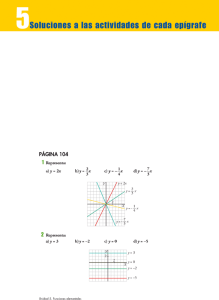
estructura módulo termodinámica avanzada
Anuncio

TERMODINÁMICA AVANZADA LECTURAS BÁSICAS MÓDULO DEL CURSO 201000 INTRODUCCIÓN UNIDAD 1. FUNDAMENTOS DE TERMODINÁMICA CLÁSICA CAPÍTULO 1. CONCEPTOS BÁSICOS LECCIÓN 1. DEFINICIÓN DE SISTEMA Y TIPOS DE SISTEMA LECCIÓN 2. DESCRIPCIÓN MACROSCÓPICA DEL ESTADO DE UN SISTEMA LECCIÓN 3. EQUILIBRIO Y TIPOS DE EQUILIBRIO. LECCIÓN 4. GRADOS DE LIBERTAD LECCIÓN 5. PROCESOS Y TIPOS DE PROCESOS CAPÍTULO 2. PRIMERA LEY DE LA TERMODINÁMICA Y TERMOQUÍMICA LECCIÓN 6. LEY CERO DE LA TERMODINÁMICA LECCIÓN 7. TERMOMETRÍA Y ESCALAS EMPÍRICAS DE TEMPERATURA LECCIÓN 8. TRABAJO, ENERGÍA, CALOR Y PRIMERA LEY LECCIÓN 9. CAPACIDADES CALORÍFICAS, SISTEMAS ABIERTOS LECCIÓN 10. TERMOQUÍMICA CAPÍTULO 3. SEGUNDA LEY DE LA TERMODINÁMICA LECCIÓN 11. SEGUNDA LEY Y CONCEPTO DE EFICIENCIA LECCIÓN 12. TEOREMA DE CARNOT Y MÁQUINA DE CARNOT LECCIÓN 13. ESCALA TERMODINÁMICA DE TEMPERATURA LECCIÓN 14. ENTROPÍA, IRREVERSIBILIDAD Y CAMBIOS EN UN GAS IDEAL LECCIÓN 15. VARIACIÓN DE LA ENTROPÍA CON TEMPERATURA, PRESIÓN Y VOLÚMEN UNIDAD 2. FUNDAMENTOS DE FISICOQUÍMICA CAPÍTULO 4. POTENCIALES QUÍMICOS Y EQUILIBRIO DE FASES, PARTE 1 LECIÓN 16. TRABAJO Y ENERGÍA LIBRE DE HELMHOLTZ LECCIÓN 17. ENERGÍA LIBRE DE GIBS LECCIÓN 18. CONDICIONES DE EQUILIBRIO LECCIÓN 19. PROPIEDADES MOLARES PARCIALES Y POTENCIAL QUÍMICO LECCIÓN 20. EQUILIBRIO DE FASES EN SISTEMAS DE UN COMPONENTE CAPÍTULO 5. EQUILIBRIO DE FASES, PARTE 2, FUGACIDAD Y ACTIVIDAD LECCIÓN 21. EQUILIBRIO DE FASES EN UN SISTEMA CON MÁS DE UN LECCIÓN 22. CONCEPTOS DE FUGACIDAD Y ACTIVIDAD COMPONENTE LECCIÓN 23. FUGACIDAD Y ACTIVIDAD EN SISTEMAS DE UN COMPONENTE LECCIÓN 24. FUGACIDAD Y ACTIVIDAD EN SISTEMAS DE MÁS DE UN COMPONENTE LECCIÓN 25. APLICACIONES DE LOS CONCEPTOS DE FUGACIDAD Y ACTIVIDAD CAPÍTULO 6. EQUILIBRIO QUÍMICO LECCIÓN 26. CONSTANTE DE EQUILIBRIO LECCIÓN 27. EQUILIBRIO EN SISTEMAS GASEOSOS HOMOGÉNEOS LECCIÓN 28. EQUILIBRIO EN SOLUCIÓN LÍQUIDA HOMOGÉNEA LECCIÓN 29. EQUILIBRIO QUÍMICO EN SISTEMAS HETEROGÉNEOS 1 LECCIÓN 30. VARIACIÓN DE LA CONSTANTE DE EQUILIBRIO CON LA PRESIÓN Y LA TEMPERATURA. INTRODUCCIÓN Es indiscutible la utilidad que tiene la termodinámica en la ingeniería y en las ciencias. El estudio de sus leyes nos lleva a fortalecer un análisis crítico y deductivo, proporcionándonos una herramienta importante para comprender los procesos de interconversión de la energía y predecir la espontaneidad o no de estos. Las leyes cero, primera y segunda, deducidas como fruto de la experiencia, nos revelan un sinnúmero de posibilidades de interés práctico y teórico a aquellos interesados en esta fascinante rama de la ciencia. El presente documento, fruto de la recopilación de varios textos de termodinámica, intenta proporcionar un camino para la comprensión de algunos tópicos relacionados con la termodinámica que serán de gran utilidad para aquellos interesados en la ingeniería de los Alimentos. El plan del documento consiste en proporcionar a Usted señor lector las bases en las cuales descansa la termodinámica, considerando algunas de sus aplicaciones y dejando de esta manera un camino recto y fácil para que pueda utilizarla en otros tipos de aplicaciones, procesos. De este modo señor lector, lo invito a introducirse en el mundo de esta ciencia, a adquirir el lenguaje termodinámico y a profundizar en esta ciencia llena de posibilidades. UNIDAD 1. FUNDAMENTOS DE TERMODINÁMICA CLÁSICA Termodinámica. De las palabras griegas calor y potencia es el estudio del calor, el trabajo, la energía y los cambios que provocan en los estados de los sistemas. En un sentido más amplio, la termodinámica estudia las relaciones entre las propiedades macroscópicas de un sistema. La temperatura es una propiedad esencial en la termodinámica, y por eso se define a veces la termodinámica como el estudio de la relación entre la temperatura y las propiedades macroscópicas de la materia. En el curso se estudiará la termodinámica del equilibrio, que trata con sistemas en equilibrio. (La termodinámica irreversible se ocupa de sistemas que no están en equilibrio y de procesos de cambio.) La termodinámica del equilibrio es una ciencia macroscópica e independiente de cualquier teoría sobre la estructura molecular. Estrictamente, la palabra «molécula» no pertenece al vocabulario de la termodinámica. La termodinámica no es aplicable a sistemas que sólo contienen unas pocas moléculas; un sistema debe constar de un gran número de moléculas para que se pueda tratar termodinámicamente. En la termodinámica, a la parte macroscópica del universo, objeto de estudio se le llama sistema. A las partes del universo que pueden interaccionar con el sistema se les denomina alrededores. Por ejemplo, para estudiar la presión de vapor del agua en función de la temperatura, se podría poner un recipiente cerrado con agua (donde se haya eliminado el aire) en un baño a temperatura constante, y conectar un manómetro al recipiente para medir la 2 presión (ver figura). En este caso, el sistema está compuesto por el agua líquida y el vapor de agua dentro del recipiente, y los alrededores son el baño a temperatura constante y el mercurio del manómetro. Un sistema abierto es aquel en el que se puede transferir materia entre el sistema y los alrededores. Un sistema cerrado es aquel en el que no es posible tal transferencia de materia. Un sistema aislado es el que no interacciona de ninguna forma con sus alrededores. Obviamente, un sistema aislado es un sistema cerrado, pero no todos los sistemas cerrados son aislados. Por ejemplo, en la figura el sistema de agua líquida más vapor en el recipiente sellado es un sistema cerrado (ya que no puede entrar ni salir materia), pero no aislado (pues puede calentarse o enfriarse con el baño que le rodea, y puede comprimirse o expandirse por medio del mercurio). En un sistema aislado no puede darse transferencia de materia ni de energía entre el sistema y los alrededores. En un sistema cerrado es posible transferir energía, pero no materia, entre el sistema y los alrededores. En uno abierto pueden intercambiarse tanto materia como energía. Un sistema termodinámico es abierto o cerrado y es aislado o no aislado. La mayor parte de las veces trataremos con sistemas cerrados. Un sistema puede estar separado de sus alrededores por varios tipos de paredes. (En la figura, el sistema está separado del baño por las paredes del recipiente.) Una pared puede ser rígida o no rígida (es decir, móvil). Puede ser permeable o impermeable, donde el término «impermeable» significa que no permite el paso de materia a través. Por último, una pared puede ser adiabática o no adiabática. En lenguaje corriente, una pared adiabática es la que no conduce en absoluto el calor, mientras que una pared no adiabática conduce el calor. Sin embargo, como aún no se ha definido el calor, para desarrollar la termodinámica de forma lógica y correcta hay que definir las paredes adiabáticas y no adiabáticas sin referirse al concepto de calor. Se tienen dos sistemas separados, A y B, en los que se observa que sus propiedades permanecen constantes a lo largo del tiempo. Se ponen A y B en contacto por medio de una pared impermeable y rígida, ver figura, si no se observa cambio temporal alguno en 3 las propiedades (por ejemplo, presiones, volúmenes) de los sistemas A y B, sean cuales sean sus valores iniciales, la pared que separa A y B se llama adiabática. Si, como es frecuente, se observan cambios con el tiempo en las propiedades de A y B tras ponerlos en contacto a través de una pared rígida e impermeable, entonces esta pared es no adiabática o térmicamente conductora. (cuando dos sistemas a diferentes temperaturas, se ponen en contacto a través de una pared térmicamente conductora, el calor pasa del sistema más caliente al más frío, cambiando así las temperaturas y otras propiedades de los dos sistemas; con una pared adiabática, cualquier diferencia de temperatura se mantiene. Como el calor y la temperatura están todavía sin definir, estas observaciones están lógicamente fuera de lugar, pero se han incluido para aclarar las definiciones de paredes adiabáticas y térmicamente conductoras.) Una pared adiabática es una idealización, pero se puede conseguir aproximadamente, por ejemplo, con las paredes dobles de un vaso Dewar o de un termo, que están separadas por un vacío casi absoluto. Un sistema rodeado por una pared rígida, impermeable y adiabática no puede interaccionar con sus alrededores, por lo que es un sistema aislado. La termodinámica que se está estudiando solo se dedica a los sistemas en equilibrio, un sistema aislado está en equilibrio, ya que sus propiedades macroscópicas permanecen constantes en el tiempo. Un sistema no aislado está en equilibrio si sus propiedades macroscópicas permanecen constantes a lo largo del tiempo y si al suprimir el contacto con sus alrededores no hay cambio alguno en sus propiedades, cuando solo se cumple la primera condición, el sistema está en estado estacionario. Un ejemplo de estado estacionario es una barra en contacto por un extremo con una masa grande a 50°C y por el otro con otra masa similar a 40°C, después de que ha pasado un tiempo suficientemente largo, la barra de metal cumple con la primera condición, se establece un gradiente uniforme de temperatura a lo largo de ella. Sin embargo, si se retira la barra del contacto con sus alrededores, la temperatura de sus partes cambia hasta que toda la barra adquiera una temperatura de 45°C. El concepto de equilibrio puede concretarse en los tres tipos siguientes, equilibrio mecánico, cuando las fuerzas externas e internas al sistema están equilibradas, en consecuencia no hay aceleración del sistema, ni turbulencias en su interior, dicho de otra manera, no hay diferencias de presión; el equilibrio material o químico se da cuando no existen reacciones químicas y como consecuencia de las cuales se da una transferencia de materia por diferencia de concentraciones y por último el equilibrio térmico se da cuando no hay diferencia de temperaturas entre el sistema y sus alrededores. El equilibrio termodinámico se da cuando se presentan simultáneamente los tres tipos de equilibrio mencionados. Ya se mencionó que el estado de un sistema está definido por sus propiedades termodinámicas, las más importantes en este caso son la composición, el volumen, la presión, la temperatura. Una propiedad termodinámica extensiva es aquella cuyo valor es igual a la suma de los valores correspondientes a diferentes partes del sistema. Así, si se divide un sistema en partes, la masa del sistema es igual a la suma de las masas de cada una de las partes; la masa es una propiedad extensiva. Lo mismo pasa con el volumen. Las propiedades que no dependen de la cantidad de materia en el sistema se denominan intensivas. La densidad y la presión son ejemplos de propiedades intensivas. Al tomar una gota de agua o una piscina llena de agua, ambos sistemas tendrán la misma densidad. 4 Si cada propiedad macroscópica intensiva es constante a lo largo de un sistema, éste es homogéneo. Cuando un sistema no es homogéneo, puede constar de una serie de partes que sí lo son. Una parte homogénea de un sistema se denomina fase. Por ejemplo, si el sistema consiste en un cristal de AgBr en equilibrio con una disolución acuosa de dicha sal, el sistema tiene dos fases: el AgBr sólido y la disolución. Una fase puede constar de varias partes separadas. Por ejemplo, en un sistema formado por varios cristales de AgBr en equilibrio con una disolución acuosa, todos los cristales son parte de la misma fase. la definición de «fase» no hace referencia a sólidos, líquidos o gases. Un sistema puede ser enteramente líquido (o enteramente sólido) y tener más de una fase. Por ejemplo, un sistema compuesto por los líquidos casi inmiscibles H2O y CCl4, tiene dos fases. Un sistema compuesto de los sólidos diamante y grafito tiene dos fases. Un sistema formado por dos o más fases es heterogéneo. Suponga que el valor de cada propiedad termodinámica de un cierto sistema termodinámico es igual al valor de la propiedad correspondiente en un segundo sistema. Entonces se dice que los sistemas están en el mismo estado termodinámico. El estado de un sistema termodinámico se define especificando los valores de sus propiedades termodinámicas. Sin embargo, no es necesario detallar todas las propiedades para definir el estado. Hay un número mínimo de ellas que determinan los valores de todas las demás. Por ejemplo, suponga que tomamos 8,66 g de H2O pura a la presión de 1 atm (atmósfera) y a 24°C. Se observa que en ausencia de campos externos todas las propiedades restantes (volumen, capacidad calorífica, índice de refracción, etc.) están fijadas. (Esta afirmación ignora la posible existencia de fenómenos de superficie) Dos sistemas termodinámicos, cada uno consistente en 8,66 g de H20 a 24°C y 1 atm, están en el mismo estado termodinámico. Los experimentos muestran que en un sistema formado por una única fase con cantidades fijas de sustancias no reactivas, la especificación de dos propiedades termodinámicas adicionales suele ser suficiente para determinar su estado termodinámico, en el supuesto de que haya campos externos y de que los efectos de superficie sean despreciables. Un sistema termodinámico en un estado de equilibrio dado tiene un valor concreto pasa cada variable termodinámica. Por eso, estas variables se llaman también funciones de estado, puesto que sus valores son función del estado del sistema. El valor de una función de estado depende sólo del estado actual de un sistema y no de su historia anterior. No importa si los 8,66 g de agua a 1 atm y 4° C se obtuvieron fundiendo hielo y calentando el agua, o condensando vapor y enfriando el agua. CAPÍTULO 1. CONCEPTOS BÁSICOS El objetivo de este capítulo es proporcionar al lector los conceptos fundamentales para abordar las leyes termodinámicas y el estudio de los distintos tipos de procesos en cuanto a la termodinámica se refiere. Por consiguiente, recomiendo estudiarla detalladamente para un buen desarrollo de las unidades posteriores. LECCIÓN 1. DEFINICIÓN DE SISTEMA Y TIPOS DE SISTEMA En general un sistema se considera como aquella parte del universo que es escogida como objeto de estudio termodinámico. Este se encuentra delimitado por paredes o superficies límites, reales o imaginarias, de los alrededores o “resto del universo”. Cuando esas paredes o superficies límites no permiten el paso de energía y de materia decimos que el sistema es un sistema aislado. Cuando esas paredes permiten solamente el 5 paso de energía pero no de materia, decimos que se trata de un sistema cerrado. Finalmente si esas paredes permiten tanto el paso de energía como de materia, hablamos de un sistema abierto. En los sistemas abiertos algunas veces suele llamarse volumen de control al sistema mismo y a las paredes que lo separan de los alrededores, superficie de control. Los sistemas pueden ser también homogéneos, es decir aquellos que son completamente uniformes, por ejemplo: mezclas de gases, líquidos o sólidos puros, soluciones sólidas o soluciones líquidas. Así mismo decimos que un sistema es heterogéneo cuando no es completamente uniformes, como lo son, sistemas que constan de dos o más fases, sistemas compuestos por líquidos inmiscibles o sistemas que no forman soluciones sólidas. LECCIÓN 2. DESCRIPCIÓN MACROSCÓPICA DEL ESTADO DE UN SISTEMA Una vez hemos seleccionado el sistema el paso a seguir es describirlo, mediante propiedades físicas adecuadas, las cuales pueden ser tanto extensivas, como intensivas. A este conjunto de propiedades que describen el sistema se denominan coordenadas termodinámicas o variables de estado. La experiencia muestra que estas variables que describen el sistema no cambian independientemente una de otra sino que por el contrario al fijar un número finito de ellas, las demás estarán determinadas. Así cuando estas variables de estado alcanzan valores constantes, hablamos de que el sistema ha alcanzado un estado particular. Consideremos como ejemplo un mol de gas ideal: la experiencia muestra que no es necesario establecer los valores de presión, temperatura y volumen para establecer el estado del sistema ya que estas propiedades se relacionan entre sí mediante la ecuación, PV = RT ( 1 ) Donde P es la presión del gas, V es el volumen ocupado por este, T es la temperatura a la que se encuentra y R es una constante, de tal forma que conociendo dos de las variables se conoce automáticamente la tercera. Este tipo de ecuaciones que relacionan variables termodinámicas se conocen como ecuaciones de estado para un sistema dado. Estas generalmente deben ser determinadas experimentalmente y en ciertas ocasiones puede ser deducidas de primeros principios mediante métodos usados en otras ramas de la física como lo es la mecánica estadística. LECCIÓN 3. EQUILIBRIO Y TIPOS DE EQUILIBRIO Un sistema se encuentra en equilibrio termodinámico cuando las coordenadas termodinámicas que describen el estado termodinámico son invariables con el tiempo. Este equilibrio termodinámico implica a su vez que deben existir tres tipos de equilibrio simultáneamente: 1. Equilibrio térmico, esto es la temperatura debe ser la misma a través de todo el sistema. 