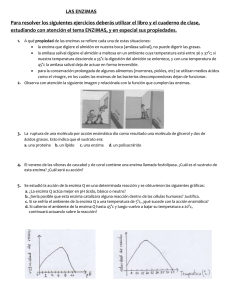
caracterización y expresión recombinante de una celulasa
Anuncio

UNIVERSIDAD DE CHILE FACULTAD DE CIENCIAS FÍSICAS Y MATEMÁTICAS DEPARTAMENTO DE INGENIERÍA QUÍMICA Y BIOTECNOLOGÍA CARACTERIZACIÓN Y EXPRESIÓN RECOMBINANTE DE UNA CELULASA DE ORIGEN ANTÁRTICO MEMORIA PARA OPTAR AL TÍTULO DE INGENIERO CIVIL EN BIOTECNOLOGÍA ROMELA MICHA MARÍN ALVARADO PROFESOR GUÍA: DRA. ORIANA SALAZAR PROFESOR CO-GUÍA: DRA. MARÍA ELENA LIENQUEO PROFESOR INTEGRANTE: DRA. ZIOMARA GERDTZEN SANTIAGO DE CHILE AGOSTO 2007 RESUMEN Las celulasas son enzimas pertenecientes al grupo de las glicosil hidrolasas y son responsables de la degradación de celulosa. Estas enzimas en la actualidad poseen variadas aplicaciones en la industria biotecnológica. Una de las aplicaciones más importantes es en la industria de detergentes. Con el fin de encontrar soluciones a la búsqueda de detergentes más eficientes y económicos en el proceso del lavado, se plantea la producción de celulasas activas a bajas temperaturas en forma recombinante, lo que busca disminuir la temperatura de lavado utilizada actualmente y permitir por lo tanto un ahorro, tanto energético como económico. En el presente Trabajo de Memoria de Título se presenta la purificación, caracterización y producción en forma recombinante de una celulasa de origen marino antártico, denominada Cel3. La enzima se purificó desde el organismo productor nativo mediante un método de tres etapas: precipitación con sulfato de amonio al 90 %, cromatografías de intercambio aniónico y de filtración en gel. En la caracterización se obtuvo que la enzima pura es estable hasta pH 11 y que su pH óptimo de reacción es 8. Su temperatura óptima de reacción es de 40 ºC aprox., decayendo la actividad drásticamente sobre los 50 ºC; es estable frente a un amplio rango de temperaturas (20-45 ºC). Se concluye que esta enzima es adecuada para ser utilizada en procesos de lavado a temperatura ambiente, ya que a 20 ºC retiene un 80 % de actividad, temperatura a la cual las enzimas de organismos mesofílicos prácticamente no poseen actividad. La enzima posee un P.M de 35,7 kDa y un P.I de aproximadamente 4,5. Los parámetros cinéticos KM, VMAX y Kcat son 10,8 mg/mL, 357 U/L y 5,6x10-3 s-1, respectivamente. Con las características encontradas, la celulasa estudiada resulta prometedora para utilizarla como aditivo a detergentes. Por esta razón se procedió a su expresión recombinante, paso importante para una futura y posible comercialización. La secuencia de Cel3 fue obtenida mediante la técnica de Genome Walking, la cual permitió obtener la secuencia completa del gen. Se encontró un marco de lectura abierto (ORF) de 1486 pb, que codifica para un péptido de 494 aa, en donde se identificó la presencia de un péptido señal de 32 aa, un dominio catalítico y un dominio de unión a celulosa, separados por una secuencia aminoacídica no estructurada. 2 Para la expresión recombinante se utilizó el sistema E. coli BL21(DE3)/pET22b(+). Con dicho sistema se obtuvo una celulasa del tipo endoglucanasa, que mostró ser activa a 37 ºC en ensayos en medio sólido. Los objetivos de esta memoria se cumplieron en su totalidad. Los resultados de la purificación y caracterización de Cel3 desde el organismo nativo y su producción en forma recombinante en E. coli son un aporte al conocimiento de las endoglucanasas de origen antártico y sus potenciales aplicaciones biotecnológicas. 3 INDICE DE CONTENIDOS RESUMEN…………... .................................................................................................... 2 INDICE DE CONTENIDOS............................................................................................. 4 INDICE DE FIGURAS .................................................................................................... 6 INDICE DE TABLAS ...................................................................................................... 6 LISTA DE ABREVIATURAS.......................................................................................... 7 LISTA DE ABREVIATURAS.......................................................................................... 7 1 INTRODUCCIÓN ....................................................................................... 8 1.1 ANTECEDENTES GENERALES........................................................................ 8 1.2 ANTECEDENTES BIBLIOGRÁFICOS ............................................................... 9 1.2.1 1.2.2 1.2.3 1.2.4 1.2.5 1.2.6 Características de la Enzima Celulasa. ................................................................ 9 Aplicaciones Industriales de celulasas. .............................................................. 11 Características de microorganismos y enzimas psicrofílicas. ............................. 13 Aplicaciones biotecnológicas de enzimas adaptadas a bajas temperaturas..... 14 Celulasas activas a bajas temperaturas. ............................................................ 15 Expresión de genes psicrofílicos en Escherichia coli.......................................... 16 1.3 DESCRIPCIÓN DEL PROYECTO Y SU JUSTIFICACIÓN .............................. 18 1.4 OBJETIVOS...................................................................................................... 19 1.4.1 1.4.2 2 Objetivo General. ............................................................................................... 19 Objetivos Específicos. ........................................................................................ 19 METODOLOGÍA ...................................................................................... 20 2.1 ESTRATEGIA DE TRABAJO............................................................................ 20 2.2 MATERIALES Y MÉTODOS............................................................................. 20 2.2.1 2.2.2 2.2.3 2.2.4 Cepas Bacterianas. ............................................................................................ 20 Reactivos. .......................................................................................................... 20 Enzimas de Restricción. ..................................................................................... 21 Vectores de clonamiento y expresión. ................................................................ 21 2.3 MÉTODOS........................................................................................................ 22 2.3.1 2.3.1.1 2.3.1.2 2.3.2 2.3.3 2.3.3.1 2.3.4 2.3.4.1 2.3.4.2 2.3.4.3 2.3.4.4 2.3.4.5 2.3.4.6 2.3.5 2.3.6 2.3.7 2.3.8 2.3.8.1 2.3.8.2 2.3.9 2.3.10 2.3.11 3 Obtención de la celulasa producida por bacterias antárticas.............................. 22 Cultivo a 4 y 20 ºC de las bacterias antárticas.................................................... 22 Purificación de la celulasa secretada por la bacteria antártica. .......................... 23 Determinación de Actividad celulasa. ................................................................. 24 Caracterización de la enzima celulasa producida por las bacterias antárticas. .. 25 Determinación de Parámetros Cinéticos. ........................................................... 26 Búsqueda de secuencias río arriba y río abajo del gen codificante para celulasa mediante la tácnica de Genome Walking. .......................................................... 27 Diseño de partidores para la obtención de las secuencias río arriba y río abajo del gen de Cel3. ................................................................................................. 27 Digestión del DNA genómico y elongación......................................................... 28 Construcción del oligo-adaptador. ...................................................................... 28 Construcción de la genoteca-adaptador. ............................................................ 28 Amplificación de los extremos 3’ y 5’ del gen de Cel3. ....................................... 29 Secuenciación y análisis de los extremos 3’ y 5’ del gen codificante para Cel3. 31 Diseño de partidores para el clonamiento del gen codificante para Cel3. .......... 31 Amplificación de DNA mediante PCR................................................................. 32 Digestión de producto de PCR y linearización del vector de expresión pET22b(+). ......................................................................................................... 33 Ligación de fragmentos de DNA a vectores de clonamiento y expresión. .......... 33 Ligación en el vector de clonamiento pGEM-T Easy. ......................................... 33 Ligación en el vector de expresión pET22b(+). .................................................. 34 Clonamiento de los constructos en células E. coli DH5α................................... 34 Clonamiento de los plasmidios recombinantes en células de E. coli BL21(DE3).35 Pruebas de actividad celulasa extracelular......................................................... 35 RESULTADOS Y DISCUSION ................................................................ 36 3.1 CARACTERIZACIÓN BÁSICA DE LA CELULASA EN EL MICROORGANISMO PRODUCTOR.............................................................................................................. 36 4 3.1.1 3.1.1.1 3.1.1.2 3.1.2 3.1.2.1 Obtención de la celulasa producida por bacterias antárticas.............................. 36 Cultivo a 4 y 20 ºC de las bacterias antárticas.................................................... 36 Purificación de la celulasa secretada por bacterias antárticas........................... 37 Caracterización de la enzima celulasa producida por las bacterias antárticas. .. 41 Determinación de Parámetros Cinéticos. ........................................................... 47 3.2 BÚSQUEDA DE SECUENCIAS FALTANTES DEL GEN CODIFICANTE PARA CELULASA ....................................................................................................... 48 3.2.1 3.2.2 3.2.3 3.2.4 3.2.5 Diseño de partidores para la obtención de las secuencias río arriba y río abajo del gen de celulasa. ........................................................................................... 49 Digestión del DNA genómico y elongación......................................................... 50 Construcción del oligo-adaptador y de la genoteca-adaptador........................... 50 Amplificación de los extremos 3’ y 5’ del gen de Cel3. ....................................... 51 Secuenciación y análisis de los extremos 3’ y 5’ del gen codificante para celulasa. ............................................................................................................. 52 3.3 CLONACIÓN DEL GEN CODIFICANTE PARA CELULASA............................ 56 3.3.1 3.3.2 Diseño de partidores para el clonamiento del gen codificante para Cel3. .......... 56 Clonamiento del gen codificante para celulasa en el vector de expresión pET22b(+). ......................................................................................................... 57 3.4 EXPRESIÓN DEL GEN DE LA CELULASA EN EL MICROORGANISMO RECOMBINANTE ............................................................................................. 59 3.4.1 4 5 6 Prueba de actividad celulasa extracelular. ......................................................... 59 A CONCLUSIONES .................................................................................... 62 BIBLIOGRAFÍA ....................................................................................... 64 ANEXOS .................................................................................................. 67 METODOLOGÍA ADICIONAL ........................................................................... 67 A.1 A.2 A.3 A.4 A.5 A.6 A.7 A.8 A.9 Determinación de la concentración de proteínas.......................................................... 67 Extracción de DNA Genómico desde bacterias............................................................ 67 Electroforesis de DNA .................................................................................................. 68 PCR de Colonias.......................................................................................................... 68 Electroforesis de proteínas SDS-PAGE ....................................................................... 69 Zimograma de Celulasas ............................................................................................. 69 Purificación de DNA a partir de geles de agarosa ........................................................ 69 Minipreparación de DNA plasmidial ............................................................................. 70 Preparación células electrocompetentes...................................................................... 70 B Medios de Cultivos y Buffers............................................................................. 71 B.1 Medios Líquidos ........................................................................................................... 71 B.2 Medios Sólidos............................................................................................................. 72 B.3 Soluciones y Buffers..................................................................................................... 73 C Curvas de Calibración....................................................................................... 75 C.1 C.2 C.3 C.4 Curva de Calibración BSA ........................................................................................... 75 Curva de Calibración DNS ........................................................................................... 75 Curva de Calibración Determinación Peso Molecular mediante Filtración por Gel....... 76 Curva Calibración de Crecimiento de Biomasa en Medio Marino al 0,5 % (p/v) CMC 77 D Curva de Linealización según Dixon de curva de saturación ............................ 78 5 INDICE DE FIGURAS Figura 1: Modelo de degradación de celulosa por un complejo natural multicompetente de celulasa. ................................................................................................................... 10 Figura 2: Vectores de clonamiento y expresión utilizados en el trabajo de tesis...................... 22 Figura 3: Diagrama de la técnica de “Genome Walking” empleada para la obtención de la secuencia 5’ y 3’ del gen codificante para la celulasa............................................... 30 Figura 4: Región de Clonamiento del vector de expresión pET22b(+). .................................... 32 Figura 5: Curvas de crecimiento microorganismo productor de la celulasa. ............................ 36 Figura 6: Cromatógrama de la etapa de Intercambio Anionico. ............................................... 38 Figura 7: Cromatógrama de la etapa de Intercambio Anionico. ............................................... 38 Figura 8: Análisis electroforético de las distintas etapas de la purificación de la celulasa........ 39 Figura 9: Zimograma de las distintas etapas de la purificación de la celulasa. ........................ 40 Figura 10: Efecto de la temperatura de reacción en la actividad específica de la celulasa.. .... 42 Figura 11: Estabilidad de la celulasa frente a la Temperatura. ................................................ 43 Figura 12: Efecto del pH de reacción en la actividad específica de la celulasa........................ 44 Figura 13: Estabilidad de la celulasa frente al pH. ................................................................... 45 Figura 14: Cromatógrama de filtración por gel de la celulasa para la determinación de su peso molecular. ................................................................................................................. 46 Figura 15: Isolelectroenfoque de la celulasa nativa.. ............................................................... 47 Figura 16: Curva de saturación de la celulasa. ........................................................................ 48 Figura 17: Partidores para la amplificación de la secuencia 5’ y 3’ del gen codificante para celulasa. ................................................................................................................... 49 Figura 18: AdaptHindIIITotal. ................................................................................................... 50 Figura 19: Análisis electroforético de los productos de PCR para la obtención del extremo 5’ y 3’ del gen codificante para celulasa. ......................................................................... 52 Figura 20: Secuencia Aminoácidica de la celulasa. ................................................................. 53 Figura 21: Análisis electroforético de los productos de PCR para la amplificación del gen codificante para celulasa e inserción de sitios de corte. ........................................... 57 Figura 22: Análisis electroforético de las digestiones de las minipreparaciones. ..................... 58 Figura 23: Ensayo de actividad de celulasa extracelular. ........................................................ 60 Figura 24: Curva de Calibración BSA. ..................................................................................... 75 Figura 25: Curva calibración DNS para la detección de azúcares reductores.......................... 76 Figura 26: Curva calibración P.M en cromatografía de filtración por gel. ................................ 76 Figura 27: Curva de calibración de Biomasa. .......................................................................... 77 Figura 28: Linealización de curva de saturación según Lineweaver-Burk................................ 78 INDICE DE TABLAS Tabla 1: Reactivos utilizados durante el trabajo de tesis.......................................................... 21 Tabla 2: Enzimas de restricción utilizadas durante el trabajo de tesis...................................... 