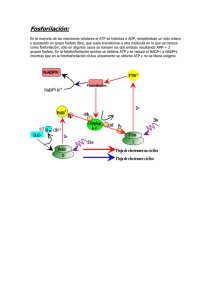
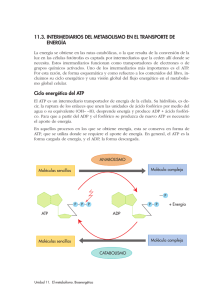
Tesis - Universidad de Colima
Anuncio

Universidad de Colima Facultad de Medicina Centro Universitario de Investigaciones Biomédicas ANÁLISIS FUNCIONAL DE MUTACIONES EN EL RECEPTOR A LAS SULFONILUREAS SUR1 ASOCIADAS CON LA HIPOGLICEMIA HIPERINSULINÉMICA PERSISTENTE DE LA INFANCIA TESIS Para obtener el grado de Doctor en Ciencias Fisiológicas con especialidad en Fisiología Presenta M en C. Myrian Velasco Torres Asesor Dr. Carlos Guillermo Onetti Percello Coasesor Dra. Esperanza García Martínez Colima, Col., Noviembre de 2004 DEDICATORIA A mi familia por su apoyo incondicional. AGRADECIMIENTOS A la Dra. Esperanza García Martínez y al Dr. Carlos Guillermo Onetti Percello, por todas las enseñanzas, el apoyo y la complicidad en la realización de este trabajo. Por ayudarme a crecer en la adversidad; Gracias. A la Dra. Lydia Aguilar-Bryan, por la donación de la línea celular y los plásmidos utilizados en este trabajo y por todo el apoyo para la realización del mismo. A los doctores: Xóchitl Trujillo Trujillo, Marcia Hiriart Urdanivia, Miguel Huerta Viera, Alejandro M. Elizalde Lozano y Clemente Vásquez Jiménez por sus valiosas sugerencias y observaciones. A mis amigos: Isis Velasco, Ada Nory Cabrera, Adriana Hernández, Consuelo Hernández, Georgina Martínez, Yureli Téllez, Gerardo Alcaraz, Armando Obregón, Álvaro Muñoz y Juan José Acevedo, por estar siempre en los momentos difíciles, por su apoyo y su amistad; Gracias. ÍNDICE. INTRODUCCIÓN Secreción de insulina 1 Canales de potasio sensibles a ATP 4 Estructura de los canales K ATP pancreáticos 5 Regulación de la actividad de los canales K ATP 8 Rundown y reactivación 8 Inhibición por ATP 10 Mecanismo de inhibición por ATP 11 Activación por ADP 14 Propiedades farmacológicas de los canales K ATP pancreáticos 15 Bloqueadores de los canales K ATP 15 Abridores de los canales K ATP 17 Hipoglicemia hiperinsulinémica persistente de la infancia (PHHI) 21 Diagnostico clínico 22 Patogénesis de la PHHI 23 Tratamiento de la PHHI 25 ANTECEDENTES. Estudio funcional de mutaciones en el receptor SUR1 asociadas con PHHI 28 Dos nuevas mutaciones asociadas con PHHI, expresadas dominantemente en el receptor SUR1 32 HIPÓTESIS 37 OBJETIVO GENERAL 37 OBJETIVOS ESPECÍFICOS 37 MÉTODOS Expresión heteróloga de canales K ATP en la línea celular COSm6 38 Transfección transitoria 38 Registros electrofisiológicos 39 Pipetas 41 Soluciones 43 Análisis de resultados 43 RESULTADOS La conductancia del canal de potasio sensible a ATP en células COSm6 es resultado de la expresión heteróloga de canales recombinantes 45 La expresión de la subunidad Kir6.2 y SUR1 en células COSm6 produce canales iónicos con propiedades similares a las del canal K ATP pancreático 45 Las mutantes ∆S1387 y D310N forman canales de potasio que son sensibles a ATP 52 Inhibición de la actividad en función de la concentración de ATP 55 Sensibilidad al ATP en ausencia de Mg2+ 55 La actividad de los canales Silvestre y mutados es regulada por el AMP-PNP 57 La mutación ∆S1387 produce la pérdida de la modulación por ADP 61 La mutación D310N produce una alteración en la regulación mediada por ADP 61 Los receptores a sulfonilureas SUR1-∆S1387 y SUR1-D310N conservan el mecanismo de activación por diazóxido 66 DISCUSIÓN. Los canales mutados presentan propiedades biofísicas semejantes al canal silvestre 70 Inhibición por ATP 72 La mutación D310N produce una alteración en la regulación por ATP 73 El ATP en presencia de Mg2+ produce reactivación del canal 74 Modelo del mecanismo de alteración en la respuesta mediada por ATP para el canal mutado Kir6.2/SUR1-D310N 77 El canal mutado Kir6.2/SUR1- ∆S1387 pierde su regulación por ADP 80 Los canales mutados son activados por diazóxido 81 CONCLUSIONES 82 PERSPECTIVAS 83 REFERENCIAS 85 ÍNDICE DE FIGURAS. Figura 1. Liberación de insulina en células beta de humano. 2 Figura 2. Estructura molecular de los canales K ATP pancreáticos. 6 Figura 3. Mutaciones en el receptor SUR1 asociadas con la PHHI. 31 Figura 4. Análisis de expresión de la mutación ∆S1387. 34 Figura 5. Esquema del dispositivo experimental de registro. 40 Figura 6. Células COSm6. 42 Figura 7. Las células COSm6 no presentan una conductancia endógena de K ATP. 46 Figura 8. La coexpresión de SUR1 y Kir6.2 en células COSm6 forman canales con conductancias similares al canal K ATP pancreático nativo. 47 Figura 9. Conductancia unitaria del canal Kir6.2/SUR1-D310N. 49 Figura 10. Conductancia unitaria del canal Kir6.2/SUR1-∆S1387. 50 Figura 11. Los canales KATP pancreáticos reconstituidos presentan actividad eléctrica semejante a los canales nativos. 51 Figura 12. Propiedades farmacológicas del canal K ATP silvestre Expresado en células COSm6. 53 Figura 13. Los canales mutados Kir6.2/SUR1-∆S1387 y Kir6.2/SUR1-D310N son sensibles a la aplicación de ATP. 54 Figura 14. Curva dosis respuesta al ATP en presencia de Mg2+. 56 Figura 15. Curva dosis respuesta al ATP en ausencia de Mg2+. 58 Figura 16. Efecto del análogo de ATP no hidrolizable el AMP-PNP sobre la actividad del canal silvestre. Figura 17. Curva dosis respuesta al AMP-PNP en presencia de Mg2+. Figura 18. Efecto del ADP sobre el canal silvestre Kir6.2/SUR1 59 60 expresados en células COSm6. 62 Figura 19. El canal mutado Kir6.2/SUR1-∆S1387 pierde su sensibilidad al ADP. 63 Figura 20. El canal Kir6.2/SUR1-D310N tiene una respuesta atípica al ADP. 64 Figura 21. El canal Kir6.2/SUR1-D310N disminuye su afinidad por ADP. 65 Figura 22. Efecto de diazóxido sobre la actividad del canal Kir6.2/SUR1-∆S1387. 67 Figura 23. Efecto del diazóxido sobre la actividad del canal Kir6.2/SUR1-D310N. 68 Figura 24. Esquema de un modelo de la respuesta mediada por MgATP para el canal mutado Kir6.2/SUR1-D310N. 79 TABLAS. Tabla 1. Propiedades farmacológicas de los canales K ATP. 18 Tabla 2. Resumen de los valores de la constante de disociación y el coeficiente de Hill. 69 RESUMEN Los canales KATP del páncreas están formados por un canal con rectificación entrante (Kir6.2) y un receptor con alta afinidad a las sulfonilureas (SUR1). Estos canales tienen una función primordial en la regulación de la secreción de insulina. Defectos genéticos en el receptor SUR1 y el canal Kir6.2 producen una enfermedad conocida como hipoglicemia hiperinsulinémica persistente de la infancia (PHHI), que se caracteriza por un incremento en la secreción de insulina en presencia de concentración bajas de glucosa. Este estudio presenta un análisis funcional de 2 nuevas mutaciones (∆S1387 y D310N) en el receptor SUR1, asociadas con la PHHI. Plásmidos que contenían las mutaciones D310N y ∆S1387, fueron subclonados en el vector pECE y cotransfectadas con el canal Kir6.2 silvestre, más la GFP, en células COSm6. Registros de la corriente unitaria utilizando la técnica de patch clamp en la configuración de inside-out fueron realizados dos días después de la transfección. Los resultados obtenidos muestran que ambos mutantes se insertan en la membrana y producen canales funcionales sensibles a ATP en presencia y ausencia de Mg2+. Las constantes de disociación (K D) obtenidas de la curva dosisrespuesta fueron de 22, 25 y 42 µM en presencia de Mg2+ y 24, 22 y 16 µM en ausencia de Mg2+ para el canal silvestre y los mutados Kir6.2/SUR1-∆S1387 y Kir6.2/SUR1-D310N respectivamente. Para explorar la causa de la reducción en la sensibilidad al ATP en presencia de magnesio para la mutante D310N se probó el AMP-PNP, un análogo no hidrolizable de ATP. La KD para el canal silvestre y el mutado D310N fueron similares (19 y 20 µM respectivamente). Además, el mutante ∆S1387 no respondió a la estimulación con ADP (0.5 mM), mientras que la mutante D310N presentó una respuesta atípica al ADP en presencia de Mg2+. Ambos canales mutados mostraron un aumento en su actividad en respuesta al diazóxido (0.1 mM), un fármaco hipoglucemiante que abre los canales KATP pancreáticos y detienen la secreción de insulina, y es el principal fármaco utilizado en el tratamiento de pacientes con la PHHI. Nuestros resultados contribuyen al conocimiento de los mecanismos que regulan al canal KATP pancreático y su alteración en condiciones fisiopatológicas. ABSTRACT Adenosine triphosphate-sensitive potassium channels (K ATP) from pancreas, are formed by four inward rectifier (K IR6.2) potassium channel subunits and four highaffinity sulfonylurea (SUR1) receptors. These channels play a key role in regulating insulin secretion. Genetic defects in the subunits Kir6.2 and SUR1 are associated with a disease known as persistent hyperinsulinemic hypoglycaemia syndrome of infancy (PHHI), a disorder characterized by inappropriate secretion of insulin despite severe hypoglycaemia. We analyzed the functional consequences of two novel dominant mutations (D310N y ∆S1387) in SUR1 associated with PHHI. SUR1 plasmids containing the mutations D310N and ∆S1387 were subcloned in pECE vector and cotransfected with wild type Kir6.2, plus GFP, in COSm6 cells. Single channel inside-out patch clamp recording was performed two days after transfection. Both SUR1-∆S1387 and SUR1-D310N produced functional channels when coexpressed with wild type Kir6.2, and they were ATP sensitive. The dissociation constant (K D) values obtained from the dose-response curves were of 22, 25, and 42 µM in the presence of Mg2+ and 24, 22 and 16 µM in the absence of Mg2+ for the wild type and the mutants Kir6.2/SUR1-∆S1387 and Kir6.2/SUR1-D310N respectively. To further explore the reduced sensitivity to ATP of the mutant SUR1-D310N when tested in the presence of Mg2+, a non-hydrolysable analogue of ATP, AMP-PNP, was tested. We obtained similar KD values for Kir6.2/SUR1 and Kir6.2/SUR1-D310N channels (19 µM and 20 µM respectively). Mutant SUR1-∆S1387 channels were not stimulated by ADP-Mg2+ (up to 0.5 mM), whereas mutant SUR1-D310N channels displayed an atypical response to the ADP in the presence of Mg2+. Both mutant channels showed an increase in their activity when exposed to diazoxide (0.1 mM), a hypoglycaemic drug that opens pancreatic KATP channels and stops insulin secretion; this observation has clinical relevance because diazoxide is commonly used in the treatment of patient with PHHI. Overall, our results contribute to a better knowledge of the mechanisms that regulate the pancreatic KATP channel and the alterations in its ATP/ADP sensitivity during pathophysiological conditions. INTRODUCCIÓN. El páncreas es una glándula mixta, que posee un componente exocrino y uno endocrino. La actividad endocrina se lleva a cabo por 2 millones de células especializadas localizadas en los Islotes de Langerhans que representan el 1 % del peso total del páncreas. Los islotes de Langerhans están formados por 3 tipos de células que secretan diferentes hormonas: las células alfa secretan glucagon, las células beta secretan insulina y las células delta secretan somatostatina; cada una de estas hormonas cumple funciones importantes en nuestro organismo. La insulina y el glucagon tienen funciones importantes en la regulación del metabolismo intermedio de carbohidratos, proteínas y lípidos. Secreción de insulina Las células beta son eléctricamente activas y pueden liberar insulina cuando se exponen (ver figura 1) a concentraciones séricas de glucosa mayores a 100 mg/dl (5.5 mM). El potencial de membrana en reposo en una célula beta es determinado por los canales de potasio sensibles a ATP (K ATP) y tiene un valor cercano a -70 mV a concentraciones séricas basales de glucosa (80 a 120 mg/dl). Las células beta del páncreas transportan la glucosa al interior celular, a través de una proteína transportadora de glucosa (GLUT2), tanto a concentraciones basales, como cuando la concentración extracelular de glucosa aumenta por arriba del nivel basal (Bell et al., 1990). El metabolismo de la glucosa se inicia en el citoplasma mediante la enzima glucocinasa (GK), que forma glucosa 6 fosfato; esta molécula inicia la glucólisis, por lo que la actividad de la glucocinasa determina la velocidad con que se metaboliza la glucosa a través de la glucólisis (De Lonlay et al., 2002; Dunne et al., 1999). La glucólisis es la vía de degradación anaeróbica de la glucosa en ácido láctico, este producto entra a las mitocondrias donde forma acetilcoenzima A (acetil CoA), y a través de esta molécula se inicia el ciclo de los ácidos tricarboxílicos. En esta ruta aeróbica, se forman dos moléculas de dióxido de carbono (CO2) y cuatro pares de átomos de hidrógeno; estos últimos se 1 2 incorporan a la cadena respiratoria, constituida por una serie de transportadores electrónicos, el proceso subsiguiente corresponde a la fosforilación de la difosfato de adenosina (ADP) en trifosfato de adenosina (ATP) en el proceso llamado fosforilación oxidativa (Lehninger, 1982). La secreción de insulina también es estimulada por otra vía, en la cual se incrementa la oxidación de glutamato por la enzima glutamato deshidrogenasa (GDH) formando alfa ceto glutarato en la matriz mitocondrial, el producto entra al ciclo de los ácidos tricarboxílicos y la fosforilación oxidativa para formar ATP (Missner et al., 1999). El resultado final de ambas vías es un aumento en la relación ATP/ADP citosólica, el aumento de ATP produce el cierre de los canales KATP, lo cual conduce a una despolarización de la membrana y la subsecuente apertura de canales de Ca+2 dependientes de voltaje tipo L. El aumento en la concentración de calcio inicia la secreción de gránulos que contienen insulina, mediante un proceso de exocitosis (Aguilar-Bryan et al., 2001; De Lonlay et al., 2002; Dunne et al., 1999). La insulina es una hormona polipeptídica que ejerce innumerables acciones biológicas, entre las cuales se incluyen la regulación de la captura y uso de glucosa, de la síntesis de proteínas y lípidos, y de la transcripción de genes (Rosen, 1987). Esta es la principal hormona involucrada en la homeostasis de la glucosa, así que la pérdida parcial o absoluta de esta hormona conduce a un daño en el metabolismo de la glucosa, que puede resultar en una disminución o un aumento en la concentración de glucosa en sangre (Aguilar-Bryan et al., 2001; Srivastava, 1998). Estas alteraciones en el metabolismo de la glucosa producen enfermedades como la diabetes mellitus, un síndrome en el que se altera la acción de la insulina en la captación de glucosa, lo cual produce un aumento en la concentración de glucosa en sangre. Por otra parte, un aumento en la secreción de insulina puede resultar en hipoglucemia. En adultos, la hipoglucemia ha sido asociada con hiperinsulinismo debido a la presencia de un tumor benigno en las células beta del páncreas (insulinoma). En los casos de hipoglucemia asociada a un hiperinsulinismo no tumoral, no se conocen los factores que lo originan. Fue en los últimos seis años que el estudio de las bases genéticas de un síndrome generado por una alteración en la secreción de insulina, llamado 3 hiperinsulinismo congénito (HI) o hipoglucemia hiperinsulinémica persistente de la infancia (PHHI), comenzó a proporcionar un entendimiento de la regulación normal en la secreción de insulina, y el tipo de defectos metabólicos que produce este desorden (Aguilar-Bryan et al., 2001). El síndrome de PHHI se caracteriza por una elevada secreción de insulina aun en presencia de concentraciones bajas de glucosa. Fue hasta 1995 que se determinó la patogénesis de la PHHI y se sabe que la causa más común de este síndrome es la mutación en los genes que codifican tanto para el receptor a las sulfonilureas SUR1 y el canal de potasio con rectificación entrante Kir6.2, ambas proteínas forman los canales KATP pancreáticos (Thomas et al., 1995b). Canales de potasio sensibles a ATP Los canales de potasio sensibles a ATP (K ATP) fueron originalmente descubiertos en miocitos cardíacos por Noma en 1983; posteriormente fueron descritos en otros tejidos entre los que se incluyen el músculo esquelético (Spruce et al., 1985), músculo liso (Standen et al., 1989), cerebro (Ashford et al., 1988), glándula pituitaria (Bernardi et al., 1993), riñón (Hunter and Giebisch, 1988), en las células beta del páncreas (Ashcroft et al., 1984; Cook et al., 1984; Rorsman and Trube, 1985), y organelos intracelulares como la mitocondria (Inoue et al., 1991). Estos canales deben su nombre a la regulación inhibitoria mediada por un aumento en la relación ATP/ADP citosólica. Se dice que estos canales ligan el estado metabólico y la excitabilidad de las células, precisamente porque al aumentar el metabolismo de la glucosa y por lo tanto la concentración de ATP, estos canales se cierran y producen una despolarización de la membrana. Por esta razón estos canales juegan un importante papel en la respuesta de varios tejidos cuando se encuentran en un estado metabólico alterado como son hiperglucemia, hipoglucemia, isquemia e hipoxia (Yokoshiki et al., 1998). Los canales KATP regulan diferentes funciones como la excitabilidad de neuronas y músculo esquelético, la recaptura de potasio en el epitelio renal, además de la secreción de insulina en las células beta del páncreas (Ashcroft et al., 1988; Cook et al., 1984; Misler and Giebisch, 1992; Terzic et al., 1995). El papel funcional 4 de los canales KATP ha sido ampliamente caracterizado en las células beta del páncreas, el mecanismo de secreción de insulina inducida por glucosa que se muestra en la Figura 1, es ampliamente aceptado; de acuerdo con este modelo los canales KATP actúan como sensores de los niveles citosólicos de ATP y ADP (Boyd, 1998). Estructura molecular de los canales K ATP pancreáticos Como hemos visto anteriormente los canales KATP cumplen diferentes funciones en diferentes tejidos y esto se asocia con la distribución diferencial de isoformas moleculares de las proteínas que forman el canal funcional. Los canales KATP son complejos hetero-octaméricos formados por dos subunidades distintas. Una de estas subunidades es un canal de potasio que pertenece a la familia de rectificadores entrantes Kir6.x; este tipo de canal está formado por dos dominios transmembrana unidos por una secuencia de aminoácidos (Glicina-FenilalaninaGlicina) altamente conservada que forma el filtro de selectividad (Figura 2). Existen dos subtipos denominados Kir6.1 y Kir6.2, que se diferencian por su conductancia unitaria. La otra subunidad es un receptor a las sulfonilureas SUR, el cual es miembro de la familia de proteínas que unen ATP, y del cual se conocen dos isoformas: el receptor SUR1 y el receptor SUR2 (ahora renombrado como SUR2A) (Aguilar-Bryan et al.,1995; Inagaki et al., 1996). Además existen variantes de SUR2A derivados por procesamiento postranscripcional de RNA (Splicing) como el SUR2B (Aguilar-Bryan and Bryan, 1999; Ashcroft and Gribble, 1998; Seino, 1999). Las subunidades Kir6.x y SUR se organizan en un octámero, donde 4 subunidades del canal Kir6.x forman el poro del canal y se unen a 4 subunidades del receptor SUR (Kir6.x/SUR)4, que actúa como una subunidad reguladora; ambas subunidades son necesarias para formar un canal KATP funcional (Aguilar-Bryan et al., 1995; 1998). Las propiedades biofísicas y farmacológicas de los diferentes subtipos de canales KATP están determinadas por el tipo de subunidades (Kir6.x y SUR) que los forman. Las subunidades que forman los canales KATP en diferentes tejidos ya han sido identificadas; los canales K ATP cardíacos están formados por SUR2A/Kir6.2 5 6 (Inagaki et al., 1996), mientras que los del músculo liso son del tipo SUR2B/Kir6.2 (Isomoto et al., 1996). Los canales KATP pancreáticos están formados por 4 subunidades del canal tipo Kir6.2 y por 4 subunidades del receptor SUR1. El receptor SUR1 tiene una alta afinidad a las sulfonilureas (K D ~ 10 nM para el receptor SUR1 de hamster) (AguilarBryan et al., 1995). La proteína consta de 3 dominios transmembrana llamados TMD0, TMD1 y TMD2 constituidos por cinco, seis, y seis segmentos transmembrana, (Figura 2) respectivamente. En el espacio intracelular el receptor SUR contiene dos sitios de unión a nucleótidos llamados NBF1 y NBF2 (Nucleotide Binding Fold); cada sitio de unión a nucleótidos contiene un motivo A y un motivo B y una secuencia altamente conservada que los une. El NBF1 está ubicado entre el TMD1 y TMD2, mientras que el NBF2 está ubicado en el segmento carboxilo terminal (Conti et al., 2001), este receptor también contiene dos sitios de glicosilación ubicados hacia el lado extracelular, uno en el segmento amino terminal y el otro entre el séptimo y octavo segmento transmembrana (Tusnády et al., 1997). El gen ABCC8 (Online Mendelian Inheritance in Man: OMIM # 600509) que codifica para el receptor SUR1, fue clonado y se sabe que este gen comprende 39 exones y ocupa un intervalo mayor a 100 Kb del DNA genómico. El receptor SUR1 es una proteína de 1581 aminoácidos con un peso molecular de 177 KDa. Los análisis de homología han mostrado que este gen es un miembro de la superfamilia de proteínas que unen de ATP (ATP-binding cassette o ABC), por lo tanto guardan similitud con la familia del regulador transmembrana de la conductancia de la fibrosis cística (CFTR) y con la familia de proteínas resistentes a diferentes drogas (MDPs) (Aguilar-Bryan et al., 1995). El gen KCNJ11 (OMIM # 600937) que codifica para el canal Kir6.2 fue identificado por Inagaki y colaboradores en 1995, este gen KCNJ11 consiste de un solo exón que codifica para una proteína de 390 amino ácidos (Meissner et al., 1999). Ambos genes ABCC8 y KCNJ11 son contiguos en la posición 15.1 del brazo corto del cromosoma 11 (Inagaki et al., 1995). 