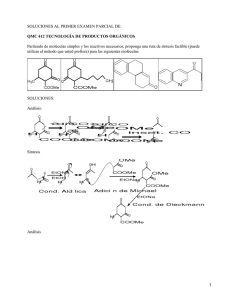
bases tecnológicas de los microarrays de proteínas
Anuncio

BASES TECNOLÓGICAS DE LOS MICROARRAYS DE PROTEÍNAS CURSO DE FORMACIÓN CONTINUADA A DISTANCIA 2009-2010 ACTUALIZACIONES EN EL LABORATORIO CLÍNICO Nº 5 I.S.S.N.- 1988-7477 Título: Actualizaciones en el Laboratorio Clínico Editor: Asociación Española de Biopatología Médica Maquetación: AEBM Fecha de Distribución: Marzo de 2010 Bases Tecnológicas de los Microarrays de Proteínas. Aplicaciones Clínicas Dr. Antonio Suárez Sanz.- Doctor en Ciencias Biológicas. Dr. José A. Suárez Redondo.- Ingeniero de Telecomunicación Introducción Los microarrays han emergido como una de las tecnologías más notables e innovadoras entre las que se dispone actualmente para el análisis de alto rendimiento de moléculas biológicas, ello se debe a que en su diseño aparecen reunidas diversas características ventajosas, como la miniaturización, la uniformidad en el tratamiento de las muestras o la posibilidad de automatización. En los últimos años se ha conseguido la inmovilización estable, sobre soportes planos (tipo porta de microscopía), de diversas biomoléculas como ácidos nucleicos (1), proteínas (2), péptidos (3), hidratos de carbono (4) o moléculas de pequeño tamaño (5), lo que ha supuesto un paso decisivo para el análisis de estos compuestos mediante arrays. Los microarrays de proteínas han surgido teniendo como modelo los de ADN, en estos la afinidad entre moléculas es la que se deriva de la complementariedad entre cadenas de ácidos nucleicos, lo que le confiere carácter predecible a las interacciones moleculares; otra de las ventajas que ofrece este tipo de plataforma es la posibilidad de incrementar la masa de los ácidos nucleicos mediante PCR. Las proteínas carecen de tales ventajas, no obstante los microarrays, diseñados específicamente para ellas, aportan ventajas que no se pueden conseguir con los de ADN, como: a) determinar la expresión proteica de forma directa, pues se ha observado que la deducida a partir de los niveles de ARNm no es concordante con ella; b) detectar la existencia de modificaciones postraduccionales de las proteínas, que frecuentemente están relacionadas con procesos biológicos vitales para la célula, como sucede con las fosforilaciones, o c) poder aplicarlos al análisis de plasma/suero, que son muestras fáciles de obtener y ricas en proteínas y que, por el contrario, tienen un bajo contenido de ácidos nucleicos. Las características de las proteínas hacen que sea más complejo su análisis pues: 1) suelen presentarse en las muestras biológicas en un abanico de concentraciones muy amplio (rango dinámico elevado); 2) poseen características fisicoquímicas muy variadas, de modo que es difícil optimizar las condiciones experimentales de ensayo simultáneamente para todos los componentes de una muestra; 3) el marcaje químico de las proteínas para su análisis pueden provocar modificaciones que alteren la afinidad de los epítopos en las interacciones antígenoanticuerpo. En un experimento con microarrays de proteínas se dan varias fases, (Fig. 1). Inicialmente se depositan las denominadas moléculas de captura (ej. anticuerpos) sobre un porta de microscopia, formando manchas circulares uniformes, de un 408 diámetro que puede variar entre 50 y 750 µm en función del tipo de experimento, dispuestas en filas y columnas mediante un dispositivo robotizado llamado arrayer. Fig 1. Estructura y funcionamiento de un array de proteínas. Curr Opin Chem Biol, (2001), 6: 76-80 Las muestras conteniendo los analitos o moléculas diana se incuban después de ponerlas en contacto con el microarray. Se eliminan por lavado las moléculas que no han reaccionado, y posteriormente se permite que interaccionen con una mezcla de anticuerpos de detección marcados con fluoróforos, que van a reconocer a las moléculas diana capturadas uniéndose a ellas a través de epítopos diferentes. La detección de la señal del complejo formado se realiza después de eliminar por lavado los anticuerpos de detección que no han reaccionado. La imagen de la superficie del microarray se obtiene por escaneado digital, él proceso consiste en barrer la superficie del microarray con una emisión de luz láser mientras que un detector capta la luz emitida por los fluoróforos, como respuesta a la excitación del láser y se traduce a intensidad píxel. Finalmente se correlacionan las intensidades de la señal con las cantidades de moléculas diana presentes en la muestra. Estas plataformas permiten identificar y cuantificar las proteínas de una muestra (obtención de perfiles de proteínas) con un rendimiento muy superior al de las técnicas clásicas de inmunoensayo como los Western immunoblotting o los ELISA, otras variantes de la técnica proporcionan información a cerca de varios aspectos relacionados con las funciones proteicas tales como su expresión en estado nativo, la existencia de interacciones proteína-proteína o la de modificaciones postraduccionales. 409 1 ELEMENTOS ESTRUCTURALES DE LOS MICROARRAYS 1.1 Moléculas de captura La disponibilidad de un número elevado de moléculas de captura específicas, y con una alta afinidad frente a sus respectivas moléculas diana es esencial para el desarrollo de la técnica de microarrays de proteínas, ya que se fundamenta en la afinidad entre ambos tipos moleculares. Como moléculas de captura se pueden emplear otras estructuras diferentes de las proteicas siempre que presenten en grado suficientemente elevado las propiedades de especificidad y afinidad entre ligandos. Las características funcionales y/o estructurales de las moléculas de captura empleadas van a variar dependiendo de la naturaleza del estudio proteómico a que se destine. Se diferencian varios grupos (Fig. 2): Antígenos Anticuerpos Proteínas (interacciones específicas proteína-proteína) Aptámeros Enzimas Receptores de membrana Fig. 2. Tipos de moléculas de captura que pueden formar parte de los microarrays de proteínas. Para participar en un análisis con microarrays, basado en interacciónes moleculares específicas, se pueden inmovilizar diferentes tipos moleculares para que actúen como moléculas de captura con diversas funciones: a) interacción antígeno-anticuerpo; b) actuar en un inmunoensayo con revelado tipo sándwich; c) una interacción específica proteína-proteína. Se pueden utilizar moléculas de captura de naturaleza química diferente a la proteica, d) moléculas tipo aptámeros; e) substrato enzimático: un substrato S se inmoviliza y se fosforila por la acción de una quinasa empleando ATP; f) moléculas de peso molecular bajo en una interacción receptor-ligando. TRENDS Biotechnol, 2002, 20: 160-166. 410 1.1.1 Anticuerpos Son moléculas de captura muy específicas y por ello herramientas muy valiosas para construir microarrays con aplicaciones al diagnóstico. El poder disponer de ellas, manteniendo sus características de especificidad y afinidad constantes indefinidamente, ha sido posible gracias a la aplicación de tecnología de obtención de anticuerpos monoclonales, pero con frecuencia el procedimiento es largo y laborioso y, como consecuencia, caro. Para eludir estos inconvenientes se pueden aplicar otras tecnologías alternativas de ingeniería genética para la obtención de anticuerpos específicos y altamente afines, como la phage-antibody display que ha demostrado ser una herramienta muy eficaz para este fin. La técnica (6) se fundamenta en la preparación de fagos híbridos que expresan fragmentos de anticuerpos en la superficie de su cápsida, y en posibilitar su selección en función de la afinidad frente a un número muy elevado de antígenos (posibles moléculas diana) en el tiempo relativamente corto de algunas semanas. Muchos de los anticuerpos así obtenidos pueden alcanzar valores de constantes de afinidad (KD) del orden pM. 1.1.2 Aptámeros Otra opción es la preparación un tipo de moléculas de captura alternativo, conocidas como aptámeros. Se trata de oligonucleotidos cuya secuencia les confiere capacidad virtual para reconocer cualquier molécula diana, con elevada afinidad y especificidad. La especificidad y afinidad de unión de estas moléculas con proteínas u otros ligandos se optimiza seleccionando las secuencias de bases por ciclos de evolución in vitro, siguiendo la técnica llamada systematic evolution of ligands by exponential enrichment (SELEX) (7-8). Estas moléculas se diseñaron inicialmente como agentes bloqueantes de proteínas participantes en diferentes estados patológicos, posteriormente se comprobó que inmovilizados en una estructura de microarray podían aplicarse al diagnóstico. El término aptámero se ha aplicado también a péptidos obtenidos mediante tecnología de mRNA display (9), presentan elevada afinidad por proteínas diana específicas, de modo que se consiguen valores de constantes de afinidad de orden nM, similares a las que presentan los complejos antígeno-anticuerpo monoclonales. 1.1.3 Affibodies Existe un tipo de proteínas microbianas llamadas receptinas, relacionadas con las interacciones huésped-parásito y con la virulencia del agente microbiano que las produce, que ofrecen como característica común presentar especificidad de unión con algunas proteínas de mamíferos. Los affibodies son moléculas de captura seleccionadas a partir de librerías combinatoriales construidas a partir de la secuencia del dominio receptor α-helice de la proteína A de Staphylococcus aureus que es una receptiva (10). Las componentes de las librerías de affibodies pueden someterse a procesos de maduración y multimerización para conseguir afinidades elevadas. 411 1.2 Tipos de soportes La elección del soporte debe ser el primer paso a dar en el diseño de un microarray de proteínas, ya que este componente va a condicionar diversos aspectos de la técnica como el formato o el tipo de detección que se habrán de emplear. Con respecto a las moléculas de captura se ha de conseguir que mantengan la funcionalidad después de su unión al substrato y que este no interfiera en el acceso a las moléculas diana. Las propiedades de un soporte han de ser: Su estabilidad química Que proporcione una buena morfología de las manchas Ausencia de uniones inespecíficas Baja señal de fondo Permitir la mayor relación superficie/volumen en las manchas Compatibilidad con los sistemas de detección Autofluorescencia baja Como soportes se pueden emplear membranas, que ofrecen gran capacidad de unión pero presentan problemas de autofluorescencia y uniones inespecíficas, otro tipo frecuente son los portas de vidrio que presentan superficies muy lisas y una fluorescencia reducida, pero poseen escasa capacidad de unión por lo que necesitan una modificación previa de su superficie que facilite la unión de proteínas. 