2. Equilibrio químico, la composición del sistema debe ser constante, no variar con el tiempo. 3. Equilibrio mecánico, no debe existir tendencia a cambios de presión con el tiempo, ya sea dentro del sistema o del sistema con relación a los alrededores, sin tener en cuenta el efecto de la gravedad. En termodinámica nos encontraremos con que algunos sistemas aunque reúnen las condiciones anteriores al actuar una perturbación de cierta magnitud el sistema cambiara su posición de equilibrio para alcanzar otra, la cual se diferencia de la primera en que la energía del sistema es menor respecto a la posición inicial. Si el estado final es 6 el estado de más baja energía que puede ser alcanzado desde el estado inicial hablamos de que el sistema se encuentra en un equilibrio termodinámico estable y al estado inicial lo conocemos como equilibrio metaestable. Ejemplos de equilibrio metaestable podemos citar agua líquida sobreenfriada es decir agua líquida a temperaturas inferiores a 0 °C, en este caso una perturbación adecuada, agitación por ejemplo, hará que el agua se congele alcanzando un estado de equilibrio estable. En general la distinción entre ambos tipos de equilibrio radica en la energía que posee el sistema en los distintos estados de equilibrio. Dentro de un sistema también pueden ocurrir varios procesos, los cuales ocurren a velocidades diferentes, en este caso podemos hablar de equilibrio respecto a los procesos más rápidos antes que el sistema alcance el equilibrio respecto a todos los procesos posibles, es decir hablamos de equilibrios parciales, por ejemplo un sistema puede alcanzar el equilibrio térmico antes de alcanzar el equilibrio químico en un proceso de hidrogenación catalítica de aceites, ya que las velocidades de los procesos son de diferentes ordenes de magnitud. LECCIÓN 4. GRADOS DE LIBERTAD Ya hemos visto que para caracterizar el estado de un sistema no es necesario fijar todas las variables de estado que describen el sistema, sino que basta fijar un número finito de estas ya que las demás quedarán determinadas. Al número mínimo de variables que son necesarias para describir el estado del sistema se conoce como grados de libertad que posee el sistema. Así por ejemplo para un gas ideal, cuya ecuación de estado esta representada por la ecuación 1, con solo fijar dos propiedades la tercera queda automáticamente determinada, por tanto un sistema compuesto por un gas ideal posee dos grados de libertad, esto es fijando presión y temperatura es posible conocer el volumen. LECCIÓN 5. PROCESOS Y TIPOS DE PROCESOS Cuando un sistema ha alcanzado un estado de equilibrio mediante una serie continua de cambios, decimos que el sistema ha realizado un proceso, dependiendo de la manera como se realizaron esos cambios podemos clasificar los procesos en: procesos reversibles, cuasiestáticos e irreversibles. Para ilustrar esta clasificación, consideremos como ejemplo la compresión de un gas, ver figura 1.1, el cual esta contenido en un cilindro y confinado mediante un pistón, el cual se desplaza de la posición 1 a la posición 2, alcanzando así el estado de equilibrio 2 del estado de equilibrio 1. Si el proceso de expansión lo realizamos infinitamente lento hablamos de un proceso cuasiestático, pero si además de realizarlo infinitamente lento el sistema esta en equilibrio en cualquier parte de la trayectoria, es decir consiste en una sucesión continua de estados de equilibrio, decimos que el proceso realizado es un proceso reversible. En la figura 1.1a y 1.1b están representados gráficamente estos dos tipos de procesos, el primero mediante una sucesión de puntos, y el segundo mediante una línea continua, ya que cualquier punto sobre al misma representará un estado de equilibrio. Las características de este proceso son bien interesantes ya que si devolvemos el pistón a su estado inicial, los alrededores serán completamente restaurados. Un proceso irreversible, será entonces un proceso en el cual el sistema una vez se remueven las fuerzas actuantes vuelve a su estado inicial sin embargo los alrededores no pueden ser restaurados. Además este proceso se realiza a velocidades finitas y durante este el sistema no se encuentra en equilibrio, gráficamente lo podemos representar como se indica en la figura 1.1c. 7 CAPÍTULO 2. PRIMERA LEY DE LA TERMODINÁMICA Y TERMOQUÍMICA El objetivo de esta unidad es desarrollar los conceptos de temperatura, el cual es deducido mediante la ley cero de la termodinámica, al igual que comprender los conceptos de trabajo y calor y de hecho el concepto de energía. Consideremos antes de introducirnos en la primera ley de la termodinámica una serie de aspectos que dieron origen a lo que conocemos como ley cero de la termodinámica y que a su vez constituyen la base para la definición de temperatura. Imaginémonos un sistema en equilibrio termodinámico, cuyas variables de estado son x , y , 1 1 z ..., el cual interactúa con los alrededores, tal que sus variables de estado cambian y 1 después de un tiempo, que depende de las características del sistema, toman valores constantes, x , y , z ..., mientras no ocurran cambios en los alrededores. Cuando esto ocurre, 2 2 2 decimos que el sistema ha alcanzado el equilibrio térmico. La velocidad con la cual el sistema alcance este tipo de equilibrio dependerá entonces de las paredes que lo separan de los alrededores. De esta forma cuando el tiempo en que el sistema alcanza el estado de equilibrio, es muy corto respecto al tiempo que observamos el sistema, decimos que las paredes que separan el sistema de los alrededores son paredes diatérmicas, o conductores térmicos ideales. Un ejemplo de este tipo de paredes son las construidas con metales que poseen alta conductividad térmica. Ahora si por el contrario, el tiempo en que se realiza el proceso se extiende por grandes periodos de tiempo, respecto al tiempo de observación del sistema, decimos que las paredes del sistema son paredes adiabáticas o aislantes térmicos ideales. LECCIÓN 6. LEY CERO DE LA TERMODINÁMICA Consideremos dos sistemas 1 y 2, ver figura 2.1, cuyos estados de equilibrio están dados por las variables x11 , y11, x12 , y12..., respectivamente. Los dos sistemas están aislados de los alrededores mediante paredes adiabáticas, figura 2.1a. Si los hacemos interactuar entre sí mediante una pared diatérmica, figura 2.1b, la experiencia nos muestra que al final las propiedades de los sistemas varían respecto a los valores iniciales y además alcanzan valores fijos x21 , y21, x22 , y22..., respectivamente. En este caso los dos sistemas están en equilibrio térmico, uno respecto al otro. 8 Consideremos ahora tres sistemas interactuando, tal y como muestra la figura 2.2, el sistema 3 esta en contacto con los sistemas 1 y 2 a través de una pared diatérmica y a su vez los sistemas 1 y 2 están separados entre sí por una pared adiabática. Después de un tiempo se encuentra que el sistema 1 estará en equilibrio con el sistema 3 y este a su vez este estará en equilibrio con el sistema 2, por consiguiente los sistemas 1 y dos deberán estar en equilibrio térmico también. En general podemos expresar: Dos sistemas en equilibrio térmico con un tercer sistema, estarán también entre sí en equilibrio térmico, este enunciado el cual es producto de la experiencia es conocido como Ley Cero de la Termodinámica. La experiencia también muestra que cuando dos o más sistemas interactuando entre sí mediante arreglos experimentales convenientes alcanzan el equilibrio térmico, todos ellos poseen una propiedad en común que es igual y constante para todos, esta propiedad se conoce como temperatura (Esta propiedad no es totalmente ajena al sentido común ya que en algunas veces decimos que una sustancia tiene más calor o más frió respecto a otra). LECCIÓN 7. TERMOMETRÍA Y ESCALAS EMPÍRICAS DE TEMPERATURA Ahora trataremos de ampliar el concepto de temperatura, no pretendiendo hacer un análisis riguroso, pero sí realizando un formalismo aceptable para luego hacer una reseña de las escalas de temperatura. Consideremos en primer lugar el concepto de temperatura: Tomemos como referencia la figura 2.2. Antes de iniciar el proceso de interacción entre ellos y considerando que todos ellos se encontraban en equilibrio térmico, podemos representar el estado de cada uno de ellos mediante las ecuaciones de estado: donde el superíndice indica el estado inicial. Una vez han alcanzado el equilibrio térmico la ley cero nos indica que existe una propiedad cuyo valor es constante para todos, ya que los valores iniciales de las variables de estado cambian respecto a los estados iniciales podemos decir que esa propiedad debe ser función de las demás variables del sistema, de tal forma que podemos escribir: lo que nos dice que esa propiedad debe ser función de las demás variables y en equilibrio térmico adquiere un valor constante T. 9 A manera de ilustración podemos considerar un gas ideal contenido en un arreglo similar al de la figura 2.2. El estado de equilibrio de cada sistema lo podemos representar por la ecuación de estado: f(P,V)=0 ( 2.3 ) Tal que cuando los tres sistemas interactúan, y alcanzan el estado de equilibro, tenemos: f(P1,V1)=g (P2,V2)=h (P3,V3)= T1 ( 2.4 ) o mejor, P1,V1= P2,V2= P3,V3 = T1 ( 2.5 ) En un diagrama de estado podríamos representar la ecuación (2.5), tal como lo presenta la figura 2.3. En este caso todos los estados cuyos valores de las variables P y V estén sobre la línea, poseerán la misma temperatura. A esta línea la llamamos isoterma. ESCALAS EMPIRICAS DE TEMPERATURA La asignación de valores numéricos a las isotermas en general obedece a la creación de distintos tipos de escalas completamente arbitrarias y dadas por conveniencia. Estas escalas en general las podemos representar por dos tipos distintos de funciones termométricas: una función que expresa una proporcionalidad dada por una relación lineal T = ax +b ( 2.6 ) o simplemente por una proporcionalidad directa, es decir, T = Ax ( 2.7 ) donde a, b y A son parámetros experimentales que dependen de los valores de referencia escogidos y X es una propiedad termométrica. En el caso de establecer una escala de temperatura de acuerdo a la ecuación (2.6), se deben escoger dos puntos fijos, tal que 10 donde T y T son los valores asignados a los dos puntos fijos, X y X son los valores de la 1 2 1 2 propiedad termométrica a T y T respectivamente y X es el valor de la propiedad 1 2 termométrica a cualquier temperatura T. En este tipo de escalas se toman generalmente como punto de referencia el punto de congelación del agua, es decir la temperatura de equilibrio entre hielo y agua saturada con aire a 1 atmósfera de presión y el punto de ebullición del agua, es decir la temperatura de equilibrio entre el vapor y el agua pura a una atmósfera de presión Así en la escala Celsius el punto de congelación T y el punto de ebullición T se toman c e como: T = 100 °C y T = 0 °C c e En la escala Fahrenheit estos valores corresponden a: T = 212 °F y T = 32 °F c e Por consiguiente: 1 ° C = 1.8 ° F De esta forma podemos escribir la ecuación (2.6): Para la escala Celsius: y para la escala Fahrenheit: Así, en el termómetro de mercurio X corresponderá a la longitud de una columna de mercurio en un tubo de vidrio; en un termómetro de gases X corresponderá al volumen o a la presión del gas; en los termómetros de resistencia X corresponderá al valor de la resistencia eléctrica de un metal; en las termocuplas X corresponderá a la diferencia de potencial cuando dos uniones permanecen a diferentes temperaturas. (Un estudio amplio sobre los distintos tipos de termómetros se encuentra en la referencia 1). Escala absoluta del gas ideal. Utilizando gases como sustancia termométrica, X corresponderá al volumen o a la presión del gas, trabajando a presión o a volumen constante respectivamente. La experiencia ha mostrado que cuando se registra la temperatura en un termómetro de gases y el gas se encuentra a presiones bajas, las lecturas son reproducibles independientemente la naturaleza del gas. Esta característica permite idear una escala que es independiente de la sustancia termométrica, esta escala se conoce como la escala absoluta del gas ideal. De este modo, sí tomamos como magnitud del grado la misma de la escala centígrada y como propiedad termométrica el volumen, podemos escribir: o también, 11 Donde A T se le conoce como temperatura absoluta y en el punto de congelación toma la forma: Experimentalmente, realizando determinaciones con distinto gases y extrapolando los resultados a presión cero, se encuentra que el valor de la ecuación (2.12) es 273.15; el valor extrapolado a presión cero, - 273.15, se conoce cero absoluto de temperatura. Esta escala conocida también como escala kelvin usa una ecuación termométrica de la forma de la ecuación (2.7), es decir toma solamente un punto de calibración, el punto triple del agua, el cual ha sido fijado por convención en T = 273.16 K, donde K significa la PT temperatura en la escala Kelvin. De este modo dependiendo de la propiedad termométrica utilizada, presión o volumen del gas, la ecuación termométrica en la escala Kelvin será: Así, la relación entre la escala centígrada y la escala Kelvin, será: La escala absoluta relacionada con la escala Fahrenheit es la escala Rankine, las cuales se relacionan entre sí mediante: T (R) = T °F + 459.67 (2.18) - Escala Internacional de Temperatura. La obtención de medidas precisas de temperatura utilizando termómetros de gases es de por sí un procedimiento bastante tedioso, en este sentido el Comité Internacional de Pesas y Medidas, adoptó lo que hoy conocemos como escala internacional de temperatura, la cual se basa en un número definido de puntos de fusión y ebullición de sustancias puras, las cuales han sido determinadas con la máxima precisión posible en centros de investigación de varios países. De esta manera las temperaturas son interpoladas de acuerdo a ecuaciones termométricas adecuadas, dependiendo de la clase de termómetros. En la tabla 1 se muestran las temperaturas de los puntos fijos, en la escala kelvin, adoptada en 1989 y revisada en 1990 por el comité Internacional de Pesas y Medidas. Tabla 2.1 12 LECCIÓN 8. TRABAJO, ENERGÍA, CALOR Y PRIMERA LEY EL CONCEPTO DE TRABAJO De forma general podemos considerar el trabajo como la acción de una fuerza a través de un desplazamiento. Matemáticamente se expresa como: Cuando la dirección del desplazamiento es la misma que la de la fuerza, la ecuación (2.19) toma la forma: Es decir, podemos representar el trabajo como la integral definida del producto de una propiedad intensiva y el cambio en una propiedad extensiva. En la tabla se encuentran las expresiones para distintos tipos de trabajo. En termodinámica es imprescindible relacionar la definición de trabajo con los conceptos termodinámico vistos anteriormente, de tal forma que cuando realizamos trabajo sobre un sistema, hemos realizado un proceso. Ilustremos esta idea mediante el siguiente ejemplo: Un objeto de masa m, nuestro sistema, sometido al campo gravitatorio de la tierra y que se encuentra en el estado inicial a una