tabla 2.2.7.1 se dan los elementos que participan en un proceso de combustión, que son: comburente, combustible y temperaturas altas. Cuando se tiene la combinación de estos tres componentes tiene lugar la combustión en un material polimérico, lo cual produce una degradación importante de sus propiedades. Los aditivos retardantes de llama o ignifugantes van a trabajar de modo que ralentizan el proceso de combustión actuando sobre cada uno de los tres elementos que participan en el proceso. Tabla 2.2.7.1.- Elementos que participan en un proceso de combustión. Mecanismo de retardo de llama. En la figura 2.2.7.1 puede verse el mecanismo de protección de los retardantes de llama. En ella se tienen los tres elementos que forman parte de un proceso de combustión: material polimérico con naturaleza combustible, atmósfera oxidante que aporta comburente y elevadas temperaturas. Al material se le puede proteger frente a la llama actuando sobre los tres elementos. Un primer mecanismo, designado como 1, actúa sobre la disminución de la naturaleza combustible del material polimérico. Así, el polietileno tiene un índice de oxígeno crítico (LOI) de 17.4 %. El índice de oxígeno crítico es una propiedad térmica que indica el tanto por ciento mínimo que debe tener la atmósfera (aire) para que se mantenga viva una reacción de combustión, como el aire tiene, aproximadamente, un 20-21 % de oxígeno, en el caso del polietileno (LOI = 17.4 %) hay suficiente oxígeno en el aire y, por tanto, arde con mucha facilidad. Figura 2.2.7.1.- Mecanismo de protección de los retardantes de llama. Sin embargo, modificando ligeramente la estructura del polietileno, como es el caso del policloruro de vinilo (PVC) donde uno de los átomos de hidrógeno se ha sustituido por uno de cloro, el un índice de oxígeno crítico aumenta de forma considerable hasta valores que oscilan entre el 45-49 %, esto implica que este material necesita casi un 50 % de oxígeno en el aire para mantener viva una llama en un proceso de combustión, con lo cual como el aire solo tiene un 20-21 % de oxígeno, no hay suficiente para mantener la combustión, el material arde pero cuando se separa el foco de la llama, se autoextingue la llama, por lo cual presenta un buen comportamiento ignifugo y no propaga la llama. El caso del politetrafluoretileno (PTFE), más conocido como teflón donde cada átomo de hidrógeno ha sido sustituido por uno de flúor, es todavía más extremo ya que tiene un índice de oxígeno crítico del 95 %, esto quiere decir que el teflón arde, prácticamente, en atmósferas de oxígeno puro, necesita un ambiente extremadamente rico en oxígeno para continuar una reacción de combustión. Por eso se emplea tanto el teflón en aplicaciones de temperaturas elevadas, ya que es muy difícil que experimente procesos de combustión. Otro de los mecanismos de protección frente al comportamiento a la llama es el que se muestra en la figura 2.2.7.2 y consiste en trabajar sobre la atmósfera oxidante (Designado como 2). En este caso se trata de formar una capa aislante que evita el contacto directo del oxígeno con la superficie del material polimérico, por lo que no tiene lugar el contacto entre combustible y comburente. Para conseguir tal objetivo, hay determinados compuestos químicos que cuando se descomponen desprenden gases no tóxicos y que generan una pequeña capa en la superficie del polímero que actúa como aislante entre el oxígeno y la superficie del material polimérico evitando o retrasando el proceso de combustión. Así, se está empleando en la actualidad grafito expandido, que está formado por una estructura laminar donde entre las láminas de grafito se interpone ácido sulfúrico, el cual en contacto con el grafito a temperaturas elevadas experimenta una reacción química en la que se desprenden CO2, H2O y SO2, los cuales van a depositarse sobre la superficie del material polimérico como unas cenizas y van a actuar como una barrera para impedir que el oxígeno entre en contacto directo con la superficie del polímero y ello retrasa el proceso de degradación. Figura 2.2.7.2.- Mecanismo de protección de los retardantes de llama. Por último, el tercer mecanismo (Designado como 3) para proteger a un material polimérico frente a la combustión es disminuir las temperaturas elevadas que se producen durante un proceso de combustión. Se trata de que entren en juego determinados compuestos químicos que contienen en su estructura agua de hidratación , que absorben energía para cambiar de estado (evaporarse) y ello provoca un efecto de refrigeración en el medio donde se está produciendo la combustión. Por ejemplo, se emplean mucho las sales hidratadas de aluminio. Figura 2.2.7.3.- Mecanismo de protección de los retardantes de llama. La combustión es una reacción en cadena que puede iniciarse y propagarse mediante radicales libres como el radical libre de hidroxilo. Como se puede ver en la serie de reacciones (2.2.7.1), los radicales de hidroxilo pueden producirse por reacción de oxígeno con radicales macroalquilo. Los radicales halógenos producidos por reacción de radicales hidroxilo con haluros, como HX, pueden servir de terminadores de la reacción. Son compuestos organoclorados, organofosforados, óxidos de antimonio o trihidrato de alúmina. (2.2.7.1) Dado que los radicales de halógenos y de fósforo se acoplan con radicales libres producidos en el proceso de combustión y terminan la reacción, muchos de los retardadores de llama son compuestos de halógenos o de fósforo. Estos compuestos pueden encontrarse como (a) aditivos, (b) retardadores externos, como el óxido de antimonio y los bromuros orgánicos, o (c) retardadores internos, como el anhídrido tetrabromoftálico que puede ser parte del polímero. En los EE.UU. se utilizan alrededor de 100000 toneladas de retardadores de llama al año. Para el proceso de combustión es necesario que haya oxígeno, combustible y alta temperatura. Por tanto, los polifluorocarbonos, fosfacenos y algunos materiales compuestos tienen propiedades de retardo de llama porque son malos combustibles. Rellenos como el trihidrato de alúmina (ATH) desprenden agua al calentarse y, por tanto, reducen la temperatura de combustión. Compuestos como el carbonato de sodio, que desprenden dióxido de carbono, aíslan los reactivos del oxígeno. La carbonilla que se forma en algunos procesos de combustión, también aísla a los reactivos del oxígeno y retrasa la difusión de productos volátiles combustibles hacia el exterior. Los polímeros aromáticos tienden a formar carbonilla, y algunos compuestos de boro y fósforo catalizan la formación de carbonilla. Los retardadores de llama sinérgicos, como las mezclas de trióxido de antimonio con un compuesto orgánico de bromo, son mucho más eficaces que los retardadores de llama aislados. Por tanto, mientras que un poliéster con un 11.5 % de anhídrido tetrabromoftálico se quema sin formar carbonilla a temperaturas altas, cuando se añade un 5 % de óxido de antimonio se observa la formación de carbonilla pero sin arder. Puesto que la combustión depende de muchas variables, los ensayos de retardo de llama no pueden pronosticar la resistencia a las llamas en condiciones anormales. Así, todos los polímeros con propiedades de retardo de llama deberán ir acompañados de un aviso que afirme que los ensayos de retraso de llama no predicen el comportamiento del material en un fuego real. Los retardadores de llama, como muchos otros compuestos orgánicos, pueden ser tóxicos o producir gases tóxicos al quemarse. Consecuentemente, deberá tenerse mucho cuidado cuando se utilicen tejidos u otros polímeros tratados con retardadores de llama. A nivel industrial se está trabajando con las denominadas formulaciones intumescentes que se basan en el empleo de diferentes componentes, cada uno de los cuales actúan de forma simultánea sobre los distintos mecanismos que se han descrito anteriormente, consiguiéndose un efecto sinérgico. Son las más empleadas a nivel industrial para la protección de los materiales poliméricos frente al fuego. Como se puede ver en la figura 2.2.7.4, las formulaciones intumescentes comprenden una base carbonosa, un formador de espuma y un agente de deshidratación, cada uno de los cuales aporta una función a la global de la protección del aditivo. La base carbonosa cuando se degrada genera un residuo (ceniza carbonosa) con grupos hidroxilo, el formador de espuma genera una gran cantidad de gases no combustibles y no tóxicos y el agente de deshidratación genera una base carbonosa con grupos ácido. Los gases dan lugar a la formación de una espuma de un residuo carbonoso que evita el contacto del oxígeno con la superficie del polímero y la base carbonosa con grupos ácido reacciona con los grupos hidroxilo que se han formado a partir de la base carbonosa inicial esterificándose los grupos hidroxilo y se desprenden moléculas de agua, que al absorber energía para evaporarse provocan un efecto de refrigeración. A nivel industrial se emplea mucho como base carbonosa el pentaeritritol (PER), como formador de espuma la melamina y como agente de deshidratación los polifosfatos amónicos. 2.2.8.- Espumantes. Los materiales poliméricos presentan unos valores de la densidad muy bajos en comparación con los otros tipos de materiales: metálicos y cerámicos. Además presentan excelentes propiedades aislantes: térmicas, eléctricas y acústicas. De hecho, las propiedades de aislamiento térmico y acústico hacen que los materiales poliméricos sean candidatos para su aplicación en el sector de la construcción. En este caso es necesario intensificar todavía más las propiedades aislantes de los materiales poliméricos, para ello lo que va hacer, básicamente, es mezclar el polímero base con un gas, que suele ser aire, para ello se necesitan agentes espumantes y procesos de espumación que permiten incrementar las posibilidades tecnológicas de los polímeros y sus posibilidades de aislamiento en distintos sectores industriales. Los polímeros con estructura celular no sólo proporcionan aislamiento y resiliencia, sino que normalmente son más resistentes que los polímeros macizos en relación a su peso. Los polímeros fluidos pueden espumarse añadiendo líquidos de bajo punto de ebullición como el pentano o los fluorocarbonos, inyectando nitrógeno gaseoso comprimido, calentando mecánicamente y añadiendo agentes de espumado. Aunque se produce algo de dióxido de carbono cuando se fabrican poliuretanos en presencia de humedad, también se añaden propulsantes auxiliares a la mezcla de prepolímero. Los mecanismos de espumación pueden ser muy diversos: 1.- Procesos físicos. 2.- Procesos químicos. A nivel industrial, los procesos químicos presentan gran interés ya que permiten controlar de forma adecuada el proceso de espumación, combinándolo con los procesos de transformación convencionales. Algunos compuestos químicos, se descomponen en determinado rango de temperaturas y desprenden gran cantidad de gases inertes. En ese sentido y a título de ejemplo el carbonato de calcio podría ser un agente espumante, ya que a una determinada temperatura tiene lugar la reacción: CaCO3 → CaO + CO2 ↑↑↑ en la que se desprende gran cantidad de gas, en este caso CO2, que puede actuar como gas para la espumación. Pero la reacción anterior tiene lugar a una temperatura en el entorno de los 900 ºC, por lo que no sería adecuada para el caso de los polímeros. Para este caso, se necesitará un compuesto químico que experimente una reacción similar, pero en un rango de temperaturas típico de los procesos de transformación de los materiales, en torno a los 200-250 ºC. Por tanto, si la temperatura de descomposición se ajusta al rango de procesado del polímero, es posible aprovechar los gases para mezclarlos con el polímero base fundido y obtener una espuma polimérica. En la tabla 2.2.8.1 se dan las características de los agentes espumantes. Tabla 2.2.8.1.- Características de los agentes espumantes El mecanismo de espumación se lleva a cabo en los procesos de transformación convencionales. En la figura 2.2.8.1 puede verse como se lleva a cabo dentro del tornillo de Arquímedes de una máquina de extrusión o de moldeo por inyección. En la tolva se añade el polímero en forma de granza y en la tolva auxiliar se incorpora el agente espumante. En el punto 3 comienza a producirse un mezclado físico de la granza de polímero y de las partículas del agente espumante, pero no se ha llegado al estado fundido. A medida que la mezcla avanza en el interior de la máquina de husillo va aumentando la temperatura y el material polimérico va pasando al estado fundido donde queda dispersado el aditivo espumante en su interior. A medida que avanza se consigue una mayor dispersión como se puede observar en el punto 5. Una vez que el polímero está fundido y el agente espumante está totalmente dispersado en el polímero en estado fundido, se llega al punto 6 donde se produce la reacción de espumación, en la que el compuesto A experimenta una degradación y se descompone en el compuesto B produciendo una gran cantidad de gas. Las moléculas del agente espumante se convierten en burbujas de gas que quedan perfectamente mezcladas en el seno del polímero fundido. Un posterior enfriamiento evita que se produzca la separación del gas y del polímero con lo cual se obtendrá una espuma de naturaleza polimérica. Figura 2.2.8.1.- Mecanismo de espumación en los procesos de transformación convencionales La selección del agente espumante más adecuado para una determinada aplicación depende de la temperatura de procesado del material que se pretende espumar. Hay que tener en cuenta que la temperatura de degradación del agente espumante coincida la temperatura de procesado del material polimérico. En la tabla 2.2.8.2 se dan las temperaturas de degradación de diversos agentes espumantes. Tabla 2.2.8.2.- Temperaturas de degradación de diversos agentes espumantes. Los agentes de espumado más utilizados son los compuestos que producen nitrógeno como la azobisformamida (ABFA). También se hallan disponibles otros tipos de espumantes que se descomponen a temperaturas diversas. Estos pueden utilizarse en la extrusión, el moldeo rotacional, el moldeo por inyección y el moldeo de lodo de los plastisoles. Los plastisoles son suspensiones de partículas de polímero en un plastificante líquido. Estos productos, como los plastisoles de PVC, se solidifican cuando la temperatura alcanza un punto en el que el plastificante penetra en las partículas del polímero. Los agentes espumantes se incorporan para conseguir materiales ligeros con buenas propiedades aislantes tanto térmicas como acústicas (espumas rígidas) o productos protectores flexibles por su capacidad de amortiguación del impacto para la industria del envase. Se forman estructuras celulares, en las que la masa de polímero retiene un determinado número de celdillas llenas de un gas. Según sea la formación de este gas espumante por simple evaporación, o a partir de un proceso químico, se denomina a los agentes de espumación físicos o químicos. Los espumantes físicos suelen ser líquidos volátiles hidrocarbonados de 5 ó 6 átomos de carbono, fluorocarbonos o gases como nitrógeno, anhídrido carbónico o aire. Los espumantes químicos en el caso de los termofijos, se generan como subproductos en el proceso de polimerización, como en el caso de los poliuretanos que producen anhídrido carbónico por reacción con agua, mientras que para los termoplásticos deben incorporarse agentes químicos como derivados de hidracina, que generan gases cuando se descomponen a la temperatura de procesado del material, desprendiendo nitrógeno. Las substancias químicas reaccionan a una temperatura conveniente en el proceso y desprenden un gas. La presión del gas expulsado se equilibra con la viscosidad del polímero fundido y se produce una espuma. Esta técnica, desde luego, se ha utilizado desde hace mucho tiempo en la cocina para elaborar pan y pasteles, donde el gas es dióxido de carbono producido por la levadura o por la descomposición del polvo de hornear que es bicarbonato de sodio. En la ciencia de los polímeros, los ejemplos más antiguos son esponja, o caucho “Sorbo”, en donde el agente espumante también es bicarbonato de sodio. Para los materiales que se funden a mayor temperatura como los termoplásticos, se han obtenido otros agentes espumantes que, por lo general, desprenden nitrógeno. 2.2.9.- Colorantes. Los colorantes se incorporan a los polímeros para comunicarles un color específico y hacerlos más atractivos (Figura 2.2.9.1). El color es un fenómeno subjetivo cuyo valor estético se aprecia desde hace muchos siglos. Dado que depende de la fuente luminosa, del objeto y del observador, el color no puede medirse directamente. Figura 2.2.9.1.- Polímeros de color. Los colorantes que proporcionan color a los polímeros pueden ser tintes solubles o pigmentos finamente divididos. Los primeros son solubles en el polímero incorporándose a su estructura molecular y se utilizan para colorear plásticos transparentes, por ejemplo polimetacrilato de metilo (PMMA), poli estireno (PS) y policarbonatos (PC). Los pigmentos son como material de relleno que no se disuelven (son insolubles), sino que permanecen como fases separadas; generalmente son partículas de pequeño tamaño, transparentes y con índice de refracción próximo al polímero base. Otros aditivos dan opacidad y color al polímero. Los colorantes suelen ser azocompuestos y antraquinonas, mientras que los pigmentos suelen ser óxidos metálicos de cadmio, cromo y especialmente titanio. Algunos artículos de materiales poliméricos, como los neumáticos de caucho, son negros debido a la presencia de grandes proporciones de negro carbón como relleno. Otros muchos productos, entre los que se incluyen algunas pinturas, son blancos debido a la presencia de dióxido de titanio, que es el pigmento inorgánico de uso más extendido. La industria americana de los polímeros consume más de 50000 toneladas anuales de colorantes. Los pigmentos se clasifican en orgánicos e inorgánicos. Los primeros son más brillantes, ligeros y de menor tamaño de partícula que los colorantes inorgánicos más ampliamente usados y más opacos. Los óxidos de hierro u ocres, que se pueden encontrar en colores amarillos, rojos, negros, marrones y bronce, son los pigmentos más usados después del dióxido de titanio. Otros pigmentos, como el cromato de plomo amarillo, el naranja de molibdato, el amarillo de cadmio y el cromato de zinc verde, son tóxicos. El cromato de zinc verde es una mezcla de cromato de plomo amarillo y ferrocianuro, de hierro(II) azul, o azul Prusia. El azul marino también es un pigmento de uso extendido. El negro de carbón es el pigmento orgánico más utilizado, aunque los azules y verdes de ftalocianina se hallan disponibles en muchos tonos y se usan también muy ampliamente. Otros pigmentos orgánicos que pueden citarse son: los colorantes azoicos, como los rojos de pirazolona, los amarillos de diarilida, el naranja de dianisidina y el naranja de tolilo. Los colorantes derivados de la quinacridona, como el violeta, el magenta y el rojo de quinacridona; los perilenos rojos. Colorantes ácidos y básicos, como el rojo de rodamina y el azul victoria; las antraquinonas, como el amarillo de flavantrono. Las dioxacinas, como el violeta de carbazol y las isindolinas, que se encuentran en rojo y amarillo. 