7 Regulación de la actividad de los canales K ATP Como se mencionó anteriormente, los canales KATP deben su nombre a la regulación mediada por los nucleótidos ATP y ADP. Los registros eléctricos realizados en células beta intactas mientras el tejido era bañado con soluciones que contenían concentraciones crecientes de glucosa, mostraron que el aumento en la liberación de insulina ocurría paralelamente a la reducción en la actividad de los canales KATP (Ashcroft et al., 1984; Misler et al., 1986). Esto experimentos ilustran que existe un acoplamiento entre el metabolismo de la glucosa, (ya que aumenta o disminuye el ATP citosólico) y la regulación de la actividad eléctrica de los canales KATP. El ATP y el ADP tienen tres efectos principales en la actividad de los canales KATP: 1) una reactivación (proceso llamado refreshment), 2) una inhibición y 3) una activación. Rundown y reactivación (refreshment) Cuando se aísla un parche de membrana que contiene canales KATP y se coloca en una solución libre de ATP los canales se abren, esto fue descrito tanto en miocitos cardíacos (Noma, 1983) como en células beta del páncreas (Cook and Hales, 1984; Trube and Hescheler, 1983). En esta condición los canales pierden gradualmente su actividad en función del tiempo, en un proceso llamado “rundown”. La actividad de los canales que caen en rundown, puede ser restablecida mediante una breve aplicación de MgATP en concentraciones milimolares; este proceso se denominó “refreshment” o reactivación. Se han propuesto varios mecanismos para explicar tanto el rundown como la reactivación de los canales KATP. En 1993 Olense y Bundgaard examinaron el papel del ATP en la regulación de los canales de potasio con rectificación entrante en células endoteliales; de acuerdo con sus resultados la reactivación de los canales una vez separado un parche de membrana, se logró al aplicar ATP (1 mM) en la cara interna de la membrana, esta reactivación no se observó cuando el parche fue bañado con un análogo no hidrolizable de ATP, el ATP-gama S, esto llevó a la conclusión que la reactivación depende de alguna manera de la fosforilación del canal. Al inhibir la síntesis de ATP mediante el uso de inhibidores metabólicos 8 (cianida e iodoacetato), se produjo el cierre de los canales en un lapso de tiempo de 1 a 6 minutos, mientras que cuando aplicaron simultáneamente iodoacetato y cianida en presencia de ATP la actividad de los canales no se alteró, pero al quitar el ATP la actividad comenzó a disminuir. Con estos resultados se propuso que el funcionamiento normal de los canales de potasio con rectificación entrante es dependiente de ATP, y que el rundown y la reactivación involucran un mecanismo que depende de fosforilación y desfosforilación (Olesen and Bundgaard, 1993). Durante el ensamble del citoesqueleto se lleva a cabo la hidrólisis del ATP (Korn et al., 1987), y se ha demostrado que varios canales en la membrana son regulados por el citoesqueleto (Galli and Defelice, 1994; Jonson and Byerly, 1994; Wang et al., 1994). Al remover el ATP o aumentar la concentración de Ca2+ se produce una alteración en los filamentos del citoesqueleto, pero también se produce el rundown de los canales KATP cardíacos (Bretscher, 1991). Con estos antecedentes, Furukawa y colaboradores (1996) propusieron un mecanismo que relaciona las proteínas del citoesqueleto con los procesos de rundown y reactivación inducida por MgATP en los canales KATP. Para esto se perturbó selectivamente la cara interna de parches de membrana de miocitos ventriculares, despolimerizando los filamentos de actina usando agentes como la citocalasina y la desoxiribonucleasa-I, con este tratamiento se aceleró el tiempo en el que los canales entran en rundown. Mientras que al utilizar la falloidina, la cual estabiliza los filamentos de actina o el fosfatidilinositol difosfato (PIP 2) que inhibe las proteínas que separan los filamentos de actina F, se inhibió el rundown espontáneo y/o el inducido por Ca2+. Cuando el rundown fue inducido por citocalasina D o por una exposición a altas concentraciones de Ca2+, la actividad del canal no pudo ser restablecida al aplicar MgATP, pero la aplicación de filamentos de actina F en presencia de MgATP si pudo restablecer la actividad. Con estos resultados se propuso que tanto el rundown y la reactivación de los canales, puede estar siendo influenciada por el ensamble y desensamble de los filamentos de actina del citoesqueleto, el cual proporciona un mecanismo regulador de estos canales. Recientemente se ha propuesto una nueva hipótesis para explicar el rundown, que involucra el extremo carboxilo de la subunidad formadora del poro (Kir6.2) de los 9 canales KATP. En primer lugar se demostró que los fosfolípidos aniónicos activan los canales KATP de las células beta, miocitos cardíacos y células del músculo esquelético. La capacidad de estos fosfolípidos en activar los canales fue proporcional al número de cargas negativas en la cabeza de los fosfolípidos aniónicos, cuando se utilizaron cationes polivalentes, los cuales capturan cargas negativas, el efecto de activación fue inhibido. Además el tratamiento enzimático con fosfolipasas que reducen la carga en los lípidos, también reduce o elimina el efecto de activación. Estos resultados sugieren que los fosfolípidos intactos con carga negativa son importantes para la activación de los canales K ATP. Se ha reportado que el fosfatidilinositol-4, 5-bifosfato (PIP 2) activa los canales KATP cardíacos nativos y reconstituidos (Hilgemann & Ball, 1996; Fan & Makielski, 1997). En la membrana plasmática PIP 2 es producido por la consecutiva fosforilación de fosfatidilinositol (PI) y fosfatidilinositol-4-monofosfato (PIP). Se propuso y se demostró que PIP 2 es la entidad que está regulando la recuperación dependiente de MgATP de los canales KATP. Esto fue demostrado utilizando la wortmannina un inhibidor de las cinasas lipídicas (cinasa PI3 y PI4), la cual bloqueó la recuperación dependiente de MgATP (Xie et al., 1999). Previamente se había reportado que mutaciones en el extremo carboxilo terminal de Kir6.2 acelera el tiempo en que los canales entran en rundown y reduce el efecto activador de los fosfolípidos, con lo cual se sugirió la participación de esta región del canal en la activación mediada por fosfolípidos (Fan and Makielski, 1997). Recientemente se ha demostrado que el PIP 2 interactúa con el extremo carboxilo terminal de Kir6.2 produciendo una marcada activación y reduciendo la sensibilidad al ATP (Baukrowitz and Fakler, 2000). Inhibición por ATP La aplicación de ATP (con o sin Mg2+), en la cara intracelular de la membrana inhibe los canales KATP. La inhibición del 50 % de la actividad (IC 50) tiene valores muy bajos en el rango de 5 a 10 µM tanto para los canales KATP pancreáticos nativos como para los canales Kir6.2/SUR1 reconstituidos en sistemas heterólogos (Inagaki et al., 1995). En condiciones basales del metabolismo la concentración citosólica del 10 ATP es cercana a 1 mM, considerando la IC 50 para los canales KATP pancreáticos bajo estas condiciones, se predice que aproximadamente el 99 % de los canales están cerrados. La alta sensibilidad al ATP ha sido motivo de confusión cuando se considera la idea, de que las fluctuaciones en la concentración citosólica del ATP regula de la actividad de los canales KATP. Varios mecanismos se han propuesto para explicar como es que los canales KATP son regulados por los cambios en la concentración de ATP citosólico, aún cuando la concentración de ATP citosólico en condiciones basales es alta y la sensibilidad al ATP es también alta (Ashcroft and Kakei, 1989). Se ha demostrado que el efecto inhibitorio de MgATP es más débil que el de ATP 4- sobre el canal KATP pancreático. En 1989 Ashcroft y Kakei propusieron que una fracción importante de ATP citosólico puede estar unido a Mg2+ en el espacio intracelular; esto significa que la concentración de ATP 4- puede ser cercana a la IC 50 determinada para los canales KATP, lo cual permite que las variaciones en ATP puedan regular la actividad de estos canales (Álvarez-Leefmans et al., 1987; Ashcroft and Kakei, 1989; Murphy, 1993). Mecanismo de inhibición por ATP Inicialmente se pensaba que el receptor SUR1 era la subunidad que mediaba el efecto de nucleótidos sobre el KATP ya que contiene dos sitios de unión a nucleótidos, NBF1 y NBF2. La contribución relativa de la subunidad Kir6.2 o del SUR1 era difícil de discernir ya que ambas subunidades SUR1 y Kir6.2 son normalmente requeridas para formar un canal KATP pancreático funcional. Recientemente se mostró que al truncar una región de 26 o 35 aminoácidos (∆C26 y ∆C35) en el segmento carboxilo terminal del canal Kir6.2 se genera una proteína que forma canales funcionales en ausencia de SUR1 (Kir6.2-∆C26 y Kir6.2-∆C35) que llegan a la membrana plasmática y conducen una corriente iónica (Babenko et al., 1999c; Tucker et al., 1997). La actividad del canal Kir6.2-∆C26, es similar al del canal Kir6.2/SUR1, lo cual demostró que SUR1 no contribuye a la formación del poro del canal. Además la aplicación de ATP en ausencia de Mg2+ inhibe la actividad de los canales Kir6.2-∆C26, lo que sugiere que el ATP ejerce un efecto inhibitorio al interactuar directamente con el canal Kir6.2 (Babenko et al., 1999a; Drain et al., 11 1999; Shyng et al., 1997b; Tanabe et al., 1999, 2000; Tucker et al., 1997). A pesar de estos hallazgos la sensibilidad del canal Kir6.2-∆C26 es mucho menor (aproximadamente 10 a 15 veces) que la del canal reconstituido por ambas subunidades Kir6.2 y SUR1, esto sugirió que aunque el ATP se una al canal Kir6.2, la subunidad SUR1 modula la inhibición por ATP (Babenko et al., 1999a, 1999c; Drain et al., 1998; Tucker et al., 1997). La primera evidencia de unión de ATP en el canal Kir6.2 se obtuvo al realizarse un ensayo de fotoafinidad utilizando como marcador el 8-azido-[γ32P] ATP (Tanabe et al., 1999, 2000). Aunque el sitio preciso de unión de ATP aun no ha sido determinado, se ha demostrado que mutaciones en Kir6.2 reducen el efecto inhibitorio de ATP (Drain et al., 1998; Koster et al., 1999; Proks et al., 1999; Reimann et al., 1999; Shyng et al., 1997; Trapp et al., 1998a; Tucker et al., 1997, 1998). Estas mutaciones pueden alterar tanto la afinidad del ATP por su sitio de unión, como la transducción de la señal después de que el ATP se ha unido o bien podría alterar la capacidad del canal para cerrarse. Se ha mostrado que la mutación de una cisteina por una serina en la posición 166 (C166S), localizada al final del segundo segmento transmembrana (TM2) del canal Kir6.2, y mutaciones en la región adyacente al segmento carboxilo terminal, tienen un efecto marcado en el mecanismo de compuerta del canal Kir6.2, y esto reduce la sensibilidad al ATP (Trapp et al., 1998a). Aunque en un principio se descartó la participación del segmento amino terminal en el mecanismo de unión de ATP, recientemente se encontró que la mutación de una arginina por una glicina en la posición 50 (R50G) en el segmento amino terminal, así como la mutación de una lisina por glutamina en la posición 185 (K185Q) en el segmento carboxilo terminal, disminuyen la sensibilidad al ATP, y sugirió que tanto la región amino como carboxilo terminal del canal Kir6.2 interactúan de manera conjunta para inhibir la actividad del canal por ATP (Tanabe et al., 2000). Trapp y colaboradores (2003), con base en datos de cristalografía (Nishida and MacKinnon, 2002) propusieron un mecanismo de inhibición por ATP, el cual sugiere la interacción física de los extremos amino y carboxilo del canal de potasio Kir6.2. De acuerdo con estos autores, los extremos amino y carboxilo terminal interactúan en regiones en las cuales se encuentran residuos importantes que 12 coordinan la unión de la molécula de ATP al canal Kir6.2, tales como una arginina en la posición 50 (R50) en el extremo amino terminal, que coordina la unión del fosfato γ del ATP al canal Kir6.2, y el residuo K185 en el extremo carboxilo terminal interactúa con el fosfato β de la molécula de ATP. Puesto que estos estudios se llevaron a cabo en ausencia de ATP, los dominios amino y carboxilo terminal parecen interactuar en ausencia de nucleótidos y es probable que cuando el ATP se encuentra presente pueda modificar esta interacción, produciendo así el cambio conformacional que se traduce a los dominios transmembranales y que tiene como resultado final el cierre del canal. Se sabe que existe un solo sitio de unión de ATP por cada monómero de Kir6.2, por lo tanto hay 4 sitios de unión a ATP por cada canal KATP, pero se ha mostrado que la unión de una sola molécula de ATP es suficiente para inducir el cierre del canal (Markworth et al., 2000). Además el efecto inhibitorio de ATP parece ser resultado de la unión directa del nucleótido y no requiere la hidrólisis de ATP ya que no requiere Mg2+ y el efecto inhibitorio de este nucleótido puede ser imitado por el AMP y ADP en ausencia de Mg2+ , así como por análogos de ATP no hidrolizables (Shyng et al., 1997). El efecto inhibitorio del nucleótido ATP sobre los canales KATP, ocurre por la unión de ATP en un sitio de unión aún no determinado en el canal Kir6.2. En el receptor SUR1 se ha demostrado que el ATP se une a NBF1 (Bokvist et al., 1991; Tucker et al., 1997) y por ello se llama sitio de inhibición, mientras que el efecto activador de MgADP involucra la participación de NBF1 y NBF2 (Gribble et al., 1997, Shyng et al., 1997b). Las propiedades de estos dos sitios de unión difieren en sus requerimientos por el catión divalente Mg2+ el cual es necesario para activar el canal, pero no es necesario para inhibirlo (Ashcroft and Kakei, 1989; Bokvist et al., 1991; Findlay, 1987). Ya que se ha demostrado que diferentes nucleótidos (MgGTP, MgADP y MgGDP) en presencia de Mg2+ pueden activar los canales KATP, recientemente se propuso que el MgATP puede activar estos canales (Trapp et al., 1997). Esta idea fue apoyada al demostrarse, que la sensibilidad al ATP de los canales KATP pancreáticos nativos aumentaba al removerse los iones Mg2+ (Dunne and Petersen, 13 1986; Kakei et al., 1986). Lo anterior se interpretó como que los canales eran inhibidos por la base libre de ATP (ATP -4) y que la concentración de esta base libre disminuía en presencia de Mg2+. Posteriormente se sugirió que al igual que MgADP, el MgATP puede tener un efecto estimulante en la actividad de los canales KATP y por lo tanto producir una disminución aparente en la sensibilidad al ATP en presencia de magnesio. En 1998 Gribble y colaboradores demostraron que el MgATP tiene un efecto estimulante sobre el canal KATP pancreático, ya que tanto el canal KATP pancreático silvestre (Kir6.2/SUR1), así como el canal Kir6.2-R50G/SUR1, que presentaba la mutación de una arginina por una glicina en la posición 50 en el canal Kir6.2, fueron activados por MgATP y por el MgADP. Como se mencionó anteriormente, la arginina en la posición 50 juega un papel importante en la unión de ATP al canal Kir6.2, así que el efecto activador de MgATP no parece depender de su unión al canal Kir6.2. Sin embargo, la activación con MgATP, fue revertida cuando se realizaron mutaciones en los sitios de unión a nucleótidos NBF1 y NBF2 del receptor SUR1. Con estos resultados se sugiere que al igual que MgADP, el MgATP estimula la actividad de los canales KATP por interacción con los sitios de unión a nucleótidos en el receptor SUR1. Activación por ADP Como se mencionó anteriormente, el ADP en presencia de Mg2+ activa al canal KATP por interactuar con los sitios de unión a nucleótidos en el receptor SUR1 (Gribble et al., 1997; Shyng et al., 1997b). Se ha demostrado que SUR1, une ATP en el sitio de unión a nucleótidos NBF1 y MgADP en el sitio de unión a nucleótidos NBF2. La activación de MgADP ocurre ya sea por la unión directa de este nucleótido o por la hidrólisis de MgATP en sitio de unión a nucleótidos NBF2. También se ha propuesto que la unión directa de MgADP o la hidrólisis de MgATP en NBF2, induce un cambio conformacional en NBF2, que resulta en la estabilización de la molécula de ATP unido en el sitio NBF1 (Ueda et al., 1997; Ueda et al., 1999a). Se ha demostrado mediante estudios bioquímicos, que el sitio de unión a nucleótidos NBF1 de SUR1 une fuertemente 8-azido-ATP y que esta unión es 14 independiente de la concentración de Mg2+, mientras que el sitio de unión a nucleótidos NBF2 une débilmente 8-azido-ATP de una manera dependiente de Mg2+, sugiriendo que el sitio NBF2 de SUR tiene actividad ATPasa, mientras que NBF1 tiene poca o ninguna (Ueda et al., 1999b). Basándose en estos hallazgos Ueda y colaboradores (1999b) propusieron un modelo para explicar la activación por ADP, que sugiere lo siguiente: cuando la relación ATP/ADP disminuye, ATP se une a NBF1 y MgADP se une a NBF2; en este estado conformacional, la interacción del receptor SUR1 con el canal Kir6.2, reduce la afinidad de Kir6.2 por ATP provocando la apertura de los canales KATP. En contraste cuando la relación ATP/ADP aumenta, el decremento en MgADP induce una disociación del MgADP unido a NBF2, y esto desestabiliza y produce la liberación del ATP unido en el sitio de unión a nucleótidos NBF1, aumentando la afinidad de Kir6.2 por ATP y cerrando el canal K ATP. Propiedades farmacológicas de los canales K ATP pancreáticos Bloqueadores de los canales KATP. Además de la regulación por nucleótidos intracelulares, los canales KATP son modulados por una gran variedad de agentes farmacológicos que pueden actuar ya sea bloqueando o activando el canal. Dentro de los fármacos que pueden bloquear la actividad se encuentra el grupo de las sulfonilureas, que son fármacos que se desarrollaron originalmente a partir de las sulfonamidas antibacterianas; las sulfonilureas producen hipoglucemia cuando se administran a dosis elevadas (Loubatieres, 1957). En el grupo de la primera generación de sulfonilureas se encuentra la tolbutamida, la gliclazida, y la tolazamida, entre otros; todos ellos tienen un grupo arilo simple substituido y una urea lipofílica substituida. La segunda generación de las sulfonilureas conserva el grupo arilsulfonilurea, pero contiene además un grupo etilamina acil aromático; dentro de este grupo se encuentran principalmente la glibenclamida (Gliburida), la glipizida y la glisindamida (Geisen et al., 1985). La tolbutamida y glibenclamida son sulfonilureas utilizadas en el tratamiento de diabetes mellitus no dependiente de insulina (Sturgess et al., 1985). El bloqueo de los canales KATP por las sulfonilureas no ocurre por una obstrucción del poro de estos canales, ni afectan su conductancia unitaria o la 15 actividad cinética dentro de las ráfagas (intraráfagas). Las sulfonilureas bloquean estos canales por acortar la duración de las ráfagas y alargar el intervalo entre las ráfagas (interráfagas), lo cual hace que los canales se mantengan en el estado cerrado por tiempo prolongado (Gillis et al., 1989). Las sulfonilureas se unen a receptores en una gran variedad de tejidos (Aguilar-Bryan and Bryan, 1999; Chutkow et al., 1999; Panten et al., 1992). La afinidad con que las sulfonilureas se unen a sus receptores SUR1 y SUR2A es diferente. Un receptor del tipo SUR1 como el que forma los canales KATP pancreáticos y cerebrales es bloqueado con una alta afinidad por tolbutamida (K D del orden micromolar (µM)) y por glibenclamida (K D del orden nanomolar (nM)). En cambio, la afinidad con que se unen estas sulfonilureas a un receptor tipo SUR2, como el que forma los canales KATP cardíacos es mucho menor (Dorschner et al., 1999). Además se ha demostrado que el bloqueo con glibenclamida es rápidamente reversible para los canales formados por SUR2A, mientras que para los receptores SUR1 el bloqueo por glibenclamida es prácticamente irreversible (Gribble et al., 1998b; Zunkler et al., 1988). Otra diferencia importante es que el bloqueo de los canales KATP pancreáticos por las sulfonilureas aumenta en presencia de concentraciones bajas de MgADP, mientras que para los canales KATP cardíacos este bloqueo se ve reducido, el mecanismo por el cual ocurre esto aún se desconoce (Gribble et al., 1998b; Virág et al., 1993). Utilizando esta diferencia en la afinidad de las sulfonilureas por los receptores SUR1 y SUR2A, se han construido proteínas quiméricas con estos dos receptores (Babenko et al., 1999b; Gribble et al., 1998; Gribble and Ashcroft, 1999) para determinar el sitio de unión a las sulfonilureas y aunque no se ha determinado el sitio preciso de unión, se han localizado regiones en el receptor que son importantes para la unión de las sulfonilureas. Se ha determinado que el dominio transmembrana 2 (TMD2) del receptor SUR1, que incluye los segmentos transmembrana 12 a 17, desempeña un papel fundamental para la unión de sulfonilureas (Ashfield et al., 1999; Babenko et al., 1999b). Se ha observado que la alta afinidad del receptor SUR1 por la tolbutamida, puede ser conferida al receptor SUR2A al substituir los segmentos transmembrana 14 a 16 de este receptor, con los correspondientes 16 segmentos del receptor SUR1. Contrariamente el receptor SUR1 se puede hacer insensible a tolbutamida al remplazar los segmentos transmembrana 13 a 16, con la región equivalente del receptor SUR2A (Ashfield et al., 1999). Estas observaciones fueron apoyadas por Babenko y colaboradores (1999b), quienes construyeron proteínas quiméricas entre SUR1 y SUR2A, y determinaron que los segmentos transmembrana de 12 al 17 de SUR1 son suficientes para conferir al receptor SUR2A una alta afinidad por la tolbutamida. Se ha demostrado el residuo serina en la posición 1237 localizada en el asa intracelular que une los segmentos transmembrana 15 y 16, es crítica para la unión de las sulfonilureas, ya que la mutación de esta serina por una tirosina (S1237Y) impide tanto el bloqueo con tolbutamida de la corriente KATP, como la unión de glibenclamida marcada radioactivamente (Ashfield et al., 1999; Babenko et al., 1999b). Debido a sus características bioquímicas la glibenclamida (una sulfonilurea y una benzamida) tiene dos sitios de unión en el receptor SUR1; además de unirse al sitio de unión a las sulfonilureas como la tolbutamida se une a una sitio de unión para la benzamida derivada de la meglitinida (Ashcroft and Gribble, 2000a). Abridores de los canales KATP. Existe un grupo de drogas que abren los canales KATP y que en conjunto se llaman abridores de canales de potasio (KCOs). Los abridores que más se han estudiado son diazóxido, pinacidil, cromakalim, sulfato de minoxidil y nicorandil. Aun cuando los KCOs son estructuralmente diferentes, todos actúan abriendo los canales KATP en diferentes tipos de células, produciendo una hiperpolarización de la membrana y por lo tanto reduciendo la actividad eléctrica (Ashcroft and Gribble, 2000b). Los canales KATP nativos en los diferentes tejidos tienen diferente sensibilidad a los KCOs (Ver Tabla 1) y esto se atribuye a los diferentes tipos del receptor SUR (SUR1, SUR2A y SUR2B). Los canales KATP pancreático (Kir6.2/SUR1) tienen propiedades farmacológicas que los diferencian de los canales KATP en otros tejidos, ya que son activados por diazóxido pero no por pinacidil, cromakalim o nicorandil a diferencia de los canales KATP cardíacos (Kir6.2/SUR2A) que son activados por pinacidil y cromakalim (bajo condiciones 17 18 particulares que se mencionarán posteriormente se ha observado una activación con diazóxido), mientras que los canales KATP del músculo liso vascular (Kir6.2/SUR2B) son activados por diazóxido, pinacidil y cromakalim (Babenko et al., 1998b; D’hahan et al., 1999a, b; Gribble et al., 1998b; Inagaki et al., 1995, 1996; Isomoto et al., 1996). Existen evidencias experimentales que muestran que los KCOs interactúan con el receptor a las sulfonilureas, ya que se ha demostrado la unión de un análogo de pinacidil marcado radioactivamente [3H]P1075 al receptor SUR2, cuando éste se expresó en la línea celular COS-7 en ausencia del canal Kir6.2 (Hambrock et al., 1998; Uhde et al., 1999); mientras que el canal Kir6.2 no es activado por los KCOs cuando se expresa en ausencia de SUR, pero si es activado cuando se coexpresa con SUR (John et al., 1998; Mikhailov et al., 1998; Tucker et al., 1997). Además cuando el canal Kir6.2 es coexpresado con diferentes tipos de SUR, los canales KATP resultantes muestran diferente sensibilidad a los abridores de los canales KATP (Inagaki et al., 1996; Okuyama et al., 1998; Shindo et al., 1998). Se ha propuesto que los KCOs se unen a una conformación específica del receptor a sulfonilureas, y la asociación con SUR1 requiere la presencia de un catión divalente (Mg2+ o Mn2+) y un nucleótido hidrolizable (Dickinson et al., 1997; Schwanstecher et al., 1998). Utilizando proteínas quiméricas entre los receptores SUR1/SUR2A y SUR1/SUR2B, se sugirió que la sensibilidad de abridores específicos para el receptor SUR2 (como el pinacidil) está determinada por dos regiones distintas del TMD2 de SUR2A, parte de la secuencia intracelular que une los segmentos transmembrana 13 y 14 y los segmentos transmembrana 16 y 17 (D’hahan et al., 1999a; Babenko et al., 2000; Uhde et al., 1999) y en el segmento 17 hay dos residuos que son especialmente importantes: una lisina en la posición 1249 y una treonina en la posición 1253 (Moreau et al., 2000). El diazóxido (DZX) es un potente abridor de los canales KATP pancreáticos, que se utiliza en el tratamiento de pacientes que presentan el síndrome de la hipoglucemia hiperinsulinémica persistente de la infancia. El diazóxido se une al receptor SUR1 y provoca la apertura de los canales KATP, esto produce una hiperpolarización de la membrana, cuando el potencial de membrana se hace más negativo los canales de Ca2+ dependientes de voltaje se mantienen cerrados, (ya que 19 estos canales se activan conforme el potencial se despolariza), impidiendo la entrada de calcio al interior celular e interfiriendo con la secreción de insulina. El diazóxido al igual que MgADP activa los canales KATP en presencia de concentraciones inhibitorias de ATP y tanto la unión de ADP como de DZX requiere Mg2+ y nucleótidos hidrolizables (Dunne et al., 1986; Findlay, 1988; Kakei et al., 1986; Larsson et al., 1993; Lederer and Nichols, 1989; Misler et al., 1986). El DZX se une tanto al receptor SUR1 como al SUR2B ya que activa canales KATP formados por estos receptores ( Gribble et al., 1997; Shyng et al., 1997b; Schwanstecher et al., 1998). Aunque el diazóxido no activa canales formados con el receptor SUR2A en presencia de MgATP, recientemente se demostró que es capaz de activar los canales formados por este receptor en presencia de MgADP (D´hanan et al., 1999b). Posteriormente se mostró mediante quimeras que los canales formados por un receptor SUR2A al que se insertan el segmento transmembrana 6-11 más NBF1 del SUR1 son activados por DZX en presencia de MgATP, lo cual sugiere que esta región del receptor SUR esta involucrada en el efecto modulador de nucleótidos (Babenko et al., 2000). Aunque el sitio de unión del diazóxido en el receptor SUR aún no ha sido determinado, se ha mostrado que la mutación de una lisina en el motivo A ya sea de NBF1 (mutación K719A) o NBF2 (mutación de K1384M), inhibió la hidrólisis del ATP (Azzaría et al., 1989; Carson et al., 1995; Ko and Pedersen, 1995) y esto provocó un bloqueo del efecto activador de MgADP, demostrando que para el efecto activador de MgADP ambos sitios de unión a nucleótidos son importantes. En este mismo estudio se demostró que el diazóxido activó los canales mutados K1384M (mutación en NBF2) pero no estimuló la actividad de los canales mutados K719A (mutación en NBF1), sugiriendo que la hidrólisis en NBF1 es crítica para la estimulación de estos canales por diazóxido (Gribble et al, 1997). En otro estudio se mostró que mutaciones en el motivo B de NBF2 y la secuencia que lo une al motivo A altera la activación del canal por diazóxido o por MgADP, mientras que mutaciones análogas en NBF1 controlan la cinética de activación. Con estos resultados se sugiere que la hidrólisis de nucleótidos en ambos sitios de unión a nucleótidos están involucrados en la respuesta de los canales K ATP al diazóxido y MgADP. 