altura x , de algún punto de referencia, lo llevamos a una 1 altura x , tal que x > x , estado final. Decimos entonces que el proceso de llevar el sistema 2 2 1 del estado inicial al estado final, es consecuencia de haber realizado trabajo sobre el sistema. Por convención el trabajo que realizan los alrededores sobre el sistema se considera negativo, mientras que el que realiza el sistema sobre los alrededores se considera positivo. Esto lo podemos representar mediante la figura 2.4 13 TRABAJO DE EXPANSION Y COMPRESIÓN REVERSIBLE E IRREVERSIBLE. Para iniciar nuestra discusión, consideremos como sistema un gas dentro de un cilindro, construido de paredes adiabáticas, y confinado mediante un pistón, tal como se muestra en la figura 2.5. Además supondremos que los efectos de la fuerza de rozamiento entre el pistón y el cilindro son despreciables respecto a los procesos que vamos a considerar. - Trabajo de expansión irreversible. De acuerdo a nuestro sistema, ver figura 2.5, el proceso de expansión se realizará al pasar del estado de equilibrio 1, cuyas variables de estado son P V , al estado de equilibrio 2, 1 1 cuyas variables de estado que caracterizan el sistema son P V . En el momento en que se 2 2 inicia el proceso la presión interna del gas, P , será mayor a la presión externa, P , int ejercida por los alrededores, el pistón, es decir: P ext int > P ; por consiguiente el sistema ext durante el proceso no estará en equilibrio hasta que se cumpla que P = P . El trabajo int ext realizado durante el proceso será entonces igual a: Pero como P es constante, además, P ext = P , por consiguiente 2 El valor entonces del trabajo de expansión irreversible será el área bajo la curva, en el diagrama P – V representado en la figura 2.5a. - Trabajo de compresión irreversible 14 En este caso el sistema se encontrará en el estado inicial 2 y mediante un proceso de compresión irreversible pasará al estado 1. En este proceso el gas se encontrará sometido a una presión externa constante e igual a P , por tanto, el trabajo realizado durante la 1 compresión será: y el valor será el área bajo la curva en el diagrama P – V representado en la figura 2.5b. - Trabajo de expansión y compresión reversible. Consideremos ahora que alcanzamos el estado 2 del estado 1 de manera reversible, en este caso el sistema pasará por una serie continua de estados de equilibrio de modo que en todo momento P = P y el trabajo realizado será: int ext En este caso, P ya no es una constante durante todo el proceso, sino que su valor estará dado por una ecuación de estado del tipo P (T,V). Además como el proceso es reversible implica que el proceso puede ocurrir en ambas direcciones por consiguiente el trabajo realizado durante la expansión debe ser igual al realizado durante la compresión: De esta forma el trabajo de expansión o compresión irreversibles será el área bajo la curva en el diagrama P-V, mostrado en la figura 2.5c. A partir de la figura 2.5, podemos sacar las siguientes conclusiones: - El Proceso de compresión irreversible requiere mayor trabajo que el proceso de expansión irreversible: - El proceso de expansión reversible requiere mayor trabajo que el proceso de expansión irreversible: - El proceso de compresión reversible requiere menor trabajo que el proceso de compresión irreversible: Finalmente podemos escribir: 15 La ecuación (2.34) nos presenta una propiedad importante del trabajo y es que no solo depende de los estado final e inicial sino también depende de la trayectoria, es decir de los detalles del proceso. Matemáticamente decimos entonces, que el trabajo es una diferencial inexacta y lo representaremos como δW : Visto ya el concepto de trabajo, podemos entrar a considerar el concepto de energía. ENERGIA DE UN SISTEMA La energía esta definida de forma general como la capacidad para realizar trabajo, esta capacidad se puede generar a partir de tres tipos básicos de energía: - Energía Potencial, E : Esta es la energía que posee un cuerpo en virtud de su posición. P Como ejemplo podemos citar un cuerpo que realiza trabajo mientras cae estando este atado a un dispositivo que consta de una cuerda y una polea. - Energía Cinética, E : Esta es la energía que posee un cuerpo en virtud de su movimiento, C así un cuerpo cayendo adquiere energía cinética la cual realizará trabajo sobre el material que cae, deformándolo. - Energía Interna, U: Es la energía asociada al sistema en reposo, el cual se encuentra a elevación constante, en ausencia de campos eléctricos o magnéticos y en ausencia de fenómenos tales como tensión superficial o capilaridad. De este modo las energías cinética y potencial las asociamos a un marco de referencia que se debe elegir, mientras que la energía interna la asociamos con el estado termodinámico del sistema. La energía total de un sistema estará dada entonces por la expresión: 16 FORMULACION DE LA PRIMERA LEY DE LA TERMODINAMICA El desarrollo de este numeral lo realizaremos estableciendo primero la relación entre la energía del sistema y el trabajo realizado y luego llegaremos al concepto de calor. Por conveniencia consideraremos sistemas cerrados. ENERGIA DEL SISTEMA Consideremos como punto de partida para nuestro desarrollo un arreglo experimental como el que se ilustra en la figura 2.6, el cual consta de un cilindro de paredes adiabáticas que contiene una serie de barreras B , B , B ,... y un pistón. Entre el pistón y la barrera B , se 1 2 3 1 encuentra un gas a P ,V ,T tal que sus valores están determinado por la ecuación de estado 1 1 1 T (P,V). Entre las otras barreras el espacio esta vacío. Realizaremos ahora dos tipos de procesos: - Primer proceso: Manteniendo el pistón en una posición constante, retiramos B , es decir 1 realizamos un proceso de libre expansión adiabática, alcanzando así el estado 2, ver diagrama 2.6a, luego alcanzamos los estados 3, 4,..., retirando las barreras B , B ,..., 2 3 respectivamente. En este tipo de proceso no se realizó ningún trabajo ya que hemos realizado un proceso de libre expansión adiabática. (Invitamos al lector a justificar esta afirmación). Si conectamos la familia de puntos que representan los estados 1,2,3..., por una línea, todos los puntos que estén sobre esta tendrán una característica en común y es que los estados 2, 3, 4..., pueden ser alcanzados del estado 1 sin realizar trabajo, a esta línea la llamamos isoenergética, ya que todos los estados que estén en ella tendrán la misma energía. - Segundo proceso: El pistón puede ahora moverse, de tal forma que por algún mecanismo puede realizar trabajo adiabático reversible, ya sea de expansión o compresión. Así ahora podremos alcanzar desde el estado A , una serie de estados B ,C ,...;y removiendo las 1 1 1 barreras también podremos alcanzar los estados A ,A ,...B ,B ,...C ,C ,..., tal como se 2 3 2 3 2 3 presenta en la figura 2.6b. Realizando entonces trabajo adiabático podremos alcanzar una u otra isoenergética, dependiendo del estado inicial. Si representamos por E la energía de la isoenergética en el 1 estado inicial y E la energía de la isoenergética en el estado final, ver figura 2.6b, la 2 cantidad de energía necesaria para pasar del estado inicial, 1, al estado final, 2, corresponderá al trabajo adiabático realizado, es decir: El signo negativo corresponde a la convención adoptada y a la vez esta de acuerdo con la idea intuitiva de que la energía del sistema aumenta (E > E ) al realizar sobre este trabajo 2 1 (W < 0), mientras que cuando el sistema realiza trabajo sobre los alrededores (W > 0) su ad ad energía disminuye (E < E ). 2 1 Consideremos ahora como el sistema podría alcanzar por ejemplo, El estado B del estado 3 A: 1 17 1. Una expansión adiabática para alcanzar el estado B y luego una libre expansión para 1 alcanzar el estado B . 3 2. Una libre expansión para alcanzar el estado A y luego un trabajo de expansión adiabática 3 para alcanzar el estado B . 3 En ambos casos el resultado es el mismo, es decir: La anterior relación es también aplicable a procesos reversibles e irreversibles (un proceso reversible seguido de un proceso irreversible constituyen un proceso neto irreversible), es decir bien podríamos haber realizado un trabajo adiabático reversible o bien podríamos haber efectuado un trabajo irreversible, el cambio de energía del sistema siempre estaría dado por la ecuación (2.38). Por consiguiente el cambio en la energía del sistema dependerá solamente de los estados inicial y final y en ningún momento de los detalles del proceso. Matemáticamente entonces podemos escribir: y así mismo podemos llegar a concluir que si el sistema realiza un ciclo, se debe cumplir con: En general se conocen como funciones de estado a las funciones que solo dependen del estado inicial y del estado final, en matemáticas se conocen como función punto. 18 EL CONCEPTO DE CALOR Para ilustrar el concepto de calor nuevamente consideremos un arreglo experimental, tal como se muestra en la figura 2.7, el cual consiste en dos sistemas A y B cada uno en equilibrio térmico y a temperaturas T1A y T1B respectivamente, tal que T1A >T1B. Luego de hacer interactuar los dos sistemas entre sí, la experiencia muestra que si T2A es la temperatura del sistema A en el estado final y T 2B la temperatura del sistema B en el estado final, que En este caso que la energía del sistema ha variado, podemos escribir: Cuando esto ocurre decimos que la energía del sistema ha cambiado debido a la transferencia de calor entre el sistema que se encontraba a mayor temperatura al sistema que se encontraba a menor temperatura. Por consiguiente la energía la podemos expresar como o Las ecuaciones (2.43) y (2.44) se conocen como la formulación de la primera ley de la termodinámica. La cual se cumple para todo cualquier tipo de proceso reversible o irreversible, realizado entre un estado de equilibrio inicial y un estado de equilibrio final. El calor al igual que el trabajo son funciones de la trayectoria. Invitamos al lector a que demuestre mediante un análisis de la ecuación (2.43) este argumento. El signo positivo del calor transferido obedece a la convención adoptada y sigue la idea intuitiva que la transferencia de calor al sistema, signo positivo, aumenta su energía interna y viceversa. Ver figura 2.8 LECCIÓN 9. CAPACIDADES CALORÍFICAS, SISTEMAS ABIERTOS A continuación consideraremos una serie de procesos con el fin de aplicar los conceptos relacionados con la primera ley de la termodinámica. Para esto consideraremos sustancias puras o mezclas de sustancias puras que formen una sola fase. 19 PROCESOS A VOLUMEN CONSTANTE, ISOCORICOS. Nuevamente consideremos dos procesos realizados a volumen constante, tal como lo muestra la figura 2.9. En el proceso representado en la figura 2.9a, el sistema construido se pone en contacto con un reservorio de calor, tal que la energía del sistema cambiará a expensas del calor transferido positivo o negativo. Así de la primera ley de la termodinámica podemos escribir: Pero como durante el proceso δW = 0, entonces dU = δQ, realizando la integración podemos escribir: Donde U y U son las energías internas en el estado inicial y final respectivamente, y Q12 1 2 representa el calor transferido durante el proceso. 20 Consideremos ahora el proceso 2.9b, en donde hemos alcanzado el estado 2 del estado 1, tal como en el proceso anterior, pero esta vez realizando trabajo sobre el sistema, por consiguiente δQ = 0, y Donde W12 representa el trabajo realizado durante el proceso. De las ecuaciones (2.46) y (2.47), se deduce que: Esto nos indica que el proceso a volumen constante lo podemos describir completamente indicando las energías internas inicial y final o indicando la cantidad de calor transferido o la cantidad de trabajo realizado, considerando un estado inicial. Ahora como este proceso esta acompañado por un cambio en la temperatura la experiencia muestra que este cambio depende del proceso y de las sustancias implicadas. Una medida de ese cambio la podemos establecer mediante la relación: donde C se conoce como capacidad calorífica a volumen constante. Para sistemas V homogéneos se suele expresar esta cantidad, refiriéndola a la unidad de masa, es decir: A cV se le conoce como calor medio especifico a volumen especifico v y entre las temperaturas T y T . Ahora cuando consideramos el límite: 1 2 21 o mejor donde c se conoce como calor específico a volumen constante. De la misma forma también V podemos expresar la capacidad calorífica a volumen constante como: Así pues mientras la capacidad calorífica es una propiedad extensiva, el calor específico es una propiedad que depende de la naturaleza de la sustancia. PROCESOS A PRESION CONSTANTE, ISOBARICOS: Nuevamente consideremos dos tipos de procesos como los presentados en la figura 2.10, en este caso es el pistón el que asegura que la presión sea constante. Consideremos esta vez en primer lugar el proceso 2.10b, en este caso podemos escribir: δW representa el trabajo realizado por las paletas y δW representa el trabajo de compresión P o expansión realizado por el pistón. Como la presión ejercida por el pistón es constante, podemos escribir la ecuación (2.54) como: Reemplazando en la expresión de la energía y realizando la integración tenemos: Donde W12es el trabajo realizado por las paletas. Ahora si consideramos el experimento de la figura 2.10a, podemos deducir que si es la cantidad de calor transferido entonces: combinando las ecuaciones (58) y (59) y haciendo P = P = P : 1 2 Donde H es una propiedad termodinámica que describe un proceso a presión constante para sustancia puras o mezclas de ellas a composición constante y en una sola fase, y se conoce como entalpía. 22 De manera de manera análoga a la ecuación (2.53): Donde C se conoce como capacidad calorífica a presión constante. Así mismo el calor P específico a presión constante, c , lo podemos definir como: P Donde H=h/m se conoce como entalpía específica. PRIMERA LEY DE LA TERMODINAMICA PARA SISTEMAS ABIERTOS Consideraremos ahora procesos en los cuales hay intercambio de masa y energía entre el sistema y los alrededores. Basemos entonces, nuestra discusión en el sistema presentado en la figura 2.11. Donde dm es la masa de sustancia que entra al sistema en un intervalo de i tiempo dt, dm es la masa de sustancia que sale del sistema en ese intervalo de tiempo, m es f t la masa de sustancia que posee en sistema en el tiempo t y m t+dt es la masa de sustancia que posee el sistema en el tiempo t+dt. Así un balance de masa sobre el sistema no resulta: De tal forma que el cambio de masa en el sistema en el intervalo de tiempo t y t+dt será: 23 Reordenando la ecuación (2.62) y dividiendo por dt, obtendremos la masa de sustancia que atraviesa el sistema por unidad de tiempo, es decir la velocidad de flujo: considerando el límite cuando dt → 0, podemos escribir la ecuación (2.63) como: donde m representa la masa total del sistema, mi la velocidad de flujo entrante a través de S una superficie de área A y mf representa la velocidad de flujo saliente a través de una superficie de área Af Si en el sistema existen diferentes superficies que permitan el flujo de sustancia, podemos generalizar la ecuación (2.64): i A la ecuación (2.65) se le conoce como ecuación de continuidad. Finalmente expresemos mf y mi y en términos de la velocidad del fluido ϑ y del área A de superficie. La cantidad de masa que cruza la superficie A estará dada por: Donde ρ es la densidad del fluido. De esta forma dividiendo por dt la expresión anterior obtendremos la expresión de la velocidad de flujo: Donde ϑ es la velocidad del fluido. 