2.2.10.- Agentes de curado (Químicos). Son aditivos que se usan para producir cambios en las propiedades. Quizá los que más se utilizan son los que se usan para generar enlaces cruzados, produciendo un polímero entrecruzado, termoestable a partir de un polímero inicialmente lineal o ramificado En este caso, las cadenas de polímero se unen químicamente una con otra en varios puntos a lo largo de su molécula. El número de estos enlaces cruzados se determina por el número y distribución de los sitios que hay en las cadenas del polímero y por las cantidades de aditivos que se emplean. Conocer este aspecto de la preparación es importante para controlarlas propiedades del producto final. El uso de agentes de curado empezó a raíz del descubrimiento casual y sorprendente de la vulcanización del caucho de hevea con azufre en 1838 por Charles Goodyear. En este procedimiento, el azufre entrelaza químicamente las cadenas de caucho. El efecto del entrelazamiento cruzado o reticulación es que incrementa la resistencia y la rigidez, y reduce la deformación permanente cuando el material se somete a cargas debido a que los enlaces cruzados impiden el deslizamiento de las cadenas. La conversión de una resina novolaca fenólica en estado A o B con hexametilenotetramina a principios de siglo es otro ejemplo relativamente temprano del uso de agentes de curado (de reticulación). Los aceleradores orgánicos o catalizadores de la vulcanización con azufre del caucho fueron descubiertos en 1912 por Oeslanger. Aunque estos aceleradores no son completamente inocuos, su toxicidad es inferior a la de la anilipa, que se utilizaba antes del descubrimiento de dichos aceleradores. Como acelerador típico puede citarse la tiocarbanilidad y el 2- mercaptobenzotiazol (Captax). El Captax se usa en proporciones de hasta un 1 % con el caucho de la hevea y da cuenta de la mayor parte de las 30000 toneladas de aceleradores que se usan en los EE.UU anualmente. Otros aceleradores típicos (cuyas fórmulas se dan a continuación) son: la sulfonamida de 2-mercaptobenzotiazol (Santocure), utilizada para la vulcanización de SBR, los dicarbamatos y los disulfuros de tiurano. Estos últimos, llamados ultraaceleradores, catalizan el curado del caucho a temperaturas moderadas y pueden usarse en ausencia de azufre. Los iniciadores como el peróxido de benzoilo se utilizan no sólo para la iniciación de polimerizaciones de reacción en cadena sino también para el curado de poliésteres, copolímeros de etilenopropileno y para el injertado de estireno en cadenas de polímeros elastoméricos. Los peróxidos como el 2,5-dimetil2,5-di(tbutilperoxi)hexino3 se utilizan para el reticulado del HDPE. Dado que estos compuestos tienen enlaces covalentes débiles, deberán tomarse medidas de precaución en su almacenamiento para evitar explosiones. También pueden obtenerse radicales libres para el reticulado mediante bombardeo con electrones, rayos y o radiación ultravioleta. Esta última es más eficaz en presencia de aditivos como el éter médico de la benzoina. Los polímeros insaturados como las resinas alquídicas pueden curarse o "secarse" en presencia de oxígeno, de una sal de un metal pesado y de un ácido orgánico que se denomina secador. Los metales que se usan normalmente son el cobalto, el plomo, y el manganeso. Los ácidos más comunes son el linoleico, abiético, naftalénico, octanoico y los ácidos grasos de sebo. Como puede verse en las reacciones (2.2.10.1), se cree que el oxígeno forma un peróxido en presencia de un secador, dando lugar a un macrorradical que es capaz de reticular. (2.2.10.1) 2.2.11- Antiestáticos. Los agentes antiestáticos, también llamados desestatizadores, se utilizan para reducir la acumulación de cargas electrostáticas en la superficie de los plásticos debido a su inherente mala conductividad eléctrica. Atraen la humedad para aumentar la conductividad superficial y así se reduce la posibilidad de que se produzca una chispa o descarga. Ejemplo: Los trajes espaciales de los astronautas contienen agentes antiestáticos para evitar las descargas eléctricas que pueden dañar los componentes electrónicos de los transbordadores espaciales. La mayoría de los polímeros, excepto los ionómeros o materiales compuestos rellenos con metales, son aislantes eléctricos que se cargan electrostáticamente con facilidad durante su procesado o manejo posterior. Esta carga eléctrica, que resulta de un exceso o déficit de electrones puede contrarrestarse utilizando barras de ionización del aire durante el procesado o mediante la adición de agentes antiestáticos. Los agentes antiestáticos pueden reducir la carga actuando como lubricantes o proporcionando un camino conductor para la disipación de cargas. La mayor parte de los agentes antiestáticos son higroscópicos y hacen que se forme una película fina de agua sobre la superficie del polímero. Los agentes antiestáticos internos se mezclan con el polímero, mientras que los externos normalmente se rocían sobre la superficie del polímero. Como ejemplos de agentes antiestáticos pueden citarse los compuestos de amonio cuaternarios, las hidroxialquilaminas, los fosfatos orgánicos, los derivados de los alcoholes polihídricos como el sorbitol y los ésteres de glicol de ácidos grasos. Al ser los plásticos malos conductores eléctricos, se forman cargas electrostáticas sobre la superficie que producen la descarga. En algunos casos, pueden producir una chispa potencialmente peligrosa, por ejemplo, donde se usan disolventes inflamables. Para evitar esto así como la fácil deposición del polvo sobre los objetos de plástico se incorporan agentes antiestáticos. Éstos se clasifican en dos categorías: los externos que se adicionan mediante sprays o los internos que se incorporan a la formulación. Su estructura química corresponde a ésteres de ácidos grasos, éteres y aminas cuaternarias. 2.2.12.- Biocidas. Aunque muchos de los polímeros sintéticos no modificados no son atacados por microorganismos, los plastificantes y otros aditivos, además de los productos de la celulosa, las proteínas, el almidón y las pinturas con base de grasa vegetal, se hallan sometidos a menudo a la degradación biológica. Los conservantes son fundamentales no sólo para evitar el ataque por parte de microorganismos, sino también para evitar el a saque del caucho y el PVC por parte de los roedores. El PVC desarrolla manchas de color rosa como resultado de la difusión de los productos: secundarios del ataque de microorganismos. Los carboxilatos de amonio cuaternario y los compuestos de tributilestaño son conservantes eficaces contra las manchas rosadas. Entre otros conservantes y biocidas pueden citarse los ésteres de ácido phidroxibenzoico, la N-(triclorometiltio)-4-ftalamida, el bis(tri-n-butilestaño) y el bis(8-quinolinato ) de cobre. Son sustancias que se incorporan para evitar ataques y crecimientos de cultivos miocrobianos. Las poliolefinas y los polímeros vinílicos son resistentes a estos ataques, no así los cauchos y derivados de la celulosa. El antimicrobiano ideal debe ser activo sólo sobre el microorganismo y no sobre otro tipo de ser vivo. 2.3.- Cargas o rellenos. Algunos materiales poliméricos como películas, fibras o plásticos se emplean sin rellenos, pero la resistencia mecánica y el precio de la mayoría de los elastómeros y materiales compuestos plásticos dependen de la presencia de rellenos apropiados. Algunos artículos de caucho como las suelas de zapatos, gomas elásticas, neumáticos y globos se fabrican sin rellenos. Sin embargo, las cubiertas de neumáticos no podrían utilizarse con éxito sin añadir negro de carbón o sílice amorfa. Por ejemplo, la incorporación de estos rellenos incrementa la resistencia a tracción del SBR de 100 psi (0.69 MPa) a 4000 psi (27.6 MPa) .De la misma manera, la mayoría de los plásticos de altas prestaciones son materiales compuestos de polímeros reforzados con fibra de vidrio. Según la norma ASTMD883 de la American Society for Testing and Materials, un relleno es un material relativamente inerte que se incorpora al plástico para modificar su resistencia mecánica, estabilidad, propiedades de uso, aumentar su procesabilidad u otras características y en el caso de los termofijos, disipar el calor de la reacción de curado. También se usan para disminuir su precio a base de reducir el coste del material. El coste del producto final disminuye porque estos materiales baratos substituyen una parte del volumen de los polímeros más caros. Las cargas pueden tener forma de platillo, esférica o esferoidal, aguja o irregular y su tamaño es pequeño (Figura 2.3.1). Las cuentas de vidrio que cumplen los requisitos de esta definición se utilizan para reducir el desgaste de los moldes y para mejorar la calidad de las piezas moldeadas. El término extendedor, utilizado algunas veces para denominar los rellenos, no es siempre adecuado, puesto que algunos rellenos son más caros que las resinas. El tamaño y la forma de la carga influyen enormemente en el material compuesto. La relación entre dimensiones de una carga es el cociente entre la longitud y la anchura. Las escamas o fibras mantienen relaciones entre sus dimensiones que las permiten resistir el movimiento o el realineamiento, por lo cual mejoran la resistencia. Figura 2.3.1.- Las esferas son isótropas pero no tienen relación entre dimensiones. Las partículas isótropas presentan propiedades mecánicas uniformes en el plano de la laminilla. Las fibras tienen relaciones entre dimensiones inferiores, pero son anisótropas. En las esferas no existe relación entre dimensiones, por lo que estos elementos se asocian a materiales compuestos con propiedades isótropas. Las laminillas metálicas se utilizan en materiales compuestos de partículas para formar una barrera o capa eléctrica en la matriz de polímero. En la tabla 2.3.1 se resumen los tipos de cargas principales y sus funciones. Entre los materiales de relleno naturales se encuentran los derivados de la celulosa, como por ejemplo el serrín, la α-celulosa, la harina de cáscaras, el almidón, y los rellenos de origen proteínico como los restos de soja. Anualmente se utilizan aproximadamente 40000 toneladas de rellenos celulósicos en la industria americana de los polímeros. El serrín, que se fabrica por molido de desgaste de restos de madera, se utiliza como relleno para las resinas fenólicas, las resinas de urea oscuras, las poliolefinas y el PVC. La harina de cáscaras, que no tiene la estructura fibrosa del serrín, se fabrica moliendo cáscaras de cacahuete y de nuez. Se usa como sustituto del serrín. La α-celulosa, que es más fibrosa que el serrín, se utiliza como relleno para los plásticos de urea y de melanina. Los platos de melanina están hechos de una estructura laminar que consta de papel impregnado con resina y moldeado. Probablemente, el formaldehído de estas resinas termoestables reacciona con los grupos hidroxilo de la celulosa para producir un material compuesto más compatible. Los derivados del almidón y de la soja son biodegradables, y la velocidad de descomposición de los materiales compuestos de resina puede controlarse con las cantidades de rellenos añadidos. También es posible añadir polímeros incompatibles para aumentar la resistencia al impacto de otros polímeros como el poli estireno. Otras resinas troceadas como las siliconas o los polifluorocarbonos se pueden añadir para aumentar la lubricidad de los plásticos. Por ejemplo, una dispersión fundida en caliente de politetrafluoroetileno en polisulfuro de fenileno se utiliza como recubrimiento antiadhesivo para utensilios y recipientes de cocina. Puesto que el butiratoacetato de celulosa es compatible con las resinas de poliéster sin curar, pero incompatible con la resina curada, es posible añadir esta sustancia a una premezcla para reducir la contracción durante el curado. Para reducir la rugosidad superficial se añade polietileno finamente dividido, que también es incompatible con las resinas de poliéster, en lo que se denomina método de las resinas de bajo perfil. El negro de carbón, que los chinos fabricaban hace ya mil años mediante el proceso de recogida de residuos de humo, es en la actualidad el relleno para polímeros más ampliamente utilizado. Gran parte de los 1.5 millones de toneladas fabricadas anualmente en los EE.UU. se utilizan como refuerzo para elastómeros. El negro de carbón más utilizado es el de horno. El tamaño de partícula de este tipo de negro de carbón es de alrededor de 0.08 mm. Su dureza en la escala de Mohs es inferior a 1. Los polímeros con negro de carbón como relleno, en especial los que están formados por negro de acetileno, son buenos conductores de la electricidad y del calor. También se han obtenido polímeros con buena conductividad embebiendo negro de carbón en las superficies los refuerzos filamentosos de nilón o poliéster. La resistencia de las poliolefinas a la radiación ultravioleta mejora mediante la incorporación de negro de carbón. Aunque las esferas de vidrio se clasifican como rellenos no reforzantes, al añadir 40 g de estas esferas a 60 g de nilón66 aumenta el módulo de flexión, la resistencia a la compresión y el índice de fundido. La resistencia a la tracción y al impacto, la fluencia y el alargamiento de estos materiales compuestos son menores que los del nilón66 sin relleno. Se constata un mayor incremento del módulo de flexión cuando la superficie del vidrio se modifica con agentes de superficie activos. Tabla 2.3.1.- Principales tipos de carga. También se han utilizado cómo relleno las fibras de vidrio molidas, los nódulos de vidrio multicelulares y las escamas de vidrio. Al añadirlos a una resina, las esferas huecas de carbón o vidrio, llamadas microglobos o microesferas, producen espumas sintácticas de densidades diversas, dependiendo de la relación de relleno y resina. Se obtienen materiales compuestos de altas prestaciones al utilizar fibra de carbono o de vidrio como aditivo. Cuando se añaden rellenos metálicos pulverizados, escamas metálicas, o rellenos con recubrimientos metálicos a una resina se obtienen materiales compuestos conductores. Estos composites (materiales compuestos) se han utilizado para fabricar herramientas de conformado para la industria aeronáutica y para superar la interferencia electromagnética en las máquinas de oficina. Los composites de olefinas con rellenos de plomo en polvo se han utilizado como escudos para las radiaciones γ y de neutrones. Se han utilizado plásticos con rellenos de zinc para fabricar electrodos de materiales compuestos y, mediante la incorporación de aleaciones de aluminio níquel o ferrita de bario en polvo, se han obtenido composites magnetizables. El óxido de zinc, que tiene una dureza de 2.5 en la escala de Mohs, se utiliza ampliamente como relleno activo del caucho y para mejorar la resistencia a la intemperie de las poliolefinas y poliésteres. La anatasa y el dióxido de titanio (rutilo) se usan como pigmentos blancos y para mejorar la resistencia a la intemperie de muchos polímeros. Las baritinas molidas (BaSO4) proporcionan plásticos opacos a los rayosX de densidad controlada. Con la mezcla de alúmina calcinada, corindón o carburo de silicio finamente divididos se obtienen materiales compuestos abrasivos. Por otro lado, el trihidrato de alúmina (ATH), que tiene una dureza en la escala de Mohs inferior a 3, se utiliza como relleno retardador de llama para los plásticos. La zircona, el silicato de zirconio y el óxido de hierro, cuyas densidades son superiores a 4.5, se utilizan para producir plásticos con altas densidades controladas. El carbonato de calcio, que tiene una dureza en la escala de Mohs igual a 3, se encuentra disponible tanto como caliza natural molida como en creta sintética. Este relleno se usa extensamente en pinturas, plásticos y elastómeros. La relación de volúmenes de carbonato cálcico a resina o el volumen de pigmento necesario para llenar los huecos en un material compuesto de resina se denomina concentración de volumen de pigmento (PIVC). La PIVC crítica (CPIVC) es el valor mínimo necesario para cubrir la demanda de la resina. A diferencia del relleno de carbonato de calcio natural de forma cúbica, el producto sintético que se obtiene al añadir dióxido de carbono a una suspensión de hidróxido de calcio tiene forma de agujas. El carbonato de calcio se usa en cantidades anuales de 700 millones de toneladas como relleno para el PVC, las poliolefinas, las espumas de poliuretano y las resinas epoxi y fenólicas (ver la tabla 2.3.2). La sílice, cuya densidad es de 2.6, se utliza en su forma natural y como sílice amorfa sintética al igual que en forma de partículas cristalinas grandes como en el caso de la arena y el cuarzo. La tierra de diatomeas, también llamada tierra de infusorios y harina de fósiles, es una sílice amorfa finamente dividida que se halla constituida por esqueletos de diatomeas. Este relleno tiene una dureza en la escala de Mohs inferior a 1.5 a diferencia de la arena de sílice fina que posee una dureza de 7 en la escala de