20 El mecanismo molecular mediante el cual el diazóxido regula la actividad de los canales KATP y por lo tanto, inhibe la secreción de insulina, aún no ha sido determinado. Se ha propuesto un mecanismo basado en observaciones de que el canal Kir6.2 truncado en su extremo carboxilo (Kir6.2-∆C26), es capaz de formar canales que llegan a la membrana plasmática y que son sensibles al ATP, con una KD de aproximadamente 100 µM, la cual cambia a ∼ 10 µM cuando el canal Kir6.2 se coexpresa con el receptor SUR1 (Nichols and Lopatin, 1993; Proks and Ashcroft, 1993). Con estos hallazgos se ha sugerido que el canal Kir6.2 tiene una sensibilidad intrínseca al ATP y que el receptor SUR1 actúa como un interruptor que aumenta la sensibilidad del canal por el ATP en ausencia de hidrólisis de nucleótidos en sus sitios de unión a nucleótidos (Nichols et al., 1996). Basándose en estas observaciones Shyng y colaboradores (1997b) propusieron un modelo para explicar la regulación del mecanismo de compuerta de los canales KATP, por el diazóxido y MgADP, en el cual sugieren que estos agentes al igual que el MgATP estabilizan al receptor SUR1 en un estado desacoplado del canal Kir6.2, esto significa que el interruptor esta apagado, favoreciendo la hidrólisis del ATP y con ello el aumento en la actividad del canal. Por esta razón el MgADP y el diazóxido aumentan la actividad del canal, ya sea porque actúan como productos de la hidrólisis (MgADP) o quizás porque simulan un estado de hidrólisis (diazóxido). Hipoglicemia hiperinsulinémica persistente de la infancia (PHHI) La hipoglucemia hiperinsulinémica persistente de la infancia, también llamada nesidioblastosis pancreática (Online Mendelian Inheritance in Man: OMIM 256450) es un desorden metabólico que se presenta en la niñez y que es caracterizado por un aumento en la secreción de insulina en presencia de concentraciones bajas de glucosa (Aguilar-Bryan and Bryan, 1999; Dunne et al., 1999; Glaser et al., 1999). Este síndrome fue descrito por primera vez en 1930 (Glaser et al., 1999), fue hasta 1981 que se sugirió un defecto en la función de las células beta, ya que se demostró que la aplicación de glucosa a diferentes concentraciones no provocó la liberación de insulina, en un tejido aislado quirúrgicamente de un niño que presentaba una alta 21 concentración de insulina en sangre cuando la concentración de glucosa en esta era baja (hiperinsulinemia en presencia de hipoglicemia)(Aynsley-Green et al., 1981). La incidencia de esta enfermedad es de 1 por cada 45000 nacimientos en el norte de Europa y de 1 por cada 2675 nacimientos en Arabia Saudita (Matheew et al., 1988; Meissner et al., 1999); la incidencia es mayor en comunidades que tienen un alto grado de consanguinidad, aproximándose a la de la fibrosis-cística en los caucásicos europeos (1 por cada 2500 nacimientos; Aynsley-Green et al., 1998). De acuerdo con la secretaria de salud, en México no existen datos sobre la incidencia de esta enfermedad. La mayoría de los recién nacidos que poseen la enfermedad, son macrosómicos al nacimiento con un peso promedio de 3.7 Kg, en un 30% de los casos se requiere cesárea (De Lonlay-Debeney et al., 1999). La hipoglicemia en estos niños es siempre severa; las concentraciones de glucosa en plasma al momento del primer síntoma son extremadamente bajas (menos de 1 mM) y estos niveles se mantienen aun después de ingerir alimentos (De Lonlay et al., 2002). Por esta razón, existe un alto riesgo a presentar convulsiones, daño cerebral y retraso mental debido a la falta de combustible para mantener el metabolismo normal en el sistema nervioso central (Dunne et al., 1999; Stanley, 1997). En los casos severos los síntomas pueden presentarse dentro de las primeras 72 horas del nacimiento y en la mitad de los casos se observan convulsiones (De Lonlay-Debeney et al., 1999). Otros síntomas son movimientos anormales como temblores, hipotonía, cianosis e hipotermia (De Lonlay et al., 2002). Diagnostico clínico El diagnóstico bioquímico de una hipoglicemia secundaria a una hipersecreción de insulina es generalmente difícil y se requiriere demostrar que se tienen niveles altos de insulina en el momento de la hipoglicemia sintomática. En un paciente con PHHI los niveles de insulina pueden fluctuar enormemente y esto hace difícil la demostración de que los niveles están elevados patológicamente, por esta razón se utilizan otros criterios para evaluar la acción de la insulina y diagnosticar PHHI: (1) Nivel de glucosa en sangre menor a 48 mg/dl; (2) Nivel de insulina elevado durante la hipoglicemia; (3) Concentraciones bajas de ácidos grasos libres y cuerpos 22 cetónicos durante la hipoglicemia; (4) Respuesta glicémica menor a 30 mg/dl después de la inyección de glucagon (0.03 mg/Kg); (5) Que se requieran más de 15 mg/Kg de glucosa para mantener los niveles de euglicemia; (6) Ausencia de cuerpos cetónicos en orina (Glaser et al., 1999). Patogénesis de la PHHI Hasta antes de 1995 poco se sabía acerca de la genética de PHHI, pero ahora se sabe que este síndrome es hereditario y que se transmite de manera autosómica principalmente en forma recesiva (Glaser et al., 1990; Thornton et al., 1991; Woolf et al., 1991) aunque también se han descrito casos de formas dominante (Thornton et al., 1998; Kukuvitis et al., 1997). En 1994 se proporcionó la primera información genética de este síndrome que se asoció a un locus en el brazo corto del cromosoma 11 en la posición 15.1 (11p15.1) (Glaser et al., 1994); en este estudió la mayoría de las familias afectadas fueron de origen judío, y posteriormente se encontró en familias de Arabia Saudita (Thomas et al., 1995a). En esta región del cromosoma se mapeo el gen que codifica para el receptor a las sulfonilureas SUR1 (ABCC8, MIM # 600509) que fue el primer gen asociado con el síndrome de PHHI (Aguilar-Bryan et al., 1995). Posteriormente se identificó el gen que codifica para el canal de potasio con rectificación entrante Kir6.2 (KCNJ11, MIM # 600937) (Inagaki et al., 1995). Ambos genes ABCC8 y KCNJ11 son contiguos en la posición 15.1 del cromosoma 11 (Inagaki et al., 1995). Además de los genes que codifican para el receptor SUR1 y el canal Kir6.2, también se ha demostrado que una alteración en el gen GCK que codifica para la enzima glucocinasa (GK) también produce PHHI (Glaser et al., 1998). La enzima glucocinasa cataliza la fosforilación de glucosa en el carbono 6; esta reacción determina la velocidad con que la glucosa es metabolizada (ver figura 1). Las alteraciones en este gen producen un aumento en la actividad de la GK un incremento en la glucólisis y, con ello, un aumento en la liberación de insulina, este incremento en la secreción de insulina es independiente de la concentración de glucosa en sangre. Esta enzima se expresa solamente en células beta pancreáticas y en hígado. El gen GCK (GenBank # M90299) fue mapeado en el brazo corto del 23 cromosoma 7 (7p15-p13), y consiste de 12 exones, que incluyen dos exones específicos, uno para las células beta (exón 1B) y otro para las células hepáticas (exón 1H) (Tanizawa et al., 1992). Por otra parte la hipoglicemia hiperinsulinémica hiperamonémica es un síndrome causado por una mutación en el gen que codifica para la enzima glutamato deshidrogenasa (GLUD1 MIM # 138130) (Stanley et al., 1998). La glutamato deshidrogenasa es una enzima de la matriz mitocondrial que cataliza de manera reversible la deaminación oxidativa de L-glutamato a α-ceto glutarato en la matriz mitocondrial; el α-ceto glutarato entra al ciclo de los ácidos cítricos y la fosforilación oxidativa formando finalmente ATP. La leucina es un activador alostérico de esta enzima que produce un incremento en la generación de ATP, y consecuentemente en la secreción de insulina a través de la vía de los canales KATP. Este defecto produce un aumento anormal en la liberación de insulina y un daño en la destoxificación de amonio en el hígado. La mayor parte de las mutaciones reportadas en la glutamato deshidrogenasa son en el dominio alostérico y son asociadas con una reducción en la sensibilidad al GTP que actúa como inhibidor alostérico en condiciones fisiológicas (Stanely et al., 1998). A pesar de que la PHHI se debe a mutaciones en los 4 genes antes descritos, el 50 - 60 % de los casos han sido asociados a mutaciones en los genes que codifican para el receptor SUR1 y el canal Kir6.2. Se han reportado aproximadamente 4 mutaciones en el canal Kir6.2 y aproximadamente 70 mutaciones en el receptor SUR1. Otro correlato que asocia los defectos en los canales KATP y la PHHI, son las características histológicas del tejido pancreático en los pacientes con este síndrome. Hasta la fecha se han descrito dos formas histopatológicas de este síndrome y son el hiperinsulinismo difuso (DiPHHI) y el hiperinsulinismo focal (FoPHHI) (Rahier et al., 2000). El hiperinsulinismo difuso es caracteriza por hipertrofia e hiperplasia de las células beta; los islotes de Langerhans tienen tamaño irregular y esta característica se distribuye por todo el páncreas. Esta forma difusa ha sido clásicamente asociada a alteraciones en los genes que codifican tanto para el receptor SUR1 (Nestorowicz et al., 1996; Thomas et al., 1995), como para el canal Kir6.2 (Nestorowicz et al., 24 1997; Thomas et al., 1996) en hiperinsulinismos heredados recesivamente, aunque también se ha asociado al gen de la enzima glucocinasa en ese caso el hiperinsulinismo es heredado en forma dominantemente (Glaser et al., 1998) Por último, la forma difusa se asocia con el gen de a l glutamato deshidrogenasa en aquellos casos en los que el hiperinsulinismo se asocia con hiperamonemia (Stanley et al., 1998). El hiperinsulinismo focal se define como una hiperplasia de las células beta en una zona definida del páncreas, que puede diferenciarse de un insulinoma. Esta forma histopatológica se debe a un fenómeno genético llamado “imprinting” el cual afecta a un número pequeño de genes, y consiste en la pérdida somática de uno de los alelos ya sea materno o paterno (Lee, 2001). Así, el hiperinsulinismo focal se produce por la pérdida somática del alelo materno en el brazo corto del cromosoma 11, que conduce a un desbalance en la expresión de genes involucrados en el control del crecimiento celular (De Lonlay et al., 1997; Verkarre et al., 1998). Los pacientes con hiperinsulinismo focal heredan una mutación recesiva provocada solo por el alelo paterno, en el gen que codifica para el receptor SUR1, entonces una parte del páncreas se reduce a homocigocidad y esto es lo que finalmente conduce al hiperinsulinismo (Ryan et al., 1998; Verkarre et al., 1998a). Las dos formas histopatológicas de PHHI se presentan con un curso temporal diferente y tienen diferente grado de severidad (Sempoux et al., 1995). La forma difusa es más severa; se desarrolla a las 48 horas después del nacimiento, y requiere una resección casi total del páncreas para controlar la hipoglicemia. La forma focal es menos severa y es identificada en pacientes que desarrollan hipoglicemia varias semanas después del nacimiento; en este caso la hipoglicemia puede ser controlada por resección de la región afectada, pero es difícil predecir su localización antes de la cirugía (Craver and Hill, 1997). Tratamiento de PHHI El tratamiento de esta enfermedad debe ser inmediato, y el principal objetivo es prevenir el daño cerebral que se puede presentar debido a la severa hipoglicemia. El tratamiento debe permitir un desarrollo psicomotor normal, se debe establecer una 25 alimentación frecuente dependiendo de la edad del niño, para asegurarse que el niño mantenga sus niveles de glucosa en sangre; para esto frecuentemente se requiere un catéter venoso central y alimentación oral continua. Además, se aplica glucosa vía intravenosa 0.5 g/Kg y alimentación naso gástrica cada 3 horas (Glaser et al., 1999). Si los requerimientos de glucosa son mayores a los 10 mg/Kg/min entonces se aplica glucagón vía intravenosa (1 a 2 mg por día); la somatostatina intravenosa (1 a 4.5 µg/Kg/h) también se puede usar sola o con el glucagon. El glucagon moviliza la glucosa almacenada como glucógeno hepático (Aynsley-Green et al., 2000). Dentro del tratamiento farmacológico el diazóxido y la clorotiazida son las drogas principalmente utilizadas y se administran en forma conjunta. El diazóxido como se mencionó anteriormente es un activador de los canales KATP, que puede unirse al receptor SUR1 y puede abrir los canales en presencia de nucleótidos intracelulares, al abrirse estos canales se invierte el mecanismo de cierre inducido por la glucosa (Shepherd et al 2000). La clorotiazida también tiene una acción en los canales KATP (Barnes et al., 2000), pero sobre todo se usa para prevenir la retención de fluidos, que es un efecto secundario del diazóxido. En el tratamiento contra la PHHI, también se han utilizado antagonistas de los canales de calcio tales como la nifedipina (Lindely et al., 1996); Sin embargo la respuesta clínica a nifedipina (0.5 a 2 mg/kg por día) es muy variable (Shepherd et al., 2000). El octreotido análogo de la somatostatina tiene múltiples acciones en las células beta, como son la alteración de la entrada de calcio e inhibición de la exocitosis de los gránulos que contienen insulina (Shepherd et al., 2000). Se puede usar este fármaco antes de una cirugía cuando no hay una respuesta al diazóxido. Las dosis de octreotido varían de 15 a 60 mg/día, que generalmente se aplican en 3 a 4 inyecciones subcutáneas (Thornton et al., 1993). Cuando los pacientes que presentan PHHI no responden al tratamiento farmacológico, se requiere realizar entonces una cirugía llamada pancreatectomía, en la cual se extirpa el páncreas para controlar los niveles sericos de insulina. Hasta hace algunos años la mayoría de los cirujanos pediátricos recomendaban una pancreatectomía en la cual se extirpaba el 95 % del páncreas, pero 26 desafortunadamente la pancreatectomía implica un alto riesgo de que los pacientes presenten diabetes mellitus, años después de la cirugía (Shilyanski et al., 1997). Recientemente se ha demostrado que los pacientes que presentan un hiperinsulinismo focal se les pueden realizar la extirpación de la zona o foco afectado sin necesidad de extirpar todo el páncreas. Por lo tanto dependiendo del tipo de lesión histológica asociada con la PHHI, ya sea hiperinsulinismo difuso o focal, es el tipo de tratamiento quirúrgico que se debe aplicar y por esto es importante diferenciar entre ambas formas histopatológicas (De Lonlay et al., 1997; De Lonlay-Debeney et al., 1999; Verkarre et al., 1998b). Generalmente en los niños que presentan un hiperinsulinismo focal se realiza una resección de la zona afectada, para ello actualmente se utiliza un examen previo a la operación, que consiste en un muestreo venoso pancreático transhepático, el cual permite identificar puntos en los cuales existe una hipersecreción de insulina (De Lonlay et al., 1997), y ayuda a diferenciar entre el hiperinsulinismo difuso y focal. Este procedimiento implica anestesia general y se toman muestras de sangre venosa recolectada en la cabeza, istmo, cuerpo y cola del páncreas, para medir concentración en sangre de glucosa, insulina y péptido C. Los pacientes con hiperinsulinismo focal presentan concentraciones plasmáticas elevadas de insulina y péptido C en las primeras muestras, y bajas concentraciones en las últimas muestras, mientras que para pacientes con hiperinsulinismo difuso la concentración plasmática de insulina y péptido C son elevadas en todas las muestras (De Lonlay et al., 2002). Otro método utilizado para diferenciar el hiperinsulinismo difuso del focal, consiste en medir la secreción de insulina, en respuesta a la aplicación intravenosa de glucosa, calcio y tolbutamida (Grimberg et al., 2001). Los niños que padecen de hiperinsulinismo difuso, no presentan una secreción de insulina después de la aplicación de tolbutamida, mientras que la aplicación intravenosa de calcio provoca la secreción de insulina; los niños que presentan el hiperinsulinismo focal, tienen respuestas diferentes a estas (Huopio et al., 2002). Shilyansky y colaboradores (1997) proponen que una pancreatectomía casi total controla la hipoglicemia en 97% de los pacientes, aunque el 57% presentaron 27 complicaciones después de la operación, y el 86% desarrollaron diabetes 3 años después de la operación. Cuando se realizaron extirpaciones del 95 % del páncreas, el 33% continuaron con hipoglicémia, el 15% presentaron complicaciones y el 69% desarrollaron diabetes en un promedio de 9.7 años después de la cirugía. Los pacientes que fueron sometidos a pancreatectomías del 85% o menores presentaron una cura del 50%. Los pacientes con hipoglicemia después de una pancreatectomía del 85% responden al tratamiento médico. El síndrome de PHHI permanece como uno de los padecimientos más difíciles de manejar dentro de la endocrinología pediátrica contemporánea. El uso del diazóxido como única alternativa farmacológica, además la heterogeneidad en la respuesta de los pacientes a esta droga limita el tratamiento de pacientes que presentan este síndrome. La cirugía como una alternativa en el tratamiento dista mucho de ser una solución al problema, no solo por la dificultad de identificar el tipo de PHHI desde el punto de vista histológico (focal y difusa), si no por el alto riesgo de presentar una síndrome igual de complejo como lo es la diabetes después de una resección del páncreas. Por lo tanto entender los mecanismos que regulan el funcionamiento de los canales KATP en las células beta, es muy importante, esto generaría las bases para el desarrollo de nuevas drogas que actúen como una alternativa terapéutica en la cura del síndrome llamado PHHI. ANTECEDENTES. Estudio funcional de mutaciones en el receptor SUR1 asociadas con PHHI Como mencioné anteriormente se han descrito aproximadamente 70 mutaciones en el receptor SUR1 asociadas con PHHI, los estudios funcionales de estas mutaciones han proporcionado información acerca de la relación estructura función de los canales KATP. Las mutaciones en el receptor SUR1 han sido clasificadas en dos categorías de acuerdo a la alteración funcional que provoca en el canal: algunas mutaciones causan una pérdida de la función, mientras que otras conducen a una alteración en la regulación metabólica defectuosa del canal. El mecanismo por el cual varias mutaciones producen una reducción funcional de los canales KATP es el bloqueó del tráfico (ensamblaje y translocación de proteínas 28 hacia la membrana) de los canales hacia la membrana plasmática (Cartier et al., 2001; Partriedge et al., 2001; Taschenberger et al., 2002). Por ejemplo la deleción de una fenilalanina (F) en la posición 1388 del receptor SUR1 (SUR1-∆F1388) resulta en la ausencia de eflujo de rubidio marcado cuando el receptor SUR1-∆F1388 se cotransfecta con el Kir6.2 en la línea celular COSm6 (Nestorowicz et al., 1996; Shyng et al., 1998). Posteriormente se demostró que la perdida de la funcionalidad de los canales formados por SUR1-∆F1388 se debe a una alteración en el plegamiento de la proteína del canal que resulta en su retención y donde las proteínas mal plegadas son retenidas en el retículo endoplásmico (Cartier et al., 2001; Shyng et al., 1998). Así, al interferir con el tráfico hacia la membrana plasmática no hay canal funcional que medie el tráfico de Rb. Otra mutación que produce una pérdida en la funcionalidad de los canales es la mutación de una arginina por una histidina en la posición 1394 (R1394H); mediante tinción con inmunofluorescencia se mostró que al coexpresar el canal Kir6.2 y el receptor SUR1-R1394H, ambas proteínas eran retenidos en el aparato de Golgi (Partridge et al., 2001). Recientemente se describieron dos nuevas mutaciones en el receptor SUR1 que previenen el tráfico de canales hacia la membrana plasmática, la mutación de una alanina por treonina en la posición 1457 (A1457T) y de una valina por ácido aspártico en la posición 1550 (V1550D) (Reimann et al., 2003). Las mutaciones ∆F1388, R1394H y A1457T (Ver figura 3) están ubicadas en el segundo sitio de unión a nucleótidos (NBF2), mientras que V1550D se ubica en el extremo carboxilo del receptor SUR1. La pérdida de la funcionalidad de los canales KATP formados por estas mutaciones guarda relación con el fenotipo clínico que muestran los pacientes; todos presentan la forma severa de la enfermedad y no responden al tratamiento con diazóxido. Por otra parte, las mutaciones que producen un defecto en la regulación de los canales KATP, alteran la interacción entre los nucleótidos de adenosina y el receptor SUR1. La primera mutación en SUR1 asociada con la PHHI, que mostraba una alteración en la regulación por nucleótidos, fue reportada por Nichols y colaboradores (1996), y consistía en la mutación puntual de una glicina por una arginina en la posición 1479 (G1479R), la cual mostró una alteración en la regulación por MgADP. 