1.3 Fijación de las moléculas de captura Las proteínas pueden inmovilizarse a través de interacciones no covalentes con la superficie del soporte, ya sea de tipo hidrofóbico (nitrocelulosa, poliestireno) o iónico (polilisina, aminosilano). Las uniones no covalentes de las moléculas de captura pueden ser demasiado débiles para impedir su pérdida durante el análisis, por este motivo la utilización de uniones covalentes tiende a generalizarse. La formación de los enlaces se consigue al reaccionar las moléculas de captura con grupos funcionales de diferente naturaleza presentes en la superficie del soporte (ej. aldehído, epoxi, esteres activos) mediante su tratamiento químico previo. También se han empleado, para inmovilizar las moléculas de captura, interacciones bimoleculares específicas del tipo estreptavidina-biotina, o la formación de complejos estables entre las moléculas de captura marcadas con ácido fenilborónico y la superficie del soporte modificada con ácido salicilhidroxámico. Para favorecer la inmovilización de las proteínas de captura es frecuente recurrir a moléculas adaptadoras, que suelen ser fundamentalmente proteínas o poliaminoácidos. Una estructura más compleja que la anterior, está formada por una molécula de afinidad anclada a la superficie del soporte, que a su vez mantienen unida la proteína de captura a través de una segunda molécula adaptadora (Fig. 3). 412 Fig. 3. Unión indirecta de una molécula de captura al soporte. El anticuerpo de captura se une al soporte a través dos moléculas una de afinidad y otra adaptadora; mediante esta estructura se optimiza la interacción con la molécula diana. Pharmacogenomics (2002), 3: 1-10. 1.4 Dispositivos para construir microarrays: arrayers La ordenación de arrays de proteínas en la superficie de los soportes se consigue mediante arrayers. Los aparatos que imprimen por contacto están dotados de agujas que aplican las moléculas de captura sobre la superficie del soporte, como lo hace el arrayer robotizado GMS 417 (Affymetrix) (Fig. 4). Hay otros tipos alternativos de arrayers (capilares, ink-jet), que no realizan la aplicación por contacto, sino que proyectan gotitas de la disolución de moléculas de captura, sobre la superficie del soporte, pertenece a este tipo el arrayer TopSpot (HSG-IMIT). También existen actualmente numerosas casas comerciales productoras de microarrays de proteínas ya formados y preparados para el uso, con aplicaciones en el campo del diagnostico o la investigación biomédica preferentemente (11). 413 Fig. 4. Construcción de un array de proteínas de fase reversa. Se utiliza un arrayer de contacto robotizado (GMS 417, Affymetrix, CA, USA). Se muestra en la imagen el array de configuración plana sobre porta de vidrio. Inserción: el cabezal del arrayer con cuatro agujas imprimiendo manchas de 750 nm de diámetro, de lisados celulares conteniendo proteínas sobre un porta de vidrio plano recubierto de nitrocelulosa. J Pathol. (2006), 208: 595-606. 1.5 Formatos de arrays de proteínas 1.5.1 Formato plano. Posiblemente sea el formato de microarrays de proteínas más extendido (Fig. 1), en él las manchas que contienen las moléculas de captura se disponen sobre superficies reactivas de un soporte de vidrio similar a un portaobjetos de los empleados en microscopia. Existen variantes de este formato, cuyas modificaciones tienen como objetivo principal disminuir las cantidades de muestra y reactivos empleados por medio de la miniaturización (11). 1.5.2 Formato de arrays en suspensión o de microesferas (suspensión array technology, SAT). En este formato el análisis se realiza aplicando citometría de flujo. Se utilizan micropartículas de polímeros codificadas ópticamente, (Fig. 5). Las manchas del formato plano tiene en este, su equivalente en una subpoblación de partículas con diferentes propiedades ópticas que porta diferentes moléculas de captura. Se puede aplicar al análisis de otras biomoléculas, además de las proteínas (12-13). Este sistema presenta la ventaja de que cada población de microesferas puede cuantificarse de forma optimizada al separarse de las demás, y el que se pueda adecuar el número de poblaciones de microesferas y sus proporciones a las características de la muestra a analizar. El procedimiento, que se ha adaptado al 414 análisis rápido de series de muestras, se perfila como una plataforma de aplicación general en la investigación y la clínica. Fig. 5. Formato de microarrays basado en la utilización de microesferas y separación por citometría de flujo. a) se dispone de diferentes poblaciones de microesferas codificadas por la proporción de fluoróforos rojo y naranja que forma parte de su composición; b) cada tipo de molécula de captura se inmoviliza sobre un tipo de microesferas diferente y se mezclan en proporciones adecuadas; c) las moléculas diana se ponen en contacto con sus anticuerpos de detección, que portan un fluoróforo, para que interaccionen; d) se unen las microesferas portando las moléculas de captura con los complejos anteriores y se produce la formación de inmunocomplejos tipo sándwich unidos a sus correspondientes microesferas; e) los análisis cuantitativo y cualitativo se realizan en un citómetro de flujo dotado de dos láseres que puede medir en cada esfera la cantidad de molécula diana captada a través de la fluorescencia emitida por el complejo tipo sándwich y separarla e identificarla a través del código de fluoróforos rojo y naranja de la microesfera y la molécula de captura que porta. Curr Opin Chem Biol. (2001), 6: 76-80. 2 DETECCIÓN DE LA SEÑAL EN MICROARRAYS DE PROTEÍNAS Una vez que han interaccionado las moléculas diana con las de captura, se ha de cuantificar el complejo formado, para ello se etiquetan previamente las moléculas diana o los anticuerpos de detección con marcadores de diferentes propiedades fisicoquímicas que condicionan las técnicas de detección a emplear. 2.1 Tipos de detección 2.1.1 Detección cromogénica: Se emplea para la detección de substancias coloreadas que se generan por la acción de determinadas enzimas sobre substratos denominados cromógenos. La detección cromogénica produce señales permanentes y 415 fáciles de visualizar para su análisis. Los cromógenos empleados con mayor frecuencia en microarrays son substratos de la fosfatasa alcalina (p-Nitrofenilfosfato), y de peroxidasa (ABTS: ácido 2,2’-azo-bis,3-etilbenzotiazolina-6-sulfónico; OPD: ofenilendiamina y TMB: 3,3’,5,5’-tetrametilbencidina). 2.1.2 Detección de quimioluminiscencia: La emisión de luz se produce por la acción de la luciferasa sobre la D-luciferina. Es una técnica muy difundida por su sensibilidad y por crear un registro permanente de la señal. 2.1.3 Detección de fluorescencia: Se utiliza para los fluoróforos que son moléculas que absorben fotones de una fuente externa de luz, generalmente un láser monocromático, lo que provoca excitación de los electrones de la molécula y una posterior emisión de luz de una longitud de onda mayor que el incidente. Esta técnica presenta incompatibilidad con los soportes que presentan autoflorescencia, ya que es difícil discriminar la señal producida por la muestra y la debida al ruido de fondo. Son fluoróforos frecuentemente empleados fluoresceína, rodamina, ficobiliproteínas, acridinas y especialmente las cianinas ( Cy2, Cy3, Cy5). Como detectores se emplean frecuentemente las cámaras denominadas charge coupled device (CCD) o escáneres láser con óptica confocal de detección. 2.2 Aspectos teóricos de la detección de la señal. Miniaturización de los arrays La elevada sensibilidad, característica de esta técnica, se basa en la miniaturización del procedimiento (14), lo que se consigue reuniendo las moléculas de captura en un área muy pequeña (mancha), de manera que al formarse el complejo molécula de captura-molécula diana, el número absoluto de moléculas aunque sea reducido estará concentrado, y, al mismo tiempo, la concentración de moléculas diana de la muestra no cambia prácticamente, incluso cuando sea baja; estas condiciones se cumplen cuando el número total de moléculas de captura en la mancha es < 0.1/KD (KD: constante de afinidad). Cuando se dan estas condiciones, el número de moléculas diana capturadas guarda relación lineal con su concentración en el medio. La sensibilidad que se consigue es alta porque: 1) la unión de ligandos se produce a la concentración mayor posible de moléculas diana y 2) el complejo molécula de captura-molécula diana se concentra en un área muy pequeña dando lugar a una señal de gran densidad. (Fig. 6). Otra consecuencia de la reducción del tamaño de las manchas, es que las propiedades del complejo formado pueden conocerse empleando volúmenes de muestra pequeños, y por ello de una menor necesidad de muestra biológica, aspecto esencial en el tratamiento de determinados grupos de pacientes. 416 Fig. 6. Señal y densidad de señal en las manchas de los microarrays. La densidad de señal (log (señal/área); escala logarítmica) (barras rojas) y la señal (intensidad total; escala logarítmica) (barras blancas) emitidas por las moléculas diana captadas (unidas a un fluoróforo) en las manchas se han representado frente a las diferentes concentraciones de moléculas de captura. Las moléculas de captura inmovilizadas tienen la misma densidad de superficie en todas las manchas. La señal total incrementa con el número de moléculas de captura al incrementar la superficie de la mancha. Cuando la mayoría de las moléculas diana posibles han sido secuestradas de la solución la señal alcanza su máximo. Por el contrario, la densidad de señal (señal/área) incrementa al disminuir la cantidad de moléculas de captura (por disminución del tamaño de la mancha), alcanzando un valor prácticamente constante mientras es < 0.1/K (siendo K la constante de asociación). En estas condiciones ambientales de las moléculas diana, su concentración en la solución se modifica mínimamente por unión a las moléculas de captura de la mancha. TRENDS Biotechnol. (2002), 20: 160-166. 