24 Visto lo anterior podemos considerar la energía del sistema: si E es la energía del sistema 1 en el estado inicial y E la energía del sistema en el estado final, entonces: 2 Donde e dm y e dm representan el aporte energético positivo o negativo debido a las i i f f cantidades de masa dm y dm que cruzan el sistema. Combinando las ecuaciones (2.68) y i f (2.69), podemos escribir: De la misma forma el trabajo positivo o negativo realizado sobre o por el sistema en el intervalo de tiempo t y t+dt será: Donde los términos entre paréntesis corresponden al trabajo de flujo y δW representa el trabajo realizado durante el proceso distinto al trabajo de flujo. Si ahora dividimos las ecuaciones (2.70) y (2.71) entre dt y luego reemplazamos en la formulación de la primera ley de la termodinámica, ecuación (2.43), obtenemos: Realizando los reemplazos respectivos, la ecuación (2.72) toma la forma: Donde Q˙ y W˙ representan la rata de calor y trabajo durante el proceso independientemente del trabajo de flujo y Z y Z representan al elevación del sistema. Si solo consideramos la i f energía interna del sistema podemos escribir: de tal forma que si no hay flujo de materia las expresiones (2.73) y (2.74), las podemos escribir: La expresión (2.75) no es otra que la expresión de la primera ley de la termodinámica. RELACION CAPACIDAD CALORÍFICA TEMPERATURA. En general se ha encontrado que para gases que contienen dos o más átomos en su molécula, la capacidad calorífica varía con la temperatura. Sin embargo la termodinámica 25 no predice la forma como se lleva a cabo esta variación. Debido a ello suelen utilizar formulas empíricas para predecir esta variación. En la tabla A3 se muestran para algunos gases la variación de su capacidad calorífica con la temperatura. EFECTO JOULE – THOMPSON Consideremos un tubo el cual posee una restricción, la cual puede ser un tapón poroso, una válvula semiabierta o un estrangulamiento, ver figura 2.12. Al hacer pasar un fluido a través de este, la experiencia muestra que debido a la restricción el fluido sufre una caída de presión. De este modo, si consideramos una mol de fluido, P y P será la presión del fluido 1 2 antes y después de pasar por la restricción respectivamente, T y T las temperaturas 1 2 correspondientes, v y v el volumen molar del fluido y ϑ y ϑ , las velocidades del fluido 1 2 1 2 antes y después de pasar por la restricción.. Este proceso cuando ocurre rápidamente y el área de la restricción es pequeña, la experiencia muestra que la transferencia de calor es muy pequeña, así que podemos suponer que el proceso es adiabático, es decir δ Q = 0. En consecuencia el trabajo efectuado por el fluido al pasar por el estrangulamiento será igual a P v – P v , ya que el volumen aumenta a v y la presión disminuye a P , De acuerdo a la 2 2 1 1 2 2 primera ley de la termodinámica, podemos escribir: Reemplazando y considerando como referencia un mol de fluido: o Así si la velocidad del fluido es constante o sus diferencias son muy pequeñas, en el proceso, la entalpía se mantiene constante. a este tipo de procesos se conocen como procesos isoentálpicos. En adelante consideraremos que el efecto Joule – Thompson es un proceso isoentálpico a menos que se especifique lo contrario. 26 Expresemos ahora la entalpía especifica en función de la temperatura y la presión; pero en el proceso dh = 0 , por consiguiente igualando la ecuación (2.79) y reordenando, podemos escribir: A la cantidad: conoce como coeficiente de Joule – Thompson, μ , y mide la velocidad de variación de la JT temperatura con la presión en un proceso de flujo a través de un tapón o un estrangulamiento. Ya que (∂h/∂T)P es igual a c , podemos escribir también el coeficiente de Joule – P Thompson como: Pero como h = e + Pv, entonces: 27 De este modo un coeficiente de Joule – Thompson positivo, significa que la temperatura disminuye durante la restricción y un coeficiente negativo implica que la temperatura aumenta durante la restricción. EJEMPLOS 0 1. Se retiran de la nevera 50 mL de leche, la cual se encuentra a una temperatura de 4 C, se 0 le agregan 100 mL de café a 92 C, ¿Cual será la temperatura de equilibrio, sabiendo que la -3 -3 densidad de la leche es 1.02 g cm ?. Suponga que la densidad del café es 1.00 g cm y que las capacidades caloríficas de los líquidos son iguales y que la capacidad calorífica del recipiente es despreciable. En este caso debemos tener en cuenta que la cantidad de calor pérdida por el café debe ser la misma ganada por la leche de este modo tenemos: Q absorbido = Q cedido Y de acuerdo a la ecuación (2.60) podemos escribir: Q = m Cp ΔT Donde m es la masa considerada, Cp es la capacidad calorífica del sistema, T y T son las f i temperaturas final e inicial de la mezcla. Reemplazando por las correspondientes cantidades, obtenemos: 51 Cp (T – 4) = 100 Cp ( 92 - T ) f f 0 Por consiguiente T = 7.44 C f 2. Un mol de gas ideal encerrado en un pistón pasa por el siguiente proceso cíclico termodinámico: 1) Una compresión adiabática; 2) una expansión debida a la transferencia de calor y trabajo sobre el pistón y 3) una compresión isotérmica para llevar al piston a su estado inicial. Exprese la cantidad total de calor transferido, suponiendo que conoce las cantidades de trabajo transferidas y las cantidades de calor transferidos en las dos primeras etapas. En este caso estamos considerando un ciclo por tanto se cumple: Si ahora representamos por: 28 Tenemos, que de acuerdo a nuestra convención: W12es negativo ya que es trabajo de compresión; Q12es cero ya que el proceso fue adiabático; W23es positivo ya que es un trabajo de expansión; Q23 es positivo ya que se le suministro al sistema; W31 es cero ya que no se realizo ningún tipo de trabajo y Q31 deberá entonces tener signo negativo ya que el hay transferencia de calor desde el sistema a los alrededores. Por tanto la respuesta al ejercicio será: 3. Un cilindro con un volumen de 2 ft3, contiene 0.5 lbm de un gas a 20 psia y 50 F. El gas se comprime en un proceso de cuasiequilibrio a 120 psia de forma tal que se cumple p (psia) = cte – 100 V, donde V esta expresado en ft3. Su temperatura final es de 20 F y la energía interna del gas cumple U (B/lbm) = T, con T en F. Dibuje un diagrama pV y calcule la transferencia de calor y el cambio de entalpía por lbm durante el proceso. En la figura E.1, representamos el proceso realizado, de esta forma, consideremos la información que tenemos. a) Calor transferido durante el proceso: De acuerdo a la primera ley de la termodinámica: Pero el trabajo realizado fue trabajo de compresión, por tanto: Donde V lo podemos conocer de la ecuación: 2 29 Así reemplazando por los correspondientes valores P y V obtenemos que la constante es: 1 1 cte = 220 psia 3 Por tanto a P = 120 psia , V = 1 ft . 2 2 Ya que conocemos U y U y por tanto ΔU y del mismo modo V y V , podemos reemplazar 1 2 1 2 y por ende realizar la integración, esto es: Reemplazando y haciendo las correspondientes conversiones obtenemos que: b) Cambio entálpico por libra La entalpía específica se relaciona con la energía interna, con la presión y con la temperatura por, ver ecuación (2.58): Reemplazando por los correspondientes valores obtenemos: Δh = 23.6 B / lbm. 0 0 4. Un compresor toma aire a 100 kPa, 40 C, y lo descarga a 690 kPa, 208 C. Los valores -1 de energía interna inicial y final para el aire son de 224 y 346 kJ kg , respectivamente. El -1 agua de enfriamiento alrededor de los cilindros elimina 70 kJ kg . Despreciando los cambios de energía cinética y potencial, calcular el trabajo realizado. Iniciemos por representar nuestro sistema. De acuerdo a la expresión general de la primera ley de la termodinámica, cuando dEp y dEc son cero, podemos escribir: Reemplazando h en términos de energía interna y presión, obtenemos la expresión: 30 Ahora, ya que no conocemos el volumen, podemos considerar que el gas se comporta como gas ideal y colocar el término PV en términos de RT, es decir: reemplazando por los correspondientes valores y teniendo en cuenta los factores de conversión, al final obtenemos que el trabajo realizado es: w = 240.5 kJ / kg 5. La velocidad de flujo a través de una turbina es de 40000lbm/h. Las velocidades de entrada y salida son de 6000 y 24000 fpm, respectivamente. Los valores inicial y final de entalpía son 1260 y 1000 B/lbm, respectivamente, y la pérdida de calor es de un total de 140000 B/h. Calcule la potencia de salida. De acuerdo a la primera ley de la termodinámica para sistemas abiertos podemos escribir. Reemplazando por los correspondientes valores obtenemos. considerando los factores de conversión y realizando las operaciones respectivas, obtenemos: w = 4043 HP AUTOEVALUACION 1. Cuando se comprime adiabáticamente una gas hasta la mitad de su volumen, se observa 0 que su temperatura aumente desde 25 a 200 C. Estime la capacidad calorífica molar del gas a presión constante. 2. Por una turbina fluye vapor a una velocidad de 40000 kg/h con valores de entalpía de entrada y salida de 2800 y 21000 kJ/kg, respectivamente. Las velocidades de entrada y salida son de 30 y 200 m/s, respectivamente. La pérdida de calor al entorno es un total de 300000 kJ/h. Calcule la potencia de salida. 31 0 3. Por un túnel de viento entra aire a 96 kPa, 20 C, con baja velocidad, y luego fluye establemente a través de una tobera hacia la sección de prueba. Si la expansión de la tobera 1.40 es tal que pv = constante, ¿A que presión debe expandirse el aire para lograr una velocidad de 220 m/s, en la sección de prueba? Suponga que el flujo es sin fricción. LECCIÓN 10. TERMOQUÍMICA En esta unidad trataremos en primer lugar los cambios energéticos cuando dos o más sustancias se mezclan, disuelven o difunden entre sí, luego cuando las sustancias reaccionan químicamente, y finalmente cuando estas cambian de fase. Iniciemos por considerar dos sustancias contenidas en dos recipientes adiabáticos separados entre sí por una pared diatérmica dispuesta de tal forma que se pueda remover, estudiando de esta manera dos tipos de procesos, uno a volumen constante, figura 3.1 y otro proceso realizado a presión constante, figura 3.2. Iniciemos nuestra discusión con el proceso a volumen constante. El sistema contiene dos sustancias, a y b, las cuales en el estado inicial se encuentran a la misma presión P y a la misma temperatura T . Cuando la pared es removida, asumiremos 1 1 que las dos sustancias se van a mezclar formando una sola fase, la presión y la temperatura en general van a cambiar (decimos que en general van a cambiar ya que cuando se mezclan gases de baja densidad o muy diluidos, el proceso de mezcla no presenta cambios en la temperatura ni en la presión), tomando los valores P y T . 2 32 2 Ya que el trabajo adiabático realizado durante este proceso es cero, la energía del sistema permanece constante, es decir: Supongamos que el sistema en el estado 2, se encuentra en un estado especial de equilibrio en el cual a pesar de que la mezcla sea potencialmente reactiva, la reacción química no se inicia, a este estado de equilibrio se le conoce como equilibrio retardado. Ahora el sistema realiza un segundo proceso el cual consiste en llevar el sistema a la temperatura inicial T , mediante transferencia de calor. De esta forma en el estado de 1 equilibrio 3, el sistema estará a una temperatura T y a una presión P . Aplicando la primera 1 3 ley de la termodinámica, tenemos para el estado 3: Donde Q3 es la cantidad de calor transferido para que el sistema alcance la temperatura Y 2V T1 manteniendo el volumen constante. Cuando la cantidad de calor transferida es positiva, T > T , el proceso de mezcla es un proceso endotérmico, pero si por el contrario la cantidad 1 2 de calor transferida es negativa T < T , el proceso es un proceso exotérmico. A la cantidad 1 2 de calor transferida pero con signo contrario se le conoce como calor de mezcla, de solución o de difusión según sea el caso de las sustancias a y b. Representaremos en adelante el calor de mezcla como Reemplazando en la ecuación (3.2), podemos escribir, Consideremos ahora un tercer proceso en el cual la reacción química tiene lugar. De este modo una vez el sistema ha alcanzado el nuevo estado de equilibrio, estado 4, se encontrará a una presión P y a una temperatura T . Ya que en este proceso no se realizó trabajo 4 4 adiabático, la energía del sistema permanecerá constante, es decir, U4 = U3 Para finalizar consideremos un último proceso en el cual el sistema alcanza la temperatura inicial T mediante transferencia de calor, Q5 , tal como se realizó en el proceso 2 → 3. Si 1 4V esta cantidad es positiva, es decir T > T , decimos que la reacción es una reacción 1 4 endotérmica, pero si por el contrario la cantidad de calor transferido es negativa, T > T , 4 1 decimos que la reacción es una reacción exotérmica. A esta cantidad de calor transferida pero con signo contrario se conoce como calor de reacción a temperatura T y volumen V . 1 1 Usualmente el calor de reacción es mayor al calor de mezcla en varios górdenes de magnitud. De acuerdo con la primera ley de la termodinámica para este proceso, podemos escribir: Representaremos el calor de reacción como: 33 Reemplazando en la ecuación (3.6), tenemos: Ya que los calores de mezcla y los calores de reacción dependen de la temperatura de referencia, en este caso T y del volumen V , suele tomarse para efectos de comparación una 1 1 temperatura de referencia, que en general es, 25 °C. Considerando ahora la misma serie de procesos pero esta vez manteniendo la presión constante, ver figura 3.2, es claro que podemos escribir ecuaciones análogas a las ecuaciones (3.1), (3.2), (3.5) y (3.6). Esto es: Al igual que en los procesos a volumen constante los calores de mezcla y los calores de reacción dependen de la temperatura de referencia T y de la presión P. En general suele 1 tomarse como estado de referencia T = 25 °C y P = 1 atm. 1 Debido a que los procesos de mezcla y reacción dependen de la cantidad de materia involucrada en el proceso, se suele especificar los calores de mezcla o reacción en términos de la masa considerada, generalmente 1 mol., por ejemplo la reacción, indica que la formación de un mol de benceno líquido a partir de 6 moles de carbono sólido y 3 moles de hidrógeno gaseoso, tiene como resultado la absorción de 11.7 kilocalorías a 298.15 K y 1 atm de presión. En 1840 G.H. Hess descubre empíricamente que el cambio térmico a presión o a volumen constante de una reacción dada es el mismo, tanto si tiene lugar en una sola etapa o si es el producto de una serie sucesiva de reacciones, mientras los reactante y productos sean los mismos. Este enunciado se conoce como la ley de Hess y se cumple también para mezclas. Esta ley es una consecuencia de la primera ley de la termodinámica ya que el calor de reacción, HMOR o EMOR, dependen únicamente de los estado inicial y final y son independientes del camino seguido. En consecuencia la ley de Hess nos permite sumar las 34 ecuaciones químicas como si fueran ecuaciones algebraicas. Ilustremos esto con el siguiente ejemplo. El haber restado la ecuación tres y por tanto conservar la cantidad del calor de reacción pero con signo contrario, también esta de acuerdo con la primera ley de la termodinámica, ya que 35 de otro modo sería posible crear energía térmica ya sea formando un compuesto y luego descomponiéndolo o viceversa, es decir el cambio térmico que acompaña una reacción química en una dirección es de magnitud exactamente igual pero de signo opuesto al que acompaña la misma reacción pero en sentido inverso. Aclaremos antes de continuar lo relacionado al estado de referencia. Este surge de la necesidad de establecer una base común para realizar cálculos consistentes, no solo con calores de formación sino también con otras propiedades de estado, como veremos en capítulos posteriores. En general para líquidos y sólidos se toma como estado de referencia el valor de la propiedad a una temperatura de 25 °C y una presión de 1 atmósfera; en el caso de los gases el estado de referencia se toma respecto al gas ideal. Ilustremos ahora el concepto de calor de formación. Para esto consideremos un reactor al cual entran los reactantes a 25 °C y 1 atm de presión, luego de ocurrir la reacción los productos salen a la misma temperatura y presión.. El proceso involucra una transferencia de calor ΔH ., para mantener la temperatura constante. De acuerdo a la primera ley de la R termodinámica esta transferencia de calor la podemos escribir como: De las expresiones (3.15) y (3.16) observamos que si la entalpía de los productos es mayor que la entalpía de los reactantes, ΔH es positivo y la reacción en consecuencia va R acompañada de absorción de energía, mientras que si sucede lo contrario, ΔH es negativo y R la reacción ira