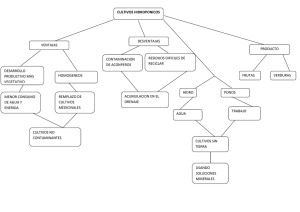
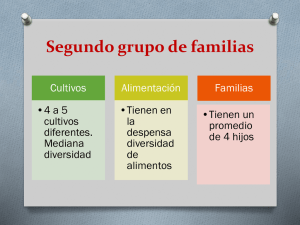
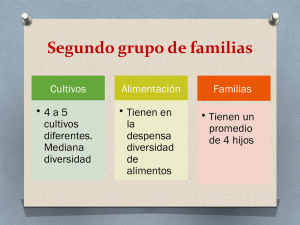
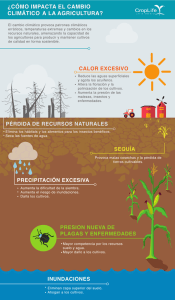
Resúmenes Clases - Facultad de Ciencias Bioquímicas y
Anuncio

Facultad de Ciencias Bioquímicas y Farmacéuticas Universidad Nacional de Rosario Curso de Posgrado: INTRODUCCIÓN AL CULTIVO DE CÉLULAS EUCARIOTAS AÑO 2015 INTRODUCCIÓN AL CULTIVO DE CÉLULAS EUCARIOTAS Directores: Dr. Esteban Serra, Dra. Virginia Perdomo. Organizadores: Dra. Victoria Alonso Dra. Virginia Perdomo Docentes: Dra. Tatiana Vega, [email protected] Dra. Silvina Villanueva, [email protected] Dra. Laura Ruiz, [email protected] Dra. Ma. Teresa Ronco, [email protected] Dra. Virginia Perdomo, [email protected] Dra. Pamela Cribb, [email protected] Dra. Victoria Alonso, [email protected] Dra. Isabel Nocito, [email protected] Dra. Romina Manarín, [email protected] Dr. Ismael Barosso, [email protected] Dra. Stella Mattaloni, [email protected] Dra. Anabela Ferretti, [email protected] Lic. Maite Arana, [email protected] Lic. Facundo Tonucci, [email protected] Lic. Luis Tavernelli, [email protected] Lic. Carla Ritagliati, [email protected] Disertantes Invitados: Dra. Guillermina Forno, Dra. Silvia Altabe, Dra. Ana Laura Cavatorta, Lic. Juliana Cicaré, Representante de ThermoFisher Scientific, Representante Instrumentalia. Nuestros colaboradores: 2 3 Índice de Temas: El laboratorio de cultivo celular. Requisitos de cultivos celulares. Fase gaseosa y temperatura. Medios de cultivo y suplementos. Material de cultivo celular. Técnicas de manipulación estéril. Trabajo en el laboratorio de cultivo celular. Manejo de cultivos celulares. Preparación de medios de cultivo e inactivación de sueros. Tipos de cultivos celulares. Cultivos en suspensión y monocapas. Líneas celulares. Cepas. Mantenimiento de líneas celulares y criopreservación. Cultivo de protozoos parásitos. Introducción al cultivo de líneas celulares. Cultivo de células animales (VERO, RAW 264.7) y parásitos. Cultivo primario. Concepto y tipos. Etapas en el establecimiento de un cultivo primario. Cultivo primario de Hepatocitos en Sandwich. Cultivo de Plantas. Transfecciones. Transfecciones de Trypanosoma cruzi. Ciclo celular y citometría de flujo. Microscopia confocal e inmunofluorescencia. Contaminaciones del cultivo celular. Fuentes, consecuencias, prevención y eliminación. Bioseguridad en el laboratorio de cultivo. Ventajas y aplicaciones de la técnica de cultivo celular. 4 Clase Teórica: LABORATORIO DE CULTIVO CELULAR Docente: Dra. Ma. Terasa Ronco [email protected] LABORATORIO DE CULTIVO CELULAR El cultivo de tejidos ha sido considerado durante mucho tiempo como una mezcla de ciencia y arte. Inicialmente se consideró como una técnica particularmente difícil de aprender. Sin embargo, estas dificultades de los comienzos están hoy en día prácticamente superadas gracias a factores como la disponibilidad de antibióticos, los medios de composición definida, las instalaciones asépticas (salas de cultivo limpias, cabinas de flujo laminar, incubadores estériles), dispositivos de cultivo. Los avances técnicos y la aparición de un buen número de compañías comerciales de suministro de medios, sueros, equipo y líneas celulares han hecho del cultivo celular una tecnología con buena reproducibilidad. Podemos dividir el cultivo de tejidos en dos grupos de técnicas: el cultivo de órganos y el cultivo de células. El cultivo de órganos se puede definir como el mantenimiento de pequeños fragmentos de tejido u órganos completos in vitro. El cultivo celular es la propagación de células dispersas tanto en suspensión como en monocapas sobre cristal o plástico. Introducción histórica. El cultivo de tejidos se desarrolló a partir de los últimos años del siglo XIX como una continuación de las técnicas de la embriología. Wilhem Roux mantuvo en el año 1885 células de embrión de pollo en solución salina durante unos dias. El zoólogo americano R.G. Harrison es considerado el iniciador de los cultivos de tejidos animales, en 1907. Harrison fue el primer autor que empleó técnicas in vitro para el estudio de fenómenos in vivo, realizando cultivos de médula espinal embrionaria de anfibios. Pudo observar el crecimiento de los axones de los neuroblastos, y estableció que el axón se formaba por expansión a partir del cuerpo neuronal y no por fusión de una cadena de células. El cultivo se realizaba en una gota de linfa del anfibio que colgaba de un cubreobjetos sobre una cámara sellada. La primera limitación para el establecimiento de cultivos era lograr un medio nutritivo adecuado. Burrows (1910) empleó plasma de pollo para nutrir los explantes de tejidos embrionarios de pollo. Este medio se reveló mucho mejor que los anteriormente probados, lo que le permitió observar el crecimiento del tejido nervioso, corazón y piel. Burrows y Carrel realizaron los primeros intentos de establecer cultivos de células de mamífero, y consiguieron mantener explantes obtenidos a partir de perros, gatos y conejos de indias, así como en el crecimiento de tumores sólidos. Demostraron que la vida del cultivo se puede prolongar mediante subcultivo. Los medios empleados fueron plasma suplementado con extractos de embrión. 5 Rous y Jones (1916) emplearon por vez primera extractos enriquecidos en tripsina para disociar las células de embriones de pollo, estableciendo el primer cultivo celular. Uno de los mayores problemas que describen para el establecimiento de los cultivos celulares es la aparición de múltiples contaminaciones, por lo que desarrollaron numerosos métodos de manipulación en condiciones de asepsia que aún hoy día se utilizan. En 1913 Carrel demostró la posibilidad de mantener en cultivo células extraídas de un animal, embrión de pollo, durante un periodo de tiempo superior al de la vida de éste. Gran parte del éxito en el mantenimiento de los cultivos se debió al desarrollo del denominado frasco de Carrel. Entre los años 1920 y 1940 se desarrollaron diferentes estrategias de obtención de cultivos y de mantenimiento de las condiciones estériles, pero sin grandes avances. A partir de los años 40, con el aislamiento de los primeros antibióticos, se desarrollaron numerosas aplicaciones de entre las que podemos destacar: Earle y col. (1948) aislaron células de la línea celular L y mostraron que eran capaces de formar clones en el cultivo de tejidos. Demostraron que para que una célula llegue a dividirse necesita ser alimentada con los nutrientes correctos. Grey y col. (1952) establecen la primera línea celular continúa, las actualmente bien conocidas células HeLa. El medio empleado era extremadamente complejo y poco definido: plasma de pollo, extracto de embrión bovino y suero de cordón umbilical humano. Rita Levi-montalcini y col. (1954) establecen que el factor de crecimiento nervioso estimula el crecimiento de los axones en tejidos en cultivo. Este trabajo supuso el Premio Nobel para Levi-Montalcini en 1986. Eagle (1955) realiza la primera investigación sistemática de los requerimientos nutritivos de las células en cultivo. Describe que las necesidades del cultivo de soluciones corporales complejas (sueros) pueden ser satisfechas por tan poco como el 1% de suero de caballo dializado en un medio definido de pequeñas moléculas (aminoácidos, azúcares) Hayflick y Moorhead (1961) usaron por primera vez antibióticos para prevenir la contaminación de los cultivos de fibroblastos. Pudieron mantener estos cultivos durante unos 12 pases, pero no consiguieron establecer líneas estables. Ham (1965) introduce el primer medio definido libre de suero capaz de mantener algunas células de mamífero en cultivo indefinidamente (Ham, 1965). Augusti-Tocco y Sato (1969) establecen la primera línea celular estable de neuroblastoma aislando clones que establecían procesos nerviosos y que eran eléctricamente excitables. Se empiezan a establecer las primeras líneas celulares diferenciadas. Kohler y Milstein (1975) establecen la primera línea celular productora de anticuerpos monoclonales. El establecimiento de la tecnología de obtención de anticuerpos monoclonales les valió el Premio Nobel. Sato y col. (1976) publicaron sus trabajos en los que demuestran que las diferentes líneas celulares requieren mezclas distintas de hormonas y factores de crecimiento para crecer en medios libres de suero. (Sato y col., 1982). 6 En los últimos años la aplicación de la tecnología del cultivo celular ha permitido grandes avances en la comprensión de los mecanismos implicados en los procesos intracelulares e intercelulares con el establecimiento de co-cultivos. A pesar de que el primer animal del que se establecieron cultivos celulares era un anfibio, poiquilotermo, rápidamente el interés se centró en el cultivo de células de animales homeotermos, especialmente humanas, por su gran interés médico. Sin embargo en los últimos años, y especialmente debido a la problemática del control de plagas e infecciones en agricultura y piscicultura, se ha desarrollado notablemente el establecimiento de líneas celulares de poiquilotermos e invertebrados. Conceptos actuales de cultivo celular: Actualmente se entiende por cultivo celular al conjunto de técnicas que permiten el mantenimiento de las células 'in vitro', manteniendo al máximo sus propiedades fisiológicas, bioquímicas y genéticas. Se pueden cultivar células a partir de tejidos humanos, animales y vegetales. Un cultivo celular implica la previa disgregación (mecánica o enzimática) del tejido original y que las células son cultivadas en monocapa (sustrato sólido) o en suspensión. Dependiendo del grado de preservación de la estructura del tejido o del órgano de origen y de su duración hablaremos de diferentes tipos de cultivos: de órganos, explantes, primarios o secundarios. El laboratorio de cultivo celular: La característica principal que define al laboratorio de cultivo celular es el mantenimiento de la asepsia. La tasa de crecimiento de las células en cultivo es muy inferior a la de los contaminantes habituales: hongos, levaduras, bacterias y micoplasmas, y por ello para el mantenimiento del cultivo será vital evitar la aparición en éste de cualquier microorganismo indeseado. El área de trabajo para realizar cultivos debe instalarse en una parte del laboratorio tranquila, alejada de las vías de paso y de ser posible dedicada exclusivamente al cultivo de células. La solución ideal e s disponer de una habitación aislada. Las aparición de cabinas de flujo laminar redujo las necesidades de aislamiento del área de trabajo pero aún así es recomendable mantener un gradiente de esterilidad, desde el medio exterior o laboratorio general al interior de las cabinas de flujo donde se manipularán los cultivos y del incubador donde se mantendrán. Un laboratorio de cultivo celular debe contar con una infraestructura básica en la cual se disponga de áreas independientes para llevar a cabo la preparación y esterilización de medios y reactivos, un espacio independiente para el proceso de lavado y preparación de material y un área propiamente destinada al trabajo con cultivos celulares. Dentro de la infraestructura física básica se debe contar con sistemas de refrigeración y congelación, incubadoras, centrífugas, balanzas, microscopios y cabinas de flujo laminar. 7 Adicionalmente, se debe disponer de un buen suplemento de material plástico y de vidrio y con sistemas de esterilización apropiados para los diferentes tipos de reactivos y material utilizados. Modelo de laboratorio para cultivo celular: Objetivo: proteger al operador y al producto. Tener en cuenta: Diseño Ubicación Equipamiento Entrenamiento del personal encargado Áreas de un laboratorio a) Área estéril o de cultivo: tener cámaras de flujo laminar o cerradas con UV. Requerimiento: Sin corrientes de aire Filtros de aire Desinfecciones periódicas Luz UV en períodos de descanso Área, equipos y materiales usados sólo para cultivo celular b) Incubación: estufas humidificadas a 37ºC, gaseadas con CO2 o termostáticamente controlados. c) Preparación Los medios se filtran en cámaras de flujo laminar pHímetros y agitador magnético guantes, barbijo y guardapolvo d) Almacenamiento: tanques de N2 para los cultivos celulares e) Lavado y esterilización: preparación del material lejos del área de cultivo cuartos Tipos de laboratorio de cultivo celular: Las necesidades de asepsia dependen del tipo de material que se va a procesar en el laboratorio. Esto define dos tipos de contención y de área de trabajo diferentes: 1. el laboratorio de cultivo de tejidos general, para la manipulación de cultivos no patógenos se instalará preferentemente en una sala aislada y a la cual se le suministrará aire filtrado (normalmente mediante un equipo de filtración y regulador de la temperatura). Esto produce un aumento de presión atmosférica en el interior del laboratorio (típicamente entre 15 y 20 mm de Hg) que impide la entrada de aire no filtrado al área limpia. 2. el laboratorio de cultivo con patógenos supone un nivel de contención superior. A fin de evitar la salida accidental de éstos agentes se filtra el aire que sale de la sala, generando un déficit de presión en el interior de ésta. El aire sale siempre estéril. 8 La instrumentación del laboratorio de cultivo celular: En el laboratorio de cultivo celular propiamente dicho se encuentran los siguientes tipos de instrumentos: 1. cabinas de flujo laminar (áreas de trabajo y de contención estéril). 2. incubadores : baños termostatizados, incubadores de CO2, Incubadores 'roller'. 3. instrumentos ópticos de observación del cultivo : microscopio invertido de contraste de fases, de fluorescencia. 4. instalación de criogenia (tanques de N2 líquido) y congeladores 5. equipo de esterilización: autoclave, equipos de filtración. 6. otros instrumentos: centrífugas, contadores electrónicos (Cell Counters), equipo de purificación de agua, balanzas, pH-metro, pipeteadores. Las Cabinas de Flujo Laminar. Su función es la de mantener un área libre de partículas, especialmente de posibles contaminantes (bacterias, levaduras) que puedan acceder al cultivo. Esto se consigue mediante un dispositivo mecánico que fuerza el paso del aire a través de un filtro de gran superficie (filtro HEPA) situado o bien en el techo (flujo vertical) o en la pared frontal (flujo horizontal) y que con una eficiencia del 99.999 % retiene las partículas por debajo de un cierto calibre que es en general de 0.2 um. El flujo del aire es laminar, sin turbulencias en las que puedan quedar retenidas partículas contaminantes. El flujo laminar se asegura tanto por la gran superficie del filtro HEPA como por la velocidad constante del aire, como por la ausencia de fuentes intensas de calor (mecheros bunsen) en el interior de las cabinas, generadores de intensas corrientes de convección. Las cabinas de flujo laminar pueden ser de dos tipos: flujo vertical y flujo horizontal, dependiendo de la posición del filtro HEPA (High Efficiency Particulate Air) y por ello de la dirección del flujo laminar Los diferentes tipos de cabinas de flujo laminar se diseñan con diferentes propósitos: a. Protección personal: protección del personal de los posibles agentes dañinos del interior de la cabina. b. Protección del producto, experimento o cultivo que se encuentra en el interior de la cabina de los contaminantes exteriores o de la contaminación cruzada con otros productos o cultivos situados en la misma cabina. c. Protección medioambiental: evitar la salida al medio ambiente de productos o agentes contaminantes. Las cabinas de flujo laminar horizontal son muy adecuadas para una buena protección del producto, pero no son adecuadas para el trabajo con materiales peligrosos o con algún tipo de riesgo pues el operador queda completamente expuesto. En las cabinas de flujo vertical, más sofisticadas, se segura una buena protección del producto, y, dependiendo de su diseño se puede asegurar una protección total del operador. Son por ello más adecuadas para el trabajo con agentes peligrosos. Dependiendo de la importancia que se le conceda a cada uno de estos factores en el diseño de la cabina se trata de cabinas de clase I, II o III. 9 Cabina de clase I. Se trata de una unidad de contención parcial adecuada para la manipulación de agentes de bajo riesgo, donde existe una necesidad de protección del operario y del medio pero no del producto. Este es el tipo de cabinas normalmente denominadas "de gases", y no son de uso común en el laboratorio de cultivo de tejidos. Figura 1: Cabina de seguridad biológica Clase I Cabina de clase II. Este tipo de cabinas protegen el producto, al personal y al medio ambiente. En general las cabinas de clase II se describen como un equipo de protección con un panel frontal de acceso y que mantienen un flujo laminar estable en el interior, con una filtración HEPA para el aire recircularizado en cada ciclo y una filtración HEPA del aire exhausto (de salida al medio). Los sucesivos diseños que se han realizado para cubrir necesidades diferentes permiten clasificar asimismo las cabinas de tipo II en tres subclases: a. Tipo A. En este tipo el 30 % del aire es eliminado en cada ciclo y el 70 % es recircularizado. El escape al medio de los agentes potencialmente peligrosos se previene mediante una corriente de aire entrante en una rejilla frontal. 10 b. Tipo B. Se trata de una cabina de flujo laminar para uso general (figura 2.3.C), y en ella se recirculariza sólo el 30 % del aire en cada ciclo, eliminándose el 70 % del aire restante. 11 c. Tipo 100 % exhausto. Se trata de una cabina en la que el 100 % del aire de cada ciclo es eliminado hacia dispositivos que puedan retener los posibles agentes peligrosos. Este tipo de cabinas se emplean fundamentalmente en laboratorios de toxicología en los que se requieren áreas de contención limpias y eliminación del aire posiblemente contaminado. Las diferencias entre estos tres subtipos se refieren especialmente a la protección del usuario, superior en el caso del tipo A, y a la eliminación de la cabina de posibles contaminaciones de tipo aerosol, no retenibles por el filtro HEPA. La tipo A, que es la más adecuada para el trabajo con agentes patógenos particulados (filtrables) es la menos adecuada para el trabajo con vapores o aerosoles peligrosos. Asimismo se ha de tener en cuenta que el funcionamiento correcto para la protección del producto y del personal depende de una correcta calibración del instrumento, especialmente de las velocidades respectivas de entrada de aire en el sistema y de flujo vertical. El punto de calibración o ajuste es dependiente del instrumento y debe ser realizado por personal especializado cada 6 a 9 meses de funcionamiento para asegurar la protección al usuario. Cabina de clase III. Se trata de una cabina de seguridad, estanca, para el trabajo con agentes biológicos de alto riesgo. Permite mantener al agente patógeno en un ambiente completamente estanco. Permiten controlar tanto los contaminantes particulados como aerosoles y contaminantes gaseosos mediante sistemas de filtración y disolución de éstos. 12 A. Puertos de guantes, con junta tórica para la fijación de los guantes del brazo de longitud al gabinete. B. faja, C. filtro HEPA de escape, D. filtro HEPA de suministro, E. autoclave de dobleterminación o caja de paso. Nota: Un tanque químico puede ser instalado, localozandolo debajo de la superficie de trabajo del BSC con acceso desde arriba. El gabinete de escape necesita ser conectado al sistema de salida del edificio. Incubadores: Se trata de equipos comunes a la mayor parte de los laboratorios de biología (estufas, baños termostatizados) con la excepción del incubador de CO2, y del incubador con "roller". Las células en cultivo son capaces de soportar sin daños importantes variaciones de temperatura, siempre que sean por debajo de la temperatura corporal del animal del que proceden. Así pues las células humanas soportan incubaciones a 4 ºC durante días y pueden ser congeladas a -196 ºC durante años (con sustancias preservantes). Sin embargo no sobreviven más de unas pocas horas a variaciones de 2 ºC por encima de 37ºC. Las células procedentes de animales poiquilotermos soportan mejor un amplio rango de temperaturas de incubación. Las células de homeotermo suelen crecer bien entre 33 y 37 ºC, pero en ese margen presentan importantes variaciones en su tasa de crecimiento. En un incubador de células es por tanto más importante la consistencia de la temperatura (invariabilidad durante prolongados periodos de tiempo, con oscilaciones inferiores a 0.5 ºC) que su precisión a fin de mantener a las células en condiciones en las que las tasas de crecimiento sean constantes. Por ello se deben usar incubadores con movimiento de aire forzado en el 13 interior, y colocar los contenedores de células sobre estanterías agujereadas y nunca sobre el fondo o las paredes del incubador. Las necesidades de asepsia en el incubador son considerablemente menores que en el área de trabajo (cabina de flujo laminar) pero sin embargo es muy recomendable mantener una estricta limpieza, desinfección periódica del incubador, y especialmente no introducir, o eliminarlos inmediatamente, cultivos contaminados en el incubador. Para determinar el número y tipo de los incubadores requeridos se tendrá en cuenta: - el tipo de cultivos a realizar: primarios o de líneas celulares estables. Es conveniente no cultivar en el mismo incubador cultivos primarios con líneas celulares estables, pues los primeros son una importante fuente de contaminación. - la especie y el tipo de células a cultivar. Es importante la determinación exacta de la temperatura de incubación para la que se tendrá en cuenta la temperatura corporal del animal del que proceden las células, así como cualquier variación regional de temperatura (la piel suele tener una temperatura inferior). Incubador de CO2: El mantenimiento de la temperatura en una atmósfera con una tensión controlada de CO2 y adicionalmente de humedad elevada es el objetivo del incubador de CO2. Un incubador moderno dispone de : 1. dispositivos de control de temperatura, con un termostato de seguridad que desconecta la función en caso de anomalía. La estabilidad de la temperatura es una característica esencial del incubador, y por ello suelen estar equipados con camisas de agua ('water jacketed') que incrementan notablemente la inercia térmica. Como consecuencia la estabilización de un incubador puede tardar varios días. 2. dispositivo de inyección de una mezcla de aire y CO2, en la proporción deseada, entre el 4 y el 7 %. El CO2 de elevada pureza se suministra en botellas presurizadas, y se mezcla en el dispositivo de inyección. El control de la mezcla se realiza fundamentalmente mediante un dispositivo IRGA ("infra-red gas analyzer"). En incubadores modernos este dispositivo analiza constantemente el porcentaje de CO2 presente en la cámara de incubación, y realiza las correcciones necesarias. Un problema usual del dispositivo de medida de CO2 es la pérdida de calibración que se produce periódicamente. Para evitarlo algunos fabricantes dotan al sistema de un mecanismo de autocalibración periódica (basado en la toma de una muestra de aire exterior que se considera tiene un valor x de CO2). 3. dispositivo de control de la humedad ambiente. Para mantener el cultivo se requiere una humedad ambiente elevada, a fin de reducir la evaporación de agua del medio de 14 cultivo. En los incubadores menos sofisticados esto se consigue mediante bandejas de agua en el fondo del incubador. Este recurso es peligroso pues son una fuente importante de contaminaciones al convertirse a los pocos días en caldos de cultivo. En los instrumentos más modernos se dispone de dispositivos que controlan la humedad atmosférica, inyectando agua estéril y filtrada. 4. dispositivo de recircularización de aire. Es importante una recirculación del aire en el interior del incubador, a fin de homogeneizar la temperatura en su interior. Si además en el circuito de circulación de aire se intercala un filtro HEPA se consiguen eliminar las posibles partículas contaminantes, y se asegura la esterilidad del ambiente. Las condiciones del cultivo: temperatura, humedad atmosférica y niveles de CO 2 son característicos de cada línea celular. El nivel de CO2 se establece para mantener el equilibrio carbonato-bicarbonato en el medio de cultivo, habitualmente al 5%. Incubador "roller": Se trata de un incubador o estufa, en el interior del cual se ha instalado un "rotor" de baja velocidad. Se utiliza para mantener cultivos en botellas cerradas (no requieren CO2). La finalidad es disponer de una gran superficie para la adhesión de las células. En estos instrumentos se incuban las células en el interior de 'roller bottles', botellas con una gran superficie de crecimiento. Instrumentos ópticos de observación: microscopio de contraste de fases invertido: El control morfológico del cultivo se realiza mediante el uso de un microscopio. El hecho de que las muestras a observar se encuentren en recipientes de un cierto grosor (más de 3 cm en el caso de frascos de Roux de 250 ml) hace que un microscopio convencional no sea adecuado (por su pequeña distancia frontal), por lo que se han desarrollado microscopios de diseño original, en los cuales la fuente de iluminación y los objetivos se encuentran invertidos respecto a la platina de un microscopio óptico convencional. La segunda característica que condiciona el instrumento óptico es su ausencia de color, es decir se trata de muestras vivas y con poco contraste. El microscopio se equipa con el dispositivo de contraste de fases (diafragmas anulares a nivel del condensador y placa de fases entre las lentes del objetivo). De esta forma el contraste de la imagen aumenta y la calidad obtenida es muy superior. Es de gran utilidad disponer asimismo de un dispositivo de captación de imágenes, fotográfico o de video acoplado al microscopio a fin de documentar el estado de los cultivos. 15 Congeladores e instalación de criogenia (depósito de N2 líquido): Es recomendable que los congeladores y la instalación de criogenia estén en recintos separados a la unidad de cultivo propiamente dicha pues los ventiladores y compresores son una fuente importante de turbulencias y suciedad en el laboratorio. Es preciso el almacenamiento de soluciones y células a diferentes temperaturas, para lo que se requiere: 1. heladeras (4 ºC) para el almacenamiento de medios, PBS. 2. congeladores de -20 ºC para el almacenamiento de suero, aditivos (glutamina, antibióticos) y soluciones enzimáticas (tripsina, colagenasa). 3. congeladores de -80 ºC para el almacenamiento a largo plazo de los aditivos del medio (suero, glutamina, antibióticos) y de sustancias especialmente sensibles (factores de crecimiento, mitógenos, inductores). 4. unidad de almacenamiento en nitrógeno líquido (-196 ºC), para el almacenamiento de las líneas celulares. Equipo de esterilización: autoclave y equipo de filtración. La necesidad de asepsia para el cultivo de tejidos se extiende no sólo al medio en que se realiza el trabajo sino muy especialmente a los recipientes en que se realiza el cultivo, a los medios líquidos o sólidos, y a los instrumentos que puedan entrar en contacto con éste en algún momento de su manipulación (pipetas, puntas de pipeta automática, pinzas, tubos, material variado de vidrio). Para esterilizar todo este material, de variada naturaleza se emplean una serie de métodos; irradiación con radiación gamma o rayos X, esterilización por gas, autoclavado, filtración. En el laboratorio general de cultivo de tejidos se suele disponer de: equipo de filtración y autoclave. La esterilización por gas, y la irradiación son técnicas propias de grandes instalaciones, por ejemplo hospitalarias, o industriales. Equipos de filtración: En la actualidad se dispone de unidades de filtración estéril muy recomendables para el trabajo rutinario. Estos equipos de filtración constan o bien de una fuente de vacío conectada a un kitasato dotado de una unidad de filtración esterilizada por autoclavado, o bien de una bomba peristáltica que fuerza el flujo de solución a través de una unidad de filtración hermética. En ambos casos la esterilización se produce al atravesar la solución un filtro de poro 0.22 um. Autoclave: El autoclave es un instrumento que permite la esterilización por calor tanto de sólidos como de líquidos. La esterilización se realiza habitualmente a 121 ºC, 1 atmósfera de sobre presión durante un tiempo superior a 20 min. Un autoclave de uso normal en el laboratorio de cultivo de tejidos suele disponer de temporizador y regulador de presión, y 16 en algunos casos de un ciclo de secado para permitir secar el material sólido (material de vidrio, instrumentos quirúrgicos, tubos. Otros instrumentos: centrífugas, contador de células electrónico ("cell counter"), equipo de purificación de agua, balanzas, pH-metro, pipeteadores Además del equipo antes citado el laboratorio de cultivo de tejidos deberá estar equipado con otros instrumentos que, o bien no son imprescindibles para un laboratorio de tamaño pequeño (por ej. el contador electrónico de células), o bien son instrumentos que forman parte de un laboratorio de uso más general (centrífugas, equipo de purificación de agua, balanzas, pH-metro, pipeteadores, etc.). Equipo de purificación de agua: Es de gran importancia en la preparación de los medios, de los aditivos, o en cualquier solución que pueda estar en contacto con el cultivo, una absoluta esterilidad y ausencia de sustancias que puedan provocar alguna alteración al cultivo. Por ello se utiliza siempre agua de la máxima calidad (tipo MilliQ) obtenida mediante equipos de doble destilación o de intercambio iónico y filtración. Pipeteadores: Se trata de dispositivos o instrumentos para el trasvase o medición de fluidos. Los de uso común en el laboratorio como las pipetas automáticas y las pipetas, pero con la salvedad del uso de bombas acopladas a la pipeta, manuales o eléctricas que permiten la aspiración mecánica del fluido. Importante para mantener la asepsia y al mismo tiempo para la protección del operador. El aire bombeado es filtrado previamente. 17 Clase Teórica: REQUERIMIENTOS DE CULTIVOS CELULARES Docente: Dra.Isabel Nocito [email protected] REQUISITOS DE CULTIVOS CELULARES. Fase gaseosa y temperatura. Medios de cultivo y suplementos El crecimiento celular in vitro necesita de un medio ambiente tan cercano como aquel donde se desarrolla in vivo. Para que esto se cumpla es importante tener en cuenta factores ambientales como el sustrato donde crece, el medio que rodea las células y la temperatura. El cultivo celular se realiza en medios artificiales preparados mediante la mezcla de componentes purificados o de soluciones orgánicas complejas, en el interior de instrumentos que mantienen las condiciones físico-químicas adecuadas y sobre soportes o recipientes que los contienen y aíslan del medio exterior. Para ello consideraremos que el medio de cultivo está formado por cuatro elementos: 1) la naturaleza del sustrato o fase en que crecen las células. 2) las condiciones fisicoquímicas y fisiológicas del medio. 3) la naturaleza y composición de la fase gaseosa. 4) las condiciones de incubación, especialmente la humedad y la temperatura. Sustrato de cultivo: La mayor parte de las líneas celulares crecen en forma de monocapa unidas a un soporte más o menos sólido. Según la célula necesite un sustrato o no para el anclaje hablamos de cultivos en suspensión o independiente del anclaje y cultivos en adhesión o dependiente del anclaje. El crecimiento en suspensión, está usualmente restringido a algunas líneas celulares como las células hematopoyéticas y tumores ascíticos. Materiales usados como sustratos: Vidrio: Empleado usualmente como sustrato de cultivo tiene como ventajas su escaso costo y su facilidad de limpieza y esterilización. Plástico: Muy empleado en la actualidad como material desechable estéril por irradiación. El plástico más usado es el poliestireno, de buena calidad óptica. Otros plásticos que se emplean son polivinil-cloruro, policarbonato (PVC), politetrafluoretileno (teflón, PTFE), thermanox (TPX). Microsoportes ("microcarriers"): Se trata de soportes plásticos (poliestireno), de sephadex o poliacrilamida en forma de pequeñas bolas a las que se unen las células dependientes de anclaje. 18 Otros sustratos que se usan son: metálicos, de paladio o en discos de acero. Han sido desarrolladas técnicas para crecer células de glía en sustratos. Superficies tratadas: La adherencia y crecimiento de las células en un frasco mejora en muchos casos si la superficie ha sido tratada con el medio de crecimiento de otro cultivo, debido a la presencia de colágeno o fibronectina liberada por las células, o bien sobre superficies recubiertas de proteínas de matriz extracelular (fibronectina, colágeno, vitronectina, Matrigel, etc.). Otros tratamientos que se han usado han sido: gelatina (músculo), poli-D-lisina (algunos teratomas de ratón). "Feeder layers": Se ha descrito previamente que algunos tipos celulares para crecer en cultivo y expresar sus características diferenciadas precisan de suplementos específicos. Una manera de obtener estos suplementos es la de hacerlos crecer sobre los restos de monocapas de otros tipos celulares. Una de las monocapas más empleadas son los fibroblastos de ratón 3T3 irradiados. Matrices tridimensionales: Son sustratos en los que las células penetran, estableciendo una distribución tridimensional, entre ellos están los geles de colágeno, esponja de celulosa sola o recubierta de colágeno o "gelfoam". Sustratos no adherentes: Son sustratos que no permiten la adhesión celular, por ejemplo agar, agarosa, o methocel (metilcelulosa de alta viscosidad). Interfases líquido-gel o líquido-líquido: Se han observado en algunas situaciones la proliferación celular y adhesión a las interfases líquido-líquido o líquido-gel, a pesar de que se desconocen exactamente los mecanismos. Haces microcapilares permeables (“hollow fiber”): Son cámaras de crecimiento de células formadas por un recipiente cilíndrico en el que se siembra, y en el interior del cual hay un haz de capilares plásticos permeables adecuados para que las células se adhieran. Tal como se ha descrito previamente, el material más utilizado como sustrato es el plástico desechable, en forma de diferentes tipos de recipientes. Los más comunes son: a) Placas de Petri (ventiladas): Disponibles en 3 tamaños: 3,5, 6,0 y 10 cm de diámetro son las más empleadas cuando se trata de crecer las células para usar directamente en experimentos. b) Multiplacas: Es una variante de las placas de Petri. Placas de varios pocillos, desde 6 a 96 pocillos. c) Frascos de Roux (botellas ventiladas o no). Roller bottles La elección del recipiente depende de: 1- Producción celular, 2- Crecimiento celular en suspensión, 3- Ventilación, 4- Frecuencia del muestreo, 5- Tipo de análisis 6- costo. 