Mohs. La tierra de diatomeas se usa para evitar que los rollos de película fotográfica se peguen (antibloqueo) y para aumentar la resistencia a compresión de las espumas de poliuretano. El trípoli, o arena en panal, es una sílice porosa formada por la desintegración de piedras arenosas. La sílice pirogénica o ahumada es un relleno finamente dividido que se obtiene haciendo reaccionar tetracloruro de silicio en una atmósfera de hidrógeno y oxígeno. Este relleno se usa como material tixotrópico para incrementar la viscosidad de las resinas líquidas. También se fabrican sílices finamente divididas mediante la acidificación de soluciones de silicato sódico y por evaporación de soluciones alcohólicas de ácido silícico. La arena de sílice angulosa se utiliza como relleno para los morteros de cemento resinosos. También se usan las cenizas de dice reactivas producidas quemando cáscaras de arroz y la novaculita, que es un relleno laminar extraído de las elevaciones novaculíticas de Arkansas, como rellenos de dice para los polímeros. Como refuerzos para elastómeros también se utilizan sílices hidratadas finamente divididas que contienen grupos silanol en la superficie. El manejo de la sílice deberá realizarse con extremo cuidado para evitar la silicosis. La industria de los polímeros utiliza aproximadamente 25 millones de toneladas de rellenos de sílice al año. Tanto los silicatos naturales como los sintéticos se utilizan ampliamente como rellenos. El silicato de aluminio hidratado o caolín tiene una densidad de 2,6 y una dureza de 2.5 en la escala de Mohs. El caolín y otras arcillas pueden disolverse en ácido sulfúrico y regenerarse añadiendo silicato de sodio. Las arcillas se utilizan como rellenos en la fabricación de papel sintético, caucho y productos bituminosos. La mica, que tiene una densidad de 2.8 y una dureza en la escala de Mohs de 3, es un relleno natural laminar con una relación de aspecto de 30. Sin embargo, se pueden obtener relaciones de aspecto aun mayores por delaminación ultrasónica. El talco es un silicato de magnesio hidratado natural fibroso cuya dureza en la escala de Mohs es 1 y cuya densidad es 2.4. Puesto que el polipropileno con relleno de talco es más resistente al calor que el PP normal, se utiliza en los accesorios de la industria del automóvil que están sometidos a altas temperaturas. Anualmente se utilizan más de 40 millones de toneladas de talco como rellenos. La sienita de nefelina, que es un relleno natural de silicato potásicosódicoalumínico, y la wollastonita, que es un relleno de metasilicato de calcio en forma de agujas, se utilizan como refuerzos para muchos plásticos. También se usan fibras de rocas fundidas o escorias (PMF) como refuerzos para polímeros. El asbesto es un silicato de magnesio natural que a pesar de su toxicidad se ha utilizado durante más de 250 años como fibra resistente al fuego. Anualmente se utilizan alrededor de 180 millones de toneladas como aditivo para los polímeros, aunque esta cantidad va disminuyendo. Las cargas más utilizadas suelen ser harina de madera, sílice, arena, vidrio, arcilla (caolín), talco, caliza, mica e incluso polímeros sintéticos, todos ellos finamente pulverizados. La mica también se utiliza para modificar las propiedades eléctricas y aislantes del material. El caolín tiene algunas veces un efecto ligeramente reforzador si es bastante fino. Los tamaños de las partículas van de 10 nm a dimensiones macroscópicas. Tabla 2.3.2..-Tipos de rellenos para polímeros. 2.4.- Refuerzos. Los refuerzos son ingredientes que se añaden a las resinas y polímeros. Estos ingredientes no se disuelven en la matriz de polímero. Por consiguiente, el material pasa a ser compuesto. Un plástico reforzado es un plástico con propiedades de resistencia muy superiores a la de la resina básica que resultan de la presencia de rellenos de alta resistencia embebidos en la composición. Según la ASTM, los laminados de plástico son los plásticos reforzados más comunes y resistentes. Es frecuente confundir los refuerzos con las cargas. Éstas son partículas pequeñas que favorecen sólo ligeramente su firmeza. En cambio, los refuerzos son ingredientes que aumentan la solidez, la resistencia al impacto y la rigidez. Una de las principales razones por las que se confunden es que algunos materiales (el vidrio, por ejemplo) pueden actuar como carga, refuerzo o ambas cosas. De acuerdo con la definición de las normas ASTM, los rellenos son relativamente inertes mientras que los refuerzos mejoran las propiedades de los plásticos. En realidad, se usan pocos rellenos que no mejoran las propiedades, pero las fibras de refuerzo dan lugar a mejoras espectaculares de las propiedades físicas de los materiales compuestos. Existen muchos refuerzos fibrosos, pero la mayoría de las teorías que se han desarrollado han sido el resultado de los estudios sobre la fibra de vidrio, que es el refuerzo para polímeros más ampliamente utilizado. Los materiales de refuerzo se adicionan a los polímeros con el fin de incrementar las propiedades mecánicas (módulo de elasticidad, resistencias a la tracción, a la compresión y a la abrasión, la tenacidad), la estabilidad dimensional y térmica y otras propiedades. El ejemplo más importante es el del negro de carbono que se añade a los cauchos con ese objetivo. Muchos cauchos, incluyendo el natural y el estirenobutadieno, que son los dos ejemplos de mayor uso, se refuerzan gracias a la adición de grandes cantidades de negro carbono. El efecto más importante es una marcada mejoría en la resistencia a la abrasión, la cual es importante en muchos de los usos del caucho, por ejemplo, llantas y bandas transportadoras. El reforzamiento depende de un tamaño pequeño de partícula del material de relleno. El otro ejemplo importante es la sílice en partículas finas. Existen seis variables generales que influyen en las propiedades de los materiales y estructuras compuestas reforzadas. 1.-Unión de interfaz entre la matriz y los refuerzos. La matriz sirve para transmitir la mayor parte de la tensión a los refuerzos (mucho más fuertes). Para cumplir este objetivo, debe existir una adherencia excelente entre la matriz y el refuerzo. 2.- Propiedades del refuerzo. Se parte de la base de que el refuerzo es mucho más fuerte que la matriz. Las propiedades reales de cada refuerzo pueden variar en composición, forma, tamaño y número de defectos. La producción, manipulación, tratamiento, perfeccionamiento de la superficie o hibridación pueden determinar asimismo las propiedades de cada tipo de refuerzo. 3.- Tamaño y forma del refuerzo. Algunas formas y tamaños pueden favorecer la manipulación, carga, tratamiento, orientación de empaquetado o adherencia a la matriz. Algunas fibras son tan pequeñas que se manejan en haces, mientras que otras crean un tejido. Es más probable que las partículas se distribuyan al azar, al contrario que las fibras largas. 4.- Carga del refuerzo. Generalmente, la resistencia mecánica del material compuesto depende del agente de refuerzo que contenga. Una pieza que contengan 60 % de refuerzo y 10 % de matriz de resina es prácticamente seis veces más resistente que una pieza que contenga las cantidades contrarias de estos dos materiales. Algunos materiales compuestos tejidos con filamentos de vidrio pueden tener hasta un 80 % (en peso) de carga por orientación unidireccional del filamento. La mayoría de los materiales termoplásticos reforzados contienen menos de un 40 % (en peso) de refuerzos. 5.- Técnica de tratamiento. Algunas técnicas de tratamiento permiten una alineación u orientación más precisa de los refuerzos. Durante el tratamiento, se pueden romper o dañar los refuerzos, dando como resultado propiedades mecánicas inferiores. Dependiendo de la técnica de tratamiento, es más probable que los refuerzos en partículas y las fibras cortas se organicen en la matriz más bien al azar que orientadas. 6.- Alineamiento o distribución del refuerzo. El alineamiento o distribución del refuerzo permite una versatilidad en los materiales compuestos. El procesador puede alinear u orientar los refuerzos para proporcionar propiedades direccionales. En la figura 2.4.1, el alineamiento paralelo (anisótropo) de hebras continuas proporciona la máxima resistencia, el alineamiento bidireccional (tela) proporciona un intervalo de resistencia medio y el alineamiento al azar (fieltro) ofrece la resistencia más baja. A diferencia de los materiales compuestos isótropos de resina y rellenos, el esfuerzo en un material compuesto reforzado con fibra de vidrio se concentra en los extremos de las fibras. Por tanto, dado que la resistencia mecánica de la fibra es mayor que la de la resina, las propiedades del material compuesto serán anisótropas y dependerán de la dirección de aplicación del esfuerzo. El módulo de elasticidad transversal (MT) y muchas otras propiedades de los materiales compuestos de fibra larga y resina pueden estimarse mediante la ley de mezclas. El módulo de elasticidad longitudinal (ML) puede estimarse con la ecuación de Kelly Tyson. El módulo longitudinal es proporcional a la suma del módulo de la fibra (MF) y el de la matriz de resina (MM). Cada uno de los módulos se basa en la fracción de volumen (c). La constante k es igual a 1 para filamentos paralelos continuos y disminuye para los filamentos cortos distribuidos aleatoriamente. M L kM F cF M M cM (2.4.1) Puesto que la contribución de la matriz de resina es pequeña en un material compuesto resistente, el segundo término de la ecuación de Kelly Tyson es despreciable. Así, el módulo longitudinal depende del módulo del refuerzo, que es independiente del diámetro de la fibra reforzante. En los materiales compuestos fabricados por enrollamiento de fibra o por pultrusión se utilizan filamentos continuos. En cada caso, la resina de impregnación forma parte del material compuesto terminado. Aunque se realizaron intentos fallidos para utilizar hilos de algodón como refuerzos, los primeros refuerzos logrados fueron los de fibra de vidrio. Este producto puede hilarse a partir de vidrio de tipo E o tipo C de calizas baratas. En el proceso de hilado, se fabrican filamentos haciendo pasar vidrio fundido a través de una serie de orificios de las boquillas. Además de los métodos de enrollado de hilo y de pultrusión, se puede utilizar fibra de vidrio picada como refuerzo para aplicar por soplado una mezcla de fibras picadas impregnadas con resina, y para compuestos de moldeo en masa (BMC) y compuestos de moldeo en lámina (SMC). El SMC es el material compuesto de resina y fibra de vidrio más ampliamente utilizado. Figura 2.4.1.Relación entre la resistencia y el alineamiento del refuerzo y el volumen de la fibra. En muchos procesos, como en el de SMC, las fibras no son continuas. Cuando su longitud es superior a un valor crítico (LC), es necesario modificar el primer término de la ecuación de Kelly Tyson multiplicando por un factor L LC 2 en el que L es igual a la longitud de fibra real. La constante k tiende a 0.5 para las fibras orientadas bidireccionalmente. Dentro del grupo de refuerzos fibrosos se incluyen seis subgrupos: 1.-Vidrio 2.-Carbonáceo 3.-Polímero 4.-Inorgánico 5.-Metal 6.-Híbridos Fibras de vidrio. Uno de los materiales de refuerzo más importantes es la fibra de vidrio (Tablas 2.4.1 y 2.4.2 ). Por la firmeza con la que el vidrio refuerza el plástico, muchas de las piezas antes hechas con metal han sido sustituidas por plástico. En la figura 2.4.2 se muestra un alojamiento de engranaje de plástico reforzado, más ligero y resistente que el de metal al que ha reemplazado. Las fibras de vidrio se obtienen por distintos métodos. Uno de los más comunes consiste en sacar una hebra de vidrio fundido una vez formada por un pequeño orificio. El diámetro de la hebra se controla por la acción de tracción. Los materiales compuestos de resina reforzados con fibra de vidrio fueron introducidos en 1940 y su uso no ha hecho más que aumentar desde entonces. Su utilización se limitaba al principio a las resinas termoestables, pero los termoplásticos reforzados con fibra de vidrio, como, por ejemplo, el nilón, son materiales compuestos de gran importancia en la actualidad. En los Estados Unidos se fabrican más de 500000 toneladas de materiales compuestos reforzados con fibra de vidrio al año. Tabla 2.4.1.- Propiedades de los termoplásticos: materiales reforzados y sin reforzar. Tabla 2.4.2.- Propiedades de plásticos termoestables: resinas reforzadas con fibra de vidrio. Figura 2.4.2.- El alojamiento de engranaje se ha sustituido por uno de boro – epoxi. Los productores de plástico adquieren los diferentes tipos de vidrio de varias formas. Las mechas son hebras largas de vidrio fibroso que se pueden cortar fácilmente y aplicar a las resinas. Una mecha se compone de muchas hebras de vidrio parecidas a una cuerda retorcida o trenzada (Figura 2.4.3). Las fibras cortadas (Figura 2.4.4) se encuentran entre las formas más baratas de refuerzo con vidrio. Las hebras cortadas tienen una longitud que oscila entre 3 y 50 mm. Figura 2.4.3.-Mecha de fibra de vidrio Figura 2.4.4.Hebras cortadas de fibra de vidrio. En la figura 2.4.5, se muestra la producción de hebras cortadas de mechas. Las fibras trituradas tienen una longitud inferior a 1.5 mm y se producen por triturado con martillo de hebras de vidrio (Figura 2.4.6). Se añaden las fibras trituradas a la resina como una mezcla previa para aumentar la viscosidad y la resistencia del producto. Figura 2.4.5.- Hebras de vidrio cortadas Figura 2.4.6.- Fibras de vidrio trituradas. Los hilos son similares a las mechas pero están retorcidos como una cuerda (Figura 2.4.7). Los hilos de refuerzo se utilizan para fabricar contenedores grandes para tanques de líquidos. Figura 2.4.7.- Hilos. Además de las formas de fibra e hilo, los refuerzos de vidrio pueden presentarse también como tela y fieltro. Los fieltros consisten en piezas no direccionales de hebras cortadas, que se mantienen juntas mediante un aglutinante resinoso o por puntadas mecánicas que se denominan hilvanado (Figura 2.4.8). El tejido plano puede proporcionar la mayor resistencia física de todas las formas fibrosas, pero es casi el doble de costosa que las demás formas. Las mechas convencionales pueden estar tejidas en forma de tela (conocidas como mechas tejidas) y se utilizan para refuerzos gruesos. Figura 2.4.8.- Fieltros de fibra de vidrio. Existen varios tipos de tejidos de vidrio planos. Los hilos de fibra de vidrio se entrelazan según varios modelos básicos, tal como se muestra en la figura 2.4.9. En la figura 2.4.10 se recogen tres formas diferentes de refuerzos de vidrio fibroso. (A) Tejido (tela) plano(Cuadrado) (B) Tejido unidireccional Figura 2.4.9.- Modelos de tejido y mecha. (A) Mecha tejida (B) Fieltro de hebra fina (C) Combinación de mecha tejida y fieltro Figura 2.4.10.- Diversas formas de refuerzos de fibras de vidrio. Fibras carbonosas. Las fibras de carbono se obtienen normalmente por oxidación, carbonización y grafitización de una fibra orgánica. El rayón y el poliacrilonitrilo (PAN) son las utilizadas actualmente. Las fibras bituminosas se pueden producir también directamente a partir de aceite y carbón. En general, las breas tienen carácter isótropo y se deben orientar para ser útiles como agentes de refuerzo. Las palabras carbono y grafito se emplean indistintamente, aunque poseen significados diferentes. Las fibras de carbono (PAN) tienen aproximadamente un 95 % de carbono, mientras que las de grafito se grafitizan a temperaturas mucho más altas y, en un análisis elemental de carbón, dan como resultado un 99 %. Una vez que se han extraído los materiales orgánicos (pirolizado y estirado en filamentos), el resultado es una fibra de alta resistencia, alto módulo y baja densidad. Fibras de polímero. Durante años se han empleado filamentos de algodón y seda como refuerzos en cinturones, neumáticos, engranajes y otros productos. Actualmente, se utilizan polímeros sintéticos de poliéster, poliamida (PA), poliacrilonitrilo (PAN), poliacetato de vinilo (PVA) y acetato de celulosa (CA), entre otros. La aramida kevlar es una fibra de polímero de poliamida aromática que posee casi el doble de rigidez y aproximadamente la mitad de densidad del vidrio. Kevlar es una marca registrada de DuPont, y aramida es el nombre genérico que denomina a una serie de fibras kevlar. Al contrario que las fibras de carbono, las de kevlar no conducen la electricidad ni tampoco son eléctricamente opacas a las ondas de radio. Las fibras kevlar 29 se utilizan para protección balística, trajes, cascos de soldado, tejidos revestidos y diversas aplicaciones de materiales compuestos. Kevlar 49 se utiliza en barcas, cascos de hidroaviones, volantes, tirantes en v, mangueras, armaduras compuestas y estructuras aeronáuticas. El kevlar 49 tiene una resistencia equivalente, pero un módulo bastante superior al del kevlar 29. Una matriz de polímero común de alta resistencia es el epóxido. En este marco se emplean poliésteres, poliimida fenólica y otras resinas y sistemas de polímero. Las fibras termoplásticas a base de poliéster y poliamida encuentran aplicación en compuestos de moldeo en volumen (BMC), compuestos de moldeo en lámina (SMC) y gruesos (TMC), colocación de chapas, pultrusión, enrollamiento de filamentos, moldeo de transferencia de resina (RTM), por inyección de reacción reforzado (RRIM) y de transferencia de resina por expansión térmica (TERTM) y operaciones de moldeo por inyección. Fibras inorgánicas. Las fibras inorgánicas conforman una clase de fibras cristalinas cortas que, a veces, reciben el nombre de fibras filamentosas de cristal. Estas fibras están hechas de óxido de aluminio, óxido de berilio, óxido de magnesio, titanato de potasio, carburo de silicio, boruro de titanio y otros materiales. Los filamentos de titanato de potasio se utilizan en grandes cantidades para reforzar materiales compuestos de matrices termoplásticas. Las fibras de boro continuas inorgánicas son más fuertes que el carbono y se pueden usar en una matriz de polímero y aluminio. El boro en una matriz de epóxido se utiliza para fabricar muchas piezas de materiales compuestos para aeronáutica militar y civil. La fabricación de estas fibras resulta muy cara con la tecnología actual; no obstante, presentan resistencias a la tracción superiores a 40 GPa. Las investigaciones sobre el uso de estos refuerzos en empastes plásticos dentales, aspas de compresores de turbina y equipo de inmersión especial han demostrado unos resultados alentadores. En la figura 2.4.11 se muestra el eje del rotor de la cola de un helicóptero hecha de epóxido reforzado con boro. Las fibras de carbono y grafito pueden superar al vidrio en términos de resistencia. Tiene muchas aplicaciones como materiales autolubricantes, cuerpos de reentrada termorresistentes, aspas para turbinas y helicópteros y compuestos de obturación de válvulas. En la figura 2.4.12 se muestran fibras de carbono y vidrio utilizadas en combinación para reforzar una raqueta de nilón moldeada por inyección. Figura 2.4.11.