2.3 Amplificación de la señal Existen factores que pueden reducir las posibilidades de aplicación de los microarrays de anticuerpos al análisis proteómico: a) la disponibilidad limitada de anticuerpos (moléculas de captura) con afinidad y especificidad elevadas; b) la variabilidad de la afinidad antígeno-anticuerpo (Ag-Ab) para los diferentes componentes de un microarray y c) el elevado rango dinámico (concentración del analito mayoritario/concentración del analito más diluido) de las muestras biológicas, especialmente del plasma/suero. Los resultados de un análisis múltiple han de ser independientes de la variabilidad que puede derivarse de estos factores (15). El recurso más generalizado para conseguir que en un análisis con microarrays la señal producida por cada complejo Ag-Ab formado, entre en el rango de detección y cuantificación consiste en poner en el microarray una serie gradual de concentraciones de molécula de captura que incremente la probabilidad de que alguna concentración esté dentro del rango de medida (measurable concentration range: MCR) 417 Pero existen otros medios para eliminar estas limitaciones, de forma total o parcial, mediante: a) Técnicas de amplificación de la señal. b) Sistemas competitivos de detección. 2.4. Técnicas de amplificación La amplificación tiene como consecuencia ampliar el MCR hacia concentraciones menores de proteína de la curva de unión antígeno-anticuerpo (Fig. 7), la amplificación de la señal es, pues, una ventaja, pero se ha de tener en cuenta que si es necesario elevar el número de pasos de la técnica, se incrementará el tiempo de realización y las posibilidades de error. Fig. 7. Curva de unión antígeno-anticuerpo. La señal emitida por el complejo antígeno-anticuerpo formado se estabiliza a partir de la cantidad de proteína adicionada con la que se ha rebasado la capacidad de unión del anticuerpo y ya no refleja la cantidad de proteína añadida; por otro lado las concentraciones bajas no pueden detectarse por debajo del umbral de sensibilidad de detección, de modo que el rango de concentraciones de proteína en que es posible detectarla y medirla es limitado. Proteomics. (2003), 4: 3717-3726. 2.4.1 Sistema de amplificación tipo sándwich Se trata de una técnica muy generalizada, característica del método ELISA (Fig. 8). Se diferencian varias fases en el proceso de aplicación: a) formación del complejo Ag-Ab por unión de las molécula diana (Ag) a las de captura (Ab) inmovilizadas; b) un anticuerpo de detección, que lleva conjugada una molécula de biotina, se une por un segundo epítopo a la molécula diana, creando así un sándwich de anticuerpos; c) se procede a la visualización del complejo utilizando estreptavidina conjugada con una molécula fluorescente Cy3 o horse radish peroxidase (HRP) que puede actuar sobre múltiples moléculas de cromógeno. 418 Fig. 8. Protocolo general del método sándwich de ELISA. Se ha de disponer de dos anticuerpos diferentes frente a la misma molécula diana, uno actuará como molécula de captura que se ha de fijar al pocillo de reacción y el otro como anticuerpo de detección, que se ha de marcar. La muestra conteniendo la molécula diana se adiciona al pocillo y se incuba; las moléculas que no han reaccionado se eliminan mediante lavados; al complejo formado se adiciona el anticuerpo de detección que puede tener unido un fluoróforo y se puede leer directamente la señal emitida o bien una enzima que incrementará la señal al actuar sobre un sustrato que genere un producto coloreado. http://64.202.120.86/upload/image/articles/2006/biopen/biopenelisa-schematic.jpg El procedimiento posee ventajas notables: 1) se trata de una técnica bien conocida y aplicada frecuentemente en protocolos de revelado; 2) las muestras no se han de derivatizar, lo que evita el enmascaramiento de epítopos; 3) los analitos de baja concentración se pueden detectar con varios ciclos de amplificación; 4) el uso de dos anticuerpos incrementa la especificidad de la prueba. También presenta algunos inconvenientes; a) son necesarios dos anticuerpos para cada proteína ensayada (de captura y detección); b) el anticuerpo de detección se ha de conjugar con la biotina y en este paso se puede afectar su afinidad por el antígeno. 2.4.2 Amplificación por rolling circle amplification (RCA) Es un método que incorpora a la detección de proteínas las posibilidades de amplificación de la PCR (Fig. 9) (16), para ello, se une covalentemente al anticuerpo de detección un oligonucleotido, en presencia de ADN circular y de un sistema PCR que produce un ADNc formado por numerosas copias de la secuencia del ADN circular, que permanece unido al anticuerpo. Posteriormente se hace interaccionar con un oligonucleotido decorador complementario que porta un fluoróforo. Con esta técnica se consiguen amplificaciones de la señal varios órdenes de magnitud mayores que la proporcionada por la amplificación tipo sándwich. 419 Fig. 9. Esquema de la amplificación de la señal por circulo rodante aplicada a microarrays. Se compone de varios elementos que actúan de forma coordinada; a) anticuerpos de captura inmovilizados sobre el soporte del microarray; b) un anticuerpo de detección biotinilado; c) unión de un anticuerpo antibiotina que porta un oligonucleotido primer complementado con polinucleotido cíclico; d) replicación del polinucleotido circular empleando PCR, lo que dará ADNc de una sola hebra que se híbrida con un oligonucleotido decorador. J Biomed Biotechnol. (2003), 5: 299-307. 2.2.3 Nanocristales (Quantum dots: Qdots) No se trata de una técnica de amplificación propiamente dicha, sino de nanoestructuras aplicables a sistemas de amplificación como sondas fluorescentes, que poseen características que aventajan en gran medida a las de los fluoróforos clásicos. En su estructura (Fig. 10) existe un núcleo formado por algunos centenares de átomos de semiconductores: Cd+Se, rodeado por una corteza del también semiconductor sulfáto de zinc y finalmente una cubierta de moléculas anfífilas que relacionan la partícula con el medio biológico. Su tamaño total se acerca al de las proteínas (10-20 nm), y presenta como propiedades, a) tener una intensidad de fluorescencia varias veces mayor que la de los fluoróforos orgánicos; b) una elevada fotoestabilidad (la fluorescencia persiste durante mucho tiempo) y c) tener como propiedad la “tuneability”, es decir que se puede modificar la longitud de onda de emisión en respuesta a una misma longitud de onda, modificando el tamaño de las nanopartículas. Estas características hacen que los nanocristales sean idóneos para marcar moléculas de proteínas, células y tejidos. 420 Fig. 10. Estructura de un nanocristal Qdot. En el esquema se representan los diferentes elementos estructurales a escala aproximada de tamaños. Qdot® nanocrystal technology. Invitrogen. www.invitrogen.com 2.5 Sistemas de detección competitivos El carácter competitivo de los procedimientos de detección elimina las diferencias derivadas de la heterogeneidad de los anticuerpos, que es una de las limitaciones mayores de los ensayos comparativos basados en la afinidad. Este tipo de análisis no mide concentraciones de proteínas, sino que proporciona una medida relativa de la abundancia de cada proteína en las muestras comparadas. 2.5.1 Detección de un marcaje doble Los anticuerpos, inmovilizados por unión covalente, se usan para capturar los antígenos marcados con un fluoróforo. Las dos muestras a comparar se marcan con fluoróforos diferentes y compiten por unirse a los mismos anticuerpos (Fig. 11). Esta técnica presenta algunos inconvenientes: a) la reacción con los marcadores de fluorescencia, a través de los grupos laterales de las proteínas, puede afectar su afinidad por la molécula de captura; b) al ser distintos los fluoróforos que reaccionan con cada muestra, pueden presentar diferencias en el rendimiento de la reacción y dar lugar a resultados sesgados; c) se marcan todas las proteínas de la muestra y se genera un fondo inespecífico. Una forma de eliminar los errores derivados de las diferencias de reactividad de los fluoróforos se basa en la repetición del marcaje intercambiando los marcadores destinados a cada muestra. 2.5.2 Detección de un solo color y desplazamiento competitivo Para aplicar este procedimiento se necesita una muestra de referencia (R), marcada con un fluoróforo, que puede tener una composición idéntica a las experimentales o puede ser una mezcla de proteínas, péptidos, aptámeros, polímeros sin actividad biológica, etc, siempre que estos componentes presenten afinidad por los 421 anticuerpos fijos y puedan ser desplazados por las proteínas presentes en las muestras experimentales que no están marcadas. Se ha de hacer un ensayo de desplazamiento para cada muestra. Fig. 11. Perfil cuantitativo de proteínas usando microarrays. (A) Detección de dos colores aplicada a microarrays de anticuerpos. Para compensar las diferencias que han podido producirse al marcar cada muestra con un fluoróforo diferente, cada una de las dos muestras a comparar se derivatiza por separado con cada fluoróforo (en gris y negro). Son pues necesarias cuatro reacciones de derivatización y dos ensayos de unión para comparar las dos muestras experimentales. (B) Protocolo de desplazamiento competitivo usando un único color. Se requiere una única muestra de referencia marcada, las muestras experimentales no se marcan. La muestra de referencia marcada puede ser idéntica a las experimentales o solamente similar siempre que muestren afinidad por los anticuerpos,(puede ser una mezcla de protínas, aptámeros, polímeros sin función biológica etc) y puedan ser desplazados por las proteínas presentes en la muestra experimental. En el ensayo se comparan las posibilidades de desplazamiento de las muestras experimentales no marcadas frente a los anticuerpos. Proteomics. (2003), 4: 3717-3726. Los ensayos permiten comparar las respectivas capacidades de desplazamiento de las muestras en su interacción con los anticuerpos (Fig. 11). Este método presenta ventajas notables; a) al no ser necesaria la derivatización de las muestras, se eliminan las alteraciones de afinidad de los antígenos y el background provocado por el marcaje; b) el carácter comparativo de la técnica reduce las exigencias sobre especificidad y afinidad de los anticuerpos de captura. 