19 1- El rendimiento celular es proporcional a la superficie a la superficie. 2- Las células podrán crecer en distintos recipientes no tratados o en botellas de agitación según los requerimientos. 3- Las placas y policubetas permiten una atmósfera húmeda y una tensión de CO 2 controlada. Las tapas con filtro mantienen un equilibrio con la fase gaseosa. 4- Dependerá del diseño de lo los experimentos con cultivos 5- Se usará distintos recipientes de acuerdo al tipo de solventes empleados en el procedimiento. 6- El plástico es el más usado por su calidad y claridad óptica. Propiedades fisicoquímicas: Propiedades físicas: Las características del medio son: pH, osmolaridad, temperatura, viscosidad y tensión superficial. pH y capacidad tamponadora. El pH óptimo de crecimiento para la mayoría de tipos celulares es de 7,4, aunque existen pequeñas variaciones. Algunas líneas normales de fibroblasto crecen mejor entre pH 7,4 y 7,7 y células transformadas lo hacen en el margen de pH de 7,0 a 7,4, células epidérmicas pueden ser mantenidas a pH 5,5. El indicador de pH que se suele emplear es rojo fenol. El medio de cultivo debe estar tamponado, a fin de evitar los cambios bruscos de pH. La solución tamponadora más empleada sigue siendo el tampón bicarbonato, que equilibra el CO2 atmosférico, a pesar de su escasa capacidad tamponadora. HEPES es la solución tamponadora de elección por su elevada capacidad tamponadora en el rango 7,2 a 7,6, y se emplea en concentraciones de 10 a 20 mM. Osmolaridad. Muchas células en cultivo tienen una amplia tolerancia frente a la osmolaridad del medio, creciendo bien en el rango de 260 a 320 mOsm/kg, con pequeñas variaciones dependiendo de la especie considerada Temperatura. La temperatura tiene gran influencia en la tasa de crecimiento de las células. La temperatura recomendada es 37ºC. Las células toleran varios días a 4ºC o pueden ser congeladas pero no toleran temperaturas 2ºC por encima de la temperatura normal. Viscosidad. La viscosidad del medio viene determinada fundamentalmente por el contenido en suero y tiene poca influencia sobre el crecimiento. Tensión superficial. La tensión superficial se ha de mantener baja, y en general sólo se ve alterada por la aparición de espumas en los cultivos en suspensión donde se burbujea CO2. La fase gaseosa: Los componentes más significativos de la fase gaseosa son el oxígeno y el dióxido de carbono: 20 a) Oxígeno. Las necesidades de oxígeno para la mayor parte de los cultivos celulares se cubre con la tensión atmosférica, aunque existen cultivos, especialmente los cultivos de órganos que requieren una tensión de oxígeno superior (del 95%) posiblemente debido a la geometría del órgano y a las dificultades de difusión del gas en su interior. b) Dióxido de carbono. El dióxido de carbono juega un complejo papel en el medio - debido a que influye la cantidad de CO2 disuelto, el pH y la cantidad de iones HCO3 . De modo que para establecer un pH determinado se debe tener en cuenta especialmente los niveles de bicarbonato sódico y la tensión de CO2. Cada medio tiene una concentración recomendada de bicarbonato y tensión de CO2 para alcanzar el pH correcto. Sin embargo, en la actualidad se emplean otras sustancias tamponadoras en la formulación de muchos medios, ya que permite una estabilidad superior del pH en el medio, así como una mayor capacidad tamponadora (por ej. los denominados Good's buffers: HEPES, Tricina). Condiciones fisiológicas: Las condiciones fisiológicas hacen referencia a la composición del medio. Inicialmente los medios de cultivos fueron realizados en medios naturales basados en extractos de tejidos, por ejemplo extracto de embrión de pollo, hidrolizados de proteínas o fluidos corporales como suero o linfa. Posteriormente la demanda de grandes cantidades de medio de mejor calidad guió a la introducción de medios definidos químicamente, tales como el medio basal de Eagle (1955) y el medio esencial mínimo de Eagle (1959) suplementado con suero de caballo, carnero o humana. Después de años de investigación en la composición de los medios la elección de éstos sigue siendo empírica. Los principales medios empleados son: Medio Basal de Eagle (BME) Medio Mínimo Esencial de Eagle (MEM) RPMI 1640 Medio MEM modificado por Dulbeco (DMEM) Modificación de Iscove del medio DMEM (IMDM) McCoy 5A Medio L-15 de Leibovitz Medio F-10 de Ham Medio F-12 de Ham. Medio 199 Todo medio de cultivo está formado por los siguientes elementos: 21 Soluciones salinas equilibradas (BSS). Una solución salina equilibrada es una mezcla de sales inorgánicas, incluyendo usualmente bicarbonato sódico y suplementada con glucosa. Se usan para diluir medios más completos, como medio de disección o lavado, o para incubaciones cortas que requieren un medio isotónico no completo nutricionalmente. Ejemplos de BSS: Hanks, Eagle, Doulbeco. Debido a su escasa capacidad tamponadora se recomienda suplementarlo con HEPES para pH en el rango 7,2 a 7,8 y Tricina en el rango 7,4 a 8,0. Aminoácidos. Es necesario suplementar el medio basal con los aminoácidos esenciales porque no son sintetizados por las células y su falta limita el crecimiento de las mimas. Un suplemento común es el de glutamina, a 2 mM final, aunque hay algunas líneas celulares pueden usar el glutamato. Vitaminas. El medio MEM suplementa con vitaminas del grupo B, siendo los demás grupos aportado por el suplemento de suero. La limitación de vitaminas se manifiesta en la supervivencia de las células y en la reducción de la tasa de crecimiento más que en la densidad celular. Glucosa. Es la fuente de energía en muchos medios. Se metaboliza preferentemente vía glucólisis hacia piruvato y este es convertido en lactato o acetoacetato e ingresa al ciclo de Krebs. Otros suplementos orgánicos de bajo peso molecular. Dependiendo del medio, encontramos a nucleósidos, intermediarios del ciclo de Krebs, piruvato, lípidos, etc. Hormonas y factores de crecimiento. No son específicos en la mayoría de los medios, aunque frecuentemente se adicionan en medios libres de suero. Inhibidores del crecimiento de los contaminantes (antibióticos y antifúngicos). A fin de evitar el crecimiento de contaminantes en el cultivo se suele suplementar éste con sustancias antibióticas de diferente espectro de acción. La adición de antibióticos ha de ser estrictamente controlada para evitar efectos nocivos sobre el cultivo. Suero: El suero contiene factores de crecimiento que promueven la proliferación celular, factores de adhesión y actividad antitripsina. Es fuente de minerales, lípidos y hormonas. Los sueros más usados son: suero de ternera ("calf serum", CF), suero bovino fetal ("fetal calf serum", FCS), suero de caballo ("horse serum", HS) y suero humano ("human serum", HuS). El más usado es el suero de ternera, mientras que el suero bovino fetal es usado en líneas más exigentes, y el suero humano en líneas humanas. Componentes del suero: Proteínas. Si bien es el componente mayoritario su función no es clara in vitro. Algunas de ellas son necesarias y otros actúan como transportadoras de minerales, ácidos grasos y hormonas. Además las proteínas aumentan la viscosidad del medio reduciendo el stress celular durante el pipeteo y agitación. 22 Factores de crecimiento. El mayor factor de crecimiento en suero es el factor de crecimiento derivado de las plaquetas (PDGF) con actividad mitogénica y estimula el crecimiento de fibobrasto y células de la glía. Otros factores son factor de crecimiento de fibroblasto, factor de crecimiento de células endoteliales. Hormonas como la insulina e hidrocortisona. Inhibidores de la proliferación celular, pero el calentamiento del suero destruye el complemento y reduce la acción citotóxica de las inmunoglobulinas. Entre otros, el suero contiene distintos nutrientes, lípidos y minerales. Desventajas en el uso de medios con suero: a) Variabilidad fisiológica: a pesar de que la composición del suero es conocida, existen gran cantidad de componentes presentes en cantidades variables y esto puede influir notablemente en el cultivo (hormonas, factores de crecimiento, etc.). b) Consistencia y vida media: el suero varía de lote a lote. La duración es de un año, pero puede deteriorarse a lo largo de ese tiempo y el ser reemplazado por otro se selecciona como similar pero no son iguales. c) Calidad de control: cada cambio de lote de suero requiere realizar una serie de controles tediosos y costosos. d) Especificidad: si se cultivan varios tipos celulares cada uno puede requerir un lote diferente, lo que complica el almacenamiento e incrementa los costos. e) Disponibilidad: crea una dependencia importante de un suministro que no siempre puede mantenerse con la periodicidad y calidad requerida. f) Procesos posteriores: si se han de purificar productos del medio de cultivo la presencia de los componentes del suero dificulta notablemente estos procesos. g) Contaminación: el suero está contaminado con frecuencia por virus y micoplasmas y puede adicionar un factor no conocido al control. h) Costo: el suero representa el principal gasto de los medios de cultivo pero el reemplazo de cada constituyente puede ser mayor que el suero. i) Inhibidores: el suero posee factores de crecimiento y tiene también inhibidores del crecimiento. Por ejemplo, el factor de crecimiento plaquetario (PDGF) estimula la proliferación de fibroblastos, lo que puede ser un problema en el establecimiento de cultivos primarios especializados. Medios libre de suero: Si bien la mayoría de las líneas celulares son suplementadas con suero en muchas instancias los cultivos pueden propagarse en medios sin suero. Para esto es necesario hacer modificaciones nutricionales y hormonales. Ventajas 1) Medio selectivo 2) Regula la proliferación y diferencia Desventajas 1) Multiplicidad de medios 23 2) 3) 4) 5) Selectividad Pureza de reactivos Proliferación celular Disponibilidad Para conseguir reemplazar el suero completamente de un medio será necesario reemplazar: 1) los factores de adhesión como la fibronectina o bien el tratamiento de las placas con poli-lisina antes de la siembra. 2) inhibidores de proteasas como el inhibidor de tripsina de soja. 3) hormonas como hormona de crecimiento, T3, hidrocortisona, dexametasona, FSH. 4) factores de crecimiento peptídico como FGF ("Fibroblast Growth Factor"), EGF ("Epidermal Growth Factor"), PDGF ("Platelet Derived Growth Factor") y MSA. 5) nutrientes como Fe, Cu, Se, y otros minerales 6) proteínas y poliaminas. Si bien existan razones del uso de medios libres de suero como, por ejemplo promover el crecimiento selectivo de una línea particular de células o evitar contaminaciones o presencia de determinadas proteínas presentes en el suero o por recomendaciones de un medio apropiado para el crecimiento celular, es importante recurrir a tablas que indiquen la disponibilidad, selección o sugerencias de su uso. Bibliografía: “Culture of animal cells. A manual of Basic technique. R. Ian Freshney. “Cultivos celulares”. www.cultek.com. 24 Clase Teórica: MATERIAL DE CULTIVO CELULAR. TÉCNICAS DE MANIPULACIÓN ESTÉRIL. Docente: Dra. Victoria Alonso [email protected] MATERIAL DE CULTIVO CELULAR. Técnicas de manipulación estéril. Las condiciones de cultivo varían ampliamente según el tipo de célula utilizado, pero el ambiente artificial donde las células se cultivan consiste invariablemente de: un contenedor apropiado con un sustrato o medio que provea los nutrientes esenciales (aminoácidos, carbohidratos, vitaminas, minerales), factores de crecimiento, hormonas y gases. La mayoría de las células son adherentes y deben ser cultivadas adheridas a un sustrato sólido o semisólido, mientras que otras pueden crecer flotando en medio de cultivo (en suspención). Los requerimientos específicos de un laboratorio de cultivo celular dependen principalmente del tipo de investigación que se lleve a cabo. Pero todos ellos tienen requerimientos comunes, siendo el más importante estar libres de microorganismos patógenos (asepsis), y además utilizan basicamente el mismo equipamiento. Los materiales más comunes en un laboratorio de cultivos son: Vidrio: Prácticamente todos los Medios se preparan y esterilizan para su almacenamiento y clasificación, en botellas de vidrio. También se utilizan diferentes tipos de pipetas de cristal, probetas, vasos y matraces para el trabajo rutinario del Laboratorio. Para preparar medios en general se utilizan botellas de boro-silicato con tapa de plástico. El lavado, preparación y esterilización de dicho material se realiza en los cuartos destinados a dicho fin. Tanto en cultivos celulares de células eucariotas, así como de microorganismos es imprescindible que todo el material que se utilice esté estéril. 25 Métodos físicos de esterilización Calor directo Flameado Calor seco Horno Pasteur Calor húmedo Autoclave Filtración Unidad de filtración o filtros de jeringa (en general con membranas de 0. 22 m) Asas, varillas, placas, tubos Material de vidrio. Medios de cultivo, material de vidrio, tubos. Líquidos: medios de cultivo, antibióticos, etc. Plástico: Todo el trabajo que se desarrolla en un laboratorio de cultivos se hace en plástico. El material de plástico descartable es muy variado y depende de la aplicación final pero, en general, para cultivo de células eucariotas necesitamos: Botellas o flasks Las mismas vienen de distintos tamaños: 25 cm2, 75 cm2, 150 cm2, 225 cm2 de área de crecimiento. También tienen distintas formas: triangular, rectangular o circular (botellas roller). Triangular: buen acceso de la pipeta y scraper a los ángulos. Su base ancha provee mayor estabilidad. Rectangular: pueden tener una rampa desde el fondo del cuello angular para facilitar el volcado y el acceso a pipetas. Otra alternativa son las tradicionales con cuello angular o recto las cuales permiten que se utilice toda el área para el crecimiento celular. Su diseño ahorra espacio y reduce el contacto del medio con el cuello y la tapa. 26 Roller: Se utilizan para la producción de vacunas o proteínas recombinantes con fines terapéuticos. Se fabrican de poliestireno o polietileno, vienen de diferentes tamaños y pueden tener surcos radiales para aumentar la superficie de crecimiento. Por otro lado podemos elegir botellas con diferentes tipos de cuello y diferentes tapas. Cuello recto: ideal para volúmenes grandes de medio ya que disminuye el contacto del medio con la tapa. Cuello inclinado: permite volcar el contenido de la botella y hace más sencillo el pipeteo o scraping. Cuello angular: mejora el acceso de la pipeta y reduce el contacto del medio con la tapa. Tapas selladas: para usar en sistemas cerrados, no permiten el paso de líquidos ni gases. Tapas tipo fenólico: cuando se dejan flojas permiten el intercambio de gases. Se usan en sistemas abiertos que requieren la entrada de gases. Tapas ventiladas: contienen una membrana impermeable de 0.2 m sellada en la tapa que provee un intercambio gaseoso consistente pero 27 minimizando los riesgos de contaminación. Son recomendadas para usar en incubadores de CO2, especialmente para uso prolongado. Placas de petri Placas con pocillos Existen placas con cantidades diferentes de pocillos, que admiten diferentes volumenes de cultivo. 28 Si se desea medir fluorescencia o luminiscencia es necesario utilizar placas que minimicen la señal cruzada entre pocillos. Para esto se utilizan placas con paredes negras o blancas. Se pueden elegir además distintos tipos de fondos según el uso final. Spining flask De vidrio (reutilizables) o de plastico (descartables). Se utilizan para crecer 29 cantidades grandes de células no-adherentes (1-2 x106 celulas/ml). Existen de distintos volúmes, desde 125 ml hasta varios litros. Una desventaja es que necesitan de un agitador magnético, lo cual requeire un mayor espacio en el incubador. La versión de vidrio son dificiles de decontaminar, limpiar, re-esterilizar y re-ensamblar. Erlenmeyers Se utilizan especialmente para crecer células de insectos que necesitan altos niveles de oxigeno. Vienen de distintos tamaños y con distintas tapas. Solamente para células adherentes y requieren un shaker. Scrapers Para remover mecanicamente las célula adherentes sin romperlas. Pipetas y tips Las pipetas Pasteur son prácticas para transferir líquidos rápidamente. Pueden ser estériles o noestériles. 30 Los tips con filtro previenen contaminaciones con líquidos o aerosoles durante el pipeteo. Crioviales Tubos de centrifuga o falcons 31 Técnicas de manipulación estéril: Área de trabajo: El mayor requerimiento de una laboratorio de cultivo celular es la necesidad de mantener el área de trabajo aséptica y restringida al trabajo con cultivo celular. En general es preferible tener un especio separado para el cultivo de celular pero si no es posible también puede designarse una área particular dentro de un laboratorio grande para la manipulación estéril, incubación y almacenado de células, medios y otros reactivos de cultivo. La manera más simple es trabajar en una cabina de bioseguridad, la cual debe ubicarse lejos de corrientes de aire de puertas, ventanas y otros equipamientos y alejada de áreas de paso. La misma debe ser lo suficientemente grande con para ser usada por una persona a la vez, deber ser fácil de limpiar por dentro y por fuera y debe tener iluminación adecuada. Es de suma importancia mantener el espacio dentro de la cabina limpio y liberado, además todos los materiales deben estar a la vista. No debe usarse para guardar o almacenar cosas. Es obligatorio lavarse las manos antes y después del trabajo de laboratorio, para evitar contaminación del usuario con material biológico, y la potencial dispersión de éste. Se debe usar material de protección (guardapolvo, guantes, barbijo, gafas, etc). Tanto el plástico desechable como el vidrio que haya estado en contacto con material biológico ha de ser inactivado antes de tirarlo, o disponerlo para su lavado y esterilización. Los residuos acumulados en vasos de precipitados han de ser inactivados con hipoclorito antes de tirarlos, y los vasos se dejarán con un poco de hipoclorito hasta su próximo uso. La mesa de trabajo y las campanas se descontaminarán antes y después de trabajar con material biológico. Cualquier material biológico derramado ha de ser descontaminado inmediatamente con etanol 70%. Cada material que ingrese a la cabina debe ser rociado con etanol 70% y secado con papel descartable. Antes de comenzar se debe apagar la luz UV, encender la luz, desinfectar todo y dejar funcionando el ventilador por 5-10 minutos hasta que se estabilice el flujo de la cabina. En general, los ítems dentro de la campana se ubican de la siguiente manera: Área amplia en el centro donde se colocan las células a trabajar, ya sea en botellas o placas. Pipeta automática a la derecha. Pipetas de vidrio esterilizadas o de plástico estériles a la izquierda. La idea es que sean fáciles de alcanzar. Reactivos y medios atrás a la derecha para facilitar el pipeteo. Contenedor atrás y al medio con hipoclorito para descartar líquidos, pipetas y tips. No es necesario ni recomendable utilizar mecheros dentro de la cabina. 32 Reactivos y medios: Los reactivos y medios comerciales pasan controles estrictos para asegurar su esterilidad pero pueden contaminarse al manipularlos, por lo cual deben extremarse las precauciones. Los medios y soluciones preparadas en el laboratorio deben esterilizarse por calor o filtrado según corresponda. Como lineamiento general al trabajar con líquidos: Evitar volcar medios directamente de las botellas. Usar pipetas de vidrio o de plástico descartables y una pipeta automática para manejar líquidos, usar cada pipeta solo una vez para evitar contaminaciones cruzadas. No sacar las pipetas estériles de su envoltorio cerrado hasta que sean usadas. Siempre tapar las botellas y frascos después de usarlos. Lo mismo para las placas, mantenerlas tapadas para prevenir contaminaciones. Nunca destapar los frascos estériles, botellas, placas, etc hasta que sea necesario usarlas y nunca dejarlas destapadas. Si al sacar una tapa es necesario apoyarla en la mesada, siempre ubicarla con la abertura hacia abajo. Siempre usar material de vidrio estéril. En lo posible no hablar, cantar o silbar mientras se trabaja con material estéril. Realizar los experimentos lo más rápido posible para minimizar contaminaciones. 33 Medidas especiales de esterilidad al congelar y descongelar células La manipulación de células durante la congelación y descongelación es un proceso que conlleva riesgo de contaminar las células. Habrá de prestarse especial atención a no tocar las roscas de los tapones de los criotubos, y tras descongelar las células en el baño, es esencial esterilizar el exterior del tubo con abundante etanol, antes de abrirlo y manipularlo. Bilbliografia: https://www.gbo.com http://www.corning.com Gibco Cell Culture Basic Handbook 2015 (www.lifetechnologies.com/cellculturebasics) Cell Culture Tissue Culture Lab Procedures. Alan Doyle. Cell Culture of Animal Cells. Ian Freshney. ANEXO LAVADO Y ESTERILIZACIÓN DE MATERIAL MATERIAL PLÁSTICO Suele utilizarse poliestireno por la baja toxicidad LAVADO: Detergente no iónico SECADO: Estufa a 50ºC ESTERILIZACIÓN: - Calor húmedo (vapor): autoclave u olla a presión - 121ºC durante 20 minutos - Luego secar el material a 50ºC. MATERIAL DE GOMA Son aconsejables tapones de silicona y material de caucho natural LAVADO: - No conviene hacerlo con detergente - Hervir en álcali diluido (NaHCO3 5%) - Enjuagar exhaustivamente con agua corriente - Enjuagar con agua destilada y luego bidestilada SECADO: Estufa a 50ºC ESTERILIZACIÓN: - Calor húmedo (Vapor) - 121 ºC durante 20 min - Secar a 37ºC 24 hs 34 MATERIAL DE CIRUGÍA Debe lavarse inmediatamente luego del uso. No dejar más de 30 minutos en solución de hipoclorito de sodio. LAVADO: Utilizar detergente y cepillos SECADO: Estufa a 80-100 ºC ESTERILIZACIÓN: Calor seco, 180ºC Material nuevo: Eliminar anticorrosivos con una esponja en hipoclorito, secar con toallas de algodón y colocarlo en estufa hasta el secado completo. MATERIAL DE VIDRIO OPCIONES: 1- DETERGENTES no iónicos 2- ÁLCALIS 3- ÁCIDOS OXIDANTES 4- ULTRASONIDO 1abc- LIMPIEZA CON DETERGENTES: Quitar los rótulos de tinta con alcohol Transferir el material a una bandeja con una dilución de detergente Cepillar vigorosamente ENJUAGUE a- 10 veces con agua corriente b- 2 veces con agua destilada c- 1 o 2 veces con agua bidestilada 2abcd- LIMPIEZA CON ÁLCALIS: (Jabón, Trifosfato de sodio, Metasilicato de Sodio) Hervir 20 minutos con solución 1X Metasilicato de Sodio Enjuagar 2 veces en agua corriente Transferir a HCl 0.01 N unas horas Enjuagar como en el lavado con detergente 3- LIMPIEZA CON ÁCIDOS:(Sulfúrico, Sulfocrómico, Nítrico) Ventaja: Corroe menos el vidrio que los álcalis Desventaja: Son difíciles de eliminar con los enjuagues 4- LIMPIEZA POR ULTRASONIDO: (Frec. Superior a 18000 ciclos/s) Ventaja: Penetra en cavidades poco accesibles Desventaja: se necesita un equipo especial y no supera a los otros métodos descriptos. 35 Clase Teórica: TIPOS DE CULTIVOS CELULARES Docente: Dra. Romina Manarin [email protected] TIPOS DE CULTIVOS CELULARES. Líneas celulares. Cepas. Generalidades. Hoy en día se entiende por cultivo celular al conjunto de técnicas que permiten el mantenimiento de las células “in vitro”, conservando al máximo sus propiedades fisiológicas, bioquímicas y genéticas. Existen tres formas principales de iniciar un cultivo celular: - Cultivo de órganos: se coloca el órgano sobre una rejilla situada en la interfase líquido-gas de un medio del que obtiene los nutrientes y al que puede liberar los desechos. Este tipo de cultivo representa una buena réplica del tejido de origen porque se trata de mantener la arquitectura característica del tejido “in vivo” al conservar las interacciones histológicas, por lo que se mantienen los tipos celulares diferenciados. Sin embargo, su crecimiento es limitado, la proliferación celular se limita a las células embrionarias de la periferia y no se pueden propagar. Se necesita un nuevo explante para cada experimento lo que supone mucho más trabajo y una limitada reproducibilidad de la muestra. Cuantificar es difícil y la cantidad de material que se puede cultivar es reducida. - Explantes primarios: Se coloca un fragmento de tejido o de órgano en la interfase sólido-líquido de un soporte de vidrio o de plástico. Las células se adhieren a la superficie y las células de la periferia del explante pueden migrar y proliferar por la superficie del soporte. Cultivo celular primario: Es el tipo de cultivo más utilizado. Se puede obtener a partir de explantes primarios o de suspensiones de células disgregadas. La disgregación celular se realiza por métodos enzimáticos o mecánicos. En estos cultivos se pierden las interacciones célula-célula y las interacciones de la célula con la matriz extracelular. Sin embargo, las células son capaces de proliferar y la población celular crece notablemente. Cuando las células ocupan toda la superficie disponible se dice que han alcanzado la confluencia. En esta etapa, las células establecen contactos entre ellas que inhiben su proliferación y el crecimiento se detiene. Por eso, al cabo de un tiempo hay que “transplantar” las células a un nuevo soporte. Esta operación se denomina subcultivo o pasaje. Los cultivos celulares primarios pueden ser de dos tipos: • Cultivos en monocapa: las células crecen adheridas sobre un soporte sólido (plástico o vidrio). El anclaje al sustrato es un prerrequisito para la proliferación celular. Es el método utilizado para la mayoría de las células excepto para las hematopoyéticas. 36 • Cultivos en suspensión: las células se encuentran dispersas en el medio de cultivo. Su crecimiento no depende del anclaje. Este tipo de cultivo se restringe a las células hematopoyéticas, células madre, líneas celulares transformadas y células tumorales. Alcanzan la confluencia cuando el número de células es grande y los nutrientes son insuficientes. Establecimiento de un cultivo celular: El cultivo primario consiste en general de un mayor o menor número de células de diferentes tipos con diferentes capacidades de crecimiento. Sin embargo, para la mayor parte de los experimentos es necesario utilizar un único tipo celular. Por lo tanto, se deberá seleccionar el tipo celular necesario. Para ello podremos: - Dejar que las células crezcan. Las células de crecimiento más rápido tomarán la dominancia del cultivo. - Controlar la composición del medio de crecimiento. La adición de factores de crecimiento o de inhibidores del crecimiento podrá seleccionar ciertos tipos celulares. - Separar las células mediante gradientes de densidad. Una vez seleccionado el tipo celular podremos mantener el cultivo durante un período de tiempo variable que depende del tipo y del origen de las células. La capacidad de crecimiento del cultivo está relacionada con el origen de las células, así, las células derivadas de tejidos embrionarios tienen un límite de crecimiento mayor que las procedentes de tejidos adultos. 37 En este proceso de establecimiento de un cultivo celular se procede a la selección de un tipo celular determinado que: ha debido de superar los procesos de disgregación enzimática y/o mecánica, posee la capacidad de crecer adherido a un substrato o de crecer en suspensión, es capaz de responder a factores estimulantes o inhibitorios del crecimiento añadidos al medio y tiene una tasa de crecimiento alta. Por lo tanto, cambiando estos factores variarán también las células seleccionadas. Como mencionamos arriba, el crecimiento de las células se detiene también tras la confluencia y es necesario dividir, propagar o subcultivar las células. Generalmente en cultivos primarios la dilución es aproximadamente de ½ a ¼. Para ello se despegan las células del substrato, se cuentan, se ajusta la concentración y se depositan sobre el nuevo substrato con la cantidad de medio ajustada a la concentración de células. Subcultivos, origen de la línea celular: Con los sucesivos pasajes o subcultivos, un cultivo primario forma una línea celular. Este término implica la presencia de varios linajes celulares de fenotipos similares o distintos. A pesar de la gran similitud que las células de una línea tienen entre sí, no son idénticas. La uniformidad genética de una línea celular puede mejorarse por clonado celular, a través del cual se aísla una sola célula, la que prolifera para formar una colonia de células clonales. Así, al seleccionar uno de los linajes celulares por clonado o por separación celular física o por cualquier otra técnica de selección, para tener ciertas propiedades específicas que han sido identificadas en la masa de células del cultivo, esta línea celular se transforma en lo que se conoce como cepa celular (o cell strain). Normalmente, las líneas celulares tienen una vida finita que, según el tipo de célula, se puede prolongar entre 20 y 80 generaciones. Superado ese límite, las células entran en una etapa que se denomina senescencia en la que pierden su capacidad de proliferar, supuestamente por el acortamiento de los telómeros, y mueren. Sin embargo, algunas células (como las células de roedores y las células tumorales) evitan la senescencia y dan lugar a líneas celulares continuas, que crecen indefinidamente. Estas células pueden surgir de forma espontánea (exposición a radiaciones ionizantes o a carcinógenos químicos) o inducida (infección vírica o transfección de ADN) y son el resultado de un cambio genotípico denominado transformación. Las células transformadas se caracterizan porque son inmortales (crecen indefinidamente); su crecimiento es aberrante (se pierde la inhibición por contacto, la limitación de la densidad celular durante la proliferación y la dependencia del anclaje); son malignas (invaden tejidos y dan lugar al crecimiento de tumores); son genéticamente inestables (son heteroploides (varía el número de cromosomas) y presentan aberraciones cromosómicas. Cuando esa línea celular continua se selecciona o clona y caracteriza, es conocida como una cepa celular continua. Es vital en este punto confirmar la identidad de las líneas celulares y excluir la posibilidad de contaminación cruzada. Sin embargo, las líneas celulares continuas tienen muchas ventajas. En la siguiente tabla se describen las ventajas y desventajas relativas de las líneas celulares finitas y continuas. 38 En 1952, se obtuvo la primera línea celular continua humana, son las células denominadas HeLa, extraídas a partir de un tumor de cuello de útero de una paciente afroamericana que se llamaba Henrietta Lacks. Bancos y colecciones de líneas celulares: Para muchos experimentos pueden obtenerse diferentes tipos de líneas celulares de colecciones o bancos que poseen centenares de líneas perfectamente caracterizadas. Generalmente es más fácil adquirir una determinada línea celular que establecerla experimentalmente. Las dos colecciones o bancos de líneas celulares más importantes son: - American Type Culture Collection (ATCC) - European Collection of Cell Cultures (ECACC) En Argentina: - Asociación Banco Argentino de Células (ABAC) - Asociación Argentina de Bancos de Tejidos (para transplantes) - Centro de Investigaciones en Química Biológica de Córdoba (ciquibic) A continuación se detallan los servicios ofrecidos por ABAC en nuestro país: - Venta de líneas celulares: los aranceles para obtener líneas celulares son de aproximadamente U$S 200 para los no socios de ABAC, con agregado de gastos de envío a su cargo y para los socios de ABAC se realiza un descuento del 10 % sobre el arancel estipulado para no socios. (Los pagos se efectuarán contra entrega cuando las líneas celulares se retiren personalmente del lugar de distribución. En caso de ser requerido el envío postal, el material será despachado aproximadamente 72 horas luego de recibida la suma total. Esta será abonada por depósito o transferencia bancaria extendido a nombre de Asociación Banco 39 - - - Argentino de células, en Banco Francés. Nº de Cuenta Corriente en Pesos: 1063584/5). Control de Micoplasma en Muestras de Células: el costo para No socios de ABAC es de U$S 100 y para los Socios de ABAC se realiza un descuento del 10 % sobre el arancel estipulado para no socios. (Para coordinar el envío de muestras los interesados deberán contactarse con la Dra. Saavedra por carta, telefónicamente, Fax o e-mail). Depósito de Ampollas en Nitrógeno Líquido: el depósito de ampollas con células congeladas en nitrógeno líquido tiene un arancel anual de U$S 100 por caña , conteniendo 6 ampollas, siendo responsabilidad de la ABAC la custodia de los materiales en depósito y el mantenimiento del frío. Este servicio no involucra ningún tipo de control de calidad ni manipulación alguna de las líneas celulares en depósito. Cada estudio sobre el material depositado se realizará a requerimiento específico del interesado, contra el pago de un arancel adicional. Cuando el depósito de ampollas en nitrógeno líquido es con fines de patentamiento tendrá un arancel anual de U$S 300 por caña. Manual de ATCC: el costo de cada ejemplar es de U$S 10 o su equivalente en $, con agregado de gastos de envío a su cargo. Las órdenes de compra deberán remitirse a los destinos detallados en referencia a los servicios de la ABAC. Almacenamiento de células: Las células procedentes de cultivos primarios o de líneas celulares establecidas pueden almacenarse durante largos períodos de tiempo en temperaturas bajo cero. Con la criopreservación se favorece: el mantenimiento de células sin tener que utilizar tejidos animales primarios, evitar la pérdida de la línea por contaminación y evitar la pérdida de la línea por cambios genéticos. Designación de líneas celulares: Las nuevas líneas celulares deberían tener un código o designación (por ejemplo: normal human brain, NHB); une clase celular o número de línea celular (si varias líneas celulares fueron obtenidas de la misma fuente (por ejemplo: NHB1, NHB2, etc.); y, si es un clon, un número de clon (por ejemplo: NHB2-1, NHB2-2, etc.). para líneas celulares finitas, el número de duplicaciones de población debería ser estimado e indicado después de una barra (por ejemplo: NHB2/2). Cuando se trata de una línea celular continua un número “p” al final es a menudo usado para indicar el número de pasaje desde el último descongelamiento (por ejemplo: HeLa-S3/p4). Es esencial que la designación de la línea celular sea única, para evitar confusiones en distintos trabajos de la literatura. Los bancos de células eliminan este inconveniente dando a cada línea celular un número de acceso. Consideraciones al elegir una línea celular: Además de los requerimientos funcionales específicos, existe una serie de parámetros a considerar a la hora de elegir una línea celular: 40 1) Línea celular finita o continua: ¿Existe una línea celular continua que exprese o tenga las funciones que necesito? Una línea celular continua generalmente es más fácil de mantener, crece rápidamente, el clonado es más fácil, producen mayor rendimiento celular por botella y se adaptan más fácilmente al medio libre de suero. 