- Eje del rotor de un helicóptero, con accesorios extremos y complementos de soporte. Figura 2.4.12.-Combinación de fibra de vidrio y de carbono utilizada para reforzar una raqueta de nilón moldeada por inyección. Las fibras cerámicas presentan una alta resistencia a la tracción y una baja expansión térmica. Algunas fibras pueden alcanzar una resistencia a la tracción de 14 GPa. Entre las aplicaciones actuales de fibras cerámicas se incluyen empastes dentales, aplicaciones de electrónica especiales e investigación aeroespacial. (Ver resistencia a la tracción de fibras filamentosas en la figura 2.4.13). Figura 2.4.13.- Resistencias a la tracción de varios materiales en volumen, forma de fibra y forma de filamento. Fibras metálicas. El acero, el aluminio y otros metales se estiran en filamentos continuos. No se pueden comparar en resistencia, densidad y otras propiedades con las demás fibras. Las fibras de metal se utilizan para lograr una mayor resistencia, transmisión calorífica y conductividad eléctrica. Fibras híbridas. Las fibras híbridas se refieren a una forma especial de estos elementos. Es posible combinar dos o más fibras (hibridación) para adaptar el refuerzo a las exigencias del diseñador. Las fibras híbridas confieren distintas propiedades y permiten muchas combinaciones de material. Pueden favorecer al máximo el rendimiento, reducir al mínimo el coste o compensar las deficiencias de otro componente de fibra (efecto sinérgico). Las fibras de vidrio y carbono se utilizan en combinación para aumentar la resistencia al impacto y la tenacidad, así como prevenir la acción galvánica y reducir el coste de un material compuesto de carbono al 10 por cien. Cuando se colocan estas fibras en una matriz, el material compuesto, no las fibras, se denomina híbrido. El material compuesto de hojas de metal o pliegues de material compuesto de metal apiladas en una orientación y secuencia específicas se denomina superhíbrido (véase lamelar). Para la fabricación de materiales compuestos de alta resistencia se han utilizado con éxito fibras de vidrio cortas orientadas aleatoriamente, y también de nilón, aramidas, poli(alcohol vinilico), poliacrilonitrilo y poliésteres. Puesto que estas fibras orgánicas cristalizan, sirven como agentes de nucleación cuando se utilizan para reforzar polímeros cristalizables como el polipropileno. El fieltro o la mecha de fibra de vidrio impregnada con resina de poliéster se utilizan como SMC y BMC. El primero de ellos se usa como polvo de moldeo y el segundo se moldea a presión y en caliente para adaptarse a la forma deseada, como por ejemplo en el caso de las dos partes de una maleta. La mecha de fibras de vidrio picada puede impregnarse con resina y proyectarse en forma de spray. El fieltro de fibra de vidrio puede impregnarse con resina justo antes del curado. Los materiales compuestos de mayor resistencia están hechos con filamentos continuos impregnados de resina antes de curar. Estos filamentos continuos se enrollan en un mandril en el proceso de enrollado de filamento, o se juntan y se hacen pasar a través de un orificio en el proceso de moldeo por pultrusión. Los primeros filamentos continuos utilizados eran de rayón, y éstos además de las fibras de acrilonitrilo han sido pirolizados para obtener fibra de carbono. Se han fabricado también filamentos de refuerzo de alto módulo elástico por deposición de átomos de boro, de vapores de tricloruro de boro, sobre filamentos de tungsteno o carbono. Los monocristales pequeños como los del titanato de potasio se vienen utilizando en cantidades anuales de más de 10 000 toneladas para reforzar nilón y otros termoplásticos. Estos materiales compuestos PMRN han sustituido a los metales de fundición en muchas aplicaciones. Otra microfibra, el hidroxicarbonato de sodio (llamada Dawsonita), también mejora las propiedades físicas y la resistencia a las llamas de muchos polímeros. Muchos otros monocristales, denominados whiskers, como pueden ser la alúmina, cromia y el carburo de boro, se han utilizado para fabricar materiales compuestos de altas prestaciones. Para reforzar termoplásticos y termofijos se utilizan fibras en continuo, fibras cortas, fibras tejidas bien de vidrio o sintéticas como pueden ser poliamidas aromáticas (Kevlar, Nomex), de carbono, etc. En la tabla 2.4.3 se exponen las propiedades de los agentes de refuerzo de fibra más utilizados. Tabla 2.4.3.- Propiedades de los agentes de refuerzo de fibra más utilizados (Metálicos y no metálicos) 3.- Propiedades de los materiales poliméricos. 3.1.- Introducción. La selección adecuada de un material polimérico para una determinada apli cación requiere el conocimiento, por una parte, de los requisitos de toda índole exigidos a las piezas o artículos a fabricar y, por otra, de las propiedades y características de los distintos materiales brutos o semielaborados que se encuentran disponibles en el mercado. También debe conocerse la posibilidad de modificar las características de los materiales básicos mediante mezcla con deter minados aditivos, cargas, colorantes, pigmentos, etc. y entre sí. En ocasiones hay que recurrir a preparar copolímeros específicos que presenten comporta mientos muy superiores a los de cada componente en determinadas condicio nes. En la actualidad, sin embargo, la tecnología y la industria de los materiales plásticos están suficientemente desarrolladas como para ofrecer una extensa gama de materias primas y de productos semielaborados que satisfacen las habituales necesidades del transformador final. La evolución de esta tecnología está impulsada, precisamente, por la pretensión de satisfacer requisitos cada vez más exigentes. Se va a completar el estudio de las propiedades mecánicas resistentes de los polímeros industriales en estado sólido, ya expuesto en el tema 4 considerando su comportamiento frente al impacto y al roce. Se tratarán así mismo las propiedades eléctricas, ópticas, térmicas y otras de gran importancia práctica, como son la permeabilidad a los gases y vapores, la estabilidad a altas temperaturas, el comportamiento en el fuego y, finalmente, la resistencia a los disolventes y reactivos químicos. 3.2.- Rozamiento y desgate. En el rozamiento no lubricado entre superficies sólidas se suele admitir como una primera aproximación simplificadora que la fuerza que se opone al movimiento F es proporcional al esfuerzo normal N entre las dos superficies e independiente del área de contacto. Dicha fuerza viene dada por la expresión: F = μN (3.2.1) donde μ es el coeficiente de rozamiento y N es la fuerza normal. El valor del coeficiente de rozamiento, μ, depende de la naturaleza de las dos superficies, de tal modo que el que corresponde a las condiciones límites previas a la iniciación del deslizamiento (coeficiente de rozamiento estático) es siempre superior al que se verifica cuando ya hay movimiento (coeficiente de rozamiento dinámico), resultando, además, este último independiente de la velocidad relativa. Tanto en aplicaciones industriales como en la vida cotidiana,* el grado de rugosidad de las superficies es importante, en ocasiones es deseable tener rugosidad “alta” y en otras ocasiones esta condición es indeseable. En algunos casos se busca que la superficie del producto terminado presente un mínimo de rugosidad, ya que esto le da brillo, mejor apariencia y disminuye la fricción de la superficie al estar en contacto con otra, reduciendo el fenómeno de desgaste y la corrosión o erosión de dichos materiales. La cuantificación de la rugosidad es uno de los problemas que aborda la topometría. Por otra parte, la fricción entre dos superficies es lo que permite sujetar un objeto sin que este resbale. Es la rugosidad de los neumáticos de los automóviles lo que favorece la fricción entre ellos y el suelo, permitiendo de esta manera el agarre y el avance controlado y seguro. En ocasiones se busca maximizar el área superficial, lo que se consigue incrementando la rugosidad, como en el caso de los catalizadores, cuya eficiencia es mejor entre mayor sea la superficie de contacto con los reactivos. Es la rugosidad de los “acetatos”, principalmente, lo que determina si pueden usarse en una impresora láser o en impresoras de inyección de tinta. La rugosidad también es un factor biológico, ya que a escala molecular afecta el modo en que las bacterias se adhieren a las superficies. Los materiales dentales deben presentar una superficie con el mínimo de rugosidad posible, para evitar la acumulación de placa bacteriana y para conseguir un mejor efecto estético. Se admite que el contacto entre los sólidos se establece principalmente en los resaltes de las superficies que, a nivel microscópico, son totalmente irregu lares, apareciendo importantes esfuerzos de cortadura ya antes de iniciarse el movimiento y que pueden llegar a arrancar el material y/o deformarlo irreversible o elásticamente, según que su naturaleza propicie un comportamiento frágil, dúctil o elástico. Iniciado el movimiento, las superficies en contacto sufren un proceso de limado, reduciéndose las asperezas y los resaltes y disminuyendo la resultante de los esfuerzos de cortadura. En los materiales cerámicos la rotura de las crestas de las irregularidades superficiales es frágil. En los metales se producen fluencias plásticas y en los materiales poliméricos, cuando la temperatura es superior a la de transición vítrea, fluencias viscoelásticas (en otro caso roturas frágiles, dependiendo de la dureza relativa de los dos materiales en contacto). En los elastómeros siempre se producen importantes deformaciones con recuperación elástica. Tan diferentes comportamientos han obligado a generalizar la ley del ro zamiento, que ahora se expresa mediante la expresión: F= KNx (3.2.2) resultando el coeficiente de rozamiento: μ = KNx-1 (3.2.3) siendo: x = 0.66 para los elastómeros x = 0.66-1 para los plásticos x = 0.85 para el PTFE x = 1 para la cerámica, madera, metales, etc. La dependencia del valor de μ con la temperatura es muy importante por el carácter viscoelástico del fenómeno del rozamiento en los plásticos y, puesto que la disipación del calor está muy restringida en estos materiales malos conductores, resulta inevitable que también dependa del valor de la velocidad relativa, puesto que el calor generado en el rozamiento y, en consecuencia, la temperatura local de las asperezas sometidas a tensiones cortantes depende del va lor de dicha velocidad. En definitiva, para cada plástico en rozamiento con otro material de referencia (por ejemplo, vidrio) existe una interdependencia coeficiente de rozamiento-velocidad de deslizamientotemperatura ambiente, mucho más notable que la que se advierte en otros materiales, y que se puede representar gráficamente en curvas isotermas como las de la figura 3.2.1, en las que se comprueba que, a cada temperatura, el coeficiente de rozamiento presenta un máximo a una determinada velocidad y que este máximo se desplaza a mayores valores de la velocidad, cuando aumenta la temperatura: Figura 3.2.1.- Coeficiente de rozamiento en función de la velocidad de deslizamiento Grosch ha demostrado el carácter viscoelástico del rozamiento en los plásticos y comprobado la aplicabilidad de la ecuación de Williams, Laundel y Ferry (WLF) obteniendo una única curva maestra come se muestra en la figura 3.2.2 a partir de las curvas de la figura 3.2.1. La ecuación de Williams, Laundel y Ferry viene dada por : log aT C10 (T T0 ) C20 (T T0 ) (3.2.4) donde C10 y C20 son constantes y T0 es una temperatura de referencia y el factor de traslación, aT, se puede definir como el cociente: (T ) (3.2.5) aT (Tr ) donde : η(T) Tiempo de relajación a la temperatura T. η(Tr)Tiempo de relajación a la temperatura de referencia Tr. Figura 3.2.2.- Curva maestra del coeficiente de rozamiento en función de la velocidad de deslizamiento y del factor de traslación. En la tabla 3.2.1 se recopilan los coeficientes típicos de rozamiento de los polímeros más usados, poniéndose de manifiesto la gran amplitud del margen de valores que se encuentran para un mismo material en función, tanto de sus propias características (grado de cristalinidad, densidad, etc.), como de las circunstancias del deslizamiento (dureza y aspereza de la superficie compañera, velocidad relativa, etc.). Tabla 3.2.1.- Coeficientes típicos de rozamiento de los polímeros más usados. El PTFE («TEFLON») y el PEAD tienen unos coeficientes de rozamiento ex traordinariamente bajos (0.06-0.08), mientras el PEBD lo tiene muy superior (0.3). A altas velocidades y temperaturas el de los dos primeros aumenta hasta 0.15-0.20 e, incluso, llega a 0.3, mientras el del PEBD alcanza valores de 0.8. Los elastómeros presentan coeficientes de rozamiento muy altos, considerándose que en ellos las fuerzas de adherencia tienen una extraordinaria importancia. Evidentemente el rozamiento en los polímeros produce un desgaste que resulta indeseable en cuanto supone una pérdida de material afectando a sus dimensiones originales. La medida de la resistencia al desgaste o a la abrasión se realiza sistemáticamente en ensayos específicos y no bien normalizados, de difícil cuantificación. Para reducir el rozamiento y el desgaste en piezas en las que ambos efectos deben minimizarse (como es el caso de los cojinetes, clavijas, engranajes, etc.) se pueden adicionar al polímero ciertos materiales con muy pequeña resistencia a los esfuerzos cortantes y bajo coeficiente de rozamiento, en forma de polvo de granulometría relativamente gruesa, que reciben el nombre de lubricantes sólidos. Los más usados en la actualidad son el grafito, el «TEFLON» (PTFE) y el disulfuro de molibdeno. Los gránulos de estos materiales actúan como micro -rodillos que se interponen entre las superficies que rozan y disminuyen su rugosidad, facilitando así el deslizamiento entre ambas. Deben distinguirse estos lubricantes sólidos de los lubricantes externos que se utilizan normalmente en los procesos de transformación para reducir el ro zamiento entre la masa del material polimérico fundido y las superficies metálicas de las máquinas y de los lubricantes internos o plastificantes a altas temperaturas que facilitan el reblandecimiento del material y que se consideran en el próximo capítulo. La resistencia relativa que opone un material a ser rayado o penetrado por efecto de una carga concentrada en una pequeña área de su superficie se denomina dureza o microdureza, según la cuantía del esfuerzo aplicado y las dimensiones de la huella obtenida. Su valor se obtiene relacionando la dimensión de la huella (medida al cabo de cierto tiempo) con la intensidad y la duración de la carga aplicada, en igualdad de condiciones del elemento penetrador (aguja o bola, etc.) y otras circunstancias; por consiguiente, la dureza debe estar siempre referida a la norma y al tipo de durómetro o microdurómetro utilizados (norma HV, VICKERS; norma HK, KNOOP; SHORE A/D, etc.). A diferencia de los metales, el carácter viscoso-elástico de los plásticos produce una rápida recuperación de la huella, tanto más, cuanto más alta sea la temperatura. Las características morfológicas, como el grado de cristalinidad y la orientación de los cristales, afectan a la medida de la dureza de un mismo material, estando estrechamente relacionada con sus restantes características mecánicas. Por esta razón y por la facilidad con que se realizan estos ensayos de carácter no destructivo, la dureza se utiliza en algunos casos, como en los cauchos y en algunas resinas duroplásticas, para caracterizar distintos grados de vulcanización, reticulación o curado. El desgaste producido en los plásticos por abrasión de sólidos granulares, generalmente mucho más duros que aquéllos, se reduce más fácilmente au mentando sus características elásticas que aumentando su dureza. Por eso, los elastómeros se utilizan sistemáticamente en la fabricación de protecciones antidesgaste en condiciones extremadamente duras y se mezclan con otros polí meros («blending»), formando las llamadas aleaciones, para mejorar su resis tencia a la abrasión y, simultáneamente, su tenacidad. 3.3.- Propiedades eléctricas. Las propiedades eléctricas de los materiales ponen de manifiesto su respuesta a la acción de un campo eléctrico aplicado. Desde un punto de vista eléctrico, los materiales se suelen clasificar en conductores (por ejemplo, metales), aislantes y semiconductores. Un material se considera aislante cuando la corriente que pasa a través de él por la acción de un campo eléctrico es muy pequeña, prácticamente despreciable. Consecuentemente, un aislante puede acumular cargas eléctricas y por tanto, energía eléctrica. La palabra «dieléctrico», especialmente si se utiliza como adjetivo, cubre un amplio rango de materiales que, al ofrecer una elevada resistencia al paso de corriente, presentan fenómenos de polarizabilidad. Estos fenómenos a su vez están relacionados con una gran variedad de aplicaciones de los materiales dieléctricos (la piezo y la ferroelectricidad, por ejemplo, la absorción de la luz, etc.). En la figura 3.3.1 puede verse la conductividad eléctrica de los tres tipos de materiales. Figura 3.3.1.