21 Tabla 3: Tabla de Purificación de la Celulasa producida por el microorganismo nativo. .......... 40 Tabla 4: Partidores para la obtención de la secuencia 5’ y 3’ para el gen codificante para celulasa. ................................................................................................................... 50 Tabla 5: Diferencias de residuos entre CelG y la Celulasa....................................................... 55 Tabla 6: Partidores para el clonamiento del gen codificante para celulasa en el vector de expresión pET22b(+).. .............................................................................................. 57 Tabla 7: Buffers utilizados para el ensayo de actividad celulasa.............................................. 74 Tabla 8: Proteínas utilizadas para la construcción de la curva de calibración de peso molecular y sus respectivos pesos moleculares........................................................................ 76 6 LISTA DE ABREVIATURAS 1Kpb Marcador de peso molecular 1Kbp Amp Ampicilina DNA Ácido desoxirribonucleico RNA Ácido ribonucleico RNAr Ácido ribonucleico ribosomal BSA Seroalbúmina de bovino dNTPs Deoxiribonucleótidos trifosfato OD Densidad óptica EDTA Etilendiaminotetraacetato CMC Carboximetil Celulosa CAPS (3-[Cyclohexylamino]-1-propanesulfonic) g Fuerza de gravedad gr Gramo h Hora IPTG Isopropil tio-β-D-galactósido KDa Kilodalton LB Medio Luria-Bertani M Molar MES (2-[N-Morpholino] ethanesulphonic acid) min Minutos mL Mililitros mM Milimolar MOPS (3-[N-Morpholino] propanesulfonic acid) PAGE Electroforesis en gel de poliacrilamida. pb Pares de bases PCR Reacción en cadena de la polimerasa pH Potencial hidrógeno rpm Revoluciones por minuto RNasa Ribonucleasa s Segundo SDS Dodecil sulfato de sodio TAPS (N-tris[Hidroxymethyl]methyl-3-aminopronesulfonic acid) TEMED N, N, N’, N’-tetrametiletilendiamina Tris (TRISMA) Tris-(hidroximetil)-aminoetano U Unidad enzimática UV Radiación ultravioleta V Voltio X-GAL 5-bromo-4-cloro-3-indolil-β-D-galactopiranósido 7 1 1.1 INTRODUCCIÓN ANTECEDENTES GENERALES El extraordinario progreso de la biotecnología industrial desde hace 10 a 20 años, particularmente en biología molecular, ingeniería de proteínas y tecnología de fermentación, ha resultado en el desarrollo de nuevas aplicaciones enzimáticas. Actualmente las áreas industriales en donde se utilizan las enzimas son diversas, tales como en la de los detergentes, la del papel y pulpa, en manufacturera textil, industria del cuero, para la producción de combustible, producción de farmacéuticos, industria de alimentos y alimenticia, cosmética, productos médicos y como herramientas para investigación y desarrollo [1]. La tecnología enzimática es una de las tecnologías básicas de la biotecnología – el gasto mundial de enzimas se cifra en varios miles de millones de dólares anuales. Los procesos además de buscar la rentabilidad industrial y económica, buscan la reducción de los costos sobre el medio ambiente, al disminuir el uso de productos químicos contaminantes [2]. Sumándose a lo anterior, la búsqueda de la disminución de costos en la eficiencia de los pasos de calentamiento/enfriamiento en los procesos biotecnológicos y aumentos en la recuperación de productos a partir de reacciones enzimáticas, hace aumentar el interés en las enzimas de los organismos extremófilos, por ser funcionales en condiciones en que los otros no lo son; por consiguiente, su utilización podría incrementar el rendimiento de los procesos industriales, reducir su costo económico y seguir mejorando las condiciones del medio ambiente [3]. El uso de enzimas como aditivos en los detergentes representa la mayor aplicación de la industria enzimática, ya sea en valor como en volumen [4]. Las enzimas contribuyen a la remoción altamente eficiente de manchas de origen proteico, grasa y almidón, mejorando el rendimiento del proceso de lavado a tiempos de ciclos menores. Entre las enzimas utilizadas como aditivos para detergentes, se enfoca el interés en las celulasas, del tipo endoglucanasas, las cuales marcaron un punto decisivo en la historia de los detergentes por tener la habilidad de no digerir las hebras principales de los tejidos, como el resto de las hidrolasas, si no que digieren la microfibrillas de éstos, dándoles un aspecto a nuevo más duradero y colores nítidos por más tiempo [5]. 8 Actualmente las enzimas utilizadas en los detergentes son del tipo mesófilo, las cuales tienen temperaturas óptimas de catálisis de alrededor de los 60 ºC, lo que obliga un aumento de temperatura del lavado para su óptimo funcionamiento [1]. Con el fin de mejorar este proceso, en términos de una disminución de la temperatura y por consiguiente un ahorro energético, nace el interés en las enzimas producidas por organismos psicrofílicos, microorganismos que habitan en ambientes con temperaturas cercanas a la de congelamiento. En particular los mares antárticos resultan ser una atractiva fuente de organismos que produzcan celulasas eficientes a bajas temperaturas (20 ºC) y resistentes a las altas concentraciones salinas. Para una óptima aplicación de las enzimas adaptadas a bajas temperaturas en la industria de los detergentes, es necesario, su expresión en forma recombinante. La técnica del DNA recombinante es actualmente la principal tecnología para el desarrollo y manufactura de nuevas enzimas, con alta producción de enzimas purificadas a gran escala y a costos competitivos, además de entregar enzimas mejoradas técnicamente en términos de especificidad, pureza, estabilidad y propiedades bioquímicas [1]. Con dichos antecedentes se centrará la investigación de este proyecto en la producción en forma recombinante de una celulasa producida por microorganismos aislados de la región antártica chilena. 1.2 1.2.1 ANTECEDENTES BIBLIOGRÁFICOS Características de la Enzima Celulasa. La celulasa es una glicosil hidrolasa, producida por hongos y bacterias, así como también algunos animales. Esta enzima posee un rol muy importante en la biosfera, ya que es responsable de la degradación de la celulosa, la que comprende la fuente de carbono más abundante en la tierra. Este polímero es un homopolisacárido (compuesto por un solo tipo de monómero) rígido e insoluble, que contiene hasta cientos de miles de unidades de glucosa, unidas mediante enlaces del tipo β-1,4glucosídico. Estas enzimas han sido clasificadas según su actividad en: endoglucanasas, las cuales hidrolizan los enlaces 1,4-β –glucosidicos en forma aleatoria en el interior de la cadena; celobiohidrolasas o exoglucanasas, encargadas de hidrolizar los enlaces 1,4β- glucosidicos, liberando celobiosa desde los extremos no reducidos de la cadena; y β-glucosidasas, que catalizan la hidrólisis de la celobiosa liberando β-D-glucosa. Los 9 dominios catalíticos de las celulasas se encuentran en 16 familias de glicosil hidrolasas, entre las 108 familias existentes en la base de datos de CAZY (Carbohydrate Active Enzymes, www.cazy.org) [6,7]. El proceso de degradación de la celulosa parece ser diferente para distintas especies, especialmente entre microorganismos aeróbicos y anaeróbicos. Microorganismos aeróbicos como Trichoderma reesei secreta una combinación de endoglucanasas y celobiohidrolasas, las cuales atacan su sustrato individualmente pero sinergísticamente. Por otro lado, microorganismos anaeróbicos como Clostridium cellulovorans, Clostridium cellulolyticum, Clostridium Josui, y Clostridium thermocellum, producen complejos multienzimáticos extracelulares que tienen alta actividad degradando celulosa cristalina [8]. La hipótesis más aceptada (Internacional Union of Biochemistry and Molecular Biology, 1992) sugiere que la hidrólisis comienza con endoglucanasas que hidrolizan los enlaces 1,4-β-glucosidicos en la sección amorfa de la celulosa (Figura 1). Esto reduce significativamente el grado de polimerización del sustrato y crea nuevos extremos de cadenas que facilitan el acceso para las celobiohidrolasas que las separan en unidades de celobiosa. Seguido a lo anterior, la β-glucosidasa hidroliza la celobiosa en unidades de glucosa [9]. Figura 1: Modelo de degradación de celulosa por un complejo natural multicompetente de celulasa. CBH I y CBH II (representado como círculos y triángulos, respectivamente) actúan en las extremidades de la cadena de celulosa, mientras que la EG (mostrada como flechas) actúa en el polímero en la zona amorfa de la celulosa (mostrada como las zonas zigzag). β-Glucosidasa (rombos) hidroliza la celobiosa (círculos dobles) a glucosa (círculos simples). 10 La mayoría de las celulasas exhiben una organización multimodular, donde dos dominios funcionales son separados por un enlace flexible. Esta organización resulta importante por aumentar la sinergia entre el dominio catalítico y el dominio de unión a la celulosa (Carbohydrate Binding Domain, CBD) con su sustrato natural, ya que la eliminación del enlace disminuye drásticamente la actividad enzimática sobre la celulosa cristalina [10]. El rol específico de este dominio no está del todo claro pero se le han atribuido un par de funciones: a) participar en la destrucción de la celulosa interfiriendo con el empaquetamiento regular de las fibrillas; b) y/o aumentar la concentración local del dominio catalítico al que están unidos. Se cree también, por resultados experimentales, que dicho dominio es necesario para la secreción de la enzima al medio extracelular [11]. Se han reportado estudios de estructuras tridimensionales que demuestran que en celobiohidrolasas, provenientes de Trichoderma reesei (CBHI y CBHII) existen túneles formados por hojas β paralelas en donde se encuentra el sitio activo, de esa forma la celulosa puede acceder al sitio activo sólo desde uno de sus extremos de la cadena, desde donde la enzima actúa progresivamente, liberando así las unidades de celobiosa. La presencia de este túnel permite una interacción más cercana entre el dominio catalítico y los extremos de la cadena que se encuentran en la superficie de la celulosa cristalina. En contraste a las celobiohidrolasas, las estructuras de las endoglucanasas, en particular el dominio catalítico de EGI, se asemeja más a un pliegue abierto, donde se une el sustrato. Esto les permite interactuar con las zonas amorfas o desordenadas de la celulosa cristalina en forma más eficiente [9]. 1.2.2 Aplicaciones Industriales de celulasas. Las celulasas y enzimas relacionadas son utilizadas en industrias alimenticias, de cervecería, de vino, alimentación animal, textil, de detergentes, del papel, así como también en la agricultura y en investigaciones [12]. Esta gran gama de aplicaciones hacen a estas enzimas muy atractivas y demuestran su enorme valor comercial en la industria biotecnológica. Por ejemplo en la industria del papel, que alcanza ventas de millones de dolares en Estados Unidos y Europa [2], las celulasas y hemicelulasas usadas en etapas de pretratamiento permiten una mejor deslignificación y ahorro en reactivos químicos en la etapa posterior de blanqueamiento [13]. Las celulasas también mejoran la remoción de 11 tintas de papeles de diario viejos, dando mejores resultados que cuando se utilizan químicos [14]. Por otro lado la industria del textil, se ha convertido en los últimos 14 años en una de las principales áreas para las aplicaciones industriales de enzimas. Las celulasas representan el mayor grupo de enzimas en esta industria, considerando el hecho que cerca del 70 % de las fibras procesadas por esta industria son de celulosa. Las aplicaciones más relevantes en esta área son: destinción de prendas para dar apariencia de gastado, tratamiento de las telas para eliminar imperfecciones de las fibras, preparación de la tela previa a tinciones o impresiones para obtener mayor afinidad con los pigmentos y desfibrilación realizada antes y después del teñido [1]. Por otro lado las celulasas poseen diversas aplicaciones en la industria de la comida, entre las cuales se destacan la habilidad de hidrolizar componentes de las paredes celulares, con lo que se disminuye la viscosidad y se mantiene la textura en los jugos de frutas. Además liberan antioxidantes de la fruta, lo que ayuda a controlar enfermedades coronarias. Así mismo, se utilizan celulasas del tipo endoglucanasas junto con xilanasas para la separación de gluten y almidón de la harina, gracias a la hidrólisis de arabinoxilano y almidón [12]. En la industria de la cerveza y el vino las celulasas tienen diversas y atractivas aplicaciones, una de ellas es en el proceso de producción de la cerveza, en donde las celulasas del tipo endoglucanasa son utilizadas para la hidrólisis del enlace 1,4-βglucosídicos de las semillas de cebada, lo que reduce la viscosidad y libera azúcares reducidos durante la primera fermentación. Con esto se obtiene un aumento de eficiencia en dicha etapa, y en la etapa de filtración, y un mejoramiento en la calidad de la cerveza. En la industria del vino, celulasas del tipo β-glucosidasa son utilizadas para el mejoramiento del aroma del vino. Así mismo el uso de endoglucanasas es conocido para el mejoramiento en las eficiencias de las etapas de maceración, clarificación y filtración, así como también para a la obtención de un vino de mejor calidad y estabilidad [12]. El uso de aditivos en los detergentes representa la mayor aplicación en la industria de las enzimas. Las hidrolasas han sido un conjunto representativo de esta industria por más de treinta años, entre ellas se pueden nombrar las proteasas, amilasas, lipasas y celulasas. Éstas últimas han sido utilizadas desde hace 15 años en esta industria. Primero fue una endoglucanasa alcalina de Bacillus sp. KSM-635, incluida en un 12 detergente de formulación tipo compacto por la compañía japonesa KAO en 1987 y más tarde la endoglucanasa V de Humicola insolens, clonada y expresada en Aspergillus oryzae, fue desarrollada como una enzima para detergente con el nombre comercial de Carezyme®. Contrariamente a las otras enzimas utilizadas en detergentes, la celulasa no degrada manchas, si no que es activa en las fibras de celulosa de la ropa, alcanzando una o más de los siguientes efectos: • Remoción de manchas de suciedad atrapada en las fibrillas de celulosa; • Suavidad permanente de la ropa; • Mejoramiento del color por la remoción de las pelusas formadas por las fibrilación; • Desfibrilación con efecto antidepositante en la suciedad. Así, los detergentes principalmente explotan una gran característica del complejo celulasa, esto es como su capacidad de remover fibrillas de celulosa que deshilachan, devolviendo el color y la apariencia original, incluso después de reiterados lavados. Adicionalmente se le suma la cualidad de no dañar el algodón u otros materiales de celulosa de la ropa, manteniendo sin cambios la resistencia a la tensión de la tela. La celulasa más comúnmente utilizada en esta aplicación es la del tipo endoglucanasa, por ser capaz de actuar en las zonas amorfas de las fibras de algodón, por lo que se asume que además contribuye indirectamente a la remoción de suciedad atrapada en la región amorfa de las fibras de algodón. Las celulasas de detergentes deben actuar en un amplio rango de pH, yendo desde pH 6 a hasta alrededor de 10, deben también ser resistentes a surfactantes aniónicos, condiciones de blanqueamiento, durante el tiempo que dura el lavado, así como también durante el periodo de almacenamiento [1,5]. 1.2.3 Características de microorganismos y enzimas psicrofílicas. Organismos adaptados a ambientes fríos o psicrofílicos crecen a temperaturas cercanas al punto de congelamiento y han colonizado exitosamente habitats fríos, tales como regiones polares, mares profundos, zonas montañosas altas, etc. Este tipo de zonas constituyen más de tres cuartos de la superficie terrestre y están expuestas a temperaturas en forma permanente de alrededor a 5 ºC [15]. El poder crecer en este 13 tipo de ambiente le atribuye a los microorganismos psicrofílicos la ventaja de haber enfrentado dos desafíos: primero, la baja temperatura, ya que cualquier disminución en la temperatura afecta exponencialmente la tasa de reacciones bioquímicas (Ecuación de Arrhenius, k=Ae-Ea/RT, donde A es el factor pre-exponencial, Ea es la energía de activación, R la constante de los gases y T la temperatura absoluta en Kelvin); y segundo, la viscosidad de ambientes acuosos, que aumenta por un factor mayor que el doble entre 37 y 0 ºC [16]. A pesar del efecto negativo de las bajas temperaturas en las reacciones bioquímicas, estos organismos respiran, crecen y se mueven a tasas similares a aquellos que viven en ambientes temperados. Ellos, por lo tanto, han desarrollado varias adaptaciones del tipo estructural al nivel de, por ejemplo, sus membranas, proteínas y enzimas constitutivas, permitiéndoles compensar los efectos negativos de la temperatura [17]. Las enzimas activas a bajas temperaturas poseen una característica en común: todas tienen una eficiencia catalítica alta, asociada sin embargo, a una baja estabilidad frente a la temperatura. En las mayorías de los casos, la adaptación al frío es alcanzada a través de una reducción en la energía de activación que posiblemente origina un aumento en la flexibilidad, de ya sea una área en particular, como de toda la proteína. Esta plasticidad mejorada parece inducir una estabilidad termal débil de las enzimas adaptadas a bajas temperaturas. Este balance entre estabilidad y flexibilidad representa uno de los puntos cruciales en la adaptación de la proteína a diferentes temperaturas [18]. La flexibilidad es alcanzada por una combinación de factores estructurales, entre los cuales se pueden incluir una reducción de centros hidrofóbicos, un menor número de interacciones iónicas y electroestáticas, un aumento de residuos cargados en la superficie que promueven el aumento de la interacción con el solvente, loops en las superficies adicionales, sustitución de residuos de prolina por glicinas en los loops de las superficies, una disminución en la razón arginina/lisina, menos interdominios con interacciones entre subunidades y menos interacciones aromáticas [19]. 