422 2.6 Escáneres de microarrays Existen una amplia gama de escáneres láser para microarrays como ScanArrayTM GM de Perkin-Elmer, DNA Microarray Scanner from Agilent Technologies. Los de la casa Tecan y GenePixR ofrecen modelos aplicables al escaneado de microarrays de proteínas. 3 TIPOS DE MICROARRAYS APLICADOS AL ANÁLISIS PROTEÓMICO Las características de las moléculas de captura inmovilizadas permiten clasificar los arrays de proteínas en diferentes grupos (17), siendo los más usuales: Microarrays de proteínas, también llamados de anticuerpos o de fase directa forward phase arrays (FPA). De fase inversa o reverse phase arrays (RPA). De tejidos o tissue microarrays (TMA). 3.1 Microarrays de proteínas o de anticuerpos (forward phase arrays: FPAs) Las moléculas de captura son fundamentalmente proteínas con función de anticuerpos, de ahí su nombre, pero también pueden serlo otro tipo de moléculas como aptámeros o affibodies. Las moléculas de captura se fijan por uniones no covalentes o covalentes sobre diferentes tipos de superficies tales como vidrio modificado o membranas (Fig. 12). Para la detección y cuantificación de este tipo de microarrays se suelen emplear alguna de las siguientes estratégias: a) ensayos tipo sándwich; b) marcaje previo de las moléculas diana; c) el sistema de doble marcaje y d) diferentes sistemas de amplificación en función de la cuantía de la muestra. La información que aporta el análisis con este tipo de microarrays permite identificar y obtener las proporciones relativas de un número elevado de proteínas pertenecientes a una misma muestra, es lo que se conoce como búsqueda de biomarcadores, procedimiento de aplicación frecuente en la investigación sobre el cáncer. También se puede aplicar al estudio de modificaciones postraduccionales; por ejemplo para conocer el grado de fosforilación de la muestra, en este caso realizando un revelado tipo sándwich se pueden seleccionar los anticuerpos de detección de modo que solo interaccionen con proteínas fosforiladas, aportando así información exclusiva sobre el fosfoproteóma. Otra aplicación de especial interés la tienen en los análisis de citoquinas, cuyas interrelaciones juegan un papel relevante en el desarrollo de numerosas enfermedades autoinmunes (18), y en los que las bajas concentraciones de estos compuestos hacen necesario aplicar los sistemas de amplificación más eficaces para su detección. Al tratarse de una técnica de alto rendimiento, su utilidad no se deriva meramente de la información aportada por la suma de datos obtenidos de los biomarcadores investigados, sino que además aporta el conocimiento de las interrelaciones existentes entre ellos, es decir que el conjunto de datos obtenidos simultáneamente aporta más información que la suma de estos datos obtenidos por separado. 423 La construcción de este tipo de microarrays necesita disponer de anticuerpos con suficiente especificidad y afinidad, y cuando su revelado se hace por un sistema tipo sándwich se ha de disponer de dos por cada componente de la muestra que se analice. También se ha de tener en cuenta que la unión de marcadores a las moléculas de proteína que han de interaccionar en el proceso de revelado (dianas o anticuerpos de revelado) puede modificar su afinidad y que el marcaje de las proteínas, en muestras con un rango lineal de concentraciones elevado, como ocurre en el suero/plasma, redunda en el incremento de fondo en el revelado. 3.2 Microarrays de Fase Inversa (reverse phase arrays: RPAs) o de Lisados de Tejidos En este tipo de array son las muestras las que se inmovilizan, posteriormente se hacen interaccionar con anticuerpos específicos de los componentes de interés de la muestra, disueltos (Fig. 12). Fig. 12. Clases de microarrays de proteínas. En los microarrays de fase directa se inmoviliza una molécula de captura, frecuentemente un anticuerpo, diseñada para captar proteínas específicas (moléculas diana) que forman parte de una muestra de naturaleza proteica. Las moléculas diana se detectan generando una señal, mediante un sistema de revelado tipo sándwich o por marcaje directo. En los microarrays de fase reversa se inmovilizan, sobre el soporte, las proteínas presentes en una muestra, frecuentemente un lisado de células o tejidos. Un anticuerpo en solución, específico para una proteína fijada, se aplica al microarray. Los anticuerpos fijados se detectan por marcaje y amplificación de la señal. Moll Cell Proteomics. (2005), 4: 346-355. 424 Frecuentemente las muestras son lisados de tejidos o celulares que se imprimen directamente sobre la superficie del soporte mediante arrayer. La superficie puede corresponder a membranas de nitrocelulosa sobre soporte de vidrio. La nitrocelulosa tiene una capacidad elevada de unión de proteínas a través de interacciones que no alteran la estructura de estas, y el fondo fluorescente que genera es relativamente bajo y compatible con la detección fluorimétrica. Las manchas se suelen disponer en series de dilución creciente para facilitar su cuantificación precisa (Fig. 13). Los RPAs se caracterizan por que a) cada mancha contiene el proteoma completo de la muestra de que procede; b) los microarrays están formados por numerosas muestras diferentes; c) es necesario utilizar un porta para cada proteína que se quiera analizar. En el proceso de detección y cuantificación es esencial que se utilicen técnicas de amplificación, pues los componentes de las muestras experimentales pueden presentar rangos dinámicos elevados. Fig. 13. Array de fase reversa. a) El array está impreso sobre un porta de vidrio cubierto de acetato de celulosa de un tamaño similar a los empleados en histopatologia clínica. Se ha teñido de forma automatizada empleando un sistema de amplificación de la señal por acción de una enzima unida por biotina. b) Mancha de 750 µm de diámetro. c) Manchas pertenecientes a un mismo lisado, impresas por duplicado, formando una curva de dilución desde la original del lisado hasta 1:16 a la derecha. d) El array porta cuatro controles, entre ellos una mezcla de todas las muestras en estudio, y un lisado de línea celular, con objeto de que permitan correlacionar todos los arrays utilizados en el estudio. J Pathol. (2006), 208: 595-606. 425 Para eludir la interferencia de la autoflorescencia de los soporte de nitrocelulosa, se ha descrito el uso de nanocristales quantum dots (Qdot) como el Qdot 655 Sav conjugado con estreptavidina que comparado con las técnicas clásicas ha incrementado el rango dinámico de concentración de las proteínas sobre las que puede actuar (19), mostrando sensibilidad por debajo de 50 fg/µl, y con una reproducibilidad intra- e interarray que se manifiesta como valores de CV<10%. Este tipo de microarrays presenta ventajas notables: 1) el análisis simultáneo de muchas muestras incrementa el rendimiento y facilita la comparación cuantitativa entre ellas. 2) las muestras inmovilizadas no se han de derivatizar para incorporarles marcadores, evitando así su posible desnaturalización; 3) al ser una técnica robusta y al mismo tiempo sensible, se puede utilizar en combinación con técnicas de aislamiento celular del tipo laser capture microdissection (LCM), pudiendo obtenerse información a partir de muestras tan reducidas como las formadas solo por 10 células. La aplicación de este tipo de microarrays está especialmente indicada para el estudio de rutas de señalización (20), por ejemplo para la obtención del perfiles de fosforilación de proteínas de señalización celular, esta aplicación se ha visto favorecida por la obtención de anticuerpos frente a proteínas fosforiladas muy específicos y con elevada afinidad, y por la posibilidad de analizar muestras reducidas procedentes de biopsias o cultivos celulares. También tienen múltiples aplicaciones en Inmunología, pues en los estados autoinmunes, los circuitos de transmisión de señales se encuentran frecuentemente alterados y ello conduce a respuestas inadecuadas. Otro campo de aplicación es el dirigido a explorar la progresión tumoral (21). Existen también algunos problemas técnicos de cuya resolución depende obtener la máxima eficacia en el empleo de este tipo de arrays: a) disponer de anticuerpos con especificidad y afinidad altas; b) conseguir una estandarización experimental, que permita comparar los diferentes ensayos dentro de un mismo experimento, de distintos experimentos entre si, y los realizados en centros diferentes, mediante la utilización de patrones internos y controles de referencia. 3.3 Microarrays de tejidos (TMAs). Los microarrays de tejidos son estructuras ordenadas de cientos de cortes de tejidos procedentes de bloques de inclusión. Se diferencian por ello de los anteriores, en que las moléculas de captura fijas a la superficie del soporte han sido substituidas por cortes histológicos (22). Este método permite analizar simultáneamente numerosas muestras de tejido mediante sistemas de detección de proteínas, de forma similar a como ocurre con los arrays de fase inversa. La construcción de los TMAs (Fig 14) se basa en la obtención de cilindros de tejido de 600 µm de diámetro a partir de piezas de tejido previamente fijadas, las denominadas formalin-fixated paraffin-embedded (FFPE). Los cilindros se incluyen ordenadamente en un bloque de parafina del que se hacen cortes histológicos para su análisis, la posterior tinción se hace mediante técnica inmunohistoquímica. La preparación de un TMA aparece en la Fig. 14. 