acompañada de liberación de energía. Sin embargo los valores absolutos no nos proporcionan información termodinámica (Invitamos al lector a justificar esta afirmación). Por convención entonces, arbitrariamente se ha tomado la entalpía de todos los elementos como cero en el estado de referencia T = 25°C y P = 1 atm (Esto en ningún momento es extraño a nuestra experiencia, ya que cuando hablamos de temperatura tomábamos puntos de referencia para determinar la temperatura de un cuerpo). De este modo las ecuaciones (3.15) y (3.16) as podemos escribir como: 36 Citemos par finalizar dos tipos especiales de calores de reacción, el calor de combustión y el calor de hidrogenación. El primero se relaciona con el calor de reacción que acompaña reacciones donde intervienen combustibles, bien sea carbón o hidrocarburos, en presencia de oxígeno para producir dióxido de carbono y agua, de tal forma que podemos definir el calor de combustión como el calor de reacción que acompaña la combustión de 1 mol de compuesto a una temperatura T y a una presión P. Por otra parte el calor de hidrogenación será el calor de reacción que acompaña a la hidrogenación de 1 mol de compuesto no saturado a una temperatura T y a una presión P. En las tablas A1 y A2 se encuentran registrados valores de formación y combustión para diversas sustancias. De hecho es evidente que a partir de calores de formación y combustión nos es posible determinar los calores de reacción de reacciones que no es posible determinarlos directamente. En los ejercicios ilustraremos estos conceptos. La experiencia muestra también que cuando ocurre un cambio de fase, vaporización de un líquido, fusión o sublimación de un sólido o la transición de una modificación cristalina a otra; los procesos van acompañados de un cambio energético. Este cambio energético se conoce como calor latente de vaporización, fusión, etc. De este modo el calor latent tomarse también como referencia un mol de sustancia. Para ilustrar el anterior concepto citemos lo siguientes ejemplos: e representa la diferencias de entalpía o energía interna de las fases que se consideran en el proceso, a la temperatura y presión a la que tiene lugar el cambio de fase. En este caso suele tomarse también como referencia un mol de sustancia. Para ilustrar el anterior concepto citemos lo siguientes ejemplos: EJEMPLOS 1. El calor de hidrogenación del etileno, C H , para dar etano, C H , es de –32.6 Kcal/mol 2 4 2 6 0 a 25 C. Utilizando los siguientes datos de calores de combustión: presentados en la tabla siguiente, determinar el cambio entálpico que acompaña a reacción de cracking: 37 ΔH reacción = + 54.7 kcal/mol. 2. Encuentre el calor de combustión por mol de propano, sabiendo que en la combustión de 0 5 gramos de este, se requieren 60.36 kcal a 25 C, encontrar el calor de combustión por mol de propano. El proceso de combustión lo puedo describir por la reacción: C H + 5 O → 3 CO + 4 H O 3 8(g) 2(g) 2(g) 2 (l) En esta se puede observar que debe ocurrir una disminución en la presión del sistema debido a que en un principio se tenían 6 moles de gas y al final de la combustión se tienen solo 3. Esto implicara entonces que el sistema realizará una cantidad adicional de trabajo, es decir: ΔH = ΔE + VΔP Si considero que los gases implicados en la reacción se comportan como gases ideales, puedo escribir: ΔH = ΔE + ΔnRT donde Δn es el cambio de moles gaseosos implicados en el proceso. Ahora de acuerdo al ejercicio tengo que son necesarias - 60.09 kcal/5g o equivalentemente 12.02 kcal/g o también – 528.82 kcal/mol, entonces: ΔH = - 528820 + (3-6)(1.99)(298) ΔH =530.6 kcal / mol propano 0 3. El calor de combustión del metano es –212.80 kcal/mol a 250 C, el calor de 0 vaporización del agua es de 10.52 kcal/mol a 25 C. Derive la expresión para calcular la temperatura final del sistema cuando se quema metano en aire, compuesto por 1mol O / 4 2 0 mol N , a 25 C, a presión constante y suponiendo que la reacción es completa. 2 En este caso , nuestro proceso es: CH 4(g) +2O 2(g) → CO 2(g) + 2 H O ΔH = - 212 kcal/mol 2 (l) 2 H O → 2 H O ΔH = 2 x 10.52 kcal/mol 2 (g) 2 (l) Por tanto: 38 CH 4(g) +2O 2(g) → CO 2(g) + 2 H O ΔHr = - 171.96 kcal/mol 2 (g) De esta forma tenemos que la cantidad de energía, ΔH , que le debemos suministrar al 1 0 sistema para elevar la temperatura desde 25 C hasta una temperatura T , será la de igual 2 magnitud al calor de reacción, pero de signo opuesto. Es decir se debe cumplir: ΔH = - ΔHr 1 Peso como ΔH = n Cp ΔT, donde n es el número de moles del componente, Cp su 0 capacidad calorífica, entonces la energía necesaria par llevar el sistema desde 25 C hasta la temperatura final, T , vendrá dada por: 2 Ahora ya que Cp no es constante con la temperatura, entonces debemos buscar la expresión que me relacione Cp con temperatura, la cual generalmente vienen de la forma: 2 3 Cp = α + β T + χT + δT ... En la tabla A3 se presenta una tabla de Cp en función de la temperatura para distintas sustancia. De este modo la expresión que buscamos tendrá la forma: AUTOEVALUACION 1. Calcular la cantidad de calor requerida para calentar 10 moles de SO desde 200 a 1100 2 0 C a presión atmosférica. 0 2. Estime el calor estándar de formación para el etilbenceno líquido a 25 C 3. Se coloca en una bomba calorímetrica una pequeña cantidad de etanol líquido junto con 0 el doble de la cantidad teórica de oxígeno a 25 C y 1 atm de presión. Tomando 9.5 kcal -1 0 mol como el calor de vaporización del alcohol a 25 C calculese la temperatura máxima del sistema, asumiendo que la combustión del etanol fue completa y el proceso se realizó en condiciones adiabáticas. 4. Los calores de combustión del n – butano y del isobutano son – 688.0 y – 686.3 Kcal, 0 respectivamente, a 25 C. Calcúlese el calor de formación de cada uno de estos isómeros a partir de sus elementos, y también el calor de isomerización, n – butano → isobutano a 25 0 C. CAPÍTULO 3. SEGUNDA LEY DE LA TERMODINÁMICA En este capítulo estableceremos una función que nos será de gran utilidad en el momento de establecer la direccionalidad y espontaneidad de un proceso, esta función es la entropía. LECCIÓN 11. SEGUNDA LEY Y CONCEPTO DE EFICIENCIA 39 FORMULACIÓN DE LA SEGUNDA LEY DE LA TERMODINAMICA La primera ley de la termodinámica nos señala que existe una equivalencia exacta entre las distintas formas de energía comprometidas en un proceso. Sin embargo no nos proporciona ninguna información sobre la direccionalidad del mismo, es decir la probabilidad de que el proceso ocurra. Así por ejemplo, si consideramos un ciclo la primera ley de la termodinámica nos indica que la cantidad de trabajo realizada, es equivalente a la cantidad de calor transferido, pero este principio en ningún momento establece la direccionalidad del ciclo ni tampoco su factibilidad. Así a la pregunta si un proceso es factible o no y de hecho cual es su direccionalidad, la segunda ley de la termodinámica tiene la respuesta. El segunda Ley de la termodinámica se ha establecido de diferentes maneras, sin embargo mencionaremos aquí dos formas tradicionales en que ha sido enunciada. En 1850 R. Clausius enuncia la segunda ley de la termodinámica en los siguientes términos: El calor no puede pasar espontáneamente desde un cuerpo de baja temperatura a un cuerpo de mayor temperatura. Estableciendo así, que el flujo neto de calor ocurre solamente en una dirección, del cuerpo de mayor temperatura al de menor temperatura. Otra forma de enunciar la segunda ley de la termodinámica se relaciona con el proceso de transformación de calor en trabajo y se debe a W. Thompson (Lord Kelvin) en 1851, quién enuncia: Es imposible construir una máquina la cual extrae calor de una fuente dada y la transforma en energía mecánica, sin que ocurran cambios adicionales en los sistemas que toman parte. Planck a su vez formula este mismo enunciado en los siguientes términos: Es imposible construir una máquina que realiza trabajo periódica y continuamente a expensas del calor extraído por una sola fuente Estos enunciados como se puede intuir, no son más que fruto de la experiencia y muestran la improbabilidad de invertir procesos espontáneos sin que ocurran una serie de cambios, los cuales hacen que una vez el sistema alcance el estado inicial, los alrededores no sean restaurados. EFICIENCIA TERMICA La realización de trabajo esta sujeta a que exista una diferencia de potencial, es decir a que exista una fuerza directora. La conversión de calor en trabajo no es la excepción, ya que la transferencia de calor se efectúa cuando existe una diferencia de temperaturas tal que la transferencia ocurre del cuerpo de mayor temperatura al de menor temperatura. En estos términos se define la eficiencia térmica como la fracción de calor transferido por una máquina térmica que puede transformarse en trabajo, matemáticamente se expresa la eficiencia como: 40 Donde W es el trabajo neto ejecutado y Q es la cantidad de calor transferido por la fuente H de calor que posee mayor temperatura. De esta forma Q expresa de alguna manera el costo H del proceso, por ejemplo el del combustible y W el trabajo útil logrado en el proceso. LECCIÓN 12. TEOREMA DE CARNOT Y MÁQUINA DE CARNOT EL TEOREMA DE CARNOT En 1824 S. Carnot, realiza un descubrimiento relacionado con el rendimiento de las máquinas térmicas, las cuales convierten calor en trabajo. El principio expuesto por Carnot se puede expresar de la siguiente manera: Todas las máquinas térmicas que operen de manera reversible, entre dos temperaturas dadas, tienen la misma eficiencia. Tratemos de demostrar esta afirmación considerando dos máquinas térmicas reversibles A y B las cuales trabajan entre las mismas temperaturas T y T , pero con eficiencias diferentes, 0 ver figura 4.1. Supongamos entonces, que el reservorio 2 el cual esta a T, le transfiere una cantidad de calor Q a la máquina A y esta a su vez convierte una parte de ese calor transferido W en trabajo y cede el restante al reservorio 1, que esta a temperatura T . Por otro 0 lado la máquina B convierte una cantidad menor W’ del calor transferido desde el reservorio 1 y cede una cantidad al reservorio 2. Supongamos ahora que acoplamos las dos máquinas tal que la maquina A funcione tomando Q del reservorio 2 y ejecutando trabajo W y cediendo al reservorio una cantidad Qá y a su vez la maquina B funciona de 0 manera inversa, esto es como refrigerador, tomando del reservorio 1, consumiendo trabajo W’ y cediendo el calor resultante al reservorio 2. Si consideramos que el sistema realiza un ciclo reversible, entonces: 41 Por lo anterior podemos deducir que: , lo que nos indica que las dos máquinas combinadas y funcionando cíclicamente convierten en trabajo la totalidad del calor transferido desde el reservorio 2. esto es contrario a la segunda ley de la termodinámica y en consecuencia las dos máquinas deben tener la misma eficiencia. De hecho entonces el teorema de Carnot es consecuencia de la segunda ley de la termodinámica. EL CICLO DE CARNOT De acuerdo al teorema de Carnot todas las máquinas térmicas reversibles que operan entre dos temperaturas poseen la misma eficiencia, por consiguiente podremos considerar cualquier máquina como objeto de estudio. El interés de estudiar este tipo de máquinas radica en la importancia que de sus propiedades se infieren para el desarrollo de la segunda ley de la termodinámica. Ya que es imposible producir trabajo continuamente mediante la transferencia de calor desde una sola fuente, se hace indispensable considerar al menos dos fuentes de calor. Así el ciclo más sencillo será cuando el sistema intercambia calor con dos fuentes de calor, las cuales se encuentran a temperaturas diferentes. Un ciclo con estas características se conoce como ciclo de Carnot, el cuál describiremos a continuación. Consideremos un fluido el cual esta contenido en un cilindro, el cual posee un pistón. La fuerza de rozamiento entre las paredes del cilindro y el pistón las consideraremos despreciables con el fin de que se cumpla la reversibilidad del proceso, el diagrama del experimento se presenta en la figura 4.2a. Supondremos además que es posible efectuar procesos adiabáticos colocando paredes adiabáticas sobre el cilindro cuando esto se requiera. Así podremos realizar el ciclo que consta de los siguientes cuatro procesos reversibles: Primer proceso: Se realiza una transferencia de calor Q desde el reservorio 1 al fluido, gracias a la pared diatérmica del cilindro, ocasionando entonces una expansión isotérmica reversible del fluido a temperatura T. 42 Segundo proceso: Se separa el cilindro del reservorio y se cubre con paredes adiabáticas. Se realiza luego un trabajo de expansión adiabática reversible W23, hasta que la temperatura del fluido descienda a la temperatura del reservorio 2, es decir T . 0 Tercer proceso: Retiramos las paredes adiabáticas del cilindro y lo colocamos en contacto con el reservorio 2, para luego realizar una compresión isotérmica reversible sobre el fluido. En este proceso habrá una transferencia de calor Q0 desde el sistema al reservorio. Cuarto proceso: adiabáticas para trabajo,W42 , de temperatura de T 0 Separamos el cilindro del reservorio, luego lo recubrimos con paredes comprimir el fluido de forma adiabática mediante la realización de tal forma que el fluido alcance el estado inicial, aumentando así su a T. Ilustremos el ciclo de Carnot, considerando ahora como fluido un gas ideal, el proceso se representa en el diagrama presión – volumen de la figura 4.2b. - Primer Proceso, camino 1 → 2 sobre la isoterma de la figura 4.2b: Ya que el proceso se realiza a temperatura constante, podemos escribir: - Segundo proceso, camino 2 → 3 sobre la adiabática de la figura 4.2b: El trabajo de expansión adiabático estará dado por: W23 = dE . Sin embargo, para el gas ideal se cumple: Por consiguiente: 43 - Tercer proceso: camino 3 → 4 sobre la isoterma de la figura 4.2b. El calor transferido al reservorio 1, será: - Cuarto proceso: camino 4 → 1 sobre la adiabática de la figura 4.2b. De forma similar al segundo proceso, podemos escribir: Así el trabajo total realizado en el ciclo será: Pero de acuerdo a las ecuaciones (4.4) y (4.6) los trabajos de expansión y compresión adiabáticos son de la misma magnitud pero de signo contrario, por consiguiente podemos escribir: De acuerdo a la definición de eficiencia, ecuación (4.1), la eficiencia de una máquina térmica que opera reversiblemente entre dos temperaturas será: Sustituyendo en la ecuación (4.9) las expresiones (4.2) y (4.5), obtenemos: Las ecuaciones (4.9) y (4.10) nos indican que siempre que las máquinas térmicas funcionen de manera reversible, su rendimiento será independiente de la naturaleza de las sustancias y solamente dependerá de la temperatura de las fuentes. Este último aspecto nos pone también de manifiesto que mientras más baja sea la temperatura de la fuente de menor temperatura, es decir la que actúa como refrigerante, mayor será la eficiencia de la máquina térmica. Sin embargo en la práctica no es conveniente que esta fuente se encuentre a temperaturas inferiores a la temperatura del ambiente, por tanto es conveniente elevar la temperatura de la otra fuente de mayor temperatura. De otro lado ya que el trabajo total realizado en el ciclo de Carnot es la suma de los trabajos isotérmicos reversibles, ya que los trabajos adiabáticos reversibles se cancelan, se deduce entonces que la eficiencia máxima que puede tener una máquina térmica se obtiene cuando esta opera de manera reversible (Invitamos al lector a justificar esta afirmación). 44 De otro lado ya que el ciclo de Carnot se efectúa de manera reversible, es posible entonces operarlo de manera inversa es decir la máquina puede tomar calor de al fuente de menor temperatura y cederlo a la fuente de mayor temperatura a expensas de realizar trabajo sobre el sistema, este tipo de máquinas se conocen como máquinas de refrigeración o bombas de calor. En este caso el trabajo realizado debe ser de magnitud igual al trabajo efectuado en el ciclo de Carnot pero de signo contrario. Por otra parte mientras que la eficiencia de una máquina térmica se define de acuerdo a la ecuación (4.9), en el caso de las máquinas de refrigeración a la razón entre el calor absorbido de la fuente de menor temperatura y el trabajo efectuado por la máquina se conoce como coeficiente de funcionamiento, ∈, el cual matemáticamente se expresa como: De forma análoga a la ecuación (4.10), podemos escribir también: De esta manera, el coeficiente de funcionamiento representa la cantidad mínima de trabajo necesario para extraer una cantidad de calor Q de una fuente de calor a T y transportarla a 0 0 una fuente de temperatura T, donde T >T . 