1.2.4 Aplicaciones biotecnológicas de enzimas adaptadas a bajas temperaturas. La síntesis de productos farmacéuticos, químicos especializados, agroquímicos y otros, está usualmente amparada por procesos de producción de alto costo, y que además sufren de baja selectividad y de la generación de productos secundarios no 14 deseados. Las enzimas mesofílicas no se adaptan, debido a las duras condiciones requeridas en los procesos industriales, esto último debido a su falta de estabilidad. Las enzimas adaptados al frío producidas por microorganismos psicrofílicos despliegan una eficiencia catalítica alta a bajas temperaturas (20 ºC) que ofrece un potencial considerable para la industria biotecnológica, como por ejemplo, en la industria de detergentes y de comida, así como también en la producción químicos finos [20]. Las dos propiedades de las enzimas psicrofílicas que tienen aplicación biotecnológica más obvia, son su alta actividad catalítica a temperatura ambiente y su baja termoestabilidad a temperaturas elevadas (sobre los 45 ºC). El principal beneficio es que el uso de este tipo de enzima conlleva un ahorro energético, ya que no requieren etapas de calentamiento, las cuales resultan ser costosas; funcionan en ambientes fríos y durante la época de invierno; además de lo anterior proveen rendimientos de reacción mejorados, se acomodan a altos niveles de estereoespecifidad, minimizan reacciones químicas no deseadas que pueden ocurrir a temperaturas más altas y al ser termolábiles se pueden inactivar fácilmente cuando el proceso lo requiera. La propiedad de inactivación al calor tiene particular importancia en la industria alimenticia, donde es importante prevenir cualquier modificación del sustrato, el que puede ser sensible a cambios de temperatura, así como también para etapas de proceso en secuencia (biología molecular), donde la acción de enzimas necesita ser terminada antes de la siguiente etapa. Con enzimas adaptadas a bajas temperaturas esto puede ser llevado a cabo por inactivación por calor en lugar de usar el método de extracción con químicos. Ejemplos de aplicaciones comerciales y aplicaciones biotecnológicas han sido listados y mencionados en literatura relacionada [19]. 1.2.5 Celulasas activas a bajas temperaturas. Hasta el momento se han publicado sólo dos celulasas activas a bajas temperaturas del tipo endoglucanasa. Ambas son provenientes de bacterias identificadas como Pseudoalteromonas [21, 22].La primera de ellas fue aislada de una bacteria antártica, la cual fue identificada como Pseudoalteromonas haloplanktis. El gen codificante para esta enzima (CelG) fue clonado, secuenciado y expresado en E. coli [21]. El segundo caso corresponde a una celulasa, denominada CelX, que fue aislada desde una bacteria psicotrópica, identificada como Psudoalteromonas sp. DY3, la cual fue extraída de sedimentos profundos del océano pacífico. Al igual que el caso anterior el 15 gen codificante para dicha celulasa fue clonado, secuenciado y expresado en E. coli [22]. Se encontró que ambas enzimas están fuertemente relacionadas en estructura con la celulasa Cel5 de Erwinia chrysanthemi. Los resultados experimentales muestran a ambas enzimas como celulasas neutras, pero siguen siendo óptimamente estables a valores de pHs entre 8 y 9,5 para el caso de CelG, y 9 y 10 en el caso de CelX. La actividad de ambas enzimas aumenta exponencialmente en temperaturas hasta los 40 ºC, pero sobre los 45 ºC se desvanece [21, 22]. En cuanto a la semejanza de las secuencias a nivel de aminoácidos, CelX presenta un 95 % de identidad con CelG. Las dos enzimas poseen 5 residuos en el dominio catalítico que son estrictamente conservados entre las glicosil hidrolasas de la familia 5, por lo que sus dominios catalíticos fueron clasificados dentro de ésta familia. Además de estos dos casos, existe la publicación de una cellobiohidrolasa (cbh 1) aislada de Penicillium chrysogenum FS010, extraido del mar Huanghai. El gen codificante se secuenció, se clonó y se expresó en Saccharomyces cerevisiae H158. Se encontró homología con celobiohidrolasas de hongos pertenecientes a las glicosil hidrolasas de la familia 7 y se determinó que su actividad óptima de reacción en el extracto crudo fue de 35 ºC, ya que no se ha caracterizado en forma purificada [23]. A pesar de que los usos y aplicaciones de celulasas, así como también la producción en forma recombinante de genes relacionados, han sido ampliamente investigados y reportados, queda de manifiesto que el caso particular de celulasas adaptadas a bajas temperaturas todavía está poco estudiado, y sigue abierto a nuevas investigaciones. Es una razón más para ahondar en este tema, en donde las aplicaciones asociadas son de gran importancia. 1.2.6 Expresión de genes psicrofílicos en Escherichia coli. La fermentación a gran escala de microorganismos psicrofílicos sufre de dos grandes inconvenientes: la baja tasa de producción por las cepas salvajes y los altos costos de crecer microorganismos a bajas temperaturas. Una alternativa a lo anterior, es sobreexpresar el gen codificante para una proteína activa a baja temperaturas en un huésped mesofílico. 16 Se ha encontrado que las propiedades de una enzima adaptada a bajas temperaturas son preservadas cuando es expresada en un huésped mesofílico, siempre y cuando se provea una temperatura lo suficientemente baja para permitir un correcto plegamiento y evitar una denaturación irreversible, y a su vez una temperatura compatible con el apropiado crecimiento del huésped y funcionamiento de la maquinaria de expresión del mismo. Para llevar a cabo lo anterior se propone como organismo huésped a la bacteria Gram-negativa Escherichia coli, debido a que es capaz de producir en forma rápida y económica proteínas recombinantes [24]. Uno de los ejemplos de expresión recombinante de proteínas psicrofílicas en un huésped mesofílico es el caso de α-amilasa de Pseudoalteromonas Haloplanktis, la cual fue expresada en E. coli. El sistema de expresión fue cultivado a una temperatura de 18 ºC [25]. Además de lo anterior, como se mencionó en el punto 1.2.5 las dos únicas celulasas activas a bajas temperaturas del tipo endoglucanasa descritas a la fecha fueron expresadas en E. coli. Es por esto, que se cree que el utilizar como organismo huésped E. coli, promete resultados exitosos en la expresión de una celulasa aislada de bacterias antárticas. 17 1.3 DESCRIPCIÓN DEL PROYECTO Y SU JUSTIFICACIÓN En los últimos años diversos grupos de investigación han concentrado el interés en enzimas adaptados a bajas temperaturas (eficiencia catalítica alta a temperatura ambiente, 20 ºC) producidas por microorganismos psicrofílicos, las cuales ofrecen un potencial considerable en la industria biotecnológica. Una de las aplicaciones más importantes es la industria de los detergentes, en donde el aporte de este tipo de enzimas se asocia a una disminución de gasto energético, ya que su eficiencia a baja temperatura permite la remoción selectiva de manchas en la ropa a temperaturas de lavado menores. Por ello producir una celulasa activa a baja temperatura en forma recombinante, enzima ampliamente utilizada en los detergentes, resulta un problema de vanguardia, en donde este proyecto pretende aportar a la solución. En este proyecto se pretende purificar, caracterizar, clonar y expresar una celulasa psicrofílica producida por microorganismos aislados de la antártica chilena, etapas que resultan ser indispensables para una posterior producción a nivel industrial. A partir de una muestra de microorganismos de origen antártico, se han realizado pruebas en el CIByB (Centro de Ingenieria Bioquímica y Biotecnología), que muestran la producción por parte de ellos de enzimas con actividad de celulasa, en particular actividad del tipo endoglucanasa, de diversas características. A partir de dichos resultados se identificó una cepa con potenciales características compatibles para una posterior aplicación en detergentes. Como primer paso se propone una purificación y caracterización breve de la celulasa nativa. Gracias a la caracterización se comprobará si la celulasa tiene efectivamente un perfil adecuado para utilizarla como aditivo a los detergentes, de ser así pasa a convertirse en una enzima atractiva para producir en mayores cantidades. Para ello es necesario obtener una celulasa expresada en forma recombinante en un sistema de fácil manejo y que permite producir grandes cantidades de la enzima en forma más económica. Con el fin de obtener al gen de la celulasa en un sistema recombinante, se considera la secuenciación del gen codificante para celulasa mediante PCR, su posterior clonamiento en el vector de expresión pET22b(+) y su respectiva expresión recombinante en células E.coli BL21(DE3). 18 Con las actividades contempladas en el proyecto se espera ofrecer una celulasa recombinante activa a bajas temperaturas compatible para el uso en la industria de detergentes, y así contribuir en la meta global de ofrecer a la sociedad detergentes comerciales eficientes a temperaturas menores de lavado. 1.4 OBJETIVOS 1.4.1 1.4.2 Objetivo General. Producir una celulasa activa a bajas temperaturas en forma recombinante. Objetivos Específicos. Estudiar las propiedades de una celulasa proveniente de un microorganismo psicrofílico. Diseñar una estrategia de clonación. Llevar a cabo la clonación de los genes de la enzima. Expresar el gen de la celulasa en el microorganismo recombinante. 19 2 METODOLOGÍA 2.1 ESTRATEGIA DE TRABAJO Se trabajó con una cepa bacteriana, aislada desde la región antártica chilena, que crece eficientemente a 4 ºC. La cepa fue seleccionada por expresar una celulasa en el sobrenadante del cultivo, del tipo endoglucanasa, con características de actividad a pH alcalinos y con buena termoestabilidad, características que fueron encontradas en ensayos preliminares. Dichas características resultan atractivas para una celulasa recombinante que se quiera comercializar en la industria de los detergentes. Esta enzima será denominada Cel3, ya que es la celulasa secretada por la cepa 3 de los microorganismos existentes en el laboratorio. La estrategia de trabajo, consistió en encontrar las condiciones que mejor permitan producir volúmenes adecuados para una producción de enzima celulasa nativa, para luego purificarla y caracterizarla. Se buscó el gen que codifica para la enzima mediante la técnica Genome Walking. El gen encontrado que codifica para la enzima celulasa fue clonado en el vector de expresión pET22b (+) y la enzima se expresó en las células BL21 (DE3) de E. coli. A continuación se muestra con mayor detalle la metodología utilizada: 2.2 MATERIALES Y MÉTODOS 2.2.1 Cepas Bacterianas. E. coli cepa DH5α, genotipo: F- Φ80dlacΔM15 Δ(lacaya-argF) U169 recA1 endA1 hsdR178rk-,mk+) phoA supE44 λ- thi-1 gyrA96 relA1. 2.2.2 E. coli BL21 (DE3), genotipo: F- ompT hsdSB (rB-mB+) gal dcm (DE3). Reactivos. Los reactivos utilizados durante el trabajo de tesis y el laboratorio proveedor se detallan en la Tabla 1. 20 Tabla 1: Reactivos utilizados durante el trabajo de tesis. Laboratorio Promega (WI-USA) Invitrogen (CA-USA) Sigma (MO-USA) Supelco Pharmacia Biotech Fermentas Disco (DT- USA) New England Biolabs (MA-USA) Novagen, Madison (WI-USA) Qiagen Winkler J.T Backer Fermelo Merk 2.2.3 Reactivos Enzimas de restricción HindIII, BamHI y PvuII. Taq DNA polimerasa, pGEM-T Easy T4 DNA ligasa, estándar de tamaño molecular 100 Kb, dNTPs, cepa E. coli DH5α X-GAL, IPTG, Reactivos MES, MOPS, TRIZMA, TAPS, CAPS Columna cromatográfica, SigmaChromTM GFC100 Columna Q-Sepharose HR 5/5 Estándar de tamaño molecular preteñido de proteínas. Triptona, extracto de levadura, medio LB, agar. Enzima de restricción XhoI, Nde I. pET22b(+), cepa E. coli BL21 (D3E). Los sistemas de purificación de productos de PCR, de DNA plasmidial y de extracción de DNA desde geles de agarosa. Tris, SDS, Glicerol Cloruro de sodio. Agarosa El resto de las sales, ácidos, bases, bromuro de etidio y solventes de grado analítico o de grado biología molecular. Enzimas de Restricción. En la siguiente tabla se detallan las enzimas de restricción utilizadas y el tipo de corte que producen. Tabla 2: Enzimas de restricción utilizadas durante el trabajo de tesis. Enzima Tipo de Corte Sitio de Reconocimiento (5’-3’) Romo CAGCTG PvuII AAGCTT HindIII Cohesivo Cohesivo CATATG NdeI Cohesivo CTCGAG XhoI 2.2.4 Vectores de clonamiento y expresión. Con el fin de obtener un alto número de copias de fragmentos de DNA, se utilizó el vector de clonamiento pGEM-T Easy. Este vector se caracteriza por poseer una timidina en el extremo 3’ de cada una de las hebras en el sitio de inserción lo que permite la ligación de segmentos de DNA obtenidos por PCR que tienen una adenina 21 en el extremo 3’ de cada hebra debido a la acción de la Taq polimerasa. Para la expresión de la proteína recombinantes en E. coli se utilizó el vector pET22b(+), el cual produce una proteína recombinante unida por su extremo carboxi terminal a un péptido de 6 histidinas que permite su posterior purificación (Figura 2). Figura 2: Vectores de clonamiento y expresión utilizados en el trabajo de tesis: F1 ori: origen de replicación de DNA de hebra simple derivado del bacteriofago F1. Ampr: gen de resistencia a ampicilina, T7: promotor para T7 polimerasa, LacI: gen que codifica el represor lac, lacZ, región codificante para la β-galactosidasa interrumpida por el sitio de clonamiento. Ap: secuencia codificante para bla, contiene la región codificante para la β-galactosidasa interrumpida por el sitio de clonamiento. (A) pET-22b(+), contiene una secuencia codificante para un péptido de polihistidinas y el promotor y terminador T7. (B) Vector pGEM-T Easy posee una timidina en el extremo 3’ de cada una de las hebras en el sitio de inserción para la ligación de segmentos de DNA obtenidos por PCR. 2.3 MÉTODOS 2.3.1 Obtención de la celulasa producida por bacterias antárticas. 2.3.1.1 Cultivo a 4 y 20 ºC de las bacterias antárticas. Se tomó un inóculo de la reserva de la cepa 3 de bacterias antárticas de placas con medio Marino 2216 (Anexo B.2 parte c), conservadas a 4 ºC, y se traspasó a un cultivo de 5 mL de medio Marino 2216 al 0,5 % (p/v) Carboximetil Celulosa (CMC) estéril (Anexo B.1 parte d). Este cultivo se incubó con agitación orbital a 4 ºC durante 4 días. Se transfirió 100 µL de este cultivo a 2 matraces con 100 mL del mismo medio y se incubaron a 4 y 20 ºC respectivamente, ambos con agitación orbital. Para medir el crecimiento celular y la actividad celulasa del sobrenadante a cada cultivo, se tomaron muestras de 1 mL cada 2 horas en condiciones de esterilidad. Para la medición de biomasa se midió la densidad óptica a 600 nm, la cual se encuentra relacionada con la concentración de biomasa por curva de calibración (Anexo C.4). Se realizó un ensayo de actividad celulasa en medio líquido, el que consiste en la determinación de azúcares reductores liberados a partir de la hidrólisis del sustrato soluble CMC. Para la 22 cuantificación de los azúcares reductores se utilizó ácido dinitrisalisílico (DNS). El producto proveniente de la reducción de DNS (Anexo B.3 parte c) se midió por espectrofotometría a 550 nm en un lector de placas. El ensayo consistió básicamente en los siguientes pasos: 1. Se centrifugó 1 mL de cultivo a 13.000 rpm por 5 minutos. (Centrifuga Eppendorf 5403 de Eppendorf AG, Hamburgo, Alemania). 2. 50 ul de sobrenadante se mezclaron con 50 µL de carboximetil celulosa (CMC) al 1 % (p/v), disuelta en buffer Etanol Amina 25 mM pH 10. Se incubó la muestra a 37 ºC por 45 min. con 700 rpm de agitación. 3. A 50 ul de la reacción se adicionó 50 µL de DNS y se procedió a incubar a 100 ºC por 10 min. 4. Se incubó un par de minutos en hielo, para luego traspasar 50 µL de muestra a una micro placa Falcon de 96 pocillos. 5. Se leyó la absorbancia a 550 nm en un lector de placas (Anthos 2010). 2.3.1.2 Purificación de la celulasa secretada por la bacteria antártica. Se cultivó el inoculo de la bacteria de acuerdo a lo descrito en el punto 2.3.1.1. Se transfirieron 500 µL de este cultivo a 500 mL del mismo medio, los cuales se incubaron a 4 ºC durante 4 días. Posteriormente el cultivo se centrifugó por 30 min a 8.500 rpm a 4 ºC (centrifuga Sorvall® RC-28S y rotor GS-3 Sorvall®, DuPont, CT, USA). El sobrenadante se precipitó con sulfato de amonio (NH4)2SO4 al 90 % (p/v). La mezcla se agitó durante 1 hora en hielo. Se dejó reposar por 30 min y se centrifugó a 8.500 rpm por 45 min (centrifuga Sorvall® RC-28S y rotor GS-3 Sorvall®, DuPont, CT, USA). El sedimento se resuspendió en 3 mL de buffer Tris-HCl 20 mM pH7, para ser dializado durante toda la noche a 4 ºC incubado en el mismo buffer de resuspensión. La muestra dializada fue sometida a purificación cromatográfica. En una primera etapa se realizó una cromatografía de intercambio aniónico y luego una filtración por gel. Para la cromatografía de intercambio aniónico (IEX) se usó una columna Q-Sefarosa (Column HR 5/5, Pharmacia Biotech, de dimensiones 5 cm x 5 mm, Resina QSepharose Fast Flow, Code Nº 17-0510-01) de 1 mL y un flujo de 0,5 mL/min, con un gradiente escalonado creciente de Buffer B, correspondiente a Tris 20 mM 1 M NaCl 23 pH 7, en donde el primer gradiente fue hasta un 15 % con una largo de 5 volúmenes de columna; el segundo gradiente aumentó la sal hasta un 35 % en 20 volúmenes de columna y finalmente el tercero terminó en un 100 % de sal en 5 volúmenes de columna. Se buscó actividad celulasa en las fracciones, siguiendo el procedimiento antes descrito para el ensayo de actividad (sección 2.3.1.1), usando en este caso 50 µL de muestra proveniente de cada una de las fracciones cromatográficas. Las fracciones que presentaron actividad celulasa positiva, se juntaron y fueron concentradas en Centriplus (Amicom), los cuales fueron centrifugados durante 6 h a 4.800 rpm (centrífuga Sorvall® RC-28S y rotor F-24 Sorvall®, DuPont, CT, USA) lográndose una concentración de 10 veces. En la filtración por gel se utilizó una columna Supelco GFC 100 (Supelco, SigmaChromTM, Gel Filtration HPLC Column, 13,25 mL de 300 x 7,5 mm) con un flujo de elusión de 0,08 mL/min al 100 % de buffer Tris 50 mM 0,1 M NaCl pH 7,5 en 1,5 volúmenes de columna. Los peacks de interés obtenidos en las cromatografías fueron analizados en electroforesis en gel de poliacrilamida 12,5 % (p/v) en condiciones denaturantes para determinar la pureza de las fracciones, según el protocolo descrito en el Anexo A.5 [26]. También se analizaron mediante zimogramas, siguiendo el protocolo descrito en Anexo A.6. Para la cuantificación de la concentración de proteínas totales se utilizó el método de Bradford de acuerdo a lo descrito en Anexo A.1. Las fracciones con mayor grado de pureza se mantuvieron a 4 ºC para su posterior uso. 2.3.2 Determinación de Actividad celulasa. A partir de muestras de celulasa purificada según el protocolo descrito en 2.3.1.2, se determinó la cantidad de azúcares reductores liberados a partir de la hidrólisis de un sustrato soluble CMC. Para la cuantificación de los azúcares reductores se utilizó DNS. El producto proveniente de la reducción de DNS se midió por espectrofotometría a 550 nm. Los pasos del ensayo estándar de actividad en medio líquido son los siguientes: 1. 