426 Fig. 14. Manufactura de un TMA. a) Se extraen núcleos cilíndricos de tejido (normalmente de 0.6 mm de diámetro) de una pieza parafinada (donadora) empleando un microarrayer de tejido; b) Se introducen en huecos hechos previamente en un bloque de parafina vacio (receptor); c) Se pueden usar microtomos convencionales para cortar secciones de microarrays de tejidos; d) Se puede usar una película adhesiva facilita la transferencia de secciones de microarrays de tejido TMA al porta y evita la pérdida de tejidos, incrementado así el número de secciones que pueden obtenerse de cada bloque de TMA; e) Estructura de una de las secciones obtenidas teñida con hematoxilina-eosina. Nat Rev Drug Discov. (2003), 2: 962-972. Este tipo de microarrays presenta como características más destacadas que: a) la información molecular se obtiene in situ, dentro de la arquitectura del tejido y la morfología celular; b) la información obtenida de las preparaciones, está sujeta a la estimación del observador, de modo que los resultados que son de tipo cualitativo o semicuantitativo. Estos arrays presentan ventajas claras frente a las técnicas inmunohistoquímicas clásicas: a) menos tiempo de preparación y de costo por muestra; b) las condiciones experimentales son las mismas para todas las muestras, lo que facilita la comparación y mejora la estadística de los resultados; c) reducen la cantidad de muestra utilizada, permitiendo obtener un mayor rendimiento de las piezas de tejido disponibles; d) se pueden utilizar las muestras FFPE existentes en bancos de tumores. A pesar de estas ventajas la plataforma presenta algunas limitaciones: 1) la dudosa representatividad de una muestra de 600 µm de diámetro procedente de un tumor potencialmente heterogéneo, especialmente si se compara con la obtenida a partir de un corte convencional de mayor extensión; 2) la subjetividad y falta de reproducibilidad de la valoración visual; 3) posible variabilidad de los resultados debida a cambios en la especificidad y afinidad de los anticuerpos utilizados en los métodos de IHC y a la falta de estandarización de los protocolos. Es un formato muy utilizado en Oncología con diversas aplicaciones: 1) el seguimiento de la progresión de tumores; 2) la obtención de perfiles proteicos asociados a fenotipos tumorales; 3) la detección de marcadores tumorales para fundamentar el diagnóstico. 427 4 ANALISIS COMPUTACIONAL DE LOS DATOS DE MICROARRAYS El desarrollo de las técnicas analíticas de alto rendimiento como los microarrays, ha hecho necesaria la incorporación de tratamientos bioinformáticos avanzados para manejar los abundantes datos que generan. En términos generales se puede considerar que este tratamiento informático/estadístico se aplica en dos fases del proceso completo de análisis de la información procedente de los microarrays: i) En primer lugar se ha de realizar un preprocesado de la información obtenida en la lectura de los microarrays para eliminar las alteraciones y sesgos derivados de defectos de la tecnología, mediante operaciones como la corrección de background, la normalización de la intensidad, o la normalización espacial. ii) Posteriormente se han de obtener los resultados evaluando la significación estadística y biomédica de los datos normalizados, con la ayuda de algoritmos matemáticos. 4.1 TRATAMIENTO PREVIO DE LOS DATOS. NORMALIZACIÓN 4.1.2 Obtención informatizada de los datos Los microarrays de anticuerpos presentan una estructura y un funcionamiento muy similares a los de DNA, pues estos han servido como modelo para su desarrollo. Dada la similitud de ambas plataformas, se puede utilizar los mismos escáneres y software para ambas, también los ficheros de datos sin tratar que proporcionan son técnicamente muy similares, esto supone una ventaja pues algunos protocolos de computación y los interfaces desarrollados para los microarrays de DNA se transfieren frecuentemente al análisis de microarrays de anticuerpos. El resultado del escaneado de un microarray que ha interaccionado con una muestra marcada con un único fluoróforo, es una imagen digital monocromática (típicamente guardada como un fichero TIFF) de la superficie del microarray. Los puntos más intensos de la imagen corresponden a manchas en las que se ha producido hibridación de ácidos nucléicos. Si dos muestras diferentes se han marcado con dos fluoróforos distintos, y se han hibridado con el mismo microarray, se obtendrán como resultado del escaneado dos imágenes digitales, una para cada longitud de onda de lectura. Las imágenes de los microarrays generadas por un escaneado láser deben procesarse con un software de análisis de imagen. En primer lugar para identificar las manchas dentro de la imagen, es decir distinguir entre los píxeles que forman parte de manchas y los que pertenecen a zonas de fondo; este proceso recibe el nombre de segmentación. Después, se ha de realizar la alineación de gradillas, que es el proceso que permite identificar las manchas. El software para la alineación de gradillas suele venir incluido al adquirir el equipo, aunque los hay de libre disposición como el TIGR Spotfinder (23) o el ScanAlyze (http://rana.lbl.gov/EisenSoftware.htm). Para la mayoría de los arrays, la gradilla ha de ser definida por el usuario, bien manualmente o semi-manualmente, mientras que para los microarrays de alta densidad, como los Affymetrix, la alineación de la imagen del microarray con la gradilla se hace automáticamente a través del software. Esta automatización se hace posible reservando algunas manchas del microarray exclusivamente para la alineación de gradillas. 428 Después de alinear la gradilla el software del análisis de imagen proporciona cierto número de datos estadísticos para cada mancha identificada, que suelen incluir la media y la mediana de la intensidad en pixeles de cada mancha, su desviación estándar, y a veces también otro tipo de información, como las relaciones de intensidad cuando se trata de un experimento con dos canales (dos longitudes de onda). El área de cada mancha (en número de píxeles) también se suministra con frecuencia. Es importante que si el software considera una mancha determinada aberrante -por ejemplo que es de forma irregular o que su intensidad es inferior que la del fondo de su entorno- la mancha pueda ser considerada como irregular. Las manchas irregulares normalmente se excluyen de los análisis estadísticos posteriores. El resultado final es un fichero de texto plano que contiene los datos sin tratar de cada mancha dispuestos en una fila. Este formato es muy accesible para el posterior análisis de arrays importándolo al paquete de software del tipo ExpressYourself (24) o MIDAS (23) o desde luego en el propio flujo de análisis de microarrays. 4.1.3 Normalización de los datos de los microarrays. Una vez que se han obtenido los datos, el paso siguiente es su normalización, dado que a través de los procesos técnicos de un experimento de microarrays pueden producirse sesgos y artefactos que influyan en el valor de los datos que se obtienen, de modo que en un experimento de doble marcaje los valores de fluorescencia pueden estar influidos por una gran diferencia de entre la cuantía de las muestras, pueden influir también las diferencias de reactividad de los fluoróforos empleados (ej Cy3 y Cy5), o incluso la localización de una mancha dentro del array. Necesidad de la Normalización La mayoría de los estudios con microarrays examinan la relación entre dos muestras biológicas comparando sus niveles relativos de expresión. La idea por la que se rigen los experimentos con dos colores se basa en que dos muestras, una marcada con Cy3 y la otra con Cy5, se hibridan simultáneamente en un único microarray, sus abundancias relativas se obtienen a partir de las relaciones de fluorescencia en cada una de las manchas del microarray. Para una mancha dada i, la concentración relativa entre las dos muestras λi se expresa como el logaritmo de la relación de las fluorescencias: λi = log ( Ri / Gi ) (1) donde Ri y Gi son las intensidades (medias o medianas de las intensidades en píxeles) cuando se escanean con láseres rojo y verde respectivamente. Un valor de λi = 0 indica que Ri y Gi son iguales. En una serie de valores expresados así los próximos a 0 indican la igualdad de contenido de ese componente de las muestras, mientras que los valores que se desvían de 0 hacia arriba o hacia abajo son indicadores de desigualdad. Una relación logarítmica de este tipo entre dos muestras para una proteína es ya una técnica de normalización. La manufactura de los microarrays no está libre de errores. Una mancha puede imprimirse con un área reducida en un microarray y en el siguiente de forma correcta. Si se emplea este tipo de normalización ambas manchas darán valores relativos comparables independientemente de su tamaño. Esta clase de normalización no ha lugar en los experimentos con un solo fluoróforo. 429 Existen algunas estrategias comunes de normalización entre las que se encuentran Corrección de fondos Normalización por intensidad total Normalización por Gene Set Normalización mediante controles internos Corrección de desviaciones debidas a la posición en el array Corrección de Fondos En muchos tipos de microarrays, se proporciona una medida del fondo local además del valor total de la mancha. Esta medida es, como norma general, la media o mediana de los pixeles correspondientes a las zonas periféricas de las manchas. Se asume que cualquier medida de la intensidad de una mancha también lleva incorporada la intensidad del fondo correspondiente. La fluorescencia de fondo se debe, en general, a la fluorescencia del vidrio y a moléculas libres del marcador fluorescente. La intensidad de fondo no tiene un significado biológico, de modo que han de eliminarse los valores de fluorescencia a él debidos antes de procesar los datos. La forma más sencilla es restar a la lectura de cada canal la media (o mediana) del fondo a la correspondiente longitud de onda antes de calcular la relación. ^ λi = log( Ri − ρ i / Gi − γ i ) (2) De forma natural en esta ecuación se asume que ρ i < Ri y que γ i < Gi , dado que cualquier mancha que no cumpla estas condiciones se considerará una mancha irregular y se excluye del cálculo, pues con la interpretación del fondo que hemos comentado, carece de sentido que éste tenga un valor mayor que el asociado a una mancha. Normalización por intensidad total Después de la substracción del fondo, se deben normalizar las intensidades de las muestras de modo que la distribución de intensidades