0 El hecho que la máxima eficiencia de una máquina térmica se logre cuando la máquina opera de forma reversible trae como consecuencia que la eficiencia de esa misma máquina operando de forma irreversible es menor, es decir: Por consiguiente la fracción de calor transferido que puede ser convertida en trabajo será menor cuando la máquina opera de forma irreversible. Análogamente en una máquina de refrigeración que opere de forma irreversible el trabajo necesario para transportar una cantidad de calor de la fuente de menor temperatura a la fuente de mayor temperatura, aumentará, respecto a sí la máquina operara reversiblemente. Por lo anterior, podemos deducir que: LECCIÓN 13. ESCALA TERMODINÁMICA DE TEMPERATURA Al ser la eficiencia del ciclo de Carnot independiente de la sustancia considerada y depender únicamente de la temperatura se nos presenta un marco propicio para relacionar la temperatura como una medida del calor transferido. Una medida de este tipo será independiente de la sustancia termométrica y también de cualquier medida realizada con base en cualquier escala empírica de temperatura. A este tipo de escala se le denomina escala absoluta de temperatura o escala termodinámica de temperatura. Ilustremos la escala termodinámica de temperatura, considerando tres reservorios de calor a temperaturas T > T >T respectivamente y tres ciclos de Carnot operando cada uno entre 1 2 3 dos de los tres reservorios, tal como se presenta en la figura 4.3. 45 Ya que la eficiencia térmica depende del calor transferido por las fuentes y este calor transferido depende de la temperatura, podemos escribir: donde f (T, T ) es una función que relaciona la razón de lo calores transferidos con las o temperaturas de las fuentes. De manera similar entonces podemos establecer las para los tres ciclos de Carnot considerados en la figura 4.3: Y de igual modo podemos escribir: Se deduce entonces que: En este caso ya que no depende de f(T1, T3), el producto del lado derecho de la ecuación (4.18) tampoco deberá depender de T . para que se cumpla esto, entonces la forma de las 2 funciones que intervienen en el producto deben ser: 46 Por consiguiente Así podemos concluir que para cualquier ciclo de Carnot que opere entre dos temperaturas T y T , se cumple: 0 Aunque existen varias relaciones funcionales que satisfacen la ecuación (4.21), tomaremos la propuesta por Kelvin, la cual expresa. Por tanto la eficiencia de una máquina térmica la podemos expresar en términos de las temperaturas absolutas, es decir. La ecuación (4.23) nos indica que el ciclo solamente puede convertir la totalidad del calor transferido de la fuente a temperatura T en trabajo si la temperatura de la fuente que actúa como refrigerante es cero, es decir a temperatura del cero absoluto: Y si de otra parte comparamos la ecuación (4.23) con la deducida para un gas ideal, ecuación (4.10), podemos darnos cuenta que son las mismas, es decir la escala del gas ideal es la misma escala termodinámica de temperatura. Esta es la justificación por la cual se denomina escala Kelvin a la escala del gas ideal. LECCIÓN 14. ENTROPÍA, IRREVERSIBILIDAD Y CAMBIOS EN UN GAS IDEAL DEFINICION DE ENTROPÍA Tal como lo muestra la ecuación (4.23) la eficiencia térmica la podemos expresar bien sea en términos del calor transferido por las dos fuentes de calor o bien en términos de las temperaturas de las dos fuentes. Por consiguiente podemos escribir: Escribamos ahora la ecuación anterior considerando que el símbolo del calor contenga el signo correspondiente, podemos entonces escribir la ecuación (4.26) como: 47 Reordenando la ecuación: Imaginémonos ahora un ciclo reversible, este ciclo lo podemos considerar formado por una serie de ciclos de Carnot. De acuerdo a lo que ya hemos visto para, ese ciclo formado de ciclos de Carnot se debe cumplir: Si consideramos cambios infinitesimales de calor intercambiado con las fuentes a temperatura T, realizados de forma irreversible e isotérmica, podemos entonces escribir la ecuación (4.28) como: Supongamos ahora, ya no un solo ciclo, sino dos ciclos, de tal forma que para el primer ciclo mediante un proceso reversible, el estado 2 es alcanzado del estado 1 siguiendo la trayectoria A y el estado 1 es alcanzado del estado 2, siguiendo la trayectoria B, para de esta forma completarse el ciclo. En el caso del segundo ciclo el estado 2 es alcanzado del estado 1 siguiendo la trayectoria C y el estado 1 es alcanzado del estado 2 para completarse el ciclo, siguiendo la trayectoria B. De acuerdo a este arreglo experimental, podemos escribir para el primer ciclo: Donde A y B representan las trayectorias seguidas. En el caso del segundo ciclo tenemos: De las ecuaciones (4.30) y (4.31), se deduce que: De esta forma las ecuaciones (4.30) a (4.32) nos indican que la cantidad es la misma para toda las trayectorias entre los estados 1 y 2. Por consiguiente podemos concluir que esta cantidad es independiente de la trayectoria y depende únicamente de los estados final e inicial, en otras palabras es una propiedad de estado. A esta propiedad se conoce como entropía, S, y matemáticamente se expresa como: 48 De este modo el cambio de entropía estará definido por el calor intercambiado de forma isotérmica y reversible dividido entre la temperatura absoluta a la que se transfiere. De acuerdo a lo anterior se deduce entonces que la entropía al igual que la energía es una propiedad extensiva que dependerá de al cantidad de materia del sistema. De este modo si se duplica la cantidad de materia, la cantidad de calor transferida para obtener el mismo cambio en entropía deberá también ser el doble y en consecuencia el cambio entrópico aumentará en igual proporción. De hecho entonces se debe verificar para la entropía que cuando un sistema esta compuesto de varias partes, el cambio total de entropía debe ser la suma de los cambios de entropía de las partes individuales. CAMBIO DE ENTROPIA EN UN PROCRESO IRREVERSIBLE Consideremos ahora procesos reversibles, los cuales son de hecho los procesos que ocurren en la naturaleza. Para ilustrar el cambio en este tipo de procesos, consideremos como punto de partida la ecuación (4.13), , la cual nos indica que la eficiencia de una máquina térmica operando de manera reversible es mayor a la eficiencia si la misma máquina operara de manera irreversible. De otra parte consideremos que en un ciclo de Carnot, la transferencia de calor en una de las fuentes se efectúa de manera irreversible, por ejemplo en la fuente a temperatura T, en la figura 4.2. De acuerdo a las ecuaciones (4.13) y (4.26), podemos escribir: Reordenando la ecuación anterior, tenemos: Y si consideramos el caso general en el ciclo tenga una o más etapas tanto reversibles como irreversibles, podemos escribir: Sin embargo de acuerdo a la ecuación (4.29), el segundo término de la ecuación (4.36) es cero, por consiguiente para una máquina térmica que opere de manera irreversible, se cumple: De forma análoga se puede deducir la ecuación (4.37) para maquinas de refrigeración (Invitamos al lector a demostrar esta afirmación). De esta forma podemos concluir que para cualquier ciclo, se cumple: 49 A la ecuación (4.38) se conoce como desigualdad de Clausius. Consideremos ahora un ciclo general 121, formado por una trayectoria 1 → 2 compuesta por una o más etapas irreversibles y una trayectoria 2 → 1 completamente reversible. De acuerdo a las ecuaciones (4.35) y (4.36) podemos escribir: de acuerdo a la definición de entropía, ecuación (4.33), podemos escribir la ecuación anterior como: o Consideremos ahora un sistema aislado, donde 0=Qδpara cualquier fuente. En este caso la expresión (4.41) toma la forma: A la ecuación anterior se conoce como principio de incremento de la entropía, y expresa que la entropía de un sistema aislado se incrementa cuando un proceso irreversible tiene lugar. Ya que cualquier proceso lo podemos analizar en términos de un sistema aislado, imaginémonos entonces que incluimos todas las fuentes que intercambian calor durante el proceso como parte del sistema. De esta forma de acuerdo al principio de incremento de la entropía podemos realizar la siguiente clasificación: Esta clasificación nos permite entonces concluir que todos los procesos espontáneos que de hecho son irreversibles, van acompañados de un incremento en la entropía. Consideremos ahora ya no sistemas aislados sino el cambio entrópico que acompaña en un proceso a los alrededores. Para esto consideremos un proceso como el que se representa en la figura 4.5, donde una cantidad de calor δQ es transferida desde los alrededores, a una temperatura T , al sistema, el cual se encuentra a una temperatura T. Para este proceso 0 podemos escribir para el sistema: y para los alrededores: 50 De manera que el cambio total de entropía será: o Y como T > T entonces ( 1/ T - 1 / T ) y δQ son positivos, podemos entonces concluir: 0 0 Por otra parte si T > T , es decir la transferencia de calor se verifica del sistema a los 0 alrededores, tanto δQ como ( 1/ T - 1 / T ) se hacen negativos y el resultado es el mismo al 0 expresado por la ecuación (4.48). El resultado expuesto por la ecuación (4.48), lo habríamos podido intuir de las ecuaciones (4.42) y (4.43). De este modo podemos concluir que como una consecuencia de la segunda ley de la termodinámica que todos los procesos que tienen lugar en la naturaleza van asociados con una ganancia de entropía y esta ganancia se verifica para el sistema y sus alrededores. CAMBIOS DE ENTROPIA EN UN GAS IDEAL En este numeral consideraremos los cambios de entropía que se verifican bajo diversas condiciones, considerando un gas ideal y en el siguiente numeral deduciremos las expresiones generales para cualquier sustancia. De acuerdo a la primera ley de la termodinámica y considerando que el trabajo realizado es trabajo de expansión, como sucede en la mayoría de los procesos considerados en termodinámica, podemos escribir: Considerando que δQ se transfiere reversiblemente a una temperatura T, podemos determinar el cambio de entropía durante un proceso, es decir: Para una mol de gas ideal se cumple: du = cv dT y P= RT/v, reemplazando en la expresión (4.50) obtenemos: 51 por consiguiente Como par un gas ideal c no depende de la temperatura, entonces: V Es decir: Si ahora sustituimos y en la ecuación anterior, obtenemos: De este modo las expresiones (4.53) y (4.54) representan el cambio de entropía que acompaña un cambio de estado cuando se considera un mol de gas ideal. Si consideramos ahora una variación de temperatura a volumen constante, el cambio de entropía para un gas ideal será: Y para la misma variación de temperatura pero a presión constante, tenemos: Pero si el proceso es isotérmico, entonces el cambio entrópico estará dado por: Es importante anotar que aunque las ecuaciones (4.53) a (4.56) fueron deducidas a partir de procesos reversibles, los resultados obtenidos son aplicables a cualquier cambio de estado termodinámico, independientemente de que el proceso se efectué de manera reversible o irreversible. Esta característica se debe a que la entropía es una función de estado. LECCIÓN 15. VARIACIÓN DE LA ENTROPÍA CON TEMPERATURA, PRESIÓN Y VOLÚMEN VARIACION DE LA ENTROPIA CON LA TEMPERATURA Deduzcamos ahora las relaciones generales para la variación de la entropía con la temperatura. De acuerdo a al ecuación (4.50), podemos escribir: 52 Si consideramos un proceso a volumen constante, el cambio de entropía del proceso será y si derivamos ahora esta expresión con respecto a la temperatura, obtenemos: Pero como podemos deducir: Por consiguiente, De la misma forma si consideramos un proceso a presión constante y utilizamos el mismo procedimiento utilizado para deducir la ecuación (4.60), obtenemos: De este modo hemos obtenido dos ecuaciones, (4.60) y (4.61), aplicables tanto a gases, líquidos o sólidos, es decir completamente generales y cuyos resultados son independientes de sí el proceso se efectúa de manera reversible o irreversible. VARIACION DE LA ENTROPIA CON EL VOLUMEN Y LA PRESION La ecuación (4.57) la podemos escribir como: Derivando la ecuación anterior respecto al volumen, a temperatura constante y el resultado derivarlo respecto a la temperatura, obtenemos: Reemplazando las expresiones que en (4.63), podemos deducir De forma similar, si en lugar de considerar la energía interna, consideramos la entalpía, de su definición encontramos: Reemplazando la ecuación anterior en la ecuación (4.62) se deduce que: 53 De tal manera que a temperatura constante, se deduce que: Procediendo de forma similar a como se dedujo la ecuación (4.64) encontramos finalmente: Por consiguiente la variación de entropía con la presión y con el volumen la podemos expresar como: y EJEMPLOS 0 1. En un sistema cerrado se expande isotérmicamente oxígeno de 250 kPa, 80 C a 110 kPa. La masa del oxígeno es 3 kg. Determine la transferencia de calor si a) el proceso es reversible y b)el proceso es irreversible y el trabajo es el 85% del producido por el proceso reversible. Solución: a) El trabajo de expansión lo podemos calcular de la siguiente forma: Reemplazando valores obtenemos: de esta forma: b) En este caso la cantidad de trabajo realizado será menor: 54 2. Una máquina de calor que opera como un ciclo de Carnot, produce 13.5 kJ de trabajo. En 0 tanto que opere entre los límites de la temperaturas 260 y 5 C. Determinar a) la eficiencia y b) la cantidad de calor transferido al sistema. Solución: El sistema lo podemos representar mediante la siguiente figura: a) La eficiencia esta dada por: b) El trabajo realizado es: y por otra parte de acuerdo a la ecuación (4.9) se deduce que: por tanto el calor transferido será igual a: Q = 28.2 kJ 0 3. 100 g de agua se colocan en contacto con un reservorio a 60 C. Calcule el cambio de 0 entropía cuando el agua alcanza los 60 C. Solución El cambio de entropía del sistema donde: ya que Cp del agua es 4.188 kJ/kg K, encontramos que: 55 viene dado por: El cambio entrópico del reservorio será: Por tanto AUTOEVALUACION 0 0 1. 100 g de agua a temperatura de 10 C se mezcla con 1.5 kilogramos de agua a 60 C en un recipiente aislado térmicamente. Determine el cambio total de entropía. 2. Determine el cambio de entropía de un mol de cloro al calentarlo de 400 a 800 K a presión de 1 atm. Use los datos necesarios de las tablas A1 – A3. 0 3. Un trozo de 120 g de cobre a 70 C se introduce en un recipiente con 150 g de agua a 10 0 0 C. Calcule el cambio de entropía en este proceso. Calor específico del cobre, 387 J/kg C 4. En un sistema cerrado, se comprime isotérmicamente nitrógeno de 14 psia, 100 F, a 35 psia. Calcule la transferencia de calor por libra si a) si el proceso es reversible y b) existen irreversibilidades que incrementan la cantidad de trabajo requerido en un 50%. 