75 µL de celulasa (0,005 mg/mL) en buffer pH 7 (Anexo B.3 parte l) se mezclaron con 75 uL de buffer pH 7 al 1 % de CMC. 2. A tiempo 0 min se toma una alícuota de 50 µL de la reacción. Se adicionó 50 uL de DNS y se procedió a incubar a 100 ºC por 10 min. 24 3. Se incubó un par de minutos en hielo, para luego traspasar 50 uL de muestra a una micro placa Falcon de 96 pocillos. 4. Se leyó la absorbancia a 550 nm en un lector de placas (Anthos 2010). 5. Se incubó la muestra restante a 20 ºC por 40 min con 700 rpm de agitación. 6. Al tiempo 40 min a 50 uL de la reacción se adicionó 50 uL de DNS y se procedió a incubar a 100 ºC por 10 min. 7. Se incubó un par de minutos en hielo, para luego traspasar 50 uL de muestra a una micro placa Falcon de 96 pocillos. 8. Se leyó la absorbancia a 550 nm en un lector de placas (Anthos 2010). Se calculó la actividad celulasa con la siguiente relación y la curva de calibración de DNS (Anexo C.2). Actividad = ( ( Abs tiempo 40 − Abs tiempo 0 ) − n) t rx ) × 1000 m (1) En la formula (1) n y m corresponden al intercepto y la pendiente de la curva de calibración respectivamente (Anexo C.2), trx al tiempo de duración de la reacción, y se multiplica por 1000 para obtener la actividad en µmoles/(min·L). Considerando la concentración de proteínas de cada muestra se calculó la actividad específica. La unidad enzimática se definió como la cantidad de enzima necesaria para liberar 1 uMol de glucosa, desde el sustrato utilizado, por minuto, a 20 ºC, de la siguiente forma: Actividad ⋅ Específica = Actividad [Pr oteína]Total (2) De esta forma la formula (2) entrega el valor de Actividad Específica en [U/mg]. 2.3.3 Caracterización de la enzima celulasa producida por las bacterias antárticas. Se analizó el efecto de la temperatura y del pH sobre la actividad específica. El ensayo de actividad se realizó a diferentes temperaturas y distintos valores de pH (el buffer con CMC al 1 % (p/v) se preparó a diferentes pH, Anexo B.3 parte l). Además se analizó la estabilidad de la celulasa frente a la temperatura y el pH. Para ello la enzima purificada se preincubó a diferentes temperaturas durante 10 min y posteriormente se midió la actividad residual a la temperatura máxima de actividad. A 25 su vez la estabilidad frente al pH se estimó preincubando 24 horas a 4 ºC la celulasa en soluciones con diferentes pH y se realizó posteriormente el ensayo de actividad estándar al pH óptimo de actividad, para finalmente calcular la actividad residual al pH máximo de actividad. La actividad relativa se calculó de la siguiente forma: Actividad Re lativa = Actividad Re sidual ActividadMáxima (3) Donde la Actividad Residual corresponde a la actividad obtenida después del ensayo de actividad habiéndola incubado previamente, ya sea a diferentes temperaturas o a soluciones a distintos pHs. La Actividad Máxima corresponde a la mejor actividad de la enzima obtenida en el ensayo estándar con la pre-incubación. Se determinó la masa molecular con cromatografía de filtración por gel en una columna Supelco GF100, relacionando el volumen de elusión a su peso molecular con la curva de calibración (Anexo C.3), realizada con proteínas de peso molecular conocidos. El P.I se determinó mediante isoelectroenfoque en el equipo PhastSystem (Pharmacia Biotech) con un gel PhasGelTM IEF 3-9 (Amersham Pharmacia Biotech AB, Suecia), siguiendo las instrucciones del proveedor para el procedimiento “Sensitive Silver Stain” (Ficha Nº210). 2.3.3.1 Determinación de Parámetros Cinéticos. Para la determinación de los parámetros cinéticos se utilizó el ensayo de actividad descrito anteriormente, pero con diferentes concentraciones de CMC, las cuales se encontraban en el rango de 0,1 a 10 mg/mL. Se consideró que la enzima seguía un comportamiento de Michaelis Menten. Se utilizó la linealización descrita por Dixon, la cual grafica s/v vs. s, en que s es la concentración de sustrato y v corresponde a la velocidad de reacción. La ecuación de la recta queda descrita como sigue: ⎛ s ⎞ ⎛⎜ K m ⎜ ⎟=⎜ ⎝ v ⎠ ⎝ Vmax ⎞ ⎛ 1 ⎟⎟ + ⎜⎜ ⎠ ⎝ Vmax ⎞ ⎟⎟ × s ⎠ (4) 26 La pendiente de la recta queda determinada por el coeficiente 1/VMAX y el intercepto con el eje y corresponde al inverso de KM/VMAX, además la prolongación de la curva, hasta que s/v es igual a cero, corresponde a -KM. 2.3.4 Búsqueda de secuencias río arriba y río abajo del gen codificante para celulasa mediante la tácnica de Genome Walking. La técnica de Genome Walking estandarizada en el laboratorio (CIBYB, Facultad de Ciencias Físicas y Matemática, Universidad de Chile), consiste básicamente en amplificar secuencias adyacentes de DNA genómico a segmentos de secuencias conocidas. Esta técnica se puede resumir en 5 etapas importantes [26]: 1. Diseño de partidores y adaptadores para la obtención de la secuencia río arriba y río debajo de un fragmento del gen de celulasa ya secuenciado. 2. Digestión del DNA genómico con enzimas de restricción, las cuales deben producir extremos romos o cohesivos con el extremo 5’ saliente, para luego elongar los fragmentos producidos, mediante el uso de la Taq DNA polimerasa que agrega una adenina a los extremos 3’ de cada fragmento. 3. Construcción de un oligo-adaptador de doble hebra que contiene una timidita no apareada en un extremo 3’. 4. Ligación del oligo-adaptador con los fragmentos de DNA, con lo que se obtiene una genoteca-adaptador. 5. Amplificación de cada uno de los extremos. A continuación de detalla la metodología de cada uno de las etapas. 2.3.4.1 Diseño de partidores para la obtención de las secuencias río arriba y río abajo del gen de Cel3. Se diseñaron partidores específicos a partir de un segmento de gen parcialmente codificante para una celulasa descrita en el laboratorio mediante la técnica de PCR y diseño de partidores degenerados, los cuales a su vez se diseñaron en base a secuencias conservadas entre secuencias de genes de celulasa. 27 2.3.4.2 Digestión del DNA genómico y elongación. Se digirió el DNA genómico de la cepa aislada en el laboratorio, el cual se purificó según el protocolo descrito en Anexo A.2, con las enzimas de restricción PvuII y HindIII, con los respectivos buffer recomendados por el proveedor. La reacción se formó con 6 µL de DNA genómico purificado (1 µg de DNA aprox.), 2 µL de Buffer 10X y 1 U de enzima. Se completó con agua Milli-Q estéril hasta un volumen final de 20 µL. La reacción se incubó por 3 horas a 37 ºC. Para la elongación de los extremos de los segmentos obtenidos con la digestión enzimática y para la obtención de una adenina en los extremos 3’, se incubó 15 µL de DNA digerido y purificado con 10 µL buffer Taq 5X, 1,375 µL de 10 mM dNTPs, 4 µL de MgCl2, 0,8 µL de Taq Polimerasa y se completó con agua Milli-Q estéril hasta un volumen final de 50 µL. La reacción se incubó a 70 ºC por 45 minutos. 2.3.4.3 Construcción del oligo-adaptador. Se construyó un oligo-adaptador, el cual incluye 6 sitios de restricción, mediante la hibridización de los oligonucleotidos desfosforilados AdaptHindIII y AdaptHindIIIR, calentando 5 minutos a 95 ºC una solución 10 mM de cada uno, y enfriando lentamente en agua hirviendo hasta llegar a temperatura ambiente. Con el alineamiento se obtiene el adaptador de doble hebra AdaptHindIIItotal. Este adaptador posee una timidina no apareada en uno de sus extremos 3’. 2.3.4.4 Construcción de la genoteca-adaptador. Para la construcción de la genoteca-adaptador, que corresponde al DNA genómico digerido unido al oligo-adaptador en sus extremos, se procedió a ligar 6 µL de la mezcla de fragmentos de DNA elongados con la adenina no apareada en los extremos 3’ con 15 pmoles del adaptador AdaptHindIIItotal, 2 µL de buffer 5X y 1 U de T4 DNA ligasa, completando un volumen total de 10 µL. La reacción se incubó a 16 ºC por 16 horas. 28 2.3.4.5 Amplificación de los extremos 3’ y 5’ del gen de Cel3. Para la amplificación del extremo 3’, se utilizó el partidor específico Cell3R1 y para el extremo 5’ Cell3F1. La reacción de PCR se llevó a cabo en un volumen final de 20 µL, con concentraciones finales de 1,9 mM de MgCl2, 0,2 mM dNTPs, 0,5 µM de partidor especifico, 4 µL de buffer Taq 5X, 5 µL de la genoteca-adaptador diluida 10 veces y 0,3 µL de Taq Polimerasa. Las condiciones térmicas de los ciclos de PCR fueron: 1 ciclo a 94 ºC por 1 min, 25 ciclos de 94 ºC por 32 s y 68 ºC por 5 min, y un ciclo adicional de 70 ºC por 7 min. Las reacciones fueron llevadas a cabo en el termociclador Eppendorf Master Cycler Gradient. El producto de PCR fue diluido 10 veces, 3 µL de esta dilución se utilizaron como templado para el segundo PCR. La segunda reacción de amplificación se llevo a cabo en un volumen total de 20 µL, en donde las concentraciones finales fueron: 1,9 mM de MgCl2, 0,2 mM dNTPs, 0,5 µM de partidor especifico, para el extremo 3’ se utilizó el partidor específico Cell3R2 y para el extremo 5’ Cell3F2, 0,2 µM del partidor específico para el oligo-adaptador AdapTF2, 4 µL de Buffer Taq 5X, 3 µL del producto del primer PCR diluido y 0,3 µL de Taq Polimerasa. Las condiciones térmicas de los ciclos de PCR fueron: 1 ciclo a 94 ºC por 1 min, 35 ciclos de 94 ºC por 32 s y 68 ºC por 5 min, y un ciclo adicional de 70 ºC por 7 min. El producto de la amplificación fue observado en un gel de agarosa al 1 % (p/v) y fue purificado desde éste (Anexo A.7); se ligó (Sección 2.3.8) al vector de clonamiento pGEM-T Easy. El producto de ligación se utilizó para transformar células de E. coli DH5α, utilizando el mismo protocolo descrito en la Sección 2.3.9, pero con la diferencia de que en este caso, las placas utilizadas contenían Ampicilina 100 μg/mL, IPTG 0,1 μM y X-Gal 80 mg/mL. Finalmente se realizó una minipreparación de DNA plasmidial (Anexo A.8) de las colonias recombinantes que resultaron positivas en el PCR de colonias (Anexo A.4) (con inserto), para su posterior secuenciación. Una ilustración de las etapas de amplificación del procedimiento Genome Walking se puede apreciar en la Figura 3. 29 Figura 3: Diagrama de la técnica de “Genome Walking” empleada para la obtención de la secuencia 5’ y 3’ del gen codificante para la celulasa. Este esquema muestra la amplificación del extremo 5’ del gen desde un fragmento de secuencia conocida. Después de la digestión de DNA genómico con las enzimas de restricción, éste se somete a la elongación de los extremos con Taq polimerasa, la cual tiene la capacidad de dejar una adenina en el extremo 3’. Luego se liga el adaptador AdaptHindIITotal, el cual contiene una timidina desapareada. El primer PCR realiza un enriquecimiento lineal del extremo del gen con un partidor específico (Cell3R1 o Cell3F1 para el extremo 3’ y 5’ respectivamente). Luego se realiza un segundo PCR, utilizando como templado el producto del primer PCR, en el cual se realiza la amplificación exponencial del extremo del gen utilizando el primer que se encuentra más central que el primer partidor utilizado (Cell3R2 o Cell3F2 para el extremo 3’ y 5’ respectivamente) y AdaptF2, el que se une a una región en AdaptHindIIITotal. 30 2.3.4.6 Secuenciación y análisis de los extremos 3’ y 5’ del gen codificante para Cel3. Los constructos en pGEMT-Easy, con los insertos correspondientes a los extremos 3’ y 5’ del gen codificante para Cel3, se mandaron a secuenciar a Macrogen S.A. (Corea). Las secuencias obtenidas se analizaron con el software Omiga 2.0, en donde se construyó la secuencia completa del gen. Además se comparó la secuencia obtenida con secuencias de genes de celulasas existentes en la base de datos de NCBI ( http://ww.ncbi.nih.gov/) mediante los algoritmos BLASTn y BLASTp. 2.3.5 Diseño de partidores para el clonamiento del gen codificante para Cel3. Con el fin de poder insertar el gen codificante para celulasa en el vector de expresión pET22b(+), se diseñaron partidores que agregan sitios de corte para enzimas de restricción, sitios que servirán para introducir el gen en el sitio de multiple clonamiento del vector. Con dichos partidores se realizó un PCR a partir del DNA genómico para la amplificación del gen con los sitios de corte incorporados. Se diseñó dos construcciones de vectores recombinantes: • Opción 1: Incluye los sitios de restricción NdeI y XhoI en el gen, con lo que se busca que el gen conserve su peptido señal y se exprese con una cola de histidinas unida en su extremo carboxi terminal. Para ello se diseñaron los partidores CelNdeIS y CelXhoIA. • Opción 2: Incluye los sitios de restricción BamHI y XhoI en el gen, con lo que se busca que el gen se ligue al vector sin su péptido señal, y que en su lugar utilice el péptido de exportación del vector. Este caso también incluye la cola de histidinas unida en su extremo carboxi terminal. Para ello se diseñó el partidor CelBamHIS y se utilizó el partidor CelXhoIA mencionado anteriormente (Figura 4). 31 Figura 4: Región de Clonamiento del vector de expresión pET22b(+). Mapa de clonamiento del vector de expresión pET22b(+). Para la opción 1 se utilizó los sitios de corte NdeI y XhoI para dejar al gen unido a su propio peptido señal. Para la opción 2 se utilizó los sitios de corte BamHI y XhoI para dejar al gen unido al péptido de exportación del vector. Figura de Novagen. 2.3.6 Amplificación de DNA mediante PCR. Se realizaron amplificaciones del gen de Cel3 a partir del material genético mediante la técnica de PCR utilizando como enzima de polimerización Taq DNA polimerasa. La reacción de PCR se llevó a cabo en tubos eppendorf de 0,6 mL estériles en un volumen final de 300 µL con buffer de PCR 1X (40 mM Tris-HCl (pH 8.4), 100 mM KCl), donde las concentraciones finales fueron: 1,9 mM de MgCl2, 0,2 mM dNTPs, 0,5 µM de partidores (CelNdeIS y CelBamHIS como partidor sense en la opción 1 y 2 respectivamente y CelXhoIA como partidor antisence para ambos casos), 7,5 µL de DNA genómicos y 2,25 µL de Taq Polimerasa. Las condiciones del PCR fueron: 1 ciclo a 94 ºC por 5 min, 2 ciclos de 94 ºC por 32 s, 45 ºC por 40 s y 70 ºC por 2 min, 30 ciclos de 94 ºC por 32 s, 60ºC por 40 s y 70 ºC por 2 min y un ciclo adicional de 70 ºC por 7 min. De esta manera el gen quedó con los extremos requeridos para las dos opciones explicadas en la sección 2.3.5 (con sitios de corte para enzimas de restricción) para ser clonados finalmente en el vector de expresión. 32 2.3.7 Digestión de producto de PCR y linearización del vector de expresión pET22b(+). Los productos de la amplificación por PCR del gen de celulasa con los extremos modificados, se purificaron a partir de un gel de agarosa al 1 % (p/v) según el protocolo descrito en el Anexo A.7. Luego fueron digeridas con las enzimas de restricción correspondientes para cada opción (sección 2.3.5). La reacción final de digestión contenian 5 μL de tampón 5X recomendado por New Englands Biolabs para digestiones dobles, 2 μL de cada enzima, 25 μL de producto de PCR purificado en 50 μL de volumen total. Con la finalidad de ligar los fragmentos provenientes de las amplificaciones del gen de celulasa al vector de expresión, éste fue digerido con las mismas enzimas de restricción con las que fue digerido el producto de PCR purificado. Las reacciones finales de digestión contenian 5 μL de tampón 5X recomendado por New Englands Biolabs para digestiones dobles, 2 μL de cada enzima, 40 μL de vector pET22b(+) y 1 μL de agua Milli-Q estéril para completar 50 μL de volumen total. Las digestiones se incubaron durante 3 horas a 37 ºC y el resultado fue analizado mediante un gel de agarosa al 1 % (p/v). Luego se purificaron según protocolo descrito en Anexo A.7. 2.3.8 Ligación de fragmentos de DNA a vectores de clonamiento y expresión. Para todas las reacciones de ligación, se utilizó como inserto los productos de PCR, los cuales se obtuvieron mediante purificación desde geles de agarosa (Anexo A.7). Los procedimientos de ligación de los insertos a vectores de clonamiento o de expresión se detallan a continuación. 2.3.8.1 Ligación en el vector de clonamiento pGEM-T Easy. La reacción se llevo a cabo en un volumen final de 10 µL, cuyos componentes fueron 50 ng de vector pGEM-T Easy, Buffer de ligación 1X 3,5 µL de inserto y 1 µL de enzima T4 DNA ligasa, correspondiente a 1 U. La reacción se dejó toda la noche a 4 ºC. El producto de ligación se utilizó para la transformación de células E. coli DH5α 33 electrocompetentes, las cuales fueron preparadas de acuerdo al protocolo descrito en el Anexo A.9. 