- Conductividad eléctrica de los materiales conductores, aislantes y semiconductores. Los polímeros industriales en general son malos conductores eléctricos (materiales aislantes), por lo que se emplean masivamente en la industria eléctrica y electrónica como materiales aislantes. Las baquelitas (resinas fenólicas) sustituyeron con vent aja a las porcelanas y el vidrio en el aparellaje de baja tensión hace ya muchos años; termoplásticos como el PVC y los PE, entre otros, se utilizan en la fabricación de cables eléctricos, llegando en la actualidad a tensiones de aplicación superiores a los 20 KV (cables secos, sin aceite), y casi todas las carcasas de los equipos electrónicos (ordenadores, estufas, secadores, batidoras, etc.) se construyen en termoplásticos de magníficas propiedades mecánicas, además de eléctricas y de gran du ración y resistencia al medio ambiente, como son, por ejemplo, las resinas ABS. Evidentemente la principal desventaja de los materiales plásticos en estas aplicaciones está en relación a la pérdida de características mecánicas y geo métricas con la temperatura. Sin embargo ya se dispone de materiales que resisten sin problemas temperaturas relativamente elevadas (superiores a los 200 °C). Las propiedades eléctricas de los polímeros industriales están determina das, principalmente, por la naturaleza química del material (enlaces covalentes de mayor o menor polaridad) y son poco sensibles a la microestructura cristalina o amorfa del material, que afecta mucho más a las propiedades mecánicas. Su estudio se acomete mediante ensayos de comportamiento en campos eléctricos de distinta intensidad y frecuencia. Seguidamente se analizan las características eléctricas de estos materiales. Conducción eléctrica. Ley de OHM. Una de las más importantes características eléctricas de un material sólido es la facilidad con que transmite una corriente eléctrica. La ley de Ohm relaciona la corriente, I, o sea el paso de carga por unidad de tiempo, con el voltaje aplicado, V, de la manera siguiente: V IR (3.3.1) donde R es la resistencia del material a través del cual pasa la corriente. Las unidades de V, I y R son, respectivamente, voltios (J/C), amperios (C/s) y ohmios (V/A). El valor de R depende de la configuración de la muestra y en muchos materiales es independiente de la corriente. La resistividad, , es independiente de la geometría de la muestra y está relacionada con R mediante la expresión: RA (3.3.2) L donde L es la distancia entre los dos puntos en que se mide el voltaje y A es el área de la sección perpendicular a la dirección de la corriente. Las unidades de son ohmios-metro (-m). A partir de la expresión de la ley de Ohm y de la ecuación (3.3.2), se tiene: RVA IL (3.3.3) La figura 3.3.2 es un esquema de un dispositivo experimental para medir la resistividad eléctrica. Figura 3.3.2 .- Esquema del aparato utilizado para medir la resistividad eléctrica. Conductividad eléctrica. Algunas veces se utiliza la conductividad eléctrica, , para especificar el carácter eléctrico de un material. Es simplemente el recíproco de la resistividad, o sea: 1 (3.3.4) e indica la facilidad con que un material es capaz de conducir una corriente eléctrica. Las unidades de son las inversas de -m (-m)-1, o bien, mho/m. Los materiales sólidos muestran un sorprendente intervalo de conductividades eléctricas, el cual se extiende unos 27 órdenes de magnitud. Probablemente ninguna otra magnitud física experimenta tan amplia variación. De hecho, una manera de clasificar a los materiales sólidos es según la facilidad o dificultad para conducir la corriente eléctrica. Los metales son buenos conductores, típicamente tienen conductividades del orden de 107 En el otro extremo están los materiales con muy bajas conductividades que van desde 10-10 a 10-20 (-m)-1. Estos son los aisladores eléctricos. Materiales con conductividades intermedias, generalmente entre 10-6y 104 (-m)-1 se denominan semiconductores. Los polímeros industriales siguen la ley de Ohm hasta valores muy altos del campo eléctrico: E V , L V I L 1 L I S X S (3.3.5) La resistividad específica ρ suele estar comprendida entre 10 10 y 10 22 Ω.cm. (Su inversa X = 1/ρ se denomina conductividad específica). La baja conductividad de los polímeros es debida a la emigración de iones extraños (restos de monómeros o de catalizadores) ocluidos en su masa, que se desplazan movidos por el efecto del campo eléctrico. Realmente no existe un flujo libre de electrones, como ocurre en los metales. La movilidad de los iones en la masa del polímero es mayor cuando aumenta la temperatura; pudiéndose expresar el aumento de la conductividad con la temperatura mediante: b (3.3.6) log( X ) a , (a, b Ctes) T En campos alternos la resistividad y la conductividad tienen exactamente el mismo valor que en campos continuos, hasta valores muy altos de la frecuencia. Cuando el campo eléctrico supera los 100 KV/cm la conductividad aumenta con la intensidad del campo, llegándose a producir la descarga eléctrica por chispa o arco, que deteriora el material. La rigidez dieléctrica, expresada en V/cm designa los resultados de una prueba realizada según un procedimiento normalizado, en la que se provoca la perforación de un espesor del dieléctrico con una cierta diferencia de potencial, en unas determinadas condiciones (humedad, temperatura, forma de los electrodos, espesor del material, etc.). Evidentemente la capacidad de adsorción de agua que presentan los polímeros con grupos po lares en sus macromoléculas, influye notablemente en su resistividad y en su rigidez dieléctrica. Debido a su baja resistividad los plásticos se han utilizado tradicionalmente como aislantes en la industria eléctrica y en la electrónica. Sin embargo, en la actualidad se están desarrollando nuevos polímeros semiconductores, cuya particular estructura molecular les proporciona una gran movilidad electrónica. Estos plásticos están constituidos fundamentalmente por cadenas de átomos de carbono con dobles enlaces conjugados que entran en resonancia, solapándose sus orbitales a lo largo de la cadena macromolecular (como ocurre en los anillos bencénicos). De esta forma se originan anchas bandas de energía, en lugar de los orbitales específicos con niveles discretos de energía en los que quedan fijados los electrones de los polímeros aislantes. La presencia de determinadas impurezas (dopantes) altera la distribución electrónica, vaciando parcialmente las bandas energéticas con lo que se potencia considerablemente la conducti vidad del material. Así resulta que algunos polímeros lineales derivados del poliacetileno (-C=C=C=C=C=C-) y otros, cuyas cadenas principales son totalmente aromáticas (como el poliparafenileno y la polianilina), resultan casi tan buenos conductores como algunos metales a la temperatura ambiente. Sin embargo todavía presentan algunos problemas en sus aplicaciones industriales (en baterías y circuitos impresos, por ejemplo) debido a que los unos se oxidan muy fácilmen te y no pueden estar en contacto con soluciones acuosas, mientras que los otros son excesivamente rígidos y frágiles y, por ser infusibles e insolubles, no se pueden procesar mediante las técnicas convencionales de transformación. Tan pronto se superen estas dificultades, se espera encuentren una aplicación mucho más extensa en estas y otras industrias. Independientemente de la resistencia que ofrecen los polímeros al paso de la corriente eléctrica a su través, es importante considerar su comportamiento al paso de la corriente por su superficie. La presencia de iones extraños, incluso de humedad, puede hacer conduc tiva la superficie, de forma que el paso de la corriente eléctrica se produce principalmente por ella. Este fenómeno puede caracterizarse mediante una cierta resistencia (resistividad superficial se le denomina para diferenciarla de la resistividad específica o volumétrica), medida en ohmios. Para algunas aplicaciones una alta resistividad superficial ocasiona ciertos inconvenientes, debido a la presencia de cargas estáticas (electrones e iones), que aparecen cuando dos superficies se separan después de haber estado en contacto. Tal es el caso de los filmes que se adhieren fuertemente entre sí, dificultando su manipulación. Para evitar estos inconvenientes, en la fabricación del polímero se adicionan aditivos antiestáticos (agentes ionizables) que pueden emigrar desde la masa del polímero a su superficie, proporcionando, con la ayu da de la humedad atmosférica, una cierta conductividad superficial que elimina la presencia de las cargas estáticas. Este efecto superficial también tiene importancia a altos valores del campo eléctrico. La rigidez de fuga define la resistencia del material a acciones destructivas de las corrientes superficiales, de la misma forma que la rigidez dieléctrica lo hace para las que se producen a su través. Constante dieléctrica y factor de pérdida. La influencia de un dieléctrico particular sobre la capacitancia de un con densador viene determinada por la constante dieléctrica también conocida por permitividad y/o capacidad inductiva específica y se define como la relación entre la capacidad de un condensador, empleando ese material como dieléctrico y la capacidad del mismo condensador sin dieléctrico, es decir, en vacío (o, para todos los efectos prácticos, en el aire). C , C0 C Q Q , C0 0 C V (3.3.7) Supóngase un condensador sin dieléctrico. La diferencia de potencial ΔV origina una carga eléctrica Q 0 en los platillos. Al intercalar un dieléctrico cual quiera, manteniendo la diferencia de potencial, la carga en los platillos aumenta como consecuencia de la polarización del material intercalado. El efecto de la polarización depende de la estructura de las moléculas. Las pequeñas y simétricas se polarizan, desplazándose el centro de gravedad de los electrones con respecto a los núcleos atómicos (polarización electrónica). Si las moléculas son asimétricas (polares), el centro de gravedad de sus cargas eléctricas no coincide, aunque no exista campo eléctrico. El efecto de la polarización, en este caso, consiste en un giro de las moléculas que se orientan en la dirección del campo eléctrico (polarización de orientación o Bipolar). Existe una diferencia fundamental entre ambos tipos de polarización: la pri mera es instantánea, por implicar el desplazamiento de electrones; la segunda requiere tiempo, puesto que la molécula entera debe girar desplazándose con respecto a las que le rodean. Si el campo eléctrico es alterno la polarización di-polar se produce con un retraso de fase 5, denominado ángulo de pérdida, y si la frecuencia de cambio de dirección del campo eléctrico es muy grande, el efec to de la polarización disminuye. El desfase entre la tensión aplicada y la inten sidad de corriente en un dieléctrico real será Φ = 90 - δ. Se denomina factor de disipación a la tag(δ), factor de potencia la cos(Φ) = sen(δ) y factor de pérdida al producto de la constante dieléctrica por el factor de disipación ε tag(δ). La variación de la constante dieléctrica y del factor de pérdida con la fre cuencia, para un material dipolar constituido por moléculas pequeñas (metano) por ejemplo), puede verse en la figura 3.3.3, siendo característica la presencia de un «pico» en el diagrama del factor de pérdida. Figura 3.3.3.- Variación de la constante dieléctrica y del factor de pérdida con la fre cuencia, para un material dipolar. El efecto de la temperatura es importante, por disminuir la viscosidad interna del material, con lo que se facilita el movimiento de los dipolos cuando esta aumenta. En el caso del metanol sólido la constante dieléctrica es 3, en el punto de fusión 55, y para el metanol líquido se reduce a 35, ya que la mayor energía interna disponible origina un movimiento desordenado que tiende a destruir el efecto de la polarización. En el caso de los materiales poliméricos apolares (como el PE y PTFE) la polarización es exclusivamente electrónica y, dado que esta es instantánea, la constante dieléctrica e varía muy poco con la frecuencia; el factor de disipación tag(δ) es bajo y no aparecen picos de pérdidas. En consecuencia los polímeros apolares resultan adecuados para su uso en dispositivos y elementos electrónicos de alta frecuencia. En los polímeros polares (como el PVC y PVA) el factor de disipación es generalmente más alto que en los apola res y suelen aparecer picos de pérdida a determinadas frecuencias, que pueden correlacionarse con las temperaturas de transición vítrea T g y de fusión T m . El comportamiento de los polímeros polares es muy diferente según lo s grupos polares estén situados en las cadenas principales de las macromoléculas (como en los policarbonatos, poliésteres saturados PET y PVC por ejemplo) o en las cadenas laterales (como el PVA y los poliacrilatos). En el primer caso la movilidad de estos grupos está condicionada a la de la propia cadena, por lo que el efecto de la polarización es mucho menos acusado por debajo de la temperatura de transición vítrea, que por encima de ella. El PVC es buen aislante a bajas frecuencias, pero en campos eléc tricos de alta frecuencia se calienta en exceso y puede alcanzar fácilmente la temperatura de reblandecimiento. Puede observarse en la figura 3.3.4 como el polietilentereftalato (poliester saturado), que tiene un amplio margen entre la temperatura de transición vítrea (70 °C) y la de fusión (255 °C), reblandeciéndose notablemente a partir de los 200 °C, presenta un pico del factor de potencia a una temperatura intermedia. La temperatura a la que se produce este pico y el factor de pérdida correspondiente aumenta con la frecuencia. Figura 3.3.4.- Variación del factor de potencia con la temperatura para diversos plásticos. La presencia de plastificantes, que reducen la temperatura de transición ví trea, tendrá como consecuencia un aumento en la polarización, y, por lo tanto, del valor de la constante dieléctrica (Figura 3.3.5). Figura 3.3.5.- Variación de la constante dieléctrica con la temperatura y con la presencia de plastificantes En el caso de moléculas poliméricas en las que los dipolos no están en las cadenas principales, su orientación resulta mucho más fácil, incluso a tempe raturas inferiores a la de transición vítrea y, por lo tanto, no se producirá esa variación brusca en los valores de la constante dieléctrica y del factor de potencia que se pone de manifiesto para los que tienen los grupos polares en la ca dena principal. En consecuencia no se utilizan como aislantes. En la tabla 3.3.1 de da el valor de algunas propiedades eléctricas de diversos materiales poliméricos. Tabla 3.3.1.- Propiedades eléctricas de algunos materiales poliméricos. Finalmente se debe tener en cuenta que la capacidad de absorción de humedad de algunos polímeros (como las poliamidas, por ejemplo) influye en sus propiedades eléctricas por diversos motivos: (i).- La mayor conductividad eléctrica del agua origina una disminución en la resistividad del material. (ii).- La acusada polaridad del agua y su pequeño tamaño molecular eleva notablemente los valores de la constante dieléctrica y de las pérdida en el conjunto. (iii).- El agua puede actuar como plastificante, aumentando la movilidad de las cadenas moleculares de algunos polímeros, por lo que se incrementa el valor del factor de pérdida propio del material. 3.4.- Propiedades ópticas. Desde el punto de vista de su utilización, las propiedades ópticas más interesantes de los materiales plásticos son las relacionadas con su capacidad de transmitir la luz, tomar color y disponer de brillo, que proporcionan a los objetos fabricados una apariencia visual estética de alta calidad. 69 Transmisión y reflexión de la luz. La transmisión de la luz a través de una placa plana de cualquier material depende de la intensidad de luz que se refleja R y de la que queda absorbida en su interior. La primera es función del ángulo de incidencia i y del índice de refracción nD = seni/sen r, siendo r el ángulo de refracción, según queda expresado mediante la ecuación de Fresnel: R 1 sen2 (i r ) tag 2 (i r ) 2 I0 2 2 sen (i r ) tag (i r ) (3.4.1) y para el caso de que la luz incida perpendicularmente a la superficie (i = 0) n 1 I R D 2 0 nD 1 2 (3.4.2) Como se comprueba en la tabla 3.4.1, el índice de refracción de los polímeros industriales es del orden de 1.5, por lo que, considerando una placa plana dispuesta en el aire perpendicularmente a la luz incidente, ya en su superficie se refleja un 4 % del flujo de luz (R = 0.04 /o). Tabla 3.4.1.- Índice de refracción de algunos polímeros. Si el polímero fuera isótropo, ópticamente homogéneo y no absorbiera la luz, el rayo incidente se transmitiría a través del polímero sin pérdida de intensidad, pero en la realidad se produce una cierta dispersión que desvía parte de la intensidad lumínica en otras direcciones, además de producirse una cierta absorción, a medida que aumenta la profundidad o espesor del material. Se define como factor de transmisión directa T de un material a la relación entre la intensidad del flujo lumínico no desviado y la del rayo incidente. 