2) Células normales o transformadas: ¿Es importante para mi modelo si la línea es transformada malignamente o no? Si es así, entonces es posible en muchos casos obtener una línea celular inmortalizada que no es tumorigénica como las 3T3 o BKK21-C13. 3) Especie: ¿Es importante la especie? Las líneas celulares no humanas tienen menores restricciones de seguridad y tienen la ventaja de que el tejido original suele ser más fácil de obtener. 4) Características de crecimiento: ¿Qué requerimiento tiene en términos de velocidad de crecimiento, rendimiento, eficiencia de plaqueo? Se deben considerar los siguientes parámetros: a. Tiempo de duplicación de la población b. Densidad de saturación c. Fracción de crecimiento d. Habilidad de crecer en suspensión 5) Disponibilidad. Si se tiene que utilizar una línea celular finita ¿existe suficiente stock disponible o se tiene que generar una línea celular propia? Si se elige una línea celular continua: ¿existen stocks disponibles autenticados? 6) Validación. ¿Qué tan bien caracterizada es la línea celular? Si ya existe, o si no, ¿puedes hacer la caracterización necesaria?, ¿es auténtica la línea celular? Es vital eliminar la posibilidad de contaminación cruzada antes de embarcarse en un programa de trabajo con una línea celular. 7) Expresión fenotípica. ¿Puede la línea celular elegida expresar las características correctas? 8) Línea celular control. Si se está utilizando un mutante, transfectada, transformada, o línea celular anormal, ¿existe una línea celular normal equivalente disponible? 9) Estabilidad. ¿cuán estable es la línea celular?, ¿ha sido clonada? Si no, puedes clonarla, y ¿cuánto tiempo toma generar la suficiente cantidad para congelar y utilizar stocks de este clon? Dentro de las aplicaciones de los cultivos celulares animales en investigación básica se destacan: 1) la actividad intracelular: transcripción de DNA, síntesis de proteínas, metabolismo energético, ciclo celular, diferenciación, apoptosis, etc; 2) el flujo intracelular de biomoléculas: procesamiento del ARN, el movimiento del ARN desde el núcleo hacia el citoplasma, el movimiento de las proteínas hacia diversos orgánulos, el ensamblaje y desensamblaje de los microtúbulos, etc; 3) genómica y proteómica: análisis genético, infección, transformación celular, inmortalización, senescencia, expresión génica, rutas 41 metabólicas, etc; 4) ecología celular: el estudio de las condiciones ambientales responsables del mantenimiento de la funcionalidad celular y de su diferenciación, el estudio de las necesidades nutricionales, la cinética de la población celular, etc; 5) las interacciones celulares: morfogénesis, proliferación celular, adhesión celular, interacciones con la matriz, invasión celular, etc. Mientras que en investigación aplicada, las técnicas de cultivo celular se utilizan en áreas tan diversas como: 1) Virología: Cultivo de virus animales y de plantas, producción de vacunas, etc; 2) Biotecnología: producción industrial de fármacos en biorreactores (interferón, insulina, hormona de crecimiento, etc.); 3) Inmunología: Producción de anticuerpos monoclonales, señalización, fenómenos de inflamación; 4) Farmacología: Efecto de diversos fármacos, interacciones con el receptor, fenómenos de resistencia, etc; 5) Ingeniería de tejidos. Producción de tejidos artificiales (piel, cartílagos) para el tratamiento de grandes quemados, injertos o autotransplantes, desdiferenciación y diferenciación inducida, etc; 6) Toxicología: citotoxicidad, mutagénesis, carcinogénesis, etc. Ventajas y desventajas de trabajar con cultivos celulares: Como ventajas podemos citar: a) Permiten un control preciso y fino del medio ambiente. En un cultivo se pueden controlar las condiciones físico-químicas (pH, temperatura, presión osmótica, presión parcial de O2 y CO2), y fisiológicos (hormonas, factores de crecimiento, densidad celular, etc.). Para algunas líneas celulares se han definido medios definidos. Un medio definido es aquél en el que se conocen todos y cada uno de los componentes que lo forman y su concentración exacta. Para establecer un medio definido hay que conocer con precisión las necesidades nutritivas de las células en cuestión. Sin embargo en el caso de muchas líneas celulares, estas necesidades no se conocen. En estos casos se utilizan medios que se suplementan con disoluciones complejas (suero, extractos de embrión, etc.) que contienen factores hormonales y nutritivos imprescindibles para el cultivo pero cuya naturaleza se desconoce. b) Caracterización y homogeneidad de la muestra. Una muestra de tejido es siempre heterogénea. Sin embargo, al cabo de uno o dos pasajes, las líneas celulares cultivadas son homogéneas, es decir, su morfología y su composición son uniformes. La presión selectiva de las condiciones de cultivo da lugar a un cultivo homogéneo del tipo celular más vigoroso. A partir de ese momento se pueden obtener réplicas idénticas en cada subcultivo y las características de la línea celular se conservan durante varias generaciones o de forma indefinida si la línea celular se conserva en nitrógeno líquido. Esto facilita mucho el tratamiento estadístico de los resultados. c) Economía. En los cultivos celulares se emplean disoluciones con una concentración mucho menor que en el caso del animal completo. Además, se garantiza el acceso directo de la sustancia a las células sin que se diluya y sin que sufra ningún tipo de modificación metabólica. El costo de los ensayos clínicos se reduce considerablemente y se puede hacer un mayor número de pruebas. También se reducen los costos relacionados con la fabricación del posible nuevo medicamento: en lugar de fabricar cantidades del orden del gramo (para el estudio en animales) basta con que se sinteticen unos pocos miligramos. 42 d) Cuestiones éticas. La investigación biomédica supone el sacrificio cada año de muchos miles de animales de experimentación. El cultivo celular no puede reemplazar siempre al ensayo “in vivo” pero es una alternativa válida en muchas situaciones. Aunque para obtener un cultivo celular primario es posible que haya que sacrificar algún animal, pero con ellos se pueden ensayar un elevado número de condiciones experimentales que, en otras circunstancias, supondrían el sacrificio de decenas o cientos de animales de experimentación. Desventajas: a) Técnica sensible. El crecimiento de las células animales se tiene que realizar en estrictas condiciones de asepsia porque su crecimiento es mucho más lento que el de los contaminantes más habituales (hongos, levaduras, bacterias, micoplasmas, etc.). Además, las células animales son incapaces de mantenerse vivas en ausencia de la compleja mezcla de nutrientes presente en el plasma o en el fluido intersticial. Todo esto condiciona en gran medida el instrumental requerido y el grado de preparación del personal encargado de los cultivos. b) Cantidad y costo. Producir 1 gramo de células en cultivo cuesta 10 veces menos que obtenerlas a partir del tejido de un animal. En un laboratorio normal se pueden conseguir hasta 10 gramos de células sin que se resienta mucho el presupuesto de investigación. Para producir hasta 100 gramos de células hay que incrementar tanto el instrumental como el personal. Para producir cantidades mayores se necesitan instalaciones de tipo industrial. c) Inestabilidad. Muchas líneas celulares continuas son inestables y adoptan una dotación cromosómica aneuploide, lo que afecta tanto a su velocidad de crecimiento como a su capacidad para diferenciarse. Es posible, por tanto, encontrar diferencias significativas en la línea celular de una generación a la siguiente. La única manera de evitarlo es emplear líneas estables que se resiembran cada determinado tiempo o después de un determinado número de generaciones a partir de un stock congelado. d) Desdiferenciación e identificación de las células. Al propagarse la línea celular, las células pierden las características fenotípicas propias del tejido de procedencia. Este fenómeno se denomina desdiferenciación y, entre otras, cosas se hacen móviles e inician su proliferación. La relación entre las células cultivadas y las células originales del tejido puede perderse. En este caso se necesitan marcadores estables que permitan identificar la procedencia de las células. En algunos casos es posible revertir la desdiferenciación, bien por procedimientos de diferenciación inducida por hormonas, bien por compuestos químicos (ésteres de forbol) pero no está claro si el estado rediferenciado es equivalente al estado de diferenciación “in vivo”. e) Validez del modelo “in vitro”. En realidad, un cultivo celular es un disgregado celular de un tejido y se diferencia de éste en que: • se ha perdido la organización espacial tridimensional propia del tejido ya que éstas se propagan en dos dimensiones. • se han perdido las interacciones entre los distintos tipos celulares y entre las células y la matriz extracelular. Además, al formarse una línea celular sólo se conservan uno o dos tipos celulares (los que proliferan a mayor velocidad). 43 • carece de los componentes sistémicos implicados en la regulación de la homeostasis “in vivo”, especialmente los sistemas nervioso y endocrino. • La presión parcial de O2 es reducida, ya que no hay un transportador de oxígeno como la hemoglobina. Por tanto, la energía se obtiene principalmente a través de la glicólisis, ya que el ciclo de Krebs juega un papel muy reducido. Por tanto, tenemos que de ser precavidos en cuanto a la validez de los resultados obtenidos “in vitro” porque pueden diferir de lo que pueda observarse “in vivo”. Bibliografía: Culture of animal cells. A manual of basic technique. Ian Freshney. 5ta ed. Wiley. Introducción al cultivo celular. Universidad de Barcelona, España, http://www.ub.es/biocel/wbc/tecnicas/cap1.htm. “Técnicas fundamentales en el cultivo celular. Un manual de laboratorio”. Sigma, http://www.sigmaaldrich.com. 44 Clase Teórica: MANTENIMIENTO DE LÍNEAS CELULARES Y CRIOPRESERVACIÓN Docente: Dra. Virginia Perdomo [email protected] MANTENIMIENTO DE LINEAS CELULARES Y CRIOPRESERVACIÓN. CRIOPRESERVACION DE CELULAS: Introducción: La criopreservación es el proceso en el cual las células son congeladas a muy bajas temperaturas (entre -130 ºC y -196 ºC), para disminuir las funciones vitales y poderlas mantener en condiciones de vida suspendida por mucho tiempo. A esas temperaturas, cualquier actividad biológica, incluidas las reacciones bioquímicas que producirían la muerte de una célula, quedan efectivamente detenidas. Las líneas celulares en cultivo continuo son propensas a variación debido a la selección del cultivo en pasajes tempranos, a inestabilidad genética y fenotípica, y las líneas celulares finitas a la senecencia. Además, incluso los laboratorios más estrictos son propensos a fallas de equipos y contaminaciones, y a la ocurrencia de contaminación cruzada o identificación errónea. Existen muchas razones, entonces, para congelar un stock validado de células, que se resumen en: 1. Deriva genotípica (fuerza evolutiva que actúa junto con la selección natural cambiando las frecuencias alélicas de las especies en el tiempo) debido a la inestabilidad genética 2. Senecencia y extinción de la línea celular 3. Transformación de las características de crecimiento y adquisición de propiedades asociadas a la malignidad. 4. Inestabilidad fenotípica debido a la selección y desdiferenciación. 5. Contaminaciones con micoplasma 6. Contaminación con otros microorganismos 7. Contaminación cruzada con otras líneas 8. Perdida de identificación debido a la manipulación descuidada 9. Falla en el incubador 10. Minimización de gasto de tiempo y materiales en mantener una línea celular que no será utilizada a la brevedad 11. Necesidad de distribución a otros usuarios Debido a todo esto y a que una línea celular establecida es un recurso valioso y, su reemplazo es costoso y consume mucho tiempo, es de vital importancia contar con stocks de células de trabajo y preservarlas en estado criogénico para el almacenamiento a largo plazo. Dentro de las células de mamíferos, se criopreservan líneas celulares inmortalizadas, células primarias aisladas a partir de tejidos y células madre. 45 Es decir que, la preservación criogénica de los cultivos celulares es usualmente utilizada para mantener tubos de reserva de células (stocks) sin el esfuerzo asociado y el gasto del mantenimiento de los cultivos en crecimiento. Principios de la Criopreservación: Para entender cómo funciona el protocolo de congelamiento, es necesario examinar los eventos intra y extra celulares que ocurren en cultivos celulares animales durante el proceso de congelamiento. El enfriamiento inicial desde temperatura ambiente a 0°C enlentece el metabolismo celular, interrumpe rápidamente el transporte activo y el bombeo de iones. En general esta interrupción no resulta en daño celular si el medio de cultivo está osmóticamente balanceado. Mientras el enfriamiento continúa, de 0° a -20°C se empiezan a formar cristales en el ambiente extracelular lo que aumenta la concentración de solutos del medio de cultivo. Como resultado, el agua comienza a salir de la célula para ingresar al medio extracelular parcialmente congelado, comenzando el proceso de deshidratación celular y contracción. Cuando el proceso de enfriamiento es rápido, se forman cristales de hielo intracelular antes de que se complete la deshidratación de la célula. Estos cristales de hielo rompen las organelas y membranas celulares y llevan a la muerte celular durante el proceso de descongelado. Cuando el proceso de enfriamiento es lento, el agua intracelular sale por ósmosis de la célula resultando en una completa deshidratación y contracción. Esto también puede causar muerte celular pero hay poco acuerdo en el mecanismo involucrado. El estrés físico del encogimiento celular podría causar algún daño produciendo la pérdida irreparable de la membrana y la rotura de organelas y del citoesqueleto. El daño también puede ser causado por las altas concentraciones de solutos en el medio extracelular que quedó sin congelar (esencialmente una solución saturada). Estos solutos atacan la membrana externa e internamente, resultando en daño de la membrana, cambios de pH y desnaturalización general de proteínas. Sin embargo, cuando la velocidad de enfriamiento es lo suficientemente lenta para prevenir la formación de hielo intracelular pero lo suficientemente rápida para evitar serios problemas por efecto de la deshidratación, las células serán capaces de sobrevivir al proceso de congelado y descongelado. Esta ventana o zona de superviviencia es fácilmente observable en muchas bacterias y otras procariotas, pero para la mayoría de las células eucariotas no existe o es muy difícil de encontrar sin usar un agente crioprotector. Estos agentes tienen poco efecto sobre el daño causado por congelamiento rápido (formación de cristales de hielo intracelular), pero en su lugar previenen o disminuyen el daño causado por el congelamiento lento (deshidratación y encogimiento). La temperatura final de almacenamiento es también crítica para una criopreservación exitosa. Para frenar completamente el tiempo biológico las temperaturas de almacenamiento deben mantenerse por debajo de los -130°C, el punto de transición vítrea por debajo del cual no existe agua líquida y la difusión es insignificante. Mientras que muchas líneas celulares se almacenan exitosamente entre -70 y -80°C por meses y aun años, el tiempo biológico no se detiene, solo se enlentece, y se pueden acumular daño cambios celulares. 46 Criopreservación exitosa: La criopreservación de células de manera exitosa requiere seguir protocolos estandarizados y reproducibles, aunque cada protocolo puede requerir modificaciones según el tipo celular o línea a usar, para lograr la máxima viabilidad después del descongelamiento. El éxito del proceso de congelamiento depende entonces de cuatro puntos críticos: 1. Manejo adecuado y recolección suave del cultivo. 2. Uso correcto de los agentes de criopreservación. 3. Una tasa de congelamiento controlada. 4. Almacenaje en condiciones de criopreservacion apropiadas. El congelamiento óptimo de las células para una recuperación de la máxima viabilidad al descongelamiento depende de la minimización de la formación de cristales de hielo intracelulares y reducir el daño criogénico por focos de altas concentraciones de solutos formados cuando el agua intracelular se congela. Esto se consigue: a) congelando lentamente para dejar que el agua se libere de la célula pero no tan lentamente estimular el crecimiento de los cristales de hielo, b) utilizando un crioprotector hidrofóbico que secuestra el agua, c) almacenando las células a la mínima temperatura posible para minimizar los efectos que tienen las altas concentraciones de sales sobre la desnaturalización de las proteínas en micelas dentro del hielo, d) descongelando rápidamente para minimizar la generación de gradientes de solutos formados mientras el hielo intracelular residual se derrite. Preparación de células para su criopreservación: Manejo adecuado y recolección suave del cultivo. Generalmente, la preparación del criovial, se lleva a cabo bajo condiciones asépticas en una campana de cultivo de células. Las suspensiones celulares suelen ser mantenidas frías (0,5 a 4,0°C), por lo que se requiere del uso de hielo fuera de la campana o una unidad de refrigeración libre de hielo dentro de la campana. Procedimiento de examinación: Antes de congelar, las células deben ser mantenidas en un estado de crecimiento activo para asegurar que las células se encuentren saludables y con una buena recuperación. Idealmente, el medio de cultivo debe ser cambiado el día anterior. Usando un microscopio invertido, de forma rápida se puede comprobar el aspecto general del cultivo, buscando señales de contaminación microbiana. También es importante examinar el cultivo al ojo desnudo en busca de pequeñas colonias de hongos que pueden estar flotando en la interface medio - aire y por lo tanto, que no sean visibles a través del microscopio. Es mejor si los cultivos se mantienen libre de antibióticos durante al menos una semana antes de la congelación, para ayudar descubrir cualquier contaminantes en el cultivo. Significancia de la morfología celular: Cualquiera que sea el procedimiento se lleva a cabo, es vital que el cultivo ser cuidadosamente examinado para confirmar la ausencia de contaminación. También se debe comprobar, en las células, si hay signos de deterioro, como granularidad alrededor del núcleo, vacuolización citoplásmica, y redondeo de las 47 células con el desprendimiento del sustrato o soporte. Tales signos pueden implicar que el cultivo requiere un cambio de medio, o puede indicar problemas más graves, como ser: medio o suero, inadecuados o tóxicos, contaminación microbiana o la senescencia de la línea celular. Deficiencias en el medio pueden iniciar además la apoptosis. Durante el mantenimiento de rutina, la frecuencia de cambio de medio o subcultivo debería tener como objetivo prevenir el deterioro celular, que a menudo es difícil de revertir. Familiarizarse con la morfología de la célula también puede dar el primer indicio de contaminación cruzada o identificación errónea. Es útil disponer de una serie de fotografías de los tipos de células utilizadas regularmente, tomadas en diferentes densidades celulares (densidades celulares preferentemente conocidos, por ejemplo, contando el número de células/cm2) para consultar durante la manipulación de los cultivos, sobre todo cuando un nuevo miembro del personal está siendo introducido al trabajo con cultivos celulares. Recolección de células y congelación: Se debe tratar las células con suavidad durante la cosecha, ya que es muy difícil para las células dañadas durante la cosecha, sobrevivir al daño adicional que se produce durante los procesos de congelación y descongelación. Medio de congelamiento y Criopreservantes: Las células de mamíferos se congelan comúnmente con un criopreservante. Estos compuestos reducen el punto de congelación permitiendo una velocidad de enfriamiento más lenta, lo que reduce el riesgo de formación de cristales de hielo que puede dañar las células y la muerte celular a causa de ello. Una gran cantidad de químicos pueden funcionar de manera adecuada como criopreservante; sin embargo, el dimetilsulfoxido (DMSO) y el glicerol son los más conveniente y ampliamente utilizados. Estos criopreservantes permiten una disminución en la temperatura del tubo a una velocidad de 1°C/minuto para evitar los efectos perjudiciales de la formación de cristales de hielo del agua dentro de la célula. De estos dos, el DMSO parece ser el más efectivo, posiblemente debido a que penetra la célula mejor que el glicerol. Este es utilizado usualmente a una concentración final de 5 a 15 % (v/v). Sin embargo, algunas líneas celulares son afectadas de manera adversa por el contacto prolongado con DMSO, resultando ser toxico o inducir la diferenciación celular. Esto puede ser reducido o eliminado al agregar el DMSO a la suspensión celular a 4°C y removerlo inmediatamente después del descongelamiento. Si esto no ayuda, se puede disminuir la concentración de DMSO o probar con glicerol. El glicerol es utilizado generalmente a una concentración final de 5 a 20 % (v/v). A pesar de ser menos toxico que el DMSO, el glicerol puede causar problemas osmóticos, especialmente después del descongelamiento. Siempre debe ser adicionado a temperatura ambiente o mayor, y remover lentamente para lograr la dilución. Debe tenerse la precaución de que el glicerol no tenga mas de un año de antigüedad, dado que se vuelve toxico después del almacenaje prolongado. Otros criopreservantes han sido sugeridos como la polivinilpirrolidina (PVP), el polietilenglicol (PEG) y el hidroxietil almidon (HES), pero ninguno ha tenido tanta aceptación. 48 Por otra parte, altas concentraciones de suero pueden también ayudar a las células a sobrevivir durante el congelamiento. Reemplazar las mezclas estándar de criopreservantes-medio por 95% de suero y 5% de DMSO puede tener mejores resultados en algunas líneas celulares más sensibles, especialmente hibridomas. Velocidad de congelamiento: Como se mencinó anteriormente, la tasa de enfriamiento utilizada para congelar cultivos debería ser lo suficientemente lenta para permitir que las células tengan tiempo de dehidratarse, y lo suficientemente rápida para prevenir el daño por exceso de dehidratación. Una tasa de congelamiento de -1°C a -3°C por minuto es satisfactoria para la mayoría de los cultivos celulares animales. Células más grandes o que tengan membranas menos permeables pueden requerir tasas de congelamiento menores, dado que su deshidratación toma más tiempo. Sin embargo, para la mayoría de las líneas celulares, suele utilizarse una velocidad de congelamiento controlada de -1°C. La congelación se puede realizar en un congelador de reducción de temperatura programada, en un congelador de -80°C ó en un contenedor de hielo seco con un recipiente de congelamiento pasivo a velocidad controlada. La mejor forma de controlar las tasas de congelamiento es utilizar unidades de congelamiento electrónicas programables. A pesar de ser costosas, ellas permiten en control preciso de los procesos de congelamiento, dando resultados muy reproducibles y uniformes y se pueden congelar gran cantidad de viales. Muchas unidades están equipadas con registradores para permitir el registro permamente de los procesos de congelamiento. Existen una variedad de unidades de congelamiento mecanicas que proveen un control adecuado de la tasa de congelamiento y son relativamente baratas. Otra posibilidad es utilizar un contenedor lleno de alcohol destinado a congelar lentamente los viales en su interior. El contenedor lleno es colocado en el interior de un ultra-freezer donde el alcohol actua como un baño para lograr una transferencia de calor y congelamiento mas uniforme. Después de congelar las células durante toda una noche, los viales son removidos del recipiente y transferidos a su lugar de almacenamiento definitivo. Desventaja: Los contenedores de congelamiento celular a base de alcohol, requieren de un constante abastecimiento de alcohol iso-propílico al 100% para mantener una velocidad aproximada de congelación de -1°C/minuto, resultando en un elevado costo y en la eliminación de residuos peligrosos. Los anillos concéntricos que utiliza este método, impide la congelación uniforme de los crioviales, ya 49 que el calor de los crioviales ubicados en su parte interior debe pasar por los crioviales exteriores. Sin embargo, los métodos actuales, tales como los que utilizan los contenedores de congelación a base de alcohol y cajas de poliestireno no proporcionan velocidades de congelación uniforme a todos los crioviales y no son reproducibles. Existen modulos de congelación de células libre de alcohol (no requieren alcohol o de cualquier otro fluido para controlar la velocidad de congelamiento de -1°C/minuto) que proporciona un medio costo-efectivo, con las mismas características de congelación de células de un congelador a -80°C ó contenedor de hielo seco. El material exterior aislante y el núcleo interno de la aleación, junto con la colocación de los crioviales en forma radialmente simétrica, regulan la velocidad de eliminación del calor en forma uniforme para todos los crioviales. Estos proporcionan una criopreservación celular uniforme, consistente y reproducible. Almacenaje de células: Por último, no debe almacenarse células en freezers de -20°C o -80°C, debido a que su viabilidad decrece rápidamente cuando son almacenadas a estas temperaturas. Existen dos formas principales de sistemas de almacenaje en nitrógeno líquido, fase vapor y fase liquida, los cuales necesitan contenedores de almacenaje de boca ancha o boca estrecha. Los sistemas de fase vapor minimizan el riesgo de explosión al nitrógeno líquido de tubos de criopreservacion (o crioviales), y son requeridos para el almacenaje de materiales de peligrosidad biológica; mientras que, los sistemas de fase líquida usualmente tienen tiempos de mantenimiento estáticos mayores, y suelen ser más económicos. Los contenedores de boca estrecha tienen una tasa de evaporación de nitrógeno más lenta y son más económicos, mientras que los contenedores de boca ancha permiten el acceso más 50 fácilmente y tienen una mayor capacidad de almacenaje. La opción elegida finalmente se basa, a menudo, en la disponibilidad de un suministro seguro de nitrógeno líquido, la capacidad de almacenaje requerida y del presupuesto disponible. Los freezers de nitrógeno liquido permiten almacenar en la fase vapor sobre el liquido a temperaturas entre -140°C y -180°C, o sumergidos en el liquido a temperaturas de -196°C. Usando el almacenaje en la fase vapor reduce ampliamente la posibilidad de que los viales perforados (o mal cerrados) exploten durante el descongelamiento. Solo aquellos freezers con la capacidad de mantener la temperatura continuamente por debajo de los -130°C podrían ser considerados para el almacenaje criogénico durante largos periodos de tiempo. A pesar de que muchos freezers de congelamiento de nitrógeno liquido y algunos freezers mecánicamente especialmente diseñados contarían con estos requerimientos, muchos laboratorios de cultivos celulares prefieren freezers de nitrógeno liquido. Viales de almacenaje: Después de la solución crioprotectora se mezcla con la suspensión de células, se añade el inoculo resultante en pequeñas alícuotas (generalmente 1 a 2 mililitros) a cada recipiente de almacenamiento. Debido a las temperaturas extremadamente bajas encontradas durante el almacenamiento criogénico, no todos los materiales o los diseños de los tubos son adecuados o seguro. Muchos materiales se vuelven muy frágiles a estas temperaturas; los tubos hechos pueden romperse o agrietarse durante el almacenamiento o la descongelación. También es importante seleccionar el diseño del sistema de cierre o tapa que se utilizará para mantener la integridad del contenedor, especialmente para el almacenamiento en nitrógeno líquido. Si estos tubos pierden durante el almacenamiento (como muchos lo hacen) que poco a poco se llenarán de nitrógeno líquido. Cuando son eventualmente devueltos a temperatura ambiente, el nitrógeno líquido se vaporiza rápidamente causando una acumulación rápida de la presión. Los tubos pueden luego volar violentamente fuera de sus tapas o explotar para liberar la presión y liberar su contenido en la atmósfera. Esta es una situación muy peligrosa, especialmente si los tubos contenían organismos patógenos o sustancias potencialmente tóxicas o nocivas. El almacenamiento encima del nitrógeno líquido puede reducir estos riesgos potenciales. Dos tipos de contenedores se utilizan comúnmente para el almacenamiento 51 criogénico: ampollas de vidrio termo-sellables y plástico (usualmente de polipropileno) viales de tapa-tornillo. Ambos están disponibles en una variedad de tamaños (de 1 a 5 ml de capacidad) aunque se prefieren los tamaños más pequeños para el almacenamiento criogénico. Debido a problemas de sellado y etiquetado, las ampollas de vidrio ya no son ampliamente utilizados en los laboratorios de cultivo celular. Escapes de pequeños agujeros invisibles pueden ocurrir en viales durante el proceso de sellado; si estos se almacenan después sumergidos en nitrógeno líquido, pueden explotar cuando se retiran para la descongelación. Los poros generalmente pueden ser detectados antes de la congelación mediante la inmersión de ampollas selladas durante 30 minutos en una solución enfriada de etanol al 70% que contiene 1% de azul de metileno. Esta solución penetra rápidamente y manchar las ampollas con fugas. Después de aclarar con agua, ampollas defectuosas son entonces fácilmente detectados y se descartan. Debido a su mayor seguridad y comodidad, los viales de plástico han sustituido en gran medida ampollas de vidrio para el almacenamiento criogénico. La gran variedad de estilos y características especiales como las zonas de marcado impresos y las tapas de colores para facilitar su identificación también añadir a su popularidad. Varios estilos del casquillo están disponibles, algunas de ellas con un tapón roscado internamente, y otras con diseños externamente rosca que ayudan a minimizar la contaminación. Recomendaciones para la criopreservación: Algunas recomendaciones deberían ser tenidas en cuentas a la hora de congelar una línea celular: Seguir de cerca las instrucciones provistas para la línea celular con la que se esta trabajando. Tener en cuenta que las condiciones óptimas de congelación depende de la línea celular en uso. Congelar las células cultivadas en una alta concentración y al menor número de pases posible. Asegurarse que las células tienen al menos 90% de viabilidad antes de la congelación. Congelar las células lentamente mediante la reducción de la temperatura a aproximadamente 1°C/minuto, utilizando un crio-congelador de velocidad de enfriamiento controlada o un recipiente crio-congelación tales como "Mr. Frosty " (NALGENER, Nalge Nunc). Utilizar siempre el medio de congelación recomendado, conteniendo un agente crioprotector como DMSO o glicerol. Guardar las células congeladas debajo de -70°C; las células congeladas comienzan a deteriorarse por encima de -50°C. Utilizar siempre crioviales estériles para el almacenamiento de células. Las células pueden ser almacenadas sumergiéndolas en nitrógeno líquido o en la fase de gas por encima del mismo. Utilizar siempre el equipo de protección personal. 52 Todas las soluciones y equipos que entran en contacto con las células deben ser estériles. Siempre utilizar una técnica estéril apropiada y trabajar en una campana de flujo laminar. Criopreservación de parásitos y plantas: ver clases correspondientes. DESCONGELAMIENTO Y RECUPERACIÓN CELULAR: Es vital descongelar las células correctamente para mantener la vitalidad del cultivo y permitir que el cultivo se recupere lentamente. Procedimiento: 1. Usar el equipo de seguridad adecuado, retire el vial del almacenamiento ubicación y verifique cuidadosamente tanto la etiqueta y almacenamiento para asegurar que es el cultivo correcto. Coloque el recipiente en agua caliente, agitando suavemente hasta que esté completamente descongelado. La descongelación rápida (60 a 90 segundos a 37°C) proporciona la mejor recuperación para la mayoría de los cultivos de células; que reduce o impide la formación de dañar los cristales de hielo dentro de las células durante la rehidratación. 2. Debido a que algunos agentes crioprotectores pueden dañar células tras prolongada exposición, retire los agentes lo más rápido y suavemente como sea posible. Varios enfoques se utilizan en función de ambos los agentes crioprotectores y características de las células: a) La mayoría de las células recuperarse normalmente si tienen el agente crioprotector eliminado por un cambio de medio plazo de 6 a 8 horas de la descongelación. Transferencia el contenido de la ampolla o vial a un frasco T-75 u otro recipiente adecuado que contiene de 15 a 20 ml de medio de cultivo y se incuba normalmente. Tan pronto como la mayoría de las células se han fijado (generalmente 3 a 4 horas), retire el medio que contiene el agente crioprotector ahora diluido y reemplazar con un medio nuevo. b) Para las células que son sensibles a los agentes crioprotectores, quitar el medio viejo se logra fácilmente mediante centrifugación suave. Transferir el contenido del vial o ampolla a un tubo de centrífuga de 15 ml que contiene 10 ml de medio fresco y centrifugado durante 5 minutos a 100 x g. Deseche el sobrenadante que contiene el agente crioprotector y resuspender la célula sedimentar en medio fresco. A continuación, transferir la suspensión celular a un adecuado recipiente de cultivo y se incuban con normalidad. ¿Qué es el subcultivo? Subcultivar, referido normalmente como “pasaje”, es la acción de cambiar el medio de cultivo y transferir las células de un cultivo previo a medio de crecimiento fresco, un procedimiento que permite la propagación adicional de la línea celular. El número de pasaje es entonces el número de veces que el cultivo ha sido subcultivado. 