2.3.8.2 Ligación en el vector de expresión pET22b(+). La reacción se llevo a cabo en un volumen final de 10 µL, cuyos componentes fueron 100 ng de vector digerido, Buffer T4 DNA ligasa 1X, 80 ng de inserto, digerido con las mismas enzimas de restricción y 1 µL de enzima T4-DNA ligasa (250 mM Tris-HCl (pH 7,6), 50 mM MgCl2, 5 mM ATP, 5 mM DTT, 25 % (p/v), correspondiente a 1 U. La reacción se incubó a 16 ºC por 16 horas. El producto de ligación se utilizó para la transformación de células E. coli BL21(DE3) electrocompetentes, las cuales fueron preparadas de acuerdo al protocolo descrito en el Anexo A.9. 2.3.9 Clonamiento de los constructos en células E. coli DH5α. Para obtener un mayor número de copias de los constructos pET-Inserto (con y sin peptido señal), éstos fueron clonados en células electrocompetentes E. coli DH5α. Para ello células DH5α fueron transformadas con el producto de la reacción de ligación. A 20 µL de células se agregó 1 uL de mezcla de ligación. La electroporación se llevo a cabo en un equipo Cell-Porator, con 420 Volts, 330 µF, baja impendancia, una tasa de carga rápida y un tiempo máximo de 2,5 mseg. Las células transformadas se mezclaron con 1 mL de medio SOC (Anexo B.1 parte b) y se incubaron durante 1 hora a 37 ºC. Se plaqueó en placas con medio LB agar estériles que contenían ampicilina 100 µg/mL (Anexo B.2 parte a). Las placas con el producto de transformación se incubaron a 37 ºC durante toda la noche. Al día siguiente se seleccionaron colonias desde la placa y se dejó creciendo en medio LB con ampicilina 100 µg/mL a 37 ºC con agitación hasta el día siguiente. Luego se realizó una minipreparación de DNA plasmidial (Anexo A.8), la cual fue digerida con las enzimas de restricción correspondientes para confirmar las presencia de inserto y por ende identificar aquellas colonias recombinantes (siguiendo el protocolo descrito en 2.3.7, con la diferencia que ahora el volumen final de reacción fue de 20 μL. Se digirió 5 μL de la minipreparación con 1 μL de cada enzima). 34 2.3.10 Clonamiento de los plasmidios recombinantes en células de E. coli BL21(DE3). Se transformaron células competentes E. coli BL21(DE3) con los plasmidios recombinantes de la misma forma en que se explicó en la sección 2.3.9. Se seleccionaron colonias y se dejaron creciendo a 37 ºC toda la noche en placas LB ampicilina 100 µg/mL. 2.3.11 Pruebas de actividad celulasa extracelular. De la transformación en células competentes E. coli BL21(DE3) se seleccionaron colonias, las cuales se traspasaron a placas LB al 0,5 % (p/v) CMC (Anexo B.2 parte b), ampicilina 100 µg/mL e IPTG 0,1 μM y a placas LB ampicilina 100 µg/mL (ambas en gradilla númerada para una posterior identificación de las colonias). Se incubó a 37 ºC hasta el día siguiente. Luego las placas con sustrato fueron teñidas con Rojo Congo al 0,1 % (p/v) por 30 minutos y luego se destiñeron con Cloruro de Sodio 1M. Aquellas colonias que actividad (degradación del sustrato) fueron seleccionadas de las placas LB ampicilina 100 µg/mL y se dejaron creciendo a 37 ºC por 8 horas en 4 mL de medio LB y carbenicilina a una concentración de 100 µg/mL. A 900 µL de cultivo se agregó 100 µL de Glicerol 80 %. Los tubos fueron guardados a -80 ºC. 35 3 RESULTADOS Y DISCUSION 3.1 CARACTERIZACIÓN BÁSICA DE LA CELULASA EN EL MICROORGANISMO PRODUCTOR 3.1.1 Obtención de la celulasa producida por bacterias antárticas. 3.1.1.1 Cultivo a 4 y 20 ºC de las bacterias antárticas. Los cultivos de bacterias antárticas incubados durante 6 días a 4 y 20 ºC, fueron monitoreados para analizar el crecimiento y la actividad celulasa que se encontraba en el sobrenadante de los cultivos en función a las dos temperaturas de incubación. Las curvas de crecimiento y de actividad de celulasa se muestran en el Figura 5. 10,00 300 Células [g/mL] 1,00 200 150 0,10 100 50 0,01 0 20 40 60 80 100 120 140 Actividad Celulasa[U/L] 250 0 160 Tiempo [h] Figura 5: Curvas de crecimiento microorganismo productor de la celulasa. Curva para cultivo incubados a 4 ºC ( ) y 20 ºC ( ). La actividad celulasa a 4 ºC ( ) y a 20 ºC ( ). Se observa en el Figura 5 que el cultivo incubado a 4 ºC tarda más en llegar al estado estacionario comparado con su similar a 20 ºC, sin embargo su nivel final de crecimiento es mayor, llegando a un valor de 2,41 g de células/mL, comparado con 1,6 gramos de células/mL que alcanza el cultivo a 20 ºC. Se observa una diferencia en la producción de celulasa en ambos cultivos: en el caso del cultivo a 4 ºC se mantiene la actividad celulasa en un nivel bajo en toda la etapa exponencial del crecimiento hasta el comienzo de la etapa estacionaria que se produce un cambio, donde se observa el aumento de la actividad celulasa en el sobrenadante. El cultivo a 20 ºC en cambio, presenta una producción de celulasa a la par al crecimiento celular, aumenta durante la etapa exponencial y se estanca en el estacionario. 36 Se eligió el cultivo a 4 ºC para posteriores experimentos, por entregar un mayor manejo del cultivo, el cual basta con detenerlo en el comienzo de la fase estacionaria, mientras que el cultivo de 20 ºC llega a su máximo de producción de celulasa antes del inicio de la fase estacionaria, punto que resulta más difícil de determinar con el monitoreo de de la actividad celulasa. 3.1.1.2 Purificación de la celulasa secretada por bacterias antárticas. La purificación de Cel3 se realizó a partir de un cultivo de 500 mL, incubado a 4 ºC, el cual se detuvo a un valor de D.O600 de 5, equivalente a 7 miligramos de células/mL (curva de calibración Anexo C.4). El inicio del estado estacionario se identificó cuando el monitoreo arrojó tres mediciones seguidas en un mismo nivel de D.O600. A los 440 mL de sobrenadante de cultivo, se agregó 297 gramos de sulfato de amonio para la precipitación de Sulfato de Amonio, la cual después de reposar en hielo, se centrifugó 2 horas a 8.500 rpm. El pellet resuspendido en 3 mL de buffer Tris 20 mM pH 7, tomó un volumen final de 10 mL, los cuales fueron dializados en el mismo buffer durante la noche. El producto de la diálisis tuvo un volumen final de 45 mL aproximadamente. En la cromatografía las fracciones con actividad celulasa se encontraron principalmente a lo largo del segundo segmento del gradiente, es decir eluyeron entre un 15 y 30 % de buffer B (con sal), Figura 6. Las fracciones con actividad positiva con valores de D.O550 mayores a 0,4 fueron juntadas y concentradas para la etapa de purificación siguiente. 37 90 800 80 700 70 600 60 500 50 400 40 300 30 200 20 100 10 0 -100 Conductividad [mS/cm] mAU 280 y 550 nm 900 0 10 20 30 40 50 60 70 0 Ve [mL] Figura 6: Cromatógrama de la etapa de Intercambio Anionico. Se observa el perfil de Abs a 280 nm [mAU] (color azúl), de Abs a 550 nm [mAU] (color céleste) y el de conductividad [mS/cm] (color verde). Las fracciones juntas se les denominaron Ori F.G (Original de Filtración por Gel), las que sumaron un total de 1,8 mL después de la concentración. Las fracciones con actividad celulasa positiva se encontraron en el inicio del peack principal, a valores de 400 500 300 400 200 300 100 200 0 Abs 550 nm mAU Abs 280 nm mAu elusión de 6 a 7 mL (Figura 7). 100 0 2 4 6 8 10 -100 12 14 16 18 0 Ve [mL] Figura 7: Cromatógrama de la etapa de Intercambio Anionico. Se observa el perfil de Abs a 280 nm [mAU] (color azúl) y el de Abs a 550 nm [mAU] (color céleste). Para un análisis de la pureza de la muestra final se realizó una electroforesis en gel de poliacrilamida al 12,5 % (p/v), el cual se ilustra en la Figura 8. Se observa una banda única en el carril en donde se cargó la celulasa, lo que indica la pureza de la enzima. 38 Figura 8: Análisis electroforético de las distintas etapas de la purificación de la celulasa. Gel de Poliacrilamida al 12,5% en condiciones desnaturantes, teñido con azul de comasie y con plata. Carril 1: muestra proveniente directamente del sobrenadante de cultivo (0,8 μg); Carril 2: muestra proveniente del dializado del sedimento precipitación con sulfato de amonio al 90% (3 μg); Carril 3: mezcla de fracciones con actividad celulasa posterior a IEX (3,6 μg); Carril 4: mezcla de fracciones con actividad celulasa posterior a F.G (proteína purificada, 1,4 μg); M: marcador de masa molécular. Como se puede observar en la Figura 8 la proteína en la última etapa de purificación se encuentra totalmente pura. Se cuantificó mediante el método de Bradford (Anexo A.1) y se obtuvo una concentración de enzima purificada de 10 ηg/mL, teniendo una producción total de 0,14 mg de celulasa a partir de un cultivo de 500 mL. En la Figura 9, se puede observar un zimograma realizado para las mismas muestras que fueron analizadas en la electroforésis de la Figura 8. En el zimograma se puede observar en cada fracción actividad de celulasa, la cual es representada por halos amarillos, los que equivalen a la degradación de CMC inducida en el gel (Anexo A.6). Esta actividad indica la presencia de la enzima en cada una de las muestras, desde el sobrenadante hasta la muestra de la enzima purificada, por lo que el proceso de purificación fue asertivo al seguimiento de la enzima de interés. 39 Figura 9: Zimograma de las distintas etapas de la purificación de la celulasa. Gel de Poliacrilamida al 10%.Carril 1: muestra proveniente directamente del sb de cultivo; Carril 2: muestra proveniente del dializado de la precipitación con sulfato de amonio al 90%; Carril 3: mezcla de fracciones con actividad celulasa posterior a IEX; Carril 4: mezcla de fracciones con actividad celulasa posterior a F.G (proteína purificada). Para la realización de la Tabla de Purificación, fue necesario realizar otro cultivo y a su vez una nueva purificación, ya que la enzima purificada en el primer cultivo no fue suficiente para este propósito. Vale decir que en este segundo cultivo no se obtuvo ni el crecimiento de bacterias esperado, y por ende la producción de celulasa fue menor (el estado estacionario de las bacterias antárticas fue de un nivel de D.O550 de 3, lo que equivale a 4,3 miligramos de células/mL). A pesar de lo anterior se procedió a la toma de datos necesarios para la construcción de la tabla. A continuación se presenta la tabla de purificación asociada al protocolo utilizado. Tabla 3: Tabla de Purificación de la Celulasa producida por el microorganismo nativo. La actividad específica se midió con CMC como sustrato. El volumen con que se empezó fue de 500 mL. Se muestran las cuatro etapas de purificación Sobrenadante Cultivo Dializado Intercambio Aniónico Filtración por Gel Vol (mL) 430 Proteína Total (mg) 6,33 Actividad Total (U) 71,697 Actividad Especifica (U/mg) 11,320 15 7,2 2,28 1,31 6,683 3,972 6,75 0,49 2,745 Recuperación (%) Paso Total 100 100 2,933 3,027 9,32 59,43 9,32 5,54 5,591 69,11 3,83 Como se puede observar en la Tabla 3, usando este protocolo de purificación cerca del 3,8 % de la proteína fue recuperada en su totalidad. Este nivel de recuperación se debe principalmente a la etapa de precipitación con sulfato de amonio, en donde tan sólo se recupera el 9,32 % de la proteína total. Se cree que las proteínas del 40 sobrenadante se degradan. Dicha etapa es crucial en el procedimiento por permitir una concentración de alrededor de 25 veces, concentración que es necesaria para las siguientes etapas. Además no se asocia el problema a que queden proteínas en el sobrenadante de la precipitación, ya que al utilizar un 90 % de saturación, se precipita la mayoría de las proteínas del sobrenadante, y subir este nivel resulta complicado de manejar. A pesar de lo anterior, las etapas de cromatografía posteriores poseen una recuperación por etapa mayor al 50 % por lo que se consideran eficientes. Se sugiere hacer el ejercicio de precipitar con sulfato de amonio, pero en presencia de inhibidores de proteasas, lo que evitaría la degradación de proteínas debido a proteolisis, de esa forma se podría saber si la perdida de proteína en dicha etapa es debida a este motivo. Observando los porcentajes de purificación se puede inferir que es en la etapa de precipitación con sulfato de amonio en donde se pierde enzima de interés, lo que se ve reflejado en los valores de actividad específica, etapa en la cual se recupera sólo el 0,25 veces de Cel3. En las etapas posteriores los valores de actividad específica aumentan levemente, lo que demuestra que en dichas etapas se purifica la proteína (1,8 veces en la última etapa). El nivel de purificación total es de 0,49 veces, lo que significa que la proteína no fue purificada en su totalidad, sin embargo en la Figura 8 se puede observar la pureza alcanzada (única banda en el carril 4), lo que se contradice con los valores de la Tabla 3. Se puede decir entonces, que la enzima si fue purificada bajo el criterio cromatográfico. En cuanto a la tabla esto no se ve reflejado, debido a artefactos que puedan haber influido en los resultados de ésta. A pesar de lo anterior, se consideró que el protocolo seguido para la purificación de la enzima a partir del microorganismo antártico, permitió obtener la celulasa purificada, que es lo que se buscaba, lo que permite realizar una posterior caracterización, a pesar de obtener cantidades pequeñas. 3.1.2 Caracterización de la enzima celulasa producida por las bacterias antárticas. Con el fin de caracterizar a la enzima purificada, y ver su comportamiento frente a la temperatura y el pH se realizaron ensayos de actividad y estabilidad en función de estas dos variables, con ellos se podrá determinar si esta enzima es apta para una futura aplicación en la industria de los detergentes. 41 Efecto de la temperatura de reacción en la actividad de la celulasa. Se encontró que la temperatura de reacción óptima fue de 45 ºC, decayendo la actividad específica fuertemente después de dicha temperatura (Figura 10), dicha termolabilidad es una característica común de las enzimas psicrofílicas, en el caso de una enzima mesófilica su temperatura óptima de reacción puede estar cercano a los 55 ºC, temperatura en la cual ya la celulasa ha perdido prácticamente el 50 % de actividad [15,21]. Se puede observar también que entre los 15 y 20 ºC la enzima posee entre un 60 y 80 % de su actividad, temperaturas en las cuales Cel5 de Erwinia chrysanthemi, una celulasa mesófilica, prácticamente no posee actividad [22]. Esto último resulta interesante, ya que de aplicar esta enzima como aditivos a detergentes, permitiría obtener una buen comportamiento de la enzima en agua sin calentar (20 ºC), obteniendo así un mejor rendimiento en el lavado. Actividad Relativa %] 120 100 80 60 40 20 0 0 10 20 30 40 50 60 70 80 Temperatura [ºC] Figura 10: Efecto de la temperatura de reacción en la actividad específica de la celulasa. Ensayo realizado a distintas temperaturas durante 40 minutos en buffer MOPS a pH 7 (ensayo estándar), usando como sustrato CMC al 1%. Termoestabilidad. Al someter a la enzima a una preincubación de 10 minutos a diferentes temperaturas y posteriormente realizar el ensayo estándar, se observó que la actividad relativa se mantuvo alrededor de 100 % para valores de temperaturas de incubación hasta los 40 ºC, para valores más altos de temperatura hubo una caída de la actividad relativa. Este resultado indica una estabilidad frente a la temperatura atractiva para una enzima proveniente de un organismo adaptado a bajas temperaturas, ya que éstas se caracterizan por ser muy termolábiles [17] (Figura 11). Sin embargo, este 42 comportamiento se repite en el caso de CelX, una de las dos endoglucanasas activas a bajas temperaturas publicadas [22]. Con estos valores fue posible determinar la Tm de la proteína, que corresponde a la temperatura donde pierde el 50 % de la actividad. Este correspondería a un valor de 54 ºC, valor que es mayor a las Tm de α-amilasa de Alteromonas haloplanktis, de xilanasa de Cryptococcus adeliae y de la lipasa de Aspergillus ridulans WG 312, las cuales rodean los 45 ºC [19]. Actividad Relativa % 120 100 80 60 40 20 0 -20 15 25 35 45 55 65 75 T [ºC] Figura 11: Estabilidad de la celulasa frente a la Temperatura. Ensayo de actividad realizado durante 40 minutos en buffer MOPS a pH 7 a 20ºC, con previa incubación de la enzima durante 10 minutos a las diferentes temperaturas usando como sustrato CMC al 1%. Efecto del pH de reacción en la actividad de la Cel3. Se encontró que el pH óptimo es de 8 (Figura 12), valor que difiere de los pH óptimos de las endoglucanasas CelG y CelX, los cuales son de 7 y 6-7, respectivamente. Además el rango en donde la celulasa posee altas actividades es mayor que el rango que poseen CelG y CelX: 80 % a pH 8 y sin actividad a pH 10 en el caso de CelG; y 73 y 63 % de actividad residual a pH 9 y 10 respectivamente en el caso de CelX [21,22]. Con lo anterior se puede decir que esta celulasa posee un comportamiento más alcalino que las enzimas mencionadas anteriormente, y es más apta para actuar como aditivo a detergentes, ya que para ello se requiere que posean actividad en amplios rangos de pHs alcalinos, rango alcanzado por esta Cel3 (valores altos desde pH 6 a 11). 43 Actividad Relativa % 120 100 80 60 40 20 0 2 3 4 5 6 7 8 9 10 11 12 pH Figura 12: Efecto del pH de reacción en la actividad específica de la celulasa. Ensayo realizado durante 40 minutos a distintos pHs a 20ºC (ensayo estándar), usando como sustrato CMC al 1%. Estabilidad de la celulasa frente al pH de incubación. Al haber preincubado la enzima durante 24 horas en soluciones de distintos pHs, se encontró que la actividad relativa se mantuvo alrededor de 90 %, en un rango de valores de pH desde 5 hasta 11 (Figura 13). Este resultado muestra que la enzima es estable a valores de pHs alcalinos, lo que resulta muy atractivo para posibles aplicaciones de esta enzima como aditivo a detergentes, y además si se le suma la propiedad de mantener cerca del 100 % de actividad previa incubación a un rango de temperatura entre 20 y 40 ºC, le agrega una ventaja sobre típicas enzimas activas a bajas temperaturas, que se caracterizan por tener baja estabilidad frente a la temperatura. 