29 Posteriormente, se realizó un análisis funcional de nuevas mutaciones dentro de las que se incluyen H125Q, N188S, F591L, T1139M, R1215Q y G1382S (Ver figura 3). Dentro de estas mutaciones sólo G1382S está ubicada en NBF2 y las 5 restantes están fuera de los sitios de unión a nucleótidos. Todas estas mutaciones (H125Q, N188S, F591L, T1139M, R1215Q y G1382S) forman canales que llegan a la membrana plasmática al coexpresarse con Kir6.2 en células COSm6. Las mutantes F591L, T1139M, R1215Q y G1382S mostraron una reducción en su respuesta a la estimulación por MgADP. La respuesta de los canales mutados al diazóxido fue variable. Algunas de las mutantes tienen buen correlato entre las resultados funcionales y las observaciones clínicas, como es el caso de H125Q y R1215Q, mientras que la mutación N188S muestra una alteración funcional mínima in vitro pero es asociada con la forma severa de la enfermedad (Shyng et al., 1998). Otra mutación asociada con PHHI, que afecta fuertemente las propiedades bioquímicas del receptor SUR1 fue descrita por Matsou y colaboradores. Esta mutación consiste de un intercambio de una arginina por una cisteina en la posición 1420 localizada en NBF2. Esta mutación disminuye la afinidad de unión de ATP y ADP en NBF2, sin embargo, conserva su mecanismo de activación por diazóxido (Matsou et al., 2000). La mutación de una leucina por una prolina en la posición 1544 (L1544P) fue identificada en un paciente que presentaba la forma severa de PHHI y reduce fuertemente la respuesta a la estimulación por MgADP y no responde al diazóxido (Taschenberger et al., 2002). Recientemente se realizó el análisis funcional de una nueva mutación en SUR1 asociada con la PHHI, en la cual una leucina se intercambió por una valina en la posición 1551 (L1551V), la cual muestra una reducción en su sensibilidad a ATP y una pérdida en la activación tanto por MgADP como por diazóxido (Reimann et al., 2003). Tanto L1544P y L1551V están localizadas en el extremo carboxilo de SUR1. Las mutaciones L1544P, L1551V y R1420C antes descritas mostraron además una disminución en su nivel de expresión de canales funcionales en la membrana. El hecho de que estas mutaciones presentan una reducción en su respuesta al MgADP causa hiperinsulinismo ya que el canal pierde su capacidad de responder a un incremento intracelular en la concentración de ADP y, en consecuencia 30 31 disminuyen los niveles de glucosa en sangre. Estos resultados apoyan la idea de que el ADP juega un papel crítico en la regulación de la actividad de los canales KATP en condiciones fisiológicas; además se ha sugerido que la pérdida de la respuesta a la estimulación por MgADP puede ser un defecto molecular común asociada con PHHI (Shyng et al., 1998). Dos nuevas mutaciones asociadas con PHHI, expresada dominantemente en el receptor SUR1 Thornton y colaboradores en 1995, reportaron 3 familias con PHHI, en las cuales la enfermedad parecía transmitirse de manera dominante más que recesiva (Thornton et al., 1998). Posteriormente se realizó la identificación de los defectos genéticos en dos de las tres familias y los resultados mostraron la expresión dominante de mutaciones en los genes que codifican para las enzimas citosólicas glutamato deshidrogenasa (GLUD1 en el brazo largo de cromosoma 10) (Stanley et al., 1998) y glucocinasa (GCK en el brazo corto del cromosoma 7) (Glaser et al., 1998). El locus genético para la tercer familia no fue identificado, aunque en ese momento el análisis de vinculación genética, usando marcadores repetidos en tandem en el brazo corto del cromosoma 11, parecía excluir a los genes que codifican para el receptor SUR1 y canal Kir6.2. En este año Thornton y colaboradores se propusieron identificar el defecto genético en la tercer familia mediante exámenes farmacológicos, utilizando para ello un examen que mide la respuesta aguda a insulina (AIR: acute insulin responses), en respuesta a diferentes estímulos, para definir los defectos en las vías que controlan la secreción de insulina. La prueba farmacológica AIR consistió en aplicar estímulos con calcio, glucosa, leucina y tolbutamida, los cuales fueron aplicados de manera intravenosa a intervalos de una hora. La prueba AIR se definió como el incremento en la insulina plasmática medida entre 1 y 3 minutos después del estímulo. Los resultados se compararon con valores previamente determinados en adultos sanos y en niños que presentaban el síndrome PHHI debido a mutaciones en el canal KATP que se transmiten de manera recesiva, así como mutaciones en la enzima glutamato deshidrogenasa que se transmiten de manera dominante (Ferry et al., 2000; Kelly et 32 al., 2001). Las mutaciones en los canales KATP que produjeron la PHHI en estos niños, fueron mutaciones comunes en el receptor SUR1 (a 3995-9g y la ∆F1388), la mayoría de los niños que presentan estas mutaciones se asocian con la forma severa de la enfermedad. Los resultados obtenidos en 3 pacientes para la AIR fueron: anormal para calcio, negativa para leucina, normal para glucosa y una respuesta alterada a la tolbutamida. Estas anormalidades en la respuesta a AIR en los miembros de la familia afectada fueron diferentes a los reportados en pacientes con PHHI debido a mutaciones en GDH; sin embargo fueron semejantes con los resultados obtenidos en pacientes con mutaciones en SUR1 (homocigoto a3995-9g y heterocigoto a3995-9g /∆F1388). Debido a estos resultados los autores examinaron nuevamente la posibilidad de que la PHHI en esta familia estuviera asociada a mutaciones en el receptor SUR1 de los canales KATP. Así el análisis de vinculación genética utilizando polimorfismos fue consistente con una transmisión dominante de un defecto en SUR1. Mediante el análisis de mutación se descubrió que la deleción de 3 nucleótidos (4159-4161) en el exón 34 de SUR1 produce la deleción de una serina en el codón 1387 (∆S1387). Las alteraciones o anormalidades observadas en los pacientes con la mutación ∆S1387, son muy similares a las observadas en pacientes que presentan las mutaciones g3992-9a o ∆F1388. Además las anormalidades histológicas de las células beta que se observaron en el páncreas de uno de los miembros de la familia estudiada fueron idénticas a las descritas en niños que presentan una PHHI asociada con alteraciones en el canal KATP, y que se transmite de manera recesiva. Los pacientes de la familia estudiada parecen responder parcialmente a la terapia con diazóxido, sugiriendo con ello que la mutación ∆S1387 forma canales que no han perdido completamente su funcionalidad (Thornton et al., 2003). Para evaluar el efecto de la mutación ∆S1387 en la expresión, el transporte intracelular y la función de los canales KATP, la mutante de SUR1 fue reconstituida con el canal Kir6.2 silvestre en células COSm6. Utilizando fotomarcaje con el ligando azido glibenclamida [125I], se obtuvo un patrón de marcaje entre el canal silvestre y el mutado muy similares (Panel A de la figura 4). Además estudios de inmunofluorescencia mostraron que los canales formados por la mutación ∆S1387, 33 34 se incorporan en la membrana de manera similar que el canal silvestre. Sin embargo al estudiar la funcionalidad de los canales mutados midiendo el eflujo de rubidio marcado [86Rb+] en presencia y ausencia de 1 µM de glibenclamida, los canales formados por la mutante no canalizan el eflujo de rubidio en respuesta a una combinación entre inhibición metabólica, baja concentración de ATP y la presencia de diazóxido, mostrando así que los canales KATP con la mutación ∆S1387 no son funcionales (panel B de la figura 4). Los resultados de la mutación ∆S1387 son muy similares a una mutación descrita anteriormente por Huopio y colaboradores (2000), en la cual un ácido glutámico se intercambia por una lisina en la posición 1506 (E1506K), localizada en el segundo sitio de unión a nucleótidos y que se transmite de manera dominante. Todos los pacientes estudiados presentaron la forma leve de la enfermedad, que generalmente fue tratada con diazóxido. Estos hallazgos clínicos fueron congruentes con los resultados de la expresión heteróloga de canales recombinantes Kir6.2/SUR1-E1506K; estos canales fueron insensibles a la inhibición metabólica, pero respondieron parcialmente a diazóxido. Además, el MgADP fue incapaz de estimular la actividad de los canales mutados. Los resultados electrofisiológicos obtenidos con esta mutante, sugieren que la mutación conduce a una reducción pero no a una pérdida completa en la función de los canales K ATP (Huopio et al., 2000). Por otro lado los resultados obtenidos con la mutación ∆S1387 son interesantes y contradictorios cuando se comparan con la mutación ∆F1388 (Cartier et al., 2001); ambas mutaciones están ubicadas en posiciones adyacentes al segundo sitio de unión a nucleótidos. El fenotipo clínico que producen ambas mutaciones también es diferente, la ∆S1387 produce la forma leve de la enfermedad, mientras que la ∆F1388 produce la forma severa de la enfermedad. Considerando lo anterior es interesante comparar que mientras la deleción de una serina en la posición 1387 produce un hiperinsulinismo que se transmite de manera dominante la deleción de una fenilalanina en la posición 1388 se transmite de manera recesiva. Además, de acuerdo con los resultados de Thornthon y colaboradores (2003), la mutación ∆S1387 producen canales que llegan a la membrana plasmática pero 35 aparentemente no conducen corriente, mientras que la ∆F1388 forma canales que son retenidos en retículo endoplásmico. Considerando entonces que la mutación ∆S1387 a diferencia de la ∆F1388 forma canales que llegan a la membrana, pero que aparentemente son no funcionales ya que no conducen un flujo de rubidio Recientemente el grupo de Lydia Aguilar-Bryan clonó una nueva mutación asociada con la PHHI en la cual un ácido aspártico se intercambia por una asparagina en la posición 310 (D310N) de la proteína del receptor SUR1, esta mutación se localiza en el sexto segmento transmembrana del primer dominio trransmembrana (TMD1) de SUR1. De acuerdo con el análisis de vinculación genética esta mutación se transmite de manera dominante, y fenotípicamente se asocia con la forma leve de la enfermedad. Para evaluar la expresión, tráfico y función de los canales formados por la mutación D310N se realizó el mismo tipo de estudios utilizados con la mutación ∆S1387. El análisis de expresión de canales reconstituidos Kir6.2/SUR1-D310N, mostró que la mutación D310N no altera el trafico de proteínas a la membrana. Al estudiar la funcionalidad midiendo el flujo de rubidio marcado a través de estos canales, se obtuvo que el porcentaje de rubidio marcado que fluye a través del canal silvestre y el canal Kir6.2/SUR1-D310N es similar. Con estos resultados se puede decir que la mutación D310N en el receptor SUR1 forma canales que llegan a la membrana y que parecen funcionar normalmente (Resultados no publicados). A pesar de que las mutaciones D310N y ∆S1387 están ubicadas en dominios estructurales diferentes del receptor SUR1, ambas se transmiten de manera dominante y son clínicamente similares ya que producen el fenotipo leve de la enfermedad, en este trabajo nos propusimos realizar un análisis funcional utilizando técnicas electrofisiológicas para tratar de entender la relación entre la estructura y la función de los canales KATP que presentan estas mutaciones y que producen el síndrome PHHI. 36 HIPÓTESIS. 1) Si la mutación ∆S1387 asociada con la PHHI forma canales que llegan a la membrana plasmática, y son no funcionales ya que no tienen un flujo de rubidio marcado, entonces esta mutación produce una pérdida de la función de los canales KATP y por lo tanto no habrá respuesta a los moduladores fisiológicos como ATP y ADP. 2) Si la mutación D310N forma canales que se insertan en la membrana plasmática y que permiten el flujo de rubidio marcado, entonces se esperaría que hubiese una alteración en su regulación por nucleótidos como ATP o ADP, para explicar el trastorno clínico. OBJETIVO GENERAL. Realizar un análisis funcional de los canales formados por dos nuevas mutaciones en el receptor SUR1 (SUR1-∆S1387 y SUR1-D310N) asociadas con la hipoglicemia hiperinsulinémica persistente de la infancia (PHHI). OBJETIVOS ESPECÍFICOS. 1) Determinar las propiedades biofísicas del canal silvestre y las de los canales mutados Kir6.2/SUR1-∆S1387 y Kir6.2/SUR1-D310N. 2) Comparar la sensibilidad al ATP, que es el inhibidor metabólico de los canales KATP, sobre la actividad del canal silvestre y los canales mutados. 3) Evaluar el efecto del ADP en presencia de Mg2+ sobre la actividad unitaria del canal silvestre y compararla con los canales que presentan las mutaciones D310N y ∆S1387. 4) Estudiar el efecto del diazóxido sobre la actividad de los canales formados por Kir6.2/SUR1-∆S1387 y Kir6.2/SUR1-D310N y compararlo con el canal silvestre (Kir6.2/SUR1). 37 ÍNDICE DE FIGURAS. Figura 1. Liberación de insulina en células beta de humano. Figura 2. Estructura molecular de los canales K ATP pancreáticos. Figura 3. Mutaciones en el receptor SUR1 asociadas con la PHHI. Figura 4. Análisis de expresión de la mutación ∆S1387. Figura 5. Esquema del dispositivo experimental de registro. Figura 6. Células COSm6. Figura 7. Las células COSm6 no presentan una conductancia endógena de K ATP. Figura 8. La coexpresión de SUR1 y Kir6.2 en células COSm6 forman canales con conductancias similares al canal K ATP pancreático nativo. Figura 9. Conductancia unitaria del canal Kir6.2/SUR1-D310N. Figura 10. Conductancia unitaria del canal Kir6.2/SUR1-∆S1387. Figura 11. Los canales KATP pancreáticos reconstituidos presentan actividad eléctrica semejante a los canales nativos. Figura 12. Propiedades farmacológicas del canal KATP silvestre expresado en células COSm6. Figura 13. Los canales mutados Kir6.2/SUR1-∆S1387 y Kir6.2/SUR1D310N son sensibles a la aplicación de ATP. Figura 14. Curva dosis respuesta al ATP en presencia de Mg2+. Figura 15. Curva dosis respuesta al ATP en ausencia de Mg2+. Figura 16. Efecto del análogo de ATP no hidrolizable el AMP-PNP sobre la actividad del canal silvestre. Figura 17. Curva dosis respuesta al AMP-PNP en presencia de Mg2+. Figura 18. Efecto del ADP sobre el canal silvestre Kir6.2/SUR1 expresados en células COSm6. Figura 19. El canal mutado Kir6.2/SUR1-∆S1387 pierde su sensibilidad al ADP. Figura 20. El canal Kir6.2/SUR1-D310N tiene una respuesta atípica al ADP. Figura 21. El canal Kir6.2/SUR1-D310N tiene una afinidad menor por ADP. Figura 22. Efecto de diazóxido sobre la actividad del canal Kir6.2/SUR1-∆S1387. Figura 23. Efecto del diazóxido sobre la actividad del canal Kir6.2/SUR1-D310N. Figura 24. Modelo del mecanismo de alteración en la respuesta mediada por ATP para el canal mutado Kir6.2/SUR1-D310N. TABLAS. Tabla 1. Propiedades farmacológicas de los canales K ATP. Tabla 2. Resumen de los valores de la constante de disociación y el coeficiente de Hill. Figura 1. Liberación de insulina en células beta de humano. En una célula beta normal a concentraciones no estimulatorias de glucosa el potencial de membrana en reposo es de –70 mV. Cuando la concentración de glucosa aumenta este carbohidrato es transportado al interior e inicia su metabolismo a través de la enzima glucocinasa (GK), formando glucosa 6 fosfato, posteriormente diferentes eventos metabólicos producen un aumento en la relación ATP/ADP, esto produce el cierre de los canales KATP, y esto conduce a una despolarización del potencial de membrana, activando así canales de calcio dependientes de voltaje y este aumento en la entrada de calcio, inicia la liberación de insulina a través de gránulos de secreción mediante exocitosis (Modificada de Dunne et al., 1999). Figura 2. Estructura molecular de los canales KATP pancreáticos. Los canales KATP de las células beta del páncreas están formados por dos subunidades: un canal de potasio con rectificación entrante Kir6.2 que presenta 2 segmentos transmembrana y la secuencia (GYG) altamente conservada en los canales selectivos a K+. La otra subunidad es un receptor con alta afinidad a las sulfonilureas SUR1, el cual contiene 17 segmentos transmembrana, dos sitios de unión a nucleótidos (NBF1 y NBF2). Ambos subunidades se organizan en un octámero donde 4 subunidades del canal Kir6.2 forman el poro del canal y se asocian a 4 subunidades del receptor SUR1 que regula este canal. Propiedades Farmacológicas de los canales KATP Abridores de los canales KATP Pancreático (Kir6.2/SUR1) Diazóxido Pinacidil Activa No 20 - 100 µ M Cardíaco (Kir6.2/SUR2A) Debil Músculo liso (Kir6.2/SUR2B) Activa 71, 63, 16 Cromakalim No 71, 63, 16 71 72, 63, 16 Activa Activa 71, 72, 63 Activa 71, 72, 63 74, 63, 16 Activa 74, 63, 16 20 - 100 µ M 7 4 Tabla 1. Propiedades Farmacológicas de los canales KATP. En esta tabla se muestran las propiedades farmacológicas de los canales KATP pancreáticos, músculo liso y cardíacos a los abridores de estos canales (71, 72, 74, 63, 16, ver bibliografía) (Modificada de Babenko et al., 1998a) Figura 3. Mutaciones en el receptor SUR1 asociadas con la PHHI. Representación topológica del receptor SUR1 y la posición de algunas mutaciones en el receptor asociadas con la PHHI, ubicadas en diferentes dominios estructurales del receptor (Modificada de Reimann et al., 2003; Shyng et al., 1998). Figura 4. Análisis de expresión de la mutación ∆ S1387. (a) Fotomarcaje de canales KATP reconstituidos con [125I] iodoazido glibenclamida. Coexpresión de Kir6.2 con: SUR1 silvestre (Carril 1), concentraciones equimolares de SUR1-∆S1387 y SUR1 silvestre (Carril 2), SUR1-∆S1387 (Carril 3) o ∆F1388, la cual afecta el tráfico a la membrana. Las bandas marcadas corresponden a la proteína inmadura de SUR1 (140 KDa), la proteína madura de SUR1 (170 KDa) y la proteína Kir6.2 (40 KDa). (b) Actividad relativa de los canales KATP, determinada al medir el eflujo de rubidio (86Rb+), inhibido por glibenclamida en células COSm6 transfectadas en las diferentes condiciones descritas en (a). Después de cargar las células con 86Rb+, las células fueron incubadas con inhibidores metabólicos (2-deoxy-glucosa y oligomicina) y diazóxido. El eflujo se midió tanto en presencia como en ausencia de 1µM de glibenclamida. Los datos son expresados como un porcentaje del eflujo inhibido por glibenclamida normalizado con respecto al total de 86Rb+ cargado en las células (Tomada de Thornton et al., 2003). Figura 5. Esquema del dispositivo experimental de registro. Los cubreobjetos sembrados con células COSm6 transfectadas con el canal KATP, eran colocados en el centro de una cámara de acrílico de 3 compartimentos la cual estaba conectada a un electrodo conectado a tierra. Para el registro de las corrientes unitarias se utilizó una micropipeta, la cual estaba conectada a un amplificador (Axopatch 200A), a través del cual la señal amplificada era enviada a un osciloscopio donde era monitoreada durante todo el experimento. A través de un convertidor analógico digital (A/D) la señal era almacenada en una computadora para su posterior análisis. Figura 6. Células COSm6. Las células COSm6 se utilizaron como un sistema de expresión heteróloga, en el cual se expresaron canales KATP recombinantes y se utilizó como marcador de la expresión la proteína verde fluorescente (GFP). Estas células fueron visualizadas en campo de contraste de fases (A) y con fluorescencia para identificar las células que dieron una expresión positiva a la GFP (B). Figura 7. Las células COSm6 no presentan una conductancia endógena de KATP. En esta figura se muestran registros obtenidos en células COSm6 que fueron transfectadas solo con la proteína verde fluorescente a un potencial de membrana de – 50 mV y mientras el parche era bañado en la cara interna y externa con una solución potasio alto (140 mM). En la condición control tanto en A como en B no se observan cambios en la corriente de membrana. La aplicación de ADP 0.1 mM (panel A), ni la aplicación de diazóxido (DZX) 0.1 mM (panel B) provocaron la activación de corrientes de potasio en estas células. Figura 8. La coexpresión de SUR1 y Kir6.2 en células COSm6 forma canales con conductancias similares al canal KATP pancreático nativo. En A se muestra la actividad registrada en la configuración de inside-out a diferentes potenciales de membrana (Vm) en condiciones de K+ simétrico ([K]E = [K]I = 140 mM), la amplitud de la corriente es mayor a potenciales de membrana negativos con respecto al potencial del mismo valor pero de signo positivo. La línea punteada indica el nivel cerrado del canal. En B se muestra la relación corriente voltaje. La línea continua representa el ajuste de los datos a la ecuación de una recta. El valor de la conductancia fue de 60 ± 4 pS (n = 4). Se graficó la media ± el error estándar de la media (EEM). Figura 9. Conductancia unitaria del canal Kir6.2/SUR1-D310N. A, Actividad del canal mutado Kir6.2/SUR1-D310N registrada a diferentes potenciales de membrana y en condiciones simétricas de K+ ([K]E = [K]I = 140 mM). B, se muestra la relación corriente voltaje, donde después de ajustar los datos a la ecuación de una recta (línea continua) se obtuvo un valor para la conductancia de 54 ± 1 pS (n = 7). Figura 10. Conductancia unitaria del canal Kir6.2/SUR1-∆ ∆ S1387. A, Actividad del canal mutado Kir6.2/SUR1-∆S1387 registrado en condiciones de potasio simétrico ([K]E = [K]I = 140 mM) a diferentes potenciales de membrana, donde la amplitud de la corriente a potenciales de membrana negativos es mayor con respecto a la amplitud a potenciales positivos. B, Relación corriente voltaje, donde la línea continua representa el ajuste de los datos a la ecuación de una recta; el valor de la conductancia para este canal fue de 56 ± 1 pS (n = 6). Figura 11. Los canales KATP pancreáticos reconstituidos presentan actividad eléctrica semejante a los canales nativos. En A, se muestra la actividad eléctrica registrada al coexpresar las subunidades Kir6.2 y SUR1 en células COSm6, los registros se realizaron a un potencial de membrana de – 50 mV en condiciones de K+ simétrico (140 mM). La actividad característica de los canales KATP, consiste de ráfagas separadas por periodos inter ráfaga en los que se aprecian aperturas breves (trazo superior). Durante una ráfaga el canal fluctúa entre el estado abierto y cerrado (Segundo trazo). La línea punteada indica el nivel cerrado del canal. En B, se muestra la probabilidad de apertura (Po) en función del potencial de membrana, y se observa que no hay una dependencia de voltaje para el canal silvestre (Kir6.2/SUR1) reconstituido. Cada círculo corresponde a la media (n =4 ) ± EEM. Figura 12. Propiedades farmacológicas de los canales KATP silvestres expresados en células COSm6. A. La actividad de los canales Kir6.2/SUR1 reconstituidos, fue bloqueada a diferentes concentraciones de ATP en presencia de 1 mM de Mg2+, el bloqueo total se observa a una concentración de 1 mM de ATP. B. Curso temporal del protocolo experimental utilizado para evaluar el efecto de diazóxido (DZX). (1) Corresponde al control, posteriormente la actividad fue reducida casi totalmente con la aplicación de ATP (1 mM), (2) Aplicación de ATP (0.1 mM) donde la actividad se recupera parcialmente y (3) durante la aplicación de DZX 0.1 mM en presencia de ATP (0.1 mM) se observa un incremento considerable en la actividad de los canales. Las barras continuas indican el tiempo de aplicación de cada uno de los fármacos. Figura 13. Los canales mutados Kir6.2/SUR1-∆ ∆ S1387 y Kir6.2/SUR1-D310N son sensibles a la aplicación de ATP. A. En esta figura se muestra el efecto de ATP sobre el canal mutado Kir6.2/SUR1D310N, la actividad de este canal fue disminuyendo conforme se aumentaba la concentración de ATP, el bloqueo total se obtuvo al aplicar 1 mM de ATP. B. La mutación ∆S1387, forma canales sensibles a ATP, y como se observa es más sensible al bloqueo por ATP, en comparación con el canal Kir6.2/SUR1-D310N. Los registros en A y B se realizaron a un potencial de membrana de –50 mV, el ATP se agregó a la solución potasio alto. Figura 14. Curva dosis respuesta al ATP en presencia de Mg 2+. La probabilidad de apertura (NPo/NPomax) de los canales fue graficada en función de la concentración de ATP, la línea continúa representa el ajuste de los datos a la ecuación de Hill (ver texto). El canal silvestre ( ) y el mutado Kir6.2/SUR1-∆S1387 ( ) presentaron una KD de 22 y 25 µM respectivamente, mientras que para el canal Kir6.2/SUR1-D310N ( ), el valor de KD fue de 53 µM. En la tabla se muestran los valores de KD y h para los 3 canales. Los experimentos se realizaron con soluciones de K+ simétrico ([K]E = [K]I = 140 mM), en presencia de Mg2+ (1 mM) y a un potencial de membrana de –50 mV. Se graficó la media ± EEM. Figura 15. Curva dosis respuesta al ATP en ausencia de Mg 2+. Se muestra la probabilidad de apertura (NPo/NPomax) en función de la concentración de ATP en ausencia de Mg2+. Los datos fueron ajustados a la ecuación de Hill (ver texto). Se obtuvieron los valores de KD para el canal silvestre ( ) y el canal Kir6.2/SUR1∆S1387 ( ) muy semejantes, 24 y 22 µM respectivamente, mientras que para el canal Kir6.2/SUR1-D310N ( ) el valor fue de 16 ± 2 µM. La tabla muestra el valor de KD y h para cada uno de los canales. Se graficó la media ± EEM. Figura 16. Efecto del análogo de ATP no hidrolizable el AMP-PNP sobre la actividad del canal silvestre. Se muestra la corriente unitaria durante la aplicación de AMP-PNP a diferentes concentraciones, para el canal Kir6.2/SUR1. Se observa como al incrementar la concentración de AMP-PNP la actividad del canal va disminuyendo hasta bloquearse cuando la concentración del análogo es de 0.3 mM. La línea punteada indica el nivel cerrado del canal. Figura 17. Curvas dosis respuesta al AMP-PNP en presencia de Mg 2+. La actividad de los canales se graficó en función de la concentración del AMP-PNP. Los datos de las curvas dosis respuesta al AMP-PNP para el canal silvestre (A), Kir6.2/SUR1-∆S1387 (B) y Kir6.2/SUR1-D310N (C) fueron ajustados a la ecuación de Hill y los valores obtenidos para la KD y h, se muestran en la tabla (D). Los experimentos se realizaron en condiciones de K+ simétrico ([K]E = [K]I = 140 mM) y en presencia de Mg2+ (1 mM) y aun potencial de membrana de –50 mV. Se graficó media ± EEM. Figura 18. Efecto del ADP sobre el canal silvestre Kir6.2/SUR1 expresado en células COSm6. En esta figura se muestran trazos representativos registrados a un potencial de membrana de –50 mV, en condiciones de K+ simétrico ([K]E = [K]I = 140 mM). El ADP se agregó a diferentes concentraciones en presencia de Mg2+ (1 mM) y ATP (0.1 mM). La probabilidad de apertura del canal aumentó conforme aumentaba la concentración de ADP. La línea punteada indica el nivel de conductancia cero. Figura 19. El canal mutado Kir6.2/SUR1-∆ ∆ S1387 pierde su sensibilidad al ADP. En esta figura se muestra la actividad del canal mutado, registrados en presencia de Mg2+ (1 mM) y de ATP (0.1 mM). Donde se observa como la actividad del canal (NPo) no cambia de manera significativa al aumentar la concentración de ADP. Estos registros se realizaron en condiciones de K+ simétrico ([K]E = [K]I = 140 mM) a un potencial de membrana de –50 mV. La línea punteada indica el nivel de conductancia cero. Figura 20. El canal Kir6.2/SUR1-D310N tiene una respuesta atípica al ADP. En esta figura se muestra la actividad característica de esta mutante, los registros se realizaron en condiciones de K+ simétrico y a un potencial de membrana de – 50 mV. Como se puede observar no hay un aumento en la actividad del canal dependiente de la concentración pero en todos las concentraciones exploradas la actividad del canal es mayor que en la condición control. La línea punteada indica el nivel de conductancia cero. Figura 21. El Kir6.2/SUR1-D310N disminuye su afinidad por ADP. En A, se muestra la curva dosis respuesta al ADP para el canal Kir6.2/SUR1 (silvestre), los datos fueron ajustados a la ecuación de Hill donde se obtuvo un valor de KD de 19 ± 6 µM (n = 6). Como se puede observar hay un incremento en la actividad del canal hasta un valor máximo y después la actividad del canal disminuye a concentraciones mayores a 0.1 mM de ADP. En B se muestra la curva dosis respuesta para la mutante D310N, el valor de KD fue de 82 µM (n = 3). El valor de KD y h se resumen en la tabla para ambos canales. Los experimentos fueron realizados a un potencial de membrana de – 50 mV. Figura 22. Efecto del diazóxido sobre la actividad del canal Kir6.2/SUR1∆ S1387. En A, se muestra el curso temporal de la actividad del canal con el protocolo experimental señalado para evaluar el efecto de diazóxido (DZX). La actividad en condiciones control (1), corresponde a K+ simétrico sin ATP. La aplicación de ATP (1 mM) bloqueó la actividad del canal. Durante la aplicación ATP (0.1 mM), la actividad se recupera parcialmente y en esta condición se aplicó diazóxido (0.1 mM) observándose como aumenta la actividad del canal. Las barras indican el tiempo de exposición de cada una de las drogas. En B, se muestra la actividad característica a un barrido mayor, para cada una de las condiciones señaladas con números: control (1), ATP (0.1 mM) (2), ATP (0.1 mM) más DZX (0.1 mM) (3). La línea punteada indica el nivel cerrado. Los experimentos fueron realizados a un potencial de membrana de –50 mV. Figura 23. Efecto del diazóxido sobre la actividad del canal Kir6.2/SUR1-D310N. En A, se muestra el curso temporal de la actividad del canal con el experimento utilizado para evaluar el efecto del diazóxido. El diazóxido fue aplicado en presencia de ATP 0.1 mM y se puede observar como aumenta la actividad del canal de manera considerable. Las barras indican el tiempo de aplicación de cada una de las drogas. En B, se observa la actividad a un barrido mayor para cada una de las condiciones indicadas con los números en A. La línea punteada indica el nivel cerrado del canal. Tabla 2. Resumen de los valores de la constante de disociación y coeficiente de Hill. Valores de la constante de disociación KD y el coeficiente de Hill (h) más menos el error estándar obtenidos bajo las diferentes condiciones experimentales para el canal silvestre y los canales mutados. Los superíndices indican los valores significativamente diferentes para una p < 0.05 analizados con la prueba estadística t de student. 