53 Fases del cultivo: Una vez que el cultivo es iniciado, sea un cultivo primario o un subcultivo de una línea celular, necesitara un cambio de medio periódico, o “alimentación”, seguido eventuamente del subcultivo si las células están proliferando. En cultivos sin proliferación, el medio aun necesita ser cambiado periódicamente, mientras las células están todavía metabolizando y algunos constituyentes del medio de agotan o degradan espontáneamente. Cuando una línea celular es subcultivada la regeneración de las células llega al punto del siguiente subcultivo generalmente siguiendo un patrón común. Un período de latencia (“lag period”) después de la siembra es seguido por un período de crecimiento exponencial, denominada fase logarítmica o de crecimiento exponencial (“log phase”). Cuando la densidad celular (células/cm2) alcanza un nivel tal que toda la superficie disponible está ocupada, o cuando la concentración celular (Células/ml de medio) excede la capacidad del medio, cesa el crecimiento o se reduce en gran medida. Es esencial familiarizarse con el ciclo de división para cada línea celular que se maneja, ya que controla la concentración de siembra, la duración de crecimiento antes del subcultivo, la duración de los experimentos, y los tiempos adecuados de muestreo durante un experimento. Debe tenerse en cuenta que las células en diferentes fases del ciclo de crecimiento se comportan de forma diferente con respecto a la proliferación celular, la actividad enzimática, la glucólisis y la respiración, síntesis de productos especializados, y muchas otras propiedades. Figura 1. Patrón de Crecimiento característico de las células cultivadas. La representación semilogarítmica muestra la densidad celular en comparación con el tiempo de permanencia en el cultivo. Las células en cultivo generalmente proliferan siguiendo un patrón de crecimiento estándar. La primera fase de crecimiento después de que el cultivo se siembra es la fase de latencia o “fase LAG”, que es un período de crecimiento lento cuando las células se están adaptando al entorno del cultivo y se preparan para el 54 crecimiento rápido. La fase de latencia va seguida de la fase de crecimiento logarítmico o “fase LOG”, un período donde las células proliferan de manera exponencial y consumen los nutrientes en el medio de crecimiento. Cuando se depleta los nutrientes del medio de crecimiento (es decir, uno o más de los nutrientes se agota) o cuando las células ocupan todo el sustrato o superficie disponible, las células entran en la fase estacionaria (es decir, fase de meseta), donde la proliferación se reduce en gran medida o cesa por completo. Criterios para subcultivar: La necesidad de subcultivar una monocapa es determinada por los siguientes criterios: 1) Densidad del cultivo. Las células normales deben subcultivarse tan pronto como se alcanzan la confluencia. Las células normales (por ejemplo, fibroblastos diploides) por lo general dejan de dividirse en una alta densidad celular, a causa de la aglomeración celular, el agotamiento de factor de crecimiento, y otras razones. Las células se bloquean en la fase G1 del ciclo celular inhibiéndose la mitosis, y se deterioran muy poco, incluso si se deja durante dos a tres semanas o más. Esto hace que posteriormente demoren en recuperarse al ser resembradas. Sin embargo, las células transformadas, líneas celulares continuas y algunas células embrionarias, se deterioran rápidamente a altas densidades celulares a menos que el medio se cambie diariamente o que se subcultiven. Además, los cultivos a una alta concentración celular agotan el medio más rápido que los que están en una baja concentración. 2) Agotamiento del medio y descenso del pH. El agotamiento del medio de cultivo suele ser evidente en la velocidad de cambio de pH, pero no siempre. En las células de mamífero, un descenso en el pH del medio de crecimiento por lo general indica una acumulación de ácido láctico, que es un subproducto del metabolismo celular. El ácido láctico puede ser tóxico para las células, y el pH disminuido puede ser sub- óptimo para el crecimiento celular. La tasa de cambio de pH es generalmente dependiente de la concentración de células en que los cultivos, en cultivos con alta concentración celular el medio se agota más rápidamente que en cultivos de bajas concentraciones celulares. La mayoría de las células dejan de crecer cuando el pH cae de pH 7,0 a pH 6,5 y comenzar a perder viabilidad entre pH 6,5 y pH 6,0, por lo que si el medio pasa de rojo a través de naranja a amarillo, el medio debe ser cambiado. Se debería entonces subcultivar las células si observa una caída rápida del pH mayor a 0,1-0,2 unidades de pH con el incremento de la concentración celular. Tenga en cuenta que una caída repentina de pH también puede ser resultado de la contaminación, así que asegúrese de chequearlo. 3) Tiempo transcurrido desde el último subcultivo. El subcultivo de rutina se realiza mejor de acuerdo a un cronograma estricto. Si las células no han alcanzado una densidad suficientemente alta (es decir, no están confluentes) en el momento apropiado, se debe aumentar la densidad de siembra, o si la confluencia se alcanza demasiado pronto, se debe reducir la densidad de siembra. Una vez establecida esta rutina, el crecimiento recurrente debería ser consistente en duración y rendimiento celular de una densidad de siembra dada. 55 Las desviaciones de este patrón entonces significan una divergencia de las condiciones normales o indican el deterioro de las células. Idealmente, una concentración celular debería ser tal que permite a las células subcultivarlas después de 7 días, con el cambio de medio después de 3-4 días. 4) Requisitos para otros procedimientos. Cuando se requieren células para fines distintos de la propagación de rutina, que también tienen que ser subcultivadas, con el fin de aumentar el stock o cambiar el tipo de recipiente de cultivo o medio. Lo ideal sería que este procedimiento se realice en el horario habitual subcultivo, cuando se sabe que el cultivo está realizando de manera rutinaria. Sin embargo, las demandas de las células no siempre se ajustan a la rutina establecida para el mantenimiento, pero (1) las células no deben subcultivarse mientras se encuentren aún dentro del período lag, y (2) las células deben siempre ser tomadas en el medio de la fase log y el tiempo antes de que hayan entrado en la fase de meseta del subcultivo anterior (a menos que exista un requisito específico para las células en fase de meseta, en cuyo caso tendrán la alimentación frecuentemente o por perfusión continua). 5) Deterioro morfológico. Este factor podria ser prevenido por el examen regular y familiaridad con la línea celular. Si se permite que el deterioro avanzace demasiado, será irreversible, ya que las células tienden a entrar en apoptosis. Subcultivo de líneas celulares adherentes en monocapa: Para una línea celular adherente, la división de un cultivo, o subcultivo, por lo general implica la eliminación del medio y la disociación de las células en la monocapa con algún agente disociante, que rompe la unión de las células entre sí y al soporte de cultivo, que está mediada por glicoproteínas de la superficie celular y Ca+2. Otras proteínas y proteoglicanos, derivados de las células y del suero, se unen a la superficie del sustrato y de otras células, y facilitan la adhesión celular. Por tanto, el primer paso en el subcultivo de células adherentes es separarlas de la superficie del recipiente de cultivo por medios enzimáticos o mecánicos, dado que, por lo general, se requiere de quelación de Ca+2 y la degradación de la matriz extracelular y, potencialmente, de dominios extracelulares de algunas moléculas de adhesión celular. La tripsina suele ser utilizada para la disocianción de las células, aunque algunas células adherentes pueden ser subcultivadas agitando la botella, recogiendo las células en el medio y diluyendo apropiadamente con medio fresco en botellas nuevas. Excepcionalmente, algunas células en monocapas no pueden disociarse de la tripsina y requieren la acción de proteasas alternativos, tales como pronasa, dispasa, y la colagenasa. De estas proteasas, pronasa es la más eficaz, pero puede ser tóxica para algunas células. La dispasa y la colagenasa son generalmente menos tóxicos que la tripsina pero puede dar disociación incompleta de las células epiteliales. La intensidad del tratamiento requerido depende del tipo de célula, como ser la sensibilidad de las células a la proteolisis, por lo que un protocolo debe ser seleccionado de forma tal que sea compatible con la generación de una suspensión de células idividuales de alta viabilidad. Observaciones: 56 1) La tripsina se inactiva en presencia de suero. Por lo tanto, es esencial eliminar todos los rastros de suero del medio de cultivo mediante lavado de la monocapa de células con PBS sin Ca2+ / Mg2+. 2) Las células sólo deben ser expuestos a la tripsina / EDTA tiempo suficiente para separar las células. La exposición prolongada podría dañar receptores de superficie. 3) La tripsina debe ser neutralizada con suero antes de la siembra de células en nuevos frascos de células, de otra manera, las células no se adherirán al soporte. Procedimiento Sacudir Scrapear Agente de disociación Agitar o sacudir gentilmente el frasco de cultivo, o pipetear vigorosamente. Scrapper de células Tripsina Tripsina + colagenasa Disociación enzimática Dispasa Quelación EDTA, Verseno Aplicación Celulas de adhesión leve, células mitóticas. Líneas celulares sensibles a proteasas; puede dañar algunas células. Células de adhesión fuerte. Cultivos de alta densidad, cultivos que han formado múltiples capas, especialmente fibroblastos. Despegado de células epiteliales confluentes, laminas intactas de la superficie de placas de cultivo sin disociar las células. Quelación de Ca+2 de células sensibles a la disociación enzimática. Subcultivo y Propagación de células en suspensión: Existen células que crecen continuamente en suspensión, ya sea porque no son adherentes (por ejemplo, muchas leucemias y tumores murinos) o porque han sido mantenidos en suspensión mecánica o seleccionadas. Subcultivar células en suspensión es menos complicado que los pasajes de células adherentes. Debido a que las células ya se suspenden en medio de crecimiento, no hay necesidad de tratarlas enzimáticamente para separarlos de la superficie del recipiente de cultivo, y todo el proceso es más rápido y menos traumático para las células. Las células se mantienen alimentándolas cada 2 a 3 días hasta que alcanzan la confluencia. Esto se puede hacer directamente mediante la dilución de las células en el frasco de cultivo y que continúen en expansión, o mediante la retirada de una parte de las células del frasco de cultivo y diluyendo las células restantes a una densidad de siembra apropiada para la 57 línea celular. En cada caso, se producirá un ciclo de crecimiento, similar al de células en monocapa, pero con un período de latencia más corto. Las células en suspensión se pueden mantener en frascos de cultivo normales, que no tiene por qué ser tratados para el cultivo de tejidos (aunque deben ser estériles, por supuesto). Las normas relativas a la profundidad de medio en cultivos estáticos son como para monocapas, es decir, de 2-5 mm para permitir el intercambio de gases. Cuando se aumenta la profundidad del cultivo en suspensión, por ejemplo, si este se expande, se requiere la agitación del medio. Manejo de diferentes líneas celulares: Diferentes líneas celulares deberán tratarse por separado, con un conjunto independiente de medios y reactivos. Si se manejan al mismo tiempo, hay un riesgo significativo de contaminación cruzada, particularmente si una línea de rápido crecimiento, tales como HeLa, se mantiene junto a una línea de crecimiento más lento. Los usuarios individuales que requieran stocks durante un período prolongado deberían congelar sus propios stocks de usuarios, que deben ser desechados cuando el trabajo esté terminado. Los Stocks de usuarios nunca deben ser pasados a otro usuario, ya que no han sido completamente validados; los nuevos usuarios deben solicitar un cultivo o ampolla de las existentes del stock de distribución. Normalización de condiciones de cultivo: La normalización de las condiciones de cultivo es esencial para mantener la estabilidad fenotípica. Aunque algunas condiciones pueden alterarse debido a las exigencias del experimento, desarrollo y producción, el mantenimiento de rutina debe ajustarse a condiciones estándar definidos. Medio. El tipo de medio utilizado influirá en la selección de diferentes tipos de células y regulará su expresión fenotípica. En consecuencia, una vez que se ha seleccionado un medio, se debe estandarizar las condiciones en ese medio, y se mantendrá preferiblemente un mismo proveedor, si el medio se compra ya hecho. Suero. El mejor método de eliminar la variación de suero es pasar a un medio libre de suero, aunque, por desgracia, las formulaciones libres de suero todavía no están disponibles para todos los tipos celulares, y la conversión puede ser costosa y consumir mucho tiempo. Sustitutos de suero pueden ofrecen mayor consistencia y son generalmente más baratos que el suero o suplementos de factores de crecimiento, pero no ofrecen el control sobre el entorno fisiológico ofrecidos por el medio libre de suero. Si se requiere suero, se debe elegir un lote, y utilizar ese lote en cada etapa del cultivo, incluyendo la criopreservación. Plásticos. La mayoría de las principales marcas de frascos de cultivo y las placas dan resultados similares, pero puede haber variaciones menores debido al tratamiento del plástico para el cultivo de tejidos. Por lo tanto es preferible utilizar siempre el mismo tipo de frasco o placa, y el mismo proveedor. Mantenimiento de la línea celular. Las líneas celulares pueden alterar sus características si se cambias las condiciones de mantenimiento. El régimen de mantenimiento debe ser optimizado y luego seguir siendo constante durante el 58 tratamiento de la línea celular. Los cultivos deberían también ser reemplazados por stocks congelados a intervalos regulares. Uso de Antibióticos. El uso continuo de antibióticos alienta la aparición de contaminaciones, particularmente micoplasma, y el desarrollo de microorganismos resistentes a antibióticos. También puede interferir con los procesos celulares que se investigan. Sin embargo, puede haber circunstancias en las que la contaminación es particularmente frecuente o se trabaja con una línea celular particularmente valiosa; en estos casos, se puede usar antibióticos. Si se utilizan, es importante mantener cierto número de stocks libre de antibióticos, con el fin de revelar la presencia de posibles contaminaciones; estas poblaciones se pueden mantener en paralelo, y los stocks pueden ser alternados con y sin antibióticos hasta que el cultivo pueda ser mantenido libre de antibióticos. No es aconsejable adoptar este procedimiento de rutina, y si se sospecha una contaminación crónica, las células deben ser desechadas o la contaminación erradicada, pudiendose volver entonces al mantenimiento libre de antibióticos. Mantenimiento de Registros. Mantenga registro detallado del mantenimiento de rutina, incluyendo la alimentación y subcultivo, y las desviaciones o cambios debe ser añadido a la base de datos de registro, para esa línea celular (cambio de proveedor de medio, si la línea se clona, se transfectada, o se cambia a medio libre de suero). Traslado de células: Los cultivos pueden ser transferidos de un laboratorio a otro como ampollas (o viales) congeladas o como cultivos vivos. Ampollas congeladas. Las ampollas congeladas se transportan en CO2 sólido, dentro de un recipiente de espuma de poliestireno de paredes gruesas. Se debe transferir la ampolla desde el congelador de nitrógeno líquido al CO2 sólido tan rápidamente como sea posible, ya que se calentará en ~10-20°C/min, y la temperatura no deberá elevarse por encima de -50°C. Por lo general, las células permanecerán congeladas hasta 3 días si se encuentran correctamente embaladas, pero si se descongelan lentamente, perderán rapidamentre su viabilidad. El vehículo debe ser informado cuando las células son enviadas en CO2 sólido. Traslado de cultivos vivos. Alternativamente, las células pueden ser enviadas como un cultivo en crecimiento. Las células deben estar en la segunda mitad de la fase logarítmica tardía; confluente o agotarán el medio más rápido y pueden tender a separarse durante el transporte. El frasco debe llenarse hasta la parte superior con el medio, sellando el cuello del frasco con una cinta adhesiva a prueba de agua de tipo estiramiento y sellado en una pequeña bolsa de polietileno. El frasco se embala en bolsas dentro de un envase rígido lleno de embalaje absorbente. Este contenedor se sella en una bolsa de polietileno y luego en espuma de plástico o plástico de burbujas. Colocar en el envase una etiqueta que diga '' frágil '' y, en letras grandes: NO CONGELAR! Tras la recepción, el matraz se retira del embalaje, se limpia a fondo con 70% de alcohol, y se abre en condiciones estériles. La mayor parte del medio se retira, dejando sólo la cantidad normal para el cultivo. Se debe subcultivar ni bien se haya recuperado. 59 Por lo general, es mejor enviar las células a través de un servicio de mensajería, que debe ser informado sobre el contenido del paquete y la urgencia de la entrega. Envíos internacionales requerirán la negociación con los controles aduaneros, y un agente nominado competente que esté familiarizado con este tipo de importación. Bibliografía: John R. W. Masters (2000). Animal Cell Culture, A Practical Approach. Editorial Oxford University. Tercera edición, 69-102. Invitrogen. CELL CULTURE BASICS. Handbook. 26 – 42 John A. Ryan. Cryogenic Preservation and Storage of Animal Cells. Corning Incorporated, Life Sciences. 1-5. R. Ian Freshney (2005). Culture of Animal Cells - A Manual of Basic Technique. Editorial Wiley-Liss. Quinta edición.Cap. 13: 199-206. R. Ian Freshney (2005). Culture of Animal Cells - A Manual of Basic Technique. Editorial Wiley-Liss. Quinta edición.Cap. 20: 321-334. http://www.biocision.com/campaigns/cell-cryopreservation-espanol 60 Clase Teórica: CULTIVO DE PROTOZOOS PARÁSITOS Docente: Dra. Pamela Cribb [email protected] CULTIVO DE PROTOZOOS PARÁSITOS El cultivo de protozoos parásitos suele ser un proceso complejo y sujeto a un gran número de variables, algunas conocidas y otras aun no definidas. La vida parasitaria en general se asocia a ciclos de vida complejos, con diferentes formas morfológicas que pueden alternar entre hospedadores muy diferentes entre si. El cultivo in vitro de estos organismos involucra un gran número de variables como el estadío del ciclo de vida del parásito, la especie o cepa del parásito, las características asociadas al hospedador (hábitat, temperatura, respuesta inmune), si el parásito presenta algún tipo de respuesta protectora, etc. Simular el hábitat del parásito dentro de cada hospedador en un sistema in vitro puede ser dificultoso (Visvesvara & Garcia, 2002). Para que cultivar parásitos? Si bien las técnicas moleculares permiten realizar diagnóstico y otros estudios con un pequeño número de parásitos, algunos estudios requieren un número mayor de organismos sin contaminantes bacterianos o de materiales del hospedador, que solo pueden conseguirse a partir de cultivos axénicos. El cultivo in vitro de parásitos causantes de enfermedades en humanos provee información acerca del desarrollo y las necesidades nutricionales del parásito y puede contribuir al diseño de nuevos enfoques para el control de las parasitosis. Es importante para el diagnóstico, ya sea como complemento de otros métodos o por ejemplo para la producción de antígenos para la posterior producción de anticuerpos usados en ensayos inmunológicos. También es útil para identificar proteínas involucradas en la invasión, para el screening de drogas, para diferenciar aislamientos resistentes o susceptibles a determinado agente q uimioterapéutico, para estudios epidemiológicos, para la producción de vacunas, para estudiar la bioquímica, fisiología y metabolismo de los parásitos y determinar sus requerimientos nutricionales, etc. En general, podemos decir que el cultivo de estos organismos se utiliza más que nada en investigación y no tanto con fines diagnósticos (Ahmed, 2014; Visvesvara & Garcia, 2002). Cultivo de protozoos de importancia clínica Podemos distinguir 3 tipos básicos de sistemas de cultivo: xénicos, en los cuales el parásito se crece en presencia de una flora indefinida; monoxénicos, donde el parásito crece en presencia de una única especie adicional; y axénico, donde el parásito crece en ausencia de ninguna otra célula. También se puede hablar de cultivos polixénicos, donde a diferencia de los cultivos xénicos, el parásito crece junto a otros organismos, todos ellos de identidad conocida (Clark & Diamond, 2002). 61 Dependiendo del organismo y el estadío que se desea cultivar, los pasos a seguir y las dificultades de cada uno de ellos, varían notablemente. El aislamiento del organismo a partir de una muestra biológica, el establecimiento del cultivo, la eliminación de contaminantes no deseados y el mantenimiento de los cultivos una vez establecidos, se deben optimizar para cada caso. Protistas parásitos del lumen intestinal: Debido a su hábitat natural, la mayoría de estos parásitos suelen ser aislados en presencia de una gran variedad de bacterias o/y hongos, lo que implica el desafío de eliminar o controlar el crecimiento de los mismos para permitir el cultivo del protozoo de interés. Para Giardia esto suele ser sencillo estableciéndose directamente cultivos axénicos (al igual que para Trichomonas vaginalis, un parásito del tracto genital), mientras que otros protozoos como Entamoeba histolytica y Blastocystis hominis no se han logrado cultivar axénicos sin antes establecerse un cultivo con otros organismos. Más aun, otros organismos como Dientamoeba fragilis y Balantidium coli solo han logrado cultivarse en presencia de otros organismos como cultivos xenicos o monoxénicos (Clark & Diamond, 2002; Leelayoova et al., 2002). Por lo tanto, dependiendo del parásito, el establecimiento de un cultivo axénico puede llevar desde algunos días hasta meses. A pesar de las diferencias en tiempo y pasos requeridos, los medios de cultivo usados para cultivar E. histolytica, Giardia intestinalis y Trichomonas vaginalis son prácticamente idénticos. Amebas de vida libre patógenas y oportunistas: Las amebas de vida libre, normalmente presentes en el suelo o el agua, son menos importantes que las amebas parásitas del tracto gastrointestinal desde el punto de vista clínico, ya que son patógenas pero no presentan un estilo de vida parasitario. En este grupo se incluyen los géneros Naegleria, Acanthamoeba y Balamuthia y son considerados patógenos oportunistas, es decir, causan enfermedades por estar en el lugar indicado en el momento justo o tomando ventaja de un hospedador inmunodeprimido. Naegleria spp. y Acanthamoeba spp. son fácilmente aisladas de muestras del medioambiente, mientras que Balamuthia tiene un crecimiento muy lento que dificulta su aislamiento. Las 3 especies deben ser crecidas como cultivos xénicos en primer lugar, utilizando bacterias como fuente de alimento, para luego lograr la axenización de manera gradual. También es posible su cultivo en presencia de cultivo de tejido en monocapa, donde las células de tejido proveen una capa de alimento para las amebas activamente fagocíticas (Schuster, 2002a). Plamodium spp.: Las 4 especies del género Plasmodium capaces de infectar al hombre (P. falciparum, P. ovale, P. vivax, P. malariae) han sido cultivadas o mantenidas in vitro, pero solo para P. falciparum se han establecido todos los estadíos del ciclo de vida en cultivo. Existen diferencias entre distintas cepas de Plasmodium. Algunas cepas son fácilmente cultivadas in vitro, mientras que otras son refractarias al cultivo. Más aun, los parásitos sufren cambios durante el cultivo. Por ejemplo, en P. falciparum la formación de 62 gametocitos es común en cepas recientemente cultivadas, pero se pierde luego de cultivo prolongado. En este sentido, cabe destacar la importancia de la criopreservación para mantener las características de determinado aislamiento que podrían perderse en cultivos prolongados. También es importante el uso de cepas clonales en experimentos y la caracterización de cepas de laboratorio en base a sus proteínas y ADN (Desai, 2013; Maier & Rug, 2013; Schuster, 2002b). Hemoflagelados: Los hemoflagelados incluyen protozoos parásitos de importancia médica de los géneros Leishmania y Trypanosoma. Una característica de los hemoflagelados -y del orden Kinetoplastida- es una organela única formada por ADN mitocondrial conocida como kinetoplasto. Estos protozoos son pleomorfos, pasando por distintos estadíos durante sus ciclos de vida. Los estadíos difieren en su forma, ubicación del kinetoplasto y longitud del flagelo. También presentan diferencias fisiológicas, bioquímicas y metabólicas de acuerdo a sus hábitats: hospedador vertebrado o insecto vector. A su vez, los ciclos de vida también difieren entre las distintas especies del orden Kinetoplastida. Por ejemplo, Trypanosoma brucei es extra celular durante todo su ciclo mientras que T. cruzi y Leishmania presentan la forma amastigote, el estadío intracelular presente en el hospedador mamífero. El cultivo suele ser una herramienta útil para el diagnóstico de enfermedades causadas por estos parásitos, por ejemplo para algunos casos de leishmaniasis después de la enfermedad de kala-azar dérmica. Existen una variedad de medios para el cultivo hemoflagelados dependiendo del tipo de cultivo y del estadío a crecer. Los requerimientos nutricionales de estos parásitos han sido determinados a partir de técnicas de cultivo. Así, Lee y Hunter (1985) determinaron la necesidad de hemo para el cultivo de kinetoplastidos, que generalmente se agrega al medio de cultivo en la forma de hemina. El cultivo, además ha provisto bases para el diseño y selección de agentes antimicrobianos y contribuído al conocimiento de los mecanismos de variación antigénica, la definición de antígenos inmunogénicos y el desarrollo de cepas atenuadas que podrían ser usadas para proteger a humanos y animales de enfermedades causadas por estos parásitos (Ahmed, 2014; Ajoko & Steverding, 2015; Breidbach, Ngazoa, & Steverding, 2002; DuranRehbein, Vargas-Zambrano, Cuéllar, Puerta, & Gonzalez, 2014; Schuster & Sullivan, 2002). Bibliografía: Ahmed, N. H. (2014). Cultivation of parasites. Tropical Parasitology, 4(2), 80–9. http://doi.org/10.4103/2229-5070.138534 Ajoko, C., & Steverding, D. (2015). A cultivation method for growing bloodstream forms of Trypanosoma brucei to higher cell density and for longer time. Parasitology Research, 114(4), 1611–2. http://doi.org/10.1007/s00436-015-4346-x Arrowood, M. J. (2002). In vitro cultivation of cryptosporidium species. Clinical Microbiology Reviews, 15(3), 390–400. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=118076&tool=pmcentrez &rendertype=abstract 63 Breidbach, T., Ngazoa, E., & Steverding, D. (2002). Trypanosoma brucei: in vitro slender-to-stumpy differentiation of culture-adapted, monomorphic bloodstream forms. Experimental Parasitology, 101(4), 223–30. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12594963 Clark, C. G., & Diamond, L. S. (2002). Methods for cultivation of luminal parasitic protists of clinical importance. Clinical Microbiology Reviews, 15(3), 329–41. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=118080&tool=pmcentrez &rendertype=abstract Desai, S. A. (2013). Insights gained from P. falciparum cultivation in modified media. TheScientificWorldJournal, 2013, 363505. http://doi.org/10.1155/2013/363505 Duran-Rehbein, G. A., Vargas-Zambrano, J. C., Cuéllar, A., Puerta, C. J., & Gonzalez, J. M. (2014). Mammalian cellular culture models of Trypanosoma cruzi infection: a review of the published literature. Parasite (Paris, France), 21, 38. http://doi.org/10.1051/parasite/2014040 Hemphill, A., Stettler, M., Walker, M., Siles-Lucas, M., Fink, R., & Gottstein, B. (2002). Culture of Echinococcus multilocularis metacestodes: an alternative to animal use. Trends in Parasitology, 18(10), 445–51. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/12377595 Leelayoova, S., Taamasri, P., Rangsin, R., Naaglor, T., Thathaisong, U., & Mungthin, M. (2002). In-vitro cultivation: a sensitive method for detecting Blastocystis hominis. Annals of Tropical Medicine and Parasitology, 96(8), 803–7. http://doi.org/10.1179/000349802125002275 Liempi, A., Castillo, C., Cerda, M., Droguett, D., Duaso, J., Barahona, K., … Kemmerling, U. (2015). Trypanosoma cruzi infectivity assessment in “in vitro” culture systems by automated cell counting. Acta Tropica, 143, 47–50. http://doi.org/10.1016/j.actatropica.2014.12.006 Maier, A. G., & Rug, M. (2013). In vitro culturing Plasmodium falciparum erythrocytic stages. Methods in Molecular Biology (Clifton, N.J.), 923, 3–15. http://doi.org/10.1007/978-1-62703-026-7_1 Schuster, F. L. (2002a). Cultivation of pathogenic and opportunistic free-living amebas. Clinical Microbiology Reviews, 15(3), 342–54. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=118083&tool=pmcentrez &rendertype=abstract Schuster, F. L. (2002b). Cultivation of plasmodium spp. Clinical Microbiology Reviews, 15(3), 355–64. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=118084&tool=pmcentrez &rendertype=abstract Schuster, F. L., & Sullivan, J. J. (2002). Cultivation of clinically significant hemoflagellates. Clinical Microbiology Reviews, 15(3), 374–89. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=118086&tool=pmcentrez &rendertype=abstract 64 Visvesvara, G. S., & Garcia, L. S. (2002). Culture of protozoan parasites. Clinical Microbiology Reviews, 15(3), 327–8. Retrieved from http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=118078&tool=pmcentrez &rendertype=abstract (Ajoko & Steverding, 2015; Arrowood, 2002; Breidbach, Ngazoa, & Steverding, 2002; Clark & Diamond, 2002; Duran-Rehbein, Vargas-Zambrano, Cuéllar, Puerta, & Gonzalez, 2014; Hemphill et al., 2002; Leelayoova et al., 2002; Liempi et al., 2015; Maier & Rug, 2013; Schuster & Sullivan, 2002; Schuster, 2002; Visvesvara & Garcia, 2002) 65 Clase Teórica: CULTIVO PRIMARIO. Docente: Dra. Ma. Laura Ruiz [email protected] CULTIVO PRIMARIO. Concepto y tipos. Etapas en el establecimiento de un cultivo primario. Actualmente se entiende por cultivo celular al conjunto de técnicas que permiten el mantenimiento de las células in vitro, conservando al máximo sus propiedades fisiológicas, bioquímicas y genéticas. Los cultivos celulares pueden ser obtenidos a partir de explantes de biopsias (fragmento escindido desde un órgano o tejido), por perfusión de un órgano específico, por disgregación de tejidos o a partir de fluidos orgánicos como la sangre. Existen dos categorías de cultivo celular: 1- Cultivo Primario 2- Línea Celular En este resumen detallaremos algunas consideraciones sobre el cultivo primario. Cultivo primario: El cultivo primario es aquel estado luego del aislamiento de las células y antes del primer subcultivo. Las células mantienen la viabilidad un periodo de tiempo limitado y no se reproducen en cultivo. En general, en cultivo presentan una reducción en el número total de células vivas a lo largo del tiempo. Ejemplos: hepatocitos obtenidos de hígado adulto, neuronas. Cuando el cultivo a partir de tejidos u órganos se mantiene por un periodo limitado de tiempo pero con reproducción de las células en cultivo, se habla de líneas primarias y son ejemplos de las mismas los queratinocitos, fibroblastos dérmicos, las células endoteliales de cordón umbilical. El cultivo primario se utiliza con preferencia a las líneas celulares cuando se buscan condiciones experimentales más representativas del tejido in vivo. Diferencias entre Cultivo Primario y cultivo de Líneas Celulares: 66 Aislamiento de células: El primer paso para aislar células de un tejido es separar la matriz extracelular que las une. Para lograrlo, existen diferentes técnicas para disgregar tejidos. 1- Técnicas que utilizan desagregación enzimática. 2- Técnicas mecánicas puras, con o sin alguna forma de maceración. La disgregación enzimática da mejores resultados cuando hay mucha cantidad de tejido y la disgregación mecánica funciona bien con tejidos blandos o algunos tejidos más duros cuando la cantidad de células viables que se necesita no es tan importante. Disgregación enzimática: En tejidos, la adhesión célula-célula es mediada por la interacción entre una variedad de glicopéptidos homotípicos (moléculas de adhesión o CAMs), algunos de los cuales son dependientes de calcio y por lo tanto sensibles a agentes quelantes como el EDTA o EGTA. Las integrinas, que se unen a sitios arginina-glicina-aspártico (RGD) en la matriz extracelular, también tienen dominios de unión a calcio y son afectados por la depleción de calcio. La matriz intercelular y la membrana basal contienen otras glicoproteínas, tales como fibronectina y laminina, que son sensibles a proteasas, y proteoglicanos los cuales son menos sensibles pero pueden a veces ser degradados por glicanasas, tales como hialuronidasa o heparinasa. Lo más sencillo es proceder desde una simple solución de disgregación a una solución más compleja, con tripsina sola o tripsina/EDTA como punto de partida, agregando otras proteasas para mejorar la disgregación, y eliminar la tripsina 67 si es necesario para mejorar la viabilidad. En general, aumentar la pureza de una enzima nos dará un mejor control y menor toxicidad con aumento en la especificidad pero podría resultar en menor disgregación. La disgregación con tripsina puede dañar las células (por ejemplo, algunas células epiteliales) o puede no ser efectiva (por ejemplo para tejido muy fibroso como el tejido conectivo fibroso), por lo que se ha desarrollado el uso de otras enzimas. Dado que la matriz extracelular frecuentemente contiene colágeno, particularmente en tejido conectivo y músculo, la colagenasa es una buena elección. La técnica de disgregación con colagenasa, es una técnica muy simple y efectiva para muchos tejidos (embrionario, adulto, normal y maligno). La colagenasa cruda es usada frecuentemente y alguna de sus acciones va a depender de la contaminación con otras proteasas no específicas. Hay disponibles colagenasas con alto grado de pureza, en caso de querer evitar la actividad proteolítica no específica, pero ésta podría no ser tan efectiva como la colagenasa cruda. Disgregación mecánica: Al haber un riesgo de daño proteolítico durante la digestión enzimática, muchas veces conviene usar la técnica alternativa de disgregación mecánica, es decir, colectar las células que se desprenden cuando el tejido es cuidadosamente cortado en slices, presionando el tejido diseccionado a través de una serie de tamices donde la malla se reduce gradualmente en tamaño u obligando a los fragmentos de tejido a pasar través de una jeringa o simplemente pipeteando repetidamente. Este procedimiento es más rápido que la digestión enzimática, pero puede causar daños mecánicos. El raspado y tamizado son probablemente los métodos mecánicos más suaves mientras que el pipeteado y, en particular, el pasaje a través de una jeringa, son los que más probablemente generen daño. Nota: En el caso particular de aislamiento de hepatocitos, se realiza una perfusión del órgano con colagenasa (disgregación enzimática), seguido de una agitación suave con varilla de vidrio para favorecer el desprendimiento de las células (disgregación mecánica). De cualquiera de las dos formas de disgregación, se obtiene a partir del tejido, una suspensión celular generalmente conteniendo más de un tipo de células. Para separar los diferentes tipos celulares de la suspensión celular se pueden utilizar varios métodos, según sus propiedades: - La diferencia de tamaño de las células permite separarlas por centrifugación. - La capacidad que tienen algunos tipos celulares de adherirse al vidrio o al plástico, permite separarlos de otras células que se adhieren débilmente. - Algunos anticuerpos se unen específicamente a sustancias presentes en determinadas células. Luego, se emplean diferentes matrices (como colágeno, bolitas de polisacáridos, plástico) a las cuales sólo las células reconocidas por los anticuerpos podrán adherirse. Las células pueden, luego, recuperarse por agitación o por tratamiento con tripsina que digiere las proteínas que median la adhesión. 