tenga propiedades deseables. Una propiedad deseable en experimentos de dos muestras es tener una distribución de logaritmos centrados en torno a 0. Esto es razonable ya que en la mayoría de los experimentos no se espera que este centrado se produzca en torno a ningún otro valor. En un experimento de expresión diferencial, los microarrays hibridarían un número similar de moléculas marcadas procedentes de cada muestra, de modo que las señales de hibridación totales fuese la misma para ambos canales. Asumiendo este supuesto, se puede calcular un factor de escalado Ctotal, que pueda usarse para corregir cualquier desviación que se observe sobre este supuesto. Si M es el número total de manchas en un microarray, tenemos que: C total =log( M ∑ Ri i =1 M ∑G i =1 i ) (3) Se puede expresar el logaritmo de la relación normalizada como: ^ λi = λi − C total (4) El resultado es una distribución de logaritmos de relaciones que se centran en algún punto cercano a cero. Este método funciona bien en la mayoría de los experimentos estándar de microarrays con un número suficientemente grande de manchas (> 430 20000) ya que en estas condiciones los valores de señales outliers hacen una contribución despreciable a las intensidades totales. Una aproximación similar a la ecuación 3 puede emplearse para normalizar intensidades entre los componentes de un grupo de microarrays de un solo canal. En esta aplicación, cada intensidad obtenida en un canal se divide por la suma de intensidades ( ej. M ∑G i =1 i ) de las manchas de un mismo microarray. De esta manera, las intensidades normalizadas pueden usarse para comparar muestras hibridadas en diferentes microarrays. Esta situación es la característica de los Affymetrix GeneChip, y se da con mucha frecuencia en los microarrays de proteínas. Normalización por Gen Set El procedimiento anterior funciona bien para los experimentos con microarrays estándar en los que el número de manchas es elevado y en general las diferencias de expresión entre las dos muestras estudiadas no es excesiva. Sin embargo, el método debe aplicarse con precaución pues puede conducir al error de concluir que siempre es similar el número de genes superexpresados y subexpresados, lo que no es cierto en muchas circunstancias. En el gen set method se asume la existencia de un conjunto de genes que no se expresan diferencialmente en las muestras estudiadas, se trata del conjunto de los genes housekeeping. El procedimiento seguido es el mismo que en la ecuación 4, con la única diferencia de que las intensidades que se suman son sólo las correspondientes a los genes housekeeping y no a todas las manchas, a este valor se llama Cgeneset . Normalización mediante controles internos Una forma de mejorar el proceso de normalización consiste en incorporar a las muestras cantidades conocidas de controles externos antes del marcaje fluorescente. La normalización se basa en balancear las intensidades de señal empleando las moléculas de RNA que se han añadido como patrón externo. Esta técnica tiene algunas ventajas como el control que se ejerce sobre la cantidad de patrón externo que es la misma para todas las muestras, otra ventaja es la de poder adaptar las cantidades de los patrones externos a los niveles de expresión de los componentes de las muestras. Presenta también algunos inconvenientes derivados de que los patrones se incorporan en una fase avanzada del experimento, o la existencia de posibles interacciones cruzadas con diferentes manchas del microarray. Corrección de desviaciones debidas a la posición en el array Se ha comprobado que existen desviaciones espaciales debidas a condiciones de hibridación inadecuadas en determinadas zonas de la superficie de un microarray. Si no se corrige este efecto, puede alterar los resultados, por ejemplo deducir una coexpresión de genes cuando lo que sucede es que están en una zona con diferente intensidad de color. Una de las formas de normalización consiste en dividir el microarray en subzonas definidas por el procedimiento de impresión del arrayer y aplicar por separado un procedimiento de normalización como el expresado en la fórmula 4. 431 Una medida que debe tomarse al construir los arrays es la de no colocar juntas manchas que responden a modificaciones de expresión conjunta sino disponerlas al azar. Los microarrays de funcionalidad de proteínas precisan de este tipo de normalización ya que se estudia la posible interacción de cada proteína con un conjunto de ellas. 4.2 ANALISIS ESTADÍSTICO DE LOS DATOS DE MICROARRAYS DE PROTEINAS. OBTENCIÓN DE RESULTADOS. A medida que incrementa la complejidad de los microarrays de proteínas, se genera un volumen de datos lo suficientemente elevado como para que no pueda deducirse su significado sin un tratamiento bioestadístico previo. Existen numerosos métodos disponibles para el análisis de los datos, que permiten extraer la información esencial e identificar las correlaciones que pueden establecerse con los parámetros clínicos y biológicos. Los métodos de análisis computacional de los datos procedentes de los microarrays pueden dividirse en dos grupos: Supervised y Unsupervised methods. Las técnicas supervised requieren algún conocimiento previo sobre las proteínas, por ejemplo, cuales proceden de controles y cuales de enfermos; en este grupo se incluyen métodos estadísticos clásicos como ANOVA, t-test, Wilcoxon rank score, el análisis discriminante y el Mann-Whitney U-text (25). Por el contrario en los unsupervised methods se desconoce cualquier dato a cerca de las proteínas, las cuales se clasifican exclusivamente por su comportamiento en la expresión. Se incluyen en este grupo el Hierarchical clustering, K-means clustering, self-organizing maps, el principal component analisys, o las redes bayesianas que proporcionan resultados especialmente útiles para la generación de hipótesis A continuación se describen algunos de los métodos unsupervised que han demostrado su utilidad en la interpretación de los datos procedentes de microarrays de ácidos nucleicos inicialmente y que también han probado su eficacia al aplicarse a los datos procedentes de los microarrays de proteínas. 4.2.1 Análisis de clusters o conglomerados En general, este método analítico tiene por objeto reunir elementos en grupos homogéneos en función de las similitudes. Existen diferentes algoritmos para determinar la relación de los datos basados en el cálculo de una métrica entre variables similares. Hierarchical Clustering Uno de los métodos empleados más frecuentemente es el hierarchical clustering. Esta familia de algoritmos proporciona una herramienta exploratoria para identificar agrupaciones o clases no evidentes de forma inmediata dentro de un grupo de datos. La idea básica consiste en ir dividiendo el conjunto inicial de todos los datos en clases cuyos elementos presenten una mayor similitud, de acuerdo a una distancia o métrica. Posteriormente se repite esta forma de actuación sobre las clases resultantes. Una de las ventajas de esta técnica consiste en la posibilidad de elegir el punto de corte en la iteración de tal forma que las clases resultantes tengan un sentido biológico fácilmente identificable. 432 El resultado final se puede mostrar como un dendrograma o árbol, en el que la longitud de las ramas refleja el grado de similitud entre variables. De forma adicional, a partir de un hierarchical clustering puede generarse también un heat map (Fig. 15) que proporciona una representación visual del rango de niveles presentados por las variables (ej.: proteínas) y la naturaleza de la relación calculada entre ellas. Este método analítico es una herramienta robusta para la generación de hipótesis a partir de los datos de microarrays. Además de su aplicación al análisis de los perfiles de expresión génica, y al análisis de datos inmunohistoquímicos, se ha demostrado que el hierarchical clustering es una herramienta útil para obtener perfiles proteómicos. Usando un algoritmo de tipo two-way clustering, se ha logrado diferenciar el carcinoma hepatocelular de la enfermedad hepática crónica, y en un estudio posterior, utilizando datos procedentes de microarrays RPA, se ha conseguido la segregación de los casos de cáncer colorectal primario de los de enfermedad metastásica del hígado mediante un algoritmo similar. Fig. 15.Heat map. Un heat map generado por un hierarchical clustering está constituido por un reticulado con las cuadrículas coloreadas, cada color representa un valor de expresión de proteínas. En este ejemplo, las muestras del paciente se localizan en el eje vertical y consiste en 49 casos de linfoma folicular (FL) y 15 casos de hiperplasia folicular (Reactive). Las proteínas de interés se localizan en el eje horizontal y son: Bcl-2, fosfo-Akt (Ser473), fosfo-Bcl-2 (Ser 70), XIAP, fragmento de 433 PARP, y Bcl-xL. El color rojo corresponde a una expresión elevada, el negro a una intermedia, y el verde a baja con respecto a un estandar común de referencia. A la derecha se representa un dendrograma asociado. En este ejemplo, por el procedimiento de clustering han separado los casos en dos perfiles uno constituido por 38 casos de FL y 2 Reactives (escritos en verde), y el otro compuesto por 11 casos FL y 13 casos Reactive. En términos generales los casos FL presentan unos niveles de expresión mas altos para las proteínas, lo que queda indicado por el predominio de colores rojo y negro. J Pathol. (2006), 208: 595-606. Una de las limitaciones del hierarchical clustering analysis es que no siempre proporciona una medida de la probabilidad de la significación de la diferencia entre clusters, dejando los resultados a una interpretación subjetiva y por tanto variable. Por otra parte puede resultar no evidente identificar el verdadero número de clusters fiables, ya que incluso datos no relacionados pueden dar lugar a algún cluster. De ahí que esta herramienta sea más adecuada para la generación de hipótesis, que puedan probarse después por otros procedimientos. Clustering no Jerárquico Otras técnicas de clustering, no jerárquicas, se utilizan para analizar los signalling pathways de las interacciones de proteínas: entre ellas se encuentran el k-means clustering y los self-organizing maps. El clustering de k-medias es un método alternativo especialmente adecuado si se tiene conocimiento previo del número de clústeres esperados, así como el reparto de los datos entre un número fijo de grupos relacionados. Sin embargo, cuando se aplica el clustering a conjuntos de datos de gran tamaño, de genes o proteínas, puede ser difícil o inadecuado elegir a priori un número definido de clústeres. Redes Neuronales Artificiales Las redes neuronales son algoritmos generales de análisis de datos basados en un uso intensivo del ordenador, que simulan el comportamiento neuronal biológico en la resolución de problemas de clasificación. Las redes neuronales artificiales se pueden emplear en la clasificación de comportamientos complejos. Estos métodos se han empleado para discriminar, mediante perfiles proteicos de suero, los pacientes de cáncer frente a individuos sin cáncer. Entre las redes neuronales los self-organising maps son ampliamente utilizados problemas de clasificación no supervisados. Este algoritmo se diferencia de otras redes neuronales por estar orientado a facilitar la identificación de clusters adyacentes a uno dado, lo que resulta especialmente valioso para describir las relaciones entre clusters. 