1, p = 0.015; 2, p = 0.016; 3, p = 0.02 y 4, p = 0.002. Figura 24. Esquema de un modelo de la respuesta mediada por MgATP para el canal mutado Kir6.2/SUR1-D310N. Esquema del canal mutado Kir6.2/SUR1-D310N. La mutación es representada con un circulo blanco en el segmento 6 en TMD1. Cuando el parche es expuesto en su cara interna a una solución que contiene ATP (representado como: A-P-P-P) en presencia de Mg2+, el ATP se une al sitio de unión a nucleótidos NBF1, mientras que el Mg2+, se une al sitio de unión NBF2 en el receptor SUR1. Cuando esto ocurre el ATP se hidroliza, el fosfato libre (P) fosforila al fosfatidilinositol (PI) y fosfatidilinositol4-monofosfato (PIP), a través de las cinasas de fosfatidilinositol (PIPK y PI4K). Formándose así el fosfatidilinositol-4, 5-bifosfato (PIP 2). El PIP 2 se une al extremo carboxilo del canal Kir6.2 y también interactúa con el asa que une los segmentos 5 y 6 trasmembranales. La presencia de PIP 2 en este sitio impide la acción inmediata del ATP sobre el canal Kir6.2, produciéndose entonces una reactivación del canal KATP. TMD0, TMD1 y TMD2 representan los dominios 0, 1 y 2 transmembrana que forman el receptor a sulfonilureas SUR1. N y C corresponden a los extremos amino y carboxilo terminal del canal Kir6.2 y del receptor SUR1. MÉTODOS Expresión heteróloga de canales K ATP en la línea celular COSm6 En este trabajo se utilizó como sistema de expresión heteróloga la línea celular COSm6 (células de riñón de simio). En estas células se transfectaron las subunidades Kir6.2 (silvestre) y SUR1 (silvestre o mutado) para formar los canales KATP recombinantes. Utilizamos 5 plásmidos que contenían el DNA complementario (DNAc): el canal Kir6.2 de humano, el receptor SUR1 de hámster, el receptor SUR1 de hámster el cual tenía la deleción de una serina en la posición 1387 (SUR1∆S1387), receptor SUR1 de hámster con la mutación de un ácido aspártico (D) por una arginina (N) en la posición 310 (SUR1-D310N), además como marcador de la expresión se utilizó a la proteína verde fluorescente (GFP), todos estos DNAs recombinantes estaban subclonados en el plásmido pECE. El plásmido pECE es un vector para la transferencia de DNA exógeno y el cual contiene un sitio de resistencia a ampicilina. La línea celular y los plásmidos fueron donados por la doctora Lydia Aguilar-Bryan (Bayllor College of Medicine, Houston Texas). Las células COSm6 eran mantenidas en frascos para cultivo (25 mm2), en un medio de cultivo Dulbecco’s modified Eagle’s medium (DMEM) suplementado con 10 % de suero bovino fetal (FBS) y 1% de antibiótico antimicótico (penicilina G sódica 10,000 U/ml; estreptomicina 10,000 U/ml y anfotericina B 25 µg/ml).y en condiciones de 5% de CO2 a 37 º C. Transfección transitoria Las células COSm6 fueron sembradas en cajas de Petri de 33 mm de diámetro que contenían medio DMEM completo. La transfección se realizó con FuGENE 6, siguiendo la instrucción del fabricante. Brevemente consiste en lo siguiente. (1) En un tubo Eppendorff se colocaba medio mínimo esencial DMEM. (2) Posteriormente se agregaba el reactivo de FuGENE 6. (3) Después se agregaron los plásmidos pECE-Kir6.2 y pECE-SUR1 a una relación 1:3. 38 (4) Finalmente se agregaba el plásmido de la proteína verde fluorescente (GFP). Esta mezcla se dejaba reposar 15 minutos antes de agregarlo a las cajas de Petri que contenían las células. En forma paralela a la transfección del canal Kir6.2 silvestre con SUR1 silvestre, se cotransfectaba Kir6.2 silvestre con cualquiera de las mutantes del receptor pECE-SUR1-∆S1387 o pECE-SUR1-D310N, a la misma concentración. La transfección con FuGENE 6 se realizó en ausencia de suero, por lo tanto antes de agregar la mezcla anterior se cambiaba el medio completo por medio mínimo esencial DMEM (sin suero ni antibiótico), 8 horas después de haber agregado la mezcla, se cambiaba nuevamente el medio mínimo esencial por medio DMEM suplementado con suero y antibiótico. Después de la transfección teníamos células COSm6 transfectadas con el canal KATP pancreático silvestre Kir6.2/SUR1 y cualquiera de los canales mutados Kir6.2/SUR1-D310N o Kir6.2/SUR1-∆S1387. Las células eran resembradas 24 horas después de la transfección en cubre objetos de vidrio de 12 mm de diámetro. Los registros electrofisiológicos se realizaron 48 horas después de la transfección. Todas las soluciones o medios utilizados tanto en el mantenimiento de las células COSm6, así como en el proceso de transfección, fueron esterilizadas por filtración con filtros de 0.22 µm. Además ambos procesos se llevaron a cabo en condiciones estériles en una campana de flujo laminar. Registros electrofisiológicos Para los registros electrofisiológico se utilizó la técnica de patch-clamp (Hamill et al., 1981) en la configuración de inside-out. El dispositivo experimental se muestra en la figura 5, los cubre objetos en los cuales fueron sembradas las células se colocaron en el compartimiento central de una cámara de registro, en la que se colocaba un electrodo de referencia. Para visualizar las células se utilizó un microscopio invertido (Olympus modelo CK40) con óptica de contraste de fases y un sistema de fluorescencia proporcionada por una lámpara de mercurio, y un filtro de excitación con una longitud de onda en el rango de 460-490 nm. Para visualizar las 39 40 células COSm6 se utilizaba el campo de contraste de fases (panel A de la Figura 6), después de esto se disminuía la intensidad de la luz y se cambiaba al filtro de fluorescencia para seleccionar las células que tenían una expresión positiva a la proteína verde fluorescente (GFP) que fue utilizada como gen reportero (panel B de la Figura 6). En contraste de fases se colocaba la pipeta sobre la célula seleccionada. Durante todo el experimento las células fueron bañadas con la solución Ringer (ver soluciones) a través de un sistema de perfusión. Después de determinar la resistencia de la pipeta, y obtener un gigasello, se escindía el parche de membrana; la cara interna de la membrana era bañada con la solución potasio alto (ver soluciones) a través de un sistema alterno de microperfusión DAD-12 (ALA Scientific Instruments Inc). Este sistema de microperfusión consiste de una punta de cuarzo (100 µm de diámetro interno) en la cual convergen 12 micro capilares de cuarzo conectado cada uno a unas mangueras de teflón (230 µm de diámetro interno), las cuales estaban conectadas a 12 reservorios o jeringas. El flujo de las soluciones desde cada reservorio se controló con una válvula solenoide a través de una computadora. Este sistema nos permitió hacer recambio de soluciones en segundos y bañar el parche en la cara interna de la membrana. El parche de membrana era bañado con soluciones que contenían 140 mM de potasio en ambos lados de la membrana y el potencial de membrana se mantenía a -50 mV durante todo el experimento. La señal fue registrada a través de una micropipeta conectada a un amplificador (AXOPATCH 200A, Figura 5), la señal filtrada (2 KHz), amplificada y muestreada a 10 KHz, era enviada a un osciloscopio y a través de este la señal fue monitoreada durante todo el experimento. Por otro lado a través de un convertidor analógico digital (DigiData 1200, Axon Instruments, Inc.) la señal era almacenada en una computadora para su posterior análisis. Pipetas Las pipetas fueron hechas con capilares de vidrio de borosilicato (1.5 mm y 0.86 mm de diámetro externo e interno respectivamente), los cuales fueron estirados 41 42 en un puller P-97 (SUTTER INSTRUMENT) y pulidos en una micro forja. Las pipetas tenían una resistencia entre 6 a 10 MΩ al ser llenadas con la solución potasio alto (ver soluciones). Soluciones La composición de la solución de Ringer con las que las células fueron bañadas durante todo el experimento fue la siguiente (en mM): NaCl, 135; KCl, 5; CaCl2, 1; MgCl2, 1; Hepes-NaOH, 10; El pH se ajustó a 7.4 con NaOH. La pipeta se llenó con la solución potasio alto con la siguiente composición (en mM): KCl, 140; HEPESKOH, 10; y EGTA-KOH, 1. Esta solución fue ajustada a un pH de 7.3 con KOH. Una vez que se separaba el parche para formar la configuración inside-out el parche era bañado en su cara interna, con la solución potasio alto, utilizando el sistema de micro perfusión DAD-12. Análisis de resultados Se registró la corriente unitaria del canal silvestre (Kir6.2/SUR1) así como la actividad de los canales mutados Kir6.2/SUR-D310N y Kir6.2/SUR-∆S1387; y se evaluaron propiedades biofísicas como conductancia unitaria y dependencia de voltaje, además de la sensibilidad a bloqueadores y abridores específicos para estos canales La corriente unitaria fue analizada con el programa pClamp (Axon Instruments Inc.). Para determinar la conductancia unitaria y dependencia de voltaje se midió la amplitud media de la corriente unitaria, para cada potencial de membrana. Con este programa se realizó un histograma de amplitudes; la amplitud media de la corriente se calculó ajustando una función Gaussiana a los histogramas de amplitud de la corriente usando el método de mínimos cuadrados. Para calcular la conductancia unitaria, se graficó la amplitud media de la corriente obtenida para cada uno de los potenciales de membrana, y los datos fueron ajustados a la ecuación de una recta, donde la pendiente representa la conductancia unitaria (1/R = G) de acuerdo con la ley de Ohm: V = IR, donde V es el potencial de membrana, I la corriente de membrana y R la resistencia de membrana. 43 Los valores de la probabilidad de apertura (Po) en el estado estable fueron obtenidos usando el método descrito por Quayle y colaboradores (1988). Para calcular la Po se mide el tiempo (tj) que permanece en los diferentes niveles de corriente j = 0, 1, 2,.... N, para después obtener el valor de probabilidad mediante la siguiente formula Po = (Σ Nj=1 tjj)/TN, donde T es la duración del registro y N es el número de canales activos en el parche. Se usaron registros de una duración de 30 segundos. Para determinar la dependencia de voltaje, se graficó la Po calculada para cada potencial de membrana y se ajustaron a la ecuación de Boltzmann, la cual es ampliamente usada y describe la sensibilidad al voltaje de la actividad del canal. La ecuación es la siguiente: PO 1 = PΟ max 1 + exp(V0 .5 − Vm ) / k Donde Po es la probabilidad de apertura para un potencial de membrana específico (Vm). V0.5 es el potencial de membrana al cual Po corresponde al 50% de su valor máximo (Pomax), y k es una constante que nos indica la sensibilidad al voltaje. Para obtener las curvas dosis respuesta se graficó la Po (normalizada con respecto al valor máximo) en función de la concentración del ligando probado; los datos se ajustaron a la ecuación de Hill, que es la siguiente: Po 1 = Po max x h 1 + K D Donde Po, es la probabilidad de apertura del canal, X es la concentración de la droga estudiada, KD (también llamada IC 50) es la concentración a la cual se bloquea el 50 % de la actividad del canal y h corresponde al coeficiente de Hill, este valor fue interpretado como el número de moléculas que se unen al canal para bloquearlo. Todos los datos se graficaron como la media mas menos (±) el error estándar de la media (EEM). El análisis estadístico se realizó con la prueba de “t” de student, utilizando un valor de significancia menor a 0.05 (p < 0.05). 44 RESULTADOS La conductancia del canal de potasio sensible a ATP en células COSm6 es resultado de la expresión heteróloga de canales recombinantes En este trabajo se utilizaron células COSm6. Para mostrar que la actividad de los canales en estudio era resultado de la expresión heteróloga de las subunidades Kir6.2 y SUR1 y no de canales de potasio endógenos en estas células, se hicieron grupos experimentales control que fueron transfectados con el vector pECE y el gen reportero. Se realizaron registros electrofisiológicos (48 horas después de la transfección) a un potencial de membrana de -50 mV en la configuración de insideout. Los resultados obtenidos se muestran en la figura 7, los primeros registros del panel A y B de esta figura corresponden a las condiciones control, es decir cuando el parche fue bañado en la cara externa e interna con la solución potasio alto (140 mM), en esta condición no sé observó actividad de canales. Tampoco se observó respuesta alguna al aplicar adenosina difosfato (ADP) (Panel A) y diazóxido (DZX) (panel B). Con estos experimentos se descartó que las células COSm6 presenten corrientes endógenas de potasio, sensible al ATP. La expresión de las subunidades Kir6.2 y SUR1 en células COSm6 produce canales iónicos con propiedades similares a las del canal K ATP pancreático Después de mostrar que las células COSm6 no presentan una conductancia endógena del tipo KATP, se registró la actividad de estos canales Kir6.2 + SUR1 reconstituidos. Los registros se hicieron con la técnica de patch-clamp en la configuración de inside-out y en condiciones de potasio simétrico ([K]i = [K]e = 140 mM). Lo primero que se hizo fue registrar la actividad del canal a diferentes potenciales de membrana en un intervalo entre –70 a +70 mV. En la figura 8A, se muestran registros de la actividad a diferentes potenciales de membrana, para una célula que fue transfectada con el canal Kir6.2/SUR1 silvestre. Con estos datos se midió la amplitud media de la corriente correspondiente a cada potencial, graficando los valores experimentales se obtuvo la curva corriente 45 46 47 voltaje (I-V) (Figura 8B). Ajustando los datos de la curva I-V a la ecuación de una recta se obtuvo un valor para la conductancia del canal silvestre de 60 ± 4 pS (n = 4). El cálculo de la conductancia para los canales KATP mutados se hizo de la misma manera que para el canal silvestre. Así, cuando coexpresamos Kir6.2 con SUR1D310N se obtuvieron canales cuya actividad se muestra en la figura 9A, la curva I-V para este canal se muestra en el panel B de la figura 9, y el valor de la conductancia fue de 54 ± 1 pS (n = 7). En la figura 10A se muestran registros de corriente obtenidos para el canal Kir6.2/SUR1-∆S1387. La conductancia para esta mutante fue de 56 ± 1 pS (n = 6; Figura 10B), un valor semejante al de los canales silvestres y al del canal Kir6.2/Sur1-D310N. Es decir, ninguna de estas mutaciones en el receptor a las sulfonilureas altera la conductancia de los canales KATP, ya que al compararlas estadísticamente no hay cambios significativos. Se observó que la actividad basal del canal Kir6.2/Sur1-D310N es mayor en comparación con el canal silvestre y Kir6.2/SUR1-∆S1387. Los canales KATP muestran una actividad eléctrica característica, que consiste en una actividad en ráfagas separadas por periodos en los que el canal permanece cerrado y durante el cual puede haber aperturas breves (Aguilar-Bryan and Bryan, 1999). En la figura 11A se ilustra esta característica en un registro de la actividad del canal silvestre (Kir6.2/SUR1). En el registro superior se pueden observar diferentes ráfagas separadas por intervalos en los que el canal permanece cerrado (inter ráfagas); al desplegar una de las ráfagas (registro inferior) a un barrido mayor podemos ver como el canal fluctúa rápidamente entre el estado cerrado y abierto en un proceso llamado “flickering”, estos registros se hicieron en condiciones de potasio simétrico (140 mM) y a un potencial de membrana de –50 mV. Para determinar la dependencia de voltaje de estos canales KATP expresados en células COSm6, se graficó la probabilidad de apertura en estado estable en función de los diferentes potenciales de membrana, esto se muestra en la figura 11B. Como se puede ver, no hay una dependencia de voltaje en estos canales, tal como se observa en los canales K ATP nativos. 48 49 50 51 Después de estudiar algunas de sus propiedades biofísicas como la conductancia unitaria y dependencia de voltaje, se exploró la respuesta de estos canales a ATP y diazóxido. Las propiedades farmacológicas para el canal silvestre se ilustran en la figura 12; en el panel A se muestran registros de la actividad del canal a diferentes concentraciones de ATP en presencia de Mg2+ obtenidos en la configuración de inside-out a un potencial de membrana de –50 mV. Como se observa el canal fue sensible al ATP y la actividad se reduce gradualmente al aumentar la concentración de ATP, hasta bloquearse totalmente con la aplicación de 1 mM de ATP. En la figura 12B se muestra el curso temporal del protocolo experimental que utilizamos para probar el efecto de diazóxido sobre estos canales. Mediante el sistema de microperfusión DAD12 (descrito en los métodos), se bañó el parche con la solución potasio alto durante un minuto (período señalado por el número 1), después aplicamos ATP a una concentración de 1 mM, lo cual produce una disminución considerable en la actividad del canal. Posteriormente se observó una recuperación parcial de la actividad cuando se aplicó 0.1 mM de ATP (2) y mientras se aplicaba ATP a esta concentración se bañó el parche con DZX a una concentración de 0.1 mM (3) observándose que la actividad de los canales fue aumentando con una cierta latencia, alcanzando amplitudes aproximadas a 30 pA. Las mutantes ∆ S1387 y D310N forman canales de potasio que son sensibles a ATP En la figura 13 se muestra un registro representativo de la actividad de los canales reconstituidos Ki6.2/SUR1-∆S1387 y Kir6.2/SUR1-D310N en presencia de ATP. Esta figura sugiere que el canal Kir6.2/SUR1-∆S1387 (Figura 13B) es más sensible al ATP que el canal Kir6.2/SUR1-D310N ya que a concentraciones de 0.05 mM de ATP el canal con la mutación ∆S1387 ha disminuido considerablemente su actividad a diferencia del canal Kir6.2/SUR1-D310N (Figura 13A) en el que a esa misma concentración se aprecia mayor actividad del canal y esto es todavía más notorio a una concentración mayor de ATP (0.3 mM) en donde el canal ∆S1387 está prácticamente bloqueado mientras que el D310N aun muestra un canal con una 52 53 54 amplitud más pequeña. Ambos canales son totalmente bloqueados a una concentración de 1 mM de ATP. Inhibición de la actividad en función de la concentración de ATP Como se mencionó anteriormente para las mutantes y el canal silvestre se registró una actividad sensible a ATP; para cuantificar esta respuesta, se registró la actividad de estos canales a diferentes concentraciones de ATP en presencia de 1 mM de Mg2+. Las curvas dosis respuesta al ATP para cada uno de los canales se muestran en la figura 14, para ello se graficó la probabilidad de apertura (Po), normalizada con respecto a la probabilidad de apertura máxima (Pomax), en función de las diferentes concentraciones de ATP. Los datos fueron ajustados a la ecuación de Hill (Po/Pomax = 1/1+ ([ATP]/K D)h), donde KD es el valor de la constante de disociación y h corresponde al valor del coeficiente de Hill. Así se obtuvieron los valores de KD y h para el canal silvestre ( , n = 5) que fueron de 22 ± 8 µM y 0.8 ± 0.2 respectivamente. Para los canales mutados Kir6.2/SUR1-∆S1387 ( , n = 4) y Kir6.2/SUR1-D310N ( , n = 5) el valor de la KD fue de 25 ± 16 y 42 ± 4 µM, mientras que el valor de h fue de 0.6 ± 0.2 y 1.5 ± 0.1 respectivamente. La constante de disociación para el canal mutado Kir6.2/SUR1-D310N, presento un valor significativamente diferente, con respecto a la KD para los canales silvestre (p = 0.015) y mutado Kir6.2/SUR1-∆S1387 (p = 0.016). Mientras que la KD para el canal mutado Kir6.2/SUR1-∆S1387 fue similar a la del canal silvestre. Sensibilidad al ATP en ausencia de Mg 2+ Para explicar la disminución en la sensibilidad al ATP para el canal mutado Kir6.2/SUR1-D310N en presencia Mg2+, se considero que el ATP a concentraciones bajas en presencia de Mg2+ puede activar los canales KATP tal como ocurre en el proceso de reactivación (Cook y Hales, 1984; Noma A, 1983; Trube y Hescheler, 1984). Entonces a concentraciones bajas de MgATP se tiene una mezcla de los dos efectos, inhibición y activación por parte de este nucleótido. Para separar estos dos efectos y conocer la contribución del efecto activador del ATP a concentraciones 55 bajas en presencia de Mg2+, se realizaron curvas dosis respuesta al ATP en ausencia de Mg2+. Los resultados obtenido se muestran en la figura 15, donde se observa que el valor de la KD para el canal silvestre Kir6.2/SUR1 (+, n = 7) y el canal mutado Kir6.2/SUR1-∆S1387 ( , n = 5) son muy similares de 24 ± 3 y 22 ± 1 µM respectivamente, mientras que para el canal mutado D310N el valor de KD fue de 16 ± 2 µM ( , n = 8). Los valores de coeficiente de Hill fueron de 1.6 ± 0.3, 1 ± 0.06 y 1.4 ± 0.3 para los canales Kir6.2/SUR1, Kir6.2/SUR1-∆S1387 y Kir6.2/SUR1-D310N respectivamente. Comparando los valores de KD obtenidos en presencia y ausencia de Mg2+, se encontró que la mutante D310N tiene un aumento en la sensibilidad al ATP en ausencia de Mg2+, el valor de KD cambia de 42 ± 4 µM en presencia de Mg2+ a 16 ± 2 µM en ausencia de Mg2+, este cambio es significativamente diferente (ver tabla 2). El valor del coeficiente de Hill fueron similares de 1.5 ± 0.1 y 1.4 ± 0.3 en presencia y ausencia de Mg2+ respectivamente. La actividad de los canales silvestre y mutados es regulada por el AMP-PNP En la figura 16 se muestra el efecto de un análogo de ATP no hidrolizable, el adenosina 5´-(β-γ-imido) trifosfato sal de tetralitio (AMP-PNP) sobre la actividad del canal silvestre, como se puede ver al incrementar la concentración del análogo la actividad del canal va disminuyendo, hasta que se bloquea a una concentración del AMP-PNP de 0.5 mM. Para cuantificar esta respuesta se graficó la probabilidad de apertura normalizada con respecto al valor máximo, en función de las diferentes concentraciones de AMP-PNP, de esta manera se obtuvieron la curva dosis respuesta, para los canales silvestre Kir6.2/SUR1 (n = 4; figura 17A) y mutado Kir6.2/SUR1-D310N (n = 7; figura 17C). Los datos en ambas curvas fueron ajustados a la ecuación de Hill, obteniéndose valores para KD de 19 ± 3 y 20 ± 2 µM, y el coeficiente de Hill de 1.3 ± 0.2 y 0.8 ± 0.06 para los canales Kir6.2/SUR1 y Kir6.2SUR1-D310N, respectivamente. 