70 Para materiales incoloros y con poca dispersión, el valor de T disminuye exponencialmente con el espesor X, y por lo tanto log T = -ζX (3.4.3) definiéndose ζ como factor de dispersión, que suele medirse mediante ensayos en muestras de materiales de 1 mm de espesor. Un material es transparente cuando resulta posible la percepción de objetos a través suyo. La transparencia puede expresarse cuantitativamente como la fracción de la intensidad luminosa que se transmite con una desviación inferior a 0,1°, cuando la luz incide perpendicularmente a la superficie. Para que un material presente una alta transparencia es necesario que el índice de refracción del material sea constante en el recorrido de la luz. La presencia de esferulitas o zonas cristalinas, que presentan índices de refracción diferentes que el resto del material, debida a una diferencia de densidad, origina en su superficie un efecto de dispersión de la luz que produce turbidez. Por este motivo la máxima transparencia se encuentra en los polímeros amorfos, libres de cargas y otras impurezas, como sucede con el PMMA. Los polímeros cristalinos pueden ser también transparentes cuando las es ferulitas son más pequeñas que la longitud de onda de la luz incidente, por lo que no se producen las interferencias que originan la turbidez. Esto sucede, por ejemplo, en filmes de PEBD que han sido enfriados rápidamente en el curso de su fabricación. En el caso de objetos de gran espesor no es posible realizar el enfriamiento rápido de toda la masa, por lo que los cristalitos aumentarán de tamaño de tal forma que, inevitablemente, producirán interferencias al paso de la luz y los objetos no serán transparentes, a lo más serán translúcidos. Para reducir el tamaño de las esferulitas se suelen aditivar agentes nuclean tes que multiplican los gérmenes de cristalización originando cristalitos micrométricos, con lo que la transparencia del material aumenta. Si la diferencia de los índices de refracción de la fase amorfa y de la fase cristalina es pequeña, como ocurre en el PP, la presencia de esferulitas de gran tamaño es compatible con una cierta transparencia. La intensidad de la luz que atraviesa un material depende de la natur aleza del material y de su microestructura molecular. En la tabla 3.4.2 se comparan valores típicos de los factores de dispersión, para diferentes polímeros usados en ingeniería, poniéndose de manifiesto las enormes diferencias en los valores del factor de transmisión directa T para muestras de espesor normalizado (1 mm). Tabla 3.4.2.- Propiedades ópticas de algunos polímeros. 71 En cualquier caso, la existencia de rugosidades en las superficies de los objetos origina una dispersión de la luz que reduce la transparencia. Esta rugosidad es debida, generalmente, al propio proceso de transformación o fabricación del objeto, y también afecta a su apariencia superficial. Si su superficie ex terior es lisa, la mayor parte de la luz se reflejará directamente y el objeto aparecerá brillante; si es rugosa, la luz reflejada será dispersa y su aspecto será mate. El estirado bidireccional de los filmes mejora la transparencia de los mate riales termoplásticos por romper las esferulitas y orientar los cristalitos resultantes en planos perpendiculares a la dirección pormal a sus superficies exteriores (que es la que sigue la luz cuando se busca la máxima transparencia), a la vez que se eliminan los defectos superficiales. Recientemente se han detectado comportamientos ópticos no lineales en algunos polímeros transparentes que, estando iluminados intensamente por un haz luminoso, presentan un índice de refracción y de absorción muy distinto a cualquier otro haz luminoso que se haga incidir sobre su superficie en otras direcciones diferentes. Tal fenómeno, de carácter reversible, se justifica para los polímeros de características semiconductoras considerando que el primer haz luminoso excita los electrones deslocalizados por solapamiento de los orbitales a lo largo de la cadena macromolecular de modo que los haces que inciden posteriormente encuentran una estructura electrónica muy alterada, que no respon de como lo haría en condiciones normales. La importancia de este descubrimiento debe considerarse de cara a la posible fabricación de dispositivos ópticos que pudieran dar paso, o no, a la luz a su través según estuvieran excitados, o no, en otra dirección por otro rayo de luz que realizase la función de control. Tales dispositivos, junto con las fibras ópticas, darían paso a sistemas informáticos y de telecomunicación íntegramente ópticos. Absorción de la luz y color. La absorción de la luz, o de cualquier otra radiación, es generalmente selectiva en los materiales plásticos. La energía electromagnética asociada a una determinada longitud de onda que se absorbe en un plástico se disipa aumentando el nivel energético de ciertos electrones localizados en determinados grupos atómicos. Este fenómeno produce dos efectos distintos: per una parte, el efecto d el color y, por otra, el de la fotodegradación. Cuando toda la luz es absorbida por el material, el objeto aparece negro. Si sólo una parte de la luz (determinadas longitudes de onda) se absorbe y el resto se transmite sin dispersarse, el objeto aparecerá como coloreado y transparente (con el color correspondiente a las longitudes de onda no absorbidas). Si, por el contrario, la fracción de luz no absorbida se dispersa íntegramente sin llegar a transmitirse, el objeto será coloreado y opaco. Los plásticos en estado puro son, generalmente, incoloros. Para proporcio nar color, sin alterar apreciablemente su transparencia, se ceben incorporar adi tivos denominados tintes que absorban las radiaciones precisas del espectro visible, dejando pasar las demás. Es n ecesario que los tintes sean compatibles con el polímero y se disuelvan en su masa sin originar discontinuidades que produzcan la dispersión de las radiaciones no absorbidas para no reducir la transparencia. Si lo que se pretende es proporcionar color y opacidad, los aditivos deben proporcionar la absorción de las correspondientes radiaciones del espectro visible y a la vez aumentar la reflexión y dispersión de la luz. Por ello su índice de refracción debe ser mucho mayor que el del polímero y su tamaño m ayor que 72 la longitud de onda de las radiaciones no absorbidas. Tales aditivos se deno minan habitualmente pigmentos. Las partículas de los pigmentos (Figura 3.4.1), con un tamaño del orden de 1 μm, producen múltiples dispersiones de la luz que les llega, aumentando el factor dispersante ζ, con lo que a poca distancia de la superficie el factor de transmisión directa T se anula prácticamente. Figura 3.4.1.- Múltiples dispersiones de la luz producidas por las partículas de los pigmentos El uso simultáneo de tintes y de pigmentos blancos, como el carbonato cál cico y el dióxido de titanio, proporciona a los plásticos colores muy luminosos, pues los segundos no absorben ningún color, sino que producen un efecto dispersante, devolviendo la luz a la superficie. El alto índice de refracción de la va riedad rutilo del TiO 2 , que es 2.76, le hace mucho más eficaz que el carbonato cálcico (n D = 1.63). Como se pone de manifiesto en la figura 3.4.2, la eficacia en la dispersión está íntimamente unida al tamaño de la partícula, aumentando cuando esta disminuye, pero no por debajo de la longitud de onda de la luz, pues entonces se pierde el efecto dispersante. Figura 3.4.2.- Eficacia en la dispersión en función del tamaño de la partícula Fotodegradación. Como consecuencia de la absorción de la energía radiante luminosa por las estructuras químicas que forman los polímeros, se produce una cierta degradación de éstos, tanto mayor cuanto menor es la longitud de onda, que se pone de manifiesto en una fragilidad y decoloración superficial, entre otros defectos más difícilmente observables (aumento de pérdidas dieléctricas, etc.). Esta degradación es debida a que la energía absorbida puede ser superior a la de disociación de un determinado enlace. El que una radiación de una longitud de onda dañe o no a un determinado polímero depende también de las frecuencias de absorción de los enlaces existentes entre los átomos 73 de su molécula (Enlace C-C absorbe a 1950, 2300 y 2500 A, mientras un enlace C=O lo hace a 1870, 2800 y 3200 A). En la figura 3.4.3 se representa la relación entre la energía absorbida y la longitud de onda de la luz incidente, indicando la energía de disociación de varios tipos de enlaces. Figura 3.4.3.- Relación entre la energía absorbida y la longitud de onda de la luz incidente. La fotodegradación de los polímeros es mucho más importante por efecto de la radiación ultravioleta que por la visible. Sin embargo, la luz visible induce un proceso irreversible de oxidación, potenciado por la presencia de óxido de nitrógeno, la acción hidrolizante de la lluvia ácida y de la actividad microbiana, conjuntamente. La ruptura de enlaces covalentes debida a la absorción de una radiación (ra diólisis) origina radicales libres que, entre otras posibilidades, o bien dan lugar a la abertura de la cadena macromolecular por debilitamiento del enlace C-C en posición β (fisión β, característica de las reacciones químicas de «cracking» térmico) con formación de una a-olefina y un nuevo radical primario, o bien proporcionan reticulaciones entre distintas macromoléculas contiguas (efecto de vulcanización o de curado, propio de los elastómeros y duroplásticos). En cualquier caso, la estructura del material original se altera, cambian sus características mecánicas y varía su comportamiento frente a los agentes químicos. En la industria se aprovecha el efecto reticulante de las radiaciones, me diante métodos controlados, para preparar elastómeros a partir de materiales lineales o termoplásticos (vulcanización del poliisobutileno, en ausencia total de butadieno, o de copolímeros etileno-propileno). También en la fotolitografía de circuitos impresos se utilizan polímeros -fotosensibles que se insolubilizan y fij an al sustrato cuando reticulan al ser iluminados por rayos laser («resists» positivos), protegiéndole del ataque químico con que se efectúa el grabado subsiguiente del circuito. Para evitar el efecto perjudicial de la luz y de radiaciones ultravioletas, es pecialmente en los elastómeros (en los que existen dobles enlaces 1,4, que absorben con facilidad energía de la luz visible, como se ve en la figura) se adicionan pigmentos absorbentes (negro de humo, por ejemplo) o cargas que absor ben las radiaciones en las proximidades de la superficie, protegiendo así el interior. Cuando se trata de plásticos y se quiere mantener su transparencia, se usan absorbedores de UV, es decir, aditivos moleculares que, al recibir la luz ultravioleta, se convierten en otras sustancias, transformando la energía recibida en calor. Así actúan los derivados de la 2-hidroxibenzofenona, entre otros: 74 Otros aditivos estabilizadores a bajas temperaturas son los compuestos organometálicos a base de níquel, que actúan desactivando los átomos o grupos atómicos que hayan llegado a ser excitados. Los antioxidantes propios para altas temperaturas de procesado también son efectivos como aditivos que previenen el envejecimiento. 3.5.- Propiedades térmicas. Por "propiedad o característica térmica" se entiende la respuesta de un material al ser calentado. A medida que un sólido absorbe energía en forma de calor, su temperatura y sus dimensiones aumentan. La energía puede transportarse de las regiones calientes a las regiones más frías de la muestra si existe un gradiente de temperatura y, finalmente la muestra puede fundirse. La capacidad calorífica, la dilatación térmica, la conductividad térmica y la refractariedad (Resistencia piroscópica) son propiedades muy importantes en la utilización práctica de los materiales y, en particular, de los materiales. Capacidad calorífica. Calor específico. Cuando se calienta un material sólido, éste experimenta un aumento de temperatura, indicando con ello que absorbe energía. La capacidad calorífica es una propiedad que indica la capacidad de un material de absorber calor de su entorno. Representa la cantidad de energía necesaria para aumentar la temperatura del material en una unidad. Si Q es la cantidad de calor que hay que dar a un cuerpo para subir su temperatura de T1 a T2, se puede definir el cociente siguiente: Q T CT 2 1 Q T2 T1 (3.5.1) T1 T2 el cual representa la capacidad calorífica promedio del cuerpo entre las temperaturas de T1 y T2. Si las dos temperaturas T1 y T2 tienden hacia un límite común T, el cociente tiende hacia un límite: C dQ dT (3.5.2) donde dQ es la energía necesaria para producir un cambio dT en la temperatura. Normalmente, la capacidad calorífica se expresa por mol de material C (J/mol.K), obteniéndose la capacidad calorífica molar, que puede ser a volumen constante, CV o a presión constante, CP. 75 La capacidad calorífica a volumen constante se define como (En los procesos a volumen constante todo el calor tomado por el sistema se invierte en incrementar su energía interna dQV = dU): U CV T V (3.5.3) donde: U = Energía interna y T = Temperatura Por su parte, la capacidad calorífica a presión constante se define como (En los procesos a presión constante el calor absorbido equivale al incremento de entalpía del sistema dQP = dH): H CP T P (3.5.4) donde: H = U+PV = Entalpía del sistema y T = Temperatura Existen dos métodos para medir esta propiedad, según cuáles sean las condiciones del medio en que se realiza la transferencia de calor. Uno es medir la capacidad calorífica mientras se mantiene la muestra a volumen constante, en este caso se representa por CV y el otro es bajo presión constante y se representa por CP. La magnitud de CP es más fácil de medir y siempre es mayor que CV. Sin embargo, esta diferencia es muy pequeña para la mayoría de los materiales sólidos a temperatura ambiente e inferiores, como se puede deducir de la expresión: CP CV 9 2VT / K donde: = Coeficiente de dilatación lineal K = Modulo de elasticidad volumétrica= (3.5.5) P (Cociente entre el cambio de presión y la disminución V V relativa de volumen) V = Volumen T = Temperatura absoluta. Para los sólidos la diferencia entre CP y CV es muy pequeña, ya que el valor de es muy pequeño y el de K grande. A temperatura ambiente la diferencia para los sólidos es del 5 %. A veces se utiliza el calor específico (a menudo representado por ce). Este representa la capacidad calorífica por unidad de masa (J/kg.K) y se define como la cantidad de calor que hay que comunicar a la unidad de masa con el fin de elevar un grado su temperatura, es decir: ce Q mT (3.5.6) La capacidad calorífica de un material está influenciada por los efectos que la temperatura (Existencia de una cierta energía térmica) ejerce sobre: 76 1.- Las vibraciones (u oscilaciones) de los átomos o iones alrededor de sus posiciones de equilibrio, denominada energía vibracional y las rotaciones dentro del material, denominada energía rotacional. 2.- Cambios del nivel de energía de los electrones en la estructura electrónica. Los electrones absorben energía aumentando su energía cinética. Sin embargo, esto sólo es posible en el caso de electrones libres, es decir: aquellos que han sido excitados desde los estados ocupados a los estados vacíos por encima del nivel de Fermi. En los metales, solamente los electrones en estados muy cercanos a la energía de Fermi pueden sufrir estas transiciones, y esto representa únicamente una fracción muy pequeña del número total. Una fracción aún menor de electrones experimenta excitaciones en los materiales aisladores o semiconductores. Por tanto, esta contribución electrónica es generalmente insignificante, excepto a temperaturas cercanas a los 0 K. 3.- Cambios en las posiciones atómicas durante la formación de los defectos en la red cristalina (Vacantes, defectos intersticiales, etc.), transiciones orden-desorden, transformaciones polimórficas, orientaciones magnéticas (aleatorización de los espines en un material ferromagnético a medida que es calentado hasta su temperatura de Curie), etc. La tabla 3.5.1 indica los valores del calor específico de varios materiales, así como el de otras propiedades térmicas. Se puede observar que los materiales poliméricos presentan un calor específico superior al de los materiales metálicos y cerámicos. Ello pone de manifiesto que para elevar la misma masa de material una determinada temperatura es necesario suministrarles más calor. Tabla 3.5.1.- Propiedades térmicas de varios materiales. 77 Dilatación térmica. Todos (o la mayoría) de los materiales sólidos experimentan variaciones dimensionales (Dilatación o contracción) por efecto de la temperatura, así se expanden cuando son calentados y se contraen cuando son enfriados. El cambio de longitud con la temperatura para un material sólido puede expresarse de la manera siguiente: L f Lo L = l T f To o bien : (3.5.7) l T Lo Lo donde Lo y Lf representan, respectivamente, las longitudes iniciales y finales al cambiar la temperatura desde To a Tf: El parámetro l se denomina coeficiente lineal de dilatación térmica. Es una propiedad que indica el grado de dilatación de un material cuando es calentado y tiene unidades del recíproco de la temperatura [(°C)-1]. Desde luego, el calentamiento o el enfriamiento afectan a todas las dimensiones del cuerpo lo cual produce un cambio de volumen. Los cambios de volumen con la temperatura pueden calcularse a partir de: V v T Vo (3.5.8) donde, ∆V y Vo son el cambio de volumen y el volumen inicial, respectivamente, y v simboliza el coeficiente de volumen de la dilatación térmica. En muchos materiales, el valor de v, es anisotrópico, es decir, depende de la dirección cristalográfica a lo largo de la cual es medido. Para los materiales isotrópicos, v, es aproximadamente igual a 3l. El tanto por ciento de dilatación (o de contracción) puede expresarse en función del coeficiente lineal de dilatación térmica, así: L (% de exp ansión)T To 100 100T To T (3.5.9) L0 o bien : (% exp ansión)T To T T o (3.5.10) 100T En la figura 3.5.1 se da el tanto por ciento de expansión frente a la temperatura de diversos materiales metálicos, cerámicos y poliméricos. 78 Figura 3.5.1.- Expansión térmica lineal de metales, polímeros y cerámicas policristalinas típicos. Desde el punto de vista atómico, la dilatación térmica se refleja en un aumento en la distancia media de separación entre los átomos. Este fenómeno se entiende mejor consultando la curva de energía potencial frente a la separación interatómica para un material sólido, que se reproduce en la figura 3.5.2. La curva tiene forma de un pozo de energía potencial, y la distancia interatómica de equilibrio, r0, corresponde al mínimo del pozo. Calentando sucesivamente a temperaturas más altas (T1, T2, T3, etc.) aumenta la energía vibracional desde El hasta E2 y E3, y así sucesivamente. La amplitud media de la energía vibracional de un átomo corresponde a la anchura del pozo a cada temperatura y el espaciado interatómico se representa por la posición media, la cual aumenta con la temperatura desde r0 a rl y r2, y así sucesivamente. La dilatación térmica se debe realmente a la asimetría de la curva de este pozo de energía potencial, más que al aumento de las amplitudes de vibración con la temperatura. Si la curva de energía potencial fuera simétrica con respecto a la vertical que pasa por el mínimo (Figura 3.5.2), no existiría un cambio neto en la separación interatómica y, en consecuencia, no existiría dilatación térmica. 