5. Considere una máquina de Carnot que utiliza aire como sustancia de trabajo. Al inicio de la expansión isotérmica. El aire esta a 550 kPa y el volumen a 5 litros. La presión y el volumen final de la expansión adiabática son de 150 kPa y 15 litros. Determine la eficiencia de la máquina. UNIDAD 2. FUNDAMENTOS DE FISICOQUÍMICA CAPÍTULO 4. POTENCIALES QUÍMICOS Y EQUILIBRIO DE FASES, PARTE 1 POTENCIALES QUÍMICOS. Consideremos ahora dos funciones que consideran en su deducción la función entropía y cuyas aplicaciones están relacionadas con el equilibrio ya sea físico o químico al igual que con la direccionalidad de los procesos. Estas funciones son la función de trabajo o también conocida como energía libre de Helmholtz y la energía libre de Gibbs. LECIÓN 16. TRABAJO Y ENERGÍA LIBRE DE HELMHOLTZ La función de trabajo esta definida como: A = E – TS (5.1) De la ecuación anterior se puede concluir que A debe ser por una parte una propiedad de estado del sistema y a su vez es una propiedad extensiva, es decir depende de la cantidad de materia que posee el sistema. Con el fin de darnos una idea del significado de la función de trabajo, consideremos un proceso isotérmico donde el subíndice 1 indicará el estado inicial y 2 el estado final tal que: 56 de tal forma que el cambio en la propiedad debido al proceso estará dado por: Comparando la ecuación anterior con la ecuación (4.33), podemos escribir: comparando la expresión anterior con la primera ley de la termodinámica, ecuación (2.43): podemos deducir entonces que: Por consiguiente bajo condiciones isotérmicas, la disminución de la función trabajo es una medida del trabajo máximo que se puede realizar en el proceso implicado. LECCIÓN 17. ENERGÍA LIBRE DE GIBBS La energía libre de Gibbs, G, esta definida de acuerdo a la ecuación: O de forma equivalente de acuerdo a las ecuaciones (5.1) y (2.58): respectivamente. De hecho al igual que la función de trabajo, la energía libre de Gibbs es una propiedad de estado, que solo depende de los estados final e inicial, y así mismo es una propiedad extensiva. Consideremos ahora un proceso isobárico, tal que el cambio en la energía libre estará dado por: y si el proceso se realizó también en condiciones termodinámica, resulta: De este modo ya que W rev representa reversible total que se obtiene durante el proceso y PΔV representa el trabajo de expansión, W rev - PΔV representará el trabajo reversible distinto al trabajo de expansión que se puede obtener a partir de un cambio de estado. 57 LECCIÓN 18. CONDICIONES DE EQUILIBRIO De acuerdo con el segundo principio de la termodinámica, el cambio neto de entropía en un proceso termodinámico se verifica por la ecuación: Donde representa la suma de todas las etapas isotérmicas tanto del sistema como de los alrededores. De esta forma si consideramos ahora el primer principio de la termodinámica, podemos escribir, de acuerdo a la ecuación (5.10): Ahora si consideramos cualquier proceso en el que la energía se mantenga constante y además no se ejecute ningún tipo de trabajo, de acuerdo a la ecuación (5.11) se concluye: esta ecuación se puede verificar considerando un proceso donde la única fuerza actuante es la presión externa, realizándose así únicamente trabajo de expansión, PdV, de tal forma que el trabajo será cero cuando dV = 0, es decir el proceso se realiza a volumen constante, por tanto podemos escribir. De este modo si la energía y el volumen se mantienen constantes, la entropía del sistema aumentará en un proceso espontáneo, irreversible y permanecerá invariable cuando mediante un cambio pequeño en el sistema, este permanece en equilibrio. Por consiguiente la entropía de un sistema en equilibrio es máxima a energía y volumen constantes. Examinemos la función de trabajo, considerando las ecuaciones (5.1) y (5.11) se logra obtener: de tal forma que si la temperatura se mantiene constante y se considera que se realiza únicamente trabajo de expansión, PdV , se concluye que o equivalentemente: Esto es, un proceso espontáneo realizado a temperatura y volumen constantes se verifica por una disminución en la función de trabajo. Considerando un argumento similar al anterior, pero esta vez considerando condiciones isobáricas en vez de isocórico, es decir volumen constante, podemos escribir: y de acuerdo a la ecuación (5.5) logramos obtener: 58 De esta forma a presión y temperatura constantes, se logra verificar: Esto es, en un proceso espontáneo realizado a presión y temperatura constantes se evidencia una disminución de la energía libre. Ya que A y G son funciones de estado podemos escribir: donde la desigualdad es la condición para un proceso termodinámico y la igualdad verifica el estado de equilibrio. LECCIÓN 19. PROPIEDADES MOLARES PARCIALES Y POTENCIAL QUÍMICO Consideremos ahora sistemas constituidos por dos o más sustancias, esto es disoluciones y sistemas heterogéneos. Estos últimos constituidos por dos o más fases. Para esto consideraremos el concepto de propiedad molar parcial. Tomemos así, una propiedad molar termodinámica X del sistema, cuyo valor es función de las propiedades presión, temperatura y cantidad de los componentes, es decir: donde n , n ,...,n representan el numero de moles de los componentes 1,2,...,i del sistema. 1 2 i De este modo, una variación en la propiedad X, debida a la variación de las propiedades del sistema, estará representado por: donde la expresión i, y se representa por (5.23) como: se conoce como propiedad molar parcial del componente . De acuerdo a esta última expresión podemos escribir la ecuación Sí tomamos como propiedad termodinámica, la energía libre molar de Gibbs, G, y consideramos la temperatura y la presión constantes, podemos escribir: 59 Si consideramos ahora un sistema con composición constante, n sistema cerrado, e T integramos la ecuación anterior obtenemos: Diferenciando la ecuación anterior, manteniendo T y P constantes, es decir: y comparando las ecuaciones (5.26) y (5.27), se cumple entonces: Si reemplazamos ahora la expresión: , conocido como potencial químico, obtenemos finalmente Esta ecuación, (5.29) se conoce como la ecuación de Gibbs – Duhem y nos muestra que cualquier variación en el potencial químico de uno de los componentes en un sistema a presión y temperaturas constantes afecta a los demás potenciales que constituyen el sistema. Realizando razonamientos similares al anterior podemos llegar a las siguientes expresiones: Es claro entonces que en general para cualquier sistema la variación en la propiedad termodinámica, consideremos por ejemplo la energía libre molar de Gibbs, esta dad por la expresión: y en consecuencia para un sistema cerrado se cumple: 60 Expresión esta que si la comparamos con la ecuación (5.18), podemos deducir que para cualquier sistema en equilibrio con la presión externa se cumple: Así vemos que si consideramos temperatura y presión constantes obtendremos de la anterior expresión la ecuación de Gibbs – Duhem, la cual nos será de gran utilidad para describir aspectos como el equilibrio de fases, en donde si el sistema se encuentra en equilibrio, se debe cumplir: Y por tanto de acuerdo a la ecuación de Gibbs – Duhem, se debe cumplir para todas las fases que constituyen el sistema en equilibrio: EJEMPLOS 1. Calcule en cambio de energía libre para -1 el proceso: -1 Considerando que Cp CO2 (1 atm), es igual a 6.21 cal mol K , es constante en ese rango -1 -1 de temperatura y además que (CO2) = 51.06 cal mol0250CS K . Solución: La energía libre la podemos expresar como: ΔG = ΔH - Δ ( TS) Iniciemos por calcular ΔH: -1 ΔH = Cp ΔT = 6.21 * ( 343 – 298) cal mol = 279.45 cal mol 61 -1 2. Considere la reacción: C H OH 2 5 (g) +O 2 (g) → CO 2 (g) +H O 2 (g) 0 Sabiendo que el cambio entálpico de la reacción a 25 C es y que el cambio en al energía libre es de . Derive las expresiones necesarias para calcular el cambio de 0 energía de la reacción a 1000 C Solución: a) Debemos encontrar primero una expresión para la variación de ΔG con T. Derivando al expresión de ΔG con respecto a T obtenemos O de forma equivalente. Finalmente podemos escribir. Donde T es 298 K y T es 1273 K. 1 2 62 b) Conociendo la anterior expresión, debemos considerar que en un rango tan amplio Cp no puede ser constante, por consiguiente debemos encontrar las ecuaciones que nos relacionan Cp con T, las cuales generalmente tienen la forma. c) El cambio entálpico par la sustancia i estar entonces determinado por: El cambio entálpico de la reacción estará dado por: c) Finalmente obtenemos: 0 Donde es el cambio de energía libre de reacción a 1000 C. AUTOEVALUACION 0 1. La presión de vapor de equilibrio del hielo a – 10 C es 1.95 mm de Hg y la del agua sobreenfriada a la misma temperatura es 2.15 mm Hg. Calcule el cambio de energía libre en 0 calorías que acompaña al paso de 1 mol de agua sobreenfriada a hielo a – 10 C, a la presión total de 1 atm. 0 2. Calcule el calor de combustión del ácido acético a 25 C. 3. Calcular para el ejemplo 2, usando las tablas A1 a A3 4. La variación del volumen de un líquido con la presión está dada aproximadamente por V = V ( 1 – βP ), donde β es el coeficiente de compresibilidad y V0 es el volumen a 0 presión baja (virtualmente cero). Dedúzcase una expresión para el cambio de energía libre que acompaña al cambio isotérmico de un líquido desde la presión P hasta la P . 0 –6 1 –1 2 Para el agua a 25 C, β es 49 x 10 atm a presiones moderadas. Calcúlese el cambio de energía libre en calorías, que acompaña a la compresión de 1 mol de agua desde 1 0 0 atm a 10 atm a 25 C. El volumen específico del agua a 1 atm de presión y 25 C es de 3 –1 1.003 cm mol ¿Existiría una diferencia apreciable en el resultado si se tomara el agua como incompresible? LECCIÓN 20. EQUILIBRIO DE FASES EN SISTEMAS DE UN COMPONENTE EQUILIBRIO DE FASES 63 Una vez estudiada la energía libre de Gibbs, podemos considerar su aplicación al equilibrio de fases, para así establecer la espontaneidad de los procesos cuando se establecen equilibrios físicos, como es el caso que atañe a esta unidad, teniendo en cuenta que las conclusiones que se establezcan serán generales a todos los tipos de equilibrio. SISTEMAS DE UN COMPONENTE Consideraremos en esta unidad sistemas compuestos por dos o más fases de una misma sustancia en equilibrio. Así estudiaremos equilibrios como equilibrio liquido – sólido, equilibrio líquido – vapor, etc. La condición expresada por la ecuación (5.19), a presión y temperatura constantes y la ecuación de Gibbs – Duhem, nos hacen deducir que es posible que ocurra transporte de materia de una fase a otra sin que se altere la condición de equilibrio. Por consiguiente si consideramos G como la energía libre molar de la sustancia en la fase a A y G como la energía libre molar de la sustancia en la fase B, se verifica que cuando A y B B se encuentran en equilibrio se debe cumplir: GA = GB (6.2) Es decir cuando dos o tres fases de la misma sustancia se encuentran en equilibrio, la energía libre molar es la misma para todas las fases, siempre que la temperatura y la presión permanezcan constantes. Si ahora perturbamos el equilibrio, variando la temperatura y la presión, el cambio de la energía libre molar de cada fase, A y B, deberá ser el mismo es decir: y ya que al ocurrir un cambio de fase no hay trabajo distinto al trabajo de expansión podemos expresar el cambio en la energía libre molar de cada fase como: donde V , V y S , S , corresponden a los volúmenes molares y la entalpías molares de los A B A B componentes A y B respectivamente. Reemplazando la ecuación (6.4) en la ecuación (6.3) y reordenando llegamos a la expresión: donde ΔS es el cambio entrópico debido a la transferencia de una mol de sustancia de la fase A a la fase B. Pero ΔS en este caso se hace igual a ΔH / T, por tanto, 64 donde ΔH representa el calor latente molar del cambio de fase que tiene lugar a temperatura T y ΔV es la diferencia de volúmenes molares entre las dos fases. L ecuación (6.6) se conoce como la ecuación de Clapeyron y representa la variación de la presión con la temperatura para dos fases cualesquiera de un sistema dado en equilibrio. De esta forma podemos escribir de manera general: - Equilibrio líquido – sólido (fusión): (6.7) (Si se expresa la propiedad por gramo en vez de mol, se habla entonces de propiedad especifica, volumen especifico, calor especifico de fusión, etc.) - Equilibrio liquido – vapor (vaporización): (6.8) - Equilibrio sólido –vapor ( sublimación): (6.9) - Equilibrio entre dos formas estables: (6.10) Consideremos ahora que la temperatura del sistema esta alejada de la temperatura del punto critico de la sustancia. Bajo esta condición, el volumen del líquido, V l, sera pequeño en comparación al volumen del vapor, VV, a la misma presión y temperatura, es decir, . Por lo tanto podemos escribir la ecuación (6.8) como: y si además el sistema se encuentra a presiones bajas, el vapor entonces se comportará como un gas ideal, PV = RT, la ecuación (6.11) toma la forma: V 65 o de forma equivalente: La expresión (6.13) se conoce como la ecuación de Clausius – Clapeyron. Aunque la ecuación de Clausius – Clapeyron es una ecuación aproximada, ya que por una parte supone comportamiento ideal del vapor y por otra desprecia el volumen del líquido, su valor radica el hecho de poder calcular la relación dp/dT o dT/dp a partir del calor de vaporización o viceversa sin necesidad de conocer los volúmenes molares o específicos del líquido y del vapor, como si lo requiere la ecuación de Clapeyron. Integrando ahora la ecuación (6.13), suponiendo el calor de vaporización independiente de la temperatura (ya que el calor de vaporización no es estrictamente independiente de temperatura, el valor obtenido será considerado como un valor promedio en el rango de temperatura considerado), entre las temperaturas T y T y las presiones de vapor p y p , 1 2 1 2 obtenemos: Ecuación que nos permite calcular el calor de vaporización si conocemos las presiones del líquido a dos temperaturas próximas. Consideremos finalmente la integración general de la ecuación de Clausius – Clapeyron: donde C es una constante . Por consiguiente si tomamos el ln p en función del inverso de temperatura obtendremos una línea recta cuya pendiente será un valor aproximado al valor del calor molar medio de vaporización, ΔH y su valor será solo aplicable cuando se V considera un intervalo pequeño de temperaturas. Cuando se desea entonces ampliar el rango de aplicación, se hace necesario expresar ΔH V como una función de temperatura, es decir: donde ΔH , α y β son constantes. 0 Otra forma de determinar ΔH es a partir de ecuaciones empíricas de presión de vapor en V función de la temperatura, de la forma. En este caso es posible calcular el calor de vaporización a cualquier temperatura. CAPÍTULO 5. EQUILIBRIO DE FASES, PARTE 2, FUGACIDAD Y ACTIVIDAD 66 LECCIÓN 21. EQUILIBRIO DE FASES EN UN SISTEMA CON MÁS DE UN COMPONENTE Consideremos ahora un sistema cerrado en equilibrio constituido por P fases, representadas por las letras a, b, c,...,P y C componentes, indicados por los números 1, 2,...,C, a temperatura T y presión P. Para este sistema, los potenciales químicos (Invitamos al lector a justificar el uso del potencial químico en lugar de la energía libre molar) de los diversos componentes estarán dado por: y si ahora dn moles de los componentes pasan de una fase a otra, a temperaturas y presión constantes, el sistema cerrado permanecerá en equilibrio, por consiguiente se deduce que: Del mismo modo, ya que en el equilibrio la masa total de cada componente debe permanecer constante, se cumple también: y por consiguiente de las ecuaciones (6.19) y (6.20) se deduce: Es decir a presión y temperatura constantes, un sistema en equilibrio, compuesto por varios componentes y coexistiendo varias fases, el potencial químico de cada componente será el mismo en todas las fases (Invitamos al lector a comprobar que si el sistema esta compuesto de un solo componente el potencial químico se hace igual a la energía libre molar). De esta forma, es claro que si el sistema no se encuentra en equilibrio, los potenciales químicos no cumplirán la condición expresada por la ecuación (6.21) y por consiguiente existirá una tendencia espontánea de transporte de masa de la región de la fase de potencial químico mayor a la fase de potencial químico menor hasta alcanzar el estado de equilibrio. REGLA DE LAS FASES 67 Establezcamos ahora el número de variables, grados de libertad, necesarias para definir completamente el sistema. En primer lugar podemos establecer que la composición de una fase que contiene C componentes esta dada por C – 1 términos de concentración, de tal manera que si consideramos P fases , necesitaremos P ( C – 1 ) términos de concentración. En consecuencia, el número total de variables de concentración del sistema es P ( C – 1 ) y sabiendo que la presión y la temperatura son las mismas para todas las fases, el número total de variables, N, necesarias para definir el sistema serán: De acuerdo a la ecuación (6.21), podemos establecer que el número de variables que se fijan automáticamente cuando un sistema se encuentra en equilibrio es igual a P – 1 por cada componente y C ( P – 1 ) variables para los C componentes constituyentes del sistema. De este modo podemos deducir que el número de variables necesarias para definir un sistema en equilibrio esta dado por la expresión: donde F representa el número de variables independientes, presión, temperatura y concentración, necesarios para definir el sistema, o mejor el número de grados de libertad EJEMPLOS 0 - 1. Un líquido hierve normalmente a 400 C y su calor de vaporización es de 14000 cal mol 1 0 . Se desea destilar el líquido a 100 C. Calcular la presión necesaria para efectuar el proceso. Solución. Utilizando la ecuación de Clausius – Clapeyron: e integrándola obtenemos: despejando P , obtenemos: 2 -1 -1 -1 -1 ln (P / P ) = ( (1/373) - (1/673)K ) (14000 cal mol )/(1.9872 cal K mol ) 2 1 P = 6 mm Hg. 2 -1 2. La pendiente de la transición de la fase α a la fase β de un mineral, es – 21 bar K a 600 K. El Δα→βV de la transición es + 0.150 cal bar –1 y 17000 cal mol-1. ¿Cuál es el para la fase β ? 