56 57 58 59 La sensibilidad al AMP-PNP, también se realizó para el canal mutado Kir6.2/SUR1-∆S1387, el valor para la constante de disociación de 14 ± 0.03 µM y el coeficiente de Hill fue igual a 1.2 ± 0.03 (n = 6; Figura 17B). La mutación ∆ S1387 produce la pérdida de la modulación por ADP Después de evaluar la sensibilidad al ATP, se estudió el efecto de la adenosina difosfato (ADP) en cada uno de los canales, esto era importante ya que la gran mayoría de las mutaciones previamente descritas en el receptor SUR1 asociadas con PHHI muestran una respuesta alterada a la regulación por ADP (Taschenberger et al., 2002; Show-Ling et al., 1998). Se estudió el efecto activador de ADP en presencia de Mg2+ (1 mM) y ATP (0.1 mM). En la figura 18 se observa como al aplicar concentraciones crecientes de ADP incrementa la actividad del canal silvestre. Estudiar el efecto de ADP sobre el canal Kir6.2/SUR1-∆S1387, era interesante ya que esta mutación esta ubicada en NBF2, el dominio estructural del receptor SUR1 el cual modula la unión de ADP. Los resultados que se obtuvieron al aplicar diferentes concentraciones de ADP sobre el canal mutado se muestran en la figura 19 y como se aprecia no hubo un efecto de ADP sobre la actividad del canal mutado Kir6.2/SUR1-∆S1387. La mutación D310N produce una alteración en la regulación mediada por ADP El efecto de ADP sobre el canal mutado Kir6.2/SUR1-D310N se muestra en la figura 20, a diferencia de la mutación ∆S1387 si hay un efecto activador de ADP, pero esta respuesta es variable. A pesar de esto es claro también que a cualquier concentración de ADP se observa un aumento en la actividad del canal Kir6.2/SUR1D310N con respecto a la condición control. Al realizar las curvas dosis respuesta al ADP, para el canal silvestre y el canal mutado Kir6.2/SUR1-D310N, se observó que para el canal silvestre (Figura 21A) la respuesta a la aplicación de ADP es bifásica, ya que a concentraciones bajas de ADP la actividad del canal va aumentando, pero a concentraciones mayores a 0.1 mM, la actividad del canal comienza a disminuir. En esta figura se muestra solo el ajuste de los datos a una ecuación de Hill para el incremento en la actividad. Los 60 61 62 63 64 valores de KD y h para el canal silvestre fueron de 19 ± 6 µM ( ) y 1 ± 0.2 respectivamente, para un total de 6 células estudiadas. En el canal mutado D310N (Figura 21B) se observa que al aumentar la concentración de ADP, aumenta la actividad del canal, el ajuste de estos datos a la ecuación de Hill dió como resultado una KD de 82 ± µM y un valor de h de 0.7 ± 0.1 (+, n = 3). Los receptores a sulfonilureas SUR1-∆ ∆ S1387 y SUR1-D310N conservan el mecanismo de activación por diazóxido El diazóxido es el primer fármaco utilizado en el tratamiento de pacientes que presentan la PHHI, por lo tanto explorar la respuesta de los canales mutados a este fármaco tiene relevancia clínica. En la figura 22A se muestra un registro representativo con el curso temporal del protocolo experimental utilizado para probar el efecto de diazóxido sobre el canal Kir6.2/SUR1-∆S1387. El número 1 indica la condición control, es decir potasio simétrico (140 mM) y Mg2+ (1 mM), posteriormente se aplicó ATP (1 mM), y como se observa el canal fue bloqueado totalmente, al usar ATP (0.1 mM), la actividad del canal se recuperó parcialmente. Como el diazóxido requiere la presencia de un nucleótido hidrolizable (Dickinson et al., 1997; Schwanstecher et al., 1998), se aplicó el diazóxido a una concentración de 0.1 mM en presencia de ATP (0.1 mM). La aplicación de diazóxido produjo un aumento considerable en la actividad del canal Kir6.2/SUR1-∆S1387. Este aumento en la actividad del canal no es inmediato al igual que el canal silvestre tarda unos segundos en alcanzar amplitudes mayores a 40 pS. Posteriormente cuando se deja de aplicar diazóxido la actividad regresa al nivel basal. Al lavar el parche con la solución potasio alto, se puede ver la recuperación casi total de la actividad del canal. En el panel B de esa misma figura se muestran las características de la actividad a diferente barrido, para cada una de las condiciones señaladas con los números (1, 2 y 3) en el panel A. Utilizando el mismo protocolo experimental se evaluó el efecto del diazóxido sobre el canal mutado Kir6.2/SUR1-D310N (Figura 23A). Para este canal, se observó un incremento gradual de la actividad del canal (hasta 50 pA de amplitud). Posteriormente la actividad del canal regresa a su valor original cuando el parche se 65 66 67 bañó con la solución potasio alto más ATP (0.1mM). Con estos resultados se muestra que los canales mutados conservan el mecanismo de activación por diazóxido. En la tabla 2, se muestra un resumen de los valores de la constante de disociación (Kd) y el valor del coeficiente de Hill (h), obtenidas para cada uno de los canales en las diferentes condiciones experimentales, además se muestran los valores que fueron significativamente diferentes de acuerdo con la prueba estadística “t” de student. Tabla 2. Resumen de los valores de la constante de disociación y coeficiente de Hill. Valores de la constante de disociación KD y el coeficiente de Hill (h) más menos el error estándar obtenidos bajo las diferentes condiciones experimentales para el canal silvestre y los canales mutados. Los superíndices indican los valores significativamente diferentes para una p < 0.05 analizados con la prueba estadística “t” de student. 1, p = 0.015; 2, p = 0.016; 3, p = 0.02 y 4, p = 0.002. 68 DISCUSIÓN. Los canales mutados presentan propiedades biofísicas semejantes al canal silvestre Se ha mostrado que la isoforma silvestre de los canales KATP pancreáticos expresados heterólogamente en células COS-1, forma canales que presentan una conductancia unitaria de aproximadamente 65 pS en presencia de 140 mM de potasio a ambos lados de la membrana. El ATP inhibe la actividad del canal reconstituido con una constante de inhibición media (K D) de aproximadamente 10 µM en presencia de iones Mg2+. Los canales reconstituidos son casi completamente inhibidos por adenil-5- imido difosfato (AMP-PNP), un análogo de ATP no hidrolizable. Venenos metabólicos como la oligomicina y la 2-deoxi glucosa estimulan el transporte de 86 Rb+ hacia el exterior de las células COSm6 donde se coexpresaron el receptor SUR1 y el canal Kir6.2. Asimismo, una sulfonilurea como la glibenclamida bloquea la salida de 86 Rb+, mientras el diazóxido estimula el flujo iónico a través de estos canales reconstituidos. Las propiedades biofísicas y farmacológicas antes mencionadas de los canales reconstituidos son similares a las propiedades del canal KATP nativo observadas en células beta del páncreas (Aschroft et al., 1984; Cook et al., 1984; Findly et al., 1985; Rorsman y Trube, 1985). El canal Kir6.2 forma el poro de conducción de los canales KATP pancreáticos y de este canal con rectificación entrante dependen las propiedades como conductancia, dependencia de voltaje, cinética de apertura y cierre, así como la inhibición por ATP. En este trabajo el canal Kir6.2 silvestre se coexpresó con el receptor SUR1 silvestre y los receptores mutados SUR1-∆S1387 y SUR1-D310N, por lo que las propiedades biofísicas antes mencionadas fueron similares para los tres canales estudiados; las mutaciones en los receptores a sulfonilurea no alteraron dichas propiedades. Los resultados muestran que los canales Kir6.2/SUR1, Kir6.2/SUR1-D310N y Kir6.2/SUR1-∆S1387 tienen conductancias similares de 60, 54 y 56 pS, respectivamente. La actividad de los canales KATP es regulada por las concentraciones de ATP y ADP citosólicas, por ello no son dependientes de voltaje; esta característica se 70 comprobó también en todas las isoformas de los canales estudiadas. Estos resultados muestran que los canales estudiados con nuestro sistema de expresión heterólogo son canales KATP con propiedades semejantes a las descritas por otros autores para los canales K ATP reconstituidos. El poro de los canales KATP está formado por 4 subunidades del canal Kir6.2, cada una con sitios de unión al ATP; se ha propuesto que el canal conduce corriente iónica sólo cuando todas las subunidades están en una conformación abierta. Por lo tanto, en presencia del ligando se puede tener una de estas subunidades en estado cerrado y una, dos, o tres subunidades en un estado abierto; aún así, el canal no conduciría, dado que cada subunidad contribuye a generar múltiples estados cerrados del canal. De acuerdo con este modelo, la actividad característica de los canales KATP, que consiste de tiempos de apertura y cierre breves (intra ráfagas) y tiempos prolongados de cierre (inter ráfagas) puede explicarse por la cinética de las transiciones entre diferentes estados (Aguilar-Bryan and Bryan, 1999; Markworth et al., 2000) El patrón de aperturas en forma de ráfagas, característico de los canales KATP, se observó al registrar la actividad para los distintos canales expresados heterólogamente. Aunque el canal silvestre Kir6.2/SUR1 (Figura 10) y los canales mutados Kir6.2/SUR1-D310N y Kir6.2/SUR1-∆S1387 presentaron una cinética similar, los canales mutados mostraron siempre una actividad mayor con respecto a la actividad del canal silvestre. La explicación de esto aun no se sabe. Sabemos que existe una interacción entre las subunidades Kir6.2 y SUR1, que resulta en la regulación de la actividad del canal. Recientemente se demostró, utilizando proteínas quiméricas entre dos isoformas del receptor SUR (SUR1 y SUR2A), que los dominios transmembrana TMD0, TMD1 y el segmento amino terminal del receptor SUR1 contienen un determinante critico que regula la cinética en ráfagas del canal (Babenko et al., 1999). Entonces, es posible pensar que la mutación D310N (la cual está localizada en el sexto dominio transmembrana en TMD1) modifica la interacción del receptor SUR1 con el canal Kir6.2, aumentando la actividad basal del canal Kir6.2/SUR1-D310N. El mecanismo que explica el cambio en la actividad basal provocado por la asociación de Kir6.2 con el receptor mutante ∆S1387 no es claro. 71 Las propiedades biofísicas de los canales mutados Kir6.2/SUR1-D310N y Kir6.2/SUR1-∆S1387 muestran características similares al canal silvestre y a las propiedades descritas por otros autores para el canal KATP pancreático nativo y reconstituido. Inhibición por ATP Uno de los objetivos de este trabajo fue comparar la regulación por ATP entre el canal silvestre y los canales mutados, y no solamente hacer una evaluación cualitativa de esta respuesta, si no que además cuantificar esta respuesta y evaluar si la afinidad por este nucleótido tricíclico era modificada por las mutaciones D310N y ∆S1387. La actividad del canal silvestre (Kir6.2/SUR1) expresado en las células COSm6 fue suprimida por ATP en presencia de Mg2+ de manera dependiente de la concentración. El valor de KD fue similar al descrito por Gribble y colaboradores (1998) para el canal Kir6.2/SUR1, expresado en ovocitos de Xenopus. Después de evaluar el efecto de ATP sobre el canal silvestre, nos interesaba saber cual era el efecto de las mutaciones ∆S1387 y D310N sobre el mecanismo mediante el cual el receptor SUR1 regula la respuesta al ATP. Una mutación en el receptor SUR1 puede alterar el canal ya sea (1) aumentando o disminuyendo la afinidad del ATP sobre el sitio de unión, (2) alterando los mecanismos de transducción de la señal una vez que el ATP esta unido, o bien (3) dañando o alterando la habilidad del canal para cerrarse. Existen antecedentes acerca del efecto de otras mutaciones en el SUR1, asociadas con la PHHI. La mutación E1506K, descrita por Huopio y colaboradores (2000), y la mutación R1420C (Matsuo et al., 2000) que presentaron valores similares a los encontrados por otros autores para el canal silvestre (Kir6.2/SUR1) (Koster et al., 1999, y Gribble et al., 1998). Por otra parte la mutación L1551V, localizada en NBF2, produce una disminución en la sensibilidad al ATP (K D cambió de 10 a 120 µM), y en este trabajo su efecto se atribuyó a un aumento en la probabilidad de apertura del canal (Reimann et al., 2003). Las mutaciones previamente descritas E1506K, R1420C, 72 L1551V, están ubicadas en NBF2, el cual es un dominio que no está relacionado con la unión directa del ATP en el receptor SUR1, pero se ha demostrado que actúa coordinando la unión de ATP en NBF1 y que existe cooperatividad entre NBF1 y NBF2. Al evaluar la sensibilidad al ATP en presencia de Mg2+ para los canales mutados obtuvimos resultados relevantes. El valor de la KD para el canal mutado ∆S1387 fue similar a la del canal silvestre (ver Tabla 2). La mutación ∆S1387 ubicada en una región muy cercana a NBF2 tiene una sensibilidad al ATP similar al canal silvestre, esto podría explicarse porque esta mutación se localiza en un dominio estructural que no está relacionado con la unión directa del ATP al receptor SUR1. Además la mutación ∆S1387 no altera la cooperatividad de NBF2 con la unión de ATP en NBF1, porque su K D es similar a la del canal silvestre. La mutación D310N produce una alteración en la regulación por ATP Un aspecto interesante es que la mutación D310N que está localizada en el sexto dominio transmembrana en TMD1, fuera de NBF1 y NBF2, que son los dominios funcionales que regulan la unión de ATP al receptor SUR1, tiene una alteración en la regulación por ATP; su sensibilidad al ATP en presencia de Mg2+ disminuyó de manera significativa con respecto al silvestre, cambió de 22 µM para el silvestre a 42 µM para el canal mutado. Revisando el análisis funcional de otras mutaciones localizadas en TMD1, como la mutación F591L (Shyng et al., 1998), la cual se comparó con otras mutaciones localizadas en NBF2, y en regiones cercanas a este sitio (G1382S y R1394H) así como con otras mutaciones localizadas fuera de los sitios de unión NBF1 y NBF2 (H125Q, N188S, T1139M y R1215Q). De todas ellas, sólo la mutación F591L mostró una alteración, incrementando ligeramente su sensibilidad al ATP. Aunque los resultados en este trabajo resulten contradictorios a los que nosotros obtuvimos, esto de alguna manera nos indica que mutaciones ubicadas en TMD1 pueden alterar la respuesta al ATP después de que se une a NBF1. Considerando que TMD1 puede determinar el comportamiento en ráfagas del canal, es posible que la mutación D310N podría alterar la actividad cinética del 73 canal, o que tal vez existe un mecanismo adicional que aumenta la actividad del canal mutado Kir6.2/SUR1-D310N. Es decir, que el cambio en la KD para el ATP puede ser sólo aparente, generándose como resultado de un cambio en el mecanismo de transducción del acoplamiento entre las subunidades, y no de la alteración en la unión de ATP a la subunidad Kir6.2. El ATP en presencia de Mg 2+ produce reactivación del canal En la gran mayoría de los parches de membrana celular que se obtuvieron, los canales KATP perdieron su actividad al ser separados y colocados en la solución potasio alto libre de ATP, es decir, entraron en el proceso llamado “rundown”. El “rundown” sugiere que al momento de separar el parche hay una pérdida de factores intracelulares indispensables para mantener integra la funcionalidad de los canales KATP. Se ha demostrado que en la gran mayoría de los casos de rundown, la actividad es restablecida con la aplicación de MgATP (Furukawa et al., 1994), es decir hay un aumento de la actividad en presencia de MgATP. El mecanismo mediante el cual se recupera la actividad del canal en presencia de MgATP aún no ha sido entendido totalmente; y se ha propuesto la hidrólisis del ATP y la fosforilación de lípidos de la membrana como posibles mecanismos de regulación positiva. En este sentido, se ha demostrado que el PI-4,5-P2 (PIP 2) que se forma en la membrana plasmática por la fosforilación del fosfatidilinositol (PI) y fosfatidilinositol-4 monofosfato (PIP) vía cinasas de fosfatidilinositol, activa los canales KATP, (Hilgemann and Ball, 1996; Fan and Makielski, 1997). Además, la fosforilación mediada por las cinasas de fosfatidilinositol favorece la recuperación de la actividad con MgATP (Hilgemann and Ball, 1996). Se ha demostrado que la disminución del PIP 2 en la membrana y la inhibición de las cinasas de fosfatidilinositol con wortmanina (considerada por algunos autores como un inhibidor especifico de las cinasa 3 y 4 de fosfatidilinositol), evitan que se recupere la actividad de los canales KATP en presencia de MgATP después de caer en rundown (Hilgemann and Ball, 1996; Xie et al., 1999; Nakanishi et al., 1995). Como un posible mecanismo del efecto de PIP 2, se ha sugerido que este lípido actúa sobre la red de filamentos de actina del citoesqueleto, el cual puede interactuar con los canales KATP (Furukawa et 74 al., 1994). Por el contrario, Fan y Makielski (1997) mostraron que existe una interacción electrostática de las regiones aniónicas de PIP 2 y los aminoácidos cargados positivamente en el extremo carboxilo del canal Kir6.2. Como consecuencia de lo anterior, al aplicar MgATP a bajas concentraciones, se puede obtener una mezcla de los procesos de “rundown” y de reactivación de los canales KATP; esto nos hizo pensar que probablemente el proceso de reactivación es el que produce el aumento en la actividad del canal mutado generando un cambio aparente en la constante KD del ATP. Para estudiar esta hipótesis se utilizó la wortmanina (inhibidor de las cinasas PI3 y PI4) registrando la actividad de los canales Kir6.2/SUR1-D310N en la configuración de inside-out. Observamos que después de sufrir rundown, la actividad del canal fue restablecida con ATP 20 µM en presencia de Mg2+; al aplicar wortmanina 100 µM en esta condición, su efecto fue variable (datos no mostrados). En algunos parches la droga parecía inhibir, mientras que en otros aumentar la actividad del canal, o incluso parecía no tener ningún efecto. Según nuestra hipótesis, esperábamos que si había una reactivación del canal mediada por PIP 2 en presencia de MgATP, entonces debería haber una disminución en la actividad ya que la wortmanina actúa bloqueando la acción de la cinasas que favorecen la producción de PIP 2. La variabilidad en nuestros resultados indica que o bien el mecanismo involucrado es distinto, o la wortmanina no es un fármaco específico para estudiar la participación de los fosfoinosítidos de la membrana. Debido a lo anterior, retomamos el proceso de reactivación en presencia de MgATP y, considerando que la hidrólisis del ATP y el Mg2+ son necesarios para la reactivación, decidimos estudiar el efecto de Mg2+ sobre la disminución en la sensibilidad al ATP que presentó la mutante D310N. Para esto realizamos curvas dosis respuesta al ATP en ausencia de Mg2+ (Figura 16). La constante de disociación tanto para el canal Kir6.2/SUR1 y Kir6.2/SUR1-∆S1387 fueron similares 24 y 22 µM respectivamente. Además los valores de la constante de disociación fueron muy semejantes para estos canales en presencia y ausencia de Mg2+. Lo que parece interesante es que el coeficiente de Hill para el canal silvestre cambió de 0.8 en presencia a 1.6 en ausencia de Mg2+. Desde el punto de vista farmacológico, esto 75 significa que en ausencia de Mg2+ se requieren dos moléculas de ATP para tener el mismo efecto sobre el canal. Una posible explicación al aumento en el coeficiente de Hill, es que la molécula de ATP contiene 4 cargas negativas concentradas en una estructura lineal de polifosfatos (Lehninger, 1982), esta estructura geométrica de la molécula de ATP tiene gran importancia funcional porque esta disposición geométrica le permite interactuar con los motivos A y B de los sitios de unión a nucleótidos en el receptor SUR1 (Walker et al., 1982; Yoshida and Amano, 1995). Durante la unión de ATP al receptor SUR1, el magnesio juega un papel importante ya que estabiliza la disposición geométrica del ATP. La disposición lineal de los fosfatos hace que estos se puedan unir directamente a los motivos A y B del sitio NBF1, y a su vez permite que el magnesio pueda interactuar con el motivo A y con moléculas de agua en el motivo B del sitio NBF2 (Szewczyk and Pikula, 1998). Es posible que en ausencia de Mg2+ se requieran dos moléculas de ATP, que de alguna manera interactúan para lograr la misma estabilidad energética y geométrica en el sitio NBF1 que se logra en presencia de Mg2+. Pero entonces uno se podría preguntar que es lo que pasa con el canal mutado Kir6.2/SUR1-∆S1387 en el cual el coeficiente de Hill es aproximadamente el mismo en presencia o en ausencia de magnesio. La explicación más probable es que, ya que esta mutación esta ubicada en el sitio NBF2, altere la cooperatividad entre NBF1 y NBF2 y el efecto del Mg2+ es mínimo. Al evaluar la respuesta al ATP en ausencia de Mg2+, para el canal Kir6.2/SUR1-D310N se obtuvieron resultados aún más interesantes, para este canal mutado se observó que en ausencia de Mg2+ la curva dosis respuesta estaba desplazada a la izquierda en comparación con la curva obtenida en presencia de Mg2+; El valor de KD cambió de 42 µM a 16 µM en presencia y ausencia de Mg2+ respectivamente, mientras que el coeficiente de Hill no cambió. Con este resultado se pensó que la disminución en la sensibilidad al ATP para la mutación D310N dependía de Mg2+, y que este catión estaba involucrado con un mecanismo que aumentaba la actividad del canal y por lo tanto estaría produciendo una disminución aparente en la sensibilidad al ATP. Con estos resultados se plantea otras interrogantes, como saber si este efecto de Mg2+, dependía solo del Mg2+ o de un 76 mecanismo que involucraba ATP en presencia de Mg2+. Para saber si era un proceso que dependía de ATP en presencia de Mg2+, se probó el efecto de un análogo de ATP no hidrolizable, como lo es el AMP-PNP en presencia de Mg2+. El efecto del AMP-PNP sobre los canales Kir6.2/SUR1 y Kir6.2/SUR1-∆S1387, los valores de la KD fueron similares a los obtenidos para estos mismo canales cuando se aplicó ATP, tanto en presencia como en ausencia de Mg2+. Los valores del coeficiente de Hill fueron similares a los obtenidos con ATP en ausencia de Mg2+. Esto llama la atención, ya que se espera que el AMP-PNP en presencia de Mg2+ tenga un coeficiente de Hill similar al obtenido para el ATP en presencia de Mg2+. A pesar de que este cambio no puedo explicarlo, estos resultados demuestran que la mutación ∆S1387 conserva su mecanismo de regulación por ATP. Lo más interesante del efecto de AMP-PNP, se obtuvo al probar este análogo de ATP no hidrolizable, con el canal mutado Kir6.2/SUR1-D310N, el valor de KD que obtuvimos fue de 20 ± 2 µM. Este valor es muy similar al valor de KD obtenido para este canal cuando se aplicó ATP en ausencia de Mg2+. De hecho, es muy similar al valor de la KD para ATP en presencia de Mg2+ obtenida para el canal silvestre. Con este resultado pensamos que el aumento en la sensibilidad al ATP en presencia de Mg2+ (42 ± 4 µM) que se obtuvo con la mutante D310N fue solo aparente, ya que con este análogo no hidrolizable la sensibilidad al ATP aumenta, por lo tanto pensamos que la mutante D310N en presencia de MgATP aumenta la actividad a través de un mecanismo que involucra la hidrólisis del ATP, y este mecanismo no involucra la acción directa de Mg2+. Modelo del mecanismo de alteración en la respuesta mediada por ATP para el canal mutado Kir6.2/SUR1-D310N La regulación por nucleótidos para los canales KATP esta en constante estudio, sobre todo para explicar procesos complejos como la reactivación del canal en presencia de MgATP, para la cual se han propuesto varios mecanismos que relacionan tanto a las proteínas del citoesqueleto (Furukawa et al., 1996), como a la fosforilación y desfosforilación (Olesen and Bundgaard, 1993). Recientemente el mecanismo que ha despertado más interés es la interacción del PIP 2 con el extremo 77 carboxilo del canal Kir6.2, que resulta en una marcada activación del canal (Baukrowitz and Fakler, 2000). Basándonos en esta investigaciones, así como en las aportaciones conceptuales descritas para la regulación de nucleótidos por los grupos de investigación de Ashcroft, Bryan y Aguilar-Bryan, entre otros, tratamos de interpretar las diferencias observadas en el canal silvestre y en la mutante D310N al análogo no hidrolizable AMP-PNP, y al ATP, tanto en presencia como en ausencia de Mg2+ obtenidas, mediante el siguiente modelo mecanístico (Figura 24). Si consideramos que en el canal silvestre, se produce la hidrólisis de ATP cuando este se une al receptor SUR1 en el dominio NBF1 y el Mg2+ al NBF2 dicha hidrólisis favorece la formación de PIP 2, a través de la fosforilación de PI y PIP. Este último podría entonces interaccionar con el extremo carboxilo del canal Kir6.2, lo cual mantendría al canal en un estado en el que el ATP no puede unirse a la subunidad Kir6.2. Así, el canal no podría ser bloqueado por completo dando origen a una cierta reactivación del canal. Ahora bien conforme se va aumentando la concentración de ATP y los componentes intracelulares como el PI y las enzimas que favorecen la formación de PIP 2 se “gastan”, entonces el canal pasa a un estado en el que se favorece la acción de ATP en el extremo carboxilo del canal Kir6.2 y también la presencia de ATP en los sitios de unión a nucleótidos en el receptor SUR1 favorecen el estado cerrado del canal, mediante cambios alostéricos en los TMD0 y TMD1 del receptor. En el caso de la mutante D310N, la presencia de una alteración en una región muy cercana en el asa que une el TMD0 con TMD1, es probable que exista una interacción de este dominio con PIP 2 y de este último, a su vez, con el extremo carboxilo del canal Kir6.2. Esta estructura compleja del canal hace que la interacción del ATP con el canal Kir6.2 sea aun más complicada y muy probablemente exista un cambio alostérico en la macromolécula que favorezca el estado abierto del canal. A pesar de que igual durante el lavado puedan eliminarse los componentes intracelulares necesarios para la formación de PIP 2, es muy probable que las interacciones electrostáticas entre el extremo carboxilo, PIP 2 y el asa que une TMD0 y TMD1 sean tan fuertes, que PIP 2 se mantenga unido, interfiriendo con la interacción de ATP con el canal Kir6.2. Por esta razón cuando aplicamos ATP en 78 79 ausencia de Mg2+ o agregamos AMP-PNP en presencia de Mg2+, en ninguno de los dos casos se favorece la formación de PIP 2 y a pesar de que la mutación D310N produzca una alteración alostérica esta no es suficiente para impedir que el ATP se una al extremo carboxilo del canal Kir6.2. El canal mutado Kir6.2/SUR1-∆ ∆ S1387 pierde su regulación por ADP Las mutaciones en el receptor SUR1 asociadas con la PHHI provocan diferentes alteraciones en el canal, una de estas alteraciones es la regulación por nucleótidos, por lo que uno de nuestros objetivos era estudiar el efecto de ADP sobre los canales mutados. De acuerdo con los resultados obtenidos el canal Kir6.2/SUR1∆S1387 no respondió a la acción estimulante de ADP, a diferencia del canal Kir6.2/SUR1-D310N que incrementó su actividad. La mutación ∆S1387 está ubicada en una región muy cercana al NBF2 que es el sitio de unión de ADP, esto explica que el canal que presenta esta mutación pierda la regulación por ADP y por esto pensamos que el hecho de que este canal no detecte el cambio en el ADP es la alteración funcional que genera el síndrome PHHI en los pacientes que presentan esta mutación. Al no responder al ADP el canal se mantiene cerrado ya que solo detecta el incremento en el ATP, al estar cerrado la membrana se despolariza activando los canales de calcio dependientes de voltaje, y el incremento en la concentración de calcio intracelular es lo que genera la liberación no regulada de insulina. El canal mutado Kir6.2/SUR1-D310N sí fue activado por el ADP y esto se explica por el hecho de que esta mutación esta ubicada en el sexto segmento transmembrana del TMD1 y por lo tanto el sitio de unión al ADP en el NBF2, no es alterado. Sin embargo la respuesta del canal mutado al ADP fue variable en comparación con el canal silvestre, y esto quizás tenga alguna relación por el hecho de que a pesar de que esta mutante no está alterando el sitio de unión del ADP, sí está ubicada en un dominio (TMD1) que está relacionado con la regulación de la cinética del canal. 