79 Figura 3.5.2.- (a) Gráfico de la energía potencial frente a la distancia interatómica, mostrando el aumento en la separación interatómica al aumentar la temperatura. Al calentar, la separación interatómica aumenta desde ro a r1, y de aquí a r2 y así sucesivamente. (b) Para una curva simétrica energía potencial-separación interatómica, no hay aumento en la separación interatómica al aumentar la temperatura (es decir, rl = r2 = r3). Para cada clase de materiales (metales, cerámicas y polímeros), cuanto mayor es la energía del enlace interatómico, más profundo y estrecho es el pozo de energía potencial. Por consiguiente, el aumento en la separación interatómica debido a un determinado aumento de temperatura será menor y tendrá un valor de αl menor. Otro factor que influye sobre el valor de αl es el la disposición o empaquetamiento de los átomos en la estructura, que cuanto mayor sea mayor es el valor del coeficiente de dilatación térmica, pues la acumulación de las separaciones es mayor. La tabla 3.5.1 indica los coeficientes lineales de dilatación térmica de varios materiales. Algunos materiales polímeros experimentan dilataciones térmicas muy elevadas al ser calentados tal como es de esperar por los altos coeficientes de dilatación que van desde aproximadamente 50 x 10-6 hasta 300 x 10-6 (°C)-1. Los valores más altos de al se encuentran en los polímeros lineales y ramificados debido a que los enlaces intermoleculares son débiles y el entrecruzado es mínimo. Al aumentar el entrecruzamiento, la magnitud del coeficiente de dilatación disminuye. Los coeficientes menores se encuentran en los polímeros termoestables tales como la baquelita, en donde el enlace es casi completamente covalente. Conductividad térmica. La conductividad térmica es una propiedad muy importante a la hora de elegir el material más adecuado desde el punto de vista del aislamiento térmico. En general, en los materiales de construcción, y especialmente en los aislantes, se requiere una baja conductividad térmica al objeto de minimizar las pérdidas de calor por las paredes de los edificios, etc. La transmisión de calor a través de un material es un fenómeno de transporte complejo, debido a que al ser un sólido poroso intervienen en él, en mayor o menor grado, los tres mecanismos de transmisión de calor: - CONDUCCION (En el sólido y en el gas encerrado en los poros) - CONVECCION (En el gas). - RADIACION (En el gas) 80 En la figura 3.5.3 pueden verse esquematizados los tres mecanismos anteriores, así como los factores más importantes que influyen sobre ellos. Figura 3.5.3.- Transmisión de calor en los materiales conformados que presentan porosidad. La ley fenomenológica que rige la conducción del calor fue propuesta por el físico y matemático francés J. B. FOURIER. Expondremos dicha ley con ayuda del sencillo problema del flujo unidimensional de calor a través de una pared plana (p. ej., una capa de aislante). La figura 3.5.4 muestra una pared plana de área A y espesor L, cuya cara en x = 0 se mantiene a la temperatura T 1, mientras que el lado en x = L se mantiene a T2 (T1 > T2). El flujo de calor Q a través de la pared se efectúa en la dirección de la disminución de la temperatura. La ley de Fourier establece que, la densidad de flujo de calor, q, (Cantidad de calor que atraviesa la unidad de superficie en la unidad de tiempo, [W/m2],) viene dada por: Q dT = q k dx A (3.5.11) donde : T es la temperatura local [K o °C], x es la coordenada en la dirección del flujo [m] y k es la conductividad térmica de la sustancia, cuyas unidades [W/m. K]. 81 El signo menos en el segundo miembro de la expresión (3.5.11) se debe a que el calor fluye en el sentido contrario al del gradiente de temperatura, es decir, desde las regiones más calientes a las más frías. Como se ve, la conductividad térmica es un fenómeno de no equilibrio que precisa de la existencia de un gradiente de temperatura. Cuando dT/dx es negativo, el signo menos en la ecuación (3.5.11) da un valor positivo de q en la dirección de las x positivas. Figura 3.5.4.- Conducción Unidimensional estacionaria a través de una pared plana. La conductividad térmica es un parámetro que depende del tipo de material (depende de manera crucial de su estructura microscópica) y de la temperatura y representa la cantidad de calor conducido por unidad de tiempo a través de la unidad de área (Perpendicular a la dirección del transporte de calor) cuando el gradiente de temperatura a través del elemento conductor del calor es la unidad Reordenando e integrando la ecuación (3.5.11) sobre el espesor de la pared, se tiene: L T2 0 T1 q dx kdT (3.5.12) donde q y A se han sacado de la integral porque son constantes. Si se ignora la variación de k con la temperatura, se obtiene: q k T T T T T (T1 T2 ) 1 2 1 2 L L Rter Rter k (3.5.13) La comparación de la ecuación (3.5.13) con la ley de Ohm, I = V/R, sugiere que ∆T = T1 -T2 puede verse como un potencial impulsor del flujo de calor, así como el voltaje es el potencial impulsor de la corriente eléctrica. Entonces Rter= L/k puede considerarse como una resistencia térmica análoga a la resistencia eléctrica. La tabla 3.5.1 da los valores de la conductividad térmica de varios materiales. Los polímeros industriales son malos conductores del calor. Su conductividad térmica es similar a la de la madera y muy inferior a la del vidrio. 82 El fenómeno de la transmisión de calor por conducción en los materiales no metálicos, en los que no existen electrones móviles, puede considerarse como la transmisión de movimiento vibratorio de unos átomos (o de unos iones) de mayor nivel energético a los contiguos. Esta transmisión resulta mucho más fácil cuando su estructura es cristalina, estando dispuestos los átomos (o los io nes, en su caso) ordenadamente en el espacio, que en los materiales amorfos. De todas formas, la transmisión de la vibración térmica es mejor para los átomos unidos por enlaces covalentes que para los de diferentes moléculas, entre los que sólo existen elativamente débiles fuerzas de cohesión. En la tabla 3.5.2 se relacionan los valores típicos de la conductividad térmica y del calor específico de algunos materiales poliméricos. A partir de estos valores y de la densidad, ρ, puede obtenerse la difusividad térmica: k . Ce Tabla 3.5.2.-Propiedades térmicas de algunos polímeros. Polímeros celulares Conductividad Conductividad térmica, k térmica, k (kcal/h.ºC.m) (W/ºC.m) PEBD 0,30 0,35 PEAD 0,38 0,44 PP 0,21 0,24 PTFE 0,23 0,27 PVC (sin plastificar) 0,14 0,16 PS 0,14 0,16 PMMA 0,16 0,19 PA (nylon)6 0,27 0,31 PA (nylon)66 0,22 0,26 PET 0,12 0,14 PUR 0,27 0,31 CR 0,18 0,21 NR 0,16 0,19 Calor específico, ce (kcal/kg.ºC) 0,45 0,55 0,46 0,25 0,25 0,29 0,33 0,38 0,40 0,24 0,42 0,40 0,46 Calor específico, ce (kJ/kg.ºC) 1,88 2,30 1,92 1,05 1,05 1,21 1,38 1,59 1,67 1,00 1,76 1,67 1,92 Se admiten, en general los siguientes axiomas de aplicación a los polímeros industriales: (i).- Dentro de la relativa uniformidad de los valores que muestra la tabla 3.5.2, los polímeros cristalinos (PE, PP y PTFE) cuentan con una mayor conductividad térmica que los amorfos (PVC, PS y PMMA), siendo mayor ésta en los que disponen de mayor grado de cristalinidad (PEAD en comparación con el PEED, concretamente). (ii).- Los polímeros de estructura orientada presentan valores de la conducti vidad térmica mucho mayores en la dirección del estirado que en las otras, lo que se interpreta como una consecuencia de la alineación de las cadenas moleculares. (iii).- En los polímeros amorfos y a igualdad de otras circunstancias, la conductividad térmica aumenta con el peso molecular promedio y disminuye con la concentración de plastificante. 83 No se ha visto una relación clara de dependencia de la conductividad con la temperatura. En algunos casos disminuye al aumentar ésta, pero en otros sucede lo contrario. En la tabla 3.5.3 figuran los valores de la conductividad de los polímeros celulares o espumas más usuales, gasificados con aire, CO, y/o N 2 . Puede verse que no siempre corresponde menor conductividad a menor densidad; de hecho es muy importante el tamaño de los poros, pues si estos son relativamente grandes, en su interior el mecanismo de la transmisión del calor puede ser más efec tivo por convección que por conducción. Tabla 3.5.3.- Conductividad térmica y densidad de polímeros celulares. Polímeros celulares Densidad, ρ Conductividad térmica, k Conductividad térmica, k (kg/m3) (kcal/h.ºC.m) (W/ºC.m) PS 16 0.033 0,0384 PS 25 0.030 0,0349 PS 32 0.027 0,0314 PE 38 0.040 0,0465 PVC 35 0.024 0,0279 PVC 45 0.030 0,0349 PUR 16 0.034 0,0395 PUR 32 0.020 0,0232 PUR 64 0.021 0,0244 PUR 96 0.037 0,0430 Los valores muy bajos de la conductividad térmica de las espumas las hacen muy útiles como materiales aislantes. Puede reducirse aún más la conductividad utilizando hidrocarburos fluorados y/o clorados como agentes gasificantes (presentan valores de la conductividad térmica 2.5 veces menores que el aire) pero, a largo plazo, el aire desplaza a estos gases por difusión, perdiendo características paulatinamente. El calor específico de los polímeros está horquillado entre 0.24 y 0.55 Kcal/ kg.°C (1.00 y 2.30 KJ/ kg.°C), con valores extremos para el PET y el PEAD, respectivamente. Los plásticos cristalinos, que presentan una cierta fracción de su masa en forma de esferulitas o de simples cristalitos, necesitan un aporte de calor proporcional a dicha fracción para que se produzca el cambio de estado fusión-cristalización. Los sólidos amorfos no cambian de estado, por lo que no requieren ningún aporte adicional al correspondiente al calor sensible. Sin embargo, en la práctica, aunque algunos polímeros se consideren a todos los efectos como completamente amorfos, tienen siempre un pequeño grado de cristalinidad, que varía según las circunstancias. Esta característica diferencial entre los plásticos cristalinos y los amorfos da lugar a exigencias muy distintas de aporte de trabajo o calor, cuando se quiere fundir o reblandecer un material de una u otra clase, mucho mayores en el primer caso que en el segundo; lo mismo que de refrigeración, cuando se quiere solidificar el material previamente plastificado. A efectos comparativos considérese que para calentar y reblandecer un PEAD con calor latente de fusión (correspondiente a su grado de cristalinidad) de 50 Kcal/Kg, desde 20 °C hasta 220 °C, se necesitan (220-20)x0.55 + 50 = 160 Kcal/Kg, mientras que para el poliestireno sólo (220-20)x0.29 = 58 Kcal/Kg. 84 3.6.- Permeabilidad a los gases y vapores. La utilización de los plásticos en el envasado de productos que pueden deteriorarse en contacto con el oxígeno o el vapor de agua atmosféricos (como es el caso de los aceites vegetales que se enrancian si el envase no es suficientemente impermeable al oxígeno) y también de líquidos que deben mantener disueltos gran cantidad de gases (como las bebidas carbónicas) obligan a consi derar el fenómeno de la permeabilidad de los gases y vapores a través de los filmes de materiales plásticos, pues esta es la forma más generalizada de su empleo como envases. En primer lugar debe recordarse que el coeficiente de permeabilidad P está relacionado con los de difusividad D y de solubilidad S mediante: P = DS (3.6.1) definiéndose el primero mediante el flujo másico Φ, por unidad de superficie, el espesor e y la diferencia de presión parcial del gas a ambos lados del film: P p1 p2 e (3.6.2) mientras que los otros dos mediante las leyes de Fick y de Henry, respectiva mente, en términos de concentración (el subíndice i se refiere a la interfase gas-sólido): D c1 c2 e (3.6.3) ci Spi Puede concluirse que la permeabilidad puede ser alta por serlo o bien la solubilidad o la difusividad. En ambos casos se exige una buena compatibilidad del gas o vapor con las moléculas del polímero. La difusión a través de las zonas amorfas es siempre mucho más fácil que a través de las cristalinas y, para un determinado material, cuanto mayor sea su densidad, tanto menor resulta la difusividad. En consecuencia y concretando para el polietileno, la permeabilidad será menor para los filmes de mayor densidad y cristalinidad. El oxígeno y el nitrógeno son muy poco solubles en todos los polímeros, mientras que el CO 2 , y el vapor de agua lo son mucho más en los polímeros de naturaleza fuertemente polar. Los poliuretanos, el poliestireno y el HIPS, con todos los elastómeros a base de polibutadieno son malos como barreras para el oxígeno y el vapor de agua. (Especialmente malos para este último lo son el alcohol polivinílico y las celulosas tipo celofán). Buenas barreras para gases y vapor de agua son, por ejemplo, las resinas ABS, los PVDC y PVDF (policloruro- fluoruro- de vinilideno), el PVC y el polietilentereftalato. Las poliolefinas (PE, PP) y el politetrafluoretileno (PTFE) son buenas barre ras para el vapor de agua, pero resultan relativamente muy permeables al oxígeno. 85 3.7.- Estabilidad a altas temperaturas y comportamiento en el fuego. Los incendios constituyen el riesgo más grave para los ocupantes de un edificio (Figura 3.7.1), además de los bienes incluidos en el mismo, e incluso la propia edificación. Las consecuencias de un incendio se resumen en una sola palabra: perdidas. Siempre habrá pérdidas materiales de bienes familiares, sociales o empresariales. Con frecuencia, también habrá derivaciones en carencia de servicios. Sin embargo, lo más grave y doloroso, por lo irreparable, son las pérdidas de vidas humanas. La estabilidad del edificio depende del comportamiento de los elementos estructurales frente al desarrollo de un incendio, ya que el enorme calor y las elevadas temperaturas, pueden provocar el colapso de los mismos. Figura 3.7.1- Incendio de un edificio. Todos los países de nuestro entorno, conscientes del grave problema de los incendios, han legislado normas de obligado cumplimiento, para aumentar la protección de los edificios. La Norma española NBE-CPI-96 contempla los aspectos de protección pasiva y activa de los edificios o establecimientos, excluidos los de uso industrial. Aunque la norma es clara en las definiciones y aplicación, es conveniente resaltar algunos aspectos de interés para el proyectista. Reacción al fuego. La reacción al fuego es una característica propia de un material o producto y trata de significar la magnitud relativa con la que pueden favorecer el inicio y desarrollo de un incendio, es decir, es la respuesta de un material al fuego medida en términos de su contribución al desarrollo del mismo con su propia combustión, bajo condiciones definidas de ensayo. Definición de fuego. El fuego se define como un proceso de combustión caracterizado por una reacción química de oxidación (desde el punto de vista del combustible) de suficiente intensidad para emitir luz, calor y en muchos casos llamas. Esta reacción se produce a temperatura elevada y evolución de suficiente calor como para mantener la mínima temperatura necesaria para que la combustión continúe. 86 A temperaturas elevadas aumenta rápidamente la velocidad de oxidación, produciendo cantidades cada vez mayores de calor por unidad de tiempo, hasta alcanzar el nivel en que se sostiene a sí misma en el medio de reacción, por el calor que produce. Triángulo del fuego. El fuego puede ser representado por un triángulo equilátero llamado TRIANGULO DE FUEGO, en el que se simbolizan en cada uno de sus lados los factores esenciales para que el mismo exista: COMBUSTIBLE + COMBURENTE + CALOR: FUEGO El fuego se extingue si se destruye el triángulo eliminando o acortando alguno de sus lados. Aunque el triángulo de fuego se ha utilizado por años como modelo de fuego, no se pueden explicar con este ciertos comportamientos en determinados fuegos. Por tal motivo se decidió incorporar a la figura anterior un cuarto factor que contempla la naturaleza química del fuego. Algunos materiales son pirofóricos, es decir, que pueden arder espontáneamente sin necesidad de que haya una fuente de ignición exterior. Por ejemplo, el sodio metálico puede reaccionar con la humedad del aire. Esta reacción produce hidrógeno gas y el calor generado por la reacción puede ser suficiente para hacer arder el hidrógeno con el oxígeno del aire. Tetraedro del fuego. Se ha descubierto que detrás de las llamas existen una serie de especies activas (iones, radicales libres, carbón libre, etc) que son las responsables químicas en cadena que se producen. Por ello la nueva representación del fuego es el TETRAEDRO DEL FUEGO. Este mantiene la misma simbología similar que el triángulo de fuego. El cuarto elemento es la reacción en cadena. 87 Definiciones de los cuatro elementos del tetraedro del fuego. Combustible- Agente reductor. Es un combustible es en sí un material que puede ser oxidado, por lo tanto en la terminología química es un agente reductor, puesto que reduce a un agente oxidante cediéndole electrones a este último. Son ejemplos: carbón, monóxido de carbono, hidrocarburos, sustancias celulósicas, solventes, etc. Pueden estar en cualquier estado de agregación: sólido, líquido o gaseoso. Comburente- Agente oxidante. El comburente es un agente que pude oxidar a un combustible (agente reductor) y al hacer esto se reduce a sí mismo. En este proceso el agente oxidante obtiene electrones tomándolos del combustible. Son ejemplos: oxígeno y ozono (generalmente en aire), peróxido de hidrógeno (agua oxigenada), halógenos, ácidos como el nítrico y sulfúrico, óxidos metálicos pesados, nitratos, cloratos, percloratos y peróxidos, cromatos, dicromatos, permanganatos, etc. Desde el punto de vista del incendio, el oxígeno del aire es el comburente principal, agente que alimenta el fuego. Calor- Temperatura de ignición. La temperatura de ignición es el tercer factor del fuego. Es la mínima temperatura a que una sustancia (sólida o líquida) debe ser calentada a fin de iniciar una combustión que se sostenga por sí misma independientemente de fuentes externas de calor. Existen otras definiciones importantes: Temperatura de inflamación: Es la menor temperatura a la que hay que elevar un líquido (o solido volátil) combustible para que los vapores que se desprendan formen con el aire que se encuentra sobre el mismo, una mezcla que se inflama al acercársele una llama. La combustión no continúa al retira la llama o fuente de ignición. La inflamabilidad es la medida de la facilidad que presenta un gas, líquido o sólido para encenderse y de la rapidez con que, una vez encendido, se diseminarán sus llamas. Cuanto más rápida sea la ignición, más inflamable será el material. Los líquidos inflamables no lo son por sí mismos, sino que lo son debido a que su vapor es combustible. 