68 de la fase α es de – Solución. De acuerdo a la ecuación de Clapeyron: Realizando los respectivos reemplazos, tenemos: AUTOEVALUACION 0 1. El calor de vaporización del clorobenceno en su punto de ebullición, 132 C, es de 73.4 –1 cal g . Determinar la presión, aproximada, en mm de Hg, bajo la cual el líquido herviría a 0 130 C. –1 0 2. A 110 C , dP / dT para el agua es 36.14 mm de Hg K 3 –1 , los volúmenes específicos ortobaros son 1209 (vapor) y 1.05 (líquido)cm g . Calcular el calor de vaporización del 0 agua a 110. C. 0 0 3. El punto de fusión del benceno aumenta desde 5.5 C hasta 5.78 C cuando se incrementa –1 la presión exterior en 100 atm. El calor de fusión del benceno es de 30.48 cal g Determinar el cambio de volumen por gramo que acompaña a la fusión del benceno. . LECCIÓN 22. CONCEPTOS DE FUGACIDAD Y ACTIVIDAD Consideremos ahora un concepto de gran utilidad para la descripción del comportamiento de lo gases reales y que es aplicado de forma especial a equilibrios en los que intervienen gases a presiones elevadas, este concepto es la fugacidad. LECCIÓN 23. FUGACIDAD Y ACTIVIDAD EN SISTEMAS DE UN COMPONENTE Tomemos como referencia un proceso isotérmico que solo implica trabajo de expansión, de tal forma que para un sistema formado por una mol de gas que se comporta idealmente se cumple: dG = RT dln P ( 7.1 ) Sin embargo ya que esta relación se cumple únicamente a gases que se comportan de manera idea lse ha definido una función f, llamada fugacidad, la cual cumple con la condición: dG = RT dln f ( 7.2) independientemente si el gas se comporta de manera ideal o no. Integrando la ecuación anterior entre dos estado G1 y G2, a temperatura constante se cumple que para una mol de gas: donde f y f corresponden a las fugacidades de los dos estados en los cuales la presión ha 1 2 variado. De acuerdo entonces alas ecuaciones (7.1) a (7.3) podemos concluir: Esto es, la fugacidad de un gas ideal se iguala con la presión cuando esta es muy baja, permitiendo así el calculo de fugacidades a diversas presiones. 69 LECCIÓN 24. FUGACIDAD Y ACTIVIDAD EN SISTEMAS DE MÁS DE UN COMPONENTE Consideremos una mezcla de gases ideales, en donde el potencial químico queda descrito por: Integrando la expresión anterior obtenemos: Sin embargo esta ecuación es solo valida para gases ideales, por tanto para generalizarla debemos hacer uso del concepto de fugacidad, esto es: La expresión (7.7) nos expresa el cambio en el potencial químico para cualquier componente en cualquier sistema, bien sea líquido sólido o gas, puro o mezclado, ideal o no. Así de manera análoga a la ecuación (7.4) podemos escribir que para todos los sistemas, puros o mezclados se cumple: donde x es la fracción molar del componente i. i De esta forma, podemos introducir una nueva función llamada actividad, la cual se define como. donde a entonces nos proporciona un medida de la diferencia entre el potencial químico de una sustancia en el estado de interés y un estado escogido de forma arbitraria y a conveniencia denominado estado estándar. Ya que el proceso se realiza en condiciones isotérmicas la temperatura del estado estándar debe ser la misma del estado de interés aunque las composiciones y las presiones no necesariamente sean las mismas. En términos generales, podemos entonces considerar la fugacidad como una presión corregida la cual para una mezcla de gases ideales es igual a la presión parcial de cada componente. Así entonces si consideramos dos fases en equilibrio A y B podemos escribir para cada una de las fases: y 70 substituyendo las anteriores dos ecuaciones en la condición de equilibrio de fases, ecuación (6.21) encontramos: Ahora podemos suponer dos casos: el primero en el que los estados estándar de las dos fases es el mismo y un segundo caso en el que las dos fases se encuentran a la misma temperatura pero no a la misma presión y composición. En ambos caso se verifica: Esta última ecuación nos muestra que cuando dos o más fases s encuentran en equilibrio las fugacidades de cada una de ellas son iguales. Por consiguiente podemos concluir que el equilibrio de fases se cumple cuando el sistema cumple las ecuaciones (6.21) o (7.13). Finalmente ya que para un gas ideal la relación f / p es igual a la unidad, podemos i i establecer un nuevo concepto, el coeficiente de actividad, definido por la relación: donde γ nos proporciona una medida del grado de idealidad del componente en la mezcla. i LECCIÓN 25. APLICACIONES DE LOS CONCEPTOS DE FUGACIDAD Y ACTIVIDAD 0 1. La presión de vapor del agua a 25 C es 23.76 mm Hg y su volumen específico bajo esas 3 -1 condiciones es de 43400 cm g . a) Calcular la fugacidad del vapor de agua. b) calcule el 0 cambio de energía libre par el paso de 1 mol de agua líquida a 25 C al vapor a fugacidad unidad. Solución. a) Para esto consideraremos la relación aproximada (Invitamos al lector a deducir esta ecuación y justificar su uso): Reemplazando lo valores de presión, y expresando P ideal en términos de volumen específico y presión, obtenemos: b) El cambio en la energía libre estará dado por la ecuación. 71 Pero en este caso f = 1 atm, por tanto, 1 -1 -1 ΔG =RT ln f2= 1.9872 cal K mol *298 ln 23.75/760= - 2052.36 cal mol -1 0 2. La fugacidad del mercurio líquido a 100 C y 1 atm, es 0.272 mm de Hg y su densidad es -3 -1 13.4 g cm , y su peso molecular es 200.6 g mol . Encuentre la fugacidad a la misma temperatura a una presión de 120 atm, asumiendo que el mercurio líquido es incompresible. Solución. Derivando la expresión (7.2) respecto a la presión obtenemos. Sin embargo el lado derecho de la ecuación es igual a V, es decir el volumen molar, por consiguiente podemos escribir: De esta forma tenemos que: Reemplazando se obtiene: f = 0.285 mm Hg 2 AUTOEVALUACION 0 1. Calcule la fugacidad del agua líquida a 100 C, a cuya temperatura y 1 atm de presión el 3 -1 volumen específico del vapor de 1675 cm g referida al estado de referencia usual del gas a presión baja. ¿Cuál es el cambio de energía libre correspondiente al paso de 1 mol 0 de vapor de agua desde 1 atm a 100 C a la fugacidad unidad a la misma temperatura? –1 0 2. La densidad del amoniaco gaseoso a 200 C, y 50 atm de presión es 24 g L . Estime la –1 fugacidad, sabiendo que el peso molecular del amoniaco es 17.03 g mol . 3. Consulte y demuestre que el coeficiente de fugacidad puede relacionarse con el factor de compresibilidad (Para consulta ver J.M. Smith, H.C. Van Ness, and M.M. Abbott, th Introduction to Chemical Engineering Thermodynamics, 5 ed., McGraw – Hill, New York, 1996). 72 CAPÍTULO 6. EQUILIBRIO QUÍMICO En este capítulo consideraremos la forma como podemos relacionar los cambios químicos con la espontaneidad del proceso, es decir la relación entre la constante de equilibrio y la energía libre de Gibbs, como un parámetro para verificar la espontaneidad de una reacción química. Comencemos por considerar la constante de equilibrio. LECCIÓN 26. CONSTANTE DE EQUILIBRIO En general cuando ocurre una reacción química se alcanza el estado de equilibrio cuando las velocidades en ambas direcciones, de este modo el sistema contendrá cantidades de reaccionantes que en muchos casos son insignificantes. En esta unidad consideraremos entonces el concepto de energía libre para considerar este tipo de equilibrios. Iniciemos entonces considerando la constante de equilibrio. Sea una reacción representada por: Ahora, sí consideramos el sentido de la reacción de derecha a izquierda, respecto a al lector, el proceso de reacción se verificara de acuerdo a la ecuación: donde dn representa el número de moles de reactivos consumidos o equivalentemente el número de moles de reactivos formados. De acuerdo a la ecuación de Gibbs – Duhem, cuando el sistema alcanza el equilibrio se cumple: pero debido a que las cantidades dn deben ser proporcionales a los coeficientes i estequiométricos, a, b,...,m, z..., de la reacción se puede escribir la condición de equilibrio como: Pero de acuerdo a la ecuación (5.26), la parte derecha de la ecuación corresponderá al cambio de energía libre debido al proceso. Por consiguiente se concluye que bajo las condiciones de equilibrio: ΔG=0 ( 8.5 ) Así las ecuaciones (8.3) y (8.4) representan la condición fundamental para todos los tipos de equilibrio, sean físicos o químicos. Consideremos ahora el concepto de actividad introducido en la unidad anterior. Reemplazando en la ecuación (7.7) el concepto de actividad, ecuación (7.9), podemos entonces escribir: Donde representa el potencial químico de la sustancia i en el estado estándar elegido (Los estados estándar se definen generalmente en función de la fugacidad unitaria para 73 gases y 1 atm de presión para líquidos, sólidos y soluciones, por consiguiente será independiente de la presión), y a es la actividad de la sustancia i en la mezcla considerada. i Reemplazando la expresión (8.6) en la ecuación (8.4) obtendremos: Por consiguiente podemos escribir De esta forma si la temperatura se mantiene constante, cuando el sistema ha alcanzado el equilibrio la relación de actividades será una constante, es decir: y por consiguiente se deduce: donde K se conoce como la constante de equilibrio del proceso, sea físico o químico. La ecuación (8.10) se conoce algunas veces como ley de equilibrio. LECCIÓN 27. EQUILIBRIO EN SISTEMAS GASEOSOS HOMOGÉNEOS Consideremos un proceso donde intervienen gases únicamente y tomemos como estado estándar, el estado de fugacidad uno, esto es gas ideal a presión de 1 atm. Así bajo estas condiciones la actividad será igual a la fugacidad y de hecho se puede entonces expresar la constante de equilibrio como: Reemplazando en la ecuación anterior, la fugacidad por la expresión para el coeficiente de actividad, obtenemos: Expresión que podemos representar como: Expresión de gran utilidad cuando se realizan procesos a presiones bajas. LECCIÓN 28. EQUILIBRIO EN SOLUCIÓN LÍQUIDA HOMOGÉNEA Para reacciones que tienen lugar en sistemas líquidos homogéneos el coeficiente de actividad se define como: donde X representa la fracción molar del componente i en la mezcla líquida. i 74 Así mismo se suele tomar como estado estándar para cada sustancia el líquido puro a la temperatura del proceso y 1 atm de presión. De esta manera podemos expresar la constante de equilibrio para un proceso en solución líquida como: Sin embargo generalmente empleamos la constante aproximada K es decir: X Muchas veces, cuando el proceso se realice en soluciones diluidas , donde la molaridad, c, es aproximadamente proporcional a la fracción molar, podemos expresar la constante de equilibrio como: LECCIÓN 29. EQUILIBRIO QUÍMICO EN SISTEMAS HETEROGÉNEOS En los equilibrios heterogéneos, gases – sólidos, líquidos – sólidos por ejemplo, es aplicable la ecuación (8.9), sin embargo la situación se simplifica al adoptar la convención universal por la cual se toma como la unidad la actividad de sólidos y líquidos puros (Si el lector se analiza esta convención se dará cuenta que esta en concordancia con los estados estándares. Líquido puro a una atmósfera de presión y sólido puro a una atmósfera de presión). LECCIÓN 30. VARIACIÓN DE LA CONSTANTE DE EQUILIBRIO CON LA PRESIÓN Y LA TEMPERATURA. La ecuación (8.10) la podemos escribir como: derivando la anterior expresión respecto a la presión, deducimos: Resultado que no nos debe sorprender ya que el estado estándar lo hemos definido independiente de la presión, ver nota al pie de pagina 2. sin embargo cabe aclarar que aunque K no dependa de la presión las constantes Kp, K y Kc si depende de la presión. X Consideremos ahora la variación de la constante de equilibrio con la temperatura. Para esto derivando la ecuación (8.19) respecto a la temperatura obtendremos: De acuerdo a la ecuación (5.7) podemos deducir entonces: donde: 75 A la ecuación (8.22) se le conoce como la ecuación de van´t Hoff. Ecuación aplicable a todo tipo de equilibrios, físicos o químicos. De esta forma si integramos la ecuación de van´t Hoff entre dos temperaturas T y T y 1 2 0 suponemos que ΔH es independiente de la temperatura podemos escribir: Donde K y K serán las constantes de equilibrio a las correspondientes temperaturas T y T . 1 2 1 2 de esta manera podemos conocer el calor involucrado en el proceso si conocemos las constantes de equilibrio a las dos temperaturas; o también conocer la constante de equilibrio 0 a cualquier temperatura si conocemos el valor de ΔH . En este caso los resultados obtenidos serán aproximados, debido a que ΔH no es estrictamente independiente de la temperatura. Ahora sí queremos conocer los valores de debemos hacer uso de ecuaciones empíricas del tipo (6.16), es decir: donde Δα, Δβ,...se deducen de las capacidades caloríficas de las sustancias reaccionantes y productos resultantes. Ahora si reemplazamos la expresión anterior en la ecuación (8.22) obtendremos finalmente: donde K puede ser, K , Kp, K o Kc e I es una constante de integración. f X AUTOEVALUACION Para los siguientes ejercicios, utilícense sí es necesario las tablas A1 a A3. 1. Se desea obtener hidrógeno a partir de CO y agua, según la reacción: CO + H O → CO + H (g) 2 (g) 2(g) 2(g) Para lograr esto se tomo CO a 12 atm de presión y vapor de agua a 6 atm de presión en un 0 reactor a 800 C, para extraer CO e H a presiones parciales de 2 atm. Calcule el cambio de 2 2 energía libre, sabiendo que Kp = 0.73. 2. Utilícense los valores de ΔG0 a 25 0C, junto con los datos de capacidad calorífica, en la deducción de una expresión para la variación con la temperatura de la constante de equilibrio de la reacción: SO + 1/2O → SO 2 (g) 2(g) 0 ¿Cual será el valor de K a 500 C? f 76 3 (g) 3. Se deja que una mezcla de dióxido de azufre y oxígeno, en la proporción molar 2:1, 0 alcance el equilibrio a 500 C a una presión total de a) 1 atm; b) 200 atm. Estímese la composición de la mezcla final en cada caso, teniendo en cuenta las desviaciones del comportamiento ideal. 0 4. Cuando H NCOONH 2 4 (s) se introduce en un reactor evacuado a 40 C, la presión del sistema cuando alcanza el equilibrio es de 130 mm Hg, Determinar Kp si se verifica la reacción: H NCOONH ⇔ 2 NH + CO 2 4 (s) 3(g) 2(g) BIBLIOGRAFÍA G.M. Anderson, Thermodynamics of Natural Systems, John Wilwy and sons, Inc, New York, 1996. S, Glasstone, Termodinámica para Químicos, Aguilar, Madrid, 1963. J.B. Jones and R.E Dugan, Ingeniería Termodinámica, Prentice Hall, México, 1997 J. Kestin, A Course in Thermodynamics, Volumes 1 and 2, Series in thermal and fluids engineering, Hemisphere Publishing Corporation, Washington, 1979. K.S. Pitzer, Thermodynamics, 3nd edition, McGraw-Hill, Inc, New York, 1995. J. M. Smith, H.C. Van Ness and M.M. Abbott, Introduction to Chemical Engineering Thermodynamics, 5th ed, Mc. Graw – Hill, New ork, 1996. G.J. Van Wylen, R.E. Sonntang y C. Borgnakke, Fundamentos de Termodinámica, segunda edición, Limusa Wiley, Mexico, 1999. R. C Weast, M. J. Astle and W.H. Beyer (editors), CRC Handbook of Chemistry and Physics, CRC press, Boca Raton, Florida, 2000. 77

![Prueba Segundos2[1]](http://s2.studylib.es/store/data/003397536_1-3ac4e8618b6474fb10e9bb3037bc9dd2-300x300.png)