68 - Se emplean anticuerpos acoplados a colorantes fluorescentes para marcar células específicas adheridas a ellos. Las células marcadas por interacción con el anticuerpo pueden ser separadas de las no marcadas en un separador de células activado por fluorescencia o cell sorter. Una vez disgregado el tejido, y separadas las células, la mayoría de las células obtenidas no están adaptadas para vivir en suspensión, y requieren una superficie sólida en la cual crecer y dividirse. Para los cultivos primarios, en general se requiere recubrir el soporte donde crecerán las células con sustancias tales como colágeno, fibronectina, gelatina, poli-lisina. Además, pueden requerir del agregado de factores de crecimiento (insulina, factores estimulantes de colonias, factor de crecimiento epidérmico, entre otros) y mayores cantidades de suero que las líneas celulares establecidas. Previamente a la siembra se realiza el recuento de las células: Para resolver el problema de conteo celular, se ha optado, en algunos casos, por calibrar sistemas espectrofotométricos, cuyo fundamento se basa en las propiedades de absorción de la luz por parte de las células; sin embargo, tales sistemas son poco exactos y los resultados que se obtienen son difíciles de repetir. Una alternativa a este problema es el contar directamente el número de células presentes en un volumen conocido. Esto puede resultar un gran trabajo considerando el tamaño de algunas células, sin embargo, la cámara de Neubauer permite contar células de distintos tamaños de manera muy práctica. La cámara de Neubauer tiene la apariencia de un portaobjetos grueso; en una cara tiene una placa muy delgada de metal grabada con dos cuadrículas muy finas de medidas definidas y separadas una de otra por una ranura. El fondo de la cámara del campo central es usualmente 0,1 mm más bajo (= profundidad cámara) que ambos campos adyacentes. Entre campo central el cubreobjetos que se coloca sobre la misma existe por tanto una ranura de 0,1 mm. La limitación lateral del volumen a contar se establece mediante la cuadrícula de recuento. De esta manera, es posible conocer el volumen de muestra celular que se coloca en la cámara, y sólo es necesario contar el número de células presentes en ese volumen. La concentración de la suspensión de células estará dada entonces por la cantidad de células contadas sobre el volumen de la cuadrícula (superficie contada * profundidad de la cámara). Determinación de la viabilidad celular: El método más comúnmente usado es el de exclusión de azul tripán. La incubación a tiempos cortos (menores a 15 min) con azul tripán permite diferenciar por recuento en cámara de Neubauer las células viables de las no viables ya que estas últimas se tiñen de azul porque no son capaces de excluir el colorante. 69 Separación de células viables y no viables: Cuando un cultivo primario adherente se prepara de células disociadas, las células no viables son sacadas en el primer cambio de medio. En los cultivos primarios mantenidos en suspensión, las células no viables son gradualmente diluidas cuando comienza la proliferación de células. Si es necesario, sin embargo, las células no viables pueden ser sacadas del disgregado primario centrifugando las células en una mezcla de Ficoll y metrizoato de sodio. Esta técnica es similar a la preparación de linfocitos de sangre periférica. Las células viables se colectan de la interfase entre el medio y el Ficoll/Metrizoate, y las células muertas quedan en el pellet al fondo del tubo. Bibliografía: Culture of animal cells. A manual of basic techniques. Autor: R. Ian Freshney. 5° Edición. Editorial: Willey-Liss. New Jersey. USA. Año 2005. Cell Biology Protocols. Edited by J. Robin Harris, John Graham, David Rickwood. John Wiley & Sons, Ltd. ISBN: 0-470-84758-1. Año 2006. 70 Clase Teórica: TRANSFECCIONES Docente: Lic. Carla Ritagliati [email protected] TRANSFECCIONES La transfección consiste en la introducción de material genético externo en células eucariotas. Las técnicas de transfección celular , han permitido en gran medida ampliar los conocimientos acerca de la regulación génica y de la función de las protein ́ as en los sistemas celulares. Actualmente se emplean en un gran número de aproximaciones experimentales, en la generación de animales transgénicos , en la selección de lin ́ eas celulares modificadas, etc... La introducción de una construcción de ADN recombinante en la que se ha situado el CDS (secuencia codificante) de un gen reportero (luciferasa, ' green fluorescent protein ', beta-galactosidasa, cloramfenicol acetiltransferasa -CAT-, etc..) bajo una secuencia de regulación que se desea estudiar , permite medir con facilidad tasas de expresión génica en diferentes situaciones experimentales. Por el contrario, la introducción de un plásmido que contiene la secuencia codificante (CDS) de una proteína de interés bajo el control de un promotor (constitutivo o regulado ) permite la producción de la proteína deseada que puede estar o no etiquetada (tagged). En este caso se emplea la célula como una factoria ́ de síntesis de proteínas a la que se le introduce en forma de plásmido la información de la proteína que se desea sintetice. Tanto en el primero como en el segundo caso puede ser importante seleccionar las células que han adquirido el plásmido . Para facilitarlo se incluyen en éstos genes de resistencia a drogas que permiten a las células que los han adquirido sobrevivir en medios selectivos. Uno de los sistemas de selección más empleado es el de la resistencia a G418 (resistencia a neomicina ) que permite seleccionar clones celulares de expresión estable . Así diferenciamos entre la transfección temporal o transiente y la transfección estable o de larga duración. MÉTODOS DE TRANSFECCIÓN: Podemos considerar dos grandes grupos de técnicas de transfección, los llamados métodos físicos (se basan en el uso de sistemas mecánicos para lograr la inserción de material genético en las células) y los métodos químicos. También existen los métodos biológicos, como la utilización de virus. Las diferencias entre métodos químicos y físicos se encuentran a nivel metodológico, no a nivel de resultados. También hay que decir que este conjunto de técnicas sólo es funcional in vitro, ya que es necesaria la exposición de las células a medios extremos. 71 El método ideal debe mostrar alta eficiencia de transfección, baja toxicidad de las células, efectos mínimos en la fisiología normal, y además debe ser reproducible y fácil de usar. Método biológico 1. Transducción de ADN mediada por virus Es altamente eficiente, y es fácil de lograr una expresión sostenible de los transgenes in vivo debido a la naturaleza viral de la integración en el genoma huésped. Desventajas: Inmunogenicidad y citotoxicidad, Reacción inflamatoria, Inserción de mutaciones (los vectores virales se integran en el genoma huésped al azar, lo que puede causar una disrupción de genes encargados de suprimir tumores, activar oncogenes o interrumpir genes esenciales), El empaquetamiento del virus tiene un espacio limitado para un gen foráneo. Métodos químicos Basados en la formación de complejos que las células sean capaces de adquirir e incorporar, bien sea directamente mediante la ruta endocit́ ica (fosfato cálcico , DEAE dextrano) o las membranas (lipofección). 1. Fosfato de calico Basado en la obtención de un precipitado entre el cloruro de calcio y el ADN en una solución salina de fosfatos . En esta situación co -precipitan formando unos agregados que son endocitados /fagocitados por las células . Aparentemente el agregado con calcio protege al DNA de la degradación por las nucleasas celulares . El tamaño y la calidad del precipitado es crítico para el éxito del proceso , que se ve afectado por factores tales como pequeños cambios en el pH de la solución . Es uno de los métodos más baratos, se puede utilizar en un amplio espectro de células y tiene buena eficiencia. Presenta varias desventajas: la consistencia en el reactivo es crítica para la reproducibilidad, los pequeños cambios en el pH (+/-0.1) pueden comprometer la eficiencia de transformación, el tamaño y calidad del precipitado son cruciales y NO se puede usar en medio RPMI, debido a la alta concentración de fosfato en el medio. 72 2. DEAE Dextrano Basado en la obtención de complejos entre la resina DEAE y el ADN . Los polímeros de DEAE dextrano o polybreno tienen una carga que les permite unirse a las muy negativamente cargadas moléculas de ADN . El ADN acomplejado se introduce en las células mediante choque osmótico con DMSO o glicerol . Este método es económico, fácil de usar y rápido. Puede ser aplicado a una amplia variedad de células. Sin embargo, altas concentraciones de DEAE-Dextran pueden ser tóxicas para las células y su uso se limita a las transfecciones transientes. 3. Lipofección Se basa en la formación de complejos entre líp idos catiónicos y el DNA . Estos complejos se forman espontáneamente si se colocan los dos en el mismo medio. Inicialmente se empezó a trabajar con lípidos no catiónicos, ya que la organización de los lípidos en bicapa podría facilitar el paso a través de la membrana citoplasmática. Pero estos lípidos no englobaban mucho DNA. A raíz de esto se crearon los liposomas que superan el problema de la compactación debido a que el ADN se contornea como consecuencia de las cargas positivas (en los liposomas el ADN está ocho veces más condensado que en lípidos normales). El complejo tiene afinidad por la membrana y permite la entrada del ADN en el citosol. Una de las posibles vías es la incorporación de liposomas a la membrana y la entrada flip -flap. Existen un gran número de lípidos que se emplean en lipofección , aunque existe una estructura consenso de un lípido catiónico sintético, a partir de la cual se han diseñado muchas variantes (por ej . DOPE). Es esencial optimizar las condiciones específicas de transfección para obtener buenas eficiencias, que suelen ser en buenas condiciones de entre el 70 y el 90% de las células de la placa . La desventaja del método es, aparte del elevado precio de los lípidos, el hecho de que no todos los lip ́ idos funcionan en todos los tipos celulares , y que hay que optimizar el ensayo para cada tipo celular . Los parámetros a optimizar son la relación entre lip ́ ido y DNA (relación de cargas ), la cantidad de DNA empleado , el tiempo que se exponen las células al complejo y la presencia o ausencia de suero. A pesar de tener una alta eficiencia, los complejos liposomales tienen una eficiencia de 104 veces menor que los complejos víricos, presentan un alto grado de 73 interferencia/inhibición en el proceso de transcripción del gen, así como en el mantenimiento del gen (por los sistemas de recombinación homóloga, fijación en el genoma) y los problemas derivados de su regulación. Sin embargo, es fácil de usar, los pasos requeridos son mínimos y es adaptable a sistemas de métodos “high throughput”. Métodos físicos 1. Microinyección La microinyección resulta ser una técnica muy potente y eficaz, ya que 1 de cada 5 células consigue incorporar el ADN foráneo de forma permanente. El problema de esta técnica es que exclusivamente se puede inyectar una célula por vez, de modo que no es el más recomendable para una terapia médica ya que es demasiado laboriosa como para obtener el número de células indicadas (suficientes, aproximadamente un millar o más) para que el tratamiento tenga éxito. muy efectiva pero laboriosa y demanda especialización. Es costoso. Es el método que se emplea en la introducción del DNA recombinante en las células embrionarias en el proceso de obtención de animales transgénicos. 2. Electroporación La electroporación es una técnica que dota a la membrana de cierta permeabilidad al DNA durante unos pocos segundos debido a una descarga eléctrica. Si la célula o grupo de células sometidas a dicho choque eléctrico están sumergidas en un medio rico en plásmidos, alguno de ellos puede penetrar en una célula. Este método no altera la estructura biológica o la función de las células, es fácil de realizar, tiene una alta eficiencia y puede ser aplicado a un amplio número de tipos celulares. El problema que suscita este método es que los choques eléctricos no pueden producir los efectos deseados en algunas células y dañarlas o matarlas debido a esta causa. 3. Biolística (gene gun) También se basa en la inyección de material genético en las células. Originalmente diseñado para la transformación de plantas. La carga útil es una nanopartícula de un metal pesado, generalmente oro, recubierto con ADN, que es "disparada" directamente en el núcleo de la célula diana. Este dispositivo es capaz de transformar casi cualquier tipo de célula, y no se limita al material genético del núcleo: también puede transformar orgánulos, incluyendo los plástidos. 74 Clase Teórica: CULTIVO IN VITRO DE TEJIDOS VEGETALES Docente: Dra.Tatiana Vega [email protected] Cultivo in vitro de tejidos vegetales En su sentido más amplio, el cultivo de tejidos de plantas puede definirse como un conjunto de técnicas de laboratorio en el que se obtienen plantas completas a partir de una porción reducida de tejido separado de la planta. Esta porcion o explanto es cultivado en condiciones asépticas en un medio artificial de composición química definida e incubado en condiciones ambientales controladas. Según su complejidad estas técnicas pueden dividirse en tres grandes grupos: - Cultivo de órganos - Cultivo celulares - Cultivo de protoplastos Totipotencia: potencialidad inherente de las células vegetales de regenerar una planta completa, bajo los estímulos adecuados. Potencialmente la mayoría de las células somáticas con núcleo y citoplasma viable puede desdiferenciarse, proliferar y volver a diferenciarse si se encuentra en el entorno adecuado. Así, los posibles destinos que pueden tomar las células de un tejido en cultivo, dependiendo del explanto y de la composición del medio, son: Formación de órganos, como tallos o raíces (organogénesis) Formación de embriones somáticos (embriogénesis) Cada de estos destinos puede alcanzarse o bien de manera directa o bien atravesando previamente la fase de callo. Un callo es una masa indiferenciada de células, formada por una rápida proliferación celular sin diferenciación. Un fenómeno muy frecuentemente asociado a la formación de callo es la variación somaclonal, que ocurre debido al aumento de mutaciones y otros cambios en el genoma. Aspectos a tener en cuenta para el establecimiento de los cultivos: 1. Explanto. La elección del material de partida está condicionada por la especie vegetal y el objetivo del cultivo. Ejemplos de explantos son discos de hoja, porciones internodales de tallo, meristemas, ovarios y embiones inmaduros. A su vez debe tenerse en cuenta la edad ontogénica del explanto ya que por lo general los tejidos más jóvenes tienen mayor capacidad para la desdiferenciación y proliferación celular. 2. Asepsia. Las condiciones nutritivas de los medios de cultivo de plantas son también las ideales para el crecimiento de muchas especies de hongos y bacterias. 75 3. Composición del medio de cultivo. La composición básica de los medios de cultivo comprende: a. Fuente de carbono (por lo general, sacarosa, aunque también se utilizan glucosa, fructosa y mio-inositol) b. Sales minerales: macro y micro elementos, esenciales también para el crecimiento de plantas completas c. Vitaminas d. Reguladores del crecimiento. Hormonas vegetales: auxinas y citocininas. El balance entre estos dos tipos de hormonas determina el programa de desarrollo que atravesara el cultivo. e. Agente gelificante. Agar-agar o phytagel son los mas utilizados, en un rango del 0,6 al 1% f. Otros compuestos, como aminoácidos, carbón activado, inhibidores de la síntesis del etileno. Se ajustan a cada sistema en particular. 76 4. Condiciones ambientales de incubación. En general la incubación se hace en cámaras de temperatura controlada (250C) y lámparas tipo GroLux o “luz dia”, con ciclos de 16/8 o 12/12 horas de luz/oscuridad. 5. Aclimatación. Una vez obtenidas las plantas regeneradas, se las traslada gradualmente a condiciones de crecimiento en tierra en invernadero, teniendo especial cuidado en evitar la desecación. Objetivos y Aplicaciones del cultivo in vitro: Estudios básicos de procesos fisiológicos en sistemas de células indiferenciadas, o en determinados órganos o grupos de células. En este caso lo que se persigue es la obtención de callos (cúmulos de células indiferenciadas) o de órganos determinados como ovarios o tallos. 77 Obtención de plantas con sanidad controlada. En caso de la infección por virus que se dispersan por el sistema vascular, es común el cultivo de meristemas para obtener nuevas plantas completas no infectadas. Micropropagación. Es posible obtener plantas completas a partir de yemas o ápices terminales, o la obtención de embriones a partir de células somáticas que pueden ser utilizados en la producción de semillas sintéticas. Producción de híbridos sintéticos. En el caso de cruzamientos interespecíficos, en los que no se produce semilla por aborto temprano de los embriones. Fue una de las primeras aplicaciones de estas técnicas. Obtención de plantas doble haploides. Generalmente a partir del cultivo de anteras. Aumento de la variabilidad. Por un proceso conocido como variación somaclonal, se puede aumentar la tasa de variación en determinados genotipos, con la subsecuente aparición de características de interés. Se lo utiliza tanto en especies de interés agronómico como en ornamentales. Producción de compuestos químicos de interés. Conservación e intercambio de germoplasma. El cultivo y criopreservación de meristemas es la técnica de elección para el intercambio de material y la conservación a mediano y largo plazo. Transgénesis. Dentro de un protocolo de transformación, ya sea por infección con Agrobacterium o por bombardeo con micropartículas, las etapas de regeneración y selección in vitro son de importancia crítica. Esquema de los diferentes procesos morfogénicos obtenidos a partir de la siembra in vitro de diferentes explantos. Las líneas continuas expresan organogénesis o embriogénesis directa mientras que las líneas quebradas indican que la morfogénesis se obtiene por vía indirecta. Tomado de Biotecnologia y Mejoramiento Vegetal II. Levitus G, Echenique V., Rubinstein, C., Hopp E., Mroginski L. (Eds.) 2010. 78 Clase Teórica: CONTAMINACIONES DEL CULTIVO CELULAR Docente: Dra. Silvina Villanueva [email protected] CONTAMINACIONES DEL CULTIVO CELULAR: Fuentes, consecuencias, prevención y eliminación. Introducción: El cultivo de células “con éxito” depende en gran medida de mantener las células libre de contaminación. La contaminación puede ser definida como un componente del sistema de cultivo celular que tiene un impacto negativo sobre este y/o su uso. La contaminación del cultivo celular sigue siendo un problema común e importante en la investigación básica así como en la fabricación de productos biológicos. Una rápida detección de la contaminación debería reducir el riesgo de contaminación cruzada y minimizar las consecuencias de cualquier evento de contaminación. Sin embargo, frecuentemente, la contaminación es crónica dado que la mayoría de los eventos de contaminación son ignorados. La contaminación es lo que realmente pone en peligro el uso de los cultivos celulares como una herramienta válida y confiable en estudios donde se los utiliza, por lo cual los riesgos potenciales para la validez de los resultados experimentales incrementan también con la duración de cualquier tipo de contaminación no reconocida. La contaminación puede ingresar al sistema de cultivo celular como un componente de dos categorías principales: contaminantes químicos (tales como impurezas en medio de cultivo, suero y agua, endotoxinas, plastificantes y detergentes) o contaminantes biológicos (bacterias, hongos, levaduras, micoplasmas y virus) del medio ambiente. Las fuentes potenciales y consecuencias de la contaminación son únicas para el sistema de cultivo celular y el contaminante. La contaminación biológica representa la mayor amenaza al sistema de cultivo celular debido a que los organismos vivos metabolizan y se replican. Las células eucariotas en cultivo crecen lentamente, con tiempos de duplicación superiores en muchos casos a las 24 h, sin embargo, tanto las bacterias como las levaduras tienen una tasa de crecimiento superior. Por lo tanto, si se da el caso de una contaminación por estos organismos, su replicación acelerada incrementa en gran medida el título de la contaminación y, consecuentemente, puede provocar la muerte del cultivo en poco tiempo. Esto se debe a que el metabolismo de los organismos contaminantes, incrementa la variedad de efectos nocivos sobre el sistema de cultivo celular por reducir nutrientes esenciales e introducir subproductos tales como toxinas. A su ves, existe un alto riesgo de propagar la infección a otros cultivos vía contaminación cruzada. Fuentes de Contaminación: Existen varias rutas potenciales de contaminación, estas incluyen: Técnica: manipulación, pipeteo, dispensación, etc. Superficie de trabajo. 79 Operador: cabello, manos, vestimenta, etc. Materiales y reactivos: recipientes, tapones, pipetas, frascos de cultivo, suministros no estériles, medios de cultivo, reactivos. Equipamiento e instalaciones: aire de la habitación, campanas de flujo laminar, incubadores. La importación de materiales biológicos: muestras de tejidos, ingreso de líneas celulares. Aunque es imposible eliminar la contaminación por completo, es posible reducir su frecuencia y gravedad mediante el conocimiento profundo de sus fuentes y siguiendo una serie de técnicas de trabajo que suponen la máxima asepsia (“técnica aséptica”), así como la esterilización del material y el trabajo en ambientes perfectamente estériles, cabina de flujo laminar. Técnica de manipulación estéril o asepsia. La fuente más frecuente de contaminación es la “atmósfera” que rodea al cultivo celular, de acuerdo a esto, la técnica de asepsia está diseñada para proporcionar una barrera entre los microorganismos del medio ambiente y el cultivo celular estéril. Los elementos de la técnica asepsia son: Área de trabajo estéril. La forma más sencilla y más económica para reducir la contaminación de partículas en el aire y aerosoles es utilizar una campana de cultivo celular o flujo laminar. Recomendaciones: • La campana de cultivo celular debe estar correctamente configurada y colocada en un área restringida al cultivo celular que esté libre de corrientes de aire de puertas, ventanas y otros equipos, y sin tráfico. • La superficie de trabajo debe estar despejada y contener sólo los elementos necesarios para un procedimiento en particular; no debe utilizarse como un área de almacenamiento. • Antes y después de su uso, la superficie de trabajo debe desinfectarse a fondo, y las áreas y equipos circundantes deben limpiarse de forma rutinaria. • Rutinariamente, limpiar la superficie de trabajo con etanol al 70% antes y durante el trabajo, especialmente después de cualquier derrame. • Es posible utilizar la luz ultravioleta para esterilizar el aire y superficies de trabajo expuestas en la campana de cultivo celular entre los usuarios. • El uso de mechero Bunsen de llama no es necesario ni recomendable en un cultivo celular capucha. • Dejar la campana de cultivo celular funcionando en todo momento, sólo apagarla cuando no se utilice por períodos prolongados de tiempo. Buena higiene personal. Lavarse las manos antes y después de trabajar con cultivos celulares. Además de que protege de los materiales peligrosos, el uso de equipo de protección personal, también reduce la probabilidad de contaminación desde la piel, así como la suciedad y el polvo de la ropa. 80 Reactivos y Medios estériles: los reactivos comerciales y medios de cultivo deben ser sometidos a estrictos controles de calidad para garantizar su esterilidad, no obstante pueden contaminarse durante la manipulación. Para evitar contaminarlos, se deben seguir las siguientes pautas para la manipulación estéril. Siempre esterilizar cualquier reactivos, medios de cultivo o soluciones preparadas en el laboratorio utilizando el procedimiento de esterilización apropiado (por ejemplo, autoclave, filtro estéril). Manipulación estéril: • Limpie siempre las manos y el área de trabajo con etanol al 70%. • Limpie el exterior de los recipientes, frascos, placas y platos con etanol al 70% antes de colocándolos en la campana de cultivo celular. • Evite verter medios y reactivos directamente desde botellas o frascos. • Utilice pipetas de vidrio estériles o de plástico descartables y una propipeta o pipeteador para trabajar con líquidos, y utilizar cada pipeta sólo una vez para evitar contaminación cruzada. No desenvolver pipetas estériles hasta el momento de utilizarla. Mantenga sus pipetas en su área de trabajo. • Tape siempre las botellas y frascos inmediatamente después de su uso. • Nunca destapar un frasco estéril, botella, caja de Petri, etc., hasta el momento en que esté listo para usarlo y nunca dejarlo abierto al medio ambiente. • Si quita una tapa y tiene que dejarla en la superficie de trabajo, coloquela con la apertura hacia abajo. • Utilice sólo material de vidrio y otros equipamientos estériles. • •Tenga cuidado de no hablar, cantar, o silbar cuando está realizando procedimientos estériles. • Llevar a cabo sus experimentos lo más rápidamente posible para minimizar la contaminación. • Cambiar el filtro de la campana de flujo laminar regularmente, de acuerdo a las instrucciones del fabricante. Los tipos de contaminaciones biológicas más frecuentes son: 1. 2. 3. 4. bacterias hongos y levaduras micoplasmas virus Usualmente, la especie o tipo de infección no es importante, a menos que se convierta en un fenómeno frecuente. En general, los organismos de rápido crecimiento son menos problemáticos ya que suelen manifestarse y detectarse fácilmente, con lo cual el cultivo puede ser descartado inmediatamente. No obstante, las dificultades surgen cuando la contaminación es críptica (fenómeno por el que un ente presenta adaptaciones que lo hacen pasar desapercibido a los sentidos de otros), ya sea porque es demasiado pequeña como para ser vista en el microscopio, por ejemplo los micoplasmas, o de crecimiento lento tal que su nivel es tan bajo que escapa a la detección. El uso de 81 antibióticos puede ser una causa común de contaminación críptica, la cual permanece no detectable. Contaminación bacteriana, por hongos y levaduras. Los medios de cultivo proporcionan un medio ideal para el crecimiento de los contaminantes microbianos, y los procedimientos habituales de manipulación (abertura de recipientes, pipeteado, etc.) son susceptibles de provocar contaminación por dichos organismos. Las características de la contaminación microbiana son las siguientes: a- Cambio repentino en el pH, habitualmente se produce una disminución de este parámetro con la mayoría de las contaminaciones bacterianas, cambia muy poco con las levaduras y a veces aumenta con la contaminación por hongos. b- Nubosidad o turbidez en el medio, especialmente si la contaminación se encuentra en una etapa avanzada, a veces con una ligera película o espuma en la superficie, o manchas en la superficie de crecimiento que se disipan cuando el frasco se mueve. c- Bajo un microscopio con bajo aumento (~10x), los espacios entre las células se observarán granular y pueden brillar con la contaminación bacteriana. Las levaduras aparecen como esferas individuales o partículas ovoides de las que pueden estar brotando partículas más pequeñas. Los hongos producen finas micelas filamentosas y, a veces, grupos más densos de esporas. d- Bajo un microscopio con más aumento (~40x), puede ser posible observar las bacterias individualmente y distinguir entre bacilos y cocos. Algunas bacterias forman grumos o están asociadas a las células en cultivo. e- Con una preparación en portaobjeto, la morfología de la bacteria puede observarse a 100x, esto es a veces necesario dado que la contaminación microbiana puede ser confundida con precipitados de constituyentes del medio o con detritus celular, sin embargo, puede ser distinguida por su morfología regular. Los grupos de bacterias pueden ser confundidos con proteínas precipitadas, no obstante, si se agita, muchas bacterias individuales o en cadenas podrán ser visualizadas. Controles de esterilidad. Para evitar las contaminaciones por bacterias es necesario extremar las condiciones de esterilidad y realizar una serie de controles sobre las soluciones y recipientes a usar. También, es recomendable comprobar el estado de contaminación de cualquier tipo celular antes de introducirlo en las salas donde se trabaja con otros tipos celulares. Los controles están diseñados para detectar no aquellos contaminantes de crecimiento rápido y que producen cambios notables en el medio de cultivo, ya que estos se detectan fácilmente incubando alícuotas de medio en la estufa durante 24 o 48 h, sino aquellos otros de crecimiento lento y que no producen cambios apreciables en el cultivo. No existe un test único capaz de detectar todos los contaminantes. Existen diferentes procedimientos propuestos en Freshney (1987) y en R.P.L. Adams (1990) "Cell culture for biochemists”, Elsevier, p. 166. 82 Erradicación de la contaminación bacteriana, por hongos y levaduras. El método más confiable de eliminación de una contaminación microbiana es descartar el cultivo, el medio de cultivo y los reactivos usados con el, dado que el tratamiento de un cultivo o bien no tendrá éxito o puede conducir al desarrollo de microorganismos resistentes. La descontaminación debería ser utilizada únicamente en situaciones extremas, bajo cuarentena y con supervisión de un experto. Sin embargo, en cualquier evento, la descontaminación completa es difícil de lograr, particularmente con levaduras, por lo que es recomendable su prevención. A continuación se enumeran algunas de las precauciones a realizar. Micoplasmas. Los micoplasmas son células procariotas diminutas (0.3 a 0.5 µm de diámetro) que pueden formar colonias pequeñas similares a las que forma el agente productor de la pleuroneumonía bovina (de ahí su nombre de "pleuropneumonia-like organisms", PPLO). No poseen pared celular y por ello sólo son capaces de crecer en ciertos medios. Existen varios grupos serológicos de micoplasmas de dos géneros (Mycoplasma y Acholeplasma). Los contaminantes responsables de las infecciones de los cultivos celulares proceden mayoritariamente de 4 especies: M. orale (humano), M. hyorhinis (cerdo), M. arginini (bovino) y A. laidlawii (bovino y también de roedores). Los cultivos primarios suelen estar libres de micoplasmas no obstante muchas de las líneas celulares continuas están infectadas. La contaminación suele proceder de suero animal contaminado con A. laidlawii y M. arginini o de la tripsina de cerdo contaminada con M. hyorhinis. A pesar de que M. hominis, M. pharyngis y M. salivarum se pueden aislar de la boca y la garganta humana, la mayor fuente de contaminación procede del uso de cultivos contaminados. Se citan datos de que alrededor del 50 al 95 % de las células utilizadas actualmente están contaminadas con micoplasmas. Dado que la contaminación con micoplasmas no es tan evidente como la contaminación por bacterias o levaduras, es importante tener en cuenta el posible efecto de los micoplasmas sobre el cultivo, ya que pueden afectar seriamente a la mayoría de los aspectos del metabolismo celular así como la antigenicidad y las características del crecimiento, y realizar controles periódicos de su presencia. Los efectos que producen sobre el cultivo celular dependen de la especie implicada. De esta forma, las especies M. gallisepticum y M. mycoides son patógenos pero son poco frecuentes. Otras especies no producen la muerte celular pero retardan el crecimiento celular, por ejemplo M. hominis parece retardar el crecimiento del cultivo dado que consume toda la arginina presente debido a su elevada actividad arginina deaminasa, mientras que A. laidlawii destruye rápidamente los deoxiribonucleótidos, y en un cultivo infectado es realmente difícil realizar medidas de incorporación de timidina tritiada dado que ésta es rápidamente convertida a timina. Los requerimientos nutricionales de los micoplasmas son complejos y sólo crecen en medios donde hay células en crecimiento. Se piensa que requieren los productos de degradación del ADN celular, ya que los nucleósidos son factores de crecimiento esenciales para estos microorganismos. 