44 Actividad Relativa % 120 100 80 60 40 20 0 2 3 4 5 6 7 8 9 10 11 12 pH Incubación Figura 13: Estabilidad de la celulasa frente al pH. Ensayo de actividad realizado durante 40 minutos a pH 7 a 20ºC, con previa incubación de la enzima durante 24 horas en buffers con distintos pHs, a 4ºC. Se utilizó como sustrato CMC al 1%. Observando estos resultados, se puede notar una diferencia entre las celulasas provenientes de organismos psicrofílicos, de las cuales ya hay antecedentes publicados, y Cel3. En el caso de CelG, el perfil de actividad frente al pH posee tiene un comportamiento marcado en el valor de pH 7, siendo este su pH óptimo, a pH 8 conserva cerca del 80 % de actividad, mientras que a pH 10 prácticamente ha perdido la totalidad de ésta. A su vez, la estabilidad frente al pH de la solución de incubación tiene su peack en los pHs 7 y 8, y para valores mayores disminuye en forma brusca; para valores de pH 10 y 11 conserva cerca del 70 y 40 % de actividad, respectivamente [21]. La mayor alcalinidad y estabilidad a pH básicos de Cel3, se puede explicar por diferencias estructurales de ambas enzimas, para ello se procederá a analizar dichas diferencias con las secuencias aminoácidicas en la Sección 3.2.5. Peso Molecular. El peso molecular de la enzima se determinó mediante una filtración en gel en donde se cargó 50 µL de la muestra de celulasa pura concentrada (0,04 mg/mL), con ello se obtuvo un peack de elusión (Figura 14), al cual se le midió su correspondiente volumen de elusión, que correspondió a 6,35 mL, con el que se obtuvo un peso molecular de proteína nativa de 35,7 kDa según a la curva de calibración (Anexo C.3). El peso molecular de esta enzima es levemente menor al peso molecular determinado para CelG, de 38 KDa, producida por Pseudoalteromonas haloplanktis [21]. Esta mínima diferencia de peso se puede deber a diferencias estructurales a nivel 45 aminoacídico, para ello se procederá a analizar dichas diferencias con las secuencias aminoacídicas en la Sección 3.2.5. Ovalbúmina BSA Ribonucleasa A Celulasa Figura 14: Cromatógrama de filtración por gel de la celulasa para la determinación de su peso molecular, mAU 280 nm vs. volumen de elusión [mL]. Se indican los niveles de elusión de las proteínas con pesos moleculares conocidos con los que se construyo la curva de calibración: BSA, Ovalbúmina y Ribonucleasa A con 66, 43 y 13,7 kDa, respectivamente. Se destaca en color rojo el peack que corresponde a la celulasa. Punto Isoeléctrico. El punto isoeléctrico se determinó mediante un isoelectroenfoque utilizando el sistema PhasSystem, en un gel con un rango de corrida de valores de pH entre 3 y 9. Los resultados se muestran en la Figura 15. 46 Figura 15: Isolelectroenfoque de la celulasa nativa. Gel PhastSystem 3-9, teñido con plata. Carril 1: celulasa purificada (13 μg); M: marcador de punto isoéletrico. En la Figura 15 se puede observar una banda entre la altura de 4,55 y 5,2, por lo que este rango puede ser definido como el rango del P.I de la celulasa. Este valor es consistente con los resultados obtenidos en la cromatografía IEX, que se realiza a un pH 7. A este pH la proteína tiene una carga neta negativa (por poseer un P.I entre 4,55 y 5,2) que permite que ésta se adsorba a la resina de la columna (Q-sepharose), que tiene carga positiva. No hay valores de punto isoeléctricos publicados para el caso de CelG y CelX, por lo que no se puede comentar al respecto. 3.1.2.1 Determinación de Parámetros Cinéticos. Para la determinación de los parámetros cinéticos se procedió a realizar el ensayo estándar, pero con diferentes concentraciones de sustrato las cuales se encontraban en el rango de 0 a 10 mg/mL. En la Figura 16, se muestra la curva de saturación obtenida. 47 Curva de Saturación 250 200 V [U/L] 150 100 50 0 -50 0 5 10 15 20 25 CMC [mg/mL] Figura 16: Curva de saturación de la celulasa. Ensayo de actividad realizado durante 40 minutos a 20ºC, pH 7, con concentraciones de sustrato de 0 a 10 mg/mL. Se utilizó como sustrato CMC. Este ensayo se realizó con la celulasa obtenida del segundo cultivo, cultivo que no cumplió con las expectativas de crecimiento. Además se observó que la celulasa obtenida de este cultivo tuvo menor actividad específica, que la celulasa obtenida anteriormente (alrededor de 8 veces menos activa que la obtenida en el primer cultivo). Se recomienda repetir este ensayo, para obtener valores más cercanos a los reales y con una enzima en sus óptimas condiciones. A pesar de lo anterior, se procedió a calcular los parámetros cinéticos con el procedimiento descrito en la Sección 2.3.3.1. Se procedió a calcular los parámetros cinéticos, con la linealización de Dixon tomando como supuesto que la enzima sigue un comportamiento descrito por Michaelis Menten (Anexo D). Los parámetros cinéticos a 20 ºC KM, VMAX y Kcat son 10,8 mg/mL, 357 U/L y 5,6x10-3 s-1, teniendo una eficiencia catalítica KM/Kcat de 5,2x10-4 mg·s-1·mL-1. Estos valores servirán como puntos de comparación para los parámetros que se obtengan de la futura caracterización de la enzima recombinante. 3.2 BÚSQUEDA DE SECUENCIAS FALTANTES DEL GEN CODIFICANTE PARA CELULASA Utilizando el método de Genome Walking estandarizado en el CIByB que permite identificar regiones desconocidas adyacentes a regiones de DNA previamente conocidas, fue posible obtener la secuencia completa del gen de Cel3 [26]. 48 3.2.1 Diseño de partidores para la obtención de las secuencias río arriba y río abajo del gen de celulasa. Los partidores específicos se diseñaron a partir de un segmento de 400 pb, previamente identificado en el laboratorio. Este segmento fue amplificado con la técnica de PCR, en el que se utilizaron partidores degenerados, los cuales fueron diseñados a partir de las zonas altamente conservadas, que fueron identificadas gracias a un alineamiento múltiple de genes codificantes para celulasas existentes en la base de datos del GeneBank (NCBI). Con la secuencia disponible de dicho segmento fue posible diseñar cuatro partidores, los cuales fueron utilizados en la búsqueda de las secuencias río arriba y río abajo del gen codificante para celulasa. En la Figura 17 se puede visualizar un esquema ilustrativo de la secuencia previamente identificada y los partidores Cell3R1, Cell3R2, Cell3F1 y Cell3F2. Sus respectivas secuencias y Tm se detallan en la Tabla 4. Figura 17: Partidores para la amplificación de la secuencia 5’ y 3’ del gen codificante para celulasa. Las secuencias en azul corresponden a los partidores Cell3R1 y Cell3R2 , que amplifican el extremo 3’. Las secuencias en morado corresponden a los partidores Cell3F1 y Cell3F2, que amplifican para el extremo 5’. 49 Tabla 4: Partidores para la obtención de la secuencia 5’ y 3’ para el gen codificante para celulasa. Nombre Secuencia (5’-3’) Tm (ºC)* Cell3R1 Cell3R2 Cell3F1 Cell3F2 GATAACGAGCCGTTACAAATCTCTTGGG ATTGTGGTTGGAACGCCTACGTGG TCGGTAAAAGCCCAGTTAGCGTGGCT TCCCCACTCTGTGGCGAATAGTGC 68 67 70 69 *: Tm calculada por OligoCalc, Oligonucleotide Properties Calculador (http://www.basic.northwestern.edu/biotools/oligocalc.html). El diseño de los partidores permite que una vez que se tengan las secuencias de los extremos 5’ y 3’, éstas posean zonas con secuencias comunes al segmento original de 400 pb, lo que facilita el construir la secuencia entera del gen con tan sólo sobreponer los extremos con el segmento previamente conocido. 3.2.2 Digestión del DNA genómico y elongación. Se digirió el DNA genómico, el cual previamente fue extraído desde la cepa de interés siguiendo el protocolo descrito en el Anexo A.2. La enzima seleccionada para la digestión fue PvuII. Esta enzima mostró la mejor digestión, por generar segmentos de tamaño entre 4 y 10 Kb, tamaños con los que se tiene una mayor probabilidad de incluir el gen su totalidad. El DNA cromosomal digerido se procedió a purificar según el protocolo descrito en el Anexo A.7. Luego se procedió a la elongación con Taq DNA polimerasa, la cual deja una adenina no apareado en los extremos 3’, lo que posteriormente permite una ligación más óptima al oligo-adaptador. 3.2.3 Construcción del oligo-adaptador y de la genoteca-adaptador. El oligo-adaptador de doble hebra AdaptHindIIITotal fue construido mediante la hibridización de los oligonucleotidos AdaptHindIII y AdaptHindIIIR desfosforilados. El diseño de este oligo-adaptador incluye una timidina no apareada en uno de sus extremos 3’ (Figura 18). Figura 18: AdaptHindIIITotal. La hibridización de AdaptHindIII y AdaptHindIIIR da como resultado este oligoadaptador con una timidita no apareada en uno de sus extremos 3’. Luego este oligo-adaptador fue utilizado para la construcción de la genotecaadaptador, ligándolo a los extremos de DNA genómico digeridos con PvuII y elongados con Taq DNA polimerasa. 50 3.2.4 Amplificación de los extremos 3’ y 5’ del gen de Cel3. La genoteca-adaptador fue utilizada como templado para el primer PCR (ver Figura 3), con los partidores Cell3R1 y Cell3F1 para la amplificación de los extremos 3’ y 5’ respectivamente. Esta reacción de polimerización permite un enriquecimiento de forma lineal, por lo que sólo se amplifica la hebra complementaria al gen. Esto permite tener un templado para el segundo PCR con mayor cantidad del fragmento de DNA de interés. Con este paso de enriquecimiento se asegura una mayor especificidad de la reacción de amplificación del segundo PCR, por tener mayor concentración del DNA de interés, lo que da la posibilidad de diluir este templado y así evitar amplificaciones no deseadas, ya que se disminuye la probabilidad de alineamiento inespecífico de los partidores a zonas no codificantes para celulasa. El producto diluido del primer PCR se utilizó como templado para la segunda amplificación de los extremos del gen de interés. En esta etapa se utiliza como partidor Cell3R2 y Cell3F2 para la amplificación del extremo 3’ y 5’ respectivamente; como segundo partidor, para la amplificación de los dos extremos se utiliza AdaptF2, el cual es complementario al oligo-adaptador. El uso de un oligo-adaptador desfosforilado, deja después de la ligación un espacio entre el extremo 5’ del fin del oligo-adaptador y el extremo 3’ del fragmento de DNA, mientras que el extremo 5’ del fragmento del DNA y el extremo 3’ del oligo-adaptador está unido. Gracias a esta construcción cualquier otra amplificación con AdaptF2 es bloqueada y sólo la hebra complementaria generada por el primer partidor específico servirá como templado para la reacción de PCR con AdaptF2 y el segundo partidor específico, así el fragmento de DNA deseado puede ser amplificado de forma específica [26]. Los productos de la segunda ronda de PCR se muestran en la Figura 19. Se observa en el análisis electroforético bandas nítidas mayores a 1000 pb, con lo que se cree que son de suficiente tamaño para completar los extremos del gen codificante para esta celulasa. Esto es debido a que mediante un análisis de las secuencias publicadas de genes codificantes, se predice un tamaño de gen de aproximadamente 1500 pb, por lo que los fragmentos obtenidos deberían ser suficientes. 51 Figura 19: Análisis electroforético de los productos de PCR para la obtención del extremo 5’ y 3’ del gen codificante para celulasa. Gel de Agarosa al 1 %. M: Marcador de Peso Molecular A): Carril 1: Extremo 5’ del gen codificante para celulasa; B) Carril 1: Extremo 3’ del gen codificante para celulasa. Por ser bandas únicas en la amplificación, se procedió a seleccionarlas y posteriormente a purificarlas directamente desde el gel de agarosa (Anexo A.7). Los fragmentos purificados fueron ligados al vector de clonamiento pGEM-T Easy, para tener un alto número de copias del vector con el inserto. Se transformó células competentes DH5α de E. coli con dicha ligación. Se seleccionó una colonia positiva (vector ligado al inserto de interés) mediante PCR de colonias (Anexo A.4), para luego realizar una minipreparación, la cual fue enviada a secuenciar. En el caso de la banda de 2 Kpb, correspondiente al extremo 5’, no se logró tener una ligación al vector de clonamiento exitosa, por lo que se mandó a secuenciar el fragmento puro. 3.2.5 Secuenciación y análisis de los extremos 3’ y 5’ del gen codificante para celulasa. Las secuencias obtenidas por el método de Genome Walking fueron analizadas con el programa Omiga y se pudo reconstruir una secuencia de DNA de 2127 pb. En dicha secuencia se identificó un ORF (marco de lectura abierto) de 1486 pb que codifican para 494 aminoácidos. Se analizó la secuencia obtenida con la herramienta Blastp y Blastn y se encontró una muy alta semejanza a nivel de aminoácidos y de nucleótidos con la celulasa CelG de 52 Pseudoalteromonas Haloplanktis y con CelX de Pseudoaltoromonas sp. DY3, con un 97 y 94 % de identidad respectivamente. Se sabe, de experimentos realizados anteriormente en el laboratorio, que la cepa con que se trabajó también pertenece a la especie Pseudoalteromonas, lo que podría explicar el alto porcentaje de identidad entre dichas celulasas. Al alinear las tres secuencias de aminoácidos se puede hacer un paralelo entre ellas y poder identificar en Cel3, el dominio catalítico, el dominio de unión a la celulosa (CBD, Carbohydrate Binding Domain) y el linker que une dicha estructuras. Con la herramienta SignalP 3.0 y PSORTb 2.0 del sitio web Expasy, se reconoció en la secuencia un péptido señal N-terminal de 32 aminoácidos, para secreción extracelular (Figura 20). La primera herramienta predice la presencia y ubicación de sitios de corte de péptidos señal en secuencias de aminoacidos de diferentes organismos, utilizando un método que incorpora una predicción de sitios de corte y una predicción de péptido señal/péptido no señal, que se basa en una combinación de varias redes neuronales artificiales y modelos de Markov. La segunda herramienta consiste en un método probabilístico que integra información obtenida a partir de análisis de secuencia de aminoácidos, similitud de la proteína con otras de localización conocida, la presencia o no de péptidos señal, etc. Como resultado entrega una lista de cinco posibles sitios de localización con puntajes de probabilidad asociados. Figura 20: Secuencia Aminoácidica de la celulasa. Los aminoácidos destacados en verde corresponden al péptido señal, en azul al dominio catalítico, en morado al linker de unión y en naranjo al dominio de unión a la celulosa (CBM, Carbohydrate-binding domain). Considerando la secuencia de la proteína madura, sin contar el péptido señal Nterminal de 32 aminoácidos, el dominio catalítico N-terminal, puede ser clasificado dentro de la familia 5 de las glicosil hidrolasas por poseer 5 residuos estrictamente 53 conservados (Arg57, His100, His194, Glu135 y Glu222) [21]. Este dominio también es relacionado con el Clan A de las glicosil hidrolasas GH-A que se caracteriza por poseer un plegamiento del tipo (β/α)8. (CAZY, Carbohydrate-Active Enzymes, http://www.cazy.org/). El CBD se puede clasificar entre los CBD de la familia 5 por tener un 95 % de similitud con el CBD de CelG, el cual fue clasificado dentro de esa familia [21]. Se estimó su peso molecular a partir de la secuencia aminoácidica en el sitio web de Expasy, mediante la herramienta Compute pI/Mw, el que fue de 52.729 Da, el que difiere del obtenido en forma experimental. Si se hace el ejercicio de introducir sólo la secuencia del dominio catalítico, se obtiene un peso molecular de alrededor de 30 kDa, lo que se acerca al peso molecular obtenido experimentalmente. Es por esto último que se cree que la proteína purificada, a partir del sobrenadante del cultivo de la bacteria antártica, corresponde a una proteína truncada que contiene solamente el dominio catalítico, probablemente cortada por una proteasa secretada por la misma bacteria. Esta deleción se ha encontrado en otras oportunidades y se afirma que es un comportamiento común en microorganismos celulolíticos [21]. Funciones de éste dominio de unión no son del todo claras, pero de le han atribuido posibles funciones, tales como: a) participar en la destrucción de la celulosa interfiriendo con el empaquetamiento regular de las fibrillas; b) o simplemente aumentar la concentración local del dominio catalítico al que están unidos. Se cree también, por resultados experimentales, que dicho dominio es necesario para la secreción de la enzima al medio extracelular [11]. Por otro lado se tiene el antecedente que su presencia aumenta la sinergia entre el dominio catalítico y el dominio de unión a la celulosa con su sustrato natural, ya que la eliminación del enlace disminuye drásticamente la actividad enzimática sobre la celulosa cristalina [10]. Al utilizar como sustrato en este caso carboximetil celulosa (CMC), celulosa del tipo no cristalina, se cree que la actividad de la enzima no debiera verse afectada en demasía por la ausencia del dominio de unión, ya que la hidrólisis del enlace 1,4-β-glucosidicos es realizada por el dominio catalítico. Gracias a la misma herramienta se estimó el punto isoeléctrico, el cual fue de 4,5, valor que coincide con el obtenido experimentalmente, que se determinó en el rango de 4,55 y 5,2, rango que resultó ser un poco estimativo, ya que la banda correspondiente a la proteína corrió de una manera bastante deformada, por lo que el 54 valor estimado por la herramienta de Expasy, ayuda a acotar dicho rango a un valor cercano a 4,5. El gran porcentaje de identidad de las secuencias de CelG y Cel3, lleva a preguntarse el porque su diferencia frente al comportamiento del pH, mostrado en la sección 3.1.2, en donde se encontró que el pH óptimo para la celulasa estudiada tiene un valor de 8 (Figura 12), valor que difiere de los pH óptimos de las endoglucanasas CelG y CelX, los cuales son de 7 y 6-7, respectivamente. Además el rango