80 Los canales mutados son activados por diazóxido Estudiar el efecto de las mutaciones (D310N y ∆S1387) en el receptor SUR1, sobre la acción del diazóxido un abridor especifico de los canales KATP fue muy importante, porque el diazóxido se utiliza en el tratamiento de pacientes que presentan la PHHI, y porque la acción de esta droga es variable, ya que algunos pacientes responden mientras que otros no lo hacen. Esto significa que algunas mutaciones en el receptor SUR1 alteran de tal manera al canal que impiden que el diazóxido se una. Se puede pensar que la ubicación topológica de las mutaciones en el receptor SUR1 son importantes ya que a pesar de que el sitio de unión al diazóxido no se ha establecido, si se han determinado segmentos transmembrana (6-11) y los sitios de unión a nucleótidos NBF1 y NBF2 son importantes para la acción de esta droga y que por lo tanto mutaciones en estos dominios del canal provocaría la anulación del efecto de esta droga. En este trabajo nosotros mostramos dos mutaciones nuevas en el receptor SUR1, la ∆S1387 la cual está ubicada en una región muy cercana al NBF2 y la D310N ubicada en el sexto segmento transmembrana del TDM1, a pesar de la ubicación de estas mutaciones, nosotros encontramos que la aplicación de diazóxido (100 µM) provocó la activación de los canales mutados Kir6.2/SUR1-D310N y Kir6.2/SUR1-∆S1387 (Figura 23 y 24), estos resultados aún no podemos explicarlos. Considerando que la acción del diazóxido requiere la presencia de un nucleótido hidrolizable y de Mg2+, nos preguntamos como es que el canal mutado Kir6.2/SUR1-∆S1387 responde al diazóxido, a pesar de que este canal no respondió a la aplicación de ADP en presencia de magnesio. Si la mutación ∆S1387, esta ubicada en una región muy cercana al segundo sitio de unión a nucleótidos NBF2, es muy probable que este alterando la unión del MgADP en el sitio de unión a nucleótidos NBF2; sin embargo el sitio de unión a nucleótidos NBF1 esta intacto y a este sitio se une el ATP, es probable que ocurra la hidrólisis en ese sitio o que solo la unión de ATP en NBF1 sea suficiente para permitir la unión de diazóxido a su sitio de unión en el receptor y favorece el estado desacoplado del receptor SUR1 con el canal Kir6.2 y por lo tanto provoca el aumento de la actividad del canal. 81 CONCLUSIONES 1) El canal Kir6.2/SUR1 y los mutados Kir6.2/SUR1-D310N y Kir6.2/SUR1∆S1387 expresados heterólogamente presentaron propiedades biofísicas similares a los canales KATP nativos pancreáticos. Esto es consistente con la idea de que dichas características están principalmente determinadas por la subunidad Kir6.2, e indica que la línea celular COSm6 es un sistema heterólogo adecuado para estudiar la relación estructura-función en los canales sensibles al ATP. 2) El mecanismo de inhibición por ATP en el canal mutado Kir6.2/SUR1-∆S1387 es semejante al del canal silvestre. Esto significa que esta mutación está ubicada en un dominio estructural del receptor que no altera la unión de ATP. 3) La mutación D310N en el receptor SUR1, disminuyó la sensibilidad al ATP en presencia de Mg2+. 4) La disminución aparente en la sensibilidad al ATP que se obtuvo para el canal Kir6.2/SUR1-D310N, involucra un mecanismo que depende de la hidrólisis del ATP y magnesio; esto sugiere que en el canal mutante hay un aumento en la reactivación, probablemente dependiente de un mecanismo de fosforilación. 5) La mutación D310N altera el mecanismo de activación por ADP. El mecanismo subyacente no ha sido explorado pero podría asociarse a cambios alostéricos que modifican la exposición de los sitios de unión al receptor SUR1. 6) El canal Kir6.2/SUR1-∆S1387 pierde la regulación por este nucleótido, lo cual es consistente con una alteración en el dominio estructural del receptor SUR1 al cual se une este nucleótido, y explicaría la hipersecreción de insulina en los portadores de esta mutación 82 7) Los canales mutados Kir6.2/SUR1-∆S1387 y Kir6.2/SUR1-D310N conservan su mecanismo de activación por diazóxido. Esto tiene relevancia clínica ya que éste es el principal fármaco utilizado en el tratamiento de pacientes con PHHI. 83 CONCLUSIONES 1) El canal Kir6.2/SUR1 y los mutados Kir6.2/SUR1-D310N y Kir6.2/SUR1∆S1387 expresados heterólogamente presentaron propiedades biofísicas similares a los canales KATP nativos pancreáticos. Esto es consistente con la idea de que dichas características están principalmente determinadas por la subunidad Kir6.2, e indica que la línea celular COSm6 es un sistema heterólogo adecuado para estudiar la relación estructura-función en los canales sensibles al ATP. 2) El mecanismo de inhibición por ATP en el canal mutado Kir6.2/SUR1-∆S1387 es semejante al del canal silvestre. Esto significa que esta mutación está ubicada en un dominio estructural del receptor que no altera la unión de ATP. 3) La mutación D310N en el receptor SUR1, disminuyó la sensibilidad al ATP en presencia de Mg2+. 4) La disminución aparente en la sensibilidad al ATP que se obtuvo para el canal Kir6.2/SUR1-D310N, involucra un mecanismo que depende de la hidrólisis del ATP y magnesio; esto sugiere que en el canal mutante hay un aumento en la reactivación, probablemente dependiente de un mecanismo de fosforilación. 5) La mutación D310N altera el mecanismo de activación por ADP. El mecanismo subyacente no ha sido explorado pero podría asociarse a cambios alostéricos que modifican la exposición de los sitios de unión al receptor SUR1. 6) El canal Kir6.2/SUR1-∆S1387 pierde la regulación por este nucleótido, lo cual es consistente con una alteración en el dominio estructural del receptor SUR1 al cual se une este nucleótido, y explicaría la hipersecreción de insulina en los portadores de esta mutación 82 7) Los canales mutados Kir6.2/SUR1-∆S1387 y Kir6.2/SUR1-D310N conservan su mecanismo de activación por diazóxido. Esto tiene relevancia clínica ya que éste es el principal fármaco utilizado en el tratamiento de pacientes con PHHI. PERSPECTIVAS En este trabajo, gracias al uso de técnicas como la biología molecular y la electrofisiología, estudiamos la relación entre la estructura y la función de dos nuevas mutaciones (D310N y ∆S1387) en el receptor SUR1, las cuales están asociadas con el síndrome llamado PHHI. A partir de los resultados en este trabajo y con el propósito de esclarecer algunas interrogantes acerca de la regulación por nucleótidos en los canales KATP en condiciones patológicas, se desprenden líneas de investigación para cubrir los siguientes aspectos: De acuerdo con nuestros resultados la mutación D310N en el receptor SUR1 coexpresado con el canal Kir6.2, forma canales que tienen una respuesta atípica al ADP y una disminución en la sensibilidad por ATP en presencia de Mg2+. Ya que la mutación D310N esta ubicada en un dominio estructural del receptor SUR1 que no interfiere con los sitios de unión a nucleótidos, cómo es posible que el canal Kir6.2/SUR1-D310N tenga alteraciones en la regulación por nucleótidos. Para responder a esta pregunta, realizamos experimentos para explicar la disminución en la sensibilidad al ATP en presencia de Mg2+, que nos llevaron a la conclusión de que la alteración en la respuesta mediada por ATP en presencia de Mg2+, involucra un mecanismo de fosforilación probablemente a través de PIP 2. Entonces, sería importante probar alguna cinasa de fosfatidilinositol especifica para el PIP 2, para estudiar la propuesta de que el mecanismo de fosforilación involucra la participación de PIP 2. Los canales que presentan la mutación ∆S1387, tienen una mejor correlación entre el defecto genético y la alteración en la función del canal, ya que su ubicación cerca del sitio de unión al ADP, genera canales que pierden su regulación por este 83 nucleótido. Mientras que su respuesta al ATP es semejante al del canal silvestre, ya que el sitio de unión de este nucleótido permanece intacto. Aun cuando la respuesta al diazóxido requiere la presencia de ADP y el canal Kir6.2/SUR1-∆S1387 pierde su regulación por este nucleótido, no se sabe el mecanismo por el cual este canal responde al diazóxido. Esta interrogante genera otra línea de investigación, para tratar de esclarecer el mecanismo de acción del principal fármaco utilizado en el tratamiento de la PHHI, lo cual generaría mayores conocimientos acerca del mecanismo de acción de los abridores de los canales KATP, información que sería importante para la producción de nuevos fármacos alternativos al tratamiento de la PHHI. De acuerdo con nuestros resultados la actividad basal para los canales mutados fue mayor con respecto al silvestre. Aunque propusimos que la mutación D310N, aumenta la actividad del canal, porque está ubicada en un dominio estructural del receptor SUR1, que se ha demostrado regula la actividad en ráfagas de los canales KATP, es necesario investigar en ambas mutantes las alteraciones que producen y llevan al aumento en el número de canales funcionales en la membrana. 84 7) Los canales mutados Kir6.2/SUR1-∆S1387 y Kir6.2/SUR1-D310N conservan su mecanismo de activación por diazóxido. Esto tiene relevancia clínica ya que éste es el principal fármaco utilizado en el tratamiento de pacientes con PHHI. PERSPECTIVAS En este trabajo, gracias al uso de técnicas como la biología molecular y la electrofisiología, estudiamos la relación entre la estructura y la función de dos nuevas mutaciones (D310N y ∆S1387) en el receptor SUR1, las cuales están asociadas con el síndrome llamado PHHI. A partir de los resultados en este trabajo y con el propósito de esclarecer algunas interrogantes acerca de la regulación por nucleótidos en los canales KATP en condiciones patológicas, se desprenden líneas de investigación para cubrir los siguientes aspectos: De acuerdo con nuestros resultados la mutación D310N en el receptor SUR1 coexpresado con el canal Kir6.2, forma canales que tienen una respuesta atípica al ADP y una disminución en la sensibilidad por ATP en presencia de Mg2+. Ya que la mutación D310N esta ubicada en un dominio estructural del receptor SUR1 que no interfiere con los sitios de unión a nucleótidos, cómo es posible que el canal Kir6.2/SUR1-D310N tenga alteraciones en la regulación por nucleótidos. Para responder a esta pregunta, realizamos experimentos para explicar la disminución en la sensibilidad al ATP en presencia de Mg2+, que nos llevaron a la conclusión de que la alteración en la respuesta mediada por ATP en presencia de Mg2+, involucra un mecanismo de fosforilación probablemente a través de PIP 2. Entonces, sería importante probar alguna cinasa de fosfatidilinositol especifica para el PIP 2, para estudiar la propuesta de que el mecanismo de fosforilación involucra la participación de PIP 2. Los canales que presentan la mutación ∆S1387, tienen una mejor correlación entre el defecto genético y la alteración en la función del canal, ya que su ubicación cerca del sitio de unión al ADP, genera canales que pierden su regulación por este 83 nucleótido. Mientras que su respuesta al ATP es semejante al del canal silvestre, ya que el sitio de unión de este nucleótido permanece intacto. Aun cuando la respuesta al diazóxido requiere la presencia de ADP y el canal Kir6.2/SUR1-∆S1387 pierde su regulación por este nucleótido, no se sabe el mecanismo por el cual este canal responde al diazóxido. Esta interrogante genera otra línea de investigación, para tratar de esclarecer el mecanismo de acción del principal fármaco utilizado en el tratamiento de la PHHI, lo cual generaría mayores conocimientos acerca del mecanismo de acción de los abridores de los canales KATP, información que sería importante para la producción de nuevos fármacos alternativos al tratamiento de la PHHI. De acuerdo con nuestros resultados la actividad basal para los canales mutados fue mayor con respecto al silvestre. Aunque propusimos que la mutación D310N, aumenta la actividad del canal, porque está ubicada en un dominio estructural del receptor SUR1, que se ha demostrado regula la actividad en ráfagas de los canales KATP, es necesario investigar en ambas mutantes las alteraciones que producen y llevan al aumento en el número de canales funcionales en la membrana. 84 REFERENCIAS 1. Aguilar-Bryan, L., Nichols, C. G., Wechsler, S. W., Clement, J. P., Boyd, A. E., Gonzalez, G., Herrera Sosa, H., Nguy, K., Bryan, J., Nelson, D. A. 1995. Cloning of the beta cell-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 268: 423-426. 2. Aguilar-Bryan, L., Clement, J., Gonzalez, G., Kunjilwar, K., Babenko A and Bryan, J. 1998. Toward understanding the assembly and structure of KATP channels. Physiol. Rev. 78: 227-245. 3. Aguilar-Bryan, L and Bryan, J. 1999. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocrinol. Rev. 20 (2): 101-135. 4. Aguilar-Bryan, L., Bryan, J and Nakazaki, M. 2001.Of mice and Men: KATP channels and insulin secretion. The Endocrine Society. Recent Prog Horm Res. 56: 47-68. 5. Alvarez-Leefmans, F. J., Giradse, F., Gamino, S. M. 1987. Intracellular free magnesium in excitable cells: its measurement and its biologic significance. Can J Physiol Pharmacol. 65: 915-925. 6. Ashcroft, F. M., Harrison, D. E., Ashcroft, S. J. H. 1984. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature. 312: 446-448. 7. Ashcroft, FM., Kakei, M. 1989. ATP-sensitive K+ channels in rat pancreatic βcell: modulation by ATP and Mg2+ ions. J Physiol (Lond) 416: 349-367. 8. Ashcroft, F. M. 1988. Adenosine 5’-triphosphate-sensitive potassium channels. Annu. Rev. Neurosci. 11: 97-118. 9. Ashcroft, F. M and Gribble, F. M. 1998. Correlating structure and function in ATP-sensitive K + channels. Trends Neurosci. 21 (7): 288-294. 10. Ashcroft, F. M and Gribble, F. M. 2000b. New windows on the mechanism of action of K ATP channel openers. Trends Neurosci. 21 (11): 439-445. 11. Ashfield, R., Gribble, FM., Ashcroft SJH., Ashcroft FM. 1999. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the KATP channel. Diabetes, Vol. 48: 1-7. 12. Aynsley-Green, A., Polak, J., Bloom, S., Gough, M., Keeling, J., Ashcroft, S et al. 1981. Nesidioblastosis of the pancreas: definition of the syndrome and the management of the severe neonatal hyperinsulinaemic hypoglycaemia. Arch Dis Child. 56: 496-508. 13. Aynsley-Green, A., Dunne, M., James, R., Lindley, K. 1998. Ions and genes in persistent hyperinsulinaemic hypoglycaemia of infancy; a commentary on the implications for tailoring treatment to disease pathogenesis. J. Peadiatr. Endocrinol Metab. 11: 121-129. 14. Aynsley-Green, A., Hussain, K., Hall, J., Saudubray, J. M., Nihoul-Fékété, C., De Lonlay-Debeney, P., Brunelle, F., Otonkoski, T., Thornton, P & Lindley, K. J. 2000. The practical management of hyperinsulinism in infancy. Archives of Diseases in Children Fetal and Neonatal Education. 82. F98-F107. 85 15. Azzaria, M., Schurr, E and Gros, P. 1989. Discrete mutations introduce in the predicted nucleotide binding sites of the mdr-1 gene abolish its ability to confer multidrug resistance. Mol. Cell. Biol. 9: 5289-5297. 16. Babenko, A. P., Aguilar-Bryan, L., Bryan, J. 1998a. A view of SUR/K ir6.x, KATP channels. Annu Rev Physiol 60: 667-687. 17. Babenko, A. P., Gonzalez, G., Aguilar-Bryan, L., Bryan, J. 1998b. Reconstituted human cardiac KATP channels: functional identity with the native channels from the sarcolemma of human ventricular cells. Circ. Res. 83 (11): 1132-1143. 18. Babenko, A. P., Gonzalez, G., Bryan, J. 1999a. Two regions of sulfonylurea receptor specify the spontaneous bursting and ATP inhibition of KATP channel isoforms. J Biol Chem. 274(17). 11587-11592. 19. Babenko, A. P. Gonzalez, G. Bryan,J. 1999b The tolbutamide site of SUR1 and a mechanism for its functional coupling to KATP channel closure. FEBS Letters. 459. 367-376. 20. Babenko, A. P., Gonzalez, G., Aguilar-Bryan, L., Bryan J. 1999c. Sulfonylurea receptors set the maximal open probability, ATP sensitivity and plasma membrane density of K ATP channels. FEBS letters 445: 131-136. 21. Babenko, A. P., Gonzalez, G., Bryan, J. 2000. Pharmaco-topology of sulfonylurea receptors. Separate domains of the regulatory subunits of KATP channel isoforms are required for selective interaction with K+ channel openers. J. Biol. Chem. 275(2): 717-720. 22. Barnes, P. D., O´Brien, R. E., Cosgrove, K. E., Abdel-Wahab, Y. H. A., Flatt, P. R., Aynsley-Green, A., & Dunne, M. J. 2000. The hyperglycaemic effects of hydrochlorothiazide involve activation of potassium channels in insulin secreting cells. Archives of Diaseases in Childhood. 82 (Suppl. 1). P24. 23. Baukrowitz, T., Flaker, B. 2000. KATP channels gated by intracellular nucleotides and phospholipids. Eur. J. Biochem. 267, 5842-5848. 24. Bell, G. I., Kayano, T., Buse, J. B., Burant, C. F., Takeda, J et al. 1990. Molecular biology of mammalian glucose transporters. Diabetes Care. 13: 198208. 25. Bernardi, H., De Weille, J. R., Epelbaum, J., Moure, C., Amoroso, S., et al. 1993. ATP modulated K+ channels sensitive to antidiabetic sulfonylureas are present in adenohypophysis and are involved in growth hormone release. Proc. Natl. Acad. Sci. USA. 90: 1340-44. 26. Bray, K. M., Quast, U. A specific binding site for K+ channel openers in rat aorta. 1992. J. Biol. Chem. 267 (17): 11689-11692. 27. Bretscher, A. 1991. Microfilament structure and function in the cortical cytoskeleton. Annu Rev Cell Biol. 7:337-374. 28. Bokvist, K., Ämmälä, C., Ashcroft, F. M., Berggren, P. O., Larsson, O & Rorsman, P. 1991. Separate processes mediate nucleotide-induced inhibition and stimulation of the ATP-regulated K(+)-channels in mouse pancreatic betacells. Proc. R. Soc. London Ser. B. 243: 139-144. 29. Boyd, A. E. III. 1988. Sulfonylurea receptors, ion channels, and fruit files. Diabetes. 37: 847-50. 86 30. Carson, M. R., Winter, M. C., Travis, S. M, and Welsh, M. J. 1995. Pyrophosphate stimulates wild-type and mutant cystic fibrosis transmembrane conductance regulator Cl- channels. J. Biol. Chem. 270: 20466-20472. 31. Cartier, E. A., Conti, L. R., Vandenberg, C. A., Shyng, S. L. 2001. Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc. Natl. Acad. Sci. USA. 98 (5): 2882-2887. 32. Conti, L. R., Radeke, C. M., Shyng, S. L., Vandenberg, C. A., 2001. Transmembrane topology of the sulfonylurea receptor SUR1. J. Biol. Chem. 276 (44), 41270-41278. 33. Cook, D. L., Hales, C. N. 1984 Intracellular ATP directly blocks K+ channels in pancreatic β-cells. Nature 311: 271-273. 34. Cook, D. L., Stain, L. S., Ashford, M. L. J., Hales, C. N. 1988. ATP-sensitive K+ channels in pancreatic β-cells. Diabetes. 37: 495-98. 35. Craver, RD., Hill, CB. 1997. Cure of hypoglycemic hyperinsulinism by enucleation of a focal islet cell adenomatous hyperplasia. J. Pediat Surg. 32: 1526-1527. 36. D’ Hahan, N., Jacquet, H., Moreau, C., Catty, P., Vivaudou, M., 1999a. A transmembrane domain of the sulfonylurea receptor mediates activation of ATP-sensitive K+ channels by K+ channel openers. Mol Pharmacol. 56(2): 308315. 37. D’ Hahan, N., Moreau, C., Prost, A. L. Jacquet, H., Alekseev, A. E., Catty, P., Vivaudou, M., 1999b. Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. Prco. Natl. Acad. Sci. USA. 96 (21): 12162-12167. 38. De Lonlay, P., Fournet, JC., Rahier, J., et al. 1997. Somatic deletion in the imprinted 11p15 region in sporadic persistent hyperinsulinemic hypoglycemia of infancy is specific of focal adenomatous hyperplasia and endorses partial pancreatectomy. J Clin Invest. 100. 802-807. 39. De Lonlay-Debeney, P., Poggi-Travert, F., Fournet, JC., et al. 1999. Clinical aspects and course of neonatal hyperinsulinism. N. Engl. J. Med. 340. 11691175. 40. De Lonlay, P., Touati, G., Robert, J and Saudubray, J. 2002. Persistent hiperinsulinaemic hypoglycaemia. Semin Neonatal. 7: 95-100. 41. Dickinson, K. E., Bryson, C. C., Cohen, R. B., Rogers, L., Green, D. W. Atwal, K. S. 1997. Mol. Pharmacol. 52: 473-481. 42. Dorschner, H., Brekardin, E., Uhde, I., Schwanstecher, C., Schwanstecher, M. 1999. Mol. Pharmacol. 55: 1060-1066. 43. Drain, P., Li, L., Wang, J. 1998. KATP channel inhibition by ATP requieres distinct functional functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. USA. 95: 13953 – 13958. 44. Dunne, M. J and Petersen, O. H. 1986. Intracellular ADP activates K+ channels that are inhibited by ATP in an insuline-secreting cell line. FEBS Lett. 208: 5962. 87 45. Dunne, M., Yule, D. I., Gallacher, D. V. & Petersen, O. H. 1990. Stimulantevoked depolarization and increase in [Ca2+]i in insulin-secreting cells is dependent on external Na+. J. Membr. Biol., 113: 131-138. 46. Dunne, M. J and Petersen, O. H. 1991. Potassium selective ion channels in insuline-secreting cells; physiology, pharmacology and their rolein stimulus secretion coupling. Biochimica Biophysica Acta (Rev Biomembranes). 1071: 67-82. 47. Dunne, M., Cosgrove, K., Shepherd, R and Ämmälä, C. 1999. Potassium channels, sulphonylurea receptor and control of insulin release. TEM. Vol. 10, No 4. 146-152. 48. Fan, Z., Makielski, J. C. 1997. Anionic phospholipids activate ATP-sensitive potassium channels. J Biol Chem. 272: 5388-5395. 49. Ferry, R. J., Kelly, A., Grimberg, A., Koo-Mccoy, S., Shapiro, M. J., Fellows, K. E., Glaser, B., Aguilar-Bryan, L., Stanfford, D. E., Stanley, C. A. 2000. Calcium-stimulated insulin secretion in diffuse and focal forms of congenital hyperinsulinism. J. Pediatr. 137: 239-246. 