4.2.2 Análisis por Componentes Principales (Principal component analisys, PCA) Es una herramienta estadística útil para reducir el número de variables en un conjunto de datos identificando un subconjunto de éstas que sea el responsable de la mayoría de las diferencias observadas. El PCA puede indicar qué variables en un conjunto son importantes y cuales son de escasa significación. Esta técnica se ha aplicado a los datos procedentes de los arrays de proteínas, logrando separar claramente los linfomas foliculares de la hiperplasia folicular. 434 El PCA es una herramienta especialmente eficaz cuando se combina con otros algoritmos de clustering, como por ejemplo el k-means clustering o el análisis discriminante lineal (Linear discriminant analysis LDA). 4.2.3 Redes Bayesianas Se utilizan para el análisis de las interacciones proteína-proteína. El método PCA y el hierarchical clustering identifican o separan muestras (o pacientes) basado en los criterios de similitud/desigualdad en la expresión de proteínas, en diferentes subclases funcionalmente asociadas. Sin embargo, para identificar pautas de expresión de proteínas a lo largo de una ruta de regulación, deben utilizarse aproximaciones alternativas. Se ha empleado el análisis computacional de redes Bayesianas en la identificación de las rutas proteicas de señalización para deducir relaciones reguladoras entre rutas diferentes, o para descubrir la existencia de interacciones moleculares. La identificación de relaciones de dependencia probabilística es particularmente importante en el descubrimiento de nuevas dianas terapéuticas, especialmente en el conjunto del fosfoproteóma, en el que la inhibición de un producto final puede tener consecuencias significativas sobre otras rutas. Los modelos de redes Bayesianas demuestran cualitativamente las relaciones causales directas e indirectas de unos componentes de las rutas sobre otros en forma de grafo dirigido compuesto por nodos y arcos. Los nodos representan las variables medidas y los arcos de interconexión representan relaciones estadísticamente significativas entre ellos, tanto en una forma lineal como no-lineal. Esta metodología es más apropiada para detectar interacciones entre variables, y podría ser una técnica valiosa para deducir los puntos de intervención farmacológica en las complejas rutas de fosforilación de proteínas seguidas mediante microarrays de proteínas. 5 APLICACIÓNES CLÍNICAS DE LOS MICROARRAYS El análisis proteómico basado en diferentes tipos de microarrays, ha incidido positivamente sobre diversos aspectos de la clínica de enfermedades multifactoriales como el cáncer o las autoinmunes, al abordar la variabilidad y complejidad de estas con nuevas herramientas moleculares, que facilitan su diagnóstico, pronóstico, y el seguimiento de la enfermedad, y, ya en el campo farmacológico, permiten el descubrimiento de nuevas dianas terapéuticas. 5.1 Cáncer 5.1.1 Cáncer de mama. Aplicaciones de TMAs El cáncer de mama es uno de los de mayor incidencia en el mundo y de los que causan mayor mortalidad. Se trata de una enfermedad compleja en la que la acumulación de numerosas alteraciones moleculares, de origen frecuentemente desconocido, provoca proliferación celular, inestabilidad genética y la adquisición de un fenotipo progresivamente más invasivo y resistente (26). La heterogeneidad de las células malignas junto con la variabilidad de los pacientes da lugar a diversos subgrupos de tumores que se caracterizan por sus 435 fenotipos y manifestaciones clínicas diferenciales, pero utilizando criterios clínicos y patológicos la heterogeneidad se capta solo parcialmente y esto limita las posibilidades de adoptar protocolos y estrategias terapéuticas nuevas más personalizadas y por tanto mas eficaces. 5.1.2 Aplicaciones al diagnóstico Los TMAs se han utilizado ampliamente en la caracterización del cáncer de mama. La mayor parte de los estudios se ha basado en la identificación del perfil proteico que se asocia a un fenotipo dado. Las proteínas que son objeto de estudio se eligen con los siguientes criterios: 1) que jueguen un papel claro en la carcinogénesis de la mama; 2) que muestren posibilidades de actuar como discriminadores en la división en clases de los cánceres de mama; y 3) su idoneidad para identificar distintas formas de diferenciación. Las proteínas que responden a estas características tienen funciones diversas en la célula, por ejemplo, puede tratarse de receptores hormonales luminales o basales, de miembros de la familia de los epitelial grow factors receptors (EGFR), estar asociadas a genes supresores de tumores, ser moléculas de adhesión celular como las mucinas, o participar en la diferenciación apocrina, y neuroendocrina. Se han realizado numerosos estudios en los que cohortes de pacientes con cáncer de mama se han subdividido en clusteres utilizando en paralelo los criterios clínicos y/o genéticos y los moleculares obteniéndose resultados muy similares o incluso los criterios proteómicos han mostrado una mayor eficacia en la diferenciación de los subgrupos inflammatory breast cancer IBC y non inflamatory breast cancer NBC, frente a los criterios clínicos o patológicos, menos resolutivos para estos caracteres. 5.1.3 Aplicaciones al pronóstico El uso de criterios de pronóstico basados en la patología resulta insuficiente, y por lo que se hace necesario realizar una diferenciación más precisa de los pacientes basada en la respuesta a los agentes terapéuticos (26). Estudios de pronóstico en cáncer de mama, basados en TMAs, han probado que la expresión elevada de determinadas proteínas está relacionada con una evolución desfavorable del tumor, como es el caso de la COX2, la subunidad catalítica de la telomerasa (TERT), la molécula epitelial de adhesión celular (EpCAM/TACSTD1), o el factor de crecimiento placentario similar a insulina, en cuanto a la expresión de la proteina KIT, que es una diana en la terapia con imatinib, no se ha podido asociar con incremento en la supervivencia. Por el contrario se ha identificado un buen pronóstico con la expresión elevada de STAT5 fosforilada en tirosina o de BCL2 Los TMAs se han utilizado también para validar pronósticos basados en datos obtenidos con microarrays de ADN, incrementándose su eficacia al aplicar los algoritmos matemáticos de análisis de tipo supervised cuando se complementan con datos cualitativos derivados de la aplicación de metodología de tinción por métodos inmunohistoquímicos. La variación de los niveles de un conjunto de proteínas ha permitido dividir los tumores en dos grupos de pronóstico: a) bueno (90% de metastasis-free survival (MFS) a los 5 años) y b) malo (61% de MFS a los 5 años), 436 siendo valida la predicción independientemente del status del estrogen receptor (ER), lo que pone de manifiesto que la predicción mediante un perfil de proteínas es más eficaz que la basada en un solo factor. 5.1.4 Cáncer de ovario. Aplicaciones de RPAs Es un tipo de cáncer que presenta especiales dificultades para el diagnóstico y tratamiento; entre los cánceres ginecológicos es el de peor pronostico pues se manifiesta clínicamente en estadios muy avanzados, en los que el tratamiento citorreductor y la quimioterapia suelen ser escasamente eficaces, de ahí la importancia que tiene la detección de nuevos biomarcadores para el diagnóstico precoz, así como disponer de indicadores de la respuesta a la terapia, y de nuevas opciones de tratamiento para pacientes con la enfermedad avanzada o refractarios al tratamiento (17). Los procesos de crecimiento, invasión y migración de las células neoplásicas están controlados a través de rutas de señalización moleculares interconectadas que se pueden estudiar empleando lisados de células aisladas a partir de biopsias mediante LCM y analizándolas con RPAs, ello permite evaluar el estado de activación de las rutas de señalización en diferentes fases de desarrollo de la enfermedad (21). El grado de fosforilación de las quinasas, que forman parte de cascadas de señalización, es más elevado en fases avanzadas del tumor y en aquellos que presentan morfología endometrioide, mientras que los niveles de expresión tienden a ser más específicos del paciente que de la fase de desarrollo del tumor. Este hecho pone de relieve la necesidad de disponer de la información necesaria para diseñar terapias ajustadas al paciente que bloqueen la progresión neoplásica (27). Un estudio realizado con RPA empleando diferentes anticuerpos fosfoespecíficos de proteínas reguladoras de la mitogénesis, transductoras de señales y factores nucleares de transcripción, ha revelado un sorprendente grado de heterogeneidad entre los pacientes con respecto a la activación de las cascadas de señalización. El análisis de los datos mediante un unsupervised hierarchical clustering ha puesto de relieve la existencia de dos grandes grupos de pacientes caracterizados por el estado de activación de los end points de las rutas de regulación. Además se han extraído otras conclusiones: a) el proteoma del carcinoma peritoneal primario no presenta diferencias significativas con respecto al del tumor primario; b) la comparación entre las rutas de señalización de un grupo primario y otro metastásico presentó variaciones considerables respecto a la activación de las rutas de señalización; y c) los perfiles procedentes de las metástasis eran claramente diferentes de los de su correspondiente tejido tumoral primario, es decir que el comportamiento proteómico del tumor evoluciona de forma diferente cuando se extiende a una localización secundaria Respecto al tratamiento se observa que los protocolos terapéuticos aplicados pueden ser eficaces solamente en determinadas subpoblaciones de pacientes, por ello se hace necesario definir un fingerprint carcinogénico para cada paciente de forma que se pueda aplicar un tratamiento personalizado, usando nuevos agentes terapéuticos solos o en combinación con otros preestablecidos. Está claro que en el estudio de los casos antes mencionados, la tecnología de los RPAs puede proporcionar datos de interés considerable pero es necesaria la circunspección antes de extrapolar los resultados de la investigación a la práctica 437 clínica, pues se han de resolver previamente diversos problemas de reproducibilidad, estandarización y validación clínica. 