83 Detección de micoplasmas. Una observación cuidadosa de las células en cultivo puede dar un primer aviso de la presencia de micoplasmas: aparición de gránulos oscuros en el citoplasma. A continuación se detallan algunas de las técnicas usadas en la actualidad para la detección de los micoplasmas: 1. Tinción fluorescente con Hoechst 33258, 33217 o DAPI . Esta técnica detecta el ADN de los micoplasmas dado que el colorante (Hoetchst 33258 o 33532, o el DAPI) se une específicamente al ADN. Se emplea la misma técnica de tinción que para la tinción nuclear. Protocolo: El DAPI (4,6-diamino-2-fenilindol) es Sigma D-1388, y se prepara 1mgr/100 ml en PBS pH 7.4. Conservar protegido de la luz a 4 ºC. a. Crecer células en "Slide Flask" durante 24 h en ausencia de antibiótico, hasta una confluencia de 70-80%. b. Lavar la monocapa 3 veces con PBS pH 7.4. c. Fijar con metanol absoluto frio (-20ºC) durante 2 min. d. Lavar la monocapa 3 veces con PBS pH 7.4. e. Añadir 1 gota de DAPI diluido 1:1 en PBS pH 7.4 sobre la monocapa. f. Cubrir con cubreobjetos y visualizar directamente en el microscopio de fluorescencia. Cultivo negativo a micoplasmas (ATCC) Cultivo positivo a micoplasmas (ATCC) 2. Detección de micoplasmas mediante PCR. Se han propuesto diversos métodos para la detección de micoplasmas mediante PCR, y se han comercializado kits, como el producido por la ATCC ("Mycoplasma detection kit") que permite detectar 5 especies de micoplasmas responsables del 95% de las contaminaciones. En el laboratorio se emplea un método de detección mediante PCR que se basa en el descrito por Wong-Lee y Lovett (1998). Empleando una sola pareja de cebadores correspondientes al ARN 18S de 8 especies de micoplasma (M. hyorhinis, M. 84 arginini, M. pneumoniae, M. fermentans, M. orale, M. pirum, Acholeplasma laidlawii y Spiroplasma mirum). Protocolo: Secuencias de los cebadores: Mico1 : GGC GAA TGG GTG AGT AAC ACG Mico2 : CGG ATA ACG CTT GCG AC TAT G Concentración de trabajo de los cebadores 10 pmol/microl Reacción de PCR: Agua 32.7 microl MgCl2 3 microl Buffer 10x 5 microl (AmpliTaq de Perkin Elmer) dNTPs 1 microl (2.5 mM cada uno dATP, dCTP, dGTP, dTTP) Taq polimerasa 0.25 microl Micro1 2.5 microl (aprox. 25 pmoles) Micro2 2.5 microl (aprox. 25 pmoles) Muestra 2.0 microl Volumen de reacción 50 microl Condiciones de PCR: 95ºC 1 min, (95ºC 1 min, 55ºC 1 min, 72ºC 1,30 min) 35 ciclos, 4ºC mantenido. Analizar 10 microl en mini-gelagarosa 2% Tamaño esperado 0.5 kb Control positivo: medio de cultivo de células contaminadas con micoplasma. Control negativo: solución de amplificación con agua. Electroforesis de un control de detección de micoplasmas. Se incluyen muestras de 7 cultivos y dos controles, positivo y negativo. El control + lo es (el método funciona), el control - lo es (no se ha contaminado la solución de amplificación). Seis de los problemas son negativos y hay señal, tenue, en uno de ellos, lo que indica su contaminación. 3. Métodos alternativos para la detección de Micoplasmas. a- Tinción con orceína (Fogh y Fogh, 1968). - Sembrar las células sobre cubreobjetos de modo que no hayan alcanzado la confluencia en 24 h. 85 - Sumergir el cubreobjetos en una placa de Petri con 3 ml de citrato sódico 0.6 % y añadir 86entamente 1 ml de agua. Dejar 10 min y añadir lentamente 4 ml de fijador de Carnoy (1:3 acético:etanol). Sacar todo el líquido y reemplazar por 2 ml de fijador de Carnoy. - Dejar fijar durante 10 min. Dejar secar. - Teñir 10 min con orceína, y lavar 3 veces con etanol absoluto. - Montar en DPX, observar con el microscopio de contraste de fase. Las células contaminadas muestran los micoplasmas preferentemente en el borde celular y en los espacios intracelulares. b- Autoradiografía de cultivos incubados con timidina tritiada. Un cultivo contaminado muestra señal autoradiográfica fuera del núcleo por efecto de la degradación de la timidina a timina propia de muchos micoplasmas. c- Test de crecimiento para micoplasmas. El efecto letal de la contaminación por micoplasmas sobre el cultivo puede acentuarse mediante la adición al medio de 6-metilpurina deoxiribonucleósido (6-MPDR). Todos los micoplasmas presentan gran actividad de la enzima adenosina fosforilasa, capaz de convertir 6-MPDR (no tóxico) en 6-metil purina y 6 metil-purina ribonucleósido, ambos tóxicos para las células de mamífero (McGarrity y Carson, 1982). Este método es la base del kit de detección de micoplasmas "Myco-Tek" comercializado por Gibco. A su vez, existen sistemas de cultivo de micoplasmas de gran sensibilidad, que permiten detectar la presencia de pequeñas cantidades de contaminantes. Es el caso del método directo de cultivo que propone ATCC en su servicio de detección de micoplasmas. Este cultivo, sin embargo, tiene como gran limitación los 28 días que necesita para su desarrollo. d- Sonda DNA para micoplasma. Comercializada por Gen-Probe Inc. es una sonda de DNA tritiada capaz de reconocer el RNA ribosomal de micoplasma en cultivos celulares. En la actualidad se comercializa la tercera versión del kit, y es muy sensible, pero aún cuesta 10 veces más que el ensayo de Hoechst. Supone obtener un lisado celular al que se añade la sonda de DNA, se hibrida a 72 C durante 1 h, se precipita y se lava el híbrido DNA/RNA y se cuenta la cantidad de radiactividad retenida. Protocolo: Kit "Mycoplasma T.C. Rapid detection system", GenProbe Inc., #1951 86 1. Crecer el cultivo a analizar en ausencia de antibióticos durante 3 a 5 dias. 2. Preparación de la muestra: I- Centrifugar de 1.5 a 2.0 ml de medio a 13000 rpm. II- Eliminar el sobrenadante sin alterar el sedimento. III- Añadir 66.6 ul de solución de sonda en cada tubo. Vortex. 3. Hibridación: I- Incubar a 72ºC 2h. II- Durante la incubación añadir 1.5 ml de suspensión de Separación (bien homogénea por inversión) a viales de centelleo de 7 ml preparados. 4. Separación: I- Pasar al contenido del tubo de hibridación a viales de centelleo de 7 ml que contiene la suspensión de separación. Vortex. II- Incubar a 72ºC durante 5 min. III- Vortex para resuspender el sedimento. IV- Centrifugar a 500 xg 1 min. V- Eliminar el sobrenadante por decantación. VI- Lavar cada sedimento dos veces según el proceso: a. añadir 1.5 ml de solución de lavado por tubo. b. incubar a 72ºC durante 5 min. c. Vortex. d. Centrifugar 500 xg 1 min. e. Decantar el sobrenadante. 5. Medida. I- Añadir 2 ml de líquido de centelleo a cada tubo. II- vórtex hasta resuspender el sedimento. III- Esperar 5 min para eliminar fosforescencia. Contar 1 min (muestra). 6. Determinación de background: Contar 2 ml de líquido de centelleo durante 1 min (background). 7. Determinación de cuentas totales. I- Poner 1.5 ml de suspensión de separación en un vial de centelleo. II- Centrifugar a 500 xg 1 min. III- Decantar el sobrendante. IV- Añadir 16.6 ul de solución de sonda. V- Añadir 2.0 ml de líquido de centelleo. VI- Vortex hasta resuspender bien el pellet. VII- Esperar 5 min para eliminar fosforescencia. Contar 1 min (total). VIII- Calcular: cuentas totales = (total - background) x 4. 8. Cálculos. 87 Se calcula el % de hibridación: % hibridación= ((muestra-background) x 100)/cuentas totales Los controles positivos deben dar al menos 30%. Los controles negativos deben dar como máximo 0.2%. Erradicación de la contaminación con micoplasmas. Técnicas para su eliminación. En general se recomienda, si el cultivo es reemplazable, eliminarlo, esterilizar todo y volver a empezar. Sólo cuando el cultivo es irreemplazable o importante se recomienda proceder a su eliminación del cultivo celular. Para ello hay varias posibilidades: 1. Tratamientos antibióticos. Se ha descrito que la kanamicina (0.1 mgr/ml) previene el crecimiento del micoplasma (Fogh y Hacker, 1960), así como la tylocina (6.0 ug/ml). Boehringuer-Mannheim comercializa dos antibióticos (BM cycle) para la eliminación de micoplasmas del cultivo. Asimismo son efectivos contra micoplasmas las quinolonas, como ciprofloxacina (Schmitt y col., 1988), y MRA. Las quinolonas inhiben la ADN girasa procariota. También tetraiclina. 2. Tratamientos con sueros inmunes. El tratamiento del cultivo con sueros antimicoplasmas se ha determinado como efectivo, pero costoso. 3. Pase por un animal. En el caso de cultivos de líneas tumorales se ha propuesto como especialmente útil la inoculación a un animal de experimentación y la recuperación posterior. El sistema inmune del animal limpia el cultivo celulares de los micoplasmas. Virus La contaminación viral es un problema grave ya que su prevención y eliminación son realmente difíciles de realizar. Desde que se descubrió que el suero bovino podía ser una fuente importante de contaminación por diferentes virus (herpesvirus y virus parainfluenza-3) los sueros son controlados rutinariamente, aunque la validez de estos ensayos es limitada. Los virus pueden tener un efecto citopático marcado, pero los más peligrosos son aquellos que crecen a tasa baja o sólo en unas pocas células. Así, en células de individuos con linfoma de Burkitt, producido por EBV (Epstein-Barr virus), no muestran señales de EBV en cultivo hasta varios pases, cuando solo unas pocas células muestran inmunofluorecencia positiva (Henle y Henle, 1966). La manera más eficaz de detectar la infección por virus es infectar un animal sensible y seguir la aparición de efectos citopáticos. Erradicación de la contaminación viral. En el presente, no existen métodos confiables para la eliminación de los virus del cultivo celular, por lo que la única alternativa es el uso de medios libres de suero. 88 Contaminación cruzada: Aunque no es tan común como la contaminación microbiana, la contaminación cruzada extensa de muchas líneas celulares con HeLa y otras líneas de células de crecimiento rápido es un problema claramente establecido con consecuencias graves. La obtención de líneas celulares a partir de bancos de células de buena reputación, la comprobación periódica las características de las líneas celulares y la práctica de una buena técnica aséptica, son medidas que ayudarán a evitar la contaminación cruzada. Huellas de ADN, análisis de cariotipo e isotipo pueden confirmar la presencia o ausencia de contaminación cruzada en los cultivos celulares. Contaminación persistente. Contaminación que parece ser refractaria a cualquier precaución o remedio sugerido. Prestar especial atención a cambios en la técnica, el nuevo personal, nuevos proveedores, nuevos equipos, y a un mantenimiento inadecuado de las campanas de flujo laminar u otros equipos. Un aumento de la tasa de contaminación se debe a: - un deterioro en la técnica de asepsia - un aumento en el contenido de esporas en la atmósfera - incubadores mantenidos en mal estado - refrigeradores o heladeras contaminados - falla del horno de esterilización o del autoclave - un mal envasado de los elementos y reactivos a utilizar - un mal monitoreo del ciclo de esterilización Factores que contribuyen a un aumento de la tasa de contaminación: - cambio en las prácticas de rutina - nuevo personal - aumento de la actividad en el laboratorio Los procedimientos deben seguir siendo estrictos. Esto permitirá reducir la contaminación y ser detectada tempranamente. El uso constante de ATB favorece la contaminación crónica. Es esencial que los cultivos sean mantenidos en condiciones libres de ATBs durante al menos parte del tiempo, y de preferencia todo el tiempo; de lo contrario persistirá la contaminación críptica, su origen será difícil de determinar, y su eliminación imposible. CONCLUSIONES: Chequear los cultivos regularmente: para contaminación microbiana usando un microscopio común y de contraste de fase, y para micoplasma mediante el empleo de tinción fluorescente de preparaciones fijadas o por PCR. 89 No mantener todos los cultivos rutinariamente en antibióticos. Crecer al menos un set de cultivos de cada línea celular sin antibióticos por un período mínimo de dos semanas, con el fin de permitir que las contaminaciones crípticas puedan ser evidentes. No intentar la descontaminación del cultivo celular a menos que sea insustituible y, en ese caso, hacerlo sólo bajo estricta supervisión. Mantener bajo cuarentena todas las líneas celulares nuevas que ingresen al laboratorio hasta que se esté seguro de que no están contaminadas. No compartir medios de cultivo u otras soluciones entre líneas celulares u operadores y chequear las características de las líneas celulares periódicamente para protegerlas contra la contaminación cruzada. Bibliografía: Adams, R.L.P. (1990). "Cell culture for biochemists". Col. Laboratory techniques in biochemistry and molecular biology, gen. edit. R.H. Burdon y P.H. van Knippenberg. Elsevier. Amsterdam-New York. Armstrong, D. (1973). Proc. Soc. Exp. Biol. Med. 122: 475. Freshney (1987). "Culture of animal cells. A manual of basic technique". 2nd. edit. Alan R.Liss (existe actualmente una tercera edición) Fogh, J. y Hacke, C. (1960). Exp. Cell Res. 21: 242. Fogh, J. y Fogh, H. (1968). Proc. Soc. Exp. Biol. Med. 117: 899. Henle, G. y Henle, W. (1966). J. Bacteriol. 91: 1248. McGarrity, G.J. y Carson, D.A. (1982). Exp. Cell Res. 139: 199. Schmitt, K., Daeubener, W., Bitter-Suermann, D. y Hadding, U. (1988). J. Immunol. Meth. 109: 17. Cell cultura basics. Handbook, Invitrogen-Gibco. 90 Clase Teórica: BIOSEGURIDAD EN EL LABORATORIO DE CULTIVO Docente: Dra. Victoria Alonso [email protected] BIOSEGURIDAD EN EL LABORATORIO DE CULTIVO Además de los riesgos de seguridad comunes a la mayoría de los lugares de trabajo, tales como riesgos eléctricos y de fuego, un laboratorio de cultivo celular tiene una serie de riesgos específicos asociados con el manejo y manipulación de células humanas o animales y tejidos, el uso de sustancias tóxicas, corrosivas y reactivos mutagénicos. Los peligros más comunes son: la inoculación accidental con agujas de jeringas u otros objetos punzantes contaminados, derrames y salpicaduras sobre la piel y las membranas mucosas y la exposición por inhalación a aerosoles infecciosos. El objetivo fundamental de cualquier programa de bioseguridad es reducir o eliminar la exposición de los trabajadores de laboratorio y el ambiente exterior a agentes biológicos potencialmente dañinos. La medida de seguridad más efectiva en un laboratorio de cultivo celular es el cumplimiento estricto de las prácticas y técnicas microbiológicas estándar. Los reglamentos y recomendaciones para la bioseguridad en los Estados Unidos se encuentran en el documento de Bioseguridad en Laboratorios Microbiológicos Biomédicos (BMBL), preparado por el Centros para el Control de Enfermedades (CDC) y el Instituto Nacional de Salud (NIH). El documento define cuatro niveles ascendentes de contención, indicados como niveles de bioseguridad 1 a 4, y describe las prácticas microbiológicas, equipos de seguridad y garantías de las instalaciones para el correspondiente nivel de riesgo asociado a la manipulación de un agente en particular. Los mismos se detallan a continuación: Nivel de Bioseguridad 1 (BSL-1) BSL-1 es el nivel básico de protección común en la mayoría de los laboratorios de investigación y clínicos, y es apropiado para los agentes que no causan enfermedades en los seres humanos normales y saludables. Nivel de Bioseguridad 2 (BSL-2) BSL-2 incluye a los agentes de riesgo moderado que se sabe que causan enfermedades humanas de diversa gravedad por ingestión o por medio de la exposición de la membrana percutánea o mucosa. La mayoría de los laboratorios de cultivos celulares deben tener al menos este nivel, aunque los requisitos exactos dependerán de la línea celular utilizada y el tipo de trabajo realizado. Bioseguridad Nivel 3 (BSL-3) 91 BSL-3 es adecuado para los agentes de potencial transmisión por aerosoles, y los agentes que pueden causar infecciones graves y potencialmente letales. B Bioseguridad Nivel 4 (BSL-4) BSL-4 es apropiado para agentes que poseen un alto riesgo individual de enfermedad potencialmente mortal por aerosoles infecciosos y para las que no se dispone de tratamiento. Estos agentes se limitan a los laboratorios de alta contención. Las Hojas de Datos de Seguridad (Data sheets) son formularios que contienen información sobre las propiedades de una sustancia en particular, incluidos los datos físicos, tales como punto de fusión, punto de ebullición y punto de inflamación, así como información sobre su toxicidad, reactividad, efectos sobre la salud, almacenamiento y eliminación. Es importante leerlas en detalle antes de utilizar dicha sustancia o reactivo. El equipo de seguridad en un laboratorio de cultivo celular incluye barreras primarias tales como cabinas de seguridad biológica, recipientes cerrados, y otros controles diseñados para eliminar o minimizar la exposición a materiales peligrosos, así como los equipos de protección personal que se utilizan a menudo en conjunción con las barreras primarias. La cabina de bioseguridad (o campana o flujo laminar) es el equipo más importante, ya que proporcionar contención contra salpicaduras o aerosoles infecciosos generados por muchos procedimientos microbiológicos. El equipo de protección personal forma una barrera inmediata entre el personal y el agente peligroso. El equipo incluye elementos de protección personal como guantes, batas de laboratorio y batas, cubiertas para zapatos, botas, mascarillas, protectores faciales, gafas de seguridad o gafas. A menudo se utilizan en combinación con los gabinetes de seguridad biológica y otros dispositivos que contienen los agentes, animales o materiales que se manejan. Existen algunas pautas generales de trabajo dentro del laboratorio de cultivo: • Usar siempre el equipo de protección personal adecuado. Cambiar los guantes cuando están contaminados, y eliminarlos con otros residuos de laboratorio contaminados (bolsa roja). 92 • • • • • • • Lávese las manos después de trabajar con materiales potencialmente peligrosos y antes de salir del laboratorio. No comer, beber, fumar, manipular lentes de contacto o aplicar cosméticos en el laboratorio. Siga las políticas institucionales relativas a la manipulación segura de objetos punzantes (es decir, agujas, bisturís, pipetas y cristalería rota). Tenga cuidado para reducir al mínimo la creación de aerosoles y / o salpicaduras. Descontaminar todas las superficies de trabajo antes y después de sus experimentos, e inmediatamente después de cualquier derrame o salpicadura de material potencialmente infeccioso con un desinfectante apropiado. Limpiar rutinariamente el equipamiento del laboratorio, incluso si no está contaminado. Descontaminar todos los cultivos y otros materiales potencialmente infecciosos antes de su eliminación. Reportar cualquier incidente que pueda dar lugar a la exposición a materiales infecciosos al personal apropiado (por ejemplo, supervisor del laboratorio, oficial de seguridad). La campana de cultivo celular ofrece un área de trabajo aséptico mientras que permite la contención de salpicaduras o aerosoles infecciosos generados por muchos procedimientos microbiológicos. Existen tres tipos de campanas de cultivo celular, designadas como Clase I, II y III, las cuales se han desarrollado para satisfacer las diversas necesidades de investigación y clínicos. Clases de cabinas de seguridad biológica • Clase I: ofrecen niveles significativos de protección para el personal de laboratorio y el medio ambiente cuando se utiliza con buenas técnicas microbiológicas, pero no proporcionan una protección al cultivo. Son similares en diseño y características de flujo de aire a las campanas de gases. • Clase II: están diseñados para trabajar con materiales BSL-1, 2 y 3, y también proporcionan un ambiente aséptico necesario para experimentos de cultivo celular. Una cabina de bioseguridad Clase II debe usarse para la manipulación de materiales potencialmente peligrosos (por ejemplo, cultivos derivados de primates, culturas infectadas por virus, radioisótopos, reactivos cancerígenos o tóxicos). Tipo A: Son aptas para la investigación en microbiología en ausencia de productos químicos volátiles o tóxicos y radionucléicos, ya que el aire vuelve a circular dentro del gabinete. Tipo B: Se conectan al sistema de escape de la construcción y contienen plena presión negativa. Existen actividades que alteran la dirección del aire como: introducir y sacar los brazos constantemente, apertura y cierre de puertas e inadecuada colocación de materiales o equipo dentro de la cámara. 93 • Clase III: proporcionan el nivel más alto posible de protección para la muestra, el personal y el medio ambiente. Se requiere un gabinete de bioseguridad de Clase III para el trabajo con agentes patógenos humanos conocidos y otros materiales BSL-4. Controla tanto los contaminantes particulados como aerosoles y contaminantes gaseosos mediante sistemas de filtración y disolución de estos. Todas las operaciones se realizan con guantes de goma. Funciona bajo presión negativa. El suministro de aire se realiza a través de filtros HEPA y el gas de escape del gabinete se filtra a través de dos filtros HEPA en serie. 94 Características de flujo de aire de campanas de cultivo celular: Los gabinetes protegen el ambiente de trabajo del polvo y otros contaminantes aerotransportados al mantener un constante flujo unidireccional de aire utilizando un filtro de gran superficie (HEPA) sobre el área de trabajo. Los filtros HEPA retienen el 99,97% de las partículas de 0,3 um de diámetro. Esto les permite retener eficazmente todos los agentes infecciosos conocidos. El flujo puede ser horizontal, que sopla paralelo a la superficie de trabajo, o puede ser vertical, que sopla desde la parte superior de la caja sobre la superficie de trabajo. Dependiendo de su diseño, una campana de flujo horizontal proporciona protección a la muestra (si el aire que fluye hacia el usuario) o al usuario (si el aire es aspirado a través de la parte frontal del armario por presión de aire negativa en el interior). No es un gabinete de seguridad biológica. Se utiliza para preparar mezclas endovenosas, para procedimientos farmacéuticos, para preparar medios de cultivo e investigación experimental limitada. Las campanas de flujo verticales, por otro lado, proporcionan una protección significativa para el usuario y el cultivo celular. 95 Consideraciones prácticas: Existen algunos químicos específicos que se utilizan para el cultivo celular que requieren especial atención. El Dimetil sulfóxido (DMSO), el cual es un solvente potente que puede penetrar la piel y transportar muchas sustancias a través de ella. Inclusive, es capaz de traspasar algunos tipos de guantes (látex o silicona). Además, algunos agentes mutagénicos, carcinogénicos y químicos tóxicos se disuelven en DMSO, aumentando el riesgo de incorporación a través de la piel. Los guantes de nitrilio son una barrera adecuada. El DMSO es incoloro, no necesita ser esterilizado, se debe alicuotar en tubos estériles (polipropileno o vidrio) y almacenar a temperatura ambiente, en oscuridad y evitar contacto con material de goma o plástico (excepto polipropileno). Como se explico en la clase de criopreservación, es utilizado para congelar células, las cuales luego son almacenadas en tanques de nitrógeno líquido. Los principales peligros del nitrógeno líquido: Quemaduras por frío, congelación e hipotermia. Deficiencia de oxígeno en el aire y asfixia. 96 Presurización y explosión. Daños a equipos. La deficiencia de oxígeno es un peligro “silencioso” debido a que sin medios técnicos no es detectable. Además se puede presentar en las condiciones normales de funcionamiento. El nitrógeno líquido al cambiar de fase líquida a gaseosa incrementa su volumen un 683% (a 15°C y 1 atm.). Este cambio de volumen es el que genera variaciones en la concentración de oxígeno en el lugar donde se produce la manipulación. Una concentración deficiente de oxígeno provoca efectos adversos en el cuerpo humano hasta producir incluso la muerte. Concentraciones de O2 inferiores al 6% es posible obtenerlas si se produce un derrame del líquido criogénico o si respiramos en las proximidades del depósito por realizar alguna manipulación incorrecta. Es aconsejable el uso de pequeñas grúas o sistemas de poleas para elevar los contenedores de muestras incluidos en los depósitos (dewars) de nitrógeno líquido sin necesidad de aproximarse excesivamente al mismo. Otro factor que va a influir en la concentración de oxígeno en los lugares donde se manipule nitrógeno líquido son las renovaciones del aire del local. Equipos de protección individual: Estos equipos están destinados a proteger al trabajador ante un contacto accidental con el líquido criogénico, en ningún caso su misión es la inmersión o contacto prolongado. Equipos para la cara: deben de proteger contra contactos por salpicaduras durante la manipulación, podrán ser tipo gafas o protector facial. Equipos para manos: guantes especiales contra protección al frío. Preferentemente que lleguen hasta el antebrazo para evitar posibles salpicaduras en las zonas no cubiertas. Equipos para el cuerpo: se usará bata de manga larga. Debe evitarse la existencia de bolsillos u orificios donde se puedan almacenar salpicaduras. No debe manejarse nitrógeno líquido con manga corta, sandalias, pantalones cortos, faldas, etc. ya que este tipo de indumentaria deja desprotegidas grandes zonas del cuerpo. Se utilizará además un delantal criogénico para proteger el torso y las piernas de posibles salpicaduras. Equipos para los pies: No se permiten sandalias o zapatos abiertos, es necesario el uso de zapatos cerrados que no permitan quemaduras por salpicaduras. Bibliografía: • Biosafety in Microbiological and Biomedica Laboratories. 5th edition. U.S. Department of Health and Human Services. Public Health Service Centers for Disease Control and Prevention National Institutes of Health. 2009. • Gibco Cell Culture Basic Handbook 2015 (www.lifetechnologies.com/cellculturebasics). • Cell Culture of Animal Cells. Ian Freshney. • Cabinas de Seguridad Biológica: Uso, deinfección y mantenimiento. Organización Mundial de la Salud. 2002. 97 Clase Teórica: VENTAJAS Y APLICACIONES DE LA TÉCNICA DE CULTIVO CELULAR Docente: Dra. Virginia Perdomo [email protected] VENTAJAS Y APLICACIONES DE LA TÉCNICA DE CULTIVO CELULAR Ventajas e inconvenientes de los cultivos de células animales y parásitos: Como ventajas de estos cultivos podemos citar: Permiten un control preciso y fino del medio ambiente. En un cultivo se pueden controlar las condiciones físico-químicas (pH, temperatura, etc.), y fisiológicos (hormonas, factores de crecimiento, densidad celular, etc.). Caracterización y homogeneidad de la muestra. La presión selectiva de las condiciones de cultivo da lugar a un cultivo homogéneo del tipo celular más vigoroso. Esto facilita mucho el tratamiento estadístico de los resultados. Economía. Durante los tratamientos, utilizan mucho menos cantidad de drogas que en el caso del animal completo. Además, se garantiza el acceso directo de la sustancia a las células sin que se diluya y sin que sufra ningún tipo de modificación metabólica previa al contacto con la célula. El coste de los ensayos clínicos se reduce considerablemente y se puede hacer un mayor número de pruebas. También se reducen los costes relacionados con la fabricación del posible nuevo medicamento: en lugar de fabricar cantidades del orden del gramo (para el estudio en animales) basta con que se sinteticen unos pocos miligramos. Cuestiones éticas. La investigación biomédica supone el sacrificio cada año de muchos miles de animales de experimentación. El cultivo celular no puede reemplazar siempre al ensayo 'in vivo' pero es una alternativa válida en muchas situaciones, pero se pueden ensayar un elevado número de condiciones experimentales que, en otras circunstancias, supondrían el sacrificio de decenas o cientos de animales de experimentación. Los cultivos celulares también pueden suponer algunas desventajas: a) Técnica sensible. El crecimiento de las células animales se tiene que realizar en estrictas condiciones de asepsia porque su crecimiento es mucho más lento que el de los contaminantes más habituales (hongos, levaduras, bacterias, micoplasmas, etc.). Además, las células animales son incapaces de mantenerse vivas en ausencia de la compleja mezcla de nutrientes presente en el plasma o en el fluido intersticial. Todo esto condiciona en gran medida el instrumental requerido y el grado de preparación del personal encargado de los cultivos. b) Cantidad y coste. Producir 1 gramo de células en cultivo cuesta 10 veces menos que obtenerlas a partir del tejido de un animal. Sin embargo, para producir hasta 100 gramos 98 de células hay que incrementar tanto el instrumental como el personal, y cantidades mayores necesitan instalaciones de tipo industrial. c) Inestabilidad. Muchas líneas celulares continuas son inestables y adoptan una dotación cromosómica aneuploide (cambio en el número de cromosomas), lo que afecta tanto a su velocidad de crecimiento como a su capacidad para diferenciarse. Es posible, por tanto, encontrar diferencias significativas en la línea celular de una generación a la siguiente. La única manera de evitarlo es emplear líneas estables que se resiembran cada determinado tiempo o después de un determinado número de generaciones a partir de un stock congelado. d) Desdiferenciación e identificación de las células. Al propagarse la línea celular, las células pierden las características fenotípicas propias del tejido de procedencia (desdiferenciación) y, entre otras cosas, se hacen móviles e inician su proliferación. La relación entre las células cultivadas y las células originales del tejido puede perderse. En este caso se necesitan marcadores estables que permitan identificar la procedencia de las células. En algunos casos es posible revertir la desdiferenciación, bien por procedimientos de diferenciación inducida por hormonas, bien por compuestos químicos (ésteres de forbol) pero no está claro si el estado “rediferenciado” es equivalente al estado de diferenciación “in vivo”. e) Validez del modelo 'in vitro'. En realidad, un cultivo celular es un disgregado celular de un tejido y se diferencia de éste en que: se ha perdido la organización espacial tridimensional propia del tejido ya que éstas se propagan en dos dimensiones (exceptuando los cultivos histotípicos y organotípicos). se han perdido las interacciones entre los distintos tipos celulares y entre las células y la matriz extracelular. Además, al formarse una línea celular sólo se conservan uno o dos tipos celulares (los que proliferan a mayor velocidad). carece de los componentes sistémicos implicados en la regulación de la homeostasis “in vivo”, especialmente los sistemas nervioso y endocrino. La presión parcial de O2 es reducida, ya que no hay un transportador de oxígeno como la hemoglobina. Por tanto, la energía se obtiene principalmente a través de la glicolisis, ya que el ciclo de Krebs juega un papel muy reducido. Por tanto, hemos de ser precavidos en cuanto a la validez de los resultados obtenidos “in vitro” porque pueden diferir de lo que pueda observarse “in vivo”. Aplicaciones del cultivo celular: El cultivo celular es una de las principales herramientas utilizadas en biología celular y molecular, proporcionando excelentes sistemas modelo para el estudio de la fisiología normal y la bioquímica de las células (por ejemplo, estudios metabólicos, envejecimiento), los efectos de los fármacos y compuestos tóxicos sobre las células, y mutagénesis y la carcinogénesis. También se utiliza en la detección y el desarrollo de fármacos, y la fabricación a gran escala de compuestos biológicos (por ejemplo, vacunas, terapéutico, proteínas). Es decir, los cultivos celulares se utilizan tanto en la investigación básica como en la aplicada. 99 En la investigación básica, permiten estudiar fenómenos complejos como, por ejemplo: Actividad intracelular. Estudia los mecanismos implicados en los diferentes procesos intracelulares, como por ej: transcripción de DNA, síntesis de proteínas, metabolismo energético, ciclo celular, diferenciación, apoptosis, etc. Flujo intracelular de biomoléculas. Estudia los movimientos intracelulares de sustancias y señales asociadas a los diferentes procesos fisiológicos, como por ejemplo: procesamiento del ARN, el movimiento del ARN desde el núcleo hacia el citoplasma, el movimiento de las proteínas hacia diversas organélas, el ensamblaje y desensamblaje de los microtúbulos, etc. Genómica y proteómica. análisis genético, infección, transformación celular, inmortalización, senescencia, expresión génica, rutas metabólicas, etc.Ecología celular: el estudio de las condiciones ambientales responsables del mantenimiento de la funcionalidad celular y de su diferenciación, el estudio de las necesidades nutricionales, la cinética de la población celular, etc. Interacciones celulares. Estudia los procesos de inducción embrionaria, cooperación metabólica, inhibición por contacto o por adhesión, interacciones célula-célula. Ejemplo: morfogénesis, proliferación celular, adhesión celular, interacciones con la matriz, invasión celular, etc. En la investigación aplicada, las técnicas de cultivo celular utilizan en áreas tan diversas como: Virología. establecimiento de condiciones de cultivo de virus animales y de plantas, producción de vacunas antivirales, etc. Biotecnología. producción industrial de fármacos en biorreactores (interferón, insulina, hormona de crecimiento, etc.), vacunas virales y de proteínas más complejas que son glicosiladas (modificadas mediante el agregado de carbohidratos) deben producirse en células animales (ejemplo: hormona eritropoyetina). Actualmente, se están realizando investigaciones para producir tales proteínas complejas en células de insectos o de plantas superiores, debido al alto costo que implica producir tales proteínas complejas en células de mamíferos. Ingeniería de proteínas. Por la producción de proteínas en líneas celulares: interferón, insulina, hormona de crecimiento. Producción de productos biológicos mediante la tecnología del ADN recombinante en cultivo celular incluyen enzimas, hormonas sintéticas, inmunobiológicos (anticuerpos monoclonales, interleucinas, linfoquinas y agentes anticancerígenos). Inmunología. Producción de anticuerpos monoclonales, señalización, fenómenos de inflamación. Farmacología. Efecto de diversos fármacos, interacciones con el receptor, fenómenos de resistencia, etc. 100 Ingeniería de tejidos. Producción de tejidos artificiales (piel, cartílagos) para el tratamiento de grandes quemados, injertos o autotransplantes, desdiferenciación y diferenciación inducida, etc. Cultivo de tejidos e ingeniería. El cultivo celular es un componente fundamental del cultivo de tejidos y la ingeniería de tejidos, ya que establece los conocimientos básicos del crecimiento y mantenimiento de células ex vivo. Vacunas. En la actualidad las vacunas para enfermedades como el polio, sarampión, paperas, rubéola y varicela se producen en cultivos de células de mamíferos. En marzo del 2009 se usó esta técnica para producir la primer partida de vacunas (destinadas al testeo) para la influenza H1N1. También podrían producirse vacunas basadas en ADN recombinante con vectores derivados de adenovirus humanos, causantes del resfrío común. Aplicaciones médicas. Es utilizado en técnicas de alto costo y complejidad, como ser, mantenimiento y producción de tejido para trasplantes, aislamiento y terapias de células madres (transplantes de médula ósea, reemplazo de piel a partir del aislamiento de queratinocitos, células cerebrales, células pancreáticas para el tratamiento de diabetes), fertilización in vitro y transferencia embrionaria, Inyección intracitoplasmática de espermatozoides, criopreservación de embriones, Cultivo de blastocistos, cocultivo los embriones, etc. Aplicaciones diagnósticas. Por ejemplo, en medicina y farmacología destacan el análisis cromosómico de células crecidas a partir de muestras de amniocentesis, detección de infecciones virales, ensayos de toxicidad, etc. Toxicología. citotoxicidad, mutagénesis, carcinogénesis, etc. Odontología. Cultivo de células madre de ligamento periodontal y células troncales de la pulpa dental, para su utilización en regeneración periodontal como herramientas de trabajo en investigación o en medicina regenerativa. Oncología. Se utiliza para estudio in vitro de distintas células tumorales (caracterizadas o no), y son de tipo morfológicos (observación en fresco o coloraciones post fijación), funcionales (viabilidad celular, adhesión, ensayos cronogenicos, migración, actividad enzimática, citotoxicidad), mixtos (morfológicos y funcionales, como inmunofluorescencia) o cromosómicos (cariotipos, traslocaciones, regiones homogéneamente teñidas). Parasitología. Se utiliza para caracterización de cepas, estudio de resistencia a drogas, caracterización de la actividad antiparasitaria de nuevas drogas o compuestos, etc. Diagnostico de: Toxoplasma, Amebas, Giardia, Trichomonas, Leishmania, Plasmodium, aunque no de rutina. Estudios de interacción y señalización celular, en la diferenciación y en el desarrollo. Salud ocupacional. Mediante ensayos citogenéticos que permiten detectar daño genético a nivel celular, donde los cultivos de linfocitos de sangre periférica son una herramienta para aplicar ensayos como: Ensayo de Micronucleos, aberraciones cromosómicas, Intercambio de Cromatides Hermanas, de trabajadores expuestos a algún agente citotóxico. 