50. Findlay, I., Dunne, M. J and Petersen, O. H. 1985. ATP-sensitive inward rectifier and voltage and calcium activated K+ channels in cultured pancreatic islet cells. J. Membr. Biol. 88: 165-72. 51. Findlay,I. 1987. The effects of magnesium upon adenosine triphosphatesensitive potassium channels in a rat insulin-secreting cell line. J. Physiol. 391:611-629. 52. Findlay, I. 1988a. Effects of ADP upon the ATP-sensitive K+ channel in rat ventricular myocytes. J. Membr. Biol. 101:83-92. 53. Findlay, I. 1988b. Calcium-dependent inactivation of the ATP-sensitive K+ channel of rat ventricular myocytes. Biochim Biophys Acta. 943:297-304. 54. Furukawa, T., Virág, L., Furukawa, N., Sawanobori, T., Hiraoka, M. 1994. Mechanism for reactivation of the ATP-sensitive K+ channel by MgATP complexes in guinea-pig ventricular myocytes. J Physiol (Lond) 479:95-107. 55. Furukawa, T., Yamane, T., Terai,T., Katayama, Y., Hiraoka, M. 1996. Functional linkage of the cardiac ATP-sensitive K+ channel to the actin cytoeskeleton. Pflugers Arch. 431: 504-512. 56. Galli, A., DeFelice, LJ. 1994. Inactivation of L-type Ca channels in embryonic chick ventricle cell: Dependence on the cytoskeletal agents colchicine and taxol. Biophys J. 67:2296-2304. 57. Geisen, K., Hitzel, V., Okomonopoulos, R., Punter, J., Weyer, R & Summ, H. D. 1985. Inhibition of 3H-glibenclamide binding to sulfonylurea receptors by oral antidiabetics. Arzneim. Forsch., 35, 707-712. 58. Gillis, .K. D., Gee, W. M., Hammoud, A., McDaniel, M. L., Falke, L.C., and Misler, S. 1989. Effects of sulfonamides on a metabolite-regulated ATPisensitive K+ channel in rat pancreatic B-cells. Am. J. Physiol. 257, C1119C1127. 59. Glaser, B., Phillip, M., Carmin, R., Lieberman, E and Landau, H. 1990. Persistent hyperinsulinemic hypoglycemia of infancy (“nesidoblastosis”): autosomal recessive inheritance. Arch. Dis. Child. 66: 529-530. 60. Glaser, B., Kesavan, P., Heyman, M., Davis, E., Cuesta, A., Buchs, A., Stanley, C. A., Thornton, P. S., Permutte, M. A. Matschinsky, F. M., & Herold, 88 K. C. 1998. Familial hyperinsulinism caused by activating glucokinase mutation. New England Journal of Medicine. 338 (19): 226-230. 61. Glaser, B., Landau, H and Permutt, A. 1999. Neonatal Hiperinsulinism. Trends Endocrinol Metab. 10 (2): 55-61. 62. Glasser, B. et al. 1994. Familial hyperinsulinism maps to chromosome 11p1415.1, 30 cM centromeric to the insulin gene. Nat. Genet. 7: 185-187. 63. Goossens, A., Grepts, W., Saudubray, J. M., Bonnefont, J. P., Nihoul Fétéké, C., Heitz, P. U., Kloppel, G. 1989. Diffuse and focal nesidioblastosis. A clinicopathological study of 24 patients with persistent neonatal hyperinsulinemic hypoglycemia. Am J Surg Pathol 13: 766-775. 64. Gribble, F. M., Tucker S. J. and Ashcroft, F. M. 1997. The essential role of the Walker A motifs of SUR1 in KATP channel activation by Mg-ADP and diazoxide. EMBO J. 16 (6), 1145-1152. 65. Gribble, F. M., Tucker S. J., Haug, T. and Ashcroft, F. M. 1998. MgATP activates the β cell KATP channel by interaction with its SUR1 subunit. Proc. Natl. Acad. Sci. USA. 95: 7185-7190. 66. Gribble, F. M., Tucker S. J., Seino, S. and Ashcroft, F. M. 1998b. Tissue specificity of sulfonylureas: studies on cloned cardiac and beta-cell KATP channels. Diabetes. 47 (9): 1412-1418 67. Grimberg, A., Ferrey, Jr., R. J., Kelly, A., Koo-McCoy, S., Polonsky, K., Glaser, B., Permutt, M. A., Aguilar-Bryan, L., Stafford, D., Thorton, P., Baker, L., & Stanley, C. A. 2001. Dysregulation of insulin secretion in children with congenital hyperinsulinism due to sulfonylurea receptor mutations. Diabetes. 50 (2), 322-328. 68. Hambrock, A. et al. 1998. Mg2+ and ATP dependence of KATP channel modulator binding to the recombinant sulfonylurea receptor SUR2B. Br. J. Pharmacol. 125. 577-583. 69. Hamill, O. P., Marty, A., Neher, E., Sakmann, B., and Sigworth, F. J. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pfluger Arch. 391, 85-100. 70. Hilgemann, D. W., Ball, R. 1996. Regulation of cardiac Na +, Ca2+ exchange and K ATP potassium channels. Science. 272: 956-959. 71. Hunter, M., Giebisch, G. 1988. Calcium activated K-channels of Amphiuma early distal tubule: inhibition by ATP. Pflügers Arch. 412: 331-33. 72. Huopio, H., Reimann, F., Ashfield, R., Komulainen, J., Lenko, H. L., Rahier, J., Vauhkonen, I., Kere, J., Laakso, M., Ashcroft, F., Otonkoski, T. 2000. Dominantly inherited hyperinsulinism caused by a mutation in the sulfonylurea receptor type 1. J. Clin. Invest. 106: 897-906. 73. Huopio, H., Jaaskelainen, J., Komulainen, J., Miettinen, R., Karkkainen, P., Laakson, M., Tapanainen, P., Voutilainen, R., & Otonkoski, T. 2002. Acute insulin response test for the differential diagnosis of congenital hyperinsulinism. Journal of Clinical Endocrinology and metabolism. 87 (10). 4502-4507. 74. Inagaki, N., Gonoi, T., Clement, J. P (4th)., Namba, N., Inazawa, J., Gonzalez, G., Aguilar-Bryan, L., Seino, S., Bryan, J. 1995. Reconstitution of KATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 270: 11661170. 89 75. Inagaki, N., Gonoi, T., Clement, J. P., Wang, C. Z., Aguilar-Bryan, L., Bryan, J., Seino, S. 1996. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 16 (5): 1011-1017. 76. Inoue, I., Nagase, H., Kishi, K., Higuti, T. 1991. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature. 352: 244-47. 77. Isomoto, S., Kondo, C., Yamada, M., Matsumoto, S., Higashiguchi, O., Horio, Y., Matsuzawa, Y., Kurachi, Y. 1996. A novel sulfonylurea receptor forms with BIR (Kir6.2) a smooth muscle type ATP-sensitive K+ channel. J. Biol Chem. 271(40): 24321-24324. 78. John, S. A. et al. 1998. The sulfonylurea receptor SUR1 regulates ATPsensitive mouse Kir6.2 K+ channels linked to the green fluorescent protein in human embryonic kidney cells (HEK293). J. Physiol. 510. 333-345. 79. Johnson, BD., Byerly, L. 1994. Ca2+-dependent inactivation in a mammalian central neuron involves the cytoskeleton. Pflüger Arch. 429:14-21. 80. Jones, P. A., Tucker, S. J and Ashcroft, F. M. 2001. Multiple sites of interaction between the intracellular domains of an inwardly rectifying potassium channels, Kir6.2. FEBS Lett. 508, 85-89. 81. Kakei, M., Kelly, R. P., Ashcroft, S. J. H. & Ashcroft, F. M. 1986. The ATPsensitivity of K+ channels in rat pancreatic B-cells in modulated by ADP. FEBS Lett. 208, 63-66. 82. Kane, C., Shepherd, R. M., Squires, P. E., Johnson, P. R., James, R. F., Milla, P. J., Aynsley-Green, A., Lindley, K. J., Dunne, M. J. 1996. Loss of functional KATP channels in pancreatic beta-cell causes persistent hyperinsulinemic hypoglycemia of infancy. Nat. Med. 2 (12): 1344-1347. 83. Kelly, A., Ng D, Ferry, RJ, Jr., Grimberg, A., Koo-McCoy, S., Thornton, P. S., Stanley, C. A. 2001. Acute insulin responses to leucine in children with the hyperinsulinism/hyperammonemia syndrome. J Clin Endocrinol Metab. 86: 3724-3728. 84. Ko, Y. H., and Pedersen, P. L. 1995. The first nucleotide binding fold of the cystic fibrosis transmembrane conductance regulator can function as an active ATPasa. J. Biol. Chem. 270: 22093-22096. 85. Korn, ED., Carlier, MF., Pantaloni, D. 1987. Actin polymerization and ATP hydrolysis. Science. 238 (4827): 638-44. 86. Koster, J. C., Sha, Q., Shyng, S. And Nichols, C. G. 1999. ATP inhibition of KATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. J. Physiol. 515: 19-30. 87. Kozlowski, RZ., Ashford, ML. 1990. ATP-sensitive K+ channel rund-down is Mg2+ dependent. Proc R Soc Lond [Biol] 240: 397-410. 88. Kukuvitis, A., Deal, C., Arbour, L and Polychronakos, C. 1997. An autosomal dominant form of familial persistent hyperinsulinemic hypoglycemia of infancy, not linked to the sulfonylurea receptor locus. J. Clin. Endocrinol. Metab. 82: 1192-1194. 89. Larsson, O., Ammala, C., Bokvist, K., Fredholm, B and Rorsman O. 1993. Stimulation of the KATP channel by ADP and diazoxide requires nucleotide hydrolysis in mouse pancreatic beta-cells. J. Physiol. (Camb). 419:193-211. 90 90. Lederer, W. J and Nichols, C. G. 1989. Nucleotide modulation of the activity of rat heart KATP channels in isolated membrane patches. J. Physiol. (Camb). 419: 193-211. 91. Lee S. W. 2001. The role of tissue-specific imprinting as a source of phenotypic heterogeneity in human disease. Biol Psychiatry. 50. 927-931. 92. Lehninger, A. 1982. Bioquimica. Editorial Omega, S. A. 427-450 93. Light P. 1996. Regulation of ATP-sensitive potassium channels by phosphorylation. Biochim Biophys Acta 1286: 65-73. 94. Lindley, K. J., Dunne, M. J., Kane, C., Shepherd, R. M., Squires, P. E., James, R. F. L., Johnson, P. R. V., Eckhardt, S., Wakeling, E., Milla, P. J., & AynsleyGreen, A. 1996. Ionic control of the β-cell function in nesidioblastosis, a possible role for calcium channel blockade. Archives of diseases in Childhood. 74. 373-378. 95. Loubatieres, A. 1957. The hypoglycemic sulfonamides: history and development of the problem from 1942 to 1955. Ann. NY. Acad. Sci. 71: 4-11. 96. Manley, P. W., Quast, U., Andres, H., Bray, K. 1993. Synthesis of and radioligand binding studies with a tritiated pinacidil analogue: receptor interactions of structurally different classes of potassium channel openers and blockers. J. Med. Chem. 36: 2004-2010. 97. Markworth, E., Schwanstecher, C., Schwanstecher, M. 2000. ATP4-mediates closure of pancreatic beta-cell ATP-sensitive potassium channels by interaction with 1 of 4 identical sites. Diabetes 49 (9): 1413-1418. 98. Mathew, P. M., Young, Y.M., Abu-Osba, Y. K., Mulherm, B. D., Hammoudi, S., Hamdan, J. A., Sadi, A. R. 1988. Persistent neonatal hyperinsulinism. Clin Pediatr. 27: 148-151. 99. Matsou, M., Trapp, S., Tanizawa, Y., Kioka, N., Amachi, T., Oka, F., Ashcroft, F. M., Ueda K. 2000. Functional analysis of a mutant sulfonylurea receptor, SUR1-R1420C, that is responsible for persistent hyperinsulinemic hypoglycemia of infancy. The Journal of Biological Chemistry. 275(52): 4118441191. 100. Meissner, T., Beinbrech, B and Mayatepek, E. 1999. Congenital Hyperinsulinism: Molecular Basis of heterogeneous disease. Human Mutation 13: 351-361. 101. Mikhailov, M. et al. 1998. Expression of functionally active ATPsensitive K-channels in insect cells using baculovirus. FEBS Lett. 429. 390394. 102. Misler, S., Falke, L. C., Gillis, K., McDaniel, M. L. 1986. A metabolite regulated potassium channel in rat pancreatic B cell. Proc Natl Acad Sci USA. 83: 7119-7123. 103. Misler, S., Giebisch, G. 1992. ATP-sensitive potassium channels in physiology, pathophysiology, and pharmacology. Curr. Opin. Nephrol. Hypertens. 1: 21-33. 104. Murphy, E. 1993. Measurement of intracellular ionized magnesium. Miner Electrolyte Metab. 19: 250-258. 105. Nakanishi, S., Yano, H., Matsuda, Y. 1995. Novel functions of phosphatidylinositol 3-kinase in terminally differentiated cells. Cellular Signalling. 7 (6). 545-557. 91 106. Nestorowicz, A., Wilson, B. A., Schoor, K. P. 1996. Mutations in the sulfonylurea receptor gene are associated with familial persistent hyperinsulinism in Ashkenazi Jews. Hum Mol Genet. 5. 1813-1822. 107. Nestorowicz, A., Inagaki, N., Gonoi, T., Schoor, K. P., Wilson, B. A., Glaser, B., Landau, H., Stanley, C. A., Thornton, P. S., Seino, S., Permutt, M. A. 1997. A nonsense mutation in the inward rectifier potassium channel gene, Kir6.2, is associated with familial hyperinsulinism. Diabetes. 46 (11), 17431748. 108. Nichols, C. G., Lopatin, A. N. 1993. Trypsin and α-chymotrypsin treatment abolishes glibenclamide sensitivity of KATP channels in rat ventricular myocytes. Pflügers Arch. 422:617-619. 109. Nichols, C. G., Shyng, S-L., Nestorowicz, A., Glaser, B., Clement IV, J., Gonzalez, G., Aguilar-Bryan, L., Permutt, A. M., Bryan, J. 1996. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 272: 1785-1787. 110. Nishida, M and Mackinnon, R. 2002. Crystal structure and mechanism of a calcium-gated potassium channels. Cell. 111: 957-965. 111. Noma, A. 1983 ATP-regulated K+ channels in cardiac muscle. Nature 305: 147-148. 112. Okuyama, Y. et al. 1998. The effects of nucleotides and potassium channel openers on the SUR2A/Kir6.2 complex K+ channel expressed in a mammalian cell line, HEK293T cells. Pflügers Arch. 435. 595-603. 113. Olesen, S. P., Bundgaard, M. 1993. ATP-dependent closure and reactivation of inward rectifier K+ channels in endothelial cells. Circ Res. 73:492-495. 114. Panten, U., Schwanstecher, M., Schwanstecher, C. 1992. Pancreatic and extrapancreatic sulfonylurea receptors. Horm. Metab. Res. 24: 549-554. 115. Partridge, C. J., Beech, D. J., Sivaprasadarao, A. 2001. Identification and pharmacological correction of a membrane trafficking defect associated with a mutation in the sulfonylurea receptor causing familial hyperinsulinism. J. Biol. Chem. 276 (38): 35947-35952. 116. Proks, P., and Ashcroft, FM. 1993. Modification of K-ATP channels in pancreatic β-cells by trypsin. Pflügers Arch. 424: 63-72. 117. Proks, P., Gribble, F. M., Adhikari, R., Tucker, S. J and Ashcroft, F. M. 1999. Involvement of the N-terminus of Kir6.2 in the inhibition of the KATP channel by ATP. J. Physiol. 514: 19-25. 118. Quast, U., Guillon, J. M., Cavero, I. 1994. Cellular pharmacology of potassium channel openers in vascular smooth muscle. Cardiovasc. Res. 28: 805-810. 119. Quayle, J. M., Standen, N. B., Stanfield, P. R. 1988. The voltagedependent block of ATP-sensitive potassium channels of frog skeletal muscle by caesium and barium ions. J Physiol (Lond). 405: 677-697. 120. Rahier, J. 1989. Relevance of endocrine pancreas nesidioblastosis to hyperinsulinism plus hyperammonemia. J Pediatr 133: 800-801. 121. Rahier, J., Guiot, Y & Sempoux, C. 2000. Persistent hyperinsulinaemic hypoglycaemia of infancy: A heterogeneous syndrome unregulated to 92 nesidioblastosis. Archives of Diseases in Children Fetal and Neonatal Education. 82(2). F108-F112. 122. Reimann, F., Ryder, T. J., Tucker, S. J and Ashcroft, F. M. 1999. The role of lysine 185 in the Kir6.2 subunit of the channel by ATP. J Physiol. 520: 661-669. 123. Reimann, F., Huopio, H., Dabrowski, M., Proks, P., Gribble, F. M., Laakso, M., Otonkoski, T., Ashcroft, F. M. 2003. Characterisation of new KATPchannel mutations associated with congenital hyperinsulinism in the Finnish population. Diabetologia. 46:241-249. 124. Rorsman, P., Trube, G. 1985. Glucose dependent K+ channels in pancreatic β-cells are regulated by intracellular ATP. Pflügers Arch. 405: 3059. 125. Rosen, O. M. 1987. After insulin binds. Science. 237: 1452-1458 126. Ryan, F. et al. 1998. Hyperinsulinism: molecular etiology of focal disease. Arch. Dis Child. 79, 445-447. 127. Sakura, H., Ammala, C., Smith, P. A., Gribbe, F. M., Ashcroft, F. M. 1995. Cloning and functional expression of the cDNA enconding a novel ATPsensitive enconding a novel ATP-sensitive potassium channel subunit expressed in pancreatic beta-cells, brain, heart and skeletal muscle. FEBS Lett. 377 (3): 338-344. 128. Schwanstecher, M., Sieverding, C., Dorschner, H., Gross, I., AguilarBryan, L. Schwanstecher, C., Bryan, J. 1998. Potassium channel openers require ATP to bind to and act through sulfonylurea receptors. EMBO J. 17: 5529-5535. 129. Seino, S. 1999. ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annu. Rev. Physiol. 61: 337-362. 130. Sempoux, C., Poggi, F., Brunelle, F., Saudubray, JM., Fekete, C., Rahier, J. 1995. Nesdidioblastosis and persistent neonata hyperinsulinism. Diabete Metab. 21: 402-407. 131. Shepherd, R. M., Cosgrove, K. E., O´Brien, R. E., Barnes, P. D., Ammala, C., & Dunne, M. J. 2000. Hyperinsulinism of infancy: Towards an understanding of unregulated insulin release. European Network for Research into Hyerinsulinism in infancy. Archives of Diseases in Children Fetal and Neonatal Education. 82. F87-F97. 132. Shindo, T. et al. 1998. SUR2 subtype (A and B)-dependent differential activation of the cloned ATP-sensitive K+ channels by pinacidil and nicorandil. Br. J. Pharmacol. 124. 985-991. 133. Shilyanski, J., Fisher, S., Cutz, E., et al. 1997. Is 95 % pancreatectomy the procedure of choice for treatment of persistent hyperinsulinemic hypoglycemia of the neonatal? J. Pediatr. Surg. 32: 342-346. 134. Shyng, S. L., Ferrigni,T and Nichols, C. G. 1997a. Control of rectificatuion and gating of KATP channels by the Kir6.2 subunit. J. Gen. Physiol. 110:141-153. 135. Shyng, S. L., Ferrigni,T and Nichols, C. G. 1997b. Regulation of KATP channels activity by diazoxide and MgADP. Disctinct functions of the two 93 nucleotide binding folds of sulfonylurea receptor. J. Gen. Physiol. 110 (6): 643654. 136. Shyng, S., Ferrigni, T., Shepard, J., Nestorowicz, A., Glaser, B., Permutt, A and Nichols, C. 1998. Funtional analyses of novel mutations in the sulphonylurea receptor 1 associated with persistent hiperinsulinemic hipoglycemia of infancy. Diabetes. 47: 1145-1151. 137. Srivastava, A. 1998. Use of pharmacological agents in elucidating the mechanism of insulin action. TiPS. 19: 205-209. 138. Stanley, C. A. 1997. Hyperinsulinism in infants and children. Pediatr Clin North Am. 44:363-374. 139. Stanley, C. A., Lieu, Y. K., Hsu, B. Y., Burlina, A. B., Greenberg, C. R., Hopwood, N. J., Perlman, K., Rich, B. H., Zammarchi, E., & Poncz, M. 1998. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate deshydrogenase gene. New England Journal of Medicine. 338 (19): 1352-1357. 140. Sturgess, N. C., Ashford, M. L., Cook, D. L and Hales, C. N. 1985. The sulphonylurea receptor may be an ATP-sensitive potassium channel. Lancet. ii: 474-475. 141. Szewczyk, A and Pikula, S. 1998. Adenosine 5’-triphosphate: an intracellular metabolic messenger. Biochimica et Biophysica Acta. 1365: 333353. 142. Tanabe, K., Tucker, S. J., Matsuo, M., Proks, P., Ashcroft, F. M., Seino, S., Amachi, T and Ueda, K. 1999. Direct photoaffinity labeling of the kir6.2 subunit of the ATP-sensitive K+ channel by 8-azido-ATP. J. Biol. Chem. 274: 3931-3933. 143. Tanabe, K., Tucker, S. L., Ashcroft, F. M., Proks, P., Kioka, N., Amachi, T and Ueda, K. 2000. Direct photoaffinity labeling of Kir6.2 by [γ32 P] ATP-[γ]4azidoanilide. Biochem. Biophys. Res Commun. 272: 316-319. 144. Tanizawa, Y., Matsutani, A., Chiu, KC., Permutt, MA. 1992. Human glucokinase gene: isolation, structural characterization, and identification of a microsatellite repeat polymorphism. Mol Endocrinol. 6:1070-1081. 145. Taschenberger, G., Mougey, A., Shen, S., Lester, L., LaFranchi, S and Shyng, S. 2002. Identification of a familial hiperinsulinism-causing mutation in the sulfonylurea receptor 1 that prevent normal trafficking and function of KATP channels. The Journal of biological chemistry. 277 (19): 17139-17146. 146. Terzic, A., Jahangir, A., Kurachi, Y. 1995. Cardiac ATP-sensitive K+ channels: regulation by intracellular nucleotides and K+ channels-opening drugs. Am. J. Physiol.269: C525-45. 147. Thomas, P. M., Cote, G. J., Hallman, D. M and Mathew, P M. 1995a. Homozygosity mapping, to chromosome 11p, of the gene for familial persistent hyperinsulinemic hypoglycemia of infancy. Am. J. Hum. Genet. 56: 416-421. 148. Thomas, P. M., Cote, G. J., Wohllk, N., Haddad, B., Mathew, P. M., Rabl, W., Aguilar-Bryan, L., Gagel, R. F., Bryan, J. 1995b. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science. 21(268 (Part 5209)), 426-429. 94 149. Thomas, P., Ye, Y., Ligthner, E. 1996. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum Mol Genet. 5. 1809-1812. 150. Thornton, P. S., Sumner, A. E., Ruchelli, E. D., Spielman, R. S., Baker, L and Stanley, C. A. 1991. Familial and sporadic hyperinsulinism: histopathologic findings and segregation analysis support a single autosomal recessive disorder. J. Pediatr. 119: 721-724. 151. Thornton, P. S., Alter, C. A., Levitt Katz, L. E., et al. 1993. Short- and long-term use of octreotide in the treatment of congenital hyperinsulinism. J. Pediatr. 123. 637-643. 152. Thornton, P. S., et al. 1998. Familial hyperinsulinism with apparent autosomal dominant inheritance: clinical and genetic differences from the autosomal recessive variant. J. Pediatr. 132: 9-14 153. Thornton, P. S., MacMullen, C., Ganguly, A., Ruchelli, E., Steinkrauss, L., Crane, A., Aguilar-Bryan, L., Stanley, C. A. 2003. Clinical and molecular characterization of a dominant form of congenital hyperinsulinism caused by a mutation in the high-affinity sulfonylurea receptor. Diabetes. Sep;52(9):240310. 154. Trapp, S., Tucker, S. J. & Ashcroft, F. M. 1997. Activation and inhibition of K-ATP currents by guanine nucleotides is mediated by different channel subunits. Proc. Natl. Acad. Sci. USA. 94: 8872-8877. 155. Trapp, S., Proks, P., Tucker, S. J and Ashcroft, F. M. 1998ª. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J. Gen. Physiol. 112: 333-349. 156. Trapp, S., Haider, S., Jones, P., Sansom, M.S.P and Ashcroft, F. M. 2003. Indentification of residues contributing to the ATP binding site of Kir6.2. EMBO Journal. 22 (12): 2903-2912. 157. Trube, G., Hescherler, J. 1984. Inward-rectifying channels in isolated patches of the heart cell membrane: ATP-dependence and comparison with cell-attached patches. Pflugers Arch 401: 178-184. 158. Tucker, S. J., Gribble, F. M., Zhao, C., Trapp, S., Ashcroft, F. M. 1997. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 378 (6629), 179-183. 159. Tucker, S. J., Gribble, F. M., Proks, P., Trapp, S., Ryder, T. J., Haug, T., Reimann, F. And Ashcroft, F. M. 1998. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 17: 3290-3296. 160. Tusnády, G. E., Bakos, E., Varadi, A., Sarkadi, B. 1997. Membrane topology distinguishes a subfamily of the ATP-binding cassette (ABC) transports. FEBS Lett. 402: 1-3. 161. Walker, JE., Saraste, M., Runswick, MJ., Gay, NJ. 1982. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO.J. 1. 945-951. 162. Wang, W-H., Cassola, A., Giebisch, G. 1994. Involvement of actin cytoskeleton in modulation of apical K channel activity in rat collecting duct. Am J Physiol. 267: F592-598. 95 163. Yakovac, W. C., Baker, L., Hummeler, K. 1971. Beta cell nesidioblastosis in idiopathic hipoglycemia of infancy. J. Pediatr. 79: 226-231. 164. Yokoshiki, H., Sunagawa, M., Seki, T., Sperelakis, N. 1998. ATPsensitive K+ channels in pancreatic, cardiac, and vascular smooth muscle cells. Am. J. Physiol. 274 (1 Part 1), C25-37. 165. Yoshida, M and Amano, T. 1995. A common topology of proteins catalyzing ATP-triggered reactions. FEBS Lett. 359. 1-5. 166. Ueda, K., Inagaki, N., Seino, S. 1997. MgADP antagonism to Mg2+independent ATP binding of the sulfonylurea receptor SUR1. J. Biol. Chem. 272 (37): 22983-22986. 167. Ueda, K., Komine, J., Matsuo, M., Seino, S., Amachi, T. 1999a. Cooperative binding of ATP and MgADP in the sulfonylurea receptor is modulated by glibenclamide. Proc. Natl. Acad. Sci. USA 96 (4), 1268-1272. 168. Ueda, K., Matsou, M., Seino, S., Amachi, T. 1999b. Comparative aspects of the function and mechanism of SUR1 y MDR1 proteins. Biochim. Biophys. Acta. 1461 (2), 305-313. 169. Uhde, I., Toman, A., Gross, I., Schwanstecher, C., Schwanstecher, M. 1999. Identification of the potassium channel opener site on sulfonylurea receptors. J. Biol. Chem. 274 (40): 28079-28082. 170. Vanoye, C. G., MacGregor, G. G., Dong, K., Tang, L., Buschmann, A. S., Hall, A. E., Lu, M., Giebisch, G. And Hebert, S. C. 2002. The carboxyl termini of KATP channels bind nucleotides. J. Biol. Chem. 277: 23260-23270. 171. Verkarre, V. et al. 1998a. Paternal mutation of the sulfonylurea receptor (SUR1) gene and maternal loss of 11p15 imprinted genes lead to persistent hyperinsulinismin focal adenomatous hyperplasia. J. Clin. Invest. 102. 12861291. 172. Verkarre, V., Fournet, J. C., Rahier, J., et al. 1998b. Maternal allele loss with somatic reduction to homozygosity of the paternally-inherited mutation of the SUR1 gene leads to congenital hyperinsulinism in focal islet cell adenomatous hyperplasia of the pancreas. J. Clin. Invest. 102: 1286-1291. 173. Virág, L., Furukawa, T., Hiraoka, M. 1993. Modulation of the effect of gliblenclamide on KATP channels by ATP and ADP. Mol Cell Biochem.119: 209215. 174. Yokoshiki, H., Sunagawa, M., Seki, T., and Sperelakis, N. 1999. Antisense oligodeoxynucleotides of sulfonylurea receptors inhibit ATPsensitive K+ channels in cultured neonatal rat ventricular cells. Pflugers Arch. 437, 400-408 175. Yoshida,M. and Amano, T. 1995. A common topology of proteins catalyzing ATP-triggered reactions. FEBS Lett. 359. 1-5. 176. Xie, LH., Tanako, M., Kakei, M., Okamura, M., Noma, A. 1999. Wortmannin, an inhibitor of phosphatidylinositol kinases, blocks the MgATPdependent recovery of ir6.2/SUR2A channels. Journal of Physiology. 514.3, 655-665. 177. Zunkler, B.J., Lins, S., Ohno-Shosaku, T., Trube, G and Panten, U. 1988. Cytosolic ADP enhances the sensitivity to tolbutamide of ATPdependent K+ channels from pancreatic B-cells. FEBS Lett. 239, 241-244. 96