5.2 Inmunología 5.2.1 Enfermedades autoinmunes. Aplicaciones de FPAs La disfunción que conduce a la respuesta autoinmune es desconocida en gran medida a pesar de los avances realizados en este campo. En estudios hechos con gemelos monocigóticos se han observado bajas tasas de concordancia para la mayoría de las enfermedades autoinmunes, lo que parece indicar que están inducidas por factores medioambientales y existir solo una leve predisposición genética. Otra de las dificultades de su estudio se deriva de la variabilidad de los autoantígenos, así en el síndrome de Sjögren y en el systemic lupus eritematosus (SLE) se han identificado múltiples autoantígenos cuyas funciones específicas en la iniciación, perpetuación y patofisiología de la enfermedad se conocen escasamente. En otras enfermedades autoinmunes, como la artritis reumatoide (AR) y la psoriasis, los autoantígenos principales siguen sin identificarse, a pesar del intenso esfuerzo experimental realizado para ello. La disfunción de las células-B en las enfermedades autoinmunes, parece tener su origen en la presencia de niveles altos de la proteína antiapoptótica Bcl-2, por lo que no experimentan apoptosis y son capaces de producir autoanticuerpos frente a determinadas proteínas que son especialmente abundantes, altamente conservativas, y/o modificadas y pueden formar grandes complejos asociados a menudo con ARN y localizados en los blebs apoptóticos. Los microarrays de proteínas junto con otras técnicas de análisis proteómico de alto rendimiento representan herramientas robustas para el estudio de la patofisiología y la especificidad de la respuesta inmune. Mediante ellos se puede abordar la identificación de la huella antigénica de cada paciente. Esto puede ser un paso crucial para identificar el antígeno patogénico principal, sin olvidar que muchas proteínas o péptidos modificados por factores medioambientales pueden inducir determinadas enfermedades autoinmunes actuando conjuntamente con la base genética individual (22). 5.2.2 Microrrays de citoquinas La aplicación de los FPAs es especialmente prometedora en el estudio clínico de las concentraciones de citoquinas, pues las terapias dirigidas a incrementar o disminuir los niveles de estas proteínas han demostrado ser especialmente útiles en el tratamiento enfermedades autoinmunes, basta mencionar los beneficios derivados de las terapias inhibidoras de IL-1, IL-6, IL-15, y el factor de necrosis tumoral α (TNF-α), en el tratamiento de la AR. En el análisis de las citoquinas es necesaria una alta sensibilidad de detección dadas las bajas concentraciones en que aparecen en los medios biológicos. Se ha aplicado con éxito el método de detección RCA que proporciona un medio excelente de amplificación lineal de la señal, esto ha permitido la detección simultánea de 75 citoquinas, en rango femtomolar de concentraciones, en un time-course de secreción de citoquinas por células dendríticas que confirmaron y aun ampliaron resultados previamente descritos (28). Aunque el RCA es un método ligeramente más elaborado 438 que otros métodos de detección, ha permitido medir más de 180 citoquinas mediante este procedimiento (16). Además de servir de guía en el diseño de nuevas estrategias farmacológicas, la determinación de los niveles de citoquinas mejora el conocimiento de la patogénesis de la enfermedad y ayuda a la identificación de sus marcadores. Por ejemplo, en el SLE, se ha determinado el perfil de los distintos tipos de interferón, y se ha podido comprobar el incremento de los interferones tipo I, en esta patología el papel de las citoquinas es más complejo, se puede mencionar como ejemplo el carácter controvertido del TNF-α como citoquina inflamatoria o inmunomoduladora. En determinadas situaciones fisiológicas en que la función de las citoquinas aparece ambigua, su análisis múltiple puede proporcionar una mejor comprensión de la compleja biología de estos compuestos aplicando a los datos obtenidos técnicas bioinformáticas. 439 BIBLIOGRAFÍA (1) Heller MJ. DNA microarray technology: Devices, systems, and applications. Ann Rev Biomed Eng. 2002; 4: 129-153. (2) Bertone P, Snyder M. Advances in functional protein microarray technology. FEBBS J. 2005; 272: 5400-5411. (3) Uttamchandani M, Yao SQ. Peptide microarrays: next generation biochips for detection, diagnostic and high-throughput screening. Current Pharmaceuthical Desing. 2008; 14: 2428-2438. (4) Ratner DM, Seeberger PH. Carbohydrate microarrays as tools in hiv glycobiology. Curr Pharm Des. 2007; 13: 173-183. (5) Duffner JL, Clemons PA, Koehler AN. A pipeline for ligand discovery using smallmollecule microarrays. Curr Opin Chem Biol. 2007; 11:74-82. (6) Gao C, Mao S, Lo CH, Wirsching P, Lerner RA, Janda KD. Making artificial antibodies: A format for phage display of combinatorial heterodimeric arrays. Proc Natl Acad Sci. USA (1999); 96: 6025-6030. (7) Brody EN, Willis MC, Smith JD, Jayasena S, Zichi D, Gold L. The use of aptamers in large arrays for molecular diagnostic. Mol Diagn. 1999; 4: 381-388. (8) Brody EN, Gold L. Aptamers as therapeutic and diagnostic agents. J. Biotechnol. 2000; 74: 5-13. (9) Wilson DS, Keefe AD, Szostak JW. The use of mRNA display to select high-affinity protein-binding peptides. Proc Natl Acad Sci. USA (2001); 98: 3750-3755. (10) Gunneriusson E, Samuelson P, Ringdahl J, Gronlund H, Nygren PA, Stahl S. Staphylococcal surface display of immunodlobulin A (IgA)- and IgE-specific in vitroselected binding proteins (affibodies) based on Staphylococcus aureus protein A. Appl Environ Microbiol. 1999; 65: 4134-4140. (11) Lopez M, Mallorquin P, Vega M. Aplicaciones de los microarrays y biochips en salud humana. Madrid: Fundación Española para el Desarrollo de la Investigación en Genómica y Proteómica/Fundación General de la Universidad Autónoma de Madrid; 2005. (12) Joos TO, Stoll D, Templin MF. Miniaturised multiplexed immunoassays. Current Op Chem Biol. 2001; 6:76-80. (13) Nolan JP, Sklar LA. Suspensión array technology: evolution of the flat-array paradigm. TRENDS Biotechnol. 2002; 20: 9-12. 440 (14) Templin MF, Stoll D, Schrenk M, Trab PC, Vöhringer CF, Joos TO. Protein microarray technology. TRENDS Biotechnol. 2002; 20: 160-166. (15) Barry R, Soloviev M. Quantitative protein profiling using antibody arrays. Proteomics 2004; 4: 3717-3726. (16) Shao W, Zhou Z, Laroche I, Lu H, Zong Q, Patel DD, Kingsmore S, Piccoli SP. Optimization of rolling-circle amplified protein microarrays for multiplexed protein profiling. J Biomed Biotechnol. 2003; 5: 299-307. (17) Sheehan KM, Calvert VS, Kay EW, Lu Y, Fishman D, Espina V, Aquino J, et al. Use of reverse phase protein microarrays and reference standar development of metastatic ovarian carcinoma. Mol Cell Proteomics 2005; 4: 346-355. (18) Balboni I, Chan S M, Kattah M, Tenenbaum JD, Butte AJ, Utz PJ. Multiplexed Protein array platforms for analysis of autoimmune diseases. Ann Rev Immunol. 2006; 24:391-418. (19) Geho D, Lahar N, Gurnani P, Huebschman M, Herrmann P, Espina V, et al. Pegylated, streptavidin conjugated quantum dots are affective detection elements for reverse-phase protein microarrays. Bioconjug Chem. 2005; 16: 559-566. (20) Charboneau L, Scott H, Chen T, Winters M, Petricoin III EF, Liotta LA, et al. Utility of reverse phase protein arrays: Applications to signalling pathways and human body arrays. Brief Funct Genomic Proteomic 2002; 1: 305-315. (21) Petricoin E, Wulfkuhle J, Espina V, Liotta LA. Clinical proteomics: Revolutionizing disease detection and patient tailoring therapy. J Proteome Res. 2004; 3: 209-217. (22) Krenn V, Petersen I, Häupl T, Koepenik A, Blind C, Dietel M, et al. Array technology and proteomics in autoimmune diseases. Pathol. Res Pract. 2004; 200: 95-103. (23) Saeed A, Sharov V, White J, Li J, Liang W, Bhagabati N, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 2003, 34: 374-378. (24) Luscombe N, Royce T, Bertone P, Echols N, Horak C, Chang J, ExpressYourself: A modular platform for processing and visualizing microarray data. Nucleic Acids Res. 2003; 31: 3477-3482. (25) Gulmann C, Sheehan KM, Kay EW, Liotta LA, Petricoin III EF. Array-based proteomics: mapping of protein circuitries for diagnostic, prognostic, and therapy guidance in cancer. J Pathol. 2006; 208: 595-606. (26) Bertucci F, Birbaum D, Goncalves A. Proteomics of breast cancer. Principles and potential clinical applications. Moll Cell Proteomics 2006; 5: 1772-1786. 441 (27) Wulfkuhle JD, Aquino JA, Calvert VS, Fishman DA, Coukos G, Liotta LA, et al. Signal pathway profiling of ovarian cancer from human tissue specimens using reverse-phase protein microarrays. Proteomics 2003; 3: 2085-2090. (28) Schweitzer B, Roberts S, Grimwade B, Shao WP, Wang MJ, Fu Q, et al. Multiplexed protein profiling on microarrays by rolling-circle amplification. Nat Biotechnol. 2002; 20: 359-365. 442