101 Cultivos in vitro de tejidos vegetales: Se refiere al cultivo de alguna parte de la planta (también llamados “explantes”) como segmentos de hoja, tallo y raíces, además de otros tejidos u órganos vegetales, en un ambiente artificial, en los que deben de controlarse la asepsia, el crecimiento y el desarrollo de estos diferentes tejidos. Ventajas de los cultivos in vitro de plantas: El cultivo in vitro es una práctica biotecnológica que tiene muchas ventajas: favorece la reproducción y multiplicación rápida de los tejidos se obtienen cultivos sanos, libres de virus y agentes patógenes, lo cual es muy importante desde el punto de vista de la seguridad alimentaria Mejor planificación durante el año dado que es posible obtener material para la siembra a lo largo de todo el año se facilita la reproducción de especies de difícil reproducción o de crecimiento lento. Uniformidad y reproducibilidad Obtención de un producto “superior” Aplicable a un amplio espectro de especies Ahorro de espacio Alta tasa de multiplicación Disminución de costes Aplicaciones del cultivo in vitro en biotecnología: La micropropagación. Permite cultivar células, tejidos, órganos, semillas, embriones y obtener individuos selectos en forma rápida. La micropropagación (propagación clonal por cultivo in vitro) constituye uno de los métodos biotecnológicos que mayores logros ha aportado al desarrollo de la agricultura, ya que se la usa en la producción masiva de especies hortícolas, aromáticas, medicinales, frutícolas, ornamentales y forestales. El cultivo de meristemas tiene numerosas aplicaciones. Obtención de plantas libres de virus, ya que esta pequeña zona de tejido generalmente no es afectada por estos patógenos vegetales. Otra muy importante es la multiplicación vegetal. La técnica permite también multiplicar especies de plantas con reproducción lenta o dificultosa (como las orquídeas), o acelerar la producción de plantas bianuales. Cultivo de células y órganos vegetales en biorreactores. Una vez obtenidos los callos a partir de algún explanto, los mismos pueden disgregarse para obtener una suspensión de células. Esta suspensión puede utilizarse para generar embriones somáticos (la base de las semillas sintéticas), o puede directamente cultivarse para producir metabolitos secundarios, que son compuestos químicos sintetizados por las células vegetales en determinadas condiciones, con gran utilidad para las industrias farmacéutica y alimenticia, entre otras. Por ejemplo, son metabolitos secundarios el mentol y las drogas anticancerígenas, vincristina y taxol, y algunos edulcorantes. Las raíces vegetales también pueden ser cultivadas en biorreactores, especialmente aquellas transformadas por Agrobacterium 102 rhizogenes, que produce un aumento abrupto en el tamaño y ramificación de la raíz, aumentando así la biomasa, y por ende la cantidad del producto deseado. Un ejemplo de compuesto farmacológico producido por cultivo de raíces es el paclitaxel, o taxol, que es utilizado como anticancerígeno. Micropropagación: La micropropagación es un sistema de propagación asexual, a partir de un segmento de una planta madre, que da como resultado la propagación masiva de plantas genéticamente idénticas, denominadas clones. La importancia de la micropropagación está basada en las siguientes ventajas: Permite la obtención de plantas de alto registro fitosanitario, ya que al someter al tejido vegetal a este sistema de cultivo se eliminan totalmente las enfermedades de tipo bacteriano y fúngico y en algunas ocasiones las de tipo viral. Las plantas obtenidas por este sistema son réplicas exactas entre si y fieles copias de la planta progenitora. El número de individuos obtenidos por este método es muy superior al obtenido por cualquier otro método de propagación. En algunas ocasiones es posible acortar los tiempos de producción de plantas. Permite tener en espacios relativamente pequeños con un gran número de plantas. Facilita el almacenaje y transporte de plantas reales o potenciales. Elimina los problemas de largas cuarentenas a que son sometidas las plantas en las fronteras cuando se trata de introducirlas de un país a otro. Posibilita incrementar rápidamente nuevos materiales. Permite controlar las condiciones ambientales, debido a su independencia de los mismos (luz, temperatura y humedad controlada). Permite estudiar diversos procesos fisiológicos. Desventajas: Debido a lo sofisticado de la técnica, la micropropagación es restringida y aplicada solo en laboratorios de investigación o en grandes empresas que cuentan con recursos para el mantenimiento del proceso y hacerlo costeable económicamente, lo cual es un hecho debido a la gran cantidad de plantas que por esta vía se obtienen. Es justificable el método solo en plantas con reproducción natural difícil, plantas en peligro de extinción, plantas con características importantes únicas como las transgénicas y plantas de uso ornamental con valor unitario elevado. Aplicaciones de los cultivos vegetales, en general: Hoy esta técnica tiene numerosas aplicaciones: Propagación masiva de plantas. Especialmente para especies de difícil propagación por otros métodos, o en vías de extinción, por Micropropagación. 103 Clonación de individuos de características agronómicas muy deseables durante todo el año Mejora sanitaria (Microinjerto) al permitir el saneamiento del material cultivado, aumentar la seguridad en el intercambio de germoplasma, y la obtención de plantas libres de virus (cultivo de meristemas) Producción de semillas sintéticas Conservación de germoplasma (conjunto de individuos que representan la variabilidad genética de una población vegetal) Obtención de metabolitos secundarios (con gran utilidad para las industrias farmacéutica y alimenticia, ejemplos: el mentol y las drogas anticancerígenos como vincristina, paclitaxel y taxol, y algunos edulcorantes) Producción de nuevos híbridos Mejora genética de plantas (incluyendo obtención de plantas transgénicas) permitiendo el Incremento de variabilidad genética, método haplodiploide y transformación genética. Proporciona Mayor rapidez en la obtención de ciertos logros, y lograr objetivos difíciles de conseguir por los métodos clásicos y objetivos inabordables por los métodos convencionales. Germinación de semillas. Producción de haploides. Estudios fisiológicos diversos. Bibliografía: María Eugenia Segretín. Los cultivos celulares y sus aplicaciones II (cultivos de células vegetales). INGEBI-CONICET. Dpto. FBMyC, FCEyN-UBA. Invitrogen. CELL CULTURE BASICS. Handbook. 26 – 42 Stefanny Romero, Katherine Córdoba, Carlos A. Martínez Valbuena, Juan G. Gutiérrez Quintero, Juan Y Durán Riveros, Juan Carlos Munévar Niño. 2014. Marcadores candidatos, estrategias de cultivo y perspectivas de las DPSCs como terapia celular en odontología. Rev. Odont. Mex Vol. 18, Núm. 2 pp. 156-163 http://www.actionbioscience.org/esp/biotecnologia/pecorino2.html 104 Clase Práctica: LABORATORIO DE CULTIVO CELULAR Docentes: Dra. Victoria Alonso - [email protected] Dra. Virginia Perdomo - [email protected] EL LABORATORIO DE CULTIVO CELULAR. Manejo de cultivos celulares. Preparación de medios de cultivo e inactivación de sueros. Diagrama del laboratorio de cultivo (ejemplos): 105 • • • • • • • • Acceso restringido (puertas permanentemente cerradas) Trampas de aire en los puntos de ingreso. Circulación unidireccional de personas, insumos, materiales y desechos. Todas las superficies (pisos, paredes, mesadas, etc) impermeables al agua y tolerantes a los desinfectantes convencionales. Ingreso del aire a través de filtros HEPA, uni-direccionado y no recirculado Sectores críticos con presión de aire positiva Totalmente separados de instalaciones donde se manejen animales Autoclaves disponibles dentro de cada departamento Identificación del equipamiento dentro de la sala: Cabinas de flujo laminar Incubadores Instrumentos ópticos de observación de cultivo Equipos de esterilización Centrifugas, contadores, equipos de purificación de agua, refrigeración, pHmetro Bomba para aspirar líquidos Como comenzar a trabajar: Es de extrema importancia lavarse las manos antes, después, y frecuentemente durante el trabajo en cultivos, para evitar contaminaciones en los experimentos, y contaminación del usuario con material biológico, y posible diseminación de éste. Después del lavado de manos, rociar exhaustivamente las manos con etanol 70% antes de comenzar a trabajar. Se debe trabajar siempre con material estéril (pipetas, puntas, etc). El material estéril sólo puede abrirse, lógicamente, dentro de la campana de cultivos, o a la llama del mechero. Por defecto, todo aquello de lo que no se esté seguro al 100% de su esterilidad ha de ser considerado como no estéril. “Asumir” que algo está estéril cuando no lo está supone, sin ninguna duda, contaminar (esto es, destruir) el experimento. Se requiere de todos los asistentes al curso un elevado grado de responsabilidad, para no arruinar el trabajo del grupo. En caso de contaminar algo involuntariamente (o incluso sospechar que ésto ha pasado), se debe poner inmediatamente en conocimiento del profesor, para evitar males mayores. Marcar siempre cualquier tubo o recipiente que contenga algo útil, de la forma más segura posible, por ejemplo tanto en la tapa como en el tubo. Marcar siempre las placas de cultivos en la tapa y en un lateral de la base, de manera distinta para cada placa, para evitar intercambiar tapas. Si se ha de pipetear repetidamente un stock de una solución (p.ej. botella con medio de cultivo) es conveniente sacar de una vez la alícuota que se vaya a usar a una botella o tubo de menor volumen, para disminuir el número de veces que se introducen pipetas en la botella con el stock, y el consiguiente riesgo de contaminación. 106 Pasos y pautas para comenzar a trabajar en el flujo: 1. Encender UV por 10-15 minutos antes de comenzar a trabajar, apagarlo, prender la luz y encender el flujo. Esperar 10-15 minutos que se estabilice. En general cada cabina tiene instrucciones especificas de encendido y apagado, consultar los manuales antes de comenzar a trabajar en una nueva cabina. 2. Limpiar con etanol 70% toda la cabina y secarla antes de trabajar (de atrás hacia delante) 3. Colocar el material previamente rociado y secado con etanol 70% 4. Controlar que el cesto de residuos sólidos este cerca de la cabina y disponer de un descarte de líquidos con hipoclorito 20% dentro de la cabina 5. Ubicar el protocolo a utilizar en un lugar visible (NO dentro del flujo) 6. NO colocar celular, calculadora en el área de trabajo 7. Trabajar siempre evitando impedir el flujo de aire sobre los materiales sobre todo con aquellos abiertos, como frascos de medios o cultivos 8. Minimizar el dialogo, no hablar por celular. Si trabaja en equipo utilizar barbijo 9. Evitar derrames, si ocurre limpiar inmediatamente con etanol 70% 10. Rociarse las manos con etanol 70% siempre que se retoma el trabajo SI NO Pautas para el uso de la estufa de cultivo: La estufa posee doble puerta. Antes de abrir la puerta interna ubicar las placas o botellas que se van a sacar para evitar tenerla mucho tiempo abierta. No usar la estufa de cultivo para descongelar material o atemperar medios. No abandonar cultivos y eliminar todo material contaminado rápidamente. Tratar de mover lo menos posible las placas de los compañeros dentro de la estufa. Rotular adecuadamente las botellas y placas. Al sacar placas o botellas siempre controlarlas en el microscopio antes de comenzar a trabajar. 107 Preparación de medios de cultivo: MEDIO DE CULTIVO (DMEM COMPLETO): DMEM suplementado con suero fetal bovino al (2-10%), glutamina 2mM (según el tipo de medio) y antibióticos (100 unidades/ml de penicilina y 100 μg/ml de estreptomicina). El medio viene en diferentes presentaciones: listo para usar líquido o en polvo el cual requiere ser disuelto en agua destilada según las indicaciones de los fabricantes. Luego es necesario filtrarlo, ya sea utilizando unidades de filtración descartables o reutilizables. En este trabajo práctico se armará la unidad de filtración reutilizable y se explicará como esterilizarla. Unidad de Filtración reutilizable: Unidad de filtración descartable: INACTIVACIÓN DE PROTEÍNAS DEL COMPLEMENTO DEL SUERO ("descomplementación"): Descongelar una botella de suero a 37ºC, aumentar entonces la temperatura del baño (con la botella dentro) hasta 56ºC, incubar a esta temperatura 45min, distribuir en alícuotas (25-50 ml) y guardar a -20ºC. Para 108 preparar medio de cultivo, usar una alícuota recién descongelada. Tripsina/EDTA: Se utiliza para despegar las células adherentes a la botella o placa de cultivo. Puede comprarse preparado o preparar en el laboratorio y filtrar (0,25% Tripsina/1mM EDTA). TEMPERATURA DE LOS MEDIOS: Los medios y soluciones que entren en contacto con las células han de estar preferiblemente a 37ºC (o, como muy bajo, temperatura ambiente), para no alterar el metabolismo celular. Siempre desinfectar con etanol 70% las botellas que pasen por el baño antes de trabajar con ellas. ALMACENAMIENTO de distintos componentes de medios de cultivo, y otros medios/reactivos: A 4ºC, stock DMEM, PBS, PBS/EDTA, DMEM completo, suero inactivado con DMSO (para congelar). A -20ºC, suero inactivado, tripsina/EDTA, stocks de tripsina, glutamina, antibióticos, etc. 109 Clase Práctica: INTRODUCCIÓN AL CULTIVO DE LÍNEAS CELULARES Docentes: Lic. Maite Arana – [email protected] INTRODUCCIÓN AL CULTIVO DE LÍNEAS CELULARES. Objetivo: Adquirir las bases y principios necesarios para el manejo de una línea celular, utilizando como ejemplo práctico la línea de origen humano proveniente de adenocarcinoma de colon: Caco-2. Células intestinales Caco-2: La línea celular Caco-2 es de origen epitelial humano y proviene de un adenocarcinoma de colon. A pesar de su origen colónico, estas células al ser cultivadas en condiciones específicas, se diferencian y polarizan de manera de adquirir características morfológicas y funcionales similares a aquellas de los enterocitos que recubren el intestino delgado. Entre estas características se encuentran: la presencia de microvellosidades y uniones estrechas, y la expresión de enzimas y transportadores característicos (disacaridasas, peptidasas, esterasas, P-glicoproteina y otros transportadores). Esta línea celular es altamente heterogénea fundamentalmente en su morfología. Es fácil notar esto simplemente mirando el cultivo al microscopio. Por esta razón los cultivos de Caco-2 utilizados presentan características muy diversas, dificultando la comparación de datos obtenidos en distintos laboratorios. Aun así, esta línea celular, es comúnmente utilizada en forma de monocapas confluentes crecidas en membranas porosas para predecir in vitro la absorción de drogas administradas en forma oral. Como trabajar con una línea celular: Materiales y soluciones: Medio de crecimiento para el cultivo de células Caco-2: DMEM (Dulbecco`s Modified Eagle Medium) (Gibco, nº de catálogo 31600) SFB (suero fetal bovino) 10 % Aminoácidos no esenciales 1 % (Gibco, nº de catálogo 11140) L-Glutamina (200 mM) 1 % (Gibco, nº de catálogo 25030) Penicilina / Streptomicina 1 % (Gibco, nº de catálogo 15140) Anfotericina B 6 ng/ml pH 7,4 PBS 1X: 110 NaH2PO4 0,26 g/L Na2HPO4 1,03 g/L NaCl 8,8 g/L pH 7,4 Tripsina-EDTA: De origen comercial (Gibco, nº de catalogo 25200), contiene Tripsina 25 g/L y EDTA 04 g/L. Descongelado: Las células se conservan en forma indefinida en tanques de nitrógeno líquido. Para comenzar a trabajar con la línea celular Caco-2 se procederá a descongelar algunos viales. Para ello, primero se retira el criovial del tanque de nitrógeno y se lo coloca en un baño a 37°C. Para conservar la viabilidad celular, el descongelado debe realizarse lo más rápido posible, de manera de evitar el crecimiento de los cristales de hielo y la formación local de gradientes de solutos. Una vez que el criovial se encuentre descongelado, se vuelca su contenido en un frasco de cultivo estéril. Como estas células tienen un crecimiento lento luego de ser congeladas, es recomendable comenzar el cultivo utilizando un medio de crecimiento conteniendo 20% de suero fetal bovino. En general los medios de cultivo de células contienen algún tipo de suero como fuente de factores de crecimiento, que benefician la proliferación de las células. Mantenimiento y subcultivo: El mantenimiento de una línea celular incluye generalmente el reemplazo del medio de cultivo y el subcultivo. Es de gran importancia la estandarización de las condiciones de cultivo (preparación del medio, suministros utilizados, frecuencia de los cambios de medio, densidad celular, frascos y placas, etc.) para mantener la estabilidad fenotípica del mismo. Generalmente las líneas celulares crecen adheridas a una superficie (con excepción de aquellas células provenientes de tejidos líquidos). Estas células son capaces de adherirse al vidrio, a algunos plásticos tratados o a superficies pretratadas con algún componente de la matriz, como colágeno o fibronectina. Una vez adheridas las células proliferan y entran en contacto entre sí. En el caso de las células Caco-2 este contacto promueve la diferenciación de las células y el cese de la proliferación. Por lo tanto, para conservar la capacidad proliferativa del cultivo es necesario levantar las células del frasco y volver a sembrarlas con una menor densidad. Cada vez que se realiza este procedimiento aumenta el número de pasaje del cultivo. El subcultivo, en el caso de células adherentes, requiere que las mismas sean disociadas del frasco de cultivo. Existen distintos procedimientos para disociar las células: • Mecánicos: • Shake-off: consiste en agitación o pipeteo y se emplea únicamente para células con interacciones débiles. 111 • • • Scraping: consiste en utilizar un cell scraper para raspar las células del frasco. Se emplea en casos en los que no pueden utilizarse proteasas, no suele generar células individuales. Enzimáticos: • Tripsina: se usa en concentraciones entre 0,01-0,5 % (comúnmente 0,25 %). Es la enzima más utilizada para líneas celulares continuas. Generalmente combinada con EDTA, u otra enzima. Es sensible al suero por lo que el cultivo suele requerir un prelavado. • Colagenasa: se usa 200 U/mL. Menos eficiente que la tripsina. • DNasa: se usa en concentraciones entre 2-10 μg/mL, generalmente luego de un método que dañe las células y libere DNA. • Otras enzimas: dispasa, pronasa, liberasa. Agentes quelantes: EDTA o EGTA. Algunas interacciones célula-célula son dependientes de Ca++. Se utiliza para algunas células con interacciones débiles y en combinación con otro método. En este trabajo práctico se realizará un subcultivo de células Caco-2. Para ello se tomará un cultivo subconfluente (no más de 70% de confluencia) y se extraerá el medio de cultivo. Luego se realizarán 2 lavados con PBS 1X (necesario ya que el SFB inhibe la tripsina). Se agregará 1 mL de la solución comercial de tripsina-EDTA (Gibco, nº de catálogo: 25200) pura o diluida y se incubará en estufa a 37°C, chequeando a intervalos si las células se van levantando. Se debe tener en cuenta que una larga exposición a la tripsina comienza a dañar las células, por lo tanto cuando se observe que las células comienzan a despegarse se debe detener la reacción. Una vez que las células se hayan desprendido del frasco, se agrega medio de crecimiento para inactivar la tripsina. Luego, se procede a disgregar los cúmulos de células, mediante pipeteo reiterado, de manera de obtener células individuales. Conteo de células en cámara de Neubauer: La cámara de Neubauer consiste en una cuadrícula de recuento formada por 9 cuadrados grandes, cada uno con una superficie de 1 mm2. A su vez estos cuadrados grandes están divididos en 25 cuadrados medianos. Utilizando un objetivo 10X se puede visualizar el cuadrado grande con las 25 divisiones medianas. En el cuadrado grande central, los 25 cuadrados medianos se dividen a su vez en 16 cuadrados pequeños. Estos pueden visualizarse claramente utilizando un objetivo 40X. La selección de las cuadrillas a utilizar depende del tamaño de la célula, para la línea celular Caco-2 se cuentan los 4 cuadrados grandes exteriores (N1, N2, N3 y N4), procediendo de la siguiente manera: Se toman 10 µL de la suspensión celular y se los coloca en la cámara de Neubauer. N1 N2 N4 N3 112 Se cuentan todas las células del cuadrado N1, incluyendo las que se encuentran sobre los bordes izquierdo y superior (siembre y cuando estén en contacto con 2 de las 3 líneas que delimitan el cuadrado grande) y se excluyen aquellas que se encuentren en contacto con los bordes derecho e inferior (con 2 de las 3 líneas, si están en contacto solo con la línea interior se cuentan), como se muestra en la imagen, los círculos negros son los que se cuentan y los blancos los que no. Procedemos igual con los cuadrados N2, N3 y N4. La concentración de células se calcula según las siguientes ecuaciones: (N1+N2+N3+N4)/4=N N x 10000x Fd= células/ml de cultivo Fd: factor de dilución (10 en el caso de hacer una dilución 1/10) Siembra y criopreservación: Una vez obtenido el dato de la cantidad de células por mL de medio, se procederá a la siembra de 250 mil células en 3 dish de 30 mm de diámetro y se agregará 2 ml de medio de crecimiento (que contiene 10 % de suero fetal bovino). Se espera que luego de 7 días de cultivo las células alcancen total confluencia y luego de 7 días más se diferencien, llegando a una diferenciación máxima a los 30 días aproximadamente. Para congelar las células, se sembrará en un criovial la cantidad de células deseadas, se agregará medio de crecimiento para Caco-2 y SFB hasta una concentración final de 20%. Por último se colocará DMSO a una concentración final de 10%. Para evitar el daño por congelado de las células se debe permitir que el agua pueda salir de las mismas antes de formarse los cristales de hielo. Esto puede lograrse congelando lentamente las células y utilizando un criopreservante (como el DMSO o glicerol). Sin embargo, el congelamiento no debe ser tan lento que permita el crecimiento de los cristales. La velocidad de congelado más utilizada es de 1ºC/min. Cuando las células se guardan por largos periodos, es preferible guardarlas a la menor temperatura posible, de manera de reducir el efecto desnaturalizante de altas concentraciones de sales. Para lograr una velocidad de enfriamiento de 1°C/min se coloca el vial en Nunc freezing container en un freezer a -70 ºC o bien dentro de en una envase de telgopor envuelto con algodón durante 2 h a -20°C y luego 24 a -70°C. Finalmente el criovial se transfiere a un termo de nitrógeno líquido para conservar las células indefinidamente. El DMSO se usa como criopreservante, sin embargo como es tóxico, no es recomendable que las células estén expuestas a altas concentraciones de DMSO durante largos periodos de tiempo a temperatura ambiente. Por esta razón, una vez colocado el DMSO, el vial debe colocarse en el freezer lo antes posible. Asimismo, al descongelarlo debe agregarse medio fresco inmediatamente. 113 Medidas de viabilidad y diferenciación en Caco-2: Materiales y soluciones: MTT 5g/L (en PBS 1X) DMSO Reactivo de Sedmak (Coomassie G250 0,06% en HClO4) BSA 1 μg/μl PBS 1X Kit para medir fosfatasa alcalina (Wienner ALP 405 cinética optimizada) Medidas de viabilidad en Caco-2: La viabilidad de un cultivo es un parámetro medido comúnmente cuando se trabaja con drogas-fármacos, excipientes o reactivos nuevos, ya que es necesario determinar previamente si afecta la viabilidad del cultivo. Una forma de medir la viabilidad celular consiste en el ensayo de MTT (bromuro de (3-(4, 5 dimetiltiazol-2-il)-2,5difeniltetrazolium). Este ensayo se basa en que las células vivas poseen la capacidad de reducir el compuesto de tetrazolio (de color amarillo) a su derivado formazán (de color violeta). Este compuesto puede detectarse por espectrofotometría a 530 nm. En el presente práctico, se procederá a medir la toxicidad del DMSO, compuesto generalmente utilizado en cultivo celular como criopreservante o como solvente de drogas y reactivos. Se sembrarán 8500 células/pocillo en placas de 96 pocillos y se las expondrá a distintas concentraciones de DMSO (0; 0,01; 0,1; 1, 10 %) durante 24 h a 37°C (en incubador). Finalizado el tratamiento, se reemplazá el medio de cultivo por medio fresco conteniendo MTT a una concentración final de 0,50 mg/mL y se incubará por 2 h a 37°C a fin de permitir la formación de los cristales de formazán, siendo ésta proporcional a la viabilidad celular. Luego de la incubación se retirará el medio de cultivo y se disolverán los cristales de formazán en 100 μL de DMSO. Finalmente, se lee la absorbancia a 530 nm corregida respecto a la absorbancia de referencia a 630 nm, en un lector de microplacas. Medidas de diferenciación en Caco-2: La diferenciación es el proceso que lleva a la expresión de las propiedades fenotípicas características de una célula funcionalmente madura e incluye grandes cambios morfológicos y funcionales. La diferenciación puede ser evaluada como la 114 expresión de uno o más marcadores asociados con la etapa final de diferenciación. En el caso del epitelio intestinal la diferenciación incluye: • Las características asociadas a la diferenciación de células epiteliales secretorias o absortivas. Entre estas, la polaridad estructural y funcional, que puede ser evaluada por la presencia de domos cuando crecen en una superficie impermeable, la presencia de uniones estrechas o la capacidad de transporte vectorial. • Las características asociadas a la diferenciación enterocítica, como la presencia de un ribete en cepillo apical e hidrolasas asociadas a dicho ribete en cepillo (fosfatasa alcalina, disacaridasas, peptidasas), o de villina (una proteína de unión a actina que se asocia específicamente al citoesqueleto de los microvilli del ribete en cepillo). En este práctico, se medirán los cambios en la actividad de la enzima fosfatasa alcalina, como una medida de la mayor expresión de la enzima en cultivos de la línea Caco-2, con tiempos de incubación crecientes. Se trabajará con placas de 30 mm de diámetro sembradas inicialmente con 250 mil células e incubadas durante 7, 14 y 21 días. Primero, se lavará el medio de cultivo con PBS 1X, se agregará 500 μL de PBS1X, con el fin de levantar las células. Se colocará la suspensión en un tubo eppendorf y se sónicará para lisar las células, mediante 3 pulsos de 5 segundos a 30% de amplitud. Una vez obtenido el homogenado, se medirá la cantidad de proteínas por el método de Sedmak: En una placa de 96 pocillos se agregan los siguientes reactivos en el orden indicado (por triplicado): 1°-95 μl de agua destilada 2°-5 μl de homogenado 3°-100 μl de reactivo de Sedmak Se realizará simultánemente una curva de calibración con 0, 2, 4, 6, 8 y 10 μg de BSA. Se mide absorbancia a 630 nm y a 450nm y se calcula la relación A630/A450 nm para cada pocillo. Luego mediante la curva de calibración: A630/A450 nm vs. μg de BSA, se calculará la concentración de proteínas de cada muestra. Finalmente, se procederá a la medición de la actividad fosfatasa alcalina: 1-Resuspender el reactivo del kit y luego colocarlo en un baño a 37°C. 2-Agregar al reactivo el volumen de homogenado correspondiente a 30 μg de proteína y medir la absorbancia a 405 nm durante 90 segundos. 3-Extraer el dato de la pendiente inicial. Bibliografía: Freshney RI. Culture of animal cells: A manual of basic technique and specialized applications. 6th edition. Wiley & Sons, Inc., Hoboken, New Jersey, 2007. Isabelle Chantret, Alain Barbat, Elisabeth Dussaulx, et al. Epithelial Polarity, Villin Expression, and Enterocytic Differentiation of Cultured Human Colon Carcinoma Cells: A Survey of Twenty Cell Lines. Cancer Res 1988; 48:1936-1942. 115 116 Clase Práctica: CICLO CELULAR Y CITOMETRÍA DE FLUJO Docentes: Dra. Stella Mattaloni - [email protected] Dra. Anabela Ferretti - [email protected] CICLO CELULAR Y CITOMETRÍA DE FLUJO Citometría de flujo: Principios generales: La citometría de flujo es una técnica que permite la medida simultánea de múltiples características físicas de una población celular proporcionándonos una amplia variedad de parámetros que van desde el simple tamaño celular hasta medidas de densidad de epítopes o mecanismos de fluidos de membrana obteniéndose un excelente muestreo estadístico de los parámetros evaluados en células individuales los cuales pueden ser correlacionados y mostrados en una amplia variedad de formatos multidimensionales. Figura 1: En el citómetro de flujo las células en suspensión atraviesan un finísimo tubo transparente sobre el que incide un delgado rayo de luz láser. La luz emitida en todas direcciones es recogida por ópticas que dirigen la luz a una serie de filtros y espejos dicroicos que aíslan longitudes de onda particulares. Las señales de luz son detectadas por tubos fotomultiplicadores y digitalizadas para análisis computacionales. 117 Ciclo Celular: Un ciclo celular típico está compuesto por dos grandes fases (figura 2): la Interfase, que se divide en tres fases, G1, S y G2, y la Mitosis. En la Interfase, en G1 se produce la acumulación del ATP necesario para el proceso de división y el incremento de tamaño celular; la fase S se caracteriza por la replicación del ADN nuclear; finalmente, en G 2, la célula se prepara para la mitosis. El ciclo celular culmina con la mitosis, donde se divide la cromatina duplicada de modo tal que cada célula hija obtiene una copia del material genético. El final de la Mitosis da cabida a un nuevo ciclo en G 1 o puede que la célula entre en una fase quiescente, G0, que corresponde a un estado de latencia respecto al ciclo celular característica de algunas células. Como todo proceso orgánico, el ciclo celular está sujeto a regulación. Ésta es realizada en momentos específicos del ciclo, llamados puntos de control o de chequeo, que pueden frenar o disparar diversos procesos que le permiten a la célula proseguir con su ciclo normal de replicación del material genético, crecimiento y división. Existen tres puntos de control, en la entrada de la fase S, en la entrada de la fase M y en la salida de la fase G2. En estos puntos de control se examina el estado nutricional, la masa celular, el estado del ADN y el estado de otros componentes celulares necesarios para la progresión de un ciclo celular típico normal. Figura 2: Fases del ciclo celular Análisis del ciclo celular por citometría de flujo: El ciclo celular de una población celular puede ser estudiado mediante el análisis del contenido, conformación o replicación (proliferación celular) de su ADN o a través del análisis de su ARN. Existen diversas técnicas con diferentes fijadores y fluoróforos, según las necesidades propias de la población celular en estudio y del citómetro a utilizar. 118 Figura 3: El análisis del estado de replicación de una población de células puede ser llevado a cabo marcando el núcleo de las células en suspensión con un colorante fluorescente y luego analizar la fluorescencia de cada célula en la población. Las células quiescentes y en G1 tendrán una copia de ADN y por lo tanto tendrán una intensidad de fluorescencia 1X. Las células en fase G2/M del ciclo celular tendrán dos copias de ADN y por lo tanto tendrán una intensidad de 2X. Y dado que las células en fase S están sintetizando ADN, tendrán valores de fluorescencia entre las poblaciones 1X y 2X. El histograma resultante consiste en tres poblaciones: dos curvas gausianas (los picos 1X y 2X) y la población en fase S. Las poblaciones adyacentes se solapan unas con otras. Debido a esto, se requiere un programa de modelado para de-convolucionar las poblaciones y asignar valores porcentuales a cada población. Figura 4: Histograma típico obtenido mediante tinción con Ioduro de Propidio (IP). El IP, tiene fluorescencia roja y puede ser excitado a 488 nm. Este colorante tiene dos desventajas; tiñe todos los ácidos nucleicos doble hebra, por lo tanto las células deben ser incubadas con RNAsa para remover cualquier ARN doble hebra y este colorante es excluido por la membrana plasmática por lo tanto las células deben ser fijadas o permeabilizadas antes de la tinción. El método más simple de fijación es fijar las células en etanol al 70 %. 