en donde Cel3 posee altas actividades es mayor que el rango que poseen CelG y CelX: 80 % a pH 8 y sin actividad a pH 10 en el caso de CelG; y 73 y 63 % de actividad residual a pH 9 y 10 respectivamente en el caso de CelX. Analizando las secuencias aminoácidicas de la proteína madura, se tiene una diferencia de 6 aminoácidos en el dominio catalítico, los que se pueden ver involucrados en las diferencias funcionales entre ambas enzimas. En la Tabla 5, se detallan dichos residuos en detalle. Tabla 5: Diferencias de residuos entre CelG y la Celulasa. Se detallan los 6 residuos en que se diferencian las secuencias en el dominio catalítico y sus respectivas posiciones en la proteína madura. CelG Asn Leu Thr Ala Tyr Ser Celulasa Ser Ile Ser Val Asp Ala Posición 18 26 40 90 133 180 Ninguno de estos residuos está incluido en el grupo de los 20 residuos que interaccionan directamente con el sustrato [21], por lo que la diferencia no recae en la interacción con éste, si no que probablemente en la conformación del dominio durante la catálisis. Si se analizan las características de los aminoácidos de la Tabla 5, se puede ver que existen dos aminoácidos que son reemplazados por residuos con características distintas, estos son el caso del residuo en la posición 180, que en CelG corresponde a una Serina (polar), mientras que en la celulasa es una Alanina (no polar); el otro caso corresponde al residuo correspondiente a la Tirosina (no polar) de la posición 133 que en la celulasa corresponde a un ácido aspartico (cargado). La diferencia de las características de estos dos residuos puede tener efectos en la alcalinidad de ambas enzimas. Posiblemente la no polaridad de la alanina y la carga negativa del aspartato en pH básicos, puede dar una conformación estructural al dominio catalítico que favorezca la catálisis del sustrato, obteniéndose así una curva 55 de Actividad vs. pH con valores más altos en un rango más amplio. En el caso de CelG, quizás la presencia de la Serina, que es un aminoácido polar, puede estar jugando un rol en la estructura del dominio catalítico con sus cambios de carga frente a cambios de pH, repeliendo o atrayendo, según sea el caso, a aminoácidos ubicados en su cercanía, influyendo así en el mecanismo de reacción de los aminoácidos que sí interaccionan con el sustrato directamente. Observando la estructura 3D de CelG se observa que los aminoácidos ubicados en la superficie de la proteína son: Asn18, Thr40, Ala90 y Ser180. Probablemente el cambio de estos aminoácidos por lo indicados en la Tabla 5, le den mayor estabilidad a Cel3 frente a pH alcalinos, antecedente que se puede sumar a lo mencionado anteriormente. Con los antecedentes disponible (secuencias aminoácidicas) es apresurado concluir que la diferencia en los perfiles se debe a lo afirmado anteriormente. Es por ello que se recomienda analizar en forma más detallada el efecto de dichos residuos en la alcalinidad de las enzimas, mediante análisis a nivel molecular del dominio catalítico o bien experimentalmente con mutagénesis sitio-dirigida para ver que rol juegan dichos residuos en la actividad de las enzimas. 3.3 3.3.1 CLONACIÓN DEL GEN CODIFICANTE PARA CELULASA Diseño de partidores para el clonamiento del gen codificante para Cel3. Para la inserción del gen de celulasa en el vector de expresión pET22b(+), se realizó a partir del DNA genómico, un PCR con partidores, que en su diseño incluyen sitios de corte para enzimas de restricción, enzimas que también tienen sitios de corte en el sitio de múltiple clonamiento del vector de expresión. Se diseñaron dos opciones de clonamiento, una incluyendo el péptido señal de exportación al medio extracelular propio del gen codificante para celulasa, y una segunda opción que reemplaza dicha secuencia por el péptido de exportación al periplasma (pelB) del vector. Ambas opciones consideran la unión en el extremo carboxi terminal de una cola de seis histidinas. Esta última conformación permite una posterior purificación que se facilita mediante el uso de cromatografías de afinidad por columna con resina de níquel agarosa (Ni-NTA His·Bond, Novagen), con las cuales tendría afinidad la cola de histidinas. 56 Las secuencias de los partidores utilizados para la inserción en el vector de expresión se detallan en la Tabla 6. Tabla 6: Partidores para el clonamiento del gen codificante para celulasa en el vector de expresión pET22b(+). El par CelBamHIS y Cel3XhoIA se utilizó para la opción de clonamiento sin péptido señal del gen, es decir utilizando el péptido de exportación al periplasma (pelB) del vector. El par CelNdeIS y Cel3XhoI se utilizó para la opción de clonamiento del gen entero. Nombre Secuencia (5’-3’) Tm (ºC)* CelBamHIS TCAACAATGGATCCTGCCGCT 61 CelNdeIS ATTTCAATTTAAGGAAATCATATGAATAAC 60 Cel3XhoIA TAAACGCTCGAGATTACAACTATAAAG 62 *: Tm calculada por OligoCalc, Oligonucleotide Properties Calculador (http://www.basic.northwestern.edu/biotools/oligocalc.html). 3.3.2 Clonamiento del gen codificante para celulasa en el vector de expresión pET22b(+). A partir del DNA genómico de amplificó el gen codificante para celulasa, con los partidores que introducen en los extremos del gen los sitios de corte para las enzimas de restricción, requeridos para la clonación en el vector de expresión. En la Figura 21, se puede observar el análisis de la amplificación del gen codificante para celulasa, para ambas combinaciones de partidores. Figura 21: Análisis electroforético de los productos de PCR para la amplificación del gen codificante para celulasa e inserción de sitios de corte. Gel de Agarosa al 1%. M: Marcador de Peso Molecular; Carril 1: Amplificación obtenida con CelBamHIS y Cel3XhoIA; Carril 2: Amplificación obtenida con CelNdeIS y Cel3XhoIA. Las bandas que se observan en la Figura 21 corresponden al tamaño esperado. En el caso de la amplificación realizada con los partidores CelBamHIS y CelXhoIA el tamaño a obtener corresponde al gen sin su péptido señal de 32 residuos, lo que equivale a 57 una banda de 1390 pb. Y para la amplificación realizada con los partidores CelNdeIS y CelXhoIA el tamaño a obtener corresponde al gen entero, que son 1486 pb. Estos productos de amplificación fueron purificados desde el gel de agarosa según el protocolo (Anexo A.7) y digeridos con las respectivas enzimas de restricción según lo explicado en la sección 2.3.7. Posterior a eso, los insertos fueron ligados al vector de expresión, que a su vez fue linealizado con las mismas enzimas de restricción que sus respectivos insertos. Los constructos fueron transformados en células DH5α de E. coli, y posteriormente sembradas en placas LB suplementadas con ampicilina. De ambas transformaciones se eligieron colonias, las cuales se dejaron creciendo en 4 mL de medio líquido LB con ampicilina. Para identificar aquellas colonias recombinantes, se realizaron minipreparaciones de los cultivos con cada colonia, para luego hacer una reacción de digestión con las enzimas de restricción utilizadas para la clonación, y así verificar la presencia de inserto, y por ende la identificación de colonias recombinantes. El análisis de cada digestión se puede apreciar en la Figura 22. Figura 22: Análisis electroforético de las digestiones de las minipreparaciones. Gel de Agarosa al 1%. M: Marcador de Peso Molecular; Primeros carriles de izquierda a derecha (Arriba) 3, 5, 9, 11, 14,1 6 y 20: digestiones de las plasmidios provenientes de las colonias de los mismos números seleccionadas de la construcción pET-BamHI-XhoI; Carril (Arriba) 1, 3, 5, 7 , (Abajo) ,9 ,11, 13, 15, 17, 19, 20, 21, 22, 23 y 24: digestiones de las plasmidios provenientes de las colonias de los mismos números seleccionadas de la construcción pET-NdeI-XhoI. Como se puede observar, en el conjunto de digestiones correspondientes a las colonias con los constructos pET-BamHI-XhoI, no se observa una ninguna banda en la altura correspondiente al inserto (1390 pb), sin embargo se observa el vector linealizado a la altura de 5493 pb, por lo que se cree que la ligación al vector del inserto purificado y digerido no se produjo. Con lo anterior se puede decir que no hubo 58 colonias recombinantes en este caso. Una explicación a lo anterior es que el sitio de restricción para BamHI no haya sido reconocido por la enzima de restricción, lo que produciría una digestión solamente en el extremo 3’ por XhoI. Así el inserto estaría no apto para ligarse al vector de expresión linealizado, por no poseer uno de los extremos necesarios. Por lo tanto las colonias analizadas no fueron recombinantes por que no poseían el inserto. Posiblemente se produjo una autoligación del vector, lo que permitió a las colonias poder crecer en al antibiótico presente en la placa. La segunda opción, correspondiente a los constructos pET-NdeI-XhoI, muestra en todos los casos, (algunos más débiles que otros) digestiones que sí liberaron al inserto de tamaño esperado, que es de 1486 pb. Este caso por lo tanto si entregó colonias recombinantes. De estas colonias fueron seleccionadas los clones 1, 5, 9 y 20 para ser transformados a células de E.coli cepa BL21(DE3). 3.4 EXPRESIÓN DEL GEN DE LA CELULASA EN EL MICROORGANISMO RECOMBINANTE 3.4.1 Prueba de actividad celulasa extracelular. Con el fin de identificar aquellos clones con actividad celulasa obtenidos de las transformaciones en células de E.coli cepa BL21(DE3), se seleccionó un grupo de colonias para ser analizadas en placas con sustrato LB CMC 0,5 % (p/v), ampicilina 100 µg/mL e IPTG 0,1 μM. La actividad celulasa se identifica en forma visual por la formación de un halo de color amarillo alrededor de la colonia, lo que significa una degradación del sustrato presente en la placa por la enzima liberada al medio. Este ensayo revela actividad extracelular, ya que el sustrato se encuentra en el medio, por lo que de haber halo alrededor de ésta, significa que la enzima se secretó al medio extracelular exitosamente y de manera activa. 59 Figura 23: Ensayo de actividad de celulasa extracelular. Placa LB al 0,5% CMC (p/v), ampicilina 100 μg/mL e IPTG 0,1 μM. Colonias recombinantes provenientes del Clon Nº5, correspondiente a la construcción pET-NdeI-XhoI. De las cuatro placas, cada una con colonias provenientes de los cuatro clones seleccionados, una de ellas se diferenció del resto por poseer halos visiblemente más grandes y nítidos. Las otras placas o no presentaron actividad o el halo era muy pequeño o difuso. En la Figura 23, se muestra la placa con colonias provenientes del clon 5, que fue la colonia con mayor actividad. En la Figura 23 se observan halos alrededor de 19 colonias de 24 sembradas, una de las cuales fue seleccionada para almacenarla a -80 ºC para un uso posterior. El tener este resultado significa que se logró producir una celulasa activa, la cual es secretada al medio extracelular en el sistema recombinante E.coli BL21(DE3)/ pET22b(+), utilizando el péptido de exportación propio del gen. Con lo que se puede afirmar que la unión en el extremo terminal de la cola de histidinas, no afectó en el plegamiento propio de la enzima, y por ende la actividad tampoco se vio afectada. Con lo anterior, sería posible purificarla desde un cultivo en medio líquido, a mediante el uso de cromatografías de afinidad en columna con resina de níquel agarosa (NiNTA His·Bond, Novagen). Un problema que puede presentarse en esta etapa, es que la cola de histidinas no tenga acceso a la columna, por encontrarse escondida en el plegamiento de la enzima, en este caso se podría intentar con el protocolo de purificación utilizado para la enzima nativa. También puede suceder que se obtenga una proteína truncada que contenga solamente el dominio catalítico, al igual que el caso de la proteína secretada por las bacterias antárticas. En ese caso la cola de histidinas también se perdería, por encontrarse en el extremo C-terminal junto al CBD. La actividad fue obtenida previa incubación de las colonias a 37 ºC, por lo que se propone producir esta enzima en medio líquido para una posterior caracterización a 60 diferentes temperaturas, y así obtener un perfil de actividad que se pueda comparar con el de la enzima nativa purificada. También con lo anterior se podrá saber si la proteína es secretada en su totalidad al medio extracelular, o bien un porcentaje queda en el periplasma o citoplasma de las células. 61 4 CONCLUSIONES Se concluye que el protocolo de purificación consistente en tres pasos: precipitación con sulfato de amonio al 90 %, cromatografía de intercambio aniónico y por último una cromatografía de filtración en gel, es efectivo para la obtención de la enzima en forma pura, pero presenta un rendimiento bajo, el cual debe mejorarse. Con dicho procedimiento se logró obtener en forma pura una celulasa, del tipo endoglucanasa, observándose una única banda a la altura de los 36 kDa aprox., el cual correspondería al dominio catalítico de la enzima. Se concluye que la celulasa extracelular nativa, secretada por el microorganismo antártico Pseudoalteromonas es estable por al menos 16 h a pH alcalinos. La actividad máxima se alcanza a pH 8. Cel3 se puede clasificar entre las enzimas moderadamente adaptadas a bajas temperaturas, ya que su temperatura óptima de reacción es de 40 ºC aprox, mayor a las estrictamente adaptadas al frío. La enzima es estable frente a un amplio rango de temperaturas (20 a 45 ºC). Se concluye que esta enzima es adecuada para ser utilizada en procesos de lavado a temperatura ambiente, ya que a 20 ºC retiene un 80 % de actividad, temperatura a la cual las enzimas de organismos mesofílicos prácticamente no poseen actividad. Con estos resultados es posible concluir que Cel3 posee características que son atractivas para utilizarla como aditivo para detergentes. Cel3 posee un P.M de 35,7 kDa y un P.I de aproximadamente 4,5. Los parámetros cinéticos KM, VMAX y Kcat son 10,8 mg/mL, 357 U/L y 5,6x10-3 s-1. Se concluye que el método de Genome Walking, es apropiado para la obtención de la secuencia completa del gen de celulasa. Este gen de 1486 pb codifica para una proteína de 494 aminoácidos. Un análisis de la secuencia del gen de Cel3 llevó a concluir que la enzima es secretada como un precursor que posee un péptido de exportación al medio extracelular de 32 aminoácidos. La enzima posee un dominio catalítico y un dominio de unión a celulosa, separados por una secuencia aminoacídica no estructurada. El 62 dominio catalítico de Cel3 presenta similitud a dominios de la familia 5 de las glicosil hidrolasas, así como el CBD se puede clasificar entre los CBD de la familia 5. Se concluye que la celulasa producida por el microorganismo de origen antártico es muy similar, a nivel aminoácidico, a las endoglucanasas de origen antártico CelG y CelX, con un 97 y 94 % de similitud, respectivamente. Se logró clonar en forma correcta la secuencia del gen en el vector de expresión pET22b(+), la cual se expresó en forma activa a 37 ºC. Se concluye por lo tanto, que el sistema de expresión E. coli BL21(DE3)/pET22b(+) es apropiado para la expresión de una celulasa recombinante activa, secretada al medio extracelular. También se concluye que la señal de exportación del microorganismo psicrofílico es funcional en E. coli, ya que permitió la liberación de la enzima al medio. En suma, todo lo anterior lleva a concluir que se produjo en forma exitosa una celulasa de origen antártico en el sistema de expresión E. coli BL21(DE3)/pET22b(+), enzima que es muy buena candidata para su utilización en la industria de los detergentes. 63 5 BIBLIOGRAFÍA 1. Galante Y. M., Formantici C. (2003) Enzyme Application in Detergency and in Manufacturing Industries. Current Organic Chemistry, 7, 1399-1422. 2. Beilen J. B., Li Z. (2002) Enzyme Technology: an overview. Current Opinion in Biotechnology, 13, 338-344. 3. Cieslinski H., Kur J., Bialkowska A., Baran I., Makowski K., Turkiewicz M. (2005) Cloning, expression, and purification of a recombinant cold-adapted βgalactosidase from antartic bacterium Pseudoalteromonas sp. 22b. Protein Expression and Purification, 39, 27-34. 4. Kirk O., Borchert T.V., Fuglsang C.C. (2002) Industrial enzyme applications. Current Opinion in Biotechnology, 13, 345-351. 5. Shirai T., Ishida H., Noda J., Yamane T., Ozaki K., Hakamada Y., Ito S. (2001) Cristal Structure of Alkaline Cellulase K: Insight into the Alkaline Adaptaron of an Industrial Enzyme. J. Mol. Biol. 310, 1079-1087. 6. Henrissat B. (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J., 280, 309-316. 7. Lo Leggio L., Larsen S. (2002) The 1.62 Å structure of Thermoascus aurantiacus endoglucanase: completing the structural picture of subfamilies in glycoside hydrolase family 5. FEBS Letters, 523, 103-108. 8. Ohara H., Noguchi J., Karita S., Kimura T., Sakka K., Ohmiya K. (2000) Sequence of eg Vand Properties of Eg V, a Ruminococcus albus Endoglucanase Containing a Dockering Domain. Biosi. Biotechnol. Biochem., 64, 80-88. 9. Hui J.P.M., White T.C., Thibault P. (2002) Identification of glycan structure and glycosylation sites in cellobiohydrolase II and endoglucanase I and II from Trichoderma ressei. Glycobiology, 12, 837-849. 10. Violot S., Aghajari N., Czjzek M., Feller G., Sonan G., Gouet P., Gerday Ch., Haser R. and Receveur-Brechot V. (2005) Structure of Full Length Psychrophilic Cellulase from Pseudoalteromonas haloplanktis recvealed by Xray Difraction and small Angel X-ray Scatterin. J. Mol. Biol. 348, 1211-1224. 11. Brun E., Gans P., Marion D., Barras F. (1995). Overproduction, purification and characterization of the cellulose-binding domain of the Erwinia chrysanthemi secreted endoglucanase EGZ. Eur. J. Biochem. 231, 142-148. 12. Bhat, M. K. (2000) Cellulases and related enzymes in Biotechnology. Biotechnology Advances, 18, 355-383. 13. Dienes D., Egyházi A., Réczey K. (2003). Treatment of recycled fiber with Trichoderma cellulases. Industrial Crops and Products, 20, 11-21. 