88 La volatilidad de un material es un indicativo de la facilidad con que un líquido o sólido pasa al estado de vapor. Se mide mediante el punto de ebullición del material (temperatura a la cual la presión de vapor del material es igual a la presión atmosférica). El término "volatilidad" se confunde con frecuencia y se utiliza como sinónimo de "inflamabilidad". Existen algunos materiales que son volátiles pero en cambio no son inflamables, como el agua, cloroformo y mercurio. Temperatura de combustión o ignición: Si se continúa calentado el líquido combustible sobre la temperatura de inflamación se encuentra una temperatura a la cual la velocidad de desprendimiento de vapores es tal que una vez iniciada la combustión, la misma continuará sin necesidad de acercar nuevamente la llama. Temperatura de auto combustión o auto ignición: Es la mínima temperatura a la cual debe elevarse una mezcla de vapores inflamables y aire, para que se encienda espontáneamente sin necesidad de la presencia de una fuente de ignición externa. Esta temperatura suele ser muy superior a las anteriores. Reacción en cadena. Cuando una sustancias se calienta, ésta desprende vapore y gases, los cuales se combinan con el oxígeno del aire que en presencia de una fuete de ignición arden. En el momento en que estos vapores arden, se libera gran cantidad de calor. Si el calor desprendido no es suficiente para generar más vapores del material combustibles, el fuego se apaga. Si la cantidad de calor desprendida es elevada, el material combustible sigue descomponiéndose y desprendiendo más vapores que se combinan con el oxígeno, se inflaman, y el fuego aumenta, verificándose la reacción en cadena. La forma de evaluar la reacción al fuego es mediante ensayo normalizado en laboratorio homologado, por la que se determina la clase de reacción al fuego. En nuestro país, de acuerdo con la norma UNE 23727, las clases son: M0 (material incombustible), M1 (material combustible, pero no inflamable) y M2, M3 y M4, que corresponden a materiales combustibles con grado creciente de inflamabilidad. Clase Definición Ejemplo M-0 No combustible (Incombustible) M-1 Material combustible, pero no inflamable Aglomerados M-1, DM-I, Poliestireno extruido M-2 Difícilmente inflamables Aglomerados tipo M-2 M-3 Medianamente inflamables Aglomerado homogéneo. M-4 Fácilmente inflamables Plásticos. Fibras textiles. Fibrocementos Silicatos lanas minerales 89 La clasificación de los materiales en los grupos M0, M1..., M4, indica la magnitud relativa de ellos para favorecer el inicio o desarrollo de un incendio, según las normas UNE. Las clases M2, M3 y M4 significan productos con un grado de inflamabilidad creciente. Las lanas minerales son de naturaleza M0, como materiales inorgánicos. Sin embargo, las normas españolas en sus exigencias no son explicitas en dos características importantes de los materiales en caso de incendio: la carga de fuego y los humos. (a).- Los materiales orgánicos presentan valores de carga de fuego caracterizados por su PC (Poder Calorífico), independiente de su clasificación al fuego. Este valor es característico de cada material y no se reduce con la adición de componentes ignifugantes que mejoran la clasificación al fuego. Así, productos que son natural mente M4, pueden pasar a clasificación M1 con ignifugantes, pero mantienen su PC prácticamente igual. Si bien la posición relativa de los materiales en una solución constructiva supone diferente nivel de riesgo, es evidente que la colocación de productos de elevado poder calorífico en posición expuesta para un posible incendio incrementa los riesgos, con independencia de la clasificación al fuego. Dentro de las posiciones expuestas, están obviamente los elementos vistos de un local y también aquellos introducidos en cámaras de aire con circulación de este y en todo caso las cámaras de alto volumen relativo, con escasa protección de incendios en alguno de los cerramientos de las mismas. 90 (b).- La generación de humos de los materiales combustibles durante un incendio supone una problemática muy grave. Los humos representan un riesgo suplementario para la evacuación de las personas y para la lucha contra el incendio, debido a la reducción de la visibilidad (opacidad) y a la disminución del oxígeno respirable. En el límite, según el tipo de incendios y de los materiales en ignición, los humos pueden contener gases tóxicos (CO, CNH...) que son letales incluso a bajas concentraciones. El monóxido de carbono (CO) es un gas tóxico, incoloro, inodoro e insípido, que se produce en combustiones incompletas. Reacciona con la hemoglobina impidiendo el transporte de oxígeno a través de la sangre. Su inhalación puede ser mortal. El dióxido de carbono (CO2) es el gas típico de la combustión. No es venenoso, aunque desplaza el oxígeno del aire pudiendo producir la muerte por asfixia. Se utiliza en muchos sistemas de protección para extinguir incendios en espacios cerrados o semicerrados, debido a su capacidad de desplazar el oxígeno. El cianuro de hidrógeno (HCN) se produce como resultado de la combustión de materiales que contienen nitrógeno como la lana y las fibras sintéticas. El ácido clorhídrico (HCl) se desprende cuando se calientan algunos materiales plásticos como el PVC. El color de los humos depende de los materiales que estén quemándose: - Color blanco o gris pálido: indica que arde libremente. - Negro o gris oscuro: indica normalmente fuego caliente y falta de oxígeno. - Amarillo, rojo o violeta: generalmente indica la presencia de gases tóxicos. Sobre la importancia de estas características, es suficiente indicar que más del 80% de las víctimas de los incendios perecen a causa de los humos, mediante dos mecanismos diferentes: la opacidad y la toxicidad de los humos. La opacidad es la medida del oscurecimiento, que impide el paso de la luz a través del humo. En la realidad de un incendio, humos con alto grado de opacidad, impiden la visión de las personas y retardan el tiempo de evacuación de las mismas. Esto aumenta considerablemente el riesgo para aquellas y el potencial número de víctimas. La generación de humos en cuanto a opacidad está ligada a las características de composición de los materiales, siendo más intensa o rápida a mayor carga de fuego, especialmente en los materiales plásticos El efecto de toxicidad está relacionado con la capacidad de algunos gases producidos en las combustiones de los materiales orgánicos, para producir alteraciones físicas o psíquicas en el ser humano. Este tipo de alteraciones pueden suponer desde la reducción de la capacidad de movimiento, hasta casos agudos de envenenamiento. En cualquier caso, impiden la evacuación de la zona de incendios por medios propios. Los análisis de composición de humos permiten conocer la presencia cuantitativa de gases tóxicos, depen dientes de la composición de los materiales. En función de las concentraciones del análisis y de las concentraciones normalizadas máximas admisibles, algunas normas (ISO, NF...) determinan el «índice de toxicidad de los humos». Ensayos de este tipo se están estudiando para las futuras normas europeas. 91 Resistencia al fuego (RF). La resistencia al fuego es la característica que corresponde a una solución constructiva, por la cual se determina la capacidad de resistir en el tiempo, a la acción del fuego. En todo caso, la característica es el tiempo: cuanto mayor sea el tiempo disponible, será mejor para evacuar personas o luchar contra el incendio. La forma de evaluar esta característica es mediante ensayo normalizado en laboratorio homologado, por la que se determina el tiempo de: 1.- La estabilidad al fuego (EF) o capacidad portante: Esto aplicable a cualquier tipo de solución constructiva. Es la capacidad de un elemento constructivo de mantener durante un tiempo determinado la estabilidad o capacidad portante de uso para impedir el colapso del edificio en caso de incendio. Se determina en un ensayo normalizado de acuerdo con la norma UNE – EN1363-1. 2.- Ausencia de emisión de gases inflamables en la cara no expuesta al fuego. 3.- Estanqueidad al paso de llamas o gases calientes a la cara no expuesta al fuego. 4.- Resistencia térmica suficiente para impedir que en la cara no expuesta al fuego, se temperaturas superiores a las establecidas por norma. produzcan La primera condición representa la estabilidad al fuego (EF). Si se cumplen las tres primeras condiciones, será parallamas ( PF, Comportamiento de un elemento constructivo sea portante o no, que garantiza durante un tiempo determinado, la estanquidad a las llamas o gases.), y si se cumplen todas, se denominara resistencia al fuego (RF). La escala de tiempos normalizada es: 15 , 30 , 60 , 90 , 120 , 180 y 240 minutos. 92 Una limitación del uso de los materiales plásticos convencionales se debe principalmente a la pérdida de las características físicas que tiene lugar a temperaturas relativamente bajas, a veces muy por debajo de los 200 °C, para exposiciones de larga duración. A esas temperaturas ya es perceptible una cierta degradación química: las cadenas moleculares se fisionan, reduciéndose pro gresivamente el peso molecular. Se separan moléculas sencillas originadas por condensación (HCI, por ejemplo, en el PVC) y otros residuos orgánicos volátiles. En contacto con el aire se verifica una oxidación acelerada, que progresa según un mecanismo de peroxidación mediante radicales libres. A partir de los 400 °C la degradación de los polímeros orgánicos es muy rápida, con reacciones propias de pirólisis, originándose gran cantidad de residuos volátiles que, en presencia de oxígeno, arden con aspecto de llama. Simultáneamente sobre la superficie del material aparecen residuos carbonosos tipo alquitrán, que arden difícilmente porque la difusión del oxígeno a su través resulta controlante. La combustión de los plásticos produce gran cantidad de humos y gases tóxicos, como el CO, HCN (cuando en su composición entra significativamente el nitrógeno), HCI e, incluso, el fosgeno (cuando arden polímeros a base de hidrocarburos clorados en determinadas circunstancias). Ello hace que los incendios en los que se queman estos materiales resulten extremadamente peligro sos. Se denomina punto de ignición de un polímero a la temperatura a la que, en un ambiente rico en oxígeno (para que la difusión de éste en la fase gaseosa no resulte controlante), la combustión se mantiene indefinidamente. Eso sucede porque el aporte de calor generado en las múltiples reacciones de oxidación supera al que se consume en las reacciones endotérmicas de pirólisis junto con el que se pierde en el ambiente exterior. Su valor para cada material depende de su naturaleza y composición, pero, sobre todo, de las circunstancias en que se produce el fuego; por eso no es un parámetro significativo del comportamiento de estos materiales. Mayor interés presenta la medida de la facilidad o dificultad que ofrece cada material al mantenimiento y propagación del fuego una vez producida la ignición, que es muy diferente según la naturaleza del material. Así el polietileno y el polipropileno, como hidrocarburos que son, arden y propagan el fuego muy fácilmente; por el contrario, el policloruro de vinilideno (PVDC), con un conte nido en cloro del 72% en peso, se autoextingue en cuanto se retira la fuente de ignición. 93 Evidentemente el mantenimiento y propagación del fuego depende de la concentración de oxígeno presente en la atmósfera que rodea el material en combustión y se ha encontrado como parámetro más significativo del comportamiento de los plásticos en el fuego al valor mínimo de esa concentración que, en las circunstancias de un ensayo normalizado, mantiene la combustión de una varilla del material en cuestión situada en el eje de un cilindro vertical por cuyo extremo inferior se introduce una mezcla controlada de oxígeno y nitrógeno. Dicha concentración mínima de oxígeno se denomina Índice de Oxígeno Crítico (COI). En la tabla 3.7.1 se relacionan los COI de algunos de los materiales plásticos más comunes. Tabla 3.7.1.- Índice de oxígeno crítico de algunos materiales poliméricos. Índices de oxígeno crítico inferiores a 0.21 (correspondiente a la composición del aire) denotan materiales con buena propagación del fuego. Por encima de 0.27 se consideran con características de autoextinción. La adición de cargas minerales aumenta el COI del material pero, para que el efecto sea notable, la proporción del inerte debe ser muy alta. Así, por ejemplo, para un polietileno con el 50 % en peso de óxido de aluminio, el COI resulta de 0.20, frente a 0.17 para el PE puro, y de 0.30 si la carga supone el 60 % en peso, en vez del 50 %. Existen determinados productos que, presentes en mucho menores pro porciones, interfieren las reacciones en cadena mediante radicales libres de la combustión; se les denomina aditivos retardadores de llama. Uno de los más usados en la actualidad es el hexabromociclodecano (HBCD), que al alcanzar altas temperaturas se descompone, dando radicales libres de bromo que estabi lizan los radicales oxigenados, frenando así la reacción en cadena; su efecto inhibidor se potencia en presencia del óxido de antimonio. Así por ejemplo, la adición de un 3 % de HBCD con un 2 % de Sb2 03 al polipropileno aumenta el COI de 0.17 a 0.27, convirtiéndole en autoextinguible. El mismo efecto de estabilización de radicales oxigenados que, aún a temperaturas mucho menores que las que tienen lugar en los fuegos, producen efectos importantes de degradación por oxidación, se consigue mediante compuestos del tipo trialquilfenol. Este y otros aditivos que actúan de modo similar reciben el nombre de antioxidantes térmicos 3.8.- Resistencia a los disolventes y reactivos químicos. La utilización de los materiales plásticos en la fabricación de envases, reci pientes e, incluso, maquinaria (bombas, ventiladores, agitadores, etc.) para el almacenamiento y manipulación de productos químicos presenta grandes ventajas sobre los metales, el vidrio y los materiales cerámicos: Inexistencia del fe nómeno de corrosión, que tanto afecta a los metales, y mucho mejores características mecánicas que el vidrio y la cerámica, con unos costes competitivos. 94 Aún en el caso de que las solicitudes mecánicas a que debe estar sometida la pieza sean excesivas para los materiales plásticos (ejes, palas de agitadores, barriles para transporte marítimo, etc.), el empleo de estos en forma de recubrimientos de elementos metálicos, siempre que se consiga una buena adherencia entre ambos, suele ser una solución satisfactoria. El metal proporciona la resis tencia mecánica y el plástico la química. En general puede afirmarse que los grandes enemigos de los plásticos son los disolventes, por una parte, y los ácidos, las bases y los oxidantes fuertes, por otra. Los disolventes perjudican la consistencia del material, produciendo un hinchamiento (que corresponde a una solvatación) cuando no la misma disolución. En ambos casos las propiedades físicas del polímero resultan gravemente afectadas. Los disolventes no polares (hidrocarburos fundamentalmente) perjudi can especialmente a los polímeros de estructura parafínica y poco polares; mientras que los disolventes fuertemente polares (cetonas, amidas, etc.) dañan a los polímeros polares, siguiendo el conocido principio de «semejante disuelve a semejante». Otro efecto perjudicial puede ser el de la extracción de los plastificantes presentes en el material que pierde así la flexibilidad in icial. (Ver apéndice C). La resistencia a la solvatación y a la disolución es tanto mayor cuanto más elevado sea el peso molecular y el grado de entrecruzamiento del polímero. De hecho los polímeros reticulados no llegan a disolverse, pero sí pueden hincharse por solvatación con disolventes altamente compatibles. La elevación de la temperatura favorece la disolución y la solvatación. El ataque químico supone una modificación de la composición molecular. Los ácidos y bases fuertes hidrolizan los grupos internos tipo ésteres o amidas. Los oxidantes provocan la oxidación y la fisión de las moléculas poliméricas. Es conocido el efecto perjudicial del ozono y del mismo oxígeno, sobre todo cuando el material polimérico está expuesto a la luz ultravioleta. En presencia de tensiones el ataque químico es potenciado, originándose en el material pequeñas grietas, aún con productos que en otras condiciones serían inocuos, como, por ejemplo, algunos detergentes. Este fenómeno, de consecuencias catastróficas, es conocido como «Environmental Stress Cracking» (ESC) y equivale a la corrosión bajo tensión de los metales. En general se produce más en los materiales amorfos en estado vítreo que en los cristalinos, pero no existen criterios generales que sirvan para predecir el comportamiento de un determinado material en un determinado medio ambiental. Para conocer la resistencia de un material polimérico a un determinado producto químico lo más seguro es recurrir a la experimentación directa, aunque las tablas de resistencia química a diferentes reactivos y a distintas concentraciones y temperaturas, que proporcionan los fabricantes y suministradores de estos materiales, suelen proporcionar una información fiable. En la tabla 3.8.1 se indican los tipos de comportamiento (bueno, B; regular, R, y malo, M) de algunos plásticos más usuales, en distintos medios químicos, que sirven de referencia, y a temperatura ambiente. 95 Tabla 3.8.1.- Comportamiento típico de algunos plásticos en medios químicos a bajas temperaturas (< 38 ºC). 96