119 En el trabajo Práctico utilizaremos células HepG2 (subclon C3A) que fueron establecidas de una biopsia de hepatocarcinoma humano. Los hepatocitos normalmente se encuentran en estado quiescente (G0). Para entrar en el ciclo celular y replicarse, necesitan transformarse en células competentes a través de estímulos apropiados, como por ejemplo, la expresión de genes del ciclo celular (p53, p21, ciclinas y Cdks) ingresando así al estado pre-replicativo G1. A diferencia de los hepatocitos normales, las células HepG2 presentan un equilibrio entre las células que se diferencian y polarizan, y las que entran al ciclo celular. Una vez que las células entran al ciclo celular, en la fase G1, el proceso puede ser revertido o bien puede continuar a la fase S y finalmente entrar en mitosis. Las células C3A, como todas las células tumorales, realizan glucólisis anaeróbica a alta tasa, a diferencia de los hepatocitos normales en donde la producción de energía es predominantemente a través de la fosforilación oxidativa. Debido a esta alta dependencia de la vía glucolítica, estas células tumorales son muy sensibles a la falta de glucosa. Una respuesta a la restricción de glucosa es activar vías de señalización que provocan el arresto del ciclo celular en fase G0/G1. DIA 1 Fijación de las células: 1- Tripsinizar células C3A previamente incubadas en medios DMEM con y sin glucosa 25 mM y centrifugar la suspensión celular (aproximadamente 1.000.000 de células) a 400 x g durante 5 minutos. 2- Lavar los pellets con PBS y centrifugar nuevamente a 400 x g durante 5 minutos. Resuspender los últimos pellets en 300 μl de PBS. 3- Preparar tubos con 8 ml de Etanol 70 % frío. Vortexear cada tubo mientras se le agrega gota a gota los 300 μl de las células resuspendidas en el PBS (este paso es muy importante para evitar que se formen grumos). Las células fijadas en etanol se pueden conservar en la heladera hasta 1 semana o 1 mes a -20ºC. Marcación con IP: 1- Centrifugar los tubos a 400 x g durante 5 minutos 2- Lavar con PBS y volver a centrifugar a 400 x g durante 5 minutos 3- Resuspender el pellet en 100 ul de PBS 4- Agregar 400 ul de Buffer K vortexeando suavemente, es solo para mezclar, tener cuidado que no se dañen las células 5- Proteger las muestras de la luz y guardar en la heladera hasta llevarlas a analizar al citómetro de flujo. Nota: Por lo general, para asegurarnos de tener buenos histogramas, realizamos la fijación y la marcación con IP en días distintos. El tercer día pasamos las muestras por el citómetro. También pueden teñirse las células y pasarlas por el citómetro el mismo día. 120 DIA 2 Citometría: Las células teñidas serán previamente filtradas y finalmente sometidas a citometría de flujo, utilizando el citómetro BD Cell Sorter BD FACSAria II. Los datos crudos obtenidos de la citometría serán posteriormente analizados con los programas WinMDi y Cylchred. BUFFER K (100 ml) • Citrato de Sodio 0.1 % • RNAasa 0.02 mg/ml • NP-40 0.3 % • pH=7.4 • Luego agregar Ioduro de Propidio 0,05 mg/ml 0.1 g 2 mg (de calidad) 300 ul csp hasta 100 ml 5mg Bibliografía Baltimore, Lodish, Berk, Zipursky, Matsudaira, Darnell (2003). Biología celular y molecular. Editorial Panamericana, Madrid. Cuarta edición, 849-904. Fausto N. (2000) Liver regeneration. J.Hepatol. 32 (1), 19-31. Mattaloni SM, Ferretti AC, Tonucci F, Favre C, Goldenring JR, Larocca MC (2013). Centrosomal AKAP350 modulates the G1/S transition. Cell logistics; Landes Bioscience. Volume 3 (1):1-8.e26331. T. Vincent Shankey, Peter S. Rabinovitch, Bruce Bagwell, Kenneth D. Bauer, Ricardo E. Duque, David W. Hedley, Brian H. Mayall, and Leon Wheeless (1993). Guidelines for Implementation of Clinical DNA Cytometry, Inc. Cytometry 14:472-477. Software WinMDI Windows Multiple Document Interface. Análisis de datos. WinMDI is freeware provided by Joe Trotter of the Scripps Institute, La Jolla, CA. Cylchred: Software from Cardiff Cell Cycle Analysis: A Pragmatic Approach. Based on algorithms by Watson et. al. (1987) Cytometry 8,1-8. Michael Brown, Carl Wittwer (2000). Flow Cytometry: Principles and Clinical Applications in Hematology Clinical Chemistry 46:8(B),1221–1229 121 Clase Práctica: MICROSCOPIA CONFOCAL E INMUNOFLUORESCENCIA Docente: Lic. Facundo Tonucci [email protected] MICROSCOPÍA CONFOCAL E INMUNOFLUORESCENCIA La microscopia confocal es una técnica de microscopia óptica ampliamente utilizada en las ciencias biológicas para obtener imágenes de alta definición y/o reconstruir imágenes tridimensionales. Utiliza un "pinhole" espacial para eliminar la luz desenfocada o destellos de la lente en especímenes que son más gruesos que el plano focal. El pinhole es una apertura localizada delante del fotomultiplicador que evita el pasaje de fluorescencia de las regiones de la muestra que no están en foco, la luz que proviene de regiones localizadas por encima o por debajo del plano focal no converge en el pinhole y no es detectada por el fotomultiplicador. Figura 1. El haz de láser excita la muestra moviéndose rápidamente por toda su extensión. Una vez excitado, el fluorocromo empleado emite una luz con una determinada longitud de onda. La luz emitida es filtrada por el “pinhole” y separada espectralmente por fotomultiplicadores, que además sirven como sensores de luz. Gracias a ellos, el sistema de escaneo del microscopio podrá recoger la luz por intervalos de longitud de onda que nosotros elijamos, pudiendo capturar simultáneamente la emisión de tantos fluorocromos distintos como fotomultiplicadores tenga nuestro aparato. Tras la captura de la primera imagen en dos dimensiones, el ordenador capturará la siguiente imagen, correspondiente al siguiente plano focal, y así sucesivamente. De esta forma se conseguirán series de imágenes consecutivas de una muestra, en formato digital, como si se tratase de rebanadas de una barra de pan. Preparación de las muestras para Microscopía En la mayoría de los casos trabajaremos con muestras previamente fijadas, el químico más ampliamente utilizado para fijar células en cultivo es el paraformaldehido (PFA). Una vez fijadas las células se procede con distintos protocolos de tinción de la 122 muestra. Por un lado podemos partir de células que previamente expresen alguna proteína o constructo fluorescente que nos interese lo cual no requeriría ninguna tinción en particular. Cuando no es el caso utilizaremos una estrategia similar al westernblot o ELISA, es decir un anticuerpo primario contra la proteína que nos interesa analizar, y luego un anticuerpo secundario unido a un fluoróforo que emita en longitudes de onda detectables por nuestro microscopio. A su vez, existen en algunos casos determinados químicos que reconocen estructuras celulares particulares y fluorescen en longitudes de onda útiles en microscopia confocal, es el caso del DAPI (4',6-diamidinofenyl-indol) que es un marcador fluorescente que se une fuertemente a regiones enriquecidas en Adenina y Timina en secuencias de ADN y es por ello ampliamente utilizado como marcador nuclear. Finalmente se monta la muestra utilizando un compuesto que ayude a preservar la fluorescencia y disminuir el “photobleaching”. Protocolo: DIA 1: Fijaremos células C3A, que expresan un constructo con GFP previamente sembradas en cubre objetos, usando PFA. Luego bloquearemos usando una solución de albumina y un detergente suave (Tritón X-100) para permeabilizar y realizaremos tinciones con Faloidina Alexa 568, como marcador de actina, y DAPI, como marcador nuclear. 1- Lavamos 3 veces con 1ml de buffer fosfato salino (PBS). 2- Fijación: 15 minutos a temperatura ambiente en 1ml de solución PFA 4%. 3- Lavamos 3 veces 5 minutos con PBS. 4- Bloqueo: 20 minutos en 1ml de solución 1% Albumina y 0,1% Tritón X-100. 5- Lavamos 3 veces con PBS. 6- Tinción: Faloidina Alexa 568 (1:250). DAPI (1:100). Una hora en un volumen final de 60ul de solución de bloqueo en cámara humeda. 7- Lavamos 3 veces 5 minutos con PBS. 8- Montamos sobre portaobjeto utilizando 25ul de ProLong. DIA 2: Observaremos y tomaremos imágenes en los preparados utilizando el microscopio invertido Nikon TE200. Bibliografía: Onetti Muda A. Confocal laser scanning microscopy. Adv Clin Path. 2000;4:235-9. Pawley JB. Handbook of Biological Confocal Microscopy. 2006. Berlin: Springer. ISBN 038725921X. 123 Clase Práctica: CULTIVO DE CÉLULAS ANIMALES Y PARÁSITOS Docentes: Dra. Romina Manarin - [email protected] Dra. Luis Tavernelli - [email protected] CULTIVO DE CÉLULAS ANIMALES (VERO, RAW 264.7) Y PARÁSITOS Criopreservación de células: Medio de congelamiento: 90% SFB+ 10% dimetilsulfóxido (DMSO). Resuspender entre 1x106 y 1x107 de células o parásitos por mL de medio de congelamiento en criotubos. La Tº debe ir bajando gradualmente, se puede almacenar a -20 Cº hasta que se congela completamente, luego se transfiere a -80 Cº durante 24-48 hs y finalmente se transfiere a nitrógeno líquido. Descongelamiento de células: Retirar el criotubo del nitrógeno y colocar en baño de 37ºC. Transferir el contenido gota a gota a un tubo Falcon de 15 mL conteniendo 10 mL de medio fresco suplementado. Si son células Vero, centrifugar a 2200 RPM 10 min, si son parásitos a 3500 RPM 10 min a Tº amb. Descartar el SN y cuantificar la viabilidad celular en cámara de Neubauer. Cultivo de células Vero (ATTC N° CCL-81, American Type Culture Collection, Rockville, MD): Organismo: cercopithecus aethiops (monkey, african green) Tejido: riñón normal Morfología: célula epitelial adherente Tiempo de duplicación: 22 hs Medio de cultivo recomendado: Eagle’s Minimun Essencial Medium modificado por Dulbecco (DMEM) + 10% fetal bovino (SFB), 1x de Penicilina-Streptomicina a 37 ºC y 5% de CO2. 124 Mantenimiento de la línea celular: 1. Como las células son adherentes, al alcanzar el estado de confluencia descartar el SN con pipeta. 2. Lavar las células con PBS 1X (1ml bot 25 cm2; 3 ml bot 75 cm2) para retirar todo el SFB, ya que inactiva la actividad de la tripsina. 3. Adicionar Tripsina-EDTA 0.05%. i. Botella 25 cm2: 1 ml de tripsina ii. Botella 75 cm2: 2 ml de tripsina 4. Incubar 4-5 min a 37 ºC 5. Chequear en el microscopio y dar leves golpecitos en la botella para facilitar el despegue de las células. 6. Inactivar la tripsina con DMEM 10% SFB (3 veces el vol de tripsina). 7. Transferir la suspensión celular a un tubo Falcon y centrifugar a 2000-2500 RPM durante 5 min a Tº amb. 8. Descartar el sobrenadante (SN) y resuspender el pellet en medio fresco para determinar la concentración celular en cámara de Neubauer mediante el método por exclusión de azul tripán. Para mantenimiento de la línea: DMEM 10%SFB+ATB Botella 25 cm2: 300000 células, VF: 5 ml. Botella 75 cm2:700000 células, VF: 15 ml. Hacer el pasaje 2 veces por semana. Para experimentos e infección de la línea: DMEM 2 %SFB+ATB Botella 25 cm2: 800000 células Botella 75 cm2:2500000 células 125 Cultivo de células RAW 264.7 (ATCC N° TIB-71): Esta línea celular fue establecida en 1978 a partir de la inyección intraperitoneal de un tumor inducido en ratones (Mus musculus) BALB/c por el virus de leucemia de Abelson. Organismo: Mus musculus (cepa BALB/c macho) Tejido: tumor inducido por el virus murino de la leucemia de Abelson Morfología: monocitos/macrófagos Tiempo de duplicación: Medio de cultivo recomendado: DMEM suplementado con 2mM glutamina y 10% SFB a 37 ºC y 5% de CO2. Son semi-adherentes. Mantenimiento de la línea celular: La línea RAW 264.7 puede mantenerse en adherencia o en suspensión. Cuando se permite que se adhieran a botellas de plástico, el mantenimiento es similar al de las células VERO, con algunas modificaciones, como por ejemplo, el uso de scraper para levantar las células o el agregado del agente de disociación comercial VERSENO (Gibco, 0.2 g EDTA por litro de PBS) en lugar de Tripsina-EDTA, abajo se describe el protocolo para el subcultivo en adherencia: 1. 2. 3. 4. Cuando las células llegan al estado de confluencia descartar el SN con pipeta. Lavar las células con 1 ml de PBS 1X (para botella de 25 cm2). Adicionar 1 ml de VERSENO. Incubar 10-15 min a 37 ºC. 126 5. Chequear en el microscopio y dar leves golpecitos en la botella para facilitar el despegue de las células. 6. Lavar con 4 ml de PBS. 7. Transferir la suspensión celular a un tubo Falcon de 15 ml y centrifugar a 1500 RPM durante 10 min a Tº amb. 8. Descartar el sobrenadante (SN) y resuspender el pellet en medio fresco para meterminar la concentración celular en cámara de Neubauer mediante el método por exclusión de azul tripán. El mantenimiento de la línea en suspensión resulta más práctico, para esto se emplean placas de Petri de plástico y se renueva el medio cada 3 días aproximadamente. Determinación de la viabilidad celular por exclusión con Azul Tripán: La membrana plasmática de una célula viva no permite la entrada de sustancias colorantes y electrolíticas. Esta propiedad sirve para determinar la viabilidad celular mediante el uso de colorantes apropiados, como el azul tripán. De esta manera es posible distinguir, de forma visual, las células vivas no teñidas de las no viables teñidas. Luego de la tinción, deben ser contadas dentro de los 10 min siguientes, ya que luego de ese tiempo, las células vivas también comienzan a incorporar el colorante. Materiales: Solución 1: azul tripán 0.5% P/V en agua. Solución 2: solución salina 5X (NaCl 4.25% P/V). Procedimiento: - En el momento de contar, mezclar 4 partes de la solución 1 con 1 parte de la solución 2. - Mezclar 1 parte de la solución anterior con 1 parte de la suspensión celular (dil ½). - Cargar las células en la cámara de Neubauer y contar en forma separada las viables (no teñidas) de las no viables (azules). Contar las células presentes en los 4 cuadrantes grandes o en 5 cuadrados del cuadrante central. % de células viables: nº de células viables / (nº de células viables + nº de células muertas). Células viables / ml: Nº de células contadas x dilución x 50000 En este caso la dilución es 2. Cultivo de epimastigores: Los epimastigotes de las distintas cepas se mantienen en frascos de cultivo de 25 cm3 a 28ºC en medio de cultivo LIT. Durante la fase exponencial se realizan los pasajes, aproximadamente cada 5 - 6 días, y en forma periódica se procede a congelar los mismos para su conservación o para ser usados en experiencias a largo plazo. 127 Medio LIT: medio de infusión de hígado y triptosa (liver infusion tryptose) para el cultivo de epimastigotes: Infusión de hígado 5g NaCl 4g KCl 0,4 g Triptosa 5g Na2HPO4 anhidro 8g Glucosa 8g Hemina 2 mL cada 100 mL de LIT H2O destilada c.s.p. 1litro Hemina: 0,2 g de hemina en 10 mL de NaOH (0,2M). Antes de autoclavar se agrega la hemina según volumen indicado en la preparación. Se autoclava el medio 10 minutos a 1,5 atmósferas y posteriormente se suplementa con suero fetal bovino al 10%, penicilina (100 U/mL) y estreptomicina (0,1 mg/mL). Infección de células VERO con tripomastigotes de Trypanosoma cruzi: - - - - Para la infección se utiliza DMEM 2% para evitar el sobre-crecimiento celular. Infectar células Vero diluidas (entre 30 y 40 % de confluencia), que inclusive se pueden plaquear el mismo día de la infección. La cantidad de células totales a la hora de infectar para cada tamaño de frasco es aproximadamente: F25cm2 = 800 000 F75cm2 = 2,5 X 106 F150cm2 = 8 X 106 NOTA: si se plaquean en el día hay que plaquear un 100% del número final de células que se quiere y dejarlas adherirse por lo menos 5 horas. Si se plaquean el día anterior hay que plaquear el 30-40% del número final. Utilizar tripomastigotes en exceso (relación 10 a 1 por lo menos). Los tripomastigotes se pueden dejar ON o durante el día y luego lavar (mínimo 4 horas a 37º). La infección se puede realizar en forma simultánea, plaqueando las células y los parásitos al mismo tiempo, ya que se observan muy buenos resultados. Se sugiere utilizar una relación parásito:célula de 10 para dejar ON y una de 25 para 4 hs de incubación. Luego de la infección, lavar las células con PBS1X para retirar las formas no internalizadas. Agregar medio DMEM 2% SFB. Cambiar el medio cada 2-3 días. A las 24 hs ya se ven amastigotes, sin embargo, recién a los 2 días el cultivo está listo para obtener amastigotes. Para la obtención de tripomastigotes, se espera alrededor de 3-4 días. El SN que contiene los tripomastigotes se puede emplear para infectar nuevas células Vero, para hacer extractos celulares, para obtener ARN, para congelar, etc. 128 Coloración de GIEMSA: Materiales: Colorante Giemsa: se utiliza dilución 1/10 en agua corriente. Procedimiento: preparar frotis del medio de cultivo fijado en metanol, teñir durante 20-30 minutos con el colorante Giemsa diluido. Lavar el portaobjeto con agua corriente y dejar escurrir en posición vertical. Ensayo de fagocitosis: El ensayo de fagocitosis se realizó en la línea RAW 264.7 empleando levaduras S. cerevisiae muertas por calor. Las levaduras fueron procesadas por la técnica descripta por Lachmann and Hobart (Lachmann, P. J., and M. J. Hobart. 1978. Complement technology, p. 1A-5A. In D. M. Weir (ed.), Handbook of experimental immunology, vol. I.Immunochemistry. Blackwell, Oxford, United Kingdom). Para obtener las levaduras muertas por calor, se parte de un cubo de 50 g de levadura fresca (S. cerevisiae) y suspender en 220 ml de PBS (pH 7.2), autoclavar a 120°C por 30 min, y lavar con PBS hasta obtener un SN claro. El pellet se resuspende en 28 ml de una solución de 0.1 M 2-mercaptoetanol en PBS incubando 2 hs con agitación. Luego, las levaduras se lavaron nuevamente y se resuspendieron en 55 ml de una solución 0.02 M iodoacetamida en PBS. Después de incubar 2 h a T amb con agitación, se lavaron las levaduras 3 veces y se resuspendieron en 220 ml de PBS (pH 7.2). Finalmente, las levadurasb se autoclavaron de nuevo, lavaron y resuspendieron en 110 ml de buffer Veronal (pH 7.2) con azida sódica, y almacenadas a 4°C hasta su uso. En cada experimento, la suspensión de levaduras se lava con PBS, se cuantifica y se suspende en el medio de cultivo correspondiente. El método empleado para la evaluación de la capacidad fagocítica de los macrófagos está basado en la adherencia de estas células sobre los portaobjetos de vidrio utilizados para microscopía, seguido de la fagocitosis de las levaduras. Mediante microscopía se demostró que existe asociación entre las levaduras y la membrana externa de los macrófagos a los pocos minutos. Sin embargo, la mayoría de las levaduras fueron completamente internalizadas después de 1h de incubación. Para el experimento se sembraron 0.35x105 células en microplacas para microscopía (Lab-TekTM chamberslide, NUNC). Las células se dejaron adherir durante toda la noche a 37°C en atmósfera de CO2 al 5 %. Luego se incubaron durante 1 hora con 1.75x105 levaduras por pocillo a 37°C en presencia de los compuestos que se quieran ensayar. Las células se lavaron 2 veces con PBS 1X y luego fueron teñidas con May-GrünwaldGiemsa. Se contaron 10 campos microscópicos diferentes y se calculó el porcentaje de fagocitosis (Número de macrófagos con levaduras ingeridas respecto del total de macrófagos por 100). 129 Clase Práctica: TRYPANOSOMA CRUZI Y TRATAMIENTO DE DROGAS Docentes: Dra. Victoria Alonso - [email protected] Lic. Carla Ritagliati - [email protected] TRYPANOSOMA CRUZI Y TRATAMIENTO DE DROGAS En este trabajo práctico se evaluará el efecto de las dos drogas utilizadas para el tratamiento de la Enfermedad de Chagas sobre epimastigotes: el Benznidazol (BZL) (A) y el Nifurtimox (NFX) (B). El mecanismo de acción del NFX, un nitrofurano, involucra la formación de nitroaniones radicales por nitroreductasas, que en presencia de oxígeno genera intermediarios reactivos. Siendo Trypanosoma cruzi un microorganismo deficiente en mecanismos de detoxificación de radicales libres, es particularmente sensible a los mismos (Docampo, R. y Moreno, S. N. J.; 1986). La acción de BZL, un nitroimidazol, no se da por daño oxidativo sino a través de la formación de uniones covalentes u otras interacciones de intermediarios nitroreducidos con componentes del parásito (Polak, A. y Richle, R.; 1978) como DNA, lípidos y proteínas (Diaz de Toranzo, E. G. et al.; 1988). La actividad antiparasitaria se evaluará sobre epimastigotes de la cepa Dm28c cultivados en medio LIT a 28ºC. Se partirá de un inóculo inicial de 4-5 x106 parásitos/ml y se utilizarán concentraciones crecientes de cada compuesto disuelto en DMSO. BZL stock 1M: 13 mg en 50 l de DMSO preparado en el momento. Nifurtimox stock 1M: 14,36 mg en 50 l de DMSO preparado en el momento El crecimiento relativo se determinará utilizando un contador hematológico automatizado (W19 Counter AA de Weiner Lab) adaptado para contaje de epimastigotes después de 72 horas de cultivo. Los ensayos se realizarán por triplicado. DIA 1: 1- Contar el cultivo de epimastigotes de partida diluyendo 1:10 en PBS-formaldehido 3,7%. 2- Diluir en medio LIT fresco a la concentración de parásitos de partida. 3- Diluir las drogas a 1 mM en DMSO (dilución de trabajo). 130 4- Dividir el cultivo en botellitas de vidrio estériles (Vf = 0,5 ml) y agregar las drogas a las siguientes concentraciones finales: 0 – 1 – 2 – 4 – 8 – 12 M. Como control se agregará solo DMSO. 5- Dejar incubando a 28ºC durante 72 hs. DIA 2: 1- Tomar las botellitas y agregar a cada una 0,5 ml de PBS-formaldehido 8%. 2- Contar en el contador automatizado y registrar las concentraciones de parásitos en cada punto. 3- Analizar los datos mediante el programa GraphPad Prism. Se graficará la cantidad de parásitos vs las concentraciones de compuestos. Se ajustarán los datos mediante una regresión no lineal y se obtendrá el valor de concentración de inhibitoria media (CI50) para cada droga. Discusión: comparar los valores obtenidos para los compuestos utilizados en la literatura. 131 Clase Práctica: TRANSFECCIÓN DE TRYPANOSOMA CRUZI Docentes: Lic. Carla Ritagliati - [email protected] Dra. Victoria Alonso - [email protected] TRANSFECCIÓN DE TRYPANOSOMA CRUZI DIA 1 1. Colectar Mid-log late células (1-5.107 p/ml) (1-5.108 p/electroporación) a 2000 x g 10’. 2. Lavar una vez en PBS 1X y resuspender a una concentración de ~108/0.35 ml en buffer de transfección (PBS + 0.5 mM MgCl2/ 0.1 mM CaCl2). 3. Mezclar la suspensión de parásitos (0.35 ml) con 40 ul de ADN (~ 2.5 ug/ml) y agregar un control de 40 ul de TE (no superar el volumen de 0.4 ml) 4. Mantener 15’ en hielo (mínimo). 5. Electroporar en cubetas de 0.2 cm biorad en Bio-Rad Gene Pulser. Un solo pulso 400 o 450 V 500 uF. Las time ctes. varían entre 3.8 a 6. 6. Mantener a temperatura ambiente durante 10’ para recuperar. 7. Transferir a 2 ml medio LIT. Observar mortalidad al microscopio. 8. Incubar 24 hs DIA 2 1. 2. 3. 4. 5. Observar al microscopio óptico. Tomar una alícuota y fijar con formaldehído al 3.7% Adherir en portaobjetos 20’ Observar fluorescencia transientes. Agregar G418 hasta Concentración final = 250 ug/ml (125 ul del Stock G418 20mg/ml a 10 ml de cultivo). Mantener cultivando durante 24-48 hs. 6. Diluir el cultivo 1:4 en LIT + 500ug/ml G418. Mantener cultivando a 28ºC. 7. A los ~20 días post-transfección los parásitos del control con TE dejan de crecer completamente y mueren. Las células resistentes deberían aparecer. 8. Subcultivar nuevamente en medio LIT + 500ug/ml G418 durante 5 días para concluir el proceso de selección. (Ir diluyendo a ojo entre 1/3 y 1/5). 132 Clase Práctica: CULTIVO PRIMARIO DE HEPATOCITOS EN SANDWICH Docentes: Dra. Ma. Laura Ruiz - [email protected] Dr. Ismael Barosso – [email protected] CULTIVO PRIMARIO DE HEPATOCITOS EN SANDWICH El cultivo de hepatocitos en configuración sándwich de colágeno (HRCS), es un cultivo primario de hepatocitos de rata entre dos capas de colágeno gelificado, que permite que los hepatocitos desarrollen polaridad funcional, formando una red de pseudocanalículos biliares con el lumen sellado y representa una aproximación exitosa para el mantenimiento de las funciones hepáticas específicas, incluyendo la secreción de la albúmina, inducción de la enzima citocromo P-450, y la absorción de ácidos biliares. Se ha descrito que la morfología de los hepatocitos y el metabolismo de drogas se mantienen por varias semanas. Este modelo permite la diferenciación entre los procesos de transporte sinusoidal y canalicular y por lo tanto puede representar una herramienta útil para investigar la disposición hepatobiliar de sustratos. Por su larga vida útil, además son adecuados para hacer estudios de knock-down (silenciamiento) de proteínas. Material utilizado (estéril): Eppendorfs. Falcons de 50 y 15 mL. H2Od. PBS 10X. PBS 1X. Tips amarillos, azules y para p5. Frascos varios para preparar y para guardar los medios. Medios y Soluciones: DMEM 10X. DMEM c/ATB. William´s E (WE) suplementado con 5 % suero fetal bovino, solución de antibióticos (gentamicina, estreptomicina, penicilina y anfotericina D), dexametasona 0,8 mg/l e insulina 4 mg/l; y glucosa hasta alcanzar una concentración de 3 g/l. Lo llamaremos WE-Glc WE idem al anterior pero sin Glucosa. Colágeno: 10 mg en 2 mL de HAc 20 mM. NaOH 0,2N. Percoll (9V de percoll + 1V de PBS 10X). 133 Procedimiento: 1. Primer Capa de Colágeno: Se prepara una solución 10 mg/ml de colágeno utilizando ácido acético 20 mM como solvente y se deja en agitación moderada a 4 °C por 12-24 hs. En un eppendorf en hielo agregar 100 uL de DMEM 10X, luego 800 uL de la solución de colágeno y por último 100 uL de NaOH 0,2 N (chequear por el color el pH tal vez se deba agregar unos µLs más). Preparar 1 eppendorf por placa así se evita que gelifique antes el colágeno. Plaquear y distribuir el colágeno con espátula. o En placa de 6 pocillos ________ 100 uL por pocillo. o En placa de 12 pocillos _______ 80 uL por pocillo. Agregar entre los pocillos H2Od y dejar 1 hora en la estufa y 1 hora en el UV. Luego si aún no se ha terminado la operación agregar DMEM 10% c/ATB para mantener el colágeno hidratado hasta sembrar (puede ser también otro medio, el que se tenga mano y este estéril). o En placa de 6 pocillos ________ 2 mL por pocillo. o En placa de 12 pocillos _______ 1 mL por pocillo. 2. Cirugía: Los hepatocitos se aislan a partir de hígados de rata mediante la técnica de perfusión con colagenasa, utilizando una modificación del método de Berry y Friend. Se anestesia con uretano una rata Wistar. Se introduce un catéter 14G (Abbocath-T, Venisystem, Abbocath Ireland Ltd., Sligo, Irlanda) en la vena porta. El hígado se perfunde por vía porta durante 10 min en un modelo no recirculante, con una solución Hanks´ oxigenada (95% O2/5% CO2) libre de Ca2+, pH = 7,477,50, suplementada con HEPES (3 g/l) y EGTA (0,24 g/l), Luego se cambia el medio de perfusión a un medio que contenía la misma solución sin EGTA, suplementada con MgSO4 1 mM, CaCl2 2,5 mM y colagenasa tipo A (4300 U/l), durante 9 min a un flujo de 20 ml/min. Por último, se remueve el hígado, y las células se aíslan por disociación mecánica mediante agitación suave con una varilla de vidrio durante 3-4 min. Los hepatocitos se purifican de células no-parenquimatosas por centrifugación a baja velocidad (30 x g, 2 min), seguida de 3 lavados consecutivos con solución Hanks´ oxigenada conteniendo CaCl2 2,5 mM y Tris 5 mM. Para garantizar una viabilidad mayor a un 90 % (según el método de azul tripán) se puede realizar una centrifugación con una solución de Percoll a 80 g por 5 min (opcional dependiendo la calidad de extracción de cada operador). La preparación resultante consiste de 400-600 x 106 hepatocitos por hígado de elevada viabilidad (> 90%), medida por el ensayo de exclusión de azul tripán. 3. Siembra (con WE + glc) (Día 0): Aspirar el DMEM (si se dejo hidratando) y el H2Od. 134 Sembrar: o Para una placa de 6 pocillos ________ 3 mL por pocillo________ 350 mil c/mL . o Para una placa de 12 pocillos _______ 1,5 mL por pocillo _______ 350 mil c/mL. Tiempo de adherencia o Attachment: 2 -3 hs. Cambio de medio (WE + glc): o Para una placa de 6 pocillos ________ 2 mL por pocillo. o Para una placa de 12 pocillos _______ 1 mL por pocillo. 4. Segunda capa de colágeno 24 hs post sembrado (Día 1): Aspirar el medio. Se deben realizar de ahora en más con tips y siempre aspirando del mismo costado. Lavar con PBS 1X (estéril) para arrastrar bien las no pegadas. Preparar el colágeno idem anterior y agregar gota a gota (en forma de flor con pétalos), una en el medio y luego en cubrir los costados (son 5 o 6 gotas en total) hasta notar que el pocillo quede cubierto. o Para una placa de 6 pocillos ________ 150 uL por pocillo. o Para una placa de 12 pocillos _______ 100 uL por pocillo. Dejar 1 hora en la estufa. Agregar WE-Glc gota a gota en el medio del pocillo (no por los costados! Se puede levantar el colágeno). o Para una placa de 6 pocillos ________ 2 mL por pocillo. o Para una placa de 12 pocillos _______ 1 mL por pocillo. 5. Cambiar el medio cada 24 hs (WE-Glc) y dejar mínimo 3 días (comienza la polarización) o 5 días (para la polarización completa). Aspirar el medio con P1000 desde el borde, tratar de aspirar siempre del mismo borde y descartar el medio en un recipiente. Agregar WE-Glc gota a gota en el medio del pocillo. Funcionalidad de Mrp2 en CHSC: Luego de los respectivos tratamientos, las placas conteniendo los HRCS se colocan sobre una placa termostatizada a 37°C en un microscopio de fluorescencia invertido (ZeissAxiovert 25). Se saca el medio de cultivo y se agrega clorometilfluoresceína diacetato, CMFDA (2,5 µM) en medio de cultivo L-15. El CMFDA se convierte enzimáticamente en la célula en glutation metil fluoresceína, GS-MF, que es sustrato del transportador canalicular Mrp2. Inmediatamente después de agregar CMFDA, se comienza a tomar imágenes de 12 bits cada 1 min durante 8 min con una cámara digital acoplada al microscopio (Olympus QColor5). El análisis de las imágenes se realiza mediante el programa ImageJ, determinando el incremento de fluorescencia promedio en los canalículos en función del tiempo, durante la fase de crecimiento lineal de la misma (de 0 a 4 min aprox). La pendiente de las curvas de regresión lineal de fluorescencia canalicular vs tiempo es considerada como la velocidad inicial de transporte (VIT) de Mrp2. 135 Clase Práctica: CULTIVO IN VITRO DE TEJIDOS VEGETALES Docentes: Dra. Tatiana Vega - [email protected] CULTIVO IN VITRO DE TEJIDOS VEGETALES Objetivo: Adquirir destreza y conocimientos básicos en la obtención y siembra de explantos en un sistema de cultivo in vitro. Materiales y Métodos: Equipos y material de laboratorio Autoclave Pinzas y bisturíes Placas Petri estériles pHmetro Cámara de Flujo Laminar Mechero Bunsen Parafilm Fitotrón o cámara de cultivo (25ºC, 12h luz12h oscuridad) Reactivos: Base salina de Murashige y Skoog (Sigma, M5524) Sacarosa (Anedra) Agar (Bacteriologico, Britania) Acido Indol Acético (AIA) (Sigma, I5148) Acido Naftalen Acético (ANA) (Sigma, N0640) Bencilaminopurina (BAP) (Sigma, B3274) Glutamina (Sigma, G5792) Kinetina (KIN) (Sigma, K0753) Acido 2,4-diclorofenoxiacético (2,4-D) (Fluka, 76514) Material Vegetal: Como material de partida se utilizarán plántulas de girasol (Helianthus annuus L.) y de tabaco (Nicotiana tabacum) germinadas en condiciones estériles. Para la obtención de las plántulas de girasol se utilizaron aquenios maduros a los que se les retiró el pericarpio. Las semillas fueron sumergidas 1 min. en etanol 96°, luego en NaClO 5% durante 15 min. y finalmente enjuagadas tres veces con agua destilada estéril bajo cámara de flujo laminar. El medio de germinación consistió en una base salina de Murashige y Skoog (MS) al 50 %, con el agregado de 0,1% de sacarosa y solidificado con 0,9 % agar. El pH fue ajustado a 6,0 con NaOH 0,1 N previo al agregado del agar. Para su esterilización, los medios de cultivo fueron autoclavados durante 20 minutos a una temperatura de 121 °C. 136 Las semillas desinfectadas fueron sembradas en el medio de germinación, bajo cámara de flujo laminar. Las mismas fueron mantenidas en oscuridad en un cámara climática durante 2 días y luego transferidos a la luz durante los 2 días siguientes. La temperatura se mantuvo constante a 25 2ºC y el fotoperíodo utilizado fue de 12 horas con una intensidad lumínica de 50 mol m-2seg-1. Para la obtención de las plántulas de tabaco se procedió de manera similar, pero desinfectando las semillas por 5 seg en etanol 96°, luego en NaClO 5% durante 1 min. y finalmente enjuagadas tres veces con agua destilada estéril bajo cámara de flujo laminar. Medios de Cultivo in vitro: Los medios utilizados para evaluar las respuestas in vitro de los materiales se formulan con la base salina de Murashige y Skoog (MS) y el agregado de distintos reguladores vegetales: CIMg: MS con agregado de 1 mg/l 2,4-D. SIMg: MS con agregado de 1 mg/l AIA, 2mg/l KIN y 0,2 g/l Glutamina. CIMt: MS con agregado de 3 mg/l ANA. SIMt: MS con agregado de 0,5 mg/l ANA, 3 mg/l BAP Todos los reguladores vegetales se agregan a partir de soluciones stock 100 mg/l. Ambos medios fueron suplementados con 3 % sacarosa y el pH fue ajustado a 6,0 con NaOH 0,1 N previo al agregado de 0,9% agar. La esterilización fue realizada en autoclave a 121ºC durante 20 min. El medio CIMg induce la formación de callos friables mientras que el medio SIMt induce la formación de vástagos y primordios a partir de explantos cotiledonales de girasol, mientras que los medios CIMt y SIMt inducen formación de vástagos y primordios a partir de explantos de tabaco, respectivamente. Siembra de explantos Trabajando en esterilidad (cámara de flujo laminar) se separan los cotiledones de las plántulas de girasol excluyendo el meristema axilar y a su vez, cada cotiledón se corta a lo largo de su eje longitudinal y cada una de las mitades fue subdividida en tres secciones, proximal (P) central (C) y distal (D) (Fig. 1). Trabajaremos con las secciones proximal y central, que tienen mayor capacidad de regeneración. Para la obtención de explantos de tabaco se cortarán secciones (“discos”) de hojas de 1cm x 1 cm. Todos los cortes se realizan con bisturí esterilizado en alcohol y flameado Fig 1. Obtención de explantos a partir de cotiledones de plántulas. P: Proximal, C: central, D: distal. 137 a la llama. Las siembras se realizan con pinzas estériles en placas de petri con 15 ml de medio en cada uno. En cada placa se siembran hasta 10 explantos en el caso de girasol, y 5 en el caso de tabaco. Una vez sembrados, los explantos se incuban durante 28 días en cámara climática con una temperatura de 25 2 °C, fotoperíodo utilizado de 12 horas de luz y una intensidad lumínica de 50 mol m-2seg-1. Al cabo de ese tiempo se registra el porcentaje de explantos que dieron callo (en medio CIM) y vástagos (en medio SIM). Los vástagos regenerados pueden ser pasados a medio MS sólido sin hormonas, en tubos de vidrio, para la etapa de enraizamiento. 138