14. Pèlach M.A., Pastor FJ., Puig J., Vilaseca F. Mutjé P. (2002) Enzymic deinking of old newspapers with cellulase. Process Biochemistry, 38, 1063-1067. 64 15. Hoyoux A., Blaise V., Collins T. D’Amico S., Gratia E., Huston A.L., Marx J.C., Sonan G., Zeng Y., Feller G., Gerday Ch. (2004) Extreme Catalysts from LowTemperature Environments. Journal of Bioscience and Bioengineering, 98, 317330. 16. D’Amico S., Collins T.,Marx J.C., Feller G., Gerday Ch. (2006) Psychrophilic microorganisms: challenges for life. European Molecular Biology Organization, 7, 385-389. 17. Gerday Ch., Aittaleb M., Benthair M., Chessa J. P, Cleverie P., Collins T., D’Amico S., Dumont J., Garsoux G., Georlette D., Hoyoux A., Lonhienne T., Meuwis M.A., Feller., G. (2000). Cold-adpted enzymes: from fundamentals ti biotechnology. Trends in Biotechnol., 18, 103- 107. 18. D’Amico S., Claverie P., Collins T., Georlette D., Gratia E., Hoyoux A., Meuwis M.A., Feller G., Gerday G. (2002) Molecular basis of cold adaptation. Phil. Trans. R. Soc. Lond. B, 357, 917-925. 19. Cavicchioli R., Siddiqui K. S., Andrews D., Sowers R. K. (2002) Low temperatures extremophiles and their applications. Current Opinion in Biotechnology, 13, 253-261. 20. Demirjian D., Moris-Varas F., Cassidy C., (2001) Enzymes from Extremophiles. Current Opinion in Chemical Biology, 5, 144-151. 21. Garsoux G., Lamotte J., Gerday Ch. And Feller G. (2004) Kinetic and structural optimization to catalysis at low temperatures in a psychrophilic cellulase from the Antartic bacterium Pseudoalteromonas haloplanktos. J. Mol. Biol. 384, 247253. 22. Zeng R., Xiong P., Wen J. (2006) Characterization and gene cloning of a coldactive cellulase from a deep-sea psychrotrophic bacterium Pseudolateromonas sp. DY3. Extremophiles, 10, 79-82. 23. Hou Y., Wang T., Long H. Zhu H. (2007). Cloning, Sequencing and Expression Analysis of the First Cellulase Gene Encoding Cellobiohydrolase 1 from a Coldadaptive Penicillium chrysogenum FS010. Acta Biochimica et Biophysica Sinica, 39, 101-107. 24. Hewitt L., McDonell J. M. (2004). Screening and Optimizing Protein Production in Escherichia coli. Methods Mol. Biol., 278, 1-16. 25. Feller G., Le Bussy O., Gerday Ch. (1997). Expression of Psychrophilic Genes in Mesophilic Hosts: Assesment of the Folding State of a Recombinant αAmilase. Applied and Environmental Microbiology, 64, 1163-1165. 26. Bollag, D. M., Rozycky, M. D., and Edelstein, S. J. (1996) “Protein Methods” Capitulo 5 y 7, segunda ed, Wiley-Liss, New York. 27. Acevedo J.P., Reyes F., Parra L., Salazar O., Andrews B.A., Asenjo J.A. (2006) Improved Genome Walking Method not limited by restriction enzymes tested in antartic bacterial genes. Biotechniques (en revisión). 65 28. Bradford, M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-due binding. Anal. Biochem. 72, 248-254. 29. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) “Molecular Cloning: A Laboratory Manual”, segunda ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor , NY. 30. Salazar O., Descripción del proyecto Fondecyt 2006, orientado a aislamiento, optimización y caracterización de celulasas psicrofílicas de origen marino. 66 6 ANEXOS A METODOLOGÍA ADICIONAL A.1 Determinación de la concentración de proteínas Se midió la concentración de proteínas total en muestras mediante el método de Bradford [28], midiendo absorbancia a 595 y 465 nm. Para la obtención de la concentración de proteínas se utilizó una curva de calibración construida con BSA como estándar (Anexo C.1), la cual posee un rango de linealidad confiable entre una razón Abs595/Abs465 de 0,5-3,5, equivalente a 0,05 -0,5 mg/mL. A.2 Extracción de DNA Genómico desde bacterias Las bacterias se crecieron en 200 mL de medio Marino 2216, al 2 % de glucosa (Anexo B.1 parte e), a 4 ºC con agitación orbital durante 4 días. Posteriormente el cultivo se dividió en tubos falcón de 15 mL, con 10 mL de cultivo cada uno. Los tubos fueron centrifugados por 10 min a 5.000 rpm a 4 ºC. El pellet obtenido fue resuspendido en 1 mL de Buffer TE (Anexo B.1 parte a). Luego se siguió el siguiente protocolo de extracción de DNA genómico bacterial. (Protocolo descrito por Sambrook & Russell) [29]: 1. En un tubo Eppendorf estéril se centrifugó a 13.000 rpm por 1 minuto. (Centrifuga Eppendorf 5403 de Eppendorf AG, Hamburgo, Alemania). 2. Se resuspendió el pellet en 200 μL de Tritón X-100 SDS filtrado (Anexo B.1 parte b). Se agregó 200 μL de una mezcla fenol-cloroformo 1:1 más 0,3 g de perlas de vidrio (425-600 μm). Se agitó con vortex por 2 minutos. 3. Se agregó 200 μL de buffer TE y se agitó con vortex. Luego se centrifugó por 5 min a velocidad máxima (13.000 rpm). (Centrifuga Eppendorf 5403 de Eppendorf AG, Hamburgo, Alemania). 4. La fase acuosa se transfirió a otro tubo Eppendorf estéril y se repitió la extracción con 200 μL de cloroformo, se volvió a centrifugar a 13.000 rpm por 5 min (Centrifuga Eppendorf 5403 de Eppendorf AG, Hamburgo, Alemania) y se transfirió nuevamente la fase acuosa (superior) cuidadosamente a un tubo limpio estéril. 67 5. Se agregó 1 mL de etanol 100 % frío. Se mezcló por inversión muy despacio, dos veces. Se dejó incubar a 4 °C por 30 min. 6. Luego de la incubación se centrifugó a máxima velocidad (13.000 rpm) por 10 min a 4 °C. (Centrifuga Eppendorf 5804 R de Eppendorf AG, Rotor Eppendorf F45-30-11, Hamburgo, Alemania). 7. El pellet se lavó con 1 mL de etanol 70 % frío y se centrifugó por 5 minutos. 8. Se dejó secar el pellet (cerca del mechero 10 a 15 minutos) hasta que se su color fue transparente. Luego se resuspendió en buffer TE (400 μL) y se agregó 1 μL de Rnasa. 9. Finalmente se guarda a -20 °C. A.3 Electroforesis de DNA Los fragmentos de DNA fueron analizados en geles de agarosa de una concentración en función del tamaño del fragmento a analizar; para tamaños de DNA genómicos se utilizó una concertación del 0,8 a 1 % (p/v), hasta 2,5 % (p/v) para fragmentos menores a 500 pb; a los geles se agregó bromuro de etidio 5 µg/ml. Se utilizó el buffer TAE 1X (Anexo B.3 d) para la preparación de agarosa y como buffer de corrida. La electroforesis se realizó a 100 volts. Las muestras se prepararon en proporción 5:1 con buffer de carga 6X (Anexo B.3 parte e). Se observó el gel sobre un transluminador UV y se fotografió. A.4 PCR de Colonias Con el fin de comprobar la presencia de insertos en las colonias transformantes de color blanco, las colonias seleccionadas se transfirieron con el extremo de una punta estéril desde la placa de transformación hacia la placa LB agar suplementada con ampicilina con una cuadrícula numerada. La placa se incubó a 37 ºC toda la noche. También la punta con la colonia rescatada de la placa es pasada en 40 µL de agua Milli-Q estéril en un tubo eppendorf de 0,6 mL. Se incubó dicho tubo a 100 ºC durante 10 min, y se transfirió 5 µL de dicha a mezcla a un tubo eppendorf de 0,6 mL con 15 µL de mezcla de PCR. La existencia de inserto se confirmó utilizando en el PCR de colonias los partidores utilizados para la amplificación del fragmento clonado. El protocolo del PCR fue el descrito en la sección 2.3.4.5. Los productos de PCR de colonias fueron analizados en un gel de agarosa al 1 % (p/v). 68 A.5 Electroforesis de proteínas SDS-PAGE Las separaciones electroforéticas en condiciones denaturantes se realizaron en una cámara vertical MiniProtean II. Las muestras para cargar se prepararon en razón 4:1 con buffer de carga 5X, se denaturaron por ebullición durante 5 minutos y posteriormente se cargaron en geles de bis-acrilamida al 12,5 % (p/v) preparado de acuerdo a la metodología descrita en [26]. La electroforesis se realizó bajo voltaje constante de 200 V, utilizando buffer de corrida descrito [26]. Para la visualización de las proteínas en el gel de poliacrilimida, se procedió a teñirlo durante 1 h con una solución de tinción Coomasie (ver Anexo B.3 parte j) incubándolo a temperatura ambiente con agitación orbital. Luego, se procedió a desteñirlo con la solución de destinción (ver Anexo B.3 parte k) incubándolo a temperatura ambiente con agitación orbital durante 1 hora. A.6 Zimograma de Celulasas Para determinar la actividad enzimática in situ, es decir visualizar la actividad en un gel de poliacrilamida, se corrió un gel de bis-acrilamida al 10 % (p/v) preparado según la metodología descrita en [26]. Las muestras se prepararon en razón 4:1 con el buffer de carga 5X “Nativo” y se cargaron en el gel. La corrida del gel se realizo igual que la electroforesis de proteínas normal. Para la visualización de las bandas de actividad en el gel de poliacrilimida, se procedió a lavarlo durante 1 h con Tritón X-100 2,5 % (p/v) incubándolo a temperatura ambiente con agitación orbital, después de eso se lavo durante 5 minutos con agua destilada. Luego, se procedió a incubarlo durante 10 min en el buffer a pH 10, o en su defecto toda la noche a temperatura ambiente. Se tiñó el gel con Rojo congo al 0,1 % (p/v) durante 15 a 20 min para finalmente lavar el gel con Cloruro de Sodio 1M. A.7 Purificación de DNA a partir de geles de agarosa Se cortó la banda de DNA de interés desde el gel de agarosa con un bisturí limpio, tratando de minimizar lo más posible el fragmento, eliminando los excesos de agarosa. 69 Se colocó la agar en un tubo eppendorf de 1,5 mL limpio estéril, previamente pesado. Se pesó el tubo con la agarosa, y se obtuvo la masa de la agarosa. Luego se siguió el protocolo descrito por el sistema QIAEX II Gel Extratction (QIAGEN). Finalmente se eluyó el DNA en agua Milli-Q estéril pH 8,5 a 50 ºC. A.8 Minipreparación de DNA plasmidial Se purificó y se extrajo el DNA plasmidial según el protocolo propuesto por el sistema QIAprep Spin Miniprep Kit (QIAGEN). A.9 Preparación células electrocompetentes Se incoculó 10 mL de medio SOB (Anexo B.1 parte a) con una colonia de células de E. coli DH5α o BL21(DE3) conservadas a -80 ºC y se creció con agitación orbital durante toda la noche a 37 ºC. Al día siguiente se midió la densidad óptica del cultivo D.O600, utilizando como blanco medio estéril, y se calculó el volumen del inóculo necesario para 1 L de cultivo con D.O600 final igual a 0,05. Se incubó el cultivo de 1 L de 2 a 4 horas con agitación orbital a 37 ºC hasta que se obtuvo una D.O600 entre 0,6 y 0,8. Luego el cultivo fue enfriado en hielo durante 10 minutos y se centrifugó a 5.000 g por 10 minutos (centrifuga Sorvall® RC-28S y rotor GS-3 Sorvall®, DuPont, CT, USA). Las células contenidas en el pellet obtenido de la centrifugación se lavan 4 veces con glicerol 10 % (v/v) estéril. Finalmente el pellet lavado se resuspendió en el glicerol residual del último lavado. Se alicuotó en volúmenes de 50, 75 y 100 µL para ser almacenados a -80 ºC. 70 B Medios de Cultivos y Buffers B.1 Medios Líquidos a) Medio SOB (100 mL) • • • • • • 2 g triptona 0,5 g de extracto de lavadura 0,0584 g de NaCl 0,0186 g de KCl 100 mL agua destilada Autoclavar 20 min a 110 ºC. b) Medio SOC (100 mL) • • • • • • • • • 0,36 g glucosa (20mM) 0,21 g cloruro de magnesio (10 mM) 0,25 g sulfato de magnesio (10 mM) 0,018 g cloruro de potasio (2,5 mM) 0,058 g cloruro de sodio (10 mM) 4 g triptona (20 g/L) 1 g extracto de Levadura (5 g/L) 100 mL agua destilada Autoclavar 20 min a 110 ºC. c) Medio Marino 2216 Difco, contiene cada 1L: • • • • • • • • • • • • • • • • 5 g peptona 1 g extracto de lavadura 0,1 g citrato férrico 19,45 g cloruro de sodio 5,9 g cloruro de magnesio 3,24 g sulfato de sodio 1,8 g cloruro de calcio 0,16 g bicarbonato de sodio 0,55 g cloruro de potasio 0,08 g bromuro de potasio 0,034 g cloruro de estroncio 0,022 g ácido bórico 0,004 g silicato de sodio 0,0024 g fluoruro de sodio 0,0016 g nitrato de amonio 0,008 g fosfato disódico Cada 1 L de agua destilada se disolvió 37,4 g del Medio Marino. Se hirvió unos minutos y se esterilizó por autoclave por 20 minutos a 110 ºC. 71 d) Medio Marino 2216 al 0,5 % (p/v) CMC Al medio marino se agregó 0,5 gramos de CMC cada 100 mL luego de hervirlo. Se autoclavó por 20 minutos a 110 ºC. e) Medio Marino 2216 al 2 % (p/v) Glucosa (100 mL) Al medio marino se agregó 0,5 g de CMC cada 100 mL luego de hervirlo. Se autoclavó por 20 minutos a 110 ºC. f) Medio LB (Luria Bertani) contiene cada 1 L: • • • 10 g Triptona 5 g extracto levadura 5 g cloruro de sodio Se agregó 0,95 g NaCl cada 100 mL y se autoclave por 20 minutos a 110 ºC. B.2 Medios Sólidos a) Placas Petri con LB-agar (100 mL) • • • • • 1,55 g de medio LB 0,95 g NaCl 1,5 g agar 100 mL agua destilada Autoclavar 20 min a 110 ºC. Se dejó enfriar hasta que llegue aproximadamente a los 50 ºC, se agregó 100 µL de ampicilica de concentración 100 mg/mL (concentración final de 100 µg/mL), 100 µL de IPTG 0,1 M (concentración final 100 µM) y 160 µL de X-Gal a una concentración de 50 mg/mL. Se agregó esta mezcla a placas petri de 8 cm antes de enfriarse totalmente. Se esperó que se gelifique. b) Placas Petri con LB-agar con sustrato (100 mL) • • • • • • 1,55 g de medio LB 0,95 g NaCl 1,5 g agar 100 mL agua destilada 50 mg Carboximetil Celulosa (CMC) Autoclavar 20 min a 110 ºC Se dejó enfriar hasta que llegue aproximadamente a los 50 ºC, se agregó 100 µL de ampicilica de concentración 100 mg/mL (concentración final de 100 µg/mL), 100 µL de IPTG 0,1 M (concentración final 0,1 mM). Se agregó esta mezcla a placas petri de 15 cm antes de enfriarse totalmente. Se esperó que se gelifique. 72 c) Placas Petri con Medio Marino 2216 Se agregó 1,5 g de agar a 100 mL de Medio Marino 2216 líquido. Se autoclave 20 min a 110 ºC. B.3 Soluciones y Buffers a) Buffer TE • • Tris 10 mM, pH 8 EDTA 1 mM pH 8 b) Tritón X-100 SDS • • • • • Tris-HCl 10 mM pH 8 Tritón X-100 2 % (v/v) SDS (Sodio Dodecil Sulfato) 1 % (p/v) NaCl 100 mM EDTA 1 mM pH 8 c) Solución DNS (100 mL) • • • 1 g de ácido dinitrosalicílico 20 mL de NaOH (2N) 30 g de sales de Rochelle d) Buffer TAE 50X (1 L) • • • 242 g Tris base 57,1 g de ácido acético glacial 100 mL EDTA 0,5 M, pH 8 e) Buffer de Carga 6X • • Glicerol 30 % (p/v) Azul de Bromofenol 0,25 % (p/v) f) IPTG 0,1 M Se preparó una solución de IPTG 0,1 M en agua Milli-Q estéril. Se esterilizó por filtración mediante un filtro estéril con tamaño de poro de 0,2 µm. Se almacenó a -20 ºC. g) X-Gal 50 mg/ml Se preparó una solución de 5-bromuro-4-cloro-3-indolil-D-galactosido (XGal) a una concentración de 50 mg/mL en N,N’-dimetil-formamida. Se almacenó a -20 ºC. 73 h) Ampicilina 100 mg/ml Se preparó una solución de ampicilina en agua Milli-Q estéril a una concentración final de 100 mg/mL. Se esterilizó por filtración mediante un filtro con tamaño de poro de 0,2 µm. Se almacenó a -20 ºC. i) Carbenicilina 100 mg/ml Se preparó una solución de carbenicilina en agua Milli-Q estéril. Se purificó por filtración mediante un filtro con tamaño de poro de 0,2 µm. Se almacenó a -20 ºC. j) Solución Tinción de Gel Coomassie (1L) • • • • 1 g Comassie Blue R-250 450 mL Metanol 450 mL Agua destilada 100 mL Acido Acético k) Solución Destinción Gel (1L) • • • l) 450 mL Metanol 450 mL Agua destilada 100 mL Ácido Acético Buffers utilizados para ensayo de actividad celulasa Tabla 7: Buffers utilizados para el ensayo de actividad celulasa. Los reactivos de los buffers de pH 6 a 11 son SIGMA, y todos se prepararon con concentración 25 mM pH 3 5 6 7 8 9 10 11 Buffer Citrato de Sodio Acetato de Sodio MES MOPS TRIZMA TAPS CAPS CAPS 74 C Curvas de Calibración C.1 Curva de Calibración BSA Se utilizaron distintas concentraciones de BSA entre 0,5 a 0,0625 mg/mL. El blanco se realiza con agua en vez de proteína. Se determinó la concentración de (Abs 595/Abs 465)M-(Abs 595/Abs 465)B proteínas según el método de Bradford [28]. 4,0 3,5 3,0 2,5 2,0 y = 6,7689x R2 = 0,994 1,5 1,0 0,5 0,0 0 0,1 0,2 0,3 0,4 0,5 0,6 BSA [mg/ml] Figura 24: Curva de Calibración BSA C.2 Curva de Calibración DNS Se utilizaron distintas diluciones de una solución de glucosa de concentración de 3,125 mg/mL. A 50 µL de cada dilución de le agregó 50 µL de DNS en una placa de 96 pocillos. Se incubó por 10 minutos a 100 ºC, se enfrió en hielo y se leyó la absorbancia a 550 nm a 50 µL de reacción. Se utiliza como blanco agua. 75 1,0 Abs 550 nm 0,8 0,6 y = 0,0469x + 0,0021 R2 = 0,9855 0,4 0,2 0,0 0 5 10 15 20 Glucosa [mM] Figura 25: Curva calibración DNS para la detección de azúcares reductores C.3 Curva de Calibración Determinación Peso Molecular mediante Filtración por Gel Para la construcción de la curva de calibración se realizó cromatografías de filtración por gel con proteínas de peso molecular conocido, dichas proteínas y sus respectivos pesos moleculares se muestran en la Tabla 8. Tabla 8: Proteínas utilizadas para la construcción de la curva de calibración de peso molecular y sus respectivos pesos moleculares Proteína Peso [kDa] BSA Ovalbúmina Quimiotripsinógeno Ribonucleasa A 66 43 25 13,7 P.M. [kDa] 80 60 40 20 0 5 5,5 6 6,5 Ve [mL] 7 7,5 y = -22,777x + 180,42 R2 = 0,9355 Figura 26: Curva calibración P.M en cromatografía de filtración por gel 76 8 C.4 Curva Calibración de Crecimiento de Biomasa en Medio Marino al 0,5 % (p/v) CMC Una vez finalizado el cultivo, se tomó 15 mL de muestra, y se realizaron diluciones en duplicado, completando un volumen de 4 mL por dilución. Se midió densidad óptica a 600 nm. Se filtró 3 mL de cada dilución utilizando filtros de 0,2 µm, los cuales fueron pesados antes de filtrar. Se dejaron secar los filtros durante 48 horas en estufa a 60 ºC. Se usó como referencia un filtro seco y un filtro en el cual se filtró solo agua. Por diferencia de peso, se obtuvo la correlación de biomasa y D.O600 medidas. 3,0 Abs 600 nm 2,5 2,0 y = 767,47x - 0,3422 R2 = 0,9765 1,5 1,0 0,5 0,0 0,001 0,0015 0,002 0,0025 0,003 Biomasa [gr/ mL] Figura 27: Curva de calibración de Biomasa 77 0,0035 0,004 D Curva de Linealización según Dixon de curva de saturación Linealización Dixon y = 0,0028x + 0,0304 R2 = 0,9586 0,1 [CMC]/V 0,08 0,06 0,04 0,02 0 0 5 10 15 20 